около 1000 детей с редкими заболеваниями в Казахстане отмечают свой праздник
28 февраля отмечается День редких заболеваний. Всего в мире насчитывается более 7 000 различных видов редких болезней, среди которых есть как смертельные, так и те, которые при правильном и регулярном лечении позволяют прожить нормальную жизнь. В Казахстане такими недугами страдают около 1 000 детей.
Ежегодно в этот день активисты стараются привлечь внимание общественности к проблемам людей, больных орфанными заболеваниями, а также повысить осведомленность о редких болезнях и их влиянии на жизнь людей.
О том, как маленьким пациентам с редким диагнозом приходится бороться за жизнь, рассказывает директор Научного центра педиатрии и детской хирургии Риза Боранбаева.
— Редкие заболевания или как их называют орфанные, это хронически тяжелые или угрожающие жизни болезни. Они могут приводить к инвалидности, сокращению продолжительности жизни. В России и Казахстане считаются редкими заболевания с распространённостью 1 случая на 10 тысяч человек. В основном это генетические заболевания, но среди них есть болезни инфекционного, аутоиммунного и токсического характера. В целом в мире имеется около 7000 заболеваний.
В основном это генетические заболевания, но среди них есть болезни инфекционного, аутоиммунного и токсического характера. В целом в мире имеется около 7000 заболеваний.
Центр педиатрии и детской хирургии является центром координации. Мы координируем детей с лизосомными болезнями и являемся методологическим центром, где проводим обучение специалистов. В прошлом году мы определили региональных координаторов по редким детским болезням. Они берут на себя организационные вопросы. Нет такого одного специалиста, который бы занимался лечебной деятельностью, пациентами занимаются различные профили.
— Существует официальная статистика больных орфанными заболеваниями в Казахстане?
— В Казахстане около 1 000 детей с редкими заболеваниями. Наблюдает рост выявляемости лизосомных заболеваний. Если в 2011 году больных мукополисахаридозом было 6 человек, то сейчас их — 39. С болезнью Гоше было 9, сегодня уже 19. А больных муковисцидозом детей насчитывается уже 106. Это говорит не о том, что заболевания растут, это говорит о том, что улучшается диагностика. Мы наладили работу с международными клиниками, отправляем лабораторные данные и они уже ставят точный диагноз.
Мы наладили работу с международными клиниками, отправляем лабораторные данные и они уже ставят точный диагноз.
— Возможно ли вылечить ребенка или обеспечить ему нормальное существование?
— Все зависит от того, есть ли лечение у этого заболевания. У ряда болезней лечение может быть лишь симптоматическим. Редкие болезни плохо изучены. У нас встречаются болезни, которые обнаруживаются раз в 3-5 лет. Если у ребенка обнаружат редкое заболевание, то лечащий врач должен направить ребенка к региональному координатору. Если у этого заболевания есть патогенетическое лечение, то пациента направят к профильным специалистам, в научные центры. Если дадут заключение, что лечение необходимо оказывать только симптоматическое, то терапию будут оказывать по месту жительства.
— Во сколько обходится лечение детей с редким диагнозом?
— Для лечения этих болезней необходимы дорогостоящие лекарства, ни одна семья со средним достатком не сможет обеспечить больного ребенка самостоятельно. Лечение патогенетических заболеваний обеспечивается из республиканского бюджета, симптоматическое — из средств местного бюджета. При этом, ни одна страна в мире полностью не покрывает лечение детей с редкими заболеваниями. В мире практикуется помощь общественных организаций и благотворительных фондов.
Лечение патогенетических заболеваний обеспечивается из республиканского бюджета, симптоматическое — из средств местного бюджета. При этом, ни одна страна в мире полностью не покрывает лечение детей с редкими заболеваниями. В мире практикуется помощь общественных организаций и благотворительных фондов.
— Что в Казахстане делается для лечения редких заболеваний?
— Есть проблемы, но все будет решаться со временем. Наше государство сделало многое для лечения редких заболеваний. Улучшилась диагностика. Если раньше ставили диагноз в 8-10 лет, то сейчас в 2-3 года. В некоторых случаях при поздней диагностике могут развиться необратимые последствия в органах и системах. Несколько детей с болезнью Гоше уже стали старше 18 лет, и мы передали их под наблюдение во взрослую службу. Раньше такие дети до совершеннолетия не доживали. Сегодня очень многое делается, выделяются средства, обучаются врачи, повышаем грамотность родителей. За прошедший год в Казахстане реализована дорожная карта по редким заболеваниям у детей. На базе Научного центра педиатрии и детской хирургии был создан общественный совет, куда вошли специалисты, врачи, неправительственные организации и пациентские организации. В этом году министром здравоохранения был утвержден и подписан перечень орфанных заболеваний.
На базе Научного центра педиатрии и детской хирургии был создан общественный совет, куда вошли специалисты, врачи, неправительственные организации и пациентские организации. В этом году министром здравоохранения был утвержден и подписан перечень орфанных заболеваний.
Орфанные заболевания / Министерство здравоохранения Республики Башкортостан
Один из миллиона, особенный, уникальный –
эти эпитеты подойдут гениям и тем, кому повезло меньше, –
людям с редкими болезнями.
Им не помогают обычные лекарства, они редко доживают до старости
и часто выглядят не так, как другие люди.
По инициативе европейской организации по изучению редких болезней EURORDIS самый редкий день в году – 29 февраля — официально получил статус Международного дня редких заболеваний (Rare Disease Day). В невисокосные годы праздник отмечается 28 февраля.
Ежедневно, до 28 февраля мы будем знакомить вас с одним из тяжелых орфанных заболеваний.
Впервые термин «орфанные болезни» появился 1983 г. в США. Редкие болезни затрагивают небольшую часть популяции, часто являются генетическими и сопровождают человека в течение всей его жизни, даже если симптомы данной патологии появляются не сразу. В разных странах определение и перечень орфанных заболеваний принимаются на государственном уровне, единого определения для них не существует, так же как нет единого критерия отнесениям заболеваний к этой группе.
Орфанные заболевания характеризуются очень низким уровнем распространенности, большим количеством нозологий, так же существует проблема, связанная с дефицитом информации об этих болезнях, но ими страдают значительное число людей по всему миру. «Очень редкие болезни» делают пациентов и их семьи изолированными и уязвимыми. Поэтому по всему миру разрабатываются новые методы диагностики, ведутся клинические исследования, многие биотехнологические и фармацевтические компании ведут активные разработки инновационных продуктов, предназначенных для лечения и реабилитации пациентов.
К редким или «орфанным» заболеваниям в России относят нозологии с распространенностью менее 10 человек на 100 000 населения.
21 ноября 2011 года был принят Федеральный закон Российской Федерации № 323–ФЗ «Об основах охраны здоровья граждан в Российской Федерации», в котором были введены понятия, касающиеся редких болезней.
Орфанными являются заболевания, которые имеют распространенность не более 10 случаев заболевания на 1000 тысяч населения. Их перечень формируется уполномоченным федеральным органом исполнительной власти на основании статистических данных и размещается на его официальном сайте в сети «Интернет». Перечень утверждается Правительством Российской Федерации.
В целях обеспечения граждан, страдающих заболеваниями, включенными в данный перечень, лекарственными препаратами и специализированными продуктами лечебного питания осуществляется ведение Федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности (далее- Федеральный регистр).
Многие редкие заболевания являются генетическими, и, следовательно, сопровождают человека в течение всей жизни, даже если симптомы проявляются не сразу. Многие редкие болезни возникают в детстве, и около 30 % детей с редкими заболеваниями не доживают до 5 лет.
В 2012 году Правительством РФ был утвержден перечень жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидности, представленный 24 формами болезней. Этот документ также определяет правила ведения Федерального регистра лиц, страдающих орфанными заболеваниями, включенными в перечень. Постановление является необходимым для осуществления статьи 44 Федерального закона №323 от 21.11.2011, которая определяет порядок финансирования лекарственного обеспечения пациентов с редкими заболеваниями.
В 2019 году, согласно Федеральных законов от 03. 08.2018 N 299-ФЗ, от 27.12.2019 N 452-ФЗ, семь нозологий были переведены в список высокозатратных нозологий. На сегодняшний день в перечень жизнеугрожающих, хронических, редких заболеваний включает 17 нозологий.
08.2018 N 299-ФЗ, от 27.12.2019 N 452-ФЗ, семь нозологий были переведены в список высокозатратных нозологий. На сегодняшний день в перечень жизнеугрожающих, хронических, редких заболеваний включает 17 нозологий.
В Башкортостане ведётся целенаправленная работа по совершенствованию помощи пациентам, страдающим редкими (орфанными) заболеваниями. В рамках данной работы был разработан и принят Приказ Министерства здравоохранения Республики Башкортостан № 1901 – Д от 25 октября 2019 года «О совершенствовании организации оказания медицинской помощи жителям Республики Башкортостан, страдающим редкими (орфанными) заболеваниями». В структуре Республиканского медико-генетического центра создан региональный центр орфанных заболеваний (ЦОЗ), разработан алгоритм действий при подозрении на редкое заболевание и маршрутизация пациентов, разработан алгоритм выявления и постановки диагноза. В ЦОЗ, с целью оптимизации работы по вопросам орфанных заболеваний, организована консультативная деятельность, в том числе в рамках взаимодействия с региональными медико-генетическими консультациями, с использованием телемедицинских технологий, так же реализуется исследовательская, информационная, образовательная деятельность, ведётся регистр и сотрудничество с международным медицинским сообществом.
Основными задачами центра орфанных заболеваний РМГЦ являются: организация и проведение мониторинга состояния здоровья пациентов с редкими заболеваниями с участием ответственных специалистов, динамическая оценка эффективности их лечения, осуществление диетической коррекции, участие в диагностике заболеваний при амбулаторном обращении пациентов, консультирование пациентов при их стационарном лечении, организация медико-психологической реабилитации, маршрутизация, участие в работе врачебной комиссии по редким болезням, ведение регистра пациентов, страдающих орфанными заболеваниями, взаимодействие с пациентскими и благотворительными организациями, учреждениями, оказывающими паллиативную помощь, внедрение в практическое здравоохранение новых методов диагностики и лечения редких болезней, повышение уровня знаний и настороженности в части редких заболеваний, своевременного их выявления и направление к профильным специалистам, скрининг и профилактика орфанных болезней у детей.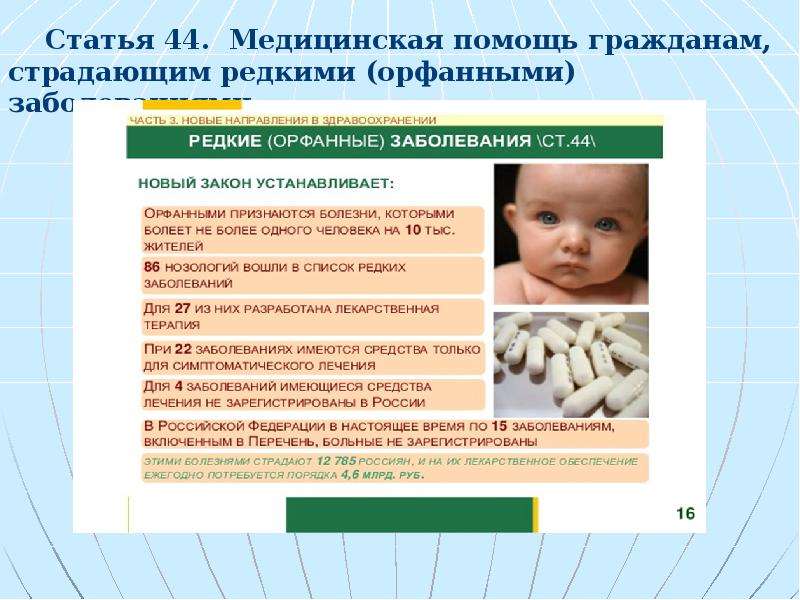
Пресс-служба Министерства здравоохранения Республики Башкортостан
(по информации РМГЦ)
Орфанные заболевания в Казахстане
В последний день февраля отмечается Международный день редких заболеваний. О том, какая работа проводится в стране для пациентов с редкими заболеваниями, мы беседуем с председателем Правления АО «Научный центр педиатрии и детской хирургии», главным детским онкогематологом МЗ РК, д.м.н. Ризой Зулкарнаевной Боранбаевой.
— Расскажите, пожалуйста, о распространенности орфанных заболеваний в мире и Казахстане.
— Редкими (орфанными) называют те заболевания, которые затрагивают небольшое количество людей от общей численности населения планеты. Сегодня в мире нет единого определения и единого перечня редких заболеваний, поэтому каждая страна индивидуально разрабатывает критерии отнесения заболеваний к этой группе. В различных странах и частях света уровень встречаемости отличается. Например, в Европе болезнь Хансена (проказа) выявляется крайне нечасто, а в центральной Африке ее диагностируют как у детей, так и взрослых. Муковисцидоз распространен в европейских странах, тогда как в Азии это генетическое заболевание является довольно редким. Поэтому в различных уголках мира уровень заболеваемости такими патологиями определяют по-разному. Так, в Японии редкими считаются те болезни, которые встречаются у 1 человека из 2,5 тыс. населения, в Америке этот показатель составляет 1 из 1,5 тыс. людей, Европе 1 из 2 тыс. и в России 5 человек на 50 тыс. населения. Во всем мире более 300 миллионов человек живут с одним или несколькими из более чем 6000 выявленных редких заболеваний. В РК такими считаются заболевания, которые имеют распространение не более 1 случая на 10 000 населения. Всего согласно Приказу МЗ РК №142 от 20 октября 2020 года в Перечень редких заболеваний входит 62 нозологии. По данным электронного регистра диспансерных больных в РК на 1.
В различных странах и частях света уровень встречаемости отличается. Например, в Европе болезнь Хансена (проказа) выявляется крайне нечасто, а в центральной Африке ее диагностируют как у детей, так и взрослых. Муковисцидоз распространен в европейских странах, тогда как в Азии это генетическое заболевание является довольно редким. Поэтому в различных уголках мира уровень заболеваемости такими патологиями определяют по-разному. Так, в Японии редкими считаются те болезни, которые встречаются у 1 человека из 2,5 тыс. населения, в Америке этот показатель составляет 1 из 1,5 тыс. людей, Европе 1 из 2 тыс. и в России 5 человек на 50 тыс. населения. Во всем мире более 300 миллионов человек живут с одним или несколькими из более чем 6000 выявленных редких заболеваний. В РК такими считаются заболевания, которые имеют распространение не более 1 случая на 10 000 населения. Всего согласно Приказу МЗ РК №142 от 20 октября 2020 года в Перечень редких заболеваний входит 62 нозологии. По данным электронного регистра диспансерных больных в РК на 1. 01.2021 г. На учете состоит всего 12 948 детей с орфанными заболеваниями.
01.2021 г. На учете состоит всего 12 948 детей с орфанными заболеваниями.
— С чем связаны эти случаи, есть ли заболевания, присущие только определенным регионам или расам?
— Примерно 72% орфанных болезней обусловлены генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте. Генетическую основу имеют практически все заболевания. Реже встречаются токсические, инфекционные или аутоиммунные болезни. Причинами их развития могут быть наследственность, ослабление иммунитета, плохая экология, высокий радиационный фон, вирусные инфекции у мамы и у самих детей в раннем возрасте.
Многие заболевания могут передаваться от одного поколения к другому, это объясняет то, почему некоторые редкие заболевания развиваются в целых семьях. Это также объясняет тот факт, что заболевание может быть редким в одном регионе и частым в другом. Например, в Турции и Азербайджане достаточно часто встречается такое заболевание, как талассемия.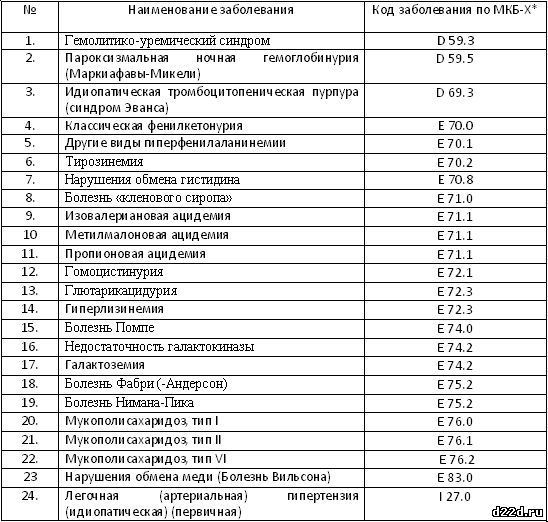 Болезнь Гоше чаще всего поражает ашкеназских евреев. А в Казахстане таких пациентов очень мало.
Болезнь Гоше чаще всего поражает ашкеназских евреев. А в Казахстане таких пациентов очень мало.
— Какие нозологии встречаются чаще всего в нашей стране? Какова их причина?
— В структуре заболеваемости детей орфанными заболеваниями лидирующие позиции в РК занимают заболевания центральной и периферической нервной системы – 7239 детей; второе место по частоте встречаемости занимают онкологические и гематологические заболевания – 3125 детей и на третьем месте заболевания, обусловленные нарушением обмена веществ – 1543 ребенка.
В основной массе – это наследственные заболевания, обусловленные генными мутациями.
Онкологические, гематологические и редкие аутоиммунные заболевания возникают в результате нарушения регуляции различный процессов в иммунной системе. Однозначной причины этому процессу нет. Орфанные болезни детей и взрослых также могут быть спровоцированы малоизученными вирусами и бактериями, а также токсическим воздействием.
— Вы являетесь руководителем главного центра страны, в котором проходят лечение дети с орфанными заболеваниями, назовите основные проблемы, с которыми сталкиваются ваши пациенты и их родители.
— АО «Научный центр педиатрии и детской хирургии» является координационным центром по редким заболеваниям у детей в РК, курирует и осуществляет консультативно-диагностическую и медицинскую помощь детям с онкологическими заболеваниями, лизосомальными болезнями (болезнь Гоше, мукополисахаридоз), наследственными нарушениями свертываемости крови, муковисцидозом и др. Одной из основных проблем, с которыми сталкиваются наши пациенты и их родители – это поздняя диагностика заболевания, когда ввиду неспецифичности симптомов многих заболеваний эти пациенты наблюдаются у специалистов разных профилей. Также важным вопросом является сложность молекулярно-генетической верификации этих заболеваний, так как в РК при многих заболеваниях эти исследования не проводятся.
Кроме первичной диагностики этих заболеваний, есть острая необходимость в мониторинге активности ферментов в процессе терапии с целью оценки эффективности лечения. Недостаточно специалистов, имеющих опыт в области лечения орфанных болезней. Немаловажным и актуальным вопросом для улучшения качества жизни детей с редкими заболеваниями является комплексный подход в лечении. То есть, на фоне проводимого лечения должны проводиться программы реабилитации.
Немаловажным и актуальным вопросом для улучшения качества жизни детей с редкими заболеваниями является комплексный подход в лечении. То есть, на фоне проводимого лечения должны проводиться программы реабилитации.
— В чем заключается сложность их диагностики?
— Сложность диагностики многих генетических заболеваний заключается в отсутствии отличительных клинических признаков, неспецифичности симптомов заболеваний и маскировкой под другие заболевания таких, как сепсис, внутриутробная пневмония, эпилепсия, нейроинфекция, перинатальная патология и другие. Поэтому не все орфанные заболевания своевременно диагностируются.
В последние годы наблюдается положительная динамика по ранней диагностике наследственных заболеваний. Это достигнуто в основном за счет обучения и повышения осведомленности врачей разных специальностей в отношении редких наследственных заболеваний, выработки маршрутов оказания помощи детям с орфанными заболеваниями и работы республиканских центров координации. Однако в РК не налажены молекулярно-генетические методы исследования по многим заболеваниям. Несмотря на большое разнообразие редких заболеваний и возможных мутаций при них, все-таки эти заболевания встречаются редко и кратность проведения исследований очень низкая, так в год диагностируется не более 20 новых случаев отдельных заболеваний. Многие реагенты для исследований не зарегистрированы в РК, их надо ввозить по разовому ввозу, что увеличивает время их приобретения (только после процедуры разрешения в МЗ), а сроки годности реактивов очень короткие. В связи с чем, ни одной лаборатории не выгодно делать большой спектр генетических заболеваний.
Однако в РК не налажены молекулярно-генетические методы исследования по многим заболеваниям. Несмотря на большое разнообразие редких заболеваний и возможных мутаций при них, все-таки эти заболевания встречаются редко и кратность проведения исследований очень низкая, так в год диагностируется не более 20 новых случаев отдельных заболеваний. Многие реагенты для исследований не зарегистрированы в РК, их надо ввозить по разовому ввозу, что увеличивает время их приобретения (только после процедуры разрешения в МЗ), а сроки годности реактивов очень короткие. В связи с чем, ни одной лаборатории не выгодно делать большой спектр генетических заболеваний.
А так как генетическая верификация диагноза не доступна в РК, исследование проводится в зарубежных молекулярно-генетических лабораториях.
— Что делается, чтобы эта ситуация изменилась, особенно в регионах? Как решается проблема при отсутствии квалифицированных специалистов?
— Известно, что чем раньше выявляешь такое заболевание и, соответственно, чем раньше начинается необходимая терапия, тем лучше результат и лучше качество жизни детей. В последние годы наблюдается положительная динамика по ранней диагностике наследственных заболеваний. Это достигнуто в основном за счет обучения и повышения осведомленности врачей разных специальностей в отношении редких наследственных заболеваний, выработки маршрутов оказания помощи детям с орфанными заболеваниями и работы республиканских центров координации.
В последние годы наблюдается положительная динамика по ранней диагностике наследственных заболеваний. Это достигнуто в основном за счет обучения и повышения осведомленности врачей разных специальностей в отношении редких наследственных заболеваний, выработки маршрутов оказания помощи детям с орфанными заболеваниями и работы республиканских центров координации.
С 2017 года в РК реализуется Дорожная карта по редким заболеваниям, в рамках которой осуществляется большая работа в виде: проведения образовательных программ в регионах, оказания консультативной и медицинской помощи детям с редкими заболеваниями, проведение информационно-разъяснительной работы (выпуск: социальных роликов, методических рекомендаций, буклетов, памяток, брошюр для родителей и пациентов), проводятся рабочие совещания по проблемным вопросам совместно с НПО и пациентскими организациями, выезды в неблагополучные регионы совместно с НПО по вопросам лекарственного обеспечения и др. В рамках магистратуры и докторантуры в нашем Центре реализуются научные проекты по темам, касающимся орфанных заболеваний у детей. На сегодня защищены 1 докторская работа и 2 магистерские диссертации, где исследованы вопросы генетической диагностики казахстанской популяции детей с мукополисахаридозом, болезнью Гоше и муковисцидозом.
На сегодня защищены 1 докторская работа и 2 магистерские диссертации, где исследованы вопросы генетической диагностики казахстанской популяции детей с мукополисахаридозом, болезнью Гоше и муковисцидозом.
Родители могут обратиться по всем вопросам с непонятными заболеваниями к координатору редких (орфанных) патологий, которые есть в каждом регионе страны.
— Существует ли регистр пациентов с орфанными заболеваниями?
— На настоящий момент единого регистра пациентов с орфанными заболеваниями в РК не существует. Но все пациенты с редкими болезнями наблюдаются динамически на уровне своих поликлиник, поэтому они должны быть зарегистрированы в электронном регистре диспансерного больного. Создание единого регистра орфанных заболеваний планируется.
— Насколько эффективна работа генетических лабораторий, исследующих риск рождения ребенка с наследственными орфанными заболеваниями?
— Диагностика наследственных заболеваний начинается обычно задолго до рождения ребенка. В стране существует сеть медико-генетических консультаций, есть кабинеты генетиков, одной из функций которых является пренатальная диагностика беременных женщин из групп риска. Если в результате пренатального скрининга выявляются факторы, говорящие о болезни у ребенка, то беременной женщине объясняются все риски и предоставляется право выбора рожать или нет ребенка. Кроме того, в Казахстане проводится неонатальный скрининг на фенилкетонурию (это заболевание, при котором ранняя диетотерапия позволяет предотвратить тяжелые осложнения и инвалидность). Планируется расширение неонатального скрининга за счет внедрения исследования тандемной масс-спектрометрии у отдельных групп новорожденных. Данная диагностика позволит выявлять 49 различных редко встречающихся наследственных болезней обмена.
В стране существует сеть медико-генетических консультаций, есть кабинеты генетиков, одной из функций которых является пренатальная диагностика беременных женщин из групп риска. Если в результате пренатального скрининга выявляются факторы, говорящие о болезни у ребенка, то беременной женщине объясняются все риски и предоставляется право выбора рожать или нет ребенка. Кроме того, в Казахстане проводится неонатальный скрининг на фенилкетонурию (это заболевание, при котором ранняя диетотерапия позволяет предотвратить тяжелые осложнения и инвалидность). Планируется расширение неонатального скрининга за счет внедрения исследования тандемной масс-спектрометрии у отдельных групп новорожденных. Данная диагностика позволит выявлять 49 различных редко встречающихся наследственных болезней обмена.
— Как государство на законодательном уровне поддерживает таких пациентов?
— Лечение детей проводится в рамках гарантированного объема бесплатно медицинской помощи как на стационарном, так на амбулаторном уровнях. В рамках Приказа Министра здравоохранения Республики Казахстан от 29 августа 2017 года №666 «Об утверждении Перечня лекарственных средств и медицинских изделий в рамках гарантированного объема бесплатной медицинской помощи и в системе обязательного социального медицинского страхования, в том числе отдельных категорий граждан с определенными заболеваниями (состояниями) бесплатными и (или) льготными лекарственными средствами, медицинскими изделиями и специализированными лечебными продуктами на амбулаторном уровне» пациенты с орфанными заболеваниями обеспечиваются лекарственными средствами и изделиями медицинского назначения бесплатно, то есть за счет средств республиканского бюджета.
В рамках Приказа Министра здравоохранения Республики Казахстан от 29 августа 2017 года №666 «Об утверждении Перечня лекарственных средств и медицинских изделий в рамках гарантированного объема бесплатной медицинской помощи и в системе обязательного социального медицинского страхования, в том числе отдельных категорий граждан с определенными заболеваниями (состояниями) бесплатными и (или) льготными лекарственными средствами, медицинскими изделиями и специализированными лечебными продуктами на амбулаторном уровне» пациенты с орфанными заболеваниями обеспечиваются лекарственными средствами и изделиями медицинского назначения бесплатно, то есть за счет средств республиканского бюджета.
По мере появления новых знаний по этиологии и патогенезу заболеваний разрабатываются новые лекарственные средства, новые технологии.
При разработке и утверждении клинических протоколов диагностики и лечения в обязательном порядке за основу берутся международные стандарты и клинические рекомендации по ведению тех или иных болезней, которые обновляются каждые 3-5 лет.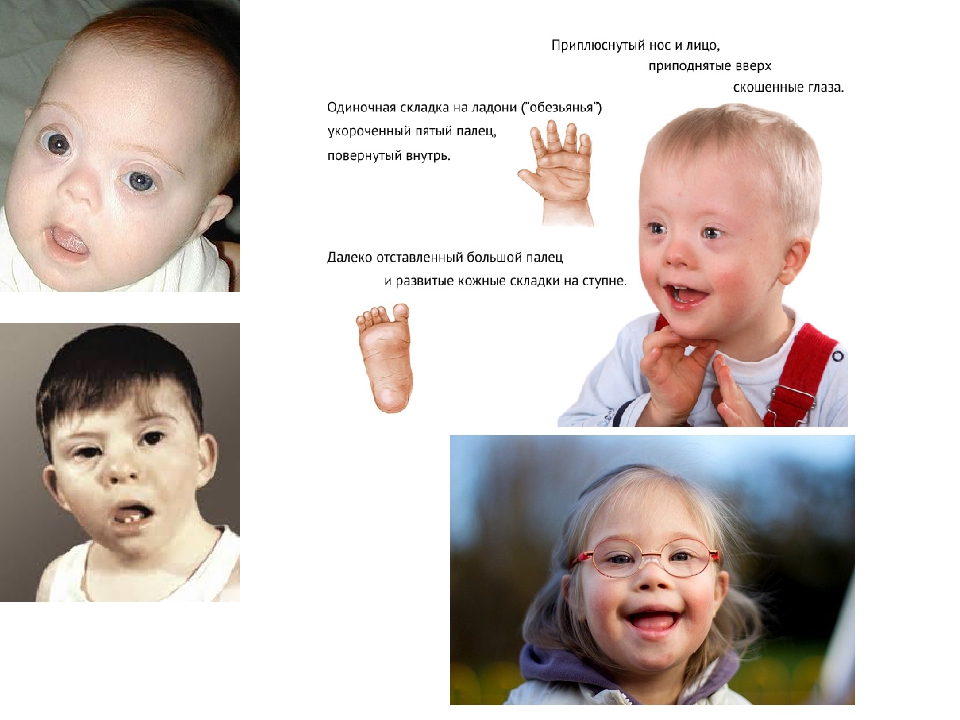
— Почему часто звучат высказывания о дефиците ЛС для терапии редких заболеваний? Как решаются эти проблемы?
— На данный момент имеется необходимость совершенствования регистра с внесением всех данных пациентов, указанием препаратов, их доз и кратностей, с отражением преемственности между службами, контроль состояния пациента при перемещении и переводе в Республиканскую медицинскую организацию. Основными проблемами лекарственного обеспечения является отсутствие регистрации отдельных препаратов в РК; отсутствие установленной предельной цены для закупа лекарственных средств. Те препараты, которые не включены в Приказ Министра здравоохранения Республики Казахстан от 29 августа 2017 года №666, но нужны пациенту, должны закупаться из местного бюджета. Поэтому в случае необходимости по рекомендации курирующих профильных специалистов препараты закупаются решением местных маслихатов.
— Есть ли у нас фонды, организации, которые оказывают помощь Вашему центру и в целом помогают таким пациентам?
— На базе НЦПДХ с 2017 г. создан Общественный совет, куда входят основные пациентские организации по вышеуказанным нозологиям и общественные фонды, осуществляющие помощь на благотворительной основе детям с редкими заболеваниями. В ноябре 2020 г. в Казахстане была создана новая «Ассоциация помощи пациентам с орфанными заболеваниями», объединившая несколько НПО с общими целями – оказание благотворительной помощи, социальной поддержки и защиты лиц, страдающих редкими заболеваниями. Только объединив усилия государственных, неправительственных организаций, бизнес-структур и широкую общественность, мы сможем совершенствовать систему помощи больным редкими заболеваниями.
создан Общественный совет, куда входят основные пациентские организации по вышеуказанным нозологиям и общественные фонды, осуществляющие помощь на благотворительной основе детям с редкими заболеваниями. В ноябре 2020 г. в Казахстане была создана новая «Ассоциация помощи пациентам с орфанными заболеваниями», объединившая несколько НПО с общими целями – оказание благотворительной помощи, социальной поддержки и защиты лиц, страдающих редкими заболеваниями. Только объединив усилия государственных, неправительственных организаций, бизнес-структур и широкую общественность, мы сможем совершенствовать систему помощи больным редкими заболеваниями.
— Благодарим за интересное интервью.
02 марта 2021
«Казахстанский фармацевтический вестник» №5 (604), март 2021 г.
Сегодня перед нами стоит задача создать все условия для обеспечения прозрачности, эффективности и справедливости принятия решений по оказанию медицинской помощи пациентам с орфанными заболеваниями – ФГБУ «ЦЭККМП» Минздрава России
[[[[«field91″,»contains_not»,»pdf»]],[[«show_fields»,»field94″]],»and»],[[[«field17″,»contains»,»\u041d\u0435\u043e\u0431\u0445\u043e\u0434\u0438\u043c\u043e\u0441\u0442\u0438 \u0432 \u0434\u0430\u043d\u043d\u043e\u043c \u043f\u0440\u043e\u0435\u043a\u0442\u0435 \u043d\u0435\u0442″]],[[«show_fields»,»field55″]],»and»],[[[«field18″,»contains»,»\u0411\u0435\u0441\u043f\u043e\u043b\u0435\u0437\u0435\u043d»]],[[«show_fields»,»field57″]],»and»],[[[«field21″,»contains»,»\u041d\u0435 \u0441\u043e\u043e\u0442\u0432\u0435\u0442\u0441\u0442\u0432\u0443\u0435\u0442″]],[[«show_fields»,»field59″]],»and»],[[[«field22″,»contains»,»\u041d\u0435 \u0441\u043e\u043e\u0442\u0432\u0435\u0442\u0441\u0442\u0432\u0443\u0435\u0442″]],[[«show_fields»,»field58″]],»and»],[[[«field23″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field60″]],»and»],[[[«field24″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field63″]],»and»],[[[«field25″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field61″]],»and»],[[[«field26″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field65″]],»and»],[[[«field27″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field64″]],»and»],[[[«field28″,»contains»,»1 \u044d\u0442\u0430\u043f. \u0424\u043e\u0440\u043c\u0438\u0440\u043e\u0432\u0430\u043d\u0438\u0435 \u043f\u0435\u0440\u0435\u0447\u043d\u044f \u0442\u0435\u043c \u0434\u043b\u044f \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0438\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440\u0430 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u0421\u043e\u0432\u0435\u0442\u043e\u043c \u043f\u043e \u043a\u0430\u0447\u0435\u0441\u0442\u0432\u0443 \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.»]],[[«show_fields»,»field73″]],»and»],[[[«field29″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field67″]],»and»],[[[«field30″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field68″]],»and»],[[[«field31″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field69″]],»and»],[[[«field32″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field70″]],»and»],[[[«field28″,»contains»,»2 \u044d\u0442\u0430\u043f.
\u0424\u043e\u0440\u043c\u0438\u0440\u043e\u0432\u0430\u043d\u0438\u0435 \u043f\u0435\u0440\u0435\u0447\u043d\u044f \u0442\u0435\u043c \u0434\u043b\u044f \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0438\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440\u0430 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u0421\u043e\u0432\u0435\u0442\u043e\u043c \u043f\u043e \u043a\u0430\u0447\u0435\u0441\u0442\u0432\u0443 \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.»]],[[«show_fields»,»field73″]],»and»],[[[«field29″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field67″]],»and»],[[[«field30″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field68″]],»and»],[[[«field31″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field69″]],»and»],[[[«field32″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field70″]],»and»],[[[«field28″,»contains»,»2 \u044d\u0442\u0430\u043f. \u0423\u0442\u0432\u0435\u0440\u0436\u0434\u0435\u043d\u0438\u0435 \u043f\u0435\u0440\u0435\u0447\u043d\u044f \u0442\u0435\u043c \u0434\u043b\u044f \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0438\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440\u0430 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u0440\u0443\u043a\u043e\u0432\u043e\u0434\u0438\u0442\u0435\u043b\u0435\u043c \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.»]],[[«show_fields»,»field75″]],»and»],[[[«field28″,»contains»,»3 \u044d\u0442\u0430\u043f. \u0424\u043e\u0440\u043c\u0438\u0440\u043e\u0432\u0430\u043d\u0438\u0435 \u0440\u0430\u0431\u043e\u0447\u0435\u0439 \u0433\u0440\u0443\u043f\u043f\u044b \u043f\u043e \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0435\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440\u0443 \u043a\u043e\u043d\u043a\u0440\u0435\u0442\u043d\u043e\u0433\u043e \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u0430 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.
\u0423\u0442\u0432\u0435\u0440\u0436\u0434\u0435\u043d\u0438\u0435 \u043f\u0435\u0440\u0435\u0447\u043d\u044f \u0442\u0435\u043c \u0434\u043b\u044f \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0438\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440\u0430 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u0440\u0443\u043a\u043e\u0432\u043e\u0434\u0438\u0442\u0435\u043b\u0435\u043c \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.»]],[[«show_fields»,»field75″]],»and»],[[[«field28″,»contains»,»3 \u044d\u0442\u0430\u043f. \u0424\u043e\u0440\u043c\u0438\u0440\u043e\u0432\u0430\u043d\u0438\u0435 \u0440\u0430\u0431\u043e\u0447\u0435\u0439 \u0433\u0440\u0443\u043f\u043f\u044b \u043f\u043e \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0435\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440\u0443 \u043a\u043e\u043d\u043a\u0440\u0435\u0442\u043d\u043e\u0433\u043e \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u0430 \u043b\u0435\u0447\u0435\u043d\u0438\u044f. «]],[[«show_fields»,»field66″]],»and»],[[[«field28″,»contains»,»4 \u044d\u0442\u0430\u043f. \u0424\u043e\u0440\u043c\u0438\u0440\u043e\u0432\u0430\u043d\u0438\u0435 \u0440\u0430\u0431\u043e\u0447\u0435\u0439 \u0433\u0440\u0443\u043f\u043f\u043e\u0439 \u0434\u043e\u0440\u043e\u0436\u043d\u043e\u0439 \u043a\u0430\u0440\u0442\u044b \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0438 \u0438 \u0432\u043d\u0435\u0434\u0440\u0435\u043d\u0438\u044f \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u0438 \u0443\u0442\u0432\u0435\u0440\u0436\u0434\u0435\u043d\u0438\u0435 \u0435\u0435 \u0440\u0443\u043a\u043e\u0432\u043e\u0434\u0438\u0442\u0435\u043b\u0435\u043c \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.»]],[[«show_fields»,»field76″]],»and»],[[[«field28″,»contains»,»5 \u044d\u0442\u0430\u043f. \u0410\u043d\u0430\u043b\u0438\u0437 \u043a\u043b\u0438\u043d\u0438\u0447\u0435\u0441\u043a\u0438\u0445 \u0440\u0435\u043a\u043e\u043c\u0435\u043d\u0434\u0430\u0446\u0438\u0439, \u0441\u0442\u0430\u043d\u0434\u0430\u0440\u0442\u043e\u0432, \u043f\u043e\u0440\u044f\u0434\u043a\u043e\u0432 \u043e\u043a\u0430\u0437\u0430\u043d\u0438\u044f \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043f\u043e\u043c\u043e\u0449\u0438 \u0432 \u0441\u043e\u043e\u0442\u0432\u0435\u0442\u0441\u0442\u0432\u0438\u0438 \u0441 \u0442\u0435\u043c\u043e\u0439 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f. «]],[[«show_fields»,»field77″]],»and»],[[[«field28″,»contains»,»6 \u044d\u0442\u0430\u043f. \u0421\u0438\u0442\u0443\u0430\u0446\u0438\u043e\u043d\u043d\u044b\u0439 \u0430\u043d\u0430\u043b\u0438\u0437 \u0441\u0442\u0435\u043f\u0435\u043d\u0438 \u0441\u043e\u043e\u0442\u0432\u0435\u0442\u0441\u0442\u0432\u0438\u044f \u0432\u043e\u0437\u043c\u043e\u0436\u043d\u043e\u0441\u0442\u0435\u0439 \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438 \u043f\u043e \u043e\u043a\u0430\u0437\u0430\u043d\u0438\u044e \u043f\u043e\u043c\u043e\u0449\u0438 \u0442\u0440\u0435\u0431\u043e\u0432\u0430\u043d\u0438\u044f\u043c \u043a\u043b\u0438\u043d\u0438\u0447\u0435\u0441\u043a\u0438\u0445 \u0440\u0435\u043a\u043e\u043c\u0435\u043d\u0434\u0430\u0446\u0438\u0439, \u0441\u0442\u0430\u043d\u0434\u0430\u0440\u0442\u043e\u0432, \u043f\u043e\u0440\u044f\u0434\u043a\u043e\u0432 \u043e\u043a\u0430\u0437\u0430\u043d\u0438\u044f \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043f\u043e\u043c\u043e\u0449\u0438.
«]],[[«show_fields»,»field66″]],»and»],[[[«field28″,»contains»,»4 \u044d\u0442\u0430\u043f. \u0424\u043e\u0440\u043c\u0438\u0440\u043e\u0432\u0430\u043d\u0438\u0435 \u0440\u0430\u0431\u043e\u0447\u0435\u0439 \u0433\u0440\u0443\u043f\u043f\u043e\u0439 \u0434\u043e\u0440\u043e\u0436\u043d\u043e\u0439 \u043a\u0430\u0440\u0442\u044b \u0440\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0438 \u0438 \u0432\u043d\u0435\u0434\u0440\u0435\u043d\u0438\u044f \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u0438 \u0443\u0442\u0432\u0435\u0440\u0436\u0434\u0435\u043d\u0438\u0435 \u0435\u0435 \u0440\u0443\u043a\u043e\u0432\u043e\u0434\u0438\u0442\u0435\u043b\u0435\u043c \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.»]],[[«show_fields»,»field76″]],»and»],[[[«field28″,»contains»,»5 \u044d\u0442\u0430\u043f. \u0410\u043d\u0430\u043b\u0438\u0437 \u043a\u043b\u0438\u043d\u0438\u0447\u0435\u0441\u043a\u0438\u0445 \u0440\u0435\u043a\u043e\u043c\u0435\u043d\u0434\u0430\u0446\u0438\u0439, \u0441\u0442\u0430\u043d\u0434\u0430\u0440\u0442\u043e\u0432, \u043f\u043e\u0440\u044f\u0434\u043a\u043e\u0432 \u043e\u043a\u0430\u0437\u0430\u043d\u0438\u044f \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043f\u043e\u043c\u043e\u0449\u0438 \u0432 \u0441\u043e\u043e\u0442\u0432\u0435\u0442\u0441\u0442\u0432\u0438\u0438 \u0441 \u0442\u0435\u043c\u043e\u0439 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f. «]],[[«show_fields»,»field77″]],»and»],[[[«field28″,»contains»,»6 \u044d\u0442\u0430\u043f. \u0421\u0438\u0442\u0443\u0430\u0446\u0438\u043e\u043d\u043d\u044b\u0439 \u0430\u043d\u0430\u043b\u0438\u0437 \u0441\u0442\u0435\u043f\u0435\u043d\u0438 \u0441\u043e\u043e\u0442\u0432\u0435\u0442\u0441\u0442\u0432\u0438\u044f \u0432\u043e\u0437\u043c\u043e\u0436\u043d\u043e\u0441\u0442\u0435\u0439 \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438 \u043f\u043e \u043e\u043a\u0430\u0437\u0430\u043d\u0438\u044e \u043f\u043e\u043c\u043e\u0449\u0438 \u0442\u0440\u0435\u0431\u043e\u0432\u0430\u043d\u0438\u044f\u043c \u043a\u043b\u0438\u043d\u0438\u0447\u0435\u0441\u043a\u0438\u0445 \u0440\u0435\u043a\u043e\u043c\u0435\u043d\u0434\u0430\u0446\u0438\u0439, \u0441\u0442\u0430\u043d\u0434\u0430\u0440\u0442\u043e\u0432, \u043f\u043e\u0440\u044f\u0434\u043a\u043e\u0432 \u043e\u043a\u0430\u0437\u0430\u043d\u0438\u044f \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043f\u043e\u043c\u043e\u0449\u0438. «]],[[«show_fields»,»field81″]],»and»],[[[«field28″,»contains»,»7 \u044d\u0442\u0430\u043f. \u0420\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0430\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.»]],[[«show_fields»,»field74″]],»and»],[[[«field28″,»contains»,»8 \u044d\u0442\u0430\u043f. \u042d\u043a\u0441\u043f\u0435\u0440\u0442\u0438\u0437\u0430 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.»]],[[«show_fields»,»field79″]],»and»],[[[«field28″,»contains»,»9 \u044d\u0442\u0430\u043f. \u0423\u0442\u0432\u0435\u0440\u0436\u0434\u0435\u043d\u0438\u0435 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u043f\u0440\u0438\u043a\u0430\u0437\u043e\u043c \u0440\u0443\u043a\u043e\u0432\u043e\u0434\u0438\u0442\u0435\u043b\u044f \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438 \u0441 \u0446\u0435\u043b\u044c\u044e \u043e\u0431\u044f\u0437\u0430\u0442\u0435\u043b\u044c\u043d\u043e\u0433\u043e \u0432\u044b\u043f\u043e\u043b\u043d\u0435\u043d\u0438\u044f \u0432 \u0434\u0430\u043d\u043d\u043e\u0439 \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438.
«]],[[«show_fields»,»field81″]],»and»],[[[«field28″,»contains»,»7 \u044d\u0442\u0430\u043f. \u0420\u0430\u0437\u0440\u0430\u0431\u043e\u0442\u043a\u0430\/\u043f\u0435\u0440\u0435\u0441\u043c\u043e\u0442\u0440 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.»]],[[«show_fields»,»field74″]],»and»],[[[«field28″,»contains»,»8 \u044d\u0442\u0430\u043f. \u042d\u043a\u0441\u043f\u0435\u0440\u0442\u0438\u0437\u0430 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.»]],[[«show_fields»,»field79″]],»and»],[[[«field28″,»contains»,»9 \u044d\u0442\u0430\u043f. \u0423\u0442\u0432\u0435\u0440\u0436\u0434\u0435\u043d\u0438\u0435 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f \u043f\u0440\u0438\u043a\u0430\u0437\u043e\u043c \u0440\u0443\u043a\u043e\u0432\u043e\u0434\u0438\u0442\u0435\u043b\u044f \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438 \u0441 \u0446\u0435\u043b\u044c\u044e \u043e\u0431\u044f\u0437\u0430\u0442\u0435\u043b\u044c\u043d\u043e\u0433\u043e \u0432\u044b\u043f\u043e\u043b\u043d\u0435\u043d\u0438\u044f \u0432 \u0434\u0430\u043d\u043d\u043e\u0439 \u043c\u0435\u0434\u0438\u0446\u0438\u043d\u0441\u043a\u043e\u0439 \u043e\u0440\u0433\u0430\u043d\u0438\u0437\u0430\u0446\u0438\u0438. «]],[[«show_fields»,»field80″]],»and»],[[[«field28″,»contains»,»10 \u044d\u0442\u0430\u043f. \u041c\u043e\u043d\u0438\u0442\u043e\u0440\u0438\u043d\u0433 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.»]],[[«show_fields»,»field78″]],»and»],[[[«field71″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field28″]],»and»],[[[«field17″,»contains»,»\u041d\u0435\u043e\u0431\u0445\u043e\u0434\u0438\u043c (\u0434\u0440\u0443\u0433\u043e\u0435)»]],[[«show_fields»,»field83″]],»and»],[[[«field18″,»contains»,»\u041f\u043e\u043b\u0435\u0437\u0435\u043d (\u0434\u0440\u0443\u0433\u043e\u0435)»]],[[«show_fields»,»field82″]],»and»]]
«]],[[«show_fields»,»field80″]],»and»],[[[«field28″,»contains»,»10 \u044d\u0442\u0430\u043f. \u041c\u043e\u043d\u0438\u0442\u043e\u0440\u0438\u043d\u0433 \u043f\u0440\u043e\u0442\u043e\u043a\u043e\u043b\u043e\u0432 \u043b\u0435\u0447\u0435\u043d\u0438\u044f.»]],[[«show_fields»,»field78″]],»and»],[[[«field71″,»contains»,»\u041d\u0435\u0442″]],[[«show_fields»,»field28″]],»and»],[[[«field17″,»contains»,»\u041d\u0435\u043e\u0431\u0445\u043e\u0434\u0438\u043c (\u0434\u0440\u0443\u0433\u043e\u0435)»]],[[«show_fields»,»field83″]],»and»],[[[«field18″,»contains»,»\u041f\u043e\u043b\u0435\u0437\u0435\u043d (\u0434\u0440\u0443\u0433\u043e\u0435)»]],[[«show_fields»,»field82″]],»and»]]
Московская медицина. НИИ организации здравоохранения. Орфанные заболевания. — НАУЧНО-ПРАКТИЧЕСКИЙ ЦЕНТР СПЕЦИАЛИЗИРОВАННОЙ МЕДИЦИНСКОЙ ПОМОЩИ ДЕТЯМ имени В.Ф.Войно-Ясенецкого
Для выстраивания грамотной тактики лечения редкого заболевания необходимо понять природу его возникновения. Клинической диагностикой и определением тактики обследования и занимается ведущий научный сотрудник, врач- генетик генетической группы научного отдела «НПЦ спец. мед.помощи детям ДЗМ», к. м. н. Светлана Сергеевна Жилина.
мед.помощи детям ДЗМ», к. м. н. Светлана Сергеевна Жилина.
Орфанными заболеваниями называются те, которые затрагивают незначительную часть популяции – с частотой 1:10000 новорожденных. К орфанным заболеваниям относится болезнь Фабри, которой болеют только мальчики, болезнь Помпе с мышечной слабостью, кардиомиопатией, ранними нарушениями дыхания и с понижением концентрации глюкозы, болезнь Гоше. Еще мукополисахаридозы – лизосомные болезни, когда продукты неполного транспорта или метаболизма накапливаются в различных тканях и влекут за собой деформации скелета, увеличение печени, селезенки, снижение остроты зрения или слуха, задержку психического развития. Эти заболевания манифестируют в раннем детском возрасте. Но есть и такие орфанные болезни, которые могут возникнуть еще при внутриутробном развитии. Например, тирозинемия с неонатальной гепатокарциномой. Мы занимаемся диагностикой наследственной синдромальной патологии при ранней детской и неонатальной онкологической патологии. В Москве успешно работает селективный скрининг на наследственные болезни обмена, который выполняется за счет бюджета города.
В Москве успешно работает селективный скрининг на наследственные болезни обмена, который выполняется за счет бюджета города.
Если у нас есть малейшее подозрение на орфанное заболевание, мы направляем биологический материал пациентов в Центр орфанных заболеваний у детей и подростков, который действует на базе Морозовской больницы. Сотрудники нашей лаборатории выполняют генетические анализы по направлениям: онкология, эпилептология, наследственные и врожденные заболевания, участвуют в исследованиях и внедрении новых генетических тестов. Лаборатория имеет самое современное оборудование, позволяющее исследовать точечные мутации и полиморфизмы, секвенировать фрагменты генов, проводить высокопроизводительное секвенирование, анализировать экспрессию генов, экспрессию микроРНК, а также проводить анализ кариотипа. В составе лаборатории работают шесть квалифицированных специалистов в области молекулярной генетики, цитогенетики и биоинформатики.
Избыточное направление на диагностику орфанных заболеваний только приветствуется. Лучше перестраховаться, чем пропустить тревожные звоночки. Если в семье выявляется один пациент, мы проводим каскадное обследование всех доступных членов родословной. Это позволяет выявить пациентов на ранней стадии заболевания. Кроме того, семья может поставить перед нами вопрос решения своих репродуктивных планов. Есть несколько вариантов развития событий. Если уже наступила естественная беременность и мы знаем, какие наследственные заболевания есть в семье, то предлагается инвазивная пренатальная диагностика. Проводится ретроспективное консультирование. Если выясняется, что плод развивается с патологией, мы информируем семью, и она принимает решение – пролонгировать беременность или прервать по медицинским показаниям. Определить орфанное заболевание у плода можно на сроке гестации 9–12 недель. Если семья решает пролонгировать беременность, то мы знаем, что родится больной ребенок. Не дожидаясь проявления этой болезни, назначается фермент-заместительная терапия, ребенку прописывают диету и образ жизни.
Лучше перестраховаться, чем пропустить тревожные звоночки. Если в семье выявляется один пациент, мы проводим каскадное обследование всех доступных членов родословной. Это позволяет выявить пациентов на ранней стадии заболевания. Кроме того, семья может поставить перед нами вопрос решения своих репродуктивных планов. Есть несколько вариантов развития событий. Если уже наступила естественная беременность и мы знаем, какие наследственные заболевания есть в семье, то предлагается инвазивная пренатальная диагностика. Проводится ретроспективное консультирование. Если выясняется, что плод развивается с патологией, мы информируем семью, и она принимает решение – пролонгировать беременность или прервать по медицинским показаниям. Определить орфанное заболевание у плода можно на сроке гестации 9–12 недель. Если семья решает пролонгировать беременность, то мы знаем, что родится больной ребенок. Не дожидаясь проявления этой болезни, назначается фермент-заместительная терапия, ребенку прописывают диету и образ жизни. Так он может вырасти здоровым, полноценным членом общества. Есть методы репродуктивной технологии с доимплантационной диагностикой, когда проводится имплантация только здорового эмбриона. Такие методики практикуют специалисты НМИЦ акушерства, гинекологии и перинатологии имени академика В. И. Кулакова Минздрава РФ, с которым мы сотрудничаем.
Так он может вырасти здоровым, полноценным членом общества. Есть методы репродуктивной технологии с доимплантационной диагностикой, когда проводится имплантация только здорового эмбриона. Такие методики практикуют специалисты НМИЦ акушерства, гинекологии и перинатологии имени академика В. И. Кулакова Минздрава РФ, с которым мы сотрудничаем.
При молекулярно-генетическом методе исследований из клеток выделяется ДНК, которая расшифровывается в секвенаторе. Это оборудование предназначено для исследования отдельных генов. Мы реализуем потребности нашего центра – исследуем ген SCN1A, который в 90–95 % ассоциирован с тяжелой эпилепсией у новорожденных. Мы проводим секвенирование последующего поколения по современной методике, где анализируем до 25 тысяч генов, кодирующих белки и ферменты. Одно исследование занимает от двух недель до 90 рабочих дней, в зависимости от объема и сложности. Много времени отнимает интерпретация, потому что доброкачественные полиморфизмы сверяются по базам данных вручную. При цитогенетическом исследовании изучается число и структура хромосом. Основной контингент такого исследования – младенцы из отделения патологии новорожденных и недоношенных детей, а также реанимации. Хромосомная патология выявляется у 5 % детей с пороками развития: с синдромом Дауна, Патау, Эдвардса. Если мы находим хромосомную патологию, то сразу исследуем и родителей, чтобы понять, откуда пришла болезнь. Это может быть не наследственная, а вновь образованная мутация.
При цитогенетическом исследовании изучается число и структура хромосом. Основной контингент такого исследования – младенцы из отделения патологии новорожденных и недоношенных детей, а также реанимации. Хромосомная патология выявляется у 5 % детей с пороками развития: с синдромом Дауна, Патау, Эдвардса. Если мы находим хромосомную патологию, то сразу исследуем и родителей, чтобы понять, откуда пришла болезнь. Это может быть не наследственная, а вновь образованная мутация.
Мы проводим информированное консультирование пациентов, впервые поступивших в наш центр. Наша цель – установить правильный диагноз, от которого зависит дальнейшее лечение. Мы консультируем ежегодно около тысячи больных. Ежедневно отрабатываем около 10 заявок, приходя к пациентам в палаты. Мы осматриваем ребенка, чтобы по внешним признакам (особенности строения лица, волос, ногтей и так далее) понять, с каким заболеванием имеем дело. Оцениваются результаты биохимических и клинических анализов, которые проводятся в общей клинической лаборатории. Более 80 % проконсультированных больных необходимы те или иные генетические тесты. Конечно же, мы отвечаем на все вопросы, которые возникают у родителей пациента. Чаще всего они спрашивают, наследственное ли у ребенка заболевание или нет, как оно наследуется и почему такое заболевание возникло, если оба родителя здоровы.
Более 80 % проконсультированных больных необходимы те или иные генетические тесты. Конечно же, мы отвечаем на все вопросы, которые возникают у родителей пациента. Чаще всего они спрашивают, наследственное ли у ребенка заболевание или нет, как оно наследуется и почему такое заболевание возникло, если оба родителя здоровы.
Генетик должен непрерывно повышать свою квалификацию, поскольку появляются новые данные о мутациях, ассоциированных с уже давно известными заболеваниями. Когда я начинала работать, мы мало знали о наследственной природе эпилепсии. Считалось, что это не наследственное заболевание, а мультифакториальное – многофакторной природы. То же самое говорили о природе сердечных аритмий. Теперь эти заболевания лучше изучены. Мы не всегда можем определить наследственную природу заболевания. Но каждый год в профессиональной литературе появляется информация о найденных и расшифрованных новых мутациях в генах. Если поступает запрос от родителей или лечащего врача, мы проводим повторный биоинформатический анализ материала пациента. Иногда переанализ инициируем сами, если у нас есть академический интерес. О результатах мы информируем родителей пациента.
Иногда переанализ инициируем сами, если у нас есть академический интерес. О результатах мы информируем родителей пациента.
Мы работаем на стыке науки и практики, исследуя генетические подходы, которые в перспективе позволят эффективнее лечить. В частности, такая работа связана с онкологическими заболеваниями, при которых так или иначе в клетках нарушаются генетические механизмы. Это приводит к тому, что клетки перестают выполнять свои функции и начинают патологически размножаться. Исследуем генетические маркеры в плазме крови. Есть спектр разных частиц, в которых содержатся ДНК, РНК, микроРНК, и по их последовательностям можно спрогнозировать эффективность терапии. Параллельно исследуются генетические маркеры, которые связаны с развитием атерогенных нарушений. Зачастую это общий порочный круг, который поддерживает онкологическое заболевание в агрессивной форме и провоцирует рецидивы, если удалось заглушить очаг. У нас в распоряжении около 120 образцов от пациентов с онкологическими заболеваниями, и наша задача – выявить основные корреляции, то есть те самые маркеры, которые играют главенствующую роль в развитии заболевания. Мы уже обнаружили микроРНК, ассоциированную с развитием атерогенных нарушений. Второй этап работы будет заключаться в сравнении динамики лечения онкологических больных на разных стадиях с разными заболеваниями и поиске корреляции у них. Цель – выбор оптимальных терапевтических стратегий. Если мы сделаем такую систему и сможем отслеживать эффективность и неэффективность терапии, то поможем врачам рекомендациями по поиску новых подходов лечения. Такие разработки также могут быть нацелены на развитие ранних подходов в диагностике онкологических заболеваний.
Мы уже обнаружили микроРНК, ассоциированную с развитием атерогенных нарушений. Второй этап работы будет заключаться в сравнении динамики лечения онкологических больных на разных стадиях с разными заболеваниями и поиске корреляции у них. Цель – выбор оптимальных терапевтических стратегий. Если мы сделаем такую систему и сможем отслеживать эффективность и неэффективность терапии, то поможем врачам рекомендациями по поиску новых подходов лечения. Такие разработки также могут быть нацелены на развитие ранних подходов в диагностике онкологических заболеваний.
«Необходимо, чтобы больные дети нашей страны имели равный доступ к одинаково качественной современной медицинской помощи» uMEDp
Орфанные заболевания – это группа редких болезней, прежде всего наследственно обусловленных. Единых подходов к их диагностике и лечению пока не существует. Разработка современных рекомендаций по лечению пациентов с орфанными заболеваниями, внедрение инновационных диагностических методов, а также обеспечение больных лекарственными препаратами – основные направления российского здравоохранения. О проблемах, связанных с диагностикой и лечением орфанных заболеваний, особенностях организации медицинской помощи пациентам рассказывает Лейла Сеймуровна НАМАЗОВА-БАРАНОВА, д.м.н., профессор, член-корреспондент РАН, заслуженный деятель науки РФ, заместитель директора Научного центра здоровья детей – директор НИИ профилактической педиатрии и восстановительного лечения Центра, заведующая кафедрой аллергологии и клинической иммунологии педиатрического факультета Первого Московского государственного медицинского университета им. И.М. Сеченова, заведующая кафедрой факультетской педиатрии педиатрического факультета Российского национального исследовательского медицинского университета им. Н.И. Пирогова.
О проблемах, связанных с диагностикой и лечением орфанных заболеваний, особенностях организации медицинской помощи пациентам рассказывает Лейла Сеймуровна НАМАЗОВА-БАРАНОВА, д.м.н., профессор, член-корреспондент РАН, заслуженный деятель науки РФ, заместитель директора Научного центра здоровья детей – директор НИИ профилактической педиатрии и восстановительного лечения Центра, заведующая кафедрой аллергологии и клинической иммунологии педиатрического факультета Первого Московского государственного медицинского университета им. И.М. Сеченова, заведующая кафедрой факультетской педиатрии педиатрического факультета Российского национального исследовательского медицинского университета им. Н.И. Пирогова.
– Лейла Сеймуровна, насколько актуальна, на Ваш взгляд, проблема орфанных заболеваний для отечественной педиатрической службы?
– Замечу, что проблема орфанных заболеваний актуальна для педиатрической службы не только России, но и большинства развитых стран, поскольку количество редких болезней, которые становятся известны человечеству, стремительно растет. Несмотря на низкую распространенность отдельных нозологических форм, общее число больных орфанными заболеваниями достаточно велико. Еще недавно раздел клинической медицины, посвященный изучению орфанных заболеваний, не входил в образовательные программы медицинских вузов. Но с развитием медицинской науки появились новые методы диагностики и терапии редких болезней. Сегодня педиатрическая общественность должна хорошо ориентироваться в этой новой, но очень сложной проблеме, ведь орфанные заболевания могут приводить к инвалидизации и смертности как в раннем детстве, так и в более позднем возрасте. Именно поэтому особое значение придается внедрению современных методов диагностики и разработке новых эффективных методов терапии редких болезней.
Несмотря на низкую распространенность отдельных нозологических форм, общее число больных орфанными заболеваниями достаточно велико. Еще недавно раздел клинической медицины, посвященный изучению орфанных заболеваний, не входил в образовательные программы медицинских вузов. Но с развитием медицинской науки появились новые методы диагностики и терапии редких болезней. Сегодня педиатрическая общественность должна хорошо ориентироваться в этой новой, но очень сложной проблеме, ведь орфанные заболевания могут приводить к инвалидизации и смертности как в раннем детстве, так и в более позднем возрасте. Именно поэтому особое значение придается внедрению современных методов диагностики и разработке новых эффективных методов терапии редких болезней.
– Какова эпидемиологическая ситуация в нашей стране в отношении орфанных заболеваний?
– В соответствии с Федеральным законом «Об основах охраны здоровья граждан в Российской Федерации» орфанными считаются заболевания, частота встречаемости которых реже чем один случай на 10 000 новорожденных. В разных странах нормы оценки распространенности редких болезней различны. Однако реальная эпидемиологическая картина в отношении орфанных заболеваний и в нашей стране, и в мире описана фрагментарно. Это связано прежде всего с тем, что в большинстве случаев редкие заболевания не удается рано диагностировать. Орфанное заболевание может оставаться невыявленным долгое время, а ребенка при этом лечат от других болезней. Поскольку большинство орфанных заболеваний имеет генетическую природу, основным диагностическим методом признан неонатальный скрининг. Однако проводить генетическое тестирование всем новорожденным слишком дорого даже для обеспеченных систем здравоохранения. Хотя, безусловно, расширение программы неонатального скрининга позволило бы улучшить ситуацию по редким заболеваниям в стране. Отмечу, что не для всех редких болезней сегодня имеются адекватные диагностические системы. На текущий момент мы можем оперировать данными только в отношении тех пациентов, которых наблюдаем.
В разных странах нормы оценки распространенности редких болезней различны. Однако реальная эпидемиологическая картина в отношении орфанных заболеваний и в нашей стране, и в мире описана фрагментарно. Это связано прежде всего с тем, что в большинстве случаев редкие заболевания не удается рано диагностировать. Орфанное заболевание может оставаться невыявленным долгое время, а ребенка при этом лечат от других болезней. Поскольку большинство орфанных заболеваний имеет генетическую природу, основным диагностическим методом признан неонатальный скрининг. Однако проводить генетическое тестирование всем новорожденным слишком дорого даже для обеспеченных систем здравоохранения. Хотя, безусловно, расширение программы неонатального скрининга позволило бы улучшить ситуацию по редким заболеваниям в стране. Отмечу, что не для всех редких болезней сегодня имеются адекватные диагностические системы. На текущий момент мы можем оперировать данными только в отношении тех пациентов, которых наблюдаем.
– Какова природа орфанных заболеваний и какие дети входят в группу риска?
– Редкие болезни условно подразделяют на две группы: наследственные и ненаследственные. Наиболее распространены наследственные орфанные заболевания, которые могут не только передаваться от родителей, но и возникать вследствие новой мутации генов. Симптомы таких заболеваний не всегда проявляются сразу после рождения ребенка. Они могут развиться через несколько месяцев или даже лет, особенно в случае болезней накопления (мукополисахаридоз, болезнь Гоше, болезнь Фабри). Родители детей и педиатры не всегда способны понять, что у ребенка редкое заболевание, поскольку проявляются неспецифические признаки болезни, например частые респираторные инфекции с первых недель и месяцев жизни, затянувшаяся желтуха, повторные грыжи. Родители, заметив у ребенка необычные симптомы, должны немедленно обратиться к педиатру. Такие симптомы могут быть стартовыми для многих редких болезней. Специалисты первичного звена, педиатры должны быть информированы о редких заболеваниях, их диагностических признаках и возможных последствиях, чтобы вовремя выявить патологические нарушения и начать своевременное лечение.
Наиболее распространены наследственные орфанные заболевания, которые могут не только передаваться от родителей, но и возникать вследствие новой мутации генов. Симптомы таких заболеваний не всегда проявляются сразу после рождения ребенка. Они могут развиться через несколько месяцев или даже лет, особенно в случае болезней накопления (мукополисахаридоз, болезнь Гоше, болезнь Фабри). Родители детей и педиатры не всегда способны понять, что у ребенка редкое заболевание, поскольку проявляются неспецифические признаки болезни, например частые респираторные инфекции с первых недель и месяцев жизни, затянувшаяся желтуха, повторные грыжи. Родители, заметив у ребенка необычные симптомы, должны немедленно обратиться к педиатру. Такие симптомы могут быть стартовыми для многих редких болезней. Специалисты первичного звена, педиатры должны быть информированы о редких заболеваниях, их диагностических признаках и возможных последствиях, чтобы вовремя выявить патологические нарушения и начать своевременное лечение.
– Каким должен быть современный алгоритм обследования детей с подозрением на орфанное заболевание?
– Каждый ребенок с подозрением на редкое заболевание нуждается в комплексном обследовании с привлечением команды специалистов. Сначала выполняют основные биохимические анализы, результаты которых могут подтвердить какие-либо изменения, например дефицит определенного фермента. Далее проводят ряд генетических исследований. Замечу, что оптимальный алгоритм обследования при подозрении на орфанное заболевание учитывает особенности патогенеза болезни, возраст и состояние ребенка. При наличии признаков какого-либо заболевания появляются основания для углубленного обследования. В силу разнообразия видов орфанных заболеваний алгоритм обследования в каждом конкретном случае, безусловно, будет индивидуальным.
– Какую тактику ведения детей с орфанными заболеваниями Вы считаете оптимальной?
– Своевременное адекватное лечение во многих случаях позволяет избежать осложнений, способствует улучшению качества и увеличению продолжительности жизни пациентов. Терапевтическая тактика лечения детей с орфанными заболеваниями зависит прежде всего от генотипа болезни. Для каждого ребенка подбирается соответствующая медикаментозная терапия. Патогенетическое лечение, корректирующее генетические нарушения при дефиците какого-либо фермента (ферментозаместительная терапия), необходимо начинать как можно раньше – до развития необратимых изменений со стороны разных органов и систем. При поздней постановке диагноза и несвоевременно начатом лечении целью терапии является коррекция патологических изменений в организме ребенка, восстановление нормального функционирования его органов с помощью определенных групп препаратов. Очень помогают нам и немедикаментозные возможности восстановления здоровья: комплексы ЛФК, кинезитерапии, физиолечение. С каждым ребенком и его семьей, конечно же, должна работать команда психологов-педагогов.
Терапевтическая тактика лечения детей с орфанными заболеваниями зависит прежде всего от генотипа болезни. Для каждого ребенка подбирается соответствующая медикаментозная терапия. Патогенетическое лечение, корректирующее генетические нарушения при дефиците какого-либо фермента (ферментозаместительная терапия), необходимо начинать как можно раньше – до развития необратимых изменений со стороны разных органов и систем. При поздней постановке диагноза и несвоевременно начатом лечении целью терапии является коррекция патологических изменений в организме ребенка, восстановление нормального функционирования его органов с помощью определенных групп препаратов. Очень помогают нам и немедикаментозные возможности восстановления здоровья: комплексы ЛФК, кинезитерапии, физиолечение. С каждым ребенком и его семьей, конечно же, должна работать команда психологов-педагогов.
– Какова в нашей стране ситуация с появлением новых препаратов для лечения редких болезней? Проводятся ли в Научном центре здоровья детей клинические исследования новых препаратов для лечения орфанных заболеваний?
– В литературе описаны тысячи видов редких болезней, причем большинство из них обусловлены генетическими поломками. Как показывает опыт, патогенетическая терапия позволяет остановить прогрессирование заболевания, способствует обратному развитию симптомов болезни и значительно улучшает качество жизни больных. Но, к сожалению, для лечения орфанных заболеваний у детей препаратов не так много. Это связано с определенными трудностями законодательного порядка. В российском законодательстве в отличие от законодательства США, стран Евросоюза, Японии нет положений, стимулирующих производство и проведение клинических исследований лекарственных препаратов именно для группы больных детского возраста. Во всем мире перед широким внедрением новых препаратов в клиническую практику обязательно проводятся строго контролируемые клинические исследования, в ходе которых подтверждается не только эффективность лекарственных средств, но и их безопасность. Но из-за несовершенства законодательства Россия не очень активно участвует в международных многоцентровых исследованиях, посвященных проблеме редких заболеваний у детей. Тем не менее на базе нашего научного центра проводятся исследования новых методов терапии, разрабатываются новые методы борьбы с орфанными болезнями.
Как показывает опыт, патогенетическая терапия позволяет остановить прогрессирование заболевания, способствует обратному развитию симптомов болезни и значительно улучшает качество жизни больных. Но, к сожалению, для лечения орфанных заболеваний у детей препаратов не так много. Это связано с определенными трудностями законодательного порядка. В российском законодательстве в отличие от законодательства США, стран Евросоюза, Японии нет положений, стимулирующих производство и проведение клинических исследований лекарственных препаратов именно для группы больных детского возраста. Во всем мире перед широким внедрением новых препаратов в клиническую практику обязательно проводятся строго контролируемые клинические исследования, в ходе которых подтверждается не только эффективность лекарственных средств, но и их безопасность. Но из-за несовершенства законодательства Россия не очень активно участвует в международных многоцентровых исследованиях, посвященных проблеме редких заболеваний у детей. Тем не менее на базе нашего научного центра проводятся исследования новых методов терапии, разрабатываются новые методы борьбы с орфанными болезнями. В Научном центре здоровья детей наблюдаются и проходят лечение большое число детей с орфанными заболеваниями.
В Научном центре здоровья детей наблюдаются и проходят лечение большое число детей с орфанными заболеваниями.
– Как складывается ситуация с лекарственным обеспечением детей с орфанными заболеваниями?
– Одна из основных проблем – высокая стоимость орфанных препаратов. Поскольку их приобретение ложится тяжелым финансовым бременем на членов семей, в которых имеются дети с орфанными заболеваниями, вопрос об обеспечении пациентов лекарственными препаратами решается на государственном уровне. Согласно действующему законодательству обеспечение лекарствами пациентов с редкими заболеваниями осуществляется за счет средств государственного бюджета. Сегодня часть пациентов с редкими болезнями получает препараты за счет федерального бюджета в рамках программы «7 нозологий». Кроме того, обеспечение лекарственными средствами больных осуществляется с помощью региональных бюджетов в рамках программы по редким болезням. Между тем ситуация с лекарственным обеспечением пациентов с орфанными заболеваниями полностью не решена. В ряде случаев пациентам отказывают в обеспечении необходимыми препаратами из-за недостатка средств регионального бюджета: иногда стоимость лечения одного орфанного больного на год превышает весь бюджет здравоохранения данного региона на всех детей на целый год. Проблему необходимо решать всесторонне, привлекая различные источники финансирования. Нужно использовать систему благотворительности. Значение благотворительных организаций в финансировании лечения тяжелых больных в мире, в том числе в России, трудно переоценить.
– Лейла Сеймуровна, в заключение нашей беседы позвольте задать последний вопрос. Каковы перспективы отечественной педиатрии в борьбе с орфанными заболеваниями?
– Мне хочется быть оптимистом. Я верю, что хороших и добрых людей больше, чем плохих. Если по каким-либо причинам у федерального или регионального бюджета сегодня нет средств на оплату лекарственных препаратов для детей с орфанными заболеваниями, может, на помощь чаще будут приходить общественность, благотворительные организации? Медицинская наука развивается, появляются новые инновационные эффективные лекарства, позволяющие детям с редкими заболеваниями вести полноценный образ жизни. Разрабатываются и внедряются новые терапевтические подходы к лечению орфанных заболеваний. В нашем научном центре разработана уникальная программа ведения маленьких пациентов с редкими болезнями, предполагающая мультидисциплинарный подход. Для ребенка подбираются соответствующая медикаментозная терапия, комплекс физической активности, физиотерапевтических процедур, с ним и его родителями работают педагоги и психологи. Мы уверены, что мультидисциплинарная тактика ведения пациентов с орфанными заболеваниями, разработанная в Научном центре здоровья детей, должна внедряться в аналогичных центрах в регионах. Необходимо, чтобы больные дети нашей страны имели равный доступ к одинаково качественной современной медицинской помощи, основанной на лучших подходах к ведению пациентов с редкими болезнями!
– Спасибо за содержательное интервью.
Страница не найдена |
Страница не найдена |
404. Страница не найдена
Архив за месяц
ПнВтСрЧтПтСбВс
12
12
1
3031
12
15161718192021
25262728293031
123
45678910
12
17181920212223
31
2728293031
1
1234
567891011
12
891011121314
11121314151617
28293031
1234
12
12345
6789101112
567891011
12131415161718
19202122232425
3456789
17181920212223
24252627282930
12345
13141516171819
20212223242526
2728293031
15161718192021
22232425262728
2930
Архивы
Июн
Июл
Авг
Сен
Окт
Ноя
Дек
Метки
Настройки
для слабовидящих
редких болезней и расстройств, которые мы лечим
Настройки
SpecialtyAdolescent MedicineAllergy и ImmunologyAnatomic и клинической PathologyAnatomic PathologyAnesthesiologyBiochemical GeneticsBlood Банки / Переливание MedicineCardiac AnesthesiologyCardiac SurgeryCardiologyChild Злоупотребление PediatricsChild и подростковая PsychiatryClinical PsychologyCongenital Сердечный SurgeryCritical CareDentistryDermatologyDevelopmental-поведенческая PediatricsDiagnostic RadiologyEmergency MedicineEpilepsyFamily PlanningFamily PracticeGastroenterologyGeneral SurgeryGynecologyHead и шеи SurgeryHematologyHepatologyInternal MedicineMedical GeneticsMinimally Инвазивные SurgeryNeonatal-Перинатальные MedicineNephrology (Почки) NeurologyNeuro-OncologyNeuroradiologyNeurosurgeryNutritionObstetrics и GynecologyOphthalmologyOrthodonticsOrthopaedic ХирургияОртопедическая травмаОтоларингология (ЛОР) ПатологияPCP (Врач первичной медицинской помощи) Педиатрия Педиатрия (Врач первичной медицинской помощи) Физическая медицина и реабилитацияПластическая хирургияПсихиатрияПсихологияПульмонологияРадиологияРевматологияСон НарушенияХирургияТоракальная хирургияХирургия трансплантатаУрологияСосудистая и интервенционная радиология
Язык
LanguageAfrikaanArabicBengaliBosnianBulgarianBurmeseCantoneseCatalanChineseChinese (мандарин) CreoleCroatianCzechDutchFarsiFilipinoFrenchGaelicGermanGreekGujaratiHebrewHindiHungarianIboIndianItalianJapaneseKannadaKashmiri KikuyuKoreanLatinLebaneseLithuanianMalaysianMarathiPersianPolishPortuguesePunjabiRomanianRussianSanskritSerbianSign LanguageSindhiSinhaleseSpanishSri-LankanSwahiliSwedishTagalogTaiwaneseTamilTeluguThaiTurkishUkrainianUrduVietnameseYiddishYoruba
Роль
Роль: Врач, Ассистент врача, практикующая медсестра,
.
Синдром СВЕЧИ | Центр терапии редких заболеваний
Что такое синдром СВЕЧИ?
Синдром СВЕЧИ — редкое аутовоспалительное заболевание.СВЕЧА означает хронический атипичный нейтрофильный дерматоз с липодистрофией и повышенной температурой. Наиболее частыми симптомами являются постоянная лихорадка, потеря жира и поражения кожи.
Синдром СВЕЧИ обычно развивается на первом году жизни ребенка. У больных детей может быть увеличена печень и повышен уровень ферментов печени. Хроническая анемия, низкий рост и вес также являются частью синдрома СВЕЧИ.
Синдром СВЕЧИ встречается очень редко. По оценкам экспертов Autoinflamasted Alliance, в мире всего 60 известных случаев.
Синдром СВЕЧИ причины
Изменения гена PSMB8 обычно являются причиной синдрома СВЕЧИ. Согласно Американскому журналу медицинской генетики, эти изменения приводят к накоплению продуктов жизнедеятельности белка и вызывают сбои в работе других клеток.
Трудно диагностировать синдром СВЕЧИ без генетического тестирования. А поскольку это аутоиммунное заболевание, тесты иногда не дают результатов.
По данным Информационного центра NIH по генетическим и редким заболеваниям (GARD), родственные заболевания включают:
- Различные синдромы протеасомной инвалидности.
- Синдром CINCA.
- Приобретенная генерализованная липодистрофия, вызванная панникулитом.
- Сладкий синдром.
- Другие кожные заболевания (похожие на острый миелолейкоз).
Профилактика синдрома СВЕЧИ и факторы риска
Поскольку он обычно генетический, вы не можете предотвратить синдром СВЕЧИ.
В настоящее время необходимы дополнительные исследования факторов риска, чтобы лучше понять это. Но диагностика и раннее вмешательство могут значительно улучшить качество жизни ребенка.И то, и другое также может увеличить продолжительность жизни, поскольку высок риск воспаления органов.
Симптомы и диагностика синдрома СВЕЧИ
Почти все случаи синдрома СВЕЧИ начинаются в раннем младенчестве. Но признаки и симптомы могут сильно отличаться от ребенка к ребенку. Тем не менее, многие симптомы синдрома СВЕЧИ могут вызывать боль и дискомфорт.
Вот некоторые из наиболее распространенных признаков и симптомов:
- Боль в суставах.
- Задержки развития.
- Рецидивирующие лихорадки.
- Пурпура.
- Опухшие веки.
- Прогрессирующая липодистрофия (похудание).
- Контракты.
- Изъятия.
Если вы заметили что-либо из следующего, немедленно позвоните своему врачу:
- Рецидивирующие или даже ежедневные лихорадки.
- Поражения кожи, особенно вокруг глаз.
- Мышечное истощение.
- Аномальная потеря жира.
Диагностика синдрома СВЕЧИ
Лечение может принести пользу вашему ребенку, даже если причина синдрома СВЕЧИ не ясна.Вот почему важны диагностика и раннее вмешательство.
Согласно статье 2017 Frontiers in Immunology , синдром СВЕЧИ обычно подозревается, когда у ребенка проявляются следующие симптомы:
- Характерные поражения кожи.
- Липодистрофия (похудание).
- Ранние лихорадки.
В настоящее время генетическое тестирование — единственный способ подтвердить диагноз. Продвинутая биопсия кожи (включающая иммуногистохимические исследования) также может быть полезна при установлении диагноза в соответствии со статьей Frontiers .
Прогноз и выживаемость
Поскольку это явление такое новое и редкое, прогноз и выживаемость синдрома СВЕЧИ неясны. Воспаление нескольких органов — наиболее серьезный и опасный для жизни симптом. Инвестиции в лечение и исследования могут дать надежду на будущее.
Лечение синдрома СВЕЧИ
Стандартного лечения синдрома СВЕЧИ пока нет. Необходимы дополнительные исследования, чтобы найти конкретное лечение для всех случаев.
Однако риск оставить синдром СВЕЧИ без лечения высок.
Врачи подбирают индивидуальный уход для каждого пациента и стремятся справиться с его болью и симптомами. Это улучшает качество жизни и предотвращает такие осложнения, как воспаление.
Физическая терапия для предотвращения контрактур суставов и терапия конкретных органов жизненно важны, как указано в статье Frontiers in Immunology . Также обязательно? Регулярные клинические наблюдения и лабораторные анализы для тщательного наблюдения за воспалительными атаками, а также регулярные осмотры суставов, глаз и кожи.
Почему выбирают UPMC Children’s?
Детям с редкими заболеваниями, такими как синдром СВЕЧИ, требуется специализированная помощь.Знающие и преданные своему делу поставщики должны быстро диагностировать каждого пациента и предоставить им передовые методы лечения. Узнайте больше о Центре терапии редких заболеваний при детской больнице UPMC в Питтсбурге.
Чтобы помочь детям с редкими аутоиммунными заболеваниями, UPMC Children’s разработала специальную программу. Она называется IDDAT: Программа диагностики и лечения иммунной дисрегуляции.
Вот некоторые из специальностей в рамках IDDAT:
Эти специалисты работают вместе, чтобы помочь детям с синдромом СВЕЧИ и связанными с ним заболеваниями.
Семьи, которые работают с командой в IDDAT, пользуются:
- Специализированная помощь пациентам.
- Доступ к инновационным исследовательским возможностям.
- Диагностическая экспертиза и поддержка.
- Доступ к полезному, спасающему жизнь лечению.
Свяжитесь с нами
Обеспокоены тем, что у вашего ребенка синдром СВЕЧИ? Обратитесь в Центр терапии редких заболеваний при UPMC Children’s.
Комбинированный иммунодефицит (CID) | Детская больница
Комбинированный иммунодефицит — также называемый комбинированным иммунодефицитом или CID — это генетическое состояние иммунной системы.Он известен как «первичный иммунодефицит».
Дети наследуют ген CID от своих родителей.
CID возникает, когда генные мутации вызывают дефекты иммунной системы. Это тот же процесс заболевания, что и тяжелый комбинированный иммунодефицит (ТКИД), но ИСД является менее серьезной и агрессивной формой.
Основная роль иммунной системы — бороться с инфекциями и всем остальным, что она считает чужеродными захватчиками для организма.
При CID тело имеет слабый иммунный ответ .Это означает, что дети с этим заболеванием подвергаются высокому риску заражения инфекциями, которые становятся тяжелыми.
Бактерии, вирусы, грибки и другие микробы, не вредные для детей с нормальным иммунитетом, могут быть фатальными для детей с CID.
Врачи видят это заболевание у очень маленьких детей. Когда дети рождаются, их системы обладают защитным иммунитетом от антител, которые они получили в утробе матери.
Когда эти материнские антитела покидают организм в течение первых нескольких месяцев, появляются симптомы CID.
CID, в отличие от SCID, ухудшается медленнее. А опасные для жизни осложнения могут не появиться несколько лет.
Симптомы комбинированного иммунодефицита
Симптомы CID во многом такие же, как и симптомы других детских болезней. Но они могут быть более продолжительными и более серьезными.
Симптомы CID:
- Повторяющиеся респираторные инфекции, включая инфекции носовых пазух и легких.
- Инфекции уха.
- Разрастание дрожжей во рту (молочница) или сильная опрелость.
- Кожные инфекции, плохо поддающиеся лечению.
- Диарея.
- Медленный рост или медленное увеличение веса.
Осложнения CID
Детей с первичным иммунодефицитом, таким как CID:
- Более подвержен инфекциям, чем другие.
- При лечении инфекции излечиваются медленнее.
Инфекции, которые не сильно повлияют на других детей, могут стать опасными для жизни у детей с CID.
Диагностика комбинированного иммунодефицита
В большинстве штатов сейчас проводится скрининг новорожденных на CID, потому что, хотя это случается редко, очень важно вылечить его на ранней стадии.
Врачи диагностируют CID с помощью анализа крови, который измеряет количество лимфоцитов (тип лейкоцитов) в крови.
Дополнительные анализы крови могут показать, какой тип CID у вашего ребенка и какие иммунные клетки не работают должным образом.
Комбинированное лечение иммунодефицита
Цель лечения CID — предотвратить заражение вашего ребенка инфекциями, которые могут стать опасными для жизни.
Заместительная ферментная терапия может лечить определенные типы CID. Это лечение не излечивает болезнь.
Наиболее распространенным методом лечения CID, который излечивает болезнь , является трансплантация костного мозга или трансплантация стволовых клеток.
Пересадка легкого также может быть целесообразной, если у вашего ребенка также серьезные повреждения легких в результате частой пневмонии или других легочных инфекций.
Центр детской трансплантации Hillman имеет единственную в мире программу, которая предлагает тандемные трансплантации легких и костного мозга.
Мы также первыми внедрили программу трансплантации костного мозга пониженной интенсивности. Программа помогает уменьшить неблагоприятное воздействие трансплантации костного мозга или стволовых клеток на органы тела.
Поскольку трансплантация — это лечение высокого риска, вашему ребенку может потребоваться принимать лекарства всю оставшуюся жизнь.
Пауль Сабольч, MD
Заведующий отделением трансплантации костного мозга и клеточной терапии
Доктор Сабольч является пионером в трансплантации несвязанной донорской пуповинной крови с пониженной токсичностью и интенсивностью при врожденных нарушениях иммунной системы.Он первым доказал возможность последовательной трансплантации легких и костного мозга от одного и того же неродственного умершего донора.
Консультации и уход за CID вашего ребенка: чего ожидать
Если врач диагностировал у вашего ребенка комбинированный иммунодефицит, мы хотим, чтобы вы знали, что вы не одиноки. Центр терапии редких заболеваний всегда готов помочь.
На номер запишитесь на прием для вашего ребенка или направьте пациента на лечение CID, свяжитесь с нами по:
Вот чего вы можете ожидать, обратившись к нам за первой консультацией CID вашего ребенка.
Как скоро я могу записаться на прием по поводу комбинированного иммунодефицита моего ребенка?
В большинстве случаев мы можем увидеть нового пациента в течение 1-2 недель .
Мы попросим лечащего врача прислать записи вашего ребенка, чтобы мы могли просмотреть их перед вашим визитом.
Чего мне следует ожидать при первом посещении моего ребенка для CID?
Ваш первый визит займет от 4 до 6 часов .
Ваш ребенок получит полную оценку от Пола Сабольча, доктора медицины, руководителя отделения трансплантации костного мозга и клеточной терапии.
Доктор Сабольч поставит или подтвердит точный диагноз и узнает, насколько прогрессировала болезнь.
Поскольку мы работаем как одна команда здесь, в центре, другие врачи и персонал могут увидеть вашего ребенка во время вашего визита.
Сюда могут входить специалисты:
- Генетические болезни
- Болезни головного мозга
- Развитие ребенка
- Слух
- Физиотерапия
Каковы следующие шаги после посещения моим ребенком центра обслуживания CID?
Перед тем, как покинуть центр, у вас будет план обслуживания CID, адаптированный к потребностям вашего ребенка .
Члены группы по уходу CID вашего ребенка расскажут вам о:
- Вероятные следующие шаги для вашего ребенка.
- Варианты лечения комбинированного иммунодефицита вашего ребенка.
- Способы ухода за ребенком дома, чтобы улучшить его или ее качество жизни.
Если операция может быть вариантом лечения CID вашего ребенка, мы рассмотрим все детали. Мы сообщим вам, как вы и ваш ребенок можете подготовиться.
Мы также назначим повторный визит через 3 месяца .
Вы познакомитесь с нашей практикующей медсестрой (NP). Вы можете связаться с нашим НП по телефону или по видеоконференции, если у вас возникнут какие-либо проблемы в период с настоящего момента до следующего приема.
Перед отъездом, пожалуйста, не стесняйтесь спросить нас о диагнозе CID вашего ребенка, лечении или о чем-нибудь еще, что вы думаете.
Как скоро я получу результаты теста CID моего ребенка?
Мы позвоним вам в течение 2 недель , чтобы обсудить результаты теста и дальнейшие действия по уходу за вашим ребенком CID.
Вы также можете найти результаты анализов своего ребенка, если вы подписались на my CHP — Детский портал для пациентов.
my CHP позволяет вам управлять здоровьем вашего ребенка в режиме онлайн. Это бесплатно для пациентов детской больницы UPMC в Питтсбурге и их родителей или опекунов
Партнеры по CID для вашего ребенка
Если у ребенка редкое заболевание, например, комбинированный иммунодефицит, оно поражает всю семью.
В Центре терапии редких заболеваний мы рассматриваем каждого члена семьи как своего партнера.
Наилучший уход достигается, когда мы объединяем наш опыт CID с вашими знаниями о том, что лучше всего для вашего ребенка.
Свяжитесь с нами, чтобы узнать больше о CID или записаться на прием для вашего ребенка:
Познакомьтесь с нашими пациентами — Центр терапии редких заболеваний
Узнайте, как семьи находят помощь и надежду в Центре терапии редких заболеваний при детской больнице Питтсбурга UPMC. Прочтите рассказы пациентов Центра терапии редких заболеваний. »
Лечение фенилкетонурии (ФКУ) | Детский Питтсбург
Фенилкетонурия — врожденная ошибка метаболизма белков.Это редкое заболевание, и дети, рожденные с этим заболеванием, наследуют его от своих родителей. Это состояние не позволяет организму правильно расщеплять белки, в частности фениланин, который содержится в белке. Когда организм не может расщеплять определенные белки, фениланин накапливается в крови и вызывает проблемы по всему телу.
Некоторые способы воздействия слишком большого количества фениланина на организм:
- Поведенческие или психические расстройства
- Задержка развития
- Изъятия
Если у беременной матери ФКУ, которая не контролируется диетой, у ее ребенка может быть более тяжелая форма этого заболевания.Это может вызвать врожденные дефекты у ребенка, такие как маленькая голова (микроцефалия) и проблемы с сердцем.
У большинства детей, рожденных с этим заболеванием, последствия могут быть незаметны, пока ребенку не исполнится несколько месяцев.
Лечение PKU
В детской больнице Питтсбурга UPMC мы лечим ФКУ следующими способами:
- Заместительная ферментная терапия
- Низкобелковая диета
- Формула
- Лекарства
Виды ПКУ
Существует четыре типа ФКУ:
- Гиперфенилаланинемия : самый низкий уровень выше нормы
- Легкая форма PKU : уровень в крови слегка повышен
- Умеренный или вариант : уровни не низкие, но не высокие
- Классическая ФКУ : высокий уровень фениланина в крови
Симптомы и диагностика фенилкетонурии
Симптомы фенилкетонурии
Дети обычно проходят тестирование на ФКУ при рождении.Если ребенок не рождается с врожденными дефектами, симптомы ФКУ могут не проявляться в течение нескольких месяцев. Эти симптомы у маленьких детей могут включать:
- Экзема, кожная сыпь
- Изъятия
- Медленный рост
- Затхлый запах тела или дыхание
Неконтролируемая ФКУ может привести к другим проблемам по мере роста ребенка, например:
- Задержка развития
- Раздражительность
- Поведенческие проблемы
- Умственная отсталость
- Проблемы с памятью
Диагностика ПКУ
Ваш ребенок будет проверен через день или два после рождения.Этот тест проводится с помощью пяточной палочки, и для этого и других стандартных тестов берется небольшое количество крови. Если тест покажет, что у вашего ребенка может быть это заболевание, будет проведено дополнительное обследование. Они могут включать другой анализ крови или анализ мочи.
Лечение PKU
Если ваш ребенок страдает этим заболеванием, ему необходимо как можно скорее начать лечение. Цель лечения — поддерживать низкий уровень фениланина в крови.
Подход к лечению будет зависеть от того, насколько тяжелое состояние вашего ребенка или насколько высок уровень его содержания в крови.
Первая линия лечения может включать диету с низким содержанием белка. Младенцам может потребоваться специальная смесь для контроля количества потребляемого белка. По мере того, как ваш ребенок становится старше, ему может потребоваться принимать пищевые добавки, чтобы гарантировать, что он получает достаточно белка.
Лекарства и заместительная ферментная терапия также могут помочь организму расщепить фениланин или помочь организму лучше его переносить.
Хотя это лечение еще не доступно, в будущем для лечения этого заболевания могут быть использованы трансплантаты печени.Пока не будет найдено лекарство, фенилкетонурия потребует пожизненного лечения и наблюдения для предотвращения симптомов и осложнений.
Свяжитесь с нами
Чтобы сделать направление или назначить встречу для вашего ребенка в Центре терапии редких заболеваний, позвоните по телефону 412-692-7273 или отправьте электронное письмо по адресу [email protected].
Джерри Фокли, MD, PhD
Начальник отдела медицинской генетики
Доктор Фокли — международный лидер в области лечения и исследований в области медицинской генетики и в области врожденных нарушений обмена веществ.Он также является основателем Североамериканской метаболической академии.
Чего ожидать
Если вашему ребенку был поставлен диагноз ФКУ, мы хотим, чтобы вы знали, что вы не одиноки — Центр терапии редких заболеваний всегда готов помочь. Вот чего вы можете ожидать, обратившись к нам за консультацией.
Как быстро мы можем записаться на прием?
В Центре терапии редких заболеваний врач, специализирующийся на генетических заболеваниях, обычно может принять нового пациента в течение 1-2 недель.Чтобы записаться на прием, позвоните Джоди Венто по телефону 412-692-7273 или отправьте электронное письмо по адресу [email protected].
Как долго мы должны ожидать нашего первого визита к врачу?
Вы можете рассчитывать, что ваше первое посещение займет от 4 до 6 часов. Ваш ребенок получит полное обследование и его могут осмотреть несколько врачей и других медицинских работников.
Что будет во время нашего первого визита?
Ваш ребенок получит полное обследование, чтобы установить или подтвердить точный диагноз и определить, насколько болезнь затронула ребенка.Поскольку мы работаем в одной команде в Центре терапии редких заболеваний, во время вашего визита вашего ребенка могут осмотреть несколько врачей и других медицинских работников, включая невролога, кардиолога, хирурга, диетолога, консультанта по генетическим вопросам и т. психолог, специалист по детскому развитию.
Мы понимаем, что вся семья страдает от редкого заболевания ребенка. Мы рассматриваем каждую семью как своего партнера в заботе о своем ребенке. Мы думаем, что лучший подход к уходу за ребенком с редким заболеванием возникает, когда мы объединяем наш глубокий опыт в лечении редких заболеваний с вашим опытом в том, что лучше всего для вашего ребенка.
Мы поговорим с вами о том, что, по нашему мнению, может произойти с вашим ребенком в ближайшем будущем, и о вариантах, которые мы можем предложить для лечения и ухода за вашим ребенком. Если для вашего ребенка возможна операция, мы поможем вам понять, что влечет за собой операция, и что вам может потребоваться для подготовки к ней. Мы также поговорим с вами о том, что вы можете делать дома, чтобы заботиться о своем ребенке и улучшать его качество жизни. Не стесняйтесь задавать нашим специалистам любые вопросы о болезни вашего ребенка, лечении и уходе за ним или обо всем, что у вас на уме.
К концу вашего визита у вас будет план ухода, адаптированный к потребностям вашего ребенка, и запись на контрольный визит через 3 месяца. Вас познакомят с нашей практикующей клинической медсестрой, с которой можно связаться по телефону или по видеоконференции, чтобы помочь вам с любыми проблемами, которые у вас возникнут в период с настоящего момента до следующего приема.
Как долго нам придется ждать результатов тестов?
В зависимости от типов тестов, которые назначают врачи, вы можете ожидать, что в течение 2 недель вам позвонят, чтобы объяснить результаты теста и обсудить рекомендуемые дальнейшие действия.Результаты тестов также доступны на Детском портале для пациентов myCHP, который бесплатно предоставляется пациентам, родителям и опекунам.
Познакомьтесь с нашими пациентами
Узнайте, как семьи находят помощь и надежду, благодаря опыту Центра терапии редких заболеваний при детской больнице Питтсбурга UPMC.
Посмотреть истории пациентов >>
Свяжитесь с нами
В Центре терапии редких заболеваний каждому ребенку, у которого диагностировано редкое заболевание, предоставляется индивидуальный план лечения и уход, ориентированный на семью.
Чтобы записаться на прием, консультацию или направление пациента к специалисту детской больницы Питтсбурга UPMC для ребенка с диагнозом ФКУ, обращайтесь:
Джоди Венто, MGC, LCGC
Телефон: 412-692-7273
Эл. Почта: [email protected]
Мантонский центр исследований орфанных болезней
О центре Manton
Основанный в 2008 году, Manton Center был одним из первых центров в мире, посвященных исключительно изучению редких заболеваний.Центр Manton надеется расширить научные знания о редких заболеваниях за счет поддержки исследований, ориентированных на пациентов, налаживания сотрудничества в сообществе редких заболеваний и финансирования существующих исследовательских усилий, направленных на редкие заболевания. Благодаря различным исследовательским проектам, финансированию и присуждению премий, а также информационно-пропагандистской деятельности Центр Мантона способствует открытию и разработке более эффективных методов диагностики и лечения редких или неизвестных состояний.
Бостонская детская больница объединилась в партнерстве с фондом Manton Foundation, чтобы создать центр, посвященный изучению «сиротских болезней» — генетических синдромов, проблем иммунной системы, ошибок метаболизма, нервно-мышечных расстройств и других малоизвестных, но важных с научной точки зрения заболеваний.В нашем центре специалисты, готовые ответить на важные вопросы, связанные с загадочными и сложными заболеваниями, могут сотрудничать, умножая влияние ключевых медицинских открытий и помогая детям и семьям, сталкивающимся с серьезными медицинскими проблемами.
Редкое или «сиротское» заболевание определяется как заболевание или расстройство, которым в любой момент времени страдают менее 200 000 человек в США. Однако слово «редкий» кажется несколько неуместным, когда мы понимаем, что 30 миллионов человек в США поражены редким заболеванием.Хотя многие люди живут с этими редкими или неизвестными состояниями, отсутствие научных знаний об их основных причинах часто может вести пациентов через путь, осложненный ошибочным диагнозом, плохими вариантами лечения и отсутствием финансирования для исследований и пропаганды.
Цели Мантонского центра
Центр Manton — это виртуальный центр для врачей и ученых Бостонской детской больницы, которые разделяют общее видение помощи семьям путем улучшения нашего понимания и осведомленности о редких заболеваниях.Цели этого сотрудничества:
- Раскройте тайны сиротских болезней , чтобы разработать более совершенные диагностические тесты и разработать новые методы лечения и лечения.
- Откройте для себя фундаментальные биологические принципы , которые имеют широкое применение и могут привести к прогрессу в нашем понимании общих заболеваний, таких как рак, болезни сердца, диабет и других серьезных проблем со здоровьем.
- Обучите новое поколение исследователей , которые вложат творческую энергию в борьбу с сиротскими болезнями.
- Распространить результаты Мантонского центра среди неспециалистов, медицинских / научных сообществ на местном, национальном и глобальном уровнях, чтобы повысить интерес и поддержку исследований и ухода за сиротами.
Эта страница последний раз обновлялась 26 ноября 2020 г.
Количество разработок лекарств от редких заболеваний, поражающих детей, увеличивается
По данным нового исследования, увеличилось количество методов лечения редких заболеваний, поражающих детей.Но федеральные стимулы, призванные стимулировать разработку лекарств для редких состояний, чаще используются для расширения использования существующих лекарств, а не для создания новых.
По оценкам, дети составляют половину пациентов с редкими заболеваниями, включая состояния, от которых страдают менее 200 000 американцев. Разработка лекарств для детей с редкими заболеваниями имеет решающее значение для обеспечения новых вариантов лечения, но не всегда выгодна производителям лекарств.
Чтобы побудить производителей разрабатывать лекарства для лечения редких заболеваний или «орфанных лекарств», федеральная политика предусматривает такие стимулы, как налоговые льготы, гранты на тестирование и семилетний период эксклюзивности, в течение которого конкуренты не могут продавать альтернативную версию лекарства. препарат от той же болезни.
ПОДРОБНЕЕ В ЛАБОРАТОРИИ: Подпишитесь на нашу еженедельную рассылку новостей
Из 402 показаний к применению орфанных препаратов, утвержденных Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США в период с 2010 по 2018 год, треть была специально предназначена для детей или для болезней, которые преимущественно затрагивали детей, согласно результатам, опубликованным в Pediatrics .
Но большинство из этих детских разрешений на орфанные препараты были новыми применениями существующих лекарств, некоторым из которых уже несколько десятилетий, и они уже были одобрены для лечения распространенных заболеваний.Двадцать человек получили статус «прорыв», который присваивается лекарствам, которые обещают усовершенствовать существующие методы лечения.
«Наше исследование выявляет причины для оптимизма и беспокойства», — говорит старший автор Као-Пинг Чуа, доктор медицины, доктор философии, педиатр и исследователь из детской больницы Michigan Medicine CS Mott и Центра оценки здоровья ребенка Susan B. Meister. Исследовательский центр.
«Многие детские сиротские показания могли быть прорывом для детей с редкими заболеваниями.В то же время большинство показаний не относились к новым лекарствам, а некоторые относились к относительно незначительному расширению их использования. Орфанные препараты обходятся обществу дорого, и важно убедиться, что эти затраты оправданы размером пользы для пациентов ».
Нравится подкасты? Добавьте Michigan Medicine News Break на свое устройство с поддержкой Alexa или подпишитесь на обновления на iTunes , Google Play и Stitcher .
Исследователи обнаружили, что 136 разрешенных педиатрических лекарств для лечения сирот нацелены на 87 уникальных заболеваний, чаще всего кистозный фиброз, острый лимфобластный лейкоз и наследственный ангионевротический отек с иммунными нарушениями.
«Хотя Закон о лекарствах для сирот оказался эффективным в стимулировании разработки лекарств, наши результаты показывают, что не все показания для лечения сирот в педиатрии имеют одинаковую ценность», — говорит ведущий автор исследования Лорен Киммел, научный сотрудник Медицинской школы Мичиганского университета и CHEAR.
«Политики должны обеспечить эффективное и действенное использование ресурсов для стимулирования разработки новых методов лечения редких заболеваний, для которых нет никаких вариантов лечения».
Цитируемая статья: «Показания для лечения орфанных педиатрических препаратов: 2010–2018 гг.», Педиатрия, . DOI: 10.1542 / peds.2019-3128
Редкие и орфанные болезни
Хотя большинство редких заболеваний имеют генетическое происхождение, многие причины до конца не изучены. Даже если причина редкого заболевания установлена, исследователи могут все еще не понять, как болезнь функционирует или как правильно ее лечить.В отчете Института медицины о редких болезнях и продуктах для сирот за 2010 год выделяются четыре потенциальных причины редких заболеваний:
- Генетические причины: 80% или более редких заболеваний имеют генетическое происхождение. Иногда один ген (как при дефиците альфа-1-антитрипсина) или несколько генов (как при синдроме Вильямса-Бёрена) способствуют проявлениям расстройства. Они могут передаваться по наследству или возникать в результате спорадических или случайных мутаций.
- Инфекционные агенты: Некоторые инфекции считаются редкими во всем мире, в то время как другие редки в богатых странах, но распространены в менее экономически развитых странах.Например, туберкулез был распространен в богатых странах до того, как были обнаружены и широко применялись профилактические меры и методы лечения, но он остается основной причиной заболеваемости и смертности в развивающихся странах. Инфекционные заболевания могут быть редкими, но широко известными и опасными (например, бешенство, ботулизм и пятнистая лихорадка Скалистых гор), в то время как другие действительно неясны (например, синдром Лемьера).
- Токсичные вещества: Воздействие природных или искусственных токсичных веществ также может вызывать редкие заболевания.Например, мезотелиома — это рак, обычно вызываемый воздействием асбеста. Многие виды отравлений можно отнести к числу редких состояний, но часто это не так. Токсичные вещества, вызывающие заболевания, такие как кадмий, хром и фосфин, обрабатываются Агентством по регистрации токсичных веществ и заболеваний (ATSDR), а морские токсины, вызванные вредоносным цветением водорослей, которые могут загрязнять пищу, отслеживаются Центрами по контролю и профилактике заболеваний (CDC). .
- Другие причины: Редкие состояния имеют множество других причин, таких как недостаточность питания, травмы, неблагоприятные последствия лечения и многие другие.
Финансовая нагрузка, связанная с сиротами и редкими заболеваниями, может повлиять как на пациентов, так и на тех, кто за ними ухаживает. Процесс диагностики может быть трудоемким и дорогостоящим. Лекарства, разработанные специально для лечения сиротских и редких заболеваний, могут стоить семьям десятки и даже сотни тысяч долларов в год. Многие редкие состояния диагностируются в детстве и продолжают поражать людей в течение десятилетий. Орфанные препараты, как правило, имеют больше ограничений по покрытию, чем неиротские лекарства, что приводит к более высокой цене за единицу.В рамках Medicaid Part D 85% планов покрывали орфанные препараты, которые были размещены на самом высоком уровне совместного несения затрат, покрывая только от 25 до 33% полной стоимости лекарства.
Потеря производительности, потеря заработной платы или отсутствие управляемой работы с важными преимуществами могут повлиять на людей, живущих с редким заболеванием. Опекуны испытывают финансовые последствия от ухода за пациентами с редкими заболеваниями: 86% сообщили, что они сократили семейные сбережения, не сделали сбережений для долгосрочных целей, использовали личные сбережения и взяли на себя больше личных долгов.
Дополнительные препятствия для доступа к адекватным медицинским услугам и вариантам лечения существуют для пациентов и лиц, ухаживающих за ними. К ним относятся более низкий доход (подвергающий их более высокому риску негативных финансовых последствий), ограниченный транспорт (затрудняющий доступ к медицинскому обслуживанию) и отсутствие доступа к вспомогательным услугам, таким как квалифицированные медицинские работники, разбирающиеся в редких заболеваниях. Только 27% опекунов заявили, что, по их мнению, их местная больница может справиться с состоянием их пациентов.
Пациенты с сиротскими и редкими заболеваниями часто обращаются за лечением в клиники, где их состояние никогда раньше не наблюдалось, а отсутствие, замаскированные, неправильно понятые или запутанные симптомы могут способствовать задержке постановки диагноза. Для трети людей с редким заболеванием может потребоваться от одного до пяти лет, чтобы получить правильный диагноз.
Раннее выявление, диагностика и вмешательство имеют решающее значение для предотвращения смерти или инвалидности при многих сиротских и редких заболеваниях. Половина всех пациентов, у которых диагностировано редкое заболевание, — дети, и до трех из десяти детей с редким заболеванием не доживут до своего пятилетия.Скрининг новорожденных, который обычно проводится 4,1 миллиона младенцев в США ежегодно, может выявить генетические, эндокринные и метаболические нарушения. Каждый год у 4000 детей диагностируется врожденное заболевание, и примерно 1000 остаются незамеченными. Рекомендуемая единообразная скрининговая панель (RUSP) включает 35 основных условий и 26 второстепенных условий. Каждый штат решает, какие условия включить в программу скрининга новорожденных; большинство из них включает тех, кто указан в RUSP. В 10 штатах в 2019 году было принято законодательство, требующее проведения скрининга на определенные заболевания, определение экономической целесообразности скрининга на дополнительные заболевания и расширение охвата скринингом в рамках Medicaid, а многие другие уже приняли законы о скрининге новорожденных.
Эффективность лечения сирот и редких заболеваний варьируется от лечения болезни до изменения ее функционирования или лечения симптомов болезни. По-настоящему лечебные методы лечения редки. Терапия, изменяющая заболевание, нацелена на основную патологию болезни, чтобы предотвратить ее ухудшение. Симптоматическое лечение направлено на смягчение симптомов или поддержание физического, эмоционального и умственного функционирования. Лечение может включать лекарства, пищевые добавки, хирургические процедуры, психотерапию, физиотерапию и трудотерапию, а также медицинские устройства.Примеры включают в себя проводники сердечного клапана, которые расширяются по мере роста ребенка, чтобы предотвратить повторные операции на протяжении всей его жизни, а также меры по питанию и диетическим добавкам, чтобы способствовать правильному росту и развитию у пациентов с врожденными нарушениями метаболизма (IEM). Задержки с разработкой, утверждением и доступностью новых лекарств для лечения орфанных и редких заболеваний могут иметь непредвиденные последствия для пациентов. Например, лечение спинальной мышечной атрофии ограничено и должно назначаться младенцам и детям в возрасте до 2 лет, что может означать разницу между жизнью и смертью для пациентов.
Многим пациентам с сиротами и редкими заболеваниями требуются медицинские устройства для диагностики и лечения их состояний, такие как генетические тесты, детские имплантаты, которые растут вместе с ребенком, и педиатрические интратекальные порты для доставки лекарств. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) и Национальный центр по продвижению трансляционных наук / Управление исследований редких заболеваний Национального института здравоохранения сообщили, что 917 врачей указали на неудовлетворенные потребности в медицинских устройствах в 2015 году, при этом многие отметили необходимость в совершенно новых диагностических и терапевтических средствах. устройств.
В 1983 году Конгресс принял Закон о орфанных лекарствах (ODA), который создал финансовые стимулы для производителей лекарств и биопрепаратов для исследования и разработки методов лечения орфанных заболеваний. К ним относятся налоговые льготы на расходы на клинические исследования, финансирование государственных грантов, помощь в клинических исследованиях и семилетний эксклюзивный маркетинговый период для первого спонсора продукта, указанного для сирот, который получает одобрение рынка от FDA. С момента принятия ODA было разработано более 250 орфанных лекарств, которые стали доступны более чем 13 миллионам американцев.
FDA управляет тремя программами по стимулированию исследований и разработок фармацевтических препаратов, биопрепаратов и медицинского оборудования для лечения редких заболеваний. Программа модернизации орфанных лекарств была создана, чтобы устранить отставание FDA от орфанных лекарств. Программа определения редких педиатрических заболеваний и ваучеров предоставляет ваучеры на приоритетное рассмотрение спонсорам заявок на получение продуктов с редкими педиатрическими заболеваниями. Программа обозначения устройств в гуманитарных целях создает альтернативный способ получения разрешения на продажу медицинских устройств, которые могут помочь людям с редкими заболеваниями или состояниями.