Мультисистемная атрофия и синдром Шая-Дрейджера
МУЛЬТИСИСТЕМНАЯ АТРОФИЯ И СИНДРОМ ШАЯ-ДРЕЙДЖЕРА
В 1960 г. два исследователя, Milton Shy и Glen Drager, описали комплекс неврологических нарушений, ассоциирующихся с вегетативными расстройствами, в настоящее время известный как мультисистемная атрофия. Это спорадическое прогрессирующее заболевание с поздним началом, характеризующееся вегетативной дисфункцией, синдромом паркинсонизма и атаксией в различных комбинациях.
Вегетативная дисфункция, проявляющаяся ортостатической гипотензией, импотенцией, нарушением мочеиспускания, обычно развивается в течение 2 лет после появления моторных симптомов. Синдром паркинсонизма и мозжечковые знаки обычно возникают в комбинации, но могут доминировать отдельные проявления.
Когда в клинической картине мультисистемной атрофии устойчиво доминирует какое-либо клиническое проявление, терминологически используют синдромальные названия (табл. 37- 1). Важно отметить, что манифестация симптомов может возникать в различных комбинациях и по-разному эволюционировать.
Мозжечковые нарушения развиваются первыми у 20% больных, приблизительно в 80% случаев дебют начинается с экстрапирамидных нарушений. При комбинации мозжечковых и экстрапирамидных симптомов часто бывает трудно оценить мозжечковый дефицит из-за яркости паркинсонических знаков.
Таблица 37- 1 . Клинические варианты мультисистемной атрофии
| Доминирующий синдром | Используемая терминология |
| Паркинсонизм | Стрионигральная дегенерация |
| Мозжечковые знаки | Спорадическая оливопонтоцеребеллярная атрофия |
| Вегетативная недостаточность | Синдром Шая-Дрейджера |
Клиническая картина
Обычно мультисистемная атрофия развивается между 5-й и 6-й декадами жизни, мужчины страдают несколько чаще женщин (соотношение составляет 1,3:1).
Самый частый моторный синдром — синдром паркинсонизма. Он обладает рядом клинических особенностей, позволяющих уже в дебюте заболевания проводить эффективную дифференциальную диагностику с болезнью Паркинсона. Синдром паркинсонизма часто начинается асимметрично, как и болезнь Паркинсона.
Он обладает рядом клинических особенностей, позволяющих уже в дебюте заболевания проводить эффективную дифференциальную диагностику с болезнью Паркинсона. Синдром паркинсонизма часто начинается асимметрично, как и болезнь Паркинсона.
Клинически заподозрить мультисистемную атрофию позволяют так называемые симптомы красного флага:
• ранние глазодвигательные нарушения;
• низкий ответ на леводопу;
• фокальная дистония (антеколлис) ;
• ранние постуральные нарушения;
• быстрое клиническое прогрессирование симптомов;
• фокальный миоклонус;
• феномен Рейно, или акроцианоз;
• дисфагия;
• усиление храпа, сонные апноэ;
• насильственный (псевдобульбарный) плач или смех;
• контрактуры.
У половины больных имеются пирамидные знаки (оживление глубоких рефлексов, симптом Бабинского) . Дизартрия и другие нарушения речи — типичный симптом мультисистемной атрофии в разгаре клинической картины. Заболевание ассоциируется с апноэ во сне (обструктивные и центральные) , способными представлять угрозу для жизни. Ночной храп и сонные апноэ обычно связаны с обструкцией верхних дыхательных путей. Умеренные когнитивные нарушения возникают приблизительно у 20% больных, страдающих мультисистемной атрофией.
Ночной храп и сонные апноэ обычно связаны с обструкцией верхних дыхательных путей. Умеренные когнитивные нарушения возникают приблизительно у 20% больных, страдающих мультисистемной атрофией.
Тяжёлая деменция не характерна для этого заболевания. По мере течения болезни прогрессивно увеличивается риск смерти от пневмонии, лёгочной эмболии, инфекции мочевого тракта или риск внезапной смерти. Прогноз неблагоприятный: от постановки диагноза до летального исхода в среднем проходит 7 лет. Считают, что наиболее плохой прогноз бывает при тяжёлом повреждении вегетативной нервной системы и меньшей вовлечённости стрионигральной системы.
Вегетативные нарушения при мультисистемной атрофии обусловлены дегенерацией центральных нейронов, в результате чего страдают симпатические и парасимпатические рефлексы. При этом постганглионарные вегетативные нейроны остаются интактными. Ослабление норадренергической рефлекторной активации приводит к ортостатической гипотензии — основному синдрому вегетативных нарушений при мультисистемной атрофии. Симптомы, связанные с нарушением рефлексов, опосредованных парасимпатической нервной системой, включают недостаточность кардиовагальных барорефлексов, запор, снижение тонуса мочевого пузыря. Дегенерация стволовых нейронов при водит к нарушению глотания, дыхания и сна.
Симптомы, связанные с нарушением рефлексов, опосредованных парасимпатической нервной системой, включают недостаточность кардиовагальных барорефлексов, запор, снижение тонуса мочевого пузыря. Дегенерация стволовых нейронов при водит к нарушению глотания, дыхания и сна.
При этом заболевании нарушение регуляции барорефлекса при интактных симпатических постганглионарных нервах клинически проявляется артериальной гипотензией на фоне положения стоя, приёма пищи («послеобеденная артериальная гипотензия») и физических усилий. При мультисистемной атрофии ортостатическая гипотензия ассоциируется с артериальной гипертензией в положении лёжа, что характерно для всех форм нейрогенной ортостатической гипотензии.
Гипертензия бывает весьма тяжёлой и может стать причиной инвалидизации больного. В отличие от здоровых людей, у которых АД ночью снижается, люди, страдающие мультисистемной атрофией, бывают нондипперами (у них ночью АД не снижается), что можно диагностировать при 24-часовом мониторировании давления. К счастью, артериальная гипертензия при мультисистемной атрофии обладает минимальным органным повреждающим эффектом. Поскольку у таких больных имеются интактные симпатические нервные терминали и барорефлекторная недостаточность, для них характерно нормальное содержание норадреналина в положении лёжа и снижение АД в ответ на лекарства, способствующие выбросу норадреналина. При индуцированной поворотным столом артериальной гипотензии у больных с мультисистемной атрофией отмечают сниженный выброс вазопрессина (антидиуретического гормона) . В то же время нейроны супраоптических и паравентрикулярных ядер гипоталамуса, синтезирующие вазопрессин, не страдают, поэтому повышение осмоляльности плазмы вызывает нормальный выброс вазопрессина. Эта диссоциация — маркёр центрального поражения барорефлекса.
К счастью, артериальная гипертензия при мультисистемной атрофии обладает минимальным органным повреждающим эффектом. Поскольку у таких больных имеются интактные симпатические нервные терминали и барорефлекторная недостаточность, для них характерно нормальное содержание норадреналина в положении лёжа и снижение АД в ответ на лекарства, способствующие выбросу норадреналина. При индуцированной поворотным столом артериальной гипотензии у больных с мультисистемной атрофией отмечают сниженный выброс вазопрессина (антидиуретического гормона) . В то же время нейроны супраоптических и паравентрикулярных ядер гипоталамуса, синтезирующие вазопрессин, не страдают, поэтому повышение осмоляльности плазмы вызывает нормальный выброс вазопрессина. Эта диссоциация — маркёр центрального поражения барорефлекса.
Ортостатическая гипотензия проявляется ощущением лёгкости в голове, общей слабостью, расплывчатостью зрения, нарушением координации, болью по задней поверхности шеи. Зрительные жалобы связаны с ретинальной ишемией и ишемией затылочных долей. Для боли в шее типично распространение на субокципитальную зону, заднюю поверхность шеи и плечи. Большинство исследователей объясняют её последствием ишемии мышц шеи. Реже больные жалуются на ортостатическое диспноэ и боли в грудной клетке, иногда принимающие стенокардический характер даже при интактных коронарных артериях. Ортостатические нарушения гемодинамики в лёгких случаях ограничиваются проявлениями липотимического состояния, в более выраженных случаях возможны обморочные состояния.
Для боли в шее типично распространение на субокципитальную зону, заднюю поверхность шеи и плечи. Большинство исследователей объясняют её последствием ишемии мышц шеи. Реже больные жалуются на ортостатическое диспноэ и боли в грудной клетке, иногда принимающие стенокардический характер даже при интактных коронарных артериях. Ортостатические нарушения гемодинамики в лёгких случаях ограничиваются проявлениями липотимического состояния, в более выраженных случаях возможны обморочные состояния.
Потеря сознания возникает градуированно или внезапно, если дополнительно включаются кардиальные причины. Ортостатическая гипотензия усиливается при уменьшении объёма циркулирующей крови, дегидратации, физическом усилии, повышении окружающей температуры, всасывании пищи. Многие лекарства, включая трициклические антидепрессанты, антигипертензивные, антипаркинсонические и другие препараты, обладают ятрогенным эффектом в отношении ортостатической гипотензии. Последняя очень вариабельна в течение дня, обычно её проявления наиболее сильно выражены в утренний период.
Дизурия — ранний симптом у больных с мультисистемной атрофией. Чаще всего это про явление связано с гипоактивностью детрузора и низким уретральным давлением. Сексуальная дисфункция манифестирует не только эректильной дисфункцией, развивающейся более чем у 60% мужчин, но также страдает сексуальное влечение, оргазм и в целом сексуальное поведение. Гастроинтестинальные симптомы включают слюнотечение, дисфагию, раннее насыщение, тошноту, вздутие живота, запор.
Лечение
Лечение вегетативных нарушений при мультисистемной атрофии симптоматическое, оно направлено на нивелирование наиболее инвалидизирующих симптомов. Возможные подходы к терапии ортостатической гипотензии описаны ниже. Наличие гастроинтестинальных симптомов в первую очередь требует коррекции диеты (увеличение приёма жидкости и предпочтение пищи, содержащей растительные волокна). Лактулоза в дозе 10-20 г помогает отдельным больным. Прогрессирующее нарушение глотания и аспирации бывает показанием к трахетомии.
Методы диагностики МСА на ранних стадиях
Мультисистемная атрофия (МСА) – это спорадическое фатальное нейродегенеративное заболевание с началом во взрослом возрасте, характеризующееся прогрессирующей вегетативной недостаточностью, паркинсонизмом, мозжечковым и пирамидным синдромами в различных комбинациях. МСА считается редким заболеванием (см. www.orpha.net) : оно встречается в 3,4 – 4,9 случаях на 100 000 населения, но для группы старше 40 лет – 7,8.
Аутопсическим индикатором становится большое скопление α-синуклеина в олигодендроцитах вкупе со стриатонигральной дегенерацией или оливопонтоцеребеллярной атаксией.
МСА выделилась в отдельную нозологическую форму в 1969 г., обобщив три ранее отдельных диагноза. До начала XX века заболевание существовало под разными названиями: стриатонигральная дегенерация (СНД), оливомостомозжечковая атрофия (ОПЦА) и синдром Шая-Дрейджера (по имени исследователей Джорджа Милтона Шая и Глена Алберта Дрейджера). Термин МСА служит отныне отдельной клинико-патоморфической единицей для разнообразных сочетаний симптомов МСА. Термин «синдром Шая-Дрейджера» более не используется.
Термин МСА служит отныне отдельной клинико-патоморфической единицей для разнообразных сочетаний симптомов МСА. Термин «синдром Шая-Дрейджера» более не используется.
На данный момент заболевание имеет два подкласса: МСА-п (паркинсонического типа, или стратонигральная дегенерация, MSA-p – англ.) и МСА-ц (оливопонтоцеребеллярная атрофия, MSA-c – англ.). Различие двух типов становится ярче по мере прогрессирования заболевания.
Диагностика МСА очень затруднительна. Как правило, начало болезни приходится на шестой десяток жизни пациента. Продолжительность жизни варьируется от 6 до 15 лет после постановки диагноза.
Симптоматика носит смешанный характер: помимо паркинсонизма, мозжечковой атаксии, двигательной атаксии, ортостатического коллапса, выявляются проблемы вегетативной системы (см. Табл. 1, и рис.1). Преобладание паркинсонизма или же мозжечковой атаксии предопределяет возможность (possible) или вероятность заболевания (probable, definite) и выбор подтипа.
Рис. 1 Мультидисциплинарное проявление МСА. Перевод на русский язык. Источник: Fanciulli, Alessandra, and Gregor K. Wenning. “Multiple-system atrophy.” New England Journal of Medicine 372.3 (2015): 249-263
В ходе лечения на первых порах можно отметить реакцию на леводопосодержащие медикаменты, но со временем их эффективность снижается. Для обоих типов МСА характерно драматически быстрое развитие болезни. Потеря автономности пациента сопровождается трудностями пищеварения, дыхания (стридор может вести к необходимости трахеостомии). По статистике, приведённой A. Фанчулли и Г. Веннингом, во время сна у 40% пациентов замечено ночное апноэ. На поздних стадиях заболевания следует избегать условий для развития пневмонии. Кроме того, МСА сопутствует гипертензия в позе лёжа. Во время сна у пациентов отмечается нарушение движений глазных яблок во время быстрого сна [5]. Также замечено уменьшение потоотделения, недержание (в т.ч. ночной полиурией), у мужчин – эректильная дисфункция.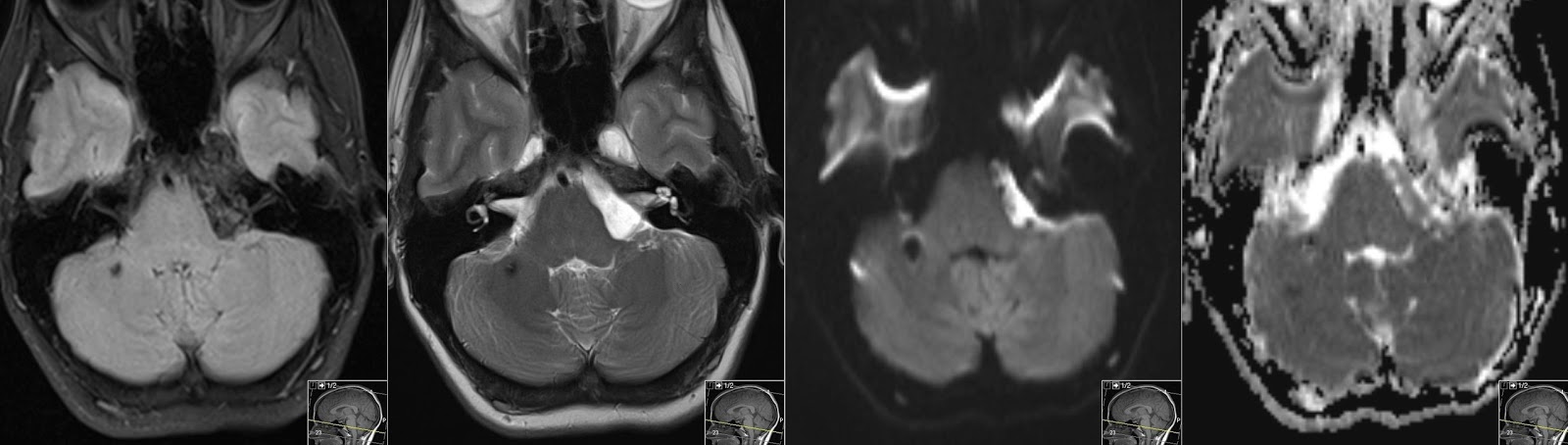 При этом следует учитывать, что последние два из перечисленных симптомов могут быть не связаны с МСА. Но в любом случае требуется превентивное и постоянное лечение инфекций мочевыводящих путей.
При этом следует учитывать, что последние два из перечисленных симптомов могут быть не связаны с МСА. Но в любом случае требуется превентивное и постоянное лечение инфекций мочевыводящих путей.
На более развитых стадиях болезни у 50% пациентов по статистике Фанчулли и Веннинга испытывают парализующую боль. Характерным условием для развития этого симптома была дистония. Симптом чаще наблюдается у женщин.
В обыденной жизни пациенты сталкиваются не только с ограничениями медицинского характера, но и с каждодневными трудностями, которые накладывает болезнь. Внешние проявления болезни выражаются в необычной походке, положении головы. Из-за спазмов на лице больных МСА иногда появляется “сардоническая улыбка”. При МСА-п непроизвольно происходит сильный наклон или вытягивание головы вперёд. Скованность и замедленность движений затрудняют выполнение рутинных задач.
Редкое заболевание известно далеко не всем, и порой вызывает непонимание окружающих. Поэтому в публичных местах из-за физических ограничений пациенты испытывают стресс, ограничивают себя в социализации, избегают людных мест, что создаёт дополнительные факторы для усугубления сопутствующей депрессии и психологического дискомфорта (см. «нейропсихологические проявления», Табл. 1).
Поэтому в публичных местах из-за физических ограничений пациенты испытывают стресс, ограничивают себя в социализации, избегают людных мест, что создаёт дополнительные факторы для усугубления сопутствующей депрессии и психологического дискомфорта (см. «нейропсихологические проявления», Табл. 1).
Таблица 1. Критерии для первичного диагностирования МСА
| Область | Критерии | Определяющий фактор |
| Вегетативная система, Урология | 1.Ортостатическая гипотензия 2. Увеличенный объём остаточной мочи, недержание Жалобы на хронический запор | Падение ортостатического давления вдвое в течение 3-х минут стоя |
| Паркинсонизм Преобладает при МСА-п | 1. Брадикинезия 2. Ригидность 3.Постуральная неустойчивость 4. Тремор покоя или движения | Брадикинезия в сочетании с двумя-четырьмя паркинсоническими симптомами и слабой реакцией на леводопу |
| Мозжечковая дисфункция Преобладает при МСА-ц | 1. Изменение походки: мелкие шажки, шаткость Изменение походки: мелкие шажки, шаткость2.Мозжечковая дизартрия 3.Окуломоторная дисфункция Заметна скованность движений | Изменения походки вкупе с несколькими проявлениями мозжечковой дисфункции |
| Дисфункция корково-спинномозгового пирамидного пути | 1.Синдром Бабинского 2. Гиперрефлексия | Не учитываются для окончательной постановки диагноза МСА, но входят в критерии возможного развития МСА |
| Дополнительно: нейропсихологические проявления | 1. Депрессия (41% случаев) 2. Галлюцинации (5%) 3. Деменция (4%) 4. Бессоница (19%) 5. Дневная сонливость (17%) 6. Неприятные ощущения в нижних конечностях (синдром беспокойных ног) | Сопутствующие проявления* |
| Gilman et al., 1999; Gilman atal., 2008; Wenning et al., 2004. *Клинические проявления МСА среди 437 пациентов, EMSA-registry. | ||
Карло Колозимо предлагает синтетическую таблицу основных и дополнительных критериев диагностики МСА (Таблица 3. 5, глава “Multiple system atrophy”, Carlo Colosimo, David E. Riley, Gregor K. Wenning, Handbook of Atypical Parkinsonism, Cambridge University Press, 2011, P. 36.)
5, глава “Multiple system atrophy”, Carlo Colosimo, David E. Riley, Gregor K. Wenning, Handbook of Atypical Parkinsonism, Cambridge University Press, 2011, P. 36.)
Основные критерии:
- Рото-лицевая дискинезия
- Диспропорциональный антеколлис
- Камптокормия (сохранение неестественного наклона вперёд тела) или «синдром Пизанской башни» (неестественный латеральный наклон тела)
- Сокращение мышц конечностей, что выражается в постоянно искривлённом положении ладоней, рук, ступней
- Затруднённое дыхание
- Серьёзные затруднения при говорении, что выражается в замедленной, обрывочной речи
- Серьёзные расстройства артикуляции
- Новоприобретённый или усиленный храп
- Холодные конечности, кроме того, бывает заметна отёчность ног
- Непроизвольный или беспричинный смех или плач
- Могут проявиться панические атаки
- Порывистый, миоклонический тремор движения или покоя
Посмотреть в нормальном размере
Дополнительные симптомы
- Покручивающие движения указательного (тремор 4-6Гц) и большого пальцев руки, наблюдаемый также при бП (pill-rolling tremor)
- История болезни, содержащая указания на другие нейропатии
- Семейная история болезней с указаниями на атаксию и паркинсонизм других родственников
- Немедикаментозные галлюцинации
- Начало болезни после 75 лет
- Деменция
- Признаки склероза
Диагностика заболевания, как уже было сказано, затруднена. Нет единого критерия или сочетания симптомов, которые могли бы однозначно указать на МСА с самого начала болезни. Выход на финальный диагноз проводится методом исключения по мере её развития. При всех общих неизвестных, первостепенным аргументом для подтверждения МСА остаётся снимок МРТ (как минимум 1,5 тесла). Но анализ МРТ на начальных стадиях не даёт точной уверенности, поэтому как правило первоначальным диагнозом становится болезнь Паркинсона (БП). Более достоверное выявление потери нейронов возможно на снимках ПЭТ (PET, позитронно-эмиссионная томография) или ОЭМТ (SPECT,однофотонная эмиссионная компьютерная томография).
Нет единого критерия или сочетания симптомов, которые могли бы однозначно указать на МСА с самого начала болезни. Выход на финальный диагноз проводится методом исключения по мере её развития. При всех общих неизвестных, первостепенным аргументом для подтверждения МСА остаётся снимок МРТ (как минимум 1,5 тесла). Но анализ МРТ на начальных стадиях не даёт точной уверенности, поэтому как правило первоначальным диагнозом становится болезнь Паркинсона (БП). Более достоверное выявление потери нейронов возможно на снимках ПЭТ (PET, позитронно-эмиссионная томография) или ОЭМТ (SPECT,однофотонная эмиссионная компьютерная томография).
Подтипы МСА изображены на фиг. 1 и 2. [1]
Фиг. 1 А : двусторонняя атрофия в области скорлупы, гиперинтенсивность края (отмечено стрелочками). В-С : гиперинтенсивность двустороннего кортикопинального тракта в кортикальной и подкорковой предцентральной извилине и за пределом тракта (С).
Фиг. 2 A : “крест” в области варолиевого моста (“hot cross bun” (англ. ) – по внешней схожести с пасхальным хлебом “мазанецем”, “крестовой булочкой”), B-C : гиперинтенсивность в области двустороннего кортикопинального тракта в подкорковой предцентральной извилине и за пределом тракта (С).
) – по внешней схожести с пасхальным хлебом “мазанецем”, “крестовой булочкой”), B-C : гиперинтенсивность в области двустороннего кортикопинального тракта в подкорковой предцентральной извилине и за пределом тракта (С).
При МСА-п (паркинсонического типа, MSA-p)(характерный снимок-см. Фиг. 1) заметны проявления брадикинезии и ригидности; гипокинетической дизартрии; постуральная неустойчивость; часто – тремор покоя.
На 2003 г. случаи паркинсонического подтипа встречались вдвое-вчетверо раз чаще, чем МСА-ц в западном полушарии. Однако МСА-ц чаще встречается в Японии. По собранной на данный момент статистике МСА-п начинает уступать по частоте МСА-ц.
МСА-ц (оливоцеребеллярная атрофия, MSA-c)(характерный снимок – см. Фиг. 2) характеризуется прежде всего мозжечковой атаксией; постепенным, но неуклонным затруднением движения, речи и походки, а также движения глазных яблок и работы верхних век. У пациентов с МСА-ц чаще наблюдается тремор действия, например, при доставании предметов. Мышечная слабость при МСА-ц может привести к невнятной речи и попёрхиванию при глотании. Яркие проявления обнаруживаются среди ортостатических расстройств кровообращения: у пациента могут быть обмороки, слабость с головокружнием, тошнота, дрожь, боль области шеи и плеч. Мозжечковая дисфункция проявляется на более ранних стадиях и сочетается более заметным затруднением дыхания во время сна.
Мышечная слабость при МСА-ц может привести к невнятной речи и попёрхиванию при глотании. Яркие проявления обнаруживаются среди ортостатических расстройств кровообращения: у пациента могут быть обмороки, слабость с головокружнием, тошнота, дрожь, боль области шеи и плеч. Мозжечковая дисфункция проявляется на более ранних стадиях и сочетается более заметным затруднением дыхания во время сна.
В статье французской исследовательской группы из Тулузы MRI Supervised and Unsupervised Classification of Parkinson’s Disease and Multiple System Atrophy приводится предварительный вывод многостороннего анализа снимков МРТ пациентов обоих подтипов МСА. На развитой стадии МСА (начало болезни наблюдаемых варьировалось между 5 и 7 годами) с высокой степенью вероятности можно отличить МСА от БП, опираясь только на МРТ-данные. Изменения отображаются на снимках в области:
1) лучистого венца верхнего отдела пирамидного пути (обе стороны),
2) верхней извилины лобной доли.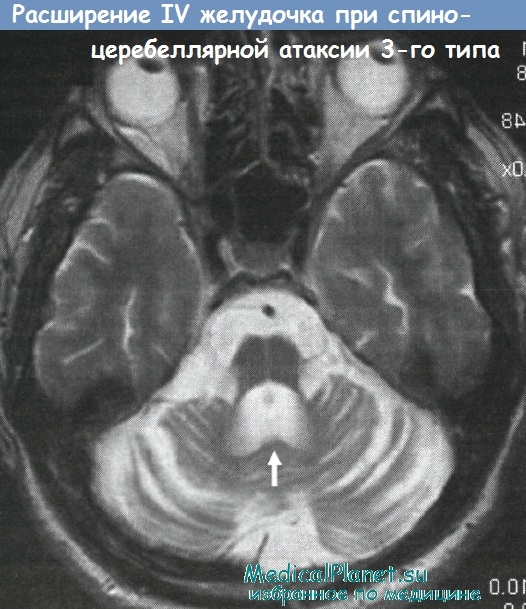
Для МСА-п характерны сокращение фракционной анизотропии в скорлупе, дополнительной моторной области и лучистом венце верхнего отдела пирамидного пути. Исходя из снимков МРТ, труднее отличить от бП МСА церебеллярного типа [7].
С помощью фтордеоксиглюкозы на снимках ПЭТ видны гипометаболизм в стриатуме, в основном в путамене, также стволе головного мозга и мозжечке при МСА-п. При МСА-ц – в основном в путамене, а также может быть заметна потеря допаминергических нейронов нигростриарного пути.
Таблица 3. Типичные результаты дополнительных тестов при МСА.
| Тест | Типичный результат |
| Кардиоваскулярные тесты | Ортостатическая гипотензия Пониженная гемодинамика Ослабленное сердцебиение Низкий коэфициент при пробе Вальсавы Слабое выделение норадреналина в лимфе, слабое сосудосужение |
| Определение уровня глюкозы | Слабый релизинг гормона роста (спорный результат) |
| Тест терморегуляции и количественный тест вызванного судомоторного аксон-рефлекса | Судомоторная дисфункция, проявляющаяся гипо- ангидрозом |
| Электрофизиологические исследования (sympathetic skin response) | Кожный симпатический потенциал либо отсутвует, либо аномальный |
| Тест цереброспинальной жидкости | Повышенный уровень нейрофиламентов |
| Электромиография наружного сфинктера заднего прохода | Либо денервация, либо раздражение (нерелевантные результаты) |
| Транскраниальная сонография | Гиперэхогенность чечевицеобразного ядра и нормальная эхогенность чёрной субстанции |
| Компьютерная томография | Не дала результатов |
| МРТ на 1,5 тесла | Аномалии в базальных ганглиях, «крест» в области варолиевого моста, атрофия мозжечка или ствола мозга |
| ДВИ (диффузно-взвешенное изображение) | Диффузность в области путамена, варолиева моста и средней ножки мозжечка |
| Волюметрия | Потеря объёма в путамене при МСА-п, потеря объёма в стволе и мозжечке при МСА-ц |
| Cканирование с радиоактивным изотопом-метайодобензилгуанидином MIBG | В норме |
| Сканирование переносчиков Иофлупаном I123 (123I-FP-CIT SPECT imaging) | Недостаток транспортёров дофамина в полосатом теле |
| Сканирование переносчиков йодобензамидом I123 123I-IBZM-SPECT | Недостаток транспортёров дофаминового рецептора D2 в полосатом теле |
| ПЭТ с флуородопой | Недостаток захвата флуорисцентной леводопы |
| ПЭТ с раклопридом | Недостаток транспортёров дофаминового рецептора D2 в полосатом теле |
| ПЭТ с изохинолиновым карбоксамидом PK-11195 | Микроглиальная активация в области базальных ганглий и ствола мозга |
| ПЭТ с использованием радиофармпрепарата фтордеоксиглюкозы | Замедленный метаболизм |
Благодаря дополнительным тестам на данный момент выявлены несколько направлений, в которых будет развиваться диагностика. Они базируются не только на исключении других болезней или изучении снимков ПЭТ (см. Табл. 3, Типичные результаты дополнительных тестов.), но и на анализе офтальмологических особенностей МСА, терморегуляции, дисфункции вегетативной системы. Остановимся на результатах некоторых из них.
Они базируются не только на исключении других болезней или изучении снимков ПЭТ (см. Табл. 3, Типичные результаты дополнительных тестов.), но и на анализе офтальмологических особенностей МСА, терморегуляции, дисфункции вегетативной системы. Остановимся на результатах некоторых из них.
Офтальмологические особенности МСА
Целевое ретроспективное наблюдение больных МСА в клинике Мэйо (Рочестер, Миннесота, США) позволило выявить основные аномалии зрения, сопутствующие заболеванию. Из 285 рассмотренных случаев были отобраны 39 пациентов с подтверждённым диагнозом. Среди пациентов с МСА-п 14 человек жаловались преимущественно на синдром сухого глаза, у 13-ти были выявлены асинхронность глазных движений. У 7-х было отмечено смещение или ограничение движения глазного яблока, у одного пациента наблюдалась монокулярная диплопия (двоение изображения для одного глаза) из-за аномального роста ресниц. Единичными случаями стали двусторонняя атрофия зрительного нерва и синдром Холмса – Эйди (парасимпатическая денервация зрачка, проявляющаяся мидриазом со снижением, а иногда и полным исчезновением способности зрачка сужаться, реагируя на свет).
Следует различать аномалии, которые по независимым причинам сопровождают МСА, и те, которые проистекают из заболевания. К последним относятся, по предположению учёных, атрофия глазного нерва и рубцевание конъюнктивы (рубцовый пемфогоид).
Среди наблюдаемых с МСА-ц офтальмологические особенности чаще всего проявляются в асинхронности движения и смещении глазного яблока. Особо выделена корреляция длительности жизни пациентов после установления диагноза и зрительными аномалиями, за исключением синдрома “сухого глаза”. В связи с этими наблюдениям медики призывают пациентов с МСА регулярно проходить офтальмологическое обследование с целью раннего выявления аномалий и предупреждения несчастных случаев по причине плохого зрения [2].
Особенности ортостатического давления и пульса при МСА
Среди критериев, указывающих на вероятное развитие болезни, отмечено падение ортостатического давления. Ортостатическая проба заключается в замере давления стоя. За три минуты в стоячем положении систолическое давление падает как минимум на 20-30 мм.рт.ст., а диастолическое – на 10-15 мм.рт.ст. при заниженном сердцебиении. Поэтому пациентам с МСА желательно носить абдоминальный бандаж, компрессионное бельё, увеличить частоту потребления воды и соли, а также медикаментов для повышения артериального давления.
За три минуты в стоячем положении систолическое давление падает как минимум на 20-30 мм.рт.ст., а диастолическое – на 10-15 мм.рт.ст. при заниженном сердцебиении. Поэтому пациентам с МСА желательно носить абдоминальный бандаж, компрессионное бельё, увеличить частоту потребления воды и соли, а также медикаментов для повышения артериального давления.
Признаки тахикардии с гипотонией являются характерной особенностью пациентов с вегетативными нарушениями. Но диапазон нарушений пока ещё не изучен. В статье Orthostatic Heart Rate Changes in Patients with Autonomic Failure caused by Neurodegenerative Synucleinopathies сообщается о диапазоне ортостатических изменений сердечного ритма у пациентов с вегетативной недостаточностью, в том числе и при МСА.
При МСА речь идёт о вегетативной нервной системе. Ортостатическая гипотензия вызвана нарушением активации симпатических вазоконстрикторных нейронов. Сердечный пульс значительно выше у пациентов с МСА в отличие от других пациентов с заболеваниями, связанными с образованием телец Леви, и в частности БП. Заметное повышение сердечного ритма при МСА объясняется тем, что постганглионарные волокна и их аксоны остаются почти незатронутыми, однако при этом констатируется потеря вегетативных нейронов головного и спинного мозга [3]. Это, например, проявляется в том, что у пациентов с МСА очень низкая температура конечностей: холодные ладони и ступни [4].
Заметное повышение сердечного ритма при МСА объясняется тем, что постганглионарные волокна и их аксоны остаются почти незатронутыми, однако при этом констатируется потеря вегетативных нейронов головного и спинного мозга [3]. Это, например, проявляется в том, что у пациентов с МСА очень низкая температура конечностей: холодные ладони и ступни [4].
Потоотделение при МСА
У пациентов с МСА, по сравнению с БП, значительно ниже показатели потоотделения ладоней и особенно ступней. Гипогидроз (пониженная потливость) или вовсе отсутствие потоотделения при МСА считается связанными с дегенерацией центральных предганлиев [6].
Авторы статьи Combined cardiovascular and sweating autonomic testing to differentiate multiple system atrophy from Parkinson’s disease подчёркивают, что сочетание трёх показателей – нарушения потоотделения, аномалии при глубоком вдохе и ортостатической гипотензии – оказались в 92% случаев верными для подтверждения диагноза МСА-п.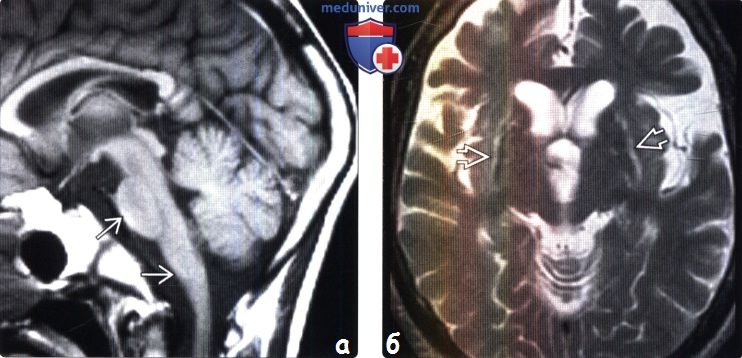 Этот результат сравним с ретроспективном анализом, проведённым в исследовании Cardiovascular autonomic testing performed with a new integrated instrumental approach is useful in differentiating MSA-P from PD at an early stage: данная работа показала, что сочетание показаний сердечно-сосудистых вегетативных тестов вкупе с оценкой наклона головы (диспропорциональный антеколлис) и пробы Вальсальвы, направленного на выявление симпатических и парасимпатических нарушений, правильно указали на наличие диагноза МСА (а не БП) в 91% случаев.
Этот результат сравним с ретроспективном анализом, проведённым в исследовании Cardiovascular autonomic testing performed with a new integrated instrumental approach is useful in differentiating MSA-P from PD at an early stage: данная работа показала, что сочетание показаний сердечно-сосудистых вегетативных тестов вкупе с оценкой наклона головы (диспропорциональный антеколлис) и пробы Вальсальвы, направленного на выявление симпатических и парасимпатических нарушений, правильно указали на наличие диагноза МСА (а не БП) в 91% случаев.
Изучение МСА затруднено редкостью заболевания, затруднённой диагностикой на начальной стадии и тем, что до сих пор не найдены способы эффективного торможения болезни. Интерес к разносторонним проявлениям болезни может в будущем привести к раннему выявлению заболевания, улучшенному лечению и повышению качества жизни пациентов.
Термины:
Проба Вальсальвы (напряжение по Вальсальве) — это форсированное выдыхание при закрытом носе и рте.
Гипокинетическая дизартрия — вид экстрапирамидной дизартрии, возникающий при поражении подкорковых узлов и их нервных связей.
Стриатонигральная дегенерация — спорадическое прогрессирующее нейродегенеративное расстройство, которое представляет собой одно из проявлений МСА. Проявляется, как правило, в снижении численности нейронов и глиозом в скорлупе, черной субстанции, стволе и мозжечке, а также в дегенерации клеток боковых рогов спинного мозга.
Оливопонтоцеребеллярные дегенерации — наследственные дегенеративные заболевания ЦНС, объединенные сходной локализацией патологического процесса в мозжечке, нижних оливах и мосте головного мозга.
Глазной рубцовый пемфигоид — заболевание, при котором происходит рубцевание конъюнктивы у пациентов пожилого возраста.
Монокулярная диплопия — ви́дение одним глазом двух или более изображений предмета.
Подготовила: Мартемьянова Е.О.
Помощь в редакции: Оськин С.
Источники
Общая литература:
Carlo Colosimo, David E. Riley, Gregor K. WenningHandbook of Atypical Parkinsonism, Cambridge University Press (2011).
Fanciulli, Alessandra, and Gregor K. Wenning. “Multiple-system atrophy.” New England Journal of Medicine 372.3 (2015): 249-263.
Приведённые статьи:
- da Rocha, Antonio José, et al. “Pyramidal tract degeneration in multiple system atrophy: the relevance of magnetization transfer imaging.” Movement disorders 22.2 (2007): 238-243.
- Garcia, Maria D., et al. “Ocular features of multiple system atrophy.” Journal of Clinical Neuroscience 47 (2018): 234-239.
- Norcliffe Kaufmann, Lucy, et al. “Orthostatic heart rate changes in patients with autonomic failure caused by neurodegenerative synucleinopathies.” Annals of neurology (2018).
- Shindo, Kazumasa, et al. “Pre-and postganglionic vasomotor dysfunction causes distal limb coldness in multiple system atrophy.
 ” Journal of the neurological sciences 380 (2017): 191-195.
” Journal of the neurological sciences 380 (2017): 191-195. - Boeve, Bradley F., Michael H. Silber, and Tanis J. Ferman. “REM sleep behavior disorder in Parkinson’s disease and dementia with Lewy bodies.” Journal of geriatric psychiatry and neurology 17.3 (2004): 146-157.
- Pavy-Le Traon, Anne, et al. “Combined cardiovascular and sweating autonomic testing to differentiate multiple system atrophy from Parkinson’s disease.” Neurophysiologie Clinique (2017).
- Péran, Patrice, et al. “MRI supervised and unsupervised classification of Parkinson’s disease and multiple system atrophy.” Movement Disorders (2018).
 ” Journal of the neurological sciences 380 (2017): 191-195.
” Journal of the neurological sciences 380 (2017): 191-195.Читайте также:
Кортикальная атрофия головного мозга у ребенка и взрослых: признаки, лечение
[24], [25], [26], [27]
Атрофия лобных долей головного мозга
В некоторых заболеваниях на первом этапе наблюдается атрофия лобных долей головного мозга с последующей прогрессией и распространением патологического процесса.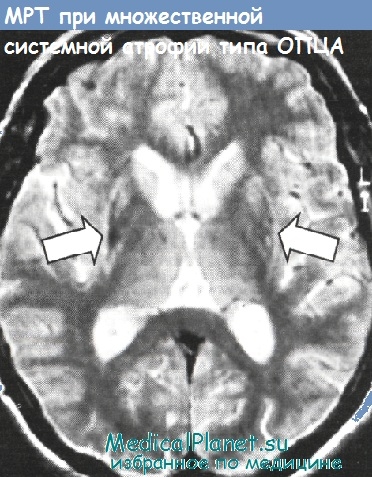 Это касается болезни Пика и Альцгеймера.
Это касается болезни Пика и Альцгеймера.
Для заболевания Пика характерно деструктивное поражение в основном нейронов лобных и височных областей, что обуславливает появление определенных клинических признаков. С их помощью врач может заподозрить болезнь и, используя инструментальные методы, поставить правильный диагноз.
Клинически повреждение данных участков encephalon проявляется изменением личности в виде ухудшения мышления и процесса запоминания. Кроме того с начала болезни можно наблюдать снижение интеллектуальных способностей. Происходит деградация человека, как личности, что выражается в угловатости характера, скрытности, отчуждения от окружающих людей.
Двигательная активность и фразы становятся вычурными и могут повторяться как по шаблону. В связи с уменьшением словарного запаса наблюдается частое повторение одной и той же информации в процессе разговора или через некоторое время. Речь становиться примитивной с использованием односложных фраз.
Атрофия лобных долей головного мозга при заболевании Альцгеймера немного отличается от патологии Пика, так как в данном случае в большей степени происходит ухудшение процесса запоминания и мышления. Что касается личностных качеств человека, то они страдают немного позже.
[28], [29], [30], [31], [32]
Атрофия мозжечка головного мозга
Дистрофические поражения могут начинаться с мозжечка, причем, не вовлекая в процесс проводящие пути. На первый план выходит атаксия и изменения тонуса мышц, не смотря на то, что причины развития и прогноз в большей степени похож на поражение нейронов полушарий.
Атрофия мозжечка головного мозга может проявляться утратой человеком способностей самостоятельного обслуживания. Поражение мозжечка характеризуется нарушениями сочетанного функционирования скелетных мышц, координации движений и поддержания равновесия.
Расстройства двигательной активности вследствие патологии мозжечка имеют несколько особенностей. Так, человек утрачивает плавность рук и ног при выполнении движений, появляется интенционное дрожание, которое отмечается в конце двигательного акта, изменяется почерк, речь и движения становятся более медленными, и возникает скандированная речь.
Атрофия мозжечка головного мозга может характеризоваться нарастанием головокружения, учащением головных болей, появлением тошноты, рвоты, сонливости и нарушений слуховой функции. Повышается внутричерепное давление, возможно появление офтальмоплегии вследствие паралича черепно-мозговых нервов, которые отвечают за иннервацию глаза, арефлексии, энуреза и нистагма, когда зрачок выполняет непроизвольные ритмичные колебания.
[33], [34], [35], [36], [37], [38]
Атрофия вещества головного мозга
Деструктивный процесс в нейронах может происходить в ходе физиологического процесса вследствие возрастных изменений после 60-ти лет или патологического – в результате какого-либо заболевания. Атрофия вещества головного мозга характеризуется постепенным разрушением нервной ткани с уменьшением объема и массы серого вещества.
Физиологическая деструкция отмечается у всех людей в преклонном возрасте, но ход которой можно лишь незначительно оказать лекарственное воздействие, замедлив деструктивные процессы. Что касается патологической атрофии вследствие негативного воздействия вредных факторов или другого заболевания, то здесь необходимо воздействовать на причину атрофии, чтобы остановить или замедлить разрушение нейронов.
Атрофия вещества головного мозга, в частности белого вещества, может развиваться вследствие различных заболеваний или возрастных изменений. Стоит выделить отдельные клинические проявления патологии.
Так, при деструкции нейронов колена появляется гемиплегия, которая представляет собой паралич мышц половины тела. Такие же симптомы отмечаются при повреждении переднего участка задней ножки.
Деструкция заднего участка характеризуется изменением чувствительности по половине участков тела (гемианестезией, гемианопсией и гемиатаксией). Поражение вещества может обуславливать также полную утрату чувствительность на одной из сторон тела.
Возможны психические расстройства в виде отсутствия узнавания предметов, выполнения целенаправленных действий и появление псевдобульбарных признаков. Прогрессирование данной патологии приводит к расстройствам речевой функции, глотания и возникновения пирамидных симптомов.
[39], [40], [41], [42], [43]
Кортикальная атрофия мозга
В связи с возрастными изменениями или в результате заболевания, оказывающего влияние на encephalon, возможно развитие такого патологического процесса, как кортикальная атрофия мозга. Чаще всего поражается лобные части, но не исключено распространение деструкции на другие области и структуры серого вещества.
Болезнь начинается незаметно и медленно начинает прогрессировать, причем нарастание симптоматики отмечается уже через несколько лет. С возрастом и при отсутствии лечения патологический процесс активно разрушает нейроны, что в конечном итоге приводит к слабоумию.
Кортикальная атрофия мозга в основном возникает у людей после 60-ти лет, но в некоторых случаях деструктивные процессы наблюдаются и в более раннем возрасте вследствие врожденного генеза развития из-за генетической предрасположенности.
Поражение двух полушарий кортикальной атрофией происходит при болезни Альцгеймера или другими словами, старческое слабоумие. Выраженная форма заболевания приводит к полному слабоумию, в то время как небольшие деструктивные очаги не оказывают существенного негативного воздействия на умственные возможности человека.
Выраженность клинических симптомов зависит от локализации и степени тяжести поражения подкорковых структур или коры. Кроме того следует учитывать скорость прогрессии и распространенность разрушительного процесса.
[44], [45], [46], [47], [48], [49], [50]
Мультисистемная атрофия мозга
Дегенеративные процессы лежат в основе развития синдрома Шая-Дрейджера (мультисистемная атрофия). В результате разрушения нейронов некоторых участков серого вещества возникают нарушения двигательной активности, и утрачивается контроль над вегетативными функциями, например, артериальным давлением или процессом мочеиспускания.
Симптоматически заболевания настолько разнообразно, что для начала можно выделить некоторые комбинации проявлений. Так, патологический процесс выражается вегетативными дисфункциями, в виде паркинсонического синдрома с развитием гипертонии с дрожанием и замедлением двигательной активности, а также в форме атаксии – неуверенной ходьбе и нарушений координации.
Начальная стадия болезни проявляется акинетически-ригидным синдромом, который характеризуется замедленными движениями и имеет некоторые симптомы болезни Паркинсона. Кроме того наблюдаются проблемы с координацией и мочеполовой системой. У мужчин первым проявлением может стать эректильная дисфункция, когда отсутствует способность достигать эрекции и удерживать ее.
Что касается мочевыделительной системы, то здесь стоит отметить недержание мочи. В некоторых случаях первым признаком патологии могут стать внезапные падения человека на протяжении года.
При дальнейшем развитии мультисистемная атрофия мозга приобретает все новые симптомы, которые можно разделить на 3 группы. К первой относится паркинсонизм, проявляющийся в замедленных неловких движениях и изменении почерка. Вторая группа включает в себя задержку мочеиспускания, недержание мочи, импотенцию, запоры и паралич голосовых связок. И, наконец, третья – состоит из дисфункции мозжечка, которая характеризуется затрудненной координацией, утратой чувства прострации, головокружением и обмороками.
Помимо когнитивных нарушений возможна и другие симптомы, такие как сухость в ротовой полости, кожных покровов, изменения потоотделения, появление храпа, одышки во время сна и двоения в глазах.
Диффузная атрофия головного мозга
Физиологические или патологические процессы в организме, в частности, в encephalon могут провоцировать запуск дегенерации нейронов. Диффузная атрофия головного мозга может возникать в результате возрастных изменений, генетической предрасположенности или под воздействием провоцирующих факторов. К ним относятся инфекционные болезни, травмы, интоксикации, заболевания других органов, а также негативное влияние окружающей среды.
Вследствие деструкции нервных клеток происходит снижение мозговой активности, утрачивается способность критического мышления и контроля над своими поступками. В пожилом возрасте человек иногда меняет поведение, которое не всегда понятно окружающим людям.
Начало заболевания может локализоваться в различных участках, что обуславливает определенную симптоматику. По мере вовлечения в патологический процесс других структур появляются новые клинические признаки. Таким образом, постепенно поражаются здоровые части серого вещества, что в конечном итоге приводит к слабоумию и потере личностных качеств.
Диффузная атрофия головного мозга вначале характеризуется появлением симптомов, сходных с корковой атрофией мозжечка, когда нарушается походка и утрачивается пространственное ощущение. В дальнейшем проявлений становится больше, так как болезнь постепенно охватывает новые участки серого вещества.
[51], [52], [53], [54], [55]
Атрофия левого полушария головного мозга
Каждый участок encephalon отвечает за определенную функцию, поэтому при его поражении человек утрачивает способность выполнения чего-либо, то ли в физическом отношении, то ли в умственном.
Патологический процесс в левом полушарии обуславливает появление речевых нарушений, так как моторной афазии. При прогрессировании болезни речь может состоять из отдельных слов. Кроме того страдает логическое мышление и развивается депрессивное состояние, особенно если атрофия локализуется большей частью в височной области.
Атрофия левого полушария головного мозга приводит к отсутствию восприятия полного изображения, окружающие объекты воспринимаются отдельно. Параллельно этому у человека нарушается способность читать, изменяется почерк. Таким образом, страдает аналитическое мышление, теряется возможность логически размышлять, анализировать поступающую информацию и манипулировать датами и числами.
Человек не может правильно воспринять и последовательно обработать информацию, что приводит к неспособности ее запоминания. Обращаемая к такому человеку речь воспринимается отдельно по предложениям и даже словам, вследствие чего отсутствует адекватная реакция на обращение.
Атрофия левого полушария головного мозга в тяжелой степени может вызывать полный или частичный паралич правой стороны с нарушением двигательной активности вследствие изменения тонуса мышц и чувствительного восприятия.
[56], [57], [58], [59], [60]
Смешанная атрофия мозга
Церебральные нарушения могут возникать в результате возрастных изменений, под воздействием генетического фактора или сопутствующей патологии. Смешанная атрофия мозга представляет собой процесс постепенной гибели нейронов и их соединений, при котором страдает кора и подкорковые структуры.
Дегенерация нервной ткани происходит большей частью у женщин, возраст которых превышает 55 лет. Вследствие атрофии развивается слабоумие, и что существенно ухудшает качество жизни. С возрастом объем и масса мозга уменьшаются вследствие постепенного разрушения нейронов.
Патологический процесс может отмечаться в детском возрасте, когда речь идет о генетическом пути передачи заболевания. Кроме того имеет место сопутствующая патология и окружающие факторы, например, радиация.
Смешанная атрофия мозга охватывает функциональные участки encephalon, отвечающие за контроль над двигательной и мыслительной активностью, планирование, анализ, а также критику своего поведения и мыслей.
Начальная стадия болезни характеризуется появлением вялости, апатии и уменьшением активности. В некоторых случаях наблюдается аморальное поведение, так как человек постепенно утрачивает самокритику и контроль над поступками.
В дальнейшем происходит уменьшение количественного и качественного состава словарного запаса, способности продуктивного мышления, утрачивается самокритика и осмысление поведения, а также ухудшается моторика, что ведет к изменению почерка. Далее человек перестает узнавать привычные для него предметы и в конечном итоге наступает маразм, когда происходит практически деградация личности.
Атрофия паренхимы головного мозга
Причинами поражения паренхимы являются возрастные изменения, наличие сопутствующей патологии, которая прямо или опосредованно воздействует на encephalon, генетические и вредные окружающие факторы.
Атрофия паренхимы головного мозга может наблюдаться вследствие недостаточного питания нейронов, так как именно паренхима наиболее чувствительна к гипоксии и недостаточному поступлению питательных веществ. В результате этого клетки уменьшаются в размере из-за уплотнения цитоплазмы, ядра и деструкции цитоплазматических структур.
Помимо качественного изменения нейронов, клетки могут вовсе исчезать, уменьшая объем органа. Таким образом, атрофия паренхимы головного мозга постепенно приводит к снижению веса мозга. Клинически поражение паренхимы может проявляться нарушением чувствительности на определенных участках тела, расстройством когнитивных функций, утратой самокритики и контроля над поведением и речевой функцией.
Течение атрофии неуклонно приводит к деградации личности и заканчивается летальным исходом. С помощью лекарственных средств можно постараться замедлить развитие патологического процесса и поддержать функционирование остальных органов и систем. Также используется симптоматическая терапия для облегчения состояния человека.
[61], [62], [63], [64], [65], [66]
Атрофия спинного мозга
Рефлекторно спинной мозг может осуществлять двигательные и вегетативные рефлексы. Двигательные нервные клетки иннервируют мышечную систему тела, в том числе диафрагму и межреберные мышцы.
Кроме того имеются симпатические и парасимпатические центры, которые отвечают за иннервацию сердца, сосудов, органов пищеварения и других структур. Например, в грудном сегменте располагается центр расширения зрачка и симпатические центры иннервации сердца. Крестцовый отдел имеет парасимпатические центры, отвечающие за функциональность мочевыделительной и половой систем.
Атрофия спинного мозга в зависимости от локализации деструкции может проявляться на рушением чувствительности – при разрушении нейронов задних корешков, или двигательной активности – передних корешков. В результате постепенного поражения отдельных сегментов спинного мозга возникают нарушения функциональности того органа, который иннервируется на данном уровне.
Так, исчезновение коленного рефлекса происходит вследствие разрушения нейронов на уровне 2-3 поясничного сегмента, подошвенного – 5 поясничного, а нарушение сокращения брюшных мышц наблюдается при атрофии нервных клеток 8-12 грудных сегментов. Особенно опасно разрушение нейронов на уровне 3-4 шейного сегмента, где располагается двигательный центр иннервации диафрагмы, что угрожает жизни человека.
Алкогольная атрофия головного мозга
Наиболее чувствительным органом к алкоголю является encephalon. Под воздействием алкоголя происходит изменение метаболизма в нейронах, в результате чего формируется алкогольная зависимость.
Вначале наблюдается развитие алкогольной энцефалопатии, обусловленной патологическими процессами в разных областях мозга, оболочках, ликворной и сосудистой системах.
Под влиянием алкоголя поражаются клетки подкорковых структур и коры. В стволе головного мозга и спинном мозге отмечается деструкция волокон. Погибшие нейроны формируются островки вокруг пораженных сосудов со скоплениями продуктов распада. В некоторых нейронах – процессы сморщивания, смещения и лизис ядра.
Алкогольная атрофия головного мозга обуславливает постепенное нарастание симптоматики, которая начинается с алкогольного делирия и энцефалопатии, а заканчивается летальным исходом.
Кроме того отмечается склероз сосудов с отложением вокруг коричневого пигмента и гемосидерина, как следствия геморрагий, и наличие кист в сосудистых сплетениях. Возможны кровоизлияния в стволе encephalon, ишемическое изменение и дистрофия нейронов.
Стоит выделить синдром Макияфавы-Биньями, который возникает в результате частого употребления алкоголя в большом количестве. Морфологически выявляется центральный некроз мозолистого тела, его отечность, а также демиелинизация и геморригии.
[67], [68], [69], [70], [71], [72], [73], [74]
Атрофия мозга у детей
Нечасто встречается атрофия мозга у детей, однако это абсолютно не означает, что она не может развиваться при наличии какой-либо неврологической патологии. Этот факт неврологи должны учитывать и предупреждать развитие данной патологии на ранних стадиях.
Для постановки диагноза они используют опрос жалоб, этапности появления симптомов, их длительности, а также выраженности и прогрессирования. У детей атрофия может развивать по окончании начальной стадии формирования нервной системы.
Атрофия мозга у детей на первой стадии может не иметь клинических проявлений, что осложняет диагностику, ведь родители со стороны не замечают отклонения, а процесс деструкции уже запущен. В данном случае поможет магнитно-резонансная томография, благодаря которой послойно исследуется encephalon, и обнаруживаются патологические очаги.
По мере прогрессии заболевания дети становятся нервными, раздражительными, происходят конфликты со сверстниками, что ведет к уединению малыша. Далее в зависимости от активности патологического процесса могут присоединять когнитивные и физические нарушения. Лечение направлено на замедление прогрессирования данной патологии, максимальное устранение ее симптомов и поддержание функционирования других органов и систем..
[75], [76], [77], [78], [79], [80], [81]
Атрофия мозга у новорожденных
Чаще всего атрофия мозга у новорожденных обуславливается гидроцефалией или водянкой мозга. Она проявляется увеличенным количеством спинномозговой жидкости, благодаря которой обеспечивается защита encephalon от повреждений.
Причин развития водянки довольно много. Она может формироваться в период беременности, когда происходит рост и развитие плода, и диагностируется с помощью УЗИ. Кроме того причиной могут стать различные сбои в закладке и развитии нервной системы или внутриутробные инфекции в виде герпеса или цитомегалии.
Также водянка и соответственно атрофия мозга у новорожденных могут возникать вследствие пороков развития головного или спинного мозга, родовых травм, сопровождающихся кровоизлиянием и возникновением менингита.
Такой малыш должен быть расположен в реанимационном отделении, так как нуждается в контроле невропатологов и реаниматологов. Эффективного лечения пока нет, поэтому постепенно данная патология приводит к серьезным нарушениям функционирования органов и систем вследствие их неполноценного развития.
[82], [83], [84], [85], [86]
Мультисистемная атрофия — Медицинский справочник
Мультисистемная атрофия
Мультисистемная атрофия — прогрессирующая дегенеративная патология головного мозга с преимущественным поражением глиальных клеток базальных ганглиев, мозжечка, вегетативных центров. Клинически проявляется сочетанием паркинсонизма с мозжечковой, вегетативной и пирамидной недостаточностью. Диагностируется преимущественно по клиническим данным, дополнительно проводится церебральная МРТ, ортостатическая проба, ЭМГ сфинктеров. Терапия мультисистемной атрофии симптоматическая (сосудистая, нейрометаболическая), большинство случаев резистентны к лечению препаратами леводопы.
Общие сведения
Термин «мультисистемная атрофия» (множественная системная атрофия, МСА) был введён в 1969 году. В 1989 году были обнаружены патогномоничные для МСА цитоплазматические включения в олигодендроглиоцитах. Понятие мультисистемная атрофия объединяет три патоморфологически сходные нозологии, клинически представляющие собой сочетание паркинсонического синдрома, вегетативной дисфункции, мозжечковой атаксии и пирамидной недостаточности. Ранее специалисты в области неврологии относили указанные заболевания в группу «паркинсонизм-плюс». На МСА приходится 10-12% случаев паркинсонизма. Встречаемость патологии в 20 раз меньше чем болезни Паркинсона. Заболеваемость составляет 3 случая на 100 тыс. населения. Дебют клинических проявлений приходится на возраст 50-60 лет. Характерно быстрое прогрессирование симптоматики.
Причины мультисистемной атрофии
Наследственный характер МСА не прослеживается, текущие наблюдения не обнаруживают семейных случаев заболевания. Однако многие исследователи предполагают генетическую детерминированность патологии как предрасположенность к развитию МСА при воздействии неблагоприятных факторов. Отдельные учёные связывают повышенный риск возникновения МСА с полиморфизмом в гене альфа-синуклеина. Этиофакторы, провоцирующие заболевание, точно не определены. Одно из проведённых исследований выявило указания на контакт с токсическими веществами (пестицидами, органическими растворителями) в анамнезе 11% пациентов с МСА.
Патогенез
Механизм развития неизвестен. Особенностью дегенеративных изменений является преимущественное поражение глиальных клеток с накоплением альфа-синуклеина, тау-протеина и ряда других нейронных белков. Патологические включения обнаруживаются в олигодендроглиоцитах надсегментарных двигательных структур (пирамидная, экстрапирамидная система, моторная область коры, мозжечок) и вегетативных центров ЦНС. Наряду с поражением чёрной субстанции происходит дегенерация дофаминовых рецепторов скорлупы, обуславливающее развитие устойчивого к дофаминергической терапии «постсинаптического» паркинсонизма. Морфологическая картина характеризуется асимметричными атрофическими изменениями белого вещества, преобладанием поражения олигодендроглиоцитов, менее выраженным повреждением нейронов. Мультисистемная дегенерация затрагивает строго определённые структуры головного мозга. Каждая клиническая форма имеет свою типичную локализацию дегенеративного процесса.
Классификация
В соответствии с современными взглядами на проблематику мультисистемная атрофия включает три нозологические формы. В основу систематизации положены клинические особенности заболевания. В зависимости от превалирующего синдрома выделяют следующие варианты:
- Стриатонигральная дегенерация (СНД). Дегенеративные изменения наиболее выражены в стриатуме и чёрной субстанции. Ведущим клиническим синдромом является паркинсонизм.
- Оливопонтоцеребеллярная атрофия (ОПЦА). Мультисистемная дегенерация распространяется на мозжечок, нижние оливы и мост. В клинической картине доминирует мозжечковый синдром. К МСА относят только спорадические случаи ОПЦА.
- Синдром Шая-Дрейджера. Превалирует прогрессирующая вегетативная недостаточность с выраженной ортостатической гипотензией. Ряд авторов предлагают не выделять синдром Шая-Дрейджера как третий вариант патологии, поскольку типичная для него вегетативная симптоматика в той или иной степени наблюдается при всех формах МСА.
Симптомы мультисистемной атрофии
Манифестация приходится на возрастной период 45-60 лет. У 60% пациентов МСА стартует нарушениями двигательной сферы, у 40% вегетативной симптоматикой. В начальном периоде в 60% случаев наблюдаются симптомы паркинсонизма: брадикинезия, замедленность движений, шаркающая походка, гипомимия, монотонность голоса. Их отличительной особенностью является изначальная симметричность проявлений. У 30% больных отмечаются мозжечковые расстройства: постуральные нарушения, дисметрия, адиадохокинез, интенционный тремор. В 10% случаев мозжечковая атаксия сочетается с паркинсонизмом.
Развёрнутая мультисистемная атрофия протекает с паркинсоническим синдромом у 90% больных. Мозжечковые нарушения слабо проявлены из-за выраженной ригидности. Об их наличии свидетельствует широкая постановка стоп при ходьбе, скандированный тип речи, усиление тремора в руке при приближении к цели (например, при попытке взять чашку). Смешанное мозжечково-паркинсоническое нарушение речи при МСА, получившее название дизартрофония, представляет собой мозжечковую дизартрию, сочетающуюся с монотонностью и приглушенностью речи. Пирамидная симптоматика характеризуется повышением сухожильных рефлексов и появлением стопных знаков, классические спастические парезы отсутствуют.
Тремор носит постурально-кинетический характер, возникает в результате сочетания дрожательного гиперкинеза и небольших миоклонических подёргиваний. Возможны дистонические проявления (спастическая кривошея, лицевой гемиспазм, фокальные дистонии конечностей), в отдельных случаях наблюдающиеся уже в дебюте заболевания. Вегетативная недостаточность проявляется ангидрозом, расстройством тазовых функций, ортостатическим коллапсом с обмороками, иногда — триадой Горнера, синдромом Рейно. Выраженные нарушения когнитивной сферы нехарактерны.
Осложнения
Тазовые нарушения осложняются присоединением вторичной инфекции с возникновением восходящего воспаления органов мочевыводящей системы: уретрита, цистита, пиелонефрита. При отсутствии своевременного лечения возможно проникновение инфекционных агентов в кровь с развитием сепсиса. Вовлечение в патологический процесс черепно-мозговых нервов приводит к прогрессирующему бульбарному параличу с характерной для него дисфагией. Последняя может осложниться попаданием пищи в дыхательные пути с последующей аспирационной пневмонией. Бульбарный паралич голосовых связок опасен появлением асфиксии, которая может стать причиной внезапной смерти.
Диагностика
Мультисистемная атрофия диагностируется на основании клинических данных, сбор которых зачастую требует наблюдения пациента в динамике. Диагноз вероятной МСА устанавливается при сочетании вегетативной недостаточности с хотя бы одним из следующих синдромов: резистентный к препаратам леводопы паркинсонизм, мозжечковая дисфункция. Против диагноза МСА выступает дебют заболевания до 30-летнего возраста, семейный анамнез, расстройство когнитивной сферы (деменция), наличие другого заболевания, являющегося причиной аналогичной симптоматики. Достоверная диагностика возможна только в результате патоморфологической экспертизы. С целью подтверждения диагноза необходимо проведение следующих исследований:
- Осмотр невролога. В неврологическом статусе выявляется паркинсонизм-плюс — сочетание признаков паркинсонизма с дополнительной симптоматикой (пирамидной, вегетативной, мозжечковой). Когнитивные расстройства, признаки очагового поражения коры (агнозия, апраксия) не определяются.
- МРТ головного мозга. В начальной стадии может соответствовать норме. В дальнейшем обнаруживаются атрофические изменения головного мозга, наиболее выраженные в мозжечке и подкорковых ганглиях. МРТ позволяет исключить рассеянный склероз, энцефалит, опухолевые процессы.
- Исследование вегетативной системы. Проводится ортостатическая проба, подтверждающая выраженное падение артериального давления при переходе в горизонтальное положение. Для диагностики тазовых нарушений осуществляется электромиография сфинктерного аппарата.
Дифференцируется мультисистемная атрофия с болезнью Паркинсона, сосудистым паркинсонизмом, спиноцеребеллярными атаксиями. Главным отличием МСА от классической болезни Паркинсона является наличие дополнительных симптомов, выходящих за рамки расстройств экстрапирамидной системы, слабая эффективность дофаминергической терапии. Сосудистый паркинсонизм отличается сопутствующими когнитивными нарушениями. Спиноцеребеллярные атаксии имеют наследственный характер, в сложных диагностических случаях исключаются при помощи ДНК-диагностики.
Лечение мультисистемной атрофии
Поскольку этиопатогенез остаётся неясным, лечение осуществляется в рамках симптоматической терапии. На начальной стадии заболевания у трети пациентов эффективны фармпрепараты леводопы, однако они усугубляют дистоническую симптоматику и течение ортостатической гипотонии. При отсутствии терапевтического эффекта, выраженных побочных явлениях леводопу отменяют. Применяют средства, улучшающие метаболизм церебральных тканей: вазоактивные, нейрометаболические препараты. Лечение ортостатической дисфункции осуществляется путём наложения компрессионных бинтов на нижние конечности, повышения содержания соли в рационе, приподнимания головного конца кровати.
Прогноз и профилактика
На сегодняшний день мультифокальная атрофия относится к неизлечимым заболеваниям. Симптоматическая терапия позволяет несколько облегчить состояние больного, но не может остановить прогрессирование дегенеративных процессов. Длительность жизни пациентов не превышает 7 лет. Летальный исход обусловлен осложнениями бульбарного синдрома, интеркуррентными инфекциями, сердечно-сосудистой недостаточностью. Профилактические мероприятия не разработаны, поскольку отсутствуют точные данные об этиологии поражения.
выбор лечения и продолжительность жизни
Ранее синдром Шая – Дрейджера, дегенерация стриатонигральная и атрофия оливопонтоцеребеллярная классифицировались как три раздельных заболевания. Однако при более глубоком их исследовании выяснилось, что проявления клинических симптомов данных болезней общие, поэтому они были объединены под одним обозначением – мультисистемная атрофия головного мозга.
Мозговая атрофия — это следствие процесса, во время которого поэтапно отмирают нервные клетки и разрушаются соединения нейронов. В ходе такого сложного отклонения в коре и подкорке мозга происходят значительные нарушения функциональности.
Важно знать! Атрофия головного мозга зачастую развивается у людей преклонного возраста. Однако в редких исключениях данное нарушение фиксируется у новорожденного.
Причины и признаки атрофии мозга
Зачастую первичные проявления мозгового атрофического нарушения выражаются в практически незаметных отклонениях личностного состояния человека. Однако существуют факторы, которые способствуют ускорению процесса заболевания, и их следует максимально исключить для продления нормального физиологичного самочувствия.
Признаки атрофии:
- Нарушение глотательного рефлекса и дыхания, расстройство сна;
- Уменьшенная интенсивность деятельности мозга;
- Значительно истощается словарный запас. Пациенту становится тяжело и требуется больше времени для того, чтобы подобрать необходимые слова при описании элементарных желаний и вещей;
- Ухудшаются двигательные функции и моторика всего тела пациента;
- Полностью отсутствует самокритика. Это часто сопровождается отклонением от нравственных принципов.
Человек, страдающий таким сложным нарушением, нуждается в своевременном лечении, уходе и контроле во избежание чрезвычайных ситуаций, в которые он способен попасть. Больной с нарушениями когнитивных функций перестает распознавать знакомые предметы и не может вспомнить, как их необходимо использовать. Также появляется нарушение ориентации в знакомом пространстве, связанное с изменением памяти. Дальнейшее прогрессирование патологии может привести к деградации и наступлению полного маразма.
При проявлении первых признаков атрофии необходимо начинать лечение пациента для поддержания нормальной функциональности его организма и мозговой деятельности. При отказе от назначенного лечения патология и атрофические отклонения мозга в конечном итоге приводят к летальному исходу.
Важно знать! В группе риска люди от 50-ти летнего возраста. Исключением могут стать люди за 45, ведущие нездоровый образ жизни.
Причины атрофии
Причины атрофии мозга до конца не изучены. Существуют предположения, что синдром обусловлен генетически. Первичные рефлексы страдают от дегенерации и отмирания центральных нейронов в головном и спинном мозге. При резком понижении артериального давления во время принятия вертикального положения наблюдается ослабление вторичных рефлексов. Развитие отклонений зависит от ряда нарушений, происходящих в головном мозге. Образованию и развитию атрофии способствуют некоторые серьезные заболевания: Паркинсона, Бехчета, Уиппла, Кушинга, Альцгеймера, Геллервордена – Шпатца. Влияют и другие факторы:
- Гидроцефалические нарушения;
- Травмы мозга;
- Наркотики, алкогольное злоупотребление и курение;
- Нарушения сосудистой системы;
- Нарушенный обмен веществ;
- Поражения инфекционного характера;
- Амиотрофический склероз.
Диагностика, лечение и профилактика атрофии головного мозга
При постановке окончательно точного диагноза не существует решающих методик диагностики. Однако исследования показали, что наиболее точные показатели атрофии мозга выявляются при исследовании МРТ.
Диагностика
В случае проявления первых признаков заболевания, при котором атрофируется головной мозг, необходимо обратиться к специалисту для диагностики и назначения эффективной терапии. Во время первого приема врач должен спросить о беспокоящих пациента жалобах, выяснить, в какое время они возникают и узнать о возможном существовании уже установленных хронических патологий.
Для выявления наличия и локализации атрофии может быть использовано несколько методов. Это делается с целью установления наиболее точного диагноза для назначения максимально эффективного лечения. С целью установления наличия патологии специалисты проводят когнитивные тесты, которые позволяют установить уровень мышления и предположить присутствие и степень патологии. Для того, чтобы исключить сосудистый генез атрофии, проводится допплерография сосудов мозга и шеи. Эта процедура дает возможность просветить сосуды, обнаружив наличие и локализацию атеросклеротических изменений.
Для более глубокого обследования изменений головного мозга применяется рентгенологическое исследование. Его целью является обнаружение возможных очагов структурного изменения и дополнительных образований в мозге пациента, таких как опухоли или гематомы.
Наличие и локализация атрофии определяется методами:
- Компьютерная томография выявляет нарушение структуры сосудов, определяет наличие новообразований и аневризм, которые затрудняют кровоток. Наиболее информативной разновидностью исследования специалистами признана мультиспиральная компьютерная томография (МСКТ). С помощью трехмерной проекции долей головного мозга она способна продемонстрировать субатрофические изменения даже самой начальной стадии.
- МРТ (магнитно-резонансная томография) признана эталоном определения структурных патологий головного мозга. Диагностика атрофии при помощи МРТ превзошла по своей эффективности и точности проведение комплекса клинически проводимых тестов. Такое обследование способно не только выявить отклонения на начальной их стадии, при помощи магнитно-резонансной томографии существует возможность следить за изменениями нарушений. Такой контроль очень важен во время наблюдения и лечения старческого слабоумия и болезни Альцгеймера.
Лечение
До нашего времени эффективное лечение атрофических отклонений не разработано, поскольку с нарушением функциональности нейронов бороться крайне сложно. Единственный способ замедлить атрофический процесс – постоянное применение назначенных врачом препаратов, направленных на улучшение церебрального кровообращения, и метаболических медикаментов. Также назначается лечение для коррекции имеющихся симптомов. Так, при ортостатической гипотензии рекомендуется увеличение употребления жидкости и поваренной соли, возможно назначение «Флудрокортизона». Также желательно поднять головной конец кровати на 10 см: это способствует уменьшению артериальной гипертензии и ночной полиурии. Также показано применение компрессионного белья для нижних конечностей. При недержании мочи назначаются препараты «Толтеродин» или «Оксибутинин» хлорида. При запорах предписывают специальную диету или клизму.
Во время лечения атрофии мозга для пациента главным образом необходимы обеспечение хорошего ухода, повышенное внимание и забота родных и близких людей. Вокруг больного необходимо создать максимально спокойную и комфортную обстановку. При этом не нужно ограждать его от выполнения привычных посильных дел по дому.
Важно знать! Для больного атрофией мозга крайне вредно содержание его в медицинских учреждениях. Это может только усугубить состояние пациента.
Также в качестве лечения назначаются успокоительные препараты, легкие транквилизаторы и антидепрессанты. Применение этих средств способствует сохранению спокойного состояния больного. Также необходимо создать условия для активного образа жизни без ограничений движения человека. Кроме всего прочего, пациенту, страдающему атрофией мозга, не противопоказан дневной сон.
Профилактика
Для предупреждения нарушений функциональности головного мозга очень важно вести здоровый и активный образ жизни. Также специалистами было установлено, что хорошее настроение и жизнелюбие значительно улучшают состояние органов и повышают продолжительность жизни.
Современными лекарствами атрофия головного мозга не лечится. Развивается это отклонение постепенно и в итоге приводит к слабоумию. Для того, чтобы предупредить болезнь и ее негативные последствия, необходимо своевременно лечить любые нарушения организма и вести здоровый образ жизни. Это поможет сохранить здоровье на многие годы.
Мультисистемная атрофия или синдром Шая-Дрейджера — MED-anketa.ru
Данная статья расскажет вам о таком недуге, как мультисистемная атрофия.
Общие сведения о заболевании, причины, признаки о диагностике, лечении о последствиях и профилактике.
Если у человека наблюдается вегетативная недостаточность, только в этом случае может развиться синдром Шая-Дрейджера, хотя на данный момент официально этого термина не существует.
Общие сведения о заболевании
Мультисистемная атрофия или по-научному Синдром Шая-Дрейджера является неврологическим недугом, тяжелого дегенеративного типа. Кроме того, ученые, считают, что спровоцировать недуг может и нарушение в работе нервных клеток, в некоторых частях головного мозга. Вследствие чего данная патология приводит к серьезным проблемам опорно-двигательного аппарата, в тяжелых случаях может быть полный паралич нижних и верхних конечностей.
Помимо проблем с опрно-двигательным аппаратом мультисистемная атрофия может приводить к нарушению и других необходимых для полноценной жизни человека функций к примеру – это может быть неконтролируемое мочеиспускание, или постоянные скачки артериального давления. По данным официальной статистики данное заболевание поражается мужчин в 55% случаях от 50-ти до 60-ти лет. Это достаточно редкое заболевание и поражает оно 4 человека из 100 тысяч.
Причины мультисистемной атрофии
Точные причины мультисистемной атрофии специалистами до конца еще не изучены. Но ряд ученых имеют версию, что всему виной ген Src гомология, она имеет сразу несколько доменов. Помимо этого Научный институт P.A Hanna et al.
Изучив 100 случаев мультисистемной атрофии, определили, что практически 20%, а это 20 человек практически всю жизнь контактировали с вредными химическими веществами (бензин, формальдегидом, пестицидами и много других) и пришли к неутешительному выводу, что всему виной нынешняя экология и работа с химикатами.
Эти важные заключения позволяют ученым предположить, что основная причина лежит в основе большой чувствительности центральной нервной системы к разным токсическим факторам. Но все, же точную причину некому выявить так и не удалось.
Симптомы заболевания
Признаки мультисистемной атрофии достаточно многообразны. Именно потому, что недуг имеет большое количество признаков, клиническая картина может, проявляется в разных комбинациях:
- В виде вегетативных нарушений.
- Могут, появляется резкие скачки артериального давления, наблюдаться замедленные реакции и движениях.
- В форме плохой координации и равновесия.
Самым частыми первичными симптомами данного заболевания являются проявление акинетической-ригидного синдрома.
Он очень сильно похож на болезнь Паркинсона. И характеризуется замедленным движением. Мультисистемная атрофия диагностируется на внешнем осмотре у врача в 65% случаях. Общие первичные симптомы заболевания следующие:
- нарушения равновесия;
- мочеполовые нарушения;
- возникновение гипертонии;
- запоры;
- нарушение речи;
- постоянное чувство жажды и сухости во рту;
- импотенция;
- громкий храп.
Важно у мужчин первыми симптомами мультистемной атрофии могут быть нарушения эрекции.
Диагностика
Диагностирование мультисистемной атрофии происходит при наличии данных после обследования. При подозрении врачи назначают:
- мрт головного мозга. Помогает выявить даже малейшие изменения в мозгу;
- клиническое обследование;
- общие анализы крови и мочи;
- сканирование МСА выявляются патологии иннервации сердца;
- вегетативные пробы. При помощи этого анализа определяют наличие вегетативную недостаточность.
Ни один способ диагностики не считается окончательным, но некоторые (например, МРТ, ядерная томография метаиодобензилгуанидином помогают подтвердить наличие мультисистемной атрофии.
Лечение
На сегодняшний день какое-либо лечение атрофических заболеваний не нашли, так как лечить нарушения работы нейронов очень сложно. Один метод, который помогает замедлить процесс дегенерации клеток мозга – это регулярный прием лекарственных препаратов, которые назначит врач.
Прежде всего, они должны быть направлены на улучшение кровообращения головного мозга и обменных процессов. Кроме того, специалисты назначают терапию, которая поможет устранить симптомы заболевания. В большинстве случаев специалисты выписывают Флудрокортизон. Помимо этого врачи советуют приподнять край кровати на 10-15 сантиметров, это поможет сократить внутричерепное давление.
Последствия мультисистемной атрофии
Мультисистемная атрофия может приводить к большому количеству последствий от небольших сокращений размеров структуры органов до их полного поражения и в последствии летального исхода
Может стать причиной частичной или полной потере зрения, бесплодия, параличу конечностей.
Профилактика
Для того, что бы предупредить возникновение атрофии очень важно соблюдать здоровый и активный образ жизни. Быть всегда в хорошем настроении — это повышает качество жизни.
Множественная системная атрофия: симптомы, лечение
Множественная системная атрофия (МСА) — изнурительное нейродегенеративное заболевание. Это редкое явление, которым страдают только четыре человека из 100 000, но его воздействие на тех, кого он касается, разрушительно. MSA имеет много патологических и клинических особенностей с более известной и более распространенной болезнью Паркинсона, за которую его часто ошибочно принимают. Фактически, аномальный белок при болезни Паркинсона (синуклеин) также играет ключевую роль в MSA.
Существует три типа MSA:
- Паркинсонизм , при котором пациенты имеют в основном тяжелые симптомы, похожие на болезнь Паркинсона, такие как замедленное движение, проблемы с ходьбой, ригидность мышц и тремор
- Cerebellar , при котором пациенты имеют проблемы в первую очередь с координацией, ходьбой и невнятной речью
- Комбинированный , в котором пациенты страдают как паркинсоническими, так и мозжечковыми симптомами
Симптомы
Помимо двигательных симптомов, характерных для каждого типа MSA, MSA может вызывать обмороки и низкое артериальное давление (а также колебания артериального давления), мышечные сокращения, недержание мочи, запоры, сексуальную дисфункцию, двоение в глазах или другие нарушения зрения. , затрудненное дыхание и глотание, нарушения сна, включая апноэ во сне, и ненормальное потоотделение.Лекарства от MSA нет, и продолжительность жизни с этим заболеванием обычно составляет от 7 до 10 лет.
Лечение
MSA быстро прогрессирует, и лечение направлено на то, чтобы попытаться контролировать симптомы, что является сложной задачей. Однако существуют эффективные методы лечения ряда симптомов MSA, и для его оптимизации важен скоординированный подход.
Дополнительные ресурсы
Проверено медицинским специалистом Cleveland Clinic.
Получите полезную, полезную и актуальную информацию о здоровье и благополучии
е Новости
Клиника Кливленда — некоммерческий академический медицинский центр.Реклама на нашем сайте помогает поддерживать нашу миссию. Мы не поддерживаем продукты или услуги, не принадлежащие Cleveland Clinic.
Политика
| AARS2: Сердечный ACAD8 : Энцефалопатия : Гипотония : Кетоацидоз : Чувствительность к EtOH : Изъятия : Энцефалопатия AMT : Энцефалопатия BCAT2 BCKDHA BCKDHB: моча кленового сиропа : Perrault 3 | D2HGDH DARS2: LBSL DECR1 DGUOK: гепатоцеребральный ДНК2: миопатия + PEO GCSH GFM1: Гепатоэнцефалопатия HADH HARS2: Perrault 2 HIBCH: подобный Leigh HSD17B10: Замедление IDh4B ISCU: Миопатия KARS: невропатия | MCCC1 MCCC2 MCEE MDh3: Энцефалопатия MGME1: PEO + Миопатия Клячи NDUFAF1: Кардиомиопатия + OTC OXCT1 PC: Атаксия + | PCCB PCK2 PDHA1: энцефалопатия RARS2: PCH6 SARS2: метаболический SUCLA2: энцефаломиопатия XPNPEP3 YARS2: миопатия + анемия |
множественная системная атрофия Википедия
Нейродегенеративное расстройство
| Множественная системная атрофия | |
|---|---|
| Иммуногистохимия головного мозга с альфа-синуклеином, показывающая множество глиальных включений | |
| Speciality | Neurology |
02 9A система множественная (MS
02 9000) редкое нейродегенеративное заболевание [1] , характеризующееся вегетативной дисфункцией, тремором, медленными движениями, ригидностью мышц и постуральной нестабильностью (все вместе известные как паркинсонизм) и атаксией.Это вызвано прогрессирующей дегенерацией нейронов в нескольких частях мозга, включая базальные ганглии, нижнее оливковое ядро и мозжечок.
Многие люди, страдающие MSA, испытывают дисфункцию вегетативной нервной системы, которая обычно проявляется в виде ортостатической гипотензии, импотенции, потери потоотделения, сухости во рту, задержки мочи и недержания мочи. Паралич голосовых связок — важное, а иногда и начальное клиническое проявление заболевания.
Модифицированная форма белка альфа-синуклеина в пораженных нейронах может вызывать МСА. [2] Около 55% случаев MSA возникает у мужчин, при этом у тех, кто поражен, первые симптомы проявляются в возрасте 50–60 лет. [3] MSA часто проявляется некоторыми из тех же симптомов, что и болезнь Паркинсона. Однако пациенты с MSA обычно плохо реагируют на препараты дофамина, используемые для лечения болезни Паркинсона, и только около 9% пациентов с MSA с тремором имели настоящий паркинсонический тремор, напоминающий пилюли. [4]
MSA отличается от мультисистемной протеинопатии, более распространенного синдрома мышечной атрофии.МСА также отличается от синдрома полиорганной дисфункции, иногда называемого полиорганной недостаточностью; из-за недостаточности полиорганной системы, часто смертельного осложнения септического шока и других тяжелых заболеваний или травм.
Признаки и симптомы []
MSA характеризуется следующими характеристиками, которые могут присутствовать в любой комбинации: [5] [6]
Также может существовать вариант с комбинированными признаками MSA и деменции с тельцами Леви. [ ненадежный медицинский источник? ] [7] Также были случайные случаи дегенерации лобно-височной доли, связанной с MSA. [8]
Первоначальная презентация []
Наиболее частым первым признаком МСА является появление «акинетико-ригидного синдрома» (то есть медленное начало движения, напоминающее болезнь Паркинсона), обнаруживаемое у 62% при первом обращении. Другие общие признаки в начале включают проблемы с равновесием (мозжечковая атаксия), обнаруженные у 22% при первом обращении, за которыми следуют мочеполовые симптомы (9%): как мужчины, так и женщины часто испытывают позывы, частоту, неполное опорожнение мочевого пузыря или неспособность мочеиспускание (задержка).Примерно у каждого пятого пациента с MSA наблюдается падение в первый год болезни. [9]
Для мужчин первым признаком может быть эректильная дисфункция. Женщины также сообщают о пониженной чувствительности гениталий. [10]
Прогресс []
По мере прогрессирования болезни преобладает одна из трех групп симптомов.
Это:
- Паркинсонизм — медленные, скованные движения, письмо становится мелким и паучьим
- Дисфункция мозжечка — нарушение координации движений и равновесия
- Дисфункция вегетативной нервной системы — нарушение автоматических функций организма, включая одно, некоторые или все из следующих:
Генетика []
Одно исследование обнаружило корреляцию между делецией генов в определенной генетической области и развитием MSA в группе японских пациентов. [14] Рассматриваемая область включает ген SHC2, который у мышей и крыс, по-видимому, выполняет некоторую функцию в нервной системе. Авторы этого исследования предположили, что может существовать связь между делецией SHC2 и развитием MSA. [ необходима ссылка ]
Последующее исследование не смогло воспроизвести этот результат у американских пациентов с MSA. [15] Авторы исследования в США пришли к выводу, что «Наши результаты показывают, что делеции гена SHC2 лежат в основе немногих, если они вообще есть, случаев хорошо охарактеризованного MSA в популяции США.Это контрастирует с японским опытом, описанным Sasaki et al., Что, вероятно, отражает гетерогенность болезни в различных генетических фонах ».
Другое исследование изучало частоту расширений интронных повторов RFC1, феномен, связанный с CANVAS; заболевание, диагностика которого частично совпадает с MSA. [16] [17] Исследование пришло к выводу, что эти повторы отсутствуют в патологически подтвержденном MSA, что указывает на альтернативную генетическую причину. [16]
Патофизиология []
Множественную системную атрофию можно объяснить потерей клеток и глиозом или пролиферацией астроцитов в поврежденных областях центральной нервной системы.Это повреждение образует рубец, который затем называют глиальным рубцом. [18] Присутствие этих телец включения, известных как тела Паппа-Лантоса, в центрах движения, баланса и вегетативного контроля мозга является определяющим гистопатологическим признаком МСА.
Основным нитчатым компонентом глиальных и нейрональных цитоплазматических включений является альфа-синуклеин. [19] Мутации в этом веществе могут играть роль в заболевании. [20] Посттрансляционно модифицированная форма белка, называемого альфа-синуклеином, может быть возбудителем болезни. [2] , вероятно, вызвано первичной олигодендроглиопатией. [21]
Тау-белки были обнаружены в некоторых глиальных цитоплазматических телец включения. [22]
Диагноз []
Клиническая []
Клинические диагностические критерии были определены в 1998 г. [23] и обновлены в 2007 г. [24] Определенные признаки и симптомы МСА также возникают при других расстройствах, таких как болезнь Паркинсона, что затрудняет диагностику. [25] [26] [27]
Радиологический []
И МРТ, и КТ могут показать уменьшение размеров мозжечка и моста у пациентов с особенностями мозжечка (MSA-C).Скорлупа гипоинтенсивна на Т2-взвешенной МРТ и может показывать повышенное отложение железа в форме болезни Паркинсона (MSA-P). В MSA-C иногда встречается знак «горячий крестик»; он отражает атрофию понтоцеребеллярных трактов, что дает сверхинтенсивный сигнал Т2 в атрофических мостах. [ необходимая ссылка ]
Изменения МРТ не требуются для диагностики заболевания, так как эти признаки часто отсутствуют, особенно на ранних стадиях заболевания. Кроме того, изменения могут быть весьма незначительными и обычно пропускаются экзаменаторами, не имеющими опыта работы с MSA.
Патологическое []
Патологический диагноз может быть поставлен только при вскрытии, обнаружив обильные GCI на гистологических образцах центральной нервной системы. [28]
В 2020 году исследователи Центра медицинских наук Техасского университета в Хьюстоне пришли к выводу, что циклическая амплификация неправильного свертывания белков может использоваться для различения двух прогрессирующих нейродегенеративных заболеваний, болезни Паркинсона и множественной системной атрофии, что является первым процессом, который поставьте объективный диагноз множественной системной атрофии вместо простого дифференциального диагноза. [29] [30]
Классификация []
MSA — одно из нескольких нейродегенеративных заболеваний, известных как синуклеинопатии: они имеют общее аномальное накопление белка альфа-синуклеина в различных частях мозга. Другие синуклеинопатии включают болезнь Паркинсона, деменцию с тельцами Леви и другие более редкие состояния. [31]
Старая терминология []
Исторически сложилось так, что для обозначения этого расстройства использовалось множество терминов, основанных на преобладающих представленных системах.Эти термины были прекращены консенсусом в 1996 году и заменены MSA и его подтипами, [32] , но знание этих старых терминов и их определений полезно для понимания соответствующей литературы до 1996 года. К ним относятся стриатонигральная дегенерация (SND), оливопонтоцеребеллярный атрофия (OPCA) и синдром Шай – Драгера. [33] Ниже приводится таблица с описанием характеристик и современных названий этих состояний:
| Историческое название | Характеристики | Современное название и сокращение |
| Стриатонигральная дегенерация | преобладающие симптомы болезни Паркинсона | MSA-P, «p» = паркинсонический подтип |
| Спорадическая оливопонтоцеребеллярная атрофия (OPCA) | характеризуется прогрессирующей атаксией (неспособностью координировать произвольные мышечные движения) походки и рук и дизартрией (затрудненное произношение слов) | MSA-C, «c» = подтип дисфункции мозжечка |
| Синдром Шай – Драгера | характеризуется паркинсонизмом плюс более выраженная недостаточность вегетативной нервной системы. [34] | Нет современного эквивалента — эта терминология вышла из моды [35] и не была указана в консенсусном документе 2007 года. [24] Более ранний консенсус 1998 года [23] относился к MSA-A, «а» = подтип вегетативной дисфункции, но этот подтип больше не используется. |
Текущая терминология []
Текущая терминология и диагностические критерии болезни были установлены на конференции экспертов 2007 года и изложены в документе с изложением позиции. [24] Это Второе согласованное заявление определяет две категории MSA, основанные на преобладающих симптомах заболевания на момент оценки. Это:
- MSA с преобладающим паркинсонизмом (MSA-P) — определяется как MSA, где преобладают экстрапирамидные признаки. Иногда это называют стриатонигральной дегенерацией, паркинсоническим вариантом.
- MSA с особенностями мозжечка (MSA-C) — определяется как MSA, при котором преобладает мозжечковая атаксия. Иногда это называют спорадической оливопонтоцеребеллярной атрофией.
Управление []
Надзор []
Рекомендуется постоянное наблюдение у невролога, специализирующегося на двигательных нарушениях, [ кем? ] , потому что сложные симптомы MSA часто не знакомы менее специализированным неврологам. Услуги хосписа / ухода на дому могут быть очень полезны при прогрессировании инвалидности.
Лекарственная терапия []
Леводопа (L-допа), препарат, используемый для лечения болезни Паркинсона, улучшает симптомы паркинсонизма у небольшого процента пациентов с MSA.Недавнее исследование показало, что только 1,5% пациентов с МСА вообще испытали какое-либо улучшение при приеме леводопы, их улучшение было менее 50%, и даже это улучшение было временным эффектом, продолжавшимся менее одного года. Плохая реакция на L-допа была предложена как возможный элемент дифференциальной диагностики MSA от болезни Паркинсона. [ необходимая ссылка ]
Рилузол неэффективен при лечении MSA или PSP. [9]
Реабилитация []
Важное значение имеет лечение трудностей с ходьбой / движением, повседневными задачами и проблемами речи со стороны специалистов по реабилитации, включая физиотерапевтов, эрготерапевтов, логопедов и других.
Физиотерапевты помогут сохранить подвижность пациента и помогут предотвратить контрактуры. [18] Инструктаж пациентов по тренировке походки поможет улучшить их подвижность и снизить риск падений. [36] Физиотерапевт может также назначить вспомогательные средства передвижения, такие как трость или ходунки, для повышения безопасности пациента. [36]
Логопеды могут помочь в оценке, лечении и поддержке речи (дизартрия) и затруднения глотания (дисфагия).Изменения речи означают, что может потребоваться альтернативное общение, например, средства общения или словарные таблицы.
Раннее вмешательство в проблемы глотания особенно полезно для обсуждения возможности зондового кормления в дальнейшем при прогрессировании заболевания. [ необходима ссылка ] В какой-то момент по мере прогрессирования заболевания может быть применена модификация жидкости и пищи.
Предотвращение постуральной гипотензии []
Одна особенно серьезная проблема — падение артериального давления при вставании (с риском обморока и, как следствие, травмы при падении), часто поддается лечению флудрокортизоном, синтетическим минералокортикоидом. [37] Другим распространенным лекарственным средством является мидодрин, агонист альфа. [38]
Немедикаментозные методы лечения включают «наклон головы вверх» (поднятие головы всей кровати примерно на 10 градусов), солевые таблетки или увеличение количества соли в рационе, обильное потребление жидкости и давление (эластичное ) чулки. Решающее значение имеют предотвращение факторов, вызывающих пониженное давление, таких как жаркая погода, алкоголь и обезвоживание. [39] Пациента можно научить двигаться и медленно переходить из положения сидя в положение стоя, чтобы снизить риск падений и ограничить эффект постуральной гипотензии. [36] Инструкции по откачиванию крови в голеностопном суставе помогают вернуть кровь из ног в системный кровоток. [36] Другими профилактическими мерами являются поднятие изголовья кровати на 8 дюймов (20,3 см), а также использование компрессионных чулок и ремней для живота. [5]
Поддержка []
Социальные работники и эрготерапевты также могут помочь справиться с инвалидностью путем предоставления оборудования и приспособлений для дома, услуг для лиц, осуществляющих уход, и доступа к медицинским услугам, как для человека с MSA, так и для членов семьи. [ необходима ссылка ]
Прогноз []
Средняя продолжительность жизни пациентов с МСА после появления симптомов составляет 6–10 лет. [3] Приблизительно 60% пациентов нуждаются в инвалидной коляске в течение пяти лет после появления двигательных симптомов, и лишь немногие пациенты выживают после 12 лет. [3] Заболевание прогрессирует без ремиссии с переменной скоростью. У тех, кто обращается в более старшем возрасте, у лиц с паркинсоническими особенностями и у пациентов с тяжелой вегетативной дисфункцией, прогноз хуже. [3] Те, у кого преобладают мозжечковые особенности, и те, у кого позже проявляется вегетативная дисфункция, имеют лучший прогноз. [3]
Причины смерти []
Наиболее частыми причинами смерти являются внезапная смерть и смерть от инфекций, в том числе инфекций, вызванных катетеризацией мочевых путей, инфекций через зонд для кормления и аспирационной пневмонии. Некоторые случаи смерти вызваны кахексией, также известной как синдром истощения. [40]
Эпидемиология []
По оценкам, множественная системная атрофия встречается примерно у 5 на 100 000 человек.При вскрытии обнаруживается, что у многих пациентов, у которых в течение жизни была диагностирована болезнь Паркинсона, действительно есть MSA, что позволяет предположить, что фактическая частота MSA выше этой оценки. [3] В то время как некоторые предполагают, что МСА поражает несколько больше мужчин, чем женщин (1,3: 1), другие предполагают, что оба пола могут быть затронуты с одинаковой вероятностью. [3] [5] [18] Заболевание чаще всего проявляется у людей в возрасте 50–60 лет. [3]
Известные случаи []
Исследования []
Терапия мезенхимальными стволовыми клетками может замедлить прогрессирование неврологического дефицита у пациентов с MSA-мозжечковым типом. a b c d e 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 902 h Fanciulli A, Wenning GK (январь 2015 г.). «Множественная системная атрофия». N. Engl. J. Med . 372 (3): 249–63. DOI: 10.1056 / NEJMra1311488. a b c Веннинг GK, Colosimo C, Geser F, Poewe W. (февраль 2004 г.). «Множественная системная атрофия». Ланцет Нейрол . 3 (2): 93–103. DOI: 10.1016 / S1474-4422 (03) 00662-8. PMID 14747001. S2CID 10162139. Множественная системная атрофия (МСА) — дегенеративное [1] неврологическое заболевание.MSA связан с дегенерацией нервных клеток в определенных областях мозга. Эта дегенерация клеток вызывает проблемы с движением, балансом и другими вегетативными функциями организма, такими как контроль мочевого пузыря или регулирование артериального давления. Причина MSA неизвестна, и никаких конкретных факторов риска выявлено не было. [2] Около 55% случаев заболевания возникают у мужчин, с типичным возрастом начала заболевания от 50 до 60 лет. [3] Общая распространенность MSA оценивается в 4 человека.6 случаев на 100000 человек. [4] [5] Это заболевание чаще встречается у мужчин, чем у женщин. Исследования показали, что соотношение варьируется от 1,4: 1 [6] до 1,9: 1. [7] MSA характеризуется комбинацией следующих элементов, которые могут присутствовать в любой комбинации: [7] [8] Когда преобладает вегетативная недостаточность, иногда используется термин синдром Шай-Драгера , хотя этот термин больше не актуален, учитывая изменения терминологии, которые объясняются ниже. [9] [10] [11] [12] Также может существовать вариант с комбинированными признаками MSA и деменции с тельцами Леви. [13] Наиболее частым первым признаком МСА является появление «акинетико-ригидного синдрома» (то есть медленное начало движения, напоминающее болезнь Паркинсона), обнаруживаемое у 62% при первом обращении. Другие общие признаки в начале включают проблемы с равновесием (мозжечковая атаксия), обнаруженные у 22% при первом обращении, за которыми следуют проблемы с мочеполовой системой (9%).Для мужчин первым признаком может быть эректильная дисфункция (неспособность достичь или поддерживать эрекцию). И мужчины, и женщины часто испытывают проблемы с мочевым пузырем, включая позывы, частоту, неполное опорожнение мочевого пузыря или невозможность мочеиспускания (задержка мочи). Примерно 1 из 5 пациентов с MSA страдает падением в первый год болезни. [3] По мере прогрессирования болезни преобладают три группы симптомов. Это: Могут возникать и другие симптомы, например двоение в глазах. [15] Не все пациенты испытывают все эти симптомы. Некоторые пациенты (20% в одном исследовании) испытывают значительные когнитивные нарушения в результате МСА. [16] MSA обычно прогрессирует быстрее, чем болезнь Паркинсона. [5] Нет ремиссии болезни. Средняя продолжительность жизни после появления симптомов у пациентов с МСА составляет 7,9 года. [5] Почти 80% пациентов становятся инвалидами в течение 5 лет после появления двигательных симптомов, и только 20% выживают в течение последних 12 лет. [17] Скорость прогрессирования различается в каждом случае, и скорость снижения может широко варьироваться у отдельных пациентов. О’Салливан и его коллеги (2008) определили раннюю вегетативную дисфункцию как наиболее важный клинический прогностический признак выживаемости при MSA. Пациенты с сопутствующей моторной и вегетативной дисфункцией в течение 3 лет после появления симптомов имели более короткую продолжительность выживания, помимо того, что они становились зависимыми от инвалидных колясок и прикованными к постели на более ранней стадии, чем те, у которых эти симптомы развились через 3 года с момента появления симптомов.Их исследование также показало, что когда у пациентов с ранней вегетативной дисфункцией развиваются частые падения, развивается зависимость от инвалидных колясок, тяжелая дисфагия или требуется стационарное лечение, от этого момента до смерти проходит более короткий интервал. [18] Иммуногистохимия альфа-синуклеина показывает множество глиальных включений. Диагностика MSA может быть сложной задачей, потому что не существует теста, который мог бы окончательно поставить или подтвердить диагноз у живого пациента.Определенные признаки и симптомы MSA также возникают при других заболеваниях, таких как болезнь Паркинсона, что затрудняет диагностику. [19] Окончательный диагноз может быть поставлен патологически только после обнаружения обильных глиальных цитоплазматических включений в центральной нервной системе. [20] Не существует известного лекарства от MSA, поэтому лечение включает устранение симптомов. Рекомендуется постоянное наблюдение у невролога, специализирующегося на «двигательных расстройствах», поскольку сложные симптомы MSA часто не знакомы менее специализированным специалистам в области здравоохранения. Одна особенно серьезная проблема — падение артериального давления при вставании (с риском обморока и, соответственно, травмы при падении), часто поддается лечению флудрокортизоном, синтетическим минералокортикоидом. Еще одним распространенным лекарственным средством является мидодрин (альфа-агонист). Немедикаментозные методы лечения включают «наклон головы вверх» (поднятие изголовья всей кровати примерно на 10 градусов), солевые таблетки или увеличение количества соли в рационе, обильное потребление жидкости и эластичные чулки. Очень важно избегать триггеров низкого кровяного давления (таких как жаркая погода, алкоголь, обезвоживание). [21] Услуги хосписа / ухода на дому могут быть очень полезны при прогрессировании инвалидности. Леводопа (L-допа), препарат, используемый для лечения болезни Паркинсона, не может улучшить симптомы паркинсонизма у большинства пациентов с MSA. Недавнее исследование показало, что только 1,5% пациентов с MSA испытали улучшение менее чем на 50% при приеме леводопы, и даже это был временный эффект, длящийся менее одного года. Плохая реакция на L-допа была предложена как возможный элемент дифференциальной диагностики MSA от болезни Паркинсона. Недавнее исследование, проведенное в Европе, не показало эффекта от препарата рилузол при лечении MSA или PSP. [3] Важное значение имеет помощь специалистов по реабилитации (физиотерапевтов, физиотерапевтов, эрготерапевтов, логопедов и др.) При проблемах с ходьбой / движением, повседневными задачами и проблемами речи. Физиотерапия может помочь сохранить подвижность пациента и предотвратить контрактуры. [6] Инструктаж пациентов по тренировке походки поможет улучшить их подвижность и снизить риск падений. [22] Физиотерапевт может также прописать вспомогательные средства передвижения, такие как трость или ходунки, для повышения безопасности пациента. [22] Другие способы, которыми физиотерапевт может помочь повысить безопасность пациента, — это научить его двигаться и медленно переходить из положения сидя в положение стоя, чтобы снизить риск падений и ограничить эффект постуральной гипотензии. [22] Инструкция по откачиванию крови в голеностопном суставе помогает вернуть кровь из ног в большой круг кровообращения. [22] Для дальнейшего контроля постуральной гипотензии может быть показано поднятие изголовья кровати на 8 дюймов (20,3 см) во время сна, а также использование эластичного компрессионного белья. [7] Социальные работники и физиотерапевты также могут помочь справиться с инвалидностью, предоставив оборудование и приспособления для дома, услуги для лиц, осуществляющих уход, и доступ к медицинским услугам, как для человека с MSA, так и для членов семьи. Мультисистемная атрофия может быть объяснена потерей клеток и глиозом или пролиферацией астроцитов в поврежденных областях центральной нервной системы.Это повреждение образует рубец, который затем называют глиальным рубцом. [6] В настоящее время подтвержденный диагноз MSA может быть поставлен только посмертно, поскольку видны глиальные цитоплазматические тельца включения. Когда ткань мозга человека с МСА исследуется под микроскопом, эти глиальные структуры видны, подтверждая диагноз. Присутствие этих включений (также известных как тела Паппа-Лантоса) в центрах движения, равновесия и автоматического контроля мозга является определяющим гистопатологическим признаком MSA.Недавние исследования показали, что основным волокнистым компонентом глиальных и нейрональных цитоплазматических включений является альфа-синуклеин. [23] Мутации в этом веществе могут играть роль в заболевании. [24] Тау-белки были обнаружены в некоторых GCI [25] Многие термины исторически использовались для обозначения этого расстройства на основе преобладающих представленных систем. К ним относятся оливопонтоцеребеллярная атрофия (OPCA), синдром Шай-Драгера (SDS) и стриатонигральная дегенерация (SND), которые когда-то считались отдельными расстройствами. [26] Эти термины и их различия были опущены в недавнем (1996 г.) медицинском использовании [10] и заменены MSA и его подтипами, но они полезны для понимания более старой литературы об этом заболевании: Текущая терминология и диагностические критерии этого заболевания были установлены на конференции экспертов по этому заболеванию в 2007 году и изложены во «Втором консенсусном заявлении по диагностике множественной системной атрофии». [28] Второе согласованное заявление определяет две категории MSA, основанные на преобладающих симптомах заболевания на момент оценки.Это: Возможная связь была идентифицирована с геном (содержащий домен Src homology 2) трансформирующим белком 2, расположенным в дистальной субтеломерной области длиной 350 kb хромосомы 19 (19p13. (PDF) Распространенность редких болезней: Библиографические данные (Отчет). a b c Веннинг GK, Colosimo C, Geser F, Poewe W (февраль 2004 г.). «Множественная системная атрофия». Ланцет Neurol 3 (2): 93–103. DOI: 10.1016 / S1474-4422 (03) 00662-8. PMID 14747001. http://linkinghub.elsevier.com/retrieve/pii/S1474442203006628. содержание сенсагента Решение для веб-мастеров Александрия Всплывающее окно с информацией (полное содержание Sensagent), вызываемое двойным щелчком по любому слову на вашей веб-странице.Предоставьте контекстные объяснения и перевод с вашего сайта ! Попробуйте здесь или получите код SensagentBox С помощью SensagentBox посетители вашего сайта могут получить доступ к надежной информации на более чем 5 миллионах страниц, предоставленных Sensagent.com. Выберите дизайн, который подходит вашему сайту. Бизнес-решение Улучшите содержание своего сайта Добавьте новый контент на свой сайт из Sensagent by XML. Сканирует продукты или добавляет Получите доступ к XML для поиска лучших продуктов. Индексирование изображений и определение метаданных Получите доступ к XML, чтобы исправить значение ваших метаданных. Напишите нам, чтобы описать вашу идею. Lettris Lettris — это любопытная игра-тетрис-клон, в которой все кубики имеют одинаковую квадратную форму, но разное содержание. На каждом квадрате есть буква. Чтобы квадраты исчезли и сэкономили место для других квадратов, вам нужно собрать английские слова (left, right, up, down) из падающих квадратов. болт Boggle дает вам 3 минуты, чтобы найти как можно больше слов (3 буквы и более) в сетке из 16 букв. Вы также можете попробовать сетку из 16 букв. Буквы должны располагаться рядом, и более длинные слова оцениваются лучше. Посмотрите, сможете ли вы попасть в Зал славы сетки! Английский словарь WordNet предоставляет большинство определений на английском языке. Английский тезаурус Перевод Измените целевой язык, чтобы найти перевод.
Веннинг GK, Colosimo C, Geser F, Poewe W. (март 2004 г.). «Опечатка». Ланцет Нейрол . 3 (3): 137. DOI: 10.1016 / S1474-4422 (04) 00695-7. Шахнаваз, Мохаммад; Мукхерджи, Абхисек; Прицкова, Сандра; Мендес, Николас; Рабадиа, Пракрути; Лю, Сянган; Ху, Бо; Шмейхель, Энн; Певец Вольфганг; Ву, банда; Цай, Ах-Лим; Ширани, Хамид; Nilsson, K. Peter R .; Low, Phillip A .; Сото, Клаудио (5 февраля 2020 г.). «Различение штаммов α-синуклеина при болезни Паркинсона и множественной системной атрофии». Природа . 578 (7794): 273–277. Bibcode: 2020Natur.578..273S. DOI: 10.1038 / s41586-020-1984-7. PMC 7066875. PMID 32025029. Ли PH, Ли Дж. Э., Ким Х.С., Сон С.К., Ли Х.С., Нам Х.С. и др. (Июль 2012 г.). «Рандомизированное исследование мезенхимальных стволовых клеток при множественной системной атрофии». Ann. Neurol . 72 (1): 32–40. DOI: 10.1002 / ana.23612. PMID 22829267. S2CID 5201446. Внешние ссылки []
определение множественной_системной_атрофии и синонимов множественной_системной_атрофии (английский)
Распространенность
Симптомы
Первоначальное представление
Симптомы по мере прогрессирования болезни
Прогноз
Диагноз
Лечение
Реабилитация
Гистопатология
Терминология
Историческое название Характеристики Аббревиатура и современное название Стриатонигральная дегенерация преобладающие симптомы болезни Паркинсона MSA-P, «p» = паркинсонический подтип Спорадическая оливопонтоцеребеллярная атрофия (OPCA) характеризуется прогрессирующей атаксией (неспособностью координировать произвольные мышечные движения) походки и рук и дизартрией (затрудненное произношение слов) MSA-C, «c» = подтип мозжечковой дисфункции Синдром Шай-Драгера характеризуется паркинсонизмом плюс более выраженная недостаточность вегетативной нервной системы. [27] MSA-A, «а» = подтип вегетативной дисфункции. (В 2007 г. не указано) Геномика
Веннинг GK, Colosimo C, Geser F, Poewe W. (март 2004 г.). «Опечатка». Ланцет Neurol 3 (3): 137. Аль-Халаби, А; Дюрр, А; Дерево, NW; Паркинсон, MH; Камузат, А; Юло, JS; Моррисон, KE; Аль-Чалаби А., Дюрр А., Вуд Н. В., Паркинсон М. Х., Камузат А., Юло Дж. С., Моррисон К. Э., Рентон А., Сассмут С. Д., Ландвермейер Б. Г., Лудольф А., Агид И., Брайс А., Ли П. Н., Бенсимон Г.; Группа генетических исследований NNIPPS. и другие. (Сентябрь 2009 г.). Хойтинк, проф. Питер. изд. «Генетические варианты гена α-синуклеина SNCA связаны с множественной системной атрофией». PLoS ONE 4 (9): e7114. DOI: 10.1371 / journal. Sasaki H, Emi M, Iijima H, et al. (2011). «Потеря числа копий гена (содержащий домен src гомологии 2) -трансформирующего белка 2 (SHC2): дискордантная потеря у монозиготных близнецов и частая потеря у пациентов с множественной системной атрофией». Mol Brain 4 : 24. DOI: 10.1186 / 1756-6606-4-24. PMC 3141657. PMID 21658278. http://www.molecularbrain.com/content/4//24. Внешние ссылки
определение мультисистемной атрофии и синонимы мультисистемной атрофии (английский)
Основные ссылки
в основном является производным от The Integral Dictionary (TID).
English Encyclopedia лицензирована Википедией (GNU).
Советы: просмотрите семантические поля (см. От идей к словам) на двух языках, чтобы узнать больше.
9039 онлайн посетителей
вычислено за 0,093 с
Множественная системная атрофия
Множественная системная атрофия (MSA) — дегенеративное [1] неврологическое заболевание.MSA связан с дегенерацией нервных клеток в определенных областях мозга. Эта дегенерация клеток вызывает проблемы с движением, балансом и другими вегетативными функциями организма, такими как контроль мочевого пузыря или регулирование артериального давления. Причина MSA неизвестна, и никаких конкретных факторов риска выявлено не было. [2] Около 55% случаев заболевания возникают у мужчин, с типичным возрастом начала заболевания от 50 до 60 лет. [3]
Общая распространенность MSA оценивается в 4 человека.6 случаев на 100000 человек. [4]
Симптомы
MSA характеризуется комбинацией следующих элементов, которые могут присутствовать в любой комбинации: [5] [6]
- вегетативная дисфункция
- Паркинсонизм (ригидность мышц + / тремор и замедленные движения)
- атаксия (плохая координация / неустойчивая ходьба)
Когда преобладает вегетативная недостаточность, иногда используется термин синдром Шай-Драгера , хотя этот термин больше не актуален, учитывая изменения в терминологии, которые объясняются ниже. [7] Этот синдром был назван в честь доктора Милтона Шай и доктора Гленна Драгера, которые идентифицировали его в 1960 году, но Американское автономное общество и Американская академия неврологии в 1996 году переопределили его как атрофию множественных систем с вегетативными явлениями. [8 ] [9] [10]
Также может существовать вариант с комбинированными признаками MSA и деменции с тельцами Леви. [11]
Первоначальное представление
Наиболее частым первым признаком МСА является появление «акинетико-ригидного синдрома» (т.е. медленность начала движения, напоминающая болезнь Паркинсона) обнаружена у 62% при первом обращении. Другие общие признаки в начале включают проблемы с равновесием (мозжечковая атаксия), обнаруженные у 22% при первом обращении, за которыми следуют проблемы с мочеполовой системой (9%). Для мужчин первым признаком может быть эректильная дисфункция (неспособность достичь или поддерживать эрекцию). И мужчины, и женщины часто испытывают проблемы с мочевым пузырем, включая позывы, частоту, неполное опорожнение мочевого пузыря или невозможность мочеиспускания (задержку мочи).Примерно 1 из 5 пациентов с MSA страдает падением в первый год болезни. [3]
Эти общие первые признаки аналогичны признакам подострой комбинированной дегенерации спинного мозга, вызванной дефицитом витамина B12: у пациентов наблюдаются слабость в ногах, руках, туловище, покалывание и онемение, которые постепенно ухудшаются. Также могут присутствовать изменения зрения и изменение психического состояния. Может развиться двусторонний спастический парез, а давление, вибрация и осязание уменьшатся.Может быть замечен положительный знак Бабинского. Длительный дефицит витамина B12 приводит к необратимому повреждению нервной системы. Витамин B12 следует проверять напрямую; его нельзя исключить общим анализом крови. В век обогащения фолиевой кислотой у пациентов не будет макроцитарной анемии. http://jnnp.bmj.com/content/65/6/822.abstract
Симптомы по мере прогрессирования болезни
По мере прогрессирования болезни преобладают три группы симптомов. Это:
- Паркинсонизм (медленные, скованные движения, письмо становится мелким и паучьим)
- Дисфункция мозжечка (нарушение координации движений и равновесия)
- Вегетативная дисфункция (нарушение автоматических функций организма) в том числе:
Могут возникать и другие симптомы, например двоение в глазах. [13] Не все пациенты испытывают все эти симптомы.
Некоторые пациенты (20% в одном исследовании) испытывают значительные когнитивные нарушения в результате МСА. [14]
Прогноз
MSA обычно прогрессирует быстрее, чем болезнь Паркинсона. [15] Нет ремиссии болезни. Распространенность MSA варьируется по данным исследования от 3 до 20 на 100 000 человек. Оставшаяся продолжительность жизни после появления симптомов у пациентов с МСА составляет 7.3 и 9,3 года. Это заболевание чаще встречается у мужчин, чем у женщин, и исследования показали, что соотношение находится в диапазоне от 1,4: 1 [16] до соотношений до 1,9: 1. [5] Некоторые полагают, что более высокое соотношение может быть связано с большим воздействием предполагаемых токсинов в окружающей среде у мужчин или различиями в эндогенных защитных факторах (гормональных) у женщин. [6] Почти 80% пациентов становятся инвалидами в течение 5 лет после появления двигательных симптомов, и только 20% выживают в течение последних 12 лет. [17] Скорость прогрессирования различается в каждом случае, и скорость снижения может широко варьироваться у отдельных пациентов.
О’Салливан и его коллеги (2008) определили раннюю вегетативную дисфункцию как наиболее важный клинический прогностический признак выживаемости при MSA. Пациенты с сопутствующей моторной и вегетативной дисфункцией в течение 3 лет после появления симптомов имели более короткую продолжительность выживания, помимо того, что они становились зависимыми от инвалидных колясок и прикованными к постели на более ранней стадии, чем те, у которых эти симптомы развились через 3 года с момента появления симптомов. Их исследование также показало, что когда у пациентов с ранней вегетативной дисфункцией развиваются частые падения, развивается зависимость от инвалидных колясок, тяжелая дисфагия или требуется стационарное лечение, от этого момента до смерти проходит более короткий интервал. [18]
Диагноз
Иммуногистохимия альфа-синуклеина показывает множество глиальных включений.
Диагностика MSA может быть сложной задачей, потому что не существует теста, который мог бы окончательно поставить или подтвердить диагноз у живого пациента. Определенные признаки и симптомы MSA также возникают при других заболеваниях, таких как болезнь Паркинсона, что затрудняет диагностику. [19]
Окончательный диагноз может быть поставлен патологически только после обнаружения обильных глиальных цитоплазматических включений в центральной нервной системе. [20]
Лечение
Не существует известного лекарства от MSA, поэтому лечение включает устранение симптомов.
Рекомендуется постоянное наблюдение у невролога, специализирующегося на «двигательных расстройствах», поскольку сложные симптомы MSA часто не знакомы менее специализированным специалистам в области здравоохранения.
Одна особенно серьезная проблема — падение артериального давления при вставании (с риском обморока и, соответственно, травмы при падении), часто поддается лечению флудрокортизоном, синтетическим минералокортикоидом.Другим распространенным лекарственным средством является мидодрин (альфа-агонист). Немедикаментозное лечение включает «наклон головы вверх» (подъем головы всей кровати примерно на 10 градусов), солевые таблетки или увеличение количества соли в рационе, обильное употребление жидкости и эластичные чулки. Очень важно избегать триггеров низкого кровяного давления (например, жаркой погоды, алкоголя, обезвоживания). [21]
Услуги хосписа / ухода на дому могут быть очень полезны при прогрессировании инвалидности.
Леводопа (L-допа), препарат, используемый для лечения болезни Паркинсона, не может улучшить симптомы паркинсонизма у большинства пациентов с MSA.Недавнее исследование показало, что только 1,5% пациентов с MSA испытали улучшение менее чем на 50% при приеме леводопы, и даже это был временный эффект, длящийся менее одного года. Плохая реакция на L-допа была предложена как возможный элемент дифференциальной диагностики MSA от болезни Паркинсона.
Недавнее исследование, проведенное в Европе, не показало эффекта от препарата рилузол при лечении MSA или PSP. [3]
Реабилитация
Важное значение имеет помощь специалистов по реабилитации (физиотерапевтов, физиотерапевтов, эрготерапевтов, логопедов и др.) При проблемах с ходьбой / движением, повседневными задачами и проблемами речи.
Физиотерапия может помочь сохранить подвижность пациента и предотвратить контрактуры. [16] Инструктаж пациентов по тренировке походки поможет улучшить их подвижность и снизить риск падений. [22] Физиотерапевт может также прописать вспомогательные средства передвижения, такие как трость или ходунки, чтобы повысить безопасность пациента. [22] Другой способ, которым физиотерапевт может помочь повысить безопасность пациента, — это научить его двигаться и медленно переходить из положения сидя в положение стоя, чтобы снизить риск падений и ограничить эффект постуральной гипотензии. [22] Инструкция по откачиванию крови в голеностопном суставе помогает вернуть кровь из ног в большой круг кровообращения. [22] Для дальнейшего контроля постуральной гипотензии может быть показано поднятие изголовья кровати на 8 дюймов (20,3 см) во время сна, а также использование эластичного компрессионного белья. [5]
Социальные работники также могут помочь справиться с инвалидностью и получить доступ к медицинским услугам как для человека с MSA, так и для членов его семьи.
Гистопатология
Множественную системную атрофию можно объяснить потерей клеток и глиозом или пролиферацией астроцитов в поврежденных областях центральной нервной системы. Это повреждение образует рубец, который затем называют глиальным рубцом. [16] В настоящее время подтвержденный диагноз MSA может быть установлен только посмертно, поскольку видны глиальные цитоплазматические включения. Когда ткань мозга человека с МСА исследуется под микроскопом, эти глиальные структуры видны, подтверждая диагноз.Присутствие этих включений (также известных как тела Паппа-Лантоса) в центрах движения, баланса и автоматического управления головного мозга является определяющим гистопатологическим признаком MSA. Недавние исследования показали, что основным волокнистым компонентом глиальных и нейрональных цитоплазматических включений является альфа-синуклеин. [23] Мутации в этом веществе могут играть роль в заболевании. [24] Тау-белки были обнаружены в некоторых GCI [25]
Терминология
Многие термины исторически использовались для обозначения этого расстройства на основе преобладающих представленных систем.К ним относятся оливопонтоцеребеллярная атрофия (OPCA), синдром Шай-Драгера (SDS) и стриатонигральная дегенерация (SND).
Эти термины и их различия были опущены в недавнем (1996 г.) медицинском использовании [26] и заменены наименование подтипа MSA, но они полезны для понимания более старой литературы об этом заболевании:
| Имя | Характеристики | Аббревиатура |
| Стриатонигральная дегенерация | преобладающие симптомы болезни Паркинсона | MSA-p, «p» = паркинсонический подтип |
| Спорадическая оливопонтоцеребеллярная атрофия (OPCA) | характеризуется прогрессирующей атаксией (неспособностью координировать произвольные мышечные движения) походки и рук и дизартрией (затрудненное произношение слов) | MSA — c, «c» = подтип дисфункции мозжечка |
| Синдром Шай-Драгера | характеризуется паркинсонизмом плюс более выраженная недостаточность вегетативной нервной системы. [27] Этот подтип упоминается в Заявлении о консенсусе 1996 года как MSA-a, «а» = подтип вегетативной дисфункции. [26] |
Текущая терминология и диагностические критерии этого заболевания были установлены на конференции экспертов по этому заболеванию в 2007 году и изложены во «Втором консенсусном заявлении по диагностике множественной системной атрофии». [28]
Второе согласованное заявление определяет две категории MSA, основанные на преобладающих симптомах заболевания на момент оценки.Это:
- MSA с преобладающим паркинсонизмом (MSA-P) MSA-P определяется как MSA, где преобладают экстрапирамидные признаки. Термин «стриатонигральная дегенерация, паркинсонический вариант» иногда используется для обозначения этой категории MSA.
- MSA с особенностями мозжечка (MSA-C). MSA-C определяется как MSA, при котором преобладает мозжечковая атаксия. Иногда это называют спорадической оливопонтоцеребеллярной атрофией.