Синдром эдвардса код по мкб 10
Рубрика МКБ-10: Q91.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Cиндром Эдвардса
Синонимы: трисомия 18
Трисомия по 18-й хромосоме встречается у новорожденных с частотой от 1:3300 до 1:10 000; у девочек бывает в 3 раза чаще, чем у мальчиков. Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Этиология и патогенез[править]
Большинство случаев связаны со свободной трисомией 18. Мозаичная трисомия 18 была обнаружена у нескольких пациентов с клинической картиной, которая варьируется от классической трисомии 18 до нормального фенотипа в зависимости от количества трисомных клеток, присутствующих в тканях. Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Клинические проявления[править]
Клиническая картина: череп необычной формы (узкий лоб и широкий выступающий затылок), низкое расположение ушей, микрогнатия, сгибательная контрактура кистей и стоп, дисплазия стоп, пороки сердца, сильная задержка психического развития. Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста. Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Эдвардса неуточненный: Диагностика[править]
Трисомию 18 можно заподозрить во время беременности по результатам ультразвукового исследования плода (задержка роста, пороки развития, множественные кисты сосудистого сплетения) и подтверждить кариотипическим анализом плода. Маркеры сыворотки (используемые для диагностики трисомии 21) также могут быть аномальными.
Дифференциальный диагноз[править]
Синдром Эдвардса неуточненный: Лечение[править]
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза, связанного с этим синдромом: 90% детей умирают в течение первого года жизни от сердечных, почечных или неврологических осложнений или от повторных инфекций.
Сообщалось о длительном выживании (в некоторых случаях до взрослого возраста), главным образом в случаях мозаичной или частичной трисомии (в результате транслокации). Большинство немозаичных пациентов имеют лишь ограниченную автономию (отсутствие речи и ходьбы).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O. Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Синдром эдвардса по мкб 10
Рубрика МКБ-10: Q91.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Cиндром Эдвардса
Синонимы: трисомия 18
Трисомия по 18-й хромосоме встречается у новорожденных с частотой от 1:3300 до 1:10 000; у девочек бывает в 3 раза чаще, чем у мальчиков.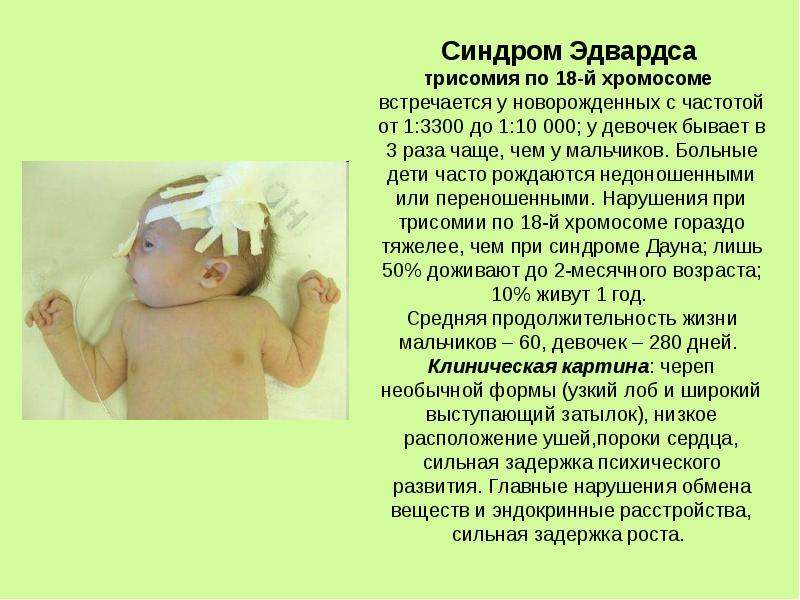 Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Этиология и патогенез[править]
Большинство случаев связаны со свободной трисомией 18. Мозаичная трисомия 18 была обнаружена у нескольких пациентов с клинической картиной, которая варьируется от классической трисомии 18 до нормального фенотипа в зависимости от количества трисомных клеток, присутствующих в тканях. Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Клинические проявления[править]
Клиническая картина: череп необычной формы (узкий лоб и широкий выступающий затылок), низкое расположение ушей, микрогнатия, сгибательная контрактура кистей и стоп, дисплазия стоп, пороки сердца, сильная задержка психического развития. Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста. Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Эдвардса неуточненный: Диагностика[править]
Трисомию 18 можно заподозрить во время беременности по результатам ультразвукового исследования плода (задержка роста, пороки развития, множественные кисты сосудистого сплетения) и подтверждить кариотипическим анализом плода. Маркеры сыворотки (используемые для диагностики трисомии 21) также могут быть аномальными.
Дифференциальный диагноз[править]
Синдром Эдвардса неуточненный: Лечение[править]
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза, связанного с этим синдромом: 90% детей умирают в течение первого года жизни от сердечных, почечных или неврологических осложнений или от повторных инфекций.
Сообщалось о длительном выживании (в некоторых случаях до взрослого возраста), главным образом в случаях мозаичной или частичной трисомии (в результате транслокации). Большинство немозаичных пациентов имеют лишь ограниченную автономию (отсутствие речи и ходьбы).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O. Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Синдром эдвардса код по мкб
Рубрика МКБ-10: Q91.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Cиндром Эдвардса
Синонимы: трисомия 18
Трисомия по 18-й хромосоме встречается у новорожденных с частотой от 1:3300 до 1:10 000; у девочек бывает в 3 раза чаще, чем у мальчиков. Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков — 60, девочек — 280 дней.
Этиология и патогенез[править]
Большинство случаев связаны со свободной трисомией 18. Мозаичная трисомия 18 была обнаружена у нескольких пациентов с клинической картиной, которая варьируется от классической трисомии 18 до нормального фенотипа в зависимости от количества трисомных клеток, присутствующих в тканях. Фенотип трисомии 18, по-видимому, связан с наличием трех копий интервала 18q11-q12.
Клинические проявления[править]
Клиническая картина: череп необычной формы (узкий лоб и широкий выступающий затылок), низкое расположение ушей, микрогнатия, сгибательная контрактура кистей и стоп, дисплазия стоп, пороки сердца, сильная задержка психического развития. Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста. Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Эдвардса неуточненный: Диагностика[править]
Трисомию 18 можно заподозрить во время беременности по результатам ультразвукового исследования плода (задержка роста, пороки развития, множественные кисты сосудистого сплетения) и подтверждить кариотипическим анализом плода. Маркеры сыворотки (используемые для диагностики трисомии 21) также могут быть аномальными.
Дифференциальный диагноз[править]
Синдром Эдвардса неуточненный: Лечение[править]
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза, связанного с этим синдромом: 90% детей умирают в течение первого года жизни от сердечных, почечных или неврологических осложнений или от повторных инфекций.
Сообщалось о длительном выживании (в некоторых случаях до взрослого возраста), главным образом в случаях мозаичной или частичной трисомии (в результате транслокации). Большинство немозаичных пациентов имеют лишь ограниченную автономию (отсутствие речи и ходьбы).
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
1. Buyse ML. Birth Defects Encyclopedia. Cambridge: Blackwell, 1990.
2. Emery A, Rimoin D. Principles and Practice of Medical Genetics (2nd ed). New York: Churchill Livingstone, 1990.
3. Gorlin RJ, et al. Syndromes of the Head and Neck (3rd ed). New York: Oxford University Press, 1990.
4. Hall JG, et al. Handbook of Normal Physical Measurements. New York: Oxford University Press, 1989.
5. Jones KL. Smith’s Recognizable Patterns of Human Malformation (4th ed). In M Markowitz (ed.), Major Problems in Clinical Pediatrics (vol VII). Philadelphia: Saunders, 1988.
6. McKusick V. Mendelian Inheritance in Man (10th ed.). Baltimore: Johns Hopkins University Press, 1992.
7. Rimoin DL. Disorders of the Endocrine Glands. In AA Dietz (ed.), Genetic Disease: Diagnosis and Treatment. Proceedings of the Fifth Arnold O.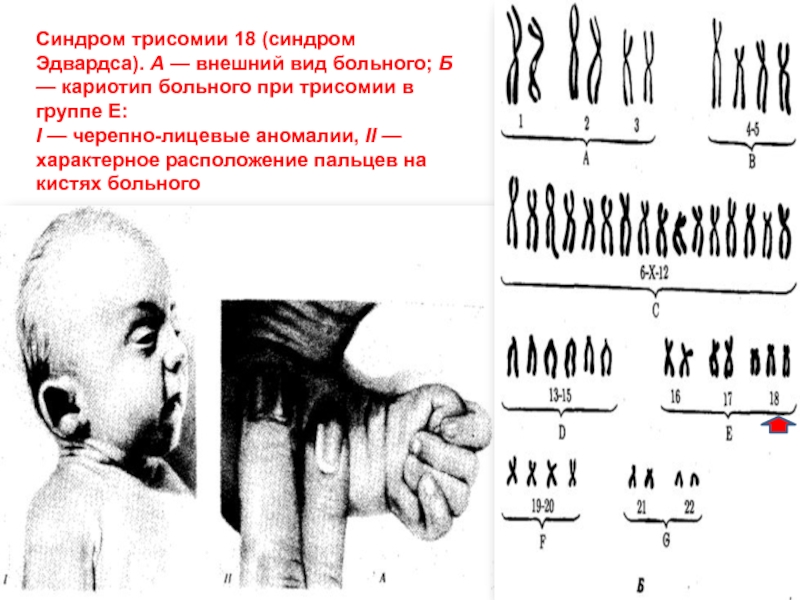 Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
Beckman Conference in Clinical Chemistry. Monterey, CA: The Association for Clinical Chemistry, 1983.
8. Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias (3rd ed). Chicago: Yearbook, 1990.
9. Vogel F, Motulsky AG. Human Genetics: Problems and Approaches (2nd ed). New York: Springer, 1986.
10. Wiedmann H-R, et al. Atlas of Clinical Syndromes—A Visual Aid to Diagnosis (2nd ed). St. Louis: Mosby, 1989.
11. Wynne-Davis R, Hall CM, Apley AG. Atlas of Skeletal Dysplasias. Edinburgh: Churchill Livingstone, 1985.
Действующие вещества[править]
Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
Названия
Синдром Эдвардса.
Симптомы при синдроме Эдвардса (трисомия 18)
Описание
Впервые трисомию по группе Е описали J. Edwads и соавт. (1960г. ). Частота синдрома Эдвардса среди новорожденных в среднем составляет 1:7000, замечено, что девочки поражаются в 3 раза чаще, чем мальчики. Ученым высказано предположение о стабилизирующем действии Х-хромосомы при абберациях пары 18, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Средний возраст матерей 32,5 года, отцов – 35 лет.
Ученым высказано предположение о стабилизирующем действии Х-хромосомы при абберациях пары 18, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Средний возраст матерей 32,5 года, отцов – 35 лет.
Симптомы
Длительность беременности превышает нормальную, и составляет в среднем 42 недели. Во время беременности диагностируют слабую активность плода и многоводие. Отмечается,что плацента имеет меньшие, чем обычно, размеры, часто обнаруживается только одна пупочная артерия; многие дети рождаются в асфиксии, с очень низкой массой тела и выраженной гипотрофией.
Фенотип больных с синдромом Эдвардса довольно характерен. Череп долихоцефалический, с низким лбом, затылок более широкий и выступающий. Часто встречается микроцефалия или гидроцефалия. Надглазничные валики сглажены, глазные щели узкие, наблюдается эпикант, птоз, часто встречается глазная патология, микрофтальмия, колобома, катаракта. Переносье вдавлено, но спинка носа тонкая, выступающая, ушные раковины расположены очень низко, диагностируется отсутствие мочки и козелка, недоразвиты завиток и противозавиток.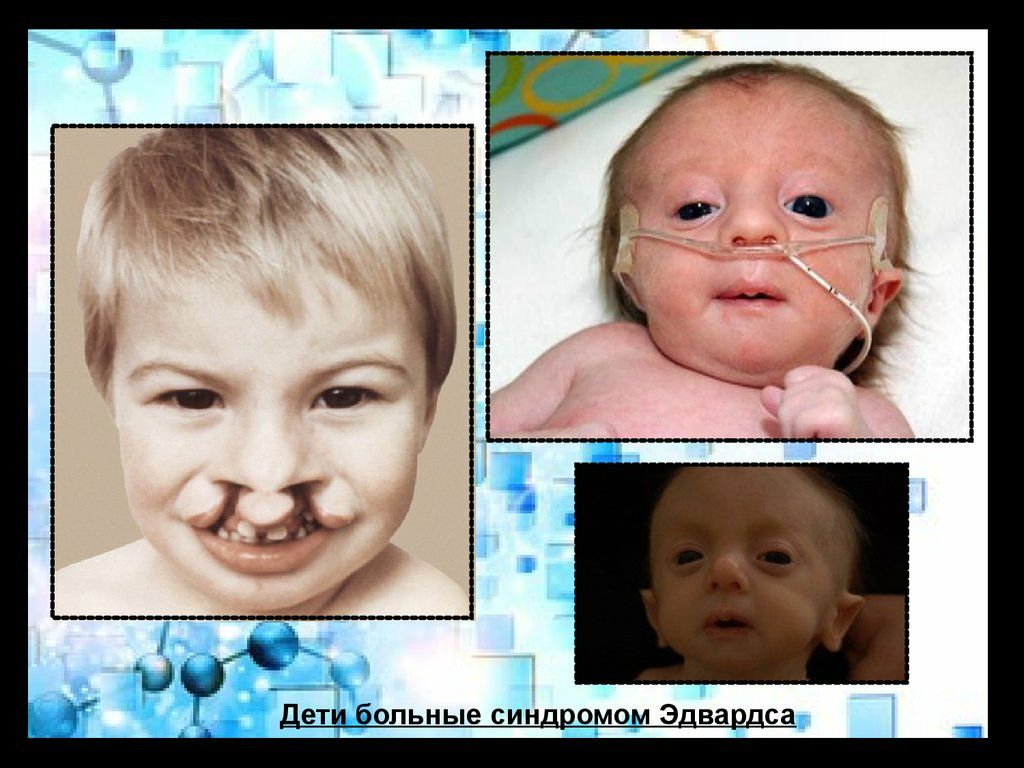 Характерна микроретрогнатия. Рот мешьше обычных размеров, имеет треугольную форму, верхняя губа короче обычного. Небо высокое, иногда с расщелиной, шея короткая, нередко с крыловидной складкой.
Характерна микроретрогнатия. Рот мешьше обычных размеров, имеет треугольную форму, верхняя губа короче обычного. Небо высокое, иногда с расщелиной, шея короткая, нередко с крыловидной складкой.
Аномалии опорно-двигательного аппарата отличаются разнообразием: расширение шрудной клетки, укорочение грудины, таз узкий, деформации конечностей, ограничена подвижность в тазобедренных суставах, описаны вывихи бедра. Кисти и пальцы короткие, клинодактилия 5 пальцев кисти, дистально расположенный, гипоплазированный 1 палец кисти, сглажен тенар. Пальцы сжаты в кулак по типу «флексорной аномалии»: 2 и 5 пальцы расположены сверху и прикрывают прижатые к ладони 3 и 4 пальцы; 1 палец стопы широкий и короткий, синдактилия 2 и 3 пальцев. Типична для трисомии 18 форма стопы в виде своеобразной «качалки». Характерна общая мышечная гипотония. У мальчиков нередко встречается крипторхизм, гипоспадия; гипертрофия клитора у девочек.
Интеллектуальный дефект соответствует олигофрении в степени идиотии ил глубокой имбецильности и только в единичных описаниях мозаичного варианта трисомии 18 умственное недоразвитие менее грубое. Нередко у больного развиваются судороги.
Нередко у больного развиваются судороги.
Внешний вид больного с синдромом Эдвардса (трисомия 18)
Диагностика
Дерматографическая картина при синдроме Эдвардса отличается своеобразными признаками: отмечается большая частота дуг на подушечках пальцев рук (примерно в 10 раз выше, чем в общей популяции), нередко дистальная сгибательная складка на пальцах не развита, у трети больных обнаруживается поперечная ладонная борозда, гребневый счет увеличен, осевой трирадиус обычно расположен дистально.
При аутопсии при синдроме Эдвардса находят разнообразные пороки развития практически всех органов и систем. С различной частотой встречаются аномалии ЦНС: аплазия или гипоплазия мозолистого тела, гипоплазия мозжечка, атрофия мозговых извилин. У 95% пациентов с трисомией 18 диагностируются пороки сердца и крупных сосудов, наиболее часто встречаются дефект межжелудочковой перегородки и незаращение артериального протока. Около половины всех случаев синдрома Эдвардса сопровождается аномалиями органов пищеварения: нарушение расположения кишечника, меккелев дивертикул, атрезия пищевода, атрезия заднего прохода. В 50% случаев отмечаются пороки развития мочеполовой системы – дольчатая или подковообразная почка, дупликация мочеточников, гипоплазия яичников.
В 50% случаев отмечаются пороки развития мочеполовой системы – дольчатая или подковообразная почка, дупликация мочеточников, гипоплазия яичников.
При цитогенетическом обследовании в 80% случаев находят трисомию 18, у 10% больных – мозайцизм. Описаны случаи транслокационного варианта, двойной анеуплодии типа 48, ХХУ +18 с участием трисомного по хромосоме 18 клона.
Лечение
Нет патогенетического лечения синдрома Эдвардса на сегодняшний день, возможна лишь симптоматическая коррекция патологий. Прогноз для жизни неблагоприятный, средняя продолжительность жизни мальчиков 2-3 месяца, девочек – 10 месяцев. 30% больных умирают в течение 1-го года жизни, до года доживает лишь 10% больных. При мозаичных вариантах прогноз для жизни несколько лучше.
Источник
Синдром Эдвардса — Википедия
Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
Edwards). Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трех вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
Дети с трисомией 18 рождаются с низким, в среднем 2177 г, весом. При этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:3000 зачатий и 1:6000 рождений живых детей. Хотя женщина в 20 или 30 лет также может родить ребёнка с синдромом Эдвардса, риск рождения больного ребёнка увеличивается с возрастом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикрепленная к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикрепленная к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
См. также
Ссылки
- ↑ база данных Disease ontology (англ.) — 2016.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
Аутизм МКБ 10. Аутизм – код и критерии по МКБ 10 у детей.
МКБ 10 – это десятый пересмотр Международной номенклатурной классификации заболеваний. По сути – справочник всех болезней и патологических состояний, принятый в официальной медицине в 1989 году и применяющийся всеми врачами.
Медиками России, эта классификация признана в 1999 году. Каждой патологии, в том числе и аутизму, присваивается по МКБ 10 свой шифр. Под ним скрываются характеристики, симптоматика болезни. Такой подход позволяет врачам разных стран синхронизировать свои научные исследования в соответствующих областях, обмениваться опытом лечения, точнее формулировать диагнозы.
Под ним скрываются характеристики, симптоматика болезни. Такой подход позволяет врачам разных стран синхронизировать свои научные исследования в соответствующих областях, обмениваться опытом лечения, точнее формулировать диагнозы.
Аутизм также имеет свой код заболевания по МКБ 10 и сформулированные критерии, определяющие, какой вариант болезни у ребенка. Благодаря этому, даже при смене страны жительства, человек получит необходимую медицинскую помощь с учетом особенностей состояния без дополнительных обследований. В зависимости от проявлений, составллено несколько шифров, которыми обозначают подвиды аутических расстройств.
Содержание статьи:
Детский аутизм F84.0
Ранний детский аутизм имеет по МКБ 10 код F84.0. Так зашифрованы проявления болезни, которые возникают с самого раннего возраста.
Критерии, на основе которых присваивается код F84.0, разделены на несколько групп.
Для диагноза, нарушения социального взаимодействия должны быть представлены хотя бы 2 пунктами из 5:
- Отсутствует социально-эмоциональная взаимность.
 Ребенок не проявляет ожидаемых ответных реакций на действия окружающих.
Ребенок не проявляет ожидаемых ответных реакций на действия окружающих. - Нарушения во время ролевых, социально-имитативных игр, отказ от подобного вида деятельности.
- Отсутствие адекватной реакции на эмоции других людей, неспособность правильно среагировать на ситуацию, агрессивность.
- Некорректное использование тональности голоса, отсутствие подкрепляющей жестикуляции.
- Отказ от речевого общения с окружающим миром, задержки речевого развития из-за нежелания коммуницировать подобным образом.
 Ребенок не проявляет ожидаемых ответных реакций на действия окружающих.
Ребенок не проявляет ожидаемых ответных реакций на действия окружающих.Узость интересов, склонность к стереотипным, повторяющимся действиям, не способствующим развитию личности, выраженные хотя бы 1 пунктом из 4:
- Ребенок стремится воспроизводить узкий круг действий. У него мало интересов, и расширять их количество он не стремится.
Чтобы психолог или психиатр поставили диагноз аутизм по коду МКБ 10, признаки атипичного поведения должны проявиться в первые 3 года жизни. При этом они не провоцируются органическим поражением участков коры головного мозга.
Атипичный аутизм F84.1
Когда признаки аутического расстройства личности проявляются позже 3 лет, не скорректированы после наступления 18 или проявляются ограниченно, врачи в диагнозе проставляют шифр, обозначающий атипичный вариант заболевания.
Такой же подход практикуется при продлении статуса инвалида по достижении совершеннолетия. Например, если человек не смог социализироваться до необходимого уровня
Нередко код атипичного аутизма по МКБ 10 (F84.1) проставляется в документах, когда специфическая симптоматика проявляется не ярко, а в одном из направлений. Как-то: отсутствие стремления к социальному взаимодействию, наличие стереотипных действий.
Также отличительными чертами диагноза являются сильные задержки речевого развития, требующие серьезной работы с логопедом, отставание в умственном развитии, детский психоз.
Синдром Ретта F84.2
Редкий синдром, регистрируемый пока что только у девочек.
Характерной чертой является изначально нормальное развитие без патологий. Однако с возраста 7 – 24 месяцев начинает проявляться и усиливаться специфическая симптоматика, быстро показывающая патологичность развития ребенка:
Однако с возраста 7 – 24 месяцев начинает проявляться и усиливаться специфическая симптоматика, быстро показывающая патологичность развития ребенка:
Позже начинается сколиоз, атаксия и другие отклонения в физическом развитии. Возможно возникновение эпилептических припадков. Поэтому люди с синдромом Ретта считаются тяжелыми инвалидами.
Включение этого синдрома в список заболеваний под общим называнием аутизм и присвоение этого кода по МКБ 10, у детей считается спорным.
Синдром Аспергера F84.5
Еще один специфический синдром, входящий в МКБ 10 по коду в расстройства аутического характера носит имя австрийского педиатра Ганса Аспергера. Его еще называют высоко функциональным аутизмом из-за отсутствия таких черт, как задержка речевого и познавательного развития.
Считается скорее мужской болезнью, так как статистика выявленных случаев показывает сильный перевес среди мальчиков: 8 пациентов мужского пола против 2 женского. Кроме того, у девочек чаще наблюдается легкая форма, ярко проявляющаяся в подростковый период и почти исчезающая по мере взросления.
Список использованной литературы
Синдром делеции 22 хромосомы (синдром ДиДжорджи) у детей. Клинические рекомендации.
Оглавление
Ключевые слова
-
Аутоиммунные осложнения
-
Велофарингеальная недостаточность
-
Врожденный порок сердца
-
Гипокальциемия
-
Гипопаратиреоз
-
Делеция 22 хромосомы
-
Дефект Т-клеточного звена
-
Задержка речевого и психомоторного развития
-
Задержка физического развития
-
Внутривенные иммуноглобулины
-
Инфекционные осложнения
-
Расщепление неба и верхней губы
-
Синдром ДиДжорджи
Список сокращений
АИГА – аутоиммунная гемолитическая анемия
АЛТ — аланинаминотрансфераза
АСТ — аспартатаминотрансфераза
ВВИГ — внутривенные иммуноглобулины
ВЗК – воспалительные заболевания кишечника
ВПС – врожденный порок сердца
ГКС — глюкокортикостероиды
ДНК — дезоксирибонуклеиновая кислота
ЖКТ — желудочно-кишечный тракт
ИТП – иммунная тромбоцитопения
КМ — костный мозг
КТ — компьютерная томография
ЛОР — ларингооторинолог
ЛПУ — лечебно-профилактическое учреждение
МЗ — Министерство здравоохранения
МКБ-10 — Международная классификация болезней 10-го пересмотра
МРТ —магнитно-резонансная томография
РКИ — рандомизированные контролируемые исследования
РНК — рибонуклеиновая кислота
РФ — Российская Федерация
Синдром del 22q11 – синдром делеции 22 хромосомы=синдром ДиДжорджи
СДД — синдром ДиДжорджи
ТГСК – трансплантация гемопоэтических стволовых клеток
УЗИ – ультразвуковое исследование
ЭКГ – электрокардиограмма
ЭхоКГ -эхокардиография
ЮРА – ювенильный ревматоидный артрит
del 22q11.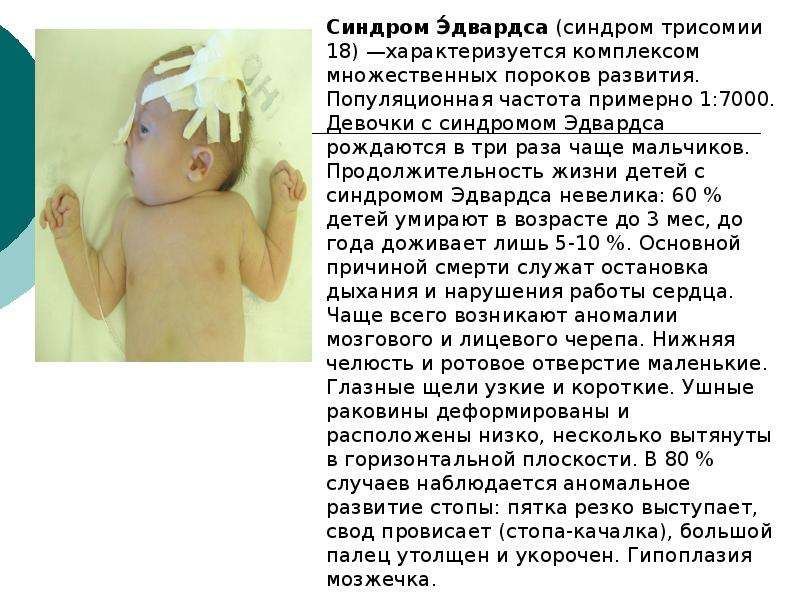 2 – делеция длинного плеча 22 хромосомы локус 11.2
2 – делеция длинного плеча 22 хромосомы локус 11.2
САТСН 22 — Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia, 22q deletion – порок сердца, лицевые аномалии, гипоплазия тимуса, расщелина неба, гипокальцемия, делеция 22
FISH –fluorescent in situ hybridization — флуоресцентная гибридизация in situ
ТВХ 1 ген –Т бокс 1 ген
TREC — T-cell Receptor Excision Circles
Термины и определения
Внутривенные иммуноглобулины – препараты, содержащие преимущественно нормальный человеческий IgG. Изготовляются из пулированной плазмы тысяч здоровых доноров, с применением специальных методов очистки и вирусинактивации.
Делеция – потеря участка хромосомы
Хромосомные аберрации – изменение числа и\или структуры хромосом
Микрогнатия — недоразвитие (гипоплазия) челюстных костей.
Ретрогнатия — смещение челюстной кости в дорзальном направлении (кзади)
Гипертелоризм — увеличенное расстояние между глазами
TREC – кольцевые фрагменты ДНК, образующиеся при развитии Т лимфоцитов в тимусе, в частности, в процессе формирования Т клеточного рецептора. Их концентрация в крови отражает эффективность тимопоэза. Используется для скрининга Т клеточных иммунодефицитов.
Трансплантация гематопоэтических стволовых клеток – метод лечения некоторых наследственных и приобретенных гематологических, онкологических и иммунных заболеваний, основанный на замене собственного, патологического кроветворения больного на нормальное кроветворение донора.
1. Краткая информация
1.1 Определение
Синдром делеции 22-й хромосомы (синдром del 22q11) или синдром ДиДжоржи (СДД) — это совокупность морфологических, иммунологических и неврологических изменений, которые являются следствием делеции длинного плеча одной копии 22-й хромосомы — del 22q11.2 [1,2].
В классическом понятии этот синдром представляет собой комплекс симптомов, состоящий из патологии лицевого скелета (расщелины твердого неба), врожденного порока сердца, иммунодефицита вследствие гипоплазии (аплазии) тимуса и гипокальциемии, как результат гипоплазии паращитовидной железы [1,3].
Как ни один другой синдром, синдром del 22q11.2 вариабелен в количестве признаков и степени их выраженности, что и объясняет тот факт, что этот синдром в литературе имеет порядка десятка различных названий, включая синдром ДиДжорджи, САТСН 22, велокардиофациальный сидром, Шпринтцена синдром, Кайлера синдром, синдром лицевых и конотрункальных аномалий и т.д.[1,3,4].
1.2 Этиология и патогенез
В основе заболевания лежит нарушение формирования органов, происходящих их третьей жаберной дуги (нижняя часть лицевого скелета, тимус, паращитовидная железа, верхние отделы сердца и магистральных сосудов). Цитогенетические и молекулярные исследования показали, что делеция 22q11.2 является ведущей причиной СДД и возникает спорадически более чем в 90% случаев [5,6,7]. В 10% случаев делеция наследуется от одного из родителей, так как наследование происходит аутосомно- доминантным путем [1,4]. В редких случаях синдром является проявлением перестроек других хромосом, а также мутации гена ТВХ1 [4].
Анализ ДНК пациентов с СДД хромосомы выявил, что в 85-90% случаев выпадающий участок является одним и тем же. Дефект находится между D22S427 на 22q11.21 и D22S801 на 22q11.23. В этом участке локализовано не менее 40 генов, что составляет около 3 млн пар нуклеиновых оснований. В 10-12% случаев встречаются более короткие делеции, которые составляют 1,5-2 млн парных оснований. Было описано несколько пациентов с синдромом делеции 22-й хромосомы, имеющих делеции за пределами наиболее часто выпадающих участков [5,6]. Результаты проведенных исследований свидетельствуют о том, что степень выраженности фенотипа не коррелирует с размером делеции, т.е. пациент с потерей 1,5 млн парных оснований может иметь такой же по тяжести фенотип, как и с делецией в 3 млн парных оснований [2,5]. Кроме того, было замечено, что вариабельность фенотипических проявлений варьирует как внутри одной семьи, так и между семьями, несмотря на идентичные участки делеции [5].
Делеция вызывает выпадение участка, включающего ген ТВХ, ген фактора транскрипции, участвующего в развитии фарингеальных дуг [5,6]. Эти изменения, в свою очередь, ведут к нарушению формирования сердца и магистральных сосудов, иммунологическим изменениям, расщеплению нёба и верхней губы, гипопаратиреоидизму, задержке умственного развития.
Несмотря на то, что ТВХ1, без сомнения, является главным геном, формирующим фенотип при синдроме делеции 22-й хромосомы, в результате исследований были выявлены и другие гены, недостаточная экспрессия которых может играть роль в формировании фенотипических проявлений [6,7].
Учитывая результаты работ по выяснению молекулярных основ заболевания, ясно, что в формировании фенотипа играет роль комплексное нарушение экспрессии и взаимодействия генов, их модификаторов и других составляющих, что приводит к дальнейшему нарушению эмбрио- и органогенеза [4,5,7].
Соответственно, при отсутствии или нарушении функции и экспрессии генов и дальнейших процессов происходит формирование пороков развития, характерных для СДД [1,5].
1.3 Эпидемиология
Синдром делеции 22-й хромосомы — одна из самых частых делеций среди других хромосомных аберраций в человеческом геноме, по частоте она уступает лишь синдрому Дауна, трисомии по 21-й хромосоме. Частота встречаемости варьирует от 1:4000 до 1:6000 новорожденных [1,2,3]. Не наблюдается ни половой, ни этнической предрасположенности к данному синдрому. Большинство пациентов с СДД имеют патологию лицевого скелета и врожденный порок сердца и развивают гипокальциемию вскоре после рождения [6]. Пациенты, не имеющие данных симптомов, зачастую диагностируются в раннем возрасте, и правильный значительно запаздывает.
1.4 Кодирование по МКБ-10
Другие иммунодефициты (D84):
D84.1 – Синдром ДиГеорга.
1.5 Классификация
Исторически сложилось, что в литературе часто используется разделение синдрома на полный и неполный (частичный) [1,3,5]:
-
Термин «Полный синдром ДиДжорджи» использовался у пациентов, имеющих полный спектр типичных проявлений, включая выраженный иммунодефицит.
-
Термин «Частичный синдром ДиДжорджи» использовался у пациентов, если они имели лишь некоторые типичные признаки, особенно без проявлений выраженного иммунодефицита. Частичный синдром делеции 22-й хромосомы в значительной степени превалирует по количеству в сравнении с полным.
2. Диагностика
2.1 Жалобы и анамнез
Спектр клинических проявлений при синдроме делеции 22-й хромосомы достаточно широк [3,6,9,11,12,14,15], поэтому жалобы и анамнез заболевания могут быть крайне разнообразными и различными по степени выраженности:
-
Врожденный порок сердца представлен не менее, чем в 80% случаев. Некоторые из пороков являются более патогномоничными: прерывание дуги аорты, общий артериальный ствол и тетрада Фалло являются наиболее частыми среди данной группы детей [6,11].
-
Гипокальциемия/гипопаратиреоз может проявляться судорожным синдромом при выраженном дефиците кальция в младенческом возрасте [6,12].
-
Поражение носоглоточного аппарата выявлено примерно в 70% случаев и проявляется в виде велофарингеальной недостаточности, расщеплении нёба, губы, раздвоении уздечки нёба, гнусавым оттенком голоса, также описано снижение обоняния, кондуктивная и/или сен- соневральная тугоухость [6,10,13].
-
Характерные черты лица (удлиненное лицо, микрог
MCB-10G-2S | Серверы и рабочие станции
Сетевая мезонинная карта 10GbE SFP + OCP
Open Compute Project
ASUS MCB-10G-2S разработан для использования с материнскими платами Open Compute Project Intel v2.0, которые имеют интерфейс PCI-E 3.0 и форм-фактор OCP. Это позволяет пользователям легко и гибко интегрировать мезонинную карту в материнские платы или системы OCP. MCB-10G-2S обеспечивает гибкость и идеально подходит для интеграции Open Compute Project.
Идти в ногу с тенденциями 10 Гбит / с
Все больше и больше приложений требуют высокой пропускной способности для передачи больших объемов данных, особенно для центров обработки данных, высокопроизводительных вычислений, веб-хостинга и корпоративного использования. Новый Ethernet 10 Гбит / с — идеальная технология для быстрого перемещения больших объемов данных, более чем достаточная для сетевых потребностей крупномасштабных виртуальных машин и сред виртуализации. Пропускная способность, которую он обеспечивает в сочетании с консолидацией серверов, очень выгодна для облачных вычислений, программного обеспечения для виртуализации, веб-кэширования, ответа приложений в реальном времени, баз данных, социальных сетей и приложений бизнес-логики, научного моделирования и профессионального моделирования.
Обеспечение оптоволоконных и медных решений для различных приложений центров обработки данных
Сетевая мезонинная карта ASUS MCB-10G-2S OCP обеспечивает гибкий, масштабируемый и доступный подход для клиентов, которые хотят повысить скорость межсоединения с нынешних 1 Гбит / с до 10 Гбит / с. С сетевой мезонинной картой MCB-10G-2S OCP пользователи могут подключаться к оптоволоконной технологии через оптические модули SFP + SR (ближний) и LR (дальний) или использовать медное решение через пассивный двухосевой кабель прямого подключения.Оптоволоконное решение SFP + подходит для связи на больших расстояниях, тогда как решение SFP + с прямым подключением для медных кабелей лучше всего при использовании на коротких отрезках, обычно 5 м или меньше, чего достаточно для связи серверных плат и коммутаторов. Применяя различные типы модулей и кабели, сетевая мезонинная плата MCB-10G-2S OCP может соответствовать широкому диапазону расстояний и различным потребностям клиентов для различной сетевой архитектуры.
синдром Эдвардса Википедия
Хромосомное заболевание, при котором присутствуют три копии хромосомы 18
Синдром Эдвардса , также известный как трисомия 18 , является генетическим заболеванием, вызванным наличием третьей копии всей или части хромосомы 18. [3] Поражены многие части тела. [3] Младенцы часто рождаются маленькими и имеют пороки сердца. [3] Другие особенности включают маленькую голову, маленькую челюсть, сжатые кулаки с перекрытием пальцев и тяжелую умственную отсталость. [3]
Большинство случаев синдрома Эдвардса возникает из-за проблем во время формирования репродуктивных клеток или во время раннего развития. [3] Заболеваемость увеличивается с возрастом матери. [3] В редких случаях дела могут передаваться по наследству от родителей. [3] Иногда не все клетки имеют дополнительную хромосому, известную как мозаичная трисомия, и симптомы в этих случаях могут быть менее серьезными. [3] Ультразвук во время беременности может усилить подозрение на заболевание, что может быть подтверждено амниоцентезом. [2]
Лечение поддерживающее. [2] После рождения одного ребенка с этим заболеванием риск рождения второго обычно составляет около одного процента. [2] Это второе по распространенности состояние, связанное с появлением третьей хромосомы при рождении, после синдрома Дауна. [4]
Синдром Эдвардса встречается примерно у 1 из 5000 живорождений. [3] Некоторые исследования показывают, что больше детей, доживающих до рождения, составляют девочки. [2] Многие из пострадавших умирают до рождения. [3] Выживаемость после года жизни составляет около 5–10%. [3] Он назван в честь английского генетика Джона Хилтона Эдвардса, который впервые описал синдром в 1960 году. [5]
Признаки и симптомы []
Сжатая рука и пальцы перекрывают друг друга: указательный палец перекрывает третий палец, а пятый палец перекрывает безымянный палец, что характерно для трисомии 18.Это вызвано врожденной контрактурой сустава. [6]
Дети, рожденные с синдромом Эдвардса, могут иметь некоторые или все следующие характеристики: пороки развития почек, структурные дефекты сердца при рождении (например, дефект межжелудочковой перегородки, дефект межпредсердной перегородки, открытый артериальный проток), кишечник, выступающий за пределы тела (омфалоцеле), атрезия пищевода. , умственная отсталость, задержка в развитии, дефицит роста, трудности с кормлением, затрудненное дыхание и артрогрипоз (мышечное заболевание, которое вызывает множественные контрактуры суставов при рождении). [7] [8]
Некоторые физические пороки развития, связанные с синдромом Эдвардса, включают маленькую голову (микроцефалия), сопровождающуюся выступающей задней частью головы (затылок), низко посаженные уродливые уши, аномально маленькую челюсть (микрогнатия) ), расщелина губы / неба, вздернутый нос, узкие веки (блефарофимоз), широко расставленные глаза (глазной гипертелоризм), опущение верхних век (птоз), короткая грудная кость, сжатые руки, кисты сосудистого сплетения, недоразвитые большие пальцы и / или ногти, отсутствие лучевой кости, перепонка на втором и третьем пальцах стопы, косолапость или коромысло ступни, а у мужчин — неопущенные яички. [7] [8]
Внутриутробно , наиболее частой характеристикой являются сердечные аномалии, за которыми следуют аномалии центральной нервной системы, такие как аномалии формы головы. Наиболее распространенной внутричерепной аномалией является наличие кист сосудистого сплетения, которые представляют собой карманы жидкости в головном мозге. Сами по себе они не представляют проблемы, но их наличие может быть маркером трисомии 18. [9] [10] Иногда наблюдается избыток околоплодных вод или многоводие. [7] Хотя синдром Эдвардса встречается нечасто, он вызывает большую часть пренатальных случаев порока развития Денди-Уокера. [11] [12]
Генетика []
Синдром Эдвардса — это хромосомная аномалия, характеризующаяся наличием дополнительной копии генетического материала на 18-й хромосоме либо полностью (трисомия 18), либо частично (например, из-за транслокаций). Дополнительная хромосома обычно возникает до зачатия. Эффекты дополнительной копии сильно различаются в зависимости от степени дополнительной копии, генетической истории и случайности.Синдром Эдвардса встречается во всех популяциях человека, но чаще встречается у потомства женского пола. [13]
Здоровая яйцеклетка и / или сперматозоид содержат отдельные хромосомы, каждая из которых вносит свой вклад в 23 пары хромосом, необходимых для формирования нормальной клетки с типичным человеческим кариотипом из 46 хромосом. Численные ошибки могут возникать на любом из двух мейотических делений и вызывать неспособность хромосомы сегрегировать в дочерние клетки (нерасхождение). Это приводит к появлению дополнительной хромосомы, что делает гаплоидное число 24, а не 23.Оплодотворение яйцеклеток или оплодотворение спермой, содержащей дополнительную хромосому, приводит к трисомии или трем копиям хромосомы, а не двум. [14]
Трисомия 18 (47, XX, + 18) вызвана событием мейотического нерасхождения. При нерасхождении гамета (, т.е. , сперматозоид или яйцеклетка) производится с дополнительной копией хромосомы 18; Таким образом, гамета имеет 24 хромосомы. В сочетании с нормальной гаметой от другого родителя у эмбриона 47 хромосом с тремя копиями 18 хромосомы.
Небольшой процент случаев возникает, когда только некоторые клетки организма имеют дополнительную копию хромосомы 18, что приводит к смешанной популяции клеток с разным числом хромосом. Такие случаи иногда называют мозаичным синдромом Эдвардса. Очень редко кусок 18 хромосомы прикрепляется к другой хромосоме (перемещается) до или после зачатия. У больных есть две копии 18-й хромосомы плюс дополнительный материал 18-й хромосомы, прикрепленный к другой хромосоме.При транслокации у человека возникает частичная трисомия по 18 хромосоме, и аномалии часто менее серьезны, чем при типичном синдроме Эдвардса.
Диагноз []
Ультразвук может усилить подозрение на заболевание, что может быть подтверждено амниоцентезом. [2]
Уровни PAPP-A, AFP, uE3, свободного β-ХГЧ, все из которых обычно снижаются во время беременности. [15]
Прогноз []
Около 95% беременностей не заканчиваются рождением живого ребенка. [13] Основные причины смерти включают апноэ и сердечные аномалии. Предсказать точный прогноз при беременности или в периоде новорожденности невозможно. [13] Половина живых младенцев не доживает до первой недели жизни. [16] Средняя продолжительность жизни составляет от пяти до 15 дней. [17] [18] Около 8–12% младенцев живут дольше 1 года. [19] [20] [ нужен лучший источник ] Один процент детей доживает до 10 лет, [13] , хотя ретроспективное канадское исследование 254 детей с трисомией 18 продемонстрировало десятилетнюю выживаемость 9.8%. [20]
Эпидемиология []
Синдром Эдвардса встречается примерно у одного из 5000 живорождений, но этот синдром влияет на большее количество зачатий, потому что большинство тех, у кого это заболевание диагностировано пренатально, не доживают до рождения. a b c d e f g h i j k l m n o p q r s t «трисомия 18». «Распространенность и частота синдрома Эдвардса». Болезни Синдром Центра-Эдвардса . Adviware Pty Ltd. 4 февраля 2008 г. Архивировано 25 июня 2004 года. Проверено 17 февраля 2008. Sindromul Edwards является детерминированным генетическим заболеванием человека, имеющего хромосомный цвет 18, плюс целлюлозы организма.Modn mod normal, fiecare celulă (cu excepția celulelor reproducătoare) au un număr de 46 de cromozomi — 22 de perechi de cromozomi autozomali și o pereche de cromozomi sexi (XX sau XY). Acesta este modul de condensed является генетической информацией для внутренних целей. O eroare în aranjarea cromozomilor implați în formarea primei celule fetale poate duce la prezența de material генетический плюс. n cazul sindromului Edwards, este vorba despre prezența în plus a unui cromozom 18 întreg (sindrom complete) sau a unei porțiuni a acestuia (sindrom parțial) în toate celulele organulza, sau une numai.Așadar, în unele sau toate celulelele organului, vor exista cei doi cromozomi 18 normali, plus încă o copie, parțială sau completetă — de aceea sindromul Edwards mai este numit și trisomia 18 . Prezența acestui set de informații genetice în plus interferează cu dezvoltarea normală a fătului. Majoritatea fetușilor cu trisomie 18 nu supraviețuiesc până la naștere. Incidența maladiei în populație este de 1 / 6.000–1 / 8.000 ноу-născuții vii , найти доуа трисомия ca și frecvență, după sindromul Down.Această mutație Genetică определяет аномалии в функциональной области inimii, rinichilor, oaselor, sistemului nervos și abilităților когнитивные. Fiind o afecțiune foarte severă, doar 5-10% dintre sugarii diagnostics cu sindrom Edwards trăiesc mai mult de un an. Nu există un tratament specific pentru această boală, dar screeningul prenatal este o opțiune cu disponibilitate largă și eficiență crescută. Trisomia 18 poate fi complete (94% dintre cazuri) sau sub formă de mozaicism генетический , atunci când doarnele celule для организма sunt afectate, cu fenotip de severitate variabilă (mai puțin de 5% dintre cazuri).Около 2% dintre cazurile de sindrom Edwards sunt repzentate de prezența в плюс сегмент unui al cromozomului 18. Cel mai frecvent, cromozomul în plus este de origine maternă i este prezent datorită un erori de disjuncie II . средний возраст матери для этого расстройства 32½
Внешние ссылки []
Синдромул Эдвардс (трисомия 18)
Cauze și factori de risc