МАТЕРИАЛЫ КОНГРЕССОВ И КОНФЕРЕНЦИЙ: VIII РОССИЙСКИЙ ОНКОЛОГИЧЕСКИЙ КОНГРЕСС
VIII РОССИЙСКИЙ ОНКОЛОГИЧЕСКИЙ КОНГРЕСС
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА РАДБОМИОСАРКОМ У ДЕТЕЙ
Д.М. Коновалов1,2, А.С. Тертычный1,2, А.Г. Талалаев1,2
1Российский государственный медицинский университет
2НИИ детской гематологии МЗ РФ
Опухоли мягких тканей составляют, по данным различных национальных регистров, от 7% до 23% всех злокачественных новообразований у детей. Международная гистологическая классификация опухолей мягких тканей содержит описания более чем 150 различных нозологических форм опухолей мягких тканей. По данным Германского регистра детских опухолей 45% всех злокачественных опухолей мягких тканей у детей приходятся на различные гистологические типы рабдомиосаркомы.
По нашим данным, у плодов и детей до 6-ти месяцев жизни рабдомиосаркома составляет 29,4% от всех злокачественных опухолей мягких тканей в данной возрастной группе.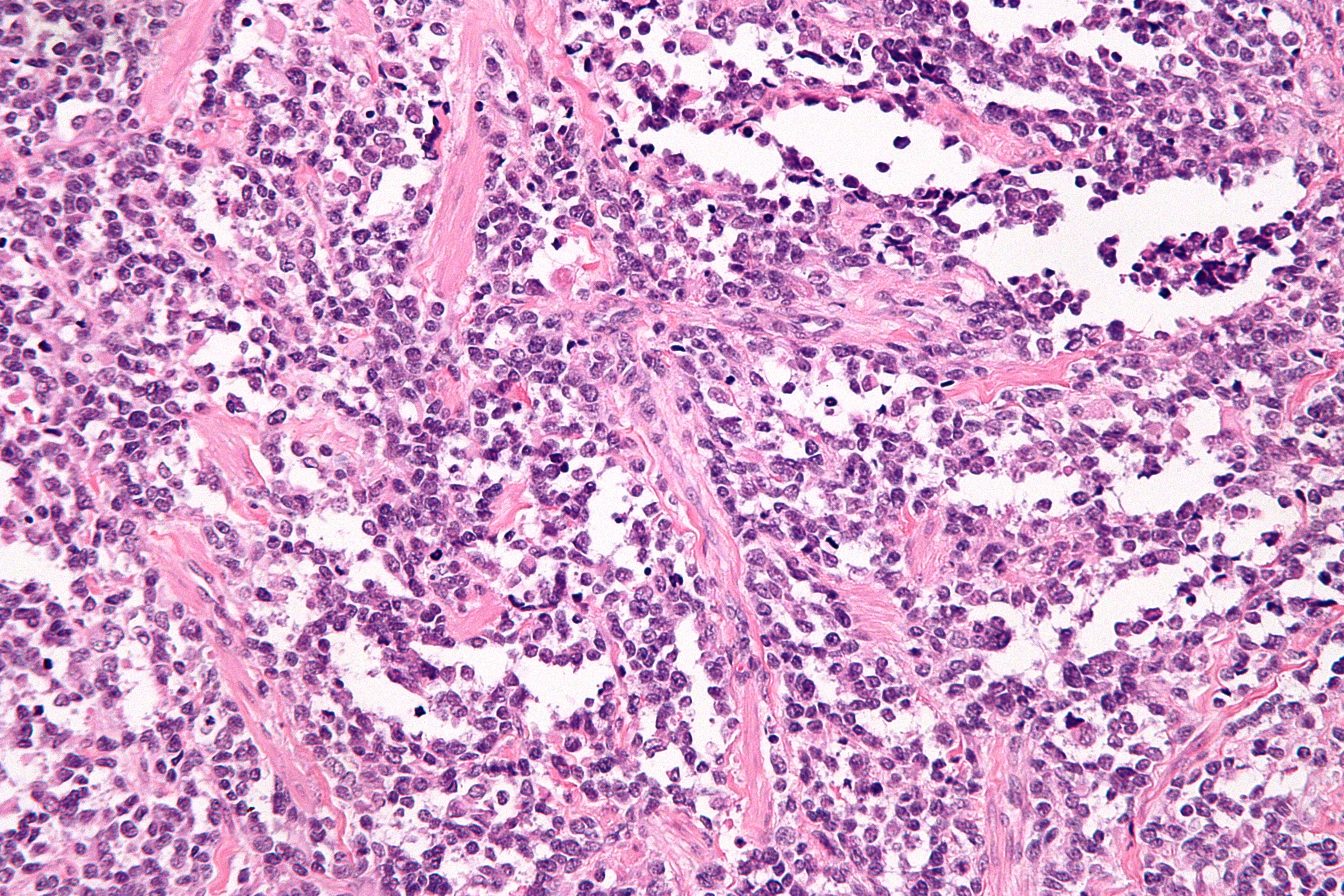
Морфологическая диагностика опухолей мягких тканей нередко вызывает затруднения из-за многообразия вариантов строения и значительного морфологического сходства опухолей различного гистогенеза. Данное обстоятельство определяется филогенетическим сходством происхождения опухолей мягких тканей: они развиваются из производных всех трех зародышевых листков, однако, наиболее часто — из мезенхимы, которая сама по себе является неоднородной. С другой стороны, часть опухолей мягких тканей с признаками той или иной терминальной дифференцировки имеют единый гистогенез, но под действием случайных соматических мутаций, эпигенетических механизмов или измененного микроокружения дифференцируются не однонаправленно.
Суммарным результатом вышеизложенного являются выраженная гетерогенность строения опухолей мягких тканей и их морфологическое сходство одновременно.
Вместе с тем в большинстве отдельных нозологических форм злокачественных опухолей мягких тканей наблюдается относительная устойчивость формальных диагностических морфологических признаков, лежащих в основе их классификации и дифференциальной диагностики, а также позволяющих определять особенности биологического поведения и прогноз клинического течения каждого конкретного образования.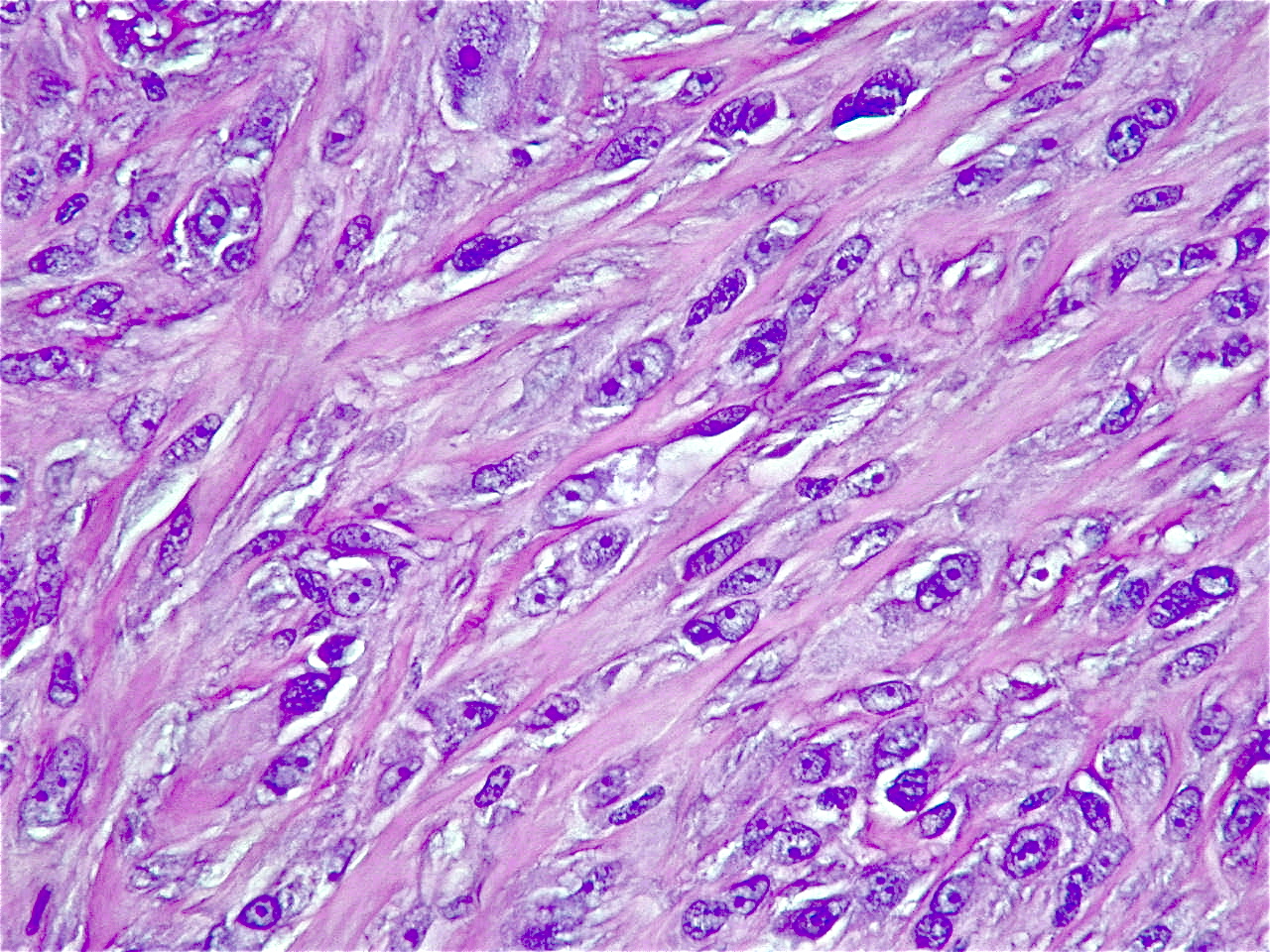
Злокачественные опухоли с признаками поперечно-полосатой мышечной дифференцировки являются структурно значимыми в аспекте терапевтических клинических программ. Учитывая важность прогностических различий, наибольший акцент в последние годы сделан на более точных и воспроизводимых дифференциально-диагностических критериях эмбрионального и альвеолярного вариантов рабдомиосаркомы. Обоснованность и целесообразность данного разграничения получена, в основном, на основании результатов цитогенетических и молекулярно-биологических исследований и подтверждена длительными клиническими наблюдениями. Морфологические признаки, позволяющие проводить данное разграничение, базируются на морфологии клеточных элементов опухоли и особенностях тканевых структур.
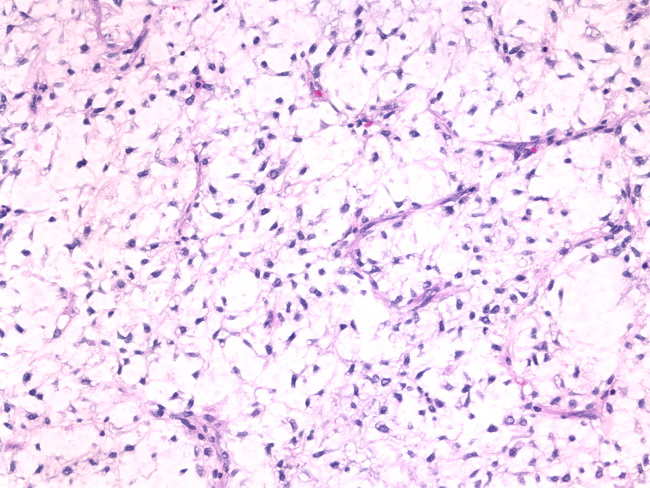
Эмбриональная рабдомиосаркома. Примитивная злокачественная опухоль мягких тканей, которая проявляет фенотипические и биологические признаки эмбриональных скелетных мышц. Эмбриональная рабдомиосаркома обычно наблюдается у детей младше 15 лет, только в 17% случаев – у больных старше этого возраста. Большая часть (46%) встречается у детей младше 5 лет, 5% — у детей до 1 года и редко – как врожденная. Гистологическое строение эмбриональной рабдомиосаркомы в целом характеризуется миксоидным характером стромы и веретеноклеточным составом. Клеточные элементы опухоли имеют вытянутую веретеновидную или звездчатую форму, неравномерно распределены среди стромального компонента. В зависимости от выраженности ядерного полиморфизма, формы и размеров клеток, количества митозов выделяют собственно веретеноклеточный и анапластический варианты эмбриональной рабдомиосаркомы. Рабдомиосаркомы, возникающие в слизистых оболочках полых органов, выделяются в отдельный вариант – ботриоидную рабдомиосаркому, которая имеет характерную макроскопическую картину в виде гроздей винограда и характеризуется преобладанием миксоидной веретеноклеточной ткани с группировкой клеточных элементов опухоли непосредственно под собственной пластинкой слизистой оболочки с формированием “камбиального слоя”.
Большая часть (46%) встречается у детей младше 5 лет, 5% — у детей до 1 года и редко – как врожденная. Гистологическое строение эмбриональной рабдомиосаркомы в целом характеризуется миксоидным характером стромы и веретеноклеточным составом. Клеточные элементы опухоли имеют вытянутую веретеновидную или звездчатую форму, неравномерно распределены среди стромального компонента. В зависимости от выраженности ядерного полиморфизма, формы и размеров клеток, количества митозов выделяют собственно веретеноклеточный и анапластический варианты эмбриональной рабдомиосаркомы. Рабдомиосаркомы, возникающие в слизистых оболочках полых органов, выделяются в отдельный вариант – ботриоидную рабдомиосаркому, которая имеет характерную макроскопическую картину в виде гроздей винограда и характеризуется преобладанием миксоидной веретеноклеточной ткани с группировкой клеточных элементов опухоли непосредственно под собственной пластинкой слизистой оболочки с формированием “камбиального слоя”.
Каждый из гистологических подтипов эмбриональной рабдомиосаркомы в соответствии с особенностями гистологического строения необходимо отличать от опухолей иного гистогенеза или иной дифференцировки.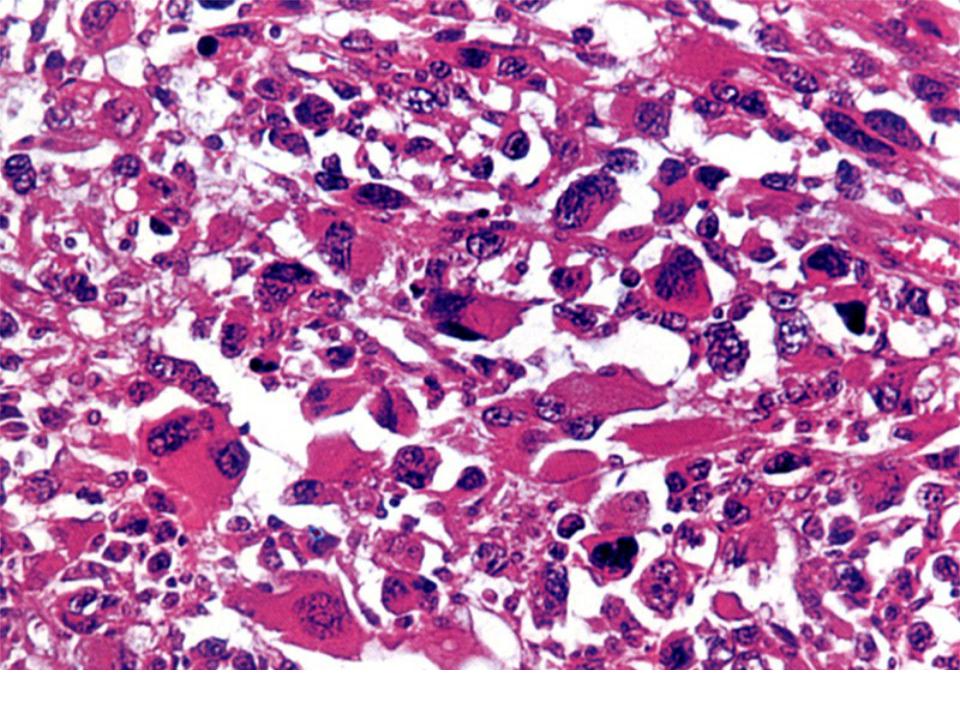 В большинстве случаев речь идет о лейомиосаркоме, монофазных синовиальных саркомах, шванномах, фибросаркомах и, в ряде случаев, о злокачественных фиброзных гистиоцитомах. Дифференциальная диагностика внутри группы веретеноклеточных опухолей на гистологическом уровне представляет значительные затруднения и чревата высокой вероятностью часто повторяющихся однотипных ошибок.
В большинстве случаев речь идет о лейомиосаркоме, монофазных синовиальных саркомах, шванномах, фибросаркомах и, в ряде случаев, о злокачественных фиброзных гистиоцитомах. Дифференциальная диагностика внутри группы веретеноклеточных опухолей на гистологическом уровне представляет значительные затруднения и чревата высокой вероятностью часто повторяющихся однотипных ошибок.
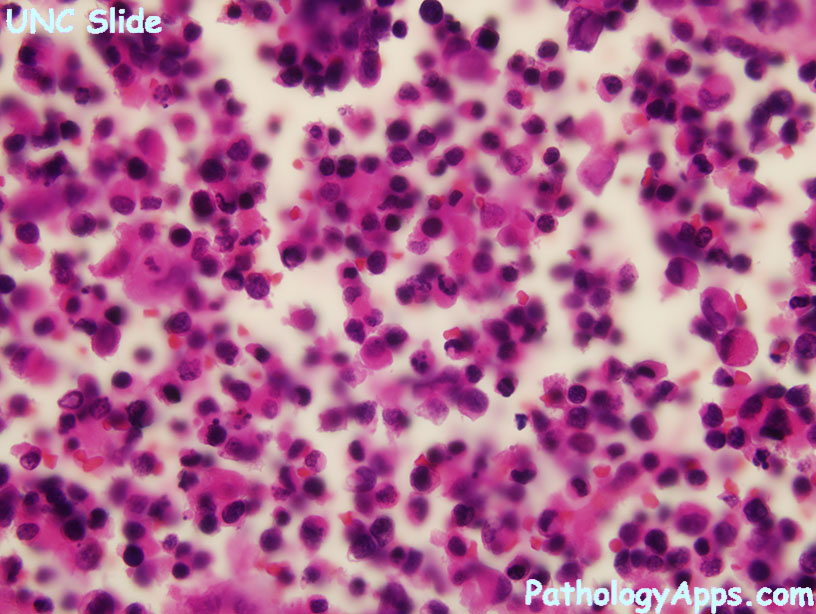
Альвеолярная рабдомиосаркома. Примитивная злокачественная круглоклеточная опухоль, которая по морфологии клеточных элементов напоминает лимфому и демонстрирует частичную поперечно-полосатую мышечную дифференцировку. Выделяют три основных гистологических варианта альвеолярной рабдомиосаркомы – типичную, солидную и смешанную альвеолярно-эмбриональная рабдомиосаркома.
Общая гистологическая характеристика альвеолярной рабдомиосаркомы, в отличие от эмбриональной, при условии сохранения признаков поперечно-полосатой дифференцировки, определяется иным, фиброзным характером стромы, образующей альвеолярные и псевдоальвеолярные структуры, выстланные опухолевыми клетками.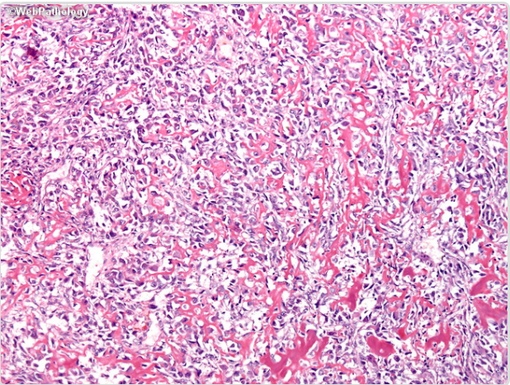 По соотношению стромального и клеточного компонентов в альвеолярной рабдомиосаркоме выделены типичный и солидный варианты. Клеточные элементы, составляющие гистологический субстрат альвеолярной рабдомиосаркомы, имеют преимущественно округлую форму с вариабельным ядерно-цитоплазматическим соотношением. Среди этих клеток часто определяются клеточные элементы с резко гиперхромными ядрами, признаками повреждения ядра, кариорексиса. Могут встречаться многоядерные клетки, клетки с эпителиоидной дифференцировкой, однако, большинство клеток опухоли не имеют признаков какой-либо дифференцировки.
По соотношению стромального и клеточного компонентов в альвеолярной рабдомиосаркоме выделены типичный и солидный варианты. Клеточные элементы, составляющие гистологический субстрат альвеолярной рабдомиосаркомы, имеют преимущественно округлую форму с вариабельным ядерно-цитоплазматическим соотношением. Среди этих клеток часто определяются клеточные элементы с резко гиперхромными ядрами, признаками повреждения ядра, кариорексиса. Могут встречаться многоядерные клетки, клетки с эпителиоидной дифференцировкой, однако, большинство клеток опухоли не имеют признаков какой-либо дифференцировки.
Гистологический тип рабдомиосаркомы с сочетанием наиболее специфичных признаков альвеолярной и эмбриональной рабдомиосаркомы принято относить к смешанному, альвеолярно-эмбриональному варианту альвеолярной рабдомиосаркомы.
Альвеолярный вариант рабдомиосаркомы необходимо дифференцировать с лимфомами, группой мелко-круглоклеточных опухолей, саркомой Юинга, десмопластической мелко-круглоклеточной опухолью и, иногда — с герминоклеточными опухолями.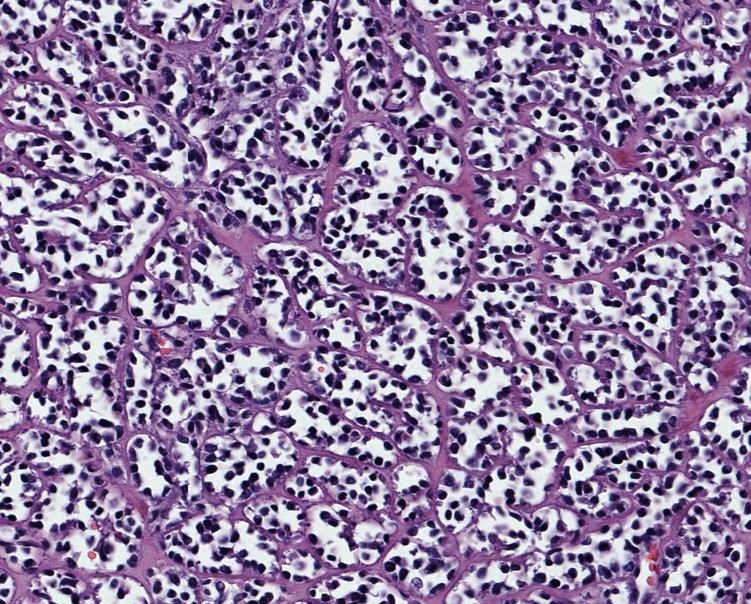 Как и в случае с эмбриональным вариантом рабдомиосаркомы, дифференциальный диагноз на уровне гистологического исследования весьма сложен.
Как и в случае с эмбриональным вариантом рабдомиосаркомы, дифференциальный диагноз на уровне гистологического исследования весьма сложен.
Отдельно описываемый вариант плеоморфной рабдомиосаркомы встречается крайне редко и описан только у взрослых. По гистологической картине этот вариант не отличается от других плеоморфных сарком, и его отношение к группе рабдомиосарком может быть установлено только с использованием специальных методов исследования. Следует отметить, что выделение различных вариантов рабдомиосаркомы возможно только на гистологическом уровне, так как иные характеристики (антигенный состав и генетические особенности) в большинстве случаев имеют групповую принадлежность и, следовательно, являются общими для всех вариантов рабдомиосарком. Выявление же молекулярно-биологических аномалий, к сожалению, не всегда доступно.
Несмотря на всю сложность и относительность гистологической субклассификации, определение гистологического типа рабдомиосаркомы является обязательной и клинически значимой характеристикой. Прогноз клинического течения определяется стадией, гистологической характеристикой, возрастом больного и локализацией образования.
Прогноз клинического течения определяется стадией, гистологической характеристикой, возрастом больного и локализацией образования.
Гистологическая классификация опухолей мягких тканей предполагает определение антигенного набора клеточных элементов опухоли. На основании данных иммуногистохимических исследований разработаны критерии, позволяющие установить гистогенез или терминальную дифференцировку опухоли. Эти критерии лежат в основе дифференциальной диагностики большинства групп опухолей, включая опухоли мягких тканей. Злокачественные опухоли поперечно-полосатой мышечной ткани характеризуются экспрессией широкого спектра мышечно-специфических иммунологических маркеров. При этом последовательность экспрессии и набор экспрессируемых антигенов коррелирует со степенью дифференцировки клеточных элементов так, как это происходит в эмбриогенезе. Наименее дифференцированные клетки экспрессируют только виментин. Об увеличении степени дифференцировки может свидетельствовать появление экспрессии десмина и мышечно-специфического актина.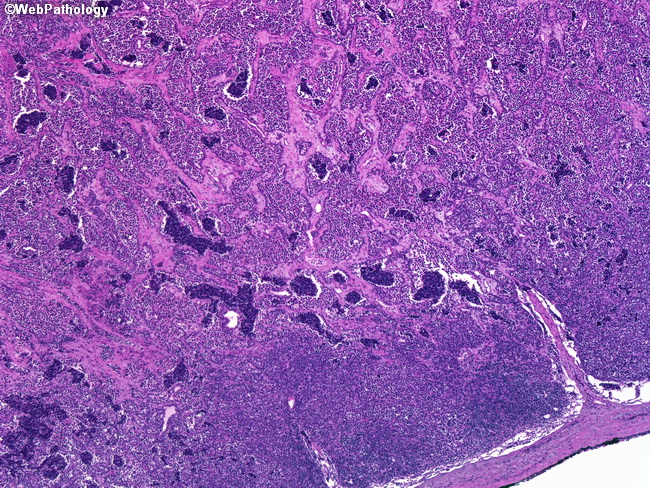 Дифференцированные клетки характеризуются позитивным связыванием с антителами к миоглобину, миозину и рецепторам креатин киназы М. Для определения мышечной дифференцировки могут быть использованы антитела к титину, дистрофину и рецепторам ацетилхолин эстеразы.
Дифференцированные клетки характеризуются позитивным связыванием с антителами к миоглобину, миозину и рецепторам креатин киназы М. Для определения мышечной дифференцировки могут быть использованы антитела к титину, дистрофину и рецепторам ацетилхолин эстеразы.
Следует помнить, что мышечные маркеры десмин и мышечно-специфический актин определяются на мышечной ткани вне зависимости от направления ее дифференцировки, т.е. в клетках с общим мышечным фенотипом, включая гладкие мышцы, сердце, миофибробласты, миоэпителиальные клетки, перициты и отдельные мезотелиальные клетки.
Антитела к MyoD1 и миогенину (Myf4) являются высокоспецифичными в отношении рабдомиосаркомы, так как соответствующие антигенные детерминанты появляются в результате приобретения специфических генетических аномалий, и на сегодняшний день считаются стандартом для постановки диагноза. Однако следует заметить, что только ядерная экспрессия может считаться специфичной, в отличие от широко встречаемой неспецифической цитоплазматической экспрессии.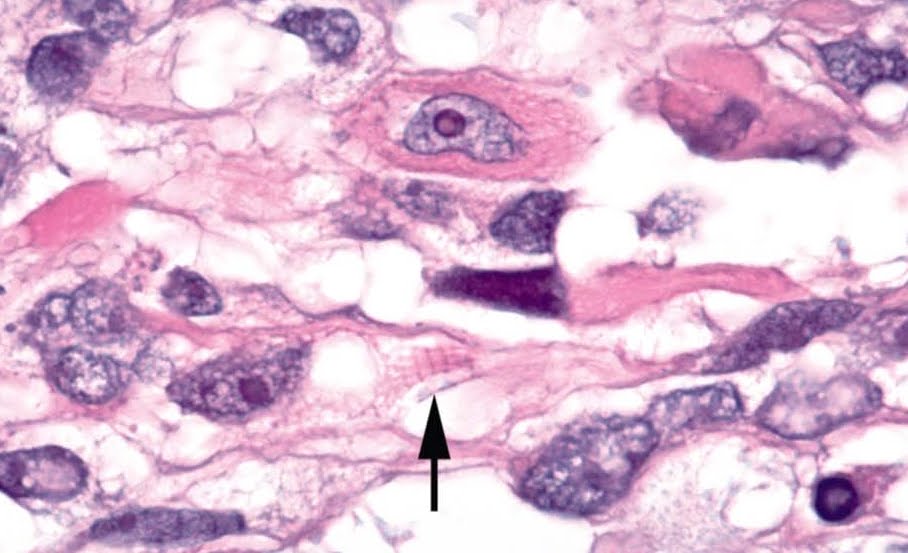
Наряду с экспрессией мышечных антигенов в части рабдомиосарком сохраняется абберантная экспрессия родственных маркеров, таких как S-100 протеин, нейрон-специфическая енолаза, синаптофизин, хромогранин А, NB84.
Таким образом, постановка диагноза рабдомиосаркомы требует тщательной гистологической и иммуногистохимической оценки патологических изменений, сравнительного анализа полученных данных с учетом особенностей гистологического строения и иммунофенотипа опухолей иного гистогенеза. Только комплексный морфологический подход с учетом возраста и локализации, а также тесное взаимодействие с врачами клинической службы может обеспечить адекватный результат.
Рабдомиосаркома | Фонд «Подари жизнь»
Суть болезни
Рабдомиосаркома (РМС) – одна из злокачественных опухолей, характерных в основном для детского возраста. Понятие «саркома» означает, что речь идет о злокачестенной опухоли соединительной ткани, а приставка «рабдомио» означает, что это опухоль возникает из клеток-предшественников поперечно-полосатых, то есть скелетных, мышц («рабдо» — палочкообразный, «мио» — мышечный).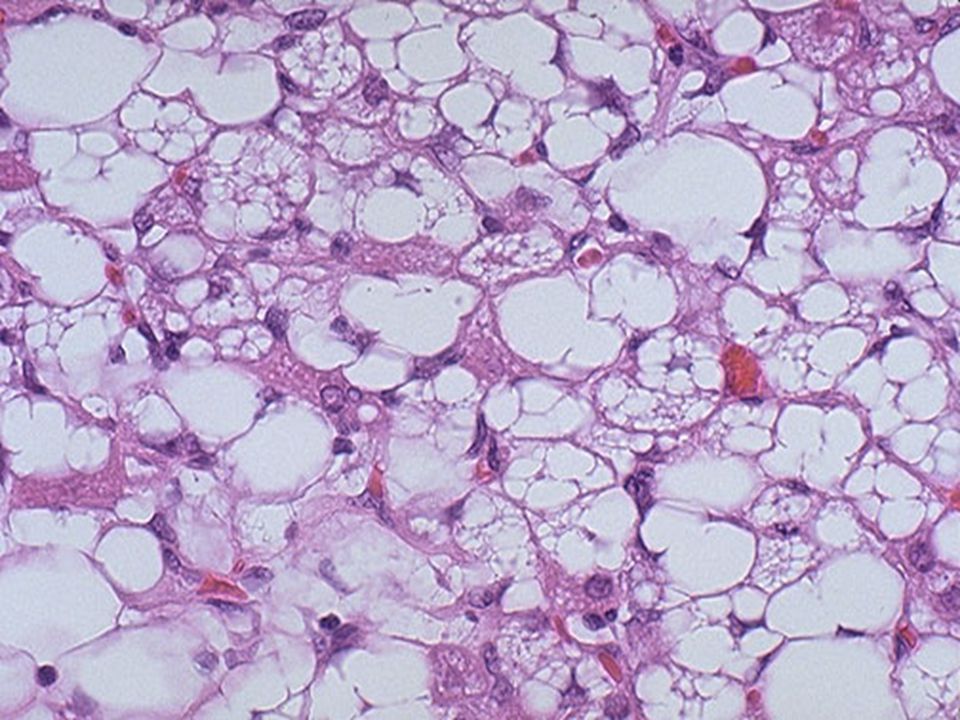
Рабдомиосаркома относится к саркомам мягких тканей. Существуют и другие мягкотканые саркомы: липосаркома, ангиосаркома, фибросаркома, синовиальная саркома и т.п. РМС выделяется среди них более частой встречаемостью в детском возрасте, поэтому именно это заболевание рассматривается здесь подробно.
РМС может обнаруживаться практически в любом участке тела, включая и области, не заполненные мышцами. Так, РМС может возникнуть в различных органах головы и шеи (включая мягкие ткани глазных орбит, носоглотку, придаточные пазухи носа, область возле шейного отдела позвоночника), в руках, ногах, мочеполовой системе, в брюшной полости и др. Метастазы могут обнаруживаться в легких, костном мозге, костях, лимфоузлах, головном мозге.
В соответствиии с международной классификацией выделяют различные типы РМС.
- Эмбриональная рабдомиосаркома – опухоль, характерная для детей раннего возраста. Большинство случаев РМС относится именно к этому варианту.
 Наиболее частые локализации опухоли при эмбриональной РМС – органы головы и шеи, органы мочеполовой системы. Выделяют также особые подтипы эмбриональной РМС – ботриоидный и веретеноклеточный; как правило, они связаны с лучшим прогнозом.
Наиболее частые локализации опухоли при эмбриональной РМС – органы головы и шеи, органы мочеполовой системы. Выделяют также особые подтипы эмбриональной РМС – ботриоидный и веретеноклеточный; как правило, они связаны с лучшим прогнозом. - Альвеолярная рабдомиосаркома (около 25% всех случаев РМС) чаще встречается в более старшем возрасте, включая подростковый. Название «альвеолярная» связано с тем, что внешний вид опухоли при ее микроскопическом исследовании напоминает вид легочных альвеол. Наиболее типичные локализации – мышцы конечностей и туловища, а также органы малого таза.
Выделяют также плеоморфную, или анапластическую РМС. Этот редкий вариант опухоли не характерен для детей (большинство заболевших – взрослые в возрасте от 30 до 50 лет) и обычно локализуется на конечностях. Также известны смешанные варианты и недифференцированные саркомы.
При РМС выделяют различные стадии опухолевого процесса и клинические группы. Так, согласно международной классификации IRS, выделяются 4 группы:
Так, согласно международной классификации IRS, выделяются 4 группы:
- Группа I: Опухоль может быть полностью удалена хирургически.
- Группа II: Опухоль может быть удалена хирургически, но опухолевые клетки обнаруживаются в окружающих тканях и/или в близлежащих лимфоузлах. Отдаленных метастазов нет.
- Группа III: Полное хирургическое удаление опухоли невозможно. Нет отдаленных метастазов.
- Группа IV: В момент установления диагноза обнаруживаются отдаленные метастазы.
Применяется также определение стадии болезни согласно общепринятой системе TNM, где T соответствует размеру опухоли, N – поражению регионарных (близлежащих) лимфатических узлов, а M – наличию отдаленных метастазов.
Частота встречаемости и факторы риска
Рабдомиосаркома составляет около 4% случаев злокачественных новообразований детского возраста; частота ее приблизительно равна 6 случаям на 1 миллион детского населения.
РМС – опухоль, характерная почти исключительно для детского возраста. После 20 лет она встречается уже крайне редко. Большинство больных младше 10 лет. Мальчики болеют несколько чаще девочек. Факторы внешней среды, которые могли бы повлиять на частоту возникновения РМС, неизвестны, как и для большинства других опухолей детского возраста.
Частота возникновения РМС, как и ряда других злокачественных опухолей, несколько повышена при определенных наследственных заболеваниях – таких как нейрофиброматоз типа I, синдром Ли-Фраумени и т.д. Однако в подавляющем большинстве случаев появление опухоли не связано ни с какими врожденными генетическими аномалиями.
Признаки и симптомы
Симптомы РМС зависят прежде всего от расположения первичной опухоли. К счастью, нередко ее удается обнаружить сравнительно рано, так как РМС часто возникает в тех областях, где она быстро становится заметной. Приблизительно в 1/3 случаев опухоль удается обнаружить в момент, когда ее еще можно практически полностью удалить хирургическим путем (хотя обычно при этом есть микрометастазы или остаточная опухоль, для лечения которых необходима химиотерапия). И менее чем у 20% больных обнаруживается опухоль уже с отдаленными метастазами.
И менее чем у 20% больных обнаруживается опухоль уже с отдаленными метастазами.
Так, опухоль, расположенную неглубоко под кожей, можно либо заметить визуально, то есть по возникновению припухлости, либо прощупать (пропальпировать). Если опухоль развивается в тканях глазной орбиты, то глаз выпячивается (экзофтальм) или начинает косить; возможны также жалобы на двоение в глазах. Если опухоль возникает в носовой полости, то болезнь может проявляться заложенностью носа, кровотечениями или кровянисто-слизистыми выделениями. Если поражен слуховой проход, то могут обнаруживаться выделения из уха и/или ухудшение слуха. При опухоли мочевого пузыря возможны затруднения с мочеиспусканием или наличие крови в моче. При опухолях в области половых органов нередко возникает отечность мошонки у мальчиков и кровянистые или слизистые выделения у девочек. При опухолях в брюшной области или области таза могут наблюдаться боли в животе, запоры, рвота. В редких случаях рабдомиосаркома развивается в области желчных протоков и приводит к желтухе.
Что касается опухолей, поражающих конечности (обычно это альвеолярная РМС у старших детей), то их не всегда сразу правильно диагностируют, поскольку часто принимают за результат ушиба. Поэтому в случае возникновения любых припухлостей и «шишек», быстро растущих или не проходящих в течение нескольких недель, следует обратиться к врачу. Следует отметить, что при РМС эти «шишки» обычно безболезненны.
При дальнейшем развитии болезни возникают симптомы общего характера: вялость, снижение аппетита, потеря веса, слабость. При метастазах в лимфоузлы наблюдается их увеличение.
Диагностика
При подозрении на РМС обязательно осуществляется открытая биопсия и анализ полученного образца ткани, при котором уточняется диагноз и устанавливается вариант РМС. Опухолевая ткань исследуется под микроскопом; производится также иммуногистохимическое исследование на клеточные маркеры, характерные для рабдомиосаркомы.
Эмбриональная и альвеолярная РМС различаются по микроскопической картине опухоли. Кроме того, они имеют свои хромосомные особенности, поэтому для уточнения диагноза применяются цитогенетические (в некоторых случаях – молекулярно-генетические) исследования. Так, для альвеолярной РМС характерна транслокация t(1;13) или t(2;13).
Кроме того, они имеют свои хромосомные особенности, поэтому для уточнения диагноза применяются цитогенетические (в некоторых случаях – молекулярно-генетические) исследования. Так, для альвеолярной РМС характерна транслокация t(1;13) или t(2;13).
Чтобы оценить размеры опухоли, степень ее проникновения в окружающие ткани, наличие и расположение метастазов, используются различные визуализирующие исследования: ультразвуковое исследование (УЗИ), компьютерная томография (КТ), магнитно-резонансная томография (МРТ). Для уточнения полученных данных может применяться также позитронно-эмиссионная томография (ПЭТ). Так как при РМС одной из обычных областей метастазирования являются легкие, производится рентгенография легких. Для выявления возможных костных метастазов используется остеосцинтиграфия с технецием (99Tc).
Поражение костного мозга устанавливается или исключается при помощи анализа его образца, взятого в ходе костномозговой пункции. Если опухоль находится близко к оболочкам головного или спинного мозга, то может также производиться пункция спинномозгового канала для исследования ликвора.
Лечение
Стратегия лечения при РМС зависит от группы риска (которая определяется многими факторами, включая стадию болезни, ее конкретный вариант, возраст больного, местоположение опухоли и т.п.), но практически всегда она включает в себя оперативное лечение и химиотерапию. Часто проводится также лучевая терапия.
Хирургическое удаление опухоли при РМС головы и шеи часто затруднено, так как опухоль окружена жизненно важными структурами. Поэтому стратегия хирургического лечения может корректироваться: так, нередко невозможна широкая резекция опухоли (с «захватом» достаточного количества здоровых окружающих тканей). Кроме того, может понадобиться помощь нейрохирургов и/или специалистов по сосудистой и пластической хирургии.
Если опухоль поражает руку или ногу, то в некоторых случаях для увеличения шансов на выживание может потребоваться ампутация, так как опухоли этой локализации часто бывают агрессивными и быстро метастазируют.
Химиотерапия при РМС может применяться до хирургического удаления опухоли с целью уменьшения ее размера (неоадъювантная химиотерапия).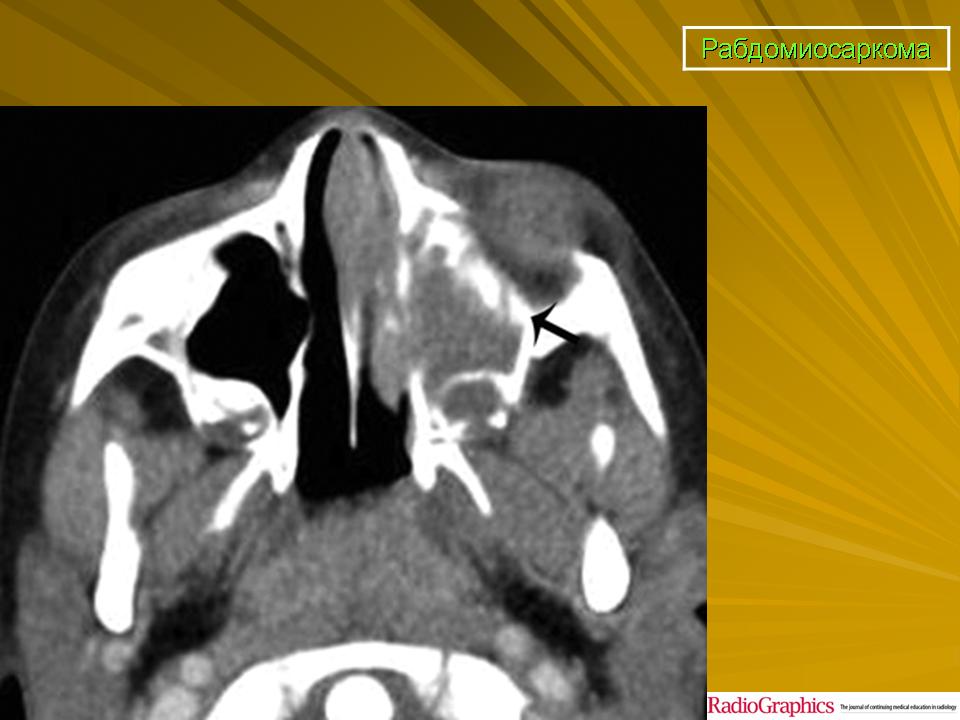 Что же касается периода после удаления опухоли, даже если оно произведено полностью, то в это время химиотерапия обязательна для уничтожения оставшихся опухолевых клеток – иначе вероятность рецидива резко повышается.
Что же касается периода после удаления опухоли, даже если оно произведено полностью, то в это время химиотерапия обязательна для уничтожения оставшихся опухолевых клеток – иначе вероятность рецидива резко повышается.
Выбор конкретных химиопрепаратов и их комбинации зависят от вида и стадии РМС. В число препаратов, которые могут применяться для химиотерапии РМС, входят винкристин, дактиномицин (космеген), циклофосфамид, а также ифосфамид, этопозид, доксорубицин, препараты платины и другие химиопрепараты. Иногда используются также препараты из числа ингибиторов топоизомеразы I: топотекан, иринотекан.
Лучевая терапия чаще всего применяется для уничтожения оставшихся злокачественных клеток после операции и последующей химиотерапии, но иногда может проводиться и до операции. Если опухоль расположена недалеко от оболочек головного или спинного мозга (параменингеальная локализация) или распространяется в кости черепа и/или центральную нервную систему, облучение может быть начато сразу после постановки диагноза.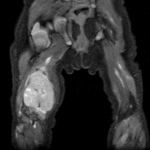
В некоторых случаях при РМС могут быть рекомендованы особые методы лучевой терапии, включая протонное облучение и брахитерапию.
При высоком риске может быть рекомендована высокодозная химиотерапия с последующей аутологичной трансплантацией костного мозга. Однако до сих пор неясно, приводит ли она к существенному повышению выживаемости.
Прогноз
Как и при большинстве злокачественных опухолей, прогноз при РМС зависит от многих факторов: стадия, на которой диагностирована болезнь; размер и локализация опухоли; возможность ее полного или почти полного хирургического удаления; возраст больного; цитогенетические характеристики клеток.
За последние десятилетия в лечении рабдомиосаркомы был достигнут серьезный прогресс. При локализованной (не метастазировавшей) опухоли прогноз достаточно хороший: примерно 80% таких больных излечивается. Прогноз при распространенной опухоли с отдаленными метастазами намного хуже: выздоравливает около 30% больных. Наилучшие результаты достигаются при лечении опухолей, расположенных в области головы и шеи, а также половых органов. Наиболее благоприятная возрастная группа – дети от 1 до 9 лет. Альвеолярная РМС в среднем имеет худший прогноз, чем эмбриональная РМС.
Наиболее благоприятная возрастная группа – дети от 1 до 9 лет. Альвеолярная РМС в среднем имеет худший прогноз, чем эмбриональная РМС.
Как и при большинстве онкологических заболеваний, пятилетняя ремиссия фактически означает выздоровление, так как вероятность возвращения болезни уже невелика. Однако возможны отдаленные последствия лечения, прежде всего лучевой терапии, применяемой при лечении маленьких детей: замедление роста облученных участков костей, ухудшение зрения или катаракта при облучении области глаз, рубцевание легочной ткани при облучении легких, проблемы с репродуктивной функцией и так далее. После лечения могут возникнуть вторичные опухоли, однако их частота низка. В случае локального рецидива РМС прогноз ухудшается, однако в ряде случаев все равно возможно эффективное лечение.
Петербургская школа магнитно-резонансной томографии
» Лучевая диагностика » ЛУЧЕВАЯ ДИАГНОСТИКА БОЛЕЗНЕЙ » Злокачественные опухоли мягких тканей и их диагностика
Описаны различные гистологические варианты злокачественных опухолей мягких тканей, их частота, клинические проявления, методы диагностики. МРТ семиотика различных злокачественных опухолей мягких тканей.
МРТ семиотика различных злокачественных опухолей мягких тканей.
При МРТ в СПб нам периодически приходится исследовать мягкие ткани разных областей на предмет опухоли.
Адипозитарные опухоли
Липосаркома – имеет несколько гистологических вариантов, из которых все, за исключением хорошо дифференцированной, относятся к высокозлокачественным. На КТ опухоли не имеют четких границ, структура их смешанная, с солидными и псевдокистозными участками. При УЗИ липосаркома также демонстрирует инфильтративный рост и смешанный тип эхогенности . При МРТ сигнал зависит от наличия жира, макроскопический жир содержится только в хорошодифференцированной липосаркоме. Соответственно, подавления сигнала от жира обычно не наблюдается.
Липосаркома мягких тканей. Т1-зависимая МРТ.
Опухоли скелетных мышц
Рабдомиосаркома – одна из самых частых злокачественных опухолей у детей, составляя 5-8% всех опухолей детского возраста. Примерно 65% рабдомиосарком встречается в возрасте до 10 лет. У лиц старше 45 лет они практически не наблюдаются. Рабдомиосаркомы могут локализоваться по всему телу, до 50% в области головы и шеи, затем мочеполового тракта и только 15% в области конечностей. Большинство рабдомиосарком относятся к эмбриональному типу. На рентгенограммах картина неспецифическая, может отмечаться деформация прилежащей длинной трубчатой кости. При УЗИ опухоль гетерогенная, четко, очерченная, низкой или смешанной эхогенности. При КТ рабдомиосаркома мягкотканой плотности, со слабым контрастированием, в 20% отмечается разрушение прилегающих костных структур. При МРТ диагностике на Т2-ВИ опухоль гиперинтенсивна, может включать сосуды, на Т1-ВИ отмечаются кровоизлияний. Контрастируется хорошо. В целом эмбриональные рабдомиосаркомы более однородны, чем альвеолярные и плеоморфные.
Примерно 65% рабдомиосарком встречается в возрасте до 10 лет. У лиц старше 45 лет они практически не наблюдаются. Рабдомиосаркомы могут локализоваться по всему телу, до 50% в области головы и шеи, затем мочеполового тракта и только 15% в области конечностей. Большинство рабдомиосарком относятся к эмбриональному типу. На рентгенограммах картина неспецифическая, может отмечаться деформация прилежащей длинной трубчатой кости. При УЗИ опухоль гетерогенная, четко, очерченная, низкой или смешанной эхогенности. При КТ рабдомиосаркома мягкотканой плотности, со слабым контрастированием, в 20% отмечается разрушение прилегающих костных структур. При МРТ диагностике на Т2-ВИ опухоль гиперинтенсивна, может включать сосуды, на Т1-ВИ отмечаются кровоизлияний. Контрастируется хорошо. В целом эмбриональные рабдомиосаркомы более однородны, чем альвеолярные и плеоморфные.
Рабдомиосаркома кисти. Т1-зависимая томограмма с контрастированием.
Недифференцированные саркомы
Недифференцированная плеоморфная саркома – одна из наиболее частых злокачественных мягкотканных опухолей у взрослых. Может локализоваться в любой части тела, но обычно в забрюшинном пространстве и проксимальных отделах конечностей. Различные гистологические подтипы недифференцированных сарком отражают клеточный состав и большую или меньшую степень агрессивности. На рентгенограммах выглядят как мягкотканое образование без четких границ, иногда с линейными или точечными включениями кальция. На КТ плотность не отличается от окружающей здоровой мышечной ткани, в опухоли присутствуют очаги некроза, кровоизлияния и могут быть кальцинаты. При МРТ в толще мышечной ткани определяется ограниченное образование промежуточного сигнала, неоднородной структуры, с хорошо контрастирующимся солидным компонентом.
Может локализоваться в любой части тела, но обычно в забрюшинном пространстве и проксимальных отделах конечностей. Различные гистологические подтипы недифференцированных сарком отражают клеточный состав и большую или меньшую степень агрессивности. На рентгенограммах выглядят как мягкотканое образование без четких границ, иногда с линейными или точечными включениями кальция. На КТ плотность не отличается от окружающей здоровой мышечной ткани, в опухоли присутствуют очаги некроза, кровоизлияния и могут быть кальцинаты. При МРТ в толще мышечной ткани определяется ограниченное образование промежуточного сигнала, неоднородной структуры, с хорошо контрастирующимся солидным компонентом.
МРТ костей и суставов адреса и цены
В частном центре ЦМРТ профессор Холин А.В. лично диагностирует на МРТ аппарате открытого типа по средам. МРТ в СПб при боязни замкнутого пространства и МРТ при большом весе. Можно сделать МРТ суставов и костей дешево и по акциям. На закрытом аппарате 1,5 Тл прием по воскресеньям и понедельникам.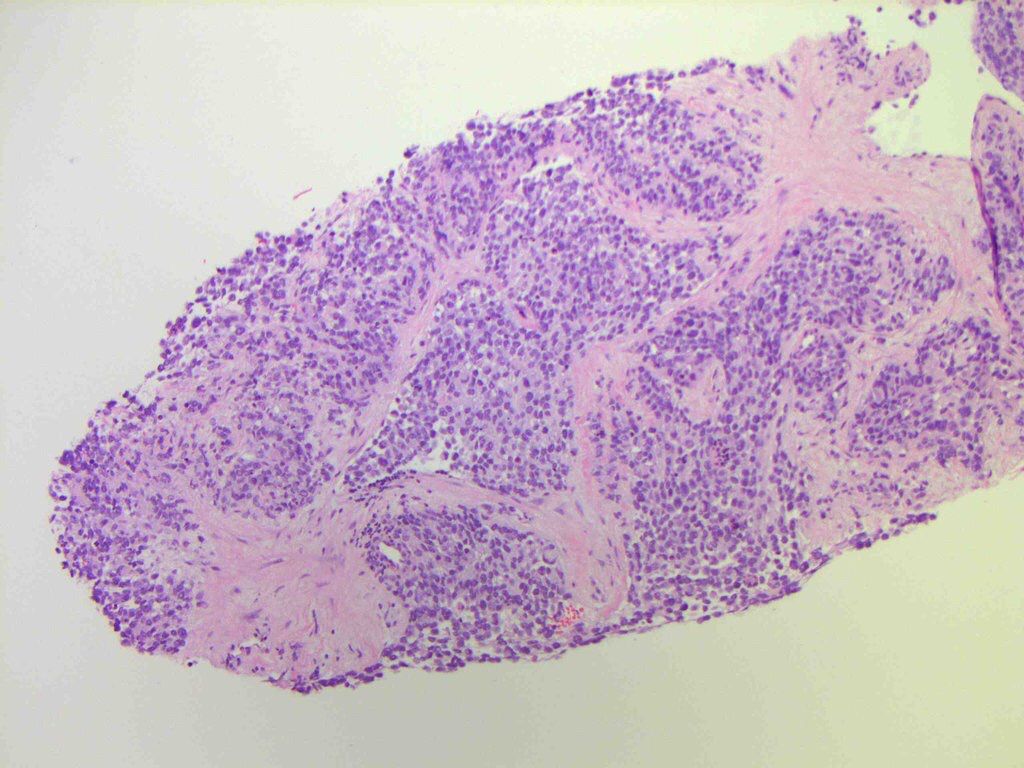
Спрашивайте МРТ цены у администратора.
Огромные МРТ учебные ресурсы на наших сайтах mrtspb.info и www.mri-kholin.ru по всем проблемам МРТ и ультразвуковой диагностики
Клинический случай химиолучевой терапии недифференцированной плеоморфной саркомы параменингеальной локализации у ребенка | Усычкина
1. Radzikowska J., Kukwa W., Kukwa A., Czarnecka A., Krzeski A. Rhabdom-yosarcoma of the head and neck in children. Contemp Oncol (Pozn) 2015; 19 (2): 98-107.
2. Chang R.C., Dave S.P., Robinson P.G. Undifferentiated pleomorphic sarcoma of the parotid gland: A rare pediatric case. Head Neck 2008; 30 (7): 970-3.
3. Clark D.W., Moore B.A., Patel S.R., Guadagnolo B.A., Roberts D.B., Sturgis E.M. Malignant fibrous histiocytoma of the head and neck region. Head Neck 2011; 33 (3): 303-8.
Head Neck 2011; 33 (3): 303-8.
4. Alaggio R., Collini P., Randall R.L., Barnette P., Million L., Coffin C.M. Undifferen-tiated high-grade pleomorphic sarcomas in children: A clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 2010; 13 (3): 209-17.
5. Paulino A.C., Fowler B.Z. Secondary neoplasms after radiotherapy for a childhood solid tumor. Pediatr Hematol Oncol 2005 Mar; 22 (2): 89-101.
6. Mohan R.P., Verma S., Siddhu V.K., Agarwal N. Malignant fibrous histiocytoma. BMJ Case Rep 2013 (31): 2013.
7. Rosenberg A.E. Malignant fibrous histiocytoma: Past, present, and future. Skeletal Radiol 2003; 32 (11): 613-8.
8. Fletcher C.D. The evolving classification of soft tissue tumours — an update based on the new 2013 WHO classification. Histopathology 2014; 64 (1): 2-11.
Fletcher C.D. The evolving classification of soft tissue tumours — an update based on the new 2013 WHO classification. Histopathology 2014; 64 (1): 2-11.
9. Ding G.X., Duggan D.M., Coffey C.W., Deeley M., Hallahan D.E., Cmelak A., Malcolm A. A study on adaptive IMRT treatment planning using kv cone-beam CT. Radiother Oncol 2007 Oct; 85 (1): 116-25.
10. Brouwer C.L., Steenbakkers R.J., Bourhis J., Budach W., Grau C., Grégoire V., et al. CT-based delineation of organs at risk in the head and neck region: DAHANCA, EORTC, GORTEC, HKNPCSG, NCIC CTG, NCRI, NRG oncology and TROG consensus guidelines. Radiother Oncol 2015 Oct; 117 (1): 83-90
Рабдомиосаркома у детей и взрослых – диагностика и лечение опухоли
На долю этого злокачественного онкологического новообразования приходится 4% всех раковых заболеваний.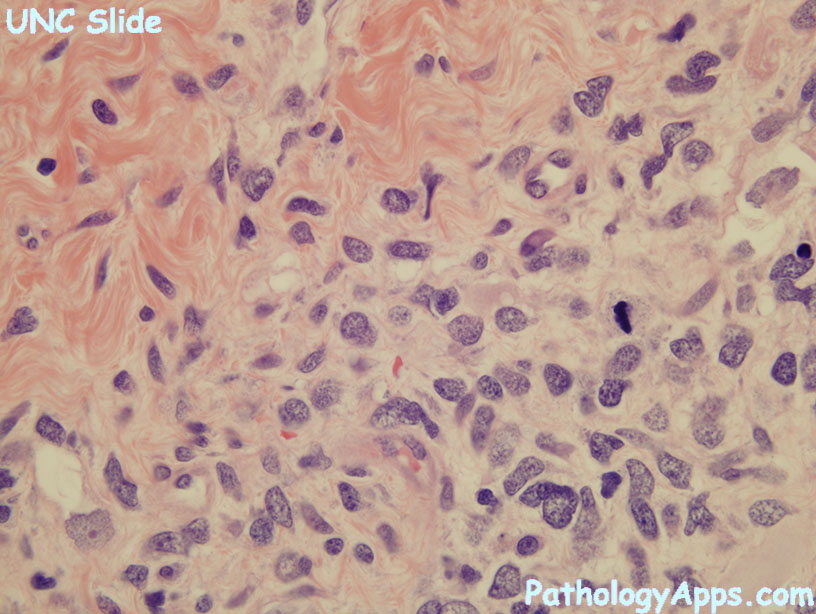 Рабдомиосаркома представляет собой атипичную мутацию тканей, при которой в них образуется постоянно растущая опухоль, способная поражать кости и метастазировать в органы. Возникает она на любых частях тела, однако особенно часто рабдомиосаркома мягких тканей поражает область таза, шеи и головы. Жертвой страшной болезни становятся дети 1-7 лет, подростки 15-20 и пожилые люди старше 50.
Рабдомиосаркома представляет собой атипичную мутацию тканей, при которой в них образуется постоянно растущая опухоль, способная поражать кости и метастазировать в органы. Возникает она на любых частях тела, однако особенно часто рабдомиосаркома мягких тканей поражает область таза, шеи и головы. Жертвой страшной болезни становятся дети 1-7 лет, подростки 15-20 и пожилые люди старше 50.
Причины возникновения опухоли
Причинами развития рабдомиосаркомы принято считать ионизирующие излучения, наследственные факторы, генетические мутации и травмы, но, увы, установить их на 100% современной медицине пока не удалось.
Рабдомиосаркома делится на эмбриональную, альвеолярную и плеоморфную. Первая развивается после рождения ребенка и преимущественно поражает шею, голову, органы мочеиспускательной системы. Вторая возникает у пациентов подросткового возраста. Свое название она получила по причине схожести с легочными альвеолами. Плеоморфная жертвами выбирает взрослых людей, поражает конечности и туловище.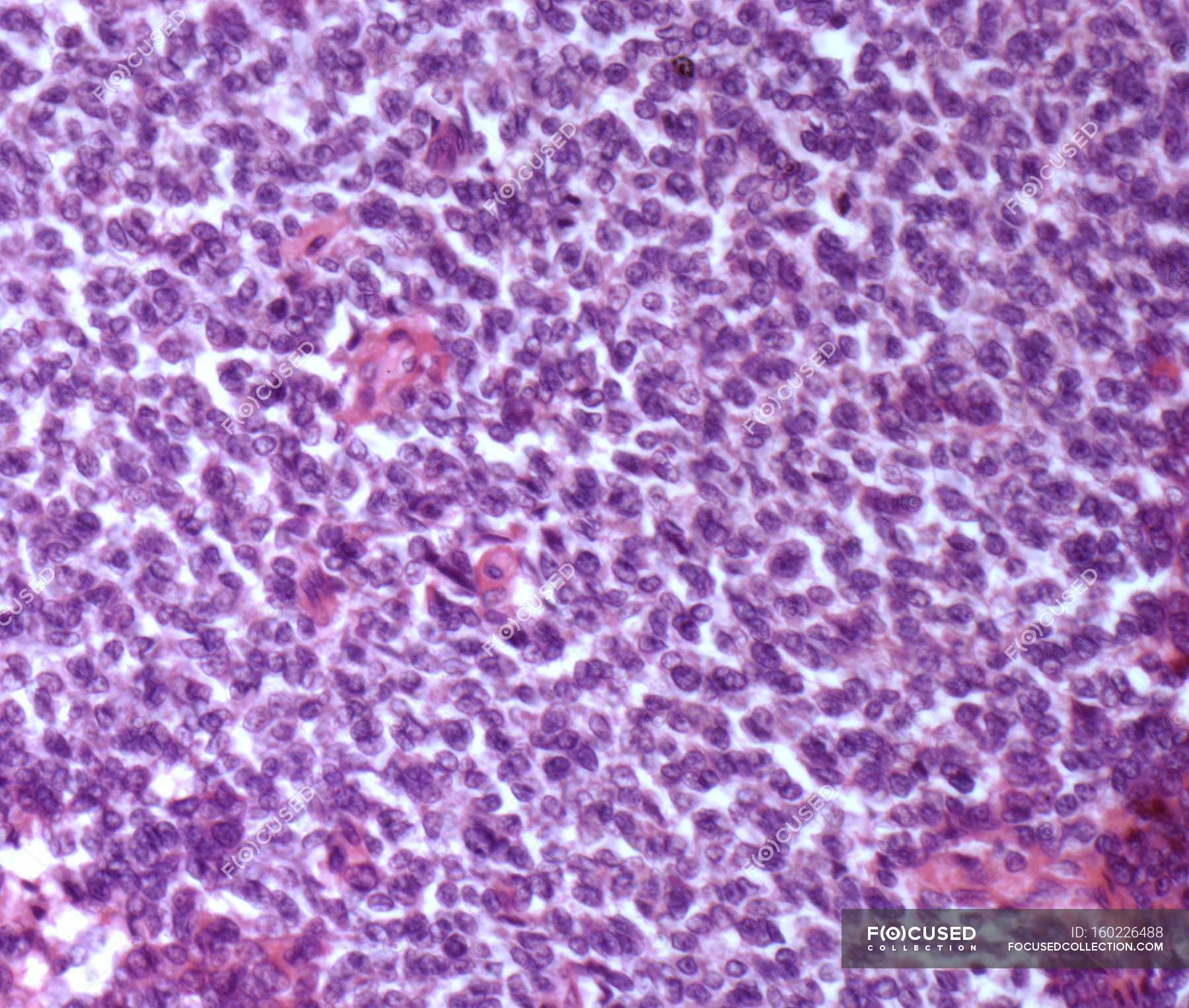
Желаете получить бесплатную консультацию — отправьте нам заявку
или обратитесь по телефону +972-77-4450-480 или +8-800-707-6168 (для жителей России бесплатно).
Симптомы рабдомиосаркомы
По своему строению ткани опухоли похожи на рабдомиопласты – клетки малышей, еще находящихся в утробе матери, которые должны превратиться в мускулы. Эмбриональная рабдомиосаркома мягких тканей поражает части тела, где нет крупных мышц. Мальчики попадают под удар болезни чаще девочек. В период развития болезни нарушается процесс обмена веществ в клетках поперечнополосатой мускулатуры; они стремительно делятся, разрастаются в количестве и образуют опухоль.
Симптомы заболевания могут различаться в зависимости от локализации и стадии. Изначально она напоминает плотную безболезненную шишку, увеличивающуюся в размерах. Процесс сопровождается вялостью, резким похудением и отсутствием аппетита.
Рабдомиосаркома глаза, расположенная в тканях позади него, делает его выпяченным. Опухоль в носу провоцирует насморк и кровотечения. В мочевом пузыре – становится причиной появления крови в моче. Опухоль давит на здоровые органы и мешает им правильно функционировать. Рабдомиосаркома в височной полости и эмбриональная среднего уха проявляется не только болью, но и потерей слуха. Пятая часть всех опухолей данного вида – патологии сердца.
Опухоль в носу провоцирует насморк и кровотечения. В мочевом пузыре – становится причиной появления крови в моче. Опухоль давит на здоровые органы и мешает им правильно функционировать. Рабдомиосаркома в височной полости и эмбриональная среднего уха проявляется не только болью, но и потерей слуха. Пятая часть всех опухолей данного вида – патологии сердца.
Лечение и диагностика рабдомиосаркомы в Израиле с D.R.A Medical
Несмотря на сложности в изучении недуга и отсутствие его полной картины, израильским врачам удалось достигнуть большого прогресса в лечении болезни, особенно если говорить о ее эмбриональном характере. Комплексный подход, высокая квалификация специалистов и наличие современного оборудования – три составляющих успеха, позволяющие в 85% случаев добиться позитивного исхода болезни.
Диагностика рабдомиосаркомы в нашем медицинском центре проводится в течение трех суток с момента обращения пациента на прием, поскольку потеря времени недопустима. Она включает:
- Первичный осмотр специалиста
- Лабораторные, инструментальные и цитогенетические исследования
- УЗИ
- Биопсию
Протокол лечения составляется на основе объемного 3D изображения опухоли после биопсии. В учет ставится риск долгосрочных осложнений от облучения растущих тканей у маленьких детей.
В учет ставится риск долгосрочных осложнений от облучения растущих тканей у маленьких детей.
Комплексное лечение рабдомиосаркомы в большинстве случае предполагает:
- Хирургическое удаление опухоли
- Химиотерапию
- Облучение
Желаете уточнить стоимость лечения — отправьте нам заявку
или обратитесь по телефону +972-77-4450-480 или +8-800-707-6168 (для жителей России бесплатно).
Хирургический метод
Основной и наиболее эффективный способ лечения рабдомиосаркомы сегодня. Опухоль удаляется полностью либо частично с последующими курсами химио/радиотерапии. В более поздних стадиях заболевания возможно также удаление пораженных лимфоузлов.
ХимиотерапияНазначается после операции для закрепления результата и в предоперационный период для приостановления размножения патологических клеток. Курс рассчитан на продолжительный срок до пяти циклов, некоторые препараты подразумевают еженедельный прием.
Лучевая терапия
Рабдомиосаркома является радиочувствительным видом рака, поэтому применение лучевой терапии дает возможность окончательно разрушить пораженные клетки после операции и химиотерапии. В начальных формах заболевания она эффективна без хирургического вмешательства и обнажения очага болезни. При необходимости частичной резекции он выделяется специальными скобами. Это позволяет организму лучше переносить химиотерапию и снижать дозы облучения. Используется в качестве подготовки к операции.
В начальных формах заболевания она эффективна без хирургического вмешательства и обнажения очага болезни. При необходимости частичной резекции он выделяется специальными скобами. Это позволяет организму лучше переносить химиотерапию и снижать дозы облучения. Используется в качестве подготовки к операции.
Целевая биологическая терапия
Включает процедуры, направленные на прерывание размножения больных клеток, рост которых подавляется с помощью антител. Это совершенно новое направление в лечении с каждым днем демонстрирует все лучшие показатели в борьбе с опухолью, особенно в в комплексе с химиотерапией.
Наряду с описанными способами лечения рабдомиосаркомы у детей и взрослых, израильские специалисты изучают возможности использования иммунной терапии, которая в будущем поможет распознавать и нейтрализовать атипичные клетки без хирургического вмешательства.
В случае тяжелой переносимости послеоперационных методик терапии возможно проведение трансплантации костного мозга для нормализации процесса выработки крови.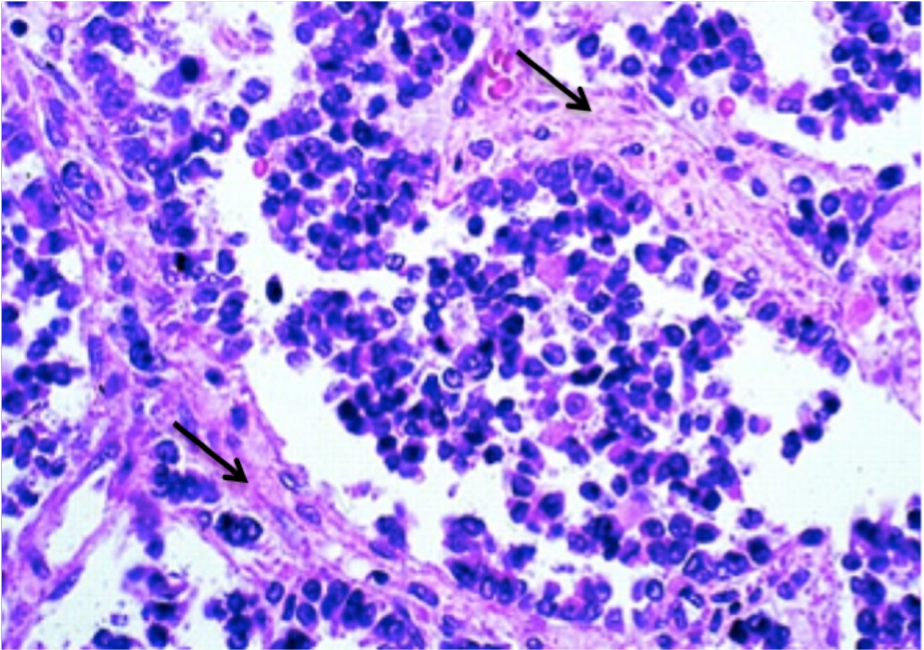
Читайте также:
Рецидив рака после операции: причины, симптомы, лечение и прогноз
D.R.A Medical работает с лучшими врачами Израиля по версии Forbes 2016
Могут ли форумы и онлайн-консультации быть полезны в лечении рака груди?
Прогноз выживаемости пациентов
Прогноз лечения рабдомиосаркомы и у детей, и у взрослых пациентов напрямую зависит от стадии заболевания и наличия метастаз. В случае детских патологий важную роль играет возраст ребенка – до семи лет чаще диагностируются менее агрессивные, эмбриональные формы, позволяющие удалить новообразование полностью.
Важно учитывать место расположения опухоли. Для орбитальных, атакующих органы зрения, прогноз более оптимистичен, чем для других видов, расположенных на шее и голове, поскольку они труднодоступы для иссечения и диагностируются на поздних стадиях.
90% рецидивов всех рабдомиосарком приходятся на первые два года после лечения. Если болезнь не проявляет себя в течение пяти лет, она считается вылеченной.
Цена лечения рабдомиосаркомы рассчитывается индивидуально для каждого пациента, в зависимости от стадии и общей картины заболевания.
Как всегда, мы не устаем напоминать: при обнаружении любых изменений в состоянии здоровья или подозрительных симптомов, не теряйте время. Обращайтесь к специалистам, ведь от этого зависит ваша жизнь!
Саркомы — Лечение в Киеве — Операция
Опухоли костей, суставов и мягких тканей
Саркомой называют группу злокачественных опухолей, которые возникают на костной, хрящевой, мышечной и жировой тканях.
Саркома является одним из редких видов злокачественного новообразования: на ее долю приходится около 1% всех онкологических заболеваний. Одной из особенностей саркомы является то, что опухоль быстро увеличивается в размерах и практически всегда есть риск послеоперационных осложнений и рецидивов злокачественного новообразования.
Важно знать, что при своевременном обращении за медицинской помощью заболевание поддается успешному лечению.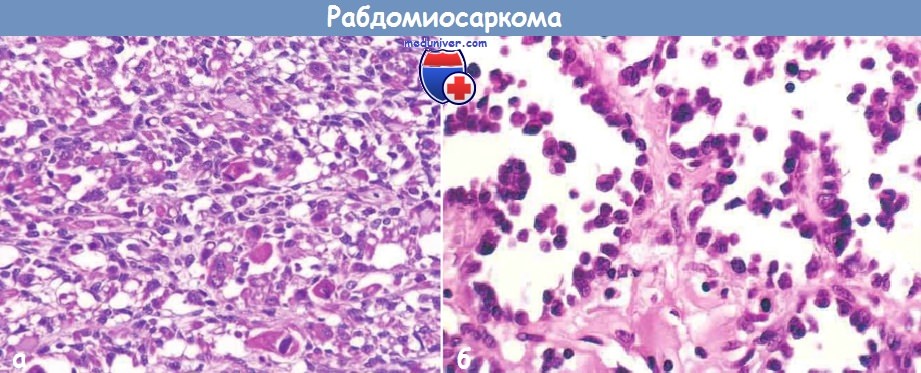
Основное отличие саркомы от других видов онкологических заболеваний в том, что в данном случае рак образовывается из эпителиальных клеток, выстилающих внутренние полости органов или из покровного эпителия. Саркомы не привязаны к каким-то конкретным органам. Чаще всего саркомы диагностируют у людей в возрасте от 18 до 35 лет.
Лечение сарком
В Клинике Спиженко к лечению каждого случая саркомы подходят индивидуально: для пациента разрабатывается персональный план лечения, в котором учитываются возраст, место локализации и размер опухоли, особенности новообразования, общее физическое состояние.
Методы лечения саркомы зависят от того, к какому типу она относится. Если диагностирована саркома костной ткани, то чаще всего используют комплексный подход. Благодаря
тому, что Клиника Спиженко располагает всеми современными инструментами терапии — малоинвазивной хирургией, лучевой терапией, радиохирургией на системе КиберНож, персонализированной химиотерапией, пациенты получают самое эффективное лечение.
В большинстве случаев, основным методом лечения костной саркомы является хирургическое удаление злокачественных новообразований. Масштаб оперативного вмешательства зависит от размеров опухоли, метастазов близлежащие органы и т.д.
Помимо операции пациенту могут назначить высокоточную лучевую терапию. Процедура может быть как однократной, так и многократной. Лучевая терапия позволяет предотвратить появление метастазов и рецидив злокачественного новообразования. Лучевая терапия костных опухолей в клинике проводится на линейном ускорителе Elekta, обладающем функцией IMRT для максимально точной доставки дозы облучения и защиты здоровых тканей вокруг новообразования.
Возможно также лечение другим комбинированным методом: хирургическое вмешательство и химиотерапия. Этот метод позволяет избавится от метастазов. Химиотерапия может быть применена как в начале комбинированного лечения, так и в комбинации с лучевой терапии, и на завершающем этапе лечения.
При лечении единичных и множественных метастазов рака в позвоночник, вызывающих компрессию спинного мозга, пациенту может быть выполнена микронейрохирургическая операция, либо же проведено радиохирургическое лечение на системе КиберНож, при отсутствии ограничений для радиохирургии.
Саркомы мягких тканей, как правило, удаляют операционным путем. Возможно также применение лучевой терапии и химиотерапии. Оба метода лечения могут быть назначены как перед операцией, так и после нее – в зависимости от размеров опухоли, агрессивности воздействия на организм.
Но, не зависимо от типа саркомы, лечение в Клинике Спиженко подбирают для каждого пациента индивидуально, после проведения междисциплинарного консилиума командой специалистов клиники.
Стоимость лечения
Каждый случай заболевания саркомой уникален. Стоимость лечения будет завесить только от того типа лечения, который выберут специалисты Клиники Спиженко, и который будет максимально подходить в каждом конкретном случае.
Важно всегда помнить, что, если вы или ваши близкие, столкнулись с онкологическим заболеванием, не стоит откладывайте борьбу с болезнью на завтра, а лучше записаться на консультацию онколога в Клинику Спиженко прямо сейчас!
Симптомы саркомы
Симптомы разных видов саркомы зависят от локализации и типа опухоли. Для большинства сарком характерны следующие признаки:
Для большинства сарком характерны следующие признаки:
- при саркоме мягких тканей — появление быстро увеличивающегося опухолевого образования
- при саркоме костей появляются боли в области пораженных костей. Одним из типичных признаков становятся боли, которые усиливаются в ночное время и не купируются стандартными дозами анальгетиков
- локальное расширение венозной сети в месте новообразования
- опухоль быстро увеличивается, становится ощутима при пальпации. На последних стадиях заболевания, в области новообразования часто возникает патологический перелом
- потеря веса до 10-15 кг без малейших усилий – диеты или тренировок, со стороны пациента
- отек температура кожи в районе опухоли повышается до субфебрильной или даже фебрильной (37 – 39 градусов)
- полное отсутствие аппетита
- при анализе крови диагностируется анемия
Важно помнить – если был обнаружен хотя бы один из вышеперечисленных признаков, необходимо обратится за консультацией к специалисту. Особенно нужно быть внимательным к потере веса, локализированной температуре тела в районе опухоли и ночным болям.
Особенно нужно быть внимательным к потере веса, локализированной температуре тела в районе опухоли и ночным болям.
Диагностика
Для того, чтобы установить точный диагноз, подтверждающий саркому, специалисты Клинике Спиженко проведут ряд исследований, среди которых рентгенологическое исследование потенциального места поражения.
Для установления окончательного диагноза пациенту в клинике проведут компьютерную томографию или магнитно-резонансную томография костей и мягких тканей.
Кроме того, выполняется ряд исследований для точной оценки характера новообразования – биопсия. По результатам исследования специалисты клиники смогут определить наличие злокачественного новообразования, тип опухоли, степень злокачественности.
Данные исследований КТ и МРТ позволят точно определить размеры новообразования, степень распространенности и поражение близлежащих тканей.
Классификация
Медицине известны более сорока видов саркомы.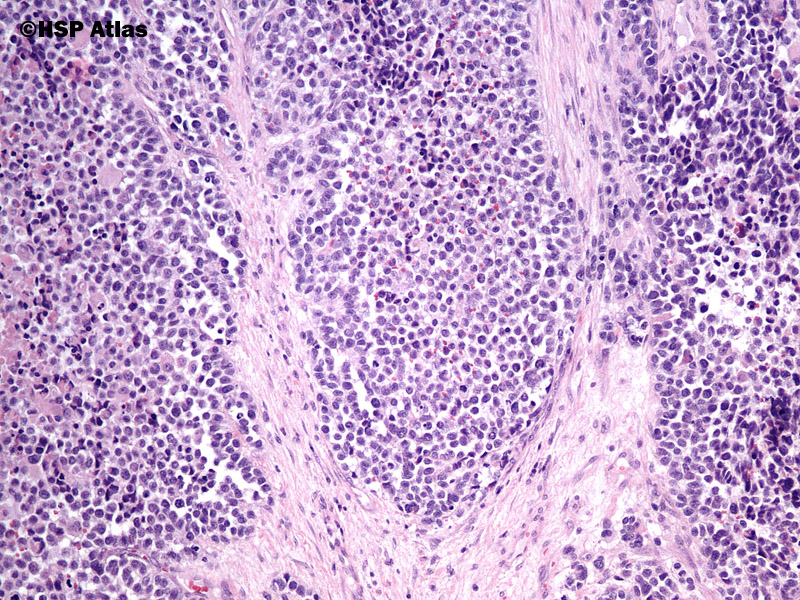 Классифицируют новообразования согласно их происхождению.
Классифицируют новообразования согласно их происхождению.
К саркомам, происходящим из костной ткани принято относить: хондросаркому, остеосаркому, саркому Юинга, паростальную саркому, ретикулосаркому. Эти виды злокачественных опухолей, как правило, поражают длинные трубчатые кости, а также область таза.
К саркомам, произрастающим из мягких тканей – мышечной и жировой, относят: эпителиоидные саркомы, плеоморфная саркому, саркому Капоши, саркомы внутренних органов, саркому кожи, нейрогенные саркомы, гемангиосаркому, альвеолярную мягкотканную саркому, ангиосаркому, цитосаркому, злокачественную фиброзную гистиоцитому, дерматофибросаркому, желудочно-кишечные стромальные опухоли, синовиальную саркому, липосаркому, лимфосаркому, лейомиосаркому, лимфангиосаркому, нейрофибросаркому, фибросаркому, рабдомиосаркому.
Примечательно, что саркома мягких тканей может быть в любом месте организма, но чаще всего все же поражает ткани конечностей.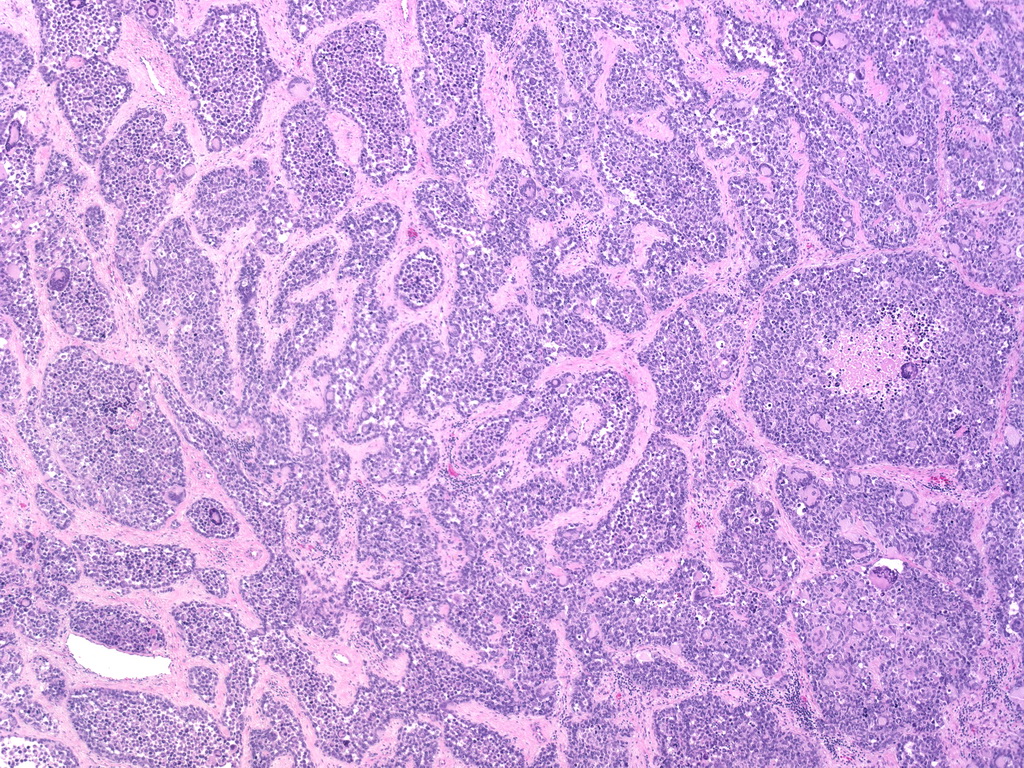
Причины появления
Причины появления саркомы могут быть разнообразными. Вот основные из них:
- предраковые заболевания
- наследственность
- радиация и экологические факторы
- онкогенные вирусы
- озлокачествление изначально доброкачественных опухолей
- механические повреждения тканей, такие как ушибы, ожоги, переломы, осколки и другие инородные тела в организме
- интенсивный рост костной ткани
Чтобы быть уверенным в своем здоровье и здоровье ваших близких, мы рекомендуем раз в год проходить обследование в Клинике Спиженко. 98% новообразований можно излечить полностью при условии ранней диагностики. Не ждите, пока болезнь даст о себе знать, обращайтесь в Клинику Спиженко уже сейчас!
Саркома мягких тканей (СМТ). Причины возникновения. Симптомы. Диагностика и лечение СМТ
Следует проверять предоставленную здесь информацию. Решение о применении определенного упомянутого здесь вещества принимает исключительно лечащий врач. Обязательно проконсультируйтесь с врачом!
Обязательно проконсультируйтесь с врачом!
Саркома мягких тканей (СМТ) – это опухоль, которая поражает ткани, развивающиеся из среднего зародышевого листка, называемого мезодерма. У человека из этого слоя формируется:
- Соединительная ткань, которая располагается между органами.
- Хрящи.
- Кости.
- Почки.
- Мышцы.
- Кровеносные сосуды.
Саркома мягких тканей затрагивает мышечные, кровеносные и соединительные волокна, жировую ткань. Чаще всего локализуется в молочных железах, конечностях, крупных сосудах и органах ЖКТ.
Опухоль считается редкой. Ее диагностируют только у 1% пациентов среди взрослых онкологических больных. В Украине за последние 13 лет зарегистрировано более 15 тыс. случаев, примерно по 1000 в год. Заболевают в основном люди старше 40 лет. Мужчины болеют немного чаще женщин (разница в 5%).
Среди детей болезнь более распространена – составляет 15% от всех видов злокачественных поражений.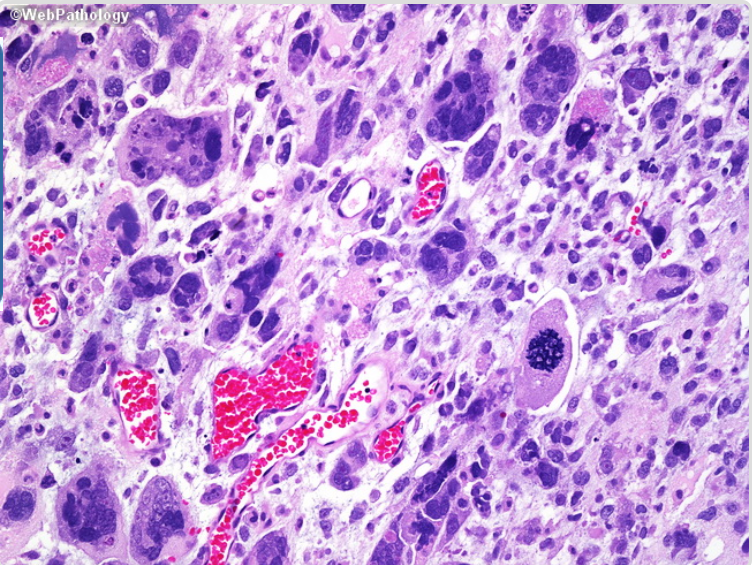
Опухоль считается опасной, потому что отличается частыми рецидивами и быстрым появлением метастазов, распространяющихся с кровью по всему организму.
Причины возникновения
Для большинства видов СМТ ученые не смогли пока определить главную причину развития, но в некоторых случаях играют роль:
- Вирусные инфекции. Речь идет о ВИЧ, Эпштейна-Барр и других вирусах герпесной группы.
- Наследственность. Некоторые заболевания, передающиеся генетически, предрасполагают к появлению онкологии. Особенно это касается наследственной ретинобластомы и синдрома Ли Фраумени. Вероятность саркомы при этом 78 и 21% соответственно. Нейрофиброматоз с появлением различных доброкачественных образований завершается онкологией в 15% случаев.
- Облучение. Наибольший риск у тех, кто проходил высокочастотное облучение при лечении рака.
В некоторых случаях пусковым механизмом становится травма.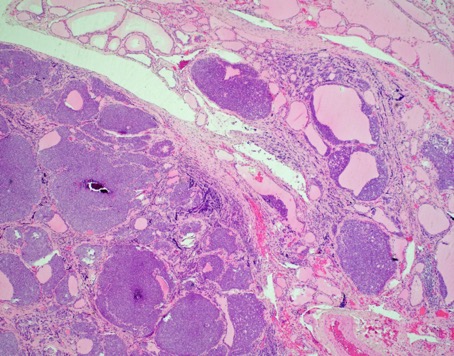
Разновидности
Существует около 70 разных гистологических видов саркомы мягких тканей. Наиболее распространенные из них:
- Липосаркома (18%) – затрагивает жировые ткани. Может появиться в любом месте, но чаще всего растет в области живота. Для этого вида характерен одновременный множественный рост.
- Фибросаркома (29%) – формируется из фибробластов, клеток соединительной ткани, вырабатывающих межклеточное вещество, в которое входят коллаген, эластин, мукополисахариды, в том числе и гиалуроновая кислота.
- Лейомиосаркома (11%) – поражает гладкие мышцы, в структуре сосудистых трубок и внутренних органов. Считается наиболее прогностически неблагоприятным образованием.
- Рабдомиосаркома (4,8%) – опухоль скелетных мышц. Включает три подтипа, которые характерны для разных возрастов, например плеоморфная поражает пожилых, а эмбриональная – маленьких детей.
- Ангиосаркомы (2,5%) – берут начало из сосудов. Известная разновидность – саркома Капоши, которая характерна для кожи. Развивается из выстилки лимфатических и кровеносных сосудов.
- Гастроинтестинальная стромальная опухоль – образование желудочно-кишечного тракта.

Внутри каждой из этих групп есть еще множественная классификация отдельных форм. Например, среди липосарком выделяют миксоидные, крупноклеточные, полиморфные разновидности.
Симптомы
Симптоматика новообразований очень разная, но все же выделяется несколько общих черт для всех сарком.
- В месте появления развивается отек. Вначале это небольшая припухлость, но по мере роста может сформироваться огромная опухоль.
- Если образование возникает близко к наружным тканям, то его видно. Заметен узелок, потом шишечка. Они мало отличаются по цвету от других участков кожи. Изначально узел мягкий и эластичный. Однако по мере роста в нем видно несколько формирующихся центров. Сдвинуть с места опухоль становится трудно. Это очень важное отличие от доброкачественных образований. Меняется цвет кожи, а при надавливании возникает болезненность. Итоговый размер может достигать 30 сантиметров в диаметре, иногда даже больше.
- Если опухоль глубоко, внешние покровы все равно видоизменяются. Может поменяться цвет, появиться сосудистый рисунок, развиться изъязвление.
- Боль не возникает сразу. Она развивается, когда опухоль затрагивает нервные ткани. Болезненность становится сильной и распространяется в разные стороны по всему телу. Усиливается по ночам. От нее не помогают обезболивающие. Но это обычно бывает на последних стадиях. Поэтому изначально ориентироваться на болевой синдром нельзя. Если заметно необычное образование, уплотнение, то следует сразу обратиться к врачу. Для активного метастазирующего развития некоторых форм, достаточно нескольких недель. Поэтому медлить нельзя.
.jpg) Однако по мере роста в нем видно несколько формирующихся центров. Сдвинуть с места опухоль становится трудно. Это очень важное отличие от доброкачественных образований. Меняется цвет кожи, а при надавливании возникает болезненность. Итоговый размер может достигать 30 сантиметров в диаметре, иногда даже больше.
Однако по мере роста в нем видно несколько формирующихся центров. Сдвинуть с места опухоль становится трудно. Это очень важное отличие от доброкачественных образований. Меняется цвет кожи, а при надавливании возникает болезненность. Итоговый размер может достигать 30 сантиметров в диаметре, иногда даже больше.

Вместе с местными симптомами появляется общее недомогание. Больные постоянно чувствуют себя разбитыми, уставшими, теряют в весе. У них пропадает аппетит, наблюдается постоянная тошнота. Это связано с интоксикацией. По этой же причине поднимается температура и после лечения антибиотиками не пропадает. В анализах крови обнаруживается анемия и признаки сильного воспаления.
Диагностирование
Поставить диагноз бывает сложно из-за большого количества форм сарком, а также вероятности доброкачественной опухоли. Поэтому используют сочетание многих методов.
- Биопсию с гистологическим анализом.
- УЗИ.
- Рентген.
- МРТ.
- Обследование с радиоизотопами.
Причем биопсия не всегда проясняет картину. Нужны все эти обследования. Они позволяют определить стадию заболевания и правильно назначить лечение.
Стадии СМТ
СМТ проходит четыре этапа развития.
- Первая стадия. Небольшое образование до 5 сантиметров. Оно еще не распространилось на другие органы и лимфоузлы. Клетки делятся медленно, их в структуре опухоли не много.
- Вторая. Повышается злокачественность, клетки начинают делиться быстро. Размер может быть меньше или больше 5 см. Опухоль активно пронизывают сосуды. Она нарушает работу органа, в котором расположена и прорастает его полностью.
- Третья. Саркома покидает пределы органа. Происходит распространение и углубление образования во внутренние ткани. Страдают все окружающие структуры.
- О четвертой стадии говорят, когда появляются метастазы. Они идут по крови, локализуются чаще всего в легких, мозге. Опухоль большого размера. Она сильно сдавливает ткани. Часто очаг распадается, кровоточит.
 Небольшое образование до 5 сантиметров. Оно еще не распространилось на другие органы и лимфоузлы. Клетки делятся медленно, их в структуре опухоли не много.
Небольшое образование до 5 сантиметров. Оно еще не распространилось на другие органы и лимфоузлы. Клетки делятся медленно, их в структуре опухоли не много.
Если затронут костный мозг, то он инициирует рост других СМТ. Очень часто речь идет о рецидивирующей саркоме.
Лечение
В первую очередь образование удаляют. Не прибегают к этому методу только, если затронуты жизненно-важные органы, больной старше 75 лет или у него тяжелые заболевания почек, сердца и печени. При удалении захватывают и некоторую часть здоровых тканей.
Не прибегают к этому методу только, если затронуты жизненно-важные органы, больной старше 75 лет или у него тяжелые заболевания почек, сердца и печени. При удалении захватывают и некоторую часть здоровых тканей.
Если образование было на 1-2 стадии, то после резекции показано облучение или химиотерапия. Если форма опухоли агрессивная, то химиотерапию проводят как до, так и после хирургии. На третьей стадии до резекции показано облучение, для уменьшения размеров новообразования.
На четвертой стадии операцию проводят не всегда. Если удаление невозможно, то используют обезболивающие, корректируют количество гемоглобина, проводят детоксикацию. Эти мероприятия облегчают жизнь больного, но не приносят выздоровление.
Прогноз
Больше всего шансов выжить у людей, которым диагноз поставлен на ранней стадии, а опухоль локализуется в конечностях. 75% из них преодолевают пятилетний рубеж. При СМТ в теле и внутренних органах выживаемость снижается и составляет 60%. Если произошло распространение онкоклеток, то удается спасти только 35% больных.
Если произошло распространение онкоклеток, то удается спасти только 35% больных.
Ошибочный диагноз первичной плеоморфной рабдомиосаркомы правого бедра у молодого взрослого: отчет о клиническом случае
Введение
Рабдомиосаркома (РМС) — очень злокачественная форма
опухоль мягких тканей с дифференцировкой скелетных мышц. В
заболеваемость RMS составляет ~ 43 случая на 10 миллионов ежегодно для
лица в возрасте до 20 лет (1).
У пациентов с локализованным заболеванием безрецидивная выживаемость
улучшилась до 70–80% (2). Тем не мение,
прогноз для пациентов с метастазами относительно плохой, с
5-летняя выживаемость всего 30% (3).Методы диагностики RMS включают:
клинико-лабораторное обследование, визуальный анализ, патологический
диагностика и иммуногистологическое обследование (4). RMS был разделен на 3 основных
подтипы: эмбриональный, альвеолярный и плеоморфный RMS (PRMS). Большинство
общие подтипы — эмбриональный и альвеолярный подтипы (5). Первичная PRMS встречается относительно редко и
Первичная PRMS встречается относительно редко и
в первую очередь поражает взрослых, с пиком заболеваемости в пятом десятилетии
жизни (6–9). Чаще всего возникает в глубоком мягком
ткани конечностей.Из-за сходства клинических
проявления и визуализационные особенности между PRMS и другими программными
опухоли тканей, PRMS часто ошибочно диагностируется (10). Настоящее исследование представляет собой случай
PRMS, который был ошибочно диагностирован как шваннома. Этот неправильный диагноз
привело к массовому прогрессированию, и только после
тонкоигольная аспирационная и гистологическая и иммуногистохимическая
анализ подтвердил, что происхождение опухоли — скелетная мышца,
поставлен окончательный диагноз PRMS правого бедра. В
Настоящее исследование было одобрено Комитетом по этике
Первая дочерняя больница Наньчанского университета (Наньчан, Китай),
и письменное информированное согласие было получено от пациента.
История болезни
В августе 2014 г. пациент 28 лет обратился с жалобой на
в ортопедическую клинику Народной больницы округа Тайхэ (Цзянь,
Китай) с основной жалобой на отек правого бедра в течение 1
месяц. По данным местной районной больницы общего профиля
По данным местной районной больницы общего профиля
рентгенолог, магнитно-резонансная томография (МРТ) выявила наличие
шванномы, и пациенту посоветовали регулярно проходить
следовать за. Однако через 6 месяцев в декабре 2014 г.
увеличение размера образования и боли в правом бедре были
отметил.Пациент направлен в ортопедическое отделение г.
Первая дочерняя больница Наньчанского университета для дальнейшего
лечение. В личном или семейном анамнезе травм или болезней не было.
записано. Общий медицинский осмотр показал, что
пассивная и активная амплитуда движений правого коленного сустава была
в норме, за исключением парестезии справа внизу
конечность. Никакой лихорадки или затрудненного дыхания не сопровождали
масса тела, и не было истории потери веса или контакта с туберкулезом.
сообщил пациент.
Дополнительный медицинский осмотр выявил
плохо очерченная, нежная и плотная масса мягких тканей справа
внутренняя поверхность бедра, но не пальпируется голова, шея, надключичные, подмышечные
или эпитрохлеарные лимфатические узлы. Маркеры воспаления,
Маркеры воспаления,
включая скорость оседания эритроцитов и С-реактивный белок,
были в пределах нормы. МРТ была проведена для оценки
массы. Осевые Т2-взвешенные изображения выявили множественные кисты
поражения разного размера с высокой интенсивностью сигнала.С подавлением жира
Т2-взвешенная МРТ также показала высокую интенсивность сигнала (рис. 1А и В). Кроме того, тонкая игла
аспирация проводилась для оценки массы и цитологического
диагноз соответствовал злокачественному новообразованию; плеоморфный
новообразование веретеноводства с выраженной ядерной атипией и выраженным
митотическая активность не наблюдалась.
На основании исключения хирургического
противопоказания, хирурги отделения
Ортопедия, Первая больница Наньчанского университета,
которые специализируются на лечении опухолей костей, провели операцию.В
пациента поместили в положение лежа на спине и после
успех эпидуральной анестезии, стерильные простыни продезинфицированы и
обычно проложили в правой нижней конечности, чтобы обнажить
операционное поле. Сначала был выполнен медиальный разрез бедра ~ 17
Сначала был выполнен медиальный разрез бедра ~ 17
см в длину. Далее подкожная клетчатка, поверхностная и глубокая.
фасция и средняя широкая мышца бедра были послойно резецированы,
пока не наблюдались бедренная артерия и вена. Суда выше
были целы. Опухоль была темно-коричневой и располагалась в обширной мышце.
промежуточная мышца.Опухоль была полностью удалена с отрицательным
поля. Интраоперационные образцы тканей были извлечены для
патологическое обследование. Была установлена дренажная трубка для раны, и каждая
слой ткани ушивали строго после полного гемостаза.
Расчетная кровопотеря составила 100 мл, переливание крови не проводилось.
требуется во время процедуры.
Опухоль была большая, гладкая, темно-коричневого цвета.
при грубой экспертизе. Масса нерегулярной ткани составила ~ 11 × 9 × 5.
см3 (рис. 2А). В
резецированную опухолевую ткань фиксировали в 10% формалине, заливали в
парафин и разрезать на срезы 5 мкм с помощью микротома.Разделы
впоследствии были окрашены гематоксилином и эозином и визуализированы
под микроскопом.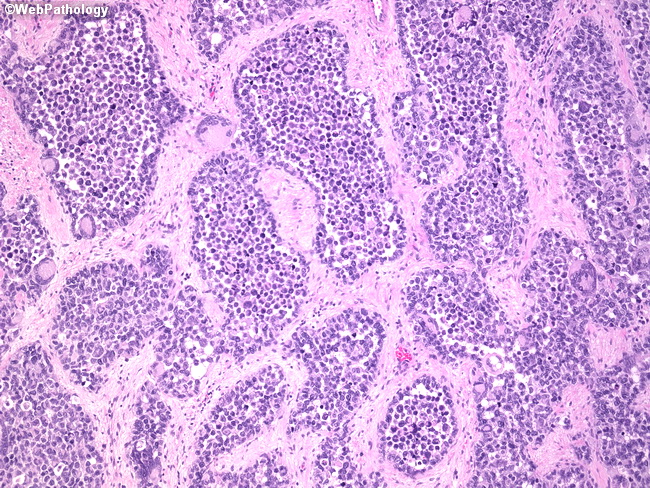 При микроскопическом исследовании обнаружена опухоль.
При микроскопическом исследовании обнаружена опухоль.
состоит из связанных между собой пучков атипичных, веретенообразных,
плеоморфные и гигантские клетки с атипичными ядерными
Особенности. Были обнаружены многочисленные аномальные и многоядерные гигантские клетки.
наблюдались, и у большинства клеток были заметные ядрышки
и обильные эозинофильные цитоплазмы (рис. 2В). Для иммуногистохимии ткани
срезы инкубировали при 25 ° C в течение 60 мин с моноклональной мышью
антитела против десмина (каталожный №, kit-0023) и миогенного
дифференцировка 1 (MyoD1; каталожный номер MAB-0119) (разведение,
1: 1000; Fuzhou Maixin Biotech Co., Ltd., Фучжоу, Китай).
Иммуногистохимический анализ показал, что клетки были положительными.
на актин, MyoD1 и десмин и отрицательный на меланому человека черный
45, кальпонин и мелан-A (рис. 2C и
D). На основании этих данных диагноз PRMS правого
бедро было обеспечено.
Больной выписан без осложнений.
1 неделя после операции. Пациенту было проведено 6 циклов
химиотерапия: доксорубицин, 90 мг / сут в течение 3 дней; 14 дней
выкл, а затем ифосфамид 3. 8 г / сут в течение 5 дней, затем 14
8 г / сут в течение 5 дней, затем 14
выходные до следующего лечебного цикла. Через 3 месяца
вверх, который состоял из простой рентгенографии и МРТ, пациент был
без симптомов и может вернуться к работе. В настоящее время пациент
на данный момент жив-здоров. Однако в случаях, подобных нынешнему,
необходимо тщательное наблюдение за пациентами из-за
высокая частота рецидивов и метастазов, связанных с ошибочно диагностированным
PRMS.
Обсуждение
RMS — очень злокачественная опухоль мягких тканей.
который возникает из поперечно-полосатых мышечных клеток, демонстрирует скелетные мышцы
дифференциация и связана с ранним и широким распространением
метастаз (11,12).RMS делится на 3 основных подтипа:
Эмбриональные и альвеолярные RMS и PRMS, согласно World 2002
Классификация мягких тканей и костей организацией здравоохранения
Новообразования (5). Эмбриональный и
альвеолярные подтипы являются наиболее распространенными и наиболее частыми сайтами
по происхождению RMS включают голову и шею, конечности и мягкие
ткани. Заболевание имеет преобладание мужчин, с переходом от мужчины к женщине.
Заболевание имеет преобладание мужчин, с переходом от мужчины к женщине.
соотношение 1,3: 1 (13).
PRMS был впервые описан Стаутом в 1946 г. (14). Первичная PRMS встречается относительно редко и
в первую очередь поражает взрослых пятого десятилетия жизни (15).Его появление у молодых людей, например
в данном случае встречается крайне редко. Примечательно, что PRMS часто
поставлен неверный диагноз или полностью пропущен, поскольку его клинические проявления
и особенности изображения аналогичны таковым для других мягких тканей
опухоли, в том числе фиброзная гистиоцитома и шваннома (9). В настоящем исследовании пациент был
ошибочно диагностировали шванному, что привело к прогрессированию
болезнь на полгода; поэтому тонкоигольная аспирация
имеет решающее значение для диагностики новообразований мягких тканей.Гистологический
проявления RMS широко варьируются, а гистопатологические
диагноз основывается на морфологическом, иммуногистохимическом и
ультраструктурные данные, которые показывают фенотип скелетных мышц
(16,17).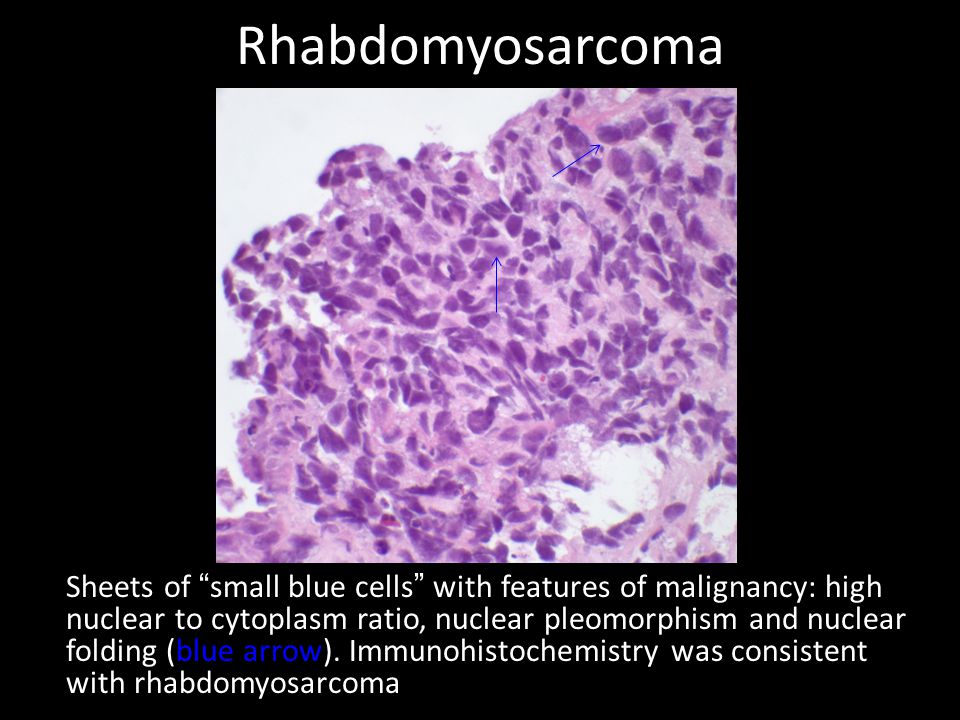 PRMS гистологически различается
PRMS гистологически различается
из двух более распространенных подтипов (эмбриональный и альвеолярный)
беспорядочное расположение ячеек, состоящих из крупных,
плеоморфные ядра и эозинофильные цитоплазмы. Клетки PRMS также могут
располагаться в пучки, что напоминает наблюдаемую структуру клеток
при лейомиосаркоме (18).Гистологический
подтипирование имеет решающее значение из-за различных прогнозов и терапевтических
подходы, используемые в PRMS в отличие от других опухолей мягких тканей.
Иммуногистохимия считается ценной для диагностики
PRMS, как серия маркеров с диапазоном специфичности и
чувствительность имеется. Первичные маркеры PRMS — MyoD1,
десмин, саркомерный актин и миозин (19,20).
Хирургическое удаление PRMS считается
предпочтительное лечение, поскольку оно снимает отек и позволяет
окончательный диагноз необходимо подтвердить гистологически.Идеальный хирургический
лечение включает полную резекцию опухоли с отрицательным
микроскопические поля (21). К
напротив, RMS имеет особую чувствительность к внешнему лучу
радиация; таким образом, полная резекция может быть отложена в зависимости от размера
уменьшение с помощью лучевой терапии.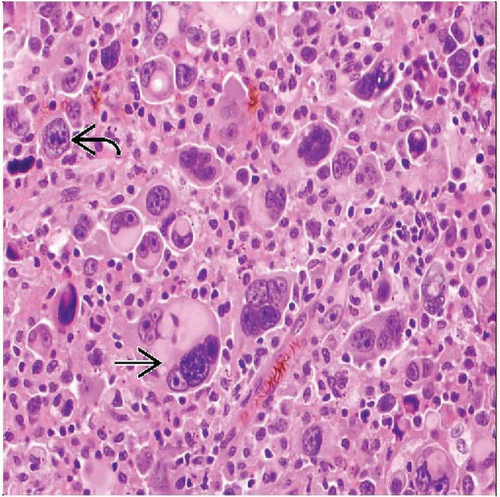 В некоторых исследованиях сообщается, что RMS
В некоторых исследованиях сообщается, что RMS
общий ответ на химиотерапию составляет 85% (22,23), в
в отличие от PRMS, при которой Ferrari et al (11) сообщили о более низком уровне ответа
6,25%. Преобладание PRMS у взрослых, а также его устойчивость к
химиотерапия, привела к тому, что PRMS часто рассматривается как отдельный
сущность из других подтипов RMS.
В заключение, настоящее дело описывает
Пациент 28 лет, страдавший первичной PRMS
правое бедро. Тонкоигольная аспирация и тотальная резекция опухоли были
выполнено, и через 3 месяца наблюдения у пациента не было
свидетельства рецидива заболевания или остаточных побочных эффектов от
терапия. Проведение лабораторных исследований и визуализации
осмотр особенно важен при дифференциальной диагностике
пациентов с опухолями мягких тканей. Хотя
Срок наблюдения за текущим пациентом был относительно коротким, в
учет высокого риска рецидива и метастазирования в
ошибочно диагностированный PRMS, часто рекомендуется долгосрочное наблюдение
такие случаи.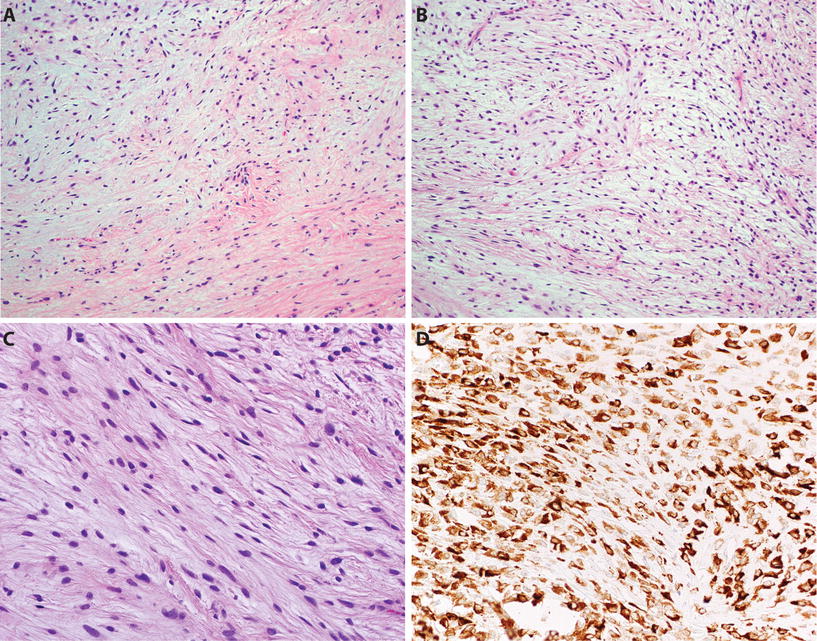
Благодарности
Настоящее исследование было поддержано Gan-Po
Проект талантов 555 провинции Цзянси, План поддержки
Департамент науки и технологий провинции Цзянси (грант №
20112BBG70020) и Фонд естественных наук Цзянси
Провинция (грант № 20132BAB205067).
Список литературы
1 | Кранмер Л.Д., Чен СС, Морган С., Мартино Дж. |
2 | Malempati S и Hawkins DS: |
3 | Бренеман Дж. |
4 | Скалл С., Амар С., Фейз-Эрфан И., Дэйв Х. и |
5 | Ли Дж. Дж., Форстнер Д. и Хендерсон К.: |
6 | Stock N, Chibon F, Binh MB, Terrier P, |
7 | Моретти Дж., Гимарайнш Р., Оливейра К. М., |
8 | Кефели М, Кандемир Б, Акполат I, Йылдырым |
9 | Хакодзаки М, Ходзё Х, Кузе Т, Таджино Т, |
10 | Mondal SK, Mandal PK, Adhikari A и Basak |
11 | Феррари А, Дилео П, Казанова М, Бертулли |
12 | Кишор Б., Кхаре П., Гупта Р. |
13 | Данг Н.Д., Тех Б.С. и Паулино А.С.: |
14 | Stout AP: рабдомиосаркома скелета |
15 | Фадаре О, Бонвичино А, Мартель М, Реншоу |
16 | Казерто Б.Г.: Сравнительный обзор собак |
17 | Яо Дж. К., Ван У. К., Ценг Х. Х. и Хван У. С.: |
18 | Атахан С., Аксу О и Экинджи С: Цитологический |
19 | Morgenstern DA, Rees H, Sebire NJ, Шипли |
20 | Cessna MH, Zhou H, Perkins SL, Tripp SR, |
21 | Ге Икс, Ма Дж, Дай Х, Рен Л, Ли Кью и Ши Дж: |
22 | Fuchs J, Dantonello TM, Blumenstock G, |
23 | Петрович Б., Джан И., Марков Б., Петрович М., |
 С., Лайден Э., Паппо А.С., член парламента по ссылке,
С., Лайден Э., Паппо А.С., член парламента по ссылке, Am J Surg Pathol. 33: 1850–1859.
Am J Surg Pathol. 33: 1850–1859. , Куалман С.Дж., Кикучи С. и Абэ М.: начальная школа.
, Куалман С.Дж., Кикучи С. и Абэ М.: начальная школа.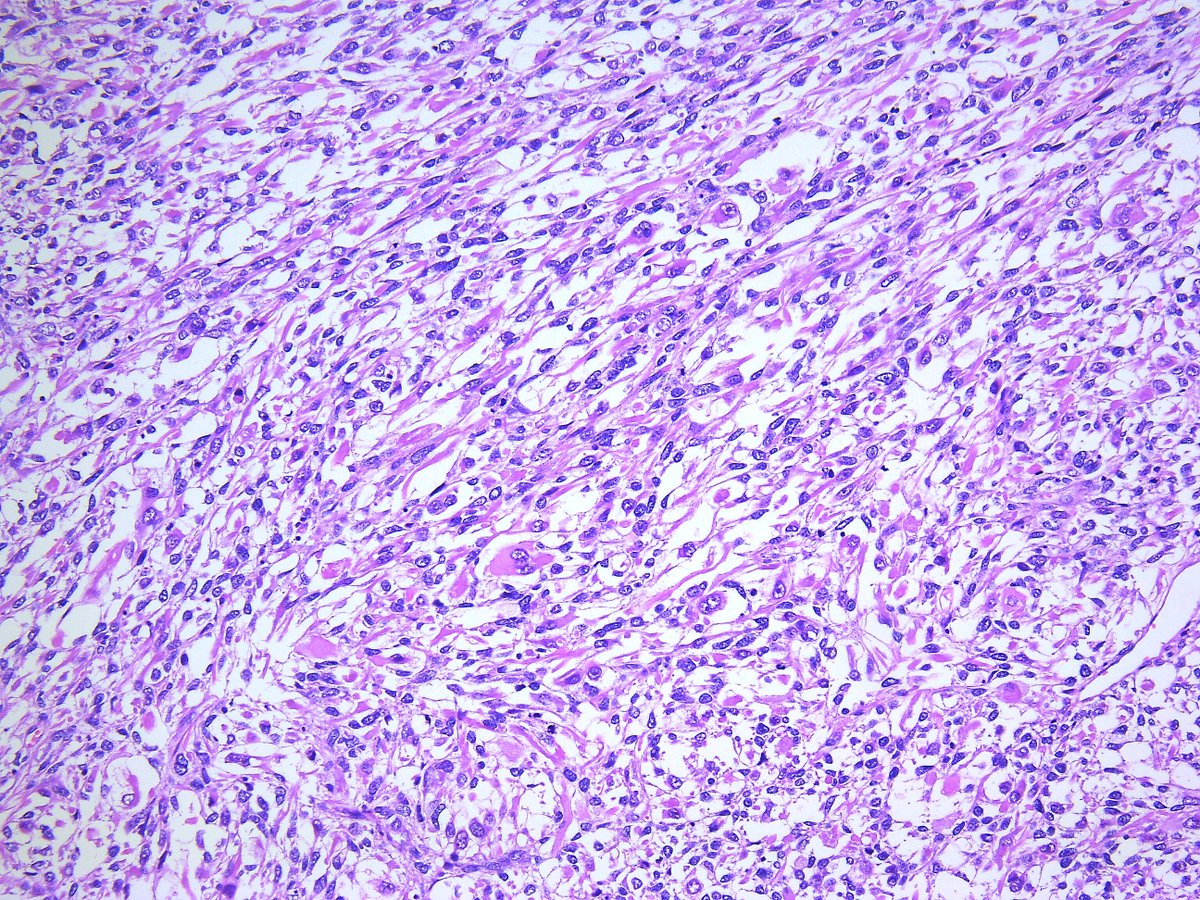 Дж., Гупта С. и
Дж., Гупта С. и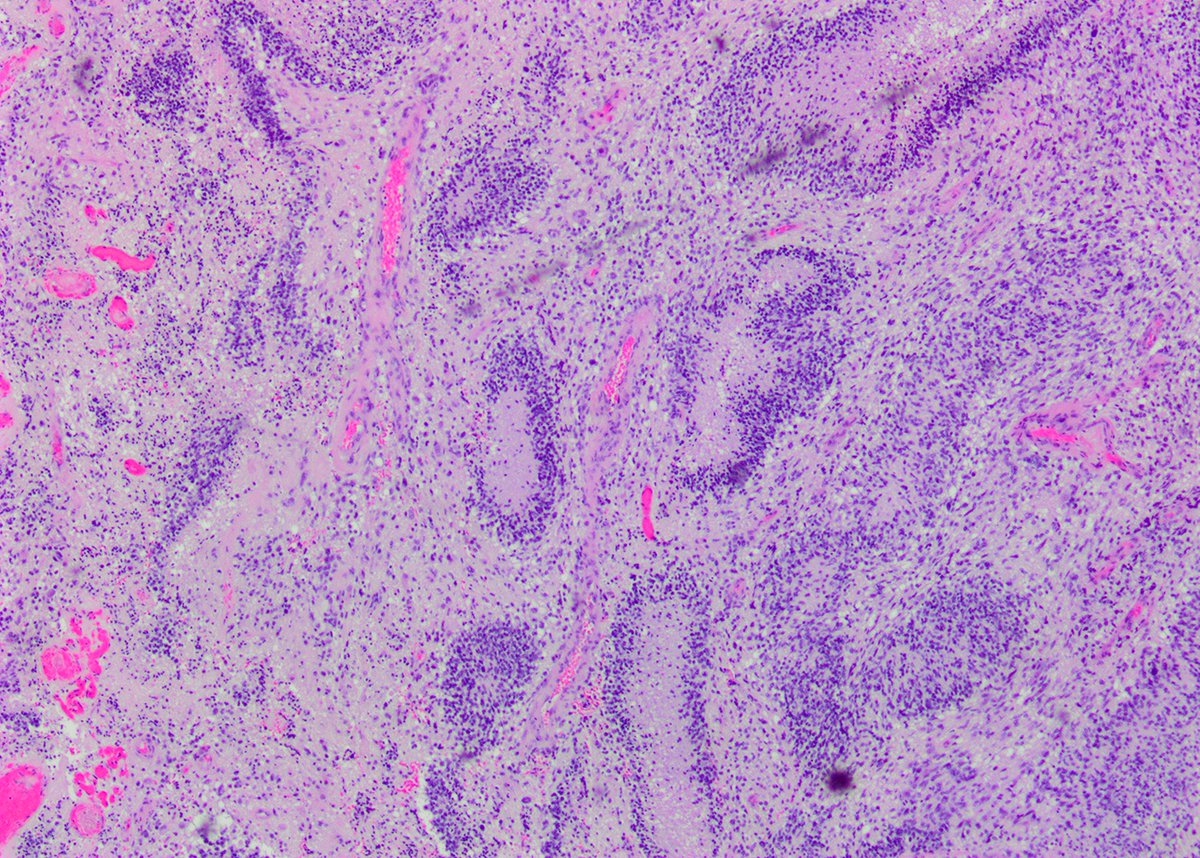 29: 122–134. 2010 г.
29: 122–134. 2010 г. И Андерсон Дж .: Подтип рабдомиосаркомы иммуногистохимическим методом.
И Андерсон Дж .: Подтип рабдомиосаркомы иммуногистохимическим методом.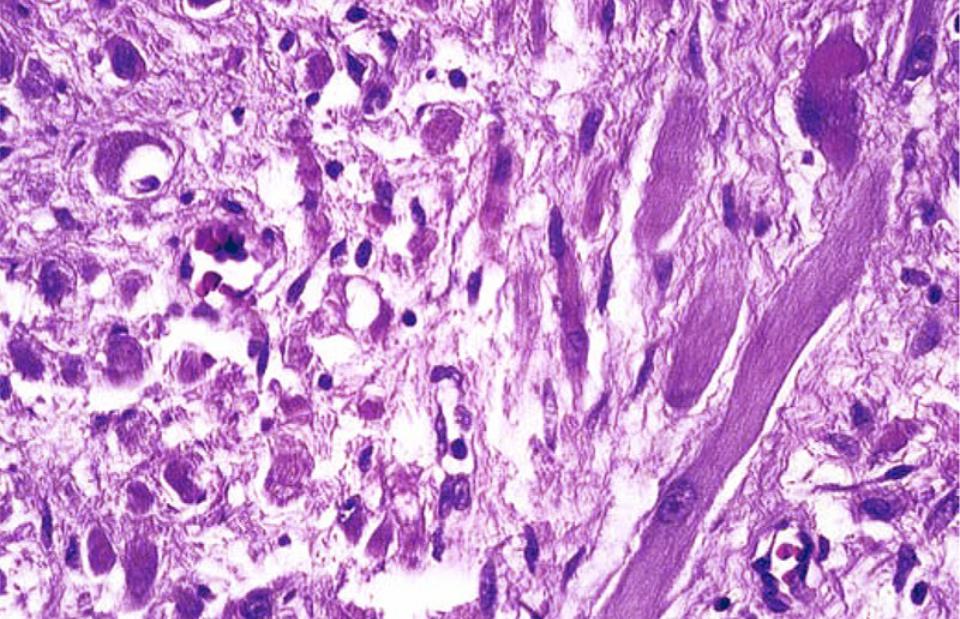 Ann Surg. 259: 1166–1172.
Ann Surg. 259: 1166–1172.Что такое рабдомиосаркома?
Рак начинается, когда клетки тела начинают бесконтрольно расти. Клетки практически в любой части тела могут стать раком и могут распространиться на другие части тела. Чтобы узнать больше о том, как рак возникает и распространяется, см. Что такое рак? Для получения информации о различиях между детским раком и взрослым раком см. Рак у детей.
Саркомы — это раковые образования, которые развиваются из соединительных тканей в организме, таких как мышцы, жир, кости, оболочки суставов или кровеносные сосуды.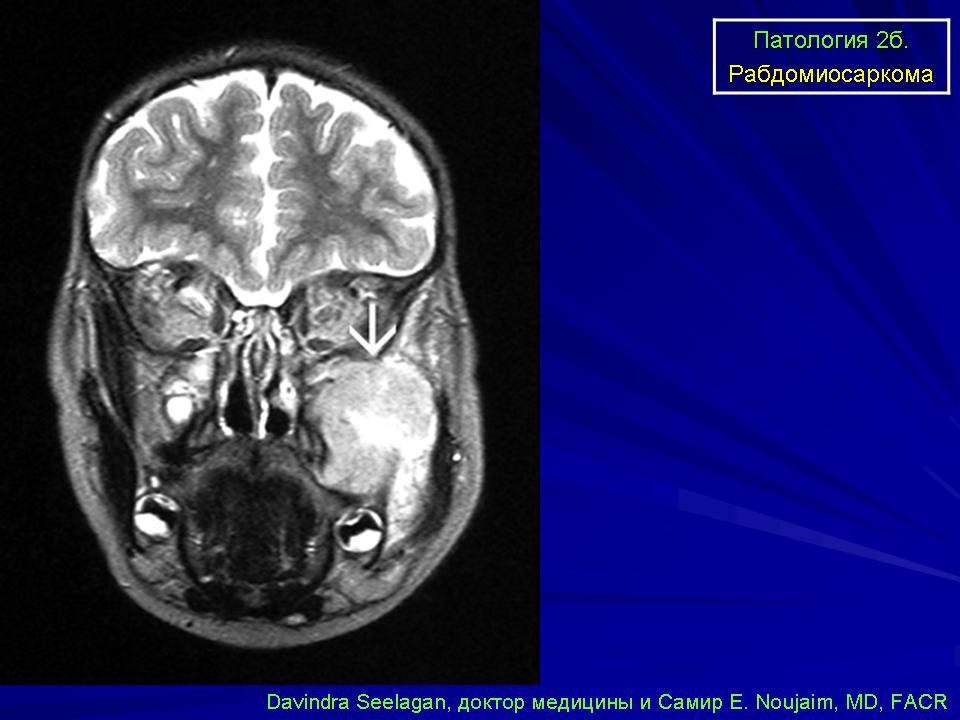 Есть много типов сарком.
Есть много типов сарком.
Рабдомиосаркома (RMS) — это тип саркомы, состоящий из клеток, которые обычно развиваются в скелетные (произвольные) мышцы. Это мышцы, которыми мы управляем, чтобы двигать частями нашего тела.
Задолго до рождения начинают формироваться клетки, называемые рабдомиобластами (которые в конечном итоге образуют скелетные мышцы).Это клетки, которые могут развиться в RMS. Поскольку это рак очень ранних форм мышечных клеток, он гораздо чаще встречается у детей, хотя иногда встречается и у взрослых.
Мы можем думать о наших скелетных мышцах, как о в основном в руках и ногах, но RMS может начинаться практически в любом месте тела, даже в некоторых частях тела, которые обычно не имеют скелетных мышц.
Общие сайты RMS включают:
- Голова и шея (например, рядом с глазом, внутри носовых пазух или в горле, или возле позвоночника в области шеи)
- Мочевые и репродуктивные органы (мочевой пузырь, предстательная железа или любой из женских органов)
- Руки и ноги
- Туловище (грудь и живот)
Типы рабдомиосарком
Существует 2 основных типа RMS, а также несколько менее распространенных типов.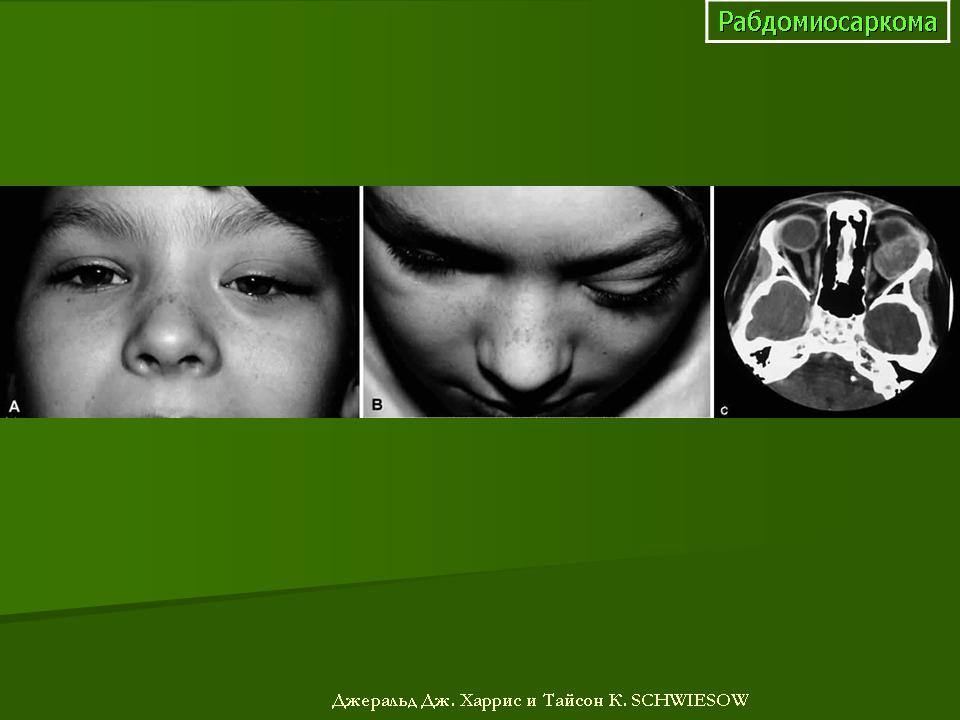
Эмбриональная рабдомиосаркома (СЭД)
ERMS обычно поражает детей первых 5 лет жизни, но может возникать и в более старшем возрасте.
ERMS, как правило, возникает в области головы и шеи, мочевого пузыря, влагалища или вокруг предстательной железы и яичек.
Два подтипа ERMS, ботриоид и веретено-клеточная рабдомиосаркома , как правило, имеют лучший прогноз (перспективы), чем более распространенная традиционная форма ERMS.
Альвеолярная рабдомиосаркома (ARMS)
ARMS обычно одинаково влияет на все возрастные группы. Он составляет большую часть RMS у детей старшего возраста, подростков и взрослых, чем у детей младшего возраста (потому что ERMS реже встречается в старшем возрасте).
ARMS чаще всего возникает в крупных мышцах туловища, рук и ног.
ARMS имеет тенденцию расти быстрее, чем ERMS, и обычно требует более интенсивного лечения. Однако в некоторых случаях ARMS в раковых клетках отсутствуют определенные изменения генов, что заставляет эти виды рака действовать больше как ERMS (и позволяет врачам проводить менее интенсивное лечение).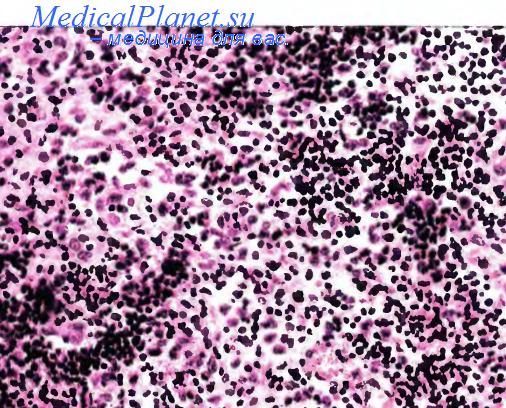
Анапластическая рабдомиосаркома и недифференцированная саркома
Анапластическая рабдомиосаркома (также называемая плеоморфной рабдомиосаркомой ) — это необычный тип, который встречается в основном у взрослых и очень редко встречается у детей.
Некоторые врачи также группируют недифференцированную саркому с рабдомиосаркомами. Используя лабораторные тесты, врачи могут сказать, что эти виды рака являются саркомами, но клетки не обладают какими-либо характеристиками, которые помогли бы их классифицировать дальше.
Оба этих необычных вида рака имеют тенденцию быстро расти и обычно требуют интенсивного лечения.
Рабдомиосаркома у взрослых
Большинство рабдомиосарком развивается у детей и подростков, но они могут возникать и у взрослых. Взрослые с большей вероятностью будут иметь более быстрорастущие типы RMS и иметь их в частях тела, которые труднее лечить. Из-за этого RMS у взрослых часто труднее лечить.
Плеоморфная рабдомиосаркома у взрослых: клинико-патологическое исследование 38 случаев с акцентом на морфологические варианты и недавние специфические для скелетных мышц маркеры
Исторически Стаут впервые представил плеоморфную рабдомиосаркому (PRMS) в литературе в 1946 году (7) как «классическую рабдомиосардомию».В течение следующих трех десятилетий диагноз основывался только на морфологии и, вероятно, включал другие саркомы (включая злокачественную фиброзную гистиоцитому и плеоморфную лейомиосаркому) с поперечными и продольными полосами (3, 5, 7, 8, 9, 10, 11, 12, 13). В 1958 году Хорн и Энтерлайн выделили четыре подтипа рабдомиосаркомы и назвали классические из них «плеоморфной рабдомиосаркомой». Популярность PRMS росла и падала с появлением электронной микроскопии (14, 15, 16, 18, 19) и злокачественной фиброзной гистиоцитомы (20) соответственно.Иммуногистохимические антитела были применены к этим опухолям в начале 1980-х годов, преимущественно с использованием миоглобина, десмина, субъединицы М креатининкиназы и различных актинов для выявления дифференцировки скелетных мышц (18, 19, 21, 22, 23, 24, 25, 26). За исключением миоглобина, белка, обнаруженного на поздних стадиях развития эмбриональных мышц и требующего опытной интерпретации, все другие антитела оказались неспецифичными для фенотипа скелетных мышц. За последнее десятилетие в нескольких сериях исследований для идентификации PRMS использовался иммуногистохимический подход (15, 17, 27, 28, 29, 30, 31).В 1993 г. в репертуар PRMS был добавлен быстрый миозин, маркер, специфичный для скелетных мышц (15). MyoD1, продукт гена, активированного на ранней стадии миогенеза, обсуждался как специфический для скелетных мышц маркер, но не применялся к плеоморфной рабдомиосаркоме до 1995 г. (31, 32, 33). Myf4, миогенин, специфичный для скелетных мышц, был изучен только в четырех случаях PRMS в литературе (27, 34). В текущем исследовании сообщается о 38 случаях PRMS у взрослых с 1970-х годов по настоящее время, диагностированных с использованием современного морфологического и иммуногистохимического подхода, последний из которых упоминал специфические и неспецифические маркеры поперечнополосатых мышц.
Клинически наше исследование показывает, что плеоморфная рабдомиосаркома — это агрессивная саркома, которая возникает преимущественно на конечностях у взрослых мужчин, средний возраст которых составляет 49 лет. Наши клинико-патологические данные аналогичны случаям, описанным в литературе (3, 5, 7, 8, 9, 10, 11, 12, 13, 15, 21, 23, 25, 27, 29, 31. Общий прогноз в литература по PRMS бедна, с показателями выживаемости от 12,5 до 50% от 1 года до 20 месяцев безрецидивной выживаемости (3, 5, 7, 8, 9, 10, 11, 13, 15, 17, 18, 29), что аналогично нашему уровню 5-летней выживаемости без болезней — 27%.По результатам нашего наблюдения 72% пациентов умерли в течение 2 лет после постановки диагноза. Для сравнения, сториформный плеоморфный тип злокачественной фиброзной гистиоцитомы, другой плеоморфной саркомы, имеет общую 5-летнюю выживаемость, варьирующуюся от 36 до 50% (35, 36). Плеоморфная липосаркома имеет 5-летнюю выживаемость 56% (37), а лейомиосаркома глубоких конечностей — 64% 5-летняя выживаемость (38).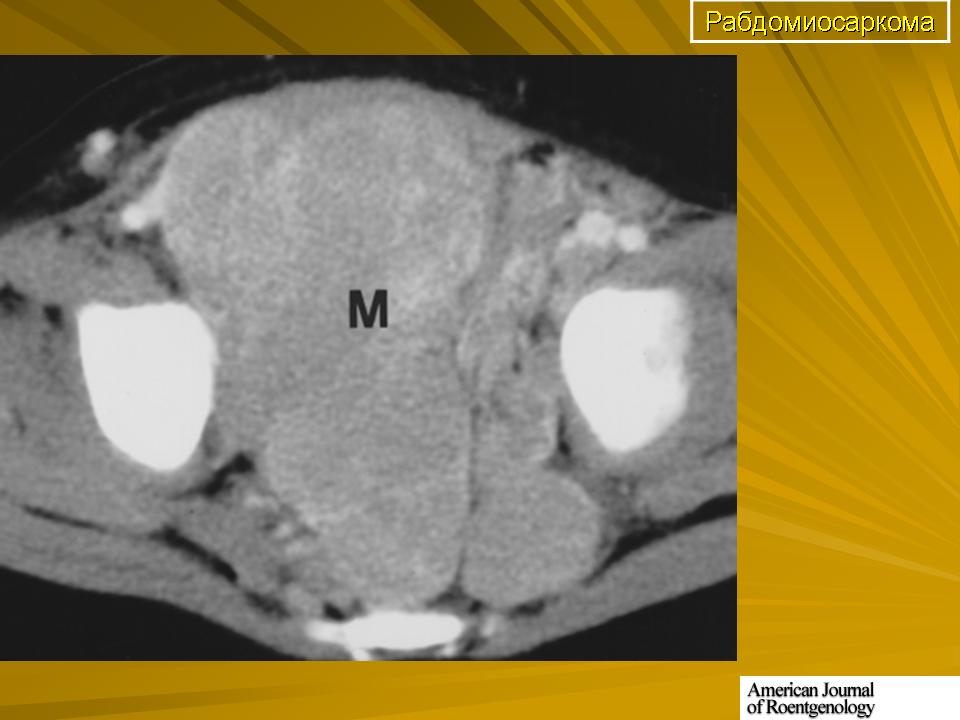 Однако не все эти опухоли были плеоморфными. Следовательно, плеоморфная рабдомиосаркома у взрослых имеет значительно худший прогноз, чем у других плеоморфных сарком.Таким образом, важен правильный диагноз плеоморфной рабдомиосаркомы.
Однако не все эти опухоли были плеоморфными. Следовательно, плеоморфная рабдомиосаркома у взрослых имеет значительно худший прогноз, чем у других плеоморфных сарком.Таким образом, важен правильный диагноз плеоморфной рабдомиосаркомы.
Морфологически все наши плеоморфные рабдомиосаркомы состояли из больших, атипичных, полигональных плеоморфных рабдомиобластов с обильной эозинофильной цитоплазмой. Эти большие рабдомиобласты часто располагаются в группах, листах или разбросанных отдельных клетках. Преобладают атипичные везикулярные ядра с выступающими ядрышками. Рабдомиобласты на заднем плане, окружающие большие плеоморфные рабдомиобласты, варьируются от округлых до веретенообразных.Морфологические особенности крупных атипичных рабдомиобластов при плеоморфной рабдомиосаркоме были ранее описаны Стаутом (7) и другими (3, 5, 8, 9, 10, 11, 12, 13, 15, 21, 29, 31). Однако, за исключением одной группы (29), которая описала фон из более крупных круглых клеток, предположительно рабдомиобластов, в литературе ранее были описаны в основном только веретенообразные и полигональные фоновые клетки (15, 16, 17, 18, 29).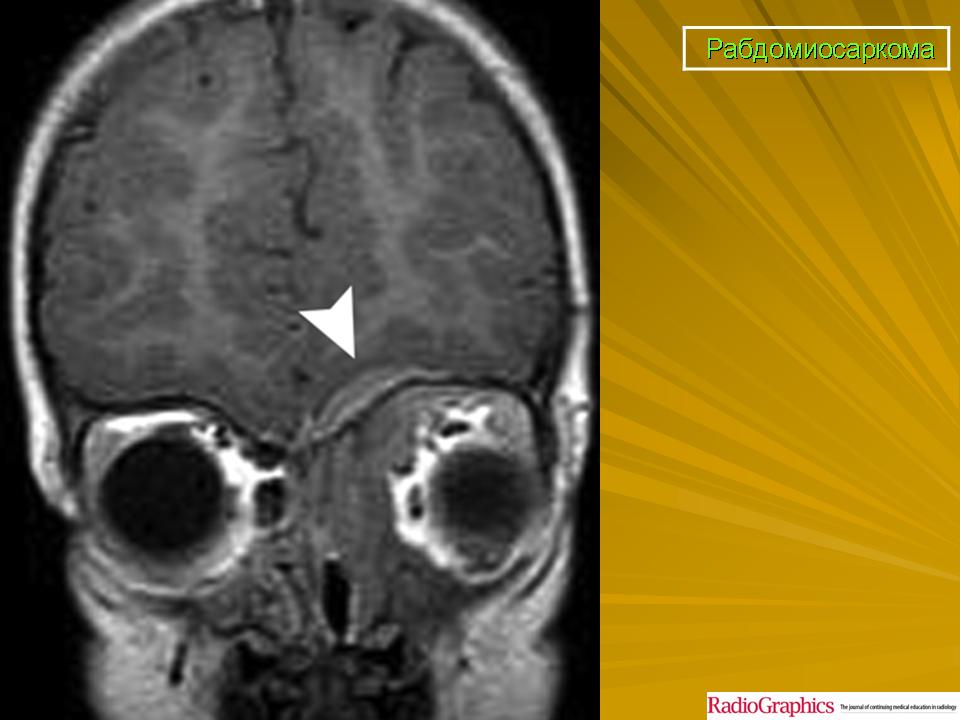 Вышеупомянутые морфологические характеристики случаев в текущем исследовании, с атипичными крупными полигональными плеоморфными рабдомиобластами и двумя дополнительными типами фоновых рабдомиобластов, различают предлагаемые нами признанные морфологические варианты плеоморфной рабдомиосаркомы как классические, круглоклеточные и варианты веретеновидных клеток соответственно.
Вышеупомянутые морфологические характеристики случаев в текущем исследовании, с атипичными крупными полигональными плеоморфными рабдомиобластами и двумя дополнительными типами фоновых рабдомиобластов, различают предлагаемые нами признанные морфологические варианты плеоморфной рабдомиосаркомы как классические, круглоклеточные и варианты веретеновидных клеток соответственно.
Иммунопрофили нашего PRMS показывают, что во всех случаях есть хотя бы один положительный маркер, специфичный для скелетных мышц (миоглобин, ядерный MyoD1, ядерный myf4 и быстрый миозин), в дополнение к положительности для неспецифических миоидных маркеров, включая десмин, миогенин (myf3) или MSA. Хотя для тестирования всех антител в каждом случае было недостаточно материала, миоглобин (95%) и миозин быстрых скелетных мышц (80%) оказались наиболее чувствительными маркерами PRMS в нашей серии. Эти результаты сопоставимы с данными о миоглобине и быстром миозине скелетных мышц на PRMS в литературе (15, 23, 28, 29). Хотя миоглобин часто трудно интерпретировать из-за высокого фона, в этом исследовании мы обнаружили, что миоглобин очень чувствителен к этим плохо дифференцированным рабдомиосаркомам, если интерпретировать его внимательно. Нам требовалась цитоплазма опухолевых клеток в центре опухоли (а не только краевой эффект) и более сильная, чем просто фоновый «чайный цвет», чтобы называть опухоль положительной. Миозин быстрых скелетных мышц был изучен только в пяти наших случаях; дополнительные случаи необходимо будет изучить с помощью этого маркера для получения дополнительных данных.
Хотя миоглобин часто трудно интерпретировать из-за высокого фона, в этом исследовании мы обнаружили, что миоглобин очень чувствителен к этим плохо дифференцированным рабдомиосаркомам, если интерпретировать его внимательно. Нам требовалась цитоплазма опухолевых клеток в центре опухоли (а не только краевой эффект) и более сильная, чем просто фоновый «чайный цвет», чтобы называть опухоль положительной. Миозин быстрых скелетных мышц был изучен только в пяти наших случаях; дополнительные случаи необходимо будет изучить с помощью этого маркера для получения дополнительных данных.
MyoD1 и миогенины (включая myf4 и myf3) принадлежат к семейству близкородственных генов, кодирующих серию ДНК-связывающих белков, которые, как считается, контролируют миогенез (27, 32, 34). По нашему опыту, MyoD1 и myf4 были менее чувствительны к PRMS. Однако мы также использовали строгие критерии для MyoD1, , т.е. , требующие ядерной реактивности. Интересно, что четырнадцать дополнительных случаев PRMS, включенных с помощью других специфичных для скелетных мышц маркеров и электронной микроскопии, показали только цитоплазматическую положительность для MyoD1. Если бы эти четырнадцать считались истинно положительными, это увеличило бы чувствительность MyoD1 с 51 до 89%. Поэтому цитоплазматический положительный результат следует использовать в качестве вспомогательного, но неспецифического признака для PRMS. Чувствительность MyoD1 в литературе достигает 90%, но были протестированы только десять случаев (27, 31). Myf4, миогенин, поддерживающий маркер скелетных мышц, по нашему опыту был только на 53% чувствителен к PRMS. Литературный опыт только по четырем случаям плеоморфной рабдомиосаркомы дает 75% -ную специфичность (27).Myf4, однако, ранее был идентифицирован как более специфичный для дифференцировки скелетных мышц, чем myf3, при рабдомиосаркомах в целом. В одном реферате (39) myf3 также окрашивал 70% лейомиосарком, 67% злокачественных фиброзных гистиоцитом, 100% опухолей Вильма, 55% других мелких круглоклеточных опухолей и 10% других опухолей. В то время как myf4 был специфичным для рабдомиосарком без другого окрашивания, за исключением одного случая опухоли Вильма, вышеупомянутых опухолей.
Если бы эти четырнадцать считались истинно положительными, это увеличило бы чувствительность MyoD1 с 51 до 89%. Поэтому цитоплазматический положительный результат следует использовать в качестве вспомогательного, но неспецифического признака для PRMS. Чувствительность MyoD1 в литературе достигает 90%, но были протестированы только десять случаев (27, 31). Myf4, миогенин, поддерживающий маркер скелетных мышц, по нашему опыту был только на 53% чувствителен к PRMS. Литературный опыт только по четырем случаям плеоморфной рабдомиосаркомы дает 75% -ную специфичность (27).Myf4, однако, ранее был идентифицирован как более специфичный для дифференцировки скелетных мышц, чем myf3, при рабдомиосаркомах в целом. В одном реферате (39) myf3 также окрашивал 70% лейомиосарком, 67% злокачественных фиброзных гистиоцитом, 100% опухолей Вильма, 55% других мелких круглоклеточных опухолей и 10% других опухолей. В то время как myf4 был специфичным для рабдомиосарком без другого окрашивания, за исключением одного случая опухоли Вильма, вышеупомянутых опухолей. Дополнительная информация о myf4 из недавней публикации показывает, что он очень чувствителен и специфичен для рабдомиосаркомы по сравнению с другими детскими опухолями (40).
Дополнительная информация о myf4 из недавней публикации показывает, что он очень чувствителен и специфичен для рабдомиосаркомы по сравнению с другими детскими опухолями (40).
Электронная микроскопия с минимальным критерием рибосомно-миозиновых комплексов подтвердила фенотип скелетных мышц во всех восьми изученных случаях, представляющих все три из наших подтипов PRMS.
Предлагаем три морфологических варианта. Все три из этих вариантов PRMS должны иметь по крайней мере один положительный специфический маркер скелетных мышц в дополнение к другим мышечным маркерам. Тип I или «классический PRMS» морфологически определяется листами больших, атипичных полигональных плеоморфных рабдомиобластов (PRMB).В нашей серии было 8 таких случаев. Тип II, также называемый «PRMS круглых клеток», морфологически состоит из этих больших PRMB среди слегка плеоморфных круглых рабдомиобластов среднего размера. Это 13 наших дел. Хотя можно считать, что этот морфологический вариант имеет сходство с эмбриональной рабдомиосаркомой, есть несколько причин, по которым эти случаи лучше классифицировать как круглоклеточный вариант плеоморфной рабдомиосаркомы.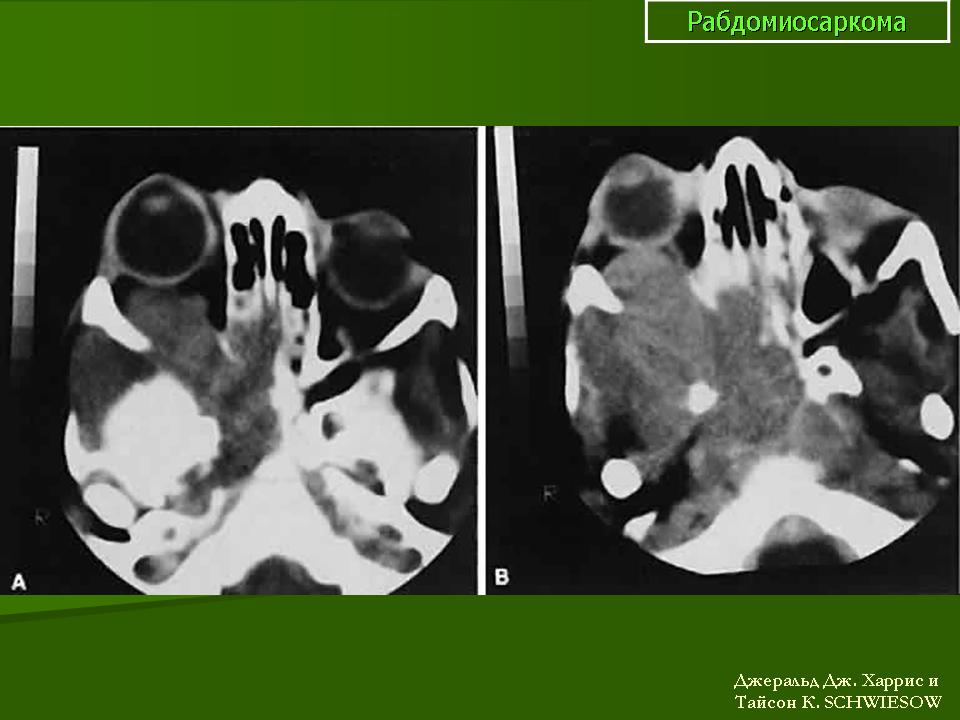 Все эти опухоли есть у взрослых. Круглые клетки больше, чем круглые клетки эмбриональной рабдомиосаркомы, и в этих опухолях более многочисленные и более атипичные плеоморфные рабдомиобласты.Подтип твердой альвеолярной рабдомиосаркомы можно отличить от этого круглоклеточного варианта PRMS по хромосомным транслокациям t (2; 13) или варианту t (1; 13). Тот же самый сайт PAX7 на хромосоме 1, по-видимому, участвует в генетических изменениях эмбриональной рабдомиосаркомы (41). Для сравнения, о генетике PRMS известно немного. Дальнейшие генетические исследования помогут нам понять взаимосвязь PRMS с другими подтипами RMS. Тип III, или PRMS веретеновидных клеток, составлял большинство наших случаев ( n = 17).Он состоял из крупных атипичных плеоморфных рабдомиобластов с сильно веретеновидным и часто сториформным фоном. Эти опухоли имитируют сториформную плеоморфную злокачественную фиброзную гистиоцитому, но имеют большие атипичные рабдомиобласты и иммунофенотипические свидетельства дифференцировки скелетных мышц в полигональные и веретенообразные клетки.
Все эти опухоли есть у взрослых. Круглые клетки больше, чем круглые клетки эмбриональной рабдомиосаркомы, и в этих опухолях более многочисленные и более атипичные плеоморфные рабдомиобласты.Подтип твердой альвеолярной рабдомиосаркомы можно отличить от этого круглоклеточного варианта PRMS по хромосомным транслокациям t (2; 13) или варианту t (1; 13). Тот же самый сайт PAX7 на хромосоме 1, по-видимому, участвует в генетических изменениях эмбриональной рабдомиосаркомы (41). Для сравнения, о генетике PRMS известно немного. Дальнейшие генетические исследования помогут нам понять взаимосвязь PRMS с другими подтипами RMS. Тип III, или PRMS веретеновидных клеток, составлял большинство наших случаев ( n = 17).Он состоял из крупных атипичных плеоморфных рабдомиобластов с сильно веретеновидным и часто сториформным фоном. Эти опухоли имитируют сториформную плеоморфную злокачественную фиброзную гистиоцитому, но имеют большие атипичные рабдомиобласты и иммунофенотипические свидетельства дифференцировки скелетных мышц в полигональные и веретенообразные клетки. Эти случаи были высоко плеоморфными и их можно было легко отделить от веретено-клеточного варианта эмбриональной рабдомиосаркомы, также описанного у взрослых (6).
Эти случаи были высоко плеоморфными и их можно было легко отделить от веретено-клеточного варианта эмбриональной рабдомиосаркомы, также описанного у взрослых (6).
Наши морфологические варианты плеоморфной рабдомиосаркомы основаны на преобладающем морфологическом паттерне и существенно не различаются в отношении прогноза, хотя для варианта с круглыми клетками прогноз несколько хуже.С другой стороны, наши цифры невелики, и поэтому статистическую значимость определить невозможно. Прогноз у всех трех групп относительно плохой. Мы считаем, что важно распознать этот морфологический спектр плеоморфной рабдомиосаркомы для диагностических целей и для того, чтобы помочь отделить плеоморфную рабдомиосаркому от других опухолей.
Наряду с интерпретацией миогенных иммуногистохимических красителей необходимо оценить широкую панель немиидных маркеров, чтобы исключить низкодифференцированные карциномы, рабдоидные опухоли, опухоли Тритона и злокачественную меланому.Хотя рабдомиосаркомы, как и другие саркомы, иногда могут экспрессировать кератин, отсутствие эпителиальных маркеров может помочь отделить PRMS от карцином и внепочечных рабдоидных опухолей. Отсутствие фокального белка S100 и GFAP и присутствие миоидных маркеров в фоновых веретенообразных рабдомиобластах помогают идентифицировать веретенообразный компонент PRMS веретенообразных клеток как отдельный от компонента злокачественной опухоли оболочки периферических нервов злокачественной опухоли тритона. Злокачественная меланома часто имеет диффузную положительную реакцию на белок S100, а также возможную положительную реакцию на маркеры меланоцитов, включая HMB-45, мелан-A и тирозиназу.
Отсутствие фокального белка S100 и GFAP и присутствие миоидных маркеров в фоновых веретенообразных рабдомиобластах помогают идентифицировать веретенообразный компонент PRMS веретенообразных клеток как отдельный от компонента злокачественной опухоли оболочки периферических нервов злокачественной опухоли тритона. Злокачественная меланома часто имеет диффузную положительную реакцию на белок S100, а также возможную положительную реакцию на маркеры меланоцитов, включая HMB-45, мелан-A и тирозиназу.
Злокачественная фиброзная гистиоцитома (MFH) (иногда называемая «плеоморфной саркомой, если не указано иное») и плеоморфная лейомиосаркома (LMS) также могут учитываться при дифференциальной диагностике. Хотя известно, что MFH иногда экспрессирует и десмин, и MSA (42), он не должен экспрессировать другие специфические маркеры скелетных мышц, такие как MyoD1, миозин быстрых скелетных мышц, myf4 или миоглобин. LMS, миоидная опухоль с экспрессией десмина, морфологически имеет пересекающиеся пучки, в ней отсутствуют крупные полигональные рабдомиобласты, а также не экспрессируются специфические маркеры для скелетных мышц.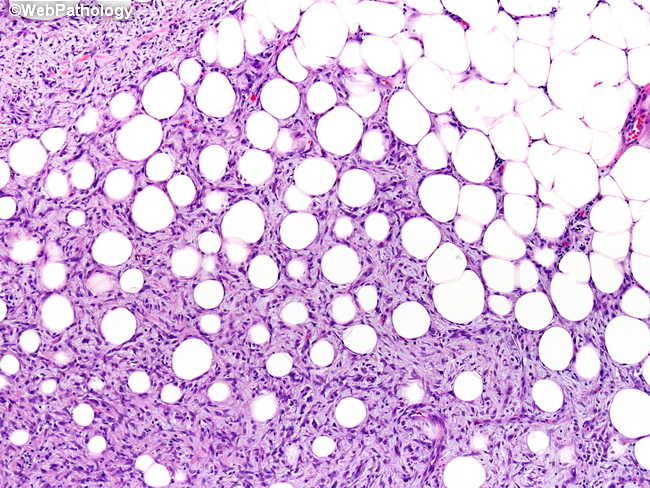
В заключение, это исследование рассматривает 38 случаев PRMS. PRMS чаще всего возникает на конечностях взрослых мужчин и обычно следует агрессивному клиническому течению с общим неблагоприятным прогнозом. Три морфологических варианта были идентифицированы на основании присутствия крупных, полигональных, атипичных плеоморфных рабдомиобластов с двумя дополнительными формами фоновых рабдомиобластов: классической, круглой клеточной и веретено-клеточной плеоморфной рабдомиосаркомой соответственно. Иммуногистохимический анализ должен включать как специфические маркеры скелетных мышц, так и неспецифические миоидные маркеры.В PRMS может быть спектр дифференцировки, о чем свидетельствует вариабельная экспрессия миорегуляторного белка, которая, по-видимому, не определяется чисто морфологически.
Плеоморфная рабдомиосаркома печени у взрослого: отчет о редком случае | BMC Surgery
RMS в печени, особенно у взрослых, трудно поддается лечению из-за отсутствия стандартных диагностических критериев или стандартного протокола лечения. На сегодняшний день зарегистрировано только 10 случаев RMS печени у взрослых, включая наш случай, которые сведены в Таблицу 1 [4,5,6,7,8,9,10,11,12].Среди этих случаев RMS чаще встречался у мужчин, чем у женщин, и в нашем случае участвовал самый старый пациент.
На сегодняшний день зарегистрировано только 10 случаев RMS печени у взрослых, включая наш случай, которые сведены в Таблицу 1 [4,5,6,7,8,9,10,11,12].Среди этих случаев RMS чаще встречался у мужчин, чем у женщин, и в нашем случае участвовал самый старый пациент.
Таблица 1 Зарегистрированные случаи рабдомиосаркомы печени у взрослых
Horn and Enterline et al. [13] сообщили о четырех подгруппах RMS: эмбриональной, альвеолярной, плеоморфной и ботриоидной. Ботриоидный RMS на самом деле является подтипом эмбрионального RMS [14]. Эмбриональный RMS является наиболее частым типом RMS у детей раннего возраста, альвеолярный RMS — наиболее частым типом у пациентов старше 10 лет, а плеоморфный RMS — наиболее частым типом у взрослых пожилых людей [3, 13].Среди взрослых пациентов, у которых RMS возник в печени, у четырех был эмбриональный RMS и у трех был плеоморфный RMS.
На сегодняшний день нет отчетов, описывающих типичные результаты визуализации и симптомы RMS.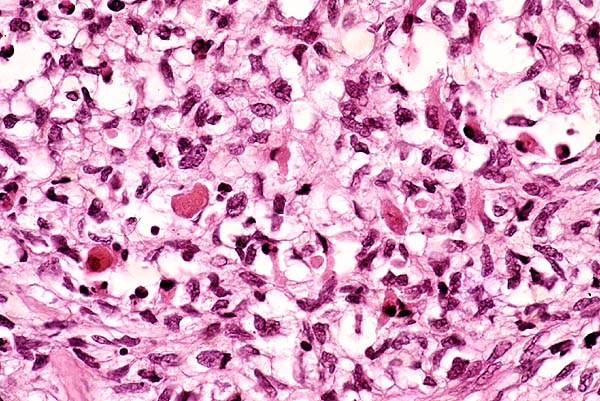 Большинство зарегистрированных случаев были обнаружены как большие образования диаметром> 10 см, занимающие долю печени. У нашего пациента была опухоль печени размером 12 см, изначально диагностированная и леченная как абсцесс печени, которая вызвала перитонеальную диссеминацию в соответствии с путём дренирования после резекции. При исследовании таких случаев важно выполнить чрескожную биопсию и включить RMS в качестве дифференциальной диагностики новообразований печени у взрослых.
Большинство зарегистрированных случаев были обнаружены как большие образования диаметром> 10 см, занимающие долю печени. У нашего пациента была опухоль печени размером 12 см, изначально диагностированная и леченная как абсцесс печени, которая вызвала перитонеальную диссеминацию в соответствии с путём дренирования после резекции. При исследовании таких случаев важно выполнить чрескожную биопсию и включить RMS в качестве дифференциальной диагностики новообразований печени у взрослых.
RMS у взрослых — это очень злокачественная опухоль с неблагоприятным прогнозом из-за отсутствия стандартного протокола лечения. Sultan et al. [15] сообщили, что RMS у взрослых имеет значительно худшие результаты, чем в детстве (средняя 5-летняя общая выживаемость, 27% ± 1,4 и 61% ± 1,4%, соответственно; P <0,0001). Среди ранее сообщенных случаев РРС, возникшего в печени, только два пациента прожили более 12 месяцев; большинство пациентов умерли в течение 12 месяцев с момента появления первых симптомов.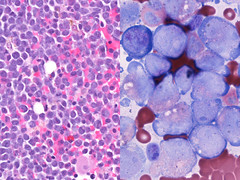 Необходимо разработать оптимальный протокол лечения и таким образом улучшить исход пациентов с этим редким, но смертельным раком.
Необходимо разработать оптимальный протокол лечения и таким образом улучшить исход пациентов с этим редким, но смертельным раком.
Радикальная резекция с отрицательными краями, химиотерапия и лучевая терапия предложены Межгрупповой исследовательской группой по рабдомиосаркоме; эти вмешательства составляют в целом оптимальный протокол лечения в детстве [16, 17]. Химиотерапевтические препараты включают актиномицин D, винкристин, доксорубицин, циклофосфамид, этопозид и ифосфамид. Мы лечили RMS нашего пациента трабектином, как и другие саркомы мягких тканей, потому что комбинированная терапия с несколькими лекарствами считается трудной из-за ухудшения состояния здоровья.Наш пациент изначально показал хороший ответ на химиотерапию; однако она не могла продолжить дальнейшую химиотерапию из-за серьезных побочных эффектов.
В данном документе мы сообщили об исключительно редком случае плеоморфного RMS печени у взрослого человека. Редкость этого случая обусловлена расположением опухоли и возрастом пациента, и его отчет поможет установить стандартный диагноз и лечение.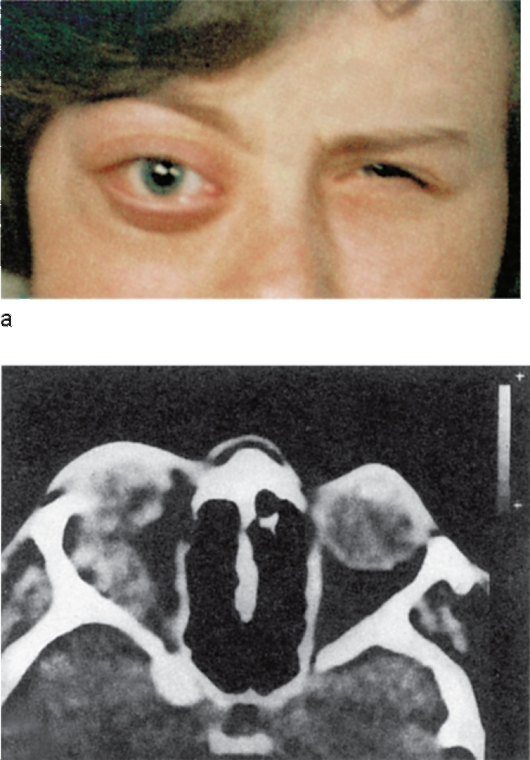
Плеоморфная рабдомиосаркома у взрослых: многоцентровое ретроспективное исследование
Abstract
Общие сведения: Плеоморфная рабдомиосаркома (RMS) является редким подтипом RMS.Оптимальное лечение остается неопределенным. Пациенты и методы: в период с 1995 по 2014 год 45 пациентов были диагностированы и пролечены в трех центрах третичной саркомы (Великобритания, Швейцария и Германия). Были проанализированы характеристики и исходы лечения. Результаты: средний возраст на момент постановки диагноза составлял 71,5 года (диапазон = 28,4–92,8 года). Медиана выживаемости для пациентов с локализованным (n = 32, 71,1%) и метастатическим заболеванием (n = 13, 28,9%) составила 12,8 месяцев (95% доверительный интервал = 8,2-34,4) и 7,1 месяца (95% доверительный интервал = 3.8-11.3) соответственно. Частота рецидивов составила 53,8% (четыре местных и 10 отдаленных). В общей сложности 14 (31,1%) пациентов получали паллиативную химиотерапию первой линии, включая схемы мультиагентной детской химиотерапии (n = 3), ифосфамид-доксорубицин (n = 4) и монотерапию доксорубицином (n = 7).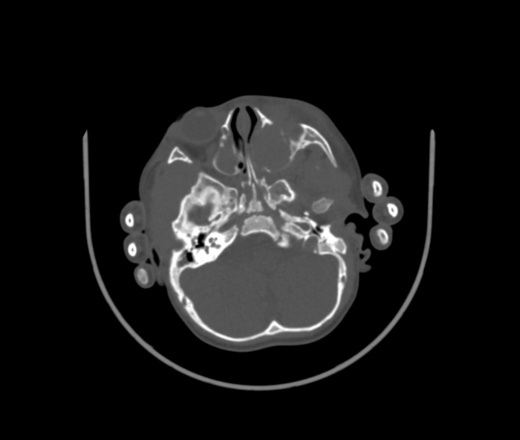 Ответ на химиотерапию был плохим (одна частичная ремиссия с винкристин-актиномицином D-циклофосфамидом и шесть случаев со стабильным заболеванием). Средняя выживаемость без прогрессирования составила 2,3 (от 1,2 до 7,3) месяца. Заключение. Плеоморфный РРС — это агрессивное новообразование, поражающее в основном пожилых пациентов, связанное с высокой частотой рецидивов, слабым и непродолжительным ответом на стандартную химиотерапию и общим плохим прогнозом как для локализованного, так и для метастатического заболевания.
Ответ на химиотерапию был плохим (одна частичная ремиссия с винкристин-актиномицином D-циклофосфамидом и шесть случаев со стабильным заболеванием). Средняя выживаемость без прогрессирования составила 2,3 (от 1,2 до 7,3) месяца. Заключение. Плеоморфный РРС — это агрессивное новообразование, поражающее в основном пожилых пациентов, связанное с высокой частотой рецидивов, слабым и непродолжительным ответом на стандартную химиотерапию и общим плохим прогнозом как для локализованного, так и для метастатического заболевания.
Рабдомиосаркомы (РМС) — это в основном детские новообразования, хотя у взрослых такие случаи могут быть нечасто диагностированы (1). Текущая классификация ВОЗ подразделяет RMS на четыре различных подтипа: эмбриональный, альвеолярный, плеоморфный и веретено-клеточный / склерозирующий (2). Плеоморфный РРС, впервые описанный Стаутом и его коллегами в 1946 г. (3), остается очень редкой болезнью, и для ее лечения недостаточно данных (4). В настоящее время это определяется как саркома высокой степени злокачественности, состоящая из недифференцированных круглых и веретенообразных клеток, которые демонстрируют дифференцировку скелетных мышц без эмбриональных или альвеолярных компонентов (2). Как правило, это агрессивное поражение, возникающее в глубоких мягких тканях конечностей с высокой склонностью к метастазированию. Из-за своей биологической и геномной сложности его клиническое поведение и чувствительность к химиотерапии больше похожи на взрослые саркомы мягких тканей высокой степени злокачественности, чем на педиатрические RMS (5, 6).
Как правило, это агрессивное поражение, возникающее в глубоких мягких тканях конечностей с высокой склонностью к метастазированию. Из-за своей биологической и геномной сложности его клиническое поведение и чувствительность к химиотерапии больше похожи на взрослые саркомы мягких тканей высокой степени злокачественности, чем на педиатрические RMS (5, 6).
Основным методом лечения локализованного заболевания является широкая хирургическая резекция. При запущенном заболевании не существует стандартной системной терапии; Пациенты проходят лечение по различным схемам химиотерапии, от схем мультиагентной детской химиотерапии до монотерапии доксорубицином (7-11).Поскольку в большинстве ретроспективных исследований различные подтипы RMS были сгруппированы вместе, нет четкого консенсуса по поводу оптимального системного лечения плеоморфного RMS. Химиочувствительность плеоморфного RMS еще предстоит определить, хотя обычно ее считают химиорезистентной.
Целью настоящего исследования было сообщить о клинических характеристиках, лечении и исходах пациентов с плеоморфным РРС, а также задокументировать реакцию на паллиативную химиотерапию для пациентов с метастатическим заболеванием, чтобы обеспечить основу для будущих исследований и дать рекомендации.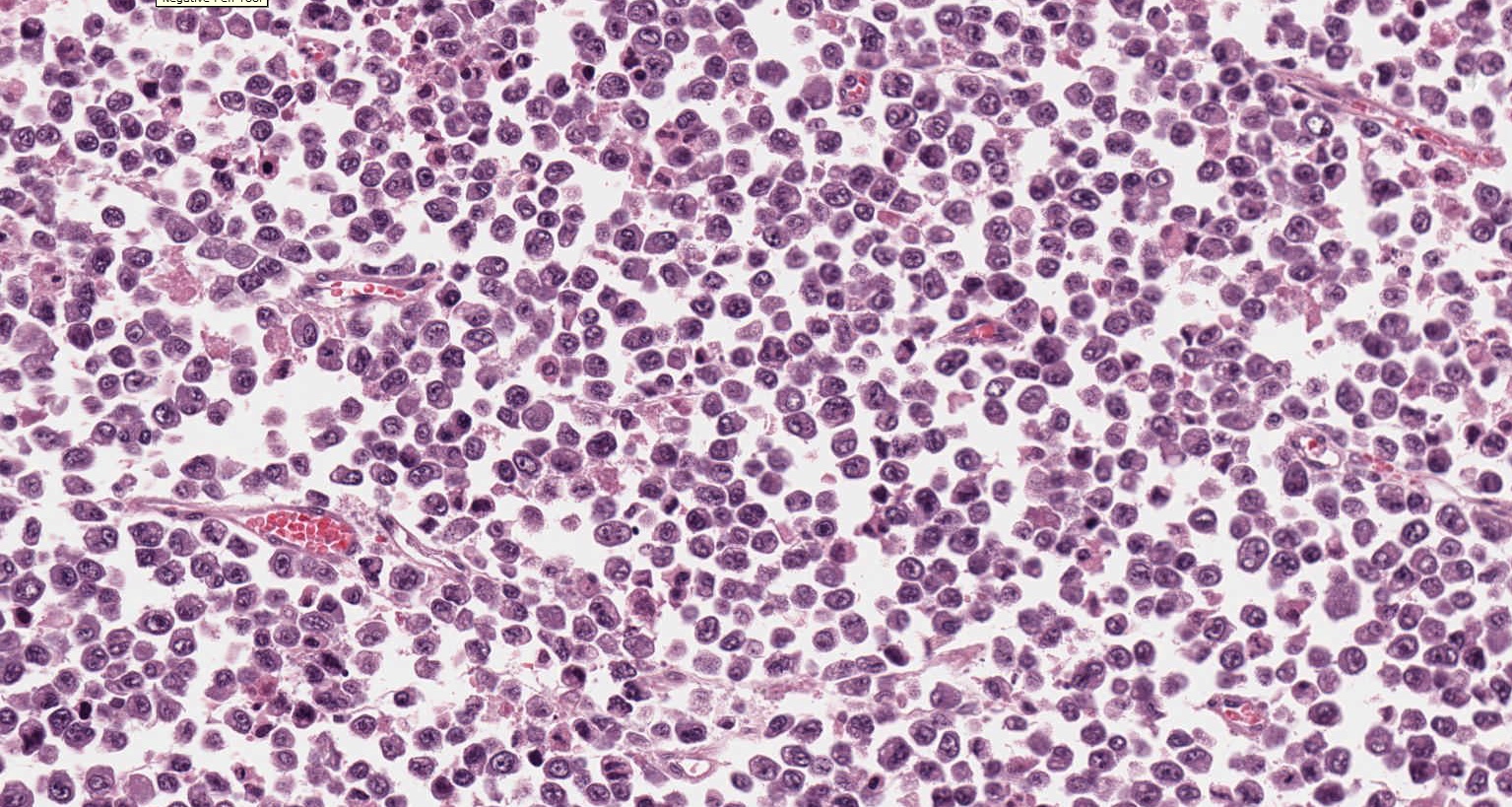 врачей и пациентов относительно выбора лечения.
врачей и пациентов относительно выбора лечения.
Пациенты и методы
Мы провели поиск в перспективных базах данных больницы Royal Marsden (Великобритания), университетской больницы Берна (Швейцария) и университетской больницы Мангейма.
(Германия) для выявления пациентов, которым в период с 1995 по 2014 год был поставлен диагноз плеоморфного RMS. Все диагнозы плеоморфного RMS были подтверждены патологами саркомы мягких тканей путем сочетания морфологических признаков, иммунопрофиля и молекулярного профилирования для исключения альтернативных диагнозов.Все пациенты с эмбриональным RMS, альвеолярным RMS и веретено-клеточным / склерозирующим RMS были исключены из исследования. Данные были собраны путем ретроспективного обзора медицинских карт. Перед началом исследования было получено одобрение этического комитета каждой больницы (номер одобрения SEVAL SE399).
Таблица I.
Исходные клинические характеристики при диагностике локализованного или метастатического заболевания.
Были зарегистрированы исходные характеристики, включая возраст, пол, расположение первичной опухоли, размер опухоли и анатомическую глубину, стадию заболевания и участки метастатического поражения.Локализованное заболевание определялось отсутствием отдаленных метастазов. Размер первичной опухоли оценивался путем макроскопического измерения хирургического образца, если он был доступен, или с помощью базовой визуализации.
Были проанализированы методы лечения, включая хирургическое вмешательство, лучевую терапию и химиотерапию, используемые для локализованных и метастатических заболеваний. Оценивали хирургические границы. Резекция R0 определялась отсутствием микроскопического поражения краев опухолью. Резекция R1 определялась наличием микроскопического поражения краев без признаков макроскопического заболевания.Резекция R2 определялась наличием остаточного макроскопического поражения. Пациенты, получавшие паллиативную химиотерапию, были повторно визуализированы с помощью компьютерной томографии или магнитного резонанса после двух или трех курсов химиотерапии в соответствии с рекомендациями местной больницы. Ответ на химиотерапию оценивался ретроспективным просмотром радиологических отчетов. В большинстве отчетов использовались критерии оценки ответа при солидных опухолях (RECIST) (12). Из-за длительного периода времени, охваченного этим исследованием, не было возможности пересмотреть радиологический ответ, поскольку электронное архивирование изображений было доступно не для всех пациентов.Окончательный результат был зарегистрирован по состоянию на март 2015 года. Пациенты были зарегистрированы как живые или мертвые.
Ответ на химиотерапию оценивался ретроспективным просмотром радиологических отчетов. В большинстве отчетов использовались критерии оценки ответа при солидных опухолях (RECIST) (12). Из-за длительного периода времени, охваченного этим исследованием, не было возможности пересмотреть радиологический ответ, поскольку электронное архивирование изображений было доступно не для всех пациентов.Окончательный результат был зарегистрирован по состоянию на март 2015 года. Пациенты были зарегистрированы как живые или мертвые.
Статистический анализ. Общая выживаемость (ОВ) определялась временем между постановкой диагноза и смертью или последним контактом с пациентом. Для пациентов с рецидивом время рецидива определялось по времени между постановкой диагноза и возникновением местного или отдаленного рецидива. Для пациентов, получающих паллиативную химиотерапию, выживаемость без прогрессирования (ВБП) рассчитывалась путем определения времени между началом лечения и прогрессированием заболевания в соответствии с критериями RECIST или смертью от любой причины.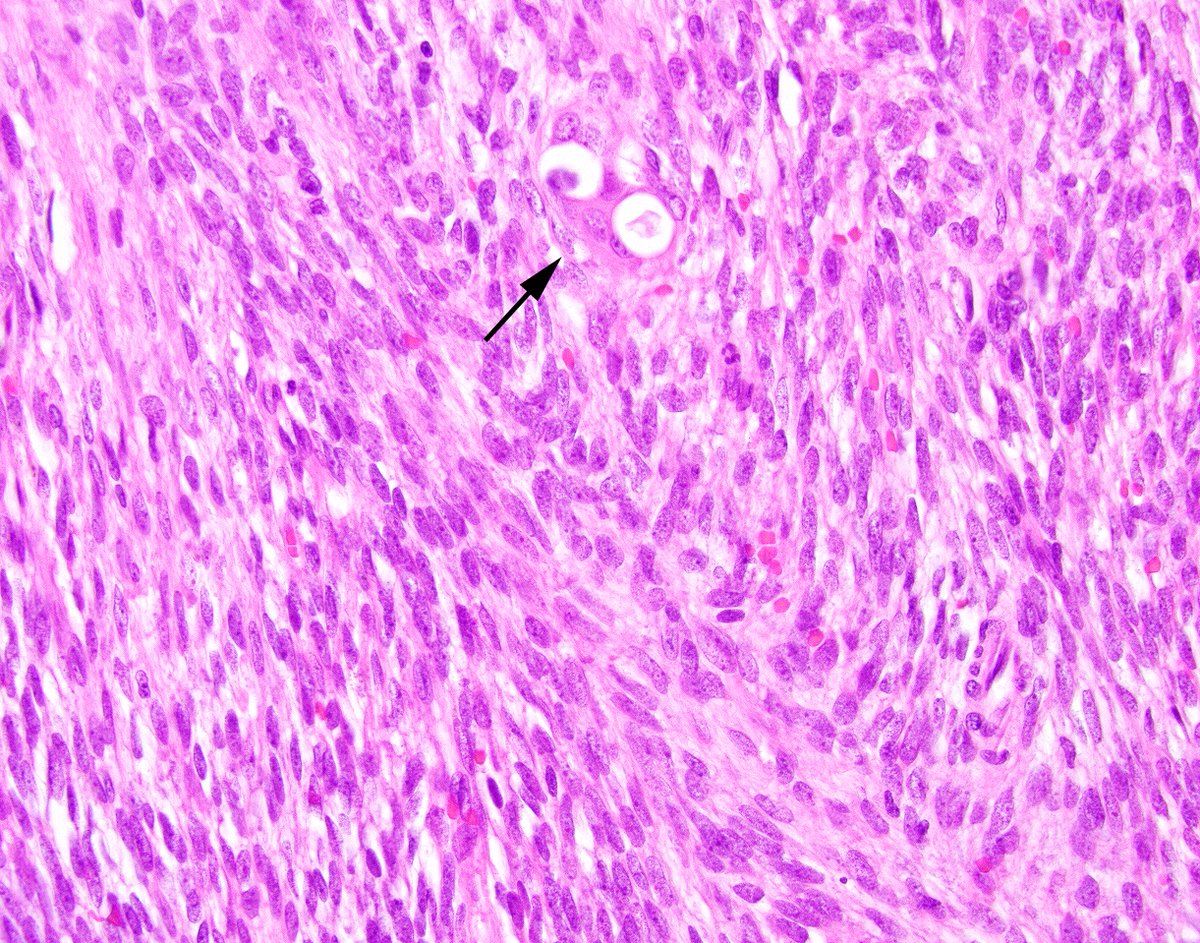 Кривые выживаемости были получены с использованием метода Каплана-Мейера. Весь статистический анализ проводился с использованием программного обеспечения MedCalc версии 14.8.1 (MedCalc Software, Остенде, Бельгия).
Кривые выживаемости были получены с использованием метода Каплана-Мейера. Весь статистический анализ проводился с использованием программного обеспечения MedCalc версии 14.8.1 (MedCalc Software, Остенде, Бельгия).
Результаты
Исходные характеристики. В период с 1995 по 2014 год у 45 пациентов (39 пациентов в Великобритании, 3 в Швейцарии и 3 в Германии) был диагностирован плеоморфный RMS. Исходные характеристики суммированы в таблице I. Общий средний возраст на момент постановки диагноза составлял 71,5 года (диапазон = 28.4-92,8 года). Чуть более двух третей пациентов были диагностированы в возрасте старше 65 лет. Соотношение мужчин и женщин составляло 1,8: 1.
Большинство пациентов (n = 32, 71,1%) имели локализованное заболевание на момент их первоначального диагноза. Первичная опухоль чаще регистрировалась в конечностях (n = 23, 51,1%). Размер опухоли был одинаковым у пациентов с локализованным и метастатическим заболеванием. У меньшинства пациентов с локализованным заболеванием была поверхностная первичная опухоль (n = 5, 16,7%).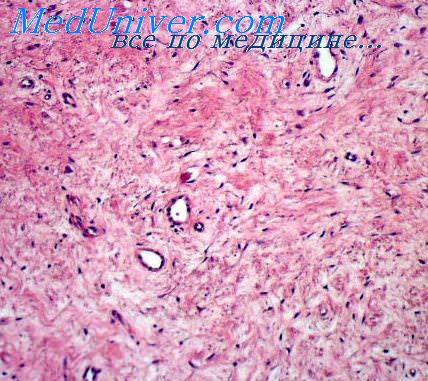 В группе с метастазами поверхностной первичной опухоли выявлено не было.Поражение лимфатических узлов было редкостью (менее 10% пациентов с локализованным заболеванием). Легкие были наиболее частым местом поражения отдаленных метастазов (n = 10, 76,9%).
В группе с метастазами поверхностной первичной опухоли выявлено не было.Поражение лимфатических узлов было редкостью (менее 10% пациентов с локализованным заболеванием). Легкие были наиболее частым местом поражения отдаленных метастазов (n = 10, 76,9%).
Таблица II.
Краткое описание лечения и исходы для пациентов с локализованным заболеванием (n = 32).
Управление локализованным заболеванием и исходом . Хирургическая резекция первичной опухоли была предпринята у 26 пациентов (81,3%) с локализованным заболеванием. Положительные границы были зарегистрированы у четырех пациентов (12,5%, одна тазовая, одна внутрибрюшная и две первичные опухоли нижних конечностей).Большинство пациентов (57,7%, 15/26 пациентов) получали послеоперационную лучевую терапию. Двое пациентов получали неоадъювантную или адъювантную химиотерапию. У одного пациента было прогрессирование заболевания после двух циклов неоадъюванта.
доксорубицин. Другой пациент прошел резекцию R1 и получил два курса ифосфамида и этопозида с сопутствующей лучевой терапией, за которыми следовали три цикла ифосфамида, этопозида и доксорубицина.
Рисунок 1.
Общая выживаемость пациентов с локализованным или метастатическим заболеванием.
Таблица III.
Краткое описание лечения и ответ на лечение пациентов, получавших паллиативную химиотерапию.
Итоги представлены в Таблице II. В ходе наблюдения рецидив был выявлен у 14 (53,8%) пациентов, первоначально оперированных по поводу локализованного заболевания. Было четыре (15,4%) местных рецидива и 10 (38,5%) метастатических рецидивов. Среднее время до рецидива составило 5,3 месяца (от 1,0 до 13,8 месяцев). Двое из четырех пациентов с локальным рецидивом не получали до- или послеоперационную лучевую терапию.У всех четырех пациентов с положительными краями (резекция R1 и R2) произошел рецидив (два местных и два метастатических). Медиана безрецидивной выживаемости составила 7,3 месяца [95% доверительный интервал (ДИ) = 4,9-25,4 месяца]. При последнем наблюдении 9 пациентов (28,1%) были живы без признаков заболевания. Медиана ОВ для пациентов с локализованным заболеванием составила 12,8 месяца (95% ДИ = 8,2–34,4 месяца) (рисунок 1).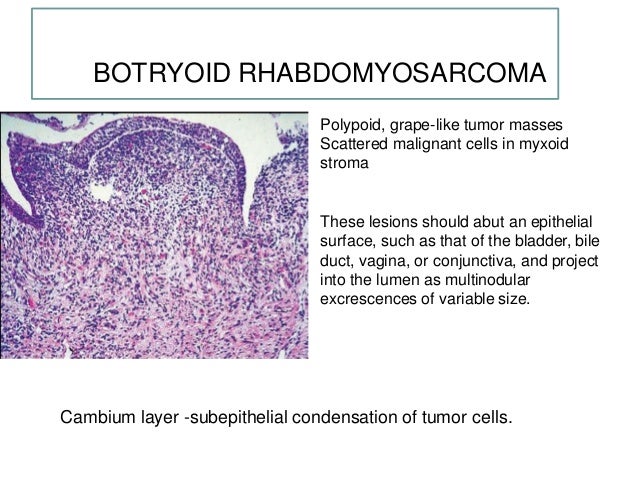 Из 14 пациентов с выявленными рецидивами 13 умерли от болезни. Один пациент, повторно прооперированный из-за местного рецидива, был все еще жив без признаков заболевания при последнем наблюдении.
Из 14 пациентов с выявленными рецидивами 13 умерли от болезни. Один пациент, повторно прооперированный из-за местного рецидива, был все еще жив без признаков заболевания при последнем наблюдении.
Лечение метастазов и исходы. Тринадцать пациентов имели метастатическое заболевание на момент обращения. В ходе лечения двум пациентам была произведена резекция первичной опухоли, шестерым была проведена паллиативная лучевая терапия, а семерым — паллиативная химиотерапия (подробности см. В следующем разделе). Остальным шести пациентам паллиативная химиотерапия не была назначена либо из-за плохой успеваемости, либо из-за преклонного возраста. Медиана ОВ пациентов с исходным метастатическим поражением составила 7.1 месяц (95% ДИ = 3,8-11,3 месяца) (рисунок 1). При последнем наблюдении 10 (76,9%) пациентов умерли от болезни, а три (23,0%) пациента были живы с признаками заболевания.
Ответ на паллиативную химиотерапию. В общей сложности 14 (31,1%) пациентов получили паллиативную химиотерапию (семь пациентов с начальным локализованным заболеванием и последующим метастатическим рецидивом и семь пациентов с начальным метастатическим заболеванием) (Таблица III). Количество линий химиотерапии варьировалось от 1 до 4 (восемь пациентов получали одну линию, четыре — две линии, один — три линии и один — четыре линии химиотерапии).В качестве терапии первой линии наиболее распространенным выбором был доксорубицин в виде монотерапии (семь пациентов). Трем пациентам была назначена мультиагентная химиотерапия, включающая винкристин, доксорубицин, циклофосфамид, чередующиеся с карбоплатином / этопозидом, (винкристин, актиномицин D, только циклофосфамид (VAC) и карбоплатин, эпирубицин, винкристин, актиномицин D, этортоубицин и ифосфамид. Комбинация ифосфамида была назначена двум пациентам: один пациент получил карбоплатин и паклитаксел из-за ошибочного первоначального диагноза аденокарциномы, а другой пациент получил доксорубицин плюс палифосфамид или плацебо в ходе клинического исследования.
Количество линий химиотерапии варьировалось от 1 до 4 (восемь пациентов получали одну линию, четыре — две линии, один — три линии и один — четыре линии химиотерапии).В качестве терапии первой линии наиболее распространенным выбором был доксорубицин в виде монотерапии (семь пациентов). Трем пациентам была назначена мультиагентная химиотерапия, включающая винкристин, доксорубицин, циклофосфамид, чередующиеся с карбоплатином / этопозидом, (винкристин, актиномицин D, только циклофосфамид (VAC) и карбоплатин, эпирубицин, винкристин, актиномицин D, этортоубицин и ифосфамид. Комбинация ифосфамида была назначена двум пациентам: один пациент получил карбоплатин и паклитаксел из-за ошибочного первоначального диагноза аденокарциномы, а другой пациент получил доксорубицин плюс палифосфамид или плацебо в ходе клинического исследования.
В первой линии клиническая активность (частичный ответ и стабильное заболевание) наблюдалась у семи (50,0%) пациентов. Частичный ответ наблюдался только у одного пациента, получавшего схему ВАК.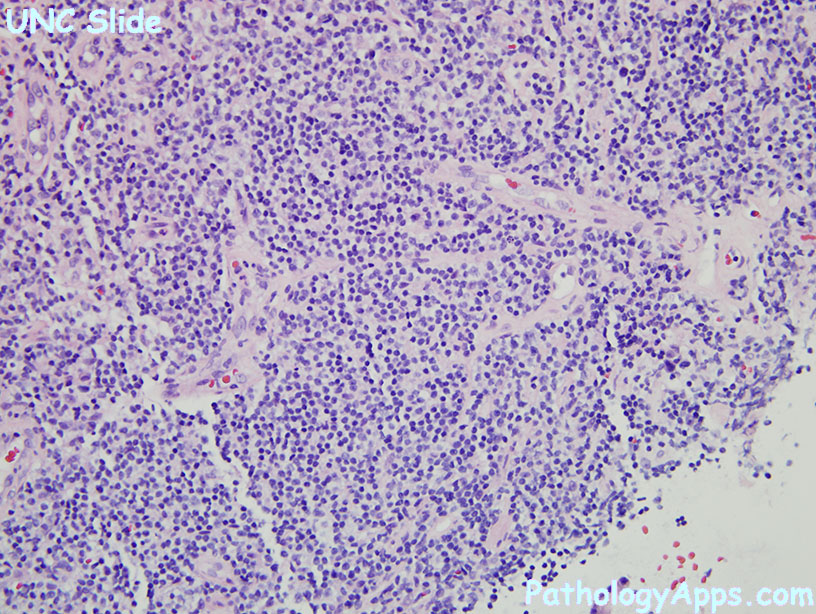 Стабильное заболевание было зарегистрировано у двух пациентов, получавших доксорубицин, у одного, получавшего комбинацию доксорубицин-ифосфамид, у одного, получавшего комбинацию доксорубицин-палифосфамид, и у двух пациентов, получавших мультиагентную химиотерапию. Респондентам было назначено в среднем четыре цикла. Единственный документально подтвержденный устойчивый ответ был достигнут при использовании режима VAC (7.3 месяца при последнем наблюдении). Медиана ВБП составила 2,3 месяца (диапазон = 1,2-7,3 месяца). Ни у одного пациента не было документально подтвержденной клинической активности помимо химиотерапии первой линии, у всех было прогрессирующее заболевание.
Стабильное заболевание было зарегистрировано у двух пациентов, получавших доксорубицин, у одного, получавшего комбинацию доксорубицин-ифосфамид, у одного, получавшего комбинацию доксорубицин-палифосфамид, и у двух пациентов, получавших мультиагентную химиотерапию. Респондентам было назначено в среднем четыре цикла. Единственный документально подтвержденный устойчивый ответ был достигнут при использовании режима VAC (7.3 месяца при последнем наблюдении). Медиана ВБП составила 2,3 месяца (диапазон = 1,2-7,3 месяца). Ни у одного пациента не было документально подтвержденной клинической активности помимо химиотерапии первой линии, у всех было прогрессирующее заболевание.
Обсуждение
Наша серия случаев, подробно описывающая методы лечения плеоморфного RMS, является самой большой когортой, о которой нам известно (4, 7-9, 11, 13-16). В соответствии с предыдущими сообщениями, плеоморфный РРС — редкое заболевание, в основном у пожилых людей, связанное с агрессивным клиническим течением.
Генетический ландшафт плеоморфных RMS часто характеризуется очень сложным кариотипом и контрастирует с менее сложными кариотипами, наблюдаемыми при альвеолярном RMS и эмбриональном RMS (17, 18).Еще одним подтверждением того, что плеоморфный RMS как отдельная сущность, является его морфология, он состоит из заметно атипичных веретен и многоугольных клеток, часто причудливых форм. Для сравнения, относительно монотонная популяция круглых и яйцевидных клеток обнаруживается в альвеолярных RMS, а яйцевидные клетки обычно описываются в эмбриональных RMS. Эти уникальные характеристики плеоморфного RMS могут предложить некоторые объяснения его агрессивного поведения и его плохой реакции на обычную химиотерапию.
При локализованном заболевании широкая хирургическая резекция с пред- или послеоперационной лучевой терапией остается стандартом лечения и может предложить возможность излечения у меньшинства пациентов.Однако, несмотря на адекватную хирургическую резекцию, во время наблюдения часто регистрировались отдаленные рецидивы. Относительно небольшое количество пациентов в нашем исследовании ограничивало возможность выявления прогностических переменных, хотя положительные пределы позволяют прогнозировать рецидив.
Старший возраст и низкая работоспособность этой когорты не позволили большинству пациентов с запущенным заболеванием получить паллиативную химиотерапию. Режимы химиотерапии сильно различались и отражают отсутствие консенсуса в отношении стандартизированного подхода к системному лечению при плеоморфном РРС.В нашей серии был только один подтвержденный частичный ответ на режим химиотерапии детского типа, однако последующее наблюдение было коротким. Это наблюдение сильно контрастирует с известной химиочувствительностью альвеолярных и эмбриональных RMS и поддерживает концепцию плеоморфных RMS как отдельного клинического объекта со своей собственной уникальной биологией опухоли. Хотя возможен некоторый ответ на химиотерапию детского типа, нельзя сделать никаких выводов из-за небольшого числа пролеченных пациентов. Более того, маловероятно, что такой уровень интенсивности химиотерапии может переноситься пожилыми пациентами, которые составляют большинство пациентов с этим заболеванием.
Основываясь на нашем опыте, плеоморфный RMS следует рассматривать как невосприимчивый к стандартной химиотерапии, обычно применяемой при лечении саркомы мягких тканей, и, следовательно, необходимо приложить усилия, чтобы лучше понять основные молекулярные особенности, ответственные за его опухолеобразование. Срочно необходимы новые эффективные методы лечения, и наша серия случаев дает основу для сравнения в будущих исследованиях.
- Получено 14 июля 2015 г.
- Исправление получено 6 сентября 2015 г.
- Принято 22 сентября 2015 г.
- Copyright © 2015 Международный институт противоопухолевых исследований (доктор Джон Г. Делинассиос), Все права защищены
Рабдомиосаркома | Саркома мягких тканей
Рабдомиосаркома — это разновидность саркомы мягких тканей. Он начинается в мышечных клетках и может возникать у детей и взрослых. Саркома мягких тканей — это разновидность рака.
Существует 3 различных типа рабдомиосаркомы. Это:
- эмбриональная рабдомиосаркома
- альвеолярная рабдомиосаркома
- плеоморфная рабдомиосаркома
Знание типа рабдомиосаркомы поможет вашему специалисту выбрать лучшее лечение для вас.
Обзор лечения
Скорее всего, вам предстоит операция, если удастся удалить саркому. Хирург удаляет опухоль вместе с каймой из здоровой ткани вокруг нее. После операции отправляют опухоль в лабораторию. Технический специалист исследует границу здоровой ткани на наличие раковых клеток.
Если там нет раковых клеток, ваш врач скажет вам, что границы тканей чистые. Насколько может сказать ваш хирург, вся ткань, содержащая раковые клетки, удалена.Наличие четких границ тканей означает меньший риск рецидива рака в том же месте.
Когда операция для вас невозможна
Если операция невозможна, вы можете пройти лучевую или химиотерапию саркомы. Иногда операция по удалению рабдомиосаркомы может сильно повлиять на вашу внешность. В подобных случаях, когда операция была бы очень уродливой, ваш врач может предложить вам лучевую терапию вместо операции.
Виды и лечение
Эмбриональная рабдомиосаркома чаще встречается у детей.Обычно это происходит в области головы и шеи, мочевого пузыря или области гениталий.
Ваше лечение зависит от того, в какой части тела находится рабдомиосаркома. Хирургия обычно является частью лечения. Химиотерапия, как правило, хорошо помогает при этом типе саркомы. Перед операцией вам может быть назначена химиотерапия, чтобы уменьшить опухоль и облегчить ее удаление. Это называется неоадъювантным лечением.
Химиотерапия после операции направлена на то, чтобы предотвратить возвращение рака. Это называется адъювантным лечением.
Если вы не можете сделать операцию
Некоторым людям может быть не удастся сделать операцию, потому что саркома находится в месте, где невозможно полностью удалить ее (например, за носом или в глазнице ). В этой ситуации вам может потребоваться комбинация лучевой терапии и химиотерапии.
Это происходит в руках или ногах у детей старшего возраста и молодых людей, но также может возникать в мышцах туловища.
Альвеолярная рабдомиосаркома обычно лечится хирургическим путем по удалению саркомы.Затем вам предстоит лучевая терапия в том месте, где была саркома.
Радиотерапия направлена на снижение вероятности возвращения опухоли в то же место. Химиотерапия обычно назначается до или после операции.
Этот тип рабдомиосаркомы обычно встречается у взрослых на руках или ногах.
Основное лечение — хирургическое. После операции вам обычно проводят лучевую терапию. Это направлено на снижение риска рецидива саркомы.
Исследования показали, что химиотерапия не очень эффективна при плеоморфной рабдомиосаркоме.Химиотерапия не входит в стандартное лечение этого типа саркомы.
Как справиться
Справиться с диагнозом редкого рака может быть особенно сложно, как практически, так и эмоционально. Если вы будете хорошо осведомлены о своей саркоме и ее лечении, вам будет легче принимать решения и справляться с происходящим.
Sarcoma UK предоставляет поддержку и информацию людям, страдающим саркомой мягких тканей и костей.
The Rare Cancer Alliance предлагает поддержку и информацию людям, страдающим редкими формами рака.
Также может помочь разговор с другими людьми, у которых есть то же самое.
Наш дискуссионный форум Cancer Chat — это место для всех, кто страдает раком. Вы можете поделиться своим опытом, историями и информацией с другими людьми, которые знают, что вы переживаете.
Ошибочный диагноз первичной плеоморфной рабдомиосаркомы правого бедра у молодого взрослого: отчет о клиническом случае
Введение
Рабдомиосаркома (РМС) — очень злокачественная форма
опухоль мягких тканей с дифференцировкой скелетных мышц.В
заболеваемость RMS составляет ~ 43 случая на 10 миллионов ежегодно для
лица в возрасте до 20 лет (1).
У пациентов с локализованным заболеванием безрецидивная выживаемость
улучшилась до 70–80% (2). Тем не мение,
прогноз для пациентов с метастазами относительно плохой, с
5-летняя выживаемость всего 30% (3). Методы диагностики RMS включают:
клинико-лабораторное обследование, визуальный анализ, патологический
диагностика и иммуногистологическое обследование (4). RMS был разделен на 3 основных
подтипы: эмбриональный, альвеолярный и плеоморфный RMS (PRMS).Большинство
общие подтипы — эмбриональный и альвеолярный подтипы (5). Первичная PRMS встречается относительно редко и
в первую очередь поражает взрослых, с пиком заболеваемости в пятом десятилетии
жизни (6–9). Чаще всего возникает в глубоком мягком
ткани конечностей. Из-за сходства клинических
проявления и визуализационные особенности между PRMS и другими программными
опухоли тканей, PRMS часто ошибочно диагностируется (10). Настоящее исследование представляет собой случай
PRMS, который был ошибочно диагностирован как шваннома. Этот неправильный диагноз
привело к массовому прогрессированию, и только после
тонкоигольная аспирационная и гистологическая и иммуногистохимическая
анализ подтвердил, что происхождение опухоли — скелетная мышца,
поставлен окончательный диагноз PRMS правого бедра.В
Настоящее исследование было одобрено Комитетом по этике
Первая дочерняя больница Наньчанского университета (Наньчан, Китай),
и письменное информированное согласие было получено от пациента.
История болезни
В августе 2014 г. пациент 28 лет обратился с жалобой на
в ортопедическую клинику Народной больницы округа Тайхэ (Цзянь,
Китай) с основной жалобой на отек правого бедра в течение 1
месяц. По данным местной районной больницы общего профиля
рентгенолог, магнитно-резонансная томография (МРТ) выявила наличие
шванномы, и пациенту посоветовали регулярно проходить
следовать за.Однако через 6 месяцев в декабре 2014 г.
увеличение размера образования и боли в правом бедре были
отметил. Пациент направлен в ортопедическое отделение г.
Первая дочерняя больница Наньчанского университета для дальнейшего
лечение. В личном или семейном анамнезе травм или болезней не было.
записано. Общий медицинский осмотр показал, что
пассивная и активная амплитуда движений правого коленного сустава была
в норме, за исключением парестезии справа внизу
конечность.Никакой лихорадки или затрудненного дыхания не сопровождали
масса тела, и не было истории потери веса или контакта с туберкулезом.
сообщил пациент.
Дополнительный медицинский осмотр выявил
плохо очерченная, нежная и плотная масса мягких тканей справа
внутренняя поверхность бедра, но не пальпируется голова, шея, надключичные, подмышечные
или эпитрохлеарные лимфатические узлы. Маркеры воспаления,
включая скорость оседания эритроцитов и С-реактивный белок,
были в пределах нормы. МРТ была проведена для оценки
массы.Осевые Т2-взвешенные изображения выявили множественные кисты
поражения разного размера с высокой интенсивностью сигнала. С подавлением жира
Т2-взвешенная МРТ также показала высокую интенсивность сигнала (рис. 1А и В). Кроме того, тонкая игла
аспирация проводилась для оценки массы и цитологического
диагноз соответствовал злокачественному новообразованию; плеоморфный
новообразование веретеноводства с выраженной ядерной атипией и выраженным
митотическая активность не наблюдалась.
На основании исключения хирургического
противопоказания, хирурги отделения
Ортопедия, Первая больница Наньчанского университета,
которые специализируются на лечении опухолей костей, провели операцию.В
пациента поместили в положение лежа на спине и после
успех эпидуральной анестезии, стерильные простыни продезинфицированы и
обычно проложили в правой нижней конечности, чтобы обнажить
операционное поле. Сначала был выполнен медиальный разрез бедра ~ 17
см в длину. Далее подкожная клетчатка, поверхностная и глубокая.
фасция и средняя широкая мышца бедра были послойно резецированы,
пока не наблюдались бедренная артерия и вена. Суда выше
были целы. Опухоль была темно-коричневой и располагалась в обширной мышце.
промежуточная мышца.Опухоль была полностью удалена с отрицательным
поля. Интраоперационные образцы тканей были извлечены для
патологическое обследование. Была установлена дренажная трубка для раны, и каждая
слой ткани ушивали строго после полного гемостаза.
Расчетная кровопотеря составила 100 мл, переливание крови не проводилось.
требуется во время процедуры.
Опухоль была большая, гладкая, темно-коричневого цвета.
при грубой экспертизе. Масса нерегулярной ткани составила ~ 11 × 9 × 5.
см3 (рис. 2А). В
резецированную опухолевую ткань фиксировали в 10% формалине, заливали в
парафин и разрезать на срезы 5 мкм с помощью микротома.Разделы
впоследствии были окрашены гематоксилином и эозином и визуализированы
под микроскопом. При микроскопическом исследовании обнаружена опухоль.
состоит из связанных между собой пучков атипичных, веретенообразных,
плеоморфные и гигантские клетки с атипичными ядерными
Особенности. Были обнаружены многочисленные аномальные и многоядерные гигантские клетки.
наблюдались, и у большинства клеток были заметные ядрышки
и обильные эозинофильные цитоплазмы (рис. 2В). Для иммуногистохимии ткани
срезы инкубировали при 25 ° C в течение 60 мин с моноклональной мышью
антитела против десмина (каталожный №, kit-0023) и миогенного
дифференцировка 1 (MyoD1; каталожный номер MAB-0119) (разведение,
1: 1000; Fuzhou Maixin Biotech Co., Ltd., Фучжоу, Китай).
Иммуногистохимический анализ показал, что клетки были положительными.
на актин, MyoD1 и десмин и отрицательный на меланому человека черный
45, кальпонин и мелан-A (рис. 2C и
D). На основании этих данных диагноз PRMS правого
бедро было обеспечено.
Больной выписан без осложнений.
1 неделя после операции. Пациенту было проведено 6 циклов
химиотерапия: доксорубицин, 90 мг / сут в течение 3 дней; 14 дней
выкл, а затем ифосфамид 3.8 г / сут в течение 5 дней, затем 14
выходные до следующего лечебного цикла. Через 3 месяца
вверх, который состоял из простой рентгенографии и МРТ, пациент был
без симптомов и может вернуться к работе. В настоящее время пациент
на данный момент жив-здоров. Однако в случаях, подобных нынешнему,
необходимо тщательное наблюдение за пациентами из-за
высокая частота рецидивов и метастазов, связанных с ошибочно диагностированным
PRMS.
Обсуждение
RMS — очень злокачественная опухоль мягких тканей.
который возникает из поперечно-полосатых мышечных клеток, демонстрирует скелетные мышцы
дифференциация и связана с ранним и широким распространением
метастаз (11,12).RMS делится на 3 основных подтипа:
Эмбриональные и альвеолярные RMS и PRMS, согласно World 2002
Классификация мягких тканей и костей организацией здравоохранения
Новообразования (5). Эмбриональный и
альвеолярные подтипы являются наиболее распространенными и наиболее частыми сайтами
по происхождению RMS включают голову и шею, конечности и мягкие
ткани. Заболевание имеет преобладание мужчин, с переходом от мужчины к женщине.
соотношение 1,3: 1 (13).
PRMS был впервые описан Стаутом в 1946 г. (14). Первичная PRMS встречается относительно редко и
в первую очередь поражает взрослых пятого десятилетия жизни (15).Его появление у молодых людей, например
в данном случае встречается крайне редко. Примечательно, что PRMS часто
поставлен неверный диагноз или полностью пропущен, поскольку его клинические проявления
и особенности изображения аналогичны таковым для других мягких тканей
опухоли, в том числе фиброзная гистиоцитома и шваннома (9). В настоящем исследовании пациент был
ошибочно диагностировали шванному, что привело к прогрессированию
болезнь на полгода; поэтому тонкоигольная аспирация
имеет решающее значение для диагностики новообразований мягких тканей.Гистологический
проявления RMS широко варьируются, а гистопатологические
диагноз основывается на морфологическом, иммуногистохимическом и
ультраструктурные данные, которые показывают фенотип скелетных мышц
(16,17). PRMS гистологически различается
из двух более распространенных подтипов (эмбриональный и альвеолярный)
беспорядочное расположение ячеек, состоящих из крупных,
плеоморфные ядра и эозинофильные цитоплазмы. Клетки PRMS также могут
располагаться в пучки, что напоминает наблюдаемую структуру клеток
при лейомиосаркоме (18).Гистологический
подтипирование имеет решающее значение из-за различных прогнозов и терапевтических
подходы, используемые в PRMS в отличие от других опухолей мягких тканей.
Иммуногистохимия считается ценной для диагностики
PRMS, как серия маркеров с диапазоном специфичности и
чувствительность имеется. Первичные маркеры PRMS — MyoD1,
десмин, саркомерный актин и миозин (19,20).
Хирургическое удаление PRMS считается
предпочтительное лечение, поскольку оно снимает отек и позволяет
окончательный диагноз необходимо подтвердить гистологически.Идеальный хирургический
лечение включает полную резекцию опухоли с отрицательным
микроскопические поля (21). К
напротив, RMS имеет особую чувствительность к внешнему лучу
радиация; таким образом, полная резекция может быть отложена в зависимости от размера
уменьшение с помощью лучевой терапии. В некоторых исследованиях сообщается, что RMS
общий ответ на химиотерапию составляет 85% (22,23), в
в отличие от PRMS, при которой Ferrari et al (11) сообщили о более низком уровне ответа
6,25%. Преобладание PRMS у взрослых, а также его устойчивость к
химиотерапия, привела к тому, что PRMS часто рассматривается как отдельный
сущность из других подтипов RMS.
В заключение, настоящее дело описывает
Пациент 28 лет, страдавший первичной PRMS
правое бедро. Тонкоигольная аспирация и тотальная резекция опухоли были
выполнено, и через 3 месяца наблюдения у пациента не было
свидетельства рецидива заболевания или остаточных побочных эффектов от
терапия. Проведение лабораторных исследований и визуализации
осмотр особенно важен при дифференциальной диагностике
пациентов с опухолями мягких тканей. Хотя
Срок наблюдения за текущим пациентом был относительно коротким, в
учет высокого риска рецидива и метастазирования в
ошибочно диагностированный PRMS, часто рекомендуется долгосрочное наблюдение
такие случаи.
Благодарности
Настоящее исследование было поддержано Gan-Po
Проект талантов 555 провинции Цзянси, План поддержки
Департамент науки и технологий провинции Цзянси (грант №
20112BBG70020) и Фонд естественных наук Цзянси
Провинция (грант № 20132BAB205067).
Список литературы
1 | Кранмер Л.Д., Чен СС, Морган С., Мартино Дж. |
2 | Malempati S и Hawkins DS: |
3 | Бренеман Дж.С., Лайден Э., Паппо А.С., член парламента по ссылке, |
4 | Скалл С., Амар С., Фейз-Эрфан И., Дэйв Х. и |
5 | Ли Дж. Дж., Форстнер Д. и Хендерсон К.: |
6 | Stock N, Chibon F, Binh MB, Terrier P, |
7 | Моретти Дж., Гимарайнш Р., Оливейра К. М., |
8 | Кефели М, Кандемир Б, Акполат I, Йылдырым |
9 | Хакодзаки М, Ходзё Х, Кузе Т, Таджино Т, |
10 | Mondal SK, Mandal PK, Adhikari A и Basak |
11 | Феррари А, Дилео П, Казанова М, Бертулли |
12 | Кишор Б., Кхаре П., Гупта Р. Дж., Гупта С. и |
13 | Данг Н.Д., Тех Б.С. и Паулино А.С.: |
14 | Stout AP: рабдомиосаркома скелета |
15 | Фадаре О, Бонвичино А, Мартель М, Реншоу |
16 | Казерто Б.Г.: Сравнительный обзор собак |
17 | Яо Дж. К., Ван У. К., Ценг Х. Х. и Хван У. С.: |
18 | Атахан С., Аксу О и Экинджи С: Цитологический |
19 | Morgenstern DA, Rees H, Sebire NJ, Шипли |
20 | Cessna MH, Zhou H, Perkins SL, Tripp SR, |
21 | Ге Икс, Ма Дж, Дай Х, Рен Л, Ли Кью и Ши Дж: |
22 | Fuchs J, Dantonello TM, Blumenstock G, |