Гормоны щитовидной железы
Щитовидная железа и ее гормоны совместно с нервной и иммунной системами принимает участие в регуляции работы всех органов человека (сердца, головного мозга, почек и т.д.). В отличии от большинства гормонов, которые действуют только на определенные клетки отдельных органов (например, для эстрадиола это половые органы), гормоны щитовидной железы необходимы для нормальной работы всем тканям и всем органам без исключения. Приникая внутрь клетки, гормон направляется в ядро, где связываясь с определенными участками на хромосомах, активирует комплекс реакций, отвечающих за процессы окисления и восстановления. Гормоны щитовидной железы являются основными регуляторами расхода энергии в организме, и поддержание их концентрации на необходимом уровне крайне важно для нормальной деятельности всех органов и систем. Для синтеза гормонов щитовидной железы необходимы два обязательных компонента — йод и аминокислота тирозин. Без йода синтез гормонов полностью прекращается, поэтому крайне важно обеспечить получение достаточного количества йода с пищей.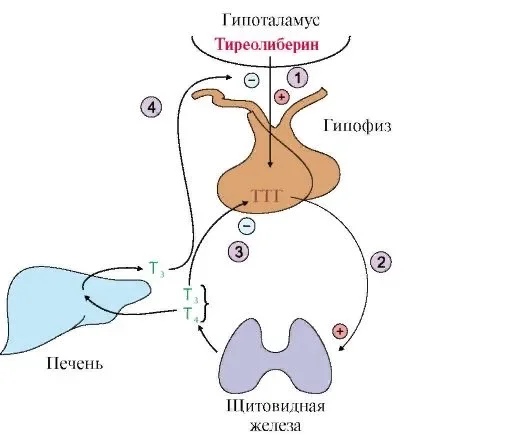 Тирозин также поступает в организм с пищей, он – основа не только гормонов щитовидной железы, но и адреналина, меланина, дофамина.
Тирозин также поступает в организм с пищей, он – основа не только гормонов щитовидной железы, но и адреналина, меланина, дофамина.
Т3 и Т4. Основные два гормона, которые вырабатывает щитовидная железа – трийодтиронин и тетрайодтиронин (тироксин). В состав трийодтиронина входят 3 молекулы йода, а в состав тироксина — 4 молекулы. Сокращённо эти гормоны называют, соответственно, Т3 и Т4. В клетках и тканях нашего организма Т4 постепенно превращается в Т3, который является главным биологически активным гормоном, непосредственно влияющим на обмен веществ. Тем не менее, тироксин (Т4) составляет около 90 % от общего количества гормонов, выделяемых щитовидной железой.
Свободный Т3 и Т4. Гормоны щитовидной железы перед попаданием в кровь должны быть связаны с транспортными белками-глобулинами (для того чтоб не «вымываться» почками), но для попадания внутрь клетки и в ткани они должны освободиться от этого «транспорта».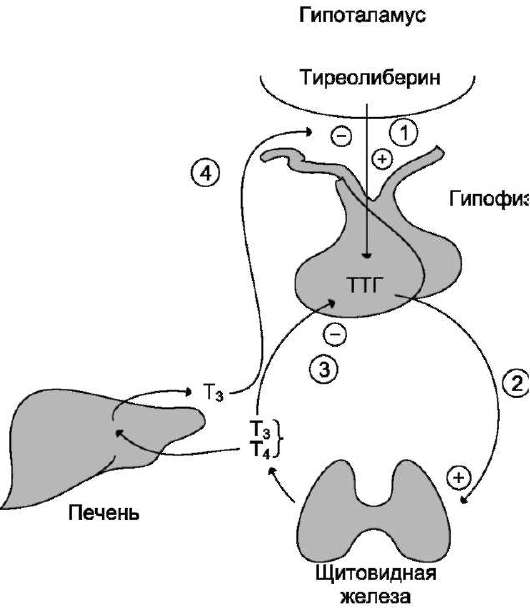 Т.о. в крови Т3 и Т4 встречаются либо в свободном, либо в связанном виде. Уровень свободных гормонов составляет менее 0,1% от общего их количества, но именно свободная фракция гормонов является наиболее биологически активной, и именно они обеспечивают все эффекты гормонов щитовидной железы.
Т.о. в крови Т3 и Т4 встречаются либо в свободном, либо в связанном виде. Уровень свободных гормонов составляет менее 0,1% от общего их количества, но именно свободная фракция гормонов является наиболее биологически активной, и именно они обеспечивают все эффекты гормонов щитовидной железы.
Анализ уровня основных гормонов Т3, Т4 и их свободных вариантов — первый и самый главный шаг в определении качества работы щитовидной железы при любых подозрениях на её заболевание.
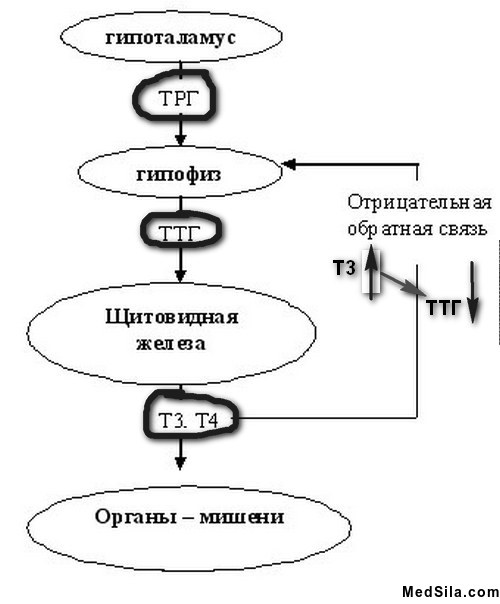
Тиреотропный гормон (ТТГ) – основной регулятор функции щитовидной железы. Вырабатывается гипофизом – небольшой железой, расположенной на нижней поверхности головного мозга. ТТГ управляет выработкой гормонов щитовидной железы (тироксина и трийодтиронина), которые, в свою очередь, регулируют процессы образования энергии в организме. Механизм обратной связи, позволяет поддерживать стабильный уровень этих гормонов — когда их содержание в крови понижается, гипоталамус определяет этот факт и даёт сигнал гипофизу на синтез ТТГ.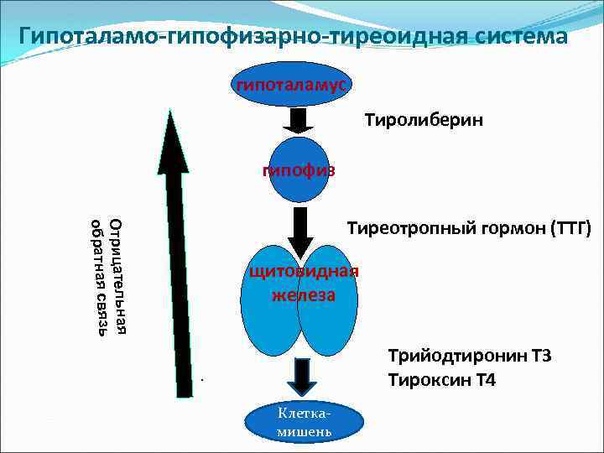 Повышение концентрации ТТГ, в свою очередь, стимулирует выработку тиреоидных гормонов щитовидной железой. Обратный процесс происходит аналогично.Дисфункция гипофиза может вызывать неуправляемое повышение или понижение уровня тиреотропного гормона, провоцируя тем самым, щитовидную железу на выработку тироксина и трийодтиронина в аномальных количествах. Повышение их концентрации становится причиной гипертиреоза, а снижение, соответственно, гипотиреоза. Заболевания гипоталамуса — регулятора секреции ТТГ гипофизом, также могут стать причиной сбоев в этой системе. Кроме того, заболевания щитовидной железы, сопровождающиеся нарушением выработкии тиреоидных гормонов, могут опосредованно (по механизму обратной связи) влиять на синтез тиреотропного гормона, вызывая понижение или повышение его концентрации.
Повышение концентрации ТТГ, в свою очередь, стимулирует выработку тиреоидных гормонов щитовидной железой. Обратный процесс происходит аналогично.Дисфункция гипофиза может вызывать неуправляемое повышение или понижение уровня тиреотропного гормона, провоцируя тем самым, щитовидную железу на выработку тироксина и трийодтиронина в аномальных количествах. Повышение их концентрации становится причиной гипертиреоза, а снижение, соответственно, гипотиреоза. Заболевания гипоталамуса — регулятора секреции ТТГ гипофизом, также могут стать причиной сбоев в этой системе. Кроме того, заболевания щитовидной железы, сопровождающиеся нарушением выработкии тиреоидных гормонов, могут опосредованно (по механизму обратной связи) влиять на синтез тиреотропного гормона, вызывая понижение или повышение его концентрации.
Антитела к тиреоглобулину (АТ-ТГ) и к тиреопероксидазе (АТ-ТПО) Вообще, антитела — это белки, синтезируемые клетками иммунной системы. Их основная функция — выявление и уничтожение чужеродных объектов (бактерий, вирусов, и т. п). Однако случается, что в результате сбоя, организм начинает вырабатывать антитела против собственных здоровых тканей. В щитовидной железе, чаще всего, объектами для выработки антител становятся фермент тиреопероксидаза (ТПО) и основа для синтеза гормонов — тиреоглобулин (ТГ).
п). Однако случается, что в результате сбоя, организм начинает вырабатывать антитела против собственных здоровых тканей. В щитовидной железе, чаще всего, объектами для выработки антител становятся фермент тиреопероксидаза (ТПО) и основа для синтеза гормонов — тиреоглобулин (ТГ).
Тиреоглобулин — это заготовка для гормонов щитовидной железы, из него клетки щитовидной железы «делают» гормоны Т3 и Т4. Вначале клетки вырабатывают тиреоглобулин (создавая т.о. запасы йода), который «складируется на будущее» в специальных емкостях — фолликулах. Потом, по мере необходимости, из тиреоглобулина синтезируются Т3 и Т4.
Тиреоидная пероксидаза – фермент щитовидной железы участвующий в образовании активной формы йода и, таким образом, играющий ключевую роль в выработке гормонов щитовидной железы.
Повреждающее действие антител может приводить к нарушению нормальной продукции гормонов щитовидной железы и негативно влиять на регуляцию её функции, что в итоге вызывает хронические патологии, связанные с гипо- или гипертиреозом. Тем не менее, важно подчеркнуть, что антитела к ТПО и ТГ не являются ключевым звеном в патогенезе аутоиммунных заболеваний щитовидной железы и начинают вырабатываться уже в ответ на её повреждение. Поэтому попытки снижения уровня антител лишены какого-либо практического смысла.
Тем не менее, важно подчеркнуть, что антитела к ТПО и ТГ не являются ключевым звеном в патогенезе аутоиммунных заболеваний щитовидной железы и начинают вырабатываться уже в ответ на её повреждение. Поэтому попытки снижения уровня антител лишены какого-либо практического смысла.
Тесты АТ-ТГ и АТ-ТПО используются для подтверждения или исключения аутоиммунной природы того или иного заболевания щитовидной железы (увеличения щитовидной железы без нарушения её функции, первичного гипо- или гипертиреоза, офтальмопатии и др.), так как это позволяет назначить наиболее эффективную терапию. Тесты также назначаются детям, рождённым от матерей с патологией эндокринных органов, для определения групп риска по развитию заболеваний щитовидной железы. Количественный анализ сыворотки крови на АТ-ТПО – наиболее чувствительный метод диагностики аутоиммунных заболеваний щитовидной железы. Анализ АТ-ТГ ценен при дифференциальной диагностике.
Цены на исследования можно узнать в разделе «Прейскурант» клинической лаборатории.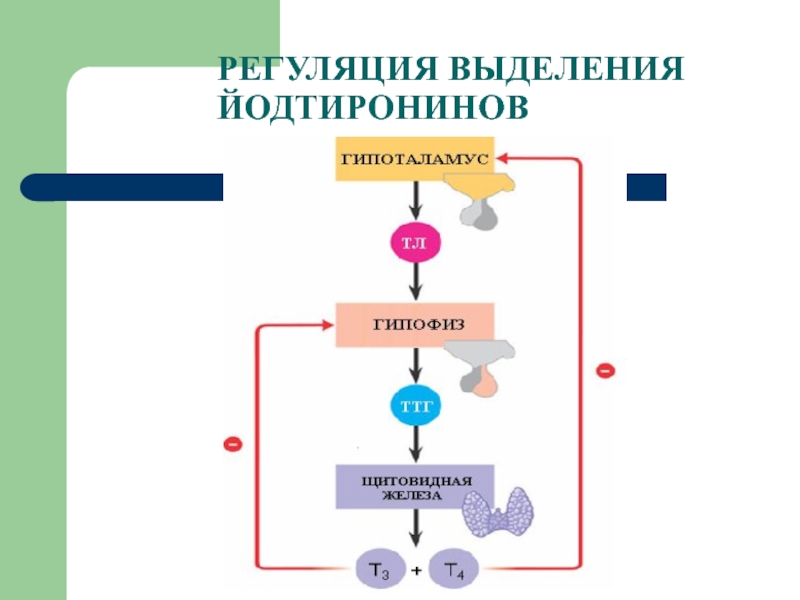 Кровь на исследования принимается ежедневно (кроме воскресенья) с 7 до 11 часов. Строго натощак.
Кровь на исследования принимается ежедневно (кроме воскресенья) с 7 до 11 часов. Строго натощак.
Нервная регуляция функции щитовидной железы Текст научной статьи по специальности «Фундаментальная медицина»
АКТУАЛЬНЫЕ ВОПРОСЫ ПАТОФИЗИОЛОГИИ
А.Э. Лычкова
Центральный научно-исследовательский институт гастроэнтерологии, Москва, Российская Федерация
Нервная регуляция функции щитовидной
железы
В обзоре рассмотрены вопросы вегетативной регуляции функции щитовидной железы. Активация центральных а-адренергических механизмов увеличивает высвобождение тиреотропного гормона гипофиза преимущественно за счет стимуляции его секреции. Дофамин угнетает секрецию этого гормона, воздействуя на D-рецепторы тиреотропоцитов. Ацетилхолин и другие холиномиметики угнетают функциональную активность тиреоцитов при участии мускариновых рецепторов. Наряду с симпатическим и парасимпатическим отделом особое внимание уделено роли серотонинергического отдела вегетативной нервной системы. Серотонин может блокировать секрецию тиреотропина гипофизом, однако оказывает прямое стимулирующее влияние на тиреоциты. Этот стимуляторный эффект серотонина опосредован 5-НТ2-рецепторами. При гипотиреозе угнетен синтез и метаболизм серотонина в мозге. Депрессия сопровождается угнетением активности фермента дейодиназы типа 2, что способствует снижению содержания серотонина при депрессии. Активация 5-НТ1-рецептора приводит к увеличению содержания внутриклеточного кальция, вызывая торможение активности промоутера кальцитонин-ген-ассоциированного пептида.
Ацетилхолин и другие холиномиметики угнетают функциональную активность тиреоцитов при участии мускариновых рецепторов. Наряду с симпатическим и парасимпатическим отделом особое внимание уделено роли серотонинергического отдела вегетативной нервной системы. Серотонин может блокировать секрецию тиреотропина гипофизом, однако оказывает прямое стимулирующее влияние на тиреоциты. Этот стимуляторный эффект серотонина опосредован 5-НТ2-рецепторами. При гипотиреозе угнетен синтез и метаболизм серотонина в мозге. Депрессия сопровождается угнетением активности фермента дейодиназы типа 2, что способствует снижению содержания серотонина при депрессии. Активация 5-НТ1-рецептора приводит к увеличению содержания внутриклеточного кальция, вызывая торможение активности промоутера кальцитонин-ген-ассоциированного пептида.
Ключевые слова: щитовидная железа, нервная регуляция.
49
Введение
Щитовидная железа (ЩЖ) — эндокринная железа позвоночных, депонирующая йод и вырабатывающая йодосодержащие гормоны (йодтиронины), участвующие в регуляции обмена веществ и росте отдельных клеток и организма в целом.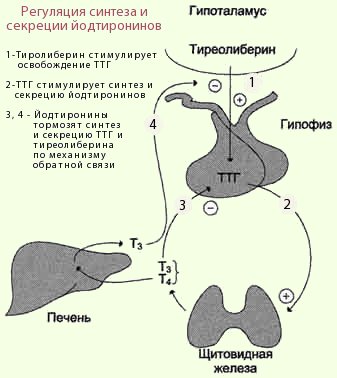 Синтез тироксина (тетрайодтиро-нина, T4) и трийодтиронина (T3) происходит в эпителиальных фолликулярных клетках (тироцитах). Кальци-тонин, пептидный гормон, синтезируется парафолликулярными С-клетками и регулирует уровень кальция в сыворотке. Транскрипты гена кальцитонина кодируют также пептид, известный как кокальцигенин (кальцито-нин ген-ассоциированный пептид, CGRP).
Синтез тироксина (тетрайодтиро-нина, T4) и трийодтиронина (T3) происходит в эпителиальных фолликулярных клетках (тироцитах). Кальци-тонин, пептидный гормон, синтезируется парафолликулярными С-клетками и регулирует уровень кальция в сыворотке. Транскрипты гена кальцитонина кодируют также пептид, известный как кокальцигенин (кальцито-нин ген-ассоциированный пептид, CGRP).
Функция ЩЖ регулируется гипоталамо—гипофизарно-тиреоидной системой, в т.ч. тиреотропин-рилизинг гормоном (ТРГ), синтезируемым гипоталамусом, тире-отропным гормоном гипофиза (ТТГ). ТРГ стимулирует синтез и секрецию ТТГ независимо от наличия или отсутствия тиреоидных гормонов. Деструкция участка ги-
поталамуса, синтезирующего ТРГ, приводит к развитию гипотиреоза. В свою очередь, синтез ТРГ регулируется уровнем гормонов ЩЖ в крови, омывающей гипоталамус, по принципу отрицательной обратной связи. ЩЖ в основном секретирует Т4.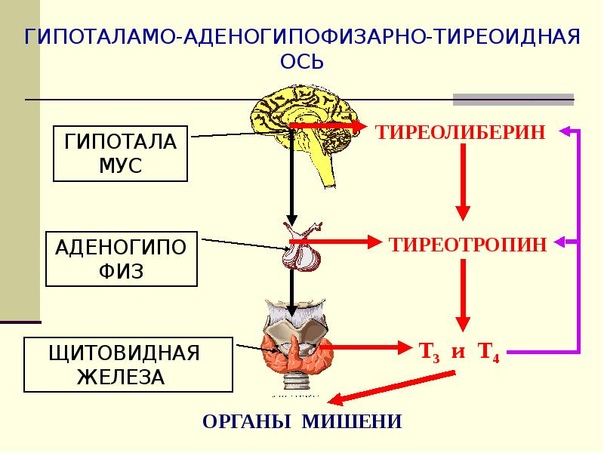 Практически весь пул (80%) циркулирующего в крови Т3 образуется за счет процессов тканевого дейодирования Т4 в печени и почках. Именно избыточное содержание Т3 подавляет синтез и секрецию ТТГ. Двойственное влияние ТРГ и тиреоидных гормонов является основным механизмом управления концентрацией ТТГ в крови. ТТГ, в свою очередь, регулирует синтез тиреоидных гормонов, пролиферацию тироцитов, кровоснабжение железы, что в совокупности обеспечивает поддержание концентрации гормонов ЩЖ на необходимом для индивида уровне [1].
Практически весь пул (80%) циркулирующего в крови Т3 образуется за счет процессов тканевого дейодирования Т4 в печени и почках. Именно избыточное содержание Т3 подавляет синтез и секрецию ТТГ. Двойственное влияние ТРГ и тиреоидных гормонов является основным механизмом управления концентрацией ТТГ в крови. ТТГ, в свою очередь, регулирует синтез тиреоидных гормонов, пролиферацию тироцитов, кровоснабжение железы, что в совокупности обеспечивает поддержание концентрации гормонов ЩЖ на необходимом для индивида уровне [1].
Особое место в регуляции функции ЩЖ занимают нейротрансмиттеры, среди которых ведущее значение придают катехоламинам, серотонину, ацетилхолину, гистамину и оксиду азота. Механизмы нейромедиаторной регуляции тиреоидной функции изучены недостаточно.
#
A.E. Lychkovа
Central Gastroenterology Research Institute, Moscow, Russian Federation
Nervous Regulation оf Thyroid Function
Review examines the autonomic regulation of thyroid function. Review examines the issues of autonomic regulation of the thyroid gland. Activation of the central а-adrenergic mechanisms increases the release of thyroid-stimulating hormone of pituitary mainly due to the stimulation of its secretion. Dopamine inhibits the secretion of this hormone, acting on D2-receptors tireotropotsitov. Acetylcholine and other cholinomimetics inhibit the functional activity of thyrocitebi with the participation of muscarinic receptors. Along the sympathetic and parasympathetic special attention paid to the role of the serotonergic division of vegetative system. Serotonin can inhibit the secretion of thyrotropin by the pituitary gland, but has a direct stimulatory effect on thyrocytes. This stimulatory effect is mediated by the serotonin 5-HT2 receptors. In hypothyroidism synthesis and metabolism of serotonin in the brain are slowed down. Depression is accompanied by inhibition of the enzyme activity deiodinase type 2, thereby reducing the concentration of serotonin.
Review examines the issues of autonomic regulation of the thyroid gland. Activation of the central а-adrenergic mechanisms increases the release of thyroid-stimulating hormone of pituitary mainly due to the stimulation of its secretion. Dopamine inhibits the secretion of this hormone, acting on D2-receptors tireotropotsitov. Acetylcholine and other cholinomimetics inhibit the functional activity of thyrocitebi with the participation of muscarinic receptors. Along the sympathetic and parasympathetic special attention paid to the role of the serotonergic division of vegetative system. Serotonin can inhibit the secretion of thyrotropin by the pituitary gland, but has a direct stimulatory effect on thyrocytes. This stimulatory effect is mediated by the serotonin 5-HT2 receptors. In hypothyroidism synthesis and metabolism of serotonin in the brain are slowed down. Depression is accompanied by inhibition of the enzyme activity deiodinase type 2, thereby reducing the concentration of serotonin.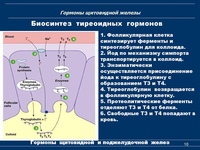 Activation of 5-HT1 receptor lead to increased levels of intracellular calcium, causing inhibition of the promoter of calcitonin gene-associated peptide.
Activation of 5-HT1 receptor lead to increased levels of intracellular calcium, causing inhibition of the promoter of calcitonin gene-associated peptide.
Key words: thyroid gland, nervous regulation.
#
50
ВЕСТНИК РАМН /2013/ № 6
К основным источникам внутриорганного обеспечения ЩЖ нейромедиаторами относят тиреоидные фолликулы, тканевые базофилы, макрофаги стромального происхождения, периваскулярный и парафолликулярный симпатический нервный аппарат, приносящие артерио-лы, перифолликулярные гемокапилляры. Парафолликулярные клетки способны секретировать простагландины и характеризуются высоким содержанием серотонина в гранулах, которые одновременно содержат кальци-тонин. При дегрануляции в ответ на гиперкальциемию происходит одновременное выделение серотонина и кальцитонина. Следовательно, при преганглионарной стимуляции эти физиологически активные вещества могут одновременно выделяться в синаптическую щель, оказывая воздействие на специфические рецепторы.
Следовательно, при преганглионарной стимуляции эти физиологически активные вещества могут одновременно выделяться в синаптическую щель, оказывая воздействие на специфические рецепторы.
Центральные и периферические отделы нервной и эндокринной системы, обеспечивающие ЩЖ регуляторными аминами, включают гипоталамо—гипофизар-ный комплекс, эпифиз, верхние шейные симпатические ганглии, энтерохромаффинные клетки, мозговое вещество надпочечников, параганглии.
В центральной нервной системе (ЦНС) тиреоидные гормоны влияют на экспрессию ряда нейрон-специфич-ных генов, контролируют синтез и метаболизм нейромедиаторов [2, 3]. Нейромедиаторы, в свою очередь, оказывают как прямое, так и опосредованное влияние на активность гипоталамо—гипофизарно—тиреоидной системы.
Вегетативная регуляция ЩЖ осуществляется при участии центральных ядер ЦНС. Визуализация нервных волокон с использованием вируса псевдобешенства (PRV) продемонстрировала существование мультсинаптических нейрональных путей, связывающих паравентрикулярное ядро (PVN) гипоталамуса с ЩЖ, вероятно, через симпатический и парасимпатический отдел вегетативной нервной системы [4].
ЩЖ имеет симпатическую и парасимпатическую иннервацию.
Симпатическая система
Ганглии ЩЖ и верхние шейные ганглии (SCG) вносят наибольший вклад в ее иннервацию, что показано с помощью флуоресцентного красителя True Blue [5]. ЩЖ также богата симпатическими нервными волокнами, но влияние прямых нервных импульсов на деятельность фолликулов существенно перекрывается гуморальными эффектами тиреотропина. Активация центральных а-адренергических механизмов увеличивает высвобождение ТТГ преимущественно за счет стимуляции его секреции [6].
Регуляторное воздействие симпатической системы на ЩЖ показано в экспериментальном исследовании, в котором электрическая стимуляция симпатического ствола вызвала появление норэпинефрина в вене ЩЖ и резкое снижение кровоснабжения ЩЖ у крыс. Как известно, интенсивность кровообращения в ЩЖ весьма высока: в пересчете на единицу веса ткани кровоток через ЩЖ значительно превышает таковой в миокарде, мозге и почках [7]. Это может свидетельствовать о том, что симпатическая система осуществляет не только прямую, но и опосредованную (путем модуляции кровоснабжения) регуляцию функции ЩЖ [8]. В частности, экзогенный норадреналин блокирует реакцию железы на ТТГ у мышей [9]. Функциональная роль симпатической иннервации ЩЖ следует из вызванного
Это может свидетельствовать о том, что симпатическая система осуществляет не только прямую, но и опосредованную (путем модуляции кровоснабжения) регуляцию функции ЩЖ [8]. В частности, экзогенный норадреналин блокирует реакцию железы на ТТГ у мышей [9]. Функциональная роль симпатической иннервации ЩЖ следует из вызванного
односторонней симпатэктомией верхних шейных ганглиев уменьшения массы ЩЖ [10].
Дофамин может ингибировать секрецию тиреотро-пина гипофизом. Базальный уровень секреции тирео-тропина, равно как и уровень секреции, стимулируемый действием ТРГ, быстро уменьшаются под влиянием дофамина и его агониста бромокриптина. Тиреоидные гормоны в сочетании с адреналином и инсулином способны непосредственно повышать захват кальция клетками, увеличивать в них концентрацию цАМФ, а также интенсифицировать транспорт аминокислот и углеводов через клеточную мембрану.
В экспериментах in vitro доказано, что эпинефрин, норэпинефрин и дофамин стимулируют органификацию йода в ЩЖ и ингибируют ТТГ-зависимую секрецию тиреоидных гормонов; в отсутствие ТТГ описанный эффект исчезал. Дальнейшие исследования показали, что ингибирующее действие эпинефрина и норэпинефрина на секрецию Т4 и Т3 связано с активацией а-адренорецепторов тироцитов. Также установлено, что полное удаление из инкубационной среды ионов Ca2+ не изменяло действия норэпинефрина. Что касается ингибирующего влияния Т-допамина на секрецию тиреоидных гормонов, то эффект, по-видимому, опосредуется активацией дофаминовых рецепторов тироцитов [11].
Дальнейшие исследования показали, что ингибирующее действие эпинефрина и норэпинефрина на секрецию Т4 и Т3 связано с активацией а-адренорецепторов тироцитов. Также установлено, что полное удаление из инкубационной среды ионов Ca2+ не изменяло действия норэпинефрина. Что касается ингибирующего влияния Т-допамина на секрецию тиреоидных гормонов, то эффект, по-видимому, опосредуется активацией дофаминовых рецепторов тироцитов [11].
Нейромедиаторы оказывают как прямое, так и опосредованное влияние на активность гипоталамо—гипофизарно—тиреоидной системы. Так, дофамин угнетает секрецию ТТГ, воздействуя на D2-рецепторы тиреотро-поцитов [2]. Стимуляция D2-рецепторов в стриатуме сопровождается снижением уровня ТРГ в этой структуре головного мозга [12]. Кроме того, указанный нейромедиатор повышает выброс соматостатина, угнетающего выделение ТТГ.
Влияние адренергической системы сказывается и на повышении активности 5-дейодиназы 2-го типа в ЦНС при стимуляции p-адренорецепторов [13].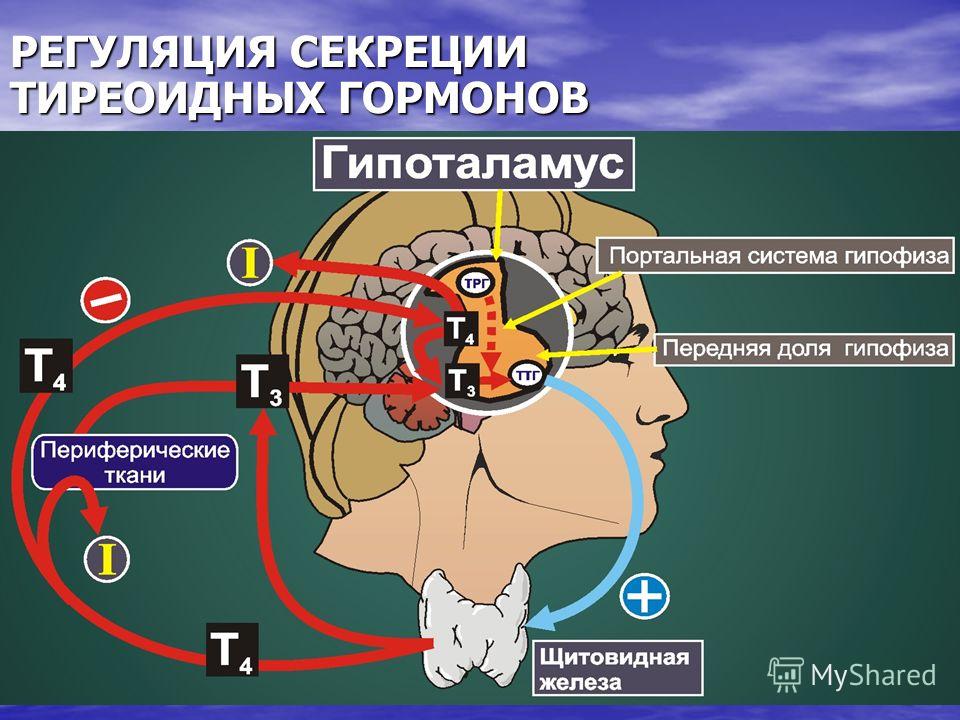 Вследствие ингибирования 5-дейодиназы 2-го типа монодейодирование Т4 преимущественно осуществляется 5-дейо-диназой 3-го типа, в результате чего образуются большие по сравнению с нормой количества ядерного рецептора гТ3 у крысы. При этом гТ3 сам по себе является мощным ингибитором 5-дейодиназы 2-го типа [2].
Вследствие ингибирования 5-дейодиназы 2-го типа монодейодирование Т4 преимущественно осуществляется 5-дейо-диназой 3-го типа, в результате чего образуются большие по сравнению с нормой количества ядерного рецептора гТ3 у крысы. При этом гТ3 сам по себе является мощным ингибитором 5-дейодиназы 2-го типа [2].
Обратное влияние гормонов щитовидной железы на симпатическую систему выражается в стимуляции транскрипции и экспрессии p-адренорецепторов. ТТГ усиливает экспрессию а-адренорецепторов на самих тиро-цитах.
Парасимпатическая система
ЩЖ хорошо иннервирована холинергическими нервными волокнами. Действие холинергической системы на парафолликулярные клетки является тормозным. ЩЖ получает парасимпатическую иннервацию ветвями блуждающего нерва, берущими начало в стволе головного мозга, а именно верхним гортанным и, в меньшей степени, возвратным гортанным нервом [5, 7]. Щитовидный нерв проецируется на щитовидный ганглий, главным образом содержащий нейроны, экспрессирующие вазоактивный кишечный пептид (VIP) и нейропептид Y (NPY).
Щитовидный нерв проецируется на щитовидный ганглий, главным образом содержащий нейроны, экспрессирующие вазоактивный кишечный пептид (VIP) и нейропептид Y (NPY).
Парасимпатическая нервная система угнетает функциональную активность тиреоцитов, однако ее тормозное влияние значительно слабее эффектов тиреостимули-рующих аутоантител. Ацетилхолин и другие холиноми-метики (пилокарпин, эзерин) угнетают функциональ-
#
#
АКТУАЛЬНЫЕ ВОПРОСЫ ПАТОФИЗИОЛОГИИ
Ф
ную активность тиреоидной паренхимы in vitro даже в случае предварительной стимуляции ТТГ [5]. Ингибирующее действие ацетилхолина на секрецию Т4 устранялось атропином, что свидетельствует об участии мускариновых рецепторов в тиреотропном действии нейротрансмиттера.
В условиях целостного организма стимуляция парасимпатического отдела вегетативной нервной системы при патологии ЩЖ приводит к противоположным результатам. Установлено, что содержание ацетилхолина и ацетилхолинэстеразы в ЩЖ и крови изменяется при тиреопатиях. Регуляторное влияние парасимпатической системы на ЩЖ осуществляется модуляцией кровотока в ней и возрастает при экспериментальном тиреотоксикозе у крыс [14]. При зобной болезни значительно увеличивается активность ацетилхолинэстеразы в паренхиме железы, превышающая активность этого фермента при тиреотоксикозе. Иными словами, зобная болезнь характеризуется активацией холинергической системы, тогда как тиреотоксикозы сопровождаются снижением парасимпатического тонуса.
Таким образом, оба отдела вегетативной нервной системы (ВНС), симпатический и парасимпатический, оказывают выраженное влияние на функцию ЩЖ. Это воздействие является прямым, направленным непосредственно на тиреоциты, а также модулирует кровоснабжение железы.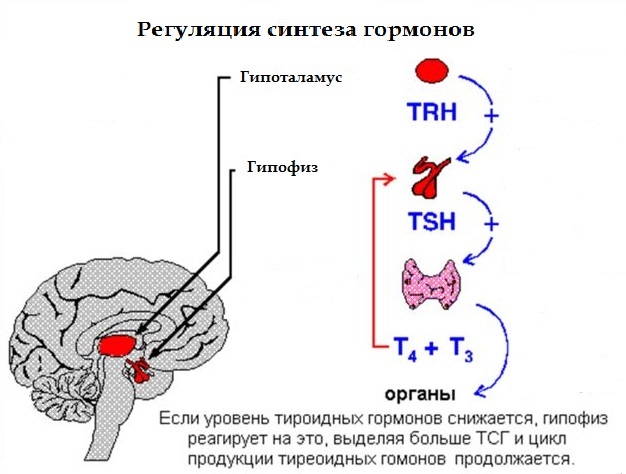
Пептидергическая система
Помимо классических нейромедиаторов ВНС, таких как норадреналин и ацетилхолин, нейроны ЩЖ содержат ряд нейропептидов, включая NPY и VIP.
NPY и VIP являются мощными вазоактивными пептидами. В изолированном кровеносном сосуде кролика NPY вызывает сужение сосудов и усиливает норадрена-лин-индуцированную вазоконстрикцию. Аналогичным образом in vivo экзогенное введение NPY сужает кровеносные сосуды ЩЖ крысы и потенцирует сходное действие норадреналина [15], что служит свидетельством непрямой регуляции функции ЩЖ за счет модуляции ее кровотока.
О самостоятельной роли регуляторных пептидов свидетельствует независимость от блокады адренергического и холинергического влияния стимулируемого пептидами накопления цАМФ клетками ЩЖ человека. VIP индуцирует высвобождение тироксина (Т4) дольками ЩЖ человека.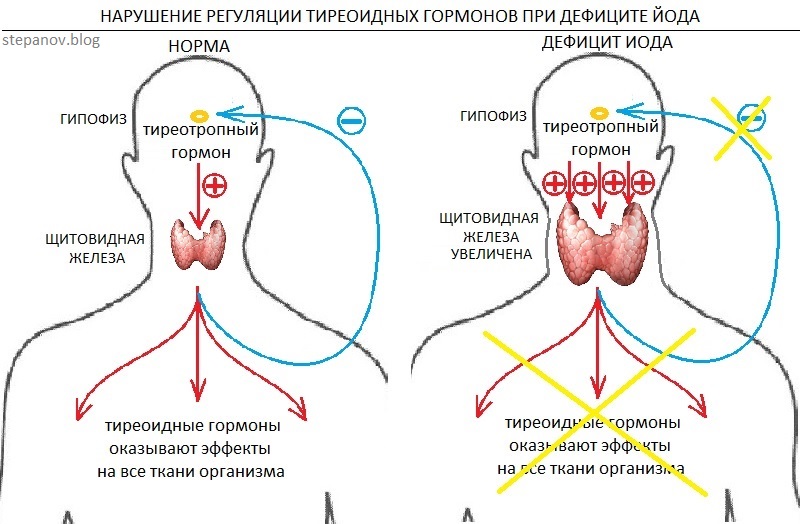 Экзогенно вводимый VIP увеличивает поглощение йода ЩЖ, приток крови и секрецию гормонов железы крысы [16].
Экзогенно вводимый VIP увеличивает поглощение йода ЩЖ, приток крови и секрецию гормонов железы крысы [16].
Насколько значима роль NPY и VIP по сравнению с нервной регуляцией функции ЩЖ? В физиологических условиях норадреналин (но не NPY) оказался основным медиатором быстрого ответа кровеносных сосудов ЩЖ на раздражение симпатического нерва [8].
Блокада VIP-ергической сигнализации не влияла на функцию ЩЖ или кровоток железы крысы [17], т.е. роль нейропептидов в регуляции ЩЖ не следует преувеличивать. Во всяком случае, она, вероятно, меньше, чем у классических нейромедиаторов.
Существует возможность взаимодействия адрен-и холинергической системы с регуляторными пептидами. Анатомической основой этого взаимодействия с нейропептидами является колокализация с ними нейротрансмиттеров. Регуляторные пептиды содержатся и высвобождаются одними и теми же нейронами, содержащими
норадреналин и/или ацетилхолин.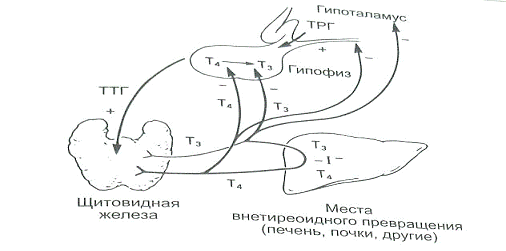 Например, NPY экспрессируется адренергическими нейронами; парасимпатические волокна щитовидного ганглия также содержат пептиды.
Например, NPY экспрессируется адренергическими нейронами; парасимпатические волокна щитовидного ганглия также содержат пептиды.
Серотонинергическая система и щитовидная железа в норме
Помимо адренергической и холинергической иннервации ЩЖ получает сигналы от разнообразных неадренергических нервных волокон центрального и периферического происхождения. Центральные волокна поступают от гипоталамуса и лимбических структур переднего и среднего мозга. Серотонинергические волокна, поступающие от ядер шва, участвуют в реакции нервной системы на воздействия стрессоров. Серотонин (5-НТ) играет важную роль в регуляции гипоталамо-гипофизарно-тиреоидной системы. Содержание серотонина в ЩЖ собаки составляет в среднем 0,03 мкг/г ткани, у кролика — 0,16 мкг/г, у крысы — 3,70 мкг/г [18]. Установлено, что с возрастом происходит снижение содержания серотонина в тканях мозга [19].
Тканевые базофилы ЩЖ не синтезируют серотонин, но путем захвата его из окружающих тканей регулируют уровень 5-НТ в микроокружении фолликулов, играя важную роль в гомеостазе органа.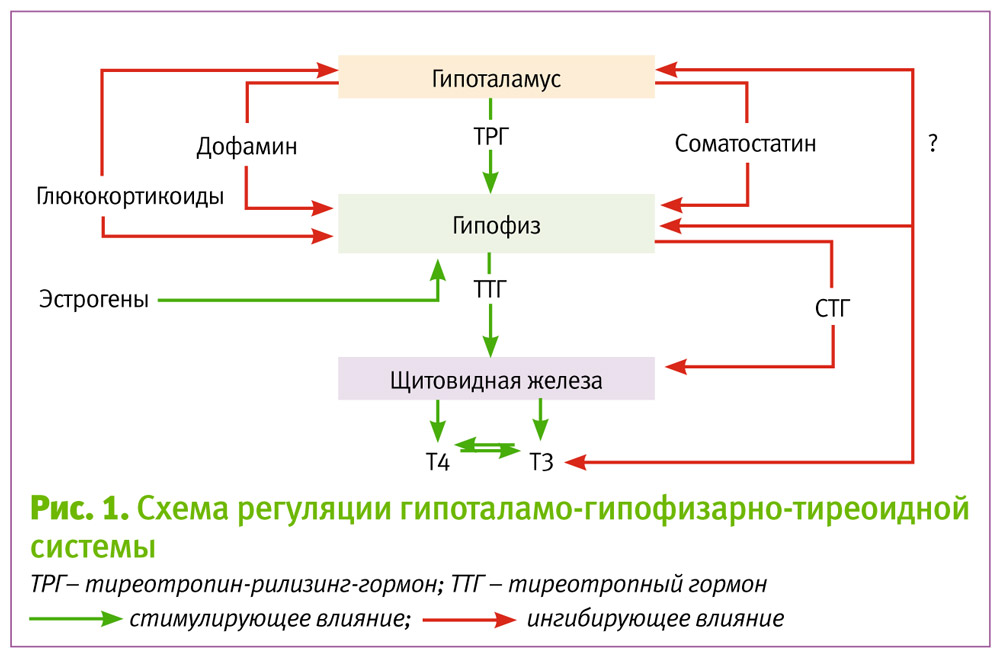 Скорость процессов накопления и выведения серотонина в интактной ЩЖ уравновешены и зависят от концентрации его в тканевых базофилах. В эксперименте установлена последовательность обмена серотонина в фолликулах ЩЖ. В первые 6 ч предшественник серотонина окситриптофан поглощается С-клетками и декарбоксилируется. Образовавшийся серотонин присоединяется к кальцитониновым гранулам и из С-клеток поступает в тироциты, которые переводят его в интрафолликулярный коллоид железы [20].
Скорость процессов накопления и выведения серотонина в интактной ЩЖ уравновешены и зависят от концентрации его в тканевых базофилах. В эксперименте установлена последовательность обмена серотонина в фолликулах ЩЖ. В первые 6 ч предшественник серотонина окситриптофан поглощается С-клетками и декарбоксилируется. Образовавшийся серотонин присоединяется к кальцитониновым гранулам и из С-клеток поступает в тироциты, которые переводят его в интрафолликулярный коллоид железы [20].
Парафолликулярные клетки расположены на наружной поверхности фолликулов, являются нейроэндокринными клетками, не поглощают йод и относятся к APUD-системе. Кроме того, парафолликулярные клетки могут располагаться в межфолликулярных прослойках соединительной ткани. В отличие от тироцитов парафолликулярные клетки совмещают образование норадреналина и серотонина путем декарбоксилирова-ния тирозина и 5-гидрокситриптофана, предшественников соответствующих нейроаминов, с биосинтезом белковых (олигопептидных) гормонов кальцитонина и соматостатина. Парафолликулярные клетки несколько крупнее фолликулярных, овальной или округлой формы, иногда отростчатые, содержащие большое ядро, митохондрии. Их цитоплазма заполнена белковыми секреторными гранулами, характеризующимися аргиро-филией или осмиофилией. В связи с обильным образованием белкового секрета в парафолликулярных клетках хорошо развиты гранулярная эндоплазматическая сеть и пластинчатый комплекс.
Парафолликулярные клетки несколько крупнее фолликулярных, овальной или округлой формы, иногда отростчатые, содержащие большое ядро, митохондрии. Их цитоплазма заполнена белковыми секреторными гранулами, характеризующимися аргиро-филией или осмиофилией. В связи с обильным образованием белкового секрета в парафолликулярных клетках хорошо развиты гранулярная эндоплазматическая сеть и пластинчатый комплекс.
Серотонин может блокировать секрецию тиреотро-пина гипофизом. Введение антител к серотонину увеличивает базальный уровень тиреотропина и потенцирует ответную реакцию повышения уровня тиреотропина на холодовой стресс. Характер серотонинергической регуляции тиреоидной функции зависит от условий действия медиатора. В опытах на изолированных тироцитах показано, что присутствие серотонина в инкубационной среде оказывает, подобно ТТГ, прямое стимулирующее действие на тироциты. Участие серотонина в стимуляции синтеза и секреции тиреоидных гормонов было под-
#
51
ВЕСТНИК РАМН /2013/ № 6
тверждено in vivo в условиях ингибирования секреции ТТГ с одновременной провокацией выброса серотонина в строму железы из тканевых базофилов. Результаты исследования позволили заключить, что серотонин повышает чувствительность тироцитов к ТТГ [21].
Результаты исследования позволили заключить, что серотонин повышает чувствительность тироцитов к ТТГ [21].
В то же время при подкожном введении крысам серотонина происходило снижение йодпоглотительной способности ЩЖ. Также имеются работы, в которых не обнаружено влияния экзогенного серотонина и 5-ги-дрокситриптофана на тиреоидную функцию [22]. Серотонин контролирует функцию гипоталамо—гипофизарно— тиреоидной системы на уровне гипоталамуса, ингибируя синтез ТРГ [23]. Экспериментально показана совместная регуляция мелатонином и серотонином функции ЩЖ. Стимуляторный эффект серотонина и блокирующее действие мелатонина на захват йода фолликулярными клетками ЩЖ выключались блокаторами 5-НТ2-рецепторов метисергидом и ципрогептадином. Это указывает на участие данных рецепторов в реализации эффекта серотонина. Серотонин и мелатонин могут взаимно предотвращать действие друг друга.
Взаимодействие серотонинергической и адренергической системы отмечено при регенерации парафолликулярных клеток, вырабатывающих кальцитонин, который находится вместе с серотонином в специфических вну-52 триклеточных гранулах, причем клетки активируются также симпатической нервной системой. Представляется важным, что активация симпатической нервной системы и проходящих в симпатическом стволе серотони-нергических волокон оказывает регуляторное влияние на функцию парафолликулярных клеток, включающую увеличение интенсивности выработки кальцитонина и рост насыщения кальцием костной ткани. Об однонаправленном влиянии серотонинергической и адренергической системы свидетельствуют результаты стимуляции верхнего шейного симпатического ганглия, которая способствовала увеличению числа и размера эпителиальных К-клеток ЩЖ и повышению содержания серотонина в ЩЖ самцов кроликов. Двусторонняя симпатэктомия привела к снижению уровня серотонина и размеров K-клеток.
Представляется важным, что активация симпатической нервной системы и проходящих в симпатическом стволе серотони-нергических волокон оказывает регуляторное влияние на функцию парафолликулярных клеток, включающую увеличение интенсивности выработки кальцитонина и рост насыщения кальцием костной ткани. Об однонаправленном влиянии серотонинергической и адренергической системы свидетельствуют результаты стимуляции верхнего шейного симпатического ганглия, которая способствовала увеличению числа и размера эпителиальных К-клеток ЩЖ и повышению содержания серотонина в ЩЖ самцов кроликов. Двусторонняя симпатэктомия привела к снижению уровня серотонина и размеров K-клеток.
Взаимодействие серотонинергической системы с регуляторными пептидами. Морфологическим субстратом данного взаимодействия является совместная локализация серотонина с пептидами (например, соматостатином) как в ЦНС, так и на периферии, в т.ч. в ЩЖ. Так, в ядрах шва и терминалях аксонов соматостатин колокализован с серотонином (а также с субстанцией Р, кальцитонин-ген-ассоциированным пептидом и энкефалинами). На локальном уровне для апудоцитов характерно сосуществование соматостатина с серотонином (и мелатонином) и рядом пептидных гормонов (субстанцией Р, мотили-ном, энкефалинами). Получены данные о совместной локализации мелатонина, эндотелина и кальретинина в париетальных клетках желудка; мелатонина и гистамина — в тучных клетках; мелатонина, соматостатина и p-эндорфинов — в естественных киллерах; мелатонина и простагландина ПГТ2 — в ретикулоэпителиальных клетках тимуса [24]. Соматостатин колокализован с серотонином, мелатонином и кальцитонином в С-клетках ЩЖ. Следовательно, при преганглионарной стимуляции эти физиологически активные вещества могут одновременно выделяться в синаптическую щель, оказывая воздействие на специфические рецепторы.
На локальном уровне для апудоцитов характерно сосуществование соматостатина с серотонином (и мелатонином) и рядом пептидных гормонов (субстанцией Р, мотили-ном, энкефалинами). Получены данные о совместной локализации мелатонина, эндотелина и кальретинина в париетальных клетках желудка; мелатонина и гистамина — в тучных клетках; мелатонина, соматостатина и p-эндорфинов — в естественных киллерах; мелатонина и простагландина ПГТ2 — в ретикулоэпителиальных клетках тимуса [24]. Соматостатин колокализован с серотонином, мелатонином и кальцитонином в С-клетках ЩЖ. Следовательно, при преганглионарной стимуляции эти физиологически активные вещества могут одновременно выделяться в синаптическую щель, оказывая воздействие на специфические рецепторы.
Серотонинергическая система и щитовидная железа при развитии патологии
Серотонин является патофизиологическим фактором заболеваний ЩЖ. При гипотиреозе угнетен синтез и метаболизм серотонина в мозге крыс. В экспериментах на животных снижение содержания серотонина в синапсах [25] и возбудимости серотониновых рецепторов при гипотиреозе компенсируется увеличением плотности 5НТ1А-рецепторов. В свою очередь введение гормонов ЩЖ животным с моделью гипотиреоза повышает уровень серотонина в коре головного мозга и снижает чувствительность 5-НТ1А-ауторецепторов в ядрах шва, что интенсифицирует освобождение серотонина корой и гиппокампом [26]. Ускорение и, возможно, усиление терапевтического эффекта антидепрессантов при их сочетанном назначении с тиреоидными гормонами может быть связано с десенсибилизацией 5-НТ1А-ауторецепторов в нейронах ядра шва головного мозга [27]. Как известно, в головном мозге млекопитающих существует 2 популяции 5-НТ1А-рецепторов: 5-НТ1А-рецепторы, расположенные пресинаптически в нейронах ядра шва среднего мозга (ауторецепторы), и постсинаптические 5-НТ1А-рецепторы, преимущественно локализованные в нейронах коры и структур лимбической системы (гетерорецепторы). Активация 5-НТ1А-рецепторов сопровождается повышением проницаемости мембраны для калия, что приводит к гиперполяризации мембраны серотонинер-гических и пирамидных нейронов коры и гиппокампа [28].
В экспериментах на животных снижение содержания серотонина в синапсах [25] и возбудимости серотониновых рецепторов при гипотиреозе компенсируется увеличением плотности 5НТ1А-рецепторов. В свою очередь введение гормонов ЩЖ животным с моделью гипотиреоза повышает уровень серотонина в коре головного мозга и снижает чувствительность 5-НТ1А-ауторецепторов в ядрах шва, что интенсифицирует освобождение серотонина корой и гиппокампом [26]. Ускорение и, возможно, усиление терапевтического эффекта антидепрессантов при их сочетанном назначении с тиреоидными гормонами может быть связано с десенсибилизацией 5-НТ1А-ауторецепторов в нейронах ядра шва головного мозга [27]. Как известно, в головном мозге млекопитающих существует 2 популяции 5-НТ1А-рецепторов: 5-НТ1А-рецепторы, расположенные пресинаптически в нейронах ядра шва среднего мозга (ауторецепторы), и постсинаптические 5-НТ1А-рецепторы, преимущественно локализованные в нейронах коры и структур лимбической системы (гетерорецепторы). Активация 5-НТ1А-рецепторов сопровождается повышением проницаемости мембраны для калия, что приводит к гиперполяризации мембраны серотонинер-гических и пирамидных нейронов коры и гиппокампа [28]. 5-НТ1А-рецепторы играют важную роль в механизмах действия антидепрессантов. Терапевтический эффект большого числа антидепрессантов связан с активацией постсинаптических 5-НТ-рецепторов. В то же время повышенный уровень серотонина в экстрацеллюлярном пространстве по принципу обратной связи активирует ауторегуляторные 5-НТ1А-рецепторы, что приводит к снижению выброса серотонина из терминалей нейронов в коре и гиппокампе и ослабляет эффект антидепрессантов [29]. Таким образом, снижение чувствительности 5-НТ1А-ауторецепторов при введении Т3 может приводить к потенцированию эффекта антидепрессантов. Повышение плотности 5-НТ2-рецепторов, наблюдаемое в префронтальной коре при введении тиреоидных гормонов, также может играть роль в механизмах их антидепрессивного действия [30].
5-НТ1А-рецепторы играют важную роль в механизмах действия антидепрессантов. Терапевтический эффект большого числа антидепрессантов связан с активацией постсинаптических 5-НТ-рецепторов. В то же время повышенный уровень серотонина в экстрацеллюлярном пространстве по принципу обратной связи активирует ауторегуляторные 5-НТ1А-рецепторы, что приводит к снижению выброса серотонина из терминалей нейронов в коре и гиппокампе и ослабляет эффект антидепрессантов [29]. Таким образом, снижение чувствительности 5-НТ1А-ауторецепторов при введении Т3 может приводить к потенцированию эффекта антидепрессантов. Повышение плотности 5-НТ2-рецепторов, наблюдаемое в префронтальной коре при введении тиреоидных гормонов, также может играть роль в механизмах их антидепрессивного действия [30].
Серотонин при сочетанном поражении щитовидной железы и психики
Принято считать, что депрессия сопровождается блокированием активности фермента дейодиназы типа 2, что способствует снижению содержания серотонина при депрессии. Введение ингибиторов обратного захвата норадреналина и серотонина дезипрамина [31] и флуоксе-тина увеличивает активность D-2; в свою очередь флуок-сетин [32] снижает активность D-3. Серотонин блокирует активность тиреолиберина и тем самым уменьшает уровень секреции тиреотропина.
Введение ингибиторов обратного захвата норадреналина и серотонина дезипрамина [31] и флуоксе-тина увеличивает активность D-2; в свою очередь флуок-сетин [32] снижает активность D-3. Серотонин блокирует активность тиреолиберина и тем самым уменьшает уровень секреции тиреотропина.
Серотониновая недостаточность. Гипотеза дефицита серотонина может объяснить низкий уровень тиреотро-пина при депрессии и то, почему пациенты с постоянно низким содержанием тиреотропина после очевидного восстановления после депрессии были подвержены ран-
#
#
АКТУАЛЬНЫЕ ВОПРОСЫ ПАТОФИЗИОЛОГИИ
Ф
ним рецидивам [33]. По мнению С. Kirkegaard и J. Faber, серотониновая недостаточность является патогенным фактором депрессии и хорошо объясняет нарушения гипоталамо—гипофизарно—тиреоидной системы при депрессии [34].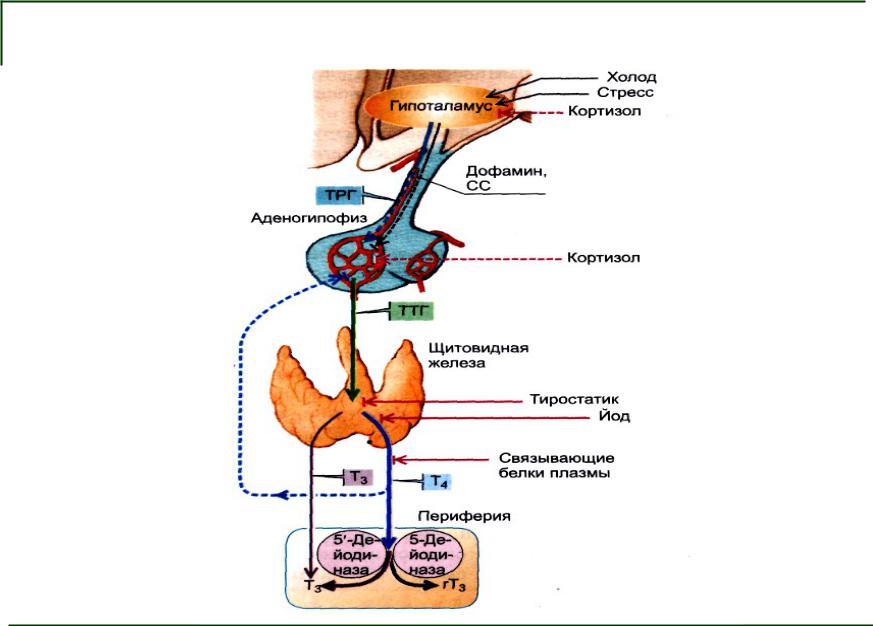
Кальцитонин
Семейство кальцитонина включает группу пептидных гормонов, структурно схожих с кальцитонином: кокальцигенин (кальцитонин ген-ассоциированный пептид, CGRP), амилин, адреномедуллин и адреноме-дуллин 2 (интермедин). Эти гормоны синтезируются различными тканями: кальцитонин — С-клетками ЩЖ C, кокальцигенин-а — в других, преимущественно нервных тканях, амилин — В-клетками островков поджелудочной железы; адреномедуллин — во многих тканях и типах клеток. После транскрипции в разных эндокри-ноцитах одних и тех же генов гормонов возможен альтернативный сплайсинг гетерогенной ядерной РНК с образованием различных мРНК, которые в дальнейшем приводят к трансляции неидентичных гормонов в различных тканях. Так, в С-клетках ЩЖ формируется преимущественно кальцитонин, а в ЦНС — кокальцигенин (CGRP), связанный с геном кальцитонина, причем в обоих случаях — на основе общего транскрипта препрокальцитонина.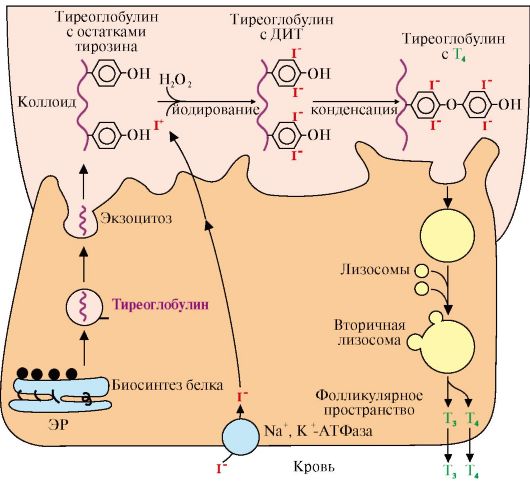 Кокальцигенин — убиквитарный пептид, синтезируемый многими нейроэндокриноцитами разной локализации. В бронхах он участвует в спазме гладких мышц, в ЦНС считается активатором симпатических центров гипоталамуса, подавляет пищевое поведение и повышает кровяное давление [35].
Кокальцигенин — убиквитарный пептид, синтезируемый многими нейроэндокриноцитами разной локализации. В бронхах он участвует в спазме гладких мышц, в ЦНС считается активатором симпатических центров гипоталамуса, подавляет пищевое поведение и повышает кровяное давление [35].
Помимо паракринных и эндокринных факторов, на образование мРНК гена кальцитонина влияет прямой контакт парафолликулярных клеток с другими рядом расположенными клетками [36].
Костная ткань, по-видимому, является общей мишенью пептидов семейства кальцитонина, хотя специфические эффекты пептидов в этой ткани варьируют. Кальцитонин вызывает быстрое снижение концентрации кальция в сыворотке крови, в основном за счет ингибирования резорбции костной ткани остеокластами. In vitro у некоторых животных амилин и кокальцигенин также эффективны в ингибировании активности остеокластов и резорбции костной ткани. Амилин, адреномедуллин и кокальцигенин, кроме того, способствуют пролиферации и стимулируют формирование костной ткани.![]()
Рецепторы пептидов семейства кальцитонина образуются путем гетеродимеризации рецептора кальцитонина (CTR) или кальцитонин рецептор-подобных рецепторов (CLR) с изменением активности рецепторных протеинов (RAMP). Анализ экспрессии рецепторов кальцитонина семьи в 16 образцах человеческих остеобластов показал высокий уровень CLR и RAMP1, низкий уровень RAMP2 и отсутствие экспрессии RAMP3 и CTR. Исследование костной ткани животных, нокаутных по гену кальцито-нина, кокальцигенина-а или амилина, показало основную физиологическую роль амилина в ингибировании костной резорбции, кокальцигенина-а — в активации формирования костной ткани, а кальцитонина — в ингибировании образования кости, не затрагивающем резорбцию кости [37].
Кальцитонин снижает концентрацию кальция в плазме, уменьшая абсорбцию Са2+ в кишечнике, тормозя активность остеокластов в костях, ослабляя реабсорбцию Са2+ в канальцах почек. В остеокластах он ингибирует ферменты, разрушающие костную ткань, в клетках почечных канальцев кальцитонин вызывает повышенный клиренс и выделение Са2+, фосфатов, Mg2+, К+, Na+ и тем самым способствует снижению концентрации Са2+ в крови. Под влиянием кальцитонина активируется деятельность остеобластов. В силу этого тормозится резорбция костного минерала оксиапатита (фосфата кальция, соединенного с гидроксильными группами) и, наоборот, усиливается его отложение в органическом матриксе кости. Наряду с этим кальцитонин предохраняет от распада органическую основу костной ткани, коллаген, стимулируя его синтез. Особо значима регуляция кальцитонином транспорта кальция через клеточные мембраны. Транспорт кальция через мембраны осуществляется путем его пассивного притока и активного оттока. Кальцитонин снижает интенсивность активного транспорта кальция из клеток, переводя его в связанное состояние. Благодаря этому он осуществляет гормональный контроль кальциевой проницаемости клеточных мембран, содержания и внутриклеточного распределения кальция; в этих процессах кальцитонин взаимодействует с паратгормоном, являясь его антагонистом.
В остеокластах он ингибирует ферменты, разрушающие костную ткань, в клетках почечных канальцев кальцитонин вызывает повышенный клиренс и выделение Са2+, фосфатов, Mg2+, К+, Na+ и тем самым способствует снижению концентрации Са2+ в крови. Под влиянием кальцитонина активируется деятельность остеобластов. В силу этого тормозится резорбция костного минерала оксиапатита (фосфата кальция, соединенного с гидроксильными группами) и, наоборот, усиливается его отложение в органическом матриксе кости. Наряду с этим кальцитонин предохраняет от распада органическую основу костной ткани, коллаген, стимулируя его синтез. Особо значима регуляция кальцитонином транспорта кальция через клеточные мембраны. Транспорт кальция через мембраны осуществляется путем его пассивного притока и активного оттока. Кальцитонин снижает интенсивность активного транспорта кальция из клеток, переводя его в связанное состояние. Благодаря этому он осуществляет гормональный контроль кальциевой проницаемости клеточных мембран, содержания и внутриклеточного распределения кальция; в этих процессах кальцитонин взаимодействует с паратгормоном, являясь его антагонистом.
Серотонинергическая система регулирует активность кокальцигенина. Анатомическим субстратом этого взаимодействия является совместная локализация серотонина и кальцитонин ген-ассоциированного пептида как в ЦНС, так и на периферии. Так, в ядрах шва и их терминалях кальцитонин-ген-ассоциированный пептид колокализован с серотонином (а также с субстанцией Р и энкефалинами). В то же время кальцитонин колока-лизован с серотонином, мелатонином и соматостатином в С-клетках ЩЖ.
Место депонирования серотонина является видоспецифичным. У крыс серотонин накапливается тучными клетками. У овцы, лошади, козы, летучей мыши и некоторых приматов серотонин содержится в ЩЖ в парафолликулярных, но не в тучных клетках [38]. Активация 5-HT1Da- и 5-НТть-рецепторов человека блокирует выделение из периваскулярных волокон тройничного нерва кокальцигенина, субстанции Р, ней-рокинина А и VIP, предотвращая развитие нейрогенного воспаления и вазодилатацию, которые являются важнейшими факторами патогенеза головной боли при мигрени [39]. Активация 5-НТ1-рецептора приводит к увеличению содержания внутриклеточного Са, вызывая торможение активности промоутера кальцитонин-ген-ассоциированного пептида [40].
Активация 5-НТ1-рецептора приводит к увеличению содержания внутриклеточного Са, вызывая торможение активности промоутера кальцитонин-ген-ассоциированного пептида [40].
Регуляторное воздействие серотонина на функцию кокальцигенина обеспечивается их колокализацией в С-клетках, которая заложена еще в онтогенезе последних. С-клетки ЩЖ отличаются от фолликулярных клеток не только конечным продуктом секреции, но и происхождением. Парафолликулярные клетки относятся к клеткам APUD-системы, которые происходят из эктодермы нервного гребня. Клетки-предшественники, мигрируя из вагальной области нервного гребня (прилежащей 1-7-му сомитам), дают начало серотонинергическим клеткам кишечника и ЩЖ млекопитающих [41]. В кишечнике серотонинергические клетки — это нейроны, которые находятся в миентеральном сплетении; в ЩЖ они распо-
#
53
ВЕСТНИК РАМН /2013/ № 6
ложены в везикулах и приобретают статус эндокринных. В нейронах кишечника и парафолликулярных клеток ЩЖ имеется один и тот же тип серотонинсвязывающего протеина (SBP). Этот факт позволяет допустить, что парафолликулярные клетки имеют те же физиологические характеристики, что и нейроны кишечника. Они вырабатывают полипептидные гормоны, которые способны к активному накоплению предшественников моноаминов и их декарбоксилированию, что позволяет относить их к APUD-системе.
В нейронах кишечника и парафолликулярных клеток ЩЖ имеется один и тот же тип серотонинсвязывающего протеина (SBP). Этот факт позволяет допустить, что парафолликулярные клетки имеют те же физиологические характеристики, что и нейроны кишечника. Они вырабатывают полипептидные гормоны, которые способны к активному накоплению предшественников моноаминов и их декарбоксилированию, что позволяет относить их к APUD-системе.
Увеличение концентрации внеклеточного Са1 2 3 4 5 6+ стимулирует парафолликулярные клетки к освобождению не только кальцитонина, но и серотонина. Высвободившись, серотонин стимулирует парафолликулярные клетки. Введение кальцитонина и серотонина овариэктомирован-ным крысам предотвращало развитие остеопороза [42]. В наших исследованиях на овариэктомированных крысах остеопороз развивался в течение полугода; последующее введение кальцийсодержащего препарата полностью восстановило структуру костной ткани и содержание в ней Са2+, Р3-, Mg2+ и фермента щелочной фосфатазы [43]. Парафолликулярные клетки экспрессируют и другой компонент серотонинергической системы — 5-НТ-рецепторы. Образуя комплексы с рецепторами, серотонин способен усиливать метаболизм фосфоино-54 зитидов и способствовать росту содержания кальция в парафолликулярных клетках. Это подтверждено в экспериментах с использованием агонистов и блокаторов серотониновых рецепторов в парафолликулярных клетках [44]. Таким образом, чувствительность этих клеток к содержанию Са2+ обеспечивает регуляцию гомеостаза кальция посредством секреции серотонина и гормонов. В парафолликулярных клетках имеется и серотониновый транспортер SERT Иными словами, регуляция серотонином активности парафолликулярных клеток опирается на серотонинергический фенотип этих клеток.
Парафолликулярные клетки экспрессируют и другой компонент серотонинергической системы — 5-НТ-рецепторы. Образуя комплексы с рецепторами, серотонин способен усиливать метаболизм фосфоино-54 зитидов и способствовать росту содержания кальция в парафолликулярных клетках. Это подтверждено в экспериментах с использованием агонистов и блокаторов серотониновых рецепторов в парафолликулярных клетках [44]. Таким образом, чувствительность этих клеток к содержанию Са2+ обеспечивает регуляцию гомеостаза кальция посредством секреции серотонина и гормонов. В парафолликулярных клетках имеется и серотониновый транспортер SERT Иными словами, регуляция серотонином активности парафолликулярных клеток опирается на серотонинергический фенотип этих клеток.
Морфологическое исследование щитовидной железы при различных функциональных состояниях
Проведено морфологическое исследование ткани ЩЖ кроликов позднестарческого возраста, у которых ранее было показано возрастное снижение активности парасимпатической системы [45, 46]. Железу окружа-
Железу окружа-
ет плотная капсула с соединительнотканными септами. В отдельных участках расположены сосуды артериального и венозного типа. Артерии сужены, с мощной стенкой, в просвете — эритроциты и свернувшаяся плазма. Коллоид сдержится в фолликулах, имеется незначительное число тиреоцитов. В перитиреоидной жировой ткани отмечаются явления отека и расширение сосудов. В мелких артериях — небольшое количество форменных элементов. В отдельных венозных сосудах наблюдается пристеночное стояние эритроцитов. Сосуды наполнены кровью. По-видимому, возрастными особенностями обусловлено увеличение содержания коллоида и уменьшение числа тиреоцитов в фолликулах. Возрастное увеличение содержания серотонина визуализируется прежде всего дилатацией и увеличением кровенаполнения сосудов ЩЖ и перитиреоидной области.
Морфологическое исследование ЩЖ в условиях двусторонней хирургической депарасимпатизации и введения серотонина было проведено на крысах. Отмечено небольшое количество коллоида в фолликулах и хорошо развитые фолликулярные эндокриноциты. Отмечается избыточное развитие перитиреоидной соединительной ткани. Показано, что серотонин, по-видимому, нормализует эндокринную функцию ЩЖ даже в условиях хронической депарасимпатизации. Кроме того, серотонин блокирует секрецию тиреотропина гипофизом, однако при гипо- и гипертиреозе содержание серотонина в мозге, соответственно, снижено или повышено с последующим увеличением или снижением плотности и чувствительности 5-НТ1-рецепторов.
Отмечено небольшое количество коллоида в фолликулах и хорошо развитые фолликулярные эндокриноциты. Отмечается избыточное развитие перитиреоидной соединительной ткани. Показано, что серотонин, по-видимому, нормализует эндокринную функцию ЩЖ даже в условиях хронической депарасимпатизации. Кроме того, серотонин блокирует секрецию тиреотропина гипофизом, однако при гипо- и гипертиреозе содержание серотонина в мозге, соответственно, снижено или повышено с последующим увеличением или снижением плотности и чувствительности 5-НТ1-рецепторов.
Заключение
Таким образом, различные отделы ВНС регулируют функцию ЩЖ с участием разных механизмов. Симпатический отдел ВНС модулирует активность ЩЖ при активации а-адренорецепторов и D2-рецепторов, а также тормозя кровоснабжение органа. Парасимпатический отдел ВНС несколько угнетает функциональную активность ЩЖ за счет возбуждения мускариновых М-рецепторов. Серотонинергический отдел ВНС стимулирует синтез и секрецию тиреоидных гормонов путем активации 5-НТ2-рецепторов; при гипотиреозе участие этого отдела ВНС выражается в увеличении плотности 5-НТ1А-ауторецепторов.
Серотонинергический отдел ВНС стимулирует синтез и секрецию тиреоидных гормонов путем активации 5-НТ2-рецепторов; при гипотиреозе участие этого отдела ВНС выражается в увеличении плотности 5-НТ1А-ауторецепторов.
#
ЛИТЕРАТУРА
1. Magner J.A. Thyroid-stimulating hormone: biosynthesis, cell biology and bioactivity. Endocr. Rev. 1990; 11 (2): 354-385.
2. Сапронов Н.С., Федотова Ю.О. Гормоны гипоталамо-гипофи-зарно-тиреоидной системы и мозг. С.Пб.: Лань. 2002. — 184 с.
3. Федотова Ю.О., Сапронов Н.С. Эффектв1 тиреоидных гормонов в центральной нервной системе. Основы нейроэндокринологии. Под ред. В.Г. Шаляпина, П.Д. Шабанова. С.Пб.: Элби-СПб. 2005. С. 204-249.
4. Klieverik L., Kalsbeek A., Fliers E. Autonomic innervation of the rhyroid gland and its functional implications. Hot Thyroidology. 2005; 1: 3-4.
Klieverik L., Kalsbeek A., Fliers E. Autonomic innervation of the rhyroid gland and its functional implications. Hot Thyroidology. 2005; 1: 3-4.
5. Sundler F., Grunditz T., Hakanson R., Uddman R. Innervation of the thyroid. A study of the rat using retrograde tracing and immunohis-tochemistry. Acta Histochem. 1989; Suppl.-Band XXXVII: 191-198.
6. Munoz-Cruzado Poce M.J., Garcia Navas A.J., Moreno Gomez M.L. Prevalence of thyroid disorders in patients diagnosed with depression. Aten. Primaria. 2000; 26 (3): 176-179.
7. Дедов И.И., Балаболкин М.И., Марова Е.И. Болезни органов эндокринной системы. М.: Медицина. 2000. 568 с.
8. Michalkiewicz M., Dey M., Huffman L., Hedge G.A. The neuropeptides, VIP and NPY, that are present in the thyroid nerves are not released into the thyroid vein.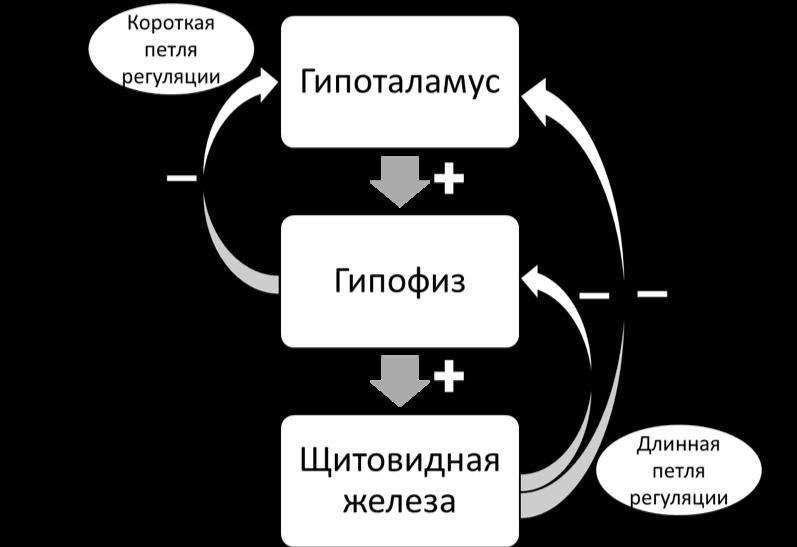 Thyroid. 1998; 8 (11): 1071-1077.
Thyroid. 1998; 8 (11): 1071-1077.
9. Ahren B., Bengtsson H.I., Hedner P. Effects of norepinephrine on basal and thyrotropin-stimulated thyroid hormone secretion in the mouse. Endocrinology. 1986; 119 (3): 1058-1062.
10. Young J.B., Burgi-Saville M.E., Burgi U., Landsberg L. Sympathetic nervous system activity in rat thyroid: potential role in goitro-genesis. Am. J. Physiol. Endocrinol. Metab. 2005; 288: 861-867.
11. Maayan M.L., Volpert E.M., Debons A.F. Neurotransmitter regulation of thyroid activity. Endocr. Res. 1987; 13 (2): 199-212.
12. Rack S.K., Makela E.H. Hypothyroidism and depression: a therapeutic challenge. Ann. Pharmacother. 2000; 34 (10): 1142-1145.
13. Gur E., Lifschytz T., Lerer B., Newman M.E. Effects of triiodothyronine and imipramine on basal 5-HT levels and 5-HT(1)
#
АКТУАЛЬНЫЕ ВОПРОСЫ ПАТОФИЗИОЛОГИИ
Ф
autoreceptor activity in rat cortex. Eur. J. Pharmacol. 2002; 457 (1): 37-43.
Eur. J. Pharmacol. 2002; 457 (1): 37-43.
14. Dey M., Michalkiewicz M., Huffman L.J., Hedge G.A. Thyroidal vascular responsiveness to parasympathetic stimulation is increased in hyperthyroidism. Am. J. Physiol. 1993; 264 (3 Pt. 1): 398-402.
15. Dey M., Michalkiewicz M., Dey M., Hedge G.A. NPY is not a primary regulator of the acute thyroid blood flow respons to sympathetic nerve stimulation. Am. J. Physiol. 1993; 265: 24-30.
16. Pietrzyk Z., Michalkiewicz M., Huffman L.J., Hedge G.A. Vasoactive intestinal peptide enhances thyroidal iodide uptake during dietary iodine deficiency. Endocr. Res. 1992; 18 (3): 213-228.
17. Michalkiewicz M., Huffman L.J., Dey M., Hedge G.A. Immunization against vasoactive intestinal peptide does not affect thyroid hormone secretion or thyroid blood flow. Am. J. Physiol. 1994; 266: 905-913.
Am. J. Physiol. 1994; 266: 905-913.
18. Consolo S., Garattini S., Ghielmetti R. Morselli P., Valzelli L. The hydroxylation of tryptophan in vivo by brain. Life Sci. 1965; 4: 625-630.
19. Reiter R.J. The pineal gland and melatonin in relation to aging: a summary of the theories and of the data. Exp. Gerontol. 1995; 30 (3-4): 199-212.
20. Виноградов С.Ю., Погорелов Ю.В. Нейромедиаторные биоамины щитовидной железы и структурно-функциональные аспекты ее гомеостаза. Архив анат. гистол. эмбриол. 1987; 1: 12-22.
21. Gershon M.D., Belshaw B.E., Nunez E.A. Biochemical, histo-chemical and ultrastructural studies of thyroid serotonin, parafollicular and follicular cells during development in the dog. Am. J. Anat. 2005; 132 (1): 5-19.
22. Brizzi G., Carella C., Foglia M.C., Frigino M. Thyroid hormone plasmatic levels in rats treated with serotonin in acute and chronic way. J. Physiol. Paris. 1997; 91 (6): 307-310.
Brizzi G., Carella C., Foglia M.C., Frigino M. Thyroid hormone plasmatic levels in rats treated with serotonin in acute and chronic way. J. Physiol. Paris. 1997; 91 (6): 307-310.
23. Broedel O., Eravci M., Fuxius S., Smolarz T., Jeitner A., Grau H., Stoltenburg-Didinger G., Plueckhan H., Meinhold H., Baumgartner A. Effects of hyper- and hypothyroidism on thyroid hormone concentrations in regions of the rat brain. Am. J. Physiol. Endocrinol. Metab. 2003; 285 (3): 470-480.
24. Райхлин Н.Т., Кветной И.М. Диффузная эндокринная система (АПУД-система). М.: Медицина. 1992.
25. Tejani-Butt S.M., Yang J., Kaviani A. Time course of altered thyroid states on 5-HT1A receptors and 5-НТ uptake sites in rat brain: An autoradiographic analysis. Neuroendocrinology. 1993; 57: 1011-1018.
26. Heal D.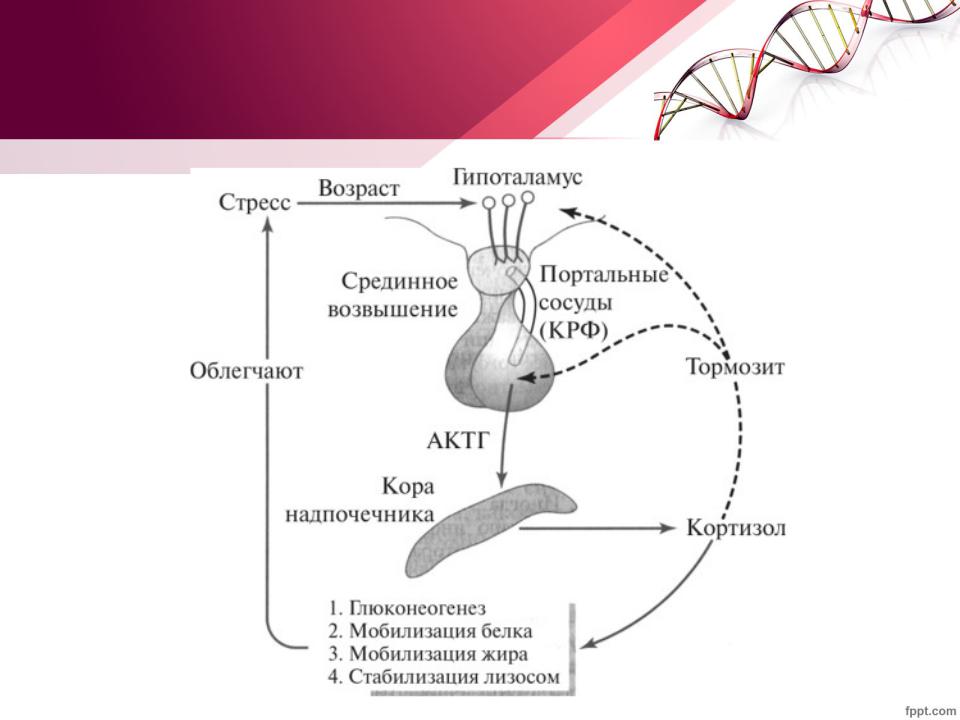 J., Smith S.L. The effects of acute and repeated administration of T3 to mice on 5-НТ1 and 5-НТ2 function in the brain and its influence on the actions of repeated eletroconvulsive shock. Neuropharmacol. 1998; 27: 1239-1248.
J., Smith S.L. The effects of acute and repeated administration of T3 to mice on 5-НТ1 and 5-НТ2 function in the brain and its influence on the actions of repeated eletroconvulsive shock. Neuropharmacol. 1998; 27: 1239-1248.
27. Haas M.J., Mreyoud A., Fishman M., Mooradian A.D. Microarray analysis of thyroid hormone-induced changes in mRNA expression in the adult rat brain .Neurosci. Lett. 2004; 365 (1):14—18.
28. Salvatore D., Low S.C., Berry M. Maia A.L., Harney J.W., Croteau W, St Germain D.L., Larsen P.R. Type 3 iodothyronine deiodinase: cloning, in vitro expression, and functional analysis of the placental selenoenzyme. J. Clin. Invest. 1995; 96: 2421-2430.
29. Crocker A.D., Overstreet D.H., Crocker J.M. Hypothyroidism leads to increased dopamine receptor sensitivity and concentration.
Pharmacol. Biochem. Behav. 1986; 24 (6): 1593-1597.
Biochem. Behav. 1986; 24 (6): 1593-1597.
30. Larisch R., Kley K., Nikolaus S., Franz M., Hautzel H., Tress W.,
Muller H.W Depression and anxiety in different thyroid function states. Horm. Metab. Res. 2004; 36 (9): 650-653.
31. Campos-Barros A., Meinhold H., Stula M., Muller F., Kohler R.,
Eravci M., Putzien O., Baumgartner A. The influence of desipra-mine on thyroid hormone metabolism in rat brain. J. Pharmacol.
Exp. Therapeut. 1994; 268: 1143-1152.
32. Baumgartner A., Dubeyko M., Campos-Barros A., Eravci M., Mein-hold H. Subchronic administration on fluoxetine to rats affects triiodothyronine production and deiodination in regions of the cortex and in the limbic forebrain.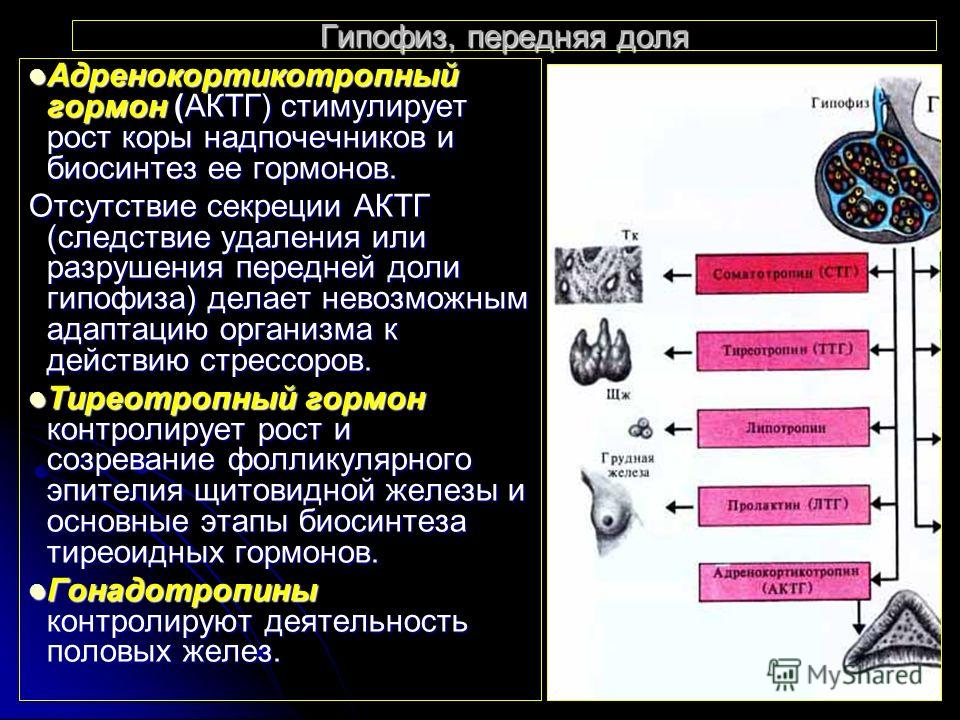 Brain Res. 1994; 635: 68-74.
Brain Res. 1994; 635: 68-74.
33. Hein M.D., Jackson I.M.D. Review: Thyroid Function in Psychiatric Illness. Gen. Hosp. Psychiatry. 1990; 12: 232-244.
34. Kirkegaard C. The thyrotropin response to thyrotropin-releasing hormone in endogenous depression. Psychoneuroendocrinol. 1981;
6: 189-212.
35. Гнилорыбов М. Нейропептиды и нейрогенные механизмы артритов. Укр. ревмат. журн. 2004; 2: 8-16.
36. Zabel M., Dietel M., Gebarowska E., Michael R. Effect of follicular cells on calcitonin gene expression in thyroid parafollicular cells in cell culture. Histochem J. 1999; 31 (3): 175-180.
37. Naot D., Cornish J. The role of peptides and receptors of the calcitonin family in the regulation of bone metabolism. Bone. 2008; 55
Bone. 2008; 55
43 (5): 813-818.
38. Falck B., Owman C. 5-hydroxytryptamine and related amines in endocrine cell systems. Adv. Pharmacol. 1968; 6 (Pt. A): 211-231.
39. Moskowitz M.A. Neurogenic versus vascular mechanism of sumatriptan and ergot alkaloids in migraine. Trends Pharmacol. Sci. 1992;
13: 307-311.
40. Durham P., Russo A. Stimulation of the calcitonin gene-related peptide enhancer by mitogen-activated protein kinases and repression by an antimigraine drug in trigeminal ganglia neurons. J .Neu-rosci. 2003; 23 (3): 807-815.
41. Russo A.F., Clark M.S., Durham P.L. Thyroid parafollicular cells.
An accessible model for the study of serotonergic neurons.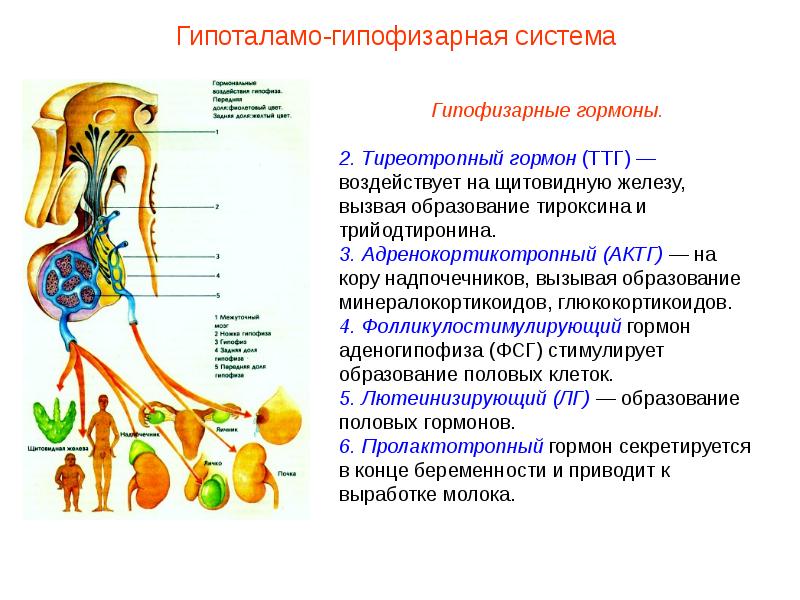 Mol.
Mol.
Neurobiol. 1996; 13 (3): 257-276.
42. Yoshimura M., Furue H., Ito A. Anti-nociceptive effect of calcitonin on chronic pain associated with osteoporosis. Clin. Calcium.
2001; 11 (9): 1153-1157.
43. Лычкова А.Э., Петраков А.В., Хомерики С.Г К вопросу о моделировании остеопороза. Вестник РАМН. 2010; 4: 31-33.
44. Tamir H., Hsiung S.C., Yu P.Y., Liu K.P., Adlersberg M.,
Nunez E.A., Gershon M.D. Serotonergic signalling between thyroid cells: protein kinase C and 5-HT2 receptors in the secretion and action of serotonin. Synapse. 1992; 12 (2): 155-168.
45. Лычкова А.Э. Серотонинергическая нервная система: градиенты нервных влияний в норме и патологии. Эксп. клин. гастроэнтерол. 2003; 6: 114-120.
Эксп. клин. гастроэнтерол. 2003; 6: 114-120.
46. Лычкова А.Э. Механизмы синергизма отделов вегетативной нервной системы. Усп. физиол. наук. 2006; 37 (1): 50-67.
#
КОНТАКТНАЯ ИНФОРМАЦИЯ
Лычкова Алла Эдуардовна, доктор медицинских наук, заведующая лабораторией клинической физиологии ЦНИИ гастроэнтерологии Департамента здравоохранения г. Москвы Адрес: 129343, Москва, шоссе Энтузиастов, д. 86; тел.: (499) 180-41-12; e-mail: [email protected]
#
ТТГ (тиреотропный гормон), сдать анализ ТТГ в Москве, цены в лаборатории Инвитро
Метод определения
Хемилюминесцентный иммуноанализ на микрочастицах
Исследуемый материал
Сыворотка крови
Доступен выезд на дом
Онлайн-регистрация
Синонимы: Тиреостимулирующий гормон, тиреотропин, ТТГ. Thyroid-stimulating Hormone, TSH, Thyrotropin.
Thyroid-stimulating Hormone, TSH, Thyrotropin.
Краткая характеристика определяемого вещества (тиреотропный гормон, ТТГ)
ТТГ – гликопротеин с молекулярной массой около 28 кДа. Синтезируется в передней доле гипофиза. Активирует продукцию и секрецию гормонов щитовидной железы (тиреоидных гормонов), инициирует клеточный рост и митотическую активность клеток щитовидной железы. Синтез и секреция ТТГ стимулируются тиротропин-рилизинг-гормоном гипоталамуса в ответ на снижение уровня циркулирующих тиреоидных гормонов. Уровень ТТГ находится в обратной логарифмической зависимости от концентрации Т4: при возрастании уровня Т4 выработка ТТГ снижается, при снижении уровня Т4 выработка ТТГ компенсаторно возрастает, что способствует поддержанию концентрации тиреоидных гормонов на необходимой высоте. Секреция ТТГ подвержена влиянию разных нейрональных механизмов и изменяется во время сна, понижения температуры, неспецифического стресса. Для ТТГ характерны суточные колебания концентрации: наивысших величин ТТГ крови достигает к 2-4 часам ночи, высокий уровень в крови сохраняется до 6-8 ч утра, минимальные значения ТТГ приходятся на 17-18 часов. Референсные значения уровня ТТГ, приводимые ниже, применимы для амбулаторных пациентов в период времени от 8 до 18 ч. Нормальный ритм секреции тиротропина нарушается при бодрствовании ночью.
Референсные значения уровня ТТГ, приводимые ниже, применимы для амбулаторных пациентов в период времени от 8 до 18 ч. Нормальный ритм секреции тиротропина нарушается при бодрствовании ночью.
С какой целью определяют ТТГ в крови
ТТГ- гормон гипофиза, регулирующий функции щитовидной железы. Один из важнейших тестов в лабораторной диагностике заболеваний щитовидной железы. Определение уровня ТТГ позволяет выявлять и субклинические стадии заболеваний щитовидной железы, когда концентрация тиреоидных гормонов еще поддерживается регуляторными механизмами в рамках референсных значений. Обычно при скрининговом исследовании функции щитовидной железы ТТГ используют в качестве единственного теста или в комплексе с определением свободного Т4.
Как изменяются показатели ТТГ в крови при нарушении функции щитовидной железы
При клинически выраженном первичном гипотиреозе (т. е. поражении на уровне щитовидной железы, которое приводит к снижению ее функции) отмечается значительное увеличение уровня ТТГ на фоне низкого уровня тиреоидных гормонов. Первичный гипертиреоз, напротив, ассоциируется со сниженным или неопределяемым уровнем ТТГ и высоким уровнем тиреоидных гормонов.
Первичный гипертиреоз, напротив, ассоциируется со сниженным или неопределяемым уровнем ТТГ и высоким уровнем тиреоидных гормонов.
При вторичном и третичном гипотиреозе, связанном с гипофизарной дисфункцией вследствие патологии гипофиза и гипоталамуса, значительно сниженные уровни Т3 и Т4 сочетаются с нормальным или слабо увеличенным уровнем ТТГ, который в этих случаях обладает редуцированной биологической активностью. Редкие клинические случаи вторичного гипертиреоза могут быть следствием ТТГ-секретирующих опухолей.
Что может повлиять на результат анализа
Прием препаратов тироксина накануне взятия крови для исследования не влияет на концентрацию ТТГ. Нормализация уровня ТТГ при проведении заместительной терапии гипотиреоза препаратами L-тироксина происходит медленно (в течение нескольких недель и месяцев), поскольку при хроническом выраженном гипотиреозе развивается гиперплазия тиреотрофов. Парадоксальное сочетание – высокий уровень ТТГ и высокий уровень свободного Т4 – в этот период является искусственно вызванным (ятрогенным) состоянием. Повторные исследования уровня ТТГ в целях контроля терапии целесообразно проводить не ранее, чем через 6 недель после изменения дозы или вида препарата.
Повторные исследования уровня ТТГ в целях контроля терапии целесообразно проводить не ранее, чем через 6 недель после изменения дозы или вида препарата.
Физиологические изменения концентрации ТТГ отмечаются во время беременности. Высокие концентрации хорионического гонадотропина, обладающего определенным структурным сходством с ТТГ, способны оказывать стимулирующее влияние на синтез тиреоидных гормонов. В I триместре беременности наблюдается временное повышение содержания Т4 и снижение уровня ТТГ. На протяжении II и III триместров уровень ТТГ возвращается к норме. Повышенный уровень ТТГ на ранних сроках беременности может говорить о скрытом гипотиреозе матери, потенциально опасном для развития плода.
Тяжелые заболевания, не связанные с патологией щитовидной железы, могут вызывать временное изменение концентрации ТТГ. Причиной может быть применение лекарственных препаратов или последствия самого заболевания. Обычно наблюдается снижение уровня ТТГ в острую фазу заболевания и некоторое повышение уровня при выздоровлении. При необходимости в таких случаях целесообразно ориентироваться на расширенный референсный диапазон ТТГ (0,02-10 мЕд/л) и использовать комплекс тестов ТТГ и Т4 (или свободный Т4).
При необходимости в таких случаях целесообразно ориентироваться на расширенный референсный диапазон ТТГ (0,02-10 мЕд/л) и использовать комплекс тестов ТТГ и Т4 (или свободный Т4).
Обратите внимание!
Пределы определения: 0,0083 мЕд/л-100 мЕд/л
Акция! Оценка функции щитовидной железы! — Сеть МЦ «Доктор Боголюбов»
Комплексное исследование направлено на выявление нарушений функции щитовидной железы (гипер- и гипотиреоза).
Исследование рекомендовано людям, входящим в группы риска по заболеваниям щитовидной железы, в том числе жителям регионов с низким природным содержанием йода в питьевой воде, и пациентам с заболеваниями щитовидной железы в анамнезе.
Т4 свободный – часть гормона щитовидной железы тироксина, которая циркулирует в крови в свободном, не связанном с белками состоянии. Именно эта фракция Т4 обеспечивает метаболическую активность гормона, т.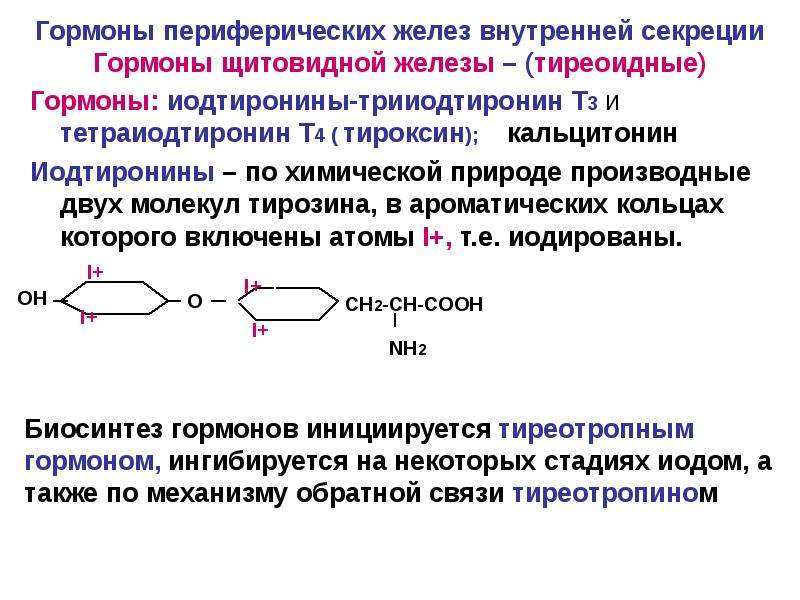 е. влияет на клетки различных органов. Повышая скорость основного обмена, он увеличивает теплопродукцию и потребление кислорода всеми тканями организма, за исключением тканей головного мозга, селезёнки и яичек. Увеличивает потребность организма в витаминах. Стимулирует синтез витамина А в печени. Снижает концентрацию холестерина и триглицеридов в крови, ускоряет обмен белка. Повышает экскрецию кальция с мочой, активирует обмен костной ткани, но в большей степени — резорбцию кости. Обладает положительным хроно- и инотропным действием на сердце. Стимулирует ретикулярную формацию и корковые процессы в центральной нервной системе.
е. влияет на клетки различных органов. Повышая скорость основного обмена, он увеличивает теплопродукцию и потребление кислорода всеми тканями организма, за исключением тканей головного мозга, селезёнки и яичек. Увеличивает потребность организма в витаминах. Стимулирует синтез витамина А в печени. Снижает концентрацию холестерина и триглицеридов в крови, ускоряет обмен белка. Повышает экскрецию кальция с мочой, активирует обмен костной ткани, но в большей степени — резорбцию кости. Обладает положительным хроно- и инотропным действием на сердце. Стимулирует ретикулярную формацию и корковые процессы в центральной нервной системе.
ТТГ (тиреотропный гормон) – это гормон, синтезирующийся передней долей гипофиза.
Главной его функцией является регуляция деятельности щитовидной железы: стимулирует синтез Т3 и Т4 клетками щитовидной железы и выделение их в кровь
Уровень ТТГ находится в обратной логарифмической зависимости от концентрации Т4: при возрастании уровня Т4 выработка ТТГ снижается, при снижении уровня Т4 выработка ТТГ компенсаторно возрастает, что способствует поддержанию концентрации тиреоидных гормонов на необходимой высоте.
Повышение уровня ТТГ в крови указывает на недостаточную продукцию гормонов щитовидной железы, т.е. гипотиреоз. При этом важным является тот факт, что данный показатель реагирует на снижение функции железы первым, нередко на субклинических стадиях заболевания, когда уровни Т3 и Т4 в сыворотке крови еще в норме.
Причинами нарушения выработки тиреотропного гормона могут быть заболевания гипоталамуса, который начинает продуцировать повышенные или пониженные количества тиреолиберина – регулятора секреции ТТГ гипофизом. Заболевания щитовидной железы, сопровождающиеся нарушением секреции тиреоидных гормонов, могут опосредованно влиять на секрецию тиреотропного гормона, вызывая понижение или повышение его концентрации в крови. Таким образом, исследование ТТГ — это один из важнейших анализов на гормоны.
Поэтому контроль содержания гормона ТТГ в крови является обязательным для пациентов с любой патологией щитовидной железы, и особенно для получающих лечение по этому поводу.
Трийодтиронин (Т3) – гормон щитовидной железы, биологическая активность которого в 3-5 раз превышает активность тироксина (Т4). Некоторое количество трийодтиронина синтезируется в щитовидной железе, однако в основном он образуется при дейодировании тироксина вне ее. Большая часть циркулирующего в крови трийодтиронина связана с белками плазмы, в частности с тироксинсвязывающим глобулином, тироксин-связывающим преальбумином и альбумином. Оставшаяся доля (менее 1 %) трийодтиронина является биологически активной (свободной) фракцией.
Гормон регулирует теплопродукцию, потребление кислорода всеми тканями организма, кроме тканей головного мозга, ретикуло-эндотелиальной системы и гонад. Также стимулирует рост костей, производство половых гормонов. синтез витамина А в печени и всасывание в кишечнике витамина B12. У детей Т3 регулирует рост и развитие центральной нервной системы. Повышает экскрецию кальция с мочой, активирует обмен костной ткани, снижает концентрацию холестерина и триглицеридов в крови, ускоряет обмен белка. Стимулирует ретикулярную формацию и корковые процессы в центральной нервной системе.
Стимулирует ретикулярную формацию и корковые процессы в центральной нервной системе.
Особые указания: При первичной проверке уровня тиреоидных гормонов следует отменить препараты, влияющие на функцию щитовидной железы за 2-4 недели до исследования (после согласования с лечащим врачом). При контроле лечения-исключить прием препаратов в день исследования и обязательно отметить это в направительном бланке (отметить также информацию о приеме других лекарств-аспирина, транквилизаторов, кортикостероидов, пероральных контрацептивов).
Если щитовидная железа не в состоянии производить необходимое количество тироксина либо вырабатывается недостаточно тиреотропного гормона для ее стимуляции, появляются симптомы гипотиреоза. У больных с пониженным уровнем Т4 увеличивается масса тела, сохнет кожа, повышается утомляемость, они становятся очень чувствительны к холоду, у женщин нарушается менструальный цикл. В случае если уровень свободного Т4 выше нормы, обменные процессы в организме и выработка в клетках энергии усиливаются, что приводит к гипертиреозу, для которого характерны учащенное сердцебиение, беспокойство, потеря веса, нарушение сна, дрожь в руках, сухость и покраснение глаз, отечность лица.
Исследование гормонов
Подготовка к исследованиям гормонов
Гормоны в организме человека выполняют очень важную роль. Это биологически активные вещества, которые вырабатываются в железах внутренней секреции. Гормоны поступают в кровь и оказываются именно в тех тканях, которые будут регулироваться ими. Количество гормонов в организме человека зависит от нескольких факторов, в том числе от возраста. Для нормальной жизнедеятельности организма необходимо определенное соотношение гормонов в крови. Чтобы определить, в норме ли количество этих биологически активных веществ, необходимо в первую очередь сдать анализы на гормоны.
В лабораторном отделении можно сдать анализы крови на гормоны щитовидной железы, гормоны репродуктивной системы, оценить эндокринную функцию поджелудочной железы, осуществляется мониторинг беременности, оценка работы гипофизарно-надпочечниковой системы и другое.
Гормоны щитовидной железы:
Щитовидная железа — один из важнейших органов эндокринной системы человека. Основная функция щитовидной железы — выработка тиреоидных гормонов. Они регулируют большинство процессов обмена веществ в организме, стимулируют рост, психическое развитие, деятельность сердечно-сосудистой и пищеварительной систем, участвуют в регуляции половой функции. Нарушение функций щитовидной железы может проявляться в виде раздражительности (или, наоборот, депрессии), повышенной утомляемости, изменении веса, непереносимости жары или холода, повышении кровяного давления, учащенном сердцебиении, нарушении менструального цикла и бесплодии у женщин. Если гормонов щитовидной железы не хватает беременной женщине, страдает не только мама, но и её будущий ребенок. У таких детей высок риск задержки умственного развития (врожденный гипотиреоз).
Тиреотропный гормон (ТТГ) – вырабатывается передней долей гипофиза и регулирует образование и секрецию гормонов щитовидной железой (Т3, Т4).
Тироксин свободный (Т4 свободный) – важнейший стимулятор синтеза белков. Вырабатывается фолликулярными клетками щитовидной железы под контролем ТТГ. Повышая скорость основного обмена, увеличивает теплопродукцию и потребление кислорода всеми тканями организма, за исключением тканей головного мозга, селезёнки и яичек. Увеличивает потребность организма в витаминах. Стимулирует синтез витамина А в печени. Снижает концентрацию холестерина и триглицеридов в крови, ускоряет обмен белка. Повышает экскрецию кальция с мочой, активирует обмен костной ткани, но в большей степени — резорбцию кости. Обладает положительным хроно- и инотропным действием на сердце. Стимулирует ретикулярную формацию и корковые процессы в центральной нервной системе.
Трийодтиронин свободный (Т3 свободный) — стимулирует обмен и поглощение кислорода тканями (активнее Т4).
Антитела к тиреоглобулину (АТ-ТГ) — Антитела к белку-предшественнику тиреоидных гормонов. Антитела к тиреоглобулину являются важным параметром для выявления аутоиммунных заболеваний щитовидной железы, таких как болезнь Хашимото, атрофический аутоиммунный тиреоидит, диффузный токсический зоб. Сочетание определения АТ-ТГ и антител к тиреоидной пероксидазе позволяет обнаружить большинство случаев болезни Хашимото и установить природу первичного идиопатического гипотиреоза.
Антитела к тиреоглобулину являются важным параметром для выявления аутоиммунных заболеваний щитовидной железы, таких как болезнь Хашимото, атрофический аутоиммунный тиреоидит, диффузный токсический зоб. Сочетание определения АТ-ТГ и антител к тиреоидной пероксидазе позволяет обнаружить большинство случаев болезни Хашимото и установить природу первичного идиопатического гипотиреоза.
Антитела к тиреоидной пероксидазе (АТ-ТПО) — аутоантитела к ферменту клеток щитовидной железы, участвующему в синтезе тиреоидных гормонов. Антитела к тиреоидной пероксидазе — показатель агрессии иммунной системы по отношению к собственному организму. Тиреоидная пероксидаза обеспечивает образование активной формы йода, которая способна включаться в процесс иодификации тиреоглобулина. Антитела к ферменту блокируют его активность, вследствие чего снижается секреция тиреоидных гормонов (T4, T3). Однако АТ-ТПО могут быть только «свидетелями» аутоиммунного процесса.
Тиреотропный гормон (ТТГ, тиротропин, Thyroid Stimulating Hormone, TSH)
Исследуемый материал
Сыворотка крови
Метод определения
Твёрдофазный хемилюминесцентный иммуноанализ. В Независимой лаборатории ИНВИТРО применяются тест-системы 3-го поколения с аналитической чувствительностью 0,002 мЕд/л (функциональная чувствительность порядка 0,004 мЕд/л). Такой уровень чувствительности особенно важен при дифференциальной диагностике пограничного гипертиреоза, а также оценке эффективности подавления гипофизарной секреции ТТГ при узловых поражениях и раке щитовидной железы после хирургического или консервативного лечения.
В Независимой лаборатории ИНВИТРО применяются тест-системы 3-го поколения с аналитической чувствительностью 0,002 мЕд/л (функциональная чувствительность порядка 0,004 мЕд/л). Такой уровень чувствительности особенно важен при дифференциальной диагностике пограничного гипертиреоза, а также оценке эффективности подавления гипофизарной секреции ТТГ при узловых поражениях и раке щитовидной железы после хирургического или консервативного лечения.
Гликопротеидный гормон, стимулирующий образование и секрецию гормонов щитовидной железы (Т3, Т4).
Вырабатывается базофилами передней доли гипофиза под контролем тиреотропного гипоталамического рилизинг-фактора, а также соматостатина, биогенных аминов и тиреоидных гормонов. Усиливает васкуляризацию щитовидной железы. Увеличивает поступление йода из плазмы крови в клетки щитовидной железы, стимулирует синтез тиреоглобулина и выщепление из него Т3 и Т4, а также прямо стимулирует синтез указанных гормонов. Усиливает липолиз.
Усиливает васкуляризацию щитовидной железы. Увеличивает поступление йода из плазмы крови в клетки щитовидной железы, стимулирует синтез тиреоглобулина и выщепление из него Т3 и Т4, а также прямо стимулирует синтез указанных гормонов. Усиливает липолиз.
Между концентрациями свободного Т4 и ТТГ в крови существует обратная логарифмическая зависимость.
Для ТТГ характерны суточные колебания секреции: наивысших величин ТТГ крови достигает к 2 — 4 часам ночи, высокий уровень в крови определяется также в 6 — 8 часов утра, минимальные значения ТТГ приходятся на 17 — 18 часов вечера. Нормальный ритм секреции нарушается при бодрствовании ночью. Во время беременности концентрация гормона повышается. С возрастом концентрация ТТГ незначительно повышается, уменьшается количество выбросов гормона в ночное время.
Секреция тиреоидного гормона — Справочник химика 21
УВЕЛИЧЕНИЕ СИНТЕЗА И СЕКРЕЦИИ ТИРЕОИДНОГО ГОРМОНА [c. 262]
262]
Биологическое действие гормонов щитовидной железы распространяется на множество физиологических функций организма. В частности, гормоны регулируют скорость основного обмена, рост и дифференцировку тканей, обмен белков, углеводов и липидов, водно-электролитный обмен, деятельность ЦНС, пищеварительного тракта, гемопоэз, функцию сердечнососудистой системы, потребность в витаминах, сопротивляемость организма инфекциям и др. Точкой приложения действия тиреоидных гормонов, как и всех стероидов (см. далее), считается генетический аппарат. Специфические рецепторы—белки —обеспечивают транспорт тиреоидных гормонов в ядро и взаимодействие со структурными генами, в результате чего увеличивается синтез ферментов, регулирующих скорость окислительновосстановительных процессов. Естественно поэтому, что недостаточная функция щитовидной железы (гипофункция) или, наоборот, повышенная секреция гормонов (гиперфункция) вызывает глубокие расстройства физиологического статуса организма. [c. 266]
266]
ТИРЕОИДНЫЕ ГОРМОНЫ (гормоны щитовидной железы) — продукты внутренне секреции щитовидной железы. До спх пор нот единой точки зрения, какое хпмич. соединение считать истинным Т. г. Наиболее обоснованным является мнение о существовании в организме человека и животных трех форм Т. г. резервной, циркулирующей и активной. [c.87]
Химическая сигнализация играет важную роль и в процессах развития животного, нередко определяя время и тип дифференцировки тех или иных клеток. В таких случаях влияние химического сигнала обычно сказывается медленно и бывает продолжительным. Например, в период полового созревания клетки яичника начинают в больших количествах секретировать стероидный женский половой гормон эстрадиол. Этот гормон вызывает изменения многих клеток в различных частях организма, что в конце концов приводит к развитию вторичных женских половых признаков, например к увеличению грудных желез. Если секреция эстрадиола прекращается, эти эффекты постепенно исчезают, но некоторые реакции, вызываемые стероидными половыми гормонами на очень раннем этапе развития млекопитающих, необратимы (см. конец табл. 13.1). Аналогичным образом десятикратное повышение концентрации тиреоидного гормона в крови вызывает ряд радикальных и необратимых изменений, приводящих к превращению головастика в лягушку (рис. 13-5). [c.251]
конец табл. 13.1). Аналогичным образом десятикратное повышение концентрации тиреоидного гормона в крови вызывает ряд радикальных и необратимых изменений, приводящих к превращению головастика в лягушку (рис. 13-5). [c.251]
Скорость основного обмена (или базальная скорость метаболизма, БСМ)-это мера потребления кислорода организмом в состоянии полного покоя спустя 12 ч после еды в расчете на единицу площади поверхности тела. Величина БСМ для больного выражается в виде отклонения (в процентах) от нормального значения БСМ, т. е. в виде процентного отклонения от нормы для индивидов того же пола, веса и роста. Измерения БСМ проводятся при диагностике заболеваний, связанных с нарушением функции щитовидной железы. Состояние, вызванное повышенной секрецией тиреоидных гормонов, называется гиперти-реоз. Скорость основного обмена при этом увеличена, нища сгорает быстрее, чем в норме, больные вьщеляют больше тепла, и им свойственна повышенная возбудимость.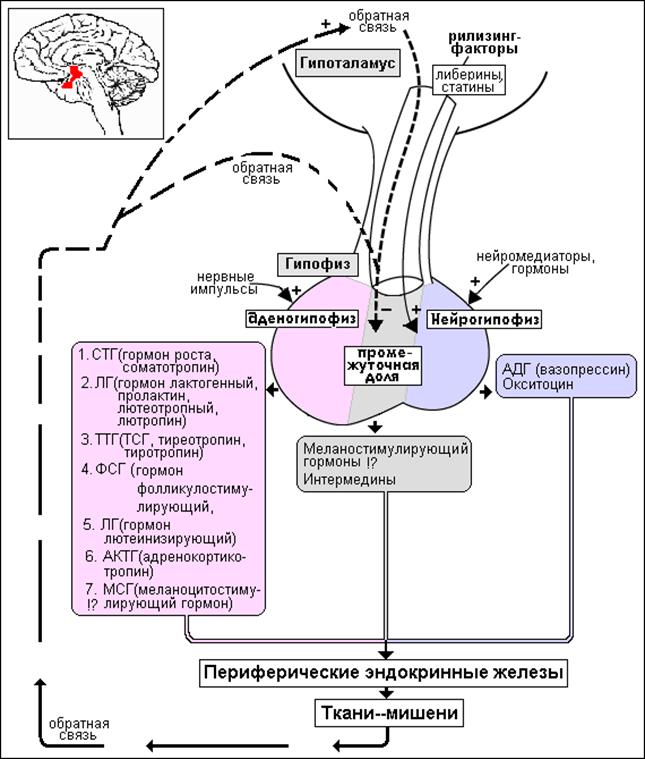 Недостаточность секреции тиреоидных гормонов, т.е. гипотиреоз, выражается в пониженной скорости основного обмена. Для больных с гипотиреозом характерны более медленное, чем в норме, сгорание пищи в организме, выделение меньшего количества тепла и некоторая общая медлительность. Недостаточность иода в пище приводит к базедовой болезни (гл. 26), сопровождающейся сильным увеличением щитовидной железы, что обусловлено накоплением очень большого по сравнению с нормой количества колловдного белка, который предназначен захватывать минимальные количества иода, циркулирующего в крови. [c.804]
Недостаточность секреции тиреоидных гормонов, т.е. гипотиреоз, выражается в пониженной скорости основного обмена. Для больных с гипотиреозом характерны более медленное, чем в норме, сгорание пищи в организме, выделение меньшего количества тепла и некоторая общая медлительность. Недостаточность иода в пище приводит к базедовой болезни (гл. 26), сопровождающейся сильным увеличением щитовидной железы, что обусловлено накоплением очень большого по сравнению с нормой количества колловдного белка, который предназначен захватывать минимальные количества иода, циркулирующего в крови. [c.804]
Так, Ю. И. Москалев и В. Н. Стрельцова, анализируя возможные механизмы развития злокачественных опухолей щитовидной железы после воздействия ионизирующих излучений, рассматривают следующую последовательность событий радиационное воздействие радиационное повреждение щитовидной железы- -снижение секреции тиреоидных гормонов- понижение уровня тиреоидных гормонов в крови- -гиперфункция гипофиза и избыток выработки тиреотропного гормона- -гиперплазия сохранившейся ткани щитовидной железы- гормонозави-симая аденома щитовидной железы гормононезависимая (злокачественная) опухоль щитовидной железы.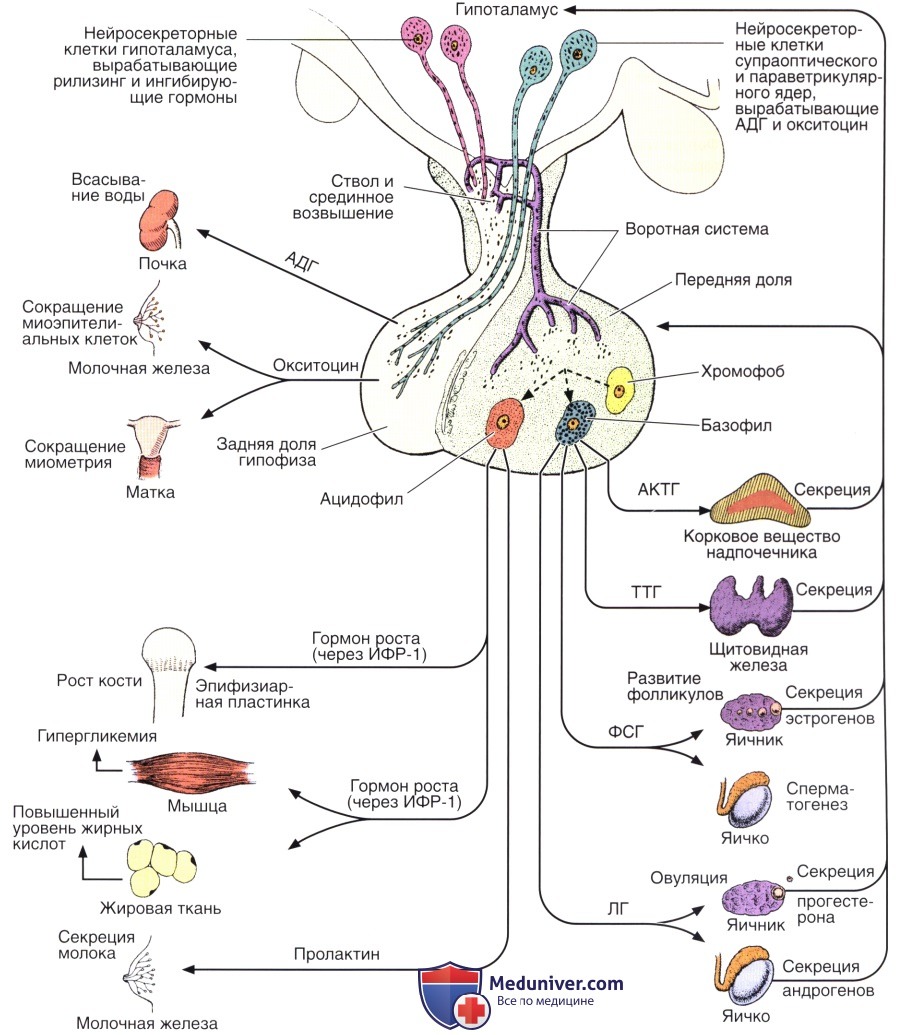 [c.67]
[c.67]
Своеобразен механизм секреции тиреоидного гормона. Сначала гормон в коллоидной форме в комплексе с крупным белком (тиреоглобулин) высвобождается в просвет фолликула щитовидной железы. Далее под влиянием тиреотропного гормона происходит быстрый эндоцитоз тиреоглобулина эпителиальными клетками. После слияния эндосом с первичными лизосомами с помощью ограниченного протеолиза образуется низкомолекуляр-ЕНЫП гормон тироксин, который диффундирует из вторичной ли-лосомы и далее путем диффузии проникает в ближайший крове-иосный капплляр. [c.72]
Гомеостаз ионов кальция регулируется сложным путем. Ключевые роли в этом процессе играют паратиреоидный гормон (ПТГ) и тиреоидный гормон кальцитонин. При уменьшении концентрации ионов Са + возрастает секреция ПТГ — пептидного гормона, содержащего 83 аминокислотных остатка. Непосредственно под влиянием этого гормона остеокласты увеличивают растворение содержащихся в костях минеральных соединений. ПТГ увеличивает также реабсорбцию ионов a + в почечных канальцах.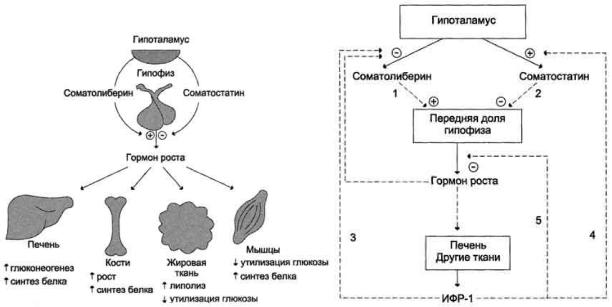 Суммарный эффект проявляется в повышении уровня кальция в сыворотке крови. В свою очередь при увеличении содержания ионов Са + сек-ретируется гормон кальцитоцин, действие которого состоит в снижении концентрации ионов Са2+засчет ускорения отложения кальция в результате деятельности остеобластов. Таким образом, эти два гормона действуют по системе пуш-пул (push-pull) с обратной связью (гл. 6, разд. Е.4). В процессе регуляции концентрации ионов кальция принимает участие также витамин D (дополнение 12-Г), который, судя по всему, требуется для синтеза Са2+-связывающих белков, необходимых для всасывания ионов Са» + в кишечнике, реабсорбции его в почках и растворения костной ткани. Своевременное поступление нужных количеств витамина D является [c.374]
Суммарный эффект проявляется в повышении уровня кальция в сыворотке крови. В свою очередь при увеличении содержания ионов Са + сек-ретируется гормон кальцитоцин, действие которого состоит в снижении концентрации ионов Са2+засчет ускорения отложения кальция в результате деятельности остеобластов. Таким образом, эти два гормона действуют по системе пуш-пул (push-pull) с обратной связью (гл. 6, разд. Е.4). В процессе регуляции концентрации ионов кальция принимает участие также витамин D (дополнение 12-Г), который, судя по всему, требуется для синтеза Са2+-связывающих белков, необходимых для всасывания ионов Са» + в кишечнике, реабсорбции его в почках и растворения костной ткани. Своевременное поступление нужных количеств витамина D является [c.374]
Тиротропин контролирует развитие и функцию щитовидной железы и регулирует биосинтез и секрецию в кровь тиреоидных гормонов. Полностью расшифрована первичная структура а- и 3-субъединиц тиротропина быка, овцы и человека а-субъединица, содержащая 96 аминокислотных остатков, имеет одинаковую аминокислотную последовательность во всех изученных ТТГ и во всех лютеинизирующих гормонах гипофиза 3-субъеди-ница тиротропина человека, содержащая 112 аминокислотных остатков, [c. 260]
260]
Биохимические функции. Тиреотропин контролирует синтез и секрецию гормонов щитовидной железы тироксина и трииодтиронина. Воздействуя по мембрано-опосредованному механизму на клетки щитовидной железы, он стимулирует образование тиреоглобулина — предшественника тиреоидных гормонов. [c.149]
Биосинтез и метаболизм. Сигнал, запускающий синтез тиреоидных гормонов, формируется в гипоталамусе в виде тиреолиберина, который, воздействуя на гипофиз, стимулирует синтез и секрецию тиреотропина. Последний взаимодействует с рецепторами на поверхности клеток щитовидной железы и опосредованно, через вторичные посредники, стимулирует синтез ряда белков, в том числе тиреоглобулин — предшественник тиреоидных гормонов. Тиреоглобулин представляет собой гликопротеин с молекулярной массой 660 кВа и необьмно большим числом тирозиновых остатков в полипептидной цепи (около 120). Углеводная часть составляет до 10% от массы тиреоглобулина. Как и все секреторные белки, тиреоглобулин синтезируется на мембран-но-связанных рибосомах, причем гликозилирование полипептидной цепи начинается в цистернах эндоплазматического ретикулума, а завершается в аппарате Гольджи. Тиреоидные гормоны являются единственной группой гормонов, для функционирования которьгх необходим микроэлемент иод. [c.151]
Тиреоидные гормоны являются единственной группой гормонов, для функционирования которьгх необходим микроэлемент иод. [c.151]
Избыточная секреция инсулина гиперин-сулинизм. При некоторых видах злокачественных опухолей поджелудочной железы происходит избыточный синтез инсулина В-клетками. У больных при этом наблюдаются следующие симптомы дрожь, слабость и утомляемость, потливость и постоянное чувство голода. Если болезнь затягивается, может происходить нарушение мозговой деятельности. Как влияет избыточная секреция инсулина на обмен углеводов, аминокислот и липидов в печени Почему развиваются описанные симптомы Объясните, почему с течением времени это состояние приводит к нарушениям мозговой деятельности. Термогенез, обусловленный тиреоидными гормонами. Гормоны щитовидной железы участвуют в регуляции скорости основного обмена (базального метаболизма). При введении избытка тироксина в печень животного возрастают скорость потребления О2 и выработка тепла (термогенез), но концентрация АТР в ткани остается на уровне нормы.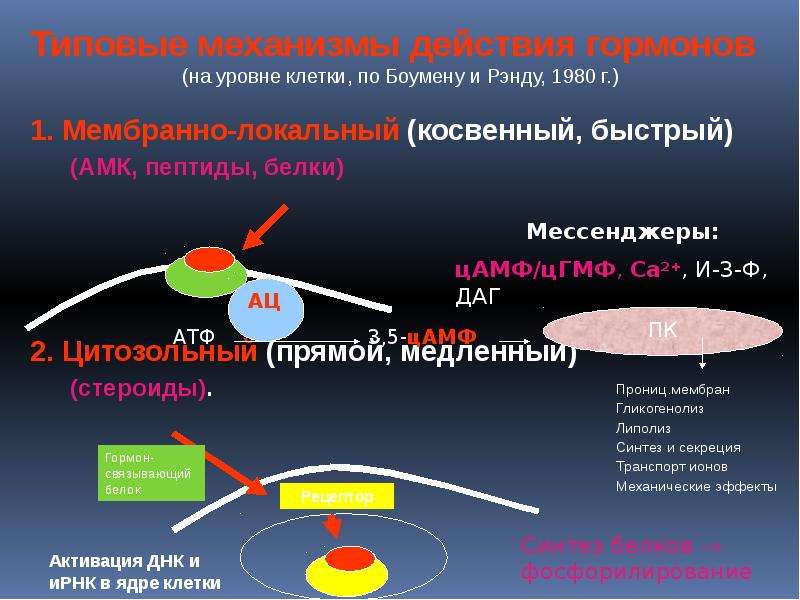 Были предложены разные объяснения термогенного действия тироксина. Одно из них состоит в том, что избыток тиреоидного гормона вызывает разобщение окислительного фосфорилирования в митохондриях. Каким образом, исходя из этого объяснения, можно понять приведенные выше наблюдения Согласно другому объяснению, термогенез обусловлен повышением скорости использования АТР в стимулируемых тироксином тканях. Считаете ли вы такое объяснение правильным Почему [c.810]
Были предложены разные объяснения термогенного действия тироксина. Одно из них состоит в том, что избыток тиреоидного гормона вызывает разобщение окислительного фосфорилирования в митохондриях. Каким образом, исходя из этого объяснения, можно понять приведенные выше наблюдения Согласно другому объяснению, термогенез обусловлен повышением скорости использования АТР в стимулируемых тироксином тканях. Считаете ли вы такое объяснение правильным Почему [c.810]
Главные компоненты, составляющие петлю отрицательной обратной связи,—это Т4, Т3, тиреотропин и тиролиберин (рис. 46.4). Т4 и Т3 тормозят свой собственный синтез по механизму обратной связи. Медиатором этого процесса может служить Т3, поскольку Т4 превращается в гипофизе в Т3. На этом уровне обратная связь ингибирует высвобождение тиреотропина. Т, (или, возможно, Т4) может подавлять высвобождение и образование тиролиберина гипоталамусом. Стимулом для повышенной секреции тиролиберина и тиреотропина служит снижение содержания тиреоидных гормонов в крови.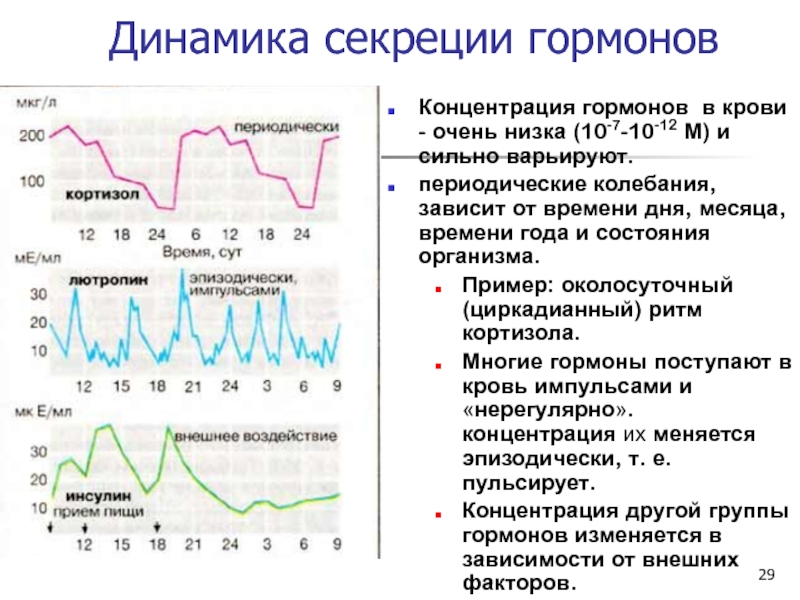 Однако даже при полной блокаде биосинтеза тиреоидных гормонов (например, при лечении антитиреоидными средствами) не происходит немедленного усиления высвобождения тиролиберина и тиреотропина. Щитовидная железа содержит запас ранее образованного гормона, обеспечивающий его поставку в течение нескольких недель имеются также внетиреоид-ные резервы гормона (в печени и в связанной с ТСГ форме), которые расходуются в первую очередь. Кроме того, при угрозе снижения биосинтеза гормона в связи с недостаточностью иода дополнительную компенсаторную роль выполняет ауторегуляторный механизм щитовидной железы. [c.190]
Однако даже при полной блокаде биосинтеза тиреоидных гормонов (например, при лечении антитиреоидными средствами) не происходит немедленного усиления высвобождения тиролиберина и тиреотропина. Щитовидная железа содержит запас ранее образованного гормона, обеспечивающий его поставку в течение нескольких недель имеются также внетиреоид-ные резервы гормона (в печени и в связанной с ТСГ форме), которые расходуются в первую очередь. Кроме того, при угрозе снижения биосинтеза гормона в связи с недостаточностью иода дополнительную компенсаторную роль выполняет ауторегуляторный механизм щитовидной железы. [c.190]
Ретроингибирование, наблюдающееся в процессах биосинтеза тиреоидных гормонов, может сказываться и на других гормональных системах организма, так как тиреолиберин стимулирует секрецию не только тиреотропного гормона, но и гормона роста (см. раздел 2.1), а кроме того, он имеет так называемые поведенческие эффекты. [c. 93]
93]
В обмене иода в организме вьщеляют три фазы пре-тиреоидный (всасывание и обмен его до включения в состав гормонов), тиреоидный (синтез гормонов, их хранение, секреция) и посттиреоидный (обмен гормоносвязанного иода). Короткоживущие изотопы иода не доживают до поздних фаз йодного обмена ( 1, 1), что приводит к смещению максимума накопления и равновесного содержания радионуклидов в различных элементах обмена и снижению поглощенной дозы на единицу активности. Эти различия имеют физическую, а не биологическую природу, но сказываются на биологической эффективности радионуклидов. [c.279]
Имеются данные о возрастном повышении порога чувствительности гипоталамуса и желез внутренней секреции к соединениям (глюкоза, кортизон, эстрогены, гонадотропины), являющимся звеньями функциональной межси-стемной регуляции по принципу прямых и обратных связей. Имеются и данные, свидетельствующие о возможном пострадиационном снижении чувствительности тканей к гормонам некоторых периферических эндокрй н ных желез (тиреоидным, кортикостероидным). Показано также возрастное снижение чувствительности семенников к гонадотропной стимуляции и понижение в старости эффекта действия тестостерона на обменные процессы и морфологию тканей. [c.109]
Показано также возрастное снижение чувствительности семенников к гонадотропной стимуляции и понижение в старости эффекта действия тестостерона на обменные процессы и морфологию тканей. [c.109]
Трансглутаминаза 2 играет противоположную роль в регуляции клеточных функций, а также в росте и гибели клеток
Fesus L, Piacentini M. Трансглутаминаза 2: загадочный фермент с разнообразными функциями. Trends Biochem Sci 2002; 27 : 534–539.
CAS
Статья
PubMed
Google ученый
Iismaa SE, Mearns BM, Lorand L, Graham RM. Трансглутаминазы и болезни: уроки генетически модифицированных моделей мышей и наследственных заболеваний. Physiol Rev 2009; 89 : 991–1023.
CAS
Статья
PubMed
Google ученый
 org/ScholarlyArticle»> 3
org/ScholarlyArticle»> 3Lorand L, Graham RM. Трансглутаминазы: сшивающие ферменты с плейотропными функциями. Nat Rev Mol Cell Biol 2003; 4 : 140–156.
CAS
Статья
PubMed
Google ученый
Iismaa SE, Chung L, Wu MJ, Teller DC, Yee VC, Graham RM.Основной домен тканевой трансглутаминазы Gh гидролизует GTP и ATP. Biochemistry 1997; 36 : 11655–11664.
CAS
Статья
PubMed
Google ученый
Лай Т.С., Бойня Т.Ф., Коропчак С.М., Харун З.А., Гринберг К.С. С-концевая делеция тканевой трансглутаминазы человека усиливает магний-зависимую активность GTP / ATPase. J Biol Chem 1996; 271 : 31191–31195.
CAS
Статья
PubMed
Google ученый
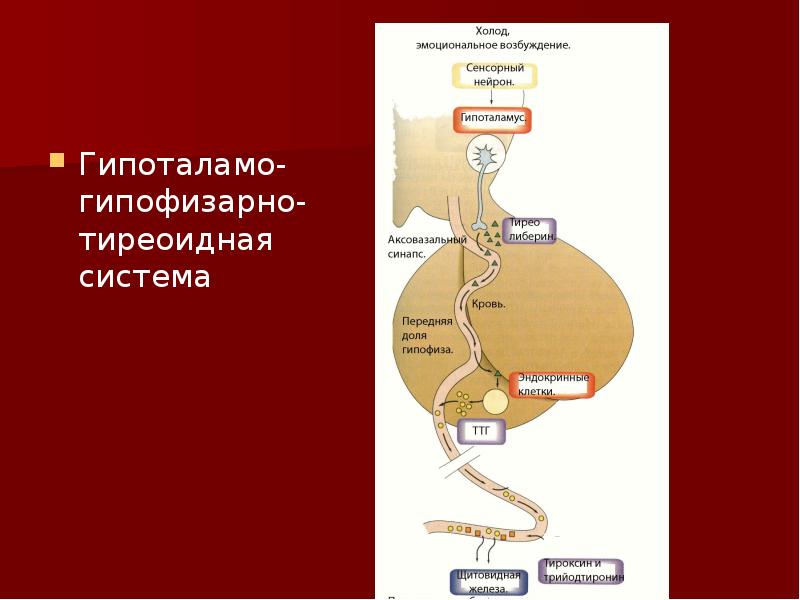 org/ScholarlyArticle»> 6
org/ScholarlyArticle»> 6Hasegawa G, Suwa M, Ichikawa Y, Ohtsuka T., Kumagai S, Kikuchi M et al .Новая функция трансглутаминазы тканевого типа: протеиндисульфидизомераза. Biochem J 2003; 373 : 793–803.
CAS
Статья
PubMed
PubMed Central
Google ученый
Мишра С., Мерфи Л.Дж. Тканевая трансглутаминаза обладает внутренней киназной активностью: идентификация трансглутаминазы 2 как протеин-3 киназы, связывающей инсулиноподобный фактор роста. J Biol Chem 2004; 279 : 23863–23868.
CAS
Статья
PubMed
Google ученый
Griffin M, Casadio R, Bergamini CM. Трансглутаминазы: природные биологические клеи. Biochem J 2002; 368 : 377–396.
CAS
Статья
PubMed
PubMed Central
Google ученый
Экерт Р.Л., Каартинен М.Т., Нурминская М., Белкин А.М., Колак Г., Джонсон Г.В. и др. .Трансглутаминазная регуляция функции клеток. Physiol Rev 2014; 94 : 383–417.
CAS
Статья
PubMed
PubMed Central
Google ученый
Канчан К., Фуксрайтер М, Фесус Л. Физиологические, патологические и структурные последствия неферментативных белок-белковых взаимодействий многофункциональной трансглутаминазы человека 2. Cell Mol Life Sci 2015; 72 : 3009–3035.
CAS
Статья
PubMed
Google ученый
Куо Т.Ф., Тацукава Х., Мацуура Т., Нагацума К., Хиросе С., Кодзима С. Свободные жирные кислоты индуцируют трансглутаминаза 2-зависимый апоптоз в гепатоцитах через ER-стресс-стимулированные пути PERK. J Cell Physiol 2012; 227 : 1130–1137.
CAS
Статья
PubMed
Google ученый
Тацукава Х., Фукая Й., Фрамптон Дж., Мартинес-Фуэнтес А., Сузуки К., Куо TF и др. .Роль трансглутаминазы 2 в повреждении печени через перекрестное связывание и подавление транскрипционного фактора Sp1. Гастроэнтерология 2009; 136 : e1710.
Артикул
CAS
Google ученый
Тацукава Х., Кодзима С. Последние достижения в понимании роли трансглутаминазы 2 при алкогольном стеатогепатите. Cell Biol Int 2010; 34 : 325–334.
CAS
Статья
PubMed
Google ученый
Хитоми К., Кодзима С., Фесус Л. (ред.).Трансглутаминазы, многофункциональные модификаторы и мишени для открытия новых лекарств, 1-е изд. Springer Japan: Токио, Япония, 2015.
Google ученый
Fesus L, Szondy Z. Трансглутаминаза 2 в балансе гибели и выживания клеток. FEBS Lett 2005; 579 : 3297–3302.
Артикул
CAS
PubMed
Google ученый
Киралы Р, Демени М, Фесус Л.Белок переамидирование по трансглютаминазе 2 в клетках: конфликтная Са2 + -зависимое действие многофункционального белка. FEBS J 2011; 278 : 4717–4739.
CAS
Статья
PubMed
Google ученый
Tee AE, Marshall GM, Liu PY, Xu N, Haber M, Norris MD et al . Противодействующие эффекты двух изоформ белков тканевой трансглутаминазы в дифференцировке клеток нейробластомы. J Biol Chem 2010; 285 : 3561–3567.
CAS
Статья
PubMed
Google ученый
Киралы Р., Барта Е., Фесус Л. Полиморфизм трансглутаминазы 2: необычно низкая частота геномных вариантов с недостаточными функциями. аминокислоты 2013; 44 : 215–225.
CAS
Статья
PubMed
Google ученый
Фенг Дж.Ф., Ри С.Г., Им МДж. Доказательства того, что фосфолипаза дельта1 является эффектором в передаче сигналов, опосредованной Gh (трансглутаминаза II). J Biol Chem 1996; 271 : 16451–16454.
CAS
Статья
PubMed
Google ученый
Саранг З., Молнар П., Немет Т., Гомба С., Кардон Т., Мелино Г. и др. . Тканевая трансглутаминаза (TG2), действующая как G-белок, защищает гепатоциты от Fas-опосредованной гибели клеток у мышей. Гепатология 2005; 42 : 578–587.
CAS
Статья
PubMed
Google ученый
Хуо Дж., Мец С.А., Ли Дж.Роль тканевой трансглутаминазы в апоптозе инсулин-секретирующих (HIT-T15) клеток, вызванном истощением ГТФ. Biochem Pharmacol 2003; 66 : 213–223.
CAS
Статья
PubMed
Google ученый
Monsonego A, Friedmann I, Shani Y, Eisenstein M, Schwartz M. GTP-зависимые конформационные изменения, связанные с функциональным переключением между Galpha и сшивающей активностью в тканевой трансглутаминазе мозга. J Mol Biol 1998; 282 : 713–720.
CAS
Статья
PubMed
Google ученый
Begg GE, Carrington L, Stokes PH, Matthews JM, Wouters MA, Husain A et al . Механизм аллостерической регуляции трансглутаминазы 2 с помощью GTP. Proc Natl Acad Sci USA 2006; 103 : 19683–19688.
CAS
Статья
PubMed
Google ученый
Эшлиманн Д., Полссон М., Манн К.Идентификация Gln726 в нидогене как акцептора амина в катализируемом трансглутаминазой перекрестном сшивании комплексов ламинин-нидоген. J Biol Chem 1992; 267 : 11316–11321.
CAS
PubMed
PubMed Central
Google ученый
Kaartinen MT, Pirhonen A, Linnala-Kankkunen A, Maenpaa PH. Катализируемое трансглутаминазой перекрестное сшивание остеопонтина ингибируется остеокальцином. J Biol Chem 1997; 272 : 22736–22741.
CAS
Статья
PubMed
Google ученый
Kleman JP, Aeschlimann D, Paulsson M, van der Rest M. Катализируемое трансглутаминазой перекрестное сшивание фибрилл коллагена V / XI в клетках рабдомиосаркомы A204. Biochemistry 1995; 34 : 13768–13775.
CAS
Статья
PubMed
Google ученый
Martinez J, Chalupowicz DG, Roush RK, Sheth A, Barsigian C.Трансглутаминаза-опосредованный процессинг фибронектина монослоями эндотелиальных клеток. Biochemistry 1994; 33 : 2538–2545.
CAS
Статья
PubMed
Google ученый
Акимов С.С., Белкин А.М. Трансглутаминаза на клеточной поверхности способствует сборке фибронектина посредством взаимодействия с желатин-связывающим доменом фибронектина: роль в TGFbeta-зависимом отложении матрикса. J Cell Sci 2001; 114 : 2989–3000.
CAS
PubMed
Google ученый
Ван З., Коллиган Р.Дж., Гросс С.Р., Данен Э.Х., Оренд Дж., Телчи Д. и др. . RGD-независимая адгезия клеток через матрикс тканевой трансглутаминазы-фибронектина способствует отложению фибрилл фибронектина и требует совместной передачи сигналов интегрина синдекан-4/2 альфа5бета1. J Biol Chem 2010; 285 : 40212–40229.
CAS
Статья
PubMed
PubMed Central
Google ученый
Ван З., Телчи Д., Гриффин М.Важность синдекана-4 и синдекана-2 в адгезии и выживании клеток остеобластов, опосредованных тканевым комплексом трансглутаминаза-фибронектин. Exp Cell Res 2011; 317 : 367–381.
CAS
Статья
PubMed
Google ученый
Song H, Chang W, Lim S, Seo HS, Shim CY, Park S и др. . Тканевая трансглутаминаза необходима для опосредованного интегрином выживания мезенхимальных стволовых клеток костного мозга. стволовых клеток 2007; 25 : 1431–1438.
CAS
Статья
PubMed
Google ученый
Ханна М., Челладурай Б., Гавини А., Ли Л., Шао М., Кортни Д. и др. . Нацеливание на адгезию опухолевых клеток яичников, опосредованную тканевой трансглутаминазой. Mol Cancer Ther 2011; 10 : 626–636.
CAS
Статья
PubMed
Google ученый
Мехта К., Кумар А., Ким Х.И.Трансглутаминаза 2: многозадачный белок в сложной цепи воспаления и рака. Biochem Pharmacol 2010; 80 : 1921–1929.
CAS
Статья
PubMed
Google ученый
Малорни В., Фаррас М.Г., Матаррезе П., Тинари А., Чиарло Л., Мусави-Шафай П. и др. . Транслокатор 1 адениновых нуклеотидов действует как субстрат трансглутаминазы 2 типа: последствия для митохондриально-зависимого апоптоза. Cell Death Differ 2009; 16 : 1480–1492.
CAS
Статья
PubMed
Google ученый
Мишра С., Мерфи Л.Дж. Онкобелок p53 является субстратом для активности киназы тканевой трансглутаминазы. Biochem Biophys Res Commun 2006; 339 : 726–730.
CAS
Статья
PubMed
Google ученый
Mishra S, Saleh A, Espino PS, Davie JR, Murphy LJ.Фосфорилирование гистонов тканевой трансглутаминазой. J Biol Chem 2006; 281 : 5532–5538.
CAS
Статья
PubMed
Google ученый
Мишра С., Мелино Дж., Мерфи Л.Дж. Активность киназы трансглутаминазы 2 способствует индуцированному протеинкиназой А фосфорилированию белка ретинобластомы. J Biol Chem 2007; 282 : 18108–18115.
CAS
Статья
PubMed
Google ученый
Beninati S, Gentile V, Caraglia M, Lentini A, Tagliaferri P, Abbruzzese A.Экспрессия тканевой трансглутаминазы влияет на метаболизм гипузина в клетках BALB / c 3T3. FEBS Lett 1998; 437 : 34–38.
CAS
Статья
PubMed
Google ученый
Beninati S, Nicolini L, Jakus J, Passeggio A, Abbruzzese A. Идентификация сайта субстрата для трансглутаминаз на факторе инициации синтеза белка человека 5 A. Biochem J 1995; 305 : 725–728.
CAS
Статья
PubMed
PubMed Central
Google ученый
Сингх США, Ли Кью, Серионе Р.Идентификация эукариотического фактора инициации 5A в качестве партнера клеточного связывания, стимулированного ретиноевой кислотой, для тканевой трансглутаминазы II. J Biol Chem 1998; 273 : 1946–1950.
CAS
Статья
PubMed
Google ученый
Curro M, Condello S, Caccamo D, Ferlazzo N, Parisi G, Ientile R. Токсичность, вызванная гомоцистеином, увеличивает экспрессию TG2 в клетках Neuro2a. аминокислоты 2009; 36 : 725–730.
CAS
Статья
PubMed
Google ученый
Николас Б., Сметерст П., Вердерио Е., Джонс Р., Гриффин М. Поперечное сшивание клеточных белков тканевой трансглутаминазой во время некротической гибели клеток: механизм поддержания целостности ткани. Biochem J 2003; 371 : 413–422.
CAS
Статья
PubMed
PubMed Central
Google ученый
Ким С.Ю.Трансглутаминаза 2 при воспалении. Front Biosci 2006; 11 : 3026–3035.
CAS
Статья
PubMed
Google ученый
Amendola A, Rodolfo C, Di Caro A, Ciccosanti F, Falasca L, Piacentini M. Экспрессия «тканевой» трансглутаминазы в ВИЧ-инфицированных клетках: фермент с противовирусным действием? Ann NY Acad Sci 2001; 946 : 108–120.
CAS
Статья
PubMed
Google ученый
Родольфо С., Мормон Е., Матаррезе П., Чиккосанти Ф., Фаррас М.Г., Гарофано Е. и др. .Тканевая трансглутаминаза — это многофункциональный белок, содержащий только Bh4. J Biol Chem 2004; 279 : 54783–54792.
CAS
Статья
PubMed
Google ученый
Szegezdi E, Szondy Z, Nagy L, Nemes Z, Friis RR, Davies PJ et al . Связанная с апоптозом регуляция in vivo тканевого промотора гена трансглутаминазы. Cell Death Differ 2000; 7 : 1225–1233.
CAS
Статья
PubMed
Google ученый
Антоняк М.А., Миллер А.М., Янсен Дж. М., Бем Дж. Э., Балкман К. Э., Вакшлаг Дж. Дж. и др. .Увеличение экспрессии тканевой трансглутаминазы и активация эпидермальным фактором роста ингибируют индуцированный доксорубицином апоптоз в клетках рака груди человека. J Biol Chem 2004; 279 : 41461–41467.
CAS
Статья
PubMed
Google ученый
Герман Дж. Ф., Мангала Л. С., Мехта К. Последствия повышенной экспрессии тканевой трансглутаминазы (TG2) в клетках лекарственно-устойчивого рака молочной железы (MCF-7). Онкоген 2006; 25 : 3049–3058.
CAS
Статья
PubMed
Google ученый
Park KS, Kim DS, Jeong KC, Kim SY. Увеличение экспрессии трансглутаминазы 2 связано с активацией NF-kappaB в тканях рака груди. Front Biosci (Landmark Ed) 2009; 14 : 1945–1951.
CAS
Статья
Google ученый
Риттер С.Дж., Дэвис П.Дж.Идентификация элемента ответа трансформирующего фактора роста-бета1 / костного морфогенетического белка 4 (TGF-beta1 / BMP4) в промоторе гена трансглутаминазы ткани мыши. J Biol Chem 1998; 273 : 12798–12806.
CAS
Статья
PubMed
Google ученый
Джонсон К., Хашимото С., Лотц М., Прицкер К., Теркельтауб Р. Интерлейкин-1 индуцирует проминерализующую активность трансглутаминазы хрящевой ткани и фактора XIIIa. Am J Pathol 2001; 159 : 149–163.
CAS
Статья
PubMed
PubMed Central
Google ученый
Суто Н, Икура К, Сасаки Р. Экспрессия, индуцированная интерлейкином-6 трансглутаминазы тканевого типа в клетках гепатобластомы человека HepG2. J Biol Chem 1993; 268 : 7469–7473.
CAS
PubMed
Google ученый
Кунцио Г.С., Цыганская М., Чжу Дж., Лю С.Л., Надь Л., Томази В. и др. .TNF-альфа модулирует экспрессию гена тканевой трансглутаминазы в клетках печени. Am J Physiol 1998; 274 : G240 – G245.
CAS
PubMed
Google ученый
Ou H, Haendeler J, Aebly MR, Kelly LA, Cholewa BC, Koike G et al . Тканевая трансглутаминаза, индуцированная ретиноевой кислотой, и апоптоз гладкомышечных клеток сосудов. Circ Res 2000; 87 : 881–887.
CAS
Статья
PubMed
Google ученый
Шимада Дж., Сузуки Ю., Ким С.Дж., Ван П.С., Мацумура М., Кодзима С.Трансактивация посредством взаимодействия RAR / RXR-Sp1: характеристика связывания между Sp1 и мотивом GC-бокса. Мол эндокринол 2001; 15 : 1677–1692.
CAS
Статья
PubMed
Google ученый
Balajthy Z, Csomos K, Vamosi G, Szanto A, Lanotte M, Fesus L. Тканевая трансглутаминаза способствует дифференцировке и функциям нейтрофильных гранулоцитов. Кровь 2006; 108 : 2045–2054.
CAS
Статья
PubMed
Google ученый
Кодзима С., Нара К., Рифкин ДБ. Потребность в трансглутаминазе для активации латентного трансформирующего фактора роста бета в эндотелиальных клетках крупного рогатого скота. J Cell Biol 1993; 121 : 439–448.
CAS
Статья
PubMed
Google ученый
Nunes I, Gleizes PE, Metz CN, Rifkin DB.Скрытые трансформирующие белковые домены, связывающие фактор роста-бета, участвуют в активации и трансглутаминазозависимом перекрестном связывании латентного трансформирующего фактора роста-бета. J Cell Biol 1997; 136 : 1151–1163.
CAS
Статья
PubMed
PubMed Central
Google ученый
Oh K, Park HB, Byoun OJ, Shin DM, Jeong EM, Kim YW et al . Эпителиальная трансглутаминаза 2 необходима для выработки Т-клетками интерлейкина-17 и последующего легочного воспаления и фиброза у мышей, получавших блеомицин. J Exp Med 2011; 208 : 1707–1719.
CAS
Статья
PubMed
PubMed Central
Google ученый
Розенталь А.К., Гор С.М., Генри Л.А., Ле М. Участие трансглутаминазы в активации латентного трансформирующего фактора роста бета1 в стареющем суставном хряще. Arthritis Rheum 2000; 43 : 1729–1733.
CAS
Статья
PubMed
Google ученый
Shweke N, Boulos N, Jouanneau C, Vandermeersch S, Melino G, Dussaule JC et al .Тканевая трансглутаминаза способствует интерстициальному фиброзу почек, способствуя накоплению фибриллярного коллагена за счет активации TGF-бета и клеточной инфильтрации. Am J Pathol 2008; 173 : 631–642.
CAS
Статья
PubMed
PubMed Central
Google ученый
Сонди З., Саранг З., Мольнар П., Немет Т., Пьячентини М., Мастроберардино П.Г. и др. . Трансглютаминаза 2 — / — мышей обнаруживают связанное с фагоцитозом перекрестное взаимодействие между макрофагами и апоптотическими клетками. Proc Natl Acad Sci USA 2003; 100 : 7812–7817.
CAS
Статья
PubMed
Google ученый
Telci D, Collighan RJ, Basaga H, Griffin M. Повышенная экспрессия TG2 может привести к индукции трансформирующего фактора роста бета1, вызывая повышенный синтез и отложение матричных белков, которые могут регулироваться оксидом азота. J Biol Chem 2009; 284 : 29547–29558.
CAS
Статья
PubMed
PubMed Central
Google ученый
Чатурведи М.М., Сун Б., Ядав В.Р., Каннаппан Р., Аггарвал Б.Б. Зависимость от NF-kappaB и ее роль в развитии рака: «один размер не подходит всем». Онкоген 2011; 30 : 1615–1630.
CAS
Статья
PubMed
Google ученый
Ким Дж. М., Волл Р. Э., Ко С., Ким Д. С., Парк К. С., Ким С.Новый регуляторный механизм активации NF-kappaB с помощью I-kappaBbeta в раковых клетках. Дж Мол Биол 2008; 384 : 756–765.
CAS
Статья
PubMed
Google ученый
Park SS, Kim JM, Kim DS, Kim IH, Kim SY. Трансглутаминаза 2 опосредует образование полимера I-kappaBalpha через С-концевой глутаминовый кластер. J Biol Chem 2006; 281 : 34965–34972.
CAS
Статья
PubMed
Google ученый
Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS.Кальций регулирует S-нитрозилирование, денитрозилирование и активность тканевой трансглутаминазы. Biochemistry 2001; 40 : 4904–4910.
CAS
Статья
PubMed
Google ученый
Сантанам Л., Тудай Е.К., Уэбб А.К., Доузики П., Ким Дж. Х., О Й. Дж. и др. . Снижение S-нитрозилирования тканевой трансглутаминазы способствует возрастному увеличению жесткости сосудов. Circ Res 2010; 107 : 117–125.
CAS
Статья
PubMed
Google ученый
Лучиани А., Виллелла В.Р., Васатуро А, Джардино I, Райя В., Петтоелло-Мантовани М. и др. . СУМОилирование тканевой трансглутаминазы как связующее звено между окислительным стрессом и воспалением. J Immunol 2009; 183 : 2775–2784.
CAS
Статья
PubMed
Google ученый
Amendola A, Gougeon ML, Poccia F, Bondurand A, Fesus L, Piacentini M.Индукция «тканевой» трансглутаминазы в патогенезе ВИЧ: свидетельство высокой скорости апоптоза CD4 + Т-лимфоцитов и дополнительных клеток в лимфоидных тканях. Proc Natl Acad Sci USA 1996; 93 : 11057–11062.
CAS
Статья
PubMed
Google ученый
Fesus L. Биохимические явления при естественных формах гибели клеток. FEBS Lett 1993; 328 : 1–5.
CAS
Статья
PubMed
Google ученый
Мелино Г., Анниккиарико-Петруцелли М., Пиредда Л., Канди Е., Джентиле В., Дэвис П.Дж. и др. .Тканевая трансглутаминаза и апоптоз: исследования смысловой и антисмысловой трансфекции с клетками нейробластомы человека. Mol Cell Biol 1994; 14 : 6584–6596.
CAS
Статья
PubMed
PubMed Central
Google ученый
Оливеро С, Амендола А, Родольфо С, Спинеди А, Пьячентини М. Ингибирование «тканевой» трансглутаминазы увеличивает выживаемость клеток, предотвращая апоптоз. J Biol Chem 1999; 274 : 34123–34128.
CAS
Статья
PubMed
Google ученый
Тухольски Дж., Джонсон Г.В. Тканевая трансглутаминаза по-разному модулирует апоптоз зависимым от стимулов образом. J Neurochem 2002; 81 : 780–791.
CAS
Статья
PubMed
Google ученый
Тацукава Х., Сано Т., Фукая Й., Ишибаши Н., Ватанабэ М., Окуно М. и др. .Двойная индукция каспазо-3- и трансглутаминазозависимого апоптоза ациклическим ретиноидом в клетках гепатоцеллюлярной карциномы. Mol Cancer 2011; 10 : 4.
CAS
Статья
PubMed
PubMed Central
Google ученый
Kweon SM, Lee ZW, Yi SJ, Kim YM, Han JA, Paik SG et al . Защитная роль тканевой трансглутаминазы в гибели клеток, вызванной TNF-альфа в клетках нейробластомы SH-SY5Y. J Biochem Mol Biol 2004; 37 : 185–191.
CAS
PubMed
Google ученый
Антоньяк М.А., Сингх США, Ли Д.А., Бем Дж. Э., Комбс С., Згола М.М. и др. . Влияние тканевой трансглутаминазы на дифференцировку клеток, индуцированную ретиноевой кислотой, и защиту от апоптоза. J Biol Chem 2001; 276 : 33582–33587.
CAS
Статья
PubMed
Google ученый
Gundemir S, Johnson GV.Внутриклеточная локализация и конформационное состояние трансглутаминазы 2: последствия для гибели клеток. PLoS One 2009 г .; 4 : e6123.
Артикул
CAS
PubMed
PubMed Central
Google ученый
Hwang IK, Yoo KY, Yi SS, Kim IY, Hwang HS, Lee KY et al . Экспрессия трансглутаминазы тканевого типа (tTG) и влияние ингибитора tTG на область CA1 гиппокампа после временной ишемии у песчанок. Brain Res 2009; 1263 : 134–142.
CAS
Статья
PubMed
Google ученый
Ientile R, Caccamo D, Marciano MC, Curro M, Mannucci C, Campisi A et al . Активность трансглутаминазы и транскрипты мРНК трансглутаминазы при ишемии мозга песчанок. Neurosci Lett 2004; 363 : 173–177.
CAS
Статья
PubMed
Google ученый
Такано К., Сираива К., Морияма М., Накамура Ю.Экспрессия трансглутаминазы 2, индуцированная стимуляцией липополисахаридом вместе с индукцией NO-синтазы в культивируемых астроцитах. Neurochem Int 2010; 57 : 812–818.
CAS
Статья
PubMed
Google ученый
Сайки Р., Парк Х, Исии I, Йошида М., Нисимура К., Тойда Т. и др. . Инфаркт мозга более тесно коррелирует с акролеином, чем с активными формами кислорода. Biochem Biophys Res Commun 2011; 404 : 1044–1049.
CAS
Статья
PubMed
Google ученый
Филиано А.Дж., Бейли С.Д., Тухольски Дж., Гундемир С., Джонсон Г.В. Трансглутаминаза 2 защищает от ишемического инсульта, взаимодействует с HIF1beta и ослабляет передачу сигналов HIF1. FASEB J 2008; 22 : 2662–2675.
CAS
Статья
PubMed
PubMed Central
Google ученый
Филиано А.Дж., Тухольски Дж., Долан П.Дж., Колак Дж., Джонсон Г.В.Трансглутаминаза 2 защищает от ишемического инсульта. Neurobiol Dis 2010; 39 : 334–343.
CAS
Статья
PubMed
PubMed Central
Google ученый
Бойд Дж. М., Мальстром С., Субраманиан Т., Венкатеш Л.К., Шапер У., Элангован Б. и др. . Белки аденовируса E1B 19 кДа и Bcl-2 взаимодействуют с общим набором клеточных белков. Cell 1994; 79 : 341–351.
CAS
Статья
PubMed
Google ученый
Ода Э, Оки Р., Мурасава Х, Немото Дж., Шибуэ Т., Ямасита Т. и др. . Noxa, Bh4-единственный член семейства Bcl-2 и кандидат в медиатор p53-индуцированного апоптоза. Science 2000; 288 : 1053–1058.
CAS
Статья
Google ученый
Jeitner TM, Pinto JT, Красников Б.Ф., Хорсвилл М., Купер А.Дж.Трансглутаминазы и нейродегенерация. J Neurochem 2009; 109 (Дополнение 1): 160–166.
CAS
Статья
PubMed
PubMed Central
Google ученый
Дудек С.М., Джонсон Г.В. Трансглутаминаза способствует образованию полимеров бета-амилоидного пептида. Brain Res 1994; 651 : 129–133.
CAS
Статья
PubMed
Google ученый
Миллер М.Л., Джонсон Г.В.Трансглутаминазное сшивание тау-белка. J Neurochem 1995; 65 : 1760–1770.
CAS
Статья
PubMed
Google ученый
Тухольски Дж., Курет Дж., Джонсон Г.В. Тау модифицируется тканевой трансглутаминазой in situ : возможные функциональные и метаболические эффекты полиаминирования. J Neurochem 1999; 73 : 1871–1880.
CAS
PubMed
Google ученый
Антоняк М.А., Янсен Дж.М., Миллер А.М., Ли Т.К., Эндо М., Церионе РА.Две изоформы тканевой трансглутаминазы опосредуют противоположные клеточные судьбы. Proc Natl Acad Sci USA 2006; 103 : 18609–18614.
CAS
Статья
PubMed
Google ученый
Citron BA, SantaCruz KS, Davies PJ, Festoff BW. Обмен интрон-экзона мРНК трансглутаминазы и агрегация нейронального тау-белка при болезни Альцгеймера. J Biol Chem 2001; 276 : 3295–3301.
CAS
Статья
PubMed
Google ученый
Вольф Дж., Ягер К., Лахманн И., Шонкнехт П., Моравски М., Арендт Т. и др. .Тканевая трансглутаминаза не является биохимическим маркером болезни Альцгеймера. Neurobiol Aging 2013; 34 : 2495–2498.
CAS
Статья
PubMed
Google ученый
Zainelli GM, Ross CA, Troncoso JC, Muma NA. Трансглутаминазные сшивки во внутриядерных включениях при болезни Хантингтона. J Neuropathol Exp Neurol 2003; 62 : 14–24.
CAS
Статья
PubMed
Google ученый
Игараси С., Коиде Р., Шимохата Т., Ямада М., Хаяси Ю., Такано Н. и др. .Подавление образования агрегатов и апоптоза ингибиторами трансглутаминазы в клетках, экспрессирующих усеченный белок DRPLA с расширенным полиглутаминовым участком. Nat Genet 1998; 18 : 111–117.
CAS
Статья
PubMed
Google ученый
Бейли С.Д., Джонсон Г.В. Тканевая трансглутаминаза способствует прогрессированию заболевания на мышиной модели болезни Хантингтона R6 / 2 посредством агрегативно-независимых механизмов. J Neurochem 2005; 92 : 83–92.
CAS
Статья
PubMed
Google ученый
Мастроберардино П.Г., Янникола С., Нардаччи Р., Бернассола Ф., Де Лауренци В., Мелино Г. и др. . Удаление «тканевой» трансглютаминазы снижает гибель нейронов и продлевает выживаемость в мышиной модели болезни Хантингтона. Cell Death Differ 2002; 9 : 873–880.
CAS
Статья
PubMed
Google ученый
Д’Элетто М., Фаррас М.Г., Фаласка Л., Реали В., Оливеро С., Мелино Г. и др. .Трансглутаминаза 2 участвует в созревании аутофагосом. Аутофагия 2009; 5 : 1145–1154.
CAS
Статья
PubMed
Google ученый
Lesort M, Lee M, Tucholski J, Johnson GV. Цистамин подавляет активность каспаз. Значение для лечения полиглутаминовых расстройств. J Biol Chem 2003; 278 : 3825–3830.
CAS
Статья
PubMed
Google ученый
Мастроберардино П.Г., Пьячентини М.Трансглутаминаза 2 типа при болезни Хантингтона: палка о двух концах с клиническим потенциалом. J Intern Med 2010; 268 : 419–431.
CAS
Статья
PubMed
PubMed Central
Google ученый
МакКонуи С.Дж., Бассо М., Ниацкая З.В., Слейман С.Ф., Смирнова Н.А., Лэнгли BC и др. . Ингибирование трансглутаминазы 2 смягчает нарушение регуляции транскрипции на моделях болезни Хантингтона. EMBO Mol Med 2010; 2 : 349–370.
CAS
Статья
PubMed
PubMed Central
Google ученый
Колби Д.В., Кэссиди Дж. П., Лин Дж. К., Инграм В.М., Виттруп К.Д. Стохастическая кинетика образования внутриклеточных агрегатов хантинтина. Nat Chem Biol 2006; 2 : 319–323.
CAS
Статья
PubMed
Google ученый
Kazemi-Esfarjani P, La Spada AR.Дежавю с изюминкой: трансглютаминазы в биоэнергетике и дисфункция транскрипции при болезни Хантингтона. EMBO Mol Med 2010; 2 : 335–337.
Артикул
PubMed
PubMed Central
Google ученый
Чен-Плоткин А.С., Садри-Вакили Г., Йорлинг Г.Дж., Бравман М.В., Бенн С.Л., Глайч К.Е. и др. . Снижение ассоциации фактора транскрипции Sp1 с генами, подавляющими активность при болезни Хантингтона. Neurobiol Dis 2006; 22 : 233–241.
CAS
Статья
PubMed
Google ученый
Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM et al . Транскрипционная активность Sp1 и TAFII130 нарушается на ранних стадиях болезни Гентингтона. Science 2002; 296 : 2238–2243.
CAS
Статья
PubMed
Google ученый
Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H et al .Взаимодействие белка болезни Гентингтона с активатором транскрипции Sp1. Mol Cell Biol 2002; 22 : 1277–1287.
CAS
Статья
PubMed
PubMed Central
Google ученый
Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L et al . Потеря опосредованной хантингтином транскрипции гена BDNF при болезни Гентингтона. Science 2001; 293 : 493–498.
CAS
Статья
PubMed
Google ученый
Коннор М.К., Иррчер I, Худ Д.А. Вызванная сократительной активностью транскрипционная активация цитохрома C включает Sp1 и пропорциональна синтезу митохондриального АТФ в мышечных клетках C2C12. J Biol Chem 2001; 276 : 15898–15904.
CAS
Статья
PubMed
Google ученый
Эванс М.Дж., Скарпулла RC.Взаимодействие ядерных факторов с множественными сайтами в промоторе соматического цитохрома с. Характеристика вышестоящих последовательностей распознавания NRF-1, ATF и интрона Sp1. J Biol Chem 1989; 264 : 14361–14368.
CAS
PubMed
Google ученый
Munsie L, Caron N, Atwal RS, Marsden I, Wild EJ, Bamburg JR и др. . Мутантный хантингтин вызывает дефектное ремоделирование актина во время стресса: определение новой роли трансглутаминазы 2 в нейродегенеративных заболеваниях. Hum Mol Genet 2011; 20 : 1937–1951.
CAS
Статья
PubMed
PubMed Central
Google ученый
Макгоф А., Папа Б, Чиу В., Сорняки А. Кофилин изменяет поворот F-актина: влияние на динамику актиновых филаментов и клеточную функцию. J Cell Biol 1997; 138 : 771–781.
CAS
Статья
PubMed
PubMed Central
Google ученый
Белкин А.М.Внеклеточный TG2: новые функции и регуляция. FEBS J 2011; 278 : 4704–4716.
CAS
Статья
PubMed
PubMed Central
Google ученый
Gaudry CA, Verderio E, Jones RA, Smith C, Griffin M. Тканевая трансглутаминаза является важным игроком на поверхности эндотелиальных клеток человека: доказательства ее экстернализации и совместной локализации с бета (1) интегрином. Exp Cell Res 1999; 252 : 104–113.
CAS
Статья
PubMed
Google ученый
Андринга Г., Лам К.Ю., Чегари М., Ван Х, Чейз Т.Н., Беннетт М.С. Тканевая трансглутаминаза катализирует образование поперечных связей альфа-синуклеина при болезни Паркинсона. FASEB J 2004; 18 : 932–934.
CAS
Статья
PubMed
Google ученый
Джунн Э., Рончетти Р.Д., Кесадо М.М., Ким С.И., Мурадян М.М.Тканевая трансглутаминаза-индуцированная агрегация альфа-синуклеина: последствия для образования телец Леви при болезни Паркинсона и деменции с тельцами Леви. Proc Natl Acad Sci USA 2003; 100 : 2047–2052.
CAS
Статья
PubMed
Google ученый
Chen CS, Wu CH, Lai YC, Lee WS, Chen HM, Chen RJ et al . Активированная NF-kappaB тканевая трансглутаминаза участвует в вызванном этанолом повреждении печени и возможной роли прополиса в предотвращении фиброгенеза. Токсикология 2008; 246 : 148–157.
CAS
Статья
PubMed
Google ученый
Grenard P, Bresson-Hadni S, El Alaoui S, Chevallier M, Vuitton DA, Ricard-Blum S. Сшивание, опосредованное трансглутаминазой, участвует в стабилизации внеклеточного матрикса при фиброзе печени человека. J Hepatol 2001; 35 : 367–375.
CAS
Статья
PubMed
Google ученый
Мирза А., Лю С.Л., Фризелл Э., Чжу Дж., Маддукури С., Мартинес Дж. и др. .Роль тканевой трансглутаминазы в повреждении печени и фиброгенезе и ее регуляция с помощью NF-kappaB. Am J Physiol 1997; 272 : G281 – G288.
CAS
PubMed
Google ученый
Strnad P, Harada M, Siegel M, Terkeltaub RA, Graham RM, Khosla C et al . Трансглутаминаза 2 регулирует образование включений мелких тел и увеличение печени, связанное с повреждением. Гастроэнтерология 2007; 132 : 1515–1526.
CAS
Статья
PubMed
PubMed Central
Google ученый
Ву Дж., Лю С.Л., Чжу Дж. Л., Нортон П.А., Нодзири С., Хук Дж. Б. и др. . Роль тканевой трансглутаминазы в индуцированном этанолом ингибировании пролиферации гепатоцитов и передаче альфа-1-адренергических сигналов. J Biol Chem 2000; 275 : 22213–22219.
CAS
Статья
PubMed
Google ученый
D’Argenio G, Amoruso DC, Mazzone G, Vitaglione P, Romano A, Ribecco MT и др. .Экстракт чеснока предотвращает вызванный CCl (4) фиброз печени у крыс: роль тканевой трансглутаминазы. Dig Liver Dis 2010; 42 : 571–577.
CAS
Статья
PubMed
Google ученый
Кури А.Дж., Хольцман Д.А., Джексон С.П., Тьянь Р. Синергетическая активация богатыми глутамином доменами человеческого фактора транскрипции Sp1. Cell 1989; 59 : 827–836.
CAS
Статья
PubMed
Google ученый
Han JA, Park SC.Трансглутаминазозависимая модуляция активности транскрипционного фактора Sp1. Mol Cells 2000; 10 : 612–618.
CAS
Статья
PubMed
Google ученый
Pascal E, Tjian R. Различные домены активации Sp1 регулируют образование мультимеров и опосредуют транскрипционный синергизм. Genes Dev 1991; 5 : 1646–1656.
CAS
Статья
PubMed
Google ученый
Хорино К., Нишюра Х., Осако Т., Сибуя Ю., Хираока Т., Китамура Н. и др. .Хемотаксический фактор моноцитов, димер рибосомного белка S19, в фагоцитарном клиренсе апоптотических клеток. Lab Invest 1998; 78 : 603–617.
CAS
PubMed
Google ученый
Тот Б., Гарабуччи Е., Саранг З., Вереб Г., Вамози Г., Эшлиманн Д. и др. . Трансглутаминаза 2 необходима для формирования эффективного фагоцитарного портала в макрофагах, поглощающих апоптотические клетки. J Immunol 2009; 182 : 2084–2092.
CAS
Статья
PubMed
Google ученый
Сух Г.Й., Хэм Х.С., Ли С.Х., Чой Дж. К., Ко В.Дж., Ким С.И. и др. . Пептид с активностью против трансглутаминазы снижает вызванное липополисахаридом воспаление легких у мышей. Exp Lung Res 2006; 32 : 43–53.
CAS
Статья
PubMed
Google ученый
Nardacci R, Lo Iacono O, Ciccosanti F, Falasca L, Addesso M, Amendola A и др. .Трансглутаминаза типа II играет защитную роль при повреждении печени. Am J Pathol 2003; 162 : 1293–1303.
CAS
Статья
PubMed
PubMed Central
Google ученый
Wu J, Zern MA. Тканевая трансглутаминаза, ключевой фермент, участвующий в заболеваниях печени. Hepatol Res 2004; 29 : 1–8.
CAS
Статья
PubMed
Google ученый
Фридман С.Л.Механизмы фиброгенеза печени. Гастроэнтерология 2008; 134 : 1655–1669.
CAS
Статья
PubMed
PubMed Central
Google ученый
Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Роль TGF-бета в фиброзе печени. Front Biosci 2002; 7 : d793 – d807.
CAS
Статья
PubMed
Google ученый
Le M, Gohr CM, Rosenthal AK.Трансглутаминаза участвует во включении латентного TGFbeta во внеклеточный матрикс стареющих хондроцитов суставов. Connect Tissue Res 2001; 42 : 245–253.
CAS
Статья
PubMed
Google ученый
Хоу XJ, Jin ZD, Jiang F, Zhu JW, Li ZS. Экспрессия Smad7 и Smad убиквитин регуляторный фактор 2 в крысиной модели хронического панкреатита. J Dig Dis 2015; 16 : 408–415.
CAS
Статья
PubMed
Google ученый
Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Мет обеспечивает важные сигналы для регенерации печени. Proc Natl Acad Sci USA 2004; 101 : 10608–10613.
CAS
Статья
PubMed
Google ученый
Хью К.Г., Фактор ВМ, Санчес А., Учида К., Коннер Э.А., Торгейрссон С.С.Фактор роста гепатоцитов / сигнальный путь c-met необходим для эффективной регенерации и восстановления печени. Proc Natl Acad Sci USA 2004; 101 : 4477–4482.
CAS
Статья
PubMed
Google ученый
Giebeler A, Boekschoten MV, Klein C, Borowiak M, Birchmeier C, Gassler N et al . c-Met обеспечивает защиту от хронического повреждения ткани печени и прогрессирования фиброза после перевязки желчных протоков у мышей. Гастроэнтерология 2009; 137 : 308 e291 – e294.
Артикул
CAS
Google ученый
Кодзима С., Куо Т.Ф., Тацукава Х., Хиросе С. Индукция перекрестного связывания и подавления Sp1 трансглютаминазой во время повреждения печени при ASH и NASH через различные пути стресса ER. Dig Dis 2010; 28 : 715–721.
Артикул
PubMed
Google ученый
Дил А.М., Абдо С., Браун Н.Дополнительный путресцин устраняет связанное с этанолом ингибирование регенерации печени. Гепатология 1990; 12 : 633–637.
CAS
Статья
PubMed
Google ученый
Hsu TC, Huang CY, Chiang SY, Lai WX, Tsai CH, Tzang BS. Ингибитор трансглутаминазы цистамин облегчает нарушения в печени мышей NZB / W F1. евро J Pharmacol 2008; 579 : 382–389.
CAS
Статья
PubMed
Google ученый
Шибли И.А. младший, Гавиган М.Д., Пеннингтон С.Н. Влияние этанола на тканевые полиамины и активность орнитиндекарбоксилазы: краткий обзор. Alcohol Clin Exp Res 1995; 19 : 209–215.
CAS
Статья
PubMed
Google ученый
Попов Ю., Свердлов Д.Ю., Шарма А.К., Бхаскар К.Р., Ли С., Фрейтаг Т.Л. и др. . Тканевая трансглутаминаза не влияет на стабильность фиброзного матрикса или регресс фиброза печени у мышей. Гастроэнтерология 2011; 140 : 1642–1652.
CAS
Статья
PubMed
PubMed Central
Google ученый
Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Экспрессия тканевой трансглутаминазы способствует прикреплению, инвазии и выживанию клеток рака груди. Онкоген 2007; 26 : 2459–2470.
CAS
Статья
PubMed
Google ученый
Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K et al .Высокоэкспрессируемые гены в аденокарциномах протоков поджелудочной железы: всесторонняя характеристика и сравнение профилей транскрипции, полученных с помощью трех основных технологий. Cancer Res 2003; 63 : 8614–8622.
CAS
PubMed
Google ученый
Kong L, Korthuis RJ. Адгезия клеток меланомы к поврежденным артериолам: механизмы стабилизированного связывания. Clin Exp Metastasis 1997; 15 : 426–431.
CAS
Статья
PubMed
Google ученый
Сюй Л., Хайнс РО. GPR56 и TG2: возможные роли в подавлении роста опухоли микросредой. Cell Cycle 2007; 6 : 160–165.
CAS
Статья
PubMed
Google ученый
Johnson TS, Knight CR, el-Alaoui S, Mian S, Rees RC, Gentile V et al .Трансфекция тканевой трансглутаминазы в высокозлокачественную фибросаркому хомяка приводит к снижению частоты роста первичной опухоли. Онкоген 1994; 9 : 2935–2942.
CAS
PubMed
Google ученый
Shrestha RT, Shrestha H, Ishibashi R, Matsuura N, Kagechika T., Kose H et al . Молекулярный механизм, с помощью которого ациклический ретиноид вызывает ядерную локализацию трансглутаминазы 2 в клетках гепатоцеллюлярной карциномы человека. Смерть клетки 2015; 6 : e2002.
CAS
Статья
PubMed
PubMed Central
Google ученый
Zhang J, Antonyak MA, Singh G, Cerione RA. Механизм повышения уровня рецепторов EGF в глиобластомах. Cell Rep 2013; 3 : 2008–2020.
CAS
Статья
PubMed
PubMed Central
Google ученый
Оливерио С., Амендола А, Ди Сано Ф., Фаррас М.Г., Фесус Л., Немес Z и др. .Тканевая трансглутаминаза-зависимая посттрансляционная модификация продукта гена ретинобластомы в промоноцитарных клетках, подвергающихся апоптозу. Mol Cell Biol 1997; 17 : 6040–6048.
CAS
Статья
PubMed
PubMed Central
Google ученый
Бём Дж. Э., Сингх У., Комбс С., Антоньяк М.А., Черионе, РА. Тканевая трансглутаминаза защищает от апоптоза путем модификации белка-супрессора опухолей p110 Rb. J Biol Chem 2002; 277 : 20127–20130.
CAS
Статья
PubMed
Google ученый
Милакович Т., Тухольски Дж., Маккой Е., Джонсон Г.В. Внутриклеточная локализация и состояние активности тканевой трансглутаминазы по-разному влияют на гибель клеток. J Biol Chem 2004; 279 : 8715–8722.
CAS
Статья
PubMed
Google ученый
Bungay PJ, Оуэн РА, Coutts IC, Гриффин М.Роль трансглутаминазы в стимулированном глюкозой высвобождении инсулина из бета-клеток поджелудочной железы. Biochem J 1986; 235 : 269–278.
CAS
Статья
PubMed
PubMed Central
Google ученый
Gomis R, Sener A, Malaisse-Lagae F, Malaisse WJ. Активность трансглутаминазы в островках поджелудочной железы. Biochim Biophys Acta 1983; 760 : 384–388.
CAS
Статья
PubMed
Google ученый
Sener A, Dunlop ME, Gomis R, Mathias PC, Malaisse-Lagae F, Malaisse WJ.Роль трансглутаминазы в высвобождении инсулина. Исследование с метиловыми эфирами глицина и саркозина. Эндокринология 1985; 117 : 237–242.
CAS
Статья
PubMed
Google ученый
Дрисколл, HK, Адкинс CD, Чертов Т.Э., Кордл МБ, Мэтьюз К.А., Чертов Б.С. Стимуляция секреции инсулина витамином А: влияние на мРНК трансглутаминазы и активность с использованием островков крыс и секретирующих инсулин клеток. Pancreas 1997; 15 : 69–77.
CAS
Статья
PubMed
Google ученый
Сибуя Х, Сакаи К., Кабир-Салмани М, Вачи Й, Ивашита М. Полимеризация белка-1, связывающего инсулиноподобный фактор роста (IGFBP-1), усиливает действие IGF-I в плаценте. J Cell Physiol 2011; 226 : 434–439.
CAS
Статья
PubMed
Google ученый
Линдси Массачусетс, Бунгей ПиДжей, Гриффин М.Участие трансглутаминазы в секреции инсулина из островков Лангерганса крыс с электропермеабилизацией. Biosci Rep 1990; 10 : 557–561.
CAS
Статья
PubMed
Google ученый
Бернассола Ф., Федеричи М., Кораццари М., Терринони А., Хрибал М.Л., Де Лауренци В. и др. . Роль трансглутаминазы 2 в толерантности к глюкозе: исследования на мышах с нокаутом и предполагаемая мутация у пациента MODY. FASEB J 2002; 16 : 1371–1378.
CAS
Статья
PubMed
Google ученый
Порцио О, Масса О, Кунсоло В, Коломбо С., Малапонти М., Бертуцци Ф. и др. . Миссенс-мутации в гене TGM2, кодирующем трансглутаминазу 2, обнаруживаются у пациентов с ранним началом диабета 2 типа. Мутации вкратце нет. 982. Онлайн. Hum Mutat 2007; 28 : 1150.
CAS
Статья
PubMed
Google ученый
Iismaa SE, Aplin M, Holman S, Yiu TW, Jackson K, Burchfield JG et al .Гомеостаз глюкозы у мышей не зависит от трансглутаминазы 2. PLoS One 2013; 8 : e63346.
Артикул
PubMed
PubMed Central
Google ученый
Дардик Р., Инбал А. Образование комплекса между тканевой трансглутаминазой II (tTG) и рецептором 2 фактора роста эндотелия сосудов (VEGFR-2): предложенный механизм модуляции ответа эндотелиальных клеток на VEGF. Exp Cell Res 2006; 312 : 2973–2982.
CAS
Статья
PubMed
Google ученый
Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Тканевая трансглутаминаза экспрессируется, активна и непосредственно участвует в заживлении кожных ран крыс и ангиогенезе. FASEB J 1999; 13 : 1787–1795.
CAS
Статья
PubMed
Google ученый
Ди Симоне Н., Де Спирито М., Ди Никуоло Ф, Терсиньи С., Кастеллани Р., Силано М. и др. .Возможные новые механизмы повреждения плаценты при целиакии: антитела против трансглутаминазы нарушают ангиогенез эндометрия человека. Биол Репрод 2013; 89 : 88.
Артикул
CAS
PubMed
Google ученый
Myrsky E, Kaukinen K, Syrjanen M, Korponay-Szabo IR, Maki M, Lindfors K. Аутоантитела к трансглутаминазе 2, специфичные для глютеновой болезни, нарушают ангиогенез. Clin Exp Immunol 2008; 152 : 111–119.
CAS
Статья
PubMed
PubMed Central
Google ученый
Nadalutti CA, Korponay-Szabo IR, Kaukinen K, Griffin M, Maki M, Lindfors K. Антитела IgA пациента с глютеновой болезнью вызывают дефекты адгезии эндотелия и поляризации клеток через внеклеточную трансглутаминазу 2. Cell Mol Life Sci 2014; 71 : 1315–1326.
CAS
Статья
PubMed
Google ученый
Джонс Р.А., Николас Б., Миан С., Дэвис П.Дж., Гриффин М.Снижение экспрессии тканевой трансглутаминазы в линии эндотелиальных клеток человека приводит к изменениям в распределении клеток, адгезии клеток и снижению полимеризации фибронектина. J Cell Sci 1997; 110 : 2461–2472.
CAS
PubMed
Google ученый
Фэй К., Шотар Э, Ольсен Б.Р., Рикар-Блюм С. Первый вариант сети взаимодействия эндостатина. J Biol Chem 2009; 284 : 22041–22047.
CAS
Статья
PubMed
PubMed Central
Google ученый
Фэй С., Инфорзато А., Биньон М., Хартманн Д. Д., Мюллер Л., Баллют Л. и др. . Трансглутаминаза-2: новый партнер эндостатина во внеклеточном матриксе эндотелиальных клеток. Biochem J 2010; 427 : 467–475.
CAS
Статья
PubMed
PubMed Central
Google ученый
Ван З., Перес М., Каха С., Мелино Дж., Джонсон Т.С., Линдфорс К. и др. .Новая внеклеточная роль тканевой трансглутаминазы в связанном с матриксом VEGF-опосредованном ангиогенезе. Смерть клетки 2013; 4 : e808.
CAS
Статья
PubMed
PubMed Central
Google ученый
Охура Н., Ямамото К., Ичиока С., Сокабе Т., Накацука Х., Баба А. и др. . Глобальный анализ генов, чувствительных к сдвигу в эндотелиальных клетках сосудов. J Atheroscler Thromb 2003; 10 : 304–313.
CAS
Статья
PubMed
Google ученый
Джонс Р.А., Котсакис П., Джонсон Т.С., Чау Д.Й., Али С., Мелино Г. и др. . Изменения матрицы, вызванные трансглютаминазой 2, приводят к ингибированию ангиогенеза и роста опухоли. Cell Death Differ 2006; 13 : 1442–1453.
CAS
Статья
PubMed
Google ученый
Beckouche N, Bignon M, Lelarge V, Mathivet T, Pichol-Thievend C, Berndt S et al .Взаимодействие гепарансульфатпротеогликанов с эндотелиальной трансглутаминазой-2 ограничивает ангиогенез, индуцированный VEGF165. Sci Signal 2015; 8 : ra70.
Артикул
CAS
PubMed
Google ученый
Csomos K, Nemet I, Fesus L, Balajthy Z. Тканевая трансглутаминаза вносит вклад в фенотип синдрома дифференцировки, индуцированного полностью транс-ретиноевой кислотой в модели NB4 острого промиелоцитарного лейкоза. Кровь 2010; 116 : 3933–3943.
CAS
Статья
PubMed
Google ученый
Лучиани А., Виллелла В.Р., Эспозито С., Брунетти-Пьерри Н., Медина Д., Сеттембре С. и др. . Дефектный CFTR вызывает образование агресом и воспаление легких при муковисцидозе за счет ингибирования аутофагии, опосредованного ROS. Nat Cell Biol 2010; 12 : 863–875.
CAS
Статья
PubMed
Google ученый
Карпуй М.В., Бехер М.В., Спрингер Дж. Э., Чабас Д., Юсеф С., Педотти Р. и др. .Увеличение выживаемости и уменьшение аномальных движений в трансгенной модели болезни Хантингтона при введении ингибитора трансглутаминазы цистамина. Nat Med 2002; 8 : 143–149.
CAS
Статья
PubMed
Google ученый
Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro анализ репрессии транскрипции, опосредованной хантингтином, выявляет множественные мишени для факторов транскрипции. Cell 2005; 123 : 1241–1253.
CAS
Статья
PubMed
Google ученый
D’Souza DR, Wei J, Shao Q, Hebert MD, Subramony SH, Vig PJ. Тканевые трансглутаминазы сшивают атаксин-1: возможная роль в патогенезе SCA1. Neurosci Lett 2006; 409 : 5–9.
CAS
Статья
PubMed
PubMed Central
Google ученый
Калем П., Грин Х, Джиан П.Действие трансглутаминазы имитирует болезнь Хантингтона: селективную полимеризацию Хантингтина, содержащего расширенный полиглутамин. Mol Cell 1998; 1 : 595–601.
CAS
Статья
PubMed
Google ученый
Гроссо Х., Ву Дж. М., Ли К. В., Им Дж. Й., Маслиа Э, Джунн Е. и др. . Трансглутаминаза 2 усиливает токсичность альфа-синуклеина у мышей и дрожжей. FASEB J 2014; 28 : 4280–4291.
CAS
Статья
PubMed
PubMed Central
Google ученый
Colak G, Johnson GV. Полная абляция трансглутаминазы 2 приводит к уменьшению ударных объемов и астроцитов, которые демонстрируют повышенную выживаемость в ответ на ишемию. Neurobiol Dis 2012; 45 : 1042–1050.
CAS
Статья
PubMed
Google ученый
Отдельные белки, которые выполняют связанные функции во внутриклеточной и внеклеточной микросреде (Журнальная статья)
Радиски, Дерек Си, Столлингс-Манн, Мелоди, Хираи, Йохей и Бисселл, Мина Дж. Отдельные белки, которые выполняют связанные функции во внутриклеточном и внеклеточном микроокружении . США: Н. П., 2009.
Интернет. DOI: 10,1038 / nrm2633.
Радиски, Дерек К., Столлингс-Манн, Мелоди, Хираи, Йохей и Бисселл, Мина Дж. Отдельные белки, которые выполняют связанные функции во внутриклеточной и внеклеточной микросреде . Соединенные Штаты.https://doi.org/10.1038/nrm2633
Радиски, Дерек Си, Столлингс-Манн, Мелоди, Хираи, Йохей и Бисселл, Мина Дж. Ср.
«Отдельные белки, которые выполняют связанные функции во внутриклеточной и внеклеточной микросреде». Соединенные Штаты. https://doi.org/10.1038/nrm2633. https://www.osti.gov/servlets/purl/966045.
@article {osti_966045,
title = {Отдельные белки, которые выполняют связанные функции во внутриклеточной и внеклеточной микросреде},
author = {Радиски, Дерек Си и Столлингс-Манн, Мелоди и Хираи, Йохей и Бисселл, Мина Дж.},
abstractNote = {Поддержание гомеостаза органов и контроль соответствующей реакции на изменения окружающей среды требует тесной координации клеточных функций и организации тканей.Важный компонент этой координации может быть обеспечен белками, которые могут выполнять различные, но связанные функции на обеих сторонах плазматической мембраны. Здесь мы представляем новую гипотезу, согласно которой неклассическая секреция может обеспечить механизм, посредством которого отдельные белки могут интегрировать сложные тканевые функции. Отдельные гены могут оказывать сложное, динамическое влияние посредством ряда различных процессов, которые действуют для умножения функции продукта (ов) гена. Альтернативный сплайсинг может создавать множество различных транскриптов, которые кодируют белки с различными, даже антагонистическими функциями из одного гена.Посттрансляционные модификации могут изменять стабильность, активность, локализацию и даже основную функцию белков. Белок может существовать в разных субклеточных локализациях. Совсем недавно стало ясно, что отдельные белки могут функционировать как внутри, так и вне клетки. Эти белки часто лишены определенных секреторных сигнальных последовательностей и проходят через плазматическую мембрану с помощью механизмов, отдельных от классического секреторного процесса ER / Golgi. Когда примеры таких белков исследуются по отдельности, их многофункциональность и отсутствие сигнальной последовательности вызывают недоумение - почему белок с хорошо известной функцией в одном контексте должен функционировать таким отличным образом в другом? Мы предполагаем, что одной из причин, по которой один белок выполняет внутриклеточные и внеклеточные роли, является координация организации и поддержания глобальной функции ткани.Здесь мы подробно описываем три конкретных примера белков, которые действуют таким образом, описывая их специфические функции во внеклеточном пространстве и во внутриклеточном пространстве, и обсуждаем, как эти функции могут быть связаны. Мы представляем эпиморфин / синтаксин-2, который может координировать морфогенез секреторных органов (как эпиморфин) с контролем секреции белка (как синтаксин-2), амфотерин / блок-1 группы высокой подвижности (HMGB1), который может связывать воспаление (как амфотерин). ) с регуляцией экспрессии гена (как HMGB1) и тканевой трансглутаминазой, которая влияет на доставку апоптотических сигналов и ответ на них, выполняя связанные функции на обеих сторонах плазматической мембраны.Поскольку примечательно, что все три из этих белков, как сообщается, проходят через плазматическую мембрану через неклассические секреторные механизмы, мы также обсудим, почему скоординированные внутренние / внешние функции могут быть обнаружены в некоторых примерах белков, которые проходят через плазматическую мембрану через неклассические секреторные механизмы. -классические механизмы и то, как эту взаимосвязь можно использовать для идентификации дополнительных белков, которые имеют эти характеристики.},
doi = {10.1038 / nrm2633},
url = {https: // www.osti.gov/biblio/966045},
journal = {Nature Reviews Molecular Cell Biology},
number =,
объем =,
place = {United States},
год = {2009},
месяц = {6}
}
Регулируемая шлифовка отделения для хранения рецепторов соматостатина регулирует секрецию гипофиза | Журнал клеточной биологии
Понимание маршрута передачи сигнального рецептора может предоставить ценную информацию о регуляции его функции, с пониманием клеточного и физиологического поведения.Ранее сообщалось, что после эндоцитоза SSTR2, основной регуляторный сигнальный GPCR в гипофизе, транспортируется в юкстануклеарный синтаксин-6-позитивный компартмент с интерпретацией, что рецептор доставляется в TGN (Csaba et al., 2007). Здесь мы подтверждаем локализацию интернализованного SSTR2 с синтаксином-6, но предоставляем убедительные доказательства того, что синтаксин-6 не является маркером TGN. Синтаксин-6 представляет собой рецептор растворимого белка прикрепления N-этилмалеимида, который опосредует слияние мембран (Bock et al., 1996). Таким образом, он выполняет разнообразные функции в экзоцитозе везикул в зависимости от типа клеток (Jung et al., 2012; Wendler and Tooze, 2001), и, таким образом, его клеточная локализация отражает его роль. Раннее исследование с помощью электронной микроскопии с одной меткой локализовало синтаксин-6 в основном в TGN и соседних пузырьках в клетках PC12 (Bock et al., 1997). С тех пор синтаксин-6 широко используется в качестве маркера TGN. Поскольку TGN показывает сложное расположение трубчатых / везикулярных мембран, его сложно различить по структуре при однократном окрашивании с помощью электронной микроскопии.Здесь иммуноцитохимические эксперименты с двойной меткой, проанализированные с помощью стандартной конфокальной микроскопии и со сверхвысоким разрешением, а также исследования биохимического фракционирования показывают, что большая часть синтаксина-6 не колокализуется с резидентными белками TGN PIST и TGN38. Более того, лечение брефельдином А было способно функционально отделить структуры, обогащенные синтаксином-6, от TGN. Грибной метаболит Брефельдин А ингибирует антероградный транспорт белка от эндоплазматического ретикулума к Гольджи, блокируя рекрутирование оболочек COPI (Cole et al., 1996). Потеря COPI вызывает слияние цистерн Гольджи с эндоплазматическим ретикулумом (Hess et al., 2000), а TGN — канальцевать в цитоплазму и сливаться с эндосомами (Reaves and Banting, 1992; Wood et al., 1991). В то время как брефельдин А вызывает иммунореактивность TGN38 и PIST для диспергирования в точечные сигналы по всей цитоплазме, что согласуется с предыдущими сообщениями о резидентных белках TGN (Chege and Pfeffer, 1990; Lippincott-Schwartz et al., 1991; Reaves and Banting, 1992; Tooze and Hollinshead, , 1992; Wagner et al., 1994), флуоресцентные сигналы SSTR2, а также синтаксин-6 остаются нетронутыми как околоядерная концентрация. Это наблюдается во многих типах клеток, включая первичные культуры нейронов. Вместе эти находки демонстрируют, что синтаксин-6 определяет функционально отличный TGN, соседний клеточный компартмент, который в клетках гипофиза получает интернализованный SSTR2.
Хотя синтаксин-6 считается маркером TGN, другие исследования описывают его как определяющий специализированный отсек для хранения и рециклинга GLUT4 в адипоцитах (Shewan et al., 2003) и мышечных клеток (Foley, Klip, 2014). Инсулин-чувствительные синтаксин-6-позитивные GSV структурно отделены как от эндосом, так и от TGN (Foley and Klip, 2014; Martin et al., 1994). Здесь мы описываем подобный субкомпартмент в эндокринных клетках гипофиза AtT20, который мы назвали GLSV. Мы обнаружили, что Rab10, функциональный маркер, который опосредует экзоцитоз GSV, пространственно и функционально ассоциируется с SSTR2-позитивными GLSV во время экзоцитоза. Рециркуляция GLSV относительно медленная, и, таким образом, рецепторы хранятся с периодом полураспада ~ 4 часа.Более того, как и GSV, GLSV мобилизуются физиологическим лигандом CRF и сигнальными компонентами ниже рецептора CRF. В этом контексте интересно, что рециклинг SSTR2 имеет гораздо более быструю кинетику в клетках HeLa, чем в клетках AtT20, предполагая, что есть факторы в клетках AtT20, которые ограничивают рециклинг, которые отсутствуют в клетках HeLa, в которых отсутствует эндогенный SSTR2. Одна из возможностей — отсутствие протеазы, необходимой для освобождения GLSV от внутриклеточной секвестрации в клетках HeLa.Другой возможностью является дифференциальная экспрессия и / или дифференциальная активность эндосомальных пептидаз, таких как эндотелин-превращающий фермент-1, которая, как было показано, необходима для деградации лиганда и высвобождения SSTR2 для рециклинга (Zhao et al., 2013).
Таким образом, это исследование демонстрирует, что концепция физиологического мобильного отсека для хранения должна быть расширена за пределы GLUT4 в мышечных и жировых клетках. Фактически, уже существует несколько таких специализированных пузырьков.Напр., Водный канал аквапорина 2 накапливается внутри клетки и мобилизуется на апикальную мембрану клеток протоков почек в ответ на передачу сигналов вазопрессина (Park and Kwon, 2015). Сходным образом рецептор AMPA хранится внутри постсинаптического компартмента и мобилизуется на синаптическую плазматическую мембрану в ответ на парную поляризацию пре- и постсинаптических нейронов, чтобы потенцировать синапсы (Choquet, 2010). Хорошо известно, что передача сигналов и транспортировка GPCRs сильно переплетены в том смысле, что локализация рецептора может формировать сигнальные способности рецепторов, а транспортировка GPCRs может регулироваться внеклеточными сигналами, которые воздействуют на рецепторы (Sorkin and von Zastrow, 2009).Напр., В последнем отношении, активация субстанцией P рецептора нейрокинина 1 усиливает μ-опиоидный рецептор после рециклинга эндоцитов зависимым от протеинкиназы C образом (Bowman et al., 2015). Опосредованное протеинкиназой А фосфорилирование β2-адренергического рецептора обеспечивает его рекрутирование в зависимый от последовательности микродомен рециклинга на эндосомах, обеспечивая более медленный регулируемый рециклинг (Vistein and Puthenveedu, 2013). Следовательно, хотя наблюдение, что активация рецептора CRF приводит к усиленному рециклированию SSTR2, является просто еще одним примером регуляции трафика GPCR, оно уникально тем, что включает мобилизацию GLSV.
Мы спросили, может ли механизм обратной связи, наблюдаемый в кортикотропах между рецептором CRF, стимулирующим высвобождение гормона, и ингибирующим высвобождение гормона SSTR2, обеспечивать механизм точной настройки эндогенных ритмов высвобождения гормона гипофиза. Мы проверили эту гипотезу в хорошо изученной системе GH из-за ее резкого и последовательного ультрадианного ритма высвобождения GH. У самцов грызунов соматотропы гипофиза имеют секреторные всплески GH (вершина) с промежуточными периодами минимума, когда уровень GH в плазме не определяется (надир).Хотя ясно, что каждый из SOM и GHRH играет решающую роль в регуляции GH, текущие теории обсуждают взаимодействие между SOM и GHRH только на уровне их высвобождения. Хорошо известно, что SOM противодействует высвобождению GH, что приводит к гипотетической модели, в которой пульсирующая секреция GH устанавливается посредством временного реципрокного высвобождения GHRH и SOM. Согласно этой модели, импульс высвобождения GHRH из гипоталамуса приведет к пику высвобождения GH, за которым следует импульс SOM, который приводит к низкому уровню высвобождения GH (Frohman et al., 1990; Соя и Сузуки, 1988 г .; Соя и др., 1990; Стахура и др., 1988; Танненбаум и Линг, 1984). Существуют убедительные доказательства того, что пики секреции GH связаны с предшествующими импульсами GHRH. Животные с поражениями вентромедиально-дугообразной области гипоталамуса демонстрируют заметное подавление амплитуды секреторных всплесков GH (Eikelboom and Tannenbaum, 1983; Martin et al., 1974; Tannenbaum et al., 1983), которое восстанавливается администрацией. GHRH (Tannenbaum et al., 1983). Кроме того, иммунонейтрализация моноклональным антителом, специфичным к GHRH гипоталамуса крысы, устраняет импульсы GH (Wehrenberg et al., 1982). Более того, прямое измерение GHRH и SOM в воротной крови овцы (Frohman et al., 1990) установило значительную связь между импульсами GHRH в воротной крови и пиками секреции GH. Однако исследование не обнаружило значительной связи между уровнями SOM в портальной крови и надиром GHRH или GH. Более того, если уровни эндогенного SOM высоки только в периоды низких уровней GH в плазме, инъекция нейтрализующей SOM антисыворотки должна повышать базальные уровни GH во время периодов надира, но не влияет на пики GH.Однако влияние антисыворотки к SOM на амплитуду пиков GH варьировалось от увеличения (Ferland et al., 1976; Varner et al., 1980) до снижения (Steiner et al., 1978) до отсутствия значительных изменений (Terry and Мартин, 1981). Кроме того, основывать регуляцию GH исключительно на времени высвобождения SOM может быть неразумным, потому что SOM участвует в регуляции других гормонов гипофиза с различными паттернами секреции (den Boon and Sarabdjitsingh, 2017; Durán-Pastén and Fiordelisio, 2013; Стейн и Нго, 2017).Таким образом, мы полагаем, что еще один уровень регуляции на уровне рецепторов — это то, что придает специфичность регуляции отдельных гормонов с помощью SOM. Согласно нашей гипотезе, GHRH связывается с рецептором GHRH на соматотропах гипофиза, активируя высвобождение GH за счет увеличения цитозольного уровня цАМФ и Ca 2+ , что приводит к пику GH в крови. Одновременно активация рецептора GHRH стимулирует восстановление поверхности SSTR2 в GLSV. Оказавшись на поверхности, связывание SSTR2 с SOM активирует передачу сигналов ингибитора G-белка, снижая уровни цитозольного цАМФ и Ca 2+ и останавливая секрецию GH.За этим следует интернализация и секвестрация SSTR2 в GLSV до следующего импульса GHRH, когда цикл начинается заново. В целом, мы предлагаем трафик SSTR2 как регуляторный механизм, вносящий вклад в тонкую настройку эндогенных ритмов различных гормонов гипофиза, задействуя ранее нераспознанный механизм для обеспечения специфичности регуляции в зависимости от типа клеток.
Этот сайт использует файлы cookie для повышения производительности.Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно. Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки своего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались.Чтобы принять файлы cookie с этого сайта, используйте кнопку «Назад» и примите файлы cookie.
- Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г.,
браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере. - Вы установили приложение, которое отслеживает или блокирует установку файлов cookie.Вы должны отключить приложение при входе в систему или уточнить у системного администратора.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу. Чтобы предоставить доступ без файлов cookie
потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файлах cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в файле cookie может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта. Например, сайт
не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к
остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
«Распространенность целиакии и антитканевой трансглутаминазы (ttg) An», Фархан Муршед
Аннотация
Введение
Целиакия и воспалительное заболевание кишечника (ВЗК) — это иммуноопосредованные хронические желудочно-кишечные расстройства, которые проявляются схожими симптомами, такими как боль в животе, диарея и нарушение роста.У взрослых одни исследования показали повышенную распространенность целиакии у пациентов с ВЗК, в то время как другие исследования этого не сделали. Нет опубликованных данных о распространенности целиакии у детей с ВЗК. Мы стремимся изучить распространенность целиакии и антител к тканевой трансглутаминазе (tTG) у детей с ВЗК.
Методы
Мы выполнили ретроспективный обзор карт детей с ВЗК, которые регулярно наблюдались в нашей клинике в течение последних 4 лет. Эти дети были сопоставлены с контрольной группой детей без ВЗК, поступивших в клинику GI с желудочно-кишечными симптомами.Все дети регулярно проходили скрининг на целиакию с использованием tTG-IgA и общего IgA. У всех детей были собраны клинические, лабораторные и гистологические данные. Аномальный tTG был определен как> 30 ед / мл.
Результаты
В исследуемую популяцию вошли 130 детей с ВЗК и 258 детей контрольной группы. Средний возраст детей составлял 14,6 ± 3 года, 58% составляли мальчики. В группу ВЗК вошли 75 детей с болезнью Крона и 55 детей с язвенным колитом. Аномальные уровни tTG были обнаружены у 6 пациентов в группе ВЗК и у 20 пациентов в контрольной группе (4.6% против 7,8%, p = 0,24). Для дальнейшей оценки наличия целиакии дети с положительным результатом tTG были протестированы с помощью эндомизиальных аутоантител (EMA) и гистологии двенадцатиперстной кишки, что привело к диагностике целиакии у одного пациента с ВЗК и 12 пациентов в контрольной группе. Распространенность целиакии была ниже среди детей с ВЗК по сравнению с контрольной группой (0,8% против 4,7%, p = 0,07). Частота ложноположительных результатов составила 3,9% и 3,3% для ВЗК и контрольной группы соответственно.
Обсуждение
У детей с ВЗК не наблюдается более высокой распространенности целиакии по сравнению с другими детьми, которые обращаются в клинику с желудочно-кишечными симптомами.Распространенность целиакии у детей с ВЗК (0,8%) аналогична зарегистрированной распространенности (1%) среди населения в целом. Частота ложноположительных антител к тТГ одинакова у детей с ВЗК и контрольной группы. Регулярный скрининг на целиакию у детей с ВЗК не требуется.
Рекомендуемое цитирование
Муршед, Фархан, «Распространенность целиакии и антител против тканевой трансглутаминазы (ttg) у детей с Ibd» (2017). Цифровая библиотека медицинских диссертаций Йельского университета .2157.
https://elischolar.library.yale.edu/ymtdl/2157
% PDF-1.7
%
510 0 объект
>
endobj
xref
510 73
0000000016 00000 н.
0000002657 00000 н.
0000002859 00000 н.
0000002895 00000 н.
0000003426 00000 н.
0000003575 00000 н.
0000003834 00000 н.
0000004373 00000 п.
0000004622 00000 н.
0000005088 00000 н.
0000005854 00000 п.
0000006284 00000 н.
0000006321 00000 п.
0000006967 00000 н.
0000007081 00000 п.
0000007193 00000 н.
0000007453 00000 н.
0000007913 00000 п.
0000009336 00000 п.
0000009468 00000 н.
0000009916 00000 н.
0000010529 00000 п.
0000010556 00000 п.
0000011242 00000 п.
0000011890 00000 п.
0000012598 00000 п.
0000013011 00000 п.
0000013536 00000 п.
0000013666 00000 п.
0000013755 00000 п.
0000014919 00000 п.
0000016073 00000 п.
0000017152 00000 п.
0000018273 00000 п.
0000019333 00000 п.
0000020475 00000 п.
0000021338 00000 п.
0000023988 00000 п.
0000024058 00000 п.
0000024172 00000 п.
0000055823 00000 п.
0000056086 00000 п.
0000056667 00000 п.
0000066056 00000 п.
0000066303 00000 п.
0000094204 00000 п.
0000099478 00000 н.
0000131238 00000 н.
0000151124 00000 н.
0000160982 00000 п.
0000161047 00000 н.
0000161139 00000 н.
0000164237 00000 н.
0000164530 00000 н.
0000164820 00000 н.
0000164847 00000 н.
0000165268 00000 н.
0000165762 00000 н.
0000166255 00000 н.
0000174867 00000 н.
0000175117 00000 н.
0000175490 00000 н.
0000175862 00000 н.
0000198306 00000 н.
0000198581 00000 н.
0000198969 00000 н.
0000199367 00000 н.
0000221267 00000 н.
0000221536 00000 н.
0000221924 00000 н.
0000258220 00000 н.
0000258259 00000 н.
0000001756 00000 н.
трейлер
] / Назад 1318245 >>
startxref
0
%% EOF
582 0 объект
> поток
h ބ S {HQ? 7H: {> k R _ $, bb, eK \ Э «LM (>? $ Ăi ڃ (10 M (ò!).{sϽ
IRE1α киназа-опосредованная секреция нетрадиционного белка восстанавливает неправильно свернутый CFTR и пендрин
Abstract
Наиболее распространенные патогенные мутации в CFTR (Δ F508 ) и SLC26A4 / pendrin / pendrin , которые вызывают кистозный фиброз и врожденную потерю слуха, соответственно, вызывают неправильную укладку белков и последующие дефекты в их перемещении на поверхность клетки. Здесь мы сообщаем, что активация пути киназы IRE1α может восстанавливать экспрессию на клеточной поверхности ΔF508-CFTR и p.H723R-пендрин посредством независимой от Гольджи нетрадиционной секреции белка (UPS). В клетках млекопитающих ингибирование киназы IRE1α, но не ингибирование эндонуклеазы IRE1α и расположенного ниже эффектора XBP1, ингибирует UPS CFTR. Обработка активатором киназы IRE1α, (E) -2- (2-хлорстирил) -3,5,6-триметил-пиразином (CSTMP), спасала экспрессию на клеточной поверхности и функциональную активность ΔF508-CFTR и p.H723R-пендрина. Обработка мышей ΔF508-CFTR нетоксичной дозой CSTMP восстанавливала поверхностную экспрессию CFTR и опосредованный CFTR транспорт анионов в толстой кишке мышей.Эти данные предполагают, что активация UPS посредством киназы IRE1α является стратегией лечения заболеваний, вызванных дефектным переносом мембранных белков на клеточную поверхность, включая ΔF508-CFTR и p.H723R-pendrin.
ВВЕДЕНИЕ
Дефицит сворачивания и транспортировки белков является распространенным патогенетическим механизмом, который нарушает гомеостаз живых систем и, следовательно, способствует возникновению множества заболеваний человека ( 1 — 3 ). В эукариотических системах секреторные белки, в том числе трансмембранные белки, синтезируются на рибосомах и котрансляционно перемещаются в эндоплазматический ретикулум (ER).ER поддерживает различные шапероны и катализаторы сворачивания, которые позволяют отличать правильно свернутые белки от неправильно свернутых ( 4 ). Секреторные белки, которые удовлетворяют механизму «контроля качества ER», экспортируются из ER, транспортируются в Golgi и, наконец, отправляются в плазматическую мембрану или внеклеточную среду. Напротив, неполностью свернутые белки сохраняются в ER и избирательно нацелены на процесс связанной с ER деградации (ERAD), чтобы предотвратить риски, которые возникают в результате неправильного функционирования белков, когда они покидают ER ( 5 ).Многие из вызывающих заболевание мутаций регулятора трансмембранной проводимости при муковисцидозе (CF) ( CFTR , ABCC7 ) и пендрина ( SLC26A4 ) приводят к нарушению фолдинга белка. Следовательно, эти неправильно свернутые белки не могут вступить в обычный путь секреции белка ER-to-Golgi, что в конечном итоге вызывает CF и врожденную потерю слуха, соответственно.
CFTR представляет собой регулируемый циклическим аденозин-5′-монофосфатом (цАМФ) транспортер с активностью анионного канала, который проводит Cl — и HCO 3 — на апикальной поверхности эпителиальных клеток секреторных органов, включая дыхательные пути, поджелудочная железа, кишечник, потовые и экзокринные железы ( 6 ).CFTR имеет два N-связанных сайта гликозилирования, которые изначально гликозилированы по ядру в ER (полоса B) и опосредуют взаимодействия с шаперонами лектинов ER как часть системы контроля качества ER ( 7 ). После транслокации в Golgi эти сайты подвергаются сложному гликозилированию (полоса C), и комплексно-гликозилированный CFTR перемещается на апикальную поверхность эпителиальных тканей ( 8 ). Среди почти 2000 идентифицированных мутаций делеция остатка фенилаланина в положении 508 (Δ F508 ) является наиболее распространенной болезненной мутацией CFTR, которая приводит к неправильной укладке белка, удержанию ER и деградации ERAD ( 9 ). .Следовательно, белки ΔF508-CFTR остаются в гликозилированной форме ядра внутри ER, и незначительные количества экспрессируются на поверхности плазматической мембраны ( 1 ).
Пендрин — это трансмембранный белок, который переносит анионы, такие как Cl — , I — и HCO 3 — , во внутреннее ухо и фолликулы щитовидной железы ( 10 ). Нарушение функции пендрина в результате генных мутаций лежит в основе несиндромной глухоты с увеличенным вестибулярным водопроводом (DFNB4) и синдромом Пендреда, который является второй наиболее частой причиной врожденной потери слуха во всем мире и наиболее частой причиной в Восточной Азии.Наиболее частая мутация пендрина, вызывающая заболевание, p.H723R (His723Arg), приводит к неправильной укладке белка, задержке ER и деградации под действием ERAD, что аналогично судьбе белков ΔF508-CFTR ( 3 ).
Большинство секреторных белков используют обычный путь секреции, опосредованный Гольджи, для перемещения от ER к плазматической мембране. В дополнение к этому классическому секреторному пути, возникающий механизм, названный «нетрадиционная секреция белка (UPS)», обеспечивает перенос различных цитозольных и трансмембранных белков на клеточную поверхность ( 11 ).Предыдущие исследования продемонстрировали, что незрелые гликозилированные в ядре CFTR и пендрин могут достигать плазматической мембраны через Гольджи-независимый путь UPS в условиях заблокированного транспорта ER-to-Golgi или стрессовых условий ER ( 12 , 13 ). После выхода на поверхность клетки гликозилированный в ядре CFTR и пендрин сохраняют свою активность по переносу анионов, хотя и на ослабленном уровне ( 12 , 13 ). Следовательно, избирательная активация UPS без инициирования значительного клеточного стресса является потенциальным терапевтическим подходом к лечению заболеваний, вызванных дефектами складывания и перемещением мембранных белков на клеточной поверхности.Однако ранее описанные методы активации UPS CFTR и пендрина (т. Е. Сверхэкспрессия GRASP55 для ΔF508-CFTR и избыточная экспрессия DNAJC14 для p.H723R-pendrin) ( 12 , 13 ) не имеют клинического значения для лечение людей с CF, DFNB4 или синдромом Пендреда.
Хотя недавние исследования выявили несколько ключевых молекул, связанных с UPS мембранных белков (например, Sec16A на выходе из ER и ранняя аутофагия / мультивезикулярные компоненты тела в везикулярном пути UPS CFTR) ( 14 , 15 ), весь комплекс регуляторных механизмов UPS остается неизвестным.В общем, большинство ИБП не является конститутивным, а вызывается стрессом ( 16 ). Напр., Блокады транспорта ER-to-Golgi индуцируют связанные со стрессом сигналы, которые участвуют в UPS мембранных белков. Блокада обычной секреции белка из ER в Golgi вызывает накопление развернутого белка в просвете ER, что затем вызывает стресс ER и адаптивный клеточный ответ, называемый ответом на развернутый белок (UPR) ( 17 ). У эукариот существует три основных ветви сенсоров UPR, в том числе инозит-требующий фермент 1α (IRE1α) и IRE1β, протеинкиназа РНК-подобная ER-киназа (PERK) и активирующий фактор транскрипции 6α (ATF6α) и ATF6β, которые локализуются в ER мембрана и трансдукция передачи сигналов стресса ER.IRE1α, наиболее эволюционно консервативная форма передачи сигналов UPR, по-видимому, играет важную роль в вызванном стрессом ER UPS мембранных белков. Истощение IRE1α устраняет вызванное блокадой ER-to-Golgi UPS CFTR и пендрина ( 13 ). Тем не менее, специфический механизм регуляции UPS с помощью IRE1α остается неясным.
IRE1 представляет собой трансмембранный белок ER типа I, который состоит из ER-просветного домена, который действует как развернутый белковый сенсор во время стресса ER, и цитозольных доменов, которые включают домен эндорибонуклеазы и домен протеинкиназы Ser / Thr.Активированный белок IRE1α млекопитающих может передавать сигналы стресса ER посредством эндонуклеолитического расщепления мРНК, кодирующих фактор транскрипции X-box-связывающий белок 1 (XBP1), в результате чего образуется активный сплайсированный XBP1 (XBP1s), который активирует связанные с UPR гены, участвующие в сворачивание белка, контроль качества белка и ERAD ( 18 ). Используя этот механизм, нагрузка развернутого белка в ER снижается, и гомеостаз ER восстанавливается. Процесс IRE1-зависимого распада (RIDD) также признан дополнительным механизмом, который ослабляет нагрузку на ER за счет разрушения мРНК различных белков ( 19 ).Кроме того, активация протеинкиназы IRE1 может запускать «пути тревожного стресса» за счет привлечения адаптерных белков, таких как фактор 2, связанный с рецептором фактора некроза опухоли (TRAF2), что приводит к активации киназы 1, регулирующей сигнал апоптоза (ASK1). и его последующий эффектор, N-концевую киназу c-Jun (JNK) ( 20 ).
В настоящем исследовании мы исследовали специфическую роль IRE1α в UPS, анализируя каждую сигнальную руку. Наши данные показали, что UPS CFTR и пендрина активируется in vitro и in vivo через сигнальный каскад, опосредованный IRE1α-киназой, но не XBP1- и RIDD-зависимые пути, которые требуют активности рибонуклеазы (RNase) IRE1α.Путь киназы IRE1α может быть потенциальной мишенью для разработки лекарств при заболеваниях, вызванных дефицитом фолдинга и транспортировки белков.
РЕЗУЛЬТАТЫ
Эндорибонуклеазная активность IRE1α избыточна для UPS CFTR
IRE1α млекопитающих сохраняется как неактивный мономер за счет связывания с белком-шапероном ER BiP ( 21 ). Олигомеризация мономеров IRE1 является самой ранней стадией активации IRE1 в ответ на накопление развернутых белков в ER ( 21 ).Ингибирование транспорта ER-to-Golgi с помощью доминантно-ингибирующей формы гуанозинтрифосфатазы Arf1, Arf1-Q71L, вызывает стресс ER из-за накопления секреторных белков в просвете ER ( 12 ). Стресс ER может быть также вызван обработкой ингибитором Ca 2+ –аденозинтрифосфатазы (АТФазы) тапсигаргином, который истощает запасы кальция в просвете ER ( 22 ). В соответствии с этими механизмами индукции стресса ER, IRE1α объединяется в олигомеры после экспрессии Arf1-Q71L (48 часов) или обработки тапсигаргином (12 часов) в анализе перекрестного связывания, проведенном на клетках 293 эмбриональной почки человека (HEK) (рис.S1A).
Олигомеризация IRE1α открывает киназный домен, чтобы инициировать его активность, и активирует РНКазный домен для процессинга субстрата мРНК ( 21 ). И Arf1-Q71L, и тапсигаргин запускали активность киназы IRE1α в клетках HEK293 и, следовательно, индуцировали аутофосфорилирование и фосфорилирование IRE1α нижележащего эффектора ASK1 (рис. 1, A и B). Лечение тапсигаргином (12 часов), которое индуцировало стресс ER за счет истощения ER Ca 2+ , вызывало сплайсинг XBP1 , в то время как длительная индукция стресса ER с избыточной экспрессией Arf1-Q71L (48 часов) не вызывала (рис.1, А и В, и рис. S1B). Поскольку и Arf1-Q71L, и тапсигаргин индуцировали UPS мембранных белков ( 12 , 13 ), это означает, что передача сигналов, опосредованная киназой IRE1α, но не субстрат РНКазы IRE1α XBP1 , преимущественно участвует в UPS, связанном со стрессом ER. . Эксперименты по поверхностному биотинилированию продемонстрировали, что истощение IRE1α, но не XBP1, устраняет UPS ΔF508-CFTR, дополнительно подтверждая гипотезу о том, что связанный со стрессом ER UPS не зависит от XBP1 (рис.1, В и Г). Иммуноблот-анализ и количественная оценка мРНК для IRE1α и XBP1 (рис. 1C и рис. S1, B и C) подтвердили нокдаун каждого гена в настоящем исследовании.
Рис. 1 Сплайсинг XBP1 и эндонуклеазная активность IRE1α не участвуют в UPS ΔF508-CFTR.
( A и B ) Arf1-Q71L-индуцированная блокада ER-to-Golgi активирует фосфорилирование ASK1, но не XBP1 сплайсинг. Клетки HEK293 трансфицировали плазмидами для Arf1-Q71L (48 часов) или обрабатывали тапсигаргином (5 мкМ, 12 часов) для индукции стресса ER.Репрезентативные иммуноблоты показаны в (A), а результаты количественной оценки нескольких экспериментов суммированы в (B) ( n = 3). Уровни фосфорилирования IRE1α и ASK1 рассчитывали как соотношение фосфо / общие белки. ( C и D ) IRE1α, но не XBP1, необходим для ИБП ΔF508-CFTR, индуцированного Arf1-Q71L. Анализы поверхностного биотинилирования проводили в клетках HEK293, трансфицированных указанными плазмидами и / или малыми интерферирующими РНК (миРНК).Репрезентативные иммуноблоты показаны в (C), а результаты нескольких экспериментов суммированы в (D) ( n = 4). Мечение белков, специфичное к клеточной поверхности, было подтверждено отсутствием цитозольной протеина альдолазы А в биотинилированной фракции. ( E и F ) Ингибирование активности эндонуклеазы IRE1α с помощью STF-083010 не блокировало UPS ΔF508-CFTR. Анализы поверхностного биотинилирования выполняли с обработкой STF-083010 (60 мкМ, 12 часов). Репрезентативные результаты показаны в (E), а результаты нескольких экспериментов суммированы в (F) ( n = 4).Данные гистограммы показаны как среднее ± SEM. ** P <0,01; n.s., не имеет значения. Данные были проанализированы с помощью одностороннего дисперсионного анализа с последующим тестом множественного сравнения Тьюки.
Из-за неожиданного открытия, что UPS ΔF508-CFTR не зависит от сплайсинга XBP1 , мы затем исследовали, может ли IRE1α опосредовать UPS в отсутствие его РНКазной активности. STF-083010 — это соединение, которое нацелено на каталитическое ядро домена РНКазы IRE1α и ингибирует эндонуклеазную активность IRE1α, не влияя на его киназную активность или общее состояние олигомеризации ( 23 ).Обработка STF-083010 (60 мкМ, 12 часов) отменяла индуцированную тапсигаргином продукцию XBP1 (фиг. S1D). Однако STF-083010 не влиял на вызванный Arf1-Q71L UPS ΔF508-CFTR (рис. 1, E и F). Эти результаты в совокупности указывают на то, что РНКазная активность IRE1α избыточна для UPS ΔF508-CFTR.
Путь киназы IRE1α – ASK1 необходим для UPS ΔF508-CFTR
Далее мы сосредоточили внимание на роли передачи сигналов, опосредованной киназой IRE1α, в UPS мембранных белков. Ингибитор киназы типа I APY29 ингибирует трансаутофосфорилирование IRE1α, конкурентно занимая аденозин-5′-трифосфат (АТФ) -связывающий карман IRE1α ( 24 ).В клетках HEK293 среднее значение ингибирующей концентрации APY29 против фосфорилирования IRE1α составило 9,6 мкМ, а обработка 100 мкМ APY29 в течение 12 часов почти полностью подавляла индуцированное Arf1-Q71L фосфорилирование IRE1α и ASK1 (рис. S2, A и Б). APY29 (100 мкМ) отменял нацеливание на поверхность ΔF508-CFTR, индуцированное эктопической экспрессией Arf1-Q71L (рис. 2, A и B). Если APY29 ингибирует синтез белка CFTR, это также может вызвать быстрое снижение ΔF508-CFTR на клеточной поверхности, поскольку известно, что его стабильность ниже, чем у дикого типа (WT) –CFTR ( 25 ).Обработка ингибитором синтеза белка циклогексимидом (0,1 мг / мл) вызвала ускоренное снижение уровней ΔF508-CFTR на клеточной поверхности (рис. S2, C — F). Однако APY29 снижал только уровни ΔF508-CFTR на клеточной поверхности, не влияя на общие уровни белка, в то время как циклогексимид вызывал быстрое снижение как клеточной поверхности, так и общих клеточных уровней ΔF508-CFTR (рис. 2, A и B). Эти данные показывают, что APY29 селективно ингибировал восстановление ΔF508-CFTR на клеточной поверхности, не влияя на синтез или разложение белка.
Рис. 2 Путь киназы IRE1α – ASK1 необходим для UPS ΔF508-CFTR.
( A и B ) Влияние ингибитора киназы IRE1α APY29 на UPS ΔF508-CFTR. Анализы поверхностного биотинилирования проводили в клетках HEK293 после индукции UPS ΔF508-CFTR с помощью Arf1-Q71L с последующим добавлением ингибитора киназы IRE1α APY29 (100 мкМ) или ингибитора синтеза белка циклогексимида (0,1 мг / мл) в течение указанного времени. периоды. Репрезентативные иммуноблоты показаны в (A), а результаты нескольких экспериментов суммированы в (B) ( n = 5).APY29 уменьшал поверхностный ΔF508-CFTR в зависимости от времени, не влияя на уровни общего белка. ( C и D ) Ингибитор ASK1 MSC2032964A (ASK1-Inh) ингибирует Arf1-Q71L-индуцированный UPS CFTR. Анализы поверхностного биотинилирования проводили в клетках HEK293, трансфицированных плазмидами, кодирующими WT-CFTR, ΔF508-CFTR и / или Arf1-Q71L. Некоторые клетки обрабатывали MSC2032964A (10 мкМ) в течение указанных периодов времени. Типичные результаты поверхностного биотинилирования WT-CFTR и ΔF508-CFTR представлены на (C).Линейный график на (D) суммирует результаты нескольких экспериментов ( n = 6). Ингибирующее действие MSC2032964A на активность ASK1 подтверждалось снижением фосфорилирования ASK1. b , гликозилированный по ядру CFTR; c , комплексно-гликозилированный CFTR. ** P <0,01 по сравнению с Arf1-Q71L, 0 часов (как для WT-CFTR, так и для ΔF508-CFTR).
Учитывая, что IRE1α активирует ASK1 посредством образования комплекса IRE1-TRAF2-ASK1 на внешней мембране ER и, таким образом, связывает сигнал стресса ER с сигнальным путем ASK1-JNK ( 20 , 26 ), мы дополнительно исследовали роль ASK1 в нетрадиционной секреции CFTR.MSC2032964A — мощный и селективный ингибитор ASK1, который блокирует индуцированное липополисахаридом фосфорилирование ASK1 ( 27 ). Результаты на фиг. 2 (C и D) показывают, что ингибирование фосфорилирования ASK1 обработкой MSC2032964A (ASK1-Inh, 10 мкМ) снижает вызванное Arf1-Q71L UPS как WT-, так и ΔF508-CFTR в зависимости от времени. Кроме того, истощение ASK1 обработкой специфическими малыми интерферирующими РНК (siRNAs) устраняет вызванные Arf1-Q71L UPS WT- и ΔF508-CFTRs (рис. S3).Вместе эти результаты показывают, что киназная активность IRE1α и ASK1 играет критическую роль в UPS гликозилированных кором CFTRs.
Повышение регуляции киназы IRE1α индуцирует UPS ΔF508-CFTR
Затем мы исследовали эффекты повышения регуляции киназы IRE1α в контексте устранения дефектов транспорта, вызванных мутацией ΔF508-CFTR. Как показано на фиг. 3 (A и B), сверхэкспрессия только IRE1α приводила к поверхностной экспрессии ΔF508-CFTR. Примечательно, что сверхэкспрессия мутанта с мертвой киназой IRE1α (K599A) ( 28 ) не индуцирует экспрессию ΔF508-CFTR на клеточной поверхности, в то время как мутант с мертвой эндонуклеазой IRE1α (N906A) ( 28 ) вызывает (рис.3, A и B), что сильно подтверждает гипотезу о том, что повышающая регуляция киназы IRE1α может спасти дефектный трафик ΔF508-CFTR.
Фиг. 3 Активация киназы IRE1α с помощью CSTMP индуцирует UPS ΔF508-CFTR.
( A и B ) Сверхэкспрессия киназы IRE1α активирует UPS ΔF508-CFTR. Клетки HEK293, экспрессирующие ΔF508-CFTR, котрансфицировали плазмидами, кодирующими WT или мутантный IRE1α (1 мкг / мл, 48 часов). Сверхэкспрессия WT и мертвой РНКазы (N906A) IRE1α, но не мертвой киназы (K599A) IRE1α, активирует UPS ΔF508-CFTR.Репрезентативные анализы поверхностного биотинилирования представлены в (А), а результаты нескольких экспериментов ( n = 3) суммированы в (В). ( C и D ) Влияние активатора киназы IRE1α CSTMP на фосфорилирование ASK1 и сигналы клеточной смерти. Клетки HEK293 обрабатывали от 3 до 100 мкМ CSTMP в течение 48 часов. Активацию сигналов клеточной смерти анализировали путем расщепления PARP и каспазы 3 (стрелка). Репрезентативные иммуноблоты показаны в (C), а результаты нескольких экспериментов суммированы в (D) ( n = 5).CSTMP в концентрации 10 мкМ активировал ASK1, но не сигнализировал о гибели клеток (красная стрелка). ( E и F ) CSTMP вызывает ИБП с ΔF508-CFTR. Анализы поверхностного биотинилирования проводили в клетках HEK293, экспрессирующих ΔF508-CFTR. Клетки обрабатывали CSTMP (10 мкМ) в течение указанных периодов времени. Репрезентативные иммуноблоты представлены в (E), а сводка нескольких экспериментов изображена в (F) ( n = 3). Мечение белков, специфичное к клеточной поверхности, было подтверждено присутствием Na- и K-зависимой АТФазы (Na, K-ATPase) протеина плазматической мембраны и отсутствием цитозольной протеиновой альдолазы A в биотинилированной фракции.( G и H ) Сравнение уровней поверхностной экспрессии WT-CFTR и ΔF508-CFTR, спасенного CSTMP. Клетки HEK293, трансфицированные WT- и ΔF508-CFTR, инкубировали с или без CSTMP (10 мкМ) в течение 24 часов. Типичные результаты поверхностного биотинилирования показаны в (G), а результаты количественной оценки множества экспериментов ( n = 3) суммированы в (H). Уровни CFTR (биотина) на клеточной поверхности были нормализованы к его уровням общего белка (лизата), как подробно описано в разделе «Материалы и методы».Данные столбчатых и линейных графиков показаны как средние значения ± SEM. ** P <0,01.
Низкомолекулярные терапевтические средства более желательны, чем подходы к сверхэкспрессии генов для лечения пациентов с заболеваниями, вызванными дефектами сворачивания и транспортировки белков. Было показано, что производное тетраметилпиразина, (E) -2- (2-хлорстирил) -3,5,6-триметил-пиразин (CSTMP) активирует комплекс IRE1α-TRAF2-ASK1 ( 29 ). Однако CSTMP может также вызывать клеточную токсичность в высоких концентрациях (> 50 мкМ), поскольку, как сообщалось, он индуцирует апоптоз клеток немелкоклеточного рака легкого человека A549 посредством активации JNK и митохондриальной дисфункции ( 29 ).Обработка клеток HEK293 различными концентрациями CSTMP в течение 48 часов показала, что 10 мкМ CSTMP в достаточной степени активировали ASK1, не вызывая апоптотических сигналов, тогда как более высокая доза CSTMP (100 мкМ) приводила к расщеплению белков-маркеров апоптоза поли (аденозин-5′-дифосфат –Рибоза) полимераза (PARP) и каспаза 3 (рис. 3, C и D).
Таким образом, мы обработали клетки 10 мкМ CSTMP и исследовали, может ли он устранить дефекты транспорта ΔF508-CFTR. Одного CSTMP было достаточно для индукции экспрессии ΔF508-CFTR на клеточной поверхности.Клетки, экспрессирующие ΔF508-CFTR, начали показывать экспрессию CFTR на клеточной поверхности после 3-часовой обработки CSTMP (10 мкМ), которая выходила на плато через 12 часов (фиг. 3, E и F). Анализ соотношения поверхность / цитозоль белков CFTR показал, что CSTMP увеличивал соотношение поверхность / цитозоль ΔF508-CFTR до 59% от уровня, измеренного в WT-CFTR (фиг. 3, G и H). Нокдаун IRE1 α ингибировал вызванное CSTMP восстановление ΔF508-CFTR на клеточной поверхности (фиг. S4, A и B). Более того, сверхэкспрессия мутанта с мертвой киназой IRE1α (K599A), но не его мутанта с мертвой эндонуклеазой (N906A), устраняет эффекты CSTMP (рис.S4, C и D). Кроме того, обработка CSTMP вызвала экспрессию на клеточной поверхности белка полосы B ΔF508-CFTR, которая ингибировалась ингибитором киназы IRE1α APY29 (фиг. S4, E и F). В контрольных экспериментах корректор CFTR VX-809 ( 25 ) индуцировал экспрессию на клеточной поверхности белка C полосы ΔF508-CFTR, на который не влиял APY29 (рис. S4, E и F). Все эти результаты показывают, что CSTMP индуцирует экспрессию ΔF508-CFTR на клеточной поверхности через активацию киназы IRE1α.
Затем мы охарактеризовали индуцированный CSTMP путь транспортировки ΔF508-CFTR на клеточную поверхность.Во-первых, статус N-гликозилирования CFTR был проанализирован путем переваривания эндогликозидазой H (Endo H), которая удаляет ER-опосредованную высокоманнозу и некоторые гибридные типы N-связанных углеводов, но не комплексные олигосахариды, опосредованные Golgi ( 11 ). Результаты на рис. S5 (A и B) показывают, что CSTMP-индуцированные, экспрессированные на клеточной поверхности формы полосы B WT- и ΔF508-CFTR были чувствительны к Endo H, тогда как полоса C WT-CFTR в контрольных условиях была Endo H устойчивой, что позволяет предположить, что CSTMP — спасенные CFTR проходят обходной путь Гольджи, чтобы достичь поверхности клетки.Во-вторых, мы исследовали зависимость Sar1 и Syntaxin 5 спасенного CSTMP ΔF508-CFTR. Доминантно-ингибирующие формы Sar1 (Sar1-T39N и Sar1-H79G) ингибируют обычный COPII-опосредованный трафик ER-to-Golgi ( 12 ). Кроме того, сверхэкспрессия синтаксина 5, целевого растворимого рецептора белка прикрепления к N -этилмалеимиду, чувствительного к факторам (SNARE) акцепторной мембраны Гольджи, ингибирует обычный транспорт ER-to-Golgi за счет ухудшения стехиометрического баланса между везикулярными и целевыми мембранами SNARE. ( 12 ).Как показано на рис. S5 (от C до F), клеточная поверхность, спасаемая ΔF508-CFTR с помощью CSTMP, была независимой от Sar1 и синтаксина 5, в то время как перемещение на клеточной поверхности комплексно-гликозилированного WT-CFTR (полоса C) блокировалось доминантно-ингибирующими мутантами Sar1 и синтаксином. 5 сверхэкспрессия. Эти результаты показывают, что спасенный CSTMP ΔF508-CFTR не проходит обычным путем ER-to-Golgi на своем пути к поверхности клетки.
Затем мы сравнили клеточную токсичность обработки 10 мкМ CSTMP с токсичностью сверхэкспрессии Arf1-Q71L.Как показано на рис. S6 (A и B), трансфекция плазмидами, экспрессирующими Arf1-Q71L, индуцировала восстановление ΔF508-CFTR на клеточной поверхности и достигала пика через 48 часов. Следует отметить, что трансфекция плазмидами, экспрессирующими Arf1-Q71L, в течение 72 часов, что вызывало длительную блокаду ER-to-Golgi и стресс ER, вызывало расщепление PARP. Напротив, обработка CSTMP (10 мкМ) до 72 часов привела к устойчивому восстановлению поверхности ΔF508-CFTR без запуска апоптотического сигнала (рис. S6, C и D).
Морфологическое и функциональное восстановление ΔF508-CFTR с помощью CSTMP
Влияние CSTMP на экспрессию ΔF508-CFTR на клеточной поверхности также исследовали с использованием иммунофлуоресцентного анализа в непроницаемых (поверхностный CFTR) и проницаемых (общий CFTR) клетках.Клетки HeLa вместо клеток HEK293 были использованы для морфологического анализа, поскольку они более прочно прикрепляются к покровным стеклам и менее уязвимы для потери и разрушения клеток во время процедур иммуноокрашивания ( 15 , 30 ). В контрольных клетках белки ΔF508-CFTR сохранялись только внутри ER (рис. 4A, вверху). Обработка CSTMP (10 мкМ) в течение 12 часов значительно индуцировала экспрессию белков CFTR на клеточной поверхности в клетках, экспрессирующих ΔF508-CFTR [фиг. 4, А (внизу) и Б].Кроме того, обработка CSTMP (10 мкМ) до 72 часов не индуцировала экспрессию маркерного белка апоптоза аннексина V, в то время как пролонгированная эктопическая экспрессия Arf1-Q71L в течение 72 часов действительно вызывала экспрессию аннексина V (рис. S7), что указывает на минимальную или отсутствие клеточная токсичность лечения CSTMP.
Фиг.4. Экспрессия на клеточной поверхности и функциональное восстановление ΔF508-CFTR с помощью CSTMP.
( A и B ) Иммунофлуоресцентные изображения ΔF508-CFTR. Клетки HeLa трансфицировали плазмидами, кодирующими внеклеточный гемагглютинин (НА) ΔF508-CFTR с меткой.Некоторые клетки инкубировали с CSTMP (10 мкМ) в течение 12 часов перед фиксацией. CFTR на клеточной поверхности иммуноокрашивали антителами против НА до пермеабилизации мембраны (зеленый), а общий CFTR окрашивали антителами против R4 CFTR после пермеабилизации (красный). Стрелки указывают на CFTR на поверхности. Морфометрическая количественная оценка интенсивности поверхностного CFTR показана на (B) ( n = 5, каждый из анализов от 20 до 30 клеток). Масштабные линейки 10 мкм. ** P <0,01 по сравнению с отсутствием лечения (первая полоса).( C — F ) CSTMP индуцировал различимые токи CFTR в клетках, экспрессирующих ΔF508-CFTR. Токи целых клеток регистрировали от клеток HEK293, трансфицированных ложной плазмидой или ΔF508-CFTR, в течение 48 часов. Токи вызывали путем подачи импульса нарастания от -100 до +100 мВ (0,8 мВ / мс; удерживающий потенциал 0 мВ) с интервалами 10 с. CFTR Cl — токи активировались цАМФ (5 мкМ форсколина и 100 мкМ IBMX) и ингибировались CFTR inh -172 (5 мкМ). Сводка плотностей тока, измеренных при -80 мВ, показана на (C) ( n = от 8 до 11).В (F) клетки обрабатывали CSTMP (10 мкМ) в течение 24 часов. Данные гистограммы показаны как средние значения ± стандартная ошибка среднего; ** P <0,01.
Чтобы проверить, сохраняет ли клеточная поверхность ΔF508-CFTR, индуцированная CSTMP, функциональную активность, клетки HEK293, экспрессирующие ΔF508-CFTR, обрабатывали CSTMP (10 мкМ) в течение 48 часов и измеряли токи Cl —, опосредованные CFTR (рис. 4, В — F). Инкубация с форсколином и 3-изобутил-1-метилксантином (IBMX), который повышает внутриклеточный цАМФ, не вызвала каких-либо токов Cl — в контрольных клетках HEK293, обработанных только реагентами для трансфекции (рис.4D). Кроме того, клетки, экспрессирующие ΔF508-CFTR, не проявляли каких-либо различимых цАМФ-активированных токов Cl — при нормальных условиях (фиг. 4E). Примечательно, что обработка CSTMP (10 мкМ) в течение 48 часов индуцировала цАМФ-активированный ток Cl — в клетках, экспрессирующих ΔF508-CFTR (фиг. 4F). Вызванные токи проявляли типичные характеристики токов CFTR, которые (i) активировались обработкой цАМФ (форсколином и IBMX), (ii) ингибировались ингибитором CFTR CFTRinh-172 и (iii) имели линейный ток-напряжение ( I ). — В ) связь (рис.4F). Амплитуда тока, индуцированная CSTMP (10 мкМ) в клетках ΔF508-CFTR, составляла 32% от амплитуды, измеренной в клетках WT-CFTR, что сопоставимо с амплитудой, индуцированной VX-809 (10 мкМ) (рис. S8). В контрольных экспериментах обработка CSTMP (10 мкМ) в течение 48 часов существенно не изменяла амплитуду или характеристики токов WT-CFTR (рис. S8, A, B и E).
Спасение p.H723R-пендрина с помощью CSTMP
Поскольку IRE1α участвует в вызванном стрессом ER UPS пендрине ( 13 ), мы затем исследовали эффекты активатора киназы IRE1α CSTMP на спасение перемещения клеточной поверхности дефекты, вызванные п.Мутантный белок H723R-пендрин. Клетки PANC-1, полученные из протоков поджелудочной железы, использовали для исследования экспрессии пендрина на клеточной поверхности ( 13 ). Сначала мы исследовали поверхностную экспрессию p.H723R-pendrin, используя анализы поверхностного биотинилирования. Как показано на фиг. 5 (A и B), p.H723R-пендрин практически не обнаруживается на плазматической мембране контрольных клеток. Для сравнения, обработка CSTMP (30 мкМ) индуцировала устойчивую экспрессию p.H723R-пендрина на клеточной поверхности. Затем мы снова исследовали природу UPS индуцированного CSTMP маршрута переноса H723R-пендрина на клеточную поверхность.Как показано на рис. S9 (A и B), p.H723R-пендрин, спасенный на клеточной поверхности с помощью CSTMP, был чувствителен к Endo H, тогда как WT-пендрин был устойчив к Endo H. Результаты на рис. S9 (от C до F) демонстрируют, что мутанты Sar1 не блокируют экспрессию p.H723R-пендрина (полоса B) на клеточной поверхности с помощью CSTMP, в то время как такая же обработка ингибирует экспрессию WT-пендрина на клеточной поверхности (полоса C). Эти результаты показали, что восстановленный CSTMP H723R-пендрин достиг клеточной поверхности путем обхода Гольджи.
Инжир.5 CSTMP восстанавливает экспрессию неправильно свернутого пендрина на клеточной поверхности.
( A и B ) Влияние CSTMP на поверхностную экспрессию p.H723R-пендрина. Клетки PANC-1, стабильно экспрессирующие p.H723R-pendrin, трансфицировали плазмидами, кодирующими Arf1-Q71L, или без них, и инкубировали с CSTMP (10 или 30 мкМ) в течение 48 часов. Репрезентативные анализы поверхностного биотинилирования показаны на (A), а результаты нескольких экспериментов суммированы в (B) ( n = 5). * P <0.05 и ** P <0,01 по сравнению с отсутствием лечения (первая полоса). # P <0,05 и ## P <0,01 по сравнению с одним только Arf1-Q71L (вторая полоса). ( C и D ) Функциональное восстановление p.H723R-пендрина с помощью CSTMP. Обменную активность Cl — o / HCO 3 — i измеряли путем регистрации внутриклеточного pH (pH i ), как подробно описано в разделе «Материалы и методы». Репрезентативные измерения анионного обмена показаны на (C), а количественная оценка нескольких экспериментов изображена на (D) ( n = от 7 до 8).CSTMP (30 мкМ) не влиял на активность WT-пендрина, но значительно увеличивал обменную активность Cl — / HCO 3 — в клетках, экспрессирующих p.H723R-пендрин. Данные гистограммы показаны как среднее ± SEM. ** P <0,01 по сравнению с одним p.H723R-пендрином.
Примечательно, что CSTMP вызывал опосредованную пендрином обменную активность Cl — / HCO 3 — в клетках, экспрессирующих p.H723R-пендрин, что было аналогично эффектам сверхэкспрессии Arf1-Q71L (рис.5, В и Г). Эти результаты предполагают, что активация киназы IRE1α обладает терапевтическим потенциалом для лечения не только заболеваний, вызванных мутантами CFTR с дефектным трафиком, но также заболеваний, вызванных другими мутантными мембранными белками, включая p.H723R-пендрин.
Обработка CSTMP in vivo восстанавливает экспрессию CFTR на клеточной поверхности и CFTR-опосредованный транспорт анионов в толстой кишке мыши
Cftr F508del
Наконец, мы оценили, может ли введение CSTMP in vivo восстановить дефекты эпителиального транспорта, вызванные Δ Мутация F508-CFTR у мышей.Мутация потери функции Δ F508-CFTR вызывает дефекты, которые преимущественно влияют на транспорт ионов и жидкости в кишечнике и, следовательно, вызывают кишечную непроходимость у мышей ( 31 ). Чтобы проанализировать эффекты CSTMP in vivo, мы сначала оценили токсичность, оценив среднюю летальную дозу (LD 50 ) CSTMP у мышей WT ( Cftr WT ). Мышам вводили перорально в течение 5 дней три различных дозы CSTMP, которые были эквивалентны от 10 до 1000 мкМ (при условии, что CSTMP равномерно распределен по всему телу).Значение LD 50 для мышей было рассчитано как 25,9 мг / кг в день (эквивалент 100 мкМ) (рис. S10). Затем 6-недельных мышей, которые были гомозиготными по WT-CFTR ( Cftr WT ) или Δ F508-CFTR ( Cftr F508del ), обрабатывали 1 20/. дозы CSTMP LD 50 (2,59 мг / кг в день, что эквивалентно 10 мкМ, один раз в день) в течение 5 дней. При такой дозировке ни одна мышь не погибла во время оценочных экспериментов LD 50 (рис.S10).
Примечательно, что эксперименты по поверхностному биотинилированию с использованием образцов, собранных из слизистой оболочки толстой кишки мыши, показали, что CSTMP восстанавливает экспрессию на клеточной поверхности гликозилированного ядра ΔF508-CFTR (фиг. 6, A и B). Иммуногистохимический анализ повторил спасительный эффект CSTMP у мышей Cftr F508del (фиг. 6C). У мышей Cftr WT CFTR экспрессируется на апикальной мембране клеток крипт толстой кишки, и обработка CSTMP не влияет на распределение WT-CFTR.В соответствии с предыдущими результатами ( 12 , 32 ) апикальная экспрессия ΔF508-CFTR отсутствовала в криптах толстой кишки мышей Cftr F508del . Примечательно, что пероральное введение CSTMP спасало экспрессию ΔF508-CFTR на апикальной мембране клеток крипт толстой кишки у мышей Cftr F508del (фиг. 6C).
Фиг. 6. Обработка CSTMP in vivo восстанавливает ΔF508-CFTR в толстой кишке мыши Cftr F508del .
( A и B ) Анализы поверхностного биотинилирования выполняли с использованием эпителиальных клеток, собранных со слизистой оболочки толстой кишки. Образцы белка из клеток HEK293 использовали в качестве контроля. WT-CFTR ( Cftr WT ) или Δ F508-CFTR ( Cftr F508del ) мыши в возрасте 6 недель получали носитель или CSTMP (2,59 мг / кг, per os, один раз в день) на 5 дней. Типичные результаты анализа поверхностного биотинилирования показаны в (A), а результаты нескольких экспериментов суммированы в (B) ( n = 4).( C ) Иммуногистохимия CFTR. Поперечные и продольные поперечные срезы крипт толстой кишки мыши иммуноокрашивали кроличьими поликлональными антителами против CFTR R4. Стрелки указывают на экспрессию CFTR на апикальной мембране крипт толстой кишки. Обработка CSTMP индуцировала экспрессию CFTR на клеточной поверхности в толстой кишке мыши Cftr F508del . Шесть независимых экспериментов показали аналогичные результаты. Масштабные линейки 10 мкм. ( D и E ) Измерения тока короткого замыкания ( I sc ) в толстой кишке мыши.Апикальная сторона толстой кишки мыши обрабатывалась амилоридом (100 мкМ) для блокирования эпителиальных каналов Na + . Нанесение активатора аденилатциклазы форсколина (10 мкМ) на апикальную сторону вызывало отрицательный по просвету I sc , который полностью ингибировался Na + -K + -2Cl — ингибитор котранспортера буметанид (100 мкМ) доставлены на базолатеральную сторону. Обработка CSTMP индуцировала CFTR-зависимый I sc в толстой кишке мыши Cftr F508del .Репрезентативные следы измерений I sc представлены в (D), а результаты нескольких экспериментов суммированы в (E) ( n = от 6 до 9). b , гликозилированный по ядру CFTR; c , комплексно-гликозилированный CFTR. Данные гистограммы показаны как среднее ± SEM. BLM, базолатеральная мембрана; LM, просветная мембрана; ** P <0,01.
Кроме того, лечение CSTMP привело к функциональному восстановлению опосредованного CFTR транспорта анионов в толстой кишке мыши Cftr F508del .Повышенные внутриклеточные уровни цАМФ, индуцированные лечением форсколином, вызвали типичный CFTR-зависимый ток короткого замыкания ( I sc ) в толстой кишке мыши Cftr WT , которая была (i) просвета отрицательной, (ii) подавлялась блокада базолатерального захвата Cl — с использованием Na + / K + / 2Cl — ингибитора котранспортера буметанида, и (iii) отсутствует в Cftr F508del толстой кишке мыши (рис. 6, D и E).Примечательно, что обработка CSTMP индуцировала активированный форсколином, отрицательный по просвету и ингибирующий буметанид I sc с амплитудой тока, которая достигла 89% уровня, измеренного у мышей Cftr WT (рис. , D и E). Вместе эти результаты предоставляют доказательства in vivo, подтверждающие терапевтический потенциал CSTMP для лечения заболеваний, вызванных мутацией CFTR, дефектной по трафику.
ОБСУЖДЕНИЕ
В течение последних двух десятилетий исследователи постепенно устанавливали концепцию и молекулярные механизмы неканонического, независимого от Гольджи UPS трансмембранных белков ( 33 , 34 ).Мутации, которые вызывают дефектную укладку мембранных белков, часто приводят к заболеваниям, связанным с потерей функции, нарушая нормальный баланс между сворачиванием, перемещением и деградацией белков. Белки с дефектами сворачивания, такие как ΔF508-CFTR и p.H723R-pendrin, могут альтернативно транспортироваться к плазматической мембране через UPS при определенных условиях ( 12 , 13 ). Однако необходимы значительные усилия для разработки терапевтических средств, активирующих UPS, которые клинически применимы для пациентов с этими заболеваниями.Например, длительная блокада ER-to-Golgi с помощью Arf1-Q71L является цитотоксической, как показано на рисунках этого исследования (фиг. S6 и S7). Кроме того, существуют технические трудности, связанные с индукцией контролируемой сверхэкспрессии генов у участников-людей. Здесь мы сообщили о первом эффективном и фармакологически приемлемом подходе для активации UPS белков мембран, дефектных по складыванию, без серьезной токсичности. В настоящем исследовании мы продемонстрировали, что киназная активность датчика стресса ER IRE1α играет ключевую роль в UPS дефектных по сворачиванию CFTR.Кроме того, мы обнаружили, что низкомолекулярный CSTMP может активировать киназу IRE1α и ее нижестоящие эффекторы для усиления UPS и устранения дефектов, вызванных мутациями ΔF508-CFTR и p.H723R-pendrin как in vitro, так и in vivo.
В физиологических условиях для компенсации стресса ЭР, вызванного чрезмерным количеством развернутого белка, активируется UPR. UPR снижает перегрузку белков в ER за счет снижения транскрипции и трансляции секреторных белков и путем направления неправильно свернутых белков в ERAD ( 5 ).В дополнение к этим классическим механизмам клетки запускают UPS для уменьшения белковой нагрузки, когда развернутые белки накапливаются в ER ( 12 ) и цитозоле ( 35 ). Три основных преобразователя UPR — IRE1, PERK и ATF6 — все являются трансмембранными белками ER и разделяют общий процесс трансдукции сигнала стресса ER путем активации факторов транскрипции, таких как XBP1s, ATF4 и расщепленный ATF6, соответственно ( 17 ). Было продемонстрировано, что IRE1α является восходящим сигналом, который регулирует UPS CFTR и пендрина в ответ на стресс ER ( 12 , 13 ).Однако механизм IRE1α-опосредованного UPS до конца не изучен.
В настоящем исследовании эктопическая экспрессия Arf1-Q71L и лечение тапсигаргином вызывали олигомеризацию IRE1α (рис. S1A) и активацию киназы (рис. 1). Наиболее известным последующим эффектом передачи сигналов IRE1α является вырезание 26-нуклеотидного интрона мРНК XBP1 в его активную форму, XBP1s , в цитозоле ( 18 , 36 ). Активные XBP1 действуют как фактор транскрипции и контролируют экспрессию ряда генов, что в конечном итоге приводит к защитному ремоделированию гомеостаза ER.Неожиданно эктопическая экспрессия с Arf1-Q71L, который является одним из наиболее эффективных активаторов CFTR UPS, не индуцирует сплайсинг XBP1 , в то время как thapsigargin вызывает (Рис. 1, A и B). Истощение XBP1 на основе РНК-интерференции также подтвердило, что XBP1 является незаменимым для UPS ΔF508-CFTR (Рис. 1, C и D). В настоящее время неясно, почему Arf1-Q71L не может индуцировать сплайсинг XBP1 . Примечательно, что было показано, что кластеризация IRE1α (образование более крупных динамических олигомеров) не обязательна и не предсказывает его РНКазную активность; напротив, кластеризация IRE1α сильно коррелирует с его киназной активностью ( 37 ).Более того, легкие формы ER стресса не вызывают эффективно сплайсинг XBP1, но все же могут индуцировать кластеризацию IRE1α ( 37 ). Ожидается, что блокада ER-to-Golgi с помощью Arf1-Q71L медленно вызывает стресс ER. Следовательно, похоже, что медленный и умеренный стресс ER, вызванный Arf1-Q71L, не может активировать эндонуклеазу IRE1α, но он все еще может индуцировать олигомеризацию IRE1α и активировать его киназную активность. Регулируемый процесс RIDD, который представляет собой независимый от XBP1 UPR выход эндонуклеазной активности IRE за счет снижения нагрузки ER белков за счет быстрой деградации специфических мРНК ( 19 ), может быть альтернативным кандидатом сигнального пути для индукции UPS.Однако эксперименты с использованием ингибитора эндонуклеазы IRE1α STF-083010 и мутанта без эндонуклеаз (IRE1α-N906A) показали, что сигнальные пути, происходящие от активности эндонуклеазы, включая RIDD, не участвуют в UPS CFTR (рис.1 и 3 и рис. S4).
В дополнение к косвенным доказательствам того, что компонент IRE1α, отличный от его эндонуклеазной активности, опосредует UPS, следующие три части прямых доказательств подтверждают мнение о том, что путь IRE1α киназа – ASK1 критически участвует в UPS мембранных белков: (i) блокада Функция киназы IRE1α с использованием APY29 (рис.2) или мертвый мутант киназы IRE1α (IRE1α-K599A; рис. 3 и рис. S4) устраняет вызванное Arf1-Q71L или CSTMP UPS CFTR, (ii) химическое ингибирование или нокдаун ASK1 снижает UPS CFTR (Рис. 2, C и D, и рис. S3), и (iii) стимуляция киназой IRE1α с помощью CSTMP вызвала UPS ΔF508-CFTR и p.H723R-пендрин (рис. 3-5). Хотя полный механизм еще предстоит установить, недавние исследования предоставили основу для понимания того, как IREα-обеспечиваемая передача сигналов может вносить вклад в UPS мембранных белков.Каскад киназы IRE1α – ASK1, как предполагается, играет роль в инициации образования аутофагосом путем модуляции Beclin-1 ( 38 ). Компоненты формирования ранних аутофагосом, такие как аутофагия 5 (ATG5) и аутофагия 7 (ATG7), участвуют в везикулярных структурах, участвующих в связанном со стрессом ER UPS CFTR ( 14 ). Кроме того, IRE1α является восходящим сигналом, который регулирует генерацию и закрепление Sec16A на сайтах выхода ER, что увеличивает экспорт ER гликозилированных по ядру CFTRs во время UPS в ответ на стресс ER ( 15 ).Сообщалось, что нокдаун IRE1 α / ERN1 индуцировал небольшое увеличение (от 10 до 20%) переноса на клеточную поверхность белка C полосы ΔF508-CFTR при скрининге РНК-интерференции киназ, предполагая, что IRE1α ингибирует Созревание по Гольджи ΔF508-CFTR ( 39 ). Хотя механизмы этого открытия неизвестны, это частично объясняется настоящими результатами, что IRE1α направляет ΔF508-CFTR в обходной путь Гольджи.
IRE1α представляет собой платформу для активации проапоптотического пути JNK в условиях полного стресса ( 20 , 26 ), что означает, что серьезная активация IRE1α может переключить принятие решения о жизни к смерти киназы IRE1α-ASK1 и вызвать клеточную токсичность .Чрезмерная активация киназы IRE1α> 100 мкМ CSTMP (фиг. 3) и длительная блокада ER-to-Golgi с помощью Arf1-Q71L в течение 72 часов (фиг. S6) индуцировали апоптотические сигналы расщепления PARP и каспазы 3 в клетках HEK293. Однако нетоксичный уровень активации киназы IRE1α 10 мкМ CSTMP селективно активировал ASK1, не активируя эти апоптотические сигналы (рис. 3). Кроме того, пероральное введение CSTMP (2,59 мг / кг в день) в течение 5 дней мышам, которое избавляет 89% цАМФ-стимулированной трансэпителиальной секреции Cl — ( I sc ) в Cftr F508del толстой кишки, не вызывало идентифицируемой токсичности (рис.6 и рис. S10). Следовательно, CSTMP, по-видимому, оказывает лечебное действие на CF, способствуя экспрессии на клеточной поверхности дефектного по фолдингу CFTR у мышей без серьезной токсичности. В настоящем пилотном исследовании мы эмпирически выбрали 1 / 10 из LD 50 CSTMP в качестве дозы и показали> 50% эффективности в восстановлении опосредованного CFTR I sc , что означает терапевтический индекс (LD 50 / ED 50 (полумаксимальная эффективная доза)] CSTMP у мышей была по меньшей мере> 10.Хотя это значение минимально соответствует безопасности лекарственного средства, разработка более безопасного и более мощного лекарственного средства с использованием крупномасштабного скрининга малых молекул, которые могут избирательно активировать киназу IRE1α, но не эндонуклеазу IRE1α и апоптотические сигналы, повысит клиническую осуществимость Активатор киназы IRE1α. Эпителиальные клетки у пациентов с МВ подвергаются различным стрессовым состояниям. Эти клеточные стрессы могут активировать киназу IRE1α и частично индуцировать UPS ΔF508-CFTR, что может повлиять на тяжесть заболевания.Следовательно, индивидуальные вариации врожденной способности UPR и последующих ответов UPS могут также вносить вклад в некоторые широкие вариации очевидной тяжести CF с мутацией Δ F508 .
При измерениях тока всей клетки комбинация CSTMP и VX-809 не показала аддитивных эффектов (рис. S8E). Амплитуда тока, индуцированная высокими концентрациями VX-809 (10 мкМ) и CSTMP (10 мкМ) в клетках ΔF508-CFTR, составляла ~ 30% от значения, измеренного в клетках WT-CFTR, и это был бы максимальный уровень, который может быть достигается в клетках ΔF508-CFTR с учетом их ускоренной деградации и изначально низкой активности ионных каналов.Для полного анализа аддитивных или потенцирующих эффектов между активатором киназы IRE1α и VX-809 требуются более сложные исследования in vitro и in vivo. Уровни вызванного CSTMP восстановления ΔF508-CFTR различались в каждом эксперименте. Например, в клетках HEK293 CSTMP увеличивал соотношение поверхность / цитозоль ΔF508-CFTR до 59% от уровня, измеренного в WT-CFTR в экспериментах по поверхностному биотинилированию (рис. 3, G и H), в то время как он индуцировал в среднем Восстановление 32% активности канала Cl — при измерении тока всей клетки (рис.S8). Однако CSTMP спасал 89% цАМФ-стимулированной трансэпителиальной секреции Cl — в толстой кишке мыши Cftr F508del (фиг. 6). Поскольку поглощение базолатерального аниона (Cl — и HCO 3 —) является лимитирующим этапом в большинстве секреторных эпителий ( 6 , 40 ), ~ 30% восстановления оттока анионов, опосредованного CFTR. на апикальной мембране, по-видимому, достаточно для восстановления общего трансэпителиального транспорта ионов в эпителии толстой кишки.
Настоящие результаты указывают на возможность применения активаторов киназы IREα в испытаниях на людях in vivo. Что касается эффективной дозы in vivo, фармакокинетические профили CSTMP, включая концентрации в тканях-мишенях, должны быть проанализированы в будущих исследованиях. Поскольку мы вводили CSTMP per os, локальная концентрация CSTMP в ткани-мишени, кишечном эпителии мыши, могла быть выше, чем в глубоких висцеральных тканях, что привело бы к сильным терапевтическим эффектам без системной токсичности.Местная ингаляционная терапия на основе аэрозолей может быть альтернативным подходом для людей с МВ, которые демонстрируют преимущественно респираторный фенотип, в случае, если концентрации CSTMP в тканях дыхательных путей человека-мишени не достигают эффективных уровней при системном введении CSTMP. В заключение мы представляем здесь активацию UPS через путь киназы IRE1α – ASK1 как многообещающую терапевтическую стратегию при заболеваниях, вызванных фолдинг-дефектными трансмембранными белками, включая CF и синдром Пендреда.
МАТЕРИАЛЫ И МЕТОДЫ
Культура клеток, плазмиды, миРНК и мыши
Клетки HEK293 и HeLa поддерживали в модифицированной Дульбекко среде с высоким содержанием глюкозы Игла (Gibco № 11995-065, Карлсбад, Калифорния) с добавлением 10% фетального коровьего молока сыворотка и 1% 100х антибиотик-антимикотик (100 единиц / мл пенициллина, 100 единиц / мл стрептомицина и 250 нг / мл амфотерицина B) (Gibco № 15240062). Клетки выращивали при 37 ° C в инкубаторе с 5% CO 2 . Плазмида экспрессии млекопитающих, кодирующая IRE1α-pcDNA3 человека.EGFP был приобретен у Addgene (идентификатор гена: 2081). Плазмиды, кодирующие человеческий pCMV-ΔF508-CFTR, pCMV-WT-CFTR (pCMVNot6.2), pCMV-GRASP55-Myc и внеклеточно-меченый гемагглютинин (HA) –ΔF508-CFTR, pClneo-HA – Syntaxo-5, pCl -Sar1-T39N, pClneo-Myc-Sar1-H79G и pcDNA3-HA-Arf1-Q71L были описаны ранее ( 12 , 30 ). ON-TARGETplus Человеческая миРНК ERN1 ( IRE1 a, ID гена: 2081) и человеческая миРНК XBP1 (ID гена: 7494) были приобретены коммерчески (SMARTpool; Dharmacon, Lafayette, CO, USA).Трансфекцию плазмидной ДНК и миРНК в клетки HEK293 или HeLa проводили с использованием системы динамической доставки TransIT-X2 (Mirus Bio LLC, Мэдисон, Висконсин, США) в соответствии с протоколом производителя. Cftr F508del мышей были получены от К. Р. Томаса (Университет штата Юта, Солт-Лейк-Сити, Юта, США) ( 31 ), как сообщалось ранее ( 12 ). Мышей разводили и содержали в соответствии с требованиями исследования животных Медицинского центра Йонсей. Все эксперименты на животных были одобрены Институциональными лабораторными ресурсами животных и Комитетом по исследованиям животных в Центре биомедицинских исследований Йонсей (разрешение №2018-0139).
Химические вещества и антитела
Тапсигаргин (Sigma-Aldrich, T9033), STF-083010 (TOCRIS, 4509), APY29 (TOCRIS, 4865), MSC 2032964A (TOCRIS, реестр 5641), VX-809 (Selleckchem, Chemical № 936727-05-8), VX-770 (Selleckchem, регистрационный номер CAS 873054-44-5) и циклогексимид (Sigma-Aldrich, C4859) были приобретены коммерчески. CSTMP был специально синтезирован Cayman Chemical (Анн-Арбор, Мичиган, США) (регистрационный номер CAS 1000672-89-8).
Были приобретены следующие антитела: анти-CFTR M3A7 (Millipore, Billerica, MA, USA), анти-IRE1α (Cell Signaling Technology, 3294), анти-фосфо-S724 IRE1α (Abcam, ab48187), анти-XBP1 ( Abcam, ab198999), anti-ASK1 (Cell Signaling Technology, 3762), anti-phospho-Thr 845 ASK1 (Cell Signaling Technology, 3765), anti-BiP (Cell Signaling Technology, 3177), anti-CHOP (Cell Signaling). Technology, 2895), анти-про / p17-каспаза 3, анти-расщепленный PARP1 (Abcam, ab136812), анти-Na- и K-зависимая АТФаза (Na, K-АТФаза) α1 (Cell Signaling Technology, 23565), анти- -HA (Cell Signaling Technology, 2367), анти-Myc (Cell Signaling Technology, 2276), анти-альдолаза A (Abcam, ab78339), анти-β-актин (Santa Cruz Biotechnology, sc47778) и антипендрин (Santa Cruz Биотехнология, sc23779).Поликлональное антитело против R4 было индуцировано против пептидов, соответствующих аминокислотам 1458-1471 человеческого CFTR, как описано ранее ( 12 ).
Количественный анализ полимеразной цепной реакции
Общую РНК экстрагировали из клеток HEK293 с использованием реагента Tri-РНК (Favorgen Biotech Corp, Тайвань) в соответствии с протоколом производителя. Для обратной транскрипции РНК в комплементарную ДНК (кДНК) очищенные образцы РНК объединяли с Премиксом РНК в кДНК EcoDry (Takara Bio Inc., Shiga, Япония), и смеси инкубировали при 42 ° C в течение 1 часа, а затем при 70 ° C в течение 10 минут.
Количественную полимеразную цепную реакцию (кПЦР) проводили с использованием системы Applied Biosystem StepOne (Applied Biosystems, Foster City, CA, USA). Реакцию ПЦР в реальном времени измеряли путем обнаружения связывания флуоресцентного красителя SYBR Green с двухцепочечной ДНК. Для ПЦР-амплификации общий реакционный объем доводили до 20 мкл водой, не содержащей РНКаз, после смешивания со 100 нг кДНК, 2 мкл наборов праймеров, 10 мкл 2 × SYBR Premix Ex Taq и 0.4 мкл эталонного красителя 50 × ROX (Takara, RR420L). Амплификацию выполняли в следующих условиях циклов: 95 ° C в течение 15 мин, затем 40 циклов при 95 ° C в течение 15 с и 60 ° C в течение 40 с. Анализы проводили в трех экземплярах для каждой кДНК. Относительные уровни экспрессии мРНК рассчитывали с использованием метода сравнительного порогового цикла ( C t ) путем нормализации экспрессии гена с помощью гена домашнего хозяйства GAPDH , и значение Δ C t рассчитывали следующим образом: Δ C t = C t ( GAPDH ) — C t (целевой ген).Среднее значение Δ C t затем вычитали из каждого описанного экспериментального условия, чтобы получить значение ΔΔ C t . Кратность изменения экспрессии гена была нормализована до GAPDH , и относительно контрольных образцов была рассчитана как 2 -ΔΔ C t . Последовательности праймеров, используемые для анализа qPCR, были следующими: hIRE1 α, прямой праймер 5′-CGG GAG AAC ATC ACT GTC CC-3 ‘и обратный праймер 5′-CCC GGT AGT GGT GCT TCT TA-3’; hXBP1 (всего), прямой праймер 5′-TTG TCA CCC CTC CAG AAC ATC-3 ‘и обратный праймер 5′-TCC AGA ATG CCC AAC AGG AT-3’; hXBP1s (сплайсинг), прямой праймер 5′-TGC TGA GTC CGC AGC AGG TG-3 ‘и обратный праймер 5′-GCT GGC AGG CTC TGG GGA AG-3’; GAPDH (глицеральдегидфосфатдегидрогеназа), прямой праймер 5′-AAT CCC ATC ACC ATC TTC CA-3 ‘и обратный праймер 5′-TGG ACT CCA CGA CGT ACT CA-3’.
Поверхностное биотинилирование, расщепление гликозидазой, анализ перекрестного сшивания и анализ иммуноблоттинга
Для биотинилирования клетки HEK293, выращенные в шестилуночных планшетах (1 × 10 6 ), инкубировали в течение 5 минут при 4 ° C и трижды промывали с охлажденным фосфатно-солевым буфером (PBS). Для биотинилирования в слизистой оболочке толстой кишки мышей ткани толстой кишки вскрывали в продольном направлении и лишали соединительных тканей и мышц. Затем трансмембранные белки плазматической мембраны культивируемых клеток или слизистой оболочки толстой кишки биотинилировали путем инкубации с осторожным встряхиванием с 1 мл раствора биотина [0.3 мг / мл сульфо-NHS-SS-биотина в охлажденном PBS (Thermo Pierce, 21331)] в течение 30 минут при 4 ° C в темноте. Затем клетки или ткани толстой кишки инкубировали с буфером для гашения, содержащим 0,5% бычий сывороточный альбумин (BSA) в PBS, в течение 10 минут при 4 ° C для удаления избытка биотина, а затем трижды промывали PBS. Затем поверхностно-биотинилированные клетки собирали в буфере для лизиса, содержащем 25 мМ трис (pH 7,4), 1% (об. / Об.) NP-40, 150 мМ NaCl, 10% глицерин и 1 мМ EDTA-Na 2 с добавками. с коктейлем ингибиторов протеаз (Roche, Германия).Лизаты клеток гомогенизировали обработкой ультразвуком в течение 20 с (импульс 1 с) с последующим центрифугированием при 16000 g в течение 20 минут при 4 ° C. Полученные супернатанты, содержащие 400 мкг общего белка, инкубировали с 200 мкл 10% стрептавидиновой агарозы (Thermo Pierce, 20347). Связанные со стрептавидином биотинилированные белки центрифугировали и пять раз промывали буфером для лизиса. Биотинилированные белки элюировали в буфере для образцов 2 × SDS с добавлением дитиотреитола (DTT) (0,02 г / мл) и разделяли электрофорезом в SDS-полиакриламидном геле (PAGE).Разделенные белки электропереносили на нитроцеллюлозную мембрану и подвергали блоттингу соответствующими первичными и вторичными антителами (в 5% обезжиренном молоке). Полосы белка детектировали с помощью усиленной хемилюминесценции, и плотность каждой полосы белка определяли количественно с использованием программного обеспечения для визуализации (Multi Gauge version 3.0; Fujifilm, Tokyo, Japan). Для количественной оценки CFTR и пендрина на поверхности клеток мы одновременно проводили эксперименты по биотинилированию поверхности и иммуноблоттингу, используя образцы из всех разработанных экспериментальных условий.Количество белка клеточной поверхности (биотина) нормализовали к каждому общему клеточному белку (лизату), анализируя интенсивности полос в тех же блотах биотинилирования поверхности и лизата.
Расщепление N-гликозилированных белков пептидом N -гликозидаза F (PNGase F) (New England Biolabs, № P0704L) и Endo H (New England Biolabs, № P0702L) выполняли с небольшой модификацией на основе данных производителя. руководство ( 12 ). Вкратце, образцы белка 50 мкг сначала денатурировали добавлением 0.5% SDS и 40 мМ DTT и инкубировали при 37 ° C в течение 10 мин. Затем образцы денатурированного белка обрабатывали PNGase F (500 Ед на реакцию, с 1% NP-40) в растворе, содержащем фосфат натрия (50 мМ, pH 7,5), или обрабатывали Endo H (1000 Ед на реакцию) в раствор, содержащий ацетат натрия (50 мМ, pH 6.0) при 37 ° C в течение 2 часов.
Анализ перекрестного связывания выполняли, как описано ранее ( 30 ). Клетки HEK293 промывали трижды PBS и инкубировали с 500 мкМ этиленгликольсукцинимидилсукцинатом (EGS; Thermo Scientific, 21565) в PBS (pH 8.2) в течение 30 мин при 37 ° С. После перекрестного связывания клетки затем инкубировали с 20 мМ трис (pH 7,4) в течение 10 минут для остановки реакции путем гашения EGS. Клетки лизировали, гомогенизировали, отбирали образцы с помощью 2 × SDS буфера для образцов, разделяли с помощью SDS-PAGE, электропереносили и иммуноблотировали с антителом против IRE1α.
Иммуноцитохимия, иммуногистохимия и морфометрический анализ
Иммунофлуоресцентное окрашивание выполняли путем незначительной модификации ранее описанных протоколов ( 12 , 15 ).Для иммуногистохимии слизистой оболочки толстой кишки ткани мышей помещали в смесь с оптимальной температурой резки (Miles, Elkhart, IN, USA), замораживали в жидком азоте и делали криосрезы на срезы размером 4 мкм. Затем криосрезы толстой кишки мыши фиксировали и повышали проницаемость путем инкубации с холодным метанолом в течение 5 минут при -20 ° C. Для иммуноцитохимии клетки HeLa культивировали на круглых покровных стеклах диаметром 18 мм и фиксировали 3,7% формальдегидом в течение 6 мин при комнатной температуре. Затем клетки были проницаемы с помощью 0.1% Triton X-100 в PBS при комнатной температуре в течение 5 мин. Срезы тканей на предметных стеклах или клетки, выращенные на покровных стеклах, трижды промывали PBS, а затем инкубировали с PBS, содержащим 1% BSA и 5% сыворотки соответствующих видов (сыворотка лошади / осла / козы), в течение 1 часа при комнатной температуре для блокирования неспецифического участок связывания. После блокирования срезы ткани или клетки окрашивали путем инкубации с соответствующими первичными антителами с последующим окрашиванием антителами, конъюгированными с вторичным флуорофором.Для достижения поверхностно-специфического мечения CFTR клетки без пермеабилизации после фиксации инкубировали с блокирующим раствором и окрашивали анти-HA антителом для обнаружения внеклеточного HA-эпитопа CFTR. Клетки на покровных стеклах помещали на предметные стекла со средой для фиксации флуоресценции (S3025; Dako, США). Флуоресцентные изображения получали с помощью лазерного сканирующего конфокального микроскопа (LSM 780; Carl Zeiss, Берлин, Германия) с масляным объективом с числовой апертурой 63 × 1,4.
Морфометрический анализ полученных конфокальных изображений был выполнен с использованием программного обеспечения для анализа микроскопии MetaMorph (версия 7.1; Molecular Devices, Саннивейл, Калифорния, США), как описано ранее ( 15 ). Вкратце, для количественной оценки изображения при каждом условии, 24-битные конфокальные изображения, включающие красный, зеленый и синий компоненты, были преобразованы в три 8-битных моноканальных изображения. Для количественной оценки поверхностной интенсивности CFTR пиксели выше порогового уровня 60 были определены как положительные по CFTR. Профиль интенсивности каждой отдельной ячейки был представлен как SD около среднего значения средней интенсивности для всей области.
Измерения активности канала Cl
— и тока короткого замыкания ( I sc )
Регистрацию целых клеток проводили на клетках HEK293, трансфицированных CFTR, как описано ранее ( 12 ). Клетки переносили в ванну, установленную на предметном столике инвертированного микроскопа (Ti2, Nikon), и цельноклеточный пластырь получали путем разрыва мембраны после гигомной герметизации. Раствор ванны перфузировали со скоростью 5 мл / мин. Регистрацию напряжения и тока проводили при комнатной температуре (от 22 до 25 ° C).Патч-пипетки с сопротивлением от 2 до 4 МОм были подключены к головному каскаду усилителя патч-зажим (Axopatch-200B; Molecular Devices, Саннивейл, Калифорния, США). Раствор ванны содержал 140 мМ N -метил-d-глюкамин хлорид (NMDG-Cl), 1 мМ CaCl 2 , 1 мМ MgCl 2 , 10 мМ глюкозу и 10 мМ Hepes (pH 7,4). Раствор для пипетки содержал 140 мМ N -метил-d-глюкамин хлорид, 5 мМ EGTA, 1 мМ MgCl 2 , 3 мМ MgATP и 10 мМ Hepes (pH 7.2). Для определения зависимости I — В режим фиксации напряжения и кривая I — В были получены путем применения импульсов линейного изменения от -100 до 100 мВ (0,8 мВ / мс; удерживающий потенциал 0 мВ). Токи CFTR активировали цАМФ (5 мкМ форсколина и 100 мкМ IBMX). Ток, генерируемый CFTR, был подтвержден применением ингибитора CFTR CFTR inh -172 (10 мкМ). Для сбора данных и подачи командных импульсов использовались pClamp 10.2 и Digidata 1550B (Molecular Devices).Кривые напряжения и тока сохраняли и анализировали с помощью pClamp 10.2 и Origin 8.0 (OriginLab Corp., Нортгемптон, Массачусетс, США). Токи фильтровались с частотой 5 кГц и дискретизировались с частотой 1 кГц. Все данные были нормированы на емкость всей клетки (пФ).
Толстая кишка мыши I sc измеряли с использованием небольших модификаций ранее описанных протоколов ( 12 ). Мышей анестезировали газом CO 2 и подвергали эвтаназии путем шейного смещения. Образцы толстой кишки от слепой до прямой кишки были взяты и вскрыты вдоль границы брыжейки.Содержимое просвета смывали буферным раствором HCO 3 —, и образцы помещали в камеру Уссинга (World Precision Instruments, Стивенидж, Великобритания) с открытой площадью поверхности 12,6 мм 2 . Ткани промывали с каждой стороны 10 мл буферного раствора HCO 3 —, содержащего 120 мМ NaCl, 5 мМ KCl, 1 мМ MgCl 2 , 1 мМ CaCl 2 , 10 мМ d-глюкозы, 5 мМ Hepes и 25 мМ NaHCO 3 (pH 7.4) при 37 ° C и 95% O 2 –5% CO 2 . На тканях фиксировалось напряжение 0 мВ с использованием зажима напряжения EVC-4000 (World Precision Instruments), и I sc непрерывно регистрировали с использованием системы сбора данных PowerLab (AD Instruments, Castle Hill, Австралия).
Измерение Cl
— / HCO 3 — обменной активности
Измерения внутриклеточного pH (pH i ) в клетках PANC-1 выполнялись с помощью pH-чувствительного флуоресцентного зонда 2 ′, 7′- бис- (2-карбоксиэтил) -5- (и-6) -карбоксифлуоресцеин (BCECF) согласно ранее опубликованным протоколам ( 13 ).Вкратце, клетки инкубировали с 2 мкМ ацетоксиметилового эфира BCECF в течение 5 минут, а затем перфузировали буферным раствором HCO 3 — [содержащим 120 мМ NaCl, 5 мМ KCl, 1 мМ MgCl 2 , 1 мМ CaCl 2 , 10 мМ d-глюкозы, 5 мМ Hepes и 25 мМ NaHCO 3 (pH 7,4)]. Флуоресценцию BCECF регистрировали при длинах волн возбуждения 490 и 440 нм с разрешением 2 / с на регистрирующей установке (Delta Ram; PTI Inc., Эдисон, Нью-Джерси, США). Активность обмена Cl — / HCO 3 — оценивалась исходя из начальной скорости повышения pH i в результате удаления Cl — из буфера, содержащего HCO 3 — (25 мМ HCO 3 — с 5% CO 2 ).Калибровку pH и проводили с использованием стандартных растворов pH, содержащих 150 мМ KCl и 5 мкМ нигерицин. Собственную буферную емкость (β i ) рассчитывали путем измерения ΔpH i в ответ на импульсы от 5 до 40 мМ NH 4 Cl в растворах, свободных от Na + . Поскольку на значения β i существенно не влияла трансфекция плазмидами, кодирующими WT-пендрин или H723R-пендрин, обменные активности Cl 2 / HCO 3 ˉ выражали как ΔpH единиц / мин без компенсации буферной емкости.
Статистика
Результаты нескольких экспериментов были представлены в виде средних значений ± SEM. Статистический анализ выполняли с использованием двусторонних тестов Стьюдента t или с односторонним дисперсионным анализом с последующим тестом множественного сравнения Тьюки, в зависимости от ситуации, с использованием GraphPad Prism 5 (GraphPad Software Inc., Ла-Холла, Калифорния, США). P <0,05 считали статистически значимым.
ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ
Дополнительные материалы к этой статье доступны по адресу http: // advance.sciencemag.org/cgi/content/full/6/8/eaax9914/DC1
Рис. S1. Сплайсинг XBP1 и эндонуклеазная активность IRE1α не участвуют в UPS ΔF508-CFTR (контрольные эксперименты на фиг. 1).
Рис. S2. Путь IRE1α киназы – ASK1 необходим для UPS ΔF508-CFTR (контрольные эксперименты на рис. 2).
Рис. S3. Истощение ASK1 ингибирует Arf1-Q71L-индуцированный UPS CFTR.
Рис. S4. CSTMP стимулирует экспрессию ΔF508-CFTR на клеточной поверхности путем активации киназы IRE1α.
Фиг.S5. CSTMP индуцирует экспрессию ΔF508-CFTR на клеточной поверхности через UPS.
Рис. S6. CSTMP (10 мкМ) не вызывает апоптотический сигнал.
Рис. S7. CSTMP индуцирует экспрессию ΔF508-CFTR на клеточной поверхности, не вызывая клеточного апоптоза.
Рис. S8. Измерения активности канала CFTR Cl — (контрольные эксперименты на рис. 4, C — F).
Рис. S9. CSTMP индуцирует экспрессию p.H723R-pendrin на клеточной поверхности через UPS.
Рис. S10. LD 50 значение CSTMP у мышей (per os) составляет 25.9 мг / кг в сутки.
Это статья в открытом доступе, распространяемая в соответствии с условиями некоммерческой лицензии Creative Commons Attribution, которая разрешает использование, распространение и воспроизведение на любом носителе, при условии, что конечным результатом будет , а не для коммерческих целей и при условии, что оригинальная работа правильно процитирована.
Благодарности: Мы благодарим Центр усовершенствованной визуализации Yonsei-Carl Zeiss за техническую помощь. Финансирование: Работа поддержана грантом 2013R1A3A2042197 Национального исследовательского фонда Министерства науки и информационных технологий Республики Корея. Вклад авторов: H.P. разработал и провел эксперименты по биохимии и иммунофлуоресценции CFTR. H.P. также написал рукопись. D.H.S. выполнили измерения патч-зажима и I sc . J.-R.S. провели анализ поверхностного биотинилирования и функционального пендрина. S.A. внесла свой вклад в сбор данных о животных. M.G.L. разработал и задумал все эксперименты, а также написал и отредактировал рукопись. Конкурирующие интересы: H.P. и M.G.L. являются изобретателями, находящимися на рассмотрении патента, связанного с этой работой, поданного Управлением исследований Университета Йонсей / Фондом университетской промышленности (№10-2019-0062327, подана 28 мая 2019 г.). Авторы не заявляют о других конкурирующих интересах. Доступность данных и материалов: Все данные, необходимые для оценки выводов в статье, представлены в статье и / или дополнительных материалах.