Перспективы исследования нейрофиброматоза I типа в Республике Башкортостан | Мустафин
1. Ponti G., Losi L., Martotana D., Priola M., Boni E., Pollio A., et al. Clinico-pathological and biomolecular findings in Italian patients with multiple cutaneous neurofi bromas. Hered Cancer Clin Pract. 2011;9(1):6. DOI: 10.1186/1897-4287-9-6
2. Voelker R. A new treatment for children with neurofi bromatosis type 1. JAMA. 2020;323(19):1887. DOI: 10.1001/jama.2020.7157
3. Chai G., Liu N., Ma J., Li H., Oblinger J.L., Prahalad A.K., et al. MicroRNA-10b regulates tumorigenesis in neurofi bromatosis type 1. Cancer Sci. 2010;101(9):1997–2004. DOI: 10.1111/j.1349-7006.2010.01616.x
4. Yap Y., McPherson J.R., Ong C., Rozen S.G., Th e B., Lee A.S., et al. Th e NF1 gene revisited — from bench to bedside. Oncotarget. 2014;5(15):5873–92. DOI: 10.18632/oncotarget.2194
Yap Y., McPherson J.R., Ong C., Rozen S.G., Th e B., Lee A.S., et al. Th e NF1 gene revisited — from bench to bedside. Oncotarget. 2014;5(15):5873–92. DOI: 10.18632/oncotarget.2194
5. Ratner N., Miller S.J. A RASopathy gene commonly mutated in cancer: the neurofi bromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15(5):290–301. DOI: 10.1038/nrc3911
6. Sabbagh A., Pasmant E., Laurendeau I., Parfait B., Barbarot S., Guillot B., et al. Unravelling the genetic basis of variable clinical expression in neurofi bromatosis 1. Hum Mol Genet. 2009;18(18):2768–78. DOI: 10.1093/hmg/ddp212
7. Melean G., Hernandez A.M., Valero M.C., Hernandez-Imaz E., Martin Y., Hernandez-Chico C. Monozygotic twins with Neurofi bromatosis type 1, concordant phenotype and synchronous development of MPNST and metastasis. BMC Cancer. 2010;10:407. DOI: 10.1186/1471-2407-10-407
BMC Cancer. 2010;10:407. DOI: 10.1186/1471-2407-10-407
8. Patil S., Chamberlain R.S. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17(1):101–16. DOI: 10.1634/theoncologist.2010-0181
9. Stevenson D., Moyer-Mileur L., Murray M., Slater H., Sheng X., Carey J.C., et al. Bone mineral density in children and adolescents with neurofi bromatosis type 1. J Pediatr. 2007;150(1):83–8. DOI: 10.1016/j.jpeds.2006.10.048
10. Biotteau M., Déjean S., Lelong S., Iannuzzi S., Faure-Marie N., Castelnau P., et al. Sporadic and familial variants in NF1: an explanation of the wide variability in neurocognitive phenotype? Front Neurol. 2020;11:368. DOI: 10.3389/fneur.2020.00368
11. Неустроева Л.М., Павлова Н.И., Соловьева Н.А., Дьяконова А.Т., Варламова М.А., Филиппова Н.П. и др. Нейрофиброматоз I типа: этиопатогенез, клиника, диагностика, лечение. Якутский медицинский журнал. 2018;1(61):69–72. DOI: 10.25789/YMJ.2018.61.21
Неустроева Л.М., Павлова Н.И., Соловьева Н.А., Дьяконова А.Т., Варламова М.А., Филиппова Н.П. и др. Нейрофиброматоз I типа: этиопатогенез, клиника, диагностика, лечение. Якутский медицинский журнал. 2018;1(61):69–72. DOI: 10.25789/YMJ.2018.61.21
12. Неробеев А.И., Голубева С.Н., Добродеев А.С., Зорин В.Л., Еремин И.И., Зорина А.И. и др. Нейрофиброматоз — возможности хирургической реабилитации. Анналы пластической, реконструктивной и эстетической хирургии. 2014;4:10–20.
13. Laycock-van Spyk S., Th omas N., Cooper D.N., Upadhyaya M. Neurofi bromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum Genomics. 2011;5(6):623–90. DOI: 10.1186/1479-7364-5-6-623
14. Мустафин Р.Н., Хуснутдинова Э. К. Роль эпигенетических факторов в патогенезе нейрофиброматоза 1-го типа. Успехи молекулярной онкологии. 2017;4(3):37–49. DOI: 10.17650/2313-805X-2017-4-3-35-49
К. Роль эпигенетических факторов в патогенезе нейрофиброматоза 1-го типа. Успехи молекулярной онкологии. 2017;4(3):37–49. DOI: 10.17650/2313-805X-2017-4-3-35-49
15. Nicita F., Torrente I., Spalice A., Bottillo I., Papetti L., Pinna V., et al. Spinal neurofi bromatosis in a family with classical neurofi bromatosis type 1 and a novel NF1 gene mutation. J Clin Neurosci. 2014;21(2):328–30. DOI: 10.1016/j.jocn.2013.01.026
16. Garcia-Linares C., Fernandez-Rodriguez J., Terribas E., Mercade J., Pros E., Benito L., et al. Dissecting Loss of Heterozygosity (LOH) in neurofi bromatosis type 1-associated neurofibromas: importance of copy neutral LOH. Human Mutation. 2011;32(1):78–90. DOI: 10.1002/humu.21387
17. De Raedt T.D., Maertens O. , Chmara M., Brems H., Heyns I., Sciot R., et al. Somatic loss of wild type NF1 allele in neurofi bromas: comparison of NF1 microdeletion and non-microdeletion patients. Genes, Chromosomes and Cancer. 2006;45(10):893–904. DOI: 10.1002 /gcc.20353
, Chmara M., Brems H., Heyns I., Sciot R., et al. Somatic loss of wild type NF1 allele in neurofi bromas: comparison of NF1 microdeletion and non-microdeletion patients. Genes, Chromosomes and Cancer. 2006;45(10):893–904. DOI: 10.1002 /gcc.20353
18. Allaway R.J., Gosline S.J.C., La Rosa S., Knight P., Bakker A., Guinney J., et al. Cutaneous neurofi bromas in the genomics era: current understanding and open questions. Br J Cancer. 2018;118(12):1539–48. DOI: 10.1038/s41416-018-0073-2
19. Burks C.A., Rhodes S.D., Bessler W.K., Chen S., Smith A., Gehlhausen J.R., et al. Ketotifen Modulates Mast Cell Chemotaxis to Kit-Ligand, but Does Not Impact Mast Cell Numbers, Degranulation, or Tumor Behavior in Neurofi bromas of Nf1-Defi cient Mice. Mol Cancer Th er. 2019 Dec;18(12):2321–30. DOI: 10.1158/1535-7163. MCT-19-0123
20. Lim U., Song M.A. DNA methylation as a biomarker of aging in epidemiologic studies. Methods Mol Biol. 2018;1856:219–31. DOI: 10.1007/978-1-4939-8751-1_12
Lim U., Song M.A. DNA methylation as a biomarker of aging in epidemiologic studies. Methods Mol Biol. 2018;1856:219–31. DOI: 10.1007/978-1-4939-8751-1_12
21. Geller M., Ribeiro M.G., Araújo A.P., de Oliveira L.J., Nunes F.P. Serum IgE levels in neurofi bromatosis 1. Int J Immunogenet. 2006;33(2):111–5. DOI: 10.1111/j.1744-313X.2006.00579.x
22. Bottillo I., De Luca A., Schirinzi A., Guida V., Torrente I., Calvieri S., et al. Functional analysis of splicing mutations in exon 7 of NF1 gene. BMC Medical Genetics. 2007;8:4–13. DOI: 10.1186/1471-2350-8-4
23. Дубова А.И., Щеголев А.И., Кармазановский Г.Г., Колокольчикова Е.Г., Степанова Ю.А., Мелихова М.В. и др. Нейрофиброматоз брюшной полости. Медицинская визуализация. 2006;(4):62–74.
24. Маратканова Т.В., Сташук Г.А., Денисова Л.Б., Шерман Л.А. К вопросу диагностики нейрофиброматоза (клинико-диагностические наблюдения). Медицинская визуализация. 2008;(6):114–23.
25. Шнайдер Н.А., Шаповалова Е.А. Нейрофиброматоз 1-го типа (болезнь Реклингхаузена). Вопросы практической педиатрии. 2011;6(1):83–8.
26. Дядькин В.Ю. Случай локализованной формы нейрофиброматоза 1 типа. Практическая медицина. 2013;1–4(73):130–1.
27. Попова А.А. Клинико-диагностические аспекты нейрофиброматоза. Университетская медицина Урала. 2016;2(2):48–50.
28. Вдовина А. С., Сажин А.А., Волков С.И. Редкий случай нейрофиброматоза с гигантскими множественными нейрофибромами туловища и конечностей (клиническое наблюдение). Тверской медицинский журнал. 2017;3:53–6.
С., Сажин А.А., Волков С.И. Редкий случай нейрофиброматоза с гигантскими множественными нейрофибромами туловища и конечностей (клиническое наблюдение). Тверской медицинский журнал. 2017;3:53–6.
29. Куракина Е.С. Болезнь Реклингаузена. Вестник современных исследований. 2018;(12.4):110–3.
30. Заричанский В.А., Притыко А.Г., Егиазарян А.К. Особенности хирургического лечения нейрофиброматоза I типа и перспективы консервативной терапии. Онкопедиатрия. 2014;3:51–2.
31. Полякова И.В. Клинический случай нейрофиброматоза 1-го типа с развитием рецидивирующей нейрофибромы левого предплечья. Здравоохранение Дальнего Востока. 2015;1:82–4.
32. Васильева И. А., Ступак В.В., Пендюрин И.В., Копылов И.С., Цегельников М.М., Селякова М.С. Двухэтапное хирургическое лечение пациента с нейрофиброматозом I типа (клиническое наблюдение). Современные проблемы науки и образования. 2017;5:60–9.
А., Ступак В.В., Пендюрин И.В., Копылов И.С., Цегельников М.М., Селякова М.С. Двухэтапное хирургическое лечение пациента с нейрофиброматозом I типа (клиническое наблюдение). Современные проблемы науки и образования. 2017;5:60–9.
33. Макурдумян Л.А. Нейрофиброматоз I типа. Проблемы диагностики и лечения. Лечащий врач. 2001;10:59–61.
34. Жуковская Е.В., Бондаренко В.П., Спичак И.И., Сидоренко Л.В. Таргетная терапия у пациентов с нейрофиброматозом. В мире научных открытий. 2017;9(4):205–18.
35. Маламашин Д.Б., Щелкунов М.М., Красников М.А., Мушкин А.Ю. Хирургическая коррекция субаксиального кифоза у ребенка с нейрофиброматозом I типа: редкое клиническое наблюдение и обзор литературы. Хирургия позвоночника. 2018;15(2):12–7. DOI: 10. 14531/ss2018.2.12-17
14531/ss2018.2.12-17
36. Бакланов А.Н., Шавырин И.А. Оперативное лечение пациента со сверхтяжелым кифосколиозом на фоне нейрофиброматоза. Хирургия позвоночника. 2013;2:28–31. DOI: 10.14531/ss2013.2.28-31
37. Матюшин А.Ф., Гаврилов В.А. Отдаленные результаты переднего спондилодеза васкуляризированным трансплантатом из ребра в лечении тяжелого кифосколиоза при нейрофиброматозе. Хирургия позвоночника. 2012;4:41–8. DOI: 14531/ss2012.4.41-48
38. Матюшин А.Ф., Гаврилов В.А. Передний спондилодез васкуляризованным трансплантатом ребра в лечении тяжелого кифосколиоза при нейрофиброматозе. Медицина и образование в Сибири. 2012;4:23.
39. Любченко Л. Н., Филиппова М.Г. Нейрофиброматоз: генетическая гетерогенность и дифференциальная диагностика. Саркомы костей, мягких тканей и опухоли кожи. 2011;4:29–36.
Н., Филиппова М.Г. Нейрофиброматоз: генетическая гетерогенность и дифференциальная диагностика. Саркомы костей, мягких тканей и опухоли кожи. 2011;4:29–36.
40. Дрозд О.В., Бабенко О.В., Семячкина А.Н., Харабадзе М.Н., Немцова М.В., Залетаев Д.В. Разработка подходов к ДНК- диагностике нейрофиброматоза 1-го типа в России. Медицинская генетика. 2005;4(7):322–6.
41. Пащенко М.С., Кузнецова Е.Б., Танас А.С., Бессонова Л.А., Матющенко Г.Н., Демина Н.А. и др. Комплексная молекулярно-генетическая диагностика нейрофиброматоза. В сб.: Молекулярная диагностика. М.: Юлис; 2017. С. 89–90.
42. Мустафин Р.Н., Бермишева М.А., Хуснутдинова Э.К. Клинико-эпидемиологическое исследование нейрофиброматоза I типа в Республике Башкортостан. Якутский медицинский журнал. 2009;2:23–5.
2009;2:23–5.
43. Мустафин Р.Н., Бермишева М.А., Хуснутдинова Э.К. Особенности нейрофиброматоза 1-го типа в Республике Башкортостан. Медицинская генетика. 2015;14(6):29–34.
44. Fahsold R., Hoff meyer S., Mischung C., Gille C., Ehlers C., Kucukceylan N., et al. Minor lesion mutational specrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. 2000;66(3):790–818. DOI: 10.1086/302809
45. Ars E., Serra E., Garcia J., Kruyer H., Gaona A., Lazaro C., et al. Mutations aff ecting mRNA splicing are the most common molecular defects in patients with neurofi bromatosis type 1. Hum Mol Genet. 2000;9(2):237–47. DOI: 10.1093/hmg/9.2.237
46. Jeong S., Park S., Kim H. Th e spectrum of NF1 mutations in korean patients with neurofi bromatosis type 1. J Korean Med Sci. 2006;21(1):107–11. DOI: 10.3346/jkms.2006.21.1.107
Jeong S., Park S., Kim H. Th e spectrum of NF1 mutations in korean patients with neurofi bromatosis type 1. J Korean Med Sci. 2006;21(1):107–11. DOI: 10.3346/jkms.2006.21.1.107
47. Messiaen, L.M., Callens, T., Mortier, G., Beysen, D., Vandenbroucke, I., Roy, N.V., et al. Exhaustive mutation analysis of NF1 gene allows identifi cation of 95% of mutations and reveals a high frequency of unusual splicing defects. Human Mutation. 2000;15(6):541–55. DOI: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N
48. Мустафин Р.Н., Бермишева М.А., Хуснутдинова Э.К. Болезнь Реклингхаузена в Республике Башкортостан, результаты и перспективы исследований. Медицинский вестник Башкортостана. 2016;11(2):9–12.
Нейрофиброматоз 1 типа | МОСМЕДПРЕПАРАТЫ
Общая информация о нейрофиброматозе 1 типа
Существуют три клинически и генетически различающихся формы нейрофиброматоза: нейрофиброматоз 1 типа (NF1), нейрофиброматоз 2 типа (NF2), шванноматоз./cafe-au-lait-skin-spot-in-neurofibromatosis-680795071-599cf32c685fbe00100f00cf.jpg) Самым распространенным является нейрофиброматоз 1 типа, также известный как болезнь фон Реклингхаузена, или периферический нейрофиброматоз. Среди отличительных признаков нейрофиброматоза 1 типа: множественные гиперпигментные пятна (макулы) на коже цвета «кофе с молоком» (café-au-lait) и связанные с ними кожные нейрофибромы. Состояние называется сегментарным нейрофиброматозом 1 типа, когда клинические признаки ограничены одной областью тела.
Самым распространенным является нейрофиброматоз 1 типа, также известный как болезнь фон Реклингхаузена, или периферический нейрофиброматоз. Среди отличительных признаков нейрофиброматоза 1 типа: множественные гиперпигментные пятна (макулы) на коже цвета «кофе с молоком» (café-au-lait) и связанные с ними кожные нейрофибромы. Состояние называется сегментарным нейрофиброматозом 1 типа, когда клинические признаки ограничены одной областью тела.
Нейрофиброматоз 1 типа, будучи одним из самых распространенных (1 случай на 2,5–3 тыс. человек) аутосомно-доминантных генетических расстройств, вызывается патогенными вариантами гена NF1, кодирующего белок нейрофибромин. Сегментарный нейрофиброматоз 1 типа отмечается в случае соматического мозаицизма ввиду постзиготной мутации гена NF1.
Типичный порядок клинической манифестации нейрофиброматоза 1 типа: макулы café-au-lait, подмышечные и/или паховые веснушки, узелки Лиша (гамартомы радужной оболочки глаза), нейрофибромы. Поражения костной ткани, если они есть, обычно появляются в первый год после рождения, а симптоматическая глиома зрительного пути — к моменту, когда исполняется три года. Другие опухоли и неврологические осложнения возникают, как правило, после первого года жизни. Гипертония и злокачественная трансформация опухолей могут дать о себе знать в подростковом периоде и взрослом возрасте.
Другие опухоли и неврологические осложнения возникают, как правило, после первого года жизни. Гипертония и злокачественная трансформация опухолей могут дать о себе знать в подростковом периоде и взрослом возрасте.
Диагностика нейрофиброматоза 1 типа основана на его характерных клинических признаках. Генетическое тестирование не требуется, хотя и полезно для подтверждения диагноза в случае не соответствия всем диагностическим критериям.
Дифференциальный диагноз нейрофиброматоза 1 типа включает: синдром Легиуса, синдром конститутивного дефицита в системе репарации (CMMR-D), нейрофиброматоз 2 типа, синдром Нунан.
Этиология и патогенез нейрофиброматоза 1 типа
Причиной развития нейрофиброматоза 1 типа является аутосомно-доминантная мутация в гене NF1. В одной половине случае она наследуется от родителей, при этом полная пенетрантность мутации завершается после окончания периода детства, то есть вероятность носительства является 100-процентной. В другой половине случаев отмечается мутация de novo.
Мутация с потерей функции гена NF1 в хромосомном сегменте 17q11.2 вызывает недостаточный синтез белка-онкосупрессора нейрофибромина. Идентифицировано свыше 1 тыс. различных мутаций NF1. Большинство мутаций приводят к сильному усечению генного продукта.
Нейрофибромин, относящийся к семейству ГТФаза-активирующих белков, отвечает за отрицательную регуляцию активности сигнального пути RAS/MAPK посредством ускорения гидролиза Ras-связанного гуанозинтрифосфата (ГТФ). Генетические поломки нарушают контроль над клеточным ростом и нейрональным развитием.
Лечение нейрофиброматоза 1 типа
Долгосрочный уход за пациентами с нейрофиброматозом 1 типа направлен на раннее выявление и симптоматическое лечение осложнений по мере их возникновения. При оказании медицинской помощи детям с нейрофиброматозом 1 типа частота посещений медицинских учреждений должна быть увеличена с целью устранения осложнений по ходу их проявлений. Решение о проведении диагностических исследований зависит от анамнеза и физических признаков. Клиническая оценка представляется более полезной для выявления осложнений, чем скрининговые исследования у бессимптомных пациентов.
Клиническая оценка представляется более полезной для выявления осложнений, чем скрининговые исследования у бессимптомных пациентов.
Подход к лечению различных опухолей, связанных с нейрофиброматозом 1 типа, зависит от типа опухоли, ее воздействия на соседние ткани и связанных с ней осложнений. Хирургическое лечение и обезболивание плексиформных нейрофибром может быть непростой задачей. Хирургическая резекция часто ограничивается циторедукцией определенного участка большого поражения.
В апреле 2010 года «АстраЗенека» (AstraZeneca) предложила «Коселуго» (Koselugo, селуметиниб) — первый лекарственный препарат, предназначенный для терапии детей в возрасте от двух лет, страдающих нейрофиброматозом 1 типа. Заболевание пациентов должно быть симптоматическим и характеризоваться наличием неоперабельных плексиформных нейрофибром.
Неврологические расстройства при нейрофиброматозе 1 типа, которые могут потребовать специфического лечения, следующие: когнитивные нарушения, нарушения обучаемости, судороги, периферическая нейропатия.
При длительной костной дисплазии и сколиозе при нейрофиброматозе 1 типа может потребоваться ортопедическое вмешательство. Иные костные отклонения, такие как остеопороз, могут не реагировать на типичные методы лечения.
Прогнозы нейрофиброматоза 1 типа
С пациентами и их семьями следует проводить всеобъемлющее консультирование, предоставляя информацию о наследственной природе нейрофиброматоза 1 типа, прогнозах и психосоциальной адаптации. Необходимо учитывать прогрессирующий характер нейрофиброматоза 1 типа и его клинические манифестации и осложнения.
Данные о влиянии нейрофиброматоза 1 типа на смертность носят ограниченный характер, хотя ожидаемая продолжительность жизни пациентов, по-видимому, сокращается. Злокачественные новообразования, особенно злокачественные опухоли оболочек периферических нервов, являются основной причиной снижения выживаемости. Продолжительность жизни при нейрофиброматозе 1 типа может сократиться на 8–20 лет.
Нет сильной корреляции между генотипом и тяжестью заболевания, за исключением небольшого числа специфических мутаций, например микроделеции гена NF1.
Плексиформные нейрофибромы чаще все начинают расти в детском и подростковом возрасте, в целом оставаясь стабильными у взрослых. Уровень их злокачественности оценивается в 4–5%.
Когнитивные проблемы — наиболее частые неврологические осложнения у пациентов с нейрофиброматозом 1 типа. Так, синдром дефицита внимания с гиперактивностью (СДВГ) или без нее возникает у 30–40% пациентов. Признаки аутизма отмечаются у 30% детей. Распространенность неспособности к обучению оценивается в 40–75%, однако что именно подразумевается под определением данного когнитивного расстройства варьирует в различных исследованиях. Проблемы с обучением и поведением могут сохраняться и во взрослой жизни, будучи чаще всего связанными с недостаточностью зрительного и пространственного восприятия, социальной компетентности или внимания.
Лечение нейрофиброматоза за рубежом | MediGlobus
Доктор Вадим Бережной
Основатель платформы MediGlobus. Медицинский эксперт, Заведующий отделением врачей-координаторов.
Медицинский эксперт, Заведующий отделением врачей-координаторов.
Время чтения – 8 минут
Нейрофиброматоз – это редкое наследственное заболевание, которое встречается у 1 человека на 25,000. Оно проявляется без симптомов и может быть опасно для жизни и здоровья. Из нашей статьи вы сможете узнать на какие признаки стоит обратить внимание, как лечат нейрофиброматозы и в каких клиниках можно провести операцию.
Слушать статью:
ЧТО ТАКОЕ НЕЙРОФИБРОМАТОЗ?
Нейрофиброматоз – это генетическая болезнь, которая передается по наследству и вызывает развитие опухолей тканей нервной системы. Они могут образовываться в головного мозге, спинном мозге или периферических нервах. Большинство опухолей – доброкачественные. Злокачественные нейрофибромы развиваются в 3% случаях.
Большинство опухолей – доброкачественные. Злокачественные нейрофибромы развиваются в 3% случаях.
Существует два типа заболевания. Нейрофиброматоз 1 типа называют болезнью фон Реклингхаузена. При этом диагнозе часто образовываются нейрофибромы – это опухоли периферических нервов. Болезнь Реклингхаузена является распространенной формой нейрофибромы и встречается у 1 человека из 3,500.
Нейрофбироматоз 2 типа вызывает развитие опухолей в клетках Шванна, которые формируют миелиновую оболочку нервных клеток. Шванноматоз является менее распространенной формой нейрофибромы и диагностируется у 1 человека на 40,000.
Причины нейрофиброматоза связаны с мутациями в генах NF1, NF2 и SMARCB1 / INI1, которые контролируют деление клеток.
СИМПТОМЫ НЕЙРОФИБРОМАТОЗА
Около трети людей не жалуются на симптомы нейрофиброматоза на начальной стадии. Тем не менее первые признаки нейрофиброматоза 1 типа можно заметить в детстве, чаще всего до 10 лет.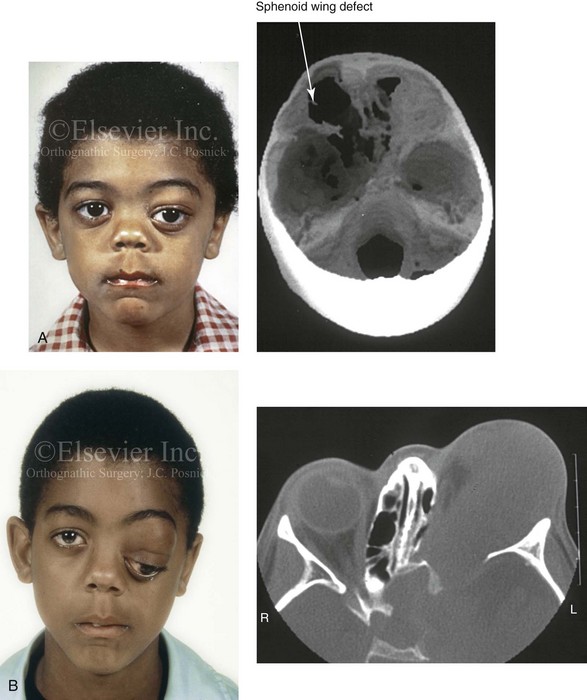 Шванномы второго типа развиваются позже, в возрасте от 20 до 24 лет. Симптомы нейрофиброматоза у взрослых редко ярко проявляются. Его можно узнать по следующим признакам:
Шванномы второго типа развиваются позже, в возрасте от 20 до 24 лет. Симптомы нейрофиброматоза у взрослых редко ярко проявляются. Его можно узнать по следующим признакам:
Внешне нейрофиброматоз легко перепутать с родимым пятном. Нейрофиброматоз с поражением головного мозга проявляется у людей в признаках нарушения внимания, координации, мышления и ориентации в пространстве. Для того чтобы убедиться в диагнозе нужно проконсультироваться у специалиста.
ОСЛОЖНЕНИЯ ПРИ НЕЙРОФИБРОМАТОЗЕ
Болезнь Реклингхаузена вызывает осложнения в работе множества органов. Если оставить заболевание нейрофиброматоза без лечения, тогда осложнения будут включать:
Нейрофиброматоз может оказывать влияние на беременность. У большинства женщин с этим видом нейрофиброматоза беременность протекает нормально. Однако, им важно наблюдаться у врача, который знаком с болезнью, так как нейрофиброматоз при беременности может провоцировать увеличение опухоли нервов.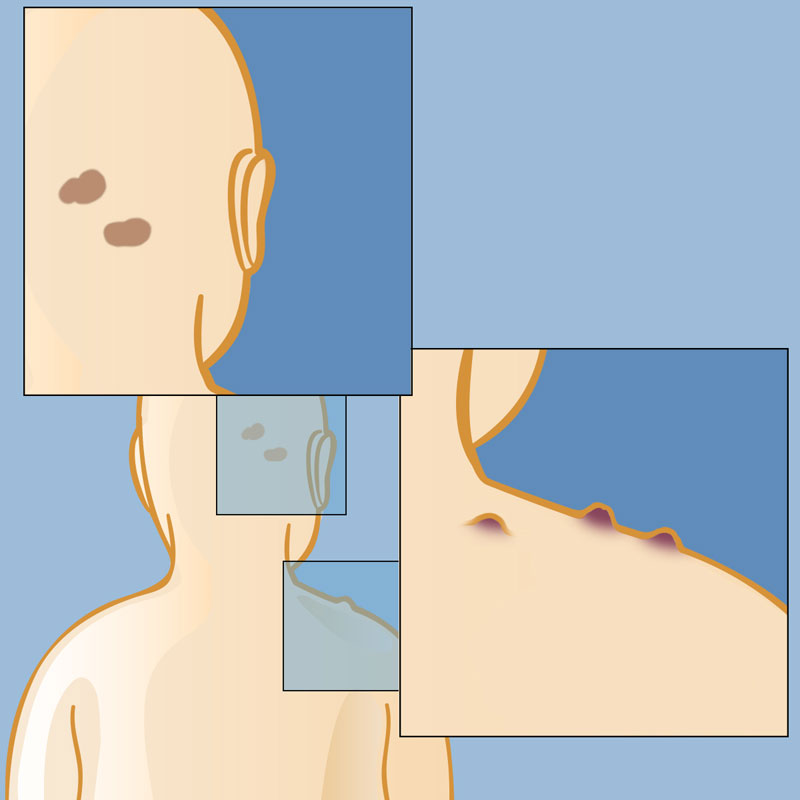
Осложнения при нейрофиброматозе второго типа включают в себя:
Шванномы 2 типа, которые развиваются в спинном мозге могут вызвать паралич, если оставить их без лечения.
Нейрофиброматоз обычно протекает безболезненно. Если человек страдает от сильных болей, ему необходимо обследоваться на шванноматоз.
ДИАГНОСТИКА НЕЙРОФИБРОМАТОЗА
Обследование при нейрофиброматозе обязательно включает консультацию с неврологом, кардиологом, онкологом, офтальмологом и ортопедом. При диагностике заболевания важна история болезни пациента и его близких родственников. Если в анамнезе семьи есть случаи нейрофиброматоза, тогда с высокой вероятностью он может быть у пациента.
Магнитно-резонансная томография (МРТ) головного мозга является основным методом диагностики нейрофиброматоза. При нейрофиброме головного мозга МРТ дает максимально точный результат. В качестве дополнительных методов обследования врач может назначить рентген или компьютерную томографию (КТ).
В зарубежных клиниках существуют пренатальные тесты для определения нейрофиброматоза Реклингхаузена. Их точность превышает 90%. Тест на определение нейрофиброматоза 2 типа до рождения ребенка также доступен за рубежом, однако его точность составляет всего лишь 65%.
КАК ЛЕЧАТ НЕЙРОФИБРОМАТОЗ В ЗАРУБЕЖНЫХ КЛИНИКАХ?
Злокачественные нейрофиброматозы 1 типа лечат химиотерапией и лучевой терапией. Лечение направлено на ослабление симптоматики и улучшение состояния пациента. Так, человеку может быть показана лучевая терапия для уменьшения размера глиомы зрительного нерв. В зарубежных клиниках возможно хирургическое лечение нейрофиброматоза, однако оно связано с рисками для здоровья пациента. В большинстве случаев операции при нейрофиброматозе назначают для облегчения симптомов. Так, многим пациентам показана хирургия для исправления сколиоза.
Нейрофиброматоз второго типа обычно лечат хирургическим путем. Если опухоль размером до 1 сантиметра, врач может полностью ее удалить и сохранить слух. При удалении шванном до 2 сантиметров хирург устанавливает кохлеарный имплант, который усилит слух пациента. Чем больше опухоль, тем труднее врачам сохранить слух. Если шваннома распространилась на значительную часть ствола мозга, операция может облегчить симптомы.
При удалении шванном до 2 сантиметров хирург устанавливает кохлеарный имплант, который усилит слух пациента. Чем больше опухоль, тем труднее врачам сохранить слух. Если шваннома распространилась на значительную часть ствола мозга, операция может облегчить симптомы.
КАК ПРОЯВЛЯЕТСЯ И ЛЕЧИТСЯ НЕЙРОФИБРОМАТОЗ У ДЕТЕЙ?
Поступление в школу ребенка с этим диагнозом можно после обследования у невролога. Он проверит уровень IQ, развитие пространственного мышления и речи. В целом, дети с нейрофиброматозом головного мозга живут так же, как их однолетки. Болезнь развивается медленно и часто не мешает нормальному развитию ребенка. Болезнь Реклингхаузена у детей проявляется в 10-летнем возрасте. Нейрофиброматоз у детей проявляется в виде симптомов:
Детский нейрофиброматоз лечит консилиум врачей. В него входят педиатр, онколог, кардиолог, дерматолог, отоларинголог, окулист и невролог. Участие междисциплинарной команды врачей важно, так как болезнь нейрофиброматоз влияет на развитие всех органов ребенка.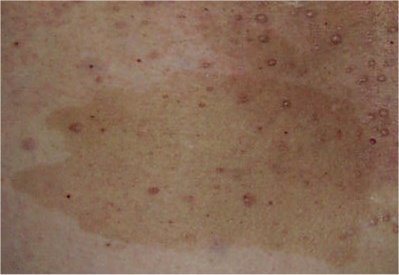
При нейрофиброматозе 1 типа у детей возможно лечение иммунотерапией. Весной 2020 года FDA одобрило препарат Коселуго. Он помогает замедлить деление клеток опухоли. Лекарство можно назначать детям после 2 лет.
ЖИЗНЬ С НЕЙРОФИБРОМАТОЗОМ
Люди с нейрофиброматозом могут получить образование, работать и жить нормальной жизнью. Заболевание важно лечить, так как его симптомы ухудшаются со временем. Те, кто живет с нейрофиброматозом испытывают трудности в обучении, так как болезнь часто приводит к развитию синдрома дефицита внимания и гиперактивности. Пациенты с подтвержденным нейрофиброматозом могут оформить инвалидность и получать материальную поддержку от государства.
ПРОДОЛЖИТЕЛЬНОСТЬ ЖИЗНИ ПРИ НЕЙРОФИБРОМАТОЗЕ
Нейрофиброматоз – это заболевание при котором прогноз жизни благоприятный. Большинство людей с нейрофибромой первого и второго типа имеют нормальную продолжительность жизни. В среднем люди с нейрофиброматозом живут больше 65 лет.
СКОЛЬКО СТОИТ ДИАГНОСТИКА И ЛЕЧЕНИЕ НЕЙРОФИБРОМАТОЗА ЗА РУБЕЖОМ?
Турция
| Процедура | Стоимость |
|---|---|
| КТ | от $400 |
| МРТ нейрофибромы | от $600 |
| Резекция нейрофибромы | от $9,000 |
Испания
| Процедура | Стоимость |
|---|---|
| УЗИ нейрофибромы | от €500 |
| Магнитно-резонансная томография | от €800 |
| Удаление опухоли | от €12,000 |
Германия
| Процедура | Стоимость |
|---|---|
| Компьютерная томография | от €700 |
| МРТ | от €800 |
| Операция при нейрофиброме | от €13,000 |
Израиль
| Процедура | Стоимость |
|---|---|
| Консультация невролога | от $680 |
| КТ мозга | от $600 |
| Удаление нейрофибромы | от $15,000 |
В КАКИХ КЛИНИКАХ ЛЕЧАТ НЕЙРОФИБРОМАТОЗ?
Лечение нейрофиброматоза в Турции самое недорогое в Европе. Сделать операцию возможно в клиниках Лив Улус, Мемориал Шишли, Аджибадем Атакент, Коч, Американском госпитале.
Сделать операцию возможно в клиниках Лив Улус, Мемориал Шишли, Аджибадем Атакент, Коч, Американском госпитале.
Лечение нейрофиброматоза в Израиле популярно среди пациентов из СНГ. Чаще всего они обращаются в медицинский центр им. Сураски (Ихилов), клинику Ассута, госпиталь им. Хаимы Шиба.
Лечение нейрофиброматоза в Испании успешно проводят в клиниках сети Quironsalud: Текнон, Кирон Мадрид, Кирон Барселона, Кирон Торревьеха. Хорошие показатели успеха лечения нейрофибром в университетской клинике Наварры.
Лечение нейрофиброматоза в Южной Корее успешно проходит в медицинском центре Асан, госпитале Сунчонхян, клинике Анам. Успешность удаления нейрофибромы в Корее может сравниться с качеством лечения нейрофиброматоза в Америке.
Лечение нейрофиброматоза в Германии проходит в университетской клинике Людвига-Максимилана, клинике Фрайбурга.
Резюме
Нейрофиброматоз – это опухоль, которая развивается в центральной или периферической нервной системе и имеет сильную наследственность.
Есть два вида нейрофиброматоза: болезнь Реклингхаузена и шваннома 2 типа. Первый тип проявляется в 10 лет, а второй тип развивается после 20 лет.
Симптомы нейрофиброматоза на начальных стадиях незаметны для человека. С возрастом они ухудшаются и могут включать пятна на коже, головную боль, деформации скелета, проблемы с сердцем. Пациенты с нейрофиброматозом испытывают трудности в обучении.
Нейрофиброматоз диагностируют при помощи МРТ, КТ, рентгена и УЗИ.
Злокачественные нейрофибромы лечат химиотерапией и лучевой терапией. Для нейрофиброматозов небольших размеров показана хирургия. На поздних стадиях заболевание не лечится, но операция может ослабить неприятные симптомы.
Нейрофиброматоз часто развивается у детей. В зарубежных клиниках детскую нейрофиброму лечат хирургией и иммунотерапией.
Средняя продолжительность жизни у людей с нейрофиброматозом составляет 65 лет. Она на 7 лет меньше, чем продолжительность жизни здоровых людей (72 года).
Вылечить нейрофиброматоз можно в клиниках Израиля, Германии, Турции, Испании и Южной Кореи.
Оставляйте свою заявку для записи на диагностику и лечение нейрофиброматоза за рубежом. Врачи-координаторы MediGlobus предложат вам удобные и недорогие варианты и помогут с организацией медицинской поездки.
Получить бесплатную консультацию
Источники:
- 1. Национальный институт неврологических болезней и инсульта (NINDS)
- 2. Американская ассоциация нейрохирургов
Доктор Вадим Бережной
Основатель платформы MediGlobus. Медицинский эксперт, Заведующий отделением врачей-координаторов.
Медицинский эксперт, Заведующий отделением врачей-координаторов.
Альбина Головина
Работает в сфере медицинского копирайтинга 2 года. Учится на аспирантуре факультета психологии. Автор нескольких научных публикаций в области клинической психологии. Дополнительно изучает физиологию ЦНС, нейропсихологию и психиатрию. В свободное время изучает практики mindfulness и проводит психологические консультации.
Похожие посты
Как работает вирусная терапия — прорыв в лечении рака кожи
Читать дальше
Какая выживаемость пациенток с раком шейки матки на разных стадиях?
Читать дальше
FDA одобрили Козела (Трилациклиб) при мелкоклеточном раке легких
Читать дальше
причины, симптомы, лечение, прогноз — клиника «Добробут»
Нейрофиброматоз у детей: типы, причины, симптомы, лечение
Что это – нейрофиброматоз? Так называют группу заболеваний с однотипными клиническими проявлениями. Причина нейрофиброматоза – мутации в определенных генах. Считается, что в половине случаев они унаследованы от родителей, а в остальных вызваны новыми мутациями (De Novo). Такие изменения происходят в половых клетках или в эмбрионах. Нейрофиброматоз – аутосомно-доминантное расстройство. Это означает, что генетический дефект может быть унаследован от любого из родителей. При этом тяжесть состояния родителя не влияет на форму расстройства ребенка.
Причина нейрофиброматоза – мутации в определенных генах. Считается, что в половине случаев они унаследованы от родителей, а в остальных вызваны новыми мутациями (De Novo). Такие изменения происходят в половых клетках или в эмбрионах. Нейрофиброматоз – аутосомно-доминантное расстройство. Это означает, что генетический дефект может быть унаследован от любого из родителей. При этом тяжесть состояния родителя не влияет на форму расстройства ребенка.
Типы и стадии нейрофиброматоза
Основные типы нейрофиброматоза:
- Нейрофиброматоз І типа (болезнь фон Реклингхаузена). Поражает кожу и мягкие ткани (пигментные пятна, нейрофибромы), кости (дисплазия). При нейрофиброматозе пятна имеют цвет кофе с молоком (так называемые кофейные пятна).
- Нейрофиброматоз ІІ типа. Характерные признаки – пигментные пятна, одно- или двусторонняя невринома слуховых (VIII пары черепно-мозговых) нервов, нейрофиброма, глиома, менингиома, шваннома.
- Шванноматоз или ІІІ тип нейрофиброматоза.
 Характерно развитие двух или более шванном в периферических нервах.
Характерно развитие двух или более шванном в периферических нервах.
 Характерно развитие двух или более шванном в периферических нервах.
Характерно развитие двух или более шванном в периферических нервах.
Опухоли при нейрофиброматозе бывают периферическими и центральными. Первые развиваются по ходу периферических нервов. Нейрофибромы состоят из шванновских, нервных и тучных клеток.
Центральные опухоли бывают нескольких форм:
- глиомы, которые обычно развиваются у детей младше пяти лет;
- невриномы слухового нерва, вызывающие головокружение, глухоту, шум в ушах;
- менингиомы.
Стадии нейрофиброматоза:
- начальная – образование пятен на спине, конечностях;
- стадия костных изменений – происходит деформация костной ткани;
- стадия опухолей – появление нейрофибром, глиом;
- стадия органических изменений – нарушение функций соседних органов.
Лечение нейрофиброматоза
Симптомы нейрофиброматоза I типа проявляются при рождении или развиваются в первые месяцы жизни.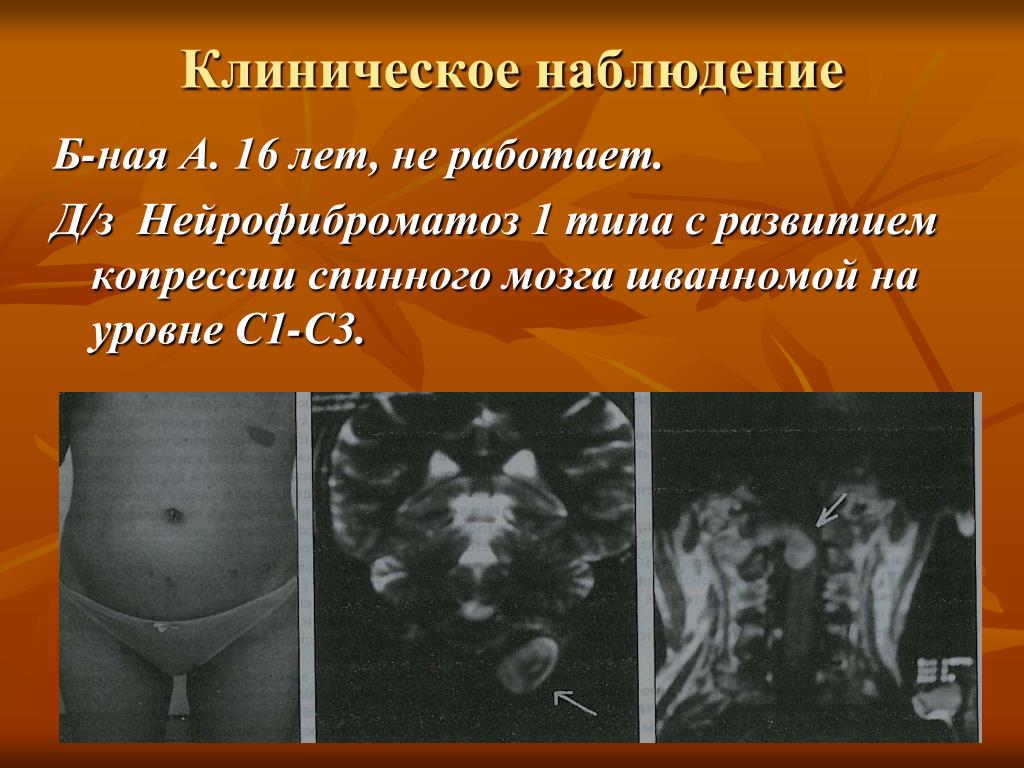 Характерные поражения кожи – веснушкоподобные или «кофейные» пятна. Они чаще расположены на туловище, в складках коленей, локтей. Позже появляются кожные опухоли. Неврологические признаки заболевания варьируются в зависимости от расположения нейрофибром. Костные изменения выражены в виде дисплазии, сколиоза (подробнее о причинах сколиоза читайте на нашем сайте Добробут.ком), ложного сустава, отсутствия большого крыла клиновидной кости с последующим пульсирующим экзофтальмом. При нейрофиброматозе у детей может развиться ювенильный миеломоноцитарный лейкоз. Злокачественные опухоли возникают редко, но чаще, чем в общей популяции.
Характерные поражения кожи – веснушкоподобные или «кофейные» пятна. Они чаще расположены на туловище, в складках коленей, локтей. Позже появляются кожные опухоли. Неврологические признаки заболевания варьируются в зависимости от расположения нейрофибром. Костные изменения выражены в виде дисплазии, сколиоза (подробнее о причинах сколиоза читайте на нашем сайте Добробут.ком), ложного сустава, отсутствия большого крыла клиновидной кости с последующим пульсирующим экзофтальмом. При нейрофиброматозе у детей может развиться ювенильный миеломоноцитарный лейкоз. Злокачественные опухоли возникают редко, но чаще, чем в общей популяции.
Лечение нейрофиброматоза сводится к хирургическому удалению опухолей (если это возможно) и химиотерапии. Терапию оптических глиом, переродившихся в злокачественные, проводят с помощью лучевой терапии или химиотерапии.
Прогноз заболевания относительно благоприятный. В большинстве случаев симптомы нейрофиброматоза выражены умеренно, и больные живут нормальной жизнью. У некоторых пациентов с диагнозом «нейрофиброматоз І типа» косметические дефекты вызывают психологические проблемы. При нейрофиброматозе ІІ типа возможно поражение черепных нервов и ствола мозга, что опасно для жизни. Многие больные, страдающие шванноматозом, испытывают боль.
У некоторых пациентов с диагнозом «нейрофиброматоз І типа» косметические дефекты вызывают психологические проблемы. При нейрофиброматозе ІІ типа возможно поражение черепных нервов и ствола мозга, что опасно для жизни. Многие больные, страдающие шванноматозом, испытывают боль.
Если у одного из родителей имеется диагноз «нейрофиброматоз», то риск передачи заболевания детям составляет 50%, поэтому перед планированием беременности целесообразна генетическая консультация.
Хирургическое лечение нейрофиброматоза I типа, ассоциированного с опухолевым поражением забрюшинного пространства
Выполнена верхнесрединная лапаротомия. При ревизии в печени пальпаторно метастазов не выявлено. В вертикальной ветви двенадцатиперстной кишки определяется опухолевидное образование с бугристой поверхностью диаметром до 12 см. Дистальнее на 3 см, в нижней горизонтальной части двенадцатиперстной кишки выявлено узловое образование размером до 3 см. В 15 см дистальнее связки Трейтца — аналогичное образование размером до 1 см. В различных отделах тонкой кишки еще 5 аналогичных образований максимальным размером до 0,5 см. При мобилизации вертикальной ветви двенадцатиперстной кишки произошел разрыв капсулы опухоли с массивным кровотечением. Кровотечение удалось остановить только после полной мобилизации двенадцатиперстной кишки по Кохеру. Выделена ножка опухолевого узла диаметром до 1 см, расположенная в дистальной части вертикальной ветви двенадцатиперстной кишки. Выполнена полнослойная резекция двенадцатиперстной кишки на расстоянии 1 см от видимых границ опухоли. Образовавшийся дефект ушит двухрядным швом. Резекция нижней горизонтальной ветви двенадцатиперстной кишки в области второго образования произведена с иссечением всех слоев кишечной стенки. Дефект ушит однорядным швом. Наибольший опухолевый узел тонкой кишки удален путем полнослойной резекции, с отступом 1 см от видимых границ опухоли. Оставшиеся 5 опухолевых узлов удалены аналогичным способом.
В 15 см дистальнее связки Трейтца — аналогичное образование размером до 1 см. В различных отделах тонкой кишки еще 5 аналогичных образований максимальным размером до 0,5 см. При мобилизации вертикальной ветви двенадцатиперстной кишки произошел разрыв капсулы опухоли с массивным кровотечением. Кровотечение удалось остановить только после полной мобилизации двенадцатиперстной кишки по Кохеру. Выделена ножка опухолевого узла диаметром до 1 см, расположенная в дистальной части вертикальной ветви двенадцатиперстной кишки. Выполнена полнослойная резекция двенадцатиперстной кишки на расстоянии 1 см от видимых границ опухоли. Образовавшийся дефект ушит двухрядным швом. Резекция нижней горизонтальной ветви двенадцатиперстной кишки в области второго образования произведена с иссечением всех слоев кишечной стенки. Дефект ушит однорядным швом. Наибольший опухолевый узел тонкой кишки удален путем полнослойной резекции, с отступом 1 см от видимых границ опухоли. Оставшиеся 5 опухолевых узлов удалены аналогичным способом. Дефекты ушиты однорядным швом.
Дефекты ушиты однорядным швом.
Длительность операции составила 100 мин. Объем кровопотери 500 мл. По данным морфологического и иммуногистохимического исследований, удаленные образования являлись ГИСО тонкой кишки (экспрессия CD117, CD34, alfa-smooth). Послеоперационный период протекал без осложнений, длительность послеоперационного периода составила 8 койко-дней. Пациентка выписана в удовлетворительном состоянии для продолжения терапии сунитинибом.
Обсуждение
ПН — нейрогенная опухоль, распространяющаяся вдоль нервных стволов и сдавливающая окружающие органы и ткани. ПН является относительно частым проявлением нейрофиброматоза I типа [12]. С учетом неэффективности консервативного лечения в случае выявления ПН показано хирургическое лечение [13, 14].
Мы не нашли в литературе публикаций, описывающих робот-ассистированное удаление ПН при болезни Реклингхаузена. В 2014 г. L. Shi и соавт. [15] опубликовали статью, посвященную лапароскопической атипичной резекции желудка при ПН, локализующейся по большой кривизне желудка. Таким образом, приведенный клинический пример удаления ПН является, по нашему мнению, первым описанием робот-ассистированного удаления опухоли данного гистологического типа.
Таким образом, приведенный клинический пример удаления ПН является, по нашему мнению, первым описанием робот-ассистированного удаления опухоли данного гистологического типа.
Шваннома — это доброкачественная опухоль, произрастающая из миелиновой оболочки нервных стволов [16]. Наиболее частой локализацией шванном являются мягкие ткани головы и шеи, средостение и мягкие ткани конечностей. Локализация опухолей данного гистологического типа в забрюшинном пространстве является крайне редкой и определяется в 0,5—3% случаев [11]. В литературе [17] описано приблизительно 60 случаев хирургического лечения шванном, локализованных в забрюшинном пространстве.
С учетом развития робот-ассистированной хирургии мы нашли описания 4 РАО по удалению шванном [11, 17—19]. Однако ни в одном из них шваннома не была ассоциирована с болезнью Реклингхаузена. В связи с этим можно считать клинический пример 2 робот-ассистированного удаления шванномы первым описанием хирургического, и в частности миниинвазивного лечения шванномы, ассоциированной с нейрофиброматозом I типа.
По данным проспективного исследования L. Képénékian и соавт. [20], феохромоцитома манифестирует при нейрофиброматозе I типа в 0,1—5,7% случаев. Описано несколько случаев лапароскопической адреналэктомии при феохромоцитоме, ассоциированной с болезнью фон Реклингхаузена [21—24].
Несмотря на доказанную эффективность и безопасность робот-ассистированной адреналэктомии [25], мы не нашли упоминаний о проведении робот-ассистированной адреналэктомии у пациента с нейрофиброматозом I типа.
В ряде публикаций (описания клинических наблюдений и короткие серии наблюдений) описывается связь между появлением ГИСО и нейрофиброматозом I типа [8, 9]. В 2016 г. Toshirou Nishida и соавт. [26] опубликовано ретроспективное мультицентровое когортное исследование по выявлению ГИСО в группе пациентов с болезнью Реклингхаузена. По данным авторов, ГИСО выявляется у 6,3% пациентов с диагностированным ранее нейрофиброматозом I типа. При этом мутаций в генах KIT и PDGFRA, присутствующих у большей части больных ГИСО, у пациентов с нейрофиброматозом I типа не выявлено. Для пациентов с болезнью Реклингхаузена также характерно появление множественных ГИСО.
Для пациентов с болезнью Реклингхаузена также характерно появление множественных ГИСО.
Клинические проявления болезни фон Реклингхаузена приведены в таблице
Частота встречаемости основных клинических проявлений нейрофиброматоза I типа [7].
Использование роботического комплекса дает хирургу ряд хорошо известных преимуществ: трехмерное изображение операционного поля, отсутствие эффекта преломления, 7 степеней свободы движения манипуляторов и т. д. Эти факторы обеспечивают высокий уровень удобства, безопасности, прецизионности и эргономичности при выполнении робот-ассистированных операций [18, 27—33]. В то же время при множественной локализации образований в различных участках брюшной полости и забрюшинного пространства возникает необходимость в интраоперационной переустановке роботических портов, а также в смене расположения роботического комплекса, что значительно увеличивает длительность и трудоемкость оперативного вмешательства [27—30].
Таким образом, проведение РАО в лечении проявлений нейрофиброматоза I типа в забрюшинном пространстве и брюшной полости безопасно и эффективно у пациентов с болезнью Реклингхаузена при наличии единичных образований. При наличии множественных опухолей, расположенных в разных отделах забрюшинного пространства или брюшной полости, целесообразнее выбирать традиционный вариант оперативного вмешательства.
При наличии множественных опухолей, расположенных в разных отделах забрюшинного пространства или брюшной полости, целесообразнее выбирать традиционный вариант оперативного вмешательства.
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflicts of interest.
Сведения об авторах
Кригер Андрей Германович — д.м.н., профессор; https://orcid.org/0000-0003-4539-9943
Берелавичус Станислав Валерьевич — д.м.н.; https://orcid.org/0000-0001-8727-6111
Стручков Владимир Юрьевич — аспирант; https://orcid.org/0000-0003-1555-1596
Сон Андрей Ильич — к.м.н.; https://orcid.org/0000-0003-0332-0360
Эффективность комплексной методики лечения больных нейрофиброматозом I типа (болезнью Реклингхаузена)
1. Бадалян JI.O., Таболин В.А., Вельтищев Ю.Е. Наследственные болезни у детей // М.: Медицина, 1971. 368с.
2. Балязин В.А., Кравченко М.И., Фомина-Черноусова Н А. Нейрокожные синдромы: клиника, диагностика // М., Элиста: АПП «Джангар». 2001.- 96 с.
3. Бочков Н.П., Мордовцев В Н., Яхно Н.Н. Нейрофиброматоз: состояние проблемы, нерешённые вопросы и пути изучения // Вестн. Дерматол. Венерол. 1993. — N4,- С. 14-18
4. Возианов А.Ф., Бутенко А К., Зак К.П. Цитокины: биологические и противоопухолевые свойства Киев, 1998.
5. Дейл М.М., Формен Дж.К. Руководство по иммунофармакологии.- М., Медицина, 1998.
6. Жибург Е.Б., Серебраная Н.Б., Каткова И.В., Дьякова В В. Цитокины в кроветворении, иммуногенезе и воспалении // Физиология. 1996. — N3. — С.38-40.
7. Каталинич Д А. Хирургическое лечение нейрофиброматоза с помощью лазера // Хирургия. 1996. — N5. — С.52-54.
8. Мордовцев В Н., Мордовцева В В., Филиппова М.Г., Мозолевский Ю.В. Ранняя диагностика нейрофиброматоза 1 типа- основа профилактики осложнений системной патологии // Русский Медицинский Журнал. 1997. — Т. 5, N11. — С.684-692.
5, N11. — С.684-692.
9. Мордовцев В.Н., Кешилева З.Б., Сергеев А С. Генетика в дерматологии. Алматы: Медицина баспасы, 2001. — 184 с.
10. Мордовцев В.Н., Суворова К.Н. Наследственные заболевания кожи. Алматы: Казахстан, 1995. — 544с.
11. Мордовцева В.В. Клиническая гетерогенность нейрофиброматоза 1 типа и прогностическая значимость доминирования отдельных признаков // Тез. докл. VII Росс, съезда дерматол. и венерол. -Казань: Медицина. 1996,- С. 161.
12. Н.Мордовцева В.В., Мордовцев В.Н., Филиппова М.Г., Старков И.В. О клиническом полиморфизме нейрофиброматоза 1 типа // Вестн. Дерматол. Венерол. 1997.-N5.- С.40-42.
13. Мордовцева В.В., Гетлинг З.М., Вавилов A.M., Мордовцев В.Н. Ультраструктурные характеристики нейрофибром на разных стадиях развития // Вестн. Дерматол. Венерол. 1998. — N2. -С.38-41.
14. Печатников J1.M., Жиляев Е.В., Золотова J1.B., Калачева И М. Об аутоиммунной патологии при нейрофиброматозе Реклингхаузена // Клин. мед. 1989. — N8. — С. 97-99.
97-99.
15. Плохинский Н И. Биометия. М.: МГУ, 1970. — 367 с.
16. Садовская Ю.Е. Компьютерная томография и электромиография в диагностике нейрофиброматоза // Сб. Всесоюзн. науч.-практ. конф. по детской неврологии. Вильнюс, 1989. — С.72-73.
17. Садовская Ю.Е. Компьютерно-томографический анализ нейрофиброматоза у детей // Клинический вестник. 1996. — N3 С.20-21.
18. Садовская Ю.Е. Нейрофиброматоз: клиническое, компьютерно-томографическое и электронейрофизиологическое исследование: Дис. . канд. мед. наук. М., 1990. — 153с.
19. Садыкова Х.К. Клиника, диагностика и патоморфология нейрофиброматоза челюстно-лицевой области: Автореф. дис. канд. мед. наук. М., 1991. — 25с.
20. Суворова К.Н., Антоньев А.А. Наследственные дерматозы М.: Медицина, 1977. — 231с.
21. Суколин Г.И. Клиническая дерматология. С.-Пб.: Гарт-Курсив,1997. 384с.
22. Табакова Л.И. Состояние ЦНС, системы метаболизма кальция при нейрофиброматозе у детей и патогенетическое обоснование путей их терапевтической коррекции: Дис. канд. мед. наук. М.,1998. 162с.
канд. мед. наук. М.,1998. 162с.
23. Турусов B.C. и др. Болезнь Фон Реклингхаузена. Экспериментальные модели и сравнительные аспекты // Арх. Патологии. 1996. — Т.58, N5. — С.3-13.
24. Фармакотерапия в дерматологии. / Под ред. Мордовцева В Н., Кешилевой З.Б. Алмааты: Казахстан, 1994. 352с.
25. Филиппова М.Г., Мазолевский Ю.В., Паренькова ТВ. Варьирующая экспрессия гена нейрофиброматоза // Генетика.-1994. Т.30. — С.166.
26. Фицпатрик Т., Джонсон Р., Вульф К. и др. Дерматология: Атлас-справочник. Пер. с англ. — М.: Практика. — 1999. — 1088с.
27. Цветкова Г.М., Мордовцева В.В., Вавилов A.M., Мордовцев В Н. Патоморфология болезней кожи. М.: Медицина, 2003. — 495с.
28. Ярилин А.А. Основы иммунологии. -М.: Медицина, 1999.
29. Barker D., Wright Е., Ngugen К. et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17 // Science. 1987. — Vol. 236,- P.1100-1102.
30. Camisa Ch., Eisenstat В., Ragaz A. Functions of some vitamins // J Amer Dermatol. -1982. Vol.6. — 620p.
-1982. Vol.6. — 620p.
31. Carlessimo O.A., Bottoni U., Innocenzi D. et al. Modulating activity of retinoids on «cutaneous homing» phenomena // Current aspects of retinoid therapy. Proc. the satellite symp. I Congr. Europ. Acad. Dermatol. 1989. — 17.
32. Catalogue. Novocastra Laboratories Ltd 2002. 184p.
33. Catalogue. DAKO. 2002. 310p.
34. Croix B.S., Rak J.W., Kapitain S. Reversal by hyaluronidase of adhesion-dependent multicellular drug resistance in mammary carcinoma cells // J. Natl. Cancer Inst. 1996. — Vol.88, N18. -P. 1285-1296.
35. Farram E., Nelson D.S. Mouse mast cells as anti-tumor effector cells II Cell Immunol. 1980. — Vol.55. — P.294-301.
36. Filion M.C., Philips N.C. // J Pharm Pharmacol. 2001. — Vol.53, N4. — P.555-561.
37. Fontana J.A., Burrows-Mezu A., Clemmons D.R., LeRoith D. Retinoid modulation of insulin-like growth factor-binding proteins and inhibition of breast carcinoma proliferation // Endocrinology. -1991. Vol.128, N2. — P. 1115-1122.
— P. 1115-1122.
38. Gerosa P.L., Bizzozero L., Fontana A. Immune reaction in von Recklinghausen’s neurofibromatosis II Minerva Medica. 1991. -Vol.82, N10. — P.613-625
39. Gerosa P.L., Spinelli M., Tannarelli S. et al. Tumour suppressor genes, immunology and local manifestations of neurofibromatosis phenotypes II Panminerva Med. 1996. — Vol.38, N3. — P.157-163.
40. Giorno R., Claman H.N. Mast cells and neurofibromatosis // Neurofibromatosis. 1988. -Vol.1, N2. — P.100-104.
41. Giorno R., Lieber J., Claman H.N. Ultrastructural evidence for mast cell activation in a case of neurofibromatosis // Neurofibromatosis. 1989. — Vol.2, N1. — P.35-41.
42. Gutmann D.H., Wood D.L., Collins F.S. Identification of the neurofibromatosis type 1 gene product // Proc. Nat. Acad. Sci. -1991. Vol.88. — P.9658-9662.
43. Goldberg N.S., Collins F.S. The hunt for the neurofibromatosis gene // Arch. Dermatol. Res. 1991. — Vol.127, N11. — P. 11705-11707.
44. Goldberg Y., Dibbern K. , Klein J. et al. Neurofibromatosis type 1 -an update and review for the primary pediatrician // J. Clinical Pediatrics. November 1996. — P.545-561
, Klein J. et al. Neurofibromatosis type 1 -an update and review for the primary pediatrician // J. Clinical Pediatrics. November 1996. — P.545-561
45. Hirota S., Nomurra S., Asada H. et al. Possible involvement of c-kit receptor and its ligand in increase of mast cells in neurofibroma tissue // Arch Pathol Lab Med. — 1993. — Vol.117, N10. — P.996-999,
46. Jaakkola S., Peltonen J., Riccardi V. et al. Type 1 neurofibromatosis: selective expression of extracellular matrixgenes by Schwann cells, perineuria! cells, and fibroblasts in mixed cultures // J. Clin. Invest. 1989. — Vol.84, N1. — P.253-261.
47. Johnson M.D., Kamso-Pratti J., Federspiel C.F. et al. Mast cell ad lymphoreticular infiltrates in neurofibromas (comparison with nerve sheath tumors) // Arch Pathol Lab Med. — 1989. — Vol.113, N11. -P.1263-1270.
48. Jurecka W. Plexiforme neurofibroma of the skin // Am. J. Dermatopathol. 1988.-Vol.10, N.3. — P. 209-216.
49. Katayama I., Yokozeki H., Nishioka K. Mast-cell-derived mediators induce epidermal cell proliferation: clue for lichenified skin lesion formation in atopic dermatitis // Int. Arch. Allergy Immunol. 1992 Vol.98, N4. — P.410-414.
Arch. Allergy Immunol. 1992 Vol.98, N4. — P.410-414.
50. Kaufmann D., Wiandt S., Veser J., Krone W. Increased melanogenesis in cultured epidermal melanocytes from patients with neurofibromatosis 1 (NF 1) // Hum. Genet. 1991. — Vol.87, N2. -P.144-150.
51. Korf B.R. Diagnostic outcome in children with multiple cafe-au-lait spots // Pediatrics. — 1992. — Vol.909, N6. — P.924-927.
52. Lazaro C., Gaona A., Ravella A. et al. Prenatal diagnosis of neurofibromatosis type 1: from flanking RFLPs to intragenic microsatellite markers // Prenat. Diagn. 1995. — Vol.15, N2. -P.129-134.
53. Ledbetter D.H., Rich D.C., O’Connell P. et al. Precise localization of NF1 to 17q 11.2 by balanced translocation // Am. J. Hum. Genet. 1989. — Vol.44, N1. — P.20-24.
54. Lee Choi K., Claman H.N. Mast cells, fibroblasts and fibrosis: New clues to the riddle of mast cells // Immunol Res. 1987. — Vol.6. -P.145-152.
55. Marchuk D.A., Saulino A.M., Tavakkol R. et al., cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1 gene product // Genomics. — 1991. N.ll. — P. 931-940.
— 1991. N.ll. — P. 931-940.
56. Matsui I., Tanimura M., Kobayashi N. et al. Neurofibromatosis type 1 and childhood cancer 11 Cancer.- 1993. Vol.72, N.3. -P.2746-2754.
57. McKusick V.A. Mendelian inheritance in man. 10-th Ed. -Baltimore, 1992.- Vol. 1-2. — 2320 p.
58. Memorandum WHO/NNFF // Bulletin of the World Health Organization. 1992. — Vol.70, N2 — P.173-82.
59. Mordovtseva V.V., Mordovtsev V.N., Filippova M.G. Neurofibromatosis: un desafio clinicopatogenico que persiste // Dermatologia & Cosmetica. 1997. — V.U. — P.297-304.
60. Mori A., Nakayama K., Suzuki J. et al. Analysis of stem cell factor for mast cell proliferation in the human myometrium // Mol. Hum. Reprod. 1997. — Vol.3, N5. — P411-418.
61. North K. Neurofibromatosis type 1: review of the first 200 patients in an Australian clinic // J. Child Neurol. 1993. — Vol.8, N4. P.395-402.
62. Norton K.K., Mahadeo D.K., Geist R.T., Gutmann D.H. Expression of the neurofibromatosis 1 (NF1) gene during growth arrest // Neuroreport. 1996. Vol.7, N2. P.601-604.
1996. Vol.7, N2. P.601-604.
63. Pauli B.U., Schwartz D.E., Thonar E.J., Kuettner K.E. Tumor invasion and host extracellular matrix // Cancer Metastasis Rev. -1983. Vol.2, N2. — P.129-152.
64. Pigatto P.D., Fiorini A., Riva E. Vitamin A // Dermatology-1983. -Vol.167. P.16-18.
65. Rasouli I., Mehta L.N. Reaction of peripheral nerves to vascular and bacterial injuries // Indian J. Lepr. 1992. — Vol.64, N1. — P. 14-27.
66. Riccardi V.M Cutaneous manifestation of neurofibromatosis: cellular interaction, pigmentation, and mast cells. // Birth Defects. 1981. V. 17. N2. Pp. 129-145.
67. Riccardi V.M. Mast-cell stabilization to decrease neurofibroma growth: preliminary experience with ketotifen // Arch. Dermatol. -1987. Vol.123, N8. — P.1011-1016.
68. Riccardi V.M., Powell P.P. Neurofibrosarcoma as a complication of von Reclinghausen neurofibromatosis // Neurofibromatosis. 1989. — Vol.2, N.3. — P.152-165.
69. Riccardi V.M., Eichner J.E. Neurofibromatosis: Phenotype, Natural History and Pathogenesis. Baltimore. Johns Hopkins University Press, 1992.
Baltimore. Johns Hopkins University Press, 1992.
70. Riccardi V.M. A controlled multiphase trial of ketotifen to minimize neurofibroma associated pain and itching // Arch. Dermatol. 1993.- Vol.129, N.5. P.577-581.
71. Riccardi V.M. Skin, blood, nerve cells, and heritability. New lessons from neurofibromatosis type 1 // Arch-Dermatol. 1995. -Vol.131, N.8. — 944p.
72. Roche W.R. Mast cells and tumour angiogenesis: the tumour-mediated release of an endothelial growth factor from mast cells // Int. J. Cancer. 1985. — Vol.36, N.6. — P.721-728.
73. Ryan J.J. Stem cell factor in mast cell and Schwann cell proliferation and hyperplasia // Diss. Abstr. Int. B. 1993. -Vol.54, N. 1. — 39p.
74. Sanguinetti C., Greco F., De Palma L. The ultrastructure of peripheral neurofibroma: the role of mast cells and their interaction with perineurial cells // Ital. J. Orthop. Traumatol. 1992. -Vol.18, N.2. — P.207-216.
75. Sempowski G.D., Derdak S., Phipps R.P. Interleukin-4 and interferon-gamma discordantly regulate collagen biosynthesis by functionally distinct lung fibroblast subsets // J. Cell Physiol. -1996. Vol.167, N.2. — P.290-296.
Cell Physiol. -1996. Vol.167, N.2. — P.290-296.
76. Scheithauer W., Temsch E. M., Stefenelli N., Lathan B. In vitro evaluation of the anticancer drug modulatory effect of hyaluronidase in human gastrointestinal cell lines // Anticancer Res.- 1988. Vol. 8, N.3. — P.391-395.
77. Schwartz D.M., Shuster S., Jumper M.D. et al., Human vitreous hyaluronidase: isolation and characterization // Curr. Eye Res. -1996,- Vol.15, N. 12,- P.1156-1162.
78. Smith T.W.,Bhawan J. Tactile-like structures in neurofibromas, an ultrastructural study // Acta Neuropathol. 1980. — Vol.50, N.3 -P.233-236.
79. Thiruvengadam R., Moran E. Hyaluronidase on chemoresistant malignancies // Proc. Annu. Meet. Am. Soc. Clin. Oncol 1995,-N. 14. — P.A1385.
80. Tobler A., Dawson M.I., Koeffler H.P. Retinoids. Structure-function relationship in normal and leukemic hematopoiesis in vitro // J. Clin. Invest. 1986. — Vol.78, N.l. — P.303-309.
81. Trautmann A., Feuerstein В., Ernst N., Brocker E. B., Klein C.E. Heterotypic cell-cell adhesion of human mast cells to fibroblasts // Arch. Dermatol. Res. 1997. — Vol.298, N.4. — P. 194-203.
B., Klein C.E. Heterotypic cell-cell adhesion of human mast cells to fibroblasts // Arch. Dermatol. Res. 1997. — Vol.298, N.4. — P. 194-203.
82. Zwarthoff E C. Neurofibromatosis and associated tumour suppressor genes // Pathol. Res. Pract. 1996. — Vol.192, N.7. — P.647-57.
| Краткое содержание | Задний план: Нейрофиброматоз 1 типа (NF1) — это генетическое заболевание, при котором пациенты находятся в группе повышенного риска. развивающихся опухолей (обычно незлокачественных) центральной и периферической нервной системы. Заболевание поражает практически все системы органов. Естественное течение NFI с течением времени плохо изучено. Для большинства пациентов единственный вариант лечения — хирургическое вмешательство. Лучшее понимание NF1 может быть полезно для разработки будущие исследования лечения. Цели: Оценить людей с NF1 более 10 лет, чтобы лучше понять естественную историю болезни. | Подробное описание | ЗАДНИЙ ПЛАН: Нейрофиброматоз 1 типа (NF1) — аутосомно-доминантное прогрессирующее генетическое заболевание. характеризуется разнообразными клиническими проявлениями. Пациенты с NF1 имеют повышенный риск развивающиеся опухоли центральной и периферической нервной системы, в том числе плексиформные нейрофибромы (PN), нейрофибромы дермы, опухоли зрительного пути, опухоли головного мозга, злокачественные опухоли оболочек периферических нервов (MPNST), ювенильный миеломоноцитарный лейкоз и феохромоцитомы. | Приемлемость | Метод отбора проб: Невероятностная выборка Критерии: — КРИТЕРИИ ПРИЕМЛЕМОСТИ ПАЦИЕНТА КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. возраст: — Возраст менее или равный 35 годам для новых пациентов, обследованных в NIH. — Нет верхнего возрастного ограничения для пациентов, ранее участвовавших в клинических испытаниях в NIH или для пациентов с диагнозом MPNST, или с клиническими проблемами для MPNST, или с нечастые или необычные проявления, связанные с NF1. Пол: Все Минимальный возраст: Нет данных Максимальный возраст: Нет данных Здоровые волонтеры: Нет |
|---|
 Чтобы охарактеризовать популяцию пациентов и изучить, как NFI влияет на качество пациентов. жизнь и функции. Право на участие: Дети, подростки и взрослые с NF1. Дизайн: Участники проходят комплексную базовую оценку, включая генетическое тестирование, опухоль. визуализация, оценка боли и качества жизни, нейропсихологические, моторные и эндокринные оценки. Пациенты наблюдаются каждые 6 месяцев — каждые 3 года, в зависимости от их индивидуальных особенностей. результаты базового исследования. При необходимости тесты могут включать следующее: — История болезни, медицинский осмотр и анализы крови. — Фотосъемка всего тела и лица для отслеживания видимых деформаций. — Нейропсихологическое тестирование, оценка качества жизни, тесты двигательных функций, эндокринологические исследования, функциональные тесты сердца и легких, тесты на слух, плотность костей сканирование и другие оценки костей. — МРТ и ПЭТ для обнаружения и оценки плексиформных нейрофибром (опухолей, возникающих из нервы и могут вызвать серьезные проблемы), параспинальные нейрофибромы (опухоли, возникающие из нервы вокруг позвоночника и могут вызывать проблемы, сдавливая спинной мозг), и злокачественные опухоли оболочек периферических нервов (тип рака, возникающий из нерв или вовлекает оболочку, покрывающую нерв).
Чтобы охарактеризовать популяцию пациентов и изучить, как NFI влияет на качество пациентов. жизнь и функции. Право на участие: Дети, подростки и взрослые с NF1. Дизайн: Участники проходят комплексную базовую оценку, включая генетическое тестирование, опухоль. визуализация, оценка боли и качества жизни, нейропсихологические, моторные и эндокринные оценки. Пациенты наблюдаются каждые 6 месяцев — каждые 3 года, в зависимости от их индивидуальных особенностей. результаты базового исследования. При необходимости тесты могут включать следующее: — История болезни, медицинский осмотр и анализы крови. — Фотосъемка всего тела и лица для отслеживания видимых деформаций. — Нейропсихологическое тестирование, оценка качества жизни, тесты двигательных функций, эндокринологические исследования, функциональные тесты сердца и легких, тесты на слух, плотность костей сканирование и другие оценки костей. — МРТ и ПЭТ для обнаружения и оценки плексиформных нейрофибром (опухолей, возникающих из нервы и могут вызвать серьезные проблемы), параспинальные нейрофибромы (опухоли, возникающие из нервы вокруг позвоночника и могут вызывать проблемы, сдавливая спинной мозг), и злокачественные опухоли оболочек периферических нервов (тип рака, возникающий из нерв или вовлекает оболочку, покрывающую нерв).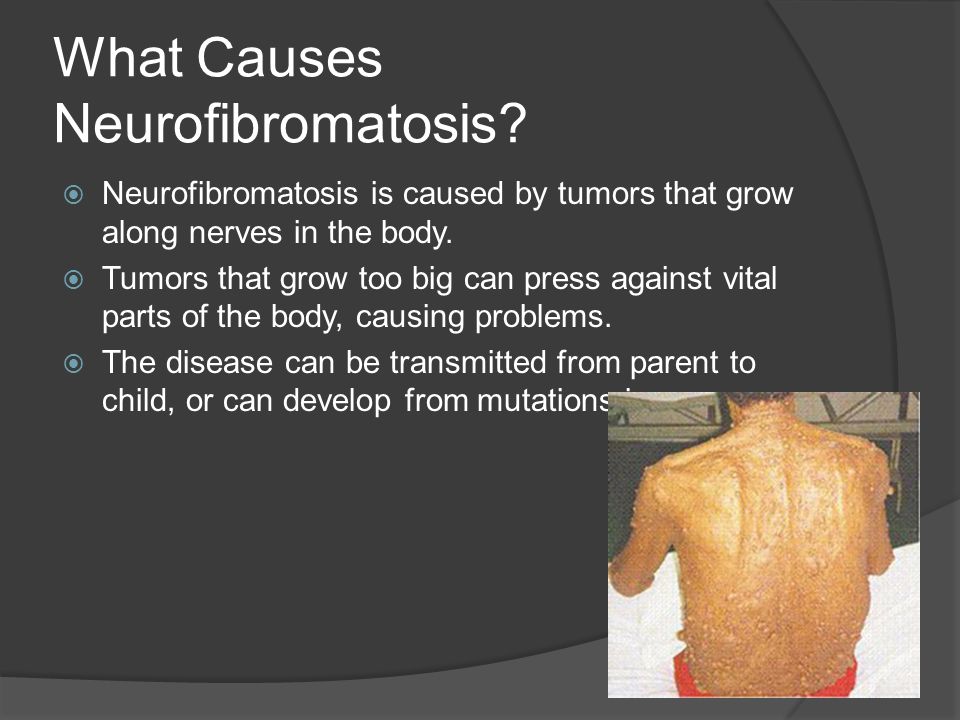 — Осмотр глаз, МРТ и ПЭТ для оценки глиом зрительного пути (возникающих опухолей от зрительных нервов или областей мозга для зрения) и химических веществ в опухоли и мозг. — Осмотр глаз и фотографии для оценки развития узелков Лиша (незлокачественные опухоли на глазу). — Фотографии кожных нейрофибром (опухолей кожи), пятен с молоком (темные или пигментированные участки на коже, которые часто являются первыми признаками NF1) и другой кожи проблемы. — Оценка боли для отслеживания различных типов боли, которую испытывают пациенты, причин боль, как часто возникает боль, влияние боли на качество жизни и что эффективны обезболивающие и альтернативные методы лечения, такие как иглоукалывание.
— Осмотр глаз, МРТ и ПЭТ для оценки глиом зрительного пути (возникающих опухолей от зрительных нервов или областей мозга для зрения) и химических веществ в опухоли и мозг. — Осмотр глаз и фотографии для оценки развития узелков Лиша (незлокачественные опухоли на глазу). — Фотографии кожных нейрофибром (опухолей кожи), пятен с молоком (темные или пигментированные участки на коже, которые часто являются первыми признаками NF1) и другой кожи проблемы. — Оценка боли для отслеживания различных типов боли, которую испытывают пациенты, причин боль, как часто возникает боль, влияние боли на качество жизни и что эффективны обезболивающие и альтернативные методы лечения, такие как иглоукалывание.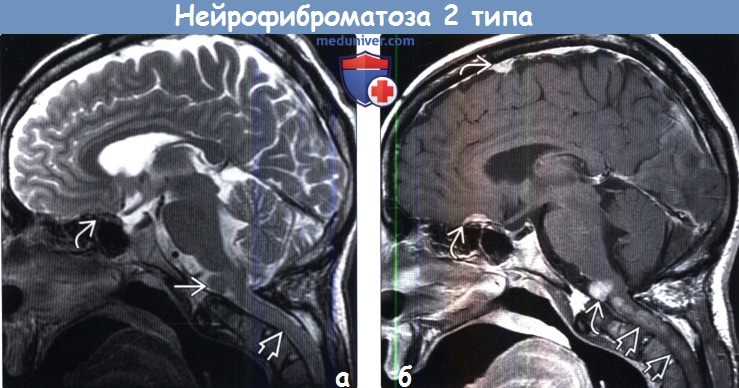 Кроме того, NF1 проявляется практически во всех системах органов, с например, скелетные и сосудистые аномалии и когнитивные нарушения. Таким образом, забота о людям с NF1 требуется мультидисциплинарный подход. Естественная история NF1 связанные опухоли и другие проявления плохо изучены, и для большинства опухолей, связанных с NF1 проявлений единственный стандартный вариант лечения — хирургическое вмешательство. Клинический центр NIH обеспечивает идеальную инфраструктуру для оценки естественного течения редких заболеваний. А лучшее понимание естественной истории опухоли, связанной с NF1, и других проявлений будет полезно для разработки исследований лечения. В НКИ, ПОБ действует активная клиническая программа испытаний опухолевых проявлений, связанных с NF1, включая PN, MPNST и в сотрудничестве с доктором Дугласом Стюартом из NHGRI, кожные нейрофибромы. В отличие от людей с рефрактерные солидные злокачественные опухоли, люди с NF1 имеют ожидаемую продолжительность жизни, близкую к нормальной, а их доброкачественные опухоли прогрессируют медленнее, чем солидные опухоли.
Кроме того, NF1 проявляется практически во всех системах органов, с например, скелетные и сосудистые аномалии и когнитивные нарушения. Таким образом, забота о людям с NF1 требуется мультидисциплинарный подход. Естественная история NF1 связанные опухоли и другие проявления плохо изучены, и для большинства опухолей, связанных с NF1 проявлений единственный стандартный вариант лечения — хирургическое вмешательство. Клинический центр NIH обеспечивает идеальную инфраструктуру для оценки естественного течения редких заболеваний. А лучшее понимание естественной истории опухоли, связанной с NF1, и других проявлений будет полезно для разработки исследований лечения. В НКИ, ПОБ действует активная клиническая программа испытаний опухолевых проявлений, связанных с NF1, включая PN, MPNST и в сотрудничестве с доктором Дугласом Стюартом из NHGRI, кожные нейрофибромы. В отличие от людей с рефрактерные солидные злокачественные опухоли, люди с NF1 имеют ожидаемую продолжительность жизни, близкую к нормальной, а их доброкачественные опухоли прогрессируют медленнее, чем солидные опухоли. Таким образом, лица с NF1 могут участвовать в нескольких испытаниях лечения. ЗАДАЧИ: Общая цель этого описательного исследования естественной истории NF1 — служить зонтиком протокол продолжающейся программы клинических испытаний NF1, позволяющий проводить продольную оценку лица с NF1 для опухоли, связанной с NF1, и неопухолевых проявлений, независимо от того, в настоящее время они участвуют в исследовании лечения или нет, и для разработки лучшего понимание биологии проявлений, связанных с NF1. После этих пациентов продольное исследование позволит исследователям лучше понять естественные история этих проявлений, обеспечивают основу для разработки конечных точек для клинические испытания и потенциально разработать более эффективные методы лечения. Проявления NF1, которые будут отслеживаться в долгосрочном плане, включая PN, MPNST, опухоли зрительного пути, кожные нейрофибромы, боль, связанная с NF1, и нейропсихологические, моторные и эндокринные функции. А будет разработан комплексный план лечения и рекомендации, которые будут доведены до сведения пациенты и лица, осуществляющие первичный уход.
Таким образом, лица с NF1 могут участвовать в нескольких испытаниях лечения. ЗАДАЧИ: Общая цель этого описательного исследования естественной истории NF1 — служить зонтиком протокол продолжающейся программы клинических испытаний NF1, позволяющий проводить продольную оценку лица с NF1 для опухоли, связанной с NF1, и неопухолевых проявлений, независимо от того, в настоящее время они участвуют в исследовании лечения или нет, и для разработки лучшего понимание биологии проявлений, связанных с NF1. После этих пациентов продольное исследование позволит исследователям лучше понять естественные история этих проявлений, обеспечивают основу для разработки конечных точек для клинические испытания и потенциально разработать более эффективные методы лечения. Проявления NF1, которые будут отслеживаться в долгосрочном плане, включая PN, MPNST, опухоли зрительного пути, кожные нейрофибромы, боль, связанная с NF1, и нейропсихологические, моторные и эндокринные функции. А будет разработан комплексный план лечения и рекомендации, которые будут доведены до сведения пациенты и лица, осуществляющие первичный уход. ПРАВО: Дети, подростки и взрослые с подтвержденным клиническим диагнозом NF1 или подтвержденным Мутация NF1. ДИЗАЙН: Будут предприняты попытки провести комплексную базовую оценку для всех людей. включая клиническое фенотипирование, генотипирование, визуализацию проявлений опухоли и боли, качество жизни, нейропсихологические, моторные и эндокринные оценки. Проявления NF1 будет проводиться долгосрочный мониторинг с периодичностью от одного года до трех лет, с объем и сроки последующих оценок в зависимости от исходных данных.
ПРАВО: Дети, подростки и взрослые с подтвержденным клиническим диагнозом NF1 или подтвержденным Мутация NF1. ДИЗАЙН: Будут предприняты попытки провести комплексную базовую оценку для всех людей. включая клиническое фенотипирование, генотипирование, визуализацию проявлений опухоли и боли, качество жизни, нейропсихологические, моторные и эндокринные оценки. Проявления NF1 будет проводиться долгосрочный мониторинг с периодичностью от одного года до трех лет, с объем и сроки последующих оценок в зависимости от исходных данных. 2.Диагностика: пациенты, которым был поставлен диагноз NF1 с помощью консенсусной конференции NIH критериев или иметь подтвержденную мутацию NF1 с анализом, проведенным в сертифицированном CLIA Лаборатория.Тест на мутацию NF1 для подтверждения соответствия критериям отбора не будет. протоколом, но в рамках отдельного скринингового исследования. Гистологическое подтверждение NF1. родственных доброкачественных опухолях не требуется при наличии устойчивых клинических и рентгенографические данные, но требуется для людей с MPNST, которые записываются на это исследование. Для клинического диагноза NF1 у всех испытуемых должно быть не менее двух или более диагностические критерии для NF1, перечисленные ниже (Консенсусная конференция NIH): 1. Шесть или более пятен кофе с молоком (больше или равно 0,5 см в препубертатном периоде). субъектов или больше или равно 1,5 см в постпубертатном периоде). 2. Больше или равно 2 нейрофибромам или 1 плексиформной нейрофиброме. 3. Шорох в подмышечной впадине или паху.
2.Диагностика: пациенты, которым был поставлен диагноз NF1 с помощью консенсусной конференции NIH критериев или иметь подтвержденную мутацию NF1 с анализом, проведенным в сертифицированном CLIA Лаборатория.Тест на мутацию NF1 для подтверждения соответствия критериям отбора не будет. протоколом, но в рамках отдельного скринингового исследования. Гистологическое подтверждение NF1. родственных доброкачественных опухолях не требуется при наличии устойчивых клинических и рентгенографические данные, но требуется для людей с MPNST, которые записываются на это исследование. Для клинического диагноза NF1 у всех испытуемых должно быть не менее двух или более диагностические критерии для NF1, перечисленные ниже (Консенсусная конференция NIH): 1. Шесть или более пятен кофе с молоком (больше или равно 0,5 см в препубертатном периоде). субъектов или больше или равно 1,5 см в постпубертатном периоде). 2. Больше или равно 2 нейрофибромам или 1 плексиформной нейрофиброме. 3. Шорох в подмышечной впадине или паху.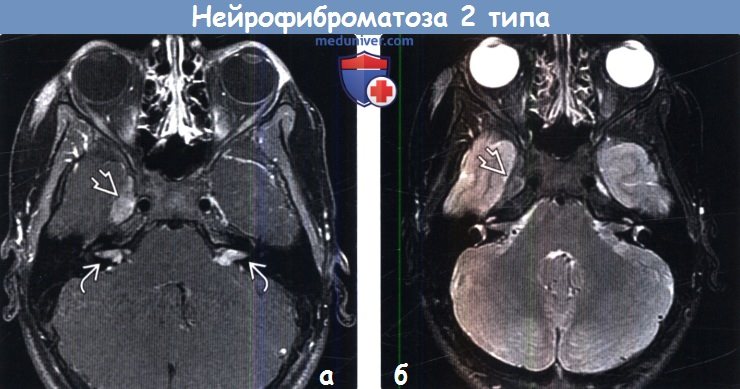 4. Оптическая глиома. 5. Два или более узелка Лиша. 6. отчетливое костное поражение (дисплазия клиновидной кости или дисплазия или истончение коры длинных костей). 7. родственник первой степени с NF1. 3. Предварительная и текущая терапия: для проявлений доброкачественных опухолей, связанных с NF1, нет стандартное эффективное лечение, и хирургическое вмешательство является единственным стандартным лечением. Химиотерапия и лучевая терапия — дополнительные варианты лечения злокачественного NF1. связанные опухоли. Для целей данного исследования субъекты, которые ранее не получившие медицинское или хирургическое лечение, пациенты, которые ранее получали медицинское или хирургическое лечение, и пациенты, которые в настоящее время проходят лечение и / или облучение для проявления, связанного с NF1, будет иметь право. лечение проявлений, связанных с NF1, будет записано при входе в исследование и на протяжении всего исследования. 4. Статус выполнения: ECOG меньше или равен 3. Субъекты, привязанные к инвалидной коляске.
4. Оптическая глиома. 5. Два или более узелка Лиша. 6. отчетливое костное поражение (дисплазия клиновидной кости или дисплазия или истончение коры длинных костей). 7. родственник первой степени с NF1. 3. Предварительная и текущая терапия: для проявлений доброкачественных опухолей, связанных с NF1, нет стандартное эффективное лечение, и хирургическое вмешательство является единственным стандартным лечением. Химиотерапия и лучевая терапия — дополнительные варианты лечения злокачественного NF1. связанные опухоли. Для целей данного исследования субъекты, которые ранее не получившие медицинское или хирургическое лечение, пациенты, которые ранее получали медицинское или хирургическое лечение, и пациенты, которые в настоящее время проходят лечение и / или облучение для проявления, связанного с NF1, будет иметь право. лечение проявлений, связанных с NF1, будет записано при входе в исследование и на протяжении всего исследования. 4. Статус выполнения: ECOG меньше или равен 3. Субъекты, привязанные к инвалидной коляске. из-за паралича будут считаться амбулаторными, когда они находятся в своем Инвалидная коляска. Субъекты должны иметь возможность поехать в NIH для оценки. 5. Информированное согласие: все пациенты или их законные опекуны (если пациент младше 18 лет) должен подписать одобренный IRB документ об информированном согласии для демонстрации их понимание исследовательского характера и рисков этого исследования до проводятся любые исследования, связанные с протоколом. При необходимости педиатрические субъекты участвовать во всех обсуждениях. 6. Долговременная доверенность (DPA): все субъекты старше или равные 18 лет. будет предложена возможность назначить DPA, чтобы другой человек мог сделать решения об их медицинском обслуживании, если они станут недееспособными или умственно ослаблен. 7. Кроме того, субъекты, участвующие в оценке вариаций в экспрессии генов должен: — Иметь хотя бы 1 плексиформную нейрофиброму и иметь возможность пройти МРТ-анализ плексиформная нейрофиброма (ы). — Если возможно, но не обязательно, иметь одного или обоих биологических родителей (NF1 затронуты или нет) готовы сдать кровь, мазок из щеки или образец жидкости для полоскания рта для Выделение ДНК.
из-за паралича будут считаться амбулаторными, когда они находятся в своем Инвалидная коляска. Субъекты должны иметь возможность поехать в NIH для оценки. 5. Информированное согласие: все пациенты или их законные опекуны (если пациент младше 18 лет) должен подписать одобренный IRB документ об информированном согласии для демонстрации их понимание исследовательского характера и рисков этого исследования до проводятся любые исследования, связанные с протоколом. При необходимости педиатрические субъекты участвовать во всех обсуждениях. 6. Долговременная доверенность (DPA): все субъекты старше или равные 18 лет. будет предложена возможность назначить DPA, чтобы другой человек мог сделать решения об их медицинском обслуживании, если они станут недееспособными или умственно ослаблен. 7. Кроме того, субъекты, участвующие в оценке вариаций в экспрессии генов должен: — Иметь хотя бы 1 плексиформную нейрофиброму и иметь возможность пройти МРТ-анализ плексиформная нейрофиброма (ы). — Если возможно, но не обязательно, иметь одного или обоих биологических родителей (NF1 затронуты или нет) готовы сдать кровь, мазок из щеки или образец жидкости для полоскания рта для Выделение ДНК. Отдельное информированное согласие будет получено от биологической лаборатории. родители. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. По мнению исследователя, пациент не может вернуться для наблюдения. посещений или получения необходимых последующих исследований. 2. По мнению исследователя, пациент не может получить МРТ. 3. Лица, которые беременны, кормят грудью или забеременели во время регистрации на этом испытании не будут исключены из участия, но не пройдут рентгенографические оценки или МРТ, запрошенные для исследовательских целей, или другие исследования, которые могут негативно повлиять на беременность. КРИТЕРИИ ПРИЕМЛЕМОСТИ ПАЦИЕНТА НА ОПЦИОНАЛЬНУЮ ОПУХОЛЬ / ТКАНЬЮ БИОПСИЮ ДЛЯ ИССЛЕДОВАНИЯ КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. возраст старше 12 лет и нейрофиброма, макула с молоком, ксантогранулема или другой участок кожи, который легко доступен и достаточно удален от жизненно важных структуры для биопсии. 2. количество планшетов должно быть больше или равно 100000 / мкл, а PT и PTT должны иметь быть в пределах нормы в течение 1 недели после каждой биопсии.
Отдельное информированное согласие будет получено от биологической лаборатории. родители. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. По мнению исследователя, пациент не может вернуться для наблюдения. посещений или получения необходимых последующих исследований. 2. По мнению исследователя, пациент не может получить МРТ. 3. Лица, которые беременны, кормят грудью или забеременели во время регистрации на этом испытании не будут исключены из участия, но не пройдут рентгенографические оценки или МРТ, запрошенные для исследовательских целей, или другие исследования, которые могут негативно повлиять на беременность. КРИТЕРИИ ПРИЕМЛЕМОСТИ ПАЦИЕНТА НА ОПЦИОНАЛЬНУЮ ОПУХОЛЬ / ТКАНЬЮ БИОПСИЮ ДЛЯ ИССЛЕДОВАНИЯ КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. возраст старше 12 лет и нейрофиброма, макула с молоком, ксантогранулема или другой участок кожи, который легко доступен и достаточно удален от жизненно важных структуры для биопсии. 2. количество планшетов должно быть больше или равно 100000 / мкл, а PT и PTT должны иметь быть в пределах нормы в течение 1 недели после каждой биопсии.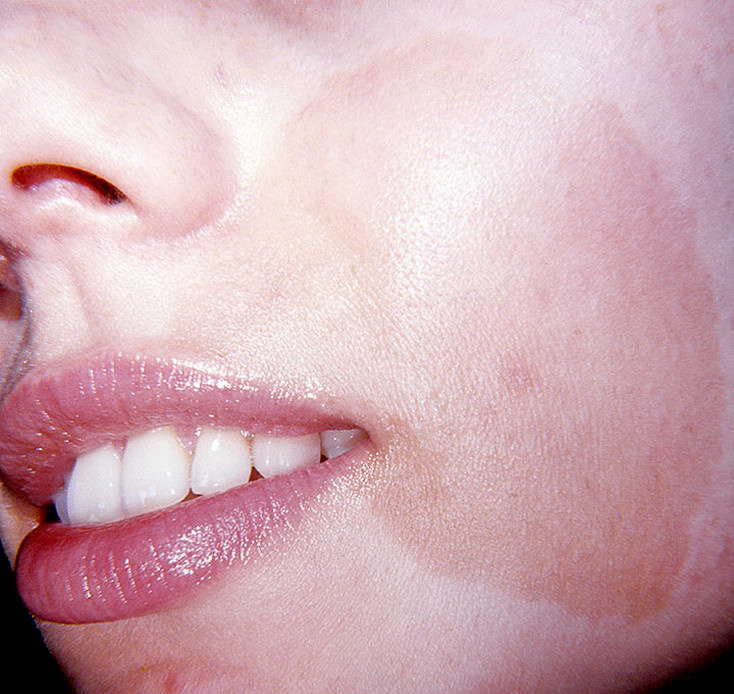 3. Субъект или родитель / опекун должен подписать отдельное согласие на биопсию, и участник, если он несовершеннолетний, должен подписать отдельное согласие с описанием биопсии. 4. Отсутствие медицинского лечения, специально направленного на опухоль, связанную с NF1, в течение шести недель до к сбору образцов. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. Биопсия не будет выполняться, если участнику требуется общая анестезия. 2. потребность в лекарствах, нарушающих функцию тромбоцитов, таких как аспирин, которую нельзя прекратить в течение 1 недели до биопсии. КРИТЕРИИ ПРАВОМОЧНОСТИ НЕЗАВИСИМОЕ ОТНОСИТЕЛЬНО БРАКОВ (НЕЙРОКОГНИТИВНАЯ ОЦЕНКА И ОЦЕНКА КАЧЕСТВА) КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. доступность родственника или сестры, не затронутого NF1, для продольной оценки нейрокогнитивная функция и оценка качества жизни. будет подготовлена форма согласия незатронутые несовершеннолетние братья и сестры, и письменное информированное согласие будет получено от братьев и сестер 18 лет и старше. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. заболевание, которое не позволяет брату или сестре участвовать в оценка нейрокогнитивной функции или качества жизни.
3. Субъект или родитель / опекун должен подписать отдельное согласие на биопсию, и участник, если он несовершеннолетний, должен подписать отдельное согласие с описанием биопсии. 4. Отсутствие медицинского лечения, специально направленного на опухоль, связанную с NF1, в течение шести недель до к сбору образцов. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. Биопсия не будет выполняться, если участнику требуется общая анестезия. 2. потребность в лекарствах, нарушающих функцию тромбоцитов, таких как аспирин, которую нельзя прекратить в течение 1 недели до биопсии. КРИТЕРИИ ПРАВОМОЧНОСТИ НЕЗАВИСИМОЕ ОТНОСИТЕЛЬНО БРАКОВ (НЕЙРОКОГНИТИВНАЯ ОЦЕНКА И ОЦЕНКА КАЧЕСТВА) КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. доступность родственника или сестры, не затронутого NF1, для продольной оценки нейрокогнитивная функция и оценка качества жизни. будет подготовлена форма согласия незатронутые несовершеннолетние братья и сестры, и письменное информированное согласие будет получено от братьев и сестер 18 лет и старше. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. заболевание, которое не позволяет брату или сестре участвовать в оценка нейрокогнитивной функции или качества жизни. КРИТЕРИИ ПРИЕМЛЕМОСТИ РОДИТЕЛЯ (РОДИТЕЛЕЙ) ПАЦИЕНТА (ИССЛЕДОВАНИЯ ГЕНЕТИЧЕСКИХ МОДИФИКАТОРОВ) КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. биологические родители (один или оба) пациентов с NF1 будут иметь право на участие, если они того пожелают. предоставить образец крови, мазка из щеки, слюны или жидкости для полоскания рта для выделения ДНК для анализа генных модификаторов. Эти люди могут быть любого пола и этнической принадлежности. согласие будет получено от каждого родителя, желающего участвовать в этой части исследования. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. Состояние здоровья, которое не позволяет родителям предоставить биологический образец. КРИТЕРИИ ПРИЕМЛЕМОСТИ РОДИТЕЛЯ (РОДИТЕЛЕЙ) ПАЦИЕНТА (АНКЕТЫ) КРИТЕРИИ ВКЛЮЧЕНИЯ: Родители (один или оба) пациентов с NF1 будут иметь право на участие, если они хотят заполнить анкеты для оценки NF1, перечисленные в разделе 1.2.8. .
КРИТЕРИИ ПРИЕМЛЕМОСТИ РОДИТЕЛЯ (РОДИТЕЛЕЙ) ПАЦИЕНТА (ИССЛЕДОВАНИЯ ГЕНЕТИЧЕСКИХ МОДИФИКАТОРОВ) КРИТЕРИИ ВКЛЮЧЕНИЯ: 1. биологические родители (один или оба) пациентов с NF1 будут иметь право на участие, если они того пожелают. предоставить образец крови, мазка из щеки, слюны или жидкости для полоскания рта для выделения ДНК для анализа генных модификаторов. Эти люди могут быть любого пола и этнической принадлежности. согласие будет получено от каждого родителя, желающего участвовать в этой части исследования. КРИТЕРИЙ ИСКЛЮЧЕНИЯ: 1. Состояние здоровья, которое не позволяет родителям предоставить биологический образец. КРИТЕРИИ ПРИЕМЛЕМОСТИ РОДИТЕЛЯ (РОДИТЕЛЕЙ) ПАЦИЕНТА (АНКЕТЫ) КРИТЕРИИ ВКЛЮЧЕНИЯ: Родители (один или оба) пациентов с NF1 будут иметь право на участие, если они хотят заполнить анкеты для оценки NF1, перечисленные в разделе 1.2.8. .Нейрофиброматоз 1 — NORD (Национальная организация редких заболеваний)
TEXTBOOKS
Short MP.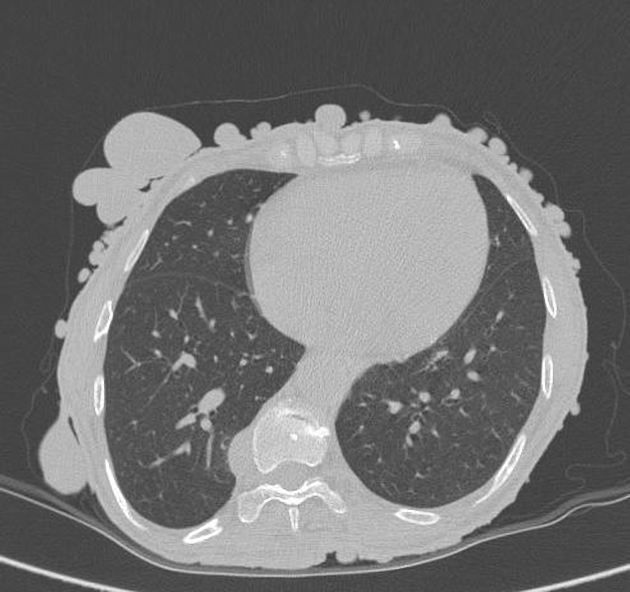 Нейрофиброматоз, тип 1. В: Справочник НОРД по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 563-64.
Нейрофиброматоз, тип 1. В: Справочник НОРД по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 563-64.
Фридман Дж.М., Гутман Д.Х., Макколлин М., Риккарди В.М. Нейрофиброматоз: фенотип, естественная история и патогенез (3-е издание). Балтимор, Мэриленд, и Лондон, Англия: издательство Университета Джона Хопкинса; 1999.
ОБЗОР СТАТЬИ
Abramowicz A, Gos M.Нейрофибромин при нейрофиброматозе 1 типа — мутации в гене NF1 как причина заболевания. Dev Period Med. 2014 июль-сентябрь; 18 (3): 297-306.
Тадини Дж., Милани Д., Менни Ф., Пеццани Л., Сабатини С., Эспозито С. Пришло время изменить диагностические критерии нейрофиброматоза 1? Eur J Intern Med. 2014 июль; 25 (6): 506-10.
Thway K, Fisher C. Злокачественная опухоль оболочки периферических нервов: патология и генетика. Ann Diagn Pathol. 2014 Apr; 18 (2): 109-16.
Кассиман К. Кегиус Э. Спилирс В. и др.Офтальмологическая оценка детей с нейрофиброматозом 1 типа. Eur J Pediatr. 2013 Октябрь; 172 (10): 1327-33.
Eur J Pediatr. 2013 Октябрь; 172 (10): 1327-33.
Фернер Ре, Гутманн DH. Нейрофиброматоз 1 типа (NF1): диагностика и лечение. Handb Clin Neurol. 2013; 115: 939-55.
Темплер AK, Titus JB, Gutmann DH. Нейропсихологический взгляд на проблемы внимания при нейрофиброматозе 1 типа. J Atten Disord. 2013 Aug; 17 (6): 489-96.
Вранчану А.М., Меркер В.Л., Парк Е. и др. Качество жизни среди взрослых пациентов с нейрофиброматозом 1, нейрофиброматозом 2 и шванноматозом: систематический обзор литературы.J Neurooncol. 2013 сен; 114 (3): 257-62.
Патель Н.Б., Стейси Г.С. Скелетно-мышечные проявления нейрофиброматоза 1 типа. AJR Am J Roentgenol. 2012 июл; 199 (1): 99-106.
Шнур РЭ. Нейрофиброматоз I типа: гено-окуло-дерматологические обновления. Curr Opin Ophthalmol. 2012 сен; 23 (5): 364-72.
Jouhilahti EM, Peltonen S, Heape AM, Peltonen J. Патоэтиология нейрофиброматоза 1. AM J Pathol. 2011 May; 178 (5) 1932-9.
Коэн Р., Шупер А. Проявления развития у детей с нейрофиброматозом 1 типа. Harefuah. 2010 Янв; 149 (1): 49-52, 61.
Harefuah. 2010 Янв; 149 (1): 49-52, 61.
Feldman DS, Jordan C, Fonseca L. Ортопедические проявления нейрофиброматоза 1 типа. J Am Acad Orthop Surg. 2010 июн; 18 (6): 346-57.
Джетт К., Фридман Дж. М.. Клинико-генетический аспект нейрофиброматоза 1. Genet Med. 2010 Янв; 12 (1): 1-11.
Альберс AC. Gutmann DH. Глиомы у пациентов с нейрофиброматозом 1 типа. Expert Rev Neurother. 2009 Apr; 9 (4): 535-9.
Элефтериус Ф, Коланчик М., Шинделер А. и др.Скелетные аномалии при нейрофиброматозе 1 типа: подходы к вариантам лечения. Am J Med Genet A. 2009 октябрь; 149А (10): 2327-38.
Лу-Эмерсон С, Плоткин СР. Нейрофиброматозы. Часть I: NF1. Rev Neurol Dis. 2009 Весна; 6 (2): 47-53.
Парсонс К.М., Кантер Р.Дж., Хатри В.П. Хирургическое лечение нейрофиброматоза. Surg Oncol Clin N Am. 2009 Янв; 18 (1): 175-96.
ИНТЕРНЕТ
Friedman JM. Нейрофиброматоз 1. 2 октября 1998 г. [Обновлено 4 сентября 2014 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al. , редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2016 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1109/ По состоянию на 3 января 2017 г.
, редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2016 гг. Доступно по адресу: https://www.ncbi.nlm.nih.gov/books/NBK1109/ По состоянию на 3 января 2017 г.
McKusick VA, Ed. Интернет-Менделирующее наследование в человеке (OMIM). Университет Джона Хопкинса. Нейрофиброматоз I типа; NF1. Регистрационный номер; 162200. Доступно на http://omim.org/entry/162200: Дата последнего редактирования 15.11.2016. По состоянию на 3 января 2017 г.
Pinson S. Neurofibromatosis type I. Orphanet. Доступно на: www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=636 Последнее обновление: июль 2014 г. По состоянию на 3 января 2017 г.
Нейрофиброматоз 1 и 2: симптомы, лечение, причины
Что такое нейрофиброматоз?
Нейрофиброматоз — генетическое заболевание нервной системы. Опухоли образуются на нервных тканях. В основном нейрофиброматозные нарушения влияют на рост и развитие ткани нервных клеток. Заболевания известны как нейрофиброматоз 1 типа (NF1) и нейрофиброматоз 2 типа (NF2).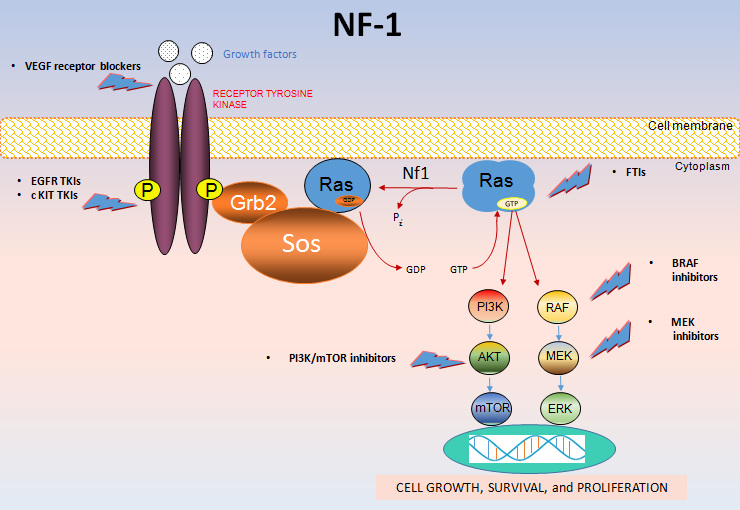 NF1 — более распространенный тип нейрофиброматоза. Шванноматоз недавно был идентифицирован как третий и более редкий тип нейрофиброматоза, но врачи еще мало о нем знают.
NF1 — более распространенный тип нейрофиброматоза. Шванноматоз недавно был идентифицирован как третий и более редкий тип нейрофиброматоза, но врачи еще мало о нем знают.
Вы также можете услышать NF1, называемый болезнью Реклингхаузена, болезнью фон Реклингхаузена, факоматозом фон Реклингхаузена, нейрофиброматозом фон Реклингхаузена, нейрофибромой (множественной), нейрофиброматозом-феохромоцитомой-дуоденальным карциноидным синдромом. Он вызывает появление нескольких пятен с молоком (участки коричневой или светло-коричневой кожи) и нейрофибромы (мягкие, мясистые наросты) на вашей коже или под ней.Это также может вызвать увеличение или деформацию костей и искривление позвоночника (сколиоз). Иногда опухоли могут развиваться в головном мозге, черепных нервах или спинном мозге. Примерно от 50% до 75% людей с NF1 также имеют проблемы с обучением.
NF2 также называют двусторонним акустическим нейрофиброматозом, вестибулярным нейрофиброматозом шванномы или центральным нейрофиброматозом. Он встречается гораздо реже, чем NF1, и характеризуется множественными опухолями черепных и спинномозговых нервов. Опухоли, поражающие как слуховые нервы, так и потерю слуха в подростковом возрасте или в возрасте 20 лет, как правило, являются первыми симптомами NF2.
Он встречается гораздо реже, чем NF1, и характеризуется множественными опухолями черепных и спинномозговых нервов. Опухоли, поражающие как слуховые нервы, так и потерю слуха в подростковом возрасте или в возрасте 20 лет, как правило, являются первыми симптомами NF2.
Симптомы нейрофиброматоза
Симптомы нейрофиброматоза 1 (NF1)
У людей с NF1 проявляются следующие симптомы:
- Несколько (обычно 6 или более) пятен с молоком
- Множественные веснушки в области подмышек или паха
- Крошечные наросты на радужной оболочке (цветной области) глаза; они называются узелками Лиша и обычно не влияют на зрение.
- Нейрофибромы, возникающие на коже или под ней, иногда даже глубоко внутри тела.Это доброкачественные (безвредные) опухоли. Но в редких случаях они могут стать злокачественными или злокачественными.
- Деформации костей, включая искривление позвоночника (сколиоз) или искривление ног
- Опухоли вдоль зрительного нерва, которые могут вызвать проблемы со зрением
- Боль, связанная с нервом
- Высокое кровяное давление
- Остеопороз
- Нарушения обучаемости
- Большая голова размер
- Низкий рост
Симптомы нейрофиброматоза 2 (NF2)
У людей с NF2 часто проявляются следующие симптомы:
- Потеря слуха
- Слабость лицевых мышц
- Головокружение
- Плохое равновесие
- Несогласованная ходьба
- Катаракта (помутнение хрусталика глаза), которая развивается в необычно раннем возрасте
- Головные боли
Симптомы шванноматоза
У людей с шванноматозом могут быть следующие симптомы:
Причины нейрофиброматоза
Нейрофиброматоз часто передается по наследству (передается n членами семьи через ваши гены). Но около 50% людей, которым впервые был поставлен диагноз, не имеют семейного анамнеза этого заболевания. Это потому, что это может быть результатом внезапной мутации (изменения) в ваших генах. Как только это изменение произойдет, вы сможете передать мутантный ген будущим поколениям. Мутации, которые приводят к нейрофиброматозу, включают:
Но около 50% людей, которым впервые был поставлен диагноз, не имеют семейного анамнеза этого заболевания. Это потому, что это может быть результатом внезапной мутации (изменения) в ваших генах. Как только это изменение произойдет, вы сможете передать мутантный ген будущим поколениям. Мутации, которые приводят к нейрофиброматозу, включают:
- Нейрофиброматоз 1 (NF1): Ген NF1 на хромосоме 17 производит белок, называемый нейрофибромином, который контролирует рост ваших клеток. Мутация этого гена вызывает потерю нейрофибромина и неконтролируемый рост клеток.
- Нейрофиброматоз 2 (NF2): Ген NF2 на хромосоме 22 вырабатывает белок, называемый мерлин или шванномин. Подавляет опухоли. Изменения этого гена вызывают потерю мерлина и неконтролируемый рост клеток.
- Шванноматоз: Мутации двух известных генов, связанных с шванноматозом, SMARCB1 и LZTR1, которые подавляют опухоли, связаны с этим типом нейрофиброматоза.

Диагностика нейрофиброматоза
Нейрофиброматоз диагностируется с помощью ряда тестов, в том числе:
Продолжение
Чтобы получить диагноз NF1, у вас должны быть две из следующих характеристик:
- Шесть или более пятен кофе с молоком, которые 1.5 сантиметров или больше у людей, переживших половое созревание, или 0,5 сантиметра или больше у людей, у которых нет
- Две или более нейрофибромы (опухоли, которые развиваются из клеток и тканей, покрывающих нервы) любого типа или одна или несколько плексиформных нейрофиброма (нерв, который стал толстым и деформировался из-за ненормального роста клеток и тканей, покрывающих нерв)
- Веснушки в подмышечной впадине или паху
- Глиома зрительного нерва (опухоль зрительного пути)
- Два или более узелка Лиша
- Характерное костное поражение, дисплазия клиновидной кости или дисплазия или истончение коры длинных костей
- Родственник первой степени с NF1
Чтобы поставить диагноз NF2, у вас должны быть:
- Двусторонний (на обоих стороны) вестибулярные шванномы, также известные как акустические невриномы. Это доброкачественные опухоли, которые развиваются из нервов равновесия и слуха, питающих внутреннее ухо.
 Это доброкачественные опухоли, которые развиваются из нервов равновесия и слуха, питающих внутреннее ухо.
Это доброкачественные опухоли, которые развиваются из нервов равновесия и слуха, питающих внутреннее ухо.Продолжение
или
- Семейный анамнез NF2 (родственник первой степени родства) плюс односторонние (с одной стороны) или двусторонние вестибулярные шванномы, или любые два из следующих состояний здоровья:
- Множественные менингиомы (возникающие опухоли в мозговых оболочках, мембранах, которые покрывают и защищают головной и спинной мозг)
- Глиома (рак головного мозга, который начинается в глиальных клетках, которые окружают и поддерживают нервные клетки)
- Любые нейрофибромы
- Шваннома
- Ювенильная катаракта
- Постепенная потеря слуха
- Звон в ушах
- Проблемы с равновесием
- Головные боли
Диагностика шванноматоза основывается на следующих критериях:
- Если вам 30 лет и старше, у вас нет признаков вестибулярных опухолей. МРТ, отсутствие известной мутации гена NF2, наличие двух или более шванном в пределах или между слоями кожи, одна из которых подтверждена биопсией.
- У вас есть одна шваннома, подтвержденная биопсией, и родственник первой степени родства, который также соответствует критериям.
- Если у вас тип, называемый сегментарным шванноматозом, у вас есть опухоли, ограниченные одной областью тела, например рукой, ногой или позвоночником.

Лечение нейрофиброматоза
Не существует лекарства от нейрофиброматоза. Лечение сосредоточено на контроле симптомов. Стандартного лечения ЯН не существует, и многие симптомы, такие как пятна от кофе с молоком, не нуждаются в лечении.Когда необходимо лечение, варианты могут включать:
- Операция по удалению проблемных новообразований или опухолей
- Лечение, которое включает химиотерапию или облучение, если опухоль стала злокачественной или злокачественной
- Операция по поводу проблем с костями, таких как сколиоз
- Терапия (включая физиотерапия, консультирование или группы поддержки)
- Операция по удалению катаракты
- Агрессивное лечение боли, связанной с заболеванием
- Стереотаксическая радиохирургия
- Слуховой ствол мозга и кохлеарные имплантаты
Осложнения нейрофиброматоза
Осложнения нейрофиброматоза могут варьироваться от человека к человеку .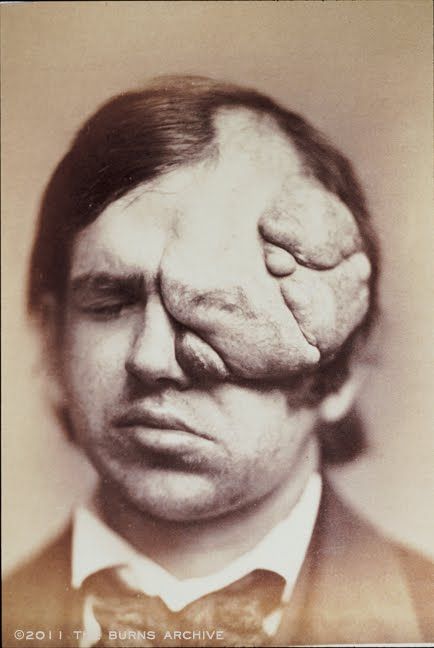 Обычно они возникают из-за растущей опухоли, прижимающейся к нервной ткани или внутреннему органу.
Обычно они возникают из-за растущей опухоли, прижимающейся к нервной ткани или внутреннему органу.
Возможные осложнения NF1 включают:
- Неврологические проблемы, такие как проблемы с обучением или мышлением
- Беспокойство или дистресс в связи с изменениями вашего внешнего вида, например, большое количество пятен в кафе с молоком
- Проблемы со скелетом, такие как искривление ног, сколиоз, переломы которые не заживают, или низкая плотность костей, повышающая риск остеопороза
- Проблемы со зрением из-за давления опухоли на зрительный нерв
- Проблемы во время гормональных изменений, таких как половое созревание или беременность
- Сердечно-сосудистые проблемы, такие как высокий уровень крови давление
- Проблемы с дыханием
- Повышенный риск некоторых видов рака, например рака груди, лейкемии, колоректального рака, опухолей головного мозга и некоторых видов рака мягких тканей
- Доброкачественная опухоль надпочечников, которая может вызвать высокое кровяное давление
Продолжение
Возможно Осложнения NF2 включают:
- Частичная потеря слуха или глухота
- Доброкачественные опухоли кожи или шванномы
- Проблемы со зрением
- Слабость или онемение конечностей
- Доброкачественные опухоли головного мозга или позвоночника, которые необходимо удалить хирургическим путем
90 074 Повреждение нерва на лице
Одним из возможных осложнений шванноматоза является сильная боль, которая требуется лечение у специалиста или даже операция.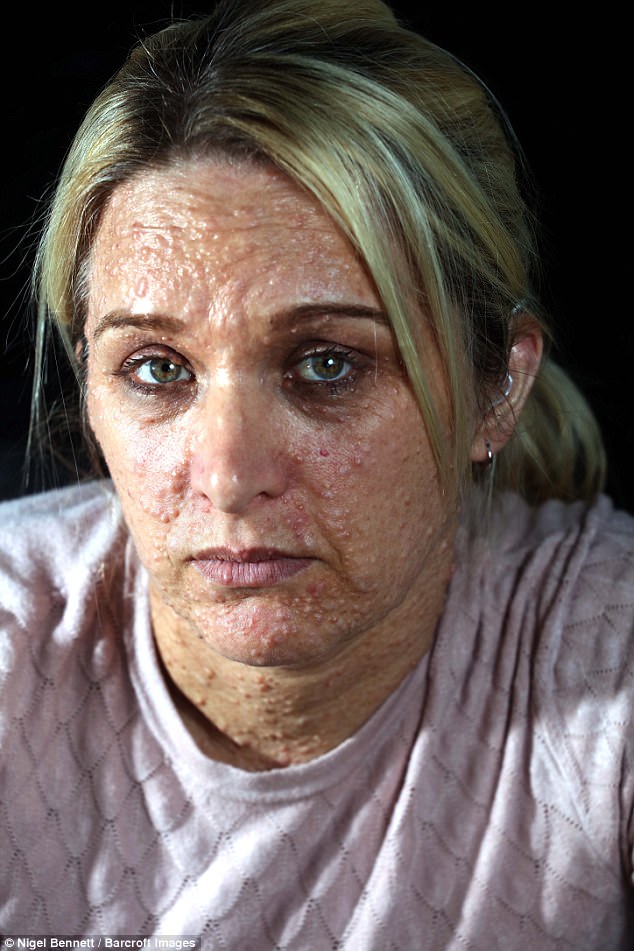
Нейрофиброматоз Перспективы
Ваш прогноз зависит от типа НФ. Часто симптомы NF1 легкие, и люди, у которых он есть, могут вести полноценную и продуктивную жизнь. Иногда боль и деформация могут привести к значительной инвалидности. Прогноз NF2 зависит от вашего возраста в начале заболевания, а также от количества и расположения опухолей. Некоторые из них могут быть опасными для жизни.
Нейрофиброматоз 1 типа — NHS
Нейрофиброматоз типа 1 (NF1) — это генетическое заболевание, которое вызывает рост опухолей вдоль нервов.Опухоли обычно не злокачественные (доброкачественные), но могут вызывать ряд симптомов.
Нейрофиброматоз 2 типа (NF2) встречается гораздо реже, чем NF1. Он рассматривается отдельно, так как имеет разные симптомы и причины.
Симптомы нейрофиброматоза 1 типа
NF1 — это заболевание, с которым вы родились, хотя некоторые симптомы развиваются постепенно в течение многих лет. Тяжесть состояния может значительно варьироваться от человека к человеку.
Тяжесть состояния может значительно варьироваться от человека к человеку.
В большинстве случаев поражается кожа, вызывая такие симптомы, как:
- пятна бледного кофейного цвета (пятна с молоком)
- мягкие доброкачественные опухоли на коже или под ней (нейрофибромы)
- скопления веснушек в необычных местах, таких как подмышки, пах и под грудью
- Проблемы с костями, глазами и нервной системой
Определенные проблемы со здоровьем часто связаны с NF1, например, трудности с обучением.Реже NF1 связан с типом рака, известным как злокачественные опухоли оболочки периферических нервов.
Подробнее о симптомах нейрофиброматоза 1.
Причины нейрофиброматоза 1 типа
NF1 вызван дефектным геном. Если ген NF1 неисправен, это приводит к неконтролируемому росту (опухолям), развивающимся в нервной системе.
В половине всех случаев NF1 дефектный ген передается от родителя к ребенку.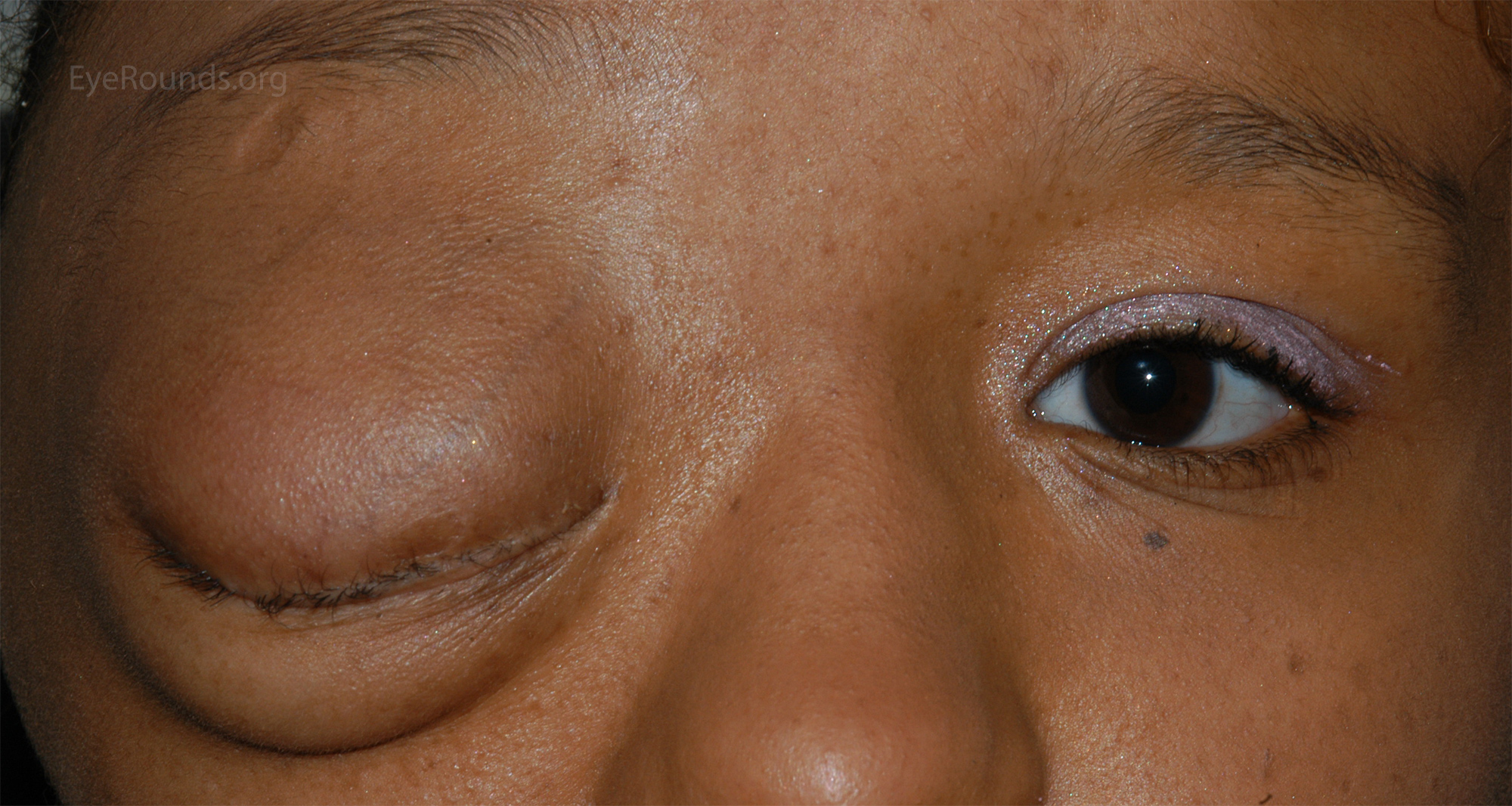 Только один родитель должен иметь дефектный ген, чтобы их ребенок подвергался риску развития этого заболевания.
Только один родитель должен иметь дефектный ген, чтобы их ребенок подвергался риску развития этого заболевания.
Если у матери или отца есть дефектный ген, вероятность того, что у каждого ребенка разовьется NF1, составляет 1 из 2.
В других случаях дефектный ген, по-видимому, развивается спонтанно. Непонятно, почему это происходит. Если у вас есть ребенок, у которого спонтанно развивается NF1, маловероятно, что у других детей также разовьется это заболевание.
Однако человек, у которого спонтанно развивается NF1, может передать это заболевание своим детям.
Диагностика нейрофиброматоза 1 типа
Обычно NF1 легко диагностировать у взрослых и детей старшего возраста, проверив типичные симптомы.
Может быть диагностирован у младенцев, у которых с рождения проявляются симптомы NF1. Однако не всегда возможно поставить точный диагноз в раннем детстве, потому что для развития некоторых симптомов требуются годы.
Если есть подозрение на NF1, могут быть рекомендованы дополнительные тесты, такие как сканирование, анализы крови или биопсия.Это необходимо для оценки наличия у вашего ребенка других симптомов или состояний, связанных с NF1.
Если есть сомнения относительно диагноза, ваш ребенок может сдать анализ крови, чтобы определить, есть ли у него дефектный ген NF1. Однако этот тест не является полностью надежным: около 5% детей с отрицательным результатом теста на дефектный ген все еще развивают NF1.
До и во время беременности
Пары с семейным анамнезом NF1 могут пожелать рассмотреть свои варианты, прежде чем родить ребенка.Ваш терапевт может направить вас к генетическому консультанту, чтобы обсудить возможные варианты.
Сюда могут входить:
- наличие ребенка с донорской яйцеклеткой или спермой
- усыновление ребенка
- пройти тест во время беременности — либо взятие пробы ворсин хориона, либо амниоцентез, чтобы узнать, будет ли у вашего ребенка NF1
- предимплантационная генетическая диагностика — яйцеклетки оплодотворяются в лаборатории и проверяются на отсутствие гена NF1 перед имплантацией в матку.

Лечение нейрофиброматоза 1 типа
В настоящее время нет лекарства от NF1.Лечение включает регулярный мониторинг и устранение любых проблем по мере их возникновения.
Лечение может включать:
- хирургия — удаление опухолей и исправление костных аномалий
- лекарство — для контроля вторичных состояний, таких как высокое кровяное давление
- физиотерапия
- психологическая поддержка
- обезболивание
Тщательное наблюдение и лечение могут помочь людям с NF1 жить полноценной жизнью. Однако существует риск развития серьезных проблем, таких как определенные виды рака, которые могут сократить продолжительность жизни.
Подробнее о лечении нейрофиброматоза 1 типа.
NF1 и беременность
Большинство женщин с NF1 имеют здоровую беременность.
Однако количество нейрофибром может увеличиваться из-за гормональных изменений. Убедитесь, что за вами ухаживает акушер со знанием NF1, или поговорите со своим специалистом по NF1.
Убедитесь, что за вами ухаживает акушер со знанием NF1, или поговорите со своим специалистом по NF1.
Поддержка от опухолей нервов, Великобритания
Nerve Tumors UK — это благотворительная организация, цель которой — улучшить жизнь людей с любым типом нейрофиброматоза.
Для получения дополнительной информации вы можете посетить веб-сайт Nerve Tumors UK, позвонить в службу поддержки по телефону 07939 046 030 или по электронной почте [email protected].
NF1 и рак груди
Женщины до 50 лет с NF1 имеют повышенный риск рака груди, и им следует начинать проходить обследование груди, когда им исполнится 40 лет.
Информация о вас
Если у вас или вашего ребенка есть NF1, ваша клиническая бригада передаст информацию в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Узнать больше о реестре.
Последняя проверка страницы: 7 июня 2018 г.
Срок следующей проверки: 7 июня 2021 г.
Нейрофиброматоз 1 типа — Симптомы
Симптомы нейрофиброматоза 1 типа (NF1) часто легкие и не вызывают серьезных проблем со здоровьем.Но у некоторых людей будут тяжелые симптомы.
Симптомы NF1 могут влиять на множество различных частей тела, но маловероятно, что у кого-то разовьются все они.
Скин
Нашивки кофейного цвета
Наиболее частым симптомом NF1 является появление на коже безболезненных пятен кофейного цвета, называемых пятнами кофе с молоком. Однако не у всех, у кого есть пятна с кофе с молоком, есть NF1.
Пятна могут присутствовать при рождении или развиваться к 3 годам.
В детстве у большинства детей с NF1 будет не менее 6 пятен с молоком диаметром около 5 мм. В зрелом возрасте они вырастают примерно до 15 мм.
Количество пятен не зависит от тяжести состояния. Например, человек с 10 пятнами имеет такие же шансы на развитие дальнейших проблем, как и человек со 100 пятнами.
Веснушки
Другой распространенный симптом NF1 — скопления веснушек в необычных местах, таких как подмышки, пах и под грудью.
Опухоли на коже или под кожей
По мере взросления ребенка, обычно в подростковом или раннем взрослом возрасте, на коже или под ней появляются опухоли (нейрофибромы).
Они вызваны доброкачественными опухолями, которые развиваются на оболочках нервов. Они могут различаться по размеру, от размером с горошину до опухолей немного большего размера. Некоторые нейрофибромы фиолетовые.
Количество нейрофибром может быть разным. У некоторых людей их всего несколько, в то время как у других они находятся на больших участках тела.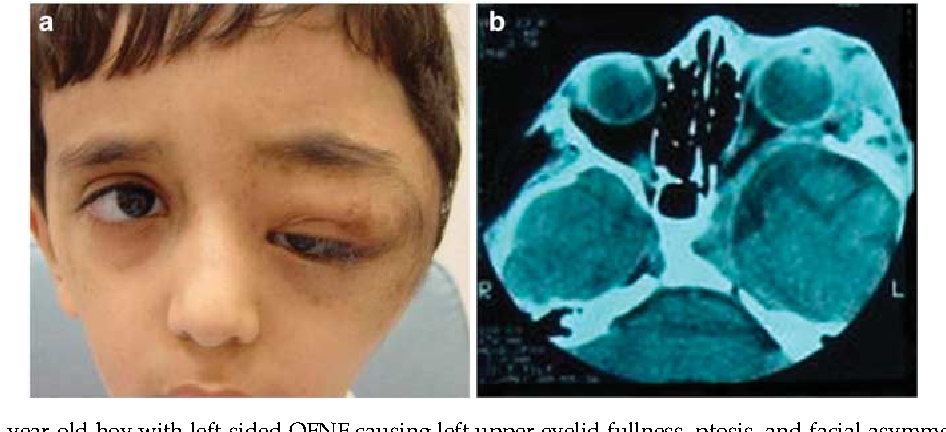
Большинство нейрофибром не особенно болезненны, но они могут быть видимыми, цепляться за одежду и иногда вызывать раздражение и покалывание.
Однако, если нейрофибромы развиваются в месте соединения нескольких ветвей нервов (плексиформные нейрофибромы), они могут вызывать большие опухоли.
Плексиформные нейрофибромы иногда возникают на коже, но могут также развиваться на более крупных нервах, расположенных в глубине тела. Иногда они могут вызывать симптомы, включая боль, слабость, онемение, кровотечение, изменения мочевого пузыря или кишечника.
Обучение и поведение
У некоторых детей с NF1 возникают проблемы с обучением и поведением. Непонятно, почему это происходит.
Дети с NF1, у которых есть трудности с обучением, могут иметь нормальный или немного ниже среднего интеллекта, но их обычно можно обучать в обычной школе.
Синдром дефицита внимания и гиперактивности (СДВГ) поражает примерно половину всех детей с NF1 и вызывает проблемы с объемом внимания, концентрацией и контролирующими импульсами.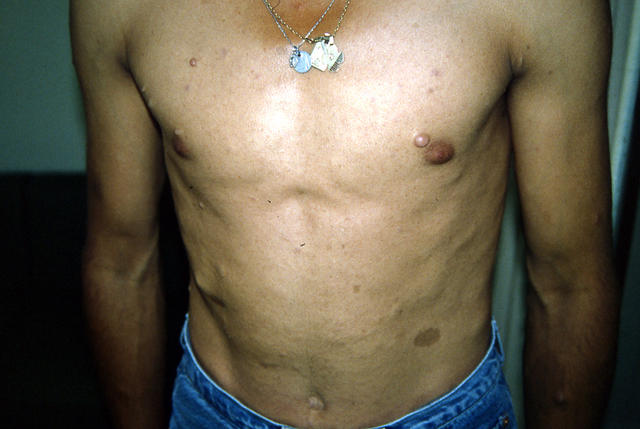
NF1 также был связан с расстройством аутистического спектра (РАС), и у некоторых детей могут быть трудности в социальном общении.
Глаза
Около 15% детей с NF1 заболевают опухолью зрительного пути.
Оптический путь расположен в задней части каждого глаза и передает информацию от глаз к мозгу. Этот тип опухоли известен как глиома оптического пути (OPG).
Известно, что дети в возрасте до 7 лет имеют самый высокий риск развития этого типа опухоли.Эти опухоли часто небольшие, медленно растут и не вызывают каких-либо заметных симптомов.
Дети с быстрорастущими ОПГ могут иметь проблемы со зрением, в том числе:
- Объекты становятся размытыми
- изменения в том, как они видят цвета
- уменьшенное поле зрения
- косоглазие
- один глаз выглядит более заметным, чем другой
Дети младшего возраста могут быть не в состоянии объяснить, что у них проблемы со зрением. Вы должны знать о любых признаках, которые ваш ребенок плохо видит, например о проблемах с поднятием мелких предметов или столкновении с ними.
Вы должны знать о любых признаках, которые ваш ребенок плохо видит, например о проблемах с поднятием мелких предметов или столкновении с ними.
Лучший способ обнаружить эти опухоли — проходить проверку зрения не реже одного раза в год, пока вашему ребенку не исполнится 7 лет.
Еще одна общая черта NF1 — появление крошечных выступающих коричневых пятен в цветной центральной части глаза (радужной оболочке). Они известны как узелки Лиша и обычно не вызывают каких-либо заметных симптомов или проблем со зрением.
Высокое кровяное давление
У некоторых детей с NF1 развивается высокое кровяное давление.
Высокое кровяное давление может быть вызвано сужением артерии к почке (стеноз почечной артерии). Это требует лечения у специалиста, если установлено, что оно вызывает у вашего ребенка высокое кровяное давление.
Высокое кровяное давление также может быть вызвано феохромоцитомой. Это опухоль надпочечников, обычно доброкачественная. Это чаще встречается у взрослых, чем у детей.
Это чаще встречается у взрослых, чем у детей.
Высокое кровяное давление может быть связано с потенциально серьезными осложнениями, такими как инсульт или сердечный приступ, если его не лечить.
Детям и взрослым с NF1 следует регулярно проверять артериальное давление, обычно не реже одного раза в год.
Физическое развитие
Многие дети с NF1 имеют одну или несколько проблем, влияющих на их физическое развитие, в том числе:
- искривленный позвоночник (сколиоз) — считается, что им подвержено около 10% детей с NF1
- голова больше среднего — это встречается примерно у половины всех детей
- меньший размер и меньший вес, чем обычно — это часто встречается у многих людей с NF1
Около 2% детей с NF1 заболевают псевдоартрозом.Это когда аномальное развитие костей вызывает искривление (искривление) конечности, обычно в большеберцовой кости голени. Кость может сломаться после небольшой травмы. Перелом не заживает полностью, что влияет на нормальное движение ноги.
Перелом не заживает полностью, что влияет на нормальное движение ноги.
Мозг и нервная система
Симптомы, поражающие мозг и нервную систему, относительно часто встречаются при NF1.
Многие люди с NF1 страдают мигренью.
У некоторых людей развиваются опухоли головного мозга, хотя это случается редко.Опухоли могут не вызывать заметных симптомов. Однако опухоли в определенных частях мозга иногда вызывают такие симптомы, как:
- изменения личности
- слабость с одной стороны корпуса
- трудности с равновесием и координацией
У некоторых детей с NF1 развивается эпилепсия, при которой у человека повторяются припадки или припадки. Это, как правило, легкая форма эпилепсии, которую обычно легко контролировать с помощью лекарств.
Злокачественная опухоль оболочки периферических нервов
Одной из наиболее серьезных проблем, которые могут поразить человека с NF1, является злокачественная опухоль оболочки периферических нервов (MPNST).
MPNST — это тип рака, который развивается в плексиформной нейрофиброме. Подсчитано, что у людей с NF1 вероятность развития MPNST в течение жизни составляет около 15%.
Большинство случаев заболевания впервые возникают у людей в возрасте от 20 до 30 лет, но они могут возникать в любом возрасте.
Симптомы MPNST включают:
- Текстура существующей нейрофибромы меняется с мягкой на твердую
- существующая нейрофиброма внезапно стала намного больше
- постоянная боль, длящаяся более месяца или будящая по ночам
- внезапно возникли проблемы с нервной системой, которых у вас не было раньше, например слабость, онемение или покалывание в руках и ногах
- потеря контроля над мочевым пузырем или кишечником
- затрудненное дыхание или глотание
Если у вас есть какие-либо из этих симптомов, как можно скорее обратитесь к лечащему врачу.
Опухоль желудочно-кишечного тракта
У некоторых людей с NF1 может развиться опухоль желудочно-кишечного тракта (GIST), которая может вызывать такие симптомы, как:
- Боль в животе
- изменения в привычках кишечника, такие как диарея или запор
- кровотечение снизу
Опухоли желудочно-кишечного тракта требуют лечения в специализированном центре.
Специализированные центры
Если вам требуется специализированное лечение, вас следует направить в специализированный центр с опытом диагностики и лечения МПНПП.
В настоящее время существует два центра: один в фонде NHS Foundation Trust Гая и Сент-Томаса в Лондоне, а другой — в фонде NHS Foundation больниц Центрального Манчестерского университета.
Последняя проверка страницы: 7 июня 2018 г.
Срок следующей проверки: 7 июня 2021 г.
Наследование и генетика нейрофиброматоза типа 1 (NF1) — Школа медицины — Программа нейрофиброматоза
Понимание мутации NF1
Нейрофиброматоз 1 типа (NF1) — это наследственное заболевание, вызванное изменением, называемым мутацией, в гене NF1 , который расположен на хромосоме 17.Ген NF1 содержит код инструкций по созданию белка, называемого нейрофибромином, который вырабатывается во многих клетках, включая нервные клетки и специализированные клетки, окружающие нервы (клетки Шванна). Белок нейрофибромина действует как супрессор опухолей, предотвращая слишком быстрый рост и деление клеток. Мутация гена NF1 приводит к выработке нефункционального или отсутствующего белка нейрофибромина, который не может регулировать рост и деление клеток, что приводит к росту нейрофибром (опухолей кожи) вдоль нервов по всему телу.
Белок нейрофибромина действует как супрессор опухолей, предотвращая слишком быстрый рост и деление клеток. Мутация гена NF1 приводит к выработке нефункционального или отсутствующего белка нейрофибромина, который не может регулировать рост и деление клеток, что приводит к росту нейрофибром (опухолей кожи) вдоль нервов по всему телу.
Унаследованные мутации
У всех людей есть две копии каждого гена — по одной копии, унаследованной от каждого родителя. Мутация гена NF1 является доминантной, что означает, что только одна из двух копий гена должна иметь мутацию, чтобы вызвать нарушение. Родитель с NF1 имеет 50% шанс передать аномальную копию гена ребенку. Ребенок, унаследовавший измененный ген, также будет иметь заболевание.
Спонтанные мутации
В то время как половина случаев NF1 передается по наследству от родителей, 50% детей с диагнозом NF1, по-видимому, являются первыми членами своей семьи, у которых это расстройство.В таких случаях генетическое изменение или мутация происходили в сперматозоиде или яйцеклетке, которые сформировали ребенка. Это называется спонтанной или новой мутацией. Человек со спонтанной мутацией гена NF1 имеет 50% шанс передать аномальную копию гена ребенку.
Это называется спонтанной или новой мутацией. Человек со спонтанной мутацией гена NF1 имеет 50% шанс передать аномальную копию гена ребенку.
Определение источника мутации NF
Важно определить, является ли заболевание наследственным или является результатом спонтанной мутации, поскольку человек с мутацией NF1 имеет 50% шанс передачи заболевания каждый раз, когда у него или нее появляется ребенок.Один из способов добиться этого — тщательное обследование каждого родителя, у ребенка которого был диагностирован NF1, для определения наличия пятен с молоком или узелков Лиша на радужной оболочке глаз. Если ни у одного из родителей не обнаружено признаков NF1, заболевание ребенка, скорее всего, является результатом спонтанной или новой мутации.
В настоящее время также доступно генетическое тестирование для подтверждения наличия мутации гена NF1 с 95% чувствительностью и может быть целесообразным в некоторых случаях.У некоторых людей особенности NF1 ограничены только одной частью или одной стороной тела. Это называется мозаичным NF1 (также называемым сегментным NF1). Мозаичный NF1 вызывается мутацией гена, возникшей после зачатия, во время раннего развития человека как эмбриона. Генетическое тестирование для людей с типом NF, хотя и возможно, может быть более сложным, чем для людей, у которых нет этой формы расстройства.
Это называется мозаичным NF1 (также называемым сегментным NF1). Мозаичный NF1 вызывается мутацией гена, возникшей после зачатия, во время раннего развития человека как эмбриона. Генетическое тестирование для людей с типом NF, хотя и возможно, может быть более сложным, чем для людей, у которых нет этой формы расстройства.
Что вызывает мутацию NF1?
В случаях, когда определяется, что NF1 у ребенка является результатом спонтанной мутации, родители часто задаются вопросом, не сделали ли они что-то, что вызвало мутацию, например, подвергли своего ребенка воздействию радиации, лекарств, алкоголя или других веществ в окружающей среде.Важно понимать, что конкретная причина мутации гена NF1 в настоящее время неизвестна, и никакого воздействия окружающей среды не предполагается. Также важно знать, что генетические мутации не редкость. Клетки в организме непрерывно делятся, и каждый раз, когда они это делают, необходимо правильно копировать огромный объем генетической информации.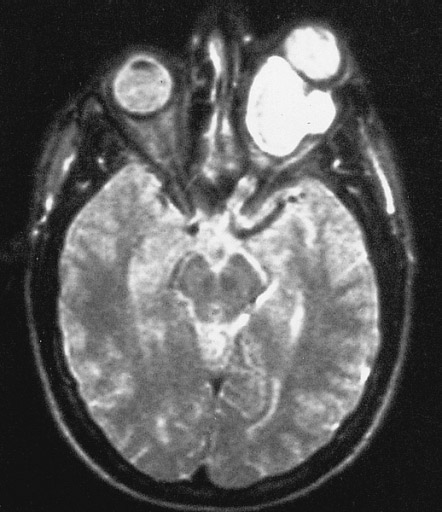 Случайные ошибки неизбежно возникают в процессе копирования и могут быть причиной мутаций, приводящих к NF1.
Случайные ошибки неизбежно возникают в процессе копирования и могут быть причиной мутаций, приводящих к NF1.
Симптомы и причины нейрофиброматоза 2 типа
Диагноз нейрофиброматоз 2 типа (NF2) вызывает множество вопросов и опасений как у молодых людей, так и у их семей.Хотя большинство людей с NF2 ведут нормальный образ жизни, многие аспекты расстройства по-прежнему трудно предсказать.
Вас может утешить то, что в рамках программы нейрофиброматоза в Бостонской детской больнице мы уже помогли тысячам детей и молодых людей успешно справиться с их НФ. Наши сострадательные и опытные врачи готовы помочь вам на каждом этапе вашего пути.
Что такое NF2?
NF2 — это генетическое заболевание, характеризующееся определенными типами опухолей, которые образуются в теле или мозге человека.
Наиболее распространенными типами опухолей, связанных с NF2, являются:
- Вестибулярная шваннома : также называемая слуховым нейромом a, вестибулярная шваннома — это доброкачественная (незлокачественная) опухоль, которая развивается на восьмом черепном нерве (нерв во внутреннем ухе, отвечающий за слух и равновесие).
- Менингиома : обычно доброкачественная, менингиома — это тип опухоли, развивающейся из мозговых оболочек (мембраны, окружающей головной и спинной мозг).Менингиомы могут вызывать судороги, гемипарез (слабость на одной стороне тела), изменения зрения и головные боли, которые со временем усиливаются.
- Эпендимома : тип опухоли головного мозга, которая развивается из клеток эпендимы, выстилающих желудочки (заполненные жидкостью пространства в головном мозге) и центральный канал спинного мозга. Эти опухоли часто вызывают такие проблемы, как онемение, слабость и боль. Эпендимомы излечиваются на 50 процентов.

Существуют ли другие типы НФ?
Да.Помимо NF2, существуют две другие различные формы нейрофиброматоза: нейрофиброматоз типа 1 (NF1) и шванноматоз.
Что такое нейрофиброматоз 1 типа (NF1)?
NF2 часто путают с NF1. Как и NF2, NF1 также является генетическим заболеванием, характеризующимся наличием опухолей, которые образуются вдоль нервов в организме. Однако нарушения вызываются двумя разными генами, расположенными на двух разных хромосомах. Крайне редко кто-то имеет и NF1, и NF2.
Однако нарушения вызываются двумя разными генами, расположенными на двух разных хромосомах. Крайне редко кто-то имеет и NF1, и NF2.
Вот некоторые заметные клинические различия между NF1 и NF2:
- NF2 встречается реже, чем NF1, и встречается у 1 из каждых 3500 рождений.
- Симптомы NF2 обычно выявляются в возрасте от 18 до 24 лет, тогда как NF1 диагностируется в младенчестве или раннем детстве.
- При NF2 доброкачественные опухоли, называемые шванномами, растут на нервах по всей нервной системе и часто вызывают нарушения слуха и зрения. При NF1 доброкачественные опухоли, называемые нейрофибромами, покрывают периферический нерв и аналогичным образом могут вызывать боль или специфические неврологические симптомы.Поскольку один тип нейрофибромы обычно растет на коже или рядом с ней, также могут наблюдаться изменения кожи от легких до тяжелых.
- NF2 вызывается мутацией на хромосоме 22 и включает белок, называемый мерлином, который, как считается, участвует в форме и структуре клеток. NF1 вызывается мутацией на хромосоме 17 и включает белок, называемый нейрофибромином, который связан с ростом и делением клеток.
- Люди с NF2 не имеют нарушений обучаемости, осложнения, которое очень часто наблюдается у людей с NF1.
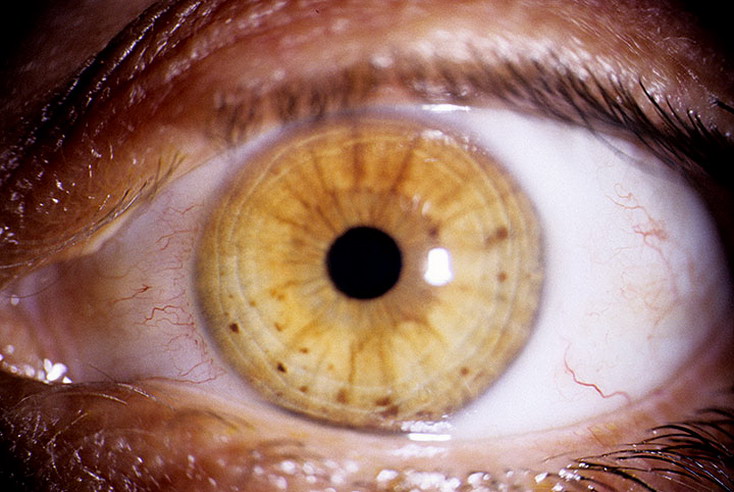 NF1 вызывается мутацией на хромосоме 17 и включает белок, называемый нейрофибромином, который связан с ростом и делением клеток.
NF1 вызывается мутацией на хромосоме 17 и включает белок, называемый нейрофибромином, который связан с ростом и делением клеток.Что такое шванноматоз?
Шванноматоз — чрезвычайно редкая форма НФ, которая поражает примерно 1 из 40 000 человек. Хотя шванноматоз имеет некоторые клинические сходства с другими формами НФ, это отдельное заболевание.
Вот важные отличительные признаки шванноматоза:
- Это генетическое заболевание, но, в отличие от NF1 и NF2, не имеет четкой схемы наследования. Вероятность передачи шванноматоза своему ребенку от больного родителя составляет 15 процентов, по сравнению с 50 процентами в NF1 и NF2.
- Шванноматоз характеризуется наличием множественных шванном (доброкачественных опухолей) на черепных, спинных и периферических нервах. Однако, в отличие от NF2, у людей с шванноматозом не развиваются вестибулярные шванномы и не наблюдается нарушения слуха.
- Если у человека имеются множественные шванномы, перед постановкой диагноза шванноматоза необходимо исключить возможность NF2. Отсутствие доказательств вестибулярных опухолей или семейного анамнеза NF2 — два диагностических критерия, используемых для этого различия.
Есть ли какие-либо медицинские осложнения, связанные с NF2?
Поскольку NF2 влияет на нервную систему, большинство осложнений связаны с проблемами, связанными со зрением, слухом и равновесием. Также может возникнуть онемение или слабость в лице, руках или ногах. Однако нет никаких доказательств того, что NF2 вызывает интеллектуальные нарушения и нарушения обучаемости, которые очень часто встречаются у людей с NF1.
Причины
Что заставило моего ребенка заболеть NF2?
В 50 процентах случаев NF2 наследуется от родителя:
- Во время зачатия каждый родитель передает эмбриону копию гена NF2.Поскольку NF2 является аутосомно-доминантным генетическим заболеванием, ребенок унаследует это заболевание, если только в одном из этих генов будет иметь мутацию.
- Это означает, что при каждой беременности родители, пораженные NF2, сами имеют 50-процентный шанс передать его своему ребенку.
- NF2 в равной степени влияет на людей всех рас, полов и национальностей.
В остальных 50 процентах случаев NF2 возникает в результате спонтанной мутации:
- Спонтанная мутация возникает, когда происходит внезапное изменение генетического материала во время зачатия.
- Спонтанные мутации возникают в ситуациях, когда нет предшествующего семейного анамнеза НФ.
Есть ли разница между унаследованным NF2 и NF2, возникающим в результате спонтанной мутации?
Нет. Помимо происхождения, нет никакой разницы между унаследованным NF2 и NF2, который был вызван спонтанной мутацией.
Сделал ли я что-нибудь, чтобы вызвать NF2 у моего ребенка?
Нет, нет никаких доказательств того, что NF2 вызывается факторами окружающей среды или чем-то, что мать сделала (или не сделала) во время беременности.
Признаки и симптомы
Когда появляются симптомы NF2?
NF2 обычно выявляется в раннем взрослом возрасте, при этом средний возраст появления симптомов составляет около 20 лет.
Каковы первые признаки NF2?
Большинство опухолей NF2 растут на восьмом черепном нерве. Расположенный во внутреннем ухе восьмой черепной нерв отвечает за передачу в мозг информации о звуке и балансе.
В результате первые симптомы NF2 обычно вызваны поражением нерва:
- потеря слуха
- звон в ушах (тиннитус)
- проблемы с балансом, начиная с подросткового возраста или в начале двадцатых годов
Другие симптомы NF2 могут включать:
- слабость лица
- головная боль
- изменения зрения
- катаракта
- Боль в спине (вызванная поражением спинного мозга)
- кожные изменения (вызванные шванномами на коже, что чаще встречается у детей)
Прогрессируют ли симптомы NF2?
Хотя почти невозможно точно предсказать, как будет развиваться NF2, вестибулярные шванномы растут медленно и обычно со временем вызывают ухудшение равновесия и слуха.К счастью, существуют хирургические вмешательства, позволяющие сохранить слух. Посетите нашу вкладку «Лечение», чтобы узнать больше об этих вариантах.
Вопросы к врачу
Диагноз NF2 вызывает множество вопросов и опасений как у пациентов, так и у их семей. Во время ваших первых встреч с врачом может быть легко оказаться перегруженным информацией и забыть о том, что вы хотели спросить.
Многие родители и молодые люди считают полезным записывать вопросы заранее или записывать их по мере их возникновения.Таким образом, когда вы поговорите с врачами, вы можете быть уверены, что все ваши проблемы будут решены.
Вот некоторые вопросы, которые вы можете задать:
- Как можно справиться с симптомами этого расстройства?
- Какие варианты хирургического вмешательства?
- Каков долгосрочный план лечения?
- На какие ресурсы вы можете указать мне для получения дополнительной поддержки и информации?
Нейрофиброматоз | DermNet NZ
Автор: Ванесса Нган, штатный писатель, 2003 г.Обновлено доктором Эбтисамом Эльгблави, дерматологом, Триполи, Ливия. Главный редактор DermNet NZ: A / Prof Amanda Oakley, дерматолог, Гамильтон, Новая Зеландия. Октябрь 2017.
Что такое нейрофиброматоз?
Нейрофиброматоз (НФ) — это генетическое заболевание, поражающее кости, мягкие ткани, кожу и нервную систему. Клинические проявления со временем нарастают.
Идентифицировано по крайней мере 8 различных клинических фенотипов NF. Он подразделяется на 2 различных типа:
- Нейрофиброматоз 1 (NF1)
- Нейрофиброматоз 2 (NF2).
Нейрофиброматоз 1
NF1 встречается примерно у 1 из 3000 родов. Он также известен как болезнь фон Реклингхаузена. Характеризуется наличием:
- 6 или более пятен с молоком — плоских светло-коричневых родинок
- Веснушки в кожных складках
- Узелки Лиша в радужной оболочке глаза.
- Множественные нейрофибромы — опухоли, свисающие с кожи
Нейрофиброматоз 2
NF2 встречается примерно у 1 из 50 000 родов.Он также известен как двусторонний акустический нейрофиброматоз или центральный нейрофиброматоз. Для него характерны множественные опухоли и поражения головного и спинного мозга.
Солитарная нейрофиброма не связана с NF1 или NF2.
Что вызывает нейрофиброматоз?
NF1 и NF2 возникают в результате дефектов в разных генах.
- NF1 вызывается мутацией гена нейрофибромина, расположенного в перицентромерной области хромосомы 17.
- NF2 вызван мутацией на 22 хромосоме.
Мутировавший ген может быть унаследован от родителя, у которого есть NF, путем аутосомно-доминантной передачи, или он может быть геном-основателем из-за спонтанной мутации. Родитель с NF имеет 50% шанс передать ген каждому из своих детей.
Генетика нейрофиброматозов 1 и 2 *
* Изображение предоставлено Genetics 4 Medics
Каковы признаки и симптомы нейрофиброматоза?
Степень и тяжесть проявлений НФ сильно различаются от человека к человеку и варьируются в пределах одной семьи.
Нейрофиброматоз 1
Макулы Café-au-lait
Макулы Café-au-lait определяются, овальные или неправильной формы, светло-коричневые пятна более 0,5 см в диаметре. Они могут присутствовать при рождении и увеличиваться в количестве в течение первых нескольких лет жизни.
Хотя изолированные пятна «кофе с молоком» можно найти у многих людей без НФ, люди с более чем 5 из них имеют хорошие шансы также иметь НФ1, особенно если они появляются на коже в течение первых 5 лет жизни.
Более 5 пятен с молоком обнаруживаются у 1,8% новорожденных, 25–40% детей и 14% взрослых с NF1.
Freckling
Веснушки в подмышечных впадинах также известны как признак Кроу и характерны для нейрофиброматоза 1 типа. Веснушки появляются в период полового созревания, после появления пятен с молоком и до появления нейрофибром. Они также могут появиться в других складках кожи, например в паху.
Нейрофиброматоз
Узелки Лиша
Узлы Лиша — это крошечные опухоли на радужной оболочке глаза.После полового созревания узелки Лиша присутствуют у 97–100% пациентов с NF1. Клинически они не вызывают никаких проблем, но помогают подтвердить диагноз.
Нейрофибромы
Нейрофибромы состоят из шванновских клеток, фибробластов, тучных клеток и сосудистых компонентов и могут развиваться в любой точке нерва. Они бывают трех типов: кожные, подкожные и плексиформные. Кожные и подкожные нейрофибромы не специфичны для нейрофиброматоза, но плексиформные нейрофибромы наблюдаются только при NF1.
- Кожные нейрофибромы представляют собой ограниченные, поверхностные, мягкие, похожие на пуговицы коричневые, розовые или кожные поверхностные узелки мягкой или твердой консистенции. При нажатии пальцем на них может появиться патогномоничная инвагинация петлицы. У них нет злокачественного потенциала.
- Подкожные нейрофибромы похожи на кожные нейрофибромы, за исключением более глубокого происхождения. Они могут вызвать локальную боль или болезненность.
- Плексиформные нейрофибромы представлены в коже в виде мешкообразных масс.Узловые плексиформные нейрофибромы поражают корешки спинных нервов, а диффузные плексиформные нейрофибромы представляют собой инвазивные опухоли, которые могут поражать все слои кожи, мышц, костей и кровеносных сосудов.
Нейрофиброматоз
См. Другие изображения нейрофиброматоза …
Другие особенности NFI
Степень поражения кожи при NF1 не является показателем степени заболевания, поскольку внутренние проявления являются обычными и часто более серьезными.Проблемы в других частях тела включают:
- Порок развития длинных костей (ниже колена и локтя) и искривление позвоночника (сколиоз)
- Низкий рост и дефицит гормона роста
- Проблемы с обучением (проблемы с речью) и поведенческие проблемы (25–40% имеют проблемы с обучением, 5–10% могут иметь умственную отсталость)
- Опухоли зрительного нерва; они могут вызвать потерю зрения
- Высокое кровяное давление и другие проблемы с кровью
- Опухоли позвоночника и головного мозга; они увеличивают риск эпилепсии
- Опухоли или поражения желудочно-кишечного тракта, которые могут вызвать кровотечение или непроходимость
- Нарушения слуха.
Хотя большинство опухолей являются доброкачественными (не злокачественными), по оценкам, у человека с NF1 риск развития раковых опухолей повышается на 3–15%.
Нейрофиброматоз 2
У NF2 не так много внешних признаков, как у NF1. Для него характерны множественные опухоли и поражения головного и спинного мозга. Первым симптомом обычно является потеря слуха из-за опухоли, растущей на одном или обоих слуховых нервах. Часто это не проявляется до позднего подросткового возраста или до 20 лет.
Опухоли в NF2 обычно доброкачественные. Но доброкачественное увеличение опухоли головного и спинного мозга может нарушить жизненно важные функции.
Наблюдение за пациентами с нейрофиброматозом
Основная роль наблюдения — наблюдение за развитием опухолей и вмешательство при необходимости.
- Здоровые дети с НФ должны наблюдаться и осматриваться каждые 6–12 месяцев педиатром.
- Пациенты с глазными симптомами и признаками должны находиться под наблюдением офтальмолога.
- Нарушения слуха должны оцениваться и наблюдаться отоларингологом (ЛОР).
- Неврологу может потребоваться осмотреть пациента с любыми неврологическими симптомами и признаками, такими как неуклюжесть, онемение или слабость.
Визуализация с помощью ультразвукового сканирования, компьютерной томографии и МРТ может быть организована для определения местоположения любой предполагаемой опухоли.
Какое лечение доступно?
Нет лекарства от НФ.
Нейрофибромы, которые становятся большими и болезненными, можно вырезать, чтобы снизить риск злокачественных новообразований и других осложнений.Деформацию лица можно уменьшить с помощью эстетической (пластической) хирургии.
Генетическое консультирование и обучение НФ очень важны. Одна проблема, которую не следует упускать из виду, — это риск изоляции или одиночества у людей с НФ. Люди с НФ часто беспокоятся о будущих осложнениях, и иногда обезображивающие поражения могут привести к уходу из общества.