Дефицит альфа-1-антитрипсина – серьезная наследственная болезнь. Недуг возникает из-за того, что в организме недостает белкового фермента, который предотвращает разрушающее действие на ткань легких протеаз. Со временем клинические симптомы становятся все многообразнее, а состояние здоровья понемногу осложняется, делается хуже.
Описание
Основная функция а1-антитрипсина заключается в том, чтобы защищать легочные ткани от повреждений ферментами, которые нужны организму. Без них невозможно его полноценное развитие и работа. К примеру, протеазы помогают выводить микробы из легких, тем самым участвуют в борьбе с инфекциями. Альфа-1-антитрипсин регулирует активность ферментов протеазы. Нехватка антипротеазы нередко протекает с поражением легких у взрослых людей и печени у детей. У 20% новорожденных с таким заболеванием возникает холестатическая желтуха. Примерно у 2% больных из-за дефицита развивается обструктивная хроническая болезнь легких.
При редком генетическом заболевании формируются два патологических процесса: печеночный и легочный. В печени, где наблюдается избыток альфа1-антитрипсина, формируется рубцовая ткань.
У больных во взрослом возрасте развивается хроническая печеночная недостаточность и цирроз печени.
При легочном аномальном процессе происходит разрушение межальвеолярных перегородок, которые образуют легочную ткань. Это негативно сказывается на газообмене. По мере развития патологии после 30-летнего возраста нередко развивается эмфизема легких.
Симптомы болезни
От степени нехватки альфа1-антитрипсина зависит выраженность и сочетание признаков. Уже у новорожденных при существенном дефиците можно увидеть проявления холестатического синдрома. У младенца становятся желтушными склеры и покровы кожи. В отдельных случаях у них появляется не воспалительная сыпь и рвота. Обычно застой печени заканчивается к 3-4 месяцам, порой процесс прогрессирует, формируется печеночная недостаточность.
Больной во взрослом возрасте устает, его беспокоит слабость. Поверхность кожи становится желтого цвета и чешется. Со временем начинают развиваться осложнения присущие циррозу печени.
У человека возникают проблемы с дыханием, оно становится свистящим, случаются приступы удушья, мучает одышка. В брюшной полости скапливается жидкость, из-за нее увеличивается живот. У больного возникает варикозное расширение вен кишечника и пищевода.
В позднем возрасте могут появиться симптомы поражения гепатобилиарной системы, отвечающей за экскрецию и пищеварение, за выведение продуктов метаболизма из организма.
Симптомы дефицита альфа1-антитрипсина:
- боли в правом подреберье;
- снижение аппетита;
- вздутие.
Человек жалуется на тошноту и рвоту. В отдельных случаях может случиться обильное кровотечение и летальный исход.
Классификация
У детей и взрослых заболевание проявляется немного по-разному. От характера генетического недостатка зависит клиника, но нарушения системы дыхания и повреждения печени наблюдаются у всех. Дефицит альфа1-антитрипсина делится на: с преимущественным поражением легких и гепатобилиарным поражением.
Первую аномалию выявляют у взрослых людей и детей с диагнозом – бронхиальная астма. Второй вид в основном у детей, причем симптомы можно увидеть с самого рождения и до 4-х месяцев. Еще одна разновидность – с сочетанным поражением, появляется в случае, когда наблюдается серьезная ферментативная недостаточность. Специфические симптомы появляются рано в детстве.
Диагностика
Больные с таким заболеванием должны обследоваться у гепатолога и пульмонолога. Первым делом врач занимается сбором анамнеза. Некоторые пациенты должны проконсультироваться у генетика, к ним относятся те, у которых выявлен:
- идиопатический цирроз печени;
- бронхоэктаз неясной этиологии;
- некротизирующий панникулит;
- эмфизема;
- устойчивая к терапевтическому лечению бронхиальная астма;
- обструктивная хроническая болезнь легких.
Есть несколько видов диагностических мероприятий:
- Инструментальная.
Рентгенография грудной клетки показывает степень поражения легких. На основании результатов процедуры врач обращает внимание на сосредоточение повышенной прозрачности ткани легких.
- Лабораторная.
При помощи анализа крови удается определить уровень а1-антитрипсина в крови.
Во время проведение осмотра обращают внимание на то, в каком состоянии находятся гепатобилиарная и дыхательная системы. Врач осматривает кожные покровы.
Лечение заболевания
Редкие наследственные заболевания вылечить невозможно. Лечение дефицита альфа1-антитрипсина включает в себя применение различных способов воздействия, которые помогают уменьшить разрушительные факторы. Для устранения симптомов врачом назначаются лекарства, которые помогают улучшить работу печени – гепатопротекторы.
Рекомендуется выполнение лечебной дыхательной гимнастики, в которую входят такие физические упражнения, как приседания и наклоны. При их выполнении больной должен совершать установленный дыхательный ритм, например, при наклонах вперед обязательно громко и сильно выдыхать.
Если состояние ухудшилось назначается кислородотерапия. В тяжелых случаях человеку при помощи аппарата искусственной вентиляции легких или кислородной маски подается кислород.
В отдельных странах лечат заместительной терапией – белок, выделенный из крови донора, вводят внутривенно. Этот вид лечения существенно нарушает состояние печени. В органе количество а1-антитрипсина накапливается еще больше.
Если при недуге развивается эмфизема легких, цирроз печени (когда погибают ее клетки) и альвеолы (полностью нарушающие работу органа), продлить жизнь поможет операция по пересадке этих органов.
Госпитализация
Показаниями для проведения плановой госпитализации является растущая эмфизема и усиливающее поражение легких и печени. Больного следует экстренно госпитализировать при развитии тяжелой дыхательной и печеночной недостаточности, обострении инфекции дыхательных путей.
Осложнения
Если у пациента наблюдается существенное поражение системы дыхания, то дефицит а1-антитрипсина осложняется тем, что формируется легочное сердце. От генотипа зависят сроки его возникновения. Легочно-сердечную недостаточность можно наблюдать у детей, подростков и молодых людей при большой нехватке фермента.
На людей с не сильно выраженным дефицитом, которые не курят, не подвергаются систематическому воздействию различных химических веществ, присутствующих в атмосфере, заболевание на продолжительность жизни не влияет.
В отдельных случаях из-за этого генетического дефекта больной заболевает гепатоцеллюлярным раком и карциномой легкого.
Профилактика
От генетических особенностей больного и принятых терапевтических мер во многом зависит, каков будет прогноз. Независимо от того как протекает заболевание, если его не лечить, то ждать позитивного прогноза нет смысла.
Дыхательная или печеночная недостаточность спустя короткое время сделают человека глубоким инвалидом. Больные, которым поставлен диагноз – легочная форма заболевания, должны знать, что для них огромное значение имеет второстепенная профилактика.
Чтобы не дать болезни прогрессировать, врачи советуют в первую очередь делать все, чтобы уберечь печень и легкие от вредного воздействия. Следует сторониться загрязненного воздуха, включая вдыхание строительной и дорожной пыли. Обязательно отказаться от курения, некурящим также нельзя находиться рядом с теми, кто курит. Обязательно отказаться от употребления спиртных напитков.
Больным, которые живут в городах, где экологическая обстановка неблагоприятная, будет рекомендовано сменить место проживания. Как один из отличных вариантов – переезд в село. Люди с дефицитом альфа1- антитрипсина должны соблюдать определенную диету, в которой предусмотрено ограниченное употребление некоторых продуктов, например, жареной и жирной пищи.
Кроме лечебной физкультуры остальные физические нагрузки должны быть ограничены. Обязательным является систематический прием препаратов, которые улучшают функцию печени. Один лечебный курс проводят в 3-6 месяцев.
Болезнь, при которой не хватает легочной протеазы, а в печени накапливается патологический антитрипсин, встречается довольно редко. Но как отмечают специалисты ежегодно больных с подобными признаками становится больше. Предотвратить его развитие невозможно.

Автор:
Глушко Раиса
Терапевт, пульмонолог, иммунолог
Дефицит альфа—1—антитрипсина. Основная информация по редкому заболеванию.
Диагноз хроническая обструктивная болезнь легких
Хроническая обструктивная болезнь легких (ХОБЛ) — состояние, при котором воздушные пути (трубочки, по которым воздух попадает в ваши легкие) частично блокируются, что вызывает затруднения при вдохе и выдохе. ХОБЛ также называют хроническая обструкция дыхательных путей или хроническое обструктивное заболевание легких. ХОБЛ может означать эмфизему, хронический бронхит или оба состояния. У некоторых людей с ХОБЛ отмечается как хронический бронхит, так и эмфизема. Наиболее частой причиной ХОБЛ является курение, но бывают другие ситуации.
Одной из причин может быть наследственное заболевание, которое называется недостаточность альфа1-антитрипсина (А1АТН). А1АТН — это снижение уровня или отсутствие белка альфа1-антитрипсина, который блокирует повреждающее действие определенных ферментов. Недостаток данного белка может вызвать разрушение ткани легких и развитие хронического обструктивного заболевания легких, такого как эмфизема. Воздушные мешочки (альвеолы) в увеличенном виде. Эмфизема: Ослабленные и спавшиеся воздушные мешочки с избыточным количеством слизи. Нормальные воздушные мешочки у здорового человека.
Что такое Дефицит альфа—1—антитрипсина?
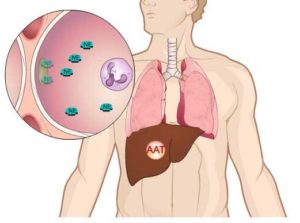 Дефицит альфа—1—антитрипсина (А1АТН), также известная как недостаточность альфа1-ингибитора протеиназы, — одно из самых распространенных серьезных наследственных заболеваний в мире. Данное состояние может привести к развитию угрожающего жизни заболевания легких или печени у детей и взрослых.
Дефицит альфа—1—антитрипсина (А1АТН), также известная как недостаточность альфа1-ингибитора протеиназы, — одно из самых распространенных серьезных наследственных заболеваний в мире. Данное состояние может привести к развитию угрожающего жизни заболевания легких или печени у детей и взрослых.
Альфа1-антитрипсин (А1АТ) — белок-ингибитор (антипротеаза), основной функцией которого является защита ткани легких от разрушения под действием протеаз. Большая часть альфа1-антитрипсина синтезируется клетками печени и моноцитами и через кровь попадает в легкие; некоторое количество белка образуется альвеолярными макрофагами и эпителиальными клетками. Люди с А1АТН унаследовали два поврежденных гена альфа1-антитрипсина.
Один поврежденный ген получен от матери и один — от отца. Существует множество типов повреждений гена альфа1-антитрипсина. Наиболее частые варианты дефектных генов называются S и Z. Нормальный ген называется M. Человек без недостаточности А1АТ имеет два гена M (MM). У людей с выявленной А1АТН как правило имеется два гена Z (ZZ). Частота заболевания у населения составляет от 1 на 1500 до 1 на 5000 населения. Большая часть случаев отмечается у лиц европеоидной расы североевропейского происхождения. Аллель Z реже встречается у лиц монголоидной и негроидной расы. А1АТН определяется по снижению уровня альфа1-антитрипсина в крови ниже 80 мг/дл 11 мкмоль/л).
У взрослых она может проявляться как заболевание легких, а у детей может быть связана с заболеваниями печени. Эластаза нейтрофилов — белок, выделяемый лейкоцитами, который переваривает бактерии и другие чужеродные объекты в легких. Связывая эластазу, А1АТ защищает тонкую ткань легких.
Если человек с Дефицит альфа-1-антитрипсина вдыхает раздражающие вещества или контактирует с инфекционными агентами, нейтрофилы выбрасывают в легкие эластазу, которая бесконтрольно продолжает переваривать раздражающие вещества, в конце концов разрушая здоровую ткань легких. Конечным результатом разрушения здоровой ткани легких под действием эластазы нейтрофилов является эмфизема.
Эмфизема — заболевание легких, вызванное разрушением тонкой структуры стенок маленьких воздушных мешочков (альвеол). При таком разрушении воздушные мешочки теряют свою эластичность и образуют более крупные, неэффективные мешочки, которые не могут должным образом обменивать кислород и углекислый газ между кровью и воздухом. Также затрудняется дыхание, так как каждый вдох растягивает легкие, но они не возвращаются к нормальному состоянию после выдоха. При этом воздух в легких попадает в ловушку, что приводит к чрезмерному наполнению воздушных мешочков.
Эмфизема, вызванная Дефицит альфа-1-антитрипсина, — прогрессирующее заболевание, которое обычно проявляется в возрасте 20-40 лет. Симптомы А1АТН включают одышку при физической нагрузке, свистящее дыхание и кашель. Дефицит альфа-1-антитрипсина часто ошибочно диагностируют как бронхиальную астму, вызванную курением эмфизему, хронический бронхит или хроническую обструктивную болезнь легких (ХОБЛ). Без лечения эмфизема в конце концов вызывает прогрессирующее снижение функции легких, которое может в значительной степени влиять на повседневную жизнь и продолжительность жизни. У некоторых людей с Дефицит альфа-1-антитрипсина также отмечается хронический бронхит, при котором выстилка легких отекает и покрывается слизью, что ограничивает поток воздуха. Мышцы бронхов (воздушных путей) часто сокращаются (бронхоспазм), что приводит к дальнейшему снижению воздушного потока и вызывает хронический кашель. У взрослых первыми симптомами Дефицит альфа-1-антитрипсина является одышка при умеренной физической активности, снижение переносимости физической нагрузки и свистящее дыхание. Такие симптомы обычно проявляются в возрасте 20-40 лет. Другие признаки и симптомы могут включать повторные инфекции дыхательных путей, усталость, учащенное сердцебиение при вставании, проблемы со зрением и непреднамеренное снижение массы тела.
Подтверждение диагноза недостаточность альфа1-антитрипсина может вызывать беспокойство и приводить в замешательство. Также как для других хронических заболеваний, следует узнать больше о самой болезни и ее лечении.
Медицинский уход с Дефицит альфа-1-антитрипсина
Надлежащий медицинский уход для пациентов с Дефицит альфа-1-антитрипсина включает профилактику заболеваний легких и печени, если это возможно, лечение инфекций в кратчайшие сроки и соблюдение режима, который вам порекомендует лечащий врач. Важной является ежегодная вакцинация против гриппа и вакцинация против пневмококковых инфекций каждые 5-6 лет. Пациентам с заболеваниями печени рекомендуется иммунизация против гепатита А и В. Легочные инфекции следует лечить незамедлительно. В случае лихорадки, озноба, усиления одышки, кашля или изменения цвета мокроты следует немедленно обратиться к врачу. Стандартные препараты для лечения симптомов со стороны легких включают бронхолитики, глюкокортикостероиды и ингаляции кислорода. Для лечения эмфиземы ваш врач также может назначить терапию, направленную на увеличение уровня альфа1-антитрипсина.
Бронхолитики — лекарственные препараты, которые расширяют воздушные пути насколько это возможно, с целью достижения максимальной функции легких. Зачастую используют ингаляционные бронхолитики, с помощью ручных распылителей, называемых «дозирующий ингалятор», или с помощью небулайзера, который распыляет жидкие препараты. Другие бронхолитики иногда назначают в форме таблеток. Кислород назначают, если его уровень в крови снижается ниже критических значений.
В таком случае рекомендуется вдыхать кислород круглосуточно, насколько это возможно. Действительно, было показано, что ингаляция кислорода почти круглосуточно повышает продолжительность жизни у пациентов с эмфиземой. Другим важным моментом в лечении эмфиземы является легочная реабилитация. Участвуя в программах, включающих инструктаж, организованные упражнения и группы поддержки, пациенты с эмфиземой могут повысить переносимость физических нагрузок.
Специфическая заместительная терапия при Дефицит альфа-1-антитрипсина
Помимо ранее указанных методов общей терапии эмфиземы, для Дефицит альфа-1-антитрипсина существует специфическая терапия. Данная терапия называется заместительной и включает регулярное применение очищенного белка А1АТ еженедельно путем инфузии, с целью повышения содержания А1АТ в крови выше определенного «защитного уровня». По данной причине такую терапию можно считать сходной с инсулинотерапией при диабете – состояние, которое приводит к недостатку вещества, и терапия, которая направлена на восстановление нормального уровня отсутствующего вещества путем его введения. вершина айсберга
Диагностируют менее 10% случаев А1АТН, более 90% не диагностируют A1AT недостаточность остается не диагностированной у многих пациентов, имеется большой разрыв между проявлением легочной симптоматики и постановкой диагноза. Большую часть из них можно выявить среди пациентов с диагнозом ХОБЛ, где примерно у 1-4,5% пациентов отмечается недостаточность А1АТ с генотипом PiZZ 2. Количество носителей PiMZ среди пациентов с ХОБЛ достигает 17,8%. Существующие в настоящее время рекомендации Всемирной организации здравоохранения включают анализ А1АТ у всех пациентов с ХОБЛ.
Бросайте курить
Первым и самым важным шагом для вас является отказ от курения. Курение вызывает разрушение А1АТ в легких. Также курение притягивает лейкоциты в легкие, что может усиливать их повреждение. избегайте пыли, пыльцы и дыма Вам следует избегать пыли, пыльцы и дыма, также как «пассивного курения» и аэрозолей. На рабочем месте это может быть сложно, поэтому, вероятно, вам потребуется обсудить с вашим работодателем качество воздуха в помещениях. Дома избегайте бытовой химии, дровяных печей, пыли, и шерсти животных.
Снижайте риск инфекций
Инфекции особенно опасны для ваших легких. По возможности избегайте контакта с больными простудой, гриппом и др.
Занимайтесь ежедневно
Упражнения могут улучшить ваше настроение и физическую форму. Для всех пациентов с А1АТН очень важны регулярные физические нагрузки, даже при минимальном проявлении или при отсутствии симптомов. При наличии проблем со стороны легких рекомендуются программы легочной реабилитации. Данные программы включают физические упражнения, дыхательную гимнастику, обучение пациентов, коррекцию питания и программы отказа от курения. Независимо от тяжести А1АТН, вам следует с лечащим врачом разработать подходящий режим упражнений.
Измените питание
Контроль питания также является важным для пациентов с Дефицит альфа-1-антитрипсина. Правильное питание помогает сохранить активность легких. Исследования показали, что пациентам с заболеваниями легких необходимо потреблять больше калорий, чем пациентам без таких заболеваний. Дополнительные калории должны поступать со здоровой пищей. Диетолог поможет вам развить навыки здорового питания.
Избегайте стрессов
Уменьшение стресса может быть полезным. Существует множество техник по снижению стресса, в том числе дыхательные упражнения, улучшение качества сна, релаксация, самоконтроль функционального состояния организма, йога.
Полезная информация:
Наблюдение пациентов в Москве осуществляется на базе МБУЗ «Городская клиническая больница №57» и НИИ пульмонологии ФМБА России (Москва, ул. 11-я Парковая, д. 32/61, контактная информация: Карчевская Наталья Анатольевна, +79264324744. Бесплатная диагностика проводится: ФГБНУ «Медико-генетический научный центр» по адресу: 115478, Москва, ул. Москворечье, д. 1; Тел: +74993248772 (регистратура). Информацию о бесплатной диагностике вы можете уточнить в общественной организации «Союз пациентов и пациентских организаций по редким заболеваниям», горячая линия + 7 499 270-35-20, e-mail: office@ spiporz.ru.
Дефицит альфа 1 антитрипсина у детей рекомендации
Дефицит альфа 1 антитрипсина у детей рекомендации клинические
Дефицит альфа 1 антитрипсина
Дефицит альфа 1 антитрипсина, альфа (один)-Антитрипсин — низкомолекулярный протеазный ингибитор, подавляющий активность многих протеолитических знзимов: трипсина, химотрипсина, плазмина, тромбина, эластазы, гиалуронидазы, протеаз лейкоцитов, макрофагов, микроорганизмов и др. В основе ряда наследственных заболеваний лежит дефицит а,-антитрипсина — гликопротеина, синтезируемого в печени. У 0,03- 0,015% (т. е. 1 на 3000-6000) новорожденных активность альфа (один) -антитрипсина резко снижена.
Дефицит и, -антитрипсина приводит к повышенному накоплению протеолитических энзимов и последующему повреждению тканей. Известно, однако, что при дефиците и,-анти-трипсина поражения легких и печени не всегда бывают тяжелыми и необратимыми. Видимо, данный дефицит может быть компенсирован другими механизмами.
Клиническая картина. Уже в неонатальном периоде отмечается увеличение печени, развитие желтухи, обесцвечивание кала, потемнение мочи вследствие холестаза. Холестаз может быть неполным, и тогда выраженность клинической картины варьирует. Лабораторные исследования указывают на наличие гипербилирубинемии конъюгирован-ного типа, гиперхолестеринемии, увеличение щелочной фос-фатазы, умеренный подъем активности трансаминаз крови. Такая картина обычно наблюдается до 10-й недели жизни и спонтанно исчезает в конце первого полугодия. В дальнейшем могут развиться цирроз печени с типичными его проявлениями (гепатоспленомегалия, портальная гипертензия и др. ) или часто рецидивирующие желтухи с зудом и выраженной гиперхолестеринемией.
При легочной форме чаще всего имеет место картина:
прогрессирующей эмфиземы, однако могут наблюдаться рецидивирующий обструктивный синдром, рецидивирующий бронхит, повторные пневмонии.
Диагноз ставят на основании анамнеза, клинических симптомов, выявления низкого уровня к,-антитрипсина.
Дифференциальный диагноз проводят с атрезией желчевыводящих путей, желтухами различного генеза, иммунодефицитными заболеваниями.
Лечение. Специфическая терапия отсутствует. Заместительная терапия альфа (один) — фракцией глобулинов неэффективна вследствие короткого периода их полувыведения. При наличии инфекции — активная антибактериальная терапия.
Прогноз неблагоприятный.
Недостаточность альфа-1-антитрипсина
α₁-ANTITRYPSIN DEFICIENCY (AATD)
MIM#107400
Генетика: ген, кодирующий α1-АТ, располагается в 14-й хромосоме в локусе q31-32.3 и называется SERPINA1. Существует полиморфизм гена α1-АТ, и некоторые аллельные варианты приводят к снижению сывороточного уровня α1-АТ ниже нормальных значений, что, однако, не всегда сопровождается клиническими проявлениями. Сывороточная концентрация α1-АТ ниже 0,9 г/л требует дальнейшего обследования.
Наиболее часто встречающиеся и клинически значимые аллели, ведущие к недостаточности α1-АТ, – аллели Z и S. Z-аллель представляет собой точечную замену глутамина лизином в позиции 342, S-аллель – замену глутамина валином в позиции 264. Гомозиготы PiSS имеют нестабильный α1-АТ, быстро деградирующий вне гепатоцитов. Z-аллель у гомозигот ведет к формированию полимеров α1-АТ внутри гепатоцитов и моноцитов, в результате чего α1-АТ секретируется в кровь в очень небольшом количестве.
Тип наследования: аутосомно-рецессивный
Эпидемиология: в Европе распространенность генотипа PiZZ варьирует между странами, в среднем составляя 1:2500 новорожденных, а максимальна она на севере Европы (в Швеции –1:1600 новорожденных).
Патогенез: альфа-1-антитрипсин является гликопротеидом, синтезируемым печенью. Функционально он обеспечивает 90% активности, ингибирующей трипсин в крови. Этот гликопротеид тормозит действие не только трипсина, но и химотрипсина, эластазы, калликреина, катепсинов и других ферментов тканевых протеаз.
Клинические проявления: альфа-1-антитрипсиновая недостаточность — наследственное заболевание, обусловленное сниженной концентрацией альфа-1-антитрипсина (ААТ) в сыворотке крови, возникающее вследствие различных мутаций в гене Рі и проявляющееся в виде хронических неспецифических заболеваний легких с развитием эмфиземы, а также поражением печени и сосудов. Клинические проявления недостаточности α1-АТ строго коррелируют с генотипом PiZZ, однако выраженность симптомов сильно варьирует – от полного отсутствия до фатальных легочных или печеночных проявлений. Также клиническими проявлениями сопровождается нулевой генотип, т.е. полное отсутствие продукции α1-АТ.
Вследствие хронической полимеризации Z-типа протеина в гепатоцитах формируются фиброз и цирроз печени. Эти состояния могут развиться уже в детстве, но, как правило, манифестируют после 50 лет. Легочными проявлениями недостаточности α1-АТ служат эмфизема легких и хроническая обструктивная болезнь легких (ХОБЛ). У курильщиков эмфизема легких развивается на 3–4-й декаде жизни, у некурящих – на 4–5-й декаде. При нулевом варианте генотипа, т.е. при полностью отсутствующей продукции α1-АТ, и сопутствующем курении эмфизема формируется в еще более раннем возрасте.
Диагностика: исследование уровня альфа-1-антитрипсина в крови, а также проведение ДНК-диагностики с использованием ПЦР и рестрикционного анализа. БЕСПЛАТНАЯ диагностика проводится: МЕДИКО-ГЕНЕТИЧЕСКИЙ НАУЧНЫЙ ЦЕНТР РАМН по адресу 115478, Москва, Москворечье ул., д. 1; тел: +74993248772 (регистратура), +74993242004 (лаборатория наследственных болезней обмена веществ) Этот адрес электронной почты защищён от спам-ботов. У вас должен быть включен JavaScript для просмотра., http://www.labnbo.narod.ru. (по направлению лечащего врача пульмонолога). Информацию о бесплатной диагностике Вы можете уточнить в представительстве Генфа Медика С.А., Швейцария: 119421 Москва, Ленинский пр., д.99.Тел. (495) 662-50-65, факс (495) 662-50-61.
Наблюдение пациентов в Москве осуществляется на базе МБУЗ «Городская клиническая больница №57» и НИИ пульмонологии ФМБА России (Москва, ул. 11-я Парковая, д. 32/61), контактная информация: Карчевская Наталья Анатольевна, +7(926)4324744
Лечение:
- Симптоматическая терапия;
- Хирургические методы лечения;
- Заместительная терапия ингибитором альфа-1-протеиназы у пациентов с клинической картиной панацинозной эмфиземы легких или ХОБЛ и фенотипами PiZZ, PiZ(-) или Pi (-)(-).
Заместительная терапия – препарат Респикам® (альфа-1-антитрипсин), Камада, Израиль. Препарат зарегистрирован на территории Российской Федерации в 2010 году. Показан для лечения панацинозной эмфиземы легких или ХОБЛ (фенотипы
Альфа-1-антитрипсин (А1АТ) – белок печени, основная роль которого заключается в инактивации ферментов-протеаз, которые расщепляют соединительную ткань в организме.
Роль А1АТ в организме
Чтобы понять функцию А1АТ, необходимо описать патофизиологический механизм: воспалительная реакция в каком-либо органе приводит к тому, что в воспалительном очаге образуется соединительная ткань. Затем для восстановления функции органа эта соединительная ткань должна быть разрушена с помощью протеаз. Однако, соединительная ткань присутствует во всем организме, и ее необходимо защищать от побочного эффекта протеаз. Прежде всего, это касается легких, в которых содержится вещество «сурфактант». Сурфактант обволакивает внутреннюю поверхность легких в виде своеобразной «смазки» — защищает их от слипания во время выдоха. В норме А1АТ в сурфактанте предотвращает разрушительный эффект протеаз, но если количества А1АТ недостаточно, то протеазы разрушают легкие и приводят к развитию эмфиземы.
Фракция альфа-1-антитрипсина относится к острофазовым белкам, их концентрация в крови повышается в период воспаления.
Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим расширением полостей. Легкое становится объемным за счет того, что лопаются стенки альвеол (маленькие пузырьки из которых, состоит легочная ткань) и образуются большие полости с воздухом, которые не могут выполнять функцию газообмена. Основные симптомы – это одышка, затруднение дыхания, хрипы.
Роль А1АТ в патологии
Дефицит А1АТ обусловлен наследственными генетическими заболеваниями, которые приводят либо к продукции недостаточного количества А1АТ, либо к неправильной его структуре, в результате чего, он не может выполнять свою физиологическую функцию. Распространенность врожденного дефицита варьирует от 1:625 до 1:2000. Риск выше у представителей европеоидной расы. Дефицит А1АТ становиться причиной эмфиземы, хронической обструктивной болезнь легких (ХОБЛ), хронического бронхита, заболеваний печени и ANCA-позитивного васкулита.
Легкие
Примечательно, что обычно эмфизема и ХОБЛ возникают у тех пациентов, которые регулярно подвергают свои легкие воздействию вредных факторов: дыму от сигарет или вдыханию частиц промышленных отходов, которые оседают в легких и повреждают их. Но если эмфизема или ХОБЛ возникает у пациента, который не курит и живет в хороших экологических условиях – то это признак дефицита А1АТ. Химические испытания показали, что сигаретный дым окисляет аминокислоту метионин, входящий в состав А1АТ и «деформируют» его структуру, что вызывает эмфизему.
Чем ниже уровень альфа-1-антитрипсина, тем выше риск развития эмфиземы.
Печень
Дефект гена, кодирующего структуру А1АТ, способствует продукции дефектного фермента А1АТ, который не может полноценно утилизироваться клетками печени. Как следствие, в печени накапливается этот фермент и повреждает орган: развивается фиброз, а затем и цирроз печени. Данная патология больше распространена в детском возрасте, чем во взрослом.
Применение в лабораторной диагностике
Исходя из свойств альфа-1-антитрипсина, имеются определенные показания для его назначения: выявление причины эмфиземы и желтухи, особенно если пациент не подвержен факторам риска развития данных заболеваний. Если пациент, страдающий ХОБЛ, имеют сопутствующую патологию печени, то это может ухудшить течение заболевания легких. Если в крови обнаруживается дефицит А1АТ, то для эффективного лечения ХОБЛ потребуется восстановление функции печени.
Читайте также: ДЕФИЦИТ АЛЬФА-1-АНТИТРИПСИНА – ПРОБЛЕМА, КОТОРУЮ НЕ ЗАМЕЧАЮТ
Дефицит антитрипсина альфа-1 — Genetics Home Reference
Carrell RW, Lomas DA. Дефицит альфа1-антитрипсина — модель для конформационных заболеваний. N Engl J Med. 2002 янв. 3; 346 (1): 45-53. Обзор.
DeMeo DL, Silverman EK. Дефицит альфа1-антитрипсина. 2: генетические аспекты дефицита альфа (1) -антитрипсина: фенотипы и генетические модификаторы риска эмфиземы. Thorax. 2004 март; 59 (3): 259-64. Обзор.
Fairbanks KD, Tavill AS.Заболевание печени при дефиците альфа-1-антитрипсина: обзор. Am J Gastroenterol. 2008 авг; 103 (8): 2136-41; викторина 2142. doi: 10.1111 / j.1572-0241.2008.01955.x. Обзор.
Fregonese L, Stolk J. Наследственный дефицит альфа-1-антитрипсина и его клинические последствия. Orphanet J Редкий Дис. 2008 июнь 19; 3: 16. doi: 10.1186 / 1750-1172-3-16. Обзор.
Ломас Д.А., Парфри Х. Дефицит альфа-1-антитрипсина. 4: Молекулярная патофизиология. Thorax. Июнь 2004 года; 59 (6): 529-35.Обзор.
Luisetti M, Seersholm N. Дефицит альфа-1-антитрипсина. 1: эпидемиология дефицита альфа1-антитрипсина. Thorax. 2004 Фев; 59 (2): 164-9. Обзор.
Needham M, Stockley RA. Дефицит альфа 1-антитрипсина. 3: Клинические проявления и естественная история. Thorax. Май 2004 года; 59 (5): 441-5. Обзор.
Перлмуттер Д.Х., Бродский Ю.Л., Балистрери В.Ф., Трапнелл BC. Молекулярный патогенез болезни печени, связанной с дефицитом альфа-1-антитрипсина: обзор совещания.Гепатологии. Май 2007 года; 45 (5): 1313-23. Обзор.
Ranes J, Stoller JK. Обзор дефицита альфа-1 антитрипсина. Semin Respir Crit Care Med. Апрель 2005 года; 26 (2): 154-66. Обзор.
Stoller JK, Aboussouan LS. Мифы и неправильные представления о дефиците {альфа} 1-антитрипсина. Arch Intern Med. 2009 март 23; 169 (6): 546-50. doi: 10.1001 / archinternmed.2009.25.
Stoller JK, Lacbawan FL, Aboussouan LS. Дефицит альфа-1 антитрипсина. 2006 окт 27 [обновлено 2017 янв 19].В: Пагон Р.А., Адам М.П., Ардингер Х.Х., Уоллес С.Е., Амемия А., Боб Л.Дж., Берд Т.Д., Ледбеттер Н., Меффорд Х.К., Смит Р.Дж., Стивенс К., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017. Доступно по адресу: http://www.ncbi.nlm.nih.gov/books/NBK1519/
Teckman JH, Линдблад Д. Дефицит альфа-1-антитрипсина: диагностика, патофизиология и лечение. Curr Gastroenterol Rep. 2006 Feb; 8 (1): 14-20. Обзор.
Teckman JH. Дефицит альфа1-антитрипсина в детстве.Semin Liver Dis. 2007 авг; 27 (3): 274-81. Обзор.
de Serres FJ, Blanco I, Fernández-Bustillo E. Предполагаемое количество и распространенность аллелей дефицита PI * S и PI * Z альфа1-антитрипсина в Азии. Eur Respir J. 2006 Dec; 28 (6): 1091-9. Epub 2006 Sep 27.
de Serres FJ, Blanco I, Fernández-Bustillo E. Оценка риска дефицита альфа-1-антитрипсина среди пациентов с ХОБЛ: данные, подтверждающие целенаправленный скрининг. ХОЗЛ. 2006 Авг; 3 (3): 133-9.Ошибка в: ХОБЛ. 2006 дек; 3 (4): 245.
de Serres FJ, Blanco I, Fernández-Bustillo E. Последствия для здоровья дефицита альфа-1-антитрипсина в странах Африки к югу от Сахары и их эмигрантах в Европе и Новом Свете. Genet Med. 2005 март; 7 (3): 175-84. Обзор.
де Серрес FJ, Бланко I, Фернандес-Бустильо Е. PI S и PI Z альфа-1 дефицит антитрипсина альфа-1 во всем мире. Обзор существующих генетических эпидемиологических данных. Мональди Арка Сундук Дис. 2007 дек; 67 (4): 184-208.Обзор.
де Serres FJ. Дефицит альфа-1-антитрипсина — не редкое заболевание, а заболевание, которое редко диагностируется. Перспектива здоровья окружающей среды. 2003 дек; 111 (16): 1851-4. Обзор.
.
Введение
Дефицит альфа-1-антитрипсина (α1-AT) является одним из наиболее распространенных наследственных расстройств среди кавказцев, характеризующихся снижением уровня или отсутствием α1-AT в сыворотке и высоким риском развития эмфиземы легких и / или заболеваний печени (1) , Хотя α1-AT является важной причиной эмфиземы в 3 и и десятилетий жизни и обычной причиной заболеваний печени у детей, он может вызывать легочные заболевания у детей.α1-AT представляет собой гликопротеин, вырабатываемый печенью. Его основная функция заключается в защите тканей легких от протеолитических ферментов, выделяемых мертвыми бактериями или лейкоцитами в легких. Концентрация протеазы лейкоцитов может быть важным фактором тяжести заболевания легких с заданным уровнем α1-AT (2,3).
Целью этой статьи является сообщение о 13-летнем мальчике с дефицитом α1-AT, у которого было хроническое бронхолегочное поражение и повторное поступление в больницу без явно паренхиматозного нарушения печени.
Презентация кейса:
Пациент был 13-летним мальчиком, родителем второй степени, родителями второй степени, который был помещен в отделение неотложной помощи нашей больницы, у которого был респираторный дистресс, лихорадка, продуктивный кашель, преимущественно в положении лежа, с неделю назад. У него была история рецидивирующей бронхопневмонии около 5 лет назад с повторным госпитализацией и консервативным лечением с несколькими курсами антибиотикотерапии и муколитической терапии.У него была история одышки напряжения от иногда назад. Он был мальчиком с нормальной массой тела при рождении, однако в это время у него не было успеха, он был анорексией и недоедал. Не было истории стремления к инородному телу. У него не было хронической диареи или каких-либо особенностей печеночных и энтеральных проблем. Его вес был 18,300 кг, рост 111 см, окружность головы 51 см, кооперативный и психически нормальный. У него была тахипноэ (частота дыхания; 43), частота сердечных сокращений: 96, температура тела: 38,8, обследование ротоглотки было нормальным.Грудь, видимо, нормальная. При аускультации отмечались двусторонние острые кровоизлияния и генерализованные хрипы в обоих легких. Рентгенография пазух носа была нормальной, но только левая верхнечелюстная пазуха была легкой. Рентгенография грудной клетки показала диффузную эмфизему, легочную иларную инфильтрацию и муковисцидоз. HRCT показал двусторонний бронхэктаз с легочным фиброзом. Не было никаких признаков инородного тела или лимфаденопатии. На рисунках показаны некоторые изображения, которые были взяты у пациента в разное время (Рисунок 1-3).
Гемограмма
показала: WBC: 26300, количество нейтрофилов: 62%, лимфоциты: 36%, эозинофилы: 2%, BUN: 10,8, креатинин: 0,65, SGOT: 12, SGPT: 58, ALP: 230, Ca: 9,6, P: 3,8, Na: 145, K: 4, PT: 14, PTT: 46, сывороточный альбумин: 5,18 г / дл, альфа-1-глобулин: 0,05 г / дл, альфа-2-глобулин: 0,7 г / дл, бета-глобулин : 1,27 г / дл, гамма-глобулин: 2,8 г / дл, соотношение A / G: 1,08, IgG: 1300 мг / дл, IgA: 210 мг / дл, IgM: 105 мг / дл, ABG = Po 2 : 78 мм рт.ст., O 2 -Sat: 85,7%, PH: 7,31, Pco 2 : 24,6 мм рт.ст., Hco 3 : 12.1, 5 шт. Тест PPD: диаметр 4 мм, тест на хлорид пота: Na: 44, Cl: 52. Уровень α1-AT в сыворотке: 18 мг / дл. Профиль электрофореза сывороточного белка показан на рис. 4. Мокрота была отрицательной на кислотоустойчивую палочку.
Бронхоскопия показала наличие двустороннего бронхоэктаза с профузной мокротой.
Семейный анамнез был отрицательным для туберкулеза или аллергических заболеваний дыхательных путей.
Его отец долгое время курил сигареты.
Обсуждение
α-ATD был впервые идентифицирован в 1963 году вместе с его ассоциацией с унаследованной формой раннего начала тяжелой панацинарной эмфиземы нижней зоны.α1-AT синтезируется в печени и защищает альвеолярные ткани легких от разрушения нейтрофильной эластазой и другими протеазами (4,5).
α-ATD является распространенным аутосомно-рецессивным состоянием (от 1: 1600 до 1: 1800), при котором заболевание печени возникает в результате удержания аномально полимеризованного α1-AT в эндоплазматической сети гепатоцитов, а эмфизема — в результате повреждения альвеолярной стенки (6).
α1-AT составляет около 95% всей антипротеазной активности в альвеолах человека, а нейтрофильная эластаза считается протеазой, в значительной степени ответственной за разрушение альвеол (7).
Клинические последствия α1-ATD в детском возрасте могут быть представлены как желтуха новорожденных, холестаз, хроническое заболевание печени; геморрагические заболевания и иногда заболевания легких, мембрано-пролиферативный гломерулонефрит, панникулит, нектотический васкулит и ранняя эмфизема, особенно у курильщиков, являются ассоциациями с α1-ATD во взрослой жизни (8,9).
α1-ATD является одним из 3 наиболее распространенных летальных генетических заболеваний, таких как муковисцидоз и синдром Дауна. В основном это касается кавказцев (белых) из северной Европы.Были представлены новые данные, демонстрирующие, что это всемирное расовое и этническое распределение, обнаруженное среди различных групп афроамериканцев, арабов, евреев на Ближнем Востоке, в Центральной, на Дальнем Востоке и в Юго-Восточной Азии, а также среди белых в австралийцах, Европе, Новая Зеландия и Северная Америка. В общей численности населения 4,4 миллиарда человек в 58 обследованных странах насчитывается не менее 116 миллионов носителей (с фенотипами Pi PiSZ и PiMZ) и 3,4 миллиона с дефицитом комбинаций аллелей (фенотипов PiSS, PiSZ) для двух наиболее распространенных аллелей дефицита PiS и P1Z (10, 11).В нашей стране нет документального подтверждения распространенности этого расстройства (8, 12,13)
α-ATD следует учитывать при дифференциальной диагностике хронических заболеваний легких в детском возрасте, механической желтухи у новорожденных, цирроза у детей и взрослых, семейных ранних обструктивных заболеваний легких у взрослых и сочетанных заболеваний легких и печени в детском и подростковом возрасте. Это наиболее распространенная генетическая причина заболеваний печени и второй признак трансплантации печени после атрезии желчевыводящих путей у детей (14-16).α-ATD демонстрирует взаимодействие воспаления с инфекционными и токсическими факторами риска, связанными с окружающей средой, и представление о том, что хронические заболевания легких или печени у взрослых могут возникать в детстве, поэтому прогрессирование можно ненавидеть, если оценить его на ранних стадиях (13, 17).
α1-AT представляет собой гликопротеин, синтезируется гепатоцитами печени и обнаруживается в большинстве биологических жидкостей. На его долю приходится 90% площади под пиком α1 в электрофорезе сывороточного белка, и на него приходится 80 — 90% способности сыворотки ингибировать трипсин.Структура электрофореза белка у нормального субъекта и пациента с α1-ATD показана на рис. 5.
Название α1-AT, полученного из активности ингибитора протеазы (Pi), включая эластазу, протеазы, коллагеназу, химотипин, плазмин и тромбин. α1-AT является реактивом острой фазы, уровни сыворотки которого повышаются в ответ на инфекцию, воспаление и стресс (8, 18, 19).
Антиэластазная активность защищает эластиновые волокна легочной ткани от протеолитического действия эластазоподобных ферментов, которые высвобождаются из лейкоцитов и макрофагов в процессе естественного распада или воспаления в результате различных оскорблений.Дефицит или пониженный уровень активности α1-AT в сыворотке обусловлен измененной конфигурацией молекулы α1-AT, которая предотвращает ее высвобождение из гепатоцитов, способствует ускоренному и даже необратимому повреждению ткани. Не у всех людей с дефицитом α1-AT развивается заболевание легких, что явно свидетельствует о дисперсии других экзогенных и эндогенных факторов риска в этом расстройстве (15, 20).
Серологическая экспрессия (фенотип) α1-AT наследуется как два аутосомных кодоминантных аллеля (генотипа), из которых существует почти 24 аллельных варианта Pi, характеризующихся их электрофоретической подвижностью в крахмальных и полиакриламидных гелях.Уровень α1-AT в сыворотке находится под генетическим контролем, что определяется типом Pi в сыворотке. Глубокий α1-ATD (PiZZ с уровнем сыворотки 10-20% от уровня PiMM) следует подозревать, когда имеется простой или плоский альфа-участок при простом электрофорезе белка. Диагноз α1-ATD основан на измененной миграции аномальной молекулы α1-AT в образцах сыворотки, подвергнутых изоэлектрическому фокусированию и анализу (15, 18, 21). Фенотип должен быть определен во всех подозреваемых случаях.
Наиболее распространенным аллелем является М (PiM), а гомозиготные индивидуумы (ММ) продуцируют нормальные количества α1 антитрипсина (уровни в сыворотке 150-350 мг / дл).Наиболее распространенная форма α1-ATD связана с аллелем Z или гомозиготным PiZ (ZZ). Сывороточные уровни α1-AT у этих пациентов составляют около 10-15% от нормальных сывороточных уровней. Пациенты с PiSZ и пациенты с нулевым геном (не продуцирующие α1-AT) имеют высокий риск раннего разрушения легких. Уровни сыворотки менее 80 мг / дл предполагают значительный риск заболевания легких (4, 16).
В нашем случае был почти плоский α-сегмент белка с электрофоретическим профилем его сыворотки и низкий уровень (18 мг / дл) сывороточного α1-AT.
Поражение легких представляет собой одышку с медленным прогрессированием, хотя у многих пациентов первоначально появляются симптомы кашля или образования мокроты и хрипов из-за прогрессирующей панцинарной эмплиземы и бронхолегочного деструктивного воспалительного процесса, о чем свидетельствуют повышенная базилярная люценция и инфильтративные изменения на рентгенограмме грудной клетки. По мере прогрессирования заболевания хроническая непереносимость кашля, хронический бронхит и бронхоэктазия усиливают клиническую картину. Курение сигарет и воздушные пулотанты определенно способствуют раннему началу презентации.Хотя легочное поражение обычно проявляется в третьем десятилетии жизни, из-за вариабельности клинического случая и генетических вариантов, наш случай представлен легочным заболеванием в детском возрасте (22-24).
Лечение включает бронходилатацию, физическую реабилитацию, отказ от курения и внутривенную аугментационную терапию с α1-AT, агрессивное лечение легочной инфекции, рутинное использование пневмококковой вакцины, вакцины против гриппа и трансплантации легкого (4, 25-29).
Консервативное лечение рекомендовано и сделано у нашего пациента с заметным улучшением.Увеличение терапии с помощью α1-AT не было доступно в нашем наборе средств.
Фенотипирование требуется для подтверждения α1-ATD. Было идентифицировано более 100 вариантов ATD, но только один фенотип, PiZZ ответственен почти за все случаи эмфиземы ATD и заболевания печени. Ген SERPINA I, ранее известный как Pi, находится в длинном плече хромосомы 14. Рентген грудной клетки, функциональные пробы печени, тест на хлорид пота, сывороточный белок и иммунный электрофорез и КТ грудной клетки с высоким разрешением могут потребоваться для окончательной диагностики и оценки. (3, 9).
Заключение
В этой статье предполагается, что если у ребенка хронические или повторные бронхолегочные признаки и деструктивное расстройство легких, то лечение α1-AT следует проводить так же, как у детей с холестатической и / или хромовой болезнью печени.
Конфликт интересов : не объявлен
.
- CareNotes
- Дефицит альфа-1-антитрипсина у детей
Этот материал нельзя использовать в коммерческих целях или в каких-либо больницах или медицинских учреждениях. Невыполнение может привести к судебному иску.
ЧТО ВЫ ДОЛЖНЫ ЗНАТЬ:
Что такое дефицит антитрипсина альфа-1?
Альфа-1-дефицит антитрипсина (AATD) — это состояние, которое повышает риск развития у вашего ребенка повреждения легких и печени.Альфа-1 антитрипсин (ААТ) вырабатывается печенью вашего ребенка и защищает его легкие и печень от инфекций. Его тело может не иметь достаточного количества ААТ, если он родился с ненормальными генами, которые производят ААТ. Если AAT, который делает его печень, неисправен, это может вызвать воспаление, повреждение печени и привести к печеночной недостаточности. Он также может развить AATD, если табачный дым или химические пары снижают его уровень AAT.
Каковы признаки и симптомы дефицита альфа-1 антитрипсина?
- Отек или боль в животе
- Синяки
- Зуд по всему телу
- Темная моча или легкие испражнения
- Медленнее прибавка в весе и рост, чем у других детей его возраста
- пожелтение кожи или белки глаз
- Кровь в рвоте, кровавые или черные движения кишечника
Как диагностируется дефицит альфа-1 антитрипсина?
- Анализы крови: Анализы крови измеряют уровень AAT вашего ребенка.
- Генетическое тестирование: Медицинские работники проверяют наличие аномальных генов, которые могут вызвать AATD.
Как лечится дефицит альфа-1 антитрипсина?
Нет лекарства от AATD. Лечение зависит от состояния здоровья вашего ребенка и от того, какие органы повреждены. Он может нуждаться в лекарствах или других методах лечения, чтобы предотвратить проблемы со здоровьем. Ему может понадобиться лечение, чтобы помочь своему телу использовать пищу.
- Питание: Спросите поставщика здравоохранения вашего ребенка, если вам нужно изменить продукты, которые ест ваш ребенок.Ему, возможно, нужно есть продукты с высоким содержанием калорий и витаминов, чтобы получить правильное питание.
- Операция: Вашему ребенку может потребоваться операция по восстановлению органов, поврежденных AATD. Если печень вашего ребенка сильно повреждена, ему может потребоваться пересадка печени.
Как я могу помочь моему ребенку с дефицитом альфа-1 антитрипсина?
Будьте активны, чтобы помочь вашему ребенку справиться с его состоянием и предотвратить другие проблемы со здоровьем:
- Обратитесь к поставщику медицинских услуг: Отведите вашего ребенка на прием к врачу.Это поможет сохранить его здоровье. Есть много медицинских проблем, которые могут возникнуть у вашего ребенка с AATD в любое время. Регулярные посещения и анализы помогут медицинским работникам узнать, есть ли у вашего ребенка проблемы, требующие лечения.
- Сделайте прививку вашему ребенку: Отвезите ребенка к своему врачу для прививок (прививок), чтобы защитить его здоровье. Это могут быть прививки от гепатита А и В.
- Держите ребенка вдали от вредных паров: Химические пары и табачный дым могут повредить или ухудшить легкие вашего ребенка.Если кто-то в вашей семье курит табак, попросите его бросить или не курить рядом с вашим ребенком. Поговорите с ребенком о вреде курения и вредных химических веществ.
Каковы риски дефицита альфа-1 антитрипсина?
- Ваш ребенок может иметь побочные эффекты от лечения AATD. Даже при лечении его симптомы могут ухудшиться. AATD может вызвать проблемы с печенью, которые могут ухудшиться с возрастом вашего ребенка. Повреждение печени может привести к циррозу, заболеванию печени или раку.Инфекции и алкоголь могут увеличить риск заболевания печени у вашего ребенка. Во время пересадки печени ваш ребенок может заразиться инфекцией или сильно кровоточить. Существует вероятность того, что новая печень вашего ребенка не будет работать вообще. Симптомы вашего ребенка могут занять некоторое время, чтобы уменьшить или исчезнуть.
- Если AATD не лечится, печень и легкие вашего ребенка могут продолжать повреждаться, пока он вырастет. Он не может набирать вес или расти так же быстро, как другие дети его возраста. Опухоли и шрамы могут появиться на печени вашего ребенка и привести к тому, что она станет твердой и перестанет работать.Его глаза и кожа могут пожелтеть, и он может легко кровоточить и ушибаться. Почки вашего ребенка также могут быть повреждены и перестать работать. Он может иметь проблемы с дыханием и легко уставать. AATD может привести к заболеванию легких, такому как эмфизема или другое заболевание легких. Он может умереть, если его легкие слишком сильно повреждены. AATD также может повредить его почки, кожу и кровеносные сосуды.
Когда мне следует обратиться к врачу моего ребенка?
Обратитесь к врачу вашего ребенка, если:
- У вас проблемы с кормлением ребенка, и вы чувствуете, что ему не хватает еды.
- Желтуха вашего ребенка (пожелтение кожи и глаз) не проходит.
- У вас или вашего ребенка есть вопросы или сомнения по поводу его состояния или ухода.
Когда мне следует немедленно обратиться за помощью?
Немедленно обратитесь за помощью или позвоните 911, если:
- У вашего ребенка проблемы с дыханием.
- У вашего ребенка боли в животе, которые не проходят.
- У вашего ребенка кровь в рвоте или испражнениях.
Где я могу найти поддержку и дополнительную информацию?
- Alpha One Foundation
2937 SW 27th Avenue, Suite 302
Miami, FL 33133
Телефон: 1- 305 —
Телефон: 1- 877 —
Веб-адрес: http: // www.alphaone.org/
- Национальный институт сердца, легких и крови
Информационный центр здравоохранения
P.O. Box 30105
Bethesda, MD 20824-0105
Телефон: 1- 301 — 592-8573
Веб-адрес: http://www.nhlbi.nih.gov/health/infoctr/index.htm
Соглашение об уходе
Вы имеете право помочь в планировании ухода за вашим ребенком. Узнайте о состоянии здоровья вашего ребенка и о том, как его можно лечить. Обсудите варианты лечения с медицинскими работниками вашего ребенка, чтобы решить, какой уход вам нужен для вашего ребенка.Приведенная выше информация является только образовательной помощью. Он не предназначен в качестве медицинского совета для индивидуальных условий или лечения. Поговорите со своим врачом, медсестрой или фармацевтом, прежде чем следовать какой-либо медицинской схеме, чтобы узнать, является ли она безопасной и эффективной для вас.
© Copyright IBM Corporation 2020 Информация предназначена только для конечного пользователя и не может быть продана, распространена или иным образом использована в коммерческих целях. Все иллюстрации и изображения, включенные в CareNotes®, являются собственностью A.D.A.M., Inc.или IBM Watson Health
Дополнительная информация
Всегда консультируйтесь со своим врачом, чтобы информация, отображаемая на этой странице, соответствовала вашим личным обстоятельствам.
Медицинский отказ от ответственности
,
![]()
![]()
Гастроэнтерология
ПРОДОЛЖАЙТЕ УЧИТЬСЯ
НАЧИНАЙ СЕЙЧАС
ПРОДОЛЖАЙТЕ УЧИТЬСЯ
НАЧИНАЙ СЕЙЧАС

КАТЕГОРИИ

- Медицинская онлайн-библиотека Lecturio
- Pre-Med Curriculum
- Биология
- Химия
- Физика
- Статистика
- Доклиническая учебная программа
- Анатомия
- Поведенческая наука
- Биохимия
- Биомедицинские науки
- Эмбриология
- Эпидемиология и биостатистика
- Гистология
- Иммунология
- Микробиология
- Патология
- Фармакология
- Физиология
- Клинический учебный план
- Анестезиология
- Кардиология
- Дерматология
- Экстренная медицина
- Эндокринология
- Семейная медицина
- Гастроэнтерология
- Гинекология
- Гематология
- Гепатология
- Инфекционные заболевания
- Медицинская генетика
- Неврология
- Офтальмология
- Отоларингология (ЛОР)
- Онкология
- Ортопедия
- Психиатрия
- Педиатрия
- Радиология
- Ревматология
- Болезни репродуктивной системы
- Респираторная медицина
- Хирургия
- нефрология / урология
- Сосудистая медицина
- Учиться и учить медицине
- Медицинская онлайн-библиотека Lecturio
- Pre-Med Curriculum
- Биология
- Химия
- Физика
- Статистика
- Доклиническая учебная программа
- Анатомия
- Поведенческая наука
- Биохимия
- Биомедицинские науки
- Эмбриология
- Эпидемиология и биостатистика
- Гистология
- Иммунология
- Микробиология
- Патология
- Фармакология
- Физиология
- Клинический учебный план
- Анестезиология
- Кардиология
.