Миелофиброз костного мозга лечение в Израиле, стоимость и отзывы
МиелофиброзМиелофиброз, лечение в Израиле в медицинском центре ASSUTA Express Medical: лучшие врачи, инновационная аппаратура, цены без посредников.
Миелофиброз – заболевание кровеносной системы, которое характеризуется формированием рубцов на костном мозге. В зависимости от причин развития патологии, миелофиброз может быть первичным, то есть болезнь является самостоятельной, и вторичным, когда рубцевание ткани вызвано различными патологиями в организме человека.
План лечения бесплатно
Методы лечения миелофиброза в Израиле
Лечение миелофиброза подбирается израильскими врачами в индивидуальном порядке для каждого клинического случая. Основные методы лечения:
Пересадка – наиболее действенный способ лечения миелофиброза, но, несмотря на простоту проведения, бывает очень сложно подобрать подходящего донора, так как родственники не всегда совпадают. Проводится пересадка только после проведения курса химиотерапии.
Стволовые донорские клетки вводятся в организм пациента внутривенно – через укол или капельницу. До того момента, как пересаженные клетки приживутся в организме пациента, как правило, на это необходимо 1-3 недели, рекомендуется постоянное переливание крови, прием антибиотиков и препаратов противовоспалительного действия для предупреждения развития инфекционных заболеваний и других осложнений. При регулярном проведении поддерживающей терапии человек может прожить долгую, полноценную жизнь.
Трансплантация костного мозга в ИзраилеДостаточно радикальный метод, который применяется в тех случаях, если, несмотря на регулярно проводимое переливание крови, положительная динамика отсутствует, происходит постепенное увеличение интенсивности симптоматической картины, болезнь прогрессирует. Для проведения химиотерапии применяются новейшие препараты.Щадящая химиотерапия в ИзраилеПроведение лучевой терапии – назначается в тех случаях, когда не представляется возможным лечить миелофиброз хирургической операцией. Лучевая терапия способствует уменьшению диаметра увеличенной селезенки, помогает быстро купировать симптоматическую картину и поддерживать нормальную функциональность организма.Точная лучевая терапия в Израиле
Лучевая терапия способствует уменьшению диаметра увеличенной селезенки, помогает быстро купировать симптоматическую картину и поддерживать нормальную функциональность организма.Точная лучевая терапия в Израиле
Хирургическая операция – спленэктомия, подразумевает удаление селезенки, размеры и функциональность которой не представляется возможным восстановить ввиду стремительного развития заболевания.
Метод переливания крови используется для увеличения концентрации кровяных телец и замедления темпов образования рубцовой ткани. Терапия проводится регулярно, и на ранних стадиях развития патологического процесса дает положительный результат. Минус методики – пациент чувствует себя постоянно усталым, апатичным. Бороться с побочной симптоматикой можно путем постоянного употребления фолиевой кислоты.
Причины возникновения миелофиброза
Истинные причины развития миелофиброза, как и многих других заболеваний кровеносной системы, остаются до конца не выясненными. Доподлинно известно только о предрасположенности к заболеванию у людей с мутацией гена JAK2.
Симптомы заболевания
Сложности в своевременной диагностике заболевания связаны с отсутствием специфической симптоматики. На ранних стадиях развития заболевания крови признаки или вовсе отсутствуют, или же слабо выражены, а потому человек до последнего не догадывается о тяжелой патологии в организме. Общие признаки миелофиброза: постоянное чувство усталости, одышка, которая развивается без видимых причин, частые болевые приступы в животе, постоянное чувство дискомфорта в желудке, вне зависимости от количества съеденной пищи, боль в составах и костях, постепенное развитие подагры, отсутствие аппетита, внезапное снижение веса без видимых причин, чрезмерное потоотделение в ночное время, частые лихорадочные приступы.
Лучшие врачи- Онкогематологи Израиля
Диагностика миелофиброза в клинике
В Израиле существует перечень диагностических методов для обследования пациентов, страдающих миелофиброзом:
Лабораторная диагностика
Для диагностирования патологии проводится анализ крови. В большинстве случаев, анализ крови при развитии миелофиброза указывает на тяжелую стадию анемии, чрезмерную концентрацию лейкоцитов или тромбоцитов.
В большинстве случаев, анализ крови при развитии миелофиброза указывает на тяжелую стадию анемии, чрезмерную концентрацию лейкоцитов или тромбоцитов.
ПодробнееБиопсия костного мозга
Для уточнения первичного диагноза и с целью исследования возможных осложнений, проводится биопсия и ультразвуковое исследование селезенки. В случае обнаружения патологического процесса на костном мозге, будет повторно проведен анализ крови, цель которого – выявить ген, который мутировал и привел к развитию миелофиброза.
ПодробнееИндивидуальный план диагностики
Цены на лечение миелофиброза в Израиле
Чтобы узнать подробнее о лечении миелофиброза в Израиле и получить медицинскую программу достаточно указать контактные данные в форме на сайте или через чат онлайн. Пришлите нам описание состояния пациента и контактные данные. Врачи международного отдела составят индивидуальную программу лечения миелофиброза в клинике «Ассута» с указанием стоимости. Координатор позвонит в течение 6 часов. Звонок бесплатный.
Звонок бесплатный.
Как приехать к нам на лечение в Израиль
1
Подать заявку на сайте или связаться с нами любым удобным для Вас способом
Подать заявку
2
Медицинский координатор свяжется с Вами для сбора анамнеза и уточнения деталей приезда
3
Врач клиники составит для вас индивидуальную медицинскую программу
4
Координатор сообщает стоимость лечения в клинике и помогаем с заказом билетов и арендой апартаментов
5
Высылаем приглашение из клиники «Ассута» и расписание прохождения консультаций и процедур
Обзорная экскурсия по ТЕЛЬ-АВИВУВ подарок
Отзывы о лечении в клинике ассута
Василий К.
Пациент: Василий К.
Возраст:33
Страна:Россия
За время пребывания в Израиле наблюдаем профессиональный подход мед.персонала, внимание к вопросам, готовность оказать помощь, дать объективную оценку состояния здоровья.
Благодаря IEM удалось получить ответы на интересующие вопросы и разобраться в ситуации.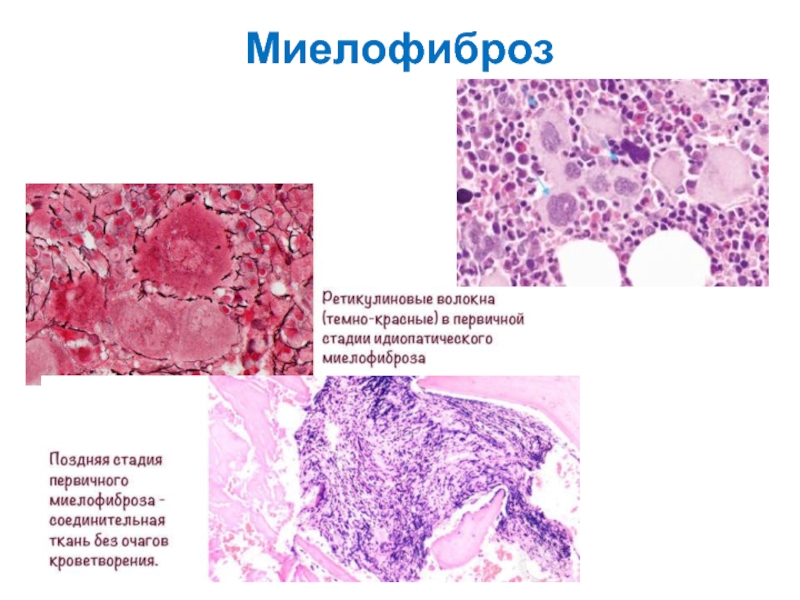
03. 05. 2018
| Губка гемостатическая |
| Азитромицин (Azithromycin) |
| Аллопуринол (Allopurinol) |
| Альбумин человека (Albumin human) |
| Амброксол (Ambroxol) |
| Амикацин (Amikacin) |
| Аминокапроновая кислота (Aminocaproic acid) |
| Аминокислоты для парентерального питания + Прочие препараты (Жировые эмульсии для парентерального питания + Декстроза + Минералы) (Aminoacids for parenteral nutrition+Other medicines (Fat emulsions + Dextrose + Multimineral)) |
| Аминофиллин (Aminophylline) |
| Амиодарон (Amiodarone) |
| Амлодипин (Amlodipine) |
| Амоксициллин (Amoxicillin) |
| Амфотерицин B (Amphotericin B) |
| Анидулафунгин (Anidulafungin) |
| Антиингибиторный коагулянтный комплекс (Antiingibitorny coagulant complex) |
| Атенолол (Atenolol) |
| Атракурия бесилат (Atracurium besylate) |
| Атропин (Atropine) |
| Ацетилсалициловая кислота (Acetylsalicylic acid) |
| Ацетилцистеин (Acetylcysteine) |
| Ацикловир (Acyclovir) |
| Бупивакаин (Bupivacaine) |
| Бусульфан (Busulfan) |
| Валацикловир (Valacyclovir) |
| Валганцикловир (Valganciclovir) |
| Ванкомицин (Vancomycin) |
| Вода для инъекций (Water for Injection) |
| Вориконазол (Voriconazole) |
| Ганцикловир (Ganciclovir) |
| Гентамицин (Gentamicin) |
| Гепарин натрия (Heparin sodium) |
| Гидроксикарбамид (Hydroxycarbamide) |
| Гидроксиэтилкрахмал (Hydroxyethyl starch) |
| Даназол (Danazol) |
| Дексаметазон (Dexamethasone) |
| Декстроза (Dextrose) |
| Деферазирокс (Deferasiroks) |
| Диазепам (Diazepam) |
| Дифенгидрамин (Diphenhydramine) |
| Добутамин (Dobutamine) |
| Допамин (Dopamine) |
| Дротаверин (Drotaverinum) |
| Имипенем (Imipenem) |
| Иммуноглобулин человеческий нормальный (Human normal immunoglobulin) |
| Интерферон альфа (Interferon alfa) |
| Итраконазол (Itraconazole) |
| Калия хлорид (Potassium chloride) |
| Кальция глюконат (Calcium gluconate) |
| Кальция хлорид (Calcium chloride) |
| Каптоприл (Captopril) |
| Каспофунгин (Caspofungin) |
| Кетопрофен (Ketoprofen) |
| Клавулановая кислота (Clavulanic acid) |
| Клотримазол (Clotrimazole) |
| Колистиметат натрия (Colistimethate sodium) |
| Комплекс аминокислот для парентерального питания (Complex of amino acids for parenteral nutrition) |
| Лактулоза (Lactulose) |
| Левофлоксацин (Levofloxacin) |
| Лидокаин (Lidocaine) |
| Лизиноприл (Lisinopril) |
| Линезолид (Linezolid) |
| Магния сульфат (Magnesium sulfate) |
| Маннитол (Mannitol) |
| Меркаптопурин (Mercaptopurine) |
| Меропенем (Meropenem) |
| Метилпреднизолон (Methylprednisolone) |
| Метилурацил (Диоксометилтетрагидропиримидин) (Methyluracil (Dioxomethyltetrahydropyrimidine)) |
| Метронидазол (Metronidazole) |
| Микафунгин (Micafungin) |
| Моксифлоксацин (Moxifloxacin) |
| Морфин (Morphine) |
| Надропарин кальция (Nadroparin calcium) |
| Натрия ацетат (Sodium acetate) |
| Натрия гидрокарбонат (Sodium hydrocarbonate) |
| Натрия хлорид (Sodium chloride) |
| Нафазолин (Naphazoline) |
| Ницерголин (Nicergoline) |
| Норэпинефрин (Norepinephrine) |
| Омепразол (Omeprazole) |
| Ондансетрон (Ondansetron) |
| Офлоксацин (Ofloxacin) |
| Пипекурония бромид (Pipekuroniyu bromide) |
| Пиперациллин (Piperacillin) |
| Повидон — йод (Povidone — iodine) |
| Преднизолон (Prednisolone) |
| Прокаин (Procaine) |
| Пропофол (Propofol) |
| Ривароксабан (Rivaroxaban) |
| Рокурония бромид (Rocuronium) |
| Руксолитиниб (Ruxolitinib) |
| Сальбутамол (Salbutamol) |
| Смектит диоктаэдрический (Dioctahedral smectite) |
| Спиронолактон (Spironolactone) |
| Сульфадиметоксин (Sulfadimethoxine) |
| Сульфаметоксазол (Sulphamethoxazole) |
| Тигециклин (Tigecycline) |
| Тикарциллин (Ticarcillin) |
| Тиопентал-натрий (Thiopental sodium) |
| Тобрамицин (Tobramycin) |
| Торасемид (Torasemide) |
| Трамадол (Tramadol) |
| Транексамовая кислота (Tranexamic acid) |
| Тримекаин (Trimecaine) |
| Триметоприм (Trimethoprim) |
| Фамотидин (Famotidine) |
| Фамцикловир (Famciclovir) |
| Фенилэфрин (Phenylephrine) |
| Фенобарбитал (Phenobarbital) |
| Фентанил (Fentanyl) |
| Филграстим (Filgrastim) |
| Флуконазол (Fluconazole) |
| Фолиевая кислота (Folic acid) |
| Фуросемид (Furosemide) |
| Хлорамфеникол (Chloramphenicol) |
| Хлоргексидин (Chlorhexidine) |
| Хлоропирамин (Chloropyramine) |
| Цефепим (Cefepime) |
| Цефоперазон (Cefoperazone) |
| Ципрофлоксацин (Ciprofloxacin) |
| Цитарабин (Cytarabine) |
| Эноксапарин натрия (Enoxaparin sodium) |
| Эпинефрин (Epinephrine) |
| Эпоэтин бета (Epoetin Beta) |
| Эритромицин (Erythromycin) |
| Эртапенем (Ertapenem) |
Первичный миелофиброз — Primary myelofibrosis
Первичный миелофиброз (ПМФ) — редкий рак крови костного мозга . Он классифицируется Всемирной организацией здравоохранения (ВОЗ) как тип миелопролиферативного новообразования , группа раковых заболеваний, при которых наблюдается аномальный рост клеток в костном мозге . Это перепроизводство чаще всего связано с соматической мутацией в маркерах генов JAK2 , CALR или MPL . В PMF здоровый костный мозг заменяется рубцовой тканью ( фиброзом ), что приводит к недостатку производства нормальных клеток крови. Симптомы включают анемию, усиление инфекции и увеличение селезенки ( спленомегалию ).
Он классифицируется Всемирной организацией здравоохранения (ВОЗ) как тип миелопролиферативного новообразования , группа раковых заболеваний, при которых наблюдается аномальный рост клеток в костном мозге . Это перепроизводство чаще всего связано с соматической мутацией в маркерах генов JAK2 , CALR или MPL . В PMF здоровый костный мозг заменяется рубцовой тканью ( фиброзом ), что приводит к недостатку производства нормальных клеток крови. Симптомы включают анемию, усиление инфекции и увеличение селезенки ( спленомегалию ).
В 2016 году префибротический первичный миелофиброз был официально классифицирован как отдельное состояние, которое прогрессирует до явного PMF у многих пациентов, причем основным диагностическим отличием является степень фиброза .
Признаки и симптомы
Основным признаком первичного миелофиброза является фиброз костного мозга, но он часто сопровождается:
Причины
Основная причина PMF неизвестна ( идиопатическое заболевание ). Существует связь между мутациями в JAK2 , CALR или MPL гена и миелофиброз. Примерно 90% людей с миелофиброзом имеют одну из этих мутаций, а 10% не имеют ни одной из этих мутаций. Эти мутации не являются специфическими для миелофиброза и связаны с другими миелопролиферативными новообразованиями, в частности истинной полицитемией и эссенциальной тромбоцитемией .
Существует связь между мутациями в JAK2 , CALR или MPL гена и миелофиброз. Примерно 90% людей с миелофиброзом имеют одну из этих мутаций, а 10% не имеют ни одной из этих мутаций. Эти мутации не являются специфическими для миелофиброза и связаны с другими миелопролиферативными новообразованиями, в частности истинной полицитемией и эссенциальной тромбоцитемией .
Мутация V617F в протеине JAK2 обнаруживается примерно у половины людей с первичным миелофиброзом. Мутация V617F представляет собой замену валина на фенилаланин в положении 617. Киназы Януса (JAK) представляют собой нерецепторные тирозинкиназы, необходимые для активации передачи сигналов, опосредованной рецепторами цитокинов, лишенных каталитической активности. К ним относятся рецепторы эритропоэтина , тромбопоэтина , большинства интерлейкинов и интерферона . Мутации JAK2 важны, потому что JAK2 играет роль в контроле производства клеток крови из гемопоэтических стволовых клеток . Мутация V617F, по-видимому, делает гемопоэтические клетки более чувствительными к факторам роста, которые нуждаются в JAK2 для передачи сигнала , включая эритропоэтин и тромбопоэтин .
Мутация V617F, по-видимому, делает гемопоэтические клетки более чувствительными к факторам роста, которые нуждаются в JAK2 для передачи сигнала , включая эритропоэтин и тромбопоэтин .
Ген MPL кодирует белок, который действует как рецептор тромбопоэтина. Мутация в этом гене, известная как мутация W515, приводит к выработке аномального рецепторного белка тромбопоэтина, что приводит к избыточной продукции аномальных мегакариоцитов . Аномальные мегакариоциты стимулируют другие клетки, фибробласты, производить коллаген в костном мозге, секретируя PDGF и TGF-β1 .
Механизм
Миелофиброз — это клональное неопластическое нарушение кроветворения , образования клеточных компонентов крови. Это одно из миелопролиферативных заболеваний , заболеваний костного мозга, при которых на определенном этапе вырабатываются избыточные клетки. Производство цитокинов, таких как фактор роста фибробластов, клоном аномальных гемопоэтических клеток (особенно мегакариоцитами ) приводит к замене кроветворной ткани костного мозга соединительной тканью посредством фиброза коллагена . Уменьшение кроветворной ткани снижает способность пациента генерировать новые клетки крови, что приводит к прогрессирующей панцитопении , нехватке всех типов клеток крови. Однако разрастание фибробластов и отложение коллагена является вторичным явлением, и сами фибробласты не являются частью аномального клеточного клона.
Уменьшение кроветворной ткани снижает способность пациента генерировать новые клетки крови, что приводит к прогрессирующей панцитопении , нехватке всех типов клеток крови. Однако разрастание фибробластов и отложение коллагена является вторичным явлением, и сами фибробласты не являются частью аномального клеточного клона.
При первичном миелофиброзе происходит прогрессирующее рубцевание или фиброз костного мозга по причинам, изложенным выше. Результатом является экстрамедуллярный гемопоэз , то есть образование клеток крови, происходящее не в костном мозге, а в других местах, поскольку гемопоэтические клетки вынуждены мигрировать в другие области, особенно в печень и селезенку . Это вызывает увеличение этих органов. В печени аномальный размер называется гепатомегалией . Увеличение селезенки называется спленомегалией , которая также способствует возникновению панцитопении, особенно тромбоцитопении и анемии . Еще одним осложнением экстрамедуллярного кроветворения является пойкилоцитоз или наличие эритроцитов неправильной формы .
Миелофиброз может быть поздним осложнением других миелопролиферативных заболеваний, таких как истинная полицитемия и, реже, эссенциальной тромбоцитемии . В этих случаях миелофиброз возникает в результате соматической эволюции аномального клона гемопоэтических стволовых клеток, вызвавшего исходное заболевание. В некоторых случаях развитие миелофиброза после этих нарушений может быть ускорено пероральным химиотерапевтическим препаратом гидроксимочевиной .
Сайты кроветворения
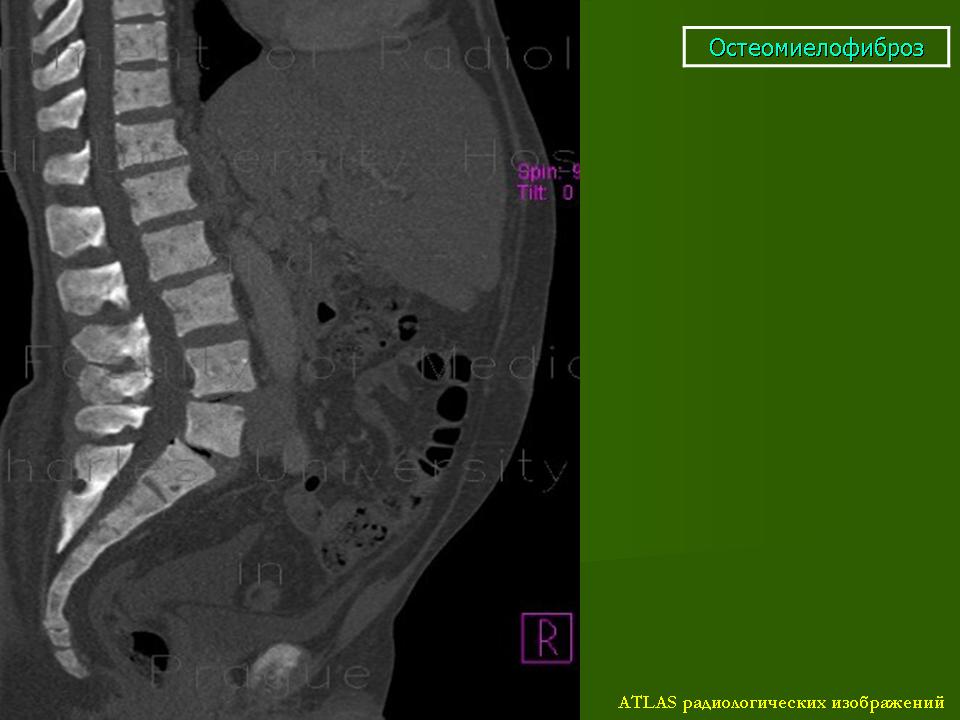
Основным участком экстрамедуллярного кроветворения при миелофиброзе является селезенка , которая обычно значительно увеличена, иногда ее вес достигает 4000 г. В результате массивного увеличения селезенки в селезенке часто возникают множественные субкапсулярные инфаркты , а это означает, что из-за прерывания подачи кислорода в селезенку происходит частичная или полная гибель ткани. На клеточном уровне селезенка содержит предшественников красных кровяных телец, предшественников гранулоцитов и мегакариоцитов , причем мегакариоциты имеют заметное количество и имеют причудливую форму.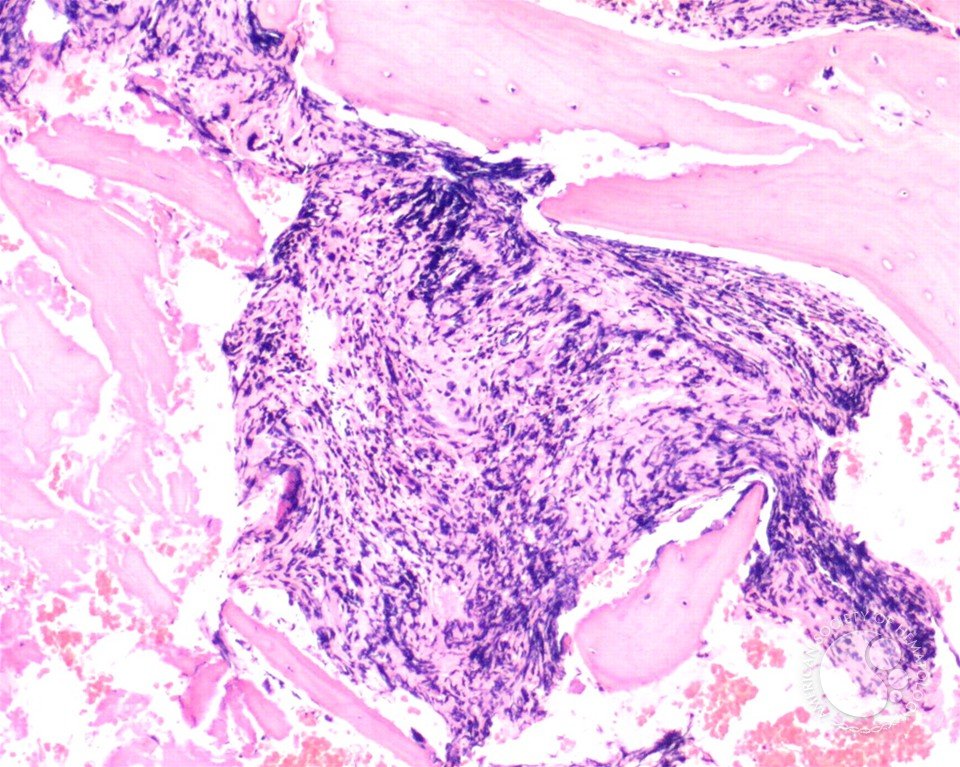 Считается, что мегакариоциты участвуют в возникновении вторичного фиброза, наблюдаемого при этом состоянии, как описано выше в разделе «Механизм». Иногда наблюдается необычная активность эритроцитов , лейкоцитов или тромбоцитов. Печень часто умеренно увеличена, с очагами экстрамедуллярного кроветворения. Микроскопически лимфатические узлы также содержат очаги кроветворения, но их недостаточно, чтобы вызвать увеличение.
Считается, что мегакариоциты участвуют в возникновении вторичного фиброза, наблюдаемого при этом состоянии, как описано выше в разделе «Механизм». Иногда наблюдается необычная активность эритроцитов , лейкоцитов или тромбоцитов. Печень часто умеренно увеличена, с очагами экстрамедуллярного кроветворения. Микроскопически лимфатические узлы также содержат очаги кроветворения, но их недостаточно, чтобы вызвать увеличение.
Есть также сообщения о гемопоэзе в легких . Эти случаи связаны с гипертонией в легочных артериях .
Костный мозг в типичном случае является гиперклеточным и диффузно фиброзным . Как на ранних, так и на поздних стадиях заболевания мегакариоциты часто видны и обычно диспластичны .
Диагностика
Эпидемиологически заболевание обычно развивается медленно и в основном наблюдается у людей старше 50 лет.
Диагноз ставится на основании биопсии костного мозга . Фиброз 2 или 3 степени определяет явную PMF, тогда как степень 0 или 1 определяет префибротический первичный миелофиброз .
Физический осмотр брюшной полости может выявить увеличение селезенки , печени или и того , и другого .
Анализы крови также используются в диагностике. Первичный миелофиброз может начаться с картины крови, аналогичной той, которая наблюдается при истинной полицитемии или хроническом миелолейкозе. Большинство людей с миелофиброзом страдают анемией от умеренной до тяжелой. Со временем развивается тромбоцитопения , уменьшение количества тромбоцитов в крови . При просмотре под микроскопом мазок крови будет выглядеть явно ненормальным с проявлением панцитопении , то есть уменьшения количества всех типов клеток крови: эритроцитов , лейкоцитов и тромбоцитов. Эритроциты могут проявлять аномалии, включая причудливые формы , такие как клетки каплевидной формы , а в мазке крови могут появиться ядросодержащие предшественники красных кровяных телец (лейкоэритробластическая реакция). Обычно зрелые эритроциты у взрослых не имеют клеточного ядра, а наличие ядерных эритроцитов предполагает, что незрелые клетки попадают в кровоток в ответ на очень высокую потребность костного мозга в производстве новых эритроцитов. . Незрелые лейкоциты и тромбоциты (большие мегакариоциты ) также обнаруживаются в образцах крови, и количество базофилов увеличивается. На поздних стадиях прогрессирования заболевания делается попытка взять образец костного мозга путем аспирации , это может привести к сухому отбору. Это означает, что там, где игла обычно может отсосать образец полужидкого костного мозга, она не дает образца, потому что костный мозг заменен коллагеновыми волокнами. Биопсия костного мозга покажет фиброз коллагена , заменяющий костный мозг, который обычно занимает это пространство.
. Незрелые лейкоциты и тромбоциты (большие мегакариоциты ) также обнаруживаются в образцах крови, и количество базофилов увеличивается. На поздних стадиях прогрессирования заболевания делается попытка взять образец костного мозга путем аспирации , это может привести к сухому отбору. Это означает, что там, где игла обычно может отсосать образец полужидкого костного мозга, она не дает образца, потому что костный мозг заменен коллагеновыми волокнами. Биопсия костного мозга покажет фиброз коллагена , заменяющий костный мозг, который обычно занимает это пространство.
Уход
Одним из известных лечебных методов лечения является трансплантация аллогенных стволовых клеток , но этот подход сопряжен со значительными рисками. Другие варианты лечения в значительной степени поддерживают и не изменяют течение заболевания (за возможным исключением руксолитиниба , как обсуждается ниже). Эти варианты могут включать регулярные переливания фолиевой кислоты , аллопуринола или крови . Дексаметазон , альфа- интерферон и гидроксимочевина (также известная как гидроксикарбамид) могут играть роль.
Дексаметазон , альфа- интерферон и гидроксимочевина (также известная как гидроксикарбамид) могут играть роль.
При его лечении можно использовать леналидомид и талидомид , хотя периферическая невропатия является частым неприятным побочным эффектом.
Также могут потребоваться частые переливания крови . Если пациент страдает диабетом и принимает сульфонилмочевину , прием следует периодически прекращать, чтобы исключить лекарственную тромбоцитопению .
Спленэктомия иногда рассматривается как вариант лечения пациентов с миелофиброзом, у которых массивная спленомегалия способствует анемии из-за гиперспленизма , особенно если они сильно нуждаются в переливании крови . Однако спленэктомия при массивной спленомегалии представляет собой процедуру с высоким риском, при этом в некоторых исследованиях риск летального исхода достигает 3%.
В ноябре 2011 года FDA одобрило руксолитиниб (Jakafi) в качестве средства для лечения миелофиброза среднего или высокого риска. Руксолитиниб служит ингибитором JAK 1 и 2. Медицинский журнал Новой Англии (NEJM) опубликовал результаты двух исследований фазы III руксолитиниба. Эти данные показали, что лечение значительно уменьшило объем селезенки, улучшило симптомы миелофиброза и было связано со значительным улучшением общей выживаемости по сравнению с плацебо. Однако недавно было поставлено под сомнение положительное влияние руксолитиниба на выживаемость.
Руксолитиниб служит ингибитором JAK 1 и 2. Медицинский журнал Новой Англии (NEJM) опубликовал результаты двух исследований фазы III руксолитиниба. Эти данные показали, что лечение значительно уменьшило объем селезенки, улучшило симптомы миелофиброза и было связано со значительным улучшением общей выживаемости по сравнению с плацебо. Однако недавно было поставлено под сомнение положительное влияние руксолитиниба на выживаемость.
В августе 2019 года FDA одобрило федратиниб для лечения взрослых с первичным или вторичным (пост-полицитемия истинная или пост-эссенциальная тромбоцитемия) миелофиброзом (МФ) со средним уровнем 2 или высоким риском.
История
Миелофиброз был впервые описан в 1879 году Густавом Хёком . Эпонимами болезни являются болезнь Хека-Ассмана или болезнь Ассманна по Герберту Ассманну , опубликовавшему описание под термином «остеосклероз» в 1907 году.
В 1951 году Уильям Дамешек охарактеризовал это как миелопролиферативное состояние .
Заболевание также было известно как миелофиброз с миелоидной метаплазией и агногенной миелоидной метаплазией . Всемирная организация здравоохранения использовала название « хронический идиопатический миелофиброз» до 2008 года, когда она приняла название первичный миелофиброз .
В 2016 году ВОЗ пересмотрела свою классификацию миелопролиферативных новообразований, чтобы определить префибротический первичный миелофиброз как отдельную клиническую единицу от явной PMF.
Рекомендации
внешняя ссылка
КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРИ МИЕЛОФИБРОЗЕ | Грибкова
Введение
Миелофиброз (МФ) — это Ph-негативное миелопролиферативное новообразование, которое патогенетически характеризуется дерегуляцией Янус киназы (Janus kinase — JAK), активаторов транскрипции JAK/STAT сигнального пути, а также гиперэкспрессией цитокинов. Руксолитиниб — ингибитор JAK киназ является препаратом для патогенетического лечения этого заболевания, одобренным на основе двух рандомизированных исследований III фазы, которые продемонстрировали клинически значимое уменьшение размеров селезенки, выраженности симптомов, улучшение качества жизни и повышение выживаемости при длительном применении [1, 2].
Однако руксолитиниб может вызывать серьезные побочные явления. Наиболее частыми негематологическими побочными эффектами (преимущественно 1-й и 2-й степени), ассоциированными с приемом руксолитиниба, являлись экхимозы, головокружение и головная боль [3]. Сообщалось, что у больных, получающих руксолитиниб, возникали инфекционные осложнения [3]. Наиболее распространенными гематологическими побочными эффектами лечения руксолитинибом были анемия и тромбоцитопения [3]. Существуют данные, что 60 % больных прекратили терапию руксолитинибом в течение 3 лет после начала лечения из-за развившихся побочных эффектов или потери ответа на лечение [4, 5]. К тому же терапия руксолитинибом не является радикальным методом лечения заболевания, поскольку она не приводит к существенному уменьшению частоты аллелей JAK2 V617F или фиброза костного мозга [6]. Таким образом, сохраняется потребность в поиске новых, эффективных стратегий лечения МФ.
В последнее время достигнуты определенные успехи в понимании молекулярного патогенеза МФ. Установлено, что, хотя активация сигнального пути JAK-STAT занимает центральное место в патогенезе заболевания, альтернативные пути также могут играть роль в развитии МФ [7, 8]. Это позволило определить дополнительные мишени для исследования. Они включают гистондеацетилазу, гипометилирующие агенты, секреторные белки семейства Hedgehog, теломеразу, пути PI3K/AKT, профиброгенные и воспалительные цитокины, а также иммунную дерегуляцию [9]. Новые препараты, влияющие на эти пути, и их рациональные комбинации с ингибиторами JAK, могут значительно улучшить результаты лечения МФ.
Установлено, что, хотя активация сигнального пути JAK-STAT занимает центральное место в патогенезе заболевания, альтернативные пути также могут играть роль в развитии МФ [7, 8]. Это позволило определить дополнительные мишени для исследования. Они включают гистондеацетилазу, гипометилирующие агенты, секреторные белки семейства Hedgehog, теломеразу, пути PI3K/AKT, профиброгенные и воспалительные цитокины, а также иммунную дерегуляцию [9]. Новые препараты, влияющие на эти пути, и их рациональные комбинации с ингибиторами JAK, могут значительно улучшить результаты лечения МФ.
В настоящее время несколько новых ингибиторов JAK (пакритиниб, момелотиниб, NS018 и LY27A45445) проходят клинические испытания [9]. Однако, учитывая отсутствие окончательного решения в отношении данных препаратов и в то же время установленную безопасность и эффективность руксолитиниба при известных его ограничениях, поиск оптимальных лекарств для комбинаций с руксолитинибом представляется наиболее логичным подходом к разработке терапии МФ [6].
Цель настоящего обзора — анализ результатов современных исследований комбинированной терапии больных МФ.
Ингибиторы гистондеацетилазы
Гистондеацетилазы (HDAC) — ферменты, модифицирующие гистоны и изменяющие конформацию хроматина, играют важную роль в регуляции экспрессии генов. В качестве монотерапии МФ в исследованиях фазы I/II были изучены различные ингибиторы HDAC [9]. Примером таких ингибиторов является панобиностат, который ингибирует сигнализацию JAK, нарушая взаимодействие между JAK2 и белком теплового шока 90. Монотерапия панобиностатом приводила к уменьшению выраженности спленомегалии и фиброзных изменений стромы костного мозга в исследованиях I/II фазы у больных МФ [10]. Комбинацию руксолитиниба и панобиностата исследовали в моделях на мышах, где она продемонстрировала синергетический эффект и уменьшение выраженности фиброза стромы костного мозга [11]. Это послужило основанием для начала клинических исследований эфф ективности комбинированной терапии ингибитором JAK с ингибитором HDAC.
Предварительные результаты одного из этих исследований [12] были обнадеживающими. Все больные получали комбинированную терапию: руксолитиниб в дозе 5—15 мг 2 раза в день и панобиностат в дозе 10—25 мг 3 раза в неделю (на 2, 4 и 6-й дни). Было показано уменьшение размеров селезенки ≥35 % у 57 и 39 % больных (из общего числа больных n = 23) на 24 и 48 неделях соответственно, что является более благоприятным по сравнению с 42 и 32 % больных, получавших монотерапию руксолитинибом и достигших той же конечной точки в исследованиях COMFORT-I и COMFORT-II [1, 2]. В исследовании [12] также упоминается об уменьшении фиброзных изменений костного мозга у 4 (33 %) из 12 больных и о ≥20 % снижении аллельной нагрузки JAK2V617F у 29 % на 48 неделе лечения. Аллельная нагрузка JAK2V617F оценивалась с помощью количественной полимеразной цепной реакции в реальном времени, проводимой на ДНК, очищенной из грануло- цитов периферической крови, в соответствии с рекомендациями, изложенными в работе [13]. Эти результаты превосходят результаты, полученные при монотерапии руксолитинибом. Наиболее распространенными нежелательными явлениями (НЯ) были анемия, тромбоцитопения и диарея. Спектр зарегистрированных НЯ и частота НЯ соответствовали тем, которые наблюдались при монотерапии руксолитинибом и панобиностатом. Для окончательных выводов необходимы более длительное наблюдение и большее число больных, исследование продолжается.
Эти результаты превосходят результаты, полученные при монотерапии руксолитинибом. Наиболее распространенными нежелательными явлениями (НЯ) были анемия, тромбоцитопения и диарея. Спектр зарегистрированных НЯ и частота НЯ соответствовали тем, которые наблюдались при монотерапии руксолитинибом и панобиностатом. Для окончательных выводов необходимы более длительное наблюдение и большее число больных, исследование продолжается.
Гипометилирующие агенты
Регулирование транскрипции в соматических клетках во многом зависит от уровня метилирования ДНК [9], аномалии метилирования часты при МФ [6]. В клиническом исследовании ингибитора метилтрансфера- зы ДНК азацитидина, применявшегося в дозе 75 мг/м2 в день в течение 7 дней, был получен ответ у 24 % больных (частичный ответ — у 3 % и клиническое улучшение — у 21 %) в когорте из 34 больных МФ (76 % из них лечились ранее). Ответы наблюдались как у больных с мутацией JAK2V617F, так и без нее [9]. Различный механизм действия свидетельствует о возможности улучшения результатов лечения при проведении комбинированной терапии руксолитинибом с азацитиди- ном в низких дозах у больных МФ [9].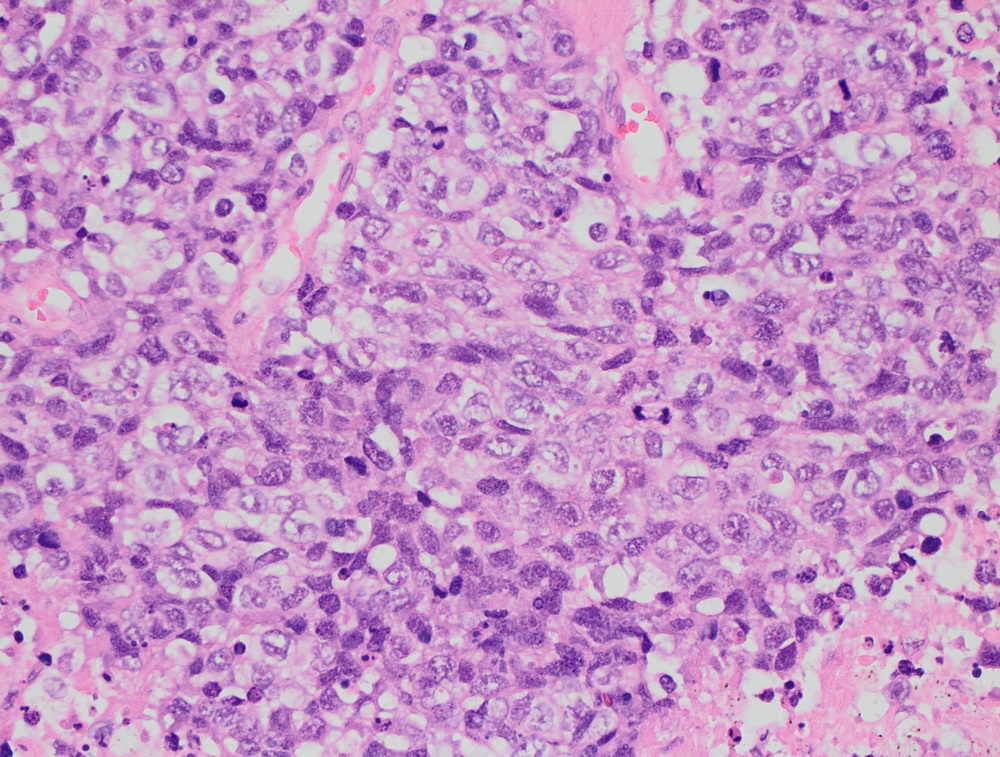
В исследовании, в котором изучали эффективность комбинации руксолитиниба с азацитидином [14], больные получали монотерапию руксолитинибом (15 мг перорально дважды в день (если количество тромбоцитов было (100—200)х109/л) или 20 мг дважды в день (если количество тромбоцитов было >200х109/л)) циклами по 28 дней в течение первых 3 месяцев, затем следовало добавление азацитидина (25 мг/м2 в дни 1—5 каждого 28-дневного цикла), начиная с 4-го цикла. Доза азацитидина могла быть постепенно увеличена до 75 мг/м2. В исследовании отмечено уменьшение размера селезенки более чем на 50 % у 48 % больных на 24-й неделе терапии, при этом в целом более 50 % уменьшения размеров селезенки в любой момент исследования достигли 79 % больных. Частота ответа на лечение по Международной шкале оценки прогноза IWG-MRT 2013 [15] составила 69 % из 41 больного. Аллельная нагрузка JAK2V617F уменьшилась у 87 % больных, имевших мутацию JAK2, а показатель фиброза в костном мозге по EUMNET [16] уменьшился у 41 % больных. Это выгодно отличается от результатов монотерапии руксолитинибом, но требуются большее количество больных и более длительные сроки наблюдения для подтверждения результатов.
Это выгодно отличается от результатов монотерапии руксолитинибом, но требуются большее количество больных и более длительные сроки наблюдения для подтверждения результатов.
Возможно, этот режим может быть полезен у больных с большим количеством бластных клеток в костном мозге. Из НЯ были выявлены анемия и тромбоцитопе- ния 3-4-й степени, но они компенсировались модификацией дозы, только один больной прекратил лечение из-за длительной цитопении.
Применение комбинированной терапии децитабина с руксолитинибом у больных, находившихся в фазе акселерации и бластной фазе МФ, было оценено в двух исследованиях. В исследовании фазы IB R. Rampal и соавт. [17] ответ на лечение был получен только у 7 (33 %) из 21 больных. Доза руксолитини- ба в этом исследовании была увеличена с 15 мг два раза в день до 50 мг два раза в день, без установления максимально переносимой дозы. В другом исследовании фазы IB [18] P. Bose и соавт. отметили ответ у 5 из 12 (42 %) больных, получавших децитабин и руксолитиниб.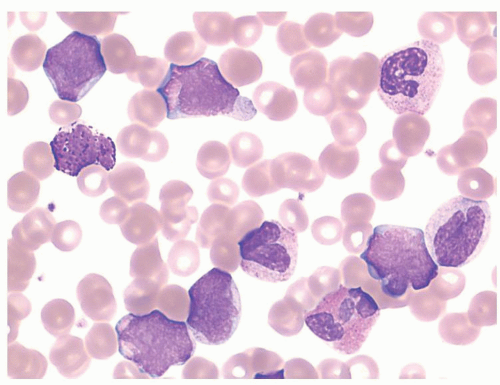 Больные получали руксолитиниб, дозу которого увеличивали от 10 до 50 мг дважды в день, а рекомендуемая доза руксолитиниба фазы II была 50 мг два раза в день в сочетании со стандартной дозой децитабина.
Больные получали руксолитиниб, дозу которого увеличивали от 10 до 50 мг дважды в день, а рекомендуемая доза руксолитиниба фазы II была 50 мг два раза в день в сочетании со стандартной дозой децитабина.
Ингибиторы секреторных белков семейства Hedgehog
Секреторные белки семейства Hedgehog играют важную роль в регуляции клеточной дифференцировки в процессе эмбриогенеза, а также стимулируют пролиферацию опухолевых клеток [19]. Таким образом, сигнальный путь Hedgehog может быть потенциальной мишенью терапии МФ.
В экспериментальной модели МФ, воспроизведенной на мышах, руксолитиниб в комбинации с ингибитором пути Hedgehog сонидегибом уменьшал выраженность спленомегалии и фиброза костного мозга по сравнению с монотерапией руксолитинибом [9]. Эти данные послужили основанием для проведения клинических испытаний IB-II фазы по изучению эффективности терапии ингибиторами пути Hedgehog в сочетании с руксолитинибом. В этом исследовании определены оптимальные дозы препаратов: сониде- гиб 400 мг 1 раз в сутки и руксолитиниб 20 мг 2 раза в сутки. Исследование сочетания сонидегиба и руксо- литиниба у 27 больных МФ промежуточного/высокого риска по Международной шкале оценки прогноза [15] показало, что у 44,4 % больных было достигнуто уменьшение объема селезенки на 35 % и более в течение 24 недель и у 55,6 % уменьшение объема селезенки этой степени достигалось в любой момент исследования [20]. Комбинированная терапия хорошо переносилась. НЯ, требующие коррекции дозы или прерывания лечения, были отмечены у 17 (63 %) больных, причем наиболее распространенными были увеличение сывороточной концентрации креатинкиназы (19 %, п = 5) и миалгии (19 % , п = 5). Среднее изменение в аллельной нагрузке JAK2 V617F составляло -9,0 % (диапазон от -56,5 до 7,0 %) от исходного уровня к концу 24-й недели лечения. При оценке выраженности фиброза костного мозга у 2 больных отмечено уменьшение (с 3-й до 2-й степени), у 8 больных выраженность фиброза не изменилась, у 3 больных к концу 24-й недели лечения выраженность фиброза стала больше. Исследование продолжается для долгосрочного наблюдения за зарегистрированными больными.
Исследование сочетания сонидегиба и руксо- литиниба у 27 больных МФ промежуточного/высокого риска по Международной шкале оценки прогноза [15] показало, что у 44,4 % больных было достигнуто уменьшение объема селезенки на 35 % и более в течение 24 недель и у 55,6 % уменьшение объема селезенки этой степени достигалось в любой момент исследования [20]. Комбинированная терапия хорошо переносилась. НЯ, требующие коррекции дозы или прерывания лечения, были отмечены у 17 (63 %) больных, причем наиболее распространенными были увеличение сывороточной концентрации креатинкиназы (19 %, п = 5) и миалгии (19 % , п = 5). Среднее изменение в аллельной нагрузке JAK2 V617F составляло -9,0 % (диапазон от -56,5 до 7,0 %) от исходного уровня к концу 24-й недели лечения. При оценке выраженности фиброза костного мозга у 2 больных отмечено уменьшение (с 3-й до 2-й степени), у 8 больных выраженность фиброза не изменилась, у 3 больных к концу 24-й недели лечения выраженность фиброза стала больше. Исследование продолжается для долгосрочного наблюдения за зарегистрированными больными.
Ингибиторы сигнального пути PI3K / AKT / mTOR
PI3K/AKT/mTOR — внутриклеточный сигнальный путь, центральными компонентами которого являются фосфатидилинозитол-3-киназа (PI3K), киназа AKT и мишень рапамицина млекопитающих (mTOR). Это — один из универсальных сигнальных путей, характерных для большинства клеток человека. Он отвечает за «уход» от апоптоза, рост, пролиферацию клеток, метаболизм [19].
Предварительные данные свидетельствуют о том, что ингибирование пути PI3K/AKT/mTOR оказывает положительные эффекты при МФ [6, 9]. Эти данные побудили начать клиническое исследование фазы IB в отношении комбинации руксолитиниба и бупарлисиба — блокатора пути PI3K. Целями этого исследования [21] были подбор максимальных переносимых доз руксолитиниба и бупарлисиба, оценка эффективности и переносимости терапии больными МФ промежуточного или высокого риска. Исследование проведено на репрезентативных выборках: 22 больных МФ не получали терапию ингибиторами JAK2 (группа А) и 20 больных получали руксолитиниб ранее (группа В). Доза руксолитиниба была 15 мг внутрь 2 раза в сутки, бупарлисиба — 60 мг внутрь ежедневно. В целом у 82 % (18 из 22) и 55 % (11 из 20) больных в группах А и В соответственно достигнуто уменьшение размеров селезенки на 50 % и более от исходных в любой момент исследования, в том числе у 15 (36 %) больных размеры селезенки нормализовались (12 — в группе А и 3 — в группе В). На 24-й неделе терапии у 55 и 20 % больных в группах А и В соответственно отмечено >50 % уменьшение размеров селезенки, определяемых пальпаторно. К 24-й неделе терапии медиана снижения аллельной нагрузки JAK2V617F по сравнению с исходными значениями составила 3,35 (диапазон 26,9-2,7) в группе А и 0,60 (диапазон 12,6-24,7) в группе В. Переносимость комбинированной терапии была удовлетворительная. Гематологическая токсичность 3-4-й степени представлена анемией (2 из 22 больных в группе А и 7 из 20 в группе В) и тромбоцитопенией (5 из 22 и 6 из 20 больных в группах А и В соответственно). После 24 недель лечения у 4 больных (п = 3 в группе A; 1 в группе B) установлено уменьшение степени выраженности фиброза костного мозга, у 19 больных (п = 9 в группе A; 10 в группе B) имела место стабилизация, у 2 больных в группе A (0 в группе B) отмечено ухудшение.
Доза руксолитиниба была 15 мг внутрь 2 раза в сутки, бупарлисиба — 60 мг внутрь ежедневно. В целом у 82 % (18 из 22) и 55 % (11 из 20) больных в группах А и В соответственно достигнуто уменьшение размеров селезенки на 50 % и более от исходных в любой момент исследования, в том числе у 15 (36 %) больных размеры селезенки нормализовались (12 — в группе А и 3 — в группе В). На 24-й неделе терапии у 55 и 20 % больных в группах А и В соответственно отмечено >50 % уменьшение размеров селезенки, определяемых пальпаторно. К 24-й неделе терапии медиана снижения аллельной нагрузки JAK2V617F по сравнению с исходными значениями составила 3,35 (диапазон 26,9-2,7) в группе А и 0,60 (диапазон 12,6-24,7) в группе В. Переносимость комбинированной терапии была удовлетворительная. Гематологическая токсичность 3-4-й степени представлена анемией (2 из 22 больных в группе А и 7 из 20 в группе В) и тромбоцитопенией (5 из 22 и 6 из 20 больных в группах А и В соответственно). После 24 недель лечения у 4 больных (п = 3 в группе A; 1 в группе B) установлено уменьшение степени выраженности фиброза костного мозга, у 19 больных (п = 9 в группе A; 10 в группе B) имела место стабилизация, у 2 больных в группе A (0 в группе B) отмечено ухудшение.
В другом исследовании [22] оценивали эффективность комбинации руксолитиниба с ингибитором пути PI3K — TGR-1202. Комбинация препаратов хорошо переносилась больными. Распространенными НЯ были анемия 2 степени и повышение сывороточных концентраций амилазы и липазы. Не было обнаружено гепатотоксичности, колита или тромбоцитопении ≥3 степени. У одного из 9 больных, участвовавших в исследовании, была достигнута полная ремиссия, у 7 — стабилизация заболевания. У 7 из 9 больных улучшились гематологические показатели, у 8 — уменьшились симптомы МФ. Данные результаты свидетельствуют о целесообразности дальнейшего исследования этой комбинации.
Антифибротические препараты
Антифибротические препараты, будучи введенными в организм, выполняют функцию эндогенных белков, которые активируются при повреждении местных тканей и стимулируют дифференцировку макрофагов и моноцитов с последующим обратным развитием фиброза [19]. В клинических исследованиях II фазы применение антифибротического препарата пентраксина-2 (PRM-151) привело к регрессии фиброза костного мозга, нормализации показателей гемограммы, уменьшению симптомов интоксикации и размеров селезенки.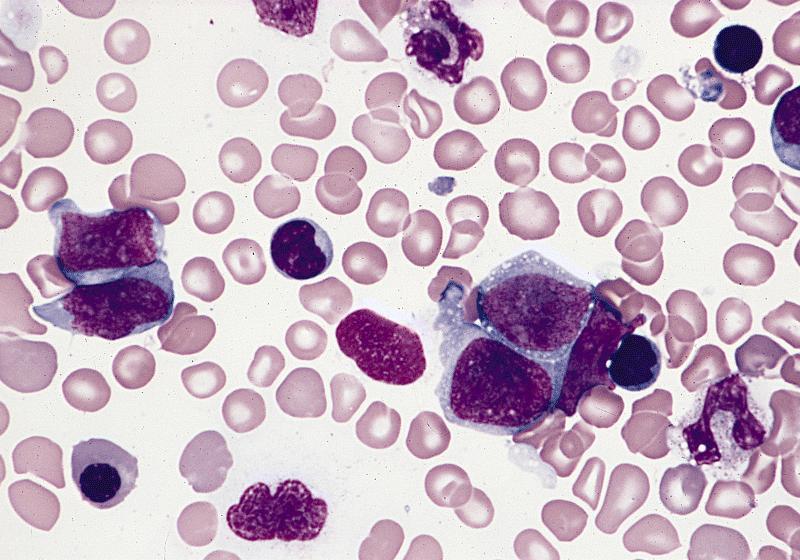 Исследование включало 27 больных МФ, получавших либо монотерапию PRM-151, либо PRM-151 в сочетании с руксолитинибом [23, 24]. Комбинация препаратов хорошо переносилась. У 13 больных, которые получали терапию не менее 72 недель, наиболее часто встречающимися побочными эффектами были: усталость (4), тошнота (3), лихорадка (3), кашель (2), диарея (2), зубная инфекция (2), головная боль (2), верхняя респираторная инфекция (2), гипергликемия (2) и гиперурикемия (2). Более чем у 70 % больных отмечено уменьшение показателя фиброза в костном мозге как минимум на 1 степень в любой момент исследования. Более чем у 60 % больных наблюдалось уменьшение выраженности спле- номегалии, а также симптомов, обусловленных заболеванием: увеличения количества тромбоцитов и концентрации гемоглобина крови, уменьшение потребности в гемотрансфузиях. Оптимальные ответы наблюдались у больных, у которых было завершено лечение в течение 72 недель, независимо от того, получали ли они монотерапию PRM-151 или в сочетании с руксолитинибом.
Исследование включало 27 больных МФ, получавших либо монотерапию PRM-151, либо PRM-151 в сочетании с руксолитинибом [23, 24]. Комбинация препаратов хорошо переносилась. У 13 больных, которые получали терапию не менее 72 недель, наиболее часто встречающимися побочными эффектами были: усталость (4), тошнота (3), лихорадка (3), кашель (2), диарея (2), зубная инфекция (2), головная боль (2), верхняя респираторная инфекция (2), гипергликемия (2) и гиперурикемия (2). Более чем у 70 % больных отмечено уменьшение показателя фиброза в костном мозге как минимум на 1 степень в любой момент исследования. Более чем у 60 % больных наблюдалось уменьшение выраженности спле- номегалии, а также симптомов, обусловленных заболеванием: увеличения количества тромбоцитов и концентрации гемоглобина крови, уменьшение потребности в гемотрансфузиях. Оптимальные ответы наблюдались у больных, у которых было завершено лечение в течение 72 недель, независимо от того, получали ли они монотерапию PRM-151 или в сочетании с руксолитинибом.
Интерферон-α
Одним из направлений терапии МФ является лечение интерфероном-α, который успешно используется на протяжении десятилетий. В основе лечебного действия интерферона-α лежит опосредованное цитокинами подавление патологической миелопролифе- рации, приводящее к уменьшению количества тромбоцитов, лейкоцитов и эритроцитов крови. Он является также ингибитором продуцируемого тромбоцитами ростового фактора (Platelet-Derived Growth Factor b — PDGF-b), поэтому можно ожидать, что он будет оказывать сдерживающее действие на развитие МФ. Доказана способность интерферона-α подавлять рост эритроидных предшественников in vivo и in vitro, а также вызывать морфологические изменения в мегакариоцитах. Использование препаратов интерферона рекомендуется для лечения молодых больных, находящихся на ранней стадии заболевания, однако 10-20 % больных вынуждены отказаться от приема препарата из-за его непереносимости или недостаточной эффективности [25]. Руксолитиниб показал высокую эффективность с точки зрения улучшения качества жизни больных МФ за счет уменьшения размеров селезенки и выраженности симптомов, опосредованных медиаторами воспаления. Поскольку сопутствующее воспаление может ослаблять эффективность терапии интерфероном-α, комбинация с руксолитинибом, сильным противовоспалительным агентом, может быть более эффективной, чем монотерапия интерфероном-α.
Поскольку сопутствующее воспаление может ослаблять эффективность терапии интерфероном-α, комбинация с руксолитинибом, сильным противовоспалительным агентом, может быть более эффективной, чем монотерапия интерфероном-α.
Результаты клинических исследований данной комбинированной терапии представлены в нескольких работах [26, 27]. Большинство включенных в исследование больных были резистентны к монотерапии интерфероном-α. На момент включения в исследование проводилась терапия интерфероном-α 45 мкг 1 раз в неделю (Pegasys®, Genentech (Roche), США) или 35 мкг 1 раз в неделю (PegIntron®, Merck Sharp & Dohme, Hertfordshire, Великобритания) + руксолитиниб по 20 мг 2 раза в сутки, доза руксолитиниба зависела от количества тромбоцитов крови. Дозы и схемы дозирования в дальнейшем были изменены в зависимости от токсичности или эффективности. У подавляющего числа больных отмечалось исчезновение симптомов опухолевой интоксикации, уменьшились размеры селезенки, определяемые пальпаторно, был достигнут контроль гематокрита без кровопусканий.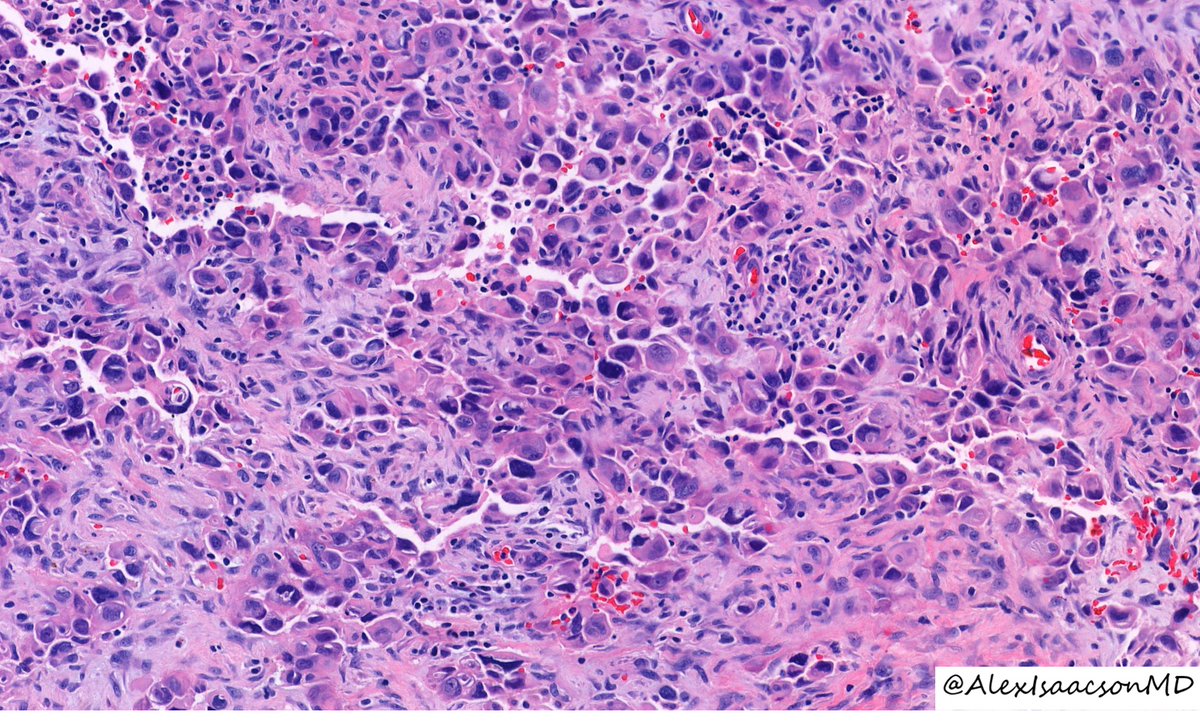 У больных МФ полная или частичная ремиссия была достигнута в 39 % случаев, а полный клинико-гематологический ответ — в 58 %. Медиана аллельной нагрузки JAK2V617F значительно уменьшилась. Наиболее распространенными были гематологические НЯ, которые корректировались снижением дозы. Авторы делают заключение о том, что комбинированная терапия интерфероном-α и руксолитинибом была эффективна и удовлетворительно переносилась больными МФ, относящимися к группам низкого/промежуточного риска, в частности при неэффективности или непереносимости монотерапии интерфероном-α.
У больных МФ полная или частичная ремиссия была достигнута в 39 % случаев, а полный клинико-гематологический ответ — в 58 %. Медиана аллельной нагрузки JAK2V617F значительно уменьшилась. Наиболее распространенными были гематологические НЯ, которые корректировались снижением дозы. Авторы делают заключение о том, что комбинированная терапия интерфероном-α и руксолитинибом была эффективна и удовлетворительно переносилась больными МФ, относящимися к группам низкого/промежуточного риска, в частности при неэффективности или непереносимости монотерапии интерфероном-α.
Однако роль руксолитиниба в популяции больных МФ из группы низкого риска еще не определена. Кроме того, существует опасность, что сочетание интерферона-α и руксолитиниба может приводить кувеличению частоты миелосупрессии и что интерферон-α может привести к ухудшению симптомов, связанных с заболеванием, и тем самым компенсировать положительный эффект руксолитиниба [28].
Уменьшение выраженности анемии
Больные МФ часто страдают от конституциональных симптомов, т.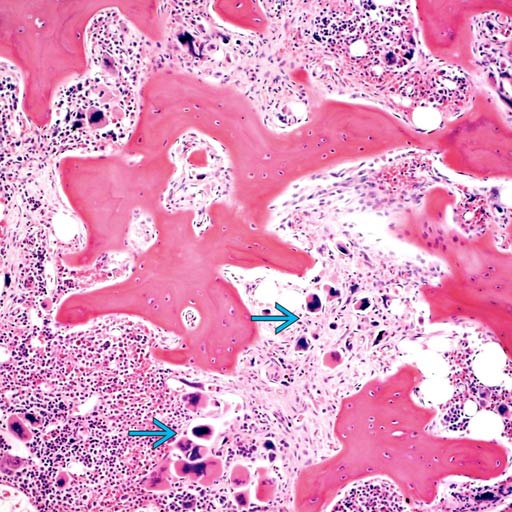 е. относящихся ко всему организму в целом, спленомегалии и цитопении. Лекарственная терапия ингибиторами JAK приводит к уменьшению выраженности спленомегалии, а также конституциональных симптомов, однако цитопения при этом остается серьезной проблемой. Для решения проблемы неэффективного эритропоэза у больных МФ и анемией необходимы альтернативные препараты [9].
е. относящихся ко всему организму в целом, спленомегалии и цитопении. Лекарственная терапия ингибиторами JAK приводит к уменьшению выраженности спленомегалии, а также конституциональных симптомов, однако цитопения при этом остается серьезной проблемой. Для решения проблемы неэффективного эритропоэза у больных МФ и анемией необходимы альтернативные препараты [9].
Анемия у больных МФ является полиэтиологич- ной, она — один из самых распространенных симптомов заболевания. Анемия является главным поводом для начала терапии [25]. Различные препараты (например, даназол, иммуномодулирующие препараты) были эффективны для лечения анемии в виде монотерапии. В связи с этим были начаты клинические исследования комбинированной терапии руксолитини- ба с этими препаратами.
В исследовании [29] оценивали эффективность и переносимость комбинированной терапии руксолитини- бом и даназолом. Исследование включало 14 больных МФ из группы среднего или высокого риска. Больные получали руксолитиниб по 10 мг 2 раза в день (при количестве тромбоцитов >75х109/л) или по 5 мг 2 раза в день (при количестве тромбоцитов <75х109/л). Руксолитиниб назначали в комбинации с даназолом: 200 мг внутрь 3 раза в день. Были получены следующие ответы по критериям IWG-MRT: стабилизация заболевания у 9 больных (64,2 %), клиническое улучшение у 3 (21,4 %) (уменьшение размеров селезенки), частичный ответ у 1 (7,1 %) и прогрессирование заболевания у 1 (7,1 %) больного. Несмотря на ограниченный ответ по IWG-MRT, была установлена стабилизация выраженности анемии и тромбоцитопении. Больные, ранее не принимавшие ингибиторы JAK (80 %), имели стабильную или увеличивающуюся концентрацию гемоглобина. У 5 (55,5 %) из 9 больных, получавших ранее терапию ингибиторами JAK, наблюдалась стабильная или повышенная концентрация гемоглобина, а у 8 больных (88,9 %) — стабильная или повышенная концентрация тромбоцитов. НЯ, связанные с гематологической токсичностью 3 степени или выше, наблюдались у 10 (71,4 %) больных, с негематологической токсичностью — у 2 (14,3 %) больных. Хотя комбинированная терапия не приводила к увеличению гематологического ответа по критериям IWG-MRT, наблюдалась гематологическая стабилизация.
Руксолитиниб назначали в комбинации с даназолом: 200 мг внутрь 3 раза в день. Были получены следующие ответы по критериям IWG-MRT: стабилизация заболевания у 9 больных (64,2 %), клиническое улучшение у 3 (21,4 %) (уменьшение размеров селезенки), частичный ответ у 1 (7,1 %) и прогрессирование заболевания у 1 (7,1 %) больного. Несмотря на ограниченный ответ по IWG-MRT, была установлена стабилизация выраженности анемии и тромбоцитопении. Больные, ранее не принимавшие ингибиторы JAK (80 %), имели стабильную или увеличивающуюся концентрацию гемоглобина. У 5 (55,5 %) из 9 больных, получавших ранее терапию ингибиторами JAK, наблюдалась стабильная или повышенная концентрация гемоглобина, а у 8 больных (88,9 %) — стабильная или повышенная концентрация тромбоцитов. НЯ, связанные с гематологической токсичностью 3 степени или выше, наблюдались у 10 (71,4 %) больных, с негематологической токсичностью — у 2 (14,3 %) больных. Хотя комбинированная терапия не приводила к увеличению гематологического ответа по критериям IWG-MRT, наблюдалась гематологическая стабилизация.
В другом исследовании [30] оценивали эффективность и безопасность комбинированной терапии руксо- литинибом и помалидомидом. В работе представлены данные о 24 больных МФ промежуточного-1, промежуточного-2 и высокого риска по прогностической шкале DIPSS. У всех больных была анемия, у 6 из них отмечалась гемотрансфузионная зависимость. Помалидомид назначали в фиксированной дозировке 0,5 мг 1 раз в день, дозу руксолитиниба, начиная с 10 мг дважды в день, изменяли с целью оптимизации эффективности и уменьшения токсичности. Средняя продолжительность лечения составляла 8 циклов по 28 дней (диапазон 1—13 циклов). За время проведения клинического исследования зарегистрировано 287 НЯ любой степени тяжести. Самые частые: увеличение степени анемии, боль в мышцах, появление конституциональных симптомов. За короткий срок наблюдения у 3 больных достигнуто клиническое улучшение в соответствии с критериями IWG-MRT: 1) уменьшение селезенки; 2) уменьшение объема селезенки + увеличение концентрации гемоглобина; 3) увеличение концентрации гемоглобина, гранулоцитов, эритроцитов.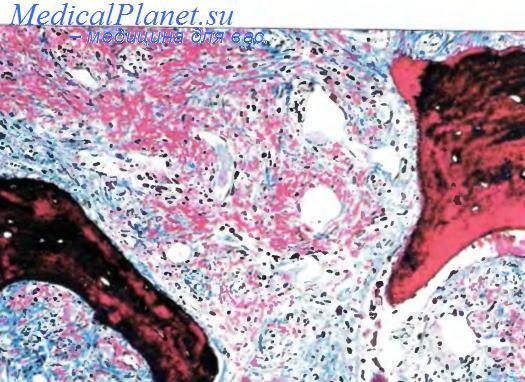 Больные оставались зависимыми от гемотрансфузий, но ни в одном случае не было зарегистрировано прогрессии заболевания. Исследование продолжается с планом проведения промежуточного анализа результатов лечения первых 37 больных, рассматривается увеличение дозы помали- домида во второй фазе исследования.
Больные оставались зависимыми от гемотрансфузий, но ни в одном случае не было зарегистрировано прогрессии заболевания. Исследование продолжается с планом проведения промежуточного анализа результатов лечения первых 37 больных, рассматривается увеличение дозы помали- домида во второй фазе исследования.
В исследовании комбинированной терапии ленали- домидом и руксолитинибом [31] больным назначали 15 мг руксолитиниба перорально два раза в день в непрерывных 28-дневных циклах в сочетании с 5 мг ле- налидомида перорально один раз в день в дни 1—21. Ответ на терапию в виде уменьшения размеров селезенки отмечен у 55 % больных, среднее время ответа составило 1,8 месяца (диапазон 0,4—31). Уменьшение выраженности фиброза костного мозга отмечено у 17 % больных. Однако данная комбинация привела к чрезмерной миелосупрессии, что явилось причиной преждевременного прекращения терапии у большинства больных. Авторы [31] полагают, что стратегия последовательной, а не совместной терапии, потенциально могла бы быть более безопасной.
PIM-киназы и циклинзависимые (CDK) киназы
Протеинкиназа PIM — проонкогенный белок, важный для патогенеза опухолей системы крови. Фермент PIM-1 (от proviral integration Moloney virus) защищает клетки от апоптоза. Этот механизм способствует злокачественной трансформации клеток. Циклинзависимые киназы (англ. cyclin-dependent kinases, CDK) — группа белков, регулируемых циклином и циклиноподобными молекулами. Большинство ци- клинзависимых киназ участвуют в смене фаз клеточного цикла; также они регулируют транскрипцию и процессинг мРНК.
Синергизм между руксолитинибом и ингибитором CDK4/6 рибоциклибом был недавно продемонстрирован в моделях in vivo [32]. Как показано в этом исследовании, он еще больше усиливается добавлением ингибитора киназы PIM — PIM447. В Европе проводится клиническое исследование эффективности этой тройной комбинации лекарств у больных МФ.
Гидроксикарбамид
Лейкоцитоз является частым осложнением у больных МФ. Нет данных об эффективности руксолитиниба в устранении лейкоцитоза. Недавнее исследование, проведенное в большой когорте больных МФ, показало эффективность терапии гидроксикарбамидом у больных с лейкоцитозом более 25х109/л [33]. В клиническом описании [34] применения комбинации руксолитиниба и гидроксикарбамида у больного МФ промежуточного риска с лейкоцитозом приводятся обнадеживающие данные. Гидроксикарбамид в дозе 500 мг/день назначался дополнительно к продолжающемуся лечению руксо- литинибом во время эпизодов лейкоцитоза. При этом достигалось уменьшение количества лейкоцитов крови до нормальных значений в течение 3—4 месяцев. Во время комбинированного лечения руксолитинибом и ги- дроксикарбамидом улучшалось клиническое состояние и уменьшалась выраженность симптомов. Не было зарегистрировано инфекционных осложнений и других побочных эффектов.
Недавнее исследование, проведенное в большой когорте больных МФ, показало эффективность терапии гидроксикарбамидом у больных с лейкоцитозом более 25х109/л [33]. В клиническом описании [34] применения комбинации руксолитиниба и гидроксикарбамида у больного МФ промежуточного риска с лейкоцитозом приводятся обнадеживающие данные. Гидроксикарбамид в дозе 500 мг/день назначался дополнительно к продолжающемуся лечению руксо- литинибом во время эпизодов лейкоцитоза. При этом достигалось уменьшение количества лейкоцитов крови до нормальных значений в течение 3—4 месяцев. Во время комбинированного лечения руксолитинибом и ги- дроксикарбамидом улучшалось клиническое состояние и уменьшалась выраженность симптомов. Не было зарегистрировано инфекционных осложнений и других побочных эффектов.
Цитарабин и меркаптопурин у больных с бластным кризом
2 клинических случая применения комбинированной терапии у больных с бластным кризом МФ приведены в статье М.С. Фоминых и соавт. [35]. Терапия руксолитинибом в комбинации с малыми дозами цитарабина (20 мг подкожно 2 раза в день в течение 5 дней, каждые 28 дней) или меркаптопурином (50 мг 1 раз в день, каждые 28 дней) приводила к улучшению состояния больных: уменьшению симптомов опухолевой интоксикации, размеров селезенки, гемотрансфузионной зависимости. Из осложнений наблюдали пневмонию, которая разрешилась после проведенной антибактериальной терапии.
[35]. Терапия руксолитинибом в комбинации с малыми дозами цитарабина (20 мг подкожно 2 раза в день в течение 5 дней, каждые 28 дней) или меркаптопурином (50 мг 1 раз в день, каждые 28 дней) приводила к улучшению состояния больных: уменьшению симптомов опухолевой интоксикации, размеров селезенки, гемотрансфузионной зависимости. Из осложнений наблюдали пневмонию, которая разрешилась после проведенной антибактериальной терапии.
Таким образом, в последнее десятилетие достигнут значительный прогресс в понимании патобиологии МФ и сигнальных путей, которые независимо или совместно с JAK-STAT играют роль в развитии этого заболевания. Проводятся клинические исследования различных рациональных комбинаций препаратов, действующих на эти пути, с руксолитинибом. Изучаются сочетания руксолитиниба с другими препаратами, способными улучшить некоторые аспекты течения болезни, на которые монотерапия руксолитинибом не влияет либо даже ухудшает их, например такие, как фиброз костного мозга и анемия. Результаты последних исследований комбинаций различных препаратов с руксолитинибом должны быть подтверждены в более крупных исследованиях и при длительном наблюдении. Ключевыми конечными точками, которые следует учитывать при оценке этих подходов, являются сокращение фиброза в костном мозге и восстановление нормального гемопоэза при одновременной оценке токсичности таких комбинаций.
Результаты последних исследований комбинаций различных препаратов с руксолитинибом должны быть подтверждены в более крупных исследованиях и при длительном наблюдении. Ключевыми конечными точками, которые следует учитывать при оценке этих подходов, являются сокращение фиброза в костном мозге и восстановление нормального гемопоэза при одновременной оценке токсичности таких комбинаций.
1. Verstovsek S., Mesa R.A., Gotlib J., et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. New Eng J Med. 2012; 366: 799–807.
2. Harrison C., Kiladjian J.J., Al-Ali H.K., et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. New Eng J Med. 2012; 366: 787–98.
3. Plosker G.L. Ruxolitinib: a review of its use in patients with myelofibrosis. Drugs. 2015; 75: 297–308. DOI: 10.1007/s40265-015-0351-8
Plosker G.L. Ruxolitinib: a review of its use in patients with myelofibrosis. Drugs. 2015; 75: 297–308. DOI: 10.1007/s40265-015-0351-8
4. Cervantes F., Vannucchi A.M., Kiladjian J.J., et al. COMFORT-II investigators. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013; 122(25): 4047–53.
5. Harrison C.N., Vannucchi A.M., Kiladjian J.J., et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016; 30(8): 1701–7.
6. Bose P., Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next? Blood. 2017; 130(2): 115–25. DOI: 10.1182/blood-2017-04-742288
7. Vannucchi A.M., Biamonte F. Epigenetics and mutations in chronic myeloproliferative neoplasms. Haematologica. 2011; 96: 1398–402.
Vannucchi A.M., Biamonte F. Epigenetics and mutations in chronic myeloproliferative neoplasms. Haematologica. 2011; 96: 1398–402.
8. Gowin K., Mesa R. Emerging therapies for the treatment of chronic Philadelphia chromosome-negative myeloproliferative neoplasm-associated myelofibrosis. Expert Opin Investig Drugs. 2013; 22: 1603–11.
9. Assi R., Verstovsek S., Daver N. ‘JAK-ing’ up the treatment of primary myelofibrosis: building better combination strategies. Curr Opin Hematol. 2017; 24(2): 115–24. DOI: 10.1097/MOH.0000000000000320
10. DeAngelo D.J., Mesa R.A., Fiskus W., et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, postessential thrombocythaemia, and postpolycythaemia vera myelofibrosis. Br J Haematol. 2013; 162: 326–35.
Br J Haematol. 2013; 162: 326–35.
11. Baffert F., Evrot E., Ebel N., et al. Improved efficacy upon combined JAK1/2 and pan-deacetylase inhibition using ruxolitinib (INC424) and panobinostat (LBH589) in preclinical mouse models of JAK2 V617F-driven disease. Blood. 2011; 118: 798.
12. Harrison C.N., Kiladjian J.J., Heidel F.H., et al. Efficacy, safety, and confirmation of the recommended phase 2 starting dose of the combination of ruxolitinib (RUX) and panobinostat (PAN) in patients (pts) with myelofibrosis (MF). Blood. 2015; 126: 4060
13. Jovanovic J.V., Ivey A., Vannucchi A.M., et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F- associated myeloproliferative neoplasms: a joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia. 2013; 27: 2032.
Leukemia. 2013; 27: 2032.
14. Daver N., Cortes J.E., Pemmaraju N., et al. Ruxolitinib (RUX) in combination with 5-azacytidine (AZA) as therapy for patients (pts) with myelofibrosis (MF). Blood. 2016; 128: 1127.
15. Tefferi A., Cervantes F., Mesa R., et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013; 122(8): 1395–8. DOI: 10.1182/blood-2013-03-488098
16. Barosi G., Bordessoule D., Briere J., et al. European Myelofibrosis Network. Response criteria for myelofibrosis with myeloid metaplasia: results of an initiative of the European Myelofibrosis Network (EUMNET). Blood. 2005; 106(8): 2849–53.
17. Rampal R., Mascarenhas J., Kosiorek H.E., et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and Post-MPN AML: results of a phase I study (myeloproliferative disorders research consortium 109 trial). Blood. 2016; 128: 1124.
18. Bose P., Verstovsek S., Gasior Y., et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results. Presented at the 58th Annual Meeting of the American Society of Hematology. 2016; 4262.
19. Меликян А.Л., Суборцева И.Н. Классические Ph-негативные ми ело пролиферативные неоплазии. Клиническая онкогематология. 2015; 8(2): 201–32.
20. Gupta V., Harrison C.N., Hasselbalch H., et al. Phase 1b/2 study of the efficacy and safety of sonidegib (LDE225) in combination with ruxolitinib (INC424) in patients with myelofibrosis. Blood. 2015; 126: 825.
Gupta V., Harrison C.N., Hasselbalch H., et al. Phase 1b/2 study of the efficacy and safety of sonidegib (LDE225) in combination with ruxolitinib (INC424) in patients with myelofibrosis. Blood. 2015; 126: 825.
21. Durrant S., Nagler A., Vannucchi A.M., et al. An Open-Label, Multicenter, 2-Arm, Dose-Finding, Phase 1b Study of the Combination of Ruxolitinib and Buparlisib (BKM120) in Patients with Myelofibrosis: Results from HARMONY Study. Blood. 2015; 126(23): 827.
22. Moyo T.K., Sochacki A., Ayers G.D., et al. Preliminary Results from a Phase I Dose Escalation Trial of Ruxolitinib and the PI3Kδ Inhibitor TGR-1202 in Myelofibrosis. Blood. 2016; 128: 1125.
23. Verstovsek S., Mesa R., Foltz L.M., et al. Phase 2 trial of PRM-151, an antifibrotic agent, in patients with myelofibrosis: stage 1 results. Blood. 2014; 124: 713.
Blood. 2014; 124: 713.
24. Verstovsek S., Mesa R.A., Foltz L.M., et al. PRM-151 in myelofibrosis: durable efficacy and safety at 72 weeks. Blood. 2015; 126: 56.
25. Меликян А.Л., Суборцева И.Н. Миелопролиферативные новообразования: новые данные. Клиническая онкогематология. 2016; 9 (2): 218–28.
26. Mikkelsen S.U., Kjaer L., Skov V., et al. Safety and Efficacy of Combination Therapy of Interferon-Alpha2 + JAK1-2 Inhibitor in the Philadelphia-Negative Chronic Myeloproliferative Neoplasms. Preliminary Results from the Danish Combi-Trial — an Open Label, Single Arm, Non-Randomized Multicenter Phase II Study. Blood. 2015; 126(23): 824.
27. Mikkelsen S.U., Kjaer L., Bjørn M.E., et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018; 7(8): 3571–81. DOI: 10.1002/cam4.1619
Mikkelsen S.U., Kjaer L., Bjørn M.E., et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018; 7(8): 3571–81. DOI: 10.1002/cam4.1619
28. Stahl M., Zeidan A.M. Management of myelofibrosis: JAK inhibition and beyond. Expert Rev Hematol. 2017; 10(5): 459–77. DOI: 10.1080/17474086.2017.1317590
29. Gowin K., Kosiorek H., Dueck A., et al. Multicenter phase 2 study of combination therapy with ruxolitinib and danazol in patients with myelofibrosis. Leuk Res. 2017; 60: 31–5. DOI: 10.1016/j.leukres.2017.06.005
30. Stegelmann F., Bangerter M., Heidel F.H., et al. A phase-Ib/II study of ruxolitinib plus pomalidomide in myelofibrosis. Blood. 2015; 126(23): 826.
31. Daver N., Cortes J., Newberry K., et al. Ruxolitinib in combination with lenalidomide as therapy for patients with myelofibrosis. Haematologica. 2015; 100: 1058–63.
32. Rampal R., Pinzon-Ortiz M., Varshini H.A.S., et al. Synergistic Therapeutic Efficacy of Combined JAK1/2, Pan-PIM, and CDK4/6 Inhibition in Myeloproliferative Neoplasms. Blood. 2016; 128(22): 634.
33. Kuykendall A.T., Talati C., Al Ali N., et al. The treatment landscape of myelofibrosis before and after ruxolitinib approval. Clin Lymphoma Myeloma Leuk. 2017; 17: 45–53.
34. Caocci G., Ghiani S., Mocci C., La Nasa G. Combination Therapy with Ruxolitinib and Hydroxyurea for the Treatment of Myeloid-Predominant Leukocytosis in a Patient with Myelofibrosis.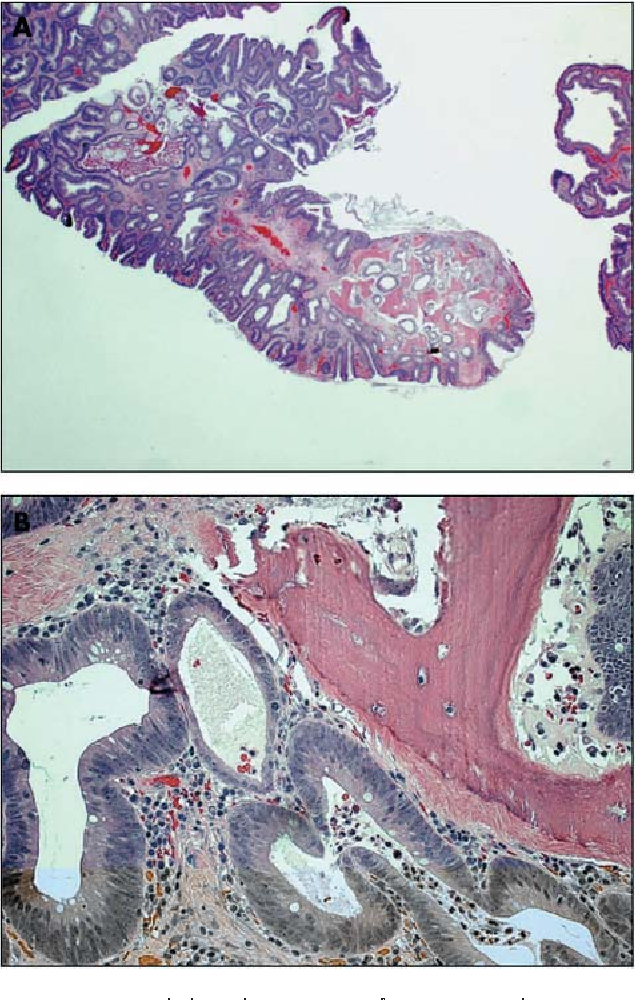 Acta Haematol. 2018; 139 (3): 164–5. DOI: 10.1159/000487582
Acta Haematol. 2018; 139 (3): 164–5. DOI: 10.1159/000487582
35. Фоминых М.С., Шуваев В.А., Мартынкевич И.С. и др. Комбинированный режим терапии руксолитинибом и малыми дозами цитозара или меркаптопурина у пациентов с бластным кризом миелофиброза. Онкогематология. 2016; 11(2): 37–9. DOI: 10.17650/1818-8346-2016-11-2-37-39
| Объект: |
| CTI Investigational Site 10002 | Scottsdale, Arizona, 85259, United States |
| CTI Investigational Site 10004 | Omaha, Nebraska, 68198, United States |
| CTI Investigational Site 10001 | Morristown, New Jersey, 07962, United States |
| CTI Investigational Site 10003 | Greenville, South Carolina, 29601, United States |
| CTI Investigational Site 61006 | Box Hill, Australia |
| CTI Investigational Site 61001 | Coffs Harbour, Australia |
| CTI Investigational Site 61005 | Geelong, Australia |
| CTI Investigational Site 61003 | Gosford, Australia |
| CTI Investigational Site 61004 | Hobart, Australia |
| CTI Investigational Site 61002 | Milton, Australia |
| CTI Investigational Site 32002 | Antwerp, Belgium |
| CTI Investigational Site 32003 | Antwerp, Belgium |
| CTI Investigational Site 32001 | Brugge, Belgium |
| CTI Investigational Site 32005 | Bruxelles, Belgium |
| CTI Investigational Site 32004 | La Louviere, Belgium |
| CTI Investigational Site 42003 | Brno, Czechia |
| CTI Investigational Site 42001 | Olomouc, Czechia |
| CTI Investigational Site 42002 | Plzen, Czechia |
| CTI Investigational Site 42004 | Prague, Czechia |
| CTI Investigational Site 33005 | Amiens, France |
| CTI Investigational Site 33006 | Caen, France |
| CTI Investigational Site 33011 | Grenoble, France |
| CTI Investigational Site 33012 | Lens, France |
| CTI Investigational Site 33007 | Lille, France |
| CTI Investigational Site 33001 | Nimes Cedex, France |
| CTI Investigational Site 33004 | Paris, France |
| CTI Investigational Site 33008 | Paris, France |
| CTI Investigational Site 33009 | Pessac, France |
| CTI Investigational Site 33010 | Pierre Benite, France |
| CTI Investigational Site 33003 | Strasbourg, France |
| CTI Investigational Site 33002 | Toulouse, France |
| CTI Investigational Site 49006 | Berlin, Germany |
| CTI Investigational Site 49007 | Berlin, Germany |
| CTI Investigational Site 49003 | Dresden, Germany |
| CTI Investigational Site 49008 | Essen, Germany |
| CTI Investigational Site 49002 | Freiburg, Germany |
| CTI Investigational Site 49001 | Koln, Germany |
| CTI Investigational Site 49005 | Mainz, Germany |
| CTI Investigational Site 49004 | Munchen, Germany |
| CTI Investigational Site 49009 | Munster, Germany |
| CTI Investigational Site 36002 | Budapest, Hungary |
| CTI Investigational Site 36005 | Debrecen, Hungary |
| CTI Investigational Site 36006 | Gyula, Hungary |
| CTI Investigational Site 36003 | Kaposvar, Hungary |
| CTI Investigational Site 36004 | Kecskemet, Hungary |
| CTI Investigational Site 36001 | Szeged, Hungary |
| CTI Investigational Site 36008 | Szolnok, Hungary |
| CTI Investigational Site 36007 | Szombathely, Hungary |
| CTI Investigational Site 39003 | Bologna, Italy |
| CTI Investigational Site 39001 | Firenze, Italy |
| CTI Investigational Site 39005 | Milano, Italy |
| CTI Investigational Site 39004 | Monza, Italy |
| CTI Investigational Site 39002 | Padova, Italy |
| CTI Investigational Site 39008 | Reggio Emilia, Italy |
| CTI Investigational Site 39006 | Rimini, Italy |
| CTI Investigational Site 31001 | Amsterdam, Netherlands |
| CTI Investigational Site 31002 | Maastricht, Netherlands |
| CTI Investigational Site 31003 | Rotterdam, Netherlands |
| CTI Investigational Site 31004 | Utrecht, Netherlands |
| CTI Investigational Site 64001 | Christchurch, New Zealand |
| CTI Investigational Site 64004 | Dunedin, New Zealand |
| CTI Investigational Site 64002 | Hamilton, New Zealand |
| CTI Investigational Site 64003 | Takapuna, New Zealand |
| CTI Investigational Site 70011 | Izhevsk, Russian Federation |
| CTI Investigational Site 70008 | Moscow, Russian Federation |
| CTI Investigational Site 70009 | Moscow, Russian Federation |
| CTI Investigational Site 70002 | Petrozavodsk, Russian Federation |
| CTI Investigational Site 70010 | Saint Petersburg, Russian Federation |
| CTI Investigational Site 70005 | Samara, Russian Federation |
| CTI Investigational Site 70006 | Sochi, Russian Federation |
| CTI Investigational Site 70001 | St. 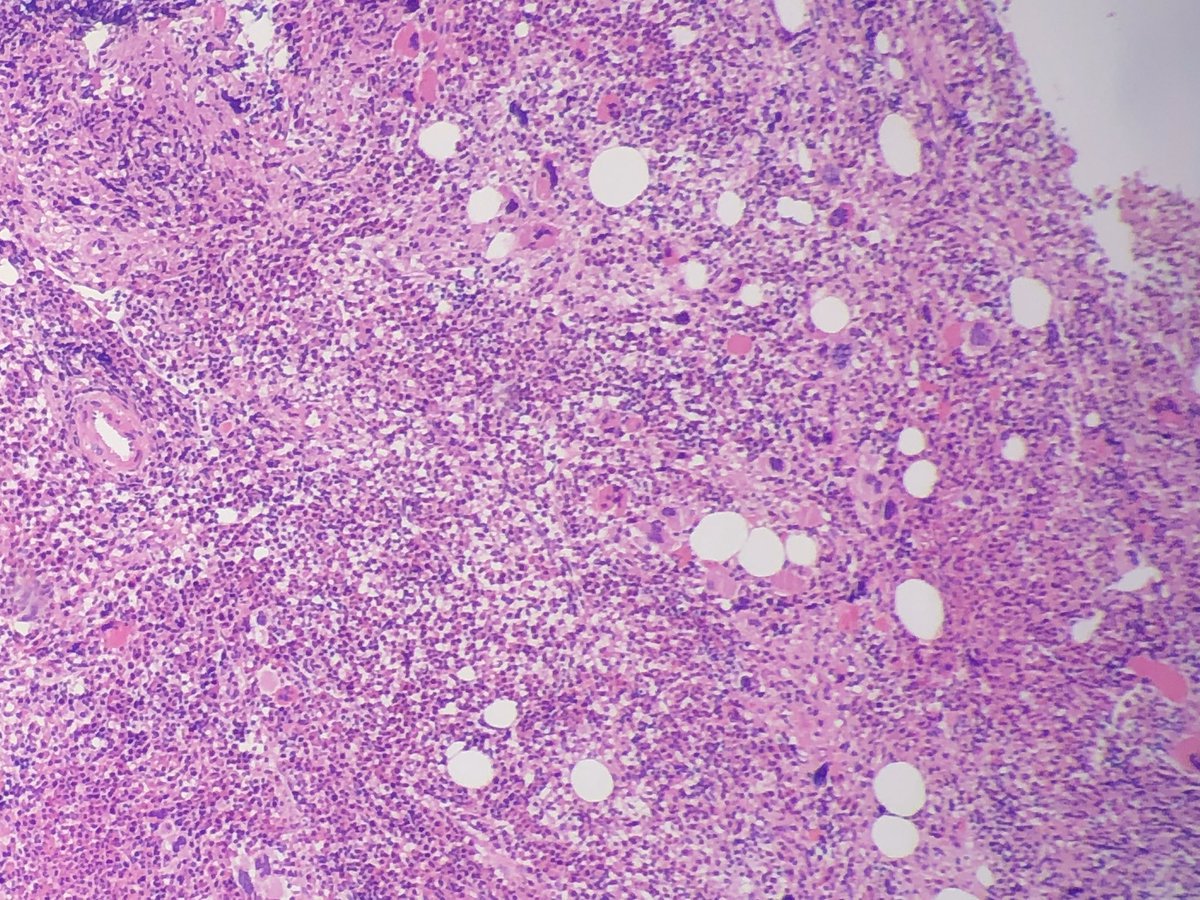 Petersburg, Russian Federation Petersburg, Russian Federation |
| CTI Investigational Site 70004 | St. Petersburg, Russian Federation |
| CTI Investigational Site 70007 | Volgograd, Russian Federation |
| CTI Investigational Site 44004 | Birmingham, United Kingdom |
| CTI Investigational Site 44008 | Bournemouth, United Kingdom |
| CTI Investigational Site 44002 | Cambridge, United Kingdom |
| CTI Investigational Site 44003 | Cardiff, United Kingdom |
| CTI Investigational Site 44001 | London, United Kingdom |
| CTI Investigational Site 44007 | London, United Kingdom |
| CTI Investigational Site 44006 | Manchester, United Kingdom |
| CTI Investigational Site 44005 | Oxford, United Kingdom |
Течение первичного миелофиброза под маской цирроза печени
Течение первичного миелофиброза под маской цирроза печени
Седых А.О., Ермолаева Н. А.
А.
Научный руководитель к.м.н. доцент Пахомова А.Л.
ФГБОУ ВО Саратовский ГМУ им. В.И. Разумовского Минздрава РФ
Кафедра терапии, гастроэнтерологии и пульмонологии Первичный миелофиброз – редкое заболевание, характеризующееся чрезмерным образованием соединительной ткани в костном мозге с потерей кроветворных элементов и усилением внекостномозгового кроветворения. Миелофиброз может протекать бессимптомно или с наличием анемии, спленомегалии, гепатомегалии, потери веса. Редкая встречаемость и неспецифическая симптоматика обусловливают трудности постановки диагноза. Поздняя диагностика и начало лечения негативно сказываются на прогнозе.
Пациентка Б., 56 лет наблюдалась в течении 10 месяцев с диагнозом: Цирроз печени неясной этиологии. Класс А. А0. Хроническая нормохромная анемия. Заболела 10.16: появилась слабость, выявлена анемия. Лечение препаратами железа без эффекта. В 01.17 диагностирована миома матки, гиперплазия эндометрия, гепатоспленомегалия, портальная гипертензия. Установлен диагноз цирроза печени. Проведена экстирпация матки, однако анемия прогрессировала. Консультирована гастроэнтерологом 03.17, далее находилась под динамическим наблюдением. Получала гепатопротекторы, симптоматическую терапию без заметного эффекта, проводились гемотрансфузии. Неоднократно консультирована гематологом, поставлен диагноз нормохромной анемии вторичного генеза. За время наблюдения обращали внимание преобладание анемического синдрома, нарастание спленомегалии, повышенные уровни ЩФ, ЛДГ, ГГТП при нормальных показателях трансаминаз и билирубина сыворотки крови. 10.17 в ОАК впервые появились миелоциты, юные клетки. Консультирована гематологом, после дообследования диагностирован первичный миелофиброз.
Установлен диагноз цирроза печени. Проведена экстирпация матки, однако анемия прогрессировала. Консультирована гастроэнтерологом 03.17, далее находилась под динамическим наблюдением. Получала гепатопротекторы, симптоматическую терапию без заметного эффекта, проводились гемотрансфузии. Неоднократно консультирована гематологом, поставлен диагноз нормохромной анемии вторичного генеза. За время наблюдения обращали внимание преобладание анемического синдрома, нарастание спленомегалии, повышенные уровни ЩФ, ЛДГ, ГГТП при нормальных показателях трансаминаз и билирубина сыворотки крови. 10.17 в ОАК впервые появились миелоциты, юные клетки. Консультирована гематологом, после дообследования диагностирован первичный миелофиброз.
Выводы: миелофиброз не имеет специфической симптоматики и может протекать под маской другой патологии; преобладание анемического синдрома, не соответствующего тяжести заболевания печени, требует настойчивого поиска гематологической патологии.
Терапия » Поражение почек при гемобластозах
Поражение почек при гемобластозах
DOI: https://dx. doi.org/10.18565/therapy.2019.2.117-122
doi.org/10.18565/therapy.2019.2.117-122
В.И. Махина, Е.В. Волошинова, Ф.Д. Голубинов
1) Кафедра госпитальной терапии лечебного факультета ФГБОУ ВО «Саратовский государственный медицинский университет им. В.И. Разумовского» Минздрава России;
2) ГУЗ «Областная клиническая больница», Саратов
Гемобластозы – группа опухолевых заболеваний, возникающих из гемопоэтических клеток и поражающих многие органы. Частота встречаемости и варианты поражения почек различаются при злокачественных заболеваниях лимфоидной и миелоидной тканей. В приведенных клинических наблюдениях описаны трудности диагностики и лечения вторичных нефропатий в рамках первичного миелофиброза и болезни отложения легких цепей. Описано два случая развития мембранопролиферативного гломерулонефрита в дебюте гематологических заболеваний, диагностированного при морфологическом исследовании почечного биоптата. Обсуждаются современные клинические и патогенетические особенности данных заболеваний, эффективность разработанных методов лечения.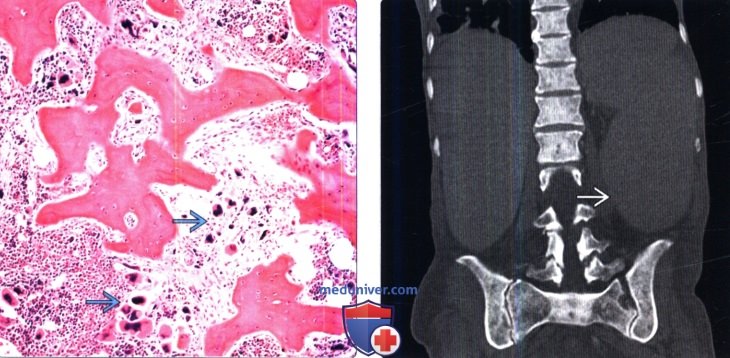
Литература
- Гематология: национальное руководство. Под ред. О.А. Рукавицына. М., 2015. 776 с.
- Arber D.A., Orazi A., Hasserjian R. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20): 2391–405.
- Меликян А.Л., Туркина А.Г., Абдулкадыров К.М., Зарицкий А.Ю., Афанасьев Б.В., Шуваев В.А., Ломаиа Е.Г., Морозова Е.В., Байков В.В., Голенков А.К., Суборцева И.Н., Соколова М.А., Ковригина А.М., Мартынкевич И.С., Грицаев С.В., Судариков А.Б., Суханова Г.А., Иванова В.Л., Капланов К.Д., Константинова Т.С., Поспелова Т.И., Агеева Т.А., Шатохин Ю.В., Савченко В.Г. Клинические рекомендации по диагностике и терапии Ph-негативных миелопролиферативных заболеваний (истинная полицитемия, эссенциальная тромбоцитемия, первичный миелофиброз). Гематология и трансфузиология. 2014; 4: 31–57.
- Klampfl T., Gisslinger H., Harutyunyan A.S., Nivarthi H.
 , Rumi E., Milosevic J.D., Them N.C.C. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013; 369(25): 2379–90.
, Rumi E., Milosevic J.D., Them N.C.C. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013; 369(25): 2379–90. - Said S.M., Leung N., Sethi S., Cornell L.D., Fidler M.E., Grande J.P., Herrmann S., Tefferi A., D’Agati V.D., Nasr S.H. Myeloproliferative neoplasms cause glomerulopathy. Kidney International. 2011; 80: 753–59. https://doi.org/10.1038/ki.2011.147.
- Захарова Е.В., Столяревич Е.С., Виноградова О.В., Тареева А.Б., Макарова Т.А., Михайлова Н.А., Ипатьева Е.И.,Тареева Е.И., Шубина А.В., Бисикало М.Л., Трапезина И.И. Поражения почек при лимфоплазмацитарных заболеваниях (обзор литературы и клинические наблюдения). Нефрология и диализ. 2014; 11(2): 68–93.
- Pant A.D., Solez K. Light chain deposition disease in kidney: a review of the literature. J. Pathol. Nepal. 2011; 1: 56–59.
- Rajasekaran A., Ngo T.T., Abdelrahim M., Glass W., Podoll A., Verstovsek S., Abudayyeh A. Primary myelofibrosis associated glomerulopathy: significant improvement after therapy with ruxolitinib. BMC. Nephrol. 2015; 16: 121.
- Del Sordo R., Brugnano R., Covarelli C., Fiorucci G., Falzetti F., Barbatelli G., Nunzi E., Sidoni A. Nephrotic syndrome in primary myelofibrosis with renal extramedullary hematopoiesis and glomerulopathy in the JAK inhibitor era. Clin. Nephrol. Case Stud. 2017; 5: 70–77.
- Виноградова О.Ю., Шуваев В.А., Мартынкевич И.С., Панкрашкина М.М., Фоминых М.С., Ефремова Е.В., Крутикова К.Ю., Полушкина Л.Б., Шаркунов Н.Н., Волошин С.В., Чечеткин А.В. Таргетная терапия миелофиброза. Клиническая онкогематология. 2017; 10(4): 471–478.
- Verstovsek S., Mesa R.A., Gotlib J. et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 2017; 10(1): 55.
- Tefferi A., Guglielmelli P., Pardanani A., Vannucchi A.M. Myelofibrosis treatment Algorithm 2018. Blood Cancer J. 2018; 8(8): 72.
- Gianelli U. , Vener C., Bossi A. et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod. Pathol. 2012; 25: 1193–202.
- Tefferi A. Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016; 91: 1262–71.
- Kaygusuz I., Koc M., Arikan H. et al. Focal segmental glomerulosclerosis associated with idiopathic myelofibrosis. Ren. Fail. 2010; 32: 273–76.
- Jimenez-Zepeda V.H. Light chain deposition disease: novel biological insights and treatment advances. Int. J. Lab. Hematol. 2012; 34: 347–55.
- Telio D., Shepherd J., Forrest D. et al. High-dose melphalan followed by auto-SCT has favorable safety and efficacy in selected patients with light chain deposition disease and light and heavy chain deposition disease. Bone Marrow Transplant. 2012; 47: 453–55.
- Toshiharu Ueno, Koichi Kikuchi, Ryo Hazue, Koki Mise, Keiichi Sumida, Noriko Hayami, Tatsuya Suwabe, Junichi Hoshino, Naoki Sawa, Kenji Arizono, Shigeko Hara, Kenmei Takaichi, Takeshi Fujii,Kenichi Ohashi, Yoshifumi Ubara. Five sequential evaluations of renal histology in a patient with light chain deposition disease. Intern. Med. 2016; 55(20): 2993–99.
- Рехтина И.Г., Бирюкова Л.С. Болезнь отложения легких цепей. В кн.: Н.А. Мухина, гл. ред. Нефрология. Национальное руководство. Краткое издание. М., 2014. С. 394–397.
 , Rumi E., Milosevic J.D., Them N.C.C. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013; 369(25): 2379–90.
, Rumi E., Milosevic J.D., Them N.C.C. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013; 369(25): 2379–90. BMC. Nephrol. 2015; 16: 121.
BMC. Nephrol. 2015; 16: 121. , Vener C., Bossi A. et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod. Pathol. 2012; 25: 1193–202.
, Vener C., Bossi A. et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod. Pathol. 2012; 25: 1193–202.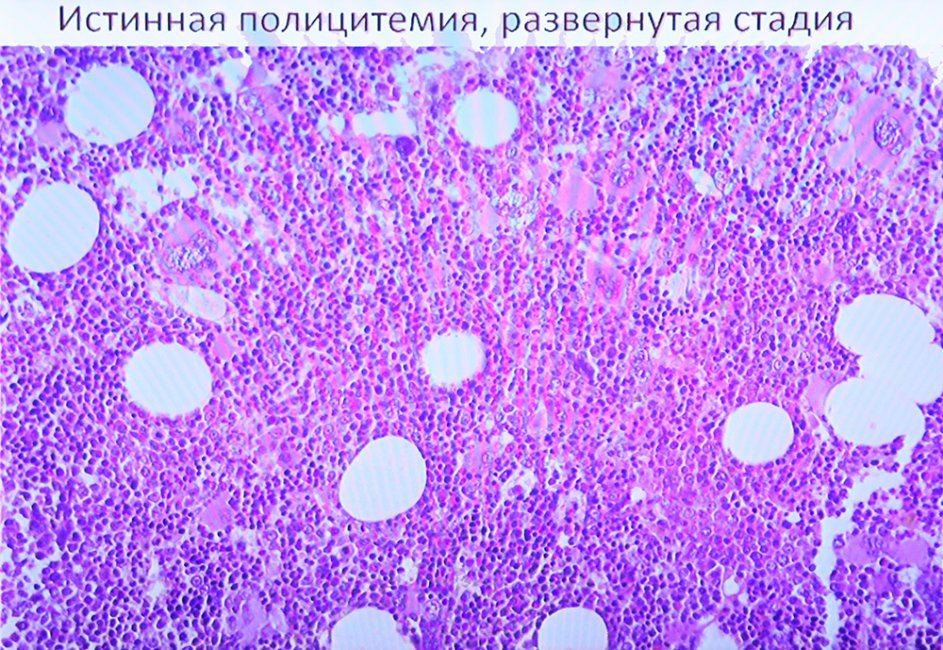 Five sequential evaluations of renal histology in a patient with light chain deposition disease. Intern. Med. 2016; 55(20): 2993–99.
Five sequential evaluations of renal histology in a patient with light chain deposition disease. Intern. Med. 2016; 55(20): 2993–99.Об авторах / Для корреспонденции
Виолетта Игоревна Махина, аспирант кафедры госпитальной терапии лечебного факультета ФГБОУ ВО «Саратовский государственный медицинский университет им. В.И. Разумовского» Минздрава России. Адрес: 410012, г. Саратов, ул. Б. Казачья, д. 112. Тел.: 8 (8452) 49-14-37. E-mail: [email protected]
Елена Викторовна Волошинова, к.м.н., доцент кафедры госпитальной терапии лечебного факультета ФГБОУ ВО «Саратовский государственный медицинский университет им. В.И. Разумовского» Минздрава России.
Адрес: 410012, г. Саратов, ул. Б. Казачья, д. 112. Тел.: 8 (8452) 49-14-37. E-mail: voloshinovaelena@mail. ru
ru
Филипп Дмитриевич Голубинов, зав. нефрологическим отделением ГУЗ «Областная клиническая больница». Адрес: 410053, г. Саратов, Смирновское ущелье, 1. Тел.: 8 (8452) 49-14-59. E-mail: [email protected]
Первичный миелофиброз: MedlinePlus Genetics
Мутации в генах JAK2 , MPL , CALR и TET2 связаны с большинством случаев первичного миелофиброза. Гены JAK2 и MPL предоставляют инструкции по созданию белков, которые способствуют росту и делению (пролиферации) клеток крови. Ген CALR предоставляет инструкции по созданию белка с множеством функций, включая обеспечение правильного сворачивания вновь образованных белков и поддержание правильных уровней сохраненного кальция в клетках.Ген TET2 предоставляет инструкции по созданию белка, функция которого неизвестна.
Белки, продуцируемые генами JAK2 и MPL , являются частью сигнального пути, называемого путем JAK / STAT, который передает химические сигналы извне клетки в ядро клетки.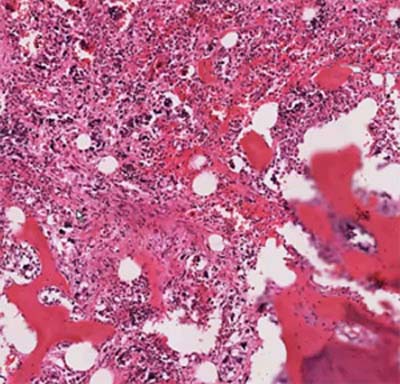 Белок, продуцируемый геном MPL , называемый рецептором тромбопоэтина, включает (активирует) этот путь, а белок JAK2 передает сигналы после активации.Посредством пути JAK / STAT эти два белка способствуют пролиферации клеток крови, особенно типа клеток крови, известных как мегакариоциты.
Белок, продуцируемый геном MPL , называемый рецептором тромбопоэтина, включает (активирует) этот путь, а белок JAK2 передает сигналы после активации.Посредством пути JAK / STAT эти два белка способствуют пролиферации клеток крови, особенно типа клеток крови, известных как мегакариоциты.
Мутации в гене JAK2 или MPL , которые связаны с первичным миелофиброзом, приводят к сверхактивации пути JAK / STAT. Аномальная активация передачи сигналов JAK / STAT приводит к перепроизводству аномальных мегакариоцитов, и эти мегакариоциты стимулируют другой тип клеток для высвобождения коллагена.Коллаген — это белок, который обычно обеспечивает структурную поддержку клеток костного мозга. Однако при первичном миелофиброзе избыток коллагена образует рубцовую ткань в костном мозге.
Хотя мутации в генах CALR и TET2 относительно распространены при первичном миелофиброзе, неясно, как эти мутации участвуют в развитии этого состояния.
У некоторых людей с первичным миелофиброзом нет мутации ни в одном из известных генов, связанных с этим заболеванием. Исследователи работают над выявлением других генов, которые могут быть вовлечены в это состояние.
Исследователи работают над выявлением других генов, которые могут быть вовлечены в это состояние.
Обзор первичного миелофиброза | MPNRF
Что такое первичный миелофиброз (MF)?
Первичный миелофиброз (MF) — это хронический рак крови, при котором в костном мозге образуется чрезмерная рубцовая ткань, которая нарушает его способность производить нормальные клетки крови.
Исследователи полагают, что причиной МФ могут быть аномальные стволовые клетки крови в костном мозге. Аномальные стволовые клетки производят зрелые клетки, которые быстро растут и захватывают костный мозг, вызывая как фиброз (образование рубцовой ткани), так и хроническое воспаление.В результате костному мозгу становится труднее создавать нормальные клетки крови, и производство клеток крови может перемещаться в селезенку (вызывая увеличение) или в другие области тела.
Классифицируемый как миелопролиферативное новообразование (MPN), MF может возникать самостоятельно или как прогрессирование истинной полицитемии (post-PV-MF) или эссенциальной тромбоцитемии (post-ET-MF). Проявления MF, post-PV-MF и post-ET-MF практически идентичны, и лечение в целом одинаково для всех трех.
Проявления MF, post-PV-MF и post-ET-MF практически идентичны, и лечение в целом одинаково для всех трех.
Щелкните здесь , если вы хотите посмотреть обзорное видео с диагнозом, описанием симптомов, течением заболевания и вариантами лечения миелофиброза для пациентов, лиц, осуществляющих уход за ними, и их близких, созданное доктором Рубеном А. Меса и доктором Робин М. Шербер из UT Health San Antonio, онкологический центр доктора медицины Андерсона.
Рекомендации по питанию для пациентов с МПН
Авторы: доктор Робин Шербер, доктор медицины; Доктор Рубен Меса, доктор медицины; Райан Эккерт, магистр медицины, Онкологический центр Мэйса в UT Health, Сан-Антонио, доктор медицины Андерсон Группа исследования качества жизни MPN; www.mpnqol
Никто точно не знает, что вызывает начало миелофиброза или других миелопролиферативных новообразований. В большинстве случаев миелофиброз не наследуется генетически — вы не можете передать болезнь своим детям или унаследовать ее от родителей (хотя в некоторых семьях наблюдается явная предрасположенность).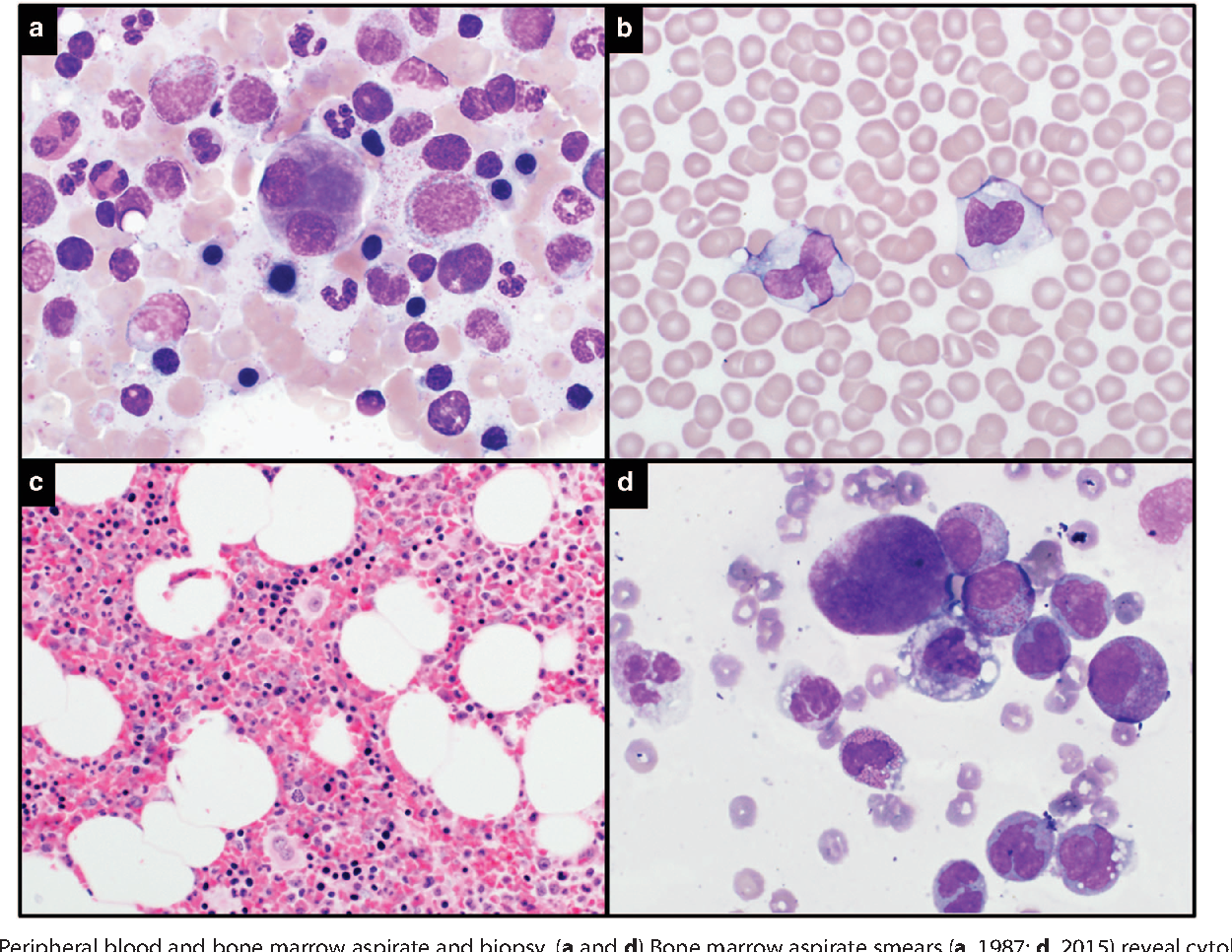
Недавно исследователи обнаружили, что эти заболевания могут быть вызваны приобретенными генными мутациями (изменениями в ДНК, которые не передаются по наследству). Некоторые из этих мутаций влияют на белки, которые работают в сигнальных путях в ваших клетках.
Факторы риска, связанные с MF, включают:
Возраст
Миелофиброз чаще всего диагностируется у людей старше 60 лет, хотя известны случаи миелофиброза у молодых.
Окружающая среда
Воздействие нефтехимических веществ (например, бензола и толуола) и ионизирующего излучения может увеличить риск развития МП.
Мутация JAK2
Приблизительно от 50% до 60% людей с MF имеют мутацию гена JAK2 в их кроветворных клетках.Мутант JAK2 заставляет клетки крови расти и делиться, даже когда организм не требует дополнительных клеток крови. От 5 до 10% пациентов будут иметь мутацию в другом гене, названном MPL, который также влияет на сигнальный путь JAK.
CALR
Около 23,5% людей с миелофиброзом и эссенциальной тромбоцитемией имеют мутацию под названием кальретикулин, или CALR. Этот генетический маркер был открыт в 2013 году двумя независимыми лабораториями, в том числе одной, финансируемой Исследовательским фондом MPN.Исследования все еще продолжаются, но есть потенциальные последствия для лечения и прогноза для людей с мутацией CALR.
Врачи рассматривают широкий спектр факторов, прежде чем поставить диагноз миелофиброза. Поскольку каждый случай МФ индивидуален, для диагностики заболевания обычно требуются анамнез, медицинский осмотр, лабораторные анализы и исследование костного мозга.
Даже если у некоторых людей, живущих с миелофиброзом, симптомы могут отсутствовать (то, что вы чувствуете), они часто проявляют признаки (то, что врач замечает во время медицинского осмотра).
Общие признаки МФ во время медицинского осмотра могут включать:
- Увеличенная селезенка (спленомегалия)
- Бледность слизистых оболочек (бледность) при наличии анемии
- Потеря мышечной массы
- Кахексия (синдром истощения, характеризующийся потерей веса, атрофией мышц, слабостью и утомляемостью)
Общие лабораторные тесты для диагностики MF включают:
Анализы крови
Общий анализ крови (ОАК) часто показывает ненормальное количество клеток крови. Красные кровяные тельца (эритроциты), белые кровяные тельца (лейкоциты), тромбоциты (тромбоциты) или другие типы клеток крови обычно поражаются МФ, и их количество может быть либо слишком высоким, либо слишком низким.
Красные кровяные тельца (эритроциты), белые кровяные тельца (лейкоциты), тромбоциты (тромбоциты) или другие типы клеток крови обычно поражаются МФ, и их количество может быть либо слишком высоким, либо слишком низким.
Биопсия костного мозга
Исследование костного мозга под микроскопом может выявить предполагающие МФ, включая образование рубцовой ткани и атипичный вид предшественников костного мозга.
Анализ мутаций генов клеток крови
Специфические мутации, связанные с некоторыми случаями MF (например,g., JAK2, CALR и MPL) можно идентифицировать с помощью анализа генных мутаций.
Визуализационные тесты
Ультразвук селезенки или другие тесты могут быть выполнены, чтобы определить, увеличена ли селезенка.
Симптомы миелофиброза часто вызваны увеличением селезенки и / или недостаточным количеством нормальных клеток крови и хроническим воспалением.
Общие симптомы и признаки MF могут включать:
- Усталость, слабость или одышка при легкой физической нагрузке
- Полнота, дискомфорт или боль в левой верхней части живота
- лихорадка
- Ночные поты
- Похудание или недоедание
- Боль в костях
- Зуд (кожный зуд)
- Легкое кровотечение или синяк
- Восприимчивость к инфекциям
- Боль в суставах или подагра
- Вздутие живота / задержка жидкости (при наличии портальной гипертензии или повышенного артериального давления в воротной вене)
- Нарушение функции печени
- Аномальный рост кроветворных клеток вне костного мозга
Регулярные медицинские осмотры, включая общий анализ крови (ОАК), важны для диагностики МФ и других МФН, поскольку у некоторых пациентов с МФ симптомы отсутствуют (особенно на ранней стадии заболевания).
Нет единого прогноза для людей, страдающих миелофиброзом — прогноз МФ индивидуален для каждого пациента. В то время как некоторые люди живут много лет без развития серьезных симптомов, другие считают, что болезнь прогрессирует быстрее.
Факторами, которые могут повлиять на прогноз МФ, являются возраст, количество лейкоцитов, количество «бластов» (незрелых кровяных телец) в крови, «конституциональные симптомы» (например, ночная потливость, потеря веса, лихорадка), анемия (низкий красный клетки крови), зависимость от переливания крови, низкий уровень тромбоцитов и аномальный анализ хромосом.
Для большинства пациентов прогноз MF требует лечения нескольких симптомов и признаков, в том числе:
- Анемия (недостаточно красных кровяных телец для переноса кислорода)
- Спленомегалия (увеличение селезенки)
- Экстрамедуллярный гемопоэз (производство клеток крови в органах за пределами костного мозга, таких как селезенка и печень)
- Тромбозы и тромбогеморрагические осложнения (осложнения свертывания крови или кровотечения)
- Лейкоцитоз (слишком много лейкоцитов)
- Тромбоцитоз (слишком много тромбоцитов) или тромбоцитопения (низкое количество тромбоцитов)
- Конституциональные и системные («все тело») симптомы (e. g., утомляемость, ночная потливость, потеря веса, зуд, лихорадка, боли в костях и суставах)
- Подагра
 g., утомляемость, ночная потливость, потеря веса, зуд, лихорадка, боли в костях и суставах)
g., утомляемость, ночная потливость, потеря веса, зуд, лихорадка, боли в костях и суставах)У небольшого числа пациентов МФ может трансформироваться в острый миелоидный лейкоз (ОМЛ), серьезный рак крови и костного мозга. Когда ОМЛ действительно возникает из-за МФ, он быстро прогрессирует и может быть трудно поддается лечению.
Не существует единого лечения, которое было бы эффективным для всех больных МФ. У каждого пациента есть уникальный набор симптомов и обстоятельств, которые требуют различных вариантов лечения, назначенных врачом.Кроме того, некоторые пациенты с МФ не имеют симптомов в течение многих лет и могут не нуждаться в немедленном лечении. Тем не менее, любой, у кого был диагностирован МФ, должен постоянно наблюдаться на предмет признаков или симптомов, указывающих на обострение болезни.
Доступные методы лечения и терапии MF включают:
Джакафи
Джакафи (руксолитиниб) — первый препарат, одобренный FDA для лечения пациентов с МФ.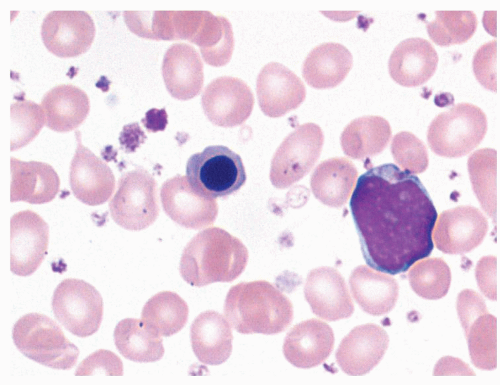 Jakafi показан для лечения пациентов с миелофиброзом (MF) среднего или высокого риска, включая первичный MF, MF после истинной полицитемии и MFA после эссенциальной тромбоцитемии. Таргетная терапия, Jakafi более специфична для аномальных клеток.При пероральном приеме Jakafi частично подавляет активность JAK2 и родственного белка JAK1. Во время клинических испытаний было показано уменьшение размера селезенки, дискомфорта в животе, раннего насыщения, боли в костях, ночного потоотделения и зуда у пациентов с МФ, а также уровня «провоспалительных цитокинов» в крови, которые могут вызывать некоторые симптомы, такие как в виде утомляемости, лихорадки, ночного потоотделения и похудания. У пациентов наблюдаются низкие показатели крови, головная боль, головокружение, синяки или инфекции. Для получения дополнительной информации о Jakafi, щелкните здесь (будет перенаправлен на веб-сайт производителя).
Jakafi показан для лечения пациентов с миелофиброзом (MF) среднего или высокого риска, включая первичный MF, MF после истинной полицитемии и MFA после эссенциальной тромбоцитемии. Таргетная терапия, Jakafi более специфична для аномальных клеток.При пероральном приеме Jakafi частично подавляет активность JAK2 и родственного белка JAK1. Во время клинических испытаний было показано уменьшение размера селезенки, дискомфорта в животе, раннего насыщения, боли в костях, ночного потоотделения и зуда у пациентов с МФ, а также уровня «провоспалительных цитокинов» в крови, которые могут вызывать некоторые симптомы, такие как в виде утомляемости, лихорадки, ночного потоотделения и похудания. У пациентов наблюдаются низкие показатели крови, головная боль, головокружение, синяки или инфекции. Для получения дополнительной информации о Jakafi, щелкните здесь (будет перенаправлен на веб-сайт производителя).
Инребич
ИНРЕБИК® (федратиниб) показан для лечения взрослых пациентов с первичным или вторичным (постполицитемия истинная или постэссенциальная тромбоцитемия) миелофиброзом (МФ) промежуточного 2 или высокого риска. Конечные точки были сосредоточены на уменьшении размера селезенки и облегчении симптомов. Это пероральный ингибитор киназы с активностью против дикого типа и мутационно активированной янус-ассоциированной киназы 2 (JAK2) и FMS-подобной тирозинкиназы 3 (FLT3). Аномальная активация JAK2 связана с миелопролиферативными новообразованиями, включая миелофиброз и истинную полицитемию.Согласно Celgene, INREBIC делает больше для подавления селективной мутации JAK2, чем другие члены семейства JAK JAK1, JAK3 и TYK2. Щелкните здесь , чтобы узнать больше.
Конечные точки были сосредоточены на уменьшении размера селезенки и облегчении симптомов. Это пероральный ингибитор киназы с активностью против дикого типа и мутационно активированной янус-ассоциированной киназы 2 (JAK2) и FMS-подобной тирозинкиназы 3 (FLT3). Аномальная активация JAK2 связана с миелопролиферативными новообразованиями, включая миелофиброз и истинную полицитемию.Согласно Celgene, INREBIC делает больше для подавления селективной мутации JAK2, чем другие члены семейства JAK JAK1, JAK3 и TYK2. Щелкните здесь , чтобы узнать больше.
Аллогенная трансплантация стволовых клеток (ASCT)
Аллогенная трансплантация стволовых клеток — единственное лечебное средство от МФ. ASCT, гемопоэтические (кроветворные) стволовые клетки передаются от донора пациенту, по сути заменяя дефектные стволовые клетки здоровыми.Перед инфузией стволовых клеток пациент получает химиотерапию и / или лучевую терапию для уничтожения пораженного костного мозга.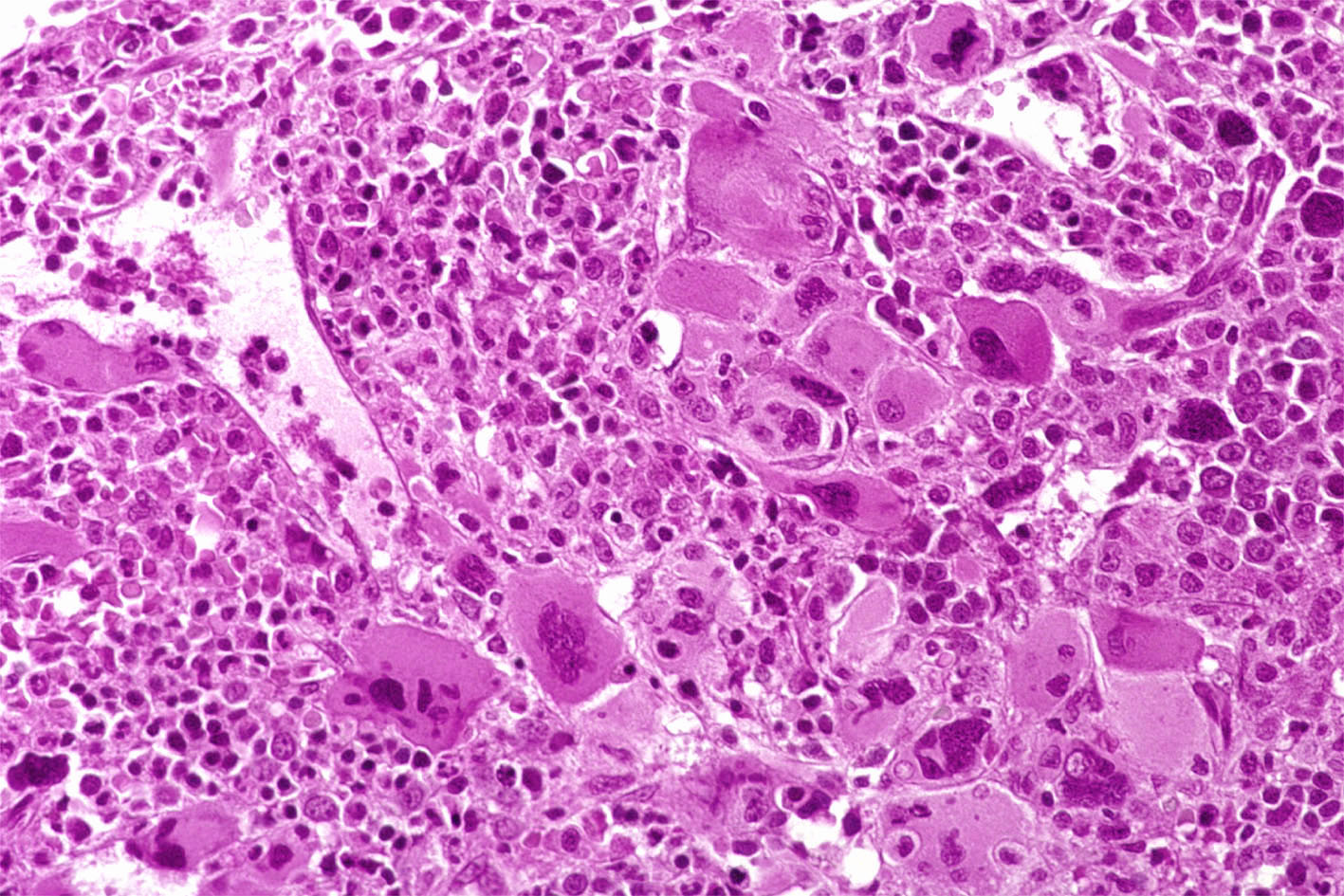 Стремясь улучшить результаты трансплантации стволовых клеток для пациентов с МФ, MPNRF создала Инструмент выбора времени для трансплантации стволовых клеток. SSTT — это портативный онлайн-инструмент, основанный на клинически подтвержденной шкале. Он обеспечивает цветной сигнал в ответ на информацию, введенную пациентом, который указывает уровень риска и среднее время выживания без трансплантации стволовых клеток. Затем пациент может взять то, что он узнал, и использовать это для облегчения конструктивного диалога между собой и своим врачом о вариантах лечения.
Стремясь улучшить результаты трансплантации стволовых клеток для пациентов с МФ, MPNRF создала Инструмент выбора времени для трансплантации стволовых клеток. SSTT — это портативный онлайн-инструмент, основанный на клинически подтвержденной шкале. Он обеспечивает цветной сигнал в ответ на информацию, введенную пациентом, который указывает уровень риска и среднее время выживания без трансплантации стволовых клеток. Затем пациент может взять то, что он узнал, и использовать это для облегчения конструктивного диалога между собой и своим врачом о вариантах лечения.
Инструмент
SCT Spectrum Transplant Timing доступен бесплатно на мобильном телефоне, планшете и ПК. Нажмите здесь , чтобы узнать больше и оценить свой уровень риска сегодня.
Лечение конкретных симптомов
Во многих случаях терапия пациентов с МФ нацелена на определенные симптомы. Эти признаки и связанные с ними методы лечения могут включать:
- Анемию можно лечить кортикостероидами, андрогенами (включая даназол и галотестин), талидомидом, леналидомидом, переливанием крови или средствами, стимулирующими эритропоэз (ESAs). В настоящее время проходят клинические испытания некоторые препараты, направленные на лечение анемии у людей с миелофиброзом.
- Спленомегалию можно лечить с помощью Джакафи, гидроксимочевины (HU), кладибрина, интерферона или, в тяжелых случаях, когда лекарственная терапия не принесла результатов, лучевой терапией или спленэктомией.
- Риск тромбоза можно контролировать с помощью низких доз аспирина или гидроксимочевины.
- Экстрамедуллярное кроветворение вне печени и селезенки можно лечить с помощью лучевой терапии.
- Конституциональные симптомы, такие как ночная потливость, кожный зуд, потеря веса и лихорадка, можно лечить с помощью Джакафи.
 В настоящее время проходят клинические испытания некоторые препараты, направленные на лечение анемии у людей с миелофиброзом.
В настоящее время проходят клинические испытания некоторые препараты, направленные на лечение анемии у людей с миелофиброзом.Новые подходы и клинические испытания
Для многих пациентов с МФ доступные подходы к лечению могут быть неэффективными, и подходящим вариантом может быть экспериментальное лечение (которое включает получение нового лекарства или лечения в рамках клинических испытаний).
На данный момент только Jakafi (руксолитиниб) был одобрен FDA для терапии MF. Но в настоящее время проходят клинические испытания другие новые методы лечения, включая несколько механизмов действия:
Но в настоящее время проходят клинические испытания другие новые методы лечения, включая несколько механизмов действия:
Ингибиторы JAK
Ряд других препаратов, ингибирующих JAK2 («ингибиторы JAK»), в настоящее время проходят клинические испытания, включая момелотиниб, пакртиниб и NS-018. Результаты этих исследований определят, могут ли эти препараты также быть эффективными для лечения пациентов с МФ.
Эпигенетические препараты
Эпигенетические препараты изменяют способ организации генов, делая их более или менее доступными для использования клеткой.Недавние исследования эпигенетических препаратов показали, что ингибитор HDAC, гивиностат, и два гипометилирующих агента, азацитидин и децитабин, были минимально эффективными при лечении MF в ранних исследованиях (в отличие от их эффективности при лечении PV). Другой ингибитор HDAC, панобиностат, находится в стадии изучения.
Помалидомид
В ранних исследованиях было показано, что помалидомид эффективно лечит анемию. Он нацелен на иммунную систему пациента, чтобы атаковать аномальные клетки, чтобы освободить место для нормальных клеток, вырабатывающих эритроциты.Обладая повышенной противораковой активностью и меньшей токсичностью по сравнению с другими препаратами этого класса, помалидомид показал себя многообещающим в первоначальных исследованиях и сейчас проходит 3 фазу клинических испытаний для его использования в качестве терапевтического средства первой линии для лечения анемии у пациентов с MF, у которых есть V617F. мутация.
Он нацелен на иммунную систему пациента, чтобы атаковать аномальные клетки, чтобы освободить место для нормальных клеток, вырабатывающих эритроциты.Обладая повышенной противораковой активностью и меньшей токсичностью по сравнению с другими препаратами этого класса, помалидомид показал себя многообещающим в первоначальных исследованиях и сейчас проходит 3 фазу клинических испытаний для его использования в качестве терапевтического средства первой линии для лечения анемии у пациентов с MF, у которых есть V617F. мутация.
Эверолимус
Эверолимус (также известный как RAD001) является ингибитором пути mTOR / AKT, который очень активен в клетках, продуцирующих кровь MF и, по-видимому, способствует аномальному росту клеток.В фазе 1 и 2 клинических испытаний эверолимус хорошо переносился и уменьшал как размер селезенки, так и системные симптомы.
Готовы изменить свой прогноз?
В Исследовательском фонде MPN мы занимаемся защитой, обучением и поддержкой пациентов с МФ и их семей. Подпишитесь, чтобы получать последние новости и обновления о лечении миелофиброза и других темах, и давайте вместе изменим ваш прогноз MF.
Подпишитесь, чтобы получать последние новости и обновления о лечении миелофиброза и других темах, и давайте вместе изменим ваш прогноз MF.
Первичный миелофиброз — NORD (Национальная организация по редким заболеваниям)
Некоторым людям с первичным миелофиброзом была проведена трансплантация аллогенных или аутологичных стволовых клеток.Стволовые клетки — это особые клетки, обнаруженные в костном мозге, которые производят различные типы клеток крови (например, эритроциты, тромбоциты). При трансплантации аутологичных стволовых клеток стволовые клетки пораженного человека удаляются после предварительного лечения, обычно с помощью лекарств. Эти здоровые стволовые клетки позже повторно вводятся в костный мозг после того, как заболевание прогрессирует. При аллогенной трансплантации стволовых клеток стволовые клетки передаются от другого человека, обычно от близкого родственника.
Трансплантаты стволовых клеток обладают потенциалом восстановления нормальной функции костного мозга у людей с первичным миелофиброзом.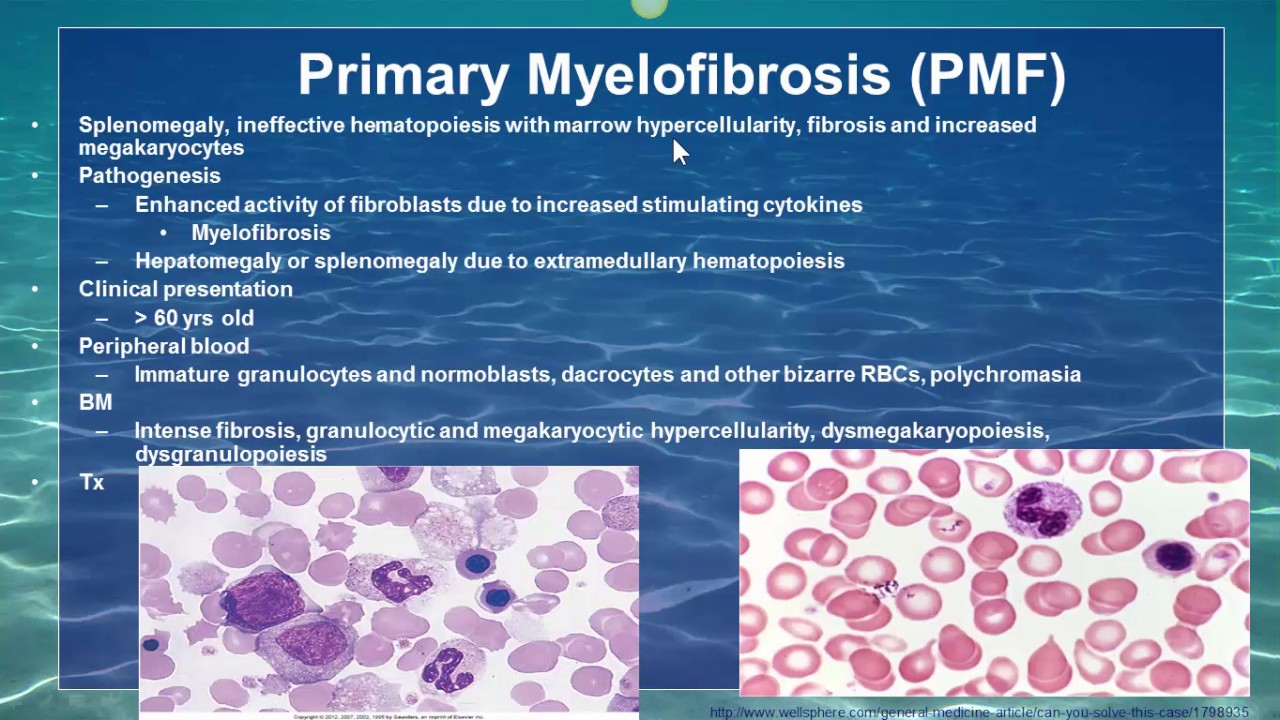 Однако, поскольку трансплантация стволовых клеток может вызвать серьезные, даже опасные для жизни осложнения, они обычно предназначены для молодых людей или людей, у которых нет других жизнеспособных вариантов лечения.
Однако, поскольку трансплантация стволовых клеток может вызвать серьезные, даже опасные для жизни осложнения, они обычно предназначены для молодых людей или людей, у которых нет других жизнеспособных вариантов лечения.
Интерферон-альфа — это лекарство, которое использовалось для лечения людей с первичным миелофиброзом. Интерферон-альфа эффективен в снижении выработки клеток крови костным мозгом. Кроме того, интерферон-альфа задерживает образование фиброзной ткани (фиброза) в костном мозге у некоторых людей с первичным миелофиброзом, которые получали лечение на ранней стадии развития заболевания.Необходимы дополнительные исследования для определения долгосрочной безопасности и эффективности альфа-интерферона для лечения первичного миелофиброза.
Исследователи вкладывают средства в использование талидомида для лечения людей с первичным миелофиброзом. Талидомид используется для лечения рака крови, известного как множественная миелома. Препарат показал себя многообещающим в улучшении некоторых симптомов, связанных с первичным миелофиброзом. Однако талидомид может вызывать определенные побочные эффекты или нежелательные эффекты, такие как увеличение количества лейкоцитов и тромбоцитов.
Однако талидомид может вызывать определенные побочные эффекты или нежелательные эффекты, такие как увеличение количества лейкоцитов и тромбоцитов.
Леналидомид относится к тому же классу препаратов, что и талидомид, но он более эффективен и обычно вызывает меньшее количество побочных эффектов. Исследования показали, что леналидомид очень эффективен при лечении аномалий периферической крови и костного мозга у некоторых людей с первичным миелофиброзом. Исследователи изучают леналидомид как одноразовое средство, средство или в комбинации с кортикостероидами. Необходимы дополнительные исследования для определения долгосрочной безопасности и эффективности леналидомида или талидомида в качестве потенциальных средств лечения людей с первичным миелофиброзом.
Дополнительные препараты, включая иматиниб, этанерцепт и бортезомиб, изучаются для использования при лечении лиц с первичным миелофиброзом. Исследователи также изучают методы сдерживания или блокирования активности фермента, продуцируемого геном JAK2 , который мутирован примерно у 50 процентов людей с первичным миелофиброзом. Исследователи считают, что этот фермент может играть роль в избыточном производстве клеток крови.
Исследователи считают, что этот фермент может играть роль в избыточном производстве клеток крови.
Информация о текущих клинических исследованиях размещена в Интернете по адресу https: // Clinicaltrials.gov /. Все исследования, финансируемые правительством США, а некоторые исследования поддерживаются частным сектором, размещены на этом правительственном веб-сайте.
Для получения информации о клинических испытаниях, проводимых в Клиническом центре NIH в Бетесде, штат Мэриленд, свяжитесь с отделом приема пациентов NIH:
(бесплатно): (800) 411-1222
TTY: (866) 411-1010
Электронная почта: [электронная почта защищена]
Некоторые текущие клинические испытания также размещены на следующей странице веб-сайта NORD:
https: // rarediseases.org / для-пациентов-и-семей / информационных-ресурсов / информация-клинические-испытания-и-исследования-исследования /
Для получения информации о клинических испытаниях, спонсируемых частными источниками, обращайтесь:
http://www. centerwatch.com /
centerwatch.com /
Для получения информации о клинических испытаниях, проведенных в Европе, обращайтесь:
https://www.clinicaltrialsregister.eu/
Контактные данные для получения дополнительной информации о первичном миелофиброзе:
Ayalew Tefferi, MD
Профессор медицины и гематологии
Отделение Гематология
Клиника Мэйо Центр трансплантологии
Клиника Мэйо
Рочестер, Миннесота
507-538-3270
[адрес электронной почты защищен]
Первичный миелофиброз — гематология и онкология
Иногда трансплантация аллогенных стволовых клеток
Иногда руксолитиниб или федратиниб
Лечение направлено на устранение симптомов и осложнений.Некоторых пациентов можно наблюдать без лечения.
Было показано, что при раннем первичном миелофиброзе пегилированный интерферон снижает фиброз костного мозга и размер селезенки и может использоваться для пациентов с низким риском.
В настоящее время при запущенном первичном миелофиброзе препаратом выбора является неспецифический ингибитор пути JAK руксолитиниб. Руксолитиниб эффективен независимо от наличия мутации JAK2 или спленомегалии. Основными побочными эффектами руксолитиниба являются анемия и тромбоцитопения.При отмене руксолитиниба необходимо соблюдать осторожность, поскольку может возникнуть синдром отмены со значительным ухудшением симптомов, частично из-за увеличения селезенки и восстановления воспалительных цитокинов. Кортикостероиды в низких дозах можно использовать в краткосрочной перспективе для контроля симптомов.
Федратиниб, также являющийся ингибитором JAK, может использоваться при наличии устойчивости или непереносимости руксолитиниба. Некоторые пациенты, у которых развивается непереносимость руксолитиниба, могут снова переносить его после периода прекращения приема препарата.
Пациентам с запущенным заболеванием может быть полезна трансплантация аллогенных стволовых клеток. Немиелоаблативная трансплантация аллогенных стволовых клеток успешно применяется у пожилых пациентов.
Немиелоаблативная трансплантация аллогенных стволовых клеток успешно применяется у пожилых пациентов.
Андрогены, эритропоэтин, спленэктомия, химиотерапия, талидомид, леналидомид, эмболизация селезенки и лучевая терапия были испытаны в качестве паллиативных средств. Из них низкие дозы талидомида и преднизона могут быть эффективными в борьбе со спленомегалией, анемией, тромбоцитопенией и циркулирующими бластными клетками.Однако другие методы имеют ограниченную эффективность или имеют серьезные побочные эффекты. По возможности следует избегать спленэктомии; Облучение селезенки имеет только временный эффект и может вызвать тяжелую нейтропению и инфекцию.
Первичный миелофиброз: основы практики, патофизиология, этиология
Теффери А. Первичный миелофиброз: обновленная информация о диагностике, стратификации риска и лечении за 2019 год. Ам Дж. Гематол . 2018 декабрь 93 (12): 1551-1560. [Медлайн]. [Полный текст].
[Медлайн]. [Полный текст].
Барози Г. Миелофиброз с миелоидной метаплазией: диагностическое определение и прогностическая классификация для клинических исследований и руководств по лечению. Дж Клин Онкол . 1999 Сентябрь 17 (9): 2954-70. [Медлайн].
Vallespí T, Imbert M, Mecucci C, Preudhomme C, Fenaux P. Диагностика, классификация и цитогенетика миелодиспластических синдромов. Haematologica . 1998, март 83 (3): 258-75. [Медлайн].[Полный текст].
Якобсон Р.Дж., Сало А, Фиалков П.Дж. Агногенная миелоидная метаплазия: клональная пролиферация гемопоэтических стволовых клеток с вторичным миелофиброзом. Кровь . 1978 Февраль 51 (2): 189-94. [Медлайн]. [Полный текст].
Mesa RA, Verstovsek S, Cervantes F, Barosi G, Reilly JT, Dupriez B, et al. Первичный миелофиброз (PMF), истинный миелофиброз после полицитемии (пост-PV MF), пост-эссенциальный миелофиброз тромбоцитемии (post-ET MF), PMF бластной фазы (PMF-BP): терминологический консенсус международной рабочей группы по исследованию и лечению миелофиброза (IWG-MRT). Лейк Рес . 2007 июн. 31 (6): 737-40. [Медлайн].
Первичный миелофиброз (PMF), истинный миелофиброз после полицитемии (пост-PV MF), пост-эссенциальный миелофиброз тромбоцитемии (post-ET MF), PMF бластной фазы (PMF-BP): терминологический консенсус международной рабочей группы по исследованию и лечению миелофиброза (IWG-MRT). Лейк Рес . 2007 июн. 31 (6): 737-40. [Медлайн].
Тэффери А., Тиле Дж., Орази А. и др. Предложения и обоснование пересмотра диагностических критериев Всемирной организации здравоохранения для истинной полицитемии, эссенциальной тромбоцитемии и первичного миелофиброза: рекомендации специальной международной группы экспертов. Кровь . 2007 15 августа. 110 (4): 1092-7. [Медлайн]. [Полный текст].
Клампфл Т., Гисслингер Х., Арутюнян А.С., Ниварти Х., Руми Э., Милошевич Дж. Д. и др.Соматические мутации кальретикулина при миелопролиферативных новообразованиях. N Engl J Med . 2013 декабрь 19, 369 (25): 2379-90. [Медлайн].
2013 декабрь 19, 369 (25): 2379-90. [Медлайн].
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Соматические мутации CALR в миелопролиферативных новообразованиях с немутантным JAK2. N Engl J Med . 2013 декабрь 19, 369 (25): 2391-405. [Медлайн]. [Полный текст].
Патель К.П., Ньюберри К.Дж., Лутра Р., Джаббур Э., Пирс С., Кортес Дж. И др.Корреляция профиля мутации и ответа у пациентов с миелофиброзом, получавших руксолитиниб. Кровь . 2015 6 августа. 126 (6): 790-7. [Медлайн]. [Полный текст].
Honda Y, Delzell E, Cole P. Обновленное исследование смертности среди рабочих на нефтеперерабатывающем заводе. Дж. Оккуп Энвирон Мед . 1995 Февраль 37 (2): 194-200. [Медлайн].
Ху Х. Миелофиброз, связанный с бензолом [письмо]. Энн Интерн Мед. .1987, январь, 106 (1): 171-2. [Медлайн].
Энн Интерн Мед. .1987, январь, 106 (1): 171-2. [Медлайн].
Visfeldt J, Andersson M. Патологоанатомические аспекты злокачественных гематологических заболеваний у датских пациентов, подвергшихся воздействию диоксида тория. АПМИС . 1995 Январь 103 (1): 29-36. [Медлайн].
Аксой М., Эрдем С., Динкол Г. Два редких осложнения хронического отравления бензолом: миелоидная метаплазия и пароксизмальная ночная гемоглобинурия. Отчет о двух случаях. Блат . 1975, 30 апреля (4): 255-60.[Медлайн].
Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. Эпидемиология миелофиброза, эссенциальной тромбоцитемии и истинной полицитемии в Европейском Союзе. Eur J Haematol . 2014 Апрель 92 (4): 289-97. [Медлайн].
Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A.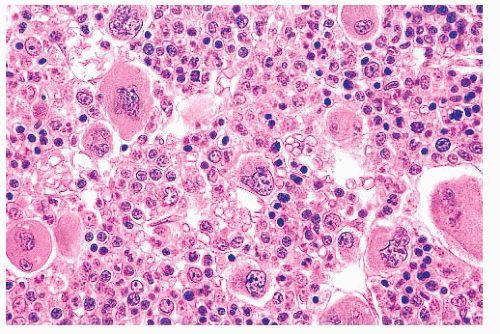 Показатели заболеваемости и выживаемости при эссенциальной тромбоцитемии и агногенной миелоидной метаплазии: исследование округа Олмстед, 1976–1995. Ам Дж. Гематол . 1999 Май. 61 (1): 10-5. [Медлайн]. [Полный текст].
Показатели заболеваемости и выживаемости при эссенциальной тромбоцитемии и агногенной миелоидной метаплазии: исследование округа Олмстед, 1976–1995. Ам Дж. Гематол . 1999 Май. 61 (1): 10-5. [Медлайн]. [Полный текст].
Tefferi A, Guglielmelli P, Lasho TL, Rotunno G, Finke C, Mannarelli C и др. Молекулярное прогнозирование первичного миелофиброза на основе мутаций CALR и ASXL1: международное исследование с участием 570 пациентов. Лейкемия . 2014 июл.28 (7): 1494-500. [Медлайн].
Розовски Ю., Верстовсек С., Маншури Т., Дембиц В., Божинович К., Ньюберри К. и др. Точная и простая прогностическая модель, состоящая из возраста, JAK2, CALR и статуса мутации MPL для пациентов с первичным миелофиброзом. Haematologica . 2017 Январь 102 (1): 79-84. [Медлайн]. [Полный текст].
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: усовершенствованная динамическая международная прогностическая система оценки первичного миелофиброза, которая включает прогностическую информацию о кариотипе, количестве тромбоцитов и статусе переливания крови. Дж Клин Онкол . 2011 г., 1 февраля. 29 (4): 392-7. [Медлайн].
DIPSS plus: усовершенствованная динамическая международная прогностическая система оценки первичного миелофиброза, которая включает прогностическую информацию о кариотипе, количестве тромбоцитов и статусе переливания крови. Дж Клин Онкол . 2011 г., 1 февраля. 29 (4): 392-7. [Медлайн].
Tefferi A, Guglielmelli P, Pardanani A, Vannucchi AM. Алгоритм лечения миелофиброза 2018. Рак крови J . 31 июля 2018 г. 8 (8): 72. [Медлайн]. [Полный текст].
Теффери А., Гульелмелли П., Николози М., Маннелли Ф., Мадиредди М., Барталуччи Н. и др. GIPSS: генетически обоснованная прогностическая система оценки первичного миелофиброза. Лейкемия . 2018 июл.32 (7): 1631-1642. [Медлайн]. [Полный текст].
Левин Р.Л., Белисл С., Уодли М. и др. Анализ клональности на основе X-инактивации и количественная оценка JAK2V617F выявляют сильную связь между клональностью и JAK2V617F в PV, но не ET / MMM, и идентифицируют подмножество JAK2V617F-негативных пациентов с ET и MMM с клональным гематопоэзом. Кровь . 2006 15 мая. 107 (10): 4139-41. [Медлайн]. [Полный текст].
Кровь . 2006 15 мая. 107 (10): 4139-41. [Медлайн]. [Полный текст].
Бакстер Э.Дж., Скотт Л.М., Кэмпбелл П.Дж. и др. Приобретенная мутация тирозинкиназы JAK2 при миелопролиферативных заболеваниях человека. Ланцет . 2005 19-25 марта. 365 (9464): 1054-61. [Медлайн].
Barosi G, Marchetti M, Massa M et al. Частота и клинический профиль мутации JAK2 V617F при миелофиброзе с миелоидной метаплазией [аннотация]. Кровь .Ноябрь 2005. 106: a256. [Полный текст].
Barosi G и др., Международная рабочая группа по исследованию и лечению миелофиброза (IWG-MRT). Предлагаемые критерии диагностики пост-полицитемии истинной и пост-эссенциальной тромбоцитемии миелофиброза: согласованное заявление Международной рабочей группы по исследованию и лечению миелофиброза. Лейкемия . 2008 22 февраля (2): 437-8. [Медлайн].
[Медлайн].
[Рекомендации] NCCN Клинические рекомендации по онкологии: миелопролиферативные новообразования.Национальная всеобъемлющая онкологическая сеть. Доступно на https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf. Версия 1.2020 — 21 мая 2020 г .; Доступ: 2 февраля 2021 г.
Kröger NM, Deeg JH, Olavarria E, Niederwieser D, Bacigalupo A, et al. Показания и лечение аллогенной трансплантации стволовых клеток при первичном миелофиброзе: согласованный процесс международной рабочей группы EBMT / ELN. Лейкемия . 2015 29 ноября (11): 2126-33. [Медлайн].
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al.Двойное слепое плацебо-контролируемое испытание руксолитиниба при миелофиброзе. N Engl J Med . 2012 г., 1. 366 (9): 799-807. [Медлайн].
 webmd.com»> Харрисон С., Киладжян Дж. Дж., Аль-Али Х. К., Гисслингер Х., Вальцман Р., Сталбовская В. и др. Ингибирование JAK руксолитинибом по сравнению с лучшими доступными методами лечения миелофиброза. N Engl J Med . 2012 г., 1. 366 (9): 787-98. [Медлайн].
webmd.com»> Харрисон С., Киладжян Дж. Дж., Аль-Али Х. К., Гисслингер Х., Вальцман Р., Сталбовская В. и др. Ингибирование JAK руксолитинибом по сравнению с лучшими доступными методами лечения миелофиброза. N Engl J Med . 2012 г., 1. 366 (9): 787-98. [Медлайн].Парданани А., Харрисон С., Кортес Дж. Э., Сервантес Ф., Меса Р. А., Миллиган Д. и др.Безопасность и эффективность федратиниба у пациентов с первичным или вторичным миелофиброзом: рандомизированное клиническое испытание. JAMA Oncol . 2015 августа 1 (5): 643-51. [Медлайн].
Гилберт Х.С. Долгосрочное лечение миелопролиферативного заболевания интерфероном-альфа-2b: возможность и эффективность. Рак . 1998 15 сентября. 83 (6): 1205-13. [Медлайн]. [Полный текст].
Меса Р.А., Стинсма Д.П., Парданани А. и др. Испытание фазы 2 комбинации низких доз талидомида и преднизона для лечения миелофиброза с миелоидной метаплазией. Кровь . 2003 г., 1. 101 (7): 2534-41. [Медлайн]. [Полный текст].
Кровь . 2003 г., 1. 101 (7): 2534-41. [Медлайн]. [Полный текст].
Merup M, Kutti J, Birgergard G, et al. Незначительные клинические эффекты талидомида у пациентов с миелофиброзом с миелоидной метаплазией. Мед Онкол . 2002. 19 (2): 79-86. [Медлайн].
Elliott MA, Mesa RA, Li CY, et al. Лечение талидомидом при миелофиброзе с миелоидной метаплазией. Br J Haematol . 2002 май. 117 (2): 288-96. [Медлайн].
Оденике О., Ховинг К., Шер Д. и др. Фаза II исследования мезилата иматиниба (IM) при миелофиброзе с миелоидной метаплазией (MMM) [аннотация]. Proc Am Soc Clin Oncol . 2003. 22: a2354.
Поццато Г., Зорат Ф., Наскимбен Ф. и др. Терапия талидомидом при компенсированном и декомпенсированном миелофиброзе с миелоидной метаплазией..jpg) Haematologica . 2001 июль 86 (7): 772-3. [Медлайн]. [Полный текст].
Haematologica . 2001 июль 86 (7): 772-3. [Медлайн]. [Полный текст].
Canepa L, Ballerini F, Varaldo R, et al.Талидомид при агногенном и вторичном миелофиброзе. Br J Haematol . 2001 ноябрь 115 (2): 313-5. [Медлайн].
Барози Г., Гросси А., Комотти Б. и др. Безопасность и эффективность талидомида у пациентов с миелофиброзом с миелоидной метаплазией. Br J Haematol . 2001 июль 114 (1): 78-83. [Медлайн].
Теффери А., Эллиот Массачусетс. Серьезные миелопролиферативные реакции, связанные с применением талидомида при миелофиброзе с миелоидной метаплазией [письмо]. Кровь . 2000 декабрь 1. 96 (12): 4007. [Медлайн]. [Полный текст].
Тэффери А., Кортес Дж., Верстовсек С. и др. Терапия леналидомидом при миелофиброзе с миелоидной метаплазией. Кровь . 2006 15 августа. 108 (4): 1158-64. [Медлайн]. [Полный текст].
Кровь . 2006 15 августа. 108 (4): 1158-64. [Медлайн]. [Полный текст].
Mesa RA, Tefferi A, Gray LA, et al. Антипролиферативная активность ингибитора фарнезилтрансферазы R115777 in vitro в гемопоэтических предшественниках пациентов с миелофиброзом с миелоидной метаплазией. Лейкемия . 2003 май. 17 (5): 849-55. [Медлайн]. [Полный текст].
Кортес Дж., Альбитар М., Томас Д. и др. Эффективность ингибитора фарнезилтрансферазы R115777 при хроническом миелоидном лейкозе и других гематологических злокачественных новообразованиях. Кровь . 1 марта 2003 г. 101 (5): 1692-7. [Медлайн]. [Полный текст].
Giles FJ, List AF, Carroll M, et al. PTK787 / ZK 222584, низкомолекулярный ингибитор тирозинкиназного рецептора фактора роста эндотелия сосудов (VEGF), обладает умеренной активностью при миелофиброзе с миелоидной метаплазией. Лейк Рес . 2007 июля 31 (7): 891-7. [Медлайн].
Джайлз Ф.Дж., Купер М.А., Сильверман Л. и др. Фаза II исследования SU5416 — низкомолекулярного ингибитора рецептора тирозинкиназы фактора роста эндотелия сосудов — у пациентов с рефрактерными миелопролиферативными заболеваниями. Рак . 2003 г. 15 апреля. 97 (8): 1920-8. [Медлайн]. [Полный текст].
Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM, et al. Экспериментальное исследование ингибитора теломеразы Иметелстат при миелофиброзе. N Engl J Med . 2015 3 сентября. 373 (10): 908-19. [Медлайн]. [Полный текст].
Tefferi A, Verstovsek S, Barosi G, et al. Помалидомид активен при лечении анемии, связанной с миелофиброзом. Дж Клин Онкол . 2009 20 сентября. 27 (27): 4563-9. [Медлайн].
Guglielmelli P, Barosi G, Rambaldi A, et al. Безопасность и эффективность эверолимуса, ингибитора mTOR, в качестве единственного агента в исследовании 1/2 фазы у пациентов с миелофиброзом. Кровь . 2011 25 августа. 118 (8): 2069-76. [Медлайн].
Koeffler HP, Cline MJ, Golde DW. Облучение селезенки при миелофиброзе: влияние на циркулирующие миелоидные клетки-предшественники. Br J Haematol . 1979 Сентябрь 43 (1): 69-77. [Медлайн].
Robin M, de Wreede LC, Wolschke C, et al. Отдаленный исход после трансплантации аллогенных гемопоэтических клеток по поводу миелофиброза. Haematologica . 2019 7 февраля [Medline]. [Полный текст].
Крегер Н., Джорджино Т., Скотт Б.Л., Дичковски М., Алчалби Н., Сервантес Ф. и др. Влияние трансплантации аллогенных стволовых клеток на выживаемость пациентов младше 65 лет с первичным миелофиброзом. Кровь . 2015 21 мая. 125 (21): 3347-50; викторина 3364. [Medline]. [Полный текст].
Салит РБ, Скотт Б.Л., Стивенс Э.А., Бейкер К.К., Гули Т.А., Диг Х.Дж. Трансплантация прегематопоэтических клеток Руксолитиниб у пациентов с первичным и вторичным миелофиброзом. Пересадка костного мозга . 2019 8 апреля [Medline].
[Рекомендации] Ваннуччи А.М., Барбуи Т., Сервантес Ф., Харрисон С., Киладжан Дж. Дж., Крегер Н. и др. Филадельфийские хромосомно-отрицательные хронические миелопролиферативные новообразования: Клинические практические рекомендации ESMO по диагностике, лечению и последующему наблюдению. Энн Онкол . 2015 26 сентября Приложение 5: v85-99. [Медлайн]. [Полный текст].
Первичный миелофиброз / агногенная миелоидная метаплазия — Консультант по терапии рака
Первичный миелофиброз / агногенная первичная миелоидная метаплазия
Что нужно знать каждому врачу:
Первичный миелофиброз (ПМФ) — хроническое злокачественное гематологическое заболевание, принадлежащее к семейству миелопролиферативных новообразований, отрицательных по филадельфийским хромосомам.
Характеризуется спленомегалией, лейкоэритробластической картиной периферического мазка, фиброзом костного мозга, цитопенией и системными симптомами.
Примерно 60% пациентов несут мутацию гена Jak2 V617F; 8% имеют активирующую мутацию гена тромбопоэтина MPL; 20% имеют вставки или делеции экзона 9 кальретикулина (CALR); а у оставшихся 10% пациентов каждая из этих мутаций отсутствует, и их заболевание считается тройным отрицательным.
Вы уверены, что у вашего пациента первичный миелофиброз? Что вы должны ожидать найти?
Диагноз PMF ставится на основании ряда клинических и лабораторных особенностей, но клинические проявления могут сильно варьироваться.
Наиболее частая жалоба — утомляемость. Несмотря на то, что часто оказывается результатом анемии, утомляемость может стать серьезным бременем даже для пациентов, не страдающих анемией. У большинства пациентов также наблюдается спленомегалия. Если спленомегалия значительна, она может привести к дискомфорту в левом верхнем квадранте живота, боли в левом плече, раннему насыщению и диарее, которые часто являются основными жалобами. У пациентов с более запущенным заболеванием часто наблюдаются системные симптомы, такие как потеря веса, боли в костях, ночная потливость и лихорадка.
Пациенты также могут иметь анемию различной степени. Представленные количества тромбоцитов и лейкоцитов также демонстрируют вариабельность, у пациентов наблюдаются как тромбоцитоз, так и тромбоцитопения, а также лейкоцитоз и лейкопения. Незрелые клетки нейтрофильного ряда всегда обнаруживаются в периферической крови, в том числе в миелобластах. Пациенты также подвергаются повышенному риску тромботических или геморрагических событий.
Тромбоцитопения обычно связана с более поздними стадиями этого заболевания.
В настоящее время Всемирная организация здравоохранения (ВОЗ) предложила следующие критерии диагностики PMF.
Основные критерии:
Наличие пролиферации и атипии мегакариоцитов, обычно сопровождающиеся ретикулиновым и / или коллагеновым фиброзом
Не соответствует критериям ВОЗ для истинной полицитемии (PV), хронического миелогенного лейкоза (CML), миелодиспластических синдромов (MDS) или других миелоидных новообразований
Демонстрация мутации Jak2 V617F или другого клонального маркера, такого как MPL515W-> L / K, или мутации CALR в отсутствие клонального маркера, отсутствие признаков фиброза костного мозга, вызванного воспалительными или другими неопластическими заболеваниями
Второстепенные критерии:
Диагностика требует соответствия всем 3 основным и как минимум 2 второстепенным критериям.
Остерегайтесь других состояний, которые могут имитировать первичный миелофиброз:
PMF следует отличать от других хронических миелопролиферативных новообразований. Миелодиспластический синдром, ХМЛ, заболевания тучных клеток и волосисто-клеточный лейкоз могут сопровождаться фиброзом костного мозга и должны быть исключены до постановки диагноза PMF.
Острый миелофиброз — это синдром, характеризующийся острым проявлением фиброза костного мозга, лихорадкой, панцитопенией и минимальным каплевидным пойкилоцитозом, а также отсутствием спленомегалии.Для костного мозга характерно появление незрелых миелоидных клеток и бластных клеток, которые часто экспрессируют мегакариоцитарные маркеры. Различие между острым миелофиброзом и PMF имеет важное значение, поскольку острый миелофиброз лечится с помощью агрессивной химиотерапии, за которой немедленно следует трансплантация стволовых клеток, что имеет мрачный прогноз.
Многие другие заболевания могут вызывать фиброз костного мозга и имитировать клинические проявления PMF. К ним относятся незлокачественные состояния, такие как туберкулез, гипопаратиреоз, системная красная волчанка (СКВ), склеродермия, болезнь Гоше и первичный аутоиммунный миелофиброз, а также другие злокачественные заболевания, такие как острый миелогенный лейкоз, множественная миелома, болезнь Ходжкина и немфкинома. волосатоклеточный лейкоз.Вторичный миелофиброз также может возникать у пациентов с метастатической карциномой в костный мозг.
Пациентов с другим миелопролиферативным новообразованием, эссенциальной тромбоцитемией, иногда можно спутать с ранними стадиями PMF, поскольку оба состояния связаны с тромбоцитозом. Некоторые, но не все, считают тщательное гистопатологическое исследование биопсии костного мозга эффективным в различении этих двух заболеваний. Префибротическая форма миелофиброза нередко переходит в более поздние стадии заболевания.Интерферон альфа был предложен в качестве лечения ранней фазы PMF, но не оценивался в строгих рандомизированных клинических испытаниях.
Какие люди наиболее подвержены риску развития первичного миелофиброза:
PMF — редкое заболевание, чаще всего поражающее пожилых людей. Средний возраст постановки диагноза PMF составляет примерно 65 лет, и большинству пациентов ставится диагноз от 50 до 69 лет. Примерно 5% пациентов получают диагноз до 40 лет.Редко сообщалось о заболевании в детской возрастной группе. Хотя это заболевание встречается редко, более высокая заболеваемость отмечена у евреев-ашкенази.
Считается, что годовая заболеваемость колеблется от 0,4 до 1,3 случая на 100 000 человек. Заболеваемость миелофиброзом среди выживших после взрыва атомной бомбы в Хиросиме была в 18 раз выше, чем в остальной части Японии, что свидетельствует о связи между чрезмерным радиационным облучением и развитием миелофиброза.Хроническое воздействие нескольких промышленных растворителей, таких как бензол и толуол, также связано с развитием PMF. Однако у большинства пациентов с диагнозом этого расстройства нет явного воздействия окружающей среды.
Какие лабораторные исследования следует заказать для постановки диагноза и как интерпретировать результаты?
Для диагностики и прогнозирования PMF показаны следующие лабораторные исследования:
Стандартные анализы крови: общий анализ крови, электролиты, функциональные пробы печени и почек, лактатдегидрогеназа, мочевая кислота, исследования железа, B12 и фолиевой кислоты, количество ретикулоцитов, уровень эритропоэтина, тиреотропный гормон
Специализированные анализы крови: полимеразная цепная реакция (ПЦР) на Jak2V617F, мутации рецептора тромбопоэтина (MPL W515L / K), BCR-Abl, мутационные анализы CALR и цитогенетические анализы с использованием периферической крови для выявления клональных цитогенетических аномалий
Инвазивные процедуры: биопсия и аспирация костного мозга
Тщательное исследование мазка периферической крови и костного мозга имеет важное значение для диагностики PMF.Лейкоэритробластоз с каплевидными эритроцитами убедительно свидетельствует об этом диагнозе. Лейкоэритробластическое состояние характеризуется наличием ядерных эритроцитов и незрелых миелоидных элементов в 96% случаев.
Анемия с гемоглобином менее 10 г / дл развивается примерно у 60% пациентов. Анемия часто бывает многофакторной по происхождению. Лейкопения встречается у 13–25% пациентов, а лейкоцитоз — у одной трети. Тромбоцитопения становится более частой по мере прогрессирования болезни.Тромбоциты могут быть аномально большими, и на периферическом мазке часто можно увидеть фрагментированные мегакариоциты.
Наличие мутации Jak2V617F, MPL W515L / K или мутации CALR являются клональными маркерами, которые помогают подтвердить наличие миелопролиферативного новообразования. Одна из этих мутаций присутствует примерно у 90% пациентов с PMF. Также распространены дополнительные лабораторные отклонения, такие как повышение уровня мочевой кислоты, билирубина, лактатдегидрогеназы и щелочной фосфатазы. Уровень B12 часто повышен, а уровень фолиевой кислоты может быть низким из-за быстрого обмена клеток.
Успешная аспирация костного мозга необычна: в 80% случаев происходит сухая пункция. Во всех случаях необходима биопсия костного мозга, необходимо оценить остаточную кроветворную активность и фиброз костного мозга. Фиброз обычно связан с атипичной гиперплазией мегакариоцитов и остеосклерозом. Цитогенетика костного мозга или периферической крови является важной частью обследования. Пациенты с цитогенетическими аномалиями имеют худший прогноз.
Какие визуализационные исследования (если таковые имеются) будут полезны для постановки или исключения диагноза первичного миелофиброза?
Ультрасонография или компьютерная томография могут быть полезны для документирования степени сплемомегалии у тех пациентов, у которых физическое обследование затруднено.
Если вы решите, что у пациента первичный миелофиброз, какие методы лечения вам следует начать немедленно?
В большинстве случаев PMF не требуется экстренного лечения PMF, поскольку это хроническое медленно прогрессирующее заболевание. При наличии острого тромботического события, кровотечения или других осложнений заболевания может потребоваться специальное лечение этого осложнения.
Более точные методы лечения?
Оптимальное лечение PMF еще не определено.
Консервативный подход к лечению обычно принимается при тщательном наблюдении и лечении пациентов с симптомами. Активная терапия обычно показана при следующих состояниях: симптоматическая анемия, симптоматическая спленомегалия, портальная гипертензия, значительная тромбоцитопения, системные симптомы, такие как боль в костях, лихорадка, ночная потливость и потеря веса.
Трансплантация аллогенных стволовых клеток — единственный вариант лечения в настоящее время, но она предлагается пациентам моложе 70 лет с подходящим родственным братом-донором (положительный результат 70%).Успешные результаты с подобранными неродственными донорами встречаются реже (30%). Использование пуповинной крови или гаплоидентичных доноров остается экспериментальным. Успешная трансплантация в конечном итоге приводит к разрешению фиброза костного мозга и нормализации кроветворения.
С момента установления в 2005 г. роли JAK2V617F в патогенезе миелопролиферативных новообразований, отрицательных по филадельфийской хромосоме, был разработан и оценен в клинических испытаниях ряд ингибиторов JAK2. Многие из этих испытаний все еще продолжаются.Первый из этих препаратов был одобрен Управлением по контролю за продуктами и лекарствами (FDA) для лечения пациентов с миелофиброзом. Руксолитиниб (Jakafi) показан пациентам с миелофиброзом среднего или высокого риска и является отличным паллиативным средством, которое не может вызвать клинико-патологическую ремиссию. Было показано, что руксолитин приводит к снижению степени спленомегалии, улучшению системных симптомов и улучшению качества жизни у этих пациентов. Однако он не отменяет прогрессирование фиброза костного мозга, не снижает нагрузку аллеля JAK2V617F и не предотвращает клональную эволюцию до острого миелоидного лейкоза.Использование руксолитиниба может быть ограничено из-за связанной с лечением анемии и тромбоцитопении
Все другие терапевтические методы также носят паллиативный характер и включают андрогенные препараты и комбинации талидомида или леналидомида и преднизона для лечения анемии и гидроксимочевины для уменьшения спленомегалии и системных симптомов. Мелфалан и бусульфан применялись у пациентов с гиперпролиферативной фазой заболевания и могут привести к стабилизации степени лейкоцитоза и уменьшению спленомегалии.
Спленэктомия — это потенциальный метод выбора для пациентов с массивной спленомегалией, которые не реагируют или не переносят медикаментозное лечение, или у пациентов с тяжелой опасной для жизни тромбоцитопенией и анемией. Его следует рассматривать у тех пациентов, у которых после терапии руксолитинибом развивается ограничивающая дозу тромбоцитопения или которые не являются кандидатами на лечение руксолитинибом из-за исходной тяжелой цитопении. В опытных центрах смертность от этой процедуры составляет менее 10%, а частота ответа превышает 90% для облегчения абдоминальных симптомов из-за массивной спленомегалии и примерно 60% для улучшения тромбоцитопении и анемии.
В связи с хроническим и прогрессирующим характером этого злокачественного новообразования гемопоэтических стволовых клеток и отсутствием одобренных FDA методов лечения, изменяющих курс заболевания, настоятельно рекомендуется направлять пациентов с PMF, когда это возможно, в специализированные центры с программами MPN и рассматривать их для клинических испытаний. зачисление.
Какие другие методы лечения помогают уменьшить осложнения?
Пациентам может быть оказана поддерживающая терапия, при необходимости они получают переливание тромбоцитов и эритроцитов.Пациентам с анемией как единственной аномалией и низким уровнем эндогенного эритропоэтина в течение короткого периода времени могут быть полезны средства, стимулирующие эритропоэз.
Пациентам с PMF часто требуется длительная трансфузионная терапия, в результате чего может развиться синдром перегрузки железом. В этой ситуации можно использовать хелатирующие агенты с железом, но они используются редко из-за ограниченной выживаемости пациентов с запущенными формами PMF.
Что вы должны сказать пациенту и его семье о прогнозе?
PMF — это заболевание с высокой вариабельностью исходов в зависимости от степени распространенности заболевания.Пациент и его семья должны быть проинформированы о том, что PMF является гематологической злокачественной опухолью. Клиническое течение при постановке диагноза иногда трудно предсказать. Пациенты могут иметь стабильное заболевание в течение многих лет без развития каких-либо осложнений или могут быстро декомпенсироваться из-за развития системных симптомов, прогрессирования до острого миелоидного лейкоза (ОМЛ) или развития тромботических или геморрагических осложнений. Средний период общей выживаемости с момента постановки диагноза составляет примерно 6-7 лет.Заболеваемость острым лейкозом как терминальным событием колеблется от 5 до 22%. Выживание после взрыва ограничено. В серии из 91 пациента с PMF средняя выживаемость после лейкемической трансформации составила 2,6 месяца. Стандартная индукционная терапия ремиссии редко бывает успешной у этой группы пациентов.
Существует ряд прогностических моделей для оценки прогноза при PMF. Одной из самых последних разработок и широко используемых является система динамической прогностической оценки (DIPSS plus), разработанная Международной рабочей группой по исследованию и лечению миелофиброза (IWG-MRT).
Следующие неблагоприятные прогностические факторы были отмечены после многофакторного анализа:
Наличие конституциональных симптомов
Гемоглобин менее 10 г / дл
Возраст старше 65 лет
Количество лейкоцитов более 25000 / мкл
Более 1% бластных клеток в периферической крови
Количество тромбоцитов менее 100000 / мкл
Неблагоприятный кариотип (сложный кариотип или наличие +8, 7- / 7q-, множественные копии 1q, -5 / 5q-
Необходимость повторных переливаний эритроцитов
Пациенты с 0, 1, 2 или 3, 4 или более из этих 8 факторов в любое время в течение клинического курса имеют прогнозируемую медианную выживаемость 15.4, 6,5, 2,9 и 1,3 года соответственно.
Пациенты с PMF, у которых отсутствуют мутации Jak2V617F, MPL W515L / K и CALR в экзоне 9 (тройной отрицательный), имеют более высокую скорость лейкемической трансформации и более короткую общую выживаемость. Точно так же пациенты с мутацией ASXL1 также имеют более короткую выживаемость.
Напротив, пациенты с мутацией CALR, по-видимому, имеют более вялое клиническое течение, чем пациенты с JAK2 V617F. Мутация кальретикулина связана с более низким риском тромбоза и более длительной общей выживаемостью.
«Что, если» сценариев.
Каков наилучший план лечения, если пациенту назначена трансплантация стволовых клеток?
Трансплантация аллогенных стволовых клеток — единственный способ лечения этого заболевания. Все пациенты и их братья и сестры должны быть типированы по человеческому лейкоцитарному антигену (HLA) при постановке диагноза, чтобы определить, есть ли потенциальное совпадение.
Пациентам в возрасте до 70 лет, с удовлетворительным статусом работоспособности и доступным донором следует рекомендовать пройти обследование на предмет трансплантации стволовых клеток.Несмотря на то, что эта процедура все еще является процедурой с высокой смертностью и заболеваемостью, пациенты, получившие трансплантацию гемопоэтических стволовых клеток от полностью подобранного родственного донора, имеют хорошие шансы на выздоровление: 2-летняя выживаемость составляет около 75%. Напротив, те пациенты, которые получили трансплантат от подобранного неродственного донора, имеют 2-летнюю выживаемость 32%. Разница в результатах в этих двух группах, по-видимому, частично связана с увеличением первичной и вторичной несостоятельности трансплантата в несвязанной группе.
Пациентам с заболеванием высокого или среднего риска, у которых имеются доноры, следует рекомендовать пройти трансплантацию до развития изнурительных симптомов и ухудшения их работоспособности.Пациенты с заболеванием низкого риска или промежуточного уровня 1 должны находиться под тщательным наблюдением без вмешательства, но их следует рассматривать для трансплантации, если болезнь прогрессирует.
Как следует лечить пациентов с тромбоцитопенией?
Пациенты с тяжелой тромбоцитопенией представляют собой серьезную проблему при минимальном выборе лечения. Большинство стандартных методов лечения PMF, таких как руксолитиниб и гидроксимочевина, приводят к дальнейшей миелосупрессии и не могут быть использованы.
Пациенты с количеством тромбоцитов от 50 000 до 75 000 могут иметь право на участие в нескольких клинических испытаниях, таких как некоторые испытания ингибиторов JAK2.Недавно одобренный ингибитор JAK2 руксолитиниб также можно попробовать в низких дозах, если количество тромбоцитов превышает 50 000. Целью этого лечения является уменьшение размера селезенки, что приводит к увеличению количества тромбоцитов.
Для пациентов с количеством тромбоцитов менее 20 000 выбор лечения действительно ограничен. Поддерживающая терапия с переливанием тромбоцитов возможна, но, вероятно, не является устойчивой в долгосрочной перспективе. Около 30-50% пациентов с тяжелой тромбоцитопенией будут иметь значительное улучшение количества тромбоцитов после спленэктомии.Однако спленэктомия у пациентов с PMF связана с частотой послеоперационной заболеваемости 15-30% и смертностью около 10%. Эти цифры, однако, сильно зависят от опыта учреждения и оперирующего хирурга.
Патофизиология
Точный патогенез PMF неизвестен. Полагают, что он возникает в результате соматической мутации в плюрипотентной гемопоэтической стволовой клетке, приводящей к клональной пролиферации и воспалительной цитокиновой среде. Драйверные мутации в рецепторе тирозинкиназы JAK2 (JAK2V617F), тромбопоэтиновом рецепторе (MPL W 515L / L) или кальретикулине были идентифицированы у пациентов с миелопролиферативными новообразованиями.Мутации кальретикулина являются взаимоисключающими с JAK2V617F и MPL W 515L / L.
Переключение нуклеотидов с F на T в экзоне 14 псевдокиназного домена JAK2 продуцирует конститутивно активную тирозинкиназу. Это приводит к цитокин-независимой активации пути JAK / STAT. Конститутивная активация передачи сигналов JAK / STAT, по-видимому, является важным патогенетическим событием в развитии PMF и других миелопролиферативных новообразований.
Все мутации кальретикулина приводят к новому С-концевому пептиду, который содержит минимум 36 аминокислот и заменяет 27 аминокислот, которые утрачены из нормальной последовательности.Кальретикулин — это многофункциональный белок, наиболее известный как кальций-связывающий шаперон в эндоплазматическом ретикулуме. Способ, которым мутации в кальретикулине могут приводить к развитию миелопролиферативного новообразования, остается плохо определенным. Удивительно, но ингибиторы JAK2 являются эффективными препаратами для лечения пациентов с PMF с мутацией кальретикулина.
Было обнаружено, что пациенты с PMF обладают большим количеством других мутаций в генах, таких как TET2, EZh3, IDh2 и IDh3, а также ASXL1.Однако ни один из них не является специфическим для PMF и не проявляет взаимной исключительности. Ясно, что молекулярная генетика PMF очень сложна, и хотя мутации драйвера (JAK2V617F, MPL W515L / K или CALR) могут влиять на фенотип заболевания, они, вероятно, не являются инициирующим генетическим событием.
Считается, что фиброз костного мозга является вторичным процессом, который является результатом аномального отложения коллагена, полученного из нормальных фибробластов. Фибробласты в PMF индуцируются для пролиферации и производства избыточного коллагена факторами роста и цитокинами, секретируемыми мегакариоцитами.Основным фактором роста мегакариоцитов является трансформирующий фактор роста бета (TGF-бета). Другие факторы роста, также вероятно способствующие фиброзной реакции, включают: фактор роста тромбоцитов (PDGF), фактор роста эндотелия сосудов (VEGF) и основной фактор роста фибробластов (bFGF). Фиброз костного мозга не является необратимым процессом при PMF, и морфологические признаки обратимости можно увидеть через 12 месяцев после успешной трансплантации гемопоэтических стволовых клеток.
Другой ключевой характеристикой PMF является конститутивная мобилизация клеток CD34 + и эндотелиальных клеток-предшественников в периферическую кровь.Это нарушение регуляции движения стволовых клеток, как правило, в конечном итоге приводит к засеиванию экстрамедуллярных участков примитивными гематопоэтическими и эндотелиальными клетками, что приводит к экстрамедуллярному кроветворению в печени и селезенке, а также в других органах.
Какие еще клинические проявления могут помочь мне диагностировать первичный миелофиброз?
Важные или необычные вопросы / симптомы, которые следует задать в анамнезе
Сообщалось, что с PMF связаны различные иммунологические нарушения, включая прямой положительный результат теста Кумба, высокие титры антинуклеарных антител, положительный ревматоидный фактор, наличие активности волчаночного антикоагулянта и гипокомплементемию.Сообщалось также о высокой частоте моноклональных гаммопатий, при этом в некоторых сериях сообщалось до 10%.
Одним из отличительных признаков PMF является экстрамедуллярный гемопоэз (EMH). Очаги ЭМГ могут развиваться в любом органе, который может иметь вид:
Спленомегалия
Гепатомегалия
Лимфаденопатия
Перикардиальный или плевральный выпот
Прямое поражение легких, желудочно-кишечного тракта или нервной ткани.
Важные или необычные признаки или результаты медицинского осмотра
В редких случаях развитие PMF может предшествовать или быть связано с синдромом Свита, который характеризуется множественными кожными бляшками и узелками. Однако этот кожный процесс может быть связан с другими гематологическими злокачественными новообразованиями.
Какие еще дополнительные лабораторные исследования можно заказать?
Сообщается, что количество циркулирующих CD34-положительных стволовых клеток у пациентов с PMF в 300 раз выше по сравнению с нормальным контролем.Однако это обычно не используется в качестве диагностического маркера, поскольку до 15% пациентов с PMF могут иметь нормальное количество CD34 + стволовых клеток в периферической крови. Число клеток CD34 + можно использовать для мониторинга течения заболевания и ответа на терапию. Увеличение количества клеток CD34 + указывает на ускорение течения болезни.
Какие доказательства?
Кралович, Р., Пассамонти, Ф, Бузер, А.С. «Мутация увеличения функции Jak2 при миелопролиферативных заболеваниях». N Engl J Med. т. 352. 2005. С. 1779-1790. [В этой публикации обсуждается идентификация новой объединяющей мутации при миелопролиферативных заболеваниях (JAK2 V617F). Примерно в то же время были опубликованы три других исследования, подтверждающих этот вывод.]
Pikman, Y, Lee, BH, Mercher, T. «MPLW515L — новая соматическая активирующая мутация при миелофиброзе с миелоидной метаплазией». . т. 3. 2006. pp. E2708 [Отчет о мутации MPLW515L в подгруппе пациентов с миелофиброзом, отрицательным по JAK2V617F.]
Меса, Р.А., Нагорни, Д.С., Швагер, С., Оллред, Дж., Теффери, А. «Паллиативные цели, отбор пациентов и периоперационное ведение тромбоцитов: результаты и уроки трех десятилетий спленэктомии при миелофиброзе с миелоидной метаплазией в клинике Майо ». Рак. т. 107. 2006. pp. 361–370. [В этой публикации обсуждаются показания и результаты для пациентов, перенесших спленэктомию.]
Guglielmelli, P, Bartalucci, N, Rotunno, G, Vannucchi, AM. «Кальретикулин: новый горизонт тестирования и лечения миелопролиферативных новообразований». Эксперт Рев Гематол. т. 7. 2014. С. 423-425. [Эта публикация кратко резюмирует наше нынешнее понимание роли мутаций кальретикулина у пациентов с ЭТ и ПМП.]
Rondelli, D, Goldberg, JD, Isola, L. «Проспективное исследование MPD-RC 101 аллогенной трансплантации гемопоэтических стволовых клеток пониженной интенсивности у пациентов с миелофиброзом». Кровь. т. 124. 2014. С. 1183-1191. [Это исследование демонстрирует ценность кондиционирования с пониженной интенсивностью у пациентов с запущенными формами миелофиброза, которым посчастливилось иметь подходящего донора стволовых клеток.]
Сервантес, Ф., Альварес-Ларран, А., Доминго, А. «Эффективность и переносимость даназола в качестве лечения анемии миелофиброза с миелоидной метаплазией: отдаленные результаты у 30 пациентов». . т. 129. 2005. С. 771-775. [Исследование, демонстрирующее, что даназол хорошо переносится и вызывает хорошие реакции у некоторых пациентов с миелофиброзом и анемией.]
Верстовсек, С., Меса, Р.А., Готлиб, Дж. «Двойное слепое плацебо-контролируемое испытание руксолитиниба при миелофиброзе». N Engl J Med. т. 366. 2012. С. 799-807. [Этот комбинированный ингибитор JAK1 / JAK2 является единственным ингибитором JAK2, который в настоящее время одобрен FDA для лечения PMF.]
Чжоу, А, О, ST. «Прогнозирование при МЖ: от общего анализа крови к цитогенетике и молекулярным маркерам». Лучшая практика Res Clin Haematol. т. 27. 2014. С. 155–164. [Превосходный обзор использования различных клинических и лабораторных инструментов для определения прогноза пациентов с миелофиброзом.]
Tefferi, A, Lasho, TL, Finke, CM.«CALR против JAK2 против MPL-мутированного или тройного отрицательного миелофиброза: клинические, цитогенетические и молекулярные сравнения». Лейкемия. т. 28. 2014. С. 1472–1477. [Ретроспективное исследование корреляции молекулярных особенностей и клинических фенотипов, а также прогноза.]
Клампфл, Т, Гиссинджер, Х, Арулюнян, АС. «Соматические мутации кальретикулина при миелопролиферативных новообразованиях». N Engl J Med. т. 369. 2013. pp. 2379–2390. [Одна из двух первых статей, описывающих обнаружение мутации гена кальретикулина у пациентов, у которых отсутствуют мутации JAk2V617F или MPL.]
Copyright © 2017, 2013 ООО «Поддержка принятия решений в медицине». Все права защищены.
Ни один спонсор или рекламодатель не участвовал, не одобрял и не платил за контент, предоставляемый Decision Support in Medicine LLC. Лицензионный контент является собственностью DSM и защищен авторским правом.
Первичный миелофиброз (PMF) — Консультант по терапии рака
Краткий обзор
Первичный миелофиброз (PMF) — это хроническое миелопролиферативное новообразование (CMPN), характеризующееся клональной пролиферацией гранулоцитов и мегакариоцитов и склонностью к развитию выраженного фиброза костного мозга.На ранних стадиях у пациентов наблюдается лейкоцитоз и / или тромбоцитоз, но в большинстве случаев у 5-30% пациентов обычно развиваются панцитопения или даже острый миелоидный лейкоз.
При префиброзном PMF пациенты обычно имеют легкую анемию, умеренный гранулоцитоз и тромбоцитоз. На этой стадии заболевания спленомегалия присутствует в 10-20% случаев. Меньшая часть пациентов сообщает о ночном потоотделении, потере веса и лихорадке. Фактически, в некоторых исследованиях до 30% пациентов на момент обращения не имеют симптомов и диагностируются в результате обычного анализа крови.
При фиброзном PMF пациенты могут иметь лейкоэритробластный периферический мазок, который показывает циркулирующие ядросодержащие эритроциты, заметно смещенные влево миелоидные элементы и эритроциты в форме капли. В периферической крови может быть даже небольшой процент миелобластов. На этой стадии болезни у пациентов часто наблюдаются симптомы недостаточности костного мозга (усталость, кровотечение и инфекции), а также спленомегалия.
Какие тесты мне следует запросить, чтобы подтвердить мой клинический Dx? Кроме того, какие дополнительные тесты могут быть полезны?
Биопсия костного мозга с окрашиванием ретикулином на фиброз имеет важное значение для диагностики PMF.Хотя фиброз является признаком PMF в фиброзной фазе, он не является специфическим. Морфология мегакариоцитов в PMF важна для отличия PMF от других CMPN, которые также могут проявляться фиброзом. Мегакариоциты PMF обычно сгруппированы и часто показывают темный хроматин, причудливую ядерную лопастность (гиполобацию, «облачно-подобные» ядра) с широким диапазоном размеров от маленьких голых мегакариоцитов с гиперхроматическим хроматином до больших мегакариоцитов с луковичными ядрами.
Тест на мутацию
JAK2 рекомендуется для диагностики любого из CMPN.Примерно 50% случаев PMF имеют мутацию JAK2V617F, которая также может наблюдаться в большинстве случаев истинной полицитемии (PV) и почти в 50% случаев эссенциальной тромбоцитемии (ET).
Анализ кариотипа важен в диагностике PMF. Поскольку примерно половина всех случаев PMF отрицательна на аномалии JAK2, кариотипические аномалии помогают подтвердить клональный гематопоэз. Общие цитогенетические изменения в PMF включают del (13q) и трисомию 9. Следует отметить, что эти изменения не специфичны для PMF, но подтверждают диагноз, если присутствуют другие факторы.Важно отметить, что имеющиеся симптомы хронического миелолейкоза (ХМЛ) с фиброзом могут быть аналогичны имеющимся симптомам PMF. Если есть подозрения на ХМЛ, следует провести флуоресцентную гибридизацию in situ (FISH) для реаранжировки BCRABL.
Диагностические критерии Всемирной организации здравоохранения (ВОЗ) 2008 г. (требуется 3 специализации и 2 несовершеннолетних)
Основные критерии
Мегакариоцитарная пролиферация с характерной атипией и фиброзом ИЛИ при отсутствии фиброза гиперцеллюлярность с атипичными мегакариоцитами и сниженным эритропоэзом
Не соответствует критериям ЛВ, ХМЛ, миелодисплазии (МДС) или других ХМПН
Jak2 V617F или мутация экзона 12 ИЛИ морфологические изменения являются первичными
Второстепенные критерии
Какие еще болезни следует учитывать и как отличить их?
Может быть значительное перекрытие между префибротическим PMF и любым другим CMPN.Ключевым моментом в различии является отсутствие перегруппировки BCRABL, обнаруженной FISH. Присутствие JAK2V617F не является специфическим для PMF, но помогает классифицировать заболевание как CMPN.
ET и префибротический PMF могут проявляться заметно повышенными тромбоцитами, легкой анемией и умеренным лейкоцитозом, хотя PMF более вероятно проявляется лейкоцитозом. PMF можно отличить от ET при биопсии костного мозга. Костный мозг PMF обычно демонстрирует гиперцеллюлярность, которая редко встречается в костном мозге ET.PMF также показывает миелоидную гиперплазию и смещенные влево кластерные мегакариоциты с атипичной морфологией и, даже на ранней стадии, с большей вероятностью покажут фиброз 1 из 3 степени по классификации ВОЗ.
PV обычно проявляется повышенным гематокритом и гемоглобином. При биопсии костного мозга обычно наблюдается панмиелез, в отличие от миелоидной гиперплазии и мегакариоцитарной гиперплазии, как это видно при PMF.
CML с фиброзом необходимо исключить до постановки диагноза PMF, поскольку клинические проявления могут быть очень похожими.FISH для BCRABL обычно достаточно для диагностики ХМЛ.
Наконец, морфология мегакариоцитов PMF отличается как от больших, многодольчатых мегакариоцитов ET, так и от нормальных мегакариоцитов PV и от маленьких гиполобатированных мегакариоцитов CML. Мегакариоциты PMF часто сгруппированы и могут иметь размер от большого до маленького. Отношение ядра к цитоплазме часто увеличивается, и хроматин может быть гиперхроматическим или атипично сгруппированным. Ядро атипично лопастное с общими «облачными» или баллонными ядрами.Обычны «голые мегакариоциты» с гиперхроматическим хроматином, луковичными ядрами и очень скудной цитоплазмой.
Какие результаты лабораторных исследований являются полностью подтверждающими?
Не существует единственного лабораторного результата, который бы полностью подтвердил PMF. Диагноз основывается на демонстрации характерной морфологии костного мозга при наличии мутации JAK2 или других цитогенетических аномалий (трисомия 9 или del (13q)). MDS, CML и другие CMPN должны быть исключены с использованием кариотипа, FISH и морфологии костного мозга.
Какие подтверждающие тесты мне следует запросить для моего клинического Dx? Кроме того, какие дополнительные тесты могут быть полезны?
Тестирование
JAK2V617F и биопсия костного мозга занимают центральное место в постановке диагноза, когда FISH демонстрирует отсутствие слияния BCRABL. Поскольку PMF имеет более высокую скорость превращения в AML, чем любой другой BCRABL-отрицательный CMPN, важно регулярно проводить биопсию костного мозга. Даже в случаях, когда не наблюдается трансформации в острый миелоидный лейкоз (AML), выживаемость у пациентов с PMF значительно короче, чем у пациентов с PV и ET.Заболеваемость и смертность у пациентов с ПМФ обусловлены их повышенной склонностью к кровотечениям, анемии и инфекции, а также повышенной скоростью прогрессирования ОМЛ.
Каков прогноз?
пациентов с PMF имеют худший прогноз, чем другие пациенты с BCRABL-отрицательной CMPN. Трансформация в ОМЛ происходит у 5-30% пациентов в зависимости от серии. Кровоизлияние и инфекция также являются причиной значительной заболеваемости и смертности. В большинстве исследований средняя выживаемость пациентов с PMF обычно на 30-50% меньше, чем у пациентов с ET или PV, порядка 10 лет по сравнению с 15-20 годами.
Стратификация риска
Международной прогностической системы оценок (IPSS) основана на возрасте, лейкоцитах (WBC), Hct, циркулирующих бластах, конституциональных симптомах и была изменена Международной рабочей группой для включения количества тромбоцитов, трансфузионной зависимости и кариотипических аномалий. по миелопролиферативной неоплазии. Увеличение стратификации группы риска тесно связано с более короткой средней выживаемостью и, следовательно, требует более агрессивной терапии. Пациенты с низким уровнем риска могут лечиться с помощью наблюдения и симптоматической терапии и переходить на андрогены или талидомид / леналидомид и преднизон при анемии и гидроксимочевину при спленомегалии.