
Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков. Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.

мальчик с синдромом Марфана
Клиническая симптоматика синдрома весьма полиморфна. Это связано с наличием пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью.
Больные имеют диспропорциональные конечности, рост выше среднего, вытянутые пальцы, гипермобильные суставы, худощавое тело, готическое небо, неправильный прикус, глубоко расположенные глаза. Они страдают гигантизмом, миопией, эктопией хрусталика, изменением формы грудины, кифосколиозом. Клинические признаки синдрома обусловлены гиперрастяжимостью тканей.

Синдром Марфана — наследственная коллагенопатия, встречающаяся у 1 из 10000-20000 человек. Патология обнаруживается повсеместно, среди людей любой национальности, пола и места проживания – одинаково часто как среди жителей юга, так и севера. В семейных парах, где отцу больше 35 лет, чаще рождаются больные дети.
Диагностика патологии основывается на данных наследственного анамнеза, визуального осмотра, результатах дополнительных исследований. Пациенты с синдромом Марфана внешне похожи друг на друга. Специалисты по внешнему виду могут предположить наличие недуга. Лечение болезни медикаментозное и оперативное, заключающееся в устранении структурных и функциональных нарушений в сердечной мышце, зрительном анализаторе, костно-суставном аппарате. Оперативное вмешательство показано в тех случаях, когда лекарственное и физиотерапевтическое лечение не дает положительных результатов. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Этиологические факторы
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
-
 аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении; - выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.
 аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Классификация
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.

Скелет
Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:
- короткое туловище и длинные ноги,
- молоткообразные пальцы стопы,
- паучьи пальцы кисти,
- худощавое телосложение,
- гипотонус мышц,
- удлиненное и узковатое лицо,
- глубокая посадка глаз,
- отсутствие зазоров между зубами,
- большой нос,
- микрогнатия,
- недоразвитые скулы,
- большие и низко расположенные уши,
- «готическое» небо,
- прогения,
- гиперподвижность суставов,
- деформированная грудная клетка в виде воронки или киля птицы,
- искривление позвоночника,
- смещение вышележащего позвонка относительно нижележащего,
- уплощение сводов стопы,
- продавливание вертлужной впадины.

Сердце и сосуды
Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,
- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии – мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца – эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.

Глаза
У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
- страбизм,
- катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.
 страбизм,
страбизм,Прочие органы
Поражение нервной системы:
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
Поражение бронхопульмональной системы:
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.

У больных пневмотораксом воздух скапливается в плевральной полости, а легкие сжимаются. Их дыхание становится частым и поверхностным, в груди болит, кожные покровы сначала бледнеют, а затем приобретают синюшный оттенок.
Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
«Готическое» небо и микрогнатия приводят к возникновению речевых нарушений. При поражении скелета и суставов появляются артралгии и миалгии, развивается ранний остеоартрит.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Методы диагностики

проявления синдрома Марфана в младшем возрасте
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Методы лечения

Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
Медикаментозное лечение:
- β-адреноблокаторы – «Пропранолол», «Атенолол»;
- блокаторы кальциевых каналов – «Нифедипин», «Верапамил»;
- ингибиторы АПФ – «Каптоприл», «Лизиноприл»;
- коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;
- метаболики – «Рибоксин», «Милдронат»;
- поливитаминные комплексы;
- антибиотики для профилактики инфекционного эндокардита;
- антикоагулянты для профилактики тромбоза;
- антиоксиданты и антигипоксанты – «Коэнзим Q10», «Элькар»;
- ноотропные препараты – «Пирацетам», «Винпоцетин».
 коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;
коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;При снижении остроты зрения пациентам с офтальмологическими заболеваниями прописывают очки для постоянного ношения или контактные линзы.
Хирургическое лечение:
- оперативное вмешательство на сердце и аорте — протезирование аорты и клапанов сердца, реконструктивные и пластические операции,
- операции на глазах — коррекция близорукости лазером, замена хрусталика, устранение глаукомы,
- хирургическая коррекция скелета — пластика грудной клетки при ее воронкообразной деформации, стабилизация позвоночника, протезирование крупных суставов.
Людям с марфаноидным фенотипом стертой формы показано физиолечение и ЛФК.
Клинические рекомендации специалистов своим пациентам:
- ведение здорового образа жизни,
- ограничение тяжелого труда и физического перенапряжения,
- отказ от спортивных игр повышенной активности,
- регулярное посещение специалистов в области кардиологии, офтальмологии, ортопедии, неврологии,
- нахождение больных под постоянным наблюдением и контролем врачей,
- периодическое прохождение диагностического обследования.
Прогноз и профилактические мероприятия

Прогноз синдрома Марфана неоднозначный. Он зависит от состояния кардиоваскулярной системы, глаз и скелета больного. При наличии серьезных нарушений со стороны этих органов длительность жизни пациентов ограничивается 40-50 годами, и повышается риск внезапной смерти. С помощью своевременной кардиохирургической коррекции можно улучшить качество жизни больных и вернуть им трудоспособность.
Все пары, в семейном анамнезе которых имелись случаи наследственных заболеваний, должны перед беременностью посетить генетика и заранее сдать все необходимы анализы. Пренатальная диагностика — комплекс мероприятий, проводимых с целью выявления патологии на стадии внутриутробного развития. Она заключается в проведении УЗИ плода и биохимического скрининга материнских сывороточных маркеров. К ее инвазивным методам относятся: биопсия ворсин хориона, исследование околоплодных вод, пуповинной крови и клеток плаценты.
Всем больным необходимо вовремя санировать имеющиеся очаги хронической инфекции в организме — лечить кариес, тонзиллит, синусит с помощью антибиотиков. Это крайне важное мероприятие, поскольку у лиц с синдромом Марфана иммунитет ослаблен. Им следует закаляться, правильно питаться, оптимизировать режим дня, полноценно спать, подолгу гулять на свежем воздухе, бороться с вредными привычками, избегать конфликтных ситуаций и стрессов.
Видео: синдром Марфана в программе “Жить здорово!”
Видео: ток-шоу о синдроме Марфана
Синдром марфана — это наследственное заболевание с аутосомно-доминантным типом наследования. Недуг распространяется на сердечнососудистую и опорно-двигательную систему и глаза. Практически все заболевания, которые относятся к данной группе патологий, приводят к инвалидизации. Многие знаменитые люди страдали этим недугом, в том числе модели и президент США.
Причины и признаки
Появление болезни марфана объясняется мутацией гена, ответственного за выработку фибриллина. Это вещество входит в состав практически всех тканей и органов. Помимо этого болезнь сопровождается нарушением метаболизма кислых гликозаминогликанов. Были известны атипичные случаи мутаций. Кариотип при данном заболевании 47, ХХУ. Как уже было сказано, наследуется по аутосомно-доминантному типу или же выступает результатом спонтанной точечной мутации. То есть, при наличии его в генотипе недуг обязательно возникнет.
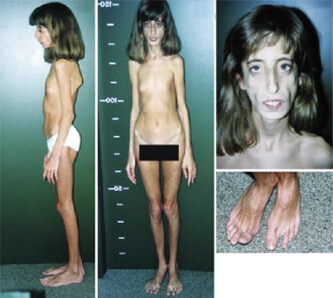
Главной отличительной чертой выступает высокорослость. Комплекция больных вытянутая, пальцы очень тонкие. Внешне пациенты очень похожи друг на друга. Их выдает удлиненная форма головы, небо в виде арки, глаза впалые, челюсть небольшая.
На лицо деформации позвоночного столба и грудной клетки, хлипкость суставов, плоскостопие. Пациенты обычно имеют несколько других диагнозов: сколиозы, кифозы, лордозы и др.
Ярко выражены и глазные проявления. Они дают о себе знать близорукостью, помутнением, увеличением глазного давления и другими расстройствами.
Страдает и дыхательная система, кожа и мягкие ткани. Поражение сердца имеет неблагоприятный прогноз, в основном приводит к смерти.
Симптомы
Клинические симптомы этого заболевания очень многогранны.Но есть общие признаки, которые указывают на наличие болезни:
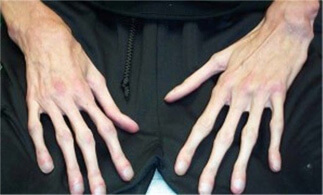
- Арахнодактилия (удлиненные пальцы ).
- Близорукость (миопия).
- Выраженная мышечная слабость.
- Деформация суставов.
- Деформация пальцев рук.
- Аритмия.
- Низкий тонус мышц.
- Частые инфекционные заболевания.
У многих лиц с таким диагнозом симптомы выражены не четко, из-за чего врачу сложно поставить диагноз.
Диагностика
При постановке диагноза во внимание берутся все клинические признаки. Подтвердить диагноз можно с помощью таких мероприятий:
- МРТ сердца.
- МРТ отделов аорты.
- КТ сердца.
- Электрокардиография.
- Эхокардиография.
- Офтальмоскопия.
- КТ с контрастированием сосудов.
- Генетический тест.
Диагностическими признаками служат патологические изменения самых разных систем и органов. Учитывается также индекс массы тела.
Немаловажным критерием служит также и возрастание в несколько раз почечной экскреции метаболитов соединительной ткани.
Лечение
Терапия представляет собой хирургическую и консервативную коррекцию сердечнососудистых патологий.
В основном, лечебные меры нацелены на профилактику развития недуга и осложнений. Больным назначают антагонисты кальция, ингибиторы АПФ и другие препараты.
По показаниям делается операция при пролапсе, недостаточности и других нарушениях.
Необходимо обязательно заниматься коррекцией зрения. Для этого пациентам рекомендованы очки и линзы, иногда прибегают к лазерному воздействию.
При скелетных расстройствах выполняют стабилизацию позвоночника хирургическим путем.
Помимо всего прочего лечебный курс состоит из приема витаминов и метаболического лечения.

Синдром марфана у детей можно легко распознать по характерным признакам, описанным выше. Неестественно длинные пальцы видны уже у новорожденных, что должно послужить поводом к обращению в медицинское учреждение.
При раннем половом созревании проводится индукция специальными гормональными препаратами. В остальном, тактика лечения точно такая же, как и у взрослых.
Пациентам обязательно нужно ходить в ортопедической обуви.
Синдром марфана и беременность — это несовместимые вещи. Заболевание относят к числу очень редких, потому при вынашивании недуг может приводить к трагическим последствиям. Довольно часто возникают выкидыши и преждевременные роды с таким заболеванием. Были даже смертельные случаи.
Прогноз при беременности крайне неблагоприятен. Это обусловлено такими причинами:
- Во-первых, появляется риск развития недуга у ребенка.
- Во-вторых, существует риск осложнений в виде аневризмы аорты и септического эндокардита.
Осложнения
На данный момент те осложнения, которые возникают, успешно лечатся медикаментами. То, что не поддается лечению лекарствами, можно исправить хирургически.
Есть число людей, которые живут и даже не догадываются о своем недуге. При этом они испытывают различные симптомы, нередко приводящие к гибели. Важно пройти обследование своевременно, иначе это приведет к ранней смерти.
Профилактика и прогноз
Исход недуга напрямую зависит от того, какие осложнения вызвало заболевание. Есть большая вероятность уменьшения продолжительности жизни и наступления резкой смерти.
Пациенты с таким диагнозом наблюдаются систематически у врачей, на постоянной основе проходят диагностику. Физическими нагрузками заниматься можно, но они должны быть дозированы. Нельзя заниматься профессиональным спортом, принимать участие в соревнованиях. Все женщины репродуктивного возраста с таким диагнозом проходят генетическую консультацию.
Синдром Марфана (по МКБ-10 Q87.4) — наследственное заболевание соединительной ткани, впервые открытое в 1896 году французским педиатром Антонио Марфаном. Люди, страдающие этим синдромом, как правило, высокого роста, имеют удлиненные конечности, аномально вытянутые пальцы (арахнодактилию), а также недоразвитие жировой клетчатки. Кроме того, у пациентов наблюдается патология в органах зрения и сердечно-сосудистой системы. Распространенность заболевания составляет примерно 1:5000.
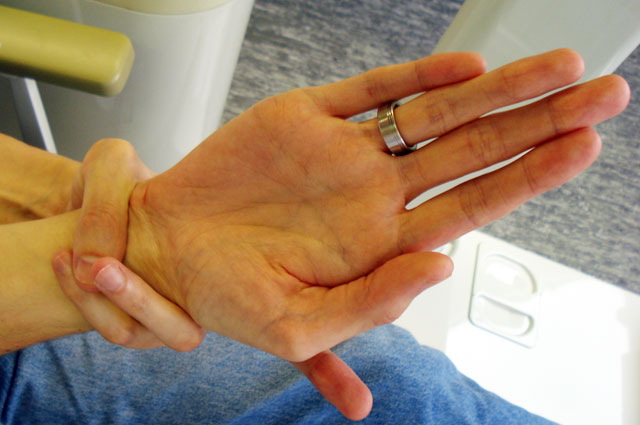
Рука человека с синдромом Марфана. Фото: Commons.wikimedia.org
Как проявляется синдром Марфана?
Как правило, рост пациентов с синдромом Марфана выше средних показателей для возраста и семьи, а размах рук превышает высоту. Также у людей, страдающих этим синдромом, наблюдается арахнодактилия (длинные, аномально вытянутые пальцы). Кроме того, у пациентов может быть килевидная грудная клетка (смещенная наружу) или воронкообразная грудь (смещенная внутрь). Может наблюдаться также гиперподвижность суставов, плоскостопие, кифосколиоз (патологическое искривление позвоночника), диафрагмальные и паховые грыжи.
Симптомы могут включать в себя проблемы со зрением: эктопию хрусталика (подвывих или смещение хрусталика вверх) и иридодонез (дрожание радужной оболочки глаза). У больных синдромом Марфана могут развиться кистозная болезнь легких и рецидивирующий спонтанный пневмоторакс. Со стороны сердечно-сосудистой системы возможна аневризма аорты (расширение участка аорты или выпячивание ее стенки) и пролапс митрального клапана (заболевание, сопровождающееся нарушением функции клапана, расположенного между левым предсердием и желудочком).

Как лечится болезнь?
Без лечения люди, страдающие синдромом Марфана, доживают максимум до 30-40 лет и умирают или из-за расслаивающейся аневризмы аорты, или от застойной сердечной недостаточности.
Лечение преимущественно симптоматическое, направленное на облегчение тех или иных симптомов заболевания. Больным требуется проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда. В случае регулярного симптоматического лечения больные могут доживать до преклонного возраста, однако их средняя ожидаемая продолжительность жизни все же ниже из-за сердечных и сосудистых осложнений.
Для симптоматического лечения синдрома Марфана в большинстве клинических исследований рекомендуется профилактическое употребление бета-адреноблокаторов с раннего возраста. Это делается для того, чтобы предотвратить расслаивающуюся аневризму аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция.
Синдром Марфана — Википедия
Материал из Википедии — свободной энциклопедии
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[1], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Наследственное заболевание.
Эпидемиология
Синдром Марфана — редкое заболевание с классическим менделевским наследованием. Распространённость в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах.
История
Впервые признаки заболевания были описаны в 1875 году американским офтальмологом Э. Вильямсом (англ. E. Williams), описавшим эктопию хрусталика у брата и сестры, которые были исключительно высокими и имели гипермобильные суставы от рождения[2]. В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном[fr], который представил в 1896 году клиническое наблюдение 5-летней девочки Габриэль с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.[3]
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.[4]
Американский генетик Виктор Маккьюсик[en] открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани.[5]
Симптомы
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.[6]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Лечение
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.[7]
Интересные факты
См. также
Примечания
- ↑ Синдром Марфана. Первый Московский Государственный Университет имени И.М. Сечнова (19 октября 2007). — Течение и прогноз. Проверено 23 августа 2012. Архивировано 18 октября 2012 года.
- ↑ Williams E. Rare Cases, with Practical Remarks // Transactions of the American Ophthalmological Societ. — 1875. — Vol. 2. — PP. 291—301. — PMC 1361735
- ↑ Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- ↑ Hecht F., Beals R. K. «New» syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. // Pediatrics. — 1972 Apr. — Vol. 49(4). — PP. 574—579. — PMID 4552107
- ↑ McKusick V. A. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. // Circulation[en]. — 1955 Mar. — Vol. 11(3). — PP. 321—341. — PMID 14352380
- ↑ Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- ↑ Goldman’s Cecil Medicine 978-1-4557-1167-3,
Литература
- Лисиченко О. В. Синдром Марфана. Новосибирск: Наука, 1986. 164 с.
Ссылки
На русском языке
На английском языке
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[1], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Наследственное заболевание.
Эпидемиология
Синдром Марфана — редкое заболевание с классическим менделевским наследованием. Распространённость в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах.
История
Впервые признаки заболевания были описаны в 1875 году американским офтальмологом Э. Вильямсом (англ. E. Williams), описавшим эктопию хрусталика у брата и сестры, которые были исключительно высокими и имели гипермобильные суставы от рождения[2]. В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном[fr], который представил в 1896 году клиническое наблюдение 5-летней девочки Габриэль с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.[3]
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.[4]
Американский генетик Виктор Маккьюсик[en] открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани.[5]
Симптомы
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.[6]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Лечение
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.[7]
Интересные факты
См. также
Примечания
- ↑ Синдром Марфана. Первый Московский Государственный Университет имени И.М. Сечнова (19 октября 2007). — Течение и прогноз. Проверено 23 августа 2012. Архивировано 18 октября 2012 года.
- ↑ Williams E. Rare Cases, with Practical Remarks // Transactions of the American Ophthalmological Societ. — 1875. — Vol. 2. — PP. 291—301. — PMC 1361735
- ↑ Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- ↑ Hecht F., Beals R. K. «New» syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. // Pediatrics. — 1972 Apr. — Vol. 49(4). — PP. 574—579. — PMID 4552107
- ↑ McKusick V. A. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. // Circulation[en]. — 1955 Mar. — Vol. 11(3). — PP. 321—341. — PMID 14352380
- ↑ Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- ↑ Goldman’s Cecil Medicine 978-1-4557-1167-3,
Литература
- Лисиченко О. В. Синдром Марфана. Новосибирск: Наука, 1986. 164 с.
Ссылки
На русском языке
На английском языке
Синдром Марфана — это… Что такое Синдром Марфана?
Синдром Марфана (Болезнь Марфана, Marfan syndrome) — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлиненные конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[1], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Наследственное заболевание, входит под номером 154700 в систему табуляции МакКьюсика OMIM.
Эпидемиология
Синдром Марфана — нередкое заболевание с классическим менделевским наследованием. Распространенность в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах.
История
Впервые признаки заболевания были описаны Вильямсом (1876), в последующие годы эта болезнь наблюдалась французским педиатром А. Марфаном (1896). В 1896 году французский профессор-педиатр Антонио Марфан представил клиническое наблюдение 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.[2]
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.[3]
Позже были описаны первые фенокопии марфаноподобных синдромов, в частности, синдром эктопии хрусталиков с аутосомно-доминантным наследованием, синдром дилатации и расслоения аорты, пролапс митрального клапана, эктазии твердой мозговой оболочки[источник не указан 901 день].
Американский генетик МакКьюсик (McKusick) открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани.[4]
Симптомы
Фенотип больных характеризуется определённой протяженностью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.[5]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое небо, недоразвитие вертлужной впадины, врожденные контрактуры локтей и пальцев, мышечная гипотония. (Бочков 2004)
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80% случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5% больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-кресцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Дифференциальная диагностика
Существует группа «марфанподобных синдромов», с состоянием больных внешне очень напоминающим синдром Марфана, которые значительно отличаются по висцеральным поражениям, осложнениям и прогнозу.[источник не указан 901 день]
Помимо триады Марфана, в других органах также наблюдаются изменения (бронхо-лёгочная система, кожа, твёрдая мозговая оболочка).[источник не указан 901 день]
Лечение
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бетта-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.[6]
См. также
Примечания
| В этой статье не хватает ссылок на источники информации. Информация должна быть проверяема, иначе она может быть поставлена под сомнение и удалена. Вы можете отредактировать эту статью, добавив ссылки на авторитетные источники. Эта отметка установлена 14 мая 2011. |

- ↑ Синдром Марфана. Первый Московский Государственный Университет имени И.М. Сечнова (19 октября 2007). — Течение и прогноз. Архивировано из первоисточника 18 октября 2012. Проверено 23 августа 2012.
- ↑ Marfan A.B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. Bull Mem Soc Med Hip (Paris) 1896; 13: 220—226.
- ↑ Hecht F., Beals R.K. «New» syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. Pediatrics 1972; 49: 574—579.
- ↑ McKusick V.A. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. Circulation 1955; 11: 321—341.
- ↑ Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- ↑ Goldman’s Cecil Medicine 978-1-4557-1167-3,
Ссылки
На русском языке
На английском языке
СИНДРОМ МАРФАНА — Справочник по болезням
мед.
Синдром Марфана — наследственное заболевание соединительной ткани с вовлечением скелетно-мышечной и сердечнососудистой систем и патологией глаз. Все доказанные случаи синдрома Марфана — следствие мутации гена фибриллина-1 (#154700, 15ql5-q21.3, ген FBN1 [434797], ЭД. Выявлено множество различных мутаций, что объясняет значительную клиническую полиморфность заболевания. Свыше 15% случаев -следствие новых мутаций. Атипичные формы синдрома Марфана могут быть вызваны мутациями в других генах, например в гене белка, трансформирующего фактор роста р (602091, 14q24, ген LTBP3, R). Частота — 1 случай на 10 000-20 000. Патоморфология
● Кистозный некроз средней оболочки аорты
● Мик-соматозная дегенерация клапанов сердца.
.
❐ Клиническая картина
● Скелетно-мышечная система
● Высокий рост
● Астеническое телосложение (длина конечностей не пропорциональна длине туловища)
● Арахнодактилия (длинные тонкие пальцы)
● Деформация грудной клетки
● Высокое арковидное нёбо
● Кифосколиоз
● Слабость связочного аппарата.
● ССС
● Дилатация корня аорты
● Аортальная регургитация
● Расслаивающая аневризма аорты
● Пролапс митрального клапана
● Регургитация крови при недостаточности митрального клапана.
● Глаза
● Иридоденез (дрожание хрусталика вследствие слабости цинновой связки)
● Подвывих хрусталика
● Отслойка сетчатки
● Близорукость высокой степени.
● Другие симптомы (редко)
● Геморрагический синдром
● Спонтанный пневмоторакс.
.
❐ Дифференциальный диагноз
● Гомоцистинурия (236200) — клиника болезни Марфана, умственная отсталость, в моче — гиперэкскреция гомоцистина и метионина
● Контрактурная арахнодактилия
● Синдром Билса (121050, ген фибриллина-2 FBN2, 5q23-q31, R) -разновидность болезни Марфана с нарушением синтеза белка фибриллина-2
● Синдром Элерса-Данло-Русакова (разные типы)
● Расслоение стенки артерий с лентигинозом (600459)
● Синдром врождённой гиперрастяжимой кожи с марфаноподобным фенотипом (* 150240, 7q31.1-q31.3, R)
● Трисомия 8 имеет костные проявления, как при синдроме Марфана, но без глазных и сердечно-сосудистых признаков
● Марфаноидное телосложение с микроцефалией и гломерулонефритом (248760, р). Клинически: умственная отсталость, микроцефалия, высокое нёбо, проптня, марфаноидный внешний вид, гломерулонефрит, почечная недостаточность, расширение IV желудочка. Лабораторные исследования неспецифичны
● Отмечают непостоянную гиперэкскрецию гликозаминогликанов и оксипролина с мочой
● Рекомендовано исследование уровня гомоцистина в моче, чтобы исключить гомоцистинурию.
.
❐ Специальные исследования
● Исследование с применением щелевой лампы — слабость циановой связки, склонность к подвывиху хрусталика
● Обзорные рентгенограммы позвоночного столба в период роста — обнаружение и оценка выраженности сколиоза
● Ежегодные скри-нинговые эхокардиограммы, начиная с пубертатного периода, -бессимптомная дилатация корня аорты и дегенерация клапанов.
.
❐ Лечение:
Тактика ведения. Наблюдение у участкового терапевта, кардиолога, офтальмолога и хирурга-ортопеда (при возможности, генетическое консультирование). Физическую активность следует ограничить.
✎ Хирургическое лечение. Реконструктивная сердечно-сосудистая операция.
✎ Лекарственная терапия:
● Пропранолол (анаприлин) может отсрочить развитие или замедлить прогрессирование расслоения аорты. Дозу подбирают до достижения желаемой ЧСС (в покое — 60 в мин, при нагрузке — не более 80 в мин)
● Эстрогены в сочетании с прогестероном для стимуляции пропорционального полового созревания у девушек (под наблюдением эндокринолога).
.
❐ Наблюдение
● Частые обследования (по крайней мере два раза в год) в период роста, особенное внимание слудет обращать на ССС и позвоночник (сколиоз)
● При поражении сердца или увеличении диаметра корня аорты более 50 мм необходимо рассмотреть возможность хирургического вмешательства
● При наличии подвывиха хрусталика — хирургическая коррекция, но учитывая высокий риск развития глаукомы, операцию нужно предлагать только тем, кто не может обойтись корригирующими линзами.
.
❐ Осложнения
● Септический эндокардит
● Расслаивающая аневризма аорты
● Недостаточность аортального или митрального клапана
● Дилатация сердца
● Отслойка сетчатки.
✎ Течение и прогноз: До широкого распространения хирургической коррекции сердечно-сосудистой патологии большинство пациентов с синдромом Марфана умирали до 35 лет. При адекватной коррекции продолжительность жизни большинства пациентов может быть нормальной.
Беременность. Беременных женщин с синдромом Марфана нужно вести как пациенток высокого риска, лучше с участием кардиолога. Результат обычно благоприятный.
✎ МКБ: Q87.4 Синдром Марфана
✎ MIM :. Синдром Марфана:
● классическая форма (154700)
● атипичная форма (6020R)
✎ Литература: Pyeritz RE, McKusick VA: The Marfan Syndrome: Diagnosis and Management. New Engi J Mod 300: 772-777, 1979; Scriver RC et al: The Metabolic Basis of Inherited Disease, 7th Ed. New York, McGraw Hill, 1995
Источник:
Справочник-путеводитель практикующего врача
на Gufo.me
Значения в других словарях
- синдром марфана —
Синдром Марфана — наследственное заболевание соединительной ткани с вовлечением скелетно-мышечной и сердечнососудистой систем и патологией глаз. Все доказанные случаи синдрома Марфана — следствие мутации гена фибриллина-1 (#154700, 15ql5-q21.
Медицинский словарь

Синдром Марфана — Genetics Home Reference
Каньядас V, Вилакоста I, Бруна I, синдром Фустера В. Марфана. Часть 1: патофизиология и диагностика. Nat Rev Cardiol. 2010 май; 7 (5): 256-65. doi: 10.1038 / nrcardio.2010.30. Epub 2010 30 марта. Обзор.
Chaffins JA. Синдром Марфана. Радиол Технол. 2007 январь-февраль; 78 (3): 222-36; викторина 237-9. Обзор.
Faivre L, Masurel-Paulet A, Collod-Béroud G, Callewaert BL, Child AH, Stheneur C, Binquet C, Gautier E, Шевалье B, Huet F, Loeys BL, Arbustini E, Mayer K, Арслан-Киршнер М, Киоцекоглу А, Комельо П, Грассо М, Хэллидей Диджей, Бериуд С, Бонитон-Копп С, Клаустр М, Робинсон П.Н., Адес Л, Де Бакер Дж, Кук П, Франк У, Де Паеп А, Буало С, Жондо Г ,Клинико-молекулярное исследование 320 детей с синдромом Марфана и родственными фибриллинопатиями I типа в серии из 1009 пробандов с патогенными мутациями FBN1. Педиатрия. 2009 янв; 123 (1): 391-8. doi: 10.1542 / peds.2008-0703.
Keane MG, Pyeritz RE. Медицинское лечение синдрома Марфана. Циркуляционный. 2008 май 27; 117 (21): 2802-13. doi: 10.1161 / CIRCULATIONAHA.107.693523. Обзор.
Киршнер Р., Хубмахер Д., Айенгар Г., Каур Ю., Фаготто-Кауфманн С., Бремме Д., Бартельс Р., Рейнхардт Д.П.Мутации классического и неонатального синдрома Марфана в фибриллин-1 вызывают дифференциальную чувствительность к протеазам и функцию белка. J Biol Chem. 2011 сент. 16; 286 (37): 32810-23. doi: 10.1074 / jbc.M111.221804. Epub 2011 Jul 22.
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P , Де Папе AM. Пересмотренная нозология Гента для синдрома Марфана. J Med Genet. 2010 Jul; 47 (7): 476-85.doi: 10.1136 / jmg.2009.072785.
Mizuguchi T, Мацумото Н. Последние достижения в области генетики синдрома Марфана и расстройств, связанных с Марфаном. J Hum Genet. 2007; 52 (1): 1-12. Epub 2006 24 октября. Обзор.
Пирсона GD, Девереукс R, Loeys B, Маслен C, Milewicz D, Pyeritz R, Рамирес F, Рифкин D, Сакаи L, Свенсон L, Вессельс A, Ван Эйк J, Dietz HC; Национальный институт сердца, легких и крови и рабочая группа Национального фонда Марфана.Доклад Национального института сердца, легких и крови и Рабочей группы Национального фонда Марфана об исследованиях синдрома Марфана и связанных с ним расстройств. Циркуляционный. 2008 Aug 12; 118 (7): 785-91. doi: 10.1161 / CIRCULATIONAHA.108.783753.
Pyeritz RE, Loeys B. 8-й международный научный симпозиум по синдрому Марфана и связанным с ним состояниям. Am J Med Genet A. 2012 Jan; 158A (1): 42-9. doi: 10.1002 / ajmg.a.34386. Epub 2011 2 декабря.
Pyeritz RE; Американский колледж медицинской генетики и геномики.Оценка подростка или взрослого с некоторыми особенностями синдрома Марфана. Genet Med. 2012 янв; 14 (1): 171-7. doi: 10.1038 / gim.2011.48. Epub 2012 5 января.
Робинсон П.Н., Артеага-Солис Е, Балдок С, Коллод-Беруд Г., Бумс П, Де Паепе, Дитц ХК, Го Г, Хэндфорд П.А., судья Д.П., Килти С.М., Лоис Б, Милевич Д., Ней А., Рамирес Ф., Рейнхардт Д. П., Тидеманн К., Вайтман П., Годфри М. Молекулярная генетика синдрома Марфана и связанных с ним расстройств. J Med Genet. 2006 окт; 43 (10): 769-87.Epub 2006 29 марта. Обзор.
Сингх К.К., Роммель К., Мишра А., Карк М., Хаверих А., Шмидтке Дж., Арслан-Киршнер М. Мутации TGFBR1 и TGFBR2 у пациентов с признаками синдрома Марфана и синдрома Лоис-Дитца. Хум Мутат. 2006 Авг; 27 (8): 770-7.
.
10 Симптомы синдрома Марфана
Синдром Марфана — это генетическое заболевание, которое влияет на соединительные ткани в организме. Серьезность того, как каждый человек затронут, варьируется. Синдром Марфана является аутосомно-доминантным состоянием, при котором наследование одного аллеля выражает фенотип. В 75% случаев синдрома Марфана это состояние связано с наследованием от родителя, в то время как остальные 25% связаны с новой мутацией. Диагноз синдрома Марфана можно поставить на основе критериев Гента.Не существует известного лечения синдрома Марфана. Тем не менее, большинство пациентов могут вести нормальную продолжительность жизни, если они получают надлежащее лечение.
Синдром Марфана часто лечится с помощью бета-блокаторов (таких как атенолол и пропранолол), блокаторов кальциевых каналов или ингибиторов ангиотензинпревращающего фермента (АПФ). В зависимости от тяжести состояния может потребоваться хирургическое вмешательство для замены клапана сердца для восстановления аорты. Упорных упражнений следует избегать. Подсчитано, что приблизительно 1 из каждых 5 000 — 10 000 человек имеет синдром Марфана.Показатели схожи между полами, расами и различными регионами мира. Состояние названо в честь французского педиатра Антуана Марфана, который впервые описал его в 1896 году.
Сколиоз — это состояние, при котором позвоночник искривлен вбок и может выглядеть как кривая в форме буквы «S» или «C». Хотя легкий сколиоз обычно не вызывает проблем, тяжелые случаи сколиоза могут привести к затруднению дыхания. Это может произойти при многих состояниях, таких как церебральный паралич, нейрофиброматоз, мышечные спазмы, синдром Марфана и другие.
Лечение сколиоза зависит от основной степени искривления позвоночника, местоположения искривления и причины. Незначительные случаи обрабатываются с помощью бдительного наблюдения и могут включать в себя готовность. Более сложные случаи потребуют хирургического вмешательства.
,
10 Симптомы синдрома Марфана
Синдром Марфана — это редкое заболевание, которое поражает скелет и многие органы тела. Он передается генетически, но может принимать разные формы у членов одной семьи. Например, некоторые люди с синдромом необычно высоки с длинными и довольно тонкими руками и ногами. В других случаях синдром может привести к плоскостопию, зубам, которые выглядят так, как будто они скучены во рту, или очень глубоко посаженным глазам. В большинстве случаев симптомы находятся на мягкой стороне, но они имеют тенденцию ухудшаться с возрастом человека.
Изогнутый позвоночник
В некоторых случаях синдром Марфана заставляет позвоночник искривляться в сторону. Врачи называют это состояние сколиозом. Степень, в которой это условие мешает нормальной жизни, зависит от степени искривления позвоночника. Повреждение позвоночника варьируется. В самых легких случаях они испытывают небольшую боль в спине, но в самых тяжелых случаях они должны справляться с сильными болями в спине в течение длительного периода. Позвоночник может начать давить на легкие и сердце и вызвать у пациента проблемы с дыханием.
Повреждение зрения
Ущерб, который синдром Марфана причиняет глазам, является одним из симптомов, который, скорее всего, влияет на качество жизни. Статистика показывает, что примерно в 50 процентах случаев это заболевание приводит к выскальзыванию хрусталика глаза из нормального положения. Некоторые люди становятся близорукими, и они могут также испытывать затуманенное зрение. В наиболее тяжелых случаях у них может развиться глаукома, которая вызывает потерю зрения, которую невозможно исправить. Любой человек с синдромом Марфана, испытывающий эти проблемы со зрением, должен проконсультироваться с окулистом и оценить, насколько безопасно продолжать движение.

Проблемы с сердцем
Повреждение сердца является еще одной областью здравоохранения, где люди с синдромом Марфана должны быть особенно обеспокоены. Некоторые люди испытывают пролапс митрального клапана, который приводит к одышке и вызывает сердечные шумы. Когда врач слушает сердце через стетоскоп, они могут быстро обнаружить неровное сердцебиение. Сам по себе шум в сердце не должен быть опасным — считается, что у каждого из 20 человек есть некоторый шум в сердце — но те, у кого синдром Марфана, могут также испытывать сильные боли в груди и другие признаки, связанные с болезнью сердца.

Воздействие на кровеносные сосуды
Возможные последствия синдрома Марфана для артерий — еще одна законная область для беспокойства. Главная артерия в теле, аорта, соединяет сердце с желудком и играет ключевую роль в передаче жизненно важного кислорода. Синдром Марфана может ослабить стенку этой артерии и даже вызвать ее увеличение. Это может привести к разрыву артерии с потенциально смертельными последствиями. Эта перспектива — еще одна веская причина, по которой любому, у кого есть такое заболевание, необходимо регулярно проходить осмотр здоровья.
Здоровье легких
Легкие могут легко повредиться из-за синдрома Марфана. Коллапс легкого может произойти из-за газа или воздуха, который накапливается между легкими и грудной клеткой. Любой, кто чувствует острую боль на одной стороне своих легких и внезапно начинает задыхаться, должен немедленно обратиться за медицинской помощью
.
Синдром Марфана
синдром Определение
Марфана является наследственным расстройством соединительной ткани, что вызывает аномалии глаз пациента, сердечно-сосудистой системы и опорно-двигательного аппарата. Он назван в честь французского педиатра Антуана Марфана (1858–1942), который впервые описал его в 1896 году. Синдром Марфана иногда называют арахнодактилией, что по-гречески означает «паукообразные пальцы», поскольку это один из характерных признаков болезни это непропорционально длинные пальцы рук и ног.По оценкам, один человек на каждые 3000-5000 человек страдает синдромом Марфана, или около 50000 человек в Соединенных Штатах. Синдром Марфана является одним из наиболее распространенных наследственных расстройств.
Описание
Синдром Марфана поражает три основные системы органов организма: сердце и систему кровообращения, кости и мышцы, а также глаза. Генетическая мутация, ответственная за Марфан, была обнаружена в 1991 году. Она влияет на выработку в организме фибриллина, который является важной частью соединительной ткани.Фибриллин является основным компонентом микрофибрилл, которые позволяют тканям многократно растягиваться без ослабления. Поскольку фибриллин пациента является ненормальным, его или ее соединительные ткани более рыхлые, чем обычно, что ослабляет или повреждает опорные структуры всего тела.
Наиболее распространенные внешние признаки, связанные с синдромом Марфана, включают чрезмерно длинные руки и ноги, при этом размах рук пациента превышает его рост. Пальцы рук и ног могут быть длинными и тонкими, со свободными суставами, которые могут быть согнуты за пределы нормы.Эта необычная гибкость называется гипермобильностью. Лицо пациента также может быть длинным и узким, и он или она может иметь заметную кривизну позвоночника. Однако важно отметить, что пациенты с синдромом Марфана сильно различаются по внешним признакам своего расстройства и степени тяжести; даже два пациента из одной семьи могут выглядеть совершенно по-разному. Большинство внешних признаков синдрома Марфана становятся более выраженными с возрастом пациента, поэтому диагностика расстройства часто легче у взрослых, чем у детей.Во многих случаях у пациента может быть мало или очень незначительные внешние признаки расстройства, и диагноз может быть пропущен, пока у пациента не возникнут проблемы со зрением или сердечные симптомы.
Синдром Марфана сам по себе не влияет на интеллект человека или способность к обучению. Однако есть некоторые клинические доказательства того, что у детей с Марфаном уровень гиперактивности и синдрома дефицита внимания (ADD) несколько выше, чем у населения в целом. Кроме того, ребенок с недиагностированной близорукостью, связанной с Марфаном, может испытывать трудности с просмотром доски или чтением печатных материалов и, следовательно, плохо учиться в школе.
Синдром Марфана одинаково влияет на мужчин и женщин и, по-видимому, распределен поровну между всеми расами и этническими группами. Однако скорость мутации гена фибриллина, по-видимому, связана с возрастом отца пациента; у старших отцов чаще появляются новые мутации в хромосоме 15.
Причины и симптомы
Синдром Марфана вызван одним геном фибриллина в хромосоме 15, который в большинстве случаев наследуется от пораженного родителя.От 15 до 25% случаев являются результатом спонтанных мутаций. Мутации гена фибриллина (FBNI) уникальны для каждой семьи, пораженной Марфаном, что делает невозможной быструю генетическую диагностику, учитывая современную технологию. Синдром является аутосомно-доминантным расстройством, что означает, что у кого-то, у кого он есть, есть шанс 50% передать его любому потомству.
Другой важной генетической характеристикой синдрома Марфана является вариабельная экспрессия. Этот термин означает, что мутированный ген фибриллина может вызывать различные симптомы очень различной степени тяжести даже у членов одной семьи.
Нарушения со стороны сердца и кровообращения
Наиболее важными осложнениями у Марфана являются поражения сердца и крупных кровеносных сосудов; некоторые из них потенциально опасны для жизни. Приблизительно у 90% пациентов с болезнью Марфана разовьются сердечные осложнения.
- Увеличение аорты. Это самое серьезное потенциальное осложнение синдрома Марфана. Из-за аномалий фибриллина пациента стенки аорты (большой кровеносный сосуд, который уносит кровь от сердца) слабее, чем обычно, и имеют тенденцию растягиваться и выпирать из формы.Это растяжение увеличивает вероятность расслоения аорты, которое представляет собой разрыв или разделение между слоями ткани, из которых состоит аорта. Расслоение аорты обычно вызывает сильную боль в животе, спине или груди, в зависимости от пораженного участка аорты. Разрыв аорты — неотложная медицинская помощь, требующая немедленной операции и приема лекарств.
- Аортальная регургитация. Ослабленная и расширенная аорта может позволить некоторой крови просачиваться обратно в сердце при каждом сердцебиении; это состояние называется аортальной регургитацией.Аортальная регургитация иногда вызывает одышку во время нормальной деятельности. В серьезных случаях это вызывает расширение левого желудочка сердца и может в конечном итоге привести к сердечной недостаточности.
- Выпадение митрального клапана. От 75 до 85% пациентов с болезнью Марфана имеют свободные или «гибкие» митральные клапаны, которые являются клапанами, которые разделяют камеры сердца. Когда эти клапаны не полностью закрывают отверстие между камерами, это состояние называется пролапсом митрального клапана.Осложнения пролапса митрального клапана включают сердечные шумы и аритмии. В редких случаях пролапс митрального клапана может привести к внезапной смерти.
- Инфекционный эндокардит. Инфекционный эндокардит — это инфекция эндотелия, ткани, которая выстилает сердце. У пациентов с Марфаном именно аномальный митральный клапан, скорее всего, заразится.
- Другие осложнения. У некоторых пациентов с Марфаном развивается кистозное заболевание легких или рецидивирующий спонтанный пневмоторакс, который представляет собой состояние, при котором воздух накапливается в пространстве вокруг легких.У многих также со временем разовьется эмфизема.
Опорно-аномалии
синдром Марфана вызывает увеличение длины костей пациента, с уменьшением поддержки со стороны связки, которые удерживают кости вместе. В результате у пациента могут развиться различные деформации скелета или расстройства, связанные с относительной рыхлостью связок.
Заболевания позвоночника
- Сколиоз. Сколиоз, или искривление позвоночника, представляет собой заболевание, при котором позвонки, составляющие позвоночник, скручиваются из линии в сторону в S-образную форму или спираль.Это вызвано сочетанием быстрого роста детей с Марфаном и ослаблением связок, которые помогают позвоночнику сохранять свою форму.
- Кифоз — это ненормальная внешняя кривизна позвоночника сзади, иногда называемая предчувствием, когда он возникает в верхней части спины. У пациентов с марфаном может развиться кифоз либо в верхнем (грудном) позвоночнике, либо в нижнем (поясничном) позвоночнике.
- Спондилолистез. Спондилолистез — это медицинский термин, обозначающий прямое смещение одного позвонка на нижний.Это вызывает боль или жесткость в нижней части спины.
- Дуральная эктазия. Твердая мозговая оболочка — это прочная, волокнистая наружная мембрана, покрывающая головной и спинной мозг. Слабая твердая мозговая оболочка у пациентов с болезнью Марфана отекает или выпирает под давлением спинномозговой жидкости. Этот отек называется эктазия. В большинстве случаев дуральная эктазия возникает в нижней части позвоночника, вызывая боль в пояснице, чувство жжения или онемение или слабость в ногах.
Расстройства грудной и нижней части тела
- Pectus excavatum.Pectus excavatum — это порок развития грудной клетки, в котором грудная клетка или грудина пациента утоплены внутрь. Это может вызвать затруднение дыхания, особенно если Марфан поразил сердце, позвоночник и легкие. Это также обычно вызывает беспокойство по поводу внешнего вида.
- Pectus carinatum. У других пациентов с Марфаном грудная клетка выталкивается наружу и сужается. Хотя pectus carinatum не вызывает затруднения дыхания, он может вызвать смущение по поводу внешнего вида. У нескольких пациентов с Марфаном может быть pectus excavatum на одной стороне их грудной клетки и pectus carinatum на другой.
- Болезни ног. Пациенты с Марфаном более склонны к развитию плоских стоп (плоскостопие) или так называемых «коготь» или «молоток» пальцев ног, чем люди в общей популяции. Они также чаще страдают от хронической боли в ногах.
- Protrusio acetabulae. Вертлужная впадина — это гнездо тазобедренного сустава. У пациентов с Марфаном, вертлужная впадина становится глубже, чем обычно во время роста, по причинам, которые еще не поняты. Хотя протрузия вертлужной впадины не вызывает проблем в детстве и подростковом возрасте, она может привести к болезненной форме артрита во взрослой жизни.
Расстройства глаз и лица
Хотя проблемы со зрением, связанные с синдромом Марфана, редко бывают опасными для жизни, они важны тем, что могут быть первыми признаками расстройства у пациента. Глазные расстройства, связанные с синдромом, включают следующее:
- Близорукость (близорукость). У большинства пациентов с Марфаном развивается близорукость, обычно в детстве.
- Ectopia lentis. Ectopia lentis — медицинский термин для вывиха хрусталика глаза.От 65 до 75% пациентов Марфана имеют вывих линзы. Это состояние является важным показанием для диагностики синдрома, потому что существует относительно немного других расстройств, которые его вызывают.
- Глаукома. Это состояние гораздо чаще встречается у пациентов с синдромом Марфана, чем среди населения в целом.
- катаракты. Пациенты с Марфаном более склонны к развитию катаракты и к развитию их значительно раньше в жизни, иногда уже в возрасте 40 лет.
- Отслойка сетчатки. Пациенты с Марфаном более уязвимы к этому расстройству из-за слабости их соединительной ткани. Без лечения отслойка сетчатки может вызвать слепоту. Опасность отслоения сетчатки является важной причиной для пациентов избегать контактных видов спорта или других видов деятельности, которые могут вызвать удар по голове или падение на землю.
- Другие проблемы с лицом. У пациентов с Марфаном иногда возникают проблемы с зубами, связанные с скученностью зубов, вызванной высоким сводчатым небом и узкой челюстью.
Другие расстройства
- стрий. Стрии — это растяжки на коже, вызванные быстрым увеличением или ростом веса; например, они часто встречаются у беременных женщин. У пациентов с марфаном часто развиваются стрии над плечами, бедрами и нижней частью спины в раннем возрасте из-за быстрого роста костей. Хотя пациент может стесняться в отношении бороздок, они не представляют опасности для здоровья.
- Обструктивное апноэ во сне. Обструктивное апноэ во сне относится к частичной обструкции дыхательных путей во время сна, вызывая нерегулярное дыхание и иногда храп.У пациентов с Марфаном обструктивное апноэ во сне вызвано необычной гибкостью тканей, выстилающих дыхательные пути пациента. Этот нарушенный тип дыхания увеличивает риск расслоения аорты.
Диагноз
В настоящее время не существует объективного диагностического теста для синдрома Марфана, отчасти потому, что расстройство не вызывает каких-либо измеримых биохимических изменений в крови или жидкостях организма пациента или клеточных изменений, которые могут быть обнаружены из образца ткани.Хотя исследователи в области молекулярной биологии в настоящее время изучают ген FBNI с помощью процесса, называемого мутационным анализом, в настоящее время он не используется в качестве диагностического теста, поскольку имеются доказательства того, что в гене фибриллина могут быть мутации, которые не продуцируют марфан. Точно так же не существует надежного пренатального теста, хотя некоторые врачи использовали ультразвук, чтобы попытаться определить длину конечностей плода при беременности, подверженной риску.
Диагноз ставится на основании семейного анамнеза и тщательного обследования глаз, сердца и костной структуры пациента.Обследование должно включать эхокардиограмму, сделанную кардиологом, осмотр глаз с помощью щелевой лампы офтальмологом и осмотр позвоночника пациента специалистом-ортопедом. С точки зрения кардиологического обследования, стандартная электрокардиограмма (ЭКГ) недостаточна для диагностики; только эхокардиограмма может обнаружить возможное расширение аорты. Важность осмотра с использованием щелевой лампы заключается в том, что он позволяет врачу выявлять вывихнутую линзу, что является существенным признаком синдрома.
Симптомы синдрома Марфана у некоторых пациентов напоминают симптомы гомоцистинурии, которая является наследственным заболеванием, характеризующимся чрезвычайно высоким уровнем гомоцистина в крови и моче пациента. Эта возможность может быть исключена анализом мочи.
В других случаях диагноз остается неопределенным из-за слабости симптомов пациента, отсутствия семейного анамнеза синдрома и других переменных. Эти пограничные состояния иногда называют марфаноидными синдромами.
Лечение
Лечение и лечение Марфаном подбирается с учетом конкретных симптомов каждого пациента. Некоторые пациенты считают, что синдром мало влияет на их общий образ жизни; другие нашли свою жизнь сосредоточенной на беспорядке.
Сердечно-сосудистая система
После того, как человеку был поставлен диагноз Марфан, ему или ей следует проводить эхокардиограмму каждые шесть месяцев, пока не станет ясно, что аорта не увеличивается. После этого пациент должен делать эхокардиограмму один раз в год.Если эхокардиограмма не позволяет врачу визуализировать все части аорты, можно использовать КТ (компьютерная томография) или МРТ (магнитно-резонансная томография). В случаях возможного расслоения аорты пациенту может быть назначена TEE (чреспищеводная эхокардиограмма).
Лекарства. Пациенту Марфана могут назначать препараты, называемые бета-адреноблокаторами, чтобы замедлить скорость увеличения аорты и снизить риск расслоения, снижая кровяное давление и уменьшая сердцебиение.Наиболее часто применяемыми бета-адреноблокаторами у пациентов с марфаном являются пропранолол (индерал) и атенолол (тенормин). Пациентам с аллергией на бета-адреноблокаторы может быть назначен блокатор кальция, такой как верапамил.
Поскольку пациенты Марфана подвергаются повышенному риску инфекционного эндокардита, они должны принять профилактическую дозу антибиотика перед стоматологической работой или незначительной операцией, поскольку эти процедуры могут позволить бактериям проникать в кровоток. Пенициллин и амоксициллин являются наиболее часто используемыми антибиотиками.
Хирургическое лечение. Операция может быть необходимой, если ширина аорты пациента быстро увеличивается или достигает критического размера (около 2 дюймов). Начиная с 2000 года, наиболее распространенное хирургическое лечение включает замену аортального клапана пациента и нескольких дюймов самой аорты композитным трансплантатом, который представляет собой протез клапана сердца, вшитый в один конец трубки Дакрона. Эта операция была широко выполнена примерно с 1985 года; большинство пациентов, у которых был сложный трансплантат, не нуждались в дополнительной операции.
Пациенты, у которых был заменен клапан, должны принимать антикоагулянтные препараты, обычно варфарин (кумадин), чтобы свести к минимуму возможность образования сгустка на протезе клапана.
Костно-мышечная система
Детей с диагнозом Марфан следует проверять на наличие сколиоза у своих педиатров при каждом ежегодном медицинском осмотре. Врач просто просит ребенка наклониться вперед, пока спина осматривается на предмет изменений кривизны. Кроме того, позвоночник ребенка должен быть подвергнут рентгеновскому исследованию, чтобы измерить степень сколиоза или кифоза.Кривая измеряется в градусах под углом между позвонками, как видно на рентгеновском снимке. Кривые 20 ° или менее вряд ли станут хуже. Кривые между 20 и 40 градусами могут увеличиться у детей или подростков. Кривые 40 градусов и более могут ухудшиться даже у взрослого, потому что позвоночник настолько сильно разбалансирован, что сила тяжести увеличит кривизну.
Сколиоз между 20 и 40 градусами у детей обычно лечится с помощью спинки. Ребенок должен носить этот прибор около 23 часов в день до завершения роста.Если искривление позвоночника увеличивается до 40 или 50 градусов, пациенту может потребоваться операция, чтобы предотвратить проблемы с легкими, боль в спине и дальнейшую деформацию. Хирургическое лечение сколиоза включает в себя выпрямление позвоночника металлическими стержнями и слияние позвонков в выпрямленном положении.
Спондилолистез лечат в небольших случаях. Если проскальзывание составляет более 30 градусов, смещенный позвонок может потребовать хирургической перестройки.
Дуральную эктазию можно отличить от других причин болей в спине при МРТ.Легкие случаи обычно не лечатся. Лекарства или шунтирование позвоночника для удаления некоторых из спинномозговой жидкости используются для лечения тяжелых случаев.
Pectus excavatum и pectus carinatum можно лечить хирургическим путем. В pectus excavatum деформированная грудина и ребра приподняты и выпрямлены металлическим стержнем. Через четыре-шесть месяцев планка снимается в амбулаторных условиях.
Protrusio acetabulae может потребоваться хирургическое вмешательство во взрослой жизни, чтобы обеспечить пациенту искусственный тазобедренный сустав, если боли в суставах сильны.
Боль в ногах или конечностях обычно лечится легким анальгетиком, таким как ацетаминофен. Пациентам с Марфаном следует надеть обувь на низком каблуке, специальные подушки или ортопедические вставки. Операция на ноге редко необходима.
Визуальные и стоматологические проблемы
Пациенты с Марфаном должны пройти тщательное обследование глаз, включая щелевую лампу, чтобы проверить смещение хрусталика, а также близорукость. Дислокацию можно лечить с помощью комбинации специальных очков и ежедневного использования одного процента глазных капель атропина сульфата или хирургическим путем.
Поскольку пациенты с Марфаном подвержены повышенному риску глаукомы, они должны измерять давление жидкости внутри глаза каждый год как часть обследования глаза. Глаукому можно лечить с помощью лекарств или хирургического вмешательства.
Катаракты лечатся с нарастающим успехом с помощью имплантации. Тем не менее, важно обратиться за помощью в медицинские центры к глазным хирургам, знакомым с возможными осложнениями операции по удалению катаракты у пациентов с синдромом Марфана.
Всех людей с Марфаном следует научить распознавать признаки отслойки сетчатки (внезапное ухудшение зрения на одном глазу становится все хуже и хуже без боли или покраснения) и немедленно обращаться за профессиональной помощью.
Дети с Марфаном должны проходить осмотр у своего стоматолога при каждом осмотре на предмет скученности зубов и возможного смещения, и при необходимости обращаться к ортодонту.
Спортивные мероприятия и профессиональный выбор. Людям с Марфаном следует избегать занятий спортом или занятий, требующих поднятия тяжестей, грубого физического контакта или быстрых изменений атмосферного давления (например, подводное плавание). Поднятие тяжестей повышает артериальное давление, что, в свою очередь, может увеличить аорту. Грубый физический контакт может вызвать отслоение сетчатки.Внезапные изменения давления воздуха могут вызвать пневмоторакс. Регулярные неконкурентные физические упражнения, однако, полезны для пациентов Марфана. Хороший выбор включает быструю ходьбу, стрельбу из корзин и медленный теннис.
Социальные проблемы и проблемы образа жизни
Курение. Курение особенно вредно для пациентов с болезнью Марфана, потому что увеличивает риск развития эмфиземы. Беременность. До недавнего времени женщинам с Марфаном рекомендовалось не забеременеть из-за риска расширения или расслоения аорты.Однако разработка бета-адреноблокаторов и эхокардиограмм позволяет теперь врачам наблюдать за пациентами на протяжении всей беременности. Рекомендуется, чтобы пациенты имели эхокардиограмму в течение каждого из трех триместров беременности. В норме вагинальные роды не обязательно являются более стрессовыми, чем кесарево сечение, но пациентам при длительных родах может быть назначен кесарево сечение для уменьшения нагрузки на сердце. Беременная женщина с Марфаном также должна получить генетическую консультацию относительно 50% риска рождения ребенка с синдромом.
Внешний вид и социальные проблемы. Дети и подростки с Марфаном могут получить пользу от поддерживающего консультирования относительно внешнего вида, особенно если их симптомы являются серьезными и побуждают их отказаться от социальной деятельности. Кроме того, семьи могут обратиться за консультацией относительно влияния синдрома на взаимоотношения в семье. Многие люди реагируют с чувством вины, страха или вины, когда в семье диагностируется генетическое заболевание, или они могут чрезмерно защитить пострадавшего члена. Группы поддержки часто являются хорошими источниками информации о Марфане; они могут предложить полезные советы о том, как жить с этим, а также эмоциональную поддержку.
Прогноз
Прогноз для пациентов с Марфаном заметно улучшился в последние годы. С 1995 года ожидаемая продолжительность жизни людей с синдромом увеличилась до 72 лет по сравнению с 48 годами в 1972 году. Это резкое улучшение объясняется новыми хирургическими методами, улучшенной диагностикой и новыми методами лечения.
Важнейшим фактором улучшения прогноза пациента является ранняя диагностика. Чем раньше пациент получит пользу от новых методов и модификаций образа жизни, тем больше вероятность того, что он или она будет иметь более продолжительную продолжительность жизни.
Ресурсы
Книги
Бирс, Марк Х. и Роберт Берков, редакторы. Педиатрия . Whitehouse Station, NJ: Merck Research Laboratories, 1999.
,
, Периц, Рид Э. и Черилл Гаснер. Синдром Марфана . New York: National Marfan Syndrome, 1999.
Ключевые термины
Arachnodactyly — Состояние, характеризующееся аномально длинными и тонкими пальцами рук и ног. Ectopia lentis — вывих хрусталика глаза.Это один из важнейших показателей в диагностике синдрома Марфана. Фибриллин — Белок, который является важной частью структуры соединительной ткани организма. При синдроме Марфана ген, ответственный за фибриллин, мутировал, заставляя организм вырабатывать дефектный белок. Гипермобильность — Необычная гибкость суставов, позволяющая им сгибаться или выходить за пределы нормального диапазона движения. Кифоз — Ненормальная внешняя кривизна позвоночника с горбом в верхней части спины. Pectus carinatum — аномалия грудной клетки, при которой грудина (грудная клетка) выталкивается наружу. Иногда ее называют «голубиная грудка». Pectus excavatum — аномалия грудной клетки, в которой грудина (грудная клетка) опускается внутрь; иногда называют «воронкой сундук». Сколиоз — ненормальное искривление позвоночника из стороны в сторону. ,