Генетические особенности синдрома ломкой Х-хромосомы
Генетические особенности синдрома ломкой Х-хромосомы
Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Самая известная и наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном заболевании, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом нарушения в гене FMR1 (fragile X mental retardation-1), который расположен на Х-хромосоме и играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота этого синдрома среди мальчиков составляет 1:4000.
Так называемая «ломкость» X-хромосомы проявляется в том, что хромосома выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1, расположенном на X-хромосоме.
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если повторов больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком состоянии заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем больше вероятность того, что у ее или его детей количество повторов будет больше 200 и заболевание разовьется.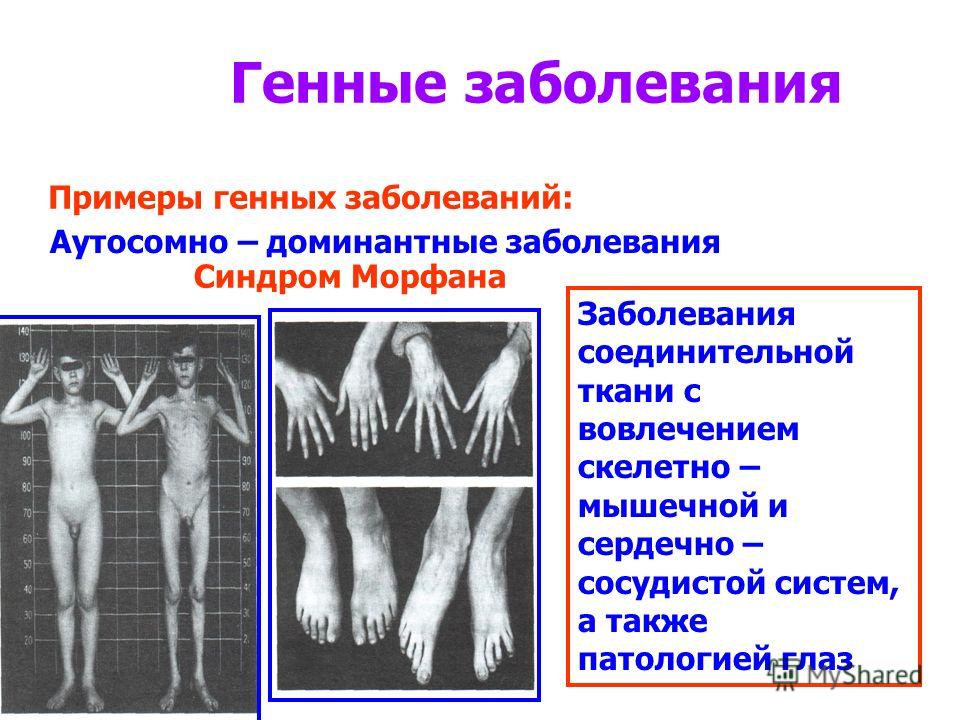 В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае, если эта одна хромосома оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому наличие одной Х-хромосомы с мутантным геном FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой может передать её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя такого гена, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние.
 Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
 Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такая диагностика позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения ребенка с этим синдромом и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Такая диагностика также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ)
2) пациентам с интеллектуальной недостаточностью и их родственникам
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием FMR1
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
В случае обнаружения бессимптомного носительства мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Массовое обследование новорожденных на наследственные болезни. Адреногенитальный синдром — Красноярский краевой медико-генетический центр
Прочтите и возьмите себе на заметку, особенно если вы молодые люди
В России уже много лет проводится массовое обследование новорожденных для выявления у них нескольких наследственных заболеваний. Такое обследование проводится во многих странах и называется скринингом новорожденных или неонаталъным скринингом.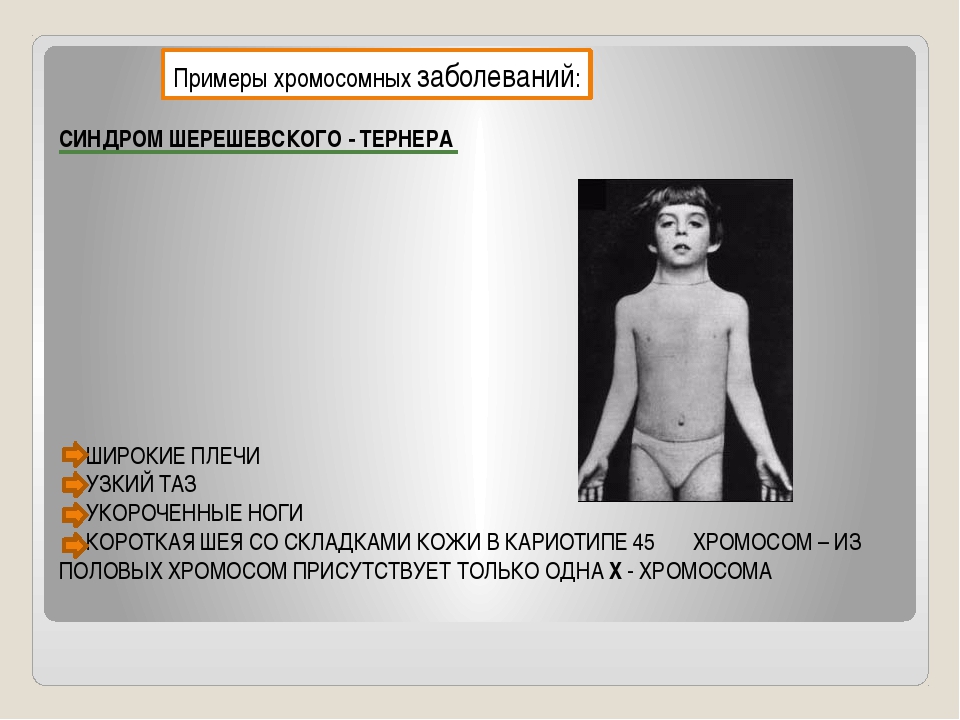
Целью скрининга новорожденных является, конечно, не само выявление новорожденных с еще не проявившимися наследственными заболеваниями, а их лечение, которое позволяет предотвратить появление клинических симптомов, во многих случаях весьма тяжелых, или даже фатальных. В результате рано начатого и аккуратно проводимого лечения вместо тяжело больных детей, а затем подростков и взрослых, получаются здоровые люди, полноценные члены общества, нередко являющиеся гордостью семьи.
Скрининг новорожденных в России ведется в отношении 5 наследственных и врожденных заболеваний: фенилкетонурии, гипотиреоза, галактоземии, адрено-гениталъного синдрома и муковисцидоза.
ЧТО ТАКОЕ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ?
Адреногенитальный синдром (сокращенно АГС) является наследственным заболеванием, которое вызывается нарушением (мутацией) в определенном гене. Такой ген называется мутантным. Синонимами АГС являются врожденная гиперплазия коры надпочечников и недостаточность 21-гидроксилазы. Скрининг новорожденных на АГС в России только начинается и поэтому его частота пока неизвестна. В других странах она колеблется от 1:10000 до 1:20000 новорожденных.
ПОЧЕМУ РЕБЕНОК МОЖЕТ ЗАБОЛЕТЬ АДРЕНОГЕНИТАЛЬНЫМ СИНДРОМОМ?
При АГС нарушается функция надпочечников. Надпочечники это железы внутренней секреции, т.е. железы, вырабатывающие гормоны, они расположены рядом с почками. Надпочечники вырабатывают различные гормоны, абсолютно необходимые для нормального развития и функционирования организма. Среди гормонов надпочечников, синтез или превращения которых оказываются нарушенными при АГС, наиболее важными являются следующие: кортизол — гормон, необходимый для того, чтобы организм нормально отвечал на стресс или проникновение инфекции; альдостерон — гормон, необходимый для поддержания нормального кровяного давления и нормальной функции почек; андрогены — гормоны, которые нужны для нормального роста и правильного формирования половых органов мужских и женских. У людей с АГС чаще всего наблюдается недостаточность фермента, который называется 21-гидроксилаза. В результате этого нарушается превращение холестерола в кортизол и альдостерон, которое контролируется этим ферментом. Одновременно происходит накопление предшественников кортизола и альдостерона, которые в норме превращаются в мужские половые гормоны — андрогены. Поскольку при АГС предшественников кортизола и альдостерона накапливается много, то образуется значительно больше, чем в норме, андрогенов, что является основной причиной возникновения клинической картины АГС.
В результате этого нарушается превращение холестерола в кортизол и альдостерон, которое контролируется этим ферментом. Одновременно происходит накопление предшественников кортизола и альдостерона, которые в норме превращаются в мужские половые гормоны — андрогены. Поскольку при АГС предшественников кортизола и альдостерона накапливается много, то образуется значительно больше, чем в норме, андрогенов, что является основной причиной возникновения клинической картины АГС.
КАКИЕ НАРУШЕНИЯ В ОРГАНИЗМЕ МОЖЕТ ВЫЗЫВАТЬ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ?
Существует несколько форм АГС, но примерно в 90% случаев он обусловлен недостаточностью фермента 21-гидроксилазы. В свою очередь, клинически различают три формы недостаточности 21-гидроксилазы. Две из них называют классическими. Одна известна как простая вирилизирующая форма, которая обусловлена избытком андрогенов, что ведет к избыточной маскулинизации, т.е. к чрезмерному проявлению мужских половых признаков. Это больше всего заметно у девочек по избыточному развитию наружных половых органов сразу после рождения. В то же время развитие внутренних половых органов (матки и яичников) у таких девочек происходит нормально. Вторая классическая форма называется соль-теряющей. Она связана с недостаточным синтезом альдостерона — гормона, необходимого для возвращения соли через почки в кровоток. При третьей форме недостаточности 21-гидроксилазы, которую называют не классической, признаки избыточной маскулинизации проявляются у девочек и мальчиков после рождения.
Самой опасной для жизни и самой частой является соль-теряющая форма АГС. Если лечение не будет назначено во время, ребенок может умереть. При других формах АГС дети быстро растут и у них очень рано появляются вторичные половые признаки, в частности, рост волос на лобке.
КАК НАСЛЕДУЕТСЯ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ?
Адреногенитальный синдром наследуется по аутосомно-рецессивному типу, т.е. больные в семье накапливаются в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок с АГС. На родословной мужчины обозначены квадратиком, а женщины — кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома из 23 пар, имеющихся у человека. Эта хромосома содержит нормальный или мутантный ген адреногенитального синдрома, последний помечен черной точкой.
На родословной мужчины обозначены квадратиком, а женщины — кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома из 23 пар, имеющихся у человека. Эта хромосома содержит нормальный или мутантный ген адреногенитального синдрома, последний помечен черной точкой.
На рисунке для простоты изображена только хромосома, содержащая ген, мутации в котором вызывают АГС. У ребенка в обеих хромосомах содержится мутантный ген, и поэтому он болен. У каждого из родителей мутантный ген содержится только в одной хромосоме, а вторая хромосома нормальная и поэтому они здоровы. Такие люди называются носителями мутантного гена. У бабки по матери мутантный ген также имеется только в одной хромосоме, как и у деда со стороны отца. Они, как и родители ребенка, здоровы, но передали хромосомы, содержащие мутантный ген, своим детям. У вторых деда и бабки обе хромосомы содержат только нормальный ген. Таким образом, при рецессивном наследовании болен только тот член семьи, который получил от своих родителей обе хромосомы, несущие мутантный ген. Все остальные члены семьи здоровы, в том числе и те, кто является носителем мутантного гена.
На представленном рисунке видно, что у родителей больного ребенка могут еще появиться больные дети. Вероятность появления больного ребенка в семьях, в которых родители являются носителями мутантного гена, составляет 1/4, или 25%. Эта вероятность не меняется от числа больных или здоровых детей в семье: для каждого следующего ребенка риск, что он будет болен, составляет 25%, Вероятность рождения здорового ребенка, обе хромосомы которого содержат только нормальный ген, составляет также 25%. Вероятность рождения детей, у которых имеется один нормальный и один мутантный ген, т.е. здоровых носителей, составляет 50% . Многие родители больных детей и их родственники, первый раз встретившись с врачом-генетиком, настойчиво повторяют, что у их ребенка не наследственное заболевание, так как в их семье ни у кого из родственников никогда не было такого заболевания.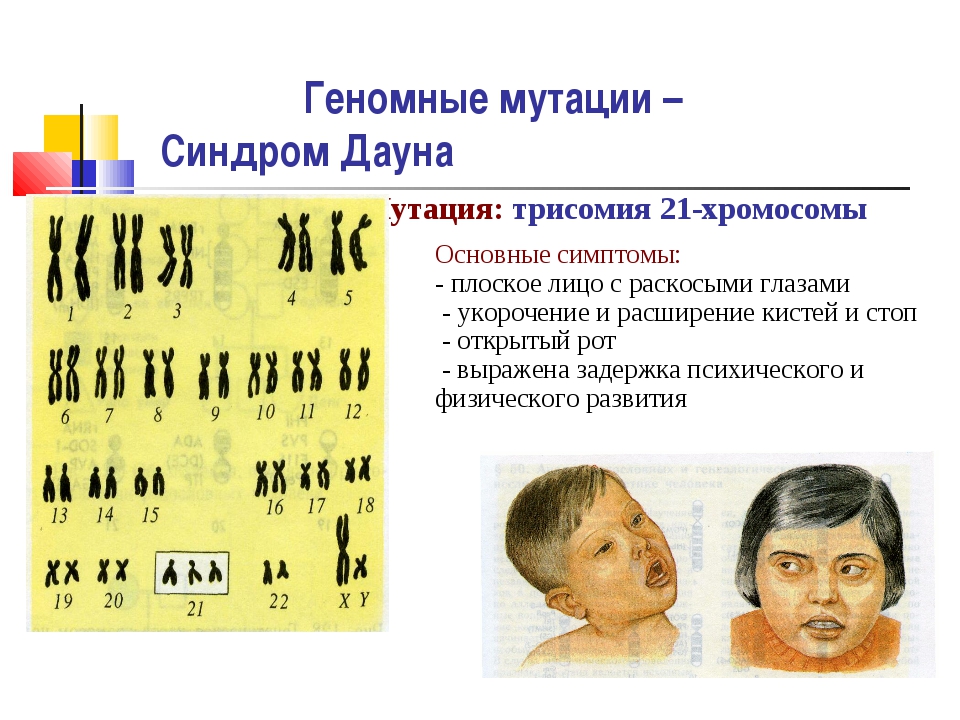 И только подробное и в доступной форме объяснение, что правила наследования бывают разные, позволяют родителям понять с какой ситуацией они столкнулись.
И только подробное и в доступной форме объяснение, что правила наследования бывают разные, позволяют родителям понять с какой ситуацией они столкнулись.
ЧТО ТАКОЕ СКРИНИНГ НОВОРОЖДЕННЫХ НА АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ?
Чтобы избежать развития таких тяжелых клинических проявлений адреногенитального синдрома надо, чтобы новорожденный был тестирован на этот синдром в первые дни после рождения. Для этого существуют программы скрининга новорожденных. Скрининг начинается с того, что у новорожденного на 4 — 5 день жизни перед выпиской из родильного дома берут из пятки несколько капель крови, которую наносят на специальную фильтровальную бумагу. Кровь высушивается, и такой бланк, на котором указана фамилия новорожденного и ряд других сведений, необходимых для его идентификации, переправляется в лабораторию региональной медико-генетической консультации. В лаборатории проводят специальное исследование, которое позволяет выявить новорожденных, у которых есть подозрение на АГС. В этом случае лаборатория запрашивает теперь уже в педиатрической службе, так как новорожденный выписан из родильного дома, повторное взятие крови. Обычно в это время родители узнают от педиатра, что первый тест на адреногенитальный синдром у их ребенка оказался ненормальным. У них появляется повод для беспокойства. Повторное тестирование образца крови у младенца является решающим. В ряде случаев при повторном исследовании уровень фермента 21-гидроксилазы оказывается нормальным. Это означает, что результат первого исследования был неверный (его называют ложноположительным). Причины этого могут быть связаны как с состоянием младенца, так и с какой-то ошибкой лаборатории. Этот результат, свидетельствующий о том, что у ребенка нет АГС, сразу сообщается родителям, и они могут успокоиться.
ЧТО ДЕЛАТЬ, ЕСЛИ ДИАГНОЗ АДРЕНОГЕНИТАЛЬНОГО СИНДРОМА НА СКРИНИНГЕ ПОДТВЕРДИЛСЯ?
Если и при втором тестировании уровень фермента 21 гидроксилазы в крови остается низким, то это означает, что ребенок болен адреногенитальным синдромом, и семья немедленно приглашается в медико-генетическую консультацию. Здесь родителям объясняют, что собой представляет это заболевание, и направляют ребенка к квалифицированному эндокринологу, который назначает лечение и наблюдает за ребенком в дальнейшем. АГС лечится ежедневным приемом кортизола и иногда альдостерона. Если лечение начато рано, то клинические симптомы АГС у ребенка не проявятся, и он будет расти здоровым, не отличаясь от сверстников. Периодически семья должна будет посещать эндокринолога, особенно при возникновении у ребенка любого заболевания, травмы и т.д., так как это все стрессовые ситуации, и, возможно, потребуется увеличение дозы кортизола, который является гормоном стресса.
Здесь родителям объясняют, что собой представляет это заболевание, и направляют ребенка к квалифицированному эндокринологу, который назначает лечение и наблюдает за ребенком в дальнейшем. АГС лечится ежедневным приемом кортизола и иногда альдостерона. Если лечение начато рано, то клинические симптомы АГС у ребенка не проявятся, и он будет расти здоровым, не отличаясь от сверстников. Периодически семья должна будет посещать эндокринолога, особенно при возникновении у ребенка любого заболевания, травмы и т.д., так как это все стрессовые ситуации, и, возможно, потребуется увеличение дозы кортизола, который является гормоном стресса.
МОЖНО ЛИ ПОМОЧЬ СЕМЬЕ, В КОТОРОЙ ПОЯВИЛСЯ БОЛЬНОЙ АДРЕНОГЕНИТАЛЬНЫМ СИНДРОМОМ, ИМЕТЬ ЗДОРОВЫХ ДЕТЕЙ?
Да, и довольно успешно. Для АГС возможна дородовая диагностика. Первым шагом в этом направлении является обращение в медико-генетическую консультацию, где врач-генетик определяет показания и возможные методические подходы к дородовой диагностике. В каждом конкретном случае решается вопрос о необходимости молекулярно-генетического обследования больного ребенка или родителей, а затем плода. Сама процедура заключается в том, что во время беременности в сроке 9-11 недель или 16-18 недель врач акушер-гинеколог проводит забор очень небольшого количества клеток плода, находящихся в околоплодной жидкости, плодных оболочках или крови плода, и направляет этот материал в специальную лабораторию пренатальной диагностики. В этой лаборатории врачи лаборанты-генетики проводят молекулярную генетическую диагностику, т.е. определяют наличие или отсутствие мутации в гене, отвечающем за АГС. В случае положительного результата семья решает вопрос о прерывании беременности больным плодом или настраивается на появление еще одного больного ребенка. Это право выбора остается за семьей.
КОМПЛЕКСНЫЙ КЛИНИКО-ГЕНЕТИЧЕСКИЙ ПОДХОД К ДИАГНОСТИКЕ СИНДРОМА РЕТТА У ДЕТЕЙ | Юров
1. Vorsanova S.G., Yurov I.Y., Yurov Y.B. Neurological, genetic and epigenetic features of Rett syndrome. J. Pediatr. Neurol. 2004; 2: 179–190.
Vorsanova S.G., Yurov I.Y., Yurov Y.B. Neurological, genetic and epigenetic features of Rett syndrome. J. Pediatr. Neurol. 2004; 2: 179–190.
2. Ворсанова С.Г., Улас В.Ю., Демидова И.А., и др. Современные представления о синдроме Ретта: клинические, цитогенетические и молекулярные исследования. Журнал неврологии и психиатрии им. С.С. Корсакова. 1999; 3: 61–69.
3. Amir R.E., Van den Veyver I.B., Wan M. et al. Rett syndrome is caused by mutations in X linked MEP2, encoding methyl pG binding protein2. Nat Genet. 1999; 23: 185–188.
4. Iourov I.Y., Vorsanova S.G., Villard L. et al. The study of X chromo some inactivation in mental retardation: comparative analysis molecular cytogenetic and polymerase chain reaction based techniques in Rett syndrome. Balkan J. Med. Genet. 2003; 6: 33–37.
5. Bienvenu T., Villard L., de Roux N. et al. Spectrum of MEP2mutations in Rett syndrome. Genet. Test. 2002; 6: 1–6.
6. Ворсанова С.Г., Демидова И.А., Улас В.Ю., и др. Цитогенетическая и молекулярноCцитогенетическая диагностика синдрома Ретта у детей. Журн. неврол. и психиатр. им. С.С. Корсакова. 1998; 4: 53–56.
7. Vorsanova S.G., Demidova I.A., Ulas V.Y. et al. Cytogenetic and molecular cytogenetic investigation of Rett syndrome. Analysis of 31 cases. Neuro Report. 1996; 7: 187–189.
8. Vorsanova S.G., Yurov Y.B., Kolotii A.D., Soloviev I.V. FISH analysis of replication and transcription of chromosome X loci: new approach for genetic analysis of Rett syndrome. Brain Dev. 2001; 23 (S1): 191–195.
Brain Dev. 2001; 23 (S1): 191–195.
9. Vorsanova S.G., Yurov Y.B., Ulas V.Y., et al. Cytogenetic and molecular cytogenetic studies of Rett syndrome (RTT): a retrospective analysis of a Russian cohort of RTT patients (the investigation of 57 girls and three boys). Brain Dev. 2001; 23 (S1): 196–201.
10. Ворсанова С.Г., Улас В.Ю., Юров Ю.Б. и др. Клинико–генетические корреляции при синдроме Ретта: изучение российской когорты больных. Журнал неврологии и психиатрии им. С.С. Корсакова. 2002; 10: 23–29.
11. Soloviev I.V., Yurov Y.B., Vorsanova S.G., Malet P. Microwave activation of fluorescence in situ hybridization: a novel method for rapid chromosome detection and analysis. Focus. 1994; 16 (4): 115–116.
12. Soloviev I.V., Yurov Y.B., Ioannou P. et al. Identification and molecular cytogenetic characterization of large subset of human plasmids, cosmids PAC and YAc clones: the search of DNA probes for pre and postnatal diagnosis. Cs. Pediat. 1997; 52: 529–538.
13. Yurov I.Y., Vorsanova S.G., Ulas V.Y. et al. Molecular genetic and epigenetic studies of Rett syndrome: insights into genotype and phenotype correlations. FENS Abstr. 2004; 198 (2А): 8 p.
14. Юров Ю.Б., Ворсанова С.Г. Молекулярно–цитогенетические исследования хромосомных аномалий и нарушений при нервно-психических заболеваниях: поиск биологических маркеров для диагностики. Вестник РАМН. 2001; 7: 26–31.
15. Diagnostic criteria for Rett syndrome. The Rett Syndrome Diagnostic Criteria Work Group. Ann. Neurol. 1988; 23: 425–428.
Ann. Neurol. 1988; 23: 425–428.
16. Hagberg B., Skjedal O.H. Rett variants: a suggested model for inclusion criteria. Peiatr. Neurol. 1994; 11: 5–11.
17. Shahbazian M.D., Zoghbi H.Y. Rett syndrome and MECP2: linking epigenetics and neuronal function. Am. J. Hum. Genet. 2002; 71: 1259–1272.
18. Villard L., Levy N., Xiang F. et al. Segregation of a totally skewed pattern of X chromosome inactivation in four familial cases of Rett syndrome without MECP2 mutation: implication for the disease. J. Med. Genet. 2001; 38: 435–442.
Синдром Ламб–Шаффера, обусловленный ранее не описанной мутацией в гене SOX5 | Шаркова
1. Ikeda T., Zhang J., Chano T. et al. Identification and characterization of the human long form of SOX5 (L-SOX5) gene. Gene 2002;298(1):59–68. DOI: 10.1016/S0378- 1119(02)00927-7. PMID: 12406576.
2. Kwan K.Y., Lam M.M., Krsnik Ž. et al. SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc Natl Acad Sci USA 2008;105(41):16021–6. DOI: 10.1073/ pnas.0806791105. PMID: 18840685.
3. Kamachi Y., Kondoh H. SОХ proteins: regulators of cell fate specification and differentiation. Development 2013;140(20):4129–44. DOI: 10.1242/ dev.091793. PMID: 24086078.
4. Lee R.W., Bodurtha J., Cohen J. et al. Deletion 12p12 involving SOX5 in two children with developmental delay and dysmorphic features. Pediatr Neurol 2013;48(4):317–20. DOI: 10.1016/j.pediatrneurol.2012.12.013. PMID: 23498568.
Pediatr Neurol 2013;48(4):317–20. DOI: 10.1016/j.pediatrneurol.2012.12.013. PMID: 23498568.
5. Lamb A.N., Rosenfeld J.A., Neill N.J. et al. Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat 2012;33(4):728–40. DOI: 10.1002/humu.22037. PMID: 22290657.
6. Nesbitt A., Bhoj E.J., McDonald Gibson K. et al. Exome sequencing expands the mechanism of SOX5-associated intellectual disability: a case presentation with review of SOX-related disorders. Am J Med Genet 2015;167A(11):2548–54. DOI: 10.1002/ ajmg.a.37221. PMID: 26111154.
7. Schanze I., Schanze D., Bacino C.A. et al. Haploinsufficiency of SOX5, a member of the SOX (SRY-related HMG-box) family of transcription factors is a cause of intellectual disability. Eur J Med Genet 2013;56(2):108–13. DOI: 10.1016/j. ejmg.2012.11.001. PMID: 23220431.
8. Lelieveld S.H., Reijnders M.R., Pfundt R. et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci 2016;19(9): 1194–6. DOI: 10.1038/nn.4352. PMID: 27479843.
9. Zech M., Poustka K., Boesch S. et al. SOX5-null heterozygous mutation in a family with adult-onset hyperkinesia and behavioral abnormalities. Case Rep Genet 2017;2017:2721615. DOI: 10.1155/2017/ 2721615. PMID: 29214085.
Сдать анализ на Синдром Жильбера
Метод определения
Фрагментный анализ
Выдаётся описание результатов врачом-генетиком!
Исследуемый материал
Цельная кровь (с ЭДТА)
Доступен выезд на дом
Исследование промоторной области гена уридиндифосфатглюкуронидазы 1 (количество TA-повторов).
Тип наследования.
Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания.
Ген расположен на хромосоме 2 в регионе 2q37.1.
Мутации в данном гене приводят также к развитию синдрома Криглера-Найяра, семейной транзиторной неонатальной гипербилирубинемии, повышенному уровню сывороточного билирубина.
Определение заболевания.
Наследственная доброкачественная неконъюгированная гипербилирубинемия.
Патогенез и клиническая картина.
Клиническая картина обусловлена мутациями в промоторной области гена UGTА1, которые приводят к снижению уровня функциональной активности фермента уридиндифосфатглюкуронидазы 1 в гепатоцитах. В результате данный фермент присутствует в гепатоцитах, но его активность снижена на 25%. Синдром Жильбера характеризуется повышением билирубина в крови, как правило, не более 80-100 мкмоль/л со значительным преобладанием непрямой фракции, остальные биохимические показатели крови и печеночные пробы остаются в пределах нормы. Основное клиническое проявление – желтушное окрашивание кожи, склер, слизистых. Эпизоды желтухи различной интенсивности у больных с синдромом Жильбера провоцируются инфекциями, приемом алкоголя, голоданием, психоэмоциональной и физической нагрузкой. Встречающийся астено-вегетативный синдром проявляется в виде: депрессии, неспособности концентрировать внимание, быстрой утомляемости, слабости, головокружения, потливости, плохом сне, неприятных ощущениях в области сердца. Диспепсия проявляется дискомфортом и незначительными болями в области правого подреберья, отсутствием аппетита, тошнотой, отрыжкой, запорами или поносами, метеоризмом. Часто больной не подозревает о том, что страдает желтухой, пока она не обнаружится при клиническом осмотре (иктеричность склер) или при проведении лабораторных исследований. У гетеро- и гомозиготных носителей мутации возможна манифестация с развитием токсических реакций при приеме некоторых лекарственных препаратов: анаболических стероидов, глюкокортикоидов, андрогенов, рифампицина, циметидина, хлорамфеникола, стрептомицина, салицилата натрия, ампициллина, кофеина, этинилэстрадиола, парацетамола, иринотекана (используется при лечении рака толстой кишки). В связи с этим рекомендуется проведение генетического анализа перед началом лечения с использованием лекарственных препаратов, обладающих гепатотоксическими эффектами.
В связи с этим рекомендуется проведение генетического анализа перед началом лечения с использованием лекарственных препаратов, обладающих гепатотоксическими эффектами.
Частота встречаемости:
Заболевание частое. Встречается среди европейцев с частотой 2-5%, среди азиатов -3% и среди африканцев — 36%.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Дрепа О.И., Карабанов А.В., Федин П.А., Иванова-Смоленская И.А., Маркова Е.Д., Шулятьев И.С., Тверская С.М., Ильченко Л.Ю. Изучение проводящей функции периферических нервов у больных с эссенциальным тремором, несущим изменения в промоторной области UGT1 в гомозиготном или гетерозиготном состоянии, с увеличением непрямого билирубина в сыворотке крови. // Материалы Национального конгресса «Человек и лекарство», Москва, 2005, с.613.
- Шулятьев И.С., Карабанов А.В., Ильченко Л.Ю., Дроздов В.Н., Петраков А.В., Иванова- Смоленская И.А., Маркова Е.Д., Тверская С.М., Куропаткина Ю.В. Сочетание экстрапирамидных расстройств с синдромом Жильбера./ /Материалы XXXII сессии ЦНИИГ. – Сб. – 2005 – с.320-321.
- Куропаткина Ю.В., Карабанов А.В., Шулятьев И.С., Тверская С.М., Поляков А.В. Инсерция в промоторной области гена UGT1 и непереносимость лекарственных препаратов.// Медицинская генетика, 2005, т.4, № 5, с.216.
- Polyakov A., Kuropatkina J., A. Karabanov, I. Shulyatiev, Tverskaya S. The correlation between insertion in the promoter region of the UGT1 gene and the tolerance of some xenobiotics. //Europ. J. Hum. Genet. 2005. — V.13, suppl. 1. — P. 273.
- Sugatani, J., Yamakawa, K., Yoshinari, K., Machida, T., Takagi, H., Mori, M., Kakizaki, S., Sueyoshi, T., Negishi, M., Miwa, M. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem. Biophys. Res. Commun. 292: 492-497, 2002
- OMIM
причины возникновения, симптомы, диагностика, классификация
В числе хромосомных патологий особое место занимает синдром Дауна – одно из наиболее распространенных генетических нарушений у новорожденных. Его основная причина – случайная генетическая мутация, вследствие которой в 21 паре хромосом появляется третья лишняя хромосома. Частота явления – приблизительно 1 случай на 600-800 малышей. Случайная мутация накладывает свой отпечаток на внешность ребенка уже на этапе внутриутробного развития, что значительно облегчает диагностику синдрома Дауна методом УЗИ.
Его основная причина – случайная генетическая мутация, вследствие которой в 21 паре хромосом появляется третья лишняя хромосома. Частота явления – приблизительно 1 случай на 600-800 малышей. Случайная мутация накладывает свой отпечаток на внешность ребенка уже на этапе внутриутробного развития, что значительно облегчает диагностику синдрома Дауна методом УЗИ.
Основные причины возникновения
Современная медицина называет сразу две причины болезни:
- Возраст матери. Это основной фактор риска для синдрома Дауна. Чем старше беременная женщина, тем выше риск рождения ребенка со случайной генетической патологией. В возрасте 30-40 лет риск генетического сбоя составляет 1/1000, после 42 лет – 1/60. Основной фактор – старение яйцеклеток, которые закладываются еще в период внутриутробного развития девочки и постепенно утрачивают способность к образованию генетически здорового плода. Также имеет значение возраст отца – до или после 45лет, когда вероятность рождения малыша с синдромом Дауна резко возрастает.
- Наследственный фактор. Причиной развития синдрома могут стать близкородственные браки, наличие заболевания у одного из родных ребенка. Также значение имеет возраст бабушки, в котором она родила дочь. Чем он выше, тем больше риск рождения внука с синдромом.
Важно помнить: синдром Дауна признан специалистами всех стран мира как случайная генетическая мутация. Она не зависит от экологической обстановки, уровня радиации, наличия вредного производства и прочих посторонних факторов.
Характерные внешние и прочие симптомы
Люди, являющиеся носителями лишней хромосомы, имеют характерную внешность:
- плоская переносица;
- монголоидный разрез глаз, из-за которого патология имеет второе название «монголизм»;
- плоское лицо и затылок.
Также в числе особенностей– некоторое отставание в развитии и сниженный иммунитет, не позволяющий организму сопротивляться внешним инфекциям. Все перечисленное не является ограничивающим фактором. Сегодня для них разработаны специальные методики обучения с первых месяцев жизни. При условии активности родителей дети с симптомами синдромома Дауна могут получить среднее образование и профессию, стать полноценными членами общества и завести собственную семью.
Все перечисленное не является ограничивающим фактором. Сегодня для них разработаны специальные методики обучения с первых месяцев жизни. При условии активности родителей дети с симптомами синдромома Дауна могут получить среднее образование и профессию, стать полноценными членами общества и завести собственную семью.
Осложнения
В зависимости от сложности формы синдрома Дауна у пациента могут отмечаться:
- врожденные пороки сердца;
- частые инфекционные заболевания;
- лейкемия;
- раннее наступление болезни Альцгеймера;
- остановка дыхания во сне;
- ожирение и т.д.
Диагностика
Выявление генетической аномалии возможно на ранних сроках беременности:
- УЗ-скрининг в период 11-13 недель оценивает размер воротникового пространства и размеры носовой кости плода;
- одновременно проводится анализ крови с уточнением количества хорионического гормона и плазменного протеина;
- на более поздних сроках беременности осуществляется забор тканей плода на предмет их генетического исследования: амниоцентез, биопсия волокон хориона или кордоцентез.
Т.к. речь идет о генетическом сбое, лечение синдрома Дауна заключается лишь в наблюдении за состоянием здоровья пациента и корректировкой осложнений основного заболевания.
Прогноз для пациентов
Сегодня средняя продолжительность жизни с генетической патологией приближается к 55-60 лет, тогда как еще несколько десятков лет назад они доживали только до 25 лет из-за неблагоприятных условий жизни.
Вероятность рождения ребенка с генетической аномалией у человека с синдромом Дауна составляет около 35-50%. Кроме того, при формировании плода у беременной женщины с заболеванием у будущего малыша могут возникнуть другие генетические сбои.
При этом риск онкологических заболеваний у таких больных сведен к нулю. Кроме того, родители отмечают радушие и неизменно хорошее настроение таких детей, их ласку, отзывчивость, способность легко идти на контакт и не обижаться на окружающих.
Кроме того, родители отмечают радушие и неизменно хорошее настроение таких детей, их ласку, отзывчивость, способность легко идти на контакт и не обижаться на окружающих.
Профилактика
Полностью исключить риск рождения ребенка с генетической патологией 21-й пары хромосом не представляется возможным. Однако в силах будущих родителей сделать все возможное, чтобы укрепить собственное репродуктивное здоровье и исключить хромосомный сбой:
- следить за своим здоровьем, своевременно обращаться за медицинской помощью для лечения выявленных заболеваний;
- вести здоровый и активный образ жизни, заниматься спортом, чтобы в яйцеклетки поступало достаточное количество кислорода;
- правильно питаться, обогащая рацион здоровыми продуктами с высоким содержанием витаминов и микроэлементов;
- поддерживать иммунную систему;
- следить за весом, т.к. его отклонение в любую сторону может стать причиной гормонального сбоя и нарушения процесса созревания половых клеток;
- своевременно проходить УЗ-обследование во время беременности, чтобы выявить у плода генетический сбой на первых неделях внутриутробного развития.
Наблюдение за пациентами с синдромом Дауна в АО «Медицина» (клиника академика Ройтберга) в Москве
В АО «Медицина» (клиника академика Ройтберга) функционирует Центр по работе с особыми детьми. Педиатры и профильные специалисты клиники в ЦАЛ Москвы готовы к работе с пациентами с синдромом Дауна независимо от их возраста и общего состояния организма.
Пациентам гарантировано внимательное и ответственное отношение, индивидуальный подход, конфиденциальность личных данных и достижение видимых результатов назначенного лечения или профилактики синдрома Дауна. Все необходимые диагностические мероприятия можно пройти в клинике, чтобы получить быстрые и достоверные результаты.
Для записи на прием и дополнительных консультаций вы можете позвонить по телефону +7 (495) 775-73-60.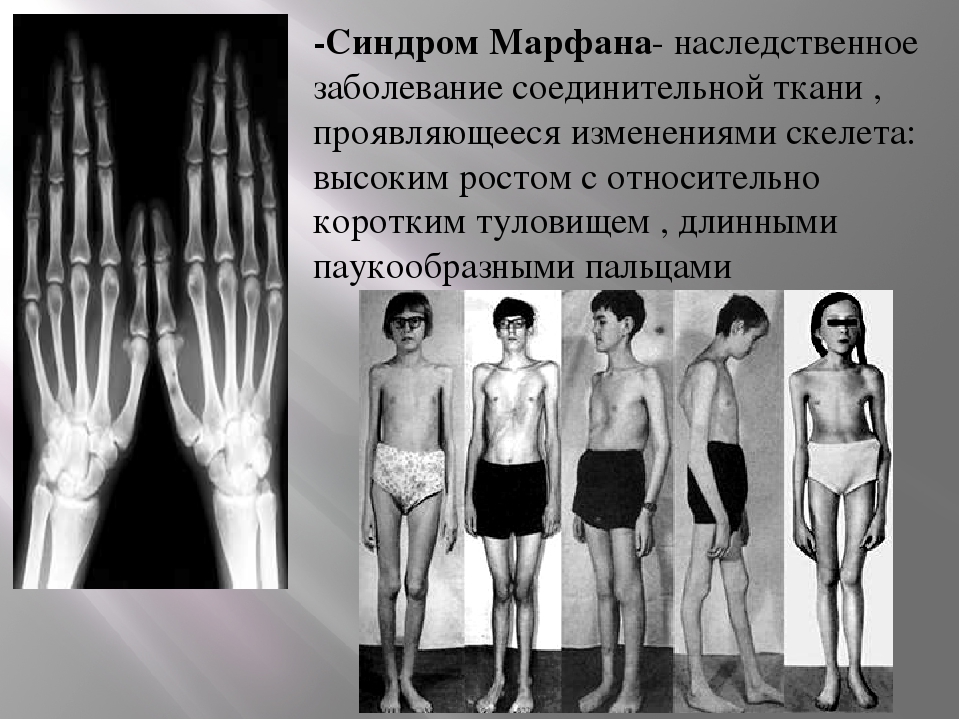
Частые вопросы о заболевании
Сколько живут люди с синдромом Дауна?
Средняя продолжительность жизни пациентов с синдромом Дауна составляет сегодня около 50 лет, увеличившись более чем в 2 раза относительно статистики середины прошлого столетия. На срок жизни влияет наличие осложнений в организме, вызванных генетическим сбоем, условия жизни, наследственность и т.д. При условии квалифицированного медицинского наблюдения продолжительность жизни пациентов удается продлить и сделать ее более комфортной.
Как определить заболевание во время беременности?
Уточнить риск развития плода с генетической аномалией можно с помощью ультразвукового исследования на ранних сроках беременности, а также при лабораторном исследовании крови на предмет содержания белков и гормонов в организме будущей матери. При наличии серьезных подозрений на генетический сбой на более поздних сроках исследуется биоматериал плода, полученный путем биопсии или забора околоплодной жидкости.
Лечится ли синдром Дауна?
Исправить последствия образования третьей хромосомы современная медицина не в состоянии. Поэтому перед ней стоит несколько задач: наблюдение за беременностью, своевременное выявление осложнений пациента с синдромом и их коррекция. Частично устранить последствия заболевания удается с помощью специальных обучающих программ и физической активности ребенка с синдромом Дауна, организм которого склонен к образованию лишнего веса.
| 1 | X-сцепленная адренолейкодистрофия |
| 2 | ААА синдром, Оллгрова синдром (ахалазия, алакримия, недостаточность надпочечников |
| 3 | Аарскога-Скотта cиндром |
| 4 | Абиотрофия сетчатки, тип Франческетти |
| 5 | Адреногенитальный синдром (врожденная гиперплазия коры надпочечников) |
| 6 | Азооспермия |
| 7 | Айкарди-Гутьереса синдром |
| 8 | Акродерматит энтеропатический |
| 9 | Аксенфельда-Ригера синдром |
| 10 | Алажиля синдром |
| 11 | Александера болезнь |
| 12 | Альбинизм глазокожный |
| 13 | Алькаптонурия |
| 14 | Альстрема синдром |
| 15 | Аменорея |
| 16 | Альфа-1-антитрипсина недостаточность |
| 17 | Ангельмана синдром |
| 18 | Андерсена синдром |
| 19 | Анемия Даймонда-Блекфена |
| 20 | Анеуплоидии |
| 21 | Аниридия |
| 22 | Антли-Бикслера синдром |
| 23 | Апера синдром |
| 24 | Арта cиндром |
| 25 | Артрогрипоз дистальный (синдром Фримена-Шелдона) |
| 26 | Атаксия Фридрейха |
| 27 | Атаксия, хорея, судороги и деменция |
| 28 | Атрофия зрительного нерва Лебера |
| 29 | Атрофия зрительного нерва с глухотой |
| 30 | Аутоиммунный лимфопролиферативный синдром |
| 31 | Аутоиммунный полиграндулярный синдром I типа |
| 32 | Аутоимунный полиэндокринный синдром |
| 33 | Афазия первичная прогрессирующая |
| 34 | Ахондроплазия |
| 35 | Баллера-Герольда синдром |
| 36 | Банаян-Райли-Рувалькаба cиндром |
| 37 | Барде-Бидля (Ларенса-Муна) синдром |
| 38 | Барта cиндром |
| 39 | Барттера синдром |
| 40 | Бёрта-Хога-Дьюба синдром |
| 41 | Бесплодие |
| 42 | Беста болезнь |
| 43 | Биотинидазы недостаточность |
| 44 | Блефарофимоз, обратный эпикант и птоз |
| 45 | Блоха-Сульцбергера синдром |
| 46 | Блума синдром |
| 47 | Боковой амиотрофический склероз |
| 48 | Боуэна-Конради синдром |
| 49 | Бранхио-окуло-фациальный синдром |
| 51 | Брахидактилия |
| 53 | Бьёрнстада синдром |
| 54 | Ваарденбурга синдром |
| 55 | Ваарденбурга-Шаха синдром |
| 56 | Ван дер Вуда синдром |
| 57 | Велокардиофациальный синдром |
| 58 | Вернера синдром |
| 59 | Видеманна-Беквита синдром, спорадическая нефробластома |
| 60 | Виллебранда болезнь |
| 61 | Вильсона-Коновалова болезнь |
| 62 | Вильямса cиндром |
| 63 | Вискотта-Олдрича cиндром |
| 64 | Вольмана болезнь, болезнь накопления эфиров холестерина |
| 65 | Вольфа-Хиршхорна синдром |
| 67 | Врожденная нечувствительность к боли с ангидрозом (врожденная сенсорная нейропатия с ангидрозом, HSAN4, CIPA) |
| 68 | Врожденной центральной гиповентиляции синдром |
| 69 | Вульгарный ихтиоз |
| 70 | Галактоземия тип I |
| 71 | Галактоземия тип II |
| 72 | Галактоземия тип III |
| 73 | Галактосиалидоз |
| 74 | Галлервордена-Шпатца болезнь |
| 75 | Ганглиозидоз GM1 тип 1,2,3 |
| 76 | Гастроинтестинальный полипоз |
| 77 | Гелеофизическая дисплазия |
| 78 | Гемофилия |
| 79 | Гемохроматоз наследственный |
| 80 | Генитопателлярный синдром |
| 81 | Германски-Пудлака синдром |
| 82 | Герстманна-Штреусслера-Шейнкера болезнь |
| 83 | Гидроцефалия, обусловленная врожденнным стенозом Сильвиева водопровода |
| 84 | Гипер-IgD синдром |
| 85 | Гипер-IgM синдром |
| 86 | Гиперкалиемический периодический паралич |
| 87 | Гипероксалурия тип I |
| 88 | Гиперорнитинемии-гипераммониемии-гомоцитрулинурии синдром (ННН синдром) |
| 89 | Гипертрофическая кардиомиопатия |
| 90 | Гиперфенилаланинемия с дефицитом тетрагидробиоптерина |
| 91 | Гиперхолестеринемии |
| 92 | Гипогонадизм |
| 93 | Гипокалиемический периодический паралич |
| 94 | Гипоспадия |
| 95 | Гипотрихоз |
| 96 | Гипофосфатазия |
| 97 | Гипофосфатемический рахит |
| 98 | Гипохондроплазия |
| 99 | Гиппеля-Линдау синдром |
| 100 | Глазо-зубо-пальцевой синдром |
| 101 | Глаукома врожденная |
| 102 | Глаукома ювенильная открытоугольная |
| 103 | Гликогеноз 0 тип |
| 104 | Гликогеноз III типа |
| 105 | Гликогеноз IV типа |
| 106 | Гликогеноз IX типа |
| 107 | Гликогеноз Iа тип |
| 108 | Гликогеноз Iв тип |
| 109 | Гликогеноз V типа |
| 110 | Гликогеноз VI типа |
| 111 | Гликогеноз XI типа, Фанкони-Бикеля синдром |
| 112 | Гломеруоцитоз почек гипопластического типа |
| 113 | Глутаровая ацидурия тип 1 |
| 114 | Глутаровая ацидурия тип 2 |
| 116 | Гомоцистинурия |
| 117 | Гоше болезнь тип 1,2,3 |
| 118 | Грейга cиндром |
| 119 | Грисцелли cиндром |
| 120 | Дауна cиндром |
| 121 | Делеции хромосомы 1p36 синдром |
| 122 | Десмоидные опухоли |
| 123 | Дефицит гормона гипофиза, комбинированный |
| 124 | Дефицит иммуноглобулина A |
| 125 | Дефицит карнитина системный первичный |
| 126 | Дефицит фактора F12 |
| 127 | Джексона-Вейсса cиндром |
| 128 | Ди Джорджи cиндром |
| 129 | Диастрофическая дисплазия |
| 130 | Дисгенезия гонад |
| 131 | Дисплазия де ля Шапеля (Ателостеогенез) |
| 132 | Дисплазия Книста |
| 133 | Дистальная моторная нейропатия |
| 134 | Дистальная спинальная амиотрофия врожденная с параличом диафрагмы |
| 135 | Дисхондростеоз Лери-Вейлля |
| 136 | Дорфмана-Чанарина синдром |
| 137 | Жильбера cиндром |
| 138 | Жубер cиндром |
| 139 | Задержка полового созревания |
| 140 | Зандхоффа болезнь |
| 141 | Изовалериановая ацидемия |
| 142 | Инверсия пола |
| 143 | Ихтиоз буллезный |
| 144 | Ихтиоз врожденный аутосомно-рецессивный |
| 145 | Ихтиоз вульгарный |
| 146 | Ихтиоз, спастическая квадриплегия и умственная отсталость |
| 147 | Кампомелическая дисплазия |
| 148 | Канавана болезнь |
| 149 | Карбамолфосфатсинтетазы недостаточность |
| 150 | Карпентера cиндром |
| 151 | Кератита-ихтиоза-тугоухости cиндром |
| 152 | Кернса-Сейра синдром |
| 153 | Клайнфельтера cиндром |
| 154 | Клиппеля-Фейля cиндром |
| 155 | Коккейна cиндром |
| 156 | Комбинированный дефицит витамин K-зависимых факторов свертывания крови |
| 157 | Косолапость врожденная с или без дефицита длинных костей и/или зеркальной полидактилией |
| 158 | Костелло cиндром |
| 159 | Костная гетероплазия прогрессирующая |
| 160 | Коудена болезнь |
| 161 | Коффина-Лоури синдром |
| 162 | Кошачьего глаза синдром |
| 163 | Кошачьего крика синдром |
| 164 | Краббе болезнь |
| 165 | Краниометафизарная дисплазия |
| 166 | Краниосиностоз |
| 167 | Краниофациальной дисморфии-глухоты-ульнарной девиации кистей синдром |
| 168 | Крейтцфельда-Якоба болезнь |
| 169 | Криглера-Найара синдром |
| 170 | Крипторхизм |
| 171 | Крузона с черным акантозом синдром |
| 172 | Крузона синдром |
| 173 | Куррарино синдром |
| 174 | Ларинго-онихо-кутанный синдром |
| 175 | Лейкодистрофия с гипомиелинизацией |
| 176 | Лейкоэнцефалопатия с «исчезающим» белым веществом, детская атаксия с гипомиелинизацией |
| 177 | Лейкоэнцефалопатия с пораженим ствола мозга и высоким уровнем лактата при спектроскопии |
| 178 | Лейкоэнцефалопатия с субкортикальными кистами |
| 179 | Лейциноз (болезнь «с запахом кленового сиропа мочи» |
| 180 | Лермитт-Дуклос болезнь |
| 181 | Леша-Найяна синдром |
| 182 | Ли синдром |
| 183 | Ли-Фраумени синдром |
| 184 | Линча синдром (наследственный неполипозный рак толстой кишки) |
| 185 | Липодистрофия врожденная генерализованная |
| 186 | Липодистрофия семейная частичная |
| 187 | Липопротеин липазы недостаточность |
| 188 | Лоу синдром |
| 189 | Люджина — Фринса синдром |
| 190 | Макла-Уэллса синдром |
| 191 | Маклеода синдром |
| 192 | Малан синдром |
| 193 | Мандибулоакральная дисплазия с липодистрофией |
| 194 | Маннозидоз альфа |
| 195 | Маринеску-Шегрена синдром |
| 196 | Мартина-Белл, УО FRAXA Синдром |
| 197 | Маршалла-Смита синдром |
| 198 | Мевалоновая ацидурия |
| 200 | Метатропная дисплазия (OMIM 156530) |
| 201 | Метахроматическая лейкодистрофия |
| 202 | Метгемоглобинемия |
| 203 | Метилмалоновая ацидурия |
| 204 | Микрофтальм изолированный |
| 205 | Микрофтальм с катарактой |
| 206 | Микроцефалии с капиллярными мальформациями синдром |
| 207 | Миллера-Дикера синдром |
| 208 | Милроя болезнь (лимфедема наследственная) |
| 209 | Миоклоническая дистония |
| 210 | Миоклоническая эпилепсия Лабофа |
| 211 | Миопатия Броди |
| 212 | Миопатия Миоши |
| 213 | Миотоническая дистрофия |
| 214 | Миотония Томсена/Беккера |
| 215 | Митохондриальные гепатопатии |
| 216 | Митохондриальные заболевания, связанные с мутациями в гене POLG |
| 217 | Митохондриальные энцефаломиопатии, связанные с мутациями мтДНК |
| 218 | Митохондриальные энцефаломиопатии, связанные с мутациями ядерных генов |
| 219 | Множественная сульфатазная недостаточность |
| 220 | Множественной эндокринной неоплазии второго типа (МЭН2) cиндром |
| 221 | Множественные вывихи суставов, задержка роста, черепно-лицевые аномалии и врожденные пороки сердца |
| 222 | Множественных птеригиумов синдром |
| 223 | Множественных синостозов синдром |
| 224 | Молибденового кофактора недостаточность |
| 225 | Монилетрикс |
| 226 | Моуат-Вильсон cиндром |
| 227 | Муковисцидоз |
| 228 | Муколипидоз II, III типа |
| 229 | Мукополисахаридоз I типа |
| 230 | Мукополисахаридоз II типа |
| 231 | Мукополисахаридоз III А, В, С, D типа |
| 232 | Мукополисахаридоз IV A, B типа |
| 233 | Мукополисахаридоз VI типа |
| 234 | Мукополисахаридоз VII типа |
| 235 | Мышечная дистрофия врождённая, интегрин А7 негативная |
| 236 | Мышечная дистрофия врожденная, мерозин-негативная |
| 237 | Мышечная дистрофия врожденная, тип 1C |
| 238 | Мышечная дистрофия Дюшенна/Беккера |
| 239 | Мышечная дистрофия поясноконечностная |
| 240 | Мышечная дистрофия тип Фукуяма |
| 241 | Мышечная дистрофия Эмери-Дрейфуса |
| 242 | Мюнке синдром |
| 243 | Накопление нейтральных липидов с миопатией |
| 244 | Нарушение формирования пола |
| 245 | Нанизм MULIBRAY |
| 246 | Нарушения гликозилирования тип 1a, синдром Жакена |
| 247 | Нарушения гликозилирования тип Ib (ген MPI) |
| 248 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I |
| 249 | Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип II |
| 250 | Наследственная нейропатия с подверженностью параличу от сдавления |
| 251 | Наследственная оптическая нейропатия Лебера |
| 252 | Наследственные глаукомы, аномалия Петерса, дермоид роговицы |
| 253 | Наследственный амилоидоз |
| 254 | Наследственный ангионевротический отек |
| 255 | Наследственный панкреатит |
| 256 | Невынашивание беременности |
| 257 | Наследственный рак желудка |
| 258 | Недостаточность N-ацетилглютаматсинтазы |
| 259 | Недостаточность длинноцепочечной 3-гидроксиацил-КоА-дегидрогеназы жирных кислот |
| 260 | Недостаточность короткоцепочечной ацил-КоА-дегидрогеназы жирных кислот |
| 261 | Недостаточность очень длинноцепочечной ацил-КоА дегидрогеназы жирных кислот |
| 262 | Недостаточность синтетазы голокарбоксилаз |
| 263 | Недостаточность среднецепочечной ацил-КоА-дегидрогеназы жирных кислот |
| 264 | Недостаточность сукцинил-КоА:3-кетоацил-КоА трансферазы |
| 265 | Незаращение родничков |
| 266 | Нейроаксональная дистрофия |
| 267 | Нейродегенерация с накоплением железа 4 |
| 268 | Нейромиотония и аксональная нейропатия |
| 269 | Нейрональный цероидный липофусциноз тип 1 |
| 270 | Нейрональный цероидный липофусциноз тип 2 |
| 271 | Нейросенсорная несиндромальная тугоухость |
| 272 | Нейрофиброматоз 1 и 2 типов |
| 273 | Нейтропения тяжёлая врождённая |
| 274 | Некетотическая гиперглицинемия |
| 275 | Некомпактного левого желудочка cиндром |
| 276 | Немалиновая миопатия |
| 277 | Нефронофтиз |
| 278 | Нефротический синдром |
| 279 | Ниймеген cиндром |
| 280 | Ниманна-Пика тип А и В болезнь |
| 281 | Ниманна-Пика тип С болезнь |
| 282 | Ногтей-надколенника синдром |
| 283 | Норри болезнь |
| 284 | Нунан синдром |
| 285 | Олигозооспермия тяжелой степени |
| 286 | Окулофарингеальная мышечная дистрофия |
| 287 | Опица GBBB синдром |
| 288 | Опица-Каведжиа синдром |
| 289 | Опухоль Вильмса |
| 290 | Орнитинтранскарбамилазы недостаточность |
| 291 | Ослера-Рендю-Вебера cиндром |
| 292 | Остеолиз карпотарзальный, мультицентрический |
| 293 | Остеопетроз рецессивный (мраморная болезнь костей) |
| 294 | Паллистера-Киллиана cиндром |
| 295 | Паллистера-Холла cиндром |
| 296 | Палочко-колбочковая дистрофия |
| 297 | Пантотенат киназы недостаточность |
| 298 | Парамиотония Эйленбурга |
| 299 | Патау cиндром |
| 300 | Пейтца-Егерса синдром |
| 301 | Пелицеуса-Мерцбахера болезнь |
| 302 | Пендреда Синдром |
| 303 | Первичная аутосомно-рецессивная микроцефалия, тип 5 |
| 304 | Первичная гипертрофическая остеоартропатия (пахидермопериостоз) |
| 305 | Первичная легочная гипертензия |
| 306 | Периодическая болезнь |
| 307 | Пигментная дегенерация сетчатки |
| 308 | Пикнодизостоз |
| 309 | Пирсона синдром |
| 310 | Пневмоторакс первичный спонтанный |
| 311 | Подколенного птеригиума cиндром |
| 312 | Полидактилия |
| 313 | Поликистоз почек |
| 314 | Помпе болезнь |
| 315 | Понтоцеребеллярная гипоплазия |
| 316 | Потоцки-Лупски cиндром |
| 317 | Почечная адисплазия |
| 318 | Прадера-Вилли Синдром |
| 319 | Преждевременная недостаточность яичников |
| 320 | Прогерия Хатчинсона-Гилфорда |
| 321 | Прогрессирующая наружная офтальмоплегия, АД и АР |
| 322 | Пропионовая ацидемия |
| 323 | Псевдоахондроплазия |
| 324 | Псевдоксантома эластическая |
| 325 | Пфайффера cиндром |
| 326 | Рабдомиолиз (миоглобинурия) |
| 327 | Рак молочной железы |
| 328 | Рак почки |
| 329 | Рак щитовидной железы. Синдром множественной эндокринной неоплазии второго типа (МЭН2) |
| 330 | Рак яичников |
| 331 | Ретинобластома |
| 332 | Ретиношизис |
| 333 | Ретта синдром |
| 334 | Рефсума болезнь |
| 335 | Ригидного позвоночника cиндром |
| 336 | Робинова синдром |
| 337 | Ротмунда-Томсена синдром |
| 338 | Рубинштейна-Тейби синдром |
| 339 | Семейная периодическая лихорадка |
| 340 | Семейный аденомоматозный полипоз, полипозный рак толстой кишки |
| 341 | Семейный внутрипеченочный холестаз 1 типа |
| 342 | Семейный внутрипеченочный холестаз 2 типа ( Баллера болезнь) |
| 343 | Семейный внутрипеченочный холестаз 3 типа |
| 344 | Семейный гемофагоцитарный лимфогистиоцитоз |
| 345 | Семейный медуллярный рак щитовидной железы |
| 346 | Семейный рак толстой кишки |
| 347 | Семейный холодовой аутовоспалительный синдром |
| 348 | Сениора-Локена синдром |
| 349 | Сенсорная полинейропатия (врожденная нечувствительность к боли) |
| 350 | Септо-оптическая дисплазия |
| 351 | Сетре-Чотзена синдром |
| 352 | Сиалидоз тип 1,2 |
| 353 | Сильвера-Рассела Синдром |
| 354 | Симпсона-Голаби-Бемель синдром |
| 355 | Синдром CADASIL, энцефалопатия с субкортикальными инфарктами |
| 357 | Синдром CINCA (холодовая лихорадка, синдром Мукле-Велса) |
| 358 | Синдром CRASH |
| 359 | Синдром ESC |
| 360 | Синдром LEOPARD |
| 361 | Синдром MASA |
| 362 | Синдром MNGIE |
| 363 | Синдром Ohdo, SBBYSS вариант |
| 364 | Синдром RAPADILINO |
| 365 | Синдром TAR |
| 366 | Синдром TRAPS (злокачественная гипертермия, амилоидоз почек) |
| 367 | Синдром тугоухости и атрофии зрительных нервов |
| 368 | Скапулоперонеальная миопатия |
| 370 | Смита-Лемли-Опитца синдром |
| 371 | Смит-Магенис синдром |
| 372 | Сотоса синдром |
| 373 | Спастическая параплегия Штрюмпеля |
| 374 | Спинальная амиотрофия типы I, II, III, IV |
| 375 | Спинальная и бульбарная амиотрофия Кеннеди |
| 376 | Спиноцеребеллярная атаксия |
| 377 | Спонгиоформная энцефалопатия с нейропсихическими проявлениями |
| 378 | Спондилокостальный дизостоз |
| 379 | Спондилоэпифизарная дисплазия (SEDT) |
| 380 | Стиклера синдром |
| 381 | Суперактивность фосфорибозилпирофосфат синтетазы |
| 382 | Талассемия beta |
| 383 | Тестикулярной феминизации синдром |
| 384 | Тея-Сакса болезнь |
| 385 | Тирозингидроксилазы недостаточность |
| 386 | Тирозинемия тип I |
| 387 | Торсионная дистония |
| 388 | Транспортера глюкозы недостаточность |
| 389 | Трихоринофалангеальный синдром |
| 390 | Тричера Коллинза-Франческетти синдром |
| 391 | Тромбоцитопения врожденная |
| 392 | Туберозный склероз |
| 393 | Умственная отсталость моногенная |
| 394 | Унферрихта-Лундборга болезнь |
| 395 | Уокера-Варбург синдром |
| 396 | Ушера синдром |
| 397 | Фабри болезнь |
| 398 | Фатальная семейная инсомния |
| 399 | Фацио-Лонде болезнь |
| 400 | Фелан-МакДермид синдром |
| 401 | Фенилкетонурия |
| 402 | Фибродисплазия оссифицирующая прогрессирующая |
| 403 | Фокальная кожная гипоплазия (Горлина-Гольца синдром) |
| 404 | Фокально-кортикальная дисплазия Тейлора |
| 405 | Фон Хиппель-Линдау Синдром |
| 406 | Фруктозо1,6 дифосфотазы недостаточность |
| 407 | Фукозидоз |
| 408 | Хайду-Чейни синдром |
| 409 | Хондродисплазия метафизарная тип Мак-Кьюсика |
| 410 | Хондродисплазия точечная Конради-Хюнермана |
| 411 | Хондрокальциноз |
| 412 | Хореоатетоз, гипотиреоидизм и неонатальная дыхательная недостаточность |
| 413 | Хорея Гентингтона |
| 414 | Хорея доброкачественная наследственная |
| 415 | Хороидермия |
| 416 | Хромосомные болезни |
| 417 | Хроническая гранулематозная болезнь |
| 418 | Х-сцепленная агаммаглобулинемия |
| 419 | Х-сцепленный лимфопролиферативный синдром (болезнь Дункана, синдром Пуртильо) |
| 420 | Х-сцепленный моторный нистагм |
| 421 | Х-сцепленный тяжелый комбинированный иммунодефицит |
| 422 | Целвегера синдром |
| 423 | Центронуклеарная миопатия |
| 424 | Цереброокулофациоскелетный синдром |
| 425 | Цистиноз |
| 426 | Цистиноз нефропатический |
| 427 | Цитруллинемия тип 1 |
| 428 | Шварца-Джампела синдром |
| 429 | Швахмана-Даймонда синдром |
| 430 | Шегрена-Ларссона синдром |
| 431 | Шерешевского-Тернера синдром |
| 432 | Широкого водопровода преддверия синдром |
| 433 | Шпринтцена-Гольдберга синдром |
| 434 | Штаргардта болезнь |
| 435 | Эдвардса синдром |
| 436 | Экзостозы множественные |
| 437 | Эксудитивная витреохореорстинальная дистрофия |
| 438 | Эктодермальная ангидротическая дисплазия |
| 439 | Эктодермальная гидротическая дисплазия |
| 440 | Эктопия хрусталика |
| 442 | Эллерса-Данло синдром |
| 443 | Эпилепсия прогрессирующая миоклоническая |
| 444 | Эпифизарная дисплазия, множественная |
| 445 | Эритрокератодермия |
| 446 | Эритроцитоз рецессивный |
| 447 | Эскобара cиндром |
Генетические, метаболические и хромосомные расстройства
Синдром — это медицинский термин, который описывает людей, которые имеют определенные физические, возрастные и / или поведенческие характеристики, которые возникают из-за одной основной причины (например, неисправного гена или набора генов). Человек, у которого диагностирован определенный синдром, может иметь некоторые или все характеристики этого синдрома. Исследования показывают, что вызывающее или проблемное поведение чаще встречается при определенных генетических синдромах, чем можно было бы ожидать, исходя из уровня умственной отсталости (ИД) человека и других характеристик, таких как трудности в общении.
Синдром Прадера-Вилли (PWS):
PWS — это генетическое заболевание, связанное с дефектом хромосомы 15. Сообщаемая распространенность PWS широко варьировала, но чаще всего упоминается, что PWS поражает примерно 1 из 15 000 рождений. Как и в случае с большинством генетических синдромов, у людей с диагнозом СПВ есть общие характеристики. Например, в раннем возрасте им обычно ставят диагноз «неспособность развиваться» из-за плохого приема пищи. Однако примерно в возрасте 18 месяцев люди с СПВ начинают страдать от гиперфагии (чрезмерного потребления пищи), которая, если ее не остановить, может привести к патологическому ожирению и связанным с этим проблемам со здоровьем.Люди с PWS часто идут на все, чтобы добыть (или украсть) еду, и может казаться, что у них есть одержимость едой. В этой популяции обычно сообщают о проблемах с поведением. Помимо гиперфагии, кражи еды и истерик, самоповреждение кожи, по-видимому, чаще встречается в популяции PWS по сравнению с общей популяцией ID. Люди также могут проявлять агрессию, разрушительное / деструктивное поведение и убегать (бегство). Многие из этих форм поведения могут возникать при попытке получить доступ к пище или в ответ на отказ в пище.
Синдром Дауна:
Синдром Дауна — наиболее часто диагностируемое хромосомное заболевание, которым страдает каждый 691 ребенок, рожденный в Соединенных Штатах. У людей с синдромом Дауна дефект хромосомы 21. Национальное общество синдрома Дауна предполагает, что люди с синдромом Дауна могут иметь общие поведенческие проблемы, включая блуждание / побег (также называемое бегством) и упрямое / оппозиционное поведение (также называемое несоблюдением).
Синдром Вильямса:
Синдром Вильямса — это генетическое заболевание, вызванное делецией генов в хромосоме 7.Синдром Вильямса поражает примерно 1 из 10 000 человек и встречается у мужчин и женщин в равной степени. Большинство людей с синдромом Вильямса имеют некоторый уровень задержки когнитивных функций — от легкой до умеренной. Вехи развития (например, разговоры, приучение к туалету) часто происходят несколько позже, чем обычно. Маленькие дети могут испытывать трудности с кормлением из-за низкого мышечного тонуса, сильных рвотных рефлексов и плохого сосания и глотания. У младенцев также могут быть периоды раздражительности и трудности с установлением типичного режима сна.Люди с синдромом Вильямса, пожалуй, наиболее известны своим излишне общительным и дружелюбным характером, веселым поведением и сильными вербальными и социальными навыками; тем не менее, им может потребоваться помощь, чтобы обеспечить уместность социального взаимодействия. Кроме того, люди с синдромом Вильямса могут с большей вероятностью иметь фобии, которые могут повлиять на их способность полноценно участвовать в жизни сообщества. Поведенческие вмешательства могут быть полезны при фобиях и для тренировки социальных навыков (например, сдерживание неуместных проявлений привязанности к незнакомцам).
Синдром ломкой Х-хромосомы:
Fragile X является наиболее частой известной причиной наследственной (генетической) умственной отсталости. Он поражает как мужчин, так и женщин, но поскольку пораженный ген находится на X-хромосоме, мужчины поражаются чаще и тяжелее, чем женщины. Распространенность Fragile X составляет примерно от 1 из 3600 до 1 из 4000 среди мужчин и примерно от 1 из 4000 до 1 из 6000 среди женщин. Мужчины с ломкой Х-хромосомой обычно имеют умственную отсталость от умеренной до тяжелой; пораженные женщины имеют более легкие когнитивные нарушения.Примерно у 40 процентов людей с ломкой X также диагностирован аутизм; эти люди, как правило, имеют более серьезные интеллектуальные нарушения, чем те, у кого также не диагностирован аутизм. Поведенческие характеристики могут включать в себя трудности с вниманием и гиперактивностью, социальную тревогу и / или избегание, плохой зрительный контакт, трудности с переходами, кусание и / или хлопанье руками и агрессивное поведение. Хотя было показано, что проблемное поведение возникает по разным причинам (например,g., чтобы получить доступ к вниманию, для сенсорной стимуляции), люди с Хрупкой X могут быть более склонны к проблемному поведению, чтобы уйти от социальных взаимодействий, потому что они имеют тенденцию находить эти взаимодействия отталкивающими.
Синдром Смита-Магениса:
Smith-Magenis — это генетическое заболевание, чаще всего вызываемое делецией генетического материала из хромосомы 17. Сообщаемая распространенность Smith-Magenis составляет 1 на 25 000 человек; однако исследователи полагают, что это заболевание недооценивается, и истинная распространенность, вероятно, ближе к 1 из 15 000 человек.Люди с синдромом Смита-Магениса имеют различные уровни умственной отсталости от легкой до умеренной. Хотя людей с диагнозом Смит-Магенис часто описывают как милых / привлекательных личностей, существуют также общие поведенческие трудности, включая задержку речи, хроническое нарушение сна, скрежетание зубами, задержку приобретения навыков пользования туалетом, гиперактивность, импульсивность, чрезмерное привлечение внимания и т. Д. отвлекаемость, длительные истерики, агрессивное и деструктивное поведение.Кроме того, распространены определенные типы стереотипного и самоповреждающего поведения. При возбуждении люди могут обнимать и / или сжимать руки; они также могут «облизывать и переворачивать» (например, быстро перелистывать страницы книг / журналов, облизывая палец или не облизывая его). Самоповреждающее поведение может включать удары головой, кусание рук, ковыряние кожи и / или ногтей и вставку предметов в отверстия тела. Хотя членовредительство может быть восприимчивым ко многим типам вознаграждений, в некоторых случаях было показано, что членовредительство, нанесенное людьми с Smith-Magenis, более вероятно в условиях с низким уровнем внимания взрослых; Было высказано предположение, что генетическая предрасположенность к социальному контакту с другими может повысить вероятность того, что СИБ возникнет для доступа к вниманию.
Синдром Ангельмана:
Синдром Ангельмана — это генетическое заболевание, вызванное потерей гена, расположенного на хромосоме 15. Сообщаемая распространенность варьирует, но наиболее часто упоминаемая оценка составляет 1 на каждые 15 000 человек. Он одинаково влияет на мужчин, женщин и все расовые группы. У большинства больных наблюдаются повторяющиеся приступы (эпилепсия), небольшой размер головы (микроцефалия), а также нарушения ходьбы и равновесия. Многие дети также испытывают трудности со сном и нуждаются в меньшем количестве сна, чем обычно, хотя есть некоторые предположения, что проблемы со сном могут улучшиться с возрастом.Люди с синдромом Ангельмана также имеют серьезные интеллектуальные нарушения, и хотя они не говорят, они обычно понимают некоторый язык. Несмотря на свои проблемы, люди с Ангелманом часто ведут себя счастливыми, часто смеются и улыбаются; исследования показали, что они склонны искать социальные взаимодействия, предполагая, что им это очень нравится. При синдроме Ангельмана часто встречается агрессивное поведение, и было высказано предположение, что у некоторых людей такое поведение может инициировать или продлевать социальное взаимодействие со взрослыми.Люди с Ангельманом могут быть возбудимыми, что может проявляться в частых взмахах руками. Кроме того, обычны гиперактивность, непродолжительное внимание и увлечение водой.
Синдром Смита-Лемли-Опица:
Синдром Смита-Лемли-Опица (SLOS) — это генетическое заболевание, вызванное дефектным геном на хромосоме 7, который отвечает за выработку холестерина, необходимого для клеток организма. Синдром Смита-Лемли-Опица поражает примерно 1 из 20 000–60 000 новорожденных.Это заболевание чаще всего встречается у кавказцев (белых) европейского происхождения, особенно у выходцев из центральноевропейских стран, таких как Словакия и Чешская Республика. Это очень редко встречается среди африканского и азиатского населения.
Признаки и симптомы SLOS сильно различаются. У людей с легким поражением могут быть только незначительные физические отклонения, связанные с обучением и поведенческими проблемами. Тяжелые случаи могут быть опасными для жизни и включать глубокую умственную отсталость и серьезные физические отклонения.Один из распространенных симптомов SLOS — задержка речи и трудности в общении в целом. Кроме того, у людей с SLOS может быть нетипичный режим сна.
Один из наиболее сложных аспектов SLOS — это постоянная борьба с поведением. Почти все люди с SLOS демонстрируют по крайней мере некоторые из проблемных форм поведения, характерных для людей с аутизмом. Поведение может варьироваться от крика, ударов, щипков и укусов до ударов головой и членовредительства.
Синдром Леша-Нихана:
Синдром Леша-Найхана является результатом перепроизводства мочевой кислоты, продукта жизнедеятельности организма, который содержится в крови и моче.Ген, связанный с этим заболеванием, расположен на Х-хромосоме; в результате Lesch-Nyhan встречается почти исключительно у мужчин. Lesch-Nyhan встречается примерно у 1 из 380 000 человек с одинаковой частотой во всех расовых / этнических популяциях.
Lesch-Nyhan характеризуется непроизвольными мышечными движениями, включая напряжение и подергивание мышц, а также раскачивание конечностей. В результате люди с Lesch-Nyan обычно привязаны к инвалидной коляске.
Самоповреждающее поведение наблюдается почти у всех людей с Lesch-Nyhan.Дети начинают кусать пальцы, губы и внутреннюю часть рта уже в 2–3 года; они также могут трясти головой. Частота и серьезность самоповреждений часто требует ограничений, и некоторым семьям пришлось прибегать к удалению зубов. Кроме того, люди с Lesch-Nyhan могут также проявлять агрессию, например, бить или плевать на других.
Корнелия де Ланге:
Корнелия де Ланге вызывается мутацией в одном из нескольких различных генов, расположенных на X-хромосоме или хромосомах 5 или 10.Примерно в 35% случаев причина синдрома Корнелии де Ланге неизвестна. Корнелия де Ланге встречается примерно у 1 из 10 000–30 000 новорожденных. Он одинаково влияет на оба пола и проявляется одинаково среди расовых и этнических групп. Почти все случаи являются результатом мутаций новых генов и возникают у людей, у которых в семье не было этого заболевания.
Лица с Корнелией де Ланж обычно имеют интеллектуальную недостаточность от легкой до глубокой, причем у большинства людей она бывает от легкой до умеренной.Режим сна и питания часто бывает нерегулярным, а проблемы с пищеварением могут привести к сложному поведению во время еды. Отмечались как тревога, так и трудности с удержанием внимания, а речь может отсутствовать или быть минимальной. Кроме того, люди с Корнелией де Ланге могут демонстрировать поведение, подобное тому, которое наблюдается у людей с аутизмом, например, повторяющееся поведение и самоповреждение (например, SIB, направленный на глаза).
Синдром Ретта:
Синдром Ретта вызывается мутациями гена Х-хромосомы.Он поражает примерно 1 из 10 000–1 из каждых 23 000 родов женского пола и проявляется одинаково во всех расовых / этнических группах. Симптомы синдрома Ретта появляются после раннего периода явно нормального или почти нормального развития до шести-восемнадцати месяцев жизни, когда наблюдается замедление или стагнация навыков. Затем следует период регресса, когда ребенок теряет коммуникативные навыки и целенаправленное использование рук.
Хотя раньше синдром Ретта классифицировался как одно из расстройств аутистического спектра, теперь он считается отдельным расстройством, имеющим множество симптомов, схожих с аутизмом.Дети с аутизмом и дети с синдромом Ретта демонстрируют нарушение социального взаимодействия, нарушение коммуникации и необычное поведение или движения. Сходство симптомов является причиной того, что синдром Ретта часто ошибочно принимают за аутизм. Однако по мере взросления ребенка с синдромом Ретта некоторые из симптомов аутизма могут уменьшаться или даже исчезать; например, люди с синдромом Ретта часто восстанавливают интерес к социальным взаимодействиям в более позднем возрасте.
Лица с синдромом Ретта обычно имеют широкий диапазон умственных нарушений от тяжелой до глубокой и имеют ограниченные выразительные языковые способности.Стереотипные движения рук, включая заламывание или «мытье» рук, также являются обычным явлением, а также кусаются и / или глотают руки и плечи. Кроме того, могут возникать задержка дыхания и раскачивание тела.
Синдром Кри-дю-Шат:
Синдром кри-дю-шат (5P-) — это генетическое заболевание, вызываемое отсутствием генов на хромосоме 5. Он поражает примерно 1 из 20 000–1 из 50 000 новорожденных. Одна из самых отличительных черт младенцев с кри-дю-чат — их пронзительный кошачий крик.
Люди с кри-дю-чатом обычно имеют серьезные интеллектуальные нарушения и трудности в общении с другими людьми. Часто встречаются проблемы с кормлением в раннем возрасте, такие как гиперактивность, навязчивая привязанность к объектам, повторяющиеся движения (например, стереотипия) и истерики, которые могут включать агрессию и самоповреждающее поведение (например, битье головой, выщипывание кожи).
Синдром Сотоса:
Синдром Сотоса вызывается дефектом хромосомы 5 и, как сообщается, встречается у 1 из 10 000–14 000 новорожденных.Однако многие случаи этого расстройства не диагностируются, поэтому истинная частота может быть ближе к 1 из 5000. Около 95 процентов случаев синдрома Сотоса возникают у людей, не страдающих этим заболеванием в семье.
Люди с синдромом Сотоса часто имеют интеллектуальные нарушения; однако у некоторых людей с Сотосом синдром только изменяет время развития, и, несмотря на ранние тенденции, взрослые могут иметь нормальный диапазон интеллектуальных способностей. Большинство детей с синдромом Сотоса имеют проблемы с поведением.Часто сообщаемые поведенческие проблемы включают нарушения сна, синдром дефицита внимания и гиперактивности (СДВГ), фобии, навязчивые идеи и компульсии, раздражительность, импульсивное поведение и истерики. Также распространены проблемы с речью и языком.
Синдром Ларсена — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Hennekam RCM, Allanson J, Krantz I, eds. Синдромы Горлина головы и шеи. 5-е изд. Нью-Йорк, Нью-Йорк: издательство Оксфордского университета; 2010: 984-987.
Лахман RS. Радиология синдромов, нарушений обмена веществ и скелетной дисплазии Тайби и Лахмана. 5-е изд. Филадельфия, Пенсильвания: Mosby Elsevier Co .; 2007: 450-452.
Джонс К.Л., Крэндалл-Джонс М., Дель Кампо, М. Распознаваемые модели пороков развития человека Смита, 7-е издание, Филадельфия, Elsevier-W.B. Saunders Co, 2013; 564-567.
СТАТЬИ ИЗ ЖУРНАЛА
Арунрут Т., Саббадини М., Джайн М. и др. Помутнение роговицы, катаракта и колобомы с новой миссенс-мутацией в B4GALT7 — обзор глазных аномалий при синдромах линкеропатии.Am J Med Genet A. 2016 Октябрь; 170 (10): 2711-2718.
Бэнкс Дж. Т., Веллонс Дж. К., Таббс Р. С. и др. Поражение шейного отдела позвоночника при синдроме Ларсена: случайная иллюстрация. Педиатрия. 2003; 111: 199-201.
Беккер Р., Вегнер Р.Д., Кунце Дж. И др. Клиническая вариабельность синдрома Ларсена: диагноз у отца после ультразвукового обнаружения сильно пораженного плода. Clin Genet. 2000; 57: 148-50.
Бернкопф М., Хант Д., Келлинг Н. и др. Количественная оценка риска передачи у пациента мужского пола с мозаичной мутацией ЛНБ, вызывающей синдром Ларсена: значение для генетического консультирования в случаях постзиготного мозаицизма.Hum Mutat. 2017 Октябрь; 38 (10): 1360-1364.
Бикнелл Л.С., Фаррингтон-Рок С., Шафегати И. и др. Молекулярное и клиническое исследование синдрома Ларсена, вызванного мутациями в FLNB. J Med Genet. 2007; 44: 89-98.
Cartault F, Munier P, Jacquemont ML и др. Расширение клинического спектра дефицита B4GALT7: гомозиготная мутация p.R270C с эффектом основателя вызывает синдром Ларсена с острова Реюньон. Eur J Hum Genet. 2015 23: 49-53.
Кричли Л.А., Чан К. Общая анестезия у ребенка с синдромом Ларсена.Анаэст Интенсивная терапия. 2003; 31: 217-20.
Debeer P, De Borre LO, De Smet L, Fryns JP. Асимметричный синдром Ларсена у молодой девушки: второй пример соматического мозаицизма при этом синдроме. Genet Couns. 2003; 14: 95-100.
Гириша К.М., Бидчол А.М., Грауль-Нойман Л. и др. Фенотип и генотип у пациентов с синдромом Ларсена: клиническая однородность и аллельная гетерогенность у семи пациентов. BMC Med Genet. 2016 6 апреля; 17:27.
Huber C, Oules B, Bertoli M et al: Идентификация мутаций CANT1 при дисплазии Desbuquois.Am J Hum Genet 2009; 85: 706–710.
Джонстон CE, 2-й, Берч Дж. Дж., Дэниэлс Дж. Л.. Шейный кифоз у пациентов с синдромом Ларсена. J Bone Joint Surg Am. 1996; 78: 538-545.
Краков Д., Робертсон С.П., Кинг Л.М. и др. Мутации в гене, кодирующем филамин B, нарушают сегментацию позвонков, формирование суставов и скелетогенез. Нат Жене. 2004; 36: 405-410.
Larsen LJ, Schottstaedt Er, Bost FC. Множественные врожденные вывихи, связанные с характерной аномалией лица.J Pediat. 1950. 37: 574-581.
Malik P, Choudhry DK. Синдром Ларсена и соображения его анестезии. Педиатр Анаест. 2002; 12: 632-6.
Маркес Л.Х.С., Мартинс Д.В., Хуарес Г.Л. и др. Отологические проявления синдрома Ларсена. Int J Pediatr Otorhinolaryngol. 2017 Октябрь; 101: 223-229.
Мэй Х., Хе Р., Лю К. и др. Предполагаемый синдром Ларсена у ребенка: наблюдение через 12 лет. J Pediatr Orthop B. 2015 24: 268-273.
Мизумото С., Ямада С., Сугахара К. Мутации в биосинтетических ферментах в области белкового линкера хондроитина / дерматана / гепарансульфата вызывают дисплазию скелета и кожи.Biomed Res Int. 2015; 2015: 861752. DOI: 10.1155 / 2015/861752. Epub 2015 25 октября.
Патель Н., Шамселдин Х.Э., Сакати Н. и др. Мутации GZF1 увеличивают генетическую неоднородность синдрома Ларсена. Am J Hum Genet. 2017 4 мая; 100 (5): 831-836.
Petrella R, Rabinowitz JF, Steinmann B, Hirschhorn K. Долгосрочное наблюдение за двумя братьями и сестрами с синдромом Ларсена, возможно, из-за мутации родительской зародышевой линии. Am J Med Genet. 1993; 47: 187-197.
Rock MJ, Green CG, Pauli RM, Peters ME. Трахеомаляция и бронхомаляция, связанные с синдромом Ларсена.Педиатр Пульмонол. 1988; 5: 55-59.
Салиан С., Шукла А., Шах Х. и др. Семь дополнительных семей с синдромом спондилокарпотарзального синостоза с новыми двуаллельными вредоносными вариантами в ЛНБ. Clin Genet. 2018 июль; 94 (1): 159-164.
Солтер К.Г., Дэвис Дж. Х., Мун Р. Дж. И др. Дальнейшее определение фенотипического спектра мутаций B4GALT7. Am J Med Genet A. 2016 июнь; 170 (6): 1556-1563.
Унгер С., Лауш Э., Росси А. и др. Фенотипические особенности дефицита углеводсульфотрансферазы 3 (CHST3) у 24 пациентов: врожденные вывихи и изменения позвонков как основные диагностические признаки.Am J Med Genet A. 2010; 152A: 2543-2549.
Vujic M, Hallstensson K, Wahlstrom J, et al. Локализация гена аутосомно-доминантного синдрома Ларсена в области хромосомы 3p21.1-14.1 рядом с локусом COL7A1, но отличается от него. Am J Hum Genet. 1995, 57: 1104-1113.
Winer N, Kyndt F, Paumier A, David A, Isidor B, Quentin M, Jouitteau B, Sanyas P, Philippe HJ, Hernandez A, Krakow D, Le Caignec C.Пренатальная диагностика синдрома Ларсена, вызванного мутацией в ген филамина В.Prenat Diagn. 2009; 29: 172-4.
Zhang, Herring JA, Swaney SS, et al. Мутации, ответственные за синдром Ларсена, группируются в белке FLNB. J Med Genet. 2006; 43: e24.
ИНТЕРНЕТ
Робертсон С. Расстройства, связанные с ЛНБ. 9 октября 2008 г. [Обновлено 17 октября 2013 г.]. В: Адам М.П., Ардингер Х.Х., Пагон Р.А. и др., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2018 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK2534/ По состоянию на 14 августа 2018 г.
Онлайн-Менделирующее наследование в человеке (OMIM).Университет Джона Хопкинса. Синдром Ларсена; LRS. Запись №: 150250. Последнее изменение 22.06.2018. Доступно по адресу: http://omim.org/entry/150250 По состоянию на 14 августа 2018 г.
Синдром
KBG — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Доулинг, штат Пенсильвания, Флеминг, Р. Синдром КБГ. В: Справочник НОРД по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 210.
Горлин Р.Дж. и др., Ред. Синдромы головы и шеи, 3-е изд.Нью-Йорк, Нью-Йорк: издательство Оксфордского университета; 1990: 840-1.
СТАТЬИ В ЖУРНАЛЕ
Low K, Ashraf T, Canham N, et al. Клинические и генетические аспекты синдрома KBG. Am J Med Genet A. 2016 [Epub перед печатью]. http://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.37842/full
Goldenberg A, Riccardi F, Tessier A, et al. Клинические и молекулярные данные у 39 пациентов с синдромом KBG, вызванным делецией или мутацией ANKRD11. Am J Med Genet A. 2016; [Epub перед печатью].https://www.ncbi.nlm.nih.gov/pubmed/27605097
Ockeloen CW, Willemsen MH, de Munnik S, et al. Дальнейшее определение синдрома KBG, вызванного аберрациями ANKRD11. Eur J Hum Genet. 2015; 23: 1270. http://www.ncbi.nlm.nih.gov/pubmed/26269249
Reynaert N, Ockeloen CW, Savedahl L, et al. Низкий рост при синдроме KBG: первые ответы на лечение гормоном роста. Horm Res Paediatr. 2015; 83: 361-364. http://www.ncbi.nlm.nih.gov/pubmed/25833229
Sirmaci A, Spiliopoulos M, Brancati F, et al.Мутации в ANKRD11 вызывают синдром KBG, характеризующийся умственной отсталостью, пороками развития скелета и макродонтией. Am J Hum Genet. 2011; 89: 289-294. http://www.ncbi.nlm.nih.gov/pubmed/21782149
Альмандей А.Х., Антонаппа Р.П., Кинг Н.М., Фунг С.В. Синдром KBG: клинические особенности и стоматологические особенности. Педиатр Дент. 2010; 32: 439-444. http://www.ncbi.nlm.nih.gov/pubmed/21070713
Бранкати Ф., Саркози А., Даллапиккола Б. Синдром КБГ. Orphanet J Rare Dis. 2006; 1:50. http: // www.ncbi.nlm.nih.gov/pmc/articles/PMC1764006/
Tekin M, Kavaz A, Berberoglu M, et al. Синдром KBG: подтверждение аутосомно-доминантного наследования и дальнейшее определение фенотипа. Am J Med Genet A. 2004; 130A: 284-287. http://www.ncbi.nlm.nih.gov/pubmed/15378538
Доулинг П.А., Флеминг П., Горлин Р.Г. и др. Синдром KBG, характерные стоматологические находки: история болезни. Int J Paediatr Dent. 2001; 11: 131-34. http://www.ncbi.nlm.nih.gov/pubmed/11310136
Сахаров С., Ли Д., Фан Ис, Текин М.Семейная микроделеция 16q24.3 с участием ANKRD11 вызывает KBG-подобный синдром. Am J Med Genet A. 2012; 158A: 547-552. http://www.ncbi.nlm.nih.gov/pubmed/22307766
Матье M, Helou M, Morin G и др. Синдром KBG: дополнительный спорадический случай. Genet Couns. 2000; 11: 33-35. http://www.ncbi.nlm.nih.gov/pubmed/10756425
Soekarman D, Volcke P, Fryns JP. Синдром KBG: данные наблюдения за тремя больными братьями. Clin Genet. 1994; 46: 283-86. http://www.ncbi.nlm.nih.gov/pubmed/7834892
Золларион М., Батталья А., Д’Аванцо М.Г. и др.Шесть дополнительных случаев синдрома KBG: клинические отчеты и описание диагностических критериев. Am J Med Genet. 1994; 52: 302-07. http://www.ncbi.nlm.nih.gov/pubmed/7810561
Fryns JP, Haspeslagh M. Умственная отсталость, низкий рост, незначительные аномалии скелета, черепно-лицевой дисморфизм и макродонтия у двух сестер и их матери. Другой вариантный пример синдрома КБГ? Clin Genet. 1984; 26: 69-72. http://www.ncbi.nlm.nih.gov/pubmed/6467660
Hermann J, Pallister PD, Tiddy W., Optiz JM.Синдром KBG — синдром низкого роста, характерного лица, умственной отсталости, макродонтии и аномалий скелета. Врожденные дефекты. 1975; 11: 7-18. http://www.ncbi.nlm.nih.gov/pubmed/1218237
ИНТЕРНЕТ
McKusick VA., ed. Интернет-Менделирующее наследование в человеке (OMIM). Балтимор. MD: Университет Джона Хопкинса; Запись №: 148050; Последнее обновление: 06.07.2018. Доступно по адресу: http://omim.org/entry/148050 По состоянию на 5 августа 2019 г.
Brancati F, Dallapiccola B, Sarkozy A.Синдром КБГ. Энциклопедия Orphanet, декабрь 2006 г. Доступно по адресу: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=2332 Проверено 5 августа 2019 г.
Genetics Home Reference. Синдром КБГ. Январь 2018 г. Доступно по адресу: https://ghr.nlm.nih.gov/condition/kbg-syndrome Проверено 5 августа 2019 г.
Семейные онкологические синдромы
Рак — распространенное заболевание, поэтому неудивительно, что во многих семьях есть хотя бы несколько членов, которые болели раком.
Иногда кажется, что определенные виды рака передаются в некоторых семьях.В некоторых случаях это может быть связано с тем, что члены семьи разделяют определенные виды поведения или воздействия, которые повышают риск рака, например курение. На риск рака также могут влиять другие факторы, такие как ожирение, которые, как правило, присущи некоторым семьям.
Но в некоторых случаях рак вызывается аномальным геном, который передается из поколения в поколение. Хотя это часто называют унаследованным раком , наследуется аномальный ген, который может привести к раку, а не сам рак.Только от 5% до 10% всех случаев рака возникают непосредственно из-за генных дефектов (называемых мутациями , ), унаследованных от родителей. Эта информация о раковых опухолях.
Гены, мутации и рак
Рак — это болезнь, при которой клетки растут бесконтрольно. Это происходит из-за изменений некоторых генов внутри клеток. Гены — это фрагменты ДНК, которые контролируют, как клетки вырабатывают белки, необходимые организму для функционирования, а также то, как клетки поддерживаются в равновесии. Ваши гены влияют на такие вещи, как цвет волос, цвет глаз и рост.Они также могут повлиять на ваши шансы заболеть определенными заболеваниями, такими как рак.
Почти каждая клетка вашего тела имеет все гены, с которыми вы родились. Хотя все клетки имеют одни и те же гены, разные клетки (или типы клеток) могут использовать разные гены. Например, в мышечных клетках используются гены, отличные от генов клеток кожи. Гены, в которых клетка не нуждается, отключаются и не используются. Гены, которые использует клетка, активируются или включаются.
Аномальное изменение гена называется мутацией .Мутации в гене могут повлиять на его работу. Например, мутация может остановить работу гена. Или он может постоянно держать ген включенным (даже когда он не нужен). В любом случае это может вызвать проблемы внутри клетки.
Генные мутации могут передаваться по наследству или приобретаться.
- В яйцеклетке или сперматозоиде, из которых сформировался ребенок, присутствует мутация унаследованного гена . Когда яйцеклетка оплодотворяется спермой, она создает одну клетку, которая затем многократно делится и в конечном итоге становится ребенком.Поскольку все клетки происходят из этой первой клетки, этот вид мутации присутствует в каждой клетке (включая яйцеклетки или сперматозоиды) и поэтому может передаваться следующему поколению.
- Приобретенная (соматическая) мутация происходит не от родителя, а приобретается спустя некоторое время. Он начинается в одной ячейке, а затем передается во все новые ячейки, созданные из этой ячейки. Такого рода мутации нет в яйцеклетках или сперматозоидах, поэтому они не передаются следующему поколению. Приобретенные мутации встречаются гораздо чаще, чем наследственные.Большинство видов рака вызвано приобретенными мутациями.
Многие семейные синдромы рака вызваны унаследованными мутациями генов-супрессоров опухолей . Это гены, которые обычно держат клетки под контролем, замедляя частоту их деления (для создания новых клеток), исправляя ошибки ДНК или приказывая клеткам умереть в нужное время.
У вас есть 2 копии большинства генов — по одной от каждого родителя. Когда кто-то наследует аномальную копию гена, его клетки уже начинают с одной мутации.Часто это не проблема, поскольку другая копия гена все еще работает. Но если другая копия гена перестает работать (например, из-за приобретенной мутации), функция гена может быть полностью потеряна. Когда ген, который перестает работать, является геном-супрессором опухоли, клетки могут бесконтрольно расти, что может привести к раку.
Человек, родившийся с наследственной мутацией в копии гена-супрессора опухоли, должен был бы только приобрести мутацию в другой копии этого гена, чтобы она не сработала.Это более вероятно, чем приобретение мутаций в обеих копиях гена, поэтому этот человек будет иметь более высокий риск рака, чем тот, кто родился без мутации гена.
Для получения дополнительной информации об изменениях генов, которые могут привести к раку, см. Гены и рак.
Как распознать синдром семейного рака?
Важно помнить, что рак — обычное дело. Фактически, примерно у 1 из 3 человек в США в течение жизни развивается рак, поэтому нередко бывает много раковых заболеваний в семье.
Когда в семье возникает много случаев рака, чаще всего это происходит случайно или потому, что члены семьи подверглись воздействию общего фактора риска, такого как курение.
Иногда может иметь место взаимодействие между определенными генами и воздействиями. Например, некоторые люди наследуют изменения генов, которые затрудняют избавление их организма от токсинов, содержащихся в табачном дыме. У этих людей может быть больше шансов заболеть раком, если они курят, чем у тех, у кого нет этих генетических изменений.
Реже раковые заболевания в семье прочно связаны с наследственной мутацией гена, которая является частью синдрома семейного рака .
Некоторые факторы повышают вероятность того, что рак в семье вызван семейным онкологическим синдромом, например:
- Многие случаи рака одного и того же типа (особенно если это необычный или редкий тип рака)
- Раковые заболевания, возникающие в более молодом возрасте, чем обычно (например, рак толстой кишки у 20-летних)
- Более одного типа рака у одного человека (например, у женщины с раком груди и яичников)
- Рак, поражающий оба органа (например, оба глаза, обе почки или обе груди)
- Более одного детского рака у братьев и сестер (например, саркома у брата и сестры)
- Рак, встречающийся у мужчин, которым обычно не подвержен (например, рак груди у мужчин)
- Рак, передаваемый из поколения в поколение (как у дедушки, отца и сына)
Пытаясь определить, может ли рак передаваться в вашей семье, сначала соберите некоторую информацию.Для каждого случая рака смотрите:
- У кого рак? Как вы связаны? На какой стороне семьи они (матери или отца)?
- Какой это тип рака? Это редко?
- Сколько лет было этому родственнику, когда им поставили диагноз?
- Заболел ли этот человек более чем одним типом рака?
- Были ли у них какие-либо известные факторы риска их типа рака (например, курение при раке легких)?
Рак у близкого родственника, например, у родителя или брата или сестры (брата или сестры), является более серьезным поводом для беспокойства, чем рак у более дальнего родственника.Даже если рак возник в результате генной мутации, вероятность его передачи вам снижается с более дальними родственниками.
Также важно смотреть на каждую сторону семьи отдельно. Наличие двух родственников, больных раком, вызывает большее беспокойство, если они находятся на одной стороне семьи. Например, если оба родственника — братья вашей матери, это больше беспокоит, чем если бы один был братом вашего отца, а другой — братом вашей матери.
Тип рака тоже имеет значение.Это больше беспокоит, если у многих родственников один и тот же тип рака, чем если у них несколько разных видов рака. Тем не менее, при некоторых синдромах семейного рака кажется, что несколько типов рака сочетаются друг с другом. Например, рак груди и рак яичников сочетаются в семьях с наследственным синдромом рака груди и яичников (HBOC). Рак толстой кишки и эндометриальный рак, как правило, сочетаются друг с другом при синдроме Линча (также известном как наследственный неполипозный колоректальный рак или HNPCC).
Точно так же, более одного случая одного и того же редкого рака вызывает большее беспокойство, чем случаи более распространенного рака.Для некоторых редких видов рака риск развития синдрома семейного рака относительно высок даже в одном случае.
Возраст человека, у которого был диагностирован рак, также важен. Например, рак толстой кишки обычно редко встречается у людей моложе 30 лет. Наличие близких родственников младше 30 лет с раком толстой кишки может быть признаком наследственного ракового синдрома. С другой стороны, рак простаты очень распространен у пожилых мужчин, поэтому, если и у вашего отца, и у его брата был обнаружен рак простаты, когда им было за 80, это с меньшей вероятностью было связано с наследственным изменением гена.
Некоторые виды доброкачественных (не раковых) опухолей и заболеваний иногда также являются частью синдрома семейного рака. Например, люди с множественной эндокринной неоплазией, синдромом II типа (MEN II) имеют высокий риск развития определенного вида рака щитовидной железы. У них также могут развиваться доброкачественные опухоли паращитовидных желез, а также опухоли надпочечников, называемые феохромоцитомами, которые обычно доброкачественные.
Если у многих родственников один и тот же тип рака, важно отметить, может ли рак быть связан с таким фактором риска, как курение.Например, рак легких обычно вызывается курением, поэтому несколько случаев рака легких в семье заядлых курильщиков с большей вероятностью связаны с курением, чем с наследственным изменением гена.
Примеры семейных онкологических синдромов
Есть много синдромов семейного рака. Некоторые из них кратко обсуждаются здесь в качестве примеров, но это не полный список. Для получения дополнительной информации о конкретном типе рака и его генетических компонентах, диагностике и лечении см. Наши материалы по этому конкретному типу рака.
Синдром наследственного рака груди и яичников (HBOC)
В некоторых семьях у многих женщин развивается рак груди и / или рак яичников. Часто эти виды рака обнаруживаются у женщин моложе обычного возраста, эти виды рака обнаруживаются, и у некоторых женщин может быть более одного рака (например, рак обеих молочных желез или рак груди и яичников). Это известно как синдром наследственного рака груди и яичников (HBOC).
Чаще всего HBOC вызывается наследственной мутацией гена BRCA1 или BRCA2 .(В некоторых семьях HBOC обнаружен на основании анамнеза рака, но у них нет мутаций ни в одном из этих генов. Ученые полагают, что могут быть и другие гены, которые могут вызывать HBOC.)
Риск рака груди и яичников очень высок у женщин с мутациями BRCA1 или BRCA2 , но, как правило, он выше при мутациях BRCA1 . Наряду с раком груди и яичников этот синдром также может привести к раку маточной трубы, первичному раку брюшины, раку груди у мужчин, раку поджелудочной железы и раку простаты, а также некоторым другим.Рак груди у мужчин, рак поджелудочной железы и рак предстательной железы можно увидеть при наличии мутаций в любом из генов, но чаще встречаются у людей с мутациями BRCA2 . В США мутации в генах BRCA чаще встречаются у людей еврейского происхождения ашкенази, чем среди населения в целом.
Женщины с сильным семейным анамнезом рака груди и / или рака яичников могут пройти генетическое консультирование, чтобы оценить свой риск мутации в одном из генов BRCA .Специалист-генетик может оценить риск, основываясь на истории болезни пациента и его семье. Если у них высокий риск, они могут решить пройти тестирование (см. Общие сведения о генетическом тестировании на рак). Если присутствует мутация, у женщины высок риск развития рака груди и рака яичников (а также некоторых других видов рака). Затем она может рассмотреть меры по раннему обнаружению рака и даже снизить риск заболевания раком.
Поскольку рак груди у мужчин встречается редко, мужчинам с этим раком часто предлагают генетическое консультирование и тестирование на мутации BRCA .Хотя наличие мутации с меньшей вероятностью повлияет на будущее здоровье мужчины, чем здоровье женщины, она может повлиять на его риск развития некоторых видов рака, например рака простаты и поджелудочной железы. Близким родственникам мужчины также может быть полезно знать, что у него мутация и что они могут оказаться в группе риска.
Если у кого-то есть мутация BRCA , это означает, что его близкие родственники (родители, братья, сестры и дети) также имеют 50% -ную вероятность мутации. Они могут захотеть пройти тестирование на мутацию или даже без тестирования могут пожелать начать скрининг на определенные виды рака на ранней стадии или принять другие меры предосторожности, чтобы снизить риск рака.
HBOC — не единственный семейный онкологический синдром, который может вызвать рак груди или яичников. Для получения информации о других генах и синдромах, повышающих риск этих видов рака, см. Рак молочной железы и рак яичников.
Синдром Линча (наследственный неполипозный колоректальный рак)
Наиболее распространенным наследственным синдромом, повышающим риск рака толстой кишки, является синдром Линча, также называемый наследственным неполипозным колоректальным раком (HNPCC).Люди с этим синдромом подвержены высокому риску развития колоректального рака. Большинство этих видов рака развиваются до 50 лет.
Синдром Линча также приводит к высокому риску рака эндометрия (рака слизистой оболочки матки), а также рака яичников, желудка, тонкой кишки, поджелудочной железы, почек, головного мозга, мочеточников (трубок, по которым моча выводится из почек). в мочевой пузырь) и желчный проток.
Синдром Линча может быть вызван мутацией в любом из нескольких генов восстановления несоответствия (MMR), включая MLh2 , MSh3 , MSH6 , PMS1 и PMS2. Эти гены обычно участвуют в восстановлении поврежденной ДНК. Когда один из этих генов не работает, в ДНК клетки могут возникать ошибки, которые могут привести к мутациям других генов и, в конечном итоге, к раку.
Врачи и генетики могут проверить вероятность синдрома Линча на основании вашего личного и семейного онкологического анамнеза, используя определенные критерии. Они, известные как Амстердамские критерии и пересмотренные рекомендации Bethesda, подробно обсуждаются в книге «Генетическое тестирование, скрининг и профилактика для людей с сильным семейным анамнезом колоректального рака».Мутации в генах, вызывающих синдром Линча, затем можно проверить с помощью генетического тестирования.
Для людей с колоректальным раком или раком эндометрия опухолевую ткань можно проверить на изменения гена MMR или другие изменения, которые могут быть вызваны неисправностью одного из этих генов, что известно как микросателлитная нестабильность (или MSI). Нормальные результаты (отсутствие изменений гена MMR или MSI) подразумевают, что у человека, вероятно, нет синдрома Линча. Но если один из них присутствует, у человека может быть синдром Линча, и его направляют на генетическую консультацию и возможное тестирование.Дополнительные сведения о генетическом тестировании см. В разделе «Генетика и рак».
Кто-то, у кого известна мутация гена, связанная с синдромом Линча, может начать скрининг на колоректальный рак, когда они моложе (например, в возрасте около 20 лет), или предпринять другие шаги, чтобы попытаться предотвратить возникновение рака (более подробно обсуждается в Колоректальный рак). Женщины с синдромом Линча могут начать обследование на рак эндометрия или предпринять другие шаги, чтобы попытаться предотвратить этот рак. Более подробно они обсуждаются в Раке эндометрия.
Если кто-то страдает синдромом Линча, это означает, что его близкие родственники (родители, братья, сестры и дети) также имеют 50% шанс мутации. Они могут захотеть пройти тестирование или даже без тестирования они могут пожелать начать скрининг на определенные виды рака раньше или принять другие меры предосторожности, чтобы снизить риск рака.
Синдром Ли-Фраумени
Синдром Ли-Фраумени — это редкий наследственный синдром, который может привести к развитию ряда видов рака, включая саркомы (например, остеосаркомы и саркомы мягких тканей), лейкоз, рак головного мозга (центральной нервной системы), рак коры надпочечников. и рак груди.Эти виды рака часто развиваются в относительно молодом возрасте.
Люди с Li-Fraumeni также могут заболеть более чем одним раком за свою жизнь. Они также имеют более высокий риск заболеть раком от лучевой терапии, поэтому врачи, лечащие этих пациентов, могут попытаться избежать облучения, когда это возможно.
Этот синдром чаще всего вызывается наследственными мутациями в гене TP53 , который является геном-супрессором опухоли. Нормальный ген TP53 вырабатывает белок, который помогает остановить рост аномальных клеток.
Синдром
Ли-Фраумени также может быть вызван мутациями в гене-супрессоре опухоли, называемом CHEK2 , который также обычно помогает остановить рост клеток с повреждением ДНК.
Если кто-то страдает синдромом Ли-Фраумени, его близкие родственники (особенно дети) также имеют повышенный шанс мутации. Они могут захотеть пройти тестирование или даже без тестирования они могут пожелать начать скрининг на определенные виды рака как можно раньше или принять другие меры предосторожности, чтобы снизить риск рака.
Генетическое консультирование и тестирование
Людям с сильным семейным анамнезом онкологических заболеваний может потребоваться изучить свой генетический код. Это может помочь этому человеку или другим членам семьи спланировать свое медицинское обслуживание на будущее. Поскольку наследственные мутации затрагивают все клетки человеческого тела, их часто можно обнаружить с помощью генетического тестирования, проведенного на образцах крови или слюны (слюны). Тем не менее генетическое тестирование полезно не для всех, поэтому важно сначала поговорить с генетическим консультантом, чтобы выяснить, подходит ли вам тестирование.Для получения дополнительной информации см. Общие сведения о генетическом тестировании на рак.
Другие распространенные семейные онкологические синдромы
Вы можете узнать больше о семейных онкологических синдромах, перечисленных выше, а также о других наследственных синдромах и генных мутациях, которые могут повлиять на риск рака у человека, прочитав:
Генетическое консультирование и тестирование на предмет риска рака молочной железы: наследственный синдром рака молочной железы и яичников (HBOC), BRCA1 и BRCA2 мутации , и другие специфические генные мутации
Генетическое тестирование, скрининг и профилактика для людей с сильным семейным анамнезом колоректального рака: синдром Линча, семейный аденоматозный полипоз (FAP) и другие специфические генные мутации
Генетическое консультирование и тестирование для людей с высоким риском меланомы: специфические генные мутации
Факторы риска рака яичников: наследственный синдром рака груди и яичников (HBOC), синдром Линча, синдром Пейтца-Егерса, полипоз, связанный с MUTYH, BRCA1 и BRCA2 мутации , и другие специфические генные мутации
Синдром
Линча | Рак.Net
Что такое синдром Линча?
Синдром Линча, также известный как наследственный неполипозный колоректальный рак (HNPCC), представляет собой тип наследственного ракового синдрома, связанный с генетической предрасположенностью к различным типам рака. Это означает, что люди с синдромом Линча имеют более высокий риск определенных типов рака.
Рак начинается, когда нормальные клетки начинают изменяться и бесконтрольно расти, образуя массу, называемую опухолью. Опухоль может быть доброкачественной (незлокачественной) или злокачественной (злокачественной), что означает, что она может распространяться на другие части тела.Доброкачественная опухоль означает, что опухоль может расти, но не распространяется.
Люди с синдромом Линча имеют значительно повышенный риск развития колоректального рака. Существует также повышенный риск развития других видов рака, таких как рак эндометрия (матки), желудка, груди, яичников, тонкой кишки (кишечника), поджелудочной железы, предстательной железы, мочевыводящих путей, печени, почек и желчных протоков.
Синдром Линча является одним из наиболее распространенных наследственных онкологических синдромов, и по оценкам, до 1 из каждых 300 человек может быть носителем изменения в гене, связанном с синдромом Линча.Признаки наличия синдрома Линча в семье включают диагноз колоректального рака и / или рака эндометрия у нескольких родственников с одной стороны семьи. Кроме того, рак, связанный с синдромом Линча, чаще диагностируется в молодом возрасте. Люди с синдромом Линча также подвергаются повышенному риску развития нескольких типов рака в течение своей жизни.
ASCO рекомендует проводить тестирование опухолей на синдром Линча у всех людей с диагнозом колоректальный рак, а недавние руководящие принципы рекомендуют также тестирование опухолей на все виды рака эндометрия.Подробнее об этих рекомендациях и рекомендациях по скринингу, перечисленным ниже, читайте на сайте www.asco.org/endorsements/HereditaryCRC. (Обратите внимание, что эта ссылка ведет на отдельный веб-сайт ASCO.)
Каковы признаки синдрома Линча?
Набор критериев, помогающих врачам решить, кого следует тестировать на синдром Линча, называется пересмотренными рекомендациями Bethesda, которые перечислены ниже:
Развитие колоректального рака или рака эндометрия моложе 50 лет
Развивающийся колоректальный рак, рак эндометрия или другой тип рака * с дефицитом восстановления несоответствия (MMR-D) или микросателлитной нестабильностью высокого уровня (MSI-H), обнаруженный при тестировании образца опухоли
Развитие колоректального рака и других типов рака *, связанных с синдромом Линча по отдельности или одновременно
Колоректальный рак у 1 или более родственников первой степени родства, которые также болеют или переболели другим раком, связанным с синдромом Линча * , причем 1 из этих видов рака развился в возрасте до 50 лет.Фраза «родственники первой степени» включает родителей, братьев, сестер и детей.
Колоректальный рак у 2 или более родственников первой или второй степени родства с другим раком, связанным с синдромом Линча *. «К родственникам второй степени родства» относятся тети, дяди, бабушки и дедушки, внуки, племянники и племянницы.
* Категория включает колоректальный рак, рак эндометрия, рак яичников, рак желудка, рак тонкой кишки, рак мочеточника или почечной лоханки, рак мочевого пузыря, рак желчных протоков, рак поджелудочной железы или сальные аденомы кожи
Определение синдрома Линча все еще развивается.Многие люди все еще могут иметь синдром Линча, даже если их семейный анамнез не полностью соответствует пересмотренным рекомендациям Bethesda. Поэтому людям, чей семейный анамнез предполагает возможность синдрома Линча, рекомендуется встретиться со специалистом в области здравоохранения, имеющим подготовку в области генетических заболеваний и состояний.
Другие формы синдрома Линча
Мьюир-Торре — это еще одно название синдрома Линча, при котором у людей развиваются необычные кожные поражения или опухоли, включая сальные аденомы, сальные эпителиомы, сальные карциномы и кератокантомы.Поскольку эти редкие кожные поражения чаще встречаются у людей с синдромом Линча, рекомендуется, чтобы человек, у которого диагностировали эти поражения, прошел генетическую оценку у квалифицированного медицинского работника.
Синдром Тюрко — это состояние, при котором у людей развиваются опухоли головного мозга и колоректальный рак. В некоторых случаях в прошлом каждый человек с синдромом Линча и / или другим генетическим синдромом, называемым семейным аденоматозным полипозом (FAP), упоминался как имеющий синдром Тюрко.Однако синдром Тюрко больше не считается отдельным генетическим синдромом.
Что вызывает синдром Линча?
Синдром Линча — это генетическое заболевание. Это означает, что риск рака может передаваться в семье из поколения в поколение. Эти типы мутаций называются наследственными или зародышевыми мутациями. Изменения в нескольких генах, участвующих в репарации несоответствия ДНК, которые были связаны с синдромом Линча. Они включают гены MLh2 , MSh3 , MSH6 , PMS2, и EPCAM .Мутация (изменение) в любом из этих генов увеличивает риск развития колоректального рака и других родственных ему видов рака на протяжении всей жизни. У женщин также есть повышенный риск развития рака эндометрия и яичников.
Не все семьи с синдромом Линча будут иметь идентифицируемые изменения в MLh2 , MSh3 , MSH6 , PMS2, или EPCAM . Исследования продолжаются, чтобы идентифицировать другие гены, связанные с синдромом Линча.У некоторых людей в этих генах разовьются изменения, которые не передаются по наследству, но происходят из-за процесса старения организма и других причин, которые не совсем понятны. Эти типы мутаций называются приобретенными. Если обнаруживается, что опухоль имеет изменения в этих генах, кровь человека также будет проверена на наличие этого аномального гена. Если и кровь, и опухоль имеют измененный ген, заболевание передается по наследству, а не приобретается, что означает, что могут пострадать другие члены семьи.
Как наследуется синдром Линча?
Обычно каждая клетка имеет по 2 копии каждого гена: 1 унаследован от матери и 1 унаследован от отца.Синдром Линча следует по аутосомно-доминантному типу наследования, при котором мутация должна произойти только в 1 копии гена, чтобы человек имел повышенный риск заболеть этим заболеванием. Это означает, что родитель с мутацией гена может передать копию своего нормального гена или копию гена с мутацией. Следовательно, ребенок, у которого есть родитель с мутацией, имеет 50% шанс унаследовать эту мутацию. Брат, сестра или родитель человека, у которого есть мутация, также имеют 50% шанс иметь ту же мутацию.Однако, если тест родителей отрицательный на мутацию (это означает, что результаты тестирования каждого человека не выявили мутации), риск для братьев и сестер значительно снижается, но их риск все равно может быть выше среднего риска.
Существуют варианты для людей, заинтересованных в рождении ребенка, когда будущий родитель несет генную мутацию, повышающую риск этого наследственного онкологического синдрома. Преимплантационная генетическая диагностика (ПГД) — это медицинская процедура, проводимая в сочетании с экстракорпоральным оплодотворением (ЭКО).Это позволяет людям, несущим конкретную известную генетическую мутацию, снизить вероятность того, что их дети унаследуют эту мутацию. Яйцеклетки женщины удаляются и оплодотворяются в лаборатории. Когда эмбрионы достигают определенного размера, одна клетка удаляется и проверяется на предмет рассматриваемого наследственного состояния. Затем люди могут перенести эмбрионы, не имеющие генетической мутации. ПГД используется уже более двух десятилетий и используется для лечения нескольких синдромов наследственной предрасположенности к раку. Тем не менее, это сложная процедура, которую необходимо учитывать перед началом работы с финансовыми, физическими и эмоциональными факторами.Для получения дополнительной информации обратитесь к специалисту по вспомогательной репродукции в клинике репродуктивной медицины.
Насколько распространен синдром Линча?
В большинстве случаев колоректальный рак носит спорадический характер, то есть возникает случайно без какой-либо известной причины. Считается, что примерно от 3% до 5% всех случаев колоректального рака и от 2% до 3% всех случаев рака эндометрия связаны с синдромом Линча.
Как диагностируется синдром Линча?
Синдром Линча можно подтвердить с помощью анализа крови. Тест может определить, несет ли кто-либо мутацию, которая может передаваться (называемая наследственной) в 1 из генов, связанных с синдромом Линча.В настоящее время доступно тестирование для генов MLh2 , MSh3 , MSH6, PMS2 и EPCAM . Однако не все семьи с синдромом Линча будут иметь идентифицируемую мутацию в одном из этих генов.
Скрининговые тесты также могут проводиться на опухолевой (раковой) ткани, чтобы определить, вероятен ли синдром Линча. Предлагаются 2 скрининговых теста: тест на микросателлитную нестабильность (MSI) и иммуногистохимическое тестирование (IHC). Результаты этих тестов могут указать, следует ли рассматривать более конкретное генетическое тестирование.
Каковы предполагаемые риски рака, связанные с синдромом Линча?
Общие пожизненные риски рака у людей с синдромом Линча
Риск рака у женщин с синдромом Линча
Последние данные показывают, что люди с синдромом Линча и мутациями зародышевой линии PMS2 могут иметь значительно более низкий риск рака, чем приведенные выше оценки.
Какие варианты скрининга на рак при синдроме Линча?
ASCO рекомендует следующий скрининг для людей с синдромом Линча.Важно обсудить эти варианты со своим врачом, так как каждый человек индивидуален:
Общие рекомендации по скринингу и снижению риска
Колоноскопия каждые 1-2 года, начиная с 20-25 лет или на 5 лет моложе самого раннего возраста на момент постановки диагноза в семье, в зависимости от того, что наступит раньше
Верхняя эндоскопия каждые 3-5 лет в дополнение к тестированию на инфекцию Helicobacter pylori при исходном обследовании с лечением в случае положительного результата
Ежегодное обследование кожи всего тела
Рассмотрение ежедневного приема аспирина, связанного со снижением риска колоректального рака и, возможно, других видов рака у людей с синдромом Линча
Скрининг других видов рака, связанных с синдромом Линча, может быть рекомендован в зависимости от семейного анамнеза человека, хотя эффективность такого скрининга остается недоказанной.
Обследование для женщин
- Ежегодное обследование органов малого таза, УЗИ органов малого таза, биопсия эндометрия, в возрасте от 30 до 35 лет. Женщины, у которых закончились роженицы, могут захотеть провести профилактическую операцию по удалению матки и яичников, тем более что скрининг на рак эндометрия и яичников никогда не доказал свою эффективность. быть эффективным у женщин с синдромом Линча.
Варианты скрининга могут меняться со временем по мере развития новых технологий и получения дополнительных сведений о синдроме Линча и других его формах.Важно обсудить со своим врачом соответствующие скрининговые тесты.
Узнайте больше о том, чего ожидать от стандартных тестов и процедур.
Вопросы, которые следует задать бригаде здравоохранения
Если вас беспокоит риск развития колоректального рака или других видов рака, поговорите со своим лечащим врачом. Может быть полезно пригласить кого-нибудь на встречи, чтобы делать заметки. Вы можете задать своей медицинской бригаде следующие вопросы:
Каков мой риск развития колоректального рака или других видов рака?
Что я могу сделать, чтобы снизить риск рака?
Какие у меня есть варианты скрининга на рак?
Если вас беспокоит история вашей семьи и вы считаете, что в вашей семье может быть синдром Линча, подумайте о том, чтобы задать следующие вопросы:
Повышает ли мой семейный анамнез риск развития колоректального рака или других видов рака?
Увеличивает ли мой семейный анамнез риск рака кожи или других кожных проблем?
Проводились ли тесты MSI или IHC на моей опухолевой ткани?
Следует ли мне встретиться с генетическим консультантом?
Стоит ли рассматривать генетическое тестирование?
Связанные ресурсы
Генетика рака
Генетическое тестирование
Чего ожидать при встрече с консультантом по генетике
Сбор семейного анамнеза рака
Обмен результатами генетического тестирования с семьей
Вопросы и ответы по семейному генетическому тестированию
Дополнительная информация
Международный синдром Линча
Альянс рака толстой кишки
C3: Коалиция по борьбе с колоректальным раком
Наследственный рак толстой кишки поражает кишки
Чтобы найти консультанта по генетическим вопросам в вашем районе, обратитесь к врачу или посетите следующий веб-сайт:
Национальное общество консультантов по генетике
журналов открытого доступа по генетическим синдромам и генной терапии
О журнале
NLM ID: 101574143
Index Copernicus Value 2016: 84.15
Genetic Syndromes & Gene Therapy — официальный рецензируемый журнал для быстрой публикации инновационных исследований, охватывающих все аспекты генного картирования и генной терапии. Генетические синдромы, генное картирование и генная терапия с наивысшим импакт-фактором предлагают вариант открытого доступа для удовлетворения потребностей авторов и максимальной видимости статьи, а также создают платформу для авторов, чтобы они могли внести свой вклад в журнал, а редакция обещает процесс рецензирования для присланные рукописи на качество публикации.
Genetic Syndromes & Gene Therapy Journal — один из лучших журналов с открытым доступом, целью которого является публикация наиболее полного и надежного источника информации об открытиях и текущих разработках в виде оригинальных статей, обзорных статей, историй болезни, коротких сообщений и т. Д. в полевых условиях и предоставить онлайн-доступ исследователям по всему миру без каких-либо ограничений или подписок.
Журнал генетических синдромов и генной терапии охватывает непрерывный охват всех биологических и медицинских аспектов потенциальной генной терапии врожденных дефектов наряду с генетическими нарушениями, включая лечение рака, артрита, инфекционных заболеваний, наследственных заболеваний, таких как муковисцидоз и болезнь Хантингтона. а также генетические аномалии или недостатки, которые лечатся путем включения определенных сконструированных генов в инфицированные клетки тела пациента, чтобы люди в электронных формах сразу же стали доступны для чтения, загрузки и обмена, чтобы улучшить девиз открытого доступа.Журнал генетических синдромов и генной терапии предоставляет онлайн-зрителям надежную информацию о модифицированных методах и последних достижениях в области генной терапии различных генетических заболеваний.
Этот журнал Genetics использует систему Editor Manager для онлайн-подачи, рецензирования и отслеживания рукописей. Члены редакционной коллегии журнала Genetic Syndromes & Gene Therapy или сторонние эксперты рецензируют рукописи; Для принятия любой цитируемой рукописи требуется одобрение как минимум двух независимых рецензентов с последующим одобрением редактора.
8.6: Генетические заболевания — Biology LibreTexts
Полли Кто?
У каждой руки на рисунке \ (\ PageIndex {1} \) есть дополнительный мизинец. Это состояние называется полидактилия, что буквально означает «многозначность». У людей с полидактилией могут быть лишние пальцы рук и / или ног, и это заболевание может поражать только одну руку или ногу или обе руки и ноги. Полидактилия часто имеет генетическое происхождение и может быть частью генетического заболевания, связанного с другими состояниями.
Рисунок \ (\ PageIndex {1} \): (CC BY 2.0; Авторы: Baujat G, Le Merrer M. через Wikimedia Commons).
Что такое генетические заболевания?
Генетические нарушения — это заболевания, синдромы или другие состояния, которые вызваны мутациями в одном или нескольких генах или хромосомными изменениями. Генетические нарушения обычно присутствуют при рождении, но их не следует путать с врожденными нарушениями, которые представляют собой любые нарушения, независимо от причины, которые присутствуют при рождении. Некоторые врожденные нарушения не вызваны генетическими мутациями или хромосомными изменениями.Вместо этого они вызваны проблемами, возникающими во время эмбрионального или внутриутробного развития или в процессе родов. Примером негенетического врожденного порока является алкогольный синдром плода. Это совокупность врожденных дефектов, включая лицевые аномалии и умственную отсталость, вызванных употреблением алкоголя матерью во время беременности.
Генетические заболевания, вызванные мутациями
Таблица \ (\ PageIndex {1} \) перечисляет несколько генетических нарушений, вызванных мутациями только в одном гене.Некоторые расстройства вызваны мутациями в аутосомных генах, другие — мутациями в генах, сцепленных с Х-хромосомой. Какие расстройства, по вашему мнению, будут чаще встречаться у мужчин, чем у женщин?
Очень немногие генетические нарушения контролируются доминантными мутантными аллелями. Доминантный аллель выражен у каждого человека, который наследует хотя бы одну его копию. Если это вызывает серьезное заболевание, пораженные люди могут умереть молодыми и не иметь потомства. Следовательно, мутантный доминантный аллель, вероятно, исчезнет из популяции.
Рецессивный мутантный аллель, такой как аллель, вызывающий серповидно-клеточную анемию или кистозный фиброз, не экспрессируется у людей, которые наследуют только одну его копию. Этих людей называют носителями. Сами они не страдают этим заболеванием, но несут мутантный аллель, и их потомки могут унаследовать его. Таким образом, аллель, скорее всего, перейдет к следующему поколению, а не исчезнет.
| Генетическое заболевание | Прямой эффект мутации | Признаки и симптомы заболевания | Режим наследования |
|---|---|---|---|
| Синдром Марфана | Дефектный белок в соединительной ткани | Дефекты сердца и костей и необычно длинные тонкие конечности и пальцы | аутосомно-доминантный |
| Серповидно-клеточная анемия | Атипичный белок гемоглобина в эритроцитах | серповидные эритроциты, которые закупоривают крошечные кровеносные сосуды, вызывая боль и повреждая органы и суставы | аутосомно-рецессивный |
| Витамин D-устойчивый рахит | Отсутствие вещества, необходимого костям для поглощения минералов | мягкие кости, которые легко деформируются, что приводит к искривлению ног и другим деформациям скелета | Х-сцепленный доминантный |
| Гемофилия A | снижение активности белка, необходимого для свертывания крови | внутреннее и внешнее кровотечение, которое возникает легко и трудно остановить | Х-сцепленный рецессивный |
Генетические заболевания, вызванные хромосомными изменениями
Как мы узнали из главы «Воспроизведение клеток», во время мейоза могут возникать ошибки, которые приводят к нерасхождению .Это неспособность реплицированных хромосом должным образом разделиться во время мейоза. В некоторых из полученных гамет не будет всей хромосомы или ее части, в то время как у других будет дополнительная копия всей хромосомы или ее части. Если такие гаметы оплодотворяются и образуют зиготы, они обычно не выживают. Если они выживут, у этих людей, вероятно, будут серьезные генетические нарушения.
Таблица \ (\ PageIndex {2} \) перечисляет несколько генетических нарушений, которые вызваны атипичным количеством хромосом.Большинство хромосомных нарушений связано с Х-хромосомой. Х- и Y-хромосомы — единственная пара хромосом, в которой две хромосомы сильно различаются по размеру. Это объясняет, почему нерасхождение половых хромосом имеет тенденцию происходить чаще, чем нерасхождение аутосом.
| Генетическое заболевание | Генотип | Фенотипические эффекты |
|---|---|---|
| Синдром Дауна | дополнительная копия (полная или частичная) хромосомы 21 (см. Рисунок ниже) | задержка в развитии, характерный внешний вид лица и другие физические условия и нарушения развития (см. Рисунок ниже) |
| Синдром Тернера | одна Х-хромосома, но нет другой половой хромосомы (ХО) | Хромосомная женщина с низким ростом и бесплодием (неспособность к размножению) |
| Синдром тройной X | три X-хромосомы (XXX) | Хромосомная женщина с легкой задержкой развития и нарушением менструального цикла |
| Синдром Клайнфельтера | одна хромосома Y и две или более хромосомы X (XXY, XXXY) | Хромосомный мужчина с проблемами полового развития и пониженным уровнем мужского гормона тестостерона |
Диагностика и лечение генетических заболеваний
Генетическое заболевание, вызванное мутацией, может передаваться по наследству.Следовательно, люди с генетическим заболеванием в семье могут быть обеспокоены тем, что у них есть дети. Консультант-генетик может помочь им понять, насколько опасны последствия для их детей. Если они решат завести детей, им могут посоветовать пройти дородовое обследование («до рождения»), чтобы узнать, есть ли у плода какие-либо генетические нарушения. Один из методов пренатального тестирования — амниоцентез. В этой процедуре из жидкости, окружающей плод внутриутробно , извлекается несколько эмбриональных клеток и исследуются хромосомы плода.Таким образом можно обнаружить синдром Дауна и другие хромосомные изменения.
Рисунок \ (\ PageIndex {2} \): Трисомия 21 (синдром Дауна) Кариотип. Кариотип — это изображение хромосом клетки. Обратите внимание на дополнительную хромосому 21. Ребенок с синдромом Дауна с характерной внешностью. (CC BY-NC 3.0 через CK-12 Foundation).
Симптомы генетических нарушений иногда можно вылечить или предотвратить. Например, при генетическом заболевании, называемом фенилкетонурия (PKU) , аминокислота фенилаланин накапливается в организме до вредных уровней.ФКУ вызывается мутацией в гене, который обычно кодирует фермент, необходимый для расщепления фенилаланина. Когда человек с фенилкетонурией употребляет продукты с высоким содержанием фенилаланина (в том числе многие продукты с высоким содержанием белка), накопление фенилкетонурии может привести к серьезным проблемам со здоровьем. У младенцев и маленьких детей накопление фенилаланина может вызвать умственную отсталость и задержку развития, а также другие серьезные проблемы. Младенцы в США и многих других странах проходят скрининг на ФКУ вскоре после рождения.Если диагностирована фенилкетонурия, младенца можно кормить диетой с низким содержанием фенилаланина. Это предотвращает накопление фенилаланина и связанные с ним проблемы со здоровьем, включая умственную отсталость. Если соблюдать диету с низким содержанием фенилаланина на протяжении всей жизни, большинство симптомов заболевания можно предотвратить.
Лечение генетических заболеваний
Лекарства от генетических заболеваний все еще находятся на ранней стадии разработки. Одно из возможных лекарств — генная терапия. Генная терапия — это экспериментальный метод, который использует гены для лечения или предотвращения заболеваний.В генной терапии нормальные гены вводятся в клетки, чтобы компенсировать мутировавшие гены. Если мутировавший ген приводит к тому, что необходимый белок становится нефункциональным или отсутствует, генная терапия может ввести нормальную копию гена для производства необходимого функционального белка.
Ген, который вставляется непосредственно в клетку, обычно не функционирует, поэтому носитель, называемый вектором , генетически сконструирован для доставки гена (рисунок \ (\ PageIndex {3} \)). Определенные вирусы, такие как аденовирусы, часто используются в качестве переносчиков.Они могут доставить новый ген, заражая клетки. Вирусы модифицированы, поэтому при использовании у людей они не вызывают болезней. Если лечение будет успешным, новый ген, доставляемый вектором, позволит синтезировать функционирующий белок. Исследователям еще предстоит преодолеть множество технических проблем, прежде чем генная терапия станет практическим подходом к лечению генетических заболеваний.
Рисунок \ (\ PageIndex {3} \): Генная терапия — это экспериментальный метод лечения генетического заболевания путем изменения генетической структуры пациента.Обычно генная терапия включает введение нормальной копии мутантного гена в клетки пациента. (Общественное достояние; Дэррил Лея из NHGRI через Wikimedia Commons).
Характеристика: Биология человека в новостях
Синдром Дауна — наиболее частая генетическая причина умственной отсталости. Это происходит примерно у 1 из 700 живорождений, и в настоящее время им страдают почти полмиллиона американцев. До недавнего времени ученые считали, что изменения, ведущие к умственной отсталости у людей с синдромом Дауна, происходят еще до рождения.
Исследователи недавно обнаружили генетическое заболевание, которое влияет на развитие мозга у людей с синдромом Дауна в детстве и во взрослом возрасте. Недавно обнаруженное генетическое заболевание изменяет связь между нервными клетками в головном мозге, что приводит к более медленной передаче нервных импульсов. Это открытие может в конечном итоге позволить разработать стратегии, способствующие функционированию мозга у пациентов с синдромом Дауна, а также может быть применимо к другим нарушениям развития, таким как аутизм.Результаты этого многообещающего исследования были опубликованы в выпуске научного журнала Neuron от 16 марта 2016 года.
Сводка
- Генетические нарушения — это заболевания, синдромы или другие атипичные состояния, которые вызваны мутациями в одном или нескольких генах или хромосомными изменениями.
- Примеры генетических нарушений, вызванных мутациями одного гена, включают синдром Марфана (аутосомно-доминантный), серповидно-клеточную анемию (аутосомно-рецессивный), устойчивый к витамину D рахит (X-сцепленный доминантный) и гемофилию A (X-сцепленный рецессивный).Очень немногие генетические нарушения вызваны доминантными мутациями, потому что эти аллели с меньшей вероятностью передаются последующим поколениям.
- Нерасхождение — это неспособность реплицированных хромосом должным образом разделиться во время мейоза. Это может привести к генетическим нарушениям, вызванным атипичным числом хромосом. Примером может служить синдром Дауна, при котором человек наследует дополнительную копию хромосомы 21. Большинство хромосомных нарушений связано с Х-хромосомой. Примером может служить синдром Клайнфельтера (XXY, XXXY).
- Пренатальное генетическое тестирование, например, с помощью амниоцентеза, может обнаружить хромосомные изменения в утробе матери. Симптомы некоторых генетических нарушений можно вылечить или предотвратить. Например, симптомы фенилкетонурии (ФКУ) можно предотвратить, соблюдая диету с низким содержанием фенилаланина на протяжении всей жизни.
- Лекарства от генетических заболеваний все еще находятся на ранних стадиях разработки. Одним из возможных способов лечения является генная терапия, при которой нормальные гены вводятся в клетки вектором, таким как вирус, для компенсации мутировавших генов.
Обзор
- Определите генетическое заболевание.
- Определите три генетических нарушения, вызванных мутациями в одном гене.
- Почему генетические нарушения с одним геном чаще контролируются рецессивными, чем доминантными мутантными аллелями?
- Что такое нерасхождение? Почему это может вызвать генетические нарушения?
- Объясните, почему генетические нарушения, вызванные определенным числом хромосом, чаще всего связаны с Х-хромосомой.
- Как определяется синдром Дауна внутриутробно ?
- Используйте пример ФКУ, чтобы проиллюстрировать, как иногда можно предотвратить симптомы генетического заболевания.
- Объясните, как работает генная терапия.
- Сравните и сопоставьте генетические нарушения и врожденные нарушения.
- Объясните, почему родители, у которых нет синдрома Дауна, могут иметь ребенка с синдромом Дауна.
- Гемофилия А и синдром Тернера связаны с проблемами с Х-хромосомой. В чем основное различие между этими двумя типами заболеваний с точки зрения воздействия на Х-хромосому?
- Можете ли вы быть носителем синдрома Марфана и не иметь этого расстройства? Поясните свой ответ.
- Верно или неверно. Люди не могут иметь более трех копий одной хромосомы.
- Верно или неверно. Ген серповидноклеточной анемии находится на половой хромосоме.
Узнать больше
Ученые обещали, что генная терапия станет следующим большим шагом в медицине, но что это такое? Узнайте больше здесь:
Представьте себе, если бы мы могли исправить собственные клетки пациента и вылечить его генетическое заболевание раз и навсегда, вместо того, чтобы лечить его хроническое заболевание медленно с течением времени.