Происхождение энзимопатии — Моногенные (молекулярные) болезни
Раздел предназначен исключительно для медицинских и фармацевтических работников! Если Вы не являетесь медицинским и фармацевтическим работником — покиньте раздел! Условия использования
Наиболее обширную и значимую группу молекулярных болезней составляют наследственные ферментопатии (энзимопатии), являющиеся результатом полного выпадения активности фермента. В зависимости от качественной и количественной структуры мутации ферментная активность может быть повышена, понижена или полностью отсутствовать. Частичная недостаточность — это частая причина индивидуальной непереносимости некоторых пищевых продуктов, медикаментов.
Первичным основным механизмом в происхождении энзимопатии является ферментный блок, т. е. прекращение реакции контролируемой данным ферментом с накоплением веществ-предшественников и недостатком последующих. Если первые токсичны или вторые важны для нормальной деятельности того или иного органа, появляются нарушения или симптомы первичного дефекта. Однако далеко не всегда наблюдается прямой эффект накопления или недостатка. Он может нивелироваться побочными путями обмена или пополнением недостающего продукта обмена иным путем. С другой стороны, именно продукты побочного обмена могут стать источником прямой интоксикации или ингибиции других ферментов. Иными словами, образуются сложные цепи вторичных обменных расстройств. Вместе взятые, первичные и вторичные обменные нарушения составляют сущность наследственных энзимопатии. В зависимости от значимости того или иного вида нарушенного обмена для нормальной деятельности органов и систем возникают болезни разной тяжести, классифицируемые прежде всего в зависимости от преимущественного страдания тех или иных систем (гемоглобинопатии, миопатии, наследственные атаксии, разные виды слабоумия и др.).
Следующая глава:
Моногенные заболевания
Предыдущая глава:
Моногенные (молекулярные) болезни
Энзимодиагностика и энзимотерапия.
 Достижения и перспективы развития медицинской энзимологии. Первичные и вторичные энзимопатии, примеры
Достижения и перспективы развития медицинской энзимологии. Первичные и вторичные энзимопатии, примеры
Работа добавлена: 2016-05-14
Энзимодиагностика и энзимотерапия. Достижения и перспективы развития медицинской энзимологии. Первичные и вторичные энзимопатии, примеры.
Основные направления:
1).Энзимопатология
2).Энзимодиагностика
3).Энзимотерапия
Энзимодиагностика. использование ферментов в качестве реагентов для открытия и количественного определения нормальных и анормальных химических в-в в крови, моче, желчи и т.д. Так же изучение количества и активности ферментов при патологии.
Оказалось что ряд ферментов появляется в сыворотке крови при распаде клеток; для диагностики органических и функциональных поражений органов и тканей широко применяются ферментные тесты. Известно множество тестов для количественного определения активности ферментов в организме. например уровень липазы, амилазы, трипсина резко увеличен при диабетеЮ поражения печени и поджелудочной железы.
Энзимотерапия. Использование ферментов в качестве лекарственных средств.например применяют пепсин, трипсин, химотрипсин при заболеваниях ЖКТ. Важной областью является применение ингибиторов ферментов. Например естественные ингибиторы протеиназ используются при тяжелых панкреатитах, артритов.
Энзимопатии:
1).Первичные – нарушение активности ферментов, обусловленное генетическим дефектом
Варианты:
А) фермент может не синтезироваться
Б)может синтезироваться с пониженной активностью или в недостаточном количестве
В) может быть лишен своих регуляторных свойств
2) Вторичная- нарушение активности ферментов, развив вследствие того или иного патологического состояния.
Возможно эти работы будут Вам интересны.
1. Применение сведений о ферментах в медицинской практике составляет раздел медицинской энзимологии
2. Психодиагностика нарушений развития. Первичный дефект, его вторичные последствия и компенсаторные перестройки
3. Взрывчатка. Перспективы развития взрывчатых веществ
4. Современные геополитические реалии и перспективы их развития
5. Основные ограничения и перспективы развития IPO в России
6. Перспективы развития центров обслуживания населения
7. Перспективы развития отношений России и Европейского Союза
8. Сущность организационных структур управления и перспективы их развития
9. История и перспективы развития вычислительной техники применительно к задачам АЭС
10. АНАЛИЗ И ПЕРСПЕКТИВЫ РЕАЛИЗАЦИИ СТРАТЕГИИ ИННОВАЦИОННОГО РАЗВИТИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
Страница не найдена |
Страница не найдена |
404. Страница не найдена
Архив за месяц
ПнВтСрЧтПтСбВс
567891011
12131415161718
19202122232425
2627282930
12
12
1
3031
12
15161718192021
25262728293031
123
45678910
12
17181920212223
31
2728293031
1
1234
567891011
12
891011121314
11121314151617
28293031
1234
12
12345
6789101112
567891011
12131415161718
19202122232425
3456789
17181920212223
24252627282930
12345
13141516171819
20212223242526
2728293031
15161718192021
22232425262728
2930
Архивы
Май
Июн
Июл
Авг
Сен
Окт
Ноя
Дек
Метки
Настройки
для слабовидящих
Новые технологии системной энзимотерапии в комплексном лечении ювенильных артритов | #02/07
Лечение ювенильных хронических артритов (ЮХА) является одной из актуальных проблем современной педиатрической ревматологии.
Несмотря на значительные достижения в совершенствовании методов диагностики и терапии этих заболеваний, специалисты c тревогой отмечают значительное нарастание удельного веса ЮХА в последние десятилетия. Согласно данным литературы, этот процесс обусловлен усилением негативных влияний целого комплекса экзогенных и эндогенных патогенных факторов, в том числе недостаточно четко проводимой вакцинацией, часто необоснованным назначением целого комплекса препаратов, имеющих много побочных эффектов [1, 2].
В последние годы сформированы основные принципы терапии ЮХА, представляющие собой комплекс мер по многокомпонентному противоревматическому лечению с применением различных по своему механизму действия препаратов, обладающих наряду с лечебным эффектом побочными действиями, которые, в свою очередь, требуют проведения дополнительных курсов восстановительного лечения и реабилитации больного ребенка [3].
В связи с этим крайне актуальным для оптимизации традиционных методов терапии ЮХА представляется поиск качественно новых лекарственных средств, которые могли бы, эффективно влияя на основные звенья патогенеза, контролировать патологический процесс, одновременно обладая минимальным количеством побочных эффектов и снижая медикаментозную нагрузку на организм больного ребенка [3].
Достоверно установлено, что любой патологический процесс на клеточно-мембранном уровне сопровождается дисфункцией всех энзимных систем рецепторного аппарата, вследствие чего изменяется его чувствительность (ингибиция, неадекватная гиперактивность, парадоксальная реакция). Это относится не только к эндогенным физиологическим индукторам (гормоны, ферменты, другие активные субстанции, включая токсические метаболиты), но и к экзогенным ксенобиотикам (вирусные и микробные агенты, первичные и вторичные токсические продукты) [4, 5, 6]. Кроме того, нельзя не учитывать наличие большого количества групп риска с наследственно детерминированными или врожденными формами энзимопатии. Естественно, эти расстройства негативно сказываются на функционировании общей системы гомеостаза организма больного ребенка, отсюда же вытекают свойственные всем патологическим процессам вторичные эндокринные, иммунохимические, иммунологические и метаболические сдвиги, которые сопровождаются вторичными нарушениями продукции ферментов, гормонов, цитокинов, иммуноглобулинов, других биологически активных веществ, нарушением функционирования системы фагоцитоза и другими расстройствами, требующими дополнительной коррекции [8, 9]. Кроме того, необходимо отметить, что одним из ведущих звеньев системной энзимопатии является дефицит ферментов желудочно-кишечного тракта (трипсина, химотрипсина, панкреатина, липазы, амилазы и др.), принимающих участие как в переваривании и всасывании питательных веществ, так и в защите организма от негативного воздействия окружающей среды и различных видов ксенобиотиков [7, 8, 9].
Естественно, эти расстройства негативно сказываются на функционировании общей системы гомеостаза организма больного ребенка, отсюда же вытекают свойственные всем патологическим процессам вторичные эндокринные, иммунохимические, иммунологические и метаболические сдвиги, которые сопровождаются вторичными нарушениями продукции ферментов, гормонов, цитокинов, иммуноглобулинов, других биологически активных веществ, нарушением функционирования системы фагоцитоза и другими расстройствами, требующими дополнительной коррекции [8, 9]. Кроме того, необходимо отметить, что одним из ведущих звеньев системной энзимопатии является дефицит ферментов желудочно-кишечного тракта (трипсина, химотрипсина, панкреатина, липазы, амилазы и др.), принимающих участие как в переваривании и всасывании питательных веществ, так и в защите организма от негативного воздействия окружающей среды и различных видов ксенобиотиков [7, 8, 9].
Установлено, что использование ферментных (энзимных) препаратов давно является стандартным методом терапии во многих областях медицины, особое место занимают в этом перечне перорально применяемые системные комбинации энзимов, все шире внедряемые в медицинскую практику с начала последнего десятилетия ХХ в. [10].
Системная энзимотерапия (СЭТ), впервые предложенная М. Вольфом и К. Рансбергом в 1954 г., представляет собой метод терапевтического воздействия с помощью перорально вводимых смесей энзимов растительного происхождения, воздействующих на основные патофизиологические процессы. СЭТ, ввиду универсальности оказываемых ею противовоспалительного и иммуномодулирующего эффектов, нашла широкое применение при различных заболеваниях внутренних органов [11]. Благодаря влиянию на ключевые физиологические и патофизиологические процессы СЭТ обладает целым рядом важных свойств: противовоспалительным, иммуномодулирующим, противоотечным, анальгезирующим воздействием, способностью улучшать микроциркуляцию и реологические свойства крови.
Энзимы, входящие в состав препаратов СЭТ, всасываются в тонком кишечнике, связываясь в процессе резорбции со специфическими для них антипротеазами, главным образом с α2-макроглобулином. Энзимы, входящие в такой комплекс, оказываются защищены от распознавания гуморальными и клеточными компонентами иммунной системы, поэтому даже при длительном применении они не проявляют антигенных свойств; ферментативная активность протеиназ в комплексах необратимо не подавляется. Комплексы протеаза-антипротеаза повышают цитотоксическую и фагоцитарную активность ряда иммуноцитов — гранулоцитов, макрофагов, Т-лимфоцитов, естественных киллеров; кроме того, они способны тормозить продукцию и повышать выведение циркулирующих иммунных комплексов и иммунных депозитов из тканей, восстанавливать способность лейкоцитов продуцировать интерферон. Помимо этого, они модулируют активность важнейших провоспалительных цитокинов, являющихся ключевыми факторами в патогенезе иммуновоспалительных заболеваний (фактор некроза опухоли a, интерлейкин (ИЛ-1β, ИЛ-2, ИЛ-6, ИЛ-8 и др.). Все вышеизложенное является основанием к применению препаратов СЭТ в комплексном лечении аутоиммунных заболеваний, в том числе ЮХА [12, 13, 14, 15].
Энзимы, входящие в такой комплекс, оказываются защищены от распознавания гуморальными и клеточными компонентами иммунной системы, поэтому даже при длительном применении они не проявляют антигенных свойств; ферментативная активность протеиназ в комплексах необратимо не подавляется. Комплексы протеаза-антипротеаза повышают цитотоксическую и фагоцитарную активность ряда иммуноцитов — гранулоцитов, макрофагов, Т-лимфоцитов, естественных киллеров; кроме того, они способны тормозить продукцию и повышать выведение циркулирующих иммунных комплексов и иммунных депозитов из тканей, восстанавливать способность лейкоцитов продуцировать интерферон. Помимо этого, они модулируют активность важнейших провоспалительных цитокинов, являющихся ключевыми факторами в патогенезе иммуновоспалительных заболеваний (фактор некроза опухоли a, интерлейкин (ИЛ-1β, ИЛ-2, ИЛ-6, ИЛ-8 и др.). Все вышеизложенное является основанием к применению препаратов СЭТ в комплексном лечении аутоиммунных заболеваний, в том числе ЮХА [12, 13, 14, 15].
Препараты вобэнзим и флогэнзим (Мукос Фарма, Германия) широко используются в лечении различных иммунопатологических заболеваний. Вобэнзим и флогэнзим выпускаются в виде драже, покрытых растворимой в кишечнике оболочкой, и содержат в одном драже вобэнзима: панкреатин — 100 мг, химотрипсин — 1 мг, трипсин — 24 мг, амилаза — 10 мг, липаза — 10 мг, бромелаин — 45 мг, папаин — 60 мг, рутин — 50 мг; в одной таблетке флогэнзима: трипсин — 48 мг, бромелаин — 90 мг, рутин — 100 мг. Флогэнзим и вобэнзим разрешены к применению в педиатрической практике.
Описанный выше механизм действия препаратов СЭТ позволяет применять их в лечении ЮХА, так как они положительно воздействуют на основные патогенетические механизмы этих заболеваний [16, 17].
Детская клиника Института ревматологии впервые в педиатрической практике осуществила открытое клиническое испытание препарата вобэнзим применительно к лечению ЮХА (А. В. Шайков, 1996). Исследование носило характер открытого 6-месячного испытания, в которое были включены 10 пациентов с ЮХА. Проведенная работа наглядно продемонстрировала благоприятное воздействие препарата, что было подтверждено клиническими и лабораторными показателями. Очень важным с практической точки зрения явилось отсутствие каких-либо побочных эффектов.
Исследование носило характер открытого 6-месячного испытания, в которое были включены 10 пациентов с ЮХА. Проведенная работа наглядно продемонстрировала благоприятное воздействие препарата, что было подтверждено клиническими и лабораторными показателями. Очень важным с практической точки зрения явилось отсутствие каких-либо побочных эффектов.
Обнадеживающие результаты пилотного исследования позволили приступить к проведению 6-месячного двойного слепого плацебо-контролируемого испытания, соoтветствующего требованиям GCP.
На проведение этого исследования было получено разрешение этического Комитета при Институте ревматологии РАМН.
В первый этап исследования были включены 10 детей с ЮХА (5 мальчиков и 5 девочек) в возрасте до 16 лет со следующими нозологическими формами: ювенильный ревматоидный артрит — 5 человек, псориатический артрит — 2 ребенка, артрит неуточненной этиологии — 3 человека.
Вобэнзим назначался по 5 таблеток 3 раза в день в комплексе со стандартной терапией. Согласно условиям испытания, в комплексном лечении разрешалось использование одного из нестероидных противовоспалительных препаратов (НПВП), доза которого на протяжении всего исследования должна была оставаться стабильной. Проведение внутрисуставных инъекций не разрешалось на протяжении всего периода исследования.
На втором этапе исследования, проведенном в соответствии с требованиями GCP, принимали участие 60 пациентов, при этом 30 из них получали флогэнзим, еще 30 — плацебо.
Критериями включения в исследование служили наличие активного суставного синдрома, возраст от 3 до 15 лет. Пациенты и их родители были информированы о проводимой работе и дали письменное согласие на включение в исследование (подписанное родителями или другим ответственным лицом). В исследование не включались пациенты, имеющие сопутствующий инфекционный процесс, заболевания желудочно-кишечного тракта в анамнезе, указания на непереносимость любого из компонентов исследуемого препарата. Не допускалось также одновременное участие больного в другом клиническом испытании, а также участие в таковом в течение последних 3 мес.
Не допускалось также одновременное участие больного в другом клиническом испытании, а также участие в таковом в течение последних 3 мес.
Все таблетки (флогэнзим и плацебо) имели маркировку «фирменные таблетки». Каждый пациент получал 1 «фирменную таблетку» на каждые 10 кг массы тела (дети весом до 60 кг получали по 1 таблетке 3 раза в день, весом 60 кг и выше — по 2 таблетки 3 раза в день). Согласно протоколу исследования, флогэнзим или плацебо применялись в сочетании с традиционной терапией, при этом доза НПВП не должна была меняться на протяжении всего периода исследования, расчетная доза глюкокортикостероидов не должна была превышать 0,2 мг на 1 кг массы в сутки. На период испытания не разрешалось применение внутрисуставных лекарственных препаратов.
Критерии оценки эффективности лечения
Нами осуществлялась клиническая оценка состояния элементов опорно-двигательного аппарата, при этом учитывались следующие показатели: количество активных суставов, количество пораженных суставов, количество припухших суставов, количество суставов, болезненных при пальпации.
У части пациентов проводилось УЗИ суставов, что позволило документировать и объективизировать клинические данные. Кроме того, оценивалась динамика лабораторных показателей: скорости оседания эритроцитов (СОЭ), количество тромбоцитов, лейкоцитов, С-реактивного белка, антинуклеарного фактора, циркулирующих иммунных комплексов (ЦИК), уровень иммуноглобулинов.
Эффективность и переносимость вобэнзима и флогэнзима и плацебо изучались отдельно в сравнительном аспекте.
Дети, получавшие указанные препараты, были сопоставимы по демографическим показателям, так во всех исследуемых группах преобладали девочки школьного возраста с суставной формой заболевания, олигоартикулярным вариантом поражения, умеренной степенью активности патологического процесса, негативные по ревматоидному фактору. По нозологической принадлежности у большинства детей диагноз верифицирован как ЮХА.
Мониторинг изучаемых показателей проводился через 2, 4 и 6 мес, а также в катамнезе.
Проведенная на первом этапе работа наглядно продемонстрировала достаточно высокую эффективность вобэнзима. Так, последний положительно влиял на суставной синдром и лабораторные параметры, динамику припухших, активных и пораженных суставов.
Как следует из представленных данных, количество активных суставов уменьшилось в 2 раза уже через 3 мес лечения. Не столь отчетливая положительная динамика имела место со стороны пораженных суставов. Принципиально важным с практической точки зрения представляется отсутствие побочных эффектов.
Проведенная на втором этапе сравнительная оценка эффективности флогэнзима и плацебо позволила констатировать, что данный энзимный препарат продемонстрировал свое явное преимущество перед плацебо в плане воздействия на изучаемые показатели суставного синдрома.
Известно, что о наличии воспалительного процесса свидетельствуют активные суставы. Если в контрольной группе (дети получали обычную противовоспалительную терапию и плацебо) за 6-месячный период испытания количество активных суставов снизилось со 118 до 86 (р > 0,05) — учитывается общее количество активных суставов во всей группе, то у пациентов, принимавших флогэнзим, количество активных суставов за тот же промежуток времени снизилось со 120 до 25 (р < 0,001). При этом лечебный эффект проявлялся уже начиная со 2-го месяца лечения. В контрольной группе отчетливые результаты были достигнуты только к 6-му мес лечения.
Согласно нашим данным, флогэнзим оказывал положительное воздействие на динамику припухших суставов, при этом процесс уменьшения их количества носил постепенный характер и достигал своего максимума к 6-му месяцу лечения — со 120 до 25 (р < 0,01). Положительная динамика активных и припухших (отечных) суставов находилась в прямой пропорциональной зависимости и свидетельствовала о стихании воспалительных процессов в элементах опорно-двигательного аппарата.
На фоне лечения флогэнзимом имело место положительное воздействие испытуемого препарата на динамику болезненных при пальпации суставов. Так, за 6 мес лечения количество суставов, болезненных при пальпации, снизилось со 178 до 28 (р < 0,001).
Следует отметить, что болезненность в суставах исчезала в более поздние сроки — через 4–5 мес.
Менее четкая динамика, как на фоне терапии флогэнзимом, так и в контрольной группе, наблюдалась в отношении числа пораженных суставов. В основной группе эти цифры снизились со 189 до 80 (р < 0,05), в контрольной — со 178 до 140 (р > 0,05).
При анализе результатов лабораторных показателей было отмечено, что наиболее значимое снижение уровня СОЭ (с 27,5 до 11,9 мм/ч), концентрация глобулиновых белковых фракций и иммуноглобулина M имели место у детей, получавших энзимные препараты. Кроме того, у них отмечалось снижение уровня ЦИК в сыворотке крови по истечении 3-месячного курса терапии.
Суммируя результаты проведенных исследований, можно достоверно констатировать положительное воздействие СЭТ на воспалительный процесс в суставах, что, скорее всего, связано с влиянием системной энзимотерапии на отдельные патогенетические звенья заболевания, в том числе активность провоспалительных цитокинов.
Системность и многоплановость положительных эффектов лечения вобэнзимом и флогэнзимом определили их влияние на внесуставные проявления ЮХА — кожный васкулит и кожные проявления при псориатическом артрите. У одного ребенка полностью исчез кожный васкулит, существовавший длительное время и с трудом поддававшийся терапии. У другого ребенка, страдающего псориатическим артритом с выраженными кожными высыпаниями, удалось значительно снизить интенсивность последних на фоне лечения флогэнзимом через 4 мес от начала терапии.
Клиническая эволюция суставного синдрома была документирована посредством УЗИ пораженных суставов (коленных и тазобедренных). Так, на фоне лечения флогэнзимом заметно уменьшались, а в некоторых случаях полностью нивелировались УЗИ-признаки воспалительного процесса, такие как периартикулярный отек, наличие жидкости в полости сустава, утолщение синовиальной оболочки, подколенные кисты. У одного ребенка, имевшего подколенные кисты, они исчезли на 4-м месяце терапии флогэнзимом (как уже указывалось, согласно протоколу исследования внутрисуставные инъекции не разрешались).
Так, на фоне лечения флогэнзимом заметно уменьшались, а в некоторых случаях полностью нивелировались УЗИ-признаки воспалительного процесса, такие как периартикулярный отек, наличие жидкости в полости сустава, утолщение синовиальной оболочки, подколенные кисты. У одного ребенка, имевшего подколенные кисты, они исчезли на 4-м месяце терапии флогэнзимом (как уже указывалось, согласно протоколу исследования внутрисуставные инъекции не разрешались).
Кроме того, нами было отмечено, что на фоне лечения флогэнзимом у детей улучшался аппетит, вирусные заболевания протекали легче, с более низкой температурой, быстрее наступало выздоровление.
Мы располагаем данными об отдаленной эффективности лечения флогэнзимом у 24 пациентов. Эти дети и подростки, получавшие препарат флогэнзим, неоднократно осматривались в катамнезе. Сроки наблюдения составили от 1 года до 6 лет.
Результаты катамнестического наблюдения свидетельствуют о том, что у 6 пациентов (25%) наступила ремиссия, стабилизация имела место у 13 (54%), прогрессирование заболевания отмечено у 5 больных. Нозологические формы заболевания, с достигнутой ремиссией, были представлены олигоартикулярным вариантом ЮХА (5 больных) и псориатическим артритом (1 пациентка).
Давность заболевания была небольшой и исчислялась 1–4 годами. Эти дети сейчас посещают учебные заведения, соблюдают общий режим, занимаются спортом. Качество их жизни они сами и их родители оценивают как вполне удовлетворительное.
Стабилизация патологического процесса ЮХА произошла у 13 пациентов. По нозологическим формам они были распределены следующим образом: 1 — системный вариант, 8 — полиартикулярный вариант, 3 — олигоартикулярный вариант, 1 — псориатический артрит. Продолжительность заболевания была более длительной и колебалась от 4 до 6 лет, активность не превышала 1–2 степень. Длительность стабилизации составила от 1 года до 4 лет. Рецидивирование заболевания в этой группе больных наблюдалось 1–3 раза в год, однако при этом прогрессирования суставного синдрома не отмечалось. Согласно субъективному мнению пациентов, прием препарата принес им значительное облегчение, заметно повысив качество жизни. Однако после окончания приема препарата это состояние сохранялось непродолжительное время, в силу чего больные нуждались в повторном приеме флогэнзима.
Согласно субъективному мнению пациентов, прием препарата принес им значительное облегчение, заметно повысив качество жизни. Однако после окончания приема препарата это состояние сохранялось непродолжительное время, в силу чего больные нуждались в повторном приеме флогэнзима.
У остальных 5 пациентов (21%) несмотря на лечение имело место прогрессирование суставного синдрома. Это были пациенты с более тяжелым течением заболевания. Из них трое страдали системной формой ЮХА и двое больных имели полиартикулярный вариант заболевания. У этих пациентов обострения патологического процесса наблюдались 3–4 раза в год и сопровождались прогрессированием суставного синдрома, что потребовало коррекции лечения.
Все вышеизложенное позволяет констатировать: 6-месячное применение при ЮХА препаратов системной энзимотерапии (вобэнзим, флогэнзим) свидетельствует от том, что оба препарата оказывают явное положительное воздействие на общее состояние детей и суставной синдром. При этом, как правило, терапевтический эффект начинал реализовываться на 2-м месяце лечения и максимально проявлялся к 4-му месяцу, сохраняясь в течение длительного времени. Несомненно, лучшие результаты получены при использовании энзимных препаратов в ранние сроки заболевания. Необходимо подчеркнуть, что на фоне приема энзимов практически у всех больных повышался аппетит, уменьшались симптомы хронической интоксикации, нивелировались астеновегетативные проявления.
Препараты хорошо переносятся, побочных эффектов ни у одного больного не зарегистрировано.
Катамнестические данные свидетельствуют о положительных отдаленных результатах лечения. Так, у преобладающего большинства детей нам удалось достичь ремиссии (25%) или стабилизации (54%) патологического процесса. Все вышеизложенное позволяет рекомендовать прием энзимов в качестве весомого компонента комплексной терапии хронических воспалительных заболеваний суставов у детей, особенно при олигоартикулярных вариантах заболевания и на ранних его сроках.
Литература
- Алексеева Е. И., Шахбазян И. Е. Принципы патогенетической терапии тяжелых системных вариантов ювенального ревматоидного артрита. М., 2002.
- Шахбазян И. Е. Ювенильный ревматоидный артрит: рук-во по детской ревматологии. М., 2002.
- Кузьмина Н. Н., Никишина И. П., Салугина С. О. Современная стратегия и тактика фармакотерапии ювенильных артритов// Русский медицинский журнал. 2003. 7 (11).
- Штаудер Г. Фармакологические эффекты пероральных комплексных энзимных препаратов// Материалы II Международной конференции по системной энзимотерапии. М., 1996.
- Рансбергер К. Перспективы развития системной энзимотерапии. Новейшие факторы в лечении аутоиммунных заболеваний и профилактике метастазирования// Материалы по II Международной конференции по системной энзимотерапии. М., 1996.
- Лысикова М., Вальд М., Масиновски З. Механизмы воспалительной реакции и воздействие на них с помощью протеолитических энзимов. Цитокины и воспаление. 2004. 3 (3): 48–6.
- Новые аспекты системной энзимотерапии/ под ред. проф. В.А. Виссарионова. М.: Триада-фарм, 2001. 160 с.
- Системная энзимотерапия: Практическое руководство для врачей/ под ред. В. А. Насоновой. СПб.: Интермедика, 2003. 32 с.
- Минаев С. В. Значение цитокинов в патогенезе острой хирургической патологии. Цитокины и воспаление. 2004. 2 (3). С. 41–45.
- Системная энзимотерапия. Новые подходы и перспективы. СПб.: Некоммерческое Партнерство издателей Санкт-Петербурга, 1999. 224 с.
- Рансбергер К. Новый взгляд на механизмы и перспективы системной энзимотерапии. Системная энзимотерапия. Опыт и перспективы/ под ред. В. И. Кулакова, В. А. Насоновой, В. С. Савельева. СПб.: Интер-Медика, 2004.
- Зборовский А. Б., Стажаров М. Ю., Мозговая Е. Э. Применение метода системной энзимотерапии в лечении ревматических заболеваний// Научно-практическая ревматология.
 2003. № 1. С. 64–66.
2003. № 1. С. 64–66. - Системная энзимотерапия. Опыт и перспективы/ под ред. В. И. Кулакова, В. А. Насоновой, В. С. Савельева. СПб.: Интер-Медика, 2004. 264 с.
- Desser L., Rehberger A., Kokron E. et al. Cytokine synthesis in human peripheral blood mononuclear cells after oral administration of polyenzyme preparations. Oncology. 1993. 5 (50): 403–4.
- Шайков А. В., Мовсисян Г. Р., Столярова А. В. Вобэнзим в комплексном лечении ювенильного хронического артрита// Детская ревматология. 1997. № 1.
- Кузьмина Н. Н., Шайков А. В., Столярова А. В. и др. Изучение переносимости и терапевтической эффективности вобэнзима в комплексном лечении больных ювенильным хроническим артритом// Мат. науч.-практ. конф. «Новые аспекты системной энзимотерапии». М., 2001.
 2003. № 1. С. 64–66.
2003. № 1. С. 64–66.
Г. Р. Мовсисян
Н. Н. Кузьмина, доктор медицинских наук, профессор
Институт ревматологии РАМН, Москва
Изменения печени при энзимопатиях тонкой кишки Текст научной статьи по специальности «Клиническая медицина»
■ И. А. Зайцева, С. Е. Прохорова. Факторы, влияюшие на вертикальный путь перелачи ВИЧ-инфекции
У 12 детей в возрасте до 18 месяцев, находящихся под наблюдением, антитела к антигенам ВИЧ стойко сохраняются, а качественный анализ методом ПЦР у 8 детей положительный, в то время как 6 детей (13,3%) остаются серонегативными.
Во второй группе, где беременность женщин протекала без осложнений, диагноз ВИЧ-инфекции поставлен 1 ребенку, не получавшему никакой химиопро-филактики. Все дети, рожденные от ВИЧ-инфицированных матерей, рождены в срок, лишь один ребенок с недоношенностью I степени. Из 33 детей 7 (21,2%) снято с учета в связи с исключением ВИЧ-инфекции. При исследовании методом ПЦР 18 детей в возрасте 6 и 9 месяцев качественный анализ был отрицательным. При обследовании методом ИфА и ИБ антитела к ВИЧ у 6 детей исчезли к 9 месяцам, лишь у одного ребенка сохранился «сомнительный» иммуноблот.
При обследовании методом ИфА и ИБ антитела к ВИЧ у 6 детей исчезли к 9 месяцам, лишь у одного ребенка сохранился «сомнительный» иммуноблот.
Заключение
Результаты исследования позволяют заключить, что от 78 беременных ВИЧ-инфицированных женщин родилось 8 детей с диагнозом ВИЧ-инфекция, что составило 10,3%. У 12 (15,3%) детей, находящихся под наблюдением, антитела к антигенам ВИЧ стойко сохраняются, а качественный анализ методом ПЦР у 8 детей положительный. Оценить возможность передачи вируса иммунодефицита человека от матери ребенку пока не представляется возможным в связи с тем, что дети находятся в возрасте до 18 месяцев и их ВИЧ-статус еще не уточнен.
В группе женщин, беременность которых протекала с осложнениями, вирус иммунодефицита человека передается детям в 15,5% случаев. В 26% случаев антитела к антигенам ВИЧ стойко сохраняются, однако
решение о постановке диагноза ВИЧ-инфекция будет принято только в возрасте 18 месяцев.
При отсутствии осложнений у женщины в течение беременности, инфицирование ВИЧ произошло в 3% случаев.
Предупреждение и своевременное лечение осложнений в течение беременности у женщин не менее важно, чем антиретровирусная терапия.
Литература:
1. Бобкова М. Р. Лабораторная диагностика ВИЧ-инфекции у детей первого года жизни // Клиническая лабораторная диагностика. — 2001. — № 2. — С. 25—32.
2. Определение концентрации РНК ВИЧ для оценки эффективности комбинированной терапии ВИЧ-инфекции / Е. В. Богословская и др. // Клиническая лабораторная диагностика. — 2001. — № 2. nce on global strategies for the prevention of HIV transmission from mother to infants. September 3—7, 1997, Washington, DC. — P. 10—12.
nce on global strategies for the prevention of HIV transmission from mother to infants. September 3—7, 1997, Washington, DC. — P. 10—12.
7. Center of Desease Control and Prevention: report of interagency recommendations // MMWR. — 1983. — N 32. — P. 101—104.
8. Division on AIDS. NIH. Zidovudin for the prevention of HIV transmission from mother to infant // MMWR. — 1994. — Vol. 343. — N. 8909. — P. 285—288.
Изменения печени
при энзимопатиях тонкой кишки
И. О. Уварова, А. Т. Камилова, А. Н. Арипов
Отдел гастроэнтерологии, отдел биохимии научно-исследовательского института педиатрии Минздрава Республики Узбекистан
Проблема заболеваний тонкой кишки является одной из актуальных в современной медицине, а в условиях Центральной Азии, где они распространены как среди взрослых, так и детей, их относят к краевой патологии.
Среди патогенетических механизмов заболеваний тонкой кишки существенную роль играют нарушения функционирования интестинальных ферментов, большинство из которых участвуют в гидролизе углеводов и белков. Интестинальные энзимопатии или энзимопатии тонкой кишки (ЭТК) являются наиболее широко распространенными гастроэнтерологическими заболеваниями среди детей раннего и дошкольного возраста в Республике Узбекистан [1].
Известно, что хронические заболевания тонкой кишки сопровождаются нарушениями кишечного всасывания,
отличаются длительностью течения и формируют тяжелые расстройства обмена веществ. Значительные метаболические изменения часто обусловлены недостаточным поступлением определенных нутриентов в организм ребенка.
У большинства больных внекишечные проявления при нарушении кишечного всасывания развиваются на фоне ярко выраженных кишечных симптомов. Но иногда заболевание тонкой кишки может проявляться в основном отставанием в физическом развитии, анемией, лихорадкой, геморрагиями, остеопорозом, эндокринными или иными нарушениями, затушевывающими скудную кишечную симптоматику (периодическая диарея, стеаторея, неопределенные боли в животе и т. д.) [2].
Печень является органом, обеспечивающим энергетические и пластические функции организма, а также
Таблица 1. Показатели общего белка и альбуминов сыворотки кроки у больных с энзимопатиями тонкой кишки
в значительной степени выполняющим детоксикацион-ную функцию, то есть защиту организма от «экологической агрессии среды», в том числе и эндогенной, связанной с течением и лечением многих заболеваний, так как метаболизм большинства лекарственных препаратов происходит в печени.
Даже если острое повреждение печени не переходит в хронический процесс, а заканчивается восстановлением функционального состояния органа, все равно бывшие повреждения напоминают о себе той или иной степенью дистрофии, более высокой чувствительностью печени к повреждающим агентам, ферментной несостоятельностью, ведущей к ее функциональной неполноценности [3].
Вопрос о вовлечении в патологический процесс печени при ЭТК мало изучен. Вместе с тем трудно себе представить, что печень остается интактной, хотя бы потому, что отягощающими факторами при ЭТК является развитие патологического процесса в самом кишечнике, функциональные нарушения в других органах пищеварения, в том числе в печени, нарушение ферментативной активности пищеварительных желез, дис-биотические изменения кишечника, расстройства обмена веществ, нарушение иммунитета, которые вторично могут поддерживать кишечные нарушения, создавая «порочный круг» с постоянной сменой причинно-следственных отношений [4].
Учитывая тесную функциональную и анатомическую взаимосвязь кишечника и печени, целью данной работы было изучение функционального состояния печени у детей при энзимопатиях тонкой кишки.
Материалы и методы исследования
Под наблюдением находилось 70 больных в возрасте от 1 года до 11 лет. Из них первичная энзимопатия — целиакия (Ц) установлена у 30 детей, у остальных имела место вторичная ферментопатия в виде синдрома целиакии (СЦ) (у 20) и синдрома ди-сахаридазной недостаточности (СДН) — у 20 детей.
Диагноз различных форм энзимопатий тонкой кишки ставился на основании генеалогического анамнеза, клинической картины заболевания, комплекса методов исследования, включающих биохимические, иммунологические, рентгенологические.
Биохимическая диагностика поражения печени была основана на определении активности ряда ферментов сыворотки крови, показателей, характеризующих состояние желчеобразующей, синтетической функции печени, а также степень ее повреждения. Определя-
лась активность трансаминаз сыворотки крови (АлАТ, АсАТ), гаммаглутамилтрансферазы (ГГТф), билирубин, тимоловая проба, протромбин, общий белок, альбумин унифицированными методами с помощью финских наборов аппарата фирмы «Кони» на биохимическом анализаторе «Оптима».
Заболевание печени вирусной этиологии исключалось путем констатации отсутствия маркеров гепатита В (HBsAg) и гепатита С (анти-HCV) иммунофермент-ным методом в сыворотке крови.
Ультразвуковое исследование печени и ее гемодинамики проводилось на сканерах, работающих в реальном масштабе времени, дающих изображение в оттенках серого, SSD-630 «Aloka» (Япония) и STERLING «Phili ps» (Голландия).
Результаты и их обсуждение
Ведущим признаком при всех формах энзи-мопатий тонкой кишки явилась хроническая диарея, характеризующаяся длительностью течения, клиническими симптомами непереносимости продуктов, содержащих те или иные углеводы, выраженным синдромом мальабсорбции, отсутствием эффекта от элими-национной диеты. При всех формах заболевания отмечались признаки астенизации, диспепсии, симптомы гипополивитаминоза, но при Ц и при СЦ они протекали в более тяжелой форме, чем при СДН. Нарушение трофики в виде дефицита массы тела (25,0— 30,0% и более), проявление гипополивитаминоза встречались чаще у больных с Ц.
В клинической картине заболевания были отмечены изменения со стороны печени. Так увеличение размеров печени мы регистрировали примерно с одинаковой частотой у больных с Ц, а при СЦ и СДН преобладало увеличение границ печени по срединной линии (30,0%). Уплотнение печени и пальмарная эритема были выявлены у больных с Ц в 26,6%. Значительное увеличение размеров печени, наличие плотности и болезненности при пальпации заставляли думать о хроническом поражении печени. Такие же изменения наблюдаются довольно часто и при СЦ (10,0 и 25,0%). Данное явление расценивается нами как реакция организма на интоксикационный синдром, свойственный детям с постинфекционным энтеритом, а также свидетельствует о выраженных биохимических нарушениях у больных с данной патологией.
При УЗИ печени такие признаки, как повышение эхогенности, «нечеткий» сосудистый рисунок наблюдались у 2 детей с Ц, что служило одним из критериев обеднения васкуляризации печеночной ткани, являющимся прогностически неблагоприятным показателем состояния печени.
Мы четко диагностировали жировую дистрофию печени у 8 обследуемых больных с Ц, которая проявлялась участками диффузной гиперэхогенности.
Со значительной частотой выявлялись при изучаемой патологии изменения в билиарной системе. Так, признаки хронического холецистита на УЗИ, характеризуемые уплотнением стенок желчного пузыря, чаще преобладали в группах детей с Ц (60,0%), по сравнению с СЦ и СДН (35,5 и 40,0%). О преимущественно гипотоническом типе дискинезии желчного пузыря
Наименование заболевания Общий белок Альбумин
Целиакия, п = 30 63,9 ± 1,0 38,0 ± 0,5
Синдром полиферментной недостаточности, п = 20 63,8 ± 1,1 37,0 ± 1,1
Синдром дисахаридазной недостаточности, п = 20 66,3 ± 1,4 36,8 ± 1,5
контроль 74,0 ± 0,6 61,2 ± 1,6
76
Детские инфекции 3 • 2004
Таблица 2. Показатели ферментов и билирубина сыворотки крови у больных с энзимопатиями тонкой кишки
Биохимические показатели контроль Ц, п = 30 СЦ, п = 20 СДН, п = 20
АлАТ (мкмоль/л) 0,32 ± 0,04 0,63 ± 0,004*** 0,56 ± 0,04 0,54 ± 0,04***
АсАТ (мкмоль/л) 0,22 ± 0,03 0,34 ± 0,02* 0,34 ± 0,04 0,37 ± 0,02*
Общий билирубин (мкмоль/л) 10,0 ± 0,7 9,9 ± 0,06* 11,48 ± 0,06 12.9 ± 0,3***
Прямой билирубин (мкмоль/л) 1,0 ± 0,35 3,8 ± 0,1*** 3,1 ± 0,1 4,7 ± 0,66***
Непрямой билирубин (мкмоль/л) 9,0 ± 0,5 5,8 ± 0,4*** 5,6 ± 0,41 9,7 ± 0,1*
Тимоловая проба (ед. ) 3,5 ± 0,2 4,3 ± 0,3* 4,4 ± 0,04 3,5 ± 0,1*
) 3,5 ± 0,2 4,3 ± 0,3* 4,4 ± 0,04 3,5 ± 0,1*
Щелочная фосфатаза (ед/л) 298,5 ± 10,5 265,0 ± 17,0** 274,0 ± 17,8 279,5 ± 13,8*
Гаммаглутамилтрансфераза (ед/л) 19,9 ± 1,6 27,0 ± 1,2*** 26,4 ± 1,4 27,6 ± 1,4***
Холестерин (ммоль/л) 4,9 ± 0,07 2,9 ± 0,1*** 2,7 ± 0,2 3,9 ± 0,2***
* — р < 0,05, ** — р < 0,01, *** — р < 0,001
свидетельствовали результаты холецистографии с желчегонным завтраком в больных с Ц, что можно объяснить уменьшением продукции холецистокинина и других интестинальных гормонов, что свойственно для данного заболевания [5]. В остальных группах отмечалось чередование двух типов дискинезии.
При исследовании показателей общего белка и альбумина при всех формах заболевания средние показатели их достоверно снижались, по сравнению с таковыми у здоровых (р < 0,001). Изучая количественные сдвиги уровня общего белка, альбумина в сыворотке крови мы могли отметить, что направленность этих изменений у детей с различной формой и тяжестью ЭТК сохранялась одна и та же. Таким образом, специфические изменения, свойственные детям с различными формами ЭТК, отсутствовали (таблица 1).
Исследование ферментов и билирубина в сыворотке крови у больных с ЭТК позволило нам выявить следующие изменения. Увеличение прямого билирубина было отмечено во всех группах, но эти значения находились в пределах возрастной нормы. Высокие показатели АлАТ и АсАТ были зарегистрированы во всех группах, однако более высокие значения последних констатировались у больных с Ц (таблица 2). Найденная гипертрансаминаземия может рассматриваться как внекишечное проявление целиакии при исключении всех известных причин заболевания печени, и классифицируется как криптогенная гипертрансаминаземия. Гипертрансаминаземия выявлена у больных с Ц, причем некоторые авторы указывали на прямую корреляционную связь обнаруженных изменений с повышенной проницаемостью тонкой кишки [6].
Гипертрансаминаземия выявлена у больных с Ц, причем некоторые авторы указывали на прямую корреляционную связь обнаруженных изменений с повышенной проницаемостью тонкой кишки [6].
В группе детей с Ц и СЦ нами было выявлено недостоверное повышение уровня тимоловой пробы крови, по сравнению с таковыми контрольной группы и у детей с СДН. Значения щелочной фосфатазы во всех группах детей не превышали нормальный уровень.
Характерным было снижение холестерина в сыворотке крови, достоверно отличающиеся от показателей у здоровых детей. Значительное снижение холес-
терина у больных с Ц и СЦ можно объяснить снижением поступления его с пищей, нарушением процессов обратного всасывания в кишечнике вследствие атрофи-ческих процессов, свойственных этим заболеваниям, стеатореи. Гипохолестеринемия отрицательно сказывается на синтезе желчных кислот, так как основной фактор, определяющий доступность холестерина как субстрата для синтеза желчных кислот — это уровень холестерина в плазме крови, что в свою очередь усугубляет нарушения процессов пищеварения [6].
Выводы
1. У детей с энзимопатиями тонкой кишки выявлены отчетливые клинико-биохимические и ультразвуковые изменения в печени: у 43,3% больных с Ц, у 35,0% — с СЦ, у 30,0% — с СДН.
2. Несмотря на идентичность клинических нарушений со стороны печени при энзимопатиях тонкой кишки наиболее серьезные изменения обнаружены у больных с Ц, из них у 26,6% выявлены ультразвуковые признаки жирового гепатоза.
3. Полученные данные необходимо учитывать при назначении рациональной дието- и медикаментозной терапии у детей с энзимопатиями тонкой кишки.
Литература:
1. камилова А. Т. Энзимопатии тонкой кишки у детей (эпидемиология, дифференциальный диагноз, лечение): Автореф. дис… д.м.н. — Т., 2001. — 37 с.
камилова А. Т. Энзимопатии тонкой кишки у детей (эпидемиология, дифференциальный диагноз, лечение): Автореф. дис… д.м.н. — Т., 2001. — 37 с.
2. Парфенов А. И. Системные проявления болезней кишечника // клин. мед. — 2001. — № 4. — С. 9—12.
3. Минушкин О. Н. Некоторые гепатопротекторы в лечении заболеваний печени // Леч. врач. — 2002. — № 6. — С. 55—58.
4. Логинов А. С. Хронический энтерит / А. С. Логинов, А. И. Парфенов, Н. И. Екисенина // Тер. архив. — 1989. — № 4. — С. 143—149.
5. Фролькис А. В. Энтеральная недостаточность. — Л., 1989. — 69 с.
6. Prevalence and clinical importance of hypertransaminasaemia in celiac disease / G. Nevaeck, W. Michseer, F. Wrba, P. Ferenci // Eur. J. Castroenterol. Hepatol. — 1999. — Vol. 11, N 3. — P. 283—288.
Митохондриальный вид наследования
В последнее время выделяется еще один тип наследования — митохондриальный. Митохондрии передаются с цитоплазмой яйцеклеток. Спермии не имеют митохондрий, поскольку цитоплазма элиминируется в процессе созревания мужских половых клеток. В яйцеклетке содержится около 25000 митохондрий. Каждая митохондрия содержит кольцевую хромосому. Болезни, обусловленные данным типом наследственности, передаются от матери и дочерям, и сыновьям в равной степени. Больные отцы болезнь не передают ни дочерям, ни сыновьям.
Вопрос 38. Моногенные болезни. Энзимопатии. Пример
Первичные энзимопатии
Первичные энзимопатии связаны с генетически обусловленной недостаточностью одного или нескольких ферментов. При этом дефектные ферменты наследуются, в основном, по аутосомно-рецессивному типу. Гетерозиготы, как правило, не имеют фенотипических отклонений. Первичные энзимопатии относят к метаболическим болезням, так как происходят нарушения определенных метаболических путей. При этом развитие заболевания может протекать по одному из трех ниже перечисленных вариантов.
Первичные энзимопатии относят к метаболическим болезням, так как происходят нарушения определенных метаболических путей. При этом развитие заболевания может протекать по одному из трех ниже перечисленных вариантов.
Нарушение образования конечных продуктов. Недостаток конечного продукта определенного метаболического пути может приводить к развитию клинических симптомов, характерных для данного заболевания. В качестве примера можно рассмотреть альбинизм. При альбинизме нарушен синтез в меланоцитах пигментов-меланинов. Возникновение альбинизма связано с недостаточностью фермента тирозиназы (тирозингидроксилазы) – одного из ферментов, участвующих в синтезе меланинов.
Накопление субстратов – предшественников. При недостаточности определенного фермента будут накапливаться метаболиты, а также во многих случаях и предшествующие соединения, которые в цепи метаболических превращений образуются до уровня расположения поврежденного энзима. Увеличение концентрации субстратов – предшественников дефектного фермента является ведущим звеном развития данных заболеваний. В качестве примера можно привести алкаптонурию. При этом заболевании нарушено окисление в тканях гомогентизиновой кислоты – промежуточного метаболита катаболизма тирозина. У таких больных наблюдается недостаточность фермента диоксигеназы гомогентизиновой кислоты. В результате этого увеличивается концентрация гомогентизиновой кислоты и её выведение с мочой. моча таких больных на воздухе окрашивается в черный цвет.
Нарушение образования конечных продуктов и накопление субстратов- предшественников. Отмечают заболевания, когда одновременно недостаток продукта и накопление исходного субстрата формируют клиническую картину. Их примером является болезнь Гирке, при которой наблюдается гипогликемия в перерывах между приемами пищи. Это связано с нарушением распада гликогена в печени и выходом из нее глюкозы вследствие дефекта фермента глюкозо-6-фосфатазы. Одновременно у таких пациентов увеличиваются размеры печени (гепатомегалия) вследствие накопления в ней гликогена.
Вторичные энзимопатии
Вторичные энзимопатии являются следствием тех или иных патологических процессов, сопровождающихся нарушением активности ферментов. Они наблюдаются при многих заболеваниях. Так например, причиной развития вторичной лактазной недостаточности могут являться: кишечные инфекции вирусной и бактериальной этиологии, паразитарные заболевания (лямблиоз и др.), синдром короткой кишки .
Одним из вариантов вторичных энзимопатий являются алиментарные энзимопатии – патологические состояния, обусловленные стойкими нарушениями активности ферментов в связи с характером питания. Алиментарные энзимопатии могут быть обусловлены длительным дефицитом белка в питании, нарушением биосинтеза коферментов при витаминной недостаточности. К алиментарным энзимопатиям относят и так называемые токсические энзимопатии, связанные с угнетением активности или биосинтеза отдельных ферментов различными естественными компонентами пищевых.
Нарушение метаболизма пуринов — обзор
Синдром Леша – Найхана
Синдром Леша – Найхана, Х-сцепленное рецессивное нарушение пуринового обмена, характеризуется умственной отсталостью, хореоатетоидными движениями и членовредительством. 29 Состояние вызвано дефицитом гипоксантин-гуанинфосфорибозилтрансферазы (HPRT), 30 , что приводит к гиперпродукции мочевой кислоты и клиническим признакам, связанным с этим заболеванием (таблица 24.1).Пациенты выглядят нормально при рождении и могут нормально развиваться в течение 6–8 месяцев. Первым распознаваемым признаком заболевания часто являются оранжевые кристаллы мочевой кислоты (напоминающие песчинки) в подгузнике или гематурия в первые месяцы жизни.
Начало мозговых проявлений может быть незаметным, с трудностями при сидении или стоянии без посторонней помощи, непроизвольными движениями, дистонией, спастичностью и усилением глубоких сухожильных рефлексов. Хотя умственная отсталость может различаться по степени, она обычно бывает тяжелой, и ненормальное поведение остается яркой характеристикой болезни. Основным клиническим признаком этого расстройства является потеря ткани вокруг рта или пальцев, которая возникает не из-за неспособности чувствовать боль, а в результате привычки ребенка к навязчивому саморазрушающему укусу этих областей. Со временем, без адекватных ограничений, вся нижняя губа, доступная зубам, может быть отгрызена. Лицо, пальцы и запястья также могут быть искалечены, и, поскольку маленькие дети часто кусают других, следует соблюдать осторожность при обращении с детьми с этим заболеванием.К другим деструктивным формам поведения относятся удары головой, вытягивание рук при проезде через дверные проемы, опрокидывание инвалидных колясок, тыкание глаз, пальцы в спицах инвалидной коляски и поведение трения. Другие проявления гиперурикемии, такие как кровянистые отложения, нефропатия и подагрический артрит, возникают позже.
Основным клиническим признаком этого расстройства является потеря ткани вокруг рта или пальцев, которая возникает не из-за неспособности чувствовать боль, а в результате привычки ребенка к навязчивому саморазрушающему укусу этих областей. Со временем, без адекватных ограничений, вся нижняя губа, доступная зубам, может быть отгрызена. Лицо, пальцы и запястья также могут быть искалечены, и, поскольку маленькие дети часто кусают других, следует соблюдать осторожность при обращении с детьми с этим заболеванием.К другим деструктивным формам поведения относятся удары головой, вытягивание рук при проезде через дверные проемы, опрокидывание инвалидных колясок, тыкание глаз, пальцы в спицах инвалидной коляски и поведение трения. Другие проявления гиперурикемии, такие как кровянистые отложения, нефропатия и подагрический артрит, возникают позже.
Не путать с семейной дизавтономией (Райли-Дей) и врожденной нечувствительностью к боли с ангидрозом (см. Гл. 8), другими состояниями, характеризующимися укусом и членовредительством, синдромом Леша – Найхана можно подтвердить клиническими проявлениями. и лабораторные демонстрации повышенного уровня мочевой кислоты в крови и моче. 31 Гетерозиготные самки протекают бессимптомно. Лечение включает использование аллопуринола (в дозировках 100–300 мг / день в разделенных дозах) для контроля уровня мочевой кислоты. Физические ограничения (перевязки для рук и шины на локтях) могут использоваться, чтобы помочь контролировать поведение пациентов с нанесением себе увечий. 29 При прикусывании губ может потребоваться удаление временных зубов, но постоянные зубы следует беречь, поскольку прикусывание губ обычно уменьшается с возрастом.
Патофизиология редких гемолитических анемий
Наследственная гемолитическая анемия — это разнородная группа заболеваний, многие из которых редки или очень редки.Грубо говоря, эти заболевания можно разделить на дефекты, влияющие на основной компонент эритроцитов (гемоглобинопатии), мембрану и цитоскелет (мембранопатии), метаболизм (энзимопатии) или продукцию . ..
..
Наследственная гемолитическая анемия — это разнородная группа заболеваний, многие из которых редки или очень редки. Грубо говоря, эти заболевания можно подразделить на дефекты, влияющие на основной компонент эритроцитов (гемоглобинопатии), мембрану и цитоскелет (мембранопатии), метаболизм (энзимопатии) или продукцию (гипорегенеративная анемия).Интенсивные исследования недавнего прошлого выявили многочисленные мутации в генах, вызывающих эти заболевания, и инициировали открытие других новых генов. Это расширило наши знания и привело к лучшему пониманию молекулярной основы этих заболеваний.
Несмотря на значительный прогресс, остается ряд вопросов, на которые нет ответов. Например, мы все еще далеки от понимания механизма (ов), участвующих в распознавании метаболически измененных эритроцитов и их последующего преждевременного разрушения, вторичных изменений, вызванных первичным дефектом, и их вклада в снижение выживаемости клеток, точной роли ионного канала (дис) функции в регуляции объема эритроцитов.Более того, для многих дефектов информация о корреляции генотип-фенотип скудна. А с появлением панельного анализа генов с помощью Next Generation Sequencing расшифровка сложного взаимодействия различных мутаций в разных генах становится все более актуальной. Наконец, для многих из этих редких заболеваний правильное лечение все еще неудовлетворительно.
Целью данной темы исследования является усиление биохимических, молекулярных и клинических исследований в области редких наследственных гемолитических анемий с конкретной целью дальнейшего улучшения нашего понимания патофизиологии и молекулярного фона этих заболеваний, что в конечном итоге может привести к новые терапевтические подходы.Тема исследования включает оригинальные исследования, иллюстративные отчеты о случаях, методы, обзоры и перспективы.
Ключевые слова :
(гемолитическая) анемия, мембранопатии, энзимопатии, дизеритропоэз, каннелопатии, патофизиология
Важное примечание :
Все материалы по данной теме исследования должны находиться в рамках того раздела и журнала, в который они были отправлены, как определено в их заявлениях о миссии. Frontiers оставляет за собой право направить рукопись, выходящую за рамки объема, в более подходящий раздел или журнал на любом этапе рецензирования.
Frontiers оставляет за собой право направить рукопись, выходящую за рамки объема, в более подходящий раздел или журнал на любом этапе рецензирования.
Утеряны неэнергетические эритроциты: ферментативные нарушения гликолиза эритроцитов | Кровь
В эритроците 2,3-ДПГ синтезируется и дефосфорилируется в шунте Рапопорта-Люберинга (рис. 1). Этот гликолитический обход является уникальным для эритроцитов млекопитающих и представляет собой важное физиологическое средство для регулирования сродства гемоглобина к кислороду. 106 На сродство гемоглобина к кислороду также влияют небольшие изменения pH крови, и соответствующая чувствительность к pH существует в шунте Rapoport-Luebering, который, опять же, позволяет изменять содержание 2,3-DPG для точной настройки кислородное сродство гемоглобина. Количественно 2,3-DPG является основным промежуточным продуктом гликолиза в красных кровяных тельцах, и его уровни примерно равны сумме других промежуточных продуктов гликолиза. 1 Дефицит ферментов проксимальнее стадии 2,3-DPG (т.е. дефицит PGK и PK) показывает повышенные уровни 2,3-DPG в результате соответствующих метаболических блоков и ретроградного накопления продуктов гликолиза.Повышенные уровни 2,3-DPG приводят к снижению сродства гемоглобина к кислороду, так что кислород легче переносится в ткани. Таким образом, уменьшается анемия и улучшается переносимость физических нагрузок. Этот положительный эффект отсутствует в дистальных дефектах гликолитического фермента HK, GPI, PFK, альдолазы и дефиците TPI, которые все вызывают снижение уровней 2,3-DPG. Некоторые из дистальных гликолитических ферментов (например, HK и PFK) ингибируются 2,3-DPG.
Обе реакции в шунте Рапопорта-Люберинга катализируются эритроцит-специфическим многофункциональным ферментом бисфосфоглицератмутазой (BPGM), который вырабатывает синтазу (образование 2,3, BPG) и фосфатазу (гидролиз 2,3-DPG до 3-фосфоглицерата). ) активность (рисунок 1). 107 BPGM представляет собой гомодимер с 30-кДа субъединицами, состоящими из 258 аминокислот. Фермент тесно связан с гликолитическим домашним ферментом MPGM. 108 Недавно была определена кристаллическая структура BPGM человека (запись PDB 1T8P), что подтверждает концепцию, что BPGM и MPGM являются структурно гомологичными ферментами, и дает обоснование для конкретных остатков, которые имеют решающее значение для синтазы, мутазы и фосфатазы. активность. 109
) активность (рисунок 1). 107 BPGM представляет собой гомодимер с 30-кДа субъединицами, состоящими из 258 аминокислот. Фермент тесно связан с гликолитическим домашним ферментом MPGM. 108 Недавно была определена кристаллическая структура BPGM человека (запись PDB 1T8P), что подтверждает концепцию, что BPGM и MPGM являются структурно гомологичными ферментами, и дает обоснование для конкретных остатков, которые имеют решающее значение для синтазы, мутазы и фосфатазы. активность. 109
Ген BPGM ( BPGM ) был картирован на хромосоме 7q31-34 и состоит из 3 экзонов, охватывающих более 22 т.п.н. 110 Дефицит BPGM (OMIM 222 800) — очень редкое аутосомно-рецессивное заболевание, и были описаны только 2 затронутые семьи. В первой семье у пациентов резко снизился уровень 2,3-ДПГ и увеличился уровень АТФ. 111 Клинически нормальные, гемолитическая анемия отсутствовала. Вместо этого они проявили эритроцитоз, который, вероятно, был результатом снижения уровней 2,3-DPG и, как следствие, повышенного сродства гемоглобина к кислороду. Позднее было обнаружено, что эти пациенты являются составными гетерозиготными по однонуклеотидному изменению в BPGM , который предсказывает замену высококонсервативного аргинина на цистеин по остатку 89 (BPGM Créteil I) и однонуклеотидную делецию, которая вводит преждевременный стоп-кодон. и, следовательно, кодирует усеченный пептид (BPGM Créteil II). 112 Во второй семье с дефицитом BPGM бессимптомный пробанд также проявил вторичный эритроцитоз. Он продемонстрировал заметно сниженный уровень активности фермента BPGM, тогда как его кровные родители показали нормальные уровни 2,3-DPG, но уровни активности BPGM составляли приблизительно 50% от нормы. Кроме того, у всех членов семьи заметно снизилась активность глюкозо-6-фосфатдегидрогеназы, хотя лабораторных доказательств гемолиза не было. Секвенирование ДНК гена BPGM показало, что пропозитус был гомозиготным по точечной мутации в экзоне 2, которая предсказывала замену Arg62 на Gln. 113
113
Добавка L-карнитина как потенциальная терапия при подозрении на гипераммонемическую энцефалопатию
Введение
Аммиак естественным образом вырабатывается как часть метаболического процесса, включая бактериальный гидролиз мочевины в кишечнике, цикл пуриновых нуклеотидов и трансаминирование аминокислот в скелетных мышцах. а также метаболические процессы, протекающие преимущественно в почках и печени. Обычно он выводится печенью как часть цикла мочевины.
Нарушение нормального метаболизма аммиака может привести к гипераммонемии, определяемой у взрослых как уровень аммиака> 50 г / дл. Клинический спектр проявления варьирует от летаргии, тошноты и рвоты, головной боли, атаксии, эпилепсии до энцефалопатии.1
Гипераммониемию можно разделить на первичную или вторичную. Первичные причины, как правило, проявляются в более раннем возрасте и связаны с энзимопатиями, влияющими либо на цикл мочевины, либо на окисление жирных кислот.2 Вторичная гипераммонемия может быть связана с основной патологией печени или непеченочными причинами, включая лекарственные препараты (вальпроат, 3-салицилат4 и 5- фторурацил5), желудочный обходной анастомоз после операции или использование полного парентерального питания.
Описание клинического случая
44-летняя женщина с церебральным параличом, неспособностью к обучению и ухудшением эпилептического контроля, обратилась в летаргию, уменьшила прием внутрь и усилилась одышка. При поступлении у нее случился длительный тонико-клонический приступ, который был купирован лоразепамом. После этого ее оценка комы в Глазго (GCS) снизилась до 6/15. На компьютерной томографии головы острой патологии не выявлено. Газы артериальной крови показали умеренную респираторную ацидемию. У пациента были взяты стандартные образцы крови (таблица 1).
Таблица 1 Результаты крови от поступления до постановки диагноза
Альбумин (г / л) | 31 * | 30 * | 27 * | |||
Щелочная фосфатаза (МЕ / л) | 244 * | 521 * | 584 * | |||
Аланин трансаминаза (IU / l) 63 | 197 * | 223 * | ||||
Общий билирубин (мкмоль / л) | 4 | 4 | 02 0 Время900 с) | 12. | 11,7 | — |
Активированное частичное тромбопластиновое время (с) | 40,1 * | 37,7 * | 02 0E / л) | 141 | 140 | 141 |
Калий (мЭкв / л) | 4,6 | 5.1 | 5,1 | |||
Мочевина (ммоль / л) | 3,6 | — | 4,1 | |||
Креатинин 9 (ммоль / л) | 27 | 27 |
 3
3Переведена в реанимационное отделение для респираторной поддержки. Несмотря на установление постоянного положительного давления в дыхательных путях и коррекцию респираторного ацидоза, ее сознательный уровень не улучшился.
Эмпирическое лечение энцефалита было начато в ожидании люмбальной пункции, которая не была проведена из-за технических трудностей. В течение следующих 4 дней функциональные тесты ее печени показали постепенное ухудшение. На УЗИ печени патологий не выявлено.
Не было улучшений в сознании через 96 часов после поступления, поэтому были определены уровни аммиака в плазме, которые были повышены до 100 мкг / дл (нормальный диапазон: 16-40 мкг / дл). Был поставлен подозреваемый диагноз гипераммониемической энцефалопатии, вызванной вальпроатом, вальпроат натрия был отменен, и пациентке было начато лечение лактулозой и леветирацетамом.К сожалению, определение уровня вальпроата для подтверждения диагноза не проводилось.
Не было существенных изменений в GCS, который оставался на уровне 7/15, в течение следующих 24 часов, несмотря на отмену вальпроата. Таким образом, внутривенное введение L-карнитина было начато с болюсной дозы 6 г, затем через 6 часов с последующими тремя дозами по 1 г каждые 4 часа. На следующий день уровень аммиака остался неизменным — 98 мкг / дл, с небольшим улучшением ее когнитивного уровня. Поэтому поддерживающая доза увеличивалась до 3 г каждые 6 часов.Через 48 часов после этого были измерены уровни аммиака. Они показали снижение уровня аммиака в плазме до 50 мкг / дл и возвращение когнитивного уровня к исходному уровню (рис. 1).
Поэтому поддерживающая доза увеличивалась до 3 г каждые 6 часов.Через 48 часов после этого были измерены уровни аммиака. Они показали снижение уровня аммиака в плазме до 50 мкг / дл и возвращение когнитивного уровня к исходному уровню (рис. 1).
Рис. 1 График, отображающий уровни аммиака и шкалу комы Глазго (GCS) на протяжении госпитализации, после диагностики гипераммонемии, вызванной вальпроатом, и отмены вапроата натрия, с указанием сроков вмешательства.
Обсуждение
Через 2 месяца после госпитализации лица, осуществляющие уход за пациентом, отметили, что снижение когнитивного уровня было очевидным с момента начала приема вальпроата натрия, за 6 месяцев до госпитализации с увеличением дозы через 3 месяца после начала.Разница в когнитивном уровне полностью исчезла после прекращения приема лекарств. Эта клиническая корреляция поддерживает диагноз гипераммониемической энцефалопатии, вызванной вальпроатом, при этом необходимо учитывать дальнейшую оценку частичных энзимопатий.
Часто обращаются в больницу при резком ухудшении психического статуса. В случаях, когда общая органическая причина этого снижения когнитивного статуса не была идентифицирована, следует рассмотреть вопрос о непеченочных причинах гипераммонемии.
Вальпроат натрия в настоящее время является противоэпилептическим препаратом первой линии при генерализованных судорогах, за исключением женщин детородного возраста, из-за потенциальных тератогенных эффектов. Это исключение имело место только с апреля 2018 года.6
Уровни аммиака проводятся редко, возможно, из-за недостаточной осведомленности, а также из-за сложности обращения с образцом. Поэтому анализ откладывается, что приводит к неточным результатам.7 Было высказано предположение, что добавление карнитина будет полезно в случаях тяжелой гипераммонемии, вызванной вальпроатом, которая определяется наличием любого из следующих факторов: кома, гепатотоксичность, сыворотка с вальпроевой кислотой. концентрация> 450 мкг / мл (> 3120 мкмоль / л) или гипераммониемическая энцефалопатия.Внутривенная терапия карнитином была связана с заметным увеличением выживаемости по сравнению с пероральной терапией8. Лечение 50–100 мг / кг было основано на серии тематических исследований и обзоре литературы, поскольку в настоящее время нет рандомизированных контрольных исследований. .9
концентрация> 450 мкг / мл (> 3120 мкмоль / л) или гипераммониемическая энцефалопатия.Внутривенная терапия карнитином была связана с заметным увеличением выживаемости по сравнению с пероральной терапией8. Лечение 50–100 мг / кг было основано на серии тематических исследований и обзоре литературы, поскольку в настоящее время нет рандомизированных контрольных исследований. .9
Восстановление могло быть связано с удалением вальпроата натрия. L-карнитин был начат после 1–3 периодов полувыведения, исходя из сообщенных 7–13-часовых периодов полувыведения вальпроата натрия, и, следовательно, не отвечал общепринятым критериям 4 периодов полураспада, прежде чем считался незначительной концентрацией внутри тела.10 После инфузии L-карнитина наблюдалось быстрое снижение уровня аммиака и глюкокортикостероидов, что нашло отражение в других отчетах.11
Заключение
В настоящем деле подчеркивается, что гипераммониемию следует рассматривать как дифференциал среди пациентов с необъяснимым диагнозом. резко снизился психический статус. Поскольку вальпроат натрия является широко используемым препаратом, клиницисты должны помнить о гипераммонемии как о потенциальном осложнении.
Ссылки
1 Chawla J. Клиническая презентация гипераммонемии.2017. https://emedicine.medscape.com/article/1174503-clinical#b1 (дата обращения 23.03.28).
2 Weng TI, Shih FF, Chen WJ. Необычные причины гипераммониемии в отделении неотложной помощи. Am J Emerg Med 2004; 22: 105–7.
3 Tsai MF, Chen CY. Гипераммониемическая энцефалопатия, вызванная вальпроатом, лечится гемодиализом. Ren Fail 2008; 30: 822–4.
4 Макела А.Л., Ланг Х., Корпела П. Токсическая энцефалопатия с гипераммонемией во время терапии высокими дозами салицилата. Acta Neurol Scand 1980; 61: 146–56.
5 Nott L, Price TJ, Pittman K et al.Гипераммониемическая энцефалопатия: важная причина неврологического ухудшения после химиотерапии. Лимфома Лейка 2007; 48: 1702–11.
6 КРАСИВЫЙ. Эпилепсия: диагностика и лечение. 1.8 Менеджмент. https://www.nice.org.uk/guidance/cg137/chapter/1-Guidance#management-2 (по состоянию на 23.06.2019).
Эпилепсия: диагностика и лечение. 1.8 Менеджмент. https://www.nice.org.uk/guidance/cg137/chapter/1-Guidance#management-2 (по состоянию на 23.06.2019).
7 Upadhyay R, Bleck TP, Busl KM. Гипераммониемия: что необходимо знать о мочевине: описание случая тяжелой нецирротической гипераммонемической энцефалопатии и обзор литературы. Case Rep Med 2016; 2016: 8512721.
8 Bohan TP, Helton E, McDonald I et al. Влияние лечения L-карнитином на гепатотоксичность, вызванную вальпроатом. Неврология 2001; 56: 1405–9.
9 Перротт Дж., Мерфи Н.Г., Зед П.Дж. L-карнитин при острой передозировке вальпроевой кислоты: систематический обзор опубликованных случаев. Ann Pharmacother 2010; 44: 1287–93.
10 Medscape. Вальпроевая кислота (Rx). https://reference.medscape.com/drug/depakene-stavzor-valproic-acid-343024#showall (дата обращения 23.06.2019).
11 Каттанео К.И., Рессико Ф., Вальсезия Р. и др.Внезапная гипераммониемия, вызванная вальпроатом, купированная с помощью L-карнитина у здорового пациента с биполярным расстройством: существенный обзор литературы и описание случая. Медицина (Балтимор) 2017; 96: e8117.
Лекарственные гематологические синдромы
Цель . Лекарства могут вызывать почти весь спектр гематологических нарушений, поражая лейкоциты, эритроциты, тромбоциты и систему свертывания крови. Эта статья призвана подчеркнуть широкий спектр гематологических синдромов, вызванных лекарственными средствами, и выделить некоторые из новейших лекарств и синдромов. Методы . Был проведен обзор литературы Medline по лекарственным гематологическим синдромам. Большинство отчетов и обзоров посвящены отдельным препаратам или цитопении. Результатов . Синдромы, вызванные лекарственными средствами, включают гемолитические анемии, метгемоглобинемию, аплазию эритроцитов, сидеробластную анемию, мегалобластную анемию, полицитемию, апластическую анемию, лейкоцитоз, нейтропению, эозинофилию, иммунную тромбоцитопению, микроангиопатические синдромы и синдромы гиперпоагистологии кровообращения, синдромы кровообращения, миопоагистопатические синдромы, циркулирующую гиперпоагистопатию, синдромы кровообращения. Некоторые из классических лекарств, которые, как известно, вызывают гематологические отклонения, были заменены более новыми лекарствами, включая биопрепараты, с собственными синдромами и непреднамеренными побочными эффектами. Выводы . Лекарства могут вызывать токсичность, охватывающую множество гематологических синдромов, опосредованную множеством механизмов. Врачи должны быть внимательны к потенциальным гематологическим осложнениям, вызванным ятрогенными препаратами.
Некоторые из классических лекарств, которые, как известно, вызывают гематологические отклонения, были заменены более новыми лекарствами, включая биопрепараты, с собственными синдромами и непреднамеренными побочными эффектами. Выводы . Лекарства могут вызывать токсичность, охватывающую множество гематологических синдромов, опосредованную множеством механизмов. Врачи должны быть внимательны к потенциальным гематологическим осложнениям, вызванным ятрогенными препаратами.
1. Введение
Гематологические нарушения возникают по разным механизмам и этиологии.Гематологические нарушения, вызванные лекарственными препаратами, могут охватывать почти весь спектр гематологии, затрагивая эритроциты, лейкоциты, тромбоциты и систему свертывания крови. Самые последние обзоры гематологических нарушений, вызванных лекарственными препаратами, сосредоточены на конкретных лекарствах или цитопении. Цель этого обзора — выделить широкий спектр гематологических синдромов, вызванных лекарственными препаратами, и выделить некоторые из новых описанных лекарств и синдромов. Однако из-за нехватки места этот обзор не претендует на исчерпывающий охват всех лекарственно-индуцированных гематологических дискразий.
2. Иммунная гемолитическая анемия
Иммунная гемолитическая анемия (IHA) характеризуется разрушением эритроцитов антителами, действующими против антигенов на мембране эритроцитов. Опосредованное антителами IgG или IgM, ИГА может быть идиопатическим или вторичным по отношению к инфекциям, аутоиммунным заболеваниям, лимфопролиферативным нарушениям или лекарствам. Пациенты поступают с анемией, ретикулоцитозом, непрямой гипербилирубинемией, повышенным уровнем ЛДГ с положительным результатом теста Кумбса.
ИГА, индуцированное лекарствами, может быть связано с лекарственно-зависимыми или лекарственно-независимыми антителами [1].Другие препараты могут вызывать адсорбцию неиммунологического белка на эритроцитах, обработанных лекарством. С лекарственно-независимыми аутоантителами, типичным примером которых является альфа-метил ДОФА, IHA может сохраняться в течение длительного времени даже после отмены препарата. ИГА была описана с цефалоспоринами, нестероидными противовоспалительными средствами, левакином, оксалиплатином и тейкопланином, среди других [1, 2].
ИГА была описана с цефалоспоринами, нестероидными противовоспалительными средствами, левакином, оксалиплатином и тейкопланином, среди других [1, 2].
Внутривенный иммуноглобулин Rh (D), используемый для лечения иммунной тромбоцитопенической пурпуры у пациентов, не подвергшихся спленэктомии, Rh (D) -позитивным, намеренно вызывает умеренный гемолиз, что, вероятно, объясняет его механизм действия.Однако тяжелый гемолиз с почечной недостаточностью, диссеминированным внутрисосудистым свертыванием и летальным исходом был зарегистрирован в небольшом количестве случаев [3].
Сообщалось, что флударабин, химиотерапевтический агент пуриновых нуклеозидов, ускоряет или обостряет аутоиммунную гемолитическую анемию, связанную с хроническим лимфолейкозом. Однако сочетание флударабина с ритуксимабом и циклофосфамидом может снизить этот риск [4].
3. Неиммунные гемолитические анемии
Дефицит G6PD — наиболее частая энзимопатия эритроцитов, связанная с гемолизом.Гемолиз может быть спровоцирован инфекцией, бобами и лекарствами. Чувствительность к различным лекарствам зависит от наследственной мутации и связанной с ней степени дефицита. В большинстве случаев лекарственный гемолиз проходит самостоятельно. Дефицит является Х-сцепленным, поэтому чаще и тяжелее проявляется у мужчин. Примахин, феназопиридин, нитрофурантоин и некоторые сульфаты связаны с гемолизом [5].
Рибавирин, используемый с пегинтерфероном для лечения гепатита С, был связан с анемией.Рибавирин концентрируется в красных кровяных тельцах, истощает АТФ и способствует гемолизу за счет окислительного повреждения мембран. В то время как анемия улучшается при отмене или снижении дозы рибавирина, такие стратегии могут поставить под угрозу эффективность противовирусной терапии. Сообщается, что эритропоэтин помогает смягчить анемию [6].
4. Метгемоглобинемия
Примерно в 3% гемоглобина в организме двухвалентное железо в геме окисляется при деоксигенации с образованием метгемоглобина. Большая часть этого встречающегося в природе метгемоглобина восстанавливается до гемоглобина через ферментную систему метгемоглобинредуктазы. Метгемоглобинемия, характеризующаяся избыточной выработкой метгемоглобина, вызывает нарушение транспорта кислорода. Метгемоглобинемия может быть врожденной (из-за дефектов ферментативного восстановления гемоглобина) или приобретенной. Пациенты обращаются с симптомами аноксии, цианоза, пониженного насыщения кислородом и артериальной крови шоколадно-коричневого цвета. Подтверждение диагноза производится путем измерения метгемоглобина в пробе газов артериальной крови.
Большая часть этого встречающегося в природе метгемоглобина восстанавливается до гемоглобина через ферментную систему метгемоглобинредуктазы. Метгемоглобинемия, характеризующаяся избыточной выработкой метгемоглобина, вызывает нарушение транспорта кислорода. Метгемоглобинемия может быть врожденной (из-за дефектов ферментативного восстановления гемоглобина) или приобретенной. Пациенты обращаются с симптомами аноксии, цианоза, пониженного насыщения кислородом и артериальной крови шоколадно-коричневого цвета. Подтверждение диагноза производится путем измерения метгемоглобина в пробе газов артериальной крови.
Лекарства, вызывающие метгемоглобинемию, либо непосредственно окисляют гемоглобин, либо метаболически активируются до окисляющих веществ [7]. Феназопиридин, используемый для купирования цистита, может вызывать окислительный гемолиз [8]. Дапсон, используемый при лепре, герпетиформном дерматите и профилактике пневмоцистной инфекции, метаболизируется до производного гидроксиламина [9]. Это была наиболее частая причина метгемоглобинемии в одной недавней серии исследований [10]. Примахин и местные анестетики, такие как бензокаин для местного применения или спрей (используемый перед эндоскопическими операциями на верхних отделах) и прилокаин, могут вызывать метгемоглобинемию [11–13].Также замешаны амилнитрит и изобутилнитрит [7]. Лечение включает прекращение действия возбудителя, кислорода и метиленового синего.
5. Мегалобластная анемия
Мегалобластная анемия характеризуется наличием гиперклеточного костного мозга с большими аномальными гематопоэтическими клетками-предшественниками (мегалобластами). Также встречаются лейкопения и тромбоцитопения. Мегалобластные анемии могут быть врожденными или приобретенными и чаще всего связаны с дефицитом витаминов (кобаламина) и фолиевой кислоты.Хотя они обычно являются результатом недоедания или неправильного всасывания, они также могут быть вызваны лекарственными препаратами.
Лекарства, влияющие на синтез ДНК, такие как антиметаболиты и алкилирующие агенты, некоторые антинуклеозиды, используемые против ВИЧ и других вирусов [14], могут вызывать мегалобластную анемию. Триметоприм (в высоких, расширенных дозах) и пириметамин, которые связываются с бактериальной дигидрофолатредуктазой с большей аффинностью, чем дигидрофолатредуктаза человека, были связаны с мегалобластной анемией, в первую очередь среди пациентов, уже находящихся в группе риска дефицита фолиевой кислоты.Антибиотики, такие как сульфасалазин, и противосудорожные средства, такие как фенитоин, связаны с изменениями, связанными с фолиевой кислотой, которые вызывают мегалобластную анемию, возможно, связанные с нарушением абсорбции.
Триметоприм (в высоких, расширенных дозах) и пириметамин, которые связываются с бактериальной дигидрофолатредуктазой с большей аффинностью, чем дигидрофолатредуктаза человека, были связаны с мегалобластной анемией, в первую очередь среди пациентов, уже находящихся в группе риска дефицита фолиевой кислоты.Антибиотики, такие как сульфасалазин, и противосудорожные средства, такие как фенитоин, связаны с изменениями, связанными с фолиевой кислотой, которые вызывают мегалобластную анемию, возможно, связанные с нарушением абсорбции.
Сообщалось о снижении уровней кобаламина при длительном применении антагонистов гистаминовых 2 -рецепторов и ингибиторов протонной помпы (например, омепразола) [15, 16]. Хотя эти агенты могут нарушить абсорбцию связанного с белком B 12 , клинически значимый дефицит кажется редким, несмотря на широкое использование.
6. Сидеробластная анемия
Сидеробластная анемия (СА) характеризуется кольцевидными сидеробластами (эритробластами, содержащими железоположительные гранулы, расположенные вокруг ядра) в костном мозге. При сидеробластных анемиях, которые могут передаваться по наследству или приобретаться, наблюдается нарушение биосинтеза гема в предшественниках эритроидов. Большинство сидеробластных анемий приобретаются как клональные нарушения эритропоэза. Кроме того, кольчатые сидеробласты могут быть обнаружены у истощенных пациентов, злоупотребляющих алкоголем [17].
Медикаментозная сидеробластная анемия связана с приемом изониазида [18]. Анемия купируется пироксидином или отменой изониазида. Хлорамфеникол, который в настоящее время используется редко, вызывает обратимое подавление эритропоэза и приводит к образованию кольцевых сидеробластов [17]. Линезолид, пеницилламин и дигидрохлорид триэтилентетрамина (хелатирующий агент, используемый для лечения болезни Вильсона) вызывают обратимую СА [19–21]. Миелодисплазии и вторичные острые лейкозы, вызванные химиотерапией, обсуждаемые ниже, могут первоначально проявляться как сидеробластная анемия [22].
7. Апластическая анемия
Апластическая анемия (АА), характеризующаяся панцитопенией с гипоцеллюлярным костным мозгом, может передаваться по наследству или приобретаться. Приобретенная апластическая анемия чаще всего является идиопатической, но может быть вторичной из-за воздействия токсинов, облучения, вирусов и лекарств. АК может развиваться как прямой ответ на воздействие, но также может развиваться косвенно, через иммуноопосредованные механизмы. Исторически сложилось так, что лекарственно-индуцированная АА нелегко отличить от идиопатических форм заболевания, поскольку с помощью редких сообщений о случаях трудно установить причинно-следственную связь [23].С иммунологической точки зрения отсутствие антител при апластической анемии предполагает, что лекарства не служат простыми гаптенами в инициации апластической недостаточности костного мозга.
Лекарства, вызывающие АК, включают противоревматические препараты, антитиреоидные препараты, противотуберкулезные препараты, НПВП и противосудорожные препараты. Указанные конкретные лекарственные средства включают хлорамфеникол, бутазон, сульфонамид, соли золота, пеницилламин, амидопирин, триметоприм / сульфаметоксазол, метимазол и фелбамат [24–27].
Многие препараты, которые, как сообщается, вызывают апластическую анемию, также могут чаще вызывать легкое подавление костного мозга, предполагая, что предварительное повреждение может иногда (возможно, связанное с метаболизмом хозяина) прогрессировать до более серьезного повреждения. Лечение и прогноз лекарственно-индуцированной апластической анемии похожи на идиопатические случаи [23].
8. Чистая аплазия красных клеток
Чистая аплазия красных клеток (PRCA) характеризуется нормоцитарной анемией, ретикулоцитопенией и отсутствием предшественников зрелых эритроидных клеток костного мозга.PRCA отличается от апластической анемии относительно нормальным количеством лейкоцитов и тромбоцитов. Он может быть врожденным или приобретенным. Приобретенная PRCA может быть идиопатической или вторичной, острым самоизлечивающимся заболеванием или стойкой хронической рефрактерной анемией. PRCA может возникать в связи с тимомой, лимфоидным раком, парвовирусом, ревматоидным артритом, беременностью и приемом лекарств.
Приобретенная PRCA может быть идиопатической или вторичной, острым самоизлечивающимся заболеванием или стойкой хронической рефрактерной анемией. PRCA может возникать в связи с тимомой, лимфоидным раком, парвовирусом, ревматоидным артритом, беременностью и приемом лекарств.
PRCA может быть приобретен при воздействии ряда лекарств, включая иммунодепрессанты (азатиоприн, FK506, антитимоцитарный глобулин), антибактериальные препараты (линезолид, изониазид, рифампин, хлорамфеникол), противовирусные препараты (интерферон-альфа, ламивударабин), зидовдарабин. (дифенилдрантоин, карбамазепин, вальпоровая кислота), а также хлорохин, аллопуринол, рибавирин и золото [28, 29].
Кроме того, сообщалось, что PRCA развивается после продолжительного воздействия рекомбинантного человеческого эритропоэтина (rHuEPO), особенно марки Eprex, преимущественно используемой в Европе [30–33]. Отмена rHuEPO с последующим лечением иммунодепрессантами (циклоспорин A) в течение нескольких месяцев сделало пациентов отрицательными к антителам против EPO и независимыми от переливания крови. PRCA, по-видимому, возникает преимущественно при подкожном введении у пациентов с почечной недостаточностью. Частота этого осложнения снизилась, по-видимому, в результате изменений в составе и обращении, которые могли снизить иммуногенность [33].
9. Иммунная тромбоцитопения
При иммунной пурпурной тромбоцитопении (ИТП) деструкция тромбоцитов вызывается связыванием антител с тромбоцитами, что приводит к их удалению ретикулоэндотелиальной системой (RES), а также за счет некоторой степени снижения продукции. ИТП может быть идиопатическим или связанным с вирусными инфекциями, аутоиммунными нарушениями, лимфопролиферативными нарушениями или лекарствами [34, 35].
Классическими причинами лекарственной тромбоцитопении являются хинин и хининоподобные препараты [36].Тромбоцитопения обычно бывает внезапной, тяжелой и может сопровождаться кровотечением. Тромбоцитопения, индуцированная этими лекарствами, вызывается антителами, которые не реагируют в отсутствие лекарства, но связываются с эпитопами на мембране тромбоцитов, гликопротеинами IIb / IIIa или Ib / IX, когда присутствует сенсибилизирующее лекарство. Ванкомицин также может быть связан с выраженной тромбоцитопенией и выявлением лекарственно-зависимых антител в сыворотке [37]. Длительная тромбоцитопения может возникать у пациентов с почечной недостаточностью, вероятно, из-за замедленного выведения препарата.Другие препараты, связанные с иммунной тромбоцитопенией, включают противомикробные препараты (сульфаномиды, рифампин, линезолид), противовоспалительные препараты, противоопухолевые препараты, антидепрессанты, бензодиазепины, противосудорожные средства (карбамазепин, фенитоин, вальпроевая кислота), а также сердечные и гипотензивные препараты [34, 35]. Хотя ИТП обычно развивается быстро, обычно она проходит после прекращения лечения и зависит от препарата.
Ванкомицин также может быть связан с выраженной тромбоцитопенией и выявлением лекарственно-зависимых антител в сыворотке [37]. Длительная тромбоцитопения может возникать у пациентов с почечной недостаточностью, вероятно, из-за замедленного выведения препарата.Другие препараты, связанные с иммунной тромбоцитопенией, включают противомикробные препараты (сульфаномиды, рифампин, линезолид), противовоспалительные препараты, противоопухолевые препараты, антидепрессанты, бензодиазепины, противосудорожные средства (карбамазепин, фенитоин, вальпроевая кислота), а также сердечные и гипотензивные препараты [34, 35]. Хотя ИТП обычно развивается быстро, обычно она проходит после прекращения лечения и зависит от препарата.
Хорошо известно, что гепарин связан с тромбоцитопенией, иногда с артериальным или венозным тромбозом, что обычно представляет собой гораздо большую угрозу, чем риск кровотечения [38, 39].Гепарин-индуцированная иммунная тромбоцитопения вызывается антителами против комплекса гепарина и фактора тромбоцитов 4 (PF4), что может привести к активации тромбоцитов и возникновению тромбозов. Хотя гепарин также может вызывать более легкую, не опосредованную иммунной системой тромбоцитопению, иммунная версия потенциально более серьезна. Гепарин-индуцированная тромбоцитопения возникает после воздействия как нефракционированного, так и низкомолекулярного гепарина, но менее распространена у последних. Обычно задержка составляет 5–10 дней для вновь подвергшихся воздействию пациентов, но тромбоцитопения может возникнуть в течение нескольких часов у пациентов, недавно перенесших гепарин, у которых еще есть антитела к PF4, или в течение нескольких дней для пациентов, ранее подвергшихся воздействию, у которых развивается анамнестический ответ.Иногда могут возникать венозная гангрена, некроз кожи и реакции анафилактического типа на гепарин. В соответствующих клинических условиях диагноз подтверждается свидетельством наличия антител к гепарину, которые могут быть обнаружены с помощью ряда анализов. К ним относятся более чувствительные серологические анализы (например, с помощью ELISA) и функциональные, более специфические анализы, такие как измерение высвобождения тромбоцитов серотонина C-14 в присутствии гепарина и сыворотки. Помимо прекращения использования гепарина, лечение включает антикоагулянтную терапию для снижения риска тромбоза, обычно сначала с аргатробаном, бивалрудином или лепирудином, с переходом на варфарин.Антикоагулянтную терапию следует продолжать в течение нескольких недель даже после нормализации количества тромбоцитов из-за высокого риска тромбоза в это время.
К ним относятся более чувствительные серологические анализы (например, с помощью ELISA) и функциональные, более специфические анализы, такие как измерение высвобождения тромбоцитов серотонина C-14 в присутствии гепарина и сыворотки. Помимо прекращения использования гепарина, лечение включает антикоагулянтную терапию для снижения риска тромбоза, обычно сначала с аргатробаном, бивалрудином или лепирудином, с переходом на варфарин.Антикоагулянтную терапию следует продолжать в течение нескольких недель даже после нормализации количества тромбоцитов из-за высокого риска тромбоза в это время.
Абциксимаб (химерный Fab-фрагмент), эптифибатид и тирофибан (ингибиторы лиганд-миметиков) часто используются после коронарной ангиопластики для уменьшения тромбоза за счет нарушения функции тромбоцитов за счет ингибирования взаимодействия GP IIb / IIIa-фибриноген. Однако, помимо индукции желаемой дисфункции тромбоцитов, они могут вызывать тяжелую тромбоцитопению у небольшого процента пациентов, вероятно, через лекарственно-зависимый механизм, опосредованный антителами [40].Это может начаться в течение нескольких часов или дней и обычно проходит спонтанно через 2–5 дней. Это может произойти при начальных или последующих инфузиях. Большинство пациентов выздоравливают без осложнений, хотя иногда могут возникать сильные кровотечения. Переливание тромбоцитов может быть выполнено при сильном кровотечении.
10. Тромботические микроангиопатии
Тромботические микроангиопатии (ТМА) проявляются тромбоцитопенией, микроангиопатической гемолитической анемией и симптомами окклюзии микрососудов.Они возникают из-за избыточной агрегации тромбоцитов. ТМА часто ассоциируется с низким уровнем металлопротеиназы ADAMTS13, которая индуцирует расщепление фактора фон Виллебранда (vWF). Тромботическая тромбоцитопеническая пурпура (ТТП) и гемолитико-уремический синдром (ГУС) являются основными тромботическими микроангиопатиями. ТМА может быть семейным, идиопатическим или приобретенным вследствие токсинов, беременности, инфекций (например, ВИЧ, некоторых шигелл и кишечной палочки) и лекарств .
Хотя лекарственно-индуцированный ТМА хорошо задокументирован, его механизм недостаточно изучен [41–44].Чаще всего предлагаются иммуноопосредованные или прямые факторы токсичности. У некоторых пациентов с лекарственно-индуцированным ТМА присутствуют аутоантитела к протеазе ADAMTS13, у некоторых пациентов заболеваемость ТМА, по-видимому, зависит от дозы, и во многих случаях вообще нет «намеков» на механизм.
Одним из препаратов, обычно участвующих в индукции ТМА, является иммунодепрессант циклоспорин А (СуА). CyA-индуцированный ТМА часто наблюдается у пациентов с трансплантатами (твердыми или жидкими), но также и у пациентов, получающих лечение от ревматоидного артрита и увеита.Считается, что этот механизм связан с дозозависимой токсичностью. ТМА обычно проходит после уменьшения или прекращения лечения СуА.
Другие препараты, связанные с ТМА, включают химиотерапевтические препараты митомицин-С, гемцитабин и цисплатин, а также α, -интерферон и такролимус [41–43]. У пациентов с метастатической аденокарциномой может быть трудно отличить ТМА, вызванную лекарственными средствами, от анемии, тромбоцитопении и микроангиопатии, связанной с карциноматозом. Митомицин-C — это известный нефротоксин, и есть доказательства того, что его механизм индукции ТМА является дозозависимым, прямым токсическим действием на эндотелий.
Тиенопиридины, антиагрегационные агенты тиклопидин и, реже, клопидогрель, также участвуют в индукции ТМА [44]. Связанная с тиклопидином TPA с большей вероятностью возникнет после как минимум двух недель терапии, будет связана с низкими уровнями ADAMTS13 с очевидными аутоантителами и получит пользу от плазмообменной терапии. ТМА, индуцированная клопидогрелом, как правило, возникает в течение первых двух недель лечения, с меньшей вероятностью связана с низким уровнем ADAMTS13 и аутоантителами и с меньшей вероятностью получит пользу от плазмафереза.
Хинин также может вызывать ТМА. Механизм иммуноопосредованный. У пациентов с ТМА, индуцированным хинином, были обнаружены антитела против эндотелиальных клеток, лимфоцитов и гранулоцитов, а также хинин-зависимые антитела, включая IgG или IgM, реактивные с гликопротеином тромбоцитов Ib / IX или IIb / IIIa. ТМА обычно проходит при отмене хинина вместе с плазмообменом [45].
У пациентов с ТМА, индуцированным хинином, были обнаружены антитела против эндотелиальных клеток, лимфоцитов и гранулоцитов, а также хинин-зависимые антитела, включая IgG или IgM, реактивные с гликопротеином тромбоцитов Ib / IX или IIb / IIIa. ТМА обычно проходит при отмене хинина вместе с плазмообменом [45].
11. Дисфункция тромбоцитов
Нарушения функции тромбоцитов могут быть обнаружены у пациентов с длительным временем кровотечения, но нормальным количеством тромбоцитов.Хотя индукция дисфункции тромбоцитов для снижения риска тромбоза часто является желаемой целью некоторых лекарств (таких как аспирин, клопидогрель и ингибиторы анти-GP IIb / IIIa), это также может быть нежелательным побочным эффектом [46].
Ацетилирование циклооксигеназы 1 (ЦОГ 1) приводит к нарушению синтеза важного агониста тромбоцитов тромбоксана. Аспирин необратимо ацетилирует ЦОГ 1, поэтому его действие сохраняется даже после того, как лекарство перестает циркулировать. Это контрастирует с неселективными нестероидными противовоспалительными препаратами, которые обратимо ацетилируют ЦОГ-1.Есть некоторые свидетельства того, что аспирин оказывает дозозависимое действие на агрегацию тромбоцитов [47].
Флуоксетин и некоторые трициклические антидепрессанты вызывают дисфункцию, подавляя захват серотонина. Некоторые препараты могут влиять на адгезию или агрегацию тромбоцитов, в том числе высокие дозы пенциллинов и других -лактамных антибиотиков, химиотерапевтические препараты, такие как митрамицин и даунорубицин, иммунодепрессанты и фенотиазины [46].
12. Гиперкоагуляция
Гиперкоагуляция со склонностью к артериальному и венозному тромбозу может передаваться по наследству или приобретаться.Наследственные тромбофильные состояния включают, среди прочего, фактор Лейдена, мутацию протромбина G20210A и дефицит белков или антитромбина III. Приобретенные состояния гиперкоагуляции могут быть вторичными по отношению к неподвижности, хирургическому вмешательству, травме, беременности, антифосфолипидному синдрому, раку и лекарствам.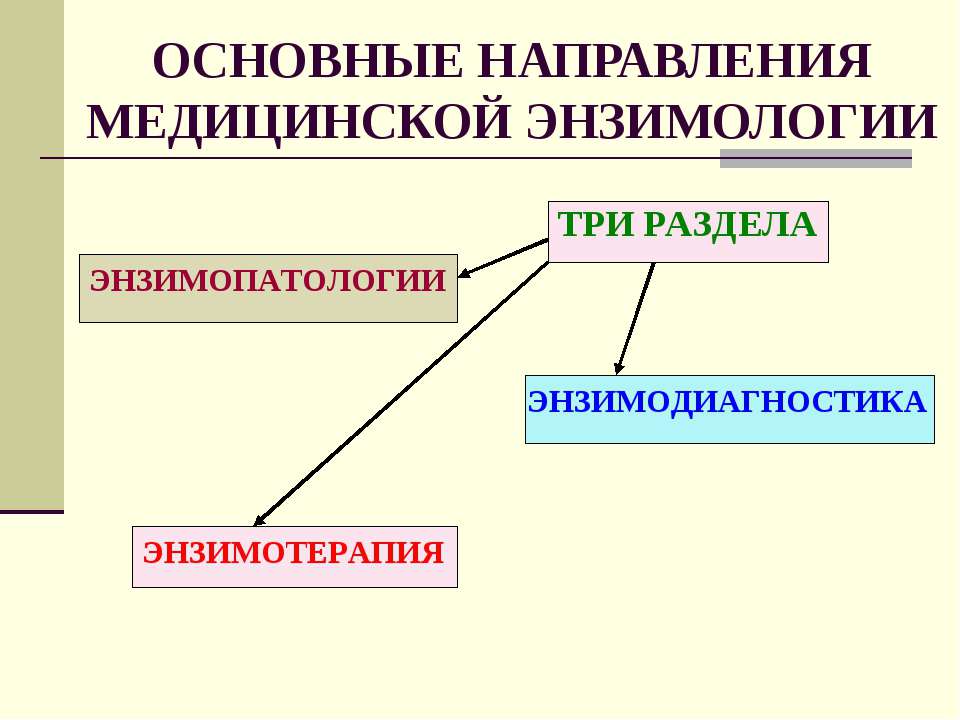
Селективные ингибиторы ЦОГ-2 с меньшим потенциалом кровотечения и желудочно-кишечной токсичности, чем традиционные ингибиторы ЦОГ, стали широко использоваться в качестве анальгетиков и противовоспалительных средств, а также были исследованы на предмет их потенциального эффекта по снижению риска полипов и колоректального рака.Однако в нескольких исследованиях было обнаружено, что целекоксиб, рофекоксиб и вальдекоксиб связаны с увеличением тромботических сердечно-сосудистых событий [48–50]. Эти результаты привели к добровольному изъятию рофекоксиба с мирового рынка и сосредоточили пристальное внимание на политике в области фармацевтики и FDA.
Эритропоэтин связан с повышенным риском тромботических заболеваний. Хотя эритропоэтин заметно улучшает анемию при почечной недостаточности, доводя уровни гемоглоблина до высоких нормальных значений, он ассоциируется с более высокими сердечно-сосудистыми заболеваниями и смертностью, чем при более низких целевых уровнях [51].Также были подняты вопросы о безопасности эритропоэтина у онкологических больных. Мета-анализ действительно показал повышенный риск тромбоза у пролеченных пациентов, а некоторые исследования показали повышенный риск смерти [52]. Рекомендации по более ограниченному и безопасному применению этих агентов у онкологических больных претерпевают изменения.
Гормональная терапия, включая оральные контрацептивы, заместительную гормональную терапию и тамоксифен (селективный модулятор рецепторов эстрогена с некоторой агонистической активностью), все связаны с повышенным риском тромбов.При применении оральных контрацептивов риск артериального и венозного тромбоза может увеличиваться с возрастом, генетическими тромбофилиями, курением и использованием определенных типов связанного прогестина [53]. Исследование Women’s Health Initiative показало, что использование комбинированного эстрогена и прогестина удваивает риск венозного тромбоза по сравнению с контролем плацебо [54]. Было обнаружено, что для химиопрофилактики рака молочной железы ралоксифен имеет более низкий риск тромбоза, а также рака матки, чем тамоксифен, и, таким образом, может быть более безопасным лекарством в этих условиях [55]. Аналогичным образом, ингибиторы ароматазы, такие как анастразол, летрозол или экземестан, используемые для лечения раннего или запущенного рака груди, демонстрируют более низкий тромботический риск, чем тамоксифен [56].
Аналогичным образом, ингибиторы ароматазы, такие как анастразол, летрозол или экземестан, используемые для лечения раннего или запущенного рака груди, демонстрируют более низкий тромботический риск, чем тамоксифен [56].
Адъювантная химиотерапия рака молочной железы с CMF (циклофосфамид, метотрексат, фторурацил), старая схема, редко используемая в настоящее время, была связана с гиперкоагуляцией [57–59]. Пациенты, получавшие CMF, могут иметь пониженные уровни белков-ингибиторов и [57]. Тромбоз у пациентов, получающих адъювантную химиотерапию, возможно, стал менее распространенным отчасти из-за изменений в типах используемой химиотерапии, более короткой продолжительности химиотерапии, менее частого использования тамоксифена у пациентов в постменопаузе и менее частого использования сопутствующего (в отличие от последовательного) ) химиотерапия и тамоксифен [58].
Тромботические осложнения были связаны с применением аспарагиназы, используемой для лечения острого лимфобластного лейкоза [60–62]. Возможны артериальные и венозные тромбозы (включая венозные синусы головного мозга). L-аспарагиназа подавляет синтез белка за счет гидролиза незаменимой аминокислоты аспарагина, вызывает снижение синтеза антитромбина III, белка и белка, что приводит к увеличению тромботического риска.
Талидомид и леналидомид, используемые при множественной миеломе, были связаны с повышенным риском тромбоза при использовании в сочетании с глюкокортикоидами [63].Рекомендованы различные профилактические меры, включая варфарин, гепарин и аспирин. Использование менее интенсивной схемы приема дексаметазона один раз в неделю снижает риск тромбоза по сравнению со стандартной четырехдневной схемой приема высоких доз в комбинации с леналидомидом [63].
Лекарственный тромбоз варфарина или гепарина может быть связан с некрозом кожи. Некроз кожи, вызванный варфарином, часто связан с уже существующими тромбофильными состояниями, такими как дефицит белка или антитромбина III [64]. Терапия состоит из отмены варфарина, введения витаминов и антикоагуляции гепарином. Однако гепарин также может вызывать некроз кожи, который может быть частью гепарин-индуцированного синдрома тромбоцитопении [65].
Терапия состоит из отмены варфарина, введения витаминов и антикоагуляции гепарином. Однако гепарин также может вызывать некроз кожи, который может быть частью гепарин-индуцированного синдрома тромбоцитопении [65].
Бевацизумаб, моноклональное антитело против фактора роста эндотелия сосудов, используется при метастатическом раке толстой кишки, легких и молочной железы, что связано с повышенным риском артериальных тромбов, особенно у пожилых людей, уже предрасположенных к сердечно-сосудистым событиям [66].
13. Циркулирующие антикоагулянты
Циркулирующие антикоагулянты подавляют факторы свертывания крови, вызывая чрезмерное кровотечение. Аутоантитела, такие как приобретенные ингибиторы фактора VIII, могут быть идиопатическими или вторичными по отношению к наследственной гемофилии, послеродовому состоянию, другим аутоиммунным нарушениям, злокачественным новообразованиям или лекарствам. У пациентов наблюдается кровотечение как синдром приобретенной гемофилии, низкий уровень F VIII: уровни и очевидные ингибиторы F VIII. Подразумеваемые лекарственные средства включают антибиотики, психотропы, флударабин и интерферон [67].Активность антител исчезает при прекращении приема препарата или при применении иммунодепрессантов. Приобретенный ингибитор фактора XIII, который перекрестно связывает и стабилизирует фибрин, был связан с изониазидом [68].
Волчаночные антикоагулянты и антифосфолипидные антитела могут быть вызваны такими лекарствами, как хлорпромазин, гидралазин, фенитоин, хинин и прокаинамид [69, 70]. В этих случаях существует связь с гиперкоагуляцией, а не с кровотечением.
14.Гипопротромбинемия
Гипопротомбинемия с удлинением ПВ / МНО чаще всего возникает из-за дефицита витаминов или заболевания печени. Некоторые лекарства, такие как антибиотики широкого спектра действия, связаны с гипопротромбинемией, обычно у пациентов, которые также страдают от недоедания. Сообщения связывают сульфаниламиды, ампициллин, хлорамфеникол, тетрациклины и цефокситин с дефицитом витаминзависимых факторов свертывания крови [71]. Цефалоспорины могут быть связаны с гипопротромбинемией, особенно с боковой цепью N-метилтиотетразола (NMTT) (например,g., моксалактам, цефоперазон), хотя они больше не используются [72].
Цефалоспорины могут быть связаны с гипопротромбинемией, особенно с боковой цепью N-метилтиотетразола (NMTT) (например,g., моксалактам, цефоперазон), хотя они больше не используются [72].
У пациентов, принимающих варфарин, многие лекарства, особенно антибиотики, вызывают усиление кровотечений. Отмечается, что систематической работы по этому вопросу было мало, а основными источниками были отчеты о случаях [73]. Тем не менее, очевидно, что многие лекарства влияют на кумадин из-за изменения фармакокинетики или динамики (например, антибиотики, особенно хинолоны, макролиды и азолы), в то время как другие увеличивают риск кровотечения за счет своих собственных механизмов (например, антибиотики, особенно хинолоны, макролиды и азолы).g., аспирин, гепарин, тиклопидин и НПВП). При назначении этих препаратов пациентам, принимающим варфарин, необходим тщательный контроль и корректировка дозы.
15. Агранулоцитоз / нейтропения
Медикаментозная нейтропения может возникать в сочетании с различными анальгетиками, психотропами, противосудорожными, антитиреоидными препаратами, антигистаминными, противоревматическими, желудочно-кишечными препаратами, противомикробными препаратами, сердечно-сосудистыми препаратами и лекарствами, как и ожидалось, с химиотерапией. [74–76]. Иммуноопосредованные механизмы связаны с некоторыми лекарствами, такими как пенициллины, которые действуют как гаптены, индуцируя образование антител против нейтрофилов.Клозапин ускоряет апоптоз нейтрофилов, а пропитиоурацил вызывает опосредованное комплементом разрушение нейтрофилов. Такие препараты, как β-лактамные антибиотики, карбамазепин и вальпроат, обладают дозозависимым ингибированием гранулопоэза. Лекарства, оказывающие прямое токсическое действие на предшественники миелоидов, включают тиклопидин, бульсуфан, метамизол, этосуксимид и хлорпромазин.
Лечение лекарственной нейтропении включает отмену препарата, охват антибиотиками, когда это необходимо, и все более частое введение рекомбинантного человеческого гранулоцитарного колониестимулирующего фактора (rhG-CSF). Использование CSF может уменьшить продолжительность нейтропении, частоту инфекции и, возможно, смертность, особенно у пациентов с глубокой нейтропенией [76]. Смертность, хотя и ниже, чем в прошлом, остается около 5%.
Использование CSF может уменьшить продолжительность нейтропении, частоту инфекции и, возможно, смертность, особенно у пациентов с глубокой нейтропенией [76]. Смертность, хотя и ниже, чем в прошлом, остается около 5%.
Ритуксимаб, антитело к CD20, используемое при лечении лимфопролиферативных нарушений В-клеток и доброкачественных аутоиммунных заболеваний, может вызывать нейтропению, обычно с отсроченным началом [77].
16. Нейтрофилия
Нейтрофилия может быть связана с миелопролиферативными заболеваниями, но чаще всего возникает в результате инфекции или воспаления.Лекарства также могут вызывать лейкоцитоз. Глюкокортикоиды вызывают нейтрофилию, вызывая высвобождение нейтрофилов из костного мозга [78]. Хотя глюкокортикоиды изменчивы, они обычно не вызывают лейкоцитоза или сдвига влево. Такое повышение количества лейкоцитов или увеличение полос может указывать на наличие инфекции [79]. Адренергические агонисты и адреналин вызывают нейтрофилию, высвобождая нейтрофилы из ограниченного пула [78]. Литий вызывает умеренную нейтрофилию и использовался для лечения нейтропении до появления спинномозговой жидкости [80].
Лейкоцитоз обычно наблюдается при приеме G / GM-CSF, который часто назначают для уменьшения тяжести и продолжительности нейтропении после химиотерапии. Синдром Свита (острый фебрильный нейтрофильный дерматоз), характеризующийся болезненными эритематозными поражениями кожи, лихорадкой и нейтрофилией, может быть вызван, среди прочего, такими лекарствами, как триметоприм-сульфаметоксазол, другими антибиотиками и гранулоцитарным колониестимулирующим фактором [81]. Синдром также может быть идиопатическим или паранеопластическим.
17. Эозинофилия
Эозинофилия может быть результатом как внутренних гематологических нарушений, так и вторичных по отношению к различным системным заболеваниям и аллергенам, включая лекарственные препараты [82].Наиболее часто ассоциированные препараты включают пенициллины, сульфаты, аллопуринол, фенитоин, карбамазепин и золото. Лекарственная сыпь с эозинофилией и системными симптомами (синдром DRESS) описывает связь эозинофилии с сыпью, лихорадкой и поражением внутренних органов, например пневмонитом, гепатитом, нефритом, аденопатией или кардитом [83]. ПЛАТЬЕ чаще всего ассоциируется с антибиотиками и противосудорожными средствами, но также могут быть задействованы и другие агенты.
Лекарственная сыпь с эозинофилией и системными симптомами (синдром DRESS) описывает связь эозинофилии с сыпью, лихорадкой и поражением внутренних органов, например пневмонитом, гепатитом, нефритом, аденопатией или кардитом [83]. ПЛАТЬЕ чаще всего ассоциируется с антибиотиками и противосудорожными средствами, но также могут быть задействованы и другие агенты.
18. Полицитемия
Полицитемия может быть первичной (истинная полицитемия) или вторичной вследствие курения, хронической гипоксии, определенных опухолей или лекарств.Лекарственная полицитемия может наблюдаться при чрезмерном использовании rHuEPO или анаболических стероидов. Злоупотребление спортсменами обоими типами препаратов может быть связано с повышенным риском тромботических заболеваний [84]. Использование диуретиков с сокращением объема может способствовать псевдополицитемии, когда гематокрит повышается из-за гемоконцентрации, но истинная масса эритроцитов не увеличивается.
19. Миелодисплазия и острый лейкоз
Миелодиспластические синдромы (МДС) и острый миелоидный лейкоз (ОМЛ) представляют собой клональные гемопоэтические нарушения, связанные с цитопенией, дефектным созреванием костного мозга и, в конечном итоге, нерегулируемой пролиферацией бластов.Многие случаи МДС перерастают в ОМЛ, и, хотя эти два заболевания отличаются друг от друга, они имеют непрерывный спектр.
Большинство случаев МДС и ОМЛ являются идиопатическими, но воздействие токсинов и радиации может увеличить риск. Лекарства также могут вызывать МДС [85, 86]. Алкилирующие агенты (такие как азотистый иприт, циклофосфамид, мелфалан, бусуфлан, хлорамбуцил) являются наиболее часто упоминаемыми преступниками. Риск связан с общей дозой, продолжительностью и конкретным типом алкилирующего агента. Прокарбазин и нитрозомочевины также связаны с миелодисплазией и лейкемией.До развития -MDS или AML может существовать латентный период от 2 до 8 лет. Лейкозу, вызванному этими агентами, обычно предшествует миелодиспластический синдром. Эти лейкозы обычно морфологически относятся к FAB M1 или M2. Типичны сложные хромосомные аномалии, обычно с делециями хромосом 5 и 7 и трисомией 8.
Эти лейкозы обычно морфологически относятся к FAB M1 или M2. Типичны сложные хромосомные аномалии, обычно с делециями хромосом 5 и 7 и трисомией 8.
Отдельный синдром вторичного лейкоза связан с ингибиторами топоизомеразы II, которые включают эпиподофиллотоксины (этопозид и тенипозид), антрациклины (дауномицин, эпирубицин и др.) доксорубицин.) и митоксантрон. Лейкоз с этими агентами имеет более короткий латентный период, чем у лейкоза, ассоциированного с алкилирующими агентами, часто менее 2 лет и обычно проявляется без предшествующего МДС. Морфологически часто наблюдаются острые миеломоноцитарные лейкозы с кариотипической аномалией, связанной с 11q23.
Описан также острый промиелоцитарный лейкоз, связанный с лечением, чаще всего в сочетании с ингибиторами topo II [87]. Они также имеют относительно короткое время ожидания от лечения без предшествующей предлейкемической фазы.Подобно de novo APL, (15; 17) с PML-RAR альфа-перестройки являются обычными. Результаты лечения относительно благоприятны при лечении как de novo APL, в отличие от плохого прогноза, наблюдаемого при других типах -AML.
У пациентов, получающих адъювантную химиотерапию по поводу рака груди, риск острого лейкоза увеличивается с возрастом, с интенсивностью терапии и с использованием лучевой терапии груди [88]. Многие из этих пациентов получали как алкилирующие агенты, так и антрациклины, оба из которых являются лейкемогенными.Кроме того, повышенный риск AML был отмечен у пациентов с раком молочной железы, получавших G-CSF вместе с адъювантной химиотерапией в некоторых [89], но не во всех исследованиях [90].
Трансплантация стволовых клеток, используемая при высоком риске, рецидивах и рефрактерных гематологических злокачественных новообразованиях, связана с риском вторичного МДС и лейкемии. Остается неясным, вызвано ли заболевание предтрансплантационной терапией или сам трансплантат.
Радиоиммунотерапия, разработанная для лечения неходжкинской лимфомы, может быть связана с некоторым риском лейкемогенеза. Однако, как и в случае с пациентами после трансплантации, некоторые риски могут быть связаны с повреждением стволовых клеток в результате предшествующего лечения или, возможно, с повышенным риском, связанным с самим основным заболеванием. Небольшое количество пациентов, получавших только радиоиммунотерапию, продемонстрировало, казалось бы, низкий риск лейкемии [93].
Однако, как и в случае с пациентами после трансплантации, некоторые риски могут быть связаны с повреждением стволовых клеток в результате предшествующего лечения или, возможно, с повышенным риском, связанным с самим основным заболеванием. Небольшое количество пациентов, получавших только радиоиммунотерапию, продемонстрировало, казалось бы, низкий риск лейкемии [93].
20. Выводы
Широкий спектр вызванных лекарствами гематологических синдромов опосредуется множеством механизмов, включая иммунные эффекты, взаимодействия с ферментативными путями и прямое ингибирование кроветворения.В таблице приведены гематологические синдромы, вызванные лекарственными средствами, которые обсуждались выше. Доказать, что лекарство вызывает определенный гематологический синдром, зачастую невозможно. Многие пациенты одновременно получают несколько препаратов, что затрудняет определение причинно-следственной связи. Повторное назначение препарата, подозреваемого в токсичности, обычно не рекомендуется. Для некоторых лекарств, таких как гепарин, хинидин и ванкомин, было проведено тестирование in vitro и выяснены механизмы цитопении. Однако такое тестирование не всегда возможно, учитывая, что для большинства нет стандартизированных, коммерчески доступных анализов и что реакции могут быть связаны с метаболитами, а не с более легко тестируемыми исходными соединениями.
По мере развития медицины старые лекарства устаревают и заменяются новыми лекарственными формами. Многие препараты, ранее связанные с гематологической токсичностью (например, пенициллин, альфа-метилдопа, хинидин, золото и хлорамфеникал), больше не используются. Однако обнаружено, что новые препараты связаны с их собственной потенциальной гематологической токсичностью (например, клопидогель, линезолид, рибавирин, ритуксимаб и ингибиторы GPIIb / IIIa). Кроме того, помимо классических цитопений, вызванных лекарственными препаратами, мы все чаще рассматриваем тромбоз как общую тему с рядом различных агентов (например,g., гепарин, ингибиторы ЦОГ-2, бевацизумаб, заместительная гормональная терапия, тамоксифен, эритропоэтин, талидомид и леналидомид). Врачам самых разных специальностей необходимо понимать гематологические последствия приема лекарств и быть готовыми к возникновению и коррекции этих явлений у своих пациентов.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||