Врождённые аномалии развития верхней конечности.
Врождённые аномалии развития верхней конечности.
В настоящее время используется классификация врожденной аномалии верхнее конечности разработанная The American Society for Surgery of the Hand (ASSH) и International Federation of Societies for Surgery of the Hand (IFSSH).
I. Нарушение строения скелета верхней конечности.
А. Поперечный дефект верхней конечности:
Тип 1 — отсутствие плеча и предплечья- кисть прикрепляется к плечевому суставу;
Тип 2 — отсутствие предплечья;
Тип 3 — отсутсвие кисти- acheiria;
Тип 4- адактилия- отсутствие пальцев кисти.
Б. Продольные дефекты в строении верхней конечности:
— фокомелия- аплазия верхней конечности
— врождённая лучевая косорукость— дефект лучевой кости;
— врождённая локтевая косорукость— недоразвитие локтевой кости;
— расщепленная кисть— аплазия одного или нескольких лучей кисти;
II. Нарушение дифференциации сегментов верхней конечности
А. Нарушение дифференциации мягких тканей:
— синдактилия;
— камптодактилия;
-стенозирующий лигаментит;
— врождённая сгибательно- приводящая контрактура первого пальца.
Б. Нарушение дифференциации костной ткани:
— клинодактилия;
— симфалангизм;
— радиоульнарный синостоз;
— артрогриппоз;
— windblown кисть.
III. Удвоение сегментов верхней конечности — полидактилия.
Тип удвоения:
— преаксиальный тип- удвоение I пальца;
— центральный тип – удвоение II, III, IV пальцев;
— постаксиальный тип — удвоение V пальца; удвоение кисти- зеркальная кисть.
IV. Чрезмерное развитие сегментов верхней конечности.
— макродактилия;
— истинная макродактилия.
V. Недоразвитие сегментов верхней конечности.
— гипоплазия I пальца кисти.
VI. Врождённые перетяжки.
VII. Системная патология скелета.
Врожденная лучевая косорукость.
Эта аномалия развития характеризуется недоразвитием лучевой кости. Эпидемиология. Частота встречаемости составляет 1 на 30 тысяч новорожденных в общей популяции. Может ассоциироваться с другими аномалиями и входит в состав многих генетических синдромов: VATER-синдрома, TAR-синдрома, Hold Oram –синдрома, анемии Fanconi.
Классификация врождённой лучевой косорукости.
Тип I — Характеризуется частичным недоразвитием дистального отдела лучевой кости.
Тип II – Характеризуется гипоплазией дистального или проксимального конца лучевой кости, до 1\ 3 от её исходной длины, что приводит к нестабильности в лучезапястном суставе.
Тип III — Гипоплазия лучевой кости, когда имеется только проксимальный отдел лучевой кости.
Тип IV — Полное отсутствие лучевой кости — аплазия.
Клиническая картина разнообразная и зависит от степени недоразвития лучевой кости. Чем больше дефект лучевой кости, тем выражение деформация предплечья и ограничение функции лучезапястного и локтевого суставов. Лучевая косорукость проявляется деформацией и укорочением предплечья, лучевой девиацией кисти, нарушением функции локтевого и лучезапястного сустава, в сочетании с различными аномалиями развития кисти.
Лечение врожденной лучевой косорукости представляет собой комплекс консервативных и хирургических методов. Зависит от типа косорукости, а также от возраста пациента. Консервативное лечение начинается с рождения и проводится на протяжении всего периода лечения и роста ребёнка.
Зависит от типа косорукости, а также от возраста пациента. Консервативное лечение начинается с рождения и проводится на протяжении всего периода лечения и роста ребёнка.
Достаточно сложным и многоэтапным является лечение пациентов с IV типом лучевой косорукости. На первом этапе проводится реконструкция и стабилизация кисти, что достигается центрацией кисти на локтевую кость. Следующим этапом с целью восстановления длины предплечья, а также при наличии деформации локтевой кости, является коррекция деформации и удлинение её с использованием управляемых систем АВФ.
Врождённая локтевая косорукость.
Данная аномалия верхней конечности характеризуется разной степенью недоразвитием локтевой кости.
Эпидемиология. Встречается в 7 раз реже, чем лучевая косорукость, является спородической аномалией. Может являться частью некоторых генетических синдромов: синдром Cornelia- Lange, синдром Schinzel, FFU- синдром (недоразвитие бедренной кости, малоберцовой и локтевой костей).
Рентгенограмма пациента с плечелучевым синостозом.
Классификация врожденной локтевой косорукости:
Тип I — Характеризуется частичным недоразвитием дистального отдела локтевой кости;
Тип II – Характеризуется гипоплазией локтевой кости, укорочение её составляет до 25 %;
Тип III — Гипоплазия локтевой кости до 50 %, так же может иметь место наличие плечелучевого синостоза;
Тип IV — Аплазия локтевой кости, наличие плечелучевого синостоза.
Внешний вид пациента с плечелучевым синостозом.
Клиническая картина. Локтевая косорукость проявляется деформацией и укорочением предплечья, локтевой девиацией кисти, нарушением функции локтевого и лучезапястного сустава, может также сочетаться с различными аномалиями кисти — синдактилией, аплазией пальцев.
При плечелучевом синостозе отмечается костное сращение плечевой и лучевой кости, отсутствие локтевой кости. При этом предплечье находится в положении пронации и разгибания, что резко ограничивает функцию верхней конечности.
Лечение врожденной локтевой косорукости представляет собой комплекс консервативных и хирургических методов. Зависит от типа косорукости, а также от возраста пациента.
Консервативное лечение начинается с рождения ребёнка и проводится на протяжении всего периода роста и развития ребёнка.
Хирургическое лечение.
При сочетании локтевой косорукости с аномалией развития кисти первостепенным является реконструкция кисти, которая проводится в возрасте пациента 1-2 года.
С целью коррекции деформации костей предплечья и восстановления длины локтевой кости, а в некоторых случаях и лучевой кости, выполняется удлинение костей предплечья с использованием аппаратов внешней фиксации в различной модификации.
Очень сложным является лечение детей с наличием плечелучевого синостоза. С целью устранения порочного положения предплечья, проводится остеотомия плечелучевого синостоза, ротация предплечья в срединное положении. При выраженном укорочении предплечья также проводится его удлинение.
Расщеплённая кисть
Этот тип аномалии характеризуется отсутствием нескольких лучей кисти. Выделяют типичную и атипичную формы расщёплённой кисти.
Эпидемиология. В популяции частота врожденной расщеплённой кисти составляет 4 на 100 тыс. новорожденных. В 70 % случаев является результатом генетических мутаций и составляющей некоторых синдромов: EEC-синдрома (Electrodactyly-ectodermal dysplasia-clefting), SHSF расщеплённая стопа и кисть, Cornelia- Lange синдрома, Oculodigital complex, Orodigital complex, Wildervank синдрома, Silver-Russell синдрома. В 30 % случаев данная аномалия является спородической.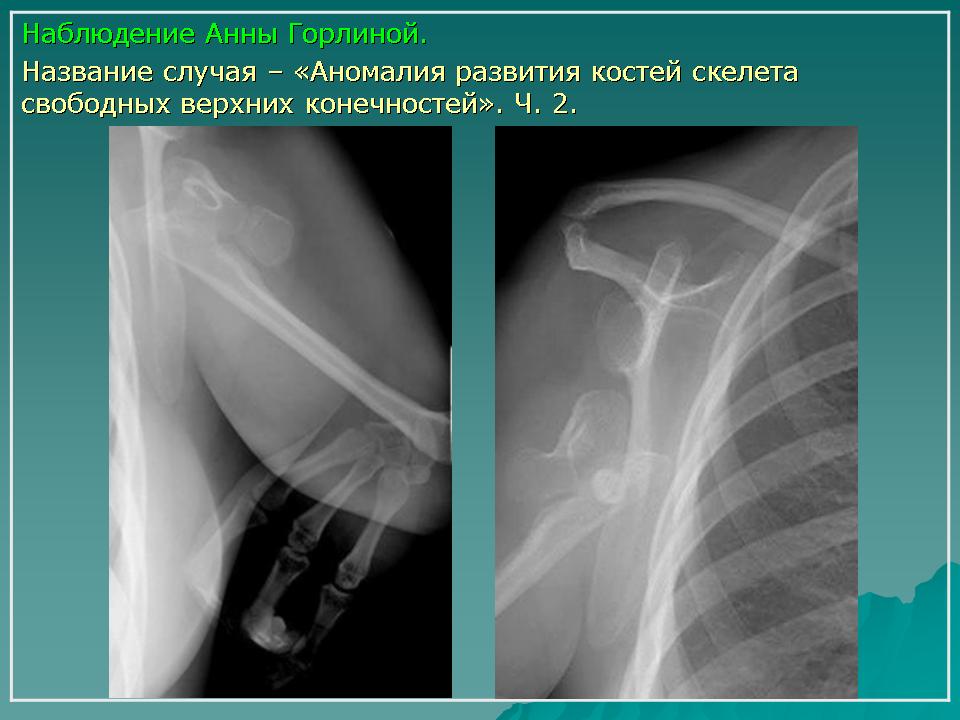
Классификация.
Типичная форма характеризуется V-формой дефекта, отсутствием центральных лучей, а конкретно пястных костей и фаланг пальцев. Как правило, это всегда двухсторонняя патология, сочетающаяся с расщеплённой стопой и имеет наследственный характер. Особенностью данного типа является расщепление кисти до костей запястья, как правило, отсутствует II-III-IV пальцы кисти. Может иметь место синдактилия пальцев, поперечное положение одной из пястных костей, может сочетаться с деформацией пальцев, гипоплазией первого пальца, сгибательными контрактурами в межфаланговых суставах.
Атипичная форма расщеплённой кисти характеризуется U- формой дефекта. При этом типе присутствуют пястные кости, но имеет место адактилия. Как правило, это односторонний процесс, отсутствует наследственный характер, стопа имеет нормальное строение.
Клиническая картина разнообразна. Проявляется адактилией, наличием расщелины- углубления между пальцами, синдактилией присутствующих пальцев, клинодактилией, симфалангизмом, контрактурой межфаланговых суставов.
Лечение только хирургическое лечение. Возраст начала лечения с 6 месяцев
Устранение расщелины осуществляется по методики Snow и Littler.
Синдактилия
Это врождённая аномалия кисти, характеризующаяся тем, что расположенные рядом пальцы сращены между собой. Сращение может как кожное, так и костное.
Эпидемиология. Частота встречаемости в общей популяции составляет 1 на 2000- 2500 новорожденных.
Классификация.
· Тотальная форма, характеризуется сращением пальцев на всём протяжении.
· Базальная форма, характеризуется наличием кожной перемычки на уровне проксимальных и средних фаланг пальцев.
· Простая форма, характеризуется только кожным сращением пальцев.
· Сложная форма, характеризуется также наличием костного сращения фаланг пальцев, с развитием деформации и контрактур пальцев.
· Акросиндактилия, форма синдактилии, характеризующаяся сращением только на уровне дистальных фаланг.
Клиника. Кожа на уровне сращения нормальной структуры, эластичная, подвижная. Пальцы всегда имеют нормальную ногтевую пластинку. Движения в межфаланговых суставах в полном объёме, сочетанные. При наличии сращения пальцев разной длины 3-4, 2-3, формируется с возрастом выраженные деформации пальцев с развитием контрактур, что резко ограничивает функции кисти. Является составляющей некоторых генетических синдромов: синдром POLAND, APERT, синдром множественных врождённых перетяжек.
Лечение только хирургическое. Возраст начала лечения 6- 12 месяцев жизни ребёнка, в зависимости от степени функциональных нарушений кисти и наличия сопутствующей патологии. Основные принципы хирургического лечения:
— формирование межпальцевого промежутка собственными тканями,
— все кожные разрезы вдоль синдактилированных пальцев должны быть фигурными — зигзагообразными, волнообразными,
— устранение деформации пальцев,
— сформировавшиеся кожные дефекты укрываются полнослойными свободными кожными лоскутами, взятыми из области паховой складки.
Обязательным условием является фиксация кисти гипсовой повязкой в положении разгибания пальцев в течение 5 недель.
Наиболее часто используемая методика Gilbert, Бауэра.
Врождённая сгибательно-приводящая контрактура первого пальца.
Данная аномалия характеризуется прижатием к ладони первого пальца и крайней степени его сгибания в пястно-фаланговом суставе.
Эпидемиология. Чаще встречается у девочек и является двухсторонним процессом.
Классификация E. Weckesser, J. Reed, K. Heiple, 1968 г. :
:
I группа- характеризуется дефицитом разгибания, контрактура отсутствует.
II группа- характеризуется дефицитом разгибания и наличием сгибательной контрактуры.
III группа- характеризуется дефицитом разгибания, гипоплазией первого пальца.
IV группа- смешанные формы.
Классификация McCarroll, 1985 год:
— гибкая деформация, характеризуется слабостью или аплазией разгибателей первого пальца.
— смешанная форма- характеризуется слабостью или аплазией разгибателей первого пальца + нарушения со стороны суставов, связок, мышц и кожи первого пальца.
Клиника. Проявляется в возрасте 2 месяцев. Характеризуется тем, что первый палец прижат к ладони («палец на ладони»)- приводящая контрактура первого пальца. Может сочетаться с гипоплазией мышц тенора, контрактурой межфалангового сустава. Всегда нарушена функция двухстороннего захвата кисти. Является составляющей генетического синдрома Фримана-Шелдона, артрогриппоза.
Лечение. С рождения начинается комплекс консервативных методов лечения в виде корригирующих гипсовых лонгет и ежедневной гимнастики. После 1 года проводится хирургическое лечение. Обязательная фиксация оперированной конечности ладонной гипсовой шиной в течении 6 недель. Курс физиотерапии, ЛФК, массажа.
Камптодактилия.
Врождённая аномалия кисти, характеризующаяся сгибательной контрактурой на уровне проксимального межфалангового сустава пальца кисти.
Эпидемиология. Мультифакториальное заболевание. В общей популяции составляет до 1%, в 2/3 случаев двухсторонний процесс, с вовлечением 5 пальца кисти. Чаще встречается у женщин.
Классификация.
I тип. Встречается у детей, в процесс вовлечён только 5 палец, поддаётся консервативному лечению.
II тип. Встречается у взрослых.
III тип. Проявляется с рождения, с вовлечением нескольких пальцев, является проявлением генетического синдрома (орофациодигитальный синдром, Фримана-Шелдона синдром (Freeman-Sheldon), окулодентодигитальная дисплазия, мукоплисахоридоз, Aarskog-Scott синдром, цереброгепаторенальный синдром (синдром Zellweger)).
Клиника. Основным проявлением камптодактилии является сгибательная контрактура пальца на уровне проксимального межфалангового сустава, что является следствием дисбаланса между сухожилиями сгибателей и разгибателей пальца. По клиническому проявлению различают восстанавливающуюся, или эластичную форму, которая поддаётся консервативному лечению. Фиксированную форму — неподдающуюся лечению, когда, при длительно существующей сгибательной контрактуре формируются фиброзные изменения в мягких тканях: в подкожной клетчатке, фасции, связках, сухожилиях.
Лечение. В 80 % случаев деформация не прогрессирует, поэтому необходимо проведение только консервативного лечения. Деформация в 30-40º не приводит к нарушению функции кисти. Сначала необходимо устранить имеющуюся деформацию с использованием статических жестких лонгет. Затем продолжают пользоваться динамическими шинами в ночное время.
При фиксированных жёстких деформациях показано хирургическое лечение.
Стенозирующий лигаментит.
Стенозирующий лигаментит- это затруднения нормальных скользящих движений сухожилий сгибателей внутри сухожильного влагалища, с формированием сгибательной контрактуры в межфаланговых суставах. Как правило, изменения происходят на уровне кольцевидных связок.
Эпидемиология. Встречается у 3 новорождённых из 1000. Это спородическое заболевание, не связанное с другими аномалиями. Встречается в возрасте 1-2 лет, в 25% случаев выявляется у новорожденных. В 25% случаев поражается 1 палец кисти, с вовлечением в процесс 1 пальцев обеих кистей.
Клиника. Проявляется у новорождённых в виде наличии узелка в проекции сухожилий сгибателя, который затрудняет движения пальца. При активных движениях происходит щелчок на уровне первой кольцевидной связки А1. У детей в 31 % случаев наступает спонтанное самоизлечение в возрасте до 1 года. С возраста 1-3 года при отсутствии лечения формируются жёсткие контрактуры межфаланговых и пястно-фаланговых суставах.
Лечение: если заболевание выявляется в период новорожденности – показано консервативное лечение в виде специальных укладок. При отсутствии эффекта от консервативного лечения показано хирургическое лечение.
Клинодактилия
Это врождённая аномалия, характеризующаяся девиацией пальца во фронтальной плоскости за счёт деформации средней фаланги, или наличии дельта-фаланги.
Эпидемиология. Часто встречается деформация 5 пальца на уровне дистального межфалангового сустава с отклонением его в лучевую сторону. В 19,5% случаев встречается у здоровых детей с двух сторон. Так же ассоциируется с некоторыми генетическими синдромами и хромосомными аномалиями. При синдроме Down встречается в 35% — 79% случаев. Клинодактилия 1- пальца встречается при синдроме Apert, Rubinstein-Taybi.
Классификация:
I тип- характеризуется минимальной угловой деформацией, длина фаланги нормальная.
II тип- характеризуется минимальной угловой деформацией, наличием деформации и укорочения средней фаланги пальца.
III тип- характеризуется выраженной угловой деформацией пальца, наличием дельта-фаланги (наличие продольно расположенной зоны роста фаланги, что приводит к нарушению роста фаланги).
Клиника. Эта патология характеризуется отклонением пальца в локтелучевую сторону. Причиной является деформация дистального конца средней фаланги пальца или наличие дельто-фаланги. Типичным является наличие деформации 5 пальца на уровне дистального межфалангового сустава и отклонением пальца в лучевую сторону. При этом функция пальца не страдает. Движения в межфаланговых суставах в полном объёме. Деформация ригидная, не устраняется при ручной коррекции и не поддаётся консервативному лечению.
Лечение. При минимальной деформации пальца до 10º и нормальной функции кисти, лечение не показано. При значительной деформации пальца показана корригирующая остеотомия средней фаланги с фиксацией спицей Киршнера. Затем иммобилизация гипсовой шиной.
Затем иммобилизация гипсовой шиной.
Симфалангизм
Врожденная аномалия, которая характеризуется изменением межфаланговых суставов пальцев кисти с отсутствием движений в них, отсутствием сухожилий сгибателей и разгибателей кисти, отсутствием кожных складок на уровне межфаланговых суставов кисти. Часто сочетается с синдактилией, генетическими синдромами (Apert, Poland). Встречается при артрогриппозе.
Клинически проявляется укорочением пальцев кисти, отсутствием сгибания в межфаланговых суставах.
Радиоульнарный синостоз
Характеризуется костным сращением проксимальных участков локтевой и лучевой кости. Это ведёт к резкому ограничению просупинационных движений предплечья в локтевом суставе. В более тяжёлых случаях предплечье пронировано, что резко ограничивает функцию верхней конечности и самообслуживание.
Диагностика. Важным является клиническое обследование, которое определяет степень ограничения просуспинационых движений. Рентгенография в двух стандартных проекциях, а также компьютерная томограмма.
Лечение только хирургическое, которое заключается в выполнение деротационной остеотомии обеих костей предплечья. После выполнения остеотомии локтевой и лучевой костей, предплечье выводится в среднее положение. Остеосинтез осуществляется либо накостными пластинами и последующей гипсовой иммобилизацией в течении 5-6 недель. Либо аппаратом внешней фиксации, что позволяет в послеоперационном периоде проводить раннюю разработку движений в локтевом и лучезапястном суставе. Фиксация аппаратом внешней фиксации осуществляется до наступления полной консолидации зон остеотомий костей предплечья.
III. Удвоение
Эпидемиология. Это наиболее часто встречающаяся мальформация кисти. Спородическая аномалия, может сочетаться с синдактилией, входить в некоторые генетические синдромы, иметь наследственный характер.
Спородическая аномалия, может сочетаться с синдактилией, входить в некоторые генетические синдромы, иметь наследственный характер.
Классификация. Тип удвоения:
— преаксиальный, или радиальный. Удвоение первого пальца;
— центральный тип. Удвоение 2-3-4 пальцев;
— постаксиальный, или ульнарный тип. Удвоение пятого пальца.
— зеркальная кисть (mirror-hand), когда имеется удвоение кисти по локтевой стороне лучезапястного сустава.
Клиника и классификация удвоения первого пальца (Wassell, 1969).
I тип – имеется расщепление дистальной фаланги.
II тип — удвоение дистальной фаланги на уровне дистального межфалангового сустава.
III тип- расщепление проксимальной фаланги.
IV тип — удвоение первого пальца на уровне пястно-фалангового сустава.
V тип – удвоение первого пальца на уровне первой пястной кости.
VI тип — удвоение первого пальца, начинающееся от пястно-многоугольного сустава.
VII тип – характеризуется удвоением трёхфалангового первого пальца.
Лечение только хирургическое. Так добавочный 5 палец подлежит удалению в возрасте до 1 года.
При удвоении I пальца проводится его реконструкция в зависимости от типа деформации. Основные принципы хирургического лечении: резекция добавочного пальца; восстановления оси пальца; моделирующая резекция суставных поверхностей; восстановление коллатеральных связок; изменение места прикрепления сухожилий; корригирующая остеотомия фаланг с фиксацией в корригированном положении спицей. Иммобилизация гипсовой лонгетой в течение 6-8 недель. После удаление спицы фиксация шиной в ночное время в течении 3 месяцев.
IV. Чрезмерное развитие сегмента- макродактилия.
Данная патология проявляется увеличением в объёме пальцев кисти. Эпидемиология. Может быть спородической патологией, а также проявлением болезни Recklinghausen, Proteus- синдрома, Klippel- Trenaunay- синдрома.
Клиническая классификация.
— статическая форма, когда увеличенный палец растет пропорционально росту всего тела.
— прогрессирующая форма, когда увеличенный палец растёт интенсивней, чем весь организм.
Гистологическая классификация:
— I тип – липофиброматоз, увеличение объёма сегмента за счёт подкожножировой клетчатки.
— II- нейрофиброматоз, характеризуется увеличением в объёме нервных стволов, чаще срединного и локтевого нервов. Является проявлением болезни Recklinghausen.
-III тип — истинный, характеризуется увеличением в объеме сегмента за счёт костного скелета, мягких тканей, исключая нервные элементы.
Клинические проявления.
Липофиброматоз встречается в 0,5% среди всех аномалий кисти. Макродактилия пальца может быть симметричной и асимметричной, что связано с равномерностью распределения подкожно-жирового слоя на протяжении. Может иметь место контрактура межфаланговых суставов, переразгибание ногтевой фаланги. В 90 % случаев это односторонний процесс и только в 8% случаев может быть связан с генетической мальформацией и сочетаться с синдактилией.
Нейрофиброматоз, или болезнь Recklinghausen- это системное заболевание, которое характеризуется гиперплазией и неоплазией соединительных волокон нервной системы. Это заболевание проявляется наличием пигментных пятен по всему телу, опухолевидных образований периферических нервных стволов. Это аутосомно- доминантное заболевание, но может иметь место и спорадичские случаи. Выявляется у 1 : 3000 новорождённых. Чаще всего вовлекается в процесс срединный нерв.
При III типе имеет место увеличение в объёме фаланг пальцев, нервы никогда не вовлечены в процесс.
Лечение. Зависит от типа макродактилии, возраста пациента, степени прогрессирования заболевания. Необходимо хирургическое лечение, заключающееся в экономной резекции мягких тканей и фаланг, коррекции костных деформаций, закрытии функционирующих зон роста.
Гипоплазия первого пальца.
Это врожденная аномалия, проявляющаяся в различной степени недоразвития первого пальца или его аплазией.
Эпидемиология. Встречается в 3,5% случаев всех врождённых аномалий верхней конечности. В 59 % случаев сочетается с врождённой лучевой косорукостью. В 63% случаев двухсторонний процесс.
Классификация Blauth, (1967), дополненная Manske в 1977 году и Paul Smith в 2002 году. Эта классификация базируется на степени недоразвития костного скелета первого луча.
I степень – первый палец уменьшен в размере, кости имеют нормальную структуру, все компоненты присутствуют.
II A степень- первый палец уменьшен в размере, костная структура нормальная, гипоплазия мышц тенора, одноплоскостная нестабильность пястно-фалангового сустава, узкий первый межпальцевой промежуток.
IIB степень- первый палец уменьшен в размере, фаланги пальца уменьшены в размере, гипоплазия мышц тенора, многоплоскостная нестабильность пястно-фалангового сустава. узкий первый межпальцевой промежуток,
IIIA степень – гипоплазия костного скелета первого пальца, аплазия мышц тенора, пястная кость нормальной длины, пястно-запястный сустав нормальный, изменение сухожилий, узкий первый межпальцевой промежуток.
IIIB степень — гипоплазия костного скелета первого пальца, аплазия мышц тенора, аплазия проксимальной части первой пястной кости, пястно-запястный сустав нестабильный, изменение сухожилий, узкий первый межпальцевой промежуток.
IV степень- болтающийся палец, когда первый палец расположен на кожной ножке, где проходит собственный сосудисто-нервный пучок, узкий первый межпальцевой промежуток.
V степень- аплазия первого пальца.
Клинические проявления. Характеризуется уменьшением в размере первого пальца, слабостью мышц, снижением функции кисти, нестабильностью суставов первого луча. Наиболее сложным является лечение детей с аплазией первого пальца.
Лечение. Консервативное лечение показано только в том случае, если имеется только уменьшение размера пальца без ограничения функции.
При I, IIA, IIB, IIIA степени гипоплазии первого пальца показана его реконструкция. Различные виды оппонентной пластики.
При IIIB, IV, V показана поллицизация II пальца кисти.
При раннем лечении в возрасте 1- 3 года функциональный результат хороший. Чем позже выполняются реконструктивные вмешательства, тем хуже прогноз и функциональный исход.
Врождённые перетяжки.
Эпидемиология. Встречается у 1 новорождённого из 15 тыс. характеризуется наличием фиброзного тяжа, который сдавливает мягкие ткани.
Клиника. Проявляется наличием втянутых циркулярных перетяжек на уровне фаланг пальцев кисти, предплечье, голени. Глубина и протяжённость перетяжек различная. При глубоких, циркулярных перетяжках со временем наступает нарушение лимфооттока, кровообращения. Поэтому данная патология требует раннего хирургического лечения.
Лечение заключается в иссечение перетяжек и кожной пластикой.
Клинико-рентгенологические особенности и Классификация проксимальных форм эктромелии верхних конечностей (обзор литературы) Текст научной статьи по специальности «Клиническая медицина»
КЛИНИКО-РЕНТГЕНОЛОГИЧЕСКИЕ ОСОБЕННОСТИ И КЛАССИФИКАЦИЯ ПРОКСИМАЛЬНЫХ ФОРМ ЭКТРОМЕЛИИ ВЕРХНИХ КОНЕЧНОСТЕЙ (ОБЗОР ЛИТЕРАТУРЫ)
A.A. Кольцов
ФГУ «Санкт-Петербургский научно-практический центр медико-социальной экспертизы, протезирования и реабилитации инвалидов им. Г.А. Альбрехта Росздрава»,
генеральный директор — д.м.н. профессор И.В. Шведовченко Санкт-Петербург
Проксимальная эктромелия — это один из наиболее редких и тяжёлых вариантов недоразвития верхних конечностей, при котором затронуты все их сегменты, а степень поражения уменьшается в дистальном направлении. Впервые этот термин был предложен L. Henkel и H.G. Willert в 1969 году по отношению к аномалиям нижних конечностей, при которых наиболее выраженная редукция наблюдалась со стороны бедренной кости при относительной сохранности голени и стопы [24]. Применительно к патологии рук данный термин начал использоваться совсем недавно: И.В. Шведовченко с соавторами впервые предложили его для обозначения такого варианта недоразвития, при котором поражены преимущественно проксимальные сегменты [11, 13]. Частота данного порока развития составляет не более 1,4 % всех аномалий верхних конечностей [11], а по данным A.E. Flatt [21] — менее 1%. В наиболее тяжёлых случаях недоразвития внешний вид укороченной и деформированной конечности напоминает ласту тюленя, с чем связано более распространённое название данной патологии — фокомелия (от греч. «forns» — тюлень, «melos» — конечность). Этот термин был впервые предложен Isidore Geoffroy-Saint-Hilaire в 1836 г. [23].
Впервые этот термин был предложен L. Henkel и H.G. Willert в 1969 году по отношению к аномалиям нижних конечностей, при которых наиболее выраженная редукция наблюдалась со стороны бедренной кости при относительной сохранности голени и стопы [24]. Применительно к патологии рук данный термин начал использоваться совсем недавно: И.В. Шведовченко с соавторами впервые предложили его для обозначения такого варианта недоразвития, при котором поражены преимущественно проксимальные сегменты [11, 13]. Частота данного порока развития составляет не более 1,4 % всех аномалий верхних конечностей [11], а по данным A.E. Flatt [21] — менее 1%. В наиболее тяжёлых случаях недоразвития внешний вид укороченной и деформированной конечности напоминает ласту тюленя, с чем связано более распространённое название данной патологии — фокомелия (от греч. «forns» — тюлень, «melos» — конечность). Этот термин был впервые предложен Isidore Geoffroy-Saint-Hilaire в 1836 г. [23].
Цель данной работы — на основании результатов предшествующих исследований определить наиболее типичные клинические и рентгенологические особенности верхних конечностей при их проксимальной эктромелии; терминологию, наиболее точно характеризующую суть патологических изменений; установить необходимость разработки классификации данного варианта недоразвития рук. Задачами работы являются изучение и анализ литературных данных, посвя-щённых клинико-рентгенологическим признакам проксимальной эктромелии; терминологии, применяемой для её описания; имеющихся классификаций данной патологии.
В отечественной литературе публикации, по-свящённые данной патологии, крайне редки и представляют собой описания результатов протезирования или хирургического лечения одного или нескольких больных, либо общую характеристику группы пациентов с различными врождёнными аномалиями верхних конечностей и фокомелией в том числе, причём число последних, как правило, не превышает 3 — 5 человек. Авторами таких публикаций являются в основном сотрудники Центрального и Ленинградского научно-исследовательских институтов протезирования. Оригинальные статьи, посвящённые изучению каких-либо аспектов данной патологии, в том числе клинико-рентгенологических особенностей и классификации, отсутствуют.
Авторами таких публикаций являются в основном сотрудники Центрального и Ленинградского научно-исследовательских институтов протезирования. Оригинальные статьи, посвящённые изучению каких-либо аспектов данной патологии, в том числе клинико-рентгенологических особенностей и классификации, отсутствуют.
Л.М. Воскобойникова и Т.В. Агуреева [9] в обзорной статье, посвящённой проблемам протезирования детей с различными врождёнными недоразвитиями рук, описали только один случай фокомелии левой верхней конечности у девочки 14 лет, при котором имели место рудименты плечевой кости и костей предплечья, двухлу-чевая кисть с трёхфаланговыми пальцами.
В работе А.Н. Витковской и Л.Е. Войновой [7], обобщающей опыт атипичного протезирования 57 детей дошкольного возраста с врождёнными дефектами верхних конечностей за 4 года, только у одного ребёнка — девочки трёх лет -была недоразвита правая верхняя конечность по типу фокомелии. Анатомические особенности заключались в резкой асимметрии надплечий, приподнятости ключицы, двухпалой рудиментарной кисти, прилежащей к области плечевого сустава. Объём движений в пальцах был резко ограничен, однако девочка могла захватывать и удерживать мелкие предметы за счёт сведения пальцев, а также дотягиваться пальцами до рта с помощью движений плечевым поясом и компенсаторных движений головой.
С.Г. Вербина [5] описала группу из 6 детей в возрасте от 4 до 13 лет, у которых плечевой сустав не был сформирован, а «зачаток плечевой кости» находился в состоянии анкилоза с более сохранной локтевой костью, к дистальному концу которой примыкала недоразвитая двух-, трёх — или четырёхпалая кисть с отсутствием I пальца. У всех больных практически полностью отсутствовали активные движения на уровне сочленения конечности с грудной клеткой, наблюдалось умеренное ограничение пассивных движений в этой зоне (до 90°).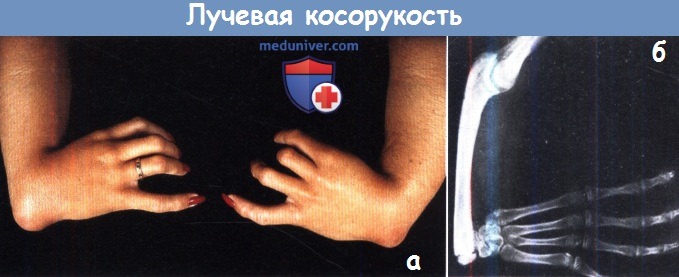 Интересно, что в этой же статье упоминается девочка 7 лет, верхняя конечность которой была представлена трёхпалой кистью, прилежащей к резко деформированному и укороченному надпле-чью, длина которого составляла 45% от длины противоположного здорового. Было возможно активное сгибание и отведение кисти до 25°, пассивное -до 90°. Годом позже тот же автор [6] привела описание мальчика 9 лет, правая верхняя конечность которого находилась в зоне сочленения с лопаткой в положении сгибания под углом 35° и представляла собой рудимент плечевой кости длиной 3,5 см, дугообразно изогнутой локтевой и значительно укороченной лучевой костями, пятипалой кистью с гипоплазированным I пальцем и резким ограничением движений в кистевом суставе и суставах пальцев. На уровне сочленения конечности с туловищем была отмечена возможность активного сгибания до 90° (т.е. объём сгибания составлял 55°), отведения — до 40°, разгибания конечности — до 15° от её исходного положения. В данной работе С.Г. Вербина отмечала, что при недоразвитии проксимального отдела плечевой кости, как правило, имеет место недоразвитие плечевого пояса — деформация лопатки, искривление и недоразвитие ключицы.
Интересно, что в этой же статье упоминается девочка 7 лет, верхняя конечность которой была представлена трёхпалой кистью, прилежащей к резко деформированному и укороченному надпле-чью, длина которого составляла 45% от длины противоположного здорового. Было возможно активное сгибание и отведение кисти до 25°, пассивное -до 90°. Годом позже тот же автор [6] привела описание мальчика 9 лет, правая верхняя конечность которого находилась в зоне сочленения с лопаткой в положении сгибания под углом 35° и представляла собой рудимент плечевой кости длиной 3,5 см, дугообразно изогнутой локтевой и значительно укороченной лучевой костями, пятипалой кистью с гипоплазированным I пальцем и резким ограничением движений в кистевом суставе и суставах пальцев. На уровне сочленения конечности с туловищем была отмечена возможность активного сгибания до 90° (т.е. объём сгибания составлял 55°), отведения — до 40°, разгибания конечности — до 15° от её исходного положения. В данной работе С.Г. Вербина отмечала, что при недоразвитии проксимального отдела плечевой кости, как правило, имеет место недоразвитие плечевого пояса — деформация лопатки, искривление и недоразвитие ключицы.
В 1980 г. Е. Cherstvoy с соавторами [33] описали случай мёртворождения ребёнка с двухсторонней фокомелией верхних конечностей. Справа имела место аплазия плечевой кости, костей предплечья, а трёхлучевая кисть с гипоплазированным большим пальцем и тотальным мягкотканным сращением II и III пальцев сочленялась с туловищем. Слева отмечалась гипоплазия плечевой и лучевой костей, отсутствие локтевой, аналогичная аномалия кисти с недоразвитием II и III пальцев по типу эктро дактилии.
И. В. Шведовченко и В. С. Прокопович [11] описали результаты реконструкции плечевой кости у трёх больных, наблюдавшихся в ФГУ РНИДОИ имени Г.И. Турнера. У всех пациентов проксимальная эктромелия была диагностирована слева, при этом определялись выраженная гипоплазия дельтовидной мышцы вплоть до её отсутствия, гипоплазия лопатки с уменьшением её линейных размеров на 10 — 15% при сохранении суставной поверхности. Плечевая кость была представлена в основном
Плечевая кость была представлена в основном
её дистальиой половиной, укорочена на 60 — 80 %, при этом у одного из трёх детей остаток плеча и предплечье были смещены на переднюю поверхность грудной клетки с проецированием на уровне средней трети ключицы. Амплитуда пассивных движений в сохранённом, но гипоплазированном локтевом суставе составляла 30 — 50°. Предплечье у всех трёх больных состояло из единственной локтевой кости, кисть была смещена в лучевую сторону, у двух детей отмечалась аплазия первого и второго лучей кисти, у одного отсутствовали первый, второй и третий лучи.
Большой вклад в изучение данной проблемы внесла И.А. Барабаш [3, 4, 8], в течение ряда лет занимавшаяся проблемой протезирования верхних конечностей у детей с фокомелией в Ленинградском институте протезирования. В соавторстве с И. П. Беловой и П.Н. Уваровым ею был проведён анализ анатомо-функциональных особенностей недоразвития верхних конечностей у 493 детей за 15 лет. Из всего контингента больных фокомелия была выявлена только у 12 человек [3]. Она подчёркивала, что отличительной особенностью фокомелии верхних конечностей является наличие активных сгибания и отведения кисти в зоне сочленения её с надплечьем [4]. Наибольшее число наблюдений в отечественной литературе принадлежит именно И.А. Барабаш в соавторстве с Л.Е. Войновой [8], которые в 1987 г. упоминают о 28 пациентах с фокомелией и о 15 пациентах с пороками развития проксимального отдела плеча (рис. 1), но не проводят анализа данной группы с целью подробной кли-нико-рентгенологической оценки, создания тератологического ряда и классификации.
Рис. 1. Фокомелия (проксимальная эктромелия) левой верхней конечности: схема строения конечности: конечность представлена четырехпалой кистью, сочленяющейся с единственной рудиментарной костью предплечья.
В зарубежной литературе удалось обнаружить большее количество работ по данной проблеме. Так, краткие упоминания о фокомелии были сделаны A. Church и F. Peterson в 1911 г. [16], L. Aschoff и E. Kaufmann в 1928 и 1929 гг. [15]. Наиболее представительным сообщением была публикация B.M. Patten в 1946 г. [30], в которой он привёл серию наблюдений с описанием больных и их фотографиями. Известно, что до 1944 г. было описано всего 44 случая фокомелии [18]. Большинство сообщений относятся к 1960 — 1970 годам, что во многом связано с «талидомидной» катастрофой, постигшей многие страны, из которых наиболее сильно пострадали Англия, Австралия и Япония. Талидомид был синтезирован в Западной Германии в 1954 г. и после многочисленных тестов, показавших его низкую токсичность, допущен к применению в качестве мягкого седативного и снотворного средства сначала в ФРГ в 1957 г., затем в других странах [17]. С 1959 г. резко возросло количество детей, рождающихся с грубыми пороками развития конечностей, в том числе с фокомелией. По оценкам разных авторов [17, 25, 29, 34, 36], в 1960-1961 гг. в Западной Германии родились от 3000 до 6500, в Англии — 500, в Японии — 700 детей с грубыми аномалиями конечностей, в том числе с фокомели-ей. С 1963 г. частота врождённых аномалий конечностей резко снизилась, хотя относительное увеличение числа новорождённых с недоразвитиями конечностей сохранялось ещё в течение ряда лет.
Так, краткие упоминания о фокомелии были сделаны A. Church и F. Peterson в 1911 г. [16], L. Aschoff и E. Kaufmann в 1928 и 1929 гг. [15]. Наиболее представительным сообщением была публикация B.M. Patten в 1946 г. [30], в которой он привёл серию наблюдений с описанием больных и их фотографиями. Известно, что до 1944 г. было описано всего 44 случая фокомелии [18]. Большинство сообщений относятся к 1960 — 1970 годам, что во многом связано с «талидомидной» катастрофой, постигшей многие страны, из которых наиболее сильно пострадали Англия, Австралия и Япония. Талидомид был синтезирован в Западной Германии в 1954 г. и после многочисленных тестов, показавших его низкую токсичность, допущен к применению в качестве мягкого седативного и снотворного средства сначала в ФРГ в 1957 г., затем в других странах [17]. С 1959 г. резко возросло количество детей, рождающихся с грубыми пороками развития конечностей, в том числе с фокомелией. По оценкам разных авторов [17, 25, 29, 34, 36], в 1960-1961 гг. в Западной Германии родились от 3000 до 6500, в Англии — 500, в Японии — 700 детей с грубыми аномалиями конечностей, в том числе с фокомели-ей. С 1963 г. частота врождённых аномалий конечностей резко снизилась, хотя относительное увеличение числа новорождённых с недоразвитиями конечностей сохранялось ещё в течение ряда лет.
Несмотря на кажущееся огромное количество характерных недоразвитий конечностей, в подавляющем большинстве статей приводятся результаты наблюдения отдельных пациентов и ближайшие результаты их абилитации. Так, F.J. Coodin [17] упоминает о нескольких мальчиках с двухсторонней фокомелией верхних конечностей и приводит подробную рентгенологическую характеристику. В частности, у одного пациента отмечалось наличие истончённых плечевых костей с невыраженным проксимальным отделом и сращённых друг с другом костей предплечий, лучевой косорукости четырёхпалых кистей с отсутствующим I пальцем, у второго — похожая деформация справа и не упоминаемая до сих пор нами деформация слева: наличие единой неясной формы рудиментарной кости вместо костей плеча и предплечья, трёхпалая кисть в положении ладонной девиации с отсутствующим первым пальцем.
P.M. Dunn с соавторами [19] в 1962 г. описали 4 детей с различными степенями недоразвития рук: от крайне тяжёлого порока — единственного трёхфалангового луча кисти, прилежащего к надплечью, до наличия у мальчиков-близнецов симметричного двухстороннего поражения, при котором каждая из порочных конечностей была
образована единой костью плеча-предплечья, сочленяющейся с трёх- или четырёхлучевой кистью. В 9 месяцев оба близнеца предпринимали активные попытки использования рудиментарных конечностей.
В первом сообщении о тератогенном влиянии талидомида в Финляндии I. Vaananen с соавторами [36] привёл описание двух случаев фоко-мелии верхних конечностей с различной клини-ко-рентгенологической картиной. У одной из больных имело место двухстороннее недоразвитие, выражающееся в наличии рудиментов дисталь-ных отделов плечевых костей длиной до 2,5 см, сращённых костях предплечья, лучевой косорукости, трёхпалых кистях, гипоплазии I пальца III степени по Blauth слева. У другой было выявлено только левостороннее поражение: рудиментарная плечевая кость длиной 2,0 см, резко недоразвитая локтевая кость кубовидной формы длиной 4,0 см, сращение относительно сохранной лучевой кости с первым лучом кисти.
P.S. Dignan с соавторами [18] опубликовали результаты наблюдений за 5 больными, у которых были выявлены пятипалые кисти, прилежащие к резко недоразвитому плечевому поясу. Подвижность пальцев у всех пациентов была ограничена в разной степени, отмечалась характерная мягко-тканная синдактилия пальцев. Рентгенологически у части пациентов обнаружены небольшие костные рудименты между гипоплазированной суставной поверхностью лопатки и кистью.
Девочку с похожей клинико-рентгенологичес-кой характеристикой (пятипалой кистью, сочленяющейся напрямую с надплечьем) описал в 1990 г. A. Schinzel [31].
A. Schinzel [31].
Наибольшее количество наблюдений, проанализированных с точки зрения клинико-рентгенологи-ческих особенностей, принадлежит G. Tytherleigh-Strong и G. Hooper [35], которые в 2003 г. описали 24 пациента (44 конечности).
При изучении литературных данных стало очевидно, что существуют несколько основных терминов, употребляемых для характеристики этого типа недоразвития верхних конечностей. Сам термин «проксимальная эктромелия» применительно к патологии рук впервые был применен только в отечественной литературе в работах И.В. Шведовчен-ко с соавторами [11, 13]. В зарубежной литературе такой диагноз встречается крайне редко и только при описании недоразвития нижних конечностей [24]. Более того, некоторые авторы указывают на невозможность его использования по отношению к аномалиям развития рук в связи с тем, что, в отличие от пороков развития ног, недоразвитие проксимальных сегментов верхних конечностей не бывает изолированным, а всегда сопровождается поражением других отделов конечности [24].
Термин «недоразвитие всех сегментов конечности» [6, 9], часто встречающийся в отечественной литературе, не совсем точно соответствует истинным анатомическим особенностям при изучаемой патологии, так как сегменты всегда гипоплазирова-ны в разной степени, что никак не отражается в данном диагнозе. Иногда авторы [5] вообще избегают точных формулировок диагноза заболевания, обозначая его просто как «аномалию развития» или «врождённое недоразвитие», приводя клинико-рен-тгенологическое описание.
В зарубежной литературе достаточно распространённым термином является «дизмелия». Он впервые был введён в употребление в 1962 г. H.-R. Wiedemann в отношении недоразвитий конечностей при талидомидной эмбриопатии и используется для определения группы заболеваний, при которых имеет место гипоплазия и частичная или тотальная аплазия трубчатых костей конечностей, варьирующая от изолированной периферической гипоплазии до полного отсутствия конечности [38], в том числе для характеристики проксимальных форм эктро-мелии.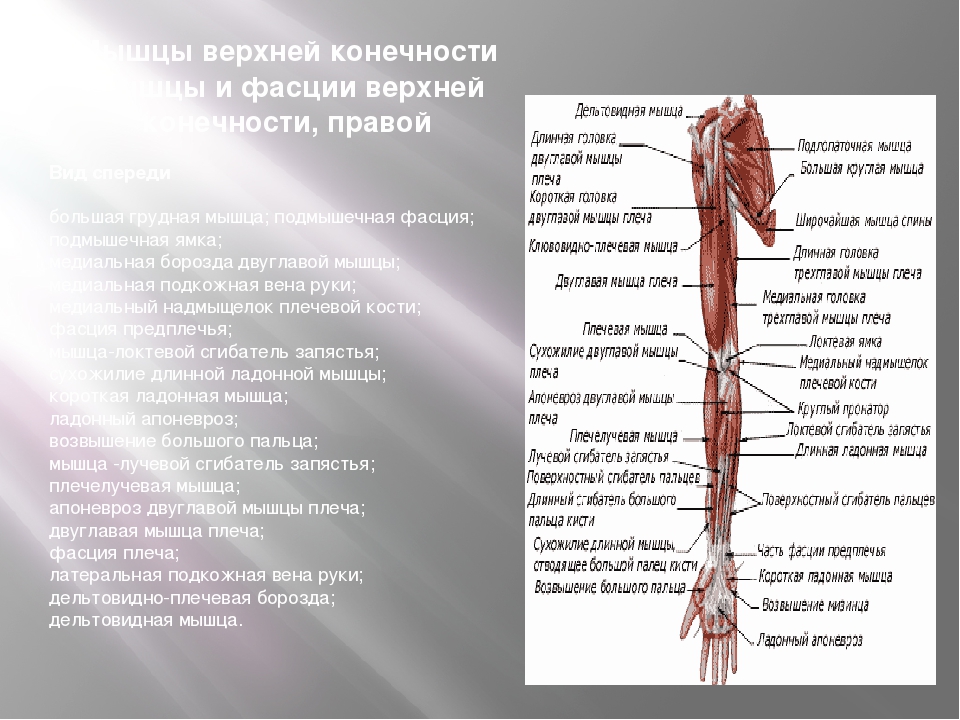 Однако очевидно, что данное понятие, ха-растеризующее целую группу заболеваний, сопровождающихся грубыми анатомическими и функциональными дефектами, не может применяться для обозначения конкретного варианта недоразвития.
Однако очевидно, что данное понятие, ха-растеризующее целую группу заболеваний, сопровождающихся грубыми анатомическими и функциональными дефектами, не может применяться для обозначения конкретного варианта недоразвития.
Наиболее распространённым термином является «фокомелия». Интересно, что как отечественные, так и зарубежные авторы дают различные определения данного понятия. Так, ещё в 1960 г. В.А. Штурм определял фокомелию как «недоразвитие проксимального сегмента конечности» [14], в то время как в современных отечественных руководствах по медицинской генетике [2] даётся более развёрнутая характеристика: «Фокомелия — врождённое отсутствие или значительное недоразвитие проксимальных частей конечностей, вследствие чего нормально развитые кисти кажутся прикреплёнными непосредственно к туловищу». Таким образом, если в первом случае в определении напрямую указывается на поражение, в первую очередь, сегмента плеча, то во втором — как сегмента плеча, так и предплечья, причём без уточнения степени их недоразвития. Несмотря на такие несоответствия, анализ отечественной литературы показал, что во всех случаях [3, 4, 7, 8] данный термин употреблялся именно по отношению к наиболее тяжёлым степеням проксимальной эктромелии.
Совершенно другая, гораздо более запутанная ситуация, наблюдается в зарубежной литературе, где авторы трактуют этот диагноз принципиально по-разному. Например, M.A. Entin [20] указывал, что для фокомелии характерно отсутствие всех структур предплечья, т.е. кисть сочленяется прямо с плечевой костью. L. Henkel и H.G. Willert
[24] применяли это название для тех степеней дизмелии, при которых нет остатков длинных трубчатых костей между поясом верхней конечности и её периферической частью (кистью). S.H. Kozin [27] определяет фокомелию как «отсутствие промежуточных сегментов конечности, то есть интеркал арную, или промежуточную, аплазию плеча, предплечья или их обоих».
Все описанные выше представления о фокомелии нашли отражение в её первой классификации, предложенной C.H. Frantz и R.O’Rahilly в 1961 г. в рамках разработанной ими классификации пороков развития конечностей [22]. Авторами были выделены 3 типа патологии: I тип — полная, когда кисть или пальцы прилежат прямо к туловищу; II — проксимальная, когда между кистью и туловищем расположены кости предплечья; III — дистальная, при которой кисть прилежит прямо к плечевой кости. При этом фо-комелия в целом была расценена как интерка-ларный (межсегментарный) порок развития.
A.B. Swanson с соавторами [32] разработали одну из наиболее известных и применяемых в мировой практике классификаций аномалий развития конечностей, принятую Американским обществом хирургии кисти и положенную в основу классификации Международной федерации обществ хирургии кисти (IFSSH). Они поддержали принцип разделения фокомелии на 3 типа: полную, проксимальную и дистальную, однако усомнились в правомочности выделения интер-каларных недоразвитий в отдельную группу в связи с наличием поражения и дистальных сегментов, в частности, кисти. Поэтому в классификации IFSSH был выделен интеркаларный (межсегментарный) тип продольного недоразвития конечностей, в который и была включена фоко-мелия [32].
Наиболее полная и продуманная попытка классифицировать фокомелию верхних конечностей была предпринята G. Tytherleigh-Strong и G. Hooper [35] на основе наблюдения за 24 пациентами с 44 недоразвитыми конечностями. Первоначально авторы попытались разделить все порочные конечности на три группы в соответствии с классификацией C.H. Frantz и R. O’Rahilly [22], однако только в 11 случаях это было успешным: 9 конечностей были недоразвиты по типу полной фокомелии, а 2 — по типу дистальной. Оставшиеся 33 конечности не укладывались ни в одну из трёх групп, так как были образованы рудиментами и плеча, и предплечья. Анализ клинических и рентгенологических особенностей этих конечностей позволил выделить 3 подгруппы аномалий на основании анатомических соотношений сегментов плеча и предплечья (рис. 2):
Рис. 2. Классификация фокомелии: тип А — недоразвитая плечевая кость и единственная кость предплечья; В — недоразвитые плечевая кость и обе кости предплечья; С — синостоз рудимента плечевой кости с костью (костями) предплечья.
— тип А, характеризующийся недоразвитой плечевой костью и единственной костью предплечья;
— В — недоразвитые плечевая кость и обе кости предплечья;
— С — рудимент плечевой кости образует единый костный блок с костью или костями предплечья.
Авторы указывают, что ни одна из порочных верхних конечностей, наблюдаемых ими, не может быть отнесена к чистым интеркаларным недоразвитиям в связи с наличием признаков поражения всех сегментов и соглашаются с A.B. Swanson [32] в том, что фокомелия представляет собой вариант продольной эктромелии.
Итак, по представлениям большинства западных исследователей, фокомелия является единым, но очень полиморфным заболеванием. Однако в некоторых отечественных и зарубежных публикациях указывается на то, что она объединяет в себе принципиально разные патологические состояния. В частности, современные данные, полученные И.В. Шведовченко и А.В. Минькиным [10, 12], свидетельствуют о том, что так называемая дистальная форма фокомелии, при которой порочная кисть сочленяется с недоразвитой плечевой костью (рис. 3), представляет собой не что иное, как одну из форм плечелу-чевого синостоза (ПЛС).
Рис. 3. Плечелучевой синостоз слева, третья форма: рентгенограмма.
Таким образом, во многих работах под данным термином скрываются совершенно иные формы недоразвития рук. Например, P.S. Dignan с соавторами [18] приводит данные о 5 пациентах с фокомелией при талидомидной эмбриопа-тии, однако при тщательном изучении описаний недоразвитых конечностей удалось установить, что только у 4 больных отмечалась проксимальная эктромелия, в то время как у одного — ПЛС. Аналогичным образом в работе R.L. Van Der Horst [37], содержащей описание аномального расположения подключичной артерии при фокомелии правой руки у мальчика народности банту, кли-нико-рентгенологическая характеристика конечности соответствует именно плечелучевому синостозу.
С другой стороны, из трёх случаев фокомелии верхних конечностей, описанных F.J. Coodin с соавторами [17], только в двух имела место проксимальная эктромелия, в то время как клинико-рентгенологическое описание третьего больного указывало на наличие у него тяжёлой степени двухсторонней лучевой косорукости.
Наконец, иногда встречаются абсолютно курьёзные публикации, в которых авторы называют фокомелией состояния, даже отдалённо её не напоминающие. В частности, H.A. Johnson [26] при описании способа создания I пальца у больного с врождённой культёй кисти на уровне костей запястья характеризовал данное недоразвитие именно как фокомелию.
Таким образом, термин «фокомелия» обозначает определённый внешний вид руки, резко укороченной, с невыраженными сегментами плеча и предплечья, кистью, отходящей от туловища и состоящей из нескольких лучей, и вовсе не характеризует качественный и количественный состав скелета конечности. В то же время, как видно из приведённых выше данных, во многих случаях так называют некоторые формы проксимального недоразвития конечностей, которые клинически совсем не похожи на ласты тюленей [33, 35, 36].
Некоторые зарубежные авторы вместо термина «проксимальная» использовали такой диагноз, как «аксиальная» эктромелия [21]. Объяснением такого подхода являлось то, что при подобных формах недоразвития, как правило, порочны все сегменты -плечо, предплечье и кисть, то есть вся ось конечности, поэтому термин «аксиальная», или осевая (поражающая всю ось конечности), наиболее приемлем. Однако в данной ситуации возникает точно такое же несоответствие формулировки диагноза принципиальным анатомическим особенностям строения руки, как и в случае использования в отечественной литературе термина «недоразвитие всех сегментов»: ни в том, ни в другом случае не отражается превалирующая роль недоразвития проксимальных отделов. Вместе с тем именно L. Henkel и H.G. Willert [24], предложившие термин «аксиальная эктромелия», предложили разделять её в зависимости от степени редукции плечевой кости на 6 типов, которые приведены ниже (рис. 4).
Рис. 4. Классификация аксиальной эктромелии верхних конечностей:
а — длинный аксиальный тип, характеризующийся гипоплазией или частичной аплазией плечевой кости с частичной аплазией лучевой кости и лучелоктевым синостозом;
Ь — длинный аксиальный тип, характеризующийся аналогичным недоразвитием плечевой и тотальной аплазией лучевой костей;
с — промежуточный аксиальный тип, для которого типичны субтотальная аплазия плечевой и частичная лучевой костей, лучелоктевым синостозом;
d — промежуточный аксиальный тип, для которого типичны субтотальная аплазия плечевой и тотальная — лучевой костей;
е — короткий аксиальный тип, при котором отмечаются тотальная аплазия плечевой и частичная аплазия лучевой костей, лучелоктевой синостоз;
{ — короткий аксиальный тип, при котором отмечаются тотальная аплазия плечевой и лучевой костей.
Очевидно, что из всех упомянутых ранее классификаций данная наиболее детально отражает анатомические особенности при различных формах проксимальной эктромелии. Однако она не получила широкого практического применения, в связи с чем не отражена в современной литературе.
Заключение
Таким образом, наиболее распространённые термины («фокомелия», «дизмелия», «недоразвитие всех сегментов конечности», «аксиальная эктромелия») характеризуют целую группу грубых аномалий рук. При этом фокомелия указывает только на внешний вид конечности, не учитывая её анатомическое строение; дизмелия констатирует нарушение формирования конечности, повлёкшее за собой значимое изменение её формы и функции, тогда как оставшиеся две формулировки указывают только на наличие недоразвития всех сегментов руки без уточнения их степени. Это означает, что перечисленные выше термины не отражают истинного характера клинических и рентгенологических изменений при описываемой в данной работе патологии, в связи с чем их использование не целесообразно.
Термин «проксимальная эктромелия» наиболее точно отражает суть анатомических и соответствующих им функциональных изменений и является оптимальным.
В отечественной и зарубежной литературе описаны различные формы проксимальной экт-ромелии рук: от наиболее лёгкой, когда конечность образована всеми тремя сегментами с преимущественным недоразвитием плечевой кости, до крайне тяжёлой, для которой характерно наличие рудиментарной кисти, прилежащей напрямую к туловищу. Все формы данного заболевания сопровождаются выраженными анатомическими и функциональными нарушениями.
Предыдущими исследователями было сделано всего несколько попыток классифицировать данную патологию. Предлагаемые классификации не охватывают весь спектр проксимальных форм недоразвития верхних конечностей. Кроме того, в имеющихся вариантах классификаций типы аномалии выделяются либо по принципу отсутствия плеча или предплечья, или обоих этих сегментов, что де-
лает подобное разделение очень грубым и принципиально неверным, либо по принципу степени редукции плечевой кости и костей предплечья, что усложняет классификацию и приводит к её неудобству в практическом применении.
Таким образом, можно заключить, что изучение проксимальных форм эктромелии верхних конечностей, создание рабочей классификации, простой и легко применимой практическими врачами в процессе абилитации пациентов, является актуальной задачей.
Литература
1. Абальмасова, Е.А. Врождённые деформации опорно-двигательного аппарата и причины их происхождения / Е.А. Абальмасова, Е.В. Лузина. — Ташкент : Медицина, 1976.
2. Аномалии развития : иллюстрированное пособие для врачей. — СПб. : Фолиант, 2007. — 323 с.
3. Барабаш, И.А Характеристика врождённых дефектов верхних конечностей у детей с точки зрения последующего протезирования / И.А. Барабаш, И.П. Белова, П.Н. Уваров // Протезирование и протезостроение : сб. трудов. — М., 1984. — Вып. 69. — С. 4-10.
4. Барабаш, И.А. Сенсорный способ управления протезами у детей с врождённой аномалией верхних конечностей / И.А. Барабаш // Протезирование и протезостроение : сб. трудов. — М., 1986. — Вып. 76. — С. 32-37.
5. Вербина, С.Г. Клиническая характеристика детей с ампутационными дефектами и аномалией развития верхних конечностей / С.Г. Вербина // Протезирование и протезостроение : сб. трудов. — М., 1982.
— Вып. 62. — С. 35-40.
6. Вербина, С.Г. Атипичное протезирование при аномалии развития сегментов верхних конечностей / С.Г. Вербина // Протезирование и протезостроение : сб. трудов. — М., 1983. — Вып. 64. — С. 42-45.
7. Витковская, А.Н. Опыт атипичного протезирования детей дошкольного возраста с врождёнными дефектами верхних конечностей / А.Н. Витковская, Л.Е. Войнова // Протезирование и протезостроение : сб. трудов. — М., 1973. — Вып. XXX. — С. 146-151.
8. Войнова, Л.Е. Результат хирургического лечения больного с фокомелией верхней конечности / Л.Е. Войнова, И.А. Барабаш // Протезирование и про-тезостроение : сб. трудов. — М., 1987. — Вып. 78.
— С. 161-164.
9. Воскобойникова, Л.М. Протезирование детей с врождённым недоразвитием верхних конечностей / Л.М. Воскобойникова, Т.В. Агуреева // Протезирование и протезостроение : сб. трудов. — М., 1969. — Вып. XXII. — С. 94-98.
10. Минькин, А.В. Медицинские аспекты реабилитации детей с врождённым плечелучевым синостозом : дис. … канд. мед. наук / Минькин А.В. — СПб., 2005. — 178 с.
11. Шведовченко, И.В. Оперативное лечение проксимальных форм врождённой эктромелии верхних конечностей у детей / И.В. Шведовченко, В.С. Про-копович // Вестн. травматологии и ортопедии им. Н.Н. Приорова. — 2002. — № 4. — С. 73-76.
12. Шведовченко, И.В. Клинико-рентгенологическая характеристика детей с врождённым плечелучевым
синостозом / И.В. Шведовченко, A.B. Минькин // Вестн. гильдии протезистов-ортопедов. — 2004. — № 2 (16). — С. 20-26.
13. Шведовченко И.В. Лечение детей с врождёнными пороками развития верхних конечностей // Травматология и ортопедия : руководство для врачей / под ред. Н.В. Корнилова. — СПб., 2005. — Т. 2. -С. 634-664.
14. Штурм, B.A. цит. по A. Абальмасовой, Е.В. Лузиной, 1976.
15. Aschoff, L., Kaufmann, E. цит. по W. Lenz, 1962.
16. Church, A., Peterson, F. цит. по F.E. Kratter, 1960.
17. Coodin, F.J. Phocomelia: report of three cases / F.J. Coodin, I.A. Uchida, C.H. Murphy // Clin. Med. Assoc. J. — 1962. — N 87. — P. 735-739.
18. Dignan, P.S. Phocomelia with congenital hypoplastic thrombocytopenia and myeloid leukemoid reactions / P.S. Dignan, A.M. Mauer, C. Frantz // J. Pediatr. -1967. — Vol. 70, N 4. — P. 561-573.
19. Dunn, P.M. Phocomelia / P.M. Dunn, A.M. Fisher, H.G. Kohler // Am. J. Obstetr. Gynecology. — 1962.
— Vol. 84, N 1. — P. 348-354.
20. Entin, M.A. Reconstruction of congenital abnormalities of the upper extremities / M.A. Entin // J. Bone Joint Surg. — 1959. — Vol. 41-A, N 4. — P. 681-701.
21. Flatt, A.E. The care of congenital hand anomalies / A.E. Flatt. — 2nd edn. — St. Louis : Quality Medical Publishing, 1994.
22. Frantz, C.H., o’Rahilly, R. цит. по G. Tytherleigh-Strong, G. Hooper, 2003.
23. Geoffroy-Saint-Hilaire, I. цит. по P.M. Dunn, 1962.
24. Henkel, L. Dysmelia. A classification and a pattern of malformation in a group of congenital defects of the limbs / L. Henkel, H.G. Willert // J. Bone Joint Surg.
— 1969. — Vol. 51-B, N 3. — P. 399-414.
25. Hirsch, W. Phocomelia / W. Hirsch // J. Int. College Surg. — 1963. — Vol. 39. — P. 238-251.
26. Johnson, H.A. Formation of a functional thumb post with sensation in phocomelia / H.A. Johnson // J Bone Joint Surg. — 1967. — Vol. 49-A, N 2. — P. 327-332.
27. Kozin, S.H. Upper-extremity congenital anomalies / S.H. Kozin // J. Bone Joint Surg. — 2003. — Vol. 85-A, N 8.
— P. 1564-1576.
28. Kratter, F.E. A case of phocomelia associated with severe mental deficiency / F.E. Kratter // Br. Med. J.
— 1960. — N 5169. — P. 328-329.
29. Lenz, W. Thalidomide and congenital anomalies / W. Lenz // Lancet. — 1962. -N 1. — P. 45.
30. Patten, B.M. цит. по W. Lenz, 1962.
31. Schinzel, A. Phocomelia and additional anomalies in two sisters / A. Schinzel // Hum. Genet. — 1990. -Vol. 84, N 6. — P. 539-541.
32. Swanson, A.B. A classification for congenital limb malformation / A.B. Swanson, G.D. Swanson, K. Tada / / J. Hand Surg. — 1983. — Vol. 8, N 5. — P. 693-702.
33.Syndrome of multiple congenital malformations including phocomelia, thrombocytopenia, encephalocele, and urogenital abnormalities / E. Cherstvoy [et al.] // Lancet.
— 1980. -Vol. 8192, N 2. — P. 485.
34.Taussig, H.B. A study of the German outbreak of phocomelia. The Thalidomide syndrome / H.B. Taussig // JAMA. — 1962. — June, 30. — P. 80-88.
35.Tytherleigh-Strong, G. The classification of phocomelia / G. Tytherleigh-Strong, G. Hooper // J. Hand Surg.
— 2003. — Vol. 28-B, N 3. — P. 215-217.
36.Vaananen, I. Two cases of phocomelia in Finnish Lapland / I. Vaananen, T. Joki //Ann. Paediatr. Fenn. — 1963. — Vol. 9. — P. 65-71.
37.Van der Horst, R.L. Anomalous origin of the subclavian
artery associated with phocomelia / R.L. Van der Horst, M.S. Gotsman // S. Afr. Med. J. — 1971. — Vol. 45, N 48. — P. 1397-1399. 38.Wiedemann, H.-R. n,m\ no L. Henkel, H.G. Willert, 1969.
РОССИЙСКИЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ ТРАВМАТОЛОГИИ И ОРТОПЕДИИ имени Р.Р. ВРЕДЕНА
в?гзеиовские чжгнил
7-8 декабря 2007 года
АКТУАЛЬНЫЕ ПРОБЛЕМЫ ЭНДОПРОТЕЗИРОВАНИЯ ТАЗОБЕДРЕННОГО И КОЛЕННОГО СУСТАВОВ
I. Первичная артропластика тазобедренного сустава: сложные случаи и новые технологии
II. Ревизионная артропластика ТБС
III. Первичная артропластика коленного сустава
С ДОКЛАДАМИ ВЫСТУПЯТ ВЕДУЩИЕ ОТЕЧЕСТВЕННЫЕ И ЗАРУБЕЖНЫЕ СПЕЦИАЛИСТЫ
По вопросам участия обращаться:
по тел.: 556-08-38 Шубняков И.И.
740-78-62 Локтионова В.В.
Рентген лучезапястного сустава (запястья)
directions
Лучезапястный сустав играет важную роль в движении руки, являясь одним из самых подвижных сочленений человеческого тела. Лучезапястный сустав – это опорно-связочный аппарат, соединяющий кости кисти и предплечья и отвечающий за движение, моторику кисти, ее вращение, сгибание-разгибание руки. Лучезапястный сустав состоит из большого количества мелких костей, что делает его довольно эластичным, но при этом подвергающимся частым травмам и заболеваниям. Любое нарушение и повреждение этого участка верхней конечности негативно сказывается на общем состоянии человека и ограничивает активность.
Врачи-специалисты
Рентгенолаборант
В настоящее время на сайте ведутся работы по изменению прайс-листа, актуальную информацию уточняйте по тел: 640-55-25 или оставьте заявку, с Вами свяжется оператор.
Цены на услуги
-
Рентгенография лучезапястного сустава в прямой ладонной проекции
900a
-
Рентгенография лучезапястного сустава в боковой ульнарной проекции
900a
-
Рентгенография лучезапястного сустава в косой ладонной проекции
900a
Информация и цены, представленные на сайте, являются справочными и не являются публичной офертой.
Наши клиники в Санкт-Петербурге
Медицентр Юго-Запад
Пр.Маршала Жукова 28к2
Кировский район
- Автово
- Проспект Ветеранов
- Ленинский проспект
Получить подробную информацию и записаться на прием Вы можете по телефону
+7 (812) 640-55-25
Рентгенография, как одно из самых информативных и наглядных диагностических исследований, помогает выявить различного рода повреждения, патологии развития, травмы, воспалительные процессы лучезапястного сустава, состояние костей, хрящей, мягких тканей нижней части предплечья, запястья. Самыми распространенными заболеваниями этой области верхней конечности являются артрит, артроз, тендинит, некроз, туннельный синдром, травмы, остеохондропатии.
Причины травм и заболеваний лучезапястного сустава
Падая, как правило, человек выставляет вперед руки для защиты тела и лица от удара, поэтому лучезапястный сустав травмируется чаще всего при падении на вытянутую руку с упором на ладонь и кисть. От механического воздействия и нагрузки массы тела кости легко подвергаются травмированию. В результате неудачных падений, падений с высоты, аварий и других несчастных случаев человек получает травмы различной степен тяжести, в том числе повреждения лучезапястного сустава.
Кроме того, переломам лучезапястного сустава подвержены пациенты, страдающие остеопорозом. Это заболевание вызывает повышенную хрупкость костей и снижение их плотности, что приводит к частым травмам.
Помимо механических повреждений причинами заболеваний лучезапястного сустава могут быть неправильные занятия спортом, старение организма, плохое питание, чрезмерные мышечные нагрузки, гиподинамия и т.д.
Быстрое и качественное рентгенологическое исследование лучезапястного сустава в Петербурге Вы можете сделать в травматологическом отделении сети клиник «Медицентр». Травмпункт «Медицентра» оборудован новейшим итальянским цифровым аппаратом Clinomat, многорежимным и практически безопасным, позволяющим производить полную рентгенологическую диагностику тела человека в кратчайшие сроки. Аппарат дает возможность специалистам «Медицентра» исследовать снимки в различных режимах обработки, быстро и точно устанавливать диагноз и назначать грамотное и эффективное лечение.
Показания к рентгенологическому исследованию лучезапястного сустава:
- переломы лучевой кости, перелом Галеацци, костей запястья;
- вывихи запястья;
- ушибы, трещины;
- острая боль;
- отек, гематомы;
- опухолевые, дегенеративно-воспалительные процессы;
- аномалии развития лучезапястного сустава и т.д.
Кроме того, рентгенографию лучезапястного сустава назначают при жалобах пациента на скованность движения кисти, хруст, боль и другие неприятные симптомы. Рентгенография лучезапястного сустава необходима и в период лечения для контроля его эффективности, либо перед проведением операции.
Противопоказания к рентгенологическому исследованию лучезапястного сустава
При назначении рентгенологического обследования каждый случай рассматривается индивидуально. Как правило, врачи стараются избегать рентгенографии лучезапястного сустава в случае беременности пациентки, общего тяжелого состояния больного. В этих случаях рентген рекомендуется заменять такими методами диагностики как УЗИ и МРТ.
Рентгенография или МРТ лучезапястного сустава?
Рентгенография является быстрым, доступным и достаточно точным диагностическим методом. Такой способ позволяет увидеть костную структуру, проанализировать ее состояние, обнаружить патологии и травмы, смещения и нарушения.
Но в случаях, когда надо оценить состояние мягких тканей, мышц и связок могут назначить МРТ. СКТ или МРТ лучезапястного сустава назначают при подозрениях на некоторые заболевания, такие как: туннельный синдром, онкологические болезни, аномалии развития кисти и т.д. Как правило, лечащий врач самостоятельно решает вопрос о необходимости назначения того или иного метода диагностического обследования пациента, учитывая характер заболевания, травмы и нарушения, особенности организма пациента, его возраст, состояние и другие индивидуальные особенности.
Подготовка к проведению рентгенологического исследования лучезапястного сустава
Перед рентгенографией лучезапястного сустава нужно снять металлические предметы с руки и ювелирные украшения, пластырь, масляные и йодные повязки. При наличии гипса, необходимо уточнить у врача, требуется ли его удаление.
Процедура проведения рентгенологического исследования лучезапястного сустава
Рентгенография лучезапястного сустава проводится в двух проекциях. На самом снимке должны быть одновременно видны нижние трети костей предплечья, кости запястья, область лучезапястного сустава, отделы пястных костей, т.к. часто повреждение лучезапястного сустава путают с повреждением запястья.
Вся процедура занимает около 5 минут, а доза излучения очень мала. После окончания рентгенологического обследования пациент получает снимок и его описание и направляется на консультацию и дальнейшее лечение к соответствующему специалисту.
1299,832,811,840,1305,1269
Дашкина Альфия Рашитьевна
26.12.2020
18:55
medi-center.ru
Хочу поблагодарить терапевта Дерешовского Александра Сергеевича за внимательность, ответственность, и главное, за эффективность лечения! Александр Сергеевич был на связи все время, подробно и доступно все объяснял и отвечал на вопросы.
Спасибо, Александр Сергеевич, за Ваш профессионализм и отношение к пациентам!
Иванова Ольга Сергеевна
18.12.2020
23:15
medi-center.ru
Огромная благодарность Шаговой Любови Сергеевне! Три недели не могла решить свою проблему со здоровьем, но тут, по совету знакомой, записалась к Любовь Сергеевне, и это любовь с первого взгляда! И все лечение очень помогло, и сама врач очень помогла. Очень приветливая и вежливая доктор!
Здравствуйте!
Выражаю благодарность всем сотрудникам медцентра и лично Поддубной Анастасии Михайловне за работу в такое непростое время, хорошие и результативные рекомендации по лечению. Переболел COVID, благодаря вам иду на поправку!
Хочу выразить благодарность Агамурату Оразмамедовичу, за отзывчивость и профессиональную помощь моему сыну, спасибо вам за нашу ручку!!!
Хочу выразить большую благодарность клинике на аллее Поликарпова 6к2, всегда вежливые, доброжелательные девушки на ресепшн. Помогли удобно состыковать время приёма врачей. А также отдельно поблагодарить замечательнейшего доктора Гиндрюк Василия Васильевича за профессионализм, заботу и внимание! Наблюдаться у него одно удовольствие!!!
Самое быстрое обслуживание в жизни. 20 минут экг+рентген. Минимум бумаг. Максимум результата.
Для занятых людей- оптимально
Множественные мальформации сердца у пациента с синдромом Холта—Орама | Сойнов
1. Vanlerberghe C., Jourdain A.S., Ghoumid J., Frenois F., Mezel A., Vaksmann G. et al. Holt-Oram syndrome: clinical and molecular description of 78 patients with TBX5 variants. Eur J Hum Genet 2019; 27(3): 360-368. DOI: 10.1038/s41431-018-0303-3
2. Van der Linde D, Konings E.E., Slager M.A., Witsenburg M., Helbing W.A. et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 2011; 58: 2241-2247. DOI: 10.1016/j.jacc.2011.08.025
3. Naiche L.A., Harrelson Z., Kelly R.G., Papaioannou V.E. T-box genes in vertebrate development. Annu Rev Genet 2005; 39: 219-239. DOI: 10.1146/annurev.genet.39.073003.105925
4. Barisic I., Boban L., Greenlees R, Garne E., Wellesley D., Calzolari E. et al. Holt Oram syndrome: a registry-based study in Europe. Orphanet J Rare Dis 2014; 9: 156. DOI: 10.1186/s13023-014-0156-y
5. Arkoumanis P.T., Gklavas A., Karageorgou M., Gourzi P., Mantzaris G., Pantou M., Papaconstantinou I. Holt-Oram Syndrome in a Patient with Crohn’s Disease: a Rare Case Report and Literature Review. Med Arch 2018; 72(4): 292-294. DOI: 10.5455/medarh.2018.72.292-294
6. Borozdin W., Bravo Ferrer Acosta A.M., Bamshad M.J., Bot-zenhart E.M., Froster U.G., Lemke J. et al. Expanding the spectrum of TBX5 mutations in Holt-Oram syndrome: detection of two intragenic deletions by quantitative real time PCR, and report of eight novel point mutations. Hum Mutat 2006; 27: 975-976. DOI: 10.1002/humu.9449
7. Heinritz W., Moschik A., Kujat A., Spranger S., Heilbron-ner H., Demuth S. et al. Identification of new mutations in the TBX5 gene in patients with Holt-Oram syndrome. Heart Br Card Soc 2005; 91: 383-384. DOI: 10.1136/hrt.2004.036855
8. Spiridon M.R., Petris A.O., Gorduza E.V., Petras A.S., Popescu R., Caba L. Holt-Oram Syndrome With Multiple Cardiac Abnormalities. Cardiol Res 2018; 9(5): 324-329. DOI: 10.14740/cr767w
9. Newbury-Ecob R.A., Leanage R., Raeburn J.A., Young I.D. Holt-Oram syndrome: a clinical genetic study. J Med Genet 1996; 33: 300-307. DOI: 10.1136/jmg.33.4.300
10. Li Q.Y., Newbury-Ecob R.A., Terrett J.A., Wilson D.I., Curtis A.R., Yi C.H. et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 1997; 15: 21-29. DOI: 10.1038/ng0197-21
11. Greulich F., Rudat C., Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res 2011; 91: 212-222. DOI: 10.1093/cvr/cvr112
12. Hasson P., DeLaurier A., Bennett M., Grigorieva E., Naiche L.A., Papaioannou V.E. et al. Tbx4 and Tbx5 acting in connective tissue are required for limb muscle and tendon patterning. Dev Cell 2010; 18: 148-56. DOI: 10.1016/j.dev-cel.2009.11.013
13. Singh G.K. Congenital Aortic Valve Stenosis. Children (Basel) 2019; 6(5). DOI: 10.3390/children6050069
14. Kulyabin Y.Y., Soynov I.A., Zubritskiy A.V., Voitov A.V., Nichay N.R., Gorbatykh Y.N. et al. Does mitral valve repair matter in infants with ventricular septal defect combined with mitral regurgitation? Interact Cardiovasc Thorac Surg 2018; 26(1): 106-111. DOI: 10.1093/icvts/ivx231
15. Voitov A., Omelchenko A., Gorbatykh Y., Zaitsev G., Arkhipov A., Soynov I. et al. Outcomes of perventricular off-pump versus conventional closure of ventricular septal defects: a prospective randomized study. Eur J Cardiothorac Surg 2017; 51(5): 980-986. DOI: 10.1093/ejcts/ezx002
Запись на допплерографию артерий и вен верхних конечностей в Санкт
При проведении дуплексного сканирования артерий и вен верхних конечностей используется одновременно два вида
ультразвукового исследования. Традиционный ультразвук позволяет дать оценку структуры артерий и вен, а допплеровское
ультразвуковое исследование показывает кровоток, а также его направление и скорость. При дуплексном сканировании
обычно формируется цветное изображение.
Дуплексное сканирование артерий и вен верхних конечностей включает исследование подключичных,
подмышечных, плечевых, локтевых и лучевых вен и артерий, а также латеральных и медиальных подкожных вен и ладонных
артерий.
При сканировании методом допплерографии главным образом оценивается проходимость сосудов, выявляются внутрипросветные
образования, а также исследуются показатели кровотока в просветах сосудов. Благодаря проведению дуплекса сосудов можно
выявлять различные заболевания, которые сложно диагностировать другими методам, но предпочтительнее пройти МРТ
Обследование проводят
Крейзер Анастасия Игоревна
— врач ультразвуковой диагностики
— врач-рентгенолог
Опыт работы: с 2012 года
Образование:
– Санкт-Петербургская Государственная Педиатрическая Медицинская Академия — педиатр
– СЗГМУ им. И.И. Мечникова, профессиональная переподготовка на врача УЗД
Основными показаниями к проведению дуплекса артерий и вен верхних конечностей
- Отеки и онемение конечностей
- Зябкость конечностей
- Изменения на коже (гиперпигментация, бледность, язвы)
- Тромбоз вен
- Варикозные изменения вен
- Болезнь Рейно
- Синдром верхней грудной апертуры
- Аномалии вен и артерий
Преимущества допплерографии
Преимуществом допплерографии является то, что данный метод не имеет противопоказаний и возрастных ограничений, поэтому
применять его можно так часто, как того требует каждый конкретный случай.
Доплер артерий и вен верхних конечностей не требует никакой предварительной подготовки и не вызывает
каких-либо осложнений или же побочных эффектов. Время сканирования составляет в среднем 30 минут, после чего пациент
может сразу же вернуться к обычному ритму жизни. К достоинствам данного метода можно отнести безболезненность,
безопасность, неинвазивность и невысокую стоимость исследования.
Детская ортопедия: деформация верхних конечностей | 161.ru
Приобретенные деформации верхних конечностей возникают, как правило, в результате перенесенных заболеваний (полиомиелит и остеомиелит), травм, ДЦП, родового паралича верхней конечности.
Родовой паралич (утрата подвижности) верхней конечности чаще всего возникает во время трудных родов, когда приходится прибегать к активному вмешательству в виде акушерских приемов. Другим фактором повреждения нервных стволов может быть их прижатие вовремя родов ключицей к первому ребру. К повреждению плечевого сплетения могут привести перелом ключицы.
Страдают при параличе преимущественно мышцы верхнего плечевого пояса. В результате нарушается сгибание или разгибание в локтевом суставе, возникают контрактуры (тугоподвижность) лучезапястного сустава и пальцев кисти, пациент не в состоянии поднять и отвести руку.
Посттравматические деформации являются результатом неправильно сросшихся переломов. Неправильное распределение нагрузки приводит к разрушению смежных суставов. Выраженная деформация сопровождается укорочением конечности, варусным и вальгусным искривлением.
Лечение деформаций верхних конечностей следует начинать как можно раньше.
Отделение детской ортопедии МБУЗ ГБ№20 готово предложить широкий спектр различных медицинских услуг для лечения пациентов – это физиопроцедуры, массаж, лечебная физкультура, электростимуляция, а также использование ортопедических фиксаторов и корректоров.
При наличии определенных показаний врачи отделения готовы провести оперативные вмешательства на высоком уровне.
Консультативный прием завотделения в отделении детской ортопедии МБУЗ ГБ №20.
Руководитель отделения детской травматологии и ортопедии для детей – главный детский ортопед Ростова-на-Дону, к.м.н., Мурадьян Владимир Юрьевич.
адрес: г. Ростов-на-Дону, пр. Коммунистический, 39, 4 этаж;
часы приема: по субботам, с 10:00 до 13:00;
контактный телефон: 271-97-20.
Заболевания
Врожденное заболевание, основными проявлениями которого является деформация мозгового и лицевого черепа, сращение и недоразвитие пальцев кистей и стоп.
Также может наблюдаться недоразвитие верхней челюсти, аномалии зубов, врожденные пороки сердца, расщелина неба, аномалии желудочно-кишечного тракта и мочевыделительной сиситемы.
Диагностика
Предварительный диагноз ставится при рождении на основании клинической картины. Постановка окончательного диагноза устанавливается на основании детекции мутации в геноме. Известна конкретная локализация поломки — ген FGFR2 в хромосоме 10q26. Также существуют варианты пренатальной (дородовой) ДНК- диагностики.
Чем обусловлено изменение формы черепа при синдроме Apert
Обычно при рождении на черепе есть незакрытые костью участки, обеспечивающие мозгу ребенка достаточное пространство для нормального роста и развития. Смыкаются они в норме к 1 году, образуя при этом сплошную кость. При синдроме Apert происходит их преждевременное сращение, до того как мозг ребенка полностью сформировался. При этом процесс роста продолжается, но идет он уже в том направлении где сопротивление минимально, то есть в высоту. Это и приводит к формированию формы головы, называемой — «башенный череп». Этот же механизм приводит к изменениям лицевого черепа, таким как глубокие кожные складки над бровями, большие глаза, антимонголоидный разрез глаз, маленький нос, высокий лоб, широкая переносица.
Синдактилия пальцев кистей и стоп, какие особенности бывают при данном заболевании?!
Более грамотным описанием данной синдактилии будет «симбрахидактилия»,. Этот термин говорит о том, что помимо наличия сращений (от лат. Syn — вместе), имеется укорочение пальцев ( от лат. Brahy — короткий) и недоразвитие межфаланговых суставов. Это означает то что в дальнейшем, даже после разделения пальцев, подвижность в этих суставах будет ограничена или отсутствовать. Движения будут осуществляться преимущественно за счет пястно-фаланговых суставов.
Выделяют три основных типа синдактилии при синдроме Apert.
При 1 типе кисть напоминает по форме лопату. При нем большой палец имеет правильную ось, или незначительное отклонение в лучевую сторону. В первом и четвертом межпальцевом промежутке как правило синдактилия неполная. Для устранения такой синдактилии пересадка дополнительной кожи чаще всего не требуется. Синдактилия между остальными пальцами является простой, что означает отсутствие костного компонента в сращении. При этом концевые отделы 2-5 пальцев могут быть не спаяны. Но не смотря на это при разделении 2-4 пальцев пересадка дополнительной кожи все равно потребуется.
При 2 типе кисть напоминает по форме ложку. В данном случае первый палец укорочен и имеет отклонение в лучевую сторону, также наблюдается тотальная (полная) синдактилия во всех межпальцевых промежутках, когда сращение доходит до концевых отделов пальцев со сросшимися ногтевыми пластинами. При этом типе сращение является простым, но часто может наблюдаться костное сращение ногтевых фаланг 3-4 пальцев. Без пересадки дополнительной кожи разделить пальцы не удастся.
При 3 типе кисть похожа на бутон. Данный тип является самым сложным в техническом плане. Первый и пятый пальцы относительно своей оси развернуты друг к другу. Сращение между всеми пальцами в области ногтевых пластин является костным, дальше пальцы расходятся веерообразно что и создает сходство с бутоном цветка.
Эти нюансы являются определяющими в тактике хирургического лечения.
Сращение пальцев стоп очень плотное, чаще костное со слиянием ногтевых пластин. При таких условиях требуется много дополнительных операций с пересадкой большого количества донорской кожи. Что в большинстве случаев не имеет смысла, поскольку синдактилии пальцев стоп редко влияют на функцию.
Основным показанием для операций на стопе является сгибательная деформация второй плюсневой кости. Это проявляется наличием выступающей « шишки » по подошвенной поверхности стопы, у основания второго пальца. Очень часто на этом месте формируется натоптыш.
Этапность лечебного процесса
Преимущественным является устранение сращения костей черепа путем установки специальных аппаратов раздвигающих кости. Своевременность оказания данного хирургического вмешательства оказывает непосредственное влияние на дальнейшее развитие центральной нервной системы и как следствие уровень интеллектуального развития.
Далее после оценки общего соматического статуса, необходимо приступать к устранению сращений пальцев кистей. В зависимости от типа синдактилий очередность хирургических вмешательств на кисти может меняться.
Но существуют общие принципы:
— Отделение и выпрямление 1 пальца играет важное значение для функции. Полноценный большой палец дает 50-70% к функции кисти. Поэтому данное вмешательство является первичным.
— Далее устранение сращения 4-5 пальцев, так как 5 палец как правило имеет более развитые межфаланговые суставы что уже даст возможность более полноценного двухстороннего схвата, чем это было бы со вторым пальцем.
— Этап операции с заимствованием донорской кожи и пересадкой на области ее потребности, является обязательным при любом типе сращения. Донорская кожа берется с области паховой складки. Швы накладываются внутрикожно, снятие швов не требуется.
Рекомендации по диспансерному наблюдению
Пациент с диагнозом синдром Apert должен наблюдаться у ряда специалистов по причине большого количества сопутствующих заболеваний:
— челюстно-лицевого хирурга
— ортодонта
— нейрохирурга
— невролога
Эпидемиология врожденных аномалий верхних конечностей в Корее: общенациональное популяционное исследование
Abstract
Это исследование было направлено на анализ эпидемиологии врожденных аномалий верхних конечностей (CULA) в Корее. Мы оценили частоту каждого типа CULA, наличие сопутствующих аномалий и статус хирургического лечения у пациентов с CULA. Мы провели ретроспективное когортное исследование пациентов в возрасте <1 года в период с 2007 по 2016 год, которые были зарегистрированы в CULA в Службе обзора и оценки медицинского страхования Кореи.Всего 10 704 пациента имели CULA, в том числе 6 174 мальчика (57,7%) и 4530 девочек (42,3%). Среднегодовая заболеваемость CULA составила 23,5 случая на 10 000 живорождений; у мальчиков он был значительно выше, чем у девочек (26,3 против 20,5, p <0,001). Среди четырех категорий CULA - полидактилии, синдактилии, дефицита конечностей и других аномалий - полидактилия была наиболее распространенной. В общей сложности у 4149 пациентов (38,8%) были другие врожденные аномалии, и сосуществующие аномалии системы кровообращения (24,9%) были наиболее распространенными.Всего 4 776 пациентов (44,6%) прошли оперативное лечение по поводу CULA в течение как минимум трех лет с момента постановки диагноза. Доля пациентов, перенесших хирургическое лечение, была значительно выше при полидактилии (73,4% против 16,8%, p <0,001) и синдактилии (65,3% против 41,5%, p <0,001), но была значительно ниже при дефиците конечностей (27,6%). % против 45,4%, p <0,001) и других аномалий (10,0% против 69,8%, p <0,001), чем у остальных пациентов с CULA. Среди оперированных 21 человек.5% перенесли несколько операций. Доля пациентов, перенесших несколько операций, была значительно выше при синдактилии (35,6% против 18,1%, p <0,001), но была значительно ниже при полидактилии (4,0% против 95,5%, p <0,001) и других аномалиях (17,9%). % против 21,9%, p <0,001), чем у остальных пациентов с CULA. Эти результаты могут стать основой для оценки национальных затрат на здравоохранение для CULA и необходимого количества специалистов CULA.
Образец цитирования: Shin YH, Baek GH, Kim YJ, Kim MJ, Kim JK (2021) Эпидемиология врожденных аномалий верхних конечностей в Корее: общенациональное популяционное исследование.PLoS ONE 16 (3):
e0248105.
https://doi.org/10.1371/journal.pone.0248105
Редактор: Лейла Хархаус, BG Trauma Center Ludwigshafen, GERMANY
Поступила: 17 ноября 2020 г .; Принята к печати: 20 февраля 2021 г .; Опубликован: 9 марта 2021 г.
Авторские права: © 2021 Shin et al. Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Доступность данных: Все соответствующие данные находятся в рукописи и вспомогательных информационных файлах.
Финансирование: Инициалы авторов, получивших каждую награду: Номера грантов YHS, присуждаемые каждому автору: 2020IT0003-1 Полное имя каждого спонсора: Медицинский центр Асан Детская больница URL-адрес веб-сайта каждого спонсора: www.amc.seoul.kr/asan/departments/deptListTypeF.do Спонсоры не играли никакой роли в дизайне исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Конкурирующие интересы: Авторы заявили, что никаких конкурирующих интересов не существует.
Введение
Понимание эпидемиологии врожденных аномалий важно для общественного здравоохранения. Эта информация дает основу для оценки национальных затрат на здравоохранение и количества необходимых специалистов. Кроме того, мониторинг изменений в частоте и характере врожденных аномалий может предупредить нас о появлении новых тератогенов, таких как талидомид, в 1960-х годах [1].
Эпидемиология врожденных аномалий верхних конечностей (CULA) изучалась ранее, но имелось несколько ограничений, чтобы рассматривать ее как определенную эпидемиологическую информацию. Некоторые исследования были сосредоточены на региональном, а не на национальном населении [2–5], или оценивали распространенность CULA, а не заболеваемость [3–5], или изучали только одну конкретную аномалию, такую как недостаточность конечности [6, 7]. Кроме того, существует только два старых региональных исследования, в которых оценивалась эпидемиология CULA в азиатской популяции [8, 9], и нет национальных исследований.
Корея внедряет систему медицинского страхования для всех граждан с 1989 года. Поскольку медицинские данные всего населения в Корее обрабатываются Службой обзора и оценки медицинского страхования (HIRA), набор данных HIRA упрощает получение и анализ данных чтобы понять медицинское состояние всей страны. Кроме того, благодаря широкому охвату национальной системы страхования доступ к медицинскому обслуживанию для граждан Кореи является лучшим среди стран Организации экономического сотрудничества и развития (ОЭСР) [10].Таким образом, мы могли оценить эпидемиологию CULA среди населения всей страны, проанализировав данные HIRA. Целью этого исследования был анализ эпидемиологии CULA в Корее. Более конкретно, мы оценили частоту каждого типа CULA, наличие сосуществующих аномалий и статус хирургического лечения у пациентов с CULA.
Материалы и методы
Источник данных
В Корее Национальная служба медицинского страхования (NHIS) покрывает 100% населения; 97% имеют медицинскую страховку и 3% получают медицинскую помощь [11].Все поставщики медицинских услуг представляют в HIRA данные о заявках на лечение в стационарных и амбулаторных условиях для возмещения медицинских расходов. К ним относятся диагностические коды (классифицированные в соответствии с Международной классификацией болезней, 10-й пересмотр [МКБ-10]), коды процедур и демографическая информация. HIRA предоставляет некоторые из этих национальных данных для поддержки разработки государственной политики и исследовательской деятельности по запросу. Этот протокол исследования был исключен для рассмотрения Экспертным советом учреждения Медицинского центра Асан (No.2020–0124) в соответствии с критериями освобождения.
Сбор данных
Мы провели ретроспективное когортное исследование пациентов с CULA в период с 2007 по 2016 годы. Во-первых, пациенты в возрасте <1 года с CULA были идентифицированы с использованием кодов ICD-10 (Таблица 1). Коды МКБ-10 для CULA были разделены на четыре категории: полидактилия, синдактилия, дефицит конечностей и другие аномалии [12]. Для пациентов, которые идентифицировались несколько раз с одним и тем же кодом, время постановки первого диагноза было критерием, используемым для расчета ежегодной заболеваемости.Если у одного пациента было несколько кодов CULA, каждый код учитывался отдельно для первоначального анализа, но при расчете годовой заболеваемости всеми CULA и каждой категорией аномалии это рассматривалось как отдельный случай. Например, если один пациент был зарегистрирован с тремя разными кодами (например, дополнительный палец (-и) (Q690.), Дополнительный (-е) палец (-и) (Q691.) И другие врожденные пороки развития верхней (-ых) конечности (-ей), включая плечевой пояс) (Q740). .)), его или ее подсчитывали отдельно для каждой кодовой заболеваемости, но как единичный случай для годовой заболеваемости CULA.Для некоторых диагностических кодов верхние и нижние конечности не различались. Например, неуточненная полидактилия (Q699.), Полисиндактность (Q704.) И врожденное отсутствие неуточненных конечностей (Q730.) Считались кодами CULA, когда они были зарегистрированы с кодами процедур для рентгенограмм верхней конечности из ключицы. к пальцу (ключица: G3101–3105; лопатка: G3201–3205; плечо: G33013305; акромиально ключичный сустав: G3901–3905; предплечье: G6101–6105; локоть: G6201–6205; плечевая кость: G6301–530640; запястье: G6301–6305–6; запястье: рука: G6501–6505; кость запястья: G6601–6605; палец: G8101–8105).Годовая заболеваемость CULA определялась как доля населения, у которого впервые был диагностирован CULA в возрасте <1 года, среди живорожденных в течение этого года. Ежегодные данные о рождении живых детей, включая количество и пол, были получены от Корейской статистической информационной службы [13]. Кроме того, была получена другая демографическая информация, включая пол и тип страхования (имел ли пациент медицинскую страховку или медицинскую помощь, что косвенно отражало социально-экономический статус) каждого пациента.
Во-вторых, у пациентов с CULA были проанализированы другие сопутствующие врожденные аномалии. Пациенты, у которых были диагностированы и зарегистрированы как имеющие другие аномалии в течение одного года после рождения, считались имеющими другие врожденные аномалии, классифицированные по основному классификационному уровню кодов МКБ-10 (Таблица 2). Для врожденных аномалий опорно-двигательного аппарата, были включены пациенты с другими, чем CULA аномалий. Количество пациентов с другими аномалиями оценивалось для каждого типа CULA.Если у одного пациента было несколько других аномалий, каждая аномалия подсчитывалась отдельно в соответствии с основным уровнем классификации кодов МКБ-10 для первоначального анализа, но при расчете частоты других сопутствующих аномалий у пациентов с CULA и каждой категории CULA это было рассматривается как единичный случай. Например, если один пациент с полидактилией большого пальца был зарегистрирован с двумя различными аномалиями, включая систему кровообращения (Q20–28) и пищеварительную систему (Q38–45), в течение одного года после рождения, он или она учитывались отдельно для оценки каждой из них. аномалия, но учитывается как единичный случай частоты сопутствующих других аномалий у пациентов со всеми CULA и полидактилией.
В-третьих, был проанализирован статус хирургического лечения пациентов с CULA. Поскольку данные собирались до конца 2019 года, были включены хирургические процедуры, проведенные в течение как минимум трех лет после первоначального диагноза CULA. Пациенты, зарегистрированные с кодами операции под кодами CULA, были определены как перенесшие операцию по CULA. В операционных кодах HIRA только три кода были кодами, определяемыми заболеванием для CULA, включая операцию полидактилии, требующую реконструкции сухожилия и кости (N0251), операцию полидактилии, требующую других процедур (N0252), и операцию синдактилии (N0260).Остальные коды были для общих процедур с костями и мягкими тканями. Таким образом, мы определили все коды операций, которые были возможны при хирургическом лечении CULA (Таблица 3). Частота хирургического лечения, время до начала операции с момента постановки диагноза и количество операций были проанализированы для каждого типа CULA.
Статистический анализ
Непрерывные данные были представлены как среднее ± стандартное отклонение (SD) или медиана и межквартильный размах (IQR), а категориальные данные представлены в виде чисел и процентов.Мы рассчитали ежегодную заболеваемость CULA (на 10 000 живорождений) у мальчиков и девочек, исходя из распределения Пуассона. Регрессионный анализ Пуассона был использован для анализа тенденций ежегодной заболеваемости CULA, CULA у каждого пола и в четырех категориях CULA. Тест хи-квадрат использовался для сравнения доли пациентов в каждой категории, у которых были другие сопутствующие врожденные аномалии, и тех, кто прошел оперативное лечение и несколько операций, с остальными пациентами CULA.P <0,05 интерпретировалось как статистически значимое. Все статистические анализы были выполнены с использованием программного обеспечения SAS Enterprise Guide версии 7.1 (SAS Institute, Inc., Кэри, Северная Каролина, США).
Результаты
Годовая частота врожденных аномалий верхних конечностей
С 2007 по 2016 год в CULA было зарегистрировано 10 704 пациента, в том числе 6 174 мальчика (57,7%) и 4530 девочек (42,3%), и за тот же период было зарегистрировано 4550 102 живорожденных (Таблица 4).Среднегодовая частота CULA составляла 23,5 на 10 000 живорождений, и была значительно выше у мальчиков (26,3 на 10 000 живорождений), чем у девочек (20,5 на 10 000 живорождений) (p <0,001) (Таблица 5). Из общего числа 10 704 пациентов 10 561 пациент (98,7%) имели медицинскую страховку и 143 пациента (1,3%) получали медицинскую помощь. Регрессионный анализ Пуассона показал, что годовая частота CULA увеличивалась в течение периода исследования (коэффициент заболеваемости (IRR), 1,017; 95% ДИ, 1,009–1,025; p <0,001).Это увеличение наблюдалось как у мальчиков (IRR, 1,016; 95% ДИ, 1,006–1,026; p = 0,021), так и у девочек (IRR, 1,018; 95% CI, 1,006–1,031; p = 0,036) (рис. 1).
Среди четырех категорий кодов МКБ-10 полидактилия (5264 пациента, 49,2%) была наиболее распространенной, за ней следовали другие аномалии (4507 пациентов, 42,1%), синдактилия (1405 пациентов, 13,1%) и дефицит конечностей (490 пациентов, 4,6%). Регрессионный анализ Пуассона показал, что годовая заболеваемость полидактилией (IRR, 1.000; 95% CI, 0.988–1.013; p = 0.951) и дефицит конечностей (IRR, 1,004; 95% ДИ, 0,974–1,034; p = 0,810) не претерпели значительных изменений в течение периода исследования. Однако годовая частота синдактилии (IRR, 1,017; 95% ДИ 1,003–1,031; p = 0,018) и других аномалий (IRR, 1,033; 95% ДИ, 1,013–1,052; p = 0,001) значительно увеличилась в течение периода исследования. (Рис 2).
Другие сопутствующие врожденные аномалии у пациентов с врожденной аномалией верхних конечностей.
Среди 10704 пациентов с впервые диагностированной CULA 4149 пациентов (38.8%) имели другие врожденные аномалии. Врожденные аномалии системы кровообращения были наиболее распространенными (2670 пациентов, 24,9%), а затем врожденных аномалий опорно-двигательного аппарата, кроме CULA (2130 пациентов, 19,9%) и врожденные аномалии органов пищеварения (2103 больных, 19,6%) (Таблица 2). Доля пациентов с другими сопутствующими врожденными аномалиями представлена на рис. 3 по категориям. Среди четырех категорий другие врожденные аномалии были значительно выше при дефиците конечностей (61.6% против 37,7%, p <0,001), синдактилии (43,7% против 38,0%, p <0,001) и других аномалий (41,8% против 36,5%, p <0,001), но значительно ниже при полидактилии (35,0%). против 42,4%, p <0,001), чем у остальных пациентов с CULA.
Рис. 3. Другие сопутствующие врожденные аномалии, которые сопровождались врожденными аномалиями верхних конечностей (CULA) в Южной Корее с 2007 по 2016 год по категориям: (A) полидактилия, (B) синдактилия, (C) дефицит конечностей и (D) другие аномалии.
https: // doi.org / 10.1371 / journal.pone.0248105.g003
Статус хирургического лечения врожденной аномалии верхних конечностей.
Среди 10 704 пациентов с впервые диагностированной CULA 4 776 пациентов (44,6%) прошли оперативное лечение CULA в течение как минимум трех лет после постановки диагноза. Доля пациентов, которым проводилось хирургическое лечение, описана в таблице 6. Среди четырех категорий частота хирургического лечения была значительно выше при полидактилии (73,4% против 16,8%, p <0,001) и синдактилии (65.3% против 41,5%, p <0,001), но значительно ниже по дефициту конечностей (27,6% против 45,4%, p <0,001) и другим аномалиям (10,0% против 69,8%, p <0,001), чем у остальных участников CULA. пациенты.
Таблица 6. Доля пациентов, перенесших оперативное лечение по поводу врожденной аномалии кисти и верхней конечности (CULA) в Южной Корее с 2007 по 2016 год в течение как минимум трех лет после постановки диагноза.
https://doi.org/10.1371/journal.pone.0248105.t006
Для 4776 пациентов, перенесших хирургическое лечение, среднее время до первой операции с момента постановки диагноза составило 5.1 месяц (IQR, 0,9–9,4 месяца). Среди четырех категорий он был самым продолжительным при дефиците конечностей, медианное значение 11,7 месяца (IQR, 6,4–19,1 месяца) и самым коротким при полидактилии, медианное значение 4,4 месяца (IQR, 0,7–8,4 месяца) (рис. 4). Из 4776 пациентов, перенесших операцию, 3750 пациентов (78,5%) перенесли одну операцию, но 1026 пациентов (21,5%) перенесли несколько операций по поводу CULA. Среди четырех категорий доля пациентов, перенесших несколько операций, была значительно выше в синдактилии (35.6% против 18,1%, p <0,001), но значительно ниже при полидактилии (4,0% против 95,5%, p <0,001) и других аномалиях (17,9% против 21,9%, p <0,001), чем у остальных пациентов с CULA ( Рис 5).
Рис. 4. Среднее время от постановки диагноза до начальной операции от постановки диагноза для пациентов, перенесших хирургическое лечение врожденных аномалий верхних конечностей (CULA) в Южной Корее с 2007 по 2016 год в соответствии с категориями; пустой синий кружок: среднее время.
https://doi.org/10.1371 / journal.pone.0248105.g004
Рис. 5. Количество операций у пациентов, перенесших хирургическое лечение врожденных аномалий верхних конечностей (CULA) в Южной Корее с 2007 по 2016 год по категориям: (A) количество пациентов и (B) доля пациентов в соответствии с количеством операций.
https://doi.org/10.1371/journal.pone.0248105.g005
Обсуждение
Общая частота CULA в нашем исследовании (23,5 на 10 000 живорождений) была аналогична той, о которой сообщалось в предыдущих исследованиях.В общем исследовании населения Западной Австралии, охватывающем 11 лет, Giele et al. сообщили о распространенности CULA на уровне 19,8 на 10 000 живорождений [2]. В общем исследовании населения Стокгольма, Швеция, за 11 лет Ekblom et al. сообщили, что частота CULA составляет 21,5 на 10 000 живорождений [4]. В исследовании реестра врожденных пороков развития Нью-Йорка за 18 лет Goldfarb et al. сообщили, что распространенность составляет 27,2 на 10 000 живорождений [3]. Эти различия могут возникать из-за различий в источниках данных, расовом составе изучаемых популяций и от того, измерялась ли заболеваемость или распространенность.В двух предыдущих исследованиях [2, 4] у мальчиков было больше, чем у девочек, однако в нашем исследовании впервые была выявлена статистически значимая разница. Годовой коэффициент живорождения в Корее немного снизился, но годовое количество CULA было постоянным или немного увеличилось в течение периода исследования. Это привело к статистически значимому увеличению ежегодной заболеваемости CULA независимо от пола. Увеличение среднего возраста матери (30,6 года в 2007 году и 32,4 года в 2016 году) и снижение уровня смертности новорожденных (34 на 10 000 живорождений в 2007 году и 28 на 10 000 живорождений в 2016 году) [13], что означает более высокую выживаемость младенцев с множественные аномалии могут быть связаны с ростом заболеваемости CULA [2].Кроме того, эта тенденция к увеличению может отражать улучшение обнаружения и сообщения о CULA или увеличение экзогенных тератогенов в нашей среде [6].
Полидактилия была наиболее распространенной категорией CULA, но данные не позволили идентифицировать местоположение аномалии. Хотя категория «другие аномалии» была второй по распространенности категорией, включающей неуказанные различные аномалии, синдактилия была второй по распространенности как единая аномалия. Этот результат является общепринятым рейтингом заболеваемости CULA [3, 14].Частота дефицита верхних конечностей была ниже, чем в когортах Северной Европы [4, 6, 7], при этом у половины пациентов отсутствовала рука и палец (пальцы). Мы думаем, что эти различия в составе CULA происходят из-за этнических различий каждой когорты.
Среди пациентов с впервые диагностированной CULA 38,8% имели другие врожденные аномалии в нашей когорте. Эта доля ниже, чем 46% в когорте Западной Австралии [2], но выше, чем 23,1% в когорте Швеции [4].Что касается типа сопутствующих врожденных аномалий, то в предыдущих исследованиях синдромальные аномалии были самой частой или второй по частоте причиной [2, 4]; однако в нашей когорте это было менее распространено. Мы думаем, что эти расхождения связаны с методами сбора данных. В предыдущих исследованиях все доступные медицинские записи и рентгенограммы были просмотрены специалистами, и в случаях, когда CULA была частью синдромных аномалий, она классифицировалась как «сопровождающая синдромную аномалию», а не «другие специфические аномалии органа».Однако в нашей когорте анализировались только зарегистрированные данные, и мы не могли просмотреть подробные медицинские записи или диагноз для пациентов с CULA. Поскольку все поставщики медицинских услуг могли предоставить данные о пациентах в нашей когорте, существует вероятность того, что определенные аномалии органов были зарегистрированы одновременно с синдромной аномалией. Среди четырех категорий у большей части пациентов с дефицитом конечностей были другие врожденные аномалии, чем у других категорий CULA. Поэтому, когда мы лечим пациентов с недостаточностью конечностей, мы должны учитывать возможность других аномалий и их общее состояние здоровья.
Поскольку большинство хирургических вмешательств по поводу CULA начинается в течение двух или трех лет после рождения [15, 16], мы думаем, что период наблюдения этого исследования (минимум три года после постановки диагноза) будет включать большинство пациентов с CULA, подвергшихся хирургическому лечению. Хотя доля пациентов, подвергшихся хирургическому лечению, была выше по полидактилии и синдактилии, чем у остальных пациентов с CULA, она не превышала 90% в обеих категориях. Это может быть связано с занижением сведений о полидактилии рудиментарного типа, которую можно было бы удалить с помощью перевязки или простого иссечения в амбулаторной клинике или отделении новорожденных вместо официальной операции под наркозом [17].Кроме того, некоторые случаи частичной синдактилии, которые не показывают функциональной инвалидности, могут наблюдаться без операции, в то время как в некоторых сложных случаях синдактилии операция может быть противопоказана, если существует риск дальнейшего функционального нарушения [3, 18]. В отношении дефицита конечностей и других аномалий доля пациентов, подвергшихся хирургическому лечению, была ниже, чем у остальных пациентов из группы CULA. Этот феномен можно объяснить ограниченной ролью хирургического лечения у пациентов с дефицитом конечностей [6, 7], а «другие аномалии» включают аномалии, которые не нарушают функцию и, следовательно, не требуют хирургического лечения, такие как клинодактилия, брахидактилия, незначительный тип. сложенный или гипоплазированный большой палец [16].Доля пациентов, перенесших несколько операций, была значительно выше в синдактилии, чем у остальных пациентов из группы CULA. Это может быть связано с более высокой частотой повторных операций синдактилии из-за ползучести полотна и отклонения разделенной цифры [19], а также с множественными операциями, необходимыми для нескольких полотен или для операций на обеих руках [20].
У нашего исследования есть несколько ограничений, аналогичных тем, которые встречаются в любом исследовании реестра. Во-первых, это несовершенный реестр для правильного определения всех CULA, поскольку подтверждение CULA часто требует клинической и радиологической оценки со стороны таких специалистов, как хирурги по врожденным кистам [3].Таким образом, некоторые пациенты могли регистрироваться под разными кодами в разное время. Мы считаем, что данные о полидактилии, синдактилии и дефиците конечностей надежны, поскольку эти случаи было легко идентифицировать. Кроме того, мы уверены, что общие данные о заболеваемости CULA точны, поскольку мы удалили повторяющиеся данные для одного и того же пациента. Напротив, пациенты с «другими аномалиями» являются наименее надежными данными из-за различных и менее однозначных диагнозов. Во-вторых, большинство исследований расслоили свои случаи с помощью известных систем классификации CULA, таких как классификация Международной федерации обществ хирургии кисти (IFSSH) [2, 21] или классификация Оберга, Манске и Тонкина (OMT) [4, 5] .Мы не могли применить эти системы классификации к нашей когорте, потому что требуемая информация не была внесена в нашу базу данных. Это ограничивает прямое сравнение результатов эпидемиологических исследований. Однако, поскольку это исследование охватило все население страны и большинство диагнозов CULA будут регистрироваться неспециалистами, регистрация и анализ данных МКБ-10 подходят для этого типа исследования, поскольку они знакомы всем поставщикам медицинских услуг. В-третьих, временные ограничения этого исследования могут недооценивать частоту CULA и их хирургическое лечение.Некоторые CULA, такие как клинодактилия, брахимезофалангия и деформация Шпренгеля, могут быть обнаружены после одного года жизни. Ограничение времени на операцию до трех лет с момента постановки диагноза может быть слишком коротким, чтобы включать в себя все несколько операций.
Выводы
Заболеваемость CULA должна составлять 23,5 на 10 000 в течение 10 лет, а заболеваемость незначительно увеличилась за 10-летний период. Среди четырех категорий: полидактилия, синдактилия, дефицит конечностей и другие аномалии, полидактилия была наиболее распространенным типом CULA.В общей сложности 38,8% пациентов с CULA имели другие врожденные аномалии, наиболее часто связанные с аномалиями системы кровообращения. В общей сложности 44,6% пациентов с CULA подверглись оперативному лечению по поводу CULA, и доля пациентов с полидактилией и синдактилией была значительно выше, чем у остальных пациентов с CULA. Среди оперированных пациентов 21,5% пациентов перенесли множественные операции. Доля пациентов, перенесших несколько операций, была значительно выше по синдактилии, чем остальные пациенты из группы CULA.Эти результаты могут облегчить понимание эпидемиологии CULA среди азиатского населения и предоставить основу для оценки национальных затрат на здравоохранение для CULA и количества специалистов, необходимых для лечения CULA.
Благодарности
В этом исследовании использовались данные исследования HIRA (Служба проверки и оценки медицинского страхования) (M20200213307), проведенного HIRA в Республике Корея. Выраженные взгляды принадлежат автору (авторам) и не обязательно принадлежат HIRA и Министерству здравоохранения и социального обеспечения Республики Корея.
Ссылки
- 1.
Меллин Г.В., Каценштейн М. Сага о талидомиде. От невропатии к эмбриопатии с сообщениями о случаях врожденных аномалий. N Engl J Med. 1962; 267: 1238–44, заключ. pmid: 13934700 - 2.
Giele H, Giele C, Bower C, Allison M. Заболеваемость и эпидемиология врожденных аномалий верхних конечностей: исследование общей популяции. J Hand Surg Am. 2001. 26 (4): 628–34. pmid: 11466636 - 3.
Гольдфарб К.А., Шоу Н., Штеффен Дж. А., Уолл Л. Б.Распространенность врожденных аномалий кистей рук и верхних конечностей на основе реестра врожденных пороков развития Нью-Йорка. J Pediatr Orthop. 2017; 37 (2): 144–8. pmid: 27078227 - 4.
Экблом А.Г., Лорелл Т., Арнер М. Эпидемиология врожденных аномалий верхних конечностей в Стокгольме, Швеция, с 1997 по 2007 год: применение классификации Оберга, Манске и Тонкина. J Hand Surg Am. 2014. 39 (2): 237–48. pmid: 24480684 - 5.
Goldfarb CA, Wall LB, Bohn DC, Moen P, Van Heest AE.Эпидемиология врожденных аномалий верхних конечностей у населения Среднего Запада США: оценка с использованием классификации Оберга, Манске и Тонкина. J Hand Surg Am. 2015; 40 (1): 127–32 e1-2. pmid: 25534840 - 6.
Koskimies E, Lindfors N, Gissler M, Peltonen J, Nietosvaara Y. Врожденные пороки развития верхних конечностей и связанные с ними пороки развития в Финляндии: популяционное исследование. J Hand Surg Am. 2011; 36 (6): 1058–65. pmid: 21601997 - 7.
Klungsøyr K, Nordtveit T.I, Kaastad TS, Solberg S, Sletten IN, Vik AK.Эпидемиология дефектов редукции конечностей, зарегистрированная в Медицинском регистре рождений Норвегии, 1970–2016 годы: популяционное исследование. PLoS One. 2019; 14 (7): e0219930. pmid: 31314783 - 8.
Леунг П.К., Чан К.М., Ченг Дж. Врожденные аномалии верхней конечности у китайского населения Гонконга. J Hand Surg Am. 1982. 7 (6): 563–5. pmid: 6294174 - 9.
Огино Т., Минами А., Фукуда К., Като Х. Врожденные аномалии верхней конечности у японцев в Саппоро. J Hand Surg Br.1986. 11 (3): 364–71. pmid: 3025320 - 10.
Организация экономического сотрудничества и развития. Статистика здравоохранения ОЭСР, 2020 г. Париж, Франция: Организация экономического сотрудничества и развития; 2020 [обновлено 1 июля 2020 г .; цитируется 13 ноября 2020 г.]. http://www.oecd.org/els/health-systems/health-data.htm. - 11.
Jo YH, Lee KH, Kim SJ, Kim J, Lee BG. Национальные тенденции в хирургии болезни вращательной манжеты плеча в Корее. J Korean Med Sci. 2017; 32 (2): 357–64. pmid: 28049250 - 12.Всемирная организация здоровья. Международная классификация болезней, 10-е издание (МКБ-10) Женева, Швейцария: Всемирная организация здравоохранения; 2020 [цитируется 13 ноября 2020 года]. https://www.who.int/classifications/icd/icdonlineversions/en/.
- 13.
Статистическое управление Кореи. Статистическая информационная служба Кореи (КОСИС) Тэджон, Республика Корея: Статистическое управление Кореи; 2020 [обновлено 13 ноября 2020 г .; цитируется 13 ноября 2020 г.]. https://kosis.kr/. - 14.
Персиваль Нью-Джерси, Сайкс П.Дж.Синдактилия: обзор факторов, влияющих на хирургическое лечение. J Hand Surg Br. 1989. 14 (2): 196–200. pmid: 2545804 - 15.
Козин Ш., Злотолов Д.А. Распространенные педиатрические врожденные патологии кисти. Plast Reconstr Surg. 2015; 136 (2): 241e – 57e. pmid: 26218399 - 16.
Уотерс PM, Bae DS. Детская хирургия кисти и верхних конечностей: практическое руководство. 1-е изд. Филадельфия: Wolters Kluwer Health / Lippincott Williams & Wilkins; 2012. х, 657 с. - 17.Абзуг Ж.М., Козин Ш. Лечение постаксиальной полидактилии типа B. J Hand Surg Am. 2013. 38 (6): 1223–5. pmid: 23540414
- 18.
Браун Т.Л., Трост Дж. Г., Педерсон В. Синдактильный релиз. Semin Plast Surg. 2016; 30 (4): 162–70. pmid: 27895538 - 19.
Ferrari BR, Werker PMN. Поперечное исследование долгосрочного удовлетворения после операции по поводу врожденной синдактилии: влияет ли пересадка кожи на удовлетворенность? J Hand Surg Eur Vol. 2019; 44 (3): 296–303. pmid: 30376761 - 20.Ван А.А., Хатчинсон Д.Т. Синдактильное высвобождение: сравнение кожных трансплантатов и методов без трансплантата у одного и того же пациента. J Hand Surg Eur Vol. 2019; 44 (8): 845–9. pmid: 31096828
- 21.
Экблом А.Г., Лорелл Т., Арнер М. Эпидемиология врожденных аномалий верхних конечностей у 562 детей, рожденных в 1997–2007 годах: общее популяционное исследование из Стокгольма, Швеция. J Hand Surg Am. 2010. 35 (11): 1742–54. pmid: 20961708
Меромелия верхней конечности с олигодактилией и брахимезофалангией стопы: необычная ассоциация
Меромелия — редкая аномалия скелета, характеризующаяся частичным отсутствием по крайней мере одной конечности.Было предложено несколько механизмов для объяснения этиопатогенеза расстройства. Сообщается, что в большинстве случаев меромелия носит спорадический характер. Это может произойти как изолированно, так и при других врожденных пороках развития. Связь VACTERL, гастрошизис, дефект межпредсердной перегородки, фокальная недостаточность проксимального отдела бедренной кости и фибулярная гемимелия — это врожденные аномалии, которые, как сообщается, связаны с меромелией. Однако на сегодняшний день никаких других врожденных аномалий, связанных с меромелией, зарегистрировано не было.Здесь мы представляем необычный случай двусторонней меромелии верхней конечности, сопровождающейся односторонней олигодактилией и брахимезофалангией стопы.
1. Введение
Амелия означает полное отсутствие хотя бы одной конечности, а меромелия характеризуется частичным отсутствием хотя бы одной конечности. Меромелию также называют «конечной тарзверской гемимелией». Фактически, амелия и меромелия — это две формы одного и того же заболевания, продолжающие друг друга по степени тяжести [1, 2].Следовательно, термин «последовательность амелия-меромелия» также используется для определения редукции конечностей этого типа [3]. Это редкое отклонение с распространенностью 1,41 на 100 000 рождений. Было описано несколько механизмов, с помощью которых могут возникать дефекты конечностей, чтобы объяснить этиологию заболевания. Развитие верхних конечностей начинается на 26-й день после оплодотворения. Инсульт в этот период или генетические ошибки в координации развития конечностей могут привести к нарушению их формирования на очень ранней стадии эмбриональной жизни.Кроме того, внутриматочная ампутация как причина образования околоплодных вод или ухудшения артериального кровоснабжения конечности также может вызвать сокращение конечности [4]. Сообщается, что большинство случаев носят спорадический характер и не являются передаваемыми. Меромелия может возникать изолированно или вместе с другими врожденными пороками развития [3]. В литературе очень мало сообщений о меромелии в сочетании с другими врожденными аномалиями [3, 5, 6]. Здесь мы представляем необычный случай меромелии, сопровождающейся врожденной деформацией стопы.
2. История болезни
20-летний мужчина с ограниченными возможностями обеих верхних конечностей поступил в нашу больницу для обязательного медицинского осмотра перед военной службой. У него не было никаких жалоб на здоровье, кроме патологии скелета верхних конечностей. В ходе подробного допроса он заявил, что на левой ноге у него было четыре пальца. Он последний из шестерых детей, рожденных от неродных здоровых родителей. Все его братья и сестры полностью здоровы. В расширенной семье нет семейного анамнеза каких-либо врожденных аномалий скелета.Его матери было 32 года, когда она родила нашу пациентку. В анамнезе отсутствуют какие-либо наркотики, дым, алкоголь или радиационное воздействие во время беременности. Наша пациентка родилась без происшествий доношенным путем нормальных вагинальных родов. Никакой другой серьезной проблемы со здоровьем в истории его детства не было.
При медицинском осмотре все элементы скелета за пределами его левого локтя и правого запястья и пятого пальца левой стопы были обнаружены отсутствующими (рис. 1). У него была олигодактилия левой стопы (рис. 2).Переднезадняя рентгенограмма правой руки показала отсутствие кисти с хорошо развитой лучевой и локтевой костью. В медиальном районе дистального отдела локтевой кости была рудиментарная кость размером около 1 см (рис. 3 (а)). Переднезадняя рентгенограмма левой руки показала, что проксимальный сегмент предплечья, участвующий в структуре локтевого сустава, небольшой, но присутствует, в то время как лучевая и локтевая кости дистальнее этой точки отсутствуют (Рисунок 3 (b)). Переднезадняя, боковая косая и медиолатеральная рентгенограммы левой стопы показали отсутствие пятого пальца и боковой клинописи.От второго до четвертого пальцы были короткие. Количество, размер и соотношение суставов других костей, образующих стопу, были нормальными (рис. 4). Увеличенные переднезадние и боковые косые рентгенограммы левой стопы показали, что средние фаланги второго и третьего пальцев короче дистальных фаланг, а средняя фаланга четвертого пальца отсутствует (рис. 5).
Затем пациент был направлен в отделение ортопедической хирургии для определения наилучшего варианта протезирования и обеспечения удобного режима тренировок.
3. Обсуждение
В недавнем эпидемиологическом исследовании, которое проводилось путем оценки большого набора данных, собранных 20 программами эпиднадзора за врожденными аномалиями, было показано, что другие врожденные аномалии сопровождают 66,9% случаев амелии. Наиболее частыми аномалиями, связанными с амелией, были другие типы скелетно-мышечных аномалий, кишечные, почечные, генитальные дефекты, дефекты сердечной перегородки, расщелины ротовой полости и анэнцефалия. По результатам комплексного исследования, врожденные деформации стоп были у 13 пациентов.8% всех случаев с амелией [4]. Однако сообщения о случаях меромелии, сопровождающихся другими врожденными аномалиями, в современной литературе крайне редки. Аномалии позвоночной, анальной, сердечной, трахео-пищеводной, лучевой и конечности (VACTERL) ассоциации [5], гастрошизис [6] и дефект межпредсердной перегородки [3] — это врожденные аномалии, которые, как сообщается, связаны с меромелией. И, мышечно-скелетные аномалии сообщили сопровождать meromelia проксимальны бедренная фокальная недостаточность и малоберцовой гемимелия [5].Врожденных деформаций стоп в сочетании с меромелией на сегодняшний день не зарегистрировано.
Врожденная олигодактилия стопы — это отсутствие одного или нескольких пальцев стопы. Сообщается, что боковые лучи поражаются чаще, чем большие пальцы. Это может быть связано с гемимелией малоберцовой кости, тарзальной коалицией и другими аномалиями стопы [7]. Брахимезофалангия — это редкая аномалия скелета, которая характеризуется отсутствием или гипоплазией средней фаланги. Этот термин используется, когда средние фаланги имеют одинаковую длину или короче дистальных фаланг.Это один из пяти основных типов брахидактилии по классификации Белла [8]. Существует шесть подтипов брахимезофалангии, все из которых являются аутосомно-доминантными наследственными заболеваниями с хорошо описанными фенотипическими характеристиками [9]. Наш пациент не входит в эту классификацию, так как все члены его расширенной семьи здоровы, и у него нет фенотипических характеристик какого-либо подтипа брахимезофалангии. Заболевание редко можно обнаружить изолированно или при других аномалиях конечностей, таких как синбрахидактилия [10, 11].
Хотя это первое сообщение о случае меромелии, связанной с аномалией стопы, мы думаем, что это не редкость. Принимая во внимание тот факт, что все пациенты с меромелией, о которых ранее сообщалось, находились на неонатальном этапе жизни, и принимая во внимание сложность диагностики незначительной аномалии скелета, такой как брахимезофалангия, у крошечного индивидуума, связь между двумя аномалиями могла быть легко упущена из виду. в ранее описанных случаях.Обследование незначительных аномалий скелета после достижения определенного уровня созревания костей кистей и стоп может быть уместным в случаях новорожденных с амелией и меромелией.
Брахимезофалангия — это незначительная аномалия скелета, которая обычно не вызывает функциональных нарушений. Аналогичным образом, при полном использовании оставшихся пальцев олигодактилия стопы не вызывает затруднений в повседневной деятельности. Однако двусторонняя меромелия верхних конечностей, вызывающая не только значительный физический недостаток, но и серьезный косметический дискомфорт, требует комплексного подхода к лечению.Дополнение протезов или ортезов показано при реабилитации детей с дефектами конечностей. Протез заменяет части конечностей, а ортез стабилизирует существующую конечность [12]. Нашему пациенту в детстве несколько раз предлагали протезирование. Однако из-за финансовых трудностей и неспособности его семьи проявлять достаточную заботу, он никогда не проходил соответствующее обучение и не проходил протезирование. Он испытывал серьезные проблемы при выполнении повседневных дел.У него даже не было возможности самостоятельно одеваться и раздеваться. Другие вопросы, которые необходимо решить при ведении пациентов с редукцией конечностей и которые не менее важны, чем протезирование, — это необходимость обучения и психологическая ситуация ребенка. Следовательно, лечение этих случаев должно осуществляться в междисциплинарных и специализированных клинических условиях. Командный подход, в котором участвуют врач, ортопед, протезист, физиотерапевт, эрготерапевт, психолог и мать ребенка, показал себя как наиболее успешный подход к повышению качества жизни ребенка-инвалида [13–15]. ].
Согласие
Информированное согласие на публикацию было получено от пациента.
Раскрытие информации
Это исследование не получало какого-либо специального гранта от финансирующих агентств в государственном, коммерческом или некоммерческом секторах.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Авторские права
Авторские права © 2019 Meltem Özdemir et al. Это статья в открытом доступе, распространяемая под лицензией Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии правильного цитирования оригинальной работы.
% PDF-1.7
%
217 0 объект
>
эндобдж
xref
217 77
0000000016 00000 н.
0000002449 00000 н.
0000002691 00000 н.
0000002749 00000 н.
0000002785 00000 н.
0000003328 00000 н.
0000003985 00000 н.
0000004022 00000 н.
0000004277 00000 н.
0000004391 00000 п.
0000004923 00000 н.
0000005631 00000 н.
0000005880 00000 н.
0000006458 00000 п.
0000007760 00000 н.
0000007872 00000 н.
0000008021 00000 н.
0000009237 00000 п.
0000009369 00000 н.
0000009801 00000 п.
0000010376 00000 п.
0000010403 00000 п.
0000011867 00000 п.
0000012286 00000 п.
0000012865 00000 п.
0000013051 00000 п.
0000013158 00000 п.
0000013768 00000 п.
0000014440 00000 п.
0000014790 00000 п.
0000015231 00000 п.
0000016695 00000 п.
0000017929 00000 п.
0000019132 00000 п.
0000020253 00000 п.
0000021269 00000 п.
0000023919 00000 п.
0000023989 00000 п.
0000024089 00000 п.
0000032380 00000 п.
0000036143 00000 п.
0000045313 00000 п.
0000045560 00000 п.
0000075190 00000 п.
0000075660 00000 п.
0000075923 00000 п.
0000113459 00000 н.
0000140643 00000 п.
0000140708 00000 н.
0000140801 00000 п.
0000143907 00000 н.
0000144200 00000 н.
0000144494 00000 н.
0000144521 00000 н.
0000144942 00000 н.
0000165170 00000 н.
0000165426 00000 н.
0000165890 00000 н.
0000181090 00000 н.
0000181359 00000 н.
0000181656 00000 н.
0000182149 00000 н.
0000182640 00000 н.
0000204149 00000 н.
0000204418 00000 н.
0000204806 00000 н.
0000226652 00000 н.
0000226927 00000 н.
0000227303 00000 н.
0000227703 00000 н.
0000236906 00000 н.
0000237156 00000 н.
0000237545 00000 н.
0000237925 00000 н.
0000274254 00000 н.
0000274293 00000 н.
0000001836 00000 н.
трейлер
] / Назад 475666 >>
startxref
0
%% EOF
293 0 объект
> поток
hb«b«`P) ef @
Синдром Холта-Орама — NORD (Национальная организация по редким заболеваниям)
Симптомы и физические данные, связанные с синдромом Холта-Орама, сильно различаются от человека к человеку, даже в пределах та же семья.Уродства верхних конечностей варьируются от аномально длинных больших пальцев, похожих на пальцы (трехфалангию), до отсутствия костей большого пальца или большого пальца. Могут также присутствовать другие типы пороков развития верхних конечностей, включая недоразвитие или отсутствие костей предплечья (лучевая и локтевая), сращение или аномальное развитие костей большого пальца и запястья (тенара и запястья), а также неправильное положение большого пальца, предплечья или плечи. Больные могут быть не в состоянии полностью разогнуть руки, повернуть руки внутрь к телу ладонями вниз (пронация) или повернуть руки наружу ладонями вверх (супинация).Более серьезные случаи включают порок развития, при котором руки прикреплены к плечу, а руки отсутствуют или укорочены (фокомелия). У некоторых больных аномальная кость запястья (запястья) является единственным признаком заболевания.
Семьдесят пять процентов пострадавших имеют врожденный порок сердца. Наиболее частыми пороками развития сердца являются дефект межпредсердной перегородки (ASD) и дефект межжелудочковой перегородки (VSD). ДМПП характеризуется аномальным отверстием в фиброзной перегородке (перегородке), разделяющей две верхние камеры (предсердия) сердца.У младенцев с дефектом межпредсердной перегородки вторичного типа отверстие во второй перегородке сердца плода не закрывается должным образом или вторичная перегородка может формироваться ненормально во время внутриутробного развития плода. В результате отверстие между предсердиями сохраняется долгое время после того, как оно должно быть закрыто, что приводит к увеличению нагрузки на правую часть сердца и чрезмерному притоку крови к легким. У многих детей с РАС симптомы отсутствуют. В некоторых случаях у детей с тяжелыми заболеваниями наблюдается замедленный рост, одышка, легкая утомляемость при физических нагрузках и / или нерегулярное сердцебиение (аритмии).
ДМЖП характеризуется аномальным отверстием в перегородке, разделяющей две нижние камеры (желудочки) сердца. Некоторые младенцы и дети с синдромом Холта-Орама имеют один или несколько межжелудочковых перегородок, возможно, в сочетании с РАС. ДМЖП может возникнуть в любой части межжелудочковой перегородки. Размер и расположение дефекта определяют серьезность симптомов. Небольшой межжелудочковый переход может закрыться сам по себе или стать менее значимым по мере взросления и роста ребенка. Дефект умеренного размера может повлиять на способность сердца эффективно перекачивать кровь к легким и остальным частям тела (застойная сердечная недостаточность).Симптомы, связанные с сердечной недостаточностью, включают ненормально учащенное дыхание (тахипноэ), хрипы, необычно быстрое сердцебиение (тахикардия), неспособность расти с ожидаемой скоростью (задержка развития) и / или другие признаки. Большой ДМЖП может вызвать опасные для жизни осложнения в младенчестве. Постоянное повышение давления в артерии, по которой кровь от сердца поступает в легкие (легочная артерия), может вызвать необратимое повреждение легких.
Лица с синдромом Холта-Орама подвержены риску нарушения сердечной проводимости, даже если врожденного порока сердца нет.Когда происходит прерывание нормального потока электрических импульсов (блокада сердца) через сердце, две верхние камеры сердца (предсердия) могут биться нормально, а две нижние камеры (желудочки) могут сокращаться реже или «отставать» предсердные сокращения. При легкой форме сердечной блокады может наблюдаться небольшая задержка между сокращениями предсердий и желудочков (удлиненный интервал P-R). В некоторых случаях могут возникать и другие степени блокады сердца (например, блокада правой ножки пучка Гиса).В некоторых случаях задержка между сокращениями предсердий и желудочков может продолжать увеличиваться до тех пор, пока сердцебиение желудочков не будет полностью прекращено. В других, более тяжелых случаях сердечной блокады только процент (например, половина, четверть и т. Д.) Предсердных сокращений может передаваться в желудочки. Когда происходит полная блокада сердца, желудочки и предсердия сокращаются независимо друг от друга.
Эффекты дефектов проводимости у людей с синдромом Холта-Орама сильно различаются: от отсутствия явных симптомов до потенциально серьезных осложнений.Например, у тех, кто демонстрирует длительные интервалы P-R, могут не проявляться какие-либо связанные с этим симптомы. Наблюдаемые симптомы могут также не возникать у пораженных людей, у которых наблюдается снижение ритма. В более тяжелых случаях блокады сердца недостаточный кровоток из желудочков может вызвать у пораженных людей одышку из-за неспособности сердца эффективно перекачивать кровь (сердечная недостаточность), боли в груди или эпизоды обморока (обмороки). В редких случаях, если сердцебиение желудочков резко замедляется или прекращается, больные могут потерять сознание, иметь судороги или симптомы, угрожающие жизни.У некоторых людей с легкими симптомами синдром Холта-Орама диагностируется только в среднем возрасте, когда появляются симптомы болезни сердечной проводимости.
Синдром Холта-Орама: MedlinePlus Genetics
Синдром Холта-Орама характеризуется аномалиями скелета кистей и рук (верхних конечностей) и проблемами с сердцем.
Люди с синдромом Холта-Орама имеют аномально развитые кости в верхних конечностях. По крайней мере, одна аномалия в костях запястья (кости запястья) присутствует у пораженных людей.Часто эти аномалии костей запястья можно обнаружить только с помощью рентгена. У людей с синдромом Холта-Орама могут быть дополнительные костные аномалии, включая отсутствие большого пальца, длинный большой палец, похожий на палец, частичное или полное отсутствие костей в предплечье, недоразвитую кость плеча и аномалии ключицы или кости. Лопатки. Эти аномалии скелета могут поражать одну или обе верхние конечности. Если поражены обе верхние конечности, костные аномалии могут быть одинаковыми или разными с каждой стороны.В случаях, когда аномалии скелета на обеих сторонах тела неодинаковы, левая сторона обычно поражается сильнее, чем правая.
Около 75 процентов людей с синдромом Холта-Орама имеют проблемы с сердцем (сердечными), которые могут быть опасными для жизни. Наиболее частая проблема — дефект мышечной стенки (перегородки), разделяющей правую и левую стороны сердца. Отверстие в перегородке между верхними камерами сердца (предсердиями) называется дефектом межпредсердной перегородки (ДМПП), а отверстие в перегородке между нижними камерами сердца (желудочками) называется дефектом межжелудочковой перегородки (ДМЖП). .У некоторых людей с синдромом Холта-Орама есть нарушение сердечной проводимости, которое вызвано аномалиями в электрической системе, которая координирует сокращения камер сердца. Заболевание сердечной проводимости может привести к таким проблемам, как более медленная, чем обычно, частота сердечных сокращений (брадикардия) или быстрое и нескоординированное сокращение сердечной мышцы (фибрилляция). Заболевание сердечной проводимости может возникать вместе с другими пороками сердца (такими как ASD или VSD) или как единственная проблема с сердцем у людей с синдромом Холта-Орама.
Признаки синдрома Холта-Орама схожи с признаками состояния, называемого синдромом лучевого луча Дуэйна; однако эти два расстройства вызваны мутациями в разных генах.
Деформации и травмы верхних и нижних конечностей у детей в RI
Многие состояния верхних и нижних конечностей можно исправить неоперативными методами лечения, включая физиотерапию. Если требуется хирургическое вмешательство, лечение включает новейшие методы артроскопической хирургии, восстановление переломов, декомпрессию нерва и реконструктивную хирургию при травмах плечевого сплетения.
Плечевое сплетение — это набор нервов, которые посылают сигналы в мозг для управления мышцами плеча и руки. Если эти нервы повреждены, у ребенка будет паралич плечевого сплетения, который может повлиять на всю руку, включая запястье и кисть, или только на плечо и локоть. Физическая терапия помогает некоторым детям оправиться от паралича плечевого сплетения, но некоторым детям может потребоваться операция для восстановления поврежденных нервов.
Болезнь Блаунта — это порок развития большеберцовой кости (голени), при котором кость выгибается и заставляет голень поворачиваться внутрь.В отличие от изогнутой ноги, которая обычно исправляется со временем, болезнь Блаунта ухудшается, если ее не лечить. Варианты лечения могут быть безоперационными или хирургическими, в зависимости от тяжести состояния.
Мы лечим следующие состояния:
- Паралич плечевого сплетения
- Болезнь Блаунта и деформация конечностей
- Несоответствие длины ног и нарушения роста
Заболевания и травмы рук у детей
Отделение детской ортопедии занимается лечением врожденных заболеваний рук, а также проблем, возникших в результате травм, травм или неврологических состояний.Врожденные нарушения включают лишние пальцы, перепончатые пальцы, отсутствие пальцев, аномальные большие пальцы и проблемы с суставами. Мы также лечим такие состояния, как травмы нервов, травмы костей, нарушения роста и артрит. Джулия Катаринчич, доктор медицины, является одним из немногих детских хирургов-хирургов, прошедших стажировку в Новой Англии.
Мы лечим следующие состояния:
- Врожденная деформация кисти
- Синдактилия (неполное разделение пальцев) и полидактилия (лишние пальцы)
- Большой палец спускового механизма
- Деформации ногтей
- Опухоли кисти и запястья
- Переломы запястья и кисти
- Детская ручная терапия и шинирование
Заболевания стоп
косолапость является общей педиатрической костно-мышечной состояние.Хотя косолапость не вызывает боли, она мешает ребенку правильно ходить. Косолапость можно исправить с помощью гипсовой повязки Понсети или другого менее инвазивного лечения. В некоторых случаях может потребоваться операция.
Врожденная вертикальная таранная кость — это порок развития небольшой таранной кости стопы, вызывающий тяжелую форму плоскостопия, которая может привести к затруднению ходьбы. Мы также лечим плоскостопие, лишние пальцы и деформации пальцев стопы.
Мы лечим следующие состояния:
- Косолапость
- Врожденная вертикальная таранная кость
- Спастическая деформация стопы при ходьбе на пальцах стопы
Подробнее о детской ортопедии в Детской больнице Хасбро
дефектов верхних конечностей | Врожденный дефект
Многие будущие родители с нетерпением ждут рождения своего ребенка и сопутствующих ему радостей родительской жизни.Когда у ребенка врожденный дефект, родители могут быть брошены в вихрь эмоций и медицинских проблем, когда они спешат выяснить, как ухаживать за своим новорожденным.
Например, в редких случаях дети могут родиться с неправильно сформированными ручками или ножками. Эти типы врожденных дефектов известны как дефекты верхних и нижних конечностей.
Примерно 1500 младенцев в год страдают дефектами конечностей, которые возникают, когда ручки или ножки ребенка не формируются должным образом в утробе матери.У младенцев с дефектами верхних конечностей руки или кисти могут быть меньше обычных или отсутствовать.