Хромосомные болезни(геномные и структурные) Синдромы Дауна, Патау,Эдвардса. Регулярные трисомии. Моносомия. Синдром кошачьего крика.
⇐ ПредыдущаяСтр 11 из 20Следующая ⇒
Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом. К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом.
Синдром Дауна- одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями
Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала 21-й хромосомы, либо целой хромосомы (трисомия), либо её участков (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от количества дополнительного генетического материала, генетического окружения и чистой случайности.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка[7].
Диагностика
Клинодактилия
Беременная женщина может пройти обследование на выявление нарушений плода. Многие стандартные дородовые обследования способны обнаружить синдром Дауна у плода. Например, имеются специфические УЗИ-признаки синдрома. Генетические консультации с генетическими тестами (амниоцентез, биопсия хориона, кордоцентез), как правило, предлагаются семьям, риск рождения в которых ребёнка с синдромом Дауна наиболее велик.
Генетические консультации с генетическими тестами (амниоцентез, биопсия хориона, кордоцентез), как правило, предлагаются семьям, риск рождения в которых ребёнка с синдромом Дауна наиболее велик.
Амниоцентез и биопсия хориона считаются инвазивными обследованиями, так как при них в матку женщины вводят различные инструменты, что несёт в себе некоторый риск повреждения стенки матки, плода или даже выкидыша.
Несколько неинвазивных пренатальных тестов, которые включают секвенирование фрагментов ДНК плода в материнской крови, были разработаны.
На данный момент амниоцентез считается самым точным обследованием. Для получения результатов у женщины требуется взять на анализ амниотическую жидкость, в которой позже выявляют клетки плода. Лабораторные работы могут занять несколько недель, но вероятность правильного результата — 99,8 %. Ложноположительный показатель очень низок.
Характерные черты, обычно сопутствующие синдрому Дауна
Обычно синдрому Дауна сопутствуют следующие внешние признаки (согласно данным из брошюры центра «Даунсайд Ап»):
«плоское лицо» — 90 %
брахицефалия (аномальное укорочение черепа) — 81 %
кожная складка на шее у новорожденных — 81 %
эпикантус (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) — 80 %
гиперподвижность суставов — 80 %
мышечная гипотония — 80 %
плоский затылок — 78 %
короткие конечности — 70 %
брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) — 70 %
катаракта в возрасте старше 8 лет — 66 %
открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65 %
зубные аномалии — 65 %
клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
аркообразное нёбо — 58 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая также «обезьяньей») — 45 %
короткая широкая шея — 45 %
ВПС (врождённый порок сердца) — 40 %
короткий нос — 40 %
страбизм (косоглазие) — 29 %
деформация грудной клетки, килевидная или воронкообразная — 27 %
пигментные пятна по краю радужки = пятна Брушфильда — 19 %
эписиндром — 8 %
стеноз или атрезия двенадцатиперстной кишки — 8 %
врождённый лейкоз — 8 %.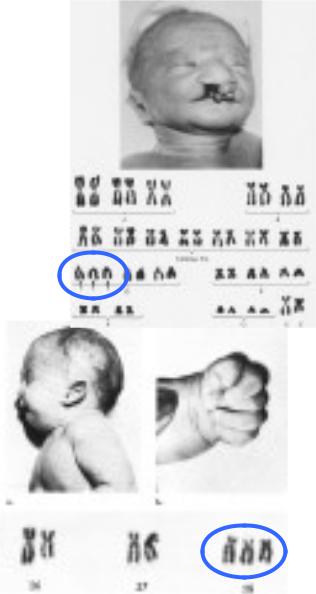
Точная диагностика возможна на основании анализа крови на кариотип. На основании исключительно внешних признаков постановка диагноза невозможна.
Синдром Пата́у (трисомия 13) — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13.
Проявления заболевани
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжёлыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врожденных пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
Синдром Эдвардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
Причины заболевания[править | править исходный текст]
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Содержание [убрать]
1 Причины заболевания
2 Проявления синдрома
3 Прогноз
4 Частота появления
5 Вариации
6 См. также
7 Ссылки
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма.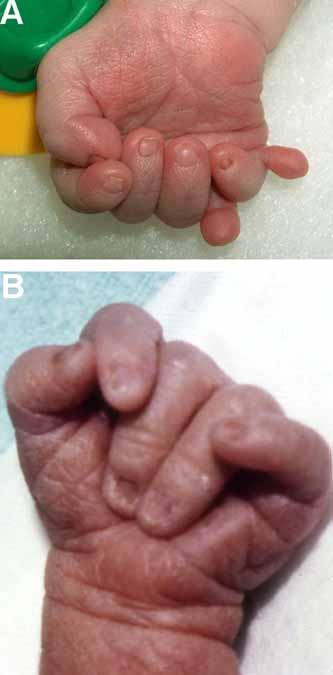 Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
Дети с трисомией 18 рождаются с низким, в среднем 2177 г. весом. При этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
ПрогнозПродолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появленияЧастота появления синдрома Эдвардса составляет ~ 1:3000 зачатий и 1:6000 рождений живых детей. Хотя женщина в 20 или 30 лет также может родить ребёнка с синдромом Эдвардса, риск рождения больного ребёнка увеличивается с возрастом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия.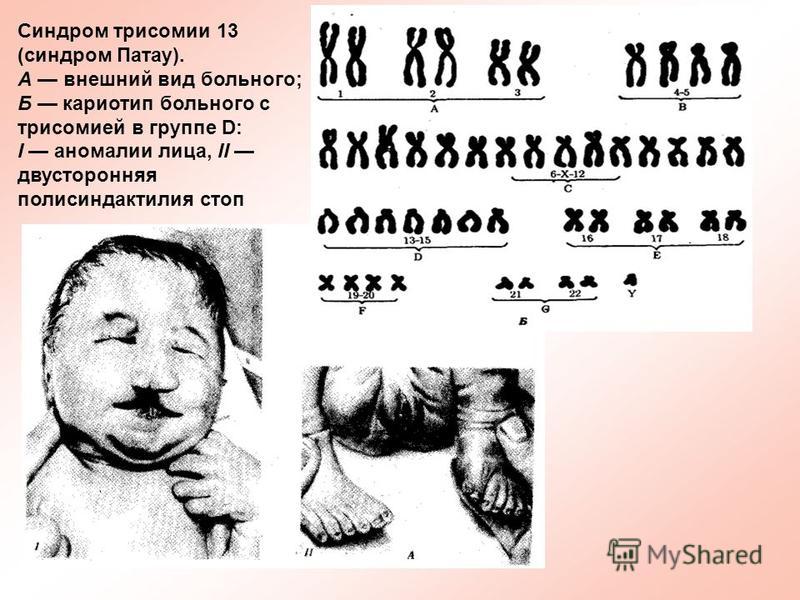 наследуются по аутосомно-рецессивному типу (ФКУ, тирозиноз, цистиноз и др.).
наследуются по аутосомно-рецессивному типу (ФКУ, тирозиноз, цистиноз и др.).
Фенилкетонурия — врождённое заболевание, вызванное нарушением перехода фенилаланина в тирозин и приводящее к задержке психического развития. неврологические и психические расстройства-• Умственная отсталость (олигофрения, идиотия или имбецильность, глубокая психическая инвалидность).• При отсутствии лечения коэффициент интеллектуального развития за каждые 10 недель снижается на 5 пунктов.• Повышенная психомоторная возбудимость в детстве• Специфическая походка• специфическая осанка и поза при сидении• Необычное положение конечностей («поза портного»).• Стереотипные движения• Повышение сухожильных рефлексов• Судороги• Раннее закрытие большого родничка• МикроцефалияИзменения кожи-• Гипопигментация кожи и волос• Сухость• Экзема• Дерматит• СклеродермияДругие клинические проявления• Рвота в периоде новорождённости (настолько сильная, что может имитировать пилоростеноз)• Светлые радужки, катаракта• Специфический «мышиный», «заплесневелый» запах тела и мочиВ клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония, спастический тетрапарез. Течение болезни прогрессирующее и нередко приводит к смерти в 2- 3- летнем возрасте.Клиническая картина ФКУ III напоминает ФКУ II и включает тяжелую умственную отсталость, микроцефалию, спастический тетрапарез.ЛечениеРежим амбулаторный, госпитализация показана для коррекции диеты в случае нестабильной концентрации фенилаланина плазмы.При атипичных формах фенилкетонурии (2 и 3) даже строгое соблюдение диеты с низким содержанием фенилаланина плазмы не приводит к предупреждению тяжёлых неврологических нарушений.Главным способом лечения ФКУ является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности.Диета с резким ограничением содержания фенилаланина вводится с момента подтверждения диагноза классической ФКУ. Лекарственная терапия• Ноотропные препараты• Белковые гидролизаты и аминокислотные смеси• При атипичных формах может быть эффективно введение дигидробиоптерина внутрь• Необходима дополнительная коррекция вторичных нейромедиаторных расстройств.
Течение болезни прогрессирующее и нередко приводит к смерти в 2- 3- летнем возрасте.Клиническая картина ФКУ III напоминает ФКУ II и включает тяжелую умственную отсталость, микроцефалию, спастический тетрапарез.ЛечениеРежим амбулаторный, госпитализация показана для коррекции диеты в случае нестабильной концентрации фенилаланина плазмы.При атипичных формах фенилкетонурии (2 и 3) даже строгое соблюдение диеты с низким содержанием фенилаланина плазмы не приводит к предупреждению тяжёлых неврологических нарушений.Главным способом лечения ФКУ является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности.Диета с резким ограничением содержания фенилаланина вводится с момента подтверждения диагноза классической ФКУ. Лекарственная терапия• Ноотропные препараты• Белковые гидролизаты и аминокислотные смеси• При атипичных формах может быть эффективно введение дигидробиоптерина внутрь• Необходима дополнительная коррекция вторичных нейромедиаторных расстройств. наследуются по аутосомно-рецессивному типу (ФКУ, тирозиноз, цистиноз и др.).
наследуются по аутосомно-рецессивному типу (ФКУ, тирозиноз, цистиноз и др.).
Фенилкетонурия — врождённое заболевание, вызванное нарушением перехода фенилаланина в тирозин и приводящее к задержке психического развития. неврологические и психические расстройства-• Умственная отсталость (олигофрения, идиотия или имбецильность, глубокая психическая инвалидность).• При отсутствии лечения коэффициент интеллектуального развития за каждые 10 недель снижается на 5 пунктов.• Повышенная психомоторная возбудимость в детстве• Специфическая походка• специфическая осанка и поза при сидении• Необычное положение конечностей («поза портного»).• Стереотипные движения• Повышение сухожильных рефлексов• Судороги• Раннее закрытие большого родничка• МикроцефалияИзменения кожи-• Гипопигментация кожи и волос• Сухость• Экзема• Дерматит• СклеродермияДругие клинические проявления• Рвота в периоде новорождённости (настолько сильная, что может имитировать пилоростеноз)• Светлые радужки, катаракта• Специфический «мышиный», «заплесневелый» запах тела и мочиВ клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония, спастический тетрапарез. Течение болезни прогрессирующее и нередко приводит к смерти в 2- 3- летнем возрасте.Клиническая картина ФКУ III напоминает ФКУ II и включает тяжелую умственную отсталость, микроцефалию, спастический тетрапарез.ЛечениеРежим амбулаторный, госпитализация показана для коррекции диеты в случае нестабильной концентрации фенилаланина плазмы.При атипичных формах фенилкетонурии (2 и 3) даже строгое соблюдение диеты с низким содержанием фенилаланина плазмы не приводит к предупреждению тяжёлых неврологических нарушений.Главным способом лечения ФКУ является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности.Диета с резким ограничением содержания фенилаланина вводится с момента подтверждения диагноза классической ФКУ. Лекарственная терапия• Ноотропные препараты• Белковые гидролизаты и аминокислотные смеси• При атипичных формах может быть эффективно введение дигидробиоптерина внутрь• Необходима дополнительная коррекция вторичных нейромедиаторных расстройств.
Течение болезни прогрессирующее и нередко приводит к смерти в 2- 3- летнем возрасте.Клиническая картина ФКУ III напоминает ФКУ II и включает тяжелую умственную отсталость, микроцефалию, спастический тетрапарез.ЛечениеРежим амбулаторный, госпитализация показана для коррекции диеты в случае нестабильной концентрации фенилаланина плазмы.При атипичных формах фенилкетонурии (2 и 3) даже строгое соблюдение диеты с низким содержанием фенилаланина плазмы не приводит к предупреждению тяжёлых неврологических нарушений.Главным способом лечения ФКУ является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности.Диета с резким ограничением содержания фенилаланина вводится с момента подтверждения диагноза классической ФКУ. Лекарственная терапия• Ноотропные препараты• Белковые гидролизаты и аминокислотные смеси• При атипичных формах может быть эффективно введение дигидробиоптерина внутрь• Необходима дополнительная коррекция вторичных нейромедиаторных расстройств.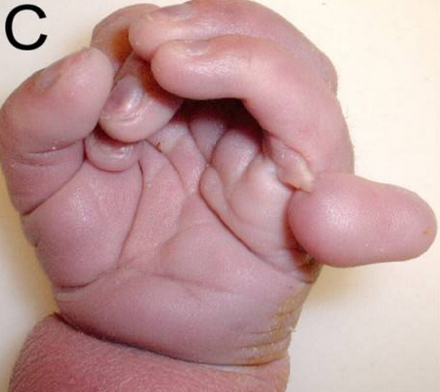 В связи с этим целесообразно в комплексе лечения детей включать лекарства с промедиаторным действием (ДОФА, наком, мадопар) Обычно, для выявления ФКУ используют высокоэффективную жидкостную хроматографию (HPLC), но в некоторых клиниках еще используют тест Гатри (который ранее применялся в рамках национальной программы биохимического скрининга).
В связи с этим целесообразно в комплексе лечения детей включать лекарства с промедиаторным действием (ДОФА, наком, мадопар) Обычно, для выявления ФКУ используют высокоэффективную жидкостную хроматографию (HPLC), но в некоторых клиниках еще используют тест Гатри (который ранее применялся в рамках национальной программы биохимического скрининга).
Читайте также:
1я беременность ( Синдром Патау у плода)
Привет всем! Хочу рассказать о своей истории.
Начну с того, что муж ходил в море, долгие разлуки, а потом жаркие встречи. Поженились в апреле 2014, и беременность не наступали перед отходом в море. в августе с сестрой поехали в Египет на 2 недели. там было пищевое отравление у меня, пила антибиотик местный, т.к наши таблетки не помогали. Вернулись домой, и бац сбился цикл. Пошла к Г, она отправила на анализы на гормоны. Сдала, оказались нарушения.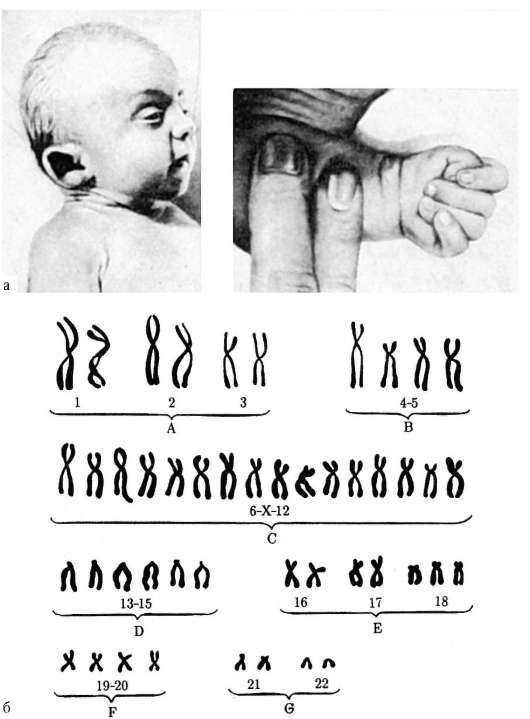 Она выписала мне дюфастон. Выравняешь цикл. Я его не пила, побоялась. Цикл сам собой стабилизировался сразу. Муж приехал в ноябре, мы к врачу, что хотим детей. Она говорит, что сбой продолжается. Пьем дюфастон по 2 т /2 раза в день с 16 дц 10 дней. После окончания ждем месячных и в первый день пьем контрацептивы Димиа 6 мес., а только потом будем планировать ляльку. мужа на спермограмму отправила, я вышла в слезах, расстроенная, что сейчас муж опять в море, а детей нет. Он анализы отказался сдавать. Говорит, если сперматозоиды живые, то всё будет. Нет — так потом переживать начнем. Мы сняли гостиницу у нас в Зелике, расслабились, забыли обо всем.
Она выписала мне дюфастон. Выравняешь цикл. Я его не пила, побоялась. Цикл сам собой стабилизировался сразу. Муж приехал в ноябре, мы к врачу, что хотим детей. Она говорит, что сбой продолжается. Пьем дюфастон по 2 т /2 раза в день с 16 дц 10 дней. После окончания ждем месячных и в первый день пьем контрацептивы Димиа 6 мес., а только потом будем планировать ляльку. мужа на спермограмму отправила, я вышла в слезах, расстроенная, что сейчас муж опять в море, а детей нет. Он анализы отказался сдавать. Говорит, если сперматозоиды живые, то всё будет. Нет — так потом переживать начнем. Мы сняли гостиницу у нас в Зелике, расслабились, забыли обо всем.
Должны прийти «дела», а начинается мазня коричневая. Тянет низ живота. Я контрацептивы не пью, потому, что думаю, что это ещё не месячные. проходит 3 дня, я по магазинам пошла, и понимаю, что на меня ни один бюстгальтер не налазит! Грудь увеличилась в 2 раза.
И тут я подумала, а может беременна?
Делаю тест: одна полоска яркая, вторая бледная.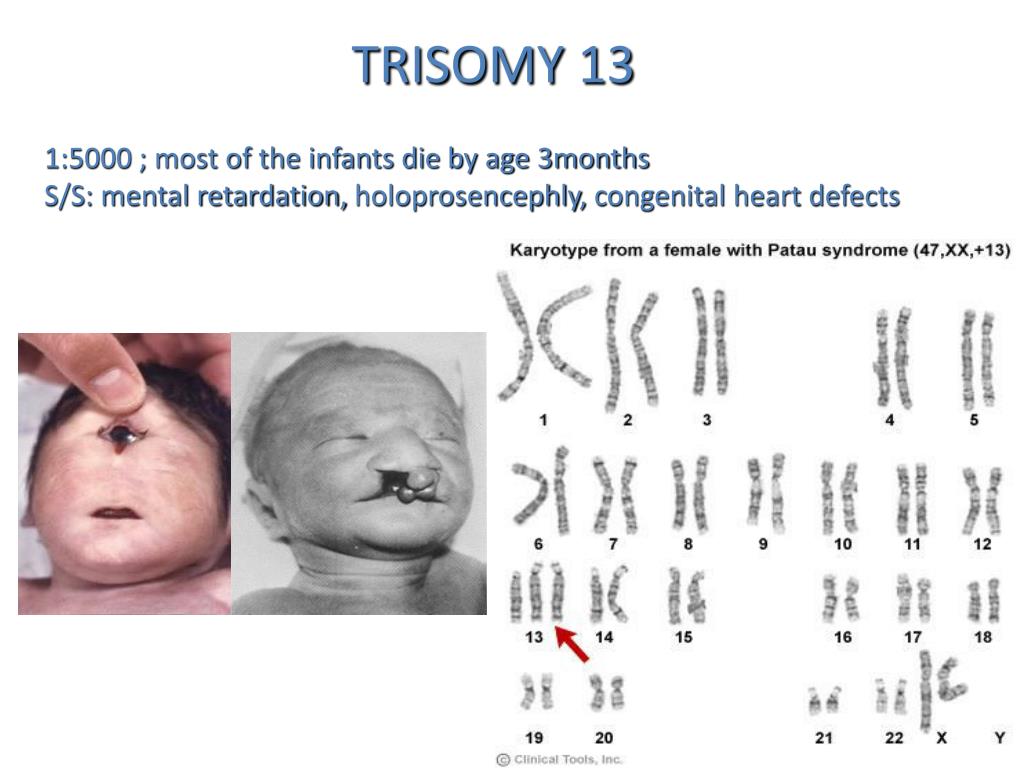 Я в испуге, еду к Г без записи. Она смотрит на УЗИ и говорит: ПОЗДРАВЛЯЮ!!! Срок 2 недели что ли. Это было 8 декабря 2014.
Я в испуге, еду к Г без записи. Она смотрит на УЗИ и говорит: ПОЗДРАВЛЯЮ!!! Срок 2 недели что ли. Это было 8 декабря 2014.
Моему счастью не было границ!!!
Но сказала, что тонус. Пьем дальше дюфастон, и свечи папаверин на ночь. Настраиваемся на беременность и снимаем каблуки)Явка перед НГ чтобы подтвердить беременность. Пришла, всё ок. Она сделал снимок. и отправила становится на учет до 12 недель.пришла в жен.консультацию на учет, всё оформили, гинеколог отправила на УЗИ узнать точный срок и сердцебиение проверить. 20.01.2015 было УЗИ. всё ок. 2,61 см и сердцебиение есть. Вроде была 7-8 неделя. Сдавала все анализы, которые просили. Всё было замечательно, до…
Записались на 1йскрининг в центр планирования. Меня очень долго смотрели. Позвали генетика на консультацию. Она сказала, что на моем сроке есть опред.маркеры и они должны соответствовать норме. так вот на УЗИ у плода носовая кость не вуализируется, воротниковое пространство ниже нормы, и сердцебиение большое. Сказали, что мои анализы крови ускорят и прийти на консультацию.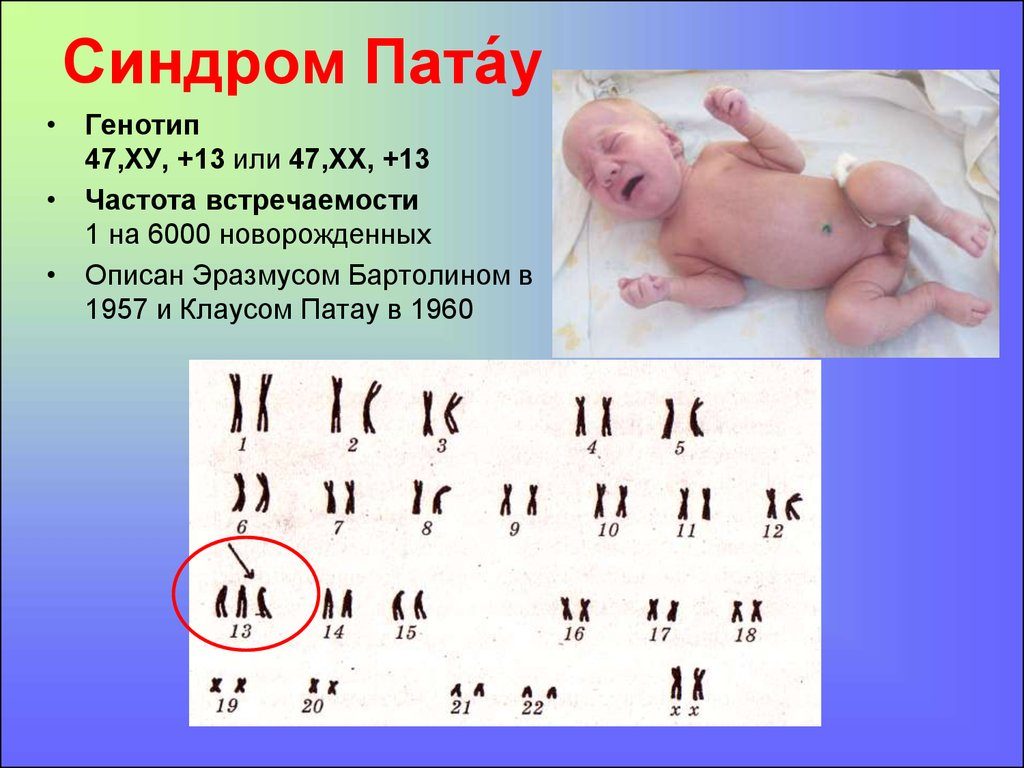 То. что творилось внутри меня не описать словами. Муж успокоил, сказал, всё хорошо будет. генетик мне подробно объяснила, что по анализам и УЗИ попадаю в зону риска, скорее всего синдром Пату у плода. т.е.лишняя 13я хромосома. Предложили подумать, и в понед.дать ответ, что я решила. Предложили сделать пункцию, взять ворсины хориона на анализ, чтобы подтвердить диагноз. Мы побежали по врачам, делать УЗИ ещё. Посоветовали клинику «Надежда». Мы приехали, я рассказала обо всём, посмотрели и сказали, что да есть дисфункции, четко выраженные. врач оказалась работником центра планирования, и настояла, чтобы сделали прокол живота. Я согласилась. Процедура не долгая была. Обезболили область живота. вставили иголку, длинную, тонкую трубочку. одни врач учила другую как правильно, но я не переживала. Мне скорей бы узнать ответ.
То. что творилось внутри меня не описать словами. Муж успокоил, сказал, всё хорошо будет. генетик мне подробно объяснила, что по анализам и УЗИ попадаю в зону риска, скорее всего синдром Пату у плода. т.е.лишняя 13я хромосома. Предложили подумать, и в понед.дать ответ, что я решила. Предложили сделать пункцию, взять ворсины хориона на анализ, чтобы подтвердить диагноз. Мы побежали по врачам, делать УЗИ ещё. Посоветовали клинику «Надежда». Мы приехали, я рассказала обо всём, посмотрели и сказали, что да есть дисфункции, четко выраженные. врач оказалась работником центра планирования, и настояла, чтобы сделали прокол живота. Я согласилась. Процедура не долгая была. Обезболили область живота. вставили иголку, длинную, тонкую трубочку. одни врач учила другую как правильно, но я не переживала. Мне скорей бы узнать ответ.
Позвонили. Подтвердили. Пол женский. Синдром Патау.
Я ревела, ревела, и ревела. сложно было прийти в себя. если бы синдром Дауна, а этот… я как почитала… и врачи настояли сделать аборт… т.к. 13 недель, если тянуть, то потом это роды… Муж успокаивал, свекровь… решили, что если подтвердился диагноз, зачем мучать бедного малыша и себя терзать. Мы решились на прерывание по мед.показаниям.
Сейчас, я бы шла до конца. Рожала. Тогда… это не передать словами, когда ребенок желанный, первый, в любящей семье, девочка….
Я не наговариваю на врачей. Но мы все люди и нам свойственно ошибаться. Я столько после читала форумов, где похожие жизненные ситуации, и девушки оставляли. Потом рождались здоровые дети… Они не исключения. Я понимаю, что наши врачи обязаны предупреждать о рисках, о самом страшном, но выбор за нами. Когда пришло вскрытие: Плод с дистрофией. И Всё.
Т.к. у меня маленький срок, это наверно сложно определить.
Я лежала в больнице с девочками здоровыми, благополучные семьи у всех. Никто не курит, не пьет. Синдром Патау, синдром Дауна, синдром Эдвардса, ребенок с пороком сердца, с пороком легких. Все диагнозы очень страшные. Только у меня был маленький срок, а остальные рожали(искусственные роды).
Мне никогда не было так страшно, так больно, так жалко всех нас….
Каждая пережила всё это по своему. Я сейчас держу связь, с некоторыми очень близко дружим. самое страшное, что когда девочки получили вскрытие плода (сроки 4-6 мес), то там не было подтверждающих слов о их диагнозах….
Не прошло и дня, чтобы я не вспоминала об этом.
Мы повенчались с мужем в июле этого года, в море он больше не ходит. Не представляю, как пережила бы это одна…
Я долго не решалась написать это. Но может кому-то моя история поможет. Умоляю вас девченки, идите до последнего, едите в другие города, делайте УЗИ. И непрестанно молитесь БОГУ о спасении.
Я молюсь каждый день, стала ходить в церковь. батюшка сказал, что нужно чаще нам исповедоваться и причащаться. И что все наши молитвы будут услышаны. Молюсь за всех девушек, кто планирует лялек!!! Девченки! Мы дождемся своих // полосочек!
Также интересно про синдром Патау
Синдром Эдвардса
Впервые
синдром был описан в 1960 г.
Частота
встречаемости
1:4500-6500 новорожденных. В основном
поражаются девочки (3:1). Большая часть
мальчиков погибает внутриутробно.
Причина:
трисомия по 18-й паре хромосом.
Клиника:
при доношенной беременности больные
рождаются с малой массой (обычно до
2500г). Новорожденные ослабленные, имеют
скошенный подбородок за счет недоразвития
нижней челюсти, выступающий затылок,
низко посаженные деформированные уши.
Отмечаются также аномалии скелета:
пятый палец накладывается на четвертый,
а второй — на третий, короткая грудина,
гипоплазия (недоразвитие) тазобедренных
суставов, стопа — «качалка» с
выступающей пяткой. Часто встречаются
пороки сердца и почек. Большая часть
больных погибает в первые 6 месяцев или
до 1 года. Только 1% таких больных доживает
до 10 лет, обнаруживая при этом глубокую
умственную отсталость.
Диагностика:
кариотипирование — лишняя 18-я хромосома,
47 + 18.
Дерматоглифика
— одна поперечная складка на ладони.
Синдром Патау
Впервые
был описан в 1960 г. группой ученых под
руководством К. Патау (Англия).
Частота
встречаемости
1:5000 – 1:7000 новорожденных, большая часть
погибает внутриутробно.
Причина:
трисомия по 13-й паре хромосом.
Клиника:
дети рождаются, как правило, преждевременно
и имеют множественные пороки развития:
расщелина мягкого и твердого неба,
недоразвитие глаз (микрофтальмия,
анофтальмия), недоразвитие мозга
(микроцефалия), атрофия обонятельных
долей мозга и зрительного тракта. В 77%
имеются дефекты сердца и мочеполовой
системы. Нередки судороги. Глубокая
умственная отсталость. Продолжительность
жизни меньше года. Однако некоторые
больные (15%) живут до 5 лет.
Диагностика:
кариотипирование – лишняя 13-я хромосома,
47
+ 13.
Дерматоглифика
— увеличение угла atd
до 108°.
Хромосомные болезни, обусловленные гетероплоидией половых хромосом Синдром Клайнфельтера
Описан
Г. Клайнфельтером в 1942 г. (Англия).
Частота
встречаемости
1:1000 новорожденных, по некоторым данным
1:500.
Причина:
лишняя одна или более Х-хромосом в
кариотипе мужчины.
Клиника:
после периода полового созревания
евнухоидное телосложение (узкая грудная
клетка, непропорционально длинные
нижние конечности, скудный рост волос
на лице). Отсутствие сперматогенеза
и бесплодие. Снижение интеллекта от
средней степени до глубокой дебильности.
Встречаются также лица с практически
нормальным интеллектом, но они
неинициативны и малоспособны к творческой
деятельности. Замечено, что глубина
умственной отсталости усиливается с
увеличением количества лишних Х-хромосом
(две или три лишние X-хромосомы).
В детском возрасте до периода полового
созревания у этих больных отмечается
лишь снижение интеллекта, пониженная
жизнеспособность и нарушенная
коммуникабельность.
Диагностика:
определение полового Х-хроматина
показывает присутствие в соматических
клетках телец Барра; кариотипирование
– одна или более лишние Х-хромосомы(47,
XXY;
48, XXXY).
Синдром лишней y-хромосомы
Впервые
эта аномалия была описана в 1962 г. Хаушком.
Частота
встречаемости
1:1000 новорожденных.
Клиника:
мужчины высокого роста (в среднем рост
равен 186 см), иногда имеют место черты
акромегалоидности – несколько увеличенная
нижняя челюсть. Интеллект бывает либо
нормальным, либо незначительно сниженным.
Лица с данным синдромом часто встречаются
среди заключенных, поскольку при
соответствующих условиях склонны к
асоциальным поступкам, излишне агрессивны.
Репродуктивная функция у них в основном
не страдает, их дети обычно имеют
нормальный кариотип. Однако у их младенцев
следует отметить повышенную внутриутробную
смертность. В отдельных случаях были
описаны сыновья ХYY
от отцов XYY.
Диагностика:
определение у -хроматина флюоресцентным
методом; кариотипирование — одна или
более лишние Y-хромосомы(47,
XYY)
40. Хромосомные болезни, обусловленные аномалиями аутосом: синдромы Дауна, Эдвардса, Патау.
синдром
Патау-Трисомия 13 (СП). Синдром П выделен
в самостоятельную нозологическую форму
в 1960 году в результате генетического
исследования, проведенного у детей с
врожденными пороками развития. Частота
СП среди новорожденных равна 1:5000 –
1:7000. Цитогенетические варианты этого
синдрома: простая полная трисомия 13 как
следствие нерасхождения хромосом в
мейозе у одного из родителей (гл. образом
у матери) встречается у 80-85% больных.
Остальные случаи обусловлены в основном
передачей дополнительной хромосомы
(ее длинного плеча). В робертсоновских
транслокациях типа D/13,G/13.
Обнаружены и другие цитогенетические
варианты (мозаицизм, изохромосома,
неробертсоновские транслокации), но
они встречаются крайне редко. Клиническая
и патологоанатомическая картина простых
трисомных форм и транлокационных не
различаются. Соотношения полов при СП
близко к 1:1. Дети с СП рождаются с истинной
пренатальной гипоплазией, которую
нельзя объяснить небольшой недоношенностью.
Характерное осложнение беременности
при вынашивании плода с СП — многоводие:
оно встречается в 50% случаев СП. Для СП
характерны множественные врожденные
пороки развития головного мозга и лица.
Это патагеническая единая группа ранних
нарушений формирования головного мозга,
глазных яблок, мозговой и лицевой части
черепа. Окружность черепа обычно
уменьшена, встречается и тригоноцефалия.
Лоб скошенный, низкий; глазные щели
узкие, переносье запавшее, ушные раковины
низко расположены и деформированы.
Типичный признак СП – расщелины верхней
губы и неба. Всегда обнаружатся пороки
нескольких внутренних органов в разной
комбинации: дефекты перегородок сердца,
незавершенный поворот кишечника, кисты
почек, аномалия внутренних половых
органов, дефекты поджелудочной железы.
Как правило, наблюдается полидактилия
и флексорное положение кистей. Клиническая
диагностика СП основывается на сочетании
характерных пороков развития. При
подозрении на СП показано УЗИ всех
внутренних органов. В связи с тяжелыми
врожденными пороками развития большинство
детей с СП умирают в первые недели/месяцы.
Но некоторые живут несколько лет. Более
того, в развитых странах отмечается
тенденция увеличения продолжительности
жизни с СП до 5 лет. Дети с СП имеют
практически всегда глубокую идиотию.
Синдром
Эдвардса – трисомия 18. Почти во всех
случаях СЭ обусловлена простой трисомной
формой (гометическая мутация у одного
из родителей). Встречается и мозаичные
формы (не расхождение на ранних стадиях
дробления). Траслокационные формы крайне
редки и, как правило, это частичные, а
не полные трисомии. Клинических различий
между цитогенетически различающимися
формами трисомии нет. Частота СЭ
составляет1:5000 – 1:7000 новорожденных.
Соотношение мальчиков и девочек = 1:3.
При СЭ отмечается выраженная задержка
пренатального развития при полной
продолжительности беременности (роды
в срок). В первую очередь это множественные
врожденные пороки развития лицевой
части черепа, сердца, костной системы,
половых органов. Череп долихоцефалической
формы; нижняя челюсть и отверстие рта
маленькие; глазные щели узкие и короткие;
ушные раковины деформированы и низко
расположены. Из других внешних признаков
отмечается флексорное положение кистей,
аномально развитая стопа, первый палец
стоп короче второго. Спинномозговая
грыжа и расщелина губ встречаются редко.
Дети с СЭ умирают в раннем возрасте от
осложнений, обусловленных врожденными
пороками развития.
Синдром
Дауна. СД – хромосомная болезнь,
обусловленная трисомией по хромосоме
21, как правило, вследствие нарушения
восхождения хромосом во время мейоза
яйцеклетки. Частота 1:650 живорожденных.
Частота увеличивается с увеличением
возраста матери, что подтверждено
данными амниоцентеза. После 20 лет у всех
пациентов в головном мозге обнаруживают
бляшки, характерные для болезни
Альцгеймера. Монголоидный разрез глаз,
брахицефалия, эпикантус, мышечная
гипотония, макроглоссия, пятна Брашфильда
на радужке, аномалия ушей, сходящееся
косоглазие, широкая переносица, маленький
подбородок, короткая шея, врожденный
порок сердца, дерматоглифика,
четырехпальцевая ладонная складка,
отсутствие подошвенных завитков
(подушечки пальцев стопы). Сближение
2-3 сгибательных складок мизинцев. Стеноз
или атрезия 12перстной кишки. Отсутствие
заднепроходного отверстия. Болезнь
Гиршспрунга у 2-3% детей. Задержка
психомоторного развития. Повышенная
восприимчивость к инфекциям.
Синдром Эдвардса: диагностика, признаки, прогнозы
Хромосомные патологии относятся к одним из самых серьезных поломок в клетках, за счет удвоения одной из пар хромосом возникают особые синдромы, имеющие типичные внешние проявления и изменения метаболизма. Трисомия по одной из пар хромосом (когда вместо двух, имеется три хромосомы) встречается наиболее часто в трех вариантах – синдром Дауна, который наиболее распространен и известен, а также Патау и Эдвардса. Свои названия они получили за счет открытия и описания их учеными.
Оглавление: Общие данные История изучения патологии Что такое трисомия Причины развития синдрома Эдвардса Типы синдрома Эдвардса: как зависит тяжесть проявлений Основа патологии: в чем вина генов? Факторы риска синдрома Эдвардса Характеристики синдрома Эдвардса в период новорожденности Развитие детей: особенности синдрома Эдвардса Методы диагностики синдрома Эдвардса Лечение и прогнозы при синдроме Эдвардса
Общие данные
Синдром Эдвардса (или синдром трисомии 18) стоит на втором месте по встречаемости трисомий, после синдрома Дауна, и он возникает при нарушении деления 18-ой пары хромосом. Патология характеризуется различными нарушениями в формировании еще внутриутробно систем и органов плода, имеет внешние типичные изменения и неблагоприятные для здоровья и жизни прогнозы.
Важно
Дети относятся к категории тяжелых инвалидов, требуют к себе особого отношения, ухода и заботы родителей и медиков.
По распространенности данная хромосомная аномалия считается достаточно частой, до 0.01-0.02% детей, рожденных в мире, имеют данное хромосомное отклонение, и нет зависимости от определенной расы или страны проживания, встречается повсеместно. По данным статистики девочки страдают чаще мальчиков в 3-4 раза, хотя аргументированных научных данных этому феномену не найдено. Точная причина формирования трисомии у плода не определена, но выделен ряд определенных факторов риска, которые повышают вероятность ее развития.
Наряду с другими хромосомными и генными поломками, на сегодняшний день синдром Эдвардса относят врожденным и неизлечимым патологиям, методы особого ухода и медикаментозная поддержка, реабилитационные мероприятия позволяют определенным образом поддерживать жизнь детей и формировать некоторые успехи в его развитии — физическом и психическом. Но в силу резкого разнообразия нарушений, которые придает лишняя хромосома, единых методик лечения и ухода нет.
История изучения патологии
В медицинских трактатах и художественной литературе можно найти единичные сведения о больных данным синдромом еще в древности, но описан подробно синдром был в начале прошлого века. Развитие технологий того времени и ранее не позволяло подробно изучить данную патологию, поэтому причин ее не знали. Кроме того, большинство рожденных с этим синдромом детей гибло в раннем возрасте в силу слабого развития медицины и своевременного оказания им помощи. Причина синдрома – трисомия по 18-ой паре хромосом была выявлена только в 1960 году Д. Эдвардсом, и его именем и была названа новая патология. По данным медицинской статистики встречаемость патологии в среднем составляет 1 случай на 3000 зачатий, но до родов остается гораздо меньше плодов. Это объясняется тем, что подобная аномалия нередко в самом начале заканчивается выкидышем или гибелью плода внутриутробно. Но нередко при таких выкидышах никаких хромосомных исследований не проводится.
Что такое трисомия
Если говорить с точки зрения медицины, трисомия является вариантом хромосомных мутаций, когда клетки пациента имеют не 46 хромосом собранных в гомологичные 23 пары, одна пара из которых определяет половые отличия, а выявляется 47 хромосом.
Сегодня врачи выделяют три типа трисомии, которые относятся к нелетальным мутациям – это синдром Дауна (страдает 21-ая пара), синдром Патау (страдает 13-я пара) и синдром Эдвардса (поражение выявляется по 18-ой паре). Все другие варианты трисомии изначально несовместимы с жизнью, такие зародыши сразу же гибнут и беременность не развивается. Но рождение детей с этими тремя трисомиями – это всегда проблемы их здоровья, физического развития и интеллекта, психических функций.
Причины развития синдрома Эдвардса
Как и две других патологии, указанные выше, синдром Эдвардса относят к генетическим патологиям, при которых обнаруживается дополнительная хромосома в аутосомах (не половые хромосомы). У здорового человека 46 хромосом – 22 пары – аутосомы (неполовые), и одна пара – половые, определяющие – девочка это или мальчик. В хромосомах компактно упакованы нити ДНК, в которой записана вся генетическая информация об организме, начиная с процесса его зачатия и до смерти. Вся программа жизни записана именно в нитях ДНК.
При синдроме Эдвардса организм страдает не от генных дефектов, а хромосомных, возникает дефект в делении половой клетки, которая затем дает ребенку жизнь, и в ней остается лишняя хромосома. При классическом варианте формирования синдрома Эдвардса имеется удвоение 18-ой хромосомы, и родиться могут с подобным синдромом как мальчик, так и девочка. Во всех клетках такого ребенка существует избыток генетической информации. Подобная избыточность ДНК приводит к целому ряду нарушений, которые приводят как к внешним дефектам, так и проблемам в работе систем и органов. Из-за наличия третьей, лишней хромосомы и возникло научное название – трисомия.
Типы синдрома Эдвардса: как зависит тяжесть проявлений
Исходя из того, какого рода хромосомный дефект имеется при синдроме, выделяют три типа течения:
- Полная трисомия по 18-ой паре.
- Частичная трисомия по 18-ой паре
- Мозаичная форма патологии.
При полной форме (она же относится к классическому синдрому) предполагается, что совершенно во всех клетках имеется дополнительная хромосома. Подобный вариант наиболее тяжелый и встречается наиболее часто, прогнозы при нем самые неблагоприятные.
Частичная трисомия относится к редким вариантам патологии, возникает в 3% случаев, при такой проблеме содержится не полная третья хромосома, а только ее кусок. Подобный дефект может образовываться в результате проблем с делением клеток (в особенности их хромосом), что возникает крайне редко. Может быть и вариант того, что лишняя часть 18-ой хромосомы крепится к отдельным молекулам ДНК, приводя к их удлинению и изменению. В таких клетках потом остается две 18-ых хромосомы и часть лишнего материала. Тогда врожденный синдром будет не таким тяжелым, так как в клетках появляется дополнительная информация не всей информации, а только ее части. Прогноз при таком состоянии хотя и неблагоприятный, но течение синдрома гораздо легче, чем при полной форме.
Развитие мозаичной формы проявляется примерно у 5-8% детей с подобной аномалией. Но по механизмам подобный тип синдрома отличается. Интересно, что подобная форма развивается после того, как сперматозоид сливается с яйцеклеткой, и обе получившиеся клетки изначально обладают нормальным набором хромосом – 46ХХ или 46XY. Но при первых же делениях в образованной зиготе образовался сбой и одна из клеток приобрела в силу этого дополнительную хромосому. В результате зародыш приобрел клетки с обычными хромосомами – их 46, другая же часть из них имеет лишнюю хромосому – то есть 47. Количество клеток с лишней хромосомой при таких проблемах не превысит 50%.
Обратите внимание
Чем выше количество дефектных клеток у зародыша и ребенка, тем хуже прогнозы, и зависит их объем от того времени, когда произошел сбой при делении. Чем позже это произошло, тем меньший объем дефектных клеток имеет кроха, и снижено количество проблем развития.
Данная форма получила название мозаичной в силу того, что нормальные клетки перемешаны с дефектными. Атипичные клетки с лишней хромосомой разбросаны хаотично по всему организму, и нет способа, чтобы можно было их каким-либо образом удалить. Эта форма протекает несколько легче, длительность жизни выше.
Основа патологии: в чем вина генов?
Всего одна хромосома у ребенка имеет много серьезных и тяжелых последствий для здоровья и жизни. Это связано с тем, что во всех клетках имеется универсальная система считывания информации, и при делении клеток – удвоения той информации, что запрограммирована в молекулах ДНК. Если есть дефект хотя бы одного гена – это уже отражается на организме в виде какой-либо болезни или проблемы, а если имеется целый кусок лишней хромосомы или даже целая хромосома – это множественные пороки развития и проблемы здоровья.
На 18-ой хромосоме содержится до 2.5% всей генетической информации о работе организма, и нарушения, которые воспроизводятся за счет синдрома – очень значительные. Лишние гены на дефектной хромосоме дают начало синтезу лишних белков, что приводит к аномальному развитию и функционированию целого ряда органов и систем ребенка. При синдроме Эдвардса подвергаются поражению скелет, определенные зоны нервной системы, мочеполовая система и сердечно-сосудистая, они имеют как аномалии в строении, так и затем – по мере развития плода — в функционировании.
Соответственно – основная и единственная причина подобного синдрома – образование в половой клетке матери или отца лишней хромосомы. При производстве половых клеток происходит не обычное деление с последующим удвоением 23 пар хромосом до 46 штук (для каждой идентичная копия в пару), а всего 23 штук. Если в процессе деления 18-я пара по каким-то причинам правильно не разделилась, в половую клетку уйдет сразу две хромосомы, и будет в ней не 23 штуки, а 24-ре. Если такая клетка даст начало новой жизни (будь то папин сперматозоид или мамина яйцеклетка), это приведет к появлению ребенка с синдромом Эдвардса.
Факторы риска синдрома Эдвардса
Чтобы половые клетки родителей неправильно делились, и образовалась та, что имеет избыточный набор хромосом, необходим различные негативные влияния, как извне, так и внутри организма. Ведущими из них можно считать:
- возраст матери и отца к моменту зачатия. Есть реальные доказательства возрастного фактора на вероятность развития трисомии. По мере возраста число аномальных клеток в репродуктивной системе увеличивается, что связано с ослаблением контроля со стороны организма за процессами деления. Риск рождения детей с хромосомными патологиями после 30 лет увеличивается в 2-3 раза по сравнению с молодым возрастом, а после 40 лет шансы увеличиваются еще в 6-8 раз. Зависимость от возраста отца не так очевидна, но тоже вполне возможны хромосомные дефекты.
- наличие вредных привычек, особенно если это никотиновая и алкогольная зависимость. Привычные интоксикации нарушают процессы деления клеток, в том числе и в области половой сферы, что приводит увеличению риска дефектных спермиев и яйцеклеток в несколько раз. Еще опаснее прием наркотических препаратов и токсикомании, они еще сильнее нарушают процессы клеточного деления.
- прием некоторых лекарственных средств, влияющих на процессы клеточного деления, превышение дозировок и длительности приема, неблагоприятные сочетания препаратов при лечении, формирующие токсические их концентрации.
- инфекции соматические и половые, которые приводят к поражению репродуктивных органов и клеток, формируют хроническое течение, влияют на процессы деления (цитомегалия, герпес, папиллома и другие).
- профессиональные вредности, облучения разного типа – ионизирующие, магнитные и многие другие. Они влияют на процесс деления клеток, обладают повреждающим действием в отношении ДНК, приводят к образованию в клетках свободных радикалов и мутациям.
Обратите внимание
Окончательные причины и все факторы риска, которые приводят к образованию клеток с трисомией, могут быть выяснены по мере открытий молекулярной биологии и генетики. Все перечисленные факторы только увеличивают риски, но не говорят о том, что обязательно родится такой ребенок.
Не исключена и предрасположенность к образованию трисомии в половых клетках, что доказывается тем фактором, что при рождении в семье одного ребенка с синдромом Эдвардса, шансы на рождение еще одного малыша повышаются в 200 раз в сравнении со среднестатистическими – с 0.04% до 2-3% и более.
Характеристики синдрома Эдвардса в период новорожденности
Хотя сегодня существует возможность диагностики синдрома до рождения, зачастую патологию обнаруживают уже при родах. Новорожденный с подобной аномалией хромосом имеет типичные и ярко выраженные пороки в развитии, их наличие может помочь в постановке диагноза даже без дополнительных исследований хромосом – генетического анализа (но его проводят в любом случае, без этих данных диагноз не окончательный).
При рождении выявляются:
- аномалии в строении формы черепа. Скелет при подобном синдроме страдает наиболее выражено – это проявляется в изменении прежде всего формы черепа с образованием долихоцефалии (резко вытянутый, узкий череп – «башенный»). Подобная аномалия не типична только для этой патологии, но указывает на имеющиеся отклонения в развитии. Наиболее выражено изменение соотношений по данным измерений в области теменных костей и длины от переносицы до затылочных бугров. Более тяжелой и выраженной аномалией черепа может стать микроцефалия, непропорционально маленький размер мозгового черепа относительно тела.
- специфически измененная форма ушной раковины. Аномальное строение ушных раковин у новорожденных выражено сильно и наблюдаются у 95% малышей, особенно при полном типе синдрома. Расположены ушные раковины гораздо ниже обычного, относительно глазниц они резко опущены, хрящи выпуклые и нет четко сформированных завитков. Мочка и козелок в зачаточном состоянии или полностью отсутствуют, слуховой проход резко сужается и у четверти детей может отсутствовать вовсе.
- аномальное строение неба с нижней челюстью (формирование микрогнатии). У новорожденных, когда подозревают синдром Эдвардса, не имеется полного срастания небных отростков в зоне верхней челюсти, что образуют продольную щель. Могут быть варианты с тотальным незаращением только мягкого неба или частичным его сращением, иногда с переходом на губы. При полной форме порока незаращение может быть двустороннее, верхняя губа расщеплена с двух сторон, дает начало патологическим расщелинам в челюстях и небе, что не дает ребенку полностью закрывать рот. В случае тяжелых патологий возможны сообщения между ротоглоткой и носовой полостями, и в том случае, если рот закрыт. Частота таких аномалий достигает 20-25% и выше.
- Могут возникать и другие проблемы – готическое небо (чрезмерно высокий свод костей) и изменения в строении нижней челюсти. Их регистрируют примерно у 70% детей. Типична микрогнатия – сильная втянутость подбородка и нижней челюсти назад, что не дает младенцам полностью закрывать рот и слюна подтекает изо рта. При подобной аномалии возникают трудности с кормлениями, особенно прикладыванием к груди.
- деформация стоп с образованием «стопы-качалки», характерные аномалии длины пальцев конечностей. Изменение стопы типично для 75% и более детей на фоне данного синдрома, возникает изменение положения косточек – таранной, пяточной либо ладьевидной, что приводит к формированию сильно выдающейся пятки на фоне отсутствия свода стопы. Он может быть выгнут вперед, придавая стопам вид ножек от кресла-качалки. Также выявляются и диспропорции в длине пальцев на ногах, особенно большого. Он короче второго пальца на ноге, что видно при распрямленных пальчиках.
- кисти в флексорном положении и сращения между пальцами, особые дерматоглифические рисунки. Синдактилия (этот термин обозначает сращение пальцев) выявляют в 45% случаев, обычно это пальцы на ногах, но могут затрагиваться и руки. При легкой степени кожа создает перепонки, в тяжелых могут образовываться костные перемычки. Также могут быть флексорные аномалии кистей – положение пальцев в аномальном, постоянно напряженном положении, прикрывая мизинцем и большим пальцем все остальные, плотно прижатые к ладошке. Типичны подобные расстройства для 90% детей с синдромом.
- Кроме всего прочего, при трисомии типичны особые узоры на пальцах и ладошках с особой формой кожных складок. При развитии этого синдрома отклонения типичны для 60% малышей. Это очень частые дуги на подушечках, отсутствие кожной складки между крайней и предпоследней фалангами, развитие поперечный бороздки – она имеет термин «обезьянья» линия.
- пороки гениталий. У мальчиков это недоразвитие пениса и мошонки, у девочек увеличение размеров клитора. Аномалии сочетаются обычно с пороками мочеполовой системы, но внешне остальные признаки не увидеть.
Помимо этих проявлений есть и более мелкие признаки и аномалии развития детей, которые могут в предварительном диагностировании синдрома. Важно отметить, что один из перечисленных признаков и даже два еще не говорят о диагнозе, при синдроме пороки множественные и имеют различные сочетания.
Важно
Большинство из описанных проявлений могут встречаться как единичные варианты и диагноза не подтверждают, они могут быть изолированным вариантом отклонений в течении беременности или проявлением иных патологий. Диагноз только по внешним порокам ставить нельзя!
Более того, на фоне неполного или мозаичного типов синдрома внешних признаков может быть немного – один-два, при этом есть серьезные пороки внутренних органов с частыми отклонениями в хромосомах. Именно они и определяют дальнейшие прогнозы на жизнь и течение синдрома.
Развитие детей: особенности синдрома Эдвардса
Только внешним видом при появлении на свет и проявлениями пороков в развития, аномалия Эдвардса не ограничивается, по мере роста и развития возможны проявления все новых и более тяжелых отклонений. Первые из них можно выявить через несколько недель с момента рождения. Нередко по ним впервые можно заподозрить диагноз при мозаичном и неполном типе патологии, если внешних дефектов мало или практически нет. Описанные выше пороки и внешние изменения становятся по мере роста более резкими и выраженными, постепенно добавляются новые проблемы и аномалии, которые при рождении не были заметны. Так, сюда стоит отнести:
- Резкое и выраженное отставание в уровне физического развития. При рождении масса таких детей не превышает 2-2.5 кг, и причиной тому являются хромосомные дефекты, которые не дают полноценно развиваться системам и органам. По мере развития показатели роста и массы тела по месяцам существенно отличаются от норм, также слабо прибывают и окружности груди. Размеры головы могут прибывать но норме или выше нее, что может указывать на скопление внутри полости черепа избытка жидкости, формируя гидроцефалию.
- Проблемы со стопами. По мере роста формируется косолапость в силу аномалий строения костей и связок, мышечного гипертонуса. Страдает и контроль за развитием стопы и тонусом от нервной системы. Это угрожает тем, что дети поздно начинают ходить или не могут этого делать вообще в силу общего тяжелого состояния. Внешне косолапость проявляется в выраженной деформации стопы и ненормальной ее установке в покое.
- Нарушение мышечного тонуса. При рождении гипертонус типичен для кистей, формируя их флексорное положение, но по мере развития распространяется на соседние групп мышц, приводя к вычурным позам. Но чаще тонус мышц снижен, дети вялые и атоничные, конечности весят плетьми. Могут также быть состояния дистонии с резким сокращением отдельных мышечных групп на фоне гипотонии других. При этом руки могут быть согнуты, а ноги – переразгибаются. Это приводит к нарушению координации и хаотичности движений, вывихам и ненормальному положению конечностей.
- Атипичные реакции нервной системы, проблемы эмоциональности. Некоторые отделы мозга у детей с синдромом Эдвардса недоразвиты, в связи с чем формируются проблемы эмоциональности и заторможенности, неадекватные реакции на окружающее. Это связано обычно с мозолистым телом и мозжечковыми гипоплазиями, что формирует также отставание умственного развития. Внешне это проявляется в отсутствующем взоре, неэмоциональности и невозможности установления контакта, отсутствии слежения за объектами. Дети не реагируют на звуки в силу проблем с ушами и нервной системой, подобные аномалии выявляют в первые же месяцы жизни.
В целом, в силу тяжелых и комбинированных поражений дети не доживают до периода совершеннолетия. Часть их при мозаичной форме может достигать возраста подростков с глубокими степенями имбецильности и внешними проявлениями пороков.
Методы диагностики синдрома Эдвардса
На сегодняшний день существует три типа диагностик синдрома, которые принципиально отличаются друг от друга. Учитывая тот факт, что патология неизлечима и летальна в раннем возрасте, важно обращать внимание на пренатальную диагностику, которая не допускает рождения детей с данными аномалиями. Полезными будут и консультации врачей-генетиков в семьях, где имели место случаи хромосомных патологий. Можно провести:
- Диагностику половых клеток еще до зачатия. К сожалению, подобный метод применим еще очень ограниченно и только в протоколах ЭКО при предварительном изучении половых клеток. Если же это естественное зачатие, врачи могут прогнозировать более высокие риски рождения таких детей, но это не более, чем прогнозы. Точно узнать – родится ли такой ребенок или нет – невозможно. Исследуется семейный анамнез с учетом всех имеющихся заболеваний, а также оцениваются факторы риска, а также проводится генетический анализ обоих родителей с применением сложной аппаратуры и анализов. За счет определения кариотипа родителей могут делаться определенные выводы.
- Выявление синдрома в период гестации. При развитии плода существует несколько методов прямого или косвенного подтверждения синдрома Эдвардса, изначально для этих целей применяют УЗИ-скрининг и анализы крови с изучением особых показателей, а при высоких рисках проводятся уже инвазивные процедуры – амниоцентез, либо кордоцентез и биопсия ворсинок хориона. Проводят скрининги в строго определенные сроки, когда изменение данных УЗИ и показателей крови будет наиболее значимым.
Важно
Получение непосредственно клеток плода или его крови позволяет провести исследование его хромосом и подтвердить или опровергнуть у него синдром Эдвардса. Это помогает в принятии решения о пролонгации или прерывании беременности по медицинским показаниям.
- Подтверждение синдрома после рождения. Его проводят по результатам внешних данных в комбинации с рядом генетических анализов, подтверждающих наличие трисомии по 18-ой паре хромосом. Помимо этого важно оценить состояние ребенка и дать прогнозы в отношении его жизни и состояния здоровья, чтобы понять, какой объем помощи ему требуется для выживания. Без медицинской помощи при подобном синдроме дети могут быстро погибать поле рождения в ближайшие недели. Для выявления пороков во внутренних органах проводят УЗИ и ЭХО-КГ, рентгенографию и ЭЭГ, целый ряд анализов крови и мочи, дополнительные исследования вплоть до МРТ и КТ.
Лечение и прогнозы при синдроме Эдвардса
К сожалению, данный синдром на сегодняшний день неизлечим, и врачи могут помочь ребенку только лучше справляться с различными проблемами за счет оперативных вмешательств и консервативной терапии. Обычно дети доживают до нескольких месяцев или года, только при частичной или мозаичной форме могут прожить более длительно. Гибель наступает от множественных пороков развития и аномалий в работе органов, а также присоединения сопутствующих осложнений. Радикально исправить дефекты хромосом в клетках тела ребенка на сегодняшнем этапе развития медицины невозможно.
Парецкая Алена, педиатр, медицинский обозреватель
8,373 просмотров всего, 4 просмотров сегодня
Загрузка…
Трисомий 13 и 18 лет. — Материнский возраст. (Синдром Патау и Эдварда)
1 Трисомии 13 и 18 (синдром Патау и Эдварда) Трисомия 21 (синдром Дауна) — наиболее частое хромосомное заболевание при рождении, которое подробно рассматривалось в предыдущих годовых отчетах 23.Другие относительно распространенные трисомии включают трисомию 13 (синдром Патау) и трисомию 18 (синдром Эдварда). Хотя эти два состояния не так распространены, как синдром Дауна, они по-прежнему представляют собой серьезные проблемы как для родителей, так и для служб здравоохранения. ЧТО ТАКОЕ ТРИСОМИЯ? Клетки человека обычно имеют 46 хромосом, которые можно сгруппировать в 22 пары аутосом (пронумерованные от 1 до 22 в порядке их размера) плюс пару половых хромосом (XX у женщин и XY у мужчин). Нормальная человеческая клетка называется диплоидной, поскольку она содержит по 2 набора каждой из 23 хромосом.Если это не так, и клетка содержит ненормальное количество хромосом, ее называют анеуплоидом. Трисомия подразумевает наличие дополнительного материала от конкретной хромосомы в дополнение к тому, что содержится в нормальной паре. Таким образом, общий хромосомный набор составляет 47 вместо 46. Таким образом, в случаях синдрома Патау имеется дополнительная хромосома 13 (рис. 27), а в случаях синдрома Эдварда — дополнительная хромосома 18 (рис. 28). В клетке может присутствовать дополнительный хромосомный материал. из-за наличия целой дополнительной хромосомы (настоящая трисомия) или в результате присоединения дополнительной части хромосомы к другим хромосомам в ядре (транслокация).Мозаицизм описывает ситуацию, когда одни клетки имеют хромосомные дефекты, а другие нормальны. В этой ситуации степень проблем зависит от относительных пропорций нормальных и аномальных клеток, которые могут различаться между органами одного и того же человека. Факторы риска Выявлен ряд факторов риска, которые являются общими для многих хромосомных заболеваний, включая трисомии 18 и 13. К ним относятся: — Возраст матери Воздействие окружающей среды, такое как ионизирующее излучение и некоторые тяжелые металлы.Недавние исследования выявили возможность того, что проживание рядом со свалками может также увеличить риск хромосомных нарушений (среди других врожденных дефектов) 24. В этой области необходимо провести гораздо больше работы, прежде чем риски станут яснее. Валлийские данные из базы данных CARIS показывают, как частота аномалий увеличивается с возрастом матери для синдромов Патау и Эдварда. Для сравнения также показаны показатели синдрома Дауна (Рисунок 29). Рисунок 27: хромосомы из случая трисомии 13 синдрома Патау Рисунок 28: хромосомы из случая трисомии синдрома Эдварда Ежегодные отчеты CARIS 1998 и Vrijheid M, Dolk H, Armstrong B, Abramsky L, Bianchi F et al.Хромосомные врожденные аномалии и проживание вблизи свалок опасных отходов. Lancet (2002) 26 января; 359 (9303):
2 20 Коэффициент трисомии 13 (Патау) на 1000 LB / SB / TOP Трисомия 18 (Эдвардс) Трисомия 21 (Даунс) 0 <возрастная группа матери Рисунок 29: Частота трисомии 13, 18 и 21 по матери Возрастная группа: данные CARIS СИНДРОМ ПАТУ ТРИСОМИЯ 13 Синдром Патау (рис. 30), как сообщается, встречается при 1: 4000–1: 10 000 живорожденных / мертворожденных, хотя эти цифры меняются по мере увеличения возможностей дородового обнаружения, что дает родителям выбор прерывания беременности. беременности.Больные младенцы, как правило, маленькие при рождении с микроцефалией, покатым лбом, лицевым дисморфизмом, дефектами глаз и аномально низко посаженными ушами. Другие особенности включают дряблую кожу на задней части шеи, гемангиомы, единственную ладонную складку, полидактилию, согнутые пальцы и заднюю выступающую часть пятки (ступни с качалкой). Ряд серьезных врожденных аномалий возникает в связи с трисомией 13, в том числе: голопрозэнцефалия (нарушение правильного разделения переднего мозга в 65% случаев) миеломенингоцеле (50% случаев) сколиоз - левая губа и / или нёбо (почти во всех случаях) Врожденные пороки сердца (80% случаев), особенно ДМЖП, ДМПП или декстрокардия Экзомфалос (10% случаев) Дефекты почек Дефекты мочеполовой системы Рис. 30: Новорожденный ребенок с трисомией 13 Перспективы для детей с трисомией 13 очень плохие.45% живорожденных младенцев умирают в неонатальном периоде, а к концу первого года жизни смертность составляет 75%. Те младенцы, которые выживают, страдают серьезной неспособностью к обучению, глухотой и нарушением зрения. Апноэ, трудности с кормлением, рефлюкс пищевода и медленный рост очень распространены у младенцев. CARIS получил сообщения о 36 случаях трисомии 13, завершившейся беременностью, что дает общий показатель 2,8 на 10 000 живорожденных и мертворожденных. Из них 4 были спонтанной гибелью плода. Из оставшихся 32 случаев 24 (75%) закончились прерыванием беременности и 8 родились живорожденными, из которых только 1 дожил до конца первого года жизни.Для 5 случаев CARIS подробности дополнительных аномалий неизвестны. Оставшийся 31 случай включал: 17 случаев (55%) с пороками сердца, включая 12 ДМЖП (39%), 2 ДМПП и 1 ОАП 6 случаев (19%) голопрозэнцефалии 17 случаев (55%) расщелины губы и / или неба 2 случаев (7%) дефекта нервной трубки 12 случаев (39%) с аномалиями почек или мочевыводящей системы -20 случаев (65%) с деформациями конечностей, в основном полидактилиями, 4 случая трахео-пищеводной атрезии / свища и 4 случая врожденной диафрагмальной грыжи Процент дополнительных аномалий в случаях CARIS обычно ниже, чем можно было ожидать.Это может быть связано с качеством данных с меньшим количеством связанных аномалий, зарегистрированных в случаях, которые не умерли и не подверглись вскрытию. 33
3 ТРИСОМИЯ СИНДРОМА ЭДУАРДА 18 Синдром Эдварда (рис. 31) встречается примерно у 2 из 1000 живорожденных или мертворожденных. Что касается трисомии 13, эта частота падает по мере улучшения антенатального обнаружения. Рисунок 31: Младенец с трисомией 18 с синдромом Эдварда Плоды, как правило, маленькие для гестационного возраста с плохой подвижностью.Многоводие, малая плацента и единственная пупочная артерия — общие черты беременности. После рождения младенцы, как правило, маленькие и слабые, издают слабый плач. У них проблемы с кормлением, и часто бывает неспособность нормально развиваться. У заболевших младенцев обычно есть микроцефалия, маленький рот и челюсть, лицевой дисморфизм, дефекты глаз и аномально низко посаженные уши. Избыточная кожа на задней части шеи, контрактуры суставов и многочисленные аномалии пальцев рук и ног. Сжатые кулаки с перекрытием указательных пальцев 3-го и 4-го пальцев почти характерны для этого состояния (рис. 32).У детей есть значительные трудности в обучении и задержка в развитии. Основные сопутствующие аномалии не так распространены, как при трисомии 13, но включают: врожденные пороки сердца (90% случаев), особенно ДМЖП, ДМПП и ОАП. Расщелина губы и / или неба Миеломенингоцеле (6% случаев) Лучевая аплазия (5-10% случаев) Дефекты почек и связанная с ними гипертензия — сколиоз 20-30% случаев умирают в течение месяца после рождения, и только 10% живорожденных детей доживают до 1 года жизни. Несмотря на это, некоторые дети, как сообщается, доживают до второго десятилетия.CARIS известно о 56 случаях трисомии 18 с завершением беременности, что дает общий показатель 4,4 на 10 000 живорождений и мертворожденных. Из них 5 были спонтанной гибелью плода. Из оставшегося 51 случая 31 (61%) закончился прерыванием беременности, 2 были мертворожденными и 18 живорожденными, из которых 6 (33% случаев живорожденных) дожили до конца первого года жизни. Для 5 случаев CARIS подробности дополнительных аномалий неизвестны. Остальные 51 случай включают: 33 случая (65%) с пороками сердца, включая 18 ДМЖП (35%), 5 ДМПП и 3 ОАП 6 случаев (12%) расщелины губы и / или неба 2 случая (4%) нервной трубки дефект -17 случаев (33%) с аномалиями почек или мочевыводящей системы 33 случая (65%) с деформациями конечностей, в том числе — 5 случаев отсутствия большого пальца и 2 случая лучевой аплазии, 4 случая атрезии / свища трахео-пищевода и 4 случая врожденная диафрагмальная грыжа Рис. 32: Сжатый кулак при синдроме Эдварда. Синдром Дауна.Антенатальное ультразвуковое исследование. Ряд результатов антенатального ультразвукового исследования связаны с различными хромосомными аномалиями. Недавний обзор этих результатов, сделанный Николаидесом и др. 25, показывает, что, хотя многие результаты ультразвукового исследования связаны с хромосомными аномалиями, отличными от синдрома Дауна, им не хватает чувствительности и специфичности, чтобы их можно было использовать в качестве самостоятельных скрининговых тестов. В недавнем обзоре опубликованных исследований Wald et al. Были рассмотрены маркеры трисомии 13, и в обзоре рекомендовалось провести амниоцентез, чтобы исключить эти 2 трисомии, если при антенатальном ультразвуковом сканировании было выявлено какое-либо из следующего: агенезия мозолистого тела диафрагмальная грыжа, расщелина губы Обструктивная уропатия экзомфалоса неба — дефекты уменьшения конечности двойного выходного отверстия правого желудочка (только трисомия 18) Анализ данных CARIS показывает, что 26 из 36 случаев синдрома Патау имели сообщения о антенатальном сканировании.Из них 11 (42%) имели признаки мягких ультразвуковых маркеров и -21 (81%) имели признаки структурных дефектов. В 41 из 56 случаев синдрома Эдварда результаты антенатального сканирования были переданы в CARIS. Из них у 32 (74%) были обнаружены мягкие маркеры. 32 (74%) имели признаки структурных аномалий. (Рисунок 33). Рисунок 33: Результаты антенатального сканирования, представленные в CARIS для трисомии 13 и трисомии 18 Трисомия 13 / Патау (n = 26) Трисомия 18 / Эдвард s (n = 41) Мягкие маркеры Структурные дефекты Мягкие маркеры Структурные дефекты 11 любой мягкий маркер 21 любой структурный дефект 32 любой мягкий маркер 32 любой структурный дефект 5 увеличенная затылочная кость 8 без 4 камеры 16 кисты сосудистого сплетения 10 кистозная гигрома вид на прозрачность или другое сердце 4 эхогенная кишка 8 косолапость 2 клубничные аномалии черепа 6 затылочная прозрачность 7 экзомфалос 1 эхогенная кишка 7 с орофациальной щелью 12 недель) 7 водянка / отек 1 эхогенная почка 6 голопрозэнцефалия 2 толщина затылочной кости 3 вентрикуломегалия 1 расширенная почечная лоханка 6 вентрикуломегалия недель) 4 дефекты нервной трубки 1 головка лимона 4 кистозная гигрома 3 расширенные почечные лоханки 3 заячья губа 1 банановый мозжечок 4 полидактилия 5 головок лимона 2 аномалии головы / черепа 1 костлявые ступни 1 экзомфалосы 2 банановые мозжечки 1 врожденный 1 двойной пузырек желудка 1 дефект нервной трубки 3 коромысла ступней диафрагмы c грыжа 1 нет пузыря желудка 1 атрезия пищевода 1 атрезия двенадцатиперстной кишки 25 Snijders RJM, Farrias M, von Kaisenberg C, Nicolaides KH.Аномалии плода (Глава 1). Ультразвуковые маркеры хромосомных дефектов плода (Эд Снейдерс и Николаидес) Границы в серии медицины плода. Издательская группа «Парфенон». 1996, Лондон 26 Wald N, Kennard A, Donnenfeld A, Leck I. Ультразвуковое сканирование для выявления врожденных аномалий (Глава 18) Антенатальный и неонатальный скрининг (2-е изд.) Редакторы Wald N, Leck I OU Press
5 Маркеры материнской сыворотки 2-й триместр Скрининговый тест материнской сыворотки на трисомию 18 был впервые предложен в начале 1990-х годов после наблюдения, что сывороточные уровни ряда маркеров, используемых для выявления синдрома Дауна, также были низкими в случаях фетального синдрома Эдварда.Был предложен ранний скрининговый тест, включающий возраст матери, уровень альфа-фетопротеина в сыворотке матери и свободный бета-хорионический гонадотропин человека. С опубликованным уровнем выявления 50% для ложноположительных 1%, это было предложено как сопоставимое с сывороточным скринингом на синдром Дауна 27. Однако синдром Эдварда встречается гораздо реже, чем синдром Дауна, поэтому многие другие амниоцентезы будут иметь для каждого выявленного случая, чем в случае синдрома Дауна. Было высказано предположение, что программа скрининга на состояние, которое может оказаться смертельным во время беременности или младенчества, вызовет значительную тревогу у родителей и приведет к аборту, по крайней мере, у такого же числа нормальных плодов, как и при обнаружении случаев трисомии 18 28,29.Уже считалось, что ультразвуковое сканирование дает больше шансов обнаружить трисомию, а скрининг сыворотки впоследствии развился только в отдельных программах. Недавно было показано, что усиленный скрининг сыворотки во 2-м триместре, связанный с возрастным риском матери, альфафетопротеином, неконъюгированным эстриолом и хорионическим гонадотропином человека, может выявлять ряд хромосомных аномалий, включая трисомию 18. Программы антенатального скрининга сыворотки для трисомии 13 или 18 не существует. Однако интересно отметить, что в CARIS есть отчеты о 12 матерях младенцев с синдромом Эдварда, которые выбрали сывороточный скрининг.Из них у 2 был высокий риск синдрома Дауна. Точно так же 1/5 матерей младенцев с синдромом Патау, которые выбрали скрининг сыворотки, были зарегистрированы как женщины с высоким риском Дауна. Амниоцентез Кровь младенца 6 Получение образца клеток плода для хромосомного анализа показано, если скрининг сыворотки или дородовое ультразвуковое исследование предполагают возможность хромосомной аномалии плода. В качестве альтернативы это может быть выбрано в качестве первого направления исследования для матерей старшего возраста или матерей с повышенным риском вынашивания плода с хромосомным заболеванием.Образцы обычно получают путем амниоцентеза или взятия проб ворсинок хориона. Хромосомный анализ может быть проведен с помощью кариотипирования, полимеразной цепной реакции (ПЦР) или флуоресцентной гибридизации in situ (FISH). Эти две последние процедуры имеют то преимущество, что они выполняются намного быстрее, чем обычное кариотипирование, но их можно использовать только для выявления заранее определенных хромосомных нарушений. В настоящее время они обычно не доступны для NHS. Данные CARIS включают результаты кариотипа 34 из 36 случаев синдрома Патау и 55 из 56 случаев синдрома Эдварда.Метод (и, следовательно, стадия беременности / младенчества), с помощью которой были получены эти образцы, показан на рисунке 34. Это говорит о том, что около 64% случаев синдрома Патау и 55% случаев синдрома Эдварда были обнаружены антенатально. post mortem 5 CVS 4 post mortem 10 CVS 9 27 Mallard SK, Coombes EJ, Maeri JN. Пренатальный скрининг на трисомию 18 со свободным бета-хорионическим гонадотропином человека в качестве маркера BMJ 1993; 307: (4 декабря) 28 Макинтош МСМ, Чард Т. Большинство умирает до или сразу после рождения.Синдром Патау, амниоцентез 19 Кровь младенца 14 Амниоцентез при синдроме Эдварда 22 [письмо] BMJ 1994; 308: 471 (12 февраля) 29 Дэвис Т. Пренатальный скрининг на трисомию 18: не следует рассматривать [письмо] BMJ 1994; 308: 471 (12 февраля) 30 Harding K. Ультразвуковое сканирование обнаружило отклонения [письмо] BMJ 1994; 308: 471 (12 февраля) Рис. 34: Метод сбора образцов кариотипа, представленных в CARIS для синдромов Патау и Эдварда,
синдром Патау | определение синдрома Патау в Медицинском словаре
трисомия
[tri´so-me]
Наличие дополнительной (третьей) хромосомы одного типа в диплоидной клетке (2n +1).прил., прил. тр.
синдром трисомии 8 синдром, связанный с дополнительной хромосомой 8, обычно мозаичный (трисомия 8 / нормальный), характеризующийся умственной отсталостью от легкой до тяжелой, выступающим лбом, глубоко посаженными глазами, толстыми губами, выступающими ушами и камптодактилией (аномально согнутые пальцы). синдром трисомии 13 голопрозэнцефалия из-за дополнительной хромосомы 13, при которой дефекты центральной нервной системы связаны с умственной отсталостью, расщелиной губы и неба, полидактилией (лишние пальцы рук или ног) и аномалиями кожного рисунка, а также аномалиями сердца , внутренности и гениталии.Вызывается также синдромом Патау. Информацию для семей, страдающих этим расстройством, можно получить в Организации поддержки трисомии 18, 13 и связанных с ней расстройств (SOFT), 2982 S. Union St., Rochester, NY 14624. синдром трисомии 18 состояние, связанное с наличием дополнительной хромосомы 18, характеризующейся неонатальным гепатитом, умственной отсталостью, скафоцефалией или другими аномалиями черепа, небольшой опущенной нижней челюстью, блефароптозом (опущенными веками), низко посаженными ушами, помутнением роговицы, глухотой, перепончатой шеей, короткими пальцами, дефектами межжелудочковой перегородки, Дивертикул Меккеля и другие деформации.Вызывается также синдромом Эдвардса. Информацию для семей, страдающих этим заболеванием, и специалистов, ухаживающих за больными, можно получить в Организации поддержки трисомии 18, 13 и связанных с ней заболеваний (SOFT), 2982 S. Union St., Rochester, NY 14624.
Синдром трисомии 22 синдром, связанный с лишней хромосомой 22, обычно характеризующийся задержкой умственного развития и роста, низкорослой головой, низко посаженными или деформированными ушами, небольшой опущенной нижней челюстью, длинным желобком на верхней губе, преаурикулярной кожной биркой или синусом и врожденным пороком сердца.У мужчин небольшой пенис или неопущенные яички.
Энциклопедия и словарь Миллера-Кина по медицине, сестринскому делу и смежным вопросам здравоохранения, седьмое издание. © 2003 Saunders, принадлежность Elsevier, Inc. Все права защищены.
Расстройства трисомии — Better Health Channel
Гены — это план нашего тела. Почти каждая клетка тела имеет копию чертежа, хранящуюся в мешочке, называемом ядром. Гены расположены на хромосомах, которые представляют собой плотно связанные цепи химического вещества дезоксирибонуклеиновой кислоты (ДНК).У людей обычно 23 пары хромосом, из которых две половые хромосомы определяют пол, а 44 хромосомы определяют другие факторы, такие как рост и функции.
Состояние хромосом вызвано изменением числа или генетической структуры хромосом. Трисомия («три тела») означает, что у пострадавшего человека есть три копии одной из хромосом вместо двух. Это означает, что у них 47 хромосом вместо 46.
Синдром Дауна, синдром Эдварда и синдром Патау являются наиболее распространенными формами трисомии.Дети, страдающие трисомией, обычно имеют ряд врожденных аномалий, в том числе задержку развития и умственную отсталость.
Факторы риска трисомии
Добавление дополнительной хромосомы обычно происходит спонтанно во время зачатия. Причина этого неизвестна, и предотвратить это невозможно. Наиболее важным фактором риска трисомии является возраст матери. У женщин в возрасте от 30 до 40 лет выше вероятность возникновения трисомии.
Трисомия 21 — синдром Дауна
В Виктории синдромом Дауна страдает примерно одна из 300 беременностей.Синдром Дауна также известен как трисомия 21, потому что у человека есть три копии хромосомы 21 вместо двух.
Существует три типа синдрома Дауна. Наиболее распространенной является стандартная трисомия 21, при которой сперматозоид отца или яйцеклетка матери содержат дополнительную хромосому. При синдроме Мозаики Дауна дополнительная хромосома появляется спонтанно по мере развития эмбриона. Транслокационный синдром Дауна, на который приходится примерно пять процентов случаев, передается по наследству.
Некоторые физические характеристики синдрома Дауна могут включать:
- небольшой наклон глаз вверх — почти у всех людей с синдромом Дауна глаза слегка наклонены вверх.Также может быть небольшая складка кожи на внутренней стороне глаза (так называемая «эпикантическая складка») и небольшие белые пятна на краю радужной оболочки глаза (известные как пятна Брашфилда).
- Характерная форма лица — лицо человека с синдромом Дауна часто округляется и имеет тенденцию к плоскому профилю
- Меньший рост — младенцы с синдромом Дауна обычно меньше и весят при рождении, чем другие. Дети с синдромом Дауна обычно растут медленнее и обычно меньше других детей своего возраста.Взрослые с синдромом Дауна обычно меньше взрослых, у которых нет синдрома Дауна.
Все люди с синдромом Дауна будут испытывать некоторую задержку в своем развитии и некоторый уровень неспособности к обучению.
Узнайте больше о синдроме Дауна.
Трисомия 18 — синдром Эдварда
В Виктории синдром Эдварда поражает примерно одну из 1100 беременностей. Синдром Эдварда также известен как трисомия 18, потому что у человека есть три копии хромосомы 18 вместо двух.
Некоторые характеристики синдрома Эдварда могут включать:
- Нарушение физического состояния почек, мочеточников, сердца, легких и диафрагмы
- расщелина губы или неба
- маленький череп (микроцефалия)
- Пороки развития кистей и стоп, включая отсутствие больших пальцев рук, косолапость и перепонки между пальцами рук и ног (синдактилия)
- Дефект нервной трубки, при котором спинной мозг, мозговые оболочки и кровеносные сосуды выступают через щель в позвонках (миеломенингоцеле)
- пороки развития половых органов.
Дети с синдромом Эдварда не выживают после неонатального периода.
Трисомия 13 — синдром Патау
В Виктории синдром Патау поражает примерно одну из 3000 беременностей. Синдром Патау также известен как трисомия 13, потому что у человека есть три копии хромосомы 13 вместо двух.
Некоторые характеристики синдрома Патау могут включать:
- маленький череп (микроцефалия)
- аномальное отверстие в черепе
- Пороки развития части головного мозга
- структурные дефекты глаза
- расщелина губы или неба
- дополнительные пальцы рук (полидактилия)
- врожденные пороки сердца, такие как дефект межжелудочковой перегородки
- Дефект нервной трубки, при котором спинной мозг, мозговые оболочки и кровеносные сосуды выступают через щель в позвонках (миеломенингоцеле)
- пороки развития половых органов.
Дети с синдромом Патау редко выживают после неонатального периода.
Признаки трисомии при беременности
Иногда признаки трисомии могут быть очевидны во время беременности. Некоторые из этих знаков могут включать:
- Слишком много околоплодных вод вокруг ребенка (многоводие)
- только одна артерия пуповины
- плацента меньше ожидаемой
- ребенок мал для срока беременности
- ребенок менее активен, чем ожидалось
- врожденный дефект, в том числе расщелина неба или аномалии сердца, выявляются во время ультразвукового сканирования.
Диагностика состояния трисомии
Пренатальные тесты, которые могут помочь обнаружить трисомию, включают:
Генетическое консультирование и условия трисомии
Если вашему ребенку был поставлен диагноз трисомии, может быть полезно поговорить с генетическим консультантом.
Консультанты-генетики — это специалисты в области здравоохранения, имеющие квалификацию как в области консультирования, так и в области генетики. Помимо эмоциональной поддержки, они могут помочь вам понять состояние вашего ребенка, его причины и значение диагноза для здоровья и развития вашего ребенка.Консультанты по генетическим вопросам обучены предоставлять информацию и оказывать поддержку с учетом семейных обстоятельств, культуры и убеждений.
Сеть генетической поддержки штата Виктория (GSNV) связана с широким кругом групп поддержки по всей Виктории и Австралии и может связать вас с другими людьми и семьями, страдающими трисомией.
Куда обратиться за помощью
Контент-партнер
Эта страница была подготовлена после консультаций и одобрена:
Служба клинической генетики штата Виктория (VCGS)
определение patau_syndrome и синонимы patau_syndrome (английский)
Синдром Патау , также известный как трисомия 13 и трисомия D , является хромосомной аномалией, синдромом, при котором у пациента появляется дополнительная 13 хромосома из-за нерасхождение хромосом при мейозе.Некоторые из них вызваны Робертсоновскими транслокациями, а другие — мозаичным синдромом Патау. Дополнительная хромосома 13 нарушает нормальный ход развития, вызывая пороки сердца и почек, среди других особенностей, характерных для синдрома Патау. [ неясно ] Как и все состояния, не связанные с расхождением (такие как синдром Дауна и синдром Эдвардса), риск этого синдрома у потомства увеличивается с возрастом матери при беременности, в среднем около 31 года. [1] Синдром Патау поражает примерно от 1 из 10 000 до 1 из 21 700 живорождений. [2]
Причины
Синдром
Патау чаще всего является результатом трисомии 13, что означает, что каждая клетка тела имеет три копии хромосомы 13 вместо обычных двух. Небольшой процент случаев возникает, когда только некоторые клетки тела имеют лишнюю копию; такие случаи называют мозаичными Патау.
Синдром
Патау также может возникать, когда часть хромосомы 13 присоединяется к другой хромосоме (перемещается) до или во время зачатия в результате Робертсоновской транслокации.У больных людей есть две копии 13-й хромосомы плюс дополнительный материал из 13-й хромосомы, прикрепленный к другой хромосоме. При транслокации у человека наблюдается частичная трисомия по хромосоме 13, и часто физические признаки синдрома отличаются от типичного синдрома Патау.
Большинство случаев синдрома Патау не передаются по наследству, а возникают как случайные события во время образования репродуктивных клеток (яйцеклеток и сперматозоидов). Ошибка в делении клеток, называемая неразъединением, может привести к образованию репродуктивных клеток с ненормальным количеством хромосом.Например, яйцеклетка или сперматозоид могут получить дополнительную копию хромосомы. Если одна из этих атипичных репродуктивных клеток вносит свой вклад в генетический состав ребенка, у ребенка будет дополнительная хромосома 13 в каждой клетке тела. Синдром мозаики Патау также не передается по наследству. Это происходит как случайная ошибка во время деления клеток на ранних этапах развития плода.
Синдром Патау вследствие транслокации может передаваться по наследству. Незатронутый человек может нести перестройку генетического материала между хромосомой 13 и другой хромосомой.Эта перестройка называется сбалансированной транслокацией, потому что нет лишнего материала от хромосомы 13. Хотя у них нет признаков синдрома Патау, люди, которые несут этот тип сбалансированной транслокации, подвергаются повышенному риску иметь детей с этим заболеванием.
Проявления и физические данные
Младенец мужского пола гестационного возраста 37 2/7 недель с синдромом Патау, демонстрирующий полидактилию
Из тех плодов, которые выживают до беременности и последующих родов, наиболее часто встречаются следующие аномалии:
- Скелетно-мышечная и кожная
Риск рецидива
Если только один из родителей не является носителем транслокации, вероятность того, что у пары родится еще один ребенок с трисомией 13, составляет менее 1% (меньше, чем при синдроме Дауна).
История
Трисомия 13 была впервые обнаружена Томасом Бартолином в 1657 году, [5] , но хромосомная природа болезни была установлена доктором Клаусом Патау в 1960 году. [6] Болезнь названа в его честь. Синдром Патау был описан и у тихоокеанских островных племен. Считалось, что эти сообщения были вызваны излучением в результате испытаний атомной бомбы. Племена были временно перемещены до и во время испытания на расстояние x пикселей. Затем их вернули туда, где они были увезены; все это произошло до того, как стало известно, как долго, и даже если радиация еще сохраняется после ядерного взрыва. [ необходима ссылка ]
В Англии и Уэльсе в 2008–09 годах было поставлено 172 диагноза синдрома Патау (трисомия 13), причем 91% диагнозов были поставлены пренатально. Было 111 плановых абортов, 14 случаев мертворождения / выкидыша / смерти плода, 30 исходов неизвестны и 17 живорождений. Приблизительно 4% синдрома Патау с неизвестными исходами могут привести к рождению живого ребенка, поэтому общее число живорождений оценивается в 18. [7] Небольшой процент детей с полным синдромом Патау, которые выживают после рождения и В раннем младенчестве может дожить до взрослого возраста, а у детей с мозаичной или частичной формой этой трисомии прогноз может быть совершенно другим и гораздо более обнадеживающим. [8]
Лечение
Ведение детей с трисомией 13 планируется в индивидуальном порядке и зависит от индивидуальных особенностей пациента. Лечение синдрома Патау сосредоточено на конкретных физических проблемах, с которыми рождается каждый ребенок. Многим младенцам трудно выжить в первые несколько дней или недель из-за серьезных неврологических проблем или сложных пороков сердца. Для устранения пороков сердца или заячьей губы и неба может потребоваться хирургическое вмешательство.Физическая, профессиональная и логопедическая терапия поможет людям с синдромом Патау полностью раскрыть свой потенциал развития.
Прогноз
Более 80% детей с синдромом Патау умирают в течение первого года жизни. [9]
Ссылки
Внешние ссылки
| |||||||||||||||||||
Синдром
Патау.Синдром Патау — наименее распространенная аутосомная трисомия (синдром Дауна и синдром Эдвардса). Это происходит из-за наличия доп.
Презентация на тему: «Синдром Патау. Синдром Патау — наименее распространенная аутосомная трисомия (синдром Дауна и синдром Эдвардса). Возникает при наличии лишнего». — Стенограмма презентации:
1
Синдром Патау
2
Синдром Патау — наименее распространенная аутосомная трисомия (синдром Дауна и синдром Эдвардса).Это происходит из-за наличия дополнительной копии хромосомы 13. Это также может произойти, когда часть хромосомы 13 прикрепляется к другой хромосоме во время оплодотворения.
3
Симптомы Люди с синдромом Патау могут иметь одну или несколько из следующих проблем: Аномальные физические особенности, например. Маленькая голова, наклонный лоб, заячья губа и нёбо Дефекты сердца Дефекты почек Отсутствие кожи на коже черепа Основные структурные проблемы мозга Дополнительные пальцы рук и ног Задержка развития и умственные нарушения
4
Физические характеристики
5
Ожидаемая продолжительность жизни Около 90% младенцев умирают от синдрома Патау в возрасте 15 лет. 5-10% людей доживают до своего первого года жизни.В некоторых редких случаях они доживают до подросткового возраста.
6
Лечение. Если ребенок все-таки выжил, ему может быть сделана операция по устранению пороков сердца или физических проблем. Им также могут быть назначены определенные формы терапии, такие как логопедия или физиотерапия.
7
Вероятность поражения. Подобно другим подобным состояниям, таким как синдром Дауна и синдром Эдвардса, риск заражения ребенка этим состоянием увеличивается с возрастом матери.От 1 из 10 000 до 1 из 21 700 живорождений страдают синдромом Патау. Это чаще встречается у женщин, чем у мужчин, так как у мужчин меньше шансов дожить до рождения.
Синдром Патау Википедия
Хромосомное расстройство, при котором присутствуют три копии хромосомы 13
| Синдром Патау | |
|---|---|
| Другие названия | Трисомия 13, трисомия D, T13 [1] |
| Женщина 16 лет с полной трисомией 13 и соответствующими чертами лица. | |
| Произношение | |
| Специальность | Медицинская генетика |
Синдром Патау — синдром, вызванный хромосомной аномалией, при которой некоторые или все клетки тела содержат дополнительный генетический материал из хромосом. 13. Дополнительный генетический материал нарушает нормальное развитие, вызывая множественные и сложные дефекты органов.
Это может происходить либо потому, что каждая клетка содержит полную дополнительную копию хромосомы 13 (нарушение, известное как трисомия 13, или трисомия D, или T13 [1] ), либо потому, что каждая клетка содержит дополнительную частичную копия хромосомы или потому что есть две разные линии клеток — одна здоровая с правильным числом 13 хромосом и другая, содержащая дополнительную копию хромосомно-мозаичного синдрома Патау.Полная трисомия 13 вызвана нерасхождением хромосом во время мейоза (мозаичная форма вызвана нерасхождением во время митоза).
Как и все состояния, не связанные с расхождением (такие как синдром Дауна и синдром Эдвардса), риск этого синдрома у потомства увеличивается с возрастом матери при беременности, в среднем около 31 года. [2] Синдром Патау поражает от 1 из 10 000 до 1 из 21 700 живорожденных. [3]
Признаки и симптомы []
Хромосома 13, хромосома, пораженная этим заболеванием.Шесть пальцев у ребенка с синдромом Патау. Такой же ребенок, как показано ниже.
37-недельный гестационный мужчина, умерший вскоре после рождения с синдромом Патау, демонстрирующий алобарную голопрозэнцефалию с циклопией. Черты лица включали покатый лоб с носом выше единственной центральной глазной щели.
Из тех плодов, которые действительно выживают до беременности и родов, общие аномалии могут включать:
- Нервная система
- Скелетно-мышечная и кожная
- Урогенитальный
- Другое
Причины []
Синдром
Патау является результатом трисомии 13, что означает, что каждая клетка в организме имеет три копии хромосомы 13 вместо обычных двух.Небольшой процент случаев возникает, когда только некоторые клетки тела имеют лишнюю копию; такие случаи называют мозаичными Патау.
Синдром Патау может также возникать, когда часть хромосомы 13 присоединяется к другой хромосоме (перемещается) до или во время зачатия в результате Робертсоновской транслокации. У больных людей есть две копии 13-й хромосомы плюс дополнительный материал из 13-й хромосомы, прикрепленный к другой хромосоме. При транслокации у человека наблюдается частичная трисомия по хромосоме 13, и часто физические признаки синдрома отличаются от типичного синдрома Патау.
Большинство случаев синдрома Патау не наследуются, а возникают как случайные события во время образования репродуктивных клеток (яйцеклеток и сперматозоидов). Ошибка в делении клеток, называемая неразъединением, может привести к образованию репродуктивных клеток с ненормальным количеством хромосом. Например, яйцеклетка или сперматозоид могут получить дополнительную копию хромосомы. Если одна из этих атипичных репродуктивных клеток вносит свой вклад в генетический состав ребенка, у ребенка будет дополнительная хромосома 13 в каждой клетке тела.Синдром мозаики Патау также не передается по наследству. Это происходит как случайная ошибка во время деления клеток на ранних этапах развития плода.
Синдром Патау вследствие транслокации может передаваться по наследству. Незатронутый человек может нести перестройку генетического материала между хромосомой 13 и другой хромосомой. Эта перестройка называется сбалансированной транслокацией, потому что нет лишнего материала от хромосомы 13. Хотя у них нет признаков синдрома Патау, люди, которые несут этот тип сбалансированной транслокации, подвергаются повышенному риску иметь детей с этим заболеванием.
Риск рецидива []
Если один из родителей не является носителем транслокации, вероятность того, что у пары родится еще один ребенок с трисомией 13, составляет менее 1% (меньше, чем при синдроме Дауна). Наиболее частыми характеристиками этого синдрома являются такие проблемы, как позднее развитие, умственная отсталость, множественные пороки развития, кардиомиопатия и почечные аномалии. Наиболее частыми физическими признаками синдрома Патау являются снижение мышечного тонуса, маленькие руки, маленькие уши, маленькая голова и рот, а также широкие и короткие руки с короткими пальцами.У детей с синдромом Патау физическое развитие происходит медленнее, чем у детей без синдрома Патау. Тем не менее, дети, страдающие синдромом Патау, по-прежнему должны регулярно заниматься физической активностью, даже если развитие мышц может происходить медленнее.
Диагноз []
Диагноз обычно основывается на клинических данных, хотя исследование хромосом плода покажет трисомию 13. Хотя многие физические данные похожи на синдром Эдвардса, есть несколько уникальных черт, таких как полидактилия.Однако, в отличие от синдрома Эдвардса и синдрома Дауна, квадроэкран не обеспечивает надежных средств диагностики этого расстройства. Это связано с вариабельностью результатов, наблюдаемых у плодов с Патау. [6]
Лечение []
Ведение детей с трисомией 13 планируется в индивидуальном порядке и зависит от индивидуальных особенностей пациента. Лечение синдрома Патау сосредоточено на конкретных физических проблемах, с которыми рождается каждый ребенок.Многим младенцам трудно выжить в первые несколько дней или недель из-за серьезных неврологических проблем или сложных пороков сердца. Для устранения пороков сердца или заячьей губы и неба может потребоваться хирургическое вмешательство. Физическая, профессиональная и логопедическая терапия поможет людям с синдромом Патау полностью раскрыть свой потенциал развития. Выжившие дети описываются как счастливые, и родители сообщают, что они обогащают свою жизнь. [7] Цитируемое исследование сгруппировало синдром Эдвардса, который иногда остается выжившим после раннего детства, вместе с Патау, следовательно, средний возраст на момент сбора данных составлял 4 года.
Прогноз []
Примерно 90% младенцев с синдромом Патау умирают в течение первого года жизни. [8] Те дети, которые выживают более одного года жизни, обычно имеют тяжелую форму инвалидности с умственной отсталостью, судорогами и психомоторными проблемами. Дети с мозаичной вариацией обычно страдают в меньшей степени. [9] В ретроспективном канадском исследовании 174 детей с трисомией 13 средняя продолжительность жизни составила 12,5 дней. Выживаемость через один и десять лет составила 19.8% и 12,9% соответственно, в том числе перенесшие агрессивное хирургическое вмешательство. [10]
История []
Трисомия 13 была впервые обнаружена Томасом Бартолином в 1657 году, [11] , но хромосомная природа болезни была установлена доктором Клаусом Патау в 1960 году. [12] Болезнь названа в его честь.
В Англии и Уэльсе в 2008–09 годах было поставлено 172 диагноза синдрома Патау (трисомия 13), причем 91% диагнозов были поставлены пренатально. «Годовые отчеты Национального Цитогенетического Регистра Синдрома Дауна за 2008/09». Архивировано 15 апреля 2010 года.
Внешние ссылки []
.