Амавротическая идиотия — причины, симптомы, диагностика и лечение
Амавротическая идиотия — это понятие, объединяющее группу генетически детерминированных заболеваний, обусловленных дисметаболизмом ганглиозидов с их накоплением в церебральных клетках. Ведущими симптомами патологии являются прогрессивное снижение интеллекта и потеря зрения. Диагностические мероприятия включают неврологический осмотр, офтальмоскопию, церебральную МРТ, генетическую консультацию, исследование липидного состава крови, гистохимический анализ мозгового биоптата. Лечение симптоматическое: противосудорожные, психотропные фармпрепараты, переливания свежезамороженной плазмы.
Общие сведения
Впервые амавротическая идиотия была описана в 1881 году Уорреном Теем — офтальмологом из США, изучавшим изменения глазного дна пациентов. В 1887 году невролог Бернард Сакс представил подробное описание клинической картины заболевания. В последующем он провёл ряд наблюдений аналогичных случаев и сделал вывод о семейном характере патологии. Так была открыта самая известная амавротическая идиотия — болезнь Тея-Сакса. Затем исследователями в области педиатрии и клинической неврологии были описаны другие варианты заболевания, имеющие более поздний дебют. Распространённость патологии в общей популяции составляет 1 случай на 500 000 населения, среди лиц еврейской национальности — на 6 000 населения. Гендерные различия в заболеваемости отсутствуют.
В последующем он провёл ряд наблюдений аналогичных случаев и сделал вывод о семейном характере патологии. Так была открыта самая известная амавротическая идиотия — болезнь Тея-Сакса. Затем исследователями в области педиатрии и клинической неврологии были описаны другие варианты заболевания, имеющие более поздний дебют. Распространённость патологии в общей популяции составляет 1 случай на 500 000 населения, среди лиц еврейской национальности — на 6 000 населения. Гендерные различия в заболеваемости отсутствуют.
Амавротическая идиотия
Причины
Семейный характер заболевания обусловлен генными мутациями, наследуемыми по аутосомно-рецессивному механизму с высокой пенетрантностью дефектного гена. Амавротическая идиотия развивается при наследовании патологического гена от обоих родителей. Вероятность рождения больного ребёнка у гетерозиготных носителей дефекта составляет 25%. Наиболее изучен генетический субстрат болезни Тея-Сакса, представляющий собой вариативные мутации гена HEXA, расположенного на 15-й хромосоме в локусе q23-q24. Указанный ген кодирует фермент, отвечающий за катаболические реакции с расщеплением ганглиозидов. Ферментная недостаточность приводит к накоплению ганглиозидов в ганглиозных клетках головного мозга и сетчатки, что сопровождается их дегенерацией.
Указанный ген кодирует фермент, отвечающий за катаболические реакции с расщеплением ганглиозидов. Ферментная недостаточность приводит к накоплению ганглиозидов в ганглиозных клетках головного мозга и сетчатки, что сопровождается их дегенерацией.
Патогенез
Амавротическая идиотия возникает вследствие метаболических нарушений с отложением липидов в церебральных и ретинальных клетках, в меньшей степени в соматических органах (печени, селезёнке). При идиотии Тея-Сакса в результате нарушений в гене HEXA, отвечающем за синтез лизосомного фермента гексозаминидазы А, не осуществляется катаболизм GM2 ганглиозидов, происходит их постепенное накопление. Аналогичными дисметаболическими сдвигами сопровождаются другие формы заболевания. В церебральных нейронах и клетках сетчатки скапливаются различные по своему составу липидные включения. Инфантильная форма отличается мелкозернистыми включениями с содержанием холестерола, при позднем детском и ювенильном вариантах заболевания продукт накопления наряду с липидами имеет большое количество белка, холестерол отсутствует.
Макроскопически амавротическая идиотия проявляется увеличением объёма мозга, обширными атрофическими изменениями затылочных долей, мозжечка, истончением зрительных трактов. Микроскопическая картина в головном мозге представлена набуханием ганглиозных клеток и их отростков, заполнением внутриклеточного пространства липоидными включениями, сморщиванием ядер, распадом тигроидной субстанции. Аналогичные изменения отмечаются в ретинальных клетках, наиболее выражены в области жёлтого пятна.
Классификация
Амавротическая идиотия включает несколько клинических вариантов, отличающихся возрастом манифестации симптоматики, характером течения, продолжительностью жизни больных. Вероятно, они обусловлены различными генетическими дефектами, определёнными особенностями метаболических нарушений. Уточнение клинической формы патологии необходимо для понимания прогноза заболевания, выбора оптимальной лечебной тактики. В соответствии с клиническими особенностями выделяют четыре основные формы:
- Ранняя детская (инфантильная форма Тея-Сакса).
 Наиболее изученный вариант заболевания. Характерна манифестация в 4-6 месяцев. Течение быстропрогрессирующее, летальный исход наблюдается до 3-летнего возраста.
Наиболее изученный вариант заболевания. Характерна манифестация в 4-6 месяцев. Течение быстропрогрессирующее, летальный исход наблюдается до 3-летнего возраста. - Поздняя детская (форма Янского-Бильшовского). Дебют приходится на возраст 3-4 года. Клиника идентична ранней детской форме, имеет немного замедленное течение. Длительность болезни составляет 4-6 лет.
- Юношеская (форма Шпильмейера-Фогта). Начинается в 6-9 лет, отличается относительно медленным прогрессированием симптомов. Пациенты доживают до 20 лет.
- Поздняя (форма Куфса). Манифестирует в пубертатном периоде или в старшем возрасте. Имеет наиболее благоприятное замедленное течение. Летальный исход наступает в среднем спустя 10-15 лет с момента дебюта симптоматики.
 Наиболее изученный вариант заболевания. Характерна манифестация в 4-6 месяцев. Течение быстропрогрессирующее, летальный исход наблюдается до 3-летнего возраста.
Наиболее изученный вариант заболевания. Характерна манифестация в 4-6 месяцев. Течение быстропрогрессирующее, летальный исход наблюдается до 3-летнего возраста.
В отдельных случаях наблюдается врождённая амавротическая идиотия, получившая название форма Нормана-Вуда. Симптомы возникают в первые недели жизни ребёнка, основное проявление — прекращение нервно-психического развития.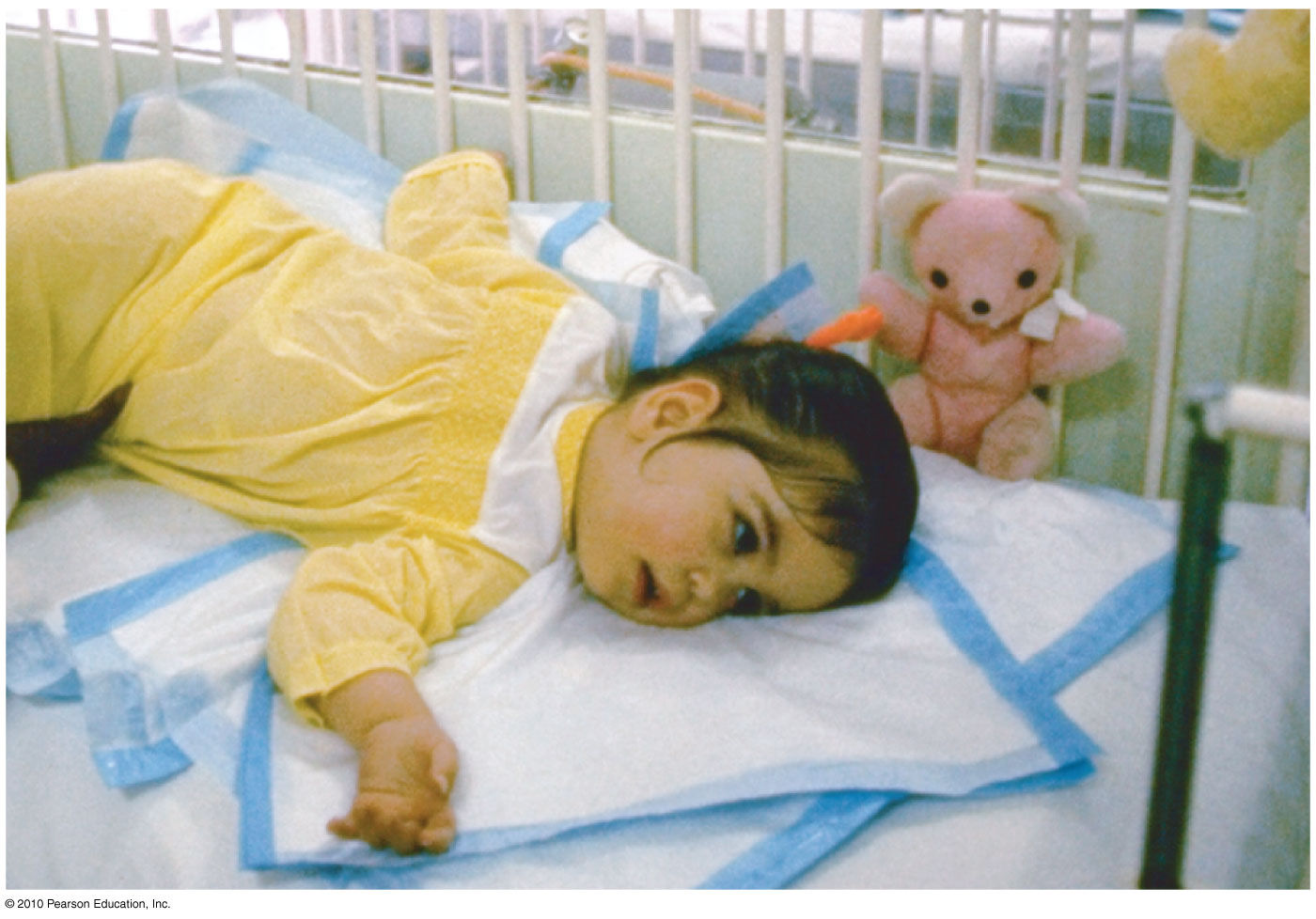 Некоторые авторы предлагают внести вариант Нормана-Вуда в классификацию в качестве отдельной нозологии.
Некоторые авторы предлагают внести вариант Нормана-Вуда в классификацию в качестве отдельной нозологии.
Симптомы амавротической идиотии
Основу клинической картины составляют прогрессирующие зрительные расстройства, постепенная утрата приобретённых интеллектуальных способностей. Сопутствующими проявлениями являются глухота, судорожный синдром, экстрапирамидные нарушения, мозжечковая атаксия. Тяжесть симптоматики зависит от формы заболевания.
Ранняя детская амавротическая идиотия манифестирует снижением двигательной активности ребёнка, утратой способности сидеть, держать головку. В последующем продолжается угасание двигательных и статических функций вплоть до тетрапареза с полной обездвиженностью. В начальном периоде типична гиперакузия, выражающаяся вздрагиванием ребёнка при звуковом раздражении. Нарушения зрения приводят к отсутствию слежения за игрушками, фиксации взгляда на блестящих предметах. В короткие сроки наступает полная слепота (амавроз). Отмечаются судороги с преобладанием тонического компонента. Возможен бульбарный синдром, вегетативные расстройства (гипергидроз, акроцианоз, гипотермия, лабильность пульса). Выявляется мышечная гипотония, прогрессирующая в терминальной стадии до децеребрационной ригидности.
Отмечаются судороги с преобладанием тонического компонента. Возможен бульбарный синдром, вегетативные расстройства (гипергидроз, акроцианоз, гипотермия, лабильность пульса). Выявляется мышечная гипотония, прогрессирующая в терминальной стадии до децеребрационной ригидности.
Амавротическая идиотия Янского-Бильшовского возникает на четвёртом году жизни. На начальном этапе наряду со снижением зрения и когнитивными нарушениями отмечаются мозжечковые расстройства: шаткость походки, грубая дискоординация, нистагм. Постепенно больной утрачивает способность ходить, стоять, сидеть. Интеллектуальная сфера деградирует до уровня идиотии, развивается амавроз. Юношеская форма отличается более мягким течением: интеллект редко деградирует до идиотии, зрительная дисфункция не доходит до амавроза, двигательная сфера остаётся более сохранной. Типичны экстрапирамидные расстройства (атетоз, хорея, тремор), мозжечковая дисфункция. Возможна глухота, возникновение эпилептических приступов. В ряде случаев обнаруживается микседематозное ожирение.
В ряде случаев обнаруживается микседематозное ожирение.
Амавротическая идиотия Куфса наблюдается крайне редко, протекает без выраженных двигательных, зрительных, интеллектуальных расстройств. Манифестирует изменениями личности, ограничением круга интересов, пониженной активностью. Со временем возникает интеллектуальное снижение, редко достигающее уровня выраженной органической деменции. Типичны различные психические расстройства: галлюцинации, бред, кататония, аффективные психозы. Возможны экстрапирамидные, мозжечковые симптомы, эпилептические пароксизмы, тугоухость.
Осложнения
Диагностика
Характерное сочетание триады симптомов (когнитивная деградация, резкое ухудшение зрения, двигательные нарушения) позволяет предположить клинический диагноз. Сложности возникают при определении поздней формы. Подтверждение диагноза возможно после проведения ряда исследований:
- Исследование неврологического статуса. Результаты осмотра невролога зависят от формы и периода заболевания, возраста пациента. Отмечается резкое несоответствие интеллектуального развития возрасту. Выявляются спастические парезы, мозжечковая дисфункция, гиперкинезы, мышечная дистония, вегетативно-трофические нарушения.
- Консультация офтальмолога. У грудничков наблюдается отсутствие реакции слежения, у пациентов старшего возраста визометрия подтверждает резкое снижение остроты зрения. Офтальмоскопия диагностирует двустороннюю атрофию зрительного нерва. Патогномоничным признаком является наличие в макулярной зоне вишнёвого пятнышка. Поздняя амавротическая идиотия отличается отсутствием данного симптома, картиной пигментного ретинита на глазном дне.
- Лабораторные анализы. В крови обнаруживаются вакуолизированные лимфоциты. При проведении биохимического исследования крови могут определяться изменения липидного профиля (увеличение уровня фосфолипидов, холестерола), повышенная концентрация отдельных ферментов.
- Нейровизуализация. МРТ головного мозга визуализирует атрофию церебральных тканей, более заметную в мозжечке, затылочной области. Выраженность атрофических изменений максимальна при раннем дебюте, минимальна при поздней форме. Наряду с атрофией выявляется истончение зрительных нервов.
- Церебральная биопсия. Гистохимическое и цитологическое исследование материала из атрофических участков позволяет достоверно установить диагноз «амавротическая идиотия» по наличию характерных внутриклеточных липидных включений. По цвету окрашивания включений можно судить о форме заболевания.
- Генетический анализ. Консультация генетика включает сбор семейного анамнеза и составление генеалогического древа. Дает возможность подтвердить семейный характер, уточнить тип наследования патологии.
 Отмечается резкое несоответствие интеллектуального развития возрасту. Выявляются спастические парезы, мозжечковая дисфункция, гиперкинезы, мышечная дистония, вегетативно-трофические нарушения.
Отмечается резкое несоответствие интеллектуального развития возрасту. Выявляются спастические парезы, мозжечковая дисфункция, гиперкинезы, мышечная дистония, вегетативно-трофические нарушения.
 Выраженность атрофических изменений максимальна при раннем дебюте, минимальна при поздней форме. Наряду с атрофией выявляется истончение зрительных нервов.
Выраженность атрофических изменений максимальна при раннем дебюте, минимальна при поздней форме. Наряду с атрофией выявляется истончение зрительных нервов.
Амавротическую идиотию дифференцируют от иных наследственных дисметаболических и дегенеративных болезней с поражением ЦНС. Ранний детский вариант необходимо отличать от болезни Ниманна-Пика, протекающей с выраженным поражением соматических органов. Поздняя форма требует дифференцировки от ряда заболеваний с ведущей мозжечковой и экстрапирамидной симптоматикой (рассеянного склероза, болезни Лафоры, лейкодистрофии).
Лечение амавротической идиотии
Специфическая терапия не разработана. Проводится симптоматическое поддерживающее лечение. При наличии эпилептических пароксизмов применяют антиконвульсанты, психических отклонений — психотропные препараты (седативные, транквилизаторы, нейролептики). Отдельные клиницисты указывают на эффективность переливания плазмы, препаратов крови. Рекомендована витаминотерапия, общеукрепляющие мероприятия. На поздних стадиях больной нуждается в тщательном уходе, профилактике инфекционных осложнений. Перспективным направлением в поиске эффективных методов лечения является генная инженерия, однако существенные результаты в этой области пока не достигнуты.
Прогноз и профилактика
Амавротическая идиотия завершается летальным исходом. Длительность жизни пациентов зависит от возраста дебюта патологических изменений. Наиболее неблагоприятный прогноз имеет инфантильная форма — больные погибают спустя 1,5-2 года. Самое медленное доброкачественное течение наблюдается при позднем варианте. Профилактические меры направлены на исключение родственных браков. Имеющим больного ребёнка родителям рекомендуется отказаться от дальнейшего деторождения. В случае беременности гетерозиготной пары возможно исследование концентрации гексозаминидазы-А в амниотической жидкости. Значительное снижение является показанием к прерыванию беременности.
Профилактические меры направлены на исключение родственных браков. Имеющим больного ребёнка родителям рекомендуется отказаться от дальнейшего деторождения. В случае беременности гетерозиготной пары возможно исследование концентрации гексозаминидазы-А в амниотической жидкости. Значительное снижение является показанием к прерыванию беременности.
АМАВРОТИЧЕСКАЯ ИДИОТИЯ — Большая Медицинская Энциклопедия
Амавротическая идиотия (греческий amauros — темный, слепой и idiōteia — невежественность) — группа наследственных заболеваний, характеризующихся прогрессирующим снижением зрения и интеллекта в сочетании с другими неврологическими симптомами. Различают следующие формы амавротической идиотии: врожденная форма Нормена—Вуда; ранняя детская (инфантильная) форма — болезнь Тея—Сакса; поздняя детская форма Билыновского—Янского; юношеская форма Баттена—Шпильмейера—Фогта; поздняя форма болезни Куфса.
Амавротическая идиотия — наследственные заболевания с аутосомно-рецессивным типом наследования (см. Наследование). Наиболее часто встречается амавротическая идиотия Тея—Сакса. Многими авторами замечено, что эта форма амавторической идиотии чаще обнаруживается среди евреев. По данным Мириантопулоса (N. С. Myrianthopoulos, 1962), частота заболевания этой формой детей в США составляла 1 на 6000 среди евреев и соответственно 1 на 500 000 среди нееврейского населения.
Наследование). Наиболее часто встречается амавротическая идиотия Тея—Сакса. Многими авторами замечено, что эта форма амавторической идиотии чаще обнаруживается среди евреев. По данным Мириантопулоса (N. С. Myrianthopoulos, 1962), частота заболевания этой формой детей в США составляла 1 на 6000 среди евреев и соответственно 1 на 500 000 среди нееврейского населения.
Патологическая анатомия
При всех формах амавротической идиотии имеются сходные патоморфологические изменения в мозге. Макроскопически отмечается увеличение мозга и наряду с этим диффузная атрофия отдельных областей, чаще мозжечка, затылочных долей больших полушарий. Гистологически обнаруживается картина генерализованного распада ганглиозных клеток нервной системы, что особенно выражено при амавротической идиотии Тея—Сакса. Дегенерация нейронов проявляется увеличением их размера и характерным набуханием, баллонообразным вздутием клеток и их отростков. При этом клетки заполняются мелкозернистым веществом липоидного характера. Ядра клеток сморщиваются, теряют свою форму и сдвигаются к периферии. Нейрофибриллы и глыбки базофильного «тигроидного» вещества полностью распадаются. Дегенерация захватывает и глиозные элементы. В тяжелых случаях происходит демиелинизация и разрушение многих аксонов, что, по-видимому, имеет вторичный характер вследствие накопления липидов.
Ядра клеток сморщиваются, теряют свою форму и сдвигаются к периферии. Нейрофибриллы и глыбки базофильного «тигроидного» вещества полностью распадаются. Дегенерация захватывает и глиозные элементы. В тяжелых случаях происходит демиелинизация и разрушение многих аксонов, что, по-видимому, имеет вторичный характер вследствие накопления липидов.
Патогенез
Установлено, что при амавротической идиотии имеется глубокое нарушение липидного обмена. Наиболее полно изучена амавротическая идиотия Тея—Сакса, которую отнесли к группе внутриклеточных липоидозов. Кленк (Е. Klenk, 1939, 1942) отметил при болезни Тея—Сакса увеличение в мозге гликолипида-ганглиозида. Нормен (R. М. Norman, 1963), Шнек (L. Schneck, 1969), Судзуки (Y. Suzuky, 1971) выявили увеличение содержания ганглиозидов в печени, селезенке, эритроцитах крови, что свидетельствовало о генерализованном нарушении обмена ганглиозидов. Нарушение метаболизма ганглиозида GM2 связывают с отсутствием или резким снижением содержания энзима гексозаминидазы-А (Шнек, 1970; Судзуки, 1971). Отсутствие этого фермента отмечается уже у эмбриона на 18—20-й неделе. У гетерозиготных носителей гена амавротической идиотии Тея—Сакса отмечается умеренное снижение гексозаминидазы-А.
Отсутствие этого фермента отмечается уже у эмбриона на 18—20-й неделе. У гетерозиготных носителей гена амавротической идиотии Тея—Сакса отмечается умеренное снижение гексозаминидазы-А.
Особенности нарушений обмена веществ при других формах амавротической идиотии и, в частности, является ли для них нарушение биохимических процессов первичным, точно еще не установлено. Предполагается, что при врожденной форме Нормена—Вуда имеет место отложение ганглиозида типа GM3 [Осетовская (E. Osetowska, 1968)]. При ювенильной форме Баттена—Шпильмейера—Фогта, по мнению ряда авторов, накапливаются ганглиозиды в смеси с холестерином и липопигмент типа липофусцина.
Клиническая картина
Врожденная форма Нормена—Вуда проявляется в первые дни или недели после рождения. У ребенка обнаруживаются гидроцефалия или микроцефалия, судороги, параличи, расстройства дыхания, глотания. Летальный исход обычно наступает в первые месяцы жизни.
Ранняя детская (инфантильная) форма — болезнь Тея—Сакса — начинается в возрасте 4—6 месяцев. Часто заболевание носит семейный характер. Рано обнаруживается снижение зрения. Ребенок не может фиксировать взгляд, не следит за игрушками. Довольно рано на глазном дне обнаруживается симптом «вишневой косточки» — вишнево-красное пятнышко в макулярной области, окруженное серовато-белым ободком. В последующем развивается атрофия зрительных нервов и полная слепота. Исчезают ориентировочные и защитные реакции. Двигательные нарушения приводят к полной обездвиженности. При болезни Тея—Сакса наблюдается симптом повышенной реакции на звуковые раздражения — дети резко вздрагивают от обычного звука, могут отмечаться судороги, носящие преимущественно тонический характер. В конечной стадии болезни развивается кахексия и состояние децеребрационной ригидности. Смерть наступает в среднем через 1,5—2 года после начала заболевания.
Часто заболевание носит семейный характер. Рано обнаруживается снижение зрения. Ребенок не может фиксировать взгляд, не следит за игрушками. Довольно рано на глазном дне обнаруживается симптом «вишневой косточки» — вишнево-красное пятнышко в макулярной области, окруженное серовато-белым ободком. В последующем развивается атрофия зрительных нервов и полная слепота. Исчезают ориентировочные и защитные реакции. Двигательные нарушения приводят к полной обездвиженности. При болезни Тея—Сакса наблюдается симптом повышенной реакции на звуковые раздражения — дети резко вздрагивают от обычного звука, могут отмечаться судороги, носящие преимущественно тонический характер. В конечной стадии болезни развивается кахексия и состояние децеребрационной ригидности. Смерть наступает в среднем через 1,5—2 года после начала заболевания.
Поздняя детская форма Бильшовского—Янского начинается в 3—4 года. Течение прогрессирующее, с ремиссиями. Нарастающая органическая деменция сочетается с общими судорожными припадками, атаксией, экстрапирамидными расстройствами. На глазном дне выявляется атрофия зрительных нервов. Некоторые авторы отрицают нозологическую самостоятельность поздней детской формы Бильшовского—Янского и рассматривают случаи этого заболевания как проявление рано начинающейся ювенильной формы Баттена—Шпильмейера—Фогта пли как более поздние варианты болезни Тея—Сакса. Летальный исход в большинстве случаев наступает в конце первого десятилетия жизни.
На глазном дне выявляется атрофия зрительных нервов. Некоторые авторы отрицают нозологическую самостоятельность поздней детской формы Бильшовского—Янского и рассматривают случаи этого заболевания как проявление рано начинающейся ювенильной формы Баттена—Шпильмейера—Фогта пли как более поздние варианты болезни Тея—Сакса. Летальный исход в большинстве случаев наступает в конце первого десятилетия жизни.
Юношеская форма Баттена—Шпильмейера—Фогта начинается в возрасте 6—10 лет, характеризуется медленно прогрессирующим течением. Картина глазного дна часто соответствует пигментному ретиниту. Заболевание начинается с постепенного падения зрения и нарастающей деменции. Изменения в двигательной системе могут быть различными и непостоянными: наблюдаются нерезко выраженные тетрапарезы, экстрапирамидные и бульбарные нарушения. Нередок эпилептиформный синдром. Заболевание заканчивается смертельным исходом на 2—3-м десятилетии жизни.
Поздняя амавротическая идиотия Куфса наблюдается крайне редко, возникает в зрелом возрасте и характеризуется вялым течением. Развиваются изменения личности по типу органического психического синдрома. На глазном дне выявляется картина пигментного ретинита с атрофией зрительных нервов. В поздней стадии заболевания развиваются параличи и эпилептиформный синдром. Больные обычно погибают в течение 10—15 лет от начала заболевания.
Развиваются изменения личности по типу органического психического синдрома. На глазном дне выявляется картина пигментного ретинита с атрофией зрительных нервов. В поздней стадии заболевания развиваются параличи и эпилептиформный синдром. Больные обычно погибают в течение 10—15 лет от начала заболевания.
Психические расстройства при разных формах амавротической идиотии имеют различия, отчасти связанные с возрастными особенностями.
При врожденной форме Нормена—Вуда в первые недели жизни наблюдается остановка нервно-психического развития.
При ранней детской форме Тея—Сакса нормально развивавшиеся до того дети относительно быстро становятся вялыми, утрачивают интерес к окружающему, перестают узнавать близких. Происходит постепенное исчезновение приобретенных двигательных навыков и речи. В течение нескольких месяцев, реже до 2 лет, развивается глубокое слабоумие типа идиотии.
При поздней детской форме Бильшовского—Янского психическая деградация происходит более медленно, чем при ранней детской форме. Может наблюдаться волнообразное течение с периодами ухудшения (в виде вялости, апатии, оглушения) и улучшения (с частичным восстановлением активности). В конечной стадии имеет место глубокое слабоумие с утратой речи и навыков самообслуживания.
При юношеской форме Баттена—Шпильмейера—Фогта наблюдаются медленно нарастающие вялость, апатия, обеднение речи. Все более заметно интеллектуальное снижение, однако редко достигающее степени идиотии, нарушения памяти. Присоединяется дизартрия, нарушаются навыки письма и чтения. В конечной стадии наблюдается выраженная органическая деменция.
При поздней форме Куфса психические расстройства отличаются наибольшей вариабельностью. Для нее характерен наиболее медленный темп прогрессирования. Начальные проявления выражаются изменениями личности в виде снижения активности, сужения круга интересов. Спустя несколько лет все более заметным становится прогрессирующее снижение интеллекта вплоть до органической деменции с апатией или легкой эйфорией, потеря навыков самообслуживания. Описываются случаи с галлюцинаторно-бредовыми и кататоническими расстройствами, а также форма с относительно сохранным интеллектом.
Диагноз
Диагноз и дифференциальный диагноз основывается на типичной клинической картине и специфических изменениях глазного дна.
Инфантильная форма Тея—Сакса имеет некоторые общие черты с болезнью Ландинга (системным позднедетским GM1-ганглиозидозом) и мукополисахаридозом типа Гурлер (гаргоилизм). Болезнь Ландинга описана под названиями «биохимически особая форма инфантильной амавротической идиотии», «болезнь Тея—Сакса с поражением внутренних органов», «вариант синдрома Гурлер», «семейный нейро-висцеральный липоидоз». Болезнь Ландинга начинается в первые месяцы жизни и заканчивается смертью в возрасте 2—3 лет. Вишнево-красное пятнышко на глазном дне бывает в 20% случаев, амавроз и гиперакузия встречаются редко, отмечается гепатоспленомегалия, психические и двигательные нарушения. Ювенильную амавротическую идиотию Баттена—Шпильмейера—Фогта иногда необходимо дифференцировать с синдромом Лоренса—Муна—Бидля, при котором наблюдаются резкое ожирение, полидактилия, выраженные вегетативно-трофические нарушения и часто отсутствуют изменения в двигательной сфере. Полиморфизм симптоматики поздней формы Куфса затрудняет прижизненную диагностику поздней формы, которую следует дифференцировать с атаксией Фридрейха, рассеянным склерозом, болезнью Альцгеймера, болезнью Пика, прогрессивным параличом, а иногда с шизофренией. В ряде случаев отдельные формы амавротической идиотии необходимо дифференцировать с лейкодистрофиями (см.).
Лечение
Специфическое лечение амавротической идиотии не разработано. Применяются седативные средства, нейролептики, противосудорожные средства, общеукрепляющие. Некоторый эффект может быть получен от применения тканевых экстрактов, переливания крови и плазмы.
Профилактика
Возможно установление гетерозиготного носительства мутантного гена у родителей больных. При болезни Тея—Сакса гетерозиготное состояние выявляется путем исследования липидного спектра эритроцитов (снижение сфингомиелина и кефалина) и исследования фермента гексозаминидазы-А, у гетерозиготных носителей гена болезни Баттена—Шпильмейера—Фогта — исследованием крови (вакуолинизированные лейкоциты) и исследованием липидов эритроцитов (снижение сфингомиелина). В случае беременности женщины, гетерозиготной по гену амавротической идиотии Тея—Сакса, целесообразно исследование гексозаминидазы-А в амниотической жидкости, полученной на 18—20-й неделе беременности. При значительном снижении гексозаминидазы-А показано искусственное прерывание беременности.
Библиография: Бадалян Л. О., Таболин В. А. и Вельтищев Ю. Е. Наследственные болезни у детей, с. 82, М., 1971; Гербер Э. Л. и Мошковская Р. Д. Поздняя форма амавротической идиотии, Сб. науч. трудов, посвящ. 150-летию Моск. психоневрол. б-цы № 3, в. 4, с. 365, 1963; Мнухин С. С. Амавротическая идиотия, Многотомн. руководство по неврол., под ред. С. Н. Давиденкова, т. 7, с. 366, Л., 1960; Bergener M. u. Jungklaass F. К. Beitrag zur Klinik und Genetik der amaurotischen Idiotie (Spätinfantile und Spätform) unter besonderer Beriicksichtigung diagnostischer und differentialdiagnostischer Probleme, Nervenarzt, S. 316, 1968; Cerebral sphingolipidoses, ed. by M. A. Stanley a. B. W. Volk, N. Y.—L., 1962, bibliogr.; Jatzkewitz H. Evidence for metabolic blocks in sphingolipidoses, Proc. 5-th int. congr. neuropathol., p. 417, Amsterdam a. o., 1966; Schettler G. Gangliosidosen, Handb. inn. Med., hrsg. v. G. Bergmann u. a., Bd 7, T. 2, S. 657, В. u. a., 1955, Bibliogr.; Schneck L. Volk B. W. a. Saifer A. The gangliosidoses, Amer. J. Med., v. 46, p. 245, 1969, bibliogr.; Stanbury J. В. а. о. The metabolic basis of inherited disease, N. Y. a. o., 1966; Suzuku J. Y. a. o. Gm2-gangliosidosis with total hexosaminidase deficiency, Neurology (Minneap.), v. 21, p. 313, 1971, bibliogr.
Л. О. Бадалян; В. В. Ковалев (психиат.).
Болезнь Тея-Сакса (ранняя детская амавротическая идиотия): Обзор заболевания
Болезнь Тея — Сакса – что это такое?
Ранняя детская амавротическая идиотия — это редкое заболевание, которое передаётся по наследству. Оно поражает мозг и нервные клетки. Существуют две формы данной болезни:
-
Первая (наиболее распространённая) начинает развиваться почти сразу после рождения ребёнка, и длительность жизни малыша в таком случае составляет 4-5 лет.
-
Болезнь Тея-Сакса с поздним началом проявляется между периодом полового созревания и возрастом 23-26 лет. Продолжительность жизни больного зависит в первую очередь от тяжести симптомов заболевания. Такие пациенты могут прожить тот же промежуток времени, что и здоровый человек.
Что может вызывать эту болезнь?
Болезнь Тея-Сакса может развиваться, если родители передали ребёнку поражённый (мутировавший) ген.
| Гены
Они входят в состав клеток организма и содержат генетическую информацию или дезоксирибонуклеиновую кислоту (ДНК), которая отвечает за физические характеристики человека. Гены (по отдельности или в совокупности) определяют какие генетические черты ребёнок унаследует от родителей, на пример, группу крови, цвет волос, глаз, а также вероятность развития того или иного заболевания. Определённое количество генов составляют хромосому. Поражения как гена, так и хромосомы, вызывают изменения в процессах и функциях организма. Эти дефекты могут практически не проявляться, однако иногда они становятся причиной развития таких серьёзных заболеваний как гемофилия или синдром Дауна. Такие проблемы со здоровьем как болезнь Стилла (форма ревматоидного артрита у детей) или депрессия также могут быть следствием отклонений на генетическом уровне. Мутировавший ген переходит от родителей к детям. Болезни, передающиеся по наследству, обычно имеют генетический характер. Человек может родиться с генетическим фоном, который будет влиять на повышенную восприимчивость или риск проявления определённого заболевания. |
Когда малыш унаследует искажённый ген от обоих родителей, он заболевает.
Если происходит передача поражённого гена от одного из родителей – ребёнок становится носителем дефекта. Это означает, что у него будет присутствовать данный ген в организме, однако сам он не заболеет.
|
Носитель заболевания Это человек, который может передать своим детям наследственную (генетическую) болезнь, хотя сам и не является поражённым недугом. Кроме этого, существует также риск передачи статуса носителя заболевания. Некоторые недуги вызваны именно отклонениями на генном и хромосомном уровнях. Каждый человек унаследует 23 хромосомы от каждого из родителей. Таким образом, у него получается 23 пары хромосом в организме. Если одна или обе хромосомы из пары поражены, возникает генетическое заболевание. В большинстве случаев, у человека с вышеуказанным дефектом обе хромосомы в паре являются повреждёнными. Такое расстройство называется болезнью с аутосомно-рецессивным типом наследования. Определённые генетические заболевания возникают из-за поражения X и Y хромосом, которые отвечают за определение пола. Если хотя бы одна хромосома из пары с отклонением, человек может быть носителем заболевания. |
Мутировавший ген препятствует выработке в организме такого фермента как гексозаминидаза А, который отвечает за расщепление ганглиозидов (сложных природных липидов) в клетках. В случае их накопления происходит блокирование работы мозга и повреждение нервных клеток, что и вызывает болезнь Тея-Сакса.
|
Ферменты Это белок, который вырабатывается для ускорения протекания определённой химической реакции. В нашем организме существует много ферментов для обеспечения различных процессов жизнедеятельности, таких как пищеварение и свёртывание крови. Некоторые генетические заболевания напрямую связаны с выработкой определённых ферментов. Чтобы установить диагноз (на пример, проблемы с почками) врач измеряет количество необходимого фермента в крови пациента. |
При болезни Тея-Сакса с поздним началом в организме производится пониженное количество вышеуказанного фермента. Люди с таким заболеванием унаследуют ген гексозаминидаза А с поздним развитием от обоих родителей или же по одному гену с поздним развитием и неспособностью к активности. Риск появления мутировавшего гена возрастает у людей с корнями, которые уходят к евреям-ашкенази, так как 1 из 30 представителей этой национальности является носителем данного заболевания. Кроме того, к развитию болезни Тея-Сакса также предрасположены люди французско-канадского происхождения, проживающие в восточной части долины реки Святого Лоренса провинции Квебек и каджуны (франкоязычные жители штата Луизиана).
Болезнь Тея-Сакса – симптомы
После рождения ребёнок с таким пороком выглядит абсолютно здоровым. Однако вы можете заметить следующее:
-
В 3-6 месяцев малыш начинает слабо зрительно реагировать на действия вокруг него, и ему сложно сфокусироваться на предмете. Врач может обнаружить красные точки на сетчатке глаза ребёнка.
-
В 6-10 месяцев у ребёнка происходит спад активности. Ему становится сложно сидеть и переворачиваться. Кроме этого, возникают проблемы со слухом и зрением.
-
После 10 месяцев болезнь активно прогрессирует. У ребёнка возможны приступы, потеря зрения и атрофия мышц.
|
Приступ Это внезапные вспышки аномальной электрической активности мозга, которые влияют на функционирование мышечной системы, контроль двигательного аппарата, искажают речь, зрительные образы и психологическое восприятие действительности. Степень поражения у каждого индивидуальная. Это также зависит от типа, частоты и тяжести припадков. В некоторых случаях человек падает на пол и начинает биться в конвульсиях, при которых мышцы тела сильно сокращаются, и возникает непроизвольное подёргивание конечностей. Другие входят в состояние транса, у них остаются подвижными лишь несколько мышц. Иногда они ощущают запахи или видят образы, нехарактерные для здорового человека. Часто припадок является симптомом других проблем со здоровьем, таких как возникновение высокой температуры тела (особенно у детей), нарушение мозгового кровообращения, инфекция, низкий уровень глюкозы в крови (гипогликемия), пониженное давление или опухоль мозга. |
В ходе развития у малыша болезни Тея-Сакса с поздним началом такие симптомы как неуклюжесть и перепады настроения могут не восприниматься серьёзно. К более поздним признакам заболевания относятся судорожное подёргивание и слабость мышц, невнятная речь и проблемы с мыслительными процессами. Характер осложнений зависит от того, какое количество гексозаминидазы А вырабатывается в организме.
Болезнь Тея-Сакса – система распознавания заболевания
Если у лечащего врача есть подозрение, что ребёнок поражён болезнью Тея-Сакса, он прежде всего проведёт обследование пациента и возьмёт анализ крови, чтобы проверить уровень гексозаминидазы А в организме. Генетический тест необходим для подтверждения диагноза.
В чём заключается лечение?
Основным лечением болезни Тея-Сакса является контроль симптомов и обеспечение ребёнку комфорта в повседневной жизни, так как панацеи от этого недуга на сегодняшний день не существует. В данном случае полезным будет пройти генетическое консультирование и посетить сеансы терапии, где можно найти поддержку и пообщаться с людьми, у которых есть аналогичная проблема. Если у вас развивается болезнь Тея-Сакса с поздним началом, лечение также основывается на мониторинге симптомов. В зависимости от симптомов вы можете получать определённые препараты, на пример, от депрессии. Несомненно, известие о том, что у вашего ребёнка обнаружили болезнь Тея-Сакса, может повергнуть вас в шок. В такой ситуации кроме заботы о ребёнке, следует помнить и о своём здоровье. Необходимо разговаривать с лечащим врачом на такие темы:
-
Ваши переживания и помощь в уходе за малышом.
-
Группа поддержки в вашем регионе.
-
Генетическое консультирование для всей семьи, чтобы каждый принял сложившуюся ситуацию.
По мере развития заболевания ребёнку может понадобиться более тщательный уход. Подбадривайте его и внушайте, что нужно бороться, подарите свою любовь и ласку. Возможно, у вас возникнет необходимость в дополнительной помощи по уходу за малышом. Проконсультируйтесь с лечащим врачом о том, куда можно обратиться по этому вопросу.
Болезнь Тея-Сакса — зачем нужно проходить обследование на наличие заболевания?
Носители данного порока могут передать детям заболевание даже если они сами не больны. Если вы и партнёр являетесь носителями болезни Тея-Сакса, риск развития аналогичного дефекта у ваших детей повышается до 1 из 4 случаев (25%). Когда вы планируете заводить ребёнка, Американская корпорация акушёров и гинекологов рекомендует сделать следующее:
Оба партнёра должны пройти скрининг тесты, если они относятся к таким категориям, как евреи-ашкенази, каджуны (франкоязычные жители штата Луизиана), имеют французско-канадское происхождение, либо же они имеют семейный анамнез по данной болезни. В случае если обследование покажет, что вы позитивны, последующие решения поможет принять прохождение генетического консультирования.
Если хотя бы один из партнёров соответствует вышеуказанным категориям, он также должен пройти процедуру тестирования. Один положительный результата на носителя заболевания предполагает также обследование другого партнёра.
|
Семейный анамнез Из семейного анамнеза врач может узнать, есть ли у пациента кровные родственники с аналогичным заболеванием. Это поможет специалисту установить степень риска передачи порока по наследству. К кровным родственникам относятся как живые, так и умершие члены семьи. Это могут быть:
В некоторых случаях семейный анамнез бывает довольно сложным. Это зависит от нескольких факторов:
Генетическое консультирование Генетическое консультирование – это пояснения медицинского работника (консультанта по генетическим вопросам или генетика), специализация которого, помогать людям разобраться в опасности передачи генетических заболеваний, а также объяснять возможность унаследования ребёнком такого вида болезни (серповидно-клеточная болезнь, кистозный фиброз, гемофилия). Генетическое консультирование включает:
|
Болезнь Тея-Сакса (ранняя детская амавротическая идиотия): Обзор заболевания
Болезнь Тея — Сакса – что это такое?
Ранняя детская амавротическая идиотия — это редкое заболевание, которое передаётся по наследству. Оно поражает мозг и нервные клетки. Существуют две формы данной болезни:
-
Первая (наиболее распространённая) начинает развиваться почти сразу после рождения ребёнка, и длительность жизни малыша в таком случае составляет 4-5 лет.
-
Болезнь Тея-Сакса с поздним началом проявляется между периодом полового созревания и возрастом 23-26 лет. Продолжительность жизни больного зависит в первую очередь от тяжести симптомов заболевания. Такие пациенты могут прожить тот же промежуток времени, что и здоровый человек.
Что может вызывать эту болезнь?
Болезнь Тея-Сакса может развиваться, если родители передали ребёнку поражённый (мутировавший) ген.
| Гены
Они входят в состав клеток организма и содержат генетическую информацию или дезоксирибонуклеиновую кислоту (ДНК), которая отвечает за физические характеристики человека. Гены (по отдельности или в совокупности) определяют какие генетические черты ребёнок унаследует от родителей, на пример, группу крови, цвет волос, глаз, а также вероятность развития того или иного заболевания. Определённое количество генов составляют хромосому. Поражения как гена, так и хромосомы, вызывают изменения в процессах и функциях организма. Эти дефекты могут практически не проявляться, однако иногда они становятся причиной развития таких серьёзных заболеваний как гемофилия или синдром Дауна. Такие проблемы со здоровьем как болезнь Стилла (форма ревматоидного артрита у детей) или депрессия также могут быть следствием отклонений на генетическом уровне. Мутировавший ген переходит от родителей к детям. Болезни, передающиеся по наследству, обычно имеют генетический характер. Человек может родиться с генетическим фоном, который будет влиять на повышенную восприимчивость или риск проявления определённого заболевания. |
Когда малыш унаследует искажённый ген от обоих родителей, он заболевает.
Если происходит передача поражённого гена от одного из родителей – ребёнок становится носителем дефекта. Это означает, что у него будет присутствовать данный ген в организме, однако сам он не заболеет.
|
Носитель заболевания Это человек, который может передать своим детям наследственную (генетическую) болезнь, хотя сам и не является поражённым недугом. Кроме этого, существует также риск передачи статуса носителя заболевания. Некоторые недуги вызваны именно отклонениями на генном и хромосомном уровнях. Каждый человек унаследует 23 хромосомы от каждого из родителей. Таким образом, у него получается 23 пары хромосом в организме. Если одна или обе хромосомы из пары поражены, возникает генетическое заболевание. В большинстве случаев, у человека с вышеуказанным дефектом обе хромосомы в паре являются повреждёнными. Такое расстройство называется болезнью с аутосомно-рецессивным типом наследования. Определённые генетические заболевания возникают из-за поражения X и Y хромосом, которые отвечают за определение пола. Если хотя бы одна хромосома из пары с отклонением, человек может быть носителем заболевания. |
Мутировавший ген препятствует выработке в организме такого фермента как гексозаминидаза А, который отвечает за расщепление ганглиозидов (сложных природных липидов) в клетках. В случае их накопления происходит блокирование работы мозга и повреждение нервных клеток, что и вызывает болезнь Тея-Сакса.
|
Ферменты Это белок, который вырабатывается для ускорения протекания определённой химической реакции. В нашем организме существует много ферментов для обеспечения различных процессов жизнедеятельности, таких как пищеварение и свёртывание крови. Некоторые генетические заболевания напрямую связаны с выработкой определённых ферментов. Чтобы установить диагноз (на пример, проблемы с почками) врач измеряет количество необходимого фермента в крови пациента. |
При болезни Тея-Сакса с поздним началом в организме производится пониженное количество вышеуказанного фермента. Люди с таким заболеванием унаследуют ген гексозаминидаза А с поздним развитием от обоих родителей или же по одному гену с поздним развитием и неспособностью к активности. Риск появления мутировавшего гена возрастает у людей с корнями, которые уходят к евреям-ашкенази, так как 1 из 30 представителей этой национальности является носителем данного заболевания. Кроме того, к развитию болезни Тея-Сакса также предрасположены люди французско-канадского происхождения, проживающие в восточной части долины реки Святого Лоренса провинции Квебек и каджуны (франкоязычные жители штата Луизиана).
Болезнь Тея-Сакса – симптомы
После рождения ребёнок с таким пороком выглядит абсолютно здоровым. Однако вы можете заметить следующее:
-
В 3-6 месяцев малыш начинает слабо зрительно реагировать на действия вокруг него, и ему сложно сфокусироваться на предмете. Врач может обнаружить красные точки на сетчатке глаза ребёнка.
-
В 6-10 месяцев у ребёнка происходит спад активности. Ему становится сложно сидеть и переворачиваться. Кроме этого, возникают проблемы со слухом и зрением.
-
После 10 месяцев болезнь активно прогрессирует. У ребёнка возможны приступы, потеря зрения и атрофия мышц.
|
Приступ Это внезапные вспышки аномальной электрической активности мозга, которые влияют на функционирование мышечной системы, контроль двигательного аппарата, искажают речь, зрительные образы и психологическое восприятие действительности. Степень поражения у каждого индивидуальная. Это также зависит от типа, частоты и тяжести припадков. В некоторых случаях человек падает на пол и начинает биться в конвульсиях, при которых мышцы тела сильно сокращаются, и возникает непроизвольное подёргивание конечностей. Другие входят в состояние транса, у них остаются подвижными лишь несколько мышц. Иногда они ощущают запахи или видят образы, нехарактерные для здорового человека. Часто припадок является симптомом других проблем со здоровьем, таких как возникновение высокой температуры тела (особенно у детей), нарушение мозгового кровообращения, инфекция, низкий уровень глюкозы в крови (гипогликемия), пониженное давление или опухоль мозга. |
В ходе развития у малыша болезни Тея-Сакса с поздним началом такие симптомы как неуклюжесть и перепады настроения могут не восприниматься серьёзно. К более поздним признакам заболевания относятся судорожное подёргивание и слабость мышц, невнятная речь и проблемы с мыслительными процессами. Характер осложнений зависит от того, какое количество гексозаминидазы А вырабатывается в организме.
Болезнь Тея-Сакса – система распознавания заболевания
Если у лечащего врача есть подозрение, что ребёнок поражён болезнью Тея-Сакса, он прежде всего проведёт обследование пациента и возьмёт анализ крови, чтобы проверить уровень гексозаминидазы А в организме. Генетический тест необходим для подтверждения диагноза.
В чём заключается лечение?
Основным лечением болезни Тея-Сакса является контроль симптомов и обеспечение ребёнку комфорта в повседневной жизни, так как панацеи от этого недуга на сегодняшний день не существует. В данном случае полезным будет пройти генетическое консультирование и посетить сеансы терапии, где можно найти поддержку и пообщаться с людьми, у которых есть аналогичная проблема. Если у вас развивается болезнь Тея-Сакса с поздним началом, лечение также основывается на мониторинге симптомов. В зависимости от симптомов вы можете получать определённые препараты, на пример, от депрессии. Несомненно, известие о том, что у вашего ребёнка обнаружили болезнь Тея-Сакса, может повергнуть вас в шок. В такой ситуации кроме заботы о ребёнке, следует помнить и о своём здоровье. Необходимо разговаривать с лечащим врачом на такие темы:
-
Ваши переживания и помощь в уходе за малышом.
-
Группа поддержки в вашем регионе.
-
Генетическое консультирование для всей семьи, чтобы каждый принял сложившуюся ситуацию.
По мере развития заболевания ребёнку может понадобиться более тщательный уход. Подбадривайте его и внушайте, что нужно бороться, подарите свою любовь и ласку. Возможно, у вас возникнет необходимость в дополнительной помощи по уходу за малышом. Проконсультируйтесь с лечащим врачом о том, куда можно обратиться по этому вопросу.
Болезнь Тея-Сакса — зачем нужно проходить обследование на наличие заболевания?
Носители данного порока могут передать детям заболевание даже если они сами не больны. Если вы и партнёр являетесь носителями болезни Тея-Сакса, риск развития аналогичного дефекта у ваших детей повышается до 1 из 4 случаев (25%). Когда вы планируете заводить ребёнка, Американская корпорация акушёров и гинекологов рекомендует сделать следующее:
Оба партнёра должны пройти скрининг тесты, если они относятся к таким категориям, как евреи-ашкенази, каджуны (франкоязычные жители штата Луизиана), имеют французско-канадское происхождение, либо же они имеют семейный анамнез по данной болезни. В случае если обследование покажет, что вы позитивны, последующие решения поможет принять прохождение генетического консультирования.
Если хотя бы один из партнёров соответствует вышеуказанным категориям, он также должен пройти процедуру тестирования. Один положительный результата на носителя заболевания предполагает также обследование другого партнёра.
|
Семейный анамнез Из семейного анамнеза врач может узнать, есть ли у пациента кровные родственники с аналогичным заболеванием. Это поможет специалисту установить степень риска передачи порока по наследству. К кровным родственникам относятся как живые, так и умершие члены семьи. Это могут быть:
В некоторых случаях семейный анамнез бывает довольно сложным. Это зависит от нескольких факторов:
Генетическое консультирование Генетическое консультирование – это пояснения медицинского работника (консультанта по генетическим вопросам или генетика), специализация которого, помогать людям разобраться в опасности передачи генетических заболеваний, а также объяснять возможность унаследования ребёнком такого вида болезни (серповидно-клеточная болезнь, кистозный фиброз, гемофилия). Генетическое консультирование включает:
|
Болезнь Тея-Сакса | Медицинский портал EUROLAB
- Новости и блоги
- Главная страница
- Новости медицины
- Здравоохранения
- Спецтема
- Новости клиник
- Блоги
- Инфографика
- Гиды по здоровью
- Аллергии
- Анемии
- Артериальная гипертензия
- Бессонница и расстройства сна
- Болезни артерий, вен и лимфатических сосудов
- Болезни глаз
- Болезни желудочно-кишечного тракта
- Болезни зубов
- Болезни легких, бронхов и плевры
- Болезни ног и стоп
- Болезни сердца
- Болезни уха, горла и носа
- Болезни щитовидной железы
- Боль в спине
- Бронхиальная астма
- Витамины и микроэлементы
- ВИЧ / СПИД
- Восстановительная медицина
- Генитальный герпес
- Гепатит А
- Гепатит В
- Гепатит С
- Головная боль и мигрень
- Грипп
- Заболевания, передающиеся половым путем (ЗППП)
- Изжога и гастроэзофагеальная рефлюксная болезнь
- Лейкемии
- Остеоартрит
- Пищевые расстройства
- Простуда
- Приготовление здоровой пищи
- Псориаз
- Рак
- Рак кожи и меланома
- Рак лёгких
- Рассеянный склероз
- Ревматоидный артрит
- Рецепты здорового питания
- Сахарный диабет
- Синдром раздраженного кишечника
- Трансплантация органов и тканей
- Фибромиалгия
- Холестерин
- Экзема
- Физиотерапия
- Обязательное медицинское страхование в России
- Здоровье от А до Я
- Энциклопедия
- Ангиология
- Боли
- Венерология
- Врожденные пороки (тератология человека)
- Гастроэнтерология
- Гематология
- Генетика
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Комунальная гигиена
- Косметология
- Маммология
- Научные статьи
- Неврология
- Онкология
- Паразитарные болезни
- Патологическая анатомия
- Педиатрия
- Первая медицинская помощь
- Психическое здоровье
- Радиология
- Ревматология
- Скорая и неотложная медицинская помощь
- Стоматология
- Токсикология и Наркология
- Урология
- Эндокринология
- Другое о здоровье
- Видеогалерея А-Я
- Первая помощь от А до Я
- Тесты от А до Я
- Заболевания А-Я
- Лечение заболеваний А-Я
- Анатомия человека А-Я
- Врачебные специальности А-Я
- Анализы А-Я
- Медицинские термины А-Я
- Советы астролога
- Сексуальный гороскоп
- Лекарства
- Справочник лекарств
- Комплексные биологические препараты
- Витамины и БАДы
- АТХ (АТС) — Классификация
- Каталог производителей
- Ароматерапия (ароматы и эфирные масла)
- Лекарственные растения и гомеопатия
- Здоровая жизнь
- Женское здоровье
- Беременность и роды
- Бесплодие и репродуктивный статус
- Контрацепция (контроль рождаемости)
- Экстракорпоральное оплодотворение (ЭКО)
- Красная волчанка
- Рак молочной железы (рак груди)
- Рак яичников
- Недержание мочи у женщин
- Гинекология
- Маммология
- Диеты
- Рецепты здоровья и красоты от звезд
- Мужское здоровье
- Выпадение волос (облысение)
- Рак предстательной железы
- Подагра
- Импотенция (эректильная дисфункция)
- Недержание мочи у мужчин
причины заболевания и его проявления
Болезнь Тея Сакса — наследственное заболевание, характеризующееся быстрым развитием и поражением мозга и центральной нервной системы ребенка.
Впервые это заболевание было описано в 19 веке. Название свое болезнь приобрела от двух ученых, внесших свой вклад в исследование этой болезни — англичанина Уоррена Тея и американца Бернарда Сакса. Это заболевание в большей степени типично для определенных этнических групп. В основном им страдают евреи, родившиеся в Восточной Европе и французы, проживающие в Канаде, районах Квебек и Луизиана. Частота заболевания в мире 1 на 250 тысяч человек.
Что провоцирует это заболевание?
Болезнь Тея Сакса развивается только в том случае, если родители вместе являются носителями гена — мутанта, а ребенок унаследует два гена с дефектом. Если же носителем гена является один из родителей, то вероятность заболеть у ребенка практически равна нулю. Но, скорее всего, он также будет носителем этого заболевания.
Что происходит при этом заболевании в организме? Причина развития недуга состоит в том, что из-за наличия у человека измененного гена, его организм перестает вырабатываться определенный фермент — гексозаминидазу, а так же отвечающего за расщепление сложных природных липидов — ганглиозидов в клетках. У здорового организма происходит неизменный процесс синтеза и распада ганглиозидов, а этот процесс очень точный и любой его сбой ведет к необратимым последствиям. В результате генетического сбоя, ганглиозиды не расщепляются, а накапливаются в организме малыша, вызывая при этом поражение мозга и всей нервной системы. К сожалению, выводить их из организма невозможно, и у человека появляются определенные симптомы болезни.
Какова клиническая картина этого недуга?
При рождении малыш, имеющий диагноз синдром Тея Сакса, выглядит абсолютно здоровым. Первые признаки болезни проявляются к шести месяцам. До этого возраста вполне здоровый на вид малыш, развивается, как положено: держит голову, агукает, ползает. Но, прогрессируя и накапливая в организме ганглиозиды, ребенок постепенно начинает лишаться приобретенных навыков. Малыш перестает реагировать на окружение, его взор устремляется в одну точку, развивается апатия. Через время умственное развитие приостанавливается и развивается слепота. Со временем лицо ребенка становится словно кукольное. Итог заболевания — инвалидность и ранняя смертность.
Рассмотрим симптомы по мере взросления ребенка:
- К 6 месяцам ребенок начинает терять контакт с внешним миром, перестает узнавать родных, реагирует только на очень громкие звуки, не может сфокусироваться на висящей игрушке, глаза вздрагивают и зрение ухудшается.
- К 10 месяцам малыш становится менее активным, нарушаются двигательные функции: малышу трудно сидеть, переворачиваться и ползать. Притупляется слух и зрение, малыш становится апатичным.
- После 1 года жизни болезнь прогрессирует очень быстро. По ребенку заметна его умственная отсталость, он быстро теряет зрение, слух, ухудшается мышечная активность, появляются трудности в дыхании, начинаются приступы.
Это важно! Дети, у которых симптомы болезни Тея Сакса появились в младенчестве, доживают до пятилетнего возраста.
Позднее появление симптомов
Иногда симптомы заболевания проявляются не сразу. Есть еще две формы этого заболевания.
Ювенильный дефицит гексазаминидазы типа А
Эта форма тяжелого недуга появляется у детей от 2 до 5 лет. Развитие болезни замедленное, в отличие от клинической формы. В этом случае симптомы можно заметить не сразу. Перепады настроения и какая-то неуклюжесть в движения не сильно привлекают к себе внимание. К тому же, в этом возрасте капризы — нормальное явление.
Но более поздние симптомы привлекают внимание:
- у ребенка развивается слабость мышц;
- судорожные подергивания;
- нарушение мыслительных способностей и нечленораздельная речь.
К сожалению, все эти признаки ведут к инвалидности. Смерть наступает примерно к 15 — 16 годам.
Хроническая форма недостатка гексазаминидазы
Как правило, возникает у людей в возрасте 30 лет. Течение болезни слабое. Развивается постепенно, протекает сравнительно легко: могут быть заметны перепады настроения, неуклюжесть, появляется невнятная речь, психические отклонения, снижается интеллект, возникает мышечная слабость, приступы. По причине того, что именно эта форма болезни была открыта недавно, прогноз на будущее делать невозможно. Ясно только одно, что эта форма болезни неизбежно приведет к инвалидности. Продолжительность жизни зависит от очень многих факторов.
Как проходит диагностика болезни?
Сегодня медицина далеко ушла вперед, и диагностировать болезнь Тея Сакса можно как у младенца, так и еще до его рождения.
Если есть подозрение на развитие у малыша синдрома Тея Сакса, в первую очередь, нужно обратиться к окулисту.
Первый признак заболевания можно диагностировать по глазному дну. Если ребенок болен, то можно увидеть вишнево-красное пятно. Это характерное для заболевания — скопление ганглиозидов в клетках сетчатки глаза.
Дальнейшее обследование включает в себя:
- обширный анализ крови — скрининг — тест;
- а так же микроскопический анализ нейронов.
Скрининг-тест — показывает, вырабатывается в организме ребенка белок — гексазаминидаза А или нет. Этот тест проводят при любой форме заболевания.
Микроскопический анализ нейронов — это выявления ганглиозидов в нейронах. При большом их количестве нейроны становятся вытянутыми.
Если родители относятся к одной из этнических групп или в роду были генетические заболевания, лучше всего пройти скрининг — тест на 10−12 неделе беременности. Тест покажет, унаследовал ли плод мутированные гены от обоих родителей или нет. Анализ берется с помощью образца крови из плаценты.
В чем заключается лечение болезни Тея Сакса?
К несчастью, медицина пока не нашла способов лечения этой болезни. Оно сводится только к поддерживающей терапии и тщательному уходу. Большинство противосудорожных препаратов не дают положительного эффекта. Так как у малышей отсутствует глотательный рефлекс, требуется кормление через зонд. Также в терапию входит лечение текущих заболеваний из-за ослабленного иммунитета ребенка. Обычно причиной смерти является вирусная инфекция.
Профилактика заболевания сводится к обследованию семейной пары на отсутствие мутации в генах на болезнь Тея Сакса. При обнаружении у обоих супругов дефектного гена, следует рекомендация не иметь детей.
Оцените статью:
Loading …
Записи по теме:
Информация о болезни Тея Сакса, изображения, симптомы и лечение
Вы знаете ребенка, у которого проявляются признаки затворничества? Он меньше смотрит вам в глаза? У ребенка может быть болезнь Тея-Сакса, поскольку это ее самые верные признаки. Читайте информацию о болезни Тея Сакса, определение болезни Тея Сакса, ее причины, симптомы, диагностику и лечение.
Что такое болезнь Тея Сакса?
Рисунок 1 — Нейрон, пораженный Тай-Саксом
Источник — allhealthsite
Болезнь Тея Сакса (TSD) — редкое заболевание, поражающее нервы головного мозга.Состояние также известно как дефицит гексозаминидазы А или ганглиозидоз GM2.
Заболевание проявляется в трех формах. Это:
Классическая детская болезнь Тея Сакса
Это наиболее распространенный тип болезни Тея Сакса. Симптомы этой формы TSD обычно наблюдаются у детей старше 6 месяцев. В худших случаях ребенок, страдающий этим заболеванием, может ослепнуть.
Ювенильная болезнь Тея-Сакса
Это состояние вызвано дефицитом фермента гексозаминидазы-A (Hex-A).Симптомы этой формы болезни Тея Сакса проявляются у детей в возрасте от 2 до 5 лет.
Хроническая болезнь Тея Сакса
Это также известно как болезнь Тея Сакса с поздним началом. Это редкая форма TSD, вызванная полным отсутствием фермента гексозаминидазы-A (Hex-A). Развивается у детей чуть старше возраста.
История болезни Тея Сакса
Болезнь Тея Сакса названа в честь Уоррена Тея, британского офтальмолога, который первым сообщил о симптомах этого состояния в 1881 году.Он сообщил о пациенте с вишнево-красным пятном в глазу, которое является одним из основных признаков этого заболевания. Частично он обязан своим названием неврологу из Нью-Йорка Бернарду Саксу, который сообщил о заболеваемости младенцами еврейского происхождения из Восточной Европы.
Заболеваемость болезнью Тея Сакса
Заболевание возникает у людей любого этнического происхождения. Однако это чаще встречается у людей асхеназского еврейского и восточноевропейского происхождения. К другим группам повышенного риска относятся французские канадцы, пенсильванские голландцы и каджунцы Луизианы.
Симптомы болезни Тея Сакса
Признаки болезни обычно не проявляются на начальных стадиях. Ребенок, рожденный с этим заболеванием, выглядит совершенно нормальным. Эффекты болезни Тея Сакса проявляются только через некоторое время.
При классической детской форме заболевания симптомы болезни Тея Сакса начинают проявляться в возрасте 3-6 месяцев. Ребенок меньше двигается. Улыбка, зрительный контакт и действия уменьшаются. Социальные навыки задерживаются.Также страдает способность ребенка воспринимать свое окружение и реагировать на него. Ребенку не хватает изюминки, он становится раздражительным. Голова может вырасти больше, чем остальное тело. Нервные клетки расширяются за счет жирового материала.
На поздних стадиях ребенок может испытывать трудности с приемом пищи. Нарушение моторики. Мышцы развиты недостаточно хорошо, также страдает интеллект.
На заключительном этапе ребенок может ослепнуть или потерять слух. Несмотря на хорошее лечение, большинство детей с болезнью Тея Сакса (TSD) рано или поздно умирают.
Болезнь Тея Сакса Ожидаемая продолжительность жизни
Больные этой болезнью живут недолго. Многие умирают, не дожив до 4-6 лет, а некоторые доживают до 12 или 13 лет.
Причины болезни Тея Сакса
Болезнь Тея Сакса обычно вызывается дефицитом или отсутствием фермента, известного как фермент гексозаминидаза-А (Hex-A). Отсутствие или низкий уровень этого фермента вызывает накопление липидного или жирного вещества, называемого ганглиозидом GM2. Этот липид откладывается в нервных клетках.У пациентов с болезнью Тея-Сакса основным причинным фактором является отсутствие фермента.
Процесс накопления липидов известен как «субстрат». Это вызывает прогрессирующее повреждение нервных клеток. Этот жирный материал вызывает у больного психическое и физическое ухудшение.
Рисунок 2 — Болезнь Тея Сакса
Источник — wikispaces
Для болезни Тея Сакса генетика играет важную роль. Болезнь Тея Сакса возникает в результате генетической мутации.Ребенок может заболеть этим заболеванием, если у обоих родителей есть мутировавший ген. Как правило, 1 человек из 250 является носителем этого заболевания. Но в некоторых общинах, таких как евреи-ашкенази, каждый 27 человек несет эту хромосомную мутацию. Дети, рожденные вне брака в такой общине, подвергаются высокому риску развития этого состояния.
Диагностика болезни Тея Сакса
Самая распространенная диагностика болезни Тея Сакса включает анализ крови. Анализ крови позволяет сразу определить это заболевание.
Для правильной диагностики болезни Тея Сакса глаза часто проверяют.Врачи наблюдают за сетчаткой пациентов с помощью офтальмоскопа. У всех пациентов с болезнью Тея Сакса появляются вишнево-красные пятна в области сетчатки. Обнаружение красного пятна на сетчатке — один из надежных способов диагностики этого состояния.
Микроскопический анализ нейронов также часто проводится, чтобы увидеть, есть ли избыточное накопление ганглиозида.
Лечение болезни Тея-Сакса
Лечение болезни Тея-Сакса невозможно. На данный момент от этого состояния здоровья нет лекарства.Лекарства используются только для того, чтобы контролировать симптомы. На начальных стадиях приступы контролируются с помощью противосудорожных препаратов.
Правильное питание необходимо для поддержания хорошего здоровья больного. Гидратация и аналогичные методы также необходимы, чтобы дыхательные пути оставались открытыми.
На поздних стадиях детям может потребоваться зонд для кормления. Это поможет детям, страдающим от проблем с питанием.
Терапия также необходима для контроля симптомов этого состояния.Генная терапия, терапия стволовыми клетками и фармакологическая терапия шаперонами — это некоторые широко известные методы лечения, используемые для лечения этого заболевания.
Профилактика болезни Тея Сакса
Профилактика этой болезни становится возможной благодаря скринингу на болезнь Тея Сакса. Тестирование носителя и пренатальное тестирование используются для проверки наличия любой дефектной копии гена. Эти шаги используются в парах, которые принадлежат к группам повышенного риска, таким как евреи-ашкенази. Установлено, что евреи-ашкенази подвержены высокому риску этого состояния.
Фотографии болезни Тея Сакса
Посмотрите эти фотографии болезни Тея Сакса, чтобы узнать о ней больше.
Изображение 3 — Tay Sachs Image
Source — allhealthsite
Picture 4 — Tay Sachs Disease Photo
Source — genetics.com.au
Tay Sachs Disease Tay Sachs Disease пациенты обычно умирают к 4 годам из-за повторных случаев инфекции. Несмотря на лучшую заботу, шансы на выживание очень низки.В некоторых случаях пациенты доживают до 12 или 13 лет. Вы не можете найти болезнь Тея Сакса у взрослых. Люди с болезнью Тея Сакса не доживают до совершеннолетия.
Если вы являетесь членом сообщества, подверженного этому заболеванию, например евреев ашкенази, рекомендуется обратиться за медицинской помощью, прежде чем создавать семью. Профилактика, как говорится, лучше лечения. И это наиболее применимо, чем неизлечимая болезнь, такая как болезнь Тея Сакса.
Ссылки :
http: // en.wikipedia.org/wiki/Tay%E2%80%93Sachs_disease
http://www.ntsad.org/S02/S02tay_sachs.htm
http://kidshealth.org/parent/medical/genetic/tay_sachs.html
http://www.ninds.nih.gov/disorders/taysachs/taysachs.htm
http://www.wisegeek.com/what-is-tay-sachs-disease.htm
Диагностика, тесты, управление и лечение
Что такое болезнь Тея-Сакса?
Болезнь Тея-Сакса (наиболее тяжелая форма дефицита гексозаминидазы A) — это прогрессирующее фатальное генетическое заболевание, поражающее нервные клетки головного мозга.У людей с синдромом Тай-Сакса не хватает специфического белка (фермента), называемого гексозаминидаза А. Этот дефицит фермента вызывает накопление в мозге жирного вещества, ганглиозида GM2. Именно это скопление вызывает симптомы Тай-Сакса.
Что вызывает болезнь Тея-Сакса?
У всех людей есть две копии гена Тай-Сакса (HEXA). Тай-Сакс возникает, когда ни один из генов HEXA человека не работает должным образом из-за мутации (вредного изменения). Если у человека есть одна рабочая копия гена HEXA, а другая копия имеет мутацию, его / ее называют «носителем».«У носителей нет симптомов, но когда у двух носителей есть дети вместе, у каждого ребенка (мальчика или девочки) будет 25% (1/4) шанс развития Тея-Сакса. Если только один родитель передаст дефектный ген ребенку (Вероятность 50% при каждой беременности), у ребенка не разовьется болезнь, но он будет носителем и может передать болезнь своим детям.
Хотя любой может быть носителем болезни Тай-Сакса, эта болезнь гораздо чаще встречается среди людей ашкеназского (восточноевропейского) еврейского происхождения.Каждый 30 человек еврейского происхождения ашкенази является носителем. Другие группы населения с таким же (или более) количеством носителей включают французских канадцев, каджунов (из Луизианы) и амишей старого порядка в Пенсильвании. Некоторые исследования утверждают, что у людей ирландского происхождения шанс стать носителем составляет 1 из 50. Примерно 1 из каждых 300 человек, не принадлежащих к этой категории, является носителем Tay-Sachs.
Каковы симптомы болезни Тея-Сакса?
Симптомы Тай-Сакса обычно развиваются в возрасте 3-6 месяцев, когда у ребенка появляется мышечная слабость, низкий мышечный тонус, повышенная реакция вздрагивания и внезапные сокращения крупных мышц при засыпании (миоклонические подергивания).
В возрасте от 6 до 10 месяцев ребенок не будет достигать двигательных навыков и может потерять способность выполнять задания (например, сидеть), которым он / она ранее научился. Снижение движений глаз и контакта, а также внимательность также наблюдаются вместе со специфическим изменением в глазах, называемым вишнево-красным пятном, которое можно увидеть во время осмотра глаз.
В возрасте от 8 до 10 месяцев ребенок будет меньше двигаться и станет менее отзывчивым. Будет потеряно зрение, и к году у многих начнутся судороги.Размер головы человека начинает увеличиваться примерно в 18 месяцев, а когда ребенку исполняется 2 года, у него обычно возникают проблемы с глотанием, и он переходит в вегетативное состояние без реакции. Возраст смерти обычно составляет от 2 до 4 лет, часто от пневмонии.
В дополнение к классическому Tay-Sachs, существуют другие формы дефицита гексозаминидазы A, которые иногда называют формами Tay-Sachs:
- Дефицит ювенильной гексозаминидазы начинается с проблем с ходьбой (атаксии) и нарушения координации движений в раннем детстве.Симптомы аналогичны симптомам классического Тея-Сакса, хотя вишнево-красное пятно встречается не так часто. Возраст смерти обычно наступает в подростковом возрасте, обычно от инфекций, хотя некоторые умирают гораздо раньше.
- Хронический дефицит гексозаминидазы A обычно развивается в возрасте до 10 лет, но люди не теряют столько же моторных навыков, как те, у кого есть Tay-Sachs. Позже в ходе курса будут затронуты когнитивные и вербальные навыки.
- Дефицит гексозаминидазы А у взрослых вызывает медленную, но прогрессирующую мышечную слабость и истощение, а также нарушение четкости речи, когнитивные проблемы и слабоумие.До 40% людей имеют психические проблемы (которые могут присутствовать без деменции). Серьезность, даже в семье, очень разная. Ожидаемая продолжительность жизни может сильно различаться и не может быть сокращена.
Далее: Диагностика и тесты
Последний раз проверял медицинский работник Cleveland Clinic 10.09.2014.
Получите полезную, полезную и актуальную информацию о здоровье и благополучии
е Новости
Клиника Кливленда — некоммерческий академический медицинский центр.Реклама на нашем сайте помогает поддерживать нашу миссию. Мы не поддерживаем продукты или услуги, не принадлежащие Cleveland Clinic.
Политика
Два любовника (2008) — Часто задаваемые вопросы
После разрыва со своей невестой после того, как у них обоих был обнаружен ген, вызывающий болезнь Тея Сакса, Леонард Крадитор (Хоакин Феникс) пытается покончить жизнь самоубийством, утопившись, но в самый последний момент его спасают.Все еще подавленный, он возвращается к своим родителям, которые знакомят его с Сандрой Коэн (Винесса Шоу), дочерью близкого друга семьи. Сандра и Леонард поладили, пока Леонард не замечает Мишель Рауш (Гвинет Пэлтроу), восхитительную и волнующую женщину, которая недавно переехала в квартиру в доме его семьи на деньги, которые она получает от своего женатого любовника Рональда Блатта (Элиас Котеас). Леонард становится одержимым Мишель, но ее личные проблемы в конечном итоге снова приводят его к депрессии и суицидальным мыслям, в конце концов заставляя его сделать выбор между непостоянной Мишель или стабильной и заботливой Сандрой.редактировать
«Два любовника» основаны на сценарии сценариста Ричарда Менелло и сценариста-режиссера Джеймса Грея, которые в некоторой степени основывают свой сценарий на рассказе русского писателя Федора Достоевского [1821–1881] «Белые ночи», первоначально опубликованном в 1848 году.
редактировать
Тай Сакс — смертельное дегенеративное заболевание, вызванное генетической мутацией, которая предотвращает образование в крови определенного химического вещества, необходимого для расщепления жирных кислот (ганглиозидов) в головном мозге.Эти жирные кислоты накапливаются в нервных клетках, которые вскоре закупориваются, в результате чего вся нервная система перестает работать. Младенцы, страдающие болезнью Тея Сакса, сначала становятся здоровыми, пока им не исполнится около шести месяцев, а затем постепенно становятся слепыми, глухими, парализованными и неспособными глотать. Смерть обычно наступает до достижения ребенком трех-четырех лет. Генетическая мутация, вызывающая Тэя Сакса, особенно распространена у французских канадцев, каджунцев Луизианы и евреев ашкенази (Леонард — еврей).Из-за разрушительного воздействия на младенцев, рожденных с этой болезнью, двум взрослым, обладающим мутациями, обычно рекомендуется не вступать в брак или, по крайней мере, не заводить детей вместе.
редактировать
Да. White Nights больше не защищена авторским правом и доступна для онлайн-чтения здесь. Просто нажмите на текст, чтобы перелистать страницы.редактировать
Вопросы и ответы, приведенные ниже, могут выдать важные моменты сюжета.
Home — Болезнь Тея-Сакса
Краткое содержание темы
Эта вики посвящена болезни Тея-Сакса (TSD). Он объясняет диагностику, лечение, профилактику и текущие исследования, возможные для этого заболевания.
Цели обучения
- Чтобы получить общее представление о болезни Тея-Сакса.
- Чтобы понять важность диагностики, прогноза, лечения и профилактики.
- Чтобы понять текущие исследования различных типов возможного терапевтического лечения этого заболевания.
Описание вики
Эта вики посвящена проекту нашего класса в Genetic. Прочитав этот сайт, вы получите представление о болезни Тея-Сакса, исследованиях и методах лечения.
Что такое болезнь Тея-Сакса (TSD)?
По данным Национального института неврологических заболеваний и инсульта (NINDS), «болезнь Тея-Сакса (TSD) — это фатальное генетическое нарушение накопления липидов, при котором вредные количества жирного вещества, называемого ганглиозидом GM2, накапливаются в тканях и нервных клетках мозг.» NINDS заявили, что это аутосомно-рецессивное генетическое заболевание, очень редкое, поскольку поражает маленьких детей и младенцев. В первые несколько месяцев жизни младенцы выглядят нормально, но симптомы появляются, когда нервные клетки набухают из-за ганглиозида GM2.Резко ухудшаются умственные и физические способности. Признаки включают глухоту, слепоту и трудности с глотанием пищи.
Болезнь Тея-Сакса вызывается генетической мутацией в геноме HEX A на хромосоме 15, как показали исследователи в конце 20 века на веб-сайте «News medical».
http://www.ninds.nih.gov/disorders/taysachs/taysachs.htm
http://www.news-medical.net/health/What-is-Tay-Sachs-Disease.aspx
История болезни Тея Сакса
Др.Уоррен Тэй (1843-1927) был первым офтальмологом, который сообщил о симптомах болезни Тея-Сакса, и он сообщил, что вишнево-красное пятно в глазу является признаком болезни Тея-Сакса.
Д-р Бернард Сакс (1855-1944), невролог из Нью-Йорка, сообщил, что в восточноевропейском еврействе наблюдаются в основном младенцы.
http://www.ntsad.org/index.php/tay-sachs/history
Вишнево-красное пятно на глазу — признак болезни Тея-Сакса
Самая тяжелая форма болезни Тай-Сакса поражает младенцев, когда им всего несколько месяцев.Вначале они начинают терять зрение и ненормально реагировать, их развитие задерживается.
Изображение 1. http://upload.wikimedia.org/wikipedia/commons/thumb/0/0f/Tay-sachsUMich.jpg/230px-Tay-sachsUMich.jpg
Изображение 2: http://www.cleveland.com/taysachs/images/nerve1105.gif
http://www.cleveland.com/taysachs/wide/index.ssf?nerve1105.html
Изображение 3:
На этом изображении лизосомы Tay Sachs не могут переваривать ганглиодид GM2
Изображение 4: http: // www.utm.utoronto.ca/~w3bio315/picts/lectures/lecture15/LysosomeTaySachs1.jpg
Видео о болезни Тай-Сакса
Болезнь проявляется в трех формах
1.) Классическая инфантильная болезнь Тея Сакса
Эта форма часто встречается у детей старше 6 месяцев. Ребенок с тяжелым заболеванием ослеп.
2.) Ювенильная болезнь Тея
Симптом проявляется у детей в возрасте от 2 до 5 лет из-за недостатка фермента гексозаминидазы-А (Hex-A).
3.) Хроническая болезнь Тея / болезнь Тея с поздним началом
Это необычная форма TSD. Дети с этой формой обычно старше, и у них полностью отсутствует фермент гексозаминидаза А (HEX-A).
http://en.wikipedia.org/wiki/Tay%E2%28%93Sachs_disease
http://www.news-medical.net/health/Tay-Sachs-Disease-Symptoms.aspx
Диагностика и обнаружение
Скрининг до зачатия
Потенциальные родители проверяют наличие мутации.
Антенатальный скрининг
Плод проверяется на наличие двух унаследованных копий мутации HEX A, которая вызывает болезнь Тея-Сакса.
Тестирование носителя
Потенциальные пары учитывают риск и возможность заболевания.
Пренатальное тестирование
Чтобы определить, унаследовал ли плод мутация от обоих родителей.
Этот тест используется, когда оба родителя не уверены, являются ли они носителями болезни. На сайте «Новости медицины» говорится, что «пренатальное тестирование может быть выполнено путем анализа активности фермента HEX A в клетках плода путем хронического отбора проб или аминоцентеза.»(Новости медицины)
http://www.news-medical.net/health/Tay-Sachs-Disease-Diagnosis.aspx
Диагностика болезни Тея Сакса
Анализ крови обычно используется для проверки на болезнь, поскольку он быстро определяет результаты.
Врачи используют офтальмоскоп для диагностики и выявления красных глазных пятен у пациентов. Микроскопический анализ нейрона также используется, чтобы увидеть, есть ли дополнительное накопление ганглиозида.
Лечение болезни Тея-Сакса
Противосудорожные препараты используются для борьбы с приступом.
Страдающим детям необходимо соответствующее питание.
Прогноз болезни Тея Сакса
Пациенты с болезнью Тея Сакса обычно умирают в возрасте 4 лет из-за инфекций.
Шансы на более длительное выживание сомнительны.
Немногие пациенты доживут до возраста 12 или 13 лет, но большинство из них не доживут до взрослого возраста.
Профилактика болезни Тея Сакса
Профилактика TSD может быть осуществлена только путем тестирования на носительство и пренатального тестирования, чтобы проверить существование любых копий мутировавшего гена.Кроме того, эти тесты используются для пар, относящихся к группе повышенного риска, например евреев-ашкенази.
Выше прогноз, лечение и профилактика из
http://en.wikipedia.org/wiki/Tay%E2%80%93Sachs_disease
Болезнь Тея-Сакса вызывается генетическим дефектом одного гена, одна копия которого унаследована от обоих родителей. Согласно веб-сайту News Medical, это аутосомно-рецессивное заболевание.
Эта мутация в гене HEXA на хромосоме 15 вызывает болезнь Тея-Сакса.
Ген HEXA играет важную роль в головном и спинном мозге и обеспечивает инструкции по созданию части фермента бета-гексозаминидазы A. Этот фермент находится в лизосомах. В клетках лизосомы расщепляют токсичные вещества и действуют как центры переработки. В лизосомах бета-гексозаминидаза А помогает расщеплять жирное вещество, называемое ганглиозидом GM2.
В результате накопление этого вещества достигает токсического уровня, особенно в мозге.
Поскольку болезнь Тея-Сакса нарушает функцию лизосомального фермента и включает накопление ганглиозида GM2, это состояние иногда называют лизосомным расстройством накопления или GM2-ганглиозидозом.
http://en.wikipedia.org/wiki/Tay%E2%80%93Sachs_disease
http://www.ninds.nih.gov/disorders/taysachs/taysachs.htm
Изображение 5:
Изображение: http://www.sfn.org/siteobjects/published/0000BDF20016F63800FD712C3158BA55/0000BDF2000006250110C68C45857663/file/bb_feb2007_large.jpg
http://sites.google.com/site/geneticdisorders6/tay-saches
«В здоровом нейроне, вверху, лизосомы действуют как центр переработки отходов клетки. При болезни Тея-Сакса генетические недостатки затрудняют работу лизосомных ферментов, которые расщепляют продукты жировых клеток, также известные как ганглиозиды, которые накапливают и разрушают клетку. .»(Morgan.M)
Изображение 6
Хромосома 15 с Tays Sachs
http://www.google.com/imgres?q=tay+sachs&um=1&hl=en&sa=N&biw=1280&bih=822&tbm=isch&tbnid=9PIpIvsSoabric8eEBM:&imgrefurl=http://www.hinsstase/dale/ru/hinsdalekg/hinsdalekg/v/ /Tay%2520Sachs%2520Disease-Hexodaminidase%2520A/Conclusion.htm&docid=tkdDbos2O29EcM&imgurl=http://www.hinsdale86.org/staff/kgabric/Disease11/Tay%252520Sachs%imageHexception/Disease11/Tay%252520Sachs%image-250020Disease /gif & w = 313 & h = 317 & ei = ApapT4ztM-Hv0gGGhKS9BQ & zoom = 1 & iact = hc & vpx = 281 & vpy = 470 & dur = 63 & hovh = 226 & hovw = 223 & tx = 91 & tynds = 117 & sig = 10034800btn & amp; tx = 91 & tynds = 117 & sig = 10034800btn & page: 0, я: 170
КАК РЕБЕНОК РОДИЛСЯ С ЗАБОЛЕВАНИЕМ СЕКСА?
TSD — это наследственное заболевание, поэтому, когда оба родителя несут ген Тея-Сакса, каждый родитель передает дефектный ген своему ребенку. Ребенок, унаследовавший два гена Тея-Сакса (по одному от каждого родителя), не производит функционального фермента Hex-A и заболеет болезнью Тея-Сакса.
National Tay-Sachs & Allied Disease, ассоциация Delaware Valley заявила, что, когда оба родителя являются носителями Tay-Sachs, вероятность того, что их ребенок заболеет, составляет 25%, а вероятность рождения ребенка — 50%. перевозчик. С другой стороны, когда только один родитель является носителем, у ребенка нет шансов заболеть. Кроме того, ассоциация Delware Valley заявила, что вероятность иметь ребенка, который будет носителем Tay-Sachs, составляет 50%.
Самая тяжелая форма болезни Тея-Сакса начинает поражать младенцев, когда им всего несколько месяцев. Изначально их развитие затягивается. Они начинают терять зрение и ненормально реагировать. Постепенно они парализованы. Симптомами болезни являются потеря слуха, судороги, неспособность глотать пищу и затрудненное дыхание.
http://www.tay-sachs.org/taysachs_disease.php
Аутосомно-рецессивное генетическое заболевание.
Изображение 7: http: // media.Commercialappeal.com/mca/content/img/photos/2007/12/15/16eggs.jpeg]]
РЕБЕНОК, РОДИЛСЯ С БОЛЕЗНЬЮ СЕЧКА
Рис. 8
http://media.commercialappeal.com/mca/content/img/photos/2007/12/15/16eggs.jpeg
Исследования и терапия
Поскольку до сих пор не существует эффективного лечения этого заболевания, исследователи пробуют несколько потенциальных методов лечения.