Кариотипирование пары (детей) | Клиника «Надия»
Цитогенетические исследования
Анализ спермы, FISH-метод
2 850,00
Анализ спермы, FISH-метод с исследованием индивидуальной хромосомы
4 980,00
Определение кариотипа амниоцитов развивающейся беременности
2 950,00
Определение кариотипа развивающейся беременности, FISH-метод
3 600,00
Определение кариотипа ворсин хориона в абортивном материале
2 880,00
Определение кариотипа ворсин хориона беременности, которая развивается
2 740,00
Определение кариотипа ворсин хориона беременности, которая развивается, FISH-метод
3 110,00
Определение кариотипа пациента
2 200,00
Доплата за срочное определение кариотипа пациента
1 290,00
Определение кариотипа плода из пуповинной крови
2 430,00
Предимплантационный генетический скрининг по 5 хромосомам (13, 18, 21, X, Y): биопсия бластомера, PGD
19 410,00
Предимплантационный генетический скрининг по 5 хромосомам (13, 18, 21, X, Y), без биопсии бластоцисты/бластомера, до 5 образцов включительно
6 900,00
Предимплантационный генетический скрининг по 5 хромосомам (13, 18, 21, X, Y), без биопсии бластоцисты/бластомера, до 10 образцов включительно
13 800,00
Доплата за 1 дополнительный эмбрион больше 10 образцов по 5 хромосомам (13, 18, 21, X, Y)
1 900,00
Предимплантационный генетический скрининг по 8 хромосомам (13, 15, 16, 18, 21, 22, X, Y), без биопсии бластоцисты/бластомера, до 5 образцов включительно
9 500,00
Предимплантационный генетический скрининг по 8 хромосомам (13, 15, 16, 18, 21, 22, X, Y), без биопсии бластоцисты/бластомера, до 10 образцов включительно
19 000,00
Доплата за 1 дополнительный эмбрион больше 10 образцов по 8 хромосомам (13, 15, 16, 18, 21, 22, X, Y)
1 400,00
Предимплантационный генетический скрининг по 9 хромосомам (13, 15, 16, 17, 18, 21, 22, X, Y), без биопсии бластоцисты/бластомера, до 5 образцов включительно
9 700,00
Предимплантационный генетический скрининг по 9 хромосомам (13, 15, 16, 17, 18, 21, 22, X, Y), без биопсии бластоцисты/бластомера, до 10 образцов включительно
19 400,00
Доплата за 1 дополнительный эмбрион больше 10 образцов по 9 хромосомам (13, 15, 16, 17, 18, 21, 22, X, Y)
1 400,00
Fish-анализ лимфоцитов крови (1 хромосома)
1 670,00
Fish-анализ лимфоцитов крови (2 хромосомы)
2 430,00
Исследование кариотипа | Полезное от клиники «Геном» в Томске
Генетические причины лежат в основе любого заболевания человека, включая нарушения репродуктивной функции. Поэтому, генетическим исследованиям при бесплодии уделяется особое внимание. Кариотипирование — одно из них. Оно позволяет выявить несоответствие в наборах хромосом мужчины и женщины, что является частой причиной бездетности пары.
Поэтому, генетическим исследованиям при бесплодии уделяется особое внимание. Кариотипирование — одно из них. Оно позволяет выявить несоответствие в наборах хромосом мужчины и женщины, что является частой причиной бездетности пары.
Кариотип – это хромосомный набор, совокупность признаков хромосом (их количество, форма, размеры, детали микроскопического строения и пр.). Для каждого биологического вида характерен свой хромосомный набор.
Кариотип человека состоит из 46 хромосом. Из них 44 — аутосомы (22 пары), которые имеют одинаковое строение у женщин и мужчин. Есть также пара половых хромосом, которые определяют половую принадлежность человека. Мужская пара хромосом — XY, женская — XX. В каждой хромосоме находятся гены, ответственные за наследственность.
Человек может быть носителем хромосомных перестроек, даже не подозревая об этом — видимых признаков и явных симптомов при этом обычно не бывает. Сложности возникают на этапе планирования семьи – это могут быть проблемы с зачатием, вынашиванием беременности, появлением врождённых пороков развития у плода. Кариотипирование позволяет не только выявить наличие генетических нарушений, но и определить предрасположенность человека к развитию наследственной патологии.
Сложности возникают на этапе планирования семьи – это могут быть проблемы с зачатием, вынашиванием беременности, появлением врождённых пороков развития у плода. Кариотипирование позволяет не только выявить наличие генетических нарушений, но и определить предрасположенность человека к развитию наследственной патологии.
Кариотипирование достаточно сделать один раз в жизни, ведь набор хромосом, а также их характеристики, не изменяются с возрастом.
Для проведения кариотипирования берется кровь из вены. Исследование кариотипа проводится с помощью цитогенетических и молекулярно-цитогенетических методов.
Показанием к проведению анализа крови на кариотип является:
— Планирование беременности. Для полноценного вынашивания и рождения здорового ребенка кариотипирование необходимо провести каждой паре до наступления беременности. Крайне важно скорректировать нежелательные состояния прежде зачатия. При необходимости, исследование проводят уже беременной женщине и ее будущему ребенку.
При необходимости, исследование проводят уже беременной женщине и ее будущему ребенку.
— Поздний репродуктивный возраст будущих родителей. С возрастом вероятность хромосомных изменений увеличивается.
— Наличие наследственных заболеваний у мужчины, женщины или их родственников
— Близкородсвенные отношения будущих родителей
— Постоянное взаимодействие с вредными факторами – это могут быть химические, радиационные, механические воздействия, неблагоприятная экологическая обстановка
— Безуспешные попытки ЭКО в анамнезе
Расшифровка анализа показывает следующие риски и отклонения:
1) Мозаицизм. Данное явление означает присутствие в одном организме клеток с различными хромосомными наборами (кариотипами).
Мозаицизм способствует мутациям в делящейся клетке. По данным ряда исследований, в первые дни эмбрионального развития, на каждое деление на одну клетку приходится три мутации.Некоторые из них не оказывают серьёзного влияния на организм, поэтому какие то клетки остаются нормальными, а другие изменяются. Обычно мозаицизм приводит к негативными последствиями. В частности, на мозаичную трисомию 21 приходится 2–4% случаев синдрома Дауна.
 Некоторые из них не оказывают серьёзного влияния на организм, поэтому какие то клетки остаются нормальными, а другие изменяются. Обычно мозаицизм приводит к негативными последствиями. В частности, на мозаичную трисомию 21 приходится 2–4% случаев синдрома Дауна.
Некоторые из них не оказывают серьёзного влияния на организм, поэтому какие то клетки остаются нормальными, а другие изменяются. Обычно мозаицизм приводит к негативными последствиями. В частности, на мозаичную трисомию 21 приходится 2–4% случаев синдрома Дауна.
2) Транслокации (фрагменты разных хромосом меняются местами или хромосомы объединяются).
Встречаются транслокации довольно часто – 1 случай к 500, но только не всегда это приводит к проблемам. Если при транслокации общее количество хромосомного материала не изменилось, то она является сбалансированной. Если происходит потеря или переизбыток генетического материала — это несбалансированная транслокация. Она может стать причиной остановки развития эмбриона на разных сроках беременности женщины, появления генетических нарушений у плода или рождения ребенка с тяжёлым заболеванием.
Носитель сбалансированной транслокации не страдает от этого, но у него с высокой долей вероятности может родится ребёнок с транслокацией несбалансированной.
Кроме того, анализ на кариотип показывает:
— Отсутствие одного из фрагментов хромосомы.
— Отсутствие одной из парных хромосом.
— Присутствие лишней хромосомы.
— Развернутость одного из фрагмента хромосомной цепи.
— Генные мутации, которые приводят к отклонениям у новорожденного.
Гены формируют цепочку молекулы ДНК, которая несёт наследственную информацию. На этапе формирования эмбриона человек получает генетическую информацию от матери и отца. Изменения в генах (мутации) опасны тем, что могут наследоваться, не давая о себе знать, а проявиться только через несколько поколений.
Исследование кариотипа перед планируемой беременностью должно стать «золотым стандартом» в комплексе мероприятий, направленных на рождение здорового ребенка.
Пройти комплексное обследование на этапе планирования семьи можно в клинике «Геном-Томск».
Поделитесь информацией:
Кариотипирование пары в Evaclinic IVF
Каждый живой организм характеризуется уникальным набором хромосом. В них заключена наследственная информация.
Кариотипом называют все морфологические особенности набора в одном организме в совокупности. Если в нем наблюдаются аномалии, это ведет к генетическим патологиям и синдромам.
Кариотип человеческого организма состоит из 46 хромосом, 44 из которых – или 22 пары, — аутосомы, то есть имеют идентичное строение и у мужчин, и у женщин. Также в мужском организме имеется пара хромосом XY, а у женщин XX.
Таким образом, кариотип 46 XX — норма женского кариотипа, 46 XY — норма мужского кариотипа. Такой набор остается неизменным на протяжении жизни.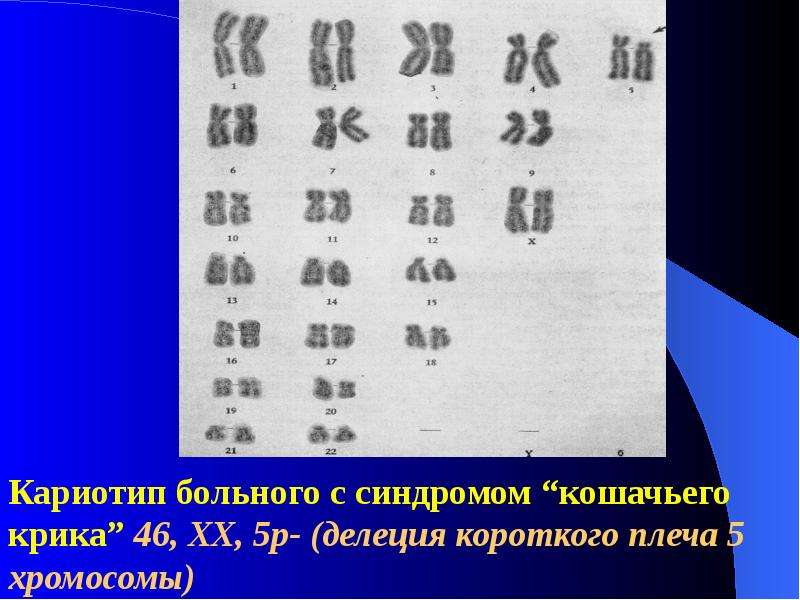
Кариотипирование – для чего проводится исследование?
Кариотипирование, или анализ крови на кариотип — подсчет количества и оценка размера и формы хромосом с помощью светового микроскопа с применением окрашивания. Кариотипирование — многоступенчатое исследование, которое проводится при вхождении клеток в фазу непрямого деления.
В результате проведения анализа выдается описание кариотипа в виде формулы, где пишется общее число хромосом, их перестройка (аберрация) и набор половых хромосом.
Кариотипирование проводят для определения причин врожденных болезней ребенка, для выявления поврежденных хромосом у плода. При определении причины выкидыша у женщины также выполняется анализ определения кариотипа, что помогает понять, стали ли хромосомные проблемы причиной внутриутробной гибели плода.
Это исследование проводят и для выяснения, не являются ли хромосомы человека аномальными, как это воздействует на его здоровье и здоровье будущих детей.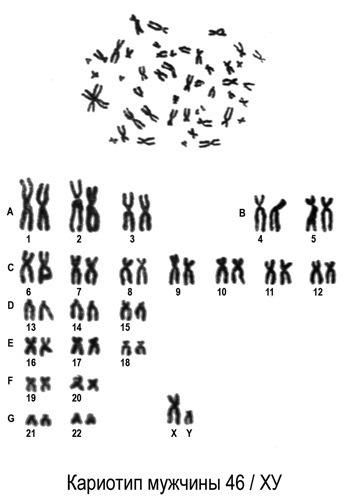
Хромосомные патологии
Иногда в наследственной информации появляются ошибки. Это происходит и в отдельных генах, и на уровне крупных объединений. Есть моменты, которые отрицательно воздействуют на формирование половых клеток, а также на их развитие после оплодотворения:
- наследственные генетические заболевания;
- работа или проживание в неблагоприятных условиях;
- неправильный рацион;
- образ жизни родителей, опасный для потомства;
- болезни родителей, которые оказывают влияние на формирование половых клеток;
- возраст старше 35 лет;
- стрессы на ранних сроках беременности.
Нарушения в наборе хромосом становятся причиной следующих генетических заболеваний:
- бесплодие;
- повторяющиеся выкидыши;
- рождение малыша с патологиями развития.
Чтобы провести цитогенетическое исследование хромосом, используют препараты культуры крови.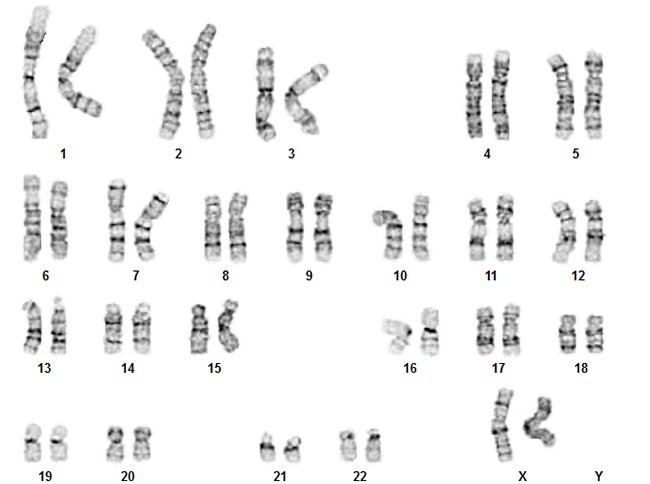
Показания к сдаче анализа на кариотип
Анализ кариотипирования назначают и детям, и взрослым. Детей обследуют в следующих ситуациях:
- врожденные пороки развития;
- нарушение развития;
- задержка в психомоторном развитии;
- родственники с умственной отсталостью;
- аномалии полового развития;
- выраженные нарушения в росте и размере головы.
Взрослым сдать анализ на кариотип необходимо в следующих случаях:
- аномалия спермограммы;
- бесплодие неясного происхождения;
- ранняя менопауза;
- самопроизвольные выкидыши;
- родители пациента со структурными аномалиями хромосом;
- неудачное ЭКО;
- повторное рождении малышей с аномалиями хромосом;
- прогноз здоровья будущих детей.
Как проводится кариотипирование в Минске?
Алгоритм проведения анализа:
- постановка культуры, рост клеток 72 часа;
- обработка культуры лимфоцитов;
- приготовление хромосомных препаратов на стекле;
- одноцветное и дифференцированное окрашивание;
- анализ хромосом.

Анализ кариотипа супругов в Минске способствует выявлению ошибок в количестве и строении хромосом. Также это дает возможность оценки вероятного появления отклонений.
Подготовка к анализу
Исследование проводится в лаборатории. За 2 недели до анализа нужно исключить курение, алкоголь, прием лекарств. При невозможности отказаться от приема лекарственных препаратов, об этом предупреждают лаборанта заранее.
Кровь забирают из вены. Последний прием пищи должен быть не позднее 2 часов до сдачи биоматериала. Анализ и подготовка результатов длится 30 дней.
Для того, чтобы сдать анализ на кариотип – просто обратитесь в нашу клинику. EVACLINIC IVF проведет кариотипирование в Минске по доступной цене.
Консультацию генетика можно получить в клинике EvaClinic.
ФГБНУ НЦПЗ. ‹‹Общая психиатрия››
Цитогенетика — это область генетики, связанная с изучением хромосом. Исследование хромосом при психической патологии показало, что диагностическое значение этот метод имеет в основном при умственной отсталости. При неврозах, эндогенных психозах, алкоголизме и других видах психической патологии хромосомные изменения, как правило, не обнаруживаются. Среди новорожденных частота хромосомной патологии равна примерно 0,6 %. Хромосомные болезни были известны еще до открытия самих хромосом (например, синдромы Клайнфельтера и Шерешевского — Тернера). Использование термина «болезнь» для хромосомной патологии не совсем верно, поскольку для любой болезни характерен тот или иной тип ее развития, т.е. закономерная смена симптомов и синдромов во времени. В случае же хромосомных аномалий совокупность специфических признаков больного — его фенотип является врожденным и практически не меняющимся. Большинство случаев хромосомных нарушений возникает спорадически в половых клетках здоровых родителей или на стадии первых делений зиготы. Хромосомные нарушения в половых клетках приводят к изменению кариотипа, т.
Исследование хромосом при психической патологии показало, что диагностическое значение этот метод имеет в основном при умственной отсталости. При неврозах, эндогенных психозах, алкоголизме и других видах психической патологии хромосомные изменения, как правило, не обнаруживаются. Среди новорожденных частота хромосомной патологии равна примерно 0,6 %. Хромосомные болезни были известны еще до открытия самих хромосом (например, синдромы Клайнфельтера и Шерешевского — Тернера). Использование термина «болезнь» для хромосомной патологии не совсем верно, поскольку для любой болезни характерен тот или иной тип ее развития, т.е. закономерная смена симптомов и синдромов во времени. В случае же хромосомных аномалий совокупность специфических признаков больного — его фенотип является врожденным и практически не меняющимся. Большинство случаев хромосомных нарушений возникает спорадически в половых клетках здоровых родителей или на стадии первых делений зиготы. Хромосомные нарушения в половых клетках приводят к изменению кариотипа, т. е. совокупности количественных и качественных признаков хромосом, во всех клетках организма. Если же хромосомные изменения возникают на ранних стадиях развития эмбриона, то они являются причиной развития мозаицизма. О мозаицизме говорят в тех случаях, когда имеются клетки с нормальным и измененным кариотипом. В этом случае индивиды имеют более «стертые» проявления заболевания, и чем больше клеток с аномальным кариотипом, тем больше выражены клинические проявления заболевания.
е. совокупности количественных и качественных признаков хромосом, во всех клетках организма. Если же хромосомные изменения возникают на ранних стадиях развития эмбриона, то они являются причиной развития мозаицизма. О мозаицизме говорят в тех случаях, когда имеются клетки с нормальным и измененным кариотипом. В этом случае индивиды имеют более «стертые» проявления заболевания, и чем больше клеток с аномальным кариотипом, тем больше выражены клинические проявления заболевания.
Материалом для микроскопического исследования хромосом служат в основном лейкоциты крови, реже для этой цели используются культуры клеток кожи или костного мозга. Хромосомные аномалии могут возникать в половых и соматических клетках. Они представляют собой изменение числа хромосом — наличие добавочных хромосом или отсутствие хромосомы, или их перестройку, т.е. структурные изменения. Структурные изменения бывают внутрихромосомными — такими как делеция (утрата части хромосомы) или дупликация (удвоение участка хромосомы), а также межхромосомными — транслокация (обмен участками между хромосомами) и др.
При описании кариотипа индивида указывают общее число хромосом, затем состав половых хромосом, наличие транслокации или мозаицизма и т.д. Например, запись 46XY означает нормальный мужской кариотип; 46ХХ — нормальный женский кариотип. Если указано 47XXY, то это кариотип синдрома Клайнфельтера, когда имеется дополнительная Х-хромосома. Кариотип 45X0 соответствует синдрому Шерешевского — Тернера, обусловленному отсутствием одной Х-хромосомы. Добавочная аутосомная хромосома указывается своим номером и знаком плюс, например, 47ХХ, 21+ обозначает кариотип девочки с лишней 21-й хромосомой (болезнь Дауна), а утрата хромосомы обозначается соответствующим номером и знаком минус. Транслокация обозначается буквой «t» с указанием номеров хромосом, которые обменялись участками, например 45XY, t(14+21). Наличие мозаичных клеток обозначается соответствующими кариотипами с использованием знака дроби, например 45X0/46, XX — мозаичность по синдрому Шерешевского — Тернера (45X0). При описании кариотипов используются также и другие символы, с правилами применения которых можно ознакомиться в специальных руководствах.
В случае хромосомной патологии почти все клинические проявления сопровождаются множественными нарушениями в строении тела и психики, причем их выраженность сильно варьирует при одних и тех же хромосомных аномалиях. Например, при болезни Дауна поражение психики проявляется слабоумием от легкой до тяжелой степени. Было также отмечено, что у больных с аномалиями аутосомных хромосом интеллект нарушается в большей степени, чем при аномалиях половых хромосом.
Генетические исследования, исследование кариотипа в Краснодаре
Хромосомные аномалии могут негативно влиять на исходы применения методов вспомогательной репродукции, провоцировать выкидыши, стать причиной наследственных заболеваний ребенка.
При бесплодии выявленные генетические отклонения могут явиться показанием для ПГД (предимплантационной генетической диагностики) в программах ВРТ.
Установление генетической причины бесплодия дает возможность подобрать оптимальную схему лечения. Также генетическое обследование может потребоваться для выявления рисков врожденной и наследственной патологии у будущего потомства и определения возможных мер по предупреждению заболеваний у детей.
Какие генетические исследования могут быть назначены женщине
Репродуктолог может дать пациентке направление на следующие генетические исследования:
— кариотипирование — его женщина проходит вместе с партнером, в ходе исследования определяется структура хромосом и их число;
— анализ на полиморфизмы генов системы свертывания крови (наследственная тромбофилия).
Исследование на кариотип
Кариотип — совокупность признаков полного набора хромосом (их размер, форма, количество, строение). Чаще всего репродуктолог назначает это исследование при привычном невынашивании беременности, бесплодии неясного генеза, пациенткам старше 40 лет. Нормальным результатом для женщин является 46 XX. Интерпретацию отклонений от принятой нормы может осуществлять только врач-генетик, основываясь также на результатах других обследований.
Чаще всего репродуктолог назначает это исследование при привычном невынашивании беременности, бесплодии неясного генеза, пациенткам старше 40 лет. Нормальным результатом для женщин является 46 XX. Интерпретацию отклонений от принятой нормы может осуществлять только врач-генетик, основываясь также на результатах других обследований.
Подготовка к исследованию
Кровь для кариотипирования не рекомендуется сдавать натощак, лучше делать это в состоянии сытости. Если пациентка принимала антибиотики, то необходимо выждать месяц после прохождения медикаментозного лечения. Также желательно не проходить кариотипирование параллельно с анализами, для которых необходима строгая диета.
Исследование на полиморфизмы генов системы свертывания крови (склонность к тромбофилиям)
Исследование полиморфизма генов, например, протромбина, имеет прогностическое значение, которое позволяет определить риск развития микротромбов — одну из потенциальных причин невынашивания беременности и отсутствия имплантации эмбрионов в программах ВРТ.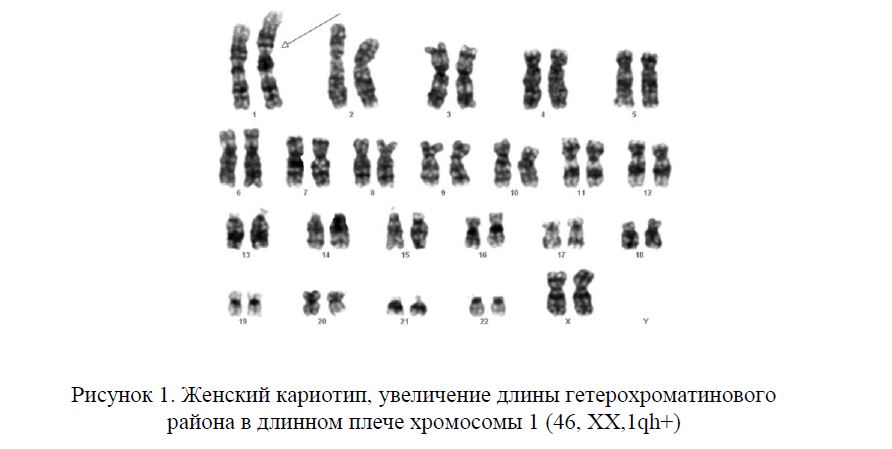 Это происходит из-за нарушений в свертывающей системе крови и требует медикаментозной подготовки к планируемой беременности.
Это происходит из-за нарушений в свертывающей системе крови и требует медикаментозной подготовки к планируемой беременности.
Исследование не требует специальной подготовки.
Исследование кариотипа (Количественные и структурные аномалии хромосом)
Исследуемый материал
Цельная кровь (с гепарином, без геля)
Метод определения
Культивирование лимфоцитов периферической крови, микроскопия дифференциально окрашенных метафазных хромосом.
ИССЛЕДОВАНИЕ НЕ ЯВЛЯЕТСЯ АНАЛОГОМ АНА-ТЕЛОФАЗНОГО МЕТОДА АНАЛИЗА ХРОМОСОМНЫХ АБЕРРАЦИЙ (100 клеток)!
КАРИОТИПИРОВАНИЕ ВХОДИТ В СОСТАВ ИССЛЕДОВАНИЙ:
Репродуктивное здоровье женщины
Репродуктивное здоровье мужчины
Кариотип — это совокупность признаков полного набора хромосом соматических клеток организма на стадии метафазы (III фаза деления клетки) – их количество, размер, форма, особенности строения.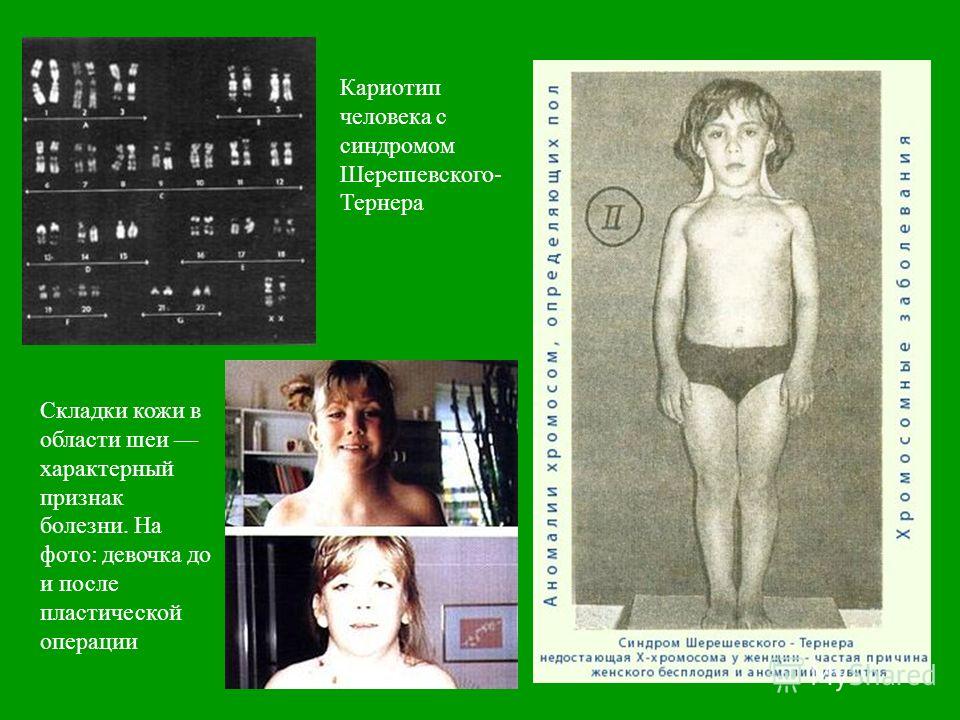 Исследование кариотипа проводят методом световой микроскопии с целью выявления патологии хромосом. Чаще всего это исследование проводят у детей для выявления заболеваний, обусловленных нарушениями в хромосомах и у супругов при бесплодии или привычном невынашивании беременности. Выявление хромосомных перестроек в этом случае позволяет установить причину бесплодия и прогнозировать риск рождения в данной семье детей с хромосомной патологией.
Исследование кариотипа проводят методом световой микроскопии с целью выявления патологии хромосом. Чаще всего это исследование проводят у детей для выявления заболеваний, обусловленных нарушениями в хромосомах и у супругов при бесплодии или привычном невынашивании беременности. Выявление хромосомных перестроек в этом случае позволяет установить причину бесплодия и прогнозировать риск рождения в данной семье детей с хромосомной патологией.
Вне процесса деления клетки хромосомы в её ядре расположены в виде «распакованной» молекулы ДНК, и они трудно доступны для осмотра в световом микроскопе. Для того, чтобы хромосомы и их структура стали хорошо видны используют специальные красители, позволяющие выявлять гетерогенные (неоднородные) участки хромосом и проводить их анализ – определять кариотип. Хромосомы в световом микроскопе на стадии метафазы представляют собой молекулы ДНК, упакованные при помощи особых белков в плотные сверхспирализованные палочковидные структуры. Таким образом, большое число хромосом упаковывается в маленький объём и помещается в относительно небольшом объёме ядра клетки.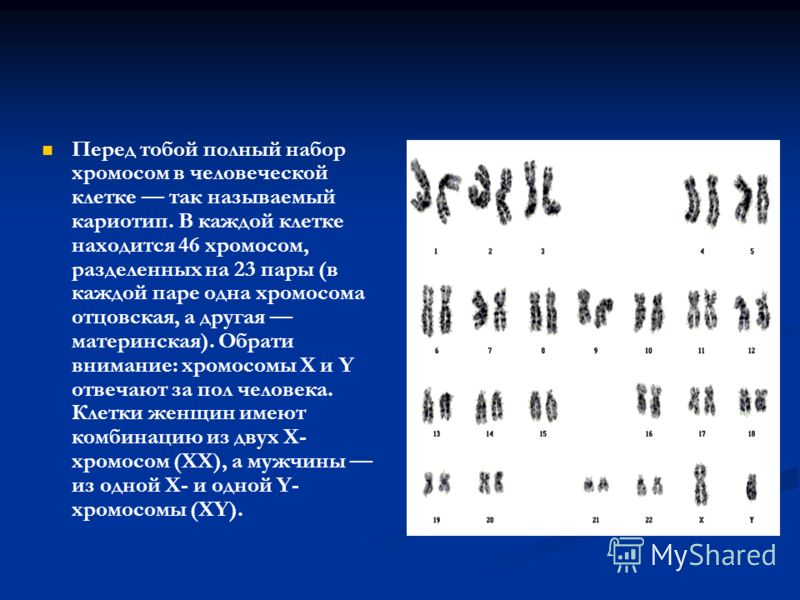 Расположение хромосом, видимое в микроскопе, фотографируют и из нескольких фотографий собирают систематизированный кариотип — нумерованный набор хромосомных пар гомологичных хромосом. Изображения хромосом при этом ориентируют вертикально, короткими плечами вверх, а их нумерацию производят в порядке убывания размеров. Пару половых хромосом помещают в самом конце изображения набора хромосом.
Расположение хромосом, видимое в микроскопе, фотографируют и из нескольких фотографий собирают систематизированный кариотип — нумерованный набор хромосомных пар гомологичных хромосом. Изображения хромосом при этом ориентируют вертикально, короткими плечами вверх, а их нумерацию производят в порядке убывания размеров. Пару половых хромосом помещают в самом конце изображения набора хромосом.
Современные методы кариотипирования обеспечивают детальное обнаружение хромосомных аберраций (внутрихромосомных и межхромосомных перестроек), нарушения порядка расположения фрагментов хромосом — делеции, дупликации, инверсии, транслокации. Такое исследование кариотипа позволяет диагностировать ряд хромосомных заболеваний, вызванных как грубыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множественностью клеточных кариотипов в организме.
Нарушения нормального кариотипа у человека возникают на ранних стадиях развития организма.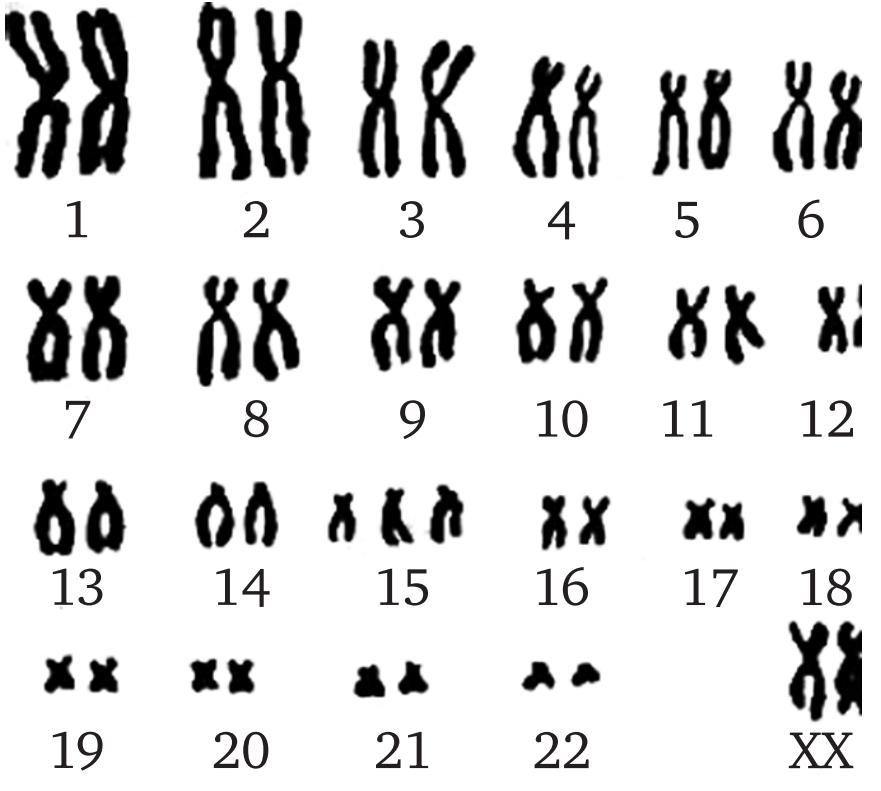 Если это происходит в половых клеток будущих родителей (в процессе гаметогенеза), то кариотип зиготы (см.), образовавшейся при слиянии родительских клеток, также оказывается нарушенным. При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма окажутся с одинаково аномальным кариотипом. Однако, нарушения кариотипа могут возникнуть и на ранних стадиях дробления зиготы. Развившийся из такой зиготы организм содержит несколько линий клеток (клеточных клонов) с разными кариотипами. Такое многообразие кариотипов во всём организме или только в некоторых его органах называют мозаицизмом.
Если это происходит в половых клеток будущих родителей (в процессе гаметогенеза), то кариотип зиготы (см.), образовавшейся при слиянии родительских клеток, также оказывается нарушенным. При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма окажутся с одинаково аномальным кариотипом. Однако, нарушения кариотипа могут возникнуть и на ранних стадиях дробления зиготы. Развившийся из такой зиготы организм содержит несколько линий клеток (клеточных клонов) с разными кариотипами. Такое многообразие кариотипов во всём организме или только в некоторых его органах называют мозаицизмом.
Как правило, нарушения кариотипа у человека сопровождаются различными, в том числе комплексными, пороками развития, и большинство таких аномалий несовместимо с жизнью. Это приводит к самопроизвольным абортам на ранних стадиях беременности. Однако достаточно большое число плодов (~2,5%) с аномальными кариотипами донашивают до окончания беременности.
Ниже приведена таблица, в которой представлены заболевания, обусловленные нарушениями в кариотипе.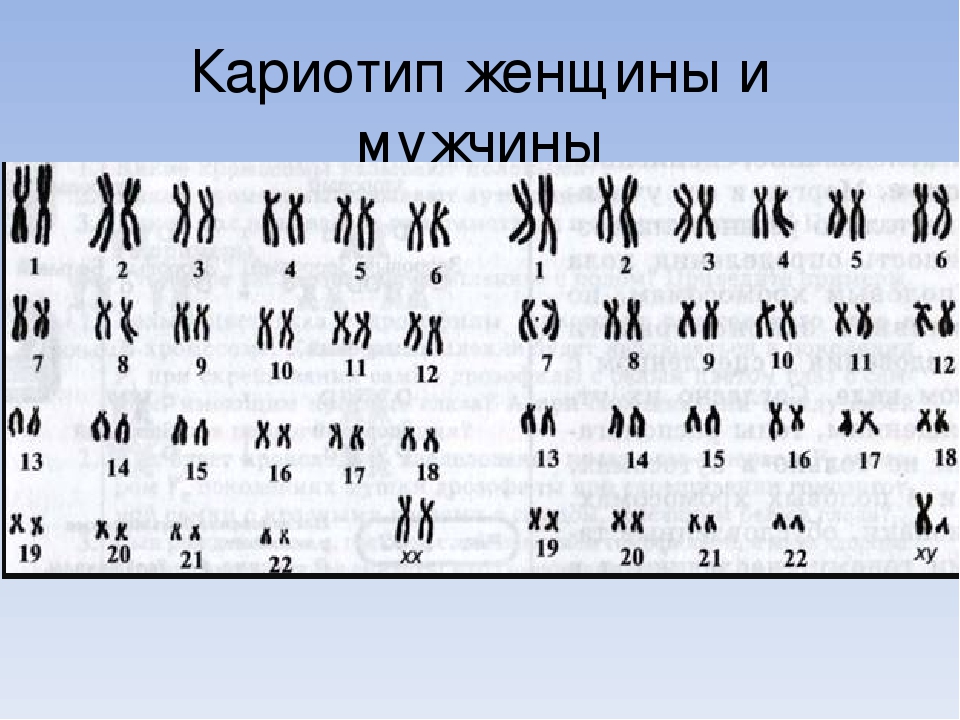
| Кариотипы | Болезнь | Комментарии |
| 47,XXY; 48,XXXY | Синдром Клайнфельтера | Полисомия по
X-хромосоме у мужчин |
| 45X0; 45X0/46XX;
45,X/46,XY; 46,X iso (Xq) | Синдром
Шерешевского — Тернера | Моносомия по
X-хромосоме, в т. ч. и мозаицизм |
| 47,ХХX; 48,ХХХХ;
49,ХХХХХ | Полисомии по
X хромосоме | Наиболее часто —
трисомия X |
| 47,ХХ,+21; 47,ХY,+21 | Болезнь Дауна | Трисомия по
21-й хромосоме |
| 47,ХХ,+18; 47,ХY,+18 | Синдром Эдвардса | Трисомия по
18-й хромосоме |
| 47,ХХ,+13; 47,ХY,+13 | Синдром Патау | Трисомия по
13-й хромосоме |
| 46,XX, 5р- | Синдром кошачьего крика | Делеция короткого плеча
5-й хромосомы |
Смотрите также:
Что такое «хромосомный паспорт» — Клиника ISIDA Киев, Украина
Бровко Антон Александрович
Биолог
07 октября 2013
Вступая в брак, супруги мечтают о долгой счастливой жизни в окружении здоровых деток.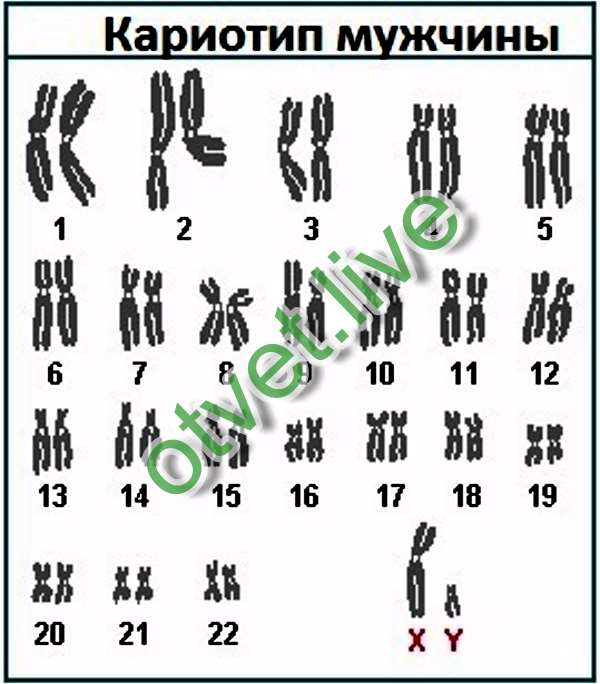 К сожалению, не всем удается стать родителям. Если у пары возникают проблемы с появлением потомства – не удается зачать ребенка или наступившая беременность прерывается, то врач может порекомендовать провести цитогенетическое исследование – кариотипирование.
К сожалению, не всем удается стать родителям. Если у пары возникают проблемы с появлением потомства – не удается зачать ребенка или наступившая беременность прерывается, то врач может порекомендовать провести цитогенетическое исследование – кариотипирование.
Также его называют «хромосомным паспортом». Что это такое и зачем это нужно, журналисту интернет-издания «Обозреватель. Женский день» объясняет Антон Бровко – биолог, цитогенетик клиники «ИСИДА».
Антон Александрович, что такое хромосомный паспорт?
Мы рождаемся с хромосомным набором, который не меняется в течение всей нашей жизни. Нормальный хромосомный набор представляет собой 46 хромосом (23 пары), две из которых – половые хромосомы. У мужчин это Х и Y, у женщин – Х и Х. Если с хромосомами все в порядке – их правильное количество, у них нормальная структура, не изменена какая-то генная последовательность, то это означает, что кариотип нормальный. Если в кариотипе присутствуют нарушения, это может отразиться на здоровье человека.
Хромосомный паспорт – это заключение цитогенетической лаборатории, в котором зафиксировано визуальное изображение хромосомного набора человека (кариотип) и его описание в виде формулы, дана интерпретация результата, все ли в порядке, и в случае наличия отклонения от нормы прописаны рекомендации. Нормальный мужской кариотип записывается как 46, ХY, нормальный женский – 46,ХХ.
В кариотипе могут быть обнаружены отклонения. Например, синдром Дауна возникает из-за того, что 21-я пара хромосом имеет одну дополнительную 21-ю хромосому, т.е. их не 2, а 3 копии. Соответственно, запись в хромосомном паспорте будет выглядеть как 47,ХХ,+21 или 47,ХY,+21.
Как происходит анализ хромосом?
Проводится цитогенетическое исследование, целью которого является анализ количества и структуры хромосом. Специалист с помощью компьютерной программы собирает хромосомы, запечатленные на снимке, попарно, и сравнивает, все ли полоски на месте, нет ли лишних или недостающих участков. Если обнаруживается какое-то несоответствие, появляется повод заподозрить, что произошла какая-то хромосомная мутация (аберрация).
Если обнаруживается какое-то несоответствие, появляется повод заподозрить, что произошла какая-то хромосомная мутация (аберрация).
Хромосомы могут разрываться в разных местах (локусах) и обмениваться участками, поэтому формулы, описывающие изменения в кариотипе, бывают очень сложными. В таких случаях на них номерами указываются точки, где произошел разрыв и куда переместился какой участок хромосомы.
Для кого актуально получение хромосомного паспорта?
Кариотипирование показано людям, имеющим проблемы со здоровьем. Нередко это касается детей, у которых наблюдаются пороки развития. Данная процедура может проводиться пренатально (до рождения) и постнатально (после рождения).
Каким образом можно провести кариотипирование будущему ребенку?
Если врач подозревает отклонения в развитии плода, то он может направить женщину на инвазивную процедуру, во время которой у беременной берут на анализ околоплодные воды или оболочки, окружающие зародыш.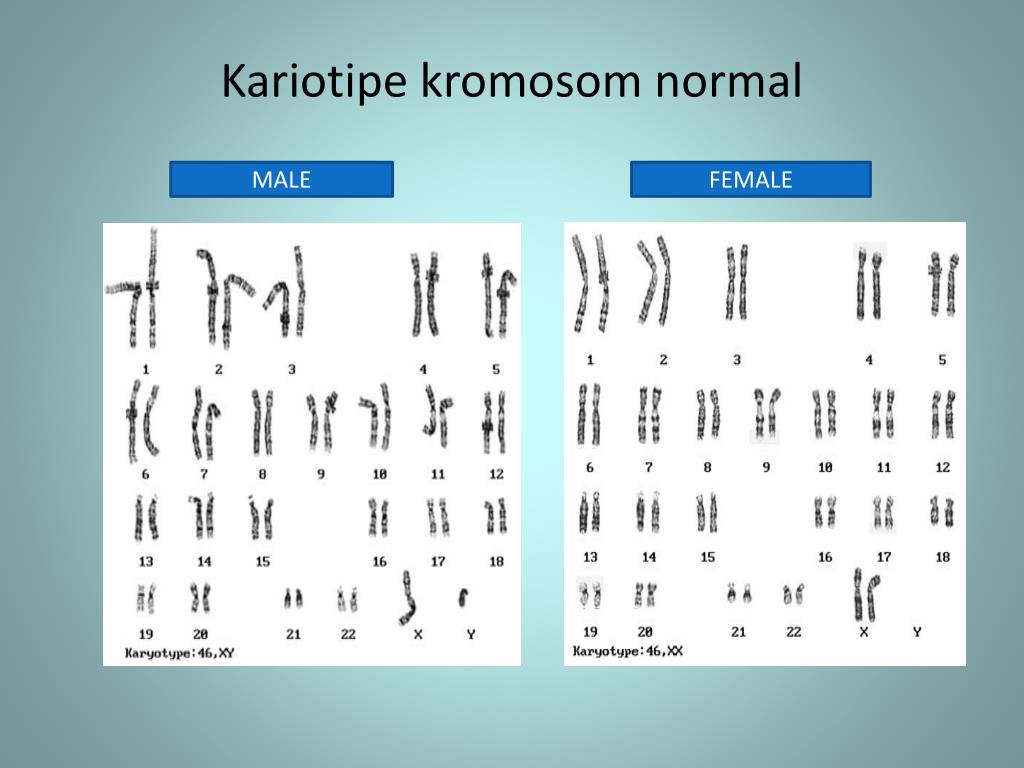 Эти биологические материалы содержит клетки плода, из которых можно получить хромосомы, чтобы составить кариотип еще до рождения ребенка. Другой вариант – уже после рождения младенца врач, который его осматривает, может заметить нарушения внешних или физиологических показателей и заподозрить какой-то хромосомный синдром. В этом случае он также направит ребенка на кариотипирование.
Эти биологические материалы содержит клетки плода, из которых можно получить хромосомы, чтобы составить кариотип еще до рождения ребенка. Другой вариант – уже после рождения младенца врач, который его осматривает, может заметить нарушения внешних или физиологических показателей и заподозрить какой-то хромосомный синдром. В этом случае он также направит ребенка на кариотипирование.
Нарушения могут возникнуть и в более позднем возрасте, например, отставание в умственном развитии, аутизм. В еще более зрелом возрасте могут обнаружиться проблемы в половой сфере – отсутствие менструаций у девочек или неправильное половое созревание у мальчиков. Все это – поводы обратиться к врачу-генетику, а он направляет пациента на кариотипирование.
Показано кариотипирование также супругам, которые длительное время не могут иметь детей. Нередко бесплодие связано с нарушением в кариотипе. Этим же бывают вызваны замирания и срывы беременности. Если это повторяется, то проводится кариотипирование абортного материала. До 65% замерших беременностей возникают из-за нарушений в хромосомном наборе зародыша, возникшим de novo (впервые) или же унаследованным от супругов.
До 65% замерших беременностей возникают из-за нарушений в хромосомном наборе зародыша, возникшим de novo (впервые) или же унаследованным от супругов.
Кроме того, кариотипирование может проводиться в случае злокачественных новообразований, например, лейкемий (рака крови), причиной возникновения которых могут быть нарушения в структуре хромосом. В этом случае на анализ берется костный мозг – в нем можно увидеть хромосомные мутации, которые приводят к развитию опухоли.
Хромосомные изменения могут наблюдаться не во всем организме, а только в конкретном органе. Например, в гонадах (половых железах), яичниках или яичках. Тогда во всем теле человека будет нормальный хромосомный набор, а в тех тканях, которые формируют половые клетки, хромосомный набор измененный, в результате здоровый человек продуцирует нездоровые половые клетки.
Все ли генетические дефекты видны при кариотипировании?
Кариотипирование проводится с помощью светового микроскопа, в который видны хромосомы, но не видны гены. Диагностика генных нарушений проводится только молекулярными методами, и это уже называется не хромосомный паспорт, а генетический паспорт. В генетический паспорт вписываются предрасположенности человека к тем или иным заболеваниям, устойчивости или неустойчивости к тем или иным медикаментам и т.д.
Диагностика генных нарушений проводится только молекулярными методами, и это уже называется не хромосомный паспорт, а генетический паспорт. В генетический паспорт вписываются предрасположенности человека к тем или иным заболеваниям, устойчивости или неустойчивости к тем или иным медикаментам и т.д.
Сколько времени делается хромосомный паспорт?
Минимум 5-7 дней. Для проведения кариотипирования у пациентов берут кровь из вены, затем лейкоциты крови в пробирке стимулируют, заставляя делиться, а спустя несколько дней (72 часа) культуру обрабатывают специальным реактивом (колцемидом), останавливающим деление клеток на стадии, когда уже видны хромосомы. Когда приходит время анализа, цитогенетик капает на предметное стекло взвесь клеток, и когда капля фиксатора растекается по стеклу, оболочка клетки лопается, и хромосомы «выпадают» на стекло в абсолютно произвольном порядке, после чего их фотографируют и анализируют при помощи специальной программы, расставляя попарно и сравнивая структуру полосок.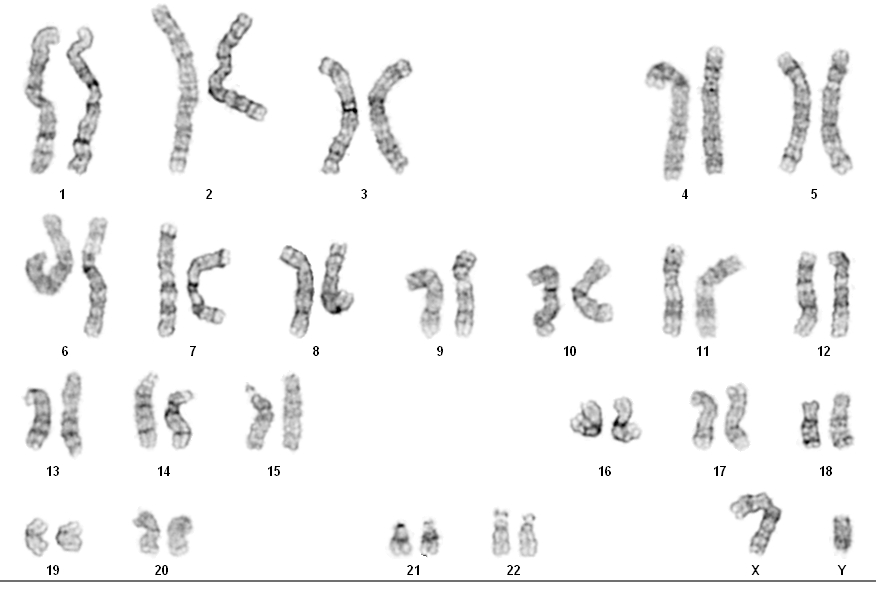 Каждая хромосома имеет свой уникальный рисунок, спутать их нельзя. По этому рисунку и размеру их классифицируют согласно международной номенклатуре ISCN (An International System for Human Cytogenetic Nomenclature).
Каждая хромосома имеет свой уникальный рисунок, спутать их нельзя. По этому рисунку и размеру их классифицируют согласно международной номенклатуре ISCN (An International System for Human Cytogenetic Nomenclature).
Кариотип 46, XX — обзор
1.2.3 Нарушения развития гонад
У женщин с кариотипом 46, XX отсутствие развития гонад не приведет к каким-либо очевидным клиническим признакам. И внутренние, и внешние половые структуры развились у женщин. Большинство пациентов привлекают внимание из-за недостаточного полового развития из-за гонадной недостаточности в подростковом возрасте, и в это время медицинское обследование покажет гипергонадотропный гипогонадизм. Были установлены ассоциации с множеством генов, и следует искать точный диагноз. 35,36
В очень редких случаях развитие яичек может быть запущено у людей с 46, XX кариотипом. 37,38 Это в основном вызвано транслокацией гена SRY на другую хромосому, но другие нижестоящие гены также могут регулироваться ненормально, чтобы начать развитие яичек. В этих случаях часто присутствуют нормальные наружные и внутренние гениталии мужчин и эндокринная функция яичек в норме. Однако мужчины 46, XX обычно бесплодны, так как половые клетки дегенерировали.В классификации DSD это называется 46, XX тестикулярный DSD.
В этих случаях часто присутствуют нормальные наружные и внутренние гениталии мужчин и эндокринная функция яичек в норме. Однако мужчины 46, XX обычно бесплодны, так как половые клетки дегенерировали.В классификации DSD это называется 46, XX тестикулярный DSD.
В некоторых случаях с кариотипом 46, XX развитие яичек является частичным, что приводит к овотестикулярной дифференцировке гонад. 39 В этих случаях одна гонада дифференцируется как яичко, а другая как яичник, но в большинстве случаев одна сторона содержит ткань яичка и яичника, а другая сторона — яичник. В зависимости от количества тестикулярной ткани и эндокринной активности у детей может быть различное развитие матки, часто проявляющееся в виде полуматки со стороны яичников.Наружные гениталии могут быть частично вирилизованы в зависимости от уровня и активности тестостерона. Эта форма DSD называется овотестикулярной DSD, и она может иметь кариотип 46, XX и 46, XY, хотя последний встречается гораздо реже. 40
Чаще 46, может возникать дисгенезия гонад XY. 41 У этих пациентов описан широкий клинический спектр, обычно называемый частичной или полной дисгенезией гонад. В 46, XY полная дисгенезия гонад фенотип полностью женский, и его нельзя отличить от 46, XX гонадная дисгенезия.У женщин нормальная матка и анатомия гениталий. Таким образом, эти пациенты снова привлекут к себе внимание в подростковом возрасте из-за отсутствия полового развития и будет диагностирован гипергонадотропный гипогонадизм. Выявление кариотипа важно только из-за возможности развития опухоли из остатков гонад и выявления возможных связанных клинических признаков.
41 У этих пациентов описан широкий клинический спектр, обычно называемый частичной или полной дисгенезией гонад. В 46, XY полная дисгенезия гонад фенотип полностью женский, и его нельзя отличить от 46, XX гонадная дисгенезия.У женщин нормальная матка и анатомия гениталий. Таким образом, эти пациенты снова привлекут к себе внимание в подростковом возрасте из-за отсутствия полового развития и будет диагностирован гипергонадотропный гипогонадизм. Выявление кариотипа важно только из-за возможности развития опухоли из остатков гонад и выявления возможных связанных клинических признаков.
Генетические причины дисгенезии гонад 46, XY многочисленны и часто еще не выяснены.Одной из причин может быть делеция или вредоносная мутация гена SRY . Также описаны мутации SOX9 и в других генах каскада развития яичек. 42 Важное значение имеет обнаружение мутаций в гене Вильмса-опухоли 1, поскольку этот ген также участвует в развитии почек. 43 У пациентов с 46, XY полной дисгенезией гонад, также называемой синдромом Фрейзера, разовьется прогрессирующая почечная недостаточность, и им обычно требуется диализ в возрасте до 20 лет. Кроме того, у них высок риск развития опухолей гонад, в основном дисгерминомы, в раннем возрасте. Но и другие генетические причины дисгенезии гонад 46, XY могут иметь связанные заболевания. У пациентов с мутациями SOX9 было описано проявление костной ткани, называемое кампомелической дисплазией. Мутации Desert Hedge Hog связаны с сенсорной полинейропатией и т. Д. 44 Таким образом, для пациента важно выяснить генетическую основу 46, XY дисгенезии гонад.
Кроме того, у них высок риск развития опухолей гонад, в основном дисгерминомы, в раннем возрасте. Но и другие генетические причины дисгенезии гонад 46, XY могут иметь связанные заболевания. У пациентов с мутациями SOX9 было описано проявление костной ткани, называемое кампомелической дисплазией. Мутации Desert Hedge Hog связаны с сенсорной полинейропатией и т. Д. 44 Таким образом, для пациента важно выяснить генетическую основу 46, XY дисгенезии гонад.
Частичная дисгенезия гонад 46, XY является основным дифференциальным диагнозом при DSD. 40,41 У этих детей очевидна частичная андрогенизация наружных половых органов с высокой вариабельностью. Кроме того, оценка внутренних гениталий может выявить различное развитие матки и фаллопиевых труб. Это происходит из-за пониженной секреции AMH частично дисгенетическими яичками. Однако в широком спектре пациентов с частичной дисгенезией гонад у некоторых пациентов есть структуры Мюллера, которые не видны макроскопически, но могут быть обнаружены при гистологическом исследовании.![]() Фенотипический спектр настолько разнообразен, что некоторые дети однозначно воспитываются как девочки, а другие, даже с мутациями в том же гене, могут быть отнесены к мужскому полу. Остальные могут быть откровенно двусмысленными. Генетические причины так же распространены, как и при полной дисгенезии гонад. Совсем недавно было описано довольно много пациентов с мутациями в NR5A1 , ведущими к дефициту SF-1. 45 Не существует корреляции генотип-фенотип. Редко описывается дополнительная недостаточность надпочечников. 46
Фенотипический спектр настолько разнообразен, что некоторые дети однозначно воспитываются как девочки, а другие, даже с мутациями в том же гене, могут быть отнесены к мужскому полу. Остальные могут быть откровенно двусмысленными. Генетические причины так же распространены, как и при полной дисгенезии гонад. Совсем недавно было описано довольно много пациентов с мутациями в NR5A1 , ведущими к дефициту SF-1. 45 Не существует корреляции генотип-фенотип. Редко описывается дополнительная недостаточность надпочечников. 46
Другие гены связаны с дисгенезом гонад, а также участвуют в генезе других органов.
Типы расстройств / различия в половом развитии | Детская больница CS Mott
В момент зачатия «мы все начинаем одинаково». Расстройства (или различия) полового развития (DSD) — это общий термин для обозначения различий в типичном пути полового развития от зачатия до рождения. На эти разные пути может влиять расположение половых хромосом, функционирование гонад (т.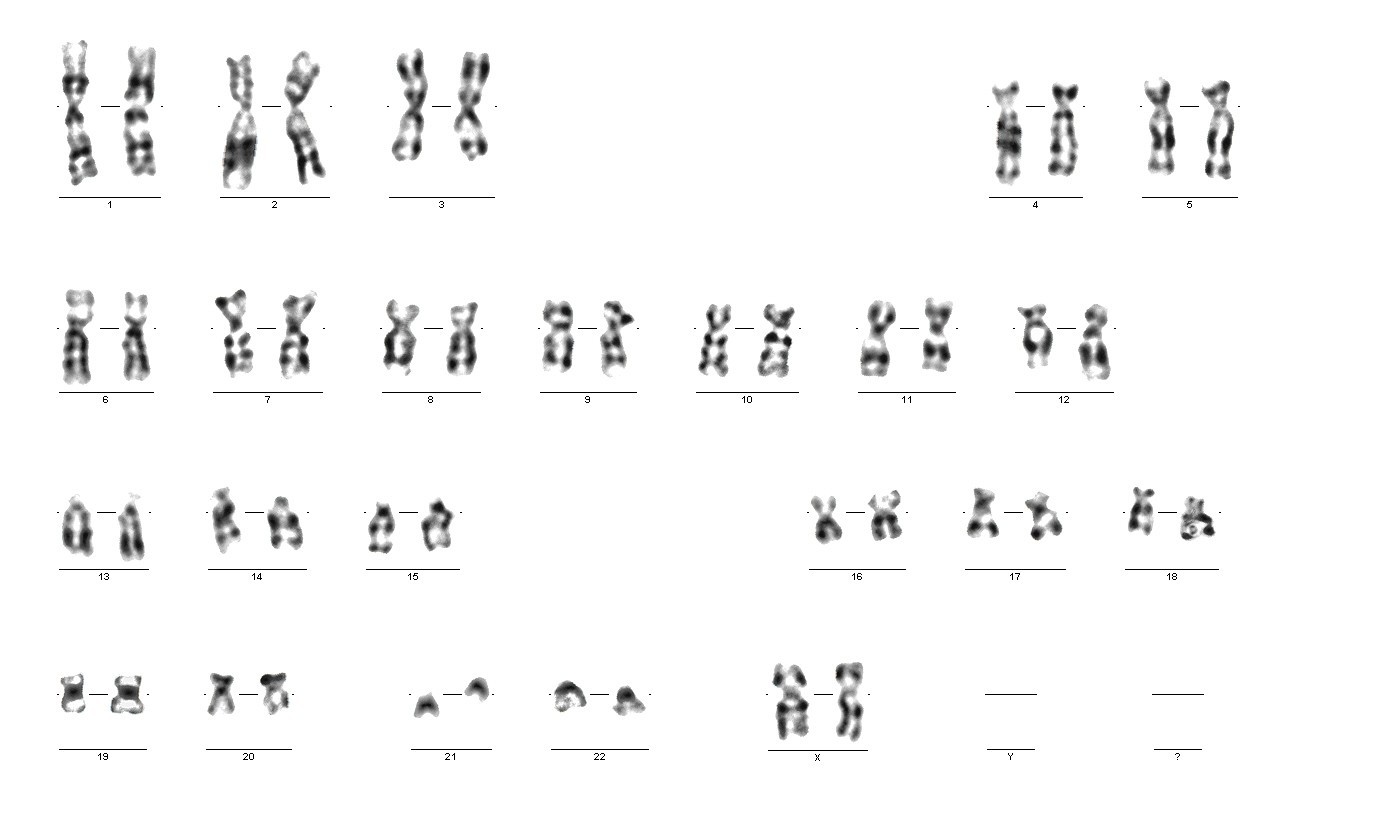 е. яички, яичники), а также реакция нашего организма на гормоны. DSD может встречаться как у мальчиков, так и у девочек, и их можно разделить на три основные категории в зависимости от половых хромосом человека:
е. яички, яичники), а также реакция нашего организма на гормоны. DSD может встречаться как у мальчиков, так и у девочек, и их можно разделить на три основные категории в зависимости от половых хромосом человека:
- Половая хромосома DSD
- 46, XY DSD
- 46, XX DSD
Обычно люди рождаются с 46 хромосомами в каждой клетке. Хромосомы представляют собой нитевидные структуры плотно упакованной ДНК, обычно состоящие из 23 пар. Одна пара хромосом, половые хромосомы, помогает определить, разовьются ли у человека мужские или женские физические половые характеристики.Есть два типа половых хромосом, X и Y. У женщин обычно есть две X-хромосомы и маркируются как (46, XX), а у мужчин обычно одна X и одна Y-хромосома (46, XY).
Половая хромосома DSD
Эти DSD включают любое состояние, при котором имеется атипичное расположение половых хромосом. Например, синдром Тернера или (45, X) — это состояние, которое возникает, когда ребенок рождается только с 45 хромосомами, потому что одна из половых хромосом отсутствует.![]() Для некоторых людей клетки их тела включают более одного типа расположения половых хромосом.Например, некоторые клетки могут иметь одну X- и Y-хромосому, а другие клетки — одну X-хромосому (45, X / 46, XY). Смесь клеток называется мозаицизмом.
Для некоторых людей клетки их тела включают более одного типа расположения половых хромосом.Например, некоторые клетки могут иметь одну X- и Y-хромосому, а другие клетки — одну X-хромосому (45, X / 46, XY). Смесь клеток называется мозаицизмом.
46, XY DSD
Дети, рожденные с одной Х-хромосомой и Y-хромосомой (46, XY), обычно развивают мужские половые признаки. Однако некоторые дети, рожденные с одной X- и Y-хромосомой, имеют недоразвитые гонады или не могут вырабатывать половые гормоны или реагировать на них для развития типичных мужских физических характеристик.Вместо этого у них может развиться внешний вид гениталий, который будет казаться более женственным или нетипичным.
46, XX DSD
Дети, рожденные с двумя Х-хромосомами (46, XX), обычно развивают женские физические половые признаки. Однако некоторые дети, рожденные с двумя Х-хромосомами, до рождения подвергались избыточному воздействию мужских половых гормонов, что привело к появлению атипичных гениталий.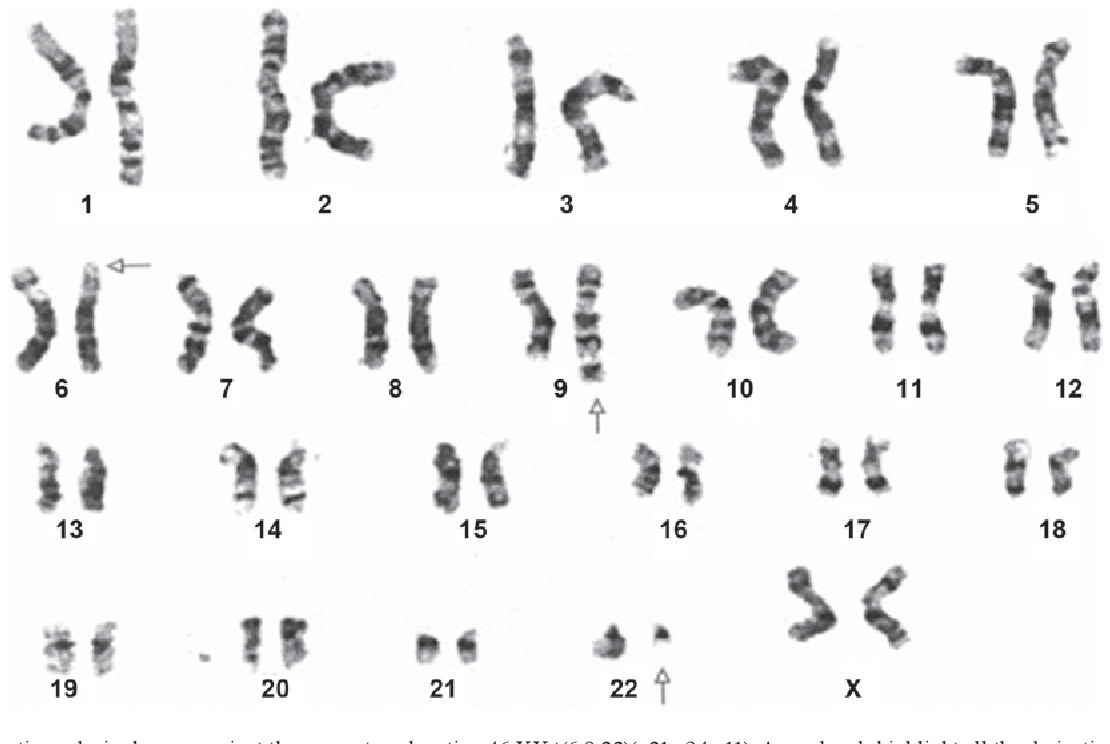
Члены группы DSD Университета Мичигана в детской больнице C.S. Mott понимают, что каждый ребенок с DSD уникален, и что у каждой семьи разные заботы и потребности.Наша миссия — сотрудничать с нашими пациентами и их семьями для предоставления комплексного скоординированного ухода, который отвечает долгосрочным физическим, социальным и эмоциональным потребностям.
Сделайте следующий шаг:
Чтобы узнать больше о программе развития сексуальных расстройств в детской больнице им. С.С. Мотта или записаться на прием, позвоните по телефону по телефону 734-232-0436 .
Мозаичный синдром Тернера с кариотипом 46, XY
Хотя синдром Тернера чаще всего ассоциируется с генотипом 45, X, другие мозаичные генотипы присутствуют примерно в половине всех случаев.Мы описываем случай синдрома Тернера с генотипом 46, XY с помощью обычного 5-клеточного кариотипа, у которого впоследствии было обнаружено мозаичный генотип 18% 45, X и 82% 46, XY с помощью 50-клеточного FISH-анализа. Лица с мозаичным генотипом 45, X / 46, XY имеют множество фенотипических представлений от мужского до женского, которые не коррелируют с процентом мозаицизма. Наш случай представляет собой крайний пример, где преобладает генотип 46, XY и фенотип, типичный для синдрома Тернера.
Лица с мозаичным генотипом 45, X / 46, XY имеют множество фенотипических представлений от мужского до женского, которые не коррелируют с процентом мозаицизма. Наш случай представляет собой крайний пример, где преобладает генотип 46, XY и фенотип, типичный для синдрома Тернера.
1. Введение
Синдром Тернера диагностируется у женщин на основании клинических проявлений в сочетании с генотипом, состоящим из одной нормальной Х-хромосомы и полного или частичного отсутствия другой Х-хромосомы [1]. Пациенты с мозаицизмом 45, X / 46, XY имеют множество фенотипов, начиная от наиболее часто смешанной дисгенезии гонад до других, таких как фенотипические признаки мужского пола, генитальная неоднозначность, синдром Тернера и женщины с нормальными женскими вторичными половыми признаками [2, 3].Синдром Тернера проявляется двусторонней полоской гонад, тогда как смешанная гонадная дисгенезия описывает те, которые проявляются отсутствующей или брюшной полоской гонады с одной стороны и нормальным или дисгенным яичком с другой. Фенотип у пациента с мозаикой 45, X / 46, XY, вероятно, зависит от распределения процента мозаицизма в различных тканях, которое, как было показано, различается между кровью и тканью гонад [4]. Этот случай является примером, когда доминирующий мозаичный генотип в крови (46, XY) не согласуется с фенотипом синдрома Тернера.
Фенотип у пациента с мозаикой 45, X / 46, XY, вероятно, зависит от распределения процента мозаицизма в различных тканях, которое, как было показано, различается между кровью и тканью гонад [4]. Этот случай является примером, когда доминирующий мозаичный генотип в крови (46, XY) не согласуется с фенотипом синдрома Тернера.
2. Изложение клинического случая
Российская женщина, 38 лет, беременная 1 пара 0010, поступила с нерегулярными менструациями каждые 2-3 месяца и 15-летним анамнезом бесплодия. Перед обращением в наше учреждение она была осмотрена специалистом по репродуктологии в России, где был проведен анализ кариотипа. Копия результата не была доступна для просмотра нашими врачами, но пациентка считала, что у нее был обнаружен кариотип 46, XY. Пациент не знал о каких-либо других соответствующих лабораторных результатах.У пациентки была менархе в возрасте 15 лет, с тех пор менструации были нерегулярными каждые 2-3 месяца. У нее был самопроизвольный аборт в первом триместре беременности, который был выявлен с помощью положительного теста на беременность в домашней моче без клинического ультразвукового исследования или патологического подтверждения.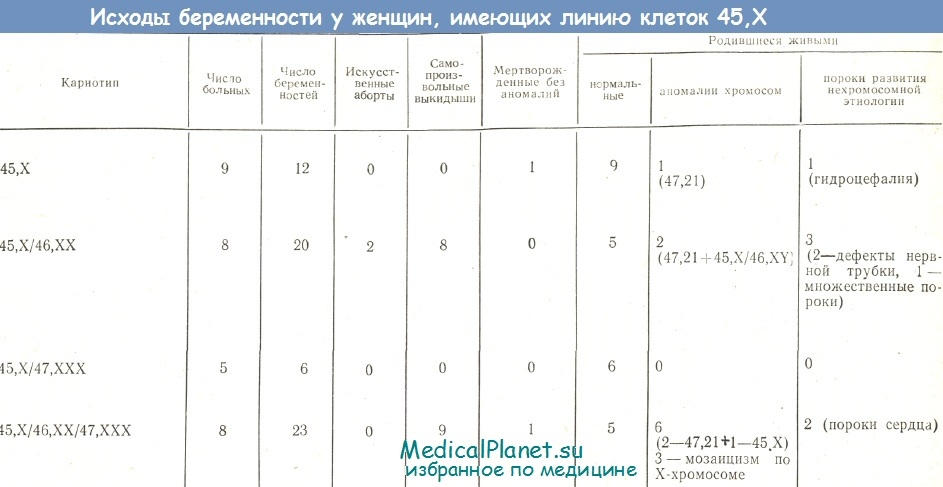 В анамнезе она перенесла лапароскопическую аппендэктомию с одновременной сальпингэктомией справа. У нее не было другого значимого медицинского или семейного анамнеза. В частности, у нее не было семейного анамнеза нерегулярных менструаций, бесплодия или преждевременной недостаточности яичников.
В анамнезе она перенесла лапароскопическую аппендэктомию с одновременной сальпингэктомией справа. У нее не было другого значимого медицинского или семейного анамнеза. В частности, у нее не было семейного анамнеза нерегулярных менструаций, бесплодия или преждевременной недостаточности яичников.
На экзамене она была ростом 160 см и весила 55 кг с ИМТ 23. Ее жизненные показатели были в норме, и у нее были нормальные женские вторичные половые признаки с развитием груди V стадии Таннера, ростом волос на лобке V стадии Таннера, нормальным влагалищем. и шейка матки, и никакого гирсутизма или клиторомегалии. У нее не было низкого роста, сколиоза, высокого неба, потери слуха, короткой или перепончатой шеи, щитовой грудной клетки, вальгусного локтевого сустава, укороченных четвертых пястных или плюсневых костей, genu valgum или varum или деформации Маделунга предплечья и запястья.
Лабораторные исследования показали преждевременную недостаточность яичников с уровнем фолликулостимулирующего гормона 104,9 мМЕ / мл, уровнем лютеинизирующего гормона 35,5 мМЕ / мл, уровнем эстрадиола <5 пг / мл и общим уровнем тестостерона <12 нг / мл. дл. Функции печени и функции щитовидной железы были в пределах нормы. Анализ кариотипа периферической крови 5 клеток при разрешении полос 400-550 показал нормальный мужской кариотип 46, XY (кровь хромосомного анализа, Quest Diagnostics). Хотя этот кариотип соответствует полному дисгенезу гонад (синдром Свайера), история болезни пациентки, связанная с развитием груди и менструацией, не соответствовала этому диагнозу.FISH-анализ проводился на 50 клетках для оценки SRY и X-центромеры для оценки возможного синдрома Свайера или мозаицизма низкого уровня. Это показало 41 клетку с 46, XY и 9 клеток с 45, X (FISH SRY / X Centromere, Quest Diagnostics), что клинически коррелировало с диагнозом мозаичного синдрома Тернера.
дл. Функции печени и функции щитовидной железы были в пределах нормы. Анализ кариотипа периферической крови 5 клеток при разрешении полос 400-550 показал нормальный мужской кариотип 46, XY (кровь хромосомного анализа, Quest Diagnostics). Хотя этот кариотип соответствует полному дисгенезу гонад (синдром Свайера), история болезни пациентки, связанная с развитием груди и менструацией, не соответствовала этому диагнозу.FISH-анализ проводился на 50 клетках для оценки SRY и X-центромеры для оценки возможного синдрома Свайера или мозаицизма низкого уровня. Это показало 41 клетку с 46, XY и 9 клеток с 45, X (FISH SRY / X Centromere, Quest Diagnostics), что клинически коррелировало с диагнозом мозаичного синдрома Тернера.
Сонографическое исследование выявило небольшую матку размером 4,4 × 2,3 × 1,2 см, правый яичник размером 1,4 × 1,2 × 0,9 см с двумя простыми кистами размером 8 мм и 9 мм, левый яичник размером 1.3 × 0,9 × 0,8 см и эхокомплекс эндометрия 6 мм. КТ брюшной полости и таза показала нормальные почки. Эхокардиограмма не показала анатомических аномалий сердца. Двухэнергетическая рентгеновская абсорбциометрия (DEXA) показала поясничный остеопороз с Т-баллом -3,5.
Эхокардиограмма не показала анатомических аномалий сердца. Двухэнергетическая рентгеновская абсорбциометрия (DEXA) показала поясничный остеопороз с Т-баллом -3,5.
В связи с повышенным риском развития гонадобластомы пациенту была предложена и проведена двусторонняя лапароскопическая гонадэктомия и левая сальпингэктомия (правая маточная труба хирургически отсутствовала) с промыванием таза.При патологическом обзоре было обнаружено, что двусторонние гонады содержат гипопластическую ткань яичника (рис. 1) с двумя небольшими серозными кистами правого яичника (рис. 2) и не имеют признаков злокачественности. По поводу остеопороза ей прописали добавки с кальцием и витамином D, и она предпочла принимать циклические комбинированные пероральные контрацептивы, а не стандартную заместительную гормональную терапию. Ей посоветовали, что беременность — это вариант для нее посредством экстракорпорального оплодотворения донорскими яйцеклетками, и она намерена продолжить, когда будет готова к созданию семьи. Ей посоветовали, что бисфосфонаты не рекомендуются женщинам с учетом будущей беременности, и направили в медицинскую эндокринологию для лечения остеопороза другими препаратами, не являющимися бисфосфонатами.
Ей посоветовали, что бисфосфонаты не рекомендуются женщинам с учетом будущей беременности, и направили в медицинскую эндокринологию для лечения остеопороза другими препаратами, не являющимися бисфосфонатами.
3. Обсуждение
Синдром Тернера связан с множественными хромосомными аномалиями, включая 45, X и 45, X / 46, XX и 45, X / 47, XXX и 45, X / 46, XY. На генотип 45, X / 46, XY приходится примерно 10–12% случаев синдрома Тернера [1]. В сообщении о 76 пренатально диагностированных случаях мозаицизма 45, X / 46, XY, у 75 были мужские наружные гениталии и только у одного были женские гениталии [5].В серии из 27 постнатально диагностированных случаев мозаицизма 45, X / 46, XY 18 были мужчинами (11 со смешанной дисгенезией гонад) и 9 имели синдром Тернера [2]. Синдром Тернера с мозаицизмом низкого уровня 45, X / 46, XY не может быть обнаружен при стандартном кариотипе, и FISH-анализ большего числа клеток может быть полезен для диагностики.
Американский колледж медицинской генетики (ACMG) предоставляет рекомендации по процедуре кариотипирования, специфичной для синдрома Тернера. Колледж рекомендует кариотипировать как минимум 30 клеток из-за высокой частоты мозаицизма, если мозаицизм не встречается в первых 20 клетках.При высоком клиническом подозрении на синдром Тернера у пациента с кариотипом 46, XX рекомендуется цитогенетическое исследование ткани второго типа (например, биопсия кожи для клеточной культуры или буккальный мазок для FISH). Кроме того, учитывая риск гонадобластомы с скрытым мозаицизмом Y-хромосомы, ACMG рекомендует 200-клеточный FISH-анализ для исследования центромер X и Y, когда 30-клеточный кариотип приводит к немозаичному 45, X кариотипу [6].
Колледж рекомендует кариотипировать как минимум 30 клеток из-за высокой частоты мозаицизма, если мозаицизм не встречается в первых 20 клетках.При высоком клиническом подозрении на синдром Тернера у пациента с кариотипом 46, XX рекомендуется цитогенетическое исследование ткани второго типа (например, биопсия кожи для клеточной культуры или буккальный мазок для FISH). Кроме того, учитывая риск гонадобластомы с скрытым мозаицизмом Y-хромосомы, ACMG рекомендует 200-клеточный FISH-анализ для исследования центромер X и Y, когда 30-клеточный кариотип приводит к немозаичному 45, X кариотипу [6].
Женщины с синдромом Тернера, обладающие материалом Y-хромосомы, имеют повышенный риск развития половых опухолей, таких как гонадобластома и дисгерминома.Национальное когортное исследование, в которое вошли 211 из этих пациентов, показало, что к 25 годам совокупный риск гонадобластомы составляет 7,9% (95% ДИ 3,1–19,0) [7]. Хотя частота гонадобластомы у пациентов с синдромом Тернера с материалом Y-хромосомы варьируется в зависимости от исследования от 4% до 60%, текущие данные свидетельствуют о том, что этот показатель составляет примерно 10%. Профилактическая гонадэктомия рекомендуется во время постановки диагноза пациентам с синдромом Тернера и материалом Y-хромосомы, таким как мозаицизм 45, X / 46, XY [1] .
Профилактическая гонадэктомия рекомендуется во время постановки диагноза пациентам с синдромом Тернера и материалом Y-хромосомы, таким как мозаицизм 45, X / 46, XY [1] .
Пациенты с синдромом Тернера с любым генотипом должны пройти стандартное обследование и лечение сердечно-сосудистых, почечных, метаболических, эндокринных нарушений, нарушений зрения, слуха и минеральной плотности костей [1]. Если диагностирована преждевременная недостаточность яичников, показана заместительная гормональная терапия до типичного возраста менопаузы, чтобы вызвать половое созревание и вторичные половые признаки, стимулировать рост матки и предотвратить потерю костной массы. Обычно рекомендуется начинать лечение с низкой дозы E 2 (3-7 мк г / день трансдермального E 2 или 0.25 мг перорально E 2 в день) в возрасте 11 или 12 лет и постепенно увеличивайте дозу до доз для взрослых (50-150 мкг г / день трансдермального E 2 или 2-4 мг перорально E 2 в день) от 2 до 3 лет. Предпочтителен трансдермальный эстрадиол, а затем пероральный или внутримышечный эстрадиол. Пероральный прием этинилэстрадиола не рекомендуется, за исключением случаев, когда другие варианты недоступны или из-за проблем, связанных с предпочтениями или комплаентностью пациента, как это было в случае пациента, представленного здесь. Прогестин добавляется при появлении прорывного кровотечения или через 2 года после начала E 2 , чтобы снизить риск гиперплазии эндометрия.Прогестин можно вводить перорально по циклической схеме с E 2 , перорально непрерывно с E 2 или в форме внутриматочной спирали, содержащей прогестин [8].
Предпочтителен трансдермальный эстрадиол, а затем пероральный или внутримышечный эстрадиол. Пероральный прием этинилэстрадиола не рекомендуется, за исключением случаев, когда другие варианты недоступны или из-за проблем, связанных с предпочтениями или комплаентностью пациента, как это было в случае пациента, представленного здесь. Прогестин добавляется при появлении прорывного кровотечения или через 2 года после начала E 2 , чтобы снизить риск гиперплазии эндометрия.Прогестин можно вводить перорально по циклической схеме с E 2 , перорально непрерывно с E 2 или в форме внутриматочной спирали, содержащей прогестин [8].
Плодовитость в будущем — важный фактор для пациентов с синдромом Тернера. Точная и ранняя диагностика мозаицизма 45, X / 46, XY может позволить проконсультироваться о репродуктивном потенциале и проведении беременности с экстракорпоральным оплодотворением донорской яйцеклеткой и / или гестационным суррогатным материнством. Успешные исходы беременности имели место у пациенток с мозаицизмом 45, X / 46, XY, а также с дисгенезом гонад 46, XY после донорства ооцитов и экстракорпорального оплодотворения, хотя большинство зарегистрированных случаев были доставлены путем кесарева сечения [9–13]. Хотя на УЗИ размер матки этой пациентки составил всего 4,4 × 2,3 × 1,2 см, противопоказаний к беременности из-за размера матки нет. Размер матки, скорее всего, является результатом низкого уровня эстрогена, а не признаком того, что матка не может вынашивать беременность.
Хотя на УЗИ размер матки этой пациентки составил всего 4,4 × 2,3 × 1,2 см, противопоказаний к беременности из-за размера матки нет. Размер матки, скорее всего, является результатом низкого уровня эстрогена, а не признаком того, что матка не может вынашивать беременность.
Таким образом, этот случай демонстрирует, что синдром Тернера с мозаицизмом низкого уровня может быть пропущен традиционным кариотипом. У некоторых женщин с диагнозом синдром Свайера на самом деле может быть синдром Тернера с низким уровнем мозаицизма.Примерно 70-80% пациентов с диагнозом синдрома Свайера не имеют мутаций SRY [14, 15], и синдром Тернера с мозаицизмом низкого уровня может быть фактической причиной дисгенезии гонад у некоторых из этих пациентов. В случаях, когда результаты обычного кариотипа не полностью соответствуют клинической картине, анализ FISH на мозаицизм низкого уровня может быть информативным.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Кариотипирование и пренатальная диагностика 47, XX, + 8 [67] / 46, XX [13] Мозаицизм: описание случая и обзор литературы | BMC Medical Genomics
Синдром трисомии — наиболее распространенный тип хромосомной (Chr) аномалии у людей.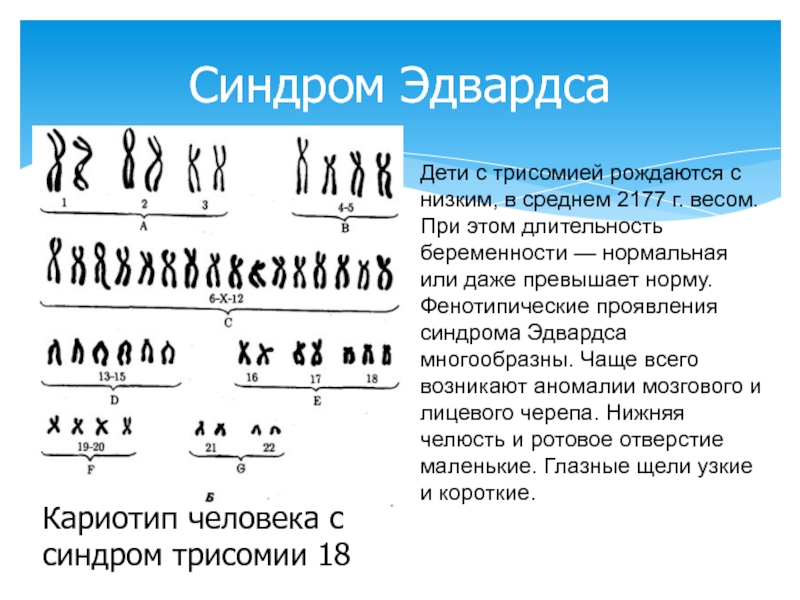 Многочисленные исследования показали, что 90% случаев синдрома трисомии связаны с Chr2, 13, 15, 16, 21, 18, 22 и X, и что более чем в 95% случаев происходит прерывание беременности до рождения; кроме того, многочисленные цитогенетические отчеты показали, что трисомии, связанные с хромосомами 2, 16 и 22, почти всегда влияют на ткань плаценты, а не на плод (ограниченный плацентарный мозаицизм) [1]. Среди них синдром мозаицизма конституциональной трисомии 8 (T8MS), также известный как синдром Варкани, является редким жизнеспособным заболеванием, о котором сообщается у 1/25 000–50 000 живорожденных, и он чаще встречается у мужчин, чем у женщин (5: 1) [2] .Трисомия 8 — редкое заболевание у людей, составляющее 0,7% самопроизвольных абортов, и, по оценкам, встречается примерно в 0,1% признанных беременностей. Chr. 8 полностью или частично дублируется при различных гематологических заболеваниях, особенно миелоидных заболеваниях, и относительно часто встречается при M4, M5 и CMML [3]. Помимо миелоидных заболеваний крови, клинические характеристики трисомии 8 включают заторможенность, языковые барьеры, перепончатую шею, двигательные нарушения, варусную подковообразную стопу, дисплазию черепа, язвы в полости рта, повторяющиеся случаи язв толстой кишки, артрит, косоглазие, маленькую ушную раковину, полную ладонь , и аномалии развития сердца, костей, почек и центральной нервной системы [4].
Многочисленные исследования показали, что 90% случаев синдрома трисомии связаны с Chr2, 13, 15, 16, 21, 18, 22 и X, и что более чем в 95% случаев происходит прерывание беременности до рождения; кроме того, многочисленные цитогенетические отчеты показали, что трисомии, связанные с хромосомами 2, 16 и 22, почти всегда влияют на ткань плаценты, а не на плод (ограниченный плацентарный мозаицизм) [1]. Среди них синдром мозаицизма конституциональной трисомии 8 (T8MS), также известный как синдром Варкани, является редким жизнеспособным заболеванием, о котором сообщается у 1/25 000–50 000 живорожденных, и он чаще встречается у мужчин, чем у женщин (5: 1) [2] .Трисомия 8 — редкое заболевание у людей, составляющее 0,7% самопроизвольных абортов, и, по оценкам, встречается примерно в 0,1% признанных беременностей. Chr. 8 полностью или частично дублируется при различных гематологических заболеваниях, особенно миелоидных заболеваниях, и относительно часто встречается при M4, M5 и CMML [3]. Помимо миелоидных заболеваний крови, клинические характеристики трисомии 8 включают заторможенность, языковые барьеры, перепончатую шею, двигательные нарушения, варусную подковообразную стопу, дисплазию черепа, язвы в полости рта, повторяющиеся случаи язв толстой кишки, артрит, косоглазие, маленькую ушную раковину, полную ладонь , и аномалии развития сердца, костей, почек и центральной нервной системы [4]. Характерные фенотипические признаки мозаичной трисомии 8 широко варьируют, а глубокие борозды на подошвах стоп очень характерны [5,6,7].
Характерные фенотипические признаки мозаичной трисомии 8 широко варьируют, а глубокие борозды на подошвах стоп очень характерны [5,6,7].
Мозаицизм трисомии 8 — необычное заболевание, и пренатальное обнаружение мозаицизма трисомии 8 может привести к проблемам при генетическом консультировании. Итак, в настоящем исследовании мы сообщаем об одном редком случае беременной женщины, у которой был диагностирован мозаицизм трисомии 8, и мы использовали различные методы для проведения тщательной пренатальной цитогенетической диагностики для нее и получения дополнительных знаний, которые могли бы указать на T8 и пренатальная диагностика для врачей.
Изучение клинического случая
Это исследование было одобрено этическим комитетом Далянской больницы по охране здоровья матери и ребенка, а информированное согласие подписано пациенткой.
Пациентка была женского пола, возрастом 37 лет, из провинции Ляонин в Китае, обследовалась в нашей больнице на 13 неделе беременности. Результаты цветного ультразвука показали, что значение затылочной прозрачности (NT) составляло 2,4 мм, и была проведена НИПТ с использованием секвенатора Ion Proton (CapitalBio Technology Inc., Пекин) на 16 неделе беременности.Однако Z-показатель (102,835) для Chr8 был вне нормального диапазона и наводил на мысль о трисомии 8. Поскольку в большинстве случаев полная трисомия 8 прерывается спонтанно на ранних сроках беременности, мы сильно подозревали, что трисомия 8 была от матери и эта трисомия 8 была мозаикой. Дальнейшее обследование с помощью трехмерного цветного ультразвука показало, что у единственного живого плода нет аномалий. Пациент отрицал курение, питье или облучение и не подвергался химическому воздействию. Мы оценили развитие мозга и репродуктивную систему плода с помощью УЗИ, явных патологических клинических проявлений не было, плод развивался нормально.Пациентка ранее была беременна трижды, в результате чего было проведено одно кесарево сечение, один самопроизвольный аборт и один род у девочки, которой в настоящее время 8 лет и которая имеет хорошее здоровье.
Результаты цветного ультразвука показали, что значение затылочной прозрачности (NT) составляло 2,4 мм, и была проведена НИПТ с использованием секвенатора Ion Proton (CapitalBio Technology Inc., Пекин) на 16 неделе беременности.Однако Z-показатель (102,835) для Chr8 был вне нормального диапазона и наводил на мысль о трисомии 8. Поскольку в большинстве случаев полная трисомия 8 прерывается спонтанно на ранних сроках беременности, мы сильно подозревали, что трисомия 8 была от матери и эта трисомия 8 была мозаикой. Дальнейшее обследование с помощью трехмерного цветного ультразвука показало, что у единственного живого плода нет аномалий. Пациент отрицал курение, питье или облучение и не подвергался химическому воздействию. Мы оценили развитие мозга и репродуктивную систему плода с помощью УЗИ, явных патологических клинических проявлений не было, плод развивался нормально.Пациентка ранее была беременна трижды, в результате чего было проведено одно кесарево сечение, один самопроизвольный аборт и один род у девочки, которой в настоящее время 8 лет и которая имеет хорошее здоровье.
Периферическая кровь пациента была взята для анализа классификации гемоцитов и микроскопии мазка. Мы не обнаружили аномальных ювенильных клеток в периферической крови при микроскопии мазков. Путем определения иммунного типирования лейкоцитов мы обнаружили, что клетки CD117 + / CD34 + составляли 0.30% ядерных клеток, что все эти клетки были нормальными миелоидными примитивными клетками, и что на гранулоциты приходилось 41,46% ядерных клеток. Незрелые моноциты занимали 0,02% ядерных клеток (0,53% моноцитов). Стволовые клетки костного мозга, В-клетки, Т-клетки и NK-клетки были нормальными, и в этом клиническом отчете не наблюдались аномальные миелоидные примитивные клетки (<10 — 4 ). Кроме того, модель развития клеток была нормальной (таблицы 1 и 2).
Таблица 1 Результаты анализов крови пациента
Таблица 2 Результаты свертывающей функции у пациентов
Затем периферическая кровь пациентки, ее мужа и дочери, а также околоплодные воды пациентки были собраны для анализа G-диапазона хромосом. Из этих результатов мы знали, что кариотип периферической крови пациента был 47, XX, + 8 [67] / 46, XX, а кариотип околоплодных вод — 46, XX; кариотипы ее мужа и дочери были 46, XY и 46, XX соответственно. Кроме того, мозаицизм трисомии 8 был подтвержден с использованием секвенатора Ion Proton (CapitalBio Technology Inc., Пекин) при 400 потоках в соответствии с инструкциями производителя. Образцы околоплодных вод, глотки, щеки, слюны и опадающих клеток шейки матки от пациента подверглись секвенированию следующего поколения.Сохраненные считывания сравнивали с эталонными последовательностями генома человека (hg19) с использованием BWA. Концентрация ДНК плода рассчитывалась для контроля качества, как описано в статье Инь [8]. Для выявления аутосомной анеуплоидии плода для трисомии использовались комбинированные методы GC-коррекции и Z-балла, как описано в статье Ляо [9]. Диапазон Z-баллов от -3 до 3 считался показателем низкого риска трисомии хромосомы, а если Z-балл был> 3, образец находился в зоне высокого риска.
Из этих результатов мы знали, что кариотип периферической крови пациента был 47, XX, + 8 [67] / 46, XX, а кариотип околоплодных вод — 46, XX; кариотипы ее мужа и дочери были 46, XY и 46, XX соответственно. Кроме того, мозаицизм трисомии 8 был подтвержден с использованием секвенатора Ion Proton (CapitalBio Technology Inc., Пекин) при 400 потоках в соответствии с инструкциями производителя. Образцы околоплодных вод, глотки, щеки, слюны и опадающих клеток шейки матки от пациента подверглись секвенированию следующего поколения.Сохраненные считывания сравнивали с эталонными последовательностями генома человека (hg19) с использованием BWA. Концентрация ДНК плода рассчитывалась для контроля качества, как описано в статье Инь [8]. Для выявления аутосомной анеуплоидии плода для трисомии использовались комбинированные методы GC-коррекции и Z-балла, как описано в статье Ляо [9]. Диапазон Z-баллов от -3 до 3 считался показателем низкого риска трисомии хромосомы, а если Z-балл был> 3, образец находился в зоне высокого риска. Результат рассматривался как трисомия, когда Z-оценка была намного выше, чем нормальное значение, и когда средняя оценка черной кривой составляла +20.Когда Z-оценка была намного ниже нормального значения и когда средняя оценка кривой черного значения составляла -20, рассматривался мономер. Хромосомный химеризм считался 1) когда Z-оценка была больше или меньше плюс-минус 3, но намного меньше Z-оценки полного трисомера или больше Z-оценки полного и 2) когда средняя оценка черная кривая была больше или меньше 0, но намного меньше среднего значения полного трисомера или больше среднего значения полного мономера.Химерная пропорция (%) = диапазон Z-баллов / 0,2/100 (самки) или химерные пропорции (%) = диапазон Z-баллов / 0,2/100/ концентрация выборки (самцы). Более того, результат NGS показал, что пропорции трисомии 8 в разных тканях явно различались (рис. 1). Пропорции трисомии 8 в образцах клеток опущения щек, слюны и шейки матки составляли 5, 60 и 100% соответственно. Кроме того, амниотическая жидкость составляла 0%, что свидетельствовало о том, что плод был нормальным (рис.
Результат рассматривался как трисомия, когда Z-оценка была намного выше, чем нормальное значение, и когда средняя оценка черной кривой составляла +20.Когда Z-оценка была намного ниже нормального значения и когда средняя оценка кривой черного значения составляла -20, рассматривался мономер. Хромосомный химеризм считался 1) когда Z-оценка была больше или меньше плюс-минус 3, но намного меньше Z-оценки полного трисомера или больше Z-оценки полного и 2) когда средняя оценка черная кривая была больше или меньше 0, но намного меньше среднего значения полного трисомера или больше среднего значения полного мономера.Химерная пропорция (%) = диапазон Z-баллов / 0,2/100 (самки) или химерные пропорции (%) = диапазон Z-баллов / 0,2/100/ концентрация выборки (самцы). Более того, результат NGS показал, что пропорции трисомии 8 в разных тканях явно различались (рис. 1). Пропорции трисомии 8 в образцах клеток опущения щек, слюны и шейки матки составляли 5, 60 и 100% соответственно. Кроме того, амниотическая жидкость составляла 0%, что свидетельствовало о том, что плод был нормальным (рис.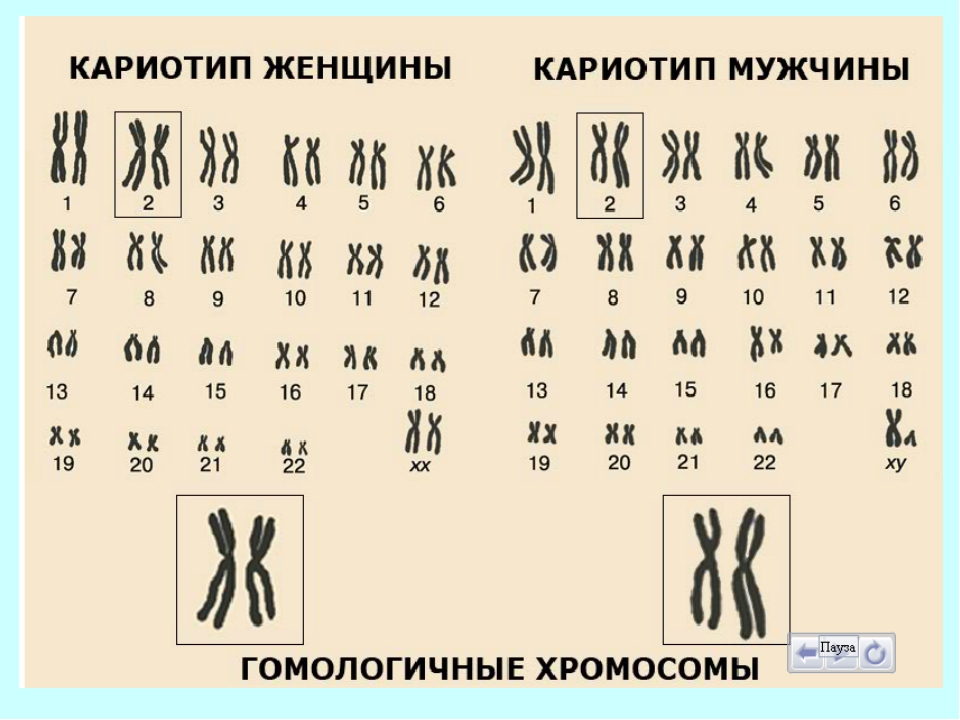 1). Наконец, хромосомы пациентки и ее ребенка были дополнительно исследованы с помощью анализа хромосомных микроматриц (CMA) с платформой CytoScan® HD с более высоким разрешением (Affymetrix, Санта-Клара, Калифорния) (рис.2). Анализ CMA показал, что результат обнаружения chr.8 в плазме пациентки был arr [GRCh47] (8) × 3,11p15.5p13 (230750–33,455,733) × 2 hmz, и результат ее ребенка был нормальным.
1). Наконец, хромосомы пациентки и ее ребенка были дополнительно исследованы с помощью анализа хромосомных микроматриц (CMA) с платформой CytoScan® HD с более высоким разрешением (Affymetrix, Санта-Клара, Калифорния) (рис.2). Анализ CMA показал, что результат обнаружения chr.8 в плазме пациентки был arr [GRCh47] (8) × 3,11p15.5p13 (230750–33,455,733) × 2 hmz, и результат ее ребенка был нормальным.
Рис. 1
Результаты НИПТ и NGS для плода и пациента. — анализ кариотипа плода с помощью НИПТ, Z-оценка составила 102,835; b анализ кариотипа околоплодных вод плода с помощью NGS, оценка Z составила -0,051, а прогнозируемое значение T8M было 0%; c о кариотипе крови пациента по NGS, Z-балл составил 158.487, а прогнозируемое значение T8M было 100%; d Хромосомный анализ образцов буккальной ткани пациента, оценка Z составила 8,246, а прогнозируемое значение T8M — 5%; e Хромосомный анализ образцов слюны пациента, оценка Z составила 108,81, а прогнозируемое значение T8M — 60%; f Хромосомный анализ отслоившихся клеток шейки матки пациента, оценка Z составила 161,646, а прогнозируемое значение T8M было 100%
Рис. 2
2
Результаты анализа CMA для пациентки и ее ребенка. — результат КМА беременной женщины, обр [GRCh47] (8) × 3,11p15.5p13 (230750–33,455,733) × 2 hmz. b CMA результат ее ребенка, нормальный
Случай партеногенетической химеры 46, XX / 46, XY с неоднозначными гениталиями
Gartler SM, Waxman SH, Giblett E. Человеческий гермафродит XX / XY, возникший в результате двойное оплодотворение. Proc Natl Acad Sci USA. 1962; 48: 332–5.
CAS
Статья
Google ученый
Laursen RJ, Alsbjerg B, Vogel I, Gravholt CH, Elbaek H, Lildballe DL, et al. Случай успешного лечения ЭКО олигоспермического мужчины с химеризмом 46, XX / 46, XY. J Assist Reprod Genet. 2018; 35: 1325–8.
CAS
Статья
Google ученый
Sheets KM, Baird ML, Heinig J, Davis D, Sabatini M, Starr DB. Случай смешения отцовства, вызванного химеризмом: что могут сделать практикующие ВРТ, чтобы предотвратить будущие бедствия для семей.J Assist Reprod Genet. 2018; 35: 345–52.
Артикул
Google ученый
Малан В., Гесни Р., Моричон-Дельваллез Н., Обри М.С., Беначи А., Санлавиль Д. и др. Пренатальный диагноз и нормальный исход химеры 46, XX / 46, XY: отчет о болезни. Hum Reprod. 2007; 22: 1037–41.
CAS
Статья
Google ученый
Малан В., Векеманс М., Турло К. Химера и другие ошибки оплодотворения.Clin Genet. 2006; 70: 363–73.
CAS
Статья
Google ученый
 org/ScholarlyArticle»> 6.
org/ScholarlyArticle»> 6.Шин С.И., Ю Х.В., Ли Б.Х., Ким К.С., Со Э.Дж. Идентификация механизма, лежащего в основе химеры человека, с помощью анализа массива SNP. Ам Дж. Мед Генет А. 2012; 158: 2119–23.
CAS
Статья
Google ученый
Ли К.Ф., Сюй С.С., Куо П.Л., Чен Дж.Л., Цзян Ю.Х., Лю И.Ю. Идентификация спонтанного химерного плода 47, XX, + 21/46, XY с мужскими гениталиями.BMC Med Genet. 2012; 13: 85.
Артикул
Google ученый
Ямазава К., Накабаяси К., Кагами М., Сато Т., Сайто С., Хорикава Р. и др. Партеногенетический химеризм / мозаицизм с фенотипом, подобным синдрому Сильвера-Рассела. J Med Genet. 2010; 47: 782–5.
CAS
Статья
Google ученый
 org/ScholarlyArticle»> 9.
org/ScholarlyArticle»> 9.Гилтай Дж. К., Брант Т., Бимер Ф. А., Вит Дж. М., ван Амстел Х. К., Пирсон П. Л. и др.Полиморфное определение партеногенетического материнского и двойного отцовского вклада в гермафродит 46, XX / 46, XY. Am J Hum Genet. 1998; 62: 937–40.
CAS
Статья
Google ученый
Kaiser-Rogers KA, McFadden DE, Livasy CA, Dansereau J, Jiang R, Knops JF, et al. Андрогенетический / двупародительский мозаицизм вызывает мезенхимальную дисплазию плаценты. J Med Genet. 2006; 43: 187–92.
CAS
Статья
Google ученый
Giurgea I, Sanlaville D, Fournet JC, Sempoux C, Bellanné-Chantelot C, Touati G, et al. Врожденный гиперинсулинизм и мозаичные аномалии плоидности. J Med Genet. 2006. 43: 248–54.
J Med Genet. 2006. 43: 248–54.
CAS
Статья
Google ученый
Габбетт М.Т., Лапорт Дж., Секар Р., Нандини А., МакГрат П., Сапкота Ю. и др. Молекулярная поддержка гетерогонеза, приводящего к сесквизиготному двойникованию. N Engl J Med. 2019; 28: 842–9.
Артикул
Google ученый
Davidsson J, Johansson B. Анализы метилирования и экспрессии синдрома Паллистера-Киллиана показывают частичную дозовую компенсацию тетрасомии 12p и гипометилирования бедных генами областей на 12p. Эпигенетика. 2016; 3: 194–204.
Артикул
Google ученый

Конлин Л.К., Тиль Б.Д., Боннеманн К.Г., Медне Л., Эрнст Л.М., Закай Е.Х. и др. Механизмы мозаицизма, химеризма и монородительской дисомии идентифицированы с помощью анализа массива однонуклеотидных полиморфизмов.Hum Mol Genet. 2010; 19: 1263–75.
CAS
Статья
Google ученый
Biesecker LG, Spinner NB. Геномный взгляд на мозаицизм и болезни человека. Nat Rev Genet. 2013; 14: 307–20.
CAS
Статья
Google ученый
Химия клетки и генетика
Определение пола
Удивительно, но только за последние 50 лет мы начали
понять природу биологических событий, которые определяют наш пол (и для
это важно, почему мы вообще беспокоимся о сексе и почему два пола лучше, чем
три и более).Не так давно женщин обвиняли, если они не
произвести на свет сына для своего мужа, и явно считалось, что сила секса
решимость лежала в теле женщины. В этом столетии
В этом столетии
хромосомные основы определения пола человека были продемонстрированы и в
за последние несколько лет были идентифицированы некоторые из ответственных генов.
Сексуальная идентичность человека определяется на нескольких уровнях,
хромосомный пол, гонадный пол, соматический пол и сексуальная ориентация.
половых хромосом
Хромосомная основа определения пола у людей была признана, когда
метафазные хромосомы от делящихся мужских и женских клеток могут быть изучены и
посчитал. Нормальный
кариотип содержит 46 хромосом, включая либо две Х-хромосомы (46XX,
женщины) или одна Х-хромосома и одна Y-хромосома (46XY, мужчины). Физическим лицам
с 45X или
47XXX
кариотипы — женские, особи с кариотипом 47XXY
мужские. Следовательно, можно сделать вывод, что Y-хромосома половая.
определение
определение пола
Эксперименты по удалению эмбриональной гонады показали, что
у млекопитающих, независимо от хромосомного пола соматических клеток, тело
разовьется как самка, если не будут присутствовать мужские гонады, выделяющие мюллериан
ингибирующее вещество и тестостерон. Это можно частично воспроизвести в
Это можно частично воспроизвести в
генетическое состояние яичек
feminisation , в котором ген, кодирующий рецептор андрогенов, не
выражается так, что, хотя яичко у особи XY секретирует
тестостерон, соматические ткани не могут на него реагировать. Следовательно,
тело человека развивается как женщина, но с внутренними яичками вместо
яичники. В 1990 году был открыт ген SRY, кодируемый Y.
который (по крайней мере, у мышей) способен трансформировать пол эмбриона XX с
от женщины к мужчине.Люди с мутациями в этом гене развиваются как самки.
несмотря на хромосомную конституцию XY. Примерно один мужчина из 10000 не
похоже, имеет Y-хромосому, но вместо этого имеет две X-хромосомы. Эти XX самцы
часто оказывается, что они унаследовали от своих отцов Х-хромосму на
которым была перенесена небольшая часть Y-хромосомы, несущей SRY
путем «незаконного» перехода. XX самцы совершенно нормальны, за исключением того, что они
бесплодны, и их рост в норме для женщин, а не
мужчина.
У других организмов все происходит иначе. И Drosophila , и
И Drosophila , и
нематода Caenorhabditis elegans использует механизм, в котором каждая клетка
измеряет относительное количество Х-хромосом по сравнению с количеством
аутосомы. Однако гены, участвующие в процессе подсчета и в его
интерпретация, похоже, не связана у двух видов.
Даже у позвоночных существует множество механизмов определения пола.
- Птицы используют хромосомную систему, однако, в отличие от системы млекопитающих, самцы
являются гомогаметными ZZ, а самки гетерогаметными ZW. - Аллигаторы хромосомно одинаковые у обоих полов, они определяют пол по
температура, при которой эмбрионы могут развиваться. Если тепло, то самцы
образуются и, если прохладно, образуются самки.
сексуальная идентичность
Это очень обсуждаемый вопрос, является ли наше собственное чувство сексуальной идентичности
имеет генетическое или экологическое происхождение. Как и большинство сложных фенотипов, он, вероятно,
может быть любым, но обычно и тем и другим! Недавние разногласия касались результатов, полученных
Хамер, который показал доказательства одного такого гена, рецессивного гена на X
хромосома, которая, будучи мутантной, может сделать ее носителя, если он мужской, с большей вероятностью
гомосексуалист.
Секс связь
рецессивные гены, сцепленные с полом
Гены, переносимые на Х-хромосоме, имеют отличительный образец наследования.
Поскольку самцы гемизиготные , то есть у них есть только одна копия X
хромосома, и потому что Y-хромосома несет очень мало генов (хотя те
которые он несет, часто гомологичны X сцепленным генам), то рецессивный
мутации проявляются в фенотипе самцов. Если мутантный ген
смертельный (например, мышечный
Дистрофия), то для рождения пораженной женщины требуется необычное событие.
Родословная, связанная с X
Типичная родословная покажет группы пораженных самцов (каждый брат будет
имеют 50% шанс быть затронутым) подключены через незатронутого оператора связи
самки. Не будет случаев прямой передачи от мужчины к мужчине
потому что мужчины передают свои Х-хромосомы своим дочерям, а не своим
сыновья.
Следующий отрывок цитируется из Истории гемофилии.
доктора П.Л.Ф. Giangrande
История королевы Виктории
Иногда упоминается гемофилия
к как Королевская болезнь.
но вскоре после рождения ее восьмого ребенка, Леопольда, в 1853 г.
очевидно, что у него была гемофилия. Королева Виктория была примером того, как
Состояние может возникнуть как спонтанная мутация. Состояние здоровья Леопольда было
сообщалось в British Medical Journal в 1868 году, и очевидно, что он был
беспокоят кровотечения, происходящие не реже одного раза в месяц. Он умер в возрасте 31 года.
в 1884 г. от внутримозгового кровоизлияния после падения.Леопольд женился на двух
лет до его смерти. Его дочь Алиса была обязательной носительницей, а также
у него родился сын-гемофилий. Руперт, виконт Трематон, родился в 1907 году.
и умер в возрасте 21 года, также от внутримозгового кровоизлияния. Это также
впоследствии выяснилось, что две собственные дочери королевы Виктории, Алиса и
Беатрис были переносчиками гемофилии. Состояние было передано через
их несколько королевских семей в Европе, включая Испанию и Россию.
Самым известным пострадавшим человеком был сын российского царя Николая II.История юного цесаревича Алексия, родившегося в 1904 году, была
сюжетом голливудского фильма, а также романа Дороти Сэйерс («У него
туша »: 1932). Было предположение, что болезнь привела к тяжелым
напряжения в королевской семье, и позволил Распутину получить влияние на
семья. Алексис и его семья были убиты большевиками в 1917 году.
гемофильный ген теперь вымер в этих королевских семьях, подчеркивая
тяжесть состояния при отсутствии эффективного лечения.Таким образом
мы до сих пор не знаем, было ли это состояние гемофилией А или
Б.
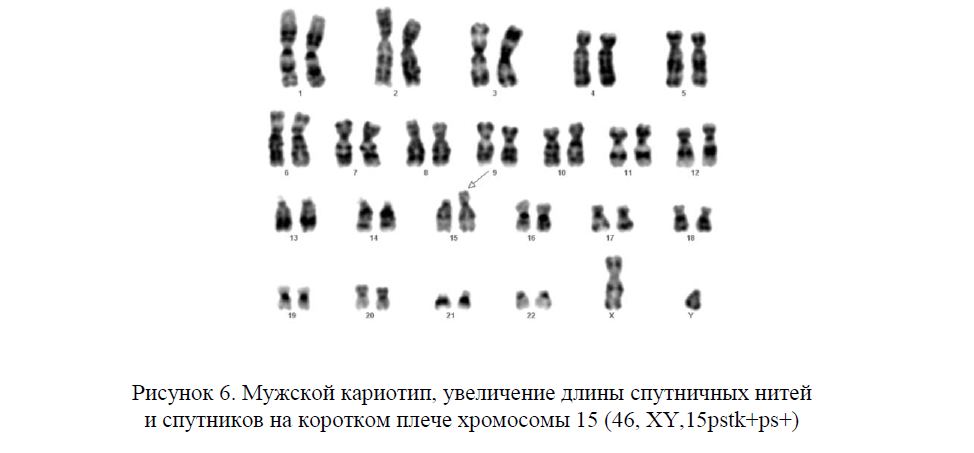 У королевы Виктории не было предков с этим заболеванием
У королевы Виктории не было предков с этим заболеванием Возможно
Возможночастота мутаций X сцепленных генов
Одна треть всех Х-хромосом присутствует у мужчин и, следовательно, одна треть всех
мутантные Х-хромосомы присутствуют у мужчин. Следовательно, если условие
летальный, то треть мутантных Х-хромосом будет потеряна из
население каждого поколения. Если частота заболевания не меняется, тогда
потерянные мутантные хромосомы должны быть заменены новой мутацией. Следовательно, частота мутаций летального X-сцепленного рецессивного заболевания составляет одну
Следовательно, частота мутаций летального X-сцепленного рецессивного заболевания составляет одну
треть частоты заболевания.
доминантных генов, сцепленных с полом
Доминантные состояния, связанные с полом, чрезвычайно редки, например, недержание мочи.
pigmenti (смертельный для мужчин) и врожденный генерализованный
гипертрихоз (синдром человека-волка).
X инактивация
Все случаи аномального кариотипа, в которых отсутствует одна аутосома (аутосомная моносомия), являются летальными во время эмбриогенеза даже для
самые маленькие аутосомы.Тем не менее, самцы с одной Х-хромосомой (средний размер
хромосома) являются (сравнительно!) нормальными. Как это достигается? Ответ
(как было предложено Мэри Лайон в 1961 году) происходит путем инактивации одного из двух X
хромосом у женщин, так что нормальное состояние клетки — наличие двух активных
наборы аутосом и только одна активная Х-хромосома. Другая Х-хромосома
конденсированный и неактивный и виден как вытесненное темное пятно «тело Барра»
против ядерной мембраны. Следующее изображение клеток с женской щеки
Следующее изображение клеток с женской щеки
тампон был «позаимствован» из США
Армия.
Можно увидеть три ядра, каждое с характерным темным пятном (которое
особенно отчетливо видно в центральном ядре).
Мэри Лайон предположила, что инактивация X произошла случайным образом в начале
развитие так, что каждая самка состоит из двух популяций клеток. В одной
в популяции экспрессируется одна Х-хромосома, а в другой — вторая Х-хромосома.
хромосома выражена. Таким образом, самки представляют собой мозаику , , т.е. состоят из двух генетически различных
клеточные популяции.Для гомозиготных генов это не имеет значения.
но для генов, по которым самка гетерозиготна, две популяции клеток
будут противоположных фенотипов. Таким образом она объяснила узоры прически
окраска, например, черепаховый кот, который всегда самка
за исключением очень редкого случая появления самца XXY, что свидетельствует о
правило!
Признаки ограничения пола
Как мы видели, черты, связанные с полом, обычно гораздо чаще выражаются в
самцов, чем самок. Однако вы должны знать, что некоторые черты, влияющие на
Однако вы должны знать, что некоторые черты, влияющие на
один пол больше, чем другой, не обязательно связаны с полом. Примерами являются случаи
экспрессия ограничена полом, которая может включать гены, влияющие на рост бороды или
размер груди и (у крупного рогатого скота) рост рогов и удой. Эти гены не имеют
видимые аффекты у одного пола, потому что необходимого механизма для их выражения нет
настоящее время.
Черты характера, влияющие на секс
На той же общей линии, что и черты, ограниченные полом, находятся черты под влиянием пола.Типичное облысение — это состояние, которое преобладает у мужчин, но рецессивно у них.
женщины.
Печать
Некоторые гены инактивируются при передаче через один пол. Синдром ангельмана
и Прадер-Вилли
синдром — это два разных состояния, оба из которых кажутся вызванными очень
похожие делеции небольшой части хромосомы 15.
На этой диаграмме два гена показаны в критической области. Каждый
инактивированный импринтинг , ген синдрома Ангельмана выключен
хромосома, унаследованная от отца, в то время как ген Прадера-Вилли превращен
выключен на хромосоме, передающейся по материнской линии. Когда удаление, охватывающее
Когда удаление, охватывающее
регион наследуется на другой хромосоме, тот или иной синдром
полученные результаты.
Страница, первоначально найденная по адресу: Химия клетки и генетика, Лекция 4
Синдром Свайера — NORD (Национальная организация по редким заболеваниям)
СТАТЬИ В ЖУРНАЛЕ
McElreavey K, Jorgensen A, Eozenou C, et al. Патогенные варианты DEAH-бокса РНК-геликазы DHX37 являются частой причиной дисгенезии гонад 46, XY и 46, синдрома регрессии яичка XY.Genet Med. 24 июля 2019 г. doi: 10.1038 / s41436-019-0606-y.
Перлман А., Локе Дж., Ле Кайнек С. и др. Мутации в MAP3K1 вызывают 46, XY нарушения полового развития и участвуют в общем пути передачи сигнала в определении семенников человека. Am J Hum Genet. 2010; 87 (6): 898-904.
Михала Л., Госвами Д., Крейтон С.М., Конвей Г.С. Синдром Свайера: проявления и исходы. BJOG. 2008; 115: 737-741.
Колер Б., Линь Л., Ферраз-де-Соуза Б. и др. Пять новых мутаций в стериодогенном факторе 1 (SF1, NR5A1) у 46 пациентов XY с тяжелой недостаточной рандрогенизацией, но без надпочечниковой недостаточности. Хум Мутат. 2008; 29: 59-64.
Хум Мутат. 2008; 29: 59-64.
Зелинска Д., Заячек С., Жепка-Горска И. Опухоли дисгенетических гонад при синдроме Свайера. J Pediatr Surg. 2007; 42: 1721-1724.
Бехташ Н., Карими Зарчи М. Дисгерминома у трех пациентов с синдромом Свайера. Мир J Surg Oncol. 2007; 5: 71.
Ли П.А., Хоук С.П., Ахмед С.Ф. и др. Консенсусное заявление о лечении интерсекс-расстройств. Педиатрия. 2006; 118: e488-e500.
Чен MJ, Ян Дж.Х., Мао Т.Л., Хо Х.Н., Ян Ю.С. Успешная беременность у женщины после гонадэктомии с дисгенезией гонад 46, XY и гонадобластомой.Ферил Стерил. 2005; 84: 217.
Canto P, Soderlund D, Reyes E, Mendez JP. Мутации в гене Desert hedgehog (DHH) у пациентов с 46, XY завершают чистый дисгенез гонад. J Clin Endocrinol Metab. 2004; 89: 4480-4483.
Сарафоглу К., Острер Х. Смена пола в семье: обзор. J Clin Endocrinol Metab. 2000; 85: 483-493.
ИНТЕРНЕТ
Mohnach L, Fechner PY, Keegan CE. Несиндромные нарушения развития яичек.