Воспалительные миопатии: дерматомиозит/полимиозит — ФГБНУ НИИР им. В.А. Насоновой
Воспалительные миопатии: дерматомиозит/полимиозит
Что такое воспалительные миопатии: дерматомиозит/полимиозит?
Воспалительные миопатии (дерматомиозит, полимиозит) – это группа редких аутоиммунных болезней, для которых характерно воспаление поперечно-полосатых мышц с развитием слабости в плечевом и тазовом поясе, туловище; возможно развитие нарушения глотания. Также возможно поражение кожи, суставов, внутренних органов.
При дерматомиозите/полимиозите могут иметь место следующие симптомы
-
Прогрессирующая в течение нескольких недель или месяцев мышечная слабость (трудно ходить вверх по лестнице, вставать со стула или корточек, причесываться, снимать одежду через голову, менять положение тела из горизонтального в вертикальное).
-
Мышечные боли (похожи на боли после интенсивной физической нагрузки).
-
Сыпь на лице, на тыле кистей (над мелкими суставами), а также над крупными суставами (коленными, локтевыми), в зоне декольте и шали, на наружных поверхностях бедер. На аналогичных участках возможно образование язвочек. Трещины на боковых поверхностях пальцев кистей.
-
Отек вокруг глаз.
-
Нарушение глотания: поперхивание при приеме жидкой или вязкой пищи, попадание жидкости в нос, застревание пищи в глотке.
-
Изменение голоса – осиплость и/или гнусавость.
-
Одышка при физической нагрузке, сухой кашель или кашель с небольшим количеством вязкой мокроты.
-
Боли и припухлость суставов. Изменение цвета пальцев на холоде.
-
Лихорадка.
Хорошо известно, что у части пациентов с дерматомиозитом и, реже, с полимиозитом, причиной развития аутоиммунной болезни может быть злокачественная опухоль. Поэтому больным проводится тщательное обследование для исключения злокачественной опухоли.
Поэтому больным проводится тщательное обследование для исключения злокачественной опухоли.
Для подтверждения диагноза назначаются лабораторные тесты – биохимический анализ крови, для выявления повышенного уровня мышечных ферментов, в первую очередь креатинфосфокиназы (КФК). Примерно у 1/3 пациентов выявляются миозит-специфические антитела – a-Jо-1, которые также имеют важное диагностическое значение. Проводится игольчатая электромиография для выявления признаков повреждения мышцы и оценки активности процесса. В сложных случаях может потребоваться биопсия мышцы. Также пациентам с воспалительными миопатиями часто проводится компьютерная томография органов грудной клетки для выявления признаков интерстициальной пневмонии.
Кому показана консультация ревматолога?
-
Прогрессирующая слабость в мышцах плечевого и тазового пояса.
-
Боли в мышцах.
-
Нарушение глотания.
-
Наличие периорбитального отека, розово-лиловой сыпи на лице, над мелкими и крупными суставами, в зоне декольте и шали, усиливающейся на солнце.
-
Наличие интерстициальной пневмонии и мышечной слабости и/или боли в суставах и /или кожных изменений – покраснений над суставами и трещин на пальцах.
-
Повышение уровня «мышечных» ферментов, в первую очередь КФК.
Лечение воспалительных миопатий
Целью лечения воспалительных миопатий является восстановление мышечной силы, улучшение и стабилизации функции легких (в случае их поражения), исчезновение кожной сыпи и заживление кожных дефектов, купирование артрита. При дерматомиозите и полимиозите мышечная сила восстанавливается очень медленно, в течение нескольких месяцев.
Залогом успеха лечения ВМ является сотрудничество пациента и врача, строгое соблюдение рекомендаций, а также внимательное отношение к своему самочувствию, наблюдение за анализами и ведение дневника.
Специалисты, занимающиеся диагностикой и лечением в институте: сотрудники лаборатории микроциркуляции и воспаления
Запишитесь на приём к специалисту:
Идиопатическая воспалительная миопатия требует бдительности врача в первый год после постановки диагноза
Взрослые пациенты с идиопатической воспалительной миопатией (ИВМ) имеют повышенный в три раза риск смерти, который является наиболее высоким в течение первого года после постановки диагноза. По результатам исследования, опубликованного в журнале Annals of the Rheumatic Diseases, врачам рекомендуется проявлять особую бдительность при наблюдении за такими пациентами в указанный период.
Исследователи проанализировали данные более 7 тыс. человек из Национального реестра пациентов в Швеции и идентифицировали когорту из 716 человек с недавно диагностированной ИВМ. Около 55% участников составляли женщины, средний возраст которых на момент начала исследования составил 61,4 года. Медианное время наблюдения составило 4,6 года в когорте ИВМ и 6,0 лет среди населения в целом.
Результаты показали, что ИВМ связана с увеличением смертности и наибольший риск был отмечен в течение первого года после постановки диагноза. Кроме того, показатели смертности оставались более чем в два раза выше, чем у общей популяции, в течение более 10 лет.
Смертность за время наблюдения составила 31% в группе пациентов с ИВМ и 12% среди населения в целом. Отмечаются различия в показателях смертности среди представителей обоих полов в группе общей популяции (выше у мужчин), но не среди пациентов с ИВМ.
Кумулятивная смертность через год после постановки диагноза составила 9% в группе ИВМ по сравнению с 1% в общей популяции, через 5 лет – 23 и 8% соответственно. Через 10 лет показатели кумулятивной смертности составили 31 и 12%, что не было статистически значимым различием.
Отмечается, что основными причинами смерти у пациентов с ИВМ были заболевания опорно-двигательного аппарата и болезни соединительной ткани, злокачественные новообразования, сердечно-сосудистые заболевания и болезни дыхательной системы.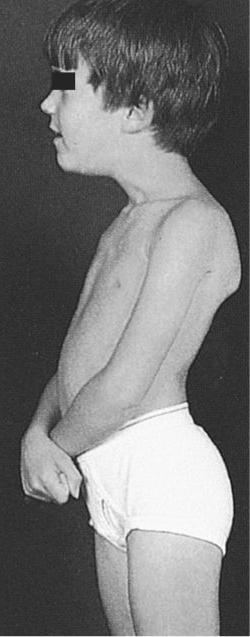 Среди населения в целом наиболее частыми причинами летальных исходов стали злокачественные новообразования, заболевания органов кровообращения и дыхательной системы.
Среди населения в целом наиболее частыми причинами летальных исходов стали злокачественные новообразования, заболевания органов кровообращения и дыхательной системы.
Миопатия: причины, симптомы, лечение. в Солнцево
Миопатией называют совокупность симптомов, возникающих в результате повреждения мышц. В отличие от заболеваний периферической нервной системы, (при которых мышцы также повреждены, но это связано с предшествующим повреждением снабжающих их нервов), при миопатиях болезненный процесс локализуется в самой мышце.
Беспокоит слабость в конечностях? Записывайтесь к неврологу!
Миопатии представляют собой очень большую группу заболеваний с различными причинами, разным течением и прогнозом. Общей чертой является мышечная слабость — поражение мышц бедер и рук, хотя мышцы лица также могут быть ослаблены. Сенсорные расстройства при миопатиях не встречаются. Мышечная слабость обычно двусторонняя и симметричная с самого начала, то есть сходна с обеих сторон тела — левой и правой.
Миопатии могут быть генетическими или приобретенными. Генетически обусловленные миопатии включают, например, мышечные дистрофии, которые характеризуются аномальной структурой мышечных клеток (например, из-за врожденного недостатка любого из его компонентов). С другой стороны, приобретенные миопатии имеют воспаление (воспалительные миопатии), могут сопровождать эндокринные заболевания (например, гипотиреоз), а также могут возникать в результате повреждения мышц некоторыми лекарственными средствами или токсичными соединениями (например, алкоголем).
Насколько распространена миопатия?
Термин «миопатия» очень широк и охватывает множество различных заболеваний. Поэтому общую заболеваемость миопатией трудно определить. Как правило, миопатии являются редкими заболеваниями.
Как проявляется миопатия?
Основным симптомом миопатии является мышечная слабость. Чаще всего это касается мышц бедер и рук — пациентам, например, трудно подниматься по лестнице, вставать из положения сидя, сидеть или выполнять действия с поднятыми руками (например, повесить шторы или делать прическу).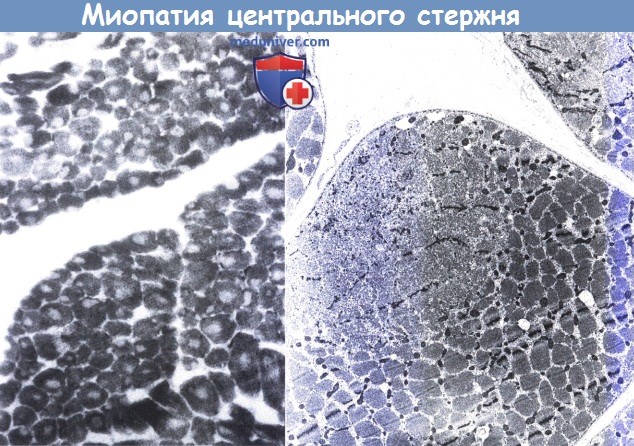 Мышечная слабость может сопровождаться болью. При некоторых миопатиях участвуют мышцы лица или горла. Может быть опущение век, снижение подвижности глазных яблок, невнятная речь, затрудненное глотание. Иногда мышцы шеи слабые — тогда не держится голова. При некоторых миопатиях может возникнуть слабость дыхательных мышц, приводящая к одышке. Иногда при миопатиях наблюдаются проблемы с расслаблением мышц, например, пациент не может быстро разжать пальцы после сжимания кулака. При прогрессирующих и длительных миопатиях возникает атрофия мышц. Эти симптомы могут сопровождаться признаками повреждения других тканей и органов вне мышечной системы.
Мышечная слабость может сопровождаться болью. При некоторых миопатиях участвуют мышцы лица или горла. Может быть опущение век, снижение подвижности глазных яблок, невнятная речь, затрудненное глотание. Иногда мышцы шеи слабые — тогда не держится голова. При некоторых миопатиях может возникнуть слабость дыхательных мышц, приводящая к одышке. Иногда при миопатиях наблюдаются проблемы с расслаблением мышц, например, пациент не может быстро разжать пальцы после сжимания кулака. При прогрессирующих и длительных миопатиях возникает атрофия мышц. Эти симптомы могут сопровождаться признаками повреждения других тканей и органов вне мышечной системы.
Что делать, если появились симптомы миопатии (слабости в конечностях)?
Если замечены какие-либо симптомы, которые могут указывать на миопатию, нужно обратиться к врачу общей практики или терапевту, который затем направит пациента к специалисту, обычно неврологу. Если симптомы длятся недолго (к примеру, несколько недель) и быстро нарастают, следует как можно скорее прийти к врачу.
Как врач диагностирует миопатию?
Врач собирает историю болезни пациента и проводит тест, который обычно показывает парез мышц (бедра, руки, иногда лица), иногда также ослабление рефлексов, вызванных при постукивании с помощью неврологического молотка. Затем он заказывает дополнительные тесты. Обычно это лабораторные анализы крови, в том числе уровни электролита, ТТГ, воспаления и креатинкиназы. Это чувствительный показатель разрушения мышц, и его уровень повышается у большинства пациентов с миопатией. Он также может быть увеличен при заболеваниях, когда повреждение мышц вызвано повреждением нервов, таких как боковой амиотрофический склероз или полинейропатия, а иногда и у здоровых людей, например, после физической нагрузки или травмы мышц. Электромиография (ЭМГ) является еще одним дополнительным тестом, важным для диагностики. ЭМГ позволяет подтвердить миопатию или указывает на другое заболевание, которое вызывает аналогичные симптомы. Окончательный диагноз миопатии, то есть определение ее типа и причины, чаще всего возможен после теста среза мышечной ткани (для этой цели берут маленький фрагмент ткани под местной анестезией для микроскопического исследования) или генетического анализа из образца крови, взятого у пациента. Генетическое тестирование проводится только для некоторых генетических миопатий.
Окончательный диагноз миопатии, то есть определение ее типа и причины, чаще всего возможен после теста среза мышечной ткани (для этой цели берут маленький фрагмент ткани под местной анестезией для микроскопического исследования) или генетического анализа из образца крови, взятого у пациента. Генетическое тестирование проводится только для некоторых генетических миопатий.
Как лечить миопатию?
Как и в случае с полиневропатией, можно выделить причинно-следственное и симптоматическое лечение. Возможно причинное лечение, например, при воспалительных миопатиях. В этих случаях используются глюкокортикоиды (стероидные гормоны), иммунодепрессанты или внутривенные иммуноглобулины. Причинное лечение миопатии, возникающей при эндокринных заболеваниях (например, заболевание щитовидной железы, гиперпаратиреоз или гиперадренокортицизм), включает лечение гормональных нарушений. При лекарственной или токсической миопатии нужно попытаться перестать принимать лекарство или прекратить воздействие токсического соединения, которое вызвало повреждение мышц. В редких случаях генетической миопатии, суть которой заключается в отсутствии одного из ферментов, необходимых для правильного функционирования мышц, возможно причинное лечение в виде введения недостающего фермента.
Симптоматическое лечение — это прежде всего реабилитация. В случае, если для пациента с быстрым мышечным расслаблением возникают обременительные трудности, фармакологическое лечение используется для смягчения этого симптома. При некоторых заболеваниях мышц поражаются другие органы, такие как сердечная мышца. Тогда для пациентов важно регулярно проходить осмотры у кардиолога. Кроме того, генетическое консультирование следует давать пациентам с генетической миопатией.
Можно ли полностью вылечить миопатию?
При генетических миопатиях полное выздоровление невозможно. Прогноз для приобретенных миопатий зависит от их причины: например, при воспалительных миопатиях мышечная сила обычно улучшается после начала лечения, а в миопатии, вызванной гормональными нарушениями, мышечные симптомы обычно исчезают после излечения основного заболевания.
Обращайтесь в поликлинику Медсемья в Солнцево — наш врач-невролог проведет диагностику и назначит правильное лечение!
Идиопатические воспалительные миопатии (вопросы клиники и этиопатогенеза) Текст научной статьи по специальности «Фундаментальная медицина»
ВЕСТНИК САНКТ-ПЕТЕРБУРГСКОГО УНИВЕРСИТЕТА
Сер. 11 2007 Вып. 4
УДК 616-021.3
Т. М. Алексеева, Н. М. Жулев, Е. В. Карпцова, В. И. Михайлов, Л. А. Сайкова, Л. П. Чурилов ИДИОПАТИЧЕСКИЕ ВОСПАЛИТЕЛЬНЫЕ МИОПАТИИ (вопросы клиники и этиопатогенеза)
1 Санкт-Петербургская медицинская академия последипломного образования
2 Санкт-Петербургский государственный университет, медицинский факультет
Воспалительные заболевания мышечной ткани являются одним из вариантов нервно-мышечной патологии в группе системных заболеваний соединительной ткани. Однако наряду с интенсивно изучаемыми заболеваниями соединительной ткани, такими как ревматодный артрит, системная красная волчанка и другие, клинические и эти- опатогенетические данные иммунозависимой воспалительной патологии мышц остаются мало изученными и редко публикуемыми в современных российских изданиях.
Нозология и эпидемиология воспалительных миопатий. Идиопатические воспалительные миопатии (ИВМ) — группа аутоаллергических заболеваний, которые характеризуются хроническим негнойным воспалением скелетной мускулатуры, типичным поражением кожи при дерматомиозите (ДМ), системными проявлениями и ассоциируются с другими диффузными болезнями соединительной ткани в рамках «перекрестных синдромов» [1, 2]. Идиопатические воспалительные миопатии включают следующие варианты: дерматомиозит, полимиозит (ПМ), миозит с тельцами включений, эозино- фильный миозит, гигантоклеточный миозит и очаговый (фокальный) миозит, полимиозит в ассоциации с системной красной волчанкой, ревматоидным артиритом, системной склеродермией. ИВМ входят в обширную группу воспалительных миопатий, к которой относятся инфекционные миозиты, острые вирусные миозиты, паразитарные миозиты, гранулематозный миозит (на фоне саркоидоза, тиротоксикоза, туберкулеза и др.), миозит при васкулитах и т. д. [3, 4].
ИВМ входят в обширную группу воспалительных миопатий, к которой относятся инфекционные миозиты, острые вирусные миозиты, паразитарные миозиты, гранулематозный миозит (на фоне саркоидоза, тиротоксикоза, туберкулеза и др.), миозит при васкулитах и т. д. [3, 4].
Дерматомиозит как заболевание впервые распознал E. Wagner, а описан он был в 1891 г. H. Unverricht [5, 6] . Полимиозит как нозологическая единица появился на 75 лет позднее, когда Walton и Adams опубликовали монографию [7]. Наблюдения Л. В. Догель включали 106 больных дермато- и полимиозитом [8]. В клинике неврологии СПб. МАПО с 1974 по 2005 г. наблюдалось более 200 пациентов различными формами ИВМ [9].
По данным В.А. Насоновой, в год на 1 млн жителей встречается 2-10 новых случев заболевания, что подтверждают и другие авторы — около 2,18-7,7 х 10-6 [10, 11]. Распространенность идиопатической воспалительной миопатии — 11/100 тыс. [12]. Заболеваемость и смертность остаются высокими без соответствующего лечения [13-15]. Полимиозит является наименее частой из всех форм и очень редким как самостоятельная нозологическая единица [16]. Типичным для данной аутоаллергической патологии считается соотношение женщин и мужчин 3:1. Об уровне выживаемости и факторах смертности в катамнезе заболевания говорит проведенное исследование пациентов
© Т. М. Алексеева, Н. М. Жулев, Е. В. Карпцова, В. И. Михайлов, Л. А. Сайкова, Л. П. Чурилов, 2007
с ИВМ. Выживаемость составила 92,8 и 71 % после 1,5 и 10 лет болезни соответственно. Основными причинами смерти служили наличие и прогрессирование новообразования, а также вовлечение сердечно-сосудистой и легочной систем [17].
Клиническая картина ИВМ. Клинические проявления дерматомиозита отличаются характерной сыпью и мышечной слабостью. Кожные проявления включают гелиотропную сыпь (сине-фиолетового цвета) на верхних веках с отеком, плоскую красную сыпь на лице и верхней части туловища, эритему на суставах пальцев с приподнимающимися фиолетовыми чешуйчатыми возвышениями (сыпь Готтрона) [2, 1820]. Эритематозная сыпь может также быть на других поверхностях тела, включая колени, локти, лодыжки, шею и переднюю поверхность грудной клетки, спину и плечи (симптом шали). Смешанная и венозная гиперемия с расширенными капиллярными петлями в основании ногтей также является характерной для дерматомизита. При дерматомиозите помимо кожного синдрома выявлялся также мышечный синдром в виде атрофий и парезов проксимальных и дистальных отделов конечностей. При выраженной мышечной слабости заболевание принимает форму миопатии с преимущественно проксимальным вовлечением мышц [2, 16, 18-21, 23, 28].
Кожные проявления включают гелиотропную сыпь (сине-фиолетового цвета) на верхних веках с отеком, плоскую красную сыпь на лице и верхней части туловища, эритему на суставах пальцев с приподнимающимися фиолетовыми чешуйчатыми возвышениями (сыпь Готтрона) [2, 1820]. Эритематозная сыпь может также быть на других поверхностях тела, включая колени, локти, лодыжки, шею и переднюю поверхность грудной клетки, спину и плечи (симптом шали). Смешанная и венозная гиперемия с расширенными капиллярными петлями в основании ногтей также является характерной для дерматомизита. При дерматомиозите помимо кожного синдрома выявлялся также мышечный синдром в виде атрофий и парезов проксимальных и дистальных отделов конечностей. При выраженной мышечной слабости заболевание принимает форму миопатии с преимущественно проксимальным вовлечением мышц [2, 16, 18-21, 23, 28].
В дополнение к первичному повреждению скелетных мышц и кожи клиника дерматомиозита может включать лекарственно-зависимый пневмонит или интерстици- альную болезнь легких. Интерстициальное заболевание легких предшествует миопатии или развивается в начале заболевания, особенно у пациентов, которые имеют анти-Jo-t антитела. Возможно появление язвенной болезни желудка и кишечника вследствие васкулита и инфекций, контрактур суставов, общих системных проявлений, таких как лихорадка, беспокойство, снижение веса, артралгия и феномен Рейно [16, 24].
Второй формой иммунозависимых воспалительных миопатий является полимиозит, который чаще ставится как диагноз исключения [2, 16, 18, 25-28]. Полимиозит — это воспалительная миопатия подострого начала (недели и месяцы) с непрерывным прогрессированием у взрослых и отсутствием кожных изменений. Клиническая картина характеризуется синдромом поражения мышц в виде миалгий, мышечных атрофий, ретракций мышц, патологической мышечной утомляемостью, поражением периферической нервной системы, вегетативно-эндокринными нарушениями. Преимущественно вовлекается передняя группа мышц шеи (сгибатели), проксимальные отделы верхних и нижних конечностей, дистальные мышцы рук (разгибатели кистей и пальцев рук, приводящие и отводящие мышцы кистей) [16, 18, 25, 26, 28].
Преимущественно вовлекается передняя группа мышц шеи (сгибатели), проксимальные отделы верхних и нижних конечностей, дистальные мышцы рук (разгибатели кистей и пальцев рук, приводящие и отводящие мышцы кистей) [16, 18, 25, 26, 28].
Общие системные расстройства, связанные с острофазным ответом, такие как лихорадка, беспокойство, снижение веса, артралгия, феномен Рейно, возможны в клинике ИВМ. Поражение сердца вследствие миокардита встречается при полимиозите у небольшого числа больных, а чаще манифестирует как нарушение атриовентрику- лярного проведения, тахиаритмии, дилятационная кардиомиопатия или сердечная недостаточность на фоне легочной гипертензии или длительного приема стероидов [16, 18, 21, 29]. Интерстициальное заболевание легких случается более чем у 10 % пациентов с ИВМ, половина из которых имеют анти-Jo-! антитела или антитела к различным рибонуклеопротеинам [2, 16, 18, 21, 29, 30].
В клинической картине ИВМ можно встретить ассоциацию ДМ или ПМ с другими заболеваниями соединительной ткани в рамках перекрестных синдромов. Перекрестные синдромы дерматомиозита с системной склеродермией и смешанными заболеваниями соединительной ткани проявляются в виде клиники дерматомиозита, склеротического уплотнения эпидермиса, микроангиопатии и депозитов кальция [2, 31]. Полимиозит встречается как синдром при различных заболеваниях в ассоциации с системными аутоиммунными заболеваниями, вирусными инфекциями или заболеваниями соединительной ткани. Полимиозит может обнаруживаться у 20 % пациентов с заболеваниями соединительной ткани, такими как системная красная волчанка, синдром Шегрена или ревматоидный артрит [2, 21, 29, 32, 33].
Наиболее ранним и частым симптомом следующей формы ИВМ, миозита с тельцами включений, является слабость четырехглавых мышц бедер. Заболевание может начинаться и со слабости дистальных мышц рук, что проявляется затруднением мелких движений в кистях. Затем вовлекаются подвздошно-поясничные, передние большеберцовые мышцы, двухглавые, трехглавые мышцы и глубокие сгибатели пальцев. Возраст прогрес- сирования симптомов этой формы миозита—после 60 лет, и болеют чаще мужчины [21].
Затем вовлекаются подвздошно-поясничные, передние большеберцовые мышцы, двухглавые, трехглавые мышцы и глубокие сгибатели пальцев. Возраст прогрес- сирования симптомов этой формы миозита—после 60 лет, и болеют чаще мужчины [21].
Эозинофильный полимиозит, характеризующийся эозинофилией в периферической крови и эозинофильными инфильтратами в эндомизиальной ткани, выделен R. B. Lay- zer в 1977 г. для описания случаев заболевания, в которых эозинофилы были основными воспалительными клетками в эндомизиальных инфильтратах.
Клиническая классификация и диагностика ИВМ. Предложен ряд систематиза- ций данной группы заболеваний, которые изменялись в ходе развития учения об этой патологии. Классификация A. Bohan и Y. Peter (1975) включает первичный (идиопатический) полимиозит, первичный (идиопатический) дерматомиозит, паранеопластический дермато-миозит (полимиозит), детский дерматомиозит (полимиозит) в сочетании с васкулитом и полимиозит или дерматомиозит в сочетании с другими заболеваниями соединительной ткани [34, 35]. В 1973 г. Л. В. Догель на материале больных с хроническим полимиозитом выделила 6 клинических форм и предложила систематизацию, которая до настоящего времени применяется в клинической практике: форма Вагнер-Унферрихта, псевдомиопатиче- ская форма, псевдомиастеническая, миосклеротическая, псевдоамиотрофическая, миалги- ческая форма. Позднее была выделена форма с синдромом Мак-Ардля [8, 9].
В работе Е. Л. Насонова рассмотрены формы полимиозита в зависимости от выявленных антител: антисинтетазный синдром, анти-SRP-синдром, анти-Mi-2-синдром, опухолевый дерматомиозит, миозит с включениями, «перекрестные» синдромы [36]. Принято выделять группу диффузных болезней соединительной ткани, которая включает системную красную волчанку, системную склеродермию, дерматомиозит, полимиозит и смешанное заболевание соединительной ткани [37].
Диагностика ИВМ основывается на пяти показателях: мышечная сила, электромиографические данные, мышечные ферменты, результаты мышечной биопсии, наличие сыпи или кальциноза. Повышение ферментов (креатинкиназы и лактатдегидрогеназы) может быть в 50 раз и более в острой фазе, а вне обострения уровень креатинкиназы и лактатдегидрогеназы обычно не превышает нормальные показатели. Электромиографические миопатические изменения включают потенциалы короткой продолжительности, низкоамплитудные, полифазные, высокую спонтанную активность с фибрилляциями, позитивные острые волны, комплексы повторных разрядов. Мышечная биопсия при ИВМ обнаруживает
участки некроза и регенерации мышечной ткани, атрофии волокон, облитерацию каппиляров. Важным отличием является то, что при полимиозите инфильтраты чаще располагаются в эндомиозии (в фасции), а при дерматомио- зите — периваскулярно или в септах фасции.
При миозите с тельцами включений выявляются признаки эндомизиального воспаления с базофильными гранулами, распределенными вокруг краев щелеподобных вакуолей («окаймленные вакуоли») и эозинофильные цитоплазматические включения. Различают 3 подтипа миозита с тельцами включений: спорадический s-IBM — это воспалительная миопатия с типичным клиническим фенотипом IBM; семейный (воспалительный) f-IBM — это типичный миозит с включениями с фенотипом, идентичным s-IBM, появляющийся у членов семьи одного поколения; гередитарный (невоспалительный) h-IBM—различают рецессивный наследственный вариант с сохранением четырехглавых мышц, связан с хромосомой 9p1q и вариант с доминантным наследованием, генетически не идентифицирован [25].
Изучение ИВМ требует научного подхода при неврологическом осмотре, рассмотрении мышечной гистопатологии, иммунопатологических и биохимических данных для того, чтобы правильно провести дифференциальную диагностику с токсиче- сикими, метаболическими, митохондриальными мышечными заболеваниями и миозитом с тельцами включений [2, 25-28, 38, 39]. Критерии A. Amato для идиопатических воспалительных миопатий более подробно описывают данные диагностики, дифференцирования типов миозита с различным патогенезом и рекомендованы для использования в рандомизированных клинических исследованиях [40].
Критерии A. Amato для идиопатических воспалительных миопатий более подробно описывают данные диагностики, дифференцирования типов миозита с различным патогенезом и рекомендованы для использования в рандомизированных клинических исследованиях [40].
Вопросы этиологии воспалительных миопатий. Инфекционная теория развития воспалительных миопатий является одной из первых и основана на отдельных клинических наблюдениях, где инфекционный процесс предшествовал или сочетался с дерматомиозитом. Обнаруживали включения, напоминающие нуклеопротеид пара- миксовируса, миксовируса, пикорнавируса, вируса Coxsackie и вируса herpes Zoster [49]. Другие исследования находят связь вирусов иммунодефицита (HIV, HTLV I) с клиникой полимиозита или миозита с тельцами включений [32, 41, 42]. Инфекции часто бывают провокаторами аутоаллергии у генетически предрасположенных лиц. При другом аутоаллергическом заболевании — инсулинзависимом сахарном диабете (ИЗСД) — диа- бетогенными вирусами, оказывающими свое действие через индукцию аутоиммунного цитолиза островковых В-клеток, считаются вирусы краснухи, кори, энтеровирусы, цитомегаловирусы. Наиболее вероятным цитоплазматическим антигеном островковых клеток при ИЗСД 1 -го типа и аутоиммунном инсулите является фермент глутаматде- карбоксилаза, представленная в В-клетках и в других тканях, например, в нейронах, нефроцитах, стероидпродуцирующих клетках, гепатоцитах.
Аутоаллергическая патология не сводится к повреждению клеток-мишеней ау- тоантителами и лимфоцитами. Большое значение имеет действие аутоантител как сигналов, блокирующих или стимулирующих рецепторы и ферменты. Мы полагаем, что невоспалительные проявления иммуноопосредованных миозитов, как и при других аутотоиммунных заболеваниях с противорецепторными и противоферментативными аутоантителами, могут вызываться действием подобных цитостимулирующих и цито- блокирующих продуктов иммунной системы, например, аутоантитела к амино-ацил-т- РНК-синтетазам могут нарушать трансляцию [43]. Сходство этиологии и патогенеза ряда болезней аутоаллергической природы доказывает факт, что и при полимиозите, и при инсулите биопсией выявляются преимущественно Т-лимфоцитарные инфильтраты с преобладанием CD8 Т-клеток и повышенная продукция белков главного комплекса гистосовместимости (ГКГС) 1-го класса [44].
Сходство этиологии и патогенеза ряда болезней аутоаллергической природы доказывает факт, что и при полимиозите, и при инсулите биопсией выявляются преимущественно Т-лимфоцитарные инфильтраты с преобладанием CD8 Т-клеток и повышенная продукция белков главного комплекса гистосовместимости (ГКГС) 1-го класса [44].
Некоторые паразиты, такие как простейшие (Toxoplasma, Trypanosoma), цестоды (Cysticerci) и нематоды (Trichinae) могут продуцировать фокальную или диффузную воспалительную миопатию, известную как «паразитарный полимиозит». В тропиках, гнойный миозит известен как «тропический полимиозит» или «пиомиозит», который может быть вызван Staphylococcus aureus, Yersinia, Streptococcus. Некоторые бактерии, такие как Borrelia burgdorferi при болезни Лайма и Legionella pneumonophila при болезни легионеров, могут также быть причиной полимиозита [45, 46]. Паразитарные полисерозиты, фасцииты и полимиозиты зачастую сопровождаются гиперэозинофилией, и в их механизме усматривается миотоксический эффект некоторых эозинофильных антибиотических белков [47].
Генетическая теория сформировалась несколько позднее инфекционной и находит свое подтверждение в неоднократно описанных семейных случаях заболевания, а также в обнаружении аутоиммунных заболеваний у родственников пациентов [48, 49]. По данным литературы, встречаются исследования о взаимосвязи антигенов главного комплекса гистосовместимости с полимиозитом. Установлена ассоциация дерма- томиозита с антигенами В14 и DR3 в европейской популяции и В7 и DRW6 у лиц негроидной расы. В 1981 г. ряд исследователей выявили, что все антиЛЛ-положительные пациенты полимиозитом были также DR3- или DRWg-положительны [50-52].
Влияние факторов внешней среды подтверждают примеры возникновения дер- матомиозита под воздействием некоторых лекарств: D-пеницилламина, сульфаниламидов, зидовудина, ртути, витамина В1, инсулина, аминазина, свинца, фенотиазина, а также после инсоляции. Имеются доказательства, что некоторые гиполипидемические препараты могут быть миотоксическими и являться причиной заболевания, напоминающего дерматомиозит [53-55]. Не исключается и роль психоэмоциональной травмы в инициировании аутоиммунного процесса [48]. Во всяком случае доказано, что хронический стресс способствует изменению направления дифференцировки Т-хелперов в пользу ТЬ2-субпопуляции, что способствует развитию ГНТ, в том числе — аутоиммунных [56]. Связь злокачественных новообразований и дерматомиозита выявлена некоторыми исследованиями [57, 58]. Рак яичников наиболее частый, затем следует кишечник, грудная клетка, легкие и рак печени в ассоциации с дерматомиозитом. Локализации новообразований соответствуют тем, что случаются более часто в пожилом возрасте [59]. Более часто новообразования ассоциированы с дерматомиозитом, например, описаны случаи ДМ и рака пищевода, ДМ и карциномы молочной железы, яичников [60].
Имеются доказательства, что некоторые гиполипидемические препараты могут быть миотоксическими и являться причиной заболевания, напоминающего дерматомиозит [53-55]. Не исключается и роль психоэмоциональной травмы в инициировании аутоиммунного процесса [48]. Во всяком случае доказано, что хронический стресс способствует изменению направления дифференцировки Т-хелперов в пользу ТЬ2-субпопуляции, что способствует развитию ГНТ, в том числе — аутоиммунных [56]. Связь злокачественных новообразований и дерматомиозита выявлена некоторыми исследованиями [57, 58]. Рак яичников наиболее частый, затем следует кишечник, грудная клетка, легкие и рак печени в ассоциации с дерматомиозитом. Локализации новообразований соответствуют тем, что случаются более часто в пожилом возрасте [59]. Более часто новообразования ассоциированы с дерматомиозитом, например, описаны случаи ДМ и рака пищевода, ДМ и карциномы молочной железы, яичников [60].
Патогенез идиопатических воспалительных миопатий. Аутоаллергический механизм возникновения полимиозита подтверждается многочисленными данными о нарушении механизмов иммуноглобулинопосредованного и Т-клеточноопосредованного иммунитета. Повышение экспрессии различных цитокинов в мышечной ткани наблюдается при ИВМ [61-63].
Антитела к скелетной мускулатуре у здоровых лиц встречаются часто, но в низких титрах — до 1:30, и направлены практически всегда против актина и других белков цитоскелета, а у пациентов с воспалительной миопатией титры, как
правило, более 1:60 и специфичность отличается, затрагивая аутоантигены, перечисленные ниже [64]. Различные антитела против антигенов клеточных ядер и цитоплазматических антигенов обнаруживаются у 20 % от всех пациентов с воспалительными миопатиями [2, 19-21, 28, 38, 65-67]. Антитела к цитоплазматическим антигенам направлены против рибонуклеопротеинов, которые вовлекаются в трансляцию и синтез белков — это различные амино-ацил-т-РНК-синтетазы и факторы трансляции. Антитела, направленные против гистидил-РНК-синтетазы, называемые анти-Jo-!, составляют 75 % от всех синтетаз. Более чем у 80 % пациентов полимиозитом с анти-Jo-! антителами развивается интерстициальное заболевание легких. Антитела к Jo-1 определяются в сыворотке 30-45 % пациентов [68], по данным других авторов — от 28,1 до 79 % [1, 65, 69]. Пациенты с антисинтетазным синдромом и наличием рибосомальных РНК-протеидов (anti-Ro/SSA) имеют тяжелую интерстициальную патологию легких и прогноз [38, 70, 71]. Пациенты с перекрестными синдромами дерматомизита и системной склеродермией могут иметь аутоантитела неясной диагностической значимости, включающие антиполимиозит/SCL, направленные против ядерного протеинового комплекса, анти- Ku, анти-и2РНК и др. [72, 73].
Антитела, направленные против гистидил-РНК-синтетазы, называемые анти-Jo-!, составляют 75 % от всех синтетаз. Более чем у 80 % пациентов полимиозитом с анти-Jo-! антителами развивается интерстициальное заболевание легких. Антитела к Jo-1 определяются в сыворотке 30-45 % пациентов [68], по данным других авторов — от 28,1 до 79 % [1, 65, 69]. Пациенты с антисинтетазным синдромом и наличием рибосомальных РНК-протеидов (anti-Ro/SSA) имеют тяжелую интерстициальную патологию легких и прогноз [38, 70, 71]. Пациенты с перекрестными синдромами дерматомизита и системной склеродермией могут иметь аутоантитела неясной диагностической значимости, включающие антиполимиозит/SCL, направленные против ядерного протеинового комплекса, анти- Ku, анти-и2РНК и др. [72, 73].
Иммуноопосредованное воспаление при дерматомиозите начинается следующим образом: предполагаемые антитела или иммунные комплексы, клиренс которых нарушен, на эндотелии сосудов активируют комплемент С3, формирующий C3b и C4b фрагменты. Это ведет к формированию C3bNEO и мембранолитического атакующего комплекса С5-С9, затем оба образуют депозиты на микрососудах эндомизия [16, 2426]. Скопления мембранолитического атакующего комплекса на внутримышечных капиллярах приводят к осмотическому лизису клеток эндотелия и некрозу капилляров, что способствует значительному уменьшению числа капилляров мышечных волокон и дилятации оставшихся сосудов для усиления компенсации уменьшенной перфузии. Большие межмышечные сосуды также вовлекаются в эту модель, приводящую к де -струкции мышечного волокна и воспалению. Перифасцикулярная атрофия, часто обнаруживаемая в хронической стадии, является отражением эндофасцикулярной гипо- перфузии. Специфические патогенные антитела против клеток эндотелия еще не идентифицированы [74, 75]. Антитела исчезают после успешного лечения внутривенным иммуноглобулином, очевидно, содержащим антиидиотипы, что ведет к клиническому улучшению [75]. Активация комплемента индуцирует высвобождение цитокинов, которые повышают экспрессию молекул клеточной адгезии VCAM-I и ICAM-I на клетках эндотелия [76]. Эти молекулы служат лигандами для интегринов VLA-4, LFA-I и Mac-I, экспрессируемых на Т-клетках, и облегчают их вход через кровеносную стенку в перимизиальное и эндомизиальное пространство. Иммунофенотипический анализ лимфоцитарных инфильтратов показывает В-клетки и CD4+ клетки в перимизии и пе- риваскулярном пространстве—это подтверждает взгляд, что гуморально-опосредованные механизмы играют главную роль при дерматомиозите [77].
Эти молекулы служат лигандами для интегринов VLA-4, LFA-I и Mac-I, экспрессируемых на Т-клетках, и облегчают их вход через кровеносную стенку в перимизиальное и эндомизиальное пространство. Иммунофенотипический анализ лимфоцитарных инфильтратов показывает В-клетки и CD4+ клетки в перимизии и пе- риваскулярном пространстве—это подтверждает взгляд, что гуморально-опосредованные механизмы играют главную роль при дерматомиозите [77].
В отличие от дерматомиозита, основным звеном полимиозита и миозита с тельцами включений является Т-клеточно-опосредованная ГЗТ, направленная против мышечного антигена. Этот вывод подтверждается присутствием CD8+ лимфоцитов, которые вместе с макрофагами вначале окружают здоровые, не некротизированные мышечные волокна, а затем инвазируют и разрушают их [78-80]. Мышечные волокна, как находящиеся рядом, так и отдаленные от области воспаления, экспрессируют антиген 1 -го класса ГКГС, который отсутствует в сарколемме нормальных мышечных волокон [19]. Цитотоксические Т-клетки узнают антигенные мишени в ассоциации с 1 классом антигенов комплекса гистосовместимости. Таким образом, при полимиозите первично иммунопатологическим механизмом является реакция гиперчувствтельности замедленного типа и ограниченный антигенами 1 -го класса ГКГС процесс.
В экспериментальных исследованиях было показано, что Т-лимфоциты, циркулирующие у пациентов, специфичны по отношению к аутоантигенам миотрубочек и оказывают на них разрушительное действие [80]. Сравнение Т-клеточного рецеп- торного репертуара при полимиозите и дерматомиозите со спектротипом подтвердило, что изменения семейства Т-клеточных рецепторов наблюдаются в лимфоцитах периферической крови, и они специфичны для полимиозита и миозита с тельцами включений [81-84]. Среди циркулирующих Т-клеток клональная экспансия происходит только у цитотоксических CD8+ клеток, которые экспрессируют гены перфорина и инфильтрируют мышечные волокна, на которых имеются антигены главного комплекса гистосовместимости 1-го класса [85, 86]. Для антигенной презентации и распознавания Т-клеток мышечные волокна и аутоинвазивные CD8+ Т-клетки нуждаются в ко-экспрессии костимуляторных молекул (B-7, B7-2, BB1, CD40 или ICOS-L) и соответстствующих контррецепторов (CD28, CTLA-4 (цитотоксические Т-лимфоцитарные антигены 4), CD40L или ICOS-L). Несколько исследований подтвердили, что мышечные волокна, экспресси- рующие белки ГКГС 1 -го класса, экспрессируют и ВВ1 (CD80), осуществляя межклеточные контакты с их CD28 или CTLA-4 лигандами на аутоинвазивных CD8+ Т-клетках [87-89]. Это отражает процесс аутопрезентации антигенов мышечных волокон.
Для антигенной презентации и распознавания Т-клеток мышечные волокна и аутоинвазивные CD8+ Т-клетки нуждаются в ко-экспрессии костимуляторных молекул (B-7, B7-2, BB1, CD40 или ICOS-L) и соответстствующих контррецепторов (CD28, CTLA-4 (цитотоксические Т-лимфоцитарные антигены 4), CD40L или ICOS-L). Несколько исследований подтвердили, что мышечные волокна, экспресси- рующие белки ГКГС 1 -го класса, экспрессируют и ВВ1 (CD80), осуществляя межклеточные контакты с их CD28 или CTLA-4 лигандами на аутоинвазивных CD8+ Т-клетках [87-89]. Это отражает процесс аутопрезентации антигенов мышечных волокон.
В исследованиях показано, что экспрессия м-РНК интерлейкина-1, интерлей- кина-2, фактора некроза опухоли-альфа и его рецептора, фактора некроза опухоли и его рецептора, интерферона-гамма, трансформирующего фактора роста бета, гранулоцит- макрофагального колониестимулирующего фактора, интерлейкина-6 и интерлейкина- 10 была усилена в мышцах большинства пациентов полимиозитом и миозитом с тельцами включений [90, 91]. При полимиозите и миозите с тельцами включений повышена продукция хемокинов: интерлейкин-8, RANTES (направленный на активацию экспрессированных и секретированных Т-клеток), моноцитарный хемоаттрактантный протеин 1, макрофагальный воспалительный протеин 1a (MIP-1a) и IP-10 [92, 93]. Адгезия лимфоцитов к мышцам может быть облегчена металлопротеиназами, семейством кальций-зависимых цинковых эндопептидаз, вовлеченных в ремоделирование экстроцеллюлярного матрикса. Активность металлопротеиназы-9 и -2 повышена в ненекротических мышечных волокнах с повышенной экспрессией белков 1-го класса ГКГС у пациентов с полимиозитом и миозитом с тельцами включений [94, 95].
На основе данных патогенеза ИВМ в настоящее время выделены два направления в лечении 4воспалительных миопатий — иммуносупрессивная и иммуномодулирующая терапия. К иммуномодулирующей медикаментозной терапии относится применение интерферона, внутривенного иммуноглобулина и различных вариантов плазмафереза [96, 97]. К иммуносупрессивным методам относят терапию глюкокортикостероидами, цитостатические препараты и ряд других веществ [98].
К иммуносупрессивным методам относят терапию глюкокортикостероидами, цитостатические препараты и ряд других веществ [98].
Summary
Alekseeva T. M., Zhulev N. M., Karptsova E. V., Mikhailov V. I., Saikova L.A., Churilov L. P.
The article contains modern data on clinical aspects, diagnostic criteria, classification of inflammatory myopathies. The data on aetiology and immunopathology of dermatomyositis, polymyositis and myositis with inclusion bodies are presented. The roles of T-cells and cytokines in immune responds of inflammatory myopathy are described.
Key words: idiopathic inflammatory myopathies, dermatomyositis, polymyositis, inclusion body myositis, cytokines, cytotoxic T-lymphocytes, autoantibody.
Литература
1. Насонов Е. Л., Самсонов М. Ю., Штутман В. З. Идиопатические воспалительные миопатии // Клинич. ревматология. 1996. № 4. С. 1013.
2. DalakasM. C. Polymyositis, dermatomyositis, and inclusion-body myositis // New Engl. J. Med. 1991. Vol. 325. P. 1487-1498.
3. Вест С.Дж. Секреты ревматологии: Пер. с англ. М.; СПб., 1999.
4. Леманн-Хорн Ф., ЛудольфА. Лечение заболеваний нервной системы: Пер. с нем. / Под ред. О. С. Левина. М., 2005. С. 309-327.
5. WagnerE. Fall liner selt nen // Muskelkrankhet Arch. Heilkd. 1863. N 4. S. 288.
6. UnverrichtH. Dermatomyositis acuta // Dtsch Med. Wochenschr. 1891. Vol. 17. S. 41-44.
7. Walton J. N., Adams R. D. Polymyositis. London, 1938.
N., Adams R. D. Polymyositis. London, 1938.
8. Догель Л. В. Полимиозит: Дис. … д-ра мед. наук. Л., 1973.
9. СайковаЛ.А., Алексеева Т. М. Хронический полимиозит. СПб., 2000.
10. Ревматические болезни: Руководство для врачей / Под ред. В. А. Насоновой, Н. В. Бунчука. М., 1997. С. 172-182.
11. Christopher-Stine L., Plotz P. H. Adult inflammatory myopathies // Best Practice and Research Clinical Rheumatology. 2004. Vol. 18. N 3. P.
331-344.
12. Ahlstrom G., Gunnarsson L. G., Leissner P., Sjoden P. O. Epidemiology of neuromuscular deseases, including the postpolio sequelae, in a Swedish country // Neuroepidemiol. 1993. Vol. 12. P. 262-269.
13. Riddoch D., Morgan-Hughes J. A. Prognosis in adult polymyositis // J. of Neurol. Sci. 1975. Vol. 26. P. 71-80.
14. CarpenterS., Karpati G., Rothman S., Walters G. The childhood type of dermatomyosi- tis // Neurol. 1976. N 26. P. 952-962.
15. Joffe M. M., Love L.A., Leff R. L. et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine and methotrexate and a comparison of their efficacy // Amer. J. of Med. 1993. Vol. 94. P. 379-387.
16. DalakasM. C., HohlfeldR. Polymyositis and dermatomyositis // Lancet. 2003. Vol. 362. P. 971-982.
17. Torres C., Belmonte R., Carmona L. et al. Survival, mortality and causes of death in inflammatory myopathies // Autoimmunity. 2006. Vol. 39. N 3. P. 205-215.
18. DalakasM. C. Inflammatory myopathies // Curr. Opin. Neurol. Neurosurg. 1990. Vol. 3. P. 689-696.
DalakasM. C. Inflammatory myopathies // Curr. Opin. Neurol. Neurosurg. 1990. Vol. 3. P. 689-696.
19. Karpati G., Carpenter S. Pathology of inflammatory myopathies // Bailliere s clinical neurology / Ed. By F. L. Mastaglia. London, 1993. P.
527-556.
20. Engel A. G., Hohlfeld R., Banker B. Q. The polymyositis and dermatomyositis syndrome // Myology / Ed. by A. G. Engel, C. FranziniArmstrong. New York, 1994. P. 1335-1383.
21. Dalakas M. C. Inflammatory myopathies: recent advances in pathogenesis and therapy // Neu- romuscular diseases / Ed. by R. Pourmand, Y. Harati. Philadelphia, 2001. P. 636-659.
22. Whitaker J. N., Engel W. K. Vascular deposits of immunoglobulin and complement in id- iopathic inflammatory myopathy // New Engl. J. Med. 1972. Vol. 286. P. 333-338.
23. SontheimerR. D. Dermatomyositis: an overview of recent progress with emphasis on der- matologic aspects // Dermatol. Clin. 2002. Vol. 20. N 3. P. 387-408.
24. DalakasM. C. Images in clinical medicine. Calcifications in dermatomyositis // New Engl. J. Med. 1995. Vol. 333. N 15. P. 978.
25. DalakasM. C. Progress in inflammatory myopathies: good but not good enough. Editorial // J. Neurol. Neurosurg. Psychiatry. 2001. Vol. 70. P. 569-573.
26. DalakasM. C. The molecular and cellular pathology of inflammatory muscle diseases // Curr. Opin. Pharmacol. 2001. N 1. P. 300-306.
27. RowlandL. P. Polymyositis, inclusion body myositis and related myopathies // Merrits textbook of neurology. 9th ed. Philadelphia, 1995. P. 798-802.
9th ed. Philadelphia, 1995. P. 798-802.
28. DalakasM. C., Karpati G. The inflammatory myopathies // Disorders of voluntary muscle. 7th ed. Cambridge, 2001. P. 636-659.
29. Dalakas M. C. Polymyositis, dermatomyosisis and inclusion body myositis // Harrisons principles of internal medicine. 15th ed. New York, 2001. P. 2524-2529.
30. Cottin V., Thivolet-Bejui F., Reynaud-Gaubert M. et al. Interstitial lung disease in amyo- pathic dermatomyositis // Eur. Respir. J. 2003. Vol. 22. N 2. P. 245-250.
31. Rosenberg N. L., Carry M. R., Ringel S. P. Association of inflammatory myopathies with other connective tissue disorders and malignancies // Polymyositis and dermatomyositis. Boston, 1988. P. 37-69.
32. DalakasM. C. The molecular pathophysiology in inflammatory myopathies // La revue de medecine interne. 2004. Vol. 25. P. 14-16.
33. Merritts textbook of neurology / Ed. by L. P. Rowland. Philadelphia, 1995.
34. Bohan A., Peter J. B. Polymyositis and dermatomyositis (first of two parts) // New Engl. J. Med. 1975. Vol. 292. N 7. P. 344-347.
35. Bohan A., Peter J. B. Polymyositis and dermatomyositis (second of two parts) // New Engl. J. Med. 1975. Vol. 292. N 8. P. 403-407.
36. Насонов Е. Л., Штутман В. З., Саложин К. В. и др. Клинико-иммунологическая гетерогенность идиопатических воспалительных миопатий // Клинич. медицина 1995. № 2. С. 4-8.
37. Клиническая ревматология: Руководство для врачей / Под ред. В. И. Мазурова. СПб., 2001. C. 208-281.
38. DalakasM. C., HohlfeldR. Polymyositis and dermatomyositis // Lancet. 2003. Vol. 362. (9388). P. 1762-1763.
DalakasM. C., HohlfeldR. Polymyositis and dermatomyositis // Lancet. 2003. Vol. 362. (9388). P. 1762-1763.
39. Mastaglia F. L., Garlepp M. J., Phillips B. A., Zilko P. J. Inflammatorymyopathies: clinical, diagnostic and therapeutic aspects // Muscle Nerve. 2003. Vol. 27. P. 407-425.
40. Hoogendijk J. E., Amato A.A., Lecky B. R. et al. Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis // 119th ENMC international workshop. 2003. 10-12 Oct. Naarden, The Netherlands: Neuromascular Disorders. 2004. № 14. P. 337-345.
41. DalakasM. C. Retroviruses and inflammatory myopathies // Baillieres. Clin. Neurol. 1993. N 2. P. 658-691.
42. Cupler E. J., Leon-Monzon M., Miller J. et al. Inclusion body myositis in HIV-1 and HTLV-1 infected patients // Brain. 1996. Vol. 119. P. 1887-1893.
43. Зайчик А. Ш., Чурилов Л. П. Основы патохимии: Учебник для студентов медицинских вузов. СПб., 2001.
44. Atkinson M. A., Eisenbarth G. S. Type 1 diabetes: new perspectives on disease pathogenesis and treatment // Lancet. 2001. Vol. 358. P. 221229.
45. AtlasE., NovakS. N, Duray P. H., Steere A. C. Lyme myositis: muscle invasion by Bor- relia burgdorferi // Ann. Intern. Med. 1988. Vol. 109. P. 245-246.
46. Warner C. L., FayadP. B., HefnerR. R Jr. Legionella myositis // Neurology. 1991. Vol. 41. P. 750-752.
47. Зайчик А. Ш., Чурилов Л. П. Патофизиология. Т. III. Механизмы развития болезней и синдромов. Вып. 1. Патофизиологические основы гематологии и онкологии. СПб., 2002.
48. Соловьева А. П. Дерматомиозит. М., 1980.
49. Сигидин Я.А., ГусеваН. Г., ИвановаМ. М. Диффузные болезни соединительной ткани. М., 1994.
50. Алексеева Т. М. Хронический полимиозит: Дис. … канд. мед. наук. СПб., 1998.
51. PlotzP. The place of autoimmunity in myositis // Autoimmunity Rev. 2004. Vol. 3. N 1. P. 36.
52. Nagaraju K., Casciola-Rosen L., Lundberg I. et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction // Arthritis Rheum. 2005. Vol. 52 N 6. P. 1824-1835.
53. SchalkeB. B., SchmidtB., ToykaK., HartungH. P. Pravastatin-associated inflammatory myo- pathy // New Engl. J. Med. 1992. Vol. 327. P. 649.
54. KhattakF. H., Morris I. M., BranfordW. A. Simvastatin-associated Dermatomyositis // Br. J. Rheumatol. 1994. Vol. 33. P. 199.
55. NoelB., Cerottini J. P., PanizzonR. G. Atorvastatin-induced dermatomyositis // Amer. J. Med. 2001. P. 670.
56. Elenkov I. J., Chrousos G. P. Stress Hormones, Th2/Th3 patterns, Pro/Anti-inflammatory Cytokines and Susceptibility to Disease // Trends Endocrinol. Metab. 1999. Vol. 10. N 9. P. 359-368.
57. Buchbinder R., Forbes A., Hall S. et al. Incidence of malignant disease on biopsy-proven inflammatory myopathy: A population-based cohort study // Ann. Intern. Med. 2001. Vol. 134. P. 1087-1095.
58. Hill C. L., Zhang Y., Sigurgeirsson B. et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study // Lancet. 2001. Vol. 357. P. 96-100.
2001. Vol. 357. P. 96-100.
59. Callen J. P. Dermatomyositis: diagnosis, evaluation and management // Minerva Med. 2002. Vol. 134. P. 1087-1095.
60. Iftikhar I., Abdelmannan D., Daw H.A. Dermatomyositis and esophageal cancer // South. Med. J. 2006. Vol. 99. N 7. P. 777-779.
61. Lundberg I., Brengman J. M., Engel A. G. Analysis of cytokine expression in muscle in inflammatory myopathies, dystrophy and non-weak controls // J. Neuroimmunol. 1995. Vol. 63. P. 9-16.
62. TewsD. S., GoebelH. H. Cytokine expression profile in idiopathic inflammatory myopathies // J. Neuropathol. Exp. Neurol. 1996. Vol. 55. P. 342-347.
63. DalakasM. C. Molecular immunology and genetics of inflammatory muscle diseases // Arch. Neurol. 1998. Vol. 55. P. 1509-1512.
64. Потехин О. Е., Малышев В. С. Современное состояние иммунологической диагностики аутоиммунных заболеваний // Иммунол. аллергол. и инфектол. 2000. № 1. С. 44-49.
65. DalakasM. C. Inflammatory myopathies // Handbook of clinical neurology. Vol. 18. Myopathies. Amsterdam, 1992. P. 369-390.
66. DalakasM. C. Immunopathogenesis of inflammatory myopathies // Ann. Neurol. 1995. Vol. 37. P. 74-86.
67. Dalakas M. C. Polymyositis // Med. Link. Neurology. San Diego, 1997.
68. VenablesP. J. W. Polymyositis — associated overlap syndrome // Br. J. Rheum. 1996. Vol. 35. N 4. P. 305-308.
69. DalakasM. C. Inflammatory myopathies: pathogenesis and treatment // Neuropharmacol. 1992. N 5. P. 327-351.
1992. N 5. P. 327-351.
70. Hengstman G. J., van Engelen B. G., Vree Egberts W. T., van Venrooij W. J. Myositis-specific autoantibodies: overview and recent developments // Curr. Opin. Rheumatol. 2001. Vol. 13. N 6. P. 476-482.
71. La Corte R., Lo Mo Naco A., Locaputo A. et al. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease // Autoim- munity. 2006. Vol. 39. N 3. P. 249-253.
72. Jury E. C., D’Cruz D., Morrow W. J. Autoantibodies and overlap syndromes in autoimmune rheumatic disease // J. Clin. Pathol. 2001. Vol. 54. P. 340-347.
73. KuboM., Ihn H., KuwanaM. et al. Prevalence in myositis of antibodies recognizing anti- U3 RNA probably in a novel complex with 22/25 kD protein and not fibrillarin // Clin. Exp. Immunol. 2001. Vol. 126. P. 339-344.
74. Cervera R., Ramires G., Fernandez-Sola J. et al. Antibodies to endothelial cells in dermatomyositis: association with interstitial lung diseases // Br. Med. J. 1991. Vol. 301. P. 880-882.
75. Stein D. P., Jordan S. C., ToyodaM. et al. Anti-endothelial cell antibodies (AECA) in der- natomyositis (DM) // Neurology. 1993. Vol. 43. P.
356.
76. Stein D. P., Dalakas M. C. Intercellular adhesion molecule-I expression is upregulated in patients with dermatomyositis (DM) // Ann. Neurol. 1993. Vol. 34. P. 268.
77. Gallardo E., De Andres I., Illa I. Cathepsins are upregulated by IFN-gamma/STAT1 in human muscle culture: a possible active factor in dermatomyositis // J. Neuropath. Exp. Neurol. 2001. Vol. 60. P. 847-855.
Neurol. 2001. Vol. 60. P. 847-855.
78. Arahata K., Engel A. G. Monoclonal antibody analysis of mononuclear cells in myopathies. II. Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis // Ann. Neurol. 1984. Vol. 16. P. 209-215.
79. Arahata K., Engel A. G. Monoclonal antibody analysis of mononuclear cells in myopathies. V. Identification and quantitation of T8+ cytotoxic and T8 supressor cells // Ibid. 1988. Vol. 23. P. 493-499.
80. Hohlfeld R, Engel A. G. The immunobiology of muscle // Immunol. Today. 1994. Vol. 15. P. 269-274.
81. Mantegazza R., Andreetta F., Bernasconi P. et al. Analysis of T cell receptor repertoire of muscle-infiltrating T lymphocytes in polymyositis:restricted V alpha/beta rearrangements may indicate antigen-driven selection // J. Clin. Invest. 1993. Vol. 91. P. 2880-2886.
82. OHanlon T. P., Dalakas M. C., Plotz P. H., Miller F. W. Predominant TCR-alpha-beta variable and joinig gene expression by muscle-infiltrating lymphocytes in the idiopathic inflammatory myopathies // J. Immunol. 1994. Vol. 152. P. 2569-2576.
83. Bender A., Ernst N., Iglesias A. et al. T cell receptor repertoire in polymyositis: clonal expansion of autoaggressive CD8+ T cells // J. Exp. Med. 1995. Vol. 181. P. 1863-1868.
84. Benveniste O., Cherin P., Maisonobe T. et al. Severe perturbations of blood T cell repertoire in polymyositis, but not dermatomyositis patients // J. Immunol. 2001. Vol. 167. P. 3521-3529.
85. Nishio J., Suzuki M., Miyasaka N., Kohsaka H. Clonal biases of peripheral CD8 T cell repertoire directly reflect local inflammation in polymyositis // J. Immunol. 2001. Vol. 167. P. 4051-4058.
Immunol. 2001. Vol. 167. P. 4051-4058.
86. HofbauerM., Wiesener S., Babbe H. et al. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR and CDR3 spectratype analysis // Proc. Natl. Acad. Sci. USA. 2003. Vol. 100. N 7. P. 4090-4095.
87. Sugiura T., Kawaguchi Y., HarigaiM. et al. Increased CD40 expression on muscle cells of polymyositis and protein-1 production // J. Immunol. 2000. Vol. 164. P. 6593-6600.
88. WiendlH., Mitsdoerffer M., Hofmeister V. et al. The nonclassical MHC molecule HLA-G protects human muscle cells from immune-mediated lysis: omplications for myoblast transplantation and gene therapy // Brain. 2003. Vol. 126. P. 176-185.
89. Schmidt J., Rakocevic G., Raju R., Dalakas M. C. Upregulated inducible costimulator and ICOS-ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity // Ibid. 2004. Vol. 127. Pt. 5. P. 1182-1190.
90. De Bleecker J. L., Meire V. I., DeclercqA W., Van Aken H. E. Immunolocalization of tumor necrosis factor-alpha and its receptors in inflammatory myopathies // Neuromusc. Disord. 1999. № 9. P. 239.
91. Kalovidouris A. E., Plotkin Z. Synergistic cytotoxic effect of interferon gamma and tumor necrosis factor alpha on cultured human muscle cells // J. Rheumatol. 1995. Vol. 22. P. 1698-1703.
92. Confalonieri P., Bernasconi P., Megna P. et al. Increased expression of beta-chemokines in muscle of patients with inflammatory myopathies // J. Neuropathol. Exp. Neurol. 2000. Vol. 59. P. 164-169.
93. Raju R., Vasconcelos O. M., Semino-Mora C. et al. Expression of interferon-gamma induc- ible chemoines in the muscles of patients with inclusion body myositis // J. Neuroimmunol. 2003. Vol. 141. P. 125-131.
et al. Expression of interferon-gamma induc- ible chemoines in the muscles of patients with inclusion body myositis // J. Neuroimmunol. 2003. Vol. 141. P. 125-131.
94. Choy Y C., Dalakas M. C. Expression of matrix metalloproteinases in the muscle of patients with inflammatory myopathies // Neurology. 2000. Vol. 54. P. 65-71.
95. KieseierB. C., Schneider C., Clements J. M. et al. Expression of specific matrix metalloproteinases in inflammatory myopathies // Brain. 2001. Vol. 124. P. 341-351.
96. Choy E. M. H., Isenberg D. A. Treatment of dermatomyositis and polymyositis // Rheumatology. 2002. Vol. 41. P. 7-13.
97. Dalakas M. C. Therapeutic approaches in patients with inflammatory myopathies // Semin. Neurol. 2003. Vol. 23. N 2. P. 199-206.
98. Mastaglia F. L., Zilko P. J. Inflammatory myopathies: how to treat the difficult cases // J. Clin. Neurosc. 2003. Vol. 10. N 1. P. 99-101.
Статья принята к печати 20 июня 2007 г.
Применение магнитно-резонансной томографии в диагностике идиопатических воспалительных миопатий | Хелковская-Сергеева
1. Bohan A, Peter JB. Polymyositis and der-matomyositis (second of two parts). N Engl J Med. 1975 Feb 20;292(8):403-7.
2. Leclair V, Lundberg IE. New Myositis Classification Criteria —What We Have Learned Since Bohan and Peter. Curr Rheumatol Rep. 2018 Mar 17;20(4):18. doi: 10.1007/s11926-018-0726-4.
3. Michelle EH, Mammen AL. Myositis Mimics. Curr Rheumatol Rep. 2015 Oct; 17(10):63. doi: 10.1007/s11926-015-0541-0.
Myositis Mimics. Curr Rheumatol Rep. 2015 Oct; 17(10):63. doi: 10.1007/s11926-015-0541-0.
4. Mammen AL. Which nonautoimmune myopathies are most frequently misdiagnosed as myositis? Curr Opin Rheumatol. 2017 Nov; 29(6):618-622. doi: 10.1097/BOR.0000000000000441.
5. Schiffenbauer A. Imaging: Seeing Muscle in New Ways. Curr Opin Rheumatol. 2014 Nov; 26(6):712-6. doi: 10.1097/BOR.0000000000000105.
6. Влодавец ДО, Казаков ДО. Диагностические возможности МРТ мышц при нервно-мышечных заболеваниях. Неврологический журнал. 2014;(3):4-13.
7. Mercuri E, Pichiecchio A, Allsop J, et al. Muscle MRI in inherited neuromuscular disorders: past, present and future. JMagn Reson Imaging. 2007 Feb;25(2):433-40.
8. Huang ZG, Gao BX, Chen h2, et al. An efficacy analysis of whole-body magnetic resonance imaging in the diagnosis and followup of polymyositis and dermatomyositis. PLoS One. 2017 Jul 17;12(7):e0181069. doi: 10.1371/journal.pone.0181069.e Collection 2017.
9. Miranda SS, Alvarenga D, Rodrigues JC, Shinjo SK. Different aspects of magnetic resonance imaging of muscles between dermato-myositis and polymyositis. Rev Bras Reumatol. 2014 Jul-Aug;54(4):295-300. doi: 10.1016/j.rbr.2014.04.004. Epub 2014 Aug 26.
10. Del Grande F, Carrino JA, Del Grande M, Mammen AL, Christopher Stine L. Magnetic resonance imaging of inflammatory myopathies. Top Magn Reson Imaging. 2011 Apr;22(2):39-43. doi: 10.1097/RMR.0b013e31825b2c35.
11. Ukichi T, Yoshida K, Matsushima S, et al. Magnetic Resonance Imaging of Skeletal Muscles in Patients with Dermatomyositis and Polymyositis: Novel and Distinctive Characteristic Findings [abstract]. Arthritis Rheumatol. 2017;69(suppl 10):1999.
Ukichi T, Yoshida K, Matsushima S, et al. Magnetic Resonance Imaging of Skeletal Muscles in Patients with Dermatomyositis and Polymyositis: Novel and Distinctive Characteristic Findings [abstract]. Arthritis Rheumatol. 2017;69(suppl 10):1999.
12. Pinal-Fernandez I, Casal-Dominguez M, Carrino JA, et al. Thigh muscle MRI in immune-mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann Rheum Dis. 2017 Apr;76(4): 681-687. doi: 10.1136/annrheumdis-2016-210198. Epub 2016 Sep 20
13. Tasca G, Monforte M, De Fino C, et al. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve. 2015 Dec;52(6):956-62. doi: 10.1002/mus.24661. Epub 2015 Aug 31.
14. Dobloug C, Garen T, Bitter H, et al. Prevalence and clinical characteristics of adult polymyositis and dermatomyositis; data from a large and unselected Norwegian cohort. Ann Rheum Dis. 2015 Aug;74(8): 1551-6. doi: 10.1136/annrheumdis-2013-205127. Epub 2014 Apr 2.
15. Subhawong TK, Wang X, Machado AJ, et al. 1H Magnetic resonance spectroscopy findings in idiopathic inflammatory myopathies at 3 T: feasibility and first results. Invest Radiol. 2013 Jul; 48(7): 509-16. doi: 10.1097/RLI.0b013e3182823562.
16. Noto Y, Shiga K, Tsuji Y, et al. Contrasting echogenicity in flexor digitorum profundus-flexor carpi ulnaris: a diagnostic ultrasound pattern in sporadic inclusion body myositis. Muscle Nerve. 2014 May;49(5): 745-8. doi: 10.1002/mus.24056.
17. Pipitone N, Versari A, Zuccoli G, Levrini G, Macchioni P, Bajocchi G, Salvarani C. 18F-Fluorodeoxyglucose positron emission tomography for the assessment of myositis: a case series. Clin Exp Rheumatol. 2012 Jul-Aug;30(4):570-3. Epub 2012 Aug 29.
18F-Fluorodeoxyglucose positron emission tomography for the assessment of myositis: a case series. Clin Exp Rheumatol. 2012 Jul-Aug;30(4):570-3. Epub 2012 Aug 29.
18. Tanaka S, Ikeda K, Uchiyama K, et al. [18F]FDG uptake in proximal muscles assessed by PET/CT reflects both global and local muscular inflammation and provides useful information in the management of patients with polymyositis/dermatomyositis. Rheumatology (Oxford). 2013 Jul;52(7): 1271-8. doi: 10.1093/rheumatology/ket112. Epub 2013 Mar 11.
Идиопатические воспалительные миопатии > Клинические рекомендации РФ (Россия) 2013-2017 > MedElement
Лечение ПМ/ДМ
Основные цели фармакотерапии ПМ/ДМ
· достижение полного клинического ответа (отсутствия клинико-лабораторной активности в течение, не менее чем 6 месяцев на фоне терапии) или ремиссии (отсутствия клинико-лабораторной активности в течение, не менее чем 6 месяцев на фоне отмены терапии) (уровень доказательности В),
· снижение риска комарбидных инфекций ГК) (уровень доказательности С)
· выявление и своевременное лечение пациентов с наибольшим риском ИПЛ
Общие рекомендации по лечению
· Лечение пациентов ПМ/ДМ должно проводиться врачами-ревматологами.
· В случае наличия ИПЛ с ФА при АСС – с привлечением пульмонологов и основываться на тесном взаимодействии врача и пациента
· Следует рекомендовать пациентам избегать факторов, которые могут спровоцировать обострение болезни: отказаться от пребывания на солнце, от курения, от контактов и инфекционными больными, избегать физических и психо-эмоциональных перегрузок.
· Следует рекомендовать пациентам исключить факторы, повышающие риск развития побочных эффектов терапии ГК: не употреблять в пищу сладкие продукты, включая мед и сладкие фрукты, повышающие риск развития стероидного сахарного диабета, также, исключение острой пищи, применение гастропротекторов с целью предотвращения язвенных осложнений (уровень доказательности С)
· Все пациенты нуждаются в активной профилактике и лечении глюкокортикоидного остеопороза. Подбор антиостеопоретической терапии зависит от результатов денситометрического исследования и оценки дополнительных факторов риска остеопороза (менопауза, эндокринные заболевания). В зависимости от исходных данных минеральной плотности костной ткани назначаются препараты кальция в сочетании с витамином Д, или эти же препараты в сочетании с бисфосфанатами.
Подбор антиостеопоретической терапии зависит от результатов денситометрического исследования и оценки дополнительных факторов риска остеопороза (менопауза, эндокринные заболевания). В зависимости от исходных данных минеральной плотности костной ткани назначаются препараты кальция в сочетании с витамином Д, или эти же препараты в сочетании с бисфосфанатами.
· У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая мышечную ткань, может способствовать как формированию постинъекционных кальцинатов, так быть причиной ложноположительных результатов уровня креатинфосфокиназы (КФК).
Ведущая роль в лечении ПМ/ДМ отводится ГК. Основные принципы лечения ГК:
· Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов) ассоциируется с благоприятным прогнозом.
· Адекватная инициальная доза: в зависимости от тяжести заболевания начальная доза колеблется от 1 до 2 мг/кг/сут.
· Ежедневный прием ГК.
· Суточную дозу ГК в начале лечения следует делить на 3 приема (оценивая ее переносимость), однако в течение первой половины дня; затем перевести пациента на прием полной дозы ГК в утренние часы.
· Оценка эффективности терапии проводиться через 2-4 недели от начала терапии ГК. Положительный эффект терапии расценивается при начавшемся снижении уровня КФК, АСТ, АЛТ, уменьшении интенсивности кожных проявлений, нарастании мышечной силы.
Отсутствие положительной динамики в течение 4 недель требует повторного проведения дифференциального диагноза с фенотипически схожими нозологиями, включающими в клинической картине миопатический синдромам, в т.ч., пересмотра морфологического материала.
· Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
· Снижение дозы ГК начинается при нормализации уровня КФК в сыворотке крови, исчезновении спонтанной активности при и-ЭМГ, нарастании мышечной силы, объема движений и проводиться под строгим клинико-лабораторным контролем. Доза ГК постепенно снижается по ¼ дозы от исходной в месяц, в среднем, по ½ — ¼ таблетки в 5-7-10 дней до достижения поддерживающего уровня. Темп снижения зависит от исходной дозы ГК и степени активности болезни. Чем ниже доза ГК, тем медленнее ее снижение.
Доза ГК постепенно снижается по ¼ дозы от исходной в месяц, в среднем, по ½ — ¼ таблетки в 5-7-10 дней до достижения поддерживающего уровня. Темп снижения зависит от исходной дозы ГК и степени активности болезни. Чем ниже доза ГК, тем медленнее ее снижение.
· Поддерживающая доза ГК индивидуальна: 5-10, реже 15 мг/сутки и зависит от клинико-иммунологического подтипа болезни, возраста больного. При ЮДМ известны случаи клинико-лабораторной ремиссии на фоне длительной отмены терапии. Полная отмена ГК у взрослых пациентов, как правило, ведет к обострению болезни, даже если они несколько лет находились в состоянии полного клинического ответа.
Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и не служит поводом для применения меньших (не адекватных) доз ГК назначаемых внутрь, как в острый период болезни, так и при ее обострении.
Потенциальные показания к подключению иммуносупрессивной терапии
· Принадлежность больных к клинико-иммунологическим подтипам ПМ/ДМ, особенностью которых является заведомо «плохой ответ» на терапию ГК: АСС c ФА, у пациентов антител к SRP
· Язвенно-некротический васкулит
· Обострение заболевания при снижении дозы ГК
· Стероидрезистентность у больных, ране получавших неадекватно малые дозы ГК
· Неэффективность ГК в течение 3-х месяцев
· Тяжелые побочные эффекты ГК, лимитирующие назначение адекватной дозы ГК (неконтролируемые сахарный диабет или артериальная гипертензия, острая язва желудка, множественные остеопоретические переломы)
Рекомендации по лечению ПМ/ДМ соответственно наиболее тяжелым синдромам ИПЛ с синдромом ФА при АСС
· Наиболее тяжелым и плохо контролируемым монотерапией ГК при ПМ/ДМ синдромом является АСС. Плохой прогноз определяется вовлечением в патологический процесс легочной ткани — ИПЛ с развитием ФА.
· Объем терапии и выбор препарата (в сочетании с ГК) определяется тяжестью ИПЛ (по данным КТ и ФЛТ: форсированной ЖЕЛ, DLCO и с учетом анамнеза (ранее применяемые иммуносупрессивные препараты).
· Основное место в лечении ИПЛ занимает ЦФ, назначаемый внутривенно в дозе 500 мг/м2 -750 мг/м2 мг в месяц в сочетании с ГК (уровень доказательности А)
· Длительность ЦФ должна быть не менее 6 месяцев (уровень доказательности С)
· Контроль эффективности ЦФ осуществляется по динамической оценке (1 раз в 6 месяцев) форсированной ЖЕЛ, показателей DLCO (уровень доказательности А), а также данных КТВР легких.
· При агрессивном течении СФА при выраженном снижении ЖЕЛ и DLCO, а также, в случае неэффективности ранее применяемой терапии ЦФ, целесообразно применение РТМ.
· Применение ММФ рассматривается в качестве терапии «второго» ряда при ИПЛ в случае невозможности применения ЦФ или РТМ
Дисфагия
· Дисфагия является фактором риска аспирационной пневмонии, течение и терапия которой осложняется иммуноскомпроментированностью пациентов, связанной с терапией высокими дозами ГК и цитостатиков.
· Рекомендовано проведение пульс-терапии ГК (метипред 1000мг) N 3 в сочетании с пероральным приемом ГК в адекватной дозе.
· Тяжелая дисфагия является потенциальным показанием ВВИГ.
Наличие дисфагии у больных ПМ/ДМ служит поводом для проведения более активного онкопоиска (уровень доказательности Д).
Язвенно-некротический васкулит
Наличие язвенно-некротического васкулита является показанием для проведения пульс-терапии циклофосфамидом в дозе 600-800-1000 мг в месяц в сочетании метилпреднизолоном 500-1000мг.
Кожный синдром при ДМ в сочетании с проксимальной мышечной слабостью отражает активность болезни и, как правило, контролируется ГК в адекватных дозах в острый период болезни.
При резистентном кожном синдроме, сохраняющемся на фоне восстановления мышечной силы, рекомендуется применение антималярийных препаратов (гидроксихлорохин по 200–400 мг/сут), ММФ, топических стероидов.
Наличие резистентного кожного синдрома и/или язвенно-некротического васкулита у больных ПМ/ДМ служит поводом для проведения более активного онкопоиска (уровень доказательности С).
Лихорадка или субфебрилитет встречаются редко, главным образом при АСС с острым началам болезни.
· Контролируется ГК и не требует дополнительной терапии (уровень доказательности В).
При появлении субфебрилитета (или лихорадки) у пациентов на фоне лечения ГК в период клинико-лабораторной положительной динамики – исключение присоединения сопутствующей инфекции. Необходимо учитывать атипизм течения инфекционных осложнений на фоне иммуносупрессивной терапии.
Поражение суставов
· Наличие артрита при ПМ/ДМ может присутствовать в начале болезни. Артриты входят в состав симптомокомплекса АСС, хорошо контролируются ГК и не требуют дополнительного лечения.
· Сгибательные контрактуры, как правило, локтевых, реже коленных суставов, развиваются в острый период ПМ/ДМ и обусловлены воспалительным поражением мышечной ткани, а не непосредственным поражением суставов. Дополнительного медикаментозного лечения не требуется (уровень доказательности С).
Кальциноз мягких тканей
· Кальциноз мягких тканей наиболее часто присутствует (и более агрессивен) при ЮДМ
· Появление множественных кальцинатов, как правило, сопутствует острому течению ПМ/ДМ. Кальцинаты сохраняются на фоне снижения активности болезни, даже при достижении клинико-лабораторной ремиссии и наиболее выражении при ЮДМ.
· При ЮДМ, с целью снижения риска развития кальциноза и его распространенности применяется пульс-терапия ГК в дозе 1-2 мг/кг/сут.
· Хирургическое лечение малоэффективно, поскольку повышает риск присоединения вторичной инфекции и может спровоцировать появление новых кальцинатов.
· В качестве медикаментозной терапии применяютт бисфосфонаты (ксидифон, фосамакс, фосаванс и др.), однако полного контроля над процессом гетеротопического кальцийобразования не достигается.
· Для лечения кальциноза применяется, также динатриевая соль этилендиаминотетрауксусной кислоты (Na2ЭДТА), образующей комплексные соединения с различными катионами, в т.ч.с ионами Са2+ и способствует выделению их с мочой.
· Имеются данные об эффективном предотвращении прогрессирования кальциноза при применении ВВИГ в течение 2 дней каждый месяц в сочетании с метилпреднизолоном (уровень доказательности С).
Традиционные иммуносупрессивные препараты, применяемые в лечении ПМ/ДМ
· Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности или плохой переносимости перорального приема препарата, особенно в высоких дозах).
· Азатиоприн по 2–3 мг/кг/сут (100–200 мг/сут)
· Циклоспорин А по 2,5–5.,0 мг/кг/cутки назначают пациентам с резистентными к ГК формами заболевания, в т.ч. при хроническом течении болезни, связанной с неадекватно малой инициальной дозой ГК (уровень доказательности С).
· ММФ. Имеются данные об эффективности ММФ при ИПЛ и резистентном кожном синдроме. Прием начинают с дозы 1000 мг/сут (в 2 приема), постепенно титруя дозу до 2000 мг/сут под контролем показателей общего и биохимического анализов крови (уровень доказательности С).
Общие принципы лечения иммуносупрессивными препаратами:
— титрование дозы: назначение с небольшой дозы и постепенное ее повышение под контролем переносимости.
— контроль переносимости: оценка уровня гемоглобина, числа лейкоцитов, тромбоцитов, азота мочевины, креатинина, активности АСТ, АЛТ. При уменьшении числа лейкоцитов менее 2,5х109/л и/или тромбоцитов – менее 100 х 109/л и повышении концентрации АСТ, АЛТ более чем в 3 раза от верхней границы нормы, лечение необходимо прекратить до устранения симптомов токсичности.
— при присоединении интеркурренотой инфекции, в т.ч. герпетической – временная отмена иммуносупрессивных препаратов до исчезновения ее признаков.
Применение ВВИГ 2 г/кг 1 раз в месяц в течение 3 месяцев является эффективным методом лечения ПМ/ДМ (особенно ЮДМ), резистентного к стандартной терапии. Потенциальным показанием для ВВИГ является тяжелая дисфагия. (уровень доказательности В)
Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным к другим метода лечения ПМ/ДМ в сочетании с ГК и цитотоксическими препаратами.
Новые направления терапии ПМ/ДМ. Биологические препараты
В настоящее время активно изучается роль и место биологической терапии в терапии ПМ/ДМ.
— Применение в терапии ПМ/ДМ ингибиторов фактора некроза опухоли α TNF-a (инфликсимаба) не принесло желаемых результатов: поскольку он не способен контролировать активность болезни, в том числе ИПЛ, а также, увеличивают риск оппортунистических инфекций [10, 44].
— Имеются данные об успешном применении этанерцепта в качестве стероидсберегающей терапии. (Уровень доказательности Д)
— Применение блокаторов ко-стимуляции Т-лимфоцитов (абатацепта) в сочетании с тиосульфат натрия при ЮДМ с язвенно-некротичнским васкулитом и прогрессирующим кальцинозом оказало положительный эффект в виде нарастания мышечной силы, восстановления целостности кожный покровов, снижения прогрессирования кальциноза, что позволило снизить поддерживающую дозу ГК. (Уровень доказательности Д)
— Особое место среди биологических препаратов, на сегодняшний день, применяемых при ПМ/ДМ, занимает использование анти В-клеточной терапии. Накоплен положительный опыт по применению РТМ у пациентов с тяжелым мышечным поражением и при АСС с СФА, резистентных к ГК и применяемой ранее традиционной цитостатической терапии (уровень доказательности Д)
Практически все авторы описывают высокую эффективность РТМ при ПМ/ДМ. Так, на фоне терапии РТМ (в сочетании с ГК) наблюдается положительная клинико-лабораторная динамика (уменьшение выраженности кожного синдрома, нарастание мышечной силы).
— В случае применения РТМ при АСС с СФА, позитивный эффект наблюдался более, чем у 70% больных в виде увеличения показателей функции внешнего дыхания: увеличения показателей ЖЕЛ и DLCO, а также уменьшения инфильтратов по КТ грудной клетки.
— Максимальный эффект развивался через 12 недель после первой инфузии и коррелировал со снижением CD 20 + В клеток.
Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой инициальной дозой ГК
· Сложность ведения таких пациентов обусловлена развитием поствоспалительной фиброзной и жировой инволюции мышечной ткани (при назначении не адекватной инициальной дозы ГК). В этом случае отсутствует (или минимален) воспалительный компонент (миозит), являющийся субстратом для проведения противовоспалительной терапии ГК.
· Клинически — сохраняется проксимальная мышечная слабость, однако показатели активности болезни (уровень КФК, данные и-ЭМГ, биоптата мышечной ткани) не свидетельствуют в пользу текущего воспалительного процесса.
· Присутствие фиброзной и жировой инволюции мышечной ткани подтверждается при МРТ исследовании проксимальных отделов конечностей.
· Повышение дозы ГК целесообразно при наличии, хотя бы минимальных, признаков воспаления мышечной ткани.
· Хроническое течение болезни, связанное с неадекватно малой инициальной дозой ГК является потенциальным показанием для подключения иммуносупрессивной терапии (Циклоспорин А, ММФ, метотрексат, азатиоприн) .
Реабилитационные мероприятия и обучение пациентов.
Проводятся в зависимости от стадии заболевания
· В острую фазу противопоказаны ЛФК и физические нагрузки, проводимые пациентами «через силу»; допускаются только пассивные упражнения
· В стадию выздоровления — изометрические, а затем изотонические упражнения
· В хронической стадии — анаэробные упражнения
Профилактика ГК- остеопороза
Препараты кальция в сочетание с витамином Д3, бисфосфанаты.
Профилактика язвенных осложнений
Гастропротекторы (миозпростол, ранитидин, омепразол).
Профилактика стероидного диабета
Строгое исключение потребления продуктов, содержащих глюкозу, в т.ч., сладких фруктов, соков и йогуртов.
Предосторожности: Исключение контакта с инфекционными больными – во избежание присоединения вторичной инфекции.
Избегание физических перегрузок (в острый период ЛФК противопоказана, только пассивные движения).
Заболевания мышц воспалительные
Заболевания мышц воспалительные
Воспалительные заболевания мышц (миозиты) — общее название патогенетически разнородных заболеваний мышц, характеризующихся нарушением сократительной способности мышечных волокон и проявляющихся мышечной слабостью, уменьшением объёма активных движений, снижением тонуса, атрофией.
Различают следующие типы воспалительных заболеваний мышц: идиопатические воспалительные заболевания мышц, инфекционные миопатии, лекарственные миопатии, очаговый миозит, гигантоклеточный миозит, эозинофильный миозит. К идиопатическим воспалительным заболеваниям мышц относят: полимиозит, дерматомиозит, полимиозит как перекрёстный синдром в клинической картине системных заболеваний соединительной ткани, паранеопластические полимиозит и ДМ, полимиозит с внутриклеточными включениями.
Идиопатические воспалительные заболевания мышц
• Полимиозит и дерматомиозит — см. Полимиозит и дерматомиозит.
• Полимиозит как перекрёстный синдром при системных заболеваниях соединительной ткани наблюдают в клинической картине системной склеродермии, СКВ, ревматоидном артрите, синдроме Шёгрена, системных васкулитах. Представляет собой трудность в диагностике, если является дебютным синдромом. По сравнению с полимиозитом/дерматомиозитом при перекрёстных синдромах часто наблюдают синдром Рейно, в крови — высокие титры АНАТ, отсутствие миозитспецифических АТ, в то время как при исследовании биоптата мышц отмечают сходные гистологические изменения. В лечении системных заболеваний соединительной ткани, имеющих в клинической картине как перекрёстный синдром полимиозит, необходимы активная тактика (высокие дозы ГК), применение цитостатических иммунодепрессантов.
• Паранеопластический полимиозит/дерматомиозит. При дерматомиозите частота выявления опухолей колеблется от 15–20%. Наиболее частая локализация опухолей (50%) — назофарингеальный рак. Наибольший риск обнаружения опухоли сопряжён со следующими факторами: возраст старше 45 лет, мужской пол, высокий уровень КФК, яркие клинические симптомы миозита. Устранение опухоли приводит к постепенному обратному развитию дерматомиозита как паранеопластического синдрома.
• Миозит с внутриклеточными включениями получил свое название ввиду характерной гистологической картины: наличия крупных внутриядерных и внутрицитоплазматических базофильных включений, выявляемых при световой микроскопии. Частота миозита с включениями составляет 15–28% всех идиопатических воспалительных миопатий. К клиническим особенностям миозита с включениями относят: асимметричность поражения, поражение не только проксимальной, но и дистальной мускулатуры (сгибателей пальцев, разгибателей стоп), лёгкое или умеренное повышение КФК, отсутствие миозитспецифических АТ, резистентность к лечению ГК и цитостатическими иммунодепрессантами, пожилой возраст больных (в среднем 60 лет), более частое развитие болезни у мужчин, чем у женщин (2:1).
Инфекционные воспалительные заболевания мышц
• Токсоплазмоз • Миозит в рамках Лаймской болезни • Трихинеллёз • ВИЧ-ассоциированные миопатии •• характерна мышечная слабость в сочетании с диареей и снижением массы тела •• на поздних стадиях возможны мышечные абсцессы •• на фоне приёма зидовудина возможна лекарственная дозозависимая миопатия.
Гигантоклеточный миозит • При саркоидозе развивается в 25% случаев. Диагноз подтверждают обнаружением в мышечном биоптате гранулём, содержащих клетки Пирогова–Лангханса • При myasthenia gravis (тимоме) наблюдают иногда гранулематозный или гигантоклеточный полимиозит и миокардит.
Очаговый миозит. Редкое заболевание, которое проявляет себя единичными или множественными болезненными очагами в мышцах. При очаговом миозите с единичными фокусами необходим дифференциальная диагностика с опухолями мышц (саркомой, рабдомиосаркомой), а множественный очаговый миозит необходимо отдифференцировать от инфарктов мышц при узелковом панартериите.
Эозинофильные воспалительные заболевания мышц • Миопатия как синдром при эозинофильном фасциите • Рецидивирующий эозинофильный полимиозит — очень редкое заболевание, проявляющееся болью и напряжённостью, но не слабостью, мышц шеи и нижних конечностей. Имеет характерную гистологическую картину эозинофильной инфильтрации перимизия • Синдром эозинофилии-миалгии связан с приёмом L-триптофана, в настоящее время практически не выявляют.
Воспалительные заболевания мышц, индуцированные приёмом ЛС. Общие черты: связь с приёмом ЛС, постепенное обратное развитие после отмены ЛС • Стероидные миопатии. Характерные черты: нормальный уровень КФК, положительная динамика на фоне снижения дозы ГК, отсутствие признаков мышечного воспаления при гистологическом исследовании мышечной ткани • Миопатии на фоне антималярийных препаратов (редко) • Миопатии на фоне применения статинов. Описаны случаи рабдомиолиза, чаще возникающего при комбинации статинов с фибратами • Миопатии как побочный эффект пеницилламина: птоз век, затруднения при глотании, прогрессирование при продолжении приёма препарата до поражения дыхательных мышц • Миопатии как побочный эффект зидовудина — см. выше.
МКБ • M60 Миозит • M63* Поражения мышцы при болезнях, классифицированных в других рубриках
ДИАГНОСТИКА И ЛЕЧЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ МЫШЦ
Воспалительные миопатии встречаются редко. Точных данных о заболеваемости или распространенности нет, но если взять два наиболее распространенных состояния, дерматомиозит и миозит с тельцами включения, их совокупная годовая заболеваемость, вероятно, составит менее 200 новых случаев в год в Великобритании (население ~ 60 миллионов). Учитывая, что пациенты с воспалительной миопатией могут обращаться в одну из нескольких специальностей (например, дерматологию, ревматологию, неврологию, общую медицину) и лечиться в ней, и что в Великобритании насчитывается около 250 неврологов, очевидно, что наиболее общие неврологи будут видеть эти состояния в среднем, вероятно, не чаще, чем раз в пару лет.Возможно, в 70% случаев диагностика и лечение просты, и успешный ранний опыт может стимулировать чувство компетентности, которое только поколебляется, когда дела идут не так, как ожидалось. Нет сомнений в том, что пациентам лучше всего обслуживает кто-то, обладающий конкретным опытом и интересом в этих редких состояниях, и в идеале в каждом регионе должен быть такой специалист, к которому они могут быть направлены.
В этой статье будут рассмотрены классификация воспалительных миопатий, клинические особенности так называемых идиопатических воспалительных миопатий, а также подходы к их диагностике и лечению.Что касается медикаментозного лечения, существует абсолютная нехватка рандомизированных контролируемых исследований, и лучший совет, который можно предложить, основан на «мнении экспертов». Крайне необходимы многоцентровые исследования. Акцент будет сделан на подводные камни в диагностике и лечении. Будут отмечены текущие взгляды на патогенез, но остаются многие области невежества, и вполне вероятно, что в следующие несколько лет мнения будут существенно пересмотрены.
Некоторые фундаментальные проблемы и полезные сообщения приведены во вставке 1.
Вставка 1
Десять важных моментов, над которыми стоит задуматься
Боль и дискомфорт при миозите редко бывают заметными
Нормальный верхний предел креатинкиназы сыворотки выше, чем вы думаете
Неспособность учесть пункты 1 и 2 приводит к тому, что многим пациентам ставится неправильный диагноз и лечится полимиозит
Чистый полимиозит — редкое заболевание
Воспалительные инфильтраты при биопсии мышц могут быть вторичным явлением, и непонимание этого является частой причиной неправильного диагноза и неправильного лечения
Отсутствие воспалительных инфильтратов при биопсии не исключает миозит
Несмотря на их клиническое сходство, дерматомиозит и полимиозит являются принципиально разными заболеваниями с точки зрения патогенеза
Миозит с включенными тельцами, наиболее распространенная приобретенная миопатия в возрасте старше 50 лет, часто ошибочно принимают за полимиозит.Действительно ли это «миозит», остается предметом горячих споров
Дерматомиозит может быть паранеопластическим заболеванием, особенно у пожилых людей
Долгосрочная заболеваемость и смертность связаны с интерстициальным заболеванием легких, поражением миокарда и ассоциированными злокачественными новообразованиями
КЛАССИФИКАЦИЯ
Вкратце, воспалительные миопатии (вставка 2) — это просто те расстройства, при которых основным патологическим процессом является воспаление в мышцах.В большинстве случаев основным клиническим последствием является слабость, гораздо реже — боль. Таким образом, это определение исключает миопатии со вторичным воспалением в мышцах, такие как некоторые мышечные дистрофии, и состояния, при которых воспалительный процесс происходит в связанных тканях, а не в самой мышце, например ревматическая полимиалгия. Эти последние состояния особенно важны, поскольку их можно неправильно диагностировать и неправильно лечить как формы миозита. При некоторых состояниях, таких как саркоидоз, ревматоидный артрит и синдром Шегрена, воспалительные инфильтраты в мышцах, вероятно, относительно распространены, но клинические последствия, такие как слабость, редки.Макрофагический миофасциит, недавно определенное заболевание, наблюдаемое в основном во Франции, вероятно, является вторичным по отношению к алюминию, используемому в качестве адъюванта в некоторых вакцинах.
Вставка 2
Классификация воспалительных миопатий
Этот обзор будет ограничен четырьмя расстройствами из-за их относительной частоты и важности для общего невролога. Дерматомиозит (DM) — наиболее распространенная форма классической воспалительной миопатии.Изолированный полимиозит (ПМ) встречается редко, но часто ставится неверный диагноз. Но ПМ относительно часто ассоциируется с различными проявлениями заболевания соединительной ткани, ситуация, для которой многие используют термин «синдром перекрытия». Некоторые утверждают, что существует разница между «перекрытием» и «ассоциацией», но на самом деле мы в настоящее время не знаем истинной взаимосвязи между этими различными условиями. Продолжаются дискуссии о том, является ли миозит с телец включения (IBM) первичной воспалительной миопатией или воспалительные изменения являются вторичным явлением.Одним из самых веских аргументов в пользу последнего является то, что IBM плохо реагирует, если вообще реагирует на лечение иммунодепрессантами / противовоспалительными средствами. IBM часто изначально ошибочно диагностируется и рассматривается как ПМ, обычно из-за неспособности оценить конкретные клинические и патологические особенности состояния.
ПАТОГЕНЕЗ
Долгое время DM и PM считались практически идентичными заболеваниями, различающимися только наличием или отсутствием поражения кожи.В настоящее время есть неопровержимые доказательства того, что это фундаментально разные расстройства с точки зрения патогенеза и что их клиническое сходство просто отражает ограниченный набор мышечных реакций на заболевание. Вероятно, что в будущем их различные патогенезы приведут к специфическому иммуномодулирующему лечению для каждого расстройства, но в настоящее время это не так, и лечение, как и для других аутоиммунных заболеваний, принимает форму «мушкетона» подхода к иммуносупрессии.
DM представляет собой гуморально опосредованное аутоиммунное заболевание.Атака, зависимая от комплемента, приводит к разрушению капилляров в мышцах и других тканях. В мышцах возникающая микроангиопатия приводит к характерным патологическим признакам инфаркта и периферифасцикулярной атрофии. Неизвестно, вызывает ли это отложение циркулирующих иммунных комплексов или связывание антитела с эндотелиальным антигеном, которое запускает этот литический путь комплемента.
PM вызвано клеточным иммунным феноменом. Аутоинвазивные CD8 + Т-клетки, узнавая неизвестный мышечный антиген, проникают в ненекротические мышечные волокна, экспрессирующие антиген главного комплекса гистосовместимости класса 1 (MHC-1), и приводят к их разрушению.
В IBM, хотя есть некоторое сходство с иммуноцитологическими данными, полученными при PM, есть доказательства того, что фундаментальный патологический процесс отличается и что по крайней мере некоторые воспалительные и иммунные изменения, наблюдаемые при IBM, могут быть эпифеноменами. Сходства были выявлены между патологическими находками в мышцах при IBM и головном мозге при болезни Альцгеймера, но, опять же, еще предстоит определить, являются ли они первичными или вторичными явлениями.
Обнаружение так называемых миозит-специфических антител в крови многих пациентов с СД, ПМ и, реже, IBM вызывает дополнительные вопросы об иммунопатогенезе, хотя нет никаких доказательств того, что эти антитела являются патогенными.Постоянным обнаружением при всех трех расстройствах является экспрессия антигена MHC-1 (который не экспрессируется конститутивно) на поверхности неповрежденных мышечных волокон, и действительно, это может быть использовано в качестве указателя на диагноз идиопатической воспалительной миопатии даже при ее отсутствии. воспалительных инфильтратов. Потенциально объединяя эти два наблюдения вместе, недавно на животной модели было показано, что индукции поверхностной экспрессии MHC 1 в мышечных волокнах достаточно для запуска воспалительной миопатии, хотя и не совсем параллельно с PM или DM, и продукции циркулирующей антисинтетазы. антитела.
Очевидно, что нам еще многое предстоит узнать об афферентных и эфферентных звеньях иммунного процесса при каждом из этих расстройств, и пока мы этого не сделаем, должна существовать неопределенность в отношении наилучшего подхода к классификации, не говоря уже о конкретном лечении и будут и дальше использоваться неудовлетворительные термины, такие как «синдром перекрытия».
КЛИНИЧЕСКИЕ ХАРАКТЕРИСТИКИ
Многие наследственные миопатии (например, мышечные дистрофии) характеризуются высокоселективным поражением определенных мышц, при этом физически прилегающие мышцы демонстрируют заметно разную степень поражения.Таким образом, при фасциально-лопаточно-плечевой мышечной дистрофии истощение и слабость проксимального отдела верхней конечности влияет на перискапулярные и плечевые (т. Е. Двуглавые и трехглавые) мышцы, но не на дельтовидную. Может даже быть различное поражение внутри мышцы, так что при некоторых мышечных дистрофиях конечностей и поясов можно увидеть области как атрофии, так и гипертрофии внутри четырехглавой мышцы. Эта избирательность, которая является мощным диагностическим инструментом для опытного клинициста, не является признаком DM или PM, но очевидна во многих случаях IBM.
При СД и ПМ, как и при многих приобретенных миопатиях (например, эндокринных и лекарственно-индуцированных миопатиях), характерным признаком является генерализованное истощение и слабость проксимальных мышц, при этом мускулатура тазового пояса почти всегда поражается сильнее, чем мышцы плечевого пояса. (необычно проявление слабости плечевого пояса). Типичные симптомы включают затруднения при подъеме по лестнице и вставании с низкого стула, а также трудности с выполнением задач на уровне плеч и выше, таких как самостоятельный уход и подъем предметов на полки.Дистальная слабость — это поздний признак, который никогда не бывает таким серьезным, как проксимальная слабость.
Напротив, IBM часто демонстрирует исключительно избирательное поражение мышц, наиболее характерным из которых является истощение и слабость четырехглавой мышцы (и почти наверняка это наиболее частая причина того, что раньше называлось «изолированной миопатией четырехглавой мышцы») и длинного пальца. сгибатели. Клинические последствия — падение из-за подгибания колен и слабости хватки. Другими словами, наиболее важные функции нижних и верхних конечностей, соответственно, нарушены, и заболевание может вызвать глубокую функциональную инвалидность.Нам еще предстоит найти удовлетворительные механические средства или другие подходы для решения этих проблем.
В дополнение к этим общим наблюдениям о характере поражения мышц, каждое заболевание также имеет дополнительные клинические характеристики и ассоциации.
Дерматомиозит
DM может поражать людей всех возрастов, но заболевание у детей несколько отличается от такового у взрослых; Характерной чертой может быть общее страдание, а не очевидная слабость, чаще встречается подкожный кальциноз, лицо может покраснеть без более специфических характеристик сыпи, наблюдаемых у взрослых, и может быть вовлечен кишечник.Сопутствующее злокачественное новообразование в детстве встречается редко.
У взрослых заболевание обычно проявляется подостро, симптомы развиваются в течение нескольких недель, но реже начало может быть очень острым с обширным мышечным и подкожным отеком. У пациентов с тяжелым заболеванием может развиться дыхательная недостаточность. Дисфагия с риском аспирации также часто встречается при тяжелом течении болезни.
Около 20% случаев, чаще в пожилом возрасте, связаны с лежащей в основе злокачественной опухолью, и, как и в случае с другими паранеопластическими заболеваниями, новообразование может не проявляться в течение значительного времени (возможно, через 2–3 года) после первого обращения.В отличие, например, от миастенического синдрома Ламберта-Итона, здесь нет тесной связи с одним конкретным местом или типом опухоли.
Кожная сыпь очевидна у большинства пациентов и часто является первым симптомом. Он может отсутствовать на всем протяжении (дерматомиозит синусный дерматит), тогда диагноз основывается на характерных данных биопсии мышц при СД, быть кратковременным и довольно неспецифическим (например, эритема лица или груди) или его трудно увидеть у темнокожих людей. (и обратите внимание, что заболеваемость СД выше у лиц афро-карибского происхождения).Дерматологи могут видеть СД без явной мышечной слабости (дерматомиозит синусит), но у большинства этих пациентов биопсия мышц выявляет характерные отклонения.
Сыпь имеет много общего с сыпью, наблюдаемой при системной красной волчанке, и действительно есть общие патологические особенности, включая наличие волнообразных канальцев в эндотелиальных клетках капилляров. Оба проявляют светочувствительность. Типичные кожные признаки DM включают эритему на светлых открытых щеках (скуловое распространение), верхней передней части грудной клетки (знак V), верхней задней части груди (знак платка) и суставах.Веки могут быть отечными и проявлять пурпурное (гелиотропное) изменение цвета, но это менее постоянный признак, чем менее специфическая эритема и ручные знаки. Наряду с эритемой может наблюдаться чешуйчатая сыпь (признак Готтрона) над суставами пальцев, но фаланги сохраняются. У основания ногтей можно увидеть расширенные капилляры. Сухие, потрескавшиеся руки называют «руками механика»; это часто, но не всегда, связано с присутствием антисинтетазных антител, включая анти-Jo-1.
Интерстициальное заболевание легких встречается примерно в 10% случаев и иногда является настоящей проблемой. Здесь есть сильная связь с присутствием антител к синтетазе, особенно к Jo-1. Его можно спутать с пневмонитом, вызванным метотрексатом, когда этот препарат используется для лечения миозита. Могут наблюдаться миокардит и нарушения проводимости, особенно при тяжелом остром заболевании. Заболеваемость и смертность при СД в основном связаны с интерстициальным заболеванием легких, поражением миокарда и осложнениями дыхательной недостаточности, вторичными по отношению к слабости дыхательных мышц.
Полимиозит
Развитие слабости происходит медленнее, чем в DM, обычно за месяцы, но обычно быстрее, чем в IBM. Дисфагия и слабость лица возникают нечасто. Это болезнь взрослой жизни. Неясно, связан ли PM со злокачественным новообразованием, но если это так, связь менее сильна, чем для DM, и, согласно имеющимся данным, обширный поиск лежащей в основе злокачественной опухоли не оправдан. Неопределенность связана с тем, что в более ранних исследованиях использовались устаревшие критерии для различения PM и DM.
PM никогда не ассоциируется с кожными признаками DM. Как и при СД, интерстициальное заболевание легких связано с анти-Jo-1 и другими специфическими для миозита антителами. Возможно поражение миокарда.
«Чистые» ТЧ встречаются редко, но, как указано ниже, ТЧ могут быть связаны с другими проявлениями заболевания соединительной ткани. Диагностика PM часто выполняется ошибочно (вставка 3). «Устойчивые к лечению» ПМ обычно являются результатом неправильного диагноза, чаще всего IBM, и действительно, многим пациентам с IBM, наконец, был поставлен правильный диагноз только после того, как они не ответили на иммуносупрессию.Неспособность диагностировать ИБМ на начальном этапе обычно происходит из-за того, что не осознает специфический паттерн мышечной слабости, в частности, отсутствует слабость сгибания пальцев, и не может оценить специфические патологические особенности, особенно окаймленные вакуоли и нитчатые включения. Мышечную дистрофию можно принять за ПМ, чаще всего, когда продолжительность симптомов непродолжительна. Образец слабости может быть очень полезен при проведении различия. Вторичные воспалительные инфильтраты могут вызывать патологическую спутанность сознания, особенно при дисферлинопатии и фациально-плечевой мышечной дистрофии.При эндокринных миопатиях обычно проявляются другие особенности эндокринопатии. Миопатия, индуцированная статинами, становится все более распространенным явлением при более широком использовании этих препаратов. Дефицит кислотной мальтазы часто первоначально ошибочно диагностируется как мышечная дистрофия конечностей и поясов или ПМ, если не присутствует характерное раннее поражение диафрагмы. Болезнь Макардла, особенно при наличии фиксированной проксимальной слабости и отсутствии четкого анамнеза индуцированного обострения, вызванного физической нагрузкой, может быть ошибочно диагностирована как ПМ.Нейрогенные расстройства могут вызывать путаницу, если не осознавать, что активная денервация может сопровождаться повышением уровня креатинкиназы в сыворотке (SCK) и что нейрофизиологические данные могут сбивать с толку.
Вставка 3
Болезни, часто ошибочно диагностируемые как полимиозит
Наконец, диагноз ПМ часто ошибочно ставится в довольно распространенной ситуации пациента с болью, симптоматической, но не объективной слабостью и умеренным повышением SCK.Биопсия мышц может выявить незначительные «отклонения», которые были ошибочно приняты для подтверждения диагноза ПМ. Стероиды могут предложить короткий период улучшения состояния в медовый месяц. При более внимательном рассмотрении таких пациентов можно заметить, что их боль не только в мышцах, но также затрагивает их суставы, а иногда и кожу и кости. То, что они называют слабостью, — это скорее трудность в поддержании усилий. Первоначальный осмотр может указывать на слабость, но при поощрении и функциональных тестах, таких как подъем из приседа, становится очевидным, что истинной слабости нет.Большинство лабораторий указывают слишком низкую верхнюю нормальную концентрацию SCK. Ценности выше у мужчин, чем у женщин, и у чернокожих, чем у белых. У нормального мужчины, выполняющего умеренную физическую активность, концентрация может достигать 600 МЕ / л. У кого-то, занимающегося более энергичными упражнениями, особенно у чернокожих, могут быть значения до 1000 МЕ / л. Трудность, конечно, состоит в том, чтобы знать, актуальна ли концентрация, скажем, 450 МЕ / л у человека, жалующегося на мышечную боль, или нет. Пациентам с таким типом проблем никогда не следует назначать стероиды без биопсии мышц.Но они есть, и когда их симптомы не исчезнут, а затем будет проведена биопсия, может быть невозможно интерпретировать результаты. Правильный диагноз у этой довольно распространенной группы пациентов может оказаться неуловимым. Ревматологи отправляют их к неврологам с диагнозом «полимиозит», и им возвращают ярлык «синдром, подобный полимиалгии», «фибромиалгия» или синдром хронической усталости.
Миозит с включенными тельцами
Это наиболее распространенная приобретенная миопатия у людей старше 50 лет.Это значительно чаще встречается у мужчин, что необычно для аутоиммунного заболевания и еще один фактор, который вызывает некоторые сомнения относительно того, является ли это истинной первичной воспалительной миопатией. Нечасто может присутствовать уже в третьем десятилетии. Семейные случаи зафиксированы. Его более правильно обозначать как спорадический IBM, чтобы отличить его от гораздо более редкого унаследованного IBM. Сюда входят доминантные и рецессивные формы, которые имеют общие клинические и патологические черты со спорадическими ИБГ, за заметным исключением отсутствия воспалительных инфильтратов.
Основные клинические особенности IBM уже обсуждались, но стоит повторить, чтобы подчеркнуть, что наличие дистальной слабости, которая является столь же серьезной или более выраженной, чем проксимальная слабость в той же конечности, очень характерно. Обычно это наиболее очевидно для сгибателей пальцев, но также может быть выражена слабость при тыльном сгибании голеностопного сустава. Другой особенностью IBM, но не PM или DM, является асимметричное вовлечение мышц, иногда выраженное. Легкая слабость лица может наблюдаться даже тогда, когда слабость конечностей относительно легкая (редко при СД и ПМ), а дисфагия может быть ранним или поздним признаком.
Скорость прогрессирования обычно ниже, чем в PM. Пациенты пожилого возраста часто не могут легко определить дату возникновения проблемы, поскольку они связывают ранние симптомы с нормальными последствиями старения. Существенное истощение и слабость четырехглавой мышцы часто проявляются при первом обращении. Основная практическая проблема — падение из-за невозможности зафиксировать колени.
Специфические антитела к миозиту гораздо реже встречаются в IBM по сравнению с DM и PM. Точно так же сопутствующие состояния встречаются реже, но есть сообщения о связи ИБМ с синдромом Шегрена, гепатитом С, инфекцией HTLV-1 и саркоидозом.
Перекрывающиеся / ассоциированные синдромы
Вместо того, чтобы пытаться определить конкретные ассоциации («расщепление»), на основе наших текущих несколько ограниченных знаний, возможно, лучше всего просто отметить, что у пациентов с миозитом могут также быть обнаружены признаки других заболеваний соединительной ткани («образование комков» »). Многие из этих ассоциаций были описаны выше. СД может быть связан с признаками склеродермии (часто с циркулирующими антителами против PM-Scl) и смешанным заболеванием соединительной ткани.ТЧ связаны со многими системными аутоиммунными заболеваниями, и действительно, изолированные ТЧ редки. DM и PM могут быть связаны с неспецифическими симптомами, такими как лихорадка и артралгия, а также с феноменом Рейно. Эти различные явления вместе с интерстициальным заболеванием легких, когда они связаны с антисинтетазными антителами, образуют основные компоненты антисинтетазного синдрома . У таких пациентов миозитный компонент может быть незначительным и изначально упускаться из виду. Наконец, чтобы избежать диагностической путаницы, стоит отметить, что СД часто ассоциируется с присутствием антиядерных антител (АНА), часто в значительном титре, но без других клинических признаков или специфических иммунологических результатов, связанных с СКВ.С другой стороны, пациенты с СКВ могут иметь ассоциированный миозит.
ДИАГНОСТИКА
Квалифицированная клиническая оценка, учитывающая все уже обсужденные факторы, возможно, является самым мощным диагностическим инструментом, но в большинстве случаев требуется лабораторное подтверждение диагноза. «Золотой стандарт» — биопсия мышц. Электромиография, оценка SCK и обнаружение антител, специфичных для миозита, могут быть полезны. Возможно, пациенту со всеми классическими клиническими признаками СД, повышением SCK и типичными нейрофизиологическими изменениями не требуется подтверждающая биопсия мышцы, но в целом несколько случаев подозрения на миозит должны избежать биопсии.Если возникают трудности с лечением, часто возникает сожаление, если перед началом лечения не была проведена биопсия.
Креатинкиназа сыворотки
Несмотря на отсутствие специфичности, это чрезвычайно полезный тест как для диагностики, так и для лечения. У подавляющего большинства пациентов с СД или ПМ концентрация повышена, как правило, до нескольких тысяч МЕ / л. Он, как правило, выше у пациентов с острым началом и значительной слабостью и ниже у пациентов с более хроническими заболеваниями, но существует значительная вариабельность.По необъяснимым причинам SCK иногда остается совершенно нормальным. У большинства пациентов с ИБГ SCK повышен, но обычно ниже, чем при СД и ПМ, часто около 1000 МЕ / л. При всех этих заболеваниях могут наблюдаться значительные колебания изо дня в день, даже без лечения.
Электромиография
Электромиография обычно показывает спонтанную «раздражающую» активность (потенциалы фибрилляции и положительные острые волны) и миопатический паттерн потенциалов двигательных единиц.В ИБМ часто наблюдаются «нейрогенные» изменения в виде потенциалов большой амплитуды длительных двигательных единиц.
Биопсия мышцы
Требуется осторожность при выборе подходящей мышцы для биопсии. Обычный выбор, в основном из-за удобства и знакомства с нормальными результатами, — между дельтовидной и четырехглавой мышцами (латеральная широкая мышца бедра). Идеальная мышца для биопсии — умеренно слабая. Очень слабая атрофическая мышца может просто показать изменения конечной стадии (фиброз и замещение жировой ткани), не имеющие диагностической ценности.Это может быть особой проблемой для IBM, когда квадрицепсы уже могут быть сильно истощены при представлении, а дельтовидная мышца может не показывать слабости. Дельтовидная мышца все еще может показывать типичные патологические изменения, но если это не так и диагностические сомнения остаются, возможно, потребуется взять образец другой мышцы.
Ключевыми вопросами для интерпретации результатов биопсии мышц являются надлежащее обращение с образцами и их обработка, а также наличие опытного специалиста по оценке. В настоящее время в Великобритании неадекватная подготовка патологов в области миологии.Кроме того, за исключением специализированных центров, небольшой объем образцов мало что добавляет к их и без того ограниченному опыту. Сочетание клинициста и патолога, которые мало интересуются или не имеют опыта в области нервно-мышечных заболеваний, является частой причиной неправильного диагноза.
Основные патологические особенности DM, PM и IBM приведены в таблице 1.
Стол 1
Результаты биопсии основных мышц
Антитела
Они имеют ограниченную ценность в диагностике и лечении.Присутствие антитела против Jo-1 может предупредить о возможности развития интерстициального заболевания легких, но эту возможность в любом случае следует учитывать, и она может иметь место в отсутствие детектируемых антител. Anti-PM-Scl может указывать на возможность развития склеродерматозных изменений, а также связано с интерстициальным заболеванием легких. Антитела против SS-A / Ro связаны с синдромом Шегрена.
УПРАВЛЕНИЕ
DM и PM будут рассматриваться вместе, IBM — отдельно.Что касается лечения наркозависимости, а также, в некоторой степени, общих вопросов управления, необходимо повторить, что в литературе мало рекомендаций в виде рандомизированных контролируемых испытаний, и что многое из того, что здесь представлено, основано на личном опыте и « мнение эксперта».
Несколько общих, но часто игнорируемых принципов применимы к лечению всех этих расстройств. Упражнения важны не только для поддержания остаточной силы мышц, но и для восстановления мышц после подавления воспалительного процесса.Кроме того, как активные, так и пассивные упражнения могут снизить риск развития контрактур. Таким образом, требуется консультация достаточно опытного физиотерапевта. В зависимости от степени тяжести и характера мышечной слабости пациенту также могут потребоваться услуги эрготерапевта и ортопеда. Это основные проблемы IBM, болезнь которых со временем будет неуклонно прогрессировать. Питание важно. Недостаточное потребление калорий приведет к катаболизму и дальнейшей потере мышечной массы.Это особенно важная проблема при остром СД, связанном с дисфагией, когда может потребоваться зондовое питание, и при ИБГ, связанном с дисфагией.
Дерматомиозит и полимиозит
Хотя их лечение лекарствами будет рассматриваться вместе, есть два вопроса, специфичных для лечения DM. Во-первых, за исключением детей, необходимо исключить возможность сопутствующего злокачественного новообразования, особенно у пожилых пациентов и пациентов с более высоким риском злокачественных новообразований (например, курильщик, семейный анамнез, другие предрасполагающие заболевания).При необходимости обследование должно включать обследование груди, влагалища и ректального исследования. Визуализация должна включать грудную клетку, живот и таз. Анамнез может предложить более подробные исследования — например, колоноскопию у кого-то с измененными привычками кишечника. Переоценка необходима в течение 1–2 лет. Во-вторых, хотя кожная сыпь, скорее всего, отреагирует на терапию системными иммунодепрессантами, бывают ситуации, когда местное лечение также может оказаться полезным, например, при сильной местной кожной сыпи.Кроме того, сыпь является светочувствительной, и солнцезащитный крем может быть очень эффективным для уменьшения кожных проявлений.
Кортикостероиды — основа лечения. Вопросы, на которые нет ответа, относятся к конкретным схемам приготовления и дозировке, выбору, применению и срокам введения других иммунодепрессантов («стероидсберегающих агентов»), а также к месту внутривенного введения иммуноглобулинов.
Кортикостероиды
Подавляющее большинство пациентов реагируют на стероиды.Отсутствие реакции чаще всего вызвано неправильной начальной дозировкой или продолжительностью лечения, реже — несоблюдением режима лечения из-за беспокойства о побочных эффектах. Некоторые пациенты действительно резистентны, но реагируют на другие схемы иммунодепрессантов. Опыт показывает, что раннее агрессивное лечение, как правило, связано с более быстрым ответом и лучшим исходом, поэтому всем, кроме самых вялотекущих случаев, вводят внутривенно метилпреднизолон 500 мг в день в течение пяти дней, а затем перорально преднизолон 1 мг / кг массы тела в день, учитывая в виде однократной утренней дозы.Как только SCK вернется в норму и состояние пациента улучшится, дозу уменьшают на 5 мг через день, так что через 1-2 месяца пациент будет принимать 1 мг / кг массы тела через день. После этого доза медленно снижается, в зависимости от клинического ответа и SCK (см. Ниже), чтобы определить минимальную поддерживающую дозу.
Открытые исследования показали, что другие схемы приема стероидов могут предложить лучшее соотношение польза / побочный эффект, но ни одно из них не было доказано в ходе соответствующего рандомизированного контролируемого исследования.К ним относятся пульсирующие режимы перорального и внутривенного введения высоких доз, а также использование дексаметазона, а не преднизолона.
Профилактика остеопороза (мы используем кальций, витамин D и еженедельно алендронат) должна использоваться с самого начала, а сканирование плотности костной ткани должно выполняться в качестве исходного уровня, а затем с интервалами, пока пациент остается на стероидах.
Иммунодепрессанты
Имеется достаточно доказательств из литературы и личного опыта, чтобы не оставлять сомнений в том, что такие препараты могут быть эффективными.В настоящее время нет достаточных данных, чтобы предположить, что один препарат лучше другого, и выбор в значительной степени определяется личным опытом, часто от использования препарата для лечения других заболеваний. Таким образом, ревматологи и дерматологи склонны использовать метотрексат (до 30 мг в неделю), поскольку у них есть опыт его применения при артрите и псориазе соответственно, тогда как многие неврологи отдают предпочтение азатиоприн (2,5 мг / кг массы тела в день), имея опыт применения его использование при миастении и иммунных невропатиях.Метотрексат может вызвать пневмонит, и, возможно, его можно спутать с интерстициальным заболеванием легких, связанным с миозитом. Циклоспорин (до 5 мг / кг массы тела в день) рекомендуется использовать при детском СД, но он также используется при взрослой форме заболевания. Микофенолят мофетил (2 г в день) в настоящее время в моде. Циклофосфамид вводили в виде внутривенных импульсов (до 1 г / м 2 площадь поверхности тела) и в виде перорального лечения (до 2 мг / кг веса тела в день). Есть некоторые свидетельства того, что он особенно полезен при лечении ассоциированного интерстициального заболевания легких.
Еще один вопрос касается сроков введения этих препаратов. Часто предлагают использовать их, если пациент не отвечает должным образом на преднизолон, имеет серьезные побочные эффекты от преднизолона или необходимая поддерживающая доза стероидов неприемлемо высока. Практическая проблема состоит в том, что может пройти много месяцев, возможно, продолжающееся ухудшение, прежде чем пациент будет идентифицирован как попадающий в одну из этих категорий. Все перечисленные выше иммунодепрессанты действуют медленнее, чем преднизолон.Азатиоприн, вероятно, действует медленнее всего — опыт миастении показывает, что для того, чтобы он стал эффективным, требуется 9–12 месяцев. Все другие лекарства, вероятно, действуют быстрее, чем это, но даже в этом случае, вероятно, потребуется много месяцев, чтобы проявить их максимальный эффект. Из-за этих проблем и параллельно с нашим опытом лечения миастении, разумно начать прием иммунодепрессантов (наш выбор — азатиоприн) одновременно с преднизолоном, в надежде, что это в конечном итоге позволит снизить поддерживающую дозу преднизолона.Эта гипотеза еще предстоит подтвердить в рандомизированном контролируемом исследовании.
Иммуноглобулин внутривенный
Было проведено достаточно исследований, чтобы сделать вывод, что внутривенный иммуноглобулин эффективен как при СД, так и при ПМ, но недостаточно данных, чтобы определить его точное положение по сравнению со стероидами и иммунодепрессантами. В настоящее время его, вероятно, лучше всего использовать для людей с заболеваниями, устойчивыми к лечению стероидами и иммунодепрессантами.
Ответ на мониторинг
Это включает в себя как элементы искусства, так и науки. Цель состоит в том, чтобы уменьшить дозу преднизолона, чтобы найти минимальную дозу, необходимую для поддержания ремиссии заболевания, которая у некоторых пациентов, принимающих дополнительный иммунодепрессант, может быть нулевой. Хотя часто подчеркивается, что лечить пациента следует, а не сдавать анализ крови, повышение SCK является поводом для беспокойства и может предвещать рецидив. С другой стороны, рецидив с нарастающей слабостью может произойти без увеличения SCK.Миопатия, индуцированная стероидами, является теоретической проблемой, но, по-видимому, редко возникает при приеме через день. Как и в случае со многими другими аутоиммунными заболеваниями, болезнь может удерживаться в стадии фармакологической ремиссии, но «излечение» труднее. По нашему опыту, длительная ремиссия без лекарств чаще встречается при СД, чем при ПМ.
Миозит с включенными тельцами
У большинства пациентов с типичным ИБГ отсутствует полезный ответ на стероиды, иммунодепрессанты или внутривенный иммуноглобулин.Если после информированного обсуждения они захотят попробовать лекарственное лечение, я бы использовал 18–24-месячное испытание преднизолона и азатиоприна (или метотрексата), как описано выше. Преднизолон будет снижаться до тех пор, пока SCK не начнет повышаться. Лечение будет продолжаться только в том случае, если будет однозначная польза. Не следует недооценивать потенциальные побочные эффекты, и в прошлом году мы стали свидетелями смерти от цитомегаловирусной пневмонии и инфекции сальмонеллы в искусственном бедре.
Нетипичные случаи побуждают меня предлагать пробное лечение, но пока для этого нет доказательной базы.Признаки могут включать начало в раннем взрослом возрасте, выраженные воспалительные инфильтраты при биопсии, исключительно высокий SCK и связанные иммунные нарушения.
СВОДКА
Наиболее распространенными воспалительными миопатиями являются дерматомиозит, полимиозит (который редко возникает изолированно, чаще связан с другими признаками заболевания соединительной ткани) и миозит с тельцами включения. Каждое из них представляет собой диагностические и терапевтические проблемы, и их лучше всего лечить в отделении, имеющем особый интерес и опыт лечения этих заболеваний.Большинству пациентов требуется длительное наблюдение специалиста. Для определения наилучших подходов к лечению наркозависимости крайне необходимы многоцентровые исследования. Немедикаментозные аспекты лечения очень важны, но им часто пренебрегают.
КЛЮЧЕВЫЕ ССЫЛКИ
Askanas V , Engel WK. Миозит с включенными тельцами: новейшие концепции патогенеза и связи со старением и болезнью Альцгеймера. Журнал Neuropathol Exp Neurol2001; 60: 1–14. ▸Обсуждает сходство между патологическими открытиями IBM и болезнью Альцгеймера.Не все согласны с предложенными патологическими механизмами.
Cheverel G , Goebels N, Hohlfeld R. Миозит — диагностика и лечение. Практическая неврология 2002; 2: 4–11. ▸Очень свежий обзор. Полезный контраст с настоящим обзором, демонстрирующим различия в личном подходе.
Далакас М , Карпати Г. В: Карпати Г., Хилтон-Джонс Д., Григгс Р. Заболевания произвольной мышцы . Кембридж: Издательство Кембриджского университета, 2001: 636–59. ▸Самый свежий обзор стандартных учебников. Полезный источник ссылок.
Далакас MC . Прогресс в воспалительных миопатиях: хорошо, но недостаточно. Журнал Neurol Neurosurg Psychiatry, 2001; 70: 569–73. ▸ Название говорит само за себя.
Dalakas MC , Koffman B, Fujii M, и др. .Контролируемое исследование внутривенного иммуноглобулина в сочетании с преднизоном при лечении IBM. Неврология, 2001; 56: 323–7. ▸После нескольких открытых исследований, которые, очевидно, продемонстрировали некоторую пользу от внутривенного введения иммуноглобулина, это правильно контролируемое исследование не показало никакой пользы.
Hill CL , Zhang Y, Sigurgeirsson B и др. Частота конкретных типов рака при дерматомиозите и полимиозите: популяционное исследование.Ланцет 2001; 357: 96–100. ▸ Лучшее на сегодняшний день исследование населения, изучающее сопутствующие риски рака, но не дающее всех ответов.
Хилтон-Джонс Д . Воспалительные заболевания мышц. Курр Опин Neurol2001; 14: 591–6. ▸Недавний обзор, содержащий самые свежие ссылки.
Масталья FL . Лечение аутоиммунных воспалительных миопатий.Курр Опин Neurol2000; 13: 507–9. ▸И снова само собой разумеющееся название и мнение другого опытного клинициста о лечении наркозависимости.
Nagaraju K , Raben N, Loeffler L и др. Условное повышение регуляции MHC класса I в скелетных мышцах приводит к самоподдерживающемуся аутоиммунному миозиту и специфическим для миозита аутоантителам. Proc Natl Acad Sci U S. A 2000; 97: 9209–14. ▸ Очень интересная статья, в которой было показано, что активация MHC 1 у мышей вызывает воспалительную миопатию и выработку специфичных для миозита антител.Пока неясно значение этого для идиопатических воспалительных миопатий у человека.
Peng A , Koffman BM, Malley JD, и др. . Прогрессирование заболевания при спорадическом миозите с тельцами включения: наблюдения у 78 пациентов. Неврология 2000; 55: 296–8. ▸Полезное естествознание. Важно при рассмотрении терапевтических испытаний для этого расстройства.
Воспалительная миопатия — Ревматологическая клиника — Рочестер, Нью-Йорк
Что такое воспалительная миопатия?
Миопатия — это название, которое врачи используют для описания болезни мышц.При воспалительной миопатии наблюдается отек (воспаление) мышц. Есть две воспалительные миопатии — полимиозит и дерматомиозит. Оба являются аутоиммунными заболеваниями. Работа вашей иммунной системы — бороться с инфекциями. Иногда он может атаковать ваши здоровые ткани, вызывая болезнь.
Кто болеет полимиозитом и дерматомиозитом?
- Оба редкие
- Они могут возникать в любом возрасте, хотя у взрослых они чаще встречаются в среднем возрасте
- Они вдвое чаще встречаются у женщин, чем у мужчин
- Они могут возникать у людей с другим аутоиммунным заболеванием, таким как волчанка, ревматоидный артрит, синдром Шегрена или склеродермия
Каковы симптомы полимиозита и дерматомиозита?
Воспалительный миозит поражает мышцы в области плеча / плеча и в области бедра / бедра с обеих сторон тела, вызывая слабость, но не боль.Люди с воспалительным миозитом часто замечают:
- Проблемы при вставании из положения сидя
- Проблемы при подъеме по лестнице
- Проблемы с расчесыванием или мытьем волос
- Не удается добраться до потолка для объектов
- Усталость
Иногда он также может поражать мышцы грудной клетки, вызывая затруднения при глотании или одышку.
У людей, больных дерматомиозитом, также появляется сыпь.Это может быть любое или все:
- Чешуйчатая красная сыпь на суставах, коленях и локтях.
- Сыпь пурпурно-красная с припухлостью на верхних веках
- Красная сыпь, которая образует «шаль», поскольку она возникает на задней части шеи, верхней части спины, плечах, плечах и верхней части груди
Как диагностируют полимиозит и дерматомиозит?
Ваш врач спросит вас о ваших симптомах, осмотрит вас и назначит анализы крови.Вашему врачу также необходимо будет «осмотреть» ваши мышцы. Это можно сделать несколькими способами. Тест, называемый ЭМГ, позволяет изучить электрические сигналы в мышцах. МРТ может помочь определить, какие мышцы поражены. Ваш врач может захотеть провести биопсию мышцы, чтобы ее можно было исследовать под микроскопом.
Как лечат полимиозит и дерматомиозит?
- Преднизон: Преднизон — очень мощное противовоспалительное средство, но со временем может иметь множество побочных эффектов.
- Иммунодепрессанты: Есть несколько лекарств, подавляющих иммунную систему. Эти препараты также используются при других аутоиммунных заболеваниях, таких как волчанка и ревматоидный артрит
- Физиотерапия: физическая терапия может помочь восстановить силы
Когда мне звонить врачу?
Позвоните своему врачу, чтобы задать вопросы о своем заболевании или лечении, или если:
- Вы не поправляетесь после лечения
- У вас появились новые симптомы
- У вас есть побочные эффекты от лекарств
Назначить встречу
(585) 486-0901
Наше местонахождение
Ревматологическая клиника в Ред-Крик
400 Red Creek Drive
Корпус 400, офис 240
Rochester, NY 14623
Карта Red Creek Drive
UR Медицина / аллергия, иммунология и ревматология
125 Lattimore Road
Люкс G-110
Rochester, NY 14620
Карта Lattimore Road
Ревматологическая клиника Канандаигуа Офис
Томпсон, профессиональное здание
395 Западная улица,
Люкс 007
Canandaigua, NY 14424
Карта офиса Canandaigua
Что такое идиопатические воспалительные миопатии?
Автор
Мифили Ситхараман, доктор медицины Консультант-ревматолог, ортопедическая ассоциация Аллентауна, при университетской больнице Св. Луки
Мифили Ситараман, доктор медицины, является членом следующих медицинских обществ: Американского колледжа ревматологии, Американской медицинской ассоциации, Общества ревматологов Пенсильвании Раскрытие информации: нечего раскрывать.
Главный редактор
Герберт С. Даймонд, доктор медицины Приглашенный профессор медицины, отделение ревматологии, Медицинский центр Нижнего штата Нью-Йорка; Почетный председатель отделения внутренней медицины больницы Западной Пенсильвании
Герберт С. Даймонд, доктор медицины, является членом следующих медицинских обществ: Alpha Omega Alpha, Американский колледж врачей, Американский колледж ревматологии, Американская медицинская ассоциация, Phi Beta Kappa
Раскрытие информации: нечего раскрывать.
Благодарности
Майкл С. Бисон, доктор медицины, магистр делового администрирования, FACEP, Профессор неотложной медицины, Колледж медицины и фармации Северо-Восточного Университета Огайо; Лечебный факультет, Акронский общий медицинский центр
Майкл С. Бисон, доктор медицинских наук, магистр делового администрирования, FACEP является членом следующих медицинских обществ: Американского колледжа врачей неотложной помощи, Совета директоров резидентур по неотложной медицине, Национальной ассоциации врачей скорой медицинской помощи и Общества академической неотложной медицины
Раскрытие: Ничего не нужно раскрывать.
Thomas H Brannagan III, MD, Адъюнкт-профессор клинической неврологии и директор Центра периферической невропатии Колумбийского университета, Колледж врачей и хирургов; Со-директор лаборатории ЭМГ, Нью-Йоркская пресвитерианская больница, Колумбийский кампус, Нью-Йорк
Томас Браннаган III, доктор медицины, является членом следующих медицинских обществ: Американской академии неврологии, Американской ассоциации нейромышечной и электродиагностической медицины и Общества периферических нервов
Раскрытие: Ничего не нужно раскрывать.
Лоуренс Х. Брент, доктор медицины, Адъюнкт-профессор медицины, Медицинский колледж Джефферсона Университета Томаса Джефферсона; Заведующий, программный директор, Департамент медицины, Отделение ревматологии, Медицинский центр Альберта Эйнштейна
Лоуренс Х. Брент, доктор медицины, является членом следующих медицинских обществ: Американской ассоциации развития науки, Американской ассоциации иммунологов, Американского колледжа врачей и Американского колледжа ревматологии
.
Раскрытие информации: Genentech Honoraria Выступление и обучение; Грант Genentech / исследовательские фонды Другое; Амджен Гонорария Выступление и преподавание; Pfizer Honoraria Устная речь и обучение; Abbott Immunology Honoraria Выступление и преподавание; Такеда Хонорария Выступление и преподавание; UCB Устная речь и обучение; Гонорар Omnicare Consulting Консультации; Гонорар Centocor Консультации Консультации
Зайнеб Дауд, доктор медицины , консультант, кафедра неврологии, Медицинский колледж Пенсильванского университета Ганемана
Раскрытие информации: Нет
Джино А. Фарина, доктор медицины, FACEP, FAAEM, Доцент кафедры неотложной клинической медицины Медицинского колледжа Альберта Эйнштейна; Программный директор, отделение неотложной медицины, Еврейский медицинский центр Лонг-Айленда
Джино А. Фарина, доктор медицины, FACEP, FAAEM является членом следующих медицинских обществ: Американской академии неотложной медицины, Американского колледжа врачей неотложной помощи и Общества академической неотложной медицины
Раскрытие: Ничего не нужно раскрывать.
Франсиско де Ассис Акино Гондим, доктор медицины, магистр наук, доктор философии, Доцент неврологии, кафедра неврологии и психиатрии, Медицинский факультет Университета Сент-Луиса
Франсиско де Ассис Акино Гондим, доктор медицинских наук, магистр наук, доктор философии является членом следующих медицинских обществ: Американской академии неврологии, Американской ассоциации нервно-мышечной и электродиагностической медицины и Общества двигательных расстройств
Раскрытие: Ничего не нужно раскрывать.
Аамир Хашмат, доктор медицины , консультант, лаборатория неврологии и нейродиагностики, отделение неврологии, Региональный медицинский центр Джеффа Андерсона
Аамир Хашмат, доктор медицины, является членом следующих медицинских обществ: Американской академии неврологии, Американского общества эпилепсии; Американская медицинская ассоциация, Фонд АО
Раскрытие информации: Нет
Milind J Kothari, DO, Профессор и заместитель председателя кафедры неврологии Медицинского колледжа Государственного университета Пенсильвании; Консультанты, Отделение неврологии, Медицинский центр им. Милтона С. Херши Государственного Пенсильвании,
Milind J Kothari, DO является членом следующих медицинских обществ: Американской академии неврологии, Американской ассоциации нервно-мышечной и электродиагностической медицины и Американской неврологической ассоциации
.
Раскрытие: Ничего не нужно раскрывать.
Кристин М. Лор, доктор медицинских наук, Профессор кафедры внутренней медицины Центра улучшения женского здоровья и отделения ревматологии, директор учебной программы ревматологии Медицинского колледжа Университета Кентукки
Кристин М. Лор, доктор медицины, магистр медицины, является членом следующих медицинских обществ: Американского колледжа врачей, Американского колледжа ревматологии и Американской ассоциации женщин-медиков
Раскрытие: Ничего не нужно раскрывать.
Гленн Лопейт, доктор медицины, Доцент кафедры неврологии отделения нервно-мышечных заболеваний Медицинской школы Вашингтонского университета; Директор неврологической клиники St Louis ConnectCare; Консультант отделения неврологии, Еврейская больница Барнс
Гленн Лопейт, доктор медицины, является членом следующих медицинских обществ: Американской академии неврологии, Американской ассоциации нервно-мышечной и электродиагностической медицины и Phi Beta Kappa
.
Раскрытие: Ничего не нужно раскрывать.
Николас Лоренцо, доктор медицины, Консультанты, специалисты по неврологии и консультанты
Николас Лоренцо, доктор медицины, является членом следующих медицинских обществ: Alpha Omega Alpha и Американской академии неврологии
Раскрытие: Ничего не нужно раскрывать.
Генри Розенкранц , доктор медицины, FAAEM, FACEP, отделение неотложной медицины, больница Норвуд
Генри Розенкранц, доктор медицины, FAAEM, FACEP является членом следующих медицинских обществ: Американской академии неотложной медицины и Американского колледжа врачей неотложной помощи
Раскрытие: Ничего не нужно раскрывать.
Erik D Schraga, MD, Штатный врач, Отделение неотложной медицины, Mills-Peninsula Emergency Medical Associates
Франсиско Талавера, фармацевт, доктор философии, Адъюнкт-профессор, Фармацевтический колледж Медицинского центра Университета Небраски; Главный редактор Medscape Drug Reference
Раскрытие информации: Medscape Reference Salary Employment
Идиопатические воспалительные миопатии и легкие
Интерстициальная болезнь легких
Эпидемиология
ILD — частое проявление дерматомиозита / полимиозита с распространенностью клинически значимых ILD от 17% до 36% [9, 47–51].В проспективной серии пациентов с систематической оценкой поражения легких частота составила 65% [9]. ILD возникает после диагноза полимиозита или дерматомиозита примерно у 40% пациентов и предшествует диагнозу явной CTD в 20–30% случаев [47, 52]. В больших группах пациентов с JDM / JPM, ILD встречается у 8–13% пациентов и, следовательно, встречается реже, чем при дерматомиозите у взрослых [53, 54]. Факторы риска развития ILD у пациента с дерматомиозитом / полимиозитом включают генетическую предрасположенность [55, 56], некоторые клинические признаки, такие как артралгию [47], и аутоиммунные особенности (таблица 2) [22, 56, 57].После поражения мышц вторым по значимости фактором заболеваемости являются ИЛЗ, связанные с дерматомиозитом / полимиозитом [47]. До половины пациентов с IIM-ассоциированными ILD могут в конечном итоге умереть от дыхательной недостаточности [13]. Смертность особенно высока при острых формах ВЗЛ, несмотря на немедленное и агрессивное лечение [48, 58].
Клинические проявления
Обычное представление
Признаки и симптомы, относящиеся к ILD в IIM, не отличаются от идиопатических ILD, и включают одышку при физической нагрузке, кашель, снижение толерантности к физической нагрузке, дискомфорт и астению.Врачи, диагностирующие ВЗЛ, должны уделять особое внимание внелегочным особенностям IIM, чтобы отличить вторичные ILD от первичных. Оценка таких пациентов включает тщательный общий (потеря веса и лихорадка), кожный (сыпь, склеродактилия, феномен Рейно, кальциноз, кожные язвы и руки механика), мышечный (потеря мышечной массы, мышечная проба и миалгия) и ревматологический (артралгия). , артрит и деформация Жакку) обследование. Подкожный кальциноз присутствует примерно в 25% случаев ЮДМ, в отличие от его редкости при дерматомиозите с дебютом у взрослых.
Помимо клинической оценки, для диагностики гипомиопатических форм миозита требуется тестирование креатинкиназы и альдолазы. В случаях высокого подозрения на ИМЗ, связанные с ИЛЗ ( например, сыпь, напоминающая дерматомиозит или положительный МСА), дополнительные исследования, адаптированные для каждого пациента, включают электромиографию, магнитно-резонансную томографию мышц и биопсию мышц.
Тест на наличие антинуклеарных аутоантител завершает клиническую оценку; дальнейшее тестирование на MSA или MAA в большинстве случаев зависит от характера ядерной флуоресценции.Клинический спектр, связанный с аутоантителами против ОРС, весьма неоднороден и включает артрит (62%), руку механика (28%), лихорадку (43%), феномен Рейно (47%) (рис.1), миозит (57%). и ILD (70%) в его классическом описании [8, 15, 23]. Из-за высокой частоты неспецифических признаков, связанных с аутоантителами против ОРС (, т.е. феномен Рейно , лихорадка, гиперкератоз и растрескивание кожи на пальцах), мы предпочитаем пересмотренное определение синдрома против ОРС, которое было предложено Коннорсом и другие. [51] на основе основных (артрит, ILD и миозит) и второстепенных критериев (таблица 3). В метаанализе клинических проявлений частота ILD и лихорадки была увеличена на 20–30% у пациентов с аутоантителами против ARS, отличными от аутоантител против Jo1 (особенно антител против PL7 и PL12) по сравнению с таковыми. с аутоантителами против Jo1 [8]. Напротив, частота механических рук и суставных и мышечных повреждений была увеличена на 50% у пациентов с аутоантителами против Jo1 по сравнению с другими антителами против ARS [8].У пациентов с антителами против PL7 или против PL12 часто наблюдаются изолированные ILD, поэтому их чаще всего осматривают специалисты по респираторным заболеваниям, тогда как другие признаки отсутствуют [15]. Пациенты с двойной положительной реакцией на анти-ARS и анти-U1RNP антитела часто испытывают проявления дерматомиозита (55%) и феномена Рейно (82%) [15]. У пациентов с антителами против ARS наличие рук механика может быть связано с более высоким риском развития ILD [59].
РИСУНОК 1
Феномен Рейно у пациента, положительного по антителам против PL7.
ТАБЛИЦА 3
Предлагаемые критерии миозита, связанного с антителом к тРНК-синтетазе
Клинические признаки, описанные у пациентов с антителами против PM / Scl, в значительной степени совпадают с таковыми у пациентов с аутоантителами против ARS [19, 32]. Действительно, пациенты с положительной реакцией на PM / Scl обычно страдают артралгией (74%), феноменом Рейно (63%), ILD (38%), лихорадкой (33%) и механическими руками (11%) [8]. Кожные проявления дерматомиозита и склеродактилии регистрируются в 49% и 76% случаев соответственно [8].Сыпь обычно легкая и преходящая, с изолированной гелиотропной сыпью или папулами Готтрона [11].
В случаях наличия аутоантител к MDA5 язвы пальцев локализуются в основном над папулами Готтрона, пульпами пальцев или околоногтевыми областями, но могут возникать и в других местах дерматомиозитной сыпи, , т.е. на тыльной стороне локтей, коленях. , ушные раковины или ягодицы (рис. 2) [57, 58]. Одно европейское исследование сообщило о повышенной частоте панникулита [56]. Артрит (80%), феномен Рейно (45%) и руки механика (80%) (рис.3) встречаются часто [55]. Аутоантитела против MDA-5 теперь были добавлены к дифференциальному диагнозу, который необходимо учитывать, особенно когда клинические признаки могут указывать на аутоантитела против ARS или против PM / Scl у пациентов с дерматомиозитом [55]. Важно отметить, что скрининг на антинуклеарные аутоантитела может быть отрицательным у пациентов с аутоантителами против MDA5, которые, следовательно, должны определяться специально при наличии клинических подозрений [56].
РИСУНОК 2
Клинические признаки, связанные с аутоантителами против MDA5, включают тяжелые кожные язвы a, b) пальцев и c) тыльной стороны локтя.
РИСУНОК 3
Руки механика у пациента с анти-MAD5-положительным дерматомиозитом. Изображение любезно предоставлено Себастьяном Дебарбье (Отделение дерматологии, Госпитальный центр Lyon Sud, Лион, Франция).
Быстро прогрессирующий ILD
Быстро прогрессирующая интерстициальная пневмония определяется острой интерстициальной пневмонией, прогрессирующей в течение нескольких недель или нескольких месяцев. Среди всех CTD это агрессивное проявление ILD указывает на IIM и особенно на дерматомиозит, CADM или гипомиопатический дерматомиозит [60, 61].Таким образом, врачи должны тщательно искать минимальные изменения кожи или легкое поражение мышц в условиях быстро прогрессирующей ILD.
Присутствие аутоантител против ARS кажется относительно защищающим от быстро прогрессирующей ILD, хотя об этом сообщалось [62, 63]. Обнаружение аутоантител против MDA5 позволяет выявлять пациентов с дерматомиозитом с высоким риском развития быстро прогрессирующей ILD, с совокупной чувствительностью 77% и специфичностью 86% в метаанализе 631 пациента с дерматомиозитом или полимиозитом [64, 65].Фульминантные презентации с аутоантителами к ILD, CADM и анти-MDA-5 могут быть особенно частыми у японских пациентов по сравнению с другими восточноазиатскими или неазиатскими пациентами [35, 64], хотя нельзя исключить предвзятость публикации. В японской ретроспективной серии 54 пациентов с JDM [66], у 10 были быстро прогрессирующие ILD, а у 19 были хронические ILD, что иллюстрирует особенно частое поражение легких в азиатской популяции.
Патогенез
Помимо недавних достижений в лечении пациентов с аутоантителами против MDA5, патогенез связанных с дерматомиозитом или полимиозитом ILD малоизвестен, в отличие от SSc [67].Иммуногистохимия при биопсии мышц дает некоторые ключи к разгадке иммуно-опосредованных механизмов. CD8 + T-клетки обычно обнаруживаются при полимиозите с диффузным цитотоксическим эффектом, приводящим к некрозу мышечных клеток [68], тогда как B-клетки и CD4 + T-клетки преобладают в периваскулярных областях при дерматомиозите и могут быть ответственны за мышечные васкулит [67, 69]. В образцах биопсии легких пациентов, страдающих ИЛЗ, ассоциированных с дерматомиозитом или полимиозитом, обнаруживаются многочисленные активированные лимфоциты CD8 + как в пораженной, так и в относительно сохраненной ткани.Доля этих клеток в жидкости бронхоальвеолярного лаважа (БАЛ) может предсказать исход после лечения кортикостероидами, поскольку количество Т-лимфоцитов CD8 + значительно увеличивается в резистентных к кортикостероидам случаях ВЗЛ, связанных с дерматомиозитом или полимиозитом [70]. Однако это наблюдение требует подтверждения, прежде чем его можно будет применить в клинике.
Кроме того, появляются корреляции между некоторыми генотипами и клиническим фенотипом или прогнозом. Например, полиморфизм в промоторе гена фактора некроза опухоли-α повышен у пациентов с дерматомиозитом / полимиозитом с ILD (OR 2.1, 95% ДИ 1,0–4,3) [71]. Гаплотип HLA-DRB1 * 03, определяющий фактор развития ILD среди пациентов с CTD [72, 73], предсказывает более высокий риск ILD при дерматомиозите / полимиозите [74], независимо от подтипа миозита или наличия аутоантител. [56]. Антитела против PM / Scl и против ARS также имеют одинаковую распространенность гаплотипов HLA-DRB1 * 03, DQA1 * 05 и DQB1 * 02 [75, 76].
Недавно сообщалось об усилении функциональных мутаций в гене IFIh2 , кодирующем белок MDA5, в связи с различными воспалительными состояниями, включая кожную сыпь, напоминающую поражения кожи при JDM [77].Анти-MDA5 представляет собой цитозольный рецептор двухцепочечной РНК, и мутации усиления функции IFIh2 приводят к усиленному продуцированию интерферона I типа. Аналогичным образом, мутации усиления функции в гене TMEN173 , кодирующем ДНК-сенсор STING (стимулятор генов интерферона), который отвечает за усиленную продукцию интерферона I типа, были обнаружены у пациентов с поражением сосудов и тяжелой формой ILD [78]. Взятые вместе, эти результаты предполагают, что интерферон типа I может быть ключевым цитокином, связывающим аутоантитела против MDA5 с поражением легких и ILD.
Радиология
Компьютерная томография высокого разрешения (КТВР) необходима для оценки ВЗЛ. HRCT-сканирование легких — это неинвазивный и чувствительный тест, демонстрирующий признаки легочных изменений у всех пациентов, страдающих ILD. Эти изменения преимущественно локализуются в нижних долях, как сообщалось в предыдущих исследованиях [47, 79]. Линейные помутнения, затухание матового стекла (гиператтенуированные области, в которых остаются видимыми бронхи и сосуды), ретикуляция и утолщение перибронховаскулярных систем — наиболее частые аномалии КТВР на начальном этапе визуализации ВЗЛ.Консолидация, соответствующая участкам с повышенным затуханием и пониженной видимостью стенок и сосудов бронхов, обычно встречается и патологически соответствует участкам организуемой пневмонии. Комбинация пятнистых областей консолидации и ретикуляции является частой и наводит на мысль о диагнозе миозита у субъектов с ILD, особенно с подострым или острым началом. Реже наблюдаются соты (участки небольших кистозных пространств с утолщенными стенками), тракционные бронхоэктазы (расширение бронхов из-за тракции фиброзной тканью) и субплевральные тяжи [80].
Патологические корреляции и корреляции изображений в IIM в целом аналогичны таковым в идиопатических условиях. В целом, различные паттерны HRCT могут наблюдаться при IIM-ассоциированной ILD, особенно при организующей пневмонии, неспецифической интерстициальной пневмонии (NSIP) (рис. 4) или смешанной паттерне NSIP-организующей пневмонии, тогда как обычная интерстициальная пневмония (UIP) паттерн (рис. 5) встречается реже [19]. Однако исследования, которые коррелируют визуализацию с патологией в контексте IIM, ограничены и страдают от систематической ошибки отбора, поскольку клиницисты склонны находить биопсию легкого показанной в менее типичных случаях.Следовательно, следует проявлять осторожность при интерпретации изображений, и картины HRCT не могут быть напрямую перенесены в гистологические исследования.
РИСУНОК 4
Компьютерная томография с высоким разрешением пациента с анти-Jo1-положительным полимиозитом, показывающая преобладающую базилярную ретикуляцию и матовое помутнение без сотов, что свидетельствует о неспецифической интерстициальной пневмонии.
РИСУНОК 5
Компьютерная томография с высоким разрешением пациента с полимиозитом, на которой видны соты, характерные для обычной интерстициальной пневмонии.
Результаты
HRCT существенно не различаются между различными проявлениями полимиозита или дерматомиозита-ILD [81]; однако острое и подострое начало ILD, по-видимому, связано с более частой и более обширной консолидацией, которая является отличительной чертой этого подтипа ILD. Другие особенности HRCT включают диффузную пятнистую непрозрачность матового стекла, нерегулярные базальные линии и некоторую ретикуляцию [63]. Картины HRCT при постановке диагноза могут помочь предсказать прогноз пациентов с полимиозитом или дерматомиозитом-ILD [82], а консолидация (в основном связанная с гистологической картиной организации пневмонии) обычно обратима при лечении.
Полимиозит-ILD может иметь лучший прогноз, чем другие формы. Паттерны HRCT дерматомиозита-ILD можно разделить на три группы, чтобы помочь в принятии клинических решений [83]. Преобладающая аттенюация матового стекла и / или ретикулярная непрозрачность без сотов, субплевральных полос и тракционного бронхэктаза часто связаны с тяжелой формой интерстициальной болезни легких и возможным летальным исходом. Выраженная консолидация типична для острого начала ILD, которое хорошо поддается кортикостероидам и часто иммуносупрессивной терапии [84].Результат паттерна КТВР с ослаблением матового стекла, преобладающей ретикуляцией, бронхоэктазами и дополнительными находками менее предсказуем.
У пациентов с аутоантителами против ОРС преобладают паттерны визуализации, указывающие на НПВП и организующуюся пневмонию, а консолидация наблюдается в половине случаев, которые разрешаются наиболее часто [85]. ILD, связанный с аутоантителами против PL12, чаще ассоциируется с ретикуляцией и тракционными бронхоэктазами, чем с аутоантителами против Jo1.Как и ожидалось, пациенты с аутоантителами против Jo1, у которых наблюдается тяжелая форма ILD, чаще имеют картину UIP при визуализации, чем пациенты с менее прогрессирующим заболеванием (54% против 18%, OR 5,02, 95% CI 1,05–243) [59], предполагая, что картина визуализации ILD может иметь прогностическое значение в этих условиях, подобно тому, что наблюдается при идиопатической интерстициальной пневмонии.
Функциональные пробы легких
ILD обычно ассоциируется с рестриктивным нарушением функции легких (общая емкость легких (TLC) и диффузионная способность легких по монооксиду углерода ( D LCO ) <80% от прогнозируемого). D LCO обычно снижается у пациентов с ILD, часто на более ранней стадии заболевания, чем TLC и форсированная жизненная емкость легких (FVC). Тесты легочной функции добавляют важную информацию в HRCT для оценки степени тяжести, поскольку они лишь частично коррелируют с оценками и паттерном HRCT. Кроме того, легочная функция более чувствительна к изменениям после терапии, чем отклонения от нормы HRCT.
Интерпретация тестов функции легких может быть затруднена в IIM из-за потенциального сосуществования ILD, слабости дыхательных мышц и / или ЛАГ, со всеми тремя нарушениями D LCO .Более того, ограничительный паттерн может быть вызван ILD, но также и слабостью дыхательных мышц, оценку которой часто трудно интерпретировать. И наоборот, псевдостабилизация ограничительного паттерна может быть ошибочно приписана улучшенной диафрагмальной прочности [51]. В одном исследовании параметры легочной функции улучшились на ранней стадии заболевания у некоторых пациентов, получавших комбинацию высоких доз кортикостероидов и иммунодепрессантов, и даже нормализовались у некоторых пациентов, несмотря на сохраняющиеся рентгенологические изменения [80].Клиническое значение сохраняющихся аномалий на КТВР, несмотря на нормальную функцию легких, в настоящее время неясно. Клиницисты должны знать о возможном несоответствии между визуализацией и функцией легких у пациентов с дерматомиозитом или полимиозитом-ILD, чтобы интерпретировать ответ на терапию и результат.
Ячеистый профиль BAL
В сочетании с результатами тщательного анамнеза, физикального обследования и визуализации грудной клетки анализ дифференциального числа клеток ЖБАЛ может способствовать диагностике ILD.Однако дифференциальное количество клеток в БАЛ само по себе не предсказывает патологию и не предсказывает ответ на терапию. Таким образом, основная роль БАЛ — исключить заражение. БАЛ имеет решающее значение для пациентов с острым или подострым началом ILD, что может соответствовать бактериальной инфекции или ILD, связанными с IIM. Это еще более актуально для субъектов с прогрессирующим заболеванием, несмотря на лечение кортикостероидами, для диагностики редких скрытых инфекций, включая атипичные микобактерии, Pneumocystis jirovecii , Nocardia , внутриклеточные бактерии, цитомегаловирус и грибы, особенно у пациентов с ослабленным иммунитетом.
Были предприняты попытки соотнести клеточные паттерны ЖБАЛ с изображениями, гистологическими паттернами или клиническими проявлениями. Например, в одном исследовании увеличение лимфоцитов и эозинофилов в БАЛ было выше при дерматомиозите-ILD, чем при полимиозите-ILD [86]. Отношение CD4 + / CD8 + было больше у пациентов с CADM, чем у пациентов с классическим дерматомиозитом и быстро прогрессирующим ILD [62, 70, 87]. Повышенное количество нейтрофилов в БАЛ было связано с худшим исходом при дерматомиозите или полимиозите-ВЗЛ в двух исследованиях [47, 63].Однако клеточные паттерны ЖБАЛ не воспроизводятся и не имеют четкого клинического значения или последствий для лечения. Кроме того, клеточный профиль ЖБАЛ субнормален или не может быть отнесен к категории в большинстве случаев, несмотря на наличие ILD [88].
Патология
ILD может иметь различные гистологические паттерны в условиях IIM. Хотя доступные исследования страдают от систематической ошибки отбора, гистопатологические паттерны включают в себя в порядке убывания частоты NSIP (65%), UIP (15%), организуемую пневмонию (9%) и диффузное альвеолярное повреждение (8%) согласно объединенным данным шести независимых серия (таблица 4) [13, 47, 79, 86, 89, 90], тогда как лимфоцитарная интерстициальная пневмония присутствовала только у 1% пациентов.Схема организации пневмонии может быть недостаточно представлена в этих сериях, поскольку клиницисты могут идентифицировать консолидацию как признак, связанный с относительно хорошим ответом на лечение и прогнозом, и реже обращаться за хирургической биопсией. Два паттерна могут сосуществовать в различных количествах у пациента, например, со смешанными паттернами организации пневмонии и NSIP. Обнаруженные паттерны очень похожи на паттерны, наблюдаемые в идиопатических условиях, с небольшими различиями, отмеченными, включая меньшее количество фибробластных очагов, меньшие сотовые пространства, более высокие зародышевые центры и общие показатели воспаления при UIP-ассоциированном с CTD, чем при идиопатическом UIP (идиопатическом фиброзе легких) [ 91].В одном исследовании сообщалось о корреляции между патологией и ответом на лечение, с хорошим прогнозом в случаях организуемой пневмонии, в отличие от тех, которые имели характер диффузного альвеолярного повреждения или UIP, как ожидалось [86].
ТАБЛИЦА 4
Результаты гистопатологии легких у пациентов с интерстициальным заболеванием легких, ассоциированным с дерматомиозитом или полимиозитом
В целом, патология IIM-ILD очень похожа на патологию идиопатической ILD. Однако в настоящее время биопсия легкого редко выполняется у пациентов с ЗСТ и ИЛЗ, поскольку обычно она не влияет на лечение по сравнению с решениями, основанными на менее инвазивных процедурах, e.грамм. клиническая оценка, легочные функциональные пробы, КТВР и, возможно, БАЛ. Биопсия легкого обсуждается в индивидуальном порядке в редких случаях с атипичными особенностями визуализации или когда рассматривается злокачественная опухоль.
Лечение
Не проводилось рандомизированных контролируемых испытаний для оценки эффективности и переносимости кортикостероидов и иммуносупрессивных препаратов у пациентов с ILD, ассоциированным с IIM. Об эффективности иммунодепрессантов в основном сообщалось в неконтролируемых небольших ретроспективных сериях, которые предполагают пользу лечения, но не позволяют сделать твердые выводы.Таким образом, показание к применению иммунодепрессанта и выбор молекулы в основном основываются на мнении экспертов, знании и опыте клинициста с конкретным лекарством, а также на профиле переносимости. Лечение необходимо начинать в случае симптоматических или прогрессирующих ВЗЛ. При постановке диагноза необходимо внимательно следить за бессимптомной ИЛЗ, чтобы определить развитие болезни. Быстро прогрессирующая ИЛЗ слабо чувствительна только к кортикостероидам и неизменно требует комбинации с иммуносупрессивной терапией.Учитывая уровень смертности, близкий к 50%, цель терапии — предотвратить прогрессирование до острой дыхательной недостаточности и смерти [51, 52].
Кортикостероиды остаются краеугольным камнем и препаратом первой линии лечения ILD, ассоциированных с дерматомиозитом или полимиозитом [13]. Пероральные схемы включают 0,5–1 мг / кг –1 преднизона ежедневно в течение 3–8 недель с последующим постепенным снижением дозы. В тяжелых случаях назначают 1–3 пульса в день по 1000 мг метилпреднизолона внутривенно [13]. Ответ на терапию кортикостероидами первой линии считается адекватным у 90–100% пациентов с хроническими ILD (таблица 5) [59, 63, 100], с гораздо более низкими показателями ответа (7–44%) при быстро прогрессирующих ILD, которые обычно требуется дополнительная иммуносупрессивная терапия (таблица 6) [58, 62, 63].В дополнение к рефрактерному заболеванию иммуносупрессивная терапия может использоваться в случаях рецидива заболевания или в качестве стероидсберегающих средств. Был использован ряд лекарств, включая азатиоприн, метотрексат, циклофосфамид, циклоспорин А, такролимус, ритуксимаб и в / в. иммуноглобулина [13, 19, 47, 48, 58, 94]; однако никаких крупных исследований по их использованию в этом контексте опубликовано не было.
ТАБЛИЦА 5
Исследования, описывающие лечебные эффекты (улучшение / стабилизация) кортикостероидов и иммунодепрессантов для хронических форм СД или PM-ассоциированных ILD
ТАБЛИЦА 6
Исследования, описывающие лечебные эффекты (выживаемость) кортикостероидов и иммунодепрессантов при быстро прогрессирующих ILD с DM или PM
Авторы рассматривают иммуносупрессивную терапию в дополнение к кортикостероидам при постановке диагноза у пациентов с наиболее тяжелыми проявлениями, а также у пациентов, которые, как ожидается, плохо переносят кортикостероиды из-за сопутствующих заболеваний, особенно ожирения.Другие пациенты обычно получают пероральные кортикостероиды в течение первых 2–3 месяцев; Затем могут быть добавлены иммунодепрессанты, чтобы помочь снизить дозу кортикостероидов, если постепенное снижение дозы затруднено, а также у пациентов с недостаточным ответом на кортикостероиды.
Азатиоприн
Азатиоприн, производное 6-меркаптопурина, является ингибитором синтеза пурина, тем самым истощая клетки аденозиновых и гуанозиновых нуклеотидов и инактивируя Т-клетки, что приводит к иммуносупрессии [105].Обычно его вводят перорально в дозе 2–3 мг · кг -1 в день. Азатиоприн широко используется у субъектов с аутоантителами против ARS и другими формами дерматомиозита / полимиозита с ILD в качестве кортикостероидсберегающего средства или в качестве поддерживающей терапии, только или иногда после индукции циклофосфамидом [59, 79, 102]. Опубликованные данные предполагают улучшение ILD у ~ 75% пациентов с хроническим заболеванием, когда они используются в качестве терапии первой линии (таблица 4) [59, 86, 92, 100, 102].В то время как результаты азатиоприна в качестве терапии второй линии были неутешительными [102], в очень небольшой серии (семь пациентов) лечение циклофосфамидом с последующим приемом азатиоприна привело к ремиссии в 58% случаев [59, 102], с большей эффективностью в случаях с аутоантитела против ARS. Азатиоприн применялся у очень небольшого числа пациентов с быстро прогрессирующими ВЗЛ, но безуспешно (таблица 6) [58]. Однако следует иметь в виду, что опубликованные ретроспективные данные обычно страдают от предвзятости публикации и могут не отражать более широкий опыт.
Метотрексат
Метотрексат — антагонист фолиевой кислоты, который ингибирует дигидрофолатредуктазу. Поскольку этот фермент необходим для синтеза пуриновых нуклеотидов, введение метотрексата приводит к снижению развития и активации Т-лимфоцитов. Обычно его вводят перорально в дозе 0,2–0,3 мг · кг -1 еженедельно. В отчетах о случаях и ретроспективных исследованиях было высказано предположение, что метотрексат может быть эффективным при лечении хронических ВЗЛ, хотя подробные сведения об эффекте лечения не приводятся (таблица 5) [13, 79, 92].В целом опыт применения метотрексата у пациентов с IIM-ILD очень ограничен, и врачи-респираторы часто не хотят использовать этот препарат из-за возможного лекарственного пневмонита.
Циклоспорин А
Циклоспорин A — это ингибитор кальциневрина, который подавляет транскрипцию интерлейкина-2 и других цитокинов [105], в основном нацеленных на Т-клетки. Циклоспорин А можно назначать в виде единственного препарата или в сочетании с другими иммунодепрессантами в тяжелых случаях [58, 93, 97].По-видимому, он эффективен в качестве терапии как первой, так и второй линии при быстро прогрессирующей ИЛЗ [103, 106]. Сообщается об эффективности 55% при хронических ILD при использовании в качестве лечения первой линии (таблица 5) [86, 92, 97, 100]; все пациенты ответили на лечение, когда циклоспорин А использовался в качестве второй линии в трех небольших сериях [93, 102, 103]. При быстро прогрессирующей ИЛЗ циклоспорин А можно использовать в качестве средства первого или второго ряда с диапазоном ответа 56–80% и 23–80% соответственно (таблица 5) [62, 58, 93, 96, 97 , 100].Однако использование циклоспорина А ограничено эндотелиальной и почечной токсичностью (которая потенциально смертельна) [103]. Более того, опубликованный опыт применения циклоспорина А в основном исходит от групп в Азии, с гораздо более ограниченным опытом в европейских и американских центрах, и поэтому соотношение риска и пользы и место этого препарата в алгоритме лечения все еще неясны.
Такролимус
Такролимус — это ингибитор кальциневрина второго поколения с большей эффективностью по сравнению с циклоспорином А.Несколько ретроспективных исследований предполагают его полезность в качестве второй [94] или третьей линии лечения хронических ILD (таблица 5) [93, 95]. Его использование в качестве терапии первой линии или в рефрактерных случаях быстро прогрессирующего ИЛЗ требует дальнейшего изучения, при этом в трех исследованиях у четырех пациентов были описаны отдельные ответы [62, 93, 94].
Микофенолят мофетил
Микофенолятмофетил, пролекарство микофеноловой кислоты, является ингибитором инозинмонофосфатдегидрогеназы, которая является ограничивающим скорость ферментом в синтезе de novo гуанозиновых нуклеотидов, контролирующих развитие Т- и В-лимфоцитов [107].Ремиссия или стабилизация ILD была достигнута у 80% пациентов с хронической ILD при использовании в качестве первой или второй линии лечения (таблица 5) [59, 101, 105, 108]. Благодаря относительно благоприятному профилю безопасности микофенолятмофетил все чаще используется у пациентов с дерматомиозитом или полимиозитом-ILD, которым требуется иммунодепрессивный препарат в сочетании с кортикостероидами.
Циклофосфамид
Алкилирующий агент циклофосфамид использовался для лечения пациентов с дерматомиозитом / полимиозитом и быстро прогрессирующим или хроническим ILD в ежемесячной дозе 0.6–0,7 мг · м −2 i.v . Из-за риска вторичных злокачественных новообразований применение циклофосфамида должно быть ограничено случаями рефрактерных ILD [96] или быстро прогрессирующих ILD [62, 92]. Сообщаемый уровень эффективности колеблется от 38% до 100% при хронических ILD [59, 86, 96, 102] и быстро прогрессирующих ILD (таблицы 5 и 6) [92].
Ритуксимаб
Ритуксимаб представляет собой химерное моноклональное антитело, нацеленное на белок CD20, который в основном обнаруживается на клеточной мембране В-лимфоцитов.Ритуксимаб может быть эффективным при быстро прогрессирующей ILD и при хронической ILD с аутоантителами против ARS. Его вводят двумя дозами по 700 мг · м −2 или 1000 мг с интервалом в 2 недели, или с четырьмя еженедельными инфузиями 375 мг · м −2 . Для достижения ремиссии часто одновременно назначают кортикостероиды. Поддерживающая терапия назначается в течение 3–6 месяцев после индукции ритуксимабом, состоящим из азатиоприна [97] или, возможно, циклофосфамида [98, 109]. У семи пациентов с аутоантителами против ОРС в качестве поддерживающей терапии была проведена вторая инфузия 1000 мг через 6 месяцев после индукции (таблица 6) [99].Однако опыт применения ритуксимаба в IIM-ILD все еще очень ограничен, и его нельзя рассматривать как терапию первой линии.
Ассоциация иммунодепрессантов или терапевтических средств
Некоторые авторы сообщили об использовании двух комбинированных иммунодепрессантов, таких как микофенолятмофетил плюс циклофосфамид, микофенолятмофетил плюс метотрексат или циклофосфамид плюс такролимус, с обоснованием аддитивных эффектов и, как сообщается, хорошей безопасностью [110]. В некоторых схемах не используются кортикостероиды [110], которые, тем не менее, считаются обязательными в большинстве случаев.
Внутривенные иммуноглобулины и плазмаферез применялись у пациентов с опасными для жизни легочными поражениями. Несмотря на сообщенное преимущество иммуноглобулинов в поражении мышц, эти методы лечения, по-видимому, имеют очень ограниченное воздействие на быстро прогрессирующие ILD [96, 104] и поэтому редко используются для лечения заболеваний легких.
Результаты
Средний уровень смертности от ILD составляет 14% [47]. В некоторых исследованиях сообщалось о смертности до 45% у пациентов с дерматомиозитом, причем смерть в основном была вызвана ILD [86]; однако это, вероятно, не соответствует большинству пациентов.ILD — основная причина смерти при JDM. Смертность пациентов с хроническими ВЗЛ, связанными с ИБС, аналогична смертности пациентов с ВЗЛ, связанных с типичным дерматомиозитом [13, 51].
Прогностический балл для лечения ИБЗ, связанных с IIM, крайне недостаточен. Средним фактором прогноза является клиническая картина ВЗЛ, ассоциированных с дерматомиозитом или полимиозитом. Быстро прогрессирующее проявление ILD может привести к острой дыхательной недостаточности с диффузным поражением альвеол (подобно острой идиопатической интерстициальной пневмонии, ранее называвшейся синдромом Хаммана – Рича) [47], смертность от которой составляет 50–75% [48, 58].Течение миозита схоже у пациентов с ILD или без них [47], хотя распространенность миозитов, резистентных к терапии кортикостероидами, была выше у пациентов с полимиозитом / дерматомиозитом с ILD, чем у пациентов без ILD в одной серии, возможно, из-за более высокой частоты аутоантител к тРНК-синтетазе у больных ИБП [47].
Прогноз может варьироваться в зависимости от наличия MSA. Риск острого развития ИЛБ и смертности у пациентов с антителами против ОРС, как правило, не увеличивается по сравнению с другими аутоантителами [52, 81, 96, 100].Среди пациентов с антителами против ARS риск ухудшения состояния легких и смерти, по-видимому, схож с пациентами с антителами против Jo1 и не-Jo1, но выводы исследования расходятся [4, 8, 15, 111]. Прогноз ILD, ассоциированного с аутоантителами против PM / Scl, аналогичен прогнозу других ILD, ассоциированных с дерматомиозитом или полимиозитом [19, 32]. Пациенты с анти-PM / Scl имеют симптоматическое поражение легких с глобально благоприятным прогнозом, сравнимым с таковым у пациентов с аутоантителами против ARS [19].Смертность от ILD низкая у пациентов с аутоантителами к PM / Scl и колеблется от 0% до 11% в двух небольших ретроспективных когортах [19, 32]. Выживаемость ILD, ассоциированных с аутоантителами против MDA5, ниже, чем у ILD против ARS [56].
Легочная артериальная гипертензия
Эпидемиология
Прекапиллярная легочная гипертензия, связанная с дерматомиозитом, была впервые описана в 1956 году [112], а совсем недавно о ней сообщалось в клинических случаях [113, 114] и в коротких сериях [5].Расчетная частота легочной гипертензии составляет 8% у пациентов с аутоантителами против ARS [5] и может достигать 29% у пациентов с аутоантителами против PL7 [115]. Легочная гипертензия может предшествовать диагнозу миозита [116]; однако в большинстве, если не во всех, случаях это связано с ILD (группа легочной гипертензии 3), часто после нескольких лет продолжительности заболевания [5]. У этих пациентов часто наблюдаются клинические признаки ССД, такие как склеродактилия, склероз кожи или язвы пальцев [5].В одной серии исследований артралгии при постановке диагноза встречались реже у пациентов с прекапиллярной легочной гипертензией, чем у тех, у кого этого не было (38% против 65%) [5].
Патогенез и патология
Изолированная легочная гипертензия очень редко встречается при дерматомиозите / полимиозите при отсутствии ILD. Хроническая гипоксическая вазоконстрикция легких является одним из основных факторов, способствующих возникновению легочной гипертензии у пациентов с IIM-ILD; однако вовлечение легочных сосудов диффузным инфильтративным и воспалительным процессом, вероятно, вносит свой вклад в патогенез, а также другие, менее идентифицированные факторы [5, 117].Фактически, легочная гипертензия существует в отсутствие значительной гипоксии, вторичной по отношению к прогрессирующей ILD [5, 117]. Кожные особенности ССЗ, включая феномен Рейно, язвы пальцев, склеродактилию и опухшие пальцы, часто наблюдаются у пациентов с аутоантителами против ОРС и чаще встречаются при явной легочной гипертензии [8, 118]. Два сообщения о случаях явного и гипомиопатического полимиозита описывают тяжелую гипертрофию правого желудочка и тяжелый плексогенный фиброз интимы, согласующийся с легочной артериопатией при вскрытии [113, 117].Облитерирующая легочная васкулопатия (возникающая в результате пролиферации эндотелиальных и гладкомышечных клеток сосудов), аналогичная таковой при SSc, но с вовлечением более проксимальных легочных артериол, часто сосуществует [117]. Как и при других формах тяжелой ЛАГ, ремоделирование сосудов затрагивает все три слоя стенки легочной артерии с утолщением интимы и медиальной гипертрофией, что напоминает изменения, наблюдаемые при идиопатической ЛАГ. Роль в патогенезе механизмов, описанных при ССД, ИЛЗ и легочной гипертензии, а именно сосудистого воспаления, периваскулярного фиброза и тромботической ангиопатии, в IIM неизвестна [119].Аналогичным образом влияние факторов роста (трансформирующий фактор роста, фактор роста эндотелия сосудов, основной фактор роста фибробластов, фактор роста тромбоцитов и интерлейкин-6) и относительный вклад вазоактивных факторов эндотелия (эндотелин, оксид азота и простаноиды) ) не исследовались в патогенезе легочной гипертензии, связанной с аутоантителами против ОРС.
Лечение
Неспецифические методы лечения легочной гипертензии, включая ингибиторы фосфодиэстеразы-5 (силденафил и тадалафил), антагонисты рецепторов эндотелина (бозентан и амбризентан) и производные простациклина ( i.v. эпопростенол, подкожный трепростинил и распыленный илопрост) можно рассматривать у пациентов с дерматомиозитом или полимиозитом, связанным с легочной гипертензией [120–122]. Однако лечение должно быть зарезервировано для тех редких пациентов с ЛАГ 1-й группы без значительного нарушения дыхания в результате ВЗЛ, тогда как лечение не рекомендуется для пациентов с легочной гипертензией 3-й группы, считающейся вторичной по отношению к ИБП [119]. Неизвестно, могут ли иммунодепрессанты и кортикостероиды (которые могут улучшить легочную гипертензию у пациентов с системной волчанкой) быть полезными у пациентов с IIM и легочной гипертензией [123, 124].
Результат
Наличие как ВЗЛ, так и легочной гипертензии связано с чрезвычайно низкой долгосрочной выживаемостью, при 3-летней выживаемости 58% [5]. Прогноз изолированной легочной гипертензии при дерматомиозите / полимиозите, по-видимому, лучше, с 3-летней выживаемостью 100%, зарегистрированной у семи пациентов [125]; однако опыт очень ограничен.
Взгляд пациентов на жизнь с идиопатической воспалительной миопатией и ограничения методов измерения активности болезни — качественное исследование | BMC Rheumatology
В интервью приняли участие восемнадцать (61% женщин) участников с подтвержденным диагнозом IIM.Четырех участников сопровождал друг или партнер; Только одна цитата партнера способствовала формированию темы. Средний возраст когорты составлял 52 года (IQR 44, 56) со средней продолжительностью IIM болезни 5 лет (IQR 2, 6). Анализ данных базового интервью выявил четыре основные темы. Каждая тема будет обсуждаться по очереди с соответствующими цитатами (Таблица 1).
Таблица 1 Цитаты участников
Усталость
Усталость, как проявление снижения физической выносливости, была наиболее частым и заметным симптомом, о котором сообщили все участники (цитаты 1 и 2 в текстовом поле).Часто сообщалось, что на повседневную деятельность (ADL) влияет усталость. В частности, часто сообщалось, что страдают ADL, требующие длительного отведения плеча, такие как сушка / укладка волос и использование телефона (цитата из текстового поля 3). Напротив, участники сообщили о меньшем влиянии на более короткие задачи, такие как одевание; некоторые также сообщили, что адаптировали свои ADL для целенаправленного включения таких краткосрочных потребностей в физических нагрузках (цитата из текстового поля 4). Влияние усталости на их способность мыть, одеваться или ухаживать за детьми вызывало серьезную обеспокоенность у ряда участников (цитата из текстового поля 5).
Ряд участников сообщили о стратегиях побуждения, которые помогают справиться с усталостью или уменьшить их, которые включают планирование «дней отдыха» (цитаты из текстового поля 6 и 7).
Сообщалось, что утомляемость является более важным симптомом по сравнению с ощущаемой мышечной слабостью, которая не была связана с воспринимаемыми вариациями активности заболевания или фактором, который напрямую влияет на качество жизни (цитаты 2 и 8 из текстового поля). Несколько участников объяснили, что усталость обычно не оценивается и не решается во время клинических консультаций.Снижение осведомленности врачей об утомляемости как о симптоме, связанном с ММВ, было предложено в качестве возможной причины этого упущения (цитата из текстового поля 9).
Путешествие, как сообщалось, было особенно утомительным занятием, и ряд участников сообщили, что ограничили свои поездки только самым важным. Кроме того, необходимость планировать все аспекты поездки, такие как наличие ступенек или спусков, расположение помещений для отдыха и уверенность в том, что будет доступен контакт для экстренной помощи, была источником беспокойства и ощущения ограничений для ряда участников. , их партнеры и семьи (цитата из текстового поля 10).Потребность в тщательном планировании всех путешествий, даже на короткие расстояния, по-видимому, усиливала чувство потери независимости, а их состояние определяло многие аспекты их жизни.
Боль
Ключевым симптомом после усталости была боль. О характере боли сообщали по-разному, включая «спазм», «жжение в мышцах», «ощущение, будто вы пробежали марафон» и настолько сильную, что «она действительно доводит вас до слез», однако это было в целом сообщили, что передать характер боли было трудно (цитата из текстового поля 11).
Было отмечено, что боль связана с выполнением физических нагрузок даже низкой интенсивности. Появление боли при выполнении повседневных дел было особенно неприятным симптомом (цитата из текстового поля 12). Ряд участников признали, что боль может возникать только тогда, когда они превышают определенный уровень продолжительности физических нагрузок, обычно связанных с определенными ADL. Таким образом, участники сообщили о необходимости быть более целеустремленными и осознанными в планировании и выполнении ADL, выбирая, какие из них были важными, а какие можно отложить, пока они не почувствуют себя способными.
Ежедневные вариации симптомов — характеристика хороших и плохих дней
Большинство участников сообщили об изменении симптомов. Многие участники сообщили, что симптомы, особенно усталость и боль, менялись ото дня к дню или даже от часа к часу (цитаты из текстового поля 14 и 15). Многие участники признали наличие «хороших» и «плохих» дней, однако они варьировались между участниками и по-разному влияли на их функции, в зависимости от их образа жизни.Хорошие дни характеризовались повышенной физической выносливостью, что позволяло лучше выполнять повседневную деятельность (ADL), улучшать способность ходить, воспринимать более высокие уровни энергии и уменьшать или отсутствовать боль (цитата из текстового поля 16). Напротив, «плохие дни» характеризовались более высоким уровнем боли и утомляемости, а также наличием недомогания, что приводило к трудностям при выполнении ADL (цитата из текстового поля 17). Участники обычно отмечали способность определять, будет ли у них плохой день, по симптомам во время пробуждения (цитата из текстового поля 18).Некоторые участники описали более серьезные симптомы, такие как боль и усталость, в дни после «хорошего дня» и предположили, что это может быть связано с перенапряжением (цитата из текстового поля 19).
Рационирование энергии, то есть сохранение собственной энергии за счет выполнения только определенных физических нагрузок, обычно упоминалось, особенно участниками с более длительным периодом болезни IIM, как стратегия выживания, направленная на предотвращение изнурительной усталости, связанной с перенапряжением. Ряд участников заявили, что каждый день им доступно только определенное количество энергии, превышение которой приведет к последующим «плохим» дням, характеризующимся ухудшением симптомов, включая усталость и боль.Кроме того, была признана сложность количественного определения этого количества энергии (цитата из текстового поля 20). Конечное количество энергии, воспринимаемое как доступное в течение определенного дня, было описано одним участником (P4) как «кубик сахара», при этом они «постоянно пытались компенсировать» физические нагрузки.
Сообщалось, что колебания симптомов недооцениваются как членами семьи / друзьями, так и клиницистами (цитаты из текстового поля 21 и 22). Ряд участников объяснили, что такое непризнание симптомов и их вариации может быть связано с отсутствием четких визуальных индикаторов болезни или симптомов.Это привело к восприятию IIM как «невидимой болезни» (цитата 23 из текстового поля). Участники объяснили последующее разочарование, которое может вызвать непризнание IIM, отчасти из-за низкого уровня осведомленности общественности и редкости этого состояния (цитата из текстового поля 24).
Сочетание вариативности симптомов, нечастого клинического осмотра и ощущения трудности передачи широкого спектра симптомов было источником беспокойства, о котором сообщали многие участники (цитата из текстового поля 25).В частности, ряд участников признали, что оценка на приеме в клинику может не отражать колебания активности заболевания с момента предыдущей оценки (цитата из текстового поля 26). Еще одним сообщаемым ограничением нечастых клинических обзоров является сложность вспомнить, возможно, множественные колебания симптомов, которые могли иметь место несколько месяцев назад, с предпочтительным вспоминанием «хороших дней» и улучшенных симптомов, что потенциально ограничивает понимание врачом опыта пациента 27).Небольшое количество участников сообщили, что ведут дневник симптомов, возникновения вспышек и предполагаемых причин, что улучшает запоминание вариаций симптомов во время приема (цитата из текстового поля 28).
Ограничения уровней CK и MMT как измерения активности заболевания
Многие участники представили подробные мнения о своем восприятии способности уровней CK и MMT оценивать активность заболевания IIM. Большинство участников объяснили свой опыт изменений уровня КФК, не соответствующих колебаниям симптомов (цитата из текстового поля 29).Даже небольшое количество участников, которые считали, что КК был связан с их активностью заболевания, чувствовали, что он недооценивает степень их симптомов (цитата из текстового поля 30). Сравнение индивидуального уровня КК с нормальным лабораторным диапазоном подверглось критике, при этом участники посчитали, что сравнение с их собственным исходным уровнем, возможно, более уместно (цитата из текстового поля 31). Кроме того, ряд участников считали, что клиницисты могут основывать свою оценку активности заболевания IIM исключительно на уровне CK, не принимая во внимание внемышечные проявления (цитата из текстового поля 32).Ряд участников также предположили, что изменения уровня КК по-разному интерпретируются разными врачами (цитата из текстового поля 33).
Многие участники также сообщали о недостатках ММТ как показателя активности заболевания, в том числе о различиях между врачами, неспособности ММТ оценивать утомляемость при длительном использовании мышц и неспособности фиксировать ежедневные колебания силы ( Цитата из текстового поля 34). Участники также отметили неспособность ММТ оценить выносливость, которая, как описано ранее, является основным источником функциональных ограничений.Также сообщалось, что оценка MMT может сильно различаться в зависимости от других факторов, в частности, мотивации пациента и предшествующего знания клиницистом слабости (цитата из текстового поля 35). Было выражено мнение о том, что врачи полагаются преимущественно на ММТ для оценки активности заболевания, при этом участники полагали, что скрытые симптомы, такие как усталость, будут упущены, как описано ранее (цитата из текстового поля 36).
Воспалительные миопатии, которые сложно диагностировать и лечить
Ревматологи часто имеют разный уровень комфорта при оценке и лечении мышечных заболеваний.Для многих эти заболевания представляют собой сложную задачу из-за трудностей с точной диагностикой миопатий и установлением эффективных терапевтических режимов. Мы выделяем три ключевых вопроса в диагностике и лечении воспалительных миопатий.
Нет Золотого стандарта диагностики
Поскольку не существует золотого стандарта для диагностики идиопатических воспалительных миопатий, мы вынуждены полагаться на критерии. Хотя было предложено более дюжины наборов критериев, первый набор, опубликованный в 1975 году Боханом и Питером, остается основным. 1,2 Для диагностики полимиозита они предложили комбинацию слабости проксимальных мышц, повышенных уровней сывороточных ферментов, полученных из скелетных мышц, классической триады электромиографических (ЭМГ) аномалий и наличия воспаления при биопсии мышц. Диагноз дерматомиозита можно было поставить с добавлением характерной кожной сыпи. Эти параметры кажутся ясными до тех пор, пока мы не углубимся в детали, потому что каждый критерий сам по себе очень неспецифичен.Например, фиксированная проксимальная мышечная слабость может быть признаком воспалительной миопатии, но этот симптом может быть вызван множеством других состояний (см. Таблицу 1).
При повышении фермента, который считается признаком миозита, является креатинфосфокиназа (КФК). Иногда ошибочно полагают, что КФК необходимо повышать хотя бы один раз в течение полимиозита, но это не всегда так. У некоторых пациентов уровень КФК остается нормальным на протяжении всего курса лечения. Кроме того, существует несколько других причин повышенного уровня КФК, включая упражнения (анаэробные и аэробные), травмы (тупые или острые), наркотики (статины, колхицин, алкоголь, кокаин) и токсины.У носителей мышечной дистрофии или некоторых метаболических миопатий может быть повышенный уровень КФК в сыворотке. У здоровых афроамериканцев уровень КФК обычно выше верхних пределов нормы, сообщаемых для населения в целом. Наконец, в крупных клинических испытаниях с использованием статинов более 30% людей имели повышенный уровень КФК. Интересно, что процент тех, кто принимал плацебо, был таким же, как и у тех, кто принимал исследуемые препараты. 3 Таким образом, полагаться на этот тест как на способ скрининга мышечного воспаления может быть проблематично.
нажмите, чтобы увидеть большую версию
Таблица 1: Причины слабости проксимальных мышц, которые могут имитировать воспалительную миопатию
Классическая триада описанных изменений ЭМГ включает: 1) фибрилляцию в покое, повышенную инсерционную активность, а также спонтанные и положительные резкие волны; 2) причудливые высокочастотные повторяющиеся разряды; 3) полифазные потенциалы малой длительности и малой амплитуды. 1,2 Однако эта триада не является диагностической и обычно присутствует только у 40% пациентов с миозитом, в то время как 10% пациентов с этими заболеваниями имеют нормальные результаты ЭМГ. 4
Воспалительная миопатия (миозит) | Хьюстон Методист
Миопатия — это термин, используемый для описания нервно-мышечного расстройства, при котором мышечные волокна не работают должным образом из-за нескольких причин, и в результате возникает мышечная слабость, непроизвольное движение или спазм. Воспалительная миопатия (миозит) возникает, когда мышечная слабость вызвана воспалением по ряду причин.
Миопатии могут передаваться по наследству или приобретаться. Миопатии сгруппированы по ряду факторов.
- Врожденные миопатии часто проявляются задержкой развития двигательных навыков с самого рождения; вы можете заметить аномалии скелета и лица. Искривление позвоночника или сколиоз являются представителями этой группы.
- Мышечные дистрофии проявляются как прогрессирующая слабость произвольных мышц, которая иногда проявляется при рождении.
- Митохондриальные миопатии вызываются генетическими аномалиями в клеточных митохондриях, структурах, контролирующих энергию в мышечных клетках.Синдром Кернса-Сейра является представителем этой группы.
- Болезни накопления гликогена в мышцах вызываются мутациями в генах, которые контролируют ферменты, метаболизирующие гликоген и глюкозу (сахар в крови). Болезнь Кори является представителем этой группы.
- Миоглобинурия возникает при нарушении метаболизма мышечного топлива (миоглобина). Болезнь Димауро является примером этого типа состояния.
- Дерматомиозит вызывает воспалительную реакцию кожи и мышц.
- Оссифицирующий миозит вызывается кальцификацией мышечной ткани.
- Семейные периодические параличи — это наследственные неврологические нарушения, вызванные мутациями в генах, регулирующих натриевые и кальциевые каналы в нервных клетках.
- Полимиозит и миозит с тельцами включения могут быть вызваны травмой, инфекцией, побочными эффектами лекарств, аллергией или аутоиммунными заболеваниями.
- Нейромиотония, также известная как синдром Исаакса, характеризуется чередованием эпизодов подергивания и скованности.Точная причина неизвестна.
- Тетания, непроизвольное сокращение мышц, обычно вызывается недостатком кальция или избытком калия.
При большинстве миопатий вы можете испытывать слабость, затрагивающую мышцы, расположенные вблизи ядра вашего тела, а также судороги, скованность, спазм, боль и усталость в этих мышцах.
Диагностика миопатии
Многие миопатии трудно диагностировать до тех пор, пока симптомы не прогрессируют до наблюдаемой точки. Наша команда запросит у вас историю болезни и проведет медицинский осмотр.
Чтобы определить, какая форма миопатии наиболее вероятна для вашего состояния, мы проведем неврологическое обследование, чтобы определить, насколько хорошо вы можете вставать со стула и ходить, а также проверим вашу координацию и реакцию коленного рефлекса. Мы можем проводить визуализацию, неврологические анализы и анализы крови, чтобы определить, есть ли у вас какие-либо эндокринные нарушения, проблемы с сердцем, психическая дисфункция, мышечная атрофия или кожная сыпь; наша задача будет заключаться в том, чтобы определить, следует ли ваша мышечная слабость какому-либо конкретному образцу.На раннем этапе вашего состояния мы можем предложить тест на ферменты сыворотки, чтобы измерить, сколько мышечного белка циркулирует в вашей крови.
С помощью электромиографа наша команда может определить, какие мышцы ослаблены и в какой степени; биопсия мышечной ткани может предоставить информацию о клеточных и белковых аномалиях.
Варианты лечения миопатии
Различные типы миопатий имеют множество различных причин, поэтому единого метода лечения миопатии не существует. Способы лечения варьируются от тех, которые направлены на устранение симптомов, до тех, которые направлены на конкретные причины состояния.