Прогерия поможет понять старение
Старение населения сегодня не сравнить ни с одним известным этапом развития человечества.
При этом о самом процессе старения известно слишком мало: людям просто не хватает жизни, чтобы исследовать его. И вот теперь ученые из Института биологических исследований Солка воспроизвели преждевременное старение в лаборатории, что позволит им исследовать связанные с ним заболевания под микроскопом.
В издании Nature от 23 февраля 2011 года профессор Хуан Карлос Изписуа Бельмонте с коллегами сообщили, что им удалось успешно вырастить индуцированную плюрипотентную стволовую клетку из клеток кожи пациентов с синдромом прогерии Хатчинсона-Гилфорда. Люди с этим расстройством стареют в 8-10 раз быстрее остальных. Полученные клетки ученые дифференцировали на клетки гладкой мышечной ткани, демонстрирующие контрольные признаки сосудистого старения.
«Медленное развитие и сложность старения делают трудным изучение патогенеза сердечно-сосудистых и других связанных со старением расстройств», сообщил Изписуа Бельмонте. «Наличие человеческой модели ускоренного старения будет способствовать развитию потенциальных методов лечения прогерии и позволит понять старение. Кроме того, исследование поможет в профилактике и лечении сердечных заболеваний».
Уникальные особенности прогерии напоминают процесс старения: люди с синдромом редко доживают до 13 лет. Почти все пациенты умирают от осложнений артериосклероза — засорения или укрепления стенок артерий и сосудов из-за бляшек, что в итоге приводит к инфарктам и инсультам.
Особенно ученые интересуются прогерией в надежде, что она даст подсказки к нормальному процессу человеческого старения. Однако заболевание это чрезвычайно редкое. В настоящее время в мире лишь у 64 детей есть этот страшный диагноз.
Синдром прогерии Хатчинсона-Гилфорда вызывается одноточечной мутацией гена, кодирующего ламин А, который формирует подложку белка на внутреннем крае ядра, что помогает поддерживать структуру хроматина и организовывать процессы, такие как синтез ДНК и РНК.
Мутация создает альтернативный участок сращивания, что приводит к выработке усеченной версии белка прогерина. В отличие от цельного белка прогерин не интегрируется должным образом в ядерную ламину, и в итоге ядерная подложка разрушается, приводя к проблемам.
«Есть признаки, что дефектный ламин А накапливается в процессе старения вследствие спорадического использования альтернативного участка сращивания», пояснил Изписуа Бельмонте. «Поэтому нам необходимо при использовании клеточной модели в пробирке идентифицировать новые маркеры старения и исследовать другие аспекты преждевременного физиологического старения человека».
По сравнению с нормальными фибробластами кожи у клеток пациентов с прогерией деформированы ядра, а также есть целый диапазон других ядерных дефектов, включая дефектную ядерную ламину, отсутствие сверхсжатой ДНК, укороченные теломеры и нестабильность генома. И все же, несмотря на все эти особенности, такие клетки легко преобразовываются в индуцированные плюрипотентные стволовые.
«В результате перепрограммирования были стерты все ядерные и эпигенетические дефекты, а помолодевшие плюрипотентные клетки вели себя совсем как нормальные здоровые клетки», сообщил постдокторант Гуан-Хью Лю.
После того, как стало ясно, что ламин А выражается лишь в дифференцированных клетках, но отсутствует в эмбриональных стволовых, ученый задался вопросом, вырабатывают ли индуцированные плюрипотентные стволовые клетки ламин А и/или прогерин, обладающий теми же свойствами. В ходе эксперимента не удалось обнаружить ни одного подтверждения этому. «Биологические часы в этих клетках перезагружены, а ламин А заглушен», сказал Лю.
Как только исследователи дифференцировали индуцированные плюрипотентные стволовые клетки пациента с прогерией, экспрессия прогерина была реактивирована. «Это обратимое подавление экспрессии прогерина, повторное программирование и последующее оживление в процессе дифференцирования обеспечивает уникальную образцовую систему для изучения человеческих патологий преждевременного старения», сказал Изписуа Бельмонте.
Главным образом прогерин накапливается в клетках гладкой мышечной ткани, содержащихся в пределах стенок артерий и кровеносных сосудов. Дегенерация таких клеток — один из симптомов прогерии.
Источник: innovanews.ru
Прогерия (Синдром Вернера) — санатории где лечат, цены и отзывы
Прогерия (Синдром Вернера) — это генетическое заболевание, характеризующееся преждевременным старением больного. У него отмечаются поражения различных систем организма, появление атеросклероза и злокачественных опухолей. Человек очень быстро превращается в старика. Чаще прогерией болеют мужчины.
Причины
Синдром Вернера передается по наследству, это аутосомно-рецессивный тип заболевания. В данном случае ребенок рождается от родителей, имеющих дефектный ген WRN, что приводит к нарушению обмена соединительной ткани. Если подобная мутация есть только у одного родителя, болезнь не возникает, если же у обоих, то у ребенка развивается прогерия.
Симптомы
Признаки заболевания обычно появляются в 14-18 лет. В этом возрасте наблюдается седина, выпадение волос. Кожа становится бледной и плотной, с пигментными пятнами, появляются морщины. Мышцы и подкожная жировая клетчатка атрофируются. Руки и ноги становятся непропорционально тонкими. Там, где под кожей выступают кости возникают язвы.
На третьем десятилетии после начала болезни у пациента меняется голос, он становится высоким. Возникают следующие симптомы:
- Катаракта.
- Сухость кожи.
- Мозоли на подошвах.
- Язвы на нижних конечностях.
- Низкий рост.
- Лунообразное лицо с выступающим подбородком и узким ртом.
- Остеопороз.
- Гиперпигментация.
- Деформация кистей.
- Остеоартриты.
- Плоскостопия, стерильность и так далее.
В зрелом возрасте больные выглядят как очень пожилые люди. На четвертом десятилетии после возникновения прогерии у них диагностируется сахарный диабет, атеросклероз, дисфункции щитовидной железы. В ряде случаев возможно развитие раковых заболеваний.
В ряде случаев возможно развитие раковых заболеваний.
Виды/формы
Синдром Вернера является подвидом прогерии, он возникает у людей в период полового созревания и даже позже. Другой подвид прогерии — синдром Хатчинсона-Гилфорда. Эта болезнь возникает у маленьких детей. Она также приводит к преждевременному старению, больные обычно не доживают до совершеннолетия.
Стадии
Болезнь развивается постепенно, в течение нескольких десятилетий. На начальной стадии наблюдается признаки раннего старения: седина в волосах и их впадание. С течением времени больной теряет способность вести полноценную жизнь. Заканчивается прогерия смертью больного. Обычно это наступает в возрасте 50 лет.
Методы лечения
Данная болезнь является хронической и постоянно прогрессирует, поэтому лечение сводится к профилактике осложнений прогерии. Больные регулярно наблюдаются у эндокринологов, кардиологов и так далее. Это поможет замедлить развитие патологических изменений в организме. Излечиться от синдрома Вернера невозможно, пациент обычно умирает от раковых опухолей или последствий атеросклероза.
Профилактика в санаториях России
Больным с синдромом Вернера будет полезно санаторно-курортное лечение. В России есть немало санаториев с комфортными условиями проживания, прекрасным оснащением, где пациенты смогут приостановить проявления болезни и укрепят свой организм. В этом им помогут опытные врачи и новые методики лечения.
Прогерия на молекулярном уровне — vechnayamolodost.ru
Ученые выяснили причины раннего старения детей
Алексей Евглевский, Naked Science
Синдром Хатчинсона-Гилфорда – это редчайший генетический дефект, который вызывает признаки раннего старения у детей. Из-за этого синдрома дети умирают в подростковом возрасте, зачастую не достигнув совершеннолетия. Генетики из Сент-Луисского университета выяснили причины, из-за которых возникает этот дефект.
Группа ученых во главе с Сусаной Гонзало назвала механизм, вызывающий синдром Хатчинсона-Гилфорда, «репликационным стрессом». Он появляется благодаря мутации в гене LMNA, который кодирует белок ламин А. Ламин А отвечает за устойчивое формирование новых клеток. При правильной работе этого белка новые клетки сохраняют прежнюю форму и структуру. Укороченная версия этого белка называется прогерином. Она вызывает неправильное деление клеток, из-за которого ядро клетки копируется с ошибкой.
Поскольку ядро хранит в себе ДНК, то неверная репликация ядра ведет к мутациям внутри ДНК, что приводит к образованию раковых клеток и старению.
Команда Гонзало обнаружила два механизма, которые наносят урон организму. Первый проявляется в нарушении слаженной работы репликации. Обычно клетки находят способ обойти препятствие и продолжают делиться, но прогерин заставляет этот процесс остановиться. Клетки, неспособные делиться, вызывают преждевременное старение. Второй механизм связан с выходом ДНК за пределы ядра. О нем рассказала
Сусана Гонзало:
«Когда фрагменты ДНК выходят за пределы ядра и попадают в цитоплазму, они воспринимаются организмом как чужеродный материал. Это активирует иммунную реакцию, известную как интерфероновый ответ. В этом случае клетка считает, что внутрь проникла бактериальная инфекция и с ней надо бороться. Этот процесс мы наблюдали в клетках прогерии, что уменьшало деление клеток».
Генетики обнаружили, что процесс можно затормозить, используя витамин D: «Когда мы блокируем этот путь витамином D, клетки омолаживаются. Иммунный ответ активируется прогерином, а затем все восстанавливается витамином D».
Гонзало надеется, что их открытие поможет не только бороться с синдромом Хатчинсона-Гилфорда, но и откроет новые перспективы в понимании самого процесса старения. Недавно ученые нашли основную причину старения мышц человека.
Портал «Вечная молодость» http://vechnayamolodost.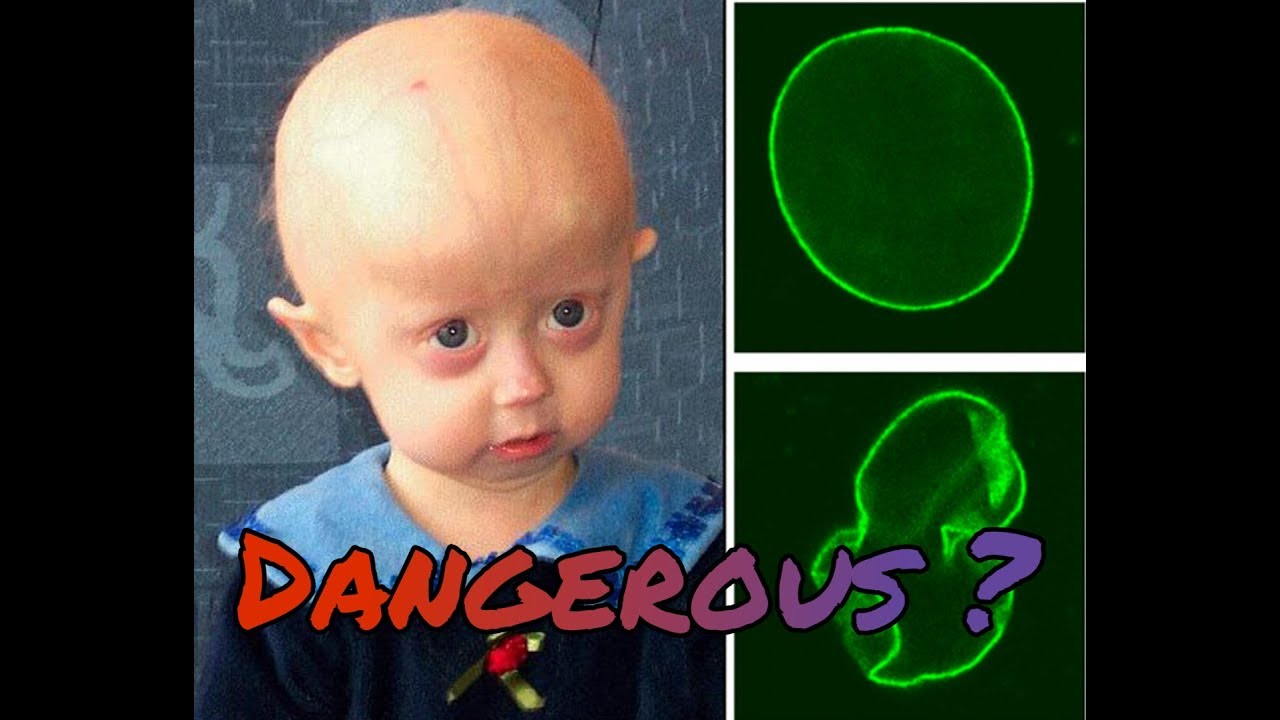 ru
ru
Ген LMNA кодирует предшественник ламина А HGPS возникает из-за мутации одного гена LMNA, кодирующего преламин А/C, приводящей к делеции 50 аминокислот в предшественнике белка ламинаА -преламине А, который является структурным белком ядерной мембраны (рис.2, 3). ]]>]]> рис 2. Схема строения гена LMNA ]]>]]> рис 3. Схема строения мРНК и белка преламина А. UTR — нетранслируемый регион, NLS — сигнал ядерной локализации, CAAX — мотив пренилирования. рис 4. Мутантный ламин Наиболее часто HGPS происходит при точечных мутациях экзона 11 LMNA, мутация происходит в кодоне 608 и активирует загадочный сайт сплайсинга, приводящий к делеции в 50 аминокислот в рамке считывания преламина А (606-656 аминокислоты), но при этом остается С-концевой CAAX мотив. При этом теряется сайт по которому происходит расщепление белка преламина во время созревания. |
 CAAX мотив запускает три последовательных ферментативных реакции посттрансляционной модификации, приводящие к зрелому ламину А, структурному белку ядерной мембраны:
CAAX мотив запускает три последовательных ферментативных реакции посттрансляционной модификации, приводящие к зрелому ламину А, структурному белку ядерной мембраны: В нормальных клетках преламин обнаружить не удается, так как он сразу же переводится в зрелый ламин А. Мутантный преламин А, образующийся при HGPS, который называют также прогерин, вмещает CAAX мотив, запускающий фарнезилирование, но делеция в 50 аминокислот предотвращает последующий процессинг в зрелый ламин А, что приводит к накоплению прогерина в нуклеоплазме ядра. Это приводит к нестабильности мембраны: нарушается ее
В нормальных клетках преламин обнаружить не удается, так как он сразу же переводится в зрелый ламин А. Мутантный преламин А, образующийся при HGPS, который называют также прогерин, вмещает CAAX мотив, запускающий фарнезилирование, но делеция в 50 аминокислот предотвращает последующий процессинг в зрелый ламин А, что приводит к накоплению прогерина в нуклеоплазме ядра. Это приводит к нестабильности мембраны: нарушается ееПреждевременное старение начинается из-за «беспорядка» в ДНК
Чем больше хромосомной ДНК находится в распакованном, активном виде, тем раньше и быстрее клетка начинает стареть.
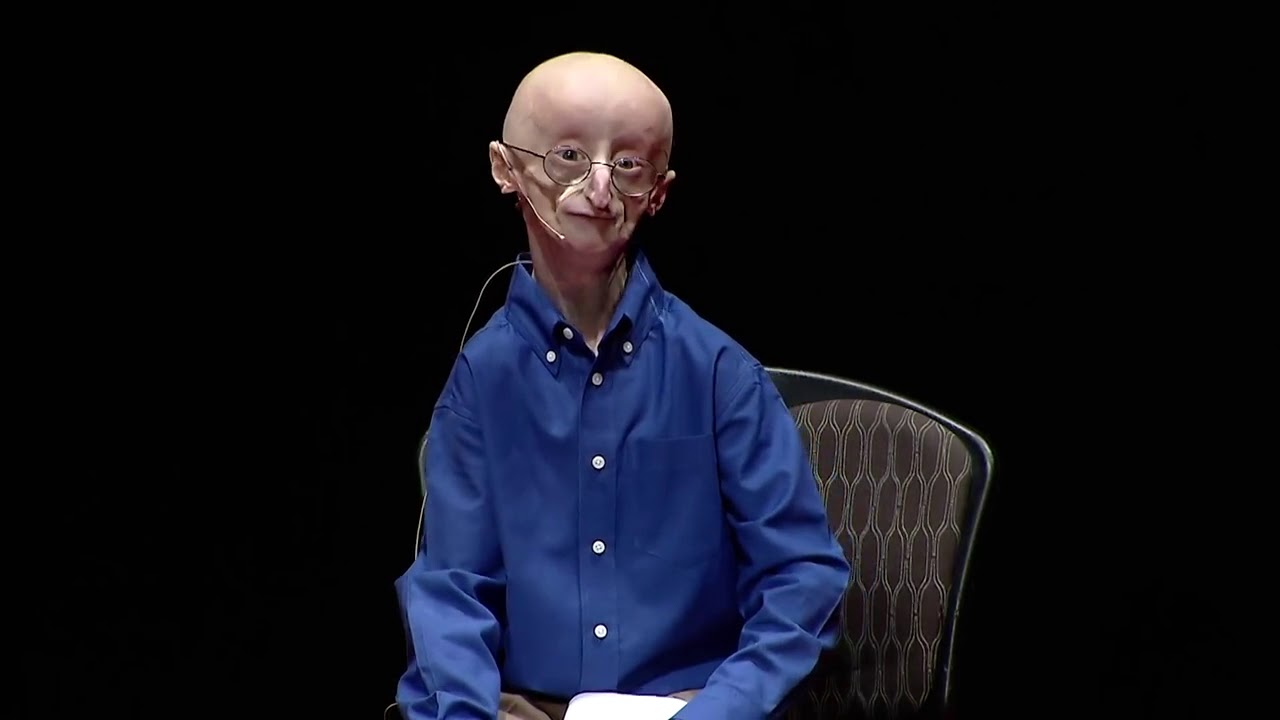
Все люди стареют по-разному, кто-то быстрее, кто-то медленнее, но, если брать в целом, признаки старости появляются у всех примерно в одном возрасте. За одним исключением: в том случае, если человек болен прогерией, стареть он начинает исключительно рано. У этой болезни есть два варианта, детский и взрослый, детский называется синдромом Хатчинсона-Гилфорда, взрослый – синдромом Вернера. Дети с прогерией страдают от заболеваний, характерных для преклонного возраста: истончение и морщинистость кожи, облысение, сердечно-сосудистые болезни, нарушения жирового обмена, атеросклероз, проблемы с суставами и т. п. У них резко замедляется рост и развивается характерный внешний вид: большая голова, маленькое заострённое лицо, недоразвитая нижняя челюсть. В среднем больные детской формой прогерии живут не дольше 12–13 лет.
Нормальный ребёнок и ребёнок того же возраста с прогерией. (Фото Ian Tomey / Flickr.com.)
Нормальные стволовые клетки (слева) и клетки с синдромом преждевременного старения (справа), одним из характерных признаков которых является увеличение размера. (Фото Salk Institute for Biological Studies.)
Фото © Кекяляйнен Андрей / Фотобанк Лори.
‹
›
Люди со взрослым вариантом прогерии живут дольше, однако и у них возрастные изменения случаются намного раньше обычного – в 20 лет начинают седеть и выпадать волосы, к 30 годам развиваются катаракта, остеопороз, и другие заболевания, например, диабет, и обычно человек с синдромом Вернера не доживает до 60 лет. Известно, что, по крайней мере, в случае тяжёлой формы в клетках происходят во многом те же молекулярные изменения, что и при обычном старении, так что, если мы найдём способ тормозить прогерию, это, возможно, даст нам инструмент против старения вообще.
Секреты болезни можно было бы понять, понаблюдав за стволовыми клетками, которые получили от больных людей. Некоторое время назад исследователи из Института биологических исследований Солка смогли превратить кожные клетки детей с синдромом Хатчинсона-Гилфорда в аналог эмбриональных стволовых клеток, так называемые индуцированные плюрипотентные стволовые клетки. Далее с ними можно было ставить опыты, выясняя, что не так в стволовых процессах у больных прогерией. Но когда то же самое попытались сделать с клетками больных синдромом Вернера, ничего не вышло – их клетки оказались слишком повреждёнными болезнью, чтобы выдержать возврат в стволовое, недифференцированное состояние. Тогда Хуан Карлос Изписуа Бельмонте (Juan Carlos Izpisua Belmonte) вместе с коллегами из Китайской академии науки и Пекинского университета пошли по другому пути – они смоделировали прогерию в изначально здоровых клетках.
Известно, что синдром Вернера сопровождается мутациями в гене WRN, который задействован в процессах копирования и репарации ДНК. И вот, чтобы создать модель болезни, исследователи попросту поломали этот ген в стволовых клетках из эмбриона человека. Эмбриональные клетки по ходу развития превращаются в более специализированные разновидности, которые в дальнейшем могут дать начало той или иной ткани – например, в мезенхимальные стволовые клетки, «родоначальники» жировой ткани, хрящей и костей. В статье в Science авторы пишут, что, когда стволовые клетки с неработающим геном WRN превращались в мезенхимальные, они тут же начинали резко стареть: в их ДНК накапливалось много повреждений, они переставали делиться, и, наконец, у них сильно укорачивались теломеры. Так называют концы хромосом, которые при копировании ДНК защищают гены от повреждений, связанных с особенностями работы белковой копировальной машины. Длина теломер уменьшается с каждым делением клетки, и потому их считают чем-то вроде молекулярных часов, отмеряющих время жизни.
И вот, чтобы создать модель болезни, исследователи попросту поломали этот ген в стволовых клетках из эмбриона человека. Эмбриональные клетки по ходу развития превращаются в более специализированные разновидности, которые в дальнейшем могут дать начало той или иной ткани – например, в мезенхимальные стволовые клетки, «родоначальники» жировой ткани, хрящей и костей. В статье в Science авторы пишут, что, когда стволовые клетки с неработающим геном WRN превращались в мезенхимальные, они тут же начинали резко стареть: в их ДНК накапливалось много повреждений, они переставали делиться, и, наконец, у них сильно укорачивались теломеры. Так называют концы хромосом, которые при копировании ДНК защищают гены от повреждений, связанных с особенностями работы белковой копировальной машины. Длина теломер уменьшается с каждым делением клетки, и потому их считают чем-то вроде молекулярных часов, отмеряющих время жизни.
Однако у клеток с синдромом Вернера была ещё одна особенность, которая более всего привлекла внимание авторов работы. Известно, что ДНК в клеточном ядре находится в комплексе с белками. Некоторые из них выполняют какие-то текущие работы на тех или иных генах (например, синтезируют РНК), другие же играют структурную роль, поддерживая в упакованном состоянии довольно обширные фрагменты хромосом. Упакованная, структурированная часть ДНК называется гетерохроматином. И вот оказалось, что у больных клеток гетерохроматина очень мало – иными словами, ДНК при синдроме Вернера приходит в свободное, «растрёпанное» состояние.
То же самое можно наблюдать и при обычном старении: когда состояние хромосом сравнили у нескольких людей разного возраста, то увидели, что чем старше человек, тем хуже у него упакована ДНК в ядрах. Очевидно, при прогерии тот же процесс происходит быстрее и начинается раньше – возможно, что уже на ранних стадиях индивидуального развития. Почему неупорядоченное, неупакованное состояние хромосом может приводить к таким последствиям? Если какой-то ген находится в гетерохроматиновом виде, это значит, что он неактивен, выключен, находится в спящем состоянии. Если же упаковка слабеет, то у нас начнут включаться гены, которые должны молчать. Как раз такая ненужная активность может в совокупности приводить к старению. С другой стороны, известно, что в гетерохроматиновом, запечатанном виде находятся мобильные генетические элементы, которые прыгают в ДНК с места на место, вызывая тем самым нежелательные мутации.
Если же упаковка слабеет, то у нас начнут включаться гены, которые должны молчать. Как раз такая ненужная активность может в совокупности приводить к старению. С другой стороны, известно, что в гетерохроматиновом, запечатанном виде находятся мобильные генетические элементы, которые прыгают в ДНК с места на место, вызывая тем самым нежелательные мутации.
Действительно ли общая распаковка и беспорядок в ДНК влечёт за собой все те изменения, характерные для стареющих клеток, и происходит ли так во всех случаях прогерии, как детской, так и взрослой, покажут дальнейшие эксперименты. Но, если всё и впрямь так, биологи смогут сосредоточиться на упаковке ДНК как потенциальной мишени для лекарств, которые помогли бы задержать старение – как преждевременное, так и обычное.
Синдром Хатчинсона-Гилфорда (прогерия детей) | Надент
Рекомендация
Студентам
Вы можете использовать данную статью как часть или основу своего реферата или даже дипломной работы или своего сайта
Просто перейдите по ссылке ниже, редактируйте статью, все картинки тоже доступны, все бесплатно
Редактировать статью?!
Скачать статью в формате PDF
Сохраните результат в MS Word Docx или PDF, делитесь с друзьями, спасибо 🙂
Категории статей
Синдром Хатчинсона-Гилфорда (прогерия детей)
Синдром Хатчинсона-Гилфорда, или прогерия детей, — крайне редкое заболевание. Его частота составляет 1 на 1 000 000 человек. Именно этот синдром занесен под названием прогерии в OMIM (Online Mendeltan inheritance in Man).
Фенотип пациентов чрезвычайно характерный: маленький рост, «птичье лицо» с клювообразным профилем, преобладание размеров мозговой части черепа над лицевой, выступающая венозная сеть на коже мозговой части, как правило, обнаженной вследствие аллопеции, часто тотальной, с выпадением бровей и ресниц. Наблюдается резкая гипоплазия ключиц, дефекты формы и числа зубов, сухая истонченная кожа, практически полное отсутствие подкожной жировой клетчатки, отставание в развитии, особенно физическом. Больные бесплодны, хотя в литературе описан случай рождения ребенка у пацентки с синдромом Хатчинсона-Гилфорда. В крови повышен уровень Средняя продолжительность жизни описанных носителей синдрома- 13,4 лет (как редкое наблюдение описан единственный 45-летний пациент). Причиной смерти, как правило, служит инфаркт миокарда, с выявлением на аутопсии генерализованного атеросклероза и фиброза миокарда, а также отложения жироподобного вещества в тканях мозга и паренхиматозных органов.
Наблюдается резкая гипоплазия ключиц, дефекты формы и числа зубов, сухая истонченная кожа, практически полное отсутствие подкожной жировой клетчатки, отставание в развитии, особенно физическом. Больные бесплодны, хотя в литературе описан случай рождения ребенка у пацентки с синдромом Хатчинсона-Гилфорда. В крови повышен уровень Средняя продолжительность жизни описанных носителей синдрома- 13,4 лет (как редкое наблюдение описан единственный 45-летний пациент). Причиной смерти, как правило, служит инфаркт миокарда, с выявлением на аутопсии генерализованного атеросклероза и фиброза миокарда, а также отложения жироподобного вещества в тканях мозга и паренхиматозных органов.
Репарация ДНК при синдроме Хатчинсона-Гилфорда нарушена: установлено, что клетки его носителей не способны избавляться от вызываемых химическими агентами сшивок ДНК-белок. Но главная диагностическая особенность клеток больных с данным синдромом состоит в резко сниженном, по сравнению с нормой, количестве делений, которое способны пройти клетки в культуре (так называемый лимит, или число Хейфлика). В 1971 г. А.М. Оловников высказал предположение об укорочении хромосомных теломер в процессе развития клеток. А в 1992 г. было показано, что для клеток пациентов с синдромом Хатчинсона-Гилфорда характерно врожденное укорочение теломер. Анализ взаимосвязи между лимитом Хейфлика, длиной теломер и активностью теломеразы (фермента, способного наращивать конец теломерной ДНК) дает возможность соотнести естественное старение и процесс формирования клинической картины при синдроме Хатчинсона-Гилфорда.
Крайне низкая частота встречаемости данной формы прогерии позволяет лишь высказывать гипотезы о типе наследования. Аутосомно-рецессивный тип предполагается по аналогии с синдромом Коккейна, имеющим отдельные черты преждевременного старения. Но есть и предположение о развитии синдрома Хатчинсона-Гилфорда вследствие доминантной аутосомной мутации, возникшей de novo. Оно получило косвенное подтверждение на основе измерения теломер у носителей синдрома, их родителей и здоровых доноров. Было показано, существенное укорочение теломер у больных по сравнению с другими двумя испытуемыми группами.
Оно получило косвенное подтверждение на основе измерения теломер у носителей синдрома, их родителей и здоровых доноров. Было показано, существенное укорочение теломер у больных по сравнению с другими двумя испытуемыми группами.
Источник: www.eurolab.ua
Редактирование оснований помогло в лечении синдрома преждевременнного старения
Исследователи из США с помощью метода генетического редактирования смогли значительно продлить срок жизни мышей с генетической вариацией, ассоциированной с прогерией – редким наследственным заболеванием, вызывающем у детей резкое преждевременное старение. Статья исследователей опубликована в журнале Nature.
Прогерия, или синдром Хатчинсона — Гилфорда, вызывается мутацией в гене LMNA (кодирующем белок ламин А), при которой азотистое основание С заменяется на T. Это приводит к усиленной выработке прогерина — более «короткой» версии ламина A, — который и вызывает ускоренное старение. Прогерия — редкая болезнь: она диагностируется у одного из четырех миллионов детей в возрасте до двух лет. Практически у каждого из подверженных синдрому в детском и подростковом возрасте начиная с года возникают проблемы со здоровьем, характерные для пожилого возраста: замедление роста, развитие сердечно-сосудистых заболеваний, потеря волос, слуха и подкожного жира, а также загрубление кожи. Такие дети обычно умирают в возрасте от 14 до 15 лет — обычно от атеросклероза или сердечного приступа. В ноябре 2020 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) США одобрило использование лонафарниба — первого препарата против прогерии. Однако он лишь на некоторое время продлевает жизнь пациента.
Исследователи из Национального научно-исследовательского института генома человека Национальных институтов здоровья США, Института Броудов и Университета Вандербильта решили изучить, как методика редактирования генома, известная как редактирование оснований, — при которой «буквы» ДНК меняются без повреждения последней, — повлияет на мышей со сходными с прогерией симптомами.
Для начала ученые протестировали метод на полученных от пациентов с прогерией тканях — он исправил мутацию в 90% клеток. Затем исследователи внутривенно ввели четырнадцатидневным мышатам с вызывающей синдром мутацией смесь из редактирующих белков и доставляющих их синтетических лентивирусов. Генетическому редактору удалось восстановить правильную последовательность гена LMNA в значительном числе органов, в том числе сердце и аорте. В последней отредактированные клетки смогли даже полностью заместить клетки, несущие мутацию. Как результат, продолжительность жизни мышей возросла почта в два раза, с семи месяцев до полутора лет — при том что средний возраст использованных в исследовании мышей составляет два года.
«В конечном счете нашей целью будет разработка этого (метода лечения — Indicator.Ru) для людей, однако остается еще ряд первостепенных проблем, который надо решить на модельных системах», — признал один из исследователей, доцент факультета сердечно-сосудистых заболеваний Медицинского центра Университета Вандербильта Джонатан Браун.
FDA одобрило первое лечение синдрома прогерии Хатчинсона-Гилфорда и некоторых прогероидных ламинопатий
- Для немедленного выпуска:
Сегодня Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило капсулы Zokinvy (лонафарниб) для снижения риска смерти из-за синдрома прогерии Хатчинсона-Гилфорда и для лечения некоторых прогероидных ламинопатий с недостаточной обработкой у пациентов в возрасте от одного года и старше.Zokinvy не одобрен для использования у пациентов с другими прогероидными синдромами или ламинопатией.
«Синдром прогерии Хатчинсона-Гилфорда и прогероидная ламинопатия являются редкими генетическими заболеваниями, которые вызывают преждевременное старение и смерть и оказывают изнурительное влияние на жизнь людей», — сказал Хилтон В. Джоффе, доктор медицины, MMSc, директор Управления редких заболеваний, педиатрия , Урологическая и репродуктивная медицина в Центре оценки и исследований лекарственных средств FDA. «С сегодняшнего одобрения Zokinvy является первым лекарством от этих разрушительных болезней, одобренным FDA.FDA продолжит работу с заинтересованными сторонами, чтобы продвигать разработку дополнительных новых, эффективных и безопасных методов лечения для этих пациентов ».
Джоффе, доктор медицины, MMSc, директор Управления редких заболеваний, педиатрия , Урологическая и репродуктивная медицина в Центре оценки и исследований лекарственных средств FDA. «С сегодняшнего одобрения Zokinvy является первым лекарством от этих разрушительных болезней, одобренным FDA.FDA продолжит работу с заинтересованными сторонами, чтобы продвигать разработку дополнительных новых, эффективных и безопасных методов лечения для этих пациентов ».
Пациенты с синдромом прогерии Хатчинсона-Гилфорда и прогероидной ламинопатией испытывают ускоренное сердечно-сосудистое заболевание из-за накопления в клетках дефектного прогерина или прогерин-подобного белка. Большинство пациентов умирают в возрасте до 15 лет от сердечной недостаточности, сердечного приступа или инсульта. До сегодняшнего утверждения единственные варианты лечения включали поддерживающую терапию и терапию, направленную на устранение осложнений, связанных с заболеванием.
Zokinvy, ингибитор фарнезилтрансферазы, представляет собой пероральный препарат, который помогает предотвратить накопление дефектного прогерина или прогерин-подобного белка. Эффективность Zokinvy для лечения синдрома прогерии Хатчинсона-Гилфорда была продемонстрирована у 62 пациентов в двух исследованиях с одной группой, которые сравнивались с подобранными, нелеченными пациентами из отдельного исследования естественной истории. По сравнению с нелеченными пациентами продолжительность жизни пациентов с синдромом прогерии Хатчинсона-Гилфорда, получавших Zokinvy, увеличилась в среднем на три месяца в течение первых трех лет лечения и в среднем на 2.5 лет при максимальном сроке наблюдения 11 лет. При одобрении Зокинви лечения некоторых прогероидных ламинопатий с недостаточным процессингом, которые встречаются очень редко, были приняты во внимание сходства в генетическом механизме заболевания и другие доступные данные.
Наиболее частые побочные эффекты включали тошноту, рвоту, диарею, инфекции, снижение аппетита и утомляемость.
Zokinvy противопоказан для одновременного приема с сильными или умеренными ингибиторами и индукторами CYP3A, а также с мидазоламом и некоторыми лекарствами, снижающими уровень холестерина.У некоторых пациентов, получавших Zokinvy, развились отклонения лабораторных тестов, такие как изменения уровня натрия и калия в крови, снижение количества лейкоцитов и повышение анализов крови печени. Периодически следует проводить регулярные лабораторные анализы крови. Глазная токсичность наблюдалась у животных, поэтому рекомендуется периодически осматривать глаза и при появлении новых визуальных изменений.
FDA присвоило этой заявке статус приоритетного рассмотрения. Зокинви получил статус орфанного лекарства, который обеспечивает стимулы для помощи и поощрения разработки лекарств от редких заболеваний, а также статус прорывной терапии.Кроме того, производитель получил ваучер на приоритетную проверку редких детских заболеваний. Ваучерная программа FDA на приоритетное рассмотрение редких педиатрических заболеваний предназначена для поощрения разработки новых лекарств и биопрепаратов для профилактики и лечения редких заболеваний у детей. FDA предоставило одобрение Zokinvy компании Eiger BioPharmaceuticals, Inc.
FDA, агентство в составе Министерства здравоохранения и социальных служб США, защищает общественное здоровье, обеспечивая безопасность, эффективность и безопасность лекарственных и ветеринарных препаратов, вакцин и других биологических продуктов для использования человеком, а также медицинских устройств.Агентство также отвечает за безопасность и сохранность продуктов питания, косметики, пищевых добавок, продуктов, излучающих электронное излучение, а также за регулирование табачных изделий.
###
Текущее содержание с:
Прогероидные синдромы Вернера и Хатчинсона-Гилфорда: механистическая основа прогероидных заболеваний человека
 org/ScholarlyArticle»> 1
org/ScholarlyArticle»> 1Martin, G.М. Генетические синдромы у человека, потенциально имеющие отношение к патобиологии старения. Врожденные дефекты Ориг. Artic. Сер. 14 , 5–39 (1978).
CAS
PubMed
Google ученый
Friedberg, E.C. et al. Ремонт ДНК и мутагенез (ASM Press, Вашингтон, 2006).
Google ученый
Вернер, О. в Синдром Вернера и старение человека (ред. Солк, Д., Фудзивара, Ю. и Мартин, Г. М.) 1–14 (Plenum Press, Нью-Йорк, 1985).
Google ученый
Эпштейн, К. Дж., Мартин, Г. М., Шульц, А. Л. и Мотульский, А. Г. Вернер, обзор его симптоматологии, естественного течения, патологических особенностей, генетики и связи с естественным процессом старения. Медицина (Балтимор) 45 , 177–221 (1966). Подробное описание клинических, патологических, клеточных и генетических особенностей синдрома Вернера.
Артикул
CAS
Google ученый
Толлефсбол, Т. О. и Коэн, Х. Дж. Синдром Вернера: недиагностируемое заболевание, напоминающее преждевременное старение. Возраст 7 , 75–88 (1984).
Артикул
Google ученый
Гото, М. Иерархическое ухудшение систем организма при синдроме Вернера: последствия для нормального старения. мех.Aging Dev. 98 , 239–254 (1997).
Артикул
CAS
PubMed
Google ученый
Huang, S. et al. Спектр мутаций WRN у пациентов с синдромом Вернера. Гм. Мутат. 27 , 558–567 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Гото, М. Клинические характеристики синдрома Вернера и других синдромов преждевременного старения: характер старения при прогероидных синдромах. Gann. Монография. Cancer Res. 49 , 27–39 (2001).
Google ученый
Uhrhammer, N.A. et al. Синдром Вернера и мутации генов WRN и LMNA во Франции. Гм. Мутат. 27 , 718–719 (2006).
Артикул
PubMed
Google ученый
Yu, C.E. et al. Позиционное клонирование гена синдрома Вернера. Наука 272 , 258–262 (1996).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Бахрати, К. З. и Хиксон, И. Д. Геликазы RecQ: супрессоры онкогенеза и преждевременного старения. Biochem. J. 374 , 577–606 (2003).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Опреско, П.Л., Ченг, В. Х. и Бор, В. А. Соединение биохимии геликазы RecQ и болезней человека. J. Biol. Chem. 279 , 18099–18102 (2004).
Соединение биохимии геликазы RecQ и болезней человека. J. Biol. Chem. 279 , 18099–18102 (2004).
Артикул
CAS
PubMed
Google ученый
Goto, M. et al. Иммунологическая диагностика синдрома Вернера по продуктам подавленных и усеченных генов. Гм. Genet. 105 , 301–307 (1999).
Артикул
CAS
PubMed
Google ученый
Мозер, М.J. et al. Экспрессия геликазы WRN в клеточных линиях синдрома Вернера. Nucleic Acids Res. 28 , 648–654 (2000).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Kawabe, T. et al. Дифференциальная регуляция геликаз семейства RecQ человека в клеточной трансформации и клеточном цикле. Онкоген 19 , 4764–4772 (2000).
Артикул
CAS
PubMed
Google ученый
Суонсон, К., Saintigny, Y., Emond, M.J. и Monnat, R.J. мл. Белок синдрома Вернера имеет раздельные функции рекомбинации и выживания. Восстановление ДНК (Amst) 3 , 475–482 (2004).
Артикул
CAS
Google ученый
Moser, M. J. et al. Генетическая нестабильность и риск гематологических заболеваний у пациентов с синдромом Вернера и гетерозигот. Cancer Res. 60 , 2492–2496 (2000).
CAS
PubMed
Google ученый
 org/ScholarlyArticle»> 18
org/ScholarlyArticle»> 18Огберн, К.E. et al. Генотоксин, индуцирующий апоптоз, отличает гетерозиготных носителей мутаций геликазы Вернера от мутантов дикого типа и гомозиготных мутантов. Гм. Genet. 101 , 121–125 (1997).
Артикул
CAS
PubMed
Google ученый
Poot, M. et al. Клетки с синдромом Вернера чувствительны к сшивающим ДНК препаратам. FASEB J. 15 , 1224–1226 (2001).
Артикул
CAS
PubMed
Google ученый
Шелленберг, Г.D., Miki, T. Yu, CE и Nakura, J. in The Metabolic and Molecular Basis of Inherited Disease (eds Scriver, CR, Beaudet, AL, Sly, WS & Valle, D.) 785–797 (McGraw -Хилл, Нью-Йорк, 2001).
Google ученый
Agrelo, R. et al. Эпигенетическая инактивация гена синдрома Вернера преждевременного старения при раке человека. Proc. Natl Acad. Sci. USA 103 , 8822–8827 (2006). Недавние доказательства того, что эпигенетическое подавление экспрессии WRN дает клеточные фенотипы, аналогичные мутациям WRN , и что подавление WRN при раке толстой кишки может иметь прогностическое значение.
Артикул
CAS
PubMed
Google ученый
Гото М., Миллер Р. В., Исикава Ю. и Сугано Х. Избыток редких видов рака при синдроме Вернера (прогерия у взрослых). Cancer Epidemiol. Биомаркеры Пред. 5 , 239–246 (1996).
CAS
PubMed
PubMed Central
Google ученый
Мушегян А. Р., Бассетт Д.E. Jr, Boguski, M. S., Bork, P. & Koonin, E. V. Позиционно клонированные гены болезней человека: паттерны эволюционной консервации и функциональные мотивы. Proc. Natl Acad. Sci. USA 94 , 5831–5836 (1997).
Артикул
CAS
PubMed
Google ученый
Mian, I. S. Сравнительный анализ последовательности рибонуклеаз HII, III, II PH и D. Nucleic Acids Res. 25 , 3187–3195 (1997).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Prince, P. R., Emond, M. J. и Monnat, R. J. Jr. Потеря функции белка синдрома Вернера способствует аберрантной митотической рекомбинации. Genes Dev. 15 , 933–938 (2001).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Сентиньи, Ю., Макиенко, К., Суонсон, К., Эмонд, М. Дж. И Моннат, Р. Дж. Дефект разрешения гомологической рекомбинации при синдроме Вернера. Мол. Клетка. Биол. 22 , 6971–6978 (2002). Ссылки 25 и 26 определяют роль WRN в гомологически зависимой рекомбинации в соматических клетках человека.
Артикул
CAS
PubMed
PubMed Central
Google ученый
Стюарт, Э., Чепмен, К. Р., Аль-Ходейри, Ф. , Carr, A. M. & Enoch, T. rqh2 + , ген делящихся дрожжей, связанный с генами синдромов Блума и Вернера, необходим для обратимой остановки S-фазы. EMBO J. 16 , 2682–2692 (1997).
, Carr, A. M. & Enoch, T. rqh2 + , ген делящихся дрожжей, связанный с генами синдромов Блума и Вернера, необходим для обратимой остановки S-фазы. EMBO J. 16 , 2682–2692 (1997).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Pichierri, P., Franchitto, A., Mosesso, P. & Palitti, F. Белок синдрома Вернера необходим для правильного восстановления после остановки репликации и повреждения ДНК, индуцированного в S-фазе клеточного цикла. Мол. Биол. Ячейка 12 , 2412–2421 (2001).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Опреско, П. Л. и др. Хеликаза и экзонуклеаза синдрома Вернера взаимодействуют для разрешения теломерных петель D способом, регулируемым TRF1 и TRF2. Мол. Ячейка 14 , 763–774 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Чанг, С.и другие. Существенная роль лимитирующих теломер в патогенезе синдрома Вернера. Nature Genet. 36 , 877–882 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Du, X. et al. Укорочение теломер раскрывает функции генов синдрома Вернера и Блума у мышей. Мол. Клетка. Биол. 24 , 8437–8446 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Крабб, Л., Verdun, R.E., Haggblom, C.I. & Karlseder, J. Дефектный синтез отстающей цепи теломер в клетках, лишенных активности геликазы WRN. Наука 306 , 1951–1953 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Лауд, П. Р. и др. Повышенная рекомбинация теломер-теломер в WRN-дефицитных теломерных дисфункциональных клетках способствует уходу от старения и вовлечению пути ALT. Genes Dev. 19 , 2560–2570 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Crabbe, L., Jauch, A., Naeger, C. M., Holtgreve-Grez, H. & Karlseder, J. Дисфункция теломер как причина геномной нестабильности при синдроме Вернера. Proc. Natl Acad. Sci. США 104 , 2205–2210 (2007).
Артикул
CAS
PubMed
Google ученый
Опреско, П.L. et al. POT1 стимулирует RecQ геликазы WRN и BLM раскручивать субстраты теломерной ДНК. J. Biol. Chem. 280 , 32069–32080 (2005). Ссылки 29–35 определяют роль WRN в поддержании теломер в соматических клетках человека и у Wrn -дефицитных мышей.
Артикул
CAS
PubMed
Google ученый
Eller, M. S. et al. Роль WRN в ответах на повреждение ДНК на основе теломер. Proc. Natl Acad. Sci. США 103 , 15073–15078 (2006).
Артикул
CAS
PubMed
Google ученый
Bai, Y. & Murnane, J. P. Нестабильность теломер в линии опухолевых клеток человека, экспрессирующих доминантно-отрицательный белок WRN. Гм. Genet. 113 , 337–347 (2003).
Артикул
CAS
PubMed
Google ученый
Моннат, Р.J., Jr & Saintigny, Y. Синдромный белок Вернера — функция раскрутки для объяснения болезни. Sci. Aging Knowledge Environ. 2004 , re3 (2004).
Артикул
PubMed
Google ученый
Dhillon, K. K. et al. Функциональная роль геликазы RecQ синдрома Вернера в фибробластах человека. Ячейка старения 6 , 53–61 (2007).
Артикул
CAS
PubMed
Google ученый
Киплинг, Д., Дэвис, Т., Остлер, Э. Л. и Фарагер, Р. Г. Что прогероидные синдромы могут рассказать нам о старении человека? Наука 305 , 1426–1431 (2004).
Артикул
CAS
PubMed
Google ученый
Франк, С. А. и Новак, М. А. Проблемы соматической мутации и рака. Bioessays 26 , 291–299 (2004).
Артикул
CAS
PubMed
Google ученый
Кампизи, Дж.Старение клеток, подавление опухолей и старение организма: хорошие граждане, плохие соседи. Cell 120 , 513–522 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Ломбард, Д. Б. и др. Мутации в гене Wrn у мышей ускоряют смертность на фоне p53 и нулевого уровня. Мол. Клетка. Биол. 20 , 3286–3291 (2000). Первоначальное описание создания и характеристики модели мышей с дефицитом Wrn , которая воспроизводит генетические и биохимические дефекты, наблюдаемые в клетках пациентов с WS.
Артикул
CAS
PubMed
PubMed Central
Google ученый
Lebel, M. & Leder, P. Делеция в геликазе мышиного синдрома Вернера вызывает чувствительность к ингибиторам топоизомеразы и потерю клеточной пролиферативной способности. Proc. Natl Acad. Sci. USA 95 , 13097–13102 (1998).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Ван, Л.и другие. Клеточные фенотипы Вернера у мышей, экспрессирующих предполагаемый доминантно-отрицательный ген WRN человека. Генетика 154 , 357–362 (2000).
CAS
PubMed
PubMed Central
Google ученый
Хеннекам, Р. С. Хатчинсон-Гилфорд синдром прогерии: обзор фенотипа. Am. J. Med. Genet. А 140 , 2603–2624 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Хатчинсон, Дж.Врожденное отсутствие волос и молочных желез при атрофическом состоянии кожи и ее придатков. Trans Med. Чир. Soc. Эдинбург 69 , 473–477 (1886).
Артикул
CAS
Google ученый
Эрикссон, М. и др. Рецидивирующие точечные мутации de novo в ламине А вызывают синдром прогерии Хатчинсона-Гилфорда. Nature 423 , 293–298 (2003).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Де Сандре-Джованноли, А.и другие. Ламин является усечением при прогерии Хатчинсона – Гилфорда. Наука 300 , 2055 (2003).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Cao, H. & Hegele, R.A. LMNA мутирует при прогерии Хатчинсона-Гилфорда (MIM 176670), но не при прогероидном синдроме Видеманна-Раутенштрауха (MIM 264090). J. Hum. Genet. 48 , 271–274 (2003). Ссылки 48–50 описывают первоначальную идентификацию LMNA как гена, мутировавшего в HGPS.
Артикул
CAS
PubMed
PubMed Central
Google ученый
Verstraeten, V. L. et al. Сложная гетерозиготность по мутациям в LMNA вызывает синдром прогерии без накопления преламина А. Гм. Мол. Genet. 15 , 2509–2522 (2006).
Артикул
CAS
PubMed
Google ученый
МакКеон, Ф.Д., Киршнер, М. В. и Капут, Д. Гомологии первичной и вторичной структуры между белками ядерной оболочки и промежуточными филаментами. Nature 319 , 463–468 (1986).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Стурман, Н., Хейнс, С. и Эби, У. Ядерные ламины: их структура, сборка и взаимодействия. J. Struct. Биол. 122 , 42–66 (1998).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Бриджер, Дж. М., Килл, И. Р., О’Фаррелл, М. и Хатчисон, С. Дж. Внутренние ламинатные структуры в ядрах G1 дермальных фибробластов человека. J. Cell Sci. 104 , 297–306 (1993).
CAS
PubMed
Google ученый
Кеннеди, Б. К., Барби, Д. А., Классон, М., Дайсон, Н. и Харлоу, Э. Ядерная организация репликации ДНК в первичных клетках млекопитающих. Genes Dev. 14 , 2855–2868 (2000).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Spann, T. P., Goldman, A. E., Wang, C., Huang, S. & Goldman, R. D. Изменение организации ядерного ламина ингибирует зависимую от РНК-полимеразы II транскрипцию. J. Cell. Биол. 156 , 603–608 (2002).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Ivorra, C. et al. Механизм подавления AP-1 через взаимодействие c-Fos с ламином A / C. Genes Dev. 20 , 307–320 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Frock, R. L. et al. Ламин A / C и эмерин имеют решающее значение для дифференцировки сателлитных клеток скелетных мышц. Genes Dev. 20 , 486–500 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Нитта, Р. Т., Джеймсон, С. А., Кудлоу, Б. А., Конлан, Л. А. и Кеннеди, Б. К. Стабилизация белка ретинобластомы ядерными ламинами А-типа необходима для остановки клеточного цикла, опосредованной INK4A. Мол. Клетка. Биол. 26 , 5360–5372 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Смит, Э.Д., Кудлоу Б. А., Фрок Р. Л. и Кеннеди Б. К. Ядерные ламины А-типа, прогерии и другие дегенеративные заболевания. мех. Aging Dev. 126 , 447–460 (2005).
Артикул
CAS
PubMed
Google ученый
Sullivan, T. et al. Потеря экспрессии ламина А-типа нарушает целостность ядерной оболочки, что приводит к мышечной дистрофии. J. Cell Biol. 147 , 913–920 (1999).
CAS
PubMed
PubMed Central
Google ученый
Николова В. и др. Дефекты ядерной структуры и функции вызывают дилатационную кардиомиопатию у мышей с дефицитом ламина A / C. J. Clin. Вкладывать деньги. 113 , 357–369 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Де Сандре-Джованноли, А. и др.Гомозиготные дефекты в LMNA , кодирующем белки ядерной оболочки ламина A / C, вызывают аутосомно-рецессивную аксональную невропатию у человека (болезнь Шарко-Мари-Тута типа 2) и мыши. Am. J. Hum. Genet. 70 , 726–736 (2002).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Chen, L. et al. LMNA мутации при атипичном синдроме Вернера. Ланцет 362 , 440–445 (2003).
Артикул
CAS
PubMed
Google ученый
Русинол, А. Э. и Синенски, М. С. Фарнезилированные ламины, прогероидные синдромы и ингибиторы фарнезилтрансферазы. J. Cell Sci. 119 , 3265–3272 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Pendas, A. M. et al. Дефектный процессинг преламина А и изменения в мышцах и адипоцитах у мышей с дефицитом металлопротеиназы Zmpste24 . Nature Genet. 31 , 94–99 (2002).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Даль, К. Н. и др. Отчетливые структурные и механические свойства ядерной пластинки при синдроме прогерии Хатчинсона – Гилфорда. Proc. Natl Acad. Sci. USA 103 , 10271–10276 (2006).
Артикул
CAS
PubMed
Google ученый
Скаффиди, П.& Мистели, Т. Обращение клеточного фенотипа при болезни преждевременного старения. Синдром прогерии Хатчинсона – Гилфорда. Nature Med. 11 , 440–445 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Лю Б. и др. Геномная нестабильность при преждевременном старении на основе ламинопатии. Nature Med. 11 , 780–785 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Скаффиди, П.& Мистели, Т. Ламин A-зависимые ядерные дефекты при старении человека. Наука 312 , 1059–1063 (2006). Доказательства того, что механизмы, лежащие в основе HGPS, также могут играть роль в нормальном старении человека.
Артикул
CAS
PubMed
PubMed Central
Google ученый
Bergo, M.O. et al. Zmpste24 дефицит у мышей вызывает спонтанные переломы костей, мышечную слабость и дефект обработки преламина А. Proc. Natl Acad. Sci. USA 99 , 13049–13054 (2002).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Янг, С. Х. и др. Ингибитор фарнезилтрансферазы улучшает фенотип заболевания у мышей с мутацией синдрома прогерии Хатчинсона-Гилфорда. J. Clin. Вкладывать деньги. 116 , 2115–2121 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Ян, С.H. et al. Блокирование протеина фарнезилтрансферазы улучшает ядерный блеббинг в фибробластах мыши с целевой мутацией синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. USA 102 , 10291–10296 (2005). Ссылка 73, а также 87 и 88 описывают три мышиные модели HGPS, которые были созданы путем мутации или изменения экспрессии Lmna .
Артикул
CAS
PubMed
Google ученый
Фонг, Л.G. et al. Ингибитор протеин-фарнезилтрансферазы облегчает течение болезни у мышей с прогерией. Наука 311 , 1621–1623 (2006). Ссылки 72 и 74 демонстрируют, что FTI заметно снижают фенотипы заболевания на мышиных моделях HGPS.
Артикул
CAS
PubMed
Google ученый
Маллампалли, М. П., Хьюер, Г., Бендейл, П., Гелб, М. Х. и Михаэлис, С. Ингибирование фарнезилирования устраняет дефект ядерной морфологии в модели клеток HeLa для синдрома прогерии Хатчинсона – Гилфорда. Proc. Natl Acad. Sci. США 102 , 14416–14421 (2005).
Артикул
CAS
PubMed
Google ученый
Glynn, M. W. & Glover, T. W. Неполный процессинг мутантного ламина A при прогерии Хатчинсона-Гилфорда приводит к ядерным аномалиям, которые устраняются ингибированием фарнезилтрансферазы. Гм. Мол. Genet. 14 , 2959–2969 (2005).
Артикул
CAS
PubMed
Google ученый
Тот, Дж.I. et al. Блокирование протеина фарнезилтрансферазы улучшает форму ядра в фибробластах людей с прогероидными синдромами. Proc. Natl Acad. Sci. США 102 , 12873–12878 (2005).
Артикул
CAS
PubMed
Google ученый
Capell, B.C. et al. Ингибирование фарнезилирования прогерина предотвращает характерное ядерное блеббирование синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci.США 102 , 12879–12884 (2005).
Артикул
CAS
Google ученый
Goldman, R.D. et al. Накопление мутантного ламина А вызывает прогрессивные изменения ядерной архитектуры при синдроме прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. USA 101 , 8963–8968 (2004).
Артикул
CAS
Google ученый
Дельбарре, Э.и другие. Укороченный prelamin A при синдроме прогерии Hutchinson-Gilford изменяет сегрегацию гомополимеров ламина A-типа и B-типа. Гм. Мол. Genet. 15 , 1113–1122 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Варела И. и др. Ускоренное старение у мышей, дефицитных по протеазе Zmpste24 , связано с активацией передачи сигналов р53. Природа 437 , 564–568 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Huang, S. et al. Коррекция клеточных фенотипов клеток прогерии Хатчинсона – Гилфорда с помощью РНК-интерференции. Гм. Genet. 118 , 444–450 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Csoka, A. B. et al. Профилирование экспрессии в масштабе генома синдрома прогерии Хатчинсона-Гилфорда выявляет широко распространенную неправильную регуляцию транскрипции, ведущую к мезодермальным / мезенхимальным дефектам и ускоренному атеросклерозу. Ячейка старения 3 , 235–243 (2004).
Артикул
CAS
PubMed
Google ученый
Sephel, G. C., Sturrock, A., Giro, M. G. и Davidson, J. M. Повышенная продукция эластина фибробластами кожи с прогерией контролируется стабильными уровнями мРНК эластина. J. Invest. Дерматол. 90 , 643–647 (1988).
Артикул
CAS
PubMed
Google ученый
Лемир, Дж.M. et al. Экспрессия аггрекана существенно и аномально повышена в дермальных фибробластах при синдроме прогерии Хатчинсона-Гилфорда. мех. Aging Dev. 127 , 660–669 (2006).
Артикул
CAS
PubMed
Google ученый
Ламмердинг, Дж. И др. Дефицит Lamin A / C вызывает нарушение ядерной механики и механотрансдукции. J. Clin. Вкладывать деньги. 113 , 370–378 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Маункес, Л.К., Козлов, С., Эрнандес, Л., Салливан, Т. и Стюарт, С. Л. Прогероидный синдром у мышей вызывается дефектами ламинов А-типа. Природа 423 , 298–301 (2003).
Артикул
CAS
PubMed
Google ученый
Varga, R. et al. Прогрессирующие дефекты гладкомышечных клеток сосудов на мышиной модели синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl Acad. Sci. США 103 , 3250–3255 (2006).H
Артикул
CAS
PubMed
Google ученый
Фонг, Л. Г. и др. Преламин А и ламин А, по-видимому, являются незаменимыми в ядерной ламине. J. Clin. Вкладывать деньги. 116 , 743–752 (2006).
CAS
PubMed
PubMed Central
Google ученый
Миллер Р.А. «Ускоренное старение»: путь к пониманию примулы? Ячейка старения 3 , 47–51 (2004).
Артикул
CAS
PubMed
Google ученый
Мартин, Г. М. Генетическая модуляция стареющих фенотипов у Homo sapiens . Cell 120 , 523–532 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Niedernhofer, L. J. et al. Новый прогероидный синдром показывает, что генотоксический стресс подавляет соматотрофную ось. Природа 444 , 1038–1043 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
van der Pluijm, I. et al. Поддержание генома подавляет ось 1 гормона роста-инсулиноподобного фактора роста у мышей с синдромом Кокейна. PLoS Biology 5 , e2 (2007).
Артикул
CAS
PubMed
Google ученый
Моннат, Р.J. Jr. От сломанного к старому: повреждение ДНК, подавление эндокринной системы IGF1 и старение. DNA Repair (в печати).
Кеньон, С. Пластичность старения: идеи долгоживущих мутантов. Cell 120 , 449–460 (2005).
Артикул
CAS
PubMed
Google ученый
Kamath-Loeb, A. S., Welcsh, P., Waite, M., Adman, E. T. и Loeb, L. A. Ферментативная активность белка синдрома Вернера блокируется аминокислотным полиморфизмом R834C. J. Biol. Chem. 279 , 55499–55505 (2004).
Артикул
CAS
PubMed
Google ученый
Дэвис, Т., Бэрд, Д. М., Хотон, М. Ф., Джонс, К. Дж. И Киплинг, Д. Предотвращение ускоренного старения клеток при синдроме Вернера с помощью ингибитора митоген-активируемой протеинкиназы p38. J. Gerontol. Биол. Sci. Med. Sci. 60 , 1386–1393 (2005).
Артикул
PubMed
Google ученый
Скадден, Д.Т. Ниша стволовых клеток как объект действия. Природа 441 , 1075–1079 (2006).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Коппе, Дж. П., Каузер, К., Кампизи, Дж. И Босежур, К. М. Секреция фактора роста эндотелия сосудов первичными фибробластами человека при старении. J. Biol. Chem. 281 , 29568–29574 (2006).
Артикул
CAS
PubMed
Google ученый
Чока, А.B. et al. Новые мутации гена ламина A / C ( LMNA ) при атипичных прогероидных синдромах. J. Med. Genet. 41 , 304–308 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Пласилова М. и др. Гомозиготная миссенс-мутация в гене ламина A / C вызывает аутосомно-рецессивный синдром прогерии Хатчинсона – Гилфорда. J. Med. Genet. 41 , 609–614 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Novelli, G. et al. Мандибулоакральная дисплазия вызвана мутацией в LMNA -кодирующем ламине A / C. Am. J. Hum. Genet. 71 , 426–431 (2002).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Гарг, А., Когулу, О., Озкинай, Ф., Onay, H. & Agarwal, A.K. Новая гомозиготная мутация Ala529Val LMNA у турецких пациентов с мандибулоакральной дисплазией. J. Clin. Эндокринол. Метаб. 90 , 5259–5264 (2005).
Артикул
CAS
PubMed
Google ученый
Navarro, C. L. et al. Дефекты ламина А и ZMPSTE24 (FACE-1) вызывают ядерную дезорганизацию и определяют рестриктивную дермопатию как летальную неонатальную ламинопатию. Гм. Мол. Genet. 13 , 2493–2503 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Agarwal, A.K., Fryns, J.P., Auchus, R.J. & Garg, A. Металлопротеиназа цинка, ZMPSTE24, мутирует при дисплазии мандибулоакральной области. Гм. Мол. Genet. 12 , 1995–2001 (2003).
Артикул
CAS
PubMed
Google ученый
Наварро, К.L. et al. Потеря ZMPSTE24 (FACE-1) вызывает аутосомно-рецессивную рестриктивную дермопатию и накопление предшественников ламина А. Гм. Мол. Genet. 14 , 1503–1513 (2005).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Fukuchi, K. et al. Мутация LMNA у 45-летнего японца с синдромом прогерии Хатчинсона-Гилфорда. J. Med. Genet. 41 , e67 (2004).
Артикул
CAS
PubMed
PubMed Central
Google ученый
Cao, K. et al. Изоформа ламина А, сверхэкспрессируемая при синдроме прогерии Хатчинсона-Гилфорда, препятствует митозу в прогерии и нормальных клетках. Proc. Natl Acad. Sci. USA 104 , 4949–4954 (2007).
Артикул
CAS
PubMed
Google ученый
Дечат, Т.и другие. Изменения митоза и развития клеточного цикла, вызванные мутантным ламином А, который, как известно, ускоряет старение человека. Proc. Natl Acad. Sci. США 104 , 4955–4960 (2007).
Артикул
CAS
PubMed
Google ученый
Синдром прогерии Хатчинсона-Гилфорда: редкое генетическое заболевание
Синдром прогерии Хатчинсона-Гилфорда (HGPS) — редкий педиатрический генетический синдром с частотой один на восемь миллионов живорожденных.Расстройство характеризуется преждевременным старением, обычно приводящим к смерти примерно в 13,4 лет. Это последующее исследование 9-летнего мужчины с клиническими и рентгенологическими признаками, сильно свидетельствующими о HGPS, и представлено здесь с описанием дифференциальной диагностики и стоматологического рассмотрения. Это первый отчет о HGPS, который показал структуру грудной клетки pectus carinatum.
1. Введение
Синдром прогерии Хатчинсона-Гилфорда (HGPS) — чрезвычайно редкое, но разрушительное заболевание, характеризующееся карликовостью и преждевременным старением [1].Это происходит спорадически с зарегистрированной заболеваемостью один из восьми миллионов и преобладанием мужчин с соотношением M: F 1,5: 1 и сильной расовой восприимчивостью у кавказцев, которые составляют 97% пациентов [2]. Тип наследования неясен, хотя были предложены как аутосомно-доминантный, так и аутосомно-рецессивный режимы [3, 4]. Последние достижения в области генетики определили LMNA как ген, вызывающий HGPS. LMNA кодирует ламинины A и C, которые являются основными компонентами промежуточной нитчатой ламины, функционируют как структурная поддержка и необходимы для репликации ДНК и транскрипции мРНК [5].Хотя клиническая картина типична, обычные радиологические и биохимические исследования помогают подтвердить диагноз. Мы представляем случай прогерии, который показал классические физические и радиологические изменения HGPS.
2. История болезни
В клинику обратился мальчик 12 лет с основной жалобой на кариес в области верхних и нижних передних зубов. Анамнез истории болезни показал, что первые два года его жизни были нормальными, после чего последовала неспособность набрать рост и вес, а затем последовала потеря волос на коже головы и бровей.Затем у него появилось растяжение кожи и неспособность правильно стоять или ходить; однако умственное развитие было нормальным. В анамнезе установлено, что пациент проходил лечение от острого гепатита (см. Рисунки 1 и 2).
В стоматологическом анамнезе установлено, что пациентка раньше обращалась в больницу для лечения кариеса в переднем отделе верхней челюсти 2 года назад. Но лечение было невозможно, так как пациент отказывался от сотрудничества. У этого второго родившегося ребенка от некровного брака не было происшествий до рождения, а другой родной брат был нормальным (см. Рисунки 3 и 4).
Никто из других членов семьи не пострадал от подобных жалоб. При общем осмотре молодой пациент напоминал «сморщенного маленького старичка».
Он был худощавым, плохо питался, невысокого роста с ненормальной походкой, тонкой атрофической кожей, потерей подкожно-жировой клетчатки вокруг конечностей, а кожа была грубой, растянутой, блестящей и утолщенной. На груди обнаружена структура pectus carinatum (см. Рисунки 5 и 6).
Системное обследование показало, что у пациента было нарушение зрения, невнятная речь, потеря памяти, одышка, сердцебиение и ограничение движений в суставах с невозможностью стоять или ходить.
При дополнительном осмотре ротовой полости у пациента была небольшая выпуклость во лбу, нос с клювом, выпученные глаза, высокий голос, гипоплазия верхней и нижней челюсти с умеренной деформацией средней части лица, придающей «вид ощипанной птицы». Открытие рта было ограничено (межрезцовое расстояние 21 мм), боковые движения височно-нижнечелюстного сустава также были ограничены.
При внутриротовом осмотре зубы были нормального размера по сравнению с небольшим размером челюсти, сломанной коронкой до 11, культи корня 12, 15, 16, 21, 22 и кариесом зубов 13, 14, 23, 24, 25, 31, 32, 41, 42, 45.Наблюдались высокое арочное небо и частичная анодония.
На основании анамнеза и клинических данных был поставлен предварительный диагноз прогерии. Для подтверждения диагноза ребенку было проведено рентгенологическое и биохимическое обследование.
Биохимические исследования показали повышенный уровень холестерина в сыворотке, который составлял 228 мг%, и увеличение экскреции гиалуроновой кислоты с мочой.
ОПГ: гипоплазия верхней челюсти, гипоплазия нижней челюсти с инфантильным углом и множественные отсутствующие зубы.Морфология мыщелка изменилась (см. Рис. 7).
Сопоставляя историю болезни, клинические признаки, рентгенологические данные и лабораторные исследования, результаты соответствовали синдрому HGP.
После тщательного системного мониторинга было запланировано удаление сильно разрушенных зубов с покрытием антибиотиками.
3. Обсуждение
Прогерия — редкое генетическое заболевание, фенотипически характеризующееся признаком преждевременного старения, впервые описанное Хатчинсоном в 1886 году [6].Термин прогерия был введен Гилфордом в 1904 году и происходит от греческого слова «гериос», означающего «старый». ДеБуск в 1972 году переименовал это состояние в «синдром прогерии Хатчинсона-Гилфорда» [2]. Скорость старения у пораженного человека ускоряется в семь раз по сравнению с нормальной. Средняя продолжительность жизни составляет 13 лет (от 7 до 27 лет) со случайной продолжительностью жизни до 45 лет. Это связано с различными аномалиями мезодермальных тканей и уменьшением времени выживания фибробластов.Сообщалось о мутации de novo LMNA, которая кодирует основной компонент внутренней мембранной пластинки [5]. Больные дети нормальны при рождении и нормально растут примерно до конца первого года жизни, когда и нормальный рост, и прибавка в весе замедляются.
В данном случае был обнаружен типичный фенотип HGPS, проявляющийся начальными симптомами на первом году жизни, выраженной недостаточностью роста, выраженной липодистрофией, выпадением волос на коже черепа и бровей, а также склеродерматозными изменениями, приводящими к характерному внешнему виду «сорванной птицы». .
Вены кожи головы становятся заметными из-за потери подкожного жира и выпадения волос. Эти пациенты обычно невысокого роста и худощавы, их средний рост составляет 100 см, а средний вес — 12–15 кг или даже меньше. Также распространены отсроченное прорезывание зубов и гиподонтия [7].
Пациент в нашем случае имел классические черты лица, которые включали черты лица, алопецию, задержку роста, плохое половое созревание и нормальный интеллект.
Обследование скелета выявляет следующие радиологические особенности: свод черепа тонкий и относительно большой, а диплоическое пространство отсутствует или очень мелкое.Лицо маленькое, с непропорционально маленькой нижней челюстью, сохраняющей инфантильный тупой угол и короткие восходящие ветви. Ключицы небольшого калибра и при рождении разрежены [8]. Этот случай обычно представлен вышеупомянутыми рентгенографическими признаками, подтверждающими предварительный диагноз.
При прогерии гиперлипидемия часто проявляется повышенным содержанием липопротеинов низкой плотности и повышенным уровнем холестерина в сыворотке, как это наблюдалось у нашего пациента. Также наблюдается повышение уровня гиалуроновой кислоты, ответственной за склеродерматозные изменения и сердечно-сосудистые изменения.
Смерть в основном наступает из-за сердечно-сосудистых осложнений, таких как инфаркт миокарда или застойная сердечная недостаточность. Отсутствие развития у пациентов с прогерией может быть связано с биоактивной формой гормона роста и отсутствием васкулогенеза, вызванным чрезмерной секрецией гиалуроновой кислоты [9].
Дифференциальный диагноз, который можно рассматривать в этих случаях, включает синдром Вернера, акрогерию, синдром Ротмунда-Томсона и синдром Кокейна [10].
На сегодняшний день эффективного лечения не существует.Единственно доступный подход к симптоматическому лечению и своевременному выявлению и оперативному лечению осложнений.
Синдром прогерии Хатчинсона – Гилфорда: болезнь преждевременного старения
Хатчинсон Дж. (1886) Врожденное отсутствие волос и молочных желез с атрофическим состоянием кожи и ее придатков у мальчика, мать которого была почти полностью лысой из-за алопеции. areata с шести лет. Medicochir Trans 69: 473–477
CAS
Google ученый
Gilford H (1897) При смешанном преждевременном и незрелом развитии. Мед Чирург Транс 80: 17–45
CAS
Статья
Google ученый
Гилфорд Х. (1904) Прогерия: форма старения. Практик 73: 188–217
Google ученый
Меридет М.А., Гордон Л.Б., Клаусс С., Сачдев В., Смит АКМ, Перри М.Б., Брюер С.К., Залевски С. и др. (2010) Фенотип и течение синдрома прогерии Хатчинсона – Гилфорда.N Engl J Med 358 (6): 592–604. doi: 10.1056 / NEJMoa0706898 Опубликован в окончательной отредактированной форме как: N Engl J Med. 2008 7 февраля
Hennekam RC (2006) Синдром прогерии Хатчинсона – Гилфорда: обзор фенотипа. Am J Med Genet A 140A: 2603–2624. DOI: 10.1002 / ajmg.a.31346
CAS
Статья
Google ученый
Sternberg S (2003) Обнаружен ген быстрого старения у детей. USA Today.http://www.usatoday.com/news/science/2003-04-16-agin-gene_x.htm. Проверено 13 декабря 2006 г.
Стив Роуч Э., Миллер В.С. (2004) Cambridge University Press, том 36, стр. 150
Ракха П., Гупта А., Дхингра Г., Нагпал М. (2011) Синдром прогерии Хатчинсона-Гилфорда: обзор. Der Pharmacia Sinica 2 (1): 110–117
CAS
Google ученый
Hsiao K-J (1998) Adv Clin Chem 33:10
Google ученый
Факты по состоянию на 1 апреля 2017 года. Исследовательский фонд Progeria. https://www.progeriaresearch.org/quick-facts/. По состоянию на 14 июня 2017 г.
Де Буск, Флорида (1972) Синдром прогерии Хатчинсона-Гилфорда. J Pediatr 90: 697–724
Статья
Google ученый
Beauregard S, Gilchrest BA (1987) Синдромы преждевременного старения. Dermatol Clin 5: 109–121
CAS
PubMed
Google ученый
Brown WT, Kieras FJ, Houck GE Jr, Dutkowski R, Jenkins EC (1985) Сравнение прогерии у взрослых и детей: синдром Вернера и синдром прогерии Хатчинсона-Гилфорда. Adv Exp Med Biol 190: 229–244
CAS
Статья
PubMed
Google ученый
Brown WT (1987) Синдромы преждевременного старения. Curr Probl Dermatol 17: 152–165
CAS
Статья
PubMed
Google ученый
Korf B (2008) N Engl J Med 358 (6): 552–555
CAS
Статья
PubMed
Google ученый
Rakha P et al (2011) Der Pharmacia Sinica 2 (1): 110–117
CAS
Google ученый
Балин А.Д. (ред.) (1989) Вклад исследований фибробластов кожи in vitro у людей с генетическими заболеваниями, которые предрасполагают к явлениям ускоренного старения, в наше понимание процесса старения.Raven Press, New York, pp. 93–9l 19
Google ученый
Исследовательский фонд Прогерии (2016) https://www.progeriaresearch.org/meet-the-kids/. Доступ 14 июня 2017 г.
Пеше К., Роте MJ (1996) Синдромы преждевременного старения. Clin Dermatol 14: 161–170
CAS
Статья
PubMed
Google ученый
Dyer CAE, Sinclair AJ (1998) Синдромы преждевременного старения: понимание процесса старения. Возраст Старение 27: 73–80
CAS
Статья
PubMed
Google ученый
Bennett GCJ, Ebrahim S (1995) Основы здравоохранения в пожилом возрасте, 2-е изд. Oxford University Press, Нью-Йорк, стр. 3–10
Google ученый
Plasilova M, Chattopadhyay C, Pal P, Schaub NA, Buechner SA, Mueller H, Miny P, Ghosh A et al (2004) Гомозиготная миссенс-мутация в гене ламина A / C вызывает аутосомно-рецессивный процесс Хатчинсона-Гилфорда. синдром прогерии.J Med Genet 41: 609–614
Smith ED, Kudlow BA, Frock RL, Kennedy BK (2005) Ядерные ламины A-типа, прогерии и другие дегенеративные расстройства. Mech Aging Dev 126: 447–460
CAS
Статья
PubMed
Google ученый
Кирквуд ТБ (2005) Понимание необычной науки о старении. Ячейка 120: 437–447
CAS
Статья
PubMed
Google ученый
Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW (2005) Ремонт ДНК, стабильность генома и старение. Ячейка 120: 497–512
CAS
Статья
PubMed
Google ученый
Decker ML, Chavez E, Vulto I, Lansdorp PM (2009) Длина теломер при синдроме прогерии Хатчинсона-Гилфорда. Mech Aging Dev 130 (6): 377–383
Silvera VM, Gordon LB, Orbach DB, Campbell SE, Machan JT, Ullrich NJ (2013) Характеристики изображений цереброваскулярной артериопатии и инсульта при синдроме прогерии Хатчинсона-Гилфорда .Am J Neuroradiol 34 (5): 1091–1097
Goss JR, Stolz DB, Robinson AR, Zhang M, Arbujas N, Robbins PD, Glorioso JC, Niedernhofer LJ (2010) Связанная с преждевременным старением периферическая нейропатия в мышиная модель прогерии. Mech Aging Dev. DOI: 10.1016 / j.mad.2011.04.010
Google ученый
Саркар П.К., Шинтон Р.А. (2001) Синдром прогерии Хатчинсона-Гилфорда. Postgrad Med J 77: 312–317
CAS
Статья
PubMed
PubMed Central
Google ученый
Olteanu I, Crisan M, Crisan D, Kozan A (2009) Синдром Хатчинсона-Гилфорда. Медицинский журнал Aradean 02: 13–18
Google ученый
Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I (1998) Х-связанный врожденный дискератоз вызывается мутациями в высококонсервативном гене с предполагаемыми ядрышковыми функциями . Nat Genet 19 (1): 6–7
Статья
Google ученый
DeBusk FL (1972) Синдром прогерии Хатчинсона-Гилфорда: отчет о 4 случаях и обзор литературы. J Pediatrics 80 (4): 697–724
CAS
Статья
Google ученый
Uitto J (2001) В поисках ключей к преждевременному старению. Семинар по синдрому Хатчинсона-Гилфорда прогерии был проведен в Бетесде, Мэриленд, США, 28 и 29 ноября
Кумар С., Кумар А., Сингла М., Сингх А. (2010) Синдром Хатчинсона-Гилфорда (прогерия).Int J Pharm Bio Sci 1 (3)
Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB et al (2008) Фенотип и течение синдрома прогерии Хатчинсона-Гилфорда. N Engl J Med 358: 592–604
CAS
Статья
PubMed
PubMed Central
Google ученый
Домингес-Герпе Л., Араихо-Вилар Д. (2008) Дети недоношенного возраста; молекулярные изменения, приводящие к прогерии Хатчинсона-Гилфорда и синдромам Вернера.Текущее старение Sci 1: 202–212
CAS
Статья
Google ученый
Hennekam RC (2006) Синдром прогерии Хатчинсона-Гилфорда: обзор фенотипа. Am J Med Genet A 140: 2603–2624
Статья
PubMed
Google ученый
Ackerman J, Gilbet-Barness E (2002) Синдром прогерии Хатчинсона-Гилфорда: патологическое исследование. Педиатр Патол Мол Мед 21: 1–13
Статья
PubMed
Google ученый
Ishii T (1976) Progeria: отчет о вскрытии одного случая с обзором патологических данных, опубликованных в литературе. J Am Geriatr Soc 24: 193–202
CAS
Статья
PubMed
Google ученый
Corcoy R, Aris A, de Leiva A (1989) Фертильность в случае прогерии. Am J Med Sci 297: 383–384
CAS
Статья
PubMed
Google ученый
Sadeghi-Nejad A, Demmer L (2007) Терапия гормоном роста при прогерии.J Pediatr Endocrinol Metab 20 (5): 633–637
CAS
Статья
PubMed
Google ученый
Hennekam RC (2006) Синдром прогерии Хатчинсона-Гилфорда: обзор фенотипа. Am J Med Genet 140: 2603–2624
Статья
PubMed
Google ученый
Arlan L (1971) Am Heart J 82: 287–289
Артикул
Google ученый
Горлин Р.О., Седано Х.О. (1968) Синдром Прогерии Хатчинсона-Гилфорда. Мод Мед 46:62
Google ученый
Batstone MD, Macleod AW (2002) Оральные и челюстно-лицевые хирургические аспекты в случае прогерии Хатчинсона-Гилфорда. Int J Paediatr Dent 12: 429–432
CAS
Статья
PubMed
Google ученый
Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB et al (2008) Фенотип и течение синдрома прогерии Хатчинсона-Гилфорда.N Engl J Med 358: 592–604
CAS
Статья
PubMed
PubMed Central
Google ученый
Shiraishi I, Hayashi S, Hirai E, Onouchi Z, Hamaoka K (2001) Смертельная легочная гипертензия, связанная с атипичным случаем прогерии Ханчинсона-Гилфорда. Педиатр Кардиол 22: 530–533
CAS
Статья
PubMed
Google ученый
Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J (1999) Гистологические и ультраструктурные особенности атеросклероза при прогерии.Cardiovasc Pathol 8: 29–39
CAS
Статья
PubMed
Google ученый
Аткинс Л. (1954) Прогерия: отчет о случае с результатами вскрытия. N Engl J Med 250: 1065–1069
CAS
Статья
PubMed
Google ученый
Исии Т. (1976) Прогерия: отчет о вскрытии одного случая с обзором патологических данных, описанных в литературе.J Am Geriatr Soc 24: 193–202
CAS
Статья
PubMed
Google ученый
Corcoy R, Aris A, de Leiva A (1989) Фертильность в случае прогерии. Am J Med Sci 297: 383–384
CAS
Статья
PubMed
Google ученый
DeBusk FL (1972) Синдром прогерии Хатчинсона-Гилфорда. Отчет о 4 случаях и обзор литературы. J Pediatr 80: 697–724
CAS
Статья
PubMed
Google ученый
Габр М., Хашем Н., Хашем М., Фахми А., Сафу М. (1960) Прогерия, патологическое исследование. J Pediatr 57: 70–77
CAS
Статья
PubMed
Google ученый
Dyck JD, David TE, Burke B, Webb GD, Henderson MA, Fowler RS (1987) Управление ишемической болезнью сердца при синдроме Хатчинсона-Гилфорда. J Pediatr 111: 407–410
CAS
Статья
PubMed
Google ученый
Халифа М.М. (1989) Синдром прогерии Хатчинсона – Гилфорда: отчет о ливийской семье и доказательства аутосомно-рецессивного наследования. Clin Genet 35: 125–132
CAS
Статья
PubMed
Google ученый
Maciel AT (1988) Доказательства аутосомно-рецессивного наследования прогерии (Hutchinson Gilford). Am J Med Genet 31: 483–487
CAS
Статья
PubMed
Google ученый
Brown WT (1979) Мутации человека, влияющие на старение — обзор. Mech Aging Dev 9: 325–336
CAS
Статья
PubMed
Google ученый
Delgado Luengo W, Rojas Martinez A, Ortiz Lopez R et al (2002) Del (1) (q23) у пациента с прогерией Хатчинсона-Гилфорда. Am J Med Genet 113: 298–301
Статья
PubMed
Google ученый
Brown WT, Adbenur J, Goonewardena P et al (1990) Синдром прогерии Хатчинсона – Гилфорда: клинические и метаболические аномалии (аннотация).Am J Hum Genet 47: A50
Google ученый
Де Сандре-Джованноли А., Бернард Р., Кау П и др. (2003) Усечение ламина А в прогерии Хатчинсона – Гилфорда. Наука 300: 2055
CAS
Статья
PubMed
Google ученый
Cao H, Hegele RA (2003) Lmna мутирует при прогерии Хатчинсона-Гилфорда (MIM 176670), но не при прогероидном синдроме Видеманна-Раутенштрауха (MIM 264090).J Hum Genet 48: 271–274
CAS
Статья
PubMed
Google ученый
Шанкер П., Вишисант П., Виджай Натх Д., Навин С., Киран Кумар Ю., Венкатешварлу П. (2010) Прогерия. Краткий обзор. Intr J Pharma & Bio Sciences 1 (2)
Plasilova M, Chattopadhyay C, Pal P, Schaub NA, Buechner SA, Mueller H et al (2004) Гомозиготная миссенс-мутация в гене ламина A / C вызывает аутосомные рецессивный синдром прогерии Хатчинсона-Гилфорда.J Med Genet 41: 609–614
CAS
Статья
PubMed
PubMed Central
Google ученый
Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA (2004) Мутации нового гена ламина A / C (LMNA) при атипичных прогероидных синдромах. J Med Genet 41: 304–308
CAS
Статья
PubMed
PubMed Central
Google ученый
Fukuchi K, Katsuya T, Sugimoto K, Kuremura M, Kim HD, Li L et al (2004) Мутация LMNA у 45-летнего японского субъекта с синдромом прогерии Хатчинсона-Гилфорда.J Med Genet 41: e67
CAS
Статья
PubMed
PubMed Central
Google ученый
Verstraeten VL, Broers JL, van Steensel MA, Zinn-Justin S, Ramaekers FC, Steijlen PM et al (2006) Гетерозиготность соединений для мутаций в LMNA вызывает синдром прогерии без накопления преламина A. Hum Mol Genet 15: 2509–2522
CAS
Статья
PubMed
Google ученый
Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y et al (2003) Мутации LMNA при атипичном синдроме Вернера. Ланцет 362: 440–445
CAS
Статья
PubMed
Google ученый
Cox LS, Faragher RG (2007) От старых организмов к новым молекулам: интегративная биология и терапевтические цели при ускоренном старении человека. Cell Mol Life Sci 64: 2620–2641
CAS
Статья
PubMed
PubMed Central
Google ученый
Scaffidi P, Misteli T (2006) Lamin A-зависимые ядерные дефекты при старении человека. Наука 312 (5776): 1059–1063
CAS
Статья
PubMed
PubMed Central
Google ученый
Уилсон К.Л., Берк Дж. М. (2010) Краткий обзор ядерной оболочки. J Cell Sci 123: 1973–1978
CAS
Статья
PubMed
PubMed Central
Google ученый
Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K (2004) Ламин B1 необходим для развития мышей и целостности ядра.Proc Natl AcadSci USA 101: 10428–10433
CAS
Статья
Google ученый
Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL et al (2006) Ламины A и C, но не ламин B1, регулируют ядерную механику. J Biol Chem 281: 25768–25780
CAS
Статья
PubMed
Google ученый
Lin F, Worman HJ (1993) Структурная организация человеческого гена, кодирующего ядерный ламин A и ядерный ламин C.J Biol Chem 268: 16321–16326
CAS
PubMed
Google ученый
Weber K, Plessmann U, Traub P (1989) Созревание ядерного ламина A включает специфическое обрезание карбокси-конца, которое удаляет сайт полиизопренилирования из предшественника; последствия для структуры ядерной пластинки. FEBS Lett 257: 411–414
CAS
Статья
PubMed
Google ученый
Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N. et al (2006) Преламин A и ламин A, по-видимому, не обязательны в ядерной пластинке. J Clin Invest 116: 743–752
CAS
Статья
PubMed
PubMed Central
Google ученый
Worman HJ, Ostlund C, Wang Y (2010) Болезни ядерной оболочки. Cold Spring Harb Perspect Biol 2: a000760
Статья
PubMed
PubMed Central
Google ученый
Worman HJ, Bonne G (2007) «Ламинопатии»: широкий спектр болезней человека. Exp Cell Res 313: 2121–2133
CAS
Статья
PubMed
PubMed Central
Google ученый
База данных мутаций генов человека (в Институте медицинской генетики в Кардиффе). http://www.hgmd.cf.ac.uk/ac/all.php. По состоянию на 14 июня 2017 г.
Qin Z, Kalinowski A, Dahl KN, Buehler MJ (2011) Структура и стабильность хвостового домена ламина A и мутанта HGPS.J. Struct Biol. DOI: 10.1016 / j.jsb.2011.05.015
PubMed Central
Google ученый
Sullivan T et al (1999) Потеря экспрессии ламина А-типа нарушает целостность ядерной оболочки, что приводит к мышечной дистрофии. J Cell Biol 147: 913–920
CAS
Статья
PubMed
PubMed Central
Google ученый
Николова В. и др. (2004) Дефекты ядерной структуры и функции способствуют дилатационной кардиомиопатии у мышей с дефицитом ламина A / C.J Clin Invest 113: 357–369
CAS
Статья
PubMed
PubMed Central
Google ученый
De Sandre-Giovannoli A et al (2002) Гомозиготные дефекты в LMNA , кодирующем белки ядерной оболочки ламина A / C, вызывают аутосомно-рецессивную аксональную невропатию у человека (расстройство Шарко – Мари – Тута 2 типа) и мышь. Am J Hum Genet 70: 726–736
CAS
Статья
PubMed
PubMed Central
Google ученый
Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ (2006) Ядерные ламины: ламинопатии и их роль в преждевременном старении. Physiol Rev 86: 967–1008
CAS
Статья
PubMed
Google ученый
Fong LG, Ng JK, Meta M, Cote N, Yang SH, Stewart CL et al (2004) Гетерозиготность по дефициту Lmna устраняет прогериеподобные фенотипы у мышей с дефицитом Zmpste24. Proc Natl Acad Sci U S A 101: 18111–18116
CAS
Статья
PubMed
PubMed Central
Google ученый
Scaffidi P, Misteli T (2005) Обращение клеточного фенотипа при болезни преждевременного старения. Синдром прогерии Хатчинсона-Гилфорда. Nat Med 11: 440–445
CAS
Статья
PubMed
PubMed Central
Google ученый
Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL et al (2005) Блокирование протеина фарнезилтрансферазы улучшает форму ядер в фибробластах людей с прогероидными синдромами. Proc Natl Acad Sci U S A 102: 12873–12878
CAS
Статья
PubMed
PubMed Central
Google ученый
Gruber J, Lampe T, Osborn M, Weber K (2005) RNAi протеазы FACE1 приводит к ингибированию роста человеческих клеток, экспрессирующих ламин A: последствия для синдрома прогерии Хатчинсона-Гилфорда. J Cell Sci 118: 689–696
CAS
Статья
PubMed
Google ученый
Субба Р.К. (2007) Механизмы заболевания: дефекты репарации ДНК и неврологические заболевания. Nat Clin Pract Neurol 3: 162–172
Статья
Google ученый
Лю Б., Ван Дж., Чан К.М., Тиа В.М., Дэн В., Гуан Х и др. (2005) Геномная нестабильность при преждевременном старении на основе ламинопатии. Nat Med 11: 780–785
CAS
Статья
PubMed
Google ученый
Манджу К., Мураликришна Б., Парнаик В.К. (2006) Экспрессия болезнетворных мутантов ламина А нарушает формирование очагов репарации ДНК. J Cell Sci 119: 2704–2714. DOI: 10.1242 / jcs.03009
CAS
Статья
PubMed
Google ученый
Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR et al (2006) Мутантный ядерный ламин A приводит к прогрессивным изменениям эпигенетического контроля при преждевременном старении. Proc Natl Acad Sci U S A 103: 8703–8708
CAS
Статья
PubMed
PubMed Central
Google ученый
Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM et al (2005) Ускоренное старение у мышей, дефицитных по протеазе Zmpste24, связано с активацией передачи сигналов p53.Природа 437: 564–568
CAS
Статья
PubMed
Google ученый
Atadja P, Wong H, Garkavtsev I, Veillette C, Riabowol K (1995) Повышенная активность p53 в стареющих фибробластах. Proc Natl AcadSci USA 92: 8348–8352
CAS
Статья
Google ученый
Кудлоу Б.А., Кеннеди Б.К., Моннат Р.Дж.-младший (2007) Синдромы прогерии Вернера и Хатчинсона-Гилфорда: механистическая основа прогероидных заболеваний человека.Nat Rev Mol Cell Biol 8: 394–404
CAS
Статья
PubMed
Google ученый
Therizols P, Fairhead C, Cabal GG, Genovesio A, Olivo-Marin JC, Dujon B et al (2006) Привязка теломер на периферии ядра важна для эффективной репарации двухцепочечных разрывов ДНК в субтеломерной области. J Cell Biol 172: 189–199
CAS
Статья
PubMed
PubMed Central
Google ученый
Bridger JM, Kill IR (2004) Старение фибробластов с синдромом прогерии Хатчинсона-Гилфорда характеризуется гиперпролиферацией и повышенным апоптозом. Exp Gerontol 39: 717–724
CAS
Статья
PubMed
Google ученый
Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB et al (2004) Накопление мутантного ламина A вызывает прогрессивные изменения ядерной архитектуры при синдроме прогерии Хатчинсона-Гилфорда.Proc Natl AcadSci USA 101: 8963–8968
CAS
Статья
Google ученый
Hutchison CJ, Alvarez-Reyes M, Vaughan OA (2001) Ламины при болезни: почему повсеместно экспрессируемые белки ядерной оболочки вызывают тканеспецифические фенотипы заболевания? J Cell Sci 114: 9–19
CAS
PubMed
Google ученый
Worman HJ, Gundersen GG (2006) А вот и СУН: недостающее звено нуклеоцитоскелета.Trends Cell Biol 16: 67–69
CAS
Статья
PubMed
Google ученый
Eriksson M et al (2003) Рецидивирующие точечные мутации de novo в ламине А вызывают синдром прогерии Хатчинсона-Гилфорда. Nature 423 (6937): 293–398
CAS
Статья
PubMed
Google ученый
Скаффиди П., Мистели Т. (2005) Обращение клеточного фенотипа при болезни преждевременного старения. Синдром прогерии Хатчинсона-Гилфорда.Nat Med 11 (4): 440–445
CAS
Статья
PubMed
PubMed Central
Google ученый
Cao K et al (2007) Изоформа белка ламина А, сверхэкспрессируемая при синдроме прогерии Хатчинсона-Гилфорда, препятствует митозу в прогерии и нормальных клетках. Proc Natl Acad Sci U S A 104 (12): 4949–4954
CAS
Статья
PubMed
PubMed Central
Google ученый
McClintock D et al (2007) Мутантная форма ламина А, вызывающая прогерию Хатчинсона-Гилфорда, является биомаркером клеточного старения в коже человека. PLoS One 2 (12): E1269
Артикул
PubMed
PubMed Central
Google ученый
Eisch V, Lu X, Gabriel D, Djabali K (2016) Прогерин нарушает поддержание хромосом за счет истощения CENP-F из метафазных кинетохоров в фибробластах прогерии Хатчинсона-Гилфорда. Oncotarget 7 (17): 24700
Артикул
PubMed
PubMed Central
Google ученый
Holzenberger M (2004) Ось GH / IGF-I и долговечность. Eur J Endocrinol 151 (Дополнение 1): S23 – S27
CAS
Статья
PubMed
Google ученый
Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E et al (2006) Новый прогероидный синдром показывает, что генотоксический стресс подавляет соматотрофную ось. Nature 444: 1038–1043
CAS
Статья
PubMed
Google ученый
Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM et al (2012) Клиническое испытание ингибитора фарнезилтрансферазы у детей с синдромом прогерии Хатчинсона-Гилфорда. Proc Natl Acad Sci USA 109 (41): 16666–16671
Gordon LB, Massaro J, D’Agostino RB, Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW (2014) Влияние фарнезилирования ингибиторы выживаемости при синдроме прогерии Хатчинсона-Гилфорда. Тираж 130 (1): 27–34 CIRCULATION AHA-113
CAS
Статья
PubMed
PubMed Central
Google ученый
Gordon LB, Kleinman ME, Massaro JM, D’Agostino RB, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM et al (2016) Клиническое испытание ингибиторов фарнезилирования белков лонафарниба, правастатина и золедроновой кислоты у детей с Hutchinson- Синдром прогерии Гилфорда. Циркуляция 134 (2): 114–125 CIRCULATION AHA-116
Эверолимус и лонафарниб. Одна рука. Фаза I: лонафарниб с возрастающими дозами эверолимуса для определения МПД. Фаза II: лонафарниб плюс эверолимус при MTD (оценка эффективности).https://clinicaltrials.gov/ct2/show/NCT02579044?term=progeria+everolimus&rank=1. По состоянию на 14 июня 2017 г.
Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS (2011). Рапамицин меняет клеточные фенотипы и увеличивает клиренс мутантного белка в клетках синдрома прогерии Хатчинсона-Гилфорда. Sci Transl Med 3 (89): 89ra58. DOI: 10.1126 / scitranslmed.3002346
Прогерия стволовых клеток: истощение стволовых клеток при синдроме прогерии Хатчинсона-Гилфорда | Журналы геронтологии: серия A
Аннотация
Синдром прогерии Хатчинсона-Гилфорда (HGPS) — редкое фатальное генетическое заболевание, которое характеризуется сегментарным ускоренным старением.Основная причинная мутация, связанная с HGPS, запускает аномальный сплайсинг информационной РНК гена ламина А, что приводит к изменениям в ядерной архитектуре. На сегодняшний день предложены две модели, объясняющие, как мутации в гене ламина А могут приводить к HGPS, структурной хрупкости и изменению экспрессии гена. Мы предпочитаем совместимую модель, которая связывает HGPS с регенерацией тканей, управляемой стволовыми клетками. В этой модели ядерная хрупкость клеток с дефицитом ламина А увеличивает апоптотическую гибель клеток до уровней, которые истощают способность тканей к управляемой стволовыми клетками регенерации.Специфичные для тканей различия в клеточной гибели или регенеративном потенциале, или в обоих случаях, приводят к тканеспецифическому сегментарному паттерну старения, наблюдаемому в HGPS. Мы предполагаем, что паттерн связанных со старением состояний, присутствующих или отсутствующих в HGPS, может дать представление о генетических факторах и факторах окружающей среды, которые способствуют нормальному старению.
Синдром прогерии ХАТЧИНСОНА-ГИЛФОРДА (HGPS) — редкое генетическое заболевание, для которого характерно очень раннее начало признаков, связанных с нормальным старением (1). У пораженных людей фенотипы, связанные со старением, по-видимому, развиваются примерно в 7 раз ускоренными темпами, оставляя маленьких детей с внешним видом и состоянием здоровья их бабушек и дедушек.HGPS поражает примерно 1 из 8 миллионов детей, при этом чуть более 100 случаев зарегистрировано в различных группах населения по всему миру.
Пациенты с HGPS обычно кажутся нормальными в раннем младенчестве. В возрасте от 9 до 24 месяцев пораженные дети начинают испытывать глубокую задержку роста, что приводит к низкому росту и низкой массе тела. Характерные черты HGPS включают характерный внешний вид лица из-за мандибулоакральной дисплазии (MAD), выпадения волос и подкожной жировой ткани, вывихов бедра, дефектов скелета и других аномалий (1–3).Патологически дети с HGPS страдают генерализованным атеросклерозом и сердечно-сосудистыми заболеваниями и умирают от инфаркта миокарда или инсульта в среднем в возрасте 13 лет (4,5).
Клинические особенности HGPS поразительно напоминают определенные аспекты естественного старения (2). Однако пациенты с HGPS не отображают все атрибуты, связанные со старостью. HGPS и другие заболевания, при которых ускоряются только некоторые аспекты нормального старения, называются сегментарными прогероидными синдромами (6).Метаболические, эндокринные и иммунологические обследования пациентов с HGPS не выявляют никаких общих отклонений. Признаков преждевременного старения мозга не наблюдается; люди имеют нормальный интеллект и эмоциональное развитие. Не наблюдается поддающейся измерению когнитивной дегенерации или снижения нейросенсорных функций, а также образования катаракты, диабета или гиперлипидемии. Наиболее поразительно то, что злокачественные новообразования не связаны с HGPS.
Белки ламина и ядерная оболочка
Ядерные ламины подразделяются на ламины типа A или B на основе их биохимических свойств и поведения во время митоза (7).Эти белки, которые составляют класс промежуточных филаментов, являются компонентами ядерной пластинки, самого внутреннего слоя ядерной оболочки. Ядерная пластинка — это белковая сеть, которая поддерживает структурную целостность ядерной оболочки и взаимодействует с хроматином (8). Ламины B-типа экспрессируются во всех клетках во время развития и необходимы для жизнеспособности клеток. Четыре ламинных белка A-типа возникают из одного гена (LMNA) путем альтернативного сплайсинга матричной РНК, при этом ламин A и ламин C являются основными белковыми продуктами.Ламин A кодируется экзонами 1–12 гена LMNA; Незрелый белок ламина А фарнезилируется около С-конца, который впоследствии отщепляется специфической протеиназой ZMPSTE24 с образованием зрелого белка. Ламин C получается путем использования альтернативного сайта сплайсинга в интроне 10 для получения более короткого белка (9). Ламины A и C образуют либо гомодимеры, либо гетеродимеры, чтобы создать нитевидную структуру ядерной пластинки (7,10).
HGPS и LMNA
Мутации в LMNA связаны с многочисленными спорадическими или наследственными заболеваниями человека, включая мышечную дистрофию Эмери-Дрейфуса 2 и 3 типов, мышечную дистрофию пояса конечностей, болезнь Шарко-Мари-Тута типа 2B1, семейную частичную липодистрофию типа Даннигана (FPLD). 2 и другие (11), для которых были установлены отношения генотипа и / или фенотипа (12).HGPS является результатом специфических de novo мутаций в LMNA [обзор в (3)] (13,14). Повторяющаяся доминантная мутация de novo G608G, обнаруженная примерно у 90% пациентов с HGPS, активирует скрытый сайт сплайсинга в интроне 11, что приводит к аберрантному транскрипту, который кодирует мутантный белок LMNA с внутренней делецией из 50 аминокислот. Этот мутантный белок сохраняет сайт фарнезилирования, но не имеет сайта протеолитического расщепления. У гетерозиготных пациентов с HGPS этот неправильно обработанный белок, как полагают, аберрантно взаимодействует с ламином C и с нормальными молекулами ламина A из аллеля дикого типа, что приводит к ядерной нестабильности.
Исследования Скаффиди и Мистели (15) обобщают вклад аберрантной ядерной архитектуры в нормальное старение. Они показали, что секретный сайт сплайсинга, вызывающий HGPS, время от времени используется в клетках здоровых людей, и продемонстрировали возрастные аберрации в морфологии ядра. Они предлагают общие механизмы, включающие ламин А и ядерную архитектуру, между HGPS и физиологическим старением (15) [обзор в (16)]. Недавно были достигнуты успехи в вариантах лечения HGPS, включая использование ингибиторов фарнезилтрансферазы (17) и модифицированных антисмысловых олигонуклеотидов для блокирования скрытого сайта сплайсинга LMNA (18).
Фибробласты HGPS демонстрируют поразительно измененные формы и размеры ядер, иногда сопровождающиеся экструзией хроматина (14). Ядра, дефектные по ламинам, механически хрупкие и подвержены ядерному повреждению и гибели клеток (19,20). Фибробласты эмбрионов мыши с дефицитом ламина A / C, которые подвергаются механической нагрузке, демонстрируют повышенную ядерную деформацию, дефектную механотрансдукцию и нарушение жизнеспособности (21). Эта хрупкость может объяснить патологии сердечных мышц и скелетных мышц у пациентов с HGPS в результате механического повреждения во время сокращения мышц.
Ядерная оболочка также участвует в регуляции паттернов экспрессии генов путем организации гетерохроматина внутри ядра (22). Считается, что белки ламина A / C регулируют активность тканеспецифических факторов транскрипции, и также предполагается, что эти белки связывают гистоны ядра и, следовательно, влияют на тканеспецифические паттерны экспрессии (11). Кроме того, было обнаружено, что ламины A и C напрямую связываются с несколькими регуляторами транскрипции, включая белок ретинобластомы (pRB) (23).Несогласованное изменение программ экспрессии генов мутантным ламином А может иметь пагубные последствия (11). Для мутаций Lmna, приводящих к мышечной дистрофии у мышей, было высказано предположение, что как структурная слабость клеток, так и нарушение дифференцировки сателлитных клеток из-за измененной экспрессии генов способствуют прогрессированию заболевания (24). При HGPS механическая слабость клеток, измененная экспрессия генов или другие механизмы могут способствовать истощению клеток и истощению стволовых клеток.
Анализ моделей мышей
Трансгенные мыши, несущие ген LMNA человека, содержащий обычную мутацию HGPS, демонстрируют прогрессирующую потерю гладкомышечных клеток сосудов в крупных артериях и раннюю смерть от атеросклероза (25).Как отметили Варга и его коллеги, наблюдение атеросклероза как основного признака заболевания у этих мышей может отражать исключительно высокую механическую нагрузку на сердечно-сосудистую систему, которая может сделать эту ткань наиболее восприимчивой к дефициту ламина А; это наблюдение согласуется с атеросклерозом, который является наиболее частой причиной смерти пациентов с HGPS. Мыши, гомозиготные по аутосомно-рецессивной мутации гена Lmna (Lmna L530P / L530P ), который заменяет нормальный ген мыши геном, вызывающим сложную аномалию сплайсинга, демонстрируют симптомы, похожие на симптомы пациентов с HGPS, а также преждевременную смерть терминально дифференцированных мезенхимальные клетки (26).Различные аспекты различных фенотипов нескольких моделей мышей отражают либо отсутствие как ламина А, так и ламина С, что у мышей Lmna — / — вызывает симптомы, подобные мышечной дистрофии Эмери-Дрейфуса (22), либо наличие аллеля который продуцирует прогерин (27), предполагая, что мутации ламина А, приводящие к прогерии, являются доминантными аллелями усиления функции (28). Недавно Фонг и его коллеги (29) показали, что мыши, у которых отсутствует ламин А, но продуцирует ламин С, кажутся нормальными [обзор в (30)], подразумевая, что недостаток белков ламина С может быть более критическим, чем предполагалось ранее.
Другие идеи относительно ламина А получены из исследований на мышах, лишенных протеиназы, расщепляющей фарнезилированный белок. Нокаут гена Zmpste24 вызывает фенокопию большинства дефектов, наблюдаемых у мышей Lmna HG / + , наряду с фенотипами заболевания, соответствующими HGPS и ламинопатиям человека (31–33).
Экспрессия LMNA и стволовые клетки
Хотя ламины B-типа повсеместно экспрессируются во всех типах клеток, экспрессия ламинов A-типа регулируется в процессе развития.Как правило, ламины A-типа отсутствуют в ранних эмбрионах, ранних эмбриональных стволовых клетках и стволовых клетках кроветворной и нейроэндокринной систем (34,35). Следовательно, эмбриональное развитие мышей Lmna — / — не зависит от отсутствия ламина А. Новорожденные мыши Lmna — / — неотличимы от своих братьев и сестер дикого типа и гетерозиготных (22), как и Дети с HGPS выглядят нормальными в младенчестве. Однако клетки постнатальных и взрослых тканей мышей Lmna — / — дегенерируют, и животные умирают преждевременно, так же как преждевременное старение HGPS начинается с задержки роста до возраста 2 лет и приводит к преждевременной смерти.
HGPS и истощение стволовых клеток: модель сегментарного старения
Поскольку пациенты с HGPS переживают лишь около одной седьмой средней продолжительности нормальной жизни, мы предполагаем, что обновление клеток и их гибель также ускоряются примерно в 7 раз. Это мнение подтверждается исследованиями фибробластов HGPS in vitro, в которых сообщалось о 4-8-кратном увеличении апоптоза (36). Ядра клеток, несущие мутации LMNA, хрупкие, нестабильные и очень чувствительные к механическому стрессу.Накопление структурных изменений и ухудшение состояния хроматина, вероятно, увеличивает уровень повреждения ДНК (37) и может привести к апоптозу. Хотя значительная работа была проделана для характеристики моделей мышей с прогерией, остается неясным, вызваны ли наблюдаемое повреждение ткани и преждевременная потеря клеток запрограммированной гибелью клеток.
Несколько авторов, включая Prolla (38) и Warner (39–41) ранее предполагали участие клеток-предшественников или неадекватную замену клеток в HGPS. Уорнер (41) утверждал, что мутации ламина А нарушают ядерную оболочку и приводят клетки к апоптозу.В подробном обзоре моделей ускоренного старения Warner и Sierra (39) обсудили возможные связи между повышенной потерей клеток и неспособностью замещения клеток у мышей группы D (XPD) с дефицитом комплементации xeroderma pigmentosum или мышей p53 + / m . Мутантные мыши с дефектами функции XPD, p53 или Ku-80 демонстрируют высокие показатели апоптоза и преждевременно умирают (42–44). Эти мыши имеют общие черты с хорошо изученными синдромами преждевременного старения человека, особенно синдромами Вернера и Блума, врожденным дискератозом и пигментной ксеродермой (45).
В обзоре анализов экспрессии генов при прогерии Prolla (38) предположил, что регенеративная способность тканей с высоким обновлением клеток может быть снижена из-за истощения клеток-предшественников. Мы далее расширяем эту идею, предполагая, что эта гипотеза объясняет не только фенотип преждевременного старения HGPS, но и специфическую сегментарную природу этой прогерии.
Почему у пациентов с HGPS наблюдается выпадение волос и подкожной жировой ткани, вывихи бедра и дефекты скелета, но не старение мозга (болезнь Альцгеймера, когнитивная дегенерация, снижение нейросенсорных функций), катаракта, диабет 2 типа, гиперлипидемия или рак (1, 46)? Некоторые из связанных с возрастом состояний, которые наблюдаются или не наблюдаются при HGPS, перечислены в таблицах 1 (47–55) и 2 (56–62), соответственно.Мы предлагаем, чтобы ткани, для которых стволовые клетки являются предпосылкой для регенерации и восстановления продолжающихся повреждений, те, которые подвергаются постоянному механическому воздействию (например, кровеносные сосуды и суставы), или те, которые необходимы для поддержания непрерывного роста (волосяные фолликулы, ногти), соответствуют те ткани, которые дегенерируют у пациентов с HGPS. Напротив, заболевания, связанные с тканями, которые защищены от механического стресса (мозг) или для которых основное воздействие требует десятилетий воздействия (образование катаракты, диабет 2 типа, гиперлипидемия), в HGPS отсутствуют.Интересно, что преждевременное сокращение скелетных мышц не наблюдается при HGPS. Наблюдение за тем, что этот тип клеток, который явно подвергается механическому стрессу, не подвержен влиянию, может отражать менее частую нагрузку на произвольную мышцу, чем на гладкую мышцу сердечно-сосудистой системы, подвергающуюся более постоянной нагрузке, особенно в течение короткого периода жизни пациента с HGPS.
Отсутствие рака в HGPS особенно информативно, поскольку хрупкость ядер клеток HGPS вызывает повышенный апоптоз.Эта высокая потеря апоптотических клеток может истощить пулы стволовых клеток и предотвратить злокачественную трансформацию. Этот аргумент также подтверждается показателями заболеваемости раком при нормальном старении, которые значительно увеличиваются в возрасте от 40 до 80 лет, но выходят на плато или даже падают выше этого уровня (56,57). Роль стволовых клеток в инициации и прогрессировании опухоли в настоящее время широко обсуждается, и были опубликованы первые доказательства рака груди и мозга (63,64). Мы предполагаем, что по прошествии 80 лет пулы взрослых стволовых клеток в значительной степени исчерпаны, что снижает риск рака.Хотя недавно сообщалось о существовании нейрональных стволовых и / или клеток-предшественников (58,59), развитый мозг поддерживает высокий уровень тканевого гомеостаза, и деления клеток происходят редко. Мы предполагаем, что ткань мозга подвергается минимальному механическому воздействию и хорошо защищена от преждевременного старения у пациентов с HGPS.
Катаракта, диабет 2 типа и гиперлипидемия вызываются механизмами, не связанными с регенерацией тканей, что согласуется с их отсутствием в HGPS. Липодистрофия или дегенерация адипоцитов, напротив, связана с мутациями ламина А при FPLD и MAD (65–67).Инсулинорезистентность и диабет 2 типа связаны с FPLD и MAD, но неясно, является ли этот фенотип первичным эффектом генетического дефекта LMNA или вторичным эффектом быстрой дегенерации жировой ткани у этих пациентов. В то время как кластерные белки затуманивают хрусталик глаза при катаракте, диабет и липидные нарушения в основном вызваны нездоровым образом жизни. Мы утверждаем, что эти заболевания возникают независимо от регенерации, связанной со стволовыми клетками, и поэтому не наблюдаются у пациентов с HGPS.
Какие патологии HGPS могут быть объяснены отсутствием регенерации тканей?
Состояния, которые демонстрируют четкую корреляцию с регенерацией тканей под действием стволовых клеток и проявляются у пораженных детей, включают атеросклероз, сердечно-сосудистые заболевания, липодистрофию, алопецию (выпадение волос), дефекты ногтей, жесткость суставов и пороки развития зубов (Таблица 1) . Из этих состояний основными причинами смерти пациентов с HGPS являются атеросклероз и сердечно-сосудистые заболевания (68,69).Сердечно-сосудистая система постоянно находится под высоким механическим стрессом, что может привести к повышенной гибели хрупких клеток с дефицитом ламина А. Традиционные взгляды на регенерацию сосудов были поставлены под сомнение недавней идентификацией эндотелиальных клеток-предшественников (EPC), которые вносят вклад в поддержание и восстановление эндотелия и гладких мышц. Пациенты с сердечно-сосудистыми заболеваниями и пациенты с HGPS демонстрируют продолжающуюся потерю EPC, что приводит к снижению способности к восстановлению эндотелия и регенерации сосудов (47,48).Однако в целом атеросклероз в основном вызван отложением липидов, связанным с образом жизни. Ускоренная потеря механически вызываемых сердечно-сосудистых клеток с дефицитом ламина А может истощить пулы EPC. Такие заболевания, как выпадение волос и липодистрофия, можно объяснить высоким обменом клеток и нарушением замещения из-за истощения пула стволовых клеток.
В настоящее время подробные знания о биологии стволовых клеток в значительной степени недоступны для многих ниш взрослых стволовых клеток. Дальнейшее понимание механизмов регенерации тканей, управляемой стволовыми клетками, необходимо для подтверждения преждевременного истощения стволовых клеток в определенных тканях при HGPS.
Заключение
HGPS отличается от других прогероидных синдромов тем, что ламин А не является прямым компонентом белковых комплексов репарации ДНК. У пациентов с синдромом Блума, Вернера и Ротмунда-Томсона наблюдается дефицит ДНК-геликаз, а у пациентов с анемией Фанкони и пигментной ксеродермией наблюдается дефицит других аспектов репарации ДНК (2). Вместо того, чтобы вносить вклад в нестабильность генома, которая приводит к повышенному риску рака при этих других синдромах, мутации ламина А, лежащие в основе HGPS, вероятно, вызывают преждевременное истощение стволовых клеток, которое истощает определенные ткани (при сохранении других), создавая высокосегментарный тканеспецифичный образец преждевременного старение, не связанное с повышенным риском рака.Напротив, системный сбой механизмов восстановления или поддержания ДНК проявляется во всем организме и вызывает более глобальное ускорение старения.
В HGPS, это внутренние аспекты старения, которые ускоряются, тогда как те, которые вызваны в основном внешними воздействиями, остаются неизменными. Если эта гипотеза окажется верной, это будет полезно для определения аспектов старения, которых можно избежать путем изменения образа жизни.
Редактор решений: Хубер Р.Уорнер, доктор философии
Таблица 1.
Распространенных возрастных заболеваний, ПРИСУТСТВУЮЩИХ при синдроме прогерии Хатчинсона-Гилфорда (HGPS).
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина наличия заболевания в HGPS . | Список литературы . | |
|---|---|---|---|---|---|
| Атеросклероз | Отложения продуктов жизнедеятельности клеток и кальция во внутренней оболочке артерии | Эндотелиальные клетки-предшественники (ЭПК) способствуют регенерации и восстановлению сосудов, заменяя поврежденные клетки сердечной мышцы и создавая новые кровеносные сосуды | эндотелиальных клеток-предшественников; механическая нагрузка на клетки увеличивает апоптоз и регенерацию тканей | 47,48 | |
| Сердечно-сосудистые заболевания | Дисфункциональные состояния сердца, артерий и вен | 49,50 | |||
| перераспределение жировых отложений, потеря подкожной жировой ткани | Жировая ткань человека является богатым источником мультипотентных стволовых клеток, полученных из жировой ткани (hMADS) | Снижение регенерации ткани из-за истощения стволовых клеток, полученных из жировой ткани | 51 | ||
| Алопеция | Волосяные фолликулы становятся спящими, а оставшиеся волосы становятся тоньше и реже | Фолликулярные стволовые клетки волос могут давать начало как волосяному фолликулу, так и эпидермису | Облысение возникает, когда рост волос, который зависит от стволовых клеток волосяного фолликула, не удается чтобы справиться с выпадением волос | 52 | |
| Дефекты ногтей | Повреждение стволовых клеток ногтя и их временного потомства амплифицирующих клеток | Пул стволовых клеток поставляет клетки для быстро растущих ногтей | Высокий оборот клеток приводит к истощению быстро делящихся стволовых клеток ногтя | 53 | |
| Жесткость сустава | Ухудшение связок и сухожилий ограничивает движение сустава без трения | Клетки-предшественники хрящей (хондроцитов) отвечают за регенерацию суставов | Снижение регенерации тканей из-за истощения клеток-предшественников хрящей | 54 | |
| 54 | |||||
| Возможные дефекты развития при миграции или активации зубных стволовых клеток | 55 | ||||
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина наличия заболевания в HGPS . | Список литературы . | |
|---|---|---|---|---|---|
| Атеросклероз | Отложения продуктов жизнедеятельности клеток и кальция во внутренней оболочке артерии | Эндотелиальные клетки-предшественники (ЭПК) способствуют регенерации и восстановлению сосудов, заменяя поврежденные клетки сердечной мышцы и создавая новые кровеносные сосуды | эндотелиальных клеток-предшественников; механическая нагрузка на клетки увеличивает апоптоз и регенерацию тканей | 47,48 | |
| Сердечно-сосудистые заболевания | Дисфункциональные состояния сердца, артерий и вен | 49,50 | |||
| перераспределение жировых отложений, потеря подкожной жировой ткани | Жировая ткань человека является богатым источником мультипотентных стволовых клеток, полученных из жировой ткани (hMADS) | Снижение регенерации ткани из-за истощения стволовых клеток, полученных из жировой ткани | 51 | ||
| Алопеция | Волосяные фолликулы становятся спящими, а оставшиеся волосы становятся тоньше и реже | Фолликулярные стволовые клетки волос могут давать начало как волосяному фолликулу, так и эпидермису | Облысение возникает, когда рост волос, который зависит от стволовых клеток волосяного фолликула, не удается чтобы справиться с выпадением волос | 52 | |
| Дефекты ногтей | Повреждение стволовых клеток ногтя и их временного потомства амплифицирующих клеток | Пул стволовых клеток поставляет клетки для быстрорастущих ногтей | Высокий оборот клеток приводит к истощению быстро делящихся стволовых клеток ногтя | 53 | |
| Жесткость сустава | Ухудшение связок и сухожилий ограничивает движение сустава без трения | Клетки-предшественники хрящей (хондроцитов) отвечают за регенерацию суставов | Снижение регенерации тканей из-за истощения клеток-предшественников хрящевых зубов | 54 | |
| 54 | |||||
| Возможные дефекты развития при миграции или активации зубных стволовых клеток | 55 | ||||
Таблица 1.
Распространенные возрастные заболевания, ПРИСУТСТВУЮЩИЕ при синдроме прогерии Хатчинсона-Гилфорда (HGPS).
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина наличия заболевания в HGPS . | Список литературы . | |
|---|---|---|---|---|---|
| Атеросклероз | Отложения продуктов жизнедеятельности клеток и кальция во внутренней оболочке артерии | Эндотелиальные клетки-предшественники (ЭПК) способствуют регенерации и восстановлению сосудов, заменяя поврежденные клетки сердечной мышцы и создавая новые кровеносные сосуды | эндотелиальных клеток-предшественников; механическая нагрузка на клетки увеличивает апоптоз и регенерацию тканей | 47,48 | |
| Сердечно-сосудистые заболевания | Дисфункциональные состояния сердца, артерий и вен | 49,50 | |||
| перераспределение жировых отложений, потеря подкожной жировой ткани | Жировая ткань человека является богатым источником мультипотентных стволовых клеток, полученных из жировой ткани (hMADS) | Снижение регенерации ткани из-за истощения стволовых клеток, полученных из жировой ткани | 51 | ||
| Алопеция | Волосяные фолликулы становятся спящими, а оставшиеся волосы становятся тоньше и реже | Фолликулярные стволовые клетки волос могут давать начало как волосяному фолликулу, так и эпидермису | Облысение возникает, когда рост волос, который зависит от стволовых клеток волосяного фолликула, не удается чтобы справиться с выпадением волос | 52 | |
| Дефекты ногтей | Повреждение стволовых клеток ногтя и их временного потомства амплифицирующих клеток | Пул стволовых клеток поставляет клетки для быстро растущих ногтей | Высокий оборот клеток приводит к истощению быстро делящихся стволовых клеток ногтя | 53 | |
| Жесткость сустава | Ухудшение связок и сухожилий ограничивает движение сустава без трения | Клетки-предшественники хрящей (хондроцитов) отвечают за регенерацию суставов | Снижение регенерации тканей из-за истощения клеток-предшественников хрящей | 54 | |
| 54 | |||||
| Возможные дефекты развития при миграции или активации зубных стволовых клеток | 55 | ||||
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина наличия заболевания в HGPS . | Список литературы . | |
|---|---|---|---|---|---|
| Атеросклероз | Отложения продуктов жизнедеятельности клеток и кальция во внутренней оболочке артерии | Эндотелиальные клетки-предшественники (ЭПК) способствуют регенерации и восстановлению сосудов, заменяя поврежденные клетки сердечной мышцы и создавая новые кровеносные сосуды | эндотелиальных клеток-предшественников; механическая нагрузка на клетки увеличивает апоптоз и регенерацию тканей | 47,48 | |
| Сердечно-сосудистые заболевания | Дисфункциональные состояния сердца, артерий и вен | 49,50 | |||
| перераспределение жировых отложений, потеря подкожной жировой ткани | Жировая ткань человека является богатым источником мультипотентных стволовых клеток, полученных из жировой ткани (hMADS) | Снижение регенерации ткани из-за истощения стволовых клеток, полученных из жировой ткани | 51 | ||
| Алопеция | Волосяные фолликулы становятся спящими, а оставшиеся волосы становятся тоньше и реже | Фолликулярные стволовые клетки волос могут давать начало как волосяному фолликулу, так и эпидермису | Облысение возникает, когда рост волос, который зависит от стволовых клеток волосяного фолликула, не удается чтобы справиться с выпадением волос | 52 | |
| Дефекты ногтей | Повреждение стволовых клеток ногтя и их временного потомства амплифицирующих клеток | Пул стволовых клеток поставляет клетки для быстрорастущих ногтей | Высокий оборот клеток приводит к истощению быстро делящихся стволовых клеток ногтя | 53 | |
| Жесткость сустава | Ухудшение связок и сухожилий ограничивает движение сустава без трения | Клетки-предшественники хрящей (хондроцитов) отвечают за регенерацию суставов | Снижение регенерации тканей из-за истощения клеток-предшественников хрящевых зубов | 54 | |
| 54 | |||||
| Возможные дефекты развития при миграции или активации зубных стволовых клеток | 55 | ||||
Таблица 2.
Общие возрастные заболевания ОТСУТСТВУЮТ при синдроме прогерии Хатчинсона-Гилфорда (HGPS).
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина отсутствия заболевания в HGPS . | Список литературы . |
|---|---|---|---|---|
| Рак | Неконтролируемый и инвазивный рост клеток | Теория стволовых клеток опухоли: рак возникает из стволовых клеток (грудных и нейрональных стволовых клеток) | Раннее истощение пулов стволовых клеток защищает от злокачественных стволовых клеток рака | 56 , 57 |
| Старение мозга (деменция, болезнь Альцгеймера, когнитивные нарушения и нарушения памяти) | Накопление амилоидных бляшек, потеря нейрональных клеток, нарушение передачи сигналов нейротрансмиттера | Нейрональные предшественники и стволовые клетки | Низкое количество делений взрослых клеток , низкая механическая нагрузка.Достаточное количество нейрональных стволовых клеток для поддержания тканевого гомеостаза | 58,59 |
| Катаракта | Помутнение хрусталика глаза из-за скопления белков | Образование катаракты не зависит от регенерации стволовых клеток; К факторам риска окружающей среды относятся курение, диабет, УФ-излучение | Биохимический процесс, который усиливается с возрастом, но не зависит от регенерации на клеточном уровне | 60 |
| Сахарный диабет 2 типа | Инсулиннезависимый диабет, инсулинорезистентность | Инсулинорезистентность не зависит от регенерации стволовых клеток | Сахарный диабет 2 типа часто связан с нездоровым образом жизни, ожирением и гипертонией | 61 |
| Гиперлипидемия | Повышение уровня липидов (холестерина, холестериновых эфиров, | Гиперлипидемия не зависит от регенерации стволовых клеток | Гиперлипидемия вызвана привычками образа жизни (ожирение, курение) или поддающимися лечению заболеваниями (диабет, заболевание почек) | 62 |
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина отсутствия заболевания в HGPS . | Список литературы . |
|---|---|---|---|---|
| Рак | Неконтролируемый и инвазивный рост клеток | Теория стволовых клеток опухоли: рак возникает из стволовых клеток (грудных и нейрональных стволовых клеток) | Раннее истощение пулов стволовых клеток защищает от злокачественных стволовых клеток рака | 56 , 57 |
| Старение мозга (деменция, болезнь Альцгеймера, когнитивные нарушения и нарушения памяти) | Накопление амилоидных бляшек, потеря нейрональных клеток, нарушение передачи сигналов нейротрансмиттера | Нейрональные предшественники и стволовые клетки | Низкое количество делений взрослых клеток , низкая механическая нагрузка.Достаточное количество нейрональных стволовых клеток для поддержания тканевого гомеостаза | 58,59 |
| Катаракта | Помутнение хрусталика глаза из-за скопления белков | Образование катаракты не зависит от регенерации стволовых клеток; К факторам риска окружающей среды относятся курение, диабет, УФ-излучение | Биохимический процесс, который усиливается с возрастом, но не зависит от регенерации на клеточном уровне | 60 |
| Сахарный диабет 2 типа | Инсулиннезависимый диабет, инсулинорезистентность | Инсулинорезистентность не зависит от регенерации стволовых клеток | Сахарный диабет 2 типа часто связан с нездоровым образом жизни, ожирением и гипертонией | 61 |
| Гиперлипидемия | Повышение уровня липидов (холестерина, холестериновых эфиров, | Гиперлипидемия не зависит от регенерации стволовых клеток | Гиперлипидемия вызывается привычками образа жизни (ожирение, курение) или поддающимися лечению заболеваниями (диабет, заболевание почек) | 62 |
Таблица 2.
Общие возрастные заболевания ОТСУТСТВУЮТ при синдроме прогерии Хатчинсона-Гилфорда (HGPS).
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина отсутствия заболевания в HGPS . | Список литературы . |
|---|---|---|---|---|
| Рак | Неконтролируемый и инвазивный рост клеток | Теория стволовых клеток опухоли: рак возникает из стволовых клеток (грудных и нейрональных стволовых клеток) | Раннее истощение пулов стволовых клеток защищает от злокачественных стволовых клеток рака | 56 , 57 |
| Старение мозга (деменция, болезнь Альцгеймера, когнитивные нарушения и нарушения памяти) | Накопление амилоидных бляшек, потеря нейрональных клеток, нарушение передачи сигналов нейротрансмиттера | Нейрональные предшественники и стволовые клетки | Низкое количество делений взрослых клеток , низкая механическая нагрузка.Достаточное количество нейрональных стволовых клеток для поддержания тканевого гомеостаза | 58,59 |
| Катаракта | Помутнение хрусталика глаза из-за скопления белков | Образование катаракты не зависит от регенерации стволовых клеток; К факторам риска окружающей среды относятся курение, диабет, УФ-излучение | Биохимический процесс, который усиливается с возрастом, но не зависит от регенерации на клеточном уровне | 60 |
| Сахарный диабет 2 типа | Инсулиннезависимый диабет, инсулинорезистентность | Инсулинорезистентность не зависит от регенерации стволовых клеток | Сахарный диабет 2 типа часто связан с нездоровым образом жизни, ожирением и гипертонией | 61 |
| Гиперлипидемия | Повышение уровня липидов (холестерина, холестериновых эфиров, | Гиперлипидемия не зависит от регенерации стволовых клеток | Гиперлипидемия вызвана привычками образа жизни (ожирение, курение) или поддающимися лечению заболеваниями (диабет, заболевание почек) | 62 |
| Болезнь . | Механизм . | Релевантность стволовых клеток . | Предлагаемая причина отсутствия заболевания в HGPS . | Список литературы . |
|---|---|---|---|---|
| Рак | Неконтролируемый и инвазивный рост клеток | Теория стволовых клеток опухоли: рак возникает из стволовых клеток (грудных и нейрональных стволовых клеток) | Раннее истощение пулов стволовых клеток защищает от злокачественных стволовых клеток рака | 56 , 57 |
| Старение мозга (деменция, болезнь Альцгеймера, когнитивные нарушения и нарушения памяти) | Накопление амилоидных бляшек, потеря нейрональных клеток, нарушение передачи сигналов нейротрансмиттера | Нейрональные предшественники и стволовые клетки | Низкое количество делений взрослых клеток , низкая механическая нагрузка.Достаточное количество нейрональных стволовых клеток для поддержания тканевого гомеостаза | 58,59 |
| Катаракта | Помутнение хрусталика глаза из-за скопления белков | Образование катаракты не зависит от регенерации стволовых клеток; К факторам риска окружающей среды относятся курение, диабет, УФ-излучение | Биохимический процесс, который усиливается с возрастом, но не зависит от регенерации на клеточном уровне | 60 |
| Сахарный диабет 2 типа | Инсулиннезависимый диабет, инсулинорезистентность | Инсулинорезистентность не зависит от регенерации стволовых клеток | Сахарный диабет 2 типа часто связан с нездоровым образом жизни, ожирением и гипертонией | 61 |
| Гиперлипидемия | Повышение уровня липидов (холестерина, холестериновых эфиров, | Гиперлипидемия не зависит от регенерации стволовых клеток | Гиперлипидемия вызвана привычками образа жизни (ожирение, курение) или поддающимися лечению заболеваниями (диабет, заболевание почек) | 62 |
Эта работа финансировалась грантом New Emerging Team Grant от Канадский институт Ute of Health Research (CIHR) А.Б.-В. и другие. J.H.-W. поддерживается стипендией Эрвина-Шредингера Австрийского научного фонда (FWF).
Список литературы
1
Hennekam RC. Синдром прогерии Хатчинсона-Гилфорда: обзор фенотипа. Am J Med Genet A .
Опубликовано июль
1
:
2,
2006
; .2
Brown WT. Синдром прогерии Хатчинсона-Гилфорда. В: Hisama F, Weissman SM, Martin GM. Синдром прогерии Хатчинсона-Гилфорда. Нью-Йорк: Марсель Деккер, инк .; 2003: 245–261.
3
Pollex RL, Hegele RA. Синдром прогерии Хатчинсона-Гилфорда.
Clin Genet.
2004
;
66
:
375
-381,4
Baker PB, Baba N, Boesel CP. Сердечно-сосудистые нарушения при прогерии. Клинический случай и обзор литературы.
Arch Pathol Lab Med.
1981
;
105
:
384
-386,5
Stables GI, Morley WN. Синдром Хатчинсона-Гилфорда.
J R Soc Med.
1994
;
87
:
243
-244,6
Мартин Г. Генетические синдромы у человека, потенциально имеющие отношение к патобиологии старения.
Врожденные дефекты Ориг. Сер.
1977
;
14
:
5
-39,7
Стурман Н., Хейнс С., Эби У. Ядерные ламины: их структура, сборка и взаимодействия.
J Struct Biol.
1998
;
122
:
42
-66,8
Берк Б., Стюарт КЛ. Жизнь на грани: ядерная оболочка и болезнь человека.
Nat Rev Mol Cell Biol.
2002
;
3
:
575
-585,9
Лин Ф, Ворман Х.Дж. Структурная организация гена человека, кодирующего ядерный ламин A и ядерный ламин C.
J Biol Chem.
1993
;
268
:
16321
-16326,10
Йе Кью, Ворман Х.Дж. Белково-белковые взаимодействия между ядерными ламинами человека, экспрессируемыми в дрожжах.
Exp Cell Res.
1995
;
219
:
292
-298.11
Smith ED, Kudlow BA, Frock RL, Kennedy BK. Ядерные ламины А-типа, прогерии и другие дегенеративные заболевания.
Mech Aging Dev.
2005
;
126
:
447
-460.12
Hegele R. Положение мутации LMNA предсказывает участие систем органов в ламинопатиях.
Clin Genet.
2005
;
68
:
31
-34,13
Эрикссон М., Браун В. Т., Гордон Л. Б. и др. Рецидивирующие точечные мутации de novo в ламине А вызывают синдром прогерии Хатчинсона-Гилфорда.
Природа.
2003
;
423
:
293
-298,14
Де Сандре-Джованноли А., Бернар Р., Кау П. и др. Ламин — усечение в прогерии Хатчинсона-Гилфорда.
Наука.
2003
;
300
:
2055
.15
Скаффиди П., Мистели Т. Ламин A-зависимые ядерные дефекты при старении человека.
Наука.
2006
;
312
:
1059
-1063,16
Warner HR, Sierra F. Ядерная архитектура и болезни.
J Gerontol A Biol Sci Med Sci.
2006
;
61
:
461
-462,17
Фонг LG, Frost D, Meta M и др. Ингибитор протеин-фарнезилтрансферазы облегчает течение болезни у мышей с прогерией.
Наука.
2006
;
311
:
1621
-1623,18
Скаффиди П., Мистели Т. Обращение клеточного фенотипа при болезни преждевременного старения. Синдром прогерии Хатчинсона-Гилфорда.
Nat Med.
2005
;
11
:
440
-445.19
Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Ламины при болезни: почему повсеместно экспрессируемые белки ядерной оболочки вызывают тканеспецифические фенотипы заболевания?
J Cell Sci.
2001
;
114
:
9
-19.20
Моррис Г.Е., Манилал С. Сердце к сердцу: от ядерных белков до мышечной дистрофии Эмери-Дрейфуса.
Hum Mol Genet.
1999
;
8
:
1847
-1851.21
Ламмердинг Дж., Шульце П.К., Такахаши Т. и др.Дефицит Lamin A / C вызывает нарушение ядерной механики и механотрансдукции.
J Clin Invest.
2004
;
113
:
370
-378,22
Салливан Т., Эскаланте-Алькальд Д., Бхатт Х. и др. Потеря экспрессии ламина А-типа нарушает целостность ядерной оболочки, что приводит к мышечной дистрофии.
J Cell Biol.
1999
;
147
:
913
-920,23
Манчини М.А., Шан Б., Никерсон Дж. А., Пенман С., Ли У. Продукт гена ретинобластомы представляет собой зависимый от клеточного цикла белок, связанный с ядерным матриксом.
Proc Natl Acad Sci U S. A.
1994
;
91
:
418
-422,24
Фрок Р.Л., Кудлоу Б.А., Эванс А.М., Джеймсон С.А., Хаушка С.Д., Кеннеди Б.К. Ламин A / C и эмерин имеют решающее значение для дифференцировки сателлитных клеток скелетных мышц.
Genes Dev.
2006
;
20
:
486
-500,25
Varga R, Eriksson M, Erdos MR, et al. Прогрессирующие дефекты гладкомышечных клеток сосудов на мышиной модели синдрома прогерии Хатчинсона-Гилфорда.
Proc Natl Acad Sci U S. A.
2006
;
103
:
3250
-3255,26
Маункес Л.С., Козлов С., Эрнандес Л., Салливан Т., Стюарт К.Л. Прогероидный синдром у мышей вызывается дефектами ламинов А-типа.
Природа.
2003
;
423
:
298
-301,27
Ян Ш., Мета М, Цяо Х и др. Ингибитор фарнезилтрансферазы улучшает фенотип заболевания у мышей с мутацией синдрома прогерии Хатчинсона-Гилфорда.
J Clin Invest.
2006
;
116
:
2115
-2121,28
Кудлоу Б.А., Кеннеди Б.К. Старение: прогерия и ламинат.
Curr Biol.
2006
;
16
:
R652
-R654.29
Fong LG, Ng JK, Lammerding J, et al. Преламин А и ламин А, по-видимому, являются незаменимыми в ядерной ламине.
J Clin Invest.
2006
;
116
:
743
-752,30
Скаффиди П., Мистели Т. Хорошие новости для ядерной оболочки: потеря ламина А может быть выгодой.
J Clin Invest.
2006
;
116
:
632
-634.31
Pendas AM, Zhou Z, Cadinanos J, et al. Дефектный процессинг преламина А и изменения в мышцах и адипоцитах у мышей с дефицитом металлопротеиназы Zmpste24.
Nat Genet.
2002
;
31
:
94
-99,32
Берго МО, Гавино Б., Росс Дж. И др. Дефицит Zmpste24 у мышей вызывает спонтанные переломы костей, мышечную слабость и дефект обработки преламина А.
Proc Natl Acad Sci U S. A.
2002
;
99
:
13049
-13054,33
Янг С.Г., Фонг Л.Г., Михаэлис С. Преламин А. Zmpste24, деформированные ядра клеток и прогерия — новые данные, свидетельствующие о том, что фарнезилирование белков может иметь важное значение для патогенеза заболевания.
J Lipid Res.
2005
;
46
:
2531
-2558,34
Маункес Л., Козлов С., Берк Б., Стюарт К. Ламинопатии: структура ядра встречается с болезнью.
Curr Opin Genet Dev.
2003
;
13
:
223
-230,35
Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Ядерные ламины: строительные блоки ядерной архитектуры.
Genes Dev.
2002
;
16
:
533
-547,36
Бриджер Дж. М., Убить IR. Старение фибробластов синдрома прогерии Хатчинсона-Гилфорда характеризуется гиперпролиферацией и усилением апоптоза.
Exp Gerontol.
2004
;
39
:
717
-724.37
Лю Б., Ван Дж, Чан К.М. и др. Геномная нестабильность при преждевременном старении на основе ламинопатии.
Nat Med.
2005
;
11
:
780
-785,38
Prolla TA. Множественные пути к фенотипу старения: понимание молекулярного расчленения прогерий с помощью анализа ДНК-микрочипов.
Mech Aging Dev.
2005
;
126
:
461
-465,39
Warner HR, Sierra F. Модели ускоренного старения могут быть информативными в отношении молекулярных механизмов старения и / или возрастной патологии.
Mech Aging Dev.
2003
;
124
:
581
-587,40
Warner HR. Дебаты между Ричардом Миллером и Полом Хэсти / Яном Виджем.
Ячейка старения.
2004
;
3
:
141
-142,41
Warner HR. Разработка программы исследований в области биогеронтологии: основные механизмы.
Sci Aging Knowledge Environ.
2005
;
2005
:
pe33
.42
de Boer J, Andressoo JO, de Wit J, et al.Преждевременное старение у мышей с дефицитом репарации и транскрипции ДНК.
Наука.
2002
;
296
:
1276
-1279,43
Тайнер С.Д., Венкатачалам С., Чой Дж. И др. Мыши с мутантом р53, проявляющие фенотипы, связанные с ранним старением.
Природа.
2002
;
415
:
45
-53,44
Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Делеция Ku86 вызывает раннее начало старения у мышей.
Proc Natl Acad Sci U S A.
1999
;
96
:
10770
-10775,45
Хисама Ф., Вайсман С.М., Мартин Г.М. Хромосомная нестабильность и старение. 1-е изд. Нью-Йорк: Марсель Деккер, инк .; 2003.
46
Мартин Г.М., Осима Дж. Уроки прогероидных синдромов человека.
Природа.
2000
;
408
:
263
-266,47
Caplice NM, Doyle B. Сосудистые клетки-предшественники: происхождение и механизмы мобилизации, дифференцировки, интеграции и васкулогенеза.
Stem Cells Dev.
2005
;
14
:
122
-139,48
Робертс Н., Джахангири М., Сюй К. Клетки-предшественники при сосудистых заболеваниях.
J Cell Mol Med.
2005
;
9
:
583
-591,49
Бельтрами А.П., Урбанек К., Кайстура Дж. И др. Доказательства того, что сердечные миоциты человека делятся после инфаркта миокарда.
N Engl J Med.
2001
;
344
:
1750
-1757,50
Джексон К.А., Майка С.М., Ван Х и др.Регенерация ишемической сердечной мышцы и эндотелия сосудов взрослыми стволовыми клетками.
J Clin Invest.
2001
;
107
:
1395
-1402,51
Zuk PA, Zhu M, Ashjian P, et al. Жировая ткань человека является источником мультипотентных стволовых клеток.
Mol Biol Cell.
2002
;
13
:
4279
-4295,52
Cotsarelis G, Sun TT, Lavker RM. Клетки, сохраняющие метку, находятся в области выпуклости волосяного покрова: последствия для фолликулярных стволовых клеток, цикла роста волос и канцерогенеза кожи.
Ячейка.
1990
;
61
:
1329
-1337,53
Hofer AC, Tran RT, Aziz OZ и др. Общие фенотипы среди сегментарных прогероидных синдромов предполагают основные пути старения.
J Gerontol A Biol Sci Med Sci.
2005
;
60A
:
10
-20,54
Куо СК, Ли В.Дж., Маук Р.Л., Туан Р.С. Инженерия хрящевой ткани: ее потенциал и использование.
Curr Opin Rheumatol.
2006
;
18
:
64
-73.55
йен AH, Шарп PT. Регенерация зубов с использованием тканевой инженерии на основе стволовых клеток.
Эксперт Opin Biol Ther.
2006
;
6
:
9
-16,56
Репетто Л., Бальдуччи Л. Случай для гериатрической онкологии.
Ланцет Онкол.
2002
;
3
:
289
-297,57
ДеПиньо РА. Возраст рака.
Природа.
2000
;
408
:
248
-254,58
Bauer HC, Tempfer H, Bernroider G, Bauer H.Нейрональные стволовые клетки у взрослых.
Exp Gerontol.
2006
;
41
:
111
-116,59
Вескови А.Л., Галли Р., Рейнольдс Б.А. Стволовые клетки опухоли головного мозга.
Nat Rev Cancer.
2006
;
6
:
425
-436,60
Asbell PA, Dualan I, Mindel J, Brocks D, Ahmad M, Epstein S. Возрастная катаракта.
Ланцет.
2005
;
365
:
599
-609,61
Винер Н., Сауэрс-младший. Эпидемиология диабета.
J Clin Pharmacol.
2004
;
44
:
397
-405,62
Menuet R, Lavie CJ, Milani RV. Важность и лечение дислипидемии при метаболическом синдроме.
Am J Med Sci.
2005
;
330
:
295
-302,63
Понти Д., Заффарони Н., Капелли С., Дайдон МГ. Стволовые клетки рака груди: обзор.
Eur J Cancer.
2006
;
42
:
1219
-1224,64
Galderisi U, Cipollaro M, Giordano A.Стволовые клетки и рак мозга.
Смерть клеток Дифференц.
2006
;
13
:
5
-11,65
Novelli G, Muchir A, Sangiuolo F и др. Мандибулоакральная дисплазия вызвана мутацией LMNA-кодирующего ламина A / C.
Am J Hum Genet.
2002
;
71
:
426
-431.66
Джейкоб К.Н., Гарг А. Ламинопатии: мультисистемные дистрофические синдромы.
Mol Genet Metab.
2006
;
87
:
289
-302.67
Caux F, Dubosclard E, Lascols O, et al. Новое клиническое состояние, связанное с новой мутацией ламинов A и C с генерализованной липоатрофией, инсулинорезистентным диабетом, диссеминированными лейкомеланодермическими папулами, стеатозом печени и кардиомиопатией.
J Clin Endocrinol Metab.
2003
;
88
:
1006
-1013.68
Stehbens WE, Delahunt B, Shozawa T., Gilbert-Barness E. Истощение клеток гладкой мускулатуры и типы коллагена в прогерических артериях.
Cardiovasc Pathol.
2001
;
10
:
133
-136.69
Аш-Шали KZ, Hegele RA. Ламинопатии и атеросклероз.
Артериосклер Thromb Vasc Biol.
2004
;
24
:
1591
-1595.
Авторские права 2007 Геронтологического общества Америки
границ | Есть ли общие механизмы между синдромом прогерии Хатчинсона – Гилфорда и естественным старением?
Введение
Прогероидные синдромы (ускоренное старение) — это группа моногенных заболеваний, которые напоминают некоторые аспекты естественного старения.Большинство, если не все, из них вызваны мутациями в генах репарации ДНК или ядерной ламины. Эти заболевания могут сыграть значительную роль в изучении механизмов естественного старения, в том числе эпигенетических (Ashapkin et al., 2017). Одним из наиболее широко изученных заболеваний ускоренного старения является синдром прогерии Хатчинсона – Гилфорда (HGPS), вызываемый мутациями гена LMNA , кодирующего ядерные фибриллярные белки ламины A и C (Baek et al., 2013). Этот синдром часто называют преждевременным старением из-за множества патологий, похожих на старение, возникающих в течение первых нескольких лет жизни.Чаще всего поражаются органы мезенхимального происхождения, такие как скелет, мышцы, жир и сердечно-сосудистая система. Продолжительность жизни у пациентов с HGPS сильно варьируется. Большинство из них умирают в возрасте 13–14 лет от инфаркта или инсульта, вызванного прогрессирующим атеросклерозом коронарных и цереброваскулярных артерий. Прогерин представляет собой усеченную версию ламина А, продуцируемого посредством альтернативного сплайсинга транскрипта LMNA и . Большинство случаев HGPS вызвано молчащей мутацией de novo 1824 C → T в клетках зародышевой линии (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003), хотя известно, что некоторые другие мутации вызывают синдромы с аналогичными характеристиками.
Нормальный ламин А является важным компонентом ядерной пластинки, белковой сети, прилегающей к внутренней ядерной мембране и обращенной внутрь ядра. Lamina участвует в поддержании ядерной структуры, организации хроматина, репликации ДНК и экспрессии генов. Он влияет на большинство генетических процессов через прикрепление ламино-ассоциированных хроматиновых доменов (LAD) к периферии ядра.Количество LAD удваивается во время репликации ДНК. Соответственно, происходит расширение пластинки, чтобы приспособиться к этому увеличенному количеству ПМЖВ (Жиронкина и др., 2016).
В фибробластах, полученных от 2-летнего пациента с HGPS во время ранних пассажей, морфология ядра кажется относительно нормальной (Goldman et al., 2004). В клетках 6 пассажа около 30% ядер имеют аномальные структурные особенности (дольки ядерной оболочки), клетки 13 пассажа — 54%, а клетки пассажа 26 — 81%.Также наблюдается увеличение толщины пластинки. В контрольных фибробластах даже на 17-м пассаже аномальные структурные особенности наблюдаются только в 8% ядер. В фибробластах, полученных от нормального 92-летнего человека, практически все ядра выглядят нормальными при пассаже 2, тогда как около 22% ядер демонстрируют аномальные особенности при пассаже 17. Тем не менее, эти аномальные особенности никогда не бывают такими серьезными, как в клетках HGPS. Ядерная аномалия, наблюдаемая только в большинстве клеток HGPS позднего пассажа, представляет собой кластеризацию комплексов ядерных пор в щелях, образованных между дольчатыми областями ядерной оболочки.В контрольных клетках на всех пассажах и в клетках HGPS на ранних пассажах ядерные поры обычно распределены по всей поверхности ядра. Частичная потеря внутреннего гетерохроматина и почти полная потеря периферического гетерохроматина происходит в дольчатых ядрах клеток HGPS позднего пассажа (Goldman et al., 2004). Вместо этого появляется необычно толстая электронно-плотная пластинка, прилегающая к внутренней ядерной мембране. Накопление пре-ламина А одновременно происходит в стареющих клетках HGPS.
Транспорт в ядра и включение в пластинку происходит вскоре после инъекции бактериально экспрессированного прогерина в цитоплазму контрольных фибробластов.Это приводит через полчаса к появлению множественных ядер с HGPS-подобной аномальной морфологией (Goldman et al., 2004). Когда вводится такое же количество нормального ламина А, наблюдается столь же быстрое включение в ядерную пластинку, но морфология ядра остается нормальной. Последующая инъекция нормального ламина А не спасает аномальный фенотип, вызванный предыдущей инъекцией прогерина. Таким образом, прогерин действует как доминирующий фактор, который напрямую и быстро нарушает морфологию ядра.
Молекулярные механизмы продукции прогерина
Зрелый белок ламина А продуцируется из предшественника, пре-ламина А, посредством ряда этапов посттрансляционного процессинга: цистеина С-концевого мотива CAAX (C — цистеин, A — алифатическая аминокислота, X — обычно метионин. или лейцин) фарнезилируется, затем концевой трипептид AAX протеолитически отщепляется, фарнезилированный C-концевой цистеин метилируется, 15 C-концевых аминокислот (647–661) отщепляются. Обе реакции протеолитического расщепления катализируются металлопротеазой цинка ZMPSTE24, единственным известным субстратом которой является пре-ламин.Молчащая мутация C → T в положении нуклеотида 1824 активирует скрытый донорский сайт сплайсинга (5’SS) в экзоне 11 транскрипта LMNA, что приводит к делеции проксимального 150-нуклеотидного сегмента 3′-конца экзона 11 и продукции усеченной на 50 аминокислот версии пре-ламина А — прогерина. Он не содержит 607–656 аминокислотных остатков пре-ламина А, но содержит С-концевой мотив CAAX. Поскольку сайт окончательного процессинга протеазы ZMPSTE24 теряется, прогерин остается постоянно фарнезилированным, что приводит к его стабильной ассоциации с ядерной оболочкой.Постоянное существование фарнезилированной формы, по-видимому, отвечает за морфологические аномалии клеточных ядер и нарушения в организации гетерохроматина, митозах, репликации и репарации ДНК и транскрипции генов.
Криптический 5’SS в экзоне 11 гена LMNA ( CAG / GT GG G C) имеет шесть из девяти нуклеотидов консенсусной последовательности 5’SS (CAG / GTAAGT), тогда как в У мутантной версии HGPS их семь ( CAGGT GG GT ) (Lopez-Mejia et al., 2011). Ожидается, что эта мутация не сильно изменит силу 5’SS, так как она влияет на конечное сильно вариабельное положение (+6). Действительно, вычислительный анализ показывает, что как дикого типа, так и мутантные версии прогерина 5’SS имеют гораздо более низкие показатели силы по сравнению с нормальным ламином A 5’SS ( CAG / GT G AGT ) на конце экзона 11. Тем не менее, прогерин продуцируется в мутантных клетках HGPS на более высоком уровне, чем ламин A. Прогерин 5’SS дикого типа, по-видимому, является частью стабильной структуры РНК, которая предотвращает его эффективное спаривание оснований с мяРНК U1 (Lopez-Mejia et al. al., 2011). Мутация прогерии дестабилизирует эту структуру, тем самым увеличивая доступность криптического 5’SS к аппарату сплайсинга. Кроме того, конкуренция между сайтами сплайсинга доноров прогерина и ламина А регулируется более надежно в пользу продукции ламина А белками SRSF1 и SRSF6 в клетках дикого типа. Оба эти механизма служат для минимизации выработки прогерина при естественном старении. С помощью анализов in vitro, и в целлюлозе , еще один белок SR, SRSF5, был идентифицирован как активатор аутентичного ламина A 5’SS (Vautrot et al., 2016). Повышенная экспрессия SRSF5 в фибробластах пациентов с HGPS способствовала использованию ламина A 5’SS против прогерина 5’SS. Стимуляция PDGF-BB приводила к повышенному уровню фосфорилирования белка SR, снижению продукции прогерина и частично восстанавливала ядерную морфологию фибробластов HGPS.
Механизмы индуцированного прогерином патогенеза
Было высказано предположение, что перманентное фарнезилирование прогерина является одной из ведущих причин фенотипа HGPS. Это предположение было подтверждено открытием, что ингибиторы фарнезилирования белков улучшают индуцированные прогерином аномалии.Однако основные фенотипы HGPS наблюдались в модели HGPS мышей, экспрессирующих нефарнезилированную версию прогерина (Yang et al., 2008). Остаток цистеина в C-концевом мотиве CAAX пре-ламина A был заменен химически подобным остатком серина, что делает невозможным фарнезилирование. Тем не менее, у мышей, экспрессирующих эту версию прогерина, развились характерные черты HGPS (замедленный рост, хрупкость костей, сокращение продолжительности жизни, уменьшение подкожного жира), хотя и менее серьезные по сравнению с мышами, экспрессирующими неизмененную версию прогерина.Эти различия можно объяснить более низкой стабильностью нефарнезилированных молекул прогерина. Соответственно, эффекты ингибиторов фарнезилирования могут быть обусловлены не ролью фарнезила per se , а скорее снижением стабильности прогерина. С другой стороны, следует учитывать, что используемая замена C на S предотвращает не только фарнезилирование прогерина, но также ингибирует две последующие стадии его процессинга, а именно, расщепление концевого трипептида и метилирование концевого остатка C.Действительно, аномалии типа HGPS не наблюдались у мышей, продуцирующих новую версию нефарнезилированного прогерина, полученного путем делеции кодона изолейцинового остатка в С-концевом тетрапептидном мотиве (CSIM> CSM) (Yang et al., 2011) .
В культивируемых клетках эктопическая экспрессия LMNA дикого типа или генов прогерина индуцировала прогероидный фенотип — уменьшение репликативной продолжительности жизни, старения и апоптоза (Candelario et al., 2008). Когда экспрессируется ген LMNA дикого типа, клетки могут быть спасены за счет сверхэкспрессии процессирующей протеазы ZMPSTE24.Вероятно, чрезмерные уровни предварительного ламината А вызывают аномалии в этом случае. Как правило, прогероидный фенотип более серьезен в клетках, экспрессирующих прогерин, по сравнению с клетками, экспрессирующими LMNA и . По-видимому, метаболизм ламина А настолько тонко сбалансирован, что даже небольшие нарушения в этой системе могут «подтолкнуть» клетку к прогероидному фенотипу. Даже в отсутствие мутаций небольших нарушений в экспрессии ламина А может быть достаточно, чтобы вызвать заметные ядерные дефекты, влияющие на старение клеток. Вероятно, такие нарушения играют роль в естественном старении клеток.
Сердечно-сосудистые заболевания считаются одними из наиболее характерных заболеваний пожилого возраста. Точно так же такие нарушения являются одними из наиболее типичных признаков ускоренного старения, вызванного прогерином, вероятно, из-за накопления прогерина в стенках артерий (Olive et al., 2010). Подобно возрастной сердечно-сосудистой патологии, в артериях HGPS присутствуют атеросклеротические бляшки и другие виды повреждений (очаги кальцификации, воспаления, эрозии). Большое количество прогерина накапливается в стенках артерий пациентов с HGPS.Некоторое производство прогерина также происходит в коронарной артерии у здоровых пожилых людей и имеет тенденцию к увеличению с возрастом. Пока неясно, представляет ли это небольшое прогрессирующее накопление прогерина небольшое хроническое повреждение, которое играет роль в сердечно-сосудистых патологиях, связанных с естественным старением. Было бы целесообразно изучить динамику накопления прогерина у лиц без HGPS с низким и высоким риском сердечно-сосудистой патологии. Вполне возможно, что постепенное накопление прогерина, даже на низких уровнях, в сосудистых клетках приводит к увеличению числа случаев гибели клеток и хронической воспалительной реакции на окислительный стресс и другие виды повреждений.Атеросклероз мог быть следствием этих процессов. Конечно, это предположение нуждается в экспериментальной проверке.
Стоит особо отметить, что сердечно-сосудистое старение, по-видимому, коррелирует не только с накоплением прогерина, но также и с накоплением пре-ламина А (Ragnauth et al., 2010). У пациентов с HGPS наиболее очевидные дефекты наблюдаются в гладкомышечных клетках сосудов (VSMC). Некоторые специфические изменения в этих клетках, такие как кальцификация, накопление липидов, фиброз и истощение VSMC, наблюдаются как при индуцированном прогерином, так и при естественном старении.Прогрессивное накопление ультраструктурных дефектов ядерной пластинки наблюдалось при старении VSMC in vivo и в культуре клеток (Ragnauth et al., 2010). Не было обнаружено сопутствующего синтеза и накопления прогерина (ОТ-ПЦР, вестерн-блоттинг), но было значительное накопление пре-ламина А (вестерн-блоттинг). Это накопление пре-ламина А происходило из-за вызванного стрессом подавления процессирующего фермента FACE1 (человеческая версия ZMPSTE24). Таким образом, аномальное накопление необработанного пре-ламина А может вызывать повреждение ДНК и митотическую дисфункцию, что в конечном итоге приводит к клеточному старению.VSMC представляют собой MSC-подобные клетки, которые могут подвергаться остеогенной и адипогенной дифференцировке, что приводит к кальцификации и накоплению липидов, патологиям, преобладающим в сосудистой сети детей с HGPS и трансгенных мышей, сверхэкспрессирующих прогерин. Накопление пре-ламина A в VSMCs, по-видимому, является причинным фактором их старения, поскольку оно предшествует старению, а его сверхэкспрессия ускоряет старение. Подобно прогерину в фибробластах HGPS, избыточный пре-ламин А, по-видимому, является доминирующим фактором, который нарушает целостность ядерной ламины, дерегулирует реакцию на повреждение ДНК и митотические контрольные точки.В контексте сердечно-сосудистых заболеваний это может привести к ускоренному старению атеросклеротических бляшек.
Как в VSMCs, накапливающих пре-ламин A, так и в фибробластах HGPS, увеличение частоты клеток с сильно фрагментированными ядрами непосредственно предшествует старению. Такое ядерное повреждение является характерной чертой митотической катастрофы, формы гибели клеток, вызываемой, когда клетки, несущие серьезные повреждения ДНК, вступают в митоз. Эти данные хорошо согласуются с редкостью пролиферирующих VSMC и их истощением, наблюдаемым в прогрессирующих атеросклеротических бляшках.Решающим событием в избыточном накоплении пре-ламина А является индуцированное окислительным стрессом подавление FACE1. Окислительное повреждение обнаруживается в ~ 90% клеток атеросклеротических бляшек. Вполне может быть, что такое индуцированное стрессом подавление FACE1 и накопление пре-ламина A приводит к увеличению повреждений ДНК, митотической катастрофе и преждевременному старению VSMC и, таким образом, вызывает возрастные сосудистые патологии.
Недавно было показано, что повышенная регуляция экспрессии прогерина происходит в сердце человека с дилатационной кардиомиопатией, где она сильно коррелирует с ремоделированием левого желудочка (Messner et al., 2018). Иммунофлуоресцентное исследование позволило легко выявить прогерин-содержащие кардиомиоциты в сердце с расширенной кардиомиопатией, но не в сердце без сердечной недостаточности. Более того, маркер апоптоза каспаза-3 и прогерин были совместно локализованы в ядрах кардиомиоцитов сердца с дилатационной кардиомиопатией, что позволяет предположить возможную роль прогерина в гибели апоптотических клеток. Таким образом, прогерин может участвовать в старении миокарда и, в конечном итоге, в сердечной недостаточности.
Играет ли прогерин роль в естественном старении?
В контрольных культурах фибробластов прогерин не мог быть обнаружен на ранних пассажах, но он появлялся и накапливался на более поздних пассажах (McClintock et al., 2007). В биоптатах кожи людей обоих полов разного возраста (22–97 лет) низкие уровни мРНК прогерина были обнаружены с помощью ОТ-ПЦР с использованием праймеров к экзонам 9 и 12 из LMNA , но корреляции с возрастом не обнаружено. Небольшие количества белка прогерина были обнаружены моноклональными антителами в образцах старых доноров, но не в образцах молодых людей. Количество прогерина у пожилых доноров имеет тенденцию к увеличению с возрастом. В первичных культурах фибробластов, полученных из кожи нормальных доноров, очень мало ядер было окрашено прогерин-специфическими антителами.Доля таких ядер была менее 0,01% у молодых доноров и 0,3–0,8% — у пожилых доноров. Эти прогерин-положительные клетки обнаруживают HGPS-подобные дефекты в морфологии ядра, такие как дольки ядерной оболочки и инвагинации, двуядерные клетки и другие. В длительных культурах фибробласт HGPS прекращал рост после 40–45 кумулятивных удвоений популяции (CPD), доля прогерин-положительных клеток достигала около 90%. В клетках, полученных от нормальных доноров в тех же условиях, произошло небольшое увеличение доли прогерин-положительных клеток.Например, в выборке, полученной от 86-летней женщины, эта доля составляла около 0,4% в более ранних пассажах и достигала примерно 0,8% в более поздних пассажах (CPD 30–35). В экстрактах первичных культур фибробластов иммунореактивный прогерин не обнаруживался, тогда как в экстрактах фибробластов HGPS его можно было легко обнаружить даже на ранних пассажах. В экстрактах культуры фибробластов, полученной от 86-летней женщины, прогерин практически не обнаруживался на более поздних пассажах. Таким образом, прогерин, по-видимому, продуцируется и накапливается в небольших количествах в фибробластах дикого типа.
В срезах кожи 9-летнего пациента с HGPS прогерин был обнаружен в ядрах дермы, кровеносных сосудах, арректорных мышцах и клетках, окружающих потовые железы. Он также был обнаружен в ядрах кератиноцитов верхних слоев эпидермиса. В коже здоровых новорожденных прогерин-положительных ядер не обнаружено. В коже груди 22-летней женщины прогерин был обнаружен в скудных клетках вблизи базальной мембраны и папиллярной дермы, тогда как у 46-летней женщины большее количество прогерин-содержащих клеток было обнаружено по всей папиллярной дерме.Кожа лба 69-летнего мужчины содержала прогерин-положительные ядра в основном в верхней части дермы, тогда как высокая плотность прогерин-положительных ядер была обнаружена во всех слоях дермы в коже лба у 93-летнего пациента. — старая женщина. Аналогичные результаты были получены с срезами кожи с других участков тела: доля прогерин-положительных ядер в верхнем слое дермы увеличивалась с возрастом, и такие ядра появлялись в более глубоких слоях в пожилом возрасте. Таким образом, градиент прогерин-положительных фибробластов возникает при естественном старении между базальной мембраной и ретикулярной дермой.Прогерин-положительные ядра не были обнаружены в большинстве образцов эпидермиса. Немногочисленные исключения составляли образцы, полученные от некоторых пожилых доноров (70–95 лет), но даже в этих образцах количество прогерин-положительных кератиноцитов было намного ниже, чем у фибробластов. По-видимому, терминально дифференцированные или стареющие клетки накапливали детектируемый прогерин. В эпидермисе эти клетки могут быть кератиноцитами в конце своей жизни, непосредственно предшествующей потере ядер и других органелл.
Все продукты сплайсинга гена LMNA были обнаружены в первичных культурах фибробластов от пациентов с HGPS, их контрольной группы дикого типа соответствующего возраста и их здоровых родителей (Rodriguez et al., 2009). МРНК ламина С была максимально экспрессирована во всех образцах (~ 1500 копий на 1 мкг общей РНК в HGPS и контроле, и ~ 2000 копий в родительских фибробластах HGPS). Концентрация мРНК ламина А была практически одинаковой во всех образцах и равной концентрации мРНК прогерина у пациентов с HGPS (~ 500 копий).Уровни мРНК прогерина в контрольных фибробластах и родительских фибробластах HGPS были намного ниже (~ 2,5 копий). При сравнении ранних и более поздних пассажей одних и тех же культур было обнаружено, что концентрация мРНК прогерина умеренно увеличивается на более поздних пассажах.
мРНК и белок прогерина были обнаружены на низких уровнях в образцах скелетных мышц человека во всех исследованных возрастах (от 16 до 71 года) (Luo et al., 2013). Количество мРНК прогерина относительно мРНК ламина А немного увеличивалось с возрастом, тогда как уровень белка прогерина сильно варьировал между людьми без явной корреляции с возрастом.В образцах скелетных мышц мышей разного возраста мРНК прогерина не обнаружена. Таким образом, сплайсинг транскриптов LMNA и в клетках скелетных мышц мышей, по-видимому, находится под более надежным контролем.
В культурах фибробластов, полученных от доноров-людей естественного возраста (81–96 лет), постоянно наблюдались ядерные аберрации, сходные с таковыми в клетках HGPS, включая снижение уровней h4K9me3 и HP1 (Scaffidi and Misteli, 2006). Частоты клеток с такими аберрациями были вполне сопоставимы между образцами естественного старения и образцами HGPS, хотя уровни истощения h4K9me3 и HP1 были намного выше в культурах HGPS.Прогрессивное накопление морфологических аномалий с пассажами наблюдалось в культурах фибробластов, полученных от нормальных доноров разного возраста. Это накопление произошло значительно раньше в клетках старых доноров. Известно, что фосфорилирование гистона h3AX маркирует локусы повреждения ДНК, которые еще не восстановлены. Было показано, что количество таких локусов повреждения ДНК намного выше в фибробластах как пожилых людей из контрольной группы, так и пациентов с HGPS, чем в фибробластах молодых доноров. Таким образом, нормальная и ускоренная формы старения имеют много общего.
мРНК прогерина была обнаружена с помощью анализа RT-PCR в культурах фибробластов, полученных от нормальных людей разного возраста. Скрытый 5’SS, по-видимому, использовался в 50 раз реже у клеток дикого типа по сравнению с клетками HGPS, независимо от возраста. Аналогичные результаты были получены с образцами тканей (печени и сердца) людей разного возраста. Количество иммунореактивного ламина A / C не меняется с возрастом, тогда как его субклеточная локализация резко меняется. В клетках молодого человека он присутствует как на периферии ядра, так и во всей нуклеоплазме.В клетках пожилого человека и клетках HGPS ламин A / C находится в основном на периферии ядра. Когда использование прогерина 5’SS было ингибировано in vivo с помощью модифицированного комплементарного олигонуклеотида, HP1 и h4K9me3 в старых донорских клетках увеличились до уровней, соответствующих молодым донорским клеткам (Scaffidi and Misteli, 2006). Более того, экспрессия маркеров старения p21, IGFBP3 и GADD45B снижалась, тогда как пролиферативная активность повышалась до уровней молодых донорских фибробластов.Очевидное предположение состоит в том, что прогерин-зависимые механизмы играют роль в естественном старении, а чрезмерная активность этих механизмов вызывает преждевременное старение в HGPS. Возрастные ядерные аномалии, вероятно, вызваны прямым доминантным действием прогерина, накопленного на периферии ядра. Постоянное присутствие прогерина даже на низких уровнях вполне может быть одной из основных причин старения дефектов ядерной структуры. Старые клетки могут быть более уязвимы к негативному влиянию прогерина, например, из-за р53-зависимой активации программы старения.
Молекулярные механизмы старения, индуцированного прогерином
Истощение теломер
Нормальные человеческие фибробласты, экспрессирующие ламин А дикого типа или прогерин, обнаруживают уменьшенную репликативную продолжительность жизни, ускоренную потерю теломер и морфологические аномалии, типичные для клеток HGPS (Huang et al., 2008). Вероятно, изменения в организации пластинки могут напрямую влиять на скорость истощения теломер и приводить к ускоренному репликативному старению и прогероидным фенотипам. Путем количественной флуоресцентной гибридизации in situ было показано, что средняя длина теломер в фибробластах HGPS меньше, чем в нормальных фибробластах (Decker et al., 2009). Длина теломер отдельных хромосом непостоянна; ни у одного из них нет неизменно более коротких или неизменно более длинных теломер. В гемопоэтических клетках, которые не экспрессируют LMNA , теломеры имеют одинаковую длину между пациентами с HGPS и нормальными людьми. Таким образом, экспрессия прогерина, по-видимому, напрямую влияет на длину теломер. Было показано, что эктопическая экспрессия прогерина в нормальных диплоидных фибробластах активирует передачу сигналов теломер-специфичного повреждения ДНК, что приводит к агрегации теломер и хромосомным аберрациям, характерным для дисфункции теломер (Benson et al., 2010). Напротив, было обнаружено, что повреждение теломер в нормальных фибробластах человека активирует продукцию прогерина и старение клеток (Cao et al., 2011).
Измененная экспрессия гена
В нескольких линиях фибробластов HGPS наблюдались двукратные или более сильные изменения уровней экспрессии для 361 гена (увеличение для 193 и снижение для 168) по сравнению с контрольными фибробластами того же возраста (Csoka et al., 2004). Гены, кодирующие фактор транскрипции, преобладали среди этих дифференциально экспрессируемых генов: всего 39, 29 из которых, как известно, участвуют в эмбриогенезе и контроле дифференцировки клеток.Значительная доля (10 из 39) дифференциально экспрессируемых факторов транскрипции участвует в развитии скелета, конечностей и мышц. Наиболее сильно пораженным геном (в 29 раз повышенным в HGPS) был MEOX2 / GAX , который кодирует гомеобоксный белок, который, как известно, играет роль в развитии мезодермы. Гены, кодирующие регуляторы хроматина ING1, TWIST2 и SALL1, были среди генов, подвергшихся отрицательному воздействию. Единственным положительно затронутым геном в этой категории был HDAC9. Вторая по величине категория содержала 30 генов, кодирующих компоненты внеклеточного матрикса (ЕСМ), такие как коллагены 4A1, 4A2 и 4A5, ламинин α5, нетрин 4 и нидоген 2.Эти данные хорошо согласуются с тем, что ткани мезодермы наиболее сильно страдают при HGPS. Что касается происхождения и функции тканей, самая большая категория (31 ген) была связана с сердечно-сосудистой системой и атеросклерозом. Вторая по величине группа (22 гена) связана с развитием и функционированием скелетной системы. Факторы транскрипции MEOX2 / GAX и GATA6, суперэкспрессированные в HGPS, являются известными репрессорами пролиферации VSMC.
Чтобы понять, действительно ли HGPS повторяет естественное старение или просто фенотипически напоминает его, клеточные сигнальные пути, затронутые естественным старением in vitro и in vivo , сравнивались с таковыми у пациентов с HGPS (Aliper et al., 2015). Полученные данные показали, что естественное старение и преждевременное старение в HGPS опосредуются сходными сигнальными путями, такими как подавление репарации ДНК, организация хроматина, каспаза и пути EGFR, а также повышенная регуляция ERK, mTOR, GH-IGF1, MAPK, TGFβ, митохондриальная дисфункция и некоторые другие пути.
Один из возможных способов положительной ко-регуляции — это конкуренция за молекулы miRNA между мРНК, имеющими общие мишени miRNA (Arancio et al., 2013). Было обнаружено, что самые длинные транскрипты гена LMNA человека имеют пять предполагаемых целевых последовательностей miRNA с транскриптами одного гена ( DICER1 ), по четыре последовательности-мишени каждая — с транскриптами четырех генов ( CDKN1A , NFκB1 , TP53 и VEGFA ), три последовательности-мишени — с транскриптами 12 генов, две последовательности-мишени — с транскриптами 51 гена и одна последовательность-мишень — с транскриптами 267 генов.Таким образом, экспрессия ламина A / прогерина, по-видимому, тесно связана с механизмом интерференции РНК, регуляцией клеточного цикла и воспалением. Возможная связь с эпигенетической регуляцией ключевых клеточных функций может частично объяснить многие последствия мутаций гена LMNA у людей.
Было показано, что индуцируемая экспрессия прогерина в TERT-иммортализованной линии фибробластов из кожи человека приводит к накоплению прогерина на 5-10 день и некоторым клеточным дефектам, характерным для клеток HGPS, таким как аберрантная морфология ядра, увеличение количества очагов повреждения ДНК и уменьшение ламин-ассоциированный полипептид 2 (Scaffidi and Misteli, 2008).Суперэкспрессия гена LMNA дикого типа приводила к аналогичным дефектам, но в гораздо более мягкой форме. Экспрессия 194 генов была изменена на 5-е сутки и 1013 генов — на 10-е сутки после индукции прогерина. Около 23% этих генов также были затронуты сверхэкспрессией ламина А, но в меньшей степени. Некоторые компоненты сигнального пути Notch активировались рано после индукции прогерина. Гены, кодирующие основные прямые эффекторы Notch, HES1 и HEY1, были одними из самых высокоактивных.Ген HES5 , кодирующий другой главный регулятор генов-мишеней Notch, и TLE1 , кодирующий эффектор репрессии транскрипции белков HES, также были активированы, в то время как несколько генов-мишеней HES были подавлены. Таким образом, активация пути Notch, по-видимому, является специфической реакцией на прогерин. Для всех затронутых генов изменения в экспрессии наблюдались уже через 5 дней после индукции, тогда как через 10 дней эти изменения достигли 10-15-кратного уровня. Экспрессия экзогенного прогерина в иммортализованных hMSCs индуцировала повышенную регуляцию HES1 и HEY1 .В этих МСК часто наблюдалась аберрантная экспрессия маркеров дифференцировки клеток, что указывает на то, что происходит спонтанная дифференцировка. Подобные, но значительно более низкие эффекты были обнаружены при чрезмерной экспрессии ламина А. Влияние прогерина на индуцированную дифференцировку hMSC было непостоянным: остеогенная дифференцировка стимулировалась, адипогенная дифференцировка — ингибировалась, хондрогенная дифференцировка — не влияла. Сходные эффекты наблюдались на экспрессии конститутивно активной версии Notch (NICD), подтверждая представление о действии прогерина через активацию сигнального пути Notch.Это Notch-зависимое нарушение регуляции дифференцировки MSC и детерминации клеточной судьбы может объяснять множественные фенотипы дисфункции мезенхимальных тканей при HGPS. Возможно, постепенное накопление прогерина с естественным старением может искажать активность пути Notch, приводя к аналогичным нарушениям в более низком масштабе.
Измененная активность сигнальных путей
Влияние прогерина на уровни и субъядерную локализацию множественных факторов регуляции транскрипции, вместе с изменениями в организации хроматина, может приводить к изменениям эпигенетических программ в прогероидных клетках.Короткий период гиперпролиферации в таких клетках обычно сопровождается остановкой митоза и апоптозом (Bridger and Kill, 2004). Одной из возможных причин таких изменений может быть активация p53, но до сих пор неясно, является ли такая активация прямым действием прогерина или одним из его вторичных эффектов (Liu et al., 2005). Было отмечено, что у мышей с нокаутом рецептора витамина D (VDR) развивается фенотип преждевременного старения, который имеет типичные черты HGPS, такие как сокращение продолжительности жизни, сердечно-сосудистая патология и некоторые другие (Bouillon et al., 2008; Кейсала и др., 2009). Было показано, что в клетках HGPS снижена экспрессия VDR и факторов репарации ДНК BRCA1 и 53BP1 (Kreienkamp et al., 2016). Добавление витамина D3 облегчило большинство функций HGPS, таких как преждевременное старение, неэффективное восстановление ДНК и другие. Важно отметить, что передача сигналов VDR влияет на экспрессию гена LMNA и генов белков репарации ДНК, снижает продукцию прогерина и стабилизирует белки BRCA1 и 53BP1 в клетках HGPS. Очевидно, прогерин может влиять на эпигенетический статус гена VDR, приводя к его репрессии, тем самым внося вклад в фенотип HGPS.Трансфекция пигментных эпителиальных клеток сетчатки прогерином вызывала остановку репликационной вилки и опосредованную нуклеазой деградацию (Kreienkamp et al., 2018). Это, вероятно, приводит к накоплению в цитоплазме однонитевых фрагментов ДНК и индукции клеточного IFN-подобного ответа. Действительно, было обнаружено, что около полусотни генов в пути ИФН / противовирусный / врожденный иммунитет активируются в HGPS и подавляются витамином D3.
Множественность сигнальных путей, затронутых в HGPS, и их взаимозависимость затрудняют различение прямых мишеней прогерина от вторичных пораженных.Было показано, что NFκB играет важную роль в регуляции экспрессии генов при старении млекопитающих (Adler et al., 2007). На моделях мышей HGPS накопление пре-ламина A или прогерина на периферии ядра, как было показано, активирует сигнальный путь NFκB, тем самым внося вклад в ассоциированный со старением секреторный фенотип (SASP) (Osorio et al., 2012). Такая активация NFκB может вызывать системное воспаление, присущее старению (воспаление) (Franceschi and Campisi, 2014). NFκB может быть основным фактором ускоренного старения, поскольку его ингибирование значительно увеличивает продолжительность жизни у мышей с моделью HGPS (Osorio et al., 2012).
Широко известно, что фактор транскрипции NRF2 связан с долголетием (Lewis et al., 2015). Он активирует экспрессию антиоксидантных генов за счет связывания с антиоксидантно-чувствительными промоторными элементами (ARE). NRF2 также был идентифицирован как основная мишень прогерина, который стимулирует его эффекты ускорения старения (Kubben et al., 2016). Было обнаружено, что более 200 предполагаемых генов-мишеней NRF2 значительно подавлены в фибробластах HGPS по сравнению с их аналогами дикого типа.Подобно прогерину, NRF2 преимущественно накапливался вблизи ядерной оболочки в клетках HGPS. Прогерин демонстрирует повышенное связывание с NRF2 по сравнению с ламином А. Таким образом, он может нарушать активность пути регуляции антиоксиданта NRF2-ARE за счет секвестрации NRF2 от его генов-мишеней.
Деацетилазные белки семейства сиртуинов SIRT1 и SIRT6, как известно, играют важную роль в старении млекопитающих (Ashapkin et al., 2017, 2018 и ссылки в них). С помощью анализов иммунопреципитации было показано, что SIRT1 взаимодействует с ламином A, но не с пре-ламином A или прогерином (Liu et al., 2012). Более того, специфическая деацетилазная активность SIRT1 в отношении р53 увеличивалась в присутствии ламина А дозозависимым образом. Сообщалось, что ресвератрол увеличивает активность SIRT1 в отношении синтетического пептида p53, конъюгированного с флуорофором, но не его нативной формы (Borra et al., 2005; Kaeberlein et al., 2005; Pacholec et al., 2010). Неожиданно оказалось, что в присутствии ламина А ресвератрол действительно увеличивал активность деацетилазы SIRT1 в отношении его нативной мишени, ацетил p53, за счет усиления ассоциации между SIRT1 и ламином A (Liu et al., 2012). Значительная часть общего SIRT1 была обнаружена во фракции ядерного матрикса в клетках дикого типа, тогда как диссоциация SIRT1 из этой фракции наблюдалась в клетках, эктопически экспрессирующих пре-ламин A или прогерин. Таким образом, прогерин, по-видимому, нарушает правильную локализацию SIRT1. В мышиной модели HGPS Zmpste24 — / — добавка ресвератрола облегчила прогероидные функции и увеличила продолжительность жизни. Интересно, что ламин А физически взаимодействует с другим связанным со старением сиртуином — SIRT6 (Ghosh et al., 2015). Это взаимодействие усиливает активность SIRT6 по деацетилированию гистона h4 в отношении h4K9ac и h4K56ac. Прогерин также связывается с SIRT6, но не влияет на его h4K9ac- и h4K56ac-специфическую активность. SIRT6, как известно, играет важную роль в репарации ДНК, рекрутируясь в локусы ДНК с двухцепочечными разрывами, где он увеличивает активность PARP1 посредством его моно-ADP-рибозилирования (Mao et al., 2011). Рекрутирование активации SIRT6 и PARP1, по-видимому, зависит от ламина А и ингибируется прогерином (Ghosh et al., 2015). Вероятно, эти эффекты прогерина ответственны за значительную часть нестабильности генома в HGPS.
Нарушение организации хроматина
Прогрессирующая потеря h4K27me3-зависимой инактивации Х хромосомы (Xi) с пассажами наблюдалась в культурах фибробластов HGPS от пациентки, но не в сопоставимых по возрасту контрольных культурах фибробластов (Shumaker et al., 2006). Потеря h4K27me3 казалась самым ранним событием, за которым последовала потеря Xi и появление аномалий в структуре ядра.Значительная потеря h4K27me3-специфической мРНК гистон-метилазы EZh3 также была обнаружена в фибробластах HGPS, но не в их нормальных аналогах. Никакой потери Xi-ассоциированной РНК XIST не наблюдали в HGPS и контрольных фибробластах. В линии клеток человека дикого типа временная экспрессия прогерина привела к быстрой потере h4K27me3, ассоциированной с Xi и ядерной пластинкой, но не было обнаружено потери Xi-ассоциированной РНК XIST . Таким образом, экспрессия эндогенного прогерина в клетках HGPS и экзогенного прогерина в нормальных клетках приводит к сходной потере эпигенетических меток h4K27me3 из факультативного гетерохроматина, вероятно, объясняя его деконденсацию.Сходная потеря др. Гетерохроматин-специфической метки метилирования гистонов h4K9me3 наблюдалась в клетках HGPS на более поздних пассажах, а также в клетках дикого типа, экспрессирующих экзогенный прогерин (Shumaker et al., 2006). В клетках дикого типа и фибробластах HGPS на ранних этапах пассажа метки h4K9me3 в основном локализованы совместно с гетерохроматиновым белком HP1α. В клетках HGPS на более поздних пассажах происходит потеря как h4K9me3, так и HP1α, а также снижение экспрессии генов, кодирующих h4K9me3-специфические гистоновые метилазы SUV39h2 и SUV39h3.Неожиданно было обнаружено, что другой маркер гетерохроматина, h5K20me3, активируется как в фибробластах HGPS, так и в клетках дикого типа, экспрессирующих экзогенный прогерин.
В контрольных фибробластах кожи человека широкие участки h4K27me3 были обнаружены в бедных генами областях, лишенных CpG-островков (CGI), тогда как более локализованное обогащение h4K27me3 наблюдалось на специфических промоторах, содержащих CGI (McCord et al., 2013). Уменьшение или даже потеря больших участков h4K27me3 в бедных генами областях часто наблюдали в фибробластах HGPS по сравнению с нормальными клетками.Эта особенность была значимой в масштабе всего генома и коррелировала с более низким уровнем мРНК, кодирующей основную h4K27me3-специфическую метилтрансферазу EZh3. Помимо этих широких изменений, наблюдалось локальное повышение или снижение уровня h4K27me3 на специфических промоторах, содержащих CGI. В глобальном масштабе среди генов, которые изменяли экспрессию по крайней мере в четыре раза между HGPS и контрольными фибробластами, гены с пониженной регуляцией преимущественно получали h4K27me3, тогда как гены с повышенной регуляцией преимущественно теряли h4K27me3, что согласуется с широко известным ингибирующим действием h4K27me3 на экспрессию генов.Корреляция наблюдалась между потерей широких пятен h4K27me3 в бедных генами регионах и потерей их ассоциации с ламином A / C, предпочтительно на ядерной периферии. Было высказано предположение, что хроматин, обогащенный h4K27me3, физически связан с ядерной пластиной. Это может объяснить возрастающую потерю периферического гетерохроматина в клетках HGPS. В нормальных клетках бедные генами неактивные области хроматина, как правило, располагаются вблизи ядерной периферии, в то время как активные, богатые генами области находятся внутри ядра.Hi-C анализ фибробластов HGPS позднего пассажа показал почти полную потерю пространственного разделения между открытыми (активными) и закрытыми (неактивными) компартментами хроматина. Глобальная потеря хромосомных компартментов, по-видимому, катастрофически происходит во всем геноме, когда клетки HGPS приближаются к стадии преждевременного старения. Некоторые изменения компартмента также наблюдались в нормальных стареющих клетках, но эти изменения никогда не были такими резкими, как в клетках HGPS. Кроме того, определенные области генома изменили идентичность своих компартментов в HGPS по сравнению с нормальными клетками, и эти изменения коррелировали с изменениями в ассоциации h4K27me3 и ламина A / C.Было обнаружено, что области, которые изменились с открытого отсека в нормальных клетках на закрытый отсек в клетках HGPS, получили связывание h4K27me3 и ламина, в то время как те, которые изменились с закрытого на открытый отсек, потеряли h4K27me3 и связывание с ламином. Описанные данные хорошо согласуются с популярным представлением о прогрессирующем разрушении репрессивной структуры хроматина, которое инициирует ложные транскрипционные события и способствует старению за счет потери ингибирующих эпигенетических меток (Ashapkin et al., 2018).
Одним из механизмов, который может объяснить нарушение структуры хроматина и ускоренное старение клеток HGPS, является измененная локализация белков-ридеров гистона h4K4me3 семейства ING.Известно, что эти белки способствуют восстановлению ДНК за счет быстрых изменений в экспрессии генов как части клеточного ответа на генотоксические воздействия (Soliman and Riabowol, 2007). Было показано, что ламин A специфически связывает белок ING1, таким образом играя важную роль в его ядерном нацеливании (Han et al., 2008). Прогерин не связывает ING1. Эти представления могут объяснить измененное субклеточное распределение ING1 в клетках HGPS. Известно, что ING1 играет роль в активном h4K4me3-направленном деметилировании ДНК (Schäfer et al., 2013). Таким образом, индуцированные прогерином изменения в распределении ING1 могут глобально влиять на метилирование ДНК. В самом деле, нарушение деметилирования ДНК у мышей с двойным нокаутом Gadd45a / Ing1 , как было показано, ведет к сегментарному фенотипу прогерии (Schäfer et al., 2018).
Метилирование измененной ДНК
Считается, что изменения метилирования ДНК
являются основной движущей силой естественного старения и могут иметь важное значение для ускоренного старения. Множественные различия в метилированных локусах были обнаружены между клетками прогероидных пациентов и их контрольными аналогами (Heyn et al., 2013). При синдроме Вернера (WS) и HGPS была общая часть дифференциально метилированных локусов, но большинство из них были специфичны для одного синдрома. Пациенты с атипичным WS и HGPS, у которых не было ни одной из известных мутаций гена WRN или LMNA , демонстрировали аналогичные изменения метилирования ДНК, хотя и меньшие по сравнению с мутантными пациентами. Индивидуальная вариабельность степени изменений метилирования ДНК была довольно высокой между пациентами с мутантом WRN . Было обнаружено, что метилирование 144 CpG постоянно изменяется как при естественном старении, так и в WS.Соответствующие гены могут быть вовлечены в естественное и ускоренное старение. В образцах атипичных WS, вызванных мутациями гена LMNA и , было идентифицировано 18480 дифференциально метилированных CpG, из которых 485 метилировались в разной степени во время естественного старения. Локус LOC149837 , кодирующий длинную некодирующую РНК (lncRNA) с неизвестной функцией и группу piRNAs, был по-разному метилирован во всех образцах прогерии. Очевидное предположение состоит в том, что его аномальное гипометилирование может изменять экспрессию этих ncRNAs, тем самым внося вклад в прогероидный фенотип.
Особо следует отметить, что использование часов метилирования пан-тканевой ДНК выявило ускорение эпигенетического возраста при сегментарных прогероидных синдромах, таких как синдром Дауна и типичный WS, но не у пациентов с HGPS и атипичным WS (Horvath et al., 2018). Напротив, применение новых часов метилирования ДНК кожи и крови показало, что фибробласты пациентов с HGPS демонстрируют ускоренное эпигенетическое старение, особенно очевидное у детей с HGPS в возрасте до 10 лет. Незначительная тенденция увеличения эпигенетического возраста была обнаружена в случаях атипичного WS, вызванного низким уровнем прогерина.
Как было отмечено выше (см. Раздел «Измененная активность сигнальных путей») добавление витамина D3 к клеткам HGPS снижает выработку прогерина и облегчает большинство функций HGPS. В рандомизированном клиническом исследовании добавление витамина D3 снижало возраст метилирования ДНК у людей с избыточным весом и ожирением с субоптимальным статусом витамина D (Chen et al., 2019). Интересно, что корреляция между возрастом по ДНК и хронологическим возрастом была выше в начале исследования, чем после теста, что свидетельствует о том, что добавка витамина D3 приводила к отклонению эпигенетического возраста от хронологического.Таким образом, активность сигнального пути витамина D, по-видимому, противодействует как преждевременному старению при HGPS, так и естественному старению.
Существуют ли общие эпигенетические механизмы между естественным и ускоренным старением?
Эпигенетическое репрограммирование соматических клеток можно рассматривать как пример полной перезагрузки возраста (Ashapkin et al., 2017, 2018 и ссылки в них). Было показано, что ИПСК, полученные из соматических клеток столетних доноров, очень похожи на эмбриональные стволовые клетки (ЭСК) по большинству маркеров молекулярного возраста, таких как профиль экспрессии генов, длина теломер и другие.Более того, подобно ЭСК, такие ИПСК имеют эпигенетический возраст, близкий к нулю, и могут быть дифференцированы в соматические полностью омоложенные клетки. Известно, что эффективность репрограммирования снижается с возрастом из-за накопления признаков старения, которые препятствуют эффективному репрограммированию (Li et al., 2009; Kim et al., 2010). Эффективность репрограммирования фибробластов HGPS на 15-20 пассажах была в 4 раза ниже по сравнению с контрольными фибробластами (Zhang et al., 2011). Тем не менее, все индивидуальные колонии ИПСК были морфологически неотличимы от ЭСК.ИПСК, происходящие из фибробластов HGPS человека, не экспрессировали прогерин и не обладали специфическими для заболевания особенностями. Однако, когда эти ИПСК дифференцировались в соматические клетки мезенхимальных клонов, такие как фибробласты, VSMC или МСК, наблюдалась повторная экспрессия прогерина, и эти клетки проявляли повышенное повреждение ДНК и ядерные аномалии. Полногеномное исследование метилирования CpG в фибробластах HGPS, контрольных фибробластах, HGPS-iPSC, контрольных iPSC и контрольных ESC человека показало, что созданные линии iPSC были намного ближе друг к другу и ESC, чем две линии фибробластов (Liu et al., 2011). Присутствие прогерина в фибробластах HGPS, по-видимому, приводило к основным эпигенетическим изменениям, которые исчезали в HGPS-iPSC одновременно с подавлением прогерина. Полногеномное профилирование мРНК также показало, что HGPS-iPSC и контрольные iPSC были тесно связаны друг с другом и ESC и сильно отличались от своих родительских фибробластов. Эти данные продемонстрировали полный сброс эпигенома и профиля экспрессии генов в клетках HGPS после перепрограммирования до плюрипотентности.Мыши модели HGPS LmnaG609G демонстрируют ускоренное проявление многих возрастных особенностей во многих органах и сокращенную продолжительность жизни (Osorio et al., 2011). Кратковременная индуцируемая экспрессия факторов Яманака (Oct4, Sox2, Klf4 и c-Myc — OSKM) в течение 2 или 4 дней в фибробластах, полученных от таких мышей, не приводила к плюрипотентности, но значительно улучшала архитектуру ядерной оболочки и уменьшала фокусы. гистона γh3AX, известного маркера повреждения ДНК, связанного со старением (Ocampo et al., 2016). Кроме того, подавленная экспрессия генов возрастной реакции на стресс в пути p53 ( p16 INK4a , p21 CIP1 , Atf3 и Gadd45B ) и связанных со старением MMP13 и IL6 Наблюдалось генов. Активность SA-β-gal и выработка митохондриальных АФК также были снижены. И последнее, но не менее важное: уровни двух гетерохроматин-специфичных эпигенетических меток, h4K9me3 и h5K20me3, которые изменяются с возрастом, были восстановлены.Важно отметить, что восстановление h4K9me3 происходит раньше, чем уменьшение фокусов γh3AX и улучшение ядерной архитектуры. Таким образом, эпигенетическое ремоделирование может быть активным фактором улучшения фенотипа старения. Поскольку известно, что индукция in vivo OSKM приводит к образованию тератомы и развитию рака, был разработан безопасный протокол циклической индукции OSKM. Интересно, что циклическая индукция OSKM в течение 6 недель у мышей LmnaG609G не изменяла экспрессию ламина A / C или прогерина, но улучшала множественные признаки старения на молекулярном, клеточном, тканевом и организменном уровнях и увеличивала продолжительность жизни.Подобно клеткам LmnaG609G , в клетках позднего пассажа дикого типа кратковременная индукция OSKM уменьшала фокусы гистона γ-h3AX, подавляла экспрессию генов возрастной стрессовой реакции в пути p53 и MMP13 , ассоциированных со старением. и IL6, гены, снижали продукцию митохондриальных АФК и восстанавливали уровни h4K9me3. В совокупности эти результаты предполагают, что циклическая индукция in vivo OSKM может замедлять старение, предотвращая эпигенетические изменения, повышение активности пути клеточного старения и другие молекулярные признаки старения.Таким образом, омоложение клеток без изменения идентичности клеток в принципе возможно. Другая возможность изменения идентичности клеток без омоложения была продемонстрирована путем индуцированного прямого преобразования человеческих фибробластов в нейроны (Mertens et al., 2015). В отличие от нейронов, полученных с помощью промежуточных ИПСК, непосредственно индуцированные нейроны сохраняли эпигенетическую подпись возраста донора. Интересно, что из более чем 200 генов, по-разному экспрессируемых между нейронами, полученными от молодых и старых доноров, только семь перекрываются между фибробластами и нейронами, тогда как 49 перекрываются между индуцированными нейронами и образцами префронтальной коры головного мозга человека.В профилях экспрессии, связанных со старением, только три гена были общими для фибробластов, индуцированных нейронов и мозга, а именно LAMA3 , PCDh20 и RANBP17 . Поскольку специфическая для фибробластов эпигенетическая подпись возраста донора каким-то образом трансформировалась в специфичную для нейронов, логично предположить, что предполагаемый главный регулятор старения был среди этих общих генов. Ген транспортного рецептора RANBP17 , связанного с ядерной порой, значительно снижается с возрастом, что указывает на возможное функциональное нарушение функции ядерной поры.Действительно, индуцированные нейроны от самых старых доноров обнаруживают значительные дефекты компартментализации нуклеоцитоплазмы по сравнению с нейронами среднего возраста и молодыми донорскими нейронами, и степень этих дефектов коррелирует с возрастом донора и подавлением RANBP17 . Кроме того, опосредованное shRNA подавление RANBP17 в молодых фибробластах изменяет экспрессию большинства генов старения фибробластов в том же направлении, что и при естественном старении. Таким образом, подавление экспрессии RANBP17 и может быть причинным фактором клеточного старения.Как и ожидалось, нейроны, полученные из ИПСК, показали существенно обновленные транскриптомы и не обнаруживали никаких нарушений компартментализации ядер-цитоплазмы. Фенотип ускоренного старения в HGPS является следствием нарушенной прогерином структуры ядерной оболочки, что может приводить к утечке ядер и нарушению компартментализации ядер-цитоплазмы. Таким образом, снижение экспрессии RANBP17 и в стареющих клетках может вносить вклад в их связанные со старением фенотипы. Широко распространено мнение, что ИПСК обладают безграничной способностью размножаться и повторно дифференцироваться в полностью омоложенные соматические клетки.Однако было показано, что длительное культивирование ИПСК приводит к метаболическим изменениям, которые препятствуют их нейрональной дифференцировке (Masotti et al., 2014). Молодые ИПСК не экспрессируют ген LMNA , тогда как старые ИПСК экспрессируют высокие уровни мРНК LMNA и имеют утолщенную ядерную пластинку, содержащую большое количество ламина A / C, очень похоже на HGPS и стареющие клетки (Petrini et al. ., 2017). Эти особенности старых ИПСК были связаны с ядерными аномалиями, напоминающими таковые у клеток HGPS.Более того, повышенная экспрессия пре-ламина А и прогерина наблюдалась в старых ИПСК, а также гиперактивация NFκB и подавление экспрессии SIRT7, которые, как известно, регулируют митохондриальный биогенез. Таким образом, при длительном размножении ИПСК накапливают возрастные изменения, характерные для нормального старения и прогероидных синдромов.
Гомозиготные WRN-нулевые линии hESC экспрессировали маркеры плюрипотентности и были способны дифференцироваться во все три клетки зародышевого листка (Zhang et al., 2015). WRN-нулевые MSC, которые были получены из WRN-нулевых ESC, экспрессировали MSC-специфические маркеры клеточной поверхности и могли быть дифференцированы в остеобласты, хондроциты и адипоциты.Эти WRN-дефицитные МСК демонстрируют основные признаки ускоренного старения, такие как снижение пролиферативного потенциала, увеличение маркеров SA-β-gal, повышенная экспрессия генов маркеров стареющего p16 Ink4a и p21 Waf1 , и активация SASP. Маркеры повреждения ДНК 53BP1, γh3AX и фосфорилированные субстраты ATM / ATR были увеличены. Более того, эти клетки обнаруживают подавление ассоциированных с гетерохроматином белков LAP2b и LBR внутренней ядерной мембраны и уменьшение периферического гетерохроматина.Наблюдалось значительное подавление конститутивной метки гетерохроматина h4K9me3, тогда как h4K27me3 обнаруживало лишь небольшое подавление. Существенных изменений в 5mC и эухроматиновой метке h4K4me3 не обнаружено. Около 70 h4K9me3-обогащенных «гор» (> 20 т.п.н. последовательных пиков h4K9me3) были идентифицированы в MSC дикого типа, 28 (38%) из них отсутствовали в WRN-дефицитных MSC. Большинство этих утраченных гор h4K9me3 располагались в субтеломерных или субцентромерных регионах. Секвенирование РНК выявило 1047 генов, которые показали дифференциальную экспрессию между MSC с дефицитом WRN и MSC дикого типа.Наиболее явно подавляемые гены кодируют белки, упаковывающие центромеры, и компоненты ядерной мембраны. Анализ иммунопреципитации показал, что WRN является частью комплекса, содержащего h4K9me3-специфическую метилтрансферазу SUV39h2, связанный с гетерохроматином белок HP1α и связанный с ламином белок LAP2b, что позволяет предположить, что WRN участвует в стабилизации гетерохроматина. Сравнение уровней гетерохроматиновых меток между МСК дикого типа, полученными от молодых (от 7 до 26 лет) и старых (от 58 до 72 лет) индивидов, показало значительное подавление белка WRN и меток h4K9me3, HP1α, SUV39h2 и LAP2b в МСК получены от старых людей.Таким образом, специфические изменения гетерохроматина лежат в основе как ускоренного, так и естественного старения МСК.
Заключение
Даже небольших нарушений в экспрессии ламина А может быть достаточно, чтобы вызвать ядерные дефекты, влияющие на старение клеток. Вероятно, такие нарушения играют роль в естественном старении клеток. Небольшое количество прогерин-положительных клеток присутствует в первичных культурах фибробластов, полученных из кожи нормальных доноров в преклонном возрасте. Эти клетки обнаруживают HGPS-подобные дефекты в морфологии ядра, такие как дольки ядерной оболочки и инвагинации, двуядерные клетки и пониженные уровни гетерохроматиновых меток h4K9me3 и HP1.Метка фосфорилирования гистона h3AX в очагах повреждения ДНК значительно повышена в фибробластах пожилых людей и пациентов с HGPS по сравнению с молодыми донорами. В клетках молодого человека ламин A / C присутствует как на периферии ядра, так и во всей нуклеоплазме, тогда как в клетках пожилого человека и в клетках HGPS он обнаруживается в основном на периферии ядра. Ингибирование продукции прогерина в клетках старых доноров, не являющихся HGPS, in vivo увеличивает их пролиферативную активность, уровни h4K9me3 и HP1 и снижает экспрессию маркеров старения p21, IGFBP3 и GADD45B до уровней молодых донорских клеток.Таким образом, при естественном старении действуют прогерин-зависимые механизмы. Чрезмерная активность этих механизмов в HGPS может быть основной причиной преждевременного старения. Истирание теломер считается одним из основных признаков старения. Нормальные человеческие фибробласты, экспрессирующие прогерин, демонстрируют ускоренную потерю теломер. Вероятно, изменения в организации пластинки могут напрямую влиять на скорость истощения теломер и приводить к ускоренному репликативному старению и прогероидным фенотипам. Интересно, что повреждение теломер в нормальных фибробластах человека активирует выработку прогерина и клеточное старение, предполагая существование порочного круга.Хронологическое старение у нормальных людей и преждевременное старение у пациентов с HGPS опосредовано аналогичными изменениями активности сигнальных путей, включая подавление репарации ДНК и организации хроматина, а также активацию ERK, mTOR, GH-IGF1, MAPK, TGFβ и митохондриальная дисфункция.
Экспрессия ламина А / прогерина, по-видимому, тесно связана с механизмом интерференции РНК, метилированием ДНК и гистонов и структурой хроматина. Связь с эпигенетической регуляцией ключевых клеточных функций может объяснить многие последствия мутаций гена LMNA у людей.Notch-зависимая дерегуляция дифференцировки MSC и детерминации клеточной судьбы может объяснять множественные фенотипы дисфункции мезенхимальных тканей при HGPS. Постепенное накопление прогерина с естественным старением может исказить активность пути Notch, что приведет к аналогичным нарушениям в более легкой форме. Накопление прогерина на периферии ядра активирует сигнальный путь NFκB, возможно, способствуя системному воспалению, присущему старению (воспаление). NFκB может быть основным фактором ускоренного старения, поскольку его ингибирование значительно увеличивает продолжительность жизни у мышей с моделью HGPS.
Связанный с долголетием фактор транскрипции NRF2 является одной из основных мишеней прогерина, которые определяют его эффекты ускорения старения. Более 200 генов-мишеней NRF2 значительно подавлены в HGPS. Благодаря своей повышенной аффинности к NRF2 прогерин может нарушать активность пути регуляции антиоксиданта NRF2-ARE за счет секвестрации NRF2 от его генов-мишеней.
Прогерин, по-видимому, нарушает правильную локализацию членов семейства сиртуинов SIRT1 и SIRT6, которые, как известно, играют важную роль в старении млекопитающих.Вероятно, эти эффекты ответственны за значительную часть нестабильности генома в HGPS.
Глобальная потеря хромосомных компартментов катастрофически происходит во всем геноме, когда клетки HGPS приближаются к стадии преждевременного старения. Подобные изменения происходят в нормальных стареющих клетках, хотя эти изменения никогда не бывают такими резкими, как в клетках HGPS. Эти данные хорошо согласуются с популярным представлением о том, что прогрессирующее разрушение репрессивной структуры хроматина происходит в естественно стареющих клетках и инициирует ложные транскрипционные события через потерю ингибирующих эпигенетических меток.ING1 играет роль в активном h4K4me3-направленном деметилировании ДНК. Прогерин изменяет свое распределение и, таким образом, может глобально влиять на метилирование ДНК. В самом деле, нарушение деметилирования ДНК у мышей с двойным нокаутом Gadd45a / Ing1 , как было показано, ведет к сегментарному фенотипу прогерии.
Считается, что изменения метилирования ДНК
являются основным признаком естественного старения и могут иметь важное значение при преждевременном старении (рис. 1; Lopez-Otın et al., 2013). Часы метилирования ДНК выявили ускорение старения при HGPS и других сегментарных прогероидных синдромах.Витамин D3 снижает выработку прогерина и облегчает большинство функций HGPS. Он также снижает эпигенетический возраст у людей с избыточным весом и ожирением с субоптимальным статусом витамина D. Таким образом, активность сигнального пути витамина D, по-видимому, противодействует как преждевременному старению при HGPS, так и естественному старению.
Рис. 1. Девять признаков старения (Lopez-Otın et al., 2013) способствуют как естественному, так и преждевременному старению. Эпигенетические изменения играют ключевую роль в воздействии на все признаки старения и на них влияют.Взаимные влияния между другими признаками, которые, вероятно, существуют, не показаны.
Cyclic in vivo Индукция OSKM может замедлить ускоренное и естественное старение, противодействуя эпигенетическим изменениям, усилению клеточного старения и другим молекулярным признакам старения.
Связанный с ядерной порами транспортный рецептор RANBP17 значительно снижается с возрастом, что указывает на возможное функциональное нарушение функции ядерной поры и значительные дефекты ядерно-цитоплазматической компартментализации.Фенотип ускоренного старения в HGPS является следствием нарушенной прогерином структуры ядерной оболочки, что может приводить к утечке ядер и нарушению компартментализации ядер-цитоплазмы.
Молодые ИПСК не экспрессируют ген LMNA , тогда как старые ИПСК, как было обнаружено, экспрессируют высокие уровни мРНК LMNA и имеют утолщенную ядерную пластинку, содержащую большое количество ламина A / C, очень похоже на HGPS и стареющие клетки. Эти особенности старых ИПСК были связаны с ядерными аномалиями, напоминающими таковые у клеток HGPS.Более того, повышенная экспрессия пре-ламина А и прогерина наблюдалась в старых ИПСК, а также гиперактивация NFκB и подавление экспрессии SIRT7, которые, как известно, регулируют митохондриальный биогенез. Таким образом, при длительном размножении ИПСК накапливают возрастные изменения, характерные для естественного старения и прогероидных синдромов.
Сердечно-сосудистые заболевания являются одними из наиболее типичных черт как естественного старения, так и HGPS. Большое количество прогерина накапливается в стенках артерий пациентов с HGPS.Повышенная выработка прогерина с возрастом также происходит в коронарной артерии нормальных людей. Специфические изменения VSMC, такие как кальцификация, накопление липидов, фиброз и истощение VSMC, наблюдаются как при индуцированном прогерином, так и при естественном старении.
В совокупности описанные выше данные показывают, что большинство признаков естественного старения обнаруживаются в HGPS. Хотя основная причина HGPS совершенно иная, вторичные причины и их последствия на уровне организма очень похожи на естественное старение (рис. 2).Таким образом, HGPS действительно представляет собой истинное ускоренное старение, а не просто видимость старения.
Рисунок 2. Эффекты прогерина, общие для HGPS и естественного старения.
Несмотря на все, что известно о HGPS и действии прогерина, еще предстоит ответить на многие вопросы (Gonzalo et al., 2017). Одна из самых загадочных особенностей HGPS заключается в том, что некоторые органы, например печень и мозг, кажутся вполне здоровыми. Некоторые характеристики, обычно наблюдаемые при нормальном старении, такие как прогрессирующая умственная отсталость, отсутствуют в HGPS.У HGPS-подобных мышей, экспрессирующих прогерин, наблюдается ускоренное старение, но функции мозга, по-видимому, в основном не затрагиваются (Yang et al., 2005; Jung et al., 2012). Одно из возможных объяснений — очень низкий уровень экспрессии гена LMNA в клетках мозга из-за посттранскрипционной подавления с помощью специфической для мозга miRNA, miR-9 (Jung et al., 2012). Ламин А не продуцируется в индуцированных плюрипотентных стволовых клетках (ИПСК), полученных из фибробластов пациентов с HGPS и контрольных субъектов (Nissan et al., 2012). Индукция экспрессии LMNA происходит во многих клетках, происходящих от ИПСК, таких как кератиноциты, МСК и некоторых других. В клетках, полученных из HGPS-iPSCs, наблюдались типичные последствия экспрессии прогерина, такие как ускоренное старение, нарушение хроматина и нарушения морфологии ядра. Не наблюдали экспрессии гена LMNA в нейронах, происходящих от ИПСК, в хорошем согласии с экспрессией miR-9. Экзогенный miR-9 подавлял продукцию ламина А и прогерина и уменьшал аномалии ядерной морфологии в MSC HGPS.Таким образом, клетки мозга, по-видимому, защищены от индуцированного прогерином повреждения у пациентов с HGPS с помощью специфической для мозга miRNA. Принудительная экспрессия человеческого прогерина в нейронах гиппокампа мышей приводит к HGPS-подобным аномалиям в структуре ядра (Baek et al., 2015). Тем не менее, структура хроматина и экспрессия генов остались неизменными (только 5 генов из 16572 исследованных показали двукратные или более сильные изменения в уровнях экспрессии), и никаких изменений в функционировании мозга обнаружено не было. Очевидно, что существуют и другие средства устойчивости мозга к прогерину, помимо его низкой экспрессии.
Учитывая широко известную связь между раком и старением, было довольно неожиданным открытием, что у прогероидных пациентов рак не развивается. Было показано, что прогерин может ингибировать трансформацию клеток путем изменения распределения общего регулятора транскрипции BRD4 на хроматине и активации клеточных путей, защищающих опухоль (Fernandez et al., 2014).
Другой интригующий вопрос: почему криптический сайт сплайсинга прогерина вообще существует? Из-за вырожденности генетического кода соответствующая последовательность экзона 11 LMNA могла потерять какое-либо сходство с консенсусной 5’SS без изменений кодируемой аминокислотной последовательности.Тем не менее, сайт сплайсинга, ответственный за продукцию прогерина, по-видимому, высококонсервативен у всех плацентарных млекопитающих (Lopez-Mejia et al., 2014). Одним из возможных объяснений может быть то, что существование прогеринов имеет некоторое эволюционное преимущество. Действительно, Lmna мышей, у которых не может происходить прогерин-специфический сплайсинг, показали увеличенную продолжительность жизни, но, как это ни парадоксально, снижение энергетического метаболизма, увеличение веса и меньшее количество митохондрий, тогда как HGPS-подобные мыши имели короткую продолжительность жизни, но повышенный митохондриальный биогенез (Lopez -Mejia et al., 2014). Более того, у старых мышей, свободных от прогерина, значительно увеличилась частота лимфоидных опухолей в брюшной полости по сравнению с мышами дикого типа. Таким образом, прогерин, по-видимому, участвует как в защите опухоли, так и в метаболической адаптации.
Надеюсь, что по мере продолжения исследования ответ на эти вопросы будет найден, и новые загадки старения будут раскрыты. Как бы то ни было, HGPS предлагает уникальную модель для выяснения роли прогерина в старении клеток.
Множественные эпигенетические изменения являются общими для преждевременного старения в HGPS и хронологического старения у нормальных людей.Недавние исследования показали, что эпигенетические системы могут играть активную роль как движущие силы обеих форм старения. Способность эпигенетических систем влиять на все другие факторы старения через изменения в экспрессии генов ставит их в уникальное положение. Можно предположить, что эти системы переводят эффекты различных внутренних и внешних факторов в универсальные молекулярные признаки, в основном общие для естественных и ускоренных форм старения (рис. 1). В отличие от других видов повреждений, возрастные эпигенетические изменения могут быть полностью или частично обращены в «молодое» состояние.Кроме того, такое эпигенетическое ремоделирование к более молодому состоянию могло бы в значительной степени улучшить эффекты других знаменателей старения. Следовательно, целенаправленное вмешательство в эпигенетические механизмы представляется наиболее многообещающей и эффективной стратегией для разработки методов лечения как естественного, так и ускоренного старения.
Авторские взносы
Все перечисленные авторы внесли существенный, прямой и интеллектуальный вклад в работу и одобрили ее к публикации.
Финансирование
Работа частично поддержана Российским научным фондом (грант ИК № 17-15-01290).
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Список литературы
Адлер, А.С., Синха, С., Кавахара, Т.Л.А., Чжан, Дж. Й., Сегал, Э. и Чанг, Х. Ю. (2007). Карта модуля мотива показывает усиление старения за счет постоянной активности NF-κB. Genes Dev. 21, 3244–3257. DOI: 10.1101 / gad.1588507
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Алипер, А.М., Чока А.Б., Буздин А., Жетка Т., Румянцев С., Москалев А. и др. (2015). Дрейф активации сигнального пути во время старения: фибробласты синдрома прогерии Хатчинсона-Гилфорда сопоставимы с нормальными клетками среднего и пожилого возраста. Aging (Олбани, штат Нью-Йорк) 7, 26–37. DOI: 10.18632 / старение.100717
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Аранчио В., Джордано К. и Пиццоланти Г. (2013). Анализ цеРНК гена LMNA с акцентом на синдром прогерии Хатчинсона-Гилфорда. J. Clin. Биоинформа. 3: 2. DOI: 10.1186 / 2043-9113-3-2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ашапкин В. В., Кутуева Л. И., Ванюшин Б. Ф. (2018). «Эпигенетика старения: накопление ошибок и многое другое», в Aging. Изучение сложного явления , изд. С. И. Ахмад (Нью-Йорк, Нью-Йорк: CRC Press), 175–205.
Бэк, Дж. Х., Маккенна, Т., и Эрикссон, М. (2013). «Синдром прогерии Хатчинсона-Гилфорда», в Genetic Disorders , Vol.1, изд. М. Пуйу (Риека: InTech), 65–87.
Google Scholar
Baek, J.-H., Schmidt, E., Viceconte, N., Strandgren, C., Pernold, K., Richard, T.J.C, et al. (2015). Экспрессия прогерина в стареющем мозге мышей выявляет структурные ядерные аномалии без обнаруживаемых значительных изменений в экспрессии генов, стволовых клетках гиппокампа или поведении. Гм. Мол. Genet. 24, 1305–1321. DOI: 10.1093 / hmg / ddu541
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Бенсон, Э.К., Ли, С. В., Ааронсон, С. А. (2010). Роль индуцированной прогерином дисфункции теломер в преждевременном клеточном старении HGPS. J. Cell Sci. 123, 2605–2612. DOI: 10.1242 / jcs.067306
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Bouillon, R., Carmeliet, G., Verlinden, L., van Etten, E., Verstuyf, A., Luderer, H. F., et al. (2008). Витамин D и здоровье человека: уроки мышей с нулевым рецептором витамина D. Endocr. Ред. 29, 726–776. DOI: 10.1210 / er.2008-0004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Бриджер, Дж. М., и Килл, И. Р. (2004). Старение фибробластов синдрома прогерии Хатчинсона-Гилфорда характеризуется гиперпролиферацией и усилением апоптоза. Exp. Геронтол. 39, 717–724. DOI: 10.1016 / j.exger.2004.02.002
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Канделарио, Дж., Судхакар, С., Наварро, С., Редди, С., и Комай, Л. (2008). Нарушение метаболизма ламина А дикого типа приводит к прогероидному фенотипу. Ячейка старения 7, 355–367. DOI: 10.1111 / j.1474-9726.2008.00393.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Cao, K., Blair, C.D., Faddah, D.A., Kieckhaefer, J.E., Olive, M., Erdos, M.R., et al. (2011). Дисфункция прогерина и теломер совместно запускает клеточное старение в нормальных фибробластах человека. J. Clin. Вкладывать деньги. 121, 2833–2844. DOI: 10.1172 / jci43578
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Чен, Л., Донг, Ю., Бхагатвала, Дж., Раед, А., Хуанг, Ю., и Чжу, Х. (2019). Влияние добавок витамина D3 на эпигенетическое старение афроамериканцев с избыточным весом и ожирением с субоптимальным статусом витамина D: рандомизированное клиническое испытание. J. Gerontol. Биол. Sci. Med. Sci. 74, 91–98. DOI: 10.1093 / gerona / gly223
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Csoka, A. B., English, S. B., Simkevich, C. P., Ginzinger, D. G., Butte, A.J., Schatten, G.P., et al.(2004). Профилирование экспрессии в масштабе генома синдрома прогерии Хатчинсона-Гилфорда выявляет широко распространенную неправильную регуляцию транскрипции, ведущую к мезодермальным / мезенхимальным дефектам и ускоренному атеросклерозу. Ячейка старения 3, 235–243. DOI: 10.1111 / j.1474-9728.2004.00105.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
De Sandre-Giovannoli, A., Bernard, R., Cau, P., Navarro, C., Amiel, J., Boccaccio, I., et al. (2003). Усечение ламина А при прогерии Хатчинсона-Гилфорда. Наука 300: 2055. DOI: 10.1126 / science.1084125
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Деккер, М. Л., Чавес, Э., Вулто, И., и Лансдорп, П. М. (2009). Длина теломер при синдроме прогерии Хатчинсона – Гилфорда. мех. Aging Dev. 130, 377–383. DOI: 10.1016 / j.mad.2009.03.001
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Эрикссон, М., Браун, В. Т., Гордон, Л. Б., Глинн, М. В., Зингерк, Дж., Скотт, Л., и другие. (2003). Рецидивирующие точечные мутации de novo в ламине А вызывают синдром прогерии Хатчинсона-Гилфорда. Природа 423, 293–298. DOI: 10.1038 / nature01629
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Фернандес, П., Скаффиди, П., Маркерт, Э., Ли, Дж. Х., Рэйн, С. и Мистели, Т. (2014). Устойчивость к трансформации при нарушении преждевременного старения определяет опухолевую функцию BRD4. Cell Rep. 9, 248–260. DOI: 10.1016 / j.celrep.2014.08.069
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Franceschi, C., и Campisi, J. (2014). Хроническое воспаление (воспаление) и его потенциальный вклад в возрастные заболевания. J. Gerontol. Биол. Sci. Med. Sci. 69, S4 – S9. DOI: 10.1093 / gerona / glu057
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Гош, С., Лю, Б., Ван, Ю., Хао, К., и Чжоу, З. (2015). Ламин A является эндогенным активатором SIRT6 и способствует опосредованной SIRT6 репарации ДНК. Cell Rep. 13, 1396–1406. DOI: 10.1016 / j.celrep.2015.10.006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Голдман, Р. Д., Шумакер, Д. К., Эрдос, М. Р., Эрикссон, М., Голдман, А. Э., Гордон, Л. Б., и др. (2004). Накопление мутантного ламина А вызывает прогрессивные изменения ядерной архитектуры при синдроме прогерии Хатчинсона-Гилфорда. Proc. Natl. Акад. Sci. США, 101, 8963–8968. DOI: 10.1073 / pnas.0402943101
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Гонсало, С., Kreienkamp, R., and Askjaer, P. (2017). Синдром прогерии Хатчинсона-Гилфорда: заболевание преждевременного старения, вызванное мутациями гена LMNA. Aging Res. Ред. 33, 18–29. DOI: 10.1016 / j.arr.2016.06.007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Хан, X., Фен, X., Раттнер, Дж. Б., Смит, Х., Бозе, П., Судзуки, К. и др. (2008). Привязка ламином А стабилизирует и нацеливает супрессор опухоли ING1. Nat. Cell Biol. 10, 1333–1340. DOI: 10.1038 / ncb1792
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Хейн, Х., Моран, С. и Эстеллер, М. (2013). Аберрантные профили метилирования ДНК при нарушениях преждевременного старения. Прогерия Хатчинсона-Гилфорда и синдром Вернера. Эпигенетика 8, 28–33. DOI: 10.4161 / epi.23366
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Хорват, С., Осима, Дж., Мартин, Г. М., Лу, А. Т., Квач, А., Коэн, Х. и др. (2018). Эпигенетические часы для кожи и клеток крови применительно к синдрому Хатчинсона-Гилфорда Прогерия и исследованиям ex vivo. Aging (Олбани, штат Нью-Йорк) 10, 1758–1775. DOI: 10.18632 / старение.101508
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Хуанг С., Рискес Р. А., Мартин Г. М., Рабинович П. С. и Осима Дж. (2008). Ускоренное укорочение теломер и репликативное старение в человеческих фибробластах, сверхэкспрессирующих мутантный ламин А и ламин A дикого типа. Exp. Cell Res. 314, 82–91. DOI: 10.1016 / j.yexcr.2007.08.004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Юнг, Х.J., Coffinier, C., Choe, Y., Beigneux, A.P., Davies, B.S.J., Yang, S.H., et al. (2012). Регулирование преламина А, но не ламина С, с помощью miR-9, специфической для мозга микроРНК. Proc. Natl. Акад. Sci. США 109, 423–431. DOI: 10.1073 / pnas.1111780109
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Kaeberlein, M., McDonagh, T., Heltweg, B., Hixon, J., Westman, E.A., Caldwell, S.D., et al. (2005). Субстрат-специфическая активация сиртуинов ресвератролом. J. Biol. Chem. 280, 17038–17045. DOI: 10.1074 / jbc.M500655200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Кейсала, Т., Минасян, А., Лу, Ю. Р., Зоу, Дж., Калуэфф, А. В., Пюйкко, И. и др. (2009). Преждевременное старение мышей с мутантными рецепторами витамина D. J. Steroid Biochem. Мол. Биол. 115, 91–97. DOI: 10.1016 / j.jsbmb.2009.03.007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ким, К., Дои, А., Вэнь, Б., Ng, K., Zhao, R., Cahan, P., et al. (2010). Эпигенетическая память в индуцированных плюрипотентных стволовых клетках. Природа 467, 285–290. DOI: 10.1038 / nature09342
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Kreienkamp, R., Croke, M., Neumann, M. A., Bedia-Diaz, G., Graziano, S., Dusso, A., et al. (2016). Передача сигналов рецептора витамина D улучшает клеточные фенотипы синдрома прогерии Хатчинсона-Гилфорда. Oncotarget 7, 30018–30031. DOI: 10.18632 / oncotarget.9065
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Kreienkamp, R., Graziano, S., Coll-Bonfill, N., Bedia-Diaz, G., Cybulla, E., Vindigni, A., et al. (2018). Внутренний интерфероноподобный ответ клетки связывает стресс репликации с клеточным старением, вызванным прогерином. Cell Rep. 22, 2006–2015. DOI: 10.1016 / j.celrep.2018.01.090
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Куббен, Н., Чжан, В., Ван, Л., Восс, Т. К., Янг, Дж., Ку, Дж. И др. (2016). Подавление антиоксидантного пути NRF2 при преждевременном старении. Cell 165, 1361–1374. DOI: 10.1016 / j.cell.2016.05.017
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Льюис, К. Н., Вейсон, Э., Эдри, Ю. Х., Кристан, Д. М., Нево, Е., и Баффенштейн, Р. (2015). Регулирование передачи сигналов Nrf2 и продолжительность жизни у долгоживущих в природе грызунов. Proc. Natl. Акад. Sci. США 112, 3722–3727. DOI: 10.1073 / pnas.1417566112
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ли, Х., Collado, M., Villasante, A., Strati, K., Ortega, S., Cañamero, M., et al. (2009). Локус Ink4 / Arf является препятствием для перепрограммирования iPS. Природа 460, 1136–1139. DOI: 10.1038 / nature08290
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лю Б., Гош С., Ян X., Чжэн Х., Лю X., Ван З. и др. (2012). Ресвератрол восстанавливает SIRT1-зависимое снижение количества взрослых стволовых клеток и смягчает прогероидные особенности прогерии, вызванной ламинопатией. Cell Metab. 16, 738–750. DOI: 10.1016 / j.cmet.2012.11.007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лю Б., Ван, Дж., Чан, К. М., Цзя, В. М., Дэн, В., Гуань, X., et al. (2005). Геномная нестабильность при преждевременном старении на основе ламинопатии. Nat. Med. 11, 780–785. DOI: 10,1038 / нм1266
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лю, Г.-Х., Бархо, Б.З., Руис, С., Дип, Д., Ку, Дж., Ян, С.-Л. и др. (2011). Резюме преждевременного старения с помощью ИПСК от синдрома прогерии Хатчинсона-Гилфорда. Природа 472, 221–225. DOI: 10.1038 / nature09879
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Lopez-Mejia, I.C, de Toledo, M., Chavey, C., Lapasset, L., Cavelier, P., Lopez-Herrera, C., et al. (2014). Антагонистические функции изоформ LMNA в расходе энергии и продолжительности жизни. EMBO Rep. 15, 529–539. DOI: 10.1002 / embr.201338126
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лопес-Мехиа, И. К., Вотро, В., де Толедо, М., Бем-Ансман, И., Буржуа, К. Ф., Наварро, К. Л. и др. (2011). Консервативный механизм сплайсинга гена LMNA контролирует преждевременное старение. Гм. Мол. Genet. 20, 4540–4555. DOI: 10.1093 / hmg / ddr385
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лопес-Отин, К., Бласко, М. А., Партридж, Л., Серрано, М., и Кремер, Г. (2013). Признаки старения. Ячейка 153, 1194–1217. DOI: 10.1016 / j.cell.2013.05.039
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ло, Ю.-B., Mitrpant, C., Johnsen, R.D., Fabian, V.A., Fletcher, S., Mastaglia, F. L., et al. (2013). Исследование возрастных изменений сплайсинга LMNA и экспрессии прогерина в скелетных мышцах человека. Внутр. J. Clin. Exp. Патол. 6, 2778–2786.
Google Scholar
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., et al. (2011). SIRT6 способствует восстановлению ДНК при стрессе, активируя PARP1. Наука 332, 1443–1446. DOI: 10.1126 / наука.1202723
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Масотти, А., Целлуцци, А., Петрини, С., Бертини, Э., Занни, Г., и Компаньуччи, К. (2014). Старые ИПСК имеют необычный митохондриальный вид и не могут пройти нейрогенез in vitro и . Aging (Олбани, штат Нью-Йорк) 6, 1094–1108. DOI: 10.18632 / старение.100708
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Макклинток, Д., Ратнер, Д., Локуге, М., Оуэнс, Д. М., Гордон, Л. Б., Коллинз, Ф. С. и др. (2007). Мутантная форма ламина А, вызывающая прогерию Хатчинсона-Гилфорда, является биомаркером клеточного старения в коже человека. PLoS One 2: e1269. DOI: 10.1371 / journal.pone.0001269
PubMed Аннотация | CrossRef Полный текст | Google Scholar
МакКорд, Р. П., Назарио-Тул, А., Чжан, Х., Чайнс, П. С., Чжан, Ю., Эрдос, М. Р. и др. (2013). Коррелированные изменения в организации генома, метилировании гистонов и взаимодействиях ДНК-ламин A / C при синдроме прогерии Хатчинсона-Гилфорда. Genome Res. 23, 260–269. DOI: 10.1101 / gr.138032.112
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Мертенс, Дж., Пакуола, А. С. М., Ку, М., Хэтч, Э., Бонке, Л., Ладжеварди, С. и др. (2015). Непосредственно перепрограммированные нейроны человека сохраняют связанные со старением транскриптомные сигнатуры и выявляют связанные с возрастом ядерно-цитоплазматические дефекты. Стволовые клетки клеток 17, 705–718. DOI: 10.1016 / j.stem.2015.09.001
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Месснер, М., Ghadge, S.K., Goetsch, V., Wimmer, A., Dorler, J., Polzl, G., et al. (2018). Повышение регуляции связанного со старением варианта сплайсинга LMNA прогерина при дилатационной кардиомиопатии. PLoS One 13: e0196739. DOI: 10.1371 / journal.pone.0196739
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Nissan, X., Blondel, S., Navarro, C., Maury, Y., Denis, C., Girard, M., et al. (2012). Уникальное сохранение нервных клеток при синдроме прогерии Хатчинсона-Гилфорда связано с экспрессией нейроноспецифической микроРНК miR-9. Cell Rep. 2, 1–9. DOI: 10.1016 / j.celrep.2012.05.015
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Окампо А., Редди П., Мартинес-Редондо П., Платеро-Луенго А., Хатанака Ф., Хишида Т. и др. (2016). Устранение возрастных признаков in vivo путем частичного перепрограммирования. Cell 167, 1719–1733. DOI: 10.1016 / j.cell.2016.11.052
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Олив, М., Хартен, И., Митчелл, Р., Бирс, Дж. К., Джабали, К., Цао, К. и др. (2010). Сердечно-сосудистая патология при прогерии Хатчинсона – Гилфорда: корреляция с сосудистой патологией старения. Артериосклер. Тромб. Васк. Биол. 30, 2301–2309. DOI: 10.1161 / atvbaha.110.209460
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Osorio, F. G., Barcena, C., Soria-Valles, C., Ramsay, A. J., de Carlos, F., Cobo, J., et al. (2012). Дефекты ядерной пластинки вызывают ATM-зависимую активацию NF-κB и связывают ускоренное старение с системной воспалительной реакцией. Genes Dev. 26, 2311–2324. DOI: 10.1101 / gad.197954.112
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Осорио, Ф. Г., Наварро, К. Л., Кадиньянос, Дж., Лопес-Мехиа, И. К., Кирос, П. М., Бартоли, К. и др. (2011). Сплайсинг-направленная терапия на новой мышиной модели ускоренного старения человека. Sci. Пер. Med. 3: 106ra107. DOI: 10.1126 / scitranslmed.3002847
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Пачолек, М., Bleasdale, J. E., Chrunyk, B., Cunningham, D., Flynn, D., Garofalo, R. S., et al. (2010). SRT1720, SRT2183, SRT1460 и ресвератрол не являются прямыми активаторами SIRT1. J. Biol. Chem. 285, 8340–8351. DOI: 10.1074 / jbc.M109.088682
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Петрини С., Борги Р., Д’Ория В., Рестальди Ф., Морено С., Новелли А. и др. (2017). Старые индуцированные плюрипотентные стволовые клетки (ИПСК) как новая клеточная модель для изучения преждевременного старения. Aging (Олбани, штат Нью-Йорк) 9, 1453–1469. DOI: 10.18632 / старение.101248
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Рагнаут, К. Д., Уоррен, Д. Т., Лю, Ю., Макнейр, Р., Тайсич, Т., Фигг, Н. и др. (2010). Преламин А ускоряет старение гладкомышечных клеток и является новым биомаркером сосудистого старения человека. Тираж 121, 2200–2210. DOI: 10.1161 / cycleaha.109.
6
PubMed Аннотация | CrossRef Полный текст
Родригес, С., Коппеде Ф., Сагелиус Х. и Эрикссон М. (2009). Повышенная экспрессия усеченного транскрипта ламина А при синдроме прогерии Хатчинсона-Гилфорда во время клеточного старения. евро. J. Hum. Genet. 17, 928–937. DOI: 10.1038 / ejhg.2008.270
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Шефер А., Карауланов Э., Стапф У., Дёдерлейн Г. и Нирс К. (2013). Ing1 участвует в деметилировании ДНК, направляя Gadd45a на h4K4me3. Genes Dev. 27, 261–273.DOI: 10.1101 / gad.186916.112
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Шефер, А., Меккер, Б., Маллик, М., Вастоло, В., Карауланов, Э., Себастьян, Д. и др. (2018). Нарушение деметилирования ДНК сайтов C / EBP вызывает преждевременное старение. Genes Dev. 32, 742–762. DOI: 10.1101 / gad.311969.118
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Шумакер, Д. К., Дечат, Т., Кольмайер, А., Адам, С. А., Бозовский, М. Р., Эрдош, М.R., et al. (2006). Мутантный ядерный ламин А приводит к прогрессирующим изменениям эпигенетического контроля при преждевременном старении. Proc. Natl. Акад. Sci. США 103, 8703–8708. DOI: 10.1073 / pnas.0602569103
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Vautrot, V., Aigueperse, C., Oillo-Blanloeil, F., Hupont, S., Stevenin, J., Branlant, C., et al. (2016). Повышенная экспрессия белка SRSF5 усиливает продукцию мРНК ламина А в клетках HeLa и фибробластах пациентов с прогерией. Гм. Мутат. 37, 280–291. DOI: 10.1002 / humu.22945
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Янг, С. Х., Андрес, Д. А., Шпильманн, Х. П., Янг, С. Г., и Фонг, Л. Г. (2008). Прогерин вызывает болезненные фенотипы прогерии у мышей, независимо от того, фарнезилирован он или нет. J. Clin. Вкладывать деньги. 118, 3291–3300. DOI: 10.1172 / jci35876
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Янг, С. Х., Берго, М. О., Toth, J. I., Qiao, X., Hu, Y., Sandoval, S., et al. (2005). Блокирование протеина фарнезилтрансферазы улучшает ядерный блеббинг в фибробластах мыши с целевой мутацией синдрома прогерии Хатчинсона-Гилфорда. Proc. Natl. Акад. Sci. США, 102, 10291–10296. DOI: 10.1073 / pnas.0504641102
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ян, С. Х., Чанг, С. Ю., Рен, С., Ван, Ю., Андрес, Д. А., Шпильманн, Х. П. и др. (2011). Отсутствие фенотипов прогерии у мышей, экспрессирующих нефарнезилированную версию прогерина. Гм. Мол. Genet. 20 436–444. DOI: 10.1093 / hmg / ddq490
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Zhang, J., Lian, Q., Zhu, G., Zhou, F., Sui, L., Tan, C., et al. (2011). Модель человеческого ИПСК прогерии Хатчинсона Гилфорда выявляет дефекты гладких мышц сосудов и мезенхимальные стволовые клетки. Cell Stem Cell 8, 31–45. DOI: 10.1016 / j.stem.2010.12.002
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Чжан, В., Li, J., Suzuki, K., Qu, J., Wang, P., Zhou, J., et al. (2015). Модель стволовых клеток с синдромом Вернера показывает, что изменения гетерохроматина являются движущей силой старения человека. Наука 348, 1160–1163. DOI: 10.1126 / science.aaa1356
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Жиронкина О.А., Курчашова С.Ю., Пожарская В., Черепанинец В.Д., Стрелкова О.С., Хозак П.и др. (2016). Механизмы роста ядерной пластинки в интерфазе. Histochem.Cell Biol. 145, 419–432. DOI: 10.1007 / s00418-016-1419-6
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Симптомы, тесты, лечение и профилактика
Обзор
Что такое прогерия?
Прогерия (синдром прогерии Хатчинсона-Гилфорда или HGPS) — очень редкое генетическое заболевание, которое вызывает быстрое старение детей.Дети с прогерией кажутся здоровыми при рождении, но обычно начинают проявлять признаки быстрого старения в первые два года жизни. Большинство умирает примерно к 13 или 14 годам, хотя некоторые доживают до 20 лет. Причиной смерти чаще всего является болезнь сердца, а иногда и инсульт.
Насколько распространена прогерия?
Прогерия поражает примерно 1 из 20 миллионов человек во всем мире. По данным исследовательского фонда Progeria Research Foundation, во всем мире в любое время живут от 350 до 400 детей с прогерией.Прогерия, кажется, одинаково влияет на мальчиков и девочек, и не чаще встречается у одной расы, чем у другой.
Симптомы и причины
Что вызывает прогерию?
Прогерия вызвана мутацией (изменением) гена ламина А (LMNA). Этот ген вырабатывает белок, который скрепляет ядро клетки.Из-за изменения гена белок становится дефектным. Это делает ядро нестабильным, что, как считается, вызывает процесс преждевременного старения.
Мутация гена LMNA не передается по наследству. Фактически, родители, братья и сестры детей с прогерией страдают редко.
Каковы симптомы прогерии?
У детей с прогерией признаки и симптомы быстрого старения появляются в течение первых двух лет жизни. К ним относятся:
- Неспособность расти такими же темпами, как у конкурентов
- Потеря жира
- Облысение
- Кожа приобретает состарившийся, морщинистый вид
- Жесткие соединения
Другие признаки и симптомы прогерии включают:
- Лица маленькие для размера головы
- Носы, которые выглядят «защемленными»
- Открытое мягкое пятно на голове
- Зубы, приходящиеся в конце
По мере развития состояния начинают развиваться симптомы, менее очевидные для родителей, включая вывих бедра, катаракту, артрит, образование бляшек в артериях и сердечные заболевания.У некоторых детей с прогерией случаются инсульты.
Несмотря на эти физические проблемы, интеллект не страдает, и дети с прогерией, как правило, так же умны, как и их сверстники.
Диагностика и тесты
Как диагностируется прогерия?
Ген, вызывающий прогерию, был идентифицирован в 2003 году, и был создан генетический тест, который может подтвердить, вызваны ли симптомы у ребенка прогерией.Для проведения теста необходимо взять образец крови у ребенка. (До того, как этот тест стал доступен, врачи могли диагностировать прогерию только на основании своих наблюдений и рентгеновских лучей.)
Если у вашего врача есть основания полагать, что у вашего ребенка может быть прогерия, он или она может связаться с Фондом исследований прогерии. Медицинский директор фонда рассмотрит случай и может организовать бесплатное обследование для семей. Результаты обычно приходят через 2–4 недели.
Ведение и лечение
Как лечить прогерию?
Нет лекарства от прогерии, но изучаются несколько препаратов для ее лечения.
Физическая терапия может помочь этим детям достичь хорошего диапазона движений, равновесия и осанки, а также уменьшить боль в бедрах и ступнях. Трудотерапия может помочь им развиваться в таких функциональных областях, как прием пищи, соблюдение личной гигиены и почерк.
Дети, страдающие прогерией, могут получить большую пользу от ухода, который поможет им жить как можно более здоровой и комфортной жизнью. Обычно сюда входят:
- Мониторинг сердечных заболеваний : Сюда входят регулярные тесты, такие как эхокардиограмма и проверка артериального давления.Терапия низкими дозами аспирина и статинов может помочь снизить некоторые риски сердечных заболеваний.
- Визуальные исследования (например, магнитно-резонансная томография или MRI ) : Их можно использовать для наблюдения за инсультами или для проверки головных болей или судорог, которые часто встречаются у этих детей.
- Регулярные осмотры зрения : У некоторых детей с прогерией есть проблемы со зрением, в том числе дальнозоркость или сухость глаз (потому что их веки не могут полностью закрыться).По мере прогрессирования состояния у них также может развиться катаракта. У этих детей могут быть более тонкие брови и ресницы, что повышает вероятность попадания раздражителей в глаза. Кроме того, некоторые дети имеют сильную чувствительность к свету, и в некоторых случаях им могут посоветовать носить солнцезащитные очки.
- Проверка слуха : У детей с прогерией может быть потеря слуха, которую можно улучшить с помощью слуховых аппаратов.
- Регулярные стоматологические осмотры : Дети с прогерией чаще имеют стоматологические проблемы, такие как кариес, сильная скученность, задержка появления зубов и углубление десен.
- Мониторинг кожных проблем : Часто это первые признаки прогерии и могут включать темные пятна или выпуклости на коже, выпадение волос, зуд и стянутость кожи, которые ограничивают движение и могут затруднять дыхание или переваривание пищи.
- Мониторинг здоровья костей : Дети с прогерией могут иметь несколько проблем, связанных с ростом и развитием костей, а также проблемами с суставами.
Детям с прогерией для роста необходимо полноценное питание.Некоторым может потребоваться дополнительное питание (включая зонд для кормления). Обеспечение достаточного увлажнения детей с прогерией может помочь снизить риск внезапных неврологических проблем. Поговорите с врачом вашего ребенка о здоровых способах поощрения вашего ребенка получать достаточно калорий и гидратации.
Что родители могут сделать, чтобы помочь своей семье, если у их ребенка прогерия?
Родители ребенка с прогерией должны стараться вести как можно более нормальную домашнюю жизнь. Постарайтесь вовлечь ребенка в как можно больше занятий и не позволяйте другим детям в семье чувствовать себя упущенными.
Будьте честны, но учитывайте возраст всей семьи, обсуждая тот факт, что ваш ребенок с прогерией доживет только до определенного возраста. Консультации могут быть полезны в разное время.
Также поговорите со своим ребенком о том, что некоторые люди вернутся, увидев его или ее, и обсудите, как ваш ребенок должен реагировать на взгляды и шепот.
Перспективы / Прогноз
Если у моего ребенка прогерия, будет ли она у моих будущих детей?
Прогерия вызывается чрезвычайно редким генетическим изменением и обычно не передается по наследству.