описание, симптомы и особенности лечения
В современной медицине принято считать синдром Дьелафуа довольно редкой патологией. Как правило, она встречается у людей относительно молодого возраста, как женского, так и мужского пола. До сих пор идут споры в медицинском мире, есть ли взаимосвязь между образом жизни пациента, его питанием и врожденной слабостью мелких подслизистых артерий в кардиальном отделе желудка (которые и являются причиной развития симптомов Дьелафуа). Триада Дьелафуа — это основной критерий для диагностики данного заболевания. Медицина относит к этим критериям три симптома: боль, мышечное напряжение и гиперестезию кожи в правой подвздошной области.
Кто такой Дьелафуа и почему его именем назван синдром
Поль Жорж Дьелафуа — известный врач из Франции, который работал в Париже. За годы своей работы он совершил множество открытий, в частности, он занимался изучением артериовенозной мальформации. Говоря простым языком, этот процесс со временем приводит к развитию желудочных или кишечных кровотечений. Это очень опасное состояние, которое требует немедленного вмешательства профессиональных медиков. Благодаря исследованиям Поля Дьелафуа перед медициной приоткрылась дверь к знаниям, которые впоследствии помогли спасти жизнь сотням тысяч пациентов.
Это очень опасное состояние, которое требует немедленного вмешательства профессиональных медиков. Благодаря исследованиям Поля Дьелафуа перед медициной приоткрылась дверь к знаниям, которые впоследствии помогли спасти жизнь сотням тысяч пациентов.
В 1890 году Поль Дьелафуа стал членом Французской Почетной Медицинской Академии, спустя 11 лет он возглавил ее.
Синдром Дьелафуа был назван так, потому что именно этот человек внес основной вклад в его изучение, предложив теорию и найдя к ней доказательную базу. Синдром появляется вследствие аномального развития сосудов слоя желудка под слизистой, при этом параллельно диагностируется эрозия крупной артерии, в итоге наблюдается печальная клиническая картина: формирование язвы, которая приводит к обильному внутреннему кровотечению, которое, в свою очередь, заканчивается летальным исходом.
Болезнь Дьелафуа (Dieulafoys disease) была впервые описана на основе исследований Поля Дьелафуа в 1884 году. Подробное описание он сделал в 1898 году, выделив ее как отдельную нозологическую форму — «простое изъязвление».
Особенности синдрома в современной гастроэнтерологии
Рассказывая о синдроме Дьелафуа, следует подчеркнуть, что в гастроэнтерологии желудочное и кишечное кровотечение — это довольно распространенный случай, и ему могут предшествовать 144 синдрома и состояния. Болезнь (язва, эрозия, синдром — патологию называют по-разному, и любое из названий будет верным, это синонимы) Дьелафуа является по сути одним из многочисленных состояний-предшественников, которые со временем приводят к разрывам сосудов и последующему кровотечению в подслизистой оболочке желудка.
О самостоятельной диагностике в домашних условиях не может идти и речи! Язва Дьелафуа в практике хирургов является одним из самых сложных и опасных состояний. Если существует хотя бы малейшее подозрение на возможность развития желудочного кровотечения, следует как можно быстрее обеспечить возможность получения квалифицированной помощи медиков.
Как проявляет себя заболевание в начальной стадии
В 82-85 % всех случаев источник, который провоцирует развитие кровотечения, дислоцируется на расстоянии, равном примерно 5-6 сантиметров от пищеводно-желудочного соустья. Обычно (в большинстве случаев) на сравнительно малой кривизне желудка. В описании клинической картины пациентов с синдромом Дьелафуа часто фигурирует также патология пищевода, поражение тонкой и толстой кишки, в редких случаях – различного рода патологии желчного пузыря, нарушение оттока желчи, в некоторых случаях поражение прямой кишки.
Обычно (в большинстве случаев) на сравнительно малой кривизне желудка. В описании клинической картины пациентов с синдромом Дьелафуа часто фигурирует также патология пищевода, поражение тонкой и толстой кишки, в редких случаях – различного рода патологии желчного пузыря, нарушение оттока желчи, в некоторых случаях поражение прямой кишки.
Диагностика синдрома часто довольно затруднительна на ранних стадиях развития. В большинстве случаев заболевание никак себя не проявляет, у больного отсутствует большинство симптомов, которые характерны для гастроэнтерологических патологий. Пациент не жалуется на отрыжку, несварение, тошноту или расстройство пищеварения. Однако следует насторожиться, если иногда больной высказывает следующие жалобы:
- покалывание в области желудка;
- рвота с примесью крови;
- диарея с примесью сукровицы;
- частые рвотные позывы после приема пищи;
- невозможность сосредоточиться на чем-то кроме ощущения дискомфорта и ноющей боли в области желудка (это состояние носит постоянный и выматывающий характер).


Однако эти симптомы являются лишь общими, и нельзя с точностью утверждать, что даже при совокупности они могут сигнализировать о развитии синдрома Дьелафуа. МКБ 10 маркирует острую кровоточащую язву желудка (а это прямое последствие синдрома) кодом К25.0. Это состояние, которое требует немедленной госпитализации.
Стадии развития заболевания
Поскольку диагностика патологии довольно сложна даже для опытных медиков (до тех пор, пока не откроется внутреннее кровотечение), то и определить стадии тяжело. Тем не менее можно разделить патологию на три стадии развития:
- на первой стадии расширяются и истончаются сосуды подслизистого слоя желудка;
- на второй начинается небольшое кровотечение, которое пациент чаще всего оставляет безо всякого внимания;
- на третьей стадии развивается сильное кровотечение, которое угрожает жизни пациента – требуется срочное хирургическое вмешательство.
Основные симптомы заболевания
Как правило, пациент с язвой анастомоза (синдром Делафуа) попадает на операционный стол внезапно даже для самого себя. Язва образуется и начинает кровоточить уже на последних стадиях болезни. До этого пациент может не испытывать каких-либо проблем со здоровьем и отрицать у себя наличие патологии.
Язва образуется и начинает кровоточить уже на последних стадиях болезни. До этого пациент может не испытывать каких-либо проблем со здоровьем и отрицать у себя наличие патологии.
Когда начинается кровотечение, симптомы таковы:
- сильная слабость, отсутствие работоспособности;
- рвотные массы с кровью;
- тошнота и рвотные позывы;
- жжение в области солнечного сплетения;
- коллапс;
- гипотензия;
- стул приобретает дегтеобразную консистенцию и цвет;
- наблюдаются общие симптомы сильной кровопотери при болезни Дьелафуа.
Голубой пузырчатый невус — это кожное образование, которое свойственно людям с наличием венозной слабости. У таких людей риск развития синдрома теоретически выше, чем у тех, у кого отсутствуют подобного рода высыпания на коже. Если пациент обнаружил у себя совокупность перечисленных выше симптомов и у него есть голубой невус — это повод обязательно пройти обследование ЭГДС.
Методы гастроэнтерологической диагностики
Выявить проблемы с сосудами можно, если регулярно обследоваться у хороших специалистов. После этого пациенту назначают курс сосудоукрепляющих препаратов, но и это не гарантия того, что у человека не разовьется последняя стадия (кровотечение) синдрома Дьелафуа. Случаи из практики показывают, что для сосудов и артерий подслизистого слоя желудка прием венотоников практически бесполезен, а стоимость их довольно высока для среднестатистического гражданина.
После этого пациенту назначают курс сосудоукрепляющих препаратов, но и это не гарантия того, что у человека не разовьется последняя стадия (кровотечение) синдрома Дьелафуа. Случаи из практики показывают, что для сосудов и артерий подслизистого слоя желудка прием венотоников практически бесполезен, а стоимость их довольно высока для среднестатистического гражданина.
Если ФГДС проводит опытный и грамотный специалист, то он может разглядеть эрозированную артерию среди нормальной окрашенной слизистой желудка. Во время активного кровотечения можно обнаружить столбик артериальной крови.
На фоне развития синдрома общий анализ крови может показать нормохромную анемию.
Причины развития заболевания
Почему развивается синдром Дьелафуа? Эндоскопическая картина (при проведении исследования ЭГДС) показывает чаще всего наличие эрозированных артерий, однако часто они настолько незначительны или их настолько мало (для развития кровотечения обычно достаточно одной или нескольких), что врач просто может их не заметить.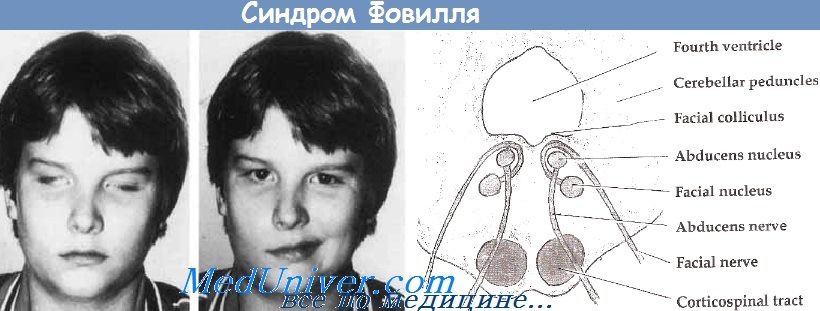 Особенно это касается медиков-новичков в гастроэнтерологии.
Особенно это касается медиков-новичков в гастроэнтерологии.
Каковы причины того, что артерии и сосуды в подслизистом слое желудка истончаются и на них образуются эрозии? Мнение официальной медицины таково, что причина кроется либо в наследственном факторе, либо в привычке пациента злоупотреблять алкогольными напитками на фоне неправильного питания. На сегодняшний день эти две причины признаны основными при развитии синдрома Дьелафуа. Если человек подозревает, что по наследственной линии ему может достаться этот недуг, то следует регулярно проходить обследование ЭГДС у опытного гастроэнтеролога, а также сдавать общий и биохимический анализ крови хотя бы раз в год.
Синдром Дьелафуа — что делать?
Если гастроэнтеролог сообщил о возможном развитии кровотечения из-за синдрома, то следует попросить его выписать препараты для профилактики. Некоторые врачи прописывают венотоники, прием которых на регулярной основе способствует укреплению стеной и артерий. Другие врачи считают, что только строгая диета и здоровый образ жизни способен предотвратить развитие язвы и кровотечения.
В первую очередь не стоит паниковать. Просто следует регулярно проходить обследование, прислушиваться к своему состоянию и полностью отказаться от вредных привычек.
Помогут ли лекарственные препараты при патологии
На сегодняшний день не существует такого лекарства, которое бы смогло с вероятностью 100 % предотвратить развитие язвы и последующего кровотечения.
Лучший «препарат» для этого — отсутствие хронического стресса, здоровое питание, глубокий сон, отказ от употребления даже незначительных доз алкогольных напитков. Прием венотоников может лишь частично снизить риск развития язвы.
Последствия желудочного кровотечения
Самое серьезное последствие — это летальный исход от кровопотери. Первое время кровь будет выделяться в небольших количествах и попросту перевариваться. Внимательные пациенты могут заметить, что со стулом происходит что-то странное, но редко кто придает этому значение.
После того как эрозии увеличиваются в размере, можно утверждать, что это терминальная стадия. Это состояние угрожает жизни и должно быть купировано в условиях стационара хирургическим путем.
Догоспитальный этап терапии
Первая помощь заключается в том, чтобы оказать влияние на уменьшение кровотечения. Для этого могут быть назначены препараты, которые способны уменьшить свертываемость крови. Они, как правило, вводятся парентерально. Точный диагноз можно поставить только в условиях стационара. Если нет возможности доставить больного в стационар, следует ввести 100 мл 6 % раствора эпсилонаминокапроновой кислоты.
Иногда с целью замедлить течение болезни применяют кальция хлорид, хотя нет доказательств того, что он может облегчить состояние хотя бы на время при синдроме Дьелафуа.
На живот больному следует положить что-то холодное, например, бутылку со льдом или влажное полотенце. Больной должен находиться в полном покое. Любые нагрузки пациенту запрещены. Если есть подозрение на внутреннее кровотечение, то следует отказаться от приема любой пищи до того момента, пока не будет проведено обследование и поставлен точный диагноз.
Язва Делафуа: современные методы лечения
После того как была точно установлена причина недомогания и ею оказалась язва Дьелафуа, пациенту следует согласиться на хирургическое вмешательство. Консервативных методов лечения кровотечения нет, и промедление может закончиться летальным исходом.
Как правило, пациент поступает в отделение в состоянии геморрагического шока. Даже опытный врач не сразу может понять, что является источником кровотечения. Поэтому перед проведением операции могут потребоваться дополнительные исследования. Иногда кровотечение сначала останавливают эндоскопическими методами.
Предполагаемое место кровотечения промывают охлажденной водой, после чего обнаруживается поврежденный сосуд, выступающий над слизистой оболочкой. Поврежденный сосуд коагулируют. Остановки крови можно добиться наложением клипс, для этой манипуляции используют вращающийся клипсаппликатор.
Взаимосвязь алкоголизма и появления заболевания
Регулярное злоупотребление спиртными напитками приносит организму большое количество хронических заболеваний. Желудок, его слизистая и подслизистый слой страдают ничуть не меньше, чем печень, которая вынуждена перерабатывать продукты распада этилового спирта.
Исследования доказали, что среди людей, которые страдают от хронического алкоголизма, процент смертности от желудочных кровотечений в несколько раз выше, чем среди людей, которые придерживаются трезвого и здорового образа жизни. Курение также крайне отрицательно влияет на состояние сосудов и артерий во всем организме.
Синдром Алажиля — причины, симптомы, диагностика и лечение
Синдром Алажиля — это редкое генетическое заболевание, при котором нарушения гепатобилиарной и сердечно-сосудистой системы сочетаются с аномалиями развития скелета. Характерные симптомы проявляются у ребенка с первых месяцев жизни и включают желтуху, кожный зуд, нутритивную недостаточность, отставание темпов психомоторного развития. Для диагностики синдрома проводят УЗИ и биопсию печени, эхокардиографию, биохимическое и генетическое тестирование. Лечение болезни Алажиля состоит из диетотерапии, приема желчегонных и антигистаминных медикаментов. По показаниям назначается хирургическое вмешательство.
Общие сведения
Синдром Алажиля (артериопеченочная дисплазия) занимает второе место (17%) среди всех заболеваний гепатобилиарного тракта у детей в первые 6 месяцев после рождения. Он выявляется с частотой 1 случай на 70 тыс. новорожденных. Патология получила свое название по имени французского педиатра Даниеля Алажиля, который подробно описал ее в 1975 году. В медицинской литературе болезнь также известна как артериопеченочная дисплазия. Полиморфизм симптоматики, сложность диагностики и серьезный прогноз обуславливают значимость синдрома в неонатологии и детской гастроэнтерологии.
Синдром Алажиля
Причины
Синдром Алажиля — наследственное заболевание, которое передается по аутосомно-доминантному типу. Он вызван мутацией на коротком плече 20 хромосомы, где расположен ген Jagged 1 (JAG1). Учитывая тип наследования, патология с одинаковой частотой встречается у мальчиков и девочек, может повторяться в каждом поколении семьи. Описаны случаи развития синдрома у младенцев с трисомией по 21 хромосоме (болезнью Дауна).
Патогенез
Ведущим патогенетическим компонентом является сужение внутрипеченочных желчных протоков, затрудняющее отток желчи с ее последующим накоплением в гепатоцитах. Желчные кислоты оказывают токсическое влияние на печеночные клетки, вызывая нарушение их функций и гибель, а также проникают в системный кровоток, провоцируя кожные признаки синдрома. Вследствие недостаточного поступления желчи в кишечник возникает мальабсорбция.
В механизме развития симптоматики важную роль играют сердечные пороки, которые сопровождаются нарушениями кровообращения и гипоксией тканей. Нарушения гемодинамики связаны с отсутствием выброса крови из правого желудочка и гипотрофией миокарда. При несвоевременной диагностики происходит дилатация правого предсердия и желудочка. При тяжелой форме синдрома может быть вторичное открытие овального окна и венозно-артериальный сброс крови.
Симптомы
Для классического синдрома Алажиля характерно сочетание 5 признаков: холестаз, стеноз легочной артерии или другие кардиальные пороки, нарушения формирования лицевого скелета, поражения глаз, незаращение тел позвонков. Для постановки диагноза достаточно наличия 3-х симптомов из этого списка. Во всех случаях развивается поражение печени, а на втором месте по частоте стоят сердечно-сосудистые патологии (до 87-95%).
Клинические признаки синдрома проявляются в первые 3 месяца жизни младенца, при менее грубых аномалиях начало болезни приходится на второе полугодие жизни. Первым симптомом является умеренная желтушность кожи, иногда с зеленоватым оттенком. Она дополняется мучительным кожным зудом, из-за чего младенец становится беспокойным, постоянно плачет, плохо спит.
Расстройства продукции и выделения желчи сопровождаются диспепсическими нарушениями, обесцвечиванием кала, но этот признак появляется намного позже желтухи. Нарушения липидного обмена могут спровоцировать появление ксантом — доброкачественных образований желтоватого цвета, которые возвышаются над уровнем кожи. В основном они локализованы на пальцах, по задней поверхности шеи, в паховой и подколенной зонах.
Для больных, страдающих синдромом Алажиля, специфичны особенности внешности, обусловленные лицевым дизморфизмом. Родители и врачи обращают внимание на слишком широкий лоб, глубоко посаженные и широко расставленные глаза, длинный прямой нос. У ребенка могут быть оттопыренные и низко расположенные уши, выступающий подбородок. Вследствие офтальмопатии у младенца возникают затруднения с узнаванием родителей, он не интересуется яркими игрушками.
Осложнения
Обострения холестаза при синдроме Алажиля зачастую сопровождаются инфекцией, что провоцирует холецистохолангит, осложняющий течение болезни. Повреждение печени желчными кислотами и хроническое воспаление приводят к перипортальному фиброзу, а у 15% детей уже в раннем возрасте формируется цирроз печени, что является прогностически неблагоприятным признаком.
На фоне хронической мальабсорбции и мальдигестии возникают белково-энергетическая недостаточность и авитаминозы. Тяжелая задержка роста и физического развития отмечаются у 50-87% детей. Также появляются сердечно-сосудистые осложнения: без своевременной коррекции кардиальный порок быстро прогрессирует. 6-летняя выживаемость пациентов с внутрисердечными поражениями составляет до 40%, а без таковых — повышается до 95%.
Диагностика
Опытный неонатолог или педиатр может заподозрить синдром Алажиля по клиническим проявлениям, но при умеренной выраженности симптомов и их позднем развитии постановка диагноза сопровождается трудностями. При физикальном обследовании врач выявляет гепатомегалию, сердечный горб, грубый систолический шум во 2 межреберье. Для подтверждения синдрома проводятся инструментальные и лабораторные исследования:
- УЗИ органов брюшной полости. Скрининговое неинвазивное исследование выполняется для визуализации печени и определения степени гепатомегалии. Сонография необходима, чтобы исключить другие причины желтухи, оценить состояние гепатобилиарной системы и желудочно-кишечного тракта ребенка.
- Эхокардиография. При визуализации сердца врач определяет сужение легочной артерии на уровне клапанов или подклапанного пространства, гипертрофию правого желудочка, дилатацию правых камер сердца. При необходимости производится рентгенологическая диагностика, которая показывает выступающую дугу легочной артерии, расширение границ сердца.
- Биопсия печени. Снижение числа внутрипеченочных протоков — патогномоничный признак синдрома Алажиля, но он может не обнаруживаться в младенческом возрасте. Биопсия рекомендована для исключения других гепатобилиарных патологий, если детский гастроэнтеролог испытывает трудности с дифференциальной диагностикой.
- Анализ крови. При биохимическом исследовании определяют повышение типичных маркеров холестаза — холестерина, щелочной фосфатазы, прямого билирубина. У ребенка наблюдается дислипопротеинемия. Ферменты цитолиза в печеночных пробах повышаются умеренно. Отклонения в протеинограмме у большинства пациентов отсутствуют.
- Генетическое тестирование. Молекулярная диагностика мутаций гена JAG1 требуется при мягких признаках синдрома или его атипичном протекании. При прямом секвенировании характерные генные нарушения удается обнаружить у 95% детей, но оно делается только на научно-исследовательской основе.
Лечение синдрома Алажиля
Консервативная терапия
Для коррекции нутритивной недостаточности ребенку назначают усиленное лечебное питание с ограничением длинноцепочечных жиров и повышением процента среднецепочечных, которые лучше усваиваются при дефиците желчи. В ежедневном рационе повышают содержание жирорастворимых витаминов. При выраженных расстройствах питания специальные составы вводятся через назогастральный зонд. Медикаментозное лечение включает:
- Желчегонные препараты. Необходимы для стимуляции оттока желчи из печеночных протоков, что способствует облегчению желтухи, улучшению пищеварения и устранения дискомфорта. Хороший эффект на гепатобилиарную систему оказывает урсодезоксихолевая кислота (УДХК).
- Антигистаминные средства. Показаны для облегчения мучительного кожного зуда у детей, однако они не всегда эффективны. При рефрактерных случаях терапию дополняют седативными средствами, транквилизаторами.
- Витамины и минералы. Для нормализации системы свертываемости крови эффективен витамин К в пероральной или парентеральной форме. Для улучшения кальциево-фосфорного обмена применяются препараты холекальциферола, а при снижении плотности костной ткани используют добавки с кальцием.
Хирургическое лечение
При кардиальных аномалиях выполняются малоинвазивные методы коррекции — вальвулопластика, баллонное расширение просвета легочной артерии и ее стентирование. При тяжелых комбинированных пороках рекомендованы открытые кардиохирургические операции. При циррозе, печеночной недостаточности, хронической портальной гипертензии рассматривается вопрос о трансплантации печени.
Прогноз и профилактика
Успех лечения зависит от степени выраженности синдрома: при незначительной артериопеченочной дисплазии удается компенсировать состояние и обеспечить нормальное развитие ребенка, а тяжелые формы болезни Алажиля коррелируют с необратимыми повреждениями печени, кардиоваскулярными расстройствами. Профилактика патологии заключается в медико-генетическом консультировании пары на этапе планировании беременности.
Синдром Прадера-Вилли — причины, симптомы, диагностика и лечение
Синдром Прадера-Вилли – это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 – 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.
Синдром Прадера-Вилли
Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% – наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом – полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек – недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Диагностика
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения – ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза – показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации – основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития – синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция – увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии – бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия – прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина – карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике – предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
Фовилля синдром | Саратовский неврологический портал
Achille Louis François Foville (1831-1877)
Син.: дорсокаудальный синдром покрышки моста; hemiplegia abducento-facialis alternans.
Один из понтинных альтернирующих синдромов ствола мозга. Возникает при поражении корешков и ядер VI и VII краниальных нервов, заднего продольного пучка и пирамидных путей, иногда – с вовлечением медиальной петли. Очаг при этом располагается в нижнем отделе моста по всей сагиттальной оси.
Поперечный срез ствола мозга на уровне моста мозга. Заштрихована область поражения при синдроме Фовилля (источник: www.medsch.wisc.edu/anatomy/bs97/text/bs/contents.htm, в собственной модификации)
Субстрат поражения и клинические симптомы:
Вовлекаемая структура | Ипсилатерально | Контрлатерально |
| Ядро отводящего нерва (VI) | Парез m.rectus lateralis:
| |
| Волокна или ядро лицевого нерва (VII) | Периферический парез/паралич мимической мускулатуры (никогда при этом не бывает нарушения вкуса) | |
| Задний продольный пучок | «Мостовой» парез взора: парез взора в сторону очага («больной смотрит на парализованные конечности»)* | |
| Пирамидные пути | Гемипарез или гемиплегия (чаще без вовлечения мимической мускулатуры), в том числе поражение XII краниального нерва по центральному типу | |
| Медиальная петля | Гемигипестезия поверхностной и/или глубокой чувствительности, чаще без вовлечения лица |
* При очагах возле шва (билатеральных и вблизи IV желудочка) развивается горизонтальный парез взора (возможны движения глаз только вверх-вниз, а также конвергенция).
Наиболее частой причиной развития синдрома является ишемический инфаркт (тромбоз) в бассейне ветвей основной артерии, реже – опухоли, воспалительные и демиелинизирующие процессы.
Возможны различные варианты сочетания синдрома Фовилля с синдромом Вебера (например, сочетанное поражение III, IV и VII нервов на стороне очага).
Описан французским неврологом, анатомом и физиологом Акиль Луи Франсуа Фовиллем, сыном невролога и психиатра Акиль Луи Фовилля, в 1858г. (Foville A.L.F. Note sur une paralysie peu connue de certains muscles de l’oeil, et la liaison avec quelques points de l’anatomie et la physiologie de la protubérance annulaire // Bulletins de la Société anatomique de Paris (1826), 1858. – Vol.33. – P.393-414). Кстати, вариантом фамилии А.Л.Ф.Фовилля был «Дефовилль» (Defoville), напр., как в его докторской диссертации, написанной в 1857г. и посвящённой эпилепсии.
Альтерниющие синдромы. Причины. Симптомы. Диагностика
А. Синдромы повреждений продолговатого мозга
Повреждения проводников на этом уровне ствола мозга может приводить к моноплегии, гемиплегии, альтернирующей (перекрёстной) гемиплегии и разнообразным сенсорным расстройствам. Может иметь место нижняя параплегия или децеребрационная ригидность. Вовлечение эфферентных волокон от обоих ядер блуждающего нерва или их компрессия могут вести к глубоким нарушениям кардиальных и респираторных функций, артериального давления и смерти.
Повреждения продолговатого мозга может быть острым, подострым или хроническим и иметь многообразную этиологию. Это могут быть опухоли, туберкулёма, саркоидоз, сосудистые повреждения (геморрагии, тромбоз, эмболия, аневризмы, мальформации), полиоэнцефалит, полиомиелит, рассеянный склероз, сирингобульбия, прогрессирующий бульбарный паралич (БАС), врождённые аномалии, инфекционные, токсические и дегенеративные процессы. Экстрамедуллярные синдромы могут быть вызваны травмой, переломами костей основания черепа, нарушениями развития скелета, острыми и хроническими воспалениями оболочек и внезапным повышением внутричерепного давления, приводящего к ущемлению продолговатого мозга в большом затылочном отверстии. К близкой картине может привести опухоль мозжечка.
I. Медиальный медуллярный синдром (передний бульбарный синдром Дежерина (Dejerine)
- Ипсилатеральный парез, атрофия и фибрилляции языка (обусловленные поражением XII нерва). Девиация языка в сторону очага. Редко функция XII нерва может быть сохранной.
- Контралатеральная гемиплегия (обусловленная вовлечением пирамиды) с сохранными функциями мышц лица.
- Контралатеральное снижение мышечно-суставной и вибрационной чувствительности (обусловленные вовлечением медиальной петли). Так как остаётся невовлечённым спиноталамический тракт, расположенный более дорсолатерально), болевая и температурная чувствительность сохранны.
Если очаг повреждения распространяется дорзально, затрагивая медиальный продольный пучок, может появиться нистагм «бьющий вверх». Иногда медиальный медуллярный синдром развивается с двух сторон (билатерально), приводя к квадриплегии (с сохранными функциями VII нерва), двусторонней плегии языка и снижению мышечно-суставной и вибрационной чувствительности во всех четырёх конечностях.
Синдром обусловлен окклюзией передней спинальной артерии или позвоночной артерии. Передняя спинальная артерия снабжает ипсилатеральную пирамиду, медиальную петлю и XII нерв с его ядром.
Вовлечение передней спинальной артерии или травма могут иногда приводить к перекрёстной гемиплегии (синдром перекреста пирамид) с контралатеральным спастическим парезом ноги и ипсилатеральным спастическим парезом руки. При этом наблюдается также вялый парез и атрофия ипсилатеральных кивательной и трапецевидной мышцы и, иногда, ипсилатеральной половины языка. Более экстенсивные повреждения над перекрестом может приводить к спастической тетраплегии.
Вариантом медиального медуллярного синдрома является синдром Авеллиса.
Медиальный медуллярный инфаркт трудно диагностировать без МРТ.
II. Латеральный медуллярный синдром Валленберга (Wallenberg) — Захарченко.
- Ипсилатеральное снижение болевой и температурной чувствительности на лице (в связи с вовлечением nucleus tractus spinalis digemini). Иногда наблюдается ипсилатеральная лицевая боль.
- Контралатеральное снижение болевой и температурной чувствительности на туловище и конечностях, обусловленное повреждением спиноталамического тракта.
- Ипсилатеральный паралич мягкого нёба, глотки и голосовой связки с дисфагией и дизартрией в связи с вовлечением nucleus ambiguus.
- Ипсилатеральный синдром Горнера (обусловленный вовлечением нисходящих симпатических волокон).
- Головокружение, тошнота и рвота (обсловленные вовлечением вестибулярных ядер).
- Ипсилатеральные мозжечковые знаки (в связи с вовлечением нижней ножки мозжечка и частично самого мозжечка).
- Иногда икота и диплопия (последняя наблюдается в случае вовлечения нижних отделов варолиева моста).
Синдром обусловлен повреждением латеральной медуллярной области и нижних отделов мозжечка. Он чаще всего развивается при окклюзии интракраниальной части позвоночной артерии или задней нижней мозжечковой артерии. Другие причины: спонтанное расслоение позвоночной артерии, злоупотребление кокаином, медуллярные опухоли (обычно метастазы), абсцессы, демиелинизирующие заболевания, радиационные повреждения, гематома (при разрыве сосудистой мальформации), манипуляции во время манульной терапии, травма.
При данном синдроме описаны также разнообразные нарушения движений глаз и зрения: косая девиация (обусловлена подъёмом контралатерального глазного яблока), ипсилатеральный наклон головы с торзией глазных яблок (ocular tilt реакция) с жалобами на двоение или наклон видимых окружающих предметов, различные виды нистагма, «нистагм век» и другие окулярные феномены.
К вариантам этого синдрома некоторые исследователи относят синдром Сестана-Шене (Chestan-Chenais) и синдром Бабинского-Нажотта (strongabinski-Nageotte) в виде комбинированного медиального и латерального инфаркта.
В то же время такие симптомокомплексы, как синдром Джексона и синдром Шмидта (как и синдромы Тэпиа, Берне, Вилларе, Колле-Сикара и другие) относят преимущественно к «невральным» синдромам (синдромам поражения краниальных нервов), при которых вовлечение вещества мозга наблюдается редко.
Что касается альтернирующего синдрома Авеллиса (Avellis), проявляющекгося поражением X пары (ипсилатеральный паралич мягкого нёба и голосовой связки), а также спиноталамического тракта и нисходящих окулосимпатических волокон (контралатеральная гемианестезия и ипсилатеральный синдром Горнера), то он относится, по-видимому, к таким раритетам, что в последнее время перестал упоминаться в неврологических монографиях и руководствах.
III. Гемимедуллярный синдром.
Редко может наблюдаться комбинированный синдром (медиальный и латеральный медуллярные синдромы (гемимедуллярный синдром), обычно обусловленный окклюзией интракраниальной позвоночной артерии.
В целом клиническая картина медуллярных инфарктов очень гетерогенна и зависит от степени распространения ишемии в продолговатом мозге; иногда они распространяется нижние отделы варолиева моста, верхние отделы спинного мозга и мозжечок. Кроме того они могут быть односторонними и двусторонними.
Повреждения каудальных отделов мозгового ствола может приводить к нейрогенному отёку лёгких.
IV. Латеральный понто-медуллярный синдром.
В этом случае наблюдается клиническая картина латерального медуллярного синдрома плюс несколько мостовых симптомов, включающих:
Ипсилатеральную слабость мимических мышц (обусловленную вовлечением VII нерва)
Ипсилатеральный шум в ухе и иногда нарушение слуха (в связи с вовлечением VIII нерва).
[7], [8], [9], [10], [11], [12], [13], [14]
В. Синдромы повреждений варолиевого моста (понтинные синдромы).
I. Вентральные понтинные синдромы.
- Мийара-Гублера (Millard-Gubler) синдром обусловлен очагом поражения в нижнем отделе моста (чаще инфаркт или опухоль). Ипсилатеральный периферический парез мимических мышц (VII краниальный нерв). Контралатеральная гемиплегия.
- Раймона (Raymond) синдром обусловлен теми же процессами. Ипсилатеральный парез m. rectus lateralis (VI краниальный нерв). Парез взора в сторону очага. Контралатеральная гемиплегия, обусловленная вовлечением пирамидного тракта.
- «Чистый» (моторный) гемипарез. Локальные повреждения в области основания варолиева моста (особенно лакунарные инфаркты), затрагивающие кортикоспинальныи тракт, могут вызвать чистый двигательный гемипарез. (Другие локализации повреждений, способных вызывать такую картину, включают: заднее бедро внутренней капсулы, ножки мозга и медуллярные пирамиды.)
- Синдром дизартрии и неловкой руки.
Локальные повреждения в основании моста (особенно лакунарные инфаркты) на границе верхней трети и нижних двух третей моста могут вызвать такой синдром. При этом синдроме слабость мимических мышц и тяжёлая дизартрия и дисфагия развиваются вместе с парезом руки, на стороне которого может быть гиперрефлексия и симптом Бабинского (при сохранной чувствительности).
(Аналогичная картина может наблюдаться при повреждениях колена внутренней капсулы или при маленьких глубоких геморра-гиях в мозжечке).
- Атактический гемипарез.
Локальные повреждения в основании моста (чаще — лакунарные инфаркты) той же локализации могут приводить к контралатеральной гемиатаксии и парезу ноги (иногда выявляется дизартрия, нистагм и парестезии) на той же стороне тела.
(Этот синдром описан также при таламокапсулярных повреждениях, процессах в области заднего бедра внутренней капсулы, красного ядра и при поверхностных инфарктах в парацентральной области.)
- Синдром «запертого человека» (Locked-in syndrome).
Билатеральное повреждение вентральных отделов моста (инфаркт, опухоль, кровоизлияние, травма, центральный понтинный миелинолиз, реже — другие причины) могут приводить к развитию этого синдрома (состояние деэфферентации). Клинические проявления включают следующие проявления:
Квадриплегия, обусловленная билатеральным вовлечением кортикоспинальных трактов в основании моста. Афония, обусловленная вовлечением кортикобульбарных волокон, идущих к ядрам нижних краниальных нервов. Иногда нарушение горизонтальных движений глазных яблок в связи с вовлечением корешков VI краниального нерва. Так как ретикулярная формация ствола мозга при этом синдроме не повреждается, больные находятся в состоянии бодрствования. Вертикальные движения глаз и моргания интактны.
Состояние деэфферентации наблюдается также при чисто периферических повреждениях (полиомиелит, полинейропатия, миастения).
II. Дорзальные понтинные синдромы
Фовилля (Foville) синдром обусловлен повреждениями в дорзальных отделах покрышки каудальной трети моста: Контралатеральная гемиплегия (гемипарез).
Ипсилатеральный периферический паралич мимических мышц (корешок или/и ядро VII нерва). Неспособность содружественно перемещать глаза в ипсилатеральную сторону из-за вовлечения парамедианной ретикулярной формации моста или ядра VI (abducens) нерва, либо того и другого.
Раймон-Сестана (Raymond-Cestan) синдром наблюдается при ростральных повреждениях дорзальных отделов моста. При этом синдроме наблюдается:
Мозжечковая атаксия с грубым «рубральным» тремором, обусловленным вовлечением верхней ножки мозжечка.
Контралатеральное снижение всех видов чувствительности за счёт вовлечения медиальной петли и спиноталамического тракта.
При вентральном распространении очага поражения может иметь место конралатеральный гемипарез (вовлечение кортикоспинального тракта) или паралич взора в сторону очага (вовлечение парамедианной ретикулярной формации моста).
III. Парамедианный понтинный синдром
Парамедианный понтинный синдром может быть представлен несколькими клиническими синдромами:
- Унилатеральный медио-базальный инфаркт: грубый фацио-брахио-круральный гемипарез, дизартрия и гомолатеральная илибилатеральная атаксия.
- Унилатеральный медио-латеральный базальный инфаркт:лёгкий гемипарез с атаксией и дизартрией, атактический гемипарез или синдром дизартрии — неловкой руки.
- Унилатеральный медио-центральный или медио-тегментальный инфаркт: синдром дизартрии — неловкой руки; атактический гемипарез с сенсорными нарушениями или нарушениямидвижений глаз; гемипарез с контралатеральным параличом мимических мышц или m. rectus lateralis (VII или VI нервы).
- Билатеральный центро-базальный инфаркт: у этих больныхразвивается псевдобульбарный паралич и билатеральные сенсо-моторные расстройства.
Самыми частыми причинами парамедианных понтинных инфарктов являются лакунарные инфаркты, вертебрально-базилярная недостаточность с инфарктами, кардиогенная эмболия.
IV. Латеральные понтинные синдромы
Мари-Фуа (Marie-Foix) синдром развивается при латеральных повреждениях моста, особенно если поражаются средние ножки мозжечка, и включает в себя:
Ипсилатеральную мозжечковую атаксию, обусловленную вовлечением связей с мозжечком. Контралатеральный гемипарез (вовлечение кориткоспинального тракта).
Вариабельную контралатеральную гемигипестезию болевой и температурной чувствительности, обусловленную вовлечением спиноталамического тракта.
С. Синдром универсальной диссоциированной анестезии
Универсальная диссоциативная анестезия является редким синдромом, описанным у больных с комбинированной окклюзией правой верхней мозжечковой артерии и левой задней нижней мозжечковой артерии. Поражение первой артерии приводит к латеральному верхнему инфаркту моста, поражение второй артерии — к левостороннему латеральному медуллярному синдрому Валленберга-Захарченко. Больной имеет снижение болевой и температурной чувствительности на лице, шее, туловище и на всех конечностях, в то время, как тактильная, вибрационная и мышечно-суставная чувствительность сохранны (диссоциированное снижение чувствительности).
Геморрагические повреждения моста сопровождаются нарушением сознания, комой и имеют несколько иную клинику.
[15], [16], [17], [18], [19], [20], [21]
Д. Синдромы повреждения мезенцефалона
I. Вентральный синдром корешка III краниального нерва Вебера (Weber).
Повреждения в ножке мозга, затрагивающие волокна пирамидного тракта и корешок III нерва, проявляются следующей картиной: Контралатеральная гемиплегия. Ипсилатеральный паралич мышц, иннервируемых Ш нервом.
II. Дорзальный синдром корешка III краниального нерва Бенедикта (Вenedikt)
Вызывается повреждением покрышки мезенцефалона с вовлечением красного ядра, верхних ножек мозжечка и корешка III краниального нерва:
Ипсилатеральный паралич мышц, иннервируемых III нервом.
Контралатеральные непроизвольные движения, включая интенционный тремор, гемихорею, гемибаллизм, обусловленные вовлечением красного ядра.
Аналогичные клинические проявления развиваются при более дорзальном повреждении покрышки среднего мозга, которое захватывает дорзальные части красного ядра и верхние ножки мозжечка и носит название синдрома Клода (Claude), в котором преобладают мозжечковые (контралатеральная гемиатаксия, гипотония) симптомы и нет гемибаллизма.
III. Дорзальный мезенцефальный синдром
Проявляется главным образом нейроофтальмологическими феноменами. Дорзальный мезенцефальный синдром (синдром силь-виева водопровода или синдром Парино (Parinaud) чаще всего выявляется на фоне гидроцефалии или опухоли гипофизарной области и включает в себя все следующие (или некоторые из них) знаки:
- Паралич взора вверх (иногда вниз).
- Зрачковые нарушения (обычно широкие зрачки с диссоциацией реакции на свет и на аккомодацию с конвергенцией).
- Конвергирующий и ретракторный нистагм при взгляде вверх.
- Патологическая ретракция век.
- Отставание век.
IV. Верхний базилярный синдром
Вызывается окклюзией ростральных отделов основной артерии (обычно вследствие эмболии), приводя к инфаркту среднего мозга, таламуса, и частично височных и затылочных долей. Синдром описан также у больных с гигантской аневризмой этой части основной артерии, при её васкулите и после церебральной ангиографии. Вариирующие проявления этого синдрома включают в себя:
- Расстройства движений глаз (унилатеральный или билатеральный паралич взора вверх или вниз, нарушения конвергенции, паралич псевдо-абдуценс, конвергирующий и ретракторный нистагм, нарушения отведения глазных яблок, отставание и ретракция верхних век, косая девиация).
- Зрачковые нарушения.
- Нарушения поведения (гиперсомния, педункулярный галлюциноз, нарушения памяти, делирий).
- Нарушения зрения (гемианопсия, корковая слепота, синдром Балинта (Вalint).
- Моторный и сенсорный дефицит.
[22], [23], [24], [25], [26], [27], [28], [29], [30]
Фовилля синдром — это… Что такое Фовилля синдром?
- Фовилля синдром
понтинный альтернирующий синдром: сочетание паралича или пареза мышц, иннервируемых лицевым и отводящим нервами, на стороне очага поражения моста мозга (варолиева моста) с гемиплегией или гемипарезом на противоположной стороне.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг.
- ФОВ
- Фовеа́льный
Смотреть что такое «Фовилля синдром» в других словарях:
Фовилля синдром — (A. L. F. Foville, 1799 1878, франц. невропатолог и психиатр) понтинный альтернирующий синдром: сочетание паралича или пареза мышц, иннервируемых лицевым и отводящим нервами, на стороне очага поражения моста мозга (варолиева моста) с гемиплегией… … Большой медицинский словарь
синдром варолиева моста — (syndromum pontis Varolii; син. синдром моста мозга) сочетание альтернирующего синдрома Мийяра Гюблера, Бриссо Сикара, Фовилля или Раймона Сестана с признаками псевдобульбарного паралича, нистагмом, иногда расширением зрачков; наблюдается при… … Большой медицинский словарь
Синдром отводящего (VI черепного) нерва — Нерв двигательный и обеспечивает иннервацию только прямой наружной мышцы глаза на своей стороне. Его поражение ведет к ограничению движения глазного яблока кнаружи. При этом возникает диплопия (двоение), особенно выраженная при попытке повернуть… … Энциклопедический словарь по психологии и педагогике
Синдром Фовилля–Вильсона — Слабость прямых наружных мышц глаз, диссоциированный нистагм одного глаза на стороне, в которую больной пытается повернуть взор. Описали как проявление рассеянного склероза французский невропатолог Foville (1799–1879) и английский невропатолог… … Энциклопедический словарь по психологии и педагогике
ФОВИЛЛЯ – ВИЛЬСОНА СИНДРОМ — (описан как проявление рассеянного склероза A. L. F. Foville, позднее – британским неврологом S. Wilson, 1878–1937) – слабость латеральных прямых мышц глаза, диссоциированный нистагм одного глаза на строне, в которую больной пытается повернуть… … Энциклопедический словарь по психологии и педагогике
МКБ-10: Класс VI — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
МКБ-10: Класс G — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
МКБ-10: Код G — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
Альтерни́рующие синдро́мы — симптомокомплексы, характеризующиеся сочетанием поражения черепных нервов на стороне очага с проводниковыми нарушениями движения и чувствительности на противоположной стороне. Возникают при поражении одной половины ствола головного мозга,… … Медицинская энциклопедия
АЛЬТЕРНИРУЮЩИЕ СИНДРОМЫ — АЛЬТЕРНИРУЮЩИЕ СИНДРОМЫ, представляют собой такую совокупность нервно патологических явлений выпадения функций, когда одна их часть внешне выражена на одной половине тела (наприм., в форме правостороннего паралича или пареза конечностей), а… … Большая медицинская энциклопедия
Лицевой нерв — Лицевой нерв … Википедия
Синдром Вильямса: особенности, причины и лечение
Синдром Вильямса — это редкое генетическое нарушение нервного развития, характеризующееся легкими проблемами в обучении или развитии, высоким уровнем кальция в крови и моче и заметно общительным характером.
Люди с синдромом Вильямса (WS) часто имеют необычную «эльфийскую» внешность с низкой переносицей. Уникальные черты личности включают высокий уровень коммуникабельности и очень хорошие коммуникативные навыки.
Высокий уровень вербальных навыков может маскировать другие проблемы развития и иногда способствовать поздней диагностике.
Проблемы, с которыми сталкивается человек с WS, включают трудности с пониманием пространственных отношений, абстрактных рассуждений и чисел, а также некоторые потенциально опасные для жизни осложнения, такие как сердечно-сосудистые проблемы и высокий уровень кальция в крови.
Синдром Вильямса (WS или WMS), или синдром Вильямса-Бёрена (WBS), возникает из-за того, что примерно 26 генов удалены из хромосомы 7.
По данным Национальной организации по редким заболеваниям (NORD), WS присутствует примерно в 10 000 и 20 000 младенцев, рожденных в Соединенных Штатах.
Люди с этим заболеванием часто нуждаются в постоянном уходе.
Поделиться в PinterestЛюди с синдромом Вильямса, как правило, обладают хорошими социальными и коммуникативными навыками.
С WS связан ряд функций, но не все с этим условием будут обладать всеми этими функциями.
Характеристики лица включают небольшой вздернутый нос, длинную верхнюю губу, широкий рот, маленький подбородок, отечность вокруг глаз и полные губы. Вокруг радужки может появиться белый кружевной узор.У взрослых может быть удлиненное лицо и шея.
Проблемы с сердцем и кровеносными сосудами может означать сужение кровеносных сосудов, включая аорту или легочные артерии. Может потребоваться операция.
Гипертония или высокое кровяное давление со временем может стать проблемой. Пациенту потребуется регулярное наблюдение.
Гиперкальциемия или высокий уровень кальция в крови может вызывать симптомы колик и раздражительность у младенцев.
Признаки и симптомы обычно уменьшаются по мере того, как ребенок становится старше, но могут быть пожизненные проблемы с уровнем кальция и метаболизмом витамина D, и могут потребоваться лекарства или специальная диета.
Режимы сна день-ночь могут занять больше времени.
Аномалии соединительной ткани увеличивают риск грыжи и проблем с суставами, мягкая дряблая кожа и хриплый голос.
Проблемы со скелетно-мышечной системой могут влиять на кости и мышцы. Суставы могут быть слабыми, а мышечный тонус может быть низким в раннем возрасте. Могут развиться контрактуры или скованность суставов.
Физиотерапия может помочь улучшить мышечный тонус, диапазон движений и силу суставов.
Проблемы с кормлением могут включать в себя тяжелый рвотный рефлекс, плохой мышечный тонус, трудности с сосанием и глотанием и тактильную защиту. Эти проблемы имеют тенденцию уменьшаться со временем.
Низкий вес при рождении может привести к диагнозу «неспособность к развитию». Врач может быть обеспокоен тем, что младенец недостаточно быстро набирает вес. У большинства взрослых с WS рост меньше среднего.
Когнитивные особенности и особенности развития могут включать от легкой до тяжелой неспособности к обучению и когнитивных проблем.Могут быть трудности с пространственными отношениями и мелкой моторикой. Задержки в развитии являются обычным явлением, и часто требуется больше времени, чем обычно, чтобы научиться ходить, разговаривать или научиться пользоваться туалетом.
Проблемы с почками немного чаще встречаются у людей с WS.
Зубья могут иметь необычный вид: широкие, немного маленькие, с более широким интервалом, чем обычно. Могут быть аномалии прикуса или совмещения верхних и нижних зубов, например, при жевании или прикусывании.
Речевые, социальные и музыкальные навыки
Речь, социальные навыки и долговременная память обычно хорошо развиты.
Личностные черты включают высокий уровень выразительных языковых навыков и желание общаться, особенно со взрослыми. Большинство детей с WS не боятся незнакомцев.
Д-р Коллин А. Моррис в книге Gene Reviews перечисляет следующие черты личности как типичные для WM: «Чрезмерное дружелюбие, сочувствие, общая тревожность, специфические фобии и синдром дефицита внимания.”
Чувствительный слух может вызывать болезненные или неприятные ощущения на определенных уровнях или частотах, но это также может быть связано с особой любовью к музыке.
Исследование, проведенное доктором Урсулой Белуджи из Института биологических наук Солка в Ла-Хойя, Калифорния, исследовало эту близость к музыке.
Исследователи отмечают, что многие дети с WS любят слушать и сочинять музыку, и у них часто есть хорошая память на песни и чувство ритма. Их слух достаточно острый, чтобы различать пылесосы разных производителей.
WM — это генетическое заболевание, при котором у человека отсутствуют 25 генов.
Один из них — ген, вырабатывающий белок эластин.
Эластин придает эластичность или эластичность кровеносным сосудам и другим тканям тела. Возможно, этот недостаток вызывает сужение кровеносных сосудов, делает кожу эластичной, а суставы гибкими.
Хотя WM является генетическим заболеванием, в большинстве случаев оно возникает случайно, но если оно есть у одного из родителей, вероятность передачи его ребенку составляет 50 процентов.
Врач изучит клинические особенности, включая черты лица и сердечно-сосудистые симптомы. Однако многие из функций не являются исключительными для WS, и не у всех людей с этим заболеванием будут одинаковые симптомы.
Можно провести анализ крови для проверки на высокий уровень кальция.
Доступен ряд генетических тестов. Более 99 процентов случаев можно диагностировать с помощью флуоресцентной гибридизации in situ (FISH) или тестирования делеции / дупликации.
Для снижения или ограничения уровня кальция может быть рекомендована специальная диета с низким содержанием витамина D. Согласно NORD, уровень кальция обычно возвращается к норме через 12 месяцев, даже без лечения.
Детям с WM не рекомендуется принимать добавки витамина D, чтобы избежать повышения уровня кальция.
Ортодонт может помочь с проблемами зубов.
Лечение включает поддерживающее вмешательство при нарушениях развития.
Может быть:
- Специальное образование и профессиональная подготовка
- Речевая и языковая, физическая, профессиональная, кормление и терапия сенсорной интеграции
- Поведенческое консультирование может сопровождаться психологическим и психиатрическим осмотром
- При дефиците внимания доступны лекарства расстройство (СДВ) и тревога
Вмешательство специалиста может потребоваться при определенных проблемах, таких как сердечно-сосудистые симптомы.
Генетическое консультирование доступно для пациента и его семьи.
У некоторых людей с WS будет нормальная продолжительность жизни, но проблемы со здоровьем могут означать, что у некоторых продолжительность жизни короче, чем обычно.
Примерно 3 из 4 человек с WS будут иметь определенную степень умственной отсталости, и большинству из них потребуется постоянный уход. Некоторые люди могут выполнять регулярную оплачиваемую работу.
В 2016 году исследователи, изучающие неврологические особенности WS, предположили, что особые структуры нейронов у людей с WS могут приводить к надсоциальному аспекту состояния.
Авторы пришли к выводу, что исследования WS могут помочь ученым понять, что делает людей социальными существами.
Что нужно знать
Что такое синдром Фрея?
Синдром Фрея — это синдром, который включает потоотделение во время еды (вкусовое потоотделение) и покраснение лица. Это вызвано повреждением нерва, называемого ушно-височным нервом, обычно после хирургической травмы околоушной железы. Этот нерв, когда он заживает, присоединяется к потовым железам вместо исходной слюнной железы (которая была удалена во время операции).
Это означает, что когда у вас должно выделяться слюноотделение, вы вместо этого потеете. Покраснение и потоотделение появляются, когда пострадавший ест, видит, мечтает, думает или говорит о продуктах, вызывающих сильное слюноотделение. У пациента покраснение и потливость в области висков, щек и верхней части шеи.
Синдром Фрея :
Синдром Фрея Анимация :
Насколько распространен синдром Фрея после операции?
Синдром Фрея, как полагают, встречается у всех пациентов, перенесших операцию на околоушной железе без реконструктивной хирургии.Симптомы могут различаться по степени тяжести, и обычно за лечением обращаются только пациенты с тяжелыми симптомами.
Опасен ли синдром Фрея?
Хотя синдром Фрея не причиняет значительного вреда, он может быть очень неудобным и неприятным для больного.
Как диагностируется синдром Фрея?
Диагноз синдрома Фрея обычно так же прост, как поговорить с пациентом и осмотреть его. Дополнительный тест, который можно использовать для определения контуров лица, называется второстепенным тестом на йод и крахмал.
В этом тесте йод наносится на сторону лица с симптомами. После высыхания наносится кукурузный крахмал. Когда пациент потеет (с пищевым раздражителем), пораженный участок темнеет.
Крахмал-йодный тест для диагностики синдрома Фрея
Получу ли я синдром Фрея после операции на околоушной железе?
У всех пациентов, перенесших операцию на слюнных железах без реконструкции, в той или иной степени будет наблюдаться вкусовое потоотделение. Степень выраженности симптомов зависит от:
- размер опухоли
- количество удаленной околоушной ткани (например,грамм. поверхностная паротидэктомия, глубокая паротидэктомия, тотальная паротидэктомия, удаление подчелюстной железы)
- длина разреза
- протяженность области рассечения
- реконструкция после удаления опухоли
Проще говоря, чем больше расслоение, тем выше вероятность и серьезность Синдром Фрея в послеоперационном периоде. Ограничение рассечения — первый шаг к успешной операции.
Однако самым важным фактором является реконструкция. Если у вашего хирурга нет опыта в профилактической реконструкции паротидэктомии, ваш риск синдрома Фрея значительно возрастет.
Какие у меня варианты лечения?
Для пациентов с более тяжелыми и беспокоящими симптомами существует несколько вариантов лечения.
Лечебные процедуры включают:
- Антихолинергические мази для местного применения (скополамин, гликопиролат)
- Антиперспиранты для местного применения (дезодорант)
- Местные альфа-агонисты (клонидин)
- Инъекции ботулотоксина
Самый простой и безопасный метод применения ботулотоксина.Он обеспечивает наиболее длительный период облегчения симптомов с наименьшими осложнениями. Однако ни одно из этих методов лечения не дает окончательного излечения; облегчение только временное.
Единственным вариантом постоянного лечения является реконструктивная хирургия. В опытных руках хирургия имеет дополнительное преимущество, так как позволяет уменьшить лицевые шрамы от разрезов и исправить деформации лица после первоначальной операции. Однако большинство хирургов не способны выполнить настоящую реконструкцию, которая одновременно предотвращает синдром Фрея и лечит деформацию лица в результате паротидэктомии.
Как проводится операция?
(1) возвышение кожи
Операция начинается с осторожного подъема кожи. Это самая важная часть, потому что необходимо соблюдать осторожность, чтобы не обнажить ветви лицевого нерва. Лицевой нерв отвечает за все движения лица. Непреднамеренное повреждение этого нерва грозит необратимым параличом.
Чтобы сделать эту процедуру безопасной, мы разработали специальные инструменты, которые повышают безопасность подъема кожи по сравнению с местом предыдущей операции.
Рис. 1. Кожа приподнята, чтобы обнажить околоушный дефект, оставшийся после предыдущей операции. Несоответствующие нервные волокна расположены внутри обозначенной области.
(2) Закройте дефект
Затем ткань собирают и ее размер выбирают для покрытия дефектной области. Это кожно-фасциальный трансплантат. Трансплантат тщательно пришивается к лицевым мышцам, что восстанавливает естественную мышечную поддержку лица. Трансплантат выполняет две задачи: изолирует потовые железы, вылечивает пациента с синдромом Фрея и поддерживает мягкие ткани щеки.
Рис. 2. Фасция / дермальный трансплантат взят и наложен шов, чтобы закрыть дефект.
(3) Наконец, любая дальнейшая реконструкция для исправления косметической деформации завершается и процедура завершается.
Рис. 3. Внешний вид трансплантата до исправления околоушного дефекта и окончательного закрытия. Обратите внимание, что трансплантат полностью покрывает область предыдущей операции на околоушной железе.
Как я могу избежать синдрома Фрея, если у меня операция на слюнной железе?
Самый эффективный способ избежать синдрома Фрея — минимизировать хирургическую травму.Единственный способ сделать это — сделать малоинвазивную операцию. Минимально инвазивная хирургия затрагивает только околоушную железу. Он не распространяется на щеки, виски и шею, как при традиционной хирургии околоушных желез.
Минимально инвазивная хирургия может быть выполнена с помощью:
После паротидэктомии необходимо восстановить барьер для предотвращения контакта слюнных нервов и потовых желез друг с другом. Если этот барьер создан, риск синдрома Фрея практически устранен.Однако это не часть большинства традиционных операций на околоушной железе.
Ключевые слова:
Вкусовые — вкусовые и / или связанные с дегустацией
Вкусное потоотделение — повышенное потоотделение в ответ на прием пищи
Люция Фрей — одна из первых женщин-неврологов в Европе, классифицировавших вкусовое потоотделение
Аурикулотемпоральный нерв — ветвь тройничного нерва, которая переносит как потовые (симпатические) волокна к потовым железам кожи головы, так и волокна слюноотделения (парасимпатические) к околоушной железе
Что такое синдром Драве?
Создание протокола чрезвычайной ситуации
Важно разработать и внедрить протокол неотложной помощи с вашим неврологом, включая бензодиазепины быстрого действия, такие как диастат, назальный препарат или лоразепам, для любого судорожного припадка продолжительностью более пяти минут.В случае крайней необходимости полезно иметь при себе письменную копию этого протокола. Этот протокол должен включать инструкции по лечению припадков и тому, когда следует вызывать экстренные службы, а также контактную информацию родителей и врача.
Мониторинг и лечение вторичных заболеваний
Помните о вторичных состояниях здоровья, общих для синдрома, чтобы правильно с ними справиться. Эти условия варьируются от пациента к пациенту и могут включать:
- Сердечно-сосудистые заболевания
- Проблемы со здоровьем зубов
- Дисавтономия
- Ортопедия и сколиоз
- Нарушения сна
- Ослабленный иммунитет
Также следует внимательно следить за ростом и весом, и родители должны знать о вариантах лечения, таких как гастростомические трубки (g-tube), когда это необходимо.
Научитесь и избегайте триггеров приступов
По возможности избегайте триггеров захвата. Общие триггеры для пациентов с синдромом Драве включают быстрые изменения окружающей среды и / или температуры тела, болезнь, стресс, перевозбуждение, паттерны и мерцающий свет. Лихорадку всегда следует лечить агрессивно, согласно плану, согласованному с вашим педиатром и / или неврологом.
Другие проблемы
Дети с синдромом Драве также часто сталкиваются с проблемами развития, такими как аутизм или аутистические характеристики, когнитивные и / или коммуникативные задержки, социальные навыки и поведенческие проблемы.Регулярные оценки развития и ранние и агрессивные методы лечения (речевые, OT, PT, развитие и т. Д.) Могут помочь в достижении общего результата для ребенка.
Оперативное управление
Дети с синдромом Драве обычно нуждаются в постоянном уходе и наблюдении, а также в помощи, чтобы избежать провоцирующих судорог. Оборудование, которое семьи сочли полезным для повседневного лечения синдрома Драве, включает видеонаблюдение, защитные шлемы, охлаждающие жилеты, пульсоксиметры, сигнализаторы припадков и очки с цветными линзами (для светочувствительности).
C как семейство
Хроническое заболевание ребенка будет иметь прямое и косвенное влияние на членов семьи и их отношения. Члены семьи нередко чувствуют отрицание, гнев, страх, шок, замешательство, самообвинение и беспомощность. Консультации по вопросам семьи и горя могут помочь семьям справиться с рождением ребенка с хроническим заболеванием.
— СИНДРОМ РОТУЛИЕНА
VOUS AVEZ UN
СИНДРОМ РОТУЛИЕНА?
QU’EST-CE QUE C’EST?
Il s’agit d’un fonctionnement anomal de votre rotule, petit os situé en avant de votre genou.Cette rotule coulisse mal dans son rail. Cela provoque un frottement anormal, à l’origine d’une
воспаление и рост хряща. Le cartilage étant la peinture qui recouvre la rotule et lui permet de glisser contre le fémur.
QUELS SONT LES SIGNES DE CE
«СИНДРОМ РОТУЛИЕНА»?
Les douleurs:
— lorsque vous êtes en position assise longée (en voiture, au cinéma par instance), votre genou s’engourdit dans une sensation d’étau.Vous sentez le besoin d’allonger la jambe. Ou de faire «
craquer votre genou ».
[pourquoi? parce que la zone enflammée de votre rotule ne support pas d’être plaquée en permanence contre le fémur. Elle vous declare alors à bouger, à changer de position pour ne plus appuyer sur
cette зона. Un peu com si quelqu’un posait son doigt sur un «bleu», un hematome, et que vous repoussiez ce doigt pour éviter la douleur.]
-dans les escaliers, notamment en Потомок, votre genou vous gène, ou flanche.Cela est parfois notable dans une randonnée en montagne, lors de la descente.
[pourquoi? parce qu’en position de descente, la rotule se plaque fortement contre le fémur pour raidir votre members inférieur et vous empêcher de tomber. Si votre rotule est enflammée par le
синдром Rotulien, elle sera d’autant plus douloureuse]
Подписчиков:
— les craquements fréquents du genou.
— L’impression que la rotule sort de son rail et se réemboîte aussitôt.
— Une sensation de crissement à l’intérieur du genou
— une sensation de lourdeur, d’échauffement du genou.
QUELLES SONT LES CAUSES DE MON SYNDROME ROTULIEN?
Il s’agit d’une forme anormale du genou, et / ou d’un glissement anormal de la rotule sur son femoral.
Normalement, lorsqu’on est debout pieds суставы, les deux genoux se touchent.
— chez 1% des gens, les genoux sont trop écartés: c’est le «genu varum». Les rotules ne glissent plus sur un rail bien droit. En pliant le genou, elles ont tendant à sortir du rail, com un
тренироваться в ун вираж.Cela engendre un frottement sur l’extérieur de la rotule qui s’use et s’enflamme.
— парфюм, плюс редкость, les genoux se touchent mais les pieds restent écartés: c’est «genu valgum», qui engendre aussi un frottement anormal.
— parfois, on a une jambe plus longue que l’autre. Cette «inégalité de longueur desmbres inférieurs» provoque aussi un dérèglement de la mécanique de glissement de la rotule.
Parfois enfin, les Muscle s’insèrent sur la rotule et la font glisser sont déséquilibrés.
-l’excès de poids: les kilos superflus appuient sur la rotule et la rendent plus sensible.
QUEL TRAITEMENT VOUS EST PROPOSE PAR LA
CLINIQUE DU GENOU?
Il s’agit d’un traitement médical. Хирургия бесполезна при вращающемся синдроме. Ce traitement comporte quatre points:
.
1 — ОРТОПЕДИЯ LES SEMELLES
Elles vont compenser une inégalité de longueur des deux members, compenser un désordre de la voûte plantaire, ré axer au mieux la rotule dans son rail, qui coulissera ainsi de manière harmonyuse.Elles amortissent en plus les chocs transmis par les pieds vers les genoux, reposant ainsi le cartilage enflammé.
Ces semelles devront être portées dans toutes les chaussures, de ville et de sport. Au mieux pour toujours. Ces semelles soulageront vos genoux, mais aussi vos hanches et votre columnsne
позвоночник.
2 — ПРЕОБРАЗОВАНИЕ ЛА
Est essentielle, minutieuse et longée. Une fois ou deux par semaine. Elle consiste en un ensemble d’exercices spécialisés, qui ont pour but de ré axer la rotule dans son rail (en rééquilibrant
les muscle qui la font travailler), et de décoller cette rotule pour diminuer son frottement (par des étirements notamment).Elle aussi un rôle antalgique au début, par l’action anti-firematoire
de la Physérapie (application d’ondes, de froid и т. д.).
3 — LA GENOUILLERE
Avec son orifice entouré de silicones, la genouillère Недавнее la rotule et l’empêche de sortir de son rail lors des activités sportives. Une journée de travail difficile, ou une longue marche
peut aussi justifier le port de cette genouillère. Elle ne doit en tout cas pas être portée en permanence, et jamais la nuit.
4 — ЛЕС МЕДИКАМЕНТЫ
Aucun médicament ni Complément alimentaire ne peut reconstituer du cartilage neuf. Определенные стабилизации Les dégâts, ce qui est déjà considérable, notamment pour les douleurs. В doit les
Prendre quasiment toute l’année, et ils mettent souvent plusieurs mois avant d’agir.
ART 50, CHONDROSULF 400, PIASCLEDINE 300 уже давно, плюс предварительные заявки.
Actuellement le Complément alimentaire de loin le plus efficace est CHONDRALINE (2 gélules chaque matin toute l’année).Ла
витамин D, содержание в CHONDRALINE, ассоциированный с гиалуроновой кислотой, глюкозамин и хондроитин, состав и состав натурального коктейля, который не вызывает никаких сомнений.
темп. C’est le seul dans son жанра.
LES ANTI INFLAMMATOIRES ne sont justifiés qu’en cas de douleur notable et très peu de temps. On s’en méfit à cause de leur effet néfaste sur l’estomac et les reins.
QUELS SPORTS POUVEZ VOUS PRATIQUER AVEC
CE СИНДРОМ РОТУЛИЕН?
Лучший спорт в NATATION.Mais pas la brasse, qui fait sortir les rotules. Le CRAWL (avec les battements de jambe) — это спортивная идея.
Les kinésithérapeutes sont des entraîneurs sportifs: ils vous guideront pour la reprise des autres sports.
Д’ОТРЕ СОВЕТАЕТ ПО ВОПРОСАМ, УЖЕ СИНДРОМ РОТУЛИЕН?
МАЙГРИР?
Les kilos superflus augmentent considérablement le travail de vos rotules. Un kilo de trop, c’est huit kilos de plus pour votre rotule, notamment dans les descentes d’escalier…
LES RANDONNEES EN MONTAGNE? Elles sont redoutables pour vos rotules: mettez les genouillères, prenez un bâton de randonneur (à tenir dans la
main opposée au genou douloureux), limit au maximum le poids de votre sac à dos.
Y A-T-IL UNE ПРЕДНАЗНАЧЕНИЕ ГЕНЕТИКА ИЗ СЕМЬИ A CE СИНДРОМ РОТУЛИЕН?
Oui. La Forme globale des genoux est souvent génétiquement transmise. Une bonne precaution pour vos enfants en fin de croissance sera de leur faire porter des semelles s’ils ont aussi un «genu varum.
»(Voir plus haut). Плюс, определенное поражение хряща плюс хрупкий que d’autres, predisposant à son usure плюс rapide.
QUELLE EST L’EVOLUTION
СИНДРОМ Д’УНА РОТУЛИЕН
МАЛ ТРАЙТ?
Les douleurs persistent.Les Frottement Anormal de la Rotule Finit avec les années par user le cartilage qui la protège: c’est l’arthrose du genou. D’où l’importance du traitement médical bien
канал.
QUE FAIRE EN CAS D’ECHEC DU TRAITEMENT MEDICAL?
Le traitement a t’il été réellement
bien suivi, des semelles faites par un podo orthésiste compétent, et réellement portées; Une rééducation bien suivie par un
kinésithérapeute atttif. Et les séances longées par votre médecin généraliste le temps nécessaire; le médicament suivi correctement (Chondraline n’agit qu’au bout de trois mois! il faut en
faire une cure de six mois минимум, voire toute l’année); les genouillères portées pour les moment difficiles.Les kilos superflus perdus?
Si les douleurs s’étaient bien améliorées mais reviennent quelques années après?
Avez-vous refait des semelles neuves régulièrement? N’avez-vous pas pris quelques kilos? Ils vaut mieux consulter à nouveau: de nouvelles séances de reéducation seront peut être nécessaires,
voire une nouvelles cure de Chondraline.
Si ce traitement a été bien suivi et que vos douleurs persistent, il faut reconsulter.
Dans de rares cas, on pourra envisager unevention visant à limiter le frottement de votre rotule (Libération et Recentrage de la Rotule sous arthroscopie par section de l’aileron rotulien
внешний).
Симптомы, причины, тесты и лечение
Что такое синдром Сусака?
Синдром Сусака — редкое аутоиммунное заболевание. «Аутоиммунный» означает, что иммунная система человека по ошибке атакует собственные ткани человека. При синдроме Сусака иммунная система атакует мельчайшие кровеносные сосуды головного мозга, сетчатки (части глаза) и внутреннего уха. Кровеносные сосуды блокируются. Проблемы с мозгом, сетчаткой и внутренним ухом возникают из-за снижения кровотока.
Кто страдает синдромом Сусака?
Синдром Сусака встречается редко, но когда он возникает, он поражает главным образом женщин в возрасте от 20 до 40 лет. Однако могут быть затронуты дети в возрасте от 8 лет и взрослые (как мужчины, так и женщины) до 50 лет.
Каковы симптомы синдрома Сусака?
Симптомы со стороны мозга:
- Сильная головная боль, часто со рвотой
- Проблемы с мышлением, такие как кратковременная потеря памяти, спутанность сознания, медленная обработка мыслей и снижение способности решать проблемы
- Неспособность сохранять бдительность или сосредоточенность
- Невнятная речь
- Изменения личности
- Психические проблемы, такие как депрессия, психоз, агрессия, беспокойство или ломка
Глазные симптомы:
- Темная зона в одной части поля зрения
- Расстройство зрения (описывается как «как будто темная тень или занавеска закрывает часть моего зрения.»)
- Потеря периферического (бокового) зрения
Симптомы внутреннего уха:
- Потеря слуха
- Головокружение (вертиго)
- Звон в ушах (тиннитус)
Все три части болезни могут не проявляться одновременно. Любой из вышеперечисленных симптомов может быть первым признаком синдрома Сусака. На проявление всех трех частей могут уйти недели, месяцы или даже годы, а у некоторых пациентов их никогда не бывает больше двух.
Что вызывает синдром Сусака?
Синдром Сусака вызывается собственной иммунной системой человека, атакующей эндотелиальные клетки — клетки, выстилающие внутренние стенки наших кровеносных сосудов — в головном мозге, сетчатке и внутреннем ухе.При атаке эндотелиальные клетки набухают и частично или полностью перекрывают кровоток по сосуду. Из-за нехватки кислорода и питательных веществ страдают пораженные органы.
Что вызывает сбой в работе иммунной системы, неизвестно.
Далее: Диагностика и тесты
Последний раз проверял медицинский работник Cleveland Clinic 26.05.2017.
Список литературы
- Susac, JO. Редакция: Синдром Сусака. Am J Neuroradiology 2004 25: 351-352.
- Информационный центр по генетическим и редким заболеваниям (GARD). Синдром Сусака Доступно 26.05.2017.
- Rennebohm R, Susac JO, Egan RA, Daroff RB. Синдром Сусака — обновленная информация. J Neurol Sci 2010 15 декабря; 299 (1-2): 86-91. Epub 19 сентября 2010 г. PMID: 20855088 [PubMed — проиндексировано для MEDLINE]
- Rennebohm RM, Egan RA, Susac JO.Лечение синдрома Сусака. Варианты лечения Curr Neurol. 2008 Янв; 10 (1): 67-74. PMID: 18325301
- Susac JO, Egan RA, Rennebohm RM, Lubow M. Susac. Синдром: 1975-2005 микроангиопатия / аутоиммунная эндотелиопатия. J Neurol Sci 2007, 15 июня; 257 (1-2): 270-2. Epub 2007 28 февраля. Обзор. PMID: 17331544 [PubMed — проиндексировано для MEDLINE] [PubMed]
- Rennebohm RM, Susac JO. Лечение синдрома Сусака. J Neurol Sci 2007, 15 июня; 257 (1-2): 215-20. Epub 2007 26 февраля. Обзор. PMID: 17324441 [PubMed — проиндексировано для MEDLINE]
- Rennebohm RM, Lubow M, Rusin J, Martin L, Grzybowski DM, Susac JO.Агрессивное иммуносупрессивное лечение синдрома Сусака у подростков: на примере лечения дерматомиозита. Pediatr Rheumatol Online J 2008, 29 января; 6: 3. PMID: 18230188 [PubMed]
Получите полезную, полезную и актуальную информацию о здоровье и благополучии
е Новости
Клиника Кливленда — некоммерческий академический медицинский центр.Реклама на нашем сайте помогает поддерживать нашу миссию. Мы не поддерживаем продукты или услуги, не принадлежащие Cleveland Clinic.