EURORDIS
Начало этого редкого заболевания означает огромные изменения не только для пациентов, но и для их семей. Дэниса Райана и Роже Пикара, которые живут в разных странах, объединяет общая беда – их женам был поставлен диагноз «болезнь Хантингтона» (HD).
Денис Райан и его жена Анна живут в Ирландии. В 1996 году у Анны был выявлен синдром Хантингтона. Ранее такой же диагноз был поставлен ее младшей сестре, а у старшей сестры также наблюдались соответствующие симптомы. Дэнис подозревал, что у его жены, скорее всего, развивается болезнь Хантингтона, но, как объясняет он сам, объявление диагноза – «это переломный момент, резко меняющий жизнь. Ты понимаешь, что бессмысленно надеяться на то, что все наладится и будет по-прежнему – прошлого уже не вернуть».
До болезни Анны они были обычной семьей – Анна присматривала за детьми, а Дэнис работал инженером.
«Пока дети подрастают, жизнь летит незаметно. Но когда они покидают родительское гнездо, родители часто задают себе вопрос «Что будет дальше?». Болезнь Хантингтона дает совершенно определенный ответ на этот вопрос…», — рассказывает Дэнис.
Болезнь Хантингтона – это наследственное заболевание, приводящее к возникновению дегенеративных процессов в определенных участках головного мозга. Эти процессы, в свою очередь, являются причиной физических, умственных и эмоциональных изменений, которые у разных людей выражены в различной степени. Как правило, симптомами болезни являются беспокойство, раздражительность, паранойя, психоз, деменция (слабоумие), двигательные и речевые расстройства, а также проблемы с жеванием и глотанием. Встречаемость заболевания составляет примерно 1 больной на 10,000-20,000 человек.
Дэнис и Анна воспользовались медленным развитием заболевания для того чтобы осуществить некоторые из своих планов на будущее, например, мечту о путешествиях.
«Сейчас о поездках уже не может быть речи, но она все еще «хвастается» тем, что мы отпраздновали 40-летний юбилей совместной жизни поездкой в Гранд-Каньон!», – говорит Дэнис. «И хотя сегодня Анна во многих отношениях совершенно беспомощна и нуждается в постоянной опеке, мы стараемся, чтобы этот факт не мешал нам по-прежнему получать максимальное удовольствие от жизни», – продолжает он.
«И хотя сегодня Анна во многих отношениях совершенно беспомощна и нуждается в постоянной опеке, мы стараемся, чтобы этот факт не мешал нам по-прежнему получать максимальное удовольствие от жизни», – продолжает он.
Разница в том, что теперь Дэнис принимает все решения в одиночку: «По мере того как болезнь изменяла личность Анны, мне тоже пришлось во многом измениться. Одно из самых важных изменений заключается в том, что я научился жить, не рассчитывая на поддержку и помощь партнера».
Жене Роже Пикара диагноз «болезнь Хантингтона» был поставлен в 1993 году, но, как и у Анны Райан, симптомы заболевания начали проявляться раньше. После 20 лет брака жизнь семьи в одночасье превратилась в настоящий кошмар. С 1992 по 1994 годы Дениз Пикар девять раз пыталась покончить жизнь самоубийством, причем во время последней попытки она подожгла дом.
«Болезнь и пожар полностью изменили нашу спокойную жизнь. Мне пришлось посвятить все свое время уходу за женой. Я был вынужден бросить работу и объявить себя банкротом», – рассказывает Роже.
Обоим мужьям пришлось научиться ухаживать за женами, попутно решая повседневные жизненные проблемы, вопросы организации необходимого лечения и получения поддержки для себя и для своих больных жен. В Европе нет единого подхода к организации помощи больным с редкими заболеваниями. Поэтому, как объяснил нам Дэнис: «Даже в маленькой Ирландии набор предоставляемых органами здравоохранения услуг отличается в зависимости от места проживания. Найти список специалистов, с которыми необходимо проконсультироваться, практически невозможно. Кроме того, поставщики услуг значительно отличаются друг от друга по степени заинтересованности, проявляемой в отношении пациентов».
Дэнис обратился за помощью в Ирландскую ассоциацию помощи пациентам с болезнью Хантингтона Disease (HDAI), которую он сейчас возглавляет. «Члены нашей ассоциации обмениваются опытом в решении конкретных проблем и делятся информацией о медицинских услугах, которые им удалось получить», – говорит Дэнис.
Роже считает, что им крупно повезло, когда в 2004 году Дениз отправили на лечение в неврологическое отделение городской больницы Кретейля, которое позднее стало клиническим центром, специализирующимся на лечении больных с синдромом Хантингтона. Вместе с тем, открытие специализированного центра не решило проблем, связанных с предоставлением пациентам социальной помощи/поддержки. «После установления диагноза они остаются наедине со всеми последствиями болезни. Нас не признают в качестве официальных опекунов и, соответственно, не считаются с нами. Я не могу рассчитывать на помощь со стороны органов здравоохранения – мне приходится всего добиваться самому».
Именно по этой причине Роже основал Фонд Дениз Пикар, в котором он исполняет обязанности президента. Фонд поддерживает усилия, направленные на создание специализированных учреждений по уходу за пациентами с нервно-дегенеративными заболеваниями.
И Роже и Дэнис подчеркивают важность поддержания собственного здоровья для обеспечения качественного ухода за своими женами, хотя часто у них на это просто не хватает времени. Однако наибольшее беспокойство у них вызывает будущее детей. Синдром Хантингтона – это наследственное заболевание, и шансы унаследовать его от одного из родителей составляют 50/50.
Как говорит Дэнис: «Вероятно, самое ужасное в этой ситуации это то, что ты понимаешь, что твой ребенок приближается к возрасту, в котором могут проявиться симптомы заболевания».
«Жизнь с пациентом, страдающим болезнью Хантингтона, может превратиться в настоящий кошмар. Больно осознавать, что человек, которого ты знал и любил, настолько изменился, что ты уже с трудом узнаешь его (а он, возможно, уже не узнает тебя!). Это все равно, что всю жизнь оплакивать смерть любимого человека».
Тем не менее, Роже и Дэнис стараются фокусироваться на положительных моментах, таких как удивительная доброта людей, появление новых друзей, работа в ассоциации помощи пациентам с синдромом Хантингтона и связанные с ней достижения.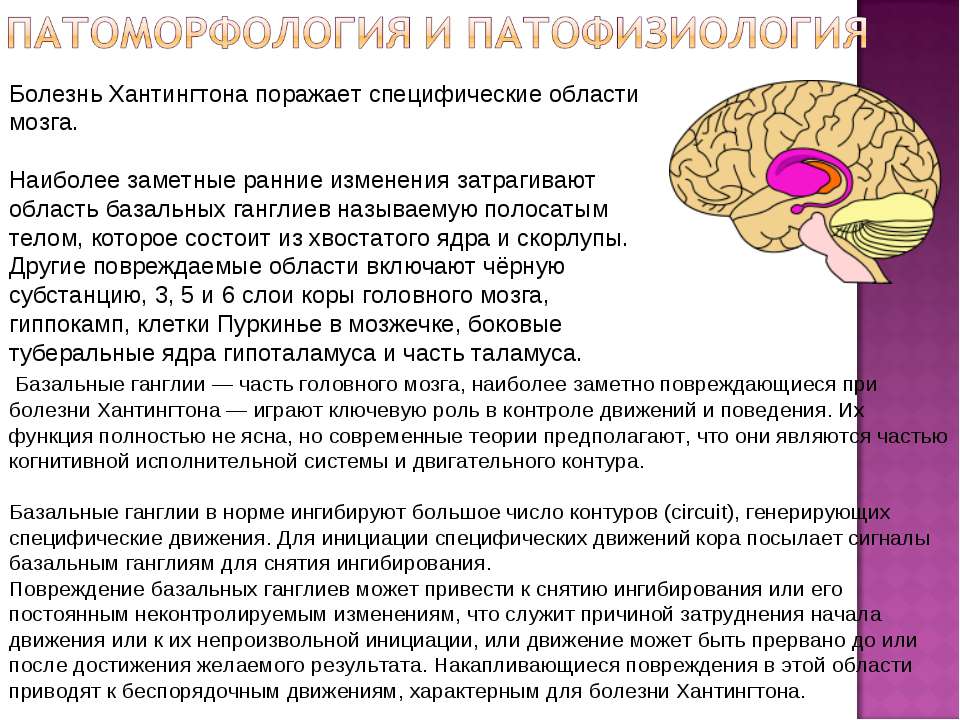 Оба они не теряют надежду, что рано или поздно будет найден способ лечения этого заболевания, который сможет вернуть здоровье, если не их женам, то, по крайней мере, их детям.
Оба они не теряют надежду, что рано или поздно будет найден способ лечения этого заболевания, который сможет вернуть здоровье, если не их женам, то, по крайней мере, их детям.
Статья была впервые опубликована в декабрь выпуске (2011 г.) информационного письма EURORDIS
Автор: Irene Palko
Фотографии: © M. R. Picard, M. D. Ryan
Page created: 03/12/2012
Page last updated: 24/01/2013
Болезнь Хантингтона | Симптомы | Диагностика | Лечение
Болезнь Хантингтона (болезь Гентингтона, болезнь Хорея Гентингтона) – заболевание нервной системы, характеризующееся появлением периодических мышечных подергиваний или спазмов. Заболевание может развиться в любом возрасте, но чем раньше проявляются первые симптомы, тем стремительнее прогрессирует болезнь.
Причины
Заболевание возникает в результате постепенной дегенерации и отмирания нервных клеток и клеток головного мозга, что вызывается белком хантингтином. Причиной болезни Гентингтона становится наследственный фактор.
Симптомы болезни Хантингтона
Характерные для болезни Хантингтона симптомы выглядят следующим образом:
Заболевание на первых стадиях протекает бессимптомно, первые признаки начинают проявляться в возрасте 35-40 лет.
Если Вы обнаружили у себя схожие симптомы, незамедлительно обратитесь к врачу. Легче предупредить болезнь, чем бороться с последствиями.
Диагностика
Чтобы определить, как лечить болезнь Хорея Гентингтона, врач — невролог проводит ряд исследований и анализов, среди которых:
Лечение болезни Хантингтона
Необходимое для болезни Гентингтона лечение направлено на уменьшение симптомов и включает в себя:
- нейролептические препараты;
- бензодиазепины;
- противопаркинсонические средства;
- вальпроевую кислоту;
- психотропные препараты;
- транквилизаторы
В настоящее время болезнь Хантингтона неизлечима.
Опасность
Если своевременно не заметить характерные для болезни Гентингтона симптомы и не начать лечение, это может привести к развитию осложнений:
- слабоумие;
- пневмония;
- ослабление иммунной системы;
- удушье;
- обезвоживание организма;
- сердечнососудистая недостаточность;
- повышенный риск травматизма.
Возможны попытки больного совершить суицид. Через 15-20 лет с момента появления первых признаков наступает летальный исход.
Группа риска
В группе риска находятся люди с генетической предрасположенностью (вероятность появления заболевания у детей больного человека составляет 50%).
Профилактика
Для профилактики заболевания беременным женщинам рекомендуется проходить перинатальную диагностику.
Данная статья размещена исключительно в познавательных целях и не является научным материалом или профессиональным медицинским советом.
причины, симптомы и лечение в статье невролога Гольченко Е. А.
Дата публикации 27 ноября 2020Обновлено 27 ноября 2020
Определение болезни. Причины заболевания
Болезнь Гентингтона (хорея Гентингтона) — это хроническое, неуклонно прогрессирующее, наследственное аутосомно-доминантное заболевание нервной системы. Болезнь проявляется нарушением речи, памяти и присутствием навязчивых вычурных движений, мешающих выполнять повседневную деятельность. Со временем пациент теряет навыки самообслуживания и нуждается в опеке, то есть инвалидизируется.
Термин «хорея» означает «танец» на греческом языке. Впервые это определение использовал известный алхимик Парацельс при описании «пляски святого Витта». Эта «пляска», возможно, была связана с религиозным экстазом, то есть имела истерический характер [1].
Позже английский врач Томас Сиденгам в процессе изучения «пляски святого Витта» описал детскую форму хореи, теперь это заболевание носит его имя. В 1872 году американский врач Джордж Гентингтон отметил наследственный вариант хореи с началом во взрослом возрасте. Впоследствии это заболевание получило название «болезнь Гентингтона» (БГ). Также его называют хореей Гентингтона (или Хантингтона), наследственной или дегенеративной хореей.
В 1872 году американский врач Джордж Гентингтон отметил наследственный вариант хореи с началом во взрослом возрасте. Впоследствии это заболевание получило название «болезнь Гентингтона» (БГ). Также его называют хореей Гентингтона (или Хантингтона), наследственной или дегенеративной хореей.
Классическим описанием патологии считается эссе «О хорее», опубликованное Джорджем Гентингтоном в «Медицинском и хирургическом вестнике» [2]. В настоящее время хорея Гентингтона является самой частой причиной наследственной хореи. Одним из самых известных людей, страдавших этим заболеванием, был американский кантри певец и композитор Вуди Гатри, который заболел в 50-е годы.
Болезнь может начинаться в разном возрасте, но чаще всего симптомы появляются в 30-50 лет и неуклонно прогрессируют [5]. Описаны две основные клинические формы болезни Гентингтона:
- гиперкинетическая, наиболее частая, характеризуется более поздним началом, постепенным прогрессированием хореических гиперкинезов (насильственных избыточных двигательных актов, возникающих помимо воли больного), а также деменцией с замедлением когнитивных функций, снижением критики, депрессией, разнообразными психозами;
- акинетико-ригидная с началом в раннем возрасте (вариант Вестфаля) наблюдается в 5-10 % случаев, характеризуется быстрым нарастанием мышечной слабости, контрактур (ограничением подвижности суставов), нарушениями поведения и психики, эпилепсией, наблюдается при передаче мутантного гена от отца.
Нейроморфологическая картина характеризуется уменьшением, то есть атрофией, полосатого тела (стриатума — глубинной структурной части мозга) уже на самой ранней стадии заболевания. На более развёрнутой стадии возможна атрофия коры головного мозга.
Распространённость хореи в Западной Европе составляет в среднем 4-10 случаев на 100 000 населения, поэтому патологию можно отнести к редким заболеваниям [3]. Наименьшая частота хореи Гентингтона установлена среди жителей Японии, Финляндии, Южной Африки и Северной Америки (7,33 на 100 000 населения). Самая высокая частота (более 17 случаев на 100 000 населения) зафиксирована в районе озера Маракайбо в Венесуэле, в Тасмании и на островах Маврикий [4]. В Российской Федерации распространённость патологии в целом не оценивалась, цифры по различным регионам разнятся.
Самая высокая частота (более 17 случаев на 100 000 населения) зафиксирована в районе озера Маракайбо в Венесуэле, в Тасмании и на островах Маврикий [4]. В Российской Федерации распространённость патологии в целом не оценивалась, цифры по различным регионам разнятся.
Мужчины и женщины болеют одинаково часто, однако существует механизм так называемой «отцовской передачи» заболевания, при котором наиболее тяжёлые варианты болезни наследуются от отца, особенно в случае передачи мутантного гена на протяжении нескольких поколений. Это объясняется нестабильностью региона, содержащего мутацию, и нарастанием числа повторов в процессе сперматогенеза [31].
Причина заболевания — мутация в гене HTT, который отвечает за образование белка гентингтина. Гены — самые маленькие единицы наследственности, которые содержатся в хромосомах. В соматических клетках человека всего одна пара половых хромосом и 22 пары аутосом. Болезнь Гентингтона явлется аутосомно-доминантным заболеванием, то есть мутантный ген расположен в аутосоме и является преобладающим. Это значит, что для развития болезни достаточно унаследовать мутантный ген от одного из родителей.
Предрасполагающими факторами развития болезни могут стать психотравмы, черепно-мозговые травмы или перенесённые инфекции.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы хореи Гентингтона
При болезни Гентингтона присутствует многолетняя стадия «предболезни», которая определяется постепенным нарастанием субклинических и биохимических изменений в структурах головного мозга. В самом начале появляются двигательные нарушения: насильственные избыточные двигательные акты, возникающие помимо воли больного — хореический гиперкинез. С развитием болезни присоединяются когнитивные или психические расстройства.
Прогрессирование заболевания в некоторых случаях может сопровождаться постепенным уменьшением выраженности непроизвольных движений, но усилением психических симптомов [9]. Однако для каждой из форм болезни выделяют превалирующие симптомы.
Однако для каждой из форм болезни выделяют превалирующие симптомы.
Клинические проявления можно распределить по следующим группам [10]:
1. Двигательные нарушения могут проявляться общим двигательным беспокойством в движениях рук и ног, возникающим при стрессе, волнении или ходьбе. Патогномоничным (характерным) и самым ранним двигательным симптомом являются глазодвигательные нарушения. По мере прогрессирования заболевания появляются грубые нарушения следящих движений глазных яблок, следовательно, пациент практически не может зафиксировать взгляд на каком-либо предмете. Постепенно к хорее присоединяются иные нарушения, например дистония (стабильные рефлекторные двигательные сокращения отдельных групп мышц) и паркинсонизм (снижение скорости движения, скованность мышц и дрожание конечностей).
Одним из основных двигательных нарушений является отсутствие контроля произвольных движений. Пациент не может удерживать какую-либо позу по причине грубых насильственных двигательных актов. На более поздних стадиях больным сложно выполнять движения, которые требуют тонкой координации. Значительно замедляется двигательная активность и нарушается ходьба, в результате человек часто падает. Больные широко расставляют ноги при ходьбе, корпус тела и шея вытянуты вперёд, корпус тела несколько повёрнут вокруг своей оси. При ходьбе появляется как бы пританцовывание, если пациент не пользуется компенсаторными приёмами, чтобы уменьшить насильственное движение (например, если не держится одной рукой за другую). Также повышается вероятность возникновения тонического напряжения мышц, приводящих бёдра. В таком случае больные передвигаются со сведёнными бёдрами и коленями.
Виды гиперкинезов:
- Хорея — проявляется неритмичным, отрывистым, кажущимся произвольным движением.
- Атетоз — это гиперкинез, который проявляется замедленным и червеобразным движением.
- Баллизм — это резкие, размашистые движения рукой или ногой.

- Дистония характеризуется медленным и неритмичным мышечным сокращением, которое часто приводит к формированию патологической позы.
- Миоклонус — это внезапные, быстрые и отрывистые движения, напоминающее удар электрического тока.

Другие двигательные проявления:
- Брадикинезия (медлительность).
- Мышечная ригидность (скованность).
- Окуломоторные (глазодвигательные) расстройства.
- Падения.
2. Когнитивные нарушения. Трудности в общении, организации собственной деятельности, упорядочивании своих мыслей и в усвоении новой информации, а также нарушения восприятия, повышенная отвлекаемость, нарушения памяти и умственной работоспособности могут появляться уже на ранних этапах заболевания. Это практически универсальные проявления хореи Гентингтона. Максимально страдают исполнительные функции, то есть способность планировать свои действия, а также оценивать агрессивное поведение. У большинства пациентов отмечается замедление психомоторных процессов, появляется апатия, снижение внимания к себе.
3. Психические нарушения: депрессия, тревога, апатия, обсессивно-компульсивные расстройства (навязчивые и бредовые идеи), раздражительность, психотические расстройства (агрессия, галлюцинации и др.), гиперсексуальность. Психические нарушения при болезни Гентингтона характеризуются в первую очередь депрессией и тревогой. Нередко можно отметить раздражительность, которая иногда становится причиной агрессивного поведения. Появление обсессивно-компульсивных расстройств значимо ухудшает качество жизни родственников, проживающих вместе с больным. Значительно реже у пациентов могут появляться психозы. Реализованные суициды и суицидальные попытки встречаются при болезни Гентингтона в 4 раза чаще, чем в общей популяции, и в 5 % случаев становятся причиной гибели при этом заболевании [30].
4. Метаболические расстройства: потеря массы тела, вплоть до кахексии (истощения) и др.
5. Прочие нарушения: расстройства сна, дизартрия (нарушение речи, при котором трудно произносить некоторые слова) и дисфагия (расстройство акта глотания). Больной испытывает трудности при разговоре, речь становится медленной, голос тихим и глухим, речь прерывается сопением, всхрапыванием, всхлипыванием, при осмотре выявляются элементы дизартрии. Тем не менее, даже на поздних стадиях пациенты доступны контакту. Из-за гиперкинезов языка, мягкого нёба, губ, диафрагмы и прямых мышц живота непроизвольные, «лишние» звуки (икота, сопение и др.) могут возникать даже когда больной молчит.
Прочие нарушения: расстройства сна, дизартрия (нарушение речи, при котором трудно произносить некоторые слова) и дисфагия (расстройство акта глотания). Больной испытывает трудности при разговоре, речь становится медленной, голос тихим и глухим, речь прерывается сопением, всхрапыванием, всхлипыванием, при осмотре выявляются элементы дизартрии. Тем не менее, даже на поздних стадиях пациенты доступны контакту. Из-за гиперкинезов языка, мягкого нёба, губ, диафрагмы и прямых мышц живота непроизвольные, «лишние» звуки (икота, сопение и др.) могут возникать даже когда больной молчит.
Патогенез хореи Гентингтона
Морфофункциональные изменения в мозге возникают намного раньше, чем появляются первые симптомы болезни Гентингтона [6][7]. Морфологические изменения в первую очередь касаются области полосатого тела. Характерным признаком при нейровизуализации является атрофия головки хвостатого ядра. В процесс вовлекаются: чёрная субстанция, кора больших полушарий, гиппокамп и мозжечок. Чуть в меньшей степени — боковые ядра гипоталамуса и таламус [8].
Патофизиологические механизмы развития болезни Гентингтона до конца не изучены. Большое значение в патогенезе этого заболевания имеет накопление и токсическое действие на нейроны мутантного белка гентингтина, в состав которого входит аминокислота глутамин.
За образование белка гентингтина отвечает ген HTT, который расположен в коротком плече 4-й хромосомы человека. При отсутствии патологии в этом участке находится определённое количество повторов последовательности тринуклеотидов цитозин-аденин-гуанин (ЦАГ) — от 6 до 35. Мутация заключается в увеличении числа повторов ЦАГ (более 40), в результате чего мутантный белок гентингтин за счёт избыточного содержания остатков аминокислоты глутамина становится токсичным в отношении определённых видов нейронов [32].
Число повторов ЦАГ:
- До 26 — норма.
- От 27 до 35 (нестабильное количество) — симптомы отсутствуют, но есть повышенный риск того, что у детей будет болезнь Гентингтона.
- 36 или больше — патогенные мутации с различной пенетрантностью (проявление гена у 100 % лиц с соответствующим генотипом называется полной пенетрантностью, в остальных случаях — неполной пенетрантностью): 36-39 — «зона неполной пенетрантности»; более 40 — полная пенетрантность.

«Неправильный» белок повреждает нервные клетки головного мозга при встраивании в процесс обмена веществ. Поэтому возникают симптомы, характерные для болезни Гентингтона. Удлинённый участок молекулы белка гентингтина способствует формированию патологических межмолекулярных связей. Таким образом, болезнь Гентингтона является «полиглутаминовым заболеванием» [11]. Одним из новых патогенетических механизмов развития патологии, особенно в ранней стадии дегенеративного процесса, является нейровоспаление, активируемое мутантным гентингтином в центральной нервной системе через моноциты и микроглиальные клетки [12].
К особенностям наследования БГ относятся:
- Антиципация — феномен, при котором увеличение ЦАГ-повторов нарастает в ряду поколений, что означает более раннее возникновение болезни и более тяжёлое течение по сравнению с предыдущими поколениями.
- Феномен «отцовской передачи». Более тяжёлая форма заболевания развивается, если патология передалась от отца, симптомы появляются в детском возрасте.
- Наличие «зоны неполной пенетрантности», или «серой зоны» (у части лиц с такими мутациями болезнь развивается, а у других заболевание может отсутствовать даже в преклонном возрасте) [33].
- Эффект геномного импринтинга. При наследовании болезни Гентингтона гены отца и матери активированы или супрессированы по‐разному.
Интересно, что генетическая нестабильность мутации при болезни Гентингтона реализуется преимущественно у лиц мужского пола (процесс образования половых клеток), и эти наблюдения объясняют эффект «отцовской передачи» — манифестацию более ранних и тяжёлых случаев болезни гентингтона у потомков больного отца [13].
Классификация и стадии развития хореи Гентингтона
Для болезни Гентингтона, как и для других нейродегенеративных заболеваний, установлено существование длительной, многолетней стадии “предболезни”. В этот период действие мутантного гена ещё не успело привести к диагностируемой клинической картине, но патохимические нарушения в нейронах уже накапливаются, и возникает ряд субклинических проявлений [17].
Этапы жизни пациента с болезнью Гентингтона (классификация профессора неврологии Раймунда Рооса) [34]:
Доклиническая стадия (как правило, длится 10-15 лет до появления первых симптомов):
- Асимптомный период. У носителя мутации заболевания не отмечается субъективных, клинических или измеряемых инструментально изменений.
- Продромальный период (период предвестников заболевания) характеризуется постепенным появлением и нарастанием невыраженных двигательных, когнитивных и поведенческих изменений. Однако такие изменения не позволяют поставить клинический диагноз болезни Гентингтона
Клиническая стадия (средняя продолжительность заболевания — 15-18 лет [35]):
- Клиническая стадия I. Появляются первые неврологические, когнитивные или психические симптомы. Основной симптом — беспорядочные, отрывистые, нерегулярные, кажущиеся произвольными движения. В повседневной жизни пациенту не требуется помощь. Летальный исход в этот период случается редко, за исключением случаев суицида.
- Клиническая стадия II. Нарастают двигательные нарушения, пациенту требуется помощь в самообслуживании. Смерть возможна в результате суицида.
- Клиническая стадия III. Тяжёлые двигательные нарушения, больной не способен на самообслуживание, полностью зависит от окружающих. Летальный исход наступает в результате осложнений заболевания.
На основании преобладания какого-либо синдрома различают несколько форм болезни Гентингтона:
1. «Классическая» гиперкинетическая форма. Характеризуется наличием навязчивых движений, нарушением походки (”танцующая походка”) и речи, а также снижением мыслительной деятельности.
«Классическая» гиперкинетическая форма. Характеризуется наличием навязчивых движений, нарушением походки (”танцующая походка”) и речи, а также снижением мыслительной деятельности.
2. Ригидная форма:
- Ювенильная (вариант Вестфаля). В отличие от классического варианта болезни, при ювенильной форме дебют происходит в возрасте до 21 года и/> наблюдается более тяжёлое течение и характер неврологической симптоматики. Проявления хореи минимальны, преобладают акинезия (отсутствие движения) и ригидность (болезненное состояние, при котором повышен тонус мышц и возникает сопротивление при попытке сделать движение). Кроме того, неврологические проявления при ювенильной форме более разнообразны: для неё характерны эпилепсия, атаксия (расстройство координации), миоклонии (молниеносные подёргивания отдельных групп мышц), дистония и другие расстройства, нетипичные для классической БГ. Давно известны генеалогические особенности этой формы: она развивается преимущественно при наследовании болезни от отца [16].
- Поздняя форма. Нередко является следствием прогрессирования «классической» формы болезни Гентингтона, при этом происходит трансформация клинической картины гиперкинетического синдрома в брадикинезию (замедленность движений) с мышечной скованностью. Примерно у 10 % взрослых пациентов болезнь начинается с акинетико-ригидного синдрома (движения скованные, трудно начать действие, медлительность во всем) с минимальными проявлениями хореи [15].
3. Психическая. Эта форма не является самостоятельной и выделяется лишь при резком преобладании психических симптомов над неврологической симптоматикой.
Чтобы оценить степень выраженности функциональных нарушений при патологии, разработана Унифицированная шкала оценки болезни Гентингтона (United Huntington’s Disease Rating Scale, UHDRS).
Компоненты UHDRS
- Двигательная оценка.
- Когнитивная оценка.
- Оценка поведенческих нарушений.
- Оценка функциональных возможностей.
- Оценка общей функциональной способности (TFC).

В зависимости от оценки по шкале общей функциональной способности (TFC), болезнь Гентингтона имеет следующую классификацию:
| Общий балл TFC | Стадия БГ | Общая характеристика |
|---|---|---|
| 11-13 | 1 | Ранние стадии: Появляются единичные двигательные нарушения: лёгкие неконтролируемые движения, незначительное изменение координации и походки, забывчивость, раздражительность. |
| 7-10 | 2 | |
| 4-6 | 3 | Стадия умеренных клинических проявлений: Присоединяются психические нарушения: нарушение речи и глотания, снижение аппетита и веса, становятся более выраженными неконтролируемые движения. |
| 1-3 | 4 | Поздние стадии, или стадии развёрнутых клинических проявлений: Появляются выраженные симптомы (двигательные и психические), пациент инвалидизируется, нуждается в посторонней помощи, появляются депрессия, галлюцинации, панические расстройства. |
| 0 | 5 |
Осложнения хореи Гентингтона
- Аспирационная пневмония — воспаление которое развивается, когда в просвет лёгких попадают инородные вещества, например рвотные массы.
- Сепсис — опасное для жизни инфекционное заболевание человека, которое развивается в ответ на проникновение в кровь инфекционных агентов или их токсинов.
- Кахексия — истощение, которое возникает на фоне отказа от еды или нарушения глотания.
- Нарушения работы сердечно-сосудистой системы из-за закупорки артерий.
- Суицид в результате психических нарушений [10].
Диагностика хореи Гентингтона
Около 90 % диагнозов болезни Гентингтона, основанных на семейном анамнезе и выявлении характерных симптомов, подтверждаются генетическим исследованием. Поэтому сбор семейного анамнеза является обязательным при осмотре пациента с хореей.
Поэтому сбор семейного анамнеза является обязательным при осмотре пациента с хореей.
Наиболее оптимальным методом диагностики является молекулярно-генетическое исследование. Для выявления мутации гена, ассоциированного с развитием хореи Гентингтона, требуется забор крови из вены. Анализ ДНК позволяет установить аномальное количество ЦАГ-повторов в гене HTT, что является непосредственной причиной развития болезни. С помощью этого метода можно подтвердить диагноз у больного и выявить носителей патологического гена среди родственников на доклинической стадии.
Аномальное количество ЦАГ-повторов в гене HTT не подтверждает диагноз, так как может быть получен задолго до появления первых симптомов. Нормальное количество ЦАГ-повторов в гене HTT говорит о том, что человек никогда не заболеет данной патологией.
При КТ головного мозга у пациентов с болезнью Гентингтона определяется расширение боковых желудочков и субарахноидальных пространств (между мозговыми оболочками, заполненными спинномозговой жидкостью). Характерно развитие гидроцефалии (избыточного скопления спинномозговой жидкости в желудочках головного мозга), при этом её степень будет зависеть от тяжести заболевания.
На МРТ можно отметить выраженную атрофию головок хвостатых ядер и расширение боковых желудочков, особенно в области передних рогов.
Однофотонная эмиссионная компьютерная томография (ОФЭКТ, англ. — SPECT) является более чувствительным и специфичным методом для постановки диагноза хореи Гентингтона: после введения радиофармпрепарата (РФП) выявляется снижение метаболизма глюкозы в хвостатых ядрах. Иногда эти изменения можно обнаружить на доклинической стадии болезни. Такие же признаки выявляются и при позитронно-эмиссионной томографии (ПЭТ) [18].
Количественная электроэнцефалография (ЭЭГ) — неинвазивный и относительно недорогой метод, в основе которого лежит регистрация электрической активности нейронов, которая представляет собой базовый механизм их взаимодействия. Ритмы ЭЭГ, в частности α- и θ-ритмы, отражают нейрофизиологические процессы, участвующие в обеспечении когнитивных функций [19][20].
Ритмы ЭЭГ, в частности α- и θ-ритмы, отражают нейрофизиологические процессы, участвующие в обеспечении когнитивных функций [19][20].
Ритмы ЭЭГ:
- α-ритмы — регистрируется у человека в условиях физического и умственного покоя при закрытых глазах и отсутствии внешних раздражений.
- β-ритмы — регистрируются при открывании глаз, умственной работе и других раздражителях.
- θ-ритмы — наблюдается в состоянии неглубокого сна, при кислородном голодании мозга и наркозе.
- δ-ритмы — отмечаются во время глубокого сна.
Изменения на ЭЭГ у пациентов с болезнью Гентингтона характеризуются значимым снижением спектральной мощности α-ритма и повышением относительной спектральной мощности β- и δ-активности. Изучению данных ЭЭГ у асимптомных носителей мутаций в гене HTT посвящено небольшое количество работ, и их результаты неоднозначны.
Лабораторные анализы при болезни Гентингтона могут существенно не отличаться от нормы. При специальном биохимическом анализе крови иногда обнаруживается снижение концентрации некоторых компонентов:
- гамма-аминомасляной кислоты (ГАМК) — важнейшего тормозного нейромедиатора центральной нервной системы;
- глутаматдекарбоксилазы — фермента, необходимого для синтеза ГАМК;
- холин-ацетилтрансферазы — фермента, участвующего в синтезе нейромедиатора нервного возбуждения — ацетилхолина [21].
При исследовании спинномозговой жидкости нередко отмечается повышение содержания белка.
Дифференциальный диагноз. Существует ряд заболеваний, которые в некоторых случаях сопровождаются хореей с когнитивными и (или) психическими нарушениями и напоминают болезнь Гентингтона: болезнь Вильсона — Коновалова, болезнь Фридрейха, хореоакантоцитоз, спиноцеребеллярные атаксии и др.
Лечение хореи Гентингтона
Болезнь Гентингтона считается неизлечимым заболеванием, поэтому терапия является симптоматической и поддерживающей. Несмотря на это, благодаря лечению можно значительно улучшить качество жизни пациентов [22].
Несмотря на это, благодаря лечению можно значительно улучшить качество жизни пациентов [22].
В настоящее время ведущую роль в лечении двигательных нарушений при болезни Гентингтона играет тетрабеназин. Это новый препарат, схожий по своим свойствам с атипичными нейролептиками [23]. Если нет выраженных психических проявлений болезни (в первую очередь депрессий, суицидальных мыслей, раздражительности, агрессивного поведения) и нарушения глотания, то этот препарат нужно рассматривать как средство первого выбора для лечения хореи при болезни Гентингтона.
В других случаях рационально рассмотреть возможность применения нейролептиков, т. е. антипсихотиков (в первую очередь атипичных — клозапин, кветиапин, рисперидон) в виде монотерапии или в сочетании с противоэпилептическими препаратами или бензодиазепинами [24].
Препараты вальпроевой кислоты (Депакин, Конвулекс и др.), известные как антиконвульсанты, применяются при наличии мышечных спазмов.
Коррекцию психических нарушений следует проводить при помощи лекарственных препаратов, применяемых в психиатрии.
По данным некоторых клинических случаев, могут быть эффективны высокие дозы клоназепама [25]. Этот короткодействующий препарат из группы бензодиазепинов чаще применяется в качестве вспомогательной терапии, если у пациента присутствует тревога. Кроме того, клоназепам назначается для коррекции миоклоний, дистонии и при нарушениях сна [26][27].
При необходимости врач назначает антидепрессанты, препаратами выбора являются ингибиторы обратного захвата серотонина благодаря небольшому числу побочных эффектов и отсутствию опасности передозировки (сертралин, эсциталопрам).
Пациентам с болезнью Гентингтона пытались проводить операции глубокой стимуляции мозга с имплантацией электродов во внутренний сегмент бледного шара (парной структуры переднего мозга, которая является перекрёстком выбора действия).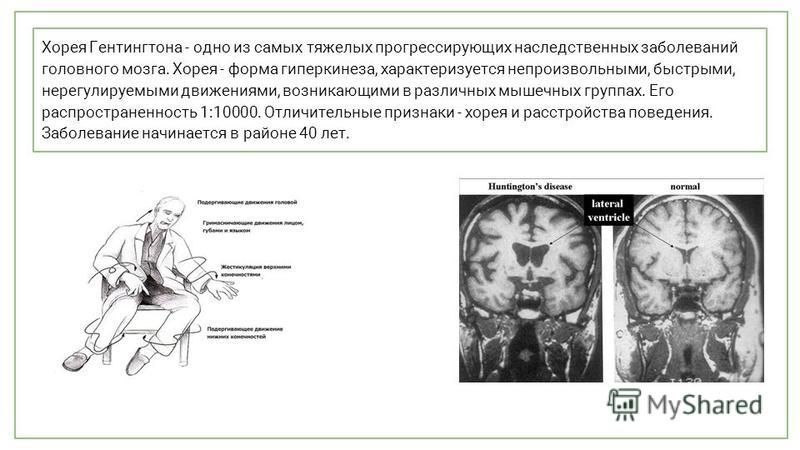 При этом наблюдалось снижение выраженности хореических гиперкинезов, но отмечалось нарастание расстройств ходьбы, брадикинезии и когнитивных нарушений [28].
При этом наблюдалось снижение выраженности хореических гиперкинезов, но отмечалось нарастание расстройств ходьбы, брадикинезии и когнитивных нарушений [28].
Есть данные о трансплантации в мозг пациентов с болезнью Гентингтона эмбриональных и стволовых клеток [29], но результаты такой экспериментальной терапии нуждаются в тщательной оценке.
В настоящее время в мире ведутся многоцентровые клинические исследования генной терапии у пациентов с хореей Гентингтона. Заболевание создаёт большие медико‐социальные проблемы, так как к моменту первых проявлений болезни люди успевают завести детей и передать им патологию, не подозревая об этом. В связи с этим необходима разработка новых методов диагностики и лечения, что делает актуальным дальнейшее изучения БГ и особенностей её течения.
Прогноз. Профилактика
Учитывая тот факт, что заболевание неуклонно прогрессирует, можно сделать вывод о неблагоприятном прогнозе. Средняя продолжительность жизни с момента появления гиперкинеза составляет 15-20 лет [15]. При ювенильной форме пациенты живут меньше.
Пациенты погибают не от самой болезни, а от её осложнений. Например, пневмония может стать смертельной из-за ослабленного иммунитета пациента. Иммунитет, в свою очередь, снижен из-за истощения пациента, который по причине проблем с глотанием мало принимает пищу. Травмы могут стать смертельными в результате нарушения движений и изменения походки. Частой причиной гибели пациентов является суицид [30].
Чем больше осведомлен пациент о своем заболевании, тем больше шансов своевременно получить необходимую терапию у врача невролога и предотвратить быстрое развитие болезни и осложнений.
Для профилактики заболевания в семьях с отягощённым анамнезом обязательно проведение медико-генетического консультирования для определения генетического статуса близких родственников и прогноза по заболеванию у потомства. Ввиду высокого риска повтора заболевания в семье с отягощённым анамнезом, в качестве профилактики рекомендуется проведение пренатальной диагностики или доимплантационного генетического тестирования эмбрионов (ПГТ-М), что обсуждается в рамках медико-генетического консультирования семьи.
Хорея Хантингтона симптомы
Хорея Хантингтона или болезнь Хантингтона, иногда ещё называют как синдром Хантингтона — это пожалуй одно из наитяжелейших нейродегенеративных прогрессирующих генных заболеваний мозга. В народе это заболевание называют «Пляска святого Витта». В некоторых источниках слово Хантингтона заменяют на Гентингтона в следствии погрешности перевода, но на английском и латыни, болезнь именуются как Huntington’s Disease или Huntington’s disorder или Huntington’s chorea. То есть, название Huntington’s одно, но из-за неточности перевода в разных русских источниках их встречается два.
Название хорея Хантингтона в точности соответствует народному наименованию заболевания. Хорея, то есть пляска в переводе с греческого («choreia»). Это одна из форм гиперкинеза, которая характеризуется быстрыми, нерегулируемыми, непроизвольными движениями, возникающими в различных группах мышц.
Хорея Хантингтона симптомы и развитие заболевания
Как правило болезнь начинает проявляться у людей к 30-35-ти годам. Бывают и более ранние и более поздние проявления. Чем раньше начинают появляться симптомы, тем стремительнее происходит и ярче развитие болезни. Чем позже проявляется клиническая картина, тем течение болезни более лёгкое и длительное. В некоторых случаях первые признаки болезни Хантингтона появляются так поздно, что полное развитие клинической картины не успевает проявится в течении жизни пациента. В итоге больной умирает своей собственной смертью и до самого конца даже и не подозревает о наличии у него этого заболевания.
Начинается заболевание исподволь. Первые проявления хореи Хантингтона обычно являются физические нарушения не воспринимаются как заболевания. Человек проявляет неусидчивость, суетливость, беспокойство и прочие признаки, на которые не сильно обращают внимание. Однако с течением времени непроизвольная моторика нарастает и становится более заметной. У больного начинают проявляться резкие непроизвольные неконтролируемые движения конечностями. Иногда судорожные неритмичные движение не только конечностей, но и всего тела.
Иногда судорожные неритмичные движение не только конечностей, но и всего тела.
Также могут возникать спазмы мышц лица и тогда кажется, что человек гримасничает. Часто у детей это так и воспринимается, что ребёнок балуется и гримасничает. Из-за непроизвольных движений лицевой мускулатуры, порой больные всхлипывают, будто они долгое время неутешно рыдали. В дальнейшем нарушается артикуляция и координация движений. Человеку становится трудно глотать, говорить, развивается невнятная речь, начинаются бесконтрольные движения мышц глаз, что нарушает сон. В дальнейшем развивается танцующая (хореическая) походка. Нарастание симптомов приводит к инвалидности. В некоторых случаях движения становятся наоборот заторможенными.
Вообще симптомы эпилептического характера проявляются в физических нарушениях, расстройстве личности и уменьшение когнитивных способностей. При чём, последнее обычно проявляется под конец заболевания, а вот физические и личностные нарушения, как правило первыми. У некоторых людей первыми могут быть физические расстройства, как написано выше, а у другие — нарушения личности. В более тяжёлой форме может наблюдаться одновременное проявление физических и личностных расстройств.
Нарушение личности начинает обычно с развития депрессий, отчуждения, апатии, периодической расторможенности и раздражительности. Иногда первыми признаками заболевания становятся навязчивые состояния или бред. В большинстве случаев при таком начале ставят ошибочный диагноз шизофрении. Эпилептические припадки при Эпилептические встречаются достаточно редко.
Нарушение когнитивной функции обычно происходит под конец, на самых последних стадиях. Но встречаются и ранние расстройства, а ещё чаще некоторые из нарушений когнитивной функции начинают проявляться на первых стадиях. Как правило это проявляется нарушении мышления, внимания или расстройстве контроля исполнительной функции. Но обычно проявляется это в достаточно лёгкой форме и прогрессирует очень медленно, по сравнению с нарастанием физических симптомов.
Также у пациента на той или иной стадии развивается расстройство абстрактного мышления. Больной теряет способность к адекватной оценке своих действий, следовать правилам или планировать свои действия. Постепенно у больного развивается эмоциональный дефицит, паника, агрессия и эгоцентризм. Нередко у пациентов начинают развиваться навязчивые идеи, гиперсексуальность и усиливаться тяга к вредным привычкам (наркомания, алкоголизм, игромания и так далее). Возникает проблема с узнаванием знакомых ему людей.
Для постановки верного диагноза очень важна дифференциальная диагностика болезни Хантингтона, особенно на первых стадиях. Также очень важно выявлять группу риска и диагностировать больных до появления первых симптомов. Но главнее всего — это диагностировать заболевание ещё в период внутриутробного развития. Это связано с тем, что пока что нет какого-то более или менее эффективного лечения болезни Хантингтона. Вернее на сегодня лечения нет вообще. А знаю ещё в период беременности, что у ребёнка есть мутантный ген, родители могут принять решение о сохранении или прерывании такой беременности.
Другие статьи по неврологии:
Компания Teva и Исследовательская группа по изучению болезни Гентингтона опубликовали результаты клинического исследования препарата Аустeдо™ (деутетрабеназин*) для лечения болезни Гентингтона в журнале JAMA Neurology
Данные говорят о возможности быстрого перехода с тетрабеназина на Аустедо для лечения хореи, связанной с болезнью Гентингтона.
Компания Teva Pharmaceutical Industries Ltd. и Исследовательская группа по изучению болезни Гентингтона (Huntington Study Group) опубликовали результаты клинического открытого неконтролируемого когортного исследования III фазы под названием «Альтернативы для уменьшения хореи при болезни Гентингтона» (Alternatives for Reducing Chorea in HD, ARC-HD) в медицинском журнале JAMA Neurology. Исследование продемонстрировало, что пациенты с хореей при болезни Гентингтона, смогли безопасно перейти с троекратного приема тетрабеназина в течение суток на двукратный прием перорального препарата АУСТЕДО™ (деутетрабеназин). Исследование было проведено компаний Teva совместно с Исследовательской группой по изучению болезни Гентингтона.
Болезнь Гентингтона – редкое и прогрессирующее нейродегенеративное заболевание, которым только в США болеют около 35 тыс. человек. Хорея, проявляющаяся неконтролируемыми, беспорядочными и насильственными движения мышц рук, ног, туловища, лица и шеи, является одним из наиболее ярких внешних проявлений этого заболевания, встречающегося примерно у 90% пациентов. Болезнь Гентингтона также может проявляться нарушением когнитивных функций, поведенческими и психическими расстройствами.
«Хорея – тяжелый симптом болезни Гентингтона, который ставит под угрозу безопасность пациентов, ухудшает качество их жизни и их способность нормально функционировать, – говорит доктор медицинских наук Майкл Хайден, президент глобального подразделения по исследованиям и разработкам и директор по научной деятельности компании Teva. – Мы рады поделиться результатами исследования ARC-HD, чтобы врачи, лечащие пациентов с болезнью Гентингтона, смогли больше узнать про препарат АУСТЕДО™».
«Исследование с участием пациентов крайне важно для более глубокого понимания заболевания и разработки новых эффективных вариантов лечения, – отмечает доктор Сэмюэль Фрэнк, руководитель клинического исследования и глава общества по изучению болезни Гентингтона при медицинском центре «Бет-Исраэль». – Мы выражаем огромную благодарность пациентам и их близким, без активного участия которых исследование было бы невозможным».
В исследовании принимали участие 37 пациентов, которые до этого принимали тетрабеназин от 8 недель и дольше. Тетрабеназин был заменен на АУСТЕДО™, суточная доза которого примерно в два раза меньше, чем суточная дозы тетрабеназина. Спустя неделю организаторы исследования могли еженедельно вносить изменения в дозировку, добиваясь тем самым оптимального контроля симптомов хореи. Эффективность препарата оценивалась на основе отклонений от исходных показателей по Унифицированной шкале оценки болезни Гентингтона (United Huntington’s Disease Rating Scale, UHDRS), Шкале общей максимальной оценки хореи (Total Maximal Chorea Score, TMC) и Шкале общей оценки моторики (Total Motor Score, TMS).
Более подробную информацию о публикации в JAMA Neurology можно получить на сайте: http://jamanetwork.com/journals/jamaneurology/fullarticle/2643174.
Об АУСТЕДО™
АУСТЕДО™ является пероральным низкомолекулярным ингибитором везикулярного транспортера моноамина-2 (VMAT2), предназначенным для регулирования уровня нейротрансмиттеров в головном мозге. АУСТЕДО™ одобрен регулирующими органами США для лечения хореи при болезни Гентингтона.
О болезни Гентингтона
Болезнь (хорея) Гентингтона (БГ) – наследственное хроническое прогрессирующее нейродегенеративное заболевание с мультисистемным поражением головного мозга, характеризующееся неконтролируемыми, беспорядочными и насильственными движениями мышц рук, ног, туловища, лица, шеи в сочетании с когнитивными и психическими расстройствами. Симптомы болезни Гентингтона могут проявиться в любом возрасте, включая детей и пожилых, но чаще в возрасте 30-50 лет. Течение заболевания отличается особой тяжестью и фатальностью.
Противопоказания:
АУСТЕДО™ противопоказан пациентам с суицидальными наклонностями, депрессией и гепатитом; пациентам, принимающим ингибиторы моноаминоксидазы в течение 14 дней после последнего приема препарата; принимающим резерпин в течение 20 дней после последнего приема препарата, и принимающим тетрабеназин. Ингибиторы VMAT2, включая АУСТЕДО™, могут вызывать ухудшение настроения и внимания, а также самочувствия в целом. АУСТЕДО™ может повысить риск возникновения акатизии, тревоги и беспокойства, и может вызывать паркинсонизм у пациентов с болезнью Гентингтона. Препарат может накапливаться в тканях после длительного применения и вызывать сонливость, диарею, сухость во рту и повышенную утомляемость.
*Деутетрабеназин на сегодняшний день не зарегистрирован на территории РФ.
Пресс-релиз
Клинический слvчай детской формы болезни Гентингтона Текст научной статьи по специальности «Клиническая медицина»
русский журнал детской неврологии
НАБЛЮДЕНИЯ ИЗ ПРАКТИКИ
КЛИНИЧЕСКИЙ СЛУЧАЙ ДЕТСКОЙ ФОРМЫ БОЛЕЗНИ ГЕНТИНГТОНА
К.Ю. Мухин1-2, МД. Тысячина1-2, ДА Саввин3, С.В. Пилия3, А.С. Петрухин1
CHILDHOOD ONSET FORM OF HUNTINGTON’S DISEASE (A CLINICAL CASE)
K.Yu. Mukhin1-2, M.D. Tysyachina1-2, DA. Savvin3, S.K Piliya3,A.S. Petrukhin1
1 — Кафедра неврологии и нейрохирургии педиатрического факультета ГОУ ВПО РГМУ Росздрава
2 — Центр детской неврологии и эпилепсии, Москва
3 — Российская детская клиническая больница, Москва
Болезнь Гентингтона — аутосомно-доминантное нейродегенеративное заболевание, характеризующееся двигательныими нарушениями (хорея у взрослых и акинетико-ригидный синдром у детей), психическими расстройствами и деменцией с прогрессирующим течением и летальным исходом. В статье рассматриваются вопросы истории изучения заболевания, этиологии и патоморфологии болезни Гентингтона, особенности клинической картины, диагностики и лечения. Представлено описание собственного наблюдения детской формы болезни Гентингтона у девочки 14 лет. Дебют заболевания зарегистрирован в возрасте 7лет, отмечено типичное течение заболевания: начало с акинетико-ригидного синдрома с дальнейшим появлением хореического и атетоидного гиперкинеза,регресс психических функций. Выявлены характерные изменения на МРТ (атрофия коры головного мозга и подкорковых структур — оград) и ЭЭГ(резкое замедление основной активности фоновой записи). Диагноз верифицирован при молекуляр-но-генетическом исследовании. Несмотря на проводимую терапию агонистами дофамина, метаболическими и ноотропными препаратами, отмечалось неуклонное прогрессирование заболевания с ухудшением моторных и когнитивных функций.
Ключевые слова: болезнь Гентингтона, этиология, клиническая картина, диагностика, лечение.
Huntington’s disease is an autosomal dominant neurodegenerative disease, characterized by motor disturbances (chorea in adults and akineticorigid syndrome in children), mental disorders, and dementia with a progressive course and a lethal outcome. The article provides an overview of research, ethiology and pathogenesis of Huntington’s disease, specific features of its clinical presentation, diagnosis, and treatment. The authors report their own observation of the childhood onset form of Huntington’s disease in a 14-year-old girl. The onset was registered at the age of 7, with a typical course starting with akineticorigid syndrome and progressing to choreal and athetoid hyperkinesis and mental regression. Typical changes were identified in MRI (atrophy of the cerebral cortex and sub-cortial structures — claustrae) and EEG (a sharp decrease of background activity). The diagnosis was verified by a molecular genetic test. In spite of the treatment with dopamine agonists, metabolic and nootropic drugs, the disease was steadily progressive, with deterioration in motor and cognitive functions.
Key words: Huntington’s disease, ethiology, clinical presentation, diagnosis, treatment.
Болезнь Гентингтона (БГ) — аутосомно-доминантное нейродегенеративное заболевание, характеризующееся двигательными нарушениями (хорея у взрослых и акинетико-ригидный синдром у детей), психическими расстройствами и деменцией с прогрессирующим течением и летальным исходом. Синонимы: хорея Гентингтона, хроническая прогрессирующая хорея, дегенеративная хорея, дементная хорея, поздняя хорея, «танцующее безумие». Заболевание впервые было описано американским врачом George Summer Huntington (1850—1916) в 1872 году (рис. 1). Джордж Гентингтон
(Хантингтон) был врачом в третьем поколении и наблюдал больных наследственной хореей еще в студенческие годы, посещая консультации своего деда и отца. Впоследствии обобщив случаи этого заболевания, Гентингтон выступил с докладом в Медицинской Академии штата Огайо, 15 февраля 1872 года [24]. Описание докладчиком клиники неизвестного заболевания было встречено медицинской общественностью с интересом, и в этом же году доктор Гентингтон опубликовал статью в медико-хирургическом журнале Филадельфии [11]. Резюме этой статьи было сразу переведено на немецкий язык
© Мухин КЮ., Тысячина МД., Саввин ДА., Пилия СВ., Петрухин А.С., 2010. Клинический случай детской формы болезни Гентингтона. Рус. жур. дет. невр.: т. V, вып. 1, 2010.
русский журнал детской неврологии
ТОМ V ВЫПУСК 1 2010
Адольфом Куссмауэлем в Германии и Карлом Нотнагелем в Австрии. В этом же году (1872 г.) было предложено называть заболевание болезнью Гентингтона.
В России наиболее подробное описание БГ и анализ собственных наблюдений представил С.Н. Давиденков в 1932 году [1]. Первое описание БГ, по-видимому, было сделано профессором В.А. Муратовым в 1908 году, который также опубликовал фотографию пациентки [2].
Этиология. Наследование БГ происходит по аутосомно-доминантному типу с полной пенетрантностью. Казуистически редко встречаются спорадические случаи (вероятно, только в случаях дебюта после 20 лет). Ген гентингтин, отвечающий за развитие БГ, расположен на хромосоме 4р1б.3. В норме он содержит от 11 до 31—34 триплетных повторов [23]. В случае мутации наблюдается экспансия тринуклеотидных повторов (САО: цито-зин—аденин—гуанозин) гентингтина [14]. Разброс при болезни Гентингтона может составлять от 35 до 121 повтора [12, 22]. Количество повторов равное 42 или выше подтверждает диагноз БГ; при показателе 34—38 повторов возможно формирование «промежуточного аллеля» (дискутируется возможность субклинического носительства), что более характерно для спорадических случаев заболевания и передачи от матерей [7, 15]. Характерен феномен антиципации, когда в последующих поколениях наблюдается более ранний дебют БГ и более тяжелое течение. Прежде всего, это относится, к случаям передачи мутантного гена от отцов, когда количество тринуклеотидных повторов увеличивается и заболевание протекает более злокачественно [12]. При детской форме БГ количество повторов всегда более 50, а в большинстве случаев — более 80; при этом, в 90% случаев заболевание передается от отцов [14].
Функция гена гентингтина, как и патогенез заболевания, пока неизвестны. Полагают, что аллельный ген необходим для нормального эмбрионального разви-
Рис. 1. George Summer Huntington (1850—1916).
тия и нейрогенеза, причем его аминокислотная последовательность отличается от любого другого белка, выделенного до настоящего времени [3]. При БГ происходит прогрессирующая потеря ГАМКерги-ческих нейронов, энкефалинов и нейронов субстанции Р в полосатом теле и черной субстанции; при этом холинергичес-кие и соматостатинергические нейроны подкорковых ядер относительно сохранны [7]. Количество рецепторов к дофамину и ацетилхолину в стриатуме существенно снижается. Уменьшение количества N-метил-Б-аспартатных рецепторов в полосатом теле коррелирует с тяжестью заболевания. Предполагается также наличие дефекта окислительной цепи митохондрий [3]. При этом степень нарушения функционирования митохондрий модифицирует фенотип БГ [5].
Патоморфология. Выявляется выраженная атрофия скорлупы и головки хво-
статого ядра с обеих сторон [10, 17, 21]. Мозг на секции выглядит сморщенным и атрофичным, особенно лобные отделы. При гистологическом исследовании коры головного мозга отмечается потеря нейронов, преимущественно, 3-го слоя [14]. Также выражены атрофические изменения в бледном шаре. Характерно разрастание астроглии, особенно в хвостатом ядре и скорлупе; полосатое тело может быть относительно интактным [26]. Дегенеративные изменения различной степени затрагивают также таламус, ствол мозга, мозжечок (особенно при детской форме) и спинной мозг.
Распространенность. Распространенность БГ около 10:100.000, при этом в Западной Европе эта цифра составляет от 3 до 7 случаев на 100.000 населения, а в Северной Америке — 5—10 на 100.000 [9, 14]. Следует отметить, что распространенность БГ выше среди белого населения, а максимальная частота встречаемости БГ во многих поколениях обнаруживается в изолятах [1, 14, 20].
Клиническая картина. Дебют БГ обычно наблюдается в возрасте 35—40 лет; описаны отдельные случаи с началом после 70 лет. Примерно в 10% случаев начало заболевания наблюдается до 20 лет [4]. Общепринятым является деление на юношескую форму (дебют в возрасте 10—20 лет) и детскую форму (дебют от 3 до 9 лет). Последняя составляет не более 5% всех больных БГ и возникает обычно в случаях, когда больны отец и дед пробанда (феномен антиципации по мужской линии) [22]. Для всех форм характерна триада признаков: двигательные нарушения, психические расстройства и прогрессирующая деменция. Чаще наблюдаются все три симптомо-комплекса; однако они могут возникать и изолированно. Болезнь начинается с двигательных или психических нарушений; признаки деменции развиваются по мере прогрессирования патологического процесса.
При классическом течение БГ у взрос-
НАБЛЮДЕНИЯ ИЗ ПРАКТИКИ
лых первые признаки заболевания могут быть незначительными: двигательная неловкость, неуклюжесть, суетливость, нервозность, игнорирование повседневных обязанностей. В дальнейшем двигательные нарушения нарастают с появлением типичных для БГ хореических гиперкинезов, выраженность которых прогрессивно нарастает. Насильственные движения вовлекают всю произвольную мускулатуру: проксимальную, дистальную, аксиальную, лицевую. В большинстве случаев сначала вовлекается мимическая мускулатура: появляется гримасничанье, движения бровями. Затем присоединяются насильственные движения в конечностях: пожимание плечами, «игра» пальцами рук. Далее вовлекается аксиальная мускулатура и нижние конечности. Походка становится «танцующей» (хореической), при этом больные отбрасывают руки в стороны, приседают, подскакивают, раскачивают туловищем, кивают головой. Речь дизартрична, голос «мягкий», слова произносятся быстро, отрывисто и нечетко [14]. Характерно усиление гипер-кинезов при волнении и исчезновение во сне. В терминальной стадии выраженность гиперкинезов уменьшается, нарастает мышечная ригидность; присоединяется нарушение жевания и глотания; могут появиться генерализованные судорожные приступы.
Отличительной особенностью клинической картины БГ с дебютом до 20 лет является начало заболевания с акинети-ко-ригидного симптомокомплекса. Появляется замедленность и скованность движений, маскообразное лицо, «шаркающая» походка. Мышечный тонус дисто-ничен с тенденцией к повышению по пластическому типу; сухожильные рефлексы не изменены или слегка оживлены. Характерен статический тремор с частотой 4—5 в секунду. Нередко появляется мозжечковая симптоматика: атаксия, интенционный тремор, нистагм, дисси-нергия, скандированная речь. Развитие
ТОМ V ВЫПУСК 1 2010
хореических гиперкинез ов при детской форме БГ наблюдается крайне редко. Возможна транзиторная хорея при назначении детям с акинетико-ригидным синдромом препаратов — агонистов дофамина [14].
Психические нарушения при классической форме проявляются эмоциональной лабильностью, суетливостью, тревожностью; могут быть симптомы депрессии. Возможно появление слуховых и зрительных галлюцинаций, спутанности, бреда. При дебюте БГ в детском возрасте в начале заболевания отмечается гиперактивность, эмоциональная лабильность; нарастает негативизм; появляются трудности в учебе. В дальнейшем происходит остановка психического развития с последующей утратой психических навыков. При этом шизофреноподобная симптоматика и депрессия развиваются редко, и на первый план сразу выступают интеллектуально-мнестические нарушения [16, 27]. По мере прогрессирования заболевания у взрослых наблюдается прогрессивное нарушение высших психических функций с исходом в глубокую деменцию.
Отдельный вопрос для обсуждения БГ с дебютом до 20 лет — нередкое развитие эпилепсии. Эпилептические приступы констатируются у 30—50% больных БГ с дебютом до 20 лет и лишь у 2% при классической форме [3, 8, 22]. Эпилепсия возникает, в среднем, спустя 2 года с момента появления первых симптомов болезни Гентингтона. Характерны следующие виды эпилептических приступов: миоклонические и миоклонически-ас-татические, атипичные абсансы, генерализованные судорожные. По мнению Р. ОепШп и соавт. (2005), наиболее типичны для детей с хореей Гентингтона миоклонические приступы [8]. А1сагШ (1998) выделяет особый «миоклоничес-кий вариант» БГ у детей. Возможно развитие эпилептического статуса миокло-нических или генерализованных тони-ко-клонических приступов [4]. По данным ЭЭГ: основная активность фоновой
записи прогрессивно замедляется [19]-Эпилептиформная активность в большинстве случаев носит диффузный характер в виде коротких разрядов пик/полипик-волна, или длительных пик-волновых разрядов частотой около 3 Гц с бифронтальным преобладанием (ЭЭГ-паттерн атипичных абсансов). M.E. Landau и K.R. Cannard (2003) выявили эпилептиформную активность у 17 (74%) из 23 пациентов с ювенильной БГ, при этом в 9 случаях были выявлены генерализованные разряды (в т.ч. поли-спайк-волновые разряды у 6 пациентов), а у 8 оставшихся отмечались мультирегиональные разряды [13]- Также могут регистрироваться региональные пик-волновые разряды в затылочных отведениях, как при болезни Лафора [9], и периодическая ритмическая диффузная дельта-активность в фоне [25]- Нередко отмечается провокация эпилептиформ-ной активности ритмической фотостимуляцией.
Диагностика. Болезнь Гентингтона диагностируется на основании положительной семейной истории заболевания (почти в 100% случаев), характерной клинической картины (специфические хореические гиперкинезы у взрослых или акинетико-ригидный синдром у детей, психические расстройства, деменция), прогрессирующего течения заболевания и типичных изменений при МРТ исследовании (атрофия головки хвостатого ядра, скорлупы, стриатума, коры больших полушарий головного мозга с вент-рикуломегалией в виде «крыльев бабочки»). Однако точная верификация диагноза, как и установление доклинических случаев заболевания, возможны только при проведении молекулярно-генетичес-кого анализа.
Молекулярно-генетическое исследование при БГ проводится методом реакции полимеразных цепей с последующим анализом фрагментов в полиакри-ламидном геле при ДНК-тестировании клеток периферической крови. Необхо-
русский журнал детской неврологии
НАБЛЮДЕНИЯ ИЗ ПРАКТИКИ
димо определение тринуклеотидных повторов: количество повторов равное 42 или выше подтверждает диагноз БГ [18]. При количестве повторов равном 34—37 диагноз остается сомнительным («пограничный генотип») [7]. В настоящее время возможна пренатальная ДНК-диагностика БГ [9].
Дифференциальный диагноз БГ следует проводить с другими заболеваниями, проявляющимися хореей, психическими расстройствами (шизофрения, экзогенные психозы, депрессия), при-обретенным
сла-боумием (сениль-ная де-менция, болезнь Альцгей-мера и др.). В детском возрасте БГ дифференцируют с наследственно-дегенеративными заболеваниями, протекающими с акине-тико-ригидным синдромом.
Лечение и прогноз. Терапия БГ исключительно паллиативная. Лечение направлено, главным образом, на уменьшение выраженности хореических гиперкинезов (или акинети-ко-ригидного синдрома у детей), а также на «сглаживание» психических нарушений. В лечении хореи применяются
Рис. 2. Пациентка Г.Ю., 14 лет. Диагноз: болезнь Гентингтона, детская форма. Астенического телосложения, резко пониженного питания; левосторонний сколиоз в грудном отделе. Маскообразное лицо, скованность движений.
Рис. 3. Пациентка Г.Ю., 14 лет. Диагноз: болезнь Гентингтона, детская форма. Атетоидный гиперкинез в кистях рук.
нейролептики (оптимально — галопери-дол), бензодиазепины (клоназепам), амантадин, резерпин, тетрабеназин [7]. Отмечен временный эффект блокатора кальциевых каналов дилтиазема [14] и убихинона (коэнзим Q10), нормализующего уровень лактата в коре и стриатуме [4]. При акинетико-ригидной форме у детей и подростков применяются агонисты дофамина, а также бромокриптин (парло-дел). В случае развития эпилепсии применяются вальпроаты (конвулекс) или леве-тирацетам (кеппра) — при преобладании миоклонических приступов. Отмечен положительный эффект высоких доз пира-цетама (ноотропил) внутривенно капель-но при выраженном эпилептическом ми-оклонусе (т.н. «миоклоническая форма» БГ) [8].
Продолжительность заболевания различна, но в среднем составляет 15 лет при классической форме, около 10 лет — при ювенильной, и при детской форме — 4— 6 лет [7]. Летальность при БГ составляет 100%. В конечной стадии заболевания пациенты находятся в состоянии глубокой инвалидизации, как в результате резко выраженных гиперкинезов, так и вследствие тяжелых психических расстройств
ТОМ V ВЫПУСК 1 2010
и деменции, требуют постоянного ухода и наблюдения. У больных БГ повышена частота суицидов [6].
Приводим историю болезни пациентки с детской формой БГ.
Пациентка Г.Ю., 14 лет, проходила обследование и лечение в ПНО-1 Российской детской клинической больницы.
Жалобы на прогрессирующее нарушение походки и координации движений, нарушение речи, хаотичные подергивания конечностей, замедленность мышления, значительные сложности в усвоении школьного материала.
Анамнез заболевания. С 1,5 лет родители обратили внимание на незначительную неловкость при ходьбе, девочка часто падала. Также отмечалась задержка развития речи: до 3 лет ребенок говорил только отдельные слова, но после активных занятий с логопедом появилась фразовая речь. Отчетливые признаки заболевания появились в возрасте 7 лет: родители заметили прогрессирующее ухудшение походки, участились падения, появилась медлительность и скованность движений; речь стала нечеткой. С 10 лет появились хаотичные подергивания конечностей и мимической мускулатуры, которые усиливались во время сна. По данным обследований, проведенных по месту жительства, на МРТ головного мозга была выявлена умеренная кортикальная субатрофия с вентрикуломегалией и легкой асимметрией желудочков. На фоне проводимой терапии (церебролизин, эн-цефабол), положительной динамики не отмечалось.
Из анамнеза жизни: ребенок от 1-ой беременности, протекавшей на фоне анемии в течение всего периода вынашивания, а также гестоза в 3-м триместре. По данным УЗИ, была диагностирована задержка внутриутробного развития плода. Роды преждевременные на 34—35 неделе, закричала сразу. Вес при рождении — 2600 г. Из роддома выписана на 4 сутки. В раннем возрасте отмечалась повышен-
ная возбудимость (девочка часто плакала, плохо засыпала). Моторное развитие по возрасту: голову держит с 3 мес., сидит с 6 мес., ходит с 10 мес. Вакцинация проводилась по возрасту. В школу пошла в 7 лет, но учиться по общей образовательной программе не смогла; сейчас обучается на дому по индивидуальной программе.
Мать осмотрена неврологом; видимых нарушений не выявлено. Отцу девочки 42 года. Отец воспитывался в детском доме, его родственники не известны. Со слов матери, примерно 4 года назад у отца появились «странности поведения». Стал замкнутым, необщительным, временами — немотивированные страхи. Перестал общаться с друзьями и родственниками, сузился круг интересов. От обследования отец отказался.
При осмотре: Состояние тяжелое. Астенического телосложения, резко пониженного питания (рис. 2). Кожные покровы со множественными akne vulgaris, преимущественно, в области лица и груди. Отмечается левосторонний сколиоз в грудном отделе. Арахнодактилия.
В неврологическом статусе: в сознании, ориентирована в месте и времени, вялая, безынициативная, правильно отвечает на вопросы и выполняет инструкции, но после некоторого латентного периода. Объем движения глазных яблок ограничен во всех направлениях. Сходящееся косоглазие. Слабость мимической мускулатуры слева: не может надуть щеки, сглаженность левой носогуб-ной складки, опущение левого угла рта. Небная занавеска подвижна. Небный и глоточный рефлексы снижены. Дизартрия. Девиация языка вправо. В рефлек-торно-двигательной сфере: тонус мышц повышен по пластическому типу, сухожильные рефлексы торпидные, мышечная сила удовлетворительная. Умеренные диффузные мышечные атрофии с преобладанием в дистальных отделах конечностей. Продольно-поперечное плоскостопие, ротация левой стопы
кнутри. Спонтанный рефлекс Бабин-ского с обеих сторон, больше слева. В координаторной сфере: в позе Ромберга неустойчива. Выявляется статический и динамический тремор. При выполнении пальце-носовой и пальце-молоточ-ковой пробы периодически отмечается хореоатетоидный гиперкинез, гиперме-трия S>D. Атетоидные гиперкинезы в кистях рук (рис. 3). Походка атактичес-кая, «шаркающая», с пропульсией (туловище наклонено кпереди), ходит с поддержкой. Периодически выявляются миоклонические гиперкинезы мимических мышц. Нарушений со стороны чувствительной сферы и тазовых органов не выявлено. Брадикинезия, брадила-лия, брадипсихия. Объем навыков и знаний снижен.
Результаты обследования. МРТголовного мозга выявила повышение МР-сигнала в Т2-изображении и в режиме FLAIR билатерально от оград. Констатирована средней степени выраженности церебральная атрофия с умеренной вторичной вентрикуломегалией. По сравнению с предыдущей МРТ — некоторое нарастание атрофических явлений.
При проведении ЭЭГ основной ритм отсутствовал, регистрировалось резкое замедление основной активности фоновой записи с доминированием уплощенных медленных форм активности тета-дельта диапазона. Предъявление функциональных нагрузок (РФС и пробы с гипервентиляцией) не оказывало значимого влияния на биоэлектрическую активность. Эпилептиформная активность не зарегистрирована.
При проведении поверхностной ЭНМГ: нарушение надсегментарных влияний. Снижение функционального состояния n. ulnaris sin. по типу аксонопатии.
Были проведены исследования крови с целью исключения ряда генетических заболеваний: церулоплазмин — 27,7 (N=22—58 мг/дл), лактат и пируват — в пределах нормы; исключены Gm1- и Ош2-ганглиозидозы.
Т
Осмотр окулиста: Ои-гиперметропи-ческий астигматизм.
Психолог: результаты нейропсихологи-ческого тестирования выявляют существенное глобальное снижение интеллекта.oraa äeTcKoro BO3pacTa. — 2004. — M.: Me,o,HU,HHa. — C. 453—457.
4. Aicardi J. Diseases of the Nervous System in Children, 2-nd edn. — Cambridge., Mac Keith Press. — 1998. — P. 334— 336.
5. Arning L., Haghikia A., Taherzadeh-Fard E., Saft C., Andrich J., Pula B., Hoxtermann S., Wieczorek S., Akkad D.A., Perrech M., Gold R., Epplen J.T., Chan A. Mitochondrial haplogroup H correlates with ATP levels and age at onset in Huntington disease // J. Mol. Med. — 2010. Jan 29. [Epub ahead of print]
6. Draper B., Peisah C., Snowdon J., Brodaty H. Early dementia diagnosis and the risk of suicide and euthanasia // Alzheimers Dement. — 2010. — V. 6(1). — P. 75—82.
7. Fahn S. Huntington disease // In: Merries Neurology 10th edition / Eds.: L.P. Rowland. — Lippincott, Philadelphia. — 2000. — P. 659—662.
8. Genton P., Malafosse A., Moulard B., Rogel-Ortiz F., Dravet Ch., Bureau M., Roger J. Progressive myoclonus epilepsies // In: Epileptic syndromes in infancy, childhood and adolescence. 4-th edition with video / Eds. J. Roger, M. Bureau, Ch. Dravet, P. Genton, C.A. Tassinari, P. Wolf — France: John Libbey, 2005. — P. 441—465.
9. Harper P.S. The epidemiology of Huntington’s disease // Hum. Genet. — 1992. — V. 89. — P. 365—376.
10. Hobbs N.Z., Barnes J., Frost C., Henley S.M., Wild E.J., Macdonald K., Barker R.A., Scahill R.I., Fox N.C., Tabrizi S.J. Onset and Progression of Pathologic Atrophy in Huntington Disease: A Longitudinal MR Imaging Study // Am. J. Neuroradiol. 2010 Feb 11. [Epub ahead of print]
11. Huntington G.S. On Chorea // Medical and Surgical Reporter. — 1872. — V. 26. — P. 317—321.
12. Kieburtz K., MacDonald M., Shih C., Feigin A., Steinberg K., Bordwell K., Zimmerman C., Srinidhi J., Sotack J., Gusella J. et al. Trinucleotide repeat length and progression of illness in Huntington’s disease // J. Med. Genet.
— 1994. — V. 31. — P. 872—874.
13. Landau M.E., Cannard K.R. EEG characteristics in juvenile Huntington’s disease: a case report and review of the literature // Epileptic Disord. — 2003. — V. 5(3). — P. 145—148.
14. Lyon G., Kolodny E.H., Pastores G.M. Neurology of Hereditary Metabolic Diseases of Children // 3-d edition, McGraw-Hill, NY, 2006. — P. 281 — 284.
15. Myers R.H., MacDonald M., Koroshetz W.J., Duyao M.P., Ambrose C.M., Taylor S.A., Barnes G., Srinidhi J., Lin C.S., Whaley W.L. et al. De novo expansion of a CAG (n) repeat in sporadic Huntington’s disease // Nat. Genet. — 1993. — V. 5. — P. 168—173.
16. Naarding P., Kremer H.P.H., Zitman F.G. Huntington’s disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena // Eur. Psychiatry. — 2001. — V. 16. — P. 439—445.
17. Osborn AG., Blaser S.I., Salzman K.L. et al. Diagnostic imaging brain // Salt Lake City, AMIRSYS, 2004. — I-9. — P. 66—69.
18. Osborne J.P., Munson P., Burman D. Huntington’s chorea. Report of 3 cases and review of the literature // Arch. Dis. Child. — 1982. — V. 57(2). — P. 99—103.
19. Painold A., Anderer P., Hodl A.K. h coaBT. EEG-mapping in patients with Huntington’s disease // Acta Neuropsychiatrica. — 2008. — V. 20. — Suppl. 1. — P. 69—70.
20. Penney J.B.Jr., Young A.B., Shoulson I., Starosta-Rubenstein S., Snodgrass S.R., Sanchez-Ramos J., Ramos-Arroyo M., Gomez F., Penchaszadeh G., Alvir J. et al. Huntington’s disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals // Mov. Disord. — 1990. — V. 5. — P. 93—99.
21. Ross C.A., Shoulson I. Huntington disease: pathogenesis, biomarkers, and approaches to experimental therapeutics // Parkinsonism Relat. Disord. — 2009. — V. 15 (Suppl. 3). — P. 135—138.
22. Ruocco H.H., Lopes-Cendes I., Laurito T.L., Li L.M., Cendes F. Clinical presentation of juvenile Huntington disease // Arq. Neuro-Psiquiatr. — 2006. — V. 64 (1). — P. 5—9.
23. Sanchez A, Mila M, Castellvi-Bel S, Calopa M, Genis D, Jimenez D, Estivill X. Molecular analysis of the IT15 gene in 79 Spanish families with Huntington’s disease: diagnostic confirmation and presymptomatic diagnosis // Med. Clin. (Barc). — 1997. — V. 108 (18). — P. 687—90.
24. Stevenson C.S. A biography of George Huntington, M.D. // Bulletin of the history of medicine, Baltimore, 1934.
— V. 2. — P. 53.
25. Ullrich N.J., Riviello J.J.Jr., Darras B.T., Donner E.J. Electroencephalographic correlate of juvenile Huntington’s disease // J. Child Neurol. — 2004. — V. 19. — P. 541 — 543.
26. Vonsattel J.P., Myers R., Stevens T. et al. Neuropathological classification of Huntington’s disease // J. Neuropathol. Exp. Neurol. — 1985. — V. 44. — P. 559—577.
27. Zdzienicka E., Rakowicz M., Mierzewska H. et al. Clinical and genetic study of juvenile form of Huntington’s disease // Neurol. Neurochir. Pol. — 2002. — V. 36 (2). — P. 245—258.
Болезнь Хантингтона
Хорея – болезнь, одинаково часто поражающая мужчин и женщин, очень часты семейные случаи болезни.
Причины
Предполагают, что в основе заболевания лежат грубые нарушения окислительного метаболизма. Обнаруживают дегенеративные изменения в коре больших полушарий, подкорковых узлах. В области черного вещества выявляют повышенное содержание железа, в эритроцитах повышен уровень магния.
Симптомы
Симптомы чаще всего появляются в возрасте старше 25 лет. Выделяют два важнейших признака: гиперкинетический синдром и изменения психики.
В основном это хореиформные гиперкинезы – быстрые, неритмичные и некоординированные сокращения различных больших мышечных групп туловища, верхних и нижних конечностей, лица. Независимо от желания заболевшего происходит нахмуривание бровей, лба, высовывается язык, совершаются беспорядочные движения руками и ногами. В отличие от ревматической хореи болезнь Хантингтона проявляется менее быстрыми гиперкинезами, и больные могут отдельные насильственные движения задержать по своей воле.
В некоторых случаях при данной патологии к хореическому гиперкинезу прибавляются проявления атетоза (медленные, червеобразные, вычурные движения в кистях и стопах, выпячивание губ, гримасничанье, прищелкивание языком, преходящие контрактуры кистей). С развитием патологического процесса расстраивается походка. Меняется речь, слова произносятся медленно, ощущение «комка во рту», речь нарушают периодические гиперкинезы артикуляционных мышц. Почерк оказывается неразборчивым, в дальнейшем больной вообще не может писать. Отмечаются эпилептиформные припадки. В разной степени нарушается интеллект.
Могут быть парестезии. Болезнь хорея Гентингтона, поражая нервную систему, проявляется не только гиперкинезами, но и разнообразными расстройствами вегетативной нервной системы, наблюдается ненасытность в поглощении пищи.
Хорея Гентингтона может проявляться следующим образом: симптомы диссоциируют, то есть у одних больных проявляются гиперкинезы, а интеллектуальных нарушений нет, у других наоборот, развивается слабоумие без гиперкинезов.
При развернутой картине заболевания, в ее завершающей стадии хореический гиперкинез ослабевает и может полностью исчезнуть, вместо него появляется экстрапирамидная ригидность – нарастает затруднение в движениях, скованность, повышается тонус мышц. В отдельных случаях болезнь Гентингтона изначально протекает в форме акинетико-ригидного синдрома.
У большого количества больных наблюдается расстройства психики. При данном заболевании легкие психопатические расстройства отмечаются раньше хореических гиперкинезов. Появляются признаки умственной ущербности, человек становится психопатом. Хореическая деменция – это снижение памяти, интеллекта, ослабление внимания. Эти симптомы нарастают постепенно. И если вначале могут отмечаться повышенное возбуждение и раздражительность, то впоследствии психическая активность угнетена, смягчаются истеричные черты и шизоидные проявления.
Наблюдения выявили, что здоровые родственники больных хореей Гентингтона могут иметь психопатические черты такие же, как и у больных хореей Гентингтона. Следовательно, присутствие таких изменений психики не всегда можно рассматривать как признак возможного развития заболевания в будущем. Считать их первыми очень ранними признаки развивающейся болезни можно лишь в том случае, если психопатия сильно выражена и сочетается с нарушением речедвигательных анализаторов, в других случаях ее симптомы расцениваются как проявление психопатии вследствие отягощенной наследственности.
Несмотря на то, что атрофические процессы, лежащие в основе слабоумия, имеют при различных патологических состояниях общие черты, деменция при хорее Гентингтона имеет свои особенности. Изменения психики нарастают медленно, поэтому способность к интеллектуальной деятельности длительно сохраняется, и больные длительно остаются трудоспособными к выполнению привычной и простой работы. Выполнение же творческих заданий становится непосильным для больного. Сохранение работоспособности и невыраженные психопатические расстройства приводят к тому, что больные попадают на лечение в поздние сроки, когда болезнь уже в определенной степени развилась. Умственная работоспособность колеблется, то повышаясь, то вновь снижаясь. В различной степени выражено расстройство памяти, однако память постепенно все-таки значительно снижается, но таких выраженных изменений, как при других атрофических процессах мозга не происходит.
Способность к самообслуживанию и интеллектуальной деятельности длительно сохраняется, так как гиперкинезы и деменция нарастают постепенно.
В начальные периоды развития заболевания отмечаются депрессивные состояния. Больные мрачные и угрюмые, злобные и раздраженные. Хорею Гентингтона от других атрофических процессов отличает возможное развитие псевдопаралитических психозов, больные бредят всемогуществом, представляют себя богатыми, их речь неумолкает, поведение отличается экспансивностью, все это выглядит нелепо. При такой клинической картине прогноз неблагоприятный. Быстро развивается глубокая деменция с тупой эйфорией. Отмечаются внезапно наступающие состояния спутанности.
Диагностика
При наличии отягощенной наследственности, т.е. наличии в роду больных, имеющих хорею Гентингтона, и появлении характерных признаков диагноз устанавливают без затруднений. В других случаях диагностика крайне затруднена.
Дифференцируют болезнь Гентингтона с заболеваниями, имеющими сходные проявления. Развитие церебрального атеросклероза обуславливает развитие старческой хореи. При гепатоцеребральной дистрофии в анализе крови отмечается снижение церулоплазмина, болезнь сопровождается наличием на роговице кольца Кайзера-Флейшера (отложение меди). Хореический гиперкинез надо дифференцировать с опухолями мозга, энцефалитами, от ревматической хореи, психических заболеваний.
Для оценки деятельности мозга проводят ЭЭГ, биоэлектрическая активность мозга снижена, альфа-ритм исчезает, присутствуют медленные волны.
Лечение
Терапия медикаментозными препаратами дает кратковременное улучшение, затем хорея Гентингтона продолжает прогрессировать. Лечение проводится препаратами, понижающими мышечный тонус, седативными и общеукрепляющими. Уменьшает гиперкинезы прием транквилизаторов – седуксен, элениум. По назначению врача с этой целью применяются артане, циклодол, леводопа. Систематически принимаются внутрь витамины группы В, глутаминовая кислота.
При психических нарушениях проводится стационарное лечение в специализированном учреждении. Ослабить гиперкинезы в некоторых случаях может оперативное лечение.
Болезнь Хантингтона — лечение в клинике Мэйо
Лечение болезни Хантингтона в клинике Мэйо
Ваша бригада по уходу в клинике Мэйо
Люди с болезнью Хантингтона будут получать помощь от врачей клиники Мэйо, прошедших подготовку в области клинической геномики, неврологии, физической медицины и реабилитации, а также психиатрии, психологии и других медицинских специальностей.
Педиатрическая экспертиза
Врачи, прошедшие специальную подготовку в области детской неврологии, работают с другими специалистами по уходу за детьми с болезнью Хантингтона в детской больнице Майо Эухенио Литта в клинике Майо в Рочестере, штат Миннесота.Специалисты по детскому образу жизни помогают вам и вашему ребенку справиться с естественными опасениями по поводу болезни, медицинских процедур и больничного ухода.
Поддерживающая терапия
Хотя болезнь Хантингтона неизлечима, врачи клиники Мэйо помогут вам и вашей семье справиться с вашим заболеванием. Персонал клиники Мэйо также предоставляет ресурсы для вас и вашей семьи, чтобы помочь вам справиться с вашим заболеванием, и у вас может быть возможность участвовать в группах поддержки.
Опыт и рейтинги
Опыт
Врачи клиники Мэйо обладают знаниями и опытом в оценке и лечении людей с болезнью Гентингтона и другими нервными (неврологическими) состояниями.Врачи, специализирующиеся на двигательных расстройствах, ежегодно проводят обследование и лечат около 65 человек с болезнью Гентингтона.
Research
Сотрудники клиники Mayo из отделения детской и подростковой неврологии изучают причины, прогрессирование, генетику и варианты лечения болезни Хантингтона. Эксперты отдела клинической геномики клиники Мэйо предоставляют доступ к новейшим методам генетического тестирования, когда это необходимо с медицинской точки зрения. Исследователи Mayo также проводят клинические испытания.
Национально признанный опыт
Клиника
Mayo в Рочестере, штат Миннесота, и клиника Mayo в Джексонвилле, штат Флорида, вошли в число лучших больниц для неврологии и нейрохирургии по версии U.S. News & World Report. Клиника Мэйо в Фениксе / Скоттсдейле, штат Аризона, получила высокую оценку в области неврологии и нейрохирургии по версии U.S. News & World Report. Клиника Мэйо также входит в число лучших детских больниц по неврологии и нейрохирургии.
Узнайте больше об опыте и рейтингах неврологического отделения клиники Мэйо.
Расположение, поездки и проживание
Mayo Clinic имеет крупные кампусы в Фениксе и Скоттсдейле, штат Аризона; Джексонвилл, Флорида; и Рочестер, Миннесота. Система здравоохранения клиники Мэйо имеет десятки отделений в нескольких штатах.
Для получения дополнительной информации о посещении клиники Mayo выберите свое местоположение ниже:
Расходы и страхование
Mayo Clinic работает с сотнями страховых компаний и является поставщиком услуг внутри сети для миллионов людей.
В большинстве случаев клиника Мэйо не требует направления к врачу. Некоторые страховщики требуют направления или могут иметь дополнительные требования для определенного медицинского обслуживания. Приоритет всех посещений определяется медицинской потребностью.
Узнайте больше о приемах в клинику Мэйо.
Пожалуйста, свяжитесь со своей страховой компанией, чтобы подтвердить медицинское страхование и получить необходимое разрешение до вашего визита. Часто номер службы поддержки вашего страховщика напечатан на обратной стороне вашей страховой карты.
Дополнительная информация о выставлении счетов и страховании:
Клиника Майо в Аризоне, Флориде и Миннесоте
Система здравоохранения клиники Мэйо
одобренных методов лечения болезни Хантингтона
Ученым еще предстоит разработать лекарство от болезни Гентингтона или лечение, которое могло бы остановить ее прогрессирование. Но есть методы лечения, которые могут помочь пациентам справиться с симптомами, включая двигательные, когнитивные и психиатрические проблемы.
И исследователи продолжают работать над более эффективными методами лечения болезни.
Лечение проблем с движением
Признак Хантингтона — проблемы с мышцами. К ним относятся мышечные сокращения, проблемы с произвольными движениями, такими как ходьба и речь, а также резкие непроизвольные движения, известные под общим названием хорея, наиболее заметный симптом болезни.
Ксеназин (тетрабеназин) — первое лекарство, одобренное Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США специально для лечения Хантингтона.Он помогает подавить резкие непроизвольные движения, но может вызвать серьезные побочные эффекты, такие как усугубление депрессии.
Антипсихотические препараты, такие как риспердал (рисперидон), галдол (галоперидол) и торазин (хлорпромазин), часто используются не по назначению для лечения болезни Гентингтона, поскольку они подавляют хорею. Терапия не по назначению — это терапия, которая помогает пациентам с состояниями, для которых препарат не был одобрен. Хотя это законно, фармацевтические компании не могут продавать лекарства для использования не по назначению.
Помимо подавления хореи, антипсихотические препараты могут помочь справиться с психиатрическими симптомами, связанными с болезнью Хантингтона, включая возбуждение и психоз. Побочные эффекты нейролептиков включают сонливость и сухость во рту. Галдол и хлорпромазин также могут усиливать непроизвольные сокращения мышц и их ригидность.
Бензодиазепины также могут уменьшить хорею. К ним относятся клонопин (клоназепам) и валиум (диазепам). Однако некоторые из этих лекарств могут ухудшить когнитивные симптомы болезни Хантингтона и могут нести высокий риск того, что пациент станет зависимым от них и будет злоупотреблять ими.
Лечение психических расстройств
Неврологический ущерб, связанный с болезнью Хантингтона, может также вызывать такие психиатрические симптомы, как депрессия, обсессивно-компульсивное расстройство, мания и биполярное расстройство.
Антидепрессанты, в основном селективные ингибиторы обратного захвата серотонина, такие как Lexapro, Prozac и Zoloft, часто используются для лечения депрессии пациентов Хантингтона. Они также могут облегчить некоторые симптомы обсессивно-компульсивного расстройства.
Врачи используют антипсихотические препараты, такие как Сероквель (кветиапин) и Риспердал (рисперидон), для лечения расстройств настроения, включая вспышки насилия или бред.
Лекарства, стабилизирующие настроение, могут предотвратить резкие взлеты и падения, характерные для биполярного расстройства. К ним относятся Карбатрол (карбамазепин), Депакон (вальпроат) и Ламиктал (ламотриджин).
Другие методы лечения
Другие виды терапии, помимо лекарств, могут облегчить симптомы Хантингтона и помочь пациентам справиться с ними.
Физиотерапия помогает поддерживать подвижность и сокращает количество падений.
Хантингтон может поражать мышцы рта и горла, поэтому речь и глотание затрудняются по мере прогрессирования болезни.Когда это происходит, может помочь логопед.
Психотерапия может помочь пациентам и их семьям разработать стратегии преодоления болезни и ее прогрессирования. Трудотерапия может помочь пациентам научиться пользоваться устройствами, которые помогают им выполнять повседневные задачи, такие как одевание и прием пищи.
Поддержка
Болезнь Хантингтона — серьезная проблема, и пациенты и лица, осуществляющие уход, нуждаются в поддержке. Некоммерческие группы, такие как Общество борьбы с болезнями Хантингтона в Америке, Общество Хантингтона в Канаде и Ассоциация по борьбе с болезнями Хантингтона в Соединенном Королевстве, являются хорошим источником информации.Они могут определить социальные услуги, доступные пациентам и их семьям, и помочь им найти группы поддержки.
***
Huntington’s Disease News — это исключительно новостной и информационный веб-сайт об этой болезни. Он не предоставляет медицинских консультаций, диагностики или лечения. Этот контент не предназначен для замены профессиональных медицинских консультаций, диагностики или лечения. Всегда обращайтесь за советом к своему врачу или другому квалифицированному поставщику медицинских услуг по любым вопросам, которые могут у вас возникнуть относительно состояния здоровья.Никогда не пренебрегайте профессиональным медицинским советом и не откладывайте его обращение из-за того, что вы прочитали на этом веб-сайте.
Болезнь Хантингтона — Лечение и поддержка
В настоящее время нет лекарства от болезни Хантингтона или какого-либо способа остановить ее ухудшение. Но лечение и поддержка могут помочь уменьшить некоторые проблемы, вызванные этим заболеванием.
Во многих районах есть клиники по лечению болезни Хантингтона, которыми руководят врач-специалист и медсестра, которые могут предложить лечение и поддержку и при необходимости направить вас к другим специалистам.
Исследования новых методов лечения продолжаются, и в последнее время были получены некоторые многообещающие результаты.
Лекарства
Лекарства могут помочь уменьшить некоторые проблемы, вызванные болезнью Гентингтона, но они не останавливают и не замедляют развитие болезни.
Сюда входят:
Некоторые из этих лекарств не лицензированы для лечения болезни Хантингтона, но было установлено, что они помогают облегчить симптомы.
Большинство этих лекарств могут вызывать неприятные побочные эффекты.Поговорите со своим врачом о возможных преимуществах и рисках их приема.
Помощь в повседневных делах
Ежедневные задания, такие как одевание, передвижение по дому и прием пищи, могут утомлять и утомлять, если у вас болезнь Хантингтона.
Эрготерапевт может взглянуть на действия, которые кажутся вам трудными, и посмотреть, есть ли другой способ их выполнения.
Они также могут порекомендовать изменения, которые можно внести в ваш дом, и оборудование, которое вы можете использовать, чтобы упростить вам жизнь.
Сюда могут входить:
- установка пандусов для доступа инвалидной коляски
- установка лестничного подъемника
- установка поручней — например, у лестницы или у кровати
- с использованием электрических консервных ножей, электрических зубных щеток и кухонных принадлежностей с большими ручками, которые легче удерживать
- фонари с голосовым управлением или программное обеспечение с голосовым управлением на компьютере
Узнайте больше о том, как трудотерапия может помочь и как ее получить.
Помощь с едой и общением
Логопед и диетолог могут помочь, если из-за болезни Хантингтона вам трудно общаться и есть.
Например, они могут проконсультировать насчет:
- альтернативные способы общения — например, электронные речевые устройства или графические карты
- высококалорийная диета для предотвращения потери веса
- способов облегчить пережевывание и глотание пищи
В какой-то момент может потребоваться зонд для кормления, который вводится прямо в желудок.
Если вы не хотите, чтобы вас кормили таким образом, вы можете подумать о том, чтобы заранее принять решение, в котором излагается, как вы хотели бы, чтобы о вас заботились на более поздних стадиях вашего состояния.
Помощь при проблемах с движением и равновесием
Если у вас болезнь Гентингтона, важно стараться оставаться как можно более активным. Это поможет вам почувствовать себя лучше как физически, так и морально.
Передвижение может быть затруднено, если у вас проблемы с координацией и равновесием, но даже регулярная ходьба с использованием вспомогательных средств, таких как трости, может быть полезной.
Физиотерапевт также может помочь с проблемами движения.
Они могут порекомендовать такие вещи, как:
- план учений
- движение и растяжка суставов (манипуляция)
- массажа
Узнайте больше о том, как физиотерапия может помочь и как ее получить.
Исследования новых методов лечения
В настоящее время ведутся исследования по поиску новых методов лечения болезни Хантингтона.
Достигнут прогресс в выявлении возможных способов замедления или остановки состояния путем «выключения» неисправного гена, который его вызывает.
Несколько препаратов сейчас проходят клинические испытания. Если они окажутся безопасными и эффективными, они могут появиться в продаже через несколько лет.
Вы можете узнать больше, посетив European Huntington’s Disease Network, HD Buzz и Huntington’s Disease Association. Вы также можете спросить своего врача-специалиста или медсестру.
Подробнее об уходе и поддержке
Ассоциация по борьбе с болезнью Хантингтона имеет дополнительную информацию о том, как получить помощь при болезни Гентингтона, в том числе советы по:
- поведенческие проблемы
- коммуникативные навыки
- сексуальные проблемы
- диета, питание и глотание
- сиденья, оборудование и приспособления
- варианты, когда требуется постоянный уход
- пособия, на которое вы можете иметь право
Вы также можете прочитать руководство NHS по адресу:
Вождение автомобиля
Вы должны обсудить любые опасения по поводу вождения со своим врачом.
Если у вас есть водительские права и у вас есть симптомы, вызванные болезнью Хантингтона, вы по закону обязаны обратиться в DVLA.
DVLA запросит у вас данные вашего врача для получения дополнительной информации. Многим людям все еще разрешено водить машину, но это будет регулярно пересматриваться.
Нет необходимости связываться с DVLA, если у вас не появились симптомы. В случае сомнений обсудите это со своим врачом.
Узнайте больше о вождении автомобиля с ограниченными физическими возможностями
Последняя проверка страницы: 13 февраля 2018 г.
Срок следующей проверки: 13 февраля 2021 г.
Лечение болезни Хантингтона
Многие агенты и хирургические процедуры были оценены при HD на предмет их эффективности в подавлении хореи, включая дофамин-истощающие агенты, антагонисты дофамина, бензодиазепины, антагонисты глутамата, ингибиторы ацетилхолинэстеразы, агонисты дофамина, антисейциниловые препараты. , глубокая стимуляция мозга и трансплантация клеток плода [45–48].Фармакологические вмешательства обычно направлены на лечение гиперкинетических двигательных расстройств, которые могут быть связаны с HD, таких как хорея, дистония, баллизм, миоклонус и тики. При выборе вмешательства медицинские работники должны учитывать положительное или отрицательное влияние агента на психиатрические проблемы, связанные с HD, такие как раздражительность, депрессия, тревога, мания, апатия, обсессивно-компульсивное расстройство или снижение когнитивных функций. Дополнительные методы лечения, альтернативные и дополнительные методы лечения, поведенческие планы и когнитивные вмешательства также могут играть роль в устранении симптомов HD, и их необходимо учитывать при выборе лекарств.
Хорея
В нескольких обзорах обобщено симптоматическое лечение хореи, ассоциированной с HD [49–61]. В целом, нет достаточных данных для руководства долгосрочным симптоматическим лечением HD, и необходимы двойные слепые и долгосрочные исследования, оценивающие различные стратегии лечения HD [55]. Несмотря на отсутствие доказательств, недавно была выпущена публикация Руководства Американской академии неврологии, в которой рекомендуется рассмотреть возможность применения тетрабеназина (TBZ), амантадина или рилузола, если хорея требует лечения [46].В Кокрановском обзоре исследований симптоматического лечения БХ было изучено 22 испытания, в которых приняли участие 1254 различных участника [56]. Девять испытаний имели кроссоверную конструкцию, а 13 были проведены параллельно. Изученные исследования были относительно короткими — от 2 до 80 недель. В число испытаний, в которых изучались различные фармакологические вмешательства, входили антидофаминергические препараты (n = 5), антагонисты рецепторов глутамата (n = 5) и энергетические метаболиты (n = 5). Основываясь на имеющихся данных, авторы Кокрановского обзора пришли к выводу, что только TBZ продемонстрировал явную эффективность для контроля хореи, но «нельзя сделать никаких заявлений относительно наилучшей медицинской практики для контроля моторных и немоторных симптомов при HD».
TBZ — единственный препарат для лечения HD, одобренный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США и предназначенный для лечения хореи, связанной с HD. В предыдущих публикациях был дан обзор химии, фармакодинамики, фармакокинетики и механизма действия [47]. Есть недавний обзор разработки и использования тетрабеназина [62]. Обратимо ингибируя центральный везикулярный переносчик моноаминов типа 2, TBZ более избирательно истощает дофамин, чем норадреналин [48, 63]. Самая высокая плотность связывания TBZ находится в хвостатом ядре, скорлупе и прилежащем ядре, областях, которые, как известно, наиболее подвержены HD [64, 65].Связывание везикулярного транспортера моноаминов типа 2 и истощение моноаминов под действием TBZ являются обратимыми, в последние часы и не изменяются длительным лечением [66, 67].
Эффективность TBZ как противохореического препарата была убедительно продемонстрирована в двойном слепом плацебо-контролируемом исследовании, проведенном Huntington Study Group [68]. TBZ титровали еженедельно с шагом 12,5 мг до максимальной 100 мг / день или до развития невыносимых побочных эффектов. По сравнению с исходным уровнем, лечение TBZ привело к снижению на 5 баллов.0 Единая шкала оценки болезни Хантингтона (UHDRS) общее максимальное количество единиц хореи по сравнению со снижением на 1,5 единицы в группе плацебо. Есть также данные, свидетельствующие о постоянной долгосрочной эффективности и переносимости TBZ у пациентов с HD [69–72].
В том же исследовании нежелательные явления, которые происходили значительно чаще в группе TBZ, включали сонливость или сонливость, бессонницу, подавленное настроение, возбуждение, акатизию и гиперкинез. Однако к завершению поддерживающей фазы, когда пациенты предположительно находились на оптимальной дозировке, не было значительных различий между TBZ и плацебо.Возможные побочные эффекты включают акатизию, депрессию, головокружение, усталость или паркинсонизм. Во время титрования пациенты могут испытывать бессонницу, сонливость или желудочно-кишечные расстройства.
В двойном слепом исследовании было совершено одно самоубийство у субъекта, принимавшего TBZ. Депрессия часто встречается при ГБ и может усугубляться ТБЗ. Однако попытки или завершенные самоубийства при HD не обязательно коррелируют с тяжестью депрессии, и необходимо учитывать другие связанные факторы, такие как импульсивность, обсессивно-компульсивное поведение и социально-экономические факторы.Тем не менее, все пациенты, принимающие TBZ, должны находиться под наблюдением на предмет наличия признаков депрессии и суицидальных мыслей. В последующем открытом расширенном исследовании не сообщалось о новых побочных эффектах, и препарат хорошо переносился при средней постоянной общей суточной дозе 62,5 мг.
При использовании TBZ в дозах выше 50 мг / день во вкладыше к препарату ксеназин говорится, что «пациенты, которым требуются дозы выше 50 мг в день, должны быть генотипированы по ферменту, метаболизирующему лекарство, CYP2D6, чтобы определить, является ли пациент слабым метаболизатором или обширным метаболизатор »[73].Однако клинически нет необходимости рассматривать генотипирование, за исключением необходимости более длительного титрования сверхбыстрых метаболизаторов, и нет никаких отличительных черт у пациентов с различными генотипами CYP2D6 [74].
Другие лекарства, которые обычно используются при лечении хореи, включают антагонисты дофамина, бензодиазепины и антагонисты глутамата. Антагонисты дофамина (нейролептики), возможно, являются наиболее часто рассматриваемыми агентами при лечении хореи и психоза у пациентов с HD, но было опубликовано мало двойных слепых плацебо-контролируемых исследований, оценивающих эффективность и безопасность этих агентов [75–77]. .В плацебо-контролируемых исследованиях не было обнаружено, что ни один из типичных нейролептиков эффективен для уменьшения хореи. Однако в исследовании галоперидола у 10 пациентов пероральные дозы 1,5–10,0 мг / день соответствовали как минимум 30% снижению хореи по сравнению с исходным уровнем [78]. Количество и качество этих данных об эффективности необходимо учитывать при рассмотрении рисков использования типичных нейролептиков, особенно поздней дискинезии. Апатия и акатизия, другие потенциальные побочные эффекты блокаторов дофаминовых рецепторов, могут быть особенно проблематичными у пациентов с HD, поскольку они могут не иметь понимания, чтобы распознать эти проблемы, или могут ошибочно приписать симптомы HD.
В связи с предполагаемой улучшенной переносимостью атипичные нейролептики недавно были протестированы при БХ. Оланзапин использовался в небольших открытых исследованиях для лечения двигательных симптомов HD [79–83]. Диапазон воздействия на хорею сократился на 0–66%. Клинических испытаний рисперидона при БХ не проводилось, но в нескольких сообщениях предполагается положительное влияние на заболевание с допустимым профилем побочных эффектов [84–87]. Кветиапин был опробован в нескольких небольших неконтролируемых нерандомизированных исследованиях БХ с некоторым успехом как в отношении моторных, так и психиатрических симптомов БХ [88–90].Клозапин изучался у пациентов с HD до дозы 150 мг / день в течение до 31 дня без пользы и значительных побочных эффектов, включая сонливость, утомляемость, антихолинергические симптомы и трудности при ходьбе [91]. Более новый атипичный агент с множественными механизмами действия, арипипразол, оказался полезным в нескольких небольших испытаниях с уменьшением хореи, эквивалентным таковому с TBZ [92–94]. Хотя арипипразол может иметь меньше побочных эффектов на настроение, чем TBZ, он может быть связан с акатизией и поздней дискинезией, как и другие типичные и атипичные нейролептики.
В контролируемых испытаниях было показано, что антагонист N-метил-D-аспартата амантадин значительно снижает хорею у пациентов с HD [95]. Для улучшения симптоматики могут потребоваться дозы в диапазоне 400 мг / день или выше, но даже в дозах, используемых для лечения гриппа, амантадин может усиливать раздражительность и агрессивность [96]. Поскольку рилузол замедляет высвобождение глутамата в полосатом теле и патологические последствия в нейротоксических моделях HD на животных, было проведено несколько крупных испытаний, чтобы определить, существует ли возможный нейрозащитный эффект.Хотя явного нейропротективного эффекта не было обнаружено, рилузол значительно уменьшал хорею в дозе 200 мг / день, но не 100 мг / день [97, 98]. Бензодиазепины также часто используются клинически у пациентов с HD для лечения тревоги и хореи, но есть ограниченные данные, позволяющие предположить, что для подавления хореи могут потребоваться более высокие дозы клоназепама (до 5,5 мг / день) [99]. В этой дозе седативный эффект становится потенциальным побочным эффектом. В нескольких краткосрочных исследованиях (часы) есть доказательства того, что агонисты дофамина могут уменьшать двигательные признаки БХ, расплывчато определяемые как «ненормальные непроизвольные движения» в 2 исследованиях, а также снижение общего моторного балла UHDRS и аномальных непроизвольных движений. шкала движений (AIMS) с инфузией апоморфина [100–102].Хотя фармакологическое обоснование использования агонистов дофамина при лечении хореи неясно, предположительно они действуют, активируя пресинаптические рецепторы дофамина, что приводит к снижению обмена дофамина. Однако более вероятно, что наблюдаемые симптоматические эффекты связаны с неспецифическими или седативными эффектами.
Цель любого лечения по уменьшению хореи должна быть обсуждена с пациентом и его семьей. Следует уменьшить хорею до уровня, приемлемого для пациента, а не для полного устранения хореи или уменьшения хореи до уровня, приемлемого для лица, осуществляющего уход, или даже для врача, лечащего пациента.Поскольку HD является прогрессирующим заболеванием, которое со временем меняется, дозу и прием лекарств необходимо пересматривать каждые несколько месяцев. Соображения при выборе средства для лечения хореи изложены в таблице.
Таблица 3
Рекомендации по фармакологическому лечению хореи
| Если у пациента есть неприятная хорея с: | Рассмотрите возможность использования: | ||
|---|---|---|---|
| Отсутствие других симптомов | 68, рубеназин (9026) | Снижение веса | Оланзапин, каннибиноиды |
| Психоз, агрессия или импульсивность | Арипипразол, галоперидол, оланзапин, рисперидон или другие нейролептики | ||
| рабочие | |||
| рабочие | |||
| Апатия | Амантадин или стимулирующие препараты и избегайте нейролептиков | ||
| Выраженная дистония | Амантадин, бензодиазепины, фокальные инъекции нейротоксина и избегайте нейролептиков | ||
| Ответ на фармакотерапию | 2 Глубокая стимуляция головного мозга |
На стадии разработки | Общество болезней Хантингтона Америки
Программа исследования болезни Хантингтона
Исследовательская «линия» — это процесс создания, тестирования и окончательного утверждения нового лекарства для использования на людях.HDSA финансирует исследователей и врачей, проводящих исследования HD на разных этапах процесса, и сотрудничает с отраслевыми партнерами, чтобы довести информацию о клинических испытаниях до общественности. Мы также финансируем исследования, ориентированные на человека, через проект HD Human Biology Project и поддерживаем молодых ученых через стипендии Дональда Кинга и стипендии Берман-Топпер по развитию карьеры.
Доклинические исследования
Фундаментальные исследования того, что происходит в головном мозге при болезни Хантингтона, по-прежнему позволяет понять, как можно лечить это заболевание.По мере определения целей проверяются существующие лекарства и добавки, которые, как известно, решают эту задачу, и предпринимаются усилия по разработке лекарств.
Перспективные лекарства тестируются на животных моделях, таких как дрозофила (плодовые мухи) и мыши, созданные для заражения болезнью Хантингтона. Если результаты будут положительными, препарат будет тщательно протестирован на одной или нескольких мышах с HD-моделями. Если лекарство эффективно и побочные эффекты допустимы, проводятся токсикологические исследования, и лекарство можно переходить к клиническим испытаниям.
Клинические Испытания лекарственных средств
После того, как лекарство готово для тестирования на людях, оно должно пройти три фазы клинических испытаний. Фаза 1 — это небольшое исследование безопасности (20-50 человек с HD). Фаза 2 — это испытание среднего размера (50-200 человек с HD), посвященное проверке безопасности и воздействия препарата на организм. Фаза 3 — это большое испытание (200-1000 человек), в котором проверяется, помогает ли препарат при симптомах. Прежде чем FDA одобрит его для использования человеком, необходимо продемонстрировать безопасность, хорошую переносимость и эффективность препарата.
Наблюдательные испытания
Наблюдательные испытания не включают в себя тестирование лекарства — они просто изучают поведение и биологию человека, чтобы узнать больше о БГ, с помощью неврологических обследований, когнитивных тестов и сдачи крови или биопроб. Наблюдая и тестируя людей с геном HD с течением времени и на разных стадиях заболевания, исследователи могут обнаружить, что меняется внутри мозга и тела до и после появления симптомов HD. Это особенно важно для планирования будущих исследований, в которых необходимо будет измерить эффективность лекарств еще до появления симптомов.
членов семьи HD — с положительным геном, группы риска, с отрицательным геном, а также лица, обеспечивающие уход, — могут добровольно принять участие в наблюдательном исследовании, таком как Enroll-HD.
Динамический процесс
Хотя кажется, что поток исследований плавно переходит от доклинических исследований к клиническим испытаниям, на практике он намного более динамичен. Чтобы получить ответы на вопросы, исследователям трансляции может потребоваться вернуться к исследователям-основателям. Препарат может показать явные признаки эффективности при доклинических испытаниях, но иметь серьезные побочные эффекты или требовать слишком больших доз для работы.В зависимости от его потенциала могут потребоваться дальнейшие усилия по разработке препарата. Препарат, который успешно лечит аналогичные симптомы при другом неврологическом расстройстве, может сразу перейти в клинические испытания для пациентов с HD. Сообществу HD важно знать, что исследования Хантингтона ведутся параллельно. Фундаментальные исследования, трансляционные исследования и клинические испытания продолжаются. Более двух десятков лекарств и добавок активно проходят через конвейер, и десятки других номинированы на рассмотрение.Когда дан ответ на один важный вопрос, усилия переключаются на другие вопросы. Когда одно лекарство терпит неудачу, ресурсы быстро перенаправляются на поиск более перспективных.
Чтобы узнать о лекарствах и добавках, которые в настоящее время проходят через исследовательский канал, обратитесь к таблице ниже.
Терапия в конвейере 2020
Тетрабеназин (ксеназин)
Лундбек: Тетрабеназин используется для лечения непроизвольных движений (хореи) при болезни Хантингтона.Тетрабеназин является ингибитором везикулярного переносчика моноаминов 2 (VMAT2) и действует путем снижения уровня нейромедиатора дофамина в синапсе (пространстве между нейронами). В 2008 году это был первый препарат HD, одобренный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
Аустедо (дейтрабеназин)
TEVA Pharmaceuticals: Дейтрабеназин использует тот же механизм действия, что и тетрабеназин, для лечения непроизвольных движений (хореи) при болезни Хантингтона.Дейтетрабеназин расщепляется организмом медленнее, чем TBZ, что может означать, что препарат можно принимать реже, в более низкой дозе или с меньшими побочными эффектами, чем у тетрабеназина.
Ingrezza (Valbenazine)
Neurocrine Biosciences: В исследовании KINECT-HD исследуется валбеназин как средство лечения хореи HD. Этот препарат уже одобрен для лечения людей с расстройством, называемым поздней дискинезией (ТД). TD вызывает движения лица и конечностей из-за лекарств, назначаемых при определенных психических состояниях.Это испытание фазы 3 набирается по всей территории Соединенных Штатов.
Придопидин (PROOF-HD)
Teva Pharmaceuticals: В предыдущем исследовании PRIDE-HD, придопидин не соответствовал ключевым целям лечения двигательных симптомов HD, но участники испытания показали небольшое улучшение показателей независимости в повседневной жизни. . PROOF-HD — это более крупное и продолжительное исследование, в котором будет проверена способность придопидина помогать пациентам с HD поддерживать их повседневную функцию.
RG6042 ASO (испытание GENERATION-HD1)
Roche / Genentech: Антисмысловые олигонуклеотиды (ASO) представляют собой препарат, снижающий уровень хантинтинга, который связывается с мРНК хантингтина (сообщение) и приводит к снижению уровень белка хантингтина.В исследовании GENERATION HD-1 тестируется ASO под названием RG6042, снижающее как расширенный, так и нормальный хантингтин, аллель-неспецифический подход. После успешного испытания на безопасность проводится крупномасштабное испытание фазы 3 с 801 участником на 90 предприятиях по всему миру.
WVE-120101/120102 ASO
Wave Life Sciences: ASO-препараты Wave используют аллель-специфический подход, снижая только вредный хантингтин путем обнаружения крошечных генетических сигналов, называемых SNP, на расширенном гене.В испытаниях PRECISION HD-1 и PRECISION HD-2 тестируются два препарата ASO примерно у 60 человек каждое. В декабре 2019 года Wave объявила, что WVE-120102 является безопасным мутантным хантингтином с пониженным содержанием мутанта и что оба испытания будут расширены, чтобы включить более высокую дозу. Эти испытания закончили набор в США.
VX15 (испытание SIGNAL)
Vaccinex: VX15 — это лекарство, которое связывается и блокирует молекулу, которая может вызвать воспаление в головном мозге людей с HD.Исследование SIGNAL было исследованием фазы II для оценки безопасности, переносимости и эффективности VX15 у людей с риском развития HD. В октябре 2020 года было объявлено, что суд не достиг своих основных целей.
SRX246 (STAIR Trial)
Azevan Pharmaceuticals: SRX246 — исследуемый препарат для лечения симптомов тревоги и агрессии у пациентов с ранней стадией HD. Он действует, блокируя рецептор вазопрессина 1a, который играет роль в регуляции эмоций.Набор участников исследования STAIR завершен.
SOM3355
SOM Biotech (Испания): SOM3355 — ингибитор везикулярного транспорта моноаминов (VMAT), который действует аналогично тетрабеназину, препятствуя передаче дофаминовых сообщений между нейронами. Эта компания специализируется на лечении редких заболеваний путем перепрофилирования уже разработанных и исследованных лекарств.
Глубокая стимуляция мозга (DBS)
Medtronic: Глубокая стимуляция мозга включает имплантацию электродов в часть мозга, называемую бледным шаром, для стимуляции нервов, которые не работают при HD.В Европе проводятся испытания, чтобы проверить, может ли устройство под названием нейростимулятор ACTIVA® PC помочь с хореей HD.
Тригептаноин
Ultragenyx Pharmaceuticals: Тригептаноин — это тип масла, называемого триглицеридом, который тестируется в качестве пищевой добавки в небольшом исследовании во Франции и Нидерландах. Основная цель — проверить, может ли он предотвратить атрофию участков мозга, пораженных HD.
Cellavita (испытание ADORE-DH)
Azidus (Бразилия): Это исследование в Бразилии тестирует долгосрочные внутривенные инъекции стволовых клеток для лечения HD.После успешного исследования безопасности на небольшом количестве людей те же участники будут набраны для приема препарата в течение более длительного периода времени, без плацебо, и все участники будут знать о лечении (известное как открытое расширенное исследование) .
Ресвератрол
Франция: Ресвератрол — это химическое вещество, которое содержится в красном вине и некоторых фруктах и может действовать как антиоксидант. Его можно приобрести в качестве добавки, но пока нет доказательств того, что он может быть полезен при HD.Французское правительство поддерживает контролируемое исследование ресвератрола, чтобы выяснить, может ли он замедлить атрофию мозга при раннем HD.
AMT-130
uniQure: AMT-130 — это препарат, снижающий хантингтин, в котором используется тип безвредного вируса, называемого вектором AAV5, который несет микро-РНК, предназначенную для подавления гена хантингтина. Терапевтическая цель — подавить производство мутантного белка путем доставки этого вируса в мозг. Это первое испытание генной терапии HD, и оно началось в 2019 году.
SAGE-718
Sage Therapeutics: Sage-718 — антагонист рецептора NMDA, который прошел ранние испытания безопасности у пациентов с болезнью Хантингтона. Цель состоит в том, чтобы лечить когнитивные изменения, связанные с HD.
Tasigna (нилотиниб)
Джорджтаунский университет: Нилотиниб, также известный как Tasigna, представляет собой химиотерапевтический препарат, используемый для лечения лейкемии. Есть некоторые свидетельства того, что он может изменять уровни дофамина, сигнальной молекулы мозга, которая нарушается при HD.Это небольшое испытание, чтобы определить, является ли нилотиниб безопасным и изменяет ли дофамин у пациентов с HD.
INT41
Vybion: INT41 — это внутрителовое лекарственное средство, предназначенное для предотвращения повреждения клеток токсичным белком хантингтина. Он доставляется с использованием вируса AAV и показал некоторые перспективы благодаря снижению хантинтина и улучшению поведения мышей HD.
P110
Mitoconix: Mitoconix разрабатывает лекарства, которые поддерживают здоровье митохондрий, источников энергии клеток, для защиты клеток мозга при HD.P110 улучшил патологию и поведение мышей HD.
Малая молекула htt-снижающая
PTC Therapeutics и Novartis: Несколько компаний работают над пероральными препаратами, снижающими хантинтин, — таблетками, принимаемыми внутрь, которые могут достичь мозга и изменить способ генерации геном HD сообщения РНК хантингтина и снизить уровень вредного белка.
Huntingtin-снижающая AAV-shRNA
Sanofi and Voyager Therapeutics: Маленькая РНК-шпилька или короткая РНК-шпилька (shRNA) могут быть использованы для вмешательства в сообщение хантингтина и предотвращения образования токсичного белка.Они доставляются в клетки с помощью безвредного вируса, называемого аденоассоциированным вирусом (AAV).
Снижение Htt ZFN
Такеда и Сангамо: Нуклеазы «цинковые пальцы» могут воздействовать на участки ДНК, чтобы модифицировать их или препятствовать образованию РНК. Такеда и Сангамо совместно работают над селективным мутантным ZFN, снижающим хантингтин, который успешно применяется в человеческих клетках в чашке.
Htt-снижающий AON
Биомарин: Биомарин разрабатывает антисмысловой олигонуклеотид (ASO, или AON), препарат, снижающий уровень хантинтина, который связывается с мРНК хантинтина (сообщение) и приводит к снижению уровень белка хантингтина.Он нацелен на повторы CAG во всех генах, не только в хантингтине.
EHP-102
Emerald Health: EHP-102 — пероральный препарат, полученный из каннабигерола, который содержится в растении каннабис. В доклинических исследованиях показано нейропротекторное действие. Emerald Health разрабатывает этот исследовательский метод лечения болезни Хантингтона.
Декстрометорфан / хинидин
Техасский университет, Хьюстон: Декстрометорфан / хинидин используется для лечения эмоциональных всплесков, которые могут произойти при определенных нейродегенеративных расстройствах.Это небольшое исследование проверяет безопасность и эффективность использования этой комбинации лекарств при раздражительности у пациентов с болезнью Хантингтона.
Одобрение FDA
Производство прекращено
Болезнь Хантингтона: причины, симптомы, лечение
Что такое болезнь Хантингтона?
Болезнь Хантингтона (БГ) — это наследственное и смертельное заболевание, при котором нервные клетки головного мозга разрушаются. Это приводит к ослаблению физических и умственных способностей, и со временем они ухудшаются.Лекарства нет. Если она начинается в раннем возрасте, это называется ювенильной болезнью Гентингтона.
Диагноз болезни Хантингтона может стать настоящим шоком. Здесь есть на что обратить внимание.
Но подключение к системе поддержки, такой как социальный работник, терапевт или группа поддержки, может сделать путешествие немного менее пугающим. С помощью медицинской бригады люди с болезнью Хантингтона могут жить самостоятельно в течение многих лет.
Симптомы болезни Хантингтона
Хотя симптомы могут впервые проявиться в среднем возрасте, болезнь Хантингтона может поразить любого от детства до пожилого возраста.Симптомы часто впервые появляются в возрасте от 30 до 40 лет. В течение 10–25 лет болезнь постепенно убивает нервные клетки головного мозга. Это влияет на тело, разум и эмоции. Если симптомы проявляются до 20 лет, это называется ювенильной болезнью Хантингтона, и она может ухудшиться быстрее.
Продолжение
Симптомы могут сильно отличаться от человека к человеку. А стресс или волнение могут ухудшить симптомы.
Некоторые симптомы обнаружить легче, чем другие. Ненормальные движения могут быть первым, что вы заметите.Похудание может вызывать беспокойство на всех этапах.
Симптомы болезни Хантингтона имеют тенденцию к развитию поэтапно.
Симптомы на ранней стадии
На ранних стадиях изменения могут быть весьма незначительными, что позволяет продолжать управлять автомобилем и работать. Вам может потребоваться небольшая дополнительная помощь.
Общие ранние симптомы включают:
Проблемы с обучением новому
Проблемы с принятием решений
Провалы памяти
Перепады настроения
Проблемы с мышцами (дистония)
Проблемы со сном (бессонница)
Потеря энергии и усталость
Неуклюжесть
4 9057 Медленные движения глаз
9057
Симптомы средней стадии
Со временем симптомы начинают больше мешать сегодняшняя жизнь.Например, вы можете начать ронять предметы или падать. Или у вас могут быть проблемы с речью или глотанием.
Сохранять организованность может быть сложно. А эмоциональные изменения могут оказать давление на отношения.
Общие симптомы средней стадии включают:
Неконтролируемые подергивания (хорея)
Проблемы при ходьбе
Путаница
Потеря памяти
Проблемы с глотанием
Проблемы с дыханием
Мысли о смерти, смерти или самоубийстве
Потеря веса
Развитие навязчивых состояний расстройство или мания
9057
изменения личности
Снижение мыслительных способностей
Продолжение
Симптомы поздней стадии
На этой стадии люди с болезнью Хантингтона должны зависеть от помощи других.Невозможно ходить и говорить. Скорее всего, вы все еще будете знать о близких, которые вас окружают. Суетливые движения могут стать серьезными или исчезнуть.
Симптомы ювенильной болезни Хантингтона
У детей и подростков болезнь Хантингтона может прогрессировать быстрее и вызывать такие симптомы, как:
Диагностика болезни Гентингтона
Семейный анамнез играет важную роль в постановке диагноза. Ваш врач задаст вам вопросы о вашем медицинском опыте и проведет медицинский осмотр.Если вы и ваш врач подозреваете болезнь Хантингтона, невролог проведет дополнительные анализы.
Невролог может проверить:
Рефлексы
Мышечная сила
Баланс
Чувство осязания
Зрение
Психическое состояние
Память
Рассуждение
Навыки мышления
Речь
Лечение и домашние средства от болезни Хантингтона
На данный момент лечение болезни Хантингтона
может помочь контролировать симптомы болезни Хантингтона
- 7. движения.Ваш врач может тесно сотрудничать с вами, чтобы контролировать любые побочные эффекты и при необходимости менять лекарства.
Некоторые антипсихотические препараты обладают побочным эффектом, контролирующим движение, и помогают некоторым людям.
Антидепрессанты также могут помочь при обсессивно-компульсивном расстройстве.
Лекарства, стабилизирующие настроение, могут облегчить симптомы расстройств настроения, но могут вызвать другие побочные эффекты.
Речевая или языковая терапия может быть полезна при любых проблемах с речью или глотанием.
Трудотерапия или физиотерапия могут помочь вам научиться лучше контролировать движения. А вспомогательные устройства, такие как поручни, могут помочь вам управлять изменяющимися физическими способностями.
Нутриционная поддержка варьируется от использования специальной посуды и сосредоточения внимания на продуктах, богатых питательными веществами, и заканчивая дополнительным питанием через зонд на более поздних этапах.
Упражнение может быть очень полезным. Люди с болезнью Хантингтона, которые остаются в хорошей форме и активны, как могут казаться, добиваются большего успеха, чем те, кто этого не делает.
Психотерапия научит вас управлять изменениями в своих эмоциях и мышлении. Такие стратегии, как разбиение задач на более простые шаги, могут значительно облегчить эти изменения для вас и вашей семьи.
Члены семьи могут помочь, внося некоторые изменения в дом:
Подавайте дополнительное питание и добавляйте высококалорийные добавки, чтобы поддерживать здоровый вес.
Не отвлекайтесь во время еды.
Выбирайте продукты, которые легче пережевывать и глотать.
Используйте вилки и другую посуду, предназначенную для людей с ограниченной моторикой.
Используйте закрытые чашки с трубочкой или носиком для питья.
Придерживайтесь обычного распорядка.
Используйте напоминания на телефоне или компьютере для выполнения задач.
Сохраняйте жизнь как можно более спокойной, простой и спокойной.
Для детей: вместе со школьным консультантом составьте план обучения.
Общайтесь с друзьями и поддерживайте социальное взаимодействие как можно чаще.
По возможности добавьте к дому пандусы и лифты для инвалидных колясок.
Добавьте защитные дуги в ванных комнатах, рядом с кроватью и на лестницах.
Используйте голосовое управление освещением и другие функции «умного» дома.
Используйте электронные речевые программы или графические таблицы для облегчения общения.
Причины болезни Хантингтона
В 1993 году исследователи обнаружили ген, вызывающий болезнь Хантингтона.У всех есть ген HD, но в некоторых семьях аномальная копия гена передается от родителя к ребенку. Если у вас есть родитель с болезнью Гентингтона, у вас есть 50% шанс получить этот ген и заболеть.
Также:
Мужчины и женщины с одинаковой вероятностью унаследуют аномальный ген.
Если у вас нет аномального гена, вы не сможете заразиться геном Гентингтона или передать его своим детям.
Болезнь не пропускает поколения.
Если вы или члены вашей семьи планируете пройти тестирование на болезнь Хантингтона, рекомендуется сначала получить профессиональную генетическую консультацию. Консультанты могут помочь объяснить, чего ожидать от результатов теста.
Зная о гене HD, ученые смогли многое узнать о том, как болезнь влияет на мозг. Что еще более важно, это открытие может помочь подготовить почву для будущего лечения.
Болезнь Хантингтона. Информация о болезни Гентингтона
Что такое болезнь Гентингтона?
Болезнь Хантингтона (БХ) названа в честь Джорджа Хантингтона, который впервые описал ее в 1872 году.Это наследственное (генетическое) заболевание, поражающее мозг и нервную систему. Это может мешать движениям вашего тела, может повлиять на ваши рассуждения, осознание, мышление и суждения (познание) и может привести к изменению вашего поведения. «Генетический» означает, что заболевание передается через семьи с помощью специальных кодов, называемых генами. Каждая клетка вашего тела содержит хромосомы, состоящие из множества генов.
Что вызывает болезнь Хантингтона?
HD вызван дефектным геном, который вы унаследовали от своих родителей.Этот дефектный ген находится на хромосоме 4. Ген отвечает за образование белка под названием «хантингтин». Генетический дефект означает, что определенные белки, необходимые для выработки химических веществ в мозге, не могут нормально вырабатываться в вашем мозгу. Считается, что это приводит к повреждению и гибели некоторых нейронов (клеток мозга) в определенных частях вашего мозга, называемых базальными ганглиями и корой. Именно это повреждение приводит к появлению симптомов ГБ. Также в мозгу накапливается химическое вещество, называемое дофамином, которое усугубляет проблемы с движением.
HD — аутосомно-доминантное заболевание. Это означает, что вы можете унаследовать HD только от одного из своих родителей. Если у одного из ваших родителей есть дефектная копия гена, есть 50:50 шанс, что каждый их ребенок унаследует дефектный ген и разовьется HD. См. Диаграмму в конце этой брошюры, которая поможет вам понять это.
Однако иногда люди с HD могут не иметь истории HD в их семье. Это может быть из-за того, что называется «новой мутацией» («мутация de novo»).Новая мутация — это мутация (или ошибка) в гене, которая впервые присутствует у одного члена семьи. Это может произойти из-за:
- дефекта в генетическом материале яйцеклетки или спермы одного из родителей пострадавшего; или
- Неисправность генетического материала яйцеклетки, оплодотворенной, когда яйцеклетка и сперматозоид встречаются (эмбрион).
Неясно, что заставляет эту неисправность (мутацию) внезапно возникать впервые.
Кроме того, если человек в вашей семье, несущий дефектный ген HD, умрет до того, как у него появятся симптомы (и, следовательно, до того, как будет диагностирован HD), родственники не будут знать об этом семейном анамнезе.Это может быть еще одной причиной отсутствия в вашей семье истории HD.
Насколько распространена болезнь Хантингтона и у кого она развивается?
HD поражает от 5 до 10 человек на 100 000 жителей Великобритании. Во всем мире он, кажется, более распространен среди белого населения, чем среди азиатских или африканских людей. HD одинаково поражает как мужчин, так и женщин. Чаще всего симптомы БГ начинают проявляться в возрасте от 30 до 50 лет. Таким образом, даже если вы унаследовали болезнь, для появления симптомов может потребоваться некоторое время.
У 5-10 из 100 человек с HD развиваются симптомы до достижения 20 лет. Это заболевание известно как ювенильное начало HD. Это может вызвать более серьезные симптомы. Большинство людей с этим типом HD наследуют заболевание от своего отца.
Каковы симптомы болезни Хантингтона?
Симптомы HD могут варьироваться от человека к человеку. Не у всех с БХ есть одинаковые симптомы или проявляются все симптомы. Симптомы имеют тенденцию прогрессировать постепенно и со временем ухудшаться.
Если HD не используется в вашей семье, иногда может потребоваться некоторое время для его диагностики.Это связано с тем, что некоторые из ранних симптомов часто можно не заметить или приписать другим проблемам — например, тонким изменениям в вашей личности или перепадам настроения.
Симптомы HD можно разделить на три основные области:
- Проблемы с движением.
- Проблемы с познанием.
- Проблемы с настроением и поведением.
Проблемы с движением
Самая распространенная проблема с движением при HD известна как хорея. Фактически, до недавнего времени HD была известна как хорея Хантингтона.Хорея означает резкие непроизвольные движения. В частности, это имеет тенденцию влиять на вашу голову, лицо, руки или ноги. Каждое движение происходит внезапно. Вначале некоторые люди могут просто подумать, что вы нервничаете; однако ваши симптомы имеют тенденцию к постепенному ухудшению. При тяжелой хорее у вас может развиться неконтролируемое раскачивание (или раскачивание) руками или ногами (это называется баллизмом). Это может помешать вашей способности передвигаться. У вас может быть больше шансов упасть и у вас возникнут трудности, например, с кормлением или одеванием.Вы можете легко уронить вещи.
Со временем, по мере прогрессирования HD, у вас может развиться проблема с движением, известная как дистония. Это имеет тенденцию постепенно заменять хорею. Дистония означает, что у вас спазм мышц, обычно мышц плеч, шеи, рук и ног. Это может привести к скручивающим движениям, повторяющимся движениям или неправильным позам. Вы также можете заметить, что со временем ваши конечности могут стать довольно жесткими или жесткими, а ваши движения замедлятся. Это замедление движений называется брадикинезией.
Проблемы с глотанием — обычное явление, если у вас HD. Это связано с тем, что HD может влиять на мышцы, контролирующие глотание, и мышцы вокруг рта. Проблема может быть в удушье, а трудности с приемом пищи также означают, что вы можете потерять вес.
Вы можете заметить проблемы с речью, и ваша речь может стать невнятной. Также могут нарушаться движения ваших глаз, из-за чего вы не можете смотреть из стороны в сторону или вверх и вниз.
Проблемы с познанием
Ваше познание (ваше восприятие, рассуждение, осознание, мышление и суждение) тесно связано с вашим поведением.Итак, изменения в вашем познании из-за потери нейронов в вашем мозгу также могут повлиять на ваше поведение (см. Ниже).
Первые симптомы, которые вы можете заметить, — это проблемы с кратковременной памятью. Недостаток концентрации, кратковременные провалы памяти и проблемы с ориентацией являются обычным явлением. Освоение новых навыков может стать трудным.
Медленное снижение когнитивных функций при HD может быть похоже на проблему типа деменции.
Проблемы с настроением и поведением
Изменения в вашем поведении могут быть одним из первых признаков HD.Они могут появиться до того, как пострадают ваши движения. Однако у некоторых людей они начинаются после появления проблем с движением.
Вы можете заметить, что становитесь раздражительным, легко возбужденным и начинаете терять интерес к вещам, которые вам раньше нравились. Люди могут комментировать, что вы становитесь неопрятным и можете начать терять интерес к уходу за собой — например, реже стираете, не заботитесь о своей внешности и т. Д.
Это может повлиять на ваше суждение о вещах.Вы можете стать агрессивным по отношению к людям, и иногда вы можете потерять свои запреты, что приведет к неловкому поведению в социальных ситуациях. Изменения личности, связанные с HD, могут быть очень трудными для вас, вашей семьи и друзей. Поведенческие изменения и антиобщественное поведение могут ухудшить ваши отношения. Возможно, вам будет трудно согласиться с тем, что ваше поведение может быть проблемой. Это часть того, как HD влияет на ваш мозг.
Подавленное настроение часто встречается у людей с HD, и существует повышенный риск самоубийства.Другие проблемы психического здоровья, включая обсессивно-компульсивное расстройство и проблемы, подобные мании и шизофрении, также чаще встречаются у людей с HD. См. Отдельные буклеты «Обсессивно-компульсивное расстройство», «Биполярное расстройство» и «Шизофрения» для получения более подробной информации.
Нужно ли мне расследование?
Если у вас уже есть симптомы
Доступно генетическое тестирование для подтверждения HD. Однако, если у вас уже есть семейный анамнез БГ и у вас есть типичные симптомы и признаки этого состояния, генетическое тестирование может не потребоваться.
КТ или МРТ вашего мозга могут показать некоторые типичные признаки HD. Однако это сканирование обычно бесполезно при попытке диагностировать HD на ранних стадиях, поскольку изменений может не быть. Сканирование в основном используется на поздних стадиях заболевания.
Если болезнь Хантингтона передается в вашей семье
Если в вашей семье есть HD, это может вызвать много беспокойства и беспокойства, так как вы можете искать возможные симптомы. Если вы подвержены риску развития HD из-за семейного анамнеза, но у вас еще нет никаких симптомов, возможно также генетическое тестирование.Это называется предсимптоматическим тестированием. Если вы унаследовали дефектный ген, у вас обязательно разовьются симптомы в какой-то момент вашей жизни. Однако невозможно предсказать, когда появятся ваши симптомы.
Пройти генетическое тестирование или нет — это выбор каждого человека в отдельности. Некоторые люди не хотят проходить испытания. Они предпочли бы подождать, чтобы увидеть, появятся ли у них какие-либо симптомы. Если вы планируете генетическое тестирование, рекомендуется пройти консультацию.Ваш терапевт сможет направить вас к специалисту, который является генетическим консультантом.
Как лечить болезнь Хантингтона?
В настоящее время лекарства от HD не существует. Кроме того, не было обнаружено лечения, которое отсрочило бы появление симптомов или замедлило прогрессирование симптомов. Таким образом, лечение направлено на то, чтобы попытаться максимально контролировать симптомы, когда они действительно развиваются.
Лечение лекарствами
Существует ряд различных лекарств, которые можно использовать для лечения хореи.В первую очередь часто используется группа лекарств, известных как бензодиазепины. Примеры этих лекарств включают клоназепам и диазепам. Также могут использоваться другие лекарства, включая тетрабеназин. Однако все эти лекарства могут иметь побочные эффекты. Например, они могут привести к замедлению ваших движений (брадикинезия), ригидности или ригидности, депрессии или седативному действию. Когда и когда начинать лечение, будет зависеть от достижения баланса между преимуществами и побочными эффектами.
Если у вас проблемы с брадикинезией и ригидностью или ригидностью конечностей, вам может помочь группа лекарств, называемых агонистами дофамина.Альтернативой этому является другое лекарство, называемое леводопа. Эти лекарства обычно используются для лечения болезни Паркинсона, при которой наблюдается аналогичное замедление движений и ригидность.
Если у вас развивается депрессия, вам могут помочь антидепрессанты. Также доступны лекарства для лечения некоторых других проблем психического здоровья, которые могут быть связаны с HD.
Другие методы лечения
Большинство людей с ГБ имеют команду медицинских специалистов, которые работают с ними. Ваш терапевт часто координирует ваше лечение.Другие члены команды могут включать специалиста по проблемам мозга, нервов и мышц (невролог), психиатра и генетического консультанта.
Вас также могут направить к физиотерапевту за помощью с упражнениями на равновесие и упражнениями, которые помогут вам легче передвигаться. Эрготерапевт может помочь вам с любой адаптацией, которая вам понадобится, чтобы облегчить вашу повседневную жизнь. Например, они могут помочь с адаптацией к вашему дому, такой как доступ для инвалидных колясок, перила и изменения в вашей спальне и ванной.
Логопед может помочь с речью и / или проблемами с глотанием. Они могут научить вас различным способам общения. Вас могут направить к диетологу, если вы сильно похудеете (из-за проблем с глотанием, а также из-за проблем с движением). Они могут посоветовать продукты, которые вам будет легче есть, потому что они требуют меньшего пережевывания. Иногда проблемы с глотанием могут означать, что вам необходимо установить назогастральный зонд. Это трубка, которая проходит через нос в желудок, чтобы пища доставлялась в желудок без необходимости глотать.
Возможные варианты лечения в будущем
Изучаются различные новые методы лечения HD. Они включают в себя генную терапию и различные виды лечения лекарствами. Например, в настоящее время проводятся испытания нового лекарства под названием придопидин. Испытания также изучают лекарства, чтобы предотвратить развитие болезни у людей с дефектным геном HD. Однако эти методы лечения все еще находятся на экспериментальном уровне. Необходимы дополнительные исследования, прежде чем мы узнаем, помогают ли такие методы лечения HD.
Каковы перспективы (прогноз) болезни Хантингтона?
HD — это состояние, которое медленно прогрессирует, так что у вас постепенно развиваются и ухудшаются симптомы. На более поздних стадиях HD вы станете полностью зависимым от других людей и вам потребуется полный уход. HD приводит к значительной инвалидности и, в настоящее время, в конечном итоге приведет к смерти.
В настоящее время большинство людей живут от 10 до 25 лет после того, как у них впервые появятся симптомы БХ. Человек с HD обычно умирает от такой инфекции, как пневмония, но самоубийства очень распространены.Помните, что новые методы лечения исследуются. Ваш специалист и Ассоциация болезней Хантингтона смогут обсудить с вами любые разработки в области лечения или пробные методы лечения.
Могу ли я передать болезнь Хантингтона своим детям?
Люди с HD часто рожают детей до того, как у них развиваются симптомы болезни. Если вы несете дефектный ген HD, вероятность того, что каждый ваш ребенок будет иметь HD, составляет 50:50. Если у вас или вашего партнера есть HD, доступно пренатальное тестирование.Это может показать, есть ли у вашего ребенка дефектный ген и, следовательно, разовьется ли у него HD. Однако тестирование не всегда бывает точным на 100%.
Аутосомно-доминантное наследование
Преимплантационная генетическая диагностика (ПГД) также доступна, если один из родителей несет дефектный ген.