Главная
12 апреля 09:54
Симптомы гастроэзофагеального рефлюкса у ребёнка: когда нужно дополнительное обследование?
14 апреля в 14:00 (мск) состоится вебинар-диалог двух педиатров: автора журнала «Доктор.Ру» Ипатовой Марии Георгиевны, к. м. н., руководителя Центра лечения аномалий развития и заболеваний гепатобилиарной системы у детей, и Мухаметовой Евгении Маратовны, к. м. н., доцента кафедры
пропедевтики детских болезней КИДЗ им. Н.Ф. Филатова ФГАОУ ВО Первый МГМУ имени
И.М. Сеченова Минздрава России (Сеченовский Университет)
9 апреля 14:40
Круп у детей: вчера, сегодня, завтра
Постоянный автор журнала «Доктор.Ру» Ревякина Вера Афанасьевна, д. м. н., профессор, заведующая отделением аллергологии ФГБУН ФИЦ питания и биотехнологии, совместно с Савенковой Мариной Сергеевной, д. м. н., профессором, ведущим научным специалистом НПЦ помощи детям им. Святого Луки, 15 апреля в 17:00 (мск) проведет вебинар, посвященный диагностике и лечению крупа у детей
9 апреля 14:37
Цервикокранииалгии: клинический полиморфизм и выбор обезболивания
14 апреля в 16:00 (мск) состоится вебинар члена редакционного совета журнала «Доктор.Ру» Табеевой Гюзяли Рафкатовны, д. м. н., профессора кафедры нервных болезней и нейрохирургии
ФГАОУ ВО Первый Московский государственный медицинский университет имени И.М.
Сеченова Минздрава России (Сеченовский Университет)
9 апреля 14:31
XXXIV межрегиональная конференция РОАГ «Женское здоровье»
13 апреля с 09:30 до 18:00 (мск) в онлайн-формате пройдет конференция с участием постоянных авторов журнала «Доктор.Ру» Аполихиной И.А., Байрамовой Г. Р., Баранова И.И., Гурьевой В.М., Кирсановой Т.В., Козлова П.В., Лединой А.В., Протасовой А.Э., Пустотиной О.А., Пекарева О.Г., Фаткуллина И.Ф., Чечневой М.А., Ших Е.В.
Р., Баранова И.И., Гурьевой В.М., Кирсановой Т.В., Козлова П.В., Лединой А.В., Протасовой А.Э., Пустотиной О.А., Пекарева О.Г., Фаткуллина И.Ф., Чечневой М.А., Ших Е.В.
9 апреля 14:28
Ингаляционная терапия у детей с бронхиальной астомй. Возрастные аспекты
Главный редактор «Доктор.Ру» Педиатрия Геппе Наталья Анатольевна, д. м. н., профессор, заведующая кафедрой детских болезней Клинического института детского здоровья им. Н.Ф. Филатова Первого МГМУ им. И.М. Сеченова (Сеченовский Университет), и Колосова Наталья Георгиевна, к. м. н., доцент кафедры детских болезней Института здоровья детей ФГБОУ ВО «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский Университет), проведут вебинар 13 апреля в 17:00 (мск)
Все новости
Телеангиэктазии, атаксия, гипермобильный синдром, гипертрофическая кардиомиопатия и сахарный диабет – новый синдром? | Кураева
Аннотация
Доля лиц с генетическими синдромами, сопровождающимися сахарным диабетом или нарушенной толерантностью к углеводам, составляет менее 1% среди всех больных сахарным диабетом. В настоящее время описано более 70 таких синдромов, в клиническом проявлении которых имеют значение нарушенная толерантность к углеводам или сахарный диабет. В качестве примеров можно привести атаксию — телеангиэктазию, миотоническую дистрофию, генерализованную или парциальную липодистрофию. В доступной литературе мы не встретили наблюдений сочетания сахарного диабета с телеангиэктазиями, атаксией, гипермобильностью суставов, гиперрастяжимостью кожи, гипертрофической кардиомиопатией. Приводим наблюдение. Больная А., 15 лет, поступила в детское отделение Института диабета Эндокринологического научного центра РАМН с жалобами на резкую слабость в ногах, невозможность самостоятельного передвижения, кровоточивость десен, носовые кровотечения, жажду, полиурию. Матери 39 лет, отцу 43 года, сестре 12 лет, все здоровы.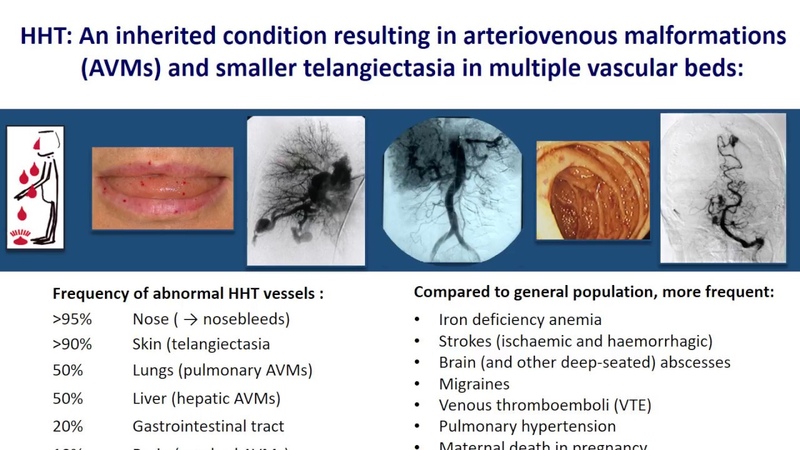 Больная от 3-й беременности, протекавшей с угрозой прерывания на протяжении всей беременности. Две предыдущие беременности у матери больной закончились выкидышами. Роды на 8-м месяце в ягодичном предлежании, с отслойкой плаценты. Масса тела при рождении 1800 г, длина тела 44 см. Больная родилась в асфиксии, с кровоизлияниями в кожу лица. В возрасте 2 лет 3 мес после перенесенной ОРВИ у ребенка появились одышка, увеличение печени до 6 см из-под края реберной дуги по среднеключичной линии, повышение систолического АД до 130 мм рт. ст. В 3 года 4 мес был установлен диагноз: идиопатический гипертрофический субаортальный стеноз. С этого возраста больную периодически беспокоили сильные боли в животе, сопровождавшиеся тошнотой и рвотой, которые расценивались как обострение хронического панкреатита. Последний приступ в 15 лет (амилаза мочи в пределах нормы). В 6 лет в связи с частыми носовыми кровотечениями, наблюдавшимися с 5-летнего возраста, а также с телеангиэктазиями сосудов кожи поставлен диагноз: болезнь Рандю—Вебера—Ослера. В связи с сохраняющейся гепатомегалией для исключения гликогеноза в 15 лет был проведен глюкозотолерантный тест. Выявлено нарушение толерантности к углеводам. Через 1 мес появились симптомы манифестации сахарного диабета. Спустя еще 1 мес в прекоматозном состоянии больная была госпитализирована в областную детскую больницу по месту жительства. При выписке суточная доза инсулина составляла 44 ЕД. Через 3 мес после манифестации сахарного диабета появилась слабость в ногах, которая быстро прогрессировала. Через 5 мес больная была госпитализирована в Эндокринологический научный центр РАМН.
Больная от 3-й беременности, протекавшей с угрозой прерывания на протяжении всей беременности. Две предыдущие беременности у матери больной закончились выкидышами. Роды на 8-м месяце в ягодичном предлежании, с отслойкой плаценты. Масса тела при рождении 1800 г, длина тела 44 см. Больная родилась в асфиксии, с кровоизлияниями в кожу лица. В возрасте 2 лет 3 мес после перенесенной ОРВИ у ребенка появились одышка, увеличение печени до 6 см из-под края реберной дуги по среднеключичной линии, повышение систолического АД до 130 мм рт. ст. В 3 года 4 мес был установлен диагноз: идиопатический гипертрофический субаортальный стеноз. С этого возраста больную периодически беспокоили сильные боли в животе, сопровождавшиеся тошнотой и рвотой, которые расценивались как обострение хронического панкреатита. Последний приступ в 15 лет (амилаза мочи в пределах нормы). В 6 лет в связи с частыми носовыми кровотечениями, наблюдавшимися с 5-летнего возраста, а также с телеангиэктазиями сосудов кожи поставлен диагноз: болезнь Рандю—Вебера—Ослера. В связи с сохраняющейся гепатомегалией для исключения гликогеноза в 15 лет был проведен глюкозотолерантный тест. Выявлено нарушение толерантности к углеводам. Через 1 мес появились симптомы манифестации сахарного диабета. Спустя еще 1 мес в прекоматозном состоянии больная была госпитализирована в областную детскую больницу по месту жительства. При выписке суточная доза инсулина составляла 44 ЕД. Через 3 мес после манифестации сахарного диабета появилась слабость в ногах, которая быстро прогрессировала. Через 5 мес больная была госпитализирована в Эндокринологический научный центр РАМН.
Доля лиц с генетическими синдромами, сопровождающимися сахарным диабетом или нарушенной толерантностью к углеводам, составляет менее 1% среди всех больных сахарным диабетом [4]. В настоящее время описано более 70 таких синдромов, в клиническом проявлении которых имеют значение нарушенная толерантность к углеводам или сахарный диабет [10]. В качестве примеров можно привести атаксию — телеангиэктазию, миотоническую дистрофию, генерализованную или парциальную липодистрофию [13, 14].
В качестве примеров можно привести атаксию — телеангиэктазию, миотоническую дистрофию, генерализованную или парциальную липодистрофию [13, 14].
В доступной литературе мы не встретили наблюдений сочетания сахарного диабета с телеангиэктазиями, атаксией, гипермобильностью суставов, гиперрастяжимостью кожи, гипертрофической кардиомиопатией [12].
Приводим наблюдение.
Больная А., 15 лет, поступила в детское отделение Института диабета Эндокринологического научного центра РАМН с жалобами на резкую слабость в ногах, невозможность самостоятельного передвижения, кровоточивость десен, носовые кровотечения, жажду, полиурию.
Матери 39 лет, отцу 43 года, сестре 12 лет, все здоровы. Больная от 3-й беременности, протекавшей с угрозой прерывания на протяжении всей беременности. Две предыдущие беременности у матери больной закончились выкидышами. Роды на 8-м месяце в ягодичном предлежании, с отслойкой плаценты. Масса тела при рождении 1800 г, длина тела 44 см. Больная родилась в асфиксии, с кровоизлияниями в кожу лица.
В возрасте 2 лет 3 мес после перенесенной ОРВИ у ребенка появились одышка, увеличение печени до 6 см из-под края реберной дуги по среднеключичной линии, повышение систолического АД до 130 мм рт. ст. В 3 года 4 мес был установлен диагноз: идиопатический гипертрофический субаортальный стеноз. С этого возраста больную периодически беспокоили сильные боли в животе, сопровождавшиеся тошнотой и рвотой, которые расценивались как обострение хронического панкреатита. Последний приступ в 15 лет (амилаза мочи в пределах нормы). В 6 лет в связи с частыми носовыми кровотечениями, наблюдавшимися с 5-летнего возраста, а также с телеангиэктазиями сосудов кожи поставлен диагноз: болезнь Рандю—Вебера—Ослера. В связи с сохраняющейся гепатомегалией для исключения гликогеноза в 15 лет был проведен глюкозотолерантный тест. Выявлено нарушение толерантности к углеводам. Через 1 мес появились симптомы манифестации сахарного диабета. Спустя еще 1 мес в прекоматозном состоянии больная была госпитализирована в областную детскую больницу по месту жительства. При выписке суточная доза инсулина составляла 44 ЕД. Через 3 мес после манифестации сахарного диабета появилась слабость в ногах, которая быстро прогрессировала. Через 5 мес больная была госпитализирована в Эндокринологический научный центр РАМН.
При выписке суточная доза инсулина составляла 44 ЕД. Через 3 мес после манифестации сахарного диабета появилась слабость в ногах, которая быстро прогрессировала. Через 5 мес больная была госпитализирована в Эндокринологический научный центр РАМН.
При осмотре: масса тела 43 кг (10-я перцентиль), длина тела 163 см (60-я перцентиль). Конституциональный тип доли- хостенический. Череп с умеренными вдавлениями и лобными буграми, гротесковые черты лица. Небо высокое. Стопы короткие с высоким сводом. Имеются распространенные телеангиэктазии сосудов кожи лица, туловища, конечностей. На левой стопе 2 очага липоидного некробиоза. Отмечаются умеренная гиперрастяжимость кожи, генерализованная гипермобильность суставов (наиболее выраженная в суставах кисти, локтевых, коленных сочленениях). Мышцы нижних конечностей субатрофичны. Мышечная сила в проксимальных отделах 3,5—4 балла, в дистальных — 4—5 баллов. При физической нагрузке небольшая одышка. Границы относительной сердечной тупости: правая — в четвертом межреберье на 1,5 см, кнаружи от края грудины, левая — в пятом межреберье по среднеключичной линии, верхняя — в третьем межреберье. Над всей поверхностью сердца выслушивается грубый голосистолический шум с максимумом в точке Боткина—Эрба. Шум проводится на сосуды шеи, межлопаточную область, брюшную аорту. Печень выступает из-под края реберной дуги по среднеключичной линии на 3,5—4 см, по передней подмышечной — на 4,5—5 см. Поверхность гладкая, край плотноэластический, слабоболезненный. Интеллект высокий. Отмечаются мышечная слабость, неустойчивость в позе Ромберга, умеренная интенция при пальценосовой пробе, положительные пробы «мимопопадания», адиадохокинеза, DCS. Наблюдается горизонтальный средней амплитуды с ротаторным компонентом нистагм, возникающий при отведении и крайных положениях глаз в стороны и вверх. Конвергенция неустойчивая. Рефлексы снижены, в руках D > S, в ногах D С S. Патологических рефлексов не выявлено. В чувствительной сфере преобладают умеренные симптомы полиневри- тического типа расстройств преимущественно поверхностной чувствительности типа высоких перчаток, носков, «лоскутные» симптомы выпадения. Выражены симптомы вегетативной дисфукции.
Выражены симптомы вегетативной дисфукции.
При общем анализе крови, мочи, анализе мочи по Нечипоренко патологических изменений не выявлено. В биохимическом анализе крови: триглицериды — 2,39 ммоль/л (норма до 1,9 ммоль/л). На рентгенограммах черепа: турецкое седло закрытого типа, с небольшим обызвествлением диафрагмы. При УЗИ печень увеличена (край на 5—6 см ниже реберной дуги), контуры ровные, структура однородная, уплотнена. Портальная вена, желчный пузырь, почки без особенностей. Поджелудочная железа не увеличена, структура однородная, уплотнена. ЭКГ: недостаточность кровоснабжения переднесептальной области, нарушение внутрижелудочковой проводимости, перегрузка левого предсердия, замедление атриовентрикулярной проводимости. ЭхоЭГ: гипертрофия межжелудочковой перегородки до 25 мм (норма 0,7—0,8 мм), стенка левого желудочка 10 мм (норма 10 мм), индекс 2,5 (норма до 1,0). На ЭЭГ умеренные диффузные изменения биоритмики с элементами резидуальной органики и указанием на вовлечение в патологический процесс преимущественно стволовых отделов мозга. Патологические знаки ирритативного характера преобладают в левом полушарии. Электронейромиография: снижение скорости проведения нервного импульса по периферическим нервам верхних и нижних конечностей от 10 до 50%. Консультация кардиолога: идиопатический гипертрофический субаортальный стеноз.
Учитывая диспластический фенотип, патологию сердечнососудистой системы, а также неврологическую симптоматику, не типичную для сахарного диабета, больную консультировали в разных медико-генетических, неврологических центрах. Дифференциальная диагностика проводилась между синдромами Фридрейха, Рефсума, Шарко—Мари—Тута, Луи—Бар (Бодер-Седжвика), Русси—Леви, Бассена—Корнцвейга, болезнью Рандю—Вебера—Ослера (см. таблицу).
Следует подчеркнуть, что в неврологическом статусе на первый план выступали симптомы мышечной слабости, сопровождающиеся диффузной амиотрофией, которая сочеталась с выраженной гипорефлексией и нарушением чувствительности по полиневритическому типу. Вместе с тем имели место статическая атаксия, умеренный интенционный тремор и мелкоразмашистый бьющий нистагм. Картина электронейромиографии соответствовала поражению периферического нейромоторного аппарата. Изменения на глазном дне соответствовали картине идиопатического отека зрительных нервов с умеренной ангиопатией сетчатки. Эпибульбарная конъюнктива была инъецирована за счет разрастания эндотелия, в петлистой перилимбальной зоне — продукты распада форменных элементов крови в виде мелкой «ржавой» пылеобразности.
Вместе с тем имели место статическая атаксия, умеренный интенционный тремор и мелкоразмашистый бьющий нистагм. Картина электронейромиографии соответствовала поражению периферического нейромоторного аппарата. Изменения на глазном дне соответствовали картине идиопатического отека зрительных нервов с умеренной ангиопатией сетчатки. Эпибульбарная конъюнктива была инъецирована за счет разрастания эндотелия, в петлистой перилимбальной зоне — продукты распада форменных элементов крови в виде мелкой «ржавой» пылеобразности.
Основные синдромы, имитирующие приведенное наблюдение
Синдром | Тип наследования | Ассоциация с сахарным диабетом или нарушением толерантности к углеводам | Ассоциация с кардиомиопатией | Основные неврологические проявления | Интеллект | Изменения стоп | Телеангиэктазии | Другие клинические проявления |
Атаксия — телеангиэктазия. Манифестация с раннего возраста | А-Р | + | Атаксия,позднее — полинейропатия, дизартрия, хореоатетоз | Умеренно снижен | Умеренные | Выражены | Ослабление конвергенции, страбизм, синопульмональ- ные инфекции, преждевременное поседение, изменения кожи по типу склеродермии, депигментация, кератоз, пигментные пятна цвета кофе с молоком, низкий рост, гипомимия | |
Атаксия Фридрейха. Манифестация с 7—15 лет | А—Д А-Р | + у 10-25% | + | Сенситивная и мозжечковая атаксия, мышечная гипотрофия, дизартрия, псевдоатетоз | Обычно сохранен | Выраженные | Снижение слуха, зрения, парез, глазодвигательных мышц | |
Синдром Рефсума. Манифестация с 7—15 лет | А-Р | + Приблизительно у 10% | + | П регрессирующая атаксия с полиневритом и дистальными парезами | Обычно сохранен | Умеренные | Атрофия зрительного и слухового нервов, пигментный ретинит, катаракта, ихтиозоподобныс кожные изменения, гипогонадизм | |
Синдром Русси—Леви. | А-Д | Мышечные гипотрофии в дистальных отделах, поли- невропатия,атаксия | Обычно сохранен | Выраженные | Катаракта, страбизм | |||
Синдром Бассе- на—Корнцвейга. Манифестация с раннего возраста | А-Р | Мышечные гипотрофии, мозжечковая атаксия | Снижен | Умеренные | Пигментная дегенерация сетчатки | |||
Синдром Шарко—Мари—Тута. Возраст манифестации может варьировать | А-Р А-Д | Мышечные гипотрофии в дистальных отделах, полиневропатия, атаксия | Обычно сохранен | Выраженные | Полиморфны | |||
Болезнь Мачадо | А-Д | + | Атаксия, дизартрия, мышечные гипотрофии | Обычно сохранен | Умеренные | Контрактуры |
 Манифестация с 7—15 лет
Манифестация с 7—15 летПримечание. А—Р — аутосомно-рецессивный, А—Д — аутосомно-доминантный тип наследования.
Отмеченные телеангиэктазии кожи, гипермобильность суставов, гиперрастяжимость кожи могли свидетельствовать о системной патологии соединительной ткани. Было предположено, что в основе развития атаксии у больной лежали сосудистые нарушения (телеангиэктазии), усугубившиеся развитием сахарного диабета.
На фоне тщательной компенсации углеводного обмена и массивной терапии ангиопротективными препаратами отмечена значительная положительная динамика. Появилась возможность самостоятельного передвижения. В дальнейшем больная переносила более значительные физические нагрузки — длительную ходьбу, танцы. Это явилось косвенным подтверждением вторичности развития атаксии вследствие сосудистой аномалии развития, носящей генерализованный характер.
Телеангиэктазии представляют собой расширение сосудов, которые выстланы эндотелием и лежат на базальной мембране при практическом отсутствии эластической и мышечной ткани [7]. Телеангиэктазии могут служить одним из клинических проявлений заболевания либо отражают суть синдрома [5]. В отдельных случаях они способны протекать по геморрагическому, псевдотуморозному типу или еще реже — с неспецифической неврологической симптоматикой, обусловленной локализацией телеангиэктазий (церебральная форма болезни Рандю—Вебера—Ослера) [3]. В связи с высокой частотой сочетаний телеангиэктазий с венозными, кавернозными мальформациями [7] такая вероятность значительно возрастает. Телеангиэктазии также могут обусловить различные патологические состояния внутренних органов (висцеральная форма болезни Рандю—Вебера—Ослера) [6]. Последнее могло быть одной из причин приступов болей в животе у больной. По мнению А. П. Зинченко и соавт. [3], изменения в размерах телеангиэктазий зависят от целого ряда физиологических или патологических факторов, таких как гормональные изменения в переходном возрасте, заболевания печени, психические травмы и пр. Вероятно, что неизбежные при сахарном диабете гормонально-метаболические изменения и явились основным фактором, усугубившим проявления телеангиэктатической болезни. Это могло привести к возникновению периферической полинейропатии и развитию атаксии. Однако мы не исключаем, что развитие полинейропатии могло быть сопряжено и с синдромом Элерса—Данло. Описанное Т. J. Farag и R. N. Schimke [11] наблюдение этого синдрома у двух сибсов в сочетании с периферической полинейропатией, рассматриваемое авторами как следствие самостоятельного генетического дефекта, подтверждает такую возможность.
Нам представляется закономерным сочетание телеангиэктатической болезни с синдромом Элерса—Данло, хотя в литературе среди 11 выделенных типов этого синдрома телеангиэктазии отсутствуют [2]. Такая сочетанная патология соединительной ткани могла быть обусловлена нарушением генетического управления синтеза одного или нескольких типов коллагена.
Имеющиеся в литературе наблюдения сочетания гипермобильного синдрома с гипертрофической кардиомиопатией [8] позволяют связать идиопатический гипертрофический субаортальный стеноз с остальными составляющими описываемого синдрома (см. рисунок).
Наследственная геморрагическая телеангиэктазия, некоторые типы синдрома Элерса—Данло и идиопатический субаортальный стеноз (по крайней мере в 50% случаев) — заболевания, наследуемые по аутосомно-доминантному типу [1, 2, 9]. Отсутствие этих недугов у родителей больной позволяет предположить мутацию de novo. Однако, учитывая привычную невынашиваемость в акушерском анамнезе матери, причиной которой могла быть непрочность плодных оболочек вследствие поражения соединительной ткани, нельзя исключить наличие рецессивного наследования описанного синдрома.
Синдромальная тетрада больной А.
Для подтверждения этой гипотезы необходимо дальнейшее накопление наблюдений данного синдрома с привычной невынащиваемостью.
В отношении сочетания описанного симптомокомплекра с сахарным диабетом возможно случайное совпадение. Вопрос об ассоциации с сахарным диабетом может быть решен при наличии повторных наблюдений.
1. Анисимова Е.Л., Бабурова Е.М. // Тер. арх. — 1987. — № 6. — С. 66 67.
2. Вельтищев Ю.Е., Казанцева Л.З., Семячкина А.И. // Наследственная патология человека / Под ред. Ю. Е. Вельтищева, Н.П. Бочкова. — М., 1992. — Т. 1. — С. 100—109.
3. Зинченко А.П., Ливандовский Ю. А., Пишель Л. В. и др. // Журн. невропатол. и психиатр. — 1980. — № 7. — С.1005-1011.
4. Керимч Н.Б. Генетика сахарного диабета с учетом возраста начала заболевания: Дис. … канд. мед. наук. — М., 1982. — С. 34.
… канд. мед. наук. — М., 1982. — С. 34.
5. Ливандовский Ю.А. // Вести, дерматол. — 1994. — № 5. — С. 21-22.
6. Логинов А.С., Сахарова Т.Н., Ткачук В.Д. и др. // Тер. арх. — 1985. — № 7. — С.112-114.
7. Мацко Д.Е. Пороки развития сосудов головного и спинного мозга: Дис. … д-ра мед. наук. — СПб., 1993. — С.57-71.
8. Морова Н.А., Шуголь С.А., Стефаненко Г.Н. и др. // Тер. арх. — 1991. — № 2. — С. 94-95.
9. Уинги Д., Браунвальд Е. // Внутренние болезни/ Под ред. Е. Браунвальда, К. Д. Иссельбахера и Р. Г. Петерсдорфа и др.: Пер. с англ. — М., 1995. — Кн. 5. — С. 332.
10. Alkolado J.С., Rees J.A., Owens D.R. // Diabetologia. — 1994. — Vol. 37, № 6. — P. 639-640.
11. Farag Т.I., Schimke R.N. // Clin. Genet. — 1989. — Vol. 35. — P.121-124.
12. McKusick V.A. Mendelian Inheritance in Man: Catalogs of Autosomal Dominant, Autosomal Recessive, and X-Linked Phenotypes. — 8-th Ed. — Baltimore, 1988.
13. Rimoin D. L. // The Genetics of Diabetes Mellitus / Eds W. Creuntzfeld, J. Koberling, J.O. Neel. — Berlin, 1976. — P. 43-64.
14. Swift M. // Ataxia — Teleangiectasia: Genetics, Neuropathology and Immunology of a Degenerative Disease of Childhood / Eds R. A. Gatti, M. Swift. — New York, 1985. — P.137—139.
A. Gatti, M. Swift. — New York, 1985. — P.137—139.
ВНУТРЕННИЕ БОЛЕЗНИ | | «РМЖ» №7 от 03.10.1996
НАСЛЕДСТВЕННАЯ ГЕМОРРАГИЧЕСКАЯ ТЕЛЕАНГИЭКТАЗИЯ (БОЛЕЗНЬ ОСЛЕРА — ВЕБЕРА — РАНДЮ): НОВОЕ О ПАТОГЕНЕЗЕ, ОСЛОЖНЕНИЯХ И ЛЕЧЕНИИ
РАСПРОСТРАНЕННОСТЬ НЕДИАГНОСТИРОВАННОЙ ПЕРНИЦИОЗНОЙ АНЕМИИ У ПОЖИЛЫХ
МИГРЕНЬ И РИСК ЦЕРЕБРАЛЬНОЙ ИШЕМИИ В МОЛОДОМ ВОЗРАСТЕ
ВЛИЯНИЕ ЗАМЕНЫ МАСЛА МАРГАРИНОМ В ПРОФИЛЬ ЛИПОПРОТЕИДОВ: РАНДОМИЗИРОВАННЫЕ ПЕРЕКРЕСТНЫЕ ИССЛЕДОВАНИЯ
АНТАГОНИСТЫ КАЛЬЦИЯ И ЖЕЛУДОЧНО-КИШЕЧНЫЕ КРОВОТЕЧЕНИЯ: СООТНОШЕНИЕ ЭФФЕКТОВ
РИСК ЖЕЛУДОЧНО-КИШЕЧНЫХ КРОВОТЕЧЕНИЙ, СВЯЗАННЫЙ С АНТАГОНИСТАМИ КАЛЬЦИЯ, У БОЛЬНЫХ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИЕЙ СТАРШЕ 67 ЛЕТ
НАСЛЕДСТВЕННАЯ
ГЕМОРРАГИЧЕСКАЯ ТЕЛЕАНГИЭКТАЗИЯ
(БОЛЕЗНЬ ОСЛЕРА — ВЕБЕРА — РАНДЮ):
НОВОЕ О ПАТОГЕНЕЗЕ, ОСЛОЖНЕНИЯХ И
ЛЕЧЕНИИ
A.J.F.A. Kerst
Наследственная
геморрагическая телеангиэктазия
(НГТ), известная также как болезнь
Ослера — Вебера — Рандю,
представляет собой редкое
наследственное нарушение,
приводящее к образованию
легкокровоточащих телеангиэктаз
на поверхности кожи и слизистых
оболочках; оно связано с наличием в
системах многих органов
артериовенозных врожденных
пороков (АВВП), которые могут
вызвать опасные осложнения.
Частота этого заболевания
составляет 1 — 2 на 1 млн человек.
Классической картиной заболевания
являются семейный характер
телеангиэктаз и наличие носовых
кровотечений и характерных
поражений от 1 до 2 мм в диаметре,
состоящих из расширенного сосуда
на поверхности кожи или слизистых
оболочек, особенно носа, губ и
языка, непосредственно
соединенного с артерией или веной.
Рецидивирующие носовые
кровотечения, наблюдаемые у 50 — 80%
пациентов, — это первая жалоба,
часто отмечаемая в возрасте до 10
лет. Переливание крови требуется
10-30% пациентов. Заболевание
наследуется по
аутосомно-доминантному типу,
вероятность проявления гена
составляет 97%. В некоторых семьях
наблюдалась мутация гена,
кодирующего рецептор к
трансформирующему фактору роста,
названный эндоглином.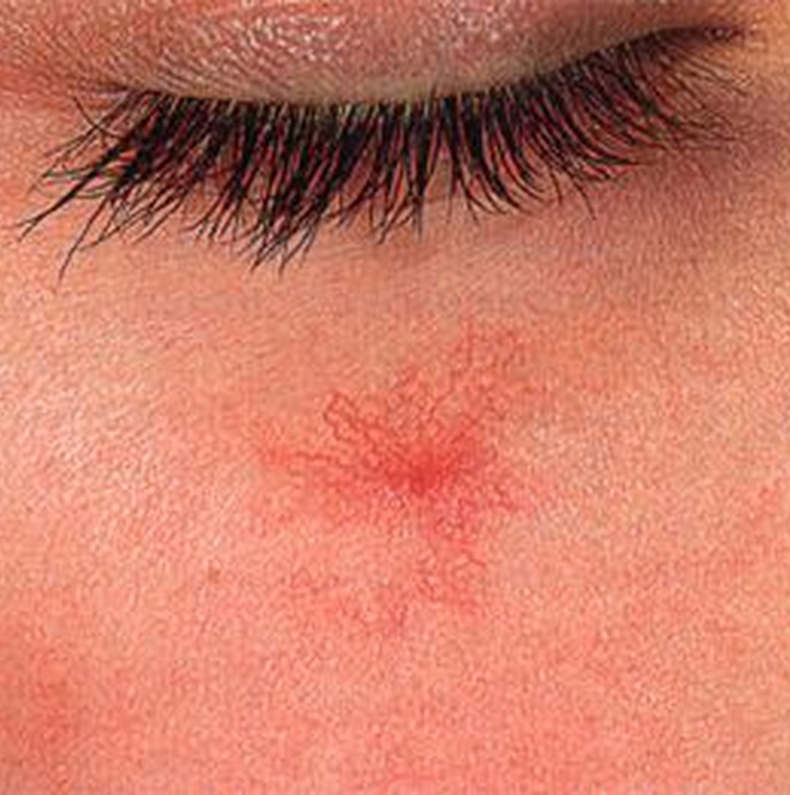 Была
Была
продемонстрирована генетическая
гетерогенность, предполагающая
участие других рецепторов к
трансформирующему фактору роста.
Это может служить объяснением
различий в клинических проявлениях
заболевания. АВВП, способные
достичь в диаметре нескольких сантиметров, состоящие
из тонкостенных васкулярных
полостей с одной или несколькими
питающими артериями, могут
появиться в любом органе, но
главным образом в легких, печени и
мозге.
АВВП легких, обнаруживаемые у 15 —
30% пациентов с НГТ, обычно питаются
легочной артерией и дренируются
через легочные вены. Они могут
привести к значительному сбросу
крови слева направо и гипоксемии.
Среди серьезных осложнений —
кровотечения с кровохарканьем,
гемотораксом с парадоксальными
эмболами или лишь слабой одышкой.
Современная практика обследования
заключается в измерении paO2 с использованием
точных референтных показателей (paO2 = 104 — 0,24 х возраст) в
сочетании с рентгенографией
грудной клетки. В случае сброса
крови с гипоксемией измерение
проводится с использованием 100%
кислородного метода. Если фракция
сброса крови ненормальная или если
возникает какое-либо подозрение
при рентгенографии грудной клетки,
то проводится внутривенная
цифровая основная ангиография.
АВВП пищеварительного тракта.
Потеря крови через телеангиэктазы
в кишке имеет место у 10 — 40%
пациентов и обычно наблюдается в
более позднем возрасте, чем носовые
кровотечения. У половины пациентов
кровоточащее пятно в виде
небольшого красного, хорошо
заметного поражения может быть
определено с помощью эндоскопии
желудка или двенадцатиперстной
кишки; телеангиэктазы в толстой
кишке встречаются редко.
Спорадические большие АВВП
наблюдаются между печеночной
артерией и веной, между воротной и
печеночной венами и между
печеночной артермией и воротной
веной, что, возможно, приводит к
сбросу крови слева направо,
сопровождающемуся сердечной
недостаточностью при высоком
минутном объеме сердца, к
печеночной энцефалопатии после
кровотечения в кишке и к портальной
гипертензии, сопровождающейся
варикозным расширением сосудов
пищевода.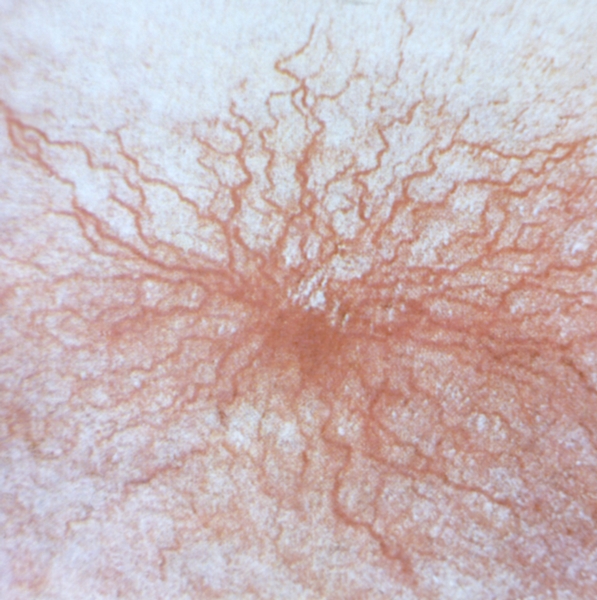 Гепатомегалия или шум над
Гепатомегалия или шум над
печенью должны побудить к
дальнейшему исследованию.
Компьютерная томография и цветная
допплерография являются
чувствительными неинвазивными
методами обнаружения.
Второй тип поражения печени,
названный циррозом НГТ, характеризуется
ненормальными расширенными
сосудами, окруженными стромой с
изменяющимся объемом,
распространенными по всей печени.
Врожденные пороки церебральных
сосудов выявляются в 5 — 10% случаев и
проявляются в виде
телеангиэктазии, кавернозных
ангиом, АВВП и аневризм; они могут
вызывать головную боль,
эпилептические припадки и ишемию
окружающих тканей в результате
синдрома обкрадывания; риск
кровотечения представляется
небольшим. Магнитно-резонансное
исследование, возможно, является
наилучшим методом скрининга.
Лечение. Раздражающие по
косметическим соображениям или
часто кровоточащие кожные
поражения можно лечить с помощью
лазерной терапии. Коагуляция
телеангиэктаз носа приносит лишь
временное облегчение. Классической
терапией для легочных АВВП была
резекция; в настоящее время методом
выбора является эмболизация
питающих сосудов с помощью
спиралей или отделяемых надувных
баллонов. Все пациенты с легочными
АВВП перед выполнением процедур,
которые могут вызвать бактериемию,
должны получить профилактическое
лечение антибиотиками.
Опасную кровопотерю при кишечных
телеангиэктазах можно лечить с
помощью комбинации низких доз
эстрогена и прогестогена, механизм
действия которой неизвестен,
однако снижение потребности в
переливании крови при ее
использовании доказано. Результаты
эндоскопического лазерного
лечения не оправдывают ожиданий, и
лечение больших АВВП в печени
должно быть консервативным. Есть
сомнение в том, следует ли лечить
бессимптомные поражения нервной
системы, так как высказываются
различные предположения
относительно соотношения риска
кровотечения и хирургических
рисков.
Для лечения АВВП могут быть
применены различные способы:
хирургия, эмболизация и
стереотактическая радиохирургия с
применением рентгеновского
излучения.
Заключение. НГТ представляет
собой редкое заболевание со
значительным риском развития
потенциально опасных осложнений.
Пациенты могут обращаться к разным
специалистам, и очень важно понять,
что это заболевание может
распространяться на области,
находящиеся за пределами той, с
которой связаны начальные жалобы. В
связи с преобладанием легочных и
церебральных АВВП все пациенты с
НГТ должны проходить скрининг на их
наличие, а родственники пациентов с
НГТ должны быть обследованы на это
заболевание.
Литература:
Haitjema T, Westermann CJJ,
Overtoom TTC, Timmer R, Disch F, Mauser H, Lammers J-WJ.
Hereditary Hemorrhagic Telangiectasia (Osler — Wеber — Rendu
Disease). Arch Intern Med. 1996;156:714-19.
РАСПРОСТРАНЕННОСТЬ
НЕДИАГНОСТИРОВАННОЙ ПЕРНИЦИОЗНОЙ
АНЕМИИ У ПОЖИЛЫХ
Е. Нурмухаметова
E. Nurmuchametova
Пернициозная анемия
обычно развивается вследствие
нарушения всасывания кобаламина
из-за сниженной секреции
внутреннего фактора в желудке.
Частота этой патологии
увеличивается с возрастом.
Проведенные ранее исследования
показали, что частота пернициозной
анемии среди населения в целом
составляет 0,1%, а среди пожилого
населения стран Северной Европы — 1%.
Однако приведенные данные
недостаточно достоверны, так как
для их получения использовались
устаревшие на сегодняшний день диагностические
методы; пациентов включали в
исследования при наличии у них
клинических проявлений. Кроме того,
большинство научных работ по
проблеме было проведено в
скандинавских странах и
Великобритании.
Автор данной статьи ставил перед
собой цель определить
распространенность
недиагностированной пернициозной
анемии среди пожилого населения
США. С одной стороны, излечимость
этого заболевания, а с другой —
возможность развития тяжелых
неврологических проявлений в тех
случаях, когда патология остается
нераспознанной, делают эту
проблему очень актуальной.
Были исследованы пробы крови от
729 человек старше 60 лет. В
В
исследование были включены лица, не
имевшие выраженных клинических
проявлений недостаточности
кобаламина. Определяли уровень
кобаламина и титр антител к
внутреннему фактору. При выявлении
каких-либо отклонений
дополнительно применялся метод
Шиллинга, проводили подсчет и
изучение морфологии клеток крови,
определение концентрации гастрина
в сыворотке, некоторые
метаболические исследования.
Распространенность
недиагностированной пернициозной
анемии составила 1,9%. Ее частота
была выше среди женщин — 2,7% против
1,4% у мужчин. Патология наиболее
распространена среди белых и
черных женщин (соответственно 4,0 и
4,3%), а у женщин из Латинской Америки
и Азии пернициозная анемия не
выявлялась.
Важно, что среди выявленных
пациентов с пернициозной анемией
лишь у некоторых она была
невыраженная; в 3 случаях
присутствовали
полисегментированные нейтрофилы, а
макроцитоз имел место лишь в 1
случае. У 3 больных имелись
незначительные неврологические
нарушения, связанные с дефицитом
кобаламина. Следовательно,
терапевты в большинстве случаев не
имеют достаточных оснований
подозревать у пациентов наличие
пернициозной анемии.
В США проживают 41,5 млн человек
старше 60 лет. При экстраполяции
результатов исследования на
население в целом получается, что
нераспознанная пернициозная
анемия имеется примерно у 800 000
пожилых американцев.
Полученные результаты имеют
также практическое значение в
связи с тем, что неоднократно
предлагалось обогащать пищевые
продукты фолатами с целью снижения
частоты рождения детей с дефектами
нервной трубки. Однако реализация
этих мер может привести к
маскировке части случаев
пернициозной анемии и
способствовать прогрессированию
неврологических нарушений у
пациентов с этой патологией, причем
не только у пожилых.
Литература:
Carmel R. Prevalence of undiagnosed pernicious anemia in the
elderly. Arch Intern Med 1996;156:1097-100.
МИГРЕНЬ
И РИСК ЦЕРЕБРАЛЬНОЙ ИШЕМИИ В
МОЛОДОМ ВОЗРАСТЕ
В. Назаренко
Назаренко
V. Nazarenko
У 308 больных в возрасте 15
— 44 лет, перенесших ишемический
инсульт или транзиторную
ишемическую атаку (ТИА), изучали
значение мигрени (М) как фактора
риска (ФР) церебральной ишемии.
Контрольная группа состояла из 591
человека без нарушений мозгового
кровообращения в анамнезе.
Пациентов наблюдали в течение 5 лет.
За этот период был выявлен только 1
повторный случай ТИА и 1 — инсульта.
В основной группе М в анамнезе была
у 14,9% (у 39 женщин и 7 мужчин), а в
контрольной — у 9,1% обследованных (р <
0,02). Только у 8 больных М протекала с
аурой. В основной группе
значительно чаще, чем в
контрольной, встречались такие ФР
церебральной ишемии, как
артериальная гипертензия, курение,
ожирение, злоупотребление
алкоголем, гиперхолестеринемия,
гипертриглицеридемия и низкий
уровень липопротеидов высокой
плотности. М оказалась ФР нарушений
мозгового кровообращения у женщин,
но не у мужчин. При этом у пациенток
с М без ауры был повышен риск ТИА, а
с аурой — инсульта. Наиболее
значимой прогностическая роль М
оказалась у женщин моложе 35 лет. В
этой группе пациенток риск ТИА при
наличии М в анамнезе был повышен в
3,7 раза (р = 0,003), риск инсульта при М с
аурой был повышен в 8,6 раза (р = 0,05).
Атерогенные ФР имели наиболее
важное значение у мужчин и у женщин
старше 35 лет. Несмотря на
существенную роль М в
происхождении ТИА и инсульта у
молодых женщин, частота их
возникновения является очень
низкой и не превышает 17 случаев на
100000 человек в год. Поэтому
профилактику нарушений мозгового
кровообращения у всех женщин
моложе 35 лет с М считают
нецелесообразной. Пациенткам с М с
аурой рекомендуют клиническое
обследование для выявления
дополнительных ФР инсульта с
последующим проведением лечебных и
профилактических мероприятий.
Литература:
Carolei A, Marini C, De Matteis G, et al. Histopy of migraine
and risk of cerebral ischemia in young adults. Lancet
1996;347:1503-6.
ВЛИЯНИЕ
ЗАМЕНЫ МАСЛА МАРГАРИНОМ В
НИЗКОЖИРОВОЙ ДИЕТЕ У ПАЦИЕНТОВ С
ГИПЕРХОЛЕСТЕРИНЕМИЕЙ НА ПРОФИЛЬ
ЛИПОПРОТЕИДОВ: РАНДОМИЗИРОВАННЫЕ
ПЕРЕКРЕСТНЫЕ ИССЛЕДОВАНИЯ
Е. Потешных
Потешных
E. Poteshnich
Диетологи большинства
стран Запада рекомендуют с целью
уменьшения риска ишемической
болезни сердца (ИБС) заменять
насыщенные жирные кислоты (ЖК) n-6
полиненасыщенными и n-9
мононасыщенными ЖК. Однако недавно
появились данные о том, что
употребление больших количеств n-6
ненасыщенных ЖК связано со
снижением концентрации
липопротеидов высокой плотности
(ЛПВП), а транс-формы ненасыщенных
ЖК могут увеличивать риск ИБС,
возможно, за счет повышения
концентрации общего холестерина
(ХС), ХС липопротеидов низкой
плотности (ЛПНП), La-липопротеидов и
снижения уровня ХС ЛПВП. Маргарины
часто предлагаются в качестве
замены содержащему большое
количество насыщенных ЖК маслу,
однако они содержат по крайней мере
несколько транс-ненасыщенных ЖК.
В исследовании, проведенном с
участием 49 больных с умеренной
гиперхолестеринемией
(концентрация ХС 5,5 — 7,9 ммоль/л,
средний возраст 46,8 года),
сравнивали влияние употребления
масла или ненасыщенного маргарина
на уровень липидов и липопротеидов
в плазме. Больные с наследственной
или вторичной
гиперхолестеринемией, а также
принимающие лекарства, влияющие на
липидный обмен, в исследование не
включались. Участники исследования
находились последовательно на
масляной и на маргариновой диете по
6 нед с 5-недельным перерывом между
курсами, в течение которого они
питались как обычно. Две
экспериментальные диеты
различались только источником
жиров. В каждой диете дневное
количество масла или маргарина
составило 17 г.
Масса тела и
артериальное давление не
изменялись в течение периода
наблюдения. При маргариновой диете
концентрация общего ХС снизилась
примерно на 5%, а уровень ХС ЛПНП и
аполипопротеина В — на 10% по
сравнению с масляной. Уровень
триглицеридов, липопротеидов Lp(a) и
ХС ЛПВП оказался сходным при обеих
диетах. Последовательность
применения диет не влияла на
результаты.
Предположение, что маргарины
могут оказывать обратное влияние
на риск ИБС и концентрацию
липопротеидов, базировалось на
эпидемиологических исследованиях.
Так, Willet обнаружил увеличение риска
ИБС в ходе 8-летнего наблюдения за
людьми, употреблявшими транс-ЖК в
количествах, в 5 раз превышающих
средние значения. Кроме того,
употребление продуктов, являющихся
главными источниками транс-ЖК
(маргарин, бисквиты, печенье и белый
хлеб), оказалось также связано с
увеличением риска ИБС. Mensik и Katan, а
также Nestel показали, что С18:1
транс-ЖК, которые составляют
большую часть транс-изомеров в
диетах, используемых на Западе,
связаны с особенно неблагоприятным
профилем
липопротеидов. Концентрация общего
ХС и ХС ЛПНП при этом по крайней
мере так же высока, как при диете,
обогащенной содержащими
повышенное количество ХС
насыщенными ЖК, а уровень ЛПВП
снижается. Однако в предлагаемой
работе более благоприятный профиль
липопротеидов наблюдали при
использовании маргариновой диеты;
концентрация транс-ЖК в маргарине,
возможно, была недостаточна для
выявления неблагоприятного
влияния на уровень липидов и
липопротеидов в плазме.
В настоящее время большинством
осознана необходимость
уменьшения употребления
насыщенных ЖК (миристиновой и
пальмитиновой). Подходящей заменой
являются масла, обогащенные
олеиновой (монозамещенная с
цисконфигурацией) и линолевой (n-6
полиненасыщенная) кислотами; при
этом n-6 полиненасыщенные ЖК не должны составлять
больше 10% от общей калорийности
диеты, так как большее количество
может быть связано со снижением
уровня ЛПВП.
Таким образом, представляется
обоснованным предпочтительное
употребление маргаринов при
дислипидемиях; возможно дальнейшее
совершенствование диет путем
уменьшения содержания в них
транс-ЖК до нуля.
Литература:
Chisholm A, Mann J, Sutherland W, et al. Effect on lipoprotein
profile of replacing butter with margarine in a low fat diet:
randomised crossover study wiht hypercholesterolaemic subjects.
BJM 1996;312:931-4.
АНТАГОНИСТЫ
КАЛЬЦИЯ И ЖЕЛУДОЧНО-КИШЕЧНЫЕ
КРОВОТЕЧЕНИЯ: СООТНОШЕНИЕ ЭФФЕКТОВ
B. Назаренко
Назаренко
V. Nazarenko
В проспективном
наблюдательном эпидемиологическом
исследовании, охватывающем три
региона США, Pahor и соавт. получили
данные, свидетельствующие о
повышенном риске
желудочно-кишечных кровотечений
(ЖКК) у больных артериальной
гипертензией (АГ) пожилого и
старческого возраста, принимающих
антагонисты кальция (АК). Несмотря
на то, что этот риск оказался выше,
чем при терапии аспирином или
нестероидными
противовоспалительными
препаратами, приведенные расчеты
показывают, что он является
достаточно низким: возникновение
одного дополнительного ЖКК можно
ожидать после 86 человеко-лет
терапии АК. Сообщение Pahor и соавт.
может иметь серьезные последствия.
Во-первых, не исключено появление
других подобных публикаций, а
во-вторых, врачи, знакомые с данной
работой, могут ограничить
назначение АК пожилым больным с АГ.
Способность АК угнетать агрегацию
тромбоцитов известна давно, с ней
связывают потенциальный
благоприятный эффект АК у больных
атеросклерозом, подобный эффекту
аспирина. Теоретически нельзя
исключить возможности некоторого
увеличения риска ЖКК при терапии
АК. Однако следует иметь в виду, что
данные, полученные Pahor и соавт.,
могут считаться лишь рабочей
гипотезой и далеко не носят
окончательного характера. Это
обусловлено рядом серьезных
методических недостатков
обсуждаемой работы, которые вообще
свойственны исследованиям
подобного рода. Диагностика ЖКК
основывалась исключительно на
данных, содержащихся в медицинской
документации, а не на собственной
оценке. О приеме того или иного
лекарственного препарата (ЛП) можно
было судить лишь приблизительно,
путем анализа упаковок ЛП,
предъявленных больными.
Приверженность лечению не
оценивалась, не было возможности
определить, принимал ли пациент тот
или иной ЛП непосредственно перед
возникновением ЖКК. Примечательно,
что за трехлетний период
наблюдения число больных,
принимающих ингибиторы
ангиотензинпревращающего
фермента, увеличилось в 10 раз, что
подчеркивает условность деления
пациентов на группы в зависимости
от приема того или иного
гипотензивного ЛП. Ответ на
Ответ на
сформулированную авторами
гипотезу может быть получен лишь в
крупных клинических исследованиях
по применению АК при АГ. В настоящее
время вопрос о целесообразности
назначения АК пациентам пожилого и
старческого возраста следует
решать индивидуально; при этом, как
и при любом другом виде лечения,
следует тщательно учитывать
соотношения между риском и пользой
терапии.
Литература:
Gordon RD. Calcium antagonists and gastro intestinal
haemorrhage: the balancing act. Lancet 1996;347:1056.
РИСК
ЖЕЛУДОЧНО-КИШЕЧНЫХ КРОВОТЕЧЕНИЙ,
СВЯЗАННЫЙ С АНТАГОНИСТАМИ
КАЛЬЦИЯ, У БОЛЬНЫХ АРТЕРИАЛЬНОЙ
ГИПЕРТЕНЗИЕЙ СТАРШЕ 67 ЛЕТ
В. Назаренко
V. Nazarenko
Антагонисты кальция
(АК), широко применяющиеся при
лечении артериальной гипертензии
(АГ), помимо сердечно-сосудистых
эффектов, обладают способностью
угнетать агрегацию тромбоцитов. В
наблюдательном эпидемиологическом
исследовании у 1636 больных АГ
(средний возраст 75,3 года),
проживающих в трех регионах США,
сравнивали частоту возникновения
желудочно-кишечных кровотечений
(ЖКК) при монотерапии АК,
b-блокаторами (ББ) и ингибиторами
ангиотензинпревращающего фермента
(ИАПФ). Пациентов обследовали
дважды с интервалом в 3 года при
посещении на дому, во время
которого просили предъявить
упаковки всех лекарственных
препаратов, принимавшихся за
последние 2 нед. О возникновении ЖКК
судили по медицинским документам:
заключительным диагнозам
стационаров, внесенным в
компьютерную систему регистрации
MEDPAR, и свидетельствам о смерти. За
период наблюдения было выявлено 120
случаев ЖКК, причем 88 — тяжелых. В
основном ЖКК возникали у пациентов
с язвенной болезнью желудка и
двенадцатиперстной кишки,
гастритом и онкологическими
заболеваниями кишечника. Риск ЖКК в
группе больных, принимавших АК, оказался в 1,86 раза
выше, чем при терапии ББ (различия
статистически значимы). При этом
ЖКК несколько чаще возникали при
лечении верапамилом и дилтиаземом,
чем нифедипином. У больных,
принимавших ИАПФ, частота
возникновения ЖКК была почти такой
же, как при терапии ББ
(относительный риск 1,23; различия
незначимы). При многовариантном
анализе, учитывающем влияние таких
факторов, как возраст, пол,
сопутствующие заболевания,
выявленные закономерности
сохранялись. В происхождении ЖКК
при терапии АК авторы считают
значимыми не только их
антиагрегантный, но и
вазодилатирующий эффекты. При
назначении АК больным АГ пожилого и
старческого возраста следует
тщательно учитывать риск ЖКК,
связанный с сопутствующей
патологией и приемом
антикоагулянтов, кортикостероидов
и нестероидных
противовоспалительных препаратов.
Авторы подчеркивают ряд
методических ограничений
проведенного исследования:
отсутствие тщательного наблюдения,
возможность ошибок при оценке
характера лечения и диагностике
ЖКК.
Литература:
Pahor M, Guralnic JM, Furberg CD, et al. Risk of
gastrointestinal haemorrage with calcium antagonists in
hypertensive persons over 67 years old. Lancet 1996;347:1061-5.
Болезнь Рандю — Ослера, синдром Ослера, семейная наследственная телеангиэктазия наследственная геморрагическая телеангиэктазия, геморрагический ангиоматоз
Пользователи также искали:
болезнь рандю — ослера красота и медицина,
Рандю,
рандю,
Ослера,
Болезнь,
болезнь,
ослера,
рекомендации,
прогноз,
презентация,
клинические,
патофизиология,
кровоточивости,
красота,
медицина,
фото,
Болезнь Рандю — Ослера,
болезнь рандю — ослера мкб -,
болезнь рандю — ослера прогноз,
болезнь рандю — ослера красота и медицина,
болезнь рандю — ослера мкб — 10,
болезнь рандю — ослера тип кровоточивости,
болезнь рандю — ослера патофизиология,
болезнь рандю — ослера фото,
болезнь рандю — ослера презентация,
болезнь рандю — ослера клинические рекомендации,
болезнь рандю — ослера,
заболевания по алфавиту. болезнь рандю — ослера,
болезнь рандю — ослера,
Болезнь Рандю — Ослера — Вебера с поражением внутренних органов и нервной системы
Болезнь Рандю — Ослера — Вебера — это заболевание с клинической картиной геморрагического диатеза. Синоним «наследственная геморрагическая телеангиэктазия» (НТГ) в полной мере отражает сущность заболевания, для которого наиболее характерной считается триада признаков: телеангиэктазии на коже, слизистых оболочках и во внутренних органах; склонность к кровотечениям; аутосомно-доминантный тип наследования (А.А. Пономарев, 1998; О.Л. Иванов и соавт., 2002).
Частота болезни Рандю — Ослера — Вебера составляет 1–2 случая на 1 млн человек. В патогенезе заболевания ведущую роль играют анатомические и функциональные изменения сосудов. Анатомическая сущность болезни заключается в поражении кровеносных сосудов различного калибра с дегенерацией и гипоплазией мышечного и эластического слоев. В результате развивается очаговое истончение стенок, затем расширение просвета микрососудов. Кровоточивость объясняется чрезвычайно легкой ранимостью сосудистой стенки в месте ангиэктазии, а также повышением фибринолитической активности крови (В.Б. Золотаревский и соавт., 1998; Г.Д. Бабушкина и соавт., 2001).
Клиническими признаками заболевания являются частые носовые кровотечения, которые возникают спонтанно или после небольшой механической травмы. Известны случаи дебюта заболевания с приступом кровавой рвоты, повторной макрогематурии. Развитие телеангиэктазий в печени может привести к формированию хронического гепатита и цирроза печени. В литературе имеется единственное сообщение о начале болезни Рандю — Ослера — Вебера плачем «кровавыми слезами».
Однажды начавшись, кровотечения склонны к рецидивированию, причем интенсивность и длительность кровотечений индивидуальны (однократная кровопотеря составляет от нескольких капель до 500 мл и более). Кроме того, для заболевания характерны и внешние проявления в виде множественных телеангиэктазий на коже головы, слизистой носа, носоглотки, гортани, пищеварительного тракта, мочеполовой системы, на конъюнктиве, в бронхах, головном мозге, печени, что может привести к грозным осложнениям — кровотечениям, железодефицитной анемии (Е.В. Казакевич и соавт., 1997; Г.В. Банченко и соавт., 2000).
По частоте развития кровотечений из внутренних органов при НТГ на первом месте стоят желудочно-кишечные кровотечения, затем легочные и почечные. Кровоизлияния в головной и спинной мозг наблюдаются у 2–3 % больных. Другие неврологические изменения включают головные боли, эпилептические припадки, парезы и нарушения зрения (Б.А. Беренбейн и соавт., 1999; O. Braun-Falco et al. 1996; A.J. Keret, 2001).
По мере прогрессирования заболевания вследствие повторных кровотечений развивается хроническая железодефицитная анемия. В отдельных случаях формируются четкие клинические проявления дефицита железа в организме (сухость кожи, ломкость ногтей, волос). При генерализованном поражении сосудов говорят об общем варикозном статусе.
Патогенетическая терапия болезни не разработана. При кровотечении используют консервативные и хирургические методы лечения.
В литературе мы не встретили описания сочетанного поражения головного и спинного мозга при болезни Рандю — Ослера — Вебера, поэтому данное сообщение будет представлять интерес для практикующих врачей. Приводим собственное клиническое наблюдение.
Больная Щ., 48 лет, обратилась в клинику нервных болезней ЛГМУ 13 февраля 2001 года с жалобами на высыпания на слизистой языка, коже спины, правой ступни и голени, частые носовые кровотечения, головокружение, шаткость при ходьбе, слабость в верхних и нижних конечностях, чувство онемения в ступнях, голенях, нарушение мочеиспускания в виде частых позывов, снижение слуха на правое ухо.
Анамнез заболевания. Считает себя больной с дошкольного возраста, когда стали появляться непродолжительные носовые кровотечения. В возрасте 18 лет после очередного носового кровотечения пациентка обратилась к отоларингологу. При осмотре слизистой носовой полости были выявлены кровоточащие телеангиэктазии.
Семейный анамнез. Двое родных сестер больны болезнью Рандю — Ослера — Вебера, старшая умерла в возрасте 45 лет от профузного маточного кровотечения, у младшей в возрасте 44 лет появились обильные носовые кровотечения. Родители больной: мать 72 года, здорова; отец в возрасте 60 лет скончался от новообразования желудка. О заболеваниях в других поколениях данных не имеется. У больной есть дочь от второй беременности, здорова. Первая беременность закончилась мертворождением в сроке 7 месяцев, осложнилась тяжелым послеродовым кровотечением.
Анамнез жизни. Венерические заболевания, болезнь Боткина, туберкулез, малярию отрицает. Вредных привычек нет. Травмы и операции: сотрясение головного мозга, резекция 2/3 желудка в связи с язвенной болезнью. Из соматических заболеваний отмечается хронический пиелонефрит, хроническая железодефицитная анемия.
Локальный статус. При осмотре слизистой языка определяются множественные телеангиэктазии в виде небольших неправильной формы пятнышек (рис. 1), на кожных покровах спины, правой голени и стопы — множество круглых и овальных узелков диаметром 5–7 мм, выступающих над поверхностью кожи на 1–2 мм (рис. 2, 3), бледнеющих при надавливании.
Соматический статус. Пониженного питания, рост 164 см, вес 57 кг. При осмотре спины разное стояние лопаток, реберный горб, выраженный кифосколиоз грудопоясничного отдела позвоночника. В легких везикулярное дыхание, хрипов нет, ЧДД — 18 в мин. Тоны сердца приглушены, ЧСС — 80 в мин, АД — 120/80 мм рт.ст. Живот мягкий, болезненный в эпигастрии; печень и селезенка не увеличены.
Неврологический статус. Сознание ясное. Перкуссия черепа безболезненна. Глазные щели S > D, зрачки S = D, единичные нистагмоидные движения при крайних отведениях глаз. Ослаблен акт конвергенции с двух сторон. Сглажена правая носогубная складка. Девиация кончика языка вправо. Снижение слуха на правое ухо. Сухожильные рефлексы с верхних конечностей оживлены S > D, коленные S > D, ахилловы S > D, высокие поликинетичные. Сила в руках снижена до 4 баллов, в нижних конечностях — 3,5 балла. Симптом Якобсона — Ласка положителен с обеих сторон. В левой нижней конечности определяются симптомы Бехтерева, Россолимо, Бабинского, Пуссепа. Суставно-мышечное чувство четко снижено в левой стопе, в правой — слегка. Левосторонняя гемигипалгезия. Снижение чувствительности по полиневритическому типу в ногах. Гиперестезия по корешковому типу Д2–Д8 справа. Атаксия кзади в позе Ромберга. Пальценосовая проба удовлетворительная, коленно-пяточная — затруднена больше слева, болезненность в окципитальных и шейных паравертебральных точках. Ограничение движений в шейном отделе позвоночника. Походка паретико-атактическая с элементами перонеальной слева.
Дополнительные исследования
Клинический анализ крови. Эр. — 3,0 х 1012/л; Hb — 93 г/л; СОЭ — 23 мм/ч; тромб. — 411,0; ЦП — 0,83; лейк. — 3,5 х 109/л; э. — 4 % , лимф. — 39 %, мон. — 10 %, п. — 2 %, с. — 45 %.
Коагулограмма. ПТИ — 73 %, фибрин плазмы — 3,6 г/л, толерантность к гепарину — 7 мин, фибр. В (–).
Печеночные пробы. Билирубин 8 мм/л.
КРЭГ. Уровень кровенаполнения полушарий резко повышен D < S, эластичность сосудистой стенки снижена. Затруднен венозный отток, умеренное повышение периферического сосудистого сопротивления преимущественно в каротидном бассейне справа.
КЭЭГ. Умеренное снижение биоэлектрической активности головного мозга диффузного характера, регистрируется θ-активность как спонтанно, так и после функциональных нагрузок преимущественно в правых лобно-височных отведениях.
ЭНМГ. Снижение амплитуды М-ответа преимущественно мышц нижних конечностей. Умеренное снижение распространения возбуждения по моторным и сенсорным волокнам малоберцовых нервов, S > D. Заключение: признаки мотосенсорной нейропатии нижних конечностей.
КТ головного мозга. Умеренное расширение конвекситальных субарахноидальных пространств. Заключение: умеренно выраженная наружная гидроцефалия.
МРТ головного мозга. Расширены боковые желудочки. Множественные субкортикальные и перивентрикулярные гиперинтенсивные очаги с нечеткими контурами, 0,5–0,7 см в диаметре. В правой лобно-височной области в проекции белого вещества визуализируется очаг ишемии без четких контуров размерами 1,5 х 1,7 х 1,5 см. Заключение: умеренно выраженная желудочковая (внутренняя) гидроцефалия, сосудистая лакунарная энцефалопатия, очаговое поражение (инсульт) правой лобно-височной области.
МРТ шейного и верхнегpудного отделов позвоночника в сагитальной проекции, с различной степенью контpастиpования тканей, в режимах Т1 и Т2 В/И + МР-миелогpафия. Визуализированы: спинной мозг от уровня большого затылочного отверстия до уровня D5, тела позвонков C1–D5, межпозвонковые диски.
Спинной мозг — контуры ровные четкие, структура гомогенная, умеренное понижение интенсивности сигнала в режиме Т1 В/И, на уровне C5 и D1 утолщен, множественные мелкие гиподенсивные участки.
Обращают на себя внимание выраженные дегенеративные изменения дисков: снижение их высоты, изменение структуры изображения за счет исчезновения высокоинтенсивного сигнала от ядер, задняя протрузия дисков С3–С4, С4–С5, С5–С6 до 3, 2 и 3 мм соответственно.
В режиме МР-миелографии ликворный сигнал прослеживается на всем протяжении, на уровне тел C7 и D1 — значительно деформирован.
Заключение: выраженные дегенеративные изменения дисков в шейном и верхнегрудном отделах позвоночника с задней протрузией дисков С3–С4, С4–С5, С5–С6, признаки ишемической миелопатии.
ЭКГ. Ритм синусовый, ЧСС — 80. ЭОС горизонтальная. Повышение биоэлектрической активности левого желудочка, неполная блокада правой ножки пучка Гиса, диффузные метаболические изменения миокарда.
Рентгенография шейного отдела позвоночника. Субхондральный склероз тел шейных позвонков С4–С7 с образованием краевых костных разрастаний, снижена высота межпозвонковых дисков С4–С5, С5–С6, С6–С7 наполовину.
УЗИ органов брюшной полости. Хронический бескаменный холецистит, мочекислый диатез, хронический пиелонефрит.
Консультации других специалистов:
— гематолог: болезнь Рандю — Ослера — Вебера, постгеморрагическая железодефицитная анемия;
— хирург: варикозная болезнь нижних конечностей, ХВН класс 1.
Основной диагноз. Рецидивирующие ишемические лакунарные инсульты (корково-подкорковой локализации) с формированием дисциркуляторной энцефалопатии (атеросклеротического и метаболического генеза) как проявление болезни Рандю — Ослера — Вебера с кохлеовестибулярным синдромом, тетрапарезом.
Сопутствующий диагноз. Остеохондроз шейного и верхнегpудного отделов позвоночника с множественными протрузиями дисков (С3–С4, С4–С5, С5–С6). Дисциркуляторная шейная миелопатия. Дисметаболическая полинейропатия нижних конечностей, сенсомоторная форма с явлениями сенситивной атаксии. Постгеморрагическая железодефицитная анемия. Варикозная болезнь нижних конечностей, ХВН класс 1.
Лечение: L-лизина эсцинат 10,0 внутривенно капельно № 10, мексидол 2,0 в/м № 10, далее переход на пероральный прием 250 мг/сутки, милдронат 5,0 в/м № 10, оксибрал по 60 мг в сутки, бетасерк по 16 мг в сутки, мильгамма 200 мг в сутки, диалипон 600 мг в сутки, вертигохеель, актиферрин.
Обсуждение
У нашей пациентки имели место патологические неврологические синдромы в виде снижения функции III, VII, VIII, ХII пар черепных нервов, комбинированных двигательных расстройств — спастический тетрапарез, более выраженный в левой половине тела, с элементами вялого в ногах, больше слева, комбинированных сенсорных расстройств — полиневритический, геми- и корешковый Д2–Д8 типы, вестибулоатактического синдрома. Указанная неврологическая симптоматика носила диффузный характер и свидетельствовала о поражении подкорковых отделов головного мозга преимущественно правой лобно-височной области, стволово-мозжечковых проводящих путей, спинного мозга на уровне шейного утолщения, периферических нервов нижних конечностей, межреберных грудных спинномозговых нервов (Д2–Д8).
Нейровизуализационные методы исследования подтвердили сосудистый характер процесса, так как обнаружили лакунарный статус и инсульт небольших размеров правой полушарно-подкорковой локализации, а также ишемические изменения в шейном отделе спинного мозга.
Развитию выявленных сосудистых нарушений у нашей больной способствовали многие факторы. Имеющийся дизрафический статус в сочетании с остеохондрозом позвоночника и множественными протрузиями дисков способствовал компрессии спинного мозга и развитию дисциркуляторной миелопатии. Кроме того, согласно литературным данным, при болезни Рандю — Ослера — Вебера формируются множественные артериовенозные анастомозы во внутренних органах, сосудистых сплетениях. Не исключена вероятность диапедезных геморрагий из артерио-венозных анастомозов, а также ишемических изменений на уровне шейных сегментов спинного мозга и подкорковых отделов головного мозга преимущественно правополушарная локализация малых инсультов препятствовала критической, адекватной оценке своего состояния здоровья со стороны больной и своевременной диагностике сосудистых осложнений.
Приведенное клиническое наблюдение свидетельствует о том, что при болезни Рандю — Ослера — Вебера наряду с висцеральными нередко развиваются неврологические осложнения. Они обусловлены множественными телеангиэктазиями интракраниальной и интраспинальной локализации, несостоятельностью их сосудистых стенок, ишемическими изменениями в прилежащей мозговой ткани.
Таким образом, НТГ могут протекать в виде цереброспинальной васкулярной формы, бессимптомных малых инсультов и способствовать прогрессирующей недостаточности церебрального и спинального кровообращения.
МКБ-10 код I78.0 | Наследственная геморрагическая телеангиэктазия
ICD-10
ICD-10 is the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD), a medical classification list by the World Health Organization (WHO).
It contains codes for diseases, signs and symptoms, abnormal findings, complaints, social circumstances, and external causes of injury or diseases.
ATC
The Anatomical Therapeutic Chemical (ATC) Classification System is used for the classification of active ingredients of drugs according to the organ or system on which they act and their therapeutic, pharmacological and chemical properties.
It is controlled by the World Health Organization Collaborating Centre for Drug Statistics Methodology (WHOCC).
DDD
The defined daily dose (DDD) is a statistical measure of drug consumption, defined by the World Health Organization (WHO).
It is used to standardize the comparison of drug usage between different drugs or between different health care environments.
Клиническая классификация ЛГ
Клиническая классификация ЛГ необходима для стандартизации диагностических подходов и лечебных мероприятий, проведения контролируемых исследований в однородных группах пациентов (таблица 1).
Таблица 1. Клиническая классификация легочной гипертензии (Российские рекомендации по диагностике и лечению ЛГ, 2007г.)
- Легочная артериальная гипертензия (ЛАГ):
- 1.1. Идиопатическая ЛГ (ИЛГ)
- 1.2. Семейная ЛАГ
- 1.3. Ассоциированная с:
- 1.3.1. коллагеновыми сосудистыми заболеваниями
- 1.3.2. врожденными пороками сердца (системно-легочные шунты)
- 1.3.3. портальной гипертензией
- 1.3.4. ВИЧ-инфекцией
- 1.3.5. лекарственными и токсическими воздействиями
- 1.3.6. другими (поражения щитовидной железы, болезнь Гошера, болезнь накопления гликогена, наследственная геморрагическая телеангиэктазия, гемоглобинопатии, миелопролиферативные болезни, спленэктомия)
- 1.4. Ассоциированная со значительным поражением вен или капилляров:
- 1.4.1. Легочная вено-окклюзионная болезнь
- 1.4.2. Легочный капиллярный гемангиоматоз
- 1.5. Персистирующая ЛАГ новорожденных
- Легочная гипертензия, ассоциированная с поражениями левых отделов сердца:
- 2.1. поражение левого желудочка
- 2.2. поражения клапанов левого желудочка
- ЛГ, ассоциированная с патологией дыхательной системы и /или гипоксемией
- 3.1.хроническая обструктивная болезнь легких
- 3.2. интерстициальные заболевания легких
- 3.3. нарушения дыхания во время сна
- 3.4. альвеолярная гиповентиляция
- 3.5.высокогорная ЛГ
- 3.6.нарушения развития легких
- ЛГ вследствие хронических тромботических или эмболических заболеваний:
- 4.1.тромбоэмболическая обструкция проксимальных ЛА
- 4.2. тромбоэмболическая обструкция дистального русла ЛА
- 4.3. нетромботические легочные эмболии (опухоли, паразитарные заболевания, инородные тела)
- Смешанные формы
- саркоидоз, гистиоцитоз Х, лимфангиоматоз, компрессия легочных сосудов (аденопатия, опухоли, фиброзирующий медиастинит)
Отдел системных гипертензий
Зав. отд. Мартынюк Тамила Витальевна +7(495) 414-64-50
Болезнь Ослера-Вебера-Ренду (наследственная геморрагическая телеангиэктазия): предпосылки, патофизиология, этиология
Джордано П., Ленато Г.М., Супресса П., Ластелла П., Дикуонцо Ф, Чиумаруло Л. и др. Наследственные геморрагические телеангиэктазии: артериовенозные мальформации у детей. J Педиатр . 2013 Июль 163 (1): 179-86.e1-3. [Медлайн].
Гутмахер А.Е., Марчук Д.А., Белый Р.И. мл. Наследственная геморрагическая телеангиэктазия. N Engl J Med .1995 Октябрь 5. 333 (14): 918-24. [Медлайн].
Нанда С., Бхатт СП. Наследственная геморрагическая телеангиэктазия: носовое кровотечение и кровохарканье. CMAJ . 2009 г., 14 апреля 180 (8): 838. [Медлайн].
Franchini M, Frattini F, Crestani S, Bonfanti C. Новые методы лечения носового кровотечения при наследственной геморрагической телеангиэктазии: систематический обзор клинического опыта применения талидомида. J Тромб Тромболизис . 2013 Октябрь, 36 (3): 355-7.[Медлайн].
Haitjema TJ, van Snippenburg R, Disch FJ, Overtoom TT, Westermann CJ. [Рецидивирующие носовые кровотечения: иногда болезнь Рендю-Ослера-Вебера]. Ned Tijdschr Geneeskd . 1996, 2 ноября. 140 (44): 2157-60. [Медлайн].
Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Возрастной клинический профиль наследственной геморрагической телеангиэктазии в популяции, набранной эпидемиологически. Am J Med Genet . 1989 марта 32 (3): 291-7.[Медлайн].
Porteous ME, Burn J, Proctor SJ. Наследственная геморрагическая телеангиэктазия: клинический анализ. J Med Genet . 1992 29 августа (8): 527-30. [Медлайн]. [Полный текст].
Ирани Ф., Касмани Р. Наследственная геморрагическая телеангиэктазия: утомляемость и одышка. CMAJ . 2009 г., 14 апреля 180 (8): 839. [Медлайн].
Шовлин К.Л., Гутмахер А.Е., Бускарини Е., Фаунан М.Э., Хайланд Р.Х., Вестерманн С.Дж. и др.Диагностические критерии наследственной геморрагической телеангиэктазии (синдром Ренду-Ослера-Вебера). Am J Med Genet . 2000 6 марта. 91 (1): 66-7. [Медлайн].
Шовлин К.Л., Хьюз Дж. М.. Наследственная геморрагическая телеангиэктазия. N Engl J Med . 1996 г., 1. 334 (5): 330-1; ответ автора 331-2. [Медлайн].
Шовлин С.Л., Летарте М. Наследственная геморрагическая телеангиэктазия и легочные артериовенозные мальформации: вопросы клинического ведения и обзор патогенетических механизмов. Грудь . 1999 г., 54 (8): 714-29. [Медлайн]. [Полный текст].
Коул С.Г., Бегби М.Э., Уоллес Г.М., Шовлин К.Л. Новый локус наследственной геморрагической телеангиэктазии (HHT3) отображается на хромосоме 5. J Med Genet . 2005 июл.42 (7): 577-82. [Медлайн]. [Полный текст].
Пригода Н.Л., Савас С., Абдалла С.А., Пиовесан Б., Рушлов Д., Вандезанде К. и др. Наследственная геморрагическая телеангиэктазия: обнаружение мутаций, чувствительность тестов и новые мутации. J Med Genet . 2006 Сентябрь 43 (9): 722-8. [Медлайн].
Zucco L, Zhang Q, Kuliszewski MA, Kandic I, Faughnan ME, Stewart DJ, et al. Дисфункция циркулирующих ангиогенных клеток у пациентов с наследственной геморрагической телеангиэктазией. PLoS One . 2014. 9 (2): e89927. [Медлайн]. [Полный текст].
Choi EJ, Chen W, Jun K, Arthur HM, Young WL, Su H. Новые мышиные модели артериовенозной мальформации мозга для наследственной геморрагической телеангиэктазии 1 типа. PLoS One . 2014. 9 (2): e88511. [Медлайн]. [Полный текст].
Браверман И.М., Кех А., Якобсон Б.С. Ультраструктура и трехмерная организация телеангиэктазий наследственных геморрагических телеангиэктазий. Дж Инвест Дерматол . 1990 Октябрь 95 (4): 422-7. [Медлайн].
van Laake LW, van den Driesche S, Post S, Feijen A, Jansen MA, Driessens MH, et al. Эндоглин играет решающую роль в восстановлении сосудов, опосредованном клетками крови. Тираж . 2006 21 ноября. 114 (21): 2288-97. [Медлайн].
Fernandez-L A, Garrido-Martin EM, Sanz-Rodriguez F, Pericacho M, Rodriguez-Barbero A, Eleno N, et al. Фингерпринт экспрессии генов при наследственной геморрагической телеангиэктазии у человека. Хум Мол Генет . 2007 г. 1. 16 (13): 1515-33. [Медлайн].
McDonald J, Bayrak-Toydemir P, Pyeritz RE. Наследственная геморрагическая телеангиэктазия: обзор диагностики, лечения и патогенеза. Генет Мед . 2011 июл.13 (7): 607-16. [Медлайн].
Дэвид Л., Маллет С., Мазебург С., Фейдж Дж., Байли С. Идентификация BMP9 и BMP10 в качестве функциональных активаторов киназы 1, подобной рецептору сиротского активина (ALK1), в эндотелиальных клетках. Кровь . 2007. 109 (5): 1953-61. [Медлайн].
Jerkic M, Sotov V, Letarte M. Окислительный стресс способствует эндотелиальной дисфункции на мышиных моделях наследственной геморрагической телеангиэктазии. Oxid Med Cell Longev . 2012. 2012: 686972. [Медлайн]. [Полный текст].
Шовлин К.Л., Хьюз Дж. М., Скотт Дж., Зайдман К. Э., Зайдман Дж. Г.. Характеристика эндоглина и выявление новых мутаций при наследственной геморрагической телеангиэктазии. Am J Hum Genet . 1997 июл.61 (1): 68-79. [Медлайн]. [Полный текст].
Гровер С., Гревал Р.С., Верма Р., Сахни Х., Муралидхар Р., Синха П. Синдром Ослера-Вебера-Ренду: клинический случай с семейной кластеризацией. Индийский J Дерматол Венереол Лепрол . 2009 Янв-Фев. 75 (1): 100-1. [Медлайн].
Ричардс-Ютц Дж., Грант К., Чао ЕС, Вальтер С.Е., Гангули А. Последние сведения о молекулярной диагностике наследственной геморрагической телеангиэктазии. Хум Генет . 2010 Июль 128 (1): 61-77. [Медлайн].
Гедж Ф., Макдональд Дж., Фансалкар А., Чоу Л.С., Кальдерон Ф., Мао Р. и др. Клиническая и аналитическая чувствительность при тестировании наследственной геморрагической телеангиэктазии и отчет о мутациях de novo. Дж Мол Диаг . 2007 апр. 9 (2): 258-65. [Медлайн]. [Полный текст].
Макаллистер К.А., Грогг К.М., Джонсон Д.В., Галлион С.Дж., Болдуин М.А., Джексон К.Э. и др. Эндоглин, белок эндотелиальных клеток, связывающий TGF-бета, является геном наследственной геморрагической телеангиэктазии типа 1. Nat Genet . 1994 Декабрь 8 (4): 345-51. [Медлайн].
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Мутации в гене активин-рецептор-подобной киназы 1 при наследственной геморрагической телеангиэктазии 2 типа. Нат Генет . 13 июня 1996 г. (2): 189-95. [Медлайн].
Галлионе С.Дж., Репетто Г.М., Легиус Э., Рустги А.К., Шелли С.Л., Тейпар С. и др. Комбинированный синдром ювенильного полипоза и наследственной геморрагической телеангиэктазии, связанный с мутациями в MADh5 (SMAD4). Ланцет . 2004 13 марта. 363 (9412): 852-9. [Медлайн].
Абдалла С.А., Летарте М. Наследственная геморрагическая телеангиэктазия: современные взгляды на генетику и механизмы заболевания. J Med Genet . 2006 Февраль 43 (2): 97-110. [Медлайн]. [Полный текст].
Ригельский К.М., Дженнингс К., Лехтонен Р., Минай О.А., Энг С., Олдред М.А. Мутация BMPR2 у пациента с легочной артериальной гипертензией и подозрением на наследственную геморрагическую телеангиэктазию. Am J Med Genet A . 1 октября 2008 г. 146A (19): 2551-6. [Медлайн].
Бегби М.Э., Уоллес Г.М., Шовлин К.Л. Наследственная геморрагическая телеангиэктазия (синдром Ослера-Вебера-Ренду): взгляд из 21 века. Postgrad Med J . 2003 января 79 (927): 18-24. [Медлайн]. [Полный текст].
Kjeldsen AD, Oxhøj H, Andersen PE, Elle B, Jacobsen JP, Vase P. Легочные артериовенозные мальформации: процедуры скрининга и легочная ангиография у пациентов с наследственной геморрагической телеангиэктазией. Сундук . 1999 августа 116 (2): 432-9. [Медлайн].
Дакеиси М., Сиоя Т., Вада Й., Синдо Т., Отака К., Манабе М. и др. Генетическая эпидемиология наследственной геморрагической телеангиэктазии в местном сообществе в северной части Японии. Хум Мутат . 2002, 19 февраля (2): 140-8. [Медлайн].
Kjeldsen AD, Vase P, Green A. Наследственная геморрагическая телеангиэктазия: популяционное исследование распространенности и смертности датских пациентов. Дж. Интерн Мед. . 1999, январь, 245 (1): 31-9. [Медлайн].
Jessurun GA, Kamphuis DJ, van der Zande FH, Nossent JC. Церебральные артериовенозные мальформации на Нидерландских Антильских островах. Высокая распространенность наследственных геморрагических телеангиэктазий, связанных с единичными и множественными артериовенозными мальформациями головного мозга. Clin Neurol Neurosurg . 1993 Сентябрь 95 (3): 193-8. [Медлайн].
Вестерманн С.Дж., Розина А.Ф., Де Фриз V, де Кото, Пенсильвания. Распространенность и проявления наследственной геморрагической телеангиэктазии у афро-карибского населения Нидерландских Антильских островов: семейный скрининг. Am J Med Genet A . 2003 г. 1 февраля. 116A (4): 324-8. [Медлайн].
Vase P, Grove O. Поражения желудочно-кишечного тракта при наследственной геморрагической телеангиэктазии. Гастроэнтерология . 1986 ноябрь 91 (5): 1079-83. [Медлайн].
Урушихара М., Фурукава С., Ота А., Иваи А., Мацумура К., Хамада Ю. Геморрагическая телеангиэктазия с тромбоцитопенией у новорожденного. Педиатр Инт . 2000 Декабрь 42 (6): 693-5. [Медлайн].
Mitchell RN. Кровеносный сосуд. Кумар В., Аббас А.К., Астер Дж.С., ред. Патологические основы болезни Роббинса и Котрана . 9 изд. Филадельфия: Эльзевьер Сондерс; 2015 г.483-522.
Sabbà C, Pasculli G, Suppressa P, D’Ovidio F, Lenato GM, Resta F, et al. Ожидаемая продолжительность жизни пациентов с наследственной геморрагической телеангиэктазией. QJM . 2006 май. 99 (5): 327-34. [Медлайн].
Fulbright RK, Chaloupka JC, Putman CM, Sze GK, Merriam MM, Lee GK и др. МР наследственной геморрагической телеангиэктазии: распространенность и спектр цереброваскулярных пороков развития. AJNR Am J Neuroradiol . 1998 Март.19 (3): 477-84. [Медлайн].
Ребейз Э.Е., Брайан Д.Д., Эрлихман Р.Дж., Шапшай С.М. Хирургическое лечение опасных для жизни носовых кровотечений при болезни Ослера-Вебера-Ренду. Энн Пласт Сург . 1995 августа 35 (2): 208-13. [Медлайн].
Хоаг Дж. Б., Терри П., Митчелл С., Рех Д., Мерло, Калифорния. Оценка тяжести носового кровотечения при наследственной геморрагической телеангиэктазии. Ларингоскоп . 2010 апр. 120 (4): 838-43. [Медлайн].
Шовлин С.Л., Чамали Б., Сантирапала В., Ливси Дж. А., Ангус Г., Мэннинг Р. и др.Ишемический инсульт у пациентов с легочными артериовенозными мальформациями и наследственной геморрагической телеангиэктазией: ассоциации с дефицитом железа и тромбоцитами. PLoS One . 2014. 9 (2): e88812. [Медлайн]. [Полный текст].
van Gent MW, Post MC, Snijder RJ, Westermann CJ, Plokker HW, Mager JJ. Реальная распространенность легочного шунта справа налево в зависимости от генотипа у пациентов с наследственной геморрагической телеангиэктазией: исследование трансторакальной контрастной эхокардиографии. Сундук . 2010 Октябрь 138 (4): 833-9. [Медлайн].
Lacombe P, Lagrange C, Beauchet A, El Hajjam M, Chinet T, Pelage JP. Диффузные легочные артериовенозные мальформации при наследственной геморрагической телеангиэктазии: отдаленные результаты эмболизации в зависимости от степени поражения легких. Сундук . 2009 апр. 135 (4): 1031-1037. [Медлайн].
Якоби П., Вайнер З., Бест Л., Ицковиц-Элдор Дж. Наследственная геморрагическая телеангиэктазия с легочными артериовенозными мальформациями. Акушерский гинекол . 2001 Май. 97 (5 Пет 2): 813-4. [Медлайн].
Cottin V, Dupuis-Girod S, Lesca G, Cordier JF. Легочно-сосудистые проявления наследственной геморрагической телеангиэктазии (болезнь Рендю-Ослера). Дыхание . 2007. 74 (4): 361-78. [Медлайн].
Берг Дж., Портеус М., Рейнхардт Д., Галлион С., Холлоуэй С., Умасунтар Т. и др. Наследственная геморрагическая телеангиэктазия: исследование на основе вопросника для определения различных фенотипов, вызванных мутациями эндоглина и ALK1. J Med Genet . 2003 г., 40 (8): 585-90. [Медлайн]. [Полный текст].
McDonald MJ, Brophy BP, Kneebone C. Синдром Ренду-Ослера-Вебера: современная перспектива церебральных проявлений. Дж. Clin Neurosci . 1998 июл.5 (3): 345-50. [Медлайн].
Poisson A, Vasdev A, Brunelle F, Plauchu H, Dupuis-Girod S, французско-итальянская сеть HHT. Острая параплегия из-за спинальной артериовенозной фистулы у двух пациентов с наследственной геморрагической телеангиэктазией. Eur J Pediatr . 2009 Февраль 168 (2): 135-9. [Медлайн].
Калхун А.Р., Болло Р.Дж., Гарбер С.Т., Макдональд Дж., Стивенсон Д.А., Хунг И.Х. и др. Спинальные артериовенозные свищи у детей с наследственной геморрагической телеангиэктазией. J Нейрохирург-педиатр . 2012 июн.9 (6): 654-9. [Медлайн].
Гуденбергер ДМ. Висцеральные проявления наследственной геморрагической телеангиэктазии. Trans Am Clin Climatol Assoc . 2004. 115: 185-99.[Медлайн]. [Полный текст].
Ремер В., Бурк М., Шнайдер В. [Наследственная геморрагическая телеангиэктазия (болезнь Ослера)]. Dtsch Med Wochenschr . 1992 24 апреля. 117 (17): 669-75. [Медлайн].
AAssar OS, Friedman CM, White RI Jr. Естественное течение носового кровотока при наследственной геморрагической телеангиэктазии. Ларингоскоп . 1991 сентябрь 101 (9): 977-80. [Медлайн].
Уайт Р.И. Младший, Линч-Найхан А., Терри П., Бюшер П.К., Фармлетт Э.Дж., Чарнас Л. и др.Легочные артериовенозные мальформации: методы и отдаленные результаты эмболотерапии. Радиология . 1988 Декабрь 169 (3): 663-9. [Медлайн].
Берг Ю.Н., Гутмахер А.Е., Марчук Д.А., Портеус М.Э. Клиническая неоднородность наследственной геморрагической телеангиэктазии: являются ли легочные артериовенозные мальформации более распространенными в семьях, связанных с эндоглином? J Med Genet . 1996 Mar.33 (3): 256-7. [Медлайн]. [Полный текст].
Коттин В., Шине Т., Лаволе А., Корре Р., Маршан Э., Рейно-Гобер М. и др.Легочные артериовенозные мальформации при наследственной геморрагической телеангиэктазии: серия 126 пациентов. Медицина (Балтимор) . 2007 январь 86 (1): 1-17. [Медлайн].
Pierucci P, Murphy J, Henderson KJ, Chyun DA, White RI Jr. Новое определение и естественная история пациентов с диффузными легочными артериовенозными мальформациями: опыт двадцати семи лет. Сундук . 2008 Март 133 (3): 653-61. [Медлайн].
Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I и др.Клинико-молекулярно-генетические особенности легочной гипертензии у пациентов с наследственной геморрагической телеангиэктазией. N Engl J Med . 2 августа 2001 г. 345 (5): 325-34. [Медлайн].
Press OW, Ramsey PG. Инфекции центральной нервной системы, связанные с наследственной геморрагической телеангиэктазией. Am J Med . 1984 июл.77 (1): 86-92. [Медлайн].
Гарсия-Монако Р., Тейлор В., Родеш Г., Альварес Н., Берроуз П., Кубес П. и др.Пиальный артериовенозный свищ у детей как проявление болезни Ренду-Ослера-Вебера. Нейрорадиология . 1995, январь, 37 (1): 60-4. [Медлайн].
Krings T, Chng SM, Ozanne A, Alvarez H, Rodesch G, Lasjaunias PL. Наследственная геморрагическая телеангиэктазия у детей: эндоваскулярное лечение сосудисто-нервных мальформаций: результаты у 31 пациента. Нейрорадиология . 2005 декабрь 47 (12): 946-54. [Медлайн].
Easey AJ, Уоллес GM, Хьюз JM, Джексон JE, Тейлор WJ, Шовлин CL.Следует ли обследовать бессимптомных пациентов с наследственной геморрагической телеангиэктазией (ГТТ) на наличие пороков развития сосудов головного мозга? Данные 22061 года жизни пациентов с HHT. J Neurol Neurosurg Psychiatry . 2003 июн. 74 (6): 743-8. [Медлайн]. [Полный текст].
Maher CO, Piepgras DG, Brown RD Jr, Friedman JA, Pollock BE. Цереброваскулярные проявления в 321 случае наследственной геморрагической телеангиэктазии. Инсульт . 2001 апр. 32 (4): 877-82. [Медлайн].
Cloft HJ. Спонтанный регресс церебральной артериовенозной мальформации при наследственной геморрагической телеангиэктазии. AJNR Am J Neuroradiol . 2002 июн-июль. 23 (6): 1049-50. [Медлайн].
Du R, Hashimoto T, Tihan T, Young WL, Perry V, Lawton MT. Рост и регресс артериовенозных мальформаций у пациента с наследственной геморрагической телеангиэктазией. История болезни. Дж Нейросург . 2007 Март 106 (3): 470-7. [Медлайн].
Леунг К.М., Агид Р., ТерБрюгге К. Спонтанный регресс церебральной артериовенозной мальформации у ребенка с наследственной геморрагической телеангиэктазией. История болезни. Дж Нейросург . 2006 ноябрь 105 (5 приложение): 428-31. [Медлайн].
Проктор Д.Д., Хендерсон К.Дж., Дзиура Д.Д., Лонгакр А.В., Уайт Р.И. мл. Энтероскопическая оценка желудочно-кишечного тракта у пациентов с симптомами наследственной геморрагической телеангиэктазии. Дж Клин Гастроэнтерол .2005 Февраль 39 (2): 115-9. [Медлайн].
Абдалла С.А., Гейстхофф Ю.В., Бонно Д., Плаучу Х., Макдональд Дж., Кеннеди С. и др. Висцеральные проявления при наследственной геморрагической телеангиэктазии 2 типа. J Med Genet . 2003 июл. 40 (7): 494-502. [Медлайн]. [Полный текст].
van Cutsem E, Rutgeerts P, Vantrappen G. Лечение кровоточащих пороков развития сосудов желудочно-кишечного тракта с помощью эстроген-прогестерона. Ланцет . 1990 21 апреля.335 (8695): 953-5. [Медлайн].
Wu JS, Saluja S, Garcia-Tsao G, Chong A, Henderson KJ, White RI Jr. Поражение печени при наследственной геморрагической телеангиэктазии: КТ и клинические данные не коррелируют у пациентов с симптомами. AJR Am J Roentgenol . 2006 Октябрь 187 (4): W399-405. [Медлайн].
Гарсия-Цао Г. Поражение печени при наследственной геморрагической телеангиэктазии (HHT). Дж Гепатол . 2007 Mar.46 (3): 499-507.[Медлайн].
Garcia-Tsao G, Swanson KL. Сосудистые мальформации печени при наследственной геморрагической телеангиэктазии: в поисках предикторов значимого заболевания. Гепатология . 2008 ноябрь 48 (5): 1377-9. [Медлайн].
Ларсон А.М. Заболевания печени при наследственной геморрагической телеангиэктазии. Дж Клин Гастроэнтерол . 2003 Февраль 36 (2): 149-58. [Медлайн].
Гарсия-Цао Дж., Корзеник Дж. Р., Янг Л., Хендерсон К. Дж., Джайн Д., Берд Б. и др.Заболевания печени у пациентов с наследственной геморрагической телеангиэктазией. N Engl J Med . 2000, 28 сентября. 343 (13): 931-6. [Медлайн].
Ализад А, Сьюард Дж.Б. Эхокардиографические особенности генетических заболеваний: часть 3. Шунты. J Am Soc Echocardiogr . 2000 марта, 13 (3): 248-53. [Медлайн].
Mohler ER 3rd, Doraiswamy V, Sibley A, Bernhardt BA, Pyeritz RE. Просвечивание пальцев при сосудистых аномалиях: новый метод оценки наследственной геморрагической телеангиэктазии. Генет Мед . 2009 Май. 11 (5): 356-8. [Медлайн].
Шовлин С.Л., Соди В., Маккарти А., Ласьяуниас П., Джексон Дж. Э., Шеппард М.Н. Оценка материнского риска беременности для женщин с наследственной геморрагической телеангиэктазией (синдром Ослера-Вебера-Ренду): предлагаемый подход для оказания акушерских услуг. БЖОГ . 2008 августа 115 (9): 1108-15. [Медлайн].
Gershon AS, Faughnan ME, Chon KS, Pugash RA, Clark JA, Bohan MJ, et al.Транскатетерная эмболотерапия легочных артериовенозных мальформаций матери при беременности. Сундук . 2001, февраль, 119 (2): 470-7. [Медлайн].
Халид С.К., Першбахер Дж., Макан М., Барзилай Б., Гуденбергер Д. Ухудшение носового кровотечения предвещает высокий сердечный выброс при наследственной геморрагической телеангиэктазии. Am J Med . 2009 август 122 (8): 779.e1-9. [Медлайн].
Нантакумар К., Грэм А.Т., Робинсон Т.И., Гранде П., Пугаш Р.А., Кларк Дж. А. и др.Контрастная эхокардиография для выявления легочных артериовенозных мальформаций. Am Heart J . 2001 февраль 141 (2): 243-6. [Медлайн].
Schneider G, Uder M, Koehler M, Kirchin MA, Massmann A, Buecker A, et al. МР-ангиография для выявления легочных артериовенозных мальформаций у пациентов с наследственной геморрагической телеангиэктазией. AJR Am J Roentgenol . 2008 апр. 190 (4): 892-901. [Медлайн].
Шовлин С.Л., Сулейман Н.Л., Говани Ф.С., Джексон Дж. Э., Бегби М.Э.Повышенный фактор VIII при наследственной геморрагической телеангиэктазии (НГТ): связь с венозной тромбоэмболией. Тромб Хемост . 2007 ноябрь 98 (5): 1031-9. [Медлайн].
De Pascalis A, Napoli M, Aprile M, Antonaci A, D’Amelio A, Buongiorno E. Макрогематурия вследствие приобретенной гемофилии при наследственной геморрагической телеангиэктазии. Фибринолиз свертывания крови . 2008 октября 19 (7): 731-3. [Медлайн].
Faughnan ME, Hyland RH, Nanthakumar K, Redelmeier DA.Скрининг пациентов с наследственной геморрагической телеангиэктазией. Сундук . 2000 августа 118 (2): 566-7. [Медлайн].
Томпсон Р.Д., Джексон Дж., Петерс А.М., Доре С.Дж., Хьюз Дж. М.. Чувствительность и специфичность радиоизотопных измерений правого-левого шунта и пульсоксиметрии для раннего выявления легочных артериовенозных мальформаций. Сундук . 1999, январь, 115 (1): 109-13. [Медлайн].
Мэнсон Д., Траубичи Дж., Мей-Захав М., Макласки И., Джон П., Стивенс Д.Узелковые помутнения в легких у детей с наследственной геморрагической телеангиэктазией. Педиатр Радиол . 2007 марта 37 (3): 264-8. [Медлайн].
Аль-Салех С., Драгулеску А., Мэнсон Д., Голдинг Ф., Траубичи Дж., Мей-Захав М. и др. Применение контрастной эхокардиографии для скрининга легочной артериовенозной мальформации при наследственной геморрагической телеангиэктазии у детей. J Педиатр . 2012 июн. 160 (6): 1039-43.e1. [Медлайн].
Milot L, Kamaoui I, Gautier G, Pilleul F.Наследственно-геморрагическая телеангиэктазия: одноэтапное магнитно-резонансное исследование для оценки поражения печени. Гастроэнтерол Clin Biol . 2008 авг-сен. 32 (8-9): 677-85. [Медлайн].
Suga K, Ishikawa Y, Matsunaga N, Tanaka N, Suda H, Handa T. Участие печени в наследственной геморрагической телеангиэктазии: оценка с помощью радионуклидной ангиографии с 99Tcm-фитатом и трансректальной портальной сцинтиграфии 123I-IMP. Br J Радиол . 2000 Октябрь 73 (874): 1115-9.[Медлайн].
Zukotynski K, Chan RP, Chow CM, Cohen JH, Faughnan ME. Оценка контрастной эхокардиографии позволяет прогнозировать легочные артериовенозные мальформации на КТ. Сундук . 2007 июл.132 (1): 18-23. [Медлайн].
Wooderchak W, Gedge F, McDonald M, Krautscheid P, Wang X, Malkiewicz J, et al. Наследственная геморрагическая телеангиэктазия: две отдельные делеции ENG в одной семье. Clin Genet . 2010 ноябрь 78 (5): 484-9. [Медлайн].
[Рекомендации] Адлер Д.Г., Лейтон Дж. А., Давила Р. Э., Хирота В. К., Якобсон BC, Куреши В. А. и др. Рекомендации ASGE: Роль эндоскопии при остром не варикозном кровотечении из верхних отделов ЖКТ. Гастроинтест Endosc . 2004 Октябрь 60 (4): 497-504. [Медлайн].
Pennazio M, Keuchel M, Jensen DM, Dulai GS. Артериовенозные заболевания. Кеучел М., Хагенмюллер Ф, Тадзири Х., ред. Видеокапсульная эндоскопия: Справочное руководство и Атлас . Берлин: Springer-Verlag; 2014 г.193-204.
Ingrosso M, Sabbà C, Pisani A, Principi M, Gallitelli M, Cirulli A и др. Доказательства вовлечения тонкой кишки в наследственную геморрагическую телеангиэктазию: капсульно-эндоскопическое исследование. Эндоскопия . 2004 Декабрь 36 (12): 1074-9. [Медлайн].
Grève E, Moussata D, Gaudin JL, Lapalus MG, Giraud S, Dupuis-Girod S, et al. Высокая диагностическая и клиническая эффективность эндоскопии капсулы тонкой кишки у пациентов с наследственной геморрагической телеангиэктазией с явным пищеварительным кровотечением и / или тяжелой анемией. Гастроинтест Endosc . 2010 Апрель 71 (4): 760-7. [Медлайн].
Рамчандани М., Редди Д. Н., Гупта Р., Лахтакиа С., Тандан М., Дарисетти С. и др. Спиральная энтероскопия: предварительный опыт среди азиатского населения. Дж Гастроэнтерол Гепатол . 2010 25 ноября (11): 1754-7. [Медлайн].
Якобсон Б.С. Наследственная геморрагическая телеангиэктазия: модель роста и расширения кровеносных сосудов. Ам Дж. Патол . 2000 Март 156 (3): 737-42.[Медлайн]. [Полный текст].
Робин Э.Д., Ламан Д., Хорн Б.Р., Теодор Дж. Платипноэ, связанное с ортодеоксией, вызванной истинным сосудистым шунтированием легких. N Engl J Med . 1976, 22 апреля. 294 (17): 941-3. [Медлайн].
Харрис П.Г., Брокбэнк М.Дж., Шекспир П.Г., Каррут Дж. Лечение наследственной геморрагической телеангиэктазии импульсным лазером на красителе. Дж Ларингол Отол . 1997 ноябрь 111 (11): 1038-41. [Медлайн].
Харрисон Д.Ф.Использование эстрогенов в лечении семейной геморрагической телеангиэктазии. Ларингоскоп . 1982 Март 92 (3): 314-20. [Медлайн].
Даллас Н.А., Самуэль С., Ся Л., Фан Фан, Грей М.Дж., Лим С.Дж. и др. Эндоглин (CD105): маркер сосудистой сети опухоли и потенциальная мишень для терапии. Clin Cancer Res . 2008 г., 1. 14 (7): 1931-7. [Медлайн].
Роберт Ф., Дерош-Кастан А., Байи С., Дюпюи-Жирод С., Фейдж Дж. Дж. Будущие методы лечения наследственной геморрагической телеангиэктазии. Орфанет J Редкие диски . 2020 7 января. 15 (1): 4. [Медлайн].
[Рекомендации] Фаунан М.Э., Магер Дж. Дж., Хеттс С.В., Палда В.А., Ланг-Робертсон К., Бускарини Э. и др. Второе Международное руководство по диагностике и лечению наследственной геморрагической телеангиэктазии. Энн Интерн Мед. . 2020 8 сентября [Medline]. [Полный текст].
Шах РК, Дхингра ДжК, Шапшай СМ. Наследственная геморрагическая телеангиэктазия: обзор 76 случаев. Ларингоскоп .2002 май. 112 (5): 767-73. [Медлайн].
Янив Э., Прейс М., Хадар Т., Шверо Дж., Хаддад М. Антиэстрогенная терапия наследственной геморрагической телеангиэктазии: двойное слепое плацебо-контролируемое клиническое испытание. Ларингоскоп . 2009 Февраль 119 (2): 284-8. [Медлайн].
Айзекс Э. Аминокапроновая кислота. Справочник по дозировке лекарственных средств для детей . 8-е изд. Оттава, Канада: Центр медицинских наук Виннипега и CSHP; 1998. 161.
Лебрин Ф., Срун С., Раймонд К., Мартин С., ван ден Бринк С., Фрейтас С. и др.Талидомид стимулирует созревание сосудов и уменьшает носовое кровотечение у людей с наследственной геморрагической телеангиэктазией. Нат Мед . 2010 Апрель 16 (4): 420-8. [Медлайн].
Harvey RJ, Kanagalingam J, Lund VJ. Влияние септодермопластики и калий-титанилфосфатной (KTP) лазерной терапии в лечении носовых кровотечений, связанных с наследственной геморрагической телеангиэктазией. Am J Rhinol . 2008 март-апрель. 22 (2): 182-7. [Медлайн].
Лесник Г.Т., Росс Д.А., Хендерсон К.Дж., Джо Дж. К., Ледер С.Б., Уайт Р.И. мл.Септэктомия и дермопластика перегородки для лечения тяжелых трансфузионных носовых кровотечений у пациентов с наследственной геморрагической телеангиэктазией и перфорацией перегородки. Am J Rhinol . 2007 май-июнь. 21 (3): 312-5. [Медлайн].
Layton KF, Kallmes DF, Gray LA, Cloft HJ. Эндоваскулярное лечение носовых кровотечений у пациентов с наследственной геморрагической телеангиэктазией. AJNR Am J Neuroradiol . 2007 май. 28 (5): 885-8. [Медлайн].
de Gussem EM, Snijder RJ, Disch FJ, Zanen P, Westermann CJ, Mager JJ.Влияние N-ацетилцистеина на носовое кровотечение и качество жизни у пациентов с HHT: пилотное исследование. Ринология . 2009 Март 47 (1): 85-8. [Медлайн].
Bose P, Holter JL, Selby GB. Бевацизумаб при наследственной геморрагической телеангиэктазии. N Engl J Med . 2009 14 мая. 360 (20): 2143-4. [Медлайн].
Massoud OI, Youssef WI, Mullen KD. Разрешение наследственной геморрагической телеангиэктазии и анемии с помощью длительной терапии альфа-интерфероном хронического гепатита С. Дж Клин Гастроэнтерол . 2004 апр. 38 (4): 377-9. [Медлайн].
Уитли-Прайс П., Шовлин С., Чао Д. Интерферон при метастатическом почечно-клеточном раке, вызывающем регресс наследственной геморрагической телеангиэктазии. Дж Клин Гастроэнтерол . 2005 апр. 39 (4): 344-5. [Медлайн].
Lupu A, Stefanescu C, Treton X, Attar A, Corcos O, Bouhnik Y. Бевацизумаб как средство экстренной помощи при тяжелом рецидивирующем желудочно-кишечном кровотечении при наследственной геморрагической телеангиэктазии. Дж Клин Гастроэнтерол . 2013 Март 47 (3): 256-7. [Медлайн].
Шовлин С.Л., Джексон Дж.Э., Бэмфорд КБ, Дженкинс И.Х., Бенджамин А.Р., Рамадан Х. и др. Первичные детерминанты риска ишемического инсульта / абсцесса головного мозга не зависят от тяжести легочных артериовенозных мальформаций при наследственной геморрагической телеангиэктазии. Грудь . Март 2008 г., 63 (3): 259-66. [Медлайн].
Шовлин К.Л., Винсток А.Р., Петерс А.М., Джексон Дж. Э., Хьюз Дж. М..Медицинские осложнения беременности при наследственной геморрагической телеангиэктазии. QJM . 1995 Декабрь 88 (12): 879-87. [Медлайн].
Buscarini E, Plauchu H, Garcia Tsao G, White RI Jr, Sabbà C, Miller F и др. Поражение печени при наследственной геморрагической телеангиэктазии: согласованные рекомендации. Печень Инт . 2006 26 ноября (9): 1040-6. [Медлайн].
Лерут Дж., Орландо Дж., Адам Р., Сабба С., Пфицманн Р., Клемпнауэр Дж. И др.Трансплантация печени при наследственной геморрагической телеангиэктазии: отчет Европейского реестра трансплантатов печени. Энн Сург . 2006 декабрь 244 (6): 854-62; обсуждение 862-4. [Медлайн]. [Полный текст].
Thevenot T, Vanlemmens C, Di Martino V, Becker MC, Denue PO, Kantelip B и др. Трансплантация печени при сердечной недостаточности у пациентов с наследственной геморрагической телеангиэктазией. Трансплантация печени . 2005 июл.11 (7): 834-8. [Медлайн].
Митчелл А., Адамс Л.А., Маккуиллан Дж., Тибболлс Дж., Ванден Дризен Р., Делривьер Л.Бевацизумаб отменяет необходимость трансплантации печени при наследственной геморрагической телеангиэктазии. Трансплантация печени . 2008 14 февраля (2): 210-3. [Медлайн].
Кюри А., Леска Дж., Коттин В., Эдери П., Беллон Дж., Фогнан М. Э. и др. Длительное наблюдение за 12 детьми с легочными артериовенозными мальформациями: подтверждение наследственной геморрагической телеангиэктазии во всех случаях. J Педиатр . 2007 Сентябрь 151 (3): 299-306. [Медлайн].
Наследственная геморрагическая телеангиэктазия: обзор диагностики, лечения и патогенеза
Sutton HG.Носовые кровотечения как признак нарушения питания и дегенерации сосудистой системы. Med Mirror 1864; 769–781.
Babington BG. Наследственное носовое кровотечение. Ланцет 1865; 2 : 362–353.
Google Scholar
Марчук Д.А., Гутмахер А.Е., Пеннер Дж. А., Гангули П. Отчет о семинаре по наследственной геморрагической телеангиэктазии, 10–11 июля 1997 г. Am J Med Genet 1998; 76 : 269–273.
CAS
PubMed
Google Scholar
Guttmacher A, Marchuck D, Pyeritz RE. Наследственная геморрагическая телеангиэктазия, 5 изд. Филадельфия, Черчилль Ливингстон, 2007.
Ассар А. Естественное течение носового кровотока при наследственной геморрагической телеангиэктазии. Am J Gastroenterol 1991; 101 : 977–980.
Google Scholar
Берг Дж., Портеус М., Рейнхардт Д. и др.Наследственная геморрагическая телеангиэктазия: исследование на основе вопросника для определения различных фенотипов, вызванных мутациями эндоглина и ALK1. J Med Genet 2003; 40 : 585–590.
CAS
PubMed Central
PubMed
Google Scholar
Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Возрастной клинический профиль наследственной геморрагической телеангиэктазии в популяции, набранной эпидемиологически. Am J Med Genet 1989; 32 : 291–297.
CAS
PubMed
Google Scholar
Porteous ME, Burn J, Proctor SJ. Наследственная геморрагическая телеангиэктазия: клинический анализ. J Med Genet 1992; 29 : 527–530.
CAS
PubMed Central
PubMed
Google Scholar
Морган Т., Макдональд Дж., Андерсон С. и др.Внутричерепное кровоизлияние у младенцев и детей с наследственной геморрагической телеангиэктазией (синдром Ослера-Вебера-Ренду). Педиатрия 2002; 109 : E12.
PubMed
Google Scholar
Kjeldsen AD, Kjeldsen J. Желудочно-кишечные кровотечения у пациентов с наследственной геморрагической телеангиэктазией. Am J Gastroenterol 2000; 95 : 415–418.
CAS
PubMed
Google Scholar
Longacre AV, Gross CP, Gallitelli M, Henderson KJ, White RI Jr, Proctor DD.Диагностика и лечение желудочно-кишечных кровотечений у пациентов с наследственной геморрагической телеангиэктазией. Am J Gastroenterol 2003; 98 : 59–65.
PubMed
Google Scholar
Ingrosso M, Sabba C, Pisani A, et al. Доказательства вовлечения тонкой кишки в наследственную геморрагическую телеангиэктазию: капсульно-эндоскопическое исследование. Эндоскопия 2004; 36 : 1074–1079.
CAS
PubMed
Google Scholar
Проктор Д.Д., Хендерсон К.Дж., Дзиура Д.Д., Лонгакр А.В., Уайт Р.И. мл., Энтероскопическая оценка желудочно-кишечного тракта у симптомных пациентов с наследственной геморрагической телеангиэктазией. J Clin Gastroenterol 2005; 39 : 115–119.
PubMed
Google Scholar
van Gent MW, Post MC, Snijder RJ, Westermann CJ, Plokker HW, Mager JJ. Реальная распространенность легочного шунта справа налево в зависимости от генотипа у пациентов с наследственной геморрагической телеангиэктазией: исследование трансторакальной контрастной эхокардиографии. Комод 2010 г .; 138 : 833–839.
PubMed
Google Scholar
Cottin V, Plauchu H, Bayle JY, Barthelet M, Revel D, Cordier JF.Легочные артериовенозные мальформации у пациентов с наследственной геморрагической телеангиэктазией. Am J Respir Crit Care Med 2004; 169 : 994–1000.
PubMed
Google Scholar
Gossage JR. Роль контрастной эхокардиографии в скрининге легочной артериовенозной мальформации у пациентов с наследственной геморрагической телеангиэктазией. Комод 2010 г .; 138 : 769–771.
PubMed
Google Scholar
Вудс Т.Д., Харманн Л., Пурат Т. и др.Шунты справа налево малого и среднего размера, выявленные с помощью контрастной эхокардиографии с физиологическим раствором, являются нормальными и не связаны с мигренозной головной болью. Комод 2010 г .; 138 : 264–269.
PubMed
Google Scholar
Lovering AT, Stickland MK, Amann M и др. Гипероксия предотвращает вызванное физической нагрузкой внутрилегочное артериовенозное шунтирование у здоровых людей. J. Physiol 2008; 586 : 4559–4565.
CAS
PubMed Central
PubMed
Google Scholar
Уайт Р.И. Младший, Легочные артериовенозные мальформации и наследственная геморрагическая телеангиэктазия: эмболотерапия с использованием баллонов и катушек. Arch Intern Med 1996; 156 : 2627–2628.
PubMed
Google Scholar
Шовлин К.Л., Соди В., Маккарти А., Ласьяуниас П., Джексон Дж. Э., Шеппард М.Н. Оценка материнского риска беременности для женщин с наследственной геморрагической телеангиэктазией (синдром Ослера-Вебера-Ренду): предлагаемый подход для оказания акушерских услуг. BJOG 2008; 115 : 1108–1115.
CAS
PubMed
Google Scholar
Cottin V, Dupuis-Girod S, Lesca G, Cordier JF.Легочно-сосудистые проявления наследственной геморрагической телеангиэктазии (болезнь Рендю-Ослера). Дыхание 2007; 74 : 361–378.
CAS
PubMed
Google Scholar
Байрак-Тойдемир П., Мао Р., Левин С., Макдональд Дж. Наследственная геморрагическая телеангиэктазия: обзор диагностики и лечения в молекулярную эру для клиницистов. Genet Med 2004; 6 : 175–191.
CAS
PubMed
Google Scholar
Kjeldsen AD, Oxhoj H, Andersen PE, Green A, Vase P.Распространенность легочных артериовенозных мальформаций (ПАВМ) и возникновение неврологических симптомов у пациентов с наследственной геморрагической телеангиэктазией (НГТ). J Intern Med 2000; 248 : 255–262.
CAS
PubMed
Google Scholar
Ианора А.А., Мемео М, Сабба С, Цирулли А, Ротондо А, Анджелелли Дж. Наследственная геморрагическая телеангиэктазия: многодетекторная спиральная компьютерная томография поражения печени. Радиология 2004; 230 : 250–259.
PubMed
Google Scholar
Buscarini E, Danesino C, Olivieri C и др. Допплеровское ультразвуковое исследование пороков развития сосудов печени при наследственной геморрагической телеангиэктазии — результаты обширного скрининга. Ultraschall Med 2004; 25 : 348–355.
CAS
PubMed
Google Scholar
Buscarini E, Danesino C, Plauchu H, et al.Высокая распространенность очаговой узловой гиперплазии печени у пациентов с наследственной геморрагической телеангиэктазией. Ultrasound Med Biol 2004; 30 : 1089–1097.
PubMed
Google Scholar
Шовлин К.Л., Гутмахер А.Е., Бускарини Е. и др. Диагностические критерии наследственной геморрагической телеангиэктазии (синдром Ренду-Ослера-Вебера). Am J Med Genet 2000; 91 : 66–67.
CAS
PubMed Central
PubMed
Google Scholar
Мей-Захав М., Летарт М., Фаунан М.Э., Абдалла С.А., Саймерман Ю., Макласки И.Б.Симптоматические дети с наследственной геморрагической телеангиэктазией: опыт педиатрического центра. Arch Pediatr Adolesc Med 2006; 160 : 596–601.
PubMed
Google Scholar
Faughnan ME, Palda VA, Garcia-Tsao G, et al. Международные рекомендации по диагностике и лечению наследственной геморрагической телеангиэктазии. J Med Genet 2011; 48 : 73–87.
CAS
Google Scholar
Кюри А., Леска Г., Коттин В. и др.Длительное наблюдение за 12 детьми с легочными артериовенозными мальформациями: подтверждение наследственной геморрагической телеангиэктазии во всех случаях. J Pediatr 2007; 151 : 299–306.
PubMed
Google Scholar
Eerola I, Boon LM, Mulliken JB и др. Капиллярная мальформация — артериовенозная мальформация, новое клиническое и генетическое заболевание, вызванное мутациями RASA1. Am J Hum Genet 2003; 73 : 1240–1249.
CAS
PubMed Central
PubMed
Google Scholar
Галлионе С.Дж., Репетто Г.М., Легиус Э и др. Комбинированный синдром ювенильного полипоза и наследственной геморрагической телеангиэктазии, связанный с мутациями в MADh5 (SMAD4). Lancet 2004; 363 : 852–859.
CAS
PubMed Central
PubMed
Google Scholar
Галлионе С.Дж., Ричардс Дж.А., Леттебоер Т.Г. и др.Мутации SMAD4 обнаружены у невыбранных пациентов с HHT. J Med Genet 2006; 43 : 793–797.
CAS
PubMed Central
PubMed
Google Scholar
Пригода Н.Л., Савас С., Абдалла С.А. и др. Наследственная геморрагическая телеангиэктазия: обнаружение мутаций, чувствительность тестов и новые мутации. J Med Genet 2006; 43 : 722–728.
CAS
PubMed Central
PubMed
Google Scholar
Шовлин К.Л., Летарте М.Наследственная геморрагическая телеангиэктазия и легочные артериовенозные мальформации: вопросы клинического ведения и обзор патогенетических механизмов. Thorax 1999; 54 : 714–729.
CAS
PubMed Central
PubMed
Google Scholar
Гарсия-Цао Г., Корзеник Дж. Р., Янг Л. и др. Заболевания печени у пациентов с наследственной геморрагической телеангиэктазией. N Engl J Med 2000; 343 : 931–936.
CAS
Google Scholar
Oxhoj H, Kjeldsen AD, Nielsen G. Скрининг легочных артериовенозных мальформаций: контрастная эхокардиография против пульсоксиметрии. Scand Cardiovasc J 2000; 34 : 281–285.
CAS
Google Scholar
Нантакумар К., Грэм А.Т., Робинсон Т.И. и др. Контрастная эхокардиография для выявления легочных артериовенозных мальформаций. Am Heart J 2001; 141 : 243–246.
CAS
Google Scholar
Olivieri C, Lanzarini L, Pagella F, et al. Эхокардиографический скрининг выявил повышенные значения систолического давления в легочной артерии у 9 из 68 невыбранных пациентов с наследственной геморрагической телеангиэктазией. Genet Med 2006; 8 : 183–190.
Google Scholar
Nawaz A, Litt HI, Stavropoulos SW, et al.Цифровая субтракционная легочная артериография в сравнении с мультидетекторной КТ в обнаружении легочных артериовенозных мальформаций. J Vasc Interv Radiol 2008; 19 : 1582–1588.
PubMed
Google Scholar
Аль-Салех С., Мей-Захав М., Фаунан М.Э. и др. Скрининг легочных и церебральных артериовенозных мальформаций у детей с наследственной геморрагической телеангиэктазией. евро Respir J 2009; 34 : 875–881.
CAS
PubMed
Google Scholar
Faughnan ME, Thabet A, Mei-Zahav M, et al. Легочные артериовенозные мальформации у детей: результаты транскатетерной эмболотерапии. J Pediatr 2004; 145 : 826–831.
PubMed
Google Scholar
Ференс Б.А., Шеннон Т.М., Уайт Р.И. младший, Завин М., Бердж СМ. Опасное для жизни легочное кровотечение с легочными артериовенозными мальформациями и наследственной геморрагической телеангиэктазией. Chest 1994; 106 : 1387–1390.
CAS
PubMed
Google Scholar
Шовлин К.Л., Уинсток А.Р., Петерс А.М., Джексон Дж. Э., Хьюз Дж. М.. Медицинские осложнения беременности при наследственной геморрагической телеангиэктазии. QJM 1995; 88 : 879–887.
CAS
PubMed
Google Scholar
Гуссоус Т., Хейнс А., Наджарян К., Даккаретт М., Дэвид С.Наследственная геморрагическая телеангиэктазия, проявляющаяся сердечной недостаточностью с высоким выбросом во время беременности. Cardiol Res Pract 2009; 2009 : 437237.
PubMed Central
PubMed
Google Scholar
Dupuis-Girod S, Giraud S, Decullier E, et al. Геморрагическая наследственная телеангиэктазия (болезнь Рендю-Ослера) и инфекционные заболевания: недооцененная связь. Clin Infect Dis 2007; 44 : 841–845.
PubMed
Google Scholar
Кэхилл Д., Баркер Ф., Дэвис К., Калва С., Сахай И., Фроск М. Случай 10-2010: 37-летняя женщина со слабостью и образованием в головном мозге. N Engl J Med 2010; 36 : 1326–1333.
Google Scholar
Gallitelli M, Guastamacchia E, Resta F, Guanti G, Sabba C. Легочные артериовенозные мальформации, наследственная геморрагическая телеангиэктазия и абсцесс головного мозга. Дыхание 2006; 73 : 553–557.
PubMed
Google Scholar
Халид С.К., Першбахер Дж., Макан М, Барзилай Б., Гуденбергер Д. Обострение кровотечения из носа свидетельствует о высоком сердечном выбросе при наследственной геморрагической телеангиэктазии. Am J Med 2009; 122 : 779.e1–779.e9.
Google Scholar
Махони Э.Дж., Шапшай С.М.Новая классификация паттернов носовой сосудистой сети при наследственной геморрагической телеангиэктазии. Am J Rhinol 2006; 20 : 87–90.
PubMed
Google Scholar
Fiorella ML, Росс Д., Хендерсон К.Дж., Уайт Р.И. Младший, Результат перегородочной дермопластики у пациентов с наследственной геморрагической телеангиэктазией. Ларингоскоп 2005; 115 : 301–305.
PubMed
Google Scholar
Hitchings AE, Lennox PA, Lund VJ, Howard DJ.Эффект лечения носового кровотечения, вторичного по отношению к наследственной геморрагической телеангиэктазии. Am J Rhinol 2005; 19 : 75–78.
PubMed
Google Scholar
Woolford TJ, Loke D, Bateman ND. Использование носового обтуратора при наследственной геморрагической телеангиэктазии: альтернатива процедуре Юнга. Дж Ларингол Отол 2002; 116 : 455–456.
CAS
PubMed
Google Scholar
Джеймсон Дж. Дж., Пещера DR.Гормональная и антигормональная терапия носовых кровотечений при наследственной геморрагической телеангиэктазии. Ларингоскоп 2004; 114 : 705–709.
PubMed
Google Scholar
Фернандес Л.А., Гарридо-Мартин Е.М., Санс-Родригес Ф. и др. Терапевтическое действие транексамовой кислоты при наследственной геморрагической телеангиэктазии (HHT): регуляция пути ALK-1 / эндоглин в эндотелиальных клетках. Thromb Haemost 2007; 97 : 254–262.
Google Scholar
Morales-Angulo C, Perez del Molino A, Zarrabeitia R, Fernandez A, Sanz-Rodriguez F, Botella LM. [Лечение носовых кровотечений при наследственной геморрагической телеангиэктазии (болезнь Ренду-Ослера-Вебера) транексамовой кислотой]. Acta Otorrinolaringol Esp 2007; 58 : 129–132.
PubMed
Google Scholar
Проктор Д.Д., Хендерсон К.Дж., Дзиура Д.Д., Уайт Р.И. Младший, Гормональная терапия для лечения желудочно-кишечного кровотечения при наследственной геморрагической телеангиэктазии. J Clin Gastroenterol 2008; 42 : 756–757.
PubMed
Google Scholar
Flieger D, Hainke S, Fischbach W. Резкое улучшение показателей наследственной геморрагической телеангиэктазии после лечения антагонистом фактора роста эндотелия сосудов (VEGF) бевацизумабом. Ann Hematol 2006; 85 : 631–632.
PubMed
Google Scholar
Trerotola SO, Pyeritz RE.Эмболизация ПАВМ: обновление. Am J Roentgenol 2010; 195 : 837–845.
Google Scholar
Ли Д.В., Уайт Р.И. младший, Эгглин Т.К. и др. Эмболотерапия крупных легочных артериовенозных мальформаций: отдаленные результаты. Ann Thorac Surg 1997; 64 : 930–939; обсуждение 939–940.
CAS
PubMed
Google Scholar
Муссуттас М., Фаяд П., Розенблатт М. и др.Легочные артериовенозные мальформации: церебральная ишемия и неврологические проявления. Неврология 2000; 55 : 959–964.
CAS
PubMed
Google Scholar
Post MC, van Gent MW, Snijder RJ, et al. Легочные артериовенозные мальформации и мигрень: новое видение. Дыхание 2008; 76 : 228–233.
CAS
PubMed
Google Scholar
Fulbright RK, Chaloupka JC, Putman CM, et al.МР наследственной геморрагической телеангиэктазии: распространенность и спектр цереброваскулярных пороков развития. AJNR Am J Neuroradiol 1998; 19 : 477–484.
CAS
PubMed
Google Scholar
Garcia-Tsao G. Поражение печени при наследственной геморрагической телеангиэктазии (НГТ). J Hepatol 2007; 46 : 499–507.
CAS
PubMed
Google Scholar
Wu JS, Saluja S, Garcia-Tsao G, Chong A, Henderson KJ, White RI Jr, Участие печени в наследственной геморрагической телеангиэктазии: КТ и клинические данные не коррелируют у пациентов с симптомами. AJR Am J Roentgenol 2006; 187 : W399 – W405.
PubMed
Google Scholar
Buscarini E, Plauchu H, Garcia Tsao G, et al. Поражение печени при наследственной геморрагической телеангиэктазии: согласованные рекомендации. Liver Int 2006; 26 : 1040–1046.
PubMed
Google Scholar
Лерут Дж., Орландо Дж., Адам Р. и др. Трансплантация печени при наследственной геморрагической телеангиэктазии: отчет Европейского реестра трансплантатов печени. Ann Surg 2006; 244 : 854–862; обсуждение 862–854.
PubMed Central
PubMed
Google Scholar
Митчелл А., Адамс Л.А., Маккуиллан Дж., Тибболлс Дж., Ванден Дризен Р., Делривьер Л. Бевацизумаб отменяет необходимость трансплантации печени при наследственной геморрагической телеангиэктазии. Liver Transpl 2008; 14 : 210–213.
PubMed
Google Scholar
Albinana V, Bernabeu-Herrero ME, Zarrabeitia R, Bernabeu C, Botella LM.Эстрогеновая терапия наследственной геморрагической телеангиэктазии (HHT): влияние ралоксифена на экспрессию эндоглина и ALK1 в эндотелиальных клетках. Thromb Haemost 2010; 103 : 525–534.
CAS
PubMed
Google Scholar
Bose P, Holter JL, Selby GB. Бевацизумаб при наследственной геморрагической телеангиэктазии. N Engl J Med 2009; 360 : 2143–2144.
CAS
PubMed
Google Scholar
Davidson TM, Olitsky SE, Wei JL.Наследственная геморрагическая телеангиэктазия / авастин. Ларингоскоп 2010; 120 : 432–435.
PubMed
Google Scholar
Лебрин Ф., Срун С., Раймонд К. и др. Талидомид стимулирует созревание сосудов и уменьшает носовое кровотечение у людей с наследственной геморрагической телеангиэктазией. Nat Med 2010; 16 : 420–428.
CAS
PubMed
Google Scholar
Абдалла С.А., Летарте М.Наследственная геморрагическая телеангиэктазия: современные взгляды на генетику и механизмы заболевания. J Med Genet 2006; 43 : 97–110.
CAS
PubMed Central
PubMed
Google Scholar
Коул С.Г., Бегби М.Э., Уоллес Г.М., Шовлин К.Л. Новый локус наследственной геморрагической телеангиэктазии (HHT3) отображается на хромосоме 5. J Med Genet 2005; 42 : 577–582.
CAS
PubMed Central
PubMed
Google Scholar
Байрак-Тойдемир П., Макдональд Дж., Акарсу Н. и др.Четвертый локус наследственной геморрагической телеангиэктазии отображается на хромосоме 7. Am J Med Genet A 2006; 140 : 2155–2162.
PubMed
Google Scholar
Ригельски К.М., Дженнингс К., Лехтонен Р., Минай О.А., Энг С., Олдред М.А. Мутация BMPR2 у пациента с легочной артериальной гипертензией и подозрением на наследственную геморрагическую телеангиэктазию. Am J Med Genet A 2008; 146A : 2551–2556.
CAS
PubMed
Google Scholar
О, С.П., Секи Т., Госс К.А. и др. Киназа 1, подобная рецептору активина, модулирует передачу сигналов трансформирующего фактора роста-бета 1 при регуляции ангиогенеза. Proc Natl Acad Sci USA 2000; 97 : 2626–2631.
CAS
PubMed
Google Scholar
Ли Д.Й., Соренсен Л.К., Брук Б.С. и др. Нарушение ангиогенеза у мышей, лишенных эндоглина. Science 1999; 284 : 1534–1537.
CAS
PubMed
Google Scholar
Манчини ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP. Эндоглин играет особую роль в рекрутировании клеток гладких мышц сосудов и регуляции артериовенозной идентичности во время ангиогенеза. Dev Dyn 2009; 238 : 2479–2493.
CAS
PubMed Central
PubMed
Google Scholar
Урнесс Л.Д., Соренсен Л.К., Ли Д.Артериовенозные мальформации у мышей, лишенных киназы-1, подобной рецептору активина. Nat Genet 2000; 26 : 328–331.
CAS
PubMed
Google Scholar
Артур Х.М., Уре Дж., Смит А.Дж. и др. Эндоглин, вспомогательный рецептор TGFbeta, необходим для внеэмбрионального ангиогенеза и играет ключевую роль в развитии сердца. Dev Biol 2000; 217 : 42–53.
CAS
PubMed
Google Scholar
Bourdeau A, Faughnan ME, Letarte M.Мыши с дефицитом эндоглина, уникальная модель для изучения наследственной геморрагической телеангиэктазии. Trends Cardiovasc Med 2000; 10 : 279–285.
CAS
PubMed
Google Scholar
Sabba C, Cirulli A, Rizzi R et al. Ангиогенез и наследственная геморрагическая телеангиэктазия. Болезнь Рендю-Ослера-Вебера. Acta Haematol 2001; 106 : 214–219.
CAS
PubMed
Google Scholar
Berg JN, Gallione CJ, Stenzel TT, et al.Ген киназы 1, подобной рецептору активина: геномная структура и мутации при наследственной геморрагической телеангиэктазии 2 типа. Am J Hum Genet 1997; 61 : 60–67.
CAS
PubMed Central
PubMed
Google Scholar
Pece N, Vera S, Cymerman U, White RI Jr, Wrana JL, Letarte M. Мутантный эндоглин при наследственной геморрагической телеангиэктазии 1 типа временно экспрессируется внутриклеточно и не является доминирующим негативом. J Clin Invest 1997; 100 : 2568–2579.
CAS
PubMed Central
PubMed
Google Scholar
Галлионе CJ, Клаус DJ, Yeh EY и др. Анализ мутации и экспрессии гена эндоглина при наследственной геморрагической телеангиэктазии выявил нулевые аллели. Hum Mutat 1998; 11 : 286–294.
CAS
PubMed
Google Scholar
Ричардс-Ютц Дж., Грант К., Чао ЕС, Вальтер С.Е., Гангули А.Обновленная информация о молекулярной диагностике наследственной геморрагической телеангиэктазии. Hum Genet 2010; 128 : 61–77.
CAS
Google Scholar
Макдональд Дж., Дамьянович К., Миллсон А. и др. Молекулярная диагностика наследственной геморрагической телеангиэктазии: результаты серии проверены одновременно с помощью секвенирования и анализа делеций / дупликаций. Clin Genet 2011; 79 : 335–344.
CAS
PubMed
Google Scholar
Галлион С., Эйлсворт А.С., Бейс Дж. И др.Перекрывающиеся спектры мутаций SMAD4 при ювенильном полипозе (JP) и синдроме JP-HHT. Am J Med Genet A 2010; 152A : 333–339.
CAS
PubMed
Google Scholar
Brusgaard K, Kjeldsen AD, Poulsen L, et al. Мутации эндоглина и активин-рецептор-подобной киназы 1 у датских пациентов с наследственной геморрагической телеангиэктазией. Clin Genet 2004; 66 : 556–561.
CAS
PubMed
Google Scholar
Letteboer TG, Zewald RA, Kamping EJ, et al.Наследственная геморрагическая телеангиэктазия: мутации ENG и ALK-1 у голландских пациентов. Hum Genet 2005; 116 : 8–16.
CAS
PubMed
Google Scholar
Schulte C, Geisthoff U, Lux A, et al. Высокая частота мутаций ENG и ALK1 / ACVRL1 у немецких пациентов с HHT. Hum Mutat 2005; 25 : 595.
PubMed
Google Scholar
Босслер А.Д., Ричардс Дж., Джордж К., Годмилов Л., Гангули А.Новые мутации в ENG и ACVRL1 идентифицированы в серии из 200 человек, проходящих клиническое генетическое тестирование на наследственную геморрагическую телеангиэктазию (HHT): корреляция генотипа с фенотипом. Hum Mutat 2006; 27 : 667–675.
CAS
PubMed
Google Scholar
Gedge F, McDonald J, Phansalkar A, et al. Клиническая и аналитическая чувствительность при тестировании наследственной геморрагической телеангиэктазы и отчет о мутациях de novo. J Mol Diagn 2007; 9 : 258–265.
CAS
PubMed Central
PubMed
Google Scholar
Lenato GM, Lastella P, Di Giacomo MC, et al. Анализ мутаций генов ENG и ALK-1 на основе DHPLC в итальянской популяции HHT. Hum Mutat 2006; 27 : 213–214.
PubMed
Google Scholar
Lesca G, Burnichon N, Raux G, et al.Распределение мутаций ENG и ACVRL1 (ALK1) у французских пациентов с HHT. Hum Mutat 2006; 27 : 598.
PubMed
Google Scholar
Olivieri C, Pagella F, Semino L, et al. Анализ генов ENG и ACVRL1 в 137 итальянских семьях HHT выявил 76 различных мутаций (24 новых). Сравнение с другими европейскими исследованиями. J Hum Genet 2007; 52 : 820–829.
CAS
PubMed
Google Scholar
Howe JR, Sayed MG, Ahmed AF, et al.Распространенность мутаций MADh5 и BMPR1A при ювенильном полипозе и отсутствие мутаций BMPR2, BMPR1B и ACVR1. J Med Genet 2004; 41 : 484–491.
CAS
PubMed Central
PubMed
Google Scholar
Sweet K, Willis J, Zhou XP и др. Молекулярная классификация пациентов с необъяснимым гамартоматозным и гиперпластическим полипозом. JAMA 2005; 294 : 2465–2473.
CAS
PubMed Central
PubMed
Google Scholar
Howe JR, Haidle JL, Lal G, et al.Мутации ENG у пациентов с ювенильным полипозом с отрицательными мутациями MADh5 / BMPR1A. Clin Genet 2007; 71 : 91–92.
CAS
PubMed
Google Scholar
Kjeldsen AD, Moller TR, Brusgaard K, Vase P, Andersen PE. Клинические симптомы в зависимости от генотипа у пациентов с наследственной геморрагической телеангиэктазией. J Intern Med 2005; 258 : 349–355.
CAS
PubMed
Google Scholar
Байрак-Тойдемир П., Макдональд Дж., Маркевиц Б. и др.Взаимосвязь генотип-фенотип при наследственной геморрагической телеангиэктазии: мутации и проявления. Am J Med Genet A 2006; 140 : 463–470.
PubMed
Google Scholar
Letteboer TG, Mager JJ, Snijder RJ, et al. Взаимосвязь генотип-фенотип при наследственной геморрагической телеангиэктазии. J Med Genet 2006; 43 : 371–377.
CAS
PubMed
Google Scholar
Харрисон Р. Э., Фланаган Дж. А., Санкело М. и др.Молекулярный и функциональный анализ определяет ALK-1 как преобладающую причину легочной гипертензии, связанной с наследственной геморрагической телеангиэктазией. J Med Genet 2003; 40 : 865–871.
CAS
PubMed Central
PubMed
Google Scholar
Trembath RC, Thomson JR, Machado RD и др. Клинико-молекулярно-генетические особенности легочной гипертензии у пациентов с наследственной геморрагической телеангиэктазией. N Engl J Med 2001; 345 : 325–334.
CAS
PubMed
Google Scholar
Наследственная геморрагическая телеангиэктазия: обзор диагностики и лечения в молекулярную эру для врачей
Вестерманн CJ, Rosina AF, De Vries V, de Coteau PA. Распространенность и проявления наследственной геморрагической телеангиэктазии у афро-карибского населения Нидерландских Антильских островов: семейный скрининг. Am J Med Genet 2003; 116A : 324–328.
PubMed
Google Scholar
Kjeldsen A, Ваза P, зеленая A. Наследственная геморрагическая телеангиэктазия: популяционное исследование распространенности и смертности датских пациентов. J Int Med 1999; 245 : 31–39.
CAS
Google Scholar
Plauchu H, Bideau A. Эпидемиология и конституция в регистре населения в связи с географической концентрацией дюн. Наследственная болезнь дюн встречается редко. Население 1984 год; 39 : 765–786.
Google Scholar
Дакеиси М., Сиоя Т., Вада Й, Синдо Т., Отака К., Манабе М. и др. Генетическая эпидемиология наследственной геморрагической телеангиэктазии в местном сообществе в северной части Японии. Hum Mutat 2002; 19 : 140–148.
CAS
PubMed
Google Scholar
Морган Т., Макдональд Дж., Андерсон С., Исмаил М., Миллер Ф., Мао Р. и др.Внутричерепное кровоизлияние у младенцев и детей с наследственной геморрагической телеангиэктазией. Педиатрия 2002; 109 : 1–7.
Google Scholar
Роман Г, Фишер М, Perl D, Позер С. Неврологические проявления наследственной геморрагической телеангиэктазии (болезнь Ренду-Ослера-Вебера): отчет о 2 случаях и обзор литературы. Ann Neurol 1978; 4 : 130–144.
CAS
PubMed
Google Scholar
Кондзиолка Д., Хамфрис Р., Хоффман Х., Хендрик Э. Б., Дрейк Дж.Артериовенозные мальформации головного мозга у детей: сорокалетний опыт. Can J Neurol Sci 1992; 19 : 40–45.
CAS
PubMed
Google Scholar
Митчелл Р., Остин Э. Легочная артериовенозная мальформация у новорожденного. J Pediatr Surg 1993; 28 : 1536–1538.
CAS
PubMed
Google Scholar
Райхман Х., Соренсен Б.Иссечение артериовенозной мальформации спинного мозга. Rocky Mt Med J 1971; 68 : 21–24.
CAS
PubMed
Google Scholar
Guttmacher A, Marchuk D, White R. Наследственная геморрагическая телеангиэктазия. N Engl J Med 1995; 333 : 918–924.
CAS
PubMed
Google Scholar
Уоллес Г., Шовлин С.Семейство наследственных геморрагических телеангиэктазий с поражением легких не связано с известными генами HHT, эндоглином и ALK-1. Thorax 2000; 55 : 685–690.
CAS
PubMed
PubMed Central
Google Scholar
Шовлин С., Гутмахер А., Бускарини Е., Фогнан М., Хайланд Р., Вестерманн С. и др. Диагностические критерии наследственной геморрагической телеангиэктазии (синдром Ренду-Ослера-Вебера). Am J Med Genet 2000; 91 : 66–67.
CAS
Google Scholar
Ассар А, Фридман С, Уайт Р. Естественное течение носовых кровотечений при наследственных геморрагических телеангиэктазах. Ларингоскоп 1991; 101 : 977–980.
Google Scholar
Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Возрастной клинический профиль наследственной геморрагической телеангиэктазии в популяции, набранной эпидемиологами. Am J Med Genet 1989; 32 : 291–297.
CAS
PubMed
Google Scholar
Портеус М., Берн Дж., Проктор С. Наследственная геморрагическая телеангиэктазия: клинический анализ. J Med Genet 1992; 29 : 527–530.
CAS
PubMed
PubMed Central
Google Scholar
Kjeldsen A, Kjeldsen J. Желудочно-кишечные кровотечения у пациентов с наследственной геморрагической телеангиэктазией. Am J Gastroenterol 2000; 95 : 415–418.
CAS
PubMed
Google Scholar
Ваза П, Холм М, Арендруп Х. Легочные артериовенозные свищи при наследственной геморрагической телеангиэктазии. Acta Med Scand 1985; 218 : 105–109.
CAS
PubMed
Google Scholar
Haitjema T, Disch F, Overtoom T, Westermann C, Lammers J.Скрининг членов семей пациентов с наследственной геморрагической телеангиэктазией. Am J Med 1995; 99 : 519–524.
CAS
PubMed
Google Scholar
Кьельдсен А, Охой Х, Андерсен П, Грин А, Ваза П. Распространенность легочных артериовенозных мальформаций (ПАВМ) и возникновение неврологических симптомов у пациентов с наследственной геморрагической телеангиэктазией (НГТ). J Int Med 2000; 248 : 255–262.
CAS
Google Scholar
Шовлин С, ЛеТарт М. Наследственная геморрагическая телеангиэктазия и легочные артериовенозные мальформации: вопросы клинического ведения и обзор патогенетических механизмов. Thorax 1999; 54 : 714–729.
CAS
PubMed
PubMed Central
Google Scholar
Белый R. Легочные артериовенозные мальформации: как их диагностировать и почему это важно. soRadiology 1992; 182 : 633–635.
Google Scholar
Ференс Б., Шеннон Т., Уайт Р., Завин М., Бердж С. Опасное для жизни легочное кровотечение с легочными артериовенозными мальформациями и наследственной геморрагической телеангиэктазией. Chest 1994; 106 : 1387–1390.
CAS
PubMed
Google Scholar
Мусуттас М., Фаяд П., Розенблатт М., Хашимото М., Поллак Дж., Хендерсон К. и др.Легочные артериовенозные мальформации: церебральная ишемия и неврологические проявления. Неврология 2000; 55 : 959–964.
CAS
PubMed
Google Scholar
Белый R, Поллак Дж, Вирт Дж. Легочные артериовенозные мальформации: диагностика и транскатетерная эмболотерапия. J Vasc Interv Radiol 1996; 7 : 787–804.
PubMed
Google Scholar
Hewes R, Auster M, Белый R.Церебральная эмболия: первое проявление легочной артериовенозной мальформации у пациентов с наследственной геморрагической телеангиэктазией. Cardiovasc Inter Rad 1985; 8 : 151–155.
CAS
Google Scholar
Шовлин С., Винсток А, Питерс А., Джексон Дж., Хьюз Дж. Медицинские осложнения беременности при наследственной геморрагической телеангиэктазии. QJ Med 1995; 88 : 879–887.
CAS
Google Scholar
Fulbright R, Chaloupka J, Putman C, Sze G, Merriam M, Lee G и др.МР наследственной геморрагической телеангиэктазии: распространенность и спектр цереброваскулярных пороков развития. AJNR Am J Neurol Radiol 1998; 19 : 477–484.
CAS
Google Scholar
Matsubara S, Manzia J, ter Brugge K, Willinsky R, Montanera W, Faughnan M. Ангиографические и клинические характеристики пациентов с артериовенозными мальформациями головного мозга, ассоциированными с наследственной геморрагической телеангиэктазией. Am J Neuroradiology 2000; 21 : 1016–1020.
CAS
Google Scholar
Виллемсе Р., Магер Дж., Вестерманн С., Овертум Т, Маузер Х., Вольберс Дж. Риск кровотечения при цереброваскулярной мальформации при наследственной геморрагической телеангиэктазии. J Neurosurg 2000; 92 : 779–784.
CAS
PubMed
Google Scholar
MontAlverne F, Musacchio M, Tolentino V, Belzile F, Riquelme C, Tournade A.Гигантский перимедуллярный свищ спинного мозга при наследственной геморрагической телеангиэктазии: диагностика, эндоваскулярное лечение и обзор литературы. Нейрорадиология 2003; 45 : 830–836.
CAS
Google Scholar
Гарсия-Цао Г., Корзеник Дж., Янг Л., Хендерсон К., Джайн Д., Берд Б. и др. Заболевания печени у пациентов с наследственной геморрагической телеангиэктазией. N Engl J Med 2000; 343 : 931–936.
CAS
PubMed
Google Scholar
Memeo M, Stabile Ianora AA, Scardapane A Buonamico P, Sabba C, Angelelli G. Поражение печени при наследственной геморрагической телеангиэктазии: Abdom Imaging 2004. Epub впереди печати.
Ианора А.А., Мемео М, Сабба С, Цирулли А, Ротондо А, Анджелелли Дж. Наследственная геморрагическая телеангиэктазия: многодетекторная спиральная Т-оценка. Радиология 2004; 230250–230259.
Saluja S, White RI. Наследственная геморрагическая телеангиэктазия печени; Гиперперслияние с относительной ишемией — бедность среди изобилия ?. Радиология 2004; 230 : 25–27.
PubMed
Google Scholar
Buscarini E, Buscarini L, Danesino C, Pinatanida M, Civardi G, Quaretti P, et al. Сосудистые мальформации печени при наследственной геморрагической телеангиэктазии: допплеровское ультразвуковое обследование в большой семье. J Hepato 1997; 26 : 111–118.
CAS
Google Scholar
Макдональд Дж., Миллер Ф., Халлам С., Нельсон Л., Марчук Д., Уорд К. Клинические проявления при большой наследственной геморрагической телеангиэктазии 2-го типа. Am J Med Genet 2000; 93 : 320–327.
CAS
PubMed
Google Scholar
Хис Д.Х., Райан Г.Ф., Хеллемс С.О. Чиран, округ Колумбия, Шейлс, Лос-Анджелес.Крупные аневризмы восходящей аорты и крупных коронарных артерий у пациента с наследственной геморрагической телеангиэктазией. Mayo Clin Proc 2003; 78 : 774–776.
Google Scholar
Tsuiki K, Tamada Y, Yasui S. Аневризма коронарной артерии без стеноза в связи с болезнью Ослера-Вебера-Ренду: отчет о клиническом случае. Ангиология 1991; 42 : 55–58.
CAS
PubMed
Google Scholar
Брант А, Шахат А, Белый Р.Глазные проявления при наследственной геморрагической телеангиэктазии. Am J Ophthalmol 1989; 107 : 642–646.
CAS
PubMed
Google Scholar
Ваза I, Ваза П. Поражения глаз при наследственной геморрагической телеангиэктазии. Acta Ophthalmol 1979; 57 : 1084–1090.
CAS
Google Scholar
Lande A, Bedford A, Schechter LS.Спектр артериографических находок при болезни Ослера-Вебера-Ренду. Ангиология 1976; 27 : 223–240.
CAS
PubMed
Google Scholar
Зиани М., Валинья С., Лопес Дж., Руффион А., Плаучу Х., Перрин П. Артериовенозная мальформация почек, требующая хирургического вмешательства при болезни Рендю-Ослера-Вебера. J Urololgy 2000; 164 : 1292–1293.
CAS
Google Scholar
Хамфрис Дж. Э., Фриерсон Х. Ф., Андервуд ПБ.Вагинальные телеангиэктазии: необычное проявление синдрома Ослера-Вебера-Ренду. Obstet Gynecol 1993; 81 : 865–866.
CAS
PubMed
Google Scholar
Барзилай Б., Вагонер А, Спессерт С, Пикус Д., Гуденбергер Д. Двумерная контрастная эхокардиография в выявлении и последующем наблюдении врожденных легочных артериовенозных мальформаций. Am J Cardiol 1991; 68 : 1507–1510.
CAS
PubMed
Google Scholar
Кьельдсен А, Охой Х, Андерсен П, Эль Б, Якобсен Дж, Ваза П. Скрининговые процедуры и легочная ангиография у пациентов с наследственной геморрагической телеангиэктазией. СУНДУК 1999; 116 : 432–439.
CAS
PubMed
Google Scholar
Нантакумар К., Грэм А., Робинсон Т., Гранде П., Пугаш Р., Кларк Дж. И др.Контрастная эхокардиография для выявления легочных артериовенозных мальформаций. Am Heart J 2001; 141 : 243–246.
CAS
PubMed
Google Scholar
Реми Дж., Реми-Жардин М., Ваттинн Л., Деффонтейн С. Легочные артериовенозные мальформации: оценка с помощью КТ грудной клетки до и после лечения. Радиология 1992; 182 : 809–816.
CAS
PubMed
Google Scholar
Белый R, Pollak J.Легочные артериовенозные мальформации: диагностика с помощью трехмерной спиральной компьютерной томографии: прорыв без контрастного вещества. Радиология 1994; 191 : 613–614.
PubMed
Google Scholar
Ли Д., Уайт Р., Эгглин Т., Поллак Дж., Фаяд П., Вирт Дж. И др. Эмболотерапия крупных легочных артериовенозных мальформаций: отдаленные результаты. Ann Thorac Surg 1997; 64 : 930–940.
CAS
PubMed
Google Scholar
Кристенсон Г.Носовое кровотечение может означать нечто гораздо более серьезное: введение в HHT. JADA 1998; 129 : 635–637.
Google Scholar
Паркин Дж., Диксон Дж. Лазерная фотокоагуляция при наследственной геморрагической телеангиэктазии. Otolaryngol Head Neck Surg 1981; 89 : 204–208.
CAS
PubMed
Google Scholar
Шапшай С, Оливер П.Лечение наследственной геморрагической телеангиэктазии методом фотокоагуляции Nd-Yag лазером. Ларингоскоп 1984; 94 : 1554–1556.
CAS
PubMed
Google Scholar
Saunders W. Дермопластика перегородки для контроля носовых кровотечений, вызванных наследственной геморрагической телеангиэктазией или перфорацией перегородки. Trans Am Acad Ophthalmol Otolaryngol 1960; 64 : 500–506.
CAS
PubMed
Google Scholar
Longacre A, Gross C, Gallitelli M, Henderson K, White R, Proctor D.Диагностика и лечение желудочно-кишечных кровотечений у пациентов с наследственной геморрагической телеангиэктазией. Am J Gastroenterology 2003; 98 : 59–65.
Google Scholar
Гершон А., Фогнан М., Чон К., Пугаш Р., Кларк Дж., Бохан М. и др. Транскатетерная эмболотерапия легочных артериовенозных мальформаций матери при беременности. СУНДУК 2001; 119 : 470–477.
CAS
PubMed
Google Scholar
Леви Э.И., Ниранджан М., Томпсон Т., Скарроу А., Кондзиолка Д., Фликингер Дж. И др.Радиохирургия при внутричерепных артериовенозных мальформациях у детей. Нейрохирургия 2000; 47 : 834–842.
CAS
PubMed
Google Scholar
Vinuela F, Dion J, Duckwiler G, Martin N, Lylyk P, Fox A и др. Комбинированная эндоваскулярная эмболизация и хирургия в лечении артериовенозных мальформаций головного мозга: опыт работы с 101 случаем. J Neurosurg 1991; 75 : 856–864.
CAS
PubMed
Google Scholar
Odorico J, Hakim M, Becker Y. Трансплантация печени как окончательная терапия осложнений после артериальной эмболизации при печеночных проявлениях наследственной геморрагической телеангиэктазии. Liver Transpl Surg 1998; 4 : 483–490.
CAS
PubMed
Google Scholar
Миллер Ф., Уайтинг Дж., Корзеник Дж., Уайт Р.С осторожностью применяйте эмболизацию печени при лечении наследственной геморрагической телеангиэктазии. Радиология 1999; 213 : 928–930.
PubMed
Google Scholar
Бойло О, Бьянко Ф. Viale J, Трансплантация печени устраняет гипердинамическое кровообращение при наследственной геморрагической телеангиэктазии с поражением печени. Гастроэнтерология 1999; 116 : 187–192.
CAS
PubMed
Google Scholar
Fernandez-Ruiz E, St-Jacques S, Bellon T, Letarte M, Bernabeu C.Отнесение гена эндоглина человека (END) к 9q34 qter. Cytogenet Cell Genet 1993; 64 : 204–207.
CAS
PubMed
Google Scholar
McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA и др. Локус заболевания наследственной геморрагической телеангиэктазии отображается на хромосоме 9q33-34. Nat Genet 1994; 6 : 197–204.
CAS
PubMed
Google Scholar
Шовлин К.Л., Хьюз Дж. М., Тудденхэм Э. Г., Темперли И., Перембелон Ю. Ф., Скотт Дж. И др.Ген наследственной геморрагической телеангиэктазии отображается на хромосоме 9q3. Nat Genet 1994; 6 : 205–209.
CAS
PubMed
Google Scholar
Винсент П., Плаучу Х., Хазан Дж., Форе С., Вайссенбах Дж., Годе Дж. Третий локус наследственной геморрагической телеангиэктазии отображается на хромосоме 12q. Hum Mol Gen 1995; 4 : 945–949.
CAS
PubMed
Google Scholar
Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Helmbold EA и др.Второй локус наследственной геморрагической телеангиэктазии отображается на хромосоме 12. Genome Res 1995; 5 : 21–28.
CAS
PubMed
Google Scholar
Roijer E, Miyazono K, Astrom AK, Geurts van Kessel A, ten Dijke P, Stenman G. Хромосомная локализация трех генов человека, кодирующих членов суперсемейства TGF-бета серин / треонинкиназных рецепторов I типа. Mamm Genome 1998; 9 : 266–268.
CAS
PubMed
Google Scholar
McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Эндоглин, белок эндотелиальных клеток, связывающий TGF-бета, является геном наследственной геморрагической телеангиэктазии типа 1. Nat Genet 1994; 8 : 345–351.
CAS
PubMed
Google Scholar
Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE и др.Ген киназы 1, подобной рецептору активина: геномная структура и мутации при наследственной геморрагической телеангиэктазии 2 типа. Am J Hum Genet 1997; 61 : 60–67.
CAS
PubMed
PubMed Central
Google Scholar
Аттисано Л., Каркамо Дж., Венчур Ф, Вайс ФМ, Массагу Дж., Врана Дж. Идентификация человеческих рецепторов активина и TGF beta типа I, которые образуют гетеромерные киназные комплексы с рецепторами типа II. Cell 1993; 5 : 671–680.
Google Scholar
Cheifetz S, BellÓn T, Calés C, Vera S, Bernabeu C, Massague J, et al. Эндоглин является компонентом рецепторной системы трансформирующего фактора роста в эндотелиальных клетках человека. J Biol Chem 1992; 267 : 19027–19030.
CAS
PubMed
Google Scholar
Беллон Т., Корби А., Ластрес П., Калес С., Себриан М., Вера С. и др.Идентификация и экспрессия двух форм человеческого трансформирующего фактора роста-бета-связывающего белка эндоглина с отдельными участками цитоплазмы. евро. J Immunol 1993; 23 : 2340–2345.
CAS
PubMed
Google Scholar
Attisano L, Wrana JL. Передача сигнала суперсемейством TGF-. Science 2002; 296 : 646–1647.
Google Scholar
ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, et al.Киназы, подобные рецепторам активина: новый подкласс рецепторов клеточной поверхности с прогнозируемой серин / треонинкиназной активностью. Онкоген 1993; 8 : 2879–2887.
CAS
PubMed
Google Scholar
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Мутации в гене киназы 1, подобной рецептору активина, при наследственной геморрагической телеангиэктазии 2 типа. Nat Genet 1996; 13 : 189–195.
CAS
PubMed
Google Scholar
Гуго А., Сен-Жак С., Гривз А., О’Коннелл П.Дж., д’Апис А.Дж., Буринг Г.Дж. и др. Идентификация отдельных эпитопов эндоглина, RGD-содержащего гликопротеина эндотелиальных клеток, лейкозных клеток и синцитиотрофобластов. Int Immunol 1992; 4 : 83–92.
CAS
PubMed
Google Scholar
Lastres P, Bellon T, Cabanas C, Sanchez-Madrid F, Acevedo A, Gougos A, et al.Регулируемая экспрессия на человеческих макрофагах эндоглина, поверхностного антигена, содержащего Arg-Gly-Asp. евро. J Immunol 1992; 22 : 393–397.
CAS
PubMed
Google Scholar
Barbara NP, Wrana JL, Letarte M. Эндоглин является дополнительным белком, который взаимодействует с сигнальным рецепторным комплексом множества членов суперсемейства трансформирующих факторов роста. J Biol Chem 1999; 274 : 584–594.
CAS
PubMed
Google Scholar
Врана Дж. Л., Аттисано Л., Каркамо Дж., Зентелла А., Дуди Дж., Лайхо М. и др. TGF-бета передает сигнал через комплекс рецепторов гетеромерной протеинкиназы. Cell 1992; 71 : 1003–1014.
CAS
PubMed
Google Scholar
Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Механизм активации рецептора TGF-бета. Nature 1994; 370 : 341–347.
CAS
Google Scholar
Wieser R, Wrana JL, Massague J. Мутации в GS-домене, которые постоянно активируют Tβ R-I, нижележащий сигнальный компонент в рецепторном комплексе TGF-β. EMBO J 1995; 14 : 2199–2208.
CAS
PubMed
PubMed Central
Google Scholar
Massagué J.Трансдукция TGF-сигнала. Annu Rev Biochem 1998; 67 : 753–791.
PubMed
Google Scholar
Derynck R, Zhang Y, Feng XH. Smads: активаторы транскрипции TGF-ответов. Cell 1998; 95 : 737–740.
CAS
PubMed
Google Scholar
Галлионе CJ, Repetto GM, Legius E, Rustig AK, Schelley SL, Tejpar S, et al.Комбинированный синдром ювенильного полипоза с мутациями в MADh5 (SMAD4). Lancet 2004; 363 : 852–859.
CAS
PubMed
Google Scholar
Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK и др. Киназа 1, подобная рецептору активина, модулирует передачу сигналов трансформирующего фактора роста-бета 1 при регуляции ангиогенеза. Proc Natl Acad Sci U S A 2000; 97 : 2626–2631.
CAS
PubMed
PubMed Central
Google Scholar
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Уравновешивание состояния активации эндотелия с помощью двух различных рецепторов TGF-бета типа I. EMBO J 2002; 21 : 1743–1753.
CAS
PubMed
PubMed Central
Google Scholar
Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al.Киназа, подобная рецептору активина (ALK) 1, является антагонистическим медиатором латеральной передачи сигналов TGFbeta / ALK5. Mol Cell 2003; 12 : 817–828.
CAS
Google Scholar
McAllister KA, Baldwin MA, Thukkani AK, Gallione CJ, Berg JN, Porteus AE, et al. Шесть новых мутаций в гене эндоглина при наследственной геморрагической телеангиэктазии 1 типа предполагают доминантно-отрицательный эффект рецепторной функции. Hum Mol Genet 1995; 4 : 1983–1985.
CAS
PubMed
Google Scholar
Pece N, Vera S, Cymerman U, White R, Wrana J, Letarte M. Мутантный эндоглин при наследственной геморрагической телеангиэктазии 1 типа временно экспрессируется внутриклеточно и не является доминирующим негативом. J Clin Invest 1997; 100 : 2568–2579.
CAS
PubMed
PubMed Central
Google Scholar
Pece-barbara N, Cymerman U, Vera S, Marchuk D, Letarte M.Анализ экспрессии четырех миссенс-мутаций эндоглина предполагает, что гаплонедостаточность является преобладающим механизмом наследственной геморрагической телеангиэктазии типа 1. Hum Mol Genet 1999; 8 : 2171–2181.
CAS
PubMed
Google Scholar
Пакет М.Э., Пес-Барбара Н., Вера С., Цимерман Ю., Карабегович А., Шовлин С. и др. Анализ нескольких мутантов эндоглина не показывает, что зрелый или секретируемый белок эндоглинов может нарушать нормальную функцию эндоглина. Hum Mol Genet 2001; 10 : 1347–1357.
CAS
PubMed
Google Scholar
Люкс А, Галлион CJ, Марчук Д.А. Анализ экспрессии миссенс-и укороченных мутаций эндоглина: понимание структуры белка и механизмов заболевания. Hum Mol Genet 2000; 9 : 745–755.
CAS
PubMed
Google Scholar
Bourdeau A, Cymerman U, Paquet ME, Meschino W, McKinnon WC, Guttmacher AE, et al.Экспрессия эндоглина снижена в нормальных сосудах, но все же обнаруживается при артериовенозной мальформации у пациентов с наследственной геморрагической телеангиэктазией 1 типа. Am J Path 2000; 156 : 911–923.
CAS
PubMed
Google Scholar
Шовлин К.Л., Хьюз Дж. М., Скотт Дж., Зейдман К. Э., Зайдман Дж. Г.. Характеристика эндоглина и выявление новых мутаций при наследственной геморрагической телеангиэктазии. Am J Hum Genet 1997; 61 : 68–79.
CAS
PubMed
PubMed Central
Google Scholar
Abdalla SA, Pece-Barbara N, Vera S, Tapia E, Paez E, Bernabeu C, et al. Анализ ALK-1 и эндоглина у новорожденных из семей с наследственной геморрагической телеангиэктазией 2 типа. Hum Mol Genet 2000; 9 : 1227–1237.
CAS
PubMed
Google Scholar
Клаус DJ, Галлионе CJ, Энтони К., Йе ЕЙ, Ю Дж, Люкс А. и др.Новые мутации миссенс и сдвига рамки считывания в гене активиноподобной киназы-1 при наследственной геморрагической телеангиэктазии. Вкратце мутаций нет. 164. Онлайн. Hum Mutat 1998; 12 : 137
CAS
PubMed
Google Scholar
Харрисон Р. Э., Фланаган Дж. А., Санкело М., Абдалла С. А., Роуэлл Дж., Мачадо Р. Д. и др. Молекулярный и функциональный анализ определяет ALK-1 как преобладающую причину легочной гипертензии, связанной с наследственной геморрагической телеангиэктазией. J Med Genet 2003; 40 : 865–871.
CAS
PubMed
PubMed Central
Google Scholar
Абдалла С.А., Саймерман У., Джонсон Р.М., Дебер С.М., Летарт М. Связанные с заболеванием мутации в консервативных остатках киназного домена ALK-1. евро J Hum Genet 2003; 11 : 279–287.
CAS
PubMed
Google Scholar
Li DY, Соренсен LK, Brooke BS, Urness LD, Davis EC, Taylor DG, et al.Нарушение ангиогенеза у мышей, лишенных эндоглина. Science 1999; 284 : 1534–1537.
CAS
PubMed
Google Scholar
Артур Х.М., Уре Дж., Смит А.Дж., Ренфорт Дж., Уилсон Д.И., Торсни Е. и др. Эндоглин, вспомогательный бета-рецептор TGF, необходим для внеэмбрионального ангиогенеза и играет ключевую роль в развитии сердца. Dev Biol 2000; 217 : 42–53.
CAS
PubMed
Google Scholar
Торсни Э., Чарльтон Р., Даймонд АГ, Берн Дж., Сомс СП, Артур Х.М.Мышиная модель наследственной геморрагической телеангиэктазии имеет генерализованную сосудистую аномалию. Тираж 2003 г .; 107 : 1653–1657.
PubMed
Google Scholar
Урнесс Л.Д., Соренсен Л.К., Ли Д. Артериовенозные мальформации у мышей, лишенных киназы-1, подобной рецептору активина. Nat Genet 2000; 26 : 328–331.
CAS
PubMed
Google Scholar
Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP, et al.Мышиная модель наследственной геморрагической телеангиэктазии (HHT) типа 2. Hum Mol Genet 2003; 12 : 473–482.
CAS
PubMed
Google Scholar
Секи Т, Юн Дж, О П. Экспрессия киназы1, специфичной для артериального эндотелия, подобной рецептору активина, предполагает ее роль в артериализации и ремоделировании сосудов. Circu Res 2003; 3 : 1–9.
Google Scholar
Bourdeau A, Dumont DJ, Letarte M.Мышиная модель наследственной геморрагической телеангиэктазии. J Clin Invest 1999; 104 : 1343–1351.
CAS
PubMed
PubMed Central
Google Scholar
Роман Б.Л., Фам В.Н., Лоусон Н.Д., Кулик М., Чайлдс С., Леквен А.С. и др. Нарушение acvrl1 увеличивает количество эндотелиальных клеток в черепных сосудах рыбок данио. Развитие 2002; 129 : 3009–3019.
CAS
PubMed
Google Scholar
Галлион С., Шесселе Э.А., Рейнхардт Д., Дуитс А.Дж., Берг Дж.Н., Вестерманн С.Дж. и др.Две распространенные мутации эндоглина в семьях с наследственной геморрагической телеангиэктазией на Нидерландских Антильских островах: свидетельства против эффекта основателя. Hum Genet 2000; 107 : 40–44.
CAS
PubMed
Google Scholar
Lesca G, Plauchu H, Coulet F, Lefebvre S, Plessis G, Odent S, et al. Молекулярный скрининг генов ALK1 / ACVRL1 и ENG при наследственной геморрагической телеангиэктазии во Франции. Hum Mutat 2004; 23 : 289–299.
CAS
PubMed
Google Scholar
Berg J, Guttmacher A, Marchuk D, Porteous M. Клиническая гетерогенность наследственной геморрагической телеангиэктазии: являются ли легочные артериовенозные мальформации более распространенными в семьях, связанных с эндоглином? Med. Genet 1996; 33 : 256–257.
CAS
Google Scholar
Heutink P, Haitjema T, Breedveld G, Janssen B, Sandkuijl L, Bontekoe C, et al.Связь наследственной геморрагической телеангиэктазии с хромосомой 9q34 и доказательства гетерогенности локуса. J Med Genet 1994; 31 : 933–936.
CAS
PubMed
PubMed Central
Google Scholar
Olivieri C, Mira E, Delu G, Pagella F, Zambelli A, Malvezzi L, et al. Идентификация 13 новых мутаций в гене ACVRL1 в группе из 52 невыбранных итальянских пациентов с наследственной геморрагической телеангиэктазией. J Med Genet 2002; 39 : E39.
CAS
PubMed
PubMed Central
Google Scholar
Абдалла С., Гейстхофф У., Бонно Д., Плаучу Х., Макдональд Дж., Кеннеди С. и др. Висцеральные проявления при наследственной геморрагической телеангиэктазии 2 типа. J Med Genet 2003; 40 : 494–502.
CAS
PubMed
PubMed Central
Google Scholar
Cymerman U, Vera S, Amna K, Abdalla S, Letarte M.Характеристика 17 новых мутаций эндоглина, связанных с наследственной геморрагической телеангиэктазией. Hum Mutat 2003; 21 : 482–492.
CAS
PubMed
Google Scholar
Piantanida M, Buscarini E, Dellavecchia C, Minelli A, Rossi A, Buscarini L. и др. Наследственная геморрагическая телеангиэктазия с обширным поражением печени не вызывается ни HHT1, ни HHT2. J Med Genet 1996; 33 : 441–443.
CAS
PubMed
PubMed Central
Google Scholar
Рекомендации Американского колледжа медицинской генетики по стандартам интерпретации вариаций последовательностей Genet. Med 2000; 2 : 302–303.
Menasha J, Schechter C, Willner J. Генетическое тестирование: взгляд врачей. Mt Sinai J Med 2000; 67 : 144–155.
CAS
PubMed
Google Scholar
Gallione C, Klaus DJ, Yeh EY, Stenzel TT, Xue Y, Anthony KB и др.Анализ мутации и экспрессии гена эндоглина при наследственной геморрагической телеангиэктазии выявил нулевые аллели. Hum Mutat 1998; 11 : 286–294.
CAS
PubMed
Google Scholar
Cymerman U, Vera S, Pece-Barbara N, Bourdeau A, White R, Dunn Jr J, et al. Выявление наследственной геморрагической телеангиэктазии 1 типа у новорожденных с помощью экспрессии протибвинов и анализа мутаций эндоглина. Pediat Res 2000; 47 : 24–35.
CAS
PubMed
Google Scholar
Ластелла П., Сабба С., Ленато Г.М., Реста Н., Латтанци В., Галлителли М. и др. Мутации и полиморфизмы гена эндоглина у итальянских пациентов с наследственной геморрагической телеангиэктазией. Clin Genet 2003; 63 : 536–540.
CAS
PubMed
Google Scholar
Ямагути Х, Адзума Х, Сигекиё Т, Иноуэ Х, Сайто С.Новая миссенс-мутация в гене эндоглина при наследственной геморрагической телеангиэктазии. Thromb Haemost 1997; 77 : 243–247.
CAS
PubMed
Google Scholar
База данных мутаций наследственной геморрагической телеангиэктазии, Эдинбург, Molecular Genetics Service 2003 Доступно по адресу: http://137.195.14.43/genisysDR/NVC/198/Display/index.htm.
Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I и др.Клинико-молекулярно-генетические особенности легочной гипертензии у пациентов с наследственной геморрагической телеангиэктазией. N Engl J Med 2001; 345 : 325–334.
CAS
PubMed
Google Scholar
Kjeldsen AD, Brusgaard K, Poulsen L, Kruse T., Rasmussen K, Green A, et al. Мутации в гене ALK-1 и фенотип наследственной геморрагической телеангиэктазии в двух больших датских семьях. Am J Med Genet 2001; 98 : 298–302.
CAS
PubMed
Google Scholar
D’Abronzo FH, Swearingen B, Klibanski A, Alexander JM. Мутационный анализ киназ рецепторов типа I и типа II активина / трансформирующего фактора роста-бета в опухолях гипофиза человека. J Clin Endocrinol Metab 1999; 84 : 1716–1721.
CAS
PubMed
Google Scholar
Геморрагическая наследственная телеангиэктазия (болезнь Ренду-Ослера) и инфекционные заболевания: недооцененная ассоциация | Клинические инфекционные болезни
Аннотация
Среди 353 пациентов с наследственной геморрагической телеангиэктазией, ретроспективно проанализированных в период 1985–2005 гг., Мы выявили 67 случаев тяжелой инфекции, у 48 пациентов (13.6%). На внемозговые инфекции приходилось 67% всех инфекций, большинство из которых были связаны с Staphylococcus aureus и были связаны с длительным носовым кровотечением. На церебральные инфекции приходилось 33% всех инфекций, в основном они были вызваны множественными и анаэробными бактериями и были связаны с наличием легочных артериовенозных мальформаций и короткой продолжительностью носовых кровотечений.
Наследственная геморрагическая телеангиэктазия (НГТ) — аутосомно-доминантное заболевание, характеризующееся рецидивирующим носовым кровотечением, кожной телеангиэктазией и висцеральными артериовенозными мальформациями (АВМ), поражающими многие органы, включая легкие, желудочно-кишечный тракт, печень и мозг.Диагноз основывается на критериях Кюрасао [1] и считается определенным, если соблюдаются по крайней мере 3 из следующих критериев: наличие спонтанного и рецидивирующего носового кровотечения, наличие телеангиэктазии, семейный анамнез заболевания и наличие висцеральные поражения.
Два гена принадлежат к разным семействам, связанным с HHT. HHT типа 1 возникает в результате мутаций в ENG на хромосоме 9 (кодирует эндоглин), а HHT типа 2 является результатом мутаций в ACRLV1 на хромосоме 12 (кодирует киназу, подобную рецептору активина [ALK] -1).Мутации любого из этих двух генов составляют большинство (но не все) клинических случаев. Кроме того, мутации в MADh5 (кодирующем SMAD4), которые вызывают ювенильный полипоз / синдром перекрытия HHT, были описаны в другом месте [2], а недавно был опубликован локус HHT3 на хромосоме 5 (5q31.3–5q32). [3].
Все 3 идентифицированных гена HHT кодируют трансмембранные белки эндотелиальных клеток, которые, по-видимому, являются компонентами рецепторных комплексов для факторов роста суперсемейства трансформирующих факторов роста β (TGF-β) [4, 5].Связываясь с рецептором TGF-β типа II, TGF-β может активировать 2 различных рецептора типа I (ALK-1 и ALK-5 в эндотелиальных клетках), каждый из которых имеет противоположные эффекты на миграцию и пролиферацию эндотелиальных клеток [6].
Многочисленные клинические случаи указывают на то, что пациенты с HHT и легочной АВМ более восприимчивы к церебральным абсцессам [7]. Однако в литературе было зарегистрировано только 4 случая опасной для жизни внемозговой тяжелой инфекции, включая остеоартрит, сепсис и спондилодискит, обычно вызванные инфекцией Staphylococcus aureus [8, 9].Иммунные функции изучены недостаточно, и недавно Cirulli et al. [10] сообщили об аномально низком уровне полиморфноядерных клеток и функций моноцитов, предполагая, что пациенты с HHT имеют более высокую предрасположенность к инфекционным осложнениям.
Для изучения инфекционных заболеваний и HHT мы ретроспективно изучили когорту пациентов с HHT, которые посещали один медицинский центр, чтобы оценить частоту внемозговых и церебральных инфекций и описать типы инфекции, бактериологические особенности и факторы риска инфицирования. .Более того, более глубокие знания о факторах, которые способствуют развитию опасных для жизни инфекционных заболеваний у пациентов с HHT, улучшили бы информацию о пациентах и лечение.
Методы . В исследование были включены все пациенты с определенным HHT (подтвержденным генетическим анализом после 2000 г.), которые были направлены во французский справочный центр Hôpital d l’Hôtel Dieu, Lyon в период 1985–2005 гг. Стандартные определения приведены в таблице 1.Тяжелая инфекция определялась как любая инфекция, требующая госпитализации пациента. Мы выделили 3 группы для сравнения данных: пациенты, никогда не переживавшие тяжелую инфекцию; пациенты с церебральной инфекцией, в том числе перенесшие церебральный абсцесс или менингит; и пациенты, перенесшие любую другую тяжелую инфекцию («группа внемозговых инфекций»). Пациенты, которых можно было разделить на 2 группы, были исключены из сравнительного анализа.
Таблица 1
Определения, используемые в исследовании геморрагической наследственной телеангиэктазии (болезнь Рендю-Ослера) и инфекционных заболеваний.
Таблица 1
Определения, используемые в исследовании геморрагической наследственной телеангиэктазии (болезнь Ренду-Ослера) и инфекционных заболеваний.
Средняя продолжительность носового кровотечения была рассчитана на основе медицинских карт пациентов за предыдущие 3 месяца. Всех пациентов, которые были направлены в наш медицинский центр, попросили заполнить таблицу с информацией об общем количестве эпизодов в день и продолжительности носового кровотечения. Поскольку на инфекционные эпизоды могут влиять повторяющиеся носовые явления, мы сравнили продолжительность носового кровотечения у пациентов, у которых развилась серьезная инфекция, с продолжительностью у тех, у кого серьезная инфекция не развивалась.
Легочные АВМ были исследованы с использованием ранее описанного алгоритма скрининга [11]. Поражение печени оценивали с помощью допплерографии [12]. Пищеварительные сосудистые мальформации оценивали эндоскопически; церебральные и спинномозговые АВМ диагностировались с помощью КТ или МРТ. Анализ мутаций был выполнен, как сообщалось ранее [13].
Программные пакеты SAS (SAS Institute) были использованы для статистического анализа. Результаты выражаются в процентах для качественных данных и средних или медианных значений для числовых данных.Факторы риска инфицирования и абсцесса головного мозга оценивали с использованием точного критерия Фишера (для качественных данных). Продолжительность носового кровотечения в зависимости от инфекционного статуса сравнивали с помощью теста Манна-Уитни.
Результаты . Из базы данных было извлечено триста пятьдесят три файла пациентов (для 204 женщин и 149 мужчин). Сто три (45,9%) из 224 обследованных пациентов имели АВМ легких, 75 (49,0%) из 153 обследованных пациентов имели АВМ печени, 23 (20.0%) из 115 обследованных пациентов имели АВМ головного или спинного мозга, а 51 (50,0%) из 100 обследованных пациентов имели АВМ пищеварительной системы. Диагноз подтвержден генетическим анализом 213 (94,2%) из 226 протестированных образцов ДНК. Шестьдесят пациентов (26,5%) имели мутацию ENG , а 153 (67,7%) имели мутацию ACRLV-1 . У остальных 13 пациентов (5,8%) мутации выявлено не было.
У 47 пациентов (13,6%) было 67 тяжелых инфекционных эпизодов; 33 пациента пережили 1 эпизод тяжелой инфекции, 10 — 2 эпизода, 2 — 3 эпизода и 2 — 4 эпизода.Пять пациентов перенесли церебральные и экстрацеребральные инфекции. Средний возраст на момент первой тяжелой инфекции составлял 44,3 года (от 12 до 80 лет). Ни один пациент в нашей когорте не умер от инфекции.
Тридцать два пациента перенесли 45 тяжелых экстрацеребральных инфекций. На внемозговые инфекции пришлось 45 (67%) из 67 тяжелых инфекций. В их число вошли сепсис (9 инфекций [13,4%]), артрит и остеомиелит (6 [8,9%]), кожные инфекции (абсцессы и рожа; 6 [8,9%]), мышечные абсцессы (5 [7,9%]).4%]), спондилодискит (4 [6,3%]), абсцессы печени (5 [7,4%]) и другие тяжелые инфекции (эндокардит, пневмония, пиелонефрит, туберкулез, риккетсиоз и острый аппендицит с перитонитом; 10 [15%] ). S. aureus было идентифицировано в 14 случаях, 1 случай сепсиса был вызван Enterococcus faecalis и Pseudomonas aeruginosa (после мочевой инфекции), а 2 случая мышечных абсцессов были вызваны Streptococcus видов и Pseudomonas. aeruginosa (в одном случае) и Streptococcus viridans и анаэробов (в другом).
У семнадцати пациентов было 19 церебральных абсцессов (у 2 пациентов было 2 отдельных эпизода церебрального абсцесса), а у 3 пациентов был менингит. Церебральные инфекции составили 22 (33%) из 67 тяжелых инфекций. Бактерии были идентифицированы в 7 случаях, а микроорганизмы, выделенные из церебральных абсцессов, в большинстве случаев были множественными и анаэробными видами ( Streptococcus видов, 3 случая; Haemophilus aphrophilus , 3 случая; Actinomyces meyeri , 2 случая;
004 Fusobacterium
). видов, 2 случая; Eikenella corrodens , 1 случай; Micromonas micros , 1 случай; Peptostreptococcus micros , 1 случай; и грамположительные кокки, 1 случай).Наблюдалось три случая менингита, но бактериальные культуры были отрицательными после анализа спинномозговой жидкости. Путь проникновения бактериальной инфекции не установлен.
Факторы риска тяжелой инфекции представлены в таблице 2. Возникновение церебрального абсцесса было достоверно связано с наличием легочной АВМ ( P <0,001) и мутации ENG ( P <0,001). Частота печеночной, неврологической и пищеварительной АВМ существенно не различалась между пациентами без тяжелой инфекции и пациентами с церебральной или экстрацеребральной инфекцией.Внецеребральные инфекции наблюдались независимо от мутировавшего гена. Обнаружена мутация гена ALK-1 в группе пациентов с экстрацеребральной инфекцией с той же частотой, что и в группе без инфекции.
Таблица 2
Факторы риска тяжелой инфекции у пациентов с геморрагической наследственной телеангиэктазией.
Таблица 2
Факторы риска тяжелой инфекции у пациентов с геморрагической наследственной телеангиэктазией.
В группе с экстрацеребральной инфекцией средняя продолжительность носового кровотечения была значительно больше, чем в группе без инфекции (60,0 против 30,5 мин / месяц). Кроме того, в группе с церебральной инфекцией продолжительность была значительно короче (5,0 против 30,5 мин / месяц).
Обсуждение . Частота тяжелой экстрацеребральной инфекции в нашей когорте пациентов с HHT была высокой: 9,0% пациентов с HHT имели по крайней мере 1 тяжелую внемозговую инфекцию, и этот тип инфекции составлял 67% всех тяжелых инфекций среди пациентов с HHT в нашем исследовании. изучение.Кроме того, этот ретроспективный анализ, вероятно, недооценивает истинную заболеваемость инфекциями. В литературе четко не описана заболеваемость такими инфекциями среди населения в целом. Однако исследование, проведенное в США, показало, что заболеваемость сепсисом среди населения в целом составляла 240 случаев на 100 000 человек в год (0,24%) [14]. В Европе данные Европейской системы надзора за устойчивостью к противомикробным препаратам предоставили ценную информацию о заболеваемости S.aureus бактериемия: они пришли к выводу, что заболеваемость составляет 1–32 случая на 100 000 человек (уровень 0,03%) [15].
Абсцессы головного мозга часто встречаются у пациентов с ГГТ, как сообщалось в другом месте [16], и считаются следствием легочных артериовенозных мальформаций (ПАВМ). Абсцессы головного мозга затронули 4% всех пациентов с HHT и 13% пациентов с HHT и PAVM, и на них приходилось 28,3% всех тяжелых инфекций в нашей когорте. Заболеваемость абсцессом головного мозга среди населения в целом не описана в литературе.Тем не менее, мы изучили частоту церебрального абсцесса, пролеченного в одном нейрохирургическом отделении больницы Пьера Вертхаймера (Лион, Франция), зона обслуживания которой составляет 1,7 миллиона человек. Мы выявили 150 случаев церебрального абсцесса за период 1998–2005 гг. Очевидно, что частота церебральных абсцессов у нашей популяции HHT выше, чем у населения в целом.
Интересно, что патогеном, идентифицированным в большинстве случаев экстрацеребральной инфекции, был S. aureus , как ранее сообщалось для 1 пациента со спондилодискитом [8] и 3 других пациентов с HHT [9].Напротив, культуры образцов церебрального абсцесса были положительными на множественные анаэробные организмы. Эти два разных типа инфекции могут отражать разные пути и механизмы проникновения. Было высказано предположение, что абсцессы головного мозга могут быть следствием ПАВМ и прямой связи между легочным и системным кровообращением с обходом капиллярного русла [17]. Наши результаты полностью согласуются с этим предположением: все пациенты с церебральными абсцессами имели ПАВМ, и ни у одного из пациентов без ПАВМ не было церебрального абсцесса.Эта ассоциация также описана в литературе для других причин шунтирования справа налево, связанных с наличием открытого овального отверстия или врожденной цианотической кардиопатией [18]. Удивительно, но средняя продолжительность носового кровотечения была значительно ниже в группе церебрального абсцесса, чем в группе пациентов без тяжелой инфекции. У нас нет четкого объяснения этому, но пациенты с ПАВМ могут часто нуждаться в лечении антибиотиками, что также приводит к лечению хронической носовой инфекции (которая, в свою очередь, играет роль в рецидивирующем носовом кровотечении).
Механизм внемозгового заражения до сих пор не выяснен. Для выявления механизмов инфицирования мы изучили факторы риска тяжелой инфекции у пациентов с ГГТ. Во-первых, мы предположили, что слизистая оболочка носа может быть входным путем для инфекции у пациентов с HHT. Действительно, носовое кровотечение требует тампонады из хлопка, и, как отметили Duval et al. [9], подобные манипуляции могли привести к травмированию слизистой оболочки носа. Присутствие инородного тела может способствовать пролиферации S. aureus в носу, что наблюдается в случаях синдрома токсического шока стафилококка, связанного с использованием тампонов во время каждого менструального цикла [19] или после тампонада носа [20].Действительно, мы обнаружили, что средняя продолжительность носового кровотечения значительно больше у пациентов с экстрацеребральной инфекцией, чем у пациентов с церебральной инфекцией или у пациентов без инфекции.
Интересно, что внемозговые инфекции были связаны как с мутациями гена ENG , так и ALK1 . Это неудивительно, потому что оба гена кодируют трансмембранные белки эндотелиальных клеток, взаимодействующие с сигнальным путем TGF-β; Ожидается, что умеренные нарушения иммунной системы будут способствовать инфекционным заболеваниям.Действительно, сообщалось, что TGF-β обладает способностью ингибировать активацию макрофагов и регулировать иммунные ответы на различные патогены [21]. В настоящее время имеется мало данных об иммунных функциях при HHT, но недавний отчет показал снижение убивающей способности нейтрофилов и моноцитов у пациентов с HHT [10]. Напротив, субпопуляции лимфоцитов были проанализированы Blanco et al. [22] в серии из 39 пациентов с HHT, и авторы не наблюдали каких-либо существенных различий в этом отношении между пациентами и здоровыми людьми.
В заключение мы показываем, что HHT ассоциируется с высокой частотой инфекционных заболеваний. Механические факторы, связанные с умеренными нарушениями иммунной системы, могут объяснить этот высокий уровень инфицирования. Проспективные клинические исследования необходимы для анализа носового кровотечения и роли местной инфекции, а также для оценки даже легких инфекций, поражающих пациентов с ГГТ. Кроме того, необходимы иммунологические исследования, чтобы выявить возможную роль аномалий иммунной системы.Действительно, осведомленность об инфекционных рисках при HHT облегчит диагностику и улучшит уход за пациентами.
Благодарности
Мы благодарим N. Berthoux и J. Lucido за техническую помощь.
Возможный конфликт интересов . Все авторы: без конфликтов.
Список литературы
1« и др.
Диагностические критерии наследственной геморрагической телеангиэктазии (синдром Ренду-Ослера-Вебера)
,
Am J Med Genet
,
2000
, vol.
91
(стр.
66
—
7
) 2« и др.
Комбинированный синдром ювенильного полипоза и наследственной геморрагической телеангиэктазии, связанный с мутациями в MADh5 (SMAD4)
,
Lancet
,
2004
, vol.
363
(стр.
852
—
9
) 3,,,.
Новый локус наследственной геморрагической телеангиэктазии (HHT3) отображается на хромосоме 5
,
J Med Genet
,
2005
, vol.
42
(стр.
577
—
82
) 4« и др.
Эндоглин, белок эндотелиальных клеток, связывающий TGF-бета, является геном наследственной геморрагической телеангиэктазии 1 типа
,
Nat Genet
,
1994
, vol.
8
(стр.
345
—
51
) 5« и др.
Участие трансформирующего фактора роста бета в патогенезе наследственной геморрагической телеангиэктазии
,
Curr Pharm Des
,
2006
, vol.
12
(стр.
1195
—
200
) 6,,,,.
Наследственная геморрагическая телеангиэктазия, сосудистая дисплазия, влияющая на сигнальный путь TGF-бета
,
Clin Med Res
,
2006
, vol.
4
(стр.
66
—
78
) 7« и др.
Абсцесс головного мозга: необходимость скрининга легочных артериовенозных мальформаций
,
Нейроэпидемиология
,
2005
, vol.
24
(стр.
76
—
8
) 8,,,,,.
Семейная телеангиэктазия, ангиоматоз Ренду-Ослера и бактериальный спондилодисцит [на французском языке]
,
Rev Med Interne
,
1998
, vol.
19
(стр.
938
—
9
) 9« и др.
Рецидивирующие Staphylococcus aureus экстрацеребральные инфекции, осложняющие наследственную геморрагическую телеангиэктазию (болезнь Ослера-Ренду-Вебера)
,
Am J Med
,
2001
, vol.
110
(стр.
671
—
2
) 10,,, et al.
Пациенты с наследственной геморрагической телеангиэктазией (HHT) демонстрируют дефицит полиморфноядерных клеток и окислительный взрыв моноцитов и фагоцитоз: возможная корреляция с измененной адаптивной иммунной реактивностью в HHT
,
Curr Pharm Des
,
2006
, vol.
12
(стр.
1209
—
15
) 11,,,,,.
Легочные артериовенозные мальформации у пациентов с наследственной геморрагической телеангиэктазией
,
Am J Respir Crit Care Med
,
2004
, vol.
169
(стр.
994
—
1000
) 12,,, et al.
A. Допплеровское ультразвуковое исследование пороков развития сосудов печени при наследственной геморрагической телеангиэктазии — результаты обширного скрининга
,
Ultraschall Med
,
2004
, vol.
25
(стр.
348
—
55
) 13« и др.
Молекулярный скрининг генов ALK1 / ACVRL1 и ENG при наследственной геморрагической телеангиэктазии во Франции
,
Hum Mutat
,
2004
, vol.
23
(стр.
289
—
99
) 14,.
Эпидемиология сепсиса: последние достижения
,
Curr Infect Dis Rep
,
2005
, vol.
7
(стр.
329
—
34
) 15,,,,,.
Staphylococcus aureus bacteremia, Европа
,
Emerg Infect Dis
,
2005
, vol.
11
(стр.
1798
—
9
) 16,,,,.
Легочные артериовенозные мальформации, наследственная геморрагическая телеангиэктазия и абсцесс головного мозга
,
Дыхание
,
2006
, vol.
73
(стр.
553
—
7
) 17,.
Легочные артериовенозные мальформации: современный обзор
,
Am J Respir Crit Care Med
,
1998
, vol.
158
(стр.
643
—
61
) 18,,,,,.
Абсцесс головного мозга, вызванный видами Capnocytophaga , видами Actinomyces и Streptococcus intermediateus у пациента с цианотическим врожденным пороком сердца
,
Eur J Clin Microbiol Infect Dis
,
2002
, vol.
21
(стр.
236
—
7
) 19,,,.
Синдром токсического шока: эпидемиологические особенности, рецидивы, факторы риска и профилактика
,
N Engl J Med
,
1980
, vol.
303
(стр.
1429
—
35
) 20,,,,.
Синдром токсического шока, связанный с тампонированием носа
,
Arch Otolaryngol
,
1983
, vol.
109
(стр.
624
—
6
) 21.
Двунаправленная регуляция функции макрофагов с помощью TGF-beta
,
Microbes Infect
,
1999
, vol.
1
(стр.
1275
—
82
) 22.
Анализ субпопуляции лимфоцитов в серии из 39 пациентов с HHT
,
Программа и тезисы 6-й научной конференции HHT (Лион, Франция)
,
2005
стр.
54
© 2007 Американское общество инфекционных болезней
Наследственная геморрагическая телеангиэктазия у пациента из Судана
Предпосылки . Наследственная геморрагическая телеангиэктазия (HHT), также известная как синдром Ослера – Вебера – Ренду, является редким аутосомно-доминантным заболеванием, которое приводит к сосудистой дисплазии, поражающей в основном висцеральные и кожно-слизистые органы.Презентация кейса . Женщина 65 лет с 12-летним анамнезом рецидивирующих спонтанных носовых кровотечений обратилась с жалобой на одышку, легкую утомляемость и двусторонний отек нижних конечностей. В ее семейном анамнезе были выявлены определенные наследственные геморрагические телеангиэктазии у родственников первой степени родства. В течение предыдущих 15 дней у нее было три эпизода рецидивирующего носового кровотечения. У нее хроническая митральная регургитация. Осмотр показал телеангиэктазии на языке, нижней губе и руке, а также признаки застойной сердечной недостаточности.Пациент соответствовал 3 \ 4 критериям Кюрасао и имел определенную ГГТ. Ее лабораторное обследование показало, что уровень гемоглобина составляет 5,4 г / дл. Эхокардиография выявила фракцию систолического выброса слева 51% с дилатацией левого предсердия и тяжелой митральной регургитацией. Рентген грудной клетки показал признаки кардиомегалии и отека легких. Ультразвуковое исследование брюшной полости показало увеличенный размер печени с однородной текстурой и скопление печеночных вен без признаков АВМ печени. Ей лечили внутривенным фуросемидом, добавкой железа, транексамовой кислотой, переливанием крови и тампонами для носа. Выводы . HTT обычно проходит незамеченным в Судане. Редкость HHT, трудности с проведением диагностических исследований изображений и низкая клиническая подозрительность среди врачей являются важными факторами, способствующими этому. Анемия, возникающая в результате повторяющихся носовых кровотечений, может играть важную роль в провоцировании острой сердечной недостаточности у пациентов с хроническим ревматическим пороком клапанов сердца.
1. Введение
Наследственная геморрагическая телеангиэктазия (НГТ) — редкое генетическое заболевание с аутосомно-доминантным типом наследования.Предполагаемая распространенность составляет 1 / 5000–1 / 8000 населения [1]. Он характеризуется диффузными телеангиэктазами (например, папулы рубинового цвета, которые бледнеют при надавливании), рецидивирующим носовым кровотечением и широко распространенными артериовенозными мальформациями (АВМ). В равной степени страдают самцы и самки.
Диагноз основан на наличии трех из четырех критериев, называемых критериями Кюрасао, которые включают рецидивирующее спонтанное носовое кровотечение, небольшую системную телеангиэктазию (из-за АВМ), вовлечение внутренних органов и сильный семейный анамнез ГГТ (Таблица 1).
| |||||||||||||||
Если они соответствуют трем или более критериям, считается, что они имеют определенный HHT. Если пациенты соответствуют 2, то считается, что у них есть возможный диагноз HHT, и «маловероятный», если присутствует 0 или 1 критерий. | |||||||||||||||
Хотя симптомы обычно различаются в зависимости от возраста, носовое кровотечение обычно является наиболее частым клиническим признаком [2].
Насколько нам известно, это первый зарегистрированный случай HHT в Судане.Редкость заболевания, трудности с предоставлением диагностических изображений и генетического тестирования, а также низкая клиническая подозрительность среди врачей — все это создает проблемы при диагностике и лечении HHT, особенно в больницах с ограниченными ресурсами.
2. Описание клинического случая
65-летняя суданская женщина обратилась в университетскую больницу в Судане с жалобами на одышку (класс 3 Нью-Йоркской кардиологической ассоциации (NYHA)), легкую утомляемость и двусторонний отек нижних конечностей.В анамнезе не было сахарного диабета или гипертонии. В течение предыдущих 15 дней у нее было три эпизода повторяющихся носовых кровотечений, которые не были связаны с какой-либо конкретной деятельностью, осанкой или сезонными колебаниями. Кровотечения из других отверстий не было. В течение многих лет эпизоды носовых кровотечений случались часто, и пациентка получила медицинскую консультацию в своей сельской больнице. Во время этих эпизодов пациенту сделали переднюю тампону носа, сделали переливание, а затем выписали, получая препараты железа.Затем пациент обратился в университетскую больницу для более тщательного обследования. Во время обследования пациента у пациентки произошел эпизод носового кровотечения, в связи с которым она перенесла переднюю тампону носа, чтобы контролировать носовое кровотечение, и получила 500 мг транексамовой кислоты в таблетках TDS, транексамовую кислоту для местного применения и упакованные клетки для переливания. Обследование показало, что у пациентки имеется известный случай хронической митральной регургитации, и ей была запланирована замена митрального клапана, которая не была проведена из-за финансовых проблем.
Семейный анамнез был значимым для определенного HHT в виде рецидивирующего спонтанного носового кровотечения и таких же небольших эритематозных поражений (телеангиэктазий) у ее бабушки, ее сына и ее дочери.
При медицинском осмотре выяснилось, что пациент выглядел больным, бледным и без желтухи. JVP был поднят. АД 110/60, пульс 88 ударов в минуту, не снижался. Насыщение кислородом составляло 90% на воздухе помещения. Осмотр ротовой полости выявил множественные небольшие эритематозные поражения, которые бледнеют при надавливании (телеангиэктазии) на ее язык (рис. 1), и одно поражение на нижней губе (рис. 2).Обследование кисти показало одну телеангиэктазию на ладонной стороне правой руки (рис. 3). Ее сердечно-сосудистое обследование выявило пан-систолический шум в апикальной области, который распространяется в подмышечную впадину, и двусторонние тонкие базальные крепитации. Печень болезненная, пальпируется на 8 см ниже правого края ребра без слышимого шума, имеется двусторонний отек нижних конечностей.
Лабораторные исследования при поступлении выявили гипохромную микроцитарную анемию (гемоглобин 5.4 г / дл). Другие лабораторные исследования описаны в таблице 2.
| ||||||||||||||||||||||||||
ЭКГ не способствовала развитию, кроме синусовой тахикардии.Рентген грудной клетки выявил отклонение верхней доли, скопившиеся тени корней и пятнистую консолидацию в нижних долях (признаки отека легких и кардиомегалии) (рис. 4). Эхокардиография показала фракцию левого систолического выброса 51% с дилатацией левого предсердия и тяжелой митральной регургитацией. Ультразвуковое исследование брюшной полости (УЗИ) показало увеличенный размер печени (19 см) с однородной текстурой и заложенность печеночных вен. На УЗИ артериовенозных мальформаций печени не выявлено (рис. 5).
Диагностические проблемы: КТ ангиография легких не проводилась из-за финансовых трудностей. Альтернативой является контрастная эхокардиография. Он был предназначен для скрининга пациента на наличие легочных АВМ. Скрининг может быть проведен с помощью контрастной эхокардиографии, а затем компьютерной томографии в случае шунта справа налево. Наиболее опасным осложнением легочной АВМ, например шунтом справа налево, является абсцесс мозга из-за парадоксальной септической эмболии, которая обычно захватывается легочными капиллярами, но может проходить через легочную АВМ.Если у пациентов с HHT не исключено наличие шунта справа налево, перед любыми нестерильными процедурами следует назначать профилактические антибиотики, чтобы предотвратить абсцесс мозга.
КТ брюшной полости с контрастированием также не была проведена из-за финансовых проблем. Он был предназначен для выявления АВМ печени и исключения сердечной недостаточности с высоким выбросом. Однако клинически признаков гипердинамического кровообращения у пациента (например, схлопывающегося пульса) не было.
Диагноз был определен как HHT, поскольку она соответствовала трем критериям Кюрасао (спонтанное, рецидивирующее кровотечение из носа, семейный анамнез у родственника первой степени родства и множественные телеангиэктазии на характерных участках, таких как язык, губы и руки).Считается, что анемия, которая была связана с повторяющимся носовым кровотечением и факторами питания, играет важную роль в ускорении сердечной недостаточности у этого пациента с хронической ревматической митральной регургитацией.
Пациент проходил лечение внутривенным фуросемидом, добавкой железа, транексамовой кислотой, переливанием крови и тампонами носа. Она была выписана на 7-й день на таблетках лизиноприла и железосодержащих добавках в дополнение к консультациям по домашней профилактике носового кровотечения.
3.Обсуждение
Наследственная геморрагическая телеангиэктазия (НГТ) — редкое аутосомно-доминантное заболевание, характеризующееся артериовенозными мальформациями (АВМ) внутренних органов и кожно-слизистыми телеангиэктазиями. У этих АВМ отсутствуют промежуточные капилляры, что приводит к прямым связям между артериями и венами. Они могут поражать кожу, слизистые оболочки, мозг, легкие, желудочно-кишечный тракт и / или печень [3]. Наиболее частыми клиническими проявлениями являются повторяющиеся спонтанные носовые кровотечения, которые наблюдаются у 50% пациентов с ГГТ в возрасте десяти лет, причем частота случаев увеличивается с возрастом.
HHT был впервые описан в 1864 году у человека с сосудистой мальформацией и рецидивирующим носовым кровотечением [4]. В 1996 году Ренду впервые заметил корреляцию между наследственным носовым кровотечением и телеангиэктазией у 52-летнего мужчины [5]. Ослер в 1901 г. и Вебер в 1907 г. затем идентифицировали связь между геморрагическими поражениями кожи и слизистых оболочек и их семейной наследственностью [6, 7]. Название «наследственная геморрагическая телеангиэктазия», описанное Ханесом в 1909 году, определяет характеристики заболевания [8].
Мутации в генах, включая гены ACVRL1, ENG и SMAD4, ответственны за наследственную геморрагическую телеангиэктазию. Мутация ENG может привести к наследственной геморрагической телеангиэктазии 1-го типа, в то время как наследственная геморрагическая телеангиэктазия 2-го типа вызывается мутациями в гене ACVRL1. Синдром ювенильного полипоза / наследственной геморрагической телеангиэктазии вызван мутацией в гене SMAD4. Было высказано предположение, что определенные мутировавшие гены HHT связаны с определенными клиническими признаками, например, мутация ACVRL1, как было установлено, больше связана с АВМ печени, АВМ спинного мозга, носовым кровотечением и легочной гипертензией.Было обнаружено, что мутации ENG больше связаны с АВМ легких и головного мозга, а мутации MADh5 связаны с ювенильным полипозом толстой кишки.
Диагноз HHT зависит от диагностических критериев, называемых критериями Кюрасао (таблица 1). Нашей пациентке был поставлен диагноз HHT, поскольку она соответствовала трем критериям Кюрасао (спонтанное, рецидивирующее кровотечение из носа, семейный анамнез у родственника первой степени родства и множественные телеангиэктазии на характерных участках, таких как язык, губы и руки).
У пациентов с HHT сердечная недостаточность обычно возникает из-за печеночной артериально-венозной мальформации, которая вызывает сердечную недостаточность с высоким выбросом. Считается, что у нашего пациента анемия, которая была связана с рецидивирующим носовым кровотечением и факторами питания, играет важную роль в ускорении сердечной недостаточности у этого пациента с хронической ревматической митральной регургитацией. Железодефицитная анемия — частое осложнение ГГТ; хроническая кровопотеря из слизистой оболочки носа или кишечника может привести к истощению запасов железа, что, в свою очередь, может привести к микроцитарной или нормоцитарной анемии.
Согласно рекомендациям ESC HF 2016, при первичной оценке пациента с недавно диагностированной сердечной недостаточностью рекомендуются тесты на насыщение ферритином и трансферрином (TSAT). Лечение рекомендуется, когда уровень ферритина <100 мкл г / л, или ферритина от 100 до 299 мкг г / л и TSAT <20%.
Не существует лечебного лечения HHT; лечение является симптоматическим, в котором основное внимание уделяется преобладающему клиническому признаку и его тяжести. Первым шагом в лечении носового кровотечения является консультирование пациента по поводу использования домашних профилактических мер, таких как увлажнение носа, солевые спреи или мазь, чтобы избежать сухости слизистой оболочки носа и снизить риск травмы носа в виде ковыряния в носу и носовое дыхание [9].Другие варианты включали тампон, фотокоагуляцию, внутрисосудистую эмболизацию и хирургическое вмешательство. Эти методы имеют высокую частоту рецидивов при ограниченной эффективности, и они недоступны во многих больницах с ограниченными ресурсами. Такие препараты, как бевацизумаб (моноклональное антитело к противоваскулярному эндотелиальному фактору роста), широко используются при лечении сердечной недостаточности с высоким выбросом при ГГТ с адекватной безопасностью и эффективностью [10]. Тамоксифен (антиэстрогеновый препарат) [11] и талидомид (антиангиогенное и иммуномодулирующее средство) [12] также исследовались при лечении HHT.
Согласно ESC, лечение сердечной недостаточности с сохраненной фракцией выброса (HFpEF) — это лечение основной причины, такой как анемия; диуретики рекомендуются пациентам с застойным флюидом с HFpEF для облегчения симптомов и признаков.
Пациенты с HHT имеют более низкую ожидаемую продолжительность жизни, чем население в целом; прогноз зависит от тяжести симптомов. Наиболее частые причины смерти при ГГТ — сердечная недостаточность и сепсис [13].
4. Заключение
В заключение, случаи HHT могли остаться незамеченными из-за редкости заболевания, что приводит к низкой клинической подозрительности среди врачей.К другим способствующим факторам относятся вариации в клинических проявлениях болезни, трудности с предоставлением диагностических визуализационных исследований, а визуализирующие исследования могут быть недоступны во многих больницах с ограниченными ресурсами. Анемия, возникающая в результате рецидивирующего носового кровотечения, может играть важную роль в провоцировании острой сердечной недостаточности у пациентов с хроническим ревматическим пороком клапанов сердца.
Аббревиатуры
| HHT: | Наследственная геморрагическая телеангиэктазия | |
| OWRD: | Болезнь Ослера-Вебера-Ренду | |
| 934-йоркская болезнь | ||
| BNP: | Натрийуретический пептид мозга | |
| HFpEF: | Сердечная недостаточность с сохраненной фракцией выброса | |
| ACVRL1: | Активин A-рецептор-подобный тип 1 | |
| TSAT: | Насыщение трансферрина. |
Доступность данных
Данные, использованные для подтверждения результатов этого исследования, включены в статью.
Этическое одобрение
Статья была одобрена Комитетом по этике исследований Университета Гезиры, медицинский факультет, Судан.
Согласие
От пациента было получено письменное согласие на публикацию.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Вклад авторов
О.Е., главный автор статьи, участвовал в подготовке пациента и отвечал за последующее наблюдение.А.А. участвовал в подготовке пациента, написании статьи и редактировании статьи. MA, OM и AE участвовали в подготовке пациента и написании статьи.
Благодарности
Авторы выражают особую благодарность Университету Гезиры, медицинскому факультету, Судан.
Симптоматические дети с наследственной геморрагической телеангиэктазией: опыт педиатрического центра | Врожденные пороки | JAMA Педиатрия
Объектив
Оценить клинико-генетические характеристики симптоматических детей с наследственной геморрагической телеангиэктазией (НГТ).
Дизайн
Перекрестное исследование.
Настройка
Клиники HHT в Торонто.
Участники
Все дети с симптоматической ГГТ получали лечение с 1 апреля 1996 г. по 31 декабря 2002 г.
Вмешательства
Участников обследовали на наличие висцеральных артериовенозных мальформаций (АВМ). Молекулярное тестирование проводилось у детей или членов их семей.
Основные показатели результатов
Распространенность носового кровотока, телеангиэктазий, легочных и церебральных АВМ, а также генетические характеристики.
Результаты
Четырнадцать детей поступили с проявлениями ВГТ. У семи были кардиореспираторные симптомы, связанные с легочными АВМ. У троих были неврологические симптомы, вторичные по отношению к кровотечению из спинномозговой или мозговой АВМ. Двое были направлены из-за кожных телеангиэктазий и 2 из-за множественных эпизодов носового кровотечения. По результатам скрининга АВМ головного мозга выявлена у 1 из 11 неврологически бессимптомных детей. Из детей без респираторных симптомов у 1 была диагностирована определенная легочная АВМ, а у 1 — подозрение на легочную АВМ.Четверо детей с легочными АВМ несли мутацию гена эндоглина (HHT тип 1), и у одного ребенка была мутация гена киназы 1, подобного рецептору активина (HHT тип 2). Двое детей с АВМ позвоночника принадлежат к одной семье HHT типа 2. Мутации не обнаружено ни у одного ребенка с легочными АВМ и у одного с церебральными АВМ.
Выводы
Висцеральные АВМ и телеангиэктазы слизистых оболочек присутствуют у детей с ГГТ и могут привести к опасным для жизни событиям. Неспособность идентифицировать мутацию, связанную с заболеванием для каждого ребенка, предполагает сложные мутации или новые гены HHT.
Наследственная геморрагическая телеангиэктазия (HHT) или синдром Ренду-Ослера-Вебера — аутосомно-доминантная сосудистая дисплазия. Хотя первоначально считалось редким, недавние сообщения предполагают распространенность от 1: 5000 до 1:10 000. 1 , 2 Наследственная геморрагическая телеангиэктазия характеризуется кожно-слизистыми телеангиэктатическими поражениями, приводящими к носовым кровотечениям у 90% пациентов 3 кровотечение из желудочно-кишечного тракта — от 20% до 30%. 4 Висцеральные артериовенозные мальформации (АВМ) преимущественно обнаруживаются в легочном, церебральном и печеночном кровообращении. 5 , 6
Клинические характеристики ГГТ у детей недостаточно хорошо описаны. Симптомы и признаки, как правило, развиваются в позднем детстве и в раннем взрослом возрасте, 2 , 5 , 6 , и у многих детей не возникает носовых кровотечений и не развиваются телеангиэктазы кожи в первое десятилетие жизни. 6 Опубликованы многочисленные отчеты 7 -12 о церебральных и легочных АВМ у детей; однако распространенность таких АВМ не установлена, и нет опубликованных серий, описывающих клинические проявления и генетику ГГТ в детстве.
Мы стремились охарактеризовать клиническую картину, наличие бессимптомных висцеральных АВМ и генотип у детей с симптомами ГГТ.
педиатрических и взрослых клиник HHT были открыты соответственно при больнице для больных детей и больнице Святого Михаила в Торонто в 1996 году. Оба центра являются специализированными центрами медицинской помощи при Университете Торонто. В настоящее исследование были включены все дети и подростки (в возрасте 0–18 лет) с симптомами ГГТ с 1 апреля 1996 г. по 31 декабря 2002 г.Дети, направленные из-за семейного анамнеза и у которых было обнаружено ГГТ, не включались.
Детей тщательно обследовали на телеангиэктазии кожи и слизистых оболочек. Также были выполнены тщательные исследования слизистых оболочек полости рта и конъюнктивы, а также передняя риноскопия на предмет носовых телеангиэктазий.
Детей обследовали на висцеральные АВМ. Легочные АВМ диагностировались с помощью эхокардиографии с контрастированием в солевом растворе и усиленной компьютерной томографии грудной клетки с последующей ангиографией легких по показаниям.Церебральные АВМ диагностировали с помощью магнитно-резонансной томографии головы с последующей церебральной ангиографией по показаниям.
Клинический диагноз HHT был основан на установленных клинических диагностических критериях HHT. Эти критерии включают рецидивирующее носовое кровотечение, множественные кожные телеангиэктазы, висцеральные АВМ или телеангиэктазы, а также наличие у члена семьи первой степени родства с ГГТ. 13 Наличие 3 критериев подтверждает диагноз, тогда как наличие 2 критериев приводит к подозрению на ГГТ. 13
Молекулярное тестирование мутаций HHT было выполнено в рамках исследовательского проекта, направленного на разработку клинически применимого диагностического теста и понимание основных механизмов заболевания. Информированное согласие было получено от участвующих пациентов и / или их родителей, в зависимости от обстоятельств, а протоколы исследования были одобрены этическим комитетом Научно-исследовательского института Детской больницы.
Геномную ДНК экстрагировали из лимфоцитов крови с использованием имеющихся в продаже наборов для экстракции ДНК (ДНКзол [Gibco BRL, Гейтерсбург, Мэриленд] или набор для выделения ДНК Puregene [Gentra Systems Inc, Миннеаполис, Миннесота]).Анализ мутаций для гена эндоглина ( ENG ; для HHT типа 1 [HHT1]) выполняли посредством количественной мультиплексной полимеразной цепной реакции и секвенирования экзонов, как описано ранее. 14 , 15 Анализ мутаций для активин-рецептор-подобной киназы 1 ( ACVRL1 ; для HHT типа 2 [HHT2]) был выполнен путем секвенирования экзонов в соответствии с опубликованными процедурами. 16 , 17
Мы обследовали 127 детей в клиниках Торонто HHT с 1 апреля 1996 г. по 31 декабря 2002 г.Сто тринадцать были направлены из-за семейного анамнеза HHT и 14 (возрастной диапазон 0-15 лет; средний возраст 9 лет) из-за симптомов или признаков HHT. В этом отчете описаны эти 14 детей. Их клиническая картина, характеристики заболевания и результаты молекулярного анализа сведены в таблицу. Девять (64%) имели семейный анамнез HHT, а мутации были обнаружены у 8.
Таблица.
Клинические и молекулярные характеристики детей с HHT
Семь детей (50%) поступили с АВМ легких; 3 (21%) — с АВМ головного или спинного мозга; и 4 (29%) — с кожно-слизистыми телеангиэктазами.Носовые кровотечения были обычным явлением, наблюдались у 12 детей (86%), но обычно были легкими; тяжелые носовые кровотечения были описаны только у 3 детей.
КЛИНИЧЕСКАЯ ПРЕЗЕНТАЦИЯ ЛЕГКИХ АВМ
Из 7 детей с легочными АВМ у 5 была длительная одышка и цианоз, начавшиеся за несколько лет до постановки диагноза, у 1 — застойная сердечная недостаточность, у 1 — кровохарканье.Легочная картина и исходы у пациентов с 1 по 5, 7 и 10 были представлены в большой серии, обобщающей результаты транскатетерной эмболотерапии легочных АВМ 18 и диффузных легочных АВМ. 19 Однако полный скрининг других АВМ и анализ мутаций HHT ранее не описывались.
Пациенту 1 в 11 месяцев был поставлен диагноз лактоацидоза и митохондриальной миопатии, вероятно, не связанных с ГГТ. Диагноз ГГТ был установлен через 3 года после направления к врачу по поводу носового кровотечения, одышки и цианоза.В левой нижней доле обнаружена большая легочная АВМ, потребовавшая эмболизации. Через 5 лет наблюдения ребенок здоров, признаков АВМ легких нет. Этот ребенок был первым в семье, у которого были признаки ГГ. Молекулярный анализ выявил мутацию сайта сплайсинга ENG (g.IVS3 + 1G> A), которая приводит к пропуску экзона 3 и экспрессии более мелкого и нестабильного мутантного белка ENG в рамке считывания. Впоследствии у бессимптомной матери ребенка были выявлены АВМ в легких и печени.Она передала мутацию своему ребенку, но не унаследовала ее от своих родителей, которые не были носителями мутации и не проявляли признаков HHT. Результаты ДНК-фингерпринта с 8 различными маркерами подтвердили, что ребенок и его мать разделяют ожидаемые аллели со своими родителями. 15 Таким образом, эта мутация возникла у матери de novo. Об этом же варианте сайта сплайсинга также сообщалось в неродственной семье с несколькими поколениями HHT, 20 , подтверждая, что одна и та же мутация может возникать в разных семьях.
Пациент 2 имел очень сложное течение, характеризовавшееся поражением многих органов. В раннем детстве ей поставили диагноз гипотиреоз и задержка развития. У нее был длительный цианоз и множественные кожно-слизистые телеангиэктазы. Однако диагноз ГГТ был установлен только в 12 лет, когда у нее были выявлены диффузные АВМ легкого и затылочная АВМ головного мозга. У нее также было заболевание печени с повышенным уровнем трансаминаз и коагулопатией, которая оставалась загадкой даже после биопсии печени.Транскатетерная эмболотерапия дала лишь временное улучшение. Предложили трансплантацию легкого, но отказались. Носовое кровотечение появилось в 17 лет. Пациент продолжал страдать тяжелым цианозом и умер в 18 лет. Была идентифицирована новая замена экзона 5 ENG (c.640G> A), которая превращает глицин в серин по кодону 214. Пациент не имел семейного анамнеза HHT. Такой же вариант последовательности был обнаружен у ее бессимптомной матери и, вероятно, связан с заболеванием, поскольку не был идентифицирован более чем у 200 человек, прошедших тестирование.Однако подтверждение того, что этот вариант последовательности связан с заболеванием, требует дальнейшего изучения.
Пациент 3 поступил в 7-летнем возрасте с множественными телеангиэктазами и длительным цианозом. В течение 13 лет наблюдения множественные легочные АВМ были эмболизированы 3 раза (рисунок, вторая эмболотерапия) с улучшением симптомов и исчезновением гипоксемии. Ранее неизвестная мутация ENG была обнаружена у пациента и 2 больных родственников: вставка из 4 оснований c.982_983insGCCT в экзоне 7, что вызывает сдвиг кадра.
Рисунок.
Легочная ангиография выявляет множественные легочные артериовенозные мальформации (стрелки) в правом (A) и левом (B) легких пациента 3 и спирали в правом легком (наконечник стрелки).
Пациенты 4 и 5 поступили с тяжелым цианозом. Диффузные легочные АВМ потребовали множественных транскатетерных эмболизов (и правой нижней лобэктомии у пациента 4) с лишь временным улучшением.
Пациент 6 вскоре после рождения обратился с жалобой на тяжелую респираторную недостаточность. В левой верхней доле была обнаружена большая легочная АВМ, вызывающая застойную сердечную недостаточность, и была выполнена лобэктомия. После 14 лет наблюдения, за исключением нескольких кожных телеангиэктазий, которые появились во время наблюдения, у нее нет симптомов легочных АВМ. Миссенс-мутация в экзоне 4 ACVRL1 (c.383C> A), которая изменяет аланин на аспарагиновую кислоту в кодоне 128, была обнаружена у этого ребенка и ее пораженного родителя. 21
Пациент 7 обратился в 11 лет с рецидивирующим кровохарканьем. Визуализация грудной клетки выявила множественные легочные АВМ, которые были эмболизированы со значительным улучшением. Рецидивирующее кровохарканье, вызванное телеангиэктазами гортани и трахеи, хорошо поддается лазерной абляции. Шесть лет спустя ей потребовалась транскатетерная эмболотерапия двусторонних АВМ легких, вызывающих гипоксию, с исчезновением симптомов. Кожные телеангиэктазы были замечены в 15-летнем возрасте. У этой пациентки и ее пораженного родителя была обнаружена дупликация ENG экзонов с 3 по 8, и можно было наблюдать большой нестабильный внутриклеточный белок, как ранее сообщалось для неродственной семьи. 12
КЛИНИЧЕСКАЯ ПРЕЗЕНТАЦИЯ АВМ головного и спинного мозга
Трое детей обратились с неврологическими симптомами. Пациентам 8 и 9, двоюродным братьям и сестрам, был поставлен диагноз АВМ позвоночника, и их первоначальное течение было описано в другом месте. 22 У пациента 8 появились новые неврологические симптомы через 3 года после перевязки питающей артерии к АВМ.Обнаружена и эмболизирована новая спинномозговая АВМ. Эти двое детей принадлежат к одной семье с HHT2, характеризующейся миссенс-мутацией ACVRL1 (c.1232G> A), которая превращает аргинин в глутамин в кодоне 411, и сообщается как вызывающая заболевание мутация в семьях, кажущихся несвязанными. 23
Пациент 10 обратился с жалобами на гемипарез справа, головную боль и судороги. Обнаружены и эмболизированы две церебральные АВМ. Полное секвенирование генов ENG и ACVRL1 не позволило идентифицировать мутацию у ее клинически пораженных родственников.
Всем 3 пациентам потребовалась реабилитация с исчезновением большинства симптомов.
Кожно-слизистые телеангиэктазы и носовое кровотечение
Только 4 ребенка были направлены из-за кожно-слизистых телеангиэктазий и носового кровотечения; однако у всех детей в период наблюдения были выявлены кожно-слизистые телеангиэктазии и / или носовое кровотечение.Носовое кровотечение появилось уже к 3 годам (средний возраст первого эпизода ± стандартное отклонение 7,5 ± 4,0 года).
Пациент 11 был направлен из-за множественных кожных телеангиэктазий, носового кровотечения и эпизода транзиторной ишемической атаки, в семейном анамнезе ГГТ не было. Результаты медицинского осмотра выявили множественные кожные телеангиэктазы на ладонях и слизистой оболочке носа. Эхокардиография с инъекцией физиологического раствора и компьютерная томография грудной клетки не продемонстрировали легочные АВМ. Легочная ангиография не проводилась, потому что вероятность обнаружения АВМ, достаточно большой, чтобы ее можно было эмболизировать, была очень низкой.Магнитно-резонансная томография головы не показала ишемических или геморрагических изменений. Церебральных АВМ не наблюдалось. Причина транзиторной ишемической атаки не может быть определена, и мигрень остается вероятным диагнозом. Однако нельзя исключать наличие мельчайших легочных АВМ. Молекулярный анализ не выявил мутации в гене ENG или ACVRL1 .
Пациент 12 обратился с жалобами на множественные телеангиэктазии на лице и конечностях, а также носовое кровотечение, начавшееся в 3-летнем возрасте, которое, как правило, было частым (до нескольких раз в неделю) и трудно контролируемым (каждый эпизод продолжался до 1 часа).Однако до обращения в нашу клинику носовое кровотечение не лечили. Пациенты 13 и 14 поступили с носовым кровотечением, и оба были успешно вылечены с помощью прижигания или лазерной абляции. У пациента 14 была обнаружена ранее описанная семейная миссенс-мутация ACVRL1 (c.1232G> A), и он не связан с пациентами 8 и 9. 17
АСИМПТОМАТИЧЕСКИЕ ПТрО, ОБНАРУЖЕННЫЕ ПРИ СКРИНИНГЕ
Тринадцать детей прошли скрининг на АВМ легких и головного мозга.Из 11 неврологически бессимптомных детей у пациента 2 была обнаружена АВМ головного мозга. Из детей без респираторных симптомов у пациента 10 были обнаружены АВМ легкого, и ему была проведена транскатетерная эмболотерапия. Пациент 14 с генетически подтвержденным HHT2 имел результаты эхокардиографии и компьютерной томографии, указывающие на небольшие легочные АВМ. За ним наблюдают консервативно.
Клинико-диагностические критерии hht
Одиннадцать из 14 детей в настоящее время соответствуют установленным диагностическим критериям определенного HHT.Однако, поскольку носовое кровотечение или телеангиэктазии появились у 5 детей после их первого обращения, 4 из них не соответствовали этим критериям при обращении. На момент написания этой статьи у 3 детей все еще не было достаточных критериев для точного диагноза ГГТ. Транзиторная ишемическая атака у пациентки 11 вызывает серьезное беспокойство по поводу наличия у нее АВМ легких; однако обширная проработка не продемонстрировала ничего. Таким образом, согласно диагностическим критериям, у нее есть подозрение на ГГТ. Пациенты 12 и 13 имеют телеангиэктазии и тяжелое носовое кровотечение без признаков висцеральных АВМ и отсутствия семейного анамнеза ГГТ.Однако в этих семьях существуют телеангиэктазы и / или носовое кровотечение, и у членов семьи не проводился скрининг на висцеральные АВМ.
В Торонто открыта детская клиника HHT для лечения детей с HHT. В недавней серии 24 , описывающей 15 детей с HHT, носовое кровотечение было обычным явлением, проявляясь уже в 4-летнем возрасте. Однако симптомы висцеральных АВМ были редкими, и скрининг висцеральных АВМ был проведен только у 6 детей.
Пятеро детей с симптомами в нашей серии поступили с острыми опасными для жизни событиями, включая инсульт, кровохарканье и застойную сердечную недостаточность. У пяти была хроническая гипоксия. Предыдущие отчеты 2 , 25 -28 установили распространенность церебральных и легочных АВМ от 5% до 10% и от 15% до 33%, соответственно, у взрослых с ГГТ. Гораздо меньше внимания уделяется детям с этим заболеванием. Также было показано, что большинство легочных АВМ растут в течение жизни, 26 , но неизвестно, все ли легочные АВМ присутствуют при рождении или некоторые развиваются позже.Сообщалось о множественных случаях летальных осложнений АВМ головного мозга у детей с ГГТ. 7 , 10 -12 Мы обследовали 127 детей, из них 113 из-за семейного анамнеза и 14 из-за симптомов. У десяти детей были выявлены симптоматические висцеральные АВМ. Учитывая аутосомно-доминантное наследование заболевания, по нашим оценкам, 50% детей действительно имеют ГГТ. Эти данные свидетельствуют о распространенности симптоматических висцеральных АВМ на уровне 16% (10/63). В нашей серии статей мы также продемонстрировали, что первый симптом ГГТ у детей может быть опасным для жизни и может возникнуть в любое время, начиная с рождения.Семейный анамнез ГГТ был известен как минимум у 9 детей, и скрининг мог выявить эти поражения раньше. Благоприятные результаты транскатетерной эмболотерапии у взрослых и детей 8 , 18 предполагают, что раннее вмешательство могло предотвратить опасные для жизни события.
Хотя носовое кровотечение в нашей серии наблюдений было обычным явлением, оно обычно было легким, в отличие от носового кровотечения у взрослых.
Применение к детям клинических диагностических критериев, установленных в основном для взрослых с HHT 13 , может вводить в заблуждение.Как уже упоминалось, симптомы и признаки ГГТ обычно развиваются в детстве и подростковом возрасте, так что отсутствие носовых кровотечений, телеангиэктазий или симптомов висцеральных АВМ у детей является обычным явлением. За время наблюдения мы отметили появление носовых кровотечений и телеангиэктазов у 5 пациентов. Мы также продемонстрировали наличие бессимптомных висцеральных АВМ, диагностированных на основании результатов скрининга, у 3 детей (пациенты 2, 10 и 14). Folz et al. 24 пришли к выводу, что результаты 15 детей, направленных на лечение из-за симптомов и признаков, указывающих на ГГ, могут установить диагноз только у 2 детей.Следовательно, клинические диагностические критерии следует использовать с осторожностью у детей, а диагноз ГГТ и скрининг на висцеральные АВМ следует рассматривать у всех детей с семейным анамнезом ГГТ независимо от симптомов.
Генетический анализ помог установить диагноз ГГТ у детей. Мы подтвердили 8 случаев, выявив мутацию, вызывающую заболевание. Однако нам не удалось найти мутации ENG или ACVRL1 в 5 случаях с клиническим диагнозом.Для пациентов с 11 по 13, у которых семейный анамнез был минимальным и не было обнаружено мутаций, можно утверждать, что диагноз сомнительный и еще предстоит подтвердить дополнительными критериями. У пациентов 5 и 10, у которых был установлен клинический диагноз, мутация может быть сложной и требовать других методов, кроме количественной мультиплексной полимеразной цепной реакции и секвенирования экзонов. Также можно предположить, что эти пациенты имеют другой тип ГГТ. Возможный HHT типа 3, теперь связанный с хромосомой 5, был предложен в семье с высокой частотой легочных АВМ. 29 , 30 Мутации в гене MADh5 , который кодирует Smad4, недавно были описаны у пациентов с комбинированным синдромом ювенильного полипоза и HHT. 31 Другой метод, показавший свою ценность в диагностике HHT, — это капиллярная микроскопия, 32 , которая может помочь в установлении диагноза HHT в будущих исследованиях.
Наше исследование также показывает, что АВМ встречаются у детей с различными типами мутаций ENG , включая замену одной пары оснований, большую и малую вставку и вариант сплайсинга, что согласуется с гаплонедостаточностью как базовой моделью HHT1. 14 , 15,20 , 33 Кроме того, у одного ребенка с HHT2 была легочная АВМ (пациент 6), а у другого подозревалась легочная АВМ (пациент 14). Хотя легочные АВМ встречаются реже, чем при HHT1, 29 они встречаются при HHT2. 17 Мы также наблюдали появление спинномозговых АВМ у 2 детей из семьи с HHT2. Спинальные АВМ редко описываются у пациентов с HHT2. 34 Анализ мутаций, таким образом, является важным инструментом в диагностике ГГТ, особенно у детей из-за различий в возрасте начала и клинических проявлений даже в пределах одной семьи.
Это исследование иллюстрирует неоднородность представления тяжелых случаев ГГТ у детей в специализированном педиатрическом центре третичного уровня. Однако у этого исследования есть ограничения. Во-первых, исследуемая группа слишком мала, чтобы представить полный клинический спектр ГГТ в детстве. Во-вторых, эти дети, вероятно, представляют собой более тяжелые случаи, потому что были включены только дети с симптомами. Чтобы установить весь спектр педиатрической ГГТ, расстройства с аутосомным типом наследования, необходимо провести большие проспективные исследования.Включение анализа мутаций в диагностические критерии увеличит вероятность выявления детей в раннем возрасте и определит распространенность АВМ у детей с обоими типами ГГТ.
Таким образом, легочные и церебральные АВМ обнаруживаются у детей с ГГТ и могут приводить к опасным для жизни осложнениям, часто как первое проявление ГГТ. Клинические диагностические критерии HHT, вероятно, недооценивают наличие этого заболевания у детей. В наших центрах проводится скрининг висцеральных АВМ у бессимптомных детей с семейным анамнезом ГГТ, чтобы оценить их распространенность и определить, может ли скрининг предотвратить серьезные осложнения, связанные с легочными и церебральными АВМ.
Корреспонденция: Меир Мей-Захав, доктор медицины, отделение респираторной медицины, отделение педиатрии, Детская больница, 555 University Ave, Торонто, Онтарио, Канада M5G 1X8 ([email protected]).
Принята к публикации: 18 декабря 2005 г.
Финансирование / поддержка: Это исследование было поддержано грантами HHT-FY-02-226 от March of Dimes (д-р Letarte) и POP-PPP-62030 от Канадских институтов исследований в области здравоохранения (д-р Letarte и Faughnan).
Благодарность: Мы благодарим HHT Solutions и Дайан Рашлоу за их вклад в анализ мутаций и Викторию Снелл за помощь в подготовке рукописи.
1. Кьельдсен
ADVase
PGreen
Наследственная геморрагическая телеангиэктазия: популяционное исследование распространенности и смертности у датских пациентов J Intern Med 1999; 24531-39PubMedGoogle ScholarCrossref 2.Plauchu
Hde Chadarevian
JPBideau
ARobert
JM Возрастной клинический профиль наследственной геморрагической телеангиэктазии в популяции, набранной эпидемиологически Am J Med Genet 1989; 32291-297PubMedGoogle ScholarCrossref 3.AAssar
О.С.Фридман
CMWhite
RI
Jr Естественное течение носового кровотечения при наследственной геморрагической телеангиэктазии Ларингоскоп 1991; 101977- 980PubMedGoogle ScholarCrossref 4. Ваза
PGrove
O Поражения желудочно-кишечного тракта при наследственной геморрагической телеангиэктазии Гастроэнтерология 1986;
9-1083PubMedGoogle Scholar5.Shovlin
CLLetarte
M Наследственная геморрагическая телеангиэктазия и легочные артериовенозные мальформации: вопросы клинического ведения и обзор патогенных механизмов Thorax 1999; 54714-729PubMedGoogle ScholarCrossref 7.Морган
TMcDonald
JAnderson
C
и другие. Внутричерепное кровоизлияние у младенцев и детей с наследственной геморрагической телеангиэктазией (синдром Ослера-Вебера-Ренду) Pediatrics 2002; 109E12PubMedGoogle ScholarCrossref 8.Lee
DWWhite
RI
JrEgglin
ТЗ
и другие. Эмболотерапия крупных легочных артериовенозных мальформаций: отдаленные результаты Ann Thorac Surg 1997; 64930- 939PubMedGoogle ScholarCrossref 9.Bennhagen
RGHolje
GLaurin
SPesonen
E Спиральная эмболизация неонатальной легочной артериовенозной мальформации Pediatr Cardiol 2002; 23235-238PubMedGoogle ScholarCrossref 10.Гарсия-Монако
RTaylor
WRodesch
грамм
и другие. Пиальный артериовенозный свищ у детей как проявление болезни Ренду-Ослера-Вебера Нейрорадиология 1995; 3760-64PubMedGoogle ScholarCrossref 11.Jessurun
Г.А.Ампуис
DJvan der Zande
FHNossent
JC Артериовенозные мальформации на Нидерландских Антильских островах: высокая распространенность наследственных геморрагических телеангиэктазий, связанных с единичными и множественными церебральными артериовенозными мальформациями Clin Neurol Neurosurg 1993; 95193-198PubMedGoogle ScholarCrossref 12.Бурдо
ACymerman
UPaquet
МЕНЯ
и другие. Экспрессия эндоглина снижена в нормальных сосудах, но все же обнаруживается при артериовенозных мальформациях у пациентов с наследственной геморрагической телеангиэктазией 1 типа Am J Pathol 2000; 156911-923PubMedGoogle ScholarCrossref 13.Shovlin
CLGuttmacher
А.Е. Бускарини
E
и другие. Диагностические критерии наследственной геморрагической телеангиэктазии (синдром Ренду-Ослера-Вебера) Am J Med Genet 2000; 9166-67PubMedGoogle ScholarCrossref 14.Cymerman
UVera
SPece-Barbara
N
и другие. Выявление наследственной геморрагической телеангиэктазии 1 типа у новорожденных с помощью экспрессии белка и анализа мутаций эндоглина Pediatr Res 2000; 4724-35PubMedGoogle ScholarCrossref 15.Cymerman
UVera
Скарабегович
AAbdalla
SLetarte
M Характеристика нескольких новых мутаций эндоглина, связанных с наследственной геморрагической телеангиэктазией Hum Mutat 2003; 21482-492PubMedGoogle ScholarCrossref 16.Абдалла
SAPece-Barbara
NVera
S
и другие. Анализ ALK-1 и эндоглина у новорожденных из семей с наследственной геморрагической телеангиэктазией 2 типа Hum Mol Genet 2000;
-1237PubMedGoogle ScholarCrossref 17. Abdalla
С.А.Кимерман
У.Джонсон
RMDeber
CMLetarte
M Мутации, связанные с заболеванием, в консервативных остатках домена киназы ALK-1 Eur J Hum Genet 2003; 11279-287 PubMedGoogle ScholarCrossref 18. Faughnan
METhabet
AMei-Zahav
M
и другие.Легочные артериовенозные мальформации у детей: результаты транскатетерной эмболотерапии J Pediatr 2004; 145826-831PubMedGoogle ScholarCrossref 19.Faughnan
MELui
YWWirth
JA
и другие. Диффузные легочные артериовенозные мальформации: характеристика и прогноз Chest 2000; 11731-38PubMedGoogle ScholarCrossref 20.Pece
NVera
Саймерман
UWhite
RI
JrWrana
JLLetarte
M Мутантный эндоглин при наследственной геморрагической телеангиэктазии 1 типа временно экспрессируется внутриклеточно и не является доминантно-негативным J Clin Invest 1997; 1002568-2579PubMedGoogle ScholarCrossref 21.Абдалла
С.А.Кимерман
URushlow
D
и другие. Новые мутации и полиморфизмы в генах, вызывающих наследственную геморрагическую телеангиэктазию Hum Mutat 2005; 25320-321 PubMedGoogle ScholarCrossref 22.Mandzia
JLterБрюгге
KGFaughnan
MEHyland
RH Артериовенозные мальформации спинного мозга у двух пациентов с наследственной геморрагической телеангиэктазией Childs Nerv Syst 1999; 1580-83PubMedGoogle ScholarCrossref 23.Bayrak-Toydemir
PMao
RLewin
SMcDonald
J Наследственная геморрагическая телеангиэктазия: обзор диагностики и лечения в молекулярную эру для клиницистов Genet Med 2004; 6175-191PubMedGoogle ScholarCrossref 24.Folz
BJZoll
Бальфке
Houssaint
AMaier
RFWerner
JA Проявления наследственной геморрагической телеангиэктазии у детей и подростков Eur Arch Otorhinolaryngol 2006; 26353-61PubMedGoogle ScholarCrossref 25.Haitjema
TWestermann
CJOvertoom
TT
и другие. Наследственная геморрагическая телеангиэктазия (болезнь Ослера-Вебера-Ренду): новые взгляды на патогенез, осложнения и лечение Arch Intern Med 1996; 156714-719PubMedGoogle ScholarCrossref 26.Ваза
PHolm
MArendrup
H Легочные артериовенозные свищи при наследственной геморрагической телеангиэктазии Acta Med Scand 1985; 218105-109PubMedGoogle ScholarCrossref 27.Haitjema
TDisch
FOvertoom
TTWestermann
CJLammers
JW Скрининг членов семьи пациентов с наследственной геморрагической телеангиэктазией Am J Med 1995; 99519-524PubMedGoogle ScholarCrossref 28.Nanthakumar
К.Грэм
ATRobinson
TI
и другие.Контрастная эхокардиография для выявления легочных артериовенозных мальформаций Am Heart J 2001; 141243-246PubMedGoogle ScholarCrossref 29.Wallace
Г.М.Шовлин
CL Семейство наследственных геморрагических телеангиэктазий с поражением легких не связано с известными генами HHT, эндоглином и ALK-1 Thorax 2000; 55685-690PubMedGoogle ScholarCrossref 30.Cole
SGBegbie
MFWallace
Г.М.Шовлин
CL Новый локус наследственной геморрагической телеангиэктазии (HHT3) отображается на хромосоме 5 J Med Genet 2005; 42577-582PubMedGoogle ScholarCrossref 31.Галлион
CJRepetto
GMLegius
E
и другие. Комбинированный синдром ювенильного полипоза и наследственной геморрагической телеангиэктазии, связанный с мутациями в MADh5 ( SMAD4 ) Lancet 2004; 363852-859PubMedGoogle ScholarCrossref 32.Mager
Дж. Дж. Вестерманн
CJ Значение капиллярной микроскопии в диагностике наследственной геморрагической телеангиэктазии Arch Dermatol 2000; 136732-734PubMedGoogle ScholarCrossref 33.Шовлин
CLHughes
JMScott
Дж.Сейдман
CESeidman
JG Характеристика эндоглина и идентификация новых мутаций при наследственной геморрагической телеангиэктазии Am J Hum Genet 1997; 6168-79PubMedGoogle ScholarCrossref 34.McDonald
Джемиллер
FJHallam
Сенельсон
Л.Марчук
DAWard
KJ Клинические проявления при большой наследственной геморрагической телеангиэктазии (HHT) 2 типа, родственной Am J Med Genet 2000;
-327PubMedGoogle ScholarCrossref
Наследственная геморрагическая телеангиэктазия — обзор
Наследственная геморрагическая телеангиэктазия
HHT, или болезнь Ослера-Вебера-Ренду, представляет собой заболевание, характеризующееся множественными телеангиэктазиями кожи и слизистых оболочек и легочными АВМ.HHT — аутосомно-доминантное заболевание, характеризующееся сосудистой дисплазией и кровотечением. Небольшое количество случаев носит спорадический характер. Типы сосудистых мальформаций у пациентов с ГГТ разнообразны. В основе нарушения лежат дефекты генов, кодирующих эндоглин и ALK-1, которые являются рецепторами TGF-β, экспрессируемыми исключительно на эндотелиальных клетках сосудов и участвующими в ангиогенезе. 155 Было обнаружено большое количество мутаций, лежащих в основе синдрома в разных семьях, включая бессмысленные, бессмысленные, сдвиговые и делеционные мутации.Заболевание одинаково поражает представителей пола. HHT демонстрирует большую генетическую гетерогенность, и его фенотипы были классифицированы на основе недавно идентифицированных мутировавших генов: эндоглина (HHT-1) и рецептора активин-подобной киназы-1 (HHT-2).
HHT поражает 1 из 5000-8000 человек и обычно проявляется в виде анемии, частых желудочно-кишечных и носовых кровотечений и характерных кожно-слизистых телеангиэктазий. Сосудистые поражения, наблюдаемые при HHT, включают АВМ и телеангиэктазии. АВМ чаще всего поражают легочное и мозговое кровообращение.АВМ могут вызывать серьезные осложнения, если они находятся в легких, печени или головном мозге. 156 Возраст обращения варьирует, и симптомы у пациентов могут проявляться первоначально в позднем взрослом возрасте. 157 Идиопатическая легочная артериальная гипертензия может возникать в подгруппе пациентов с ГГТ (≈10%). 158
Телеангиэктазии обнаруживаются на коже или слизистых оболочках желудочно-кишечного тракта, дыхательных путей и мочевыводящих путей. Носовое кровотечение, кровотечение из желудочно-кишечного тракта и мочевыводящих путей являются частыми клиническими проявлениями.Поражения сосудов могут также присутствовать в других органах, таких как печень и мозг, где они обычно являются случайными результатами вскрытия. О поражении печени сообщается до 30% людей, страдающих HHT. Большие АВМ в печени могут привести к серьезным осложнениям, включая застойную сердечную недостаточность с высоким выбросом, портальную гипертензию, печеночную энцефалопатию, ишемию желчевыводящих путей и печеночную недостаточность. Эмболизация больших АВМ в печени остается спорным вопросом; однако трансплантация печени может успешно устранить эти осложнения. 159 Пациенты с HHT могут иметь гепатомегалию или неврологические симптомы. Эмболы и абсцессы ЦНС могут быть результатом парадоксической эмболии. 160 Геморрагическая тенденция у пациентов с ГГТ усиливается с возрастом.