Синдром Эллерса-Данло тип VI, PLOD ч.м.
Метод определения
Секвенирование.
Выдаётся заключение врача-генетика!
Исследуемый материал
Цельная кровь (с ЭДТА)
Доступен выезд на дом
Исследование частых мутаций в гене PLOD.
Синдром Элерса — Данло тип VI (EDS, СЭД, OMIM225400) — гетерогенная группа наследственных заболеваний соединительной ткани, в основе которых лежит недостаточное развитие коллагеновых структур в различных системах организма. Назван в честь двух дерматологов, Эварда Элерса и Генри Данлоса. Но ещё раньше синдром был описан Николаем Черногубовым. Проявляется патологией кожи, опорно-двигательного аппарата, сердечно-сосудистой системы, глаз. Относится к моногенным заболеваниям с различными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена, одного из основных белков соединительной ткани. Конечный результат каждого из механизмов формирования патологии один и тот же — уменьшение стабильности коллагенового волокна. Генерализованность клинических проявлений при синдроме Элерса — Данло обусловлена тем, что элементы поражённой соединительной ткани присутствуют практически во всех тканях и системах организма. Диагноз устанавливают на основании анамнестических сведений (задержка моторного развития, плохая заживляемость ран, склонность к кровотечениям и экхимозам, вывихи и подвывихи), характерной клинической картины (гиперэластичносгь и хрупкость кожи, гиперподвижность суставов в сочетании с патологией сердца, глаз и др.).
Согласно классификации наследственных болезней соединительной ткани, выделяют 9 форм синдрома Элерса — Данло, различающихся особенностями клинической картины и типом наследования. Характерными признаками синдрома Элерса — Данло типа VI являются аутосмно-рецессивный тип наследования, мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии. У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
Характерными признаками синдрома Элерса — Данло типа VI являются аутосмно-рецессивный тип наследования, мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии. У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
Тип наследования.
Аутосомно- рецессивный.
Гены, ответственные за развитие заболевания.
Ген PLOD1 расположен на хромосоме 1 в регионе 1p36.22. Содержит 19 экзонов.
Патогенез и клиническая картина.
У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена — одного из основных белков соединительной ткани, приводящих к уменьшение стабильности коллагенового волокна. Элементы пораженной соединительной ткани присутствуют практически во всех тканях и системах организма, что приводит к генерализованному поражении организма при заболевании. Характерными признаками синдрома Элерса-Данло типа VI являются: мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии.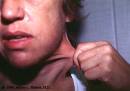
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Giunta, C., Randolph, A., Al-Gazali, L. I., Brunner, H. G., Kraenzlin, M. E., Steinmann, B. Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA). Am. J. Med. Genet. 133A: 158-164, 2005.
- OMIM.
Сосудистый тип синдрома Элерса–Данло – редкое моногенное заболевание соединительной ткани | Семячкина
1. Malfait F., Francomano C., Byers P., Belmont J., Berglund B., Black J. et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet 2017; 175(1): 8–26. DOI: 10.1002/ajmg.c.31552
2. Germain D.P. Ehlers–Danlos syndrome type IV. Orphanet J Rare Dis 2007; 2:32. DOI: 10.1186/1750-1172-2-32
3. Eagleton M.J. Arterial complications of vascular Ehlers–Danlos syndrome. J Vasc Surg 2016; 64(6): 1869–1880. DOI: 10.1016/j.jvs.2016.06.120
4. Papagiannis J. Sudden death due to aortic pathology. Cardiol Young 2017; 27(S1): S36–S42. DOI: 10.1017/S1047951116002213
5. Legrand A., Devriese M., Dupuis-Girod S., Simian C., Venisse A., Mazzella J.M. et al. Frequency of de novo variants and parental mosaicism in vascular Ehlers–Danlos syndrome. Genet Med 2019; 21(7): 1568–1575. DOI: 10.1038/s41436-018-0356-2
Genet Med 2019; 21(7): 1568–1575. DOI: 10.1038/s41436-018-0356-2
6. Shields L.B.E., Rolf C.M., Davis G.J., Hunsaker J.C. Sudden and unexpected death in three cases of Ehlers–Danlos syndrome type IV. Case Reports. J Forensic Sci 2010; 55(6): 1641–5. DOI: 10.1111/j.1556-4029.2010.01521.x
7. Park M.A., Shin S.Y., Kim Y.J., Park M.J., Lee S.H. Vascular Ehlers–Danlos syndrome with cryptorchidism, recurrent pneumothorax, and pulmonary capillary hemangiomatosis-like foci: A case report. Medicine (Baltimore) 2017; 96(47): e8853. DOI: 10.1097/MD.0000000000008853
8. Park K.Y., Gill K.G., Kohler J.E. Intestinal Perforation in Children as an Important Differential Diagnosis of Vascular Ehlers–Danlos Syndrome. Am J Case Rep 2019; 20: 1057–1062. DOI: 10.12659/AJCR.917245
9. D’hondt S., Van Damme T., Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers–Danlos syndrome: a systematic review. Genet Med 2018; 20(6): 562–573. DOI: 10.1038/gim.2017.138
Диагностика и хирургическое лечение больных с cосудистым типом синдрома Элерса—Данло
Чрезвычайные ситуации в сосудистой хирургии, связанные со спонтанными разрывами (СР) периферических артерий крупного и среднего диаметра, опасны для жизни и требуют своевременного неотложного лечения [1, 2]. Исследование Global Burden Disease [3, 4] продемонстрировало, что общее число летальных исходов по причине аневризм и расслоения аорты выросло с 2,49 на 100 000 до 2,78 на 100 000 случаев за период с 1990 по 2010 г.
Пациенты с наследственными дисплазиями соединительной ткани (НДСТ) в течение жизни могут неоднократно нуждаться в разных типах хирургических вмешательств, в первую очередь в реконструктивных вмешательствах на сосудах и сердце.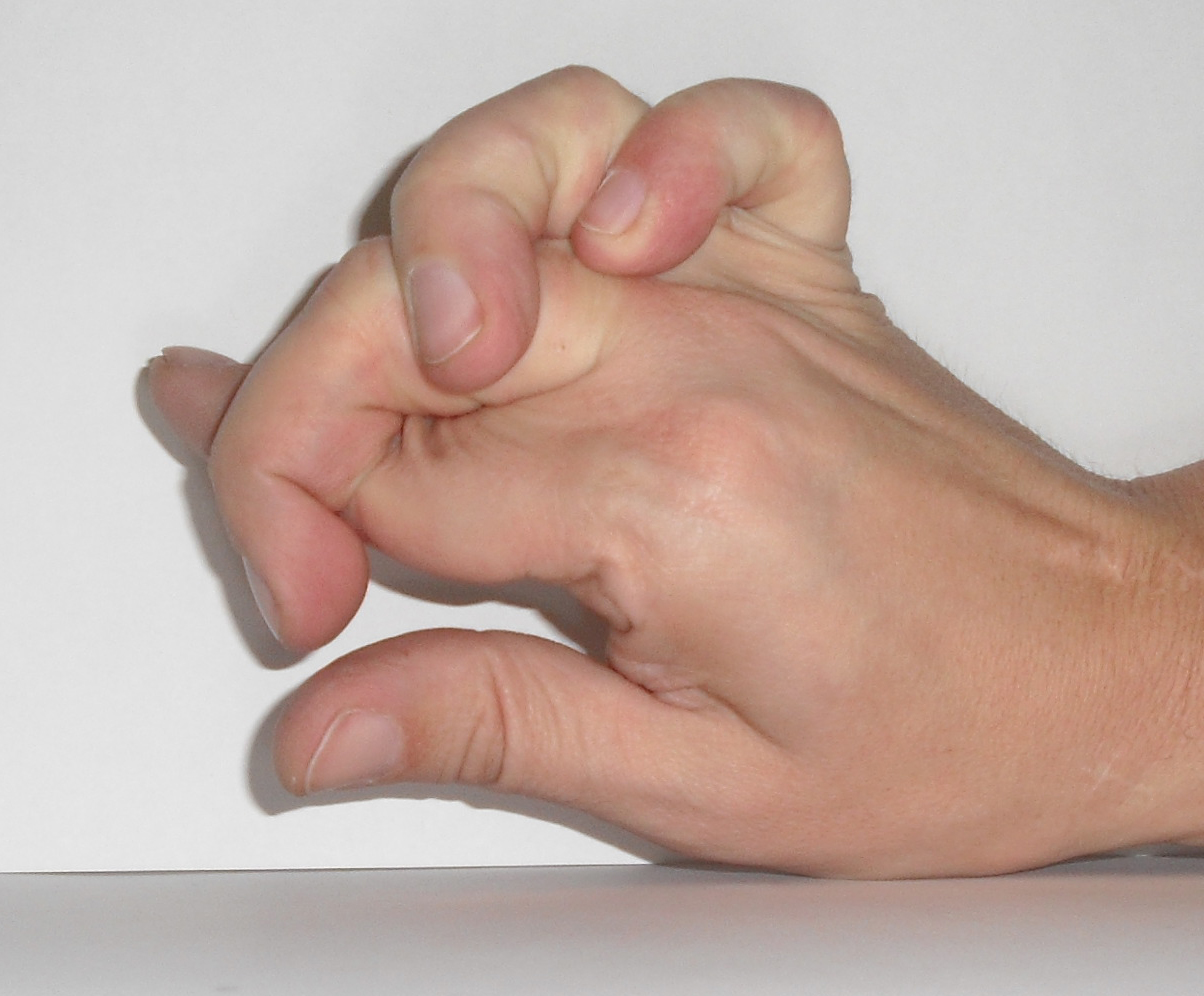 НДСТ — клинически и генетически гетерогенная группа заболеваний, связанная с наследственными нарушениями синтеза и функционирования коллагеновых и эластических белков. Клиническая дифференциальная диагностика НДСТ чрезвычайно трудна вследствие сходства симптоматики. И если «марфаноидный фенотип» хорошо известен кардиологам и сосудистым хирургам, то внешний вид пациентов с сосудистым типом синдрома Элерса—Данло (СЭД IV) редко дифференцируется в клинике. Клиническая диагностика синдрома СЭД основана на Вильфраншских критериях 1998 г. [9]. Сбор семейного анамнеза и проведение ДНК-диагностики является необходимым этапом в подтверждении диагноза сосудистого типа СЭД [27]. Лабораторное тестирование рекомендуется лицам, имеющим два и более диагностических симптома. Верификация генетической причины заболевания у пробанда позволяет проводить подтверждающую, раннюю и пресимптоматическую диагностику заболевания (в том числе пренатальную по запросу семьи) у всех родственников, доступных для обследования [16]. Заболевание является полиорганным и жизнеугрожающим, вследствие риска разрыва сосудов среднего и крупного калибра, а также полых органов (кишечник, матка, мочевой пузырь). Продолжительность жизни больных значительно снижена, при естественном течении заболевания выживаемость к 50 годам не превышает 50% [5, 6]. Частота СЭД IV (MIM 130 050) составляет 1:250 000—1:100 000 населения, однако возможна недооценка истинной распространенности этого заболевания вследствие гиподиагностики [5].
НДСТ — клинически и генетически гетерогенная группа заболеваний, связанная с наследственными нарушениями синтеза и функционирования коллагеновых и эластических белков. Клиническая дифференциальная диагностика НДСТ чрезвычайно трудна вследствие сходства симптоматики. И если «марфаноидный фенотип» хорошо известен кардиологам и сосудистым хирургам, то внешний вид пациентов с сосудистым типом синдрома Элерса—Данло (СЭД IV) редко дифференцируется в клинике. Клиническая диагностика синдрома СЭД основана на Вильфраншских критериях 1998 г. [9]. Сбор семейного анамнеза и проведение ДНК-диагностики является необходимым этапом в подтверждении диагноза сосудистого типа СЭД [27]. Лабораторное тестирование рекомендуется лицам, имеющим два и более диагностических симптома. Верификация генетической причины заболевания у пробанда позволяет проводить подтверждающую, раннюю и пресимптоматическую диагностику заболевания (в том числе пренатальную по запросу семьи) у всех родственников, доступных для обследования [16]. Заболевание является полиорганным и жизнеугрожающим, вследствие риска разрыва сосудов среднего и крупного калибра, а также полых органов (кишечник, матка, мочевой пузырь). Продолжительность жизни больных значительно снижена, при естественном течении заболевания выживаемость к 50 годам не превышает 50% [5, 6]. Частота СЭД IV (MIM 130 050) составляет 1:250 000—1:100 000 населения, однако возможна недооценка истинной распространенности этого заболевания вследствие гиподиагностики [5].
В отечественной литературе мы не обнаружили случаев хирургического лечения пациентов с генетически подтвержденным СЭД IV типа [1, 2, 11]. В настоящей работе представлен пример мультидисциплинарного подхода, обеспечившего успешное лечение 2 пациентов с сочетанием хирургического лечения, таргетной терапии, а также медико-генетическим консультированием и ДНК-диагностикой пациентов с сосудистым типом СЭД.
Материал и методы
В отделении кардиохирургии № 1 (хирургии аорты и ее ветвей) Российского научного центра хирургии им. Б.В. Петровского за последние 10 лет было только 2 пациента с генетически подтвержденным СЭД IV типа, что составило 0,2% от всех больных с различными формами дисплазий, пролеченных за этот период. Всех пациентов с подозрением на НДСТ, госпитализированных в отделение, консультировали в лаборатории медицинской генетики ФГБНУ «Российский научный центр хирургии им. акад. Б.В. Петровского», что позволяло уточнить диагноз и корригировать дальнейшее лечение.
Б.В. Петровского за последние 10 лет было только 2 пациента с генетически подтвержденным СЭД IV типа, что составило 0,2% от всех больных с различными формами дисплазий, пролеченных за этот период. Всех пациентов с подозрением на НДСТ, госпитализированных в отделение, консультировали в лаборатории медицинской генетики ФГБНУ «Российский научный центр хирургии им. акад. Б.В. Петровского», что позволяло уточнить диагноз и корригировать дальнейшее лечение.
Клинические данные пациентов приведены в таблице. Таблица 1. Клинические характеристики пациентов Примечание. ЛПклА — левая подключичная артерия; ПА — почечная артерия; ВоА — восходящая аорта; АК — аортальный клапан; СР миокарда — спонтанный разрыв миокарда.
Больной З., 16 лет, поступил в ноябре 2010 г. для оперативного лечения в отделение хирургии аорты и ее ветвей РНЦХ им. акад. Б.В. Петровского.
Из анамнеза известно, что в ноябре 2008 г. почувствовал нарастающую боль в левом плечевом суставе, позже появилась пульсирующая припухлость. Госпитализирован бригадой СМП в больницу Сыктывкара, где диагностирован разрыв аневризмы левой подключичной артерии, с прорывом в левую плевральную полость, гемоторакс объемом до 2 л. Экстренно выполнено протезирование левой подключичной артерии. Через 6 мес после вмешательства при проведении планового обследования выявлен тромбоз протеза с признаками ишемии левой верхней конечности. В апреле 2010 г. по месту жительства в отделении микрохирургии предпринята попытка репротезирования, однако операция оказалась безуспешной ввиду выраженных изменений стенки подключичной артерии (резкое истончение, отсутствие эластичности). При гистологическом исследовании отмечалась очаговая аплазия мышечного слоя и отсутствие внутренней эластической мембраны артерии. Тогда же, по данным ангиографии, диагностирована аневризма правой почечной артерии.
При поступлении предьявлял жалобы на слабость и сильные боли в левой верхней конечности. Больной истощен (рост 170 см, масса тела 45 кг). Отмечается выраженная термоасимметрия верхних конечностей, а также гипотрофия, снижение силы и тонуса в левой верхней конечности.
Отмечается выраженная термоасимметрия верхних конечностей, а также гипотрофия, снижение силы и тонуса в левой верхней конечности.
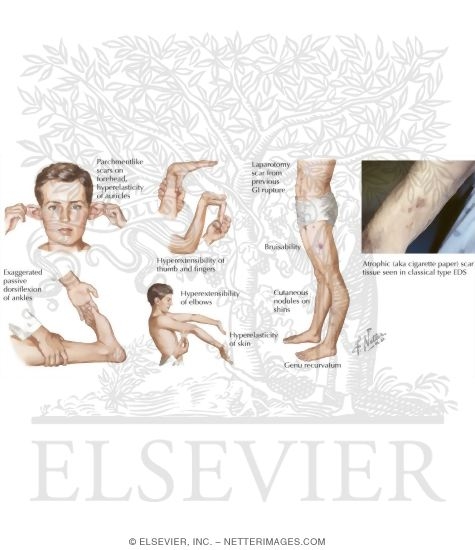
Больной проконсультирован врачом-генетиком. При осмотре были выявлены признаки дисплазии соединительной ткани (рис. 1). Рис. 1. Особенности фенотипа, характерные для сосудистого типа СЭД. а — гипермобильность суставов кистей, б — «папиросные рубцы» после травмы.
Кожа гиперэластичная, тонкая, подкожные вены ясно видны под кожей грудной клетки, несколько «папиросных» рубцов, послеоперационные келоидные рубцы, гипотония лицевых мышц, выраженная гипермобильность суставов, искривление позвоночника, небольшое воронкообразное искривление грудины, плоскостопие обеих стоп, синдактилия II—IV пальцев стоп, небо высокое, неправильный рост зубов, в анамнезе пупочная и паховая грыжи. Отмечал частое появление подкожных гематом, усилившихся в последнее время. Указаний на заболевания печени, которые могли бы послужить причиной гипокоагуляции, в анамнезе или истории болезни не обнаружено. Характерный для сосудистого типа СЭД фенотип (узкий нос, тонкие губы), обусловленный снижением подкожного жирового слоя, не отмечался [5, 10]. Семейный анамнез не известен (ребенок усыновлен).
С целью подтверждения диагноза, последующего медико-генетического консультирования семьи и оценки риска хирургических осложнений был выполнен поиск мутаций в гене COL3A1 методом прямого секвенирования по Сенгеру, кодирующих участков и прилегающих интронных областей. Была выявлена замена p. Gly183Ser в 6-м экзоне гена COL3A1 в гетерозиготном состоянии, приведшая к появлению миссенс-мутации (рис. 2). Рис. 2. ДНК-диагностика сосудистого типа синдрома СЭД у пациента З. Фрагмент прямого секвенирования 6-го экзона гена COL3A1. Стрелкой указана мутация c.547G>A (p.Gly183Ser) в гетерозиготном состоянии.
Эта мутация была неоднократно описана в качестве причины СЭД IV типа и зарегистрирована в международной специализированной базе данных мутаций Ehlers Danlos Syndrome Variant Database [9—12]. Таким образом, диагноз у пробанда был подтвержден молекулярно-генетическими методами. По данным дуплексного сканирования (ДС), левая подключичная артерия сразу после отхождения позвоночной артерии окклюзирована, дистальнее, на уровне подмышечной артерии, отмечался коллатеральный кровоток. На основании клинических, лабораторно-инструментальных и генетических данных поставлен диагноз: СЭД IV типа (сосудистый). Тромбоз протеза левой подключичной артерии. Хроническая ишемия верхней левой конечности, аневризма правой ПА.
Таким образом, диагноз у пробанда был подтвержден молекулярно-генетическими методами. По данным дуплексного сканирования (ДС), левая подключичная артерия сразу после отхождения позвоночной артерии окклюзирована, дистальнее, на уровне подмышечной артерии, отмечался коллатеральный кровоток. На основании клинических, лабораторно-инструментальных и генетических данных поставлен диагноз: СЭД IV типа (сосудистый). Тромбоз протеза левой подключичной артерии. Хроническая ишемия верхней левой конечности, аневризма правой ПА.
Пациенту выполнено аутовенозное сонно-плечевое шунтирование слева. Вследствие выраженных изменений больших подкожных вен на обеих нижних конечностях (малый диаметр, сегментарная деструкция стенки) шунт сформирован из трех фрагментов, забранных с обеих нижних конечностей. Хирургическое вмешательство усложнилось наличием грубых рубцовых деформаций в этой зоне после двух ранее перенесенных реконструкций (рис. 3, а,). Рис. 3. Интраоперационное фото: а — послеоперационный рубец в месте предыдущих реконструкций; б — дистальный анастомоз с плечевой артерией; в — проксимальный анастомоз с левой общей сонной артерией; г — конечный вид реконструкции из надключичного доступа. Левая общая сонная артерия выделена из надключичного доступа, левая плечевая артерия эксплорирована в верхней трети плеча. Выполнен анастомоз аутовенозного шунта с плечевой артерией по типу конец в бок атравматической полипропиленовой нитью 6/0. Шунт проведен под малой и большой грудными мышцами и над ключицей к левой общей сонной артерии. Сформирован анастомоз шунта с левой общей сонной артерией по типу конец в бок атравматической полипропиленовой нитью 6/0 (см. рис. 3, б, в).
Послеоперационный период протекал без осложнений. При контрольном дуплексном сканировании сонно-плечевой аутовенозный шунт слева проходим, кровоток по шунту Vs=2,8 м/с. Больной выписан на 7-е сутки после операции. Протезирование почечной артерии по поводу аневризмы выполнено в дальнейшем по месту жительства.
Больной П., 46 лет, поступил для оперативного лечения в отделение хирургии аорты и ее ветвей РНЦХ им. акад. Б.В. Петровского с жалобами на периодические боли в области сердца, давящего характера, возникающие при физической нагрузке, учащенное сердцебиение.
Из анамнеза: три сотрясения головного мозга, ишемический инсульт, перевязка правой внутренней подвздошной артерии по причине разрыва ложной посттравматической аневризмы верхней ягодичной артерии. Семейный анамнез отягощен: родители умерли от сердечно-сосудистых заболеваний, сестра страдает варикозной болезнью с 15 лет.
При физикальном осмотре отмечаются признаки дисплазии соединительной ткани: кожа гиперэластичная, бархатистая на ощупь, «положительный симптом щипка», характерные потертости на коленях, «папиросные» рубцы, экхимозы, нарушение осанки, поперечное плоскостопие обеих стоп, высокое небо, неправильный рост зубов, выраженная гипермобильность суставов.
Заключение генетика: СЭД. Был выполнен поиск мутаций в гене COL3A1 методом прямого секвенирования по Сенгеру, кодирующих участков и прилегающих интронных областей (рис. 4). Рис. 4. ДНК-диагностика сосудистого типа синдрома СЭД у пациента П. Фрагмент прямого секвенирования 45-го экзона гена COL3A1. Стрелкой указана мутация c. 3301G>A (p.Gly1101Arg) в гетерозиготном состоянии.
Выявленная замена p.Gly1101Arg в 45-м экзоне гена COL3A1 в гетерозиготном состоянии и приводит к появлению миссенс-мутации. Эта мутация зарегистрирована в международной специализированной базе данных мутаций Ehlers Danlos Syndrome Variant [14].
По данным эхокардиографии диагностирован порок аортального клапана с аортальной недостаточностью 2—3 степени, кардиомегалия (КДР 6,5 см, КДО 220 мл). При Д.С. периферических артерий выявлена патологическая извитость и деформация хода внутренних сонных артерий с обеих сторон, наружные подвздошные артерии S-образно извиты.
Пациенту выполнено протезирование аортального клапана механическим протезом. Из интраоперационных особенностей отмечается спонтанный разрыв корня аорты на 2/3 диаметра проксимальнее области аортотомного шва ввиду выраженной дисплазии тканей и повторное подключение аппарата И.К. Ткани взяты на гистологическое исследование (рис. 5). Рис. 5. Морфологическая картина дисплазии соединительной ткани аортального клапана и аорты: рыхлая соединительная ткань с явлениями дезорганизации и миксоматозом (1), истончение и неравномерное распределение эластических волокон (2), очаговый склероз (3). Окраска пикрофуксином по Ван-Гизону с докраской на эластические волокна — ×100 (а), × 50 (б).
Из интраоперационных особенностей отмечается спонтанный разрыв корня аорты на 2/3 диаметра проксимальнее области аортотомного шва ввиду выраженной дисплазии тканей и повторное подключение аппарата И.К. Ткани взяты на гистологическое исследование (рис. 5). Рис. 5. Морфологическая картина дисплазии соединительной ткани аортального клапана и аорты: рыхлая соединительная ткань с явлениями дезорганизации и миксоматозом (1), истончение и неравномерное распределение эластических волокон (2), очаговый склероз (3). Окраска пикрофуксином по Ван-Гизону с докраской на эластические волокна — ×100 (а), × 50 (б).
Произведена пластика восходящей аорты ксеноперикардом с использованием тефлоновых полосок. Ранний послеоперационный период осложнился кровотечением с тампонадой сердца и гемотораксом после удаления дренажей на 2-е сутки. При ревизии перикарда и средостения отмечались спонтанные разрывы в области передней стенки правого желудочка и задней стенки левого желудочка. Раны ушиты на тефлоновых прокладках. Пациент выписан на 12-е сутки.
В течение жизни пациенты, имеющие клинические проявления сосудистого типа СЭД (аневризмы периферических и висцеральных артерий крупного и среднего диаметра, спонтанные разрывы полых органов в анамнезе, варикозную болезнь в молодом возрасте, каротидно-кавернозные фистулы, гипермобильность суставов, гиперэластичнось кожи, стрии, папиросные и келоидные рубцы, деформации грудной клетки, позвоночника, нижних конечностей, патология глаз, грыжи и т. п.), с высокой частотой попадают в хирургический стационар для оказания различных видов хирургической помощи [11]. Зачастую первичное вмешательство является неэффективным, как показано в первом клиническом примере, что связано в первую очередь с системной патологией, и поэтому требуются повторные операции [12]. Однако в отсутствие патогномоничных биомаркеров при стертых и неспецифичных клинических проявлениях клиническая диагностика НДСТ и оценка хирургических рисков только по фенотипическим особенностям очень субъективна. Поэтому «золотым стандартом» диагностики сосудистого типа СЭД является подтверждающая ДНК-диагностика (выявление мутаций в гене COL3A1), которая была впервые разработана в лаборатории медицинской генетики РНЦХ им. акад. Б.В. Петровского.
Поэтому «золотым стандартом» диагностики сосудистого типа СЭД является подтверждающая ДНК-диагностика (выявление мутаций в гене COL3A1), которая была впервые разработана в лаборатории медицинской генетики РНЦХ им. акад. Б.В. Петровского.
Коллаген III типа является основной структурной единицей в архитектонике соединительнотканного остова дермы, связок, сухожилий, капсул суставов, стенки аорты и крупных артерий, вен, подслизистого слоя полых органов, что напрямую коррелирует с широким спектром осложнений при IV типе СЭД [17]. Коллаген III типа принадлежит к гомотримерным фибриллярным коллагенам. Он формируется при сочетании трех мономеров или α-цепей белка-предшественника протоколлагена. Последовательность аминокислот тройной спирали характеризуется повторами Gly-X-Y последовательностей, где X и Y часто представлены пролином и гидроксипролином. Для того чтобы обеспечить правильное связывание α-мономеров и стабильность коллагеновых волокон, не должно быть перерывов в повторах глицин-X-Y триплетов, а длина тройной спирали должна оставаться одинаковой для каждой α-цепи. Поэтому практически все мутации, приводящие к замене глицина на другую аминокислоту (как у представленных пациентов), являются патогенными и приводят к снижению содержания коллагена III до 10—15% от нормы [14, 15]. Другие мутации в гене COL3A1, при которых содержание нормального коллагена составляет не менее 50%, ассоциированы с более мягким течением заболевания, более поздними сосудистыми осложнениями, отсутствием кишечных осложнений и лучшим долгосрочным прогнозом [16].
Сосудистый тип СЭД обычно диагностируется после разрыва сосуда (65,4%) или органа (30,1%), либо только при патологоанатомическом исследовании. Максимальная частота спонтанных артериальных разрывов при данной форме заболевания приходится на третью—четвертую декады жизни. Часто поражается грудной и брюшной отдел аорты [19, 20]. Риск разрыва крупных артерий составляет 25% в возрасте до 20 лет, 80% — до 40 лет [18]. В исследовании, представленном G. Oderich и соавт. [7], было зарегистрировано 132 сосудистых осложнения у 24 пациентов со следующим распределением, представленным на рис. 6. Рис. 6.Локализация патологии у больных СЭД IV типа (G. Oderich и соавт. The spectrum, management and clinical outcome of Ehlers—Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005 Jul;42 (1):98−106).
В исследовании, представленном G. Oderich и соавт. [7], было зарегистрировано 132 сосудистых осложнения у 24 пациентов со следующим распределением, представленным на рис. 6. Рис. 6.Локализация патологии у больных СЭД IV типа (G. Oderich и соавт. The spectrum, management and clinical outcome of Ehlers—Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005 Jul;42 (1):98−106).
M. Pepin и соавт. [8] на когорте из 220 пациентов также доказали, что в 92% случаев причиной отсроченной смерти у пациентов с СЭД IV типа являлись сосудистые осложнения.
Варикозная болезнь часто встречается у данной категории пациентов, однако хирургическое лечение приводит в большинстве случаев к рецидивам. Разрыв кишечника чаще всего включает сигмовидную кишку.
Современная тактика лечения основана на динамическом наблюдении, снижении риска и хирургическом лечении осложнений. Выполнение ангиографии или артериальной пункции обычно противопоказано в связи с возможным образованием крупных гематом, ложных аневризм в месте доступа, в некоторых сообщениях говорится об отдаленных разрывах артерий. Однако в некоторых опубликованных статьях [21, 22] сообщается об успешном эндоваскулярном лечении данной категории пациентов. Профилактические операции не рекомендуются, несмотря на низкую хирургическую летальность у данной группы пациентов. Причиной отказа от нее является высокий процент послеоперационных и графт-ассоциированных осложнений, которые зачастую являются причиной летальности [11, 20].
Каждый пациент требует специализированного анестезиологического подхода [23]. Анестезиологические службы должны быть готовы к переливанию большого объема гемокомпонентов, сердечным аномалиям и порокам развития, требующим определенной оценки во время предоперационной подготовки [24]. Особое внимание стоит уделить нарушениям проводимости миокарда, вторичной митральной недостаточности и атриомегалии. Согласно руководству National Institute for Clinical Excellence, все постановки центральных венозных катетеров и артериальные пункции должны проводиться строго под УЗИ-контролем в целях снижения риска образования гематом, ложных аневризм, спонтанных разрывов сосудов [23]. В литературе описаны случаи эрозии сосудистой стенки в месте стояния катетера, осложненные эффузией крови в плевральную полость и перикард. Ввиду этого катетеры необходимо удалять как можно скорее [25]. Также персонал должен быть ориентирован на максимально прецизионную и бережную интубацию, ввиду склонности больных к кровоточивости и травматизации тканей. Обычная ларингоскопия может стать причиной травмы десен, слизистой полости рта, мягкого неба, вывиха височно-нижнечелюстного сустава. Рекомендовано использовать вспомогательную фиброоптическую бронхоскопию для интубации с целью минимизации травмы и определения точного положения интубационной трубки [26]. Любая фиксация катетеров, трубок, электродов может сопровождаться образованием экхимозов. При укладке больного возможна дислокация суставов. Все внутримышечные и подкожные инъекции могут сопровождаться образованием гематом [23].
В литературе описаны случаи эрозии сосудистой стенки в месте стояния катетера, осложненные эффузией крови в плевральную полость и перикард. Ввиду этого катетеры необходимо удалять как можно скорее [25]. Также персонал должен быть ориентирован на максимально прецизионную и бережную интубацию, ввиду склонности больных к кровоточивости и травматизации тканей. Обычная ларингоскопия может стать причиной травмы десен, слизистой полости рта, мягкого неба, вывиха височно-нижнечелюстного сустава. Рекомендовано использовать вспомогательную фиброоптическую бронхоскопию для интубации с целью минимизации травмы и определения точного положения интубационной трубки [26]. Любая фиксация катетеров, трубок, электродов может сопровождаться образованием экхимозов. При укладке больного возможна дислокация суставов. Все внутримышечные и подкожные инъекции могут сопровождаться образованием гематом [23].
Тип генетического повреждения и расположение мутации позволяют адекватно ориентировать врача на этапе планирования вмешательства с целью достижения максимально радикального результата хирургического лечения. Деликатное, атравматичное, прецизионное отношение к тканям, использование зажимов с мягким покрытием, баллонных окклюдеров, мягких ретракторов, тефлоновых прокладок при наложении анастомозов позволяют исключить осложнения в послеоперационном периоде.
В настоящее время разрабатывается новый подход к лечению, направленный на репрессию мутантного гена с использованием специфических аллельспецифичных малых интерферирующих РНК (siRNA). Эффективность такого подхода уже была показана в культуре кожных фибробластов больного с васкулярным типом СЭД, что может служить основой для разработки максимально персонализированной этиологической терапии этого заболевания [15].
В данном сообщении продемонстрирован пример мультидисциплинарного подхода к лечению пациентов с редкой патологией (СЭД IV типа). В представленных клинических случаях мы наблюдали истинное течение заболевания с большим количеством осложнений, зачастую требующих отклонения от сложившейся тактики его ведения. В последних рекомендациях по лечению патологии аорты отдельным пунктом вынесена стратегия и менеджмент данной категории пациентов. Выработанный подход, основанный в первую очередь на современных рекомендациях и работах современных клиник, накопленном коллективном опыте ведения пациентов с наследственными дисплазиями соединительной ткани при самой разнообразной сердечно-сосудистой патологии, позволяет существенно уменьшить количество осложнений и рецидивов даже у пациентов «очень высокого риска», таких как больные с СЭД IV типа.
В последних рекомендациях по лечению патологии аорты отдельным пунктом вынесена стратегия и менеджмент данной категории пациентов. Выработанный подход, основанный в первую очередь на современных рекомендациях и работах современных клиник, накопленном коллективном опыте ведения пациентов с наследственными дисплазиями соединительной ткани при самой разнообразной сердечно-сосудистой патологии, позволяет существенно уменьшить количество осложнений и рецидивов даже у пациентов «очень высокого риска», таких как больные с СЭД IV типа.
Подтвержденный диагноз сосудистого типа СЭД, как и почти любого системного заболевания, утяжеляет дальнейший прогноз даже при успешно выполненном хирургическом лечении и требует детального консультирования пациентов перед выпиской. Всем пациентам также рекомендовано динамическое наблюдение с ежегодными консультациями профильных специалистов. Предпочтения в диагностике стоит отдавать неинвазивным методам. Аутосомно-доминантный тип наследования сосудистого типа СЭД позволяет ожидать накопления заболевания в семье, что требует выполнения каскадного семейного скрининга. План динамического наблюдения и профилактики осложнений для родственников-носителей мутации в гене COL3A1 должен соответствовать программе мониторинга здоровья пациента.
Авторы заявляют об отсутствии конфликта интересов.
e-mail: [email protected]
Синдром Элерса-Данло — ДНК-диагностика — Центр Молекулярной Генетики
Синдром Элерса-Данло – это гетерогенная группа наследственных заболеваний соединительной ткани, в основе которых лежит недостаточное развитие коллагеновых структур в различных системах организма. Проявляется патологией кожи, опорно-двигательного аппарата, сердечно-сосудистой системы, глаз. Относится к моногенным заболеваниям с различными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена — одного из основных белков соединительной ткани.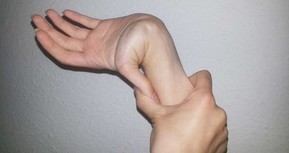 Конечный результат каждого из механизмов формирования патологии один и тот же — уменьшение стабильности коллагенового волокна. Генерализованность клинических проявлений при синдроме Элерса-Данло обусловлена тем, что элементы пораженной соединительной ткани присутствуют практически во всех тканях и системах организма. Диагноз устанавливают на основании анамнестических сведений (задержка моторного развития, плохая заживляемость ран, склонность к кровотечениям и экхимозам, вывихи и подвывихи), характерной клинической картины (гиперэластичносгь и хрупкость кожи, гиперподвижность суставов в сочетании с патологией сердца, глаз и др.).
Конечный результат каждого из механизмов формирования патологии один и тот же — уменьшение стабильности коллагенового волокна. Генерализованность клинических проявлений при синдроме Элерса-Данло обусловлена тем, что элементы пораженной соединительной ткани присутствуют практически во всех тканях и системах организма. Диагноз устанавливают на основании анамнестических сведений (задержка моторного развития, плохая заживляемость ран, склонность к кровотечениям и экхимозам, вывихи и подвывихи), характерной клинической картины (гиперэластичносгь и хрупкость кожи, гиперподвижность суставов в сочетании с патологией сердца, глаз и др.).
Согласно классификации наследственных болезней соединительной ткани, выделяют 9 форм синдрома Элерса-Данло, различающихся особенностями клинической картины и типом наследования.
Характерными признаками синдрома Элерса-Данло типа VI являются аутосомно-рецессивный тип наследования, мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии. У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
В ООО «Центр Молекулярной Генетики» проводится исследование двух частых мутаций в гене PLOD1: дупликации 8.9 KB и Arg319X.
Элерса-Данло тип VI синдром
Случай сочетания сахарного диабета и синдрома Элерса-Данло | Кононенко
1. Балаболкин М.И., Дедов И.И. // Сахар. диабет.-2000.-Ы 1 -с.2-1 0.
Балаболкин М.И., Дедов И.И. // Сахар. диабет.-2000.-Ы 1 -с.2-1 0.
2. Кураева Т.Л., Зильберман Л.И.//Сахар. диабет.-2000.-Ы 1-с.43-45.
3. Лавина Н. (ред.) Эндокринология: Пер. с англ.- М., 1999. — 1 128 с.
4. Стефани Д.В., Бельтищев Ю.Е. Иммунология и иммунопатология детского возраста. — М., 1 996.
5. Харрисон Т.Р. (ред.). Внутренние болезни: Пер. с англ.: Онкология и эндокринология (книга 8).-М.,1996.
6. Чистяков Д.А., Дедов И.И // Сахар.диабет.-1999.-N3.-с.52-56.
7. Щеглова О.С., Руденская Г.Е., Кураева Т.Л., Кюшников С.А.// Сахар.диабет. 2000. -N 4-С.51-54.
8. Beighton P., De Раере A., Steinmann В., Tsipouras P., Wenstrup R.J. // Am J of Med Genet, 1998,77,p 31 -37
9. Burch G.H., Gong Y., Liu W., Dettman R.W. // Nature Genetics, 1 997, vol 1 7, September, p 104-108
10. Burrows NP. // Clinical and Experimental Dermatology, 1999, 24, p99-l 06
11. Burrows NP et al. // J Invest Derm, 1996, 106, pi 273-1 276
12. Burrows N.P., Nicholls A.C., Richards A.J., Luccarini C., J. Harrison B.,. Yates J.R.W, Pope M.F. // Am J Hum Genet, 1998, 63, p 390-398
13. De Paepe A. et al. // Am J Hum Genet, 1997, 60, p547-554
De Paepe A. et al. // Am J Hum Genet, 1997, 60, p547-554
14. Loughlin J., Irven C., Hardwick L. J., Butcher S., Walsh S., Wordsworth P., Sykes B. // Hum Molec Genet, 1995, 4, p 1649-1651
15. Michalickova K., Susie М., Willing M.C., Wenstrup R. J., Cole W.G. // Hum Molec Genet, 1998, Vol.7, №2, p 249-255
16. Nuyting L., Freund М., Lagae L., Pierard G., Hrmanns-Le Т., De Paepe A. // Am J Hum Genet, 2000, 66, 1398-1402
17. Richards A.J. et al. // J Med Genet, 1 998, 35, p846-848
18. Schwarze U., Atkinson М., Hoffman G.G., Greenspan D.S., Byers P.H. // Am J Hum Genet, 2000, 66, pi 757-1 765
19. Sokolov BP et al. //Hum Genet, 1 991, 88, pi 25-129
20. Wenstrup R.J., Langland G. Т., Willing М. C, D’Souza V.N., Cole W.G. // Hum Molec Genet, 1996, Vol.5, № 1 1, pl733-1736
21. Schalkwijk J., Zweers М. C., Steijlen P. М., Dean, W. B., Taylor G.,van Vlijmen I. М., van Haren B., Miller W. L., Bristow J. // New Eng. J. Med. 345: 1167-1175, 2001
новые классификационные критерии 2017 г.
Клиническая диагностика синдромов Элерса–Данло: новые классификационные критерии 2017 г.
DOI: https://dx.doi.org/10.18565/therapy.2018.6.121-127
Е.Л. Трисветова
Белорусский государственный медицинский университет, 2-я кафедра внутренних болезней, Минск
Синдромы Элерса–Данло (СЭД), как проявление наследственных нарушений соединительной ткани, отличает многосистемность поражения и вариативность признаков. Диагностические критерии, разработанные в 1998 г., включающие описание 6 типов СЭД, успешно применяли в течение двух десятилетий. Однако результаты клинического наблюдения, биохимических и молекулярных исследований дополнились новыми знаниями о синдроме. В новой международной классификации 2017 г. выделены диагностические критерии 13 типов СЭД, пересмотрены признаки гипермобильного типа.
Диагностические критерии, разработанные в 1998 г., включающие описание 6 типов СЭД, успешно применяли в течение двух десятилетий. Однако результаты клинического наблюдения, биохимических и молекулярных исследований дополнились новыми знаниями о синдроме. В новой международной классификации 2017 г. выделены диагностические критерии 13 типов СЭД, пересмотрены признаки гипермобильного типа.
Ключевые слова: синдром Элерса–Данло, клиническая диагностика, большие и малые критерии
Литература
, Robert L., Rohrbach M., Sanders L., Sobey G.J., Van Damme T., Vandersteen A., van Mourik C., Voermans N., Wheeldon N., Zschocke J., Tinkle B. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017;175(1):8-26. Int. J. Clin. Pract. 2003;57(3):163-6.
Int. J. Clin. Pract. 2003;57(3):163-6.Об авторах / Для корреспонденции
Евгения Леонидовна Трисветова, д.м.н., профессор 2-й кафедры внутренних болезней учреждения образования «Белорусский государственный медицинский университет». 220116, Беларусь, Минск, пр. Дзержинского, 83. Тел.: +7 (017) 277-12-01. E-mail: [email protected]
Синдром Элерса-Данло, тип VI. Поиск частых мутаций в гене PLOD, ч. м. (Ehlers-Danlos Syndrome, Type VI, Gene PLOD, Freq. Mut.)
Исследуемый материал
Цельная кровь (с ЭДТА)
Метод определения
Секвенирование.
Выдаётся заключение врача-генетика!
Исследование частых мутаций в гене PLOD.
Синдром Элерса — Данло тип VI (EDS, СЭД, OMIM225400) — гетерогенная группа наследственных заболеваний соединительной ткани, в основе которых лежит недостаточное развитие коллагеновых структур в различных системах организма. Назван в честь двух дерматологов, Эварда Элерса и Генри Данлоса. Но ещё раньше синдром был описан Николаем Черногубовым. Проявляется патологией кожи, опорно-двигательного аппарата, сердечно-сосудистой системы, глаз. Относится к моногенным заболеваниям с различными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена, одного из основных белков соединительной ткани. Конечный результат каждого из механизмов формирования патологии один и тот же — уменьшение стабильности коллагенового волокна. Генерализованность клинических проявлений при синдроме Элерса — Данло обусловлена тем, что элементы поражённой соединительной ткани присутствуют практически во всех тканях и системах организма. Диагноз устанавливают на основании анамнестических сведений (задержка моторного развития, плохая заживляемость ран, склонность к кровотечениям и экхимозам, вывихи и подвывихи), характерной клинической картины (гиперэластичносгь и хрупкость кожи, гиперподвижность суставов в сочетании с патологией сердца, глаз и др.).
Согласно классификации наследственных болезней соединительной ткани, выделяют 9 форм синдрома Элерса — Данло, различающихся особенностями клинической картины и типом наследования. Характерными признаками синдрома Элерса — Данло типа VI являются аутосмно-рецессивный тип наследования, мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии. У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
Тип наследования.
Аутосомно- рецессивный.
Гены, ответственные за развитие заболевания.
Ген PLOD1 расположен на хромосоме 1 в регионе 1p36.22. Содержит 19 экзонов.
Патогенез и клиническая картина.
У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена — одного из основных белков соединительной ткани, приводящих к уменьшение стабильности коллагенового волокна. Элементы пораженной соединительной ткани присутствуют практически во всех тканях и системах организма, что приводит к генерализованному поражении организма при заболевании. Характерными признаками синдрома Элерса-Данло типа VI являются: мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Giunta, C., Randolph, A., Al-Gazali, L. I., Brunner, H. G., Kraenzlin, M. E., Steinmann, B. Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA). Am. J. Med. Genet. 133A: 158-164, 2005.
- OMIM.
Синдром Элерса-Данлоса: симптомы, диагностика, лечение
Синдром Элерса-Данлоса (СЭД) — это заболевание, которое ослабляет соединительные ткани вашего тела. Это такие вещи, как сухожилия и связки, которые скрепляют части вашего тела. EDS может сделать ваши суставы расшатанными, а кожу тонкой и легко получить синяки. Он также может ослабить кровеносные сосуды и органы.
Нет лекарства от EDS, но симптомы часто можно лечить и контролировать.
Типы EDS
Существует несколько типов синдрома Элерса-Данлоса, но все они делают ваши суставы слабыми и слабыми, а кожу необычно эластичной.
- Самая распространенная форма EDS заставляет ваши суставы изгибаться больше, чем они должны. Это увеличивает вероятность их вывиха или растяжения. Примерно 1 человек из 10 000 может иметь эту форму заболевания, называемую типом гипермобильности.
- При классическом типе EDS ваша кожа гладкая, очень эластичная и хрупкая. Люди с этим типом часто имеют шрамы на коже над коленями и локтями и легко образуют синяки. У них также могут быть растяжения, вывихи или состояния, такие как плоскостопие, а также проблемы с сердечным клапаном или артерией.Эта форма EDS встречается примерно у 1 из 20 000–40 000 человек. Но некоторые люди могут иметь легкую форму болезни и не подозревать об этом.
- Примерно 1 человек из 250 000 рождается с сосудистым типом EDS. Этот тип ослабляет кровеносные сосуды и увеличивает вероятность разрыва ваших органов.
Продолжение
Другие типы синдрома Элерса-Данлоса очень редки:
- Во всем мире было обнаружено около 60 случаев заболевания по типу кифосколиоза. Это когда дети рождаются со слабыми мышцами и костями.У них часто необычно длинные конечности или пальцы, а также изогнутый позвоночник, который становится хуже по мере роста. У них также часто возникают проблемы со зрением, такие как близорукость или глаукома, которые связаны со слишком сильным давлением внутри глаза.
- При артрохалазической форме EDS дети рождаются с неуместными тазобедренными суставами. Их суставы очень рыхлые, а позвоночник такой же изогнутый, как и у людей с кифосколиозом. Диагностировано около 30 случаев этого типа.
- Из всего около дюжины зарегистрированных случаев самый редкий тип EDS называется дерматоспараксис.У людей с этим очень мягкая, рыхлая кожа, на которой легко появляются синяки и рубцы. У них также чаще возникают грыжи.
Причины
EDS возникает, когда ваше тело не вырабатывает белок, называемый коллагеном, правильным образом. Коллаген помогает формировать связи, которые удерживают вместе кости, кожу и органы вашего тела. Если с этим есть проблема, эти структуры могут быть слабыми и иметь больше шансов иметь проблемы.
EDS — генетическое заболевание. Значит, это то, что вы получаете от родителей.Если у кого-то из ваших родителей есть это заболевание, вероятно, оно у вас тоже.
Диагноз
Ваш врач, скорее всего, начнет с медицинского осмотра:
- Он проверит, насколько далеко могут сгибаться ваши колени, локти, пальцы и поясница. Может ли ваш большой палец касаться предплечья? Можете ли вы согнуть мизинец более чем на 90 градусов?
- Они потянут вашу кожу, чтобы посмотреть, насколько она растянута, и будут искать шрамы, которые могла вызвать болезнь.
- Они спросят о вашей истории болезни и о том, были ли у вас или у кого-либо из членов вашей семьи подобные симптомы в прошлом.
- Если ваш врач считает, что у вас могут быть проблемы с сердцем или кровеносными сосудами, ваше обследование может включать эхокардиограмму, которая использует звуковые волны для создания изображения вашего сердца.
- Вы также можете пройти другие визуализационные тесты. Например, врач может попросить компьютерную томографию (КТ), которая снимает рентгеновские лучи под разными углами и соединяет их вместе, чтобы получить более полную картину. Или они могут попросить сделать магнитно-резонансную томографию (МРТ). Для получения детального изображения используются мощные магниты и радиоволны.
- Ваш врач может также взять биопсию. Для этого они возьмут небольшой образец кожи, чтобы найти признаки аномального коллагена под микроскопом. Они также могут провести химические тесты образца, чтобы попытаться выяснить, какой тип EDS у вас может быть. Другие виды тестов могут показать, какие гены могут вызывать проблему.
Лечение
Как только ваш врач узнает, какая у вас форма EDS, вы можете обсудить, как управлять своими симптомами. Возможно, вам потребуется обратиться к нескольким врачам, в том числе:
- Ортопеду, специализирующемуся на проблемах суставов и скелета
- Дерматологу, лечащему кожные заболевания
- Ревматологу, который занимается заболеваниями соединительной ткани
Продолжение
Некоторые варианты лечения включают:
- Физическая терапия и упражнения для повышения мышечного тонуса и улучшения координации.Более сильные мышцы могут снизить вероятность вывиха сустава. Полезные упражнения могут включать ходьбу, аэробику с малой нагрузкой, плавание или езду на велосипеде. Лечебная физкультура особенно важна для детей с СЭД.
- Подтяжки или другие вспомогательные приспособления, например инвалидное кресло или скутер, для облегчения передвижения.
- Добавки кальция и витамина D для укрепления костей.
- Безрецептурные препараты для облегчения боли в суставах. Если это не поможет, вам могут понадобиться лекарства, отпускаемые по рецепту.
- Женщинам с EDS может потребоваться особая помощь, если они беременны из-за возможных проблем с болью.
Жизнь с EDS
Некоторые вещи могут помочь облегчить повседневную жизнь:
- Используйте щетки с мягкой щетиной для чистки зубов.
- Толстые ручки или карандаши уменьшат нагрузку на пальцы.
- Надевайте защитную одежду или накладки на колени и локти, чтобы предотвратить появление синяков или порезов.
- Избегайте контактных видов спорта и высокоэффективных упражнений, таких как бег или катание на лыжах.
Кроме того, терапевт, консультант или группа поддержки могут помочь вам лучше справиться с изменениями в вашей жизни.
Синдромы Элерса-Данлоса — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Jones KL, ed. Распознаваемые модели пороков развития человека Смита. 5-е изд. Филадельфия, Пенсильвания: W. B. Saunders Co: 1997: 482-83.
Bennett JC, Plum F, ред. Сесил Учебник медицины. 20-е изд. Филадельфия, Пенсильвания: W.B. Saunders Co; 1996: 1120-22.
Буйс М.Л., изд. Энциклопедия врожденных пороков. Довер, Массачусетс: Научные публикации Блэквелла; Для: Центр врожденных дефектов Information Services Inc; 1990: 198, 610-11, 1269-70.
СТАТЬИ В ЖУРНАЛЕ
Malfait F, et al. Международная классификация синдромов Элерса-Данлоса 2017 г. Am J Med Genet C Semin Med Genet. 2017 Март; 175 (1): 8-26. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31552/full
Henderson Sr FC, et al. Неврологические и спинномозговые проявления синдромов Элерса-Данлоса. Am J Med Genet C Semin Med Genet. 2017 Март; 175 (1): 195-211. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31552/full
Bowen JM, et al. Синдром Элерса-Данлоса, классический тип.Am J Med Genet C Semin Med Genet. 2017 13 февраля, EPUB выходит в печать. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31548/full
Chopra P, et al. Обезболивание при синдромах Элерса-Данлоса. Am J Med Genet C Semin Med Genet. 2017 13 февраля, EPUB выходит в печать. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31554/full
Ericson WB Jr и Wolman R. Ортопедическое лечение синдромов Элерса-Данлоса. Am J Med Genet C Semin Med Genet. 2017 13 февраля, EPUB выходит в печать. http: // онлайн-библиотека.wiley.com/doi/10.1002/ajmg.c.31551/full
Хакин А. и др. Сердечно-сосудистая вегетативная дисфункция при синдроме Элерса-Данлоса по гипермобильному типу. Am J Med Genet C Semin Med Genet. 2017 13 февраля, EPUB выходит в печать. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31543/full
Bulbena A, et al. Психиатрические и психологические аспекты синдромов Элерса-Данлоса. Am J Med Genet C Semin Med Genet. 2017 10 февраля, EPUB выходит в печать. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31544/full
Fikree A, et al.Поражение желудочно-кишечного тракта при синдромах Элерса-Данлоса. Am J Med Genet C Semin Med Genet. 2017 10 февраля, EPUB выходит в печать. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31546/full
Burkitt Wright EM, Porter LF, Spencer HL, et al. Синдром хрупкой роговицы: распознавание, молекулярная диагностика и лечение. Журнал «Орфанет редких болезней». 2013; 8: 68. DOI: 10.1186 / 1750-1172-8-68.
Giunta, C, et al. Спондилохейро-диспластическая форма синдрома Элерса-Данлоса — аутосомно-рецессивное заболевание, вызванное мутациями в гене транспортера цинка SLC39A13.Являюсь. J. Hum. Genet. 2008; 82: 290-1305.
McDonnell et al. Эхокардиографические данные при классическом и гипермобильном синдромах Элерса-Данлоса. Am J Med Genet A. 2006, 15 января: 140 (2): 129-36.
Schalkwijk J, Zweers MC, Steijlen PM, et al. Рецессивная форма синдрома Элерса-Данлоса, вызванная дефицитом тенасцина-X. N Engl J Med. 2001; 345: 1167-75.
Пепин М. и др. Клинико-генетические особенности синдрома Элерса-Данлоса IV типа, сосудистого типа. New Engl J Med. 2000; 342: 673-80.
Pyeritz RE, синдром Элерса-Данлоса. New Engl J Med. 2000: 342: 730-32.
Beighton P, et al. Синдромы Элерса-Данлоса: пересмотренная нозология, Вильфранш, 1997. Национальный фонд Элерса-Данлоса (США) и Группа поддержки Элерса-Данлоса (Великобритания). Am J Med Genet. 1998; 77: 31-7.
Михаликова К. и др. Мутации альфа2 (V) цепи коллагена типа V нарушают сборку матрикса и вызывают синдром Элерса-Данлоса типа I. Hum Mol Genet. 1998; 7: 249-55.
Ричардс А.Дж. и др. Мутация с одним основанием в COL5A2 вызывает синдром Элерса-Данлоса типа II.J Med Genet. 1998; 35: 846-48.
Байерс PH, et al. Синдром Элерса-Данлоса типа VIIA и VIIB является результатом мутаций сплайс-соединения или геномных делеций, которые затрагивают экзон 6 в генах COL1A1 и COL1A2 коллагена типа I. Am J Med Genet. 1997; 72: 94-105.
De Paepe A, et al. Мутации в гене COL5A1 являются причиной синдромов Элерса-Данлоса I и II. Am J Hum Genet. 1997; 60: 547
Берроуз Н. П. и др. Ген, кодирующий коллаж альфа1 (V) (COL5A1), связан со смешанным синдромом Элерса-Данлоса типа I / II.J Invest Dermatol. 1996; 106: 1273-76.
Loughlin J, et al. Связь гена, кодирующего альфа-1 цепь коллагена V типа (COL5A1), с синдромом Элерса-Данло II типа (EDS II). Hum Mol Genet. 1995; 4: 1649-51.
North KN, et al. Цереброваскулярные осложнения при синдроме Элерса-Данлоса IV типа. Энн Нейрол. 1995; 38: 960-64.
Нарциси П. и др. Семья с синдромом Элерса-Данлоса типа III / синдромом гипермобильности суставов имеет замену глицина 637 на серин в коллагене III типа.Hum Mol Genet. 1994; 3: 1617-20.
Superti-Furga A, et al. Микроангиопатия при синдроме Элерса-Данлоса IV типа. Int J Microcirc Clin Exp. 1992; 11: 241-47.
Венструп Р.Дж. и др. Тип VI по Элерсу-Данлосу: клинические проявления дефицита лизилгидроксилазы коллагена. J Pediat. 1989; 115: 405-09.
Superti-Furga A, et al. Синдром Элерса-Данлоса тип IV: делеция нескольких экзонов в одном из двух аллелей COL3A1, влияющая на структуру, стабильность и процессинг проколлагена III типа.J Biol Chem. 1988; 263: 6226-32.
Рирдон В. и др. Естественная история дерматоспараксиса человека (синдром Элерса-Данлоса типа VIIC). Clin Dysmorphol. 1995; 4: 1-11 Beighton P, et al. Международная нозология наследственных заболеваний соединительной ткани, Берлин, 1986. Am J Med Genet. 1988; 29: 581-94.
Незначительный RR, et al. Дефекты преобразования процеллагена в коллаген обнаруживаются в культивируемых фибробластах пациентов с синдромом Элерса-Данлоса и несовершенным остеогенезом.J Biol Chem. 1986; 261: 10006-14.
Tsipouras P, et al. Синдром Элерса-Данлоса тип IV: косегрегация фенотипа с аллелем COL3A1 проколлагена III типа. Hum Genet. 1986; 74: 41-46.
Beighton P, et al. Х-сцепленный синдром Элерса-Данлоса тип V: следующее поколение. Clin Genet. 1985; 27: 472-78.
ИНТЕРНЕТ
Francoman C и Bloom L. Классификация EDS 2017, ответы на ваши вопросы. [Видео веб-семинар]. http://ehlers-danlos.com/wp-content/uploads/QandA-2.pdf. По состоянию на 27 сентября 2017 г.
Мальфаит Ф., Венструп Р., Де Паепе А. Синдром Элерса-Данлоса, классический тип. 29 мая 2007 г. [Обновлено 18 августа 2011 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1244/ По состоянию на 27 сентября 2017 г.
Levy HP. Синдром Элерса-Данлоса, тип гипермобильности. 22 октября 2004 г. [Обновлено 31 марта 2016 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы.GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1279/ По состоянию на 27 сентября 2017 г.
Pepin MG, Murray ML, Byers PH. Сосудистый синдром Элерса-Данлоса. 2 сентября 1999 г. [Обновлено 19 ноября 2015 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1494/ По состоянию на 27 сентября 2017 г.
Йеуэлл Х.Н., Штейнманн Б. Синдром Элерса-Данлоса, кифосколиотическая форма. 2 февраля 2000 г. [Обновлено 24 января 2013 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1462/ По состоянию на 27 сентября 2017 г.
синдромов Элерса-Данлоса — NHS
Синдромы Элерса-Данлоса (СЭД) — это группа редких наследственных состояний, поражающих соединительную ткань.
Соединительные ткани служат опорой для кожи, сухожилий, связок, кровеносных сосудов, внутренних органов и костей.
Симптомы синдромов Элерса-Данлоса (СЭД)
Существует несколько типов EDS, которые могут иметь общие симптомы.
К ним относятся:
- увеличенный диапазон движений в суставах (гипермобильность суставов)
- эластичная кожа
- хрупкая кожа, которая легко ломается или легко покрывается синяками
EDS может влиять на людей по-разному.У некоторых состояние относительно легкое, в то время как у других симптомы могут приводить к потере трудоспособности.
Различные типы EDS вызваны дефектами определенных генов, которые делают соединительную ткань более слабой.
В зависимости от типа EDS дефектный ген мог быть унаследован от одного или обоих родителей.
Иногда дефектный ген не передается по наследству, но встречается у человека впервые.
Некоторые из редких тяжелых типов могут быть опасными для жизни.
Основные типы синдромов Элерса-Данлоса (СЭД)
Существует 13 типов СЭД, большинство из которых очень редки.
Гипермобильный ЭЦП (hEDS) является наиболее распространенным типом.
Другие типы EDS включают классические EDS, сосудистые EDS и кифосколиотические EDS.
На веб-сайте EDS Support UK можно найти дополнительную информацию о различных типах EDS.
Hypermobile EDS
Люди с hEDS могут иметь:
В настоящее время нет тестов для подтверждения наличия hEDS.
Диагноз ставится на основании истории болезни человека и медицинского осмотра.
Классический EDS
Классический EDS (cEDS) встречается реже, чем гипермобильный EDS, и имеет тенденцию больше влиять на кожу.
Люди с КЭДС могут иметь:
- гипермобильность суставов
- свободные, нестабильные суставы, которые легко вывихиваются
- эластичная кожа
- хрупкая кожа, которая может легко расслаиваться, особенно на лбу, коленях, голенях и локтях
- гладкая, бархатистая кожа, на которой легко появляются синяки
- раны, которые медленно заживают и оставляют широкие рубцы
- грыжи и выпадение органов
EDS сосудов
EDS сосудов (vEDS) является редким типом EDS и часто считается наиболее серьезным.
Поражает кровеносные сосуды и внутренние органы, что может вызвать их разрыв и привести к опасному для жизни кровотечению.
У людей с vEDS могут быть:
- кожа, на которой очень легко появляются синяки
- тонкая кожа с видимыми мелкими кровеносными сосудами, особенно в верхней части груди и ног
- хрупкие кровеносные сосуды, которые могут вздуться или разорваться, что приведет к серьезному внутреннему кровотечению
- риск проблем с органами, таких как разрыв кишечника, разрыв матки (на поздних сроках беременности) и частичное коллапс легких
- гипермобильные пальцы рук и ног, необычные черты лица (например, тонкий нос и губы, большие глаза и маленькие мочки ушей), варикозное расширение вен и замедленное заживление ран
Кифосколиотическая EDS
Кифосколиотическая EDS (kEDS) встречается редко.
Люди с kEDS могут иметь:
- искривление позвоночника — это начинается в раннем детстве и часто ухудшается в подростковом возрасте
- гипермобильность суставов
- свободные, нестабильные суставы, которые легко вывихиваются
- слабый мышечный тонус с детства ( гипотония) — это может вызвать задержку в сидении и ходьбе или затруднения при ходьбе при ухудшении симптомов
- хрупкие глаза, которые можно легко повредить
- мягкая бархатистая кожа, эластичная, легко покрывается синяками и шрамами
Получение медицинской консультации
Обратитесь к терапевту, если у вас есть несколько неприятных симптомов EDS.
Обычно вам не нужно беспокоиться, если у вас всего несколько симптомов, и они не вызывают никаких проблем.
Гипермобильность суставов, например, является относительно распространенным явлением, затрагивая примерно 1 из 30 человек. Поэтому маловероятно, что это может быть вызвано EDS, если у вас нет других симптомов.
Ваш терапевт может направить вас к специалисту по суставам (ревматологу), если у вас есть проблемы с суставами, и они подозревают EDS.
Если есть вероятность, что у вас может быть один из редких типов EDS, ваш терапевт может направить вас в местную генетическую службу для оценки.
Местный генетик спросит о вашей истории болезни, семейном анамнезе, оценит ваши симптомы и может провести генетический анализ крови для подтверждения диагноза.
Если потребуется дополнительное расследование, врач вашей больницы может направить вас в специализированную диагностическую службу EDS в Шеффилде или Лондоне.
Лечение синдромов Элерса-Данлоса (EDS)
Специального лечения EDS не существует, но можно управлять многими симптомами с помощью поддержки и совета.
Людям с EDS также может быть полезна поддержка различных специалистов в области здравоохранения.
Например:
- физиотерапевт может научить вас упражнениям, которые помогут укрепить ваши суставы, избежать травм и справиться с болью
- эрготерапевт может помочь вам управлять повседневными делами и дать совет относительно оборудования, которое может помочь вам
- консультирование и когнитивные поведенческая терапия (КПТ) может быть полезна, если вы изо всех сил пытаетесь справиться с долговременной болью
- для определенных типов EDS, регулярное сканирование, проводимое в больнице, может выявить проблемы с внутренними органами
- генетическое консультирование может помочь вам узнать больше о причина вашего состояния, как оно передается по наследству и каковы риски его передачи вашим детям
Ваш терапевт или консультант может направить вас в эти службы.
Жизнь с синдромами Элерса-Данлоса (EDS)
Важно проявлять осторожность при занятиях, которые сильно нагружают суставы или могут привести к травмам.
Но также важно не проявлять чрезмерной заботы и избегать нормальной жизни в остальном.
Консультации будут зависеть от того, какой у вас тип ЭЦП и как он на вас влияет:
- вам могут посоветовать полностью избегать некоторых видов деятельности, таких как поднятие тяжестей и контактные виды спорта
- для некоторых видов деятельности вам может потребоваться соответствующее защиты и научить, как снизить нагрузку на суставы
- Можно рекомендовать занятия с меньшим риском, такие как плавание или пилатес, чтобы помочь вам оставаться в форме и быть здоровым
- Если утомляемость является проблемой, вас могут научить, как сохранить энергия и темп вашей деятельности
Как наследуются синдромы Элерса-Данлоса (СЭД)
EDS может передаваться по наследству, но в некоторых случаях это происходит случайно у кого-то, у кого это заболевание не было в семейном анамнезе.
Два основных способа наследования ЭЦП:
Лицо с ЭЦП может передавать своим детям только ЭЦП того же типа.
Например, дети человека с гипермобильной EDS не могут унаследовать сосудистую EDS.
Степень тяжести состояния может варьироваться в пределах одной семьи.
Дополнительная информация
Следующие веб-сайты предоставляют дополнительную информацию, советы и поддержку для людей с EDS и их семей:
Информация о вас
Если у вас есть EDS, ваша клиническая бригада передаст информацию о вас в Национальную службу регистрации врожденных аномалий и редких заболеваний
Это поможет ученым найти более эффективные способы профилактики и лечения этого состояния.
Вы можете отказаться от регистрации в любое время.
Последняя проверка страницы: 6 февраля 2019 г.
Срок следующего рассмотрения: 6 февраля 2022 г.
Синдром Элерса-Данлоса: загадка разгадана — Harvard Health Blog
АРХИВНОЕ СОДЕРЖАНИЕ: В качестве услуги для наших читателей Harvard Health Publishing предоставляет доступ к нашей библиотеке заархивированного содержимого. Обратите внимание на дату публикации или последнего рецензирования каждой статьи.Никакой контент на этом сайте, независимо от даты, никогда не должен использоваться вместо прямого медицинского совета вашего врача или другого квалифицированного клинициста.
Я всегда был подвержен авариям. Для меня не было ничего необычным внезапно потерять равновесие, идя по ровной асфальтированной дорожке, как будто кто-то скользнул передо мной банановой кожурой. Я сильно ударился о землю, поранив колени и локти. Однажды я упал на велосипеде с дощатого настила в болото, мое тело бросало впечатляющие очертания в камышах — как мультипликационный персонаж, пробившийся сквозь стену.Мое тело часто было покрыто синяками и струпьями, в то время как большая часть моей посуды в какой-то момент была обречена на разрушение, вызывая ужас среди моих соседей по комнате. Блюда были лишь некоторыми из жертв моей неуклюжести, за что меня иногда презирали и ругали учителя, родители и парни.
Но я никогда не связывал эту неуклюжесть с тем, что мои суставы и сухожилия казались такими же хрупкими, как стеклянная посуда, которую я иногда разбивал: лодыжки, которые искривлялись и растягивались при малейшей ошибке; запястья повреждены и воспалены в течение многих лет после первых нескольких попыток нисходящего движения собаки во время вводного урока йоги; челюсть, которая частично вывихнулась в результате простого пережевывания чипса из тортильи.Эти случаи стали менее редкими и более обычными с течением времени, а также стали более серьезными.
Около шести лет назад, после того как я помог своему тогдашнему парню перенести диван на три этажа в нашу квартиру, я целую неделю не мог встать с постели. Диски в моей спине просто выдохлись, как коробка с желейными пончиками, на которые кто-то сел. Я помню, как мой бывший говорил мне, что даже его мать могла бы выполнить такую простую задачу без травм, но я не мог. Такое неодобрение по поводу моего тела и его особенностей теперь автоматически отстраняется с моей стороны.Если я встречу кого-то, кто критически относится к моим физическим недостаткам, я не собираюсь больше с ним разговаривать.
Разъяснения причины моей неуклюжести
В последнее время мое тело мучила непрекращающаяся, широко распространенная, ноющая боль до костей. В частности, мой крестец и бедра чувствовали себя так, как будто они были пропитаны осколками битого стекла, которые ударялись о меня, когда я шел и натирал свои мягкие ткани до ожогов, слишком много сидел. Результаты визуализации показали большой разрыв губной губы в моем левом бедре и позвоночник, изобилующий поврежденными дисками и кистами, опухшими от спинномозговой жидкости.Физиотерапевты всегда спрашивали, в каком несчастном случае я попал или каким агрессивным спортом я занимался, чтобы получить так много травм, но я мог только пожать плечами и сказать, что нет никаких причин, по которым я мог бы убедиться, что жизнь просто сказывалась на мне — хотя и более тяжелый, чем казалось другим, и в более раннем возрасте, чем многие испытывали.
Тайна разгадана
Вот почему это было огромным подтверждением, когда я, наконец, посетил генетика в начале этого года, который после тщательной двухчасовой оценки обнаружил, что у меня тип III редкого заболевания соединительной ткани, известного как синдром Элерса-Данлоса или EDS.EDS характеризуется гипермобильностью суставов и дефицитом коллагена (соединительной ткани), что приводит к боли и повторным травмам. Этот диагноз не только объясняет мою неуклюжесть и большую часть телесных повреждений, которые я накопил за эти годы, он также помогает объяснить некоторые из моих мигреней и частые высыпания на коже, проблемы с регулированием температуры тела и кишечника. и проблемы с мочевым пузырем.
В то время как я испытал облегчение после диагноза, я также был возмущен. Последние несколько лет я приставал к врачам из-за своей хронической и часто инвалидизирующей боли.Мой генетик сказал мне, что для получения диагноза EDS часто требуется в среднем от 10 до 20 лет, и многие люди не получают точный диагноз до тех пор, пока им не исполнится 40 лет. Как и многие другие заболевания, EDS непропорционально сильно влияет на женщин. Это может быть причиной задержки в получении правильного диагноза и лечения, поскольку исследования неоднократно показывали, что медицинское сообщество не так хорошо осведомлено о проблемах со здоровьем, которые влияют на женщин, и, следовательно, с большей вероятностью будет неправильно диагностировать и игнорировать их.Мой генетик сказал мне, что многие женщины, которым ей поставили диагноз, в какой-то момент пытались покончить жизнь самоубийством, чтобы положить конец своим страданиям, как физическим страданиям от расстройства, так и эмоциональным страданиям из-за того, что их боль игнорировалась и даже подвергалась сомнению в течение многих лет или десятилетий.
Теперь, когда у меня, наконец, есть правильный диагноз, я взял на себя инициативу, необходимую для лучшего лечения расстройства и его симптомов, включая отказ от определенных видов деятельности и применение вспомогательных средств для стабилизации и защиты моих суставов.
Поскольку EDS не является широко изученным или известным заболеванием, я очень надеюсь, что мой опыт может послужить руководством для других пациентов, которые подозревают, что их хроническая боль — это нечто большее, чем кажется на первый взгляд.
Синдром Элерса-Данлоса: симптомы, причины, лечение
Обзор
Что такое синдром Элерса-Данлоса?
Синдром Элерса-Данлоса — это группа состояний, поражающих соединительные ткани в организме.Эти ткани включают хрящи, кости, жир и кровь. Они поддерживают органы и другие ткани по всему телу.
Врачи классифицируют синдром Элерса-Данлоса на 13 типов в зависимости от их наиболее заметных особенностей и частей тела, на которых проявляются симптомы. У людей с наиболее распространенным типом есть такие симптомы, как очень слабые суставы и хрупкая кожа, которая легко рвется.
Синдром Элерса-Данлоса может быть генетическим, то есть передается от членов семьи. По оценкам, 1 из 5000-20 000 человек страдает наиболее распространенным типом синдрома Элерса-Данлоса.
Симптомы и причины
Каковы симптомы синдрома Элерса-Данлоса?
Каждый тип синдрома Элерса-Данлоса имеет свои симптомы. Наиболее распространенным типом состояния является гипермобильность Элерса-Данлоса или гипермобильность EDS. Его симптомы включают:
- Гипермобильные (чрезмерно гибкие) суставы
- Нестабильные суставы
- Мягкая кожа, более тонкая и растягивающаяся больше, чем обычно
- Чрезмерный синяк
Что вызывает синдром Элерса-Данлоса?
Дефект коллагена (белков, придающих соединительной ткани гибкость и прочность) вызывает синдром Элерса-Данлоса.Люди с этим заболеванием имеют дефектный ген, который приводит к слабому коллагену или недостаточному количеству нормального коллагена в тканях. Эти дефекты могут повредить способность соединительной ткани поддерживать мышцы, органы и другие ткани.
Диагностика и тесты
Как врачи диагностируют синдром Элерса-Данлоса?
Врачи используют ваш семейный анамнез и несколько тестов для диагностики синдрома Элерса-Данлоса.Ваш диагноз может включать:
- Генетическое тестирование: Наиболее распространенный способ определения состояния — поиск дефектного гена.
- Биопсия : В некоторых случаях врач использует тест, называемый биопсией. В этом тесте врач берет образец кожи и исследует его под микроскопом, чтобы найти признаки заболевания, такие как определенные гены и генные мутации (аномалии).
- Осмотр: Во время осмотра врачи могут увидеть, насколько растягивается кожа и насколько могут двигаться суставы.
- Imaging: Тесты, которые предоставляют изображения внутренней части тела, могут помочь врачам выявить аномалии, включая проблемы с сердечной функцией и искривленные кости. Эти тесты включают рентгеновские снимки и компьютерную томографию (КТ).
Ведение и лечение
Каковы общие методы лечения синдрома Элерса-Данлоса?
Лечение синдрома Элерса-Данлоса направлено на предотвращение опасных осложнений.Это также может помочь защитить суставы, кожу и другие ткани от травм. Лечение человека зависит от многих факторов, включая тип расстройства и симптомы.
Для защиты кожи врачи рекомендуют использовать солнцезащитный крем и мягкое мыло. Дополнительный прием витамина С может помочь уменьшить синяки. Лечебная физкультура (упражнения для укрепления мышц, поддерживающих суставы) может помочь предотвратить травмы суставов. Подтяжки также помогают стабилизировать суставы.
Поскольку кровеносные сосуды хрупкие, врачи будут наблюдать за людьми с синдромом Элерса-Данлоса и могут использовать лекарства для поддержания низкого и стабильного артериального давления.
Вывихи суставов и другие травмы суставов часто встречаются у людей с синдромом Элерса-Данлоса. По этой причине врачи рекомендуют избегать:
- Подъем тяжелых грузов
- Упражнение с высокой ударной нагрузкой, при котором тело ударяется о землю
- Контактные виды спорта
Профилактика
Можно ли предотвратить синдром Элерса-Данлоса?
Поскольку он является генетическим, невозможно предотвратить синдром Элерса-Данлоса.
Перспективы / Прогноз
Каковы перспективы для людей с синдромом Элерса-Данлоса?
Перспективы людей с синдромом Элерса-Данлоса зависят от типа состояния и индивидуальных симптомов. Большинство форм этого состояния не влияют на продолжительность жизни.
Жить с
Каковы общие осложнения или побочные эффекты синдрома Элерса-Данлоса?
Осложнения некоторых типов синдрома Элерса-Данлоса могут быть опасными для жизни. Некоторые типы, включая сосудистый синдром Элерса-Данлоса, могут вызывать разрыв (разрыв) кровеносных сосудов.Когда это происходит, это может привести к опасному внутреннему кровотечению и инсульту.
Люди с этими типами синдрома Элерса-Данлоса также имеют более высокий риск разрыва органа. Чаще всего может разорваться кишечник или матка беременной.
Осложнения других видов синдрома Элерса-Данлоса зависят от типа. Эти осложнения могут включать:
- Проблемы с клапанами, проталкивающими кровь через сердце
- Сильное искривление позвоночника
- Истончение роговицы глаз
- Искривленные (изогнутые) конечности
- Дефекты зубов и десен
Как лучше всего жить с синдромом Элерса-Данлоса?
Врачи могут помочь вам справиться с симптомами с помощью физиотерапии и, при необходимости, обезболивания.Наблюдение за синдромом Элерса-Данлоса посредством регулярных посещений врача — лучший способ убедиться, что его состояние не мешает вести здоровый образ жизни.
Что такое EDS? — Служба поддержки Ehlers-Danlos UK
Синдромы Элерса-Данлоса (СЭД) — это группа из тринадцати индивидуальных генетических состояний, каждое из которых влияет на соединительную ткань организма. Соединительная ткань лежит между другими тканями и органами, разделяя их, соединяя их, удерживая все на месте и обеспечивая поддержку, как раствор между кирпичами.В EDS мутация гена приводит к тому, что определенный вид соединительной ткани — вид будет зависеть от типа EDS, но обычно это форма коллагена — становится хрупкой и эластичной. Эту растяжку иногда можно увидеть на коже человека с EDS; Люди с этим заболеванием также могут иметь возможность расширять свои суставы дальше, чем обычно — это известно как гипермобильность, гибкость или двусуставность. Поскольку коллаген присутствует во всем теле, люди с EDS, как правило, испытывают широкий спектр симптомов, большинство из которых менее заметны, чем различия на коже и суставах.Это сложные синдромы, поражающие сразу несколько систем организма, несмотря на то, что этот СЭД часто является невидимой инвалидностью. Симптомы обычно включают, помимо прочего, длительную боль, хроническую усталость, головокружение, учащенное сердцебиение и расстройства пищеварения. Такие проблемы и их серьезность значительно различаются от человека к человеку, даже в одном и том же типе EDS и в одной семье.
Поиска по предметным -2021 Профессиональным сериям — детей- & NBSP & NBSP & NBSP & nbspPregnancy-Условие, связанное с EDS- & NBSP & NBSP & NBSP & проблема nbspBladder & NBSP & NBSP & NBSP & nbspBrain и позвоночник & NBSP & NBSP & NBSP & проблема nbspCardiovascular & NBSP & NBSP & NBSP & nbspDental, оральные и речевые проблемы & NBSP & NBSP & NBSP & расстройства nbspDigestive & NBSP & NBSP & NBSP & проблема nbspJoint & NBSP & NBSP & NBSP & расстройство активации nbspMast клеток & NBSP & NBSP & NBSP & nbspMental Здоровье & NBSP & NBSP & NBSP & nbspPostural тахикардия синдром (POTS) & NBSP & NBSP & NBSP & nbspSkin-Диагностика — Здоровье professionals— Жизнь с EDS- & NBSP & NBSP & NBSP & nbspFatigue & NBSP & NBSP & NBSP & nbspPain & NBSP & NBSP & NBSP & nbspPhysiotherapy и самоуправление & NBSP & NBSP & NBSP & управления nbspSurgery-Я — Виды EDS- & NBSP & NBSP & NBSP & nbspAll типы & NBSP & NBSP & NBSP & nbspClassical EDS & NBSP & NBSP & NBSP & nbspHypermobile EDS и гипермобильность спектра расстройств & NBSP & NBSP & NBSP & nbspVascular EDS & NBSP & NBSP & NBSP & nbspVascular EDS и гипермобильном EDS по сравнению
Синдром Элерса-Данлоса: история вопроса, патофизиология, этиология
Castori M, Hakim A.Современный подход к лечению гипермобильности суставов и связанных с ней заболеваний. Curr Opin Pediatr . 2017 29 декабря (6): 640-649. [Медлайн].
Junkiert-Czarnecka A, Pilarska-Deltow M, Bąk A, Heise M, Haus O. Новые варианты гена COL5A1 среди польских пациентов с синдромом Элерса-Данлоса: анализ девяти случаев. Постэпы Дерматол Алергол . 2019 Февраль 36 (1): 29-33. [Медлайн].
Grahame R. Мультисистемная природа и естественная история синдрома гипермобильности суставов и синдрома Элерса-Данлоса у детей: данные нового исследования противоречат широко распространенным взглядам. Ревматология (Оксфорд) . 2017 г. 1. 56 (12): 2048-2049. [Медлайн].
Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, et al. Синдромы Элерса-Данлоса, редкие типы. Am J Med Genet C Semin Med Genet . 2017 Март 175 (1): 70-115. [Медлайн].
Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X и др. Диагностика, естественное течение и лечение сосудистого синдрома Элерса-Данлоса. Am J Med Genet C Semin Med Genet . 2017 Март 175 (1): 40-47. [Медлайн].
Eder J, Laccone F, Rohrbach M, Giunta C, Aumayr K, Reichel C, et al. Новая мутация COL3A1 при синдроме Элерса-Данлоса типа IV. Exp Dermatol . 2013 марта 22 (3): 231-4. [Медлайн].
Гали Н., Бейкер Д., Брэди А.Ф., Берроуз Н., Черви Е., Силлиерс Д. и др. Атипичные варианты COL3A1 (от глутаминовой кислоты до лизина) вызывают сосудистый синдром Элерса-Данлоса с постоянным фенотипом хрупкости тканей и повышенной эластичности кожи. Генет Мед . 2019 6 марта [Medline].
Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH. Нулевые аллели гена COL5A1 коллагена V типа являются причиной классических форм синдрома Элерса-Данло (типы I и II). Am J Hum Genet . 2000 июн.66 (6): 1757-65. [Медлайн].
Bouma P, Cabral WA, Cole WG, Marini JC. Мутация акцептора сплайсинга экзона 14 COL5A1 вызывает функциональный нулевой аллель, гаплонедостаточность альфа 1 (V) и аномальные гетеротипические интерстициальные фибриллы при синдроме Элерса-Данлоса II. Дж. Биол. Хим. . 2001, 20 апреля. 276 (16): 13356-64. [Медлайн].
Венструп Р.Дж., Флорер Дж.Б., Виллинг М.С., Джунта С., Штейнманн Б., Янг Ф. и др. Гаплонедостаточность COL5A1 — это общий молекулярный механизм, лежащий в основе классической формы EDS. Am J Hum Genet . 2000 июн.66 (6): 1766-76. [Медлайн].
Сан М., Конниццо Б.К., Адамс С.М., Фридман Б.Р., Венструп Р.Дж., Сословски Л.Дж. и др. Нацеленная делеция коллагена V в сухожилиях и связках приводит к классическому фенотипу суставов при синдроме Элерса-Данлоса. Ам Дж. Патол . 2015 май. 185 (5): 1436-47. [Медлайн].
Йеуэлл Х.Н., Уокер ЛК. Мутации в гене лизилгидроксилазы 1, которые приводят к дефициту фермента и клиническому фенотипу синдрома Элерса-Данлоса типа VI. Мол Генет Метаб . 2000 сен-окт. 71 (1-2): 212-24. [Медлайн].
Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, et al. Рецессивная форма синдрома Элерса-Данлоса, вызванная дефицитом тенасцина-X. N Engl J Med . 2001, 18 октября. 345 (16): 1167-75. [Медлайн].
Boudko SP, Engel J, Okuyama K, Mizuno K, Bächinger HP, Schumacher MA. Кристаллическая структура пептида, содержащего цистиновый узел коллагена III типа человека Gly991-Gly1032, демонстрирует тройную спиральную симметрию как 7/2, так и 10/3. Дж. Биол. Хим. . 2008 21 ноября. 283 (47): 32580-9. [Медлайн].
Окамото О., Андо Т., Ватанабэ А., Сато Ф., Мимата Х., Шимада Т. и др. Новая точечная мутация в гене коллагена III типа, приводящая к пропуску экзона 24 в случае синдрома Элерса-Данлоса сосудистого типа. Arch Dermatol Res . 2008 Октябрь 300 (9): 525-9. [Медлайн].
Viglio S, Zoppi N, Sangalli A, Gallanti A, Barlati S, Mottes M и др. Способность заживления ран в фибробластах EDS может быть восстановлена экзогенным коллагеном V типа. Научный Мир Журнала . 2008 г. 3 октября 8: 956-8. [Медлайн].
Кузьмина Н.С., Шипаева Е.В., Семячкина А.Н., Васильева И.М., Коваленко Л.П., Дурнев А.Д. и др. Полиморфизм генов детоксикации и устойчивость клеток к мутагенам у пациентов с синдромом Элерса-Данлоса. Бык Эксперимент Биол Мед . 2007 ноябрь 144 (5): 717-21. [Медлайн].
Мюррей М.Л., Ян М., Фаут С., Байерс PH. Синдром Элерса-Данлоса, связанный с FKBP14: расширение фенотипа с включением сосудистых осложнений. Am J Med Genet A . 2014 июл. 164A (7): 1750-5. [Медлайн].
Bethea BT, Fitton TP, Alejo DE, Barreiro CJ, Cattaneo SM, Dietz HC, et al. Результаты операций с сохранением аортального клапана: опыт процедур ремоделирования и реимплантации у 65 пациентов. Энн Торак Хирург . 2004 сентябрь 78 (3): 767-72; обсуждение 767-72. [Медлайн].
Ремвиг Л., Флихт Л., Кристенсен К. Б., Юул-Кристенсен Б. Отсутствие консенсуса по тестам и критериям общей гипермобильности суставов, синдром Элерса-Данлоса: гипермобильный тип и синдром гипермобильности суставов. Am J Med Genet A . 2014 Март 164A (3): 591-6. [Медлайн].
Castori M, Dordoni C, Morlino S, Sperduti I, Ritelli M, Valiante M и др.Спектр кожно-слизистых проявлений у 277 пациентов с синдромом гипермобильности суставов / синдромом Элерса-Данлоса по типу гипермобильности. Am J Med Genet C Semin Med Genet . 2015 Март 169С (1): 43-53. [Медлайн].
Шепер М.К., Николсон Л.Л., Адамс Р.Д., Тофтс Л., Пейси В. Естественное течение детей с синдромом гипермобильности суставов и типом гипермобильности Элерса-Данлоса: продольное когортное исследование. Ревматология (Оксфорд) . 18 апреля 2017 г. [Medline].
Ясин О.М., Рихани ФБ. Множественные аномалии развития зубов и синдром Элерса-Данлоса типа гипермобильности. J Clin Педиатр Дент . 2006 Лето. 30 (4): 337-41. [Медлайн].
Kapferer-Seebacher I, Oakley-Hannibal E, Lepperdinger U, Johnson D, Ghali N, Brady AF, et al. Проспективные клинические исследования детей с пародонтальным синдромом Элерса-Данлоса выявили генерализованное отсутствие прикрепленной десны как патогномоничный признак. Генет Мед . 2021, 23 февраля (2): 316-322. [Медлайн].
Кортес-Бретон Бринкманн Дж., Гарсиа-Хиль I, Лобато-Пенья Д.М., Мартинес-Мера С., Суарес-Гарсия М.Дж., Мартинес-Гонсалес Дж. М. и др. Ключевая роль практикующего стоматолога в ранней диагностике пародонтальных синдромов Элерса-Данлоса: редкий случай, связанный с братьями и сестрами. Квинтэссенция Инт . 2021. 52 (2): 166-174. [Медлайн].
van Bon AC, Kristinsson JO, van Krieken JH, Wanten GJ.Сопутствующий пелиоз селезенки и сосудистый синдром Элерса-Данлоса. Анн Васк Сург . 2009 марта, 23 (2): 256.e1-4. [Медлайн].
Гуала А., Виглио С., Оттинетти А., Анджели Г., Канова Г., Коломбо Е. и др. Кожная метапластическая синовиальная киста при синдроме Элерса-Данлоса: отчет о втором случае. Am J Dermatopathol . 2008 30 февраля (1): 59-61. [Медлайн].
Вилисаар Дж., Харикришнан С., Сури М., Константинеску Ч.С. Синдром Элерса-Данлоса и рассеянный склероз: возможная связь. Мультик Склер . 2008 май. 14 (4): 567-70. [Медлайн].
Перрино А., Матос М., Маруани А., Мондон К., Мачет Л. [Отсутствие нижней губной или язычной уздечки при синдроме Элерса-Данлоса: новый диагностический критерий?]. Энн Дерматол Венереол . 2007 ноябрь 134 (11): 859-62. [Медлайн].
Baba CS, Sharma PK, Deo V, Pal S, Sethuraman G, Gupta SD, et al. Ассоциация синдрома Элерса-Данлоса и синдрома солитарной язвы прямой кишки. Индийский J Гастроэнтерол . 2007 май-июнь. 26 (3): 149-50. [Медлайн].
Саваста С., Криспино М., Валли М., Каллигаро А., Замбеллони С., Поггиани С. Субэпендимальные перивентрикулярные гетеротопии у пациента с синдромом Элерса-Данло: новый случай. J Детский нейрол . 2007 марта 22 (3): 317-20. [Медлайн].
Майлз SC, Робинсон PD, Майлз JL. Синдром Элерса – Данлоса и нервная анорексия: опасное сочетание ?. Педиатр дерматол .2007 май-июнь. 24 (3): E1-4. [Медлайн].
Kundu AK, Chattopadhyay P, Kundu S, Choudhury S. Беременность при синдроме Элерса-Данлоса. J Assoc Physitors Индия . 2006 Декабрь 54: 938. [Медлайн].
Cortini F, Villa C, Marinelli B, Combi R, Pesatori AC, Bassotti A. Понимание основ синдрома Элерса-Данлоса в эпоху секвенирования следующего поколения. Arch Dermatol Res . 2 марта 2019 г. [Medline].
Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT.Поперечная и продольная оценка дилатации корня аорты и клапанных аномалий при гипермобильном и классическом синдроме Элерса-Данлоса. J Педиатр . 2011 Май. 158 (5): 826-830.e1. [Медлайн].
Дордони С., Рителли М., Вентурини М., Кьярелли Н., Пеццани Л., Васкелларо А. и др. Рецидивирующий и генерализованный висцероптоз при синдроме Элерса-Данлоса по типу гипермобильности. Am J Med Genet A . 2013 май. 161A (5): 1143-7. [Медлайн].
Hamonet C, Frédy D, Lefèvre JH, Bourgeois-Gironde S, Zeitoun JD.Повреждение мозга, раскрывающее синдромы Элерса-Данлоса после травмы: отпечаток волокна. Орфанет Дж. Редкий Диск . 2016 22 апреля, 11:45. [Медлайн].
Ayoub S, Ghali N, Angwin C, Baker D, Baffini S, Brady AF и др. Клинические особенности, молекулярные результаты и ведение 12 человек с редким синдромом Элерса-Данлоса артрохалазией. Am J Med Genet A . 2020 24 февраля. [Medline].
Мальфаит Ф., Франкомано С., Байерс П. и др.Международная классификация синдромов Элерса-Данлоса 2017 года. Am J Med Genet C Semin Med Genet . 2017 Март 175 (1): 8-26. [Медлайн].
Castori M, Tinkle B, Levy H, Grahame R, Malfait F, Hakim A. Основа для классификации суставной гипермобильности и связанных состояний. Am J Med Genet C Semin Med Genet . 2017 Март 175 (1): 148-157. [Медлайн].
Синибальди Л., Урсини Г., Кастори М. Психопатологические проявления гипермобильности суставов и синдром гипермобильности суставов / синдром Элерса-Данлоса, тип гипермобильности: пересмотрена связь между соединительной тканью и психологическим дистрессом. Am J Med Genet C Semin Med Genet . 2015 Март 169С (1): 97-106. [Медлайн].
Aalberts JJ, van den Berg MP, Bergman JE, du Marchie Sarvaas GJ, Post JG, van Unen H, et al. Многоликая агрессивная патология аорты: синдром Лойса-Дитца. Нет Харт J . 2008 Сентябрь 16 (9): 299-304. [Медлайн].
Синдром (ы) Кастори М. Элерса-Данлоса, имитирующий жестокое обращение с детьми: влияет ли это на клиническую практику ?. Am J Med Genet A .2016 5 мая. [Medline].
Ritelli M, Morlino S, Giacopuzzi E, Bernardini L, Torres B, Santoro G и др. Распознаваемое системное заболевание соединительной ткани с поликлапанной дистрофией сердца и дисморфизмом, связанным с мутациями TAB2. Clin Genet . 2018 Январь 93 (1): 126-133. [Медлайн].
Умекодзи А., Фукаи К., Хосоми Н., Исии М., Танака А., Мураками К. и др. Сосудистый тип синдрома Элерса-Данлоса, ассоциированный с легкой формой гемофилии A. Клин Эксп Дерматол . 2009 г., 34 (1): 101. [Медлайн].
Rombaut L, Malfait F, De Wandele I, Cools A, Thijs Y, De Paepe A, et al. Медикаментозное лечение, хирургия и физиотерапия у пациентов с синдромом Элерса-Данлоса с гипермобильностью. Arch Phys Med Rehabil . 2011 июл.92 (7): 1106-12. [Медлайн].
Китагава Т., Мацуи Н., Накайзуми Д. Программа структурированной реабилитации разнонаправленной нестабильности плеча у пациента с синдромом Элерса-Данлоса. Чехол Rep Orthop . 2020. 2020: 8507929. [Медлайн].
Reina-Bueno M, Vázquez-Bautista C, Palomo-Toucedo IC, Domínguez-Maldonado G, Castillo-López JM, Munuera-Martínez PV. Изготовленные на заказ ортезы для ног уменьшают боль и усталость у пациентов с синдромом Элерса-Данлоса. Пилотное исследование. Int J Environ Res Public Health . 2020 20 февраля. 17 (4): [Medline].
Alkadhi H, Wildermuth S, Desbiolles L, Schertler T, Crook D, Marincek B и др.Сосудистые неотложные состояния грудной клетки после тупой и ятрогенной травмы: многодетекторная рядная компьютерная томография и трехмерная визуализация. Рентгенография . 2004 сентябрь-октябрь. 24 (5): 1239-55. [Медлайн].
Monroe GR, Harakalova M, van der Crabben SN, Majoor-Krakauer D, Bertoli-Avella AM, Moll FL, et al. Семейный синдром Элерса-Данлоса с летальными артериальными событиями, вызванными мутацией в COL5A1. Am J Med Genet A . 2015 Июнь 167 (6): 1196-203. [Медлайн].
Fernández-Alcantud J, López RC, Osado IR, Castro MR.[Управление анестезией при синдроме Элерса-Данлоса сосудистого типа (тип IV)]. Rev Esp Anestesiol Reanim . 2008 май. 55 (5): 312-3. [Медлайн].
Jones TL, Ng C. Анестезия при кесаревом сечении у пациента с синдромом Элерса-Данлоса, связанным с синдромом постуральной ортостатической тахикардии. Int J Obstet Anesth . 2008 17 октября (4): 365-9. [Медлайн].
Faber P, Craig WL, Duncan JL, Holliday K. Успешное использование рекомбинантного фактора VIIa у пациента с синдромом Элерса-Данлоса сосудистого типа. Acta Anaesthesiol Scand . 2007 Октябрь, 51 (9): 1277-9. [Медлайн].
Nourissat G, Vigan M, Hamonet C, Doursounian L, Deranlot J. Диагностика синдрома Элерса-Данлоса после первого вывиха плеча. J Хирургическая установка для плечевого локтя . 2018 27 января (1): 65-69. [Медлайн].
.