Миотоническая дистрофия
Миотоническая дистрофия (МД) представляет собой мультисистемное заболевание с исключительной вариабельностью клинических проявлений, среди которых миотония, прогрессирующая мышечная слабость, поражения сердца, зндокринно-вегетативные расстройства, катаракта, снижение интеллекта. МД передается по аутосомно-доминантному типу. Заболевание начинается в любом возрасте, но наиболее часто встречается в 20-30 лет. В большинстве случаев МД носит прогрессирующее течение, приводя к социальной дезадаптации больных.
| Симптомы миотонической дистрофии: Дебютирует на 2-3 десятилетии. Появляется миотонический спазм и затруднения при разгибании пальцев рук. Характерно фронтальное облысение лба. Миотоническая реакция состоит в аномально длительном сокращении мышц. Атрофируются преимущественно мышцы лица (миопатическое лицо с птозом) и (дистальные) мышцы голеней и предплечий, в поздней стадии также мышцы плечевого пояса. Миотоническая катаракта (у 98 % пациентов), атрофия радужки, пигментные изменения сетчатки, а также кардиологические и эндокринные нарушения (атрофия яичек, недостаточность яичников, сахарный диабет). Нарушения сердечного ритма могут привести к внезапной остановке сердца . |
 Слабость мышц лица, шеи, конечностей.
Слабость мышц лица, шеи, конечностей.Причина заболевания связана с увеличением числа тринуклеотидных CTG (цитозин-тимин-гуанин) повторов в 3′- конце гена миотонинпротеинкиназы (DMPK), расположенном на коротком плече 19 хромосомы в области 13.213.3 [Brook et al., 1992; Buxton et al., 1992; Harley et al, 1992; Mahadevan et al., 1992]. Число тринуклеотидных CTG-повторов в разных популяциях полиморфно, в норме колеблется от 5 до 27-37, причем отличия показаны не только между этносами, но и субпопуляциями одного этноса [Иващенко и др., 1997; Лим-борская и др., 2002; Фатхлисламова, 1999; Попова и др., 2003; Сломинский и др., 2000; Zerylnick et al., 1995]. Количество CTG-повторов от 37 до 50 известно как премутация [Yamagata et al. , 1998]. У больных МД число повторов превышает 50 и может достигать нескольких тысяч. Предполагается, что в основе всех случаев МД лежит одна или очень малое количество древних мутаций северо-евразийского происхождения с большим количеством носителей мутации или премутации [Novelli et al., 1994]. Новые мутации при МД встречаются крайне редко [Krähe et al., 1995]. Предрасполагающим к мутации фактором многие исследователи считают нестабильность аллелей с большим числом CTG-повторов (19-30 триплетов), которые составляют резерв для мутаций [Горбунова, 2000]. Показана тесная корреляция между числом CTG-повторов и тяжестью клинического синдрома: больные с небольшой степенью экспансии триплетов (50-160 копий) имеют минимальные клинические проявления, например, только катаракту. Большее число копий триплетов имеют больные с развернутой клинической картиной. Наибольшая степень экспансии СГО-повторов до 4000 отмечена при тяжелой врожденной форме МД. В последние годы помимо классической МД, ген которой расположен на хромосоме 19, на хромосоме Зq картирован ген проксимальной миотонической миопатии (РИОММ) — МД2 [Яапиш & а1.
, 1998]. У больных МД число повторов превышает 50 и может достигать нескольких тысяч. Предполагается, что в основе всех случаев МД лежит одна или очень малое количество древних мутаций северо-евразийского происхождения с большим количеством носителей мутации или премутации [Novelli et al., 1994]. Новые мутации при МД встречаются крайне редко [Krähe et al., 1995]. Предрасполагающим к мутации фактором многие исследователи считают нестабильность аллелей с большим числом CTG-повторов (19-30 триплетов), которые составляют резерв для мутаций [Горбунова, 2000]. Показана тесная корреляция между числом CTG-повторов и тяжестью клинического синдрома: больные с небольшой степенью экспансии триплетов (50-160 копий) имеют минимальные клинические проявления, например, только катаракту. Большее число копий триплетов имеют больные с развернутой клинической картиной. Наибольшая степень экспансии СГО-повторов до 4000 отмечена при тяжелой врожденной форме МД. В последние годы помимо классической МД, ген которой расположен на хромосоме 19, на хромосоме Зq картирован ген проксимальной миотонической миопатии (РИОММ) — МД2 [Яапиш & а1. , 1998; Шскег et а1., 1999], обусловленной расширением цигозин-цитозин-тимин гуанин (ССГО) повторов.
, 1998; Шскег et а1., 1999], обусловленной расширением цигозин-цитозин-тимин гуанин (ССГО) повторов.
Распространенность МД в РС (Я) составляет 10,3 на 100 тыс. населения. МД встречается с высокой частотой (21,3 на 100 тыс.) среди коренного якутского населения с преимущественным локальным накоплением в Вилюйской и Центральной группе улусов. В семьях с МД зарегистрированы низкий уровень рождаемости и высокая частота патологических исходов беременностей и ранней детской смертности. На больную МД женщину приходится в среднем 1,4 выживших детей, тогда как на здоровую 3,6. МД в РС (Я) проявляется классической юношеской (35,8%) и взрослой (45,7%), ранней детской (12,3%), врожденной (3,7%) и минимальной (2,5%) формами. Наиболее частыми проявлениями МД определены миотония, прогрессирующая мышечная слабость, кардиальные и вегето-эндокринные нарушения. Внедрение молекулярно-генетических методов в практику медико-генетического консультирования в PC (Я) позволило не только диагностировать МД в семьях, но и проводить дифференциальную диагностику с заболеваниями со сходным фенотипом.
Литература:
- Сухомясова A.JL, Ноговицына А Н., Федорова С.А., Максимова Н.Р., Кононова С.К., Коротов М.Н., Алексеева С.П. Клинико-генетическая характеристика миотонической дистрофии в Республике Саха (Якутия) // Тез. докл. научно-практ. конф. «Вопросы формирования здоровья и патологии человека на Севере: факты, проблемы и перспективы». Якутск, 2002. С. 273-275.
- Максимова Н.Р., Ноговицына А.Н., Сухомясова AJL, Коротов М.Н. Медико-генетическое консультирование больных с наследственными заболеваниями нервной системы // Тез. докл. научно-практ. конф. «Актуальные проблемы клинической неврологии». Якутск, 2001. С.20-21.
- Федорова С.А., Кононова С К., Ноговицына АН , Сухомясова A.JI. Моле-кулярно-генетическая диагностика наследственных болезней нервной системы в МГК НЦМ PC (Я) // Тез. докл. научно-практ. конф. «Актуальные вопросы детской неврологии и педиатрии». Якутск, 2001 С.77-79.
- Сухомясова АЛ., Федорова С.А., Коротов М.Н., Максимова Н.Р.
 , Алексеева С.П., Сидорова О.Г., Николаева И.А., Кононова С.К.,Степанова С.К., Фатхлисламова Р.И., Хуснутдинова Э.К., Ноговицына А.Н. Миотоническая дистрофия в Республике Саха (Якутия): популяционные особенности и подходы к ДНК-тестированию // Якутский медицинский журнал. 2003. №2 С 12-17.
, Алексеева С.П., Сидорова О.Г., Николаева И.А., Кононова С.К.,Степанова С.К., Фатхлисламова Р.И., Хуснутдинова Э.К., Ноговицына А.Н. Миотоническая дистрофия в Республике Саха (Якутия): популяционные особенности и подходы к ДНК-тестированию // Якутский медицинский журнал. 2003. №2 С 12-17. - Федорова С.А., Кононова С.К., Сухомясова АЛ. и др. Внедрение молекулярно-генетических методов диагностики наследственных болезней в практическое здравоохранение PC (Я) // Тез докл. научно-практ. конф. «Вопросы формирования здоровья и патологии человека на Севере: факты, проблемы и перспективы». Якутск, 2002. С. 276-279.
- Сухомясова АЛ., Федорова С.А., Максимова Н.Р., Ноговицына А.Н. Миотоническая дистрофия в Якутии // Тез. докл. X Российско-Японского симпозиума. Якутск, 2003. С. 509.
- Сухомясова A. JI, Максимова Н.Р., Коротов М.Н., Алексеева С.П., Кононова С.К., Федорова С.А., Ноговицына А.Н. Клинический полиморфизм миотонической дистрофии в практике медико-генетического консультирования в Республике Саха (Якутия) // Тез. докл. научно-практ. конф. «Детское здравоохранение в Республике Саха (Якутия): Оптимизация работы и стратегия развития». Якутск, 2003. С. 150-152.
- Коротов М.Н., Сухомясова АЛ.. Максимова Н.Р., Алексеева С.П., Николаева И.А., Степанова С.К., Федорова С.А., Ноговицына А.Н. Миотония Томсе-на в Якутии и дифференциальная диагностика с миотонической дистрофией // Якутский медицинский журнал. 2004. №2. С. 16-19.
- Сухомясова A.Л. Максимова Н.Р., Алексеева С П., Сидорова О.Г., Николаева И.А., Степанова С.К., Федорова С.А., Коротов М.Н., Ноговицына А.Н. Диагностика миотонической дистрофии в PC (Я) // Тез. докл. Научно-практ. конф. «Достижения и перспективы перинатальной медицины». Якутск, 2004 С.49.
- Сухомясова АЛ., Максимова Н.Р., Коротов М.Н., Алексеева С.П., Ноговицына А.Н. Сердечно-сосудистые нарушения у больных с миотонической дистрофией // Тез. докл. Научно-практ. конф. Современные проблемы сердечно-сосудистой патологии на Крайнем Севере. Якутск, 2004. С. 57-59.
- Ноговицына А.Н, Максимова Н.Р., Сухомясова А.Л., Сидорова О.Г., Алексеева С.П. Республиканский генетический Регистр наследственной и врожденной патологии в Республике Саха (Якутия) // Якутский медицинский журнал. 2004. №4. С. 4-7.
- Коротов М.Н, Николаева И.А., Максимова Н.Р., Алексеева С П., Сухомясова А.Л., Ноговицына АН. Наследственно-дегенеративные заболевания нервной системы в Якутии // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера. — Якутский медицинский журнал. Приложение №1. Якутск, 2005. С. 30-31.
- Семенова Л И., Бурцева А.Р, Сухомясова А.Л., Козлова И.Н., Лукина Н.В , Афанасьева В.П., Куприянова М.Н., Постникова A.A. Клинический случай врожденной формы миотонической дистрофии // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера. Якутский медицинский журнал. Приложение №1. Якутск, 2005. С. 5557.
- М.Степанова С.К., Кононова С.К., Федорова С.А., Сидорова О. Г., Сухомясова А.Л., Ноговицына А Н Пренатальная ДНК-диагностика моногенных болезней в Медико-генетической консультации // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера Якутский медицинский журнал. Приложение №1. Якутск, 2005 С.68.
- Сухомясова А.Л., Назаренко Л.П., Кучер А.Н., Коротов М.Н., Максимова Н Р., Данилова А.Л., Ноговицына А.Н. Территориально-этническая характеристика миотонической дистрофии в Республике Саха (Якутия) // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера. Якутский медицинский журнал. Приложение №1. Якутск, 2005. С.69-70.
- Сухомясова АЛ., Назаренко Л.П., Кучер А.Н., Максимова Н.Р., Данилова А.Л., Ноговицына А.Н. Витальные статистики в отягощенных миотонической дистрофией семьях // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера. Якутский медицинский журнал. Приложение №1. Якутск, 2005. С. 71.
- П. Сидорова О.Г., Кононова С.К., Степанова С.К., Федорова С.А., Сухомясова А.Л. Некоторые этические проблемы пренатальной ДНК-диагностики моногенных заболеваний в МПС РБ№1-Национального центра медицины II Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера Якутский медицинский журнал. Приложение №1 Якутск, 2005. С. 114-115.
- Кучер А Н., Максимова Н.Р., Ноговицына А Н., Сухомясова АЛ. Генетико-демографическое описание сельского населения Усть-Алданского улуса Республики Саха (Якутия): миграционные процессы, брачная структура // Генетика. 2004 Т. 40. №5. С. 685-690.
- Кучер А Н, Максимова Н Р., Ноговицына А Н., Сухомясова А.Л. Генетико-демографическое описание сельского населения Усть-Алданского улуса Республики Саха (Якутия): национальный и половозрастной состав, витальные статистики, фамильная структура // Генетика. 2004. Т. 40. №5. С. 677684.
- Данилова А.Л., Кучер А.Н, Максимова Н.Р., Сухомясова АЛ., Николаева И.А., Павлов Ф. В., Ноговицына А.Н. Генетнко-демографический анализ сельских популяций Республики Саха (Якутия) // Наука и образование. 2005 №2. С. 98-103.
- Федорова С. А., Сухомясова А.Л., Николаева И.А., Куличкин С.С., Платонов Ф.А., Хуснутдинова Э.К. Аллельный полиморфизм гена миотонинпротеин-киназы в популяциях населения Республики Саха (Якутия) // Наука и образование. 2005. №2 С. 59-65.
- Федорова С.А., Хусаинова Р И, Кутуев И.А., Сухомясова А.Л., Николаева И.А., Куличкин С.С., Ахметова В.Л., Салимова А 3., Святова Г.С., Березина Г.М, Платонов Ф.А., Хуснутдинова Э.К. Полиморфизм (СТОХ-повторов гена миотонинпротеинкиназы в популяциях Республики Саха (Якутия) и Средней Азии // Молекулярная биология. 2005. Т.39. №3. С. 385-393.
- Сухомясова AJL. Максимова Н.Р., Федорова С.А., Коротов М.Н., Ноговицына А.Н. Распространенность миотонической дистрофии в PC (Я) // Мат. V съезда мед. генетиков. Ч.ГО Медицинская генетика. 2005. №6. С 272-273.
- .Максимова Н.Р., Сухомясова А. Л., Коротов М.Н., Сидорова О Г, Алексеева С П., Павлов Ф.В., Федорова С А, Степанова С К., Ноговицына А.Н. Диагностика врожденной миотонической дистрофии в Республике Саха (Якутия) // Мат. V съезда мед. генетиков. Ч.П. Медицинская генетика. 2005. №5. С. 223.
- Ноговицына АН., Максимова Н.Р, Сухомясова А.Л, Сидорова О Г., Алексеева С.П. Регистр наследственной и врожденной патологии в Республике Саха (Якутия) // Мат. V съезда мед. генетиков Ч П. Медицинская генетика. 2005. №5 С. 241.
- Fedorova S A., Sukhomyasova A.L., Maximova N.R., Nikolaeva I.A. Study of CTG-trinucleotide repeats polymorphism in the myotonic dystrophy gene in Yakut and Evenk populations // Тез. докл. науч. конф. «Genetics of Complex Diseases and isolated Populations» Tortoli, Sardinia, Italy, 2003. P. 10.
- Sukhomyasova A.L., Maximova NR., Korotov M.N, Nikolaeva IA, Sidorova O.G, Fedorova S A., Nogovitsina A.N. Congenital myotonic dystrophy in Yakutia // The Eleventh International Symposium. Niigata, Japan, 2004. P. 262.
- Sukhomyasova A.L., Maximova N.R, Nikolaeva I A., Stepanova S К , Fedorova S.A, Nogovitsina A N. Myotonic dystrophy in Yakutia H European Human Genetics conference. Munich, Germany, 2004. P. 124.
- .Sukhomyasova A.L., Maximova N.R., Korotov M.N., Fedorova S.A., Danilova A.L., Nogovicina A.N. Myotonic dystrophy in the Republic of Sakha (Yakutia) (Russia) // European Human Genetics conference. Prage, Czech Republic, 2005. P 184.
 , Алексеева С.П., Сидорова О.Г., Николаева И.А., Кононова С.К.,Степанова С.К., Фатхлисламова Р.И., Хуснутдинова Э.К., Ноговицына А.Н. Миотоническая дистрофия в Республике Саха (Якутия): популяционные особенности и подходы к ДНК-тестированию // Якутский медицинский журнал. 2003. №2 С 12-17.
, Алексеева С.П., Сидорова О.Г., Николаева И.А., Кононова С.К.,Степанова С.К., Фатхлисламова Р.И., Хуснутдинова Э.К., Ноговицына А.Н. Миотоническая дистрофия в Республике Саха (Якутия): популяционные особенности и подходы к ДНК-тестированию // Якутский медицинский журнал. 2003. №2 С 12-17. докл. научно-практ. конф. «Детское здравоохранение в Республике Саха (Якутия): Оптимизация работы и стратегия развития». Якутск, 2003. С. 150-152.
докл. научно-практ. конф. «Детское здравоохранение в Республике Саха (Якутия): Оптимизация работы и стратегия развития». Якутск, 2003. С. 150-152.
 Г., Сухомясова А.Л., Ноговицына А Н Пренатальная ДНК-диагностика моногенных болезней в Медико-генетической консультации // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера Якутский медицинский журнал. Приложение №1. Якутск, 2005 С.68.
Г., Сухомясова А.Л., Ноговицына А Н Пренатальная ДНК-диагностика моногенных болезней в Медико-генетической консультации // Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера Якутский медицинский журнал. Приложение №1. Якутск, 2005 С.68.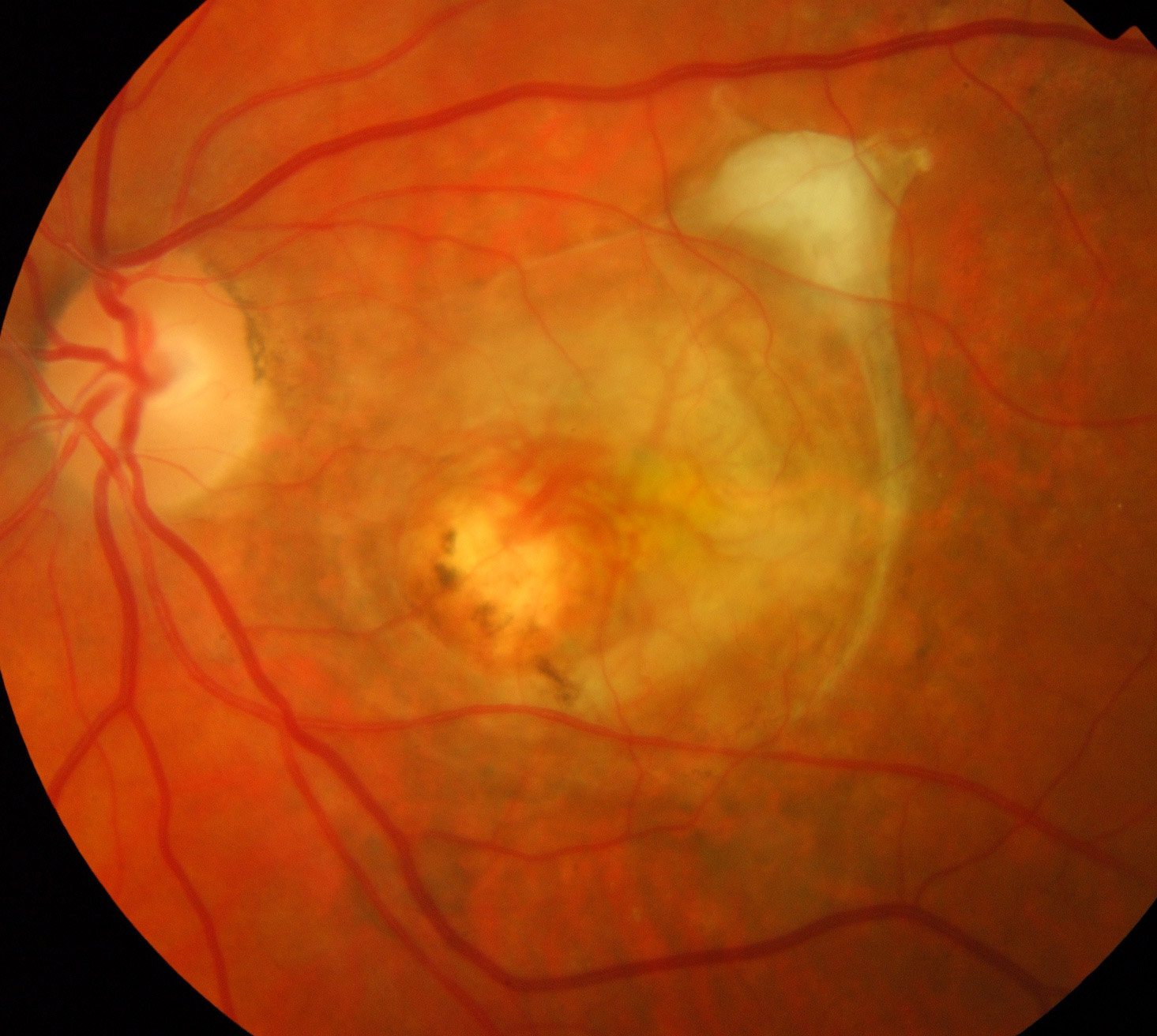 Сидорова О.Г., Кононова С.К., Степанова С.К., Федорова С.А., Сухомясова А.Л. Некоторые этические проблемы пренатальной ДНК-диагностики моногенных заболеваний в МПС РБ№1-Национального центра медицины II Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера Якутский медицинский журнал. Приложение №1 Якутск, 2005. С. 114-115.
Сидорова О.Г., Кононова С.К., Степанова С.К., Федорова С.А., Сухомясова А.Л. Некоторые этические проблемы пренатальной ДНК-диагностики моногенных заболеваний в МПС РБ№1-Национального центра медицины II Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера Якутский медицинский журнал. Приложение №1 Якутск, 2005. С. 114-115.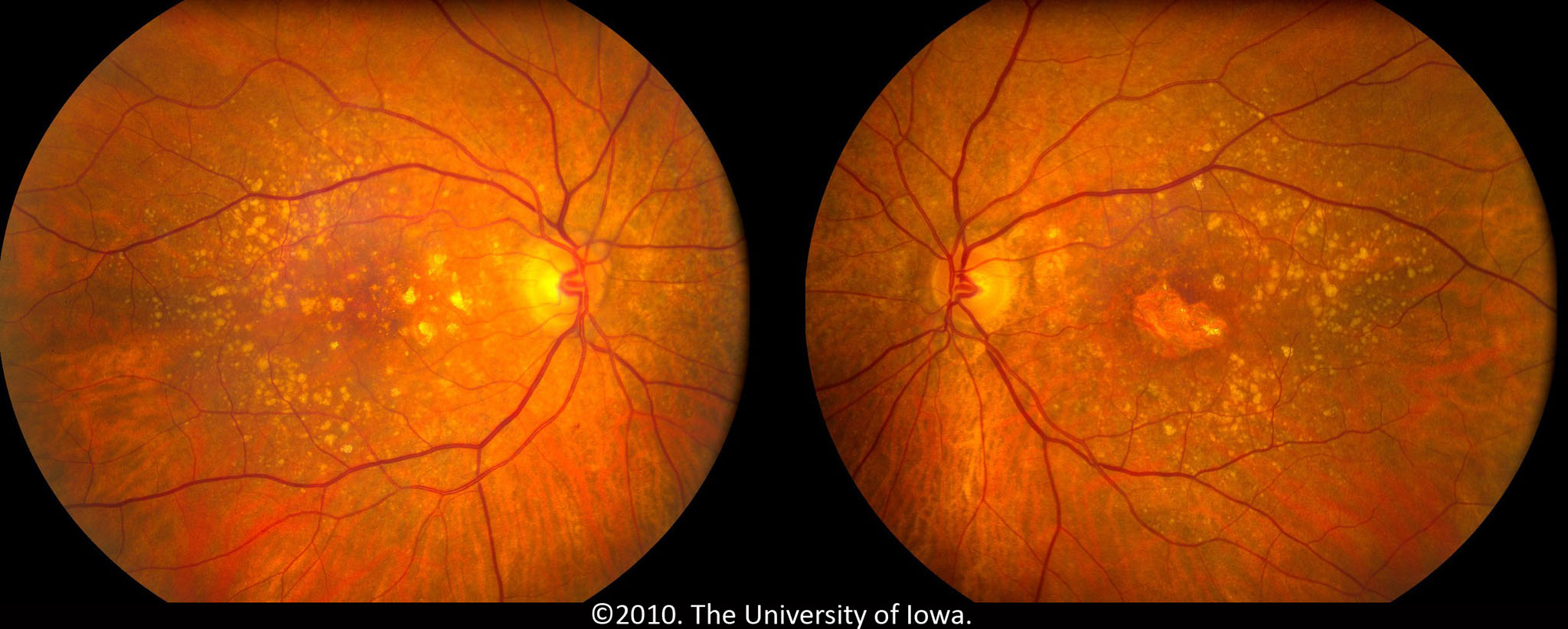 В., Ноговицына А.Н. Генетнко-демографический анализ сельских популяций Республики Саха (Якутия) // Наука и образование. 2005 №2. С. 98-103.
В., Ноговицына А.Н. Генетнко-демографический анализ сельских популяций Республики Саха (Якутия) // Наука и образование. 2005 №2. С. 98-103.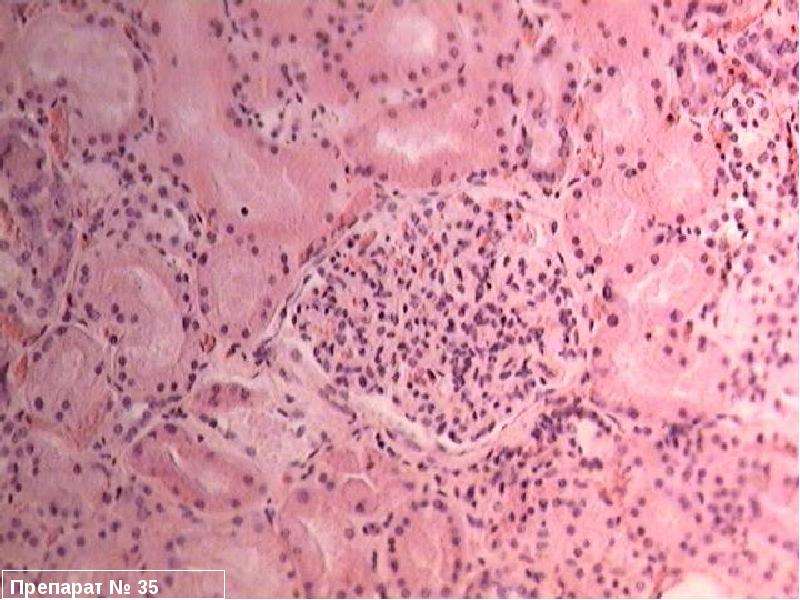 Л., Коротов М.Н., Сидорова О Г, Алексеева С П., Павлов Ф.В., Федорова С А, Степанова С К., Ноговицына А.Н. Диагностика врожденной миотонической дистрофии в Республике Саха (Якутия) // Мат. V съезда мед. генетиков. Ч.П. Медицинская генетика. 2005. №5. С. 223.
Л., Коротов М.Н., Сидорова О Г, Алексеева С П., Павлов Ф.В., Федорова С А, Степанова С К., Ноговицына А.Н. Диагностика врожденной миотонической дистрофии в Республике Саха (Якутия) // Мат. V съезда мед. генетиков. Ч.П. Медицинская генетика. 2005. №5. С. 223. P. 262.
P. 262.Список использованных сокращений:
- МД — миотоническая дистрофия
- МПС — медико-генетическая консультация
- ПААГ — полиакриламидная гель t
- ПЦР — полимеразная цепная реакция
- PC (Я) — Республика Саха (Якутия)
- CTG — цитозин-тимин-гуанин
- DMPK — миотонинпротеинкиназа
Врожденные мышечные дистрофии: классификация и диагностика | Rivier
1. Sparks S., Quijano-Roy S. , Harper A. et al. Congenital muscular dystrophy overview in: Pagon RA, Bird TD, Dolan CR, et al. Gene reviews. 1993–2001.
, Harper A. et al. Congenital muscular dystrophy overview in: Pagon RA, Bird TD, Dolan CR, et al. Gene reviews. 1993–2001.
2. Muntoni F., Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14(10):635–49.
3. Sparks S.E., Escolar D.M. Congenital muscular dystrophies. Handb Clin Neurol 2011;101:47–9.
4. Mercuri E., Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann Neurol 2012;72(1):9–17.
5. Godfrey C., Foley A.R., Clement E. et al. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011;21(3):278–85.
6. Mathews K.D., Stephan C.M., Laubenthal K. et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 2011;76(2):194–5.
Mathews K.D., Stephan C.M., Laubenthal K. et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 2011;76(2):194–5.
7. Clement E.M., Feng L., Mein R. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord 2012;22(6):522–7.
8. Hayashi Y.K., Chou F.L., Engvall E. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 1998;19(1):94–7.
9. Hara Y., Balci-Hayta B., Yoshida-Moriguchi T. A dystroglycan mutation associated with limbgirdle muscular dystrophy. N Engl J Med 2011;364(10):939–46.
10. Godfrey C., Clement E., Mein R. et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130(10):2725–35.
Brain 2007;130(10):2725–35.
11. Mercuri E., Messina S., Bruno C. et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology 2009;72(21):1802–9.
12. Ferreiro A., Quijano-Roy S., Pichereau C. et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002;71(4):739–49.
13. Mitsuhashi S., Ohkuma A., Talim B. et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am J Hum Genet 2011;88(6):845–51.
14. Tomé F.M., Evangelista T., Leclerc A. et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III 1994;317(4):351–7.
Tomé F.M., Evangelista T., Leclerc A. et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III 1994;317(4):351–7.
15. Helbling-Leclerc A., Zhan X., Topaloglu H. et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet 1995;11(2):216–8.
16. Geranmayeh F., Clement E., Feng L.H. et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20(4):241–50.
17. Lamer S., Carlier R.Y., Pinard J.M. et al. Congenital muscular dystrophy: use of brain MR imaging findings to predict merosin deficiency. Radiology 1998;206(3):811–6.
18. Okada M., Kawahara G., Noguchi S. et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69(10):1035–42.
Okada M., Kawahara G., Noguchi S. et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69(10):1035–42.
19. Allamand V., Briñas L., Richard P. et al. ColVI myopathies: where do we stand, where do we go? Skelet Muscle 2011;1:30.
20. Briñas L., Richard P., Quijano-Roy S. et al. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol 2010;68(4):511–20.
21. Nadeau A., Kinali M., Main M. et al. Natural history of Ullrich congenital muscular dystrophy. Neurology 2009;73(1):25–31.
22. Mercuri E., Lampe A., Allsop J. et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303–10.
Neuromuscul Disord. 2005;15(4):303–10.
23. Quijano-Roy S., Avila-Smirnow D., Carlier R.Y. et al. Whole body muscle MRI protocol: pattern recognition in early onset NM disorders. Neuromuscul Disord 2012;22.
24. Hicks D., Lampe A.K., Barresi R. et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology 2008;70(14):1192–9.
25. Moore C.J., Winder S.J. The inside and out of dystroglycan post-translational modification. Neuromuscul Disord 2012;22(11):959–65.
26. Wells L. The o-mannosylation pathway: glycosyltransferases and proteins implicated in congenital muscular dystrophy. J Biol Chem 2013;288(10):6930–5.
27. Kobayashi K., Nakahori Y., Miyake M. et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature, 1998;394(6691):388–92.
Kobayashi K., Nakahori Y., Miyake M. et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature, 1998;394(6691):388–92.
28. Yoshida A., Kobayashi K., Manya H. et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001;1(5):717–24.
29. Brockington M., Blake D.J., Prandini P. et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001;69(6):1198–209.
30. Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A. et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71(5):1033–43.
Am J Hum Genet 2002;71(5):1033–43.
31. van Reeuwijk J., Janssen M., van den Elzen C. et al. POMT2 mutations cause alphadystroglycan hypoglycosylation and Walker- Warburg syndrome. J Med Genet.2005 Dec;42(12):907–12.
32. Longman C., Brockington M., Torelli S et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 2003;12(21):2853–61.
33. Cirak S., Foley A.R., Herrmann R. et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain 2013;136(Pt1):269–81.
34. Barone R., Aiello C., Race V. et al. DPM2-CDG: a muscular dystrophydystroglycanopathy syndrome with severe epilepsy. Ann Neurol 2012;72(4):550–8.
Ann Neurol 2012;72(4):550–8.
35. Lefeber D.J., de Brouwer A.P., Morava E. et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PloS Genet 2011;7(12).
36. Lefeber D.J., Schönberger J., Morava E. et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 2009;85(1):76–86.
37. Willer T., Lee H., Lommel M. et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 2012;44(5):575–80.
38. Roscioli T., Kamsteeg E.J., Buysse K. et al.Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan.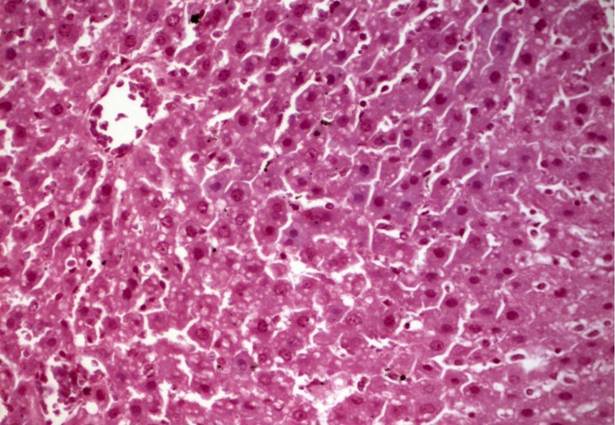 Nat Genet. 2012;44(5):581–5.
Nat Genet. 2012;44(5):581–5.
39. Manzini M.C., Tambunan D.E., Hill R.S. et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet 2012;91(3):541–7.
40. Vuillaumier-Barrot S., Bouchet-Séraphin C., Chelbi M. et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am J Hum Genet 2012;91(6):1135–43.
41. Stevens E., Carss K.J., Cirak S. et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;92(3):354–65.
42. Buysse K., Riemersma M., Powell G. et al. Missense mutations in β-1,3-Nacetylglucosaminyltransferase 1 (B3GnT1) cause Walker-Warburg syndrome. Hum Mol Genet 2013;22(9):1746–54.
Hum Mol Genet 2013;22(9):1746–54.
43. Carss K.J., Stevens E., Foley A.R. et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;93(1):29–41.
44. Yang A.C., Ng B.G., Moore S.A. et al. Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy. Mol Genet Metab 2013; 110(3):345–51.
45. Vuillaumier-Barrot S., Quijano-Roy S., Bouchet-Seraphin C. et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul Disord 2009;19(3):182–8.
46. Schara U.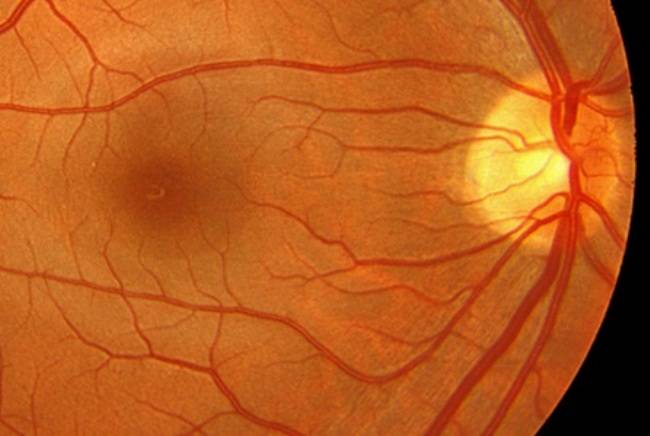 , Kress W., Bönnemann C.G. et al. The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol 2008;12(3):224–30.
, Kress W., Bönnemann C.G. et al. The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol 2008;12(3):224–30.
47. Scoto M., Cirak S., Mein R. et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76(24):2973–8.
48. Mercuri E., Pichiecchio A., Allsop J. et al. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging. 2007;25(2):433–40.
49. Quijano-Roy S., Mbieleu B., Bönnemann C.G. et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008;64(2):177–86.
50. Ben Yaou et al. Les Cahiers de myologie. 2010(3) :24–33.
2010(3) :24–33.
51. Bonne G., Quijano-Roy S. Emery–Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol 2013;113:1367–76.
52. Hattori A., Komaki H., Kawatani M. et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul Disord 2012;22(2):149–51.
53. Mercuri E., Clements E., Offiah A. et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010;67(2):201–8.
54. Makri S., Clarke N.F., Richard P. et al. Germinal mosaicism for LMNA mimics autosomal recessive congenital muscular dystrophy. Neuromuscul Disord 2009;19(1):26–8.
Мышечная дистрофия | Неврология | Заболевания
Мышечная дистрофия – это патологическое заболевание, которое характерно для людей, ведущих лежачий образ жизни. Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Виды
Мышечная дистрофия очень распространенное патологическое заболевание. Бывает детская и взрослая дистрофия мышц. Также мышечная дистрофия имеет наследственный характер (генетическая и наследственная дистрофия). По характеру и месту локализации различают:
-
инфекционную и неинфекционную; -
миотоническую; -
тазово-плечевую; -
врожденную; -
плечелопаточную;
.
Симптомы
Мышечная дистрофия прогрессирует заболевание мышечной слабости и потери трудоспособности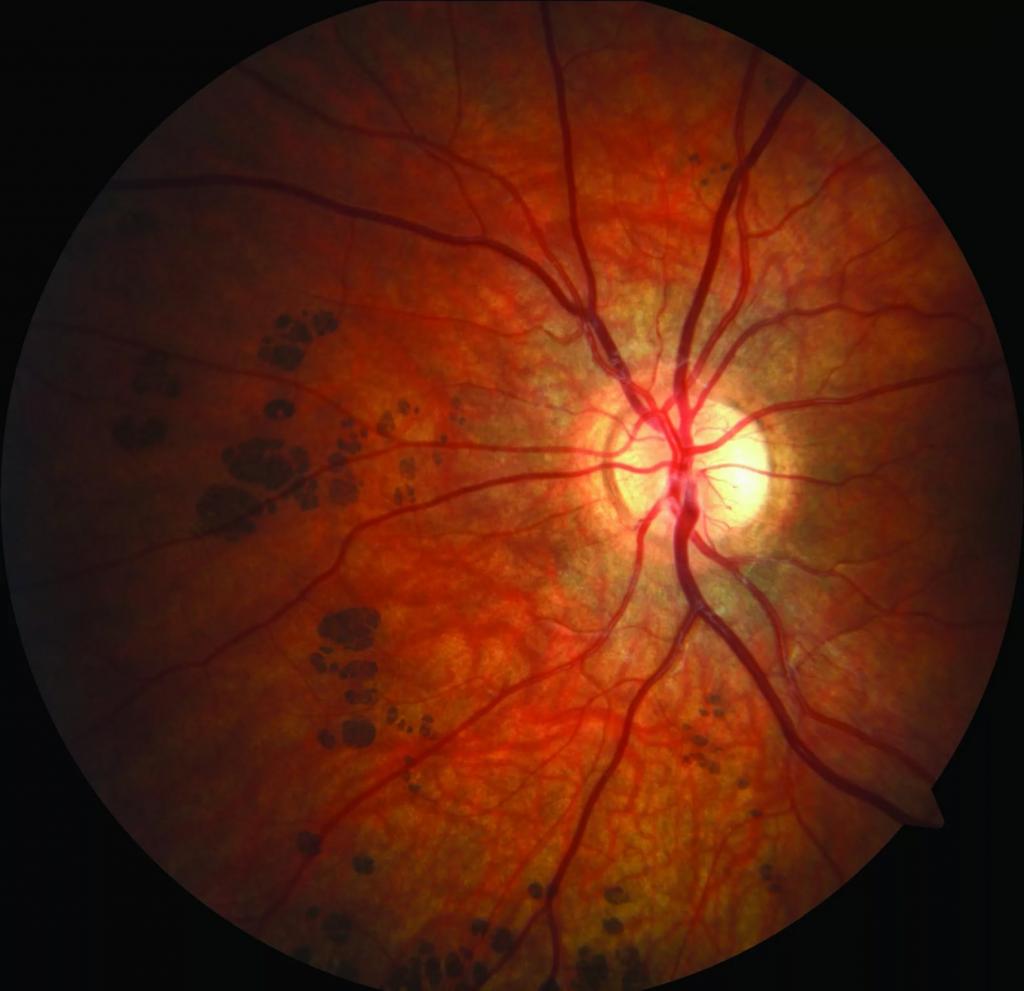 Человек, который страдает от мышечной дистрофии, начинает замечать такие характерные симптомы: быстрая усталость, вялость всего организма, тошнота, рвота, потеря аппетита, резкое снижение массы тела, в некоторых случаях потеря сознания. Мышечная дистрофия прогрессирующее заболевание. Поэтому не следует откладывать визит к доктору.
Человек, который страдает от мышечной дистрофии, начинает замечать такие характерные симптомы: быстрая усталость, вялость всего организма, тошнота, рвота, потеря аппетита, резкое снижение массы тела, в некоторых случаях потеря сознания. Мышечная дистрофия прогрессирующее заболевание. Поэтому не следует откладывать визит к доктору.
Диагностика
Обратиться к доктору следует немедленно, как только вы заметили мышечную слабость.. К методам диагностики на данном этапе развития медицины относят МРТ. Оно покажет анатомические и физиологические изменения в организме. При этом заболевании также стоит сдать общий анализ крови, мочи и кала. После проведенных диагностик доктор поставит диагноз и направит на лечение.
Лечение
Лечение проводиться с помощью комплексной терапии: консервативное лечение и физиотерапия. Еще не разработано лечение, которые бы полностью устранило это заболевание. Консервативное лечение составляет прием кортикостероидов и препаратов для улучшения мышечной массы. Физиотерапия очень распространенный метод.
Физиотерапия очень распространенный метод.
Профилактика
Лечение мышечной дистрофии продолжается его профилактикой. Очень важно после выписки с госпиталя не забыть о приписках врача. Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
МЕРОЗИНДЕФИЦИТНАЯ ВРОЖДЕННАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ВМД1А): КЛИНИЧЕСКИЙ ПРИМЕР ВРОЖДЕННОЙ МЫШЕЧНОЙ ДИСТРОФИИ С ВОВЛЕЧЕНИЕМ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | Клочкова
1. Rivier F., Meyer P., Walther-Louvie U., Mercier M., Echenne B., Quijano-Roy S. Врожденные мышечные дистрофии: классификация и диагностика. Нервно-мышечные болезни. 2014; 1: 6–20.
2. Tome F. M., Evangelista T., Leclerc A., Sunada Y., Manole E., Estournet B., Barois A., Campbell K. P. , Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994; 317 (4): 351–7.
, Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994; 317 (4): 351–7.
3. Hillaire D., Leclerc A., Faure S., Topaloglu H., Chiannilkulchai N., Guicheney P., Grinas L., Legos P., Philpot J., Evangelista T. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum Mol Genet. 1994; 3 (9): 1657–61.
4. Clement E. M., Feng L., Mein R., Sewry C. A., Robb S. A., Manzur A. Y., Mercuri E., Godfrey C., Cullup T., Abbs S., Muntoni F. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord. 2012; 22 (6): 522–7.
5. Darin N., Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord. 2000; 10 (1): 1–9.
Neuromuscul Disord. 2000; 10 (1): 1–9.
6. Norwood F. L., Harling C., Chinnery P. F., Eagle M., Bushby K., Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009; 132 (Pt. 11): 3175–86.
7. Peat R. A., Smith J. M., Compton A. G., Baker N. L., Pace R. A., Bur-kin D. J., Kaufman S. J., Lamande S. R., North K. N. Diagnosis and etiology of congenital muscular dystrophy. Neurology. 2008; 71 (5): 312–21.
8. Аверьянов Ю. Н. Врожденная мышечная дистрофия с лейкоэнцефалопатией. Журнал неврологии и психиатрии им. C. C. Корсакова. 1993; 5: 27–29.
9. Руденская Г. Е., Галкина В. А., Дунаевская Г. Н. Редкие формы наследственных прогрессирующих мышечных дистрофий с контрактурами. Теоретические и прикладные проблемы мед. генетики. 1993. С. 105–119.
Теоретические и прикладные проблемы мед. генетики. 1993. С. 105–119.
10. Руденская Г. Е., Дадали Е. Л., Ситников В. Ф. Наследственная сочетанная церебромышечная патология в детском возрасте. Организационные и клинические проблемы детской неврологии и психиатрии. 1993. С. 253–255.
11. Дадали Е. Л., Руденская Г. Е., Щагина О. А., Тибуркова Т. Б., Сухоруков В. С., Харламов Д. А., Поляков А. В. Мерозин-дефицитная врожденная мышечная дистрофия (ВМД1А). Журнал неврологии и психиатрии им. C. C. Корсакова. 2010; 110 (3): 83–89.
12. Комарова Н. В., Тибуркова Т. Б., Щагина О. А., Дадали Е. Л., Руденская Г. Е., Поляков А. В. Врожденная мышечная дистрофия, мерозин-негативная (ВМД1А) у российских больных. Материалы VI съезда Российского общества медицинских генетиков. Медицинская генетика (прил. к № 5). 2010. 88 с.
Медицинская генетика (прил. к № 5). 2010. 88 с.
13. Mendell J. R., Boue D. R., Martin P. T. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol. 2006; 9 (6): 427–43.
14. Zhang X., Vuolteenaho R., Tryggvason K. Structure of the human laminin alpha2-chain gene (LAMA2), which is affected in congenital muscular dystrophy. J Biol Chem. 1996; 271 (44): 27664–9.
15. Gawlik K. I., Durbeej M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle. 2011; 1 (1): 9.
16. Geranmayeh F., Clement E., Feng L. H., Sewry C., Pagan J., Mein R., Abbs S., Brueton L., Childs A. M., Jungbluth H., De Goede C. G., Lynch B. , Lin J. P., Chow G., Sousa Cd, O’Mahony O., Majumdar A., Straub V., Bushby K., Muntoni F. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010; 20 (4): 241–50.
, Lin J. P., Chow G., Sousa Cd, O’Mahony O., Majumdar A., Straub V., Bushby K., Muntoni F. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010; 20 (4): 241–50.
17. Philpot J., Bagnall A., King C., Dubowitz V., Muntoni F. Feeding problems in merosin deficient congenital muscular dystrophy. Arch Dis Child. 1999; 80 (6): 542–7.
18. Tezak Z., Prandini P., Boscaro M., Marin A., Devaney J., Marino M., Fanin M., Trevisan C. P., Park J., Tyson W., Finkel R., Garcia C., Angelini C., Hoffman E. P., Pegoraro E. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat. 2003; 21 (2): 103–11.
19. Jones K. J., Morgan G., Johnston H. , Tobias V., Ouvrier R. A., Wilkinson I., North K. N. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001; 38 (10): 649–57.
, Tobias V., Ouvrier R. A., Wilkinson I., North K. N. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001; 38 (10): 649–57.
20. Leite C. C., Lucato L. T., Martin M. G., Ferreira L. G., Resende M. B., Carvalho M. S., Marie S. K., Jinkins J. R., Reed U. C. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol. 2005; 35 (6): 572–9.
21. Philpot J., Pennock J., Cowan F., Sewry C. A., Dubowitz V., Bydder G., Muntoni F. Brain magnetic resonance imaging abnormalities in merosin-positive congenital muscular dystrophy. Eur J Paediatr Neurol. 2000; 4 (3): 109–14.
22. Bonnemann C. G., Wang C. H., Quijano-Roy S., Deconinck N., Bertini E., Ferreiro A. , Muntoni F., Sewry C., Beroud C., Mathews K. D., Moore S. A., Bellini J., Rutkowski A., North K. N. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014; 24 (4): 289–311.
, Muntoni F., Sewry C., Beroud C., Mathews K. D., Moore S. A., Bellini J., Rutkowski A., North K. N. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014; 24 (4): 289–311.
23. Oliveira J., Santos R., Soares-Silva I., Jorge P., Vieira E., Oliveira M. E., Moreira A., Coelho T., Ferreira J. C., Fonseca M. J., Barbosa C., Prats J., Ariztegui M. L., Martins M. L., Moreno T., Heinimann K., Barbot C., Pascual-Pascual S. I., Cabral A., Fineza I., Santos M., Bronze-da-Rocha E. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. 2008; 74 (6): 502–12.
24. Vainzof M., Richard P., Herrmann R., Jimenez-Mallebrera C., Talim B., Yamamoto L. U., Ledeuil C., Mein R., Abbs S., Brockington M., Romero N. B., Zatz M., Topaloglu H., Voit T., Sewry C., Muntoni F., Guicheney P., Tome F. M. Prenatal diagnosis in laminin alpha2 chain (merosin)-deficient congenital muscular dystrophy: a collective experience of five international centers. Neuromuscul Disord. 2005; 15 (9–10): 588–94.
B., Zatz M., Topaloglu H., Voit T., Sewry C., Muntoni F., Guicheney P., Tome F. M. Prenatal diagnosis in laminin alpha2 chain (merosin)-deficient congenital muscular dystrophy: a collective experience of five international centers. Neuromuscul Disord. 2005; 15 (9–10): 588–94.
25. Matsumura K., Yamada H., Saito F., Sunada Y., Shimizu T. Peripheral nerve involvement in merosin-deficient congenital muscular dystrophy and dy mouse. Neuromuscul Disord. 1997; 7 (1): 7–12.
26. Vigliano P., Dassi P., Di Blasi C., Mora M., Jarre L. LAMA2 stop-codon mutation: merosin-deficient congenital muscular dystrophy with occipital polymicrogyria, epilepsy and psychomotor regression. Eur J Paediatr Neurol. 2009; 13 (1): 72–6.
Нервная трофика и нейрогенные дистрофии
Трофика
(питать — trophe) — совокупность процессов
клеточного питания, обмена веществ,
направленная на сохранение структуры
и функции органа и ткани целостного
организма.
Дистрофия
— неправильное, нарушенное, измененное
питание и обмен веществ, ведущие к
качественным и количественным нарушениям
структуры и функции.
Трофика.
нейрогенная дистрофия
— типовой патологический процесс.
Нарушения нервов,
ЦНС, спинного мозга сопровождается
следующими изменениями:
кожа — атрофия
гиперемия
гиперкератоз
кости — остеопороз
(Зудковский)
мышцы — атрофия.
Примеры нейрогенных
дистрофий:
1. 1852 г. Мелатон —
тяжелое поражение стопы — перфорирующая
язва стопы:
при третичном
сифилисе
сирингомиелии
спина бифида
Нарушение
чувствительности в области стопы опоры
(и гиперкератоз). Затем язва — форма
конуса — увеличивается вглубь и вширь.
2. Норма (водяной рак
— влажная гангрена щек, наружных половых
органов, мочки уха и прямой кишки — при
повреждении нервов).
3. Поражение одного
парного органа при вовлечении в
патологический процесс другого.
Симпатическая
офтальная — слепота второго глаза при
травме первого.
Доказательства:
1824 г. — Мажанди —
перерезал 1-ю веточку тройничного нерва:
язвенный карпит
слепота
1850 г. — Самюэль —
раздражение гассерова узла — язвенный
кератит и слепота.
1856 г. — Сеченов —
поддержка анатомической, химической и
функциональной целостности ткани — это
функции нервной системы.
1878 г. — Гайденгайн —
раздражение нервов, иннервирующих
слюнную железу:
а) n. sympaticus — мало
слюны, густая слюна
б) барабанной струны
(парасимпатическая нервная система) —
много слюны, жидкая слюна.
Теории:
1. Травматическая
теория — при перерезке чувствительных
нервов ткань теряет связь с центром и
становится беззащитной перед повреждающими
воздействиями окружающей Среды.
2. Сосудистая теория
— при слабом воздействии на нерв нарушается
тонус сосудов (спазм — ишемия ткани;
парез — венозное полнокровие).
3. Павлов — нерв,
раздражение которого усиливает сокращение
сердца (усиливающий нерв Павлова —
изменяет обмен веществ в сердечной
мышце).
По Павлову любой
орган находится под контролем 3-х нервов:
1. Функционального
2. Сосудистого
3. Трофического
Павлов создал школу
исследователей:
а) Орбели (+Генецинский)
— концепция адаптационно-трофической
функции симпатической нервной системы:
Опыты:
мышца после утомления
при стимуляции симпатической нервной
системы опять работала
Вывод:
Трофическая функция
присуща определенным образованиям
нервной системы.
б) Сперанский. Его
выводы:
Не отдельные
образования н/с выполняют трофическую
функцию, а вся н/с выполняет эту функцию.
в) Вишневский —
перерезка нерва дополнялась раздражением
центрального отрезка перерезанного
нерва (нейрогенная дистрофия в 100% случаев
наблюдалась).
г) Голуб — перерезка
нерва дополнялась раздражением
периферического отрезка нерва (80%
нейрогенной дистрофии).
д) Сперанский 0
буксация мозга — в области foramen magnum иглой
извлекали ликвор из спинномозгового
канала и опять возвращали. До тех пор,
пока ликвор не опалесцирует.
Появлялись симметричные
повреждения в результате раздражения
спинного мозга.
е) Сперанский —
шариковая операция — стеклянный шарик
на область турецкого седла (полукольцо
раздражает только гипоталамус, не
затрагивая гипофиз):
100% нейрогенных
дистрофий в опыте симметричные
повреждения.
стандартная форма
нейрогенной дистрофии
Проявления стандартной
формы нейрогенной дистрофии:
облысение
язвы
изменения в легких,
костях (ломкие или размягчаются).
У собак:
Шерсть теряет блеск
— тусклая, матовая, выпадает (гнездовидное
облысение).
Ротовая полость:
гингивиты
стоматиты
кариес множественный
крошатся и выпадают
зубы
отслойка десны от
корня зуба — парадонтоз
(альвеолярная
пиорея)
ЖКТ:
кровоизлияния
множественные язвы:
pylorus
duodenum
баугиниевой заслонки
rectum
Легкие:
картина крупозной
пневмонии
Кости:
крошатся, ломаются
или размягчаются
Карташова и Маклей:
фенол, кретоновое
масло в отверстие зуба + пломба; затем
удаляли зуб — развивалась форма нейрогенной
дистрофии.
Все опыты показали,
что трофическая функция присуща:
ЦНС
эфферентному звену
н/с
афферентному звену
н/с
Повреждение
гипоталамуса и афферентного звена
наиболее часто вызывает нейрогенную
дистрофию.
1. Перерезка афферентных
нервов нарушает связь периферических
тканей с центром.
2. Центральный отрезок
при перерезке нерва испытывает
раздражающее воздействие от рубцовой
ткани — необычные импульсы в ЦНС.
3. Раздражение
периферического отрезка перерезанного
отрезка — антидромное распространение
импульсов.
4. Лишение чувствительной
иннервации — безззащитность перед
различными воздействиями.
Механизм
контролирующего влияния н/с на трофику:
Клетка — сложная
саморегулирующаяся система, процессы
в которой осуществляются по принципу
разветвленных цепных реакций с
автоматически регулированием на основных
этапах их осуществления.
Клетка — не замкнутая
система.
Ее жизнедеятельность
регулируется интегративными системами
организма.
1. Во внешнем стимуле
нуждаются не все процессы, а только
первичные инициальные звенья.
2. Внутриклеточные
процессы подлежат внешнему контролю
не на всех этапах, а на узловых этапах
разветвленных реакций.
3. Влияние интегративных
систем реализуется через внутриклеточные
и внутритканевые процессы регулирования.
4. Клетка ткани
противостоит (!) действию интегративных
систем.
Язва — далеко зашедший
дистрофический процесс.
Раньше возникает
внутриклеточная дистрофия (Крыжановский).
Предпосылка к
развитию нейрогенных дистрофий —
дисбаланс между функциональной нагрузкой
и ее трофическим обеспечением.
В гнезде этих
нарушений — изменение физиологической
регенерации внутриклеточных структур.
Если контроль
регенерации со стороны н/с хороший/ то
клетка адаптируется.
Посредник — генетический
аппарат — экспрессия определенных
структурных генов — ускоренное обновление
внутриклеточных структур.
Меняется не только
интенсивность, но и качественная сторона
обменных процессов:
белая (быстрая) мышца
— преобладает анаэробное окисление
глюкозы, перерезали нервы, идущие к
мышцам и затем перекрестно реиннервировали
мышцы.
В результате: красная
мышцы становилась белой (обмен веществ
переключался на гликолиз), белая мышцы
становилась красной.
При нарушении
иннервации ткани все больший удельный
вес имеют явления гликолиза.
Структурные
изменения при нейрогенных дистрофиях:
Митохондрии:
уменьшение количества
их
набухание и разрушение
крист
просветление
матрикса
гибель
Возврат к эмбриональному
состоянию:
обмен веществ —
гликолиз
уменьшается
количество изоферментов (изоферментное
упрощение).
Антигенная: дивергенция
реверсия
упрощение
Клетка становится
чужеродной для организма — аутоиммунные
процессы.
Механизм
трофического контроля за трофикой
тканей:
1) медиаторы могут
оказывать:
немедиаторное
действие — активное включение в обменные
процессы в клетке.
2) Комедиаторы —
нейромодуляторы регуляции процессов:
рецепторов
мембранных структур
3) Трофогены и
макромолекулярные вещества белковой,
пептидной и нуклеиновой природы,
осуществляющие собственно трофическое
влияние на нервные клетки и иннервируемые
ими ткани. В клетке трофогены путем
эндоцитоза либо:
а) ассимилируются
и участвуют в структурно-метаболических
процессах
б) либо действуют
на генетический аппарат клетки (экспрессия
или супрессия).
Трофогены образуются:
1. В нейронах и
аксоплазматическим током поступают в
периферические ткани.
2. В тканях — рецепторах,
с ретроградным аксоплазматическим
током поступают к нейрону.
3. В глиальных и
шванновский клетках.
В качестве трофогенов
могут выступать:
гормоны
эндорфины
субстанция Р.
Дистрофия может
развиться в результате следующих причин:
1. Нарушение выработки
ли нарушение поступления в ткань
медиатора
комедиатора
трофогена
2. Влияние патотрофогенов
— веществ, индуцирующих патологически
устойчивые изменения клеток.
Наследственная
дегенерация мышц (патотрофогены
образуются в нейронах, вызывают дистрофию
мышц (болезнь Дюшена).
3. Отторжение
трансплантата — также вследствие
нарушения трофики.
ЦНС влияние на метаболизм
повреждающий агент
чувствительный нерв
эфферентный нерв
1.
Нарушенная афферентация
2.
Антидромное влияние на ткани
Нарушение транспорта и выделения
медиатора и веществ медиаторной природы
(аксональный ток), вещества трофики
Изменение мембранных процессов
активности ферментов, работы, генома.
Перестройка метаболических процессов,
возврат к эмбриональному
Дедифференциация
Снижение митотической активности
Извращение чувствительности гуморальным
фактором
Нарушение функции, структуры, метаболизма,
гибель ткани
Лечебные принципы
в медицине и их научные основы:
Лечение — совокупность
мероприятий с целью восстановления
здоровья и устранения или облегчения
страданий больного человека.
Первобытнообщинный
строй:
шаманы, колдуны,
знахари
примитивизм
женьшень
лимон по форме
напоминает сердце — полезен при сердечных
болезнях
ревень, касторовое
масло, ромашка, наперстка.
Гиппократ. заслуги:
1) Заложил основы о
двойственной природе болезни.
2) Целостность
сложения организма
3) принципы:
1. Прежде всего не
вреди
2. Природа исцеляет,
врач только лечит.
3. Лечить больного,
а не болезнь
4. Прогностическая
оценка заболевания.
4 сока:
sanguis
flegma
черная желчь
желтая желчь
Правильное их
смешение (кразис) — обеспечивает здоровье.
Неправильное их
смешение (рискразия) — вызывает болезнь.
Болезни возникают
от дурных соков.
1. Ревульсивный
(аллиопатический) принцип:
кровопускание
рвотное
слабительное
мочегонные
банки
горчичники
пиявки
Заволока — продевать
кожу иглой со святой корпией.
Бюсе — кровопускание
по поводу насморка.
2. Аллопатический
принцип.
Гален — основоположник.
Противоположное
лечится противоположным (contracio contratius):
если давление
высокое, его нужно понижать
если высокая
температура — снижать температуру
если нет стула —
давать слабительное.
3. Гомеопатический
принцип.
Ганеман — основоположник.
У него была малярия, он ввел себе большую
дозу акрихина, и вместо снятия приступа,
была высокая лихорадка. Подобное лечится
подобным —
Лечить ничтожно
малыми дозами веществ, вызывающих в
больших дозах такие же эффекты, как и
проявления заболевания, которое лечится.
1988 — доказано, что
дегенерация базофилов может быть вызвана
сильным разбавлением антител. Вода
может хранить память о содержащейся в
ней когда-то БАВ.
Сарчук — диагностика
с помощью определения БАВ в жидкостях
организма.
4. Этиологический
принцип.
Листер, Кох, Пастер,
Мечников. Воздействие на причину болезни
(преимущественно на микроорганизмы).
антибиотики
сульфаниламиды
Но не все болезни
можно так лечить:
1) Не знаем причин
многих заболеваний
2) не знаем противоядий
от известных причин болезней
3) не всегда устранение
причин означает выздоровление
4) Микроорганизмы
быстро приспосабливаются к лекарственным
средствам.
Масса побочных
эффектов.
5. Патогенетический
принцип:
Лечение должно быть
направлено на основное звено в механизме
болезни — инсулин вызывает снижение
глюкозы в крови, стимулирует утилизацию
глюкозы — комиссуротомия при пороках
сердца приводит к ликвидации порока.
а) подавление,
устранение, ликвидация патологических
проявлений болезни
б) создание оптимальных
условий для развития механизмов
компенсации
6. Симптоматический
принцип.
Цель — устранить
тяжелый симптом заболевания (боль и
др.).
7. Лечение по жизненным
показаниям.
«Созвездие» — офтальмологическая клиника в Сыктывкаре
Лечение заболеваний сетчатки глаза
СЕТЧАТКА — это тонкий слой нервной ткани, расположенный с внутренней стороны задней части глазного яблока и поглощающий свет. Сетчатка глаза отвечает за восприятие изображения, которое проецируется на нее при помощи роговицы и хрусталика, и преобразование его в нервные импульсы, которые затем передаются в головной мозг. Важнейшей частью сетчатки является макула, отвечающая за самое качественное зрение. Заболевания макулы могут значительно снизить зрение (до 10% и меньше).
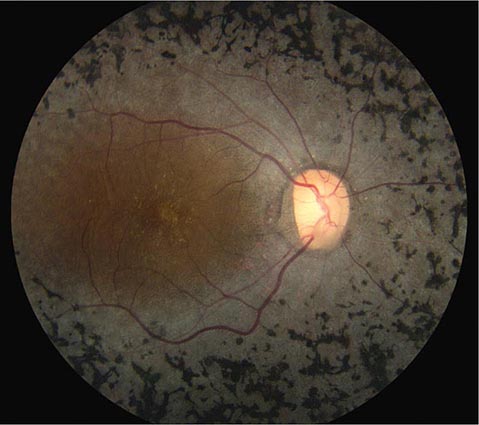
Картина глазного дна в норме: 1) Диск зрительного нерва
2) Желтое пятно (макулярная область)
ЗАБОЛЕВАНИЯ СЕТЧАТКИ
Заболевания сетчатки могут быть самыми разнообразными, так к ним относятся: острые нарушения кровообращения в центральной вене самой сетчатки, болезни Коатса и Илса, дистрофия центральных отделов сетчатки, периферическая дистрофия сетчатки. Любые заболевания сетчатки существенно влияют на состояние зрения. Наиболее серьезным из них является отслойка сетчатки, данное заболевание сетчатки характеризуется отделением сетчатой оболочки глаза от самого глазного яблока, в частности от сосудистой оболочки, расположенной непосредственно под сетчаткой глаза.
ОСНОВНЫЕ» ГРУППЫ РИСКА»
-
люди со средней и высокой степенью близорукости; -
беременные женщины; -
пожилые люди; -
пациенты, страдающие сахарным диабетом; -
пациенты, страдающие гипертонической болезнью.
ПРИЧИНЫ ЗАБОЛЕВАНИЯ СЕТЧАТКИ
Повреждение сетчатки может возникнуть в результате:
- Заболевания глаз: дальнозоркость, близорукость, дистрофические и воспалительные процессы
- Травмы головного мозга и глаза
- Врожденные изменения самой сетчатки глаза, вызванные различными родовыми травмами и имеющие наследственную предрасположенность
- Операции, отравления, стрессы
- Ряда заболеваний, которые не связаны напрямую с сетчаткой глаза: атеросклероз, сахарный диабет, заболевания почек, болезни крови, ревматизм, гипертоническая болезнь, менингит
КАК ПРОТЕКАЕТ ПРОЦЕСС ЗАБОЛЕВАНИЯ СЕТЧАТКИ
Ряд проблем с сетчаткой глаза связан с дефектами в кровообращении. Сосуды сетчатки при этом становятся расширенными и извилистыми, или же, наоборот, уплотненными, суженными и неспособными питать сетчатку. Помимо этого, нередко возникает частичная или полная закупорка вен центральной области сетчатки. В результате чего возникают небольшие кровоизлияния, сетчатка начинает отекать, зрительный нерв повреждается, и информация от глаз в мозг перестает передаваться. В некоторых случаях у молодых людей развиваются дистрофии сетчатки, так происходит гибель «колбочек» — «палочек». Кроме того существует возможность возникновения всевозможных воспалительных изменений сетчатки, сопровождающихся поражением мелких сосудов с последующими множественными кровоизлияниями. Чаще всего подобные заболевания сетчатки возникают у молодых парней. Разрывы и отслойки сетчатки возникают чаще всего в результате, каких бы то ни было других заболеваний сетчатки и травм, опухолей и физической нагрузки.
ПРИЗНАКИ ЗАБОЛЕВАНИЯ СЕТЧАТКИ.
Основными признаками заболевания сетчатки глаза являются: неуклонное падение зрения, у человека перед глазами начинает появляться «туманная завеса» и своеобразные искры, в результате повреждения сетчатки нередко выпадают отдельные поля зрения, чаще всего сбоку. Человек может страдать от мигрени, головокружений, в некоторых случаях наблюдается онемение и посинение пальцев.
ДИАГНОСТИКА ЗАБОЛЕВАНИЯ СЕТЧАТКИ
Характер заболеваний сетчатки глаза может быть установлен врачом при помощи проверки глазного дна и остроты зрения при помощи специальной лампы и зеркала. Помимо этого окулист нередко назначает ультразвуковое исследование глаз и специальную ангиографию сосудов, данные исследования являются абсолютно безболезненными и проводятся на специальных компьютерах.
ЛЕЧЕНИЕ ЗАБОЛЕВАНИЯ СЕТЧАТКИ.
Лечение любых заболеваний сетчатки в первую очередь зависит от причин возникновения изменений в ней. Чаще всего общее лечение основных заболеваний способствует улучшению самого состояния сетчатки глаза. В качестве лечения нередко назначаются дополнительные средства, способные улучшить питание сосудистой системы сетчатки, витамины и средства, участвующие в рассасывании кровоизлиянии, сосудорасширяющие и противовоспалительные препараты. Лекарства, как правило, назначаются в виде таблеток, уколов или капель. Разрывы и отслойки сетчатки требуют безотлагательного хирургического лечения, которое должно осуществляться на начальных сроках.
Лечение заболеваний сетчатки в клинике «Созвездие» осуществляется медикаментозно или с помощью лазеркоагуляции на лазере.
Выбор методики осуществляется после проведения необходимых диагностических процедур.
Процедура лазеркоагуляции сетчатки проводится амбулаторно (без госпитализации), под местной анестезией (после закапывания анестетика). На глаз устанавливается специальная линза (трехзеркальная линза Гольдмана), позволяющая сфокусировать лазерное излучение на любой участок глазного дна. Лазерное излучение подается через щелевую лампу, и хирург имеет возможность контролировать ход операции.
Лазер обладает очень высокой точностью и используется для создания сращений между сетчатой и сосудистой оболочкой глаза (т.е. укрепления сетчатки).
Сущность процедуры состоит в «приваривании» сетчатки к подлежащим тканям и создании прочной спайки, которая не даст сетчатке отслоиться. На сетчатку наносятся 3-5 рядов лазерных коагулятов вокруг патологического очага. На месте коагулята в дальнейшем формируется хориоретинальное сращение, блокирующее развитие отслойки сетчатки.
Лазеркоагуляция практически безболезненна — от пациета требуется только усидчивость, так как лазерный хирург работает при большом увеличении, тщательно контролируя нанесение коагулятов на ткань сетчатки.
Для образования прочной хориоретинальной спайки требуется время – около 10-14 дней. Отсутствие прогрессирования отслойки сетчатки, ее распространения за границу коагулятов служит основанием считать данную лазеркоагуляцию успешной.
После успешной профилактической лазеркоагуляции не реже чем 1 раз в 6 месяцев, а по рекомендации врача и чаще, необходимо производить профилактический осмотр периферии глазного дна обоих глаз с широким зрачком на предмет появления новых зон дегенераций сетчатки или возникновения истончения и разрывов сетчатки в ранее выявленных зонах. Профилактическая лазеркоагуляция таких участков позволит в несколько раз снизить риск развития отслойки сетчатки и избежать потери зрительных функций.
Записаться на диагностику, узнать цены, задать интерисующие вас вопросы, вы можете по телефонам в г.Сыктывкаре (8212) 515-866 или 575-866, либо на нашем интернет сайте www.sozvezdie11.ru
ФГБНУ НЦПЗ. ‹‹Патология психического развития››
Среди наследственных заболеваний, затрагивающих мышечную и нервную системы, очень велика доля тех форм, которые могут сопровождаться интеллектуальным дефектом. При некоторых из этих заболеваний интеллектуальный дефект имеет врожденный непрогредиентный характер, при других — проявляется очень рано и медленно прогрессирует. Итак, наследственные
неврологические и нервно-мышечные заболевания должны встречаться и встречаются в контингенте умственно отсталых детей. Только среди врожденных или рано начавшихся атаксий и параплегии насчитывается не менее 40 генных дефектов, сопровождающихся умственной отсталостью [Калмыкова Л. Г., 1976]. Однако большинство из них встречается очень редко; многие описаны только в единичных семьях, часто у потомков родителей, находящихся в кровно-родственном браке.
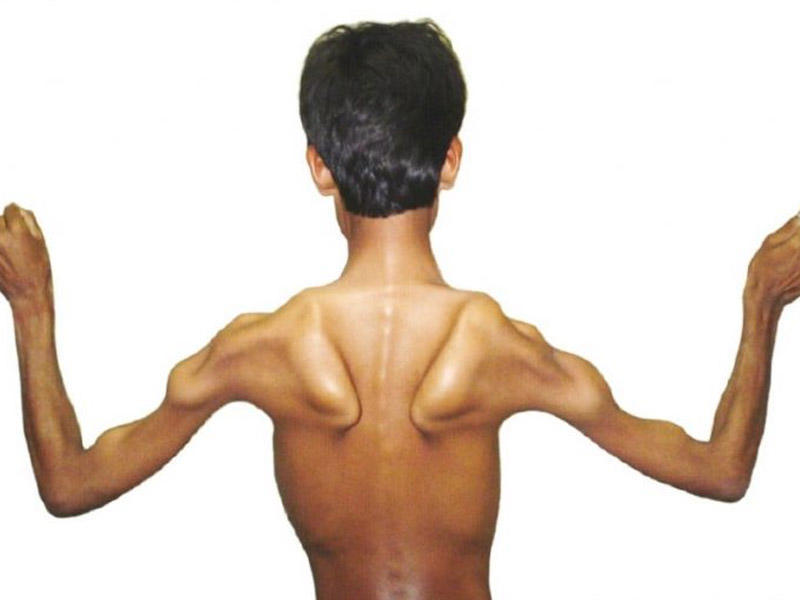
По-нашему опыту, в контингенте умственно отсталых детей, обследованных в медико-генетической консультации, вся группа прогрессирующих заболеваний нервной и мышечной систем в целом составляет небольшую долю. Самые частые из них — миопатия Дюшенна и миотоническая дистрофия. Дети, страдающие этими заболеваниями, до развития выраженных проявлений со стороны мышечной системы нередко попадают под наблюдение детского психиатра с диагнозом «олигофрения».
Миотоническая дистрофия (атрофическая миотония, дистрофическая миотония, болезнь Штейнерта — Баттона). Впервые заболевание было подробно описано в 1909 г. Его частота варьирует от 1:50 000 до 1:10 000. Необычно высокая распространенность миотонической дистрофии (1:3000) обнаружена в Северной Швеции.
Клиническая картина. Заболевание очень резко варьирует по клинической картине и срокам начала. При типичной картине оно начинается на 2—4-м десятилетии жизни и характеризуется развитием следующих симптомов: миотонии, атрофии мышц, особенно лица, шеи, дистальных отделов конечностей, катаракты, раннего облысения, атрофии гонад. Однако умственная отсталость встречается при внутриутробном начале этого заболевания.
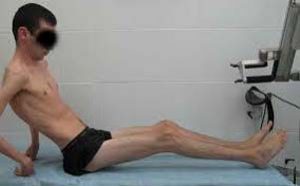
Самый характерный симптом внутриутробного поражения — резко выраженный двусторонний парез лицевой мускулатуры, так называемая лицевая диплегия: лицо амимично, рот открыт, больной не может наморщить лоб, зажмурить глаза, улыбнуться. При этом родители нередко отмечают такое лицо у ребенка с первых недель жизни. Дети очень плохо сосут, не улыбаются. Для врожденной миотонической дистрофии характерна также общая мышечная гипотония. Мышцы мягкие, тестоватые на ощупь. Сухожильные рефлексы резко снижены или отсутствуют. Дыхание вследствие поражения дыхательной мускулатуры учащенное, поверхностное. Глотание часто затруднено, крик очень слабый. Нередко встречаются врожденные уродства — косолапость, гиперостозы и асимметрия черепа, артрогрипоз; небо всегда резко сужено, оно высокое, арковидное.
Дистрофические и особенно миотонические симптомы, характерные для заболевания при его обычном, позднем начале, у детей с внутриутробной формой болезни выявляются только в более старшем (дошкольном) возрасте, но и тогда в отличие от пораженных взрослых бывают выражены слабо. Катаракты (весьма частый симптом при позднем начале заболевания) у детей, как правило, не бывает.
У детей старшего возраста уже появляются и атрофические изменения в мышцах. Можно отметить и такие характерные для взрослых больных симптомы, как западения в области височных ямок вследствие атрофии мышц, легкая атрофия мышц голеней. Иногда отмечается своеобразный рост волос на лбу — залысины с обеих сторон. Миотонические симптомы у детей школьного возраста иногда уже легко выявляются при клиническом осмотре: дети медленно, с трудом разжимают сжатые в кулак пальцы при повторном действии.
Умственная отсталость при внутриутробном начале заболевания встречается у всех пораженных: IQ от 20 до 70 ед.
Вопрос о наличии прогредиентности в интеллектуальном снижении у детей является спорным. Истинная прогредиентность на клиническом уровне отсутствует. Более того, отмечается некоторая положительная динамика в развитии моторики и речи. Больным свойственны выраженная вялость, беспомощность даже при отсутствии заметных двигательных нарушений. Как правило, они робки и послушны.
Очень характерна тяжелая дизартрия вследствие изменений лицевой мускулатуры и мышц языка. При этом речь имеет выраженный носовой оттенок из-за фарингеального пареза.
Диагноз ставят на основании клинической картины; окончательно он может быть подтвержден в сомнительных случаях электромиографией, которая выявляет изменения, характерные для миотонии, а также молекулярной диагностикой мутантного гена.
Заболевание вызывается аутосомно-доминантным геном с резко варьирующей экспрессивностью. Мутантный ген передается практически всегда матерью. Наследование дефекта от матерей, возможно, связано с резкими эндокринными нарушениями у пораженных мужчин, приводящими к бесплодию. Механизм действия мутантного гена и патогенез умственной отсталости неясны. Но определены структура и локализация мутации. Заболевание вызывается (аналогично синдрому Мартина — Белл) амплификацией, т. е. повышением числа нуклеотидов в определенном регионе одной из хромосом (19ql3,3). Число повторов нуклеотидов увеличивается при передаче мутации из поколения в поколение. Тяжесть заболевания четко коррелирует с численностью этих повторов. Наибольшее их число определяется при врожденной тяжелой форме заболевания [Brook J. D. et al., 1992]. Выявленный механизм объясняет феномен антиципации — утяжеления и все более раннего начала болезни в нисходящих поколениях.
Лечение. Специфического лечения не существует. При наличии атрофии применяют анаболические стероиды (ретаболил, неробол), общеукрепляющую терапию. В тех случаях, когда имеется значительно выраженная миотоническая симптоматика, назначают курсы дифенина по 0,03—0,05 г 3 раза в день, длительностью 2—3 нед. Предполагают, что дифенин оказывает угнетающее действие на синаптическую проводимость и снижает посттетаническую активность в мышцах [Гехт Б. М., Ильина Н. А., 1982].
В настоящее время доступна пренатальная диагностика носительства гена миотонической дистрофии.
Прогрессирующая мышечная дистрофия (миопатия Дюшенна) как самостоятельная клиническая форма в группе мышечных дистрофий достаточно хорошо изучена. Впервые она была подробно описана в 1868 г. Частота в популяции — 1:30 000.
Клиническая картина. Наблюдается почти исключительно у мальчиков. Основная симптоматика заболевания заключается в прогрессирующем нарастании дистрофических изменений с постепенным обездвиживанием больного.
Поражаются прежде всего нижние конечности. Первые симптомы появляются, как правило, в возрасте 2—4 лет, хотя до этого, уже на 1—2-м году жизни, двигательная активность больных обычно снижена. Дети позднее начинают ходить, не бегают, не влезают на стулья и т. п. В дальнейшем развивается специфическая («утиная») походка с переваливанием из стороны в сторону.
Характерны псевдогипертрофии икроножных мышц при нарастании диффузной мышечной атрофии. Икроножные мышцы становятся плотными, увеличиваются в объеме. Сухожильные рефлексы исчезают, но в ранней стадии заболевания они могут быть даже несколько повышены.
Процесс имеет восходящий характер: помимо ног, мышечная слабость распространяется на мышцы спины, грудной клетки, верхних конечностей. К 13—15 годам больные, как правило, полностью обездвижены. Смерть наступает в конце 2-го десятилетия чаще всего от острой сердечной недостаточности или от пневмонии.
По данным разных авторов, 30—70 % больных миопатией Дюшенна страдают умственной отсталостью. Последняя предшествует появлению мышечных нарушений и не нарастает, несмотря на прогрессирование самого заболевания [Dubolit V., 1965, и др.].
Степень интеллектуального недоразвития может быть различной. Преобладают больные с негрубым снижением интеллекта, но встречаются случаи олигофрении в степени имбецильности.
Диагноз. Врожденный характер интеллектуального дефекта в сочетании с симптомами миопатии, проявляющимися на ранних стадиях главным образом запаздыванием в становлении моторики, нередко является причиной диагностических ошибок при обследовании ребенка в раннем возрасте В этих случаях иногда диагностируют остаточные явления органического поражения мозга с умственной отсталостью и атонической формой детского церебрального паралича.
При дифференциальном диагнозе должен учитываться характер мышечных нарушений: более резкое поражение проксимальных отделов мышц конечностей по сравнению с дистальными, очень резко выраженная гипотония мышц. В сомнительных случаях диагноз уточняют с помощью электромиографии. На ЭКГ — изменения, характерные для поражения мышц.
Прогрессирующая мышечная дистрофия Дюшенна вызывается рецессивным геном, локализованным в Х-хромосоме.
Патогенез умственной отсталости при этом заболевании до конца полностью неясен. Анатомических изменений в мозге, как правило, не обнаруживают, хотя описывают в отдельных случаях изменения корковой цитоархитектоники с гетеротопией нейронов.
Лечение. Эффективной патогенетической терапии нет. Проводят общеукрепляющее лечение, включающее биогенные стимуляторы, аутогемотерапию, витамины.
Используются в лечении больных препараты, улучшающие синтез белка, в частности анаболические гормоны (неробол, ретаболил). Наблюдался положительный эффект от применения аллопуринола — ингибитора ксантиноксидазы, участвующей в распаде пуринов, а также препаратов из группы -адреноблокаторов.
При решении вопроса о прогнозе потомства у женщин с установленным носительством гена миопатии Дюшенна методом выбора является определение пола плода.
Мышечная дистрофия: надежда благодаря исследованиям
Введение
Что такое мышечная дистрофия?
Что вызывает MD?
Сколько человек больны MD?
Как MD влияет на мышцы?
Есть ли другие состояния, подобные MD?
Чем отличаются мышечные дистрофии?
Как диагностируют мышечные дистрофии?
Как лечат мышечные дистрофии?
Какой прогноз?
Какие исследования проводятся?
Где я могу получить дополнительную информацию?
Глоссарий
Введение
Первое историческое описание мышечной дистрофии появилось в 1830 году, когда сэр Чарльз Белл написал эссе о болезни, вызывающей прогрессирующую слабость у мальчиков.Шесть лет спустя другой ученый сообщил о двух братьях, у которых развилась общая слабость, мышечное повреждение и замещение поврежденной мышечной ткани жировой и соединительной тканью. В то время симптомы считались признаками туберкулеза.
В 1850-х годах описания мальчиков, которые становились все слабее, утратили способность ходить и умерли в раннем возрасте, стали более заметными в медицинских журналах. В следующее десятилетие французский невролог Гийом Дюшенн дал исчерпывающий отчет о 13 мальчиках с наиболее распространенной и тяжелой формой болезни (которая теперь носит его имя — мышечная дистрофия Дюшенна).Вскоре стало очевидно, что болезнь имеет несколько форм и что эти болезни поражают людей любого пола и всех возрастов.
Что такое мышечная дистрофия?
Мышечная дистрофия (МД) относится к группе из более чем 30 генетических заболеваний, вызывающих прогрессирующую слабость и дегенерацию скелетных мышц, используемых во время произвольных движений. Слово дистрофия происходит от греческого dys , что означает «трудный» или «ошибочный», и troph или «питать».«Эти расстройства различаются по возрасту начала, тяжести и типу пораженных мышц. Все формы БМ усугубляются по мере того, как мышцы постепенно дегенерируют и ослабевают. Многие люди со временем теряют способность ходить.
Некоторые типы MD также влияют на сердце, желудочно-кишечный тракт, железы внутренней секреции, позвоночник, глаза, мозг и другие органы. Могут возникнуть респираторные и сердечные заболевания, а у некоторых людей может развиться нарушение глотания. MD не заразен и не может быть вызван травмой или деятельностью.
Что вызывает MD?
Все мышечные дистрофии передаются по наследству и включают мутацию в одном из тысяч генов, которые программируют белки, критически важные для целостности мышц. Клетки организма не работают должным образом, если белок изменен или произведен в недостаточном количестве (или иногда полностью отсутствует). Многие случаи БМ возникают из-за спонтанных мутаций, которые не обнаруживаются в генах ни одного из родителей, и этот дефект может быть передан следующему поколению.
Гены подобны чертежам: они содержат закодированные сообщения, которые определяют характеристики или черты человека.Они расположены вдоль 23 стержневидных пар из хромосом *, причем половина каждой пары унаследована от каждого родителя. Каждая половина пары хромосом похожа на другую, за исключением одной пары, которая определяет пол человека. Мышечные дистрофии могут передаваться по наследству тремя способами:
- Аутосомно-доминантное наследование происходит, когда ребенок получает нормальный ген от одного родителя и дефектный ген от другого родителя. Аутосомно означает, что генетическая мутация может произойти в любой из 22 неполовых хромосом в каждой из клеток организма.Доминирующий означает, что только один родитель должен передать аномальный ген, чтобы вызвать заболевание. В семьях, где один из родителей несет дефектный ген, у каждого ребенка есть 50-процентная вероятность унаследовать этот ген и, следовательно, заболевание. Мужчины и женщины одинаково подвержены риску, и тяжесть заболевания может различаться от человека к человеку.
- Аутосомно-рецессивное наследование означает, что оба родителя должны нести и передавать дефектный ген. У каждого из родителей есть один дефектный ген, но это заболевание не распространяется.Дети в этих семьях имеют 25-процентный шанс унаследовать обе копии дефектного гена и 50-процентный шанс унаследовать один ген и, следовательно, стать носителем , способным передать дефект своим детям. Дети любого пола могут быть затронуты этим типом наследования.
- Х-сцепленный (или сцепленный с полом) рецессивный наследование происходит, когда мать несет пораженный ген на одной из своих двух Х-хромосом и передает его своему сыну (мужчины всегда наследуют Х-хромосому от своей матери и Y-хромосому. хромосома от отца, а дочери наследуют Х-хромосому от каждого родителя).Сыновья матери-носительницы имеют 50-процентный шанс унаследовать заболевание. У дочерей также есть 50-процентный шанс унаследовать дефектный ген, но обычно это не затрагивает, поскольку здоровая Х-хромосома, полученная от отца, может компенсировать дефектную хромосому, полученную от их матери. Больные отцы не могут передать Х-сцепленное заболевание своим сыновьям, но их дочери будут носителями этого заболевания. Самки-носители иногда могут проявлять более легкие симптомы MD.
* Термины, выделенные курсивом, определены в глоссарии.
Сколько человек больны MD?
MD встречается во всем мире, затрагивая все расы. Заболеваемость варьируется, поскольку одни формы встречаются чаще, чем другие. Его наиболее распространенная форма у детей, мышечная дистрофия Дюшенна, поражает примерно 1 из 3500-6000 новорожденных мальчиков ежегодно в Соединенных Штатах. ** Некоторые типы MD более распространены в некоторых странах и регионах мира. Многие мышечные дистрофии являются семейными, что означает, что у них есть семейный анамнез заболевания.Случаи Дюшенна часто не имеют предшествующего семейного анамнеза. Вероятно, это связано с большим размером гена дистрофина, который вовлечен в заболевание, что делает его мишенью для спонтанных мутаций.
** Центры по контролю и профилактике заболеваний, Национальный центр по врожденным дефектам и порокам развития, 17 июля 2013 г.
Как MD влияет на мышцы?
Мышцы состоят из тысяч мышечных волокон. Каждое волокно на самом деле представляет собой ряд отдельных клеток, которые во время развития соединились вместе и заключены в наружную мембрану.Мышечные волокна, составляющие отдельные мышцы, связаны соединительной тканью.
Мышцы активируются, когда импульс или сигнал посылается из головного мозга через спинной мозг и периферические нервы (нервы, которые соединяют центральную нервную систему с органами чувств и мышцами) к нервно-мышечному соединению (пространство между нервным волокном и нервным волокном). мышцы, которые он активирует). Там выброс химического вещества ацетилхолина запускает серию событий, которые заставляют мышцу сокращаться.
Мембрана мышечных волокон содержит группу белков, называемую комплексом дистрофин-гликопротеин , которая предотвращает повреждение при сокращении и расслаблении мышечных волокон. Когда эта защитная мембрана повреждена, мышечные волокна начинают пропускать белок , креатинкиназу (необходим для химических реакций, которые производят энергию для мышечных сокращений), и поглощают избыток кальция, что наносит дополнительный вред. Пораженные мышечные волокна в конечном итоге умирают от этого повреждения, что приводит к прогрессирующей дегенерации мышц.
Хотя МД может поражать несколько тканей и органов тела, в наибольшей степени он влияет на целостность мышечных волокон. Заболевание вызывает дегенерацию мышц, прогрессирующую слабость, гибель волокон, разветвление и расщепление волокон, фагоцитоз (при котором материал мышечных волокон разрушается и разрушается клетками-мусорщиками) и, в некоторых случаях, хроническое или постоянное укорочение сухожилий и мышц. Кроме того, общая сила мышц и сухожильные рефлексы обычно уменьшаются или теряются из-за замещения мышц соединительной тканью и жиром.
Есть ли другие состояния, похожие на MD?
Есть много других наследственных заболеваний, поражающих мышцы, нервы или нервно-мышечные соединения. Такие заболевания, как воспалительная миопатия , прогрессирующая мышечная слабость и кардиомиопатия (слабость сердечной мышцы, которая мешает насосной способности), могут вызывать симптомы, очень похожие на те, которые встречаются при некоторых формах MD), но они вызваны различными генетическими дефектами. Дифференциальный диагноз для людей с подобными симптомами включает в себя врожденную миопатию, мышечную атрофию позвоночника и врожденные миастенические синдромы.Распространенность симптомов среди множества нервно-мышечных заболеваний и распространенность спорадических случаев в семьях, ранее не пораженных МД, часто затрудняет получение быстрого диагноза для людей с МД. Генное тестирование может предоставить окончательный диагноз для многих типов MD, но были обнаружены не все гены, ответственные за некоторые типы MD. Некоторые люди могут иметь признаки MD, но не несут ни одной из признанных в настоящее время генетических мутаций. Однако исследования других родственных мышечных заболеваний могут внести свой вклад в то, что мы знаем о МД.
Чем отличаются мышечные дистрофии?
Существует девять основных групп мышечных дистрофий. Расстройства классифицируются по степени и распределению мышечной слабости, возрасту начала, скорости прогрессирования, тяжести симптомов и семейному анамнезу (включая любой тип наследования). Хотя некоторые формы MD проявляются в младенчестве или детстве, другие могут появиться только в среднем возрасте или позже. В целом частота и тяжесть заболевания различаются, но каждая из дистрофий вызывает прогрессирующее разрушение скелетных мышц, а некоторые типы поражают сердечную мышцу.
Дюшенн MD — наиболее распространенная детская форма MD, а также самая распространенная из мышечных дистрофий в целом, составляющая примерно 50 процентов всех случаев. Поскольку наследование является X-сцепленным рецессивным (вызвано мутацией X или половой хромосомы), MD Дюшенна в первую очередь поражает мальчиков, хотя у девочек и женщин, несущих дефектный ген, могут проявляться некоторые симптомы. Около одной трети случаев связаны с новыми мутациями, а остальные — в семьях. Сестры мальчиков с MD Дюшенна имеют 50-процентную вероятность нести дефектный ген.
Дюшенн MD обычно проявляется в первые годы жизни, иногда вскоре после того, как больной ребенок начинает ходить. Прогрессирующая слабость и истощение мышц (уменьшение мышечной силы и размера), вызванное дегенерирующими мышечными волокнами, начинается в верхней части ног и тазу, а затем распространяется на плечи. Другие симптомы включают потерю некоторых рефлексов, походку вразвалку, частые падения и неуклюжесть (особенно при беге), трудности при вставании из положения сидя или лежа или при подъеме по лестнице, изменение общей позы, нарушение дыхания, слабость легких и кардиомиопатию.Многие дети не умеют бегать или прыгать. Истощающие мышцы, в частности икроножные мышцы (и, реже, мышцы ягодиц, плеч и рук), могут увеличиваться за счет накопления жира и соединительной ткани, в результате чего они выглядят больше и здоровее, чем они есть на самом деле ( называется псевдогипертрофией ). По мере прогрессирования заболевания мышцы диафрагмы, которые помогают дышать и кашлять, могут ослабевать. Больные могут испытывать затрудненное дыхание, респираторные инфекции и проблемы с глотанием.Истончение костей и сколиоз (искривление позвоночника) являются обычным явлением. Некоторые затронутые дети имеют когнитивные и поведенческие нарушения разной степени. В возрасте от 3 до 6 лет у детей могут наблюдаться короткие периоды физического улучшения, за которыми следует прогрессирующая мышечная дегенерация. Дети с MD Дюшенна обычно теряют способность ходить в раннем подростковом возрасте. Без агрессивной помощи они обычно умирают в подростковом возрасте или в начале двадцатых годов от прогрессирующей слабости сердечной мышцы, респираторных осложнений или инфекции.Однако улучшение многопрофильной помощи позволило увеличить продолжительность жизни и значительно улучшить качество жизни этих детей; многочисленные люди с мышечной дистрофией Дюшенна сейчас доживают до 30 лет, а некоторые даже до 40 лет.
Дюшенн MD является результатом отсутствия дистрофина мышечного белка. Дистрофин — это белок, содержащийся в мышцах, который помогает мышцам оставаться здоровыми и сильными. Анализы крови детей с MD Дюшенна показывают аномально высокий уровень креатинкиназы; это открытие очевидно с рождения.
Becker MD менее серьезен, чем MD Дюшенна, но тесно связан с ним. Люди с MD Беккера имеют частичную, но недостаточную функцию белкового дистрофина. Клиническое течение Беккера MD более вариабельно, чем у Дюшенна. Заболевание обычно проявляется в возрасте около 11 лет, но может возникнуть и в возрасте 25 лет, и больные обычно доживают до среднего возраста или позже. Скорость прогрессирующей, симметричной (с обеих сторон тела) атрофии и слабости мышцы и слабости сильно различается среди пораженных людей.Многие люди могут ходить, пока им не исполнится 30 или более лет, в то время как другие не могут ходить после подросткового возраста. Некоторым больным никогда не нужно пользоваться инвалидной коляской. Как и в случае с Дюшенном, мышечная слабость в Беккере обычно сначала проявляется в плечах и плечах, бедрах и тазу.
Ранние симптомы Becker MD включают ходьбу на цыпочках, частые падения и трудности с подъемом с пола. Мышцы икр могут казаться большими и здоровыми, поскольку истощенные мышечные волокна заменяются жиром, а мышечная активность может вызывать судороги у некоторых людей.Сердечные осложнения не так часто присутствуют в группе Беккера по сравнению с Дюшенном, но в некоторых случаях могут быть столь же серьезными. Когнитивные и поведенческие нарушения не так распространены или серьезны, как у Дюшенна, но они все же встречаются.
Врожденный MD относится к группе аутосомно-рецессивных мышечных дистрофий, которые либо присутствуют при рождении, либо проявляются до 2 лет. Они поражают как мальчиков, так и девочек. Степень и прогрессирование мышечной слабости и дегенерации зависят от типа заболевания.Слабость может быть впервые отмечена, когда дети не соответствуют ориентирам в двигательной функции и мышечном контроле. Дегенерация мышц может быть легкой или тяжелой и ограничивается в основном скелетными мышцами. Большинство людей не могут сидеть или стоять без поддержки, а некоторые пострадавшие дети могут никогда не научиться ходить. Выделяют три группы врожденных MD:
- мерозин-отрицательные расстройства, при которых отсутствует белок мерозин (обнаруженный в соединительной ткани, окружающей мышечные волокна);
- мерозин-положительное заболевание, при котором мерозин присутствует, но отсутствуют другие необходимые белки; и
- нарушение миграции нейронов, при котором на очень ранних этапах развития нервной системы плода миграция нервных клеток (нейронов) к их надлежащему местоположению нарушается.
Дефекты белка мерозина являются причиной почти половины всех случаев врожденной БМ.
У людей с врожденной БМ может развиться контрактур (хроническое укорочение мышц или сухожилий вокруг суставов, которое препятствует их свободному движению), сколиоз, затруднения дыхания и глотания, а также деформации стопы. Некоторые люди имеют нормальное интеллектуальное развитие, в то время как другие серьезно страдают. Слабость мышц диафрагмы может привести к дыхательной недостаточности.Врожденный БМ также может влиять на центральную нервную систему, вызывая проблемы со зрением и речью, судороги и структурные изменения в головном мозге. Некоторые дети с заболеваниями умирают в младенчестве, в то время как другие могут дожить до взрослой жизни с минимальной инвалидностью.
Дистальный MD , также называемый дистальной миопатией, описывает группу, по крайней мере, из шести специфических мышечных заболеваний, которые в первую очередь поражают дистальные мышцы (наиболее удаленные от плеч и бедер) в предплечьях, руках, голенях и ступнях.Дистальные дистрофии обычно менее серьезны, прогрессируют медленнее и затрагивают меньшее количество мышц, чем другие формы MD, хотя они могут распространяться на другие мышцы, включая проксимальные, позже в ходе болезни. Дистальный МД может повлиять на сердце и дыхательные мышцы, и отдельным пациентам в конечном итоге может потребоваться использование аппарата ИВЛ. Больные могут быть не в состоянии совершать точные движения руками и с трудом разгибать пальцы. При поражении мышц ног становится трудно ходить и подниматься по лестнице, и некоторые люди не могут прыгать или стоять на пятках.Дистальная БМ, поражающая как мужчин, так и женщин, обычно возникает в возрасте от 40 до 60 лет. Известно, что при одной из форм дистального БМ комплекс белков мышечной мембраны, называемый дисферлином, отсутствует.
Хотя дистальный БМ является преимущественно аутосомно-доминантным заболеванием, у молодых людей были зарегистрированы аутосомно-рецессивные формы. Симптомы аналогичны симптомам Дюшенна, но с другим типом повреждения мышц. Сообщалось также о младенческой форме аутосомно-рецессивного дистального БМ.Медленная, но прогрессирующая слабость часто впервые замечается в возрасте около 1 года, когда ребенок начинает ходить, и продолжает очень медленно прогрессировать на протяжении всей взрослой жизни.
Emery-Dreifuss MD в первую очередь поражает мальчиков. Заболевание имеет две формы: одна является Х-сцепленной рецессивной, а другая аутосомно-доминантной.
Начало MD Эмери-Дрейфуса обычно проявляется к 10 годам, но симптомы могут появиться уже к середине двадцатых годов. Это заболевание вызывает медленное, но прогрессирующее истощение мышц плеча и голени и симметричную слабость.Контрактуры в позвоночнике, лодыжках, коленях, локтях и задней части шеи обычно предшествуют значительной мышечной слабости, которая менее серьезна, чем при Дюшенне. Контрактуры могут привести к тому, что локти будут заблокированы в согнутом положении. По мере прогрессирования болезни весь позвоночник может стать ригидным. Другие симптомы включают ухудшение состояния плеч, ходьбу на носках и легкую слабость лица. Уровень креатинкиназы в сыворотке может быть умеренно повышенным. Почти все люди с MD Emery-Dreifuss к 30 годам имеют ту или иную форму проблемы с сердцем, часто требующей кардиостимулятора или другого вспомогательного устройства.Женщины-носительницы заболевания часто имеют сердечные осложнения без мышечной слабости. Больные часто умирают в среднем возрасте от прогрессирующей легочной или сердечной недостаточности. В некоторых случаях сердечные симптомы могут быть самым ранним и наиболее значительным симптомом заболевания и могут появиться за годы до мышечной слабости.
Facioscapulohumeral MD (FSHD) первоначально поражает мышцы лица (facio), плеч (scapulo) и предплечий (плеча) с прогрессирующей слабостью.Эта третья по распространенности форма БМ, также известная как болезнь Ландузи-Дежерина, является аутосомно-доминантным заболеванием. У большинства людей нормальная продолжительность жизни, но некоторые люди становятся инвалидами. Заболевание обычно прогрессирует очень медленно, с периодическими всплесками быстрого разрушения мышц. Начало обычно в подростковом возрасте, но может произойти уже в детстве или в возрасте 40 лет. Отличительной чертой ЛЛПД является то, что она обычно вызывает асимметричную слабость. Часто сначала поражаются мышцы вокруг глаз и рта, а затем появляется слабость в области плеч, груди и предплечий.Из-за определенного типа мышечного истощения плечи кажутся наклонными, а лопатки — крылатыми. Также могут ослабеть мышцы нижних конечностей. Рефлексы ослаблены, как правило, в той же степени, что и слабость. Изменения во внешности лица могут включать в себя кривую улыбку, надутый вид, сглаженные черты лица или маску. Некоторые люди не могут сморщить губы или свистеть, могут испытывать трудности с глотанием, жеванием или речью.У некоторых людей мышечная слабость может распространяться на диафрагму, вызывая респираторные проблемы. Другие симптомы могут включать потерю слуха (особенно на высоких частотах) и лордоз , аномальную кривую колебания позвоночника. Контрактуры редки. Некоторые люди с ЛЛПД ощущают сильную боль в пораженной конечности. Сердечные мышцы обычно не поражаются, а значительная слабость тазового пояса встречается реже, чем при других формах БД. Детская форма ЛЛПД также может вызывать заболевание сетчатки и некоторую потерю слуха.
Конечностный пояс MD (LGMD) относится к более чем 20 наследственным состояниям, отмеченным прогрессирующей потерей мышечной массы и симметричным ослаблением произвольных мышц, в первую очередь плеч и бедер. Идентифицировано по крайней мере 5 форм аутосомно-доминантной БМ конечностей-пояса (известных как тип 1) и 17 форм аутосомно-рецессивных БМ поясничных конечностей (известных как тип 2). В настоящее время известно, что некоторые аутосомно-рецессивные формы заболевания возникают из-за дефицита любого из четырех белков комплекса дистрофин-гликопротеин, называемых саркогликанами.Недостаток дистрогликана, который обычно ассоциируется с врожденными мышечными дистрофиями, также может вызывать LGMD.
Рецессивные LGMD встречаются чаще, чем доминантные формы, обычно начинаются в детстве или подростковом возрасте и демонстрируют резко повышенный уровень креатинкиназы в сыворотке. Доминирующие LGMD обычно начинаются во взрослом возрасте. В целом, чем раньше появляются клинические признаки, тем быстрее прогрессирует болезнь. БД конечностей-пояса поражает как мужчин, так и женщин.Некоторые формы заболевания быстро прогрессируют, что приводит к серьезному повреждению мышц и потере способности ходить, в то время как другие развиваются очень медленно в течение многих лет и вызывают минимальную инвалидность, обеспечивая нормальную продолжительность жизни. В некоторых случаях заболевание временно прекращается, но затем возобновляется прогрессирование.
Модель мышечной слабости аналогична таковой у Дюшенна и Беккера. Слабость обычно сначала наблюдается в области бедер, а затем распространяется на плечи, ноги и шею.Люди развивают походку вразвалку и испытывают трудности при вставании со стульев, подъеме по лестнице или переноске тяжелых предметов. Они часто падают и не могут бегать. Контрактуры в локтях и коленях встречаются редко, но у людей могут развиваться контрактуры мышц спины, что придает им вид жесткости позвоночника. Проксимальные рефлексы (ближайшие к центру тела) часто нарушены. Некоторые люди также страдают кардиомиопатией и респираторными осложнениями, частично в зависимости от конкретного подтипа.Интеллект в большинстве случаев остается нормальным, хотя бывают и исключения. Многие люди с БМ конечностей становятся тяжелыми инвалидами в течение 20 лет после начала болезни.
Миотоническая дистрофия (DM1) , также известная как болезнь Штейнерта и миотоническая дистрофия, является еще одной распространенной формой MD. Миотония , или неспособность расслабить мышцы после внезапного сокращения, встречается только при этой форме MD, но также встречается при других недистрофических мышечных заболеваниях. Люди с СД1 могут прожить долгую жизнь с переменной, но медленно прогрессирующей инвалидностью.Типичное начало болезни — в возрасте от 20 до 30 лет, но начало болезни в детстве и врожденное начало хорошо задокументированы. Мышцы лица и передней части шеи обычно первыми проявляют слабость и могут привести к изможденному, «топорному» лицу и тонкой лебединой шее. Истощение и слабость заметно сказываются на мышцах предплечий. DM1 поражает центральную нервную систему и другие системы организма, включая сердце, надпочечники и щитовидную железу, глаза и желудочно-кишечный тракт. Другие симптомы включают сердечные осложнения, затрудненное глотание, опущенные веки (так называемый птоз), катаракту, плохое зрение, раннее лобное облысение, потерю веса, импотенцию, атрофию яичек, легкие умственные нарушения и повышенное потоотделение.Люди также могут чувствовать сонливость и повышенную потребность во сне. Существует вторая форма заболевания, которая похожа на классическую, но обычно сильнее поражает проксимальные мышцы. Эта форма известна как миотоническая дистрофия 2 типа (СД2).
Это аутосомно-доминантное заболевание поражает как мужчин, так и женщин. У женщин могут быть нерегулярные менструации и иногда они бесплодны. Заболевание может возникнуть раньше и в последующих поколениях протекать тяжелее. Детская форма миотонического МД может проявиться в возрасте от 5 до 10 лет.Симптомы включают общую мышечную слабость (особенно в области лица и дистальных мышц), отсутствие мышечного тонуса и умственные нарушения.
Женщина с СД1 может родить ребенка с редкой врожденной формой заболевания. Симптомы при рождении могут включать затруднение глотания или сосания, нарушение дыхания, отсутствие рефлексов, деформации скелета и контрактуры (например, косолапость) и мышечную слабость, особенно в области лица. Дети с врожденным миотоническим MD также могут испытывать умственные нарушения и задержку моторного развития.Эта тяжелая детская форма миотонического БМ встречается почти исключительно у детей, унаследовавших дефектный ген от своей матери, симптомы которой могут быть настолько легкими, что она иногда не осознает, что у нее заболевание, до тех пор, пока у нее не появится больной ребенок.
Унаследованный дефект гена, вызывающий СД1, представляет собой ненормально длинное повторение трехбуквенного «слова» в генетическом коде. У здоровых людей это слово повторяется несколько раз, но у людей с СД1 оно повторяется много раз.Этот тройной повтор становится длиннее с каждым последующим поколением. Механизм триплетного повтора в настоящее время вовлечен как минимум в 15 других заболеваний, включая болезнь Хантингтона и спиноцеребеллярную атаксию.
Окулофарингеальный MD (OPMD) обычно начинается в возрасте от 40 до 50 лет и поражает как мужчин, так и женщин. В Соединенных Штатах это заболевание чаще всего встречается в семьях франко-канадского происхождения и среди испаноязычных жителей северного Нью-Мексико. Люди сначала сообщают об опущенных веках, а затем о слабости лицевых мышц и глоточных мышц в горле, что вызывает затруднения при глотании.Язык может атрофироваться, и могут произойти изменения голоса. Веки могут опускаться настолько резко, что некоторые люди компенсируют это отклонением головы назад. У пораженных людей может быть двоение в глазах и проблемы с верхним взглядом, а у других может быть пигментный ретинит (прогрессирующая дегенерация сетчатки, которая влияет на ночное зрение и периферическое зрение) и сердечные нарушения. Мышечная слабость и истощение в области шеи и плеч — обычное явление. Также могут быть затронуты мышцы конечностей. Людям с OPMD может быть трудно ходить, подниматься по лестнице, становиться на колени или наклоняться.Те люди, которые пострадали в наибольшей степени, в конечном итоге потеряют способность ходить.
Как диагностируют мышечные дистрофии?
Следует тщательно изучить историю болезни человека и полную семейную историю, чтобы определить, является ли мышечное заболевание вторичным по отношению к заболеванию, поражающему другие ткани или органы, или является наследственным заболеванием. Также важно исключить любую мышечную слабость, возникшую в результате предшествующей операции, воздействия токсинов или текущих лекарств, которые могут повлиять на функциональное состояние человека или исключить многие приобретенные мышечные заболевания.Тщательные клинические и неврологические обследования могут исключить нарушения центральной и / или периферической нервной системы, выявить любые паттерны мышечной слабости и атрофии, проверить рефлекторные реакции и координацию, а также выявить сокращения.
Для подтверждения диагноза MD могут использоваться различные лабораторные тесты.
Анализы крови и мочи могут обнаружить дефектные гены и помочь выявить специфические нервно-мышечные расстройства. Например:
- Креатинкиназа — это фермент, который выделяется из поврежденной мышцы.Повышенный уровень креатинкиназы может указывать на повреждение мышц, включая некоторые формы MD, до того, как физические симптомы станут очевидными. Уровни значительно повышаются на ранних стадиях Дюшенна и Беккера. Тестирование также может определить, является ли молодая женщина носителем заболевания.
- Уровень сывороточной альдолазы, фермента, участвующего в расщеплении глюкозы, измеряется для подтверждения диагноза заболевания скелетных мышц. Высокий уровень фермента, который присутствует в большинстве тканей тела, отмечается у людей с MD и некоторыми формами миопатии.
- Миоглобин измеряется при подозрении на повреждение или заболевание скелетных мышц. Миоглобин — это связывающий кислород белок, содержащийся в клетках сердечных и скелетных мышц. Высокий уровень миоглобина в крови обнаруживается у людей с MD.
- Полимеразная цепная реакция (ПЦР) может обнаруживать некоторые мутации в гене дистрофина. ПЦР, также известная как молекулярная диагностика или генетическое тестирование, представляет собой метод создания и анализа нескольких копий фрагмента ДНК.
- Электрофорез сыворотки — это тест для определения количества различных белков в ДНК человека.Образец крови помещается на специально обработанную бумагу и подвергается воздействию электрического тока. Заряд заставляет разные белки образовывать полосы, которые указывают относительную долю каждого фрагмента белка.
Тесты с физической нагрузкой могут обнаруживать повышенные уровни определенных химических веществ после упражнений и используются для определения природы MD или другого мышечного расстройства. Некоторые тесты с физической нагрузкой можно проводить у постели больного, в то время как другие — в клиниках или других местах с использованием сложного оборудования.Эти тесты также оценивают мышечную силу. Они выполняются, когда человек расслаблен и находится в надлежащем положении, что позволяет техническим специалистам измерить функцию мышц против силы тяжести и обнаружить даже небольшую мышечную слабость. Если есть подозрение на слабость дыхательных мышц, дыхательную способность можно измерить, попросив человека сделать глубокий вдох и медленно считать при выдохе.
Генетическое тестирование ищет гены, которые, как известно, вызывают или связаны с наследственными заболеваниями мышц.Анализ ДНК и ферментные тесты могут подтвердить диагноз некоторых нервно-мышечных заболеваний, включая MD. Исследования генетического сцепления могут определить, наследуются ли вместе определенный генетический маркер на хромосоме и заболевание. Они особенно полезны при изучении семей, в которых есть затронутые члены разных поколений. Точный молекулярный диагноз необходим для некоторых разрабатываемых в настоящее время стратегий лечения. Достижения в области генетического тестирования включают секвенирование всего экзома и всего генома, что позволит людям одновременно проверять все свои гены на наличие болезнетворных мутаций, а не проверять только один или несколько генов за раз.Секвенирование экзома рассматривает часть генетического материала или генома человека, которая «кодирует» (или преобразует) в белки.
Генетическое консультирование может помочь родителям с семейным анамнезом MD определить, несут ли они один из мутировавших генов, вызывающих заболевание. Можно использовать два теста, чтобы помочь будущим родителям выяснить, страдает ли их ребенок.
- Амниоцентез, который обычно проводится на 14–16 неделе беременности, проверяет образец околоплодных вод в матке на генетические дефекты (жидкость и плод имеют одинаковую ДНК).Под местной анестезией тонкая игла вводится через живот женщины в матку. Около 20 миллилитров жидкости (примерно 4 чайные ложки) отбирают и отправляют в лабораторию для оценки. Результаты анализов часто занимают 1-2 недели.
- Взятие пробы ворсин хориона или CVS включает в себя удаление и тестирование очень небольшого образца плаценты на ранних сроках беременности. Образец, содержащий ту же ДНК, что и плод, удаляется с помощью катетера или тонкой иглы, вводимой через шейку матки, или тонкой иглы, вводимой через брюшную полость.Ткань проверяется на генетические изменения, выявленные у пораженного члена семьи. Результаты обычно доступны в течение 2 недель.
Диагностическая визуализация может помочь определить конкретный характер заболевания или состояния. Один из таких типов изображений, называемый магнитно-резонансной томографией (МРТ), используется для изучения качества мышц, любых атрофий или отклонений в размере и замещения жировой ткани в мышечной ткани, а также для отслеживания прогрессирования заболевания. Сканирующее оборудование МРТ создает вокруг тела сильное магнитное поле.Затем радиоволны проходят через тело, чтобы вызвать резонансный сигнал, который можно обнаружить под разными углами внутри тела. Компьютер преобразует этот резонанс в трехмерное изображение или двухмерный «срез» сканируемой ткани. МРТ — это неинвазивная безболезненная процедура. Другие формы диагностической визуализации для MD включают магнитно-резонансную спектроскопию фосфора, которая измеряет клеточную реакцию на упражнения и количество энергии, доступной мышечным волокнам, и ультразвуковую визуализацию (также известную как сонография), которая использует высокочастотные звуковые волны для получения изображений внутри тело.Отголоски звуковой волны записываются и отображаются на экране компьютера в виде визуального изображения в реальном времени. Ультразвук можно использовать для измерения мышечной массы. МРТ головного мозга может быть полезным при диагностике определенных форм врожденной мышечной дистрофии, при которых обычно присутствуют структурные аномалии головного мозга.
Биопсия мышц используется в диагностических целях и в исследовательских учреждениях, чтобы контролировать течение заболевания и эффективность лечения. Используя местную или общую анестезию, врач или хирург может взять небольшой образец мышцы для анализа.Образец может быть взят хирургическим путем через разрез в коже или с помощью иглы для биопсии , при которой тонкая полая игла вводится через кожу в мышцу. Небольшой кусок мышцы остается в полой игле, когда ее вынимают из тела. Образец мышц окрашивают и исследуют, чтобы определить, есть ли у человека мышечное заболевание, заболевание нервов ( нейропатия, ), воспаление или другая миопатия. Биопсия мышц иногда также может помочь в тестировании на носительство.С появлением точных молекулярных методов биопсия мышц становится все менее необходимой для диагностики мышечных дистрофий. Биопсия мышц по-прежнему необходима для постановки диагноза при большинстве приобретенных мышечных заболеваний.
Иммунофлуоресценция. Тестирование позволяет обнаруживать специфические белки, такие как дистрофин, в мышечных волокнах. После биопсии флуоресцентные маркеры используются для окрашивания образца, содержащего интересующий белок.
Электронная микроскопия может идентифицировать изменения в субклеточных компонентах мышечных волокон.Электронная микроскопия также может идентифицировать изменения, которые характеризуют гибель клеток, мутации в митохондриях мышечных клеток и увеличение соединительной ткани, наблюдаемое при мышечных заболеваниях, таких как MD. Изменения в мышечных волокнах, которые очевидны при редкой форме дистального MD, можно увидеть с помощью электронного микроскопа.
Нейрофизиологические исследования позволяют выявить физические и / или химические изменения в нервной системе.
- Исследования скорости нервной проводимости позволяют измерить скорость и силу, с которой электрический сигнал распространяется по нерву.Небольшой поверхностный электрод стимулирует нерв, а регистрирующий электрод обнаруживает результирующий электрический сигнал либо в другом месте того же нерва, либо на мышце, контролируемой этим нервом. Ответ можно оценить, чтобы определить, присутствует ли повреждение нерва.
- Исследования повторяющейся стимуляции включают электрическую стимуляцию двигательного нерва несколько раз подряд для оценки функции нервно-мышечного соединения. Регистрирующий электрод помещают на мышцу, контролируемую стимулированным нервом, как это делается при обычном исследовании проводимости двигательного нерва.
- Электромиография (ЭМГ) может регистрировать активность мышечных волокон и двигательных единиц. Крошечная игла с электродом вводится через кожу в мышцу. Электрическая активность, обнаруженная в мышце, может отображаться на мониторе, а также может быть услышана при воспроизведении через динамик. Результаты могут выявить электрическую активность, характерную для MD или других нервно-мышечных расстройств.
Как лечат мышечные дистрофии?
Все формы MD являются генетическими и не могут быть предотвращены в настоящее время, за исключением использования вмешательств пренатального скрининга.Однако доступные методы лечения направлены на то, чтобы сохранить независимость человека как можно дольше и предотвратить осложнения, возникающие в результате слабости, ограниченной подвижности, а также сердечных и респираторных заболеваний. Лечение может включать комбинацию подходов, включая физиотерапию, лекарственную терапию и хирургическое вмешательство. Доступные методы лечения иногда весьма эффективны и могут существенно повлиять на продолжительность и качество жизни.
Вспомогательная вентиляция легких часто требуется для лечения слабости дыхательных мышц, которая сопровождает многие формы MD, особенно на поздних стадиях.Воздух, содержащий дополнительный кислород, подается через гибкую маску (или, в некоторых случаях, через трубку, вводимую через пищевод в легкие), чтобы помочь легким полностью надуть воздух. Поскольку затруднение дыхания может быть наиболее сильным в ночное время, некоторым людям может потребоваться ночная вентиляция. Многие люди предпочитают неинвазивную вентиляцию, при которой маска, надеваемая на лицо, соединяется трубкой с аппаратом, который генерирует периодические выбросы принудительного воздуха, который может включать дополнительный кислород. У некоторых людей с MD Дюшенна, особенно у людей с избыточным весом, может развиться обструктивное апноэ во сне, и им потребуется вентиляция в ночное время.Людям, находящимся на аппарате ИВЛ, также может потребоваться использование желудочного зонда для кормления.
Медикаментозная терапия может быть назначена для задержки мышечной дегенерации. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило инъекции препаратов голодирсена и вилтоларсена для лечения пациентов с мышечной дистрофией Дюшенна (МДД), у которых есть подтвержденная мутация гена дистрофина, поддающаяся пропуску экзона 53. По оценкам, около 8% пациентов с МДД имеют эту мутацию. FDA одобрило инъекцию препарата казимерсен для лечения пациентов с подтвержденной мутацией гена DMD, которая поддается пропуску экзона 45.FDA также одобрило три применения финголимода (Gilenya) для лечения рецидивирующей формы рассеянного склероза у взрослых. Кортикостероиды, такие как преднизон, могут замедлить скорость разрушения мышц у доктора Дюшенна и помочь детям сохранить силу и продлить самостоятельную ходьбу на целых несколько лет. Однако у этих лекарств есть побочные эффекты, такие как увеличение веса, изменения лица, потеря линейного (роста) роста и хрупкость костей, которые могут особенно беспокоить детей. Иммунодепрессанты, такие как циклоспорин и азатиоприн, могут отсрочить повреждение умирающих мышечных клеток.Лекарства, которые могут обеспечить кратковременное облегчение миотонии (мышечные спазмы и слабость), включают мексилетин; фенитоин; баклофен, который блокирует сигналы, посылаемые спинным мозгом для сокращения мышц; дантролен, нарушающий процесс сокращения мышц; и хинин. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов предоставило ускоренное разрешение на применение препарата Exondys 51 для лечения пациентов с подтвержденной мутацией гена дистрофина, допускающей пропуск экзона 15. Ускоренное утверждение означает, что лекарство можно назначать отдельным лицам, которые соответствуют критериям редкого заболевания, в то время как компания работает над дополнительными испытаниями, чтобы узнать больше об эффективности препарата.(Лекарства от миотонии могут быть неэффективными при миотоническом MD, но хорошо работают при врожденной миотонии, генетическом нервно-мышечном заболевании, характеризующемся медленным расслаблением мышц.) Респираторные инфекции можно лечить антибиотиками.
Физиотерапия может помочь предотвратить деформации, улучшить движения и сохранить мышцы как можно более гибкими и сильными. Варианты включают пассивную растяжку, коррекцию осанки и упражнения. Программа разработана для удовлетворения индивидуальных потребностей.Терапию следует начинать как можно скорее после постановки диагноза, до того, как появится напряжение в суставах или мышцах.
- Пассивное растяжение может повысить гибкость суставов и предотвратить контрактуры, которые ограничивают движение и вызывают потерю функции. При правильном выполнении пассивная растяжка безболезненна. Терапевт или другой обученный медицинский работник медленно перемещает сустав как можно дальше и сохраняет это положение примерно 30 секунд. Движение повторяется несколько раз за сеанс.Пассивное растяжение у детей может быть легче после принятия теплой ванны или душа.
- Регулярные умеренные упражнения могут помочь людям с MD поддерживать диапазон движений и силу мышц, предотвратить атрофию мышц и отсрочить развитие контрактур. Люди с ослабленной диафрагмой могут научиться кашлю и упражнениям на глубокое дыхание, которые предназначены для полного расширения легких.
- Коррекция осанки используется для противодействия мышечной слабости, контрактурам и нарушениям позвоночника, которые вынуждают людей с MD принимать неудобные позы.По возможности люди должны сидеть прямо, ступни под углом 90 градусов к полу. Подушки и клинья из пенопласта помогают удерживать человека в вертикальном положении, равномерно распределять вес и выпрямлять ноги. Подлокотники должны быть на нужной высоте, чтобы обеспечивать поддержку и предотвращать наклон.
- Вспомогательные приспособления, такие как инвалидные коляски, шины и скобы, другие ортопедические приспособления и перекладины (трапеции) над головой, могут помочь сохранить подвижность. Подтяжки используются, чтобы помочь растянуть мышцы и обеспечить поддержку, сохраняя при этом возможность пациента амбулаторно.Спинальные опоры могут помочь предотвратить сколиоз. Ночные шины в сочетании с пассивным растяжением могут отсрочить контрактуры. Ортопедические устройства, такие как стоячие рамы и поворотные ходунки, помогают людям оставаться стоя или ходить как можно дольше, что способствует лучшему кровообращению и улучшает удержание кальция в костях.
- Повторяющиеся низкочастотные импульсы электростимуляции мышц бедра могут вызвать небольшое увеличение силы у некоторых мальчиков с MD Дюшенна, хотя эффективность этой терапии не доказана.
Трудотерапия может помочь некоторым людям справиться с прогрессирующей слабостью и потерей подвижности. Некоторым людям может потребоваться освоить новые рабочие навыки или новые способы выполнения задач, в то время как другим людям может потребоваться сменить работу. Вспомогательные технологии могут включать изменения в домашних условиях и на рабочем месте, а также использование моторизованных инвалидных колясок, аксессуаров для инвалидных колясок и адаптивной посуды.
Логопед может помочь людям с ослабленными мускулами лица и горла.Люди могут научиться пользоваться специальными устройствами связи, такими как компьютер с синтезатором речи
Диетические изменения не замедляют прогрессирование MD. Однако правильное питание имеет важное значение для здоровья в целом. Ограниченная подвижность или отсутствие активности в результате мышечной слабости могут способствовать ожирению, обезвоживанию и запорам. Может помочь диета с высоким содержанием клетчатки, белка и низким содержанием калорий в сочетании с рекомендуемым потреблением жидкости. Методы кормления могут помочь людям с MD, у которых есть расстройство глотания, и которым трудно перейти изо рта в желудок или жидкость из него.
Корректирующая операция часто проводится для облегчения осложнений, вызванных MD.
- Операция на сухожилиях или мышцах рекомендуется, когда контрактура становится достаточно серьезной, чтобы заблокировать сустав или сильно затруднить движение. Процедура, которая включает удлинение сухожилия или мышцы для свободного движения, обычно проводится под общим наркозом. Реабилитация включает использование подтяжек и физиотерапию для укрепления мышц и поддержания восстановленного диапазона движений.После этих ортопедических процедур часто требуется период неподвижности, поэтому преимущества процедуры следует сопоставить с риском этого периода неподвижности, поскольку последний может привести к регрессу.
- Людям с Эмери-Дрейфусом или миотонической дистрофией в какой-то момент может потребоваться кардиостимулятор для лечения сердечных заболеваний.
- Операция по уменьшению боли и постурального дисбаланса, вызванного сколиозом, может помочь некоторым людям. Сколиоз возникает, когда мышцы, поддерживающие позвоночник, начинают ослабевать и больше не могут удерживать позвоночник прямым.Слишком большой изгиб позвоночника может мешать дыханию и осанке, вызывая боль. К позвоночнику может потребоваться прикрепить один или несколько металлических стержней для увеличения силы и улучшения осанки. Другой вариант — сращение позвоночника, при котором кость вставляется между позвонками в позвоночник и дает ей возможность расти, сращивая позвонки вместе для повышения стабильности позвоночника.
- У людей с миотонической дистрофией часто развивается катаракта, помутнение хрусталика глаза, которое блокирует свет. Операция по удалению катаракты включает удаление мутного хрусталика для улучшения зрения.
Какой прогноз?
Прогноз зависит от типа MD и скорости прогрессирования. Некоторые типы являются легкими и прогрессируют очень медленно, что обеспечивает нормальную продолжительность жизни, в то время как другие являются более тяжелыми и приводят к функциональной инвалидности и потере способности передвигаться. Ожидаемая продолжительность жизни часто зависит от степени мышечной слабости, а также от наличия и тяжести респираторных и / или сердечных осложнений.
Какие исследования проводятся?
Национальный институт неврологических расстройств и инсульта (NINDS), входящий в состав Национальных институтов здоровья (NIH), поддерживает обширную программу исследований в области медицины и здравоохранения.Цели этих исследований — улучшить понимание MD и его причин, разработать более эффективные методы лечения и, в конечном итоге, найти способы лечения. NINDS и его дочерние институты, Национальный институт артрита, скелетно-мышечных и кожных заболеваний (NIAMS), Национальный институт здоровья детей и развития человека (NICHD) и Национальный институт сердца, крови и легких (NHLBI), возглавляют Исследования доктора медицины проводились в NIH и учреждениях-получателях по всей стране.
NIH поддерживает широкий спектр фундаментальных, трансляционных и клинических исследований у докторов медицины.Успехи в фундаментальных исследованиях важны для базового понимания каждого типа MD. Хотя многие гены, вызывающие мышечную дистрофию, еще предстоит идентифицировать, успехи в секвенировании генов помогли идентифицировать гены, которые могут быть задействованы в большинстве типов мышечной дистрофии. В свою очередь, новые знания о механизмах конкретных заболеваний позволяют определить потенциальные цели для разработки терапии. В последние годы исследования механизмов основных заболеваний открыли новые возможности для развития терапии почти всех типов БД.Например, достижения в области таргетной терапии привели к многообещающим усилиям в области миотонической дистрофии и фациоскапуло-плечевой мышечной дистрофии.
Федеральное финансирование через NIH и другие агентства, а также программы венчурной благотворительности, поддерживаемые группами защиты интересов пациентов, привлекли инвестиции биотехнологических и фармацевтических фирм в терапию для MD.
В настоящее время используются различные стратегии для разработки новых лекарственных и биологических методов лечения целого ряда MD.Изучаемые стратегии либо специфичны для конкретного типа БД, либо могут касаться прогрессирования заболевания, которое может относиться к нескольким типам БД.
Генная заместительная терапия
Генная терапия имеет потенциал для непосредственного устранения основной причины БМ, обеспечивая выработку недостающего белка. Препятствия, которые необходимо преодолеть, включают определение времени терапии (для возможного преодоления генетического дефекта), предотвращение или ослабление потенциальных иммунных ответов на замещающий ген, а в случае Дюшенна — большой размер заменяемого гена.Для тех, у кого есть последствия для центральной нервной системы (врожденная мышечная дистрофия и миотоническая дистрофия), исследователи разрабатывают и корректируют векторы генной терапии (способ доставки генетического материала в клетки), которые могут преодолевать защитный гематоэнцефалический барьер.
Недавний прогресс в доставке замещающих генов при MD включает в себя значительное уточнение типов вирусных векторов, которые улучшают нацеливание на скелетные мышцы и сосудистые подходы для доставки замещающего гена в большинство или все скелетные мышцы.Подходы, которые работают для скелетных мышц, могут работать или не работать для сердечной мышцы; это проблема, которую необходимо решить, поскольку многие MD вызывают кардиомиопатию. Стратегиям оценки потенциальных иммунных ответов на белки, кодируемые замещающими генами, и управления этими ответами также уделялось значительное внимание в исследованиях на животных моделях и в клинических испытаниях на людях. Наконец, для некоторых MD может потребоваться раннее выявление мутаций, вызывающих заболевание, посредством скрининга новорожденных, чтобы использовать генную заместительную терапию на достаточно раннем этапе, чтобы смягчить прогрессирование заболевания.
Клинические испытания стратегии генной терапии у MD продолжаются для лечения мышечной дистрофии Дюшенна и пояса конечностей. Инъекции векторов генной терапии в отдельные мышцы участников были сделаны в качестве первого шага для установления безопасности подхода. При поддержке обширных исследований на животных моделях, клинические испытания в настоящее время продвигаются к тестированию генной терапии всех мышц целых конечностей с использованием подхода изолированной сосудистой доставки. Если подходы к изолированной доставке конечностей окажутся безопасными и эффективными, исследования перейдут к системной доставке векторов генной терапии, чтобы можно было лечить все мышцы одновременно.
Утрофин — это белок, который тесно связан с дистрофином и не влияет на генные мутации, вызывающие MD Дюшенна. Нацеливание на повышенную экспрессию утрофина может оказаться полезным подходом при лечении MD Дюшенна. NIH поддерживает как генную терапию, так и программы разработки низкомолекулярных лекарств для увеличения мышечной продукции атрофина.
Наконец, группы, финансируемые Национальным институтом здравоохранения (NIH), обнаружили гены-модификаторы — гены, деятельность которых снижает тяжесть MD.Эти гены, в том числе латентный TGF-связывающий белок 4 и остеопонтин, представляют собой новые терапевтические мишени для потенциального снижения тяжести некоторых типов мышечной дистрофии.
Генетическая модификационная терапия для обхода наследственных мутаций
У большинства людей с Дюшенном есть мутации в гене дистрофина, которые вызывают его неправильное функционирование и прекращают производство белка дистрофина. Манипулируя процессом синтеза белка, производство гена, который либо «считывает», либо «пропускает» генетическую мутацию, может привести по крайней мере к частичному функциональному дистрофину.
В настоящее время изучаются две стратегии обхода мутаций дистрофина, одна из которых — лекарства, которые заставляют механизм синтеза белка игнорировать сигнал преждевременной остановки и продуцировать функциональный дистрофин. Эта стратегия, которая потенциально может быть полезна примерно у 15% пациентов с MD Дюшенна, в настоящее время проходит клинические испытания. Во-вторых, в более современном подходе используются антисмысловые олигонуклеотиды (короткие цепи нуклеиновой кислоты, предназначенные для блокирования передачи некоторой генетической информации в производство белка) для изменения сплайсинга и получения почти полноразмерного гена дистрофина, потенциально превращая человека с Дюшенном в очень мягче Becker MD.Две биотехнологические компании в настоящее время тестируют олигонуклеотидные препараты в расширенных клинических испытаниях для людей, которым требуется пропуск 51 экзона дистрофина. (Экзон — это кодирующая последовательность в гене белка). NINDS и NIAMS поддерживают доклиническую работу над олигонуклеотидными препаратами для людей с MD Дюшенна, которым требуется пропуск экзона 45. В то время как подход пропуска экзона требует «персонализированных лекарств» для подгрупп людей с Дюшенном, которым необходимо пропускать определенные экзоны, до 80 процентов пострадавших людей могут извлечь выгоду из этой новой технологии.
Антисмысловая олигонуклеотидная технология также оценивается для использования при миотонической дистрофии, но по другому механизму, чем в Duchenne MD. При миотонической дистрофии длинные дупликации повторяющихся последовательностей ДНК приводят к образованию токсичной РНК, которая секвестрирует регулятор сплайсинга, Muscleblind, вызывая неправильное сплайсинг многих генов в мышцах и головном мозге. Проект, поддерживаемый NINDS и NIAMS, продвигает олигонуклеотидный терапевтический препарат, предназначенный для разрушения токсичной РНК и смягчения дефектов сплайсинга.Этот подход в сотрудничестве с академическими исследователями, биотехнологическими и фармацевтическими компаниями может помочь всем людям, страдающим миотонической дистрофией, и в ближайшие несколько лет планируется пройти клинические испытания.
Медикаментозная терапия для замедления мышечного истощения путем стимулирования роста мышц или смягчения повреждений, вызванных воспалением
Прогрессирующая потеря мышечной массы в первую очередь ответственна за снижение качества и продолжительности жизни у больных MD.Стратегии медикаментозного лечения, разработанные для замедления дегенерации мышц, могут существенно повлиять на качество жизни. Точно так же скелетные мышцы обладают способностью к самовосстановлению, но их механизмы регенерации и восстановления постепенно истощаются в течение нескольких типов МД. Понимание механизмов восстановления может предоставить новые методы лечения, замедляющие и, возможно, стабилизирующие дегенерацию мышц.
Известно, что кортикостероиды продлевают способность людей с MD Дюшенна ходить до 2 лет, но стероиды имеют существенные побочные эффекты, и механизм их действия неизвестен.Поскольку используются несколько протоколов кортикостероидов, финансируемое NINDS исследование оценивает лекарственные препараты, их эффективность и переносимость в различных дозах, чтобы определить оптимальную клиническую практику для их использования в больнице Дюшенна. Кроме того, биотехнологическая компания, поддерживаемая Национальным центром развития трансляционных наук Национального института здравоохранения США, разрабатывает модифицированный стероид, чтобы повысить его эффективность при Дюшенне, одновременно уменьшая побочные эффекты, которые часто ограничивают использование кортикостероидной терапии.
В рамках доклинической разработки лекарств при поддержке NINDS и NIAMS разрабатывается пептидный терапевтический препарат, который на животных моделях обладает двойной активностью в уменьшении повреждения мышц из-за воспаления, а также в усилении регенерации мышц. Попытки сохранить мышечную массу за счет ингибирования негативного регулятора мышечного роста, миостатина, натолкнулись на некоторые препятствия, включая неудачные клинические испытания, но все еще изучаются.
Клеточная терапия
В мышечных клетках людей с MD часто отсутствует критический белок, такой как дистрофин в MD Дюшенна или саркогликан в некоторых MDs пояснично-конечностей.Ученые изучают возможность того, что недостающий белок можно заменить путем введения мышечных стволовых клеток, способных производить недостающий белок в новых мышечных клетках. Такие новые клетки будут защищены от прогрессирующей дегенерации, характерной для MD, и потенциально восстановят мышечную функцию у пораженных людей.
Естественная регенеративная способность мышц обеспечивает возможность лечения MD. Исследователи показали, что стволовые клетки можно использовать для доставки функционального гена дистрофина в скелетные мышцы мышей и собак с дистрофией.В центре внимания исследований было выявление типов клеток с наивысшим потенциалом для приживления и роста мышц, а также стратегии доставки этих клеток-предшественников мышц в скелетные мышцы человека. В целом, клеточные терапевтические подходы рассматриваются для многих типов БД.
Продвижение вперед с исследованиями в MD
До недавнего времени большинство программ развития терапии в MD было сосредоточено на Дюшенне. Благодаря значительному прогрессу в понимании механизмов заболевания, в настоящее время предпринимаются значительные усилия по разработке терапии для многих типов БМ.Финансирование NINDS поддерживает команды, работающие над механизмами заболевания при фасциально-плечевой мышечной дистрофии, вовлечением центральной нервной системы в миотоническую дистрофию и ролью фиброза в развитии болезни Дюшенна. Точно так же проекты, поддерживаемые NIAMS, определяют новые цели развития терапии, которые вызывают интерес у биотехнологических и фармацевтических компаний, и помогут перейти к программам развития терапии для всех типов MD.
Важно отметить, что параллельные усилия должны быть предприняты для подготовки к клиническим испытаниям, чтобы клинические испытания были возможны, когда кандидат в терапевтический препарат достигнет этой стадии.Реестры пациентов, исследования естественной истории, идентификация биомаркеров, разработка критериев конечных точек клинических испытаний и появление стандартов медицинской помощи — все это имеет важное значение для поддержки клинических испытаний и продвигается при нескольких типах мышечной дистрофии при поддержке партнеров как из государственного, так и из частного сектора. . NIH недавно предпринял несколько новых инициатив в области обучения, развития карьеры и исследований, нацеленных на MD. Эти достижения, наряду с ориентацией NINDS на трансляционные и клинические исследования, приведут к росту клинических испытаний и многообещающих стратегий лечения.
Закон MD CARE и федеральное обязательство по борьбе с мышечной дистрофией
В декабре 2001 года президент Джордж Буш подписал Закон 2001 года о поправках к Закону о помощи, исследованиях и образовании сообществ по лечению мышечной дистрофии (Закон о MD CARE, Public Закон 107-84). Закон о MD-CARE был повторно авторизован в 2008 году.
В ответ на Закон о MD CARE, NIH сформировал Координационный комитет по мышечной дистрофии, чтобы помочь в проведении исследований по MD. Координационный комитет MD состоит из врачей, ученых, профессиональных сотрудников NIH, а также представителей других федеральных агентств и добровольных организаций здравоохранения, специализирующихся на MD.Цель группы — помочь NIH добавить новые возможности к национальным усилиям по пониманию и лечению MD без дублирования существующих программ. Координационный комитет MD разработал широкий план действий по мышечным дистрофиям и продолжает уточнять план по улучшению фундаментальных, трансляционных и клинических исследований в области MD с целью улучшения качества жизни людей с MD. Информация о комитете и плане доступна на https://mdcc.nih.gov/.
NIH расширяет и интенсифицирует свои исследования мышечных дистрофий и учредил сенатора Пола Д.Центры совместных исследований мышечной дистрофии Wellstone для содействия фундаментальным и клиническим исследованиям этих заболеваний. Шесть центров Wellstone в настоящее время финансируются NINDS, NIAMS, NICHD и NHLBI. Закон также уполномочил Центры по контролю и профилактике заболеваний выдавать гранты на эпидемиологические исследования, сбор данных и разработку стандартов лечения для некоторых типов МД. Другие федеральные агентства вносят свой вклад в эту исследовательскую инициативу.
Исследования привели к открытию механизмов заболевания и улучшили лечение многих форм MD.Текущие исследования обещают привести к дальнейшим улучшениям. В ближайшие годы врачи и больные могут рассчитывать на новые формы терапии, разработанные на основе понимания уникальных особенностей MD.
Где я могу получить дополнительную информацию?
Для получения дополнительной информации о неврологических расстройствах или исследовательских программах, финансируемых Национальным институтом неврологических расстройств и инсульта, свяжитесь с Институтом мозговых ресурсов и информационной сети (BRAIN) по телефону:
МОЗГ
П.О. Box 5801
Bethesda, MD 20824
800-352-9424
Информацию также можно получить в следующих организациях:
Ассоциация мышечной дистрофии
Национальный офис — 222 S. Riverside Plaza
Suite 1500
Chicago, IL 60606
[email protected]
Тел .: 800-572-1717
Факс: 520-529-5300
Parent Project Muscular Dystrophy (PPMD)
401 Hackensack Avenue, 9th Floor
Hackensack, NJ 07601
[email protected]
Tel: 800-714-KIDS (5437)
Fax: 201-944-9987
Cure CMD
P.О. Box 701
Olathe, KS 66051
[email protected]
Tel: 424-265-0874
Facioscapulohumeral Muscular Dystrophy (FSH) Society
64 Grove Street
Watertown, MA 02472
[email protected]
Тел: 617-658-7877
Факс: 617-658-7879
Coalition to Cure Calpain 3 (C3)
15 Compo Parkway
Westport, CT 06880
[email protected]
Тел .: 203-221-1611
Факс: 734-668-4755
Фонд миотонической дистрофии
1004 O’Reilly Avenue
San Francisco, CA 94129
info @ myotonic.org
Tel: 86-MYOTONIC; 415-800-7777
Jain Foundation
9725 Third Avenue NE
Suite 204
Seattle, WA 98115
[email protected]; admin @ jain-foundation
Тел .: 425-882-1440
Факс: 425-658-1703
Национальный институт артрита, скелетно-мышечных и кожных заболеваний (NIAMS)
Национальные институты здравоохранения, DHHS
31 Центр доктора, Rm. 4C02 MSC 2350
Bethesda, MD 20892-2350
[email protected]
Тел .: 301-496-8190; 877-22-НИАМС (226-4267)
Национальный институт здоровья детей и развития человека (NICHD)
Национальные институты здоровья, DHHS
31 Center Drive, Rm.2A32 MSC 2425
Bethesda, MD 20892-2425
Тел .: 301-496-5133
Факс: 301-496-7101
Центры по контролю и профилактике заболеваний (CDC)
Министерство здравоохранения и социальных служб США
1600 Clifton Road
Atlanta, GA 30333
[email protected]
Tel: 800-311-3435; 404-639-3311; 404-639-3543
атрофия — уменьшение или истощение части тела или ткани.
аутосомно-доминантный — образец наследования, при котором ребенок приобретает болезнь, получая нормальный ген от одного родителя и дефектный ген от другого родителя.
аутосомно-рецессивный — схема наследования, при которой оба родителя несут и передают дефектный ген своему ребенку.
биопсия — процедура, при которой ткань или другой материал удаляется из организма и исследуется на наличие признаков заболевания.
носитель — человек, не имеющий заболевания, но имеющий один нормальный ген и один ген генетического нарушения и, следовательно, способный передать эту болезнь своим детям.
хромосом — генетические структуры, содержащие ДНК.
контрактура — хроническое укорочение мышцы или сухожилия, ограничивающее подвижность костного сустава, например локтя.
креатинкиназа — белок, необходимый для химических реакций, производящих энергию для мышечных сокращений; высокий уровень в крови указывает на повреждение мышц.
дистрофин — белок, помогающий поддерживать форму и структуру мышечных волокон.
электромиография — запись и исследование электрических свойств скелетных мышц.
гликопротеин — молекула, имеющая белковый и углеводный компонент.
исследований сцепления — тесты, проводимые среди членов семьи для определения того, как генетический признак передается из поколения в поколение.
лордоз — аномальное искривление позвоночника вперед.
мерозин — белок, содержащийся в соединительной ткани, окружающей мышечные волокна.
мышечное истощение — уменьшение мышечной силы и размера.
миоглобин — кислородсвязывающий белок в мышечных клетках, который генерирует энергию, превращая глюкозу в углекислый газ и воду.
миопатия — любое поражение мышечной ткани или мышц.
миотония — неспособность расслабить мышцы после внезапного сокращения.
невропатия — заболевание или дисфункция нервной системы, которое может вызывать симптомы, включая мышечную слабость, потерю мышечной массы, мышечные судороги и спазмы, а также боль.
псевдогипертрофия — состояние, при котором мышцы могут увеличиваться за счет накопления жира и соединительной ткани, в результате чего они выглядят больше и здоровее, чем они есть на самом деле.
сколиоз — аномальное искривление позвоночника вбок или в сторону.
Х-сцепленный рецессивный — модель наследования болезни, при которой мать несет пораженный ген на хромосоме, определяющей пол ребенка, и передает его своему сыну.
«Мышечная дистрофия: надежда благодаря исследованиям», NINDS, дата публикации август 2013 г.
Публикация NIH № 13-77
Вернуться на страницу информации о мышечной дистрофии
См. Список всех заболеваний NINDS
Publicaciones en Español
Дистрофия мышечная
Подготовлено:
Офис по связям с общественностью
Национальный институт неврологических расстройств и инсульта
Национальные институты здравоохранения
Бетесда, Мэриленд 20892
Материалы
NINDS, связанные со здоровьем, предоставляются только в информационных целях и не обязательно представляют собой поддержку или официальную позицию Национального института неврологических расстройств и инсульта или любого другого федерального агентства.Консультации по лечению или уходу за отдельным пациентом следует получать после консультации с врачом, который обследовал этого пациента или знаком с историей болезни этого пациента.
Вся информация, подготовленная NINDS, находится в открытом доступе и может свободно копироваться. Благодарность NINDS или NIH приветствуется.
| Тип | Возраст начала болезни | Симптомы, скорость прогрессирования и ожидаемая продолжительность жизни |
|---|---|---|
| Беккер | от подросткового до раннего взросления | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы почти идентичны Дюшенну, но менее серьезны; прогрессирует медленнее, чем по Дюшенну; дожить до среднего возраста.Как и в случае с Дюшенном, болезнь почти всегда ограничивается мужчинами. |
| Врожденный | рождение | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают общую мышечную слабость и возможные деформации суставов; болезнь медленно прогрессирует; сокращенная продолжительность жизни. |
| Дюшенна | От 2 до 6 лет | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают общую мышечную слабость и истощение; поражает таз, плечи и голени; в конечном итоге задействует все произвольные мышцы; выживание после 20 лет — редкость.Встречается только у мальчиков. Очень редко может повлиять на женщину, у которой гораздо более легкие симптомы и лучший прогноз. |
| Дистальный | От 40 до 60 лет | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают слабость и истощение мышц рук, предплечий и голеней; прогрессирование медленное; редко приводит к полной нетрудоспособности. |
| Эмери-Драйфус | от детства до раннего подросткового возраста | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают слабость и истощение мышц плеча, предплечья и голени; суставные деформации распространены; прогрессирование медленное; внезапная смерть может наступить из-за проблем с сердцем. |
| Лицево-лопаточно-плечевой | от детства к ранним взрослым | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают слабость и слабость лицевых мышц с некоторым истощением плеч и предплечий; прогрессирование медленное с периодами быстрого ухудшения; продолжительность жизни может составлять многие десятилетия после начала заболевания. |
| Ремень для конечностей | от позднего детства до среднего возраста | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают слабость и истощение, поражающие в первую очередь плечевой и тазовый пояс; прогрессирование медленное; смерть обычно наступает из-за сердечно-легочных осложнений. |
| Миотонический | От 20 до 40 лет | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы включают слабость всех групп мышц, сопровождающуюся замедленным расслаблением мышц после сокращения; поражает в первую очередь лицо, ступни, руки и шею; прогрессирование идет медленно, иногда от 50 до 60 лет. |
| Окулофарингеальный | От 40 до 70 лет | Возраст на момент начала заболевания Возраст на момент начала заболевания Симптомы поражают мышцы век и горла, вызывая ослабление мышц горла, что со временем приводит к неспособности глотать и истощению из-за недостатка пищи; прогрессирование идет медленно. |
Заболевания нервной системы | Определение, обследование, патология и типы
История болезни
Старая поговорка в медицине: «Слушайте пациента; он сообщает вам диагноз », — особенно верно в неврологии. Описание симптомов пациентом — ценный инструмент, который позволяет врачу узнать о природе и локализации возможного неврологического заболевания. Изучая историю болезни пациента, невролог отмечает уровень осведомленности пациента, потерю памяти, осанку и походку, манеру поведения и выражение, речь и, в некоторой степени, личность.Невролог также отмечает такие симптомы, как боль, головная боль, потеря чувствительности, слабость, нарушение координации, истощение определенных групп мышц и аномальные движения.
Способность замечать окружающую среду и реагировать на нее — это не временное явление, а континуум. Из состояния полной бдительности человек может перейти через сонливость в ступор, состояние, при котором осознанность значительно снижается, и наилучшим двигательным ответом на стимуляцию является стон или другая голосовая (но не вербальная) реакция. Более глубокие уровни бессознательного проходят через легкую кому, в которой сильная стимуляция вызывает только неуклюжий двигательный ответ, до глубокой комы, в которой есть только рефлекторное движение или нет никакой реакции.Такое угнетение сознания возникает при нарушении функций ствола мозга или коры головного мозга. Заболевания ствола мозга могут вызвать кому, если ствол мозга сдавлен другими частями мозга, опухшими из-за болезни, или если он поражен местной болезнью, такой как энцефалит, инсульт или сотрясение мозга. Заболевания коры головного мозга, вызывающие кому, включают отравление седативными препаратами, недостаток глюкозы или кислорода в крови, кровоизлияние в мозг и некоторые редкие инфильтративные расстройства, при которых снижение уровня сознания происходит в течение недель или месяцев.Кратковременные периоды потери сознания, о которых пациент может не знать, возникают при многих формах эпилепсии, нарколепсии, повторяющихся приступах низкого уровня сахара в крови и снижении кровоснабжения головного мозга, особенно ствола мозга.
Получите подписку Britannica Premium и получите доступ к эксклюзивному контенту.
Подпишитесь сейчас
Головная боль
Когда давление внутри черепа увеличивается, чувствительные к боли структуры в головном мозге и вокруг него искажаются и вызывают боль в плохо локализованной области, но часто идентифицируемой в передней или задней части головы, что называется тракционной головной болью.Тяговые головные боли могут быть вызваны отеком мозга, инфекцией, кровотечением, опухолью, стрессом или затрудненным оттоком спинномозговой жидкости. Кроме того, боль может ощущаться в области головы, хотя заболевание, вызывающее боль, находится в другом месте; Примером может служить лицевая боль, иногда ощущаемая при недостатке крови к сердцу. Местное заболевание таких структур черепа, как челюстные суставы, придаточные пазухи носа и зубы, среднее ухо и сами кости черепа, также может вызывать боль.
Головные боли напряжения вызваны длительным чрезмерным сокращением мышц, которые проходят по черепу спереди назад; эти головные боли часто вызваны стрессом.Обычно описывается постоянная давящая или тянущая боль, часто с пульсирующей составляющей. Мигрень может возникать одновременно с головной болью напряжения и характеризуется пульсирующей болью с болезненностью кожи головы, тошнотой, рвотой и чувствительностью к шуму и свету.
Когнитивные изменения
Плохая концентрация, вызванная озабоченностью, усталостью или депрессией, является наиболее частой причиной потери памяти, но также являются причинами широко распространенных заболеваний мозга, авитаминоза, эпилепсии и деменции (потери умственных способностей).Когда период потери памяти четко определен, основными причинами являются травма головы, судороги, отравление (например, алкоголем) и кратковременные эпизоды недостаточного кровоснабжения мозга. Нарушение понимания, рассуждения, логического мышления и способности планировать наперед также могут быть симптомами неврологического расстройства.
Языковой и речевой дефицит
Пациенты с афазией могут точно знать, что они хотят сказать, но они не могут выразить свои мысли устными (а часто и письменными) словами.Они также могут быть не в состоянии понимать значение устной или письменной речи, поэтому нормальная речь звучит как иностранный язык. Инсульт — самая частая причина афазии, но причиной может быть любое очаговое заболевание головного мозга.
Сходными проблемами языка и понимания речи являются апраксия и агнозия. Апраксия — это неспособность выполнять полезные или умелые действия; Пациенты с апраксией могут назвать такой предмет, как гребень или ключ, но они могут не знать, как им пользоваться. Агнозия — это неспособность понять значение неязыкового стимула; больной агнозом может быть не в состоянии распознать источник звука музыкального инструмента.
Дизартрия, или затруднение артикуляции, обычно вызвано аномалией нервов и мышц во рту и вокруг рта или в их соединениях. Проблемы с воспроизведением речевых звуков, называемые дисфонией, часто указывают на проблему, затрагивающую гортань или нервы и мышцы этой структуры. Поскольку черепные нервы, снабжающие эти области, берут начало в стволе мозга, неврологические заболевания этой области также могут быть причиной.
Найдите нервно-мышечное заболевание | Ассоциация мышечной дистрофии
Чтобы узнать больше об одном из этих нервно-мышечных заболеваний, а также об исследованиях, уходе и поддержке, которые мы предоставляем, воспользуйтесь строкой поиска или найдите заболевание в списке ниже.
Узнайте о реакции MDA на COVID-19
Мышечные дистрофии
Мышечные дистрофии — это группа заболеваний, вызывающих слабость и дегенерацию скелетных мышц.
Болезни двигательных нейронов
При заболевании двигательных нейронов нервные клетки, называемые двигательными нейронами, постепенно теряют функцию, в результате чего контролируемые ими мышцы становятся слабыми, а затем становятся нефункциональными.
Заболевания ионных каналов
Заболевания, связанные с дефектами белков, называемых ионными каналами, обычно характеризуются мышечной слабостью, отсутствием мышечного тонуса или эпизодическим параличом мышц.
Митохондриальные болезни
Митохондриальные заболевания возникают, когда структуры, вырабатывающие энергию, нарушают работу клеток.
Миопатии
Миопатия — это мышечное заболевание, при котором мышечные волокна не функционируют должным образом, что приводит к мышечной слабости.
Болезни нервно-мышечного соединения
Расстройства нервно-мышечного соединения возникают в результате разрушения, неисправности или отсутствия одного или нескольких ключевых белков, участвующих в передаче сигналов между мышцами и нервами.
Болезни периферических нервов
При заболеваниях периферических нервов поражаются двигательные и сенсорные нервы, которые соединяют головной и спинной мозг с остальными частями тела, вызывая нарушение чувствительности, движения или других функций.
Спинальная мышечная атрофия (СМА) — Заболевания
Спинальная мышечная атрофия (СМА)
Что такое мышечная атрофия позвоночника?
Спинальная мышечная атрофия (СМА) — это генетическое заболевание, поражающее центральную нервную систему, периферическую нервную систему и произвольные движения мышц (скелетные мышцы).
Большинство нервных клеток, управляющих мышцами, расположены в спинном мозге, поэтому в названии болезни содержится слово spinal . SMA — это мышечная , потому что ее основное воздействие на мышцы, которые не получают сигналы от этих нервных клеток. Атрофия — медицинский термин, обозначающий уменьшение размеров, что обычно происходит с мышцами, когда они не стимулируются нервными клетками.
SMA включает потерю нервных клеток, называемых двигательными нейронами, в спинном мозге и классифицируется как болезнь двигательных нейронов.
В наиболее распространенной форме СМА (СМА 5 хромосомы или СМА, связанная с SMN) существует большая вариабельность возраста начала, симптомов и скорости прогрессирования. Чтобы учесть эти различия, связанная с 5-й хромосомой СМА, которая часто является аутосомно-рецессивной, классифицируется на типы с 1 по 4.
Возраст, в котором появляются симптомы СМА, примерно коррелирует со степенью нарушения двигательной функции: чем раньше возраст начала, тем сильнее влияние на двигательную функцию.Дети, у которых проявляются симптомы при рождении или в младенчестве, обычно имеют самый низкий уровень функционирования (тип 1). Позднее начало СМА с менее тяжелым течением (типы 2 и 3, а у подростков или взрослых — тип 4) обычно коррелирует со все более высокими уровнями двигательной функции.
Подробнее см. Формы SMA.
Что вызывает СМА?
СМА хромосомы 5 вызывается дефицитом белка мотонейрона, называемого SMN, для «выживания мотонейрона». Этот белок, как следует из его названия, по-видимому, необходим для нормальной функции двигательных нейронов.SMN играет ключевую роль в экспрессии генов в двигательных нейронах. Его дефицит вызван генетическими дефектами (мутациями) хромосомы 5 в гене SMN1 . Наиболее частая мутация в гене SMN1 у пациентов с диагнозом SMA — это делеция целого сегмента, называемого экзоном 7. 1 Соседние SMN2 гены могут частично компенсировать нефункциональные гены SMN1, поскольку между ними 99% идентичности. эти два гена. 2
Другие редкие формы SMA (не хромосомы 5) вызываются мутациями в генах, отличных от SMN1 . 3
Для получения дополнительной информации, включая подробные сведения о редких, не связанных хромосомами 5 СМА, см. «Формы СМА» и «Причины / наследование».
Каковы симптомы СМА?
Симптомы СМА охватывают широкий спектр, от легких до тяжелых.
Основным симптомом СМА, связанной с хромосомой 5 (SMN), является слабость произвольных мышц. Наиболее поражены мышцы, расположенные ближе всего к центру тела, например, плечи, бедра, бедра и верхняя часть спины.Кажется, что нижние конечности поражены больше, чем верхние, а глубокие сухожильные рефлексы снижены. 4
Особые осложнения возникают при поражении мышц, используемых для дыхания и глотания, что приводит к нарушению этих функций. Если мышцы спины слабеют, могут развиться искривления позвоночника.
Возраст начала и уровень двигательной функции, достигнутой при СМА, связанной с хромосомой 5, сильно различаются. Они примерно коррелируют с тем, сколько функционального белка SMN присутствует в мотонейронах, что, в свою очередь, коррелирует с количеством копий генов SMN2 , которые есть у человека.Сенсорные, умственные и эмоциональные функции при СМА 5-й хромосомы полностью нормальны.
Некоторые формы SMA не связаны с хромосомой 5 или дефицитом SMN. Эти формы сильно различаются по степени тяжести и наиболее пораженным мышцам. В то время как большинство форм, таких как форма, связанная с хромосомой 5, затрагивает в основном проксимальные мышцы, существуют другие формы, которые влияют в основном на дистальные мышцы (те, что дальше от центра тела) — по крайней мере, вначале.
Подробнее см. Признаки и симптомы.
Как прогрессирует СМА?
При СМА, связанной с хромосомой 5, чем позже проявляются симптомы и чем больше в нем белка SMN, тем вероятнее будет более легкое течение заболевания. Если раньше младенцы со СМА обычно не выживали более двух лет, сегодня большинство врачей считают СМА, связанную с СМА, континуумом, и предпочитают не делать жестких прогнозов относительно продолжительности жизни или слабости, основываясь исключительно на возрасте начала.
SMA — наиболее частая генетическая причина смертности младенцев.
Каков статус исследований SMA?
Исследования были сосредоточены на стратегиях увеличения производства в организме белка SMN, отсутствующего в формах заболевания, связанных с хромосомой 5. Подходы включают методы, помогающие двигательным нейронам выжить в неблагоприятных обстоятельствах.
23 декабря 2016 г. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило Spinraza (nusinersen) для лечения СМА. Спинраза предназначена для лечения основного дефекта СМА, что означает, что он потенциально может быть эффективным для замедления, остановки или, возможно, обращения симптомов СМА.Для получения дополнительной информации см. Спинраза одобрена.
В мае 2019 года FDA одобрило Zolgensma (онасемноген abeparvovac-xioi), первую генную заместительную терапию нервно-мышечного заболевания. Золгенсма — это одноразовая внутривенная (в вену) инфузия для лечения детей младше 2 лет с СМА с биаллельными мутациями в гене SMN1 , включая тех, у которых на момент постановки диагноза наблюдаются предсимптомные симптомы. Для получения дополнительной информации прочтите, что FDA утверждает Zolgensma AveXis для лечения спинальной мышечной атрофии у педиатрических пациентов .
Для получения дополнительной информации см. Исследование SMA: впереди на полной скорости и в фокусе: мышечная атрофия позвоночника (SMA). Истории семей, живущих с SMA, можно найти в наших историях о SMA на Strongly, в блоге MDA .
В августе 2020 года FDA одобрило рисдиплам (торговая марка Evrysdi *) для лечения СМА у взрослых и детей в возрасте двух месяцев и старше. Evysdi — это пероральный препарат, предназначенный для повышения уровня белка SMN за счет увеличения выработки «резервного» гена SMN2.
Загрузите наш информационный бюллетень о спинальной мышечной атрофии
Узнайте о реакции MDA на COVID-19
Список литературы
- Огино, С. и Уилсон, Р. Б. Генетическое тестирование и оценка риска спинальной мышечной атрофии (СМА). Генетика человека (2002). DOI: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Идентификация и характеристика гена, определяющего мышечную атрофию позвоночника. Ячейка (1995).DOI: 10.1016 / 0092-8674 (95)
-3 - Даррас, Б. Т. Спинальные мышечные атрофии без 5q: буквенно-цифровой суп густеет. Неврология (2011). DOI: 10.1212 / WNL.0b013e3182267bd8
- Арнольд В. Д., Кассар Д. и Киссель Дж. Т. Спинальная мышечная атрофия: диагностика и лечение в новую терапевтическую эру. Мышцы и нервы (2015). DOI: 10.1002 / mus.24497
Детская нейроаксональная дистрофия: MedlinePlus Genetics
Детская нейроаксональная дистрофия — это заболевание, которое в первую очередь поражает нервную систему.Люди с детской нейроаксональной дистрофией обычно не имеют никаких симптомов при рождении, но в возрасте от 6 до 18 месяцев они начинают испытывать задержки в приобретении новых моторных и интеллектуальных навыков, таких как ползание или начало говорить. В конце концов они теряют ранее приобретенные навыки (регресс в развитии). В некоторых случаях признаки и симптомы детской нейроаксональной дистрофии впервые появляются позже в детстве или в подростковом возрасте и прогрессируют медленнее.
Дети с детской нейроаксональной дистрофией испытывают прогрессирующие трудности с движением.Обычно у них мышцы сначала слабые и «вялые» (гипотонические), а затем постепенно становятся очень жесткими (спастическими). В конце концов, пораженные дети теряют способность двигаться самостоятельно. Недостаток мышечной силы вызывает трудности с кормлением. Слабость мышц также может привести к проблемам с дыханием, что может привести к частым инфекциям, таким как пневмония. У некоторых детей возникают судороги.
Быстрые непроизвольные движения глаз (нистагм), глаза, которые не смотрят в одном направлении (косоглазие), и потеря зрения из-за ухудшения (атрофии) нерва, который передает информацию от глаза к мозгу (зрительный нерв) часто возникают при детской нейроаксональной дистрофии.Также может развиться потеря слуха. Дети с этим расстройством испытывают прогрессирующее ухудшение когнитивных функций (слабоумие) и в конечном итоге теряют осведомленность об окружающем.
Детская нейроаксональная дистрофия характеризуется развитием опухолей, называемых сфероидными телами, в аксонах, волокнах, которые отходят от нервных клеток (нейронов) и передают импульсы мышцам и другим нейронам. У некоторых людей с детской нейроаксональной дистрофией аномальное количество железа накапливается в определенной области мозга, называемой базальными ганглиями.Связь этих особенностей с симптомами детской нейроаксональной дистрофии неизвестна.
Роль дистрогликана в нервной системе: выводы, полученные на животных моделях мышечной дистрофии | Модели и механизмы заболеваний
Агрин: протеогликан, секретируемый нервными окончаниями, который связывается с MuSK и αDG на постсинаптической мышечной мембране. Агрин является главным инструктивным секретируемым сигналом для образования нервно-мышечных соединений.
Базальная мембрана: компактных листа полимеризованных матричных белков, обычно состоящих из ламинина, коллагена, перлекана и белков нидогена.Базальные мембраны можно разделить на три слоя на основе электронной микроскопии: (1) разреженная электронами «lamina lucida» на поверхности клетки, состоящая из связывающего клетки длинного плеча ламинина, (2) лежащая сверху «lamina densa» типа Внутривенный коллаген, перлекан, нидоген и сшивающие более короткие ветви ламинина (3) и, кроме того, «ретикулярная пластинка» фибриллярных коллагенов. Эта молекулярная решетка образует периферию многих органов, служит для закрепления отдельных клеток и обеспечивает основу для тканевой структуры у всех многоклеточных организмов.
Лиссэнцефалия булыжника: состояние развития, характеризующееся необычно гладкой поверхностью мозга с аномально сформированными извилинами, напоминающими «брусчатку», также называемое лиссэнцефалией типа II. Это происходит в результате чрезмерной миграции нейронов в субарахноидальное пространство, в результате чего образуется гладкая поверхность с мелкими булыжниками и лежащими под ней плотными складками или извилинами, напоминающими полимикрогирию.
Cre-driver line: линия мышей, генетически сконструированная с геном Cre-рекомбиназы, управляемым выбранным промотором.При скрещивании со штаммом, несущим стратегически размещенные сайты loxP в интересующем гене (флокированные экзоны), это приведет к временному и селективному по типу клеток нокауту гена (так называемая система Cre / lox ).
DAG1 : ген, кодирующий αDG и βDG, которые транскрибируются и транслируются как один и расщепляются посттрансляционно.
Электроретинограмма (ERG): электродное измерение передачи сигналов между фоторецепторами и их нижележащими биполярными и ганглиозными клетками сетчатки.На графике ЭРГ первое отклонение — это a-волна, представляющая активность фоторецепторов, а второе отклонение — это b-волна, опосредованная ганглиозными и биполярными клетками.
Эмбриоидное тело: культура эмбриональных стволовых клеток в сферических агрегатах, которые дифференцируют внутреннее эпибластоподобное ядро и экстраэмбриональную энтодерму-подобную периферию. Между этими двумя клеточными слоями образуется базальная мембрана, напоминающая мембрану, обнаруженную между эпибластом и примитивной энтодермой эмбриона млекопитающих до гаструляции.
Гликозилирование: посттрансляционный процесс добавления углеводных цепей или гликанов к белкам ферментами гликозилтрансфераз эндоплазматического ретикулума и аппарата Гольджи. Эти сахара часто опосредуют сворачивание белка или белок-белковые взаимодействия. Форма гликозилирования αDG, которая может быть нарушена при α-дистрогликанопатиях, называется O-маннозилированием, что указывает на молекулярную связь и первый добавленный сахар.
Гидроцефалия: состояние повышенного объема спинномозговой жидкости в головном мозге, которое приводит к расширению желудочков и часто черепа.
Гипогликозилирование: уменьшенное количество гликанов на гликозилированном белке. Это обнаруживается по снижению молекулярной массы белка, а в случае αDG это сопровождается сниженной способностью связывать внеклеточные лиганды, такие как ламинин.
Ламинины: массивные крестообразные гетеротримерные белки внеклеточного матрикса, которые связываются с αDG на поверхности клетки и интегрином. Наряду с коллагеном IV типа ламинины являются основными составляющими базальных мембран.Обычно они состоят из одной тяжелой α-цепи и β- и γ-легких цепей. Их номенклатура соответствует номерам цепей α, β и γ, так что гетеротример, состоящий из α2, β1 и γ1, будет обозначен как ламинин 211 или lm211.
Large : ген, кодирующий подобную ацетилглюкозаминилтрансферазу (также известную как Large), конечную бифункциональную гликозилтрансферазу, непосредственно ответственную за синтез матригликанов на αDG.
LG доменов: белковых структур семейства глобулярных доменов ламинина.Они обычно находятся во внеклеточных белках и участвуют во взаимодействии белок-белок.
Матригликан: уникальный гликан, состоящий из повторяющихся дисахаридных единиц (ксилоза и глюкуроновая кислота), который связывается с доменами LG, обнаруженными в ламинине и других белках. Это конечный результат активности Большой гликозилтрансферазы.
MuSK: рецепторная тирозинкиназа, обнаруженная в мембране мышечных клеток, которая опосредует образование нервно-мышечных соединений в ответ на связывание агрина.
Нервная дисплазия: аномальное расположение нейрональных и глиальных клеток в нервной системе в результате нарушения развития, внутри или за пределами их нормальных тканевых границ, вызывая эктопическую локализацию смешанных клеточных популяций (гетеротопия).
Нервно-мышечное соединение (НМС): синапс, образованный между двигательным нейроном и мышечным волокном, позволяющий двигательным импульсам вызывать сокращение мышц.
Узел Ранвье: участок нейрального аксона, в котором отсутствует миелиновая оболочка, обеспечивающая проницаемость для ионов и распространение потенциала действия.Эти промежутки возникают через равные промежутки времени вдоль аксона всех миелинизированных нейронов.
Pia mater: самый внутренний слой менингеальных оболочек, выстилающих мозг. Он состоит из гетерогенной популяции клеток, включая фибробласты, которые секретируют ламинин и другие матричные белки, чтобы сформировать базальную мембрану пиальной поверхности на поверхности мозга.