| 1 | Аарскога-Скотта синдром | FGD1 | Наследственные синдромы | 305400 |
| 2 | Абиотрофия сетчатки, тип Франческетти | ABCA4, ABCR, RMP | Болезни органов зрения | 248200 |
| 3 | Адреногенитальный синдром | CYP21A2, CYP21, P450C21, CYP21B, CA21H | Патология эндокринных желез | 201910 |
| 4 | Азооспермия | null | null | 415000 |
| 5 | Айкарди-Гутьереса синдром | ADAR, ADAR1, DRADA, IFI4, G1P1, RNASEh3B, DLEU8, FLJ11712, TREX1, DNase III | null | 225750 |
| 6 | Акродерматит энтеропатический | SLC39A4, ZIP4 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 201100 |
| 7 | Аксенфельда-Ригера синдром | PITX2, FOXC1, FKHL7, FREAC3, PTX2, RIEG1, RIEG, ARP1 | null | 180500 |
| 8 | Альбинизм глазокожный | TYR, OCA2 | Болезни органов зрения | 203100 |
| 9 | Альстрема синдром | ALMS1, KIAA0328 | null | 203800 |
| 10 | Андерсена синдром | null | Заболевания сердечно-сосудистой системы и легких | 170390 |
| 11 | Анемия Даймонда-Блекфена | RPS19 | Болезни крови и иммунной системы | 105650 |
| 12 | Анеуплоидии | null | Хромосомная патология | ——— |
| 13 | Антли-Бикслера синдром | null | Наследственные синдромы | 207410 |
| 14 | Апера синдром | null | Наследственные синдромы | 101200 |
| 15 | Арта синдром | PRPS1 | Нервно-мышечные болезни | 301835 |
| 16 | Артрогрипоз дистальный (синдром Фримена-Шелдона) | MYh4, MYHSE1 | Скелетные дисплазии | 193700 |
| 17 | Атаксия Фридрейха | FXN, FRDA, X25 | null | 229300 |
| 18 | Атаксия, хорея, судороги и деменция | ATN1 | 125370 | |
| 19 | Атрофия зрительного нерва Лебера | null | Болезни органов зрения | 535000 |
| 20 | Атрофия зрительного нерва с глухотой | null | Болезни органов зрения | 125250 |
| 21 | Аутоиммунный лимфопролиферативный синдром | FAS, TNFRSF6, APT1, APO1, CD95 | Болезни крови и иммунной системы | 601859 |
| 22 | Аутоиммунный полиэндокринный синдром | null | Болезни органов зрения | 240300 |
| 23 | Афазия первичная прогрессирующая | GRN, PGRN, PEPI, GEP, PCDGF, GP88 | null | 607485 |
| 24 | Ахондроплазия | FGFR3, TACC3 | null | 100800 |
| 25 | Баллера-Герольда синдром | null | null | 218600 |
| 26 | Банаян-Райли-Рувалькаба синдром | PTEN, PTEN1, MMAC1 | Наследственные синдромы | 153480 |
| 27 | Барта синдром | null | Заболевания сердечно-сосудистой системы и легких | 302060 |
| 28 | Беста болезнь | BEST1, VMD2, TU15B | Болезни органов зрения | 153700 |
| 29 | Бёрта-Хога-Дьюба синдром | FLCN, FLCL, BHD | Опухоли | 135150 |
| 30 | Блефарофимоз, обратный эпикант и птоз | null | null | 110100 |
| 31 | Блоха-Сульцбергера синдром | IKBKG, NEMO, FIP3 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 308300 |
| 32 | Блума синдром | RECQL3, RECQ2, BLM | null | 210900 |
| 33 | Боковой амиотрофический склероз | VAPB, ALSIN, FIG4, ALS2, KIAA1563, SAC3, KIAA0274, DVAP33A | Нервно-мышечные болезни | 205100 |
| 34 | Боуэна-Конради синдром | EMG1, NEP1, C2F | Наследственные синдромы | 211180 |
| 35 | Брахидактилия | HOXD13, NOG, ROR2, HOX4I, NTRKR2 | Скелетные дисплазии | 113000 |
| 36 | Бьёрнстада синдром | BCS1L | null | 262000 |
| 37 | Ваарденбурга синдром | PAX3, HUP2 | Наследственные синдромы | 193500 |
| 38 | Ваарденбурга-Шаха синдром | EDNRB, ETB, ETBR | Наследственные синдромы | 277580 |
| 39 | Ван дер Вуда синдром | IRF6 | Наследственные синдромы | 119300 |
| 40 | Велокардиофациальный синдром | TBX1 | Заболевания сердечно-сосудистой системы и легких | 192430 |
| 41 | Вильсона-Коновалова болезнь | ATP7B | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 277900 |
| 42 | Вильямса синдром | null | Наследственные синдромы | 194050 |
| 43 | Вискотта-Олдрича синдром | WAS, WASP | Болезни крови и иммунной системы | 301000 |
| 44 | Врожденная нечувствительность к боли с ангидрозом | NTRK1, TRK, TRKA | Нервно-мышечные болезни | 256800 |
| 45 | Врожденной центральной гиповентиляции синдром | PHOX2B, PMX2B, NBPHOX | Заболевания сердечно-сосудистой системы и легких | 209880 |
| 46 | Галлервордена-Шпатца болезнь | PANK2 | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 234200 |
| 47 | Гелеофизическая дисплазия | null | Скелетные дисплазии | 231050 |
| 48 | Гемофилия | F8, F9, F8C, PTC | Болезни крови и иммунной системы | 306700, 306900 |
| 49 | Генитопателлярный синдром | null | null | 606170 |
| 50 | Германски-Пудлака синдром | HPS1 | Наследственные синдромы | 203300 |
| 51 | Герстманна-Штреусслера-Шейнкера болезнь | PRNP, PRP, PRIP | null | 137440 |
| 52 | Гидроцефалия, обусловленная врожденнным стенозом Сильвиева водопровода | L1CAM, MIC5, CAML1 | null | 307000 |
| 53 | Гипер-IgD синдром | MVK | Болезни крови и иммунной системы | 260920 |
| 54 | Гипер-IgM синдром | CD40LG, CD40L, CD154, TRAP, TNFSF5 | Болезни крови и иммунной системы | 308230 |
| 55 | Гиперкалиемический периодический паралич | SCN4A, NAV1. 4 4 | Нервно-мышечные болезни | 170500 |
| 56 | Гипертрофическая кардиомиопатия | CAV3, TNNT2 | Заболевания сердечно-сосудистой системы и легких | 192600 |
| 57 | Гиперфенилаланинемия с дефицитом тетрагидробиоптерина | GCh2, PTS, QDPR, PTPS, DHPR | Наследственные болезни обмена веществ | 261640 |
| 58 | Гипокалиемический периодический паралич | null | null | 170400 |
| 59 | Гипофосфатемический рахит | PHEX | Скелетные дисплазии | 307800 |
| 60 | Гипохондроплазия | null | Скелетные дисплазии | 146000 |
| 61 | Гиппеля-Линдау синдром | VHL | null | 193300 |
| 62 | Глазо-зубо-пальцевой синдром | null | null | 164200 |
| 63 | Глаукома врожденная | CYP1B1, P4501B1 | Болезни органов зрения | 231300 |
| 64 | Глаукома ювенильная открытоугольная | MYOC, TIGR | Болезни органов зрения | 137750 |
| 65 | Гломеруоцитоз почек гипопластического типа | HNF1B, TCF2, HNF2 | Патология почек | 137920 |
| 66 | Грейга синдром | GLI3 | Наследственные синдромы | 175700 |
| 67 | Грисцелли синдром | RAB27A, RAB27, RAM | Болезни крови и иммунной системы+Наследственные синдромы+Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 607624 |
| 68 | Дауна синдром | null | Хромосомная патология | 190685 |
| 69 | Делеции хромосомы 1p36 синдром | null | Хромосомная патология | 607872 |
| 70 | Дефицит гормона гипофиза, комбинированный | null | null | 262600 |
| 71 | Дефицит иммуноглобулина A | TNFRSF13B, TACI | null | 609529 |
| 72 | Дефицит карнитина системный первичный | SLC22A5, OCTN2 | Наследственные болезни обмена веществ | 212140 |
| 73 | Дефицит фактора F12 | null | null | 234000 |
| 74 | Джексона-Вейсса синдром | FGFR2, FGFR1, TK14, FLT2, FLG | Пороки развития nullа, умственная отсталость, судороги | 123150 |
| 75 | Ди Джорджи синдром | null | Хромосомная патология | 188400 |
| 76 | Диастрофическая дисплазия | SLC26A2, DTDST | Скелетные дисплазии | 222600 |
| 77 | Дисплазия де ля Шапеля (Ателостеогенез) | null | Скелетные дисплазии | 256050 |
| 78 | Дистальная моторная нейропатия | BSCL2, GARS, HSPB8, HSPB1, IGHMBP2, TRPV4, SEIPIN, HSP22, h21, E2IG1, HSP27, CATF1, SMUBP2, VROAC, OTRPC4, TRP12 | null | 182960 |
| 79 | Дистальная спинальная амиотрофия, врождённая с параличом диафрагмы | null | Нервно-мышечные болезни | 604320 |
| 80 | Дистальная спинальная амиотрофия, врожденная, непрогрессирующая | null | Нервно-мышечные болезни | 600175 |
| 81 | Дисхондростеоз Лери—Вейлля | null | null | 127300 |
| 82 | Дорфмана-Чанарина синдром | null | null | 275630 |
| 83 | Жильбера синдром | UGT1A1, UGT1 | null | 143500 |
| 84 | Жубер синдром | NPHP1, NPh2 | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 213300 |
| 85 | Инверсия пола 46 XX | SRY, TDF, TDY | null | 400045 |
| 86 | Инверсия пола 46 XY | NR5A1, FTZ1, FTZF1, SF1, AD4BP | null | 400044 |
| 87 | Ихтиоз буллезный | KRT2, KRT2A, KRT2E | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы+Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 146800 |
| 88 | Ихтиоз врожденный аутосомно-рецессивный | TGM1, NIPAL4, ALOX12B, ICHYN, TGK | null | 242300 |
| 89 | Ихтиоз вульгарный | FLG | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 146700 |
| 90 | Ихтиоз, спастическая квадриплегия и умственная отсталость | ELOVL4 | null | 614457 |
| 91 | Кампомелическая дисплазия | SOX9 | null | 114290 |
| 92 | Карпентера синдром | RAB23 | Наследственные синдромы | 201000 |
| 93 | Кератита-ихтиоза-тугоухости синдром | GJB2, CX26 | Болезни органов зрения | 148210 |
| 94 | Клайнфельтера синдром | null | Хромосомная патология | —— |
| 95 | Клиппеля-Фейля синдром | GDF6, CDMP2 | Скелетные дисплазии | 118100 |
| 96 | Коккейна синдром | ERCC6, CSB | Болезни органов зрения | 133540 |
| 97 | Комбинированный дефицит витамин K-зависимых факторов свертывания крови | VKORC1, VKOR | Болезни крови и иммунной системы | 607473 |
| 98 | Косолапость врожденная с или без дефицита длинных костей и/или зеркальной полидактилией | null | null | 119800 |
| 99 | Костелло синдром | HRAS, HRAS1, RASh2, p21(RAS) | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы+Наследственные синдромы+Пороки развития nullа, умственная отсталость, судороги | 218040 |
| 100 | Костная гетероплазия прогрессирующая | GNAS, GNAS1 | Скелетные дисплазии | 166350 |
| 101 | Коудена болезнь | null | Тугоухость+Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы+Патология эндокринных желез+Наследственные синдромы+Опухоли+Болезни органов зрения | 158350 |
| 102 | Коффина-Лоури синдром | RPS6KA3, RSK2, ISPK1, MAPKAPK1B | Наследственные синдромы+Пороки развития nullа, умственная отсталость, судороги | 303600 |
| 103 | Кошачьего глаза синдром | null | Хромосомная патология | 115470 |
| 104 | Краниометафизарная дисплазия | ANKH, GJA1, HANK, CX43 | Скелетные дисплазии | 123000 |
| 105 | Краниосиностоз | MSX2, TWIST1 | Скелетные дисплазии | 604757 |
| 106 | Краниофациальной дисморфии-тугоухости-ульнарной девиации кистей синдром | null | Наследственные синдромы | 122880 |
| 107 | Крейтцфельда-Якоба болезнь | null | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 123400 |
| 108 | Криглера-Найяра синдром | null | null | 218800 |
| 109 | Крузона с черным акантозом синдром | null | Наследственные синдромы | 612247 |
| 110 | Крузона синдром | null | Наследственные синдромы | 123500 |
| 111 | Лермитт-Дуклос болезнь | null | null | 158350 |
| 112 | Липодистрофия семейная частичная | LMNA | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 151660 |
| 113 | Макла-Уэллса синдром | NLRP3, CIAS1, NALP3, PYPAF1 | Патология почек+Тугоухость | 191900 |
| 114 | Маклеода синдром | XK, KX | Болезни крови и иммунной системы | 300842 |
| 115 | Мандибулоакральная дисплазия с липодистрофией | null | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 248370 |
| 116 | Маринеску-Шегрена синдром | SIL1, BAP | null | 248800 |
| 117 | Маршалла-Смита синдром | NFIX, NF1A | null | 602535 |
| 118 | Мевалоновая ацидурия | null | Наследственные болезни обмена веществ | 610377 |
| 119 | Мезомелическая дисплазия Лангера | null | null | 249700 |
| 120 | Метгемоглобинемия | CYB5R3, DIA1, B5R | Болезни крови и иммунной системы | 250800 |
| 121 | Метилглутаконовая ацидурия | null | Наследственные болезни обмена веществ | 258501 |
| 122 | Микрофтальм изолированный | null | Болезни органов зрения | 613094 |
| 123 | Микрофтальм с катарактой | CRYBA4 | Болезни органов зрения | 610426 |
| 124 | Микроцефалии с капиллярными мальформациями синдром | 614261 | ||
| 125 | Миллера-Дикера синдром | PAFAh2B1, LIS1 | Пороки развития nullа, умственная отсталость, судороги | 247200 |
| 126 | Милроя болезнь (лимфедема наследственная) | FLT4, VEGFR3 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 153100 |
| 127 | Миоклоническая дистония | SGCE | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 159900 |
| 128 | Миопатия Миоши | null | null | 613319 |
| 129 | Миопатия с диспропорцией типов мышечных волокон | ACTA1, SEPN1, SELN, ASMA | Нервно-мышечные болезни | 255310 |
| 130 | Миотоническая дистрофия | DMPK, ZNF9, DMK, CNBP1 | Нервно-мышечные болезни | 160900 |
| 131 | Миотония Томсена/Беккера | CLCN1, CLC1 | Нервно-мышечные болезни | 160800, 255700 |
| 132 | Множественной эндокринной неоплазии второго типа (МЭН2) синдром | null | Патология эндокринных желез | 171400, 162300 |
| 133 | Множественные вывихи суставов, задержка роста, черепно-лицевые аномалии и врожденные пороки сердца | B3GAT3, GLCATI | Патология эндокринных желез | 245600 |
| 134 | Множественных птеригиумов синдром | null | null | 265000 |
| 135 | Множественных синостозов синдром | NOG | null | 186500 |
| 136 | Моуат-Вильсон синдром | ZEB2, ZFHX1B, SMADIP1, SIP1, KIAA0569 | Патология эндокринных желез+Болезни органов зрения | 235730 |
| 137 | Муковисцидоз | CFTR, ABCC7 | Заболевания сердечно-сосудистой системы и легких | 219700 |
| 138 | Мышечная дистрофия врождённая | ITGA7 | Нервно-мышечные болезни | 613204 |
| 139 | Мышечная дистрофия Дюшенна-Беккера | DMD | Нервно-мышечные болезни | 310200 |
| 140 | Мышечная дистрофия поясноконечностная | SGCG, SGCA, POMT1, CAPN3, | Нервно-мышечные болезни | 253600 |
| 141 | Мышечная дистрофия тип Фукуяма | null | Нервно-мышечные болезни | 253800 |
| 142 | Мышечная дистрофия Эмери-Дрейфуса | null | Нервно-мышечные болезни | 310300, 181350 |
| 143 | Мюнке синдром | null | null | 602849 |
| 144 | Накопления нейтральных липидов с миопатией болезнь | null | null | 610717 |
| 145 | Нанизм MULIBREY | TRIM37, MUL, KIAA0898 | Скелетные дисплазии | 253250 |
| 146 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II | |||
| 147 | Наследственная моторно-сенсорная нейропатия (Шарко-Мари-Тута) тип I | PMP22, EGR2, FGD4, GJB1, GDAP1, LITAF, MPZ, NDRG1, Sh4TC2, YARS, DNM2, MFN2, NEFL, GAS3, KROX20, FRABIN, CX32, P0, MPP, PROXY1, KIAA1985, TYRRS, YTS, YRS, DYN2, KIAA0214, NFL, NF68 | Нервно-мышечные болезни | 118200, 118210 |
| 148 | Наследственная нейропатия с подверженностью параличу от сдавления | null | Нервно-мышечные болезни | 162500 |
| 149 | Наследственный амилоидоз | TTR, TBPA | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 105210 |
| 150 | Наследственный ангионевротический отек | C1NH, C1INH, SERPING1 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 106100 |
| 151 | Незаращение родничков | ALX4 | Пороки развития nullа, умственная отсталость, судороги | 609597 |
| 152 | Нейромиотония и аксональная нейропатия | HINT1 | 137200 | |
| 153 | Нейросенсорная несиндромальная тугоухость | GJB3, GJB6, SLC26A4, CX31, CX30, PENDRIN, PDS | Тугоухость | 220290 |
| 154 | Нейтропения тяжёлая врождённая | ELANE, ELA2, NE, HNE, HLE | Болезни крови и иммунной системы | 202700 |
| 155 | Некомпактного левого желудочка синдром | null | Заболевания сердечно-сосудистой системы и легких | 300183 |
| 156 | Немалиновая миопатия | null | null | 161800 |
| 157 | Нефронофтиз | null | Патология почек | 256100 |
| 158 | Нефротический синдром | NPHS2, PDCN | Патология почек | 256300 |
| 159 | Ниймеген синдром | NBN, NBS1 | Болезни крови и иммунной системы | 251260 |
| 160 | Ногтей-надколенника синдром | LMX1B, LMX1. 2 2 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 161200 |
| 161 | Нормокалиемический периодический паралич | null | Нервно-мышечные болезни | 170600 |
| 162 | Норри болезнь | NDP, NORRIN | Болезни органов зрения | 310600 |
| 163 | Окулофарингеальная мышечная дистрофия | PABPN1, PAB2 | Нервно-мышечные болезни | 164300 |
| 164 | Опица GBBB синдром | MID1, MIDIN, FXY | Патология эндокринных желез | 300000 |
| 165 | Ослера-Рендю-Вебера синдром | ENG, CD105 | null | 187300 |
| 166 | Остеолиз карпотарзальный, мультицентрический | null | null | 166300 |
| 167 | Остеопетроз рецессивный (мраморная болезнь костей) | TCIRG1, ATP6V0A3 | Скелетные дисплазии | 259700 |
| 168 | Охдо синдром, SBBYSS вариант | KAT6B | 603736 | |
| 169 | Паллистера синдром | TBX3 | Патология эндокринных желез | 181450 |
| 170 | Паллистера-Холла синдром | null | Патология эндокринных желез | 146510 |
| 171 | Патау синдром | null | Хромосомная патология | —— |
| 172 | Пахионихия врожденная | KRT6B, K6B, CK6B | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 167210 |
| 173 | Пейтца-Егерса синдром | null | null | 175200 |
| 174 | Первичная гипертрофическая остеоартропатия (пахидермопериостоз) | HPGD, PGDh2, PGDH | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 259100 |
| 175 | Первичная легочная гипертензия | BMPR2 | Заболевания сердечно-сосудистой системы и легких | 178600 |
| 176 | Периодическая болезнь | MEFV, PYRIN, MARENOSTRIN | null | 249100 |
| 177 | Пигментная дегенерация сетчатки | RP2, NRL, CA4, CA IV, D14S46E | Болезни органов зрения | 268000 |
| 178 | Пикнодизостоз | CTSK | Патология эндокринных желез | 265800 |
| 179 | Пневмоторакс первичный спонтанный | null | Заболевания сердечно-сосудистой системы и легких | 173600 |
| 180 | Подколенного птеригиума синдром | null | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 119500 |
| 181 | Полидактилия | LMBR1, ZRS | Скелетные дисплазии | 174700, 174200 |
| 182 | Понтоцеребеллярная гипоплазия | VRK1 | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 607596 |
| 183 | Потоцки-Лупски синдром | null | Хромосомная патология | 610883 |
| 184 | Почечная аплазия/гипоплазия | UPK3A, UPK3, UPIII | Патология почек | 191830 |
| 185 | Проверка результатов NGS (МПС) | null | null | |
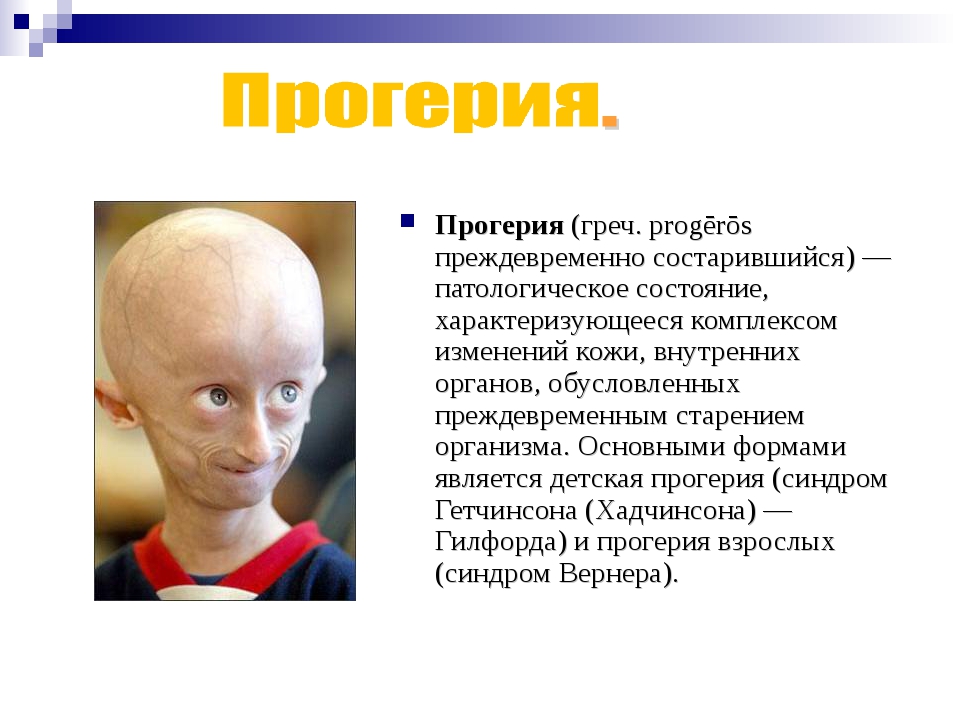
| 186 | Прогерия Хатчинсона-Гилфорда | null | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 176670 |
| 187 | Псевдоахондроплазия | COMP, THBS5 | Скелетные дисплазии | 177170 |
| 188 | Псевдоксантома эластическая | ABCC6, ARA, MRP6 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 264800 |
| 189 | Пфайффера синдром | null | Скелетные дисплазии | 101600 |
| 190 | Рабдомиолиз (миоглобинурия) | LPIN1, PAP1, KIAA0188 | Нервно-мышечные болезни | 268200 |
| 191 | Ретиношизис | RS1 | Болезни органов зрения | 312700 |
| 192 | Ретта синдром | MECP2 | Пороки развития nullа, умственная отсталость, судороги | 312750 |
| 193 | Робинова синдром | null | null | 268310 |
| 194 | Ротмунда-Томсона синдром | null | null | 268400 |
| 195 | Рубинштейна-Тейби синдром | null | Патология эндокринных желез | 180849 |
| 196 | Семейная периодическая лихорадка | TNFRSF1A, TNFR1, TNFAR | null | 142680 |
| 197 | Семейный гемофагоцитарный лимфогистиоцитоз | PRF1, STX11, STXBP2, UNC13D, PFN1, UNC18B, MUNC18-2, MUNC13-4 | Болезни крови и иммунной системы | 267700 |
| 198 | Семейный медуллярный рак щитовидной железы | null | Патология эндокринных желез+Опухоли | 155240 |
| 199 | Семейный холодовой аутовоспалительный синдром | null | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 120100 |
| 200 | Сениора-Локена синдром | null | null | 266900 |
| 201 | Сенсорная полинейропатия (врожденная нечувствительность к боли) | NGF, WNK1, NGFB, PSK, PRKWNK1, KDP | Нервно-мышечные болезни | 608654, 201300 |
| 202 | Септо-оптическая дисплазия | HESX1, RPX | Болезни органов зрения | 182230 |
| 203 | Сетре-Чотзена синдром | HESX1 | Патология эндокринных желез | 101400 |
| 204 | Симпсона-Голаби-Бемель синдром | GPC3 | Патология эндокринных желез | 312870 |
| 205 | Синдром CINCA | null | Патология эндокринных желез | 607115 |
| 206 | Синдром CRASH | null | null | 303350 |
| 207 | Синдром ESC | NR2E3, PNR | Патология эндокринных желез | 268100 |
| 208 | Синдром MASA | null | null | 303350 |
| 209 | Синдром RAPADILINO | null | null | 266280 |
| 210 | Синдром TAR | RBM8A, RBM8 | Патология эндокринных желез | 274000 |
| 211 | Скапулоперонеальная миопатия | FHL1, SLIM1 | null | 300695 |
| 212 | Смит-Магенис синдром | null | Патология эндокринных желез | 182290 |
| 213 | Смита-Лемли-Опица синдром | DHCR7 | Патология эндокринных желез | 270400 |
| 214 | Спинальная амиотрофия типы I, II, III, IV | SMN1, SMNT, SMN, T-BCD541 | Нервно-мышечные болезни | 253300 |
| 215 | Спинальная и бульбарная амиотрофия Кеннеди | AR, DHTR, NR3C4 | Нервно-мышечные болезни | 313200 |
| 216 | Спиноцеребеллярная атаксия | ATXN1, ATXN2, ATXN3, ATXN7, ATXN8, AT3, MJD1, SCA3 | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 164400 |
| 217 | Спонгиоформная энцефалопатия с нейропсихическими проявлениями | null | Пороки развития nullа, умственная отсталость, судороги | 606688 |
| 218 | Спондилокостальный дизостоз | DLL3 | Скелетные дисплазии | 277300 |
| 219 | Спондилоэпифизарная дисплазия (SEDT) | TRAPPC2, SEDL | Скелетные дисплазии | 313400, 183900 |
| 220 | Суперактивность фосфорибозилпирофосфат синтетазы | null | Наследственные болезни обмена веществ | 300661 |
| 221 | Тестикулярной феминизации синдром | null | null | 300068 |
| 222 | Торсионная дистония | PRRT2, SPR, TOR1A, DYT1 | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 128100 |
| 223 | Трихоринофалангеальный синдром | TRPS1 | Патология эндокринных желез | 190350 |
| 224 | Тричера Коллинза-Франческетти синдром | TCOF1, TREACLE | Патология эндокринных желез | 154500 |
| 225 | Тромбоцитопения врожденная | MPL, TPOR, MPLV | Болезни крови и иммунной системы | 604498 |
| 226 | Унферрихта-Лундборга болезнь | CSTB, STFB | null | 254800 |
| 227 | Уокера-Варбург синдром | null | null | 236670 |
| 228 | Ушера синдром | null | null | 276900 |
| 229 | Фатальная семейная инсомния | null | null | 600072 |
| 230 | Фенилкетонурия | PAH | Наследственные болезни обмена веществ | 261600 |
| 231 | Фибродисплазия оссифицирующая прогрессирующая | ACVR1, ACVRLK2, ALK2 | Скелетные дисплазии | 135100 |
| 232 | Фокальная кожная гипоплазия (Горлина-Гольца синдром) | null | null | 305600 |
| 233 | Х-сцепленная агаммаглобулинемия | CYBB | Болезни крови и иммунной системы | 300300 |
| 234 | Х-сцепленный лимфопролиферативный синдром | Sh3D1A, XIAP, SAP, BIRC4, API3, MIHA | Болезни крови и иммунной системы | 308240 |
| 235 | Х-сцепленный моторный нистагм | FRMD7 | Болезни органов зрения | 310700 |
| 236 | Х-сцепленный тяжелый комбинированный иммунодефицит | IL2RG, CD132 | Болезни крови и иммунной системы | 300400 |
| 237 | Хайду-Чейни синдром | null | null | 102500 |
| 238 | Хондродисплазия метафизарная тип Мак-Кьюсика | RMRPR, RMRP | Скелетные дисплазии | 250250 |
| 239 | Хондродисплазия точечная Конради-Хюнермана | EBP | Скелетные дисплазии | 302960 |
| 240 | Хондрокальциноз | null | Скелетные дисплазии | 118600 |
| 241 | Хореоатетоз, гипотиреоидизм и неонатальная дыхательная недостаточность | NKX2-1, TITF1, TTF1 | null | 610978 |
| 242 | Хорея Гентингтона | HTT, IT15 | Нейродегенеративные заболевания с поражением экстрапирамидной и мозжечковой систем и спастическая параплегия Штрюмпеля | 143100 |
| 243 | Хорея доброкачественная наследственная | null | null | 118700 |
| 244 | Хороидермия | CHM, REP1, GGTA | Болезни органов зрения | 303100 |
| 245 | Хроническая гранулематозная болезнь | CYBB, NOX2 | Болезни крови и иммунной системы | 306400 |
| 246 | Цереброокулофациоскелетный синдром | null | Скелетные дисплазии+Патология эндокринных желез | 214150 |
| 247 | Цистиноз нефропатический | null | null | 219800 |
| 248 | Швахмана-Даймонда синдром | SBDS | null | 260400 |
| 249 | Шегрена-Ларссона синдром | ALDh4A2 | null | 270200 |
| 250 | Шерешевского-Тернера синдром | null | Хромосомная патология | —— |
| 251 | Шпринтцена-Гольдберга синдром | SKI | null | 182212 |
| 252 | Штаргардта болезнь | null | null | 248200 |
| 253 | Эдвардса синдром | null | Хромосомная патология | —— |
| 254 | Экзостозы множественные | EXT1, EXT2, EXT | Болезни крови и иммунной системы | 133700 |
| 255 | Экссудативная витреохореоретинальная дистрофия | null | Болезни органов зрения | 305390 |
| 256 | Эктодермальная ангидротическая дисплазия | EDA, EDAR, EDARADD, EDA1, ED1, EDA1R | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 305100 |
| 257 | Эктодермальная гидротическая дисплазия | null | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 129500 |
| 258 | Эктопия хрусталика | null | Болезни органов зрения | 129600 |
| 259 | Элерса-Данло синдром | null | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 225400 |
| 260 | Эпилепсия прогрессирующая миоклоническая | KCTD7 | null | 611726 |
| 261 | Эпифизарная дисплазия, множественная | null | Скелетные дисплазии | 226900 |
| 262 | Эритрокератодермия | GJB4, CX30. 3 3 | Патология кожи и ее придатков, подкожной клетчатки и соединительной ткани, и лимфатической системы | 133200 |
| 263 | Эритроцитоз рецессивный | null | null | 263400 |
| 264 | Эскобара синдром | CHRNG, ACHRG | Патология эндокринных желез | 265000 |
Чтобы вылечить генетическое заболевание, нужно разбудить отцовский ген
Наш геном складывается из двух копий — отцовского и материнского набора хромосом. Каждый ген имеет своего двойника; оба могут выполнять одну функцию, но делают это по-разному. Очень часто случается, что одна из копий засыпает, а всю работу выполняет другая. Это нормально: например, у нас работает только материнская копия гена UBE3A, расположенного на 15-й хромосоме.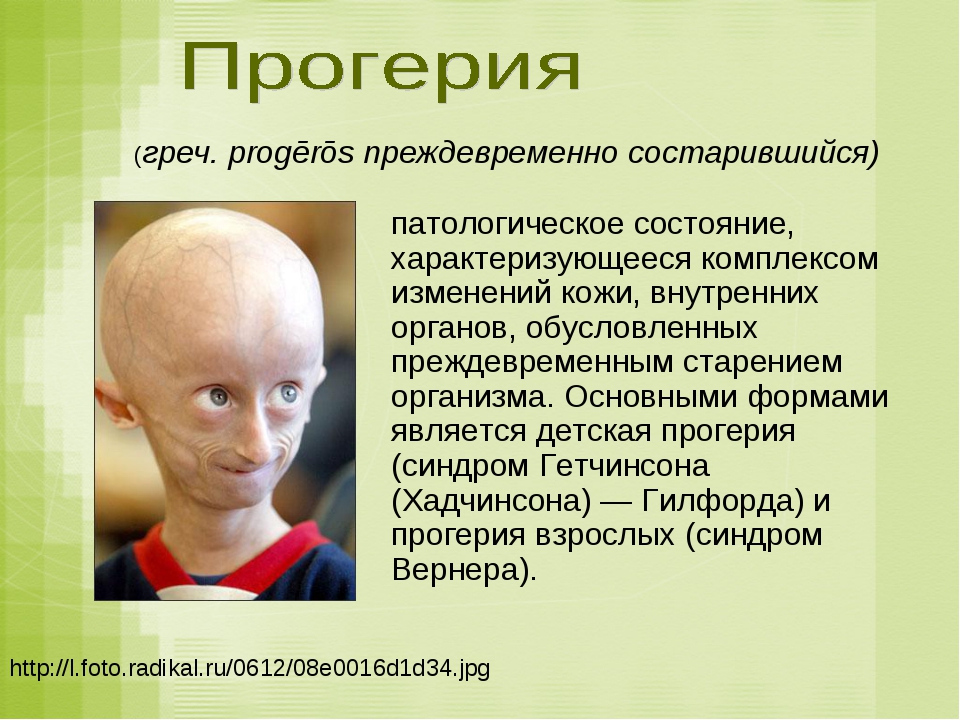 Белок, кодируемый этим геном, принимает участие в развитии нервной системы и регулирует деградацию ненужных белков. Если же этот ген по какой-то причине не работает, у человека развивается генетическое неврологическое заболевание, называемое синдромом Ангельмана. Для него характерны и задержка в развитии, и неконтролируемые резкие, хаотические движения, и приступы смеха. Недуг не такой уж редкий, встречается у одного человека из 15 тысяч.
Белок, кодируемый этим геном, принимает участие в развитии нервной системы и регулирует деградацию ненужных белков. Если же этот ген по какой-то причине не работает, у человека развивается генетическое неврологическое заболевание, называемое синдромом Ангельмана. Для него характерны и задержка в развитии, и неконтролируемые резкие, хаотические движения, и приступы смеха. Недуг не такой уж редкий, встречается у одного человека из 15 тысяч.
Итак, при синдроме Ангельмана ситуация такова: отцовская копия гена, как ей и полагается, находится в спящем состоянии, а материнская, которая в норме должна работать, сломана. Решение проблемы кажется очевидным: нужно «разбудить» отцовскую копию гена. Но это проще сказать, чем сделать. Исследователи из Университета Северной Каролины (США) перебрали около 2 300 разных соединений в надежде, что какое-нибудь из них сможет включить отцовскую копию UBE3A. В конце концов они попробовали ингибитор топоизомеразы I, известный противораковый препарат. Топоизомераза представляет собой фермент, помогающий клетке удваивать генетический материал; без неё невозможна репликация ДНК и размножение. Ингибиторы топоизомеразы делают так, что фермент остаётся постоянно связанным с ДНК, в итоге раковая клетка не может размножаться. Кроме того, ДНК часто не выдерживает столь тесных «объятий» фермента и начинает рваться.
Ингибиторы топоизомеразы делают так, что фермент остаётся постоянно связанным с ДНК, в итоге раковая клетка не может размножаться. Кроме того, ДНК часто не выдерживает столь тесных «объятий» фермента и начинает рваться.
Оказалось, что ингибитор топоизомеразы обладает ещё и генно-терапевтическими свойствами. В статье, опубликованной в журнале Nature, авторы пишут, что этот медикамент пробуждал от спячки отцовскую копию гена UBE3A. Причём эффект длился целых 12 недель после введения мышам этого ингибитора. Как противораковый препарат активирует ген? Исследователи полагают, что ингибитор топоизомеразы выключает синтез антисмысловой РНК, которая связывается с мРНК, синтезируемой на гене UBE3A, и подавляет синтез белка на ней. Так или иначе, белок Ube3a начинал синтезироваться во всех важнейших участках центральной нервной системы, включая гиппокамп, мозжечок, кору полушарий и спинной мозг.
Необходимо подчеркнуть, что все эти данные относятся к нервным клеткам, и исследователи пока не знают, как подействует ингибитор топоизомеразы на другие типы здоровых клеток. С другой стороны, хотя препарат давно используется в химиотерапии, будет ли он лечить у человека ещё и нервную систему, сказать заранее невозможно.
С другой стороны, хотя препарат давно используется в химиотерапии, будет ли он лечить у человека ещё и нервную систему, сказать заранее невозможно.
Сейчас всё лечение больных синдромом Ангельмана сводится лишь к облегчению симптомов, а ингибитор топоизомеразы позволяет надеяться на то, что можно будет устранить саму причину болезни. И, пожалуй, самым главным результатом можно назвать то, что исследователи показали реальность такого способа терапии, когда на замену неработающему гену встаёт его копия из нашего же генома. Таким образом можно лечить самые разные генетические заболевания, не прибегая к редактированию человеческой ДНК.
Наследственные заболевания и генетические синдромы
Диагностика генетических синдромов и наследственных заболеваний:
На настоящем этапе развития медицины многие генетические заболевания выявляются с помощью молекулярных диагностических методик еще до проявления первых клинических симптомов. Например, тест на предрасположенность эмбриона к синдрому Дауна делают на первом и втором триместрах беременности. Многие нарушения развития можно выявить при УЗИ плода.
Например, тест на предрасположенность эмбриона к синдрому Дауна делают на первом и втором триместрах беременности. Многие нарушения развития можно выявить при УЗИ плода.
Причинами для направления на генетическую диагностику детей могут служить:
- Выявленные пороки и аномалии развития;
- Незначительные отклонения, не оказывающие влияния на функцию органа (так называемые «стигмы дисэмбриогенеза»), в количестве более трех, например: видоизмененный копчик, деформация мочек ушей, недоразвитость крыльев носа;
- Нарушение психического, моторного развития;
- Необычное поведение, радикально выходящее за пределы возрастной нормы;
- Опережение или отставание от сверстников в физическом развитии;
- Возраст матери на время беременности и родов старше 40 лет;
- Нестандартные реакции на применяемую терапию;
- Типичные для генетических синдромов симптомы.
Женщинам, имевшим в анамнезе патологию беременности (замершие беременности, мертворождения неустановленной причины, бесплодие) также необходимо пройти генетическую диагностику.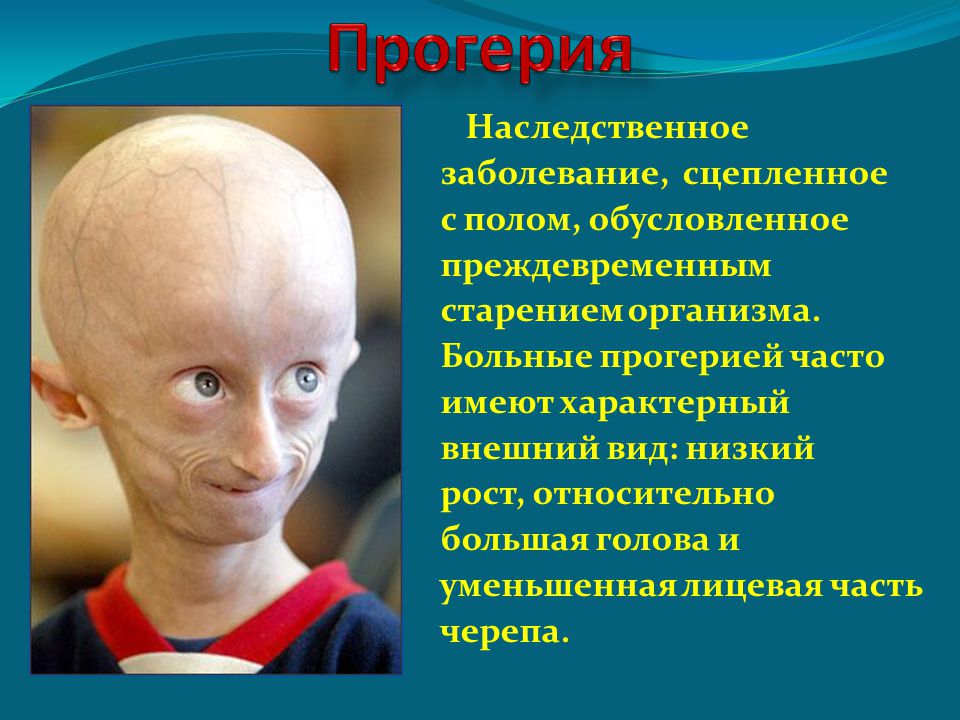
Для того, чтобы диагностика состояния ребенка была назначена корректно, важно подробно и достоверно рассказать врачу об истории тревожащих симптомах у малыша, его болезнях, применявшемся лечении и его результатах, особенностях развития и семейном генетическом анамнезе. В диагностике могут применяться лабораторные методы исследований, рентген/компьютерная томография/МРТ/УЗИ, ЭЭГ и другие.
Лечение детей с генетическими синдромами и наследственными заболеваниями:
В Центре Здоровья и Развития имени Святителя Луки ведут прием детский невролог, педиатр, детский психиатр, психолог, эпилептолог, реабилитолог, команда педагогов-дефектологов и другие специалисты. Наши сотрудники проводят коррекцию проявлений генетических заболеваний. Коррекционные педагоги работают с отстающими в развитии детьми, детьми с нарушениями зрения и слуха, нарушениями речи. Психиатр поможет в лечении психических нарушений, детский психолог – в социальной адаптации ребенка. Специалисты по двигательным расстройствам подберут комплекс упражнений для развития опорно-двигательного аппарата и моторики, при необходимости назначат массаж, мануальную терапию и/или физиотерапию.
Центр Здоровья и Развития ориентирован преимущественно на работу с детьми, имеющими отклонения в развитии. Доктора и педагоги имеют не только необходимые знания, но и релевантный опыт помощи детям с нарушениями речи, психическими расстройствами, синдромом Дауна, олигофренией, нарушением зрения или слуха и другими патологиями.
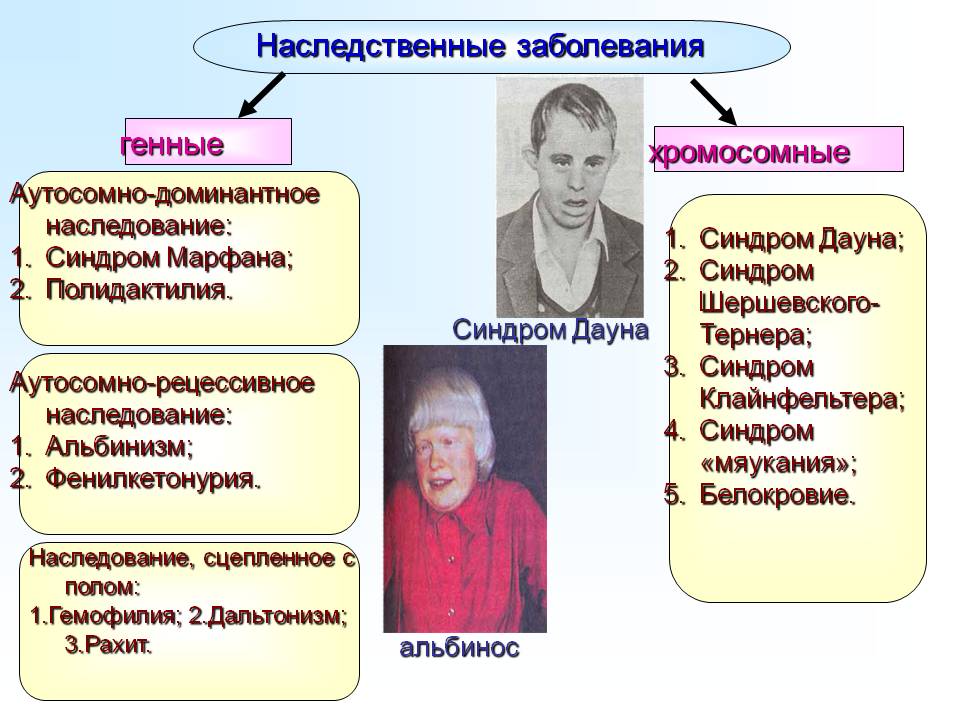
Хромосомные заболевания (изменение количества или структуры хромосом):
В норме каждая клетка человека содержит 46 хромосом – структур клетки, несущих в себе генетическую информацию. 23 хромосомы передаются от яйцеклетки, 23 – от сперматозоида. Исследование хромосом проводит врач цитогенетик, используя в качестве материала клетки крови – лимфоциты, подготовленные специальным образом. Цитогенетик при исследовании распределяет хромосомы по парам (кариотипирование) и нумерует. К примеру, последняя двадцать третья пара несет в себе информацию о поле: ХХ (Х-хромосома) – женский пол, ХУ (Y-хромосома) – мужской пол.
Существует более простое исследование, определяющее число хромосом — кариотипирование, а также сложное и подробное исследование, позволяющее выявить мельчайшие поломки хромосом – хромосомный микроматричный анализ.
Существуют хромосомные болезни, при которых вышеописанная схема нарушается.
Один из самых распространённых синдромов, в основе которых лежат хромосомные нарушения – синдром Дауна. При этом заболевании обнаруживается дополнительная 47-я хромосома. Такое же количество хромосом наблюдается при достаточно редком синдроме — болезни Клайнфельтера. В отличие от синдрома Дауна это патология возникает только у лиц мужского пола.
У лиц женского пола может наблюдаться кариотип из 45 хромосом (отсутствие одной половой хромосомы), так называемая болезнь Шерешевского-Тернера.
Другие заболевания наследственной природы:
В ядре человеческой клетки насчитывается более тридцати тысяч генов! Если в одном из них возникает изменение или мутация, говорят о моногенном синдроме, то есть связанным с 1 определенным геном. Каждому гену соответствуют белки, которые в свою очередь отвечают за работу определенных органов и систем организма. Нарушение в гене – это нарушение в синтезе белка и, как следствие, нарушение в работе клеток/органов.
Нарушение в гене – это нарушение в синтезе белка и, как следствие, нарушение в работе клеток/органов.
Примеры наиболее распространенных моногенных синдромов:
Фенилкетонурия. При этом синдроме аминокислота фенилаланин не усваивается организмом, в связи с чем фенилаланин и его токсичные продукты накапливаются, достигают токсических концентраций и начинают негативно влиять на организм, в частности на головной мозг.
Муковисцидоз. Тяжелое нарушение в работе системы дыхания и желудочно-кишечного тракта.
Гемофилия. Нарушение свертываемости крови. Заболевание проявляется у мужчин. Мутировавший ген переходит от матери (являющейся носителем патологического гена без проявлений болезни).
Современная молекулярная диагностика позволяет выявить генетические синдромы и принять меры для коррекции проявлений генетической «поломки».
Для выявления генетических заболеваний применяются различные генетические анализы – от подробного исследования отдельных генов до исследования большого числа генов, связанных с определенными заболеваниями – панели генов. Наиболее широкие исследования – полное секвенирование экзома и секвенирование генома человека – включают анализ всех известных к настоящему времени генетических аномалий.
Наиболее широкие исследования – полное секвенирование экзома и секвенирование генома человека – включают анализ всех известных к настоящему времени генетических аномалий.
В норме каждая клетка человека содержит 46 хромосом – структур клетки, несущих в себе генетическую информацию. Исследование хромосом проводит врач-цитогенетик, используя в качестве материала клетки крови – лимфоциты, подготовленные специальным образом.
Существует более простое исследование, определяющее число хромосом — кариотипирование, а также сложное и подробное исследование, позволяющее выявить мельчайшие поломки хромосом – хромосомный микроматричный анализ.
Генетические заболевания
Генетические болезни можно разделить на три группы:
В случае моногенных заболеваний мутация затрагивает только один ген. Она может присутствовать в одной или в обеих хромосомах (от каждого родителя наследуется по одной хромосоме). Примерами таких заболеваний являются кистозный фиброз, поликистозная болезнь почек, анемия. В сравнении с такими распространенными мультифакториальными болезнями, как сахарный диабет и болезни сердца, моногенные заболевания встречаются довольно редко. Моногенные заболевания делятся на «доминантные» и «рецессивные».
Примерами таких заболеваний являются кистозный фиброз, поликистозная болезнь почек, анемия. В сравнении с такими распространенными мультифакториальными болезнями, как сахарный диабет и болезни сердца, моногенные заболевания встречаются довольно редко. Моногенные заболевания делятся на «доминантные» и «рецессивные».
- Доминантные заболевания возникают из-за наличия гена болезни в одной из унаследованных хромосом. Вероятность, что ребенок заболеет доминантной болезнью, составляет 50%. Примерами таких заболеваний являются болезнь Хантингтона и синдром Марфана.
- Рецессивные заболевания возникают из-за наличия гена болезни в обеих унаследованных от родителей хромосомах. В этом случае вероятность того, что ребенок получит по наследству рецессивную болезнь, составляет 25%. Примеры рецессивных заболеваний – кистозный фиброз и болезнь Тея-Сакса.
Хромосомные болезни – это заболевания, при которых изменено количество или структура хромосом.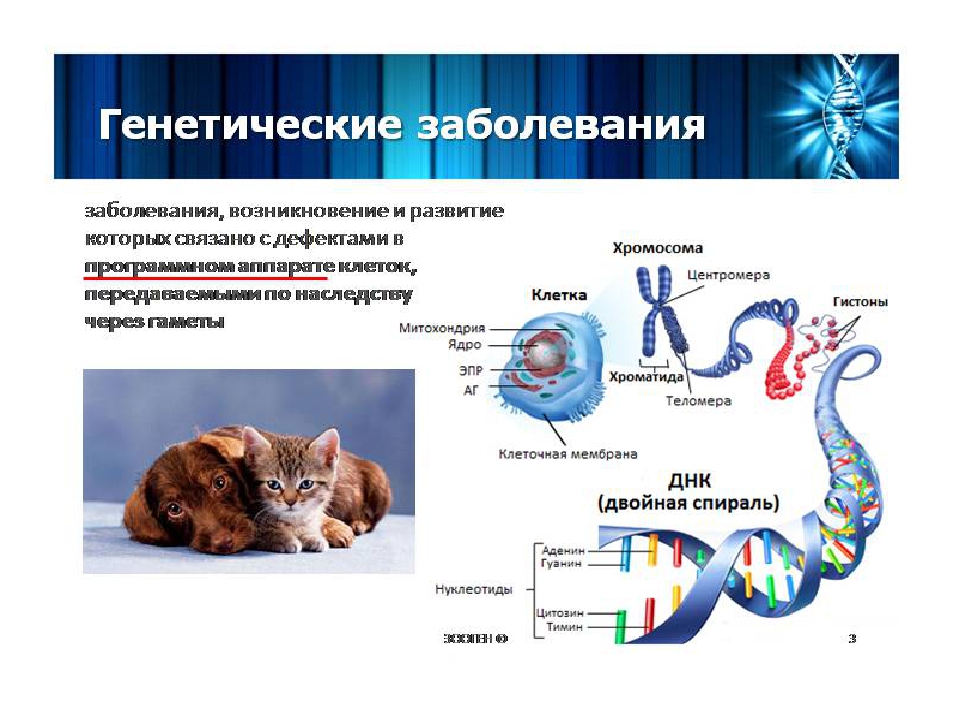 Хромосомы – это структуры, в которых находится ДНК с нашими генами. Например, болезнь Дауна возникает в том случае, когда вместо 46 хромосом появляется 47, причем ни один ген в хромосомах не изменен.
Хромосомы – это структуры, в которых находится ДНК с нашими генами. Например, болезнь Дауна возникает в том случае, когда вместо 46 хромосом появляется 47, причем ни один ген в хромосомах не изменен.
Мультифакториальные болезни – это заболевания, при которых мутируют два и более генов, но возникновение болезни и ее проявление тесно связано с факторами внешней среды и образом жизни человека. Примером таких болезней являются болезни сердца, диабет и большинство разновидностей рака. Ученые открывают все больше новых данных о влиянии генов на поведенческие расстройства, на пациентов с алкоголизмом, в случае ожирения, при психических заболеваниях и болезни Альцгеймера.
ЦЕНТР ОРФАННЫХ ЗАБОЛЕВАНИЙ
В России редкими предложено считать заболевания с распространенностью не более 10 случаев на 100 000 человек.
В список орфанных болезней специалисты Минздравсоцразвития РФ в 2012 году внесли 230 наименований.
По данным Формулярного комитета РАМН, россиян с этими болезнями насчитывается около 300 тысяч человек.
На сегодня разработаны 24 стандарта оказания помощи таким больным.
Что такое генетические заболевания, как часто они встречаются и как их предотвратить, об этом мы беседуем с
Главным специалистом по медицинской генетике МЗ Челябинской области
заведующей Медико- генетической консультацией и
заведующей центром орфанных болезней Челябинской областной детской клинической больницы
кандидатом медицинских наук
Галиной БУЯНОВОЙ.
— Галина Викторовна, что такое орфанные заболевания?
— Понятие «орфанные» так и переводится как редкие, еще их называют «болезни-сироты». Термин появился в 1983 году в США. На сегодня описано около 7 000 их разновидностей (врожденных, гематологических, онкологических, аутоиммунных и других), которыми страдает 6–8 % населения планеты. Распространенность орфанных заболеваний составляет около 1:2 000 и реже. Такая статистика весьма условна, бывают патологии – одна на миллион человек. В то же время заболевание может быть редким в одном регионе и частым в другом, например, проказа часто встречается в Индии, но редко в Европе.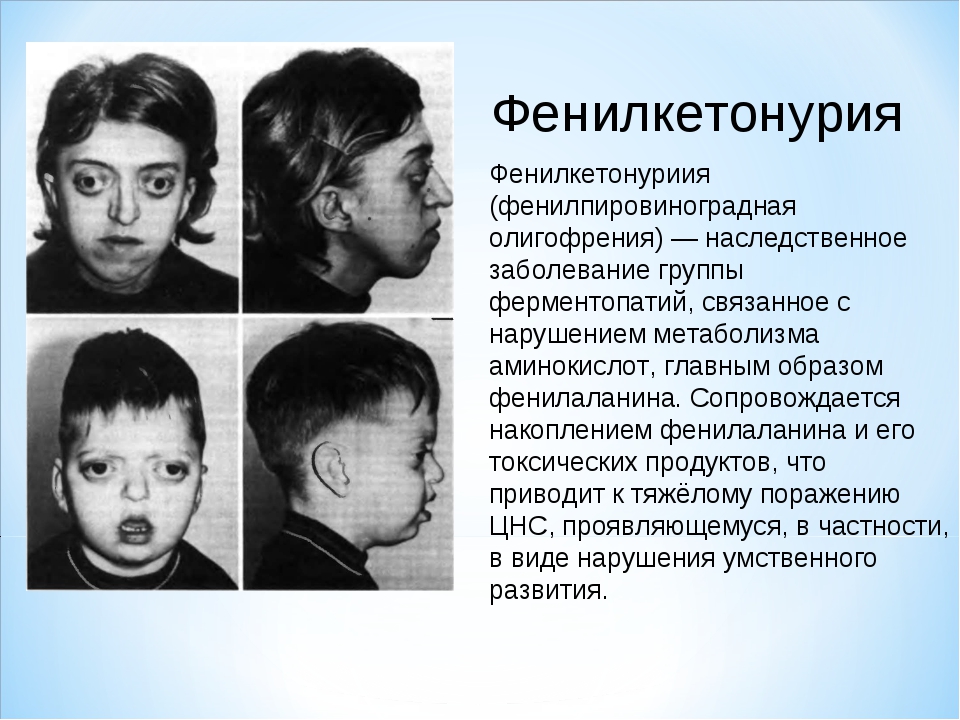 А болезнь Гоше имеет национальную доминанту и чаще всего поражает ашкеназских евреев.
А болезнь Гоше имеет национальную доминанту и чаще всего поражает ашкеназских евреев.
— Почему именно в последние десятилетия эти болезни так громко заявили о себе?
— Дело в том, что только в конце XX века был расшифрован геном человека. У нас появились совершенно фантастические технологии для диагностики и лечения орфанных заболеваний. Раньше медицина была практически бессильна помочь даже цесаревичу Алексею, императорскому сыну, страдавшему гемофилией, президенту США Джону Кеннеди, у него была болезнь Аддисона. Сегодня мы можем применить самые современные технологии обычным людям, всем нашим детям. С 1995 года в Челябинской области внедрен неонатальный скрининг на наследственные болезни обмена веществ: адреногенитальный синдром, галактоземию, муковисцидоз, фенилкетонурию, врожденный гипотериоз. В первые дни жизни еще в роддоме по капельке крови, взятой из пяточки младенца, проводится исследование на пять этих наиболее распространенных наследственных заболевания.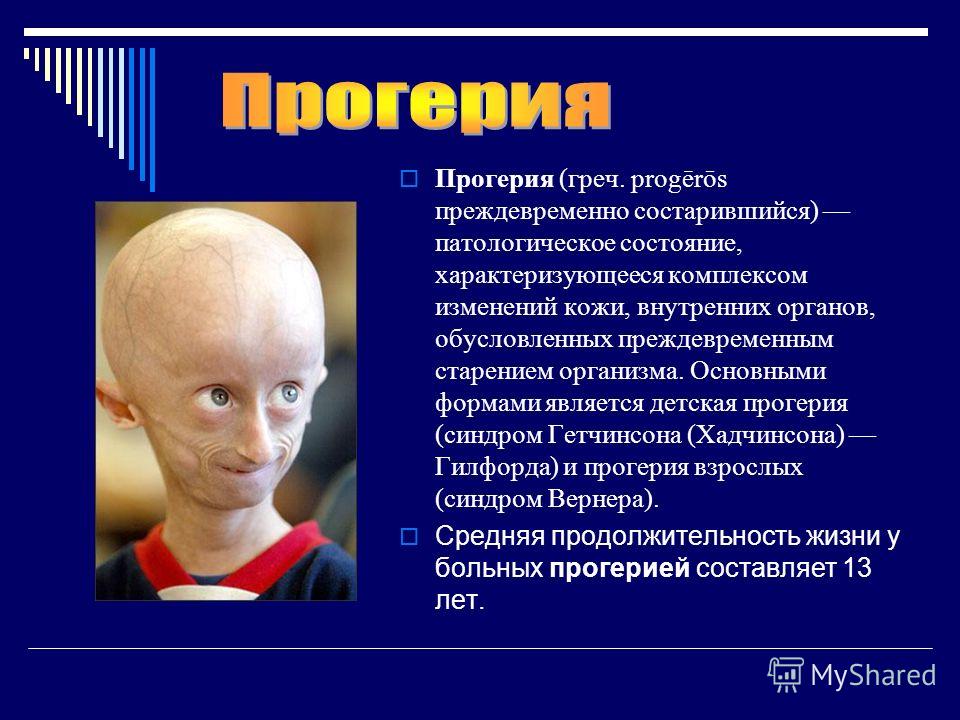
— Но вы же сами говорили, что «ваши» болезни неизлечимы, невозможно поправить генную или хромосомную мутацию.
— Они лечатся симптоматически. У нас появились возможности не допустить инвалидности ребенка. Мы выявляем много детей с галактоземией. Это очень серьезное заболевание, при котором кормление малыша грудным молоком опасно для жизни. Раньше такие дети погибали. Сейчас мы можем вовремя поставить диагноз, с первого дня обеспечить ребенка соевой безмолочной смесью, и он будет здоров.
Раньше мы не знали, как поправить не работающий из-за генетического дефекта фермент, диета не помогала. Сейчас появилась ферментозаместительная терапия: фермент доставляется в клетку и работает вместо недостающего. Это, конечно, уже научный прорыв. Трем девочкам в нашей области мы поставили диагноз мукополисахаридоз. У них нормальный интеллект, но при этом тяжелое поражение скелета и внутренних органов. Им необходимо пожизненное лечение, капельницы раз в неделю. Без него они погибнут. Но стоимость его невероятно дорогая — около несколько миллионов в год на одного больного. Их лечение оплачивает бюджет. Получая его регулярно, дети хорошо развиваются, прекрасно выглядят. Лекарство работает. И мы постоянно держим руку на пульсе.
Без него они погибнут. Но стоимость его невероятно дорогая — около несколько миллионов в год на одного больного. Их лечение оплачивает бюджет. Получая его регулярно, дети хорошо развиваются, прекрасно выглядят. Лекарство работает. И мы постоянно держим руку на пульсе.
— Расскажите о вашей службе. Сколько у нас врачей-генетиков?
— Генетиков-клиницистов по всей России всего около 250 человек. Честно говоря, мы неплохо представлены, потому что десять из них работают в нашей области: двое в Магнитогорске и восемь в Челябинске. Все они – высокопрофессиональные, опытные специалисты. Южноуральская генетика ведет свой отсчет с 1984 года: тогда в многопрофильной городской клинической больнице №1 Челябинска в составе консультации «Брак и семья» был открыт кабинет медико–генетического консультирования. Сегодня в Челябинске четыре таких консультации. Практически все исследования, которые делаются в Москве и за рубежом, сегодня можно получить здесь, не выезжая из Челябинска. У нас отработаны технологии пересылки крови, других биологических образцов. И все современные методы исследований мы применяем. В нашем распоряжении — цитогенетическая и биохимическая лаборатории детской областной больницы, а также возможности всех структурных подразделений медучреждения: диагностических, параклинических и лечебных отделений. В среднем за год в нашем центре получают консультацию врача-генетика порядка трех тысяч семей. Помимо амбулаторных консультаций генетик по показаниям осматривает детей, находящихся на обследовании и лечении в стационарах нашей больницы. Мы ведем регистр орфанных заболеваний — всего в нем 431 человек из Челябинской области, из них 239 детей.
У нас отработаны технологии пересылки крови, других биологических образцов. И все современные методы исследований мы применяем. В нашем распоряжении — цитогенетическая и биохимическая лаборатории детской областной больницы, а также возможности всех структурных подразделений медучреждения: диагностических, параклинических и лечебных отделений. В среднем за год в нашем центре получают консультацию врача-генетика порядка трех тысяч семей. Помимо амбулаторных консультаций генетик по показаниям осматривает детей, находящихся на обследовании и лечении в стационарах нашей больницы. Мы ведем регистр орфанных заболеваний — всего в нем 431 человек из Челябинской области, из них 239 детей.
— Почему так много? Откуда берутся орфанные болезни?
— Примерно 40% из них обусловлены генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте. Генетическую основу имеют практически все заболевания. Поэтому, видите, у меня в шкафу стоят книги по глазным болезням и эндокринологии, ортопедии и кардиологии… Генетики помогают поставить диагноз, находят мутацию, но наблюдают пациентов другие специалисты – нам это просто не по силам.
Реже встречаются токсические, инфекционные или аутоиммунные болезни. Причинами их развития могут быть наследственность, ослабление иммунитета, плохая экология, высокий радиационный фон, вирусные инфекции у мамы и у самих детей в раннем возрасте.
— Какие генетические заболевания встречаются на Южном Урале чаще всего?
— Больше всего меня печалит громадное количество детей с алкогольной фетопатией: беременные женщины, особенно в деревнях, пьют, причем спокойно говорят об этом. «Да, я выпивала, так тянуло на пиво!» — как ни в чем не бывало, признается мамочка. Это страшный момент, потому что такие дети обречены на умственную отсталость и физическое недоразвитие, они все на одно лицо, зачастую диагноз можно поставить с порога. В этом году мы зарегистрировали уже 15 алкогольных фетопатий. Эта грозная тенденция: за последние годы число детей с этим заболеванием выросло втрое.
Очень опасно для будущего малыша и курение его мамы, оно влияет на сосуды плаценты: дети у таких мам рождаются маловесные, слабенькие.
Второе по частоте место среди генетических заболеваний, передающихся по наследству, занимает нейрофиброматоз. Заподозрить болезнь у малышей можно по многочисленным пятнам на коже цвета кофе с молоком. Когда такого ребенка приводят на консультацию, приходится раздевать его папу и маму. Часто и у них тоже по всему телу обнаруживаются кофейные пятна. «Ну и что? – Удивляются родители. — Всю жизнь с ними живем, даже не замечаем!» Иногда бывает и так. Но чаще болезнь начинает проявляться в подростковом возрасте скелетными изменениями, мозговыми опухолями, ребенок плохо учится. Заболевание прогрессирует.
Третьего лидера я вам назвать не могу. Множественных врожденных пороков развития очень много, в этом году их уже 85, и все они – разные.
— Галина Викторовна, у вас огромный опыт, вы работаете в этой сфере 27-ой год. Но как можно поставить правильный диагноз, если генетических болезней — тысячи и большинство из них вы, слава богу, никогда не видели?
— Мы запаслись всевозможной литературой, заглядываем в Интернет, связываемся с нашими коллегами в Москве, Санкт-Петербурге, Томске, где очень сильное НИИ медицинской генетики. Некоторые анализы для уточнения диагноза генетических заболеваний приходится отправлять в столицы или даже за рубеж.
Некоторые анализы для уточнения диагноза генетических заболеваний приходится отправлять в столицы или даже за рубеж.
Вот у меня настольная книга «Наследственные синдромы» — огромный энциклопедический том с цветными фотографиями всевозможных пороков. Нередко приходится начинать с портретной диагностики. В отделении новорожденных у нас лежал ребеночек с нарушением ритма сердца. Осматривая его, мы заподозрили синдром Костелло, чрезвычайно редкое генетическое заболевание, встречается одно на миллион. Он был направлен в Томск на радиочастотную операцию. Мы дали родителям направление на генетическую консультацию у наших коллег в Сибирском отделении РАМН, и они подтвердили наш диагноз.
Мало того, вместе с этим малышом лежал еще один из дома ребенка. Когда увидела его, ахнула – те же грубоватые черты лица, избыток кожи… Неужели еще один синдром Костелло? Хромосомный микроматричный анализ подтвердил наши догадки.
Такие бывают совпадения: уникальное, крайне редкое заболевание, а у нас сразу два в одном отделении.
— Почему так важно поставить правильный диагноз?
— Во-первых, тактика лечения меняется. Ведь часть детей раньше вообще не имела конкретного диагноза. Огромное количество генетических заболеваний прячется под маской ДЦП, пороков сердца. Во-вторых, диагноз очень важен для профилактики, прогноза здоровья будущего потомства в семьях, где уже есть больной ребенок. К нам постоянно приходят беременные, мы с ними проговариваем все риски. При необходимости берем у плода материал в первом триместре и смотрим, здоров ли он, сохранять ли беременность. В этом году наши мамы, у которых первый ребенок болен муковисцидозом, поликистозом почек, спинальной амиотрофией, родили вторых здоровых детей.
— Поздравляю! Это, конечно, заслуга врачей. Так может быть всем молодым парам следует обращаться к генетику?
— Нет, конечно. Опасность в чем? Каждый человек является носителем 5-7 генетических мутаций, которые могут никогда не проявиться. Но если ваш будущий супруг имеет такую же мутацию, один с вами дефектный ген, и они совпадут, вероятность развития заболевания у ребенка очень велика.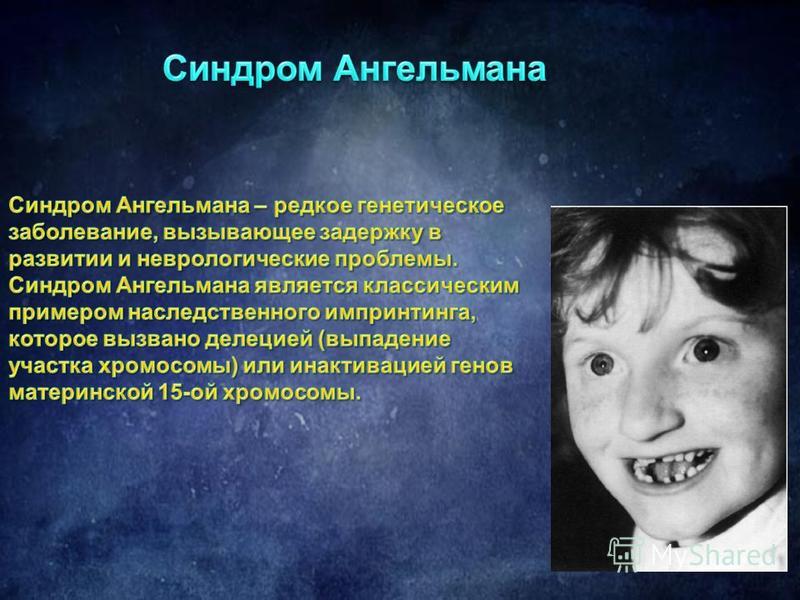 Сейчас есть программы обследования при вступлении в брак на носительство мутаций самых частых генетических заболеваний. Если ты – носитель болезни, партнер должен сдать только один анализ на ту же мутацию.
Сейчас есть программы обследования при вступлении в брак на носительство мутаций самых частых генетических заболеваний. Если ты – носитель болезни, партнер должен сдать только один анализ на ту же мутацию.
— Почему же тогда у нас все-таки рождаются дети с хромосомными мутациями, болезнью Дауна?
— Что касается болезни Дауна, то в плане диагностики это самое простое заболевание. Пренатальная (до родов) диагностика с каждым годом выявляет все больше таких случаев, за 2015 год, например, выявлено 65,8 % всех синдромов Дауна в Челябинской области. Это очень неплохая цифра!
— А 100% достигнуть нельзя?
— Нет, это нереально. Всегда, к сожалению, есть женщины, пропускающие скрининги, не встающие вовремя на учет, не делающие УЗИ, не сдающие кровь. Кроме того, 20-25% пациенток отказываются от инвазивной диагностики плода и при самом неблагоприятном прогнозе все-таки решают рожать. Вероятность появления на свет ребенка с болезнью Дауна составляет один случай на 600 родов. Сегодня акушеры отмечают тенденцию возрастных родов, а после 35 лет эта частота увеличивается в десять раз.
Сегодня акушеры отмечают тенденцию возрастных родов, а после 35 лет эта частота увеличивается в десять раз.
— Почему фактором риска становится возраст женщины, а не мужчины?
— Женщина отличается от мужчины еще и способом производства половых клеток. У мужчины каждые три месяца образуется новый генетический материал. А женщина всю жизнь живет со своими яйцеклетками. Болезни, гормональные сбои, лекарства со временем накапливаются и отражаются на качестве яйцеклетки. Чем старше женщина, тем больше вероятность хромосомных сбоев. Женщине, рожающей после 35 лет, консультация генетика просто необходима.
Нина ЧИСТОСЕРДОВА
О медико — генетической консультации более подробно можно прочитать здесь Медико – генетическая консультация
О платных услугах Платные услуги
| Наследственное заболевание | Коротко о заболевании (частота, проявление, прогноз) | Зачем нужен генетический анализ |
| Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия) | АР (Аутосомно-рецессивное)-заболевание (носительство 1 на 90 человек), нарушается обмен меди и происходит увеличение ее концентрации в различных органах: головном мозге, почках, печени и роговице. Проявляется в возрасте 10-25 лет, чаще в школьном возрасте (10-16 лет) в виде поражения печени, в более позднем – неврологическими нарушениями; б/х снижен церулоплазмин; при осмотре глаз пигментные кольца Кайзера-Флейшера на радужке. Проявляется в возрасте 10-25 лет, чаще в школьном возрасте (10-16 лет) в виде поражения печени, в более позднем – неврологическими нарушениями; б/х снижен церулоплазмин; при осмотре глаз пигментные кольца Кайзера-Флейшера на радужке. | Анализ нужен для точной диагностики (мутации в гене АТР7В, кодирующем белок медь-транспортирующую аденозинтрифосфатазу). |
| Синдром Жильбера (наследственная доброкачественная неконъюгированная гипербилирубинемия (80-100 мкмоль/л)) | Частота от 2-5% до 36%. Мутации в гене UGT1A1 приводят к снижению активности фермента уридиндифосфатглюкуронилтрансферазы 1 в гепатоцитах на 25%, что и обуславливает клиническую картину. Этот фермент (УДФ-ГТ1) участвует в метаболизме некоторых лекарственных веществ, т.е. возможна манифестация болезни с развитием токсических реакций при приеме – анаболических стероидов, глюкокортикоидов, андрогенов, рифампицина, циметидина, хлорамфеникола, стрептомицина, салицилата натрия, ампициллина, кофеина, этинилэстрадиола, парацетамола, иринотекана (используется при лечении рака толстой кишки).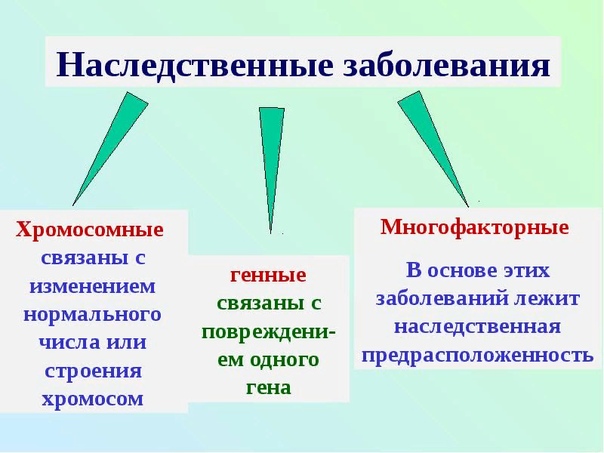 | Рекомендуется проведение анализа перед назначением препаратов, обладающих гепатотоксическими эффектами. |
| Гемохроматоз (пигментный цирроз, бронзовый диабет) | АР-заболевание (частота встречаемости 1 на 135 – 330 человек), мутации в гене HFE приводят к усилению всасывания железа в ЖКТ и его накоплению в тканях организма – печени, коже, сердце, суставах, гипофизе с последующим повреждением клеток и разрастанием соединительной ткани. Возраст начала болезни 40 – 60 лет у мужчин, в постменопаузе у женщин. Клиника: слабость, утомляемость, гиперпигментация, сахарный диабет, цирроз, кардиомиопатия. | Важна диагностика носительства для профилактики клинических проявлений (контроль уровня ферритина сыворотки крови: норма до 50 нг/мл, при необходимости терапия – кровопускание) |
| Муковисцидоз | АР-заболевание (частота 1:5000 новорожденных), мутация в гене CFTR приводит к поражению экзокринных желез: бронхолегочная система, поджелудочная железа, печень, потовые, слюнные, половые железы, кишечник (повышается вязкость секрета, концентрация хлоридов и натрия). Возраст начала: от новорожденного до взрослого. Летальность высокая, чаще от дыхательной недостаточности. Средний возраст выживания в США 33 года. Лечение симптоматическое. Возраст начала: от новорожденного до взрослого. Летальность высокая, чаще от дыхательной недостаточности. Средний возраст выживания в США 33 года. Лечение симптоматическое. | Анализ важен для адекватной терапии и прогнозирования будущего потомства в пораженных семьях. Мы проводим определение 36 наиболее частых мутаций в гене |
Генетические заболевания | Каталонский Институт Сетчатки
Генетические заболевания | Каталонский Институт Сетчатки
Генетические заболевания глаз — это заболевания, вызванные изменениями в генетическом коде, которые появляются на протяжении всей жизни и могут вызвать проблемы со зрением, в большей или меньшей степени влияющие на качество жизни пациента. Из-за своего генетического характера, они наиболее часто наследуются детьми от родителей и могут повлиять на различные ткани зрительной системы: на сетчатку в зоне макулы, на роговицу, на глазной нерв и т.д. Более 60% случаев детской слепоты связано с генетическими факторами.
Каковы основные причины глазных болезней генетического происхождения?
- Дистрофия сетчатки. Она представляет собой ряд генетических заболеваний сетчатки, которые приводят к вырождению фоторецепторных клеток (палочки и колбочки) . Основное заболевание — это пигментный ретинит, болезнь, влияющая на клетки сетчатки, главным образом на палочки, которые отвечают за периферическое зрение и зрение в условиях низкой освещённости. Также могут быть затронуты колбочки, которые являются фоторецепторными клетками, отвечающими за цветное зрение. Причинами развития этого заболевания являются генетические изменения, которые могут отличаться у разных пациентов.
- Врожденная глаукома — это редкий тип глаукомы, который развивается у младенцев и маленьких детей, наследственного характера и имеющего генетическую основу.
- Врожденная катаракта. Катаракта обычно появляется в преклонном возрасте и представляет собой помутнение хрусталика.
 Однако в случае врожденной катаракты, она присутствует уже при рождении ребёнка по генетическими причинами. Она может быть наследственной и развиться в одном или в обоих глазах.
Однако в случае врожденной катаракты, она присутствует уже при рождении ребёнка по генетическими причинами. Она может быть наследственной и развиться в одном или в обоих глазах. - Наследственное косоглазие. В некоторых случаях косоглазие может быть наследственным, если в семейном анамнезе наблюдается наличие косоглазия. Рекомендуется провести полное офтальмологическое обследование ребёнка.
- Пороки развития. Анофтальмия (полное отсутствие одного или обоих глаз), микрофтальмия (необычно маленький размера глазного яблока) и многие другие пороки развития зрительной системы.
- Дальтонизм. Генетическое заболевание, связанное с Х-хромосомой, при котором нарушается способность воспринимать цвета из-за отсутствия или сбоя в работе колбочек сетчатки — клеток, ответственных за восприятие цветов — зеленый, красный и синий.
- Дистрофии роговицы, группа заболеваний, которые вызывают потерю прозрачности роговицы.
- Атрофия и воспаления зрительного нерва наследственного характера. При атрофии зрительного нерва происходит прогрессирующая потеря зрения из-за повреждения зрительного нерва, которая может иметь наследственные причины. Ещё одной наследственной болезнью является оптическая невропатия Лебера, при которой важную роль играет митохондриальная наследственность.
- Системные заболевания, которые влияют на зрение. Некоторые заболевания системного характера, которые можно отнести к генетическим заболеваниям — заболевание Грависа или сахарный диабет, могут повлиять на зрение.
 Однако в случае врожденной катаракты, она присутствует уже при рождении ребёнка по генетическими причинами. Она может быть наследственной и развиться в одном или в обоих глазах.
Однако в случае врожденной катаракты, она присутствует уже при рождении ребёнка по генетическими причинами. Она может быть наследственной и развиться в одном или в обоих глазах.
Есть другие заболевания, очень распространеные среди населения в целом, как дегенерация макулы или жёлтого тела и глаукома, которые дают наиболее высокий риск развития у тех людей, в анамнезе семьи которых уже имеются эти заболевания. Дегенерация макулы связанная с возрастом, это дегенеративное заболевание, которое влияет на части сетчатки, ответственную за центральное зрение. Основным фактором риска здесь является возраст, и есть большой риск развития заболевания после 60 лет. Проявляется в нарушенях восприятия чёткости, формы и размера изображений. Глаукома – это заболевание, которое повреждает зрительный нерв и может привести к прогрессивной потере периферического зрения.
Основным фактором риска здесь является возраст, и есть большой риск развития заболевания после 60 лет. Проявляется в нарушенях восприятия чёткости, формы и размера изображений. Глаукома – это заболевание, которое повреждает зрительный нерв и может привести к прогрессивной потере периферического зрения.
Почему так важно проходить офтальмологические осмотры?
Офтальмологические осмотры необходимы для обнаружения любого вида генетических заболеваний . Вот почему, если у вас в семейном анамнезе есть болезни, которые могут передаваться по наследству и повлиять на зрение, очень важно пройти тщательное обследование глаз для того, чтобы исключить их присутствие или начать своевременное лечение при их наличии.
Статьи по теме
Отделение занимается диагностикой и лечением нарушений в бинокулярной функции зрения, глазомоторных нарушений, измерением отклонений при различных углах зрения, интенсивности супрессии, стереоскопическим зрением, фузией, аномальной корреспонденции сетчаток, и лечением диплопии и амблиопии (синдром ленивого глаза).
Что такое катаракта? Катаракта представляет собой помутнение хрусталика. Хрусталик – это структура внутри глаза, находящаяся за радужной оболочкой и зрачком, которая при рождении является абсолютно прозрачной. Его строение напоминает строение линзы, как по форме, так и по размеру. Помимо этого, он, так же как и линза, имеет внешнюю оболочку и внутреннее содержимое, состоящее из белков, […]
Понятие и значение глаукомы Глаукома считается важным заболеванием, так как оно является очень распространённым и потенциально серьезными. Примерно 1,5 — 2% населения старше 40 лет страдает этим заболеванием и с возрастом частота его увеличивается. Глаукома является второй ведущей причиной потери зрения в нашем обществе. Тем не менее, потерю зрения можно предотвратить путем своевременной диагностики и […]
Этот сайт использует куки-файлы для сбора статистической информации о навигации. Если вы продолжаете навегацию на сайте, мы посчитаем это за ваше согласие использования куки-файлов. Более подробная информация..
Более подробная информация..
We use cookies on our website to give you the most relevant experience by remembering your preferences and repeat visits. By clicking “Accept”, you consent to the use of ALL the cookies.
Manage consent
генов и болезней | Изучайте науку в Scitable
Эта тематическая комната посвящена механизмам возникновения болезней. При этом исследуется, почему некоторые люди страдают от определенных состояний, таких как полидактилия, расщелина позвоночника и рак. Кроме того, в нем обсуждается, что сделали ученые и какие инструменты они разработали для исследования этих состояний с целью их лучшего лечения или профилактики. Однако эта тематическая комната не ставит своей целью предоставить информацию обо всех заболеваниях человека.Скорее, его цель состоит в том, чтобы развить интерес и осознать сложные отношения между генетикой человека и различными болезненными состояниями.
В зависимости от генетического вклада болезни человека можно разделить на моногенные, хромосомные или многофакторные. Моногенные заболевания вызываются изменениями в одном гене, и они распределяются по семьям в соответствии с традиционными менделевскими принципами наследования. Хромосомные заболевания, как следует из их названия, вызываются изменениями в хромосомах.Например, в геноме индивидуума могут отсутствовать некоторые хромосомы, могут присутствовать дополнительные копии хромосом или некоторые части хромосом могут быть удалены или дублированы. Наконец, подавляющее большинство болезней человека можно отнести к категории многофакторных. Эти состояния также называются сложными заболеваниями, и на них ложится большая часть нагрузки на нашу систему здравоохранения. Примеры этих состояний включают сердечно-сосудистые заболевания, рак, диабет и ряд врожденных дефектов и психических расстройств.По определению, сложные заболевания вызваны вариациями многих генов, и на них может влиять или не влиять окружающая среда. Хотя эти состояния встречаются часто, они представляют собой самую большую проблему для исследователей-генетиков, и выявить гены, которые способствуют возникновению этих заболеваний, оказалось довольно сложно.
Моногенные заболевания вызываются изменениями в одном гене, и они распределяются по семьям в соответствии с традиционными менделевскими принципами наследования. Хромосомные заболевания, как следует из их названия, вызываются изменениями в хромосомах.Например, в геноме индивидуума могут отсутствовать некоторые хромосомы, могут присутствовать дополнительные копии хромосом или некоторые части хромосом могут быть удалены или дублированы. Наконец, подавляющее большинство болезней человека можно отнести к категории многофакторных. Эти состояния также называются сложными заболеваниями, и на них ложится большая часть нагрузки на нашу систему здравоохранения. Примеры этих состояний включают сердечно-сосудистые заболевания, рак, диабет и ряд врожденных дефектов и психических расстройств.По определению, сложные заболевания вызваны вариациями многих генов, и на них может влиять или не влиять окружающая среда. Хотя эти состояния встречаются часто, они представляют собой самую большую проблему для исследователей-генетиков, и выявить гены, которые способствуют возникновению этих заболеваний, оказалось довольно сложно. Помимо вышеупомянутых причин, ряд альтернативных генетических сценариев также может привести к заболеванию; такие сценарии подпадают под эпигенетику.
Помимо вышеупомянутых причин, ряд альтернативных генетических сценариев также может привести к заболеванию; такие сценарии подпадают под эпигенетику.
Одна из целей генетических исследований — лучше понять механизмы болезни, чтобы можно было предложить новые подходы к лечению и профилактические меры.Технологии прошли долгий путь в этом отношении, и в настоящее время возможно одновременное исследование почти одного миллиона сайтов в геномной ДНК любого человека с целью обнаружения ассоциаций между данным заболеванием и генетической вариацией. Однако технический прогресс также создал новые проблемы для ученых, например, как лучше всего обрабатывать миллионы точек данных, используемых в генетических исследованиях болезней. Математические и статистические модели должны быть улучшены, чтобы соответствовать растущему количеству данных, генерируемых сегодняшними исследованиями.Ученые также должны продолжать переосмысливать клинические описания болезней. Поскольку теперь исследователи понимают, что генетический вклад во многие болезни является сложным и что одно и то же заболевание не проявляется одинаково у всех людей, описания, которые включают градиенты болезни и здоровья, обычно более эффективны, чем те, которые классифицируют людей как «больные». «или» здоровый «.
«или» здоровый «.
Понимание роли генетики в развитии болезней стало центральной частью медицинских исследований.Соответственно, эта тематическая комната призвана служить отправной точкой для изучения этой относительно новой области медицины.
Изображение: Брайан К. Капелл / Национальный исследовательский институт генома человека.
Генетические нарушения: определение, развитие и примеры
Генетическое заболевание — это состояние, которое возникает в результате мутации ДНК. Есть несколько различных генетических нарушений.
Большинство клеток тела содержат молекулу ДНК. Эта молекула дает клетке инструкции о том, как функционировать.Изменение или мутация ДНК может привести к неправильному функционированию клетки.
В этой статье рассказывается, что такое генетические нарушения. В нем обсуждаются различные типы, основные симптомы каждого типа и способы их развития.
Генетические нарушения — это состояния, возникающие в результате изменений или мутаций ДНК в клетках организма.
Большинство клеток тела содержат длинные нити ДНК, которые снабжают клетку инструкциями. Каждая нить ДНК плотно намотана вокруг белка, называемого гистоном.Эта спиралевидная структура называется хромосомой.
Хромосомы содержат небольшие участки ДНК, называемые генами. Эти гены дают телу определенный набор инструкций. Каждая клетка человека обычно содержит 23 пары хромосом, по одной из каждой пары от каждого родителя. Следовательно, у человека есть две копии каждого гена.
Изменение или ошибка в ДНК может вызвать генетическое заболевание. Поскольку гены передаются от родителей к ребенку, эти нарушения могут передаваться по наследству. Однако не все члены семьи с генетическим заболеванием будут испытывать симптомы заболевания.
Генетические условия могут повлиять на любой ген или хромосому. Это означает, что существует широкий спектр генетических нарушений, каждое из которых вызывает различные симптомы.
Под геномом человека понимаются все гены и ДНК, необходимые для построения и содержания человека.
Проект «Геном человека» (HGP) был глобальным исследовательским проектом по картированию генома человека. В рамках проекта была установлена последовательность генома человека и функции различных генов.
По оценкам HGP, в геноме человека содержится около 20 000–25 000 генов.ДНК внутри этих генов содержит четыре химических основания, которые действуют как единицы информации. Это:
- аденин
- цитозин
- гуанин
- тимин
Каждая молекула ДНК содержит две скрученные цепи ДНК. Пары химических оснований соединяют одну цепь ДНК с другой, напоминая ступеньки лестницы. Пары оснований химических веществ между каждой цепью всегда сочетаются определенным образом. Например, аденин на одной цепи ДНК всегда соединяется с тимином на противоположной цепи ДНК.
Порядок пар химических оснований в каждой молекуле ДНК влияет на то, какие инструкции ДНК предоставляет организму. Секвенирование ДНК включает определение порядка этих пар оснований.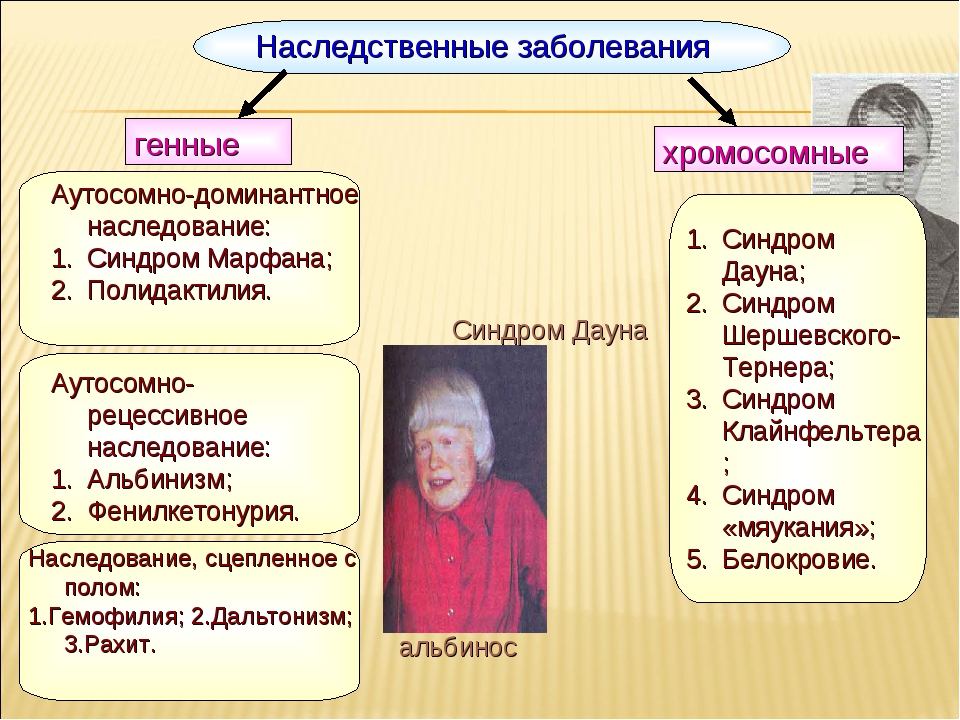
Секвенирование генома человека было важным шагом в понимании того, как гены могут вызывать заболевания.
Генетические заболевания, как правило, передаются по наследству. Родители передают гены своим детям, и некоторые из этих генов могут содержать основу генетического заболевания.
Однако каждый родитель передает только половину своих генов.Версия каждого гена, которую передает родитель, называется аллелем.
Если два аллеля от каждого родителя различаются, организм может принимать инструкции только от одного из них. Аллель, от которого клетка получает инструкции, известен как доминантный аллель. Другой известен как рецессивный аллель.
Некоторые генетические состояния передаются доминантным аллелем, а другие — рецессивным аллелем. Как правило, человек наследует конкретное генетическое заболевание, только если у него есть хотя бы один доминантный аллель для расстройства или два рецессивных аллеля для расстройства.
Единичное наследственное или моногенное заболевание — это заболевание, которое возникает в результате дефекта одного гена.
В следующих разделах будут описаны некоторые примеры условий одиночного наследования.
Болезнь Хантингтона
Болезнь Гентингтона — дегенеративное заболевание головного мозга, вызывающее:
- неконтролируемых движений
- эмоциональные расстройства
- снижение когнитивных функций
Болезнь Хантингтона развивается из-за мутации доминантного аллеля в хромосоме 4.У людей с этим аллелем со временем разовьется заболевание.
Лечение
В настоящее время нет способа остановить или замедлить прогрессирование болезни Хантингтона.
Однако некоторые лекарства могут помочь человеку справиться с симптомами. К ним относятся лекарства, которые помогают контролировать непроизвольные движения, и лекарства для лечения смены настроения, раздражительности и депрессии.
Серповидно-клеточные заболевания
Серповидно-клеточные заболевания (ВСС) — это группа состояний, которые влияют на эритроциты.
Серповидно-клеточная анемия — это разновидность ВСС, при которой красные кровяные тельца, транспортирующие кислород к тканям организма, деформированы. Их необычная форма означает, что они менее способны переносить кислород и более склонны к слипанию.
Их необычная форма означает, что они менее способны переносить кислород и более склонны к слипанию.
Сгустки этих клеток крови могут блокировать кровеносный сосуд, потенциально вызывая:
- боль
- инфекции
- острый грудной синдром
- инсульт
ВСС возникают в результате мутаций в гене HBB .Этот ген дает инструкции по производству красных кровяных телец.
ХДС рецессивные. Это означает, что человеку необходимо унаследовать два аллеля, содержащие мутацию, чтобы иметь заболевание.
Лечение
Лечение ВСС направлено на предотвращение осложнений и продление жизни.
Врач может прописать лекарство гидроксимочевину для увеличения размера красных кровяных телец, тем самым увеличивая количество кислорода, которое каждая клетка может переносить.
Мышечные дистрофии
Мышечные дистрофии — это группа генетических состояний, которые со временем вызывают повреждение и слабость мышц.Они происходят из-за мутаций гена DMD .
Мышечные дистрофии — это Х-сцепленные нарушения, что означает, что они влияют на ген на Х-хромосоме. Эти состояния чаще встречаются у мужчин. Это потому, что у мужчин одна Х-хромосома и одна Y-хромосома, тогда как у женщин две Х-хромосомы. У женщин здоровая Х-хромосома может противодействовать пораженной, но у мужчин нет другой Х-хромосомы, которая могла бы это сделать.
Лечение
В настоящее время не существует доступного лечения, чтобы остановить или обратить вспять мышечные дистрофии.
Вместо этого лечение направлено на предотвращение осложнений и улучшение качества жизни человека. Примеры таких методов лечения включают:
- физиотерапия для поддержания силы и гибкости мышц
- респираторная терапия для поддержания силы дыхательных мышц
- логопедия для людей, у которых слабость горла или лицевых мышц влияет на речь
- трудотерапия, чтобы помочь человеку использовать вспомогательные устройства, такие как инвалидные коляски
- одно или несколько из следующих лекарств, чтобы помочь замедлить или контролировать симптомы:
- глюкокортикоиды, чтобы увеличить мышечную силу и замедлить прогрессирование мышечной слабости
- иммунодепрессантов, которые могут помочь отсрочить повреждение мышечных клеток
- противосудорожные средства, чтобы помочь контролировать мышечные спазмы и судороги
Многофакторные наследственные расстройства (MID) — это состояния, которые развиваются из-за комбинации генетических факторов и факторов окружающей среды или образа жизни.
Некоторые из этих негенетических факторов могут включать:
Некоторые состояния, которые могут подпадать под категорию MID, включают:
Определенные генетические мутации могут увеличить риск этих состояний. Однако четкой схемы наследования нет.
Хромосомные аномалии — это проблемы, которые влияют на хромосому. Хромосомные аномалии могут включать:
- с отсутствующей хромосомой
- с дополнительной хромосомой
- с хромосомой с какой-либо структурной аномалией
Хромосомные аномалии обычно возникают, когда происходит ошибка при делении клетки.Эти ошибки обычно возникают в яйцеклетке или сперматозоиде, но могут возникать и после зачатия.
Возможно унаследовать хромосомную аномалию от родителя. Однако некоторые развиваются внутри человека впервые.
В следующих разделах будут описаны некоторые примеры хромосомных аномалий.
Синдром Дауна
Синдром Дауна — это тип хромосомной аномалии, которая влияет на интеллектуальное и физическое развитие.
Синдром Дауна возникает, когда человек получает дополнительную копию хромосомы 21.Это означает, что каждая клетка в теле содержит три копии хромосомы 21 вместо обычных двух.
Лечение
Синдром Дауна — это пожизненное заболевание. Однако различные виды терапии могут помочь в интеллектуальном и физическом развитии человека. Примеры включают:
- получение дополнительной помощи или внимания в школе
- логопед
- физиотерапия
- трудотерапия
Синдром Вольфа-Хиршхорна
Синдром Вольфа-Хиршхорна — это хромосомная аномалия, которая может повлиять на все тело.Основными особенностями этого состояния являются:
- задержка роста и развития
- снижение мышечного тонуса
- умственные нарушения
- судороги
Синдром Вольфа-Хиршхорна развивается из-за делеции участка хромосомы 4. Большинство случаев возникает при впервые у человека, у которого есть расстройство. Однако человек также может унаследовать заболевание от родителя, у которого есть хромосомная аномалия.
Лечение
В настоящее время нет лекарства от синдрома Вольфа-Хиршхорна.Однако следующие методы лечения могут помочь человеку справиться с симптомами и улучшить качество жизни:
- физиотерапия или профессиональная терапия
- консультирование
- лекарства, которые могут помочь при определенных симптомах, таких как судороги
Митохондрии — это биологические структуры, которые существуют внутри клеток тела. Они генерируют большую часть энергии, необходимой клеткам для проведения биохимических реакций.
Митохондриальные нарушения — это группа генетических состояний, которые влияют на ДНК внутри самих митохондрий.Эти мутации ДНК приводят к тому, что митохондрии не могут производить достаточно энергии для поддержания клеток тела.
Митохондриальные нарушения могут поражать любой орган или часть тела. Симптомы, которые испытывает человек, будут зависеть от части тела, пораженной заболеванием.
Некоторые возможные симптомы митохондриальных нарушений включают:
Мутации митохондриальной ДНК наследуются по материнской линии.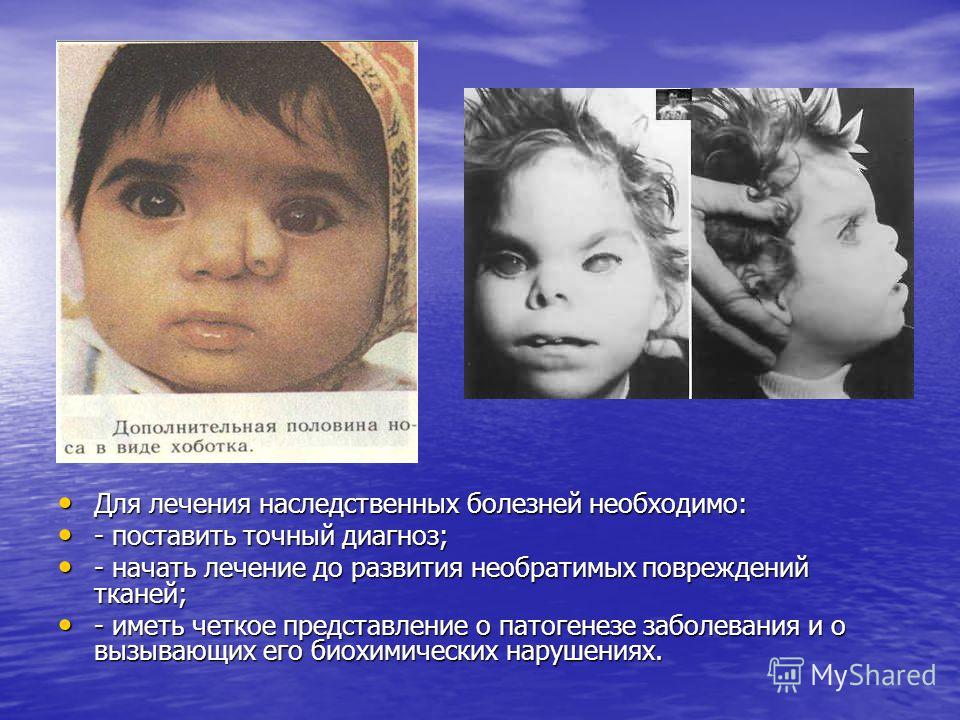 Это означает, что только мать может передавать митохондриальные нарушения.
Это означает, что только мать может передавать митохондриальные нарушения.
Лечение
В настоящее время не существует лекарства или высокоэффективного лечения митохондриальных нарушений.
Однако следующие методы лечения могут помочь человеку справиться с ними:
- Управление питанием
- витаминные добавки
- аминокислотные добавки
- лекарства, которые помогают лечить определенные проблемы, такие как мышечная слабость или судороги
Генетические нарушения возникают, в результате мутации ДНК. Эта мутация может затронуть целые хромосомы или определенные гены в хромосомах. Мутации ДНК также могут происходить в ДНК митохондрий, которые питают клетки человека.
Большинство генетических состояний передаются по наследству, но некоторые могут впервые возникнуть у человека, который страдает этим заболеванием.
Генетические заболевания — это пожизненное состояние. По этой причине лечение, как правило, направлено на то, чтобы помочь человеку справиться с симптомами, предотвратить осложнения и улучшить качество жизни.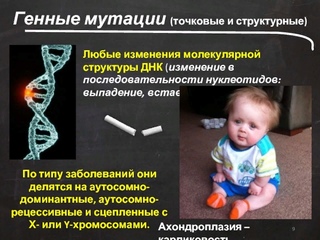
В некоторых случаях могут быть доступны лекарства, помогающие замедлить прогрессирование определенного заболевания.
Основы генетики | CDC
Генетические исследования изучают, как отдельные гены или группы генов участвуют в здоровье и болезнях.Понимание генетических факторов и генетических нарушений важно для того, чтобы больше узнать о укреплении здоровья и профилактике заболеваний.
Некоторые генетические изменения были связаны с повышенным риском рождения ребенка с врожденным дефектом или пороком развития или с развитием таких заболеваний, как рак или болезнь сердца. Генетика также может помочь нам понять, как возникают заболевания.
Как мы получаем наши гены
Люди получают (наследуют) свои хромосомы, содержащие их гены, от своих родителей.Хромосомы бывают парами, а у человека 46 хромосом, состоящих из 23 пар. Дети случайным образом получают по одной хромосоме каждой пары от матери и по одной хромосоме каждой пары от отца. Хромосомы, образующие 23-ю пару, называются половыми хромосомами. Они решают, мужчина человек или женщина. У женщины две Х-хромосомы, а у мужчины одна Х-хромосома и одна Y-хромосома. Каждая дочь получает X от матери и X от отца. Каждый сын получает X от матери и Y от отца.
Они решают, мужчина человек или женщина. У женщины две Х-хромосомы, а у мужчины одна Х-хромосома и одна Y-хромосома. Каждая дочь получает X от матери и X от отца. Каждый сын получает X от матери и Y от отца.
Генетические заболевания
Генетические нарушения могут возникать по многим причинам.Генетические нарушения часто описываются с точки зрения хромосомы, содержащей ген. Если ген находится на одной из первых 22 пар хромосом, называемых аутосомами, генетическое заболевание называется аутосомным заболеванием. Если ген находится на Х-хромосоме, заболевание называется Х-сцепленным.
Генетические расстройства также группируются по тому, как они протекают в семьях. Расстройства могут быть доминантными или рецессивными, в зависимости от того, как они вызывают заболевания и как протекают в семьях.
Доминант
Доминантные заболевания могут быть вызваны только одной копией гена с мутацией ДНК.Если один из родителей болен заболеванием, вероятность унаследовать мутировавший ген у каждого ребенка составляет 50%.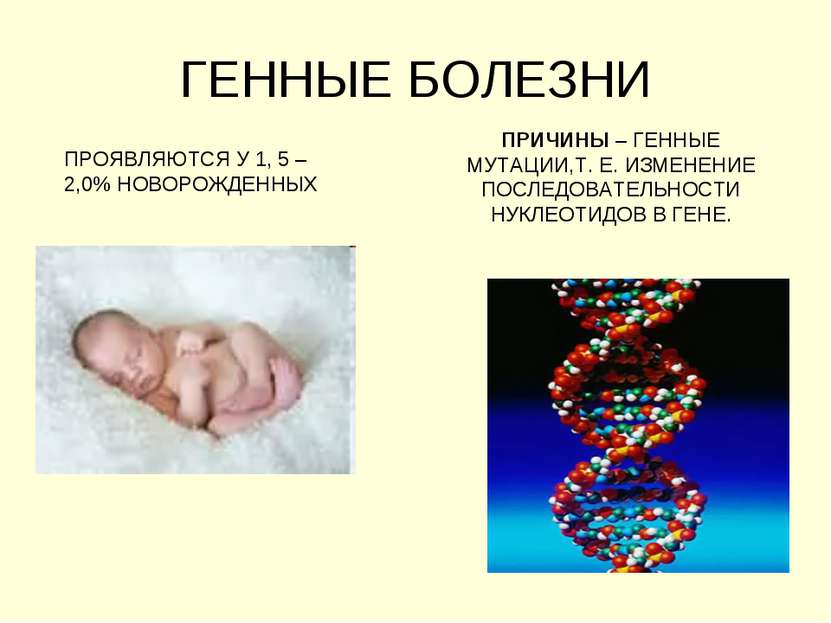
рецессивный
При рецессивных заболеваниях обе копии гена должны иметь мутацию ДНК, чтобы получить одно из этих заболеваний. Если у обоих родителей есть одна копия мутировавшего гена, вероятность заболевания каждого ребенка составляет 25%, даже если ни один из родителей не болен. В таких случаях каждого родителя называют носителем болезни. Они могут передать болезнь своим детям, но сами не болеют.
Заболевания с одним геном
Некоторые генетические заболевания вызваны мутацией ДНК в одном из генов человека. Например, предположим, что часть гена обычно имеет последовательность ТАС. У некоторых людей мутация может изменить последовательность на TTC. Это изменение в последовательности может изменить способ работы гена, например, изменив производимый белок. Мутации могут передаваться ребенку от его родителей. Или они могут произойти впервые в сперме или яйцеклетке, так что у ребенка будет мутация, а у родителей — нет.Заболевания одного гена могут быть аутосомными или Х-сцепленными.
Например, серповидно-клеточная анемия — это аутосомное заболевание с одним геном. Это вызвано мутацией в гене, обнаруженном на хромосоме 11. Серповидноклеточная анемия вызывает анемию и другие осложнения. С другой стороны, синдром ломкой Х-хромосомы — это заболевание с одним геном, сцепленным с Х-хромосомой. Это вызвано изменением гена на Х-хромосоме. Это наиболее распространенная из известных причин умственной отсталости и пороков развития, которая может передаваться по наследству (передаваться от одного поколения к другому).
Хромосомные аномалии
Различное количество хромосом
У людей обычно 23 пары хромосом. Но иногда человек рождается с другим номером. Если у человека есть лишняя хромосома, это называется трисомией. Если у человека отсутствует хромосома, это называется моносомией.
Например, у людей с синдромом Дауна есть дополнительная копия хромосомы 21. Эта дополнительная копия изменяет нормальное развитие тела и мозга и вызывает интеллектуальные и физические проблемы у человека. Некоторые расстройства вызваны разным количеством половых хромосом. Например, люди с синдромом Тернера по внешнему признаку обычно имеют только одну половую хромосому, X. Женщины с синдромом Тернера могут иметь проблемы с ростом и пороками сердца.
Некоторые расстройства вызваны разным количеством половых хромосом. Например, люди с синдромом Тернера по внешнему признаку обычно имеют только одну половую хромосому, X. Женщины с синдромом Тернера могут иметь проблемы с ростом и пороками сердца.
Изменения в хромосомах
Иногда хромосомы неполные или имеют другую форму, чем обычно. Если небольшая часть хромосомы отсутствует, это называется делецией. Если он переместился на другую хромосому, это называется транслокацией.Если он был перевернут, это называется инверсией.
Например, у людей с синдромом Вильямса внешний значок отсутствует небольшая часть хромосомы 7. Эта делеция может привести к умственной отсталости и отличительной внешности лица и личности.
Сложные условия
A Комплексное заболевание вызывается как генами, так и факторами окружающей среды. Сложные заболевания также называют многофакторными. Большинство хронических заболеваний, таких как болезни сердца, рак и диабет, представляют собой сложные состояния. Например, хотя некоторые случаи рака связаны с наследственными генетическими изменениями, например, синдромом Линча и наследственным раком груди и яичников, большинство из них, скорее всего, вызвано изменениями в нескольких генах, действующих вместе с воздействием окружающей среды.
Например, хотя некоторые случаи рака связаны с наследственными генетическими изменениями, например, синдромом Линча и наследственным раком груди и яичников, большинство из них, скорее всего, вызвано изменениями в нескольких генах, действующих вместе с воздействием окружающей среды.
Для получения дополнительной информации
Что вам нужно знать о 5 наиболее распространенных генетических заболеваниях
Генетические нарушения могут быть результатом генетических аномалий, таких как мутация гена или дополнительные хромосомы. Эффекты аномалий в ДНК человека когда-то были совершенно непредсказуемыми.Однако современная медицина разработала методы определения потенциальных последствий для здоровья генетических нарушений, о чем свидетельствуют медицинские исследования, проведенные образованными практикующими медсестрами с продвинутой квалификацией и практикующими врачами. Собрав следующие основанные на фактических данных статистические наблюдения, эти специалисты определили некоторые из текущих передовых методов выявления, лечения и потенциального предотвращения некоторых генетических нарушений.
Синдром Дауна
Обычно ядро отдельной клетки содержит 23 пары хромосом, но синдром Дауна возникает, когда 21-я хромосома копируется в дополнительное время во всех или некоторых клетках.Практикующие медсестры и врачи обычно проводят подробные пренатальные скрининговые тесты, такие как анализы крови, которые определяют количество хромосомного материала и других веществ в крови матери. Этот тип тестирования может с высокой точностью определить, родится ли ребенок с синдромом Дауна. Когда человеку ставят диагноз синдром Дауна, у него могут наблюдаться различные уровни задержки когнитивных функций от легкой до тяжелой. Другие маркеры синдрома Дауна включают более высокую предрасположенность к врожденным порокам сердца, низкий мышечный тонус, меньший физический рост и наклон вверх к глазам.По данным Центров по контролю и профилактике заболеваний (CDC), примерно один из каждых 700 новорожденных в США будет иметь синдром Дауна. Кроме того, чем старше мать на момент рождения, тем больше вероятность, что у ребенка синдром Дауна. [5]
[5]
Талассемия
Талассемия — это семейство наследственных генетических состояний, которые ограничивают количество гемоглобина, которое человек может вырабатывать естественным образом. Это состояние препятствует потоку кислорода по всему телу. Вероятность того, что дети, унаследовавшие ген талассемии от обоих родителей, родятся с талассемией, составляет 25 процентов.[7] К людям, которые с особой вероятностью могут быть носителями дефектного гена, ответственного за талассемию, относятся выходцы из Юго-Восточной Азии, Индии, Китая, Ближнего Востока, Средиземноморья и Северной Африки. Любая форма талассемии обычно сопровождается тяжелой анемией, которая может потребовать специализированной помощи, такой как регулярное переливание крови и хелатирующая терапия.
Муковисцидоз
Муковисцидоз — это хроническое генетическое заболевание, при котором у пациентов выделяется густая и липкая слизь, угнетающая их дыхательную, пищеварительную и репродуктивную системы. Как и талассемия, болезнь обычно передается по наследству с частотой 25%, когда оба родителя имеют ген муковисцидоза. В Соединенных Штатах около 30 000 человек живут с кистозным фиброзом, и у них часто возникают более серьезные проблемы со здоровьем. Например, 95 процентов мужчин с муковисцидозом бесплодны, а средний возраст выживания для всех пациентов составляет 33,4 года [9]. Образованные практикующие медсестры могут продлить типичное время выживания пациента, предлагая эффективные стратегии ухода, включающие физиотерапию, а также диетические и медицинские добавки.
Как и талассемия, болезнь обычно передается по наследству с частотой 25%, когда оба родителя имеют ген муковисцидоза. В Соединенных Штатах около 30 000 человек живут с кистозным фиброзом, и у них часто возникают более серьезные проблемы со здоровьем. Например, 95 процентов мужчин с муковисцидозом бесплодны, а средний возраст выживания для всех пациентов составляет 33,4 года [9]. Образованные практикующие медсестры могут продлить типичное время выживания пациента, предлагая эффективные стратегии ухода, включающие физиотерапию, а также диетические и медицинские добавки.
Болезнь Тай-Сакса
Генетическое заболевание, известное как Тай-Сакс, переносится примерно одним из каждых 27 евреев и примерно одним из каждых 250 представителей населения в целом. Состояние вызвано хромосомным дефектом, аналогичным синдрому Дауна. Однако, в отличие от синдрома Дауна, синдром Тея-Сакса возникает в результате дефекта, обнаруженного в хромосоме №15, и это расстройство является необратимо фатальным при обнаружении у детей.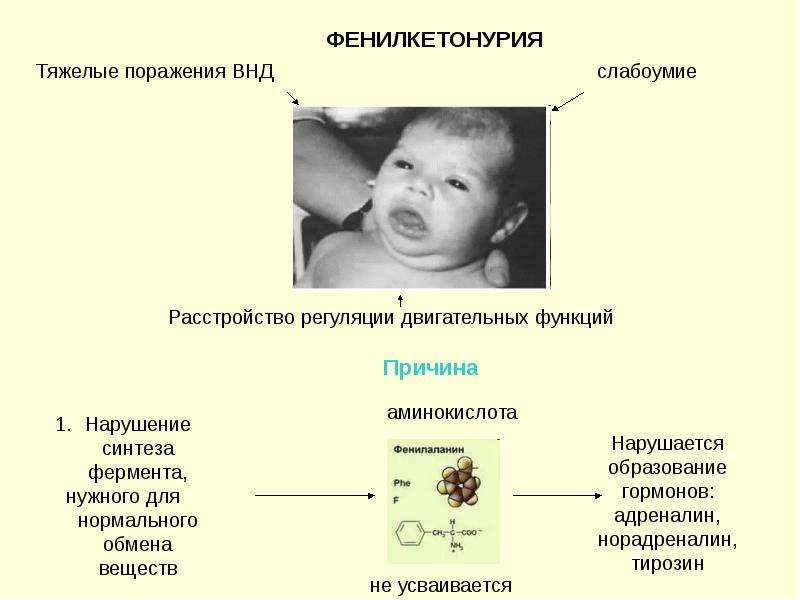 [10] Болезнь Тея-Сакса постепенно разрушает нервную систему, что часто приводит к смерти к пяти годам.У взрослых также может быть диагностирована болезнь Тея-Сакса с поздним началом, которая вызывает управляемый уровень снижения когнитивных способностей. Хотя обнаружение Tay-Sachs может быть выполнено с помощью методов ферментативного анализа или исследований ДНК, существует возможность полностью предотвратить риск. Могут быть применены методы вспомогательной репродуктивной терапии, которые позволяют тестировать эмбрионы in vitro на наличие Тея-Сакса перед имплантацией их матери. Это может позволить выбрать только здоровые эмбрионы.
[10] Болезнь Тея-Сакса постепенно разрушает нервную систему, что часто приводит к смерти к пяти годам.У взрослых также может быть диагностирована болезнь Тея-Сакса с поздним началом, которая вызывает управляемый уровень снижения когнитивных способностей. Хотя обнаружение Tay-Sachs может быть выполнено с помощью методов ферментативного анализа или исследований ДНК, существует возможность полностью предотвратить риск. Могут быть применены методы вспомогательной репродуктивной терапии, которые позволяют тестировать эмбрионы in vitro на наличие Тея-Сакса перед имплантацией их матери. Это может позволить выбрать только здоровые эмбрионы.
Серповидно-клеточная анемия
Серповидно-клеточная анемия — это пожизненное генетическое заболевание, которое может передаваться по наследству, когда оба родителя передают черту серповидной клетки своим детям.Эта черта чаще передается по наследству людям, живущим к югу от Сахары, Индии или Средиземноморью. Серповидно-клеточная болезнь приводит к тому, что эритроциты меняют свою обычную форму пончика на форму серпа. Это заставляет клетки слипаться и попадать в кровеносные сосуды, вызывая сильную боль и серьезные осложнения, такие как инфекции, повреждение органов и острый респираторный синдром. По данным CDC, серповидноклеточная болезнь поражает примерно 100 000 американцев. Кроме того, каждый 365 афроамериканский ребенок рождается с серповидно-клеточной анемией.[11] Для сравнения, у одного из 16 300 детей латиноамериканского происхождения диагностируется это заболевание. Современные достижения в медицине ограничили уровень смертности от серповидно-клеточной анемии, предоставив большее разнообразие вакцин и вариантов лечения.
Это заставляет клетки слипаться и попадать в кровеносные сосуды, вызывая сильную боль и серьезные осложнения, такие как инфекции, повреждение органов и острый респираторный синдром. По данным CDC, серповидноклеточная болезнь поражает примерно 100 000 американцев. Кроме того, каждый 365 афроамериканский ребенок рождается с серповидно-клеточной анемией.[11] Для сравнения, у одного из 16 300 детей латиноамериканского происхождения диагностируется это заболевание. Современные достижения в медицине ограничили уровень смертности от серповидно-клеточной анемии, предоставив большее разнообразие вакцин и вариантов лечения.
Рождение ребенка с генетическим заболеванием может быть проблемой для родителей, но эффективный постоянный уход со стороны обученных медсестер может значительно облегчить последствия. Благодаря программе «Доктор наук по сестринскому делу» практикующие медсестры могут расширить свои знания и практические способности для борьбы с этими расстройствами и их смягчения.Добавляя новый опыт в передовые методы обнаружения, профилактики и лечения генетических заболеваний, медсестры с продвинутой квалификацией могут сыграть ключевую роль в помощи родителям, детям, взрослым больным и обществу в целом.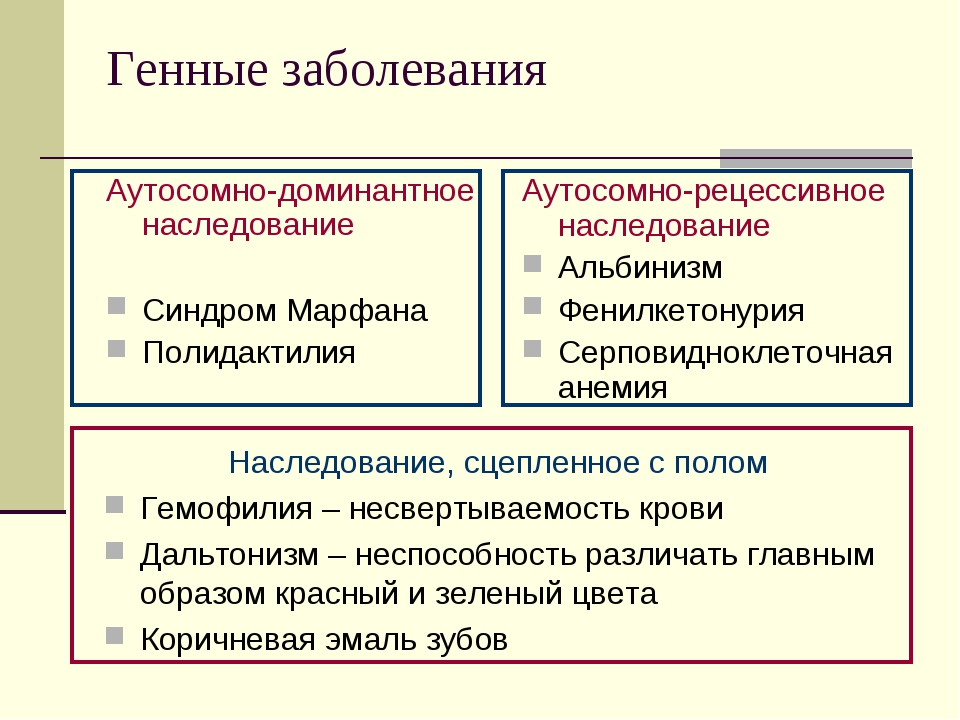
Узнать больше
Здравоохранение — это динамичная и постоянно развивающаяся область, и теперь от руководителей медсестер ожидают большего. Фактически, Американская ассоциация колледжей медсестер (AACN) призвала к тому, чтобы образование на уровне докторантуры стало требованием для медсестер с продвинутой практикой.Получение статуса доктора медсестер (DNP) в режиме онлайн ставит медсестер с сертификатом MSN, таких как вы, на передний план отрасли, готовых к лидерству, обучению медсестер, уходу за пациентами и формированию будущих политик и процедур в области здравоохранения.
Рекомендуется
Как семейные медсестры со степенью DNP расширяют возможности пациентов и их семьи
Источники
1. NIH — Как лечат или управляют генетические заболевания?
2. NIH — Какую информацию о генетическом заболевании может предоставить статистика?
3.NIH — Genetic Disorders
4. Национальный исследовательский институт генома человека
5. Национальное общество синдрома Дауна
6. Центры по контролю и профилактике заболеваний — синдром Дауна
Центры по контролю и профилактике заболеваний — синдром Дауна
7. Демография талассемии
8. NHS.uk
9. Новости муковисцидоза Сегодня
10. Национальный исследовательский институт генома человека — Изучение болезни Тея-Сакса
11. Центры по контролю и профилактике заболеваний — серповидноклеточная болезнь
12. Центры по контролю и профилактике заболеваний — Заболеваемость серповидноклеточной чертой в США
Как Наследуются ли генетические заболевания?
Как наследуются генетические заболевания?
Шаблоны наследования
Клетки организма не работают должным образом, если белок изменен или произведен в недостаточном количестве (или иногда полностью отсутствует).Некоторые нервно-мышечные заболевания могут быть вызваны спонтанной мутацией, которая не обнаруживается в генах ни одного из родителей, — дефект, который может быть передан следующему поколению.
Гены подобны планам; они содержат закодированные сообщения, которые определяют характеристики или черты человека. Они расположены вдоль 23 стержневидных пар хромосом, причем половина каждой пары наследуется от каждого родителя. Каждая половина пары хромосом похожа на другую, за исключением одной пары, которая определяет пол человека.Наследование может происходить тремя способами:
Они расположены вдоль 23 стержневидных пар хромосом, причем половина каждой пары наследуется от каждого родителя. Каждая половина пары хромосом похожа на другую, за исключением одной пары, которая определяет пол человека.Наследование может происходить тремя способами:
- Аутосомно-доминантное наследование происходит, когда ребенок получает нормальный ген от одного родителя и дефектный ген от другого родителя. Аутосомно означает, что генетическая мутация может произойти на любой из 22 неполовых хромосом в каждой из клеток организма. Доминантный означает, что только один родитель должен передать аномальный ген, чтобы вызвать заболевание. В семьях, где один из родителей несет дефектный ген, у каждого ребенка есть 50-процентная вероятность унаследовать этот ген и, следовательно, заболевание.Мужчины и женщины одинаково подвержены риску, и тяжесть заболевания может различаться от человека к человеку.
- Аутосомно-рецессивное наследование означает, что оба родителя должны нести и передавать дефектный ген. Каждый из родителей имеет по одному дефектному гену, но не страдает заболеванием. Дети в этих семьях имеют 25-процентный шанс унаследовать обе копии дефектного гена и 50-процентный шанс унаследовать один ген и, следовательно, стать носителем, способным передать дефект своим детям.Этот образец наследования может повлиять на детей любого пола.
- Х-сцепленное (или сцепленное с полом) рецессивное наследование происходит, когда мать несет пораженный ген на одной из своих двух Х-хромосом и передает его своему сыну (мужчины всегда наследуют Х-хромосому от своей матери и Y-хромосому от их отец, а дочери наследуют Х-хромосому от каждого родителя). Сыновья матери-носительницы имеют 50-процентный шанс унаследовать заболевание. У дочерей также есть 50-процентный шанс унаследовать дефектный ген, но обычно это не затрагивает, поскольку здоровая Х-хромосома, полученная от отца, может компенсировать дефектную хромосому, полученную от их матери.Больные отцы не могут передать Х-сцепленное заболевание своим сыновьям, но их дочери будут носителями этого заболевания. Самки-носители иногда могут проявлять более легкие симптомы MD.
 Каждый из родителей имеет по одному дефектному гену, но не страдает заболеванием. Дети в этих семьях имеют 25-процентный шанс унаследовать обе копии дефектного гена и 50-процентный шанс унаследовать один ген и, следовательно, стать носителем, способным передать дефект своим детям.Этот образец наследования может повлиять на детей любого пола.
Каждый из родителей имеет по одному дефектному гену, но не страдает заболеванием. Дети в этих семьях имеют 25-процентный шанс унаследовать обе копии дефектного гена и 50-процентный шанс унаследовать один ген и, следовательно, стать носителем, способным передать дефект своим детям.Этот образец наследования может повлиять на детей любого пола. Самки-носители иногда могут проявлять более легкие симптомы MD.
Самки-носители иногда могут проявлять более легкие симптомы MD.Кредит содержания: Национальный институт неврологических расстройств и инсульта (NINDS)
Генетические расстройства | Genetic Alliance UK
Генетическое заболевание — это заболевание, вызванное изменениями или мутациями в последовательности ДНК человека.Генетические нарушения можно разделить на три разные категории: одиночные генные, хромосомные или сложные нарушения.
Что такое нарушения с одним геном?
Расстройства одного гена вызваны дефектами одного конкретного гена. Существует более 10 000 заболеваний человека, вызванных изменением, известным как мутация, в одном гене.
По отдельности единичные генные расстройства очень редки, но в целом они затрагивают около одного процента населения.
Поскольку задействован только один ген, эти нарушения часто можно легко проследить в семьях, и генетики могут предсказать риск их возникновения в последующих поколениях.
Заболевания с одним геном можно разделить на различные категории: доминантные, рецессивные и Х-сцепленные.
Что такое хромосомные нарушения?
Хромосомные нарушения возникают в результате изменения числа или структуры хромосом. Изменения в количестве хромосом происходят, когда копий определенной хромосомы больше или меньше, чем обычно. Изменения в структуре хромосомы происходят, когда материал отдельной хромосомы каким-либо образом разрушается или перестраивается.Это может включать добавление или потерю частей хромосомы.
Что такое сложные расстройства?
Сложные расстройства (также известные как многофакторные или полигенные) — это расстройства, которые вызваны одновременным действием множества различных генов, часто в сложном взаимодействии с факторами окружающей среды и образа жизни, такими как диета.
Многие из распространенных заболеваний взрослой жизни, такие как сахарный диабет, гипертония, шизофрения и наиболее распространенные аномалии развития, такие как заячья губа и врожденные пороки сердца, имеют сильную генетическую составляющую и вызваны более чем одним генетическим изменением.
Поскольку полигенные болезни включают более одного гена, модели наследования разнообразны и сложны. Если родитель болен, это не обязательно означает, что у ребенка разовьется такая же болезнь. С другой стороны, человек может не родиться с болезнью, но может иметь более высокий риск ее развития. Это известно как генетическая предрасположенность или предрасположенность.
Хотя генетический состав человека нельзя изменить, некоторые изменения в образе жизни и окружающей среде (например, более частые обследования на заболевания и поддержание здорового веса) могут снизить риск заболевания у людей с генетической предрасположенностью.
Дополнительную информацию можно найти здесь или здесь.
Полезна ли эта информация? Вам нужна дополнительная информация или доступ к источникам? Есть что-нибудь непонятное?
СВЯЗАТЬСЯ С НАМИ
Генетические заболевания | ACOG
Амниоцентез: Процедура, при которой амниотическая жидкость и клетки берутся из матки для исследования.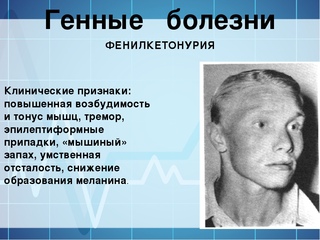 В этой процедуре используется игла для извлечения жидкости и клеток из мешочка, в котором находится плод.
В этой процедуре используется игла для извлечения жидкости и клеток из мешочка, в котором находится плод.
Анеуплоидия: Ненормальное количество хромосом.
Аутосомно-доминантные заболевания: Генетические нарушения, вызванные одним дефектным геном. Дефектный ген находится на одной из хромосом, не являющейся половой.
Аутосомно-рецессивные заболевания: Генетические нарушения, вызванные двумя дефектными генами, по одному унаследованному от каждого родителя. Дефектные гены расположены на одной из пар хромосом, которые не являются половыми хромосомами.
Врожденные дефекты: Физические проблемы, присутствующие при рождении.
Носитель: Человек, не проявляющий признаков заболевания, но способный передать ген своим детям.
Внеклеточная ДНК: ДНК плаценты, которая свободно перемещается в крови беременной женщины. Анализ этой ДНК может быть выполнен как неинвазивный пренатальный скрининговый тест.
Ячейки: Наименьшие элементы конструкции в теле. Клетки — это строительные блоки для всех частей тела.
Взятие образцов ворсинок хориона (CVS): Процедура, при которой небольшой образец клеток берется из плаценты и исследуется.
Хромосомы: Структуры, расположенные внутри каждой клетки тела. Они содержат гены, определяющие физическое состояние человека.
Муковисцидоз (CF): Наследственное заболевание, вызывающее проблемы с дыханием и пищеварением.
Диагностические тесты: Тесты для выявления болезни или причины заболевания.
ДНК: Генетический материал, передающийся от родителей к ребенку. ДНК упакована в структуры, называемые хромосомами.
Синдром Дауна (трисомия 21): Генетическое заболевание, которое вызывает аномальные черты лица и тела, проблемы со здоровьем, такие как пороки сердца, и умственную отсталость. Большинство случаев синдрома Дауна вызвано дополнительной 21-й хромосомой (трисомия 21).
Большинство случаев синдрома Дауна вызвано дополнительной 21-й хромосомой (трисомия 21).
Синдром Эдвардса (трисомия 18): Генетическое заболевание, вызывающее серьезные проблемы.Это вызывает маленькую голову, пороки сердца и глухоту.
Яйцо: Женская репродуктивная клетка, продуцируемая и высвобождающаяся из яичников. Также называется яйцеклеткой.
Оплодотворение: Многоступенчатый процесс, объединяющий яйцеклетку и сперму.
Плод: Стадия человеческого развития после 8 полных недель после оплодотворения.
Ген: Сегмент ДНК, содержащий инструкции по развитию физических качеств человека и контролю над процессами в организме.Ген является основной единицей наследственности и может передаваться от родителей к ребенку.
Консультант по генетике: Специалист в области здравоохранения со специальной подготовкой в области генетики, который может дать экспертный совет по генетическим нарушениям и пренатальному тестированию.
Гемофилия: Заболевание, вызванное мутацией Х-хромосомы. Больные обычно — это мужчины, у которых в крови отсутствует вещество, способствующее свертыванию. Люди с гемофилией подвержены риску сильного кровотечения даже при незначительных травмах.
Болезнь Хантингтона: Заболевание, которое вызывает потерю контроля над движениями тела и умственными функциями.
Моносомия: Состояние, при котором отсутствует хромосома.
Дефекты нервной трубки (ДНТ): Врожденные дефекты, возникающие в результате проблем в развитии головного, спинного мозга или их покровов.
Врач акушер-гинеколог (акушер-гинеколог): Врач со специальной подготовкой и образованием в области женского здоровья.
Синдром Патау (трисомия 13): Генетическое заболевание, вызывающее серьезные проблемы. Это касается сердца и мозга, расщелины губы и неба, а также дополнительных пальцев рук и ног.
Плацента: Орган, который обеспечивает питательными веществами плод и выводит его отходы.
Дородовое наблюдение: Программа ухода за беременной женщиной до рождения ребенка.
Скрининговые тесты: Тесты, которые ищут возможные признаки заболевания у людей, у которых нет признаков или симптомов.
Расстройства, связанные с полом: Генетические расстройства, вызванные изменением гена, расположенного на половых хромосомах.
Сперма: Клетка, вырабатываемая мужскими яичками, которая может оплодотворять женскую яйцеклетку.
Спинальная мышечная атрофия (SMA): Наследственное заболевание, вызывающее атрофию мышц и сильную слабость. СМА — ведущая генетическая причина смерти младенцев.
Трисомия: Проблема с лишней хромосомой.
Синдром Тернера: Проблема, с которой сталкиваются женщины, когда отсутствует или повреждена Х-хромосома.