Прогерия – преждевременное старение: причины, видео
Прогерия является одной из самых необычных и загадочных болезней. На самом деле ее вызывают не зловредные вирусы, а генетический дефект. Прогерия сопровождается изменением кожи и внутренних органов, а пациенты выглядят чрезмерно постаревшими. У этих людей наблюдается характерный внешний вид: невысокий рост, большая голова и уменьшенная лицевая часть.
♥ ПО ТЕМЕ: Чревовещатели (вентрологи), или как говорить и петь не раскрывая рта (видео).
Что такое «прогерия»?
Для начала расшифруем само слово «прогерия». Оно составлено из двух греческих слов – «сверх» и «старик». Таким образом больные становятся «сверхстариками», что выглядит удручающе. Прогерия может быть, как детской, так и взрослой. Впервые такой случай был описан еще в 1886 году английским врачом Хатчинсоном. Он наблюдал за шестилетним мальчиком, который начал быстро стареть, что сопровождалось отмиранием кожи. На основе этой истории и появился в медицине термин «прогерия». Аналогичное заболевание у взрослых было описано чуть позже – немецкий врач Карл Вернер на основе наблюдений за четырьмя пациентами защитил докторскую диссертацию в 1904 году. Прогерия – редчайшая болезнь, всего зафиксировано около 350 ее случаев.
Он наблюдал за шестилетним мальчиком, который начал быстро стареть, что сопровождалось отмиранием кожи. На основе этой истории и появился в медицине термин «прогерия». Аналогичное заболевание у взрослых было описано чуть позже – немецкий врач Карл Вернер на основе наблюдений за четырьмя пациентами защитил докторскую диссертацию в 1904 году. Прогерия – редчайшая болезнь, всего зафиксировано около 350 ее случаев.
♥ ПО ТЕМЕ: Что такое синдром Туретта: симптомы, причины, видео.
Причины возникновения прогерии
Сегодня науке удалось выявить причину такого заболевания. У детей прогерия появляется из-за мутации гена LMNA, отвечающего за кодировку белков-ламинов. В некоторых семьях прогерия встречается у детей одних и тех же родителей (например, Анджали Кумари и Кешав Кумари, видео в конце статьи), иногда ими являются кровные родственники, что тоже может являться причиной сбоя в наследственности.
Прогерия чаще всего не проявляется с рождения, ее признаки дают о себе знать на 2-3 году жизни. Ребенок снижает темпы роста, резко меняется состояние кожи и подкожной клетчатки. Поверхность тела становится сухой и морщинистой, начинают просвечиваться вены. У ребенка увеличивается голова, но не лицо, а нижняя челюсть остается недоразвитой. Развитие организма тоже происходит с явными нарушениями: атрофируются мышцы, выпадают зубы, волосы и ногти, слабым становится костный аппарат и суставы, прекращаются развиваться половые органы, ухудшается зрение.
Ребенок снижает темпы роста, резко меняется состояние кожи и подкожной клетчатки. Поверхность тела становится сухой и морщинистой, начинают просвечиваться вены. У ребенка увеличивается голова, но не лицо, а нижняя челюсть остается недоразвитой. Развитие организма тоже происходит с явными нарушениями: атрофируются мышцы, выпадают зубы, волосы и ногти, слабым становится костный аппарат и суставы, прекращаются развиваться половые органы, ухудшается зрение.
К сожалению, с таким набором болезней человеку трудно бороться – средняя продолжительность жизни при детской прогерии составляет всего 13 лет. Смерть наступает в возрасте от 7 до 27 лет, в истории медицины известны только 2 пациента, прожившие дольше 27 лет.
Прогерия у взрослых обусловлена нарушением гена WRN. Проявляется заболевание в период полового созревания человека, что приводит к замедлению его роста. На третьем десятке жизни начинают седеть и выпадать волосы, развивается катаракта, тонкой становится кожа. На ней появляются язвы. Ротовое отверстие и нос становятся узкими, тем самым лицо начинает быть похожим на маску. В 30-40 лет проявляет себя сахарный диабет, атеросклероз, возможны и злокачественные опухоли. К сожалению, общего лечения нет – врачи борются с конкретными осложнениями в зависимости от силы симптомов. И в данном случае прогерия заканчивается смертью из-за многочисленных осложнений сосудистой системы и злокачественных образований.
Ротовое отверстие и нос становятся узкими, тем самым лицо начинает быть похожим на маску. В 30-40 лет проявляет себя сахарный диабет, атеросклероз, возможны и злокачественные опухоли. К сожалению, общего лечения нет – врачи борются с конкретными осложнениями в зависимости от силы симптомов. И в данном случае прогерия заканчивается смертью из-за многочисленных осложнений сосудистой системы и злокачественных образований.
Уже установлено, что прогерия напрямую связана с молекулярными изменениями в организме, происходящими во время старения. Заболевание называют «синдромом Бенджамина Баттона» в честь киноперсонажа, тоже выглядевшим пожилым в детском возрасте.
К сожалению, человечество еще не нашло способа бороться со старостью, да и нужно ли это? Увы, но некоторые люди начинают стареть не душой, а телом еще в детстве. Но история Сэма Бернса показывает, что любовь к жизни все равно сильнее страха смерти. Американский подросток, который умер от прогерии в возрасте всего 17 лет, много выступал с мотивационными лекциями. Сэм публично рассказывал, как даже с физическими ограничениями пытается радоваться каждому прожитому дню и осуществлять свои мечты.
Сэм публично рассказывал, как даже с физическими ограничениями пытается радоваться каждому прожитому дню и осуществлять свои мечты.
Другие дети с заболеванием прогерия
Адалия Роуз (видео 2018 года, девочке 11 лет, ссылка на Instagram Адалии)
Байезид Хуссейн (видео 2016 года, на видео мальчику всего 4 года)
Анджали Кумари и Кешав Кумари (видео 2016 года, девочке 11 лет, мальчику 1,5 года)
Другие дети с заболеванием прогерия
Адалия Роуз (видео 2018 года, девочке 11 лет, ссылка на Instagram Адалии)
Байезид Хуссейн (видео 2016 года, на видео мальчику всего 4 года)
Анджали Кумари и Кешав Кумари (видео 2016 года, девочке 11 лет, мальчику 1,5 года)
Нихал Битла (видео 2016 года, мальчику 15 лет)
 youtube.com/embed/BPLnZB5LKYk» frameborder=»0″ allowfullscreen=»allowfullscreen»/>
youtube.com/embed/BPLnZB5LKYk» frameborder=»0″ allowfullscreen=»allowfullscreen»/>
Смотрите также:
Детская прогерия: роль эндотелия сосудов
Исследователи из Университета Дюка создали биоинженерную модель кровеносных сосудов, показывающую механизм развития редкой и во многом загадочной болезни — детской прогерии, или синдрома Гетчинсона-Гилфорда. Результаты работы опубликованы в журнале Stem Cell Reports.
Синдром Гетчинсона-Гилфорда приводит к стремительному старению организма в совсем юном возрасте (выделяют также прогерию взрослых, или синдром Вернера). Старение проявляется в выпадении волос, морщинистой коже, дистрофии мышц и подкожной жировой клетчатки, поражении суставов. Но наиболее опасная патология — быстрое развитие атеросклероза, поэтому болезнь приводит к преждевременной смерти, обычно в возрасте от 10 до 20 лет, из-за сердечно-сосудистых патологий. Детская прогерия относится к очень редким заболеваниям, считается, что в настоящее время в мире от нее страдают 250 человек.
Причина болезни — мутация в гене Lamin A, при которой образуется белок прогерин. Он накапливается в клетках кровеносных сосудов, и именно его считают ответственным за развитие атеросклероза. Исследования показали, что больше всего прогерина образуется в гладкомышечных клетках, образующих промежуточный слой стенки сосудов. Но до сих пор оставалась неясной роль клеток эндотелия — внутреннего слоя сосудистой стенки.
Чтобы разобраться в механизме развития атеросклероза при детской прогерии, ученые использовали биоинженерный подход, при котором выращивали модели кровеносных сосудов, используя индуцированные плюрипотентные стволовые клетки (ИПСК). Их получали из фибробластов, перепрограммируя их по стандартному протоколу, а затем, воздействуя на ИПСК определенными факторами роста, направляли их дифференцировку на получение клеток стенки сосудов.
Ранее команда под руководством Джорджа Труски, профессора Университета Дюка, уже разработала 3D-биоинженерную модель кровеносных сосудов, которые были сконструированы из гладкомышечных клеток, выращенных из ИПСК пациентов с прогерией, и эндотелиальных клеток, выращенных из ИПСК здоровых доноров.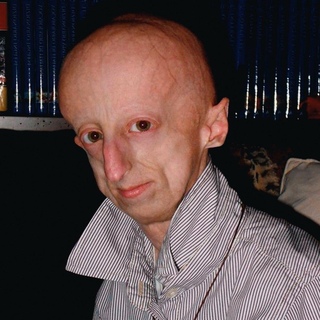 На этой модели они исследовали роль гладкомышечных клеток в развитии заболевания, но она не показывала роль эндотелиальных клеток. Теперь же ученые изменили протокол эксперимента. Они получали ИПСК, перепрограммируя фибробласты пациентов с прогерией, а затем стимулировали дифференцировку клеток по двум путям развития, в результате чего получали от одних и тех же пациентов и гладкомышечные и эндотелиальные клетки сосудов.
На этой модели они исследовали роль гладкомышечных клеток в развитии заболевания, но она не показывала роль эндотелиальных клеток. Теперь же ученые изменили протокол эксперимента. Они получали ИПСК, перепрограммируя фибробласты пациентов с прогерией, а затем стимулировали дифференцировку клеток по двум путям развития, в результате чего получали от одних и тех же пациентов и гладкомышечные и эндотелиальные клетки сосудов.
«Эндотелий накапливает токсичный белок на гораздо более низком уровне, чем слой гладкомышечных клеток сосудов, — объясняет Надя Абуталеб, соавтор статьи, специалист по биомедицинской инженерии из Университета Дюка. — Поэтому ранее исследования были в основном сосредоточены на гладкомышечных клетках, а роль эндотелия в развитии заболевания изучали только в статичной клеточной культуре. Мы впервые использовали для исследования роли эндотелия динамичную 3D модель сосудов».
В динамичной 3D-модели через биоинженерный сосуд протекал ток жидкости.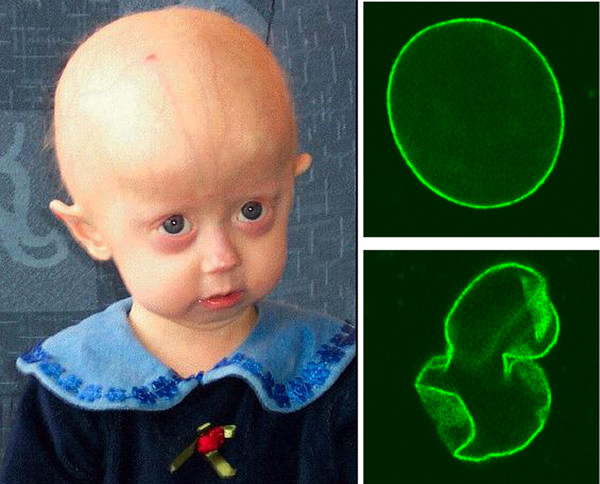 Модель, в которой оба слоя сосудистой стенки были выращены из ИПСК, полученных от пациентов с прогерией, проявляла сниженную способность к вазоконстрикции (сужению) и вазодилатации (расширению) в ответ на воздействие физиологически активных веществ, например, ацетилхолина. Если же в модели сосуда только эндотелиальные клетки были мутантными (то есть получены от пациентов с прогерией), а гладкомышечные получены от здоровых доноров, оставалась сниженной способность к вазодилатации.
Модель, в которой оба слоя сосудистой стенки были выращены из ИПСК, полученных от пациентов с прогерией, проявляла сниженную способность к вазоконстрикции (сужению) и вазодилатации (расширению) в ответ на воздействие физиологически активных веществ, например, ацетилхолина. Если же в модели сосуда только эндотелиальные клетки были мутантными (то есть получены от пациентов с прогерией), а гладкомышечные получены от здоровых доноров, оставалась сниженной способность к вазодилатации.
Ученые также нашли, что в мутантных эндотелиальных клетках прогерин накапливается так же, как и в гладкомышечных. Более того, в эндотелиальных клетках, полученных от пациентов с прогерией, появлялись молекулы, характерные для воспалительных процессов, в частности, молекулы клеточной адгезии CAM-1 и E-selectin. Все говорило о том, что эндотелиальные клетки с мутантным геном играют самостоятельную роль в атеросклеротическом поражении сосудов при прогерии.
Специалисты рассчитывают, что разработанная ими биоинженерная модель не только помогает разобраться в механизмах детской прогерии, но и может быть использована для поиска способов лечения.
«Поскольку эта болезнь очень редка, трудно набрать достаточное количество пациентов для проведения клинических исследований, — подчеркивает профессор Труски. — Мы надеемся, что наша платформа обеспечит альтернативный путь для тестирования потенциальных лекарственных средств».
Девочка-старушка: 7-летняя винничанка с редчайшей болезнью надеется на помощь американских медиков
Уже сейчас у Иринки Химич – букет болячек, на которые обычно жалуются пожилые люди.
Несколько лет назад врачи, наконец, нашли их причину – у малышки преждевременное старение. До совершеннолетия такие дети доживают редко.
«Не выявлено, но не исключено»
Диагноз «прогерия» семья Химич из пригорода Винницы узнала, когда ребенку исполнилось четыре года. До этого безуспешно лечили болезни кожи, искали причины острой реакции на солнце, болей в суставах, облысения… Но правда оказалась хуже любых догадок отечественных врачей.
|
Малышка Ириша, долгожданный первенец, родилась семь лет назад. Незадолго до этого супруги как раз закончили строить дом – взяли кредит на землю, своими руками клали кирпич. Чтобы погасить его, Дина до последних дней беременности ходила на работу.
Новорожденная весила чуть меньше, чем обычно младенцы – 2300 граммов, 52 сантиметра. Но сегодня выхаживают и полукилограммовых деток, так что ничто не могло омрачить счастья новоиспеченных родителей.
Первые звоночки болезни появились в два месяца.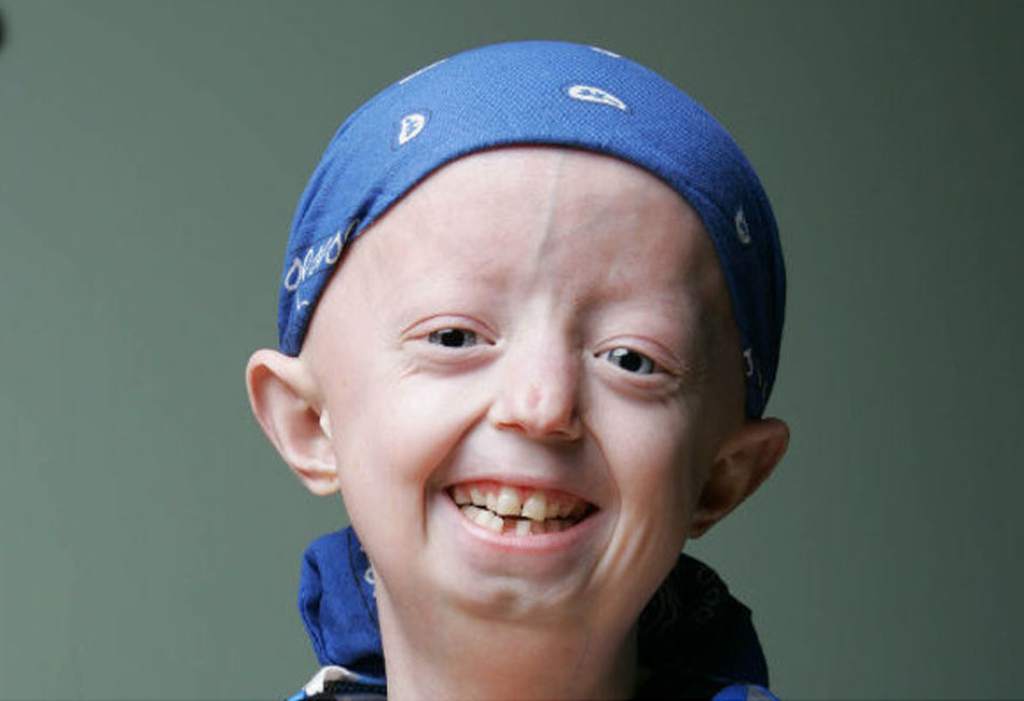 Из-за спазмов девочка не могла даже сама сходить в туалет. Еще через два месяца признаки чего-то нехорошего стали более заметны – Ириша не могла ползать из-за чрезмерно стянутой кожи, ножки выворачивало, совершенно не было подкожного жира, немного мышц и кожа — как будто обожженная, ни с того ни с чего возникали кровотечения. Позже появилась боль в глазах от солнечных лучей, боль в суставах, в два года исчезли толком не успевшие вырасти волосы, а в пять лет у малышки случился инсульт.
Из-за спазмов девочка не могла даже сама сходить в туалет. Еще через два месяца признаки чего-то нехорошего стали более заметны – Ириша не могла ползать из-за чрезмерно стянутой кожи, ножки выворачивало, совершенно не было подкожного жира, немного мышц и кожа — как будто обожженная, ни с того ни с чего возникали кровотечения. Позже появилась боль в глазах от солнечных лучей, боль в суставах, в два года исчезли толком не успевшие вырасти волосы, а в пять лет у малышки случился инсульт.
— И днем, и ночью ребенок кричал смертным криком, и даже когда засыпала – не умолкала, — рассказывает «КП» в Украине» мама девочки Дина Химич. На первый взгляд, женщина держится, для других такой диагноз у ребенка – ужас, а для нее – уже привычные будни. – Только гораздо позже, когда начала говорить, наконец сказала, где болит и что не так. А тогда же я не знала, как вообще дотянуть до утра и что делать. Кожа была красной, тоненькой, слазила, мы даже не одевали ее, чтобы не было трения об одежду. И в год впервые повезли в Берегово на термальные воды. Там Ира начала ходить, смогла самостоятельно и на горшок, суставы не так болели.
И в год впервые повезли в Берегово на термальные воды. Там Ира начала ходить, смогла самостоятельно и на горшок, суставы не так болели.
|
Что происходит с девочкой, врачи даже не могли объяснить. Одни прямо говорили – никогда такого не видели, боимся навредить. Другие измышляли что-то насчет различных заболеваний. В конце концов, посчитали, что это системная склеродермия – заболевание соединительной ткани с поражением кожи, сосудов, органов и костей. Куча анализов, ни один не подтверждался.
— Врачи писали: «Не найдено, но не исключено». Думай, что хочешь – есть это заболевание или нет, — продолжает Дина Химич. – А когда поставили системную склеродермию, я повезла дочку в Москву в дерматологическую клинику. Ей было четыре года. Там мы впервые услышали о такой болезни как прогерия. Возникли подозрения, что мы имеем дело именно с ней. Нас направили к генетикам. Не сразу, но диагноз подтвердился.
– А когда поставили системную склеродермию, я повезла дочку в Москву в дерматологическую клинику. Ей было четыре года. Там мы впервые услышали о такой болезни как прогерия. Возникли подозрения, что мы имеем дело именно с ней. Нас направили к генетикам. Не сразу, но диагноз подтвердился.
Надежда на бостонских врачей, похоронивших сына с прогерией
Прогерия, редчайший генетический дефект – это, по-простому, преждевременное старение организма. Как лечить – неизвестно. И лечится ли вообще – тоже. Несколько таких деток было в Москве, но с началом боевых действий на востоке контакты с тамошними клиниками сошли на нет. Просто не отвечали. В мире таких больных – чуть больше трех сотен.
Химичи не теряют надежды. Вышли на клинику в Бостоне, врачи которой консультировали украинских о прогерии. Сэм, сын бостонской врача Лесли Гордон, также страдал от этой болезни. Почти никакой информации о ней не было и тогда, в 1998-м. Семья медиков погрузилась в вопросы лечения и замедления процесса старения, создали свой фонд помощи таким больным и их родным, искали финансирование для исследований этой болезни. Сэм умер в 16 лет. Его смерть только утвердила родителей в решимости продолжать поиск способа лечения. Лесли Гордон прислала Дине электронную книгу о том, как ухаживать за такими больными и дома, и в клиниках. Ее женщина разослала многим специализированным и нет украинским больницам.
Сэм умер в 16 лет. Его смерть только утвердила родителей в решимости продолжать поиск способа лечения. Лесли Гордон прислала Дине электронную книгу о том, как ухаживать за такими больными и дома, и в клиниках. Ее женщина разослала многим специализированным и нет украинским больницам.
— В Бостоне медики попытались заново формировать ДНК, испытывали это на животных, экспериментально и на детях уже пару лет, — говорит Дина Химич. – Я подала анкету, чтобы нас включили в эксперимент, ее утвердили. Лесли была удивлена, что ребенок в таком состоянии только благодаря моему уходу. Когда уже были готовы лететь, оказалось, что сосуды в очень плохом состоянии. Надо подлечить, чтобы перенесла полет.
В комнату то и дело вбегает худенькая Ириша – интересуется мамиными делами. В семь лет она весит всего 10 кг 800 г. Заболеет, понервничает – 10 кг 500 г. Потом эти 300 граммов набирают месяцами. Поэтому ее болезнь при ней мама не обсуждает – девочке противопоказаны негативные эмоции, стресс. Из-за переживаний дочки в отсутствие мамы Дине пришлось совсем уйти с работы. Сейчас к Ирише как раз пришли друзья – общительная, дети ее любят. А она друзьям отдаст последнюю конфету.
Из-за переживаний дочки в отсутствие мамы Дине пришлось совсем уйти с работы. Сейчас к Ирише как раз пришли друзья – общительная, дети ее любят. А она друзьям отдаст последнюю конфету.
— Врачи не знают даже, как у нее лечить ангину, организм совершенно непредсказуемо реагирует на простые лекарства, — понижает голос Дина. – У таких деток старение за год прогрессирует, как за 5, 7, 10 лет – как у кого. Еще в четыре месяца мне говорили, что она – не жилец. Но ей уже семь, и я верю, что организм переборет болезнь.
|
«Ногти на руках спасла – а на ногах не успеха»
Муж-бухгалтер пошел вкалывать на стройку – больше платят. Они одни, родители далеко. Помощи ждать неоткуда. Все это время тянули ребенка из последних сил. Дважды в год возят то в Берегово, то на глицериновое озеро в Ровенской области. Специально для купания везут воду из Йосафатовой долины в области – после нее кожа не краснеет и не болит. Объездили здравницы и святые места. Водят на кружки, в дошкольную группу, в бассейн, к репетиторам – делают все, чтобы ребенок развивался не хуже других. За все, конечно, платят. С пенсии по инвалидности ребенка и зарплаты строителя.
Они одни, родители далеко. Помощи ждать неоткуда. Все это время тянули ребенка из последних сил. Дважды в год возят то в Берегово, то на глицериновое озеро в Ровенской области. Специально для купания везут воду из Йосафатовой долины в области – после нее кожа не краснеет и не болит. Объездили здравницы и святые места. Водят на кружки, в дошкольную группу, в бассейн, к репетиторам – делают все, чтобы ребенок развивался не хуже других. За все, конечно, платят. С пенсии по инвалидности ребенка и зарплаты строителя.
Дина, всхлипывая, пересказывает все тяготы, с которыми сталкивается ежедневно. Одежды для такого ребенка нет – перешивают. Обуви – тем более, у девочки одни тапки на все случаи жизни. Ножка просто тонет в обычной детской обувке – нет пятки, фактически кость и кожа. Ортопедическая бесплатная обувь от государства не подходит. В ней стирает ступни до крови. Носят на руках или подкладывают носок. Мама не скрывает – именно сейчас им нужна помощь.
— У таких детей очень многое зависит от времени. Ногти на руках я спасла, а на ногах не успела, — мама срывается на плач. – В бассейн ходим, чтобы работали суставы. Одна такая девочка села – и суставы заклинило на 90 градусах. Она так и ходит, на полусогнутых. Сколько у меня заняло времени, чтобы Ира смогла открыть рот и взять обычную ложку — еще недавно кормила ее десертной.
Ногти на руках я спасла, а на ногах не успела, — мама срывается на плач. – В бассейн ходим, чтобы работали суставы. Одна такая девочка села – и суставы заклинило на 90 градусах. Она так и ходит, на полусогнутых. Сколько у меня заняло времени, чтобы Ира смогла открыть рот и взять обычную ложку — еще недавно кормила ее десертной.
Ушло все, что было, что могли – продали, родители, пока могли, помогали. Хотя мы не были бедными. Мы сделали все, что могли, что от нас зависело. Но наша проблема, да и проблемы детей с другими болезнями, никого не интересуют.
Откуда деньги на дорогое лечение, обследования, поездки в Москву, планы лететь в Бостон? Все делали под залог дома. Затем отдают долги. Правда, на Бостон уже нужен спонсор, совсем другие расценки.
И все же Дина уверена, что вырвет Иринку из лап неизвестного чудовища по имени Прогерия.
КАК ПОМОЧЬ
Карта ПриватБанка мамы Химич Дина Сергеевна 4149 4978 6719 3940
Новая надежда в борьбе с редкой детской генетической болезнью
В апреле 2011 г. , в возрасте всего 10 мес уроженке штата Огайо Карли Кудзии (Carly Kudzia) был поставлен диагноз «прогерия». Это редкое и смертельное генетического заболевание, которое еще известно как болезнь преждевременного старения. Встречающаяся всего у 1 из 20 млн человек, прогерия обусловлена генетической мутацией, которая приводит к огромному избытку белка, известного как прогерин. Этот излишек вызывает быстро прогрессирующее клеточное повреждение, кульминацией которого является необратимая сердечная недостаточность. На сегодня не существует никакого специфического лечения болезни. При этом пациенты с прогерией редко доживают до своего 14-го дня рождения.
, в возрасте всего 10 мес уроженке штата Огайо Карли Кудзии (Carly Kudzia) был поставлен диагноз «прогерия». Это редкое и смертельное генетического заболевание, которое еще известно как болезнь преждевременного старения. Встречающаяся всего у 1 из 20 млн человек, прогерия обусловлена генетической мутацией, которая приводит к огромному избытку белка, известного как прогерин. Этот излишек вызывает быстро прогрессирующее клеточное повреждение, кульминацией которого является необратимая сердечная недостаточность. На сегодня не существует никакого специфического лечения болезни. При этом пациенты с прогерией редко доживают до своего 14-го дня рождения.
«Карли — по большей части — как и любой другой семилетний ребенок, — говорит Хизер Ансингер (Heather Unsinger), мать Карли. — Она остроумная, артикулирующая, счастливая, энергичная, яростно независимая и очень активная. Список вещей, которые она может сделать, настолько большой, что мы не думаем о том, чего мы не можем делать».
Доктор Лесли Гордон (Leslie Gordon), соучредитель и медицинский директор некоммерческого Фонда исследований прогерии (Progeria Research Foundation), и ее коллеги из университетов Гарварда (Harvard Universities) и Брауна (Brown Universities) провели новое небольшое клиническое исследование, результаты которого очень обнадеживают. Исследование сосредоточено на экспериментальном препарате, известном как лонафарниб. Это лекарственное средство, которое изначально разработано для борьбы с раком, ингибирует активность фермента, который имеет решающее значение для производства прогерина. При этом он перестает прикреплять прогерин к клеточным мембранам пациента, что предотвращает повреждение клетки.
Исследование сосредоточено на экспериментальном препарате, известном как лонафарниб. Это лекарственное средство, которое изначально разработано для борьбы с раком, ингибирует активность фермента, который имеет решающее значение для производства прогерина. При этом он перестает прикреплять прогерин к клеточным мембранам пациента, что предотвращает повреждение клетки.
В новом исследовании 27 пациентов с прогерией ежедневно 2 раза сутки принимали таблетки лонафарниба. В среднем лечение продолжалось чуть более 2 лет, в течение которых в этой группе умер 1 пациент. В тот же период времени 9 из 27 пациентов с прогерией, не получавших лечение лонафарнибом, умерли.
Побочные эффекты обычно возникали в течение первых нескольких месяцев лечения и включали диарею, тошноту и потерю аппетита.
Вторая группа из 63 пациентов продолжает принимать лонафарниб. До сих пор только 4 из 63 пациентов, принимающих лонафарниб, умерли, по сравнению с 17 смертями среди 63 пациентов, не получающих лечения.
«Лонафарниб — это лечение, а не панацея и не способ излечить заболевание, — подчеркнула Л. Гордон. — Но это первая поддержка, которую мы можем предоставить для влияния на продолжительность жизни детей с прогерией с помощью одного лекарства».
Л. Гордон добавила, что результаты, опубликованные в выпуске журнале «Journal of the American Medical Association» от 24 апреля, чрезвычайно обнадеживают. Она также отметила, что препараты для проведения исследования бесплатно предоставляла компания-разработчик Merck и фонд намерен «отстаивать позицию, что все семьи с данным заболеванием будут получать лекарство бесплатно». По данным фонда, в настоящее время зарегистрировано 144 случая прогерии в 45 странах.
Х. Ансингер на вопрос, что для нее представляет собой препарат лонафарниб, ответила следующее: «Надежду, что у меня будет больше времени с моей дочерью. Когда тело стареет в 8–10 раз быстрее обычного, насколько важен дополнительный год, два, пять или десять? Полученные результаты исследования переводят мои убеждения из ранга исключительно надежды в ранг частичной надежды и частичной реальности».
Эта мысль была поддержана соавтором исследования, доктором Фуки Мари Хисамой (Fuki Marie Hisama), профессором медицинской генетики на кафедре медицины Вашингтонского университета (University of Washington), США. «Эти результаты являются первыми в своем роде для этой редкой болезни и представляют собой значительный прорыв. Это исследование дает надежду детям с прогерией и их семьям на улучшение результатов в будущем», — отметила она.
По материалам www.medicinenet.com
Редкие эндокринные заболевания в СПб в клинике «Аванта».
Очень редко встречающиеся заболевания называются орфанными. Несмотря на то, что их не так много и встречаются они лишь у небольшого количество человек, среди эндокринных заболеваний их довольно много. Наиболее частыми причинами возникновения редких эндокринных заболеваний являются генетический или аутоиммунный факторы, а причины некоторых иногда просто еще неизвестны. Именно поэтому многие люди сейчас предпочитают проводить генетическое обследование близких родственников и детей, чтобы выявить факторы риска и возможные редкие или опасные заболевания.
Перечень редких эндокринных заболеваний
Министерство здравоохранения Российской Федерации выпустило полный список орфанных заболеваний известных на сегодняшний день. Среди эндокринных заболеваний названы:
- Врожденный гиперинсулинизм;
- Соматотропинома;
- Пролактинома;
- Гипопитуитаризм;
- Кортикотропинома;
- Адреногенитальный синдром;
- Гермафродитизм;
- Синдром Ларона;
- Прогерия;
- Фенилкетонурия;
- Алкаптонурия;
- Синдром Хермански-Пудлака;
- Болезнь «кленового сиропа»;
- Цистиноз;
- Орнитинемия;
- Гликогенозы;
- Галактоземия;
- Болезнь Фабри и пр.
К сожалению, перечень настолько обширный, что привести его полностью просто невозможно.
Лечение редких эндокринных заболеваний
Очень часто люди с редкими эндокринными заболеваниями испытывают особую трудность в поиске грамотного специалиста, который окажет необходимую помощь, довольно часто затруднено проведение первичной диагностики, так как для этого необходимо посетить крупные медицинские центры, не всегда науке известны способы лечения.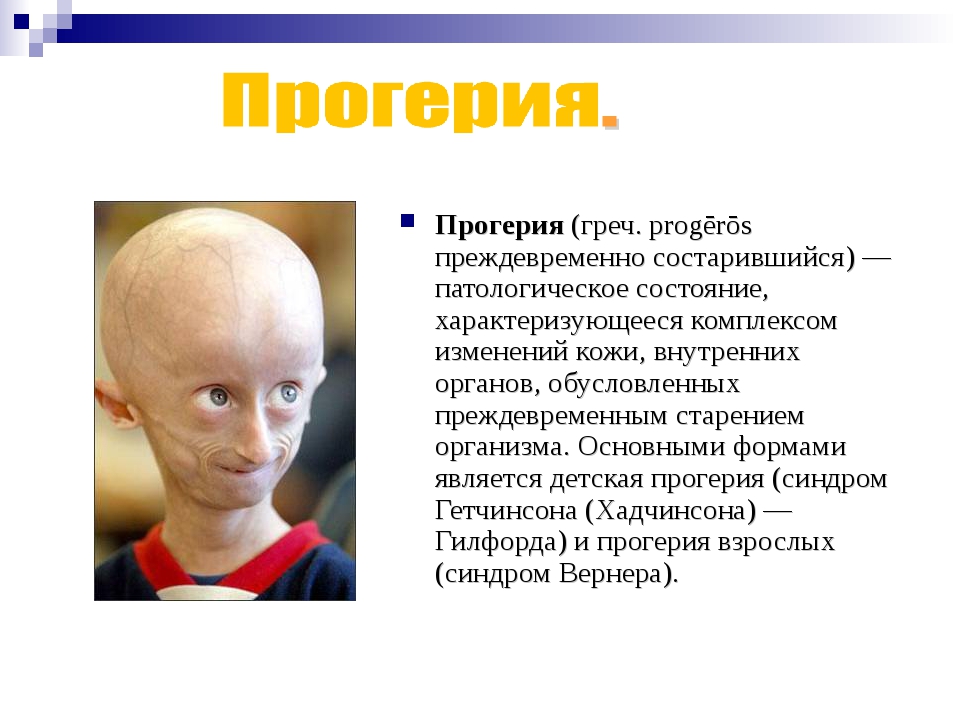 Также крайне сложно получить достоверную информацию о заболевании, просто потому, что таких случаев довольно мало.
Также крайне сложно получить достоверную информацию о заболевании, просто потому, что таких случаев довольно мало.
Среди общих способ лечения эндокринных заболеваний можно назвать:
- Хирургическое удаление гипофиза;
- Облучение;
- Гормональная терапия;
- Удаление опухолевых образований;
- Удаление надпочечников;
- Медикаментозное лечение;
- Различные виды заместительной терапии и др.
Например, синдром МЭН поддается лечению посредством удаления новообразований и восстановления гормонального баланса. А лечение гастриномы предполагает химиотерапию, оперативное вмешательство для удаления опухолей или резекцию желудка.
В медицинском центре «Аванта» Вы сможете проконсультироваться с различными специалистами, и в первую очередь с эндокринологом , сделать возможные клинические обследования и получить рекомендации по дальнейшему лечению.
Когда следует обратиться к врачу?
В подавляющем большинстве случаев редкие эндокринные заболевания дают о себя знать при рождении или в детстве.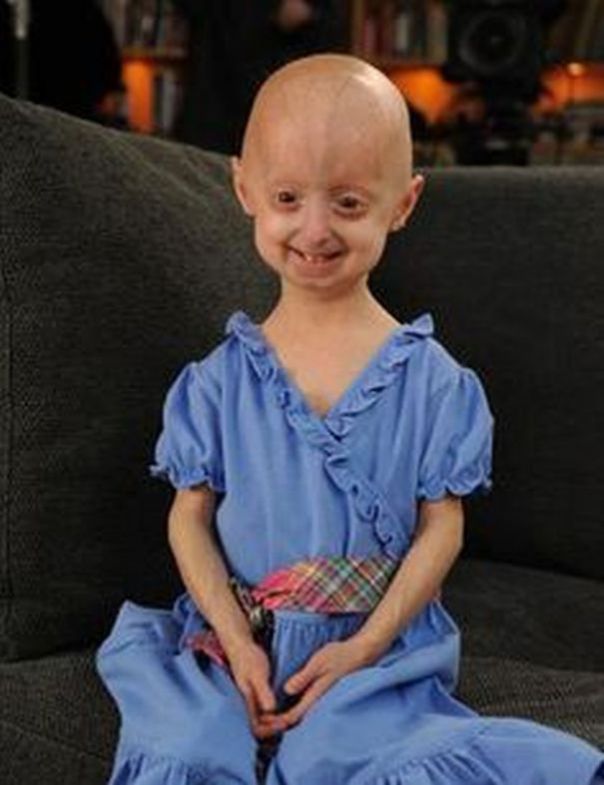 Редко так случается, что первые симптомы проявляются во взрослом возрасте. Основными симптомами редких эндокринных заболеваний становятся различные изменения во внешности или поведении, выбивающиеся из понятия нормы. Именно поэтому крайне трудно привести какие-то более конкретные симптомы.
Редко так случается, что первые симптомы проявляются во взрослом возрасте. Основными симптомами редких эндокринных заболеваний становятся различные изменения во внешности или поведении, выбивающиеся из понятия нормы. Именно поэтому крайне трудно привести какие-то более конкретные симптомы.
| 1 | X-сцепленная адренолейкодистрофия |
| 2 | ААА синдром, Оллгрова синдром (ахалазия, алакримия, недостаточность надпочечников |
| 3 | Аарскога-Скотта cиндром |
| 4 | Абиотрофия сетчатки, тип Франческетти |
| 5 | Адреногенитальный синдром (врожденная гиперплазия коры надпочечников) |
| 6 | Азооспермия |
| 7 | Айкарди-Гутьереса синдром |
| 8 | Акродерматит энтеропатический |
| 9 | Аксенфельда-Ригера синдром |
| 10 | Алажиля синдром |
| 11 | Александера болезнь |
| 12 | Альбинизм глазокожный |
| 13 | Алькаптонурия |
| 14 | Альстрема синдром |
| 15 | Аменорея |
| 16 | Альфа-1-антитрипсина недостаточность |
| 17 | Ангельмана синдром |
| 18 | Андерсена синдром |
| 19 | Анемия Даймонда-Блекфена |
| 20 | Анеуплоидии |
| 21 | Аниридия |
| 22 | Антли-Бикслера синдром |
| 23 | Апера синдром |
| 24 | Арта cиндром |
| 25 | Артрогрипоз дистальный (синдром Фримена-Шелдона) |
| 26 | Атаксия Фридрейха |
| 27 | Атаксия, хорея, судороги и деменция |
| 28 | Атрофия зрительного нерва Лебера |
| 29 | Атрофия зрительного нерва с глухотой |
| 30 | Аутоиммунный лимфопролиферативный синдром |
| 31 | Аутоиммунный полиграндулярный синдром I типа |
| 32 | Аутоимунный полиэндокринный синдром |
| 33 | Афазия первичная прогрессирующая |
| 34 | Ахондроплазия |
| 35 | Баллера-Герольда синдром |
| 36 | Банаян-Райли-Рувалькаба cиндром |
| 37 | Барде-Бидля (Ларенса-Муна) синдром |
| 38 | Барта cиндром |
| 39 | Барттера синдром |
| 40 | Бёрта-Хога-Дьюба синдром |
| 41 | Бесплодие |
| 42 | Беста болезнь |
| 43 | Биотинидазы недостаточность |
| 44 | Блефарофимоз, обратный эпикант и птоз |
| 45 | Блоха-Сульцбергера синдром |
| 46 | Блума синдром |
| 47 | Боковой амиотрофический склероз |
| 48 | Боуэна-Конради синдром |
| 49 | Бранхио-окуло-фациальный синдром |
| 51 | Брахидактилия |
| 53 | Бьёрнстада синдром |
| 54 | Ваарденбурга синдром |
| 55 | Ваарденбурга-Шаха синдром |
| 56 | Ван дер Вуда синдром |
| 57 | Велокардиофациальный синдром |
| 58 | Вернера синдром |
| 59 | Видеманна-Беквита синдром, спорадическая нефробластома |
| 60 | Виллебранда болезнь |
| 61 | Вильсона-Коновалова болезнь |
| 62 | Вильямса cиндром |
| 63 | Вискотта-Олдрича cиндром |
| 64 | Вольмана болезнь, болезнь накопления эфиров холестерина |
| 65 | Вольфа-Хиршхорна синдром |
| 67 | Врожденная нечувствительность к боли с ангидрозом (врожденная сенсорная нейропатия с ангидрозом, HSAN4, CIPA) |
| 68 | Врожденной центральной гиповентиляции синдром |
| 69 | Вульгарный ихтиоз |
| 70 | Галактоземия тип I |
| 71 | Галактоземия тип II |
| 72 | Галактоземия тип III |
| 73 | Галактосиалидоз |
| 74 | Галлервордена-Шпатца болезнь |
| 75 | Ганглиозидоз GM1 тип 1,2,3 |
| 76 | Гастроинтестинальный полипоз |
| 77 | Гелеофизическая дисплазия |
| 78 | Гемофилия |
| 79 | Гемохроматоз наследственный |
| 80 | Генитопателлярный синдром |
| 81 | Германски-Пудлака синдром |
| 82 | Герстманна-Штреусслера-Шейнкера болезнь |
| 83 | Гидроцефалия, обусловленная врожденнным стенозом Сильвиева водопровода |
| 84 | Гипер-IgD синдром |
| 85 | Гипер-IgM синдром |
| 86 | Гиперкалиемический периодический паралич |
| 87 | Гипероксалурия тип I |
| 88 | Гиперорнитинемии-гипераммониемии-гомоцитрулинурии синдром (ННН синдром) |
| 89 | Гипертрофическая кардиомиопатия |
| 90 | Гиперфенилаланинемия с дефицитом тетрагидробиоптерина |
| 91 | Гиперхолестеринемии |
| 92 | Гипогонадизм |
| 93 | Гипокалиемический периодический паралич |
| 94 | Гипоспадия |
| 95 | Гипотрихоз |
| 96 | Гипофосфатазия |
| 97 | Гипофосфатемический рахит |
| 98 | Гипохондроплазия |
| 99 | Гиппеля-Линдау синдром |
| 100 | Глазо-зубо-пальцевой синдром |
| 101 | Глаукома врожденная |
| 102 | Глаукома ювенильная открытоугольная |
| 103 | Гликогеноз 0 тип |
| 104 | Гликогеноз III типа |
| 105 | Гликогеноз IV типа |
| 106 | Гликогеноз IX типа |
| 107 | Гликогеноз Iа тип |
| 108 | Гликогеноз Iв тип |
| 109 | Гликогеноз V типа |
| 110 | Гликогеноз VI типа |
| 111 | Гликогеноз XI типа, Фанкони-Бикеля синдром |
| 112 | Гломеруоцитоз почек гипопластического типа |
| 113 | Глутаровая ацидурия тип 1 |
| 114 | Глутаровая ацидурия тип 2 |
| 116 | Гомоцистинурия |
| 117 | Гоше болезнь тип 1,2,3 |
| 118 | Грейга cиндром |
| 119 | Грисцелли cиндром |
| 120 | Дауна cиндром |
| 121 | Делеции хромосомы 1p36 синдром |
| 122 | Десмоидные опухоли |
| 123 | Дефицит гормона гипофиза, комбинированный |
| 124 | Дефицит иммуноглобулина A |
| 125 | Дефицит карнитина системный первичный |
| 126 | Дефицит фактора F12 |
| 127 | Джексона-Вейсса cиндром |
| 128 | Ди Джорджи cиндром |
| 129 | Диастрофическая дисплазия |
| 130 | Дисгенезия гонад |
| 131 | Дисплазия де ля Шапеля (Ателостеогенез) |
| 132 | Дисплазия Книста |
| 133 | Дистальная моторная нейропатия |
| 134 | Дистальная спинальная амиотрофия врожденная с параличом диафрагмы |
| 135 | Дисхондростеоз Лери-Вейлля |
| 136 | Дорфмана-Чанарина синдром |
| 137 | Жильбера cиндром |
| 138 | Жубер cиндром |
| 139 | Задержка полового созревания |
| 140 | Зандхоффа болезнь |
| 141 | Изовалериановая ацидемия |
| 142 | Инверсия пола |
| 143 | Ихтиоз буллезный |
| 144 | Ихтиоз врожденный аутосомно-рецессивный |
| 145 | Ихтиоз вульгарный |
| 146 | Ихтиоз, спастическая квадриплегия и умственная отсталость |
| 147 | Кампомелическая дисплазия |
| 148 | Канавана болезнь |
| 149 | Карбамолфосфатсинтетазы недостаточность |
| 150 | Карпентера cиндром |
| 151 | Кератита-ихтиоза-тугоухости cиндром |
| 152 | Кернса-Сейра синдром |
| 153 | Клайнфельтера cиндром |
| 154 | Клиппеля-Фейля cиндром |
| 155 | Коккейна cиндром |
| 156 | Комбинированный дефицит витамин K-зависимых факторов свертывания крови |
| 157 | Косолапость врожденная с или без дефицита длинных костей и/или зеркальной полидактилией |
| 158 | Костелло cиндром |
| 159 | Костная гетероплазия прогрессирующая |
| 160 | Коудена болезнь |
| 161 | Коффина-Лоури синдром |
| 162 | Кошачьего глаза синдром |
| 163 | Кошачьего крика синдром |
| 164 | Краббе болезнь |
| 165 | Краниометафизарная дисплазия |
| 166 | Краниосиностоз |
| 167 | Краниофациальной дисморфии-глухоты-ульнарной девиации кистей синдром |
| 168 | Крейтцфельда-Якоба болезнь |
| 169 | Криглера-Найара синдром |
| 170 | Крипторхизм |
| 171 | Крузона с черным акантозом синдром |
| 172 | Крузона синдром |
| 173 | Куррарино синдром |
| 174 | Ларинго-онихо-кутанный синдром |
| 175 | Лейкодистрофия с гипомиелинизацией |
| 176 | Лейкоэнцефалопатия с «исчезающим» белым веществом, детская атаксия с гипомиелинизацией |
| 177 | Лейкоэнцефалопатия с пораженим ствола мозга и высоким уровнем лактата при спектроскопии |
| 178 | Лейкоэнцефалопатия с субкортикальными кистами |
| 179 | Лейциноз (болезнь «с запахом кленового сиропа мочи» |
| 180 | Лермитт-Дуклос болезнь |
| 181 | Леша-Найяна синдром |
| 182 | Ли синдром |
| 183 | Ли-Фраумени синдром |
| 184 | Линча синдром (наследственный неполипозный рак толстой кишки) |
| 185 | Липодистрофия врожденная генерализованная |
| 186 | Липодистрофия семейная частичная |
| 187 | Липопротеин липазы недостаточность |
| 188 | Лоу синдром |
| 189 | Люджина — Фринса синдром |
| 190 | Макла-Уэллса синдром |
| 191 | Маклеода синдром |
| 192 | Малан синдром |
| 193 | Мандибулоакральная дисплазия с липодистрофией |
| 194 | Маннозидоз альфа |
| 195 | Маринеску-Шегрена синдром |
| 196 | Мартина-Белл, УО FRAXA Синдром |
| 197 | Маршалла-Смита синдром |
| 198 | Мевалоновая ацидурия |
| 200 | Метатропная дисплазия (OMIM 156530) |
| 201 | Метахроматическая лейкодистрофия |
| 202 | Метгемоглобинемия |
| 203 | Метилмалоновая ацидурия |
| 204 | Микрофтальм изолированный |
| 205 | Микрофтальм с катарактой |
| 206 | Микроцефалии с капиллярными мальформациями синдром |
| 207 | Миллера-Дикера синдром |
| 208 | Милроя болезнь (лимфедема наследственная) |
| 209 | Миоклоническая дистония |
| 210 | Миоклоническая эпилепсия Лабофа |
| 211 | Миопатия Броди |
| 212 | Миопатия Миоши |
| 213 | Миотоническая дистрофия |
| 214 | Миотония Томсена/Беккера |
| 215 | Митохондриальные гепатопатии |
| 216 | Митохондриальные заболевания, связанные с мутациями в гене POLG |
| 217 | Митохондриальные энцефаломиопатии, связанные с мутациями мтДНК |
| 218 | Митохондриальные энцефаломиопатии, связанные с мутациями ядерных генов |
| 219 | Множественная сульфатазная недостаточность |
| 220 | Множественной эндокринной неоплазии второго типа (МЭН2) cиндром |
| 221 | Множественные вывихи суставов, задержка роста, черепно-лицевые аномалии и врожденные пороки сердца |
| 222 | Множественных птеригиумов синдром |
| 223 | Множественных синостозов синдром |
| 224 | Молибденового кофактора недостаточность |
| 225 | Монилетрикс |
| 226 | Моуат-Вильсон cиндром |
| 227 | Муковисцидоз |
| 228 | Муколипидоз II, III типа |
| 229 | Мукополисахаридоз I типа |
| 230 | Мукополисахаридоз II типа |
| 231 | Мукополисахаридоз III А, В, С, D типа |
| 232 | Мукополисахаридоз IV A, B типа |
| 233 | Мукополисахаридоз VI типа |
| 234 | Мукополисахаридоз VII типа |
| 235 | Мышечная дистрофия врождённая, интегрин А7 негативная |
| 236 | Мышечная дистрофия врожденная, мерозин-негативная |
| 237 | Мышечная дистрофия врожденная, тип 1C |
| 238 | Мышечная дистрофия Дюшенна/Беккера |
| 239 | Мышечная дистрофия поясноконечностная |
| 240 | Мышечная дистрофия тип Фукуяма |
| 241 | Мышечная дистрофия Эмери-Дрейфуса |
| 242 | Мюнке синдром |
| 243 | Накопление нейтральных липидов с миопатией |
| 244 | Нарушение формирования пола |
| 245 | Нанизм MULIBRAY |
| 246 | Нарушения гликозилирования тип 1a, синдром Жакена |
| 247 | Нарушения гликозилирования тип Ib (ген MPI) |
| 248 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I |
| 249 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II |
| 250 | Наследственная нейропатия с подверженностью параличу от сдавления |
| 251 | Наследственная оптическая нейропатия Лебера |
| 252 | Наследственные глаукомы, аномалия Петерса, дермоид роговицы |
| 253 | Наследственный амилоидоз |
| 254 | Наследственный ангионевротический отек |
| 255 | Наследственный панкреатит |
| 256 | Невынашивание беременности |
| 257 | Наследственный рак желудка |
| 258 | Недостаточность N-ацетилглютаматсинтазы |
| 259 | Недостаточность длинноцепочечной 3-гидроксиацил-КоА-дегидрогеназы жирных кислот |
| 260 | Недостаточность короткоцепочечной ацил-КоА-дегидрогеназы жирных кислот |
| 261 | Недостаточность очень длинноцепочечной ацил-КоА дегидрогеназы жирных кислот |
| 262 | Недостаточность синтетазы голокарбоксилаз |
| 263 | Недостаточность среднецепочечной ацил-КоА-дегидрогеназы жирных кислот |
| 264 | Недостаточность сукцинил-КоА:3-кетоацил-КоА трансферазы |
| 265 | Незаращение родничков |
| 266 | Нейроаксональная дистрофия |
| 267 | Нейродегенерация с накоплением железа 4 |
| 268 | Нейромиотония и аксональная нейропатия |
| 269 | Нейрональный цероидный липофусциноз тип 1 |
| 270 | Нейрональный цероидный липофусциноз тип 2 |
| 271 | Нейросенсорная несиндромальная тугоухость |
| 272 | Нейрофиброматоз 1 и 2 типов |
| 273 | Нейтропения тяжёлая врождённая |
| 274 | Некетотическая гиперглицинемия |
| 275 | Некомпактного левого желудочка cиндром |
| 276 | Немалиновая миопатия |
| 277 | Нефронофтиз |
| 278 | Нефротический синдром |
| 279 | Ниймеген cиндром |
| 280 | Ниманна-Пика тип А и В болезнь |
| 281 | Ниманна-Пика тип С болезнь |
| 282 | Ногтей-надколенника синдром |
| 283 | Норри болезнь |
| 284 | Нунан синдром |
| 285 | Олигозооспермия тяжелой степени |
| 286 | Окулофарингеальная мышечная дистрофия |
| 287 | Опица GBBB синдром |
| 288 | Опица-Каведжиа синдром |
| 289 | Опухоль Вильмса |
| 290 | Орнитинтранскарбамилазы недостаточность |
| 291 | Ослера-Рендю-Вебера cиндром |
| 292 | Остеолиз карпотарзальный, мультицентрический |
| 293 | Остеопетроз рецессивный (мраморная болезнь костей) |
| 294 | Паллистера-Киллиана cиндром |
| 295 | Паллистера-Холла cиндром |
| 296 | Палочко-колбочковая дистрофия |
| 297 | Пантотенат киназы недостаточность |
| 298 | Парамиотония Эйленбурга |
| 299 | Патау cиндром |
| 300 | Пейтца-Егерса синдром |
| 301 | Пелицеуса-Мерцбахера болезнь |
| 302 | Пендреда Синдром |
| 303 | Первичная аутосомно-рецессивная микроцефалия, тип 5 |
| 304 | Первичная гипертрофическая остеоартропатия (пахидермопериостоз) |
| 305 | Первичная легочная гипертензия |
| 306 | Периодическая болезнь |
| 307 | Пигментная дегенерация сетчатки |
| 308 | Пикнодизостоз |
| 309 | Пирсона синдром |
| 310 | Пневмоторакс первичный спонтанный |
| 311 | Подколенного птеригиума cиндром |
| 312 | Полидактилия |
| 313 | Поликистоз почек |
| 314 | Помпе болезнь |
| 315 | Понтоцеребеллярная гипоплазия |
| 316 | Потоцки-Лупски cиндром |
| 317 | Почечная адисплазия |
| 318 | Прадера-Вилли Синдром |
| 319 | Преждевременная недостаточность яичников |
| 320 | Прогерия Хатчинсона-Гилфорда |
| 321 | Прогрессирующая наружная офтальмоплегия, АД и АР |
| 322 | Пропионовая ацидемия |
| 323 | Псевдоахондроплазия |
| 324 | Псевдоксантома эластическая |
| 325 | Пфайффера cиндром |
| 326 | Рабдомиолиз (миоглобинурия) |
| 327 | Рак молочной железы |
| 328 | Рак почки |
| 329 | Рак щитовидной железы. Синдром множественной эндокринной неоплазии второго типа (МЭН2) |
| 330 | Рак яичников |
| 331 | Ретинобластома |
| 332 | Ретиношизис |
| 333 | Ретта синдром |
| 334 | Рефсума болезнь |
| 335 | Ригидного позвоночника cиндром |
| 336 | Робинова синдром |
| 337 | Ротмунда-Томсена синдром |
| 338 | Рубинштейна-Тейби синдром |
| 339 | Семейная периодическая лихорадка |
| 340 | Семейный аденомоматозный полипоз, полипозный рак толстой кишки |
| 341 | Семейный внутрипеченочный холестаз 1 типа |
| 342 | Семейный внутрипеченочный холестаз 2 типа ( Баллера болезнь) |
| 343 | Семейный внутрипеченочный холестаз 3 типа |
| 344 | Семейный гемофагоцитарный лимфогистиоцитоз |
| 345 | Семейный медуллярный рак щитовидной железы |
| 346 | Семейный рак толстой кишки |
| 347 | Семейный холодовой аутовоспалительный синдром |
| 348 | Сениора-Локена синдром |
| 349 | Сенсорная полинейропатия (врожденная нечувствительность к боли) |
| 350 | Септо-оптическая дисплазия |
| 351 | Сетре-Чотзена синдром |
| 352 | Сиалидоз тип 1,2 |
| 353 | Сильвера-Рассела Синдром |
| 354 | Симпсона-Голаби-Бемель синдром |
| 355 | Синдром CADASIL, энцефалопатия с субкортикальными инфарктами |
| 357 | Синдром CINCA (холодовая лихорадка, синдром Мукле-Велса) |
| 358 | Синдром CRASH |
| 359 | Синдром ESC |
| 360 | Синдром LEOPARD |
| 361 | Синдром MASA |
| 362 | Синдром MNGIE |
| 363 | Синдром Ohdo, SBBYSS вариант |
| 364 | Синдром RAPADILINO |
| 365 | Синдром TAR |
| 366 | Синдром TRAPS (злокачественная гипертермия, амилоидоз почек) |
| 367 | Синдром тугоухости и атрофии зрительных нервов |
| 368 | Скапулоперонеальная миопатия |
| 370 | Смита-Лемли-Опитца синдром |
| 371 | Смит-Магенис синдром |
| 372 | Сотоса синдром |
| 373 | Спастическая параплегия Штрюмпеля |
| 374 | Спинальная амиотрофия типы I, II, III, IV |
| 375 | Спинальная и бульбарная амиотрофия Кеннеди |
| 376 | Спиноцеребеллярная атаксия |
| 377 | Спонгиоформная энцефалопатия с нейропсихическими проявлениями |
| 378 | Спондилокостальный дизостоз |
| 379 | Спондилоэпифизарная дисплазия (SEDT) |
| 380 | Стиклера синдром |
| 381 | Суперактивность фосфорибозилпирофосфат синтетазы |
| 382 | Талассемия beta |
| 383 | Тестикулярной феминизации синдром |
| 384 | Тея-Сакса болезнь |
| 385 | Тирозингидроксилазы недостаточность |
| 386 | Тирозинемия тип I |
| 387 | Торсионная дистония |
| 388 | Транспортера глюкозы недостаточность |
| 389 | Трихоринофалангеальный синдром |
| 390 | Тричера Коллинза-Франческетти синдром |
| 391 | Тромбоцитопения врожденная |
| 392 | Туберозный склероз |
| 393 | Умственная отсталость моногенная |
| 394 | Унферрихта-Лундборга болезнь |
| 395 | Уокера-Варбург синдром |
| 396 | Ушера синдром |
| 397 | Фабри болезнь |
| 398 | Фатальная семейная инсомния |
| 399 | Фацио-Лонде болезнь |
| 400 | Фелан-МакДермид синдром |
| 401 | Фенилкетонурия |
| 402 | Фибродисплазия оссифицирующая прогрессирующая |
| 403 | Фокальная кожная гипоплазия (Горлина-Гольца синдром) |
| 404 | Фокально-кортикальная дисплазия Тейлора |
| 405 | Фон Хиппель-Линдау Синдром |
| 406 | Фруктозо1,6 дифосфотазы недостаточность |
| 407 | Фукозидоз |
| 408 | Хайду-Чейни синдром |
| 409 | Хондродисплазия метафизарная тип Мак-Кьюсика |
| 410 | Хондродисплазия точечная Конради-Хюнермана |
| 411 | Хондрокальциноз |
| 412 | Хореоатетоз, гипотиреоидизм и неонатальная дыхательная недостаточность |
| 413 | Хорея Гентингтона |
| 414 | Хорея доброкачественная наследственная |
| 415 | Хороидермия |
| 416 | Хромосомные болезни |
| 417 | Хроническая гранулематозная болезнь |
| 418 | Х-сцепленная агаммаглобулинемия |
| 419 | Х-сцепленный лимфопролиферативный синдром (болезнь Дункана, синдром Пуртильо) |
| 420 | Х-сцепленный моторный нистагм |
| 421 | Х-сцепленный тяжелый комбинированный иммунодефицит |
| 422 | Целвегера синдром |
| 423 | Центронуклеарная миопатия |
| 424 | Цереброокулофациоскелетный синдром |
| 425 | Цистиноз |
| 426 | Цистиноз нефропатический |
| 427 | Цитруллинемия тип 1 |
| 428 | Шварца-Джампела синдром |
| 429 | Швахмана-Даймонда синдром |
| 430 | Шегрена-Ларссона синдром |
| 431 | Шерешевского-Тернера синдром |
| 432 | Широкого водопровода преддверия синдром |
| 433 | Шпринтцена-Гольдберга синдром |
| 434 | Штаргардта болезнь |
| 435 | Эдвардса синдром |
| 436 | Экзостозы множественные |
| 437 | Эксудитивная витреохореорстинальная дистрофия |
| 438 | Эктодермальная ангидротическая дисплазия |
| 439 | Эктодермальная гидротическая дисплазия |
| 440 | Эктопия хрусталика |
| 442 | Эллерса-Данло синдром |
| 443 | Эпилепсия прогрессирующая миоклоническая |
| 444 | Эпифизарная дисплазия, множественная |
| 445 | Эритрокератодермия |
| 446 | Эритроцитоз рецессивный |
| 447 | Эскобара cиндром |
Телу Майлза 96 лет: «Как быть со смертным приговором?» (Svenska Dagbladet, Швеция) | Общество | ИноСМИ
Майлз ходит в шестой класс, но у него уже тело старика. Несмотря на все трудности и неопределенное будущее, его родители считают, что им повезло.
«Очень легко закрыться в своем коконе и начать жалеть себя, но на самом-то деле нам невероятно повезло», — говорит отец Майлза Якоб Вернерман (Jakob Wernerman).
Майлз Вернерман сидит на диване, скрестив ноги, и гуглит свое имя.
«„Нюхетсморон» (Nyhetsmorgon), 318 тысяч. „Малу» (Malou), больше миллиона».
Он удовлетворенно улыбается из-под козырька кепки, у его видео стало еще больше просмотров, чем было, когда он смотрел в прошлый раз.
В первый раз я встречалась с Майлзом в 2012 году, когда он катался на трехколесном велосипеде в парке развлечений в стокгольмском Сёдермальме. Он только что переехал в Швецию и пошел в первый класс. Он залезал на самый верх всех детских лазалок и отказывался снимать кепку со Спайдерменом. Его новая классная руководительница согласилась сделать для него исключение и отступить от правил о головных уборах, потому что Майлзу неприятно быть единственным в классе, у кого нет волос.
Это было шесть лет назад. С тех пор тело Майлза состарилось на 48 лет.
Всего в мире известна примерно сотня случаев заболевания прогерия, и Майлз — один из таких детей. Это значит, что он стареет в восемь раз быстрее обычного человека. Средняя продолжительность жизни таких людей 13,5 лет.
Настало время полдника. Майлз и его младшие брат с сестрой садятся на кухне за чай с медом и начинают спорить, какие артисты и звезды «Ютуба» крутые, а какие — зануды.
«Я больше не могу пить чай, — говорит Майлз и делает театральную паузу, осматривая стол. — Потому что иначе я перегреюсь!»
Брат и сестра смеются. Майлз колотит ногами в тапочках из овчины по высокому стулу, на котором сидит. У него совсем нет жира на теле, в том числе и в ступнях, поэтому ему больно ходить без мягких подошв. Другие последствия болезни: у него рано появились типично возрастные недуги, нет волос, и он очень маленький — при 110 сантиметрах роста весит 14 килограмм. По его словам, это хуже всего. На его размер не делают классную одежду и кроссовки. Ему 12 лет, и он не хочет носить одежду из детских отделов, с грузовиками и динозаврами на груди.
Я спрашиваю, не странно ли ему, что все его приятели сейчас так быстро растут.
«Да нет. Я с ними почти каждый день. Так что не замечаю, что они растут и все такое прочее».
В следующем году он официально станет подростком.
«Иногда мы забываем, сколько ему лет, — говорит его мама Лея Ричардсон (Leah Richardson). — Он ведь такой маленький. Но потом приходят в гости его друзья, и они такие большие! Просто гигантские, почти взрослые мужчины!»
«Худший период в моей жизни». Так она описывает время, когда они узнали диагноз. Майлз казался здоровым, когда только родился в Италии, где Леа и Якоб жили и работали в ООН. Через три месяца врачи взяли у него анализ на эту невероятно редкую болезнь. Все результаты были отрицательными, и семья смогла вздохнуть с облегчением. Но когда Майлзу исполнилось два, его снова обследовали. Семья уже жила в Нью-Йорке, и теперь результат оказался иным: прогерия.
«Это был смертный приговор. Как быть, когда вашему ребенку выносят смертный приговор?»
Лея рассказывает, как мир для нее рушился вновь и вновь каждое утро. В первые годы с Майлзом говорил о болезни Якоб, а она изо всех сил старалась сделать каждую секунду жизни своего ребенка как можно более наполненной, и мучилась совестью, когда сердилась или когда Майлзу было скучно или грустно.
«Очень легко забиться в свою нору и горевать там, когда попадаешь в такую ситуацию, как наша», — говорит Якоб, а Леа вставляет, что именно это она и делала много лет.
Но со временем они как-то научились принимать это. И сегодня они даже благодарны за осознание, которое дал им Майлз: жизнь конечна.
«А Майлз успеет пойти в университет?»
Вся семья уселась в автомобиль, и семилетний братишка Майлза задал этот вопрос. И уточнил:
«Майлз успеет пойти в университет, прежде чем умрет?»
«Вероятно», — ответила Леа.
Майлз задумался. Потом вспомнил своего старшего товарища по прогерии: «Сэму 20, и он ходит в университет».
Больше об этом не говорили.
Раз в год проходит встреча европейских «семей с прогерией», в этом октябре ее проводили в Португалии. У Майлза на письменном столе стоит фото всей компании, и он показывает своих лучших друзей — двух больных прогерией мальчиков постарше из Бельгии и Англии. Они очень любят встречаться, ведь тогда они оказываются в среде, где их состояние — норма. Но прощаться всегда грустно. Неизвестно, кто из них еще будет жив в следующий раз.
О том, что средняя продолжительность жизни детей с прогерией составляет 13,5 лет, Майлз обычно не думает и не говорит, даже с родителями. Как и многие другие двенадцатилетние дети, он полностью занят тем, что происходит здесь и сейчас. Например, игрой в футбол и ночными посиделками с друзьями. Или перепалками с братом и сестрой. Или домашними заданиями. Или просмотром забавных видео на «Ютубе».
Но каждый день приходится принимать лекарства: замедляющие ход заболевания, понижающие холестерин, разжижающие кровь, а также сердечные препараты. В детской больнице имени Астрид Линдгрен Майлз ходит к врачам, которые занимаются его кожей, глазами, суставами и сердцем. По большей части речь идет о предотвращении типичных возрастных недугов, вроде инсульта и инфаркта, от которых обычно умирают дети с прогерией.
«Свенска дагбладет»: Чего тебе больше всего хотелось бы?
Майлз прилег на овечью шкуру на полу, он только что нам рассказал, что мягкие игрушки, которые тоже там лежат, — старые.
«Наверное… компьютер! Собственный стационарный компьютер со всем, что нужно, который будет стоять у меня в комнате. Это было бы круто».
Сейчас он в основном играет в приставку. Рассказывает, что ему разрешают «гамать» по вторникам, пятницам, субботам и воскресеньям.
«Лучше всего в субботу, я тогда могу играть и утром, и вечером».
Daily Mail Dagens Hälsa The IndependentВ списке рождественских желаний есть и щенок. Но пока что ему приходится довольствоваться палочником. У них с младшей сестренкой Клементиной есть по палочнику: одного зовут Джордж Буш, другого — Анни Лёф (Annie Lööf).
На стене висят две голубые футболки: светло-голубая с автографом Месси и вторая — его любимой команды «Юргорден» (Djurgården). На вопрос, есть ли на ней тоже автографы, он отвечает:
«Не, ну может, парочка».
Улыбается и переворачивает ее, показывая автографы от всей команды.
Когда Майлз снимался в передаче «Нюхетсморон» этой осенью, ведущая Тильде де Паула (Tilde de Paula) вручила ему подарок на день рождения: билет на домашний мачт «Юргордена» против «Гётеборга», причем он смог потом выйти на футбольное поле, поесть в вип-салоне и отпраздновать победу 2:0 вместе с футболистами. В раздевалке.
Леа просит его рассказать, что ему сказал звездный футболист Кевин Уокер (Kevin Walker), и Майлз опускает голову, чтобы скрыть улыбку.
«Нет, мама, ты расскажи».
«Он поблагодарил Майлза за победу, потому что он был их талисманом», — говорит она и тыкает его в бок.
Не в первый раз у Майлза была возможность сделать то, о чем другие дети лишь мечтают. Якоб и Лея считают, что трудно бывает поддерживать баланс: они не хотят, чтобы он избаловался, но при этом им кажется, что «нужно брать от жизни все».
Мы поужинали суши, и на улице уже темным-темно. Дети, как всегда, немного повозражали и отправились делать домашнюю работу каждый в свою комнату. Мы устроились на диване, и я спросила Якоба и Лею, как изменилось их отношение к жизни с того дня в Нью-Йорке десять лет назад. Когда врачи со своими анализами перевернули их жизнь с ног на голову.
«Никто из нас не знает, сколько мы проживем, неважно, есть у вас смертельный диагноз или нет. Однажды утром вас может переехать автобус. Так что главное — правильно использовать время, которое мы можем провести вместе, и не откладывать слишком многое на потом», — говорит Якоб.
Он упирает на то, что Майлз действительно живет здесь и сейчас.
«Он очень мало думает, что будет дальше, он сконцентрирован на том, чтобы жить как можно веселее и делать то, что нравится, иногда чуть ли не до абсурда. И это нас порядком подбадривает».
Лея тщательно подбирает слова. Подчеркивает, насколько в действительности ей тяжело принять этот образ мысли, сколько бы лет она на это ни потратила.
«Конечно, все понимают, как хорошо жить настоящим. Но совсем другое дело, когда ты знаешь, что есть определенная дата, что у тебя есть только пять или десять лет».
Давно уже главным для нее стало сделать короткую жизнь Майлза как можно более полной. Но, кроме этого, они с Якобом и сами стремятся жить как можно полнее, чтобы стать более жизнерадостными людьми и хорошими родителями.
«Это не значит, что мы всегда кричим „да», что бы дети ни попросили, скорее мы просто стараемся ничего не откладывать и не мечтать впустую. Например: „О, мы всегда хотели поехать в Австралию, но это так далеко, так дорого, и у нас трое детей…» А мы говорим: „Давай сделаем это!»»
По словам Якоба, он пришел к выводу, что на самом деле им повезло. Могло быть хуже. И он, и Лея много работали в развивающихся странах, она — в медицине катастроф, а он — в срочной международной помощи в чрезвычайных ситуациях. Они видели, в каких ужасных условиях живут многие люди.
«Возможно, это звучит ужасно, но я говорю, что нам повезло. Я, конечно, никогда бы сам такого не пожелал. Но у каждого в жизни свои проблемы. Не все они касаются продолжительности жизни. Помимо того, что у Майлза прогерия, мы — совершенно обычная, счастливая семья. За это я очень благодарен».
«Однажды, детка, мы состаримся,
О, детка, мы будем старыми,
И только подумай, какие истории,
Мы сможем тогда рассказать».
Песня Асафа Авидана (Asaf Avidan) льется из громкоговорителей на скалодроме на юге Стокгольма. Майлз пришел сюда прямо с контрольной по алгебре, на нем красная толстовка и черная кепка. Желто-голубые скалолазные туфли ему на несколько размеров велики, и он отказывается от маминой помощи, надевая их. Это его третье занятие со специалистом по физической реабилитации Осой (Åsa).
«Это отличный способ предотвратить проблемы с суставами. Ты действительно сообразил, в чем фишка, Майлз, ползи, как маленький паучок, вверх по стене — вжух! Здорово, что ты физически активен, иначе моя работа была бы гораздо труднее», — говорит она и улыбается.
Майлз в страховке, а в руке у него бутылка воды. Он уверенно идет к стене, носки немного развернуты в стороны, руки на бедрах.
Несколькими днями позже я получила письмо от Леи. Она написала, что придется перенести запланированное фотографирование на футбольном поле. Бедренная кость Майлза выскочила из сустава, когда он играл в футбол в школе. Это первый явный признак того, что его тело состарилось, и пора начинать совершенно новый курс реабилитации. «Казалось, весь мир остановился. Хотя мы знали, что когда-то это произойдет, но к такому никогда не бываешь готов», — написала Лея.
Майлз становится известным, и его семья видит в этом только хорошее. Они хотят, чтобы люди видели его и говорили: «посмотрите, это Майлз, у которого та болезнь», а не «посмотрите, как странно выглядит этот человек». Сам он привык, что на него смотрят. Мне кажется, это нормально, говорит он и пожимает плечами.
«Иногда кто-то подходит, чаще всего дети, и говорит, что видели меня по телевизору или в газете. Это круто».
«Свенска дагбладет»: Хочешь что-то передать всем, кто это будет читать?
Он улыбается очень широко и видно, что у него нет одного переднего зуба.
«Все должны приехать ко мне и сделать селфи. Все! Это будет круто».
Я задаю тот же вопрос его родителям.
«Все должны приехать к нам и сделать с нами селфи», — отвечает Лея, и они покатываются со смеху.
Затем она становится серьезной. Говорит о шведской вежливости, которая иногда некстати. Семья бывает в парке развлечений, и иногда слышит, как дети спрашивают родителей «что с ним?», а в ответ получают шиканье и строгие взгляды.
«Они не хотят об этом говорить и объяснять, чтобы ребенок понял. Наверное, они думают, что нам это было бы неприятно».
Якоб кивает и объясняет, что на самом деле все наоборот.
«Это не неприятно, это ободряет! Гораздо неприятнее слышать перешептывания. Подойдите, спросите, сфотографируйтесь — что угодно!»
Прежде чем мы прощаемся, я спрашиваю Майлза, что бы он сделал, если бы ненадолго смог колдовать? Какие большие желания он бы выполнил? Он замолкает, но лишь на пару секунд. Затем сияет.
«Я бы сделал так… чтобы колдовать всегда!»
Прогерия
Прогерия или синдром Гетчинсона-Гилфорда — крайне редкая смертельная болезнь, которая обусловлена преждевременным старением и смертью клеток. Тело стареет примерно в восемь раз быстрее, чем у здоровых людей. Прогерию вызывает генетическая мутация, которая приводит к тому, что белок преламин А работает некорректно. Преламин А нужен, чтобы мембрана клетки оставалась стабильной.
Симптомы начинают проявляться в первые два года жизни, а примерно в пять ребенок прекращает расти. Он теряет волосы и подкожный жир, суставы закостеневают, скелет становится хрупким. На интеллект все это не влияет.
Средняя продолжительность жизни таких больных — 13,5 года, но некоторые доживают до 20 с лишним. Самые частые причины смерти — инсульт и инфаркт. Сегодня примерно 100 детей в мире живут с эти диагнозом. Майлз — единственный в Швеции. Болезнь не наследственная. Сейчас от нее нет никаких лекарств, ее развитие можно только притормозить.
Синдром Вернера — менее известная форма прогерии. Из-за этой болезни у человека признаки старения проявляются раньше, чем было бы адекватно для его возраста.
Синдром Вернера, в отличие от синдрома Гетчинсона-Гилфорда, чаще всего обнаруживается не ранее 20-летнего возраста.
Материалы ИноСМИ содержат оценки исключительно зарубежных СМИ и не отражают позицию редакции ИноСМИ.
Синдром прогерии Хатчинсона-Гилфорда — PubMed
Синдром прогерии Хатчинсона-Гилфорда (HGPS) — чрезвычайно редкое, неизменно фатальное, сегментарное заболевание «преждевременного старения», при котором у детей проявляются фенотипы, которые могут дать нам представление о процессе старения как на клеточном, так и на организменном уровнях. Первоначальное проявление в раннем детстве в первую очередь основывается на результатах роста и дерматологических данных. Первичная заболеваемость и смертность детей с HGPS обусловлены атеросклеротическим сердечно-сосудистым заболеванием и инсультами, в результате которых смерть наступает в среднем в возрасте 14 лет.6 лет. Растет количество данных, подтверждающих уникальный фенотип черепно-лицевой и цереброваскулярной анатомии, который сопровождает процесс преждевременного старения. Инсульты при HGPS могут возникать после окклюзии сонной артерии и / или позвоночной артерии, стеноза и кальцификации с заметным образованием коллатеральных сосудов. Присутствуют заболевания как крупных, так и мелких сосудов, и инсульты часто клинически бессимптомны. Несмотря на наличие мультисистемного преждевременного старения, у детей с HGPS, по-видимому, не наблюдается ухудшения когнитивных функций, что позволяет предположить, что некоторые аспекты функции мозга могут быть защищены от пагубного воздействия прогерина, вызывающего болезнь белка.На основании ограниченного материала аутопсии нет никаких патологических свидетельств деменции или изменений типа Альцгеймера. В модели прогерии у трансгенных мышей с экспрессией наиболее распространенной мутации HGPS в головном мозге, коже, костях и сердце наблюдаются искажения нейронных ядер на ультраструктурном уровне с неправильной формой и тяжелыми инвагинациями, но отсутствуют доказательства включений или аберрантного тау-белка. в отделах мозга. Важно отметить, что ядерные искажения не приводили к значительным изменениям экспрессии генов в нейронах гиппокампа.В этой главе будут обсуждаться доклинические и клинические аспекты генетики, патобиологии, клинического фенотипа, клинической помощи и лечения HGPS, с особым вниманием к неврологическим и кожным результатам.
Ключевые слова:
ССЗ; HGPS; Прогерия; старение; атеросклероз; цереброваскулярный; ламин; ламинопатия; орфанная болезнь; редкое заболевание; Инсульт.
| 80-99% людей имеют эти симптомы | ||
| Отсутствие подкожно-жировой клетчатки | Отсутствие жира под кожей Отсутствие жировой ткани под кожей [ более ] | 0007485 |
| Кондуктивное нарушение слуха | Кондуктивная глухота Кондуктивная потеря слуха [ более ] | 0000405 |
| Микрогнатия | Маленькая нижняя челюсть Маленькая челюсть Маленькая нижняя челюсть [ более ] | 0000347 |
| Узкая горловина | Маленький рот | 0000160 |
| Преждевременное сморщивание кожи | 0100678 | |
| Выступающие поверхностные кровеносные сосуды | Выраженная поверхностная сосудистая сеть | 0007394 |
| Выступающий пупок | Выступающий пупок Выступающий пупок [ более ] | 0001544 |
| Пубертатная недостаточность развития у женщин | 0008647 | |
| Тяжелая потеря роста | Сильный неуверенный вес Сильная потеря веса [ более ] | 0001525 |
| Кайма тонкая киноварь | Уменьшение объема губ Тонкие губы [ более ] | 0000233 |
| Потеря веса | 0001824 | |
| 30% -79% людей имеют эти симптомы | ||
| Полная алопеция | 0007418 | |
| Анкилоглоссия | Связанный язык | 0010296 |
| Атеросклероз | Сужение и затвердение артерий | 0002621 |
| Цоколь валга | 0002673 | |
| Черепно-лицевая диспропорция | 0005461 | |
| Снижение лептина сыворотки | 0003292 | |
| Отсроченное начало менструального цикла | Отложенное начало первого периода | 0012569 |
| Дистрофические ногти | Плохое формирование ногтей | 0008391 |
| Дистрофический ноготь на ноге | Плохое формирование ногтей на ногах | 0001810 |
| Одышка при физической нагрузке | 0002875 | |
| Женский гипогонадизм | 0000134 | |
| Высокое небо | Повышенное небо Увеличенная небная высота [ более ] | 0000218 |
| Высокий голос | 0001620 | |
| Вывих бедра | Вывих бедра Вывих бедра [ более ] | 0002827 |
| Гипоплазия мужских наружных половых органов | Маленькие мужские наружные гениталии Недоразвитые мужские гениталии [ более ] | 0000050 |
| Инсулинорезистентность | Организм не реагирует на инсулин | 0000855 |
| Отсутствие эластичности кожи | 0100679 | |
| Диастолическая дисфункция левого желудочка | 0025168 | |
| Низкочастотное нейросенсорное нарушение слуха | 0008573 | |
| Узкий носовой гребень | Уменьшение ширины носового гребня. Защемленный нос Тонкий носовой гребень [ более ] | 0000418 |
| Узкий кончик носа | Узкий кончик носа Кончик носа узкий Кончик носа защемлен Защемление кончика носа Защемленный кончик носа Тонкий кончик носа Тонкий кончик носа [ более ] | 0011832 |
| Очаговая алопеция | Очаговое облысение | 0002232 |
| Относительная макроцефалия | Относительно большая голова | 0004482 |
| Ретрогнатия | Опущенный подбородок Опускающаяся нижняя челюсть Слабый подбородок Слабая челюсть [ более ] | 0000278 |
| Мелкие орбиты | Уменьшение глубины глазниц Неглубокие глазницы [ более ] | 0000586 |
| Короткая уздечка языка | 0000200 | |
| Шаркающая походка | Перемешанная прогулка | 0002362 |
| 5% -29% людей имеют эти симптомы | ||
| Отсутствие брови | Нарушение развития бровей | 0002223 |
| Регургитация аорты | 0001659 | |
| Кальциноз аортального клапана | 0004380 | |
| Стеноз аортального клапана | Сужение аортального клапана | 0001650 |
| Аваскулярный некроз | Смерть кости из-за снижения кровоснабжения | 0010885 |
| Окклюзия сонной артерии | Закупорка сонной артерии | 0012474 |
| Носовой гребень выпуклый | Клювый нос Клювовидный выступ Нос с горбинкой Деформация носа Polly Beak [ более ] | 0000444 |
| Помутнение роговицы | 0007957 | |
| Цианоз | Посинение кожи | 0000961 |
| Отсроченное прорезывание зубов | Отсроченное извержение Задержка прорезывания зубов Отсроченное прорезывание зубов Извержение с задержкой Позднее прорезывание зубов Позднее прорезывание зубов [ более ] | 0000684 |
| Стоматологическая скученность | Скученные зубы Стоматологическая скученность Скученность зубов [ более ] | 0000678 |
| Кожная атрофия | Дегенерация кожи | 0004334 |
| Высокочастотное нейросенсорное нарушение слуха | 0001757 | |
| Боль в бедре | 0030838 | |
| Гипермеланотическое пятно | Гиперпигментированные пятна | 0001034 |
| Гипертония | 0000822 | |
| Гиподонтия | Отсутствие развития от одного до шести зубов | 0000668 |
| Роликовый зуб | 0011079 | |
| Внутричерепное кровоизлияние | Кровотечение внутри черепа | 0002170 |
| Жесткость шарнира | Жесткий сустав Жесткие суставы [ более ] | 0001387 |
| Ограничение движений в области голеностопного сустава | 0010505 | |
| Ограниченное движение бедер | 0008800 | |
| Ограниченное движение плеча | 0006467 | |
| Ограниченный механизм запястья | Ограниченное движение запястья | 0006248 |
| Выпадение ресниц | Ресницы выпали Отсутствие ресниц [ более ] | 0011457 |
| Митральная регургитация | 0001653 | |
| Митральный стеноз | 0001718 | |
| Кальцификация митрального клапана | 0004382 | |
| Инфаркт миокарда | Острое сердечно-сосудистое заболевание | 0001658 |
| Ночной лагофтальм | Веки остаются открытыми на ночь Невозможность закрывать веки ночью [ более ] | 0030002 |
| Артроз | Дегенеративное заболевание суставов | 0002758 |
| Остеолитические дефекты дистальных фаланг кисти | 0009839 | |
| Папула | 0200034 | |
| Остаточные молочные зубы | Поздняя потеря молочных зубов Неспособность потерять молочные зубы Сохраненные молочные зубы [ более ] | 0006335 |
| Прогрессирующий акроостеолиз ключицы | 0000905 | |
| Выступающая спираль уха | 0009904 | |
| Феномен Рейно | 0030880 | |
| Пониженная минеральная плотность костной ткани | Низкая прочность и масса костей | 0004349 |
| Короткий подбородок | Уменьшенная высота подбородка Короткая нижняя треть лица [ более ] | 0000331 |
| Ключицы короткие | Короткая ключица | 0000894 |
| Транзиторная ишемическая атака | Мини ход | 0002326 |
| Обструкция верхних дыхательных путей | 0002781 | |
| Гипертрофия желудочков | 0001714 | |
| 1% -4% людей имеют эти симптомы | ||
| Стенокардия | 0001681 | |
| Изъязвление роговицы | 0012804 | |
| Систолическая дисфункция левого желудочка | 0025169 | |
| Легочная артериальная гипертензия | Повышенное артериальное давление в сосудах легких | 0002092 |
| Процент людей, у которых есть эти симптомы, недоступен через HPO | ||
| Облысение | Потеря волос | 0001596 |
| Аутосомно-доминантное наследование | 0000006 | |
| Аутосомно-рецессивное наследование | 0000007 | |
| Застойная сердечная недостаточность | Сердечная недостаточность Сердечная недостаточность Сердечная недостаточность [ более ] | 0001635 |
| Остеопороз генерализованный | 0040160 | |
| Задержка роста | Задержка роста Дефицит роста Нарушение роста Задержка роста Плохой рост Замедленный рост [ более ] | 0001510 |
| Уплощение скуловой кости | Скуловое уплощение | 0000272 |
| Ретрузия средней зоны лица | Уменьшен размер средней зоны лица Дефицит средней зоны лица Недоразвитие средней зоны лица [ более ] | 0011800 |
| Остеолиз | Разрушение кости | 0002797 |
| Ранний атеросклероз | 0004416 | |
| Преждевременный атеросклероз коронарной артерии | Преждевременная ишемическая болезнь сердца | 0005181 |
Симптомы, тесты, лечение и профилактика
Обзор
Что такое прогерия?
Прогерия (синдром прогерии Хатчинсона-Гилфорда или HGPS) — очень редкое генетическое заболевание, которое вызывает быстрое старение детей.Дети с прогерией кажутся здоровыми при рождении, но обычно начинают проявлять признаки быстрого старения в первые два года жизни. Большинство умирает примерно к 13 или 14 годам, хотя некоторые доживают до 20 лет. Причиной смерти чаще всего бывает болезнь сердца, а иногда и инсульт.
Насколько распространена прогерия?
Прогерия поражает примерно 1 из 20 миллионов человек во всем мире. По данным Исследовательского фонда прогерии, во всем мире в любое время живут от 350 до 400 детей с прогерией.Прогерия, кажется, одинаково влияет на мальчиков и девочек, и не чаще встречается у одной расы, чем у другой.
Симптомы и причины
Что вызывает прогерию?
Прогерия вызывается мутацией (изменением) гена ламина А (LMNA). Этот ген вырабатывает белок, который скрепляет ядро клетки.Из-за изменения гена белок становится дефектным. Это делает ядро нестабильным, что, как считается, вызывает процесс преждевременного старения.
Мутация гена LMNA не передается по наследству. Фактически, родители, братья и сестры детей с прогерией страдают редко.
Каковы симптомы прогерии?
Дети с прогерией начинают проявлять признаки и симптомы быстрого старения в течение первых двух лет жизни. К ним относятся:
- Неспособность расти такими же темпами, как у конкурентов
- Потеря жира
- Облысение
- Кожа приобретает состарившийся, морщинистый вид
- Жесткие суставы
Другие признаки и симптомы прогерии включают:
- Лица маленькие для размера головы
- Носы, которые выглядят «защемленными»
- Открытое мягкое пятно на голове
- Зубы поздние
По мере развития состояния начинают развиваться симптомы, менее очевидные для родителей, включая вывих бедра, катаракту, артрит, образование бляшек в артериях и сердечные заболевания.У некоторых детей с прогерией случаются инсульты.
Несмотря на эти физические проблемы, интеллект не страдает, и дети с прогерией, как правило, так же умны, как и их сверстники.
Диагностика и тесты
Как диагностируется прогерия?
Ген, вызывающий прогерию, был идентифицирован в 2003 году, и был создан генетический тест, который может подтвердить, вызваны ли симптомы у ребенка прогерией.Для проведения теста необходимо взять образец крови у ребенка. (До того, как этот тест стал доступен, врачи могли диагностировать прогерию только на основании своих наблюдений и рентгеновских лучей.)
Если у вашего врача есть основания полагать, что у вашего ребенка прогерия, он или она может связаться с Фондом исследований прогерии. Медицинский директор фонда рассмотрит случай и может организовать бесплатное обследование для семей. Результаты обычно приходят через 2–4 недели.
Ведение и лечение
Как лечить прогерию?
Нет лекарства от прогерии, но изучаются несколько препаратов для ее лечения.
Физическая терапия может помочь этим детям достичь хорошего диапазона движений, равновесия и осанки, а также уменьшить боль в бедрах и ступнях. Трудотерапия может помочь им развиваться в таких функциональных областях, как прием пищи, соблюдение личной гигиены и почерк.
Дети с прогерией могут получить большую пользу от ухода, который поможет им жить как можно более здоровой и комфортной жизнью. Обычно сюда входят:
- Мониторинг сердечных заболеваний : Сюда входят регулярные тесты, такие как эхокардиограмма и проверка артериального давления.Терапия низкими дозами аспирина и статинов может помочь снизить некоторые риски сердечных заболеваний.
- Визуальные исследования (например, магнитно-резонансная томография или МРТ ) : Их можно использовать для наблюдения за инсультами или для проверки головных болей или судорог, которые часто встречаются у этих детей.
- Регулярные осмотры глаз : У некоторых детей с прогерией есть проблемы со зрением, в том числе дальнозоркость или сухость глаз (потому что их веки не могут полностью закрыться).По мере прогрессирования состояния у них также может развиться катаракта. У этих детей могут быть более тонкие брови и ресницы, что повышает вероятность попадания раздражителей в глаза. Кроме того, некоторые дети имеют сильную чувствительность к свету, и в некоторых случаях им могут посоветовать носить солнцезащитные очки.
- Проверка слуха : У детей с прогерией может быть потеря слуха, которую можно улучшить с помощью слуховых аппаратов.
- Регулярные стоматологические осмотры : Дети с прогерией чаще имеют стоматологические проблемы, такие как кариес, сильная скученность, задержка появления зубов и углубление десен.
- Мониторинг кожных проблем : Часто это первые признаки прогерии и могут включать темные пятна или выпуклости на коже, выпадение волос, зуд и стянутость кожи, которые ограничивают движение и могут затруднять дыхание или переваривание пищи.
- Мониторинг здоровья костей : Дети с прогерией могут иметь несколько проблем, связанных с ростом и развитием костей, а также проблемами с суставами.
Детям с прогерией для роста необходимо полноценное питание.Некоторым может потребоваться дополнительное питание (включая зонд для кормления). Обеспечение достаточного увлажнения детей с прогерией может помочь снизить риск внезапных неврологических проблем. Посоветуйтесь с врачом вашего ребенка о здоровых способах поощрения вашего ребенка получать достаточно калорий и гидратации.
Что могут сделать родители, чтобы помочь своей семье, если у их ребенка прогерия?
Родители ребенка с прогерией должны стараться вести как можно более нормальную семейную жизнь. Постарайтесь вовлечь ребенка в как можно больше занятий и не позволяйте другим детям в семье чувствовать себя упущенными.
Будьте честны, но учитывайте возраст всей семьи, обсуждая тот факт, что ваш ребенок с прогерией доживет только до определенного возраста. Консультации могут быть полезны в разное время.
Также поговорите со своим ребенком о том, что некоторые люди вернутся, увидев его или ее, и обсудите, как ваш ребенок должен реагировать на взгляды и шепот.
Перспективы / Прогноз
Если у моего ребенка прогерия, будет ли она у моих будущих детей?
Прогерия вызывается чрезвычайно редким генетическим изменением и обычно не передается по наследству.Общая вероятность иметь ребенка с прогерией составляет примерно 1 к 4 миллионам. Однако, если у человека родился ребенок с прогерией, вероятность родить с ним еще одного ребенка возрастает на 2–3%, потому что у него может быть генетический признак прогерии, но на самом деле он не болен. Люди, у которых был ребенок с прогерией, могут пройти генетическое тестирование, чтобы узнать свои шансы на рождение еще одного ребенка с этим заболеванием.
Жить с
Может ли ребенок с прогерией посещать школу?
Многие дети с прогерией ходят в школу, хотя обычно им нужны приспособления, позволяющие им полноценно участвовать, чувствовать себя комфортно и безопасно.Родители должны регулярно встречаться со школьной администрацией, медсестрами, терапевтами, учителями и другими людьми, чтобы каждый мог работать вместе, чтобы удовлетворить потребности ребенка. Это включает в себя создание и распространение плана оказания неотложной помощи ребенку в случае необходимости в школе (например, при внезапной одышке или боли в груди).
FDA одобрило лекарство от прогерии, редкого заболевания, вызывающего быстрое старение у детей.
FDA одобрило первый препарат для снижения риска смертности от прогерии, чрезвычайно редкого заболевания, которое вызывает быстрое старение и смерть, как правило, к 15 годам.
Утверждение было предоставлено компании Eiger BioPharmaceuticals, которая в партнерстве с Progeria Research Foundation (PRF) на лонафарниб (Zokinvy). Лонафарниб — это пероральный ингибитор фарнезилтрансферазы (FTI), который показал улучшение выживаемости у детей с синдромом прогерии Хатчинсона-Гилфорда и для лечения некоторых прогероидных ламинопатий с недостаточным процессингом у пациентов в возрасте от 1 года и старше.
По заявлению FDA, препарат не одобрен для использования у пациентов с другими прогероидными синдромами или ламинопатией.
Компания Eiger начала поставлять препарат для клинических испытаний в 2015 году, а в 2018 году стала партнером PRF с целью прохождения процесса утверждения препарата FDA. Заявка получила статус приоритетного рассмотрения, а препарат был признан орфанным лекарством, а также прорывной терапией. Кроме того, Эйгер получил ваучер на приоритетную проверку лечения редких детских заболеваний.
Компания заявила, что исследования, лежащие в основе этой терапии, могут привести к другим открытиям о сердечных заболеваниях, а также о процессе старения.
Пациенты с синдромом прогерии Хатчинсона-Гилфорда и прогероидной ламинопатией испытывают ускоренное сердечно-сосудистое заболевание из-за накопления в клетках дефектного прогерина или прогерин-подобного белка. Лонафарниб помогает предотвратить накопление дефектного белка.
Эффективность была показана у 62 пациентов в 2 исследованиях с одной группой, которые сравнивались с подобранными, нелеченными пациентами из отдельного исследования естественной истории. Данные, основанные на информации из Международного реестра пациентов PRF и клинических исследований, координируемых PRF и Бостонской детской больницей, показали, что лонафарниб снижает частоту смертности на 60% ( P =.0064) и увеличили среднее время выживания на 2,5 года при максимальном сроке наблюдения 11 лет.
Без лечения дети с прогерией умирают от болезней сердца в среднем в возрасте 14,5 лет.
Утверждение приняло во внимание сходство в основном генетическом механизме этого заболевания и другие доступные данные, сообщает FDA.
«Синдром прогерии Хатчинсона-Гилфорда и прогероидная ламинопатия — редкие генетические заболевания, которые вызывают преждевременное старение и смерть и оказывают изнурительное воздействие на жизнь людей», — сказал Хилтон В.Иоффе, доктор медицинских наук, доктор медицинских наук, директор отдела редких заболеваний, педиатрии, урологической и репродуктивной медицины Центра оценки и исследований лекарственных средств Управления по санитарному надзору за качеством пищевых продуктов и медикаментов. «С сегодняшнего одобрения Zokinvy является первым лекарством от этих разрушительных заболеваний, одобренным FDA. FDA продолжит работу с заинтересованными сторонами, чтобы продвигать разработку дополнительных новых, эффективных и безопасных методов лечения для этих пациентов ».
Наиболее частыми побочными эффектами были тошнота, рвота, диарея, инфекция, снижение аппетита и утомляемость.
Зокинви противопоказан для одновременного приема с сильными или умеренными ингибиторами и индукторами CYP3A, а также с мидазоламом и некоторыми лекарствами, снижающими уровень холестерина. У некоторых пациентов, получавших Zokinvy, развились отклонения лабораторных тестов, такие как изменения уровня натрия и калия в крови, снижение количества лейкоцитов и повышение анализов крови печени.
PRF, созданная семьей Сэма Бернса после того, как ему поставили диагноз в возрасте 2 лет, также сотрудничала с Национальными институтами здравоохранения по лечению; во всем мире около 400 детей страдают этим расстройством.Бернс умер в 2014 году.
FDA одобрило первое лекарство от прогерии, болезни, вызывающей быстрое старение
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило лечение, которое может дать детям с редким генетическим заболеванием, вызывающим преждевременное старение, больше времени на жизнь.
Дети с заболеванием, известным как синдром прогерии Хатчинсона-Гилфорда, или сокращенно прогерия, часто умирают от сердечной недостаточности, сердечного приступа или инсульта в подростковом возрасте. Большинство детей с этим заболеванием умирают, не дожив до 15 лет.Недавно одобренный препарат, названный Zokinvy, является первым и единственным одобренным препаратом для лечения прогерии и некоторых родственных синдромов, объявило FDA 20 ноября.
В клинических испытаниях 62 детей, принимавших препарат, Zokinvy увеличил продолжительность жизни в среднем примерно на 3 месяца в течение первых трех лет лечения по сравнению с другим 81 ребенком, которые не принимали препарат, из отдельного исследования, в котором были собраны данные об их здоровье. Наблюдение за детьми, которые продолжали получать Зокинви до 11 лет, показало, что в среднем продолжительность жизни детей увеличивалась примерно на 2 года.5 лет.
«Это не лекарство», — предупреждает Моника Кляйнман, педиатрический врач интенсивной терапии Бостонской детской больницы, которая принимала участие в клинических испытаниях. «Мы надеемся продлить продолжительность жизни [детей] за счет замедления темпов заболевания», но, по ее словам, препарат не дает детям нормальной продолжительности жизни.
Подпишитесь на последние новости от
Science News
Заголовки и резюме последних новостей Science News статей, доставленных на ваш почтовый ящик
По оценкам, от 350 до 400 детей во всем мире страдают прогерией.Для этих детей одна мутация в их генетическом коде ухудшает их здоровье ( SN: 2/7/13 ). Эта мутация нарушает работу гена, ответственного за образование ламина А, который помогает удерживать ядра клеток вместе. Дети с прогерией в конечном итоге вырабатывают большее количество дефектного белка, называемого прогерином, который похож на ламин А, но с прикрепленным дополнительным фрагментом. По словам Клейнмана, этот белок застревает в клеточных мембранах и не может быть переработан для получения свежих белков, что приводит к преждевременному старению клеток и повышению жесткости кровеносных сосудов и соединительной ткани.
Все вырабатывают прогерин, и организм вырабатывает больше с возрастом, объясняет Клейнман, но «дети с прогерией зарабатывают очень много». Дети обычно кажутся нормальными при рождении, но начинают проявлять признаки болезни в первые два года жизни. На протяжении жизни эти дети испытывают потерю волос и жировых отложений, жесткость суставов, сердечно-сосудистые заболевания и другие симптомы ускоренного старения.
Zokinvy, производства компании Eiger BioPharmaceuticals из Пало-Альто, Калифорния., блокирует выработку прогерина, снижая его количество, которое накапливается в детских клетках. Но пероральный препарат, принимаемый в виде капсул, не полностью блокирует выработку, говорит она, и количество, которое могут получить пациенты, ограничено побочными эффектами препарата, включая рвоту, диарею и усталость.
Этот препарат является «свидетельством силы фундаментальных исследований», — говорит Том Мистели, клеточный биолог из Национального института рака в Бетесде, штат Мэриленд, который не участвовал в работе над препаратом.По его словам, Зокинви основывается на десятилетиях исследований многих аспектов белка ламина А, включая «кажущуюся эзотерической химической модификацией», которая формирует прогерин.
«Никто, изучающий этот белок или его модификацию, не мог ожидать, что он станет мишенью для лекарств», — добавляет Мистели. Но как только ген, вызывающий заболевание, был идентифицирован, исследователи сосредоточили внимание на классе лекарств, который включает Zokinvy в качестве потенциального лечения.
По словам Мистели, после одобрения нового препарата основное внимание уделяется тестированию дополнительных лекарств или терапевтических средств в сочетании с Zokinvy.Это может еще больше продлить жизнь детей с прогерией. Исследователи также изучают подходы к генной терапии с целью исправления мутации, вызывающей изнурительное заболевание.
прогерии | Определение, типы, симптомы и факты
Прогерия , любое из нескольких редких заболеваний человека, связанных с преждевременным старением. Двумя основными типами прогерии являются синдром прогерии Хатчинсона-Гилфорда (HGPS), который возникает в раннем детстве, и синдром Вернера (прогерия у взрослых), который возникает в более позднем возрасте.Третье состояние, синдром Галлермана-Штрейффа-Франсуа, характеризуется наличием прогерии в сочетании с карликовостью и другими признаками аномального роста. Прогерия встречается крайне редко; например, глобальная заболеваемость синдромом прогерии Хатчинсона-Гилфорда составляет примерно один на каждые четыре-восемь миллионов рождений.
прогерия
Ребенок с синдромом прогерии Хатчинсона-Гилфорда.
Gallo Images / Alamy
Британская викторина
44 вопроса из самых популярных викторин «Британника» о здоровье и медицине
Что вы знаете об анатомии человека? Как насчет медицинских условий? Мозг? Вам нужно будет много знать, чтобы ответить на 44 самых сложных вопроса из самых популярных викторин Britannica о здоровье и медицине.
Признаки синдрома прогерии Хатчинсона-Гилфорда появляются примерно в возрасте одного года после явно нормального младенчества. Пострадавшие люди редко превышают рост нормального 5-летнего ребенка, хотя к 10 годам они имеют физический облик 60-летнего взрослого человека. Многие из поверхностных аспектов старения, такие как облысение, истончение кожа, выступающие кровеносные сосуды волосистой части головы и сосудистые заболевания. Половые органы остаются маленькими и недоразвитыми.Некоторые люди с прогерией являются умственно неполноценными, но большинство из них имеют нормальный интеллект и могут быть даже преждевременно развиты. К 10 годам развиваются обширный артериосклероз и сердечные заболевания, и большинство пациентов умирают, не дожив до 30 лет; средний возраст смерти 13 лет. Состояние не передается по наследству. Скорее, это результат спонтанной мутации гена, известного как LMNA (ламин A / C). Только одна копия мутировавшего гена необходима в каждой клетке, чтобы вызвать заболевание; следовательно, это аутосомно-доминантное заболевание.
Синдром Вернера обычно появляется после полового созревания, причем видимые признаки старения становятся наиболее очевидными после 20 лет. Изменения в старении таковы, что больные выглядят примерно на 30 лет старше своего хронологического возраста. Люди могут не достигать своего прогнозируемого роста взрослого человека, поскольку скачок роста в подростковом возрасте может быть притуплен. Больные синдромом Вернера половозрелые, но вторичные половые признаки развиты слабо. К поверхностным признакам старения относятся преждевременное облысение или поседение волос, потеря зубов и слуха, катаракта, эпизоды острого артрита, язвы на коже и остеопороз (потеря костной ткани).Существует повышенная тенденция к развитию сердечных заболеваний, сахарного диабета и рака, а средняя продолжительность жизни составляет 47 лет. Синдром Вернера вызывается мутациями в гене WRN (синдром Вернера, RecQ-геликазоподобный). Этот тип прогерии является наследственным и передается по рецессивному признаку (две копии мутировавшего гена, по одной от каждого родителя, вызывают заболевание).
Первоначально прогерия изучалась как модель нормального старения, но, поскольку поражены не все органы, это больше не считается приемлемым.Например, нет никаких свидетельств дряхлости или старения центральной нервной системы. Есть некоторые экспериментальные данные, свидетельствующие о том, что лечение препаратом, известным как n-ацетилцистеин, может замедлить процесс старения, связанный с синдромом прогерии Хатчинсона-Гилфорда.
Получите подписку Britannica Premium и получите доступ к эксклюзивному контенту.
Подпишитесь сейчас
Прогероидные синдромы Вернера и Хатчинсона-Гилфорда: механистическая основа прогероидных заболеваний человека
Martin, G.М. Генетические синдромы у человека, потенциально имеющие отношение к патобиологии старения. Врожденные дефекты Ориг. Artic. Сер. 14 , 5–39 (1978).
CAS
PubMed
Google Scholar
Friedberg, E.C. et al. Ремонт ДНК и мутагенез (ASM Press, Вашингтон, 2006).
Google Scholar
Вернер, О. в Синдром Вернера и старение человека (ред. Солк, Д., Фудзивара, Ю. и Мартин, Г. М.) 1–14 (Plenum Press, Нью-Йорк, 1985).
Книга
Google Scholar
Эпштейн, К. Дж., Мартин, Г. М., Шульц, А. Л. и Мотульски, А. Г. Вернер синдром: обзор его симптоматологии, естественного течения, патологических особенностей, генетики и связи с естественным процессом старения. Медицина (Балтимор) 45 , 177–221 (1966). Подробное описание клинических, патологических, клеточных и генетических особенностей синдрома Вернера.
Артикул
CAS
Google Scholar
Толлефсбол, Т. О. и Коэн, Х. Дж. Синдром Вернера: недиагностируемое заболевание, напоминающее преждевременное старение. Возраст 7 , 75–88 (1984).
Артикул
Google Scholar
Гото, М. Иерархическое ухудшение систем организма при синдроме Вернера: последствия для нормального старения. мех.Aging Dev. 98 , 239–254 (1997).
Артикул
CAS
PubMed
Google Scholar
Huang, S. et al. Спектр мутаций WRN у пациентов с синдромом Вернера. Хум. Мутат. 27 , 558–567 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Гото, М. Клинические характеристики синдрома Вернера и других синдромов преждевременного старения: характер старения при прогероидных синдромах. Gann. Монография. Cancer Res. 49 , 27–39 (2001).
Google Scholar
Uhrhammer, N.A. et al. Синдром Вернера и мутации генов WRN и LMNA во Франции. Хум. Мутат. 27 , 718–719 (2006).
Артикул
PubMed
Google Scholar
Yu, C.E. et al. Позиционное клонирование гена синдрома Вернера. Science 272 , 258–262 (1996).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Бахрати, К. З. и Хиксон, И. Д. Геликазы RecQ: супрессоры онкогенеза и преждевременного старения. Biochem. J. 374 , 577–606 (2003).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Опреско, П.Л., Ченг, В. Х. и Бор, В. А. Соединение биохимии геликазы RecQ и болезней человека. J. Biol. Chem. 279 , 18099–18102 (2004).
Артикул
CAS
PubMed
Google Scholar
Goto, M. et al. Иммунологическая диагностика синдрома Вернера по продуктам подавленных и усеченных генов. Хум. Genet. 105 , 301–307 (1999).
Артикул
CAS
PubMed
Google Scholar
Мозер, М.J. et al. Экспрессия геликазы WRN в клеточных линиях синдрома Вернера. Nucleic Acids Res. 28 , 648–654 (2000).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Kawabe, T. et al. Дифференциальная регуляция геликаз семейства RecQ человека в трансформации клеток и клеточном цикле. Онкоген 19 , 4764–4772 (2000).
Артикул
CAS
PubMed
Google Scholar
Суонсон, К., Saintigny, Y., Emond, M.J. и Monnat, R.J. мл. Белок синдрома Вернера имеет раздельные функции рекомбинации и выживания. Восстановление ДНК (Amst) 3 , 475–482 (2004).
Артикул
CAS
Google Scholar
Moser, M. J. et al. Генетическая нестабильность и риск гематологических заболеваний у пациентов с синдромом Вернера и гетерозигот. Cancer Res. 60 , 2492–2496 (2000).
CAS
PubMed
Google Scholar
Огберн, К.E. et al. Генотоксин, индуцирующий апоптоз, отличает гетерозиготных носителей мутаций геликазы Вернера от мутантов дикого типа и гомозиготных мутантов. Хум. Genet. 101 , 121–125 (1997).
Артикул
CAS
PubMed
Google Scholar
Poot, M. et al. Клетки с синдромом Вернера чувствительны к сшивающим ДНК препаратам. FASEB J. 15 , 1224–1226 (2001).
Артикул
CAS
PubMed
Google Scholar
Шелленберг, Г.D., Miki, T. Yu, CE и Nakura, J. in The Metabolic and Molecular Basis of Inherited Disease (eds Scriver, CR, Beaudet, AL, Sly, WS & Valle, D.) 785–797 (McGraw -Хилл, Нью-Йорк, 2001).
Google Scholar
Agrelo, R. et al. Эпигенетическая инактивация гена синдрома Вернера преждевременного старения при раке человека. Proc. Natl Acad. Sci. США 103 , 8822–8827 (2006). Недавние доказательства того, что эпигенетическое подавление экспрессии WRN дает клеточные фенотипы, аналогичные мутациям WRN , и что WRN может иметь значимое влияние на рак толстой кишки.
Артикул
CAS
PubMed
Google Scholar
Гото М., Миллер Р. В., Исикава Ю. и Сугано Х. Избыток редких видов рака при синдроме Вернера (прогерия у взрослых). Cancer Epidemiol. Биомаркеры Пред. 5 , 239–246 (1996).
CAS
PubMed
PubMed Central
Google Scholar
Мушегян А. Р., Бассетт Д.E. Jr, Boguski, M. S., Bork, P. & Koonin, E. V. Позиционно клонированные гены болезней человека: паттерны эволюционной консервации и функциональные мотивы. Proc. Natl Acad. Sci. США 94 , 5831–5836 (1997).
Артикул
CAS
PubMed
Google Scholar
Mian, I. S. Сравнительный анализ последовательности рибонуклеаз HII, III, II PH и D. Nucleic Acids Res. 25 , 3187–3195 (1997).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Принс П. Р., Эмонд М. Дж. И Моннат Р. Дж. Младший. Потеря функции белка синдрома Вернера способствует аберрантной митотической рекомбинации. Genes Dev. 15 , 933–938 (2001).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Сентиньи, Ю., Макиенко, К., Суонсон, К., Эмонд, М. Дж. И Моннат, Р. Дж. Дефект разрешения гомологической рекомбинации при синдроме Вернера. Мол. Клетка. Биол. 22 , 6971–6978 (2002). Ссылки 25 и 26 определяют роль WRN в гомологически зависимой рекомбинации в соматических клетках человека.
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Стюарт, Э., Чепмен, К. Р., Аль-Ходейри, Ф., Carr, A. M. & Enoch, T. rqh2 + , ген делящихся дрожжей, связанный с генами синдромов Блума и Вернера, необходим для обратимой остановки S-фазы. EMBO J. 16 , 2682–2692 (1997).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Pichierri, P., Franchitto, A., Mosesso, P. & Palitti, F. Белок синдрома Вернера необходим для правильного восстановления после остановки репликации и повреждения ДНК, индуцированного в S-фазе клеточного цикла. Мол. Биол. Ячейка 12 , 2412–2421 (2001).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Опреско, П. Л. и др. Хеликаза и экзонуклеаза синдрома Вернера взаимодействуют для разрешения теломерных петель D способом, регулируемым TRF1 и TRF2. Мол. Ячейка 14 , 763–774 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Чанг, С.и другие. Существенная роль лимитирующих теломер в патогенезе синдрома Вернера. Nature Genet. 36 , 877–882 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Du, X. et al. Укорочение теломер раскрывает функции генов синдрома Вернера и Блума у мышей. Мол. Клетка. Биол. 24 , 8437–8446 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Краббе, Л., Verdun, R.E., Haggblom, C.I. & Karlseder, J. Дефектный синтез отстающей цепи теломер в клетках, лишенных активности геликазы WRN. Наука 306 , 1951–1953 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Лауд, П. Р. и др. Повышенная рекомбинация теломер-теломер в WRN-дефицитных теломерных дисфункциональных клетках способствует уходу от старения и вовлечению пути ALT. Genes Dev. 19 , 2560–2570 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Crabbe, L., Jauch, A., Naeger, C. M., Holtgreve-Grez, H. & Karlseder, J. Дисфункция теломер как причина геномной нестабильности при синдроме Вернера. Proc. Natl Acad. Sci. США 104 , 2205–2210 (2007).
Артикул
CAS
PubMed
Google Scholar
Опреско, П.L. et al. POT1 стимулирует RecQ геликазы WRN и BLM раскручивать субстраты теломерной ДНК. J. Biol. Chem. 280 , 32069–32080 (2005). Ссылки 29–35 определяют роль WRN в поддержании теломер в соматических клетках человека и у Wrn -дефицитных мышей.
Артикул
CAS
PubMed
Google Scholar
Eller, M. S. et al. Роль WRN в ответах на повреждение ДНК на основе теломер. Proc. Natl Acad. Sci. США 103 , 15073–15078 (2006).
Артикул
CAS
PubMed
Google Scholar
Bai, Y. & Murnane, J. P. Нестабильность теломер в линии опухолевых клеток человека, экспрессирующих доминантно-отрицательный белок WRN. Хум. Genet. 113 , 337–347 (2003).
Артикул
CAS
PubMed
Google Scholar
Моннат, Р.J., Jr & Saintigny, Y. Синдромный белок Вернера — функция раскрутки для объяснения болезни. Sci. Aging Knowledge Environ. 2004 , re3 (2004).
Артикул
PubMed
Google Scholar
Dhillon, K. K. et al. Функциональная роль геликазы RecQ синдрома Вернера в фибробластах человека. Ячейка старения 6 , 53–61 (2007).
Артикул
CAS
PubMed
Google Scholar
Киплинг, Д., Дэвис, Т., Остлер, Э. Л. и Фарагер, Р. Г. Что прогероидные синдромы могут рассказать нам о старении человека? Наука 305 , 1426–1431 (2004).
Артикул
CAS
PubMed
Google Scholar
Франк, С. А. и Новак, М. А. Проблемы соматической мутации и рака. Bioessays 26 , 291–299 (2004).
Артикул
CAS
PubMed
Google Scholar
Campisi, J.Старение клеток, подавление опухолей и старение организма: хорошие граждане, плохие соседи. Cell 120 , 513–522 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Lombard, D. B. et al. Мутации в гене Wrn у мышей ускоряют смертность на фоне p53 -null. Мол. Клетка. Биол. 20 , 3286–3291 (2000). Первоначальное описание создания и характеристики модели мышей с дефицитом Wrn , которая воспроизводит генетические и биохимические дефекты, наблюдаемые в клетках пациентов с WS.
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Lebel, M. & Leder, P. Делеция в мышиной геликазе с синдромом Вернера индуцирует чувствительность к ингибиторам топоизомеразы и потерю клеточной пролиферативной способности. Proc. Natl Acad. Sci. США 95 , 13097–13102 (1998).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Ван, Л.и другие. Клеточные фенотипы Вернера у мышей, экспрессирующих предполагаемый доминантно-отрицательный ген WRN человека. Генетика 154 , 357–362 (2000).
CAS
PubMed
PubMed Central
Google Scholar
Хеннекам, Р. К. Синдром прогерии Хатчинсона – Гилфорда: обзор фенотипа. г. J. Med. Genet. А 140 , 2603–2624 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Хатчинсон, Дж.Врожденное отсутствие волос и молочных желез при атрофическом состоянии кожи и ее придатков. Trans Med. Чир. Soc. Эдинбург 69 , 473–477 (1886).
Артикул
CAS
Google Scholar
Eriksson, M. et al. Рецидивирующие точечные мутации de novo в ламине А вызывают синдром прогерии Хатчинсона – Гилфорда. Природа 423 , 293–298 (2003).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Де Сандре-Джованноли, А.и другие. Ламин является усечением при прогерии Хатчинсона – Гилфорда. Наука 300 , 2055 (2003).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Cao, H. & Hegele, R.A. LMNA мутировал при прогерии Хатчинсона-Гилфорда (MIM 176670), но не при прогероидном синдроме Видеманна-Раутенштрауха (MIM 264090). J. Hum. Genet. 48 , 271–274 (2003). Ссылки 48–50 описывают первоначальную идентификацию LMNA как гена, мутировавшего в HGPS.
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Verstraeten, V. L. et al. Сложная гетерозиготность по мутациям в LMNA вызывает синдром прогерии без накопления преламина А. Хум. Мол. Genet. 15 , 2509–2522 (2006).
Артикул
CAS
PubMed
Google Scholar
МакКеон, Ф.Д., Киршнер, М. В. и Капут, Д. Гомологии первичной и вторичной структуры между белками ядерной оболочки и промежуточными филаментами. Nature 319 , 463–468 (1986).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Стурман Н., Хейнс С. и Эби У. Ядерные ламины: их структура, сборка и взаимодействия. J. Struct. Биол. 122 , 42–66 (1998).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Бриджер, Дж. М., Килл, И. Р., О’Фаррелл, М. и Хатчисон, К. Дж. Внутренние ламинатные структуры в ядрах G1 дермальных фибробластов человека. J. Cell Sci. 104 , 297–306 (1993).
CAS
PubMed
Google Scholar
Кеннеди, Б.К., Барби, Д.А., Классон, М., Дайсон, Н. и Харлоу, Э. Ядерная организация репликации ДНК в первичных клетках млекопитающих. Genes Dev. 14 , 2855–2868 (2000).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Spann, T. P., Goldman, A. E., Wang, C., Huang, S. & Goldman, R. D. Изменение организации ядерного ламина ингибирует зависимую от РНК-полимеразы II транскрипцию. J. Cell. Биол. 156 , 603–608 (2002).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Ivorra, C. et al. Механизм подавления AP-1 через взаимодействие c-Fos с ламином A / C. Genes Dev. 20 , 307–320 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Frock, R. L. et al. Ламин A / C и эмерин имеют решающее значение для дифференцировки сателлитных клеток скелетных мышц. Genes Dev. 20 , 486–500 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Нитта, Р. Т., Джеймсон, С. А., Кудлоу, Б. А., Конлан, Л. А. и Кеннеди, Б. К. Стабилизация белка ретинобластомы ядерными ламинами А-типа необходима для остановки клеточного цикла, опосредованной INK4A. Мол. Клетка. Биол. 26 , 5360–5372 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Смит, Э.Д., Кудлоу Б. А., Фрок Р. Л. и Кеннеди Б. К. Ядерные ламины А-типа, прогерии и другие дегенеративные заболевания. мех. Aging Dev. 126 , 447–460 (2005).
Артикул
CAS
PubMed
Google Scholar
Sullivan, T. et al. Потеря экспрессии ламина А-типа нарушает целостность ядерной оболочки, что приводит к мышечной дистрофии. J. Cell Biol. 147 , 913–920 (1999).
CAS
PubMed
PubMed Central
Google Scholar
Николова В. и др. Дефекты ядерной структуры и функции вызывают дилатационную кардиомиопатию у мышей с дефицитом ламина A / C. J. Clin. Вкладывать деньги. 113 , 357–369 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Де Сандре-Джованноли, А. и др.Гомозиготные дефекты в LMNA , кодирующем белки ядерной оболочки ламина A / C, вызывают аутосомно-рецессивную аксональную невропатию у человека (болезнь Шарко – Мари – Тута, тип 2) и мыши. г. J. Hum. Genet. 70 , 726–736 (2002).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Chen, L. et al. LMNA мутации при атипичном синдроме Вернера. Ланцет 362 , 440–445 (2003).
Артикул
CAS
PubMed
Google Scholar
Русинол А.Э. и Синенски М.С. Фарнезилированные ламины, прогероидные синдромы и ингибиторы фарнезилтрансферазы. J. Cell Sci. 119 , 3265–3272 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Pendas, A. M. et al. Дефектный процессинг преламина А и изменения в мышцах и адипоцитах у мышей с дефицитом металлопротеиназы Zmpste24 . Nature Genet. 31 , 94–99 (2002).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Dahl, K. N. et al. Отчетливые структурные и механические свойства ядерной пластинки при синдроме прогерии Хатчинсона – Гилфорда. Proc. Natl Acad. Sci. США 103 , 10271–10276 (2006).
Артикул
CAS
PubMed
Google Scholar
Скаффиди, П.& Мистели, Т. Обращение клеточного фенотипа при болезни преждевременного старения. Синдром прогерии Хатчинсона – Гилфорда. Nature Med. 11 , 440–445 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Лю Б. и др. Геномная нестабильность при преждевременном старении на основе ламинопатии. Nature Med. 11 , 780–785 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Скаффиди, П.& Мистели, Т. Ламин A-зависимые ядерные дефекты при старении человека. Наука 312 , 1059–1063 (2006). Доказательства того, что механизмы, лежащие в основе HGPS, также могут играть роль в нормальном старении человека.
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Bergo, M.O. et al. Zmpste24 Дефицит у мышей вызывает спонтанные переломы костей, мышечную слабость и дефект обработки преламина А. Proc. Natl Acad. Sci. США 99 , 13049–13054 (2002).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Ян, С. Х. и др. Ингибитор фарнезилтрансферазы улучшает фенотип заболевания у мышей с мутацией синдрома прогерии Хатчинсона-Гилфорда. J. Clin. Вкладывать деньги. 116 , 2115–2121 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Ян, С.H. et al. Блокирование протеина фарнезилтрансферазы улучшает ядерный блеббинг в фибробластах мыши с целевой мутацией синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. США 102 , 10291–10296 (2005). Ссылка 73, а также 87 и 88 описывают три мышиные модели HGPS, которые были созданы путем мутации или изменения экспрессии Lmna .
Артикул
CAS
PubMed
Google Scholar
Фонг, Л.G. et al. Ингибитор протеин-фарнезилтрансферазы облегчает течение болезни у мышей с прогерией. Наука 311 , 1621–1623 (2006). Ссылки 72 и 74 демонстрируют, что FTI заметно снижают фенотипы заболевания на мышиных моделях HGPS.
Артикул
CAS
PubMed
Google Scholar
Маллампалли, М. П., Хьюер, Г., Бендейл, П., Гелб, М. Х. и Михаэлис, С. Ингибирование фарнезилирования обращает дефект ядерной морфологии в модели клеток HeLa для синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. США 102 , 14416–14421 (2005).
Артикул
CAS
PubMed
Google Scholar
Glynn, M. W. & Glover, T. W. Неполный процессинг мутантного ламина A при прогерии Хатчинсона-Гилфорда приводит к ядерным аномалиям, которые устраняются ингибированием фарнезилтрансферазы. Хум. Мол. Genet. 14 , 2959–2969 (2005).
Артикул
CAS
PubMed
Google Scholar
Тот, Дж.I. et al. Блокирование протеина фарнезилтрансферазы улучшает форму ядра в фибробластах людей с прогероидными синдромами. Proc. Natl Acad. Sci. США 102 , 12873–12878 (2005).
Артикул
CAS
PubMed
Google Scholar
Capell, B.C. et al. Ингибирование фарнезилирования прогерина предотвращает характерное ядерное блеббирование синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci.США 102 , 12879–12884 (2005).
Артикул
CAS
Google Scholar
Goldman, R.D. et al. Накопление мутантного ламина А вызывает прогрессивные изменения ядерной архитектуры при синдроме прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. США 101 , 8963–8968 (2004).
Артикул
CAS
Google Scholar
Делбарре, Э.и другие. Укороченный prelamin A при синдроме прогерии Hutchinson-Gilford изменяет сегрегацию гомополимеров ламина A-типа и B-типа. Хум. Мол. Genet. 15 , 1113–1122 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Варела И. и др. Ускоренное старение у мышей, дефицитных по протеазе Zmpste24 , связано с активацией передачи сигналов р53. Природа 437 , 564–568 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Huang, S. et al. Коррекция клеточных фенотипов клеток прогерии Хатчинсона – Гилфорда с помощью РНК-интерференции. Хум. Genet. 118 , 444–450 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Csoka, A. B. et al. Профилирование экспрессии в масштабе генома синдрома прогерии Хатчинсона-Гилфорда выявляет широко распространенную неправильную регуляцию транскрипции, ведущую к мезодермальным / мезенхимальным дефектам и ускоренному атеросклерозу. Клетка старения 3 , 235–243 (2004).
Артикул
CAS
PubMed
Google Scholar
Sephel, G.C., Sturrock, A., Giro, M.G. и Davidson, J.M. Повышенная выработка эластина фибробластами кожи с прогерией контролируется стабильными уровнями мРНК эластина. J. Invest. Дерматол. 90 , 643–647 (1988).
Артикул
CAS
PubMed
Google Scholar
Лемир, Дж.M. et al. Экспрессия аггрекана существенно и аномально повышена в дермальных фибробластах при синдроме прогерии Хатчинсона-Гилфорда. мех. Aging Dev. 127 , 660–669 (2006).
Артикул
CAS
PubMed
Google Scholar
Ламмердинг, Дж. И др. Дефицит Lamin A / C вызывает нарушение ядерной механики и механотрансдукции. J. Clin. Вкладывать деньги. 113 , 370–378 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Маункес, Л.К., Козлов, С., Эрнандес, Л., Салливан, Т. и Стюарт, С. Л. Прогероидный синдром у мышей вызывается дефектами ламинов А-типа. Природа 423 , 298–301 (2003).
Артикул
CAS
PubMed
Google Scholar
Varga, R. et al. Прогрессирующие дефекты гладкомышечных клеток сосудов на мышиной модели синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. США 103 , 3250–3255 (2006).H
Артикул
CAS
PubMed
Google Scholar
Фонг, Л. Г. и др. Преламин А и ламин А, по-видимому, являются незаменимыми в ядерной пластинке. J. Clin. Вкладывать деньги. 116 , 743–752 (2006).
CAS
PubMed
PubMed Central
Google Scholar
Миллер Р.А. «Ускоренное старение»: путь к пониманию примулы? Ячейка старения 3 , 47–51 (2004).
Артикул
CAS
PubMed
Google Scholar
Мартин Г. М. Генетическая модуляция стареющих фенотипов у Homo sapiens . Cell 120 , 523–532 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Niedernhofer, L. J. et al. Новый прогероидный синдром показывает, что генотоксический стресс подавляет соматотрофную ось. Природа 444 , 1038–1043 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
van der Pluijm, I. et al. Поддержание генома подавляет ось 1 гормона роста-инсулиноподобного фактора роста у мышей с синдромом Кокейна. PLoS Biology 5 , e2 (2007).
Артикул
CAS
PubMed
Google Scholar
Моннат, Р.J. Jr. От сломанного к старому: повреждение ДНК, подавление эндокринной системы IGF1 и старение. DNA Repair (в печати).
Кеньон, К. Пластичность старения: идеи долгоживущих мутантов. Cell 120 , 449–460 (2005).
Артикул
CAS
PubMed
Google Scholar
Kamath-Loeb, A. S., Welcsh, P., Waite, M., Adman, E. T. и Loeb, L. A. Ферментативная активность белка синдрома Вернера блокируется аминокислотным полиморфизмом R834C. J. Biol. Chem. 279 , 55499–55505 (2004).
Артикул
CAS
PubMed
Google Scholar
Дэвис, Т., Бэрд, Д. М., Хотон, М. Ф., Джонс, К. Дж. И Киплинг, Д. Профилактика ускоренного старения клеток при синдроме Вернера с использованием ингибитора митоген-активируемой протеинкиназы p38. J. Gerontol. Биол. Sci. Med. Sci. 60 , 1386–1393 (2005).
Артикул
PubMed
Google Scholar
Скадден, Д.Т. Ниша стволовых клеток как объект действия. Природа 441 , 1075–1079 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Коппе, Дж. П., Каузер, К., Кампизи, Дж. И Босежур, К. М. Секреция фактора роста эндотелия сосудов первичными фибробластами человека при старении. J. Biol. Chem. 281 , 29568–29574 (2006).
Артикул
CAS
PubMed
Google Scholar
Чока, А.B. et al. Новые мутации гена ламина A / C ( LMNA ) при атипичных прогероидных синдромах. J. Med. Genet. 41 , 304–308 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Пласилова М. и др. Гомозиготная миссенс-мутация в гене ламина A / C вызывает аутосомно-рецессивный синдром прогерии Хатчинсона – Гилфорда. J. Med. Genet. 41 , 609–614 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Novelli, G. et al. Мандибулоакральная дисплазия вызвана мутацией LMNA -кодирующего ламина A / C. г. J. Hum. Genet. 71 , 426–431 (2002).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Гарг, А., Когулу, О., Озкинай, Ф., Onay, H. & Agarwal, A.K. Новая гомозиготная мутация Ala529Val LMNA у турецких пациентов с мандибулоакральной дисплазией. J. Clin. Эндокринол. Метаб. 90 , 5259–5264 (2005).
Артикул
CAS
PubMed
Google Scholar
Navarro, C. L. et al. Дефекты ламина А и ZMPSTE24 (FACE-1) вызывают ядерную дезорганизацию и идентифицируют рестриктивную дермопатию как летальную неонатальную ламинопатию. Хум. Мол. Genet. 13 , 2493–2503 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Agarwal, A.K., Fryns, J.P., Auchus, R.J. и Garg, A. Металлопротеиназа цинка, ZMPSTE24, мутирует при дисплазии мандибулоакральной области. Хум. Мол. Genet. 12 , 1995–2001 (2003).
Артикул
CAS
PubMed
Google Scholar
Наварро, К.L. et al. Потеря ZMPSTE24 (FACE-1) вызывает аутосомно-рецессивную рестриктивную дермопатию и накопление предшественников ламина А. Хум. Мол. Genet. 14 , 1503–1513 (2005).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Fukuchi, K. et al. LMNA Мутация у 45-летнего японца с синдромом прогерии Хатчинсона – Гилфорда. J. Med. Genet. 41 , e67 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Cao, K. et al. Изоформа ламина А, сверхэкспрессируемая при синдроме прогерии Хатчинсона-Гилфорда, препятствует митозу в прогерии и нормальных клетках. Proc. Natl Acad. Sci. США 104 , 4949–4954 (2007).
Артикул
CAS
PubMed
Google Scholar
Дечат Т.и другие. Изменения митоза и развития клеточного цикла, вызванные мутантным ламином А, который, как известно, ускоряет старение человека. Proc. Natl Acad. Sci. США 104 , 4955–4960 (2007).
Артикул
CAS
PubMed
Google Scholar
.