Хорея Гентингтона — это… Что такое Хорея Гентингтона?
Болезнь (или синдром, или хорея) Хантингтона (Huntington Disease) — это генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 35-50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене IT-15. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией кортекса.
CAG триплет кодирует аминокислоту глутамин. Мутация, или удлинение полиглутаминовой последовательности в белке хантингтин, приводит к изменению конформации белка, который становится токсичным для нервных клеток. Более того, удлинение полиглутаминовой последовательности приводит к агрегации хантингтина, при этом образуются так называемые внутриклеточные тельца включения. До недавнего времени считалось, что именно тельца включения ответственны за смерть нейронов. Однако, это не так: тельца включения наоборот, защищают нейрон от смерти, аккумулируя мутантный хантингтин, и именно неагрегированный белок токсичен.[1] Почему нейроны стриатумa отличаются повышенной чувствительностью к мутантному хантингтину, остаётся неизвестным.
До недавнего времени считалось, что именно тельца включения ответственны за смерть нейронов. Однако, это не так: тельца включения наоборот, защищают нейрон от смерти, аккумулируя мутантный хантингтин, и именно неагрегированный белок токсичен.[1] Почему нейроны стриатумa отличаются повышенной чувствительностью к мутантному хантингтину, остаётся неизвестным.
Мутантный ген был предположительно завезён в США в 1630 году двумя братьями, эмигрировавшими из Эссекса в Бостон [2], [3]. В настоящее время от хореи Хантингтона в США страдает около 7000 человек. Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000, и 1:1000000 среди остальных рас[4]. Название болезни дано в честь трёх поколений врачей, изучавших её в штате Коннектикут.
Симптомы
В самом начале обычно возникают проблемы из-за физических симптомов, которые выражаются в резких, внезапных и не поддающихся контролю движениях. В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Обычно происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, может возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм, агрессия, навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания.
В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Обычно происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, может возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм, агрессия, навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания.
Прогноз
С момента появления первых симптомов продолжительность жизни составляет около 15-20 лет. Смерть обычно происходит не из-за болезни Хантингтона, а из-за сопутствующих ей осложнений, включая пневмонию, заболевания сердца и травмы. Также частой причиной смерти является суицид.
Смерть обычно происходит не из-за болезни Хантингтона, а из-за сопутствующих ей осложнений, включая пневмонию, заболевания сердца и травмы. Также частой причиной смерти является суицид.
Ссылки
- ↑ Arrasate, M. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431(7010):805-10.
- ↑ Vessie, P. R. (1932) «On the transmission of Huntington’s chorea for 300 years—the Bures family group». Journal of Nervous and Mental Disease, Baltimore 76: 553—573.
- ↑ Wexler, A. (2008) «The Woman Who Walked into the Sea: Huntington’s and the Making of a Genetic Disease», Yale University Press; 1 edition
- ↑ NCBI OMIM Huntington’s Disease. Проверено 22 мая 2008.
Wikimedia Foundation.
2010.
About Huntington’s Disease — European Huntington’s Disease Network
БГ названа в честь Джорджа Гентингтона, американского врача, подробно описавшего это заболевание в 1872 г. Его описание основано на наблюдениях семей с БГ из деревни Ист-Хэмптон, расположенной на острове Лонг-Айленд (Нью-Йорк, США), где жил и работал д-р Гентингтон. БГ известна в прошлом как хорея Гентингтона и пляска святого Витта.
Его описание основано на наблюдениях семей с БГ из деревни Ист-Хэмптон, расположенной на острове Лонг-Айленд (Нью-Йорк, США), где жил и работал д-р Гентингтон. БГ известна в прошлом как хорея Гентингтона и пляска святого Витта.
Как часто встречается БГ?
БГ является редким заболеванием, распространённость которого в европейской популяции составляет около 5–10 человек на 100 000 населения. Примерно такая же распространённость и в странах, население которых имеет преимущественно европейское происхождение, например, в США. Распространённость БГ меньше в странах Азии и Африки и составляет около 1 человека на 100 000 населения. Мужчины и женщины имеют одинаковые риски унаследовать экспансию, которая приведёт к развитию БГ.
Каковы симптомы БГ?
БГ характеризуется сочетанием двигательных (моторных), поведенческих (например, изменение настроения) и когнитивных (например, трудности в понимании) нарушений, однако могут развиваться и другие симптомы. От пациента к пациенту (даже среди членов одной и той же семьи) симптомы БГ могут отличаться по своей тяжести, возрасту появления и темпу прогрессирования. У одного человека могут быть выраженные двигательные расстройства, но при этом лёгкие поведенческие и когнитивные нарушения, в то время как у другого депрессия и тревога могут наблюдаться за годы до каких-либо двигательных симптомов. Про БГ пишут, что она развивается исподволь, т.к. довольно сложно определить точную дату появления её первых симптомов.
От пациента к пациенту (даже среди членов одной и той же семьи) симптомы БГ могут отличаться по своей тяжести, возрасту появления и темпу прогрессирования. У одного человека могут быть выраженные двигательные расстройства, но при этом лёгкие поведенческие и когнитивные нарушения, в то время как у другого депрессия и тревога могут наблюдаться за годы до каких-либо двигательных симптомов. Про БГ пишут, что она развивается исподволь, т.к. довольно сложно определить точную дату появления её первых симптомов.
- Двигательные симптомы
К двигательным симптомам БГ относятся хорея, брадикинезия и дистония, которые в значительной степени нарушают поддержание физиологической позы, равновесие и ходьбу. Термин «хорея» происходит от греческого слова «choreia», означающего «танец», и используется для обозначения непроизвольных движений, часто наблюдающихся при БГ. К другим распространённым двигательным проявлениям БГ относятся трудности инициации произвольных движений (брадикинезия) и продолжительные сокращения мышц, приводящие к формированию нефизиологических поз, «скручиванию» или подёргиванию тех или иных частей тела (дистония). Кроме того, у пациентов с БГ часто отмечаются глазодвигательные расстройства, трудности при проглатывании пищи и нечёткая речь, которая с течением времени становится всё более смазанной.
Кроме того, у пациентов с БГ часто отмечаются глазодвигательные расстройства, трудности при проглатывании пищи и нечёткая речь, которая с течением времени становится всё более смазанной.
 Кроме того, у пациентов с БГ часто отмечаются глазодвигательные расстройства, трудности при проглатывании пищи и нечёткая речь, которая с течением времени становится всё более смазанной.
Кроме того, у пациентов с БГ часто отмечаются глазодвигательные расстройства, трудности при проглатывании пищи и нечёткая речь, которая с течением времени становится всё более смазанной.- Личностные и поведенческие изменения
К наиболее частым психопатологическим симптомам при БГ относятся апатия, тревога, депрессия, раздражительность, вспышки агрессии, импульсивное поведение, обсессивно-компульсивные нарушения, нарушение сна и социальное отчуждение. Реже встречаются мания и шизофреноподобные проявления, включая бред (ложные убеждения) и галлюцинации (зрительное, слуховое или иное восприятие вещей, не существующих на самом деле). У лиц с БГ могут также отмечаться суицидальные мысли (особенно на ранних стадиях заболевания). Большинство пациентов и ухаживающих лиц считают поведенческие изменения более обременительными, чем двигательные или когнитивные нарушения вследствие БГ.
- Когнитивные нарушения
БГ характеризуется постепенным нарушением способности понимания и логического суждения, а также снижением памяти. Когнитивные нарушения включают в себя замедление мышления и затруднение концентрации внимания, сложности в организации, планировании, принятии решений и в ответах на вопросы, снижение краткосрочной памяти в сочетании с затруднениями в решении проблем, понимании и усвоении новой информации.
 Когнитивные нарушения включают в себя замедление мышления и затруднение концентрации внимания, сложности в организации, планировании, принятии решений и в ответах на вопросы, снижение краткосрочной памяти в сочетании с затруднениями в решении проблем, понимании и усвоении новой информации.
Когнитивные нарушения включают в себя замедление мышления и затруднение концентрации внимания, сложности в организации, планировании, принятии решений и в ответах на вопросы, снижение краткосрочной памяти в сочетании с затруднениями в решении проблем, понимании и усвоении новой информации.- Другие симптомы БГ
По мере течения БГ у человека могут наблюдаться и другие изменения, такие как снижение аппетита, потеря массы тела, снижение самооценки, нарушение сексуального влечения, а также недержание мочи и стула.
Когда появляются симптомы БГ?
У большинства людей, являющихся носителями мутации БГ, симптомы заболевания развиваются в среднем возрасте — между 35 и 55 годами жизни. Примерно в 10 % случаев болезнь дебютирует до 20 лет (в таком случае имеет место ювенильная БГ), а в ещё 10 % — после 55 лет. В целом, БГ развивается постепенно, в связи с чем она может оставаться недиагностированной в течение многих лет. В среднем, продолжительность жизни от момента постановки диагноза составляет от 15 до 20 лет, но эти рамки значительно варьируют среди пациентов и могут сильно зависеть от качества ухода за больным.
В среднем, продолжительность жизни от момента постановки диагноза составляет от 15 до 20 лет, но эти рамки значительно варьируют среди пациентов и могут сильно зависеть от качества ухода за больным.
От чего зависит возраст появления симптомов?
Факторы, определяющие возраст начала заболевания, довольно сложны и являются предметом проводимых в настоящее время исследований. При анализе больших групп пациентов с БГ учёные выявили корреляцию между числом тринуклеотидных повторов и возрастом появления симптомов (см. рисунок ниже). Это означает, что, в целом, чем больше CAG-повторов, тем раньше появляются симптомы (см. рисунок ниже). Тем не менее, как видно на рисунке ниже, для каждого конкретного числа СAG-повторов предполагаемый возраст дебюта заболевания может варьировать в пределах 30 лет. Возможно, такая вариабельность связана с действием других генов, отличных от HTT (так называемые генетические модификаторы), а также с влиянием внешних факторов, таких как образ жизни и диета. Таким образом, точно предсказать конкретный возраст появления первых симптомов заболевания у носителя мутации БГ очень сложно.
Таким образом, точно предсказать конкретный возраст появления первых симптомов заболевания у носителя мутации БГ очень сложно.
Если в вашем гене HTT содержится 40 или более CAG-повторов, в течение жизни (при условии обычной её продолжительности) у вас разовьются симптомы БГ, но сложно сказать, когда именно это произойдёт. Как правило, чем больше у носителя мутации CAG-повторов, тем раньше появляются симптомы БГ, однако среди носителей 45 CAG-повторов возраст дебюта заболевания может варьировать в пределах 30 лет. Это говорит о том, что возраст начала БГ определяется не только числом CAG-повторов, но также и иными факторами, которые ещё предстоит определить. Рисунок адаптирован из Andrew, S. E. et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nature Genetics 4, 398–403 (1993).
Какие стадии выделяют в течении БГ?
Согласно классификации, разработанной неврологом и специалистом по БГ Айрой Шолсеном (Ira Shoulson) из Джорджтаунского университета (США), течение БГ можно разделить на пять стадий.
- Ранняя стадия: человеку выставлен клинический диагноз БГ, однако он может полноценно функционировать как дома, так и на работе.
- Ранняя промежуточная стадия: человек остаётся трудоустроенным, однако его трудоспособность снижается; несмотря на некоторые трудности, он/она может справляться с повседневными делами.
- Поздняя промежуточная стадия: человек больше не может справляться с рабочими обязанностями и делами по дому; он/она нуждается в значительной помощи
- Собственно поздняя стадия: человек нуждается в полноценной помощи во всех видах повседневной активности и, как правило, в профессиональном уходе.
- Начало поздней стадии: человек перестаёт быть самостоятельным в повседневных делах, однако сохраняет способность проживать дома при условии поддержки со стороны семьи или профессиональных ухаживающих лиц.
- Собственно поздняя стадия: человек нуждается в полноценной помощи во всех видах повседневной активности и, как правило, в профессиональном уходе.
Отличаются ли симптомы ювенильной формы БГ от симптомов
заболевания с началом во взрослом возрасте?
Если БГ начинается в раннем возрасте (до 20 лет), наиболее выраженными симптомами являются замедленность движений (брадикинезия) и скованность (дистония), а не насильственные движения (хорея). К ранним симптомам ювенильной БГ относятся поведенческие нарушения, трудности в обучении и речи, снижение успеваемости в школе. В ряде случаев могут наблюдаться эпилептические приступы, что характерно для пациентов более молодого возраста. В целом, ювенильная форма БГ прогрессирует быстрее, чем БГ с дебютом во взрослом возрасте.
Каковы симптомы БГ, если заболевание развивается в
позднем возрасте?
Если БГ начинается в позднем возрасте, наиболее выраженным симптомом является хорея, а не замедленность или скованность движений.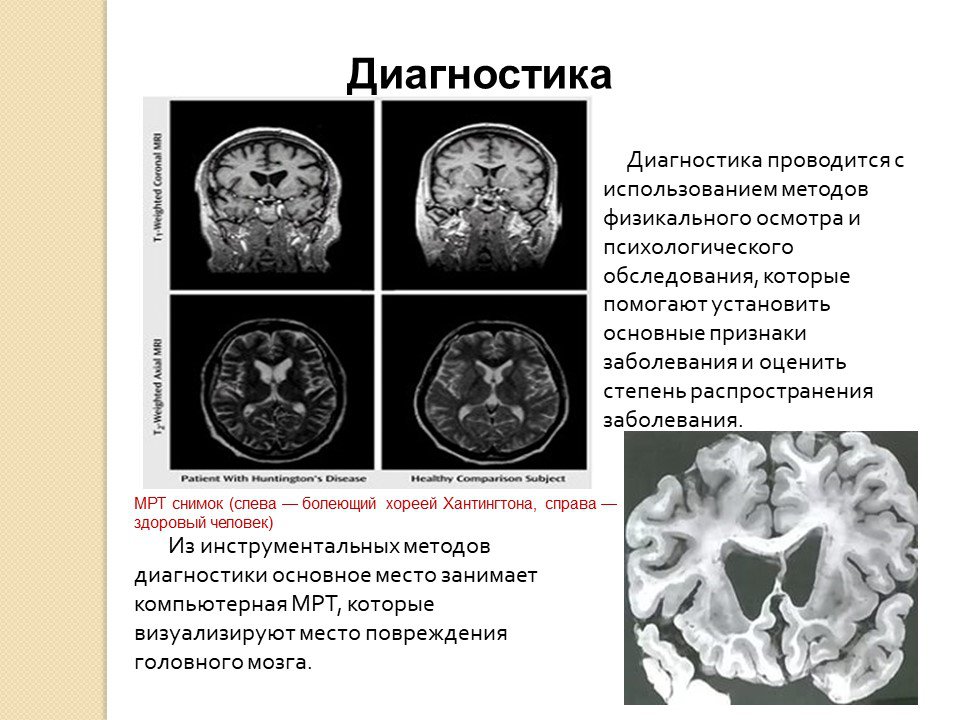 В таких случаях нередко сложно выявить семейный характер заболевания, т.к. родители пациента, чаще всего, уже скончались, по всей видимости, не дожив до того возраста, когда бы у них развились симптомы БГ.
В таких случаях нередко сложно выявить семейный характер заболевания, т.к. родители пациента, чаще всего, уже скончались, по всей видимости, не дожив до того возраста, когда бы у них развились симптомы БГ.
Причины смерти
Люди с БГ не умирают непосредственно от этого заболевания — причиной смерти являются другие медицинские состояния, являющиеся осложнениями общего истощения организма. Последние включают в себя пневмонию (она является причиной смерти при БГ в трети случаев), попёрхивание, сердечную недостаточность, травмы головы вследствие падений и нарушение питания. Высок также и риск суицида — он является причиной смерти примерно у 7 % пациентов.
Каким образом ставится диагноз БГ?
Диагноз БГ ставится на основании сочетания результатов клинического обследования и генетического теста.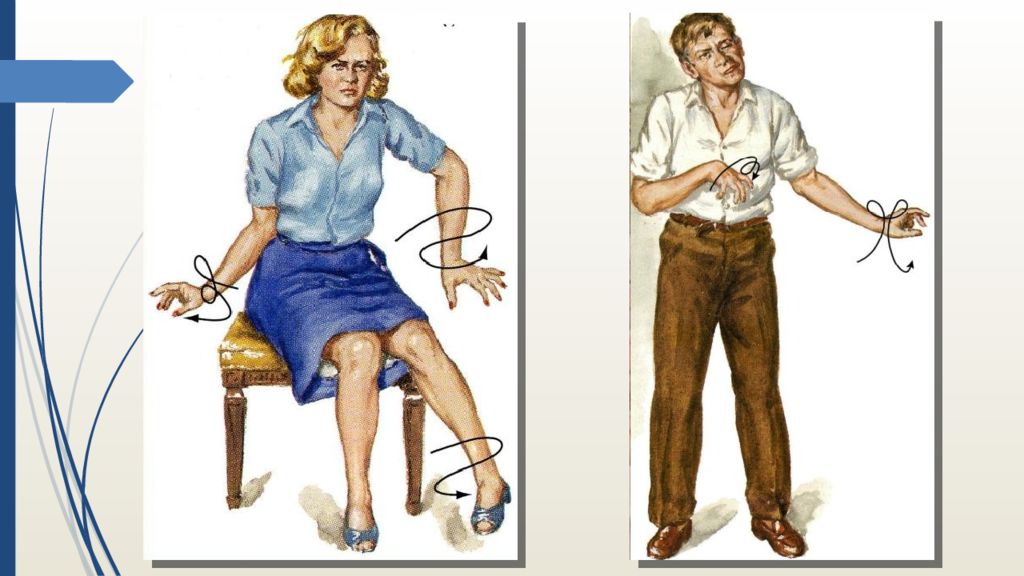 Клинический диагноз базируется на данных истории болезни и семейного анамнеза, а также на данных стандартного обследования с использованием клинических шкал, которые оценивают частоту и выраженность симптомов БГ. Клинический диагноз, как правило, подтверждается проведением генетического тестирования на наличие мутации (экспансии) в гене HTT (так называемое диагностическое, или подтверждающее, генетическое тестирование). Если у человека нет никаких симптомов, но он находится в группе риска по носительству мутации БГ, определить его генетический статус может пресимптоматическое генетическое тестирование (известное также как предиктивное генетическое тестирование).
Клинический диагноз базируется на данных истории болезни и семейного анамнеза, а также на данных стандартного обследования с использованием клинических шкал, которые оценивают частоту и выраженность симптомов БГ. Клинический диагноз, как правило, подтверждается проведением генетического тестирования на наличие мутации (экспансии) в гене HTT (так называемое диагностическое, или подтверждающее, генетическое тестирование). Если у человека нет никаких симптомов, но он находится в группе риска по носительству мутации БГ, определить его генетический статус может пресимптоматическое генетическое тестирование (известное также как предиктивное генетическое тестирование).
Какие методы клинического обследования используются для
диагностиrи БГ?
Методы клинического обследования, которые используются для диагностики БГ и оценки различных её проявлений, не являются одинаковыми во всех больницах и во всех странах. Тем не менее, наиболее часто используемой является Унифицированная шкала оценки болезни Гентингтона (англ. Unified Huntington’s Disease Rating Scale, UHDRS), имеющая подразделы для двигательной, когнитивной оценки, а также оценки поведенческих и функциональных нарушений. Кроме того, часто используется Шкала оценки поведенческих нарушений при болезни Гентингтона (англ. Problem Behaviours Assessment for Huntington’s Disease, PBA) для исследования выраженности и частоты нарушений поведения (таких как сниженное настроение, апатия и раздражительность) и ряд других тестов. Например, Краткая шкала оценки психического статуса (англ. Mini-Mental State Examination, MMSE) и Шкала Маттиса для оценки деменции (англ. Mattis Dementia Rating Scale) применяются дополнительно к подразделу UHDRS по оценке когнитивных нарушений.
Тем не менее, наиболее часто используемой является Унифицированная шкала оценки болезни Гентингтона (англ. Unified Huntington’s Disease Rating Scale, UHDRS), имеющая подразделы для двигательной, когнитивной оценки, а также оценки поведенческих и функциональных нарушений. Кроме того, часто используется Шкала оценки поведенческих нарушений при болезни Гентингтона (англ. Problem Behaviours Assessment for Huntington’s Disease, PBA) для исследования выраженности и частоты нарушений поведения (таких как сниженное настроение, апатия и раздражительность) и ряд других тестов. Например, Краткая шкала оценки психического статуса (англ. Mini-Mental State Examination, MMSE) и Шкала Маттиса для оценки деменции (англ. Mattis Dementia Rating Scale) применяются дополнительно к подразделу UHDRS по оценке когнитивных нарушений.
Какова процедура прохождения пресимптоматического генетического тестирования?
Знание того, что у вас есть риск заболеть БГ, может привести к чувству постоянного беспокойства в течение жизни. По этой причине человек может посчитать, что лучше знать наверняка, есть ли у него мутация этого заболевания или нет. В таком случае крайне рекомендуется проведение генетического консультирования с психологической поддержкой, т.к. это позволяет лучше понять возможные пути для выбора и обсудить беспокоящие вопросы. В целом, пресимптоматическое тестирование не рекомендуется для лиц младше 18 лет — возраста, в котором, как считается, человек способен воспринять информацию о том, что он является носителем мутации БГ. Тем не менее, в порядке исключения детям может проводиться подтверждающее генетическое тестирование, если, например, у ребёнка имеются симптомы ювенильной формы БГ или если женщина моложе 18 лет забеременела. Если вы решили пройти генетическое тестирование, из вены на вашей руке возьмут образец крови, чтобы извлечь из неё молекулы ДНК. В зависимости от условий лаборатории, где сдавалась кровь, результаты будут готовы через 2–8 недель. В 2012 г. рабочая группа EHDN по генетическому тестированию и консультированию обновила
По этой причине человек может посчитать, что лучше знать наверняка, есть ли у него мутация этого заболевания или нет. В таком случае крайне рекомендуется проведение генетического консультирования с психологической поддержкой, т.к. это позволяет лучше понять возможные пути для выбора и обсудить беспокоящие вопросы. В целом, пресимптоматическое тестирование не рекомендуется для лиц младше 18 лет — возраста, в котором, как считается, человек способен воспринять информацию о том, что он является носителем мутации БГ. Тем не менее, в порядке исключения детям может проводиться подтверждающее генетическое тестирование, если, например, у ребёнка имеются симптомы ювенильной формы БГ или если женщина моложе 18 лет забеременела. Если вы решили пройти генетическое тестирование, из вены на вашей руке возьмут образец крови, чтобы извлечь из неё молекулы ДНК. В зависимости от условий лаборатории, где сдавалась кровь, результаты будут готовы через 2–8 недель. В 2012 г. рабочая группа EHDN по генетическому тестированию и консультированию обновила
Рекомендации по процедуре пресимптоматического генетического тестирования.
Что определяет генетическое тестирование?
Генетическое тестирование определяет число CAG-повторов в гене HTT. Тест позволяет узнать, есть ли у человека мутация БГ или нет, однако он не даёт информации о возрасте дебюта этого заболевания, скорости его прогрессирования или о том, какие именно симптомы разовьются. Точность результата генетического тестирования близка к 100 %. Результаты анализа ДНК, как правило, дважды перепроверяются с использованием двух разных образцов крови. Кроме того, для подтверждения исходного диагноза может быть также взята на анализ кровь родителя обследуемого лица (или, в случае отсутствия такой возможности, другого члена семьи).
Возможно ли человеку, являющемуся носителем мутации БГ,
предотвратить передачу этой мутации своему ребёнку?
Да. Это можно сделать с использованием современной диагностической процедуры под названием преимплантационная генетическая диагностика (ПГД), или скрининг эмбрионов, которая применяется в сочетании с экстракорпоральным оплодотворением (ЭКО) и включает в себя скрининг эмбрионов на носительство мутации БГ до их имплантации в матку. Использование этой технологии позволяет быть уверенным в том, что имплантированы будут только эмбрионы с нормальным числом CAG-повторов. Таким образом, даже если у кого-то из родителей есть мутация БГ (вне зависимости от того, мужчина это или женщина), ПГД позволяет паре зачать ребёнка, не являющегося носителем мутации БГ. Тем не менее, в некоторых странах приняты законы о защите эмбриона, не позволяющие проводить ПГД. Также необходимо знать, что шансы на успешное завершение беременности после ПГД/ЭКО ниже, чем при «естественном» зачатии. В некоторых странах возможно тестирование нерождённого плода, зачатого естественным образом, с целью дальнейшего решения о возможном прерывании беременности на основании знания генетического статуса плода.
Это можно сделать с использованием современной диагностической процедуры под названием преимплантационная генетическая диагностика (ПГД), или скрининг эмбрионов, которая применяется в сочетании с экстракорпоральным оплодотворением (ЭКО) и включает в себя скрининг эмбрионов на носительство мутации БГ до их имплантации в матку. Использование этой технологии позволяет быть уверенным в том, что имплантированы будут только эмбрионы с нормальным числом CAG-повторов. Таким образом, даже если у кого-то из родителей есть мутация БГ (вне зависимости от того, мужчина это или женщина), ПГД позволяет паре зачать ребёнка, не являющегося носителем мутации БГ. Тем не менее, в некоторых странах приняты законы о защите эмбриона, не позволяющие проводить ПГД. Также необходимо знать, что шансы на успешное завершение беременности после ПГД/ЭКО ниже, чем при «естественном» зачатии. В некоторых странах возможно тестирование нерождённого плода, зачатого естественным образом, с целью дальнейшего решения о возможном прерывании беременности на основании знания генетического статуса плода.
Могу ли я провести генетическое тестирование своему ребёнку,
который ещё не родился?
Пренатальная (до рождения) диагностика возможна только в том случае, когда запрашивающие её люди могут показать, что их случай удовлетворяет определённым медицинским и юридическим критериям, которые варьируют в зависимости от страны. Существует две стандартные процедуры проведения пренатальной диагностики. Первая — это амниоцентез (известна также как исследование амниотической жидкости), при котором при помощи вводимой через стенку живота матери иглы собирается амниотическая жидкость, содержащая клетки плода. Как правило, амниоцентез проводится после 14-й недели беременности. Вторая процедура для пренатальной диагностики — биопсия ворсин хориона, при которой забирается образец ворсины хориона (плацентарная ткань). Исследование ворсин хориона может проводиться раньше — между 10-й и 13-й неделями беременности, однако это более опасная для плода процедура.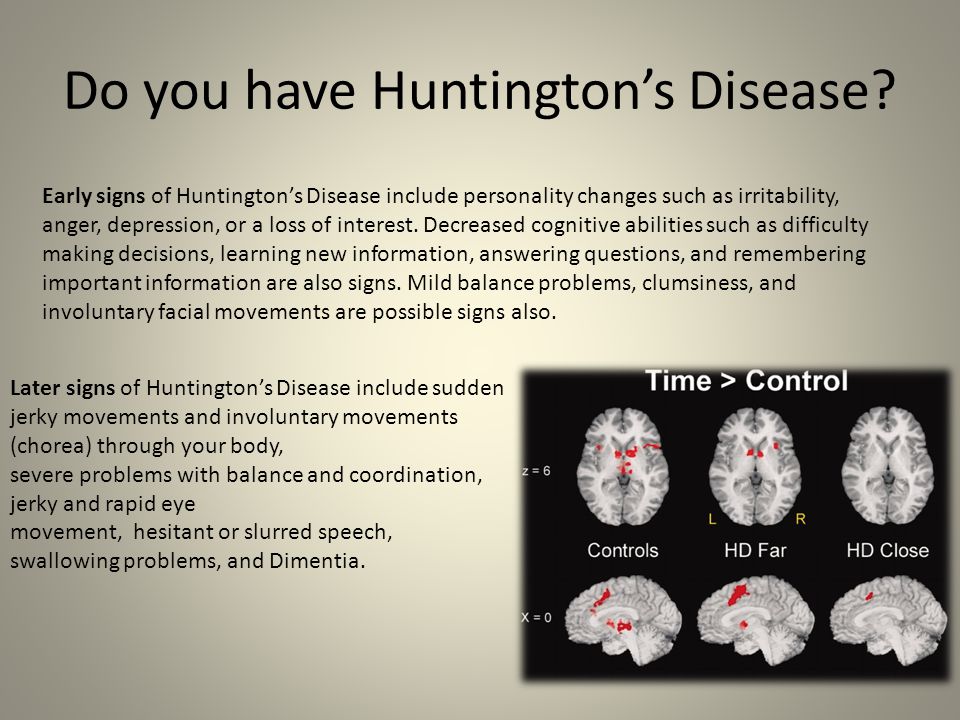
Есть ли какие-либо методы лечения БГ?
На текущий момент не существует методов, позволяющих эффективно воздействовать на причину развития БГ. Тем не менее, в течение последних лет фундаментальные и клинические исследования значительно расширили наши знания о БГ, и , направленных на изучение патогенеза этого заболевания, с целью разработки препаратов, которые смогут отсрочить начало болезни или замедлить её прогрессирование. На сегодняшний день имеются подходы, применение которых уменьшает выраженность симптомов БГ (симптоматическое лечение), что позволяет улучшить качество жизни пациентов. Эти подходы подразделяются на медикаментозное (лекарственное) и немедикаментозное (нелекарственное) лечение.
Какие существуют методы медикаментозной коррекции симптомов БГ?
Хорея, брадикинезия, раздражительность, апатия, депрессия, тревога и нарушение сна являются теми симптомами БГ, которые причиняют наибольшие трудности.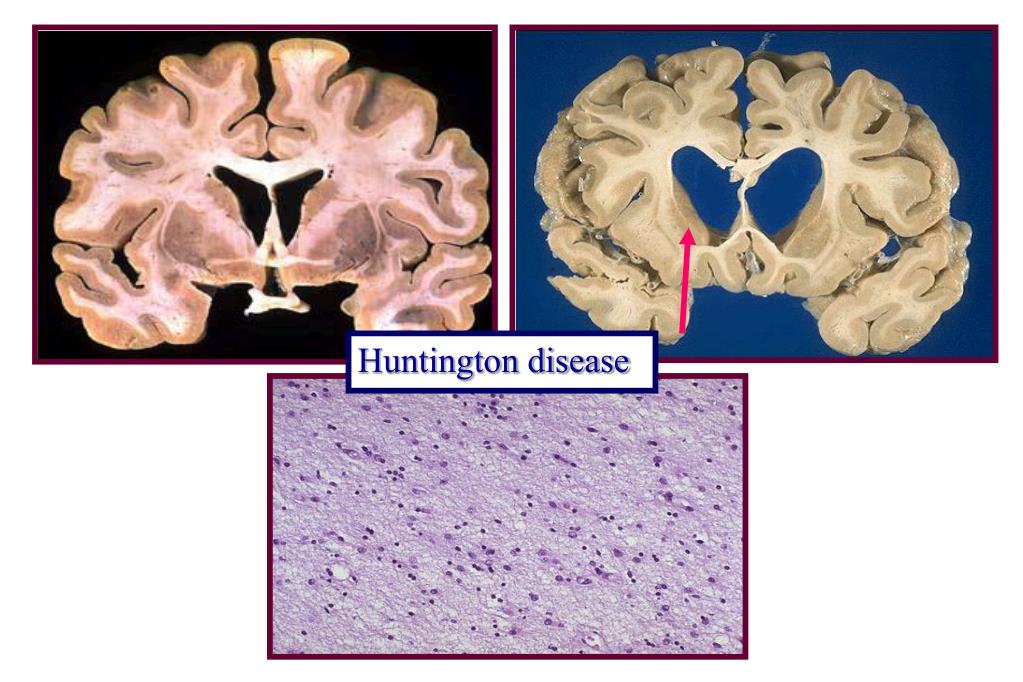 Существует несколько вариантов медикаментозной коррекции этих симптомов. Тем не менее, многие лекарства могут вызывать нежелательные реакции или уменьшать эффективность других сопутствующих препаратов. Кроме того, одно и то же лекарство у разных людей может приводить к различным эффектам. Лечение должно проводиться и быть персонализированным, строящимся с учётом имеющихся у пациента симптомов и его/её ответа на пробуемые лекарства.
Существует несколько вариантов медикаментозной коррекции этих симптомов. Тем не менее, многие лекарства могут вызывать нежелательные реакции или уменьшать эффективность других сопутствующих препаратов. Кроме того, одно и то же лекарство у разных людей может приводить к различным эффектам. Лечение должно проводиться и быть персонализированным, строящимся с учётом имеющихся у пациента симптомов и его/её ответа на пробуемые лекарства.
Как могут помочь немедикаментозные методы лечения?
Немедикаментозные методы лечения (такие как психотерапия, когнитивная терапия, лечебная физкультура, логопедические занятия, дыхательная и трудотерапия) могут способствовать уменьшению выраженности как психопатологических, так и физических симптомов БГ. В частности, показано, что после применения перечисленных подходов отмечается улучшение настроения, контроля произвольных движений, речи, равновесия, глотания и ходьбы. Хорошо известно, что физические упражнения укрепляют и психическое (включая уменьшение симптомов депрессии), и физическое здоровье, способствуя улучшению общего самочувствия. Всё больше данных накапливается в пользу того, что физические занятия помогают также замедлить прогрессирование двигательных расстройств при БГ. Например, некоторые программы лечебной физкультуры показали свою эффективность в отношении контроля движения, ходьбы и равновесия. Рабочая группа EHDN по физиотерапии опубликовала для реабилитологов, работающих с пациентами с БГ.
Хорошо известно, что физические упражнения укрепляют и психическое (включая уменьшение симптомов депрессии), и физическое здоровье, способствуя улучшению общего самочувствия. Всё больше данных накапливается в пользу того, что физические занятия помогают также замедлить прогрессирование двигательных расстройств при БГ. Например, некоторые программы лечебной физкультуры показали свою эффективность в отношении контроля движения, ходьбы и равновесия. Рабочая группа EHDN по физиотерапии опубликовала для реабилитологов, работающих с пациентами с БГ.
Может ли какая-то особая диета уменьшить выраженность
симптомов БГ?
Потенциальная польза от диеты, обогащённой витаминами, коферментами и иными веществами являлась предметом большого числа обсуждений, однако пока положительное влиянияние такой диеты в отношении БГ так и не доказано клинически. Тем не менее, для некоторых пациентов с БГ, особенно на поздних стадиях заболевания, одной из проблем является снижение массы тела, в связи с чем очень важно обеспечить в течение болезни здоровое питание. На поздних стадиях может потребоваться высококалорийная диета. Также может быть оправданным направление на консультацию к диетологу.
На поздних стадиях может потребоваться высококалорийная диета. Также может быть оправданным направление на консультацию к диетологу.
Что приводит к развитию БГ?
БГ развивается вследствие изменения (увеличения — экспансии — копий одного из участков) гена (HTT), кодирующего белок под названием гентингтин. Из-за этой экспансии с гена образуется изменённый вариант белка, что, в свою очередь, приводит к нарушению функционирования и гибели нервных клеток (нейронов) в определённых участках головного мозга. Поскольку у гентингтина имеется множество функций, точные механизмы развития заболевания сложны и многогранны. Для того, чтобы разработать методы лечения, изменяющие течение болезни, исследователи работают над подробным изучением механизмов её развития.
Что является причиной развития БГ?
В 1993 г. учёные выявили мутацию, приводящую к развитию БГ. Ген HTT расположен на 4-й хромосоме и кодирует белок под названием гентингтин. Этот ген содержит последовательность из трёх нуклеотидов (базовых компонентов ДНК) — цитозин-аденин-гуанин (CAG) — которая несколько раз повторяется. Число этих так называемых тринуклеотидных повторов может варьировать. Если у человека имеется 40 или более CAG-повторов в одной из копий гена HTT, то в течение жизни (при условии её нормальной продолжительности), т.е. в среднем возрасте, у него разовьётся БГ. Поскольку мутация, являющаяся причиной развития БГ, присутствует во всех клетках организма с самого зачатия и может передаваться последующим поколениям, это заболевание является наследственным.
учёные выявили мутацию, приводящую к развитию БГ. Ген HTT расположен на 4-й хромосоме и кодирует белок под названием гентингтин. Этот ген содержит последовательность из трёх нуклеотидов (базовых компонентов ДНК) — цитозин-аденин-гуанин (CAG) — которая несколько раз повторяется. Число этих так называемых тринуклеотидных повторов может варьировать. Если у человека имеется 40 или более CAG-повторов в одной из копий гена HTT, то в течение жизни (при условии её нормальной продолжительности), т.е. в среднем возрасте, у него разовьётся БГ. Поскольку мутация, являющаяся причиной развития БГ, присутствует во всех клетках организма с самого зачатия и может передаваться последующим поколениям, это заболевание является наследственным.
Ген гентингтина (HTT) расположен на 4-й хромосоме. В нём содержатся повторяющиеся последовательности трёх базовых компонентов ДНК — C-A-G. Если у человека имеется 40 или более повторов C-A-G, то в течение жизни (при условии её нормальной продолжительности) у него разовьётся болезнь Гентингтона.
Что означает число CAG-повторов в гене
HTT?
По мере увеличения числа CAG-повторов определённый участок ДНК становится более нестабильным. Нестабильность означает, что при передаче следующему поколению число этих повторов может либо увеличиться, либо уменьшиться. Если число CAG-повторов в гене HTT менее 27, то этот участок стабилен. Если число повторов находится между 27 и 35 (так называемое промежуточное число повторов), у человека не разовьётся БГ, т.к. это укладывается в нормальные значения. Однако число CAG-повторов в 27 и более нестабильно, и существует риск увеличения их числа в следующем поколении, что обусловливает риск развития БГ у этих детей. У лиц, имеющих от 36 до 39 CAG-повторов, БГ может развиться, но только в пожилом возрасте. Этот диапазон известен как число повторов с неполной пенетрантностью. В случае наличия у человека более 39 CAG-повторов в течение жизни (при условии её нормальной продолжительности) у него разовьётся БГ — чаще всего, в среднем возрасте. В редких случаях число CAG-повторов может быть исключительно большим, что приводит к дебюту заболевания в подростковом или детском возрасте (ювенильная БГ). У пациентов с началом заболевания до 10 лет нередко имеется более 80 CAG-повторов.
В редких случаях число CAG-повторов может быть исключительно большим, что приводит к дебюту заболевания в подростковом или детском возрасте (ювенильная БГ). У пациентов с началом заболевания до 10 лет нередко имеется более 80 CAG-повторов.
| Число CAG-повторов | Разовьётся ли болезнь? | Последствия для потомства | Название состояния |
|---|---|---|---|
| Менее 27 | Нет | Нет | Нормальное число повторов |
| 27–35 | Нет | Число CAG-повторов от 27 до 35 является нестабильным и может увеличиться при передаче следующему поколению | Промежуточное число повторов |
| 36–39 | Вероятно | Есть. Для каждого из детей риск унаследовать мутацию составляет 50 % | Увеличение числа повторов с неполной пенетрантностью |
| 40 и более | Да | Есть. Для каждого из детей риск унаследовать мутацию составляет 50 % | Увеличение числа повторов с полной пенетрантностью |
Что такое ген?
Гены находятся на хромосомах внутри каждой клетки нашего тела. Ген — это участок ДНК, содержащий код для образования определённого белка; с ДНК синтезируется матричная РНК (мРНК), с которой затем образуется белок. В большинстве случаев каждый человек наследует по две копии каждого гена: одну от мамы и одну от папы. При БГ поражается ген HTT, который кодирует белок под названием гентингтин. Если ребёнок наследует копию гена HTT с увеличенным числом CAG-повторов, у него тоже разовьётся БГ. При этом у его родителя уже могут быть симптомы заболевания либо они разовьются позже в течение жизни.
Ген — это участок ДНК, содержащий код для образования определённого белка; с ДНК синтезируется матричная РНК (мРНК), с которой затем образуется белок. В большинстве случаев каждый человек наследует по две копии каждого гена: одну от мамы и одну от папы. При БГ поражается ген HTT, который кодирует белок под названием гентингтин. Если ребёнок наследует копию гена HTT с увеличенным числом CAG-повторов, у него тоже разовьётся БГ. При этом у его родителя уже могут быть симптомы заболевания либо они разовьются позже в течение жизни.
Пояснение к рисунку. Гены располагаются вдоль хромосом, как бусинки на леске (изображены в виде разноцветных полос). В большинстве случаев каждый человек наследует по две копии каждого гена: одну от мамы и одну от папы.
Что такое белок?
соответствующем гене. Таким образом, гены выступают в качестве схемы — последовательности инструкций для клетки о том, как построить определённые белки. Ген HTT содержит инструкции о том, как построить белок гентингтин. Белки — это молекулы, которые выполняют различные функции внутри клетки — они обеспечивают множество необходимых для неё процессов, таких как ферментативные реакции и поддержание структуры. Если белок функционирует неправильно или отсутствует, например, из-за увеличения в нём числа копий какого-то участка (экспансии), то это может повлиять на клетку и, в конечном счёте, на организм в целом, приводя в ряде случаев к развитию болезни.
Ген HTT содержит инструкции о том, как построить белок гентингтин. Белки — это молекулы, которые выполняют различные функции внутри клетки — они обеспечивают множество необходимых для неё процессов, таких как ферментативные реакции и поддержание структуры. Если белок функционирует неправильно или отсутствует, например, из-за увеличения в нём числа копий какого-то участка (экспансии), то это может повлиять на клетку и, в конечном счёте, на организм в целом, приводя в ряде случаев к развитию болезни.
Белок гентингтин
Белок гентингтин довольно крупный и образуется (или, по-научному, экспрессируется) с различной интенсивностью в каждой клетке организма человека, однако в наибольшей степени это происходит в головном мозге. По всей видимости, гентингтин является очень важным белком, т.к. его отсутствие приводит к смерти мышей ещё на эмбриональном этапе. На одном из концов молекулы гентингтина есть последовательность повторяющейся аминокислоты — глутамина. Этот особенный структурный участок, называемый полиглутаминовым повтором, как правило, содержит до 35 остатков глутамина. У людей, являющихся носителями мутации БГ, этот участок гентингтина содержит не менее 36 повторов, что приводит к нарушению функционирования белка.
Этот особенный структурный участок, называемый полиглутаминовым повтором, как правило, содержит до 35 остатков глутамина. У людей, являющихся носителями мутации БГ, этот участок гентингтина содержит не менее 36 повторов, что приводит к нарушению функционирования белка.
Каким образом наследуется БГ?
БГ является доминантно наследуемым заболеванием. Это означает, что человек, рождённый с одной мутантной копией гена HTT, заболеет БГ несмотря на наличие у него и одной нормальной копии этого гена. Носитель мутации БГ, вне зависимости от того, симптомный он или нет, может передать с 50 %-ной вероятностью как нормальную, так и мутантную копию гена (при условии, что у него/неё мутантной является только одна из двух копий гена HTT). Медицинские технологии позволяют сделать так, чтобы его/её детям передалась только здоровая копия гена HTT. С другой стороны, у человека, который не унаследовал мутантный ген HTT, БГ уже никогда не разовьётся, и его/её дети тоже будут вне риска относительно этого заболевания. Мутация БГ не может перепрыгнуть поколение. Тем не менее, может случиться, что носитель мутации умер до того, как у него/неё появились бы симптомы болезни, а его его/её дети не знают, что имеют риск развития БГ.
Мутация БГ не может перепрыгнуть поколение. Тем не менее, может случиться, что носитель мутации умер до того, как у него/неё появились бы симптомы болезни, а его его/её дети не знают, что имеют риск развития БГ.
Участки головного мозга, поражающиеся при БГ
Определённые функции головного мозга, такие как контроль движений, мышление и речь, при БГ постепенно нарушаются, т.к. ответственные за них нервные клетки повреждаются и гибнут. Часть головного мозга, которая страдает в наибольшей степени при БГ, носит название полосатое тело и является частью так называемых базальных ядер — структур, расположенных глубоко в центральной части головного мозга. В первую очередь, полосатое тело отвечает за планирование и контроль движений, но оно также обеспечивает и многие другие процессы, включая мышление и эмоции. По мере прогрессирования БГ повреждается и кора головного мозга (наиболее поверхностно расположенная его часть, имеющая извилины), что усугубляет выраженность когнитивных нарушений. В целом, с течением времени БГ приводит к атрофии всего головного мозга, приводя к общей инвалидизации человека.
В целом, с течением времени БГ приводит к атрофии всего головного мозга, приводя к общей инвалидизации человека.
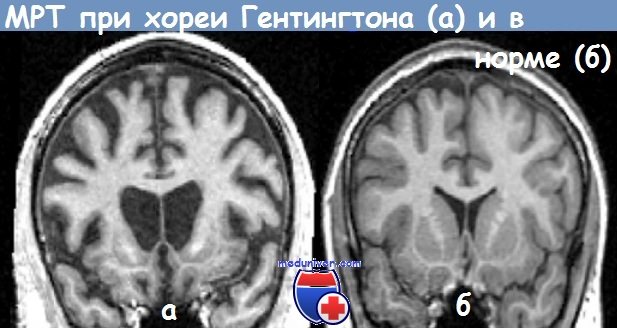
Полосатое тело представляет собой структуру, расположенную глубоко в центральной части головного мозга; при БГ оно поражается в первую очередь. Рисунок из Wikipedia.com.
Как БГ влияет на повседневную жизнь?
информирование других членов семьи о том, что они тоже имеют риск носительства мутации этого заболевания. Взрослые люди молодого возраста в особенности должны иметь в виду последствия выявления мутации БГ для образования, обучения и трудоустройства. По мере прогрессирования болезни человек постепенно теряет возможность быть жить независимо от других. Трудовая деятельность, социальная жизнь и общие повседневные дела становятся проблематичными, и пациенты становятся всё более зависимы от помощи со стороны их родственников, работников здравоохранения и социальных работников. Более подробную информацию и помощь можно получить в местных пациентских организациях и по БГ.
Более подробную информацию и помощь можно получить в местных пациентских организациях и по БГ.
Есть ли способы лучше приспособиться к жизни с БГ?
Эффективные способы приспособиться к жизни с БГ должны быть индивидуальными — они зависят от самого человека, стадии заболевания и семейного контекста. БГ развивается очень медленно, что, в целом, даёт время для приспособления к тем изменениям, что она приносит. В разработке стратегии выстраивания жизни с учётом этих изменений и в поддержании хороших отношений с человеком, страдающим БГ, ухаживающим лицами и близким людям может помочь более детальное понимание поведенческих и когнитивных нарушений, которые несёт заболевание. Полезную информацию и советы можно получить у и в пациентских организациях.
Как я могу связаться с EHDN?
На вы найдёте список языковых координаторов, которые могут вам помочь. Вы также можете воспользоваться формой для обратной связи, расположенной по этой ссылке.
Вы также можете воспользоваться формой для обратной связи, расположенной по этой ссылке.
Как я могу получить консультацию специалиста?
Вы можете записаться на консультацию, либо получив направление от вашего врача общей практики, либо связавшись с от EHDN для вашей страны, который подскажет вам, к кому можно обратиться.
Есть ли возможность поговорить со специалистом, не посещая
больницу?
Независимой мнение по БГ можно получить в пациентской организации в вашей стране.
Как я могу присоединиться к изучению БГ?
EHDN играет ключевую роль в исследовании мирового масштаба Enroll-HD. Это наблюдательное исследование, которое не подразумевает изучения действия каких-либо вмешательств. Таким образом, само по себе оно не предполагает исследования экспериментальных методов лечения. Участники исследования проходят клиническое обследование в рамках ежегодных визитов; на основании получаемых данных участники могут подходить для участия в клинических исследованиях симптоматических или болезнь-модифицирующих методов лечения, если таковые появляются.
Это наблюдательное исследование, которое не подразумевает изучения действия каких-либо вмешательств. Таким образом, само по себе оно не предполагает исследования экспериментальных методов лечения. Участники исследования проходят клиническое обследование в рамках ежегодных визитов; на основании получаемых данных участники могут подходить для участия в клинических исследованиях симптоматических или болезнь-модифицирующих методов лечения, если таковые появляются.
Участие в Enroll-HD доступно на базе многих исследовательских центров по БГ в разных странах мира. Выяснить, есть ли такой центр вблизи вашего места проживания, можно здесь либо у по вашей стране, который сможет проинформировать вас об исследованиях, проводимых в вашем регионе. Ваша местная пациентская организация(и) сможет(гут) предоставить вам общую информацию об участии в исследованиях. Для более подробной информации об изучении БГ пройдите, пожалуйста, по или посетите сайт HDBuzz, на котором вы сможете найти новости об исследовании БГ, написанные учёными понятным языком и переведённые на большинство языков.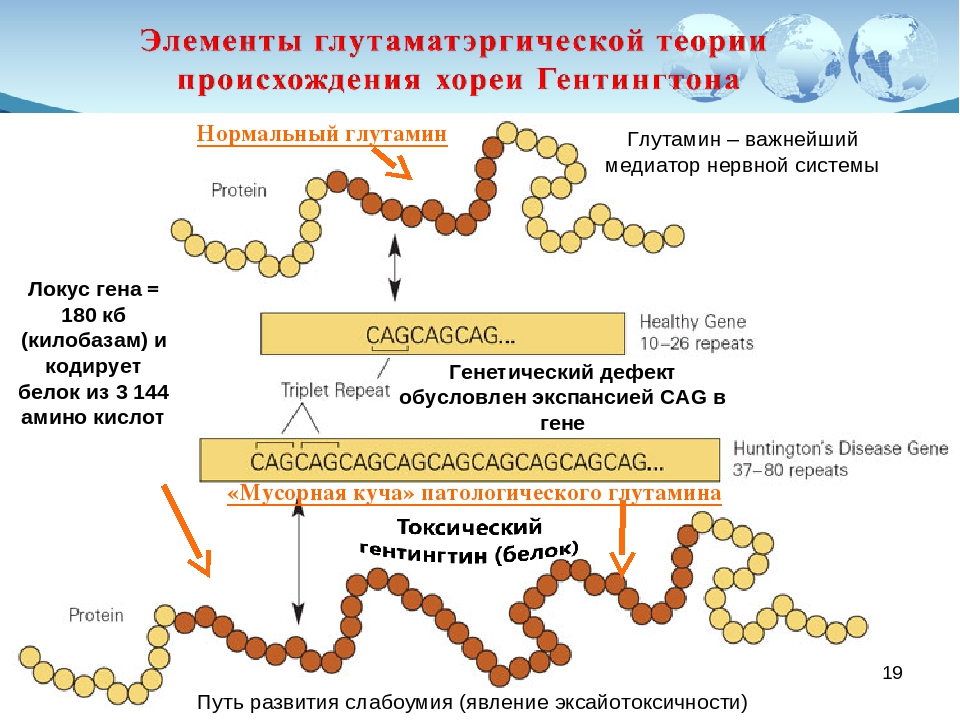
Существуют ли группы поддержки, специализирующиеся на БГ?
Да, ряд организаций по защите прав пациентов предоставляет поддержку людям и семьям, связанным с БГ. Связаться с этими организациями можно через вашего врача общей практики или специалиста по БГ; вы можете также обратиться в эти организации сами напрямую. Европейская ассоциация по болезни Гентингтона (англ. European Huntington’s disease Association, (EHA) ведёт список пациентских организаций которые могут вам помочь.
По любым другим вопросам свяжитесь, пожалуйста, с вашим от EHDN или с местной пациентской организацией.
Пляска святого Витта | Официальный сайт Научного центра неврологии
С.А. Клюшников
кандидат медицинских наук
ГУ НЦ неврологии РАМН
Первые упоминания о необычном заболевании, именуемом сейчас болезнью Гентингтона (БГ), встречаются еще в западноевропейских исторических документах XVI—XVIII веков. Многие обращали внимание на самое яркое внешнее проявление заболевания — непроизвольные движения рук, ног, туловища больных, нередко напоминающие своеобразный танец. Неврологи называют подобный вид насильственных (то есть не поддающихся произвольному контролю) движений хореей, откуда и пошло распространенное синонимичное название БГ — хорея Гентингтона. Из глубокого средневековья до наших дней дошло еще одно название заболевания — «пляска Святого Витта»; этот необычный термин известен многим людям, не имеющим отношения к истории медицины и неврологии. Святой Витт был историческим персонажем и жил на Сицилии во времена начала упадка Римской империи. Этот юный христианин был замучен римлянами в 303 году во времена гонений на христиан, развернутых императором Диоклетианом. Спустя 1200 лет (с XVI века) его имя стало ассоциироваться с «пляской». Тогда по неизвестным причинам по всей Германии распространилось поверье, что всякий, кто спляшет перед статуей святого Витта в его день (15 июня), получит заряд бодрости на весь год.
Многие обращали внимание на самое яркое внешнее проявление заболевания — непроизвольные движения рук, ног, туловища больных, нередко напоминающие своеобразный танец. Неврологи называют подобный вид насильственных (то есть не поддающихся произвольному контролю) движений хореей, откуда и пошло распространенное синонимичное название БГ — хорея Гентингтона. Из глубокого средневековья до наших дней дошло еще одно название заболевания — «пляска Святого Витта»; этот необычный термин известен многим людям, не имеющим отношения к истории медицины и неврологии. Святой Витт был историческим персонажем и жил на Сицилии во времена начала упадка Римской империи. Этот юный христианин был замучен римлянами в 303 году во времена гонений на христиан, развернутых императором Диоклетианом. Спустя 1200 лет (с XVI века) его имя стало ассоциироваться с «пляской». Тогда по неизвестным причинам по всей Германии распространилось поверье, что всякий, кто спляшет перед статуей святого Витта в его день (15 июня), получит заряд бодрости на весь год.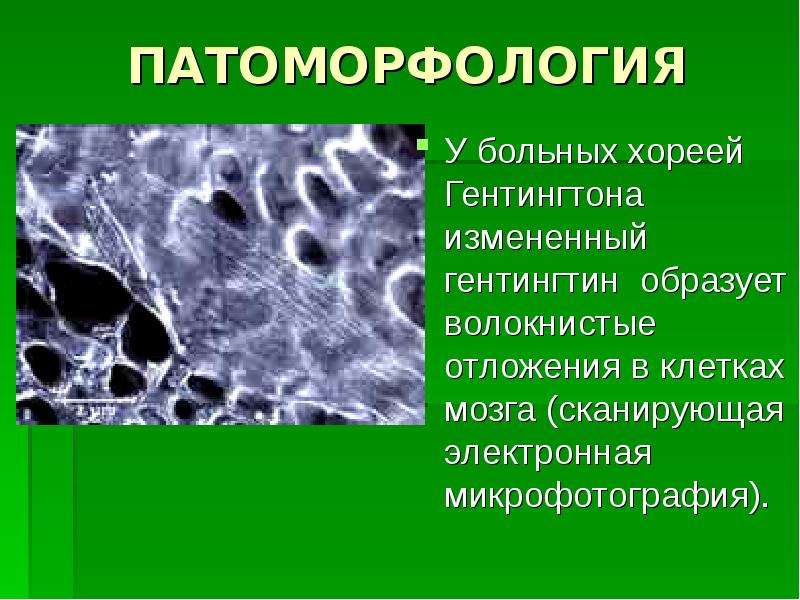 Тысячи людей толпились вокруг статуй святого в этот день, и их пляски нередко носили весьма экспансивный, эмоциональный характер. В конце концов хорею стали называть «пляской святого Витта» и даже пытались прибегать к помощи этого святого с целью излечения.
Тысячи людей толпились вокруг статуй святого в этот день, и их пляски нередко носили весьма экспансивный, эмоциональный характер. В конце концов хорею стали называть «пляской святого Витта» и даже пытались прибегать к помощи этого святого с целью излечения.
Современный научный этап в изучении БГ ведет свой отсчет с 1872 года, когда на заседании Медицинского научного общества штата Огайо (США) Джордж Генингтон (George Huntington) представил блестящее клиническое подробное описание заболевания, основанное на анализе многих собственных наблюдений. Не случайно наследственная хорея была названа именем этого американского врача. Последующие десятилетия были периодом накопления клинических фактов и результатов анализа родословных, попытками их систематизации. В 1983 году БГ стала первым наследственным неврологическим заболеванием, при котором была установлена точная локализация патологического гена на определенной хромосоме. Спустя 10 лет ученые смогли расшифровать точную структуру патологического гена заболевания. Одновременно были разработаны методы ДНК-диагностики, позволившие устанавливать носительство патологического гена БГ задолго до непосредственного появления симптомов болезни.
Одновременно были разработаны методы ДНК-диагностики, позволившие устанавливать носительство патологического гена БГ задолго до непосредственного появления симптомов болезни.
По распространенности БГ является одним из самых частых наследственных заболеваний нервной системы — в среднем 5-7 случаев на 100000 населения. БГ — наследственное прогрессирующее заболевание головного мозга, начинающееся, как правило, в среднем возрасте (около 40 лет), основным внешним клиническим проявлением которого являются непроизвольные движения рук, ног, туловища, нередко мимических мышц лица, называемые хореическими гиперкинезами. В начале заболевания они носят едва заметный, «случайный» характер. Нередко на ранних стадиях заболевания явных гиперкинезов не видно, но улавливается некоторое двигательное беспокойство человека, ему «не сидится на месте». В дальнейшем, по мере развития заболевания, хореические гиперкинезы усиливаются, затрагивают всё новые и новые группы мышц. Постоянная мышечная активность отнимает у больных много сил, что часто проявляется характерной жалобой пациентов на общую слабость. При длительном многолетнем течении заболевания интенсивность гиперкинезов нередко снижается, и избыточная двигательная активность сменяется общей заторможенностью, напоминающей таковую при болезни Паркинсона. Характерными проявлениями заболевания являются также нарушения памяти, мышления, интеллектуальной деятельности, то есть когнитивные нарушения (или расстройства познавательной функции головного мозга). В первую очередь страдает кратковременная память, нарушается концентрация внимания, человек становится рассеянным. Одновременно утрачивается аналитическая функция головного мозга, способность к абстракциям, обобщениям и логическим умозаключениям, мышление становится примитивным, наблюдается утрата привычных интересов. Страдают личностные характеристики человека, изменяется характер, появляются эмоционально-волевые и нередко психические нарушения. Течение БГ отличается медленным, но неуклонным прогрессированием на протяжении 15-20 лет. Развитие БГ неизбежно приводит к инвалидизации и необходимости посторонней бытовой помощи.
При длительном многолетнем течении заболевания интенсивность гиперкинезов нередко снижается, и избыточная двигательная активность сменяется общей заторможенностью, напоминающей таковую при болезни Паркинсона. Характерными проявлениями заболевания являются также нарушения памяти, мышления, интеллектуальной деятельности, то есть когнитивные нарушения (или расстройства познавательной функции головного мозга). В первую очередь страдает кратковременная память, нарушается концентрация внимания, человек становится рассеянным. Одновременно утрачивается аналитическая функция головного мозга, способность к абстракциям, обобщениям и логическим умозаключениям, мышление становится примитивным, наблюдается утрата привычных интересов. Страдают личностные характеристики человека, изменяется характер, появляются эмоционально-волевые и нередко психические нарушения. Течение БГ отличается медленным, но неуклонным прогрессированием на протяжении 15-20 лет. Развитие БГ неизбежно приводит к инвалидизации и необходимости посторонней бытовой помощи.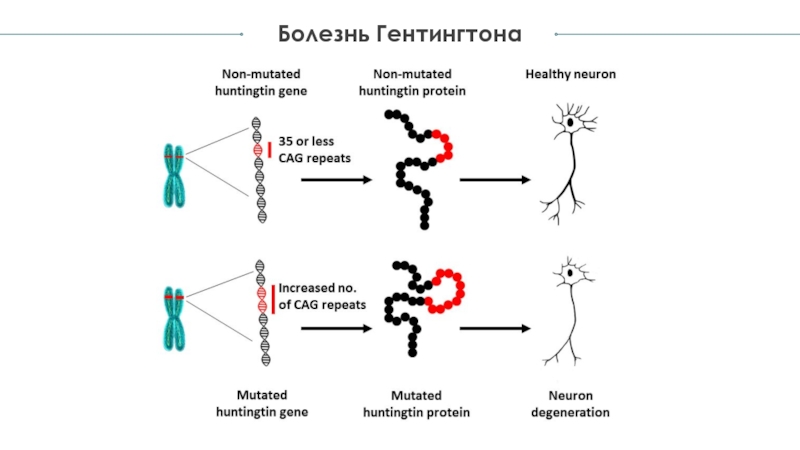
БГ передается наследственным путем по аутосомно-доминантному типу, означающему наследование от одного из больных родителей (независимо от их пола) с вероятностью 50% для каждого ребенка. Болеют также лица обоего пола. Важной характеристикой аутосомно-доминантного механизма наследования гена БГ является так называемый вертикальный путь передачи, с наличием случаев заболевания в каждом поколении, без пропусков. Если в каком-то поколении все родственники являются генетически здоровыми, то дальнейшее наследование заболевания в роду прекращается — через поколение БГ не «перескакивает». В каждом последующем поколении наблюдается более раннее начало заболевания и более тяжелое его течение. Это явление получило название «антиципация». Близко к этому феномену находится и так называемый «эффект отцовской передачи», заключающийся в том, что заболевание с более ранним началом и более тяжелым течением развивается преимущественно при передаче по отцовской линии.
Многие тонкие молекулярные механизмы развития БГ были раскрыты в течение последнего десятилетия.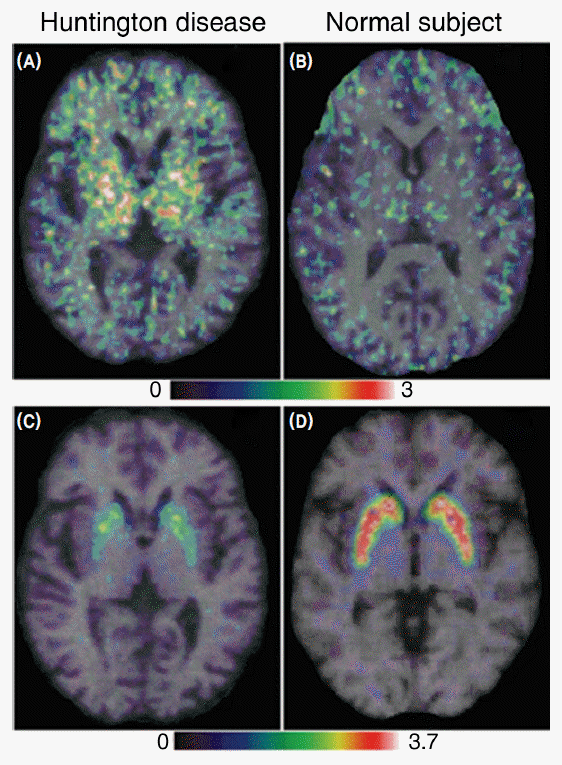 Особая динамическая мутация в гене (увеличение числа копий тринуклеотидных повторов) приводит к нарушению нормальной пространственной укладки соответствующего белкового продукта и приобретению этим мутантным белком токсических свойств. Аномальный белок накапливается в определенных клетках головного мозга, приводя к их гибели. К сожалению, в настоящее время средств прямого воздействия на механизмы развития заболевания не существует. Применяемые современные препрепараты (такие как галоперидол, тиаприд, семакс, мемантин и др.) направлены на облегчение состояния больных, уменьшение насильственных движений, смягчение психических и интеллектуальных нарушений. Однако комплексное лечение БГ позволяет в ряде случаев притормозить развитие заболевания и улучшить качество жизни пациентов.
Особая динамическая мутация в гене (увеличение числа копий тринуклеотидных повторов) приводит к нарушению нормальной пространственной укладки соответствующего белкового продукта и приобретению этим мутантным белком токсических свойств. Аномальный белок накапливается в определенных клетках головного мозга, приводя к их гибели. К сожалению, в настоящее время средств прямого воздействия на механизмы развития заболевания не существует. Применяемые современные препрепараты (такие как галоперидол, тиаприд, семакс, мемантин и др.) направлены на облегчение состояния больных, уменьшение насильственных движений, смягчение психических и интеллектуальных нарушений. Однако комплексное лечение БГ позволяет в ряде случаев притормозить развитие заболевания и улучшить качество жизни пациентов.
ДНК-диагностика (генная диагностика) БГ, как и любого другого наследственного заболевания, является основой системы медико-генетического консультирования — особого вида специализированной медицинской помощи, направленного на предупреждение появления повторных случаев наследственных заболеваний в отягощенных семьях.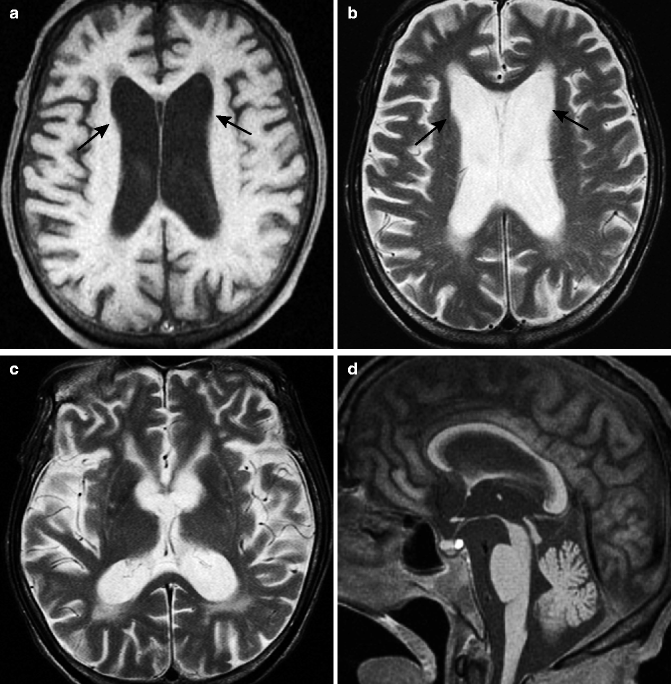 Медико-генетическое консультирование позволяет установить точный диагноз, рассчитать генетический риск у консультируемых родственников, включая точное установление их генетического статуса с помощью прогностического тестирования, определить прогноз для потомства, в том
Медико-генетическое консультирование позволяет установить точный диагноз, рассчитать генетический риск у консультируемых родственников, включая точное установление их генетического статуса с помощью прогностического тестирования, определить прогноз для потомства, в том
числе с помощью пренатальной ДНК-диагностики плода на ранних сроках беременности, а также помочь консультируемой семье в решении ряда других вопросов, касающихся планирования жизни, репродуктивного поведения и возможности деторождения, психологической поддержки, социальной адаптации.
В 1995 на базе нейрогенетического отделения Института неврологии РАМН (сейчас — Научного центра неврологии РАМН) была создана Российская ассоциация по борьбе с БГ. Она интегрирована в структуру Всемирной ассоциации по борьбе с БГ. Российская ассоциация основана специалистами-нейрогенетиками и работает в тесной связи с нейрогенетической клиникой и ДНК-лабораторией Научного центра неврологии РАМН. На основании полученного опыта Ассоциацией отработаны собственные этические и организационные принципы проведения медико-генетического консультирования в отягощенных семьях. Пациенты, страдающие БГ и находящиеся в поле зрения Ассоциации, имеют возможность периодически проходить стационарное лечение в нейрогенетической клинике Научного центра неврологии РАМН и получать амбулаторную консультативную помощь. Ассоциация по борьбе с болезнью Гентингтона России считает также своей целью всестороннюю информационно-образовательную поддержку отягощенных семей, для чего выпускаются специальные буклеты, проводятся семинары для членов семей с БГ и другие мероприятия.
Пациенты, страдающие БГ и находящиеся в поле зрения Ассоциации, имеют возможность периодически проходить стационарное лечение в нейрогенетической клинике Научного центра неврологии РАМН и получать амбулаторную консультативную помощь. Ассоциация по борьбе с болезнью Гентингтона России считает также своей целью всестороннюю информационно-образовательную поддержку отягощенных семей, для чего выпускаются специальные буклеты, проводятся семинары для членов семей с БГ и другие мероприятия.
© Журнал «Нервы», 2007, №1
наверх
Салтыков-Щедрин: биография и его сказки
Михаил Салтыков (псевдоним «Щедрин» присоединится к фамилии позже) стал шестым по счёту ребенком в старинной дворянской семье. Родился он в Калязинском уезде Тверской губернии. Там же начал получать образование – сначала с помощью сестры, батюшки местной церкви, крепостного художника, затем в Московском дворянском институте и, наконец, в знаменитом Царскосельском лицее.
В биографии этого человека удивительным образом, не препятствуя друг другу, переплетаются две основных линии – канцелярского чиновника и талантливого литератора. В разные годы Салтыков-Щедрин занимал должности старшего чиновника особых поручений (при вятском губернаторе), правителя губернаторской канцелярии, советника губернского правления. Дважды, сначала в Рязани, потом в Твери, назначался вице-губернатором.
В разные годы Салтыков-Щедрин занимал должности старшего чиновника особых поручений (при вятском губернаторе), правителя губернаторской канцелярии, советника губернского правления. Дважды, сначала в Рязани, потом в Твери, назначался вице-губернатором.
В ходе своих многочисленных служебных командировок в самые глухие места провинциальной глубинки, Салтыков близко познакомился с жизнью крестьян и помещиков. Что нашло отражение в его блестящих «Губернских очерках», которые сразу сделали автора популярным в литературной среде.
Позже из-под пера писателя-чиновника выйдут «Письма из провинции», «История одного города», «Помпадуры и Помпадурши», всем сегодня известные «Господа Головлёвы».
Отдельной строкой выделяется цикл сатирических сказок Салтыкова-Щедрина. В общей сложности их насчитывается 32, написанных Михаилом Евграфовичем за 18 лет. Писатель хотел выпустить сказки одним сборником в виде предельно дешевой брошюры, чтобы она оказалась доступной для приобретения читателями из самых разных социальных слоёв.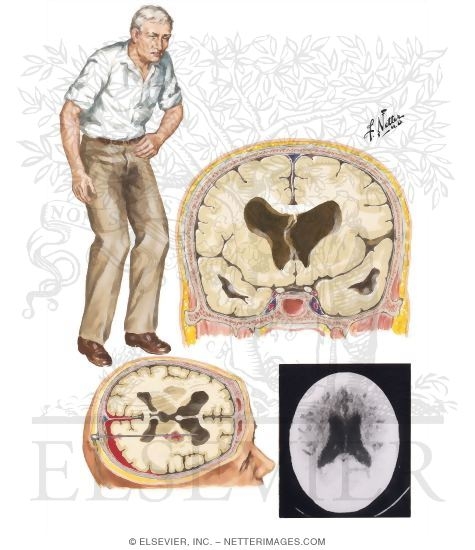
Однако цензура наложила запрет на такое издание в его полном виде, так как в сказочных героях легко угадывались их прототипы – российские самодуры, казнокрады и взяточники. Отдельные произведения из этого цикла – скажем, «Богатырь» – и вовсе увидели свет лишь после 1917 года.
Тем не менее, сказки Салтыкова-Щедрина были очень популярны, перепечатывались нелегально, распространяясь как в России, так и за рубежом.
Проблемы со здоровьем начались у писателя довольно рано: первые признаки ревматизма появились в 23 года. Уже в тридцать лет медики обнаруживают у литератора порок сердца.
Пришлось начать лечение. Сколь длительное, столь же, увы, и малоэффективное. Баден-Баден, Париж, Бад-Эльстер, Висбаден, вновь Баден-Баден – здесь Салтыкова-Щедрина пытаются выходить многие известные врачи. Лечат минеральными водами, молочной сывороткой, писатель регулярно принимает минеральные ванны.
Когда писателю не исполнилось еще и шестидесяти лет, врач С.П. Боткин отмечал у него целый «букет» болезней: острый полиартрит, эндокардию, перикардию, ревматическую пневмонию.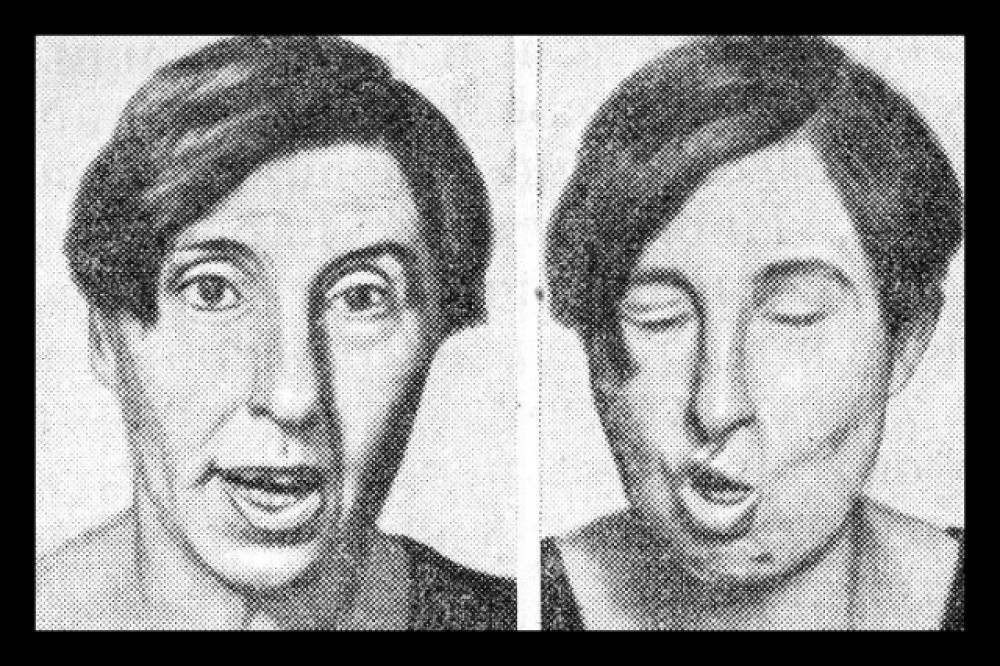
Ко всем прочим напастям прибавляется новая. Первым её замечает доктор Н.А. Белоголовый. У Салтыкова-Щедрина появляются непроизвольные подёргивания век, глаз, мышц лица, всё чаще выходят из-под контроля больного движения рук, шеи. Писатель становится раздражённым, беспокойным, его порой преследуют страхи перед безумием, посещают мысли о суициде.
Врач делает предположение о том, что писатель стал жертвой «ревматической хореи». Сегодня на современном медицинском языке это заболевание называется «хорея Гентингтона». Что это такое?
Речь идёт о генетическом заболевании нервной системы, связанным с нарушением двигательных функций. Этот недуг убивает нейроны головного мозга, отвечающие за контроль и координацию произвольных движений, а также регуляцию мышечного тонуса. Кстати, сегодня только в США насчитывается около семи тысяч таких больных.
С большой долей вероятности можно предположить, что вывод доктора Н.А. Белоголового в отношении Салтыкова-Щедрина оказался верным, так как очень уж характерна описанная врачом симптоматика.
Это подтверждается следующими признаками, обнаруженными у литератора: нервные, неконтролируемые подергивания мышц лица (можно встретить другое название хореи – «пляска святого Витта»), размашистая, плохо координируемая жестикуляция, неровная походка.
Насколько успешно сегодня диагностируется заболевание? Это делается с помощью компьютерной и магнитно-резонансной томографии. Именно такие способы позволяют выявить нарушения состояния головного мозга и своевременно начать лечение.
При заболевании человека хореей прогноз, к сожалению, не очень утешительный. Несмотря на все, даже самые современные, достижения медицины, способов полного излечения от заболевания пока не найдено. При своевременном обнаружении хореи можно лишь уменьшить степень проявления болезни.
Статистика показывает, что с момента заболевания хореей Гентингтона человек живёт еще 10-15 лет. Что, в общем, подтверждается на примере жизни, заболевания и смерти М.Е. Салтыкова-Щедрина. Он умер в 1889 году в возрасте 63 лет.
Некоторые исследователи жизни и творчества писателя склонны думать, что в «обличителя пороков современности» он превратился, преследуемый своими болезнями. Однако прямых доказательств этому – чем более, подтверждённых с медицинской точки зрения – нет.
Что же касается столь рано появившегося у писателя ревматизма, то здесь, вероятно, можно говорить о таком заболевании, как ревматоидный артрит. Каковы симптомы болезни, её диагностика и лечение у взрослых в современных условиях?
Здесь мы имеем дело с воспалением тканей суставов. Болезнь носит аутоиммунный характер: по какой-то причине лимфоциты человека в его крови входят в «конфликт» с абсолютно здоровыми клетками соединительной ткани суставов, в результате чего количество межсуставной жидкости уменьшается, а это, в свою очередь, приводит к воспалению, опухоли суставов. И острым болям.
Диагностируется заболевание посредством комплексного обследования, включающим в себя, в том числе, магнито-резонансные и компьютерные исследования.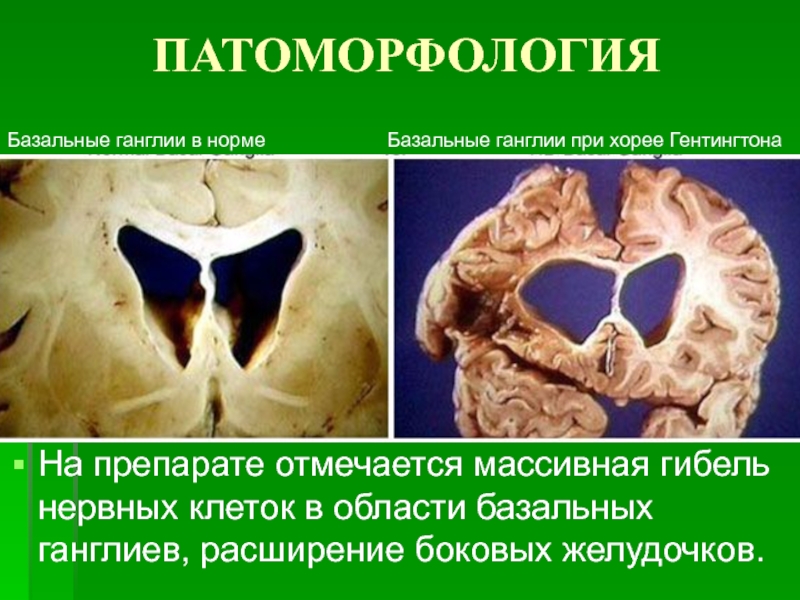 Лечение ставит главной своей целью использование специальных препаратов (противоревматические препараты для суставов, цитостатики и иммунодепрессанты), которые помогают противостоять дальнейшему развитию аутоиммунной реакции.
Лечение ставит главной своей целью использование специальных препаратов (противоревматические препараты для суставов, цитостатики и иммунодепрессанты), которые помогают противостоять дальнейшему развитию аутоиммунной реакции.
Клеточная модель хореи Гентингтона – Наука – Коммерсантъ
Исследователи Института цитологии и генетики в содружестве с коллегами из Института химической биологии и фундаментальной медицины Сибирского отделения РАН использовали редактирование генома для создания клеточной модели хореи Гентингтона, неизлечимой генетической болезни.
Мутации генов, ведущие к хорее Гентингтона, были открыты 20 лет назад, но за эти годы ученым не удалось создать лекарство, позволяющее исцелить от этого заболевания или замедлить его течение; медики могут только лишь облегчить некоторые симптомы. При хорее Гентингтона у больного постепенно гибнут определенного типа нейроны стриатума, структуры переднего мозга, в результате чего мозг начинает неизбежно терять контроль над работой организма.
Эта нейродегенеративная патология связана с увеличением числа ЦАГ-последовательностей (азотистых оснований цитозин-аденин-гуанин) в гене HTT, который кодирует белок гентингтин, названный в честь того же ученого, что и заболевание. Науке неизвестно, за что отвечает этот белок в здоровом организме, но у больного хореей Гентингтона он формирует белковые структуры в нейронах стриатума, которые своей жизнедеятельностью отравляют нервные клетки и в конце концов убивают их.
Для того чтобы лучше понять механизм развития заболевания и, возможно, найти способ исцелить его, сибирские ученые создали при помощи генного редактирования изогенные линии пар, имеющие одинаковый генетический фон и различающиеся между собой исключительно наличием или отсутствием пагубной мутации. Чистоты такого соотношения опыта и контроля практически невозможно добиться без редактирования генома, поскольку человеческие гены отличаются бесчисленным количеством полиморфизмов, которые могут влиять на общую картину.
Сибирские исследователи внесли соответствующие мутации в генетическую базу здоровых клеток. Такая методика позволит ученым рассмотреть целый спектр различных линий пар, содержащих различное число ЦАГ-повторов в гене HTT.
Петр Харатьян
Хорея — Chorea — xcv.wiki
Хорея ( иногда хорея ) — это ненормальное непроизвольное двигательное расстройство , одно из группы неврологических расстройств, называемых дискинезиями . Термин хорея происходит от древнегреческого : χορεία («танец»; см. Хорея ), поскольку быстрые движения ног или рук сравнимы с танцем.
Термин гемихорея относится к хореи одной стороны тела, такой как хорея одной руки, но не обеих (аналог гемибаллизма ).
Презентация
Хорея характеризуется короткими, полунаправленными, нерегулярными движениями, которые не являются повторяющимися или ритмичными, но кажутся переходящими от одной мышцы к другой. Эти «танцевальные» движения хореи часто возникают при атетозе , который добавляет скручивающие и извивающиеся движения. Ходьба может стать затруднительной и включать странные позы и движения ног.
Эти «танцевальные» движения хореи часто возникают при атетозе , который добавляет скручивающие и извивающиеся движения. Ходьба может стать затруднительной и включать странные позы и движения ног.
В отличие от атаксии , которая влияет на качество произвольных движений, или паркинсонизма , который является препятствием для произвольных движений, движения хореи и баллизма происходят сами по себе, без сознательного усилия. Таким образом, хорея считается гиперкинетическим двигательным расстройством.
Когда хорея серьезная, легкие движения станут толчками; эта форма тяжелой хореи называется баллизмом или баллизмом.
Причины
Болезнь Хантингтона
Болезнь Хантингтона — нейродегенеративное заболевание и наиболее частая наследственная причина хореи. Ранее это состояние называлось хореей Хантингтона, но было переименовано из-за важных нехореических особенностей, включая снижение когнитивных функций и изменение поведения.
Другие генетические причины
Другие генетические причины хореи встречаются редко. К ним относятся классические «мимические» или фенокопийные синдромы болезни Хантингтона , называемые синдромом, подобным болезни Хантингтона, типы 1, 2 и 3; наследственное прионное заболевание , спиноцеребеллярная атаксия 1, 3 и 17 типов , нейроакантоцитоз , зубочелюстно-паллидолуизиальная атрофия (DRPLA), нарушения накопления железа в мозге , болезнь Вильсона , доброкачественная наследственная хорея , атаксия Фридрейха , митохондриальная болезнь и синдром Ретта .
Приобретенные причины
Наиболее частыми приобретенными причинами хореи являются цереброваскулярные заболевания и, в развивающихся странах, ВИЧ- инфекция, обычно в связи с криптококковой болезнью .
Хорея Сиденхама возникает как осложнение стрептококковой инфекции. У 20% (20%) детей и подростков с ревматической лихорадкой в качестве осложнения развивается хорея Сиденхама. Он встречается все реже, что частично может быть связано с пенициллином, улучшением социальных условий и / или естественным сокращением количества бактерий (Streptococcus), от которых он произошел. Психологические симптомы могут предшествовать этой приобретенной хореи или сопровождать ее, а также могут быть рецидивирующими и переходящими. Более широкий спектр детских аутоиммунных нейропсихиатрических расстройств, связанных со стрептококковой инфекцией, может вызывать хорею и в совокупности называется PANDAS .
Он встречается все реже, что частично может быть связано с пенициллином, улучшением социальных условий и / или естественным сокращением количества бактерий (Streptococcus), от которых он произошел. Психологические симптомы могут предшествовать этой приобретенной хореи или сопровождать ее, а также могут быть рецидивирующими и переходящими. Более широкий спектр детских аутоиммунных нейропсихиатрических расстройств, связанных со стрептококковой инфекцией, может вызывать хорею и в совокупности называется PANDAS .
Chorea gravidarum относится к хореическим симптомам, возникающим во время беременности. Если не лечить, болезнь проходит у 30% пациентов до родов, но у остальных 70% она сохраняется. Затем симптомы постепенно исчезают в течение следующих нескольких дней после родов.
Хорея также может быть вызвана лекарствами (обычно леводопа , противосудорожные и антипсихотические препараты ).
Другие приобретенные причины включают утечку спинномозговой жидкости , системную красную волчанку , антифосфолипидный синдром , тиреотоксикоз , красную полицитемию , трансмиссивные губчатые энцефалопатии и целиакию .
лечение
Стандартного курса лечения хореи не существует. Лечение зависит от типа хореи и связанного с ней заболевания. Несмотря на то, что существует множество лекарств, которые могут контролировать это, лекарства до сих пор не найдено.
История
Исторически такие хореи, как болезнь Хантингтона и хорея Сиденхэма, назывались танцем Святого Вита , связанным с серией одноименных социальных явлений .
Смотрите также
Ноты
внешние ссылки
Как спасти Тринадцатую? (Перспективы лечения болезни Хантингтона)
Статья на конкурс «био/мол/текст»: Сейчас сложно найти человека, который никогда не слышал про болезни Альцгеймера, Паркинсона или Хантингтона. Эти недуги относятся к группе нейродегенеративных заболеваний, вызывающих гибель нейронов и постепенное разрушение головного мозга. К сожалению, все они являются неизлечимыми. Поэтому ученые активно работают над тем, чтобы раскрыть механизмы развития этих болезней и найти терапию, которая поможет спасти пациентов. В своем исследовании мы обратились к пока еще малоизученному вопросу — что происходит с синаптической связью нейронов при нейродегенеративном процессе? Результаты этой работы открывают новое направление для разработки лекарства от болезни Хантингтона и других нейродегенеративных заболеваний.
Эта работа заняла первое место в номинации «Своя работа» конкурса «био/мол/текст»-2013.
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
С увеличением средней продолжительности жизни все больше людей страдают от болезни Альцгеймера и болезни Паркинсона. К сожалению, годы исследований пока не привели ученых к открытию причин развития этих заболеваний и возможной терапии. Это связано, главным образом, с тем, что почти ничего не известно о факторах, вызывающих болезнь, а также с тем, что очень мало пациентов имеют генетическую предрасположенность. Чаще всего эти заболевания являются спорадическими, т.е. причины их возникновения не установлены. Это приводит к бесконечным спорам — никто не знает, как искусственно вызвать это заболевание у модельных животных для экспериментов и поиска лекарств. Поэтому все больше ученых обращают свое внимание на генетические заболевания нервной системы, такие как болезнь Хантингтона (БХ). Это заболевание также, как болезнь Альцгеймера и болезнь Паркинсона, относится к группе нейродегенеративных заболеваний, с которыми его объединяет ряд схожих черт: гибель нейронов центральной нервной системы, накопление амилоидоподобных агрегатов белков, когнитивные и двигательные нарушения у больных. При этом БХ имеет важное преимущество с точки зрения исследователей, т.к. известно, какая мутация вызывает это заболевание. Это дает возможность создавать точные генетические модели и исследовать их на животных. Это важно, потому что если мы поймем патогенез болезни Хантингтона, то нам легче будет разобраться и со спорадическими нейродегенеративными заболеваниями. Это мы и попытались сделать в своем исследовании.
Болезнь Хантингтона
Болезнь Хантингтона (БХ, в русскоязычной литературе также «болезнь Гентингтона») — наследственное заболевание нервной системы, которое поражает примерно 1 из 10 тыс. людей. Болезнь была впервые описана Джорджем Хантингтоном (George Huntington) в 1872, и с тех пор носит его имя, однако клинические симптомы этого заболевания были известны еще в XVI веке под названием «хорея» (от лат. choreus — танец). К признакам хореи относили непроизвольные, нескоординированные быстрые движения, похожие на судороги; именно так описывают и современные медики моторные нарушения, характерные для БХ. Болезнь может порой длиться до двадцати лет, но исход неизменно один и тот же: больной теряет способность самостоятельно передвигаться, говорить, а затем и мыслить. Как правило, симптомы болезни Хантингтона проявляются в возрасте от 30 до 50 лет, хотя у 5–10% пациентов отмечается появление симптомов в возрасте до 20 лет — так называемая ювенильная форма заболевания [1].
Первый симптом болезни Хантингтона — непроизвольные подёргивания конечностей, торса и лицевых мышц. Довольно часто они сопровождаются резкими сменами настроения, депрессией, раздражительностью, неразборчивостью речи и неуклюжестью движений. По мере прогрессирования болезни, к этим симптомам добавляются затруднения или боль при глотании, неустойчивость походки, потеря равновесия, нарушение мыслительных функций и ухудшение памяти. В конце концов, больной теряет способность передвигаться без помощи посторонних и умирает обычно от пневмонии, остановки сердца или других осложнений.
Важной для врачей и исследователей особенностью БХ является то, что это заболевание является наследственным и вызывается мутацией в одном-единственном гене. Оказалось, что к развитию БХ приводит увеличение количества повторов триплета CAG, кодирующего глутамин, в первом экзоне гена белка хантингтина. При этом, чем больше количество повторов этого триплета, тем раньше начинается развитие заболевания. В норме в человеческой популяции встречается от 10 до 35 повторов. У пациентов с БХ количество повторов может быть от 36 до 121, при ювенильной форме — от 50 и выше [2]. Благодаря выявлению генетической основы заболевания, диагностика БХ в настоящее время не представляет проблемы; кроме того, возможной стала пренатальная диагностика заболевания и проверка эмбрионов перед имплантацией при ЭКО, которая позволяет иметь здоровых детей даже носителям мутантного гена.
К сожалению, выявление точной мутации все еще не позволяет ученым определить причину развития болезни Хантингтона и найти соответствующее лечение. Появление в клетке мутантного гена и, соответственно, измененного (мутантного) белка может привести к развитию патологии двумя путями: потеря функции (loss-of-function) или приобретение функции (gain-of-functin). В первом случае мутантный белок не может выполнять ту же функцию, что белок нормальный, и это приводит к нарушению клеточных процессов. Во втором случае, мутантный белок мешает нормальной жизнедеятельности клетки, начиная выполнять какую-то «лишнюю функцию». Чтобы разобраться, что происходит при БХ, ученые интенсивно изучают как функцию нормального белка хантингтина, так и поведение его мутантной формы [3].
К сожалению, попытки определить точную клеточную функцию хантингтина пока не увенчались успехом. Различные исследования указывают на участие этого белка в широком спектре биологических процессов, включая транспорт белков и везикул (мембранных пузырьков-транспортеров), организацию цитоскелета, клатрин-опосредованный эндоцитоз, постсинаптический сигналинг, регуляцию транскрипции и анти-апоптотические процессы [4]. Если удастся доказать, что нарушение какой-либо из этих функций является ключевым для развития заболевания, то лекарственные препараты для поддержания этой функции могут спасти пациентов с болезнью Хантингтона.
Если верна гипотеза о приобретении функции, особое внимание стоит обратить на поведение мутантной формы хантингтина. Оказалось, что мутантный белок формирует агрегаты, которые являются одной из характерных черт развития БХ как у людей,так и у модельных животных (см.врезку). Сначала агрегаты были описаны только в ядре, однако последующие работы выявили их также в цитоплазме и отростках нейронов [5]. В последние годы многие авторы склоняются к тому, что образование агрегатов несет скорее протективную функцию, а основной патогенной формой мутантного хантингтина является мономерный растворимый белок [6].
Модели для изучения болезни Хантингтона
Модели БХ на животных появились более 30 лет назад. Первыми были модели, основанные на введении в стриатум нейротоксических веществ (например, хинолиновой кислоты — агониста NMDA-рецепторов), которые вызывали гибель нейронов. В настоящее время большинство исследователей работает на моделях трансгенных животных, среди которых есть не только мыши и крысы, но и беспозвоночные животные — мушка Drosophila melanogaster и червь Caenorhabditis elegans.
Мышиные модели болезни Хантингтона отличаются друг от друга количеством CAG-повторов и уровнем экспрессии трансгена — искусственно внесенного гена хантингтина. Т.к. именно от этих факторов зависит развитие БХ, разные линии мышей отличаются друг от друга скоростью развития патологий. К наиболее широко используемым моделям относят линии мышей R6/2, R6/1 и YAC128, которые были использованы и в нашей работе. У мышей этих линий симптомы заболевания наиболее выражены и проявляются достаточно быстро. Кроме того, у этих животных с возрастом прогрессируют когнитивные и моторные нарушения, развивается частичная потеря нейронов в стриатуме и коре.
Еще одним из способов моделирования БХ является использование клеточной культуры. В самом простом случае используются культуры клеток со стабильной трансфекцией гена хантингтина. Например, это клетки линии PC12, содержащие индуцибельный трансген первого экзона хантингтина или нейроны стриатума с экспрессией фрагментов хантингтина разной длины. Кроме того, можно использовать первичные культуры из нейронов трансгенных мышей или иммортализованные нейроны.
Почему мы решили исследовать параметры синаптической передачи при болезни Хантингтона
Синаптическая передача — это передача сигналов между нейронами с помощью синаптического контакта. При возбуждении одного нейрона его синаптическое окончание выделяет в синаптическую щель медиатор — химическое вещество, которое оказывает свое возбуждающее или тормозящее воздействие на синаптическое окончание второго нейрона (рис. 1). Таким образом, синапсы связывают нейроны между собой, обеспечивая нормальное функционирование нейронных сетей и всей нервной системы. Если какая-то из систем головного мозга перестает функционировать, причина может крыться либо в нарушении работы отдельных нейронов, либо в нарушении связи между ними, т.е. нарушении синаптической передачи.
Рисунок 1. Схематическое изображение устройства синапса.
При болезни Хантингона поражается специфическая область головного мозга, называемая стриатумом. Стриатум является частью важного нейронного пути — экстрапирамидной системы, которая участвует в управлении движением и поддержании мышечного тонуса. Гибель нейронов стриатума при болезни Хантингтона приводит к разрушению экстрапирамидной системы, что связано с потерей контроля над движениями у больного человека. Но когда возникают первые патологические симптомы (тремор, нарушение координации), головной мозг человека еще не поврежден: нейроны начинают погибать только через несколько лет после начала развития заболевания. Т.е. болезнь начинается, когда что-то меняется в работе самих нейронов или в синаптической передаче, и эти нарушения впоследствии ведут к гибели нейронов и необратимым последствиям.
Накопившиеся за последние годы результаты исследований заставляют многих ученых склоняться к мысли, что именно нарушение нормальной работы системы нейрональной связи, синапсов и синаптической передачи, приводит к ранним нарушениям в работе экстрапирамидной системы. Оказалось, что в нейронах с мутациями в гене, кодирующем белок хантингтин, наблюдается целый ряд патологических изменений, которые нарушают синаптическую передачу. В таких мутантных клетках нарушается формирование и обновление запаса везикул (пузырьков с медиатором), изменяется внутриклеточная концентрация кальция , который необходим для нормального высвобождения медиатора в синаптическую щель, снижается количество ряда необходимых для функционирования синапса белков и т.п. [7]. Все это приводит к сниженному выбросу медиатора в синаптическую щель, а если медиатора недостаточно, то нейроны начинают хуже «слышать» друг друга, и команды, посылаемые корой головного мозга, не будут выполняться по всей строгости.
Изучение нарушенной синаптической передачи при БХ было темой нашего исследования. Может ли быть, что неправильная работа нейронов стриатума на ранней стадии БХ вызвана тем, что они «не слышат» команды нейронов коры? Может ли ослабление синаптической связи приводить к необратимым изменениям в нейронах стриатума и вести к их гибели? О чем мы узнали во время поиска ответов на эти вопросы, рассказано ниже.
Результаты исследований: изменения в синаптической передаче при болезни Хантингтона
Изучать синаптическую передачу можно различными способами. Например, это можно делать, проникнув в нейронную цепочку с помощью методов электрофизиологии. Нейрон проявляет свою активность с помощью электрического тока, который можно измерить. Если экспериментатор возьмет цепочку из двух нейронов и, активировав один нейрон, зарегистрирует электрическую активность второго, он сможет выяснить, насколько хорошо проходит сигнал. Другой способ изучать функционирование синаптической передачи — исследовать морфологию нейрона. Дело в том, что многие нейроны (в том числе, нейроны коры и стриатума) имеют особые выросты мембраны — шипики, которые нужны им именно для образования синапсов (рис. 2). Чем более активно нейрон «общается» со своими соседями, тем больше на его поверхности шипиков. Взяв на вооружение эти два подхода, мы решили исследовать, как работает синаптическая передача при БХ.
Рисунок 2. Дендритные шипики на поверхности стриатного нейрона. Шипики — небольшие выросты на поверхности нейрональных отростков; на увеличенном изображении они отмечены стрелками.
Рисунок 3. Нейрональная культура из нейронов коры и стриатума. С помощью специфических антител нейроны коры окрашены в красный цвет, а нейроны стриатума — в желто-зеленый.
В качестве модели для изучения болезни Хантингтона была использована клеточная культура из нейронов коры и стриатума. Для приготовления культуры незрелые нейроны из изучаемых зон мозга мышей высаживаются в чашки Петри, где они формируют полноценные нейрональные отростки и нейронные цепочки (рис. 3). Использовались мыши дикого типа (без мутаций) и мыши линии YAC128, которые несут мутацию в гене белка хантингтина и являются признанной моделью БХ. На 14–15 день после высаживания нейронов в чашку Петри они достигают зрелого состояния, соответствующего состоянию нейронов в мозге взрослого человека, а на 19–20 день нейроны считаются «старыми»: в них наблюдается ряд клеточных процессов, характерных для мозга пожилых людей. Кроме того, с возрастом в нейронах мышей YAC128 происходит накопление мутантного белка хантингтина и его агрегатов, поэтому изучение нейрональной культуры на этих двух этапах дает представление о том, что происходи в мозге пациента с БХ на ранней и на поздней стадиях заболевания.
Рисунок 4. Шипики разных типов на поверхности дендрита — микрофотография и схематическое изображение.
Для начала мы исследовали морфологические отличия между двумя линиями, т.е. сравнили их внешний вид. Для нормальных нейронов стриатума характерно наличие большого количества шипиков, благодаря чему их называют средними шипиковыми нейронами (СШН). Именно шипики формируют большую часть синаптических контактов между нейронами стриатума и коры, и для осуществления нормальной синаптической передачи важно наличие определенного их количества. Так же важно и «качество» шипиков: в современной нейробиологии их разделяют на три группы согласно размеру и форме (рис. 4): грибовидные, тонкие и пеньковые. При этом шипики разных типов выполняют разные функции: считается, что только грибовидные шипики формируют активные синапсы, в то время как тонкие и пеньковые контактов с другими нейронами не образуют. Таким образом, для нормального функционирования нейронной цепочки и эффективной передачи информации по ней необходимо наличие определенного количества грибовидных шипиков.
Для того, чтобы узнать, сколько грибовидных шипиков должно быть на СШН в норме, на всех этапах морфологического анализа как контроль использовалась культура нейронов из головного мозга мышей дикого типа. Оказалось, что количество шипиков на СШН стриатума на 14 день культивирования (молодые нейроны) одинаково в культурах YAC128 и дикого типа, но на 20 день (у «старых» нейронов) наблюдаются значительные изменения (рис. 5). В «постаревшей» культуре YAC128 снижается общее количество шипиков, причем относительное количество грибовидных шипиков, которые образуют активные синапсы, уменьшается в два раза [10]. Получается, что морфологические изменения нейронов, свидетельствующие о нарушении синаптической передачи, развиваются только к «старости» (на поздней стадии заболевания). Значит, на ранних этапах корень проблемы должен лежать в другой области.
Рисунок 5. Морфологический анализ нейронов стриатума. а — шипики нейронов на 14 и 20 дни культивирования. На микрофотографиях показаны участки дендритов нейронов. Относительное количество шипиков разных типов отмечено на круговой диаграмме: зеленый — грибовидные шипики, красный — тонкие шпики, черный — тонкие шипики. На 20 день культивирования на поверхности нейронов YAC128 снижается доля грибовидых шипиков и возрастает доля пеньковых. б — Плотность дендритных шипиков (среднее количество шипиков на участке дендрита длиной 10 мкм) на нейронах дикого типа и YAC28.
Поэтому мы сравнили электрофизиологические характеристики двух линий. Это возможно благодаря электрической активности нейронов, которую можно зарегистрировать с помощью специального метода, называемого пэтч-кламп. Для этого к поверхности нейрона прикладывают тонкую стеклянную пипетку, внутри которой находится электрод. Когда нейрон активируется, на его клеточной мембране изменяется напряжение, и это изменение регистрируется электродом. Если взять два связанных синаптическим контактом нейрона и, возбуждая один, регистрировать ответную электрическую активацию на втором, можно измерить эффективность передачи сигнала через синапс (рис. 6). Если ответная активация возникает не всегда, то, вероятно, синаптическая передача ослаблена. Этот метод может выявить нарушения работы синапса до изменений морфологии нейрона.
Рисунок 6. Схема эксперимента при совместном применении оптогенетики и электрофизиологической регистрации. Нейрон коры активируется при освещении синим светом, а на контактирующем с ним нейроне стриатума производится регистрация ответной активности с помощью стеклянной пипетки с электродом.
Чтобы активировать нейрон, можно использовать несколько способов. Можно добавить в окружающую его жидкость химическое вещество, открывающее ионные каналы, что приводит к электрическому возбуждению нейрона. Можно стимулировать нейрон электрическим током. Но мы для своих экспериментов выбрали более тонкий инструмент воздействия на нейроны, а именно — оптогенетику. Этот подход основан на внесении в нейроны специальных светочувствительных белков — опсинов, в результате чего и сами нейроны становятся чувствительными к свету (рис. 7). В результате, освещая нейроны светом определенной длины волны, можно изменять их активность. Освещение синим светом возбуждает нейрон, а желтым — вызывает торможение (т.е. подавляет активность нейрона).
Рисунок 7. Опсины — светочувствительные белки, обеспечивающие движение ионов через клеточную мембрану и изменение активности нейронов. Каналородопсин (ChR2) вызывает деполяризацию мембраны и активацию нейрона, галородопсин (NpHR) вызывает гиперполяризацию мембраны и торможение нейрона.
Оптогенетика
Оптогенетика — метод, объединяющий подходы генетики и оптики для тонкого контроля электрической активности электровозбудимых клеток (нейронов и мышечных волокон) [9]. Для этого в изучаемые клетки вводят гены специальных светочувствительных белков — микробных опсинов, которые являются ионными каналами или насосами (рис. 7). Первая работа, показавшая возможность управлять электрической активностью нейронов при использовании опсина, была опубликована в 2005 году. За последующие несколько лет появился еще ряд экспериментальных работ, позволивших доработать эту методику и доказать ее применимость в различных экспериментальных условиях.
За последние годы было открыто множество различных опсинов, из которых наибольшее применение в оптогенетике нашли галородопсины и каналородопсины. При доставке гена опсина с помощью методов генной инженерии в нейрон, на плазматической мембране появляются светочувствительные каналы, а сама клетка становится светочувствительной. При действии синего света открывается пора каналородопсина (максимум поглощения — 470 нм), который вызывает движение положительно заряженных ионов внутрь клетки, обеспечивая деполяризацию мембраны нейрона и генерацию потенциалов действия. При действии желтого света активируется галородопсин (максимум поглощения — 580 нм), мембрана нейрона гиперполяризуется, вызывая торможение нейрона. Высокое временное разрешение метода оптогенетики позволяет обеспечить очень тонкую регуляцию синаптических событий и является, таким образом, важным инструментом для изучения межнейронных связей.
Совместное применение оптогенетики и классических методов электрофизиологии позволяет извлечь выгоду из положительных качеств каждого из этих подходов. Точность электрофизиологической регистрации объединяется с возможностью использовать световые стимулы разной длительности и интенсивности, что помогает ученым подробно изучать работу нейронных связей.
Электрическая активность нейронов выражается в резких скачках напряжения на клеточной мембране и появлении вследствие этого электрического тока. Эти резкие скачки называются спайками или потенциалами действия и длятся несколько миллисекунд (рис. 8). Мы выяснили, что чем дольше нейрон освещается синим светом, тем больше спайков он за это время создает. Если освещаемый нейрон связан синаптическим контактом с другим нейроном, то на втором нейроне можно зарегистрировать ответную активность — тоже в виде отдельных спайков.
Рисунок 8. Пример записи, получаемой при электрофизологической регистрации активности нейрона. Спайки (потенциалы действия) отражаются на записи в виде вертикальных линий, показывающих резкие скачки мембранного тока.
В наших экспериментах мы использовали пару молодых (14 дней) нейронов коры и стриатума, связанных синаптическим контактом. Нейрон коры активировали синим светом, а на нейроне стриатума производили регистрацию ответной активности. Оказалось, что для возникновения ответа на нейроне стриатума нужна определенная длительность освещения (порог активации). Если длительность освещения была ниже порогового значения, то спайк на нейроне стриатума возникал в ответ не на каждую вспышку света. И что самое важное: порог активации для нейронов из мозга здоровых мышей отличался от мышей YAC128 (с мутацией в гене хантингтина). Наиболее ярко эта разница была видна при 50%-активации нейрона стриатума, т.е. длительности освещения, при которой спайк возникает в ответ на каждую вторую вспышку света. Порог 50%-активации нейрона стриатума в ответ на облучение кортикального нейрона для культуры клеток YAC 128 был примерно в два раза (точнее в 2,3±0,8) выше по сравнению с положительным контролем [10].
Получается, что мутация в белке хантингтине приводит к тому, что синаптическая передача ухудшается уже в молодых нейронах, и при этом нарушения происходят на функциональном уровне (без морфологических изменений). Может ли быть так, что именно эти функциональные нарушения в дальнейшем приводят к появлению морфологических изменений, таких как исчезновение шипиков у старых нейронов стриатума?
Чтобы ответить на этот вопрос, мы снова обратились за помощью к оптогенетике, но теперь с ее помощью мы подавляли светом активность нейронов (рис. 9а). Если наше предположение верно, то временное отсутствие активирующего воздействия коры должно привести к исчезновению шипиков на нейронах стриатума в культуре YAC128. Действительно, после эксперимента количество шипиков в положительном контроле осталось неизменным, но в культуре YAC128 значительно снизилось (рис. 9б, в). Получается, что моделирующие БХ нейроны особенно чувствительны к ослаблению активирующего воздействия нейронов коры, поэтому длительное ослабление синаптической связи между этими нейронами приводит к уменьшению количества шипиков на 20 день культивирования [10].
Рисунок 9. Влияние на нейроны с помощью оптогенетики. а — подавление активности нейрона коры с помощью оптогенетики. При освещении нейрона желтым светом (оранжевая полоса) он становится неактивен — на электрофизиологической записи в этот промежуток времени почти нет спайков. б — влияние длительного оптогенетического торможения на морфологию нейронов. На микрофотографиях видно уменьшение количества шипиков на дендрите нейрона YAC128. в — изменение плотности дендритных шипиков после оптогенетического торможения. В культуре дикого типа количество шипиков не изменяется, а в культуре YAC128 плотность шипиков значительно снижается.
Таким образом, мы обнаружили, что в нашей модели БХ наблюдается нарушение синаптической передачи, которое развивается в два этапа. На ранней стадии (молодые нейроны, на 14 день культивирования нейронов) происходит функциональное ослабление сипаптической связи, которое на более поздней стадии (старые нейроны, на 20 день культивирования) приводит к морфологическим нарушениям синаптических контактов.
Вывод: нарушение синаптической передачи и нейродегенеративные заболевания
Исследование подтвердило нашу первоначальную догадку: нарушение работы стриатума при БХ связано, прежде всего, с нарушением синаптической передачи. Нейроны стриатума перестают «слышать» команды нейронов коры, и, в результате, человек начинает терять контроль над своими движениями. Но на ранней стадии заболевания эти нарушения носят функциональный характер и, вероятно, обратимы. Если выяснить причины, ведущие к нарушению работы синапсов, то возможно разработать лекарственное средство, которое поможет остановить патологические изменения, обеспечит нейронам стриатума необходимый уровень активации и, таким образом, предотвратит развитие необратимых морфологических нарушений.
Результаты этой работы подталкивают и к другому важному заключению: при изучении нейродегенеративных заболеваний ученым следует обратить больше внимания на синаптическую передачу и взаимодействие нейронов. Мы знаем достаточно много о том, как влияет накопление амилоидных агрегатов при болезни Альцгеймера на жизнедеятельность клетки, и какие нейроны погибают при развитии болезни Паркинсона, но это все еще не привело к появлению эффективной терапии. Возможная причина этих неудач — мы не учитываем последствия нарушения синаптических связей — те, которые мы открыли при исследовании болезни Хантингтона. Хочется надеяться, что наши находки подскажут исследователям направление для разработки эффективных способов лечения болезни Хантингтона, и Тринадцатая наконец-то будет здоровой!
Исследование было проведено в лаборатории молекулярной нейродегенерации СПбГПУ (зав. лаб. — проф. Техасского университета Илья Борисович Безпрозванный) под руководством к.б.н. Дмитрия Николаевича Артамонова.
- Hayden M.R. Huntington’s Chorea. New York: Springer, 1982;
- DR Langbehn, RR Brinkman, D Falush, JS Paulsen, MR Hayden, on behalf of an International Huntington’s Disease Collaborative Group. (2004). A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clinical Genetics. 65, 267-277;
- Идентифицированы белки, «слипающиеся» при болезни Гентингтона;
- Sara Imarisio, Jenny Carmichael, Viktor Korolchuk, Chien-Wen Chen, Shinji Saiki, et. al.. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochem. J.. 412, 191-209;
- Claire-Anne Gutekunst, Shi-Hua Li, Hong Yi, James S. Mulroy, Stefan Kuemmerle, et. al.. (1999). Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J. Neurosci.. 19, 2522-2534;
- Boxun Lu, James Palacino. (2013). A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. The FASEB Journal. 27, 1820-1829;
- Benjamin Ray Miller, Ilya Bezprozvanny. (2010). Corticostriatal circuit dysfunction in Huntington’s disease: intersection of glutamate, dopamine and calcium. Future Neurology. 5, 735-756;
- Нобелевская премия по физиологии и медицине (2013): везикулярный транспорт;
- Karl Deisseroth. (2011). Optogenetics. Nat Methods. 8, 26-29;
- Артамонов Д.Н., Коржова В.В., Ву Д., Рыбальченко П.Д., Им К., Красноборова В.А., Власова О.Л., Безпрозванный И.Б. (2013). Нарушения синаптической передачи при болезни Хантингтона на кортикостриатной модели культуры нейронов. «Биологические мембраны». 30, 1–13;
- Оптогенетика + голография = прозрение?.
Хорея | Психология вики | Фэндом
Оценка |
Биопсихология |
Сравнительный |
Познавательная |
Развивающий |
Язык |
Индивидуальные различия |
Личность |
Философия |
Социальные |
Методы |
Статистика |
Клиническая |
Образовательная |
Промышленное |
Профессиональные товары |
Мировая психология |
Клинический:
Подходы ·
Групповая терапия ·
Техники ·
Типы проблем ·
Области специализации ·
Таксономии ·
Терапевтические вопросы ·
Способы доставки ·
Проект перевода модели ·
Личный опыт ·
Chorea sancti viti (лат. Для «Св.«Танец Витуса») — это ненормальное непроизвольное двигательное расстройство, одно из группы неврологических расстройств, называемых дискинезиями. Термин хорея происходит от греческого слова khoreia (разновидность танца), означающего быстрые движения ног. или руки смутно сравнимы с танцами или игрой на фортепиано.
Хорея характеризуется короткими нерегулярными сокращениями, которые не являются повторяющимися или ритмичными, но кажутся переходящими от одной мышцы к другой.
Эти «танцевальные» движения хореи (от того же корня, что и «хореография») часто возникают при атетозе, который добавляет скручивающие и извивающиеся движения.
Хорея может возникать при различных состояниях и расстройствах.
Когда хорея серьезная, легкие движения станут толчками; эта форма тяжелой хореи называется баллистической. Ходьба может стать необычной и включать странные позы и движения ног. В отличие от атаксии и дистонии, которые влияют на качество произвольных движений, или паркинсонизма, который является препятствием для произвольных движений, движения хореи и баллизма происходят сами по себе, без сознательного усилия.
Стандартного курса лечения хореи не существует. Лечение зависит от типа хореи и связанного с ней заболевания.
| Причина | Лечение |
| Болезнь Хантингтона | Распространенным лечением являются дофаминергические антагонисты, хотя лечение в основном является поддерживающим. |
| Хорея Синденхэма | Обычно включает антибиотики для лечения инфекции с последующей медикаментозной терапией для предотвращения рецидива. |
| лекарственно-индуцированная хорея. | Регулировка дозировки лекарств. |
| Хореи, связанные с метаболизмом и эндокринной системой | Лечение проводится в зависимости от причины (ей) симптомов. |
- REDIRECT Шаблон: ЦНС заболевания нервной системы
Болезнь Хантингтона — NHS
Болезнь Хантингтона — это заболевание, при котором части мозга со временем перестают работать должным образом. Он передается (наследуется) от родителей человека.
Со временем состояние ухудшается и обычно заканчивается смертельным исходом через 20 лет.
Симптомы
Симптомы обычно появляются в возрасте от 30 до 50 лет, но могут появиться гораздо раньше или позже.
Симптомы болезни Хантингтона могут включать:
- трудности с концентрацией внимания и провалы в памяти
- депрессия
- спотыкание и неуклюжесть
- непроизвольные подергивания или суетливые движения конечностей и тела
- перепады настроения и изменения личности
- проблемы глотания, речи и дыхание
- затрудненное движение
На более поздних стадиях заболевания требуется постоянный медсестринский уход.
Подробнее о симптомах болезни Гентингтона.
Как она передается по наследству
Болезнь Хантингтона вызывается дефектным геном, в результате чего части мозга со временем постепенно повреждаются.
Обычно вы рискуете заболеть этим заболеванием только в том случае, если он есть у одного из ваших родителей. Его могут получить и мужчины, и женщины.
Если у одного из родителей есть ген болезни Гентингтона, существует:
- 1 из 2 (50%) вероятность того, что у каждого из их детей разовьется заболевание — затронутые дети также могут передать ген любым своим детям
- 1 из 2 (50%) вероятность того, что у каждого из их детей никогда не разовьется заболевание — здоровые дети не могут передать заболевание ни одному из своих детей
В очень редких случаях болезнь Хантингтона может развиться, не имея в анамнезе этого заболевания. в семье.Но обычно это происходит потому, что у одного из ваших родителей это заболевание никогда не диагностировалось.
Когда обращаться за медицинской помощью
Обратитесь за советом к своему терапевту, если:
- вы беспокоитесь, что у вас могут быть симптомы болезни Хантингтона, особенно если у кого-то из вашей семьи была или была она
- у вас есть история болезни в вашей семье, и вы хотите узнать, заболеете ли вы, тоже
- у вас есть история болезни в вашей семье, и вы планируете беременность
Ваш терапевт может направить вас к специалисту для проведения анализов для болезни Хантингтона.
Лечение и поддержка
В настоящее время нет лекарства от болезни Хантингтона или какого-либо способа остановить ее ухудшение.
Но лечение и поддержка могут помочь уменьшить некоторые из проблем, которые оно вызывает, например:
- лекарства от депрессии, перепадов настроения и непроизвольных движений
- трудотерапия, помогающая облегчить повседневные задачи
- речевая и языковая терапия для кормления и проблемы с общением
- физиотерапия для улучшения движения и равновесия
Узнайте больше о лечении и поддержке при болезни Хантингтона.
Дополнительная информация и советы
Жизнь с болезнью Гентингтона может быть очень тяжелой и разочаровывающей для человека с этим заболеванием, а также для его близких и лиц, осуществляющих уход.
Вы можете найти Ассоциацию болезней Хантингтона полезным источником информации и поддержки.
Они предлагают:
Видео: Болезнь Хантингтона
Посмотрите это видео, чтобы узнать о болезни Хантингтона и важности сдачи анализов.
Последний раз просмотр СМИ: 1 апреля 2021 г.
Срок сдачи обзора СМИ: 1 апреля 2024 г.
Последняя проверка страницы: 29 марта 2021 г.
Срок следующей проверки: 29 марта 2024 г.
Хорея хантингтон — zxc.wiki
Аутосомно-доминантное наследование
Болезнь Хантингтона , также известная как хорея Хантингтона или Болезнь Хантингтона (старое название: хорея , большая хорея , большая хорея ), все еще является неизлечимым наследственным заболеванием головного мозга, которое » обычно характеризуется непроизвольными, нескоординированными движениями в то же время дряблым мышечным тонусом ».Часто это HD , сокращенно обозначающее английскую болезнь Хантингтона.
Больные страдают от прогрессирующего разрушения области мозга, которая важна для мышечного контроля и основной умственной функции, — полосатого тела. Там клетки мозга разрушаются дефектным белком, который образуется в результате дефекта так называемого гена Хантингтона. Внешние симптомы болезни включают нарушения эмоциональной жизни, мышечного контроля, включая мимику (что может создать у пострадавшего впечатление, что болезнь прогрессировала намного больше, чем на самом деле) и, наконец, функцию мозга в целом (в деменция в конечной стадии).
Болезнь Хантингтона — аутосомное нейродегенеративное заболевание с доминантной наследственностью, которое обычно приводит к первым симптомам заболевания — двигательным расстройствам и психологическим симптомам — примерно в возрасте 40 лет. Болезнь всегда тяжелая и приводит к смерти в среднем через 15 лет после первого симптомы. За редким исключением, все носители признаков рано или поздно заболевают (полная пенетрантность). С 1993 года аллель, вызывающий заболевание, был обнаружен на коротком плече четвертой хромосомы (локус гена p16.3), даже у нерожденных, путем амниоцентеза или биопсии ворсин хориона.
происхождение названия
Первая страница публикации Джорджа Хантингтона
Хантингтонская хорея ( chorea , греч. Χορεία ‘танец’) была подробно описана нью-йоркским доктором Джорджем Хантингтоном в 1872 году. Он описал клиническую триаду, которая действовала долгое время:
- по наследству (по наследству)
- психические отклонения и суицидальные наклонности (безумие и самоубийство)
- тяжелые симптомы только во взрослой жизни
Последний критерий позже оказался неверным.Хантингтон изначально предполагал, что распространение Хантингтона на Лонг-Исландии (США) было ограниченным. Фактически, болезнь уже тогда была обнаружена во всем мире. Немецкое имя потомственное Veitstanz . Название «Танец Св. Вита» (особенно Великий Св. Вита Танец ), которое засвидетельствовано с 16 века, происходит от того факта, что Святой Вит (Вит) был призван как помощник. Неизвестно, зачем призывали этого святого. Возможно, название было St.Танец Вита в 15-м или 16-м веке, когда в День Святого Вита (15 июня) в Страсбурге и в других местах были взяты в большом количестве люди из «Танцвута».
Термин болезнь Хантингтона теперь распространен во всех медицинских и других специализированных кругах.
Эпидемиология
Болезнь Хантингтона — одно из самых распространенных наследственных заболеваний головного мозга. Метаанализ, опубликованный в 2012 году, показывает, что средняя распространенность составляет 2,71: 100 000. Оцененные исследования показали распространенность 5.7: 100 000 для Европы, Северной Америки и Австралии, тогда как в Азии он составляет всего 0,40: 100 000. Однако распространенность значительно варьируется от страны к стране. В Финляндии это на 2,12: 100 000 меньше, чем в остальной Европе. В Германии официально затронуты около 10 000 человек (по данным на 2014 г.). В отдельных популяциях, например в Тасмании и в регионе Сулия в Венесуэле, распространенность намного выше, что частично можно отнести к отдельным людям, иммигрировавшим из Европы и унаследовавшим ген (эффект основателя).Напротив, очень низкие показатели распространенности обнаружены в Африке к югу от Сахары, а также среди афроамериканцев и, например, в Японии, где распространенность ниже 1: 100 000. Средний уровень новых заболеваний (заболеваемость) составляет 4: 1 000 000. В равной степени страдают мужчины и женщины.
Патофизиология
генетика
Болезнь Хантингтона — аутосомно-доминантное наследственное заболевание. Это означает, что потомство пострадавшего человека также поражено с вероятностью не менее 50% — в зависимости от того, имеет ли фенотипически больной родитель один или два мутировавших аллеля (два мутировавших аллеля (гомозиготность) = 100% вероятность заболевания в потомство).Если оба родителя больны и гетерозиготны, вероятность заболевания потомства составляет 75%. Скачки поколений не происходит, мужчины и женщины страдают одинаково. В отличие от этого около 5-10 процентов пациентов имеют новую мутацию. Белок, вызывающий заболевание, называется Huntingtin , а кодирующий его ген расположен на коротком плече хромосомы 4 (локус гена p16.3). Болезнь Хантингтона — это тринуклеотидное заболевание: в репрезентативной нормальной популяции основной триплет CAG повторяется примерно от 16 до 20 раз, у больных — от 36 до 250 раз.Если повторить от 27 до 35, будет небольшое увеличение без развития болезни. У людей с количеством от 36 до 39 повторов CAG (триплетных повторов) есть исключение из полной пенетрантности болезни, то есть не у всех людей этого генотипа развивается болезнь, и никакие окончательные прогнозы невозможны даже после генетического контрольная работа. Чем чаще повторяется это повторение, тем раньше возникает заболевание (в среднем) (эффект ожидания). Ювенильная болезнь Хантингтона проявляется более чем у 60 троек CAG.Описана вспышка на четвертом году жизни.
У очень небольшого процента заболевших анализ родословной не может найти кровного родственника с болезнью Гентингтона, от которого они могли бы ее унаследовать. В этих редких случаях это новая мутация, увеличение количества повторов CAG, при которых превышается предел примерно в 36. Обычно у одного из родителей больного человека с новой мутацией уже есть от 30 до 35 повторений (премутация). При наследовании от отца количество троек CAG увеличивается чаще, чем при наследовании от матери (феномен импринтинга).Это в основном вызвано так называемым «проскальзыванием» (проскальзыванием ДНК-полимеразы во время репликации) или (что менее вероятно, но по крайней мере возможно) невзаимным (асимметричным) кроссинговером.
Молекулярная биология
мРНК, полученная в результате триплета CAG, кодирует аминокислоту глутамин. Таким образом, мутированный хантингтин имеет больше, чем обычно, количество остатков глутамина в ряду. Возможно, это «мутация с усилением функции», что означает, что нормальная функция белка HD может быть сохранена, но он также имеет другие — токсичные — свойства.Высокая экспрессия хантингтина приводит к амилоидоподобным внутриклеточным отложениям ( включений ) мутировавшего хантингтина, которые стимулируются белком, активирующим ГТФазу ARF1, вероятно, также потому, что расщепление мутированного белка протеасомой больше не работает должным образом. С другой стороны, некоторые рабочие группы также показали токсичность свободного мутантного Хантингтина, так что агрегаты Хантингтона можно рассматривать как защиту. Пораженные клетки имеют нарушенный метаболизм глюкозы.Это приводит к повышенной чувствительности к окислительному стрессу и возбуждающему нейротрансмиттеру глутамату. Эти клетки особенно богаты рецепторами глутамата и имеют много глубоких глутаматергических связей. Тем не менее, это можно объяснить только неудовлетворительным образом, почему токсичность может быть обнаружена только в описанных областях, хотя хантингтин образуется во всех ядросодержащих клетках.
Физиологическая функция хантингтина до конца не изучена. Существует множество доказательств того, что он играет важную роль во внутриклеточном транспорте везикул и органелл.
Нейроанатомия и физиология
В болезни Хантингтона, т. А. нейроны непрямого пути от полосатого тела к бледному шару.
Скорлупа, которая является частью полосатого тела в базальных ганглиях и может влиять на глобус бледный медиальный (внутренний) прямым и непрямым путем.
Непрямой противодействует прямому пути. Общий тормозящий эффект непрямого пути достигается у здоровых людей с помощью следующих станций: Блокирующие движения части полосатого тела, в свою очередь, подавляют globus pallidus lateralis (externa).Теперь это снижает его ингибирующее действие на субталамическое ядро , что увеличивает его активность. Поскольку субталамическое ядро оказывает глутаматергическое воздействие на медиальный бледный шар, оно оказывает тормозящее действие на таламус.
У людей с болезнью Гентингтона ГАМК / энкефалин -ергические нейроны в первую очередь дегенерируют; H. начало косвенного пути разрушено. В результате globus pallidus medialis более слабо ингибируется прямым путем, чем у здоровых людей.Поскольку globus pallidus medialis сам по себе обычно подавляет таламус, теперь он меньше ингибируется, то есть активируется (= растормаживание). Следствие — перевозбуждение таламуса и коры.
Так как косвенные связи обычно сначала разрушаются в ходе болезни, гиперактивация с чрезмерными движениями выходит на первый план в начале болезни. В дальнейшем прямые связи также теряются, и преобладают отсутствие движения (акинезия) и ригидность (ригидность).
Клиническая картина
курс
Первые симптомы болезни обычно появляются в возрасте от 30 до 40 лет. Симптомы описываются в возрасте от трех до 75 лет. Пациенты с ранним началом болезни часто имеют более тяжелое течение.
Проблемы с психическим здоровьем часто на несколько лет предшествуют двигательным нарушениям. Двигательные расстройства обычно начинаются с гиперкинезии (нежелательных движений) со снижением мышечного тонуса. В дальнейшем чаще появляется гипокинезия (малоподвижный образ жизни) и повышение мышечного тонуса.Форма, при которой малоподвижный образ жизни находится на переднем плане с самого начала, называется Вестфальский вариант в честь Карла Вестфала и чаще встречается с ранним началом болезни. Болезнь Хантингтона обычно прогрессирует до 30 лет с увеличением потребности в уходе. Это происходит из-за затрудненного приема пищи (дисфагия) и постоянно повышенного энергопотребления, часто приводящего к кахексии. Большинство пациентов умирают в течение 15 лет от начала болезни. Развитие болезни может быть ускорено стрессом, и наоборот, благоприятные условия жизни с активацией, соответствующей страданию, положительно влияют на течение болезни.
Психологические жалобы и психиатрические симптомы
Первые симптомы психологического изменения обычно включают расстройства аффекта и влечения. Они также могут предшествовать двигательным расстройствам. Позже может возникнуть неосторожное и импульсивное поведение и расторможенность в межличностных отношениях. Из-за неадекватного контроля над мышцами (например, лица с гримасой) может возникнуть ложное впечатление об уже запущенной утрате личности, что может вызвать смирение и депрессию у пациента.Это может привести к суицидальному поведению, особенно на ранних стадиях заболевания. Нарушения в обработке зрительной информации также возникают на ранней стадии. Б. приводит к тому, что у больных особо критичная мимика своих собратьев — таковых. Б. Раздражение — неспособность правильно воспринимать и, следовательно, неспособность адекватно реагировать. На ранних стадиях часто упускаются из виду легкие нарушения интеллектуальных способностей и нарушения памяти. На поздних стадиях заболевания у пациентов развивается подкорковая деменция, т.е.е. Х. происходит потеря своих познавательных способностей. Наблюдаются нарушения способности запоминать, связанные с дезориентацией и языковым обеднением. У некоторых пациентов развиваются бредовые идеи, которые приводят к их лечению в психиатрических клиниках (психически напряженный курс).
Двигательные расстройства
Хорея обычно начинается с первоначально едва заметного беспокойства движений рук и ног, лица, а затем головы и туловища. Это беспокойство может перерасти в сильный хореический гиперкинез.Это внезапные непроизвольные движения различных мышц, которые прерывают произвольные движения. Пострадавшие сначала пытаются скрыть движения хора, объединяя их в произвольные последовательности движений, например Б. погладить волосы после стреляющего движения руки. Однако все чаще движения мышц выходят из-под контроля. При полном заболевании возникают внезапные гримасы и покачивания (хорея) рук и ног. Становятся все труднее говорить и глотать (дизартрия и дисфагия).Обычно эти гиперкинезы начинаются в отдаленных от туловища частях конечностей (в руках) и на лице, рот широко открыт, язык вытянут и сразу отведен («язык хамелеона»). В дальнейшем поражаются и конечности около туловища. Когда срабатывает рефлекс подколенного сухожилия, колено остается разогнутым (феномен Гордона). Неугомонность движений усиливается при эмоциональном и физическом напряжении. Хотя неконтролируемые движения прекращаются во время сна, они имеют тенденцию усиливаться, когда вы устаете.Первоначально хореические гиперкинезы сменяются хореоатетозом или дистонией по мере прогрессирования заболевания, в результате чего конечности остаются в иногда болезненном смещении от минут до часов по мере увеличения мышечного напряжения (мышечного тонуса). Вместо гримасы может возникнуть анартрия, т.е. H. может быть полная неспособность выполнять речевые движения, и пациент больше не может реагировать с помощью мимики, жестов и речи. Глотание становится все труднее для пациента и может привести к опасным для жизни осложнениям, особенно потому, что пациенты расходуют больше энергии из-за гиперкинезии.На последних стадиях болезни эта скорость может увеличиваться более чем в пять раз по сравнению с нормальной скоростью основного обмена, поэтому адекватное питание возможно только при дополнительном парентеральном питании.
Независимо от хореи, люди с болезнью Гентингтона демонстрируют недостаточную двигательную настойчивость, то есть неустойчивый мышечный тонус. Это также точно описано на английском языке как доярка . Этот разрыв мышечного тонуса лучше подходит для диагностики прогрессирования заболевания, чем хорея, потому что в отличие от хореи он неуклонно увеличивается по мере прогрессирования заболевания.
Диагностика
Диагноз обычно ставится клинически на основании симптомов. Другие варианты — магнитно-резонансная томография или компьютерная томография. Они показывают атрофию полосатого тела и особенно хвостатого ядра . Эта атрофия приводит к увеличению боковых желудочков. 18 FDG — PET показывает нарушение метаболизма глюкозы в полосатом теле . Возможный дифференциальный диагноз — CJD (болезнь Крейтцфельдта-Якоба) и vCJD (вариант CJD).Также есть возможность подтвердить диагноз с помощью генетического анализа. Еще до появления первых симптомов анализ ДНК может быть проведен в институте генетики человека, в случае будущего ребенка в рамках анализа околоплодных вод.
Этические проблемы генетической диагностики человека
Теперь возможно, задолго до появления каких-либо симптомов у людей из пораженных семей, четко определить, есть ли у них генетический дефект, приводящий к болезни Хантингтона, и сколько повторов последовательности CAG присутствует.Для детей пораженного родителя, чей анализ ДНК недоступен, вероятность возникновения заболевания составляет 50%. Вы либо точно заболеете, либо никогда.
Решение о том, желателен ли такой диагноз, является очень личным и может быть принято после исчерпывающей информации. Прежде чем решить, иметь ли детей, особенно полезен анализ ДНК. Такая диагностика также дает информацию о других кровных родственниках. При положительном диагнозе у внука пострадавшего также станет ясно, что соответствующий родитель поражен, даже если у него еще не было никаких симптомов.
Дифференциальная диагностика
Следует различать синдром Хантингтона, подобный болезни, и акантоцитоз Хантингтона.
терапия
Не существует какой-либо известной терапии, которая излечивает само заболевание или останавливает ее навсегда. Различные витамины и пищевые добавки используются с разной степенью успеха для защиты клеток от окислительного стресса и, таким образом, для замедления течения болезни. Препарат рилузол снижает высвобождение глутамата и замедляет этот процесс.Исследования показывают, что паллидная глубокая стимуляция мозга (DBS) также оказывает положительное влияние, особенно на двигательные симптомы. Течение болезни невозможно остановить с помощью стимуляции, но пострадавшие сообщают о повышении качества жизни, поскольку они меньше зависят от посторонней помощи и могут продолжать активно участвовать в общественной жизни. Доклинические исследования также предполагают улучшение других немоторных симптомов, таких как когнитивные функции и настроение. Дальнейшие исследования проводились и проводятся для дальнейшего изучения положительных результатов, наблюдаемых на данный момент у отдельных пациентов.Все методы лечения сопровождаются физиотерапией, трудотерапией и логопедом для улучшения способности двигаться, говорить и глотать. В то же время пациент и его родственники должны пройти психологическое лечение, а в случае психических расстройств — психиатрическое лечение. Диета должна учитывать повышенные потребности пациента в энергии, трудности с глотанием и повышенную потребность в сахаре. При необходимости можно лечить отдельные симптомы.
Лечение двигательных нарушений
Против гиперкинезов следует также тетрабеназин — антагонисты дофамина, в основном тиаприд или сульпирид.Однако клинические исследования показывают, что прием тетрабеназина может усугубить депрессию и суицидальные наклонности. Кроме того, у некоторых людей тетрабеназин усугублял экстрапирамидные синдромы.
Когда наступает окоченение, используются агонисты дофамина или L-допа, но они могут усугубить гиперкинезы. Поэтому медикаментозную терапию начинают только тогда, когда двигательные расстройства серьезно ухудшают повседневную жизнь пациента. Глубокая стимуляция мозга (DBS) положительно влияет на хореические симптомы, которые трудно лечить с помощью лекарств, с хорошей переносимостью.Влияние на дистонию и другие двигательные расстройства проверяется в международном клиническом исследовании.
Лечение психических симптомов
В случае психотических симптомов используются атипичные нейролептики, а антидепрессанты из группы СИОЗС в основном используются при депрессивных симптомах. Бензодиазепины можно использовать при расстройствах сна и тревоге.
Нейропротекторное лечение
Есть исследования, предполагающие, что габапентин оказывает нейропротекторное действие на пациентов с болезнью Хантингтона.Цель этого препарата — снизить эксайтотоксичность глутамата для нервной клетки.
дальнейшее чтение
- Р.А. Роос: Болезнь Хантингтона: клинический обзор. В: Журнал редких заболеваний Орфанской сети. Том 5, номер 1, 2010 г., с. 40. DOI: 10.1186 / 1750-1172-5-40. PMID 21171977. PMC 3022767 (полный текст). (Рассмотрение). (Открытый доступ)
- И. Шоулсон, А.Б. Янг: вехи болезни Хантингтона. В: Двигательные расстройства. Volume 26, Number 6, May 2011, pp. 1127-1133. DOI: 10.1002 / mds.23685. PMID 21626556. (Рассмотрение).
- А.Л. Саутвелл, П.Х. Паттерсон: Генная терапия на мышиных моделях болезни Хантингтона. In: Невролог. Volume 17, Number 2, April 2011, pp. 153-162. DOI: 10,1177 / 1073858410386236. PMID 21489966. PMC 3131092 (полный текст). (Рассмотрение).
- D. Eidelberg, DJ Surmeier: Мозговые сети при болезни Хантингтона. В: Журнал клинических исследований. Volume 121, Number 2, February 2011, pp. 484-492. DOI: 10,1172 / JCI45646. PMID 21285521. PMC 3026742 (полный текст). (Рассмотрение).
- Дж. Рутисхаузер: Болезнь Хантингтона: разрушить роковое влечение. (PDF; 163 кБ). В: Switzerland Med Forum. 24, 2002, стр. 586-587.
- E. Cattaneo et al. a: Загадка болезни Хантингтона. В: Спектр науки. 2004, стр. 60-66.
- Никола Биллер-Андорно: Танец Святого Вита, хорея мажор (современность). В: Вернер Э. Герабек, Бернхард Д. Хааге, Гундольф Кейл, Вольфганг Вегнер (ред.): Enzyklopädie Medizingeschichte. De Gruyter, Берлин / Нью-Йорк 2005, ISBN 3-11-015714-4, стр. 1438 ф.
Интернет-ссылки
Индивидуальные доказательства
- ↑ Никола Биллер-Андорно: Veitstanz, Chorea major (современное время). В: Вернер Э. Герабек, Бернхард Д. Хааге, Гундольф Кейл, Вольфганг Вегнер (ред.): Enzyklopädie Medizingeschichte. De Gruyter, Берлин / Нью-Йорк 2005, ISBN 3-11-015714-4, стр.EC Wicke: попытка монографии о великом танце Св. Вита и непроизвольных мышечных движениях, лучшие замечания о танце тарантула и бербери. Лейпциг 1844 г.
- ↑ Никола Биллер-Андорно: Veitstanz, Chorea major (современное время). В: Вернер Э. Герабек, Бернхард Д. Хааге, Гундольф Кейл, Вольфганг Вегнер (ред.): Enzyklopädie Medizingeschichte. De Gruyter, Берлин / Нью-Йорк 2005, ISBN 3-11-015714-4, стр. 1438 и далее; здесь: стр. 1438.
- ↑ Т.Pringheim, K. Wiltshire, L. Day, J. Dykeman, T. Steeves, N. Jette: Заболеваемость и распространенность болезни Хантингтона: систематический обзор и метаанализ. В: Мов. Disord. Том 27, № 9, 2012 г., стр. 1083-1091. PMID 22692795
- ↑ JOSipilä, M. Hietala, A. Siitonen, M. Päivärinta, K. Majamaa: Эпидемиология болезни Хантингтона в Финляндии. In: Parkinsonism Relat. Disord. Том 21, № 1, 2015, стр. 6-49. PMID 25466405
- ↑ Veitstanz должен сообщить об этом общественности. A b F. O. Walker: Болезнь Хантингтона. В: Ланцет. , том 369, номер 9557, январь 2007 г., стр. 218-228. DOI: 10.1016 / S0140-6736 (07) 60111-1. PMID 17240289. (Рассмотрение).
- ↑ NetDoctor: Болезнь Хантингтона .
- ↑ Белок способствует патологическим отложениям в сети взаимодействия белков хореи при болезни Хантингтона, ведущей по следу . Пресс-релиз Центра молекулярной медицины Макса Дельбрюка в Ассоциации Гельмгольца, 24 сентября 2004 г., по состоянию на 8 октября 2015 г.
- ↑ JP Caviston, EL Holzbaur: Хантингтин как важный интегратор внутриклеточного везикулярного движения. В: Тенденции клеточной биологии. Том 19, номер 4, апрель 2009 г., стр. 147-155. DOI: 10.1016 / j.tcb.2009.01.005. PMID 19269181. PMC 2930405 (полный текст). (Рассмотрение).
- ↑ Махлон Р. Делонг: Базальные ганглии. В: Эрик Р. Кандел, Джеймс Х. Шварц, Томас М. Джесселл: Принципы нейронологии . 2000, ISBN 0-8385-7701-6, стр.Э. Моро, А. Э. Ланг, А. П. Страфелла и др. a .: Двусторонняя стимуляция бледного шара при болезни Хантингтона. In: Ann Neurol. 56 (2), август 2004 г., стр. 290-294. DOI: 10.1002 / ana.20183. PMID 15293283.
- ↑ Б. Биолси, Л. Сиф, Х. Э. Фертит, С. Г. Роблес, П. Кубес: Долгосрочное наблюдение за болезнью Хантингтона, леченной двусторонней глубокой стимуляцией внутреннего бледного шара. В: J Neurosurg. 109 (1), июль 2008 г., стр. 130-132. DOI: 10.М.О. Хебб, Р. Гарсия, П. Годе, И.М. Мендес: Двусторонняя стимуляция внутреннего бледного шара для лечения хореатетоза при болезни Хантингтона: отчет о техническом случае. В: Нейрохирургия. 58 (2), февраль 2006 г., стр. E383; обсуждение E383. DOI: 10.1227 / 01.NEW.0000195068.19801.18. PMID 16462466
- ↑ Л. Войтеки, С. Гройсс, С. Ферреа, С. Эльбен, С. Дж. Хартманн, С. Даннет, А. Россер, К. Сафт, М. Зюдмайер, К. Оманн, А. Шницлер, Дж. Веспер: A проспективное пилотное исследование паллидальной стимуляции глубокого мозга при болезни Хантингтона. В: Фронт. Neurol. 6, стр. 177. doi: 10.3389 / fneur.2015.00177
- ↑ Л. Войтеки, С. Дж. Гройсс, С. Дж. Хартманн, С. Эльбен, С. Омлор, А. Шницлер, Дж. Веспер: Глубокая стимуляция мозга при болезни Хантингтона — предварительные данные по патофизиологии, эффективности и безопасности. В: Brain Sci. 6 (3) .pii, 30 августа 2016 г., стр. E38, DOI: 10.3390 / brainsci6030038.
- ↑ Y. Temel, C. Cao, R. Vlamings and a .: Улучшение моторики и когнитивных функций за счет глубокой стимуляции мозга на трансгенной крысиной модели болезни Хантингтона. In: Neurosci Lett. 406 (1-2), 2 октября 2006 г., стр. 138-141. DOI: 10.1016 / j.neulet.2006.07.036. PMID 162.
- ↑ a b Клиническое исследование (фаза II): Глубокая стимуляция мозга (DBS) Globus Pallidus (GP) при болезни Хантингтона (HD) (HD-DBS) на сайте Clinicaltrials.gov Национального института здоровья
- ↑ a b Кара Дж. Вайант, Эндрю Дж. Риддер, Правин Дайалу: Обновленная информация о лечении по болезни Хантингтона. In: Current Neurology and Neuroscience Reports. 17, 2017 г., DOI: 10.1007 / s11910-017-0739-9.
- ↑ М. де Томмазо: Лечение болезни Хантингтона: роль тетрабеназина. In: Терапия и управление клиническими рисками. 7, 2011. стр. 123–129, DOI: 10.2147 / TCRM.S17152.
- ↑ Д. Бонекамп: 1H магнитно-резонансная спектроскопия при болезни Хантингтона при нейропротекторной терапии габапентином. Диссертация, Медицинский факультет Charité — Universitätsmedizin Berlin, 2004.С. Косентино, Л. Торрес, Дж. М. Куба: Габапентин для болезни Хантингтона. В: J Неврология. Том 243, 1996, S: S75-S76.
Эта статья посвящена проблеме со здоровьем. не используется, не используется для самодиагностики, а не заменяет диагноз, поставленный врачом. Обратите внимание на информацию по вопросам здоровья!
О болезни Хантингтона и связанных с ней расстройствах в больнице Джонса Хопкинса
Краткая история болезни Хантингтона
Болезнь Хантингтона (HD) названа в честь Джорджа Хантингтона, который описал ее среди жителей Ист-Хэмптона, Лонг-Айленд в 1872 году.Это наследственное нейродегенеративное заболевание. В 1993 году совместная группа исследователей открыла ген, вызывающий HD. В результате этого открытия теперь можно диагностировать HD с помощью образцов крови или тканей.
Генетическая причина HD
HD вызывается мутацией в гене, расположенном на хромосоме 4. Этот ген встречается у каждого человека и содержит последовательность повторов CAG. Мы еще не обнаружили нормальную функцию гена. В случае HD ген содержит аномально большое количество повторов CAG.Чем больше количество триплетных повторов, тем, вообще говоря, раньше в жизни разовьется HD. Более того, когда ген передается от отца к ребенку (но не от матери к ребенку), ген может удлиняться еще больше, что приводит к более раннему возрасту начала заболевания. Это явление известно как ожидание .
Гены заболеваний могут быть доминантными или рецессивными. Ген HD является доминирующим.Каждый ребенок пораженного родителя имеет шанс 50/50 получить мутантный ген и, следовательно, имеет 50% шанс унаследовать болезнь. С другой стороны, если люди, чьи родители страдают от HD, не наследуют мутантный ген, они не могут передать его кому-либо еще.
Важная информация о тестировании HD
Важно понимать, что, хотя люди рождаются с мутировавшим геном HD, в большинстве случаев у них не разовьются симптомы до более позднего возраста.Следовательно, кто-то может быть бессимптомным или бессимптомным в течение нескольких лет. Раньше не было возможности проверить аномальный ген, но теперь анализ крови может определить, несет ли человек ген HD. Этот тест может использоваться в случаях подозрения на HD, для подтверждения диагноза, или он может использоваться в качестве прогностического теста у людей, которые подвержены риску HD. Люди, входящие в группу риска, могут захотеть пройти прогностическое тестирование, чтобы успокоиться, спланировать свои медицинские потребности или до рождения детей.Решение пройти такое испытание — важное, и к нему нельзя относиться легкомысленно. Большинство центров, которые проводят прогностическое тестирование, включая наш, требуют периода консультирования до и после теста.
Каковы характеристики HD?
Начало обычно в среднем возрасте, но может произойти в любое время от детства до старости. Начальные признаки этого расстройства могут быть незаметными. HD характеризуется двигательным расстройством, слабоумием и психическими расстройствами. Дополнительные характеристики HD включают изменения личности, потерю веса (вероятно, из-за сочетания затруднений с приемом пищи и калорий, сожженных непроизвольными движениями), затрудненное глотание и трудную для понимания речь.
Ход HD
Когда у человека появляются признаки HD, течение болезни может длиться от десяти до тридцати лет. Обычно курс HD можно условно разделить на три этапа.
Ранняя стадия:
На этой стадии пациенты все еще могут выполнять большую часть своей обычной деятельности. Возможно, они все еще работают и могут водить машину. Непроизвольные движения мягкие и нечастые, речь по-прежнему ясна, а деменция, если она вообще присутствует, является легкой.
Средняя стадия:
На этой стадии пациенты в большей степени являются инвалидами и могут нуждаться в помощи в некоторых повседневных делах. Падения, потеря веса и трудности с глотанием могут стать проблемой. Для случайного наблюдателя деменция более очевидна. Непроизвольные движения более выражены.
Поздняя стадия:
Пациентам, входящим в эту стадию болезни, требуется почти полный уход, и они могут проживать в больницах или домах престарелых, хотя некоторые остаются дома.Они могут больше не ходить или говорить. Теперь они могут быть более жесткими и показывать меньше непроизвольных движений. Люди на этой стадии могут или не могут глотать пищу. На этой стадии кажется, что большинство пациентов не сильно страдают, поскольку они явно не осознают свое окружение. Когда пациент умирает, причина смерти обычно связана с недоеданием, пневмонией или сердечной недостаточностью.
Какие методы лечения доступны?
На данный момент лекарства от HD нет.Исследователи работают над рядом методов лечения, которые могут замедлить прогрессирование заболевания. Сегодня существует ряд вмешательств, которые улучшают качество жизни больных HD. На ранней и средней стадиях болезни пациентам с HD можно давать лекарства в малых дозах, чтобы помочь подавить непроизвольные движения. Депрессия и другие психические расстройства у людей с HD поддаются достаточно эффективному лечению. Правильное питание, физические упражнения и меры предосторожности в домашних условиях могут помочь свести к минимуму многие потенциальные последствия HD, такие как потеря веса, падения и удушье.Наконец, контакт с другими больными HD, членами их семей и поставщиками услуг может быть жизненно важным источником поддержки для пациентов с HD и их семей.
Future Care
Людям с HD следует обсудить свои опасения и пожелания относительно лечения / вмешательств (например, зондов для кормления, запросов на реанимацию) и вскрытия со своими семьями и врачами, пока они еще могут говорить за себя.
Was ist Huntington | Deutsche Huntington-Hilfe e.V.
Die Huntington-Krankheit
Die Huntington-Krankheit ist eine sehr seltene, vererbbare Erkrankung des Gehirns.Sie ist eine fortschreitende Erkrankung, die meist zwischen dem 35. und 45. Lebensjahr ausbricht. In seltenen Fällen kann sie auch in der frühen Kindheit oder im höheren Alter auftreten. Der Verlauf der Erkrankung ist Individual und von Patient zu Patient Verschieden. Viele Patienten leiden unter neuroologischen Störungen, beispielsweise Bewegungs-störungen oder Psychischen Veränderungen wie Verhaltensstörungen. Im fortgeschrittenen Stadium kommt ein Rückgang der intellektuellen Fähigkeiten hinzu.Die Huntington-Krankheit verläuft fortschreitend. Ursache ist ein verändertes Gen (Генмутация).
Benannt ist die Huntington-Krankheit nach dem amerikanischen Arzt Джордж Хантингтон на Лонг-Айленде (Нью-Йорк, США), der sie 1872 beschrieb. Er hatte erkannt, dass es sich um eine erbliche Krankheit handelt.
Die Huntington-Krankheit (abgekürzt HK; англ. Болезнь Хантингтона, abgekürzt HD) wird auch Chorea Huntington, Morbus Huntington genannt und war früher als Veitstanz bekannt.
Der Name Chorea (griech. Choreia = Tanz) rührt von den für die Erkrankung typischen, zeitweise einsetzenden unwillkürlichen, raschen, unregelmäßigen und nicht vorhersehbaren Bewegungen her. Zusammen mit dem unsicheren, fast torkelnden Gang und dem Grimassieren können diese Symptome sehr entfernt an einen Tanz erinnern.
In der Medizin werden heute mit «Chorea» plötzlich einsetzende, vielgestaltige unwillkürliche Bewegungen verschiedener Muskeln, besonders der Arme und Beine, bezeichnet.Da die choreatischen Bewegungen nur einen Teil der Symptome ausmachen, spricht man heute weniger von Chorea Huntington als von der Huntingtonschen Erkrankung.
Симптом Vielfältige
Meist stehen am Anfang der Erkrankung fortschreitende mentalische Auffälligkeiten im Vordergrund: Die Patienten sind depressiv or vermehrt reizbar und aggressiv or enthemmt; andere bemerken einen Verlust an geistigen Fähigkeiten oder eine zunehmende Ängstlichkeit. Später kommt es häufig zur Demenz.
Die Bewegungsstörungen bestehen in plötzlich auftretenden, unkontrollierbaren und überschießenden Bewegungen von Extremitäten oder Rumpf. Diese Störungen können в Ruhe auftreten oder andere Bewegungen beeinträchtigen. Anfangs ist es möglich, dass diese übertriebenen und ungewollten Bewegungen oft noch in scheinbar sinnvolle Bewegungsabläufe eingebaut werden. So entsteht beispielsweise eine für den Beobachter übertrieben wirkende Gestik. Auch die Zungen- und Schlundmuskulatur können betroffen sein.Die Sprache wirkt in diesen Fällen abgehackt und unverständlich, Laute werden explosionsartig ausgestoßen. Ebenso kann es zu Schluckstörungen kommen, так что dass die Nahrungsaufnahme sehr schwierig wird. Lungenentzündung aufgrund von Schluckstörungen sind eine häufige Komplikation.
In späteren Stadien steht eher eine Muskelsteifheit mit Bewegungsverminderung im Vordergrund.
Sichere Diagnose notwendig
Bei Erkrankung der Eltern oder Geschwister kann die Verdachtsdiagnose anhand der typischen Symptome und des Verlaufes gestellt werden.Trotzdem sind weitere Neurologische und Psychiatrische Untersuchungen notwendig, da auch andere Krankheiten ähnliche Symptome hervorrufen können. Diese sollten am besten von einem erfahrenen Arzt an einem der Huntington-Zentren durchgeführt werden. Bestätigt werden kann die Diagnose auch durch eine molkulargenetische Untersuchung, die das veränderte Gen nachweist.
Schwierige Behandlung
Bisher kann die Huntington-Krankheit nicht ursächlich behandelt werden. Es gibt Medikamente, die einzelne Symptome mindern können.Таким образом, действует неконтролируемый Bewegungsabläufen Neuroleptika gegeben, депрессивный Verstimmungen Antidepressiva.
Neben der medikamentösen Therapie sind aber auch Krankengymnastik, Beschäftigungstherapie sowie Sprechtraining wichtig. Da die Symptome bei den verschiedenen Patienten unterschiedlich sein können, muss die Therapie Individual angepasst werden.
Лечение поздней дискинезии и хореи Хантингтона
Утвержденное использование
AUSTEDO — это лекарство, отпускаемое по рецепту, которое используется для лечения:
- непроизвольных движений (хореи) при болезни Хантингтона.AUSTEDO не лечит причину непроизвольных движений и не лечит другие симптомы болезни Хантингтона, такие как проблемы с мышлением или эмоциями.
- движений лица, языка или других частей тела, которые невозможно контролировать (поздняя дискинезия).
Неизвестно, безопасно и эффективно ли AUSTEDO у детей.
Важная информация по технике безопасности
AUSTEDO может вызывать серьезные побочные эффекты у людей с болезнью Гентингтона, включая депрессию, суицидальные мысли или суицидальные действия. Не начинайте принимать AUSTEDO , если у вас депрессия (нелеченная депрессия или депрессия, которая плохо контролируется медициной). или имеют суицидальные мысли. Обращайте пристальное внимание на любые изменения, особенно внезапные, в настроении, поведении, мыслях или чувствах. Это особенно важно при запуске AUSTEDO и изменении дозы. Немедленно позвоните своему врачу, если вы впали в депрессию, у вас необычные изменения в настроении или поведении или возникли мысли о самоубийстве.
Не принимайте AUSTEDO, если вы:
- страдают болезнью Гентингтона и находятся в депрессии или имеют мысли о самоубийстве.
- имеют проблемы с печенью.
- принимают лекарство с ингибитором моноаминоксидазы (ИМАО). Не принимайте MAOI в течение 14 дней после прекращения приема AUSTEDO. Не запускайте AUSTEDO, если вы прекратили прием MAOI в течение последних 14 дней. Если вы не уверены, спросите своего лечащего врача или фармацевта.
- принимают резерпин. Не принимайте лекарства, содержащие резерпин (такие как Серпалан ® и Renese ® -R) с AUSTEDO. Если ваш лечащий врач планирует переключить вас с приема резерпина на AUSTEDO, вы должны подождать не менее 20 дней после приема последней дозы резерпина, прежде чем начинать прием AUSTEDO.
- принимают тетрабеназин (Xenazine ® ). Если ваш лечащий врач планирует перевести вас с тетрабеназина (Xenazine ® ) на AUSTEDO, примите первую дозу AUSTEDO на следующий день после последней дозы тетрабеназина (Xenazine ® ).
- принимают валбеназин (Ingrezza ® ).
Другие возможные серьезные побочные эффекты включают:
- Нерегулярное сердцебиение (удлинение интервала QT). AUSTEDO увеличивает вероятность определенных изменений электрической активности сердца. Эти изменения могут привести к опасному нарушению сердечного ритма. Прием AUSTEDO с некоторыми лекарствами может увеличить этот шанс.
- Злокачественный нейролептический синдром. Немедленно позвоните своему врачу и обратитесь в ближайшее отделение неотложной помощи, если у вас появятся эти признаки и симптомы, не имеющие другой очевидной причины: высокая температура, жесткие мышцы, проблемы с мышлением, очень быстрое или неравномерное сердцебиение или повышенное потоотделение.
- Беспокойство. У вас может возникнуть состояние, при котором вы почувствуете сильное желание двигаться. Это называется акатизией.
- Паркинсонизм. Симптомы включают: легкую дрожь, скованность тела, проблемы с движением, нарушение равновесия или падения.
Сонливость (седативный эффект) — частый побочный эффект AUSTEDO. Принимая AUSTEDO, не водите автомобиль и не управляйте опасными механизмами, пока не узнаете, как AUSTEDO влияет на вас. Употребление алкоголя и других лекарств, которые также могут вызывать сонливость, во время приема AUSTEDO может усилить любую сонливость, вызванную AUSTEDO.
Наиболее частые побочные эффекты AUSTEDO у людей с болезнью Хантингтона включают сонливость (седативный эффект), диарею, усталость и сухость во рту.
Наиболее частые побочные эффекты AUSTEDO у людей с поздней дискинезией включают воспаление носа и горла (ринофарингит) и проблемы со сном (бессонница).
Это не все возможные побочные эффекты AUSTEDO. Спросите у своего доктора о побочных эффектах. Вам рекомендуется сообщать в FDA о побочных эффектах рецептурных препаратов.Посетите www.fda.gov/medwatch или позвоните по телефону 1-800-FDA-1088.
Пожалуйста, прочтите сопроводительный документ.
Ювенильное начало HD | Общество болезни Хантингтона Америки
Что такое болезнь Хантингтона с юношеским началом?
Болезнь Хантингтона с юношеским началом (JHD) — это форма болезни Хантингтона (HD), которая поражает детей и подростков. Болезнь Хантингтона — это наследственное нейродегенеративное заболевание, которое характеризуется прогрессирующим ухудшением двигательных, когнитивных, поведенческих и психиатрических симптомов.JHD вызывается мутацией гена хантингтина, называемой «экспансией CAG-повторов». Мутация приводит к постепенной дегенерации нейронов в базальных ганглиях головного мозга, которые отвечают за координацию движений, мыслей и эмоций. По мере прогрессирования JHD другие области мозга претерпевают дегенерацию нейронов с диффузной и тяжелой атрофией головного мозга, которая сравнима с поздней стадией болезни Альцгеймера.
Признаки неисправности JHD
Презентация JHD включает изменения личности, координации, поведения, речи или способности к обучению.Физические изменения включают ригидность, скованность в ногах, неуклюжесть, замедленность движений, тремор или миоклонус. По сравнению с HD у взрослых, судороги и ригидность являются обычным явлением, а хорея — редкостью.
Предрасположенность и типичные начальные симптомы подросткового дебюта HD включают:
- Положительный семейный анамнез HD, обычно у отца
- Скованность в ногах
- Неуклюжесть рук и ног
- Снижение когнитивной функции
- Изменения в поведении
- Изъятия
- Изменения двигательной функции полости рта
- Хорея у подростка
- Нарушения поведения
Начало и прогрессирование
JHD обычно имеет более высокую скорость прогрессирования, чем HD с дебютом у взрослых; чем раньше наступит заболевание, тем быстрее прогрессирует JHD.Смерть часто наступает в течение 10 лет после начала JHD, в отличие от 10-25 лет при появлении HD у взрослых.
Лечение
Не существует лекарства или лечения, чтобы остановить, замедлить или обратить вспять прогрессирование JHD. Для облегчения симптомов могут быть прописаны лекарства. Детский психиатр или специалист по управлению поведением может заняться поведенческими расстройствами. Патолог речевого языка может оценить проблемы с общением и глотанием. По мере прогрессирования заболевания можно проконсультироваться с диетологом, чтобы удовлетворить потребности в питании.Вспомогательные устройства, такие как инвалидные коляски, шлемы и доски связи, могут использоваться для обеспечения безопасности и улучшения качества жизни.
Рекомендуется связаться с ближайшим к вам HDSA Center of Excellence , если у вас есть основания подозревать случай JHD.
Служба социального обеспечения
обязана быстро предоставлять пособия заявителям, состояние здоровья которых настолько серьезно, что их состояние явно соответствует стандартам инвалидности. Пособия по состраданию — это способ быстрого выявления заболеваний и других заболеваний, которые неизменно подпадают под установленный законом стандарт для инвалидности.Программа Compassionate Allowances ускоряет принятие решений об инвалидности, чтобы гарантировать, что американцы с наиболее серьезными формами инвалидности получат решения о выплате пособий в течение нескольких дней, а не месяцев или лет.
Предлагаемые медицинские свидетельства в записи для оценки
Для соответствия списку Compassionate Allowance требуется диагноз «HDU у несовершеннолетних» (JHD), а также клиническая информация, документирующая неврологические или когнитивные изменения.
Предпочтительными источниками этой информации являются истории болезни лечащего терапевта, невролога или психиатра, в том числе:
- Записи лечащего врача, документирующие прогрессирование двигательных, когнитивных и психиатрических симптомов, семейный анамнез и аномальные результаты неврологического обследования, соответствующие ювенильному началу HD
- Лабораторные испытания, показывающие экспансию полностью проникающего CAG-повтора в гене HD (> 39 CAG-повторов).
- Визуализация головного мозга, подтверждающая доказательства
- Психологические или психиатрические заключения, включая нейрокогнитивные тесты
- Школьные документы, подтверждающие.
Щелкните здесь, чтобы получить дополнительную информацию о социальных пособиях по состраданию.
Дополнительная литература
Чтобы узнать больше, нажмите на ссылки ниже, чтобы загрузить PDF-версии информативных и подробных публикаций HDSA:
Служба поддержки HDSA
.