Вторичные энзимопатии
Вторичные
энзимопатии являются следствием тех
или иных патологических процессов,
сопровождающихся нарушением активности
ферментов. Они наблюдаются при многих
заболеваниях. Так например, причиной
развития вторичной лактазной
недостаточности могут являться: кишечные
инфекции вирусной и бактериальной
этиологии, паразитарные заболевания
(лямблиоз и др.), синдром короткой кишки
(пострезекционный), целиакия, токсическое
и лекарственное поражения кишечника
(на фоне лучевой и химиотерапии,
антибиотикотерапии).
Одним
из вариантов вторичных энзимопатий
являются алиментарные
энзимопатии
– патологические состояния, обусловленные
стойкими нарушениями активности
ферментов в связи с характером питания.
Алиментарные энзимопатии могут быть
обусловлены длительным дефицитом белка
в питании (например при квашиоркоре),
нарушением биосинтеза коферментов при
витаминной недостаточности, угнетением
синтеза металлоферментов при низком
содержании в рационе соответствующих
минеральных веществ. Кроме того, они
могут возникать при несбалансированном
питании в целом. К развитию алиментарных
энзимопатий может приводить также
нарушение поступления пищевых веществ
из желудочно-кишечного тракта в кровь
при длительных поносах, атрофии или
поражении слизистой оболочки кишечника
и др. К алиментарным энзимопатиям относят
и так называемые токсические
энзимопатии,
связанные с угнетением активности или
биосинтеза отдельных ферментов различными
естественными компонентами пищевых
продуктов (ингибиторы протеолитических
ферментов, антивитамины и др.) или
чужеродными веществами (например
пестицидами), содержащимися в них.
Клинические
проявления приобретенных энзимопатий
зависят от вида фермента, функция
которого нарушена и характеризуется
нарушениями того или иного вида обмена
веществ.
Энзимопатии углеводнго обмена
Галактоземия
– возникает при нарушении обмена
галактозы, обусловленном наследственным
дефектом одного из трех ферментов,
включающим галактозу в метаболизм
глюкозы. Галактоземия, вызванная
недостаточностью
галактозо-1-фосфатуридилилтрансферазы
(ГАЛТ) наиболее хорошо изучена. Это
заболевание проявляется очень рано и
особенно опасно для детей, так как
основным источником углеводов для них
служит материнское молоко, содержащее
лактозу. Ранние симптомы дефекта ГАЛТ:
рвота, диарея, дегидратация, уменьшение
массы тела, желтуха, гепатомегалия,
катаракта, задержка психического
развития. Лабораторные исследования
при этом выявляют галактоземию,
галактозурию, галактозо-1-фосфатемию,
тенденцию к гипогликемии.
При
дефекте фермента галактокиназы у
пациентов отмечается галактоземия,
галактозурия, катаракта. Гораздо реже
причиной возникновения галактоземии
является дефект уридилфосфат-4-эпимеразы,
при котором не отмечается тяжелых
клинических проявлений.
При
диагностике галактоземии исследуют
мочу на содержание галактозы, собранную
после нескольких кормлений молоком.
При обнаружении у ребенка катаракты
его обследуют на недостаточность
галактокиназы и ГАЛТ.
Недостаточность
дисахаридаз.
Дисахаридазы локализованы в щеточной
каемке клеток слизистой тонкого кишечника
и участвуют в расщеплении дисахаридов
на моносахариды, что является необходимым
условияем их всасывания. Дисахаридазы
подразделяются на α-гликозидазы
(изомальтаза, сахараза, мальтаза) и
β-гликозидазы (лактаза, β-галактозидаза).
К
этой группе нарушений относится
отсутствие активности сахаразы и
изомальтазы. Дисахариды не расщепляются
и не могут быть утилизированы. Они
осмотически активны, связывают воду в
просвете кишечника и вызывают диарею
после пероральной нагрузки дисахаридами.
Кроме того, после такой нагрузки в крови
не удается обнаружить повышение гликемии
в интервале 30-90 минут, как это отмечается
у здоровых людей. Непереносимость
лактозы обусловлена дефектом лактазы
и проявляется также, как и вышеперечисленные
состояния.
Нарушения
метаболизма мукополисахаридов
(гликозаминогликанов).
Важнейшей составной частью соединительной
ткани являются протеогликаны, состоящие
из агрегированных мономерных субъединиц,
содержащих центрально расположенное
«белковое ядро», с которым связаны
гликозидные цепи различных
гликозаминогликанов (ГАГ). Количество
и соотношение различных протеогликанов
зависит от типа соединительной ткани.
Они образуются в специальных клетках
этой ткани – фибробластах, в лизосомах
этих же клеток они после эндоцитоза
разрушаются. Функции ГАГ – поддержание
структурной целостности соединительной
ткани и организация межклеточного
матрикса. ГАГ взаимодействуют с
компонентами клеточных мембран в таких
процессах, как рост клеток, межклеточные
коммуникации, восприятие информации,
взаимодействие некоторых плазменных
белков с сосудистой стенкой. Разрушение
ГАГ начинается с терминального
моносахарида под влиянием специфических
гликозидаз. Если какие-либо из этих
лизосомальных ферментов отсутствуют
или их активность нарушена в соединительной
ткани начинается накопление неразрушенных
или частично разрушенных ГАГ. Это
приводит к возникновению ряда заболеваний,
объединенных общим названием
мукополисахаридозы.
Мукополисахаридозы отличаются
прогрессирующим течением с различной
степенью тяжести. Общие признаки
различных форм этих заболеваний:
деформация черт лица, изменение скелета,
деформация суставов, поражение печени,
селезенки, сердца, кровеносных сосудов.
Характерна также задержка психомоторного
и умственного развития. Ниже приводится
характеристика некоторых форм
мукополисахаридозов. Синдром
Гурлера –
обусловлен дефицитом α-L-идуронидазы.
Протекает тяжело – больные умирают в
возрасте до 10 лет. У детей отмечается
деформация позвоночника, суставов,
отставание в росте, комбинированная
проводниковая и нейросенсорная глухота,
гепатоспленомегалия. С мочой экскретируются
гепаран- и дерматан-сульфаты.
Синдром
Моркио –
вызван дефектом галактозо-6-сульфатазы,
которая расщепляет связь, имеющуюся
только в кератансульфате. Это ведет к
накоплению последнего в составе хрящей
межпозвоночных дисков и роговицы, что
и определяет клинику заболевания.
Поражается преимущественно скелет –
выступают нижние ребра, наступает
х-образное искривление ног, характерно
выпирание грудины и очень короткая шея.
С мочой выделяется кератансульфат.
Дефицит
глюкуронидазы
– дефект, проявляющийся огрублением
черт лица, гепатоспленомегалией,
изменениями скелета. Лабораторно
выявляется: наличие дерматан- и
гепаран-сульфатов в моче, метахроматические
гранулы в гранулоцитах периферической
крови, снижение активности фермента в
лейкоцитах и сыворотке крови.
причины, диагностика, лечение наследственных и приобретенных энзимопатий
Энзимопатии – понятие, включающее большое количество патологий – наследственных и приобретенных. Успешная терапия основывается на своевременной диагностике и вовремя начатом и полноценном лечении.
Что такое энзимопатии
Под этим термином объединяют большую группу заболеваний, в основе возникновения которых лежат дефекты функционирования ферментов. Список патологий обширен, как и их клиническая картина.
Причины возникновения
Энзимопатии могут развиваться вследствие генетических мутаций. Также среди этиологических факторов имеются нарушения в питании, острые или хронические отравления токсическими веществами.
Классификация
Энзимопатии бывают врожденными и приобретенными. Первые делятся следующим образом:
- Нарушения метаболизма аминокислот, углеводов, липидов, нуклеиновых оснований, стероидов.
- Врожденные патологии соединительной ткани.
- Нарушения метаболизма, связанные с дефектами слизистой оболочки органов пищеварения.
В основе врожденных патологий лежат генетические мутации.
Приобретенные энзимопатии бывают алиментарными и токсическими. Алиментарные вызываются в результате соблюдения различных нерациональных диет или недостаточного поступления некоторых веществ в организм.
Возможные осложнения
Среди осложнений энзимопатий – отставание в умственном и физическом развитии. У пациентов могут наступать тяжелые нарушения функционирования внутренних органов. При острых отравлениях или авитаминозах имеется высокий риск летального исхода.
Симптомы энзимопатии
Симптоматика зависит от основного заболевания. Например:
- При нарушениях обмена аминокислот отмечается вялость ребенка, задержка умственного развития. При различных поражениях кожа и глаза могут приобретать характерный для определенного заболевания цвет. Например, при фенилкетонурии у пациентов кожный покров и глаза очень светлые.
- Липидозы проявляются болями в костях, патологическими переломами, гепато- и спленомегалией, задержкой роста, снижением зрения, нарушениями чувствительности, иногда – спастическими парезами. Для этой группы патологий характерно накопление липидов и продуктов их обмена во внутренних органах, что отражается на клинической картине. Умственное развитие таких пациентов также может задерживаться, но не всегда.
- Нарушения углеводного метаболизма проявляются желтухой, плохим набором массы тела, задержкой умственного и физического развития, диспепсией (боли в животе, поносы и запоры, повышенное газообразование).
- Для подагры характерно повышение содержания мочевой кислоты в организме, которая откладывается в суставах. Это состояние называется подагрический артрит.
- Алиментарные энзимопатии проявляются симптомами гиповитаминозов. У пациентов могут быть жалобы на сухость слизистых оболочек и кожного покрова, парезы и параличи, нарушения чувствительности, кровоточивость десен, слабость, утомляемость и эмоциональную лабильность.
- Нарушения обмена веществ в соединительной ткани проявляются большим количеством симптомов – от повышенной гибкости конечностей до пролапса сердечных клапанов.
- Токсические энзимопатии возникают в результате отравления солями тяжелых металлов. Симптоматика различная, вплоть до угнетения функционирования жизненно важных систем и органов.
Диагностика
Включает общий осмотр, консультации различных специалистов узкого профиля. Пациентам назначают лабораторные и инструментальные методы исследования:
- Общий анализ крови и мочи.
- Биохимия крови.
- Специальные диагностические тесты для выявления врожденных энзимопатий, токсических отравлений.
- Анализ крови на ревмофакторы и аутоиммунное воспаление.
- При необходимости проводится биопсия тканей.
Если требуется, пациентам назначается фиброгастроскопия, УЗИ органов брюшной полости, полостные исследования органов пищеварительного тракта.
Лечение энзимопатий
Тактика лечения зависит от основного заболевания, терапия чаще проводится в амбулаторных условиях.
- Большинство врожденных и приобретенных патологий удается успешно купировать при ранней диагностике. Эта группа энзимопатий лечится путем модификации диеты. Например, при фенилкетонурии следует исключить продукты с фениаланином – растительные и животные белки.
- При подагре назначается прием Аллопуринола – препарата, способного снизить содержание мочевой кислоты до нормы. Кроме этого, важно соблюдение диеты с исключением продуктов, богатых пуриновой кислотой.
- При аутоиммунном воспалении назначаются иммуносупрессанты – Циклоспорин, Метотрексат.
- В случае гиповитаминозов назначаются витаминные препараты с повышенным содержанием того вещества, нехватку которого испытывает организм пациента.
- При отравлениях требуется госпитализация с проведением дезинтоксикационной терапии.
- В качестве дополнительных средств назначаются препараты из группы НПВС (Нимесулид, Кетонал, Лорноксикам), витамины группы В (Мильгамма, Неуробекс), антиконвульсанты (Карбамазепин, вальпроевая кислота, Габапентин), антидепрессанты, нейролептики и седативные лекарства (Диазепам, Грандаксин, Стимулотон).
- При нарушениях обмена стероидов врачи используют гормональную терапию. При таких патологиях может потребоваться хирургическая коррекция изменений.
Первая помощь
Первая помощь при энзимопатиях не требуется, так как, чтобы дойти до критического состояния в случае данных заболеваний, нужно большое количество времени. Главное, что нужно таким больным – это своевременная диагностика и лечение.
Исключение составляют острые токсические отравления, для терапии которых требуется введение антидотов, дезинтоксикационных препаратов и сорбентов, проведение форсированного диуреза.
Профилактика
Профилактика заключается в своевременной диагностике. Что касается приобретенных энзимопатий, то предупреждение их развития заключается в правильном и полноценном питании.
Гасанова Сабина Павловна
Роль ферментов в метаболизме. Многообразие ферментов. Понятие о классификации. Наследственные первичные энзимопатии: фенилкетонурия, алкаптонурия. Другие примеры наследственных энзимопатий. Вторичные энзимопатии. Значение ферментов в медицине. — Биохимия. Ответы на билеты
В клетке постоянно происходит большое количество разнообразных химических реакций, которые формируют метаболические пути — последовательное превращение одних соединений в другие. Метаболизм — совокупность всех метаболических путей, протекающих в клетках организма. Основу всех жизненных процессов составляют тысячи химических реакций, катализируемых ферментами.
В 1961 г в Москве V Международный биохимический союз принял современную классификацию ферментов. В соответствии с этой классификацией все ферменты делятся: а) на классы по типу катализируемой реакции, б)каждый класс подразделяется на подклассы по природе атакуемой химической группы, в) подклассы делятся на подподклассы по характеру атакуемой связи или по природе акцептора. Выделяют 6 классов ферментов:
I класс Оксидоредуктазы (Ферменты этого класса катализируют окислительно-восстановительные реакции, лежащие в основе биологического окисления. Класс насчитывает 22 подкласса. Коферментами этого класса являются НАД, НАДФ, ФАД, ФМН, убихинон, глутатион, липоевая кислота).
II класс Трансферазы (Трансферазы катализируют реакции переноса различных групп от одного субстрата (донор) к другому (акцептор), участвуют в реакциях взаимопревращения различных веществ, обезвреживания природных и чужеродных соединений. Коферментами являются пиридоксальфосфат, коэнзим А, тетрагидрофолиевая кислота, метилкобаламин. Класс подразделяется на 9 подклассов в зависимости от строения переносимых групп. Примером подклассов являются ферменты, переносящие одноуглеродные фрагменты, альдегидные или кетоостатки, ацильные остатки, азотсодержащие группы, фосфорсодержащие группы. Часто встречается рабочее название трансфераз киназы. Это трансферазы, катализирующие перенос фосфата от АТФ на субстрат (моносахариды, белки и др), т.е. фосфотрансферазы. Систематическое название образуется: Донор группы : акцептор группы переносимая группа трансфераза).
III класс Гидролазы (осуществляют разрыв внутримолекулярных связей в субстрате (за исключением С-С связей) путем присоединения элементов Н2О, подразделяются на 13 подклассов. Сохранены тривиальные названия, например, пепсин, трипсин. Коферменты отсутствуют. Широко представлены ферментами желудочно-кишечного тракта и лизосомальными ферментами. Осуществляют распад макромолекул, образуя легко адсорбируемые мономеры. Примером подклассов служат группы ферментов, действующие на сложные эфиры, на простые эфиры, на пептиды, на углерод-углеродные связи. Систематическое название образуется: Гидролизуемый субстрат : отделяемая группа гидролаза. Исторически названия гидролаз складывались из названия субстрата с окончанием «аза» коллагеназа, амилаза, липаза, ДНК-аза. Наиболее часто встречаются следующие рабочие названия гидролаз: 1. Эстеразы гидролиз сложноэфирных связей. 2. Липазы гидролиз нейтральных жиров (триацилглицеролов). 3. Фосфатазы гидролиз моноэфиров фосфорной кислоты. 4. Гликозидазы гидролизуют О- и S-гликозидные связи. 5. Протеазы, пептидазы гидролиз белков и пептидов. 6. Нуклеазы гидролиз нуклеиновых кислот.).
IV класс Лиазы (ферменты, катализирующие разрыв С-О, С-С, C-N и других связей, а также обратимые реакции отщепления различных групп негидролитическим путем. Выделяют 7 подклассов. Эти реакции сопровождаются образованием двойной связи или присоединением групп к месту двойной связи. Лиазы являются сложными ферментами. Коферментами служат пиридоксальфосфат, тиаминдифосфат, участвует магний, кобальт. Примером подклассов являются ферменты, действующие на углерод-углеродные связи, углерод-кислородные связи, углерод-азотные связи. Систематическое название образуется: Расщепляемый субстрат : отделяемая группа лиаза).
V класс Изомеразы (ферменты, катализирующие изомерные превращения в пределах одной молекулы. Изомеразы сложные ферменты. К их коферментам относятся пиридоксальфосфат, дезоксиаденозилкобаламин, глутатион, фосфаты моносахаридов (глюкозо-1,6-дифосфат) и др. Выделяют 6 подклассов изомераз в зависимости от типа реакции. Например, в первый подкласс выделяют рацемазы (обратимое превращение L- и D-стереоизомеров) и эпимеразы (превращения изомеров, имеющих более одного центра асимметрии, например, α-D-глюкозу в β-D-глюкозу. Систематическое название образуется: Субстрат [ ] реакция, где [ ] обозначение, отражающее суть реакции, например, «номер изменяемого атома углерода», изменение «цис-транс», изменение «кето-енол», изменение «альдозо-кетозо»).
VI класс Лигазы (синтетазы ферменты, катализирующие присоединение друг к другу двух молекул с использованием энергии высокоэнергетических связей АТФ (или других макроэргов). Лигазы сложные ферменты. Они содержат нуклеотидные (УТФ), биотиновые (витамин Н), фолиевые коферменты. Выделяют 6 подклассов. Примером подклассов служат группы ферментов по виду образуемой связи: углерод-кислород, углерод-сера, углерод-азот, углерод-углерод. Систематическое название образуется: Субстрат 1 : субстрат 2 лигаза)
Каждому ферменту присвоен четырехзначный классификационный номер, включающий класс, подкласс, подподкласс и порядковый номер в подподклассе. Чтобы дать ферменту название существует два способа:
1. Систематическое название согласно современной классификации. Часто такое название длинно и сложно для использования, поэтому как производное систематического названия у многих ферментов имеется одно или несколько рабочих названий. 2. Тривиальное название название, сложившееся исторически. Например, пепсин, трипсин. Для некоторых ферментов (чаще для гидролаз) к названию субстрата добавляется окончание «-аза» уреаза, амилаза, липаза. Тем не менее и у таких ферментов имеется систематическое название.
В случае, если фермент не может выполнять свою функцию, говорят об энзимопатологии (энзимопатии) состояния, связанные с патологическим изменением активности ферментов. Наиболее часто встречается снижение активности и нарушение каких-либо метаболических процессов. В результате энзимопатологии клиническое значение может иметь:
-накопление субстрата реакции, например: фенилаланина при фенилкетонурии, свободного билирубина при желтухах новорожденных, некоторых жиров при болезнях лизосомального накопления (липидозы),
-недостаток продукта, например: меланина при альбинизме, катехоламинов при паркинсонизме,
-обе особенности одновременно, как при гликогенозах, сопровождающихся гипогликемией при избытке гликогена в печени.
По характеру нарушения выделяют первичные и вторичные энзимопатии.
Первичные (наследственные) энзимопатии связаны с генетическим дефектом и наследственным снижением активности. Например, фенилкетонурия связана с дефектом фенилаланин-4-монооксигеназы, которая превращает фенилаланин в тирозин. В результате накапливаются аномальные метаболиты фенилаланина, оказывающие сильный токсический эффект. Заболевание подагра связано с дефектом ферментов метаболизма пуриновых оснований и накоплением мочевой кислоты. Алкаптонурия — нарушено окисление гомогентизиновой кислоты в тканях (гомогентизиновая кислота — промежуточный метаболит катаболизма тирозина). У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты — диоксигеназы гомогентизиновой кислоты, приводящей к развитию заболевания. В результате увеличиваются концентрация гомогентизиновой кислоты и выведение её с мочой. В присутствии кислорода гомогентизиновая кислота превращается в соединение чёрного цвета — алкаптон. Поэтому моча таких больных на воздухе окрашивается в чёрный цвет. Кроме этого, распространенными первичными энзимопатиями являются галактоземия, недостаточность лактазы и сахаразы, различные липидозы.
Вторичные (приобретенные) энзимопатии возникают как следствие заболеваний органов, вирусных инфекций и т.п., что приводит к нарушению синтеза фермента или условий его работы, например, гипераммониемия при заболеваниях печени, при которых ухудшается синтез мочевины и в крови накапливается аммиак. Другим примером может служить недостаточность ферментов желудочно-кишечного тракта при заболеваниях желудка, поджелудочной железы или желчного пузыря.
Недостаток витаминов и их коферментных форм также является причиной приобретенных ферментопатий.
Использование ферментов в медицине происходит по четырем направлениям:
1. энзимодиагностика (это исследование активности ферментов плазмы крови, мочи, слюны с целью диагностики тех или иных заболеваний. Примером может служить фермент лактатДГ, определение его активности в плазме крови необходимо при заболеваниях сердца, печени, скелетной мускулатуры. Увеличение активности α-амилазы в плазме крови и моче наблюдается при воспалительных процессах в поджелудочной и слюнных железах. С другой стороны, заболевания тех или иных органов всегда сопровождаются специфичным «ферментативным профилем». Например, инфаркт миокарда сопровождается увеличением активности лактатДГ, креатинкиназы, аспартатаминотрансферазы),
2. энзимотерапия (использование ферментов в качестве лекарственных средств. Самыми распространенными ферментативными препаратами являются комплексы ферментов желудочно-кишечного тракта (Фестал, Мезим форте), содержащие пепсин, трипсин, амилазу и т.п., и используемые для заместительной терапии при нарушениях переваривания веществ в желудочно-кишечном тракте. Рибонуклеаза и дезоксирибонуклеаза входят в состав глазных капель для лечения вирусных конъюнктивитов. Трипсин ингалируют при бронхолегочных заболеваниях для разжижения густой и вязкой мокроты. Коллагеназу применяют для ускорения отторжения некротизированных тканей, для очистки трофических язв),
3. использование ферментов в медицинских технологиях и промышленности (Специфичность ферментов к определенным субстратам применяетсяв лабораторной диагностике. Многие лабораторные методы основаны на взаимодействии добавляемого извне фермента с определяемым соединением. В результате возникает специфичный продукт реакции, после определения содержания последнего судят о концентрации искомого вещества (глюкозооксидазный, холестеролоксидазный методы), иммуноферментные методы, основанные на образовании тройного комплекса фермент-антиген-антитело. Определяемое вещество не является субстратом фермента, но является антигеном. Фермент может присоединять этот антиген вблизи от активного центра. Если в среде есть антиген, то при добавлении антител и формировании тройного комплекса активность фермента изменяется. Активность фермента измеряют любым способом),
4. применение ингибиторов ферментов (Весьма широко применяются в настоящее время ингибиторы протеаз (контрикал, гордокс) при панкреатитах состояниях, когда происходит активирование пищеварительных ферментов в протоках и клетках поджелудочной железы. Ингибиторы холинэстеразы (физостигмин, прозерин) приводят к накоплению нейромедиатора ацетилхолина в синапсах и показаны при миастении, двигательных и чувствительных нарушениях при невритах, радикулитах, психогенной импотенции. Препараты, содержащие ингибиторы моноаминоксидазы (наком, мадопар), повышают выработку нейромедиаторов катехоламинов в ЦНС при лечении паркинсонизма. Подавление активности моноаминооксидазы (разрушающей катехоламины) сохраняет нормальную передачу сигналов в нервной системе.
Ингибиторы ангиотензинпревращающего фермента (каптоприл, эналаприл и т.п.) используются как антигипертензивное средство и вызывают расширение периферических сосудов, уменьшение нагрузки на миокард, снижение артериального давления. Аллопуринол ингибитор ксантиноксидазы, фермента катаболизма пуринов, требуется для снижения образования мочевой кислоты и подавления развития гиперурикемии и подагры. Ингибиторы гидроксиметилглутарил-SКоА-редуктазы (ловастатин, флувастатин, аторвастатин) применяются для снижения синтеза холестерола при атеросклерозе, заболеваниях сердечно-сосудистой системы, дислипопротеинемиях. Ингибитор карбоангидразы (ацетазоламид) используется как мочегонное средство при лечении глаукомы, отеков, эпилепсии, алкалозах и горной болезни).
Лекция 5. Детские болезни. Энзиимопатии. —
Все процессы обмена веществ в человеческом организме протекают с гораздо большей скоростью, чем соответствующие химические реакции вне организма.
Их осуществление происходит при определенных условиях включающих температуру около 38 оС и рН в пределах 7,4 и возможны только благодаря ферментам.
Заболевания обмена веществ, в патогенезе которых основную роль играет отсутствие, недостаток или дефект структуры клеточных ферментов носят названия — энзимопатии.
Выделяют наследственные и приобретенные энзимопатии. В основе наследственных энзимопатий лежат генетически детерминированные нарушения ферментов наступивших в результате генных мутаций в настоящее время известно около 600 наследственных энзимопатий. При приобретенных энзимопатиях энзимные нарушения возникают вторично в ходе патологического процесса. Выделяют нарушения обмена липидов, углеводов и аминокислот.
ФЕНИЛКЕТОНУРИЯ.
В основе этих нарушений лежит врожденное отсутствие, дефицит или дефект тех ферментов, которые участвуют в обмене аминокислот. Из этой группы наиболее распространенным заболеванием является фенилкетонурия или фенилпировиноградная олигофрения.
Это наследственное заболевание, обусловленное нарушением обмена фенилаланина. Наследуется по аутосомно-рецессивному типу. В 15% случаев отмечается кровное родство родителей больного. Частота заболевания 1:10000 новорожденных. Оно является результатом неполноценности фермента фенилаланиноксидазы. Нарушается переход фенилаланина поступающего с пищей в тирозин. Это приводит к накоплению в крови фенилаланина, уровень которого повышается также в цереброспинальной жидкости и моче. Избыток накопленного фенилаланина частично подвергается дезаминированию в результате чего образуется фенилпировиноградная, фенилмолочная и фенилуксусная кислоты, которые оказывают токсическое действие на ЦНС. Формируется дефицит тирозина и недостаточный синтез меланина и катехоламинов, что приводит к снижению пигментации кожи, волос и артериальной гипотонии. Кроме того, нарушается обмен триптофана, а значит синтез серотонина играющего важную роль в деятельности ЦНС. В мозге при фенилкетоурии обнаруживаются нарушения процессов миелинизации.
Клиническая картина
Проявляется уже в период новорожденности. Эти дети белокурые со светлой кожей и голубыми глазами. У них отмечаются дерматиты, экзема, повышенная потливость с характерным мышиным запахом. У них появляется рвота, череп чаще микроцефалический. Дети вялые, пассивные, не интересуются окружающим. Иногда они раздражительны, плаксивы. Они отстают в физическом и нервно-психическом развитии. У них прогрессивно снижается интеллект и в первые 3-4 года формируется тяжелое слабоумие. Наблюдаются эпилептиформные припадки.
В раннем возрасте у этих детей выявляется мышечная гипотония, которая постепенно сменяется мышечной гипертонией, приводящей к своеобразной позе «портного» (прижатые ноги и согнутые руки), сухожильные рефлексы повышены. Отмечаются гиперкинезы, тремор пальцев рук, атаксия, иногда центральные парезы.
Диагноз
Ставится на основании данных анамнеза, соответствующей клинической картине и лабораторных данных. Скрининг тестом является проба Феленга:
2-5мл свежей мочи + 10 капель 10% FeCl3 = сине-зеленое окрашивание
Это дает подозрение на фенилкетонурию. Необходимо определить содержание в плазме крови фенилаланина (в норме 0,09-0,1 мкмоль/л). При фенилкетонурии его содержание повышается до 3 и более мкмоль/л.
Лечение
В основном заключается в диетотерапии. Назначается диета с ограниченным содержанием фенилаланина (совсем исключить его нельзя т.к. это незаменимая аминокислота).
Разработаны специальные диеты:
Для детей до 1 года жизни – «Лофенак»
Для детей старше 1 года – «Нофенал»
Все дети обеспечиваются бесплатным питанием. Мало фенилаланина содержится в моркови, капусте, помидорах, яблоках, винограде. Лечение следует проводить под контролем уровня фенилаланина в крови. С возрастом проницаемость гематоэнцефалического барьера снижается, поэтому переносимость избытка фенилаланина повышается. Чем раньше начато лечение, тем лучше результат, поэтому очень важно распознать заболевание еще в роддоме.
ТИРОЗИНОЗ.
Это наследственное заболевание характеризуется развитием дистрофии, цирроза печени, рахитоподобных состояний косей и поражением почечных канальцев.
В основе заболевания лежит дефект в системе оксидазы парагидроксифенилпировиноградной кислоты.
Клиническая картина.
Различают острые и хронические формы. Острые формы развиваются в первые дни жизни и проявляются рвотой, частым жидким стулом, в дальнейшем задержкой физического развития, увеличением размеров печени и селезенки, возникает дыхательная недостаточность. Первое впечатление, что у ребенка кишечный токсикоз или непроходимость кишечника. Позднее у этих детей развиваются признаки печеночной недостаточности, отеки, асцит, геморрагический синдром. Они редко доживают д 5-6 лет. При хроническом течении развиваются дистрофия, гепатомегалия, геморрагический синдром, появляются значительные костные изменения (остеопороз, искривления костей, задержка появления ядер окостенения), кожа постепенно приобретает желтушный оттенок, развивается цирроз печени. Эти дети существенно отстают в физическом и нервно-психическом развитии, отмечается также увеличение размеров почек.
В крови: гипоглекимия, увеличенное содержание тирозина в плазме крови, снижено содержание 7 фактора свертывания крови.
В моче: белок, повышенное выделение тирозина и метионина, гиперфосфатурия.
Лечение.
Сводится в основном к диетотерапии: эти дети вскармливаются в основном специальными смесями из белковых гидролизатов, прогноз неблагоприятный. Диетотерапия позволяет увеличить продолжительность жизни ребенка.
ГАЛАКТОЗЕМИЯ.
Это наследственное заболевание, при котором нарушается процесс ферментативного превращения галактозы в глюкозу, что обусловлено недостаточностью фермента галактозо-1-фосфатуридилтрансферазы и галактокиназы. В итоге галактоза-1-фосфат накапливается в клетках и оказывает на них токсическое действие. Особенно тяжело поражаются клетки ЦНС, печени, почек, развивается помутнение хрусталика.
Заболевание наследуется по аутосомно-рецессивному типу.
Клиническая картина.
Проявляется в зависимости от величины ферментного дефекта и от количества получаемой с пищей галактозы. Возникает расстройство пищеварения и развивается симптоматика интоксикации: у ребенка появляются рвота, жидкий частый стул, анорексия, эксикоз. Уже в первые дни жизни проявляются и стойко сохраняются выраженная желтуха постепенно прогрессирующие признаки печеночной недостаточности.
Уже на 2-3 месяце жизни наблюдается развитие сосудистых коллатералей, асцит, обнаруживается спленомегалия, появляются кровоизлияния на коже слизистых оболочках. Рано обнаруживается задержка психомоторного развития, которая с возрастом становиться более выраженной. Появляются судороги, в тяжелых случаях уже при рождении ребенка выявляется двусторонняя катаракта, развитие которой связано с токсическим воздействием галактозы на плод в период внутриутробного развития, в более легких случаях катаракта появляется на 4-7 неделе жизни. Развивается дистрофия вплоть до кахексии. Дети погибают от наслоения интеркурентных заболеваний.
Лабораторно: повышение содержания галактозы в крови, повышенное выделение с мочой, гиперхолистеринэмия, протеинурия. Концентрация галактозы в крови 1 г/л и выше (в норме 0,2 г/л). Содержание глюкозы в крови снижено.
Диагноз.
Ставиться на основании анамнеза, клинической картины, лабораторного исследования.
Лечение.
Представляет собой сложную задачу т.к. ребенок должен быть переведен на безмолочную диету. Рекомендуется полное или частичная замена женского молока соевым молоком. В тяжелых случаях ребенок вскармливается белковыми гидролизатами.
Фруктоземия
Наследственное заболевание, связанное с нарушением обмена фруктозы. Тип наследования аутосомно-рецессивный.
Причина: недостаточная активность фермента фруктозо-1-фосфатальдолазы. Процесс превращения фруктозы задерживается на этапефруктозо-1-фосфат, который накапливается в тканях и оказывает токсическое действие. Заболевание проявляется уже в первые недели жизни, т.е. с того момента, когда ребенок начинает получать соки, сладкий чай. У ребенка появляется упорная рвота после каждого кормления, анорексия. Он становится вялым, кожа бледная, возникает акроцианоз. Развивается гипогликемическое состояние в тяжелых случаях может возникнуть гипогликемическая кома. У этих детей отмечается увеличение размеров печени, появляется желтуха. Иногда в старшем возрасте, в более легких случаях заболевание проявляется только отвращением к сладкой пище. Зубы у этих детей всегда в очень хорошем состоянии — никогда не бывает кариеса.
Лабораторно: В моче- фруктоза, белок, лейкоцитурия. Уровень сахара в крови или на нижней границе нормы или ниже ее.
Лечение.
Сводится к диетотерапии: исключают сахар, фрукты, мед. Если диагноз выставлен своевременно при проведении диетотерапии прогноз благоприятный.
Существует еще доброкачественная фруктоземия, которая не имеет никаких клинических проявлений, при которой повышенная экскреция фруктозы с мочой зависит от количества вводимой фруктозы с пищей. Патогенез этого заболевания связан с недостаточностью фермента фосфаткиназы. Вторичная фруктозурия наблюдается у больных с тяжелым поражением печени.
ГЛИКОГЕНОЗЫ
Это группа энзимопатий, при которых нарушаются процессы распада и синтеза гликогена, что сопровождается накоплением его в клетках печени, почек, мышечной ткани или генерализованно. В зависимости от того, где именно происходит накопление гликогена, выделяют несколько типов гликогенозов.
Вызывается отсутствием фермента глюкозо-6-фосфотазы в печени и слизистой кишечника. Заболевание наследуется по аутосомно-рецессивному типу.
Клиническая картина.
Обусловлена накоплением гликогена в печени, основными симптомами являются гепатомегалия, отставание в росте, гипогликемия, гиперлипидемия. Уже в первые дни жизни ребенка обнаруживается вялость адинамия, отсутствие аппетита, рвота, а затем развивается гипотрофия. Печень плотная с гладкой поверхностью, она увеличена в размерах ( ведущий клинический симптом) нарушены функции печени, а т.к. она увеличена, увеличен размер живота Значительный объем живота компенсируется лордозом в поясничном отделе позвоночника, также увеличиваются размеры почек. В связи с гипогликемией у этих детей случаются судороги и развивается гипогликемическая кома. Если ребенка вовремя не кормить он становится бледным, вялым, повышается потливость. Нервно-психическое развитие удовлетворительное. Дети имеют малый рост, полноваты, с купольно-округлым лицом и короткой шеей (болезнь «Пупса»). При присутствии интеркурентных заболеваний/ инфекций у таких детей быстро развивается кетоацидоз. При длительно сохраняющейся гиперлипидемии развиваются ксантомы. Характерен геморрагический синдром.
Лабораторно: гипогликемия, но повышенное содержание холестерина, общих липидов, белок и ацетон.
Прогноз чаще неблагоприятный. Дети первых 3 лет жизни обычно погибают от интеркурентных заболеваний. В легких случаях они выживают. Тогда в возрасте 5-7 лет у их развиваются симптомы геморрагического диатеза, а затем симптомокомплекс подагры. Интеллект, как правило, не нарушен, но редко может быть наоборот.
Причина: отсутствие кислой лизосомальной а-1,4-гликозидазы. Значительные количества гликогена откладывается в печени, почках, нервной системе, мышцах (в том числе и миокарде).
Выделяют 2 подтипа:
Сердечный вариант.
Уже с момента рождения у ребенка выявляется общий цианоз, расстройства дыхания, адинамия. Размеры сердца увеличены, оно имеет шарообразную форму, выслушивается систолический шум. На ЭКГ левограмма( а для ребенка в этом возрасте характерна правограмма) и отрицательный зубец Т во всех отведениях. Дети обычно погибают на 1 году жизни от сердечной недостаточности.
Нервно-мышечный вариант.
Характерно преобладание нервно-мышечной дистрофии. Уже с момента рождения обнаруживается гипотрофия мышц, гипорефлексия. В дальнейшем мышечная слабость прогрессирует. Ребенок не может сам удерживать голову. Постепенно нарастает неврологическая симптоматика, тремор, бульбарные расстройства, спастические параличи. Эти дети погибают от бульбарных расстройств
Наследуется по аутосомно-рецессивному типу. Причина: недостаточность фермента амило-1,4 -гликозидазы и олиго-1,4-глюкотрансферазы. В печени накапливается аномальный по своей структуре гликоген, инертный для обмена. Печень, для того чтобы поддержать глюкозный гомеостаз активирует компенсаторный механизм: усиливается распад белков и жиров, таким образом, образуется избыток кетоновых тел и ацетона. Выделяют 4 варианта этого заболевания АВС и D. При всех вариантах отмечается увеличение размеров живота (резко выраженная гепатомегалия), страдает функция печени, отмечается снижение мышечного тонуса, развивается мышечная дистрофия. Содержание сахара в крови обычно снижено, в моче обнаруживается ацетон. Прогноз более благоприятный, Особенно опасно это заболевание в 3-5 лет (часто развивается гипогликемическая кома), а затем симптомы постепенно сглаживаются.
Недостаток амило-1,4-трансгликозидазы и а-1,4гликан-6-глюкотрансферазы. Тип наследования — аутосомно-рецессивный. Заболевание выявляется с 1 недели жизни и характеризуется гепато-спленомегалией , увеличением размеров почек. Постепенно нарушаются функции печени и в дальнейшем, развивается цирроз. Прогноз неблагоприятный. Все дети поибают от цирроза на первом году жизни или несколько позже.
Обусловлена дефектом фосфорилазы мышц. Основным симптомом является прогрессирующая миопатия и болезненные судороги, после физической нагрузки. Отмечается общая слабость, быстрая утомляемость и неспособность выполнять даже самые умеренные физические упражнения, поэтому активность этих всегда резко ограничена. При выполнении физических упражнений дети испытывают резкую боль в мышцах, которая проходит после отдыха. Затем это ведет к развитию судорог и мышечных дистрофий. У некоторых больных после физической нагрузки появляется темная моча (миоглобинурия). Прогноз для жизни благоприятный.
Вызывается отсутствием фосфорилазы печени, что ведет к грубым нарушениям обмена гликогена. Характерна клиническая картина: задержка роста, гепатомегалия и повышение активности аминотрансфераз. Прогноз, как правило, благоприятный. По-видимому, эти нарушения в обмене гликогена могут компенсироваться за счет глюконеогенеза.
Недостаточная активность фосфоглюкомутазы. Клинически это заболевание проявляется нарушением походки. Эти дети ходят на пальцах, что связано с укорочением мышечных волокон икроножных мышц. Характерна мышечная слабость, повышенная утомляемость, гепатомегалия. Прогноз для жизни относительно благоприятный.
Вызывается отсутствием фермента фосфофруктокиназы. Клиническаякартина напоминает 5 тип: мышечная слабость, повышеннаяутомляемость, ригидность мышц, миоглобинурия после физической нагрузки, развитие болезненных судорог. Прогноз для жизни благоприятный
Вызывается отсутствием фосфорилазы В в печени. Клиника похожа на 6 тип гликогенозов и проявляется увеличением размеров печени, задержкой роста и повышением активности аминотрансфераз.
Вызывается дефектом протеинкиназы. Проявляется только гепатомегалией.
Обусловлен дефектом фермента фосфоизомеразы. Заболевание проявляется несфероцитарной анемией и гемолитическими кризами.
Встречается еще агликогеноз. Заболевание обусловлено недостаточностью гликогенсинтетазы печени. Ведущий клинический симптом – гипоглекимия натощак, обусловленная неспособностью организма поддерживать нормальную концентрацию глюкозы в крови после ночного или длительного голодания. В связи с гипогликемией у этих детей развивается судорожный синдром, отмечается отставание в физическом и умственном развитии. Прогноз неблагоприятный.
Лечение гликогенозов.
Придается значение диетотерапии, гормонотерапии, энзимотерапии и химиотерапии. Всем детям рекомендуется более частые приемы пищи, которая должна быть богата углеводами, но с низким содержанием жиров. В зависимости от типа гликогеноза из диеты исключаются отдельные продукты.
Заболевание:
1 тип: исключение продуктов содержащих галактозу и фруктозу
2тип: назначение тироксина
3 тип: высокобелковая диета с низким содержанием жиров
1,3,6 типы: назначение дифенина
5 тип: внутривенно глюкоза с инсулином
8тип: гепарин в малых дозах.
При остальных вариантах лечение симптоматическое.
Заболевания связанные с нарушением обмена липидов
Подгруппы:
- болезни накопления (внутриклеточные липидозы), наблюдается поражение нервных клеток и вторичное поражение проводящих путей
- болезни с повышенным содержанием липидов в плазме крови (плазматические липоидозы или семейные гиперлипидемии)
- Болезни обмена липопротеидов
- Дейкодистрофии
Чаще всего наблюдаются болезни первой подгруппы а из них болезни Нимана-Пика, Гаше и амавротическая идиотия.
Болезнь Нимана-Пика (сфингомиелиноз)
Наследственное заболевание обмена сфингомиелина в мозге, печени, селезенке, РЭС. Тип наследования аутосомно-рецессивный. Клиническая картина проявляется с первых дней жизни и характеризуется сочетанием мозговой симптоматики и висцеральной патологии. Характерно появление рвоты. Ребенок отказывается от еды. Увеличиваются размеры печени, селезенки. Отмечается задержка психического развития. Развиваются спастические парезы, слепота, ребенок теряет слух. Примерно в 30% случаев при осмотре глазного дна выявляется симптом вишневой косточки (в центре желтого пятна определяется вишневого цвета участок, окруженный сероватым ободком). Кожа серовато-желтого оттенка.
Клинические варианты: типы АВС и D.
Тип А: типичная форма болезни. Выявляется в раннем детском возрасте и характеризуется наличием висцеральной патологии и изменением со стороны ЦНС, чаще эти дети погибают в первые 3 года жизни.
Тип В: характеризуется выраженной висцеральной патологией без поражения ЦНС.
Тип С: проявляется в подростковом возрасте. Вначале отмечается увеличение печени и селезенки, затем появляются изменения со стороны ЦНС.
Тип D: протекает по типу острого гепатита с переходом в цирроз печени.
Лечение.
Лечение в основном симптоматическое. Патогенетического лечения нет. Некоторые улучшения наблюдаются после введения витаминов, плазмы, тканевых экстрактов.
Болезнь Гаше
Наследственно обусловленное нарушение обмена глюкоцереброзидов. Возникает при недостаточности фермента глюкоцереброзидазы. Это ведет к накоплению глюкоцереброзидов в клетках ретикулоэндотелиальной системы.
Клинические варианты.
1. Острая форма.
Характерна для детей раннего возраста. Уже с рождения у них наблюдается выраженная гипотрофия, бульбарные расстройства. У них нарушено глотание, беззвучный крик, имеется поражение глазодвигательного нерва, что проявляется в виде сходящегося косоглазия, появляются тонико-клонические судороги, резко снижено зрение, что связано с пигментной дистрофией сетчатки. Дети погибают на 1 году жизни от аспирационной пневмонии.
2. Подострая форма
Может быть, у детей всех возрастов. Имеется сочетание церебральной и висцеральной патологии. Характерно отставание в психомоторном развитии и наличие судорог. Отмечается гипер- или гипотония мышц. Увеличены размеры печени и селезенки с нарушением функции печени, поэтому у этих детей всегда увеличен размер живота и имеются симптомы дыхательной недостаточности, что связано инфильтрацией легких клетками Гаше, быстро развивается дистрофия вплоть до кахексии. Заболевание прогрессирует, обычно дети погибают через 1-1,5 года от начала заболевания.
3.Ювенильная форма.
Отличается хроническим доброкачественным течением. Клинически она проявляется гепатомегалией, анемией, геморрагическим синдромом и поражением трубчатых костей. У них развивается остеодистрофии, которые являются причиной деформации скелета и патологических переломов. Течение заболевания длительное т.к. у них резко снижена иммунная реактивность. Дети погибают от наслоения инфекции.
Диагноз.
Ставится на основании анамнеза, клинической картины и подтверждается лабораторно.
Лечение.
Лечение только симптоматическое, (противосудорожная и антибактериальная терапия) патогенетической терапии нет.
Амавротическая идиопатия
В этой группе заболеваний выделяют
- Врожденная форма (Нормана-Вуда)
- Раннедетская форма (Тея-Сакса)
- Позднедетская форма (Большовского-Янковского)
- Юношеская форма ( Баттена-Шпильмейстера-Фогта)
- Поздняя форма (Куфса)
Встречается чаще других, она связана с дефектом в-гексозаминидазы А. Частота в европейской популяции 1:50000 среди евреев 1:6000, то есть имеется этническая предрасположенность. Эти дети рождаются в срок с нормальной массой тела и ростом правильно развиваются до 4-7 месяцев. Болезнь часто носит семейный характер. Ребенок постепенно утрачивает интерес к окружающему, перестает играть, узнавать мать. Обнаруживается снижение остроты зрения. Ребенок не может фиксировать взгляд, не следит за игрушкой. На глазном дне характерен симптом вишневой косточки. В дальнейшем происходит атрофия зрительного нерва и наступает слепота. Одновременно снижается интеллект до степени идиотии. Возникает двигательные расстройства, приводящие к к полному обездвиживанию ребенка. Отмечается повышенная чувствительность к слуховым раздражителям, затем развиваются судорожные припадки, кахексия. Дети погибают через 1,5-2 года от начала заболевания.
Проявляется в первые дни после рождения тяжелой неврологической симптоматикой. Развивается гидроцефалия или микроцефалия, судороги, параличи, слепота. Все дети погибают
Имеет такую же клинику, как и болезнь Тея-Сакса, но заболевание начинает проявляться в 6-10 лет.
Клиника сходна с болезнью Тея-Сакса, но проявляться начинает у детей младшего школьного возраста.
Начинает проявляться в позднем школьном возрасте, а клинически повторяет болезнь Тея-Сакса.
Диагноз
Ставится на основании типичной клинической картины, характерных изменений глазного дна и дефекта фермента в-гексозаминидазы А. Специфического лечения нет. Проводится в основном симптоматическая терапия и профилактика. В случае беременности возможна перинатальная диагностика (при сроке 18-20 недель исследуется активность ферментов в амниотической жидкости, при их низкой активности показано прерывание беременности).
ГИПЕРЛИПИДЕМИИ
Для них характерно повышение уровня липидов в крови.
1 тип.
Обусловлен недостаточностью фермента липопротеидлипазы. Клиника проявляется приступообразными болями в животе, сопровождающихся лихорадкой и лейкоцитозом. При повышении концентрации липидов более 15 г/л на коже сгибательной поверхности конечностей, спине появляются ксантомы, окруженные ободком гиперемии. При снижении концентрации липидов ксантомы могут временно исчезать. Если отложения липидов происходит в области перекреста зрительного нерва, может развиться слепота.
2 тип.
Резко выраженный ксантоматоз с отложением липидов в тканях, но здесь ксантомы локализуются в области ахиловых сухожилий, на кистях, на коленях. Рано развивается задержка нервно-психического развития.
3 тип.
Ксантоматоз и раннее развитие атеросклероза, но здесь развиваются так называемые плантарные ксантомы т.к. они распологаются на подошвах, тыльных поверхностях кистей рук, кончиках пальцев. Характерно появление стенокардии и даже инфаркта миокарда. При поражении клапанного аппарата развивается хроническая недостаточность кровообращения.
4 тип.
Ксантоматоз. Больных беспокоят боли в животе нередко связанные с приемом пищи. Появляется гепато-спленомегалия в сочетании с ожирением и сахарным диабетом. Страдает сердечно-сосудистая система: наблюдается ИБС, стенокардия, инфаркт миокарда. При поражении клапанного аппарата развивается хроническая сердечная недостаточность.
5 тип.
Клиника напоминает 1 и 3 типы: ксантоматоз, абдоминальные боли, гепато-спленомегалия, панкреатит, сахарный диабет, ожирение, ИБС, стенокардия, инфаркт миокарда.
Лечение.
Лечение сводится к проведению диетотерапии. Диета сводится к ограничению количества животных жиров и замены их жирами растительного происхождения. Проводят медикаментозную терапию, назначают линетон, делипин, никотиновую кислоту. При 3 типе гиперлипидемий назначают ионообменные смолы. При 3,4,5 типах показано ограничение углеводов в диете. При 1,5 типах назначают гепарин в малых дозах, а в остальном, лечение симптоматическое.
Прогноз определяется степенью и темпами развития заболевания и мерой поражения сердечно- сосудистой системы.
(Visited 41 times, 1 visits today)
Энзимопатии. Биологическая химия
В основе многих заболеваний лежат нарушения функционирования ферментов в клетке – энзимопатии. Приобретённые энзимопатии, как и вообще протеинопатии, по-видимому, наблюдают при всех болезнях.
При первичных энзимопатиях дефектные ферменты наследуются, в основном, по аутосомно-рецессивному типу. Гетерозиготы, чаще всего, не имеют фенотипических отклонений. Первичные энзимопатии обычно относят к метаболическим болезням, так как происходит нарушение определённых метаболических путей. При этом развитие заболевания может протекать по одному из ниже перечисленных «сценариев». Рассмотрим условную схему метаболического пути:
Е1 Е2 Е3 Е4
А ? В ? С ? D ? Р
Вещество А в результате последовательных ферментативных реакций превращается в продукт Р. При наследственной недостаточности какого-либо фермента, например фермента Е3, возможны разные нарушения метаболических путей:
Нарушение образования конечных продуктов.
Недостаток конечного продукта этого метаболического пути (при отсутствии альтернативных путей синтеза) может приводить к развитию клинических симптомов, характерных для данного заболевания.
Клинические проявления. В качестве примера можно рассмотреть альбинизм. При альбинизме нарушен синтез в меланоцитах пигментов – меланинов. Меланин находится в коже, волосах, радужке, пигментном эпителии сетчатки глаза и влияет на их окраску. При альбинизме наблюдают слабую пигментацию кожи, светлые волосы, красноватый цвет радужки глаза из-за просвечивающих капилляров. Проявление альбинизма связано с недостаточностью фермента тирозингидроксилазы (тирозиназы) – одного из ферментов, катализирующего метаболический путь образования меланинов.
Накопление субстратов-предшественников.
При недостаточности фермента будут накапливаться определенные вещества, а также во многих случаях и предшествующие им соединения. Увеличение субстратов-предшественников дефектного фермента – ведущее звено развития многих заболеваний.
Клинические проявления. Известно заболевание алкаптонурия, при котором нарушено окисление гомогентизиновой кислоты в тканях (гомогентизиновая кислота – промежуточный метаболит катаболизма тирозина). У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты – диоксигеназы гомогентизиновой кислоты, приводящей к развитию заболевания. В результате увеличиваются концентрация гомогентизиновой кислоты и выведение её с мочой. В присутствии кислорода гомогентизиновая кислота превращается в соединение чёрного цвета – алкаптон. Поэтому моча таких больных на воздухе окрашивается в чёрный цвет. Алкаптон также образуется и в биологических жидкостях, оседая в тканях, коже, сухожилиях, суставах. При значительных отложениях алкаптона в суставах нарушается их подвижность.
Нарушение образования конечных продуктов и накопление субстратов-предшественников.
Отмечают заболевания, когда одновременно недостаток продукта и накопление исходного субстрата вызывают клинические проявления.
Клинические проявления. Например, у людей с болезнью Гирке (гликогеноз I типа) наблюдают снижение концентрации глюкозы в крови (гипогликемия) в перерывах между приёмами пищи. Это связано с нарушением распада гликогена в печени вследствие дефекта фермента глюкозо-6-фосфатазы. Одновременно у таких людей увеличиваются размеры печени (гепатомегалия) вследствие накопления в ней не используемого гликогена.
Данный текст является ознакомительным фрагментом.
Читать книгу целиком
Поделитесь на страничке
понятие, классификация, молекулярные причины возникновения и механизмы развития, последствия, биохимическая диагностика.
I. Энзимопатология
Энзимопатология
– это наука, которая изучает энзимопатии.
Энзимопатии
– это группа заболеваний, которые
вызваны различными дефектами ферментов.
Энзимопатий делятся на: наследственные
(первичные) и приобретенные (вторичные).
1. Наследственные энзимопатии
Наследственные
энзимопатии
– это заболевания, вызванные наследственными
нарушениями биосинтеза ферментов или
их структуры и функции.
В
норме:
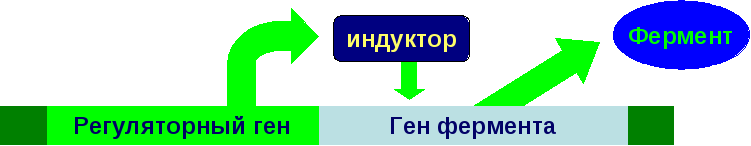
Полное
или частичное нарушения биосинтеза
ферментов вызывают дефекты генов
регуляторных белков, которые контролируют
синтез ферментов:
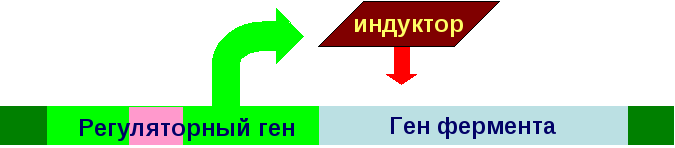
Нарушение
структуры и функции ферментов вызывают
дефекты генов этих ферментов:
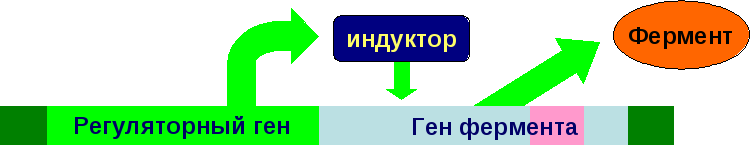
У
образовавшегося фермента наблюдаются
структурные изменения, которые проявляются
в изменении его каталитической активности
(как правило, она исчезает), чувствительности
к активаторам и ингибиторам, сродству
к субстратам, оптимумам рН, температуры.
В связи с этим изучением констант
фермента является решающим в постановке
диагноза врожденных энзимопатий.
Наследственные энзимопатии по типу нарушений метаболизма делят на:
нарушения
обмена аминокислот: фенилкетонурия,
альбинизм, алкаптонурия и др.;нарушения
углеводного обмена: галактоземия,
наследственная непереносимость
фруктозы, гликогенозы;нарушения
липидного обмена: липидозы;нарушения
обмена нуклеиновых оснований: подагры,
синдрома Леш-Нихана и др.;нарушение
обмена в соединительной ткани:
мукополисахаридозы, хондродистрофия
и др.;дефекты
ферментов в ЖКТ: муковисцидоз, целиакия,
непереносимость лактозы и др.нарушения
обмена стероидов и т.д.
В
норме метаболический путь протекает
следующим образом:
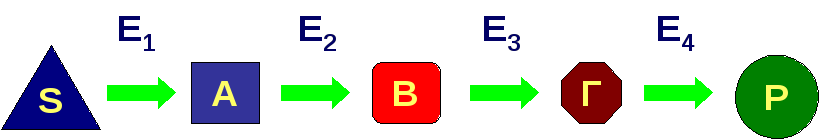
Из-за
дефекта в метаболическом пути (цикле,
шунте) одного из ферментов в организме
происходит накопление промежуточных
продуктов (часто токсичных в высоких
концентрациях) и дефицит жизненно
необходимых конечных продуктов, что
приводит к клиническим проявлениям:
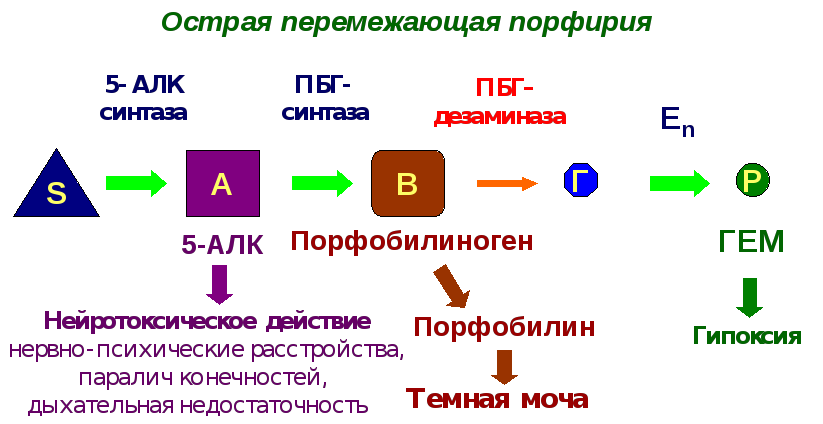
Пример:
фенилпировиноградная
олигофрения
– наследственное заболевание, приводящее
в раннем детстве к гибели ребенка или
к развитию у него тяжелой умственной
отсталости.
Причиной
заболевания является отсутствие в
печени фермента фен-4-монооксигеназы,
которая обеспечивает превращение
незаменимой аминокислоты Фен в Тир:
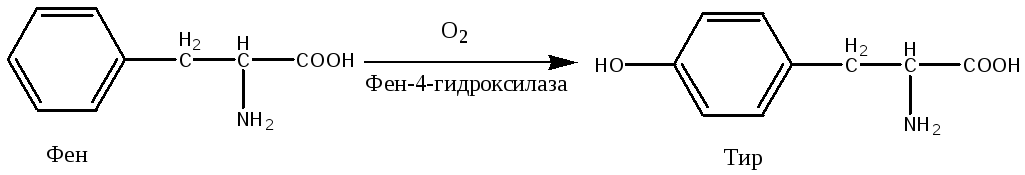
Эта
реакция необходима для катаболизма
Фен, т.е. удаления его излишков. При
отсутствии фен-4-монооксигеназы в
организме происходит накопление Фен и
превращение его в различные производные:
фенилпировиноградную, фенилмолочную
и фенилуксусную кислоты.
Фен
и его производные в высоких концентрациях
токсичны, накапливаясь в тканях, они
оказывают на них повреждающее действие.
Самой чувствительной к Фен и его
производным оказывается нервная ткань
детей, она поражается в первую очередь.
Диагноз
фенилкетонурия ставят на основании
обнаружения Фен в крови или
фенилпировиноградной кислоты на пеленках
детей. Лечение в основном сводится к
исключению из питания ребенка Фен. Для
такого ребенка Тир оказывается незаменимой
аминокислотой.
Другое
тяжелое наследственное заболевание –
галактеземия
(непереносимость молочного сахара),
связано с отсутствием синтеза в печени
ферментов, катализирующих превращение
галактозы в глюкозу. В результате в
раннем возврате происходит накопление
в тканях галактозы, приводящее к развитию
катаракты, поражению печени, мозга,
нередко вызывающее гибель ребенка.
Лечение в данном случае сводиться к
исключению из диеты молочного сахара.
5.1. Энзимопатии
Если фермент не
может выполнять свою функцию, говорят
об энзимопатологии
(энзимопатии).
Энзимопатологии
(энзимопатии)
– это состояния, связанные с патологическим
изменением активности биокатализаторов.
Наиболее часто встречается снижение
их эффективности, провоцирующее нарушение
каких-либо метаболических процессов.
Клиническое значение при этом может
иметь накопление субстрата (как при
фенилкетонурии, сахарном диабете,
гликогенозах, липидозах) или недостаток
продукта (при альбинизме, паркинсонизме),
или обе особенности одновременно. По
характеру нарушения выделяют первичные
и вторичные
энзимопатии.
Первичные
(наследственные) энзимопатии
обусловлены генетическим дефектом,
приводящим к синтезу повреждённого
фермента, что снижает его активность.
Например, фенилкетонурия
связана с мутацией гена
фенилаланин-4-монооксигеназы,
которая обычно превращает фенилаланин
в тирозин. Когда этого не происходит,
накапливаются аномальные метаболиты
фенилаланина, оказывающие сильный
токсический эффект. Непереносимость
лактозы обусловлена подавлением
активности лактазы
в желудочно-кишечном тракте, накопление
низкомолекулярного субстрата грозит
диареей, метеоризмом.
Кроме указанных,
довольно распространенными первичными
энзимопатиями являются галактоземия,
муковисцидоз,
адреногенитальный синдром,
различные липидозы.
Сюда же относят и
врождённые
энзимопатии,
связанные с заболеваниями, перенесёнными
женщинами в период беременности, которые
отражаются на плоде. Например, эндемический
зоб у мамы способен спровоцировать
развитие кретинизма у младенца.
Вторичные
(приобретенные) энзимопатии
возникают как следствие действия
различных факторов (вирусов, токсинов,
бактерий, механических повреждений и
т.д.), которые нарушают синтез того или
иного фермента, условия его работы;
например, при поражении печени
ухудшается мочевинообразование, и в
крови накапливается ядовитый аммиак
(гипераммониемия),
что служит примером токсической
энзимопатии.
Алиментарная
энзимопатия
может возникать из-за недостаточного
выделения
секретов желудочно-кишечного
тракта при
заболеваниях желудка, поджелудочной
железы или желчного пузыря. Дефицит
витаминов
и их коферментных форм также является
причиной приобретенных ферментопатий.
Энзимодиагностика
Энзимодиагностика
– это исследование активности ферментов
плазмы крови и других биологических
жидкостей, клеток тканей, используемое
в целях диагностики тех или иных
заболеваний. Примером может служить
изучение спектра изоферментов
лактатдегидрогеназы
в плазме
крови, что поможет отдифференцировать
заболевания сердца, печени, скелетной
мускулатуры. Увеличение активности
α-амилазы,
особенно в моче, наблюдается при
воспалительных процессах в поджелудочной
или слюнных железах.
С другой стороны,
для поражений тех или иных органов
характерен специфический «ферментативный
профиль». Например, инфаркт миокарда
сопровождается ростом значений ЛДГ,
креатинкиназы,
аспартатаминотрансферазы.
Специфичность
энзимов к определенным субстратам нашла
широкое применение в лабораторной
диагностике. Многие её методы основаны
на взаимодействии с определяемым
соединением энзима, добавляемого в
качестве реагента. В результате возникает
специфичный продукт реакции, после
выявления содержания последнего судят
о концентрации искомого вещества
(глюкозооксидазный,
холестеролоксидазный
методы).
В иммуноферментных
пробах
используют образование тройного
комплекса: фермент-антиген-антитело.
Энзимопатия: биоинформатика болезней: Novus Biologicals
Разместите свое изображение, связанное с болезнями, чтобы оно было рекомендовано!
Социальные сети
Разместите свою учетную запись Twitter, связанную с Enzymopathy, чтобы быть отмеченным!
Блоги
Разместите свой блог на Enzymopathy, чтобы его отметили! |
События
| Разместите свое мероприятие по Enzymopathy, чтобы оно было отмечено! |
Видео
Разместите свое видео о Enzymopathy, чтобы его отметили!
Благотворительность
Подайте заявку на участие в программе Enzymopathy, чтобы ее разместили!
Энзимопатия — это заболевание, которое приводит к отсутствию или дефекту ферментов.Заболевание часто считается генетическим заболеванием, так как большинство людей с энзимопатией рождаются с дефектными ферментами. Две наиболее распространенные формы энзимопатии являются результатом отключенных ферментов или мутировавших реакций. В первой форме фермент полностью отключен и не может участвовать в химических реакциях в организме. Вторая форма возникает, когда потребность в ферменте и кофакторе увеличивается в ходе реакции, вызывая меньшее количество реакций из-за дефицита фермента. Энзимопатию можно диагностировать, проверяя различные ферменты и определяя, какие из них не работают.Методы лечения включают диетические ограничения или добавки, замену ферментов или лечение конкретных симптомов в индивидуальном порядке.
Инструмент биоинформатики энзимопатии
Laverne — это удобный инструмент биоинформатики, помогающий облегчить научное исследование родственных генов, болезней и путей на основе совместного цитирования. Узнайте больше об энзимопатии ниже!
Для получения дополнительной информации о том, как использовать Laverne, прочтите Руководство.
Лучшие исследовательские реагенты
У нас есть 1333 продукта для изучения энзимопатии, которые можно применять для вестерн-блоттинга, проточной цитометрии, иммуноцитохимии / иммунофлуоресценции, иммуногистохимии из нашего каталога антител и наборов для ELISA.NB100-236
![Western Blot: Glucose 6 Phosphate Dehydrogenase Antibody [NB100-236] - Detection of Human and Mouse G6PD by Western Blot. Samples: Whole cell lysate (50 ug) from HeLa, 293T, and mouse NIh4T3 cells prepared using NETN lysis buffer. Antibody: Affinity purified rabbit anti-G6PD antibody NB100-236 used for WB at 1 ug/ml. Detection: Chemiluminescence with an exposure time of 30 seconds.](/800/600/https/images.novusbio.com/thumbnails2/Glucose-6-Phosphate-Dehydrogenase-Antibody-Western-Blot-NB100-236-img0007.jpg)
Кролик Поликлональный
Виды Человек, Мышь
Приложения WB, IB, IHC
| 17 Публикации | добавить в корзину |
NBP2-03621
Мышь Monoclonal
Виды Человек
Приложения WB, ICC / IF, IHC
NBP1-31470
![Immunocytochemistry/Immunofluorescence: Triosephosphate isomerase Antibody [NBP1-31470] - HeLa cells were fixed in 4% paraformaldehyde at RT for 15 min. Green: Triosephosphate isomerase stained by Triosephosphate isomerase antibody [C2C3], C-term diluted at 1:500. Blue: Hoechst 33342 staining. Scale bar= 10um.](/800/600/https/images.novusbio.com/thumbnails2/Triosephosphate-isomerase-Antibody-Immunocytochemistry-Immunofluorescence-NBP1-31470-img0019.jpg)
Кролик Поликлональный
Виды Человек, Мышь, Крыса
Приложения WB, ELISA, Flow
| 1 Обзор 7 Публикаций | добавить в корзину |
NBP1-89519
![Western Blot: APRT Antibody [NBP1-89519] - Lane 1: Marker [kDa] 230, 130, 95, 72, 56, 36, 28, 17, 11. Lane 2: Human cell line RT-41](/800/600/https/images.novusbio.com/thumbnails2/APRT-Antibody-Western-Blot-NBP1-89519-img0005.jpg)
Кролик Поликлональный
Виды Человек
Приложения WB, ICC / IF, IHC
| 1 Публикация | добавить в корзину |
NBP1-90824
![Western Blot: CD77 Synthase Antibody [NBP1-90824] - Lane 1: Marker [kDa] 250, 130, 95, 72, 55, 36, 28, 17, 10<br/>Lane 2: Negative control (vector only transfected HEK293T lysate)<br/>Lane 3: Over-expression lysate (Co-expressed with a C-terminal myc-DDK tag (~3.1 kDa) in mammalian HEK293T cells, LY402588)](/800/600/https/images.novusbio.com/thumbnails2/CD77-Synthase-Antibody-Western-Blot-NBP1-90824-img0005.jpg)
![Immunocytochemistry/Immunofluorescence: CD77 Synthase Antibody [NBP1-90824] - Staining of human cell line MCF7 shows localization to mitochondria. Antibody staining is shown in green.](/800/600/https/images.novusbio.com/thumbnails2/CD77-Synthase-Antibody-Immunocytochemistry-Immunofluorescence-NBP1-90824-img0006.jpg)
Кролик Поликлональный
Виды Человек
Приложения WB, ICC / IF
H00051776-M03
![Western Blot: ZAK Antibody (3G5) [H00051776-M03] - ZAK monoclonal antibody (M03), clone 3G5. Western Blot analysis of ZAK expression in human stomach.](/800/600/https/images.novusbio.com/thumbnails2/ZAK-Antibody-(3G5)-Western-Blot-H00051776-M03-img0001.jpg)
![Immunocytochemistry/Immunofluorescence: ZAK Antibody (3G5) [H00051776-M03] - Analysis of monoclonal antibody to ZAK on HeLa cell. Antibody concentration 10 ug/ml](/800/600/https/images.novusbio.com/thumbnails2/ZAK-Antibody-(3G5)-Immunocytochemistry-Immunofluorescence-H00051776-M03-img0004.jpg)
Мышь Моноклональная
Виды Человек, мышь, крыса
Приложения WB, ELISA, ICC / IF
| 3 Публикации | добавить в корзину |
AF6240


Овцы Поликлональные
Виды Человек
Приложения WB, ELISA, IHC
| 1 Публикация | добавить в корзину |
NBP1-85740
Кролик Поликлональный
Виды Человек, Свинья
Приложения WB, ICC / IF, IHC
| 4 Обзоры 8 Публикации | добавить в корзину |
NBP2-67503
![Western Blot: Hexokinase 1 Antibody (ST47-05) [NBP2-67503] - Analysis of Hexokinase 1 on different lysates using anti-Hexokinase 1 antibody at 1/1,000 dilution. Positive control: Lane 1: Hela Lane 2: 293 Lane 3: MCF-7 Lane 4: HepG2](/800/600/https/images.novusbio.com/thumbnails2/Hexokinase-1-Antibody-(ST47-05)-Western-Blot-NBP2-67503-img0009.jpg)
Кролик Моноклональный
Виды Человек, Мышь, Крыса
Приложения WB, Flow, ICC / IF
| 1 Обзор | добавить в корзину |
NBP2-75717
![Immunohistochemistry-Paraffin: Xanthine Oxidase Antibody (JG38-40) [NBP2-75717] - Analysis of paraffin-embedded rat liver tissue using anti-Xanthine Oxidase antibody. Counter stained with hematoxylin.](/800/600/https/images.novusbio.com/thumbnails2/Xanthine-Oxidase-Antibody-(JG38-40)-Immunohistochemistry-Paraffin-NBP2-75717-img0003.jpg)
Кролик Моноклональный
Виды Человек, мышь, крыса
Приложения WB, IHC, IHC-P
,
Статья об энзимопатии из The Free Dictionary
наследственное заболевание, вызванное врожденной ошибкой метаболизма, которая возникает в результате ферментативного расстройства.
Энзимопатия может быть вызвана отсутствием фермента, снижением активности фермента или отсутствием или неправильным синтезом кофермента. Каждое из этих отклонений приводит к определенному нарушению обмена веществ и определяет клиническую картину данной энзимопатии. Например, аномальный метаболизм углеводов может проявляться сахарным диабетом или галактоземией, аномальным метаболизмом жиров, болезнью Тея-Сакса или болезнью Ниманна-Пика, а также аномальным метаболизмом амигокислот, алкаптонурией или альбинизмом.
Существует около 500 типов энзимопатий, многие из которых являются полиморфными и гетерогенными. При гетерогенных энзимопатиях аномалии генов, регулирующих взаимодействие ферментов, могут иметь идентичные проявления, поскольку ферменты, контролирующие различные биохимические реакции, часто дают идентичный метаболический результат. Большинство энзимопатий передаются аутосомно-рецессивным путем.
Некоторые энзимопатии, такие как фенилкетонурия, могут быть обнаружены с помощью экспресс-анализа в течение первых нескольких дней жизни.Ранняя диагностика часто способствует нормализации метаболизма с помощью диеты, введения в организм необходимого вещества (заместительная терапия), с помощью гормональной терапии или путем устранения избыточных метаболитов, нарушающих обмен веществ.
Техника амниоцентеза — перспективное средство диагностики энзимопатий. Медико-генетическая консультация становится все более важным средством профилактики энзимопатий.
СПИСОК ЛИТЕРАТУРЫ
Badalian, L.O., Таболин В.А., Э. Вельтищев. Наследственные болезни у детей . М., 1971.
Харрис Х. Основы биохимической генетики человека . Москва, 1973. (Пер. С англ.)
Хауэлл Р. Р., К. М. Мур. «Пренатальная диагностика в профилактике генетических заболеваний». Техасская медицина , 1974, т. 70, нет. 5. С. 77–84.
.
Enzymopathies — Biochemistry 2 with Devendra в Тринити-школе медицины
Продолжить с Google
Чтобы войти в систему с помощью Google, включите всплывающие окна
Продолжить с Facebook
Чтобы войти в систему с помощью Google, включите всплывающие окна
или
Нет учетной записи? Зарегистрироваться
Продолжить с Google
Чтобы зарегистрироваться в Google, включите всплывающие окна
Продолжить с Facebook
Чтобы зарегистрироваться в Google, включите всплывающие окна
или
Зарегистрируйтесь по электронной почте
Зарегистрируйтесь через Google или Facebook
или
Имя
Электронная почта
Пароль
День рождения
?
Для регистрации вам должно быть не менее 13 лет.Другие люди не увидят ваш день рождения.
Месяц
январь
февраль
марш
апрель
май
июнь
июль
августейший
сентябрь
октября
ноябрь
Декабрь
День
12345678910111213141516171819202122232425262728293031
Год
зарегистрироваться
,
15. Энзимопатии обмена аминокислот — греч.доктор
Последнее обновление: 3 июля 2020 г., 10:43
Цели обучения
- Почему аммиак токсичен для мозга?
- Какой фермент является дефектом при указанных заболеваниях?
Гипераммонемия
Как указывалось ранее, слишком много аммиака — очень плохо. Он может легко преодолевать гематоэнцефалический барьер, поэтому токсичен для мозга. Если в мозге слишком много аммиака, он попытается избавиться от него двумя способами.
Первый — это реакция глутаматдегидрогеназы , которая превращает α-кетоглутарат и аммоний в глутамат. Однако это истощает α-кетоглутарат, который необходим для TCA.
Второй — путем превращения глутамата и аммония в глутамин в реакции глутаминазы . Однако это приведет к истощению запасов глутамата, который является прямым предшественником ГАМК, важного нейромедиатора.
Гипераммонемия является результатом либо дефекта цикла мочевины, так как организм не может выводить аммиак, либо постоянной активации глутаматдегидрогеназы , которая расщепляет глутамат на α-кетоглутарат и аммиак.
Дефекты ферментов цикла мочевины
| Неисправный фермент | Болезнь |
|---|---|
| Карбамоилфосфатсинтетаза I (CPS I) | Врожденная гипераммонемия I типа |
| Орнитин-транскарбамоилаза | Врожденная гипераммониемия II типа |
| Аргининосукцинатсинтетаза | Цитруллинемия |
| Аргининосукциназа | Аргининосукцинат ацидурия |
| Аргиназа | Аргининемия (дефицит аргиназы) |
| N-ацетилглутаматсинтаза | Дефицит N-ацетилглутаматсинтазы |
Генетические нарушения деградации аминокислот
| Затронутый фермент | Пораженный путь | Болезни и |
|---|---|---|
| Фермент расщепления глицина | Разложение глицина | Гиперглицинемия |
| Комплекс дегидрогеназы α-кетокислот с разветвленной цепью | Расщепление аминокислот с разветвленной цепью | Болезнь мочи кленового сиропа |
| Метилмалонил-КоА мутаза | Валин, изолейцин, треонин, расщепление метионина | Метилмалоновая ацидемия |
| Цистатионин-β-синтаза | Разложение метионина | Гомоцистеинурия I |
| Фенилаланингидроксилаза | Разложение фенилаланина | Фенилкетонурия |
| Тирозинаминотрансфераза | Разложение фенилаланина | Тирозинемия II |
| п-гидроксифенилпируват диоксигеназа | Разложение фенилаланина | Тирозинемия III |
| Гомогентизат 1,2-диоксигеназа | Разложение фенилаланина | Алкаптонурия |
| Фумарилацетоацетаза | Разложение фенилаланина | Тирозинемия I |
| Тирозиназа | Синтез меланина из тирозина | Альбинизм |
Метилмалоновая ацидемия / ацидурия
В 1991 году женщина по имени Патрисия Столлингс была признана виновной в убийстве собственного сына Райана после того, как тесты показали повышенный уровень этиленгликоля (антифриза) в крови ее сына.Она была осуждена за отравление собственного сына антифризом в 1989 году. В 1990 году она родила второго сына, который вскоре заболел, но до того, как был нанесен серьезный ущерб, у него была диагностирована метилмалоновая ацидемия.
Это сделало вероятным, что у Райана также была метилмалоновая ацидемия, но это заболевание не вызывает накопления этиленгликоля, поэтому никакой связи между ними не установлено. После того, как биохимик просмотрел эпизод на канале Unresolved Mysteries , касающийся этого события, в конечном итоге было обнаружено, что метод теста, используемый для определения этиленгликоля в крови, будет положительным в случаях метилмалоновой ацидемии, поскольку это заболевание вызывает накопление пропионовой кислоты , похожая молекула.Это доказало, что Райан также умер от метилмалоновой ацидемии, а не от отравления. Столлингс был освобожден из тюрьмы и подал в суд на больницу и лаборатории.
Фенилкетонурия
Фенилкетонурия — это генетическое заболевание, вызванное дефицитом фенилаланингидроксилазы , фермента, участвующего в превращении фенилаланина в тирозин. Это заставляет фенилаланин разлагаться альтернативным путем, который вместо этого превращает фенилаланин в другие метаболиты.
Накопление фенилаланина вредно для мозга, но вредные эффекты возникают также из-за дефицита тирозина. У этих людей тирозин становится незаменимой аминокислотой. Это проблема, поскольку тирозин является предшественником нейротрансмиттеров, меланина и гормонов щитовидной железы.
Болезнь мочи кленовым сиропом
Болезнь мочи, вызванная кленовым сиропом, — это генетическое заболевание, вызываемое дефицитом комплекса дегидрогеназы α-кетокислоты с разветвленной цепью . Это заболевание приводит к накоплению аминокислот с разветвленной цепью, а также их метаболитов кетокислот.Это вызывает характерный сладкий запах мочи, отсюда и название.
Сводка
- Почему аммиак токсичен для мозга?
- Мозг выводит аммиак двумя способами
- Первый — это реакция глутаматдегидрогеназы , которая превращает α-кетоглутарат и аммоний в глутамат. Однако это истощает α-кетоглутарат, который необходим для TCA.
- Второй — путем превращения глутамата и аммония в глутамин в реакции глутаминазы .Однако это приведет к истощению запасов глутамата, который является прямым предшественником ГАМК, важного нейромедиатора.
- Какой фермент является дефектом при указанных заболеваниях?
- См. Здесь сводку по всем болезням, которые вы должны знать
Предыдущая страница:
14. Синтез незаменимых аминокислот
Следующая страница:
16. Синтез биологически активных молекул из аминокислот
,