НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ И СИНДРОМЫ, СОПРОВОЖДАЮЩИЕСЯ ФЕБРИЛЬНЫМИ СУДОРОГАМИ: КЛИНИКО-ГЕНЕТИЧЕСКИЕ ХАРАКТЕРИСТИКИ И СПОСОБЫ ДИАГНОСТИКИ | Дадали
1. Мухин К.Ю., Петрухин А.С. Идиопатические формы эпилепсии: систематика, диагностика, терапия. М.: Арт-Бизнес-Центр, 2000. 319 с. [Mukhin K. Yu., Petrukhin А.S. Idiopathic forms of epilepsy: systematics, diagnosis, therapy. Мoscow: Art-Biznes-Tsentr, 2000. 319 p. (In Russ.)].
2. Мухин К.Ю., Петрухин А.С., Миронов М.Б. Эпилептические синдромы. Диагностика и терапия (справочное руководство для врачей). М.: Системные решения, 2008. 224 с. [Mukhin K.Yu., Petrukhin A.S., Mironov M.B. Epileptic syndromes. Diagnosis and therapy (reference manual). Moscow: Systemnye resheniya, 2008. 224 p. (In Russ.)].
3. Петрухин А.С., Мухин К.Ю., Благосклонова Н.К., Алиханов А.А. Эпилептология детского возраста. М.: Медицина, 2000. 623 с. [Petrukhin A.S., Mukhin K.Yu., Blagosklonova N.K., Alikhanov A.A. Childhood epileptology. Moscow: Meditsina, 2000. 623 p. (In Russ.)].
4. Темин П.А., Никанорова М.Ю. Эпилепсии и судорожные синдромы у детей. Руководство для врачей. М.: Медицина, 1999. 656 с. [Temin P.A., Nikanorova M.Yu. Epilepsy and convulsive syndromes in children. Guidelines for doctors. Moscow: Meditsina, 1999. 656 p. (In Russ.)].
5. Angelman H. Puppet children: a report of three cases. Dev Med Child Neurol 1965;7:681–8.
6. Baram T.Z., Shinnar Sh. Febrile seizures. Orlando: Academic Press, 2002. P. 337.
7. Bonanni P., Malcarne M., Moro F. et al. Generalized epilepsy with febrile seizures plus (GEFS+): clinical spectrum in seven italian families unrelated to SCN1A, SCN1B, and GABRG2 gene mutations. Epilepsia 2004;45(2):149–58.
Bonanni P., Malcarne M., Moro F. et al. Generalized epilepsy with febrile seizures plus (GEFS+): clinical spectrum in seven italian families unrelated to SCN1A, SCN1B, and GABRG2 gene mutations. Epilepsia 2004;45(2):149–58.
8. Capovilla G., Wolf P., Beccaria F., Avanzini G. The history of the concept of epileptic encephalopathy. Epilepsia 2013;54 Suppl 8:2–5. DOI: 10.1111/epi.12416.
9. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978:8:543–8.
10. El Achkar C.M., Olson H.E., Poduri A., Pearl P.L. The genetics of the epilepsies. Curr Neurol Neurosci Rep 2015;15(7):39. DOI: 10.1007/s11910-015-0559-8.
11. Escayg A., MacDonald B.T., Meisler M.H. et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24(4):343–5. DOI: 10.1038/74159.
12. Fiumara A., Pittalà A., Cocuzza M., Sorge G. Epilepsy in patients with Angelman syndrome. Ital J Pediatr 2010;36:31. DOI: 10.1186/1824-7288-36-31.
13. Gilissen C., Hehir-Kwa J.Y., Thung D.T. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014;511(7509):344–7. DOI: 10.1038/nature13394.
14. Guerrini R., Pellock J.M. Age-related epileptic encephalopathies. Handb Clin Neurol 2012;107:179–93. DOI: 10.1016/B978-0-444-52898-8.00011-2.
15. Hardies K., Weckhuysen S., De Jonghe P., Suls A. Lessons learned from gene identification studies in Mendelian epilepsy disorders. Eur J Hum Genet 2016;24(7):961–7. DOI: 10.1038/ejhg.2015.251.
Eur J Hum Genet 2016;24(7):961–7. DOI: 10.1038/ejhg.2015.251.
16. Hauser W.A. The prevalence and incidence of convulsive disorders in children. Epilepsia 1994;35 Suppl 2:S1–6.
17. Hildebrand M.S., Dahl H.H., Damiano J.A. et al. Recent advances in the molecular genetics of epilepsy. J Med Genet 2013;50(5): 271–9. DOI: 10.1136/jmedgenet-2012-101448.
18. https://decipher.sanger.ac.uk.
19. Khair A.M., Elmagrabi D. Febrile seizures and febrile seizure syndromes: an updated overview of old and current knowledge. Neurol Res Int 2015;2015:849341. DOI: 10.1155/2015/849341.
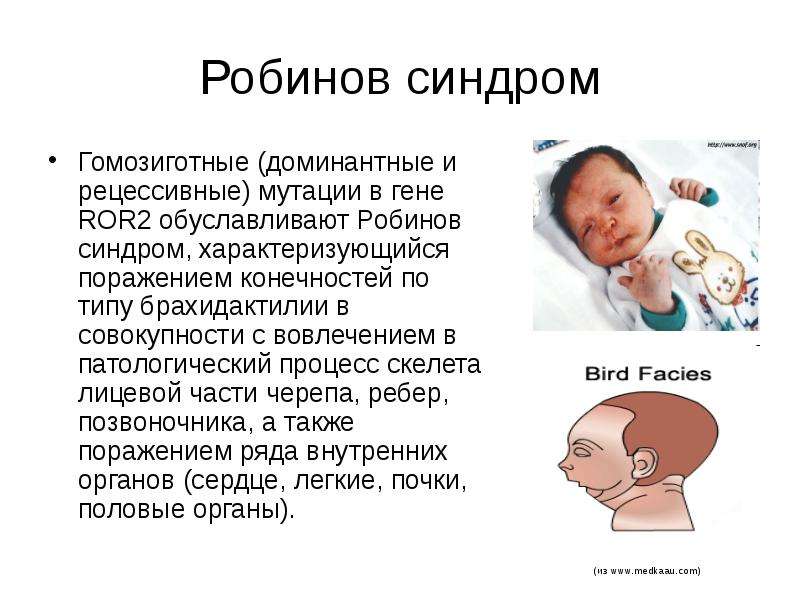
20. Lu Y., Wang X. Genes associated with idiopathic epilepsies: a current overview. Neurol Res 2009;31(2):135–43. DOI: 10.1179/174313209X393942.
21. Mercimek-Mahmutoglu S., Patel J., Cordeiro D. et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015;56(5):707–16. DOI: 10.1111/epi.12954.
22. Mowat D.R., Croaker G.D., Cass D.T. et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22q23. J Med Genet 1998;35(8):617–23.
23. Ohtahara S., Ishida T., Oka E. et al. On the specific age dependent epileptic syndrome: the early-infantile epileptic encephalopathy with suppression-burst. No to Hattatsu 1976;8:270–9.
24. Oka E., Ishida S., Ohtsuka Y., Ohtahara S. Neuroepidemiological study of childhood epilepsy by application of international classification of epilepsies and epileptic syndromes (ILAE, 1989). Epilepsia 1995;36(7):658–61.
Oka E., Ishida S., Ohtsuka Y., Ohtahara S. Neuroepidemiological study of childhood epilepsy by application of international classification of epilepsies and epileptic syndromes (ILAE, 1989). Epilepsia 1995;36(7):658–61.
25. Orhan G., Bock M., Schepers D. et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol 2014;75(3):382–94. DOI: 10.1002/ana.24080.
26. Panayiotopoulos C.P. The epilepsies: seizures, syndromes and management. Bladon Medical Publishing, 2005. 417 p.
27. Shinnar S., Glauser T.A. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
28. Scheffer I.E., Berkovic S.F. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120(Pt 3):479–90.
29. Sun Y., Ruivenkamp C.A., Hoffer M.J. et al. Next-generation diagnostics: gene panel, exome, or whole genome? Hum Mutat 2015;36(6):648–55. DOI: 10.1002/humu.22783.
30. Tsuboi T. Prevalence and incidence of epilepsy in Tokyo. Epilepsia 1988;29(2):103–10.
31. Valente K.D., Koiffmann C.P., Fridman C. et al. Epilepsy in patients with Angelman syndrome caused by deletion of the chromosome 15q11-13. Arch Neurol 2006;63(1):122–8. DOI: 10.1001/archneur.63.1.122.
32. Wakamatsu N., Yamada Y., Yamada K. et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease.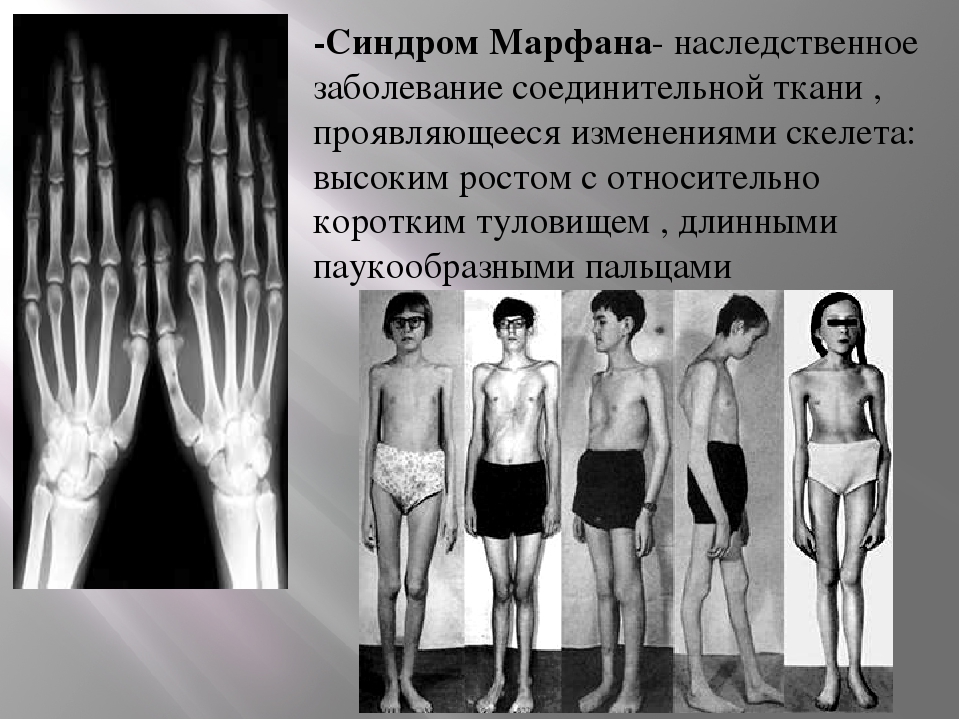 Nat Genet 2001;27(4):369–70. DOI: 10.1038/86860.
Nat Genet 2001;27(4):369–70. DOI: 10.1038/86860.
33. Zweier C., Albrecht B., Mitulla B. et al. “Mowat-Wilson” syndrome with and without Hirschsprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am J Med Genet 2002;108(3):177–81.
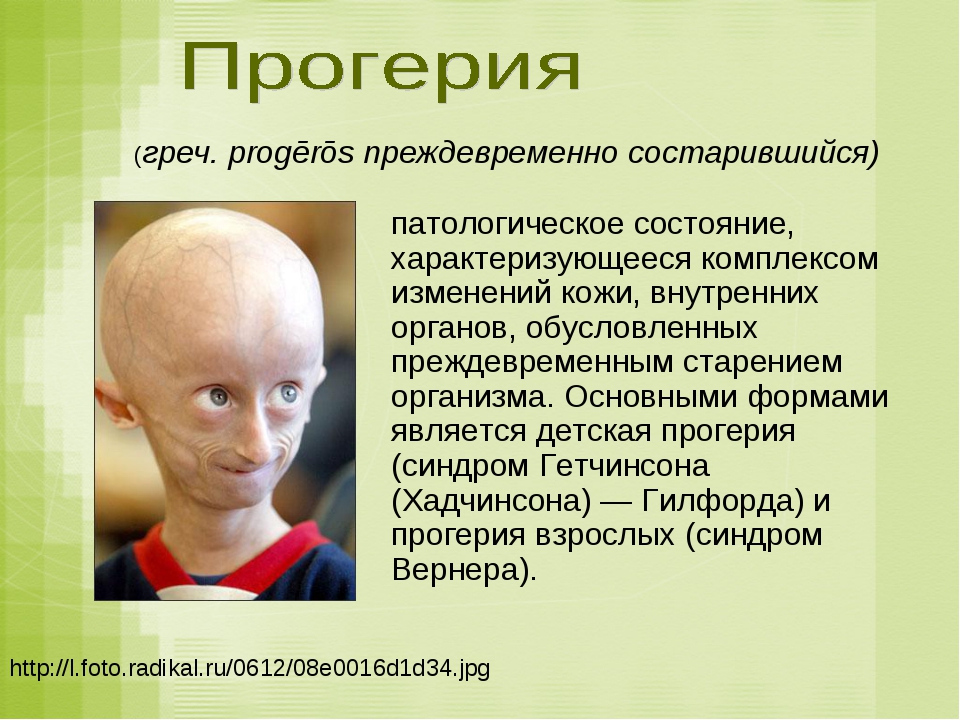
Наследственные синдромы с признаками преждевременного старения | Голоунина
1. López-Otín C, Blasco MA, Partridge L, et al. The Hallmarks of Aging. Cell. 2013;153(6):1194-1217. doi: https://doi.org/10.1016/j.cell.2013.05.039
2. Kudlow BA, Kennedy BK, Monnat RJ, Jr. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8(5):394-404. doi: https://doi.org/10.1038/nrm2161
3. Hutchinson J. Congenital Absence of Hair and Mammary Glands with Atrophic Condition of the Skin and its Appendages, in a Boy whose Mother had been almost wholly Bald from Alopecia Areata from the age of Six. Med Chir Trans. 1886;69:473-477. doi: https://doi.org/10.1177/095952878606900127
4. Gilford H. On a Condition of Mixed Premature and Immature Development. Med Chir Trans. 1897;80:17-46 25. doi: https://doi.org/10.1177/095952879708000105
5. Werner O. On cataract in conjunction with scleroderma. In: Salk D, Fujiwara Y, Martin GM, editors. Werner’s Syndrome and Human Aging. Advances in Experimental Medicine and Biology. Vol. 190. Boston: Springer; 1985. p. 1-14. doi: https://doi.org/10.1007/978-1-4684-7853-2_1.
Vol. 190. Boston: Springer; 1985. p. 1-14. doi: https://doi.org/10.1007/978-1-4684-7853-2_1.
6. Rautenstrauch T, Snigula F. Progeria: a cell culture study and clinical report of familial incidence. Eur J Pediatr. 1977;124(2):101-111. doi: https://doi.org/10.1007/bf00477545
7. Wiedemann HR. An unidentified neonatal progeroid syndrome: follow-up report. Eur J Pediatr. 1979;130(1):65-70. doi: https://doi.org/10.1007/bf00441901
8. Wambach JA, Wegner DJ, Patni N, et al. Bi-allelic POLR3A Loss-of-Function Variants Cause Autosomal-Recessive Wiedemann-Rautenstrauch Syndrome. Am J Hum Genet. 2018;103(6):968-975. doi: https://doi.org/10.1016/j.ajhg.2018.10.010
9. Paolacci S, Li Y, Agolini E, et al. Specific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome. J Med Genet. 2018;55(12):837-846. doi: https://doi.org/10.1136/jmedgenet-2018-105528
10. Paolacci S, Bertola D, Franco J, et al. Wiedemann-Rautenstrauch syndrome: A phenotype analysis. Am J Med Genet A. 2017;173(7):1763-1772. doi: https://doi.org/10.1002/ajmg.a.38246
11. Becerra CH, Contreras-Garcia GA, Perez Vera LA, et al. Wiedemann-Rautenstrauch syndrome prenatal diagnosis. J Perinatol. 2014;34(12):954-956. doi: https://doi.org/10.1038/jp.2014.156
12. Beauregard-Lacroix E, Salian S, Kim H, et al. A variant of neonatal progeroid syndrome, or Wiedemann-Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL. Eur J Hum Genet. 2020;28(4):461-468. doi: https://doi.org/10.1038/s41431-019-0539-6
Eur J Hum Genet. 2020;28(4):461-468. doi: https://doi.org/10.1038/s41431-019-0539-6
13. Gargiuli C, Schena E, Mattioli E, et al. Lamins and bone disorders: current understanding and perspectives. Oncotarget. 2018;9(32):22817-22831. doi: https://doi.org/10.18632/oncotarget.25071
14. Дадали Е.Л., Билева Д.С., Угаров И.В. Клинико-генетическая характеристика наследственных ламинопатий. // Анналы клинической и экспериментальной неврологии. — 2008. — Т. 2. — №4. — С. 28-33. [Dadaly EL, Bileva DS, Ugarov IV. Clinical and genetic characteristics of hereditary laminopathies. Annaly klinicheskoy i eksperimental’noy nevrologii. 2008;2(4):28-33. (In Russ.)]
15. Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18-29. doi: https://doi.org/10.1016/j.arr.2016.06.007
16. Ashapkin VV, Kutueva LI, Kurchashova SY, Kireev II. Are There Common Mechanisms Between the Hutchinson–Gilford Progeria Syndrome and Natural Aging? Front Genet. 2019;10. doi: https://doi.org/10.3389/fgene.2019.00455
17. Turgay Y, Eibauer M, Goldman AE, et al. The molecular architecture of lamins in somatic cells. Nature. 2017;543(7644):261-264. doi: https://doi.org/10.1038/nature21382
18. Лаврушкина С.В., Овсянникова Н.Л., Юдина А.С., и др. Канцерогенез и старение: взгляд со стороны ядерной ламины. // Цитология. — 2018. — Т. 60. — №11. — С. 892-894. [Lavrushlina SV, Ovsyannikova NL, Yudina AS, et al. Carcinogenesis and ageing: a view from nuclear lamina. Cell and tissue biology. 2018;60(11):892-894. (In Russ.)] doi: https://doi.org/10.1134/S0041377118110056.
Carcinogenesis and ageing: a view from nuclear lamina. Cell and tissue biology. 2018;60(11):892-894. (In Russ.)] doi: https://doi.org/10.1134/S0041377118110056.
19. Swahari V, Nakamura A. Speeding up the clock: The past, present and future of progeria. Dev Growth Differ. 2016;58(1):116-130. doi: https://doi.org/10.1111/dgd.12251
20. Piekarowicz K, Machowska M, Dzianisava V, Rzepecki R. Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment. Cells. 2019;8(2):88. doi: https://doi.org/10.3390/cells8020088
21. Politano L, Lattanzi G, Benedetti S, et al. Emerging perspectives on laminopathies. Cell Health Cytoskelet. 2016:25. doi: https://doi.org/10.2147/chc.s59507
22. Hamczyk MR, del Campo L, Andrés V. Aging in the Cardiovascular System: Lessons from Hutchinson-Gilford Progeria Syndrome. Annu Rev Physiol. 2018;80(1):27-48. doi: https://doi.org/10.1146/annurev-physiol-021317-121454
23. Navarro CL, Esteves-Vieira V, Courrier S, et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur J Hum Genet. 2013;22(8):1002-1011. doi: https://doi.org/10.1038/ejhg.2013.258
24. McKenna T, Sola Carvajal A, Eriksson M. Skin Disease in Laminopathy-Associated Premature Aging. J Invest Dermatol. 2015;135(11):2577-2583. doi: https://doi.org/10.1038/jid.2015.295
25. Filesi I, Gullotta F, Lattanzi G, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23(2):150-158. doi: https://doi.org/10.1152/physiolgenomics.00060.2005
Filesi I, Gullotta F, Lattanzi G, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23(2):150-158. doi: https://doi.org/10.1152/physiolgenomics.00060.2005
26. Yaou RB, Navarro C, Quijano-Roy S, et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur J Hum Genet. 2011;19(6):647-654. doi: https://doi.org/10.1038/ejhg.2010.256
27. Соркина Е.Л., Тюльпаков А.Н. Наследственные и приобретенные липодистрофии: молекулярно-генетические и аутоиммунные механизмы. // Ожирение и метаболизм. — 2018. — Т. 15. — №1. — С. 39-42. [Sorkina EL, Tyulpakov AN. Inherited and acquired lipodystrophies: molecular-genetic and autoimmune mechanisms. Obesity and metabolism. 2018;15(1):39-42. (In Russ.)] doi: https://doi.org/10.14341/OMET2018139-42
28. Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374(Pt 3):577-606. doi: https://doi.org/10.1042/BJ20030491
29. Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519-552. doi: https://doi.org/10.1146/annurev-biochem-060713-035428
30. Guo RB, Rigolet P, Ren H, et al. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35(18):6297-6310. doi: https://doi.org/10.1093/nar/gkm536
31. Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232(1):107-120. doi: https://doi.org/10.1016/j.canlet.2005.07.042
Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232(1):107-120. doi: https://doi.org/10.1016/j.canlet.2005.07.042
32. Shamanna RA, Croteau DL, Lee JH, Bohr VA. Recent Advances in Understanding Werner Syndrome. F1000Res. 2017;6:1779. doi: https://doi.org/10.12688/f1000research.12110.1
33. O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11(3):171-181. doi: https://doi.org/10.1038/nrm2848
34. Johnson JE, Cao K, Ryvkin P, et al. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 2010;38(4):1114-1122. doi: https://doi.org/10.1093/nar/gkp1103
35. Драпкина О.М., Шепель Р.Н. Теломеры и теломеразный комплекс. Основные клинические проявления генетического сбоя // Кардиоваскулярная терапия и профилактика. – 2015. Т.14. – №1. – С. 70-77. [Drapkina OM, Shepel RN. Telomeres and telomerase complex. The main clinical manifestation of genetic malfunctioning. Cardiovascular Therapy and Prevention 2015;14(1):70-77. (In Russ.)] doi: https://doi.org/10.15829/1728-8800-2015-1-70-77
36. Tang W, Robles AI, Beyer RP, et al. The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum Mol Genet. 2016;25(10):2060-2069. doi: https://doi.org/10.1093/hmg/ddw079
37. Ishikawa N, Nakamura K-I, Izumiyama-Shimomura N, et al. Accelerated <i>in vivo</i> epidermal telomere loss in Werner syndrome. Aging. 2011;3(4):417-429. doi: https://doi.org/10.18632/aging.100315
Accelerated <i>in vivo</i> epidermal telomere loss in Werner syndrome. Aging. 2011;3(4):417-429. doi: https://doi.org/10.18632/aging.100315
38. Maierhofer A, Flunkert J, Oshima J, et al. Accelerated epigenetic aging in Werner syndrome. Aging. 2017;9(4):1143-1152. doi: https://doi.org/10.18632/aging.101217
39. Zhang W, Li J, Suzuki K, et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348(6239):1160-1163. doi: https://doi.org/10.1126/science.aaa1356
40. Sarbacher CA, Halper JT. Connective Tissue and Age-Related Diseases. Subcell Biochem. 2019;91:281-310. doi: https://doi.org/10.1007/978-981-13-3681-2_11
41. Masala MV, Scapaticci S, Olivieri C, et al. Epidemiology and clinical aspects of Werner’s syndrome in North Sardinia: description of a cluster. Eur J Dermatol. 2007;17(3):213-216. doi: https://doi.org/10.1684/ejd.2007.0155
42. Yokote K, Chanprasert S, Lee L, et al. WRN Mutation Update: Mutation Spectrum, Patient Registries, and Translational Prospects. Hum Mutat. 2017;38(1):7-15. doi: https://doi.org/10.1002/humu.23128
43. Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005;307(5710):720-724. doi: https://doi.org/10.1126/science.1099593
44. Oshima J, Sidorova JM, Monnat RJ, Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev. 2017;33:105-114. doi: https://doi.org/10.1016/j.arr.2016.03.002
Ageing Res Rev. 2017;33:105-114. doi: https://doi.org/10.1016/j.arr.2016.03.002
45. Lessel D, Kubisch C. Hereditary Syndromes with Signs of Premature Aging. Dtsch Arztebl Int. 2019;116(29-30):489-496. doi: https://doi.org/10.3238/arztebl.2019.0489
46. Ozturk M, Akdeniz N, Ayakta H, Kosem M. A brother and sister with Werner’s syndrome demonstrating extensive tendon calcification and sacroiliitis. Clin Exp Dermatol. 2006;31(4):615-616. doi: https://doi.org/10.1111/j.1365-2230.2006.02130.x
47. Honjo S, Yokote K, Fujimoto M, et al. Clinical outcome and mechanism of soft tissue calcification in Werner syndrome. Rejuvenation Res. 2008;11(4):809-819. doi: https://doi.org/10.1089/rej.2007.0649
48. Leone A, Costantini AM, Brigida R, et al. Soft-tissue mineralization in Werner syndrome. Skeletal Radiol. 2005;34(1):47-51. doi: https://doi.org/10.1007/s00256-004-0792-8
49. Sickles CK, Gross GP. Progeria (Werner Syndrome). Treasure Island: StatPearls Publishing; 2020.
50. Belaya ZE, Grebennikova TA, Yashina JN, et al. Rare causes of secondary hyperparathyroidism clinical cases of Werner’s syndrome, Gitelman’s syndrome and osteopetrosis among patients referred for primary hyperparathyroidism. In: Osteoporosis International. Vol. 26. London: Springer; 2015. p. 111.
51. Lauper JM, Krause A, Vaughan TL, Monnat RJ, Jr. Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS One. 2013;8(4):e59709. doi: https://doi.org/10.1371/journal.pone.0059709
PLoS One. 2013;8(4):e59709. doi: https://doi.org/10.1371/journal.pone.0059709
52. de Renty C, Ellis NA. Bloom’s syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases. Ageing Res Rev. 2017;33:36-51. doi: https://doi.org/10.1016/j.arr.2016.05.010
53. Bloom D. congenital telangiectatic erythema resembling lupus erythematosus in dwarfs<subtitle>Probably a Syndrome Entity. Arch Pediatr Adolesc Med. 1954;88(6):754. doi: https://doi.org/10.1001/archpedi.1954.02050100756008
54. German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28(8):743-753. doi: https://doi.org/10.1002/humu.20501
55. Fares F, Badarneh K, Abosaleh M, et al. Carrier frequency of autosomal-recessive disorders in the Ashkenazi Jewish population: should the rationale for mutation choice for screening be reevaluated? Prenat Diagn. 2008;28(3):236-241. doi: https://doi.org/10.1002/pd.1943
56. Kaneko H, Kondo N. Clinical features of Bloom syndrome and function of the causative gene, BLM helicase. Expert Rev Mol Diagn. 2004;4(3):393-401. doi: https://doi.org/10.1586/14737159.4.3.393
57. Lu L, Jin W, Wang LL. Aging in Rothmund-Thomson syndrome and related RECQL4 genetic disorders. Ageing Res Rev. 2017;33:30-35. doi: https://doi.org/10.1016/j.arr.2016.06.002
58. Thomson MS. Poikiloderma Congenitale: Two Cases for Diagnosis. Proc R Soc Med. 1936;29(5):453-455.
Proc R Soc Med. 1936;29(5):453-455.
59. Colombo EA, Locatelli A, Cubells Sanchez L, et al. Rothmund-Thomson Syndrome: Insights from New Patients on the Genetic Variability Underpinning Clinical Presentation and Cancer Outcome. Int J Mol Sci. 2018;19(4). doi: https://doi.org/10.3390/ijms19041103
60. Oshima J, Kato H, Maezawa Y, Yokote K. RECQ helicase disease and related progeroid syndromes: RECQ2018 meeting. Mech Ageing Dev. 2018;173:80-83. doi: https://doi.org/10.1016/j.mad.2018.05.002
61. Hafsi W, Badri T. Poikiloderma Congenitale. Treasure Island (FL): StatPearls Publishing; 2019.
62. Araujo SJ, Kuraoka I. Nucleotide excision repair genes shaping embryonic development. Open Biol. 2019;9(10):190166. doi: https://doi.org/10.1098/rsob.190166
63. Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst). 2015;36:13-18. doi: https://doi.org/10.1016/j.dnarep.2015.09.003
64. Moriwaki S, Kanda F, Hayashi M, et al. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017;44(10):1087-1096. doi: https://doi.org/10.1111/1346-8138.13907
65. Brooks BP, Thompson AH, Bishop RJ, et al. Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology. 2013;120(7):1324-1336. doi: https://doi.org/10.1016/j.ophtha.2012.12.044
66. Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48(3):168-176. doi: https://doi.org/10.1136/jmg.2010.083022
Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48(3):168-176. doi: https://doi.org/10.1136/jmg.2010.083022
67. Karass M, Naguib MM, Elawabdeh N, et al. Xeroderma pigmentosa: three new cases with an in depth review of the genetic and clinical characteristics of the disease. Fetal Pediatr Pathol. 2015;34(2):120-127. doi: https://doi.org/10.3109/15513815.2014.982336
68. Kaliki S, Jajapuram SD, Maniar A, Mishra DK. Ocular and Periocular Tumors in Xeroderma Pigmentosum: A Study of 120 Asian Indian Patients. Am J Ophthalmol. 2019;198:146-153. doi: https://doi.org/10.1016/j.ajo.2018.10.011
69. Black JO. Xeroderma Pigmentosum. Head Neck Pathol. 2016;10(2):139-144. doi: https://doi.org/10.1007/s12105-016-0707-8
70. Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1936;11(61):1-8. doi: https://doi.org/10.1136/adc.11.61.1
71. Wilson BT, Stark Z, Sutton RE, et al. The Cockayne Syndrome Natural History (CoSyNH) study: clinical findings in 102 individuals and recommendations for care. Genet Med. 2016;18(5):483-493. doi: https://doi.org/10.1038/gim.2015.110
72. Слижов П.А., Долинина Т.И., Плескай Н.М., и др. Маркеры старения в клетках больных синдромом Коккейна. Общие и индивидуальные различия. // Цитология. — 2018. — Т. 60. — №3. — С. 188-199. [Slizhov PA, Dolinina TI, Pleskach NM, et al. Aging markers in cells of patients with Cockayne Syndrome.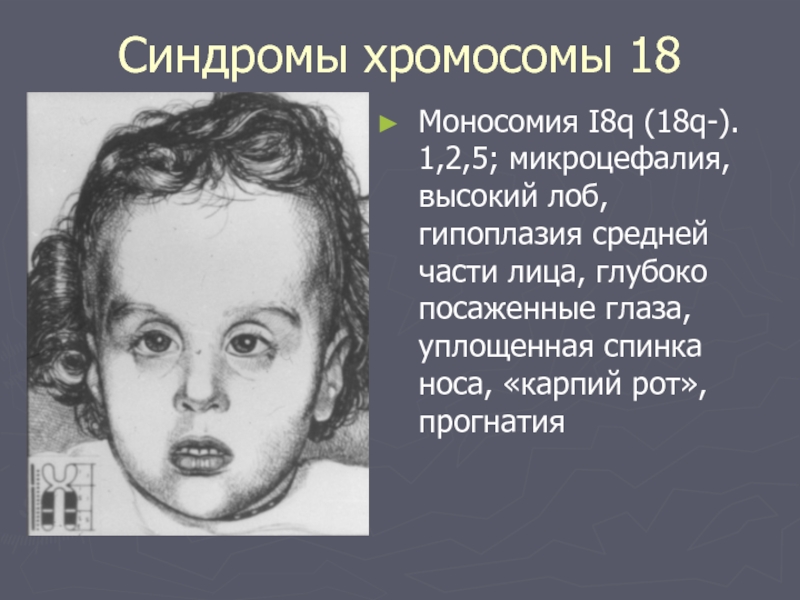 General and individual differences. Cell and tissue biology. 2018;60(3):188-199. (In Russ.)] doi: https://doi.org/10.31116/tsitol.2018.03.05
General and individual differences. Cell and tissue biology. 2018;60(3):188-199. (In Russ.)] doi: https://doi.org/10.31116/tsitol.2018.03.05
73. Kubota M, Ohta S, Ando A, et al. Nationwide survey of Cockayne syndrome in Japan: Incidence, clinical course and prognosis. Pediatr Int. 2015;57(3):339-347. doi: https://doi.org/10.1111/ped.12635
74. Karikkineth AC, Scheibye-Knudsen M, Fivenson E, et al. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res Rev. 2017;33:3-17. doi: https://doi.org/10.1016/j.arr.2016.08.002
75. Kalantaridou SN, Zoumakis E, Makrigiannakis A, et al. Corticotropin-releasing hormone, stress and human reproduction: an update. J Reprod Immunol. 2010;85(1):33-39. doi: https://doi.org/10.1016/j.jri.2010.02.005
76. Hayashi M, Miwa-Saito N, Tanuma N, Kubota M. Brain vascular changes in Cockayne syndrome. Neuropathology. 2012;32(2):113-117. doi: https://doi.org/10.1111/j.1440-1789.2011.01241.x
77. Kraemer KH, Patronas NJ, Schiffmann R, et al. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145(4):1388-1396. doi: https://doi.org/10.1016/j.neuroscience.2006.12.020
78. Pereira LB, Valente NYS, Rocha VB. Do you know this syndrome? Ichthyosis associated with neurological condition and alteration of hairs. An Bras Dermatol. 2018;93(1):135-137. doi: https://doi.org/10.1590/abd1806-4841.20187727
79. Farmaki E, Nedelkopoulou N, Delli F, et al. Brittle Hair, Photosensitivity, Brain Hypomyelination and Immunodeficiency: Clues to Trichothiodystrophy. Indian J Pediatr. 2017;84(1):89-90. doi: https://doi.org/10.1007/s12098-016-2209-9
Farmaki E, Nedelkopoulou N, Delli F, et al. Brittle Hair, Photosensitivity, Brain Hypomyelination and Immunodeficiency: Clues to Trichothiodystrophy. Indian J Pediatr. 2017;84(1):89-90. doi: https://doi.org/10.1007/s12098-016-2209-9
80. Potter H, Chial HJ, Caneus J, et al. Chromosome Instability and Mosaic Aneuploidy in Neurodegenerative and Neurodevelopmental Disorders. Front Genet. 2019;10:1092. doi: https://doi.org/10.3389/fgene.2019.01092
81. Khetarpal P, Das S, Panigrahi I, Munshi A. Primordial dwarfism: overview of clinical and genetic aspects. Mol Genet Genomics. 2016;291(1):1-15. doi: https://doi.org/10.1007/s00438-015-1110-y
82. O’Driscoll M, Ruiz-Perez VL, Woods CG, et al. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33(4):497-501. doi: https://doi.org/10.1038/ng1129
83. Qvist P, Huertas P, Jimeno S, et al. CtIP Mutations Cause Seckel and Jawad Syndromes. PLoS Genet. 2011;7(10):e1002310. doi: https://doi.org/10.1371/journal.pgen.1002310
84. Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS. Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47(6):411-414. doi: https://doi.org/10.1136/jmg.2009.076646
85. Kalay E, Yigit G, Aslan Y, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43(1):23-26. doi: https://doi.org/10.1038/ng.725
86. Sir JH, Barr AR, Nicholas AK, et al. A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet. 2011;43(11):1147-1153. doi: https://doi.org/10.1038/ng.971
Sir JH, Barr AR, Nicholas AK, et al. A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet. 2011;43(11):1147-1153. doi: https://doi.org/10.1038/ng.971
87. Dauber A, Lafranchi SH, Maliga Z, et al. Novel microcephalic primordial dwarfism disorder associated with variants in the centrosomal protein ninein. J Clin Endocrinol Metab. 2012;97(11):E2140-2151. doi: https://doi.org/10.1210/jc.2012-2150
88. Shaheen R, Faqeih E, Ansari S, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24(2):291-299. doi: https://doi.org/10.1101/gr.160572.113
89. Ogi T, Walker S, Stiff T, et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet. 2012;8(11):e1002945. doi: https://doi.org/10.1371/journal.pgen.1002945
90. Barbelanne M, Tsang WY. Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int. 2014;2014:547986. doi: https://doi.org/10.1155/2014/547986
91. Savage SA. Dyskeratosis Congenita. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2020.
92. Sharma RK, Gupta M, Sood S, Gupta A. Dyskeratosis congenita: presentation of cutaneous triad in a sporadic case. BMJ Case Rep. 2018;11(1). doi: https://doi.org/10.1136/bcr-2018-226736
93. Aplas V. Poikiloderma, parapsoriasis and atrophia cutis cum pigmentatione, dystrophia ungium et leukoplakia oris Zinsser, so-called dyskeratosis congenita. Arch Klin Exp Dermatol. 1956;202(3):224-237. doi: https://doi.org/10.1007/bf00476707
Aplas V. Poikiloderma, parapsoriasis and atrophia cutis cum pigmentatione, dystrophia ungium et leukoplakia oris Zinsser, so-called dyskeratosis congenita. Arch Klin Exp Dermatol. 1956;202(3):224-237. doi: https://doi.org/10.1007/bf00476707
94. Wang F, Du YQ, Gong W, et al. Research progress of dyskeratosis congenita. Zhonghua Kou Qiang Yi Xue Za Zhi. 2019;54(2):130-134. doi: https://doi.org/10.3760/cma.j.issn.1002-0098.2019.02.010
95. Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753-764. doi: https://doi.org/10.1097/GIM.0b013e3181f415b5
96. Dodson LM, Baldan A, Nissbeck M, et al. From incomplete penetrance with normal telomere length to severe disease and telomere shortening in a family with monoallelic and biallelic PARN pathogenic variants. Hum Mutat. 2019;40(12):2414-2429. doi: https://doi.org/10.1002/humu.23898
97. Savage SA. Beginning at the ends: telomeres and human disease. F1000Res. 2018;7. doi: https://doi.org/10.12688/f1000research.14068.1
98. Kutbay NO, Yurekli BS, Erdemir Z, et al. A case of dyskeratosis congenita associated with hypothyroidism and hypogonadism. Hormones (Athens). 2016;15(2):297-299. doi: https://doi.org/10.14310/horm.2002.1655
99. Shomali W, Brar R. Late presentation of dyskeratosis congenita. Br J Haematol. 2019;187(3):273. doi: https://doi.org/10.1111/bjh.16131
100. Du H, Guo Y, Ma D, et al. A case report of heterozygous TINF2 gene mutation associated with pulmonary fibrosis in a patient with dyskeratosis congenita. Medicine (Baltimore). 2018;97(19):e0724. doi: https://doi.org/10.1097/MD.0000000000010724
Du H, Guo Y, Ma D, et al. A case report of heterozygous TINF2 gene mutation associated with pulmonary fibrosis in a patient with dyskeratosis congenita. Medicine (Baltimore). 2018;97(19):e0724. doi: https://doi.org/10.1097/MD.0000000000010724
101. de Boer J, Andressoo JO, de Wit J, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296(5571):1276-1279. doi: https://doi.org/10.1126/science.1070174
102. Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775(1):138-162. doi: https://doi.org/10.1016/j.bbcan.2006.08.007
103. Zhang W, Li J, Suzuki K, et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348(6239):1160-1163. doi: https://doi.org/10.1126/science.aaa1356
104. Shumaker DK, Dechat T, Kohlmaier A, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103(23):8703-8708. doi: https://doi.org/10.1073/pnas.0602569103
105. Davis T, Brook AJ, Rokicki MJ, et al. Evaluating the Role of p38 MAPK in the Accelerated Cell Senescence of Werner Syndrome Fibroblasts. Pharmaceuticals (Basel). 2016;9(2). doi: https://doi.org/10.3390/ph9020023
106. Tivey HS, Brook AJ, Rokicki MJ, et al. p38 (MAPK) stress signalling in replicative senescence in fibroblasts from progeroid and genomic instability syndromes. Biogerontology. 2013;14(1):47-62. doi: https://doi.org/10.1007/s10522-012-9407-2
Biogerontology. 2013;14(1):47-62. doi: https://doi.org/10.1007/s10522-012-9407-2
107. Bagley MC, Davis T, Murziani PG, et al. Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome. Pharmaceuticals (Basel). 2010;3(6):1842-1872. doi: https://doi.org/10.3390/ph4061842
108. Yamaga M, Takemoto M, Shoji M, et al. Werner syndrome: a model for sarcopenia due to accelerated aging. Aging (Albany NY). 2017;9(7):1738-1744. doi: https://doi.org/10.18632/aging.101265
109. von Walden F, Liu C, Aurigemma N, Nader GA. mTOR signaling regulates myotube hypertrophy by modulating protein synthesis, rDNA transcription, and chromatin remodeling. Am J Physiol Cell Physiol. 2016;311(4):C663-C672. doi: https://doi.org/10.1152/ajpcell.00144.2016
110. Dormond O. mTOR in Human Diseases. Int J Mol Sci. 2019;20(9). doi: https://doi.org/10.3390/ijms20092351
111. Ou HL, Schumacher B. DNA damage responses and p53 in the aging process. Blood. 2018;131(5):488-495. doi: https://doi.org/10.1182/blood-2017-07-746396
112. Wu D, Prives C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018;25(1):169-179. doi: https://doi.org/10.1038/cdd.2017.187
113. Inoki K, Ouyang H, Li Y, Guan KL. Signaling by target of rapamycin proteins in cell growth control. Microbiol Mol Biol Rev. 2005;69(1):79-100. doi: https://doi.org/10.1128/MMBR.69.1.79-100.2005
114. Weichhart T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology. 2018;64(2):127-134. doi: https://doi.org/10.1159/000484629
Weichhart T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology. 2018;64(2):127-134. doi: https://doi.org/10.1159/000484629
115. Wiza C, Nascimento EB, Ouwens DM. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab. 2012;302(12):E1453-1460. doi: https://doi.org/10.1152/ajpendo.00660.2011
116. Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91-99. doi: https://doi.org/10.1016/j.ccr.2004.06.007
117. Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458-471. doi: https://doi.org/10.1016/j.cmet.2007.11.001
118. Zhao J, Brault JJ, Schild A, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6(6):472-483. doi: https://doi.org/10.1016/j.cmet.2007.11.004
119. Wu JJ, Liu J, Chen EB, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013;4(5):913-920. doi: https://doi.org/10.1016/j.celrep.2013.07.030
120. Vellai T, Takacs-Vellai K, Zhang Y, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426(6967):620. doi: https://doi.org/10.1038/426620a
121. Bjedov I, Toivonen JM, Kerr F, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11(1):35-46. doi: https://doi.org/10.1016/j.cmet.2009.11.010
122. Kaeberlein M, Powers RW, 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310(5751):1193-1196. doi: https://doi.org/10.1126/science.1115535
123. Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1(1):29. doi: https://doi.org/10.1186/2001-1326-1-29
124. Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8(12):1888-1895. doi: https://doi.org/10.4161/cc.8.12.8606
125. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392-395. doi: https://doi.org/10.1038/nature08221
126. Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570-578. doi: https://doi.org/10.1056/NEJMoa012437
127. Brown RJ, Oral EA, Cochran E, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018;60(3):479-489. doi: https://doi.org/10.1007/s12020-018-1589-1
128. Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666-16671. doi: https://doi.org/10.1073/pnas.1202529109
129. Gordon LB, Massaro J, D’Agostino RB, Sr., et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130(1):27-34. doi: https://doi.org/10.1161/CIRCULATIONAHA.113.008285
130. Gordon LB, Shappell H, Massaro J, et al. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA. 2018;319(16):1687-1695. doi: https://doi.org/10.1001/jama.2018.3264
131. McNally EM, Wyatt EJ. Welcome to the splice age: antisense oligonucleotide-mediated exon skipping gains wider applicability. J Clin Invest. 2016;126(4):1236-1238. doi: https://doi.org/10.1172/JCI86799
132. Lee JM, Nobumori C, Tu Y, et al. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016;126(4):1592-1602. doi: https://doi.org/10.1172/JCI85908
133. Ершова О.Б., Белова К.Ю., Дегтярев А.А., и др. Анализ летальности у пациентов с переломом проксимального отдела бедра. // Остеопороз и остеопатии. — 2015. — Т. 18. — №3. — С. 3-8. [Ershova OB, Belova KY, Degtyarev AA, et al. Analysis of mortality in patients with a fracture of the proximal femur. Osteoporosis and bone diseases. 2015;18(3):3-8. (In Russ.)] doi: https://doi.org/10.14341/osteo201533-8
134. Мельниченко Г.А., Белая Ж.Е., Рожинская Л.Я., и др. Краткое изложение клинических рекомендаций по диагностике и лечению остеопороза Российской ассоциации эндокринологов. // Остеопороз и остеопатии. — 2016. — Т. 19. — №3. — С. 28-36. [Melnichenko GA, Belaya ZE, Rozhinskaya LY, et al. Summary of clinical guidelines for the diagnosis and treatment of osteoporosis of the Russian association of endocrinologists. Osteoporosis and bone diseases 2016;19(3):28-36. (In Russ.)] doi: https://doi.org/10.14341/osteo2016328-36
135. Белая Ж.Е., Рожинская Л.Я. Витамин D в терапии остеопороза: его роль в комбинации с препаратами для лечения остеопороза, внескелетные эффекты. // Эффективная фармакотерапия. — 2013. — Т. 38. — №2. — С. 14-29. [Belaya ZY, Rozhinskaya LY. Vitamin D in the treatment of osteoporosis: its role in the combination with antiosteoporotic therapy, non-skeletal effects. Effektivnaya farmakoterapiya. 2013;38(2):14-29. (In Russ.)]
136. Geusens PP, Lems WF. Fracture prevention in postmenopausal women with osteoporosis by an annual infusion of zoledronic acid. Ned Tijdschr Geneeskd. 2007;151(26):1445-1448.
137. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357(18):1799-1809. doi: https://doi.org/10.1056/NEJMoa074941
138. Cummings SR, Lui LY, Eastell R, Allen IE. Association Between Drug Treatments for Patients With Osteoporosis and Overall Mortality Rates: A Meta-analysis. JAMA Intern Med. 2019. doi: https://doi.org/10.1001/jamainternmed.2019.2779
139. Bliuc D, Tran T, van Geel T, et al. Mortality risk reduction differs according to bisphosphonate class: a 15-year observational study. Osteoporos Int. 2019;30(4):817-828. doi: https://doi.org/10.1007/s00198-018-4806-0
140. Lee P, Ng C, Slattery A, et al. Preadmission Bisphosphonate and Mortality in Critically Ill Patients. J Clin Endocrinol Metab. 2016;101(5):1945-1953. doi: https://doi.org/10.1210/jc.2015-3467
141. Bergman J, Nordstrom A, Hommel A, et al. Bisphosphonates and mortality: confounding in observational studies? Osteoporos Int. 2019;30(10):1973-1982. doi: https://doi.org/10.1007/s00198-019-05097-1
142. Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell Metab. 2016;23(6):1060-1065. doi: https://doi.org/10.1016/j.cmet.2016.05.011
143. Anisimov VN, Berstein LM, Egormin PA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7(17):2769-2773. doi: https://doi.org/10.4161/cc.7.17.6625
144. Landman GW, Kleefstra N, van Hateren KJ, et al. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33(2):322-326. doi: https://doi.org/10.2337/dc09-1380
145. Lee MS, Hsu CC, Wahlqvist ML, et al. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC Cancer. 2011;11:20. doi: https://doi.org/10.1186/1471-2407-11-20
146. Tseng CH. Diabetes, metformin use, and colon cancer: a population-based cohort study in Taiwan. Eur J Endocrinol. 2012;167(3):409-416. doi: https://doi.org/10.1530/EJE-12-0369
147. Tosca L, Rame C, Chabrolle C, et al. Metformin decreases IGF1-induced cell proliferation and protein synthesis through AMP-activated protein kinase in cultured bovine granulosa cells. Reproduction. 2010;139(2):409-418. doi: https://doi.org/10.1530/REP-09-0351
148. Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer. 2013;13:235. doi: https://doi.org/10.1186/1471-2407-13-235
149. Zi FM, He JS, Li Y, et al. Metformin displays anti-myeloma activity and synergistic effect with dexamethasone in in vitro and in vivo xenograft models. Cancer Lett. 2015;356(2 Pt B):443-453. doi: https://doi.org/10.1016/j.canlet.2014.09.050
150. Niehr F, von Euw E, Attar N, et al. Combination therapy with vemurafenib (PLX4032/RG7204) and metformin in melanoma cell lines with distinct driver mutations. J Transl Med. 2011;9:76. doi: https://doi.org/10.1186/1479-5876-9-76
151. Colquhoun AJ, Venier NA, Vandersluis AD, et al. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15(4):346-352. doi: https://doi.org/10.1038/pcan.2012.16
152. Li L, Han R, Xiao H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014;20(10):2714-2726. doi: https://doi.org/10.1158/1078-0432.CCR-13-2613
153. Blandino G, Valerio M, Cioce M, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun. 2012;3:865. doi: https://doi.org/10.1038/ncomms1859
154. Xu Y, Lu S. Metformin inhibits esophagus cancer proliferation through upregulation of USP7. Cell Physiol Biochem. 2013;32(5):1178-1186. doi: https://doi.org/10.1159/000354517
155. Algire C, Amrein L, Zakikhani M, et al. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17(2):351-360. doi: https://doi.org/10.1677/erc-09-0252
156. Gandini S, Puntoni M, Heckman-Stoddard BM, et al. Metformin and Cancer Risk and Mortality: A Systematic Review and Meta-analysis Taking into Account Biases and Confounders. Cancer Prev Res (Phila). 2014;7(9):867-885. doi: https://doi.org/10.1158/1940-6207.capr-13-0424
157. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352(9131):854-865. doi: https://doi.org/10.1016/s0140-6736(98)07037-8
158. Lautrup S, Caponio D, Cheung H-H, et al. Studying Werner syndrome to elucidate mechanisms and therapeutics of human aging and age-related diseases. Biogerontology. 2019;20(3):255-269. doi: https://doi.org/10.1007/s10522-019-09798-2
Генетические синдромы, ассоциированные с сахарным диабетом: синдром Вольфрама | Дедов
Синдром Вольфрама — прогрессирующее нейродсгснсра- тивное заболевание, сочетающее нсаутоиммунный инсулинзависимый сахарный диабет и атрофию зрительного нерва [17].
Впервые этот редкий генетический синдром был открыт в 1938 г. американскими врачами D. Wolfram и Н. Wagener, которые описали развитие сахарного диабета и атрофии зрительного нерва у 4 из 8 сибсов [72]. В дальнейшем наряду с сахарным диабетом и атрофией зрительного нерва, которые обычно развиваются в 1 -ю декаду жизни, были описаны другие проявления этого синдрома: во 2-е десятилетие жизни у больных часто выявляли нейросенсорную тугоухость [15] и несахарный диабет [26]. Понятие «DlDMOAD-синдром» (Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy and Deafness) включает в себя наиболее часто встречающиеся проявления этого генетического заболевания: несахарный диабет, сахарный диабет, атрофию зрительного нерва и нейросенсорную тугоухость [5, 10, 28]. В 3-й и 4-й декадах жизни пациентов нередко выявляются другие проявления синдрома Вольфрама: атония мочевыводящих путей [21, 39], неврологические [21, 57, 60] и психиатрические изменения [64—66]. Обязательным и достаточным критерием для постановки диагноза является сочетание двух клинических симптомов: неаугоиммунного инсулинзависимого сахарного диабета и атрофии зрительного нерва, развившихся в возрасте до 30 лет [4, 9, 10, 17].
В этиологической классификации сахарного диабета синдром Вольфрама включен в группу других генетических синдромов, иногда сочетающихся с сахарным диабетом [1—3, 6].
Причиной развития синдрома Вольфрама является аутосомно-рецессивная мутация [22, 53, 56] в гене синдрома Вольфрама 1 (WFS1), который локализуется на коротком плече хромосомы 4 в положении 16 и состоит из 8 экзонов [63, 70].НА0Г0 СЛуЧаЯ развития диабетической неф- Р t [41]. Однако смертность у пациентов с синдромом 52
Вольфрама значительно выше, чем у больных сахарным диабетом 1-го типа; 60% больных с синдромом Вольфрама умирают в возрасте 30-35 лет [8. 41], средняя продолжительность жизни составляет 25—45 лет [8]. Смерть наиболее часто наступает в результате центральной респираторной недостаточности с атрофией ствола головного мозга [8, 41]. Кроме того, распространенными причинами смерти являются инфекционные септические осложнения [24] и осложнения со стороны мочевыделительной системы [40].
При аутопсии у этих больных часто отсутствуют р-клетки поджелудочной железы [3] или их количество значительно снижено [41]. На сегодняшний день остается неизученным вопрос о причине отсутствия микрососудистых осложнений у больных с синдромом Вольфрама, несмотря на длительность заболеваний сахарным диабетом нередко более 30 лет.
Таким образом, больные с синдромом Вольфрама имеют несколько иные клиническую картину и характер осложнений по сравнению с пациентами, страдающими сахарным диабетом 1-го типа. Следовательно, своевременная постановка диагноза имеет значение для определения тактики ведения и прогноза заболевания. Важным дифференциально-диагностическим критерием является наличие у пациентов атрофии зрительного нерва. По рекомендации Американской диабетической ассоциации офтальмоскопическое исследование пациентам с сахарным диабетом 1 -го типа необходимо проводить через 5 лет после манифестации заболевания [13]. Однако при проведении офтальмоскопического исследования в более ранние сроки у 589 больных сахарным диабетом 1-го типа у 27 из них был диагностирован синдром Вольфрама [13]. В 100% случаев родители больных с синдромом Вольфрама состояли в кровном родстве по сравнению с 19% у больных сахарным диабетом 1-го типа. У 55% больных с синдромом Вольфрама был положительный наследственный анамнез по сахарному диабету 1-го типа, тогда как среди больных сахарным диабетом без атрофии зрительного нерва менее 10% имели родственников с сахарным диабетом 1-го типа.FSl «мены гуанина на аденин в положении 2146 приводит к замене аминокислоты аланин на тирозин в положении 716. Фенотипически это проявляется более тяжелым нарушением слуха, чем наблюдаемое при синдроме Вольфрама В первые 2 декады жизни у пациентов выпадают звуки средней и высокой частот, затем звуки низкой частоты, и в возрасте 40 лет, как правило, наступает полная глухота [73].
Нссахарный диабет у пациентов с синдромом Вольфрама также обычно манифестирует во 2-й декаде жизни. При аутопсии пациентов с клинической картиной синдрома Вольфрама была исключена мутация в хромосоме 20 (причина развития аутосомно-доминантного нейрогипофизарного несахарного диабета) [36] и получены данные о том, что несахарный диабет развивается вследствие не только атрофии нейронов в супраоптическом ядре, но и дефекта в процессинге предшественника вазопрессина [24]. При иммунореактивном анализе материала аутопсии пациентов с синдромом Вольфрама нс было найдено вазопрессина и признаков его процессинга в супраоптическом и паравентрикулярных ядрах гипоталамуса, однако обнаружено значительное количество предшественника вазопрессина. Также отсутствовали прогормонконвертаза вазопрессина (РС2), которая относится к семейству эндопротсаз, и нейроэндокринный полипептид (7В2), который предотвращает преждевременную активацию предшественника прогормонконвертазы вазопрессина (РС2) [24]. Таким образом, можно наблюдать нарушение обработки сразу нескольких пептидов, участвующих в синтезе вазопрессина, при этом нс было найдено мутаций ни в одном гене кодирующем данные белки.
В 3-й декаде жизни у пациентов развиваются осложнения со стороны мочевыводящего тракта и неврологические нарушения. Дилатация мочевыводящих путей далеко нс всегда сочетается с несахарным диабетом и может существовать как независимое проявление заболевания [21, 33, 39, 551. Высказываются предположения о том, что данный симптом имеет центральное происхождение и развивается вследствие атрофии структур головного мозга [55]. Кроме того, был описан случай развития острой почечной недостаточности у 30-летнего пациента с инсулинзависимым сахарным диабетом, атрофией зрительного нерва и снижением слуха. У больного при удовлетворительной компенсации углеводного обмена на фоне относительно стабильного состояния развилась анурия, было зарегистрировано повышение уровня креатинина до 334,4 мкмоль/л. При урогра- фическом исследовании выявлена дилатация мочевыводящих путей. Через 1 нед после госпитализации в урологическое отделение уровень креатинина составил 132,5 мкмоль/л. Через 2 года содержание креатинина составляло 105 мкмоль/л, однако пациент был прикован к постели в связи с неврологическими осложнениями [55].
У обследованных также выявлено большое количество неврологических проявлений синдрома: периферическая нейропатия [21], атаксия, нистагм [57], аносмия, эпилепсия, возможно, развитие паркинсонизма, нарушение интеллекта [61]. Неврологические осложнения синдрома Вольфрама, в частности атрофия головного мозга и респираторная недостаточность, наиболее часто являются причиной смерти пациентов [8, 40]. При магнитно-резонансном томографическом исследовании были обнаружены распространенные изменения головного мозга, некоторые из них коррелировали с неврологическими чертами синдрома [57]. На магнитно-резонансных томограммах обычно отсутствует физиологический сигнал задней доли гипофиза, значительно уменьшены в размерах зрительный нерв, хиазмы и тракты, визуализируется атрофия области гипоталамуса, ствола мозга, мозжечка и коры головного мозга [25].
Нередко пациенты с синдромом Вольфрама страдают психическими нарушениями, такими как депрессии, агрессивное поведение, психозы, тревожность, панические атаки, галлюцинации, снижение памяти и интеллекта, суицидальные попытки [65]. Очевидно, что мутации в гене WFS1 могут приводить к психическим нарушениям. Пациенты с гетерозиготной мутацией обычно нс страдают всеми проявлениями DIDMOAD-син- Дрома, однако нередко имеют точно такую же психическую патологию, как и больные с гомозиготной мутацией [15]. Пациенты с гетерозиготной мутацией в гене WFS1, которые составляют приблизительно 1% в популяции [64, 66], в 26 раз чаше госпитализируются в психиатрические клиники и совершают суицида, чем люди, не несущие мутаций в этом гене 156,66].“p0M0MВольФРама из одном семьи и 4 больных из ДРУГ и (2I ]. В первой семье язвы в желудочно-кишечном тракте развились у I девочки в возрасте 14 лет, у 1 мальчика в 17 лет, У мальчика в 18 лет и у 3 детей в 20 лет. В другой семье 3 ребенка получили это осложнение в возрасте 13 лет и 1 — в возрасте 26 лет [21].
мпдп пациснтов с развернутой клинической картиной D1D- MOAD-синдрома был описан случай развития мсгалобластной, сидеробластной анемии, нейтропении и пограничной тромбоцитопении. Концентрация тиамина в крови у одного пациента была сниженной, у другого — нормальной. Однако у обоих детей была снижена активность тиаминфосфатазы и тиаминпирофосфокиназы в эритроцитах по сравнению с таковой у здоровых детей. Через 1 мес после лечения тиамином гематологические показатели восстановились до нормальных значений и улучшилась чувствительность к инсулину. Отмена тиамина приводила к повторному развитию гематологических нарушений и ухудшению чувствительности к инсулину [16].
Кроме того, характерным для данного синдрома является развитие первичного гипогонадизма [8, 31, 54]. Однако имеются также данные о благополучной беременности и родах у пациенток с DIDMOAD-синдромо.м [54, 59].
Причиной развития синдрома Вольфрама, как упоминалось выше, является аутосомно-рецессивная мутация [22, 53, 56] в гене, локализованном на коротком плече хромосомы 4 в положении 16 (ген WFS1) [63, 70]. Для развития клинической картины (минимальные диагностические критерии — инсулинзависимый сахарный диабет и атрофия зрительного нерва) мутация в гене WFS1 должна быть гомозиготной [19, 31, 70] или необходимо присутствие 2 гетерозиготных мутаций и более [19, 31]. Последовательность нуклеотидов в гене WFS1 описана, определение мутации этого гена может иметь диагностическое и прогностическое значение для пациентов, у которых выявлены сахарный диабет и слепота [70]. Наиболее часто встречаются мутации в 8-м экзоне [31, 68], однако описаны мутации, нередко с более тяжелой клинической картиной, и в других экзонах. При исследовании системы HLA генов, характерных для развития сахарного диабета 1-го типа, у пациентов с синдромом Вольфрама не найдено [47].
Продукт экспрессии гена WFS1 полипептид вольфрамин состоит из 890 аминокислот и имеет мол. массу 100 кД [63]. Вольфрамин — это трансмембранный протеин, включающий в себя 3 структурных домена: гидрофильный аминоконцевой конец, гидрофильный карбоксиконцсвой конец и гидрофобную часть, состоящую из 9 трансмембранных сегментов [35]. В процессе созревания вольфрамин подвергается N-гликозилирова- нию и включается в высокомолекулярные комплексы (400 кД) в мембране [35].
Наиболее интенсивно синтез вольфрамина происходит в тканях головного мозга и поджелудочной железы. При проведении иммуногистохимического анализа тканей различных структур мозга крысы с оценкой содержания вольфрамина и мРНК, кодирующей этот белок, наиболее высокое содержание выявлено в гиппокампе, миндалевидном теле, зрительном бугре и структурах аллокортекса, т. е. в компонентах лимбической системы [65], а также в нейронах вентральных кохлеарных ядер и нижних холмиков среднего мозга [15]. Соответственно нарушение функции вольфрамина может приводить к патологическим изменениям в эмоциональной сфере, поведении и висцеральном контроле, а также нарушениям в обработке информации от сетчатки и улитки.
В связи с некоторым сходством клинической картины синдрома Вольфрама и митохондриального диабета (сахарный диабет, глухота, неврологические проявления, почечная недостаточность [5]), обусловленного точечной мутацией в ДНК митохондрий, долгое время считались, что в основе синдрома Вольфрама также лежит мутация в митохондриальной ДНК [ 12, 34, 50, 58]. Вольфрамин в этом случае должен относиться к собственным белкам митохондрий с нарушением функции митохондрий должны быть связаны фенотипические проявления заболевания. Однако исследования, проведенные в последнее время, нс выявили изменений ДНК митохондрий у больных с клинически очевидным синдромом Вольфрама (критериями включения в исследование было наличие сахарного диабета и атрофии зрительного нерва, развившихся в возрасте до 30 лет) [11,38].
В последние годы было проведено несколько исследований, доказывающих, что вольфрамин является собственным гликопротеином эндоплазматического ретикулума [51, 67, 68]. В чавании чего можно предположить присутствие нсбольшо концентрации вольфрамина комплексе Гольджи. На фрагменте и цвет остался красным, следовательно, вольфрамин не присутствует в митохондриях, и эти органеллы нс играют роли в патогенезе синдрома Вольфрама [67]. Функция вольфрамина в эндоплазматическом ретикулуме изучена не до конца. На сегодняшний день считается, что этот пептид участвует в трансмембранном транспорте, процессинге протеинов, синтезированных в эндоплазматическом ретикулуме [67], разрушении недостаточно упакованных протеинов [30]. Кроме того, было показано, что вольфрамин играет роль в регуляции гомеостаза кальция в эндоплазматическом ретикулуме [51, 67]. Опыты, проведенные на культуре ооцитов, показали, что вольфрамин способен индуцировать большой катионселективный ионный канал, который был заблокирован кальцием или магнием. Экспрессия вольфрамина увеличивала уровень кальция в цитоплазме ооцита. Высказано предположение о том, что вольфрамин может непосредственно являться новым кальциевым каналом в эндоплазматическом ретикулуме или регулировать активность кальциевого канхта. Возможно, что опосредованная вольфрамином регуляция внутриклеточного уровня кальция играет важную протективную роль в секреторных клетках, которые зависят от кальциевого сигнала. Нарушение этой функции может приводить к увеличению риска гибели клеток и. следовательно, к развитию клинической картины синдрома Вольфрама [51].
С другой стороны, клиническая картина синдрома Вольфрама может быть результатом перенапряжения эндоплазматического ретикулума вследствие нарушения процессинга и разрушения недостаточно упакованных протеинов [30]. Синтез к клетке наиболее активно происходит в эндоплазматическом ретикулуме, а также эта органелла обеспечивает специальную среду для посттрансляционных изменений и упаковки протеинов, секретируемых трансмембранно, и протеинов различных ком- партментов внутри клетки.’ ретикулума, которые обеспечивают упаковку, экспорт и ЛРг?Г° дацию [30]. Этот механизм увеличивает возможности органы’ по обработке протеинов, однако, поскольку он требует синге новых протеинов и липидов, ему свойственна задержка У мл копитаюших сразу несколько эффекторов, активировании» стрессом эндоплазматического ретикулума, вызывают актива цию специфических генов [32, 69, 71]. Разделение матричной рибонуклеиновой кислоты и последующий сплайсинг приводят к образованию нетрадиционного фактора транскрипции и активации промоторов генов.
Третья функциональная составляющая — это запуск программы клеточной гибели [30]. На этом этапе происходит уда- ление клеток, а следовательно, повреждение, причиняемое стрессом эндоплазматического ретикулума, становится необратимым. Эта составляющая имеет самую длинную задержку и регулируется специальными эффекторами с удивительной специфичностью для стресса эндоплазматического ретикулума. Возможно, в многоклеточном организме есть польза от элиминации клеток, которые имеют высокий уровень напряжения эндоплазматического ретикулума. В настоящее время непонятно, какую роль это может играть. Вероятно, гибель нескольких клеток в состоянии стресса может быть частью цикла регенерации, который позволяет вернуть функцию органа. Считается, что в реализации программы клеточной гибели у млекопитающих принимает участие сразу несколько эффекторов. Caspase-12 локализован на мембранах эндоплазматического ретикулума и проходит активацию в клетках, которые испытывают стресс эндоплазматического ретикулума [30]. У мышей с выбитым геном Caspase-12 -/-, подвергнутых действию токсинов, вызывающих стресс эндоплазматического ретикулума, отмечалась меньшая гибель клеток [49]. Caspase-12 принадлежит к семейству Caspase- 1, которые активируют каспазы, приводящие к гибели клеток. Таким образом, представляется, что клетки развили специфическую каспазу для соединения стресса эндоплазматического ретикулума с общими путями гибели клеток. Другим медиатором программы клеточной гибели при стрессе эндоплазматического ретикулума является фактор транскрипции CHOP (С/ЕВг- гомологичный протеин, также называемый GADD153).ноМ
эндоплазматического ретикулума. У мышей с выбитым а панкреатической киназы эндоплазматического pci™
огр К -/-) также развился сходный клинический синдром [291 U пни родились с близкими к нормальным островками Лангер- пгт но в течение первых недель произошла прогрессивная де- Svkuhh р-клеток. Когда клетки от мышей PERK -/- до развита диабета выращивали в культуре, р-клетки производили гор- илнальный проинсулин и секретировали нормальный гормон Когда р-клетки были перенесены из культуры с низким содержанием глюкозы в среду с высоким содержанием глюкозы, они произвели инсулина больше, чем неизмененные клетки [29].
Мутаиия в гене WFSI приводит к изменению структуры и следовательно, нарушению упаковки и функции вольфрамина’ который является собственным белком эндоплазматического ретикулума и участвует в трансмембранном транспорте, процессинге протеинов [67] и деградации измененных протеинов [30]. Очевидно, что нарушение его функций должно приводить к накоплению большого количества необработанных протеинов, не пригодных для использования на нужды клетки и организма, что запускает механизмы ответа на стресс эндоплазматического ретикулума. Данные, подтверждающие участие стресса эндоплазматического ретикулума в патогенезе DlDMOAD-синдро- ма, представлены в недавнем исследовании материалов аутопсий гипоталамуса пациентов с синдромом Вольфрама [24]. В супраоптическом ядре гипоталамуса не было найдено сразу несколько белков, присутствующих в норме, однако обнаружены их предшественники, и не было мутаций в генах, кодирующих эти белки [24], что свидетельствует о нарушении процессинга и деградации протеинов. Очевидно, что по сходному механизму происходит гибель клеток поджелудочной железы и миелинпродуиируюших нейронов [27]. Можно предположить, что стресс эндоплазматического ретикулума быстрее приводит к гибели наиболее интенсивно секретирующих клеток, в частности р-клеток поджелудочной железы, возможно, поэтому сахарный диабет часто становится первым проявлением синдрома.
Гетерогенность клинической картины синдрома Вольфрама, наиболее вероятно, определяется тяжестью повреждения функции вольфрамина и, следовательно, характером мутации в гене WFS1. В Великобритании был проведен анализ участка ДНК, кодирующего ген WFS1, у 30 больных с синдромом Вольфрама из 19 английских семей. Были найдены 24 мутации: 8 мутаций замены, 8 бессмысленных мутаций, 3 делеции из структуры гена, 1 вставка в структуру, 4 мутации сдвига рамки генетического кода. Наиболее часто мутации регистрировались в 8-м экзоне. Большинство пациентов были гетерозиготны по двум мутациям и не было найдено каких-либо общих характерных мутаций [31].
Данные были проанализированы на взаимоотношение генотипа и фенотипа, что показало очень близкую клиническую картину у больных, несущих одинаковую мутацию. В частности,
- пациента из Африки, которые имели большую близкородственную родословную, содержали гомозиготную мутацию замены в 4-м экзоне (замену цитозина на тимин в положении 406). Это прогнозирует значительное укорочение структуры вольфрамина, удаление трансмембранного и карбокситерминального доменов. Все больные с этой мутацией страдали тяжелыми психиатрическими осложнениями. Эти осложнения развивались между 30 и 40 годами и включали в себя значительное снижение памяти, эмоциональную лабильность, галлюцинации и эндогенную депрессию. Старшая сестра из этой семьи умерла в возрасте 39 лет. В возрасте между 20 и 30 годами в госпитале ей был поставлен диагноз пресенильной деменции, эндогенной депрессии, они страдала от частых галлюцинаций. Братья имели почти все описанные проявления синдрома Вольфрама: сахарный диабет, атрофию зрительного нерва, глухоту, осложнения со стороны мочевыделительной системы, неврологические проявления; ведущими клинике были психиатрические изменения [31].
В семье с гомозиготной делецией в 8-м экзоне (удаление
- нуклеотидов в положении 2648-2651) старшая сестра умерла в возрасте 28 лет от атрофии ствола мозга и центральной респираторной недостаточности.отаДов 1611-1624) и мутацию за- адСНИН В положснии 1433). Интересно, что KOBoftLSS” клинических проявлений у больных с одина- V пепплгУ\ЦИСЙ нсрсдко различался. В частности, в этой семье У’]5рпог браТа сахаРный Диабет развился в возрасте 7 лет, ат- Р Р я зрительного нерва — в 10 лет, несахарный диабет — в глухота — в 30 лет, нарушения со стороны мочевыдели- льнои системы — в 34 года, неврологические осложнения — в эо лет. У второго брата сахарный диабет развился в возрасте 12 лет, атрофия зрительного нерва — в 15 лет, несахарный диа- оет в 12 лет, глухота — в 15 лет, нарушения со стороны мо- чевыдслительной системы — в 33 года, неврологические осложнения — в 36 лет. У обоих пациентов выявлен первичный гипогонадизм: у них низкий уровень тестостерона и они никогда не брились [31].
В другом исследовании у девушки в возрасте 28 лет была зарегистрирована гомозиготная мутация вставки цитозина в положении 1038. У этой пациентки в возрасте 7 лет развился сахарный диабет, в 11 лет — атрофия зрительного нерва, в этом возрасте — нейросенсорная тугоухость на звуки высокой частоты (более 4000 Гц), в 20 лет — несахарный диабет, в 28 лет — дилатация мочевыводящих путей, мочепузырный рефлюкс и оп- соаменорея, которая, вероятно, имела центральное происхождение [19]. У пациентки оба родителя здоровы, есть 3 здоровых сибса и 1 больной брат, у которого в возрасте 20 лет имелись сахарный диабет и атрофия зрительного нерва. Одна из бабушек пациентки страдала сахарным диабетом 2-го типа [19].
Различные мутации сравнивали у семей с ранним развитием симптомов (до 5 лет) и также у тех, у кого симптомы развились после 6 лет. Не найдено очевидных различий в пропорциональном соотношении мутаций между двумя группами [31].
Одиночная гетерозиготная мутация в гене WFS1 не приводит к развитию клинической картины синдрома Вольфрама. Наиболее часто такая мутация увеличивает риск развития нейросенсорной тугоухости [15, 44, 68, 73], психиатрических заболеваний [64, 66] и сахарного диабета [21]. В Японии было проведено исследование гена WFS1 у 185 пациентов с сахарным диабетом 1-го типа и 380 здоровых лиц, не страдающих сахарным диабетом. Частота гетерозиготной мутации в гене WFS1 была достоверно выше у больных сахарным диабетом 1 -го типа по сравнению с группой контроля. При этом аутоиммунные компоненты сахарного диабета и сопутствующее аутоиммунное поражение щитовидной железы у больных с гетерозиготной мутацией встречались достоверно реже, чем у больных без такой мутации. Следует также отметить, что фактор транскрипции CHOP играет важную роль в NO-опосредованной гибели этих клеток [52]. Эти наблюдения, сделанные на культуре клеток, позволяют предположить, что стресс эндоплазматического ретикулума может также играть роль в потере р-клеток при сахарном диабете 1-го типа. Позже в Великобритании был проведен скрининг мутации в гене WFS1 у 323 пациентов с сахарным диабетом 2-го типа (170 из них заболели до 45 лет и 157 из этих больных имели минимум 1 больного сибса). Группа контроля состояла из 357 лиц без нарушений углеводного обмена. Частота мутации в гене WFS1 оказалась выше у больных с сахарным диабетом 2-го типа по сравнению с группой контроля [48]. Весьма вероятно, что изменение структуры вольфрамина играет важную роль в неаутоиммунной гибели р-клеток с сахарным диабетом 1-го и 2-го типов [7, 48].
Таким образом, причиной развития синдрома Вольфрама являются аутосомно-рецессивная гомозиготная мутация или несколько гетерозиготных мутаций в гене WFS1, кодирующих полипептид вольфрамин. Вольфрамин относится к собственным белкам эндоплазматического ретикулума. В основе патогенеза синдрома Вольфрама лежит стресс эндоплазматического ретикулума, который после ряда компенсаторных реакций приводит к гибели клетки. Гибель клетки должна наступать тем быстрее, чем больше нагрузка на эндоплазматический ретикулум клетки со стороны организма. Возможно, именно поэтому самым ранним проявлением синдрома Вольфрама становится инсулинзависимый, нсаутоиммунный сахарный диабет. Остается непонятным, почему у пациентов с синдромом Вольфрама не развиваются или развиваются минимально микроангиопатии, характерные для больных с сахарным диабетом 1-го типа.
Одиночная гетерозиготная мутация в гене WFS1 увеличивает риск развития сахарного диабета. Вероятно, нарушение структуры вольфрамина, приводящее к стрессу эндоплазматического ретикулума, играет важную роль в общих механизмах гибели клеток поджелудочной железы при сахарном диабете 1-го и 2-го типов.
1. Балаболкин М.И. Диабетология. -М., 2000.
2. Дедов И.И., Шестакова М.В., Максимова М.А. Федераль-ная целевая программа «Сахарный диабет». -М., 2002. -С. 6-8.
3. Дедов И.И., Шестакова М.В. Сахарный диабет. -М., 2003. -С. 66.
4. Кураева Т.Л., Зильберман Л.И.//Сахарный диабет. -2000. -№ 1. -С. 43-45.
5. Лавин Н. Эндокринология. -М., 1999.
6. American Diabetes Association: Report of the Expert Commit-tee on the Diagnosis and Classification of Diabetes Mellitus//Diabetes Care. -2003. -Vol.26. -P. 5-156.
7. Awata Т., Inoue K., Kurihara S. et al.//Biochem. Biophys. Res. Commun. -2000. -Vol.268. -P. 612-616.
8. Barrett T.G., Bundey S.E., Macleod A.F.//Lancet. -1995. -Vol.346. -P. 1453-1458.
9. Barrett T.G., Bundey S.E., Fielder A.R., Good P.А.//Eye. -1997. -Vol.6. -P. 882-888.
10. Barrett T.G., Bundey S.E.//J. Med. Genet. -1997. -Vol.34. -P. 838-841.
11. Barrett T.G., Scott-Brown M., Seller A. et al.//J. Med. Genet. -2000. -Vol.37. -P. 463-466.
12. Barrientos A., Volpini V., Casademont J. et al.//J. Clin. Invest. -1996. -Vol.97. -P. 1570-1576.
13. Baz P., Azar S.Т., Medlej R. et al.//Diabetes Care. -1999. -Vol.22. -P. 1376-1378.
14. Bertolotti A., Zhang Y., Hendershot L.M. et al.//Nature Cell Biol. -2000. -Vol.2. -P. 326-332.
15. Bespalova I.N., Camp G.V., Bom S.J.H. et al.//Hum. Mol. Genet. -2001. -Vol.10. -P. 2501-2508.
16. Borgna-Pignatti C., Marradi P., Pinelli L. et al.//J. Pediatr. -1989. -Vol.114. -P. 405-410.
17. Bouslama K., Naoui A., Rezgui L. et al.//Tunisie Med. -2002. -Vol.80. -P. 714-717.
18. Delepine M., Nicolino M., Barrett T. et al.//Nature Genet. -2000. -Vol.25. -P. 406-409.
19. Eller P., Foger В., Gander R. et al.//J. Med. Genet. -2001. -Vol.38. -P. 37-42.
20. Elligaard L., Molinari M., Helenius A.//Science. -1999. -Vol.286. -P. 1882-1888.
21. El-Shanti H., Lidral A.C., Jarrah N. et al.//Am. J. Hum. Genet. -2000. -Vol.66. -P. 1229-1236.
22. Fraser F.C., Gunn T.//J. Med. Genet. -1977. -Vol. 14. -P. 190-193.
23. Friedman A.D.//Cancer Res. -1996. -Vol. 56. -P. 3250-3256.
24. Gabreels B.A.Th.F., Swaab D.F., Kleijn D.P.V. et al.//J. Clin. Endocrinol. Metab. -1998. -Vol.83. -P. 4026-4033.
25. Galluzv P., Filosomi G., Vallone I. M. et al.//Neuroradiology. -1999. -Vol.41. -P. 729-731.
26. Gossain V.V., Sugawara M., Hagen G.A.//J. Clin. Endocri-nol. Metab. -1975. -Vol.41. -P. 1020-1024.
27. Gow A., Southwood C.M., Lazzarini R.A.//J. Cell Biol. -1998. -Vol.140. -P. 925-934.
28. Gunn Т., Bortolussi R., Little J.M. et al.//J. Pediatr. -1976. -Vol.89. -P. 565-570.
29. Harding H.P., Zeng H., Zhang Y. et al.//J. Mol. Cell. -2001. -Vol.7. -P. 1153-1163.
30. Harding H.P., Ron D.//Diabetes. -2002. -Vol.51. -P. 455-461.
31. Hardy C., Khanim F., Torres R. et al.//Am. J. Hum. Genet. -1999. -Vol.65. -P. 1279-1290.
32. Haze K., Yoshida H., Yanagi H. et al.//Mol. Biol. Cell. -1999. -Vol.10. -P. 3787-3799.
33. Hernandez-Mijares A., Morillas C., Lluch I. et al.//Diabetes Care. -1999. -Vol.22. -P. 1378-1379.
34. Hofmann S., Bezold R., Jaksch M. et al.//Genomics. -1997. -Vol.39. -P. 8-18.
35. Hofmann S., Philbrook C., Gerbits K., Bauer M.F.//Hum. Mol. Genet. -2003. -Vol.12. -P. 2003-2012. 56
36. Ito M., Jameson J. L., Ito M.//J. Clin. Invest. -1997. -Vol.99. -P. 1897-1905.
37. Ito Y., Pandey P., Mishra N. et al.//Mol. Cell. Biol. -2001. -Vol.21. -P. 6233-6242.
38. Jackson M.J., Bindoff L.A., Weber K. et al.//Diabetes Care. -1994. -Vol.17. -P. 728-733.
39. Jarrah N.S., El-Shanti H., Shennak M.M., Ajlouni K.M.//Ann. Saudi Med. -1999. -Vol.19. -P. 132-134.
40. Julier С.//Nature Genet. -2001. -Vol. 29. -P. 358-359.
41. Kinsley В.Т., Swift M., Dumont R. H., Swift R.G.//Diabetes Care. -1995. -Vol.18. -P. 1566-1570.
42. Lee A.S.//Curr. Opin. Cell Biol. -1992. -Vol.4. -P. 267-273.
43. Lee A.S.//Trends Biochem. Sci. -2001. -Vol. 26. -P. 504-510.
44. Lesperance M.M., Hall J.W. 3-rd, Bess F.H. et al.//Hum. Mol. Genet. -1995. -Vol.4. -P. 1967-1972.
45. Liu C.Y., Schroder M., Kaufman R.J.//J. Biol. Chem. -2000. -Vol.275. -P. 24881-24885.
46. McCullough K.D., Martindale J.L., Klotz L.-O. et al.//Mol. Cell. Biol. -2001. -Vol.21. -P. 1249-1259.
47. Mattina Т., Li Volti S., Palmeri P. et al.//Ophthal. Paediatr. Genet. -1998. -Vol.9. -P. 25-28.
48. Minton J.A.L., Hattersley А.Т., Owen K. et al.//Diabetes. -2002. -Vol.51. -P. 1287-1290.
49. Nakagava Т., Zhu Н„ Morishima N. et al.//Nature. -2000. -Vol.403. -P. 98-103.
50. Odawara M., Sasaki K., Yamashita K.//J. Clin. Endocrinol. Metab. -1995. -Vol.80. -P. 1290-1294.
51. Osman A.A., Saito M., Makepeace C. et al.//JBC Papers in Press. Published on October 3, 2003 as Manuscript M310331200.
52. Oyadomari S., Takeda K., Takiguchi M. et al.//Proc. Natl. Acad. Sci. USA. -2001. -Vol.98. -P. 10845-10850.
53. Page M.M., Asmal A.C., Edwards C.R.//Quart. J. Med. -1976. -Vol.45. -P. 505-520.
54. Peden N.R., Gay J.D., Jung R.Т., Kuwayti K.//Quart. J. Med. -1986. -Vol.58. -P. 167-180.
55. Piccoli G.В., Mezza E., Jeantet A., Segoloni G.P.//Nephrol. Dial. Transplant. -2003. -Vol.18. -P. 206-208.
56. Polymeropoulos M.H., Swift R.G., Swift M.//Nature Genet. -1994. -Vol.8. -P. 95-97.
57. Rando T.A., Norton J.C., Layser R.B.//Neurology. -1992. -Vol.42. -P. 1220-1224.
58. Rotig A., Cormier V., Chatelain P. et al.//J. Clin. Invest. -1993. -Vol.91. -P. 1095-1098.
59. Rugolo S., Mirabella D., Palumbo M.A. et al.//Eur. J. Obstet. Gynecol. Reprod. Biol. -2002. -Vol.105. -P. 192-193.
60. Salih M.A., Tuvemo T.//Acta Paediatr. Scand. -1991. -Vol.80. -P. 567-572.
61. Scolding N.J., Kellar-Wood H.F., Shaw C. et al.//Ann. Neurol. -1996. -Vol.39. -P. 352-360.
62. Shi Y., Vattem K.M., Sood R. et al.//Mol. Cell. Biol. -1988. -Vol.18. -P. 7499-7509.
63. Strom T.M., Hortnagel K., Hofman S. et al.//Hum. Mol. Genet. -1998. -Vol.7. -P. 2021-2028.
64. Swift M., Swift R.G.//Biol. Psychiatry. -2000. -Vol.47. -P. 787-793.
65. Swift R.G., Sadler D.В., Swift M.//Lancet. -1990. -Vol.336. -P. 667-669.
66. Swift R.G., Perkins D.O., Chase C.L. et al.//Am. J. Psychi-atry. -1991. -Vol.148. -P. 775-779.
67. Takeda K., Inoue H., Tanizawa Y. et al.//Hum. Mol. Genet. -2001. -Vol.10. -P. 477-478.
68. Tanizawa Y.//Rinsho Byori. -2003. -Vol.51. -P. 544-549.
69. Tirasophon W., Welihinda A.A., Kaufman R.J.//Genes Dev. -1998. -Vol.17. -P. 1812-1824.
70. Van den Ouweland J.M.W., Cryns K., Pennings R.J.E. et al.//J. Mol. Diagn. -2003. -Vol.5. -P. 88-95.
71. Wang X.Z., Harding H.P., Zhang Y. et al.//Eur. Mol. Biol. Org. -1998. -Vol.17. -P, 5708-5717.
72. Wolfram D.J., Wagener H.P//Mayo Clin. Proc. -1938. -Vol.13. -P. 715-718.
73. Young Т., Ives E., Lynch E. et al.//Hum. Mol. Genet. -2001. -Vol.10. -P. 2509-2514.
74. Zinszner H., Kuroda M., Wang X.Z. et al.//Genes Dev. -1998. -Vol.12. -P. 982-995.
ГЕНЕТИЧЕСКИЕ ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ. Наследственные заболевания и синдромы
Код исследования Наименование Срок выполнения, дни Цена исследования, руб 648 Поиск частых мутаций в экзоне 10 гена MEFV (Периодическая болезнь) 14 7 800 11 Синдром ломкой Х хромосомы: анализ метилирования (синдром Мартина-Белл) 14 6 900 31 Врожденная гиперплазия коры надпочечников (адреногенитальный синдром). Поиск 9-ти наиболее частых мутаций в гене CYP21A2 у родительской пары при недоступности материала больного ребенка 21 12 100 12 Синдром ломкой Х хромосомы: определение числа CGG повторов 21 10 000 514 Муковисцидоз. Секвенирование гена CFTR 90 30 000 29 Синдром Драве. Секвенирование гена SCN1A 90 30 000 30 Врожденная гиперплазия коры надпочечников (адреногенитальный синдром). Поиск 9-ти наиболее частых мутаций в гене CYP21A2 21 9 500 218 Тандемная масс-спектрометрия (спектр ацилкарнитинов, аминокислот) 30 4 800 492 Газовая хроматография образцов мочи (органические ацидурии) 21 6 500 38 Альбинизм глазокожный: поиск мутаций в гене TYR 21 11 500 4 Анализ полиморфизмов, ассоциированных с функциями интерлейкина 28В 7 2 000 39 Альбинизм глазокожный: поиск мутаций в гене OCA2 90 30 000 3 Анализ полиморфизма c.-13910C>T, ассоциированного с метаболизмом лактозы 7 1 300 46 Анемия Даймонда-Блекфена: поиск мутаций в гене RPS19 21 12 100 57 Атрофия зрительного нерва Лебера: поиск трех частых мутаций митохондриальной ДНК 14 5 800 58 Атрофия зрительного нерва Лебера: поиск 12-ти частых мутаций митохондриальной ДНК 21 9 200 59 Ахондроплазия: секвенирование гена FGFR3 90 30 000 64 Болезнь Вильсона-Коновалова: поиск 12 наиболее частых мутаций в гене ATP7B 14 8 100 66 Болезнь Гиршпрунга: поиск мутаций в гене EDNRB 21 15 000 67 Болезнь Гиршпрунга: поиск мутаций в экзонах 10, 11, 13, 14, 15 гена RET 21 10 500 68 Болезнь Гиршпрунга: поиск мутаций в гене NTRK1 30 22 000 69 Болезнь Гиршпрунга: поиск мутаций в гене ZEB2 30 27 000 85 Гипертрофическая кардиомиопатия: поиск мутаций в гене TNNT2 30 22 000 102 Ихтиоз вульгарный: поиск частых мутаций в гене FLG 21 8 100 103 Ихтиоз ламеллярный: поиск мутаций в гене TGM1 21 18 000 104 Липодистрофия: поиск мутаций в «горячих» участках гена LMNA 21 6 300 105 Липодистрофия: поиск мутаций в гене LMNA 21 18 000 107 Миотония Томсена/Беккера: поиск частых мутаций в гене CLCN1 14 8 100 113 Муковисцидоз: поиск крупных делеций/дупликаций в гене CFTR 21 10 400 114 Синдром Ретта: поиск мутаций в гене MECP2 30 15 000 117 Мышечная дистрофия Дюшенна/Беккера: поиск делеций и дупликаций в гене дистрофина у мальчиков 21 17 000 122 Мышечная дистрофия Эмери-Дрейфуса: поиск мутаций в гене EMD 21 8 900 123 Мышечная дистрофия Эмери-Дрейфуса: поиск мутаций в гене LMNA 21 18 000 305 Глициновая энцефалопатия (секвенирование генов GLDC, GCSH, AMT) 90 35 000 124 Мышечная дистрофия Эмери-Дрейфуса: поиск мутаций в гене FHL1 21 15 000 125 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I: поиск дупликаций на хромосоме 17 в области гена PMP22 14 5 800 126 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I: поиск мутаций в гене GJB1 (Cx32) 21 7 500 129 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I: поиск мутаций в гене EGR2 21 10 400 132 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I: поиск частых рецессивных мутаций в генах FGD4, Sh4TC2, FIG4, GDAP1 14 8 100 133 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I: поиск частых мутаций в генах NDRG1 и Sh4TC2 14 7 000 136 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II: поиск мутаций в гене GDAP1 21 13 800 137 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II: поиск мутаций в гене NEFL 21 12 000 140 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II: поиск мутаций в гене DNM2 90 30 000 142 Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II: поиск мутаций в гене FIG4 90 30 000 143 Нейросенсорная несиндромальная тугоухость: поиск частых мутаций в гене GJB2 14 2 500 152 Прогерия Хатчинсона-Гилфорда: поиск мутаций в гене LMNA 21 21 000 157 Спастическая параплегия Штрюмпеля: поиск мутаций в гене ATL1 90 30 000 161 Синдром Прадера-Вилли/Ангельмана 14 6 900 163 Псевдоксантома эластическая: поиск частых мутаций в гене ABCC6 21 6 300 164 Псевдоксантома эластическая: поиск мутаций в гене ABCC6 30 47 000 169 Синдром Аарскога-Скотта: секвенирование гена FGD1 30 25 000 170 Синдром Ваарденбурга: поиск мутаций в гене PAX3 21 15 000 171 Синдром Вильямса: поиск делеций в регионе 7q11 21 10 400 173 Синдром Коккейна: поиск мутаций в гене ERCC6 90 30 000 175 Синдром Коффина-Лоури: поиск мутаций в гене RPS6KA3 90 30 000 178 Поиск мутаций в гене BCS1L 21 10 400 179 Синдром Марфана: поиск мутаций в гене FBN1 90 30 000 180 Синдром множественной эндокринной неоплазии второго типа (МЭН2): секвенирование гена RET 90 30 000 184 Синдром Мовата-Вильсона: поиск мутаций в гене ZEB2 30 25 500 188 Синдром Смит-Магенис: поиск делеций в регионе 17p11.2 14 15 000 192 Синдром фон Хиппеля-Линдау: секвенирование гена VHL 21 8 000 197 Спинальная амиотрофия типы I, II, III, IV: поиск делеций в гене SMN1 14 6 700 204 Туберозный склероз: поиск мутаций в гене TSC1 и TSC2 90 30 000 210 Хорея Гентингтона: поиск наиболее частых мутаций в гене HTT 14 5 500 211 Тяжелый комбинированный иммунодефицит, Х-сцепленный: поиск мутаций в гене IL2RG 21 10 400 212 Ихтиоз ламеллярный: поиск мутаций в гене ALOX12B 21 19 000 213 Экзостозы множественные: поиск мутаций в гене EXT1 и EXT2 90 30 000 225 Синдром Беквита-Видемана 14 6 900 245 Галактоземия (секвенирование гена GALT) 20 20 000 273 Синдром Блума (поиск частых мутаций в гене RECQL3 (BLM) 14 5 800 297 Мукополисахаридоз 2 типа (секвенирование гена IDS) 90 30 000 335 Миотоническая дистрофия тип 1 и 2 (секвенирование генов DMPK и ZNF9) 90 30 000 351 Синдром Жильбера 14 3 700 352 Синдром Алажилля (секвенирование гена JAG1) 90 30 000 353 Синдром удлиненного интервала QT 90 30 000 361 Гипофосфатемический витамин-D-резистентный рахит (секвенирование гена PHEX) 90 30 000 542 Синдром Цельвегера 90 30 000 544 Синдром Чедиака-Хигаши (секвенирование гена LYST) 90 30 000 546 Голопрозэнцефалия (секвенирование генов FGF8, GLI2, GLI3, PTCh2, SHH, SIX3, TGIF1, ZIC2 90 30 000 548 Врожденная дизэритропоэтическая анемия (секвенирование генов CDAN1, SEC23B) 90 30 000 550 Дистрофия роговицы (секвенирование генов TGFBI, SLC4A11) 90 30 000 561 Айкарди-Гутьерес синдром (секвенирование генов TREX1, RNASEh3B, ADAR) 90 30 000 520 Талассемия 90 30 000 545 Наследственная моторно-сенсорная демиелинизрующая нейропатия 90 30 000 579 Секвенирование гена AR 90 30 000 605 Подтверждение мутации, выявленной при NGS секвенированием по Сэнгеру 30 5 000 612 Синдром Аксенфельда-Ригера: поиск мутаций в гене FOXC1 21 9 200 613 Синдром Андерсена: поиск мутаций в гене KCNJ2 21 9 000 614 Синдром Антли-Бикслера: поиск мутаций в экзоне 9 гена FGFR2 21 6 900 616 Аутоиммунный лимфопролиферативный синдром: поиск мутаций в «горячих» участках гена TNFRSF6 (FAS) 21 6 000 617 Аутоиммунный лимфопролиферативный синдром: поиск мутаций в гене TNFRSF6 (FAS) 21 16 000 618 Первичная прогрессирующая афазия: поиск мутаций в гене GRN 21 13 000 621 Синдром Баннаян-Райли-Рувалькаба 21 15 500 623 Синдром Бёрта-Хога-Дьюба: поиск мутаций в гене FLCN 21 23 000 626 Синдром Боуэна-Конради: поиск мутаций в гене EMG1 21 10 400 627 Брахидактилия: поиск мутаций в гене HOXD13 21 10 400 628 Брахидактилия: поиск мутаций в экзонах 8 и 9 гена ROR2 21 9 200 629 Брахидактилия: поиск мутаций в гене NOG 21 5 800 630 Синдром Ван дер Вуда: поиск мутаций в гене IRF6 21 15 500 632 Синдром врожденной центральной гиповентиляции: поиск частых мутаций в гене PHOX2B 14 5 800 633 Синдром Галлервордена-Шпатца: поиск наиболее частых мутаций в гене PANK2 21 6 300 637 Синдром Германски-Пудлака: поиск частых мутаций в гене HPS1 21 7 500 657 Фенилкетонурия: расширенный поиск мутаций в гене PAH (25 шт.) 14 9 900 658 Фенилкетонурия: поиск мутаций в гене PAH 21 23 000 659 Торсионная дистония: поиск мутаций в гене TOR1A 21 11 400 785 Муковисцидоз: Расширенный поиск частых мутаций в гене CFTR (30 шт.) 14 10 400 791 Поиск наиболее частых мутаций в гене AR 21 5 800 833 Синдром Сильвера-Рассела 14 6 900 859 Анализ числа (CAG)-повторов в гене андрогенового рецептора (AR), частые делеции в AZF локусе, частые мутации в гене CFTR (22 шт.+IVS8TT) 21 9 000 864 Подтверждение мутации, выявленной при NGS секвенированием по Сэнгеру у трио 30 13 000 865 Поиск делеций в гене NF1 методом MLPA 21 6 900 962 Поиск наиболее частых мутаций в генах ATXN1, ATXN2, ATXN3 14 7 600 987 Наследственная нейропатия с подверженностью параличу от сдавления (поиск мутаций в гене PMP22) 21 10 400 988 Клиническое секвенирование экзома трио 90 140 000 1005 Частичный анализ гена NOTCh4 (CADASIL синдром) 21 10 000 1056 Лицелопаточно-плечевая мышечная дистрофия тип 1 40 45 000 1060 Определение числа копий генов SMN1, SMN2 14 8 500 1098 Поиск наиболее частых мутаций в гене ATXN7 14 5 300 1111 Поиск мутаций в гене MEFV 21 17 000 1118 Частичный анализ гена PLP методом MLPA -дупликации гена (Пелициуса-Мельцбахера) 15 7 000 1131 Дефицит карнитина системный первичный (поиск мутаций в гене SLC22A5) 21 19 000 1143 Синдром Ниймеген: Поиск наиболее частых мутаций в гене NBN 14 6 000 1147 Поиск делеций в гене NF2 методом MLPA 21 6 900 1187 Синдром Нунан 90 35 000 1210 Поиск наиболее частых мутаций в экзонах 10, 11 гена RET при МЭН2А 21 9 000 1217 Поиск частых мутаций в гене GALT 21 6 000 1290 Окулофарингеальная мышечная дистрофия: поиск наиболее частых мутаций в гене PABPN1 14 7 000 1292 Поиск частых мутаций в гене POLG (6 мутаций) методом MLPA 14 7 000 1293 Поиск мутаций в «горячих» участках гена ACVR1 21 12 000 1437 Поиск мутаций в гене GJB2 (Cx26) 21 6 000 Частоты носительства генетических Мутаций опухолевых синдромов недооценены
Более 10% всех онкологических заболеваний – это наследственные болезни. Ими занимаются врачи-генетики ФБГНУ «МГНЦ». Одна из первоочередных задач, которая стоит перед ними – налаживание тесного сотрудничества с врачами-онкологами для создания современной программы помощи пациентам.
Сегодня описано более 200 заболеваний, проявление которых связано с передачей из поколения в поколение фатальной предрасположенности к развитию рака. При этом наследственные опухолевые синдромы, как правило, проявляют себя на 10 – 15 лет раньше, чем так называемые спорадические, проще говоря, возникшие без очевидных причин.
Владимир Стрельников, доктор биологических наук, заведующий лабораторий эпигенетики ФБГНУ «МГНЦ»: «Считается, что из 1 миллиона жителей 5 500 человек являются носителями мутаций каких-либо наследственных опухолевых синдромов (или же 1 из 180 человек). Мы считаем, что частоты носительства недооценены, на самом деле они могут составлять 1: 100 – 1:150 человек. В России точные цифры еще предстоит выяснить. Чтобы уточнить частоты носительства и спектр клинических проявлений, требуется селективный (выборочный) скрининг. И анализировать нужно все возможные мутации. Популярный сегодня «экономичный подход», когда ищут изменения только в «горячих точках» генов, себя изжил! Здесь нам на помощь приходят методы высокопроизводительного секвенирования, ведь эффективная ДНК-диагностика наследственных опухолевых синдромов без них невозможна. Сегодня уже ясно, что такая диагностика должна проводиться в центрах опережающего развития, крупных медико-генетических научных центрах, таких, каким является МГНЦ».
Министерство здравоохранения России рассматривает возможность создания государственной программы профилактики онкологических заболеваний. ФБГНУ «МГНЦ» внес свои рекомендации, в частности по определению групп риска в семьях, отягощенных наследственными опухолевыми синдромами.
Как выявить генетические дефекты будущего ребенка? | Научные открытия и технические новинки из Германии | DW
То, что в крови беременной вскоре после зачатия появляются и фрагменты ДНК эмбриона, было впервые обнаружено 15 лет назад. С тех пор ученые пытаются использовать этот феномен для получения информации о здоровье, а вернее — о возможных генетических и хромосомных дефектах будущего ребенка. И не только пытаются, но уже достигли на этом пути значительных успехов.
Правда, до самого недавнего времени единственным способом пренатальной, то есть дородовой диагностики, позволяющей выявлять генетические аномалии такого рода, был так называемый амниоцентез. Это не только крайне малоприятная, но и связанная с риском выкидыша инвазивная процедура, в ходе которой врач специальной иглой через переднюю брюшную стенку беременной выполняет пункцию зародышевой оболочки с целью получения пробы околоплодных вод для их последующего лабораторного исследования.
Однако совсем недавно был разработан метод, позволяющий на основе анализа крови беременной надежно диагностировать у плода трисомии — прежде всего, трисомию по 21-й хромосоме, то есть синдром Дауна. Этот метод уже применяется в США, в июле ожидается его поступление и на немецкий рынок.
Новый успех дородовой диагностики
Но разработка такого теста на трисомию была, как теперь выяснилось, лишь первым шагом в перспективном направлении. Потому что группа американских медиков добились нового крупного успеха в сфере дородовой диагностики: им удалось на основе анализа проб крови матери и слюны отца полностью реконструировать наследственный материал будущего ребенка и проверить его на наличие так называемых моногенных заболеваний.
Руководитель проекта Джей Шендью (Jay Shendure), профессор генетики Вашингтонского университета в Сиэтле, поясняет: «Это заболевания, вызываемые дефектом какого-то одного гена и передающиеся по наследству по законам классической генетики. Каждое такое заболевание встречается весьма редко, но зато их много, так что в среднем у каждого сотого новорожденного обнаруживается такой дефект».
Учеными описано сегодня уже более трех тысяч моногенных наследственных заболеваний. Среди наиболее известных — муковисцидоз и хорея Хантингтона, однако сюда же относятся и фибродисплазия, и болезнь Гоше, и синдром Марфана, и болезнь Вильсона-Коновалова, и многие другие недуги, названия которых подавляющее большинство непричастных к медицине, да и многое врачи, к счастью, никогда в жизни даже не слышали. И все же та или иная из этих болезней встречается в среднем у одного процента новорожденных. «Один процент — это довольно высокий показатель, — говорит профессор Шендью, — тем более, что речь идет, как правило, об очень тяжелых заболеваниях. И многие из них неизлечимы».
И старые мутации, и новые…
Сегодня, даже когда беременная решается на амниоцентез, медики обычно проверяют ДНК будущего ребенка лишь на трисомии и на наличие вполне определенных опасных мутаций — тех, что ранее были обнаружены у родителей, дедушек или бабушек, — то есть выясняют, передался ли этот генный дефект по наследству следующему поколению. Но дело в том, что моногенные заболевания могут не только передаваться по наследству, но и возникать в результате новых, впервые появившихся у плода мутаций. В геноме каждого ребенка таких новых мутаций в среднем около полусотни, и среди них может оказаться и такая, что чревата развитием тяжелого заболевания.
Сегодняшние методы пренатальной диагностики не способны выявить такие мутации, но вскоре, похоже, ситуация кардинально изменится — благодаря работам профессора Шендью и его коллег. Ученый говорит: «Я думаю, здесь просматриваются два важных потенциальных преимущества. Во-первых, доступ к полной генетической информации о плоде может быть получен без рискованной инвазивной процедуры амниоцентеза. Во-вторых, похоже, нам удастся просканировать геном плода сразу на множество моногенных заболеваний. Мы сможем даже выявить новые мутации. Тут еще есть над чем поработать, но мы показали, что в принципе это функционирует».
Ученые анализировали пробы материнской крови и отцовской слюны от двух супружеских пар. Личного контакта между исследователями и подопытными не было, пробы поступили из базы данных местной клиники. Одна из женщин была на 18-й неделе беременности, другая — на восьмой. Прежде всего, ученые полностью секвенировали геномы родителей — благо, сегодняшнее оборудование позволяет сделать это очень быстро и с высокой точностью. Затем настала очередь генома ребенка. По словам исследователей, в ДНК, содержащейся в плазме материнской крови, наследственный материал плода составляет в среднем 13 процентов. Выделить его, а затем секвенировать было не так уж трудно.
39 из 44 — много это или мало?
Самой сложной и трудоемкой задачей был дальнейший сравнительный анализ генов родителей и эмбриона. «Нам удалось установить, какие варианты генов плод унаследовал от матери, а какие — от отца, — говорит профессор Шендью. — Кроме того, мы смогли выявить и новые мутации, те, что отсутствуют в геномах родителей и впервые появились в ДНК эмбриона».
После того, как дети обеих пар появились на свет, их пуповинная кровь была проанализирована непосредственно, и геномы секвенированы повторно. При контрольном сравнении результатов оказалось, что из 44 новых мутаций у одного из новорожденных ученые смогли заранее, на стадии внутриутробного развития, обнаружить 39. Неплохо для начала, говорит исследователь, но тут же добавляет: «С другой стороны, прежде чем такой анализ войдет в клиническую практику, его точность и достоверность должны быть существенно повышены».
На это уйдет не один год. Кроме того, нынешний анализ обошелся в 50 тысяч долларов — это терпимо, когда речь идет об исследовательском проекте, но совершенно неприемлемо для рутинного метода диагностики. К тому же полученная генетическая информация, при полной надежности самого метода, может быть интерпретирована очень по-разному. И, наконец, открытыми остаются многие этические вопросы. Но ответа на них бессмысленно ждать от генетиков.
Клинико-генетическая характеристика наследственных заболеваний и синдромов с глаукомой | Хлебникова
1. Аветисов Э.С., Ковалевский Е.И., Хватова А.В. Руководство по детскойофтальмологии. М.: Медицина, 1987. — 496 с.
2. Ковалевский Е.И., Смирнова Т.С., Медведева Н.И. Критерии клинической диагностики врожденных детских глауком. Профилактика слепоты и слабовидения у детей: Материалы Всерос. науч.-практ. конференции детских офтальмологов. М., 1996. с.84-86.
3. Катаргина Л.А., Хватова А.В., Коголева Л.В., Мазанова Е.В., Гвоздюк Н.А. Значение современных методов визуализации при аномалиях переднего сегмента глаза и врожденной глаукоме у детей. Российский офтальмологический журнал. 2010; 3 (2): 7-11.
4. McKusick V.A. Online Mendelian inheritance in man. Catalogs of Human Genes and Genetic Disorders. Baltimore; London: John Hopkins, Univ.press. 2010. http://www.ncbi.nlm.nih.gov/OMIM
5. Bowen, P., Lee, C. S. N., Zellweger, H., Lindenberg, R. A familial syndrome of multiple congenital defects. Bull. Johns Hopkins Hosp. 1964;114: 402-414,
6. Combarros, O., Calleja, J., Leno, C., Berciano, J. Association of an ataxia indistinguishable from Friedreich’s ataxia and congenital glaucoma in a family: a new syndrome. J. Med. Genet. 1988; 25: 44-46.
7. Furuichi, T., Dai, J., Cho, T.-J, Sakazume, S., Ikema, M., Matsui, Y., et al. CANT1 mutation is also responsible for Desbuquois dysplasia, type 2 and Kim variant. J. Med. Genet. 2011; 48: 32-37.
8. Rodriguez-Rojas, L.X., Garcia-Cruz, D., Mendoza-Topete, R., Barba, L.B., Barrios, M.T., Patino-Garcia, B., et al. Familial iridogoniodysgenesis and skeletal anomalies: a probable new autosomal recessive disorder. Clin. Genet. 2004; 66: 23-29.
9. Godfrey, C., Clement, E., Mein, R., Brockington, M., Smith, J., Talim, B. et al. Refining genotype-phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007; 130: 2725-2735.
10. an Reeuwijk, J., Janssen, M., van den Elzen, C., Beltran-Varero de Bernabe, D., Sabatelli, P., et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005; 42: 907-912.
11. Vuillaumier-Barrot, S., Bouchet-Seraphin, C., Chelbi, M., Devisme, L., Quentin, S., Gazal, S., et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am. J. Hum. Genet. 2012; 91: 1135-1143.
12. Phillips, J.C., Del Bono, E.A., Haines, J.L., Pralea, A.M., Cohen, J.S., Greff, L.J., et al. A second locus for Rieger syndrome maps to chromosome 13q14. Am. J. Hum. Genet. 1996; 59: 613-619.
13. Weisschuh, N., Wolf, C., Wissinger, B., Gramer, E. A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters’ anomaly. Clin. Genet. 2008; 74: 476-480.
14. Lowry, R.B., Gould, D.B., Walter, M.A., Savage, P.R. Absence of PITX2, BARX1, and FOXC1 mutations in De Hauwere syndrome (Axenfeld-Rieger anomaly, hydrocephaly, hearing loss): a 25-year follow up. Am. J. Med. Genet. 2007; 143A: 1227-1230.
15. Meuwissen, M.E.C., Halley, D.J.J., Smit, L.S., Lequin, M.H., Cobben, J.M., de Coo, R., et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet. Med. 2015; 17: 843-853.
16. Karadeniz, N. N., Kocak-Midillioglu, I., Erdogan, D., Bokesoy, I. Is SHORT syndrome another phenotypic variation of PITX2? Am. J. Med. Genet.2004; 130A: 406-409.
17. Reis, L.M., Tyler, R.C., Schilter, K.F., Abdul-Rahman, O., Innis, J.W., Kozel, B.A., et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 2011; 130: 495-504.
18. Liegel, R.P., Handley, M.T., Ronchetti, A., Brown, S., Langemeyer, L., Linford, A., et al. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg Micro syndrome in humans. Am. J. Hum. Genet. 2013; 93: 1001-1014.
19. Kosho, T., Miyake, N., Hatamochi, A., Takahashi, J., Kato, H., Miyahara, T., et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. 2010; 152A: 1333-1346.
20. Voermans, N.C., Bonnemann, C.G., Lammens, M., van Engelen, B.G., Hamel, B.C.J. Myopathy and polyneuropathy in an adolescent with the kyphoscoliotic type of Ehlers-Danlos syndrome. Am. J. Med. Genet. 2009; 149A: 2311-2316.
21. Hilton, E., Johnston, J., Whalen, S., Okamoto, N., Hatsukawa, Y., Nishio, J., et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Europ. J. Hum. Genet. 2009; 17: 1325-1335.
22. Bongers,E. M. H. F., Huysmans, F.T., Levtchenko, E.,de Rooy, J. W., Blickman, J. G., Admiraal, R. J. C., et al. Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Europ. J. Hum. Genet. 2005; 13: 935-946.
23. Brice, G., Ostergaard, P., Jeffery, S., Gordon, K., Mortimer, P. S., Mansour, S.A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clin. Genet. 2013; 84: 378-381.
24. Cau, M., Addis, M., Congiu, R., Meloni, C., Cao, A., Santaniello, S., et al. A locus for familial skewed X chromosome inactivation maps to chromosome Xq25 in a family with a female manifesting Lowe syndrome. J. Hum. Genet. 2006; 51: 1030-1036.
25. Florijn, R.J., Loves, W., Maillette de Buy Wenniger-Prick, L.J.J.M., Mannens, M.M.A.M., Tijmes, N., Brooks, S.P., et al. New mutations in the NHS gene in Nance-Horan syndrome families from the Netherlands. Europ. J. Hum. Genet. 2006; 14: 986-990.
26. Shirley, M.D., Tang, H., Gallione, C.J., Baugher, J.D., Frelin, L.P., Cohen, B., et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. New Eng. J. Med. 2011; 3368: 1971-979.
27. Hall, B.D., Berg, B.O., Rudolph, R.S., Epstein, C.J. Pseudoprogeria — Hallermann-Streiff (PHS) syndrome. Birth Defects Orig. Art. Ser. 1974; 10(7): 137-146.
28. Majewski, F. Caroline Crachami and the delineation of osteodysplastic primordial dwarfism type III, an autosomal recessive syndrome. Am. J. Med. Genet. 1992; 44: 203-209.
29. Platzer, K., Huning, I., Obieglo, C., Schwarzmayr, T., Gabriel, R., Strom, T. M.,et al. Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures. Am. J. Med. Genet. 2014; 164A: 1976-1980.
30. Smith, J.D., Hing, A.V., Clarke, C.M., Johnson, N.M., Perez, F.A., Park, S.S., et al. University of Washington Center for Mendelian Genomics, Cunningham, M. L. Exome sequencing identifies a recurrent de novo ZSWIM6 mutation associated with acromelic frontonasal dysostosis. Am. J. Hum. Genet, 2014; 95: 235-240.
31. Winter, R.M., Patton, M.A., Challener, J., Mueller, R.F., Baraitser, M. Moore-Federman syndrome and acromicric dysplasia: are they the same entity? J. Med. Genet. 1989; 26: 320-325.
32. Prabhu, V.G., Kozma, C., Leftridge, C.A., Helmbrecht, G.D., France, M.L. Dyssegmental dysplasia Silverman-Handmaker type in a consanguineous Druze Lebanese family: long term survival and documentation of the natural history. Am. J. Med. Genet. 1998; 75: 164-170.
33. Hoornaert, K. P., Vereecke, I., Dewinter, C., Rosenberg, T., Beemer, F.A., Leroy, J.G., et al. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Europ. J. Hum. Genet. 2010; 18: 872-880, Note: Erratum: Europ. J. Hum. Genet. 2010; 18: 881 only.
34. Huang, C.-T., Chu, S.-Y., Lee, Y.-C. Optical coherence tomography of chorioretinopathy caused by mucopolysaccharidoses. Ophthalmology. 2015; 122: 1535-1537.
35. Latkany, P.A., Jabs, D.A., Smith, J.R., Rosenbaum, J.T., Tessler, H., Schwab, I.R., et al. Multifocal choroiditis in patients with familial juvenile systemic granulomatosis. Am. J. Ophthal. 2002; 134: 897-904.
36. Al-Torki, N.A., Sabry, M.A., Al-Awadi, S.A., Al-Tarkeit, N. Lowry-Maclean syndrome with osteopenic bones and possible autosomal dominant inheritance in a Bedouin family. (Letter) Am. J. Med. Genet. 1997; 73: 491-492.
37. Rutsch, F., MacDougall, M., Lu, C., Buers, I., Mamaeva, O., Nitschke, Y., et al. A specific IFIh2 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 2015; 96: 275-282.
38. Ackerman, J. L., Ackerman, A. L., Ackerman, A. B. Taurodont, pyramidal and fused molar roots associated with other anomalies in a kindred. Am. J. Phys. Anthrop. 1973; 38: 681-694.
Синдром Аарского | Информационный центр по генетическим и редким заболеваниям (GARD) — программа NCATS
От 80% до 99% людей имеют эти симптомы Широкая стопа Широкие ноги
Широкая стопа
[ более ]
0001769
Широкая ладонь Широкие руки
Широкая рука
Широкая ладонь
[ более ]
0001169
Камптодактилия пальца Постоянное сгибание пальца
0100490
Киноварь вывернутой нижней губы Опущенная нижняя губа
Вывернутая наружу нижняя губа
[ более ]
0000232
Гипертелоризм Широко посаженные глаза
Широко расставленные глаза
[ более ]
0000316
Платок мошонка Мошонка окружает пенис
0000049
Короткая ножка Короткие ноги
Маленькие ноги
[ более ]
0001773
Короткая ладонь 0004279
Низкий рост Маленький рост
Уменьшенный рост
[ более ]
0004322
Маленькая рука Непропорционально маленькие руки
0200055
Пупочная грыжа 0001537
30% -79% людей имеют эти симптомы Антевертированные ноздри Поднятый кончик носа
Кончик носа перевернут
Вздернутые ноздри
Вздернутый нос
[ более ]
0000463
Лоб широкий Увеличенная ширина лба
Широкий лоб
[ более ]
0000337
Клинодактилия 5-го пальца Постоянное искривление мизинца
0004209
Когнитивные нарушения Нарушение познания
Когнитивная аномалия
Когнитивные дефекты
Когнитивный дефицит
Умственное нарушение
Умственное нарушение
[ более ]
0100543
Крипторхизм Неопустившиеся яички
Неопустившееся яичко
[ более ]
0000028
Нисходящие глазные щели Наклон отверстия между веками вниз
0000494
Порок развития наружного уха 0008572
Синдактилия пальцев 0006101
Высокий передний волосяной покров Высокая лобная линия роста волос
0009890
Гиперэластичная кожа Гиперэластичная кожа
Гиперэластичность кожи
Эластичная кожа
[ более ]
0000974
Паховая грыжа 0000023
Гипергибкость суставов Суставы выходят за пределы ожидаемого диапазона движений
0005692
Длинный желобок 0000343
Низко посаженные, повернутые назад уши 0000368
Птоз Опущенное верхнее веко
0000508
Широкая переносица Широкая носовая перемычка
Широкий корень носа
Расширенный носовой мост
Увеличенная ширина переносицы
Увеличенная ширина переносицы
Увеличенная ширина переносицы
Увеличенная ширина переносицы
Носовая перемычка широкая
Широкая переносица
Расширенный носовой мост
[ более ]
0000431
5% -29% людей имеют эти симптомы Аномальная сегментация и сращение позвонков 0005640
Нарушение морфологии сердечно-сосудистой системы 0030680
Синдром дефицита внимания с гиперактивностью Дефицит внимания
Синдром дефицита внимания
Синдром дефицита внимания и гиперактивности
Дефицит внимания
Детский дефицит внимания / синдром гиперактивности
[ более ]
0007018
Волчья пасть Расщелина неба
0000175
Расщелина верхней губы Заячья губа
0000204
Застойная сердечная недостаточность Сердечная недостаточность
Сердечная недостаточность
Сердечная недостаточность
[ более ]
0001635
Отсроченное прорезывание зубов Отсроченное извержение
Задержка прорезывания зубов
Отсроченное прорезывание зубов
Извержение с задержкой
Позднее прорезывание зубов
Позднее прорезывание зубов
[ более ]
0000684
Эпикантус Складки глаз
Выраженные складки глаз
[ более ]
0000286
Genu recurvatum Заднее колено
Гиперэкстензия колена
[ более ]
0002816
Гипоплазия верхней челюсти Уменьшение размера верхней челюсти
Уменьшение размера верхней челюсти
Верхнечелюстная недостаточность
Ретрузия верхней челюсти
Малая челюсть
Малая верхняя челюсть
Малые кости верхней челюсти
Недостаток верхней челюсти
Ретрузия верхней челюсти
[ более ]
0000327
Megalocornea Увеличенная роговица
0000485
Pectus excatum Сундук-воронка
0000767
Pes planus Плоскостопие
Плоскостопие
[ более ]
0001763
Круглый торцевой Круглое лицо
Круглый внешний вид лица
Круглая форма лица
[ более ]
0000311
Короткая шея Уменьшенная длина шеи
0000470
Одиночная поперечная ладонная складка 0000954
Косоглазие Косоглазый
Косоглазие
Прищуренные глаза
[ более ]
0000486
Талипы 0001883
У 1% -4% людей есть эти симптомы Широкий желобок 0000289
Глобальная задержка развития 0001263
Умственная отсталость, легкая Умственная отсталость, пограничная легкая
Легкая и непрогрессирующая умственная отсталость
Умеренная умственная отсталость
[ более ]
0001256
Пупок ромбовидной формы 0032277
Выступающий пупок Выступающий пупок
Выступающий пупок
[ более ]
0001544
Короткий 5-й палец Короткий пятый палец
Короткие пятые пальцы
Короткий мизинец
Короткий мизинец
Короткий мизинец
[ более ]
0009237
Короткий нос Уменьшение длины носа
Укороченный нос
[ более ]
0003196
Процент людей, у которых есть эти симптомы, недоступен через HPO Брахидактилия Короткие пальцы рук или ног
0001156
Гипермобильность шейного отдела позвоночника 0003318
Клинодактилия Постоянное искривление пальца
0030084
Изогнутая линейная ямка под нижней губой 0002055
Задержка полового созревания Задержка пубертатного развития
Задержка полового созревания
Задержка полового созревания
[ более ]
0000823
Отказ от роста Падающий вес
Падение веса
[ более ]
0001508
Гиперрастяжимость суставов пальцев Повышенная растяжимость суставов пальцев
Сверхрастяжимые цифры
Гиперрастяжимый палец
[ более ]
0001187
Гиперметропия Дальнозоркость
Дальнозоркость
[ более ]
0000540
Гиподонтия Отсутствие развития от одного до шести зубов
0000668
Гипоплазия зубовидного отростка 0003311
Увеличенное соотношение верхнего и нижнего сегментов 0012774
Большая мочка уха Мясистая мочка уха
Мясистые мочки ушей
Выступающие мочки ушей
выступающие мочки уха
[ более ]
0009748
Небольшой рост 0003502
Радиальное отклонение пальца 0009466
Сколиоз 0002650
Синдактилия Перепончатые пальцы рук или ног
0001159
Пик вдовы Пик линии роста волос
Точка линии роста волос
Остроконечная линия роста волос в передней части головы
V-образная линия роста волос спереди
[ более ]
0000349
Х-сцепленное рецессивное наследование 0001419
Генетические синдромы, редкие эндокринные заболевания
Генетические синдромы / редкие эндокринные заболевания
Генетические синдромы — это заболевания, вызванные наследственными мутациями в наших генах.Другими словами, они могут передаваться от родителей к детям и передаваться в семьях. Это очень редкие заболевания, которые встречаются примерно в 1 из 500 000 или 1 000 000 случаев. Благодаря большому количеству пациентов мы коллективно накопили опыт ведения пациентов со следующими генетическими синдромами, влияющими на наши эндокринные органы.
Когда пациенты с заболеваниями щитовидной железы, паращитовидных желез и надпочечников нуждаются в генетическом тестировании?
Подавляющее большинство заболеваний щитовидной железы и паращитовидных желез не имеют генетической составляющей.Каждого пациента, обследуемого по поводу заболеваний щитовидной и паращитовидных желез, спрашивают об их семейном анамнезе. Когда есть четкая картина заболевания у членов семьи или определенных подтипов заболеваний щитовидной железы или паращитовидных желез, мы направляем пациентов к нашим генетикам.
Напротив, сейчас известно, что примерно 1/3 случаев феохромоцитомы является генетической , что означает, что они имеют наследственную или семейную связь. Наиболее распространенными ассоциированными генетическими состояниями являются множественная эндокринная неоплазия (МЭН) 2 типа и синдром фон Хиппеля-Линдау, за которыми следуют нейрофиброматоз 1 типа и синдром семейной параганглиомы (который вызывается мутациями в гене сукцинатдегидрогеназы).Мы рекомендуем генетическое консультирование всем пациентам с феохромоцитомой или параганглиомой, так как это может предоставить важную информацию для прогноза пациента и более раннего обследования членов семьи.
Для получения дополнительной информации о генетическом консультировании и тестировании в UCLA>Что такое синдромы множественной эндокринной неоплазии (МЭН)?
Синдромы множественной эндокринной неоплазии (МЭН) передаются по наследству. Мутировавший ген заставляет разные эндокринные железы в организме развиваться доброкачественные и злокачественные нейроэндокринные опухоли.Эндокринные железы выделяют гормоны, поэтому опухоли, возникающие в этих железах, также могут вызывать чрезмерную выработку гормонов, что вызывает симптомы. Ранняя диагностика и лечение необходимы, чтобы уменьшить влияние этих состояний на пациента.
Существует несколько форм синдромов МЭН:
- MEN-I
- MEN-IIA
- МЕН-IIB
- Семейный медуллярный рак щитовидной железы (вариант MEN-IIA)
- МЕН-IV
Что такое множественная эндокринная неоплазия 1 типа (МЭН-I)?
Множественная эндокринная неоплазия (МЭН) I — наследственное заболевание, при котором в одной или нескольких из следующих желез образуется избыточная нормальная ткань (гиперплазия) или аденома (опухоль): паращитовидная железа, поджелудочная железа, гипофиз и (редко) надпочечники и щитовидная железа.Все это эндокринные железы, которые производят гормоны и выделяют их в кровь или лимфатическую систему.
Что вызывает MEN-I?
Причина МУЖЧИН I генетическая. Опухоли разных желез появляются у одного и того же человека, но не обязательно в одно и то же время. Заболевание передается по наследству, может возникнуть в любом возрасте и одинаково поражает мужчин и женщин.
Каковы признаки и симптомы MEN-I?
Большинство людей, страдающих этим синдромом, обращаются за медицинской помощью по одной из следующих причин: язвенная болезнь, симптомы, связанные с низким уровнем сахара в крови, симптомы, связанные с высоким уровнем кальция в сыворотке или почечными камнями, или симптомы, связанные с проблемами гипофиза (например, головная боль) .
Факторами риска являются семейный анамнез этого заболевания, предшествующая опухоль гипофиза и история синдрома Золлингера-Эллисона.
Симптомы:
Симптомы сильно различаются от человека к человеку и могут быть связаны с язвенной болезнью, гипогликемией, гиперкальциемией или дисфункцией гипофиза.
- Боль в животе
- Жжение, боль или дискомфорт от голода в верхней части живота или нижней части груди, которые облегчаются антацидами, молоком или пищей
- Черный дегтеобразный стул
- Тошнота и рвота
- Ощущение вздутия после еды
- Слабость
- Головная боль
- Проблемы со зрением
- Нарушение координации
- Беспокойство
- Психические изменения или спутанность сознания
- Кома при отсутствии лечения гипогликемии
- Потеря аппетита
- Мышечная боль
- Усталость
- Чувствительность к холоду
- Непреднамеренная потеря веса
- Низкое артериальное давление
- Выпадение подмышечных или лобковых волос
- У женщин, прекращение менструации, бесплодие или недостаточность лактата
- У мужчин: снижение сексуального интереса, потеря волос на теле или лице
Какое лечение необходимо пациентам с MEN-I?
Лечение зависит от пораженных желез внутренней секреции.Когда это возможно, методом выбора является хирургическое удаление пораженной железы. В некоторых случаях при аденомах гипофиза вместо хирургического вмешательства можно использовать лекарство под названием бромокриптин. Заместительная гормональная терапия показана при удалении желез или недостаточной секреции.
Каков прогноз для пациентов с MEN-I?
Опухоли гипофиза и паращитовидных желез обычно доброкачественные, но некоторые опухоли могут стать злокачественными (злокачественными), что в целом снижает продолжительность жизни.Симптомы язвенной болезни, гипогликемии, гиперкальциемии или дисфункции гипофиза должны поддаваться лечению.
Что такое множественная эндокринная неоплазия (МЭН) типа II?
Множественная эндокринная неоплазия II (МЭН II) — это наследственное заболевание, при котором тип рака щитовидной железы сопровождается рецидивирующим раком надпочечников.
Что вызывает MEN-II?
Причина MEN II генетическая — мутация в гене RET. Множественные опухоли могут появиться у одного и того же человека, но не обязательно в одно и то же время.Опухоль надпочечников — это феохромоцитома, а опухоль щитовидной железы — медуллярная карцинома щитовидной железы. Расстройство может возникнуть в любом возрасте и в равной степени поражает мужчин и женщин. Основным фактором риска является семейный анамнез МЭН II.
В чем разница между MEN-IIA и MEN-IIB?
Изображение большего размера>
MEN-IIA может вызывать медуллярный рак щитовидной железы, гиперпаратиреоз и феохромоцитому.
Изображение большего размера>
MEN-IIB может вызывать медуллярный рак щитовидной железы, феохромоцитому и невриномы слизистой оболочки (неровности на губах и языке).
Каковы признаки и симптомы MEN-II?
Симптомы:
- Сильная головная боль
- Учащенное сердцебиение
- Учащение пульса
- Потоотделение
- Боль в груди
- Боль в животе
- Нервозность
- Раздражительность
- Потеря веса
- Диарея
- Кашель
- Кашель с кровью
- Усталость
- Боль в спине
- Повышенный диурез
- Повышенная жажда
- Потеря аппетита
- Тошнота
- Мышечная слабость
- Депрессия
- Изменения личности
Симптомы могут различаться, но соответствуют симптомам феохромоцитомы, медуллярной карциномы щитовидной железы и иногда гиперпаратиреоза.
Какое лечение необходимо пациентам с МЭН-II?
Операция необходима для удаления медуллярной карциномы щитовидной железы и феохромоцитомы. Медуллярный рак щитовидной железы необходимо лечить с полным удалением щитовидной железы и удалением окружающих лимфатических узлов. После операции назначают заместительную гормональную терапию.
Члены семьи должны пройти скрининг на мутацию гена RET.
Каков прогноз для пациентов с МЭН-II?
Феохромоцитома обычно доброкачественная (не рак), но сопутствующая медуллярная карцинома щитовидной железы, которая характеризует это состояние, является очень агрессивным и потенциально смертельным раком.Тем не менее, ранняя диагностика и хирургическое вмешательство часто могут привести к излечению.
Что такое MEN-IV?
Множественная эндокринная неоплазия 4 типа (МЭН4) — очень редкая форма МЭН, наследственного онкологического синдрома, характеризующегося опухолями паращитовидных желез и передней доли гипофиза, возможно, связанными с опухолями надпочечников, почек и репродуктивных органов.
Что такое синдромы сукцинатдегидрогеназы (SDH)?
SDH — это ген, который помогает подавлять рост опухолей. Люди, у которых есть мутация в этом гене, более склонны к параганглиомам и феохромоцитомам, хотя это встречается не у всех.Эти опухоли могут вызывать симптомы, увеличиваясь в размерах или чрезмерно выделяя гормоны «бей или беги», вызывая эпизоды высокого кровяного давления, беспокойства и головной боли.
Что такое синдром фон Гиппеля-Линдау?
Этот синдром вызван мутацией в гене VHL, который помогает предотвратить рост опухолей. Приблизительно у 10–34% пациентов с синдромом ВХЛ могут развиваться феохромоцитомы или параганглиомы, помимо гемоангиобластом, ангиом сетчатки, почечно-клеточного рака и нейроэндокринных опухолей поджелудочной железы.
Что такое нейрофиброматоз I типа?
Нейрофиброматоз I типа возникает в результате мутации в гене NF1, который, как и многие из вышеперечисленных генов, препятствует слишком быстрому росту клеток. Без этого гена у людей развивается множество доброкачественных опухолей нервов и кожи, а также аномальные участки пигментации на коже. Пациенты с нейрофиброматозом I типа могут иметь когнитивные нарушения и подвержены риску некоторых видов рака, включая саркомы мягких тканей. До 6% пациентов с NF-1 также развиваются феохромоцитомы и параганглиомы.
Назначить встречу или телемедицинскую консультацию
Запросить встречу
Телефон: 310-267-7838
Генетические синдромы и гинекологические последствия у подростков
Номер 768 (Подтверждено в 2020 г.)
Комитет по охране здоровья подростков
Североамериканское общество детской и подростковой гинекологии одобряет этот документ. Это заключение комитета было разработано Комитетом Американского колледжа акушеров и гинекологов по охране здоровья подростков в сотрудничестве с членами комитета Мередит Лавлесс, доктором медицины, и Кимберли Гувер, доктором медицины.
РЕЗЮМЕ: По мере того как подростки с генетическим синдромом переходят на медицинское обслуживание взрослых, их могут направлять к акушерам-гинекологам для получения плановых профилактических или противозачаточных услуг, скрининга или консультирования по поводу инфекций, передаваемых половым путем, или для контроля менструального цикла. Хотя некоторые генетические синдромы не имеют физических или интеллектуальных нарушений, у других есть серьезные; поэтому образование и гинекологическая помощь должны основываться на интеллектуальных и физических возможностях пациента.Важно помнить, что подростки с генетическим синдромом или без него — сексуальные существа. Таким образом, при лечении пациентов с генетическими синдромами важно просвещение в вопросах репродуктивного здоровья, ожиданий в отношении фертильности и здоровых взаимоотношений. Акушеры-гинекологи должны уважать независимость пациента и избегать принуждения в любых обсуждениях с пациентом, включая решения о выборе контрацепции, сексуальной активности и планировании беременности. Большинство пациентов с генетическими синдромами и нейротипами могут переносить обычные гинекологические осмотры в офисе, когда это необходимо.Пациента нельзя принуждать к обследованию или удерживать для осмотра. Акушерская помощь подросткам и женщинам с генетическими синдромами может создавать проблемы и часто требует мультидисциплинарного подхода с момента предполагаемой беременности до послеродового периода. При уходе за подростком с генетическим синдромом следует учитывать индивидуальные проблемы пациента и опекуна, медицинские диагнозы, связанные с конкретными генетическими синдромами, и лекарственные взаимодействия.Акушерам-гинекологам рекомендуется искать дополнительные ресурсы и опыт при уходе за подростками с основными генетическими синдромами.
Рекомендации и выводы
Американский колледж акушеров и гинекологов предлагает следующие рекомендации и выводы:
Акушерам-гинекологам рекомендуется искать дополнительные ресурсы и знания при уходе за подростками с основными генетическими синдромами.
Хотя некоторые генетические синдромы не имеют физических или интеллектуальных нарушений, у других есть серьезные; поэтому образование и гинекологическая помощь должны основываться на интеллектуальных и физических возможностях пациента.
Акушеры-гинекологи должны уважать самостоятельность пациента и избегать принуждения в любых обсуждениях с пациентом, включая решения о выборе контрацепции, сексуальной активности и планировании беременности.
Пациентам с основным генетическим синдромом следует предложить соответствующий возрасту гинекологический скрининг и вакцинацию против вируса папилломы человека.
Когда обследование необходимо, но не срочно для пациента, который не переносит осмотр в офисе, акушер-гинеколог должен попытаться скоординировать осмотр с другими процедурами, требующими седации, такими как стоматологическая работа.
Акушеры-гинекологи должны знать о любых уникальных рисках хирургического вмешательства или анестезии, связанных с генетическим синдромом подростка.
Многие генетические синдромы имеют уникальные последствия для репродуктивного здоровья.Когда подростки с генетическим синдромом переходят на медицинское обслуживание взрослых, их могут направлять к акушерам-гинекологам для получения плановых профилактических или противозачаточных услуг, скрининга или консультирования по поводу инфекций, передаваемых половым путем (ИППП), или для ведения менструального цикла. В этом заключении Комитета подчеркиваются общие соображения в отношении репродуктивного здоровья
Таблица 1
перечислены важные гинекологические, противозачаточные и репродуктивные аспекты для пациентов с генетическими синдромами, которые могут быть обращены к акушеру-гинекологу.Акушерам-гинекологам рекомендуется искать дополнительные ресурсы и опыт при уходе за подростками с основными генетическими синдромами. Для получения информации о конкретных генетических синдромах акушер-гинеколог может запросить направление к специалистам или онлайн-информацию из таких источников, как Национальный центр развития трансляционных наук при Национальном институте здравоохранения и Национальный исследовательский институт генома человека 1. В некоторых регионах доступны варианты телемедицины с узкими специалистами. возникающий.Профилактическая гинекологическая помощь пациентам с генетическим синдромом аналогична потребностям их сверстников без генетического синдрома. Хотя некоторые генетические синдромы не имеют физических или интеллектуальных нарушений, у других есть серьезные; поэтому образование и гинекологическая помощь должны основываться на интеллектуальных и физических возможностях пациента. 2. Важно помнить, что подростки с генетическим синдромом или без него являются сексуальными существами. Таким образом, просвещение по вопросам репродуктивного здоровья, ожиданий в отношении фертильности и здоровых отношений важно при лечении пациентов с генетическими синдромами 3 4.Акушеры-гинекологи должны знать, что пациенты с физическими недостатками или нарушениями развития, или и тем, и другим, подвергаются более высокому риску сексуального насилия по сравнению со своими сверстниками без особых потребностей и должны проверять их, как и всех пациентов, на наличие в анамнезе сексуального насилия 5. Следует рассмотреть возможность скрининга на ИППП среди этой группы населения. При лечении подростков, подвергшихся сексуальному насилию, акушеры-гинекологи должны быть знакомы с законами и постановлениями, которые могут требовать обращения в правоохранительные органы.
Для тех акушеров-гинекологов и других медицинских работников, которые заботятся о сложных с медицинской точки зрения пациентах-подростках, следует подчеркнуть важность контрацепции для предотвращения нежелательной беременности. Акушеры-гинекологи могут обратиться в Центры по контролю и профилактике заболеваний.
Медицинские критерии приемлемости США для использования противозачаточных средств
6. Если остаются вопросы относительно наиболее безопасных методов контрацепции, следует запросить консультацию специалиста.Гинекологическое обследование женщин с генетическими синдромами
Как и для всех пациентов подросткового возраста, решение о проведении обследования должно основываться на показаниях. Если пациент может участвовать в разговоре, рекомендуется конфиденциальное интервью для обсуждения сексуальной активности и сексуальности. Акушеры-гинекологи должны уважать независимость пациента и избегать принуждения в любых обсуждениях с пациентом, включая решения о выборе контрацепции, сексуальной активности и планировании беременности.Перед обследованием пациентки важно оценить ее качество жизни и функциональные физические способности. Сбор анамнеза может включать посещение туалета и способность поддерживать гигиену во время менструации в школе или на работе; эта информация важна, когда помогает пациенту достичь ее целей в повседневной деятельности. Пациентам с основным генетическим синдромом должен быть предложен соответствующий возрасту гинекологический скрининг и вакцинация против вируса папилломы человека 7. Большинство пациентов с генетическими синдромами и нейротипами могут при необходимости переносить обычные гинекологические осмотры в офисе.Пациента нельзя принуждать к обследованию или удерживать для осмотра. При необходимости может быть направлено к врачу-гинекологу, имеющему опыт и знания в этой группе пациентов. При необходимости для гинекологической помощи следует проводить гинекологическое обследование, например, для оценки выделений из влагалища или тазовой боли, или для тестирования на ИППП, если показано. Также можно собрать мочу для скрининга на ИППП.
Если обследование необходимо, но не срочно (например, скрининг на рак шейки матки или установка внутриматочной спирали [ВМС]) для пациента, который не переносит осмотр в офисе, акушер-гинеколог должен попытаться скоординировать обследование с другими процедурами. которые требуют седативных средств, например при стоматологических вмешательствах.Если необходимы седативные препараты или анестезия, в этой группе пациентов может быть полезна консультация по анестезии перед процедурой. Акушеры-гинекологи должны знать обо всех уникальных рисках хирургического вмешательства или анестезии, связанных с генетическим синдромом подростка. Это может включать необходимость осторожного размещения и перевода пациентов на стол для процедур и обратно, чтобы минимизировать деформацию шейного отдела позвоночника, переломы или травмы, связанные с гипермобильностью, связанные с такими синдромами, как дисплазия скелета и нарушения соединительной ткани.
Акушерские аспекты
Акушерская помощь подросткам и женщинам с генетическими синдромами может создавать проблемы и часто требует междисциплинарного подхода с момента предполагаемой беременности до послеродового периода. Эти пациенты должны пройти предимплантационную генетическую консультацию с их акушерами-гинекологами, медицинскими генетиками, специалистами по медицине матери и плода или другими соответствующими узкими специалистами для оптимизации ухода. Некоторые из этих синдромов могут оказывать существенное влияние на здоровье женщины, ее плода или и того, и другого во время беременности, и пациентку следует проконсультировать об этих рисках до беременности.
Кардиологические аспекты
Когда генетические синдромы связаны со структурным и функциональным заболеванием сердца, акушер-гинеколог должен работать с кардиологом пациента и достичь консенсуса до начала приема лекарств или проведения процедур у пациентов из группы риска. Во многих случаях методы контрацепции и менструального регулирования или манипуляции зависят от сердечной аномалии и текущего состояния сердечно-сосудистой системы. Полезные ресурсы для оказания гинекологической помощи этой группе населения включают Американский колледж грудных врачей.
CHEST Рекомендации и консенсус
8 и
U.S. Медицинские критерии приемлемости для использования противозачаточных средств
6. Гинекологические проблемы включают возможность вагусной реакции при установке ВМС; таким образом, пациентам со сложными пороками сердца может потребоваться мониторинг сердца во время установки для выявления аритмии. Лекарства, содержащие эстроген, противопоказаны многим пациентам с сердечными заболеваниями, и их следует избегать у пациентов с повышенным риском тромбоза. Системные контрацептивы, содержащие только прогестин, считаются безопасными для большинства пациентов с врожденными пороками сердца; однако их следует использовать с осторожностью у пациентов с застойной сердечной недостаточностью из-за возможности дальнейшего удержания жидкости, что может привести к сердечному перенапряжению 9.Менструальные манипуляции
Гинеколог может проконсультироваться по поводу менструальных манипуляций для подростков и молодых женщин с генетическими синдромами. У некоторых людей с генетическими синдромами присутствует судорожное расстройство; в этих случаях можно использовать подавление менструального цикла для купирования обострения судорожной активности, вызванного гормональными изменениями. Часто встречается взаимодействие противосудорожных препаратов и гормональной контрацепции. Эти взаимодействия могут повлиять на эффективность противосудорожных средств, а также гормональной контрацепции.После того, как основная цель гинекологической помощи пациентом или опекуном установлена, гинеколог должен работать с неврологом пациентки и прийти к консенсусу, прежде чем начинать прием лекарств для управления менструальным циклом. Примечательно, что депо ацетат медроксипрогестерона может быть связан со снижением судорожной активности 10.
Медицинские критерии приемлемости США для использования противозачаточных средств
также может предоставить руководство по лекарственным взаимодействиям с судорожными расстройствами 6.При генетических синдромах, при которых присутствует задержка развития, лица, осуществляющие уход, часто стремятся к подавлению менструального цикла, чтобы улучшить менструальную гигиену, предотвратить беременность или уменьшить обострения поведенческих проблем, которые возникают после гормонального приема. шаблон.Для получения информации и рекомендаций по вариантам ведения менструального цикла для подростков с физическими и когнитивными нарушениями см. Заключение Комитета № 668.
Менструальные манипуляции для подростков с физическими недостатками и нарушениями развития
2.Заключение
При уходе за подростком с генетическим синдромом следует учитывать индивидуальные проблемы пациента и опекуна, медицинские диагнозы, связанные с конкретными генетическими синдромами, и лекарственные взаимодействия.По мере появления большего количества данных о генах и типах наследования могут появиться рекомендации по надзору и лечению пациентов с генетическими синдромами. Из-за текущей ограниченности литературы и данных по лечению гинекологических проблем у лиц с генетическим синдромом, рекомендации по скринингу и вмешательству в значительной степени основаны на мнении экспертов.
Таблица 1
является руководством и не заменяет сотрудничество с другими поставщиками медицинских услуг пациента или консультации с поставщиком медицинских услуг, который имеет опыт работы с конкретными генетическими синдромами.Опубликовано онлайн 21 февраля 2019 г.
Авторское право 2019 г. Американского колледжа акушеров и гинекологов. Все права защищены. Никакая часть этой публикации не может быть воспроизведена, сохранена в поисковой системе, размещена в Интернете или передана в любой форме и любыми средствами, электронными, механическими, путем фотокопирования, записи или иными способами, без предварительного письменного разрешения издателя.
Запросы на разрешение на изготовление фотокопий следует направлять в Центр защиты авторских прав, 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400.
Американский колледж акушеров и гинекологов 409 12th Street, SW, PO Box 96920, Вашингтон, округ Колумбия 20090-6920
Генетические синдромы и гинекологические последствия у подростков. Заключение комитета ACOG № 768. Американский колледж акушеров и гинекологов. Obstet Gynecol 2019; 133: e226–34.
Эта информация разработана как образовательный ресурс, чтобы помочь клиницистам в оказании акушерской и гинекологической помощи, и использование этой информации является добровольным.Эта информация не должна рассматриваться как исчерпывающая информация обо всех надлежащих процедурах или методах ухода или как изложение стандарта ухода. Он не предназначен для замены независимого профессионального суждения лечащего врача. Вариации на практике могут быть оправданы, когда, по разумному мнению лечащего врача, такой курс действий определяется состоянием пациента, ограниченностью доступных ресурсов или достижениями в знаниях или технологиях. Американский колледж акушеров и гинекологов регулярно проверяет свои публикации; однако его публикации могут не отражать самые последние свидетельства.Любые обновления этого документа можно найти на
www.acog.org
или позвонив в ресурсный центр ACOG.Несмотря на то, что ACOG прилагает все усилия для предоставления точной и надежной информации, данная публикация предоставляется «как есть», без каких-либо гарантий точности, надежности или иных явных или подразумеваемых условий. ACOG не гарантирует, не гарантирует и не поддерживает продукты или услуги какой-либо фирмы, организации или лица. Ни ACOG, ни его должностные лица, директора, участники, сотрудники или агенты не несут ответственности за любые убытки, ущерб или претензии в отношении любых обязательств, включая прямые, особые, косвенные или косвенные убытки, понесенные в связи с этой публикацией или доверием. по представленной информации.
Все члены и авторы комитета ACOG подали заявление о раскрытии конфликта интересов в отношении этого опубликованного продукта. Любые потенциальные конфликты были рассмотрены и урегулированы в соответствии с Политикой раскрытия информации о конфликте интересов ACOG. Политики ACOG можно найти на
acog.org
. Для продуктов, разработанных совместно с другими организациями, раскрытие информации о конфликте интересов представителями других организаций осуществляется этими организациями.Американский колледж акушеров и гинекологов не запрашивал и не принимал какое-либо коммерческое участие в разработке содержания этого опубликованного продукта.Синдромы наследственного рака | Онкологический центр им. Доктора медицины Андерсона
Хотя мы не определили генетические причины всех типов рака, мы знаем несколько изменений генов или мутаций, которые могут передаваться от родителей к ребенку и увеличивать риск развития болезни у человека. Эти изменения известны как синдромы наследственного рака.
Наследственный синдром рака груди и яичников
Наиболее распространенным типом наследственного рака груди является синдром наследственного рака груди и яичников (HBOC). HBOC вызывается мутациями в генах BRCA1 и BRCA2. Женщина с наследственной мутацией в генах BRCA имеет более высокий шанс заболеть раком груди и яичников в течение жизни, чем женщина, у которой нет мутации. Мужчина с наследственной мутацией в генах BRCA имеет более высокий шанс заболеть раком груди и простаты в течение своей жизни.Кроме того, в некоторых семьях выше заболеваемость раком поджелудочной железы, меланомой и другими видами рака.
Синдром Каудена
Синдром Каудена (CS) характеризуется множественными опухолевидными опухолями и повышенным риском некоторых видов рака. У большинства пациентов с CS развиваются небольшие незлокачественные новообразования или гамартомы на коже и слизистых оболочках, но эти новообразования также могут возникать в кишечном тракте или головном мозге. Люди с CS также имеют повышенный риск развития доброкачественных и злокачественных опухолей груди, матки и щитовидной железы.CS связан с мутациями в гене PTEN.
Синдром Линча (наследственный неполипозный синдром колоректального рака
Синдром
Линча (HNPCC) характеризуется колоректальным раком и раком матки (эндометрия) в раннем возрасте, а также другими опухолями вне толстой кишки. Синдром Линча вызывается мутациями в генах восстановления несоответствия ДНК (MLh2, MSh3, MSH6 или PMS2). Вариант синдрома Линча, называемый синдромом Мьюра Торре, связан с повышенным риском развития некоторых опухолей кожи.
Синдромы наследственного лейкоза и гематологических злокачественных новообразований
В последние годы исследователи обнаружили около десятка наследственных генетических состояний, которые могут привести к лейкемии и родственным заболеваниям крови, таким как миелодиспластический синдром, острый миелоидный лейкоз и апластическая анемия. Хотя крупномасштабные исследования больных лейкемией еще не завершены, по оценкам врачей, около 5-10% всех случаев лейкемии являются наследственными. Больным лейкемией с семейным анамнезом болезни следует рассмотреть возможность генетического тестирования, чтобы узнать, есть ли у их болезни наследственный компонент.Эта информация может повлиять на планы лечения пациента. Пациенты MD Anderson с подозрением на одно из этих заболеваний должны быть обследованы в нашей Клинике наследственной лейкемии.
Семейный аденоматозный полипоз (ФАП)
Семейный аденоматозный полипоз (FAP) или синдром Гарднера — это синдром предрасположенности к раку толстой кишки, при котором от сотен до тысяч предраковых полипов толстой кишки (называемых аденомами) развиваются по всему желудочно-кишечному тракту (в основном в толстой и прямой кишке, но также в желудке и тонком кишечнике) .
Аттенуированный FAP (AFAP) является более мягкой формой FAP и связан с повышенным риском рака толстой кишки, но меньшим количеством полипов толстой кишки. Синдром Гарднера связан с типичным количеством полипов, как при FAP, но также с остеомами (доброкачественными опухолями кости) и опухолями мягких тканей (так называемыми десмоидами). FAP связан с мутациями в гене APC. Есть и другие, недавно описанные предрасположенности к полипозу, связанные с разными генами.
Синдром Ли-Фраумени
Синдром Ли-Фраумени (LFS) — редкое генетическое заболевание, характеризующееся повышенным риском развития нескольких типов рака.Раковые заболевания, встречающиеся в LFS, можно диагностировать в детстве, подростковом или взрослом возрасте. Установлено, что у большинства людей с LFS есть мутации в гене TP53. Наиболее распространенные типы рака, связанные с LFS, включают:
- Саркомы мягких тканей (опухоль в жировой ткани, мышцах, нервах, суставах, кровеносных сосудах, костях или глубокой коже)
- Рак груди
- Лейкемия
- Рак легкого
- Опухоли головного мозга
- Рак надпочечников
Болезнь фон Гиппеля-Линдау
Болезнь фон Гиппеля-Линдау (VHL) — это мультисистемное заболевание, характеризующееся аномальным ростом кровеносных сосудов (называемое гемангиобластомами или ангиомами).Гемангиобластомы могут развиваться в сетчатке, определенных областях головного мозга, спинном мозге и других частях нервной системы. Другие типы опухолей могут развиваться в надпочечниках, почках и поджелудочной железе. Люди с ВХЛ также имеют более высокий риск развития определенных типов рака, особенно рака почки. Обнаружено, что почти все люди с VHL имеют мутации в гене VHL.
Множественные эндокринные новообразования
Синдромы множественной эндокринной неоплазии (МЭН) получили свое название потому, что они предрасполагают людей к развитию опухолей эндокринных желез.Эндокринная система состоит из желез, которые выделяют в кровоток гормоны, которые контролируют многочисленные процессы в организме. Эндокринная система играет важную роль в регулировании настроения, роста, развития и обмена веществ, а также сексуальной функции и репродуктивных процессов.
В настоящее время существует два различных синдрома МЭН: МЭН1 (обычно характеризующийся разрастанием гипофиза, паращитовидной железы и поджелудочной железы) и МЭН2 (обычно характеризующийся медуллярным раком щитовидной железы).
Когда подозревать генетический синдром
1.McCandless SE,
Брюнгер Дж. В.,
Кэссиди СБ.
Бремя генетических заболеваний на стационарном лечении в детской больнице [опубликованная поправка опубликована в Am J Hum Genet. 2004; 74 (4): 788]. Ам Дж. Хам Генет .
2004; 74 (1): 121–127 ….2. Стивенсон Д.А.,
Кэри JC.
Вклад пороков развития и генетических нарушений в смертность в детской больнице. Ам Дж. Мед Генет А .
2004. 126A (4): 393–397.3. О’Мэлли М.,
Hutcheon RG.Генетические нарушения и врожденные пороки развития в педиатрической долгосрочной помощи. J Am Med Dir Assoc .
2007. 8 (5): 332–334.4. Макинтош Р.,
Мерритт К.К.,
Ричардс MR,
Сэмюэлс MH,
Сильфон MT.
Частота врожденных пороков развития: исследование 5964 беременностей. Педиатрия .
1954. 14 (5): 505–522.5. Зеленый CR.
Частота недоразвития человека. Ам Дж. Дис Детский .
1963; 105: 301–312.6. Бэрд П.А.,
Андерсон Т.В.,
Ньюкомб HB,
Лоури РБ.
Генетические расстройства у детей и молодых людей: популяционное исследование. Ам Дж. Хам Генет .
1988. 42 (5): 677–693.7. Кэссиди С.Б., Аллансон Дж. Э. Управление генетическими синдромами. 3-е изд. Хобокен, Нью-Джерси: Уайли-Блэквелл; 2010.
8. Пагон РА.
Генетическое тестирование: когда тестировать, когда обращаться. Ам Фам Врач .
2005. 72 (1): 33–34.9. Хантер А.Г.Медицинская генетика: 2. Диагностический подход к ребенку с дисморфическими признаками. CMAJ .
2002. 167 (4): 367–372.10. Аллансон Дж. Э.,
Biesecker LG,
Кэри JC,
Hennekam RC.
Элементы морфологии: введение. Ам Дж. Мед Генет А .
2009. 149A (1): 2–5.11. Доннаи Д.
Успехи дисморфологии: от диагностики к лечению. Клин Мед .
2009. 9 (2): 154–155.12. Фальк М.Дж.,
Робин Н.Х.Подход врача первичной медико-санитарной помощи к врожденным аномалиям Prim Care .
2004; 31 (3): 605–619, х.13. Соломон Б.Д.,
Мерсье S,
Велес Джи,и другие.
Анализ корреляций генотип-фенотип при голопрозэнцефалии человека. Am J Med Genet C Semin Med Genet .
2010. 154С (1): 133–141.14. Джонс К.Л., Смит Д.В.. Распознаваемые модели человеческих пороков развития Смита. 6-е изд. Филадельфия, Пенсильвания: Эльзевье-Сондерс; 2006 г.
15. Кобринский Л.Ю.,
Салливан К.Э.
Велокардиофациальный синдром, синдром ДиДжорджи: синдромы делеции хромосомы 22q11.2. Ланцет .
2007. 370 (9596): 1443–1452.16. Судья Д.П.,
Dietz HC.
Синдром Марфана. Ланцет .
2005; 366 (9501): 1965–1976.17. Перешить БП.
Диагностика, генетика и лечение наследственных синдромов недостаточности костного мозга. Образовательная программа по гематологии и соц. Гематол .200729–39.18. Basson CT,
Бачинский Д.Р.,
Линь RC,и другие.
Мутации в TBX5 человека [исправленные] вызывают пороки развития конечностей и сердца при синдроме Холта-Орама [опубликованные исправления опубликованы в Nat Genet. 1997; 15 (4): 411]. Нат Генет .
1997. 15 (1): 30–35.19. Moeschler JB.
Генетическая оценка умственной отсталости. Семин Педиатр Нейрол .
2008; 15 (1): 2–9.20. Калер С.Г.,
Fahey MC.Нарушения обмена веществ и умственная отсталость. Am J Med Genet C Semin Med Genet .
2003. 117C (1): 31–41.21. Zschocke J, Hoffmann GF, Milupa AG. Vademecum Metabolicum: Руководство по метаболической педиатрии. 2-е изд. Фридрихсдорф, Германия: Шаттауэр; 2004.
22. Уокер Ф.О.
Болезнь Хантингтона. Ланцет .
2007. 369 (9557): 218–228.23. Парейсон Д.
Диагностика наследственных невропатий у взрослых пациентов. Дж. Neurol .2003. 250 (2): 148–160.24. Wirdefeldt K,
Адами Х.О.,
Коул П,
Трихопулос Д.,
Мандель Дж.
Эпидемиология и этиология болезни Паркинсона: обзор доказательств. Eur J Epidemiol .
2011; 26 (приложение 1): S1 – S58.25. Бленнов К.,
де Леон MJ,
Зеттерберг Х.
Болезнь Альцгеймера. Ланцет .
2006. 368 (9533): 387–403.26. Wattendorf DJ,
Хэдли DW.
Семейный анамнез: родословная в трех поколениях. Ам Фам Врач .
2005. 72 (3): 441–448.27. Беннетт Р.Л.,
Французский KS,
Реста Р.Г.,
Дойл Д.Л.
Стандартизированная номенклатура родословных людей: обновление и оценка рекомендаций Национального общества генетических консультантов. Дж. Генет Коунс .
2008. 17 (5): 424–433.28. Гутмахер А.Е.,
Коллинз Ф.С.,
Carmona RH.
Семейная история важнее, чем когда-либо. N Engl J Med .
2004. 351 (22): 2333–2336.29. Belle DJ,
Сингх Х.
Генетические факторы метаболизма лекарств. Ам Фам Врач .
2008. 77 (11): 1553–1560.30. Ама Т,
Bounmythavong S,
Blaze J,
Вейсманн М,
Мариенау М.С.,
Николсон WT.
Значение фармакогеномики для анестезиологов. AANA J .
2010. 78 (5): 393–399.31. Соломон Б.Д.,
Джек Б.В.,
Feero WG.
Клиническое содержание преждевременной помощи: генетика и геномика. Am J Obstet Gynecol .
2008; 199 (6 доп. 2): S340 – S344.32. Салливан К.Е.,
Mullen CA,
Блейз Р.М.,
Винкельштейн Я.
Многопрофильный обзор синдрома Вискотта-Олдрича. J Педиатр .
1994; 125 (6 ч. 1): 876–885.33. Cremin B,
Гудман Х,
Спрангер Дж,
Бейтон П.
Червячные кости при несовершенном остеогенезе и других нарушениях. Скелетный радиол .
1982. 8 (1): 35–38.Какие генетические синдромы увеличивают риск рака груди?
Сигель Р.Л., Миллер К.Д., Джемаль А. Статистика рака, 2019. CA Cancer J Clin . 2019 января 69 (1): 7-34. [Медлайн]. [Полный текст].
Рабочая группа США по статистике рака. Инструмент визуализации данных статистики рака США на основе данных, представленных в ноябре 2017 г. (1999-2015 гг.). Центры по контролю и профилактике заболеваний. Доступно по адресу https://nccd.cdc.gov/uscs/. Июнь 2018 г .; Доступ: 13 марта 2019 г.
Feuer EJ. Моделирование влияния адъювантной терапии и скрининговой маммографии на смертность от рака груди в США в период с 1975 по 2000 год: введение в проблему. J Natl Cancer Inst Monogr . 2006. 364 (25): 2-6. [Медлайн].
Dossus L, Benusiglio PR. Дольковый рак молочной железы: заболеваемость, генетические и негенетические факторы риска. Рак молочной железы Res . 2015 13 марта, 17:37. [Медлайн]. [Полный текст].
Морроу М., Бурштейн HJ, Харрис-младший.Злокачественные опухоли груди. ДеВита В.Т. младший, Лоуренс Т.С., Розенберг С.А. Рак ДеВиты, Хеллмана и Розенберга: принципы и практика онкологии . 10-е изд. Филадельфия, Пенсильвания: Wolters Kluwer Health; 2015. 1117-19.
Рак молочной железы в фактах и цифрах, 2017-2018 гг. Американское онкологическое общество. Доступно по адресу https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures- 2017-2018 гг.pdf. Доступ: 3 апреля 2019 г.
Ривз Г.К., Берал В., Грин Дж., Гатани Т., Бык Д. Миллион женщин-соавторов исследования. Гормональная терапия менопаузы и риска рака груди по гистологическому типу: когортное исследование и метаанализ. Ланцет Онкол . Ноябрь 2006. 7 (11): 910-8.
[Рекомендации] Национальная комплексная онкологическая сеть. Снижение риска рака груди. Руководство NCCN по клинической практике в онкологии. Доступно по адресу https: //www.nccn.org / Professional / Physician_gls / pdf / Breast_risk.pdf. Версия 1.2019 — 11 декабря 2018 г .; Доступ: 3 апреля 2019 г.
Mørch LS, Skovlund CW, Hannaford PC, Iversen L, Fielding S, Lidegaard Ø. Современная гормональная контрацепция и риск рака груди. N Engl J Med . 2017 Dec 7. 377 (23): 2228-2239. [Медлайн]. [Полный текст].
Hamajima N, et al; Совместная группа по гормональным факторам при раке молочной железы. Алкоголь, табак и рак груди — совместный повторный анализ индивидуальных данных 53 эпидемиологических исследований, в том числе 58 515 женщин с раком груди и 95 067 женщин без этого заболевания. Br J Рак . 2002, 18 ноября. 87 (11): 1234-45. [Медлайн]. [Полный текст].
Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, eds. SEER Обзор статистики рака, 1975-2014 гг. . Бетесда, Мэриленд: Национальный институт рака; Апрель 2017 г. [Полный текст].
Наследственный рак груди и яичников: рак груди и яичников и категории риска в семейном анамнезе.Центры по контролю и профилактике заболеваний. Доступно по адресу https://www.cdc.gov/genomics/resources/diseases/breast_ovarian_cancer/risk_categories.htm. 29 июля 2016 г .; Доступ: 3 апреля 2019 г.
Редакционная коллегия PDQ Cancer Genetics. Генетика рака груди и гинекологического рака (PDQ®): версия для специалистов в области здравоохранения. Обновлено 21 марта 2019 г. [Medline]. [Полный текст].
Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, et al. Популяционные частоты мутаций BRCA1 и BRCA2 и пенетрантность рака: исследование родственных когорт в Онтарио, Канада. Национальный институт рака . 2006 декабрь 6. 98 (23): 1694-706. [Медлайн]. [Полный текст].
Консорциум по связям с раком груди. Риск рака у носителей мутации BRCA2. Национальный институт рака . 1999, 4 августа. 91 (15): 1310-6. [Медлайн]. [Полный текст].
[Рекомендации] Целевая группа превентивных служб США. Рак, связанный с BRCA: оценка риска, генетическое консультирование и генетическое тестирование. USPSTF. Доступно по адресу https://www.uspreventiveservicestaskforce.org / Page / Document / RecommendedStatementFinal / brca-related-Cance-risk-оценка-генетическое-консультирование-и-генетическое-тестирование. Декабрь 2013; Доступ: 3 апреля 2019 г.
Allain DC. Генетическое консультирование и тестирование распространенных наследственных синдромов рака молочной железы: доклад с симпозиума больницы Уильяма Бомонта 2007 года по молекулярной патологии. Дж Мол Диаг . 2008 Сентябрь 10 (5): 383-95. [Медлайн]. [Полный текст].
Mangoni M, Bisanzi S, Carozzi F, Sani C, Biti G, Livi L и др.Связь между генетическими полиморфизмами генов XRCC1, XRCC3, XPD, GSTM1, GSTT1, MSh3, MLh2, MSh4 и MGMT и радиочувствительностью у пациентов с раком молочной железы. Int J Radiat Oncol Biol Phys . 2011 Сентябрь 1. 81 (1): 52-8. [Медлайн].
Заболеваемость раком груди по расе и этнической принадлежности. Центры по контролю и профилактике заболеваний. Доступно по адресу http://www.cdc.gov/cancer/breast/statistics/race.htm. Июнь 2018 г .; Дата обращения: 5 ноября 2018 г.
Hartmann LC, Sellers TA, Frost MH, Lingle WL, Degnim AC, Ghosh K и др.Доброкачественные заболевания груди и риск рака груди. N Engl J Med . 2005 21 июля. 353 (3): 229-37. [Медлайн]. [Полный текст].
Devesa SS, Diamond EL. Связь рака груди и рака шейки матки с доходом и образованием среди белых и черных. Национальный институт рака . 1980 Сентябрь 65 (3): 515-28. [Медлайн].
Совместная группа по гормональным факторам при раке молочной железы. Рак молочной железы и гормональные контрацептивы: совместный повторный анализ индивидуальных данных о 53 297 женщинах с раком груди и 100 239 женщинах без рака груди по результатам 54 эпидемиологических исследований. Ланцет . 1996, 22 июня. 347 (9017): 1713-27. [Медлайн].
Кубба А.А. Рак груди и таблетки. J R Soc Med . 2003 июн. 96 (6): 280-3. [Медлайн]. [Полный текст].
Рак груди и заместительная гормональная терапия: совместный повторный анализ данных 51 эпидемиологического исследования 52 705 женщин с раком груди и 108 411 женщин без рака. Совместная группа по гормональным факторам при раке молочной железы. Ланцет .1997, 11 октября 350 (9084): 1047-59. [Медлайн].
Ривз Г.К., Берал В., Грин Дж., Гатани Т., Бык Д., Миллион женщин-соавторов исследования. Гормональная терапия менопаузы и риска рака груди по гистологическому типу: когортное исследование и метаанализ. Ланцет Онкол . 2006 г., 7 (11): 910-8. [Медлайн].
Chlebowski RT, Anderson GL, Gass M, Lane DS, Aragaki AK, Kuller LH, et al. Заболеваемость эстрогеном плюс прогестином и раком груди и смертность у женщин в постменопаузе. ЯМА . 20 октября 2010 г. 304 (15): 1684-92. [Медлайн].
Совместная группа по гормональным факторам при раке молочной железы. Тип и время менопаузальной гормональной терапии и риск рака груди: метаанализ отдельных участников всемирных эпидемиологических данных. Ланцет . 2019 28 сентября. 394 (10204): 1159-1168. [Медлайн]. [Полный текст].
Ингрэм И. ЗГТ оказывает 20-летнее влияние на риск рака молочной железы. MedPage сегодня. Доступно по адресу https: // www.medpagetoday.com/meetingcoverage/sabcs/83882. 13 декабря 2019 г .; Дата обращения: 16 декабря 2019 г.
Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, et al. Неблагоприятные последствия для здоровья женщин, подвергшихся внутриутробному воздействию диэтилстильбестрола. N Engl J Med . 2011 Октябрь 6. 365 (14): 1304-14. [Медлайн]. [Полный текст].
Gaudet MM, Gapstur SM, Sun J, Diver WR, Hannan LM, Thun MJ. Активное курение и риск рака груди: исходные когортные данные и метаанализ. Национальный институт рака . 2013 г. 17 апреля. 105 (8): 515-25. [Медлайн]. [Полный текст].
Michels KB, Solomon CG, Hu FB, Rosner BA, Hankinson SE, Colditz GA, et al. Диабет 2 типа и последующая заболеваемость раком груди в исследовании здоровья медсестер. Уход за диабетом . 2003 июн. 26 (6): 1752-8. [Медлайн].
Chen WY, Rosner B, Hankinson SE, Colditz GA, Willett WC. Умеренное употребление алкоголя во взрослом возрасте, режимы употребления алкоголя и риск рака груди. ЯМА . 2011 г. 2 ноября. 306 (17): 1884-90. [Медлайн]. [Полный текст].
Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES. Работа в ночное время и риск рака груди: систематический обзор и метаанализ. Eur J Cancer . 2005 сентябрь 41 (13): 2023-32. [Медлайн].
Foraker RE, Абдель-Расул М., Куллер Л.Х., Джексон Р.Д., Ван Хорн Л., Сегин Р.А. и др. Сердечно-сосудистое здоровье и сердечно-сосудистые заболевания и рак: Инициатива по охране здоровья женщин. Am J Prev Med . 2016 Февраль 50 (2): 236-40. [Медлайн].
Chen Z, Arendell L, Aickin M, Cauley J, Lewis CE, Chlebowski R, et al. Плотность бедренной кости позволяет прогнозировать риск рака груди независимо от оценки Гейл: результаты инициативы Women’s Health Initiative. Рак . 2008 сен 1. 113 (5): 907-15. [Медлайн]. [Полный текст].
Grenier D, Cooke AL, Lix L, Metge C, Lu H, Leslie WD. Минеральная плотность костной ткани и риск рака груди в постменопаузе. Лечение рака молочной железы . 2011 Апрель 126 (3): 679-86. [Медлайн].
Qu X, Zhang X, Qin A, Liu G, Zhai Z, Hao Y, et al. Минеральная плотность костной ткани и риск рака груди у женщин в постменопаузе. Лечение рака молочной железы . 2013 февраль 138 (1): 261-71. [Медлайн].
Куган П.Ф. Блокаторы кальциевых каналов и рак груди: возрожденная гипотеза. JAMA Intern Med . 2013 Сентябрь 23, 173 (17): 1637-8. [Медлайн].
Li CI, Daling JR, Tang MT, Haugen KL, Porter PL, Malone KE.Использование гипотензивных препаратов и риск рака груди среди женщин в возрасте от 55 до 74 лет. JAMA Intern Med . 2013 Сентябрь 23, 173 (17): 1629-37. [Медлайн]. [Полный текст].
Келли Дж. Некоторые гипотензивные средства связаны с риском рака груди. Медицинские новости Medscape. Доступно на http://www.medscape.com/viewarticle/808935. 5 августа 2013 г .; Доступ: 1 ноября 2017 г.
Wilson LE, D’Aloisio AA, Sandler DP, Taylor JA. Долгосрочное использование препаратов, блокирующих кальциевые каналы, и риск рака груди в предполагаемой когорте женщин из США и Пуэрто-Рико. Рак молочной железы Res . 2016 г. 5. 18 (1): 61. [Медлайн]. [Полный текст].
Llanos AAM, Rabkin A, Bandera EV, Zirpoli G, Gonzalez BD, Xing CY, et al. Использование средств для волос и риск рака груди среди афроамериканок и белых женщин. Канцерогенез . 2017 г. 1. 38 (9): 883-892. [Медлайн]. [Полный текст].
Коэн Р. Краска для темных волос и химические релаксанты связаны с раком груди. Медицинские новости Medscape. Доступно по адресу https: // www.medscape.com/viewarticle/882833. 13 июля 2017 г .; Доступ: 1 ноября 2017 г.
Перманентная краска для волос и выпрямители могут повысить риск рака груди. Национальные институты здоровья. Доступно по адресу https://www.nih.gov/news-events/news-releases/permanent-hair-dye-straighteners-may-increase-breast-cancer-risk. 4 декабря 2019 г .; Дата обращения: 5 декабря 2019 г.
Lancaster JM, Powell CB, Kauff ND, Cass I, Chen LM, Lu KH и др. Заявление Комитета по образованию Общества гинекологов-онкологов по оценке риска наследственной предрасположенности к гинекологическому раку. Гинеколь Онкол . 2007 ноябрь 107 (2): 159-62. [Медлайн].
Robson ME, Storm CD, Weitzel J, Wollins DS, Offit K, Американское общество клинической онкологии. Обновление заявления о политике Американского общества клинической онкологии: генетическое и геномное тестирование на предрасположенность к раку. Дж. Клин Онкол . 2010 10 февраля. 28 (5): 893-901. [Медлайн].
[Рекомендации] Берлинер Дж. Л., Фэй А. М., Каммингс С. А., Бернетт Б., Тиллманнс Т. Практическое руководство NSGC: оценка риска и генетическое консультирование при наследственном раке груди и яичников. Дж. Генет Коунс . 2013 22 апреля (2): 155-63. [Медлайн].
[Рекомендации] Палуч-Шимон С., Кардосо Ф., Сесса С., Бальмана Дж., Кардосо М.Дж., Гилберт Ф. и др. Профилактика и скрининг носителей мутации BRCA и других наследственных синдромов рака груди / яичников: Руководство ESMO по клинической практике по профилактике и скринингу рака. Энн Онкол . 2016, 27 сентября (приложение 5): v103-v110. [Медлайн].
[Рекомендации] Национальная комплексная онкологическая сеть.Генетическая / семейная оценка высокого риска: грудь и яичники. NCCN. Доступно на https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Версия 3.2019 — 18 января 2019 г .; Доступ: 3 апреля 2019 г.
Панкрац В.С., Хартманн Л.С., Дегним А.С., Виркант Р.А., Гош К., Вачон С.М. и др. Оценка точности модели Гейл у женщин с атипичной гиперплазией. Дж. Клин Онкол . 2008 20 ноября. 26 (33): 5374-9. [Медлайн]. [Полный текст].
Адамс-Кэмпбелл Л.Л., Макамби К.Х., Фредерик В.А., Гаскинс М., Девитти Р.Л., Маккаскилл-Стивенс В.Оценка риска рака груди с использованием моделей Гейл и CARE у афроамериканок. Грудь J . 2009 сентябрь-октябрь. 15 Дополнение 1: S72-5. [Медлайн]. [Полный текст].
Берри Д.А., Иверсен Э.С. мл., Гудбьяртссон Д.Ф., Хиллер Э.Х., Гарбер Я.Э., Пешкин Б.Н. и др. Проверка BRCAPRO, чувствительность генетического тестирования BRCA1 / BRCA2 и распространенность других генов предрасположенности к раку молочной железы. Дж. Клин Онкол . 2002, 1 июня. 20 (11): 2701-12. [Медлайн]. [Полный текст].
Тайрер Дж., Даффи С.В., Кузик Дж.Модель прогнозирования рака груди, включающая семейные и личные факторы риска. Stat Med . 2004 15 апреля. 23 (7): 1111-30. [Медлайн].
Амир Э., Эванс Д.Г., Шентон А., Лаллоо Ф., Моран А., Боггис С. и др. Оценка пакетов оценки риска рака груди в программе оценки семейного анамнеза и скрининга. J Med Genet . 2003 ноябрь 40 (11): 807-14. [Медлайн]. [Полный текст].
Boughey JC, Hartmann LC, Anderson SS, Degnim AC, Vierkant RA, Reynolds CA и др.Оценка модели Тайрера-Кузика (Международное исследование рака груди) для прогнозирования риска рака груди у женщин с атипичной гиперплазией. Дж. Клин Онкол . 2010 г., 1. 28 (22): 3591-6. [Медлайн]. [Полный текст].
- Определение: генетические синдромы, вызванные микроделецией (15q11-q13) в сочетании с геномным импринтингом.
- Этиология: возникающее в результате состояние зависит от копии пораженного гена.
- Диагноз: генетические тесты
- Клинические особенности
- Лечение
- Прогноз: нормальная продолжительность жизни, если избежать крайнего ожирения
- Клинические особенности
- Задержка умственного развития и приобретения моторики у детей грудного и раннего возраста; отсутствие речевого развития
- Умственная отсталость
- Более чем в 80% случаев — выраженные эпилептические припадки
- Микроцефалия
- Атаксия, дрожащие движения конечностей
- Гипотония туловища, гипертонус конечностей, гиперрефлексия
- Проблемы со сном
- Характерное счастливое поведение с частым смехом (несоответствующий смех)
- Гипервозбудимость, непродолжительное внимание
- Очарование водой
- Лечение
- Прогноз: Продолжительность жизни обычно нормальная.
- Определение: аутосомно-рецессивный дефект гена PEX, который приводит к нарушению синтеза пероксисом [10] [11]
- Клинические особенности
- Диагноз: повышенная концентрация жирных кислот с очень длинной цепью в крови.
- Лечение
- В настоящее время не существует лекарств от синдрома Зеллвегера.
- Поддерживающее лечение, в том числе:
- Установка зонда для питания
- Консультации специалистов для лечения индивидуальных проблем (например, аудиологов, офтальмологов, ортопедов)
- Прогноз: смерть в течение одного года после рождения.
- Определение: набор аномалий, вызывающих пороки развития ротовой полости и челюстно-лицевой области у плода [12]
- Клинические особенности
- Диагноз: флуоресцентная гибридизация in situ (FISH)
- Лечение
- При умеренной одышке: симптоматическое лечение
- Неинвазивная вентиляция
- Наблюдение и помощь во время еды
- При сильной одышке: хирургическая коррекция
- Специальные вмешательства для длительной коррекции (например,д., отвлечение нижней челюсти, чтобы язык был ближе к передней части рта)
- В случаях острого опасного для жизни респираторного дистресс-синдрома → трахеостомия
- При умеренной одышке: симптоматическое лечение
- Определение: редкий синдром, вызванный аберрацией хромосомы 5 [14] [15]
- Эпидемиология: распределение по полу: ♀> ♂ (2: 1)
- Этиология: микроделеция короткого плеча на хромосоме 5 (46, XX, 5p- или 46, XY, 5p‑).
- Клинические особенности
- Лечение
- Симптоматическое лечение
- Ранняя психологическая и физическая помощь
- Прогноз: нормальная продолжительность жизни возможна, но зависит от сопутствующих симптомов и терапевтической помощи.
- Определение: Х-сцепленное расстройство с прогрессирующей потерей интеллекта и когнитивных способностей, таких как речь, передвижение и мелкая моторика [16]
- Этиология: мутация Х-сцепленного доминантного гена в гене метил-CpG-связывающего белка 2 (ген MECP2)
- Обычно это не наследственный дефект гена, а спорадическая мутация.
- Мутация обычно происходит в отцовском аллеле и, таким образом, затрагивает почти исключительно женщин (при поражении плоды мужского пола умирают внутриутробно или вскоре после рождения).
- Клинические особенности
- Нормальное развитие до появления первых симптомов, что обычно происходит в возрасте 6–18 месяцев
- Симптомы регресса нервного развития, в том числе:
- Диагноз: сочетание типичных клинических проявлений и обнаружения генных мутаций.
- Прогноз: недостаточно данных относительно ожидаемой продолжительности жизни после 40 лет, так как отсутствуют долгосрочные исследования и болезнь встречается довольно редко.
- Определение: мультисистемное нарушение развития, вызванное делецией хромосомы 7.
- Этиология: микроделеция длинного плеча хромосомы 7 (включая делецию гена эластина).
- Клинические особенности
- Диагноз: сочетание типичных клинических проявлений и обнаружения генных мутаций.
- Синдром Тричера Коллинза.
https://www.ncbi.nlm.nih.gov/pubmed/20301704 .
Обновлено: 1 января 1993 г.
Доступ: 2 июля 2020 г. - Синдром ломкой Х-хромосомы.
https: // medlineplus.gov / ency / article / 001668.htm .
Обновлено: 8 января 2015 г.
Доступ: 7 мая 2017 г. - Синдром ломкой Х-хромосомы.
https://ghr.nlm.nih.gov/condition/fragile-x-syndrome .
Обновлено: 12 декабря 2017 г.
Доступ: 16 декабря 2017 г. - Нарушения биогенеза пероксисом, спектр синдрома Зеллвегера.
https://www.ncbi.nlm.nih.gov/books/NBK1448/ .
Обновлено: 10 мая 2012 г.Доступ: 7 мая 2017 г. Карими Р., Брумфилд Т., Брамфилд Ф., Сафайян Ф., Стейн С. Синдром Зеллвегера: генетическое заболевание, которое изменяет биосинтез и метаболизм липидов. Интернет-журнал фармакологии . 2006; 5
(1).- Страница информации о синдроме Ретта.
https://www.ninds.nih.gov/Disorders/All-Disorders/Rett-Syndrome-Information-Page .
.
Доступ: 7 мая 2017 г. Mainardi PC. Синдром Кри дю Шат. Орфанет J Редкий Dis . 2006; 1
(33).
DOI: 10.1186 / 1750-1172-1-33. | Открыть в режиме чтения QxMD- Шиффер РБ, Рао С.М., Фогель Б.С. Нейропсихиатрия: всеобъемлющий учебник .
LWW
; 2003 г. Портер Ф.Д. Синдром Смита – Лемли – Опица: патогенез, диагностика и лечение. евро J Hum Genet . 2008; 16
(5): стр.535-541.
DOI: 10.1038 / ejhg.2008.10. | Открыть в режиме чтения QxMD- Синдром Смита-Лемли-Опица.
- Синдром Прадера-Вилли.
https://rarediseases.org/rare-diseases/prader-willi-syndrome/ .
Обновлено: 1 января 2018 г.
Дата обращения: 22 июня 2020 г. Маскари М.Дж., Готлиб В., Роган П.К. и др. Частота однопородной дисомии при синдроме Прадера-Вилли. N Engl J Med . 1992; 326
(24): с.1599-1607.
DOI: 10,1056 / nejm199206113262404. | Открыть в режиме чтения QxMD- Синдром Прадера-Вилли.
https://www.ncbi.nlm.nih.gov/books/NBK1330/ .
Обновлено: 4 февраля 2016 г.
Доступ: 7 мая 2017 г. - Синдром Прадера-Вилли.
https://medlineplus.gov/ency/article/001605.htm .
Обновлено: 19 апреля 2016 г.
Доступ: 7 мая 2017 г. - Робб МП. Введение: Руководство по коммуникационным наукам и расстройствам, второе издание .
Множественное издание
; 2013 - Синдром Ангельмана.
https://www.ncbi.nlm.nih.gov/books/NBK1144/ .
Обновлено: 14 мая 2015 г.
Доступ: 7 мая 2017 г. - Синдром Ангельмана.
https://ghr.nlm.nih.gov/condition/angelman-syndrome .
Обновлено: 28 ноября 2017 г.Доступ: 30 ноября 2017 г. - Изолированная последовательность Пьера Робена.
https://ghr.nlm.nih.gov/condition/isolated-pierre-robin-sequence .
Обновлено: 2 мая 2017 г.
Доступ: 7 мая 2017 г. Drescher F, Jotzo M, Goelz R, Meyer T, Bacher M, Poets CF. Когнитивное и психосоциальное развитие детей с последовательностью Пьера Робена. Acta Paediatr . 2008; 97
(5): с.
Редкие наследственные синдромы — AMBOSS
Последнее обновление: 8 марта 2021 г.
Резюме
На этой карточке представлен обзор наследственных симптомокомплексов, которые редко встречаются в общей популяции. Эти синдромы вызваны наследственными генетическими дефектами, которые возникают либо из-за хромосомных аберраций, либо из-за аутосомных / сцепленных с полом признаков.Проявления различаются для каждого синдрома, при этом большинство особенностей возникает из-за аномалий развития, функциональных или структурных аномалий различных органов. Диагноз можно подтвердить с помощью молекулярно-генетического обнаружения, флуоресцентной гибридизации in situ (FISH) или других генетических / хромосомных исследований. Лечение обычно симптоматическое.
Синдром ломкой Х-хромосомы
Хрупкой Х-хромосомы: «X-tra large» уши, яички и лицо у этих пациентов.
Синдром Прадера-Вилли и синдром Ангельмана
Обзор
«Прадер скучает по папе, а Ангел — по маме»: аллельная мутация / делеция отцовского происхождения при синдроме Прадера-Вилли и материнского при синдроме Ангельмана.
Синдром Прадера-Вилли
[5] [6] [7]
Синдром Ангельмана
[7] [8] [9]
«Ангелы на небесах»: беспечная, легко возбудимая личность, атаксия, вербальное недоразвитие, эпилептические припадки, аномальные черты лица, тяжелая умственная отсталость.
Синдром Зеллвегера (цереброгепаторенальный синдром)
Последовательность Пьера Робена (синдром Пьера Робена)
Синдром кри-дю-чат (синдром кошачьего крика)
Синдром Ретта
Регресс Ретта: за нормальным развитием вскоре следует потеря целенаправленных движений рук и умственная и вербальная инвалидность.
Синдром Вильямса
Уильям принимает ICEcream от незнакомцев (умственные отклонения, сердечно-сосудистые пороки, эльфийские черты лица, комфорт с незнакомцами).