Овотестикулярное нарушение формирования пола у пациента с кариотипом 46,XY
Овотестикулярное нарушение формирования пола (ОТ НФП) является редким вариантом патологической дифференцировки гонад. Наиболее частой причиной являются различные варианты гоносомного мозаицизма. Кариотип 46, XY имеют не более 7% пациентов с ОТ НФП. В экспериментальных работах показаны модели развития овотестис в результате нарушения соотношения аутосомных генов, регулирующих овариальные и тестикулярные пути развития [1, 2].
Классическим вариантом овотестис является двойственная абдоминальная гонада, состоящая из отдельных отсеков различной по структуре ткани. Для верификации ОТ-НФП необходимо гистологическое исследование, подтверждающее наличие овариальной и тестикулярной ткани в составе одной гонады [3]. Основными клиническими задачами при ведении пациентов с ОТ-НФП являются выбор пола и определение хирургической тактики в отношении гонад.
Описание случая
Родители пациента 1 года 3 мес обратились с жалобами на неправильное строение наружных половых органов ребенка.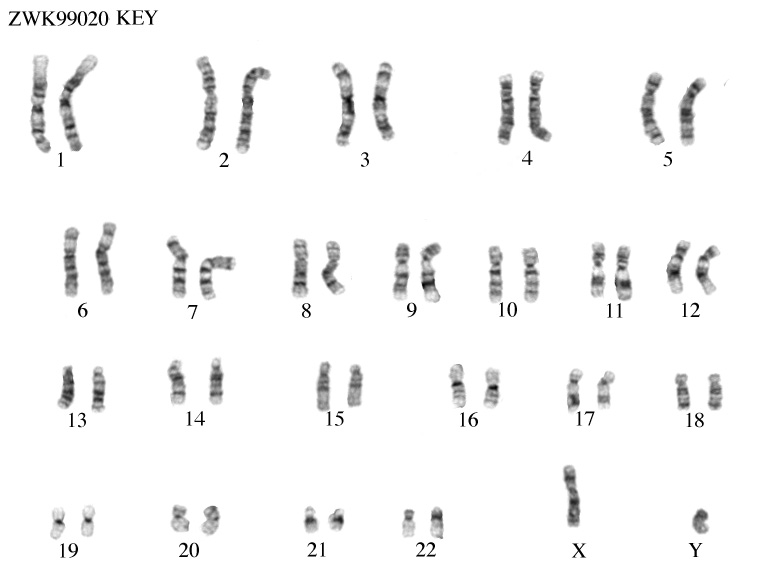 Из анамнеза известно, что при рождении ребенка фенотипический пол не определен. На основании кариотипирования (46,XY) и ДНК-диагностики (наличие SRY-гена) был зарегистрирован в мужском поле. При осмотре в возрасте 1,5 года рост 80 см, SDS роста: 0,02, масса тела 10 кг, ИМТ 15,6 кг/м2. Наружные половые органы имеют двойственное строение: лабиоскротальные складки полностью расщеплены, гонады не пальпируются; клитерофаллос длиной 3 см, кавернозные тела, головка сформированы; отверстие уретры открывается на промежности.
Из анамнеза известно, что при рождении ребенка фенотипический пол не определен. На основании кариотипирования (46,XY) и ДНК-диагностики (наличие SRY-гена) был зарегистрирован в мужском поле. При осмотре в возрасте 1,5 года рост 80 см, SDS роста: 0,02, масса тела 10 кг, ИМТ 15,6 кг/м2. Наружные половые органы имеют двойственное строение: лабиоскротальные складки полностью расщеплены, гонады не пальпируются; клитерофаллос длиной 3 см, кавернозные тела, головка сформированы; отверстие уретры открывается на промежности.
Обследование ребенка включало эхографию малого таза, лапароскопию и цистоскопию; определение тестостерона, эстрадиола и антимюллерова гормона (АМГ) в сыворотке; гистологическое исследование интраоперационного материала гонад и цитогенетическое исследование материала гонад методом ДНК-зондов.
Результаты физикального, лабораторного и инструментального исследования
При визуализации полости малого таза обнаружена матка с шейкой, влагалище. По данным ультразвукового сканирования, левая гонада была расценена как яичник объемом 3,7 см3 с фолликулами по периферии в количестве до 7—8 в одном поле сканирования. При лапароскопической визуализации левая гонада состояла из двух отделов, четко разделенных между собой прослойкой соединительной ткани; правая гонада также состояла из двух отделов, имела объем не более 1 см3. В одном из отделов каждой гонады определялись овариальные фолликулы. При проведении цистоуретроскопии на расстоянии 0,5 см от меатуса определяется вход во влагалище, которое хорошо развито, свободно проходимо для цистоскопа, имеет длину около 5 см, заканчивается шейкой матки. Рядом располагается вход в уретру, ведущую в мочевой пузырь.
По данным ультразвукового сканирования, левая гонада была расценена как яичник объемом 3,7 см3 с фолликулами по периферии в количестве до 7—8 в одном поле сканирования. При лапароскопической визуализации левая гонада состояла из двух отделов, четко разделенных между собой прослойкой соединительной ткани; правая гонада также состояла из двух отделов, имела объем не более 1 см3. В одном из отделов каждой гонады определялись овариальные фолликулы. При проведении цистоуретроскопии на расстоянии 0,5 см от меатуса определяется вход во влагалище, которое хорошо развито, свободно проходимо для цистоскопа, имеет длину около 5 см, заканчивается шейкой матки. Рядом располагается вход в уретру, ведущую в мочевой пузырь.
При гормональном обследовании уровень тестостерона на фоне стандартного стимуляционного теста хорионическим гонадотропином — 9,5 нмоль/л; уровень АМГ — 115 нг/мл, эстрадиола — 17 пг/мл.
В ходе лапароскопии правая гонада была удалена. Проведена сепарация левой гонады по линии соединительнотканной перегородки с удалением тестикулярного компонента гонады. Проведена биопсия овариального отсека левой гонады.
Гистологическое исследование интраоперационного материала гонад показало наличие ткани яичка и яичника, разделенных фиброзной прослойкой. Ткань яичника фиброзирована, деление на корковый и мозговой слой нечеткое. В корковом слое определяются примордиальные фолликулы с выраженными дистрофическими изменениями (рис. 1). Ткань яичка разбита на дольки фиброзными прослойками, единичные микрокальцинаты. В эпителии канальцев резко выраженные дистрофические и пролиферативные изменения: увеличение размеров клеток, ядер, гиперхромия ядер. Клетки сперматогенного эпителия располагаются хаотично и находятся в разной степени зрелости. Обращают на себя внимание крупные клетки с четко определяемым ядром и наличием в нем ядрышек — половые клетки (рис. 2). Такое строение сперматогенного эпителия соответствует организации, характерной для антенатального периода [4].
Рис. 1. Микроскопическая картина ткани яичника: дифференцировка на корковый и мозговой слой, в корковом слое примордиальные фолликулы с дистрофическими изменениями. Окраска гематоксилином и эозином, ув. 250.
Рис. 2. Микроскопическая картина ткани яичка: эпителий канальцев незрелый, с дистрофическими и пролиферативными изменениями. Окраска гематоксилином и эозином, ув. 250.
FISH-анализ с набором ДНК-зондов, специфичных к центромерным районам хромосом Х (DXZ1) и Y (DYZ3), был выполнен на интраоперационном материале гонад. При анализе интерфазных ядер в препаратах из некультивированных клеток овотестикулярной гонады выявлено по 1 копии последовательности, специфичной центромерному району хромосом X и Y. Подтвержден мужской кариотип, числовых отклонений по хромосомам X и Y не выявлено. Молекулярно-генетическая диагностика была проведена в Институте детской эндокринологии ЭНЦ в рамках программы «Альфа-Эндо» методом параллельнорго секвенирования на панели генов нарушения формирования пола; на данный момент генетическая причина заболевания не установлена.
На основании результатов проведенного обследования был выставлен диагноз: 46, XY, овотестикулярное нарушение формирования пола. В ходе клинического консилиума с участием родителей было принято решение о воспитании ребенка в женском поле.
На основании результатов гистологического исследования было принято решение о проведении гонадэктомии с последующим проведением заместительной терапии в период полового созревания.
Основанием для выбора женского пола воспитания у данного пациента явились фенотипическое строение наружных гениталий, хорошее развитие мюллеровых структур — матки, влагалища, определяющих возможность адаптации ребенка в период полового созревания на фоне проведения заместительной терапии. Овотестикулярные гонады пациента имели типичное макроскопическое строение: каждая гонада состояла из двух компонентов, четко разделенных между собой соединительнотканной перегородкой. Выброс тестостерона в ответ на стимуляцию хориогонином свидетельствовал о частично сохранной функции клеток Лейдига. Источником высокого уровня АМГ можно считать незрелые примордиальные фолликулы [5].
Согласно данным литературы [6—8], сохранение овариальных частей гонад у девочек с ОТ-НФП определяет возможность самостоятельного пубертата. Пациентки с овотестис могут иметь нормально развитые молочные железы, менструировать. Возможность сохранения овариальных компонентов определяется гистологическим строением гонады — присутствием дифференцированной овариальной ткани, овариальных фолликулов различной степени зрелости. Недифференцированные гонады подлежат обязательному удалению в связи с высоким риском их малигнизации.
С целью определения оптимальной тактики на первом этапе хирургического лечения была проведена сепарация правой гонады с удалением дисгенетического тестикулярного компонента, биопсией овариальной ткани и последующими диагностическими исследованиями на материале гонад. Для исключения скрытого гоносомного мозаицизма было проведено молекулярно-цитогенетическое исследование методом FISH-диагностики, в результате которого числовых отклонений по половым хромосомам не выявлено, подтвержден мужской кариотип. Развитие овотестис при кариотипе XY является редким вариантом ОТ-НФП. В 2009 г. D. Wilhelm и L. Washburn [9] показали, что развитие овотестикулярной гонады у XY-особей прослеживалось в тех случаях, когда экспрессия тестикул детерминирующих генов (SRY, SOX 9) была недостаточной для подавления овариального пути развития.
Результаты гистологического исследования гонад нашего пациента показали наличие незрелой тестикулярной ткани с выраженными пролиферативными изменениями, микрокальцинатами. В ткани яичника отсутствовало четкое деление на корковое и мозговое вещество. Примордиальные фолликулы, определяемые в овариальной части гонады, имели выраженные дистрофические изменения. На основании этих данных было принято решение о проведении гонадэктомии.
Выбор пола у пациента с ОТ-НФП проводился на основании совокупности данных: фенотипического строения гениталий, функционального состояния гонад, возможностей хирургической коррекции и перспективы адаптации пациента в период полового созревания. Хирургическую тактику для пациента с овотестикулярным нарушением формирования пола определило морфологическое строение и степень зрелости структурных компонентов овотестикулярной гонады.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Согласие пациента. Мать пациента добровольно подписала информированное согласие на публикацию медицинской информации о ребенке в обезличенной форме в журнале «Проблемы эндокринологии».
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Кариотип 46,X iso (Xq) (дети, стационар)
МКБ: Q96.1
Кариотип 46,X iso (Xq) (дети, стационар) — это нарушение здоровья, относящееся к группе хромосомные аномалии. Кариотип — совокупность признаков (число, размеры, форма и т. д.) полного набора хромосом, присущая клеткам данного биологического вида (видовой кариотип), данного организма (индивидуальный кариотип) или линии (клона) клеток.
Симптомы кариотипа 46,X iso (Xq) у детей широко варьируются от человека к человеку, от гениталий промежуточного типа до нормальных гениталий. Нормальные кариотипы человека — 46,XX (женский) и 46,XY (мужской). Нарушения нормального кариотипа у человека возникают на ранних стадиях развития организма: в случае, если такое нарушение возникает при гаметогенезе, в котором продуцируются половые клетки родителей, кариотип зиготы, образовавшейся при их слиянии, также оказывается нарушенным. При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма обладают одинаковым аномальным кариотипом.
В случае диагноза кариотип 46,X iso (Xq), чтобы узнать как вылечить кариотип 46,X iso (Xq), следует обратиться к врачам, которые указаны в стандарте оказания медицинской помощи.
Лечение кариотипа 46,X iso (Xq) у детей в данном случае подразумевает прием лекарственных препаратов из стандарта оказания медицинской помощи.
Информация предоставлена на основании приказа Министерства здравоохранения РФ от 29 декабря 2012 г. N 1659н «Об утверждении стандарта специализированной медицинской помощи детям при задержке роста»
Biogenom показывает все мероприятия для подтверждения диагноза, которые указаны в стандартах Минздрава РФ.
Точный список мероприятий может определить только Ваш лечащий врач.
Диагностика заболевания
Получите персональную консультацию врача по Вашему состоянию здоровья.
Для диагностики заболевания проводят следующие мероприятия:
Функциональные исследования
- Осмотр периферии глазного дна трехзеркальной линзой Гольдмана
- Регистрация электрокардиограммы
- Рентгенография кисти руки
- Магнитно-резонансная томография головного мозга
- Ультразвуковое исследование щитовидной железы и паращитовидных желез
- Эхокардиография
- Компьютерная томография головы
- Компьютерная томография головы с контрастированием структур головного мозга
- Магнитно-резонансная томография головного мозга с контрастированием
- Периметрия
- Ультразвуковое исследование матки и придатков трансабдоминальное
- Ультразвуковое исследование надпочечников
- Ультразвуковое исследование органов мошонки
- Ультразвуковое исследование почек и надпочечников
- Электроэнцефалография
Лабораторные исследования
- Анализ крови биохимический общетерапевтический
- Анализ мочи общий
- Исследование тиреотропина сыворотки крови
- Исследование уровня гликированного гемоглобина в крови
- Исследование уровня глюкозы в крови
- Исследование уровня инсулиноподобного ростового фактора I в крови
- Исследование уровня неорганического фосфора в крови
- Исследование уровня пролактина в крови
- Исследование уровня свободного тироксина (T4) сыворотки крови
- Исследование уровня щелочной фосфатазы в крови
- Общий (клинический) анализ крови развернутый
- Анализ крови по оценке нарушений липидного обмена биохимический
- Исследование уровня ионизированного кальция в крови
- Исследование уровня калия в крови
- Исследование уровня лютеинизирующего гормона в сыворотке крови
- Исследование уровня натрия в крови
- Исследование уровня общего кальция в крови
- Исследование уровня свободного кортизола в крови
- Исследование уровня фолликулостимулирующего гормона в сыворотке крови
- Цитогенетическое исследование (кариотип)
- Идентификация генов
- Исследование уровня инсулина плазмы крови
- Исследование уровня общего тестостерона в крови
- Исследование уровня общего эстрадиола в крови
- Исследование уровня соматотропного гормона в крови
- Определение объема мочи
- Определение удельного веса (относительной плотности) мочи
К каким специалистам следует обращаться
- Ежедневный осмотр врачом- детским эндокринологом с наблюдением и уходом среднего и младшего медицинского персонала в отделении стационара
- Прием (осмотр, консультация) врача- детского эндокринолога первичный
- Прием (осмотр, консультация) врача-офтальмолога первичный
- Составление родословной
- Прием (осмотр, консультация) врача-генетика первичный
- Прием (осмотр, консультация) врача-офтальмолога повторный
- Прием (осмотр, консультация) врача-акушера-гинеколога первичный
- Прием (осмотр, консультация) врача-акушера-гинеколога повторный
- Прием (осмотр, консультация) врача- детского кардиолога первичный
- Прием (осмотр, консультация) врача- детского кардиолога повторный
- Прием (осмотр, консультация) врача- детского уролога-андролога первичный
- Прием (осмотр, консультация) врача- детского уролога-андролога повторный
- Прием (осмотр, консультация) врача-невролога первичный
- Прием (осмотр, консультация) врача-нейрохирурга первичный
- Прием (осмотр, консультация) врача сурдолога-оториноларинголога первичный
- Прием (осмотр, консультация) врача сурдолога-оториноларинголога повторный
- Прием (осмотр, консультация) врача-травматолога-ортопеда первичный
- Прием (осмотр, консультация) врача-травматолога-ортопеда повторный
Лечение заболевания
Для лечения заболевания используются следующие группы лекарственных препаратов:
Инсулины короткого действия и их аналоги для инъекционного введения
- Инсулин растворимый [человеческий генно- инженерный]
Другие ирригационные растворы
- Декстроза (Глюкоза Браун, Глюкоза буфус, Глюкоза-Э)
Агонисты имидазолиновых рецепторов
- Клонидин (Клофелин)
Производные 3-оксоандрост-4-ена
- Тестостерон [смесь эфиров] (Омнадрен 250, Сустанон-250)
- Тестостерон (Андрогель, Небидо, Тестостерона пропионат)
Природные и полусинтетические эстрогены
- Эстрадиол (Дивигель, Климара, Эстрожель)
Производные прегнадиена
- Дидрогестерон (Дюфастон)
Соматропин и его агонисты
- Соматропин (Динатроп, Нордитропин, Нордитропин НордиЛет)
Глюкокортикоиды
- Гидрокортизон (Латикорт, Локоид Крело, СОЛУ-КОРТЕФНоваМедика)
Гормоны щитовидной железы
- Левотироксин натрия (L-Тироксин 100 Берлин-Хеми, Левотироксин, Эутирокс)
Окончательный перечень функциональных и лабораторных исследований, консультаций врачей, а также лекарственная терапия определяются лечащим врачом.
Профилактика заболевания
- Психологическая адаптация
Исследование кариотипа — новая услуга нашей лаборатории
COVID-19
Получить результат
Кариотипирование –изучение хромосомного набора человека, позволяющее обнаружить отклонения вструктуре и числе хромосом. Оно помогает выявить нарушения хромосом,вероятно, не влияющие на здоровье человека, но тем не менее важные дляпланирования будущей беременности и для здоровья будущего ребенка (патологииплода, аномалии развития).
Кариотип – это полный хромосомный набор клетки человека. В норме он состоит из 46 хромосом, из них 44 аутосомы (22пары), имеющих одинаковое строение и в мужском, и в женском организме, и одна пара половых хромосом (XY у мужчин и XX у женщин). Каждая хромосома несет гены,ответственные за наследственность. Кариотип 46, ХХ – соответствует нормальному женскому кариотипу, а кариотип 46, XY – это нормальный мужской кариотип.Кариотип остается неизменным в течение всей жизни.
Нарушения хромосомного набора могут являться причиной наследственной патологии, бесплодия, невынашивания беременности,рождения ребенка с различными пороками развития.
Подготовка:
- Исследование проводится в состоянии сытости, не рекомендуется сдавать кровь на данное исследование натощак.
- Исключить (по согласованию с врачом) прием антибактериальных и химиотерапевтических препаратов в течение 14 дней до исследования.
- Исследование рекомендуется проводить не ранее чем через 2 недели после перенесенных инфекционных/острых воспалительных заболеваний.
Цена исследования с фото — 5900р, без фото — 4900
Забор крови производится во всех филиалах клиники без предварительной записи:
ул. Мира д.9, тел. 8(4922) 777-957, 8-900-588-60-05
ул. Горького д.117, тел. 8(4922) 604-957, 8-900-588-93-91
пр-т Ленина д.25, тел. 8(4922) 601-957, 8-900-588-60-04
ул. Добросельская д. 124, 8(4922) 602-957, 8-900-588-60-02
Дополнительную информацию уточняйте у администраторов.
Возврат к списку
Случай тканевого мозаицизма по кольцевой/трисомной форме хромосомы 18 у пациента с множественными врожденными пороками развития | Никулина
1. Garcia-Castillo H, Vasquez-Velasquez AI, Rivera H, Barros-Nunez P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am J Med Genet. 2008;146A(13):1687- 1695. doi: 10.1002/ajmg. a.32315.
2. Lane AH, Aijaz N, Galvin-Parton P, Lanman J, Mangano R, Wilson TA. Mosaic variegated aneuploidy with growth hormone deficiency and congenital heart defects. Am J Med Genet. 2002;110(3):273-277.
3. Callier P, Faivre L, Cusin V, Marle N, Thauvin- Robinet C, Sandre D et al. Microcephaly is not mandatory for the diagnosis of mosaic variegated aneuploidy syndrome. Am J Med Gen. 2005;127(2):204-207.
4. Jacquemont S, Boceno M, Rival JM, Mechinaud F, David A. High risk of malignancy in mosaic variegated aneuploidy syndrome. Am J Med Gen. 2002;109(1):17-21.
5. Choo S, Teo SH, Tan M, Yong MH, Ho LY. Tissue- limited mosaicism in Pallister-Killian syndrome — a case in point. J Perinat. 2002;22(5):420-423.
6. Cormier-Daire V, Le Merrer M, Gigarel N, Morichon N, Prieur M, Lyonnet S et al. Prezygotic origin of the isochromosome 12p in Pallister-Killian syndrome. Am J Med Gen. 1997;69(2):166-168.
7. Никулина ТС, Злотина АМ, Яни НА, Сухоцкая АА, Щеголева НА, Кувшинова ЛА и др. Врожденная диафрагмальная грыжа — частный случай синдрома Паллиастера-Киллиана. Лечение и профилактика. 2013;7(3):121-125. [Nikulina TS, Zolotina AM, Yani NA, Sukhotskaya AA, Schegoleva NA, Kuvshinova LA et al. Congenital diaphragmatic hernia — a special case of Pallister-Killian syndrome. Disease Treatment and Prevention. 2013;7(3):121-125. In Russian].
8. Ohashi H, Ishikiriyama S, Fukushima Y. New diagnostic method for Pallister-Killian syndrome: detection of i(12p) in interphase nuclei of buccal mucosa by fluorescence in situ hybridization. Am J Med Gen. 1993;45 (1):123-128.
9. Wenger SL, Boone LY, Steele MW. Mosaicism in Pallister i(12p) syndrome. Am J Med Gen. 1990;35 (4) :523-525.
10. Rasmussen SA, Colman SD, Ho VT, Abernathy CR, Arn PH, Weiss L et al. Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Gen. 1998;35(6):469-471.
11. Streubel B, Latta E, Kehrer-Sawatzki H, Hoffmann GF, Fonatsch C, Rehder H. Somatic mosaicism of a greater than 1.7-Mb deletion of genomic DNA involving the entire NF1 gene as verified by FISH: further evidence for a contiguous gene syndrome in 17q11.2. Am J Med Gen. 1999;87(1):12-16.
12. Tucker ME; Garringer HJ; Weaver DD Phenotypic spectrum of mosaic trisomy 18: two new patients, a literature review, and counseling issues. Am J Med Gen. 2007;143 (5) :505-517.
ФГБНУ НЦПЗ. ‹‹Общая психиатрия››
Цитогенетика — это область генетики, связанная с изучением хромосом. Исследование хромосом при психической патологии показало, что диагностическое значение этот метод имеет в основном при умственной отсталости. При неврозах, эндогенных психозах, алкоголизме и других видах психической патологии хромосомные изменения, как правило, не обнаруживаются. Среди новорожденных частота хромосомной патологии равна примерно 0,6 %. Хромосомные болезни были известны еще до открытия самих хромосом (например, синдромы Клайнфельтера и Шерешевского — Тернера). Использование термина «болезнь» для хромосомной патологии не совсем верно, поскольку для любой болезни характерен тот или иной тип ее развития, т.е. закономерная смена симптомов и синдромов во времени. В случае же хромосомных аномалий совокупность специфических признаков больного — его фенотип является врожденным и практически не меняющимся. Большинство случаев хромосомных нарушений возникает спорадически в половых клетках здоровых родителей или на стадии первых делений зиготы. Хромосомные нарушения в половых клетках приводят к изменению кариотипа, т.е. совокупности количественных и качественных признаков хромосом, во всех клетках организма. Если же хромосомные изменения возникают на ранних стадиях развития эмбриона, то они являются причиной развития мозаицизма. О мозаицизме говорят в тех случаях, когда имеются клетки с нормальным и измененным кариотипом. В этом случае индивиды имеют более «стертые» проявления заболевания, и чем больше клеток с аномальным кариотипом, тем больше выражены клинические проявления заболевания.
Материалом для микроскопического исследования хромосом служат в основном лейкоциты крови, реже для этой цели используются культуры клеток кожи или костного мозга. Хромосомные аномалии могут возникать в половых и соматических клетках. Они представляют собой изменение числа хромосом — наличие добавочных хромосом или отсутствие хромосомы, или их перестройку, т.е. структурные изменения. Структурные изменения бывают внутрихромосомными — такими как делеция (утрата части хромосомы) или дупликация (удвоение участка хромосомы), а также межхромосомными — транслокация (обмен участками между хромосомами) и др.
При описании кариотипа индивида указывают общее число хромосом, затем состав половых хромосом, наличие транслокации или мозаицизма и т.д. Например, запись 46XY означает нормальный мужской кариотип; 46ХХ — нормальный женский кариотип. Если указано 47XXY, то это кариотип синдрома Клайнфельтера, когда имеется дополнительная Х-хромосома. Кариотип 45X0 соответствует синдрому Шерешевского — Тернера, обусловленному отсутствием одной Х-хромосомы. Добавочная аутосомная хромосома указывается своим номером и знаком плюс, например, 47ХХ, 21+ обозначает кариотип девочки с лишней 21-й хромосомой (болезнь Дауна), а утрата хромосомы обозначается соответствующим номером и знаком минус. Транслокация обозначается буквой «t» с указанием номеров хромосом, которые обменялись участками, например 45XY, t(14+21). Наличие мозаичных клеток обозначается соответствующими кариотипами с использованием знака дроби, например 45X0/46, XX — мозаичность по синдрому Шерешевского — Тернера (45X0). При описании кариотипов используются также и другие символы, с правилами применения которых можно ознакомиться в специальных руководствах.
В случае хромосомной патологии почти все клинические проявления сопровождаются множественными нарушениями в строении тела и психики, причем их выраженность сильно варьирует при одних и тех же хромосомных аномалиях. Например, при болезни Дауна поражение психики проявляется слабоумием от легкой до тяжелой степени. Было также отмечено, что у больных с аномалиями аутосомных хромосом интеллект нарушается в большей степени, чем при аномалиях половых хромосом.
Клинико-генетическое описание и анализ случая хромосомного мозаицизма mos47,XY,+8/46,XY | Нерсесян
1. Nicolaides K.H. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat. Diagn. 2011; 31 (1): 7–15. DOI: 10.1002/pd.2637.
2. Chen C.P., Chen M., Pan Y.J., Su Y.N., Chern S.R., Tsai F.J., Chen Y.T., Wang W. Prenatal diagnosis of mosaic trisomy 8: Clinical report and literature review. Taiwanese Journal of Obstetrics & Gynecology. 2011; 50(3): 331–338. DOI: 10.1016/j.tjog.2011.07.013.
3. Paulsson K., Säll T., Fioretos T., Mitelman F., Johansson B. The incidence of trisomy 8 as a sole chromosomal aberration in myeloid malignancies varies in relation to gender, age, prior iatrogenic genotoxic exposure, and morphology. Cancer Genet. Cytogenet. 2001; 130 (2): 160–165. DOI: 10.1016/s0165-4608(01)
4. Becker K., FitzGerald O., Green A.J., Keogan M., Newbury-Ecob R., Greenhalgh L. Withers S., Hollox E.J., Aldred P.M.R., Armour J.A.L. Constitutional trisomy 8 and Behcet syndrome. Am. J. Med. Genet. 2009; 149A (5): 982–986. DOI: 10.1002/ajmg.a.32756.
5. Rauen K.A., Golabi M., Cotter P.D. Fertility in a female with mosaic trisomy 8. Fertility and Sterility. 2003; 79 (1): 206–208. doi: 10.1016/s0015-0282(02)04410-2.
6. Hale N.E., Keane J.F. Piecing together a picture of trisomy 8 mosaicism syndrome. J. Am. Osteopath. Assoc. 2010; 110 (1): 21–23.
7. Giraldo G., Gómez A.M., Mora L., Suarez-Obando F., Moreno O. Mosaic trisomy 8 detected by fibroblasts cultured of skin. Colomb. Med. 2016; 47 (2): 100–104.
8. Sun S., Zhan F., Jiang J., Zhang X., Yan L., Cai W., Liu H., Cao D. Karyotyping and prenatal diagnosis of 47,XX-,+8[67]/46,XX[13] mosaicism: case report and literature review. BMC Medical Genomics. 2019; 12 (1): 197. DOI: 10.1186/s12920-019-0639-8.
9. Udayakumar A.M., Al-Kindy A. Constitutional trisomy 8 mosaicism syndrome: case report and review. Journal of Pediatric Genetics. 2013; 2 (4): 197–201. DOI: 10.3233/PGE-13069.
10. Wiśniewska M., Mazurek M. Trisomy 8 mosaicism syndrome. J. Appl. Genet. 2002; 43 (1): 115–118.
11. Paulsson K., Johansson B. Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathologie Biologie. 2007; 55 (1): 37–48. DOI: 10.1016/j.patbio.2006.04.007.
12. Saumell S., Solé F., Arenillas L., Montoro J., Valcárcel D., Pedro C., Sanzo C., Luño E., Giménez T., Arnan M., Pomares H., De Paz R., Arrizabalaga B., Jerez A., Martínez A.B., Sánchez-Castro J., Rodríguez-Gambarte J.D., Raya J.M., Ríos E., Rodríguez-Rivera M., Espinet B., Florensa L. Trisomy 8, a cytogenetic abnormality in myelodysplastic syndromes, is constitutional or not? PLoS One. 2015; 10 (6): e0129375. DOI: 10.1371/journal.pone.0129375.
13. Greenberg P.L., Tuechler H., Schanz J., Sanz G., Garcia-Manero G., Solé F. Revised international prognostic scoring system (IPSS-R) for myelodysplastic syndromes. Blood. 2012; 120 (12): 2454–2465. DOI: 10.1182/blood-2012-03-420489.
14. Shen Y., Ma H.F., Luo D., Cai J.F., Zou J., Guan J.L. High incidence of gastrointestinal ulceration and cytogenetic aberration of trisomy 8 as typical features of Behcet’s disease associated with myelodysplastic syndrome: a series of 16 consecutive Chinese patients from the Shanghai Behcet’s disease database and comparison with the literature. Biomed. Res. Int. 2018; 2018: 8535091. DOI: 10.1155/2018/8535091.
кариотип — 9 ответов на Babyblog
Евгения
Ты девочка,а он мальчик Кариотип это твой набор хромосом. У каждого нормального человека без всяких синдромов количество хромосом равно 46 штук. У женского пола ещё 2 половые хромосомы Х (девочка) и Х (девочка) Итого нормальный женский кариотип это 46 ХХ.У мужского пола Х(девочка) и У (мальчик) Итого нормальный мужской кариотип 46 ХУ.Когда происходит зачатие то вот при таком наборе они начинают делится и получается либо мальчик, либо девочка. если случается генетическая поломка,синдромы там всякие. Это называется генетическая ошибка. Происходит выкидыши и ЗБ на генетическом уровне, либо рождается ребёночек с синдромом каким нить, что к сожалению не лечится.Кариотип это твоё ДНК. Сдаётся один раз в жизни, стоит дорого. Не меняется и исправить увы его нельзя. ДНК не исправить.Бывает так, что кариотип может быть не нормой а с незначительными отклонениями, и на все вопросы ответит генетик. Зачастую это поломка на каком нибудь генетической хромосоме, или мутирование. Лучше чтобы была норма.А вообще мы все это проходили в школе учебник биологии раздел анатомия, 8-ой или 9-ый класс. ТАм целый параграф был этому посвящён.изменение числа хромосом в кариотипе человека может привести к различным заболеваниям. Наиболее частым хромосомным заболеванием у человека является синдром Дауна, обусловленный трисомией (к паре нормальных хромосом прибавляется еще одна такая же, лишняя) по 21-й хромосоме. Встречается этот синдром с частотой 1-2 на 1000. Нередко трисомия по 21 паре хромосом является причиной гибели плода, однако иногда люди с синдромом Дауна доживают до значительного возраста, хотя в целом продолжительность их жизни сокращена. Известны трисомии по 13-й хромосоме — Синдром Патау, а также по 18-й хромосоме — синдром Эдвардса, при которых жизнеспособность новорожденных резко снижена. Они гибнут в первые месяцы жизни из-за множественных пороков развития.Достаточно часто у человека встречается изменение числа половых хромосом. Среди них известна моносомия Х (из пары хромосом присутствует только одна (Х0)) — это синдром Шерешевского-Тернера. Реже встречается трисомия Х и синдром Клайнфельтера (ХХУ, ХХХУ, ХУУ и т.д.). Люди с изменением числа половых хромосом при наличии У-хромосомы развиваются по мужскому типу. Это является следствием того, что факторы, определяющие мужской тип развития, находятся в У-хромосоме. В отличии от мутаций аутосом (все хромосомы, кроме половых), дефекты умственного развития у больных выражены не столь отчетливо, у многих оно в пределах нормы, а иногда даже выше среднего. Вместе с тем у них постоянно наблюдается нарушения развития половых органов и роста. Реже встречаются пороки развития других систем.
9 лет
Ответить
Самка с 46, XY-кариотипом
Реферат
Нарушения полового развития (DSD) — врожденные патологии, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола. 46, XY DSD может быть результатом нарушений развития яичек или нарушений синтеза / действия андрогенов. Профилактическая гонадэктомия должна рассматриваться у пациентов с 46, XY DSD из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях 46, XY DSD, включая XY чистую дисгенезию гонад и синдром полной нечувствительности к андрогенам, которым была проведена профилактическая гонадэктомия.
Ключевые слова: 46, XY женский; Синдром нечувствительности к андрогенам; Дисгенезия гонад
Введение
Нарушения полового развития (ДПР) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола. DSD делится на три группы по хромосомному компоненту: 46, XX DSD; 46, XY DSD; и DSD половых хромосом. 46, XY DSD встречается редко, но может возникать в любой точке пути половой дифференцировки, который представляет собой сложный процесс взаимодействия генов, синтеза андрогенов и регуляции гормонов за счет взаимодействия лигандов с их соответствующими рецепторами [ 1 ].Диагноз пациентов с 46, XY DSD в основном клинический и обычно устанавливается при обследовании на предмет первичной аменореи или задержки полового созревания. Пациентам с DSD, имеющим Y-хромосомы, следует рассмотреть возможность профилактической гонадэктомии из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях из 46 пациентов с XY DSD, перенесших профилактическую гонадэктомию из-за риска развития злокачественных новообразований гонад: один случай — чисто гонадный дисгенез XY, связанный с нарушением развития яичек, а другой — синдром полной нечувствительности к андрогенам (CAIS). связаны с неспособностью рецептора андрогенов реагировать на андрогены, но в остальном нормальным развитием яичек и синтезом андрогенов.Мы также обсуждаем патофизиологические процессы и оптимальные сроки гонадэктомии с доказательствами риска неоплазии у пациентов с 46, XY DSD.
История болезни
1. Случай 1
В нашу клинику была направлена 17-летняя девочка для оценки первичной аменореи и задержки полового созревания. У нее не было соответствующего прошлого медицинского или семейного анамнеза. Ее вес 56 кг, рост 168 см, индекс массы тела 19,8 кг / м 2 . У пациентки была маленькая грудь, которая не была развита должным образом (стадия 2 груди по Таннеру), и отсутствовали волосы на лобке (стадия лобковых волос по Таннеру 1).Осмотр гениталий показал, что у пациентки были нормальные женские наружные гениталии с неповрежденной девственной плевелой и влагалище глубиной 5 см (). Трансабдоминальное ультразвуковое исследование и магнитно-резонансная томография (МРТ) выявили небольшую матку размером около 15 мм в длину; однако ни яичников, ни семенников не было видно. Лабораторная оценка показала заметно повышенный уровень фолликулостимулирующего гормона (65,08 мМЕ / мл) и очень низкий уровень эстрадиола (менее 5 пг / мл). Уровень лютеинизирующего гормона в сыворотке крови — 21.31 мМЕ / мл, а тестостерона — 3 нг / дл, что было в пределах нормы для женщин этого возраста. Был получен кариотип, который показал нормальный мужской кариотип 46, XY. Планировалось провести профилактическую лапароскопическую гонадэктомию на основании диагноза дисгенезия гонад XY. Лапароскопическое исследование показало наличие двусторонних полосатых гонад с маленькой маткой и нормальными маточными трубами (). Пациенту выполнена двусторонняя лапароскопическая гонадэктомия и сальпингэктомия ().Патологическое исследование образцов показало двусторонние полоски в яичниках, но не обнаружило признаков клеток Сертоли или злокачественных компонентов (). Пациенту назначена циклическая заместительная терапия эстрогенами и прогестероном.
Клинические данные пациента с XY чистой дисгенезией гонад. (A) Нормальные наружные женские гениталии с неповрежденной девственной плевелой. (B) Маленькая матка (звездочка). (C) Левая полоса яичника (стрелка) и маточная труба (наконечник стрелки). (D) Правая полоса яичника (стрелка) и маточная труба (наконечник стрелки).(E) Перерезка левой полоски яичника и маточной трубы с помощью LigaSure. (F) Правая полоса яичника и маточная труба пересекаются с помощью LigaSure. (G) Микроскопическое обнаружение полоски яичника у пациента с чисто XY дисгенезией гонад, показывающее простую кубовидную выстилку из модифицированных мезотелиальных клеток в поверхностном эпителии полоски яичника. Подчеркивающая строма состоит из параллельных веретенообразных клеток или ячеистого узора, который является характерным признаком стромы яичников (H&E, × 100).(H) Общее обнаружение паховых яичек у пациента с синдромом полной нечувствительности к андрогенам. (I) Микроскопическое исследование паховых яичек у пациента с синдромом полной нечувствительности к андрогенам, показывающее незрелые семенные канальцы с толстой гиалинизированной базальной мембраной (H&E, × 100).
2. Случай 2
23-летняя женщина прошла обследование по поводу первичной аменореи. В анамнезе хирургическая паховая грыжа в возрасте 8 лет. Ее вес 62 кг, рост 160 см, индекс массы тела 24.2 кг / м 2 . У пациентки не было волос в подмышечных впадинах, рост груди на 3-й стадии по Таннеру и рост лобковых волос на 1-й стадии по Таннеру. Осмотр ее наружных половых органов показал нормальный вид больших и малых половых губ. Однако был обнаружен короткий слепой вагинальный мешок глубиной 4 см. Паховые образования пальпировались с обеих сторон и были идентифицированы на МРТ как яичкообразные гонады, расположенные в паховом канале. Трансвагинальное УЗИ и МРТ показали отсутствие матки, маточных труб и яичников.Лабораторная оценка показала уровни фолликулостимулирующего гормона, лютеинизирующего гормона и эстрадиола в сыворотке крови 21,2 мл / мл, 22,6 мл / мл и 46,5 пг / мл соответственно. Уровень тестостерона в сыворотке был повышен до 1032 нг / дл, и хромосомный анализ подтвердил нормальный мужской кариотип 46, XY. Пациенту был поставлен диагноз CAIS. Гонадэктомия была рекомендована из-за возможности злокачественной трансформации скрытых элементов яичка. Мы выполнили двустороннюю гонадэктомию через паховый доступ, что обеспечило легкий доступ к семенникам в обоих паховых каналах.Операция была несложной, патологическое исследование гонад показало гиперплазию клеток Лейдига и только клетки Сертоли в семенных канальцах с обеих сторон (). Пациентке была начата терапия эстрогенами для развития груди и здоровья костей.
Обсуждение
46, Дисгенез чистых гонад XY, ранее известный как синдром Свайера, может быть вызван мутацией в определяющей пол области гена Y (SRY), расположенного на дистальном коротком плече Y-хромосомы (Yp11.3) [ 1 ].Мутации в других генах, таких как SOX9, DAX1, WT-1 и SF1, которые участвуют в регуляции экспрессии SRY или действуют как активатор транскрипции ниже SRY в пути, определяющем яички, также могут приводить к чистому XY дисгенезу гонад [ 2 ]. Мутации, участвующие в развитии яичек, приводят к развитию недифференцированных полосковых гонад, которые не продуцируют антимюллеров гормон или андрогены [ 1 ]. Следовательно, развиваются влагалище, шейка матки, матка и маточные трубы, а наружные гениталии принадлежат женщинам из-за отсутствия действия андрогенов ().
Схематическая диаграмма различных путей, вовлеченных в половое развитие пациентов с XY чистой дисгенезией гонад и синдромом полной нечувствительности к андрогенам (CAIS). SF1, стероидогенный фактор 1; WT1, опухоль Вильмса 1; SRY — пол-определяющая область Y; WNT4, член семейства сайтов интеграции MMTV бескрылого типа 4; FOXL2, фактор транскрипции вилки L2; DAX1, чувствительный к дозировке критический участок с изменением пола — врожденная гипоплазия надпочечников на Х-хромосоме, ген 1; RSPO1, R-спондин 1; SOX9, относящийся к SRY HMG box 9; АМГ, антимюллеров гормон; Т, тестостерон; ДГТ, дигидротестостерон; Е2, эстрадиол; AR, рецептор андрогенов.
В отличие от дисгенетических гонад в XY чистой гонадной дисгенезии, люди с CAIS имеют нормальные семенники, а клетки Лейдига в яичке обычно производят тестостерон. Однако действие андрогенов оказывается недостаточным из-за мутаций в гене рецептора андрогенов [ 1 ]. Следовательно, Вольфов проток спонтанно регрессирует в отсутствие андрогенов и не может дифференцироваться на придаток яичка и семявыносящий проток. Клетки Сертоли в семенниках выделяют антимюллеров гормон обычным образом и вызывают регресс мюллеровых структур [ 1 ].Следовательно, у пациенток с CAIS короткое, слепое влагалище без матки, яичников или маточных труб ().
У пациентов с CAIS семенники могут располагаться где угодно на пути опускания яичка эмбриона; однако они чаще располагаются в паховых каналах или больших половых губах [ 3 ]. Deeb и Hughes [ 4 ] сообщили, что более половины пациентов с CAIS имели паховую грыжу; половина из них были двусторонними, а одна треть содержала гонады.
Тщательный сбор анамнеза и физикальное обследование имеют первостепенное значение в диагностическом процессе пациентов с 46, XY DSD. В обоих наших случаях у пациентов были женские наружные гениталии. Однако пациентка с CAIS отличалась адекватным развитием груди, вероятно, из-за действия эстрогенов, которому не препятствовало действие андрогенов. Кроме того, МРТ может быть эффективным диагностическим инструментом для пациентов с 46, XY DSD, в частности, для выявления гипоплазии матки при чистом гонадном дисгенезе XY и локализации крипторхидных семенников в случаях CAIS.Кроме того, высокие уровни тестостерона в сыворотке также помогают отличить CAIS от чисто гонадной дисгенезии XY.
Клиническое ведение пациентки XY обычно включает профилактическую гонадэктомию для предотвращения злокачественной трансформации, соответствующую заместительную гормональную терапию (ЗГТ) и психологическое консультирование.
Лица, кариотипы которых содержат Y-клетки, предрасположены к опухолям гонадного гребня, включая гонадобластомы, дисгерминомы, опухоли желточного мешка и хориокарциномы [ 5 ].Этим пациентам рекомендуется профилактическая гонадэктомия, чтобы исключить риск потенциальных злокачественных изменений гонад [ 5 ]. При рассмотрении сроков проведения гонадэктомии у пациентов с чисто XY дисгенезией гонад рекомендуется ранняя гонадэктомия из-за высокого риска злокачественного новообразования, который оценивается в 46–75% [ 1 ]. В нашем случае чисто гонадальной дисгенезии XY пациенту была выполнена лапароскопическая двусторонняя гонадэктомия и сальпингэктомия вскоре после постановки диагноза.Пациенты с чисто гонадной дисгенезией XY могут забеременеть посредством оплодотворения in vitro с использованием донорских ооцитов [ 6 ]. По сравнению с чисто гонадальной дисгенезией XY, гонадэктомия рекомендуется после завершения пубертатного развития в случаях CAIS из-за низкого риска злокачественных новообразований до полового созревания и того факта, что беспрепятственный тестикулярный эстроген вызывает развитие груди [ 7 ]. В нашем случае CAIS пациенту была проведена профилактическая гонадэктомия после пубертатного развития, и не было никаких признаков злокачественности гонад.
После выполнения гонадэктомии пациентам XY женского пола следует назначить экзогенную ЗГТ для инициирования, созревания и поддержания вторичных половых признаков [ 8 ]. Профилактика ишемической болезни сердца и остеопороза является дополнительным преимуществом терапии эстрогенами [ 8 ]. Для людей с XY чисто гонадным дисгенезом терапия циклическими эстрогенами и прогестагенами, начатая после завершения индуцированного эстрогеном развития груди, предотвратит гиперплазию эндометрия, которая может возникнуть в результате беспрепятственной стимуляции эстрогенами [ 8 ].В обоих наших случаях пациенты получали ЗГТ, которая будет продолжаться до достижения ими 50-летнего возраста.
Психологическое консультирование пациента и его родителей является важным элементом лечения пациентов с 46, XY DSD. В целом, большинство людей с 46, XY DSD имеют полностью женский фенотип; следовательно, женская гендерная идентичность может быть усилена. Когда пациенты испытывают трудности с проникновением из-за гипоплазии влагалища, функциональное влагалище может рассматриваться для получения нормального сексуального удовлетворения с помощью прогрессивного расширения влагалища, традиционной вагинопластики или лапароскопического метода Веккиетти [ 9 ].
Насколько нам известно, было немного сообщений, описывающих результаты клинической визуализации у пациентов с 46 XY DSD. Результаты визуализации, включая женские наружные гениталии, полоску половых желез и матку, в нашем случае помогут клиницистам оценить пациентов с 46, XY DSD.
Самка с кариотипом 46, XY
Реферат
Нарушения полового развития (DSD) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.46, XY DSD может быть результатом нарушений развития яичек или нарушений синтеза / действия андрогенов. Профилактическая гонадэктомия должна рассматриваться у пациентов с 46, XY DSD из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях 46, XY DSD, включая XY чистую дисгенезию гонад и синдром полной нечувствительности к андрогенам, которым была проведена профилактическая гонадэктомия.
Ключевые слова: 46, XY женский; Синдром нечувствительности к андрогенам; Дисгенезия гонад
Введение
Нарушения полового развития (ДПР) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.DSD делится на три группы по хромосомному компоненту: 46, XX DSD; 46, XY DSD; и DSD половых хромосом. 46, XY DSD встречается редко, но может возникать в любой точке пути половой дифференцировки, который представляет собой сложный процесс взаимодействия генов, синтеза андрогенов и регуляции гормонов за счет взаимодействия лигандов с их соответствующими рецепторами [ 1 ]. Диагноз пациентов с 46, XY DSD в основном клинический и обычно устанавливается при обследовании на предмет первичной аменореи или задержки полового созревания.Пациентам с DSD, имеющим Y-хромосомы, следует рассмотреть возможность профилактической гонадэктомии из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях из 46 пациентов с XY DSD, перенесших профилактическую гонадэктомию из-за риска развития злокачественных новообразований гонад: один случай — чисто гонадный дисгенез XY, связанный с нарушением развития яичек, а другой — синдром полной нечувствительности к андрогенам (CAIS). связаны с неспособностью рецептора андрогенов реагировать на андрогены, но в остальном нормальным развитием яичек и синтезом андрогенов.Мы также обсуждаем патофизиологические процессы и оптимальные сроки гонадэктомии с доказательствами риска неоплазии у пациентов с 46, XY DSD.
История болезни
1. Случай 1
В нашу клинику была направлена 17-летняя девочка для оценки первичной аменореи и задержки полового созревания. У нее не было соответствующего прошлого медицинского или семейного анамнеза. Ее вес 56 кг, рост 168 см, индекс массы тела 19,8 кг / м 2 . У пациентки была маленькая грудь, которая не была развита должным образом (стадия 2 груди по Таннеру), и отсутствовали волосы на лобке (стадия лобковых волос по Таннеру 1).Осмотр гениталий показал, что у пациентки были нормальные женские наружные гениталии с неповрежденной девственной плевелой и влагалище глубиной 5 см (). Трансабдоминальное ультразвуковое исследование и магнитно-резонансная томография (МРТ) выявили небольшую матку размером около 15 мм в длину; однако ни яичников, ни семенников не было видно. Лабораторная оценка показала заметно повышенный уровень фолликулостимулирующего гормона (65,08 мМЕ / мл) и очень низкий уровень эстрадиола (менее 5 пг / мл). Уровень лютеинизирующего гормона в сыворотке крови — 21.31 мМЕ / мл, а тестостерона — 3 нг / дл, что было в пределах нормы для женщин этого возраста. Был получен кариотип, который показал нормальный мужской кариотип 46, XY. Планировалось провести профилактическую лапароскопическую гонадэктомию на основании диагноза дисгенезия гонад XY. Лапароскопическое исследование показало наличие двусторонних полосатых гонад с маленькой маткой и нормальными маточными трубами (). Пациенту выполнена двусторонняя лапароскопическая гонадэктомия и сальпингэктомия ().Патологическое исследование образцов показало двусторонние полоски в яичниках, но не обнаружило признаков клеток Сертоли или злокачественных компонентов (). Пациенту назначена циклическая заместительная терапия эстрогенами и прогестероном.
Клинические данные пациента с XY чистой дисгенезией гонад. (A) Нормальные наружные женские гениталии с неповрежденной девственной плевелой. (B) Маленькая матка (звездочка). (C) Левая полоса яичника (стрелка) и маточная труба (наконечник стрелки). (D) Правая полоса яичника (стрелка) и маточная труба (наконечник стрелки).(E) Перерезка левой полоски яичника и маточной трубы с помощью LigaSure. (F) Правая полоса яичника и маточная труба пересекаются с помощью LigaSure. (G) Микроскопическое обнаружение полоски яичника у пациента с чисто XY дисгенезией гонад, показывающее простую кубовидную выстилку из модифицированных мезотелиальных клеток в поверхностном эпителии полоски яичника. Подчеркивающая строма состоит из параллельных веретенообразных клеток или ячеистого узора, который является характерным признаком стромы яичников (H&E, × 100).(H) Общее обнаружение паховых яичек у пациента с синдромом полной нечувствительности к андрогенам. (I) Микроскопическое исследование паховых яичек у пациента с синдромом полной нечувствительности к андрогенам, показывающее незрелые семенные канальцы с толстой гиалинизированной базальной мембраной (H&E, × 100).
2. Случай 2
23-летняя женщина прошла обследование по поводу первичной аменореи. В анамнезе хирургическая паховая грыжа в возрасте 8 лет. Ее вес 62 кг, рост 160 см, индекс массы тела 24.2 кг / м 2 . У пациентки не было волос в подмышечных впадинах, рост груди на 3-й стадии по Таннеру и рост лобковых волос на 1-й стадии по Таннеру. Осмотр ее наружных половых органов показал нормальный вид больших и малых половых губ. Однако был обнаружен короткий слепой вагинальный мешок глубиной 4 см. Паховые образования пальпировались с обеих сторон и были идентифицированы на МРТ как яичкообразные гонады, расположенные в паховом канале. Трансвагинальное УЗИ и МРТ показали отсутствие матки, маточных труб и яичников.Лабораторная оценка показала уровни фолликулостимулирующего гормона, лютеинизирующего гормона и эстрадиола в сыворотке крови 21,2 мл / мл, 22,6 мл / мл и 46,5 пг / мл соответственно. Уровень тестостерона в сыворотке был повышен до 1032 нг / дл, и хромосомный анализ подтвердил нормальный мужской кариотип 46, XY. Пациенту был поставлен диагноз CAIS. Гонадэктомия была рекомендована из-за возможности злокачественной трансформации скрытых элементов яичка. Мы выполнили двустороннюю гонадэктомию через паховый доступ, что обеспечило легкий доступ к семенникам в обоих паховых каналах.Операция была несложной, патологическое исследование гонад показало гиперплазию клеток Лейдига и только клетки Сертоли в семенных канальцах с обеих сторон (). Пациентке была начата терапия эстрогенами для развития груди и здоровья костей.
Обсуждение
46, Дисгенез чистых гонад XY, ранее известный как синдром Свайера, может быть вызван мутацией в определяющей пол области гена Y (SRY), расположенного на дистальном коротком плече Y-хромосомы (Yp11.3) [ 1 ].Мутации в других генах, таких как SOX9, DAX1, WT-1 и SF1, которые участвуют в регуляции экспрессии SRY или действуют как активатор транскрипции ниже SRY в пути, определяющем яички, также могут приводить к чистому XY дисгенезу гонад [ 2 ]. Мутации, участвующие в развитии яичек, приводят к развитию недифференцированных полосковых гонад, которые не продуцируют антимюллеров гормон или андрогены [ 1 ]. Следовательно, развиваются влагалище, шейка матки, матка и маточные трубы, а наружные гениталии принадлежат женщинам из-за отсутствия действия андрогенов ().
Схематическая диаграмма различных путей, вовлеченных в половое развитие пациентов с XY чистой дисгенезией гонад и синдромом полной нечувствительности к андрогенам (CAIS). SF1, стероидогенный фактор 1; WT1, опухоль Вильмса 1; SRY — пол-определяющая область Y; WNT4, член семейства сайтов интеграции MMTV бескрылого типа 4; FOXL2, фактор транскрипции вилки L2; DAX1, чувствительный к дозировке критический участок с изменением пола — врожденная гипоплазия надпочечников на Х-хромосоме, ген 1; RSPO1, R-спондин 1; SOX9, относящийся к SRY HMG box 9; АМГ, антимюллеров гормон; Т, тестостерон; ДГТ, дигидротестостерон; Е2, эстрадиол; AR, рецептор андрогенов.
В отличие от дисгенетических гонад в XY чистой гонадной дисгенезии, люди с CAIS имеют нормальные семенники, а клетки Лейдига в яичке обычно производят тестостерон. Однако действие андрогенов оказывается недостаточным из-за мутаций в гене рецептора андрогенов [ 1 ]. Следовательно, Вольфов проток спонтанно регрессирует в отсутствие андрогенов и не может дифференцироваться на придаток яичка и семявыносящий проток. Клетки Сертоли в семенниках выделяют антимюллеров гормон обычным образом и вызывают регресс мюллеровых структур [ 1 ].Следовательно, у пациенток с CAIS короткое, слепое влагалище без матки, яичников или маточных труб ().
У пациентов с CAIS семенники могут располагаться где угодно на пути опускания яичка эмбриона; однако они чаще располагаются в паховых каналах или больших половых губах [ 3 ]. Deeb и Hughes [ 4 ] сообщили, что более половины пациентов с CAIS имели паховую грыжу; половина из них были двусторонними, а одна треть содержала гонады.
Тщательный сбор анамнеза и физикальное обследование имеют первостепенное значение в диагностическом процессе пациентов с 46, XY DSD. В обоих наших случаях у пациентов были женские наружные гениталии. Однако пациентка с CAIS отличалась адекватным развитием груди, вероятно, из-за действия эстрогенов, которому не препятствовало действие андрогенов. Кроме того, МРТ может быть эффективным диагностическим инструментом для пациентов с 46, XY DSD, в частности, для выявления гипоплазии матки при чистом гонадном дисгенезе XY и локализации крипторхидных семенников в случаях CAIS.Кроме того, высокие уровни тестостерона в сыворотке также помогают отличить CAIS от чисто гонадной дисгенезии XY.
Клиническое ведение пациентки XY обычно включает профилактическую гонадэктомию для предотвращения злокачественной трансформации, соответствующую заместительную гормональную терапию (ЗГТ) и психологическое консультирование.
Лица, кариотипы которых содержат Y-клетки, предрасположены к опухолям гонадного гребня, включая гонадобластомы, дисгерминомы, опухоли желточного мешка и хориокарциномы [ 5 ].Этим пациентам рекомендуется профилактическая гонадэктомия, чтобы исключить риск потенциальных злокачественных изменений гонад [ 5 ]. При рассмотрении сроков проведения гонадэктомии у пациентов с чисто XY дисгенезией гонад рекомендуется ранняя гонадэктомия из-за высокого риска злокачественного новообразования, который оценивается в 46–75% [ 1 ]. В нашем случае чисто гонадальной дисгенезии XY пациенту была выполнена лапароскопическая двусторонняя гонадэктомия и сальпингэктомия вскоре после постановки диагноза.Пациенты с чисто гонадной дисгенезией XY могут забеременеть посредством оплодотворения in vitro с использованием донорских ооцитов [ 6 ]. По сравнению с чисто гонадальной дисгенезией XY, гонадэктомия рекомендуется после завершения пубертатного развития в случаях CAIS из-за низкого риска злокачественных новообразований до полового созревания и того факта, что беспрепятственный тестикулярный эстроген вызывает развитие груди [ 7 ]. В нашем случае CAIS пациенту была проведена профилактическая гонадэктомия после пубертатного развития, и не было никаких признаков злокачественности гонад.
После выполнения гонадэктомии пациентам XY женского пола следует назначить экзогенную ЗГТ для инициирования, созревания и поддержания вторичных половых признаков [ 8 ]. Профилактика ишемической болезни сердца и остеопороза является дополнительным преимуществом терапии эстрогенами [ 8 ]. Для людей с XY чисто гонадным дисгенезом терапия циклическими эстрогенами и прогестагенами, начатая после завершения индуцированного эстрогеном развития груди, предотвратит гиперплазию эндометрия, которая может возникнуть в результате беспрепятственной стимуляции эстрогенами [ 8 ].В обоих наших случаях пациенты получали ЗГТ, которая будет продолжаться до достижения ими 50-летнего возраста.
Психологическое консультирование пациента и его родителей является важным элементом лечения пациентов с 46, XY DSD. В целом, большинство людей с 46, XY DSD имеют полностью женский фенотип; следовательно, женская гендерная идентичность может быть усилена. Когда пациенты испытывают трудности с проникновением из-за гипоплазии влагалища, функциональное влагалище может рассматриваться для получения нормального сексуального удовлетворения с помощью прогрессивного расширения влагалища, традиционной вагинопластики или лапароскопического метода Веккиетти [ 9 ].
Насколько нам известно, было немного сообщений, описывающих результаты клинической визуализации у пациентов с 46 XY DSD. Результаты визуализации, включая женские наружные гениталии, полоску половых желез и матку, в нашем случае помогут клиницистам оценить пациентов с 46, XY DSD.
Самка с кариотипом 46, XY
Реферат
Нарушения полового развития (DSD) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.46, XY DSD может быть результатом нарушений развития яичек или нарушений синтеза / действия андрогенов. Профилактическая гонадэктомия должна рассматриваться у пациентов с 46, XY DSD из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях 46, XY DSD, включая XY чистую дисгенезию гонад и синдром полной нечувствительности к андрогенам, которым была проведена профилактическая гонадэктомия.
Ключевые слова: 46, XY женский; Синдром нечувствительности к андрогенам; Дисгенезия гонад
Введение
Нарушения полового развития (ДПР) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.DSD делится на три группы по хромосомному компоненту: 46, XX DSD; 46, XY DSD; и DSD половых хромосом. 46, XY DSD встречается редко, но может возникать в любой точке пути половой дифференцировки, который представляет собой сложный процесс взаимодействия генов, синтеза андрогенов и регуляции гормонов за счет взаимодействия лигандов с их соответствующими рецепторами [ 1 ]. Диагноз пациентов с 46, XY DSD в основном клинический и обычно устанавливается при обследовании на предмет первичной аменореи или задержки полового созревания.Пациентам с DSD, имеющим Y-хромосомы, следует рассмотреть возможность профилактической гонадэктомии из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях из 46 пациентов с XY DSD, перенесших профилактическую гонадэктомию из-за риска развития злокачественных новообразований гонад: один случай — чисто гонадный дисгенез XY, связанный с нарушением развития яичек, а другой — синдром полной нечувствительности к андрогенам (CAIS). связаны с неспособностью рецептора андрогенов реагировать на андрогены, но в остальном нормальным развитием яичек и синтезом андрогенов.Мы также обсуждаем патофизиологические процессы и оптимальные сроки гонадэктомии с доказательствами риска неоплазии у пациентов с 46, XY DSD.
История болезни
1. Случай 1
В нашу клинику была направлена 17-летняя девочка для оценки первичной аменореи и задержки полового созревания. У нее не было соответствующего прошлого медицинского или семейного анамнеза. Ее вес 56 кг, рост 168 см, индекс массы тела 19,8 кг / м 2 . У пациентки была маленькая грудь, которая не была развита должным образом (стадия 2 груди по Таннеру), и отсутствовали волосы на лобке (стадия лобковых волос по Таннеру 1).Осмотр гениталий показал, что у пациентки были нормальные женские наружные гениталии с неповрежденной девственной плевелой и влагалище глубиной 5 см (). Трансабдоминальное ультразвуковое исследование и магнитно-резонансная томография (МРТ) выявили небольшую матку размером около 15 мм в длину; однако ни яичников, ни семенников не было видно. Лабораторная оценка показала заметно повышенный уровень фолликулостимулирующего гормона (65,08 мМЕ / мл) и очень низкий уровень эстрадиола (менее 5 пг / мл). Уровень лютеинизирующего гормона в сыворотке крови — 21.31 мМЕ / мл, а тестостерона — 3 нг / дл, что было в пределах нормы для женщин этого возраста. Был получен кариотип, который показал нормальный мужской кариотип 46, XY. Планировалось провести профилактическую лапароскопическую гонадэктомию на основании диагноза дисгенезия гонад XY. Лапароскопическое исследование показало наличие двусторонних полосатых гонад с маленькой маткой и нормальными маточными трубами (). Пациенту выполнена двусторонняя лапароскопическая гонадэктомия и сальпингэктомия ().Патологическое исследование образцов показало двусторонние полоски в яичниках, но не обнаружило признаков клеток Сертоли или злокачественных компонентов (). Пациенту назначена циклическая заместительная терапия эстрогенами и прогестероном.
Клинические данные пациента с XY чистой дисгенезией гонад. (A) Нормальные наружные женские гениталии с неповрежденной девственной плевелой. (B) Маленькая матка (звездочка). (C) Левая полоса яичника (стрелка) и маточная труба (наконечник стрелки). (D) Правая полоса яичника (стрелка) и маточная труба (наконечник стрелки).(E) Перерезка левой полоски яичника и маточной трубы с помощью LigaSure. (F) Правая полоса яичника и маточная труба пересекаются с помощью LigaSure. (G) Микроскопическое обнаружение полоски яичника у пациента с чисто XY дисгенезией гонад, показывающее простую кубовидную выстилку из модифицированных мезотелиальных клеток в поверхностном эпителии полоски яичника. Подчеркивающая строма состоит из параллельных веретенообразных клеток или ячеистого узора, который является характерным признаком стромы яичников (H&E, × 100).(H) Общее обнаружение паховых яичек у пациента с синдромом полной нечувствительности к андрогенам. (I) Микроскопическое исследование паховых яичек у пациента с синдромом полной нечувствительности к андрогенам, показывающее незрелые семенные канальцы с толстой гиалинизированной базальной мембраной (H&E, × 100).
2. Случай 2
23-летняя женщина прошла обследование по поводу первичной аменореи. В анамнезе хирургическая паховая грыжа в возрасте 8 лет. Ее вес 62 кг, рост 160 см, индекс массы тела 24.2 кг / м 2 . У пациентки не было волос в подмышечных впадинах, рост груди на 3-й стадии по Таннеру и рост лобковых волос на 1-й стадии по Таннеру. Осмотр ее наружных половых органов показал нормальный вид больших и малых половых губ. Однако был обнаружен короткий слепой вагинальный мешок глубиной 4 см. Паховые образования пальпировались с обеих сторон и были идентифицированы на МРТ как яичкообразные гонады, расположенные в паховом канале. Трансвагинальное УЗИ и МРТ показали отсутствие матки, маточных труб и яичников.Лабораторная оценка показала уровни фолликулостимулирующего гормона, лютеинизирующего гормона и эстрадиола в сыворотке крови 21,2 мл / мл, 22,6 мл / мл и 46,5 пг / мл соответственно. Уровень тестостерона в сыворотке был повышен до 1032 нг / дл, и хромосомный анализ подтвердил нормальный мужской кариотип 46, XY. Пациенту был поставлен диагноз CAIS. Гонадэктомия была рекомендована из-за возможности злокачественной трансформации скрытых элементов яичка. Мы выполнили двустороннюю гонадэктомию через паховый доступ, что обеспечило легкий доступ к семенникам в обоих паховых каналах.Операция была несложной, патологическое исследование гонад показало гиперплазию клеток Лейдига и только клетки Сертоли в семенных канальцах с обеих сторон (). Пациентке была начата терапия эстрогенами для развития груди и здоровья костей.
Обсуждение
46, Дисгенез чистых гонад XY, ранее известный как синдром Свайера, может быть вызван мутацией в определяющей пол области гена Y (SRY), расположенного на дистальном коротком плече Y-хромосомы (Yp11.3) [ 1 ].Мутации в других генах, таких как SOX9, DAX1, WT-1 и SF1, которые участвуют в регуляции экспрессии SRY или действуют как активатор транскрипции ниже SRY в пути, определяющем яички, также могут приводить к чистому XY дисгенезу гонад [ 2 ]. Мутации, участвующие в развитии яичек, приводят к развитию недифференцированных полосковых гонад, которые не продуцируют антимюллеров гормон или андрогены [ 1 ]. Следовательно, развиваются влагалище, шейка матки, матка и маточные трубы, а наружные гениталии принадлежат женщинам из-за отсутствия действия андрогенов ().
Схематическая диаграмма различных путей, вовлеченных в половое развитие пациентов с XY чистой дисгенезией гонад и синдромом полной нечувствительности к андрогенам (CAIS). SF1, стероидогенный фактор 1; WT1, опухоль Вильмса 1; SRY — пол-определяющая область Y; WNT4, член семейства сайтов интеграции MMTV бескрылого типа 4; FOXL2, фактор транскрипции вилки L2; DAX1, чувствительный к дозировке критический участок с изменением пола — врожденная гипоплазия надпочечников на Х-хромосоме, ген 1; RSPO1, R-спондин 1; SOX9, относящийся к SRY HMG box 9; АМГ, антимюллеров гормон; Т, тестостерон; ДГТ, дигидротестостерон; Е2, эстрадиол; AR, рецептор андрогенов.
В отличие от дисгенетических гонад в XY чистой гонадной дисгенезии, люди с CAIS имеют нормальные семенники, а клетки Лейдига в яичке обычно производят тестостерон. Однако действие андрогенов оказывается недостаточным из-за мутаций в гене рецептора андрогенов [ 1 ]. Следовательно, Вольфов проток спонтанно регрессирует в отсутствие андрогенов и не может дифференцироваться на придаток яичка и семявыносящий проток. Клетки Сертоли в семенниках выделяют антимюллеров гормон обычным образом и вызывают регресс мюллеровых структур [ 1 ].Следовательно, у пациенток с CAIS короткое, слепое влагалище без матки, яичников или маточных труб ().
У пациентов с CAIS семенники могут располагаться где угодно на пути опускания яичка эмбриона; однако они чаще располагаются в паховых каналах или больших половых губах [ 3 ]. Deeb и Hughes [ 4 ] сообщили, что более половины пациентов с CAIS имели паховую грыжу; половина из них были двусторонними, а одна треть содержала гонады.
Тщательный сбор анамнеза и физикальное обследование имеют первостепенное значение в диагностическом процессе пациентов с 46, XY DSD. В обоих наших случаях у пациентов были женские наружные гениталии. Однако пациентка с CAIS отличалась адекватным развитием груди, вероятно, из-за действия эстрогенов, которому не препятствовало действие андрогенов. Кроме того, МРТ может быть эффективным диагностическим инструментом для пациентов с 46, XY DSD, в частности, для выявления гипоплазии матки при чистом гонадном дисгенезе XY и локализации крипторхидных семенников в случаях CAIS.Кроме того, высокие уровни тестостерона в сыворотке также помогают отличить CAIS от чисто гонадной дисгенезии XY.
Клиническое ведение пациентки XY обычно включает профилактическую гонадэктомию для предотвращения злокачественной трансформации, соответствующую заместительную гормональную терапию (ЗГТ) и психологическое консультирование.
Лица, кариотипы которых содержат Y-клетки, предрасположены к опухолям гонадного гребня, включая гонадобластомы, дисгерминомы, опухоли желточного мешка и хориокарциномы [ 5 ].Этим пациентам рекомендуется профилактическая гонадэктомия, чтобы исключить риск потенциальных злокачественных изменений гонад [ 5 ]. При рассмотрении сроков проведения гонадэктомии у пациентов с чисто XY дисгенезией гонад рекомендуется ранняя гонадэктомия из-за высокого риска злокачественного новообразования, который оценивается в 46–75% [ 1 ]. В нашем случае чисто гонадальной дисгенезии XY пациенту была выполнена лапароскопическая двусторонняя гонадэктомия и сальпингэктомия вскоре после постановки диагноза.Пациенты с чисто гонадной дисгенезией XY могут забеременеть посредством оплодотворения in vitro с использованием донорских ооцитов [ 6 ]. По сравнению с чисто гонадальной дисгенезией XY, гонадэктомия рекомендуется после завершения пубертатного развития в случаях CAIS из-за низкого риска злокачественных новообразований до полового созревания и того факта, что беспрепятственный тестикулярный эстроген вызывает развитие груди [ 7 ]. В нашем случае CAIS пациенту была проведена профилактическая гонадэктомия после пубертатного развития, и не было никаких признаков злокачественности гонад.
После выполнения гонадэктомии пациентам XY женского пола следует назначить экзогенную ЗГТ для инициирования, созревания и поддержания вторичных половых признаков [ 8 ]. Профилактика ишемической болезни сердца и остеопороза является дополнительным преимуществом терапии эстрогенами [ 8 ]. Для людей с XY чисто гонадным дисгенезом терапия циклическими эстрогенами и прогестагенами, начатая после завершения индуцированного эстрогеном развития груди, предотвратит гиперплазию эндометрия, которая может возникнуть в результате беспрепятственной стимуляции эстрогенами [ 8 ].В обоих наших случаях пациенты получали ЗГТ, которая будет продолжаться до достижения ими 50-летнего возраста.
Психологическое консультирование пациента и его родителей является важным элементом лечения пациентов с 46, XY DSD. В целом, большинство людей с 46, XY DSD имеют полностью женский фенотип; следовательно, женская гендерная идентичность может быть усилена. Когда пациенты испытывают трудности с проникновением из-за гипоплазии влагалища, функциональное влагалище может рассматриваться для получения нормального сексуального удовлетворения с помощью прогрессивного расширения влагалища, традиционной вагинопластики или лапароскопического метода Веккиетти [ 9 ].
Насколько нам известно, было немного сообщений, описывающих результаты клинической визуализации у пациентов с 46 XY DSD. Результаты визуализации, включая женские наружные гениталии, полоску половых желез и матку, в нашем случае помогут клиницистам оценить пациентов с 46, XY DSD.
Самка с кариотипом 46, XY
Реферат
Нарушения полового развития (DSD) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.46, XY DSD может быть результатом нарушений развития яичек или нарушений синтеза / действия андрогенов. Профилактическая гонадэктомия должна рассматриваться у пациентов с 46, XY DSD из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях 46, XY DSD, включая XY чистую дисгенезию гонад и синдром полной нечувствительности к андрогенам, которым была проведена профилактическая гонадэктомия.
Ключевые слова: 46, XY женский; Синдром нечувствительности к андрогенам; Дисгенезия гонад
Введение
Нарушения полового развития (ДПР) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.DSD делится на три группы по хромосомному компоненту: 46, XX DSD; 46, XY DSD; и DSD половых хромосом. 46, XY DSD встречается редко, но может возникать в любой точке пути половой дифференцировки, который представляет собой сложный процесс взаимодействия генов, синтеза андрогенов и регуляции гормонов за счет взаимодействия лигандов с их соответствующими рецепторами [ 1 ]. Диагноз пациентов с 46, XY DSD в основном клинический и обычно устанавливается при обследовании на предмет первичной аменореи или задержки полового созревания.Пациентам с DSD, имеющим Y-хромосомы, следует рассмотреть возможность профилактической гонадэктомии из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях из 46 пациентов с XY DSD, перенесших профилактическую гонадэктомию из-за риска развития злокачественных новообразований гонад: один случай — чисто гонадный дисгенез XY, связанный с нарушением развития яичек, а другой — синдром полной нечувствительности к андрогенам (CAIS). связаны с неспособностью рецептора андрогенов реагировать на андрогены, но в остальном нормальным развитием яичек и синтезом андрогенов.Мы также обсуждаем патофизиологические процессы и оптимальные сроки гонадэктомии с доказательствами риска неоплазии у пациентов с 46, XY DSD.
История болезни
1. Случай 1
В нашу клинику была направлена 17-летняя девочка для оценки первичной аменореи и задержки полового созревания. У нее не было соответствующего прошлого медицинского или семейного анамнеза. Ее вес 56 кг, рост 168 см, индекс массы тела 19,8 кг / м 2 . У пациентки была маленькая грудь, которая не была развита должным образом (стадия 2 груди по Таннеру), и отсутствовали волосы на лобке (стадия лобковых волос по Таннеру 1).Осмотр гениталий показал, что у пациентки были нормальные женские наружные гениталии с неповрежденной девственной плевелой и влагалище глубиной 5 см (). Трансабдоминальное ультразвуковое исследование и магнитно-резонансная томография (МРТ) выявили небольшую матку размером около 15 мм в длину; однако ни яичников, ни семенников не было видно. Лабораторная оценка показала заметно повышенный уровень фолликулостимулирующего гормона (65,08 мМЕ / мл) и очень низкий уровень эстрадиола (менее 5 пг / мл). Уровень лютеинизирующего гормона в сыворотке крови — 21.31 мМЕ / мл, а тестостерона — 3 нг / дл, что было в пределах нормы для женщин этого возраста. Был получен кариотип, который показал нормальный мужской кариотип 46, XY. Планировалось провести профилактическую лапароскопическую гонадэктомию на основании диагноза дисгенезия гонад XY. Лапароскопическое исследование показало наличие двусторонних полосатых гонад с маленькой маткой и нормальными маточными трубами (). Пациенту выполнена двусторонняя лапароскопическая гонадэктомия и сальпингэктомия ().Патологическое исследование образцов показало двусторонние полоски в яичниках, но не обнаружило признаков клеток Сертоли или злокачественных компонентов (). Пациенту назначена циклическая заместительная терапия эстрогенами и прогестероном.
Клинические данные пациента с XY чистой дисгенезией гонад. (A) Нормальные наружные женские гениталии с неповрежденной девственной плевелой. (B) Маленькая матка (звездочка). (C) Левая полоса яичника (стрелка) и маточная труба (наконечник стрелки). (D) Правая полоса яичника (стрелка) и маточная труба (наконечник стрелки).(E) Перерезка левой полоски яичника и маточной трубы с помощью LigaSure. (F) Правая полоса яичника и маточная труба пересекаются с помощью LigaSure. (G) Микроскопическое обнаружение полоски яичника у пациента с чисто XY дисгенезией гонад, показывающее простую кубовидную выстилку из модифицированных мезотелиальных клеток в поверхностном эпителии полоски яичника. Подчеркивающая строма состоит из параллельных веретенообразных клеток или ячеистого узора, который является характерным признаком стромы яичников (H&E, × 100).(H) Общее обнаружение паховых яичек у пациента с синдромом полной нечувствительности к андрогенам. (I) Микроскопическое исследование паховых яичек у пациента с синдромом полной нечувствительности к андрогенам, показывающее незрелые семенные канальцы с толстой гиалинизированной базальной мембраной (H&E, × 100).
2. Случай 2
23-летняя женщина прошла обследование по поводу первичной аменореи. В анамнезе хирургическая паховая грыжа в возрасте 8 лет. Ее вес 62 кг, рост 160 см, индекс массы тела 24.2 кг / м 2 . У пациентки не было волос в подмышечных впадинах, рост груди на 3-й стадии по Таннеру и рост лобковых волос на 1-й стадии по Таннеру. Осмотр ее наружных половых органов показал нормальный вид больших и малых половых губ. Однако был обнаружен короткий слепой вагинальный мешок глубиной 4 см. Паховые образования пальпировались с обеих сторон и были идентифицированы на МРТ как яичкообразные гонады, расположенные в паховом канале. Трансвагинальное УЗИ и МРТ показали отсутствие матки, маточных труб и яичников.Лабораторная оценка показала уровни фолликулостимулирующего гормона, лютеинизирующего гормона и эстрадиола в сыворотке крови 21,2 мл / мл, 22,6 мл / мл и 46,5 пг / мл соответственно. Уровень тестостерона в сыворотке был повышен до 1032 нг / дл, и хромосомный анализ подтвердил нормальный мужской кариотип 46, XY. Пациенту был поставлен диагноз CAIS. Гонадэктомия была рекомендована из-за возможности злокачественной трансформации скрытых элементов яичка. Мы выполнили двустороннюю гонадэктомию через паховый доступ, что обеспечило легкий доступ к семенникам в обоих паховых каналах.Операция была несложной, патологическое исследование гонад показало гиперплазию клеток Лейдига и только клетки Сертоли в семенных канальцах с обеих сторон (). Пациентке была начата терапия эстрогенами для развития груди и здоровья костей.
Обсуждение
46, Дисгенез чистых гонад XY, ранее известный как синдром Свайера, может быть вызван мутацией в определяющей пол области гена Y (SRY), расположенного на дистальном коротком плече Y-хромосомы (Yp11.3) [ 1 ].Мутации в других генах, таких как SOX9, DAX1, WT-1 и SF1, которые участвуют в регуляции экспрессии SRY или действуют как активатор транскрипции ниже SRY в пути, определяющем яички, также могут приводить к чистому XY дисгенезу гонад [ 2 ]. Мутации, участвующие в развитии яичек, приводят к развитию недифференцированных полосковых гонад, которые не продуцируют антимюллеров гормон или андрогены [ 1 ]. Следовательно, развиваются влагалище, шейка матки, матка и маточные трубы, а наружные гениталии принадлежат женщинам из-за отсутствия действия андрогенов ().
Схематическая диаграмма различных путей, вовлеченных в половое развитие пациентов с XY чистой дисгенезией гонад и синдромом полной нечувствительности к андрогенам (CAIS). SF1, стероидогенный фактор 1; WT1, опухоль Вильмса 1; SRY — пол-определяющая область Y; WNT4, член семейства сайтов интеграции MMTV бескрылого типа 4; FOXL2, фактор транскрипции вилки L2; DAX1, чувствительный к дозировке критический участок с изменением пола — врожденная гипоплазия надпочечников на Х-хромосоме, ген 1; RSPO1, R-спондин 1; SOX9, относящийся к SRY HMG box 9; АМГ, антимюллеров гормон; Т, тестостерон; ДГТ, дигидротестостерон; Е2, эстрадиол; AR, рецептор андрогенов.
В отличие от дисгенетических гонад в XY чистой гонадной дисгенезии, люди с CAIS имеют нормальные семенники, а клетки Лейдига в яичке обычно производят тестостерон. Однако действие андрогенов оказывается недостаточным из-за мутаций в гене рецептора андрогенов [ 1 ]. Следовательно, Вольфов проток спонтанно регрессирует в отсутствие андрогенов и не может дифференцироваться на придаток яичка и семявыносящий проток. Клетки Сертоли в семенниках выделяют антимюллеров гормон обычным образом и вызывают регресс мюллеровых структур [ 1 ].Следовательно, у пациенток с CAIS короткое, слепое влагалище без матки, яичников или маточных труб ().
У пациентов с CAIS семенники могут располагаться где угодно на пути опускания яичка эмбриона; однако они чаще располагаются в паховых каналах или больших половых губах [ 3 ]. Deeb и Hughes [ 4 ] сообщили, что более половины пациентов с CAIS имели паховую грыжу; половина из них были двусторонними, а одна треть содержала гонады.
Тщательный сбор анамнеза и физикальное обследование имеют первостепенное значение в диагностическом процессе пациентов с 46, XY DSD. В обоих наших случаях у пациентов были женские наружные гениталии. Однако пациентка с CAIS отличалась адекватным развитием груди, вероятно, из-за действия эстрогенов, которому не препятствовало действие андрогенов. Кроме того, МРТ может быть эффективным диагностическим инструментом для пациентов с 46, XY DSD, в частности, для выявления гипоплазии матки при чистом гонадном дисгенезе XY и локализации крипторхидных семенников в случаях CAIS.Кроме того, высокие уровни тестостерона в сыворотке также помогают отличить CAIS от чисто гонадной дисгенезии XY.
Клиническое ведение пациентки XY обычно включает профилактическую гонадэктомию для предотвращения злокачественной трансформации, соответствующую заместительную гормональную терапию (ЗГТ) и психологическое консультирование.
Лица, кариотипы которых содержат Y-клетки, предрасположены к опухолям гонадного гребня, включая гонадобластомы, дисгерминомы, опухоли желточного мешка и хориокарциномы [ 5 ].Этим пациентам рекомендуется профилактическая гонадэктомия, чтобы исключить риск потенциальных злокачественных изменений гонад [ 5 ]. При рассмотрении сроков проведения гонадэктомии у пациентов с чисто XY дисгенезией гонад рекомендуется ранняя гонадэктомия из-за высокого риска злокачественного новообразования, который оценивается в 46–75% [ 1 ]. В нашем случае чисто гонадальной дисгенезии XY пациенту была выполнена лапароскопическая двусторонняя гонадэктомия и сальпингэктомия вскоре после постановки диагноза.Пациенты с чисто гонадной дисгенезией XY могут забеременеть посредством оплодотворения in vitro с использованием донорских ооцитов [ 6 ]. По сравнению с чисто гонадальной дисгенезией XY, гонадэктомия рекомендуется после завершения пубертатного развития в случаях CAIS из-за низкого риска злокачественных новообразований до полового созревания и того факта, что беспрепятственный тестикулярный эстроген вызывает развитие груди [ 7 ]. В нашем случае CAIS пациенту была проведена профилактическая гонадэктомия после пубертатного развития, и не было никаких признаков злокачественности гонад.
После выполнения гонадэктомии пациентам XY женского пола следует назначить экзогенную ЗГТ для инициирования, созревания и поддержания вторичных половых признаков [ 8 ]. Профилактика ишемической болезни сердца и остеопороза является дополнительным преимуществом терапии эстрогенами [ 8 ]. Для людей с XY чисто гонадным дисгенезом терапия циклическими эстрогенами и прогестагенами, начатая после завершения индуцированного эстрогеном развития груди, предотвратит гиперплазию эндометрия, которая может возникнуть в результате беспрепятственной стимуляции эстрогенами [ 8 ].В обоих наших случаях пациенты получали ЗГТ, которая будет продолжаться до достижения ими 50-летнего возраста.
Психологическое консультирование пациента и его родителей является важным элементом лечения пациентов с 46, XY DSD. В целом, большинство людей с 46, XY DSD имеют полностью женский фенотип; следовательно, женская гендерная идентичность может быть усилена. Когда пациенты испытывают трудности с проникновением из-за гипоплазии влагалища, функциональное влагалище может рассматриваться для получения нормального сексуального удовлетворения с помощью прогрессивного расширения влагалища, традиционной вагинопластики или лапароскопического метода Веккиетти [ 9 ].
Насколько нам известно, было немного сообщений, описывающих результаты клинической визуализации у пациентов с 46 XY DSD. Результаты визуализации, включая женские наружные гениталии, полоску половых желез и матку, в нашем случае помогут клиницистам оценить пациентов с 46, XY DSD.
Самка с кариотипом 46, XY
Реферат
Нарушения полового развития (DSD) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.46, XY DSD может быть результатом нарушений развития яичек или нарушений синтеза / действия андрогенов. Профилактическая гонадэктомия должна рассматриваться у пациентов с 46, XY DSD из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях 46, XY DSD, включая XY чистую дисгенезию гонад и синдром полной нечувствительности к андрогенам, которым была проведена профилактическая гонадэктомия.
Ключевые слова: 46, XY женский; Синдром нечувствительности к андрогенам; Дисгенезия гонад
Введение
Нарушения полового развития (ДПР) — это врожденные состояния, характеризующиеся атипичным развитием хромосомного, гонадного и фенотипического пола.DSD делится на три группы по хромосомному компоненту: 46, XX DSD; 46, XY DSD; и DSD половых хромосом. 46, XY DSD встречается редко, но может возникать в любой точке пути половой дифференцировки, который представляет собой сложный процесс взаимодействия генов, синтеза андрогенов и регуляции гормонов за счет взаимодействия лигандов с их соответствующими рецепторами [ 1 ]. Диагноз пациентов с 46, XY DSD в основном клинический и обычно устанавливается при обследовании на предмет первичной аменореи или задержки полового созревания.Пациентам с DSD, имеющим Y-хромосомы, следует рассмотреть возможность профилактической гонадэктомии из-за повышенного риска злокачественного новообразования гонад. Мы сообщаем о двух редких случаях из 46 пациентов с XY DSD, перенесших профилактическую гонадэктомию из-за риска развития злокачественных новообразований гонад: один случай — чисто гонадный дисгенез XY, связанный с нарушением развития яичек, а другой — синдром полной нечувствительности к андрогенам (CAIS). связаны с неспособностью рецептора андрогенов реагировать на андрогены, но в остальном нормальным развитием яичек и синтезом андрогенов.Мы также обсуждаем патофизиологические процессы и оптимальные сроки гонадэктомии с доказательствами риска неоплазии у пациентов с 46, XY DSD.
История болезни
1. Случай 1
В нашу клинику была направлена 17-летняя девочка для оценки первичной аменореи и задержки полового созревания. У нее не было соответствующего прошлого медицинского или семейного анамнеза. Ее вес 56 кг, рост 168 см, индекс массы тела 19,8 кг / м 2 . У пациентки была маленькая грудь, которая не была развита должным образом (стадия 2 груди по Таннеру), и отсутствовали волосы на лобке (стадия лобковых волос по Таннеру 1).Осмотр гениталий показал, что у пациентки были нормальные женские наружные гениталии с неповрежденной девственной плевелой и влагалище глубиной 5 см (). Трансабдоминальное ультразвуковое исследование и магнитно-резонансная томография (МРТ) выявили небольшую матку размером около 15 мм в длину; однако ни яичников, ни семенников не было видно. Лабораторная оценка показала заметно повышенный уровень фолликулостимулирующего гормона (65,08 мМЕ / мл) и очень низкий уровень эстрадиола (менее 5 пг / мл). Уровень лютеинизирующего гормона в сыворотке крови — 21.31 мМЕ / мл, а тестостерона — 3 нг / дл, что было в пределах нормы для женщин этого возраста. Был получен кариотип, который показал нормальный мужской кариотип 46, XY. Планировалось провести профилактическую лапароскопическую гонадэктомию на основании диагноза дисгенезия гонад XY. Лапароскопическое исследование показало наличие двусторонних полосатых гонад с маленькой маткой и нормальными маточными трубами (). Пациенту выполнена двусторонняя лапароскопическая гонадэктомия и сальпингэктомия ().Патологическое исследование образцов показало двусторонние полоски в яичниках, но не обнаружило признаков клеток Сертоли или злокачественных компонентов (). Пациенту назначена циклическая заместительная терапия эстрогенами и прогестероном.
Клинические данные пациента с XY чистой дисгенезией гонад. (A) Нормальные наружные женские гениталии с неповрежденной девственной плевелой. (B) Маленькая матка (звездочка). (C) Левая полоса яичника (стрелка) и маточная труба (наконечник стрелки). (D) Правая полоса яичника (стрелка) и маточная труба (наконечник стрелки).(E) Перерезка левой полоски яичника и маточной трубы с помощью LigaSure. (F) Правая полоса яичника и маточная труба пересекаются с помощью LigaSure. (G) Микроскопическое обнаружение полоски яичника у пациента с чисто XY дисгенезией гонад, показывающее простую кубовидную выстилку из модифицированных мезотелиальных клеток в поверхностном эпителии полоски яичника. Подчеркивающая строма состоит из параллельных веретенообразных клеток или ячеистого узора, который является характерным признаком стромы яичников (H&E, × 100).(H) Общее обнаружение паховых яичек у пациента с синдромом полной нечувствительности к андрогенам. (I) Микроскопическое исследование паховых яичек у пациента с синдромом полной нечувствительности к андрогенам, показывающее незрелые семенные канальцы с толстой гиалинизированной базальной мембраной (H&E, × 100).
2. Случай 2
23-летняя женщина прошла обследование по поводу первичной аменореи. В анамнезе хирургическая паховая грыжа в возрасте 8 лет. Ее вес 62 кг, рост 160 см, индекс массы тела 24.2 кг / м 2 . У пациентки не было волос в подмышечных впадинах, рост груди на 3-й стадии по Таннеру и рост лобковых волос на 1-й стадии по Таннеру. Осмотр ее наружных половых органов показал нормальный вид больших и малых половых губ. Однако был обнаружен короткий слепой вагинальный мешок глубиной 4 см. Паховые образования пальпировались с обеих сторон и были идентифицированы на МРТ как яичкообразные гонады, расположенные в паховом канале. Трансвагинальное УЗИ и МРТ показали отсутствие матки, маточных труб и яичников.Лабораторная оценка показала уровни фолликулостимулирующего гормона, лютеинизирующего гормона и эстрадиола в сыворотке крови 21,2 мл / мл, 22,6 мл / мл и 46,5 пг / мл соответственно. Уровень тестостерона в сыворотке был повышен до 1032 нг / дл, и хромосомный анализ подтвердил нормальный мужской кариотип 46, XY. Пациенту был поставлен диагноз CAIS. Гонадэктомия была рекомендована из-за возможности злокачественной трансформации скрытых элементов яичка. Мы выполнили двустороннюю гонадэктомию через паховый доступ, что обеспечило легкий доступ к семенникам в обоих паховых каналах.Операция была несложной, патологическое исследование гонад показало гиперплазию клеток Лейдига и только клетки Сертоли в семенных канальцах с обеих сторон (). Пациентке была начата терапия эстрогенами для развития груди и здоровья костей.
Обсуждение
46, Дисгенез чистых гонад XY, ранее известный как синдром Свайера, может быть вызван мутацией в определяющей пол области гена Y (SRY), расположенного на дистальном коротком плече Y-хромосомы (Yp11.3) [ 1 ].Мутации в других генах, таких как SOX9, DAX1, WT-1 и SF1, которые участвуют в регуляции экспрессии SRY или действуют как активатор транскрипции ниже SRY в пути, определяющем яички, также могут приводить к чистому XY дисгенезу гонад [ 2 ]. Мутации, участвующие в развитии яичек, приводят к развитию недифференцированных полосковых гонад, которые не продуцируют антимюллеров гормон или андрогены [ 1 ]. Следовательно, развиваются влагалище, шейка матки, матка и маточные трубы, а наружные гениталии принадлежат женщинам из-за отсутствия действия андрогенов ().
Схематическая диаграмма различных путей, вовлеченных в половое развитие пациентов с XY чистой дисгенезией гонад и синдромом полной нечувствительности к андрогенам (CAIS). SF1, стероидогенный фактор 1; WT1, опухоль Вильмса 1; SRY — пол-определяющая область Y; WNT4, член семейства сайтов интеграции MMTV бескрылого типа 4; FOXL2, фактор транскрипции вилки L2; DAX1, чувствительный к дозировке критический участок с изменением пола — врожденная гипоплазия надпочечников на Х-хромосоме, ген 1; RSPO1, R-спондин 1; SOX9, относящийся к SRY HMG box 9; АМГ, антимюллеров гормон; Т, тестостерон; ДГТ, дигидротестостерон; Е2, эстрадиол; AR, рецептор андрогенов.
В отличие от дисгенетических гонад в XY чистой гонадной дисгенезии, люди с CAIS имеют нормальные семенники, а клетки Лейдига в яичке обычно производят тестостерон. Однако действие андрогенов оказывается недостаточным из-за мутаций в гене рецептора андрогенов [ 1 ]. Следовательно, Вольфов проток спонтанно регрессирует в отсутствие андрогенов и не может дифференцироваться на придаток яичка и семявыносящий проток. Клетки Сертоли в семенниках выделяют антимюллеров гормон обычным образом и вызывают регресс мюллеровых структур [ 1 ].Следовательно, у пациенток с CAIS короткое, слепое влагалище без матки, яичников или маточных труб ().
У пациентов с CAIS семенники могут располагаться где угодно на пути опускания яичка эмбриона; однако они чаще располагаются в паховых каналах или больших половых губах [ 3 ]. Deeb и Hughes [ 4 ] сообщили, что более половины пациентов с CAIS имели паховую грыжу; половина из них были двусторонними, а одна треть содержала гонады.
Тщательный сбор анамнеза и физикальное обследование имеют первостепенное значение в диагностическом процессе пациентов с 46, XY DSD. В обоих наших случаях у пациентов были женские наружные гениталии. Однако пациентка с CAIS отличалась адекватным развитием груди, вероятно, из-за действия эстрогенов, которому не препятствовало действие андрогенов. Кроме того, МРТ может быть эффективным диагностическим инструментом для пациентов с 46, XY DSD, в частности, для выявления гипоплазии матки при чистом гонадном дисгенезе XY и локализации крипторхидных семенников в случаях CAIS.Кроме того, высокие уровни тестостерона в сыворотке также помогают отличить CAIS от чисто гонадной дисгенезии XY.
Клиническое ведение пациентки XY обычно включает профилактическую гонадэктомию для предотвращения злокачественной трансформации, соответствующую заместительную гормональную терапию (ЗГТ) и психологическое консультирование.
Лица, кариотипы которых содержат Y-клетки, предрасположены к опухолям гонадного гребня, включая гонадобластомы, дисгерминомы, опухоли желточного мешка и хориокарциномы [ 5 ].Этим пациентам рекомендуется профилактическая гонадэктомия, чтобы исключить риск потенциальных злокачественных изменений гонад [ 5 ]. При рассмотрении сроков проведения гонадэктомии у пациентов с чисто XY дисгенезией гонад рекомендуется ранняя гонадэктомия из-за высокого риска злокачественного новообразования, который оценивается в 46–75% [ 1 ]. В нашем случае чисто гонадальной дисгенезии XY пациенту была выполнена лапароскопическая двусторонняя гонадэктомия и сальпингэктомия вскоре после постановки диагноза.Пациенты с чисто гонадной дисгенезией XY могут забеременеть посредством оплодотворения in vitro с использованием донорских ооцитов [ 6 ]. По сравнению с чисто гонадальной дисгенезией XY, гонадэктомия рекомендуется после завершения пубертатного развития в случаях CAIS из-за низкого риска злокачественных новообразований до полового созревания и того факта, что беспрепятственный тестикулярный эстроген вызывает развитие груди [ 7 ]. В нашем случае CAIS пациенту была проведена профилактическая гонадэктомия после пубертатного развития, и не было никаких признаков злокачественности гонад.
После выполнения гонадэктомии пациентам XY женского пола следует назначить экзогенную ЗГТ для инициирования, созревания и поддержания вторичных половых признаков [ 8 ]. Профилактика ишемической болезни сердца и остеопороза является дополнительным преимуществом терапии эстрогенами [ 8 ]. Для людей с XY чисто гонадным дисгенезом терапия циклическими эстрогенами и прогестагенами, начатая после завершения индуцированного эстрогеном развития груди, предотвратит гиперплазию эндометрия, которая может возникнуть в результате беспрепятственной стимуляции эстрогенами [ 8 ].В обоих наших случаях пациенты получали ЗГТ, которая будет продолжаться до достижения ими 50-летнего возраста.
Психологическое консультирование пациента и его родителей является важным элементом лечения пациентов с 46, XY DSD. В целом, большинство людей с 46, XY DSD имеют полностью женский фенотип; следовательно, женская гендерная идентичность может быть усилена. Когда пациенты испытывают трудности с проникновением из-за гипоплазии влагалища, функциональное влагалище может рассматриваться для получения нормального сексуального удовлетворения с помощью прогрессивного расширения влагалища, традиционной вагинопластики или лапароскопического метода Веккиетти [ 9 ].
Насколько нам известно, было немного сообщений, описывающих результаты клинической визуализации у пациентов с 46 XY DSD. Результаты визуализации, включая женские наружные гениталии, полоску половых желез и матку, в нашем случае помогут клиницистам оценить пациентов с 46, XY DSD.
Мозаичный синдром Тернера с кариотипом 46, XY
Хотя синдром Тернера чаще всего ассоциируется с генотипом 45, X, другие мозаичные генотипы присутствуют примерно в половине всех случаев.Мы описываем случай синдрома Тернера с генотипом 46, XY с помощью обычного 5-клеточного кариотипа, у которого впоследствии было обнаружено мозаичный генотип 18% 45, X и 82% 46, XY с помощью 50-клеточного FISH-анализа. Лица с мозаичным генотипом 45, X / 46, XY имеют множество фенотипических представлений от мужского до женского, которые не коррелируют с процентом мозаицизма. Наш случай представляет собой крайний пример, когда преобладает генотип 46, XY и фенотип, типичный для синдрома Тернера.
1. Введение
Синдром Тернера диагностируется у женщин на основании клинических проявлений в сочетании с генотипом, состоящим из одной нормальной Х-хромосомы и полного или частичного отсутствия другой Х-хромосомы [1]. Пациенты с мозаицизмом 45, X / 46, XY имеют множество фенотипов, от наиболее часто смешанной дисгенезии гонад до других, таких как фенотипический мужской фенотип, генитальная неоднозначность, синдром Тернера и женщины с нормальными женскими вторичными половыми признаками [2, 3].Синдром Тернера проявляется двусторонней полоской гонад, тогда как смешанная гонадная дисгенезия описывает те, которые проявляются отсутствием или абдоминальной полоской гонады с одной стороны и нормальным или дисгенным яичком с другой. Фенотип у пациента с мозаикой 45, X / 46, XY, вероятно, зависит от распределения процента мозаицизма в разных тканях, которое, как было показано, различается между кровью и тканью гонад [4]. Этот случай является примером, когда доминантный мозаичный генотип в крови (46, XY) не согласуется с фенотипом синдрома Тернера.
2. Изложение клинического случая
Российская женщина с беременностью 1 пара 0010, 38 лет, поступила с нерегулярными менструациями каждые 2-3 месяца и 15-летним анамнезом бесплодия. Перед обращением в наше учреждение она была осмотрена специалистом по репродуктологии в России, где был проведен анализ кариотипа. Копия результата не была доступна для просмотра нашими врачами, но пациентка считала, что у нее был обнаружен кариотип 46, XY. Пациент не знал о каких-либо других соответствующих лабораторных результатах.У пациентки начался менархе в 15 лет, с тех пор менструации были нерегулярными каждые 2-3 месяца. У нее был самопроизвольный аборт в первом триместре беременности, который был выявлен с помощью положительного теста на беременность в домашней моче без клинического ультразвукового исследования или патологического подтверждения. В анамнезе она перенесла лапароскопическую аппендэктомию с одновременной сальпингэктомией справа. У нее не было другого значительного медицинского или семейного анамнеза. В частности, у нее не было семейного анамнеза нерегулярных менструаций, бесплодия или преждевременной недостаточности яичников.
На экзамене она была ростом 160 см и весила 55 кг с ИМТ 23. Ее жизненно важные признаки были нормальными, и у нее были нормальные женские вторичные половые признаки с развитием груди V стадии по Таннеру, ростом волос на лобке V стадии по Таннеру, нормальным влагалищем. и шейка матки, и никакого гирсутизма или клиторомегалии. У нее не было низкого роста, сколиоза, высокого неба, потери слуха, короткой или перепончатой шеи, щитовой груди, вальгусного локтевого сустава, укороченных четвертых пястных или плюсневых костей, genu valgum или varum или деформации Маделунга предплечья и запястья.
Лабораторные исследования показали преждевременную недостаточность яичников с уровнем фолликулостимулирующего гормона 104,9 мМЕ / мл, уровнем лютеинизирующего гормона 35,5 мМЕ / мл, уровнем эстрадиола <5 пг / мл и общим уровнем тестостерона <12 нг / мл. дл. Функции печени и функции щитовидной железы были в пределах нормы. Анализ кариотипа периферической крови 5 клеток при разрешении полос 400-550 показал нормальный мужской кариотип 46, XY (кровь хромосомного анализа, Quest Diagnostics). Хотя этот кариотип соответствует полному дисгенезу гонад (синдром Свайера), история болезни пациентки, связанная с развитием груди и менструациями, не соответствовала этому диагнозу.FISH-анализ был выполнен на 50 клетках для оценки SRY и X-центромеры для оценки возможного синдрома Свайера или мозаицизма низкого уровня. Это показало 41 клетку с 46, XY и 9 клеток с 45, X (FISH SRY / X Centromere, Quest Diagnostics), что клинически коррелировало с диагнозом мозаичного синдрома Тернера.
Сонографическое исследование выявило небольшую матку размером 4,4 × 2,3 × 1,2 см, правый яичник размером 1,4 × 1,2 × 0,9 см с двумя простыми кистами размером 8 мм и 9 мм, левый яичник размером 1.3 × 0,9 × 0,8 см и эхокомплекс эндометрия 6 мм. КТ брюшной полости и таза показала нормальные почки. Эхокардиограмма не показала анатомических аномалий сердца. Двухэнергетическая рентгеновская абсорбциометрия (DEXA) показала поясничный остеопороз с T-оценкой -3,5.
В связи с повышенным риском развития гонадобластомы пациенту предложили лапароскопическую двустороннюю гонадэктомию и левую сальпингэктомию (правая маточная труба хирургически отсутствовала) с промыванием таза.При патологическом обзоре было обнаружено, что двусторонние гонады обладают гипоплазированной тканью яичника (рис. 1) с двумя небольшими серозными кистами правого яичника (рис. 2) и без признаков злокачественности. По поводу остеопороза ей прописали добавки кальция и витамина D, и она предпочла принимать циклические комбинированные пероральные контрацептивы, а не стандартную заместительную гормональную терапию. Ей посоветовали, что беременность — это вариант для нее путем экстракорпорального оплодотворения донорскими яйцеклетками, и она намерена продолжить, когда будет готова к созданию семьи.Ей посоветовали, что бисфосфонаты не рекомендуются женщинам с учетом будущей беременности, и направили в медицинскую эндокринологию для лечения остеопороза другими препаратами, не являющимися бисфосфонатами.
3. Обсуждение
Синдром Тернера связан с множественными хромосомными аномалиями, включая 45, X и 45, X / 46, XX и 45, X / 47, XXX и 45, X / 46, XY. На генотип 45, X / 46, XY приходится примерно 10–12% случаев синдрома Тернера [1]. В сообщении о 76 пренатально диагностированных случаях мозаицизма 45, X / 46, XY, у 75 были мужские наружные гениталии и только у одного были женские гениталии [5].В серии из 27 постнатально диагностированных случаев мозаицизма 45, X / 46, XY 18 были мужчинами (11 со смешанной дисгенезией гонад) и 9 имели синдром Тернера [2]. Синдром Тернера с мозаицизмом низкого уровня 45, X / 46, XY не может быть обнаружен при стандартном кариотипе, и FISH-анализ большего количества клеток может быть полезен для диагностики.
Американский колледж медицинской генетики (ACMG) предоставляет рекомендации по процедуре кариотипирования, характерной для синдрома Тернера. Колледж рекомендует кариотипировать минимум 30 клеток из-за высокой частоты мозаицизма, если мозаицизм не встречается в первых 20 клетках.При высоком клиническом подозрении на синдром Тернера у пациента с кариотипом 46, XX рекомендуется цитогенетическое исследование второго типа ткани (например, биопсия кожи для клеточной культуры или буккальный мазок для FISH). Кроме того, учитывая риск гонадобластомы с скрытым мозаицизмом Y-хромосомы, ACMG рекомендует 200-клеточный FISH-анализ для исследования центромер X и Y, когда 30-клеточный кариотип приводит к немозаичному 45, X кариотипу [6].
Женщины с синдромом Тернера, обладающие материалом Y-хромосомы, имеют повышенный риск развития опухолей половых клеток, таких как гонадобластома и дисгерминома.Национальное когортное исследование, в котором участвовал 211 из этих пациентов, показало, что к 25 годам совокупный риск гонадобластомы составляет 7,9% (95% ДИ 3,1–19,0) [7]. Хотя частота гонадобластомы у пациентов с синдромом Тернера с материалом Y-хромосомы варьируется в зависимости от исследования от 4% до 60%, текущие данные свидетельствуют о том, что частота составляет примерно 10%. Профилактическая гонадэктомия рекомендуется во время постановки диагноза пациентам с синдромом Тернера и материалом Y-хромосомы, таким как мозаицизм 45, X / 46, XY [1] .
Пациенты с синдромом Тернера с любым генотипом должны пройти стандартное обследование и лечение сердечно-сосудистых, почечных, метаболических, эндокринных нарушений, нарушений зрения, слуха и минеральной плотности костей [1]. Если диагностирована преждевременная недостаточность яичников, показана заместительная гормональная терапия до типичного возраста менопаузы, чтобы вызвать половое созревание и вторичные половые признаки, стимулировать рост матки и предотвратить потерю костной массы. Обычно рекомендуется начинать лечение с низкой дозы E 2 (3-7 мк г / день трансдермального E 2 или 0.25 мг перорально E 2 в день) в возрасте 11 или 12 лет и постепенно увеличивайте дозу до доз для взрослых (50-150 мкг г / день трансдермального E 2 или 2-4 мг перорально E 2 в день) от 2 до 3 лет. Предпочтителен трансдермальный эстрадиол, а затем пероральный или внутримышечный эстрадиол. Пероральный прием этинилэстрадиола не рекомендуется, за исключением случаев, когда другие варианты недоступны или из-за проблем, связанных с предпочтениями или комплаентностью пациента, как это было в случае с пациентом, представленным здесь. Прогестин добавляется при внезапном кровотечении или через 2 года после начала E 2 для снижения риска гиперплазии эндометрия.Прогестин можно вводить перорально по циклической схеме с E 2 , перорально непрерывно с E 2 или в форме внутриматочной спирали, содержащей прогестин [8].
Будущая фертильность — важное соображение для пациентов с синдромом Тернера. Точная и ранняя диагностика мозаицизма 45, X / 46, XY может позволить проконсультироваться о репродуктивном потенциале и проведении беременности с экстракорпоральным оплодотворением донорской яйцеклеткой и / или гестационным суррогатным материнством. Успешные исходы беременности имели место у пациенток с мозаицизмом 45, X / 46, XY, а также с дисгенезом гонад 46, XY после донорства ооцитов и экстракорпорального оплодотворения, хотя в большинстве зарегистрированных случаев роды были произведены путем кесарева сечения [9–13].Хотя на УЗИ размер матки этой пациентки составлял всего 4,4 × 2,3 × 1,2 см, противопоказаний к беременности из-за размера матки нет. Размер матки, скорее всего, является результатом низкого уровня эстрогена, а не признаком того, что матка не может вынашивать беременность.
Таким образом, этот случай демонстрирует, что синдром Тернера с мозаицизмом низкого уровня может быть пропущен традиционным кариотипом. У некоторых женщин с диагнозом синдром Свайера на самом деле может быть синдром Тернера с мозаицизмом низкого уровня.Примерно 70-80% пациентов с диагнозом синдром Свайера не имеют мутаций SRY [14, 15], и синдром Тернера с мозаицизмом низкого уровня может быть фактической причиной дисгенезии гонад у некоторых из этих пациентов. В случаях, когда результаты обычного кариотипа не полностью соответствуют клинической картине, анализ FISH для мозаицизма низкого уровня может быть информативным.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Случай из практики секвенирования полного генома у самки XY: выявление новой мутации SRY и пересмотр ошибочного диагноза синдрома нечувствительности к андрогенам | BMC Endocrine Disorders
18-летняя женщина впервые обратилась за медицинской помощью из-за первичной аменореи.В то время она была высокой худой женщиной с женскими внешними гениталиями и без образования вульвы или паха, однако развитие груди и вторичные половые волосы явно отсутствовали. Кариотип 46, XY, что позволяет предположить диагноз CAIS. Из-за подозрения на крипторхию, дисгенетические половые железы с высоким потенциалом злокачественности, пациентке была проведена открытая лапаротомия, выявляя полоски гонад, которые были удалены, и инфантильную матку, оставленную на месте. Гистопатология показала двустороннюю гонадобластому и дисгерминому в правой гонаде.Получила лучевую терапию брюшно-тазовой области, рецидива опухоли не было. После гонадэктомии ей начали принимать оральные контрацептивы, а затем перешли на заместительную гормональную терапию в более низких дозах.
В возрасте 46 лет пациент переехал в Австралию и обратился за помощью в наше учреждение. Среди ее активных медицинских проблем были сахарный диабет 2 типа, дислипидемия, синдром обструктивной дефекации и полипы толстой кишки. У нее было высокое стройное телосложение с индексом массы тела 20,4 кг / м 2 , а ее пропорции были евнухоидными, рост 181 см, размах рук 182 см, длина нижнего сегмента 102 см.Лобковых волос было мало, а в подмышечных впадинах не было, но теперь грудь развивалась нормально. У нее были женские наружные гениталии и ретровертированная матка.
Результаты гормональных исследований и гематокрита наиболее соответствовали диапазонам гипогонадизма у женщин (таблица 2). Двухэнергетическая рентгеновская абсорбциометрия показала легкую остеопению с T-оценкой -1,1 в области бедра и позвоночника. Из-за меноррагии она прошла трансвагинальное УЗИ, которое показало миому внутри гипоплазированной матки (рис.1). Аномалии почечного тракта не выявлено. После неудачной гистероскопии и множественных изменений в заместительной гормональной терапии ее лечили трансдермальным эстрадиолом 25 мкг / час и пероральным норэтистероном 5 мг в день с прекращением менструаций в возрасте 48 лет.
Таблица 2 Результаты биохимических тестов Рис. 1
Трансвагинальное ультразвуковое исследование, показывающее гипоплазию матки с поперечным (до p) и продольным (снизу) сечениями, иллюстрирующими глобулярную форму. В полости матки видна миома размером 4,5 см, вероятно, способствующая меноррагии у пациентки
Именно сейчас, на пятом десятилетии жизни пациентки, ее первоначальный диагноз CAIS был поставлен под сомнение.В то время как внешний женский фенотип, кариотип XY и отсутствие вторичных половых волос соответствовали CAIS, наличие полосатых гонад и функционирующих мюллеровских структур наряду с отсутствием спонтанного развития груди способствовало диагностике CGD. Хотя мюллеровы остатки наблюдались у женщин с CAIS, нет сообщений о функционировании маточных структур, обнаруженных у нашей пациентки. Мы выполнили различные генетические исследования на местном уровне, начиная с цифрового кариотипа, подтверждающего исходный результат 46, XY.FISH продемонстрировал интактный ген SRY , исключая делецию SRY как причину атипичного DSD у пациента. Сравнительная геномная гибридизация с использованием массивов не показала изменения числа копий при среднем эффективном разрешении 0,2 МБ.
Основными генетическими возможностями на этом этапе были варианты последовательностей и вариации числа копий в известных генах CGD, таких как DHH, SRY, NR5A1 и NR0B1 [2]. В соответствии с первоначальным диагнозом пациента за границей, синергические мутации в AR и AMH или AMHR2 считались возможными, поскольку это теоретически могло привести к невирилизованному женскому фенотипу XY с интактными мюллеровыми структурами.Полное секвенирование экзома рассматривалось как следующий шаг в нашей генетической оценке, однако это было сочтено менее подходящим из-за несмежных этапов захвата и амплификации, которые могут препятствовать идентификации вариаций числа копий, которые являются важной причиной CGD [11]. Массив однонуклеотидного полиморфизма (SNP) был альтернативой, поскольку его полезность была установлена в DSD [12], однако генетическое разрешение низкое и не позволяет диагностировать точечные мутации [11]. Вместо этого мы выбрали полногеномное секвенирование (WGS), чтобы исследовать все гены восприимчивости одновременно и с высоким разрешением.WGS также получил признание из-за его способности обнаруживать вариации числа копий, поскольку весь геном непрерывно анализируется, что позволяет обнаруживать точки разрыва ДНК в случаях генетических делеций и дупликаций [11].
WGS выполняли с использованием системы секвенирования Illumina HiSeq X Ten в Институте медицинских исследований Гарвана (Приложение). Биоинформатический анализ был проведен в Лаборатории молекулярной генетики патологии SA с классификацией вариантов по пятиуровневой системе Американского колледжа медицинской генетики: доброкачественные, вероятные доброкачественные, неопределенная значимость, вероятные патогенные или патогенные [13].
Варианты, найденные на WGS, были отфильтрованы на основе мутационных последствий (влияние snpEff высокое или умеренное) и фенотипической корреляции. WGS выявил варианты в восьми генах, связанных с нарушениями полового развития, а именно: AKR1C4 , DMRT1 , MAP2K1 , MAP3K1 , NR0B1 , NR5A1 SR, POR и POR. Все варианты, кроме NR0B1 и SRY , были исключены на основании частотности популяции вариантов> 1% в базах данных 1000 геномов (1 K), Exome Sequencing Project (ESP) и Exome Aggregation Consortium (ExAC).Затем вариант NR0B1 был исключен из-за того, что популяционная частота> 1% среди лиц в базе данных ExAC той же этнической принадлежности, что и пациент.
Оставшийся гемизиготный миссенс вариант в гене SRY (NM_003140.2, p.Arg130Pro, c.389G> C) был подтвержден при секвенировании по Сэнгеру SRY . Об этом варианте ранее не сообщалось в базах данных 1K, ESP, ExAC или dbSNP, хотя в базе данных ClinVar имеется большое количество патогенных миссенс-мутаций (рис.2). Он расположен на 3’-конце боксового домена HMG в области, необходимой для ядерной локализации (UniProt Q05066), и, по прогнозам PolyPhen2 и MutationTaster, он является патогенным, но не SIFT. Он умеренно консервативен (phyloP 3.43, CADD 10.44) и имеет умеренное различие по Грэнтэму, равное 103. Моделирование комплекса белок-ДНК показало, что замещение аргинина (Arg) на пролин (Pro) вызывает стерические препятствия в структуре нативного белка SRY, что может повлиять на Связывание ДНК (рис. 3а и б).
Рис. 2
График доменов нового варианта SRY , R130P (подчеркнут), обнаруженного у пациента вместе с патогенными вариантами, ранее описанными в ClinVar
Рис. 3
a Структура SRY дикого типа человека белок в комплексе с ДНК. R130 ( розовый ) является частью области случайной спирали белка SRY дикого типа. b Замена R130P ( синий ) была показана с помощью моделирования гомологии для создания стерических препятствий ( красные пунктирные линии ) для соседних остатков
Коллективные доказательства продемонстрировали, что этот новый вариант SRY является вероятным патогенным, что согласуется с с диагнозом 46, XY DSD (OMIM 400044) и, более конкретно, CGD, таким образом изменяя диагноз пациента с CAIS на CGD через 30 лет после ее первоначального обращения.