Миссенс-мутация — Missense mutation — qaz.wiki
Генетическая точечная мутация, приводящая к замене аминокислот в белке.
В генетике , А мутация миссенса является точечной мутацией , в которой один нуклеотидном изменении приводит к кодону , который кодирует другую аминокислоту . Это разновидность несинонимичной замены .
Замена белка от мутации ДНК
На этом изображении показан пример миссенс-мутации. Один из нуклеотидов (аденин) заменен другим нуклеотидом (цитозином) в последовательности ДНК. Это приводит к включению неправильной аминокислоты (пролина) в последовательность белка.
Миссенс-мутация относится к изменению одной аминокислоты в белке, возникающему в результате точечной мутации в одном нуклеотиде. Миссенс-мутация — это тип несинонимичной замены в последовательности ДНК. Два других типа несинонимичных замен — это бессмысленные мутации, при которых кодон заменяется на преждевременный стоп-кодон, что приводит к усечению результирующего белка , и непрерывные мутации, при которых стирание стоп-кодона приводит к более длинному, нефункциональному белку. .
Миссенс-мутации могут сделать полученный белок нефункциональным, и такие мутации ответственны за такие заболевания человека, как буллезный эпидермолиз , серповидно-клеточная анемия и БАС, опосредованный SOD1 .
В наиболее общем варианте серповидно-клеточной анемией, 20 — й нуклеотид гена для бета — цепи из гемоглобина изменяется от кодона GAG до GTG. Таким образом, 6-я аминокислота глутаминовая кислота заменяется валином, обозначенным как мутация «E6V», и белок в достаточной степени изменен, чтобы вызвать серповидно-клеточную анемию.
Не все миссенс-мутации приводят к заметным изменениям белка. Аминокислота может быть заменена аминокислотой с очень похожими химическими свойствами, и в этом случае белок все еще может нормально функционировать; это называется нейтральной, «тихой», «тихой» или консервативной мутацией.![]() Альтернативно, аминокислотная замена может происходить в области белка, которая не оказывает значительного влияния на вторичную структуру или функцию белка. Когда аминокислота может кодироваться более чем одним кодоном (так называемое «вырожденное кодирование»), мутация в кодоне может не вызывать никаких изменений в трансляции; это было бы синонимичной заменой, а не миссенс-мутацией.
Альтернативно, аминокислотная замена может происходить в области белка, которая не оказывает значительного влияния на вторичную структуру или функцию белка. Когда аминокислота может кодироваться более чем одним кодоном (так называемое «вырожденное кодирование»), мутация в кодоне может не вызывать никаких изменений в трансляции; это было бы синонимичной заменой, а не миссенс-мутацией.
Пример
Дикий тип (слева) и мутировавшая (справа) форма ламина А (pdb id: 1IFR). Обычно аргинин 527 (синий) образует солевой мостик с глутаматом 537 (пурпурный), но замена R527L приводит к нарушению этого взаимодействия (лейцин имеет неполярный хвост и поэтому не может образовывать статический солевой мостик).
DNA: 5' - AAC AGC CTG CGT ACG GCT CTC - 3'
3' - TTG TCG GAC GCA TGC CGA GAG - 5'
mRNA: 5' - AAC AGC CUG CGU ACG GCU CUC - 3'
Protein: Asn Ser Leu Arg Thr Ala Leu
Миссенс-мутация LMNA (c.1580G> T), введенная в ген LMNA — положение 1580 (нуклеотид) в последовательности ДНК (CGT), вызывающая замену гуанина тимином , что приводит к CTT в последовательности ДНК. Этот результат на уровне белка в замене аргинина по лейцин в положении 527. Это приводит к разрушению солевого мостика и структуры дестабилизации. На уровне фенотипа это проявляется наложением мандибулоакральной дисплазии и синдрома прогерии .
Полученный транскрипт и белковый продукт:
DNA: 5' - AAC AGC CTG CTT ACG GCT CTC - 3'
3' - TTG TCG GAC GAA TGC CGA GAG - 5'
mRNA: 5' - AAC AGC CUG CUU ACG GCU CUC - 3'
Protein: Asn Ser Leu Leu Thr Ala Leu
Экспериментальный анализ
Связанные с раком миссенс-мутации могут привести к резкой дестабилизации получаемого белка. В 2012 году был предложен метод скрининга таких изменений, а именно быстрый параллельный протеолиз (FASTpp) .
Смотрите также
Рекомендации
внешняя ссылка
<img src=»https://en.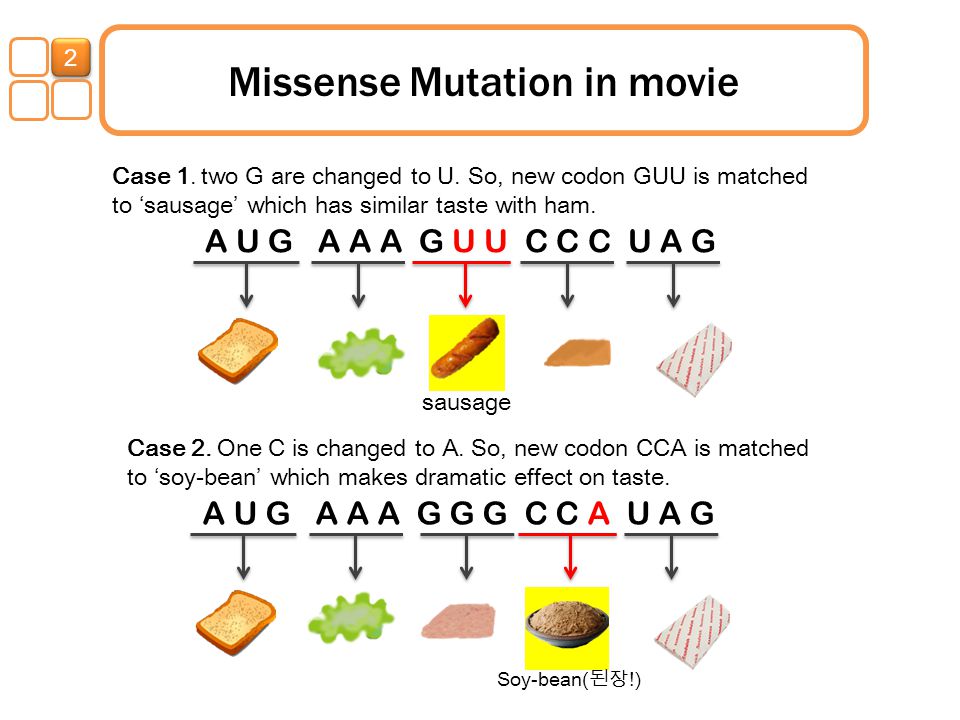 wikipedia.org//en.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1×1″ alt=»» title=»»>
wikipedia.org//en.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1×1″ alt=»» title=»»>
мутации — это… Что такое Миссенс-мутации?
- Миссенс-мутации
- Миссенс-мутации
мутации, возникающие в результате замены оснований в вирусном геноме. М.-м. можно определить по изменению белка, кодируемого мутированным геном, или по изменению функции, связанной с измененным белком.
(Источник: «Словарь терминов микробиологии»)
.
- Миокамицин
- Митоген
Смотреть что такое «Миссенс-мутации» в других словарях:
Миссенс-мутация — точечная мутация, в результате которой измененный кодон начинает кодировать другую аминокислоту. В зависимости от того, насколько различаются свойства белков, синтезированных на основе измененных кодонов, от свойств изначальных протеинов,… … Википедия
миссенс-супрессия — Форма супрессии, при которой супрессорной мутацией является миссенс мутация; при внутригенной супрессии она может, изменяя мутировавший кодон, обеспечивать включение в мутантный сайт более подходящей аминокислоты, чем у исходных мутантов… … Справочник технического переводчика
Мутации точковые — * мутацыі кропкавыя * point mutations 1. Нормально менделирующие мутации, не связанные со структурными изменениями хромосом и с нарушениями процесса кроссинговера (см.). М. т. это мутации генные, при которых происходит замена (. Трансверсия),… … Генетика. Энциклопедический словарь
МУТАЦИИ — (от лат. mutatio изменение), внезапные (скачкообразные) естественные или вызванные искусственно наследуемые изменения генетич. материала (генома), приводящие к изменению тех или иных признаков организма.
 Различают генеративные М., возникающие в… … Химическая энциклопедия
Различают генеративные М., возникающие в… … Химическая энциклопедияМутации — Эта статья о термине из области биологии, см. также Мутация (значения) Главный мутаген табачного дыма бензопирен связанный с одним из нуклеотидов молекулы ДНК. Мутация стойкое (то есть такое, которое может быть унаследовано потомками данной… … Википедия
миссенс-супрессия — missense suppression миссенс супрессия. Форма супрессии, при которой супрессорной мутацией является миссенс мутация <missense mutation>; при внутригенной супрессии она может, изменяя мутировавший кодон, обеспечивать включение в мутантный… … Молекулярная биология и генетика. Толковый словарь.
гормоны — * гармоны * hormones высокоспецифичные биологически активные органические вещества, являющиеся регуляторами важнейших жизненных процессов. Г. вырабатываются в организме высокоспециализированными клетками или органами (эндокринными железами, или… … Генетика. Энциклопедический словарь
дефицит глюкозо-6-фосфатдегидрогеназы — glucose 6 phosphate dehydrogenase deficiency, G 6 PD deficiency дефицит глюкозо 6 фосфатдегидрогеназы. Cиндром врожденной недостаточности фермента глюкозо 6 фосфатдегидрогеназы (Г6ФДГ), обусловливающий избыточный, индуцируемый лекарственными… … Молекулярная биология и генетика. Толковый словарь.
гемоглобин Констант-Спринг — hemoglobin Constant Spring гемоглобин Констант Спринг. Клиника нормальная; α цепь состоит не из 141, а из 172 аминокислотных остатков и образуется в результате миссенс мутации <missense mutation>, превращающей стоп кодон ТАА в смысловой… … Молекулярная биология и генетика. Толковый словарь.
растекающаяся мутация — leaky mutation ликовая (растекающаяся) мутация. Форма миссенс мутации <missence mutation>, при которой мутантный фермент обладает сниженной активностью либо снижен уровень его синтеза; Л.
м. в регуляторных элементах генов проявляются в… … Молекулярная биология и генетика. Толковый словарь.
 Различают генеративные М., возникающие в… … Химическая энциклопедия
Различают генеративные М., возникающие в… … Химическая энциклопедия м. в регуляторных элементах генов проявляются в… … Молекулярная биология и генетика. Толковый словарь.
м. в регуляторных элементах генов проявляются в… … Молекулярная биология и генетика. Толковый словарь.Генные мутации, задачи на генные мутации
Генные мутации, задачи на генные мутации. Генная мутация-изменение нуклеотидной последовательности одного гена.
Типы генных мутаций:
- Замена одного нуклеотида в гене на другой нуклеотид (миссенс мутация). Происходит из-за ошибки ДНК-полимеразы при репликации ДНК.
Последствия миссенс мутаций:
а) Миссенс мутация приводит к изменению первичной структуры и функции соответствующего белка, если образовавшейся в результате мутации кодон кодирует новую аминокислоту.
До мутации:
ДНК: АТГЦЦАААГГГА
иРНК: УАЦГГУУУЦЦЦУ
первичная стр-ра белка: Тир Гли Фен Про
После мутации:
ДНК: АТГЦЦАААТГГА
иРНК: УАЦГГУУУАЦЦУ
первичная стр-ра белка: Тир Гли Лей Про
В результате мутации произошла замена одной аминокислоты, следовательно, первичная структура и функция белка изменились.
б) Миссенс мутация не приводит к изменению первичной структуры и функции соответствующего белка, если образовавшейся в результате мутации кодон кодирует ту же аминокислоту, что и исходный (из-за свойства вырожденности генетического кода).
До мутации:
ДНК: АТГЦЦАААГГГА
иРНК: УАЦГГУУУЦЦ
первичная стр-ра белка: Тир Гли Фен Про
После мутации:
ДНК: АТГЦЦААААГГА
иРНК: УАЦГГУУУУЦЦУ
первичная стр-ра белка: Тир Гли Фен Про
В результате мутации произошла замена Г на А в составе последовательности ДНК. Однако эта мутация не привела к изменению структуры и функции соответствующего белка, так как новый кодон УУУ кодирует ту же аминокислоту (Фен), что и исходный – УУЦ.
- Выпадение или вставка одного или нескольких кодонов в составе нуклеотидной последовательности гена. Выпадение одного кодона происходит из-за ошибки ДНК-полимеразы при репликации ДНК и приводит к выпадению одной аминокислоты из первичной структуры белка. Соответственно, такая мутация приводит к изменению структуры и функции соответствующего белка.
До мутации:
ДНК: АТГЦЦАААГГГА
иРНК: УАЦГГУУУЦЦЦУ
первичная стр-ра белка: Тир Гли Фен Про
После мутации:
ДНК: АТГААГГГА
иРНК: УАЦУУЦЦЦУ
первичная стр-ра белка: Тир Фен Про
- Вставка или выпадение одного или 2-х нуклеотидов (Мутация со смещением открытой рамки считывания). Происходит из-за ошибки ДНК-полимеразы при репликации ДНК. Данная мутация приводит к изменению всех аминокислот в первичной структуре белка, начиная с точки мутации. Это в большинстве случаев приводит к полному нарушению структуры и функции белка.
До мутации:
ДНК: АТГЦЦЦАТААГЦ
иРНК: УАЦГГГУАУУЦГ
первичная стр-ра белка: Тир Гли Тир Сер
В результате мутации произошла вставка 1 нуклеотида
После мутации:
ДНК: АТГГЦЦЦАТААГЦ
иРНК: УАЦЦГГГУАУУЦГ
первичная стр-ра белка: Тир Арг Вал Фен
- Появление стоп-кодона в кодирующей части гена (нонсенс мутация). В результате, полипептидная цепь соответствующего белка становится короче, что приводит к значительному изменению первичной структуры и функции белка.
До мутации:
ДНК: АТГЦЦЦАТААГЦ
иРНК: УАЦГГГУАУУЦГ
первичная стр-ра белка: Тир Гли Тир Сер
Произошла замена Т на Ц в последовательности нуклеотидов соответствующего гена. В результате в кодирующей части мРНК возник стоп-кодон – УАГ, что привело к преждевременной остановке трансляции.
После мутации:
ДНК: АТГЦЦЦАЦААГЦ
иРНК: УАЦГГГУАГУЦГ
первичная стр-ра белка: Тир Гли
Одна из задач ЕГЭ на тему «Генные мутации»
Задача 6
В результате генной мутации в полипептидной цепи соответствующего белка аминокислота Про заменилась на Цис. Последовательность иРНК до мутации: ГЦУУУЦЦЦЦГАЦУЦА. Определите аминокислотный состав молекулы нормального и мутированного белка, а также возможные последовательности нуклеотидов мутированной иРНК. Ответ поясните.
До мутации:
иРНК: ГЦУУУЦЦЦЦГАЦУЦА
белок: Ала Фен Про Асп Сер
Причиной замены третьей аминокислоты Про на Цис являлась генная мутация в нуклеотидной последовательности соответствующего гена, в результате которой произошло изменение триплета в составе мРНК, кодирующего третью аминокислоту. Исходя из свойства вырожденности генетического кода, аминокислота Цис может быть закодирована двумя возможными триплетами – УГУ, УГЦ. Соответственно, в результате мутации в иРНК мог появиться любой из этих триплетов. Вероятнее всего УГЦ, так как при этом должно замениться меньше всего нуклеотидов.
Варианты мутированной последовательности иРНК: ГЦУУУЦУГУГАЦУЦА; ГЦУУУЦУГЦГАЦУЦА
После мутации:
иРНК: ГЦУУУЦУГЦГАЦУЦА
белок: Ала Фен Цис Асп Сер
Ответ: последовательности иРНК с мутацией: ГЦУУУЦУГУГАЦУЦА; ГЦУУУЦУГЦГАЦУЦА.
Первичная структура нормального белка: Ала Фен Про Асп Сер
Первичная структура белка после мутации: Ала Фен Цис Асп Сер
Мутации
Онкогеномика. Мастер-класс проф. Д.В. Залетаева
Мутации
Все изменения последовательности нуклеотидов в ДНК, независимо от локализации и влияния на жизнеспособность клетки, – мутации. Нейтральные мутации или полиморфизмы – последовательности ДНК, не приводящие к заметным нарушениям функций.
Нейтральные мутации или полиморфизмы – последовательности ДНК, не приводящие к заметным нарушениям функций.
Существуют две классификации мутаций (Strachan T., Read A., 2003). Одна базируется на функциональной характеристике и не рассматривает характер самой мутации. Вторая классифицирует мутации по структурным изменениям в ДНК и РНК.
Функциональная классификация подразделяет мутации:
- связанные с потерей функции белка;
- связанные с приобретением новой аномальной функции белка;
- в регуляторных областях гена, приводящие к количественным изменениям первичного белкового продукта.
Структурная классификация выделяет следующие типы:
- нонсенс-мутация – изменение в нуклеотидной последовательности ДНК кодирующей области гена, приводящее к возникновению стоп-кодона и преждевременному прекращению синтеза белка;
- миссенс-мутация – изменение в нуклеотидной последовательности ДНК кодирующей области гена, приводящее к изменению одной аминокислоты, что не нарушает процесс синтеза белка;
- мутации, приводящие к сдвигу рамки считывания белка и возникновению стоп-кодона на некотором расстоянии от самой мутации, что приводит к преждевременной терминации синтеза белка. Мутации сдвига рамки считывания вызываются делециями и инсерциями, не кратными трем (кодон = 3) нуклеотидам;
- мутации в сайтах сплайсинга приводят к тому, что нарушается процессинг мРНК, что ведёт к: а) делеции всего или части экзона; б) обычно удаляемые интронные области могут стать смысловыми. Такая патология приводит к сдвигу рамки считывания и появлению стоп-кодона. В результате белковый продукт гена не только укорачивается, но и может оказаться совершенно аномальным.
Для злокачественных опухолей характерны все типы мутаций. Высокоинформативными структурными ДНК-маркерами, позволяющими проводить раннюю диагностику опухолевого процесса, определять прогноз развития заболевания и подбирать наиболее эффективные варианты терапии, являются характерные нарушения нуклеотидной последовательности белок-кодирующих генов в некоторых типах опухоли.
ДНК-диагностика мутаций может быть косвенной и прямой (Strachan T., Read A., 2003).
При прямой диагностике предметом анализа являются мутации гена. Прямые методы возможны лишь при наличии информации об экзон-интронной организации или полноразмерной нуклеотидной последовательности ДНК гена.
ПЦР с использованием определенного фермента гидролиза ДНК возможна при стандартной мутации с изменением сайта рестрикции, если без изменения сайта рестрикции – аллель-специфическая ПЦР.
Определение нуклеотидной последовательности фрагмента ДНК, показавшего аномальную электрофоретическую подвижность, и заключительным этапом анализа мутаций является их секвенирование. Прямое секвенирование позволяет с 100% эффективностью определить мутацию.
В косвенной диагностике мутаций используются несколько методов. Наиболее просто при электрофоретическом анализе обнаруживаются мутации, изменяющие длину амплифицированных фрагментов ДНК.
Для выявления точковых мутаций, небольших делеций и инсерций в исследуемых генах используется множество различных подходов, основанных на методе полимеразной цепной реакции. При ПЦР возможно многократно увеличить уникальную последовательность ДНК, а затем проанализировать её на наличие мутации.
Метод конформационного полиморфизма однонитевой ДНК (SSCP) – один из наиболее простых в исполнении высокочувствительных методов поиска однонуклеотидных замен в исследуемом участке геномной ДНК. Оптимальный размер исследуемого фрагмента ДНК 200-250 п.н., при котором вероятность обнаружения мутаций составляет 70-95%.
Вероятность идентификации точковых мутаций методом гетеродуплексов достигает 80-90% при длине фрагментов ДНК не более 300 п.н. Метод основан на том, что за счет конформационных особенностей в местах несовпадения нуклеотидов электрофоретическая подвижность гетеродуплексов, образующихся при комплиментарном взаимодействии мутантной и нормальной ДНК отличается от подвижности гомодуплексов нормальных фрагментов ДНК.
Наиболее распространенным способом скрининга мутаций, позволяющим выявить точковые мутации почти в 100% случаев и не требующим больших затрат времени, считается комбинация анализа гетеродуплексов и метода однонитевого конформационного полиморфизма.
Mutations | Protocol (Translated to Russian)
13.10: Мутации
Обзор
Мутации – это изменения в последовательности ДНК. Эти изменения могут происходить спонтанно или они могут быть вызваны воздействием факторов окружающей среды. Мутации можно охарактеризовать различными способами: изменяют ли они аминокислотную последовательность белка, происходят ли они на небольшой или большой площади ДНК, и встречаются ли они в соматических клетках или зародышевых клетках.
Последствия точечных мутаций на молекулярном уровне
Мутации, происходящие при одном нуклеотиде, называются точечными мутациями. Когда точечные мутации происходят в генах, последствия могут варьироваться в степени тяжести в зависимости от того, что происходит с закодированной аминокислотной последовательностью. Тихая мутация не меняет аминокислотной идентичности и не будет иметь никакого влияния на организм. Мутация миссенса изменяет одну аминокислоту, и последствия могут быть серьезными, если изменение изменяет функцию белка. Нонсенс мутации производит стоп-кодон, который усечения белка, вероятно, что делает его нефункциональным. Мутации рама возникают, когда один или несколько нуклеотидов вставляются или удаляются из последовательности ДНК, кодирующей белок, влияя на все кодоны ниже по течению от места мутации.
Хромосомные изменения являются крупномасштабными мутациями
Самый резкий тип мутации, хромосомное изменение, изменяет физическую структуру хромосомы. Хромосомные изменения могут включать удаление, дублирование или инверсию больших участков ДНК в пределах одной хромосомы, или интеграцию части другой хромосомы. Эти мутации, как правило, гораздо серьезнее, чем точечные мутации, поскольку они охватывают многие гены и регуляторные элементы.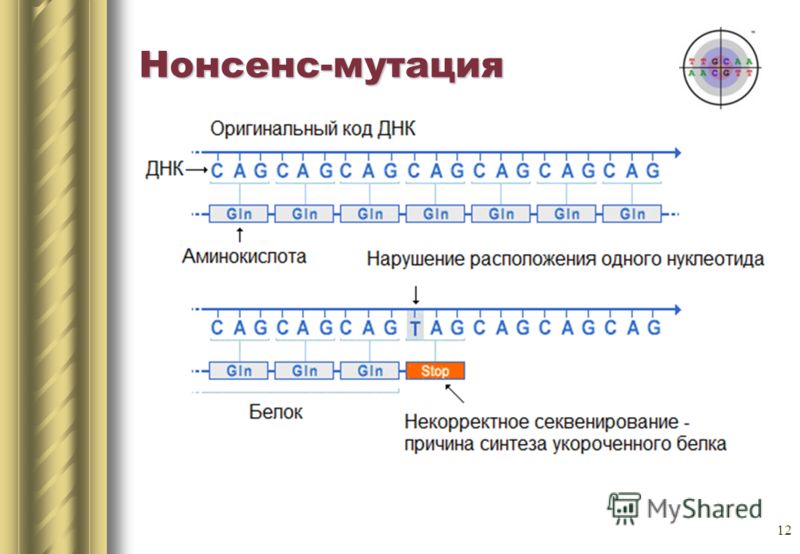 Хромосомные изменения могут быть обнаружены путем кариотипирования пораженной клетки.
Хромосомные изменения могут быть обнаружены путем кариотипирования пораженной клетки.
Наследуются только мутации зародышевой линии
Мутации могут происходить в любой клетке, но только мутации зародышевой линии — те, что присутствуют в яйцеклетках и сперматозоидах — могут передаваться потомству. Например, наследственные заболевания — это подтип генетического заболевания, вызванного вредными мутациями зародышевой линии. Они могут быть аутосомными, обнаруживаясь на хромосомах с первой по 22, или сцепленными с полом, встречаясь на X- или Y-хромосоме. Одним из примеров наследственного заболевания является муковисцидоз (МВ), заболевание, которое в первую очередь поражает легкие. Это вызвано делецией в гене CFTR , который удаляет одну аминокислоту из белка CFTR. МВ — аутосомно-рецессивное заболевание, что означает, что у человека с одной мутированной копией гена и одной нормальной копией заболевание не разовьется; другие заболевания, такие как болезнь Хантингтона, нейродегенеративное заболевание, являются аутосомно-доминантными, что означает, что для развития болезни необходима только одна мутированная копия гена.
Некоторые мутации вызваны факторами окружающей среды
Как соматические мутации—, возникающие вне зародышевой линии, так и мутации зародышевой линии— могут возникать спонтанно во время репликации ДНК, но они также могут быть вызваны воздействием радиации или химических веществ в окружающей среде. Внешние факторы, повреждающие ДНК и вызывающие мутации, называются мутагенами. Один хорошо охарактеризованный мутаген окружающей среды — ультрафиолетовое (УФ) излучение. Ультрафиолетовое излучение несет больше энергии, чем видимый свет, и повреждает ДНК, разрывая связи между парами оснований, в результате чего основания тимина на одной и той же цепи ДНК соединяются друг с другом в характерные димеры тимина. Солнце — естественный источник УФ-излучения. Наиболее опасные длины волн, УФ-С, улавливаются высоко в атмосфере, но УФ-А и УФ-В лучи достигают поверхности Земли. Искусственные источники УФ-излучения включают солярии, которые пропускают в основном УФ-А-лучи с меньшим количеством УФ-В. К счастью, у клеток есть механизмы для восстановления поврежденной ДНК, но иногда повреждение не восстанавливается до деления клеток в быстро делящихся клетках, таких как клетки кожи. Если повреждение ДНК происходит в области генома, которая важна для регулирования роста и деления клеток, это может привести к раку, если его не исправить.
Искусственные источники УФ-излучения включают солярии, которые пропускают в основном УФ-А-лучи с меньшим количеством УФ-В. К счастью, у клеток есть механизмы для восстановления поврежденной ДНК, но иногда повреждение не восстанавливается до деления клеток в быстро делящихся клетках, таких как клетки кожи. Если повреждение ДНК происходит в области генома, которая важна для регулирования роста и деления клеток, это может привести к раку, если его не исправить.
Глоссарий | EXCEMED
Этот глоссарий призван обеспечить точные определения ключевых терминов. Слова, выделенные курсивом, имеют отдельные пояснения. Если вы считаете, что некоторые термины должны быть добавлены в словарь, пожалуйста, отправьте сообщение по электронной почте [email protected] с вашим предложением.
Кликните на одно сообщение выше, чтобы перейти на страницу, начиная с этого сообщения.
B
Bh5
- Сокращенное название тетрагидробиоптерина (кофактора фенилаланингидроксилазы ), который часто используется в медицинских исследованиях.
См.: Сапроптерин
Синонимы: tetrahydrobiopterin
Перейти к началу
H
HPA
Распространенное сокращенное название гиперфенилаланинемии.
Перейти к началу
I
IQ
«Коэффициент умственного развития», широко распространенный показатель умственного развития, который определяется при помощи тестов. IQ представляет собой соотношение умственного и физического развития человека, которое выражается в процентах.
Перейти к началу
P
PEG-PAL
Аммиаклиаза фенилаланина — это фермент , получаемый из растений, который расщепляет фенилаланин , но при этом не требует тетрагидробиоптерин в качестве кофермента .
PEG-PAL — это пегилированный инъекционный состав аммиаклиазы фенилаланина, который исследуется в клинических условиях на людях с фенилкетонурией. В данный момент PEG-PAL еще не утвержден как официальное терапевтическое средство.Phe
- Сокращенное название фенилаланина.
 PEG-PAL — это пегилированный инъекционный состав аммиаклиазы фенилаланина, который исследуется в клинических условиях на людях с фенилкетонурией. В данный момент PEG-PAL еще не утвержден как официальное терапевтическое средство.
PEG-PAL — это пегилированный инъекционный состав аммиаклиазы фенилаланина, который исследуется в клинических условиях на людях с фенилкетонурией. В данный момент PEG-PAL еще не утвержден как официальное терапевтическое средство.Перейти к началу
А
Аминокислота
Аминокислоты — это маленькие молекулы с определенной структурой, с карбоксильными и аминными группами, отделенными от центрального атома углерода с боковой связью, которая позволяет различать разные аминокислоты. Все белки состоят их цепочек аминокислот, связанных вместе, некоторые аминокислоты имеют огромное значение для нервной системы. Существует около 20 аминокислот, которые важны для физиологии человека. MБольшинство из них синтезируется в теле человека, но некоторые не синтезируются, а должны поступать извне в процессе питания (« Незаменимые аминокислоты «). Люди, которые страдают фенилкетонурией и соблюдают диету с низким содержанием фенилаланина, не могут получать с пищей достаточное количество белков; для получения незаменимых аминокислот им могут потребоваться продукты лечебного питания
Аммиаклиаза фенилаланина
См.: PEG-PAL
Аспартам
Аспартам — это искусственный заменитель сахара, имеет разные коммерческие названия. Аспартам высвобождает фенилаланин в процессе обмена веществ, поэтому людям с фенилкетонурией следует избегать его употребления.
Атипичная гиперфенилаланинемия
Этот термин используется в отношении к пациентам с недостатком тетрагидробиоптерина, а также для того, чтобы охарактеризовать людей с легкой формой гиперфенилаланинемии , которая возникает в результате определенной мутации гена фенилаланингидроксилазы.
Аутосомно-рецессивная наследственность
Вид наследования мутации гена, который несущественно влияет на фенотип человека, из-за того что другая (немутированная) копия гена обеспечивает достаточную активность белка, кодированного геном . Например, у пациентов с одной мутированной и одной нормальной копией фенилаланингидроксилазы (один из родителей является носителем мутации гена) не выражается клинический фенотип фенилкетонурии, характеризуемый гиперфенилаланинемией . Наследование двух мутированных копий (по одной от каждого родителя) в результате будет выражено фенотипом фенилкетонурии.

Перейти к началу
Б
Белое вещество
В ЦНС содержится «серое вещество», которое образовано телами нервных клеток, и «белое вещество», которое состоит в основном из нервных волокон (аксонов). Недостаточная регуляция фенилаланина в крови в течение длительного времени может негативно повлиять на белое вещество в головном мозге.
Болезнь Фёллинга
Ранний термин для обозначения фенилкетонурии, связанный с именем врача Асбьёрна Фёллинга, который открыл зависимость синдрома прогрессирующей умственной отсталости , известной в наше время как фенилкетонурия , от избытка фенилаланина .
Большие нейтральные аминокислоты
- Этот термин описывает некоторые аминокислоты в соответствии с их химической структурой (большие неионизированные боковые цепи) и, как правило, относится к: фенилаланину, тирозину , триптофану, валину, изолейцину, лейцину, метионину и гистидину. Людям с фенилкетонурией иногда назначают добавки с LNAA.
Синонимы: LNAA
Перейти к началу
В
Веконосовые складки
Складки кожи верхнего века, которые закрывают внутренние уголки глаз.
Восприимчивый к тетрагидробиоптерину
Этот термин используется для описания людей с фенилкетонурией, которые положительно реагируют на лечение сапроптерином или на нагрузочную пробу на тетрагидробиоптерин . Такие люди с медицинской точки зрения потенциально подходят для долгосрочного лечения сапроптерином с целью регуляции уровня фенилаланина в крови.
Вставка
- Мутация , при которой дополнительная ДНК (одна или более пар оснований) выпадает из гена.

Перейти к началу
Г
Ген
- Последовательность ДНК, которая определяет структуру определенного белка. Количество мутаций в гене фенилаланингидроксилазы , которые, как известно, например, ухудшают способность фермента преобразовывать фенилаланин в тирозин , приводя тем самым к фенилкетонурии.
Генетическая гетерогенность
Каждый человек уникален в отношении его ДНК. Генетическая гетерогенность — это термин, который используется для описания ряда мутаций в определенном гене у разных индивидуумов одной группы. Много разных мутаций генов могут вызвать фенилкетонурию, поэтому данная численность людей обладает генетической гетерогенностью высокой степени.
Генная терапия
Метод лечения, в котором применяется изменение самого генетического кода пациента. Например, в экспериментальных исследованиях производится поиск метода генной терапии, который позволит создать рабочую копию фенилаланингидроксилазы для пациентов с фенилкетонурией, которые страдают от недостатка этого фермента . Однако, в настоящее время эти исследования являются теоретическими, а использование их результатов для лечения фенилкетонурии возможно лишь в далеком будущем.
Генотип
- Данный термин описывает образец мутаций , которые присутствуют в определенных генах на уровне ДНК индивидуума. Генотип пациентов с фенилкетонурией во многом, хотя не полностью, прогнозирует их фенотип фенилкетонурии. Например, мутации R408W или IVS-12 существенно снижают активность фенилаланингидроксилазы и приводят к среднему или тяжелому фенотипу фенилкетонурии. Другие мутации , как, например, E390G, Y414C, или A300S, не столь сильно угнетают активность фермента , поэтому питательная переносимость фенилаланина выше, а фенотип фенилкетонурии является менее тяжелым. У пациентов с фенилкетонурии часто наблюдаются разные мутации в каждой копии гена фенилаланингидроксилазы .
Гетерозиготный
- У каждого из нас есть две копии каждого гена . Организм с определенной мутацией только одного гена (при отсутствии мутации другого) называется гетерозиготным в отношении данной мутации.
Гипертония
Повышенный мышечный тонус.
Гиперфенилаланинемия
Повышенный уровень фенилаланина в крови. Уровень фенилаланина в крови, который разделяет нормальное содержание фенилаланина в крови и гиперфенилаланинемию , в соответствии с положениями рекомендаций , изменяется с возрастом.
Гиперфенилаланинемия без фенилкетонурии
У людей с гиперфенилаланинемией без фенилкетонурии наблюдается незначительное повреждение фенилаланингидроксилазы . Это часто возникает по причине мутации только одной из двух имеющихся копий гена фенилаланингидроксилазы или по причине мутаций, которые незначительно ухудшают функцию фермента . При этом наблюдается слегка повышенный уровень фенилаланина в крови, который является недостаточным для диагностирования фенилкетонурии и, как правило, не требует лечения.
Гипотония
Пониженный мышечный тонус.
Гликопротеин
Источник белка, полученный из молочной сыворотки, в которой не содержится фенилаланина .
Гликопротеин иногда используется в качестве белковой пищевой добавки ( продукта лечебного питания ) для пациентов с фенилкетонурией.Гомозиготный
- У каждого из нас есть две копии каждого гена . Организм с идентичной мутацией обоих генов называется гомозиготным в отношении данной мутации.
Губной желобок
Желобок посредине над верхней губой, который тянется от носа до верха губы.
 Генотип пациентов с фенилкетонурией во многом, хотя не полностью, прогнозирует их фенотип фенилкетонурии. Например, мутации R408W или IVS-12 существенно снижают активность фенилаланингидроксилазы и приводят к среднему или тяжелому фенотипу фенилкетонурии. Другие мутации , как, например, E390G, Y414C, или A300S, не столь сильно угнетают активность фермента , поэтому питательная переносимость фенилаланина выше, а фенотип фенилкетонурии является менее тяжелым. У пациентов с фенилкетонурии часто наблюдаются разные мутации в каждой копии гена фенилаланингидроксилазы .
Генотип пациентов с фенилкетонурией во многом, хотя не полностью, прогнозирует их фенотип фенилкетонурии. Например, мутации R408W или IVS-12 существенно снижают активность фенилаланингидроксилазы и приводят к среднему или тяжелому фенотипу фенилкетонурии. Другие мутации , как, например, E390G, Y414C, или A300S, не столь сильно угнетают активность фермента , поэтому питательная переносимость фенилаланина выше, а фенотип фенилкетонурии является менее тяжелым. У пациентов с фенилкетонурии часто наблюдаются разные мутации в каждой копии гена фенилаланингидроксилазы . Гликопротеин иногда используется в качестве белковой пищевой добавки ( продукта лечебного питания ) для пациентов с фенилкетонурией.
Гликопротеин иногда используется в качестве белковой пищевой добавки ( продукта лечебного питания ) для пациентов с фенилкетонурией.Перейти к началу
Д
Двигательные навыки
Способность групп мышц выполнять контролируемые точные движения. Измерение двигательных навыков иногда назначается в качестве нейропсихологических тестов , для того чтобы следить за развитием людей с фенилкетонурией.
Делеция
Мутация , при которой ДНК (одна или более пар оснований) выпадает из гена.
Дефицит тетрагидробиоптерина
См.: Недостаток тетрагидробиоптерина
Диагностирующий тест(ы)
- Диагностирующие тесты используются для определения болезней у населения. В большинстве стран все новорожденные дети проверяются на наличие фенилкетонурии и других наследственных нарушений обмена веществ в первые дни или недели жизни. При необходимости может быть назначено лечение с целью предотвратить долгосрочные негативные последствия болезни.
Диарея
Симптом, для которого характерны частые ежедневные испражнения и жидкий стул. Диарея может быть вызвана разными факторами: у больных фенилкетонурией причиной диареи может быть сохранение низких концентраций фенилаланина в крови в течение длительного времени. Более длительный период низких концентраций фенилаланина может привести к снижению скорости роста, атрофии кожи (которое можно ошибочно принять за экзему) и диарее, которая может свидетельствовать о слабой функции кишечника или неспособности кишечника синтезировать новые клетки.
С другой стороны, диарея, так же как рвота или повышение температуры, может стать причиной катаболизма и последующего повышения концентрации фенилаланина в крови.Диета с низким содержанием фенилаланина
Фенилкетонурия характеризуется неспособностью регулировать уровень фенилаланина в крови, при этом в обычном режиме питания данный уровень может подняться до токсического. Следовательно, пациентам требуется специальная диета с низким содержанием фенилаланина , которая обычно дополняется назначением продуктов лечебного питания.
Допустимая норма фенилаланина
- Количество фенилаланина , которое пациент может принять с пищей без риска последующего превышения допустимого уровня фенилаланина в крови.
Дофамин
- Важный нейромедиатор в ЦНС . Дофамин участвует во многих функциях ЦНС , включая точное управление движениями (недостаток дофаминергических клеток наблюдается при болезни Паркинсона) и когнитивную деятельность. По предположению, вызванное недостатком миелина нарушение функций переднего мозга, регулируемых дофамином, при долговременном заболевании гиперфенилаланинемией может стать причиной повреждения когнитивной функции .
Для осуществления биосинтеза дофамина требуется тирозин . Высокий уровень фенилаланина в крови сдерживает проникновение тирозина в мозг (для фенилаланина и тирозина требуется один и тот же переносчик больших нейтральных аминокислот в гематоэнцефалический барьер). Это может быть очередным фактом негативного воздействия гиперфенилаланинемии на функцию дофаминергических систем мозга.
 С другой стороны, диарея, так же как рвота или повышение температуры, может стать причиной катаболизма и последующего повышения концентрации фенилаланина в крови.
С другой стороны, диарея, так же как рвота или повышение температуры, может стать причиной катаболизма и последующего повышения концентрации фенилаланина в крови.Перейти к началу
З
Заменитель белка
Пациенты с фенилкетонурией, как правило, не могут принимать в пищу продукты — натуральные источники белка, в которых содержится фенилаланин . Заменители белка ( продукты лечебного питания ) являются источниками белка, в которых нет фенилаланина , но есть другие аминокислоты , необходимые для полноценного питания.

Перейти к началу
И
Интроны
- Интроны — это последовательности ДНК в гене , которые не переносятся в зрелый белок. Последовательности РНК, возникающие на основе интронов, выпадают во время процессинга начальной ДНК для формирования окончательного мессенджера РНК, который будет использоваться для трансляции в белок.
Исполнительная функция
- Термин, который описывает некоторые из высших функций головного мозга. К исполнительным функциям относятся воля, понимание, суждение, согласованное использование ресурсов (например, памяти) для достижения поставленных целей, планирование задач, разработка стратегий или создание комплекса действий для выполнения более объемной задачи, исправление ошибок, решение проблем, выработка соответствующего поведения с учетом полученной информации, реагирование на действия, которые еще не изучены, надлежащее поведение в ситуациях, в которых требуется преодолеть искушение или силу привычки. Исследования людей с фенилкетонурией выявили ухудшени исполнительной функции в сравнении с людьми, которые не страдают данным заболеванием.
Перейти к началу
К
Классическая фенилкетонурия
Этот термин обычно используется для описания пациентов с мутациями гена фенилаланингидроксилазы , который приводит к снижению активности ферментов , вплоть до отсутствия таковой. Вполне вероятно, что у таких пациентов будет развиваться тяжелая форма гиперфенилаланинемии (уровень фенилаланина в крови, как правило, выше 1200 μmol/л) и при этом будет необходима диета с низким содержанием фенилаланина . Для пациентов с классической фенрилкетонурией нехарактерна восприимчивость к лечению сапроптерином .
Кофактор
Некоторым ферментам требуется другая молекула для передачи их функции — кофактор.
Тетрагидробиоптерин является кофактором фенилаланингидроксилазы ( фермента , недостаток которого наблюдается при фенилкетонурии).Куван (Kuvan®)
- Kuvan® — это дигидрохлорид сапроптерина фармацевтическая композиция тетрагидробиоптерина(растворимые таблетки), которая официально применяется для лечения фенилкетонурии во многих странах.
 Тетрагидробиоптерин является кофактором фенилаланингидроксилазы ( фермента , недостаток которого наблюдается при фенилкетонурии).
Тетрагидробиоптерин является кофактором фенилаланингидроксилазы ( фермента , недостаток которого наблюдается при фенилкетонурии).Перейти к началу
Л
Легкая форма гиперфенилаланинемии
К легкой форме гиперфенилаланинемии относятся небольшие концентрации фенилаланина в крови, которые считаются недостаточными для диагностирования фенилкетонурии и применения диеты с низким содержанием фенилаланина .
Легкая форма фенилкетонурии
Низкий или средний уровень фенилаланина в крови, который обнаруживается у людей с мутациями гена фенилаланингидроксилазы и только частично подавляет функцию фермента.
Перейти к началу
М
Материнская фенилкетонурия
Если мать болеет фенилкетонурией, то развивающийся плод подвергается негативному воздействию высокого уровня фенилаланина в крови. Женщинам с фенилкетонурией в период беременности или во время планирования беременности следует принять меры для регуляции уровня фенилаланина в крови. Для плода последствия материнской фенилкетонурии с отсутствием регуляции (если уровень фенилаланина в крови выше 1200 μmol/л) представляют высокий риск (более 90% случаев) умственной отсталости и микроцефалии . Также возможны физические недостатки ( нарушение строения лица) или врожденный порок сердца.
Меланин
Меланин — это гормон, который вызывает потемнение кожи, например, при реакции на солнечный свет. Высокий уровень фенилаланина в крови человека, не соблюдающего лечение фенилкетонурии, подавляет выработку меланина в организме.
В результате этого у пациентов с фенилкетонурией часто бывает светлая кожа.Метаболит
Химическое вещество, которое вырабатывается в организме, обычно в результате функционирования ферментов . Обычно эти вещества используются в последующих биохимических реакциях, расщепляются дальше или выводятся с мочой. Например, птерины в моче (измеряются для диагностики дефицита тетрагидробиоптерина ) являются метаболитами тетрагидробиоптерина.
Миелин
Оболочка, которая состоит из определенных клеток (олигодендроцитов в ЦНС , шванновских клеток — в периферийной), которые покрывают и изолируют нервы, а также повышают скорость передачи нервных импульсов. Недостаточная регуляция уровня фенилаланина в крови в течение длительного времени, как известно, может вызвать повреждение миелина в головном мозге. Однако, остается неясным, каким образом данное явление связано с подавлением исполнительных и когнитивных функций, что наблюдается у пациентов с фенилкетонурией.
Миссенс-мутация
Миссенс- мутация — это мутация одного основания гена , при которой в конечном белке одна аминокислота заменяется другой. Например, миссенс- мутация R408W изменяет ДНК таким образом, что нуклеотид CGG (аргинин) заменяется на TGG (триптофан). Эта замена аминокислоты значительно подавляет активность фенилаланингидроксилазы в образовавшемся белке.
Молчащая мутация
Не все мутации ДНК меняют активность получаемого белка. Например, нуклеотид, изменяясь с ЦAA на ЦАГ, производит в итоговом белке глутамин, в любом случае, и белок будет идентичен любому генотипу. Данная молчащая мутация была описана применительно к фенилаланингидроксилазе .
 В результате этого у пациентов с фенилкетонурией часто бывает светлая кожа.
В результате этого у пациентов с фенилкетонурией часто бывает светлая кожа.Перейти к началу
Н
Нагрузочная проба
См.: Нагрузочная проба на тетрагидробиоптерин
Нагрузочная проба на тетрагидробиоптерин
Медицинское исследование с целью обнаружить у людей восприимчивость к лечению сапроптерином . Положительный результат определяется согласно количеству снижения фенилаланина в крови после лечения (обычно, но не всегда, 30%-снижение уровня фенилаланина в крови сравнивается с уровнем, измеренным непосредственно перед лечением). В данный момент в разных центрах используются разные виды нагрузочных проб.
Нарушение строения
Нарушенное физическое развитие. Распространенным последствием материнской фенилкетонурии является нарушение строения лица, для которого характерна широкая переносица, веконосовые складки, широкий губной желобок, низко расположенные уши, поднятые ноздри и меленькие губы.
Натуральный белок
Натуральный белок — это белок, который содержится в обычных продуктах питания, в отличие от искусственного белка в продуктах лечебного питания.
Недостаток тетрагидробиоптерина
Состояние, при котором генные мутации ослабляют выработку тетрагидробиоптерина, кофермента фенилаланингидроксилазы . Активность любого из нескольких ферментов может стать неполноценной (ГТФ циклогидролаза I; 6-пирувоил- тетрагидробиоптерин синтаза; сепиаптеринредуктаза 1; дигадроптеринредуктаза). Некоторые, но не все формы недостатка тетрагидробиоптерина являются признаками гиперфенилаланинемии , что требует диеты с низким содержанием фенилаланина . Около 1-2% случаев гиперфенилаланинемии обнаружены при обычном осмотре новорожденных по выявленному дефициту тетрагидробиоптерина.
Тетрагидробиоптерин также требует продуцирования ключевых нейромедиаторов в ЦНС , а некоторые формы дефицита тетрагидробиоптерина имеют симптомы, возникающие вследствие измененной активности нейромедиаторов .
Незаменимые аминокислоты
Аминокислоты , которые не синтезируются в организме и должны быть получены с питанием. Для пациентов с фенилкетонурией источником незаменимых аминокислот являются продукты лечебного питания.
Нейропсихологическое тестирование
Долговременная гиперфенилаланинемия нарушает функцию головного мозга. Для отслеживания развития головного мозга у молодых людей, страдающих фенилкетонурией, а также для измерения ряда фугкций головного мозга, как, например, когнитивная функция , память, двигательные навыки и др. необходимо проведение нейропсихологических тестов.
Нейротоксический
Вызывающий повреждение нервных клеток. Долговременное повышение уровня фенилаланина при отсутствии лечения или несвоевременном лечении фенилкетонурии является нейротоксическим для головного мозга и вызывает умственную отсталость или прочие отрицательные последствия.
Несвоевременное лечение
В контексте фенилкетонурии несвоевременное лечение — это отсутствие диеты с низким содержанием фенилаланина у людей с фенилкетонурией с первых дней или недель жизни. Несвоевременное лечение людей с фенилкетонурией почти всегда приводит к нарушениям развития, которые характерны при фенилкетонурии (как, например, тяжелая умственная отсталость).
Нонсенс-мутация
Нонсенс-мутацией называется преобразование нуклеотида аминокислоты в терминирующий кодон (стоп-кодон) . В результате этого образуется укороченный белок, у которого отсутствует биологическая активность. Мутация R111X изменяет нуклеотид ДНК с ЦГА (аргинин) на ТГА (стоп).
Носитель (мутации гена)
- У ребенка может развиться классическая фенилкетонурия , если он наследует от своих родителей (по одной копии от каждого) два мутированных гена фенилаланингидроксилазы , фермента , которого недостаточно у людей с фенилкетонурией. Люди с одной мутацией гена не страдают от фенилкетонурии, однако, они являются носителями мутации. Два носителя мутации гена фенилаланингидроксилазы (который подавляет активность фермента ) в результате получают:
— В одном случае из четырех у них рождается ребенок с фенилкетонурией (две мутированные копии гена ),
— в одном случае из четырех у них рождается ребенок без мутаций , вызывающих фенилкетонурию (нет мутированных копий гена), и
— в одном случае из двух у них рождается ребенок-носитель первой или второй мутации .
Перейти к началу
О
Обменное поглощение
См.: Обменное поглощение фенилаланина
Обменное поглощение фенилаланина
Показатели обменного поглощения фенилаланина — это информация о продуктах, которая указывает, сколько фенилаланина в них содержится. Эти показатели помогают людям с фенилкетонурией планировать свою диету и определить, какую пищу можно есть свободно (в пределах разумного), какую — только в строго регулируемых количествах, а от какой пищи вообще следует отказаться.
Перейти к началу
П
Переносчик больших нейтральных аминокислот
Головной мозг отделен от остального организма барьером (гематоэнцефалическим барьером), который выполняет многие функции, включая точную модуляцию химической среды ЦНС . Большие нейтральные аминокислоты (в том числе и фенилаланин ) необходимы для полноценной деятельности мозга, например, синтеза важных нейромедиаторов, таких как дофамин , норэпинефрин (норадреналин) или серотонин (5-окситриптамин). LNAA переносятся через гематоэнцефалический барьер в мозг при помощи особого белка-переносчика и могут затем использоваться нервными клетками.
Приверженность лечению
- Термин «приверженность лечению» помогает описать, насколько точно пациент следует программе, которую ему назначил специалист. Следовательно, люди могут хорошо или плохо соблюдать назначения врача (напр., принимать прописанные лекарства в нужном количестве в нужное время), специальную диету (напр., прием рекомендуемых продуктов питания в правильных количествах и отказ от нежелательных продуктов, и т.д.). Это касается людей с фенилкетонурией, которые соблюдают диету с низким содержанием фенилаланина или проходят сапроптериновую терапию , к примеру. Термины «приверженность лечению» и « соблюдение указаний врача » являются взаимозаменяемыми.
Проба Гатри
- Метод массового скрининга новорожденных на фенилкетонурию, который до сих пор используется в некоторых странах. Для этого теста берут пробу крови из пятки, помещают ее на фильтровальную бумагу и отправляют в лабораторию. Из бумаги внутри пятна крови выбивают маленькие круглые кусочки, которые помещают на агаровый гель с содержащимися в нем бактериями Bactilis subtilis , для роста которых требуется фенилаланин . В состав геля также входит β-2-тиенилаланин — вещество, которое подавляет рост бактерий путем сдерживания использования фенилаланина этими бактериями. Повышение локальной концентрации фенилаланина (в крови младенца, больного гиперфенилаланинемией ) позволяет преодолеть подавление роста бактерий, и в течение одного дня вокруг положительного образца наблюдается рост кольца бактерий. По диаметру бактериальной колонии можно оценить степень концентрации фенилаланина в образце.
Во многих странах проба Гатри заменена более новыми методами, например, тандемной масс-спектрометрией.
Продукты лечебного питания
Пациенты с фенилкетонурией, как правило, не могут принимать в пищу продукты — натуральные источники белка, поскольку в них содержится фенилаланин . Продукты лечебного питания (иногда их называют заменителями белка ) являются источниками белка, в которых нет фенилаланина , но есть другие аминокислоты , необходимые для полноценного питания.
Птерины (в моче)
- Группа веществ ( метаболитов ), которые вырабатываются в организме в процессе расщепления тетрагидробиоптерина . При недостатке тетрагидробиоптерина у пациентов измеряются свойства птеринов в моче, для того чтобы определить, у какого из ферментов, участвующих в выработке тетрагидробиоптерина, нарушена функция. См. тж. дефицит тетрагидробиоптерина .
См.: Недостаток тетрагидробиоптерина
Пяточная проба
Проба крови для скрининга новорожденных на фенилкетонурию, а также на другие заболевания обычно берется из пятки младенца при помощи укола иглой.
Перейти к началу
Р
Развитие
- Термин с широким значением, охватывает физические и когнитивные изменения, которые происходят вместе с ростом человека; термин обычно относится к молодым людям. Для поддержки нормального развития людей, страдающих фенилкетонурией, важна оптимизация питания — например, диета с низким содержанием фенилаланина.
Разновидность фенилкетонурии
Термин обычно используется для описания пациентов с фенилкетонурией с некоторой остаточной активностью фенилаланингидроксилазы . Уровень фенилаланина в крови обычно ниже, чем в случае классической фенилкетонурии, хотя диета с низким содержанием фенилаланинина в большинстве случаев все еще необходима. Люди с остаточной активностью фенилаланингидроксилазы в большей степени поддаются лечению с помощью сапроптерина, чем люди с классической формой фенилкетонурии.
Рекомендация
- С точки зрения медицины к рекомендациям обычно относятся документы, в которых установлены оптимальные методы и режим лечения в определенной области. В рекомендациях обычно содержатся подробные обзоры медицинских заключений, которыми подтверждаются определенные методы лечения с указаниями по их оптимальному применению. Врачи не обязаны следовать всем этим рекомендациям, в отдельных случаях они могут выносить свою клиническую оценку.
Перейти к началу
С
Сапроптерин
Химическое название формы тетрагидробиоптерина ( Bh5 ): вещество, возникающее естественным образом, необходимое для активности фенилаланингидроксилазы ( фермента , недостаток которого наблюдается при фенилкетонурии). В группе пациентов с фенилкетонурией ( восприимчивый к тетрагидробиоптерину фенотип — обычно, но не обязательно — пациенты с более легкой формой фенилкетонурии), лечение сапроптерином повышает активность фенилаланингидроксилазы и обеспечивает регуляцию фенилаланина в крови.
Своевременное лечение
В контексте фенилкетонурии своевременное лечение — это назначение и поддержка диеты с низким содержанием фенилаланина с первых дней или недель жизни человека для смягчения негативного воздействия повышенного уровня фенилаланина в крови на развитие человека.
Сдвиг рамки считывания
- В синтезе белка ДНК вначале считывается матричной РНК, созданной из цепочки нуклеотидов. В генетическом коде набор из трех смежных оснований (нуклеотидов) в цепочке РНК кодирует каждую аминокислоту белка. Нуклеотиды также обеспечивают другую информацию, например, о завершении. Рамка считывания состоит из последовательности этих нуклеотидов . Если количество пар оснований, которые вставляются или удаляются из ДНК, не кратно трем, то происходит мутация со сдвигом рамки. С этого момента происходит изменение рамки считывания. Например, рассмотрим следующую цепочку РНК, в которой основания показаны как рамка считывания шести нуклеотидов, которые завершаются стоп-кодоном.
… UUA UAC AGU AAA GCC UAG
Лейцин Тирозин Серин Лизин Аланин СТОП
Теперь предположим, что вставляется дополнительное отдельное основание
( миссенс-мутация , выделена красным):… UUC AUA CAG UAA AGC CUA
Фенилаланин Изолейцин Глутамин СТОП Не считан
Скрининг новорожденных
Процедура тестирования всех новорожденных на определенные заболевания, как, например, фенилкетонурию, для своевременного оказания медицинской помощи с целью предотвращения длительного вредного воздействия выявленного заболевания на ребенка. Также называется « скрининг новорожденных ».
Скрининг новорожденных
См.: Скрининг новорожденных
Соблюдение указаний врача
- По значению подобно приверженности лечению, употребляется для того, чтобы охарактеризовать, насколько пациент соблюдает программу лечения, согласованную между ним и его лечащим врачом. Соблюдение диеты с низким содержанием фенилаланина — главный аспект лечения фенилкетонурии, например, когда недостаточное соблюдение указаний врача зачастую может привести к гиперфенилаланинемии, и при этом повышает риск осложнений при фенилкетонурии со стороны нервной системы, как, например, ухудшение исполнительной и когнитивной деятельности.
Содержание фенилаланина/тирозина
Большая часть тирозина в организме обычно вырабатывается из фенилаланина с помощью фермента — фенилаланингидроксилазы . Если функция фенилаланингидроксилазы нарушена, как при фенилкетонурии, то уровни этих двух аминокислот могут быть нарушены (высокий уровень фенилаланина , низкий уровень тирозина ). Это имеет важное значение, в особенности для функции головного мозга, для обеспечения которой требуются и фенилаланин , и тирозин . Для некоторых людей, страдающих фенилкетонурией, достижение нормального соотношения между уровнями фенилаланина и тирозина может быть важнейшим аспектом их лечения с применением диеты с низким содержанием фенилаланина и, в некоторых случаях, продуктов лечебного питания с добавкой тирозина.
Специальная диета
- Пациенты с фенилкетонурией должны строго регулировать прием фенилаланина, им необходима специальная диета с низким содержанием фенилаланина (см. Продукты лечебного питания ).
См.: Продукты лечебного питания
Сращивание
Прерывание последовательности пар оснований ДНК для ввода новой ДНК или удаления уже существующей. Процесс мутации сращивания IVS10 распространен среди населения с фенилкетонурией, при этом активность фенилаланингидроксилазы существенно уменьшается.
Перейти к началу
Т
Тандемная масс-спектрометрия
Передовая техника для массового осмотра новорожденных, которая позволяет сделать анализ крови младенцев на фенилкетонурию, а также другие наследственные заболевания. Это наиболее продвинутая методология скрининга из всех доступных сегодня, которая во многих странах почти заменила старые методы ( проба Гатри , флуорометрическое обследование ).
Терминирующий кодон (стоп-кодон)
Последовательность трех нуклеотидов (оснований) в ДНК/РНК, которая дает команду, чтобы остановить транскрипцию ДНК в РНК или выработку белка.
Тетрагидробиоптерин
Вещество, возникающее естественным образом, необходимое для активности фенилаланингидроксилазы ( фермента , недостаток которого наблюдается при фенилкетонурии). В Японии доступна фармакологическая технология изготовления тетрагидробиоптерина.
Тир
- Сокращенное название тирозина, аминокислоты , которая главным образом вырабатывается с помощью преобразования фенилаланина ферментом фенилаланингидроксилазой.
См.: Фенилкетонурия
Триплет (кодон)
Последовательность из трех нуклеотидов в ДНК/РНК, которая кодирует отдельную аминокислоту или определяет окончание синтеза цепи ( стоп-кодон ).
Перейти к началу
У
Умственная отсталость
- Недостаточное развитие интеллектуальных способностей. Если не проводить лечение фенилкетонурии в раннем детстве, то это может привести к серьезной умственной отсталости.
См.: Cretinism
Уровень фенилаланина
Измеряемый уровень фенилаланина в крови. Показатель уровня фенилаланина в крови используется для диагностики фенилкетонурии и в случае подтверждения — для назначения диеты с низким содержанием фенилаланина или другого лечения фенилкетонурии. Рекомендуемый уровень фенилаланина в крови может изменяться в зависимости от возраста.
Перейти к началу
Ф
Фенилаланингидроксилаза
- Это фермент ( ген EC 1.14.16.1, локус 12q24.1), который преобразует фенилаланин (полученный из продуктов питания) в другую аминокислоту , тирозин . У людей с фенилкетонурией в генах фенилаланингидроксилазы имеется мутация , которая сокращает или совсем блокирует ее активность. Мутации в обеих копиях гена фенилаланингидроксилазы являются причиной клинической фенилкетонурии.
Синонимы: PAH
Фенилкетонурия
- Наследственное аутосомно-рецессивное заболевание, вызванное мутацией гена фермента фенилаланингидроксилазы , который в обычном состоянии преобразовывает фенилаланин в тирозин . Возникающая вследствие этого концентрация фенилаланина в крови является токсичной для центральной нервной системы , если уровень фенилаланина не регулируется при помощи специальной диеты или другого метода лечения (см. Сапроптерин , Аминокислоты ).
См.: Аминокислота Сапроптерин
Фенотип
Данный термин служит для характеристики внешности человека в медицинском смысле. Например, тех людей, у которых реакция на лечение сапроптерином выражается в значительном снижении уровня фенилаланина в крови, можно охарактеризовать как «фенотип с восприимчивостью к тетрагидробиоптерину при фенилкетонурии».
Ферменты
- Ферменты — это белки, которые действуют в качестве катализатора жизненно важных биохимических реакций. Фенилаланингидроксилаза — это фермент, который отвечает за преобразование фенилаланина в тирозин , недостаток которого возникает при фенилкетонурии.
Флуориметрический анализ
Этот метод применяется в некоторых странах для скрининга новорожденных на фенилкетонурию и другие наследственные нарушения обмена веществ. Подобно другим методам, с его помощью измеряется уровень фенилаланина в крови. Во многих странах данный метод был заменен тандемной масс-спектрометрией.
Перейти к началу
Ц
ЦНС
- центральная нервная система: головной и спинной мозг.
Перейти к началу
Ш
Шаперон («наставник»)
Шаперон (молекулярный шаперон) служит для сохранения структуры и функции другой молекулы, как правило, протеина. Например, тетрагидробиоптерин является коферментом для фенилаланингидроксилазы . Подгруппа пациентов (обычно, но не исключительно, с легкой формой фенилкетонурии) реагирует на лечение тетрагидробиоптерином , что выражается в повышении активности фенилаланингидроксилазы и снижении уровня фенилаланина в крови. Считается, что мутация фенилаланингидроксилазы изменяет ее активность путем изменения ее 3-мерной структуры. Тетрагидробиоптерин , как считают исследователи, помогает сохранить структуру мутантного белка фенилаланингидроксилазы таким образом, что активность фермента сохраняется. Это может также предотвратить разрушение белка фенилаланингидроксилазы или его инактивацию другими ферментами . Таким образом, тетрагидробиоптерин действует как «молекулярный шаперон».
Перейти к началу
Э
Экзема
Экзема — это состояние кожи, которое характеризуется воспалением, сухостью, шелушением и зудом. Как правило, наблюдается образование везикул (пузырьков), после чего может возникнуть эритема (покраснение), эдема (припухлости), появление бугорков на коже (папул), а затем утолщение и огрубение кожи. Экзема является широко распространенной особенностью фенилкетонурии, если при этом не соблюдается диета с низким содержанием фенилаланина с первых дней жизни.
Экзоны
Экзоны и интроны являются частями генов в их последовательности ДНК. Экзон — это участок гена, который будет представлен в образованном белке. Участки РНК, к которым относятся интроны , удаляются из гена для формирования зрелой РНК, которая переводится в белок.
Эпилепсия
Эпилепсия — это состояние, при котором несогласованная электрическая активность в мозге приводит к потере сознания и судорогам. Эпилепсия является широко распространенной особенностью фенилкетонурии, если при этом не соблюдается диета с низким содержанием фенилаланина с первых дней жизни.
Перейти к началу
Википедия — свободная энциклопедия
Избранная статья
Прохождение Венеры по диску Солнца — разновидность астрономического прохождения (транзита), — имеет место тогда, когда планета Венера находится точно между Солнцем и Землёй, закрывая собой крошечную часть солнечного диска. При этом планета выглядит с Земли как маленькое чёрное пятнышко, перемещающееся по Солнцу. Прохождения схожи с солнечными затмениями, когда наша звезда закрывается Луной, но хотя диаметр Венеры почти в 4 раза больше, чем у Луны, во время прохождения она выглядит примерно в 30 раз меньше Солнца, так как находится значительно дальше от Земли, чем Луна. Такой видимый размер Венеры делает её доступной для наблюдений даже невооружённым глазом (только с фильтрами от яркого солнечного света), в виде точки, на пределе разрешающей способности глаза. До наступления эпохи покорения космоса наблюдения этого явления позволили астрономам вычислить расстояние от Земли до Солнца методом параллакса, кроме того, при наблюдении прохождения 1761 года М. В. Ломоносов открыл атмосферу Венеры.
Продолжительность прохождения обычно составляет несколько часов (в 2004 году оно длилось 6 часов). В то же время, это одно из самых редких предсказуемых астрономических явлений. Каждые 243 года повторяются 4 прохождения: два в декабре (с разницей в 8 лет), затем промежуток в 121,5 года, ещё два в июне (опять с разницей 8 лет) и промежуток в 105,5 года. Последние декабрьские прохождения произошли 9 декабря 1874 года и 6 декабря 1882 года, а июньские — 8 июня 2004 года и 6 июня 2012 года. Последующие прохождения произойдут в 2117 и 2125 годах, опять в декабре. Во время прохождения наблюдается «явление Ломоносова», а также «эффект чёрной капли».
Хорошая статья
Резня в Благае (сербохорв. Масакр у Благају / Masakr u Blagaju) — массовое убийство от 400 до 530 сербов хорватскими усташами, произошедшее 9 мая 1941 года, во время Второй мировой войны. Эта резня стала вторым по счету массовым убийством после создания Независимого государства Хорватия и была частью геноцида сербов.
Жертвами были сербы из села Велюн и его окрестностей, обвинённые в причастности к убийству местного мельника-хорвата Йосо Мравунаца и его семьи. Усташи утверждали, что убийство было совершено на почве национальной ненависти и свидетельствовало о начале сербского восстания. Задержанных сербов (их число, по разным оценкам, составило от 400 до 530 человек) содержали в одной из школ Благая, где многие из них подверглись пыткам и избиениям. Усташи планировали провести «народный суд», но оставшаяся в живых дочь Мравунаца не смогла опознать убийц среди задержанных сербов, а прокуратура отказалась возбуждать дело против кого-либо без доказательства вины. Один из высокопоставленных усташей Векослав Лубурич, недовольный таким развитием событий, организовал новый «специальный суд». День спустя дочь Мравунаца указала на одного из задержанных сербов. После этого 36 человек были расстреляны. Затем усташи казнили остальных задержанных.
Изображение дня
Эхинопсисы, растущие на холме посреди солончака Уюни
Какие типы вариантов гена возможны ?: MedlinePlus Genetics
Последовательность ДНК гена может быть изменена несколькими способами. Варианты генов (также известные как мутации) могут по-разному влиять на здоровье в зависимости от того, где они возникают и изменяют ли они функцию основных белков. Типы вариантов включают следующие:
Замена
Вариант этого типа заменяет один строительный блок ДНК (нуклеотид) другим. Варианты замены можно дополнительно классифицировать по их влиянию на продукцию белка из измененного гена.
Миссенс
Миссенс вариант — это тип
замены, при которой изменение нуклеотида приводит к замене одного строительного блока белка (аминокислоты) другим в белке, полученном из гена. Замена аминокислоты может изменить функцию белка.
Ерунда
Бессмысленный вариант — это еще один вид подмены. Однако вместо того, чтобы вызвать изменение одной аминокислоты, измененная последовательность ДНК приводит к стоп-сигналу, который преждевременно сигнализирует клетке о прекращении создания белка.Этот тип варианта приводит к укороченному белку, который может функционировать неправильно, быть нефункциональным или разрушаться.
Вставка
Вставка изменяет последовательность ДНК, добавляя к гену один или несколько нуклеотидов. В результате белок, полученный из гена, может не функционировать должным образом.
Удаление
Делеция изменяет последовательность ДНК, удаляя по крайней мере один нуклеотид в гене. Небольшие делеции удаляют один или несколько нуклеотидов в гене, в то время как более крупные делеции могут удалять весь ген или несколько соседних генов.Удаленная ДНК может изменить функцию затронутого белка или белков.
Удаление-вставка
Этот вариант возникает, когда делеция и вставка происходят одновременно в одном и том же месте гена. В варианте с делецией-вставкой по меньшей мере один нуклеотид удален и по меньшей мере один нуклеотид вставлен. Однако изменение должно быть достаточно сложным, чтобы отличаться от простой замены. Полученный белок может не функционировать должным образом. Вариант с делецией-вставкой (делины) также может быть известен как вариант с вставкой-удалением (indel).
Дублирование
Дупликация происходит, когда участок из одного или нескольких нуклеотидов в гене копируется и повторяется рядом с исходной последовательностью ДНК. Этот тип варианта может изменить функцию белка, полученного из гена.
инверсия
Инверсия изменяет более одного нуклеотида в гене путем замены исходной последовательности той же последовательностью в обратном порядке.
Сдвиг рамы
Рамка считывания состоит из групп по три нуклеотида, каждая из которых
код для одной аминокислоты.А
Вариант смещения рамки считывания возникает, когда происходит добавление или потеря нуклеотидов, которые сдвигают группировку и изменяют код для всех последующих аминокислот. Получающийся в результате белок обычно нефункциональный. Вставки, удаления и дублирования могут быть вариантами со сдвигом кадра.
Повторить раскрытие
Некоторые участки ДНК содержат короткие последовательности нуклеотидов, которые повторяются несколько раз подряд. Например, тринуклеотидный повтор состоит из последовательностей из трех нуклеотидов, а тетрануклеотидный повтор состоит из последовательностей из четырех нуклеотидов.А
повторная экспансия — это вариант, который увеличивает количество повторений короткой последовательности ДНК. Этот тип варианта может привести к неправильному функционированию полученного белка.
Миссенс-мутация — обзор
Мутации кальциевого канала при гипокалиемическом периодическом параличе
Миссенс-мутации в остатках аргинина в сегментах датчика напряжения S4 Са V 1.1 составляют примерно 60% случаев семейной гипоПП (40,49 ). Канал скелетных мышц Ca 2+ L-типа представляет собой гетеропентамер с основной порообразующей субъединицей α 1 , Ca V 1.1, и четыре дополнительных субъединицы, β, γ, α 2 и δ. Мутации только субъединицы α 1 Ca V 1.1 были идентифицированы в семейном HypoPP. Напротив, мутации Ca V 1.1 также описаны в отношении предрасположенности к злокачественной гипертермии.
Как и в случае с другими формами периодического паралича, провоцирующим событием для острого приступа в Ca V 1.1 — HypoPP является деполяризация остатка V , которая инактивирует каналы Na + и делает волокно невосприимчивым (4).Механизм, с помощью которого вызванные деполяризацией эпизоды паралича в HypoPP могут быть причинно связаны с миссенс-мутациями в Ca V 1.1, оставался загадкой более 10 лет. Канал L-типа Ca 2+ в скелетных мышцах функционирует в первую очередь как датчик напряжения, который связывает деполяризацию TT с открытием рецептора рианодина и высвобождением Ca 2+ из SR. Канал не играет известной роли в стабилизации покоя V , и в биопсированных волокнах пациентов с HypoPP не было обнаружено никаких дефектов связи возбуждения и сокращения.Понимание патомеханизма HypoPP для мутаций Ca V 1.1 было по аналогии с дефектами, вызванными мутациями в гомологичных регионах сегментов S4 в Na V 1.4 (39,41). Было высказано предположение, что ток утечки стробирующей поры, аналогичный измеренному в мутантных каналах HypoPP — Na V 1.4, продуцируется аргининовыми мутациями в S4 Ca V 1.1. Этот аномальный входящий ток при V rest может вызвать восприимчивость к парадоксальной деполяризации с гипокалиемией в Ca V 1.1-ГипоПП. Экспериментальное подтверждение пока невозможно, потому что мембранная экспрессия Ca V 1.1 плохая в гетерологичных системах экспрессии. Гипотеза получила подтверждение в результате наблюдений, что (i) мутации аргинина, спроектированные в сегменты S4 других каналов (например, канал Shaker K), приводят к стробированию поровых токов, (ii) пять из шести известных мутаций HypoPP в Ca V 1.1. представляют собой миссенс-мутации аргининов в S4, причем выбросом является валин для глутамата в соседнем сегменте S3, который в принципе может поддерживать ток закрывающейся поры, и (iii) шесть из девяти аргининовых мутаций в S4 Na V 1.4 были испытаны экспериментально, и все шесть производят токи затворных пор; тогда как преобразование S4 аргинина в цистеин в Na V 1.4, связанное с PMC, не вызывает ток затворной поры (50).
Миссенс-мутация — обзор
Мутации кальциевого канала при гипокалиемическом периодическом параличе
Миссенс-мутации в остатках аргинина в сегментах датчика напряжения S4 Са V 1.1 составляют примерно 60% случаев семейной гипоПП (40,49 ). Канал скелетных мышц Ca 2+ L-типа представляет собой гетеропентамер с основной порообразующей субъединицей α 1 , Ca V 1.1, и четыре дополнительных субъединицы, β, γ, α 2 и δ. Мутации только субъединицы α 1 Ca V 1.1 были идентифицированы в семейном HypoPP. Напротив, мутации Ca V 1.1 также описаны в отношении предрасположенности к злокачественной гипертермии.
Как и в случае с другими формами периодического паралича, провоцирующим событием для острого приступа в Ca V 1.1 — HypoPP является деполяризация остатка V , которая инактивирует каналы Na + и делает волокно невосприимчивым (4).Механизм, с помощью которого вызванные деполяризацией эпизоды паралича в HypoPP могут быть причинно связаны с миссенс-мутациями в Ca V 1.1, оставался загадкой более 10 лет. Канал L-типа Ca 2+ в скелетных мышцах функционирует в первую очередь как датчик напряжения, который связывает деполяризацию TT с открытием рецептора рианодина и высвобождением Ca 2+ из SR. Канал не играет известной роли в стабилизации покоя V , и в биопсированных волокнах пациентов с HypoPP не было обнаружено никаких дефектов связи возбуждения и сокращения.Понимание патомеханизма HypoPP для мутаций Ca V 1.1 было по аналогии с дефектами, вызванными мутациями в гомологичных регионах сегментов S4 в Na V 1.4 (39,41). Было высказано предположение, что ток утечки стробирующей поры, аналогичный измеренному в мутантных каналах HypoPP — Na V 1.4, продуцируется аргининовыми мутациями в S4 Ca V 1.1. Этот аномальный входящий ток при V rest может вызвать восприимчивость к парадоксальной деполяризации с гипокалиемией в Ca V 1.1-ГипоПП. Экспериментальное подтверждение пока невозможно, потому что мембранная экспрессия Ca V 1.1 плохая в гетерологичных системах экспрессии. Гипотеза получила подтверждение в результате наблюдений, что (i) мутации аргинина, спроектированные в сегменты S4 других каналов (например, канал Shaker K), приводят к стробированию поровых токов, (ii) пять из шести известных мутаций HypoPP в Ca V 1.1. представляют собой миссенс-мутации аргининов в S4, причем выбросом является валин для глутамата в соседнем сегменте S3, который в принципе может поддерживать ток закрывающейся поры, и (iii) шесть из девяти аргининовых мутаций в S4 Na V 1.4 были испытаны экспериментально, и все шесть производят токи затворных пор; тогда как преобразование S4 аргинина в цистеин в Na V 1.4, связанное с PMC, не вызывает ток затворной поры (50).
Мутация и восстановление ДНК
Мутация и восстановление ДНК
Мутация ДНК и
Ремонт
Мутация, которая может
возникают во время репликации и / или рекомбинации, это постоянное изменение
нуклеотидная последовательность ДНК. Поврежденная ДНК может быть мутирована либо путем замены, либо
удаление или вставка пар оснований.Мутации по большей части безвредны
кроме случаев, когда они приводят к гибели клеток или образованию опухолей. Из-за смертельного
потенциал мутаций ДНК клетки развили механизмы для восстановления поврежденных
ДНК.
Типы мутаций
Существует три типа мутаций ДНК: замены оснований, делеции.
и прошивки.
1. Базовые замены
Одиночная база замен
называются точечными мутациями, вспомним точечную мутацию Glu ——> Val, которая
вызывает серповидно-клеточную анемию.Точечные мутации — самый распространенный тип мутаций.
и есть два типа.
Переход : это происходит
когда пурин замещен другим пурином или когда пиримидин замещен
с другим пиримидином.Трансверсия : когда пурин
замещает пиримидин или пиримидин заменяет пурин.
Точечные мутации, встречающиеся в ДНК
последовательности, кодирующие белки, либо молчащие, либо бессмысленные, либо бессмысленные.
Silent : если abase замена
встречается в третьей позиции кодона, есть большая вероятность, что синоним
кодон будет сгенерирован. Таким образом, аминокислотная последовательность, кодируемая геном, имеет вид
не изменен, и мутация молчит.
Отсутствие : При подстановке базы
приводит к генерации кодона, который определяет другую аминокислоту и
следовательно, приводит к другой полипептидной последовательности.В зависимости от типа амино
кислотная замена миссенс-мутация может быть консервативной или неконсервативной.
Например, если структура и свойства замещенной аминокислоты являются
очень похожа на исходную аминокислоту, мутация считается консервативной
и, скорее всего, мало повлияет на структуру результирующих белков.
/ функция. Если замена приводит к аминокислоте с очень другой структурой
и свойства, мутация неконсервативна и, вероятно, будет вредной
(плохо) для результирующей структуры / функции белков (т.е.е. серповидноклетка
точечная мутация).
Ерунда : При подстановке базы
приводит к стоп-кодону, в конечном итоге усекающему перевод и, скорее всего, ведущему
к нефункциональному белку.
2. Исключения
Удаление, приводящее к сдвигу кадра,
результат, когда одна или несколько пар оснований теряются из ДНК (см. рисунок выше).
Если одна или две базы удаляются, трансляционный фрейм изменяется, в результате чего
в искаженном сообщении и нефункциональном продукте.Удаление трех и более
основания оставляют рамку считывания нетронутой. Результат удаления одного или нескольких кодонов
в белке отсутствует одна или несколько аминокислот. Это может быть вредно или нет.
3. Вставки
Вставка дополнительной базы
пары могут приводить к сдвигу кадра в зависимости от того, кратно ли трем
вставлены пары оснований. Комбинации вставок и удалений, приводящие к
также возможны различные исходы.
Причины мутаций
Ошибки репликации ДНК
В очень и очень редких случаях ДНК
полимераза будет включать некомплементарное основание в дочернюю цепь.
Во время следующего раунда репликации неверно инкорпорированная база приведет к
мутация. Однако это случается очень редко, поскольку экзонуклеаза выполняет функцию корректора.
механизм распознавания несовпадающих пар оснований и их удаления.
Ошибки рекомбинации ДНК
ДНК часто перестраивается
процесс, называемый рекомбинацией, протекает через множество механизмов. Время от времени
ДНК теряется во время репликации, что приводит к мутации.
Химическое повреждение ДНК
Многие химические мутагены, некоторые экзогенные,
некоторые искусственные, некоторые из окружающей среды способны повредить ДНК. Многие химиотерапевтические
лекарства и препараты интеркалирующих агентов действуют, повреждая ДНК.
Излучение
Гамма-лучи, рентгеновские лучи, даже УФ-свет
может взаимодействовать с соединениями в клетке, генерируя свободные радикалы, которые вызывают
химическое повреждение ДНК.
Ремонт ДНК
Поврежденная ДНК может быть восстановлена несколькими
разные механизмы.
Устранение несоответствия
Иногда ДНК-полимераза включает
неправильный нуклеотид во время синтеза цепи и система редактирования от 3 ‘до 5’,
экзонуклеаза, не может исправить это.Эти несоответствия, а также одиночные базовые вставки
и удаления исправляются с помощью механизма исправления несоответствия. Исправление несоответствия
полагается на вторичный сигнал в ДНК, чтобы различать родительские
цепь и дочерняя цепь, содержащая ошибку репликации. Клетки человека
обладает системой восстановления несоответствия, аналогичной системе E. coli, которая описана
здесь. Метилирование последовательности GATC происходит на обеих цепях через некоторое время после
Репликация ДНК.Поскольку репликация ДНК полуконсервативна, новая дочь
цепь остается неметилированной в течение очень короткого периода времени после репликации.
Это различие позволяет системе исправления несоответствий определить, какая цепь
содержит ошибку. Белок, MutS распознает и связывает несовпадающее основание.
пара.
Другой белок, MutL, затем связывается
к MutS, а частично метилированная последовательность GATC распознается и связывается
эндонуклеаза, MutH.Комплекс MutL / MutS затем связывается с MutH, который разрезает
неметилированная цепь ДНК на сайте GATC. ДНК-геликаза, MutU раскручивает
Нить ДНК в направлении несовпадения и экзонуклеаза разрушает
прядь. Затем ДНК-полимераза заполняет разрыв, а лигаза закрывает его. Дефекты
гены репарации несоответствия, обнаруженные у людей, по-видимому, связаны с
развитие наследственного колоректального рака.
Эксцизионное восстановление нуклеотидов
(NER)
NER в клетках человека начинается с
образование комплекса белков XPA, XPF, ERCC1, HSSB при поражении на
ДНК.Фактор транскрипции TFIIH, содержащий несколько белков, затем связывается
к комплексу в АТФ-зависимой реакции и делает разрез. Результирующий
Затем 29-нуклеотидный сегмент поврежденной ДНК разматывается, разрыв заполняется (ДНК
полимераза) и ника (лигаза).
Прямой ремонт поврежденной ДНК
Иногда повреждение базы может быть
непосредственно восстанавливается специализированными ферментами без удаления нуклеотида.
Восстановление рекомбинации
Этот механизм позволяет ячейке
повторить повреждение и исправить его позже.
Регламент контроля повреждений
У млекопитающих регулируется репарация ДНК
клетки с помощью сенсорного механизма, который обнаруживает повреждение ДНК и активирует белок
называется p53. p53 — фактор регуляции транскрипции, который контролирует экспрессию
некоторых генных продуктов, влияющих на клеточный цикл, репликацию ДНК и репарацию ДНК.Вот некоторые из функций p53, которые только сейчас выясняются: стимуляция.
экспрессии генов, кодирующих p21 и Gaad45. Потеря функции p53 может
быть вредным, около 50% всех раковых заболеваний человека имеют мутированный ген p53.
Белок p21 связывает и инактивирует
киназа клеточного деления (CDK), которая приводит к остановке клеточного цикла. p21 также связывает
и инактивирует PCNA, что приводит к инактивации репликационных вилок. В
Комплекс PCNA / Gaad45 участвует в эксцизионной репарации поврежденной ДНК.
Некоторые примеры болезней, возникающих в результате
от дефектов в механизмах репарации ДНК.
Ксеродерма
пигментный
Кокейна
синдром
Наследственный
неполипозный колоректальный рак
© Д-р Ноэль
Штурм 2019
Заявление об ограничении ответственности: Взгляды и мнения, выраженные на неофициальных страницах штата Калифорния
Университет, преподаватели, сотрудники или студенты университета Домингес Хиллс являются строго такими же
авторы страницы.Содержание этих страниц не проверялось или
одобрен Калифорнийским государственным университетом, Домингес Хиллз.
Расшифровка болезнетворных механизмов миссенс-мутаций из супрамолекулярных структур
Сначала мы собрали информацию о миссенс-мутациях из базы данных онлайн-менделевского наследования человека (OMIM) 19 и опубликовали литературу и связали их с фенотипами. Всего в таблицу было включено 2512 пар ген-заболевание (1951 уникальный ген и 2366 уникальных заболеваний) с аутосомно-рецессивным (AR) или аутосомно-доминантным (AD) типами наследования (таблица 1 и дополнительная таблица 1).Кроме того, для 404 из 1140 заболеваний AD типы вызывающих болезнь механизмов, а именно HI, DN или GF, миссенс-мутаций были назначены вручную (дополнительная таблица 2). Наконец, всего 22 004 сайта патогенных миссенс-мутаций были картированы на 3D-структурах белков, для которых известные молекулярные взаимодействия аннотированы 20, 21 . Из этих остатков 14 164 были отнесены к упорядоченным (определяемым структурой) участкам. Несинонимичные замены, наблюдаемые у здоровых людей, обозначенные как однонуклеотидные варианты (SNV), полученные из базы данных ExAC 2 без какого-либо порогового значения для частоты аллелей, также были нанесены на карту в структурах, и локальные распределения сравнили с патогенными. миссенс-мутации.
Таблица 1 Статистика патогенных и доброкачественных мутаций.
Результаты показали, что мутации AR и AD, отнесенные к упорядоченным областям, были значительно выше по количеству, чем SNV, почти половина (47,8%) из которых были отнесены к внутренне неупорядоченным областям (рис. 1). Когда для SNV использовалось пороговое значение 1% частоты минорного аллеля (порог для SNP), это , то есть . Всего было исключено 12 215 SNV с более низкой частотой, доля остатков, расположенных в неупорядоченных областях, была немного увеличена до 49.3%, тогда как те, которые расположены на границе раздела молекул, немного уменьшились (с 13,6% до 12,7%). Это означает, что несинонимичные замены, которые не были связаны с заболеваниями, имели тенденцию избегать областей, важных для формирования структуры или молекулярных взаимодействий, как также сообщалось в предыдущих исследованиях 12, 16,17,18,19,20,21,22 .
Рисунок 1
Распределение сайтов мутаций в белковых структурах. ( a — f ) Круговые диаграммы представляют распределение миссенс-мутаций в различных структурных областях, а именно, на границе раздела молекул, скрытых, открытых или неупорядоченных областях для AD ( a ), AR ( b ) , SNV ( c ), HI ( d ), DN ( e ) и GF ( f ).
Локальное распределение мутаций AR и AD на трехмерных структурах преимущественно различалось. Для мутаций AR 43,0% были расположены в скрытой области, тогда как такое же значение для мутаций AD составляло только 21,5%. Мутации AD были в значительной степени обогащены остатками, расположенными на межмолекулярных интерфейсах комплексов белок-белок или белок-ДНК.
Затем мы сравнили структурные особенности доминантных фенотипов HI, DN и GF (рис. 1d-f). Мутации DN и GF, но не HI, были относительно обогащены межмолекулярными интерфейсами.В скрытых областях было обнаружено больше мутаций HI, чем ожидалось, по сравнению с мутациями DN и GF (дополнительный рис. 1), что указывает на то, что структурная локализация мутаций HI была подобна таковой мутаций AR. Считается, что мутации AR вызывают потерю функции продуктов гена за счет дестабилизации структуры белка, и результат предполагает, что лежащий в основе молекулярный механизм будет аналогичен фенотипам HI.
Интерфейсы были разделены на интерфейсы для белков (PPI) и ДНК (PDI), чтобы глубже изучить влияние мутаций на молекулярные взаимодействия (рис.2а, б). Мутации AD были смещены в сторону областей PPI [отношение шансов (OR) против случайного распределения составляло 1,32 с 95% доверительным интервалом (CI) от 1,25 до 1,40]. Для мутаций AR эта тенденция была противоположной (OR 0,96, 95% CI от 0,90 до 1,02). Когда мутации AD были дополнительно проанализированы, локализация мутаций DN и GF для PPI была довольно подчеркнута (OR 1,45, 95% CI от 1,28 до 1,67 для DN и OR 1,27, 95% CI от 1,04 до 1,54 для GF), тогда как для HI (OR 1.13, 95% доверительный интервал от 0,96 до 1,33). Сайты мутаций HI, а также DN были значительно обогащены PDI (OR 2,46, 95% CI от 2,09 до 2,88 для HI и OR 2,30, 95% CI от 1,88 до 2,78 для DN) по сравнению с GF (OR 1,17, 95% ДИ от 0,17 до 2,35).
Рисунок 2
Отношение шансов мутаций на интерфейсах для каждого режима наследования. ( a — d ) Распределение отношения шансов вероятностей того, что мутации в категории произошли в данном интерфейсе белков ( a ) или ДНК ( b ).( c ) Соотношения мутаций, используемых для гомо-по сравнению с гетеро-субъединичными взаимодействиями. ( d ) Соотношения мутаций, используемые для доменных взаимодействий.
Хорошо известно, что дефекты факторов транскрипции часто вызывают заболевания по механизму гаплонедостаточности 23 . Соответственно, большая часть генов, связанных с фенотипами HI (36 генов, 31,3% от общего числа) и DN (26 генов, 19,5%), кодирует факторы транскрипции по сравнению с генами с фенотипами GF (6 генов, 5.1%). Одно из возможных объяснений того, почему мутации в сайтах связывания ДНК факторов транскрипции вызывают гаплонедостаточность, состоит в том, что дефекты способности связывания ДНК отражают их собственные уровни экспрессии. Большинство таких факторов транскрипции связываются с промоторами своих генов и активируют экспрессию.
Например, миссенс-мутации GATA2 , кодирующие белок фактора транскрипции GATA2, вызывают семейные синдромы с иммунодефицитом гаплонедостаточным образом.GATA2 связывается со своим собственным промотором и активирует транскрипцию, а миссенс-мутации в сайтах связывания ДНК нарушают способность связывания промотора, что приводит к снижению транскрипции GATA2
24 . Другой пример может объяснить доминантно-отрицательные механизмы: мутации в PITX2 , кодирующем гомеодоменный белок PITX2, ответственны за синдром Аксенфельда-Ригера (MIM # 180500). Известно, что PITX2 действует как гомодимер. Саади и др. .продемонстрировали, что мутантный белок PITX2 с миссенс-мутацией Lys88Glu в сайте связывания ДНК образует довольно стабильные гетеродимеры с PITX2 дикого типа и значительно снижает связывание с промотором, вызывая, таким образом, болезнь DN 25 .
Чтобы выяснить, вносят ли взаимодействия гомо или гетеро субъединицы вклад в различия в механизмах, вызывающих заболевание, интерфейсы PPI были далее разделены на интерфейсы гомо и гетеро субъединиц (Fig. 2c). Мутации AD были слегка смещены в сторону гомоинтерфейсов (OR мутаций, наблюдаемых в гомоинтерфейсах, по сравнению с мутациями в гетероинтерфейсах против случайного распределения было 1.15, 95% доверительный интервал от 1,04 до 1,26). Мутации DN имели тенденцию быть на гомо-интерфейсах (OR 2,58, 95% CI от 2,05 до 3,28), тогда как для мутаций HI и GF не наблюдалось значительного смещения между интерфейсами гомо и гетеро субъединиц.
Обогащенный анализ Gene Ontology показал, что термин GO для «гомоолигомеризации белков» был значительно обогащен генами, связанными с фенотипами DN (12 генов, p = 9,6 × 10 −8 ) по сравнению с генами с HI ( без генов) или GF (3 гена, p > 0.05) фенотипы, предполагая, что белки, которые действуют как гомоолигомеры, сильно связаны с фенотипами DN. Хотя общее количество остатков для интерфейсов гомо-субъединиц (6865 остатков) существенно не отличалось от таковых для интерфейсов гетеро-субъединиц (6174 остатка) в белках с фенотипами DN, мутации были значительно обогащены ( p = 8,7 × 10 -7 по биномиальному тесту) в гомо (283 остатка) по сравнению с интерфейсами гетеро (110 остатков) субъединиц в тех же белках.Мутации в интерфейсах гомо субъединиц могут нарушать образование соответствующих олигомеров или вносить вклад в образование неактивных олигомеров доминантно-негативным образом, как предполагалось ранее 26 .
Мы дополнительно исследовали структурные особенности миссенс-мутаций с точки зрения доменных взаимодействий в одной белковой молекуле. Конфигурации доменов также важны для регуляции функций белков, и некоторые из остатков, не участвующих в межмолекулярных взаимодействиях, используются для междоменных взаимодействий.База данных ECOD использовалась при определении границ домена, и некоторые из мутантных сайтов, которые были назначены скрытым или неинтерфейсным областям, были переназначены для интерфейса домена. Интересно, что мутации GF были обогащены в интерфейсах домен-домен (OR против случайного взаимодействия составляло 2,0, 95% CI от 1,6 до 2,5) по сравнению с мутациями с HI и DN мутациями (Fig. 2d). Это значение мутаций AD (OR 1,5, 95% ДИ от 1,4 до 1,7) было сопоставимо с таковыми с мутациями AR (OR 1,6, 95% ДИ 1.От 5 до 1,7). Результат предполагает, что одной из основных причин фенотипа увеличения функции может быть мутация, которая ставит под угрозу регуляторную функцию, осуществляемую за счет междоменного взаимодействия внутри одной белковой молекулы.
Например, мутации GF PTPN11 , кодирующие Sh3-домен протеин-тирозинфосфатазы 2 (SHP2), которые ответственны за синдром Нунана (NS, MIM # 163950), в основном локализовались на границе раздела между N-Sh3 и PTP домены (рис. 3).Эти мутации изменяли остатки, участвующие в аутоингибиторном взаимодействии, таким образом заставляя молекулу легко принимать открытую конформацию, и, следовательно, увеличивали конститутивную каталитическую активность фосфатазы 27 . С другой стороны, описанные мутации DN SHP2 были локализованы только в домене PTP и были закрыты для связывания субстрата или каталитических сайтов SHP2. Было продемонстрировано, что эти мутации снижают активность фосфатазы, что приводит к синдрому Нунана с множественными лентиго (NS-ML, MIM # 151100, ранее называвшимся синдромом LEOPARD) из-за эффекта DN 28 .
Фиг. 3
Распределение участков мутации в белке PTPN11. ( a ) Схематическая первичная структура PTPN11 со структурными доменами и патогенными миссенс-мутациями. Мутации, связанные с разными типами наследования, окрашены по-разному (мутации DN и GF показаны зеленым и красным соответственно). ( b ) Картирование мутаций кристаллической структуры человеческого PTPN11 (код PDB: 2shp). Мутантные остатки представлены сферическими моделями.( c ) Схематические модели взаимодействия домена PTPN11 с патогенными мутациями.
Другим примером, возможно, наиболее подходящим для представления результатов этого исследования, является случай STAT1 , который кодирует фактор транскрипции STAT1, который регулирует развитие различных типов иммунных клеток. Дефекты STAT1 вызывают несколько заболеваний, например: аутосомно-рецессивный дефицит STAT1 (дефицит AR-STAT1, MIM № 613796), аутосомно-доминантный дефицит STAT1 (дефицит AD-STAT1, MIM № 614162) и аутосомно-доминантный хронический кандидоз слизистой оболочки (CMC, MIM № 614892).
Известные мутации, вызывающие заболевание, были нанесены на карту кристаллической структуры комплексов STAT1 человека (рис. 4b, c). Миссенс-мутация Leu600Pro, которая, как известно, вызывает дефицит AR-STAT1, была расположена в скрытой области домена Sh3 (рис. 4b, d). Таким образом, предполагалось, что мутация дестабилизирует домен Sh3, приводя к снижению уровня экспрессии белка за счет деградации. Мутации, связанные с дефицитом AD-STAT1, который, как известно, встречается в режиме DN 29, 30 , были локализованы в гомодимерном интерфейсе Sh3-домена или ДНК-связывающего домена (DBD) (рис.4б). Мутации DN нарушали фосфорилирование STAT1 и ДНК-связывающую активность 29 . В отличие от недостатка AD-STAT1, CMC вызывается режимом GF 31, 32 . Мутации СМС показали повышенное фосфорилирование Tyr701 из STAT1 из-за нарушения ядерного дефосфорилирования. Мутировавшие остатки в основном располагались в домене coiled-coil (CCD), который, как полагали, работает в дефосфорилировании STAT1 путем образования антипараллельного димера (Fig. 4c). Большинство мутаций GF (CMC) было локализовано в этом интерфейсе (рис.4г), и они нарушили бы образование антипараллельного димера. Однако пара мутаций GF (CMC) не наблюдалась в этом интерфейсе, а была локализована в интерфейсе домена. Например, одна из мутаций GF (CMC), Arg274Gln, была обнаружена в интерфейсе между CCD и DBD. Недавно было продемонстрировано, что Arg274 участвует в регуляции активности STAT1 посредством взаимодействия с DBD 33 .
Рисунок 4
Распределение участков мутации в белке STAT1.( a ) Схематическая первичная структура STAT1 со структурными доменами и патогенными миссенс-мутациями. ( b, c ) Мутации, связанные с разными типами наследования, окрашены по-разному. Отображение мутаций в кристаллических структурах показано для параллельного димера ( b ), код PDB: 1bg1) и антипараллельного димера ( c ), кода PDB: 1yvl). Домены раскрашены в соответствии с рисунком а. Мутантные остатки представлены сферическими моделями. ( d, e ) Схематические модели параллельного димера ( d ) и антипараллельного димера ( e ) с патогенными мутациями с фенотипами.Мутация, связанная с дефицитом AR-STAT1 (окрашена в оранжевый цвет), была похоронена в домене Sh3.
миссенс-мутаций в генах, предрасполагающих к раку: можем ли мы их понять? | Наследственный рак в клинической практике
Есть много аспектов, которые необходимо учитывать при определении того, действительно ли миссенс-мутация является причиной или просто обычным полиморфизмом, который не влияет на функциональную активность рассматриваемого гена. Определение миссенс-мутации — это «замена одной пары оснований, которая приводит к трансляции другой аминокислоты в этом положении» [1].Это отличает миссенс-изменения от молчаливого полиморфизма, при котором не наблюдается явных изменений белкового продукта гена.
При рассмотрении того, какой эффект может иметь пропущенное изменение, необходимо учитывать несколько аспектов. 1) Связано ли изменение с изменением полярности соответствующей аминокислоты, и если да, то какое влияние это может иметь на функцию белка? Если изменение не изменяет полярность аминокислоты, есть ли какой-либо наблюдаемый эффект (т.е.е. изменение функции)? 2) Насколько распространена смена населения? Разные ли группы населения подвержены разным частотам изменений, и если да, то связаны ли они с болезнью? 3) Происходит ли бессмысленное изменение в связи с другим изменением, и если да, то является ли оно явно вредным (например, бессмысленное изменение), другими бессмысленными изменениями или более загадочными изменениями, связанными с модификацией гена?
При определении влияния неверных изменений можно использовать несколько способов расследования.К ним относятся ассоциация миссенс-изменений с заболеванием (анализ сегрегации), эволюционная консервация области внутри гена и наличие потери гетерозиготности в образцах опухолей от пациентов, у которых есть изменения.
Частота миссенс-мутаций при различных наследственных предрасположенностях
Что касается наследственной предрасположенности к раку, в базу данных генома человека было внесено значительное количество миссенс-мутаций, как показано в таблице 1.Число на сегодняшний день отражает незначительную долю, но, тем не менее, появляются некоторые тенденции в отношении вероятности выявления бессмысленного изменения генов, связанных со предрасположенностью к злокачественным новообразованиям. При рассмотрении таблицы 1 кажется, что существует значительная разница между количеством миссенс-изменений, идентифицированных в гене APC, по сравнению с любым из других генов. Скорее всего, это связано с систематической ошибкой отбора причинных мутаций, выявленных с помощью теста усечения белка (PTT), который использовался многими группами для выявления мутаций в экзоне 15 гена APC из-за его большого размера (~ 6800 п.н. ).Вторая причина предвзятости связана с пониманием биологии гена APC, которое предполагает, что миссенс-мутации вряд ли связаны с заболеванием. В настоящее время основная функция гена APC, по-видимому, заключается в регуляции β-катенина. Белок APC содержит ряд сайтов связывания β-катенина и доменов связывания β-катенина и доменов подавления регуляции. Единичная миссенс-мутация вряд ли нарушит эту функцию, поскольку существует несколько доменов, которые несут эти функциональные фрагменты [2].Это мнение подтверждается исследованиями, которые показывают, что сайт первичной мутации в гене APC, по-видимому, дает селективное преимущество для определенных типов мутации в остающемся аллеле дикого типа [2]. Однако имеется мало информации о том, приводят ли миссенс-мутации в гене APC к селективным преимуществам. Различие в частотах миссенс-мутаций в других генах, перечисленных в таблице 1, также можно отнести к методам, используемым для анализа соответствующих генов.Например, и BRCA1, и BRCA2 содержат большие экзоны, состоящие из нескольких тысяч пар оснований, которые, как и APC, поддаются анализу с помощью PTT. Этот тест может распознавать только изменения, которые приводят к преждевременным кодонам терминации, и не может использоваться для выявления каких-либо миссенс-мутаций. Следовательно, относительный процент миссенс-изменений будет ниже, чем у генов, проверенных с помощью других методологий, которые исследуют ДНК более конкретно (например, прямое секвенирование или денатурирующая высокоэффективная жидкостная хроматография).Это наблюдается при наследственном неполипозном колоректальном раке (HNPCC) и синдроме Пейтца-Йегерса (PJS), где большая часть миссенс-мутаций выявляется в трех генах, перечисленных в таблице 1. В заключение, вероятность выявления миссенс-мутаций в генах, связанных с с наследственной предрасположенностью к раку зависит от того, как выполняется большинство анализов на мутации, и, в меньшей степени, от функции полученного белкового продукта.
Таблица 1 Процент вариантов миссенс-мутаций, идентифицированных в базе данных генома человека (на май 2005 г.) [7]
Можно ли классифицировать миссенс-мутации при отсутствии функциональных исследований?
При отсутствии функциональных исследований можно сделать прогноз с относительно высокой степенью точности, если принять во внимание ряд косвенных свидетельств.
Нейтральные варианты: накапливаются данные, свидетельствующие о том, что существует примерно 3 миллиона однонуклеотидных полиморфизмов (SNP), распространенных по всему геному человека, некоторые из которых можно было бы описать как миссенс-мутации, если они происходят в кодирующих последовательностях ДНК. Такие изменения можно рассматривать как нейтральные варианты, если они часто происходят в общей популяции и не могут быть связаны с болезнью. Одним из таких примеров является изменение D1822V в гене APC, которое первоначально считалось причинным, но в популяционных исследованиях происходило часто и не было связано с повышенной вероятностью колоректального рака.Действительно, в наших собственных исследованиях изменение D1822V было идентифицировано с частотой примерно 40% в неотобранной популяции [3]. Более того, чтобы подтвердить это доказательство, обширные семейные исследования не смогли выявить какой-либо сегрегации с заболеванием. Вместе эту информацию можно использовать для определения причинного статуса миссенс-мутации.
Напротив, при использовании этого подхода некоторые варианты пропуска будут рассматриваться как причинные. Например, изменения, которые наблюдаются только при редкой восприимчивости к болезням и не наблюдаются в популяционных исследованиях, с большой вероятностью указывают на причинную связь.Одним из таких изменений, например, является миссенс-мутация L285P, выявленная в семействе PJS, которая встречается редко и отделяется исключительно от болезни.
Однако этот подход не является абсолютно надежным. Возможно, существуют мутации, которые соответствуют вышеуказанным критериям, но не могут быть конкретно определены как причинное изменение. В одной семье Birtt-Hogg-Dube пораженный пробанд содержал изменение S128R, которое, по-видимому, было ответственным за почечно-клеточный рак пациента, но когда отец (также страдающий почечно-клеточным раком) пробанда был проверен на это изменение, результат был отрицательный.Вместо этого здоровая мать индивида укрывала изменение, предполагая, что изменение было нейтральным или было связано с сильно изменчивой пенетрантностью болезни.
Совместное появление в транс с другой вредной мутацией
Миссенс-мутация может быть классифицирована как нейтральная, если она наблюдается в транс с другой причинной мутацией в зародышевой линии. Это предполагает, что гомозиготность по причинной мутации или сложная гетерозиготность по двум причинным изменениям в любом из общих генов, связанных с предрасположенностью к раку, вероятно, будут летальными состояниями.Чтобы определить, что миссенс-мутация находится в транс-трансформации с другой вредоносной мутацией, требуется доступ к подходящим базам данных, которые содержат полные данные о секвенировании рассматриваемого гена. В настоящее время они ограничены, и, вероятно, одним из лучших источников такой информации в отношении генетики рака является источник Myriad Genetics ™, который обладает достаточной информацией для оценки таких изменений в BRCA1 и, возможно, BRCA2.
Одного наблюдения совместного возникновения миссенс-мутации с причинной мутацией достаточно, чтобы классифицировать миссенс-вариацию как не вызывающую.Однако классификация миссенс-мутаций как причинных с помощью этого метода более проблематична, поскольку требует изучения большого числа людей для получения достоверной оценки не встречаемости. Таким образом, такой подход к оценке релевантности миссенс-мутации, вероятно, приведет к классификации ряда миссенс-мутаций как не вызывающих причинно-следственную связь, но в целом существует низкая вероятность выявления причинной связи.
Эволюционная консервация
Эволюционная консервация, возможно, представляет собой относительно простую и эффективную меру для оценки причинности бессмысленных изменений.Преимущество этого подхода состоит в том, что он может применяться к каждому ошибочному изменению, но, что нельзя переоценить, только косвенно относится к риску заболевания и, как таковой, всегда требует дополнительной информации для обеспечения какой-либо надежности в прогнозе.
При использовании этого подхода для оценки ошибочного изменения недостаточно просто сравнивать человеческую последовательность с одним другим видом. Лучше провести сравнение нескольких гомологов, поскольку чем дальше в эволюционном времени различаются сходства последовательностей, тем больше вероятность, что последовательность абсолютно необходима для функции кодируемого белка.Отсутствие наблюдаемых эволюционных изменений может указывать на недостаточное время для изменений, а не на сохранение последовательности посредством эволюции. Одним из существенных недостатков этого подхода является доступность информации о последовательности для достаточно большого разнообразия организмов, поскольку без нее надежность результата, вероятно, будет ниже, чем требуется.
При использовании этого подхода следует помнить, что этот метод прогнозирования будет эффективен только для консервативных последовательностей, и если изменение последовательности происходит в сегменте гена, который не является решающим для его функции, то маловероятно, что этот подход будет полезно при определении причинно-следственной связи.
Функция
Оценка влияния варианта на белок и клеточную функцию, вероятно, представляет собой окончательную оценку для определения причинной связи. Причиной может быть любой эффект миссенс-мутации, который приводит к аннулированию или потере функции кодируемого белка. Однако следует проявлять осторожность при экстраполяции функциональных изменений на болезненные состояния. Необходимо учитывать взаимосвязь конкретной функции белка с заболеванием.
Несмысленные изменения, которые отличаются от тех, которые, как сообщается, изменяют функцию белка, но происходят на участках, о которых сообщалось о других изменениях, не могут автоматически считаться причинными, если не были предприняты функциональные исследования. Как упоминалось выше, не все функциональные анализы, однако, могут быть связаны с причиной рака, и поэтому необходимо проявлять осторожность при интерпретации некоторых изменений в активности белка. Примеры этого проявляются в доменах активации транскрипции в генах BRCA1, BRCA2 и способности hMLh2 или hMSh3 репарации несовпадений.
Одним из основных недостатков в оценке функции белков является отсутствие функциональных тестов для значительной части генов, связанных с предрасположенностью к раку. Разрабатываются альтернативные методы для оценки роли миссенс-изменений в заболевании, а также несколько новых алгоритмов для прогностических целей [4]. На момент написания этого обзора новый SNPWEB разрабатывается в UCSF (сервер в настоящее время находится в стадии строительства). Цель этой веб-базы — определить, как SNP влияют на структуры белковых доменов, сравнивая трехмерную модель белка дикого типа с моделью, содержащей SNP [4].Результат этого анализа обеспечивает предсказание влияния SNP на 3D-модель и то, как это может быть связано с изменением функции белка.
В заключение, функциональные исследования, которые включают вариации, вероятно, являются лучшим доказательством причинно-следственной связи, но поскольку функциональный анализ часто трудно разработать, более вероятно, что будут разработаны соответствующие алгоритмы, которые будут иметь высокую степень предсказуемости, и что они будут в будущем будет использоваться все чаще.
Современные практические подходы к присвоению причинного статуса миссенс-мутациям
Из приведенного выше описания того, что можно сделать для определения причинной связи миссенс-мутаций, очевидно, что для определения того, связаны ли они с болезнь. Лучшим подходом было бы использовать все подходы вместе для получения результата с высокой предсказательной силой. К сожалению, не все подходы применимы во всех ситуациях, и некоторые из них требуют дальнейшего совершенствования.Следующий пример из Томпсона и др. [5] показывает, как комбинированный подход может привести к точному предсказанию причинно-следственной связи.
Миссенс-мутация D2723H в BRCA2 24 раза описывалась в базе данных информационного ядра рака груди (BIC), но ни разу не было доказано, что это вредная мутация.
Однако исходные данные в базе данных BIC предполагают, что в лучшем случае шансы в пользу причинно-следственной связи составляют 2: 1. Полный анализ косегрегации с заболеванием с использованием 10 родословных дает шансы в пользу причинно-следственной связи 13 731: 1.Сочетание шансов обоих этих факторов приводит к изменению в пользу причинно-следственной связи до 57 000: 1 [5]. Когда эта информация берется вместе со знанием того, что изменение происходит в консервативной области генома и что это изменение было связано с неправильной локализацией белка [6], что приводит к снижению способности репарации ДНК, это почти наверняка что это изменение является причинным.
Этот подход ясно демонстрирует силу применения комплексного подхода к определению причинной связи данной миссенс-мутации.Однако необходимо внести существенные улучшения, чтобы можно было легко оценить причинно-следственную связь недостающего изменения без поиска в нескольких различных базах данных и без проведения функциональных исследований.
Таким образом, Голдгар и др. [6] предложили комплексный подход к классификации миссенс-мутаций, включающий следующие моменты:
- 1)
— первый шаг в разработке комплексного подхода — для уточнения модели требуется больше данных,
- 2)
Классификация
основана на: — полной нейтральности ИЛИ причинно-следственной связи, сравнимой с известными мутациями (т.е ничего промежуточного),
- 3)
Классификация
, идеально основанная на клинических наблюдениях, поскольку напрямую связана с риском рака и более поддается количественной оценке,
- 4)
геномных данных эволюционной консервации было полезным прогностическим фактором серьезность данных аминокислотных замен не было,
- 5)
обеспечивает стандартную систему оценки неклассифицированных вариантов,
- 6)
позволяет избежать чрезмерной зависимости от какого-либо одного метода.
Для оптимизации этого подхода требуются дальнейшие уточнения, и в результате использованного выше подхода уже использованные методы, которые включают косегрегацию, семейный анамнез, патологию и другие клинические наблюдения, вероятно, являются наиболее полезными. Количественное определение вышеперечисленного должно обеспечить лучшую стандартизацию и сравнение, и, наконец, геномные данные могут усилить аргумент за или против, но обычно не являются достаточным самостоятельным доказательством.
Какой бы подход ни использовался, ясно, что в настоящее время требуется более систематический подход к интерпретации миссенс-мутаций в генах, предрасполагающих к раку. Это должно включать систему оценки отношения шансов для исследований сегрегации, чтобы можно было проводить сравнения между исследованиями и отчетами о недостоверных изменениях. Это могло бы распространяться на включение этих оценок в базы данных, которые сообщают о бессмысленных изменениях, что позволяет комбинировать различные исследования, потенциально ведущие к классификации пропущенных изменений при достижении значимости.
Характеристика 19 связанных с заболеванием миссенс-мутаций в регуляторной области регулятора трансмембранной проводимости при муковисцидозе | Молекулярная генетика человека
Аннотация
Чтобы лучше понять структуру и функцию регуляторного домена (RD) белка-регулятора трансмембранной проводимости при муковисцидозе (CFTR), были функционально охарактеризованы 19 миссенс-мутаций RD, выявленных у пациентов.Девять из них (I601F, L610S, A613T, D614G, I618T L619S, H620P, G628R и L633P) привели к ошибочной обработке. Следовательно, в клетках, экспрессирующих эти мутанты, на клеточной мембране не будет или появиться очень небольшое количество функциональных белков CFTR. Эти мутации были сгруппированы в N-концевой части RD, что позволяет предположить, что этот субдомен имеет паттерн сворачивания, который очень чувствителен к аминокислотным изменениям. Мутации, не вызывающие аберрантной обработки, были дополнительно охарактеризованы на электрофизиологическом уровне.Сначала они были изучены на уровне целых клеток в ооцитах Xenopus laevis . Мутанты, которые индуцировали ток целой клетки, который значительно отличался от CFTR дикого типа, впоследствии анализировали на уровне одного канала в клетках COS1, временно экспрессирующих различные мутантные белки и белки дикого типа. Три мутантных хлоридных канала, G622D, R792G и E822K CFTR, характеризовались значительно более низкой внутренней активностью хлоридных каналов по сравнению с CFTR дикого типа. Две мутации, H620Q и A800G, привели к увеличению активности внутреннего транспорта хлоридов.Наконец, T665S и E826K CFTR имели одноканальные свойства, существенно не отличающиеся от CFTR дикого типа.
Введение
Муковисцидоз (МВ), вызванный мутациями в гене трансмембранного регулятора проводимости муковисцидоза (МВТР), является наиболее частым летальным аутосомно-рецессивным заболеванием в популяции европеоидной расы. Заболевание характеризуется аномальным транспортом соли через апикальную границу эпителиальных клеток (1). Белок CFTR (2–4) представляет собой трансмембранный белок, состоящий из двух повторяющихся мотивов, каждый из которых состоит из трансмембранного домена и нуклеотид-связывающего домена (NBD), разделенных высокозаряженным регуляторным доменом (RD).RD состоит из чередующихся кластеров положительно и отрицательно заряженных аминокислот и содержит несколько согласованных последовательностей для фосфорилирования протеинкиназой A (PKA) и протеинкиназой C (PKC) (3).
Восстановление белка CFTR в протеолипосомы доказало, что он образует линейный хлоридный канал с низкой проводимостью (5). Активация CFTR сложна и происходит в несколько этапов. Во-первых, RD фосфорилируется cAMP-зависимой PKA. Этот шаг усиливается за счет предварительной стимуляции PKC (6).На втором этапе АТФ связывается с первым NBD и гидролизуется на нем, тем самым открывая канал и позволяя ионам хлора течь вниз по электрохимическому градиенту. На третьем этапе гидролиз АТФ на NBD2 закрывает канал (7). Пока RD фосфорилируется, АТФ может повторно открыть канал. Дефосфорилирование RD фосфатазами (8) приводит к окончательному закрытию канала.
Точные механизмы, с помощью которых RD регулируют поток анионов, еще далеко не изучены. Хотя RD содержит девять консенсусных сайтов для PKA, все они избыточны, и фосфорилирования по одному сайту достаточно для активации канала (9).Учитывая тот факт, что активация CFTR может происходить только через фосфорилирование PKA, такая дегенерация сайтов фосфорилирования вызывает большие затруднения. С другой стороны, канал может быть заблокирован в активированном состоянии посредством мутагенеза фрагментов Ser в Asp на консенсусных сайтах PKA (10). Это наблюдение указывает на то, что отрицательные заряды, вносимые фосфорилированием PKA, важны для активации каналов. Удаление части RD (аминокислоты 708–835) также дает канал CFTR, который избегает регуляции PKA и является конститутивно активным (11).Поэтому была высказана гипотеза, что RD закрывает поры хлоридного канала и что он будет отталкиваться от поры при введении отрицательных зарядов. Хотя эти белки ΔR-CFTR являются конститутивно активными, их вероятность открытия ( P o ) составляет лишь одну треть от вероятности, обнаруженной для CFTR дикого типа. Более того, добавление нефосфорилированного RD к ΔR-CFTR не влияет на активность этого канала, в то время как добавление фосфорилированного RD удваивает P o .Это увеличение активности, по-видимому, является результатом более высокой скорости открытия канала из-за более высокой чувствительности NBD1 к АТФ. На основании этих данных было высказано предположение, что фосфорилирование RD изменяет чувствительность NBD1 к АТФ (12, 13).
Учитывая решающую роль РД в регулировании деятельности канала, необходимо доскональное знание его структуры и функций. В этом отношении известные миссенс-мутации RD, обнаруженные у пациентов, могут предоставить важную информацию о метаболизме и функции RD.Наши исследования 19 миссенс-мутаций покажут, что некоторые мутации влияют на созревание белка, другие приводят к снижению функции или усилению функции, в то время как третьи, по-видимому, не влияют на используемые системы анализа.
Результаты
Межвидовой анализ RD
Когда последовательности экзона 13 CFTR из девяти различных организмов были выровнены (рис. 1), наблюдалась высокая консервация N-концевой части (RD1, вплоть до аминокислоты 650).В C-концевых двух третях (RD2) консервативными были только сайты фосфорилирования PKA, и все они были локализованы в этом субдомене. RD очень заряжен, отчасти из-за присутствия консенсусных последовательностей PKA, которые являются очень основными. Были обнаружены два участка кислых аминокислот. Первая (аминокислоты 725–730) расположена в первой половине RD2 и, хотя эта область плохо консервативна, ее отрицательный заряд обнаружен у всех изученных видов. Вторая кислотная область (аминокислоты 822–828) расположена на конце RD и полностью консервативна.
Из 21 миссенс-мутации RD три (K698R, R766M и R792G) были локализованы в консенсусных сайтах узнавания PKA. Только R766M и R792G нарушают сайт узнавания PKA и, следовательно, могут мешать регуляции CFTR. Различные мутации RD затронули почти все, кроме D648V, V754M и A800G, аминокислотные части, которые были абсолютно или высококонсервативными. Высокая сохранность этих мутировавших аминокислот подчеркивает их важность для структуры и функции. Шесть из 21 болезненной мутации повлияли на заряд аминокислотного остатка, а две даже привели к появлению аминокислоты с противоположным зарядом.Последние два были локализованы в высококонсервативном С-концевом кислотном домене.
Влияние мутаций RD на процессинг CFTR
В начале этого исследования у пациентов было описано 19 различных мутаций RD (таблица 1). С тех пор было обнаружено, что мутация F693L является полиморфизмом (14, 15) и что полиморфизм I807M связан с врожденным двусторонним отсутствием семявыносящего протока (CBAVD) (наши неопубликованные данные). Были идентифицированы две дополнительные мутации заболевания, которые не были охарактеризованы на функциональном уровне в этом исследовании [K698R и V754M (15)].Однако природа, по которой все мутации RD вызывают дисфункцию CFTR, неизвестна. Известно, что некоторые мутации CFTR, такие как F508, вызывают неправильную укладку белка в эндоплазматическом ретикулуме (ER), что приводит к задержке и деградации системой контроля качества, которая работает в этом клеточном компартменте (16). Поэтому мы сначала определили характер созревания различных мутантных белков CFTR, перечисленных в таблице 1. Различные мутации вводили в кДНК CFTR дикого типа, вставляли в эукариотический вектор экспрессии pcDNA3, и мутантные конструкции трансфицировали в клетки COS1.Соответствующие белки анализировали с помощью экспериментов с отслеживанием импульсов. Когда продукты трансляции CFTR попадают в ER, они становятся гликозилированными за счет добавления двух гликозильных групп с высоким содержанием маннозы на четвертой внеклеточной петле. Это событие приводит к появлению белка 150 кДа, наиболее заметной формы CFTR дикого типа после 30-минутной погони (рис. 2, дорожка 1). Когда белок покидает ER и попадает в аппарат Гольджи, гликозильные группы становятся модифицируемыми в сложные сахарные группы, и это дает белок с более низкой подвижностью (190 кДа), как можно видеть на дорожках 2 и 3 на рисунке 2.Только белок 190 кДа будет двигаться дальше к плазматической мембране и вносить вклад в активность хлоридных каналов. Этот образец созревания был обнаружен не только для CFTR дикого типа, но также и для некоторых мутантов (таблица 2), таких как I807M CFTR (рис. 2). Некоторые из мутантов, однако, демонстрировали аномальный паттерн созревания, как это можно видеть для L610S и D614G CFTR (рис. 2). Эти мутантные белки давали только форму 150 кДа даже после 210-минутной погони, что указывает на то, что они давали неправильно свернутые белки, которые сохранялись и деградировали в ER.Для подтверждения того, что форма 150 кДа действительно была формой ER, а форма 190 кДа была комплексной гликозилированной формой, были выполнены два ферментативных переваривания (результаты не показаны). Когда иммуноочищенные белки инкубировали с эндогликозидазой H, ферментом, который распознает и отщепляет только высокоманнозные остатки, переваривались только 150 кДа формы созревающих и незрелых мутантов. N-гликозидаза F, фермент, который распознает и расщепляет высокоманнозные и сложные гликозильные группы, разрушает форму 150 кДа, а также форму 190 кДа.Мутации, которые привели к белку, который не смог перейти в форму 190 кДа (I601F, L610S, A613T, D614G, I618T, L619S, H620P, G628R и L633P; Таблица 2), таким образом, относятся к мутациям второго класса (17), где фенотип заболевания вызван отсутствием достаточного количества белка CFTR на поверхности клетки.
Таблица 1
Праймеры, используемые для мутагенеза
Таблица 1
Праймеры, используемые для мутагенеза
Рисунок 1
Сопоставление последовательностей экзона 13, полученных от разных видов.Экзон 13 получен из колючей рыбы ( Squalus acanthias , инвентарный номер GenBank 116141), африканской когтистой лягушки ( Xenopus laevis , инвентарные номера GenBank 64623 и 1617482), коровы ( Bos Taurus , инвентарный номер GenBank19 4617). овца ( Ovis aries , инвентарный номер GenBank 862652), человек ( Homo sapiens , инвентарный номер GenBank 625338), европейский кролик ( Oryctolagus cuniculuc , инвентарный номер GenBank 1100985), норвежская крыса ( Rattus Rattus) , Регистрационный номер GenBank382755) и домовую мышь ( Mus musculus , номер доступа в GenBank 192567) выравнивали с использованием программы множественного выравнивания последовательностей GENALIGN программного обеспечения Intelligenetics. Мутации, обнаруженные у пациентов (I601F, L610S, A613T, D614G, I618T, L619S, H620P, H620Q, D622G, G628R, L633P, T665S, F693L, K698R, V754M, R766M, R792K227, указаны в A8G, E8G и E8G). жирные и подчеркнутые участки фосфорилирования PKA отмечены стрелкой, а два кислых домена заключены в рамку.
Рисунок 1
Выравнивание последовательностей экзона 13, полученных от разных видов.Экзон 13 получен из колючей рыбы ( Squalus acanthias , инвентарный номер GenBank 116141), африканской когтистой лягушки ( Xenopus laevis , инвентарные номера GenBank 64623 и 1617482), коровы ( Bos Taurus , инвентарный номер GenBank19 4617). овца ( Ovis aries , инвентарный номер GenBank 862652), человек ( Homo sapiens , инвентарный номер GenBank 625338), европейский кролик ( Oryctolagus cuniculuc , инвентарный номер GenBank 1100985), норвежская крыса ( Rattus Rattus) , Регистрационный номер GenBank382755) и домовую мышь ( Mus musculus , номер доступа в GenBank 192567) выравнивали с использованием программы множественного выравнивания последовательностей GENALIGN программного обеспечения Intelligenetics. Мутации, обнаруженные у пациентов (I601F, L610S, A613T, D614G, I618T, L619S, H620P, H620Q, D622G, G628R, L633P, T665S, F693L, K698R, V754M, R766M, R792K227, указаны в A8G, E8G и E8G). жирные и подчеркнутые участки фосфорилирования PKA отмечены стрелкой, а два кислых домена заключены в рамку.
Целые клеточные токи в
ооцитах Xenopus , экспрессирующих CFTR дикого типа и мутантный тип
Поскольку все изученные мутации были идентифицированы у пациентов, ожидалось, что они будут мешать нормальной функции хлоридных каналов CFTR.Поэтому мутации, не вызывающие остановки созревания белка, анализировали на электрофизиологическом уровне. С этой целью РНК CFTR созревающих мутантов получали путем транскрипции in vitro и вводили в ооциты Xenopus laevis . Базальные и цАМФ-индуцированные токи хлоридов цельной клетки измеряли с использованием метода двухэлектродного зажима напряжения. Результаты показаны на рисунке 3. Четыре мутации (T665S, R792G, E822K и E826K) вызвали значительное снижение цАМФ-индуцированного тока хлорида.Интересно, что две мутации (H620Q и A800G) привели к возникновению хлоридных каналов со значительно более высокой активностью транспорта хлоридов. Остальные (G622D, D648V, F693L, R766M и I807M) не оказали значительного влияния на способность транспорта хлора по сравнению с каналами CFTR дикого типа.
Таблица 2
Модель созревания мутаций RD и связанный с ними фенотип, обнаруженный у пациентов с указанным генотипом (когда мутация связана с CF, указывается только статус поджелудочной железы)
Таблица 2
Модель созревания мутаций RD и их ассоциированный фенотип, обнаруженный у пациентов с указанным генотипом (когда мутация связана с МВ, указывается только статус поджелудочной железы)
Одноканальные измерения хлоридных каналов CFTR дикого типа и мутантных
Изменение активности транспорта хлоридов может быть вызвано разницей в количестве функциональных белков CFTR, включенных в апикальную клеточную мембрану, и / или различиями в проводящих и / или управляющих свойствах этих каналов.Поэтому мутанты, влияющие на токи целых клеток, изучались на уровне одного канала. Различные кДНК CFTR дикого типа и мутантные кДНК CFTR были включены в бицистронный вектор, так что после трансфекции в клетки COS1 формировался бицистронный транскрипт CFTR и зеленого флуоресцентного белка (GFP). Клетки COS1, экспрессирующие GFP и, следовательно, также CFTR, использовали для одноканальных измерений. Ток измеряли до и после добавления форсколина и IBMX к внеклеточной стороне клетки.Примеры трассировок для белков CFTR дикого типа и H620Q показаны на рисунке 4 A. Хлоридные каналы дикого типа (рис. 4 B) и мутантные каналы CFTR показали линейные отношения I – V с проводимостью одного канала, не отличающейся от проводимости канала дикого типа ( Рис. 4 C). Были определены P o различных вариантов CFTR (рис. 5). Активность одного канала хорошо коррелировала с токами всей клетки, измеренными в ооцитах для большинства изученных белков CFTR. G622D, R792G и E822K дали начало хлоридному каналу CFTR со значительно более низким значением P o , чем CFTR дикого типа; H620Q и A800G CFTR привели к появлению каналов со значительно более высоким значением P o .
Рисунок 2
Импульсная погоня и иммунопреципитация CFTR от клеток COS1, временно экспрессирующих CFTR дикого типа, L610S, D614G или I807M. Белки метили [ 35 S] метионином и [ 35 S] цистеином, гоняли в течение указанных периодов времени, и белки CFTR подвергали иммунопреципитации антителом, направленным против С-концевой части CFTR. Иммуноочищенные белки разделяли на 4–12% геле SDS и визуализировали с помощью авторадиографии.Показаны гликозилированные ядра 150 кДа и зрелые 190 кДа формы.
Рисунок 2
Импульсная погоня и иммунопреципитация CFTR от клеток COS1, временно экспрессирующих CFTR дикого типа, L610S, D614G или I807M. Белки метили [ 35 S] метионином и [ 35 S] цистеином, гоняли в течение указанных периодов времени, и белки CFTR подвергали иммунопреципитации антителом, направленным против С-концевой части CFTR. Иммуноочищенные белки разделяли на 4–12% геле SDS и визуализировали с помощью авторадиографии.Показаны гликозилированные ядра 150 кДа и зрелые 190 кДа формы.
Рисунок 3
Потоки хлоридов во всей клетке, измеренные в ооцитах Xenopus laevis , экспрессирующих белки CFTR дикого типа и мутантные. Ооциты Xenopus laevis инъецировали in vitro. транскрибировали РНК дикого типа и мутантную РНК CFTR, и через 72 часа измеряли базальный (черная полоса) и цАМФ-индуцированный (белая полоса) хлоридные токи с помощью двухэлектродной фиксации напряжения. .Мутанты, которые показали значительно отличающийся от цАМФ-зависимый хлоридный ток ( P <0,05), по сравнению с CFTR дикого типа, отмечены звездочкой и указано количество проанализированных клеток.
Рисунок 3
Потоки хлоридов во всей клетке, измеренные в ооцитах Xenopus laevis , экспрессирующих белки CFTR дикого типа и мутантные. Ооциты Xenopus laevis инъецировали in vitro. транскрибировали РНК дикого типа и мутантную РНК CFTR, и через 72 часа измеряли базальный (черная полоса) и цАМФ-индуцированный (белая полоса) хлоридные токи с помощью двухэлектродной фиксации напряжения. .Мутанты, которые показали значительно отличающийся от цАМФ-зависимый хлоридный ток ( P <0,05), по сравнению с CFTR дикого типа, отмечены звездочкой и указано количество проанализированных клеток.
Обсуждение
С момента открытия гена CFTR у пациентов была выявлена 21 миссенс-мутация RD. Мы изучили влияние 19 из этих мутаций (таблица 1) на созревание и электрофизиологические свойства CFTR.Девять мутаций вызвали аберрантный процессинг: I601F, L610S, A613T, D614G, I618T, L619S, H620P, G628R и L633P. Очень поразительным наблюдением было то, что все мутации, вызывающие дефект процессинга, были сгруппированы в N-концевой части (аминокислоты 601–619) RD, в то время как те, которые показали нормальный паттерн созревания, были сгруппированы в C-концевом конце (амино кислоты 648–826). Эти эффекты не были вызваны различными типами введенных мутаций, поскольку сходные аминокислотные изменения были обнаружены в обоих субдоменах.В области, связывающей эти две части, эффект мутации зависит от внесенной замены аминокислоты. Это проиллюстрировано для аминокислоты 620, где у пациентов с МВ были обнаружены две разные мутации. Когда His620 был заменен на Gln, никакого влияния на созревание не наблюдалось. Когда His620 был заменен на Pro, аминокислоту, которая резко влияет на вторичную структуру белка и, следовательно, на его правильную укладку, был получен незрелый белок. Однако следует отметить, что эффекты мутаций с участием Pro не столь драматичны, когда они расположены во втором субдомене CFTR, поскольку рекомбинантный белок, в котором были мутированы три остатка Pro дикого типа, все еще образовывал зрелую форму 190 кДа. (18).
RD изначально был назначен той части белка CFTR, который кодируется экзоном 13. Однако накопление доказательств указывает на то, что RD можно разделить на два субдомена. Межвидовой анализ показал, что N-концевая часть этого домена была высококонсервативной на аминокислотном уровне во время эволюции от акулы до человека, в то время как в C-концевых двух третях консервативны только сайты фосфорилирования (19). Более того, все сайты фосфорилирования RD расположены исключительно во второй части.Новая модель для NBD1 была недавно получена путем компьютерного моделирования, основанного на рентгеновской структуре
бычьей митохондриальной F1-АТФазы. Согласно этой модели, аминокислоты 453-650, которые до сих пор приписывались экзону 13 RD, на самом деле предположительно являются частью NBD1 (20). Здесь мы обнаружили, что миссенс-мутации, расположенные в этой части RD, не созревают, что характерно для большинства мутаций NBD1 (21).
Мутации, не влияющие на созревание (H620Q, G622D, D648V, T665S, F693L, R766M, R792G, A800G, I807M, E822K и E826K) впоследствии были проанализированы на электрофизиологическом уровне.
Три из них (G622D, R792G и E822K) дали начало хлоридным каналам со значительно более низким P o , чем канал дикого типа. Одна из этих мутаций, R792G, нарушает консенсусный сайт узнавания PKA. Поскольку было показано, что этот сайт фосфорилируется in vivo с помощью PKA (9) и что он является одним из самых сильных стимулирующих сайтов для активации CFTR (22), устранение этой последовательности узнавания PKA может привести к хлоридному каналу с аберрантной регуляцией. и, как следствие, более низкая активность.Поразительно, что две мутации (H620Q и A800G) показали значительно увеличенное значение P o по сравнению с CFTR дикого типа. До сих пор было показано, что только мутация P574H, локализованная в NBD1, дает белок с более высокими внутренними свойствами транспорта хлоридов. Однако этот мутант не созревает (23, 24). Описанные здесь «гиперактивные» мутации H620Q и A800G, вызывающие заболевание, действительно созревают. Остальные мутации (D648V, T665S, F693L, R766M, I807M и E826K) не вызвали значительных изменений внутренней активности хлоридных каналов.F693L оказался полиморфизмом (15), что объяснило его нормальное функционирование.
Помимо хлоридного канала, CFTR также является регулятором других каналов. Было обнаружено, что CFTR является негативным регулятором амилорид-чувствительных эпителиальных натриевых каналов (25–28) и позитивным регулятором внешне выпрямляющих хлоридных каналов (29, 30), калиевых каналов (31) и АТФ-канала, связанного с CFTR. (32). Следовательно, мутантные белки, которые обладают диким типом или повышенной активностью каналов, могут вызывать фенотип CF из-за аномальных регуляторных свойств.Ввиду того факта, что RD является уникальным для CFTR по сравнению с другими белками АТФ-связывающей кассеты, и что показано взаимодействие между RD и каналом АТФ, этот домен становится главным кандидатом на роль в регулирующая деятельность CFTR.
Рисунок 4
Активация отдельных каналов путем экспрессии дикого типа и мутантного CFTR (H620Q). ( A ) Одноканальные записи тока дикого типа и H620Q CFTR до и после добавления коктейля активации (1 мкМ IBMX и 0.1 мкМ форсколина) на внешний участок мембранного пластыря. 0, закрытое состояние; 1′3, события открытого канала. ( B ) Соответствующее соотношение тока и напряжения (I-V), полученное для CFTR дикого типа. ( C ) Одноканальные проводимости каналов CFTR дикого типа и мутантных каналов, рассчитанные на основе тока, измеренного при -60 мВ, и теоретического реверсивного потенциала при 0 мВ. Количество проанализированных клеток идентично количеству клеток на фиг. 5. Все измерения были выполнены в конфигурации с присоединенными клетками с использованием условий симметричного хлорида на клетках COS, временно экспрессирующих CFTR дикого типа и мутантный.
Рисунок 4
Активация отдельных каналов путем экспрессии дикого типа и мутантного CFTR (H620Q). ( A ) Одноканальные записи тока дикого типа и H620Q CFTR до и после добавления коктейля активации (1 мкМ IBMX и 0,1 мкМ форсколина) к внешнему участку мембранного пластыря. 0, закрытое состояние; 1′3, события открытого канала. ( B ) Соответствующее соотношение тока и напряжения (I-V), полученное для CFTR дикого типа. ( C ) Одноканальные проводимости каналов CFTR дикого типа и мутантных каналов, рассчитанные на основе тока, измеренного при -60 мВ, и теоретического реверсивного потенциала при 0 мВ.Количество проанализированных клеток идентично количеству клеток на фиг. 5. Все измерения были выполнены в конфигурации с присоединенными клетками с использованием условий симметричного хлорида на клетках COS, временно экспрессирующих CFTR дикого типа и мутантный.
Недавно мы показали, что полиморфные аллели, подобные тем, которые обнаруживаются в локусе M470V, также могут влиять на свойства CFTR как на уровне созревания, так и на электрофизиологическом уровне (33). Следовательно, «нормальные» или «гиперфункциональные» мутации могут вызывать сбои в работе CFTR, если они присутствуют на определенном фоне гаплотипа.Поскольку гаплотипы для большинства изученных мутаций не были известны, их функциональное действие могло быть замаскировано. С другой стороны, нельзя исключить, что эти пациенты несли еще не обнаруженную вторую мутацию на том же аллеле CFTR. В результате изучаемая мутация экзона 13 будет полиморфизмом.
В заключение, основываясь на исследованиях созревания, CFTR RD можно подразделить на два субдомена: N-концевую треть экзона 13, которая очень чувствительна к миссенс-мутациям, и C-концевую часть экзона 13, который предъявляет менее строгие требования к конструкции.Однако для некоторых мутаций дефекта созревания обнаружить не удалось. Более того, они дали начало хлоридным каналам с внутренней активностью, которая была подобна CFTR дикого типа или даже превышала ее. Необходимы дальнейшие эксперименты, чтобы выяснить механизм, с помощью которого последние мутации вызывают заболевание.
Материалы и методы
Конструирование мутантов RD
Фрагмент Xho I- Kpn I, содержащий кодирующую область CFTR , был выделен из прокариотического вектора pTG5960 (Transgene SA, Страсбург, Франция) и вставлен в эукариотический вектор экспрессии pcDNA3 (Invitrogen, Leek, Нидерланды). .Чтобы удалить потенциальный стартовый кодон ATG, происходящий из сайта множественного клонирования pTG5960, конструкцию расщепляли Kpn I и Eco RV, и полученную линейную плазмиду лигировали по тупым концам после обработки ДНК-полимеразой Т4. Кодирующая область CFTR , присутствующая в полученной конструкции, была охарактеризована дидезокси-секвенированием, и был идентифицирован один полиморфизм, V470. Последовательность также содержала три нейтральные аминокислотные замены (t930c, a933g и t936c), которые были введены для того, чтобы инактивировать скрытый прокариотический промотор (36).Все миссенс-мутации RD, о которых было сообщено до 23 мая 1996 г. Консорциуму по генетическому анализу кистозного фиброза (таблица 1), были внесены с использованием набора для сайт-направленного мутагенеза трансформера (Clontech Laboratories, Пало-Альто, Калифорния). Праймеры, используемые для мутагенеза, перечислены в таблице 1. Была секвенирована полная кодирующая область CFTR 20 мутантов; никаких других мутаций, кроме желаемых, обнаружено не было.
Рисунок 5
Сводка изменений открытых вероятностей ( P o ) хлоридных каналов CFTR дикого типа и мутантных, экспрессируемых в клетках COS1.Клетки COS1 трансфицировали бицистронной конструкцией кДНК GFP и кДНК CFTR дикого типа или мутантной. Клетки, экспрессирующие GFP, были отобраны и использованы для одноканальных измерений. Количество каналов оценивалось по перекрывающимся событиям развертки. Каналы мутантного хлорида CFTR, характеризующиеся значением P o , значительно отличающимся от значения CFTR дикого типа, отмечены звездочкой ( P <0,05), и указано количество исправленных клеток.
Рисунок 5
Сводка изменений открытых вероятностей ( P o ) дикого типа и мутантных хлоридных каналов CFTR, экспрессируемых в клетках COS1. Клетки COS1 трансфицировали бицистронной конструкцией кДНК GFP и кДНК CFTR дикого типа или мутантной. Клетки, экспрессирующие GFP, были отобраны и использованы для одноканальных измерений. Количество каналов оценивалось по перекрывающимся событиям развертки. Каналы мутантного хлорида CFTR, характеризующиеся значением P o , значительно отличающимся от значения CFTR дикого типа, отмечены звездочкой ( P <0.05) и указано количество исправленных ячеек.
Для одноканальных измерений различные мутантные кДНК и кДНК CFTR дикого типа были перенесены в бицистронный вектор экспрессии GFP pCINeo / IRES-GFP (37). Для этой цели кДНК CFTR дикого типа, полученная из pcDNA3-CFTR, лигировали в виде обработанного ДНК-полимеразой Т4 фрагмента Xho I- Sac I в pCINeo / IRES-GFP. Последний сначала линеаризовали с помощью Nhe I, затупляли концы ДНК-полимеразой Т4 и дефосфорилировали бычьей щелочной фосфатазой.RD и его фланкирующие последовательности были впоследствии удалены из pCINeo / wtCFTR-IRES-GFP путем полного переваривания Xba I и частичного переваривания Bst XI и были заменены соответствующими мутантными фрагментами CFTR , которые были получены путем полного расщепления. расщепление различных мутантных конструкций pcDNA3-CFTR с помощью Xba I и Bst XI.
Экспрессия, отслеживание импульсов и иммунопреципитация CFTR
Двадцать микрограмм плазмидной ДНК электропорировали (BioRad Gene Pulser; Bio-Rad, Hercules, CA) в 1.5–3 × 10 7 клеток COS1. Трансфицированные клетки культивировали в течение 48–72 ч при 37 ° C в среде DMEM F12 (Life Technologies, Inchinnan, UK) с добавлением 10% фетальной телячьей сыворотки (HyClone Laboratories, Logan, UT), а затем отбирали с 480 мг / л дисульфата G418. (Duchefa, Харлем, Нидерланды). После 2-хнедельного отбора клетки голодали в течение 30 минут в среде RPMI 1640 (Life Technologies, Inchinnan, UK) без метионина и цистеина, меченных в течение 30-минутного импульса в RPMI 1640 с добавлением 100 мкКи / мл [ 35 S] метионина. и [ 35 S] цистеин (ICN Pharmaceuticals, Коста-Меса, Калифорния) и, наконец, преследовали в течение различных периодов времени в DMEM F12 с добавлением 10% фетальной бычьей сыворотки.Клетки соскабливали и впоследствии лизировали ультразвуком в ледяном буфере IPPA (20 мМ Трис, 150 мМ NaCl, 1% дезоксихолат натрия, 1% Тритон X-100, 0,1% SDS, pH 7,4) с добавлением ингибиторов протеаз (20 мкг / мл. ингибитор трипсина сои, 1 мкг / мл лейпептина, 1 мкг / мл антипаина, 1 мкг / мл пепстатина, 1 мкг / мл химостатина и 1 мМ фенилметилсульфонилфторид). Лизат был предварительно очищен гранулами протеина A-сефарозы CL-4B (Pharmacia Biotech, Уппсала, Швеция), и белки CFTR были очищены аффинно путем инкубации в течение 1 часа с моноклональным антителом против CFTR, направленным против С-концевой части CFTR (Genzyme Диагностика, Кембридж, Массачусетс).Иммунокомплексы очищали на гранулах протеин A-сефароза CL-4B и после добавления загрузочного буфера (1% β-меркаптоэтанол, 16 мМ трис-HCl, pH 6,8, 4% SDS, бромфеноловый синий и 10% глицерин), загружали на 4–12% гель SDS (Novex, Сан-Диего, Калифорния). После высыхания гель выдерживали при −70 ° C на светочувствительной пленке в течение 3–4 ч.
Переваривание гликозидазами
Все расщепления гликозидазами проводили на белках CFTR, которые были иммунопреципитированы и очищены на гранулах белок A-сефароза CL-4B.Расщепление эндогликозидазой H проводили в течение ночи при 37 ° C с 2,5 мЕд эндогликозидазы H (Boehringer Mannheim, Mannheim, Германия) в 0,05 М цитратном буфере, содержащем 0,1% SDS, pH 6,0. Чтобы получить полное дегликозилирование, белки CFTR инкубировали в течение 15 минут при комнатной температуре в 0,3% SDS с последующей инкубацией в течение ночи при 37 ° C с 0,25 ед. N-гликозидазы F (Boehringer Mannheim) в буфере N-гликозидазы F (50 мМ Tris, 25 мМ EDTA и 1% Triton X-100, pH 8,0).
In vitro транскрипция
Различные конструкции CFTR были in vitro, транскрибированы с использованием крупномасштабной системы продуцирования РНК T7 RiboMAX (Promega, Madison, WI) в соответствии с протоколом производителя.Качество РНК определяли с помощью гель-электрофореза на формальдегидеагарозе (1%).
Инъекция РНК в ооциты
Самок жаб ( Xenopus laevis ) анестезировали в ледяной воде, содержащей 2 г / л этилового эфира 3-аминобензойной кислоты (Sigma-Aldrich, Bornem, Бельгия), а затем собирали ооциты посредством абдоминальной иссечения. Ооциты инкубировали в бескальциевом растворе, содержащем 2 мг / мл коллагеназы A (Boehringer Mannheim), в течение 1 часа.Ооциты инъецировали 50 нл , транскрибированной in vitro РНК (5 мкг / мкл).
Двухэлектродные клещи для измерения напряжения
Через три дня после инъекции РНК ооциты помещали в перфузионную камеру, протыкали двумя электродами и фиксировали напряжение. Для блокирования эндогенных потоков хлоридов, активированных Ca 2+ , перфузионный раствор (96 мМ NaCl, 5 мМ HEPES, 2 мМ KCl, 1,8 мМ CaCl 2 и 1 мМ MgCl 2 , pH 7,4) содержал 100 мкМ нифлумовая кислота.Токи CFTR активировали добавлением 10 мкМ форсколина и 1 мМ IBMX. Все данные были проанализированы с помощью теста Стьюдента t . Значение P <0,05 считалось значимым. Данные представлены как средние значения ± стандартная ошибка среднего. Хлоридные токи не активировались в неинфицированных ооцитах, ооцитах, инъецированных водой, или ооцитах, инъецированных РНК, полученной из , транскрибированной in vitro pcDNA3.
Одноканальные записи
клеток COS1 трансфицировали различными конструкциями pcINeo / GFP-CFTR (37) и через 36 часов высевали на покровные стекла.Покровные стекла помещали в установку патч-зажим (усилитель EPC-7; List, Дармштадт, Германия), содержащую микроскоп Zeiss Axiovert 100, ксеноновый источник света и эпифуоресцентную оптику (Zeiss, XBO 75 и конденсатор флуоресценции Zeiss-EPI). 48–72 ч после трансфекции. Для возбуждения GFP использовался полосовой фильтр (Zeiss BP 450–490). Возбуждение осуществлялось через дихроичное зеркало (Zeiss FT 510). Свет, испускаемый клетками, экспрессирующими GFP, проходил через длиннопроходный фильтр 520 нм (Zeiss LP 520) и обнаруживался визуально.Для одноканальных измерений использовали только клетки, излучающие зеленый флуоресцентный свет после возбуждения и, следовательно, экспрессирующие белки GFP и CFTR. Все электрофизиологические измерения проводились при комнатной температуре в присоединенной к ячейке конфигурации с сопротивлением пипетки ~ 10 МОм, токи оцифровывались при 1 кГц и фильтровались при 500 Гц. Данные собирались непрерывно с помощью программного пакета AXO Tape v.2.02 (Axon Instruments, Forster City, CA) и анализировались с помощью программного обеспечения ASCD (38).Исследовали только один пластырь на клетку; следовательно, количество пятен равно количеству исследованных клеток. Растворы для пипетки и ванны содержали 140 мМ N -метил-D-глюкамин хлорид, 10 мМ NaF, 10 мМ HEPES, 2 мМ MgCl 2 , 5 мМ CsCl, 1 мМ Mg-ATP, 1 мМ EGTA, pH 7,4. . Каналы активировали добавлением 1 мкМ IBMX и 0,1 мкМ форсколина в раствор ванны (внешний). Вероятность открытия P o была рассчитана из P o = I / i · n , где I — усредненный ток за весь период стимуляции, i — одноканальный ток, полученный из амплитудных гистограмм, и n — это количество каналов, оцененное по перекрывающимся событиям.Все данные были проанализированы с помощью теста Стьюдента t . Значение P <0,05 считалось значимым. Данные представлены как средние значения ± стандартная ошибка среднего.
Благодарности
Мы хотели бы поблагодарить Яна Эггермонта, любезно предоставившего pCINeo / IRES-GFP, и Transgene SA (Страсбург, Франция), великодушно предоставившего вектор pTG5960. Мы также очень благодарны Тило Дорку, Буркхарду Тюммлеру, Дамьяну Главаку, Клоду Ферку, Кристине Бомбьери, Джулиану Зеленски, Гарри Каттингу и Мартину Шварцу за информацию о фенотипах, связанных с различными изученными мутациями.Анн Ванкеер-Берген — научный сотрудник Het Vlaams Instituut voor de Bevordering van het Wetenschappelijk-Technologisch Onderzoek in de Industrie, а Гарри Куппенс — научный сотрудник постдокторантуры из Onderzoeksfonds KU Leuven. Эти исследования также были поддержаны грантом Альфонса и Жана Фортона от Koning Boudewijn Stichting, грантом G.0237.95 от Fonds voor Wetenschappelijk Onderzoek (для BN), COF / 96/22-A0659 (для BN) и IUAP нет. 3П4 / 23.
Сокращения
CBAVD
врожденное двустороннее отсутствие семявыносящего протока
CF
CFTR
муковисцидоз трансмембранный регулятор проводимости
NBD
нуклеотидсвязывающий домен
RD
PKA
PKC
P o
Сокращения
3 o
03 Ссылка
1,,,.,,,. ,
Метаболические и молекулярные основы наследственного заболевания, муковисцидоза
,
1995
New York, NY
McGraw-Hill
(стр.
3799
—
3876
) 2,,,,,,,,, ,,,,,.
Идентификация гена муковисцидоза: ходьба и прыжки по хромосомам
,
Science
,
1989
, vol.
245
(стр.
1059
—
1065
) 3,,,,,,,,,,,,.
Идентификация гена муковисцидоза: клонирование и характеристика комплементарной ДНК
,
Science
,
1989
, vol.
245
(стр.
1066
—
1073
) 4,,,,,,,.
Идентификация гена муковисцидоза: генетический анализ
,
Science
,
1989
, vol.
245
(стр.
1073
—
1080
) 5,,,,,,.
Очистка и функциональное восстановление регулятора трансмембранной проводимости при муковисцидозе (CFTR)
,
Cell
,
1992
, vol.
68
(стр.
809
—
818
) 6,,,.
Регулируемый фосфорилированием канал Cl — в клетках СНО, стабильно экспрессирующих ген муковисцидоза
,
Nature
,
1991
, vol.
352
(стр.
628
—
631
) 7,,.
Два нуклеотидсвязывающих домена регулятора трансмембранной проводимости при муковисцидозе (CFTR) выполняют разные функции по контролю активности канала
,
J. Biol. Chem.
,
1995
, т.
270
(стр.
1711
—
1717
) 8,,.
Регулирование канала Cl — регулятора трансмембранной проводимости муковисцидоза с помощью специфических протеинкиназ и протеинфосфатаз
,
J. Biol. Chem.
,
1993
, т.
268
(стр.
2037
—
2047
) 9,,,,,.
Фосфорилирование домена R цАМФ-зависимой протеинкиназой регулирует хлоридный канал CFTR
,
Cell
,
1991
, vol.
66
(стр.
1027
—
1036
) 10,,,,,,.
Регулирование трансмембранного регулятора проводимости при муковисцидозе Cl — посредством отрицательного заряда в домене R
,
J. Biol. Chem.
,
1993
, т.
268
(стр.
20259
—
20267
) 11,,,,,.
Эффект удаления домена R на хлоридных каналах, генерируемых CFTR
,
Science
,
1991
, vol.
253
(стр.
205
—
207
) 12,,,,.
Функция R-домена в регуляторе трансмембранной проводимости при муковисцидозе хлоридном канале
,
J.Биол. Chem.
,
1997
, т.
272
(стр.
28133
—
28141
) 13,.
Стимуляция активности CFTR его фосфорилированным доменом R
,
Nature
,
1997
, vol.
389
(стр.
294
—
296
) 14,,,,,,.
Идентификация трех новых мутаций муковисцидоза в выборке итальянских пациентов с муковисцидозом
,
Hum. Hered.
,
1993
, т.
43
(стр.
295
—
300
) 15
CF Genetic Analysis Consortium at
16,,,,,,,.
Нарушение внутриклеточного транспорта и процессинга CFTR является молекулярной основой большинства форм муковисцидоза
,
Cell
,
1990
, vol.
63
(стр.
827
—
834
) 17,.
Молекулярные механизмы дисфункции хлоридных каналов CFTR при муковисцидозе
,
Cell
,
1993
, vol.
73
(стр.
1251
—
1254
) 18,,,,,.
Структура и функция регуляторного домена или R домена CFTR
,
Pediat. Пульмонол.
,
1997
, т.
14
доп.
(стр.
171
—
172
) 19,.
Двухдоменная модель для R-домена регулятора трансмембранной проводимости при муковисцидозе, основанная на сходстве последовательностей
,
FEBSLett.
,
1994
, т.
343
(стр.
109
—
114
) 20,,,,,,,.
Новая модель первого нуклеотидсвязывающего домена регулятора трансмембранной проводимости при муковисцидозе
,
FEBS Lett
,
1997
, vol.
407
(стр.
303
—
308
) 21,,,,,,,,.
Созревание и функция муковисцидоза Варианты трансмембранного регулятора проводимости, несущие мутации в предполагаемых нуклеотидсвязывающих доменах 1 и 2
,
Mol. Клетка. Биол.
,
1991
, т.
11
(стр.
3886
—
3893
) 22,,,,,,. Активация
CFTR: аддитивные эффекты сайтов стимулирующего и ингибирующего фосфорилирования в домене R
,
Am.J. Physiol.
,
1997
, т.
17
(стр.
L127
—
L133
) 23,,,.
Механизм дисфункции двух мутаций нуклеотидсвязывающего домена в регуляторе трансмембранной проводимости при муковисцидозе, связанных с панкреатической недостаточностью
,
EMBO J.
,
1995
, vol.
14
(стр.
876
—
883
) 24,,,,,,,,,.
Изменение режима стробирования, приводящее к увеличению внутренней активности канала Cl —, компенсирует дефектный процессинг у мутанта по муковисцидозу, соответствующего легкой форме заболевания
,
EMBO J.
,
1995
, т.
14
(стр.
2417
—
2423
) 25,,,,,,.
CFTR как цАМФ-зависимый регулятор натриевых каналов
,
Science
,
1995
, vol.
269
(стр.
847
—
850
) 26,,,.
CFTR дикого типа, но не F508 CFTR ингибирует проводимость Na + при совместной экспрессии в ооцитах Xenopus
,
FEBS Lett.
,
1996
, т.
381
(стр.
47
—
52
) 27,,,,,,,.
Регулирование эпителиальных натриевых каналов с помощью регулятора трансмембранной проводимости при муковисцидозе
,
J. Biol. Chem.
,
1996
, т.
271
(стр.
4725
—
4732
) 28,,.
Регулятор трансмембранной проводимости при муковисцидозе инвертирует опосредованную протеинкиназой А регуляцию одноканальной кинетики эпителиальных натриевых каналов
,
J. Biol. Chem.
,
1997
, т.
272
(стр.
14037
—
14040
) 29,,,.
CFTR и внешне выпрямляющие каналы представляют собой разные белки с регулирующими отношениями
,
Nature
,
1993
, vol.
363
(стр.
263
—
266
) 30,,,,,,.
CFTR регулирует внешнее выпрямление хлоридных каналов посредством аутокринного механизма с участием АТФ
,
Cell
,
1995
, vol.
81
(стр.
1063
—
1073
) 31,,,,.
cAMP стимуляция CFTR-экспрессирующих ооцитов Xenopus активирует хроманол-ингибирующую проводимость K +
,
Pflügers Arch.
,
1996
, т.
432
(стр.
516
—
522
) 32,,.
CFTR Cl — канал и связанный с CFTR канал АТФ: отдельные поры регулируются общими воротами
,
EMBO J.
,
1998
, vol.
17
(стр.
898
—
908
) 33,,,,,,,,,,.
Поливариантные мутантные гены трансмембранного регулятора проводимости муковисцидоза
,
J. Clin. Вкладывать деньги.
,
1998
, т.
101
(стр.
487
—
496
) 34,,,,,,,.
Является ли врожденное двустороннее отсутствие семявыносящего протока первичной формой муковисцидоза? Анализ гена CFTR у 67 пациентов
,
Am. J. Hum. Genet.
,
1995
, т.
56
(стр.
272
—
277
) 35,,,,,.
Обнаружение более 94% мутаций муковисцидоза в выборке из бельгийской популяции и идентификация четырех новых мутаций
,
Hum. Mut.
,
1993
, т.
2
(стр.
16
—
20
) 36,,,,,,,,.
Экспрессия и характеристика регулятора трансмембранной проводимости при муковисцидозе
,
Nature
,
1990
, vol.
347
(стр.
382
—
386
) 37,,,,.
Использование бицистронного вектора экспрессии GFP для характеристики ионных каналов после трансфекции в клетках млекопитающих
,
Pflügers Arch.
,
1997
, т.
434
(стр.
632
—
638
) 38,.
Кинетические свойства сердечного кальциевого канала Т-типа у морской свинки
,
J. Physiol.
,
1989
, т.
419
(стр.
627
—
650
)
© 1998 Oxford University Press
.