РЕНИН-АНГИОТЕНЗИНОВАЯ СИСТЕМА В РЕГУЛЯЦИИ ГЕМОПОЭЗА | Канаева
1. Jokubaitis V.J., Sinka L., Driessen R. et al. Angiotensin converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal and adult hematopoietic tissues. Blood 2008;111(8):4055–63. DOI: 10.1182/blood-2007-05-091710. PMID: 17993616.
2. Tigerstedt R., Bergman P.G. Niere und Kreislauf. Scandinav Arch J Physiol 1898;8:223.
3. Bernstein K.E., Martin B.M., Bernstein E.A. et al. The isolation of angiotensin-converting enzyme cDNA. J Biol Chem 1988;263(23):11021–4. PMID: 2841312.
4. Ferrario C.M. Does angiotensin-(1–7) contribute to cardiac adaptation and preservation of endothelial function in heart failure? Circulation 2002;105(13):1523–5. DOI: 10.1161/01.CIR.0000013787.10609.DC. PMID: 11927512.
5. Коваленко В.Н., Талаева Т.В., Братусь В.В. Ренин-ангиотензиновая система в кардиальной патологии. Український кардіологічний журнал 2012;3:105–29. [Kovalenko V.N., Talaeva T.V., Bratus V.V. Reninangiotensin system in cardiac pathology. Ukrainskiy kardiologicheskiy zhurnal = Ukrainian Cardiology Journal 2012;3:105–29. (In Russ.)].
6. Haznedaroğlu I.C., Tuncer S., Gürsoy M. A local renin-angiotensin system in the bone marrow. Med Hypotheses 1996;46(6):507–10. PMID: 803932.
7. Hubert C., Savary K., Gasc J.M. et al. The hematopoietic system: a new niche for the renin-angiotensin system. Nat Clin Pract Cardiovasc Med 2006;3(2):80–5. DOI: 10. 1038/ncpcardio0449. PMID: 16446776.
1038/ncpcardio0449. PMID: 16446776.
8. Zambidis E.T., Park T.S., Yu W. et al. Expression of angiotensin-converting enzyme (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells. Blood 2008;112(9):3601–14. DOI: 10.1182/blood-2008-03-144766. PMID: 18728246.
9. Soubrier F., Alhenc-Gelas F., Hubert C. et al. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci USA 1988;85(24):9386–90. PMID: 2849100.
10. Corvol P., Williams T. A., Soubrier F. Peptidyl dipeptidase A: angiotensin I- converting enzyme. Methods Enzymol 1995;248:283–305. PMID: 7674927.
11. Кугаевская Е.В. Ангиотензин-превращающий фермент. Доменная структура и свойства. Биомедицинская химия 2005;51(6):567–80. [Kugaevskaya E.V. Angiotensin converting enzyme domain structure and properties. Biomeditsinskaya khimiya = Biomed Khim 2005;51(6): 567–80. (In Russ.)]. PMID: 16521820.
12. Paul M., Mehr A.P., Kreutz R. Physiology of Local Renin-Angiotensin Systems. Physiol 2006;86(3):787–803. DOI: 10.1152/physrev.00036.2005. PMID: 16816138.
13. Kamper A.-L., Nielsen O.J. Effect of enalapril on haemoglobin and serum erythropoietin in patients with chronic nephropathy. Scand J Clin Lab Invest 1990;50(6):611–8. DOI: 10.3109/00365519009089178. PMID: 2247767.
14. Weber H., Taylor D.S., Molloy C.J. Angiotensin II induces delayed mitogenesis and cellular proliferation in rat aortic smooth muscle cells. Correlation with the expression of specific endogenous growth factors and reversal by suramin. J Clin Invest 1994;93(2):788–98. DOI: 10.1172/JCI117033. PMID: 7509348.
Correlation with the expression of specific endogenous growth factors and reversal by suramin. J Clin Invest 1994;93(2):788–98. DOI: 10.1172/JCI117033. PMID: 7509348.
15. Mrug M., Stopka T., Julian V.A. et al. Angiotensin II stimulates proliferation of normal early erythroid progenitors. J Clin Invest 1997;100(9):2310–4. DOI: 10.1172/JCI119769. PMID: 9410909.
16. Rodgers K.E., Xiong S., Steer R., diZerega G.S. Effect of Angiotensin II on Hematopoietic Progenitor Cell Proliferation. Stem Cells 2000;18(4):287–94. DOI: 10.1634/stemcells.18-4-287. PMID: 10924095.
17. Pennefather J.N., Lecci A., Candenas M.L. et al. Tachykinins and tachykinin receptors: a growing family. Life Sci 2004;74(12):1445–63. PMID: 14729395.
18. Rameshwar P., Gascón P. Substance P (SP) mediates production of stem cell factor and interleukin-1 in bone marrow stroma: potential autoregulatory role for these cytokines in SP receptor expression and induction. Blood 1995;86(2):482–90. PMID: 7541664.
19. Harmer D., Gilbert M., Borman R., Clark K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 2002;532(1–2):107–10. PMID: 12459472.
20. Ferrario C.M., Trask A.J., Jessup J.A. Advances in biochemical and functional roles of angiotensin – converting enzyme 2 and angiotensin-(l – 7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol 2005;289(6):2281–90. DOI: 10.1152/ajpheart.00618.2005. PMID: 16055515.
21. Santos R.A., Haibara A.S., CampagnoleSantos M.J. et al. Characterization of a new selective antagonist for angiotensin-(1–7), D-pro7-angiotensin(1–7). Hypertension 2003;41(3 Pt 2):737–43. DOI: 10.1161/01.HYP.0000052947.60363.24. PMID: 12623989.
Santos R.A., Haibara A.S., CampagnoleSantos M.J. et al. Characterization of a new selective antagonist for angiotensin-(1–7), D-pro7-angiotensin(1–7). Hypertension 2003;41(3 Pt 2):737–43. DOI: 10.1161/01.HYP.0000052947.60363.24. PMID: 12623989.
22. Allen L.F., Lefkowitz R.J. Caron M.G., Cotecchia S. G-protein-coupled receptor genes as protooncogenes: constitutively activating mutation of the a1B-adrenergic receptor enhances mitogenesis and tumorigenicity. Proc Natl Acad Sci 1991;88(24):11354–8. PMID: 1662393.
23. Martin K.A., Hockfield S. Expression of the mas proto-oncogene in the rat hippocampal formation is regulated by neuronal activity. Brain Res Mol Brain Res 1993;19(4):303–9. DOI: 10.1016/0169-328X(93)90129-D. PMID: 8231733.
24. Hill C.S., Treisman R. Transcriptional regulation by extracellular signals: Mechanisms and specificity. Cell 1995;80(2):199–211. PMID: 7834740.
25. Ellefson D.D., diZerega G. S., Espinoza T. et al. Synergistic effects of co-administration of angiotensin 1–7 and Neupogen on hematopoietic recovery in mice. Cancer Chemother Pharmacol 2004;5391:15–24. DOI: 10.1007/s00280-003-0710-0. PMID: 14569417.
26. Heringer-Walther S., Eckert K., Schumacher S.M. et al. Angiotensin-(1–7) stimulates hematopoietic progenitor cells in vitro and in vivo. Haematologica 2009;94(6):857–60. DOI: 10.3324/haematol.2008.000034. PMID: 19377080.
27. Rieger K.J., Saez-Servent N., Papet M.P. et al. Involvementof human plasma angiotensin I-converting enzyme in the degradation of the haemoregulatory peptide N-acetyl-seryl-aspartyl-lysylproline. Biochem J 1993;296(Pt. 2):373–8. PMID: 8257427.
Biochem J 1993;296(Pt. 2):373–8. PMID: 8257427.
28. Azizi M., Rousseau A., Ezan E. et al. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetylseryl-aspartyl-lysyl-proline. J Clin Invest 1996;97(3):839–44. DOI: 10.1172/JCI118484. PMID: 8609242.
29. Lenfant M., Wdzieczak-Bakala J., Guittet E. et al. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci 1989;86(3):779–82. PMID: 2915977.
30. Coutton C., Guigon M., Bohbot A. et al. Photoprotection of normal human hematopoietic progenitors by the tetrapeptide N-AcSDKP. Exp Hematol 1994; 22(11):1076–80. PMID: 7925774.
31. Watanabe T., Brown G.S., Kelsey L.S. et al. In vivo protective effects of tetrapeptide AcSDKP, with or without granulocyte colony-stimulation factor, on murine progenitor cells after sublethal irradiation. Exp Hematol 1996;24(6):713–21. PMID: 8635527.
32. Deeg H.J., Seidel K., Hong D.S. et al. In vivo radioprotective effect of AcSDKP on canine myelopoiesis. Ann Hematol 1997;74(3):117–22. PMID: 9111424.
33. Fuchs S., Xiao H.D., Cole J.M. et al. Role of the N-terminal catalytic domain of angiotensin-converting enzyme investigated by targeted inactivation in mice. J Biol Chem 2004;279(16): 15946–53. DOI: 10.1074/jbc.M400149200. PMID: 14757757.
34. Bernstein K.E., Shen X.Z., GonzalezVillalobos R.A. et al. Different in vivo functions of the two catalytic domains of angiotensin-converting enzyme (ACE). Curr Opin Pharmacol 2011;11(2):105–11. DOI: 10.1016/j.coph.2010.11.001. PMID: 21130035.
Curr Opin Pharmacol 2011;11(2):105–11. DOI: 10.1016/j.coph.2010.11.001. PMID: 21130035.
35. Bonnet D., Lemoine F.M., PontvertDelucq S. et al. Direct and reversible inhibitory effect of the tetrapeptide acetylN-Ser-Asp-Lys-Pro(Seraspenide) on the growth of human CD34+ subpopulations in response to growth factors. Blood 1993;82(11):3307–14. PMID: 7694679.
36. Ni L., Feng Y., Wan H. et al. Angiotensin-(1–7) inhibits the migration and invasion of A549 human lung adenocarcinoma cells through inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol Rep 2012;27(3):783–90. DOI: 10.3892/or.2011.1554. PMID: 22089256.
37. George A.J., Thomas W.G., Hannan R.D. The renin angiotensin system and cancer: оld dog, new tricks. Nat Rev Cancer 2010;10(11):745–59. DOI: 10.1038/nrc2945. PMID: 20966920.
38. Yasumatsu R., Nakashima T., Masuda M. et al. Effects of the angiotensin-I converting enzyme inhibitor perindopril on tumor growth and angiogenesis in head and neck squamous cell carcinoma cells. J Cancer Res Clin Oncol 2004;130(10):567–73. DOI: 10.1007/s00432-004-0582-7. PMID: 15449186.
39. Kosaka T., Miyajima A., Takayama E. et al. Angiotensin II type 1 receptor antagonist as an angiogenic inhibitor in prostate cancer. Prostate 2007;67(1): 41–9. DOI: 10.1002/pros.20486. PMID: 17044086.
40. Tamarat R., Silvestre J.S., Kubis N. et al. Endothelial nitric oxide synthase lies downstream from angiotensin II-induced angiogenesis in ischemic hindlimb. Hypertension 2002;39(3):830–5. PMID: 11897773.
41. Dolley-Hitze T., Jouan F., Martin B. et al. Angiotensin-2 receptors (AT1-R and AT2-R), new prognostic factors for renal clear-cell carcinoma? Br J Cancer 2010;103(11):1698–705. DOI: 10.1038/sj.bjc.6605866. PMID: 21102591.
Dolley-Hitze T., Jouan F., Martin B. et al. Angiotensin-2 receptors (AT1-R and AT2-R), new prognostic factors for renal clear-cell carcinoma? Br J Cancer 2010;103(11):1698–705. DOI: 10.1038/sj.bjc.6605866. PMID: 21102591.
42. Ager E.I., Neo J., Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis 2008;29(9): 1675–84. DOI: 10.1093/carcin/bgn171. PMID: 18632755.
43. Паровичникова Е.Н., Ходунова Е.Е., Савченко В.Г. и др. Маркеры апоптоза в CD34-позитивных клетках при острых лейкозах. Клиническая онкогематология. Фундаментальные исследования и клиническая практика 2013;6(4):373–8. [Parovichnikova E.N., Khodunova E.E., Savchenko V.G. et al. Apoptotic markers in CD34-positive cells in acute leukemias. Klinicheskaya onkogematologiya. Fundamentalnye issledovaniya i klinicheskaya praktika = Clinical Oncohematology. Basic Research and Clinical Practice 2013;6(4):373–8. (In Russ.)].
44. Albayrak M., Celebi H., Albayrak A. et al. Elevated serum angiotensin converting enzyme levels as a reflection of bone marrow renin-angiotensin system activation in multiple myeloma. J Renin Angiotensin Aldosterone Syst 2012;13(2):259–64. DOI: 10.1177/1470320312437070. PMID: 22345095.
45. Savary K., Michaud A., Favier J. et al. Role of the renin-angiotensin system in primitive erythropoiesis in the chick embryo. Blood 2005;105(1):103–10. DOI: 10.1182/blood-2004-04-1570. PMID: 15367438.
Ренин-ангиотензиновая система при новой коронавирусной инфекции COVID-2019 | Загидуллин
1. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H et al. Genomic characterization and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi:10.1016/S0140-6736(20)30251-8
Genomic characterization and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi:10.1016/S0140-6736(20)30251-8
2. Liu Y, Gayle AA, Wilder-Smith A, Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med. 2020;27(2): taaa021. doi:10.1093/jtm/taaa021
3. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020: e200994. doi:10.1001/jamainternmed.2020.0994
4. Maksimov ML, Dralova OV, Starodubtsev AK. Angiotensin II type 1 receptor antagonists and ACE inhibitors in the regulation of hemodynamics and renin-angiotensin-aldosterone system activity: focus on the organ protection. Cardiovasc Ther Prev. 2010;9(2):115–124.
5. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE 2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87(5):1–9. doi:10.1161/01.res.87.5.e1
6. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captoprilin sensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–33243. doi:10.1074/jbc.M002615200
7. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE 2, the putative receptor of Wuhan 2019-nCov. BioRxiv 919985 [Preprint]. 2020.
BioRxiv 919985 [Preprint]. 2020.
8. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J et al. Hydrolysis of biological peptides by human angiotensinconverting enzyme-related carboxypeptidase. J Biol Chem. 2002;277(17):14838–14843. doi:10.1074/jbc.M200581200
9. Raizada MK, Ferreira AJ. ACE 2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol. 2007;50(2):112–119. doi:10.1097/FJC.0b013e3180986219
10. Alenina N, Xu P, Rentzsch B, Patkin EL, Bader M. Genetically altered animal models for Mas and angiotensin-(1–7). Exp Physiol. 2008;93(5):528–537. doi:10.1113/expphysiol.2007.040345
11. Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116(6):960–975. doi:10.1161/CIRCRESAHA.116.303587
12. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE 2/Angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res. 2016;118(8):1313–1326. doi:10.1161/CIRCRESAHA.116.307708
13. Santos R, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M et al. The ACE 2/Angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2018;98(1):505–553. doi:10.1152/physrev.00023.2016
14. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi:10.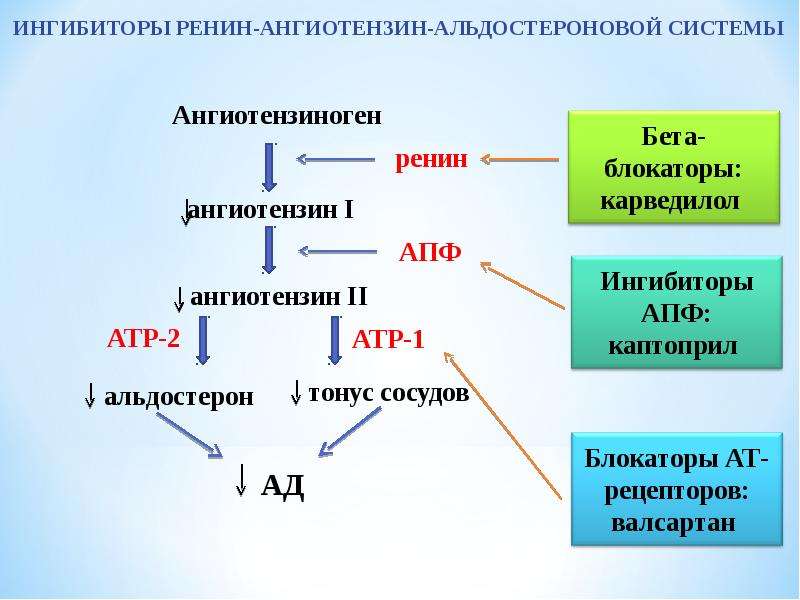 1038/nature02145
1038/nature02145
15. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi:10.1038/nature00786
16. Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J et al. Characterization of renal angiotensinconverting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41(3):392–397. doi:10.1161/01.HYP.0000060689.38912.CB
17. Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow M et al. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensinconverting enzyme Homologue ACE 2. Circulation. 2003;108(14):1707–1712. doi:10.1161/01.CIR.0000094734.67990.99
18. Brake SJ, Barnsley K, Lu W, McAlinden KD, Eapen MS, Sohal SS. Smoking upregulates angiotensin-converting enzyme-2 receptor: a potential adhesion site for novel coronavirus SARS-CoV-2 (Covid-19). J Clin Med. 2020;9(3):841. doi:10.3390/jcm9030841
19. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):26052610. doi:10.1161/CIRCULATIONAHA.104.510461
20. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
21. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE 2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt1):45–51. doi:10.1042/BJ20040634
Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE 2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt1):45–51. doi:10.1042/BJ20040634
22. Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, Ma M et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1–7/mas receptor axis. Int J Biol Sci. 2011;7(8):1077–1092. doi:10.7150/ijbs.7.1077
23. Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT. Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1–7/ Mas receptor cascade. Biochem Pharmacol. 2017;144:90–99. doi:10.1016/j.bcp.2017.07.022
24. Burchill LJ, Velkoska E, Dean RG, Griggs K, Patel SK, Burrell LM. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci (Lond). 2012;123(11):649–658. doi:10.1042/CS20120162
25. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374. doi:10.1007/s11427-020-1643-8
26. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al. A crucial role of angiotensin converting enzyme 2 (ACE 2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):8758787589. doi:10.1038/nm1267
27. Henry С, Zaizafoun М, Stock E, Ghamande S, Arroliga AC, White HD. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Proc (Bayl Univ Med Cent). 2018;31(4):419–423. doi:10.1080/08998280.2018.1499293
Henry С, Zaizafoun М, Stock E, Ghamande S, Arroliga AC, White HD. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Proc (Bayl Univ Med Cent). 2018;31(4):419–423. doi:10.1080/08998280.2018.1499293
28. Li J, Wang X, Chen J, Zhang H, Deng A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) Infection in Wuhan, China. J Am Med Assoc Cardiol. 2020; doi:10.1001/jamacardio.2020.1624
29. Guan W, Ni Z, Hu Y, Liang WH, Ou CQ, He JX et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720. doi:10.1056/NEJMoa2002032
30. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Zh et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi:10.1016/S0140-6736(20)30566-3
31. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;1–3. doi:10.1007/s00134-020-05991-x
32. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J Am Med Assoc. 2020;323(11):1061–1069. doi:10.1001/jama.2020.1585
33. Arentz M, Yim E, Klaff L, Lokhandwala Sh, Riedo FX, Chong M et al. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. J Am Med Assoc. 2020;323(16):1612–1614. doi:10.1001/jama.2020.4326
J Am Med Assoc. 2020;323(16):1612–1614. doi:10.1001/jama.2020.4326
34. ACE Inhibitor Myocardial Infarction Collaborative Group. Indications for ACE inhibitors in the early treatment of acute myocardial infarction: systematic overview of individual data from 100,000 patients in randomized trials. Circulation. 1998;97(22):2202–2212. doi:10.1161/01.cir.97.22.2202
35. Шляхто Е. В., Конради А. О., Арутюнов Г. П., Арутюнов А. Г., Баутин А. Е., Бойцов С. А. и др. Руководство по диагностике и лечению болезней системы кровообращения в контексте пандемии COVID-19. Российский кардиологический журнал. 2020;25(3):3801. doi:10.15829/1560-4071-2020-3-3801.
36. Monteil V, Kwon H, Patricia P, Hagelkrüys A, Wimmer RA, Stahl M et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE 2. Cell. 2020; S 0092–8674(20):30399–8. doi:10.1016/j.cell.2020.04.004
37. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD et al. A pilot clinical trial of recombinant human angiotensinconverting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21(1):234. doi:10.1186/s13054-017-1823-x
38. Zhang H, Baker A. Recombinant human ACE 2: acing out angiotensin II in ARDS therapy. Crit Care. 2017;21(1):305. doi:10.1186/s13054-017-1882-z
Ингибиторы ренин-ангиотензиновой системы в сравнении с другими типами лекарств при гипертензии
Вопрос обзора
Мы определили, насколько ингибиторы РАС (ренин-ангиотензиновой системы) сравнимы в качестве лекарств первой линии для лечения гипертензии с другими типами лекарств первой линии (тиазидные диуретики, бета-блокаторы, БКК, альфа-адреноблокаторы или лекарства, влияющие на центральную нервную систему (ЦНС)).
Актуальность
Гипертензия является долгосрочным медицинским состоянием, связанным с сердечно-сосудистой смертностью и такими заболеваниями, как коронарные заболевания сердца, цереброваскулярная болезнь и болезнь периферических сосудов, которые снижают качество жизни. В последние годы ингибиторы РАС стали центральными вмешательствами при гипертензии, и их широко назначают при лечении гипертензии. Однако, остается неясным, превосходят ли ингибиторы РАС другие антигипертензивные средства с точки зрения клинически значимых исходов.
Дата поиска
Мы провели поиск доказательств вплоть до ноября 2017 года.
Характеристика исследований
В настоящий обзор мы включили рандомизированные, двойные слепые контролируемые испытания с параллельным дизайном. Были включены 45 испытаний с 66 625 участниками, с периодом наблюдения от полугода до 5,6 лет. Средний возраст участников был 66 лет.
Основные результаты
Мы обнаружили, что при применении ингибиторов РАС первой линии частота сердечной недостаточности и инсультов была выше, по сравнению с тиазидами первой линии. По сравнению с блокаторами кальциевых каналов первой линии, ингибиторы РАС первой линии показали преимущества в предотвращении развития сердечной недостаточности, но были хуже в предотвращении инсультов; снижение абсолютного риска сердечной недостаточности было более выраженным по сравнению с повышением риска развития инсульта. В сравнении с бета-блокаторами первой линии, ингибиторы РАС снижали общее число сердечно-сосудистых событий и инсультов. Были обнаружены небольшие различия в эффективности в снижении артериального давления, но они, по-видимому, не были связаны с числом сердечных приступов, инсультов или заболеваний почек.
Определенность доказательств
В целом, определенность доказательств была оценена как низкая или умеренная согласно оценке по шкале GRADE. Доказательства умеренной определенности продемонстрировали преимущества тиазидов первой линии в сравнении с ингибиторами РАС первой линии в предотвращении развития сердечной недостаточности и инсульта. Определенность доказательств была оценена как умеренная для сравнения ингибиторов РАС и блокаторов кальциевых каналов. Определенность доказательств была низкой для сравнения ингибиторов РАС и бета-блокаторов в отношении общего числа сердечно-сосудистых событий и инсультов, поскольку результаты были основаны главным образом на одном большом испытании с риском смещения (систематической ошибки) от умеренного до высокого.
Определенность доказательств была оценена как умеренная для сравнения ингибиторов РАС и блокаторов кальциевых каналов. Определенность доказательств была низкой для сравнения ингибиторов РАС и бета-блокаторов в отношении общего числа сердечно-сосудистых событий и инсультов, поскольку результаты были основаны главным образом на одном большом испытании с риском смещения (систематической ошибки) от умеренного до высокого.
Ренин-ангиотензиновая и калликреин-кининовая системы простаты: роль в патогенезе гиперплазии простаты | Чибичян
1. Chapple CR, Wein AJ, Abrams P, Dmochowski RR, Giuliano F, Kaplan SA, McVary KT, Roehrborn CG. Lower urinary tract symptoms revisited: a broader clinical perspective. Eur Urol. 2008;54(3):563-56. DOI: 10.1016/j.eururo.2008.03.109
2. Gharaee-Kermani M, Kasina S, Moore BB, Thomas D, Mehra R, Macoska JA. CXC-type chemokines promote myofibroblast phenoconversion and prostatic fibrosis. PLoS One. 2012;7(11):e49278.23. DOI: 10.1371/journal.pone.0049278
3. Vignozzi L, Gacci M, Cellai I, Santi R, Corona G, Morelli A, Rastrelli G, Comeglio P, Sebastanelli A, Maneschi E, Nesi G, De Nunzio C, Tubaro A, Mannucci E, Carini M, Maggi M. Fat boosts, while androgen receptor activation counteracts, BPH-associated prostate inflammation. Prostate. 2013;73(8):789-800. DOI: 10.1002/pros.22623
4. Middleton LW, Shen Z, Varma S, Pollack AS, Gong X, Zhu S, Zhu C, Foley JW, Vennam S, Sweeney RT, Tu K, Biscocho J, Eminaga O, Nolley R, Tibshirani R, Brooks JD, West RB, Pollack JR. Genomic analysis of benign prostatic hyperplasia implicates cellular re-landscaping in disease pathogenesis. JCI Insight. 2019;4(12):e129749. DOI: 10.1172/jci.in-sight.129749
2019;4(12):e129749. DOI: 10.1172/jci.in-sight.129749
5. Коган М.И., Черногубова Е.А., Чибичян М.Б., Матишов Д.Г. Активность протеолитических ферментов и их ингибиторов в секрете простаты при доброкачественной гиперплазии и раке предстательной железы. Онкоурология. 2011;7(2):46-51. DOI: 10.17650/1726-9776-2011-7-2-46-51
6. Коган М.И., Черногубова Е.А., Чибичян М.Б., Мационис А.Э., Повилайтите П.Э., Матишов Д.Г. Роль калликреин-кининовой и ренин-ангиотензиновой систем в патогенезе рака предстательной железы. Урология. 2015;3:50-54.
7. Чибичян М.Б., Коган М.И., Черногубова Е.А., Павленко И.А., Матишов Д.Г. Роль рецепторов ангиотензина II второго типа в прогнозировании биохимического рецидива при терапии рака предстательной железы. Урология. 2016; 3:89-94.
8. Коган М.И., Черногубова Е.А., Чибичян М.Б., Матишов Д.Г. Ангиотензинпревращающий фермент — новый прогностический маркер рецидива при терапии рака предстательной железы. Онкоурология. 2016;12(2):46-51. DOI: 10.17650/1726-9776-2016-12-4-87-93
9. Горбунова Е.Н., Давыдова Д.А., Крупин В.Н. Хроническое воспаление и фиброз как факторы риска простатических интраэпителиальных неоплазий и рака предстательной железы. Соврем технол мед. 2011;(1):79-83.
10. ГОСТ Р 52379-2005 Национальный стандарт РФ «Надлежащая клиническая практика» (Good Clinical Practice; GCP), (утверждён приказом Федерального агентства по техническому регулированию и метрологии от 27 сентября 2005 г. N 232-ст). Доступно по: http://docs.cntd.ru/document/1200041147 Ссылка активна на 25.06.2018.
11. Яровая Г.А., Нешкова А.Е. Калликреин-кининовая система. Прошлое и настоящее. (к 90-летию открытия системы). Биоорганическая химия. 2015; 41(3): 275-291. DOI: 10.7868/S0132342315030112
12. Пасхина Т.С., Кринская А.В. Упрощенный метод определения калликреиногена и калликреина в сыворотке ( плазме ) крови человека в норме и при некоторых па -тологических состояниях. Вопросы медицинской химии. 1974; 20(6): 660-663
13. Пасхина, Т.С., Яровая Г.А. Калликреин сыворотки крови человека. Активность фермента и хроматографический метод определения. Биохимия. 1970;35(5):1055-1058.
14. Нартикова В.Ф., Пасхина Т.С. Унифицированный метод определения активности а1-антитрипсина и а2-макроглобулина в сыворотке (плазме) крови человека. Вопросы медицинской химии. 1979; 25(4):494-502.
15. Доценко В.Л., Нешкова Е.А., Яровая Г.А. Выявление лейкоцитарной эластазы человека из комплекса с плазменным а1-протеиназным ингибитором по ее энзиматической активности с синтетическим субстратом. Вопросы медицинской химии. 1994;40(3):20-25.
16. Парфенкова Г.А., Оглоблина О.Г., Домба Г.Ю. Клиническое значение определения активности эластазо- и химитрипсиноподобных протеиназ в плазме крови больных неспецифическим аортоартериитом и атеросклерозом. Кардиология. 1989;9:94-96.
17. Реброва О.Ю. Статистический анализ медицинских данных. Применение пакета прикладных программ STATISTICA. М.: МедиаСфера; 2002.
18. Чибичян М.Б., Мационис А.Э., Повилайтите П.Э., Коган М.И. Роль рецепторного аппарата калликреин-кинино-вой системы в пролиферативных процессах предстательной железы. Онкоурология. 2013;(1):43-50.
19. Leeb-Lundberg LM, Marceau F, MuNer-Esterl W, Peffibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27-77. DOI: 10.1124/pr.57.1.2
20. Srinivasan D, Kosaka AH, Daniels DV, Ford AP, Bhattacharya A. Pharmacological and functional characterization of bradykinin B2 receptor in human prostate. Eur J Pharmacol. 2004;504(3):155-67. PMID: 15541417
21. Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, Shen XZ, Fuchs S, Touyz RM. A modern understanding of the traditional and nontraditional biological functions of Angiotensin-converting enzyme. Pharmacol Rev. 2013:65: 1-46. DOI: 10.1124/pr.112.006809
22. Kryukova OV, Tikhomirova VE, Golukhova EZ, Evdokimov VV, Kalantarov GF, Trakht IN, Schwartz DE, Dull RO, Gusakov AV, Uporov IV, Kost OA, Danilov SM. Tissue Specificity of Human Angiotensin I-Converting Enzyme. PLoS One. 2015;10(11):e0143455. DOI: 10.1371/journal.pone.0143455
23. Dinh DT, Frauman AG, Somers GR, Ohishi M, Zhou J, Casley DJ, Johnston CI, Fabiani ME. Evidence for activation of the renin-angiotensin system in the human prostate: increased angiotensin II and reduced AT(1) receptor expression in benign prostatic hyperplasia. J Pathol. 2002;196(2):213-9. DOI: 10.1002/path.1021
24. Fabiani ME, Sourial M, Thomas WG, Johnston CI, Johnston CI, Frauman AG. Angiotensin II enhances noradrenaline release from sympathetic nerves of the rat prostate via a novel angiotensin receptor: implications for the pathophysiology of benign prostatic hyperplasia. J Endocrinol. 2001;171(1):97-108. PMID: 11572794
25. Kwon CH, Park HJ, Lee JR, Kim HK, Jeon TY, Jo HJ, Kim DH, Kim GH, Park DY. Serpin peptidase inhibitor clade a member 1 is a biomarker of poor prognosis in gastric cancer. Br J Cancer. 2014:111:1993-2002. DOI: 10.1038/bjc.2014.490
26. Zhao W, Yang Z, Liu X, Tian Q, Lv Y, Liang Y, Li C, Gao X, Chen L. Identification of a1-antitrypsin as a potential prognostic biomarker for advanced nonsmall cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors by proteomic analysis. J Int Med Res. 2013;41:573-583. DOI: 10.1177/0300060513476582
27. Kuvibidila S, Rayford W. Correlati on between serum prostate-specific antigen and alpha-1-antitrypsin in men without and with prostate cancer. J Lab Clin Med. 2006;147(4):174-81. PMCID 16581345
28. Zhang WM, Finne P, Leinonen J, Stenman UH. Characterization and determination of the complex between prostate-specific antigen and a1-protease inhibitor in benign and malignant prostatic diseases. Scand J Clin Lab Invest Suppl. 2000;60(233):51-58. DOI: 10.1080/clb.60.233.51.58
29. Kostova MB, Brennen WN, Lopez D, Anthony L, Wang H, Platz E, Denmeade SR. PSA-alpha-2-macroglobulin complex is enzymatically active in the serum of pati ents with advanced prostate cancer and can degrade circulating peptide hormones. Prostate. 2018;78(11):819-829. DOI: 10.1002/pros.23539
30. Зорин Н.А., Зорина В.Н., Зорина Р.М. Универсальный модулятор цитокинов а2-макроглобулин. Иммунология. 2004; 25(5): 302-304. 2
31. Geffins PG. Serpin structure, mechanism, and function. Chem. Rev. 2002;102(12):4751-4804. DOI: 10.1021/cr010170
Современный взгляд на роль ренин–ангиотензиновой системы в патогенезе диабетической ретинопатии | Рябина М.В., Охоцимская Т.Д.
Резюме
Ренин–ангиотензиновая система (РАС) является филогенетически старой гормональной системой, отвечающей за регуляцию артериального давления и водно–электролитного баланса. РАС также принимает участие в патогенезе многих сердечно–сосудистых заболеваний, в том числе артериальной гипертонии, атеросклероза, гипертрофии миокарда, диабетической и аутоиммунной нефропатии. Имеются убедительные экспериментальные данные, доказывающие, что локальная РАС представлена также в тканях глаза, в частности в сетчатке и ретинальных сосудах. Данные об участии РАС в патогенезе диабетической ретинопатии позволяют сделать вывод, что фармакологическая блокада РАС полезна при лечении данного заболевания. Результаты доклинических исследований продемонстрировали, что ингибиторы ангиотензин–превращающего фермента и блокаторы АТ1–рецепторов способны оказывать ретинопротективное действие. Этот вывод был подтвержден клиническими исследованиями EUORODIAB, DIRECT.
Резюме
Ренин–ангиотензиновая система (РАС) является филогенетически старой гормональной системой, отвечающей за регуляцию артериального давления и водно–электролитного баланса. РАС также принимает участие в патогенезе многих сердечно–сосудистых заболеваний, в том числе артериальной гипертонии, атеросклероза, гипертрофии миокарда, диабетической и аутоиммунной нефропатии. Имеются убедительные экспериментальные данные, доказывающие, что локальная РАС представлена также в тканях глаза, в частности в сетчатке и ретинальных сосудах. Данные об участии РАС в патогенезе диабетической ретинопатии позволяют сделать вывод, что фармакологическая блокада РАС полезна при лечении данного заболевания. Результаты доклинических исследований продемонстрировали, что ингибиторы ангиотензин–превращающего фермента и блокаторы АТ1–рецепторов способны оказывать ретинопротективное действие. Этот вывод был подтвержден клиническими исследованиями EUORODIAB, DIRECT.
Ключевые слова: ренин–ангиотензиновая система, сетчатка, диабетическая ретинопатия, ингибитор ангиотензин–превращающего фермента, блокатор АТ1–рецепторов.
Abstract
Modern glance on the role of renin–angiotensin system in pathogenesis of diabetic retinopathy. Literary review
M.V. Ryabina, T.D. Okhotsimskaya
FGU “MNII GB named after Gelmgoltsa Rosmedbiotechnology”, Moscow
Renin–angiotensin system (RAS) is a phylogenetically old hormonal system responsible for the regulation of blood pressure and electrolyte homeostasis. Beyond these general systemic effects, the RAS is also involved in the pathogenesis of many cardiovascular diseases including hypertension, atherosclerosis, myocardial hypertrophy, diabetic and autoimmune nephropathy. There is strong experimental evidence demonstrating that the local RAS is also represented in the eye tissues, particularly in the retina and retinal vessels. The evidence of the involvement of the RAS in the pathogenesis of diabetic retinopathy suggests that pharmacological blockade of the RAS is useful in the treatment of this disease. The results of preclinical studies have demonstrated that angiotensin converting enzyme inhibitors and AT1–receptor blockers are able to provide retinoprotection. This conclusion was confirmed by clinical studies EUORODIAB and DIRECT.
Key words: Renin–angiotensin system, retina, diabetic retinopathy, angiotensin converting enzyme inhibitor, AT1–receptor blocker.
История изучения РАС
В 1898 г. R. Tigerstend и P. Bergman опубликовали первые данные о гормональной системе, которую мы теперь называем ренин–ангиотензиновой. Авторами было описано вещество, выделенное из ткани почки кролика и влияющее на артериальное давление (АД): инъекция гомогената почечной ткани здорового кролика другому здоровому кролику приводила к повышению АД у реципиента. Вещество, влияющее на АД, было названо «ренином» по аналогии с местом локализации в почке [110].
Последующее изучение ренина было затруднено из–за отсутствия надежной воспроизводимой модели гипертонии на животных. Такая модель (частичная окклюзия почечной артерии серебряной клипсой на собаках) была разработана H. Goldblatt в 1934 г. [56]. Через 5 лет две независимые группы ученых одновременно обнаружили, что ренин не является самостоятельным вазоконстриктором. Будучи ферментом, он действует на специфичный субстрат, и, таким образом, генерирует вазоактивный пептид, получивший название «гипертензин» и «ангиогенин» [18,89]. В 1957 г. ученые пришли к мнению о единой сущности этого активного соединения и назвали его «ангиотензин», взяв таким образом по части от двух предыдущих названий [19].
Ангиотензин–превращающий фермент (АПФ) и его способность образовывать ангиотензин–II из ангиотензина–I была описана L.T. Skeggs в 1954 г. [103]. Открытие АПФ послужило толчком к развитию препаратов, влияющих на ренин–ангиотензиновую систему (РАС). Первым таким препаратом был ингибитор АПФ каптоприл [44].
Циркуляторная (системная) РАС
РАС является эндокринной системой, основное действие которой заключается в регуляции АД и водно–электролитного баланса [4–6,86,107].
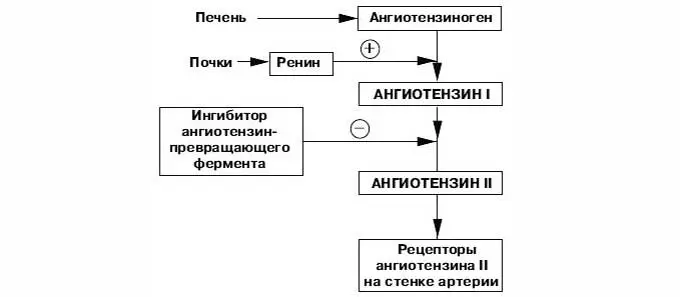
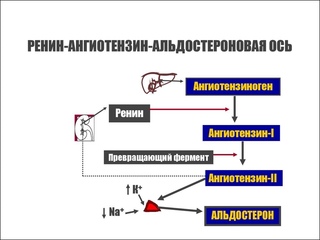
Первый этап ферментного каскада, приводящий к синтезу активного гормона ангиотензина из неактивного предшественника ангиотензиногена, осуществляется ренином. Ангиотензиноген синтезируется в печени и поступает в общий кровоток. Ренин синтезируется в юкстагломерулярном аппарате почек и выбрасывается в общий кровоток в ответ на почечную симпатическую активацию, снижение АД и понижение концентрации солей в тубулах. В крови большая часть молекул ангиотензиногена расщепляется ренином, образуя декапептид ангиотензин–I. Процесс расщепления ангиотензиногена, всегда находящегося в избытке, лимитирован количеством и активностью ренина [4–7,86,107].
Ангиотензин–II является главным эффекторным пептидом РАС. Он освобождается из ангиотензина–I посредством АПФ. Ангиотензин–II связывается с двумя основными подтипами рецепторов: АТ1 и АТ2. АТ1–рецепторы представлены в большинстве тканей взрослого организма и являются медиаторами основных действий ангиотензина–II, таких как вазоконстрикция, выработка альдостерона, задержка натрия, развитие фиброза, гипертрофии и воспалительных процессов. АТ2–рецепторы являются антагонистами АТ1–рецепторов, а следовательно, стимулируют вазодилатацию, препятствуют развитию фиброза, гипертрофии и воспаления. АТ2–рецепторы способствуют процессам репарации и регенерации, выступая в роли тканевых протекторов [3,5,36,67,106].
В последние годы были описаны новые компоненты РАС (рис. 1). Одним из них является ангиотензин 1–7, образующийся из ангиотензина–I и ангиотензина–II с помощью фермента АПФ–2 [43,112]. Ангиотензин 1–7 противостоит биологическому действию ангиотензина–II, вызывая расширение сосудов путем стимуляции секреции NO и простациклина. Кроме того, ангиотензин 1–7 не стимулирует секрецию альдостерона, не вызывает жажду, обладает натрийуретическим и диуретическими свойствами, блокирует ангиогенез и пролиферацию гладкомышечных клеток сосудов. Иными словами, ангиотензин 1–7 выступает в роли конкурентного антагониста ангиотензина–II в отношении его действия, опосредованного АТ1–рецепторами [5,59,117].
Еще одним важным событием в понимании сложности функционирования РАС стало открытие (про)ренинового рецептора. Этот рецептор связывается и с проренином, и ренином тканей, вызывая действие, сходное с действием ангиотензина–II на АТ1–рецепторы [22,84,119,120,124].
Фармакологическое воздействие на РАС может осуществляться различными путями. Действие препаратов из группы ингибиторов АПФ заключается в снижении образования ангиотензина–II. Другой класс препаратов, воздействующий на РАС и оказывающий выраженное тканевое действие, направлен на блокаду АТ1–рецепторов. Одновременная стимуляция АТ2–рецепторов приводит к дополнительному сосудорасширяющему эффекту. Блокаторы АТ1–рецепторов не влияют на процессы расщепления брадикинина, что позволяет избежать таких побочных действий, как кашель, крапивница, аллергические отеки, свойственные ингибиторам АПФ. «Эффект ускользания», заключающийся в снижении эффективности препарата при длительном его применении, может наблюдаться при приеме ингибиторов АПФ и практически отсутствует при использовании блокаторов АТ1–рецепторов [5–7,12,76,86].
Новые стратегические возможности фармакологического воздействия на РАС видятся в блокаде ферментативной активности ренина [60] и стимуляции «полезных» путей этой системы, например за счет синтеза ангиотензина 1–7 и создания непептидных агонистов АТ2–рецепторов [59,117].
Глазная локальная РАС
Первоначально РАС была описана как гормональная система, обладающая преимущественно системным эффектом. Однако в начале 1990–х гг. была высказана теория о существовании так называемых тканевых (локальных) РАС. Аргументом в пользу этого явилось обнаружение компонентов РАС в «нетрадиционных» для них местах локализации. К примеру, почечный фермент ренин был обнаружен в тканях мозга. В настоящее время признано, что органы–мишени, поражающиеся при сахарном диабете (СД) (глаз, почка), а также большинство других органов (сердце, легкие, мозг, кишечник и пр.) содержат локальные РАС, в которых ангиотензин–2 образуется независимо от циркулирующей крови [10,11,51,90,86,116].
В 1978 г. Ikemoto и Yamamoto, инкубируя образцы водянистой влаги с экзогенным ангиотензиногеном, получили данные о существовании локальной системы образования ангиотензина–I в глазах собак, кроликов и обезьян [63]. В 1989–1990 гг. аналогичные результаты были получены при исследовании глаза человека. Способность генерировать ангиотензин–I была обнаружена в стекловидном теле, сетчатке, комплексе «пигментный эпителий–хориоидея», переднем увеальном тракте [33,96,105,118]. Во всех отделах глаза были обнаружены проренин и ренин, причем количество проренина преобладало над ренином. Обнаружение концентрационного градиента (заднее стекловидное тело ⟶ переднее стекловидное тело ⟶ водянистая влага) явилось косвенным доказательством существования локального синтеза проренина в сетчатке [34,37,45,63,104].
К настоящему времени определена экспрессия компонентов РАС в сетчатке животных и человека. Согласно этим данным, все компоненты РАС (ангиотензиноген, проренин, ренин, АПФ, АТ1– и АТ2–рецепторы) представлены в сетчатке [17,52,78,97,98,100,106,111,115].
Механизмы повреждения тканей при СД
За последние годы существенно расширились наши представления о молекулярных механизмах, приводящих к повреждению тканей при СД. Установлено, что гипергликемия является первичным патогенетическим звеном в развитии диабетической ретинопатии (ДР) (рис. 2). Далее запускаются различные биохимические процессы, приводящие к нарушению клеточного гомеостаза. Наиболее важными из них являются:
• внутри– и внеклеточная продукция конечных продуктов гликирования;
• активация полиольного пути обмена;
• активация протеинкиназы С, опосредованная повышением синтеза диацилглицерола;
• активация гексозаминового пути обмена [2,5,20,21,35].
Эти биохимические процессы связаны друг с другом множеством связей и, взаимодействуя, потенцируют «вредный» эффект друг друга. Оксидативный стресс является первым событием в гипергликемической клетке (во многих тканях при СД, в том числе в сетчатке), а также «объединяющим» механизмом, который может инициировать все четыре биохимических пути, ведущих к нарушению клеточного гомеостаза [2,20,21,39,46,53].
Конечные продукты гликирования (КПГ) возникают в результате неферментативного гликирования (реакция Майларда) в условиях гипергликемии. Формирование КПГ приводит к повреждению клеток посредством нарушения функционирования гликированных клеточных белков; повреждения экстрацеллюлярного матрикса; активации нуклеарного фактора каппа В (NF–κB), являющегося важным провоспалительным медиатором [5,21].
Активизация полиольного пути обмена приводит к уменьшению количества НАДФ(Н), снижению синтеза глутатиона и, таким образом, к усилению оксидативного стресса. Однако этот путь не является основным в развитии осложнений СД [58].
Повышение уровня протеинкиназы С вызывает сосудистую дисфункцию [64] за счет повышения синтеза сосудистого эндотелиального фактора роста VEGF [123,125], вазоконстриктора эндотелина–I [129], медиатора воспаления NF–κB [82,127], снижения продукции NO [71].
Активизация протеинкиназы С и пути обмена гексозамина приводят к повышению уровня активатора ингибитора плазминогена (РАI 1) [21,41] и трансформирующего фактора роста β (TGFβ) [70].
Возникновение описанных выше обменных нарушений способствует васкулопатии и эндотелиальной дисфункции в макро– и микрососудах. В макрососудах это приводит к атеросклерозу, в микрососудистом русле – к потере перицитов, повышению проницаемости сосудистой стенки, избыточному образованию экстрацеллюлярного матрикса [2,5,7,42,55,57].
Глазная РАС при СД
В настоящее время не существует единого мнения о характере изменений активности системной РАС при СД. Одни авторы утверждают, что ДР сопровождается подавлением РАС [92], другие считают, что повышение в плазме уровня проренина является маркером прогрессирования ДР [47,122,128]. Мнения относительно изменений локальной РАС при СД не столь противоречивы. Целенаправленное изучение органов–мишеней, подверженных патологическим изменениям при СД (почка и глаз), позволило получить данные о повышении активности локальной (тканевой) РАС при СД [8–10,23,38,45].
В 1989 г. Danser et al. сообщили о том, что у пациентов с отслойкой сетчатки и пролиферативной ДР уровень проренина в стекловидном теле был выше, чем у пациентов с отслойкой сетчатки без ДР [34]. Повышенный уровень ангиотензина–2 был определен в стекловидном теле пациентов с СД, причем этот показатель коррелировал с тяжестью ДР [49].
Локальная РАС в глазу активно участвует в повреждении тканей при ДР. Повышение уровня ангиотензина–II через стимуляцию AT1–рецепторов приводит к следующим изменениям: повышению экспрессии VEGF, повреждающего внутренний гематоретинальный барьер [126]; вазоконстрикции, дополнительно ухудшающей кровоток в сетчатке [69]; активации NF–κB, стимулирующего воспалительные процессы [94]; усилению оксидативного стресса [46,99]; накоплению экстрацеллюлярного матрикса, приводящего к развитию фиброза [68].
Помимо этого, РАС вовлекается во все биохимические процессы, вызванные гипергликемией и приводящие к повреждению клеток. Гипергликемия стимулирует экспрессию гена ангиотензиногена через путь обмена гексозамина и, таким образом, увеличивает синтез ангиотензина–II [50,62]. Ангиотензин–II может повышать уровень КПГ и ретинальной протеинкиназы C, стимулировать полиольный путь обмена, создавая «порочный круг» биохимических нарушений [31,88,91,109].
Оксидативный стресс также находится под сильным влиянием ангиотензина–II [39,46,99,126]. При ДР описано влияние ангиотензина–II на оксидативный стресс, которое приводит к стимуляции экспрессии VEGF и лейкостазу [27,48,126]. Кроме того, оксидативный стресс прямо стимулирует экспрессию АТ1–рецепторов и образование ангиотензина–II [16,29].
Недавно Toma et al. была предложена новая теория, объясняющая, каким образом гипергликемия может повышать активность РАС. Был описан паракринный сигнальный путь в диабетической почке и показано, что гипергликемия стимулирует локальное накопление промежуточных продуктов обмена лимонной кислоты, которые через ряд превращений стимулируют высвобождение ренина [113]. Sapieha P. et al. было обнаружено участие этого пути обмена в развитии ретинальной неоваскуляризации [95].
Ингибирование РАС при ДР
Доклинические исследования. Во многих экспериментальных работах были получены доказательства положительного фармакологического влияния ингибиторов АПФ и блокаторов АТ1–рецепторов на течение ДР [5,32].
На моделях СД 2–го типа блокатор АТ1–рецепторов кандесартан предотвращал патологическую аккумуляцию КПГ при диабетической нефропатии и ретинопатии. В диабетической почке уменьшение содержания КПГ коррелировало с ослаблением оксидативного стресса, а в сетчатке – с уменьшением экспрессии VEGF. В обоих исследованиях лечение блокаторами АТ1–рецепторов приводило к улучшению функционирования органов: наблюдалось снижение альбуминурии и повышение электрической активности сетчатки [40,79,108].
Блокаторы АТ1–рецепторов телмисартан и лазартан уменьшали лейкостаз и воспаление при ДР в эксперименте in vivo. Предполагается, что этот эффект был связан с подавлением активности NF–κB [81,82].
Фармакологическое влияние на РАС может быть направлено на снижение активности протеинкиназы С. На клеточной культуре кардиомиоцитов было показано, что блокатор АТ1–рецепторов лазартан может ингибировать активность протеинкиназы С, вызванную гипергликемией [75]. Прием ингибитора АПФ рамиприла у крыс со стрептозотоциновым СД снижал активность протеинкиназы С в сетчатке, брызжеечной артерии и клубочковом аппарате почки, что коррелировало со снижением альбуминурии [87].
Гемодинамика сетчатки при СД изменяется в результате нарушения баланса между сосудосуживающими и сосудорасширяющими факторами, основными из которых являются вазоконстриктор эндотелин–I и вазодилататор NO [35,42]. Ингибирование РАС через влияние на протеинкиназу С улучшает ретинальный кровоток, снижает экспрессию эндотелина–I и увеличивает продукцию NО [13,61,65, 74,85].
При экспериментальном СД прием кандесартана или каптоприла значительно улучшал функции эндотелия [61]. Прием ингибитора АПФ эналаприла достоверно снижал уровень эндотелина–I в плазме крови у пациентов с гипертонической болезнью и СД 2–го типа [65]. Применение лазартана в течение 1 мес. значительно уменьшало экспрессию рецептора эндотелина, причем этот эффект был сопоставим с действием инсулина [85].
На мышах разных линий с экспериментальным СД было показано, что применение ингибиторов АПФ и блокаторов АТ1–рецепторов снижает повышенную экспрессию VEGF [54,80,83]. Прием ингибиторов АПФ рамиприла блокировал развитие эндотелиальной пролиферации в сетчатке и радужке [80]. Прием валсартана приводил к снижению образования ацеллюлярных капилляров, причем этот эффект не зависел от гипотензивного действия препарата, т.к. снижение АД до аналогичных показателей под влиянием атенолола такого эффекта не оказывало [121].
Гиперактивация РАС при СД вносит вклад в усиление оксидативного стресса, а ингибирование РАС улучшает состояние поврежденных тканей [27,77]. Положительное влияние блокаторов АТ1–рецепторов на ретинальный лейкостаз сопоставимо с действием антиоксидантов [27].
Длительное (в течение 6 мес.) лечение мышей с СД ингибитором АПФ рамиприлом значительно снижало продукцию свободных радикалов и улучшало функцию эндотелия [74]. В небольшом проспективном контролируемом исследовании, проведенном у подростков и молодых людей с СД 1–го типа с первыми признаками ангиопатии (14 пациентов) и без ангиопатии (11 пациентов), было показано, что гиперпродукция супероксида была связана со снижением выработки и активности внутриклеточных антиоксидантных ферментов, зависимых от гипергликемии. Лечение ирбесартаном на протяжении 6 мес. приводило к достоверному увеличению продукции и активности этих ферментов [30].
В эксперименте было показано, что блокатор АТ1–рецепторов кандесартан и новый сартан с низкой афинностью к АТ1–рецепторам Р–147176 имеют прямой антиоксидантный эффект, независимый от блокады АТ1–рецепторов [28,66]. Оба препарата показали протективное действие при диабетической нефропатии и ретинопатии.
Клинические исследования. Терапевтическая эффективность блокады РАС в стратегии лечения диабетических микроваскулярных осложнений была широко изучена и подтверждена на примере диабетической нефропатии [6,93]. Согласно рекомендациям Американской диабетической ассоциации в 2008 г. препараты, относящиеся к ингибиторам АПФ и блокаторам АТ1–рецепторов, являются препаратами выбора в лечении пациентов с микро– и макроальбуминурией независимо от уровня АД [12,14].
Данные о терапевтической эффективности блокады РАС при ДР малочисленны. Первым исследованием, доказавшим необходимость строгого контроля АД для профилактики прогрессирования ДР, было UKPDS. В рамках этого исследования пациенты одной группы для снижения АД применяли каптоприл [114]. В отличие от UKPDS, в котором ингибитор АПФ использовали у пациентов с артериальной гипертензией, последующие небольшие когортные исследования изучали влияние блокады РАС на течение ДР у пациентов с нормальным АД [1,8,9,24,73].
В двухгодичном рандомизированном двойном слепом плацебо контролируемом исследовании EUCLID сравнивали действие ингибитора АПФ лизиноприла и плацебо у 530 нормотензивных пациентов с CД 1–го типа [26]. ДР была второй конечной точкой данного исследования, результаты оценивались по данным 354 пациентов. В ходе анализа было выявлено, что в группе пациентов, принимавших лизиноприл, значимо снижалось прогрессирование ДР. Однако показатели АД и гликемии также были несколько лучше в группе пациентов, получавших лизиноприл, что могло повлиять на конечные результаты исследования и уменьшить скорость прогрессирования ДР [5,26].
Исследование DIRECT было запланировано для оценки эффективности блокады РАС (на примере блокатора АТ1–рецепторов кандесартана) в снижении прогрессирования ретинопатии у пациентов с СД 1–го и 2–го типа. В исследовании DIRECT было обнаружено, что у пациентов с СД 1–го типа и нормальным АД кандесартан снижал частоту возникновения новых случаев ДР, однако не влиял на прогрессирование уже имеющейся ретинопатии. У пациентов с СД 2–го типа с нормальным и повышенным АД прием кандесартана снижал прогрессирование ДР, однако не влиял на частоту появления новых случаев ДР [25,101,102].
Заключение
ДР является одним из наиболее часто встречающихся и тяжелых осложнений СД. Развитие ДР несет в себе большой риск значительного снижения зрения. В настоящее время меры по предотвращению ДР заключаются в строгом контроле уровня гликемии и АД, а лечение развитых форм заболевания включает в себя проведение лазерной коагуляции сетчатки и анти–VEGF терапии.
Во множестве экспериментальных работ были представлены данные, уверенно подтверждающие участие РАС в патогенезе ДР. Целенаправленное изучение этой проблемы выявило гиперактивацию локальной глазной РАС при СД, приводящую к повреждению сетчатки.
Действие локальной РАС осуществляется либо через каскад патологических реакций, связанный с прямым воздействием на АТ1–рецепторы (повышение экспрессии VEGF, вазоконстрикция, активация NF–κB, возрастание оксидативного стресса и накопление экстрацеллюлярного матрикса), или непрямым влиянием на патобиохимический путь, индуцированный гипергликемией (генерация конечных продуктов гликирования, активация протеинкиназы С, полиольного и гексозаминового путей обмена, гиперпродукция супероксида). Фармакологическое воздействие на разбалансированную РАС при СД изучалось во многих исследованиях, показавших эффективность ингибиторов АПФ и блокаторов АТ1–рецепторов в лечении ДР.
Литература
1. Алтынбаев У.Р. Прогнозирование и профилактика осложнений хирургического лечения пролиферативной диабетической ретинопатии: Автореф. дис. … канд. мед. наук. Уфа, 2006.
2. Балаболкин М.И., Клебанова Е.М. Роль окислительного стресса в патогенезе сосудистых осложнений сахарного диабета (лекция) // Терапевтический архив. 2000. Т. 73. № 4. С. 3–8.
3. Белова Л.С. Ангиотензин II–образующие ферменты // Биохимия. 2000. Т. 65. Вып. 12. С. 1589–1599.
4. Григорьев Ю.В. Рациональная антигипертензивная терапия // ГВМУ МО РФ, ГИУВ МО РФ: Методические рекомендации. – М., 2000.
5. Дедов И.И., Шестакова М.В. Сахарный диабет и артериальная гипертензия. М.: МИА, 2006. 345 с.
6. Демидова Т.Ю. Особенности патогенеза артериальной гипертонии и применения ингибиторов АПФ у больных с различными клиническими формами СД 2 типа: Автореф. дис. … канд. мед. наук. М., 1997. 25 с.
7. Карпов Ю.А. Артериальная гипертония у больных сахарным диабетом: основные направления лечения // Русский медицинский журнал. 2001. Т. 9. № 24. С. 11–15.
8. Нероев В.В., Рябина М.В., Охоцимская Т.Д., Зуева М.В., Цапенко И.В. О применении препарата «Периндоприл» в лечении больных диабетической ретинопатией // Вестник офтальмологии. 2006. № 4. Вып. 123(4). С. 31–33.
9. Нероев В.В., Чеснокова Н.Б., Охоцимская Т.Д., Рябина М.В., Кост О.А., Никольская И.И., Павленко Т.А. Активность ангиотензинпревращающего фермента в крови и слезе у больных с диабетической ретинопатией // Вестник офтальмологии. 2006. № 3. Вып. 122 (3). С. 11–14.
10. Павленко Т.А. Ангиотензин–превращающий фермент в тканевых структурах и жидких средах глаза в норме и патологии, пути регуляции: Автореферат дис. … канд. мед. наук. М., 2009.
11. Чеснокова Н.Б., Григорьев А.В., Павленко Т.А., Никольская И.И., Кост О.А., Казанская Н.Ф. Роль компонентов ренин–ангиотензиновой системы в тканях глаза в норме и в патологии // Вестник РАМН. 2003. № 9. С. 29–31.
12. Шестакова М.В., Чугунова Л.А., Шамхалова М.Ш. Антигипертензивная терапия при сахарном диабете в сочетании с поражением почек // Фарматека. 2004. № 9. – С. 29–31.
13. Aiello L.P., Clermont A.C., Arora V., Davis M.D., Sheetz M.J., Bursell S–E. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes–induced retinal hemodynamic abnormalities in patients // Invest Ophthalmol Vis Sci. 2006. Vol. 47. Р. 86–92.
14. American Diabetes Association. Standards of medical care in diabetes –2008 // Diabetes Care. 2008. Vol. 31(suppl 1). Р. 12–54.
15. Antonetti D.A., Klein R., Gardner T.W. Diabetic retinopathy // N Engl J Med. 2012 Mar 29. Vol. 366(13). Р. 1227–1239.
16. Banday A.A., Lokhandwala M.F. Oxidative stress–induced renal angiotensin ATI receptor upregulation causes increased stimulation of sodium transporters and hypertension // Am J Physiol Renal Physiol 2008. Vol. 295. Р. 698–706.
17. Berka J.L., Stubbs A.J., Wang D.Z., Alcorn D., Campbell D.J., Skinner S.L. Renin–containing Muller cells of the retina display endocrine features // Invest Ophthalmol Vis Sci. 1995. Vol. 36. Р. 1450–1458.
18. Braun–Mendenez E., Fasciolo J.C., Leloir L.F., Munoz J.M. The substance causing renal hypertension // J Physiol. 1940. Vol. 98. Р. 283–298.
19. Braun–Mendenez E., Page I.H. Suggested revision of nomenclature: angiotensin // Science. 1958. Vol. 127. Р. 242.
20. Brownlee M. Banting lecture 2004. The pathobiology of diabetic complications: a unifying mechanism // Diabetes. 2005. Vol. 54. Р. 1615–1625.
21. Brownlee M. Biochemistry and molecular cell biology of diabetic complications // Nature. 2001. Vol. 414. Р. 813–820.
22. Burckle C., Bader M. Prorenin and its ancient receptor // Hypertension. 2006. Vol. 48. Р. 549–551.
23. Carey R.M., Siragy H.M. The intrarenal renin–angiotensin system and diabetic nephropathy // Trends Endocrinol Metab. 2003. Vol. 14. Р. 274–281.
24. Chase H.P., Garg S.K., Harris S., Hoops S., Jackson W.E., Holmes D.L. Angiotensin–converting enzyme inhibitor treatment for young normotensive diabetic subjects: a two–year trial // Ann Ophthalmol. 1993. Vol. 25. Р. 284–289.
25. Chaturvedi N., Porta M., Klein R., Orchard T., Fuller J., Parving H.H., Bilous R., Sjolie A.K. for the DIRECT Programme Study Group: Effect of candesartan on prevention (DIRECT–Prevent 1) and progression (DIRECT–Protect 1) of retinopathy in type 1 diabetes: randomised, placebo–controlled trials // Lancet. 2008. Vol. 372. Р. 1394–1402.
26. Chaturvedi N., Sjolie A.K., Stephenson J.M., Abrahamian H., Keipes M., Castellarin A., Rogulja–Pepeonik Z., Fuller J.H., the EUCLID Study Group: Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes // Lancet. 1998. Vol. 351. Р. 28–31.
27. Chen P., Guo A.M., Edwards P.A., Trick G., Scicli A.G. Role of NADPH oxidase and ANGII in diabetes–induced retinal leukostasis // Am J Physiol Regul Integr Comp Physiol. 2007. Vol. 293. Р. 1619–1629.
28. Chen S., Ge Y., Si J., Rifai A., Dworkin L.D., Gong R. Candesartan suppresses chronic renal inflammation by a novel antioxidant action independent of AT1R blockade // Kidney Int. 2008. Vol. 74. Р. 1128–1138.
29. Cheng X.W., Murohara T., Kuzuya M., Izawa H., Sasaki T., Obata K., Nagata K., Nishizawa T., Kobayashi M., Yamada T., Kim W., Sato K., Shi G.P., Okumura K., Yokota M. Superoxide–dependent cathepsin activation is associated with hypertensive myocardial remodeling and represents a target for angiotensin II type 1 receptor blocker treatment // Am J Pathol. 2008. Vol. 173. Р. 358–369.
30. Chiarelli F., Di Marzio D., Santilli F., Mohn A., Blasetti A., Cipollone F., Mezzetti A., Verrotti A. Effects of irbesartan on intracellular antioxidant enzyme expression and activity in adolescents and young adults with early diabetic angiopathy // Diab Care. 2005. Vol. 28. Р. 1690–1697.
31. Clements R.S. Jr., Morrison A.D., Winegrad A.I. Polyol pathway in aorta: regu¬lation by hormones // Science. 1969. Vol. 166. Р. 1007–1008.
32. Clermont A., Bursell S.E., Feener E.P. Role of the angiotensin II type 1 receptor in the pathogenesis of diabetic retinopathy: effects of blood pressure control and beyond // J Hypertens Suppl. 2006. Vol. 24. Р. 73–80.
33. Danser A.H., Derkx F.H., Admiraal P.J., Deinum J., de Jong P.T., Schalekamp M.A. Angiotensin levels in the eye // Invest Ophthalmol Vis Sci. 1994. Vol. 35. Р. 1008–1018.
34. Danser A.H., van den Dorpel M.A., Deinum J., Derkx F.H., Franken A.A., Peperkamp E., de Jong P.T., Schalekamp M.A. Renin, prorenin, and immunoreac–tive renin in vitreous fluid from eyes with and without diabetic retinopathy // J Clin Endocrinol Metab. 1989. Vol. 68. Р. 160–167.
35. Das Evcimen N., King G.L. The role of protein kinase С activation and the vascular complications of diabetes // Pharmacol Res. 2007. Vol. 55. Р. 498–510.
36. De Gasparo M., Catt K.J., Inagami T., Wright J.W., Unger T. International union of pharmacology. XXIII. The angiotensin II receptors // Pharmacol Rev. 2000. Vol. 52. Р. 415–472.
37. Deinum J., Derkx F.H., DanserA.H., Schalekamp M.A. Renin in the bovine eye // J Hypertens Suppl. 1989. Vol. 7. Р. 216–217.
38. Ebrahimian T.G., Tamarat R., Clergue M., Duriez M., Levy B.I., Silvestre J.S. Dual effect of angiotensin–converting enzyme inhibition on angiogenesis in type 1 diabetic mice // Arterioscler Thromb Vase Biol. 2005. Vol. 25. Р. 65–70.
39. Ellis E.A., Grant M.B., Murray F.T., Wachowski M.B., Guberski D.L., Kubilis P.S., Lutty G.A. Increased NADH oxidase activity in the retina of the BBZ/Wor diabetic rat // Free Rad Biol Med. 1998. Vol. 24. Р. 111–201.
40. Fan Q., Liao J., Kobayashi M., Yamashita M., Gu L., Gohda T., Suzuki Y., Wang L.N., Horikoshi S., Tomino Y. Candesartan reduced advanced glycation end–prod¬ucts accumulation and diminished nitro–oxidative stress in type 2 diabetic KK/Ta mice // Nephrol Dial Transplant. 2004. Vol. 19. Р. 3012–3020.
41. Feener E.P., Xia P., Inoguchi T., Shiba T., Kunisaki M., King G.L. Role of protein kinase С in glucose– and angiotensin II–induced plasminogen activator inhibitor expression // Contrib Nephrol. 1996. Vol. 118. Р. 180–187.
42. Feke G.T., Buzney S.M., Ogasawara H., Fujio N., Goger D.G., Spack N.P., Gabbay K.H. Retinal circulatory abnormalities in type 1 diabetes // Invest Ophthalmol Vis Sci. 1994. Vol. 35. Р. 2968–2975.
43. Ferrario C.M., Trask A.J., Jessup J.A. Advances in biochemical and functional roles of angiotensin–converting enzyme 2 and angiotensin–(l–7) in regulation of cardiovascular function // Am J Physiol Heart Circ Physiol. 2005. Vol. 289. Р. 2281–2290.
44. Ferreira S.H., Bartelt D.C., Greene L.J. Isolation of bradykinin–potentiating pep¬tides from Bothrops jararaca venom // Biochemistry. 1970. Vol. 9. Р. 2583–2593.
45. Fletcher E.L., Phipps J.A., Ward M.M., Vessey K.A., Wilkinson– Berka J.L. The renin–angiotensin system in retinal health and disease: Its influence on neurons, glia and the vasculature // Prog Retin Eye Res. 2010 Jul. Vol. 29(4). Р. 284–311. Epub 2010 Apr 7.
46. Forstermann U. Oxidative stress in vascular disease: causes, defense mecha¬nisms and potential therapies // Nat Clin Pract Cardiovasc Med. 2008. Vol. 5. Р. 338–349.
47. Franken A.A., Derkx F.H., Schalekamp M.A., Man in t’Veld A.J., Hop W.C., van Rens E.H., de Jong P.T. Association of high plasma prorenin with diabetic retinopathy // J Hypertens Suppl. 1988. Vol. 6.Р. 461–463.
48. Fukumoto M., Takai S., Ishizaki E., Sugiyama T., Oku H., Jin D., Sakaguchi M., Sakonjo H., Ikeda T., Miyazaki M. Involvement of angiotensin II–dependent vascular endothelial growth factor gene expression via NADPH oxidase in the retina in a type 2 diabetic rat model // Curr Eye Res. 2008. Vol. 33. Р. 885–891.
49. Funatsu H., Yamashita H., Ikeda T., Nakanishi Y., Kitano S., Hori S. Angio¬tensin II and vascular endothelial growth factor in the vitreous fluid of patients with diabetic macular edema and other retinal disorders // Am J Ophthalmol. 2002. Vol. 133. Р. 537–543.
50. Gabriely I., Yang X.M., Cases J.A., Ma X.H., Rossetti L., Barzilai N. Hyperglycemia modulates angiotensinogen gene expression // Am J Physiol Regul Integr Comp Physiol. 2001. Vol. 281. Р. 795–802.
51. Ganten D., Minnich J.L., Granger P., Hayduk K., Brecht H.M., Barbeau A., Boucher R., Genest J. Angiotensin–forming enzyme in brain tissue // Science. 1971. Vol. 173. Р. 64–65.
52. Geng L., Persson K., Nilsson S.F. Angio¬tensin converting anzyme (ACE) activity in porcine ocular tissue: effects of diet and ACE inhibitors // J Ocul Phar–macol Ther. 2003. Vol. 19. Р. 589–598.
53. Giacco F., Brownlee M. Oxidative stress and diabetic complications // Circ Res. 2010 Oct 29. Vol. 107(9). Р. 1058–1070.
54. Gilbert R.E., Kelly D.J., Cox A.J., Wilkin¬son–Berka J.L., Rumble J.R., Osicka T., Panagiotopoulos S., Lee V., Hendrich E.C., Jerums G., Cooper M.E. Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyper–permeability in experimental diabetes // Diabetologia. 2000. Vol. 43. Р. 1360–1387.
55. Giuliari G.P. Diabetic retinopathy: current and new treatment options // Curr Diabetes Rev. 2012 Jan 1. Vol. 8(1). Р. 32–41.
56. Goldblatt H., Lynch J., Hanzal R.F. Studies on experimental hypertension // J Exp Med. 1934. Vol. 59. Р. 347–379.
57. Hammes H.P., Feng Y., Pfister F., Brownlee M. Diabetic Retinopathy: Targeting Vasoregression // Diabetes. 2011 Jan. Vol. 60(1). Р. 9–16.
58. Hammes H.P., Porta M. Experimental Approaches to Diabetic Retinopathy // Frontiers of Diabets. Vol. 20. Basel, Karger, 2010.
59. Hernandez Prada J.A., Ferreira A.J., Katovich M.J., Shenoy V., Qi Y., Santos R.A., Castellano R.K., Lampkins A.J., Gubala V., Ostrov D.A., Raizada M.K. Structure–based identification of small–molecule angiotensin–converting enzyme 2 activators as novel antihypertensive agents // Hypertension. 2008. Vol. 51. Р. 1312–1317.
60. Hershey J.C., Steiner B., Fischli W., Feuerstein G. Renin inhibitors: An antihypertensive strategy on the verge of reality // Drug Discov Today Ther Strateg. 2005. Vol. 2. Р. 181–185.
61. Horio N., Clermont A.C., Abiko A., Abiko T., Shoelson B.D., Bursell S–E., Feener E.P. Angiotensin AT1 receptor antagonism normalizes retinal blood flow and acetylcholine–induced vasodiliation in normotensive diabetic rats // Diabetologia. 2004. Vol. 47. Р. 113–123.
62. Hsieh T.J., Fustier P., Zhang S.L., Filep J.G., Tang S.S., Ingelfinger J.R., Fantus I.G., Hamet P., Chan J.S. High glucose stimulates angiotensinogen gene expression and cell hypertrophy via activation of the hexosamine biosynthesis pathway in rat kidney proximal tubular cells // Endocrinology. 2003. Vol. 144. Р. 4338–4349.
63. Ikemoto F., Yamamoto K. Renin–angiotensin system in the aqueous of rabbits, dogs and monkeys // Exp Eye Res. 1978. Vol. 27. Р. 723–725.
64. Ishii H., Jirousek M.R., Koya D., Takagi C., Xia P., Clermont A., Bursell S.E., Kern T.S., Ballas L.M., Heath W.F., Stramm L.E., Feener E.P., King G.L. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor // Science. 1996. Vol. 272. Р. 728–731.
65. Iwase M., Doi Y., Goto D., Ichikawa K., Iino K., Yoshinari M., Fujishima M. Effect of nicardipine versus enalapril on plasma endothelin–1 in hypertensive patients with type 2 diabetes mellitus // Clin Exp Hypertens. 2000. Vol. 22. Р. 695–703.
66. Izuhara Y., Sada T., Yanagisawa H., Koike H., Ohtomo S., Dan T., Ito S., Nangaku M., van Ypersele de Strihou C., Miyata T. A novel sartan derivative with very low angiotensin II type 1 receptor affinity protects the kidney in type 2 diabetic rats // Arterioscler Thromb Vase Biol. 2008. Vol. 28. Р. 1767–1773.
67. Kaschina E., Grzesiak A., Li J., Foryst–Ludwig A., Timm M., Rompe F., Sommerfeld M., Kemnitz R., Curato C., Namsolleck P., Tschope C., Hallberg A., Alterman M., Hucko T., Paetsch I., Dietrich T., Schnackenburg B., Graf C., Dahlof B., Kintscher U., Unger T., Steckelings U.M. AT2–receptor stimulation: a novel option of therapeutic interference with the renin–angiotensin–system in myocardial infarction? // Circulation. 2008. Vol. 118. Р. 2523–2532.
68. Kato H., Suzuki H., Tajima S., Ogata Y., Tominaga T., Sato A., Saruta T. Angiotensin II stimulates collagen synthesis in cultured vascular smooth muscle cells // J Hypertens. 1991. Vol. 9. Р. 17–22.
69. Kawamura H., Kobayashi M., Li Q., Yamanishi S., Katsumura K., Minami M., Wu D.M., Puro D.G. Effects of angiotensin II on the pericyte–containing microvasculature of the rat retina // J Physiol. 2004. Vol. 561. Р. 671–683.
70. Koya D., Jirousek M.R., Lin Y.W., Ishii H., Kuboki K., King G.L. Characterization of protein kinase С beta isoform activation on the gene expression of transforming growth factor–beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats // J Clin Invest. 1997. Vol. 100. Р. 115–126.
71. Kuboki K., Jiang Z.Y., Takahara N., Ha S.W., Igarashi M., Yamauchi T., Feener E.P., Herbert T.P., Rhodes C.J., King G.L. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin // Circula¬tion. 2000. Vol. 101. Р. 676–681.
72. Lang G.E. Diabetic retinopathy. Development in ophthalmology. Vol. 39. Basel, Karger, 2007.
73. Larsen M., Hommel E., Parving H.H., Lund–Andersen H. Protective effect of captopril on the blood–retina barrier in normotensive insulin–dependent diabetic patients with nephropathy and background retinopathy // Graefes Arch Clin Exp Ophthalmol. 1990. Vol. 228. Р.?505–509.
74. Liang W., Tan C.Y.R., Ang L., Granville D.J., Wright J.M., Laher I. Ramipril improves oxidative stress–related vascular endothelial dysfunction in db/db mice // J Physiol Sci. 2008. Vol. 58. Р. 405–411.
75. Malhotra A., Kang B.P.S., Cheung S., Opawumi D., Meggs L.G. Angiotensin II promotes glucose–induced activation of cardiac protein kinase С isozymes and phosphorylation of troponin I // Diabetes. 2001. Vol. 50. Р. 1918–1926.
76. Mancia G. (ed): Angiotensin II Receptor Antagonists. Abingdon, Informa Healthcare. 2006. Р. 1–207.
77. Manea A., Constantinescu E., Popov D., Raicu M. Changes in oxidative balance in rat pericytes exposed to diabetic conditions // J Cell Mol Med. 2004. Vol. 8. Р. 117–120.
78. Maruichi M., Oku H., Takai S., Muramatsu M., Sugiyama T., Imamura Y., Minami M., Ueki M., Satoh B., Sakaguchi M., Miyazaki M., Ikeda T. Measurement of activities in two different angio¬tensin II generating systems, chymase and angiotensin–converting enzyme, in the vitreous fluid of vitreoretinal diseases: a possible involvement of chymase in the pathogenesis of macular hole patients // Curr Eye Res. 2004. Vol. 29. Р. 321–325.
79. Miller A.G., Tan G., Binger K.J., Pickering R.J., Thomas M.C., Nagaraj R.H., Cooper M.E., Wilkinson–Berka J.L. Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase–I function // Diabetes. 2010 Dec. Vol. 59 (12). Р. 3208–3215. Epub 2010 Sep 17.
80. Moravski C.J, Skinner S.L., Stubbs A.J., Sarlos S., Kelly D.J., Cooper M.E., Gilbert R.E., Wilkinson–Berka J.L. The renin–angiotensin system influences ocular endothelial cell proliferation in diabetes: transgenic and interventional studies // Am J Pathol. 2003. Vol. 162. Р. 151–160.
81. Mori F., Hikichi T., Nagaoka T., Takahashi J., Kitaya N., Yoshida A. Inhibitory effect of losartan, an ATI angiotensin II receptor antagonist, on increased leucocyte entrapment in retinal microcirculation of diabetic rats // Br J Oph¬thalmol. 2002. Vol. 86. Р. 1172–1174.
82. Nagai N., Izumi–Nagai K., Oike Y., Koto T., Satofuka S., Ozawa Y., Yamashiro K., Inoue M., Tsubota K., Umezawa K., Ishida S. Suppression of diabetes–induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor–kappaB pathway // Invest Ophthalmol Vis Sci. 2007. Vol. 48. Р. 4342–4350.
83. Nagisa Y., Shintani A., Nakagawa S. The angiotensin II receptor antagonist candesartan disorders in rats // Diabetologia. 2001. Vol. 44. Р. 883–888.
84. Nguyen G., Delarue F., Burckle C., Bouzhir L., Giller T, Sraer J.D. Pivotal role of the renin/prorenin receptor in angio¬tensin II production and cellular responses to renin // J Clin Invest. 2002. Vol. 109. Р.?1417–1427.
85. Nuwayri–Salti N., Karam C.H., Al Jaroudi W.A., Usta J.A., Maharsy W.M., Bitar K.M., Bikhazi A.B. Effect of type–1 diabetes mellitus on the regulation of insulin and endothelin–1 receptors in rat hearts // Can J Physiol Pharmacol. 2007. Vol. 85. Р. 215–224.
86. Opie L.H. Angiotensin–Converting Enzyme Inhibitors: The Advanced Continues. NY., 1999. 274 p.
87. Osicka T.M., Yu Y., Lee V., Panagiotopoulos S., Kemp B.E., Jerums G. Aminoguanidine and ramipril prevent dia¬betes induced increases in protein kinase С activity in glomeruli, retina and mesenteric artery // Clin Sci. 2001. Vol. 100. Р. 249–257.
88. Otani A., Takagi H., Oh H., Koyama S., Honda Y. Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin–2, in bovine retinal endothelial cells // Diabetes. 2001. Vol. 50. Р. 867–875.
89. Page I.H., Helmer O.M. Acristalline pressor substance (angiotonin) resulting from the reaction between renin and renin activator // J Exp Med. 1940. Vol. 71. Р. 29–42.
90. Paul M., Poyan Mehr A., Kreutz R. Physiology of local renin–angiotensin sys¬tems // Physiol Rev. 2006. Vol. 86. Р. 747–803.
91. Policha A., Daneshtalab N., Chen L., Dale L.B., Altier C., Khosravani H., Thomas W.G., Zamponi G.W., Ferguson S.S.G. Role of angiotensin II type 1A receptor phosphorylation, phospholipase D, and extracellular calcium in isoform–specific protein kinase С membrane translocation responses // J Biol Chem. 2006. Vol. 281. Р. 26340–26349.
92. Price D.A., Porter L.E., Gordon M., Fisher N.D.L., De’Oliviera J.M.F., Laffel L.M.B., Passan D.R., Williams G.H., Hollenberg N.K. The paradox of the low–renin state in diabetic nephropathy // J Am Soc Nephrol. 1999. Vol. 10. Р. 2382–2391.
93. Ruilope L.M. Angiotensin receptor blockers: RAAS blockade and renopro–tection // Curr Med Res Opin. 2008. Vol. 24. Р. 1285–1293.
94. Ruiz–Ortega M., Lorenzo O., Egido J. Angiotensin II increases monocytic chemotactic protein–1 and activates nuclear transcription factor кВ and activator protein–1 in cultured mesangial and mononuclear cells // Kidney Int. 2000. Vol. 57. Р. 2285–2298.
95. Sapieha P., Sirinyan M., Hamel D., Zaniolo K., Joyal J–S., Cho J–H., Honore J–C., Kermorvant–Duchemin E., Varma DR., Tremblay S., Leduc M,. Rihakova L., Hardy P., Klein W.H., Mu X., Mamer O., Lachapelle P., Di Polo A., Beausejour C., Andelfinger G., Mitchell G., Sennlaub F., Chemtob S. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis // Nat Med. 2008. Vol. 14. Р. 1067–1076.
96. Sarlos S., Wilkinson–Berka J.L..The renin–angiotensin system and the developing retinal vasculature // Invest Ophthalmol Vis Sci. 2005. Vol. 46. Р. 1069–1077.
97. Savaskan E., Loffler K.U., Meier F., Muller–Spahn F., Flammer J., Meyer P. Immunohistochemical localization of angiotensin–converting enzyme, angiotensin II and ATI receptor in human ocular tissues // Ophthalmic Res. 2004. Vol. 36. Р. 312–320.
98. Senanayake P., Drazba J., Shadrach K., Milsted A., Rungger– Brandle E., Nishiyama K., Miura S., Karnik S., Sears JE., Hollyfield JG. Angiotensin II and its receptor subtypes in the human retina // Invest Ophthalmol Vis Sci. 2007. Vol. 48. Р. 3301–3311.
99. Seshiah P.N., Weber D.S., Rocic P., Valppu L., Taniyama Y., Griendling K.K. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators // Circ Res. 2002. Vol. 91. Р. 406–413.
100. Shiota N., Saegusa Y., Nishimura K., Miyazaki M. Angiotensin II–generating system in dog and monkey ocular tissues // Clin Exp Pharmacol Physiol. 1997. Vol. 24. Р. 243–248.
101. Sjolie A.K., Klein R., Porta M., Orchard T., Fuller J., Parving H.H., Bilous R., Chaturvedi N., for the DIRECT Pro¬gramme Study Group: Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT–Protect 2): a randomised placebo–controlled trial // Lancet. 2008. Vol. 372. Р. 1385–1393.
102. Sjolie A.K. Prospects for angiotensin receptor blockers in diabetic retinopa–thy // Diab Res Clin Prac. 2007. Vol. 76. Р. 31–39.
103. Skeggs L.T. Jr., Marsh W.H., Kahn J.R., Shumway N.P. The existence of two forms of hypertensin // J Exp Med. 1954. Vol. 99. Р.?275–282.
104. Sramek S.J., Wallow I.H., Day R.P., Ehrlich E.N. Ocular renin–angiotensin: immunohistochemical evidence for the presence of prorenin in eye tissue // Invest Ophthalmol Vis Sci. 1988. Vol. 29. Р. 1749–1752.
105. Sramek S.J., Wallow I.H., Tewksbury D.A., Brandt C.R., Poulsen G.L. An ocular renin–angiotensin system. Immunohistochemistry of angiotensinogen // Invest Ophthalmol Vis Sci. 1992. Vol. 33. Р.?1627–1632.
106. Steckelings U.M., Kaschina E., Unger T. The AT2 receptor – a matter of love and hate // Peptides. 2005. Vol. 26. Р. 1401–1409.
107. Steckelings U.M., Unger T. The renin–angiotensin–aldosterone system; in Mancia G, Grassi G, Kjeldsen S (eds): Manual of Hypertension of The Euro¬pean Society of Hypertension. Abingdon, Informa HealthCare, 2008. Vol. 14. Р. 110–116.
108. Sugiyama T., Okuno T., Fukuhara M., Oku H., Ikeda T., Obayashi H., Ohta M., Fukui M., Hasegawa G., Nakamura N. Angiotensin II receptor blocker inhibits abnormal accumulation of advanced glycation end products and retinal damage in a rat model of type 2 diabetes // Exp Eye Res. 2007. Vol. 85.Р. 406–412.
109. Thomas M.C., Tikellis C., Burns W.M., Bialkowski K., Cao Z., Coughlan M.T., Jandeleit–Dahm K., Cooper M.E., Forbes J.M. Interactions between renin angiotensin system and advanced glycation in the kidney // J Am Soc Nephrol. 2005. Vol. 16. Р. 2976–2984.
110. Tigerstedt R., Bergman P.G. Niere und Kreislauf // Skand Arch Physiol. 1898. Vol. 8. Р. 223–271.
111. Tikellis C., Johnston C.I., Forbes J.M., Burns W.C., Thomas M.C., Lew R.A., Yarski M., Smith A.I., Cooper M.E. Identification of angiotensin converting enzyme 2 in the rodent retina // Curr Eye Res. 2004. Vol. 29. Р. 419–427.
112. Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin–converting enzyme. Cloning and functional expression as a captopril–insensitive carboxypeptidase // J?Biol Chem. 2000. Vol. 275. Р. 33238–33243.
113. Toma I., Kang J.J., Sipos A., Vargas S., Bansal E., Hanner F., Meer E., Peti–Peterdi J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney // J Clin Invest. 2008. Vol. 118. Р. 2526–2534.
114. UK Prospective Study Group: Tight blood pressure control and risk of mac–rovascular and microvascular complications in type 2 diabetes. UKPDS 38 // BMJ. 1998; Vol. 317:703–713.
115. Vita J.B., Anderson J.A., Hulem C.D., Leopold I.H. Angiotensin– converting enzyme activity in ocular fluids. Invest Ophthalmol Vis Sci 1981; Vol. 20. Р. 255–257.
116. Wagner J., Danser A.H., Derkx F.H., de Jong T.V., Paul M., Mullins J.J., Schalekamp M.A., Ganten D. Demonstration of renin mRNA, angiotensino–gen mRNA, and angiotensin converting enzyme mRNA expression in the human eye: evidence for an intraocular renin–angiotensin system // Br J Ophthalmol. 1996. Vol. 80. Р. 159–163.
117. Wan Y., Wallinder C., Plouffe B., Beaudry H., Mahalingam A.K., Wu X., Johansson B., Holm M., Botoros M., Karlen A., Pettersson A., Nyberg F., Fandriks L., Gallo–Payet N., Hallberg A., Alterman M. Design, synthesis, and biological evalu¬ation of the first selective nonpeptide AT2 receptor agonist // J Med Chem. 2004. Vol. 47. Р.?5995–6008.
118. Weinreb R.N., Sandman R., Ryder M.I., Friberg T.R. Angiotensin– converting enzyme activity in human aqueous humor // Arch Ophthalmol. 1985. Vol. 103. Р. 34–36.
119. Wilkinson–Berka J.L., Campbell D.J. (Pro)renin receptor: a treatment target for diabetic retinopathy? // Diabetes. 2009 Jul. Vol. 58(7). Р. 1485–1487.
120. Wilkinson–Berka J.L., Miller A.G., Fletcher E.L. Prorenin and the (pro)renin receptor: do they have a pathogenic role in the retina? // Front Biosci (Elite Ed). 2010 Jun 1. Vol. 2. Р. 1054–1064.
121. Wilkinson–Berka J.L., Tan G., Jaworski K., Ninkovic S. Valsartan but not atenolol improves vascular pathology in diabetic Ren–2 rat retina // Am J Hypertens. 2007. Vol. 20. Р. 423–430.
122. Wilkinson–Berka J.L. Angiotensin and diabetic retinopathy // Int J Biochem Cell Biol. 2006. Vol. 38. Р. 752–765.
123. Williams B., Gallacher B., Patel H., Orme D. Glucose–induced protein kinase С activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro // Diabetes. 1997. Vol. 46. Р. 1497–1503.
124. Wruck C.J., Funke–Kaiser H., Pufe T., Kusserow H., Menk M., Schefe J.H., Kruse M.L., Stoll M., Unger T. Regulation of transport of the angiotensin AT2 recep¬tor by a novel membrane–associated Golgi protein // Arterioscler Thromb Vase Biol. 2005. Vol. 25. Р. 57–64.
125. Xu X., Zhu Q., Xia X., Zhang S., Gu Q., Luo D. Blood–retinal barrier breakdown induced by activation of protein kinase С via vascular endothelial growth fac¬tor in streptozotocin–induced diabetic rats // Curr Eye Res. 2004. Vol. 28. Р. 251–256.
126. Yamagishi S., Amano S., Inagaki Y., Okamoto T., Inoue H., Takeuchi M., Choei H., Sasaki N., Kikuchi S. Angiotensin II–type 1 receptor interaction upregulates vascular endothelial growth factor messenger RNA levels in retinal peri¬cytes through intracellular reactive oxygen species generation // Drugs Exp Clin Res. 2003. Vol. 29. Р.?75–80.
127. Yemeni K.K., Bai W., Khan B.V., Medford R.M., Natarajan R. Hyperglycemia–induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells // Diabetes. 1999. Vol. 48. Р.?855–864.
128. Yokota H., Mori F., Kai K., Nagaoka T., Izumi N., Takahashi A., Nikichi T., Yoshida A., Suzuki F., Ishida Y. Serum prorenin levels and diabetic retinopa–thy in type 2 diabetes: new method to measure serum level of prorenin using antibody activating direct kinetic assay // Br J Ophthalmol. 2005. Vol. 89. Р. 871–873.
129. Yokota T., Ma R.C., Park J–Y., Isshiki K., Sotiropoulos K.B., Rauniyar R.K., Bornfeldt K.E., King G.L. Role of protein kinase С on the expression of platelet–derived growth factor and endothelin–1 in the retina of diabetic rats and cul¬tured retinal capillary pericytes // Diabetes. 2003. Vol. 52. Р. 838–845.
.
РЕНИН-АНГИОТЕНЗИН-АЛЬДОСТЕРОНОВАЯ СИСТЕМА ПРИ ХРОНИЧЕСКОЙ БОЛЕЗНИ ПОЧЕК | Карабаева
1. Jalil JE, Janicki JS, Pick R, Weber KT. Coronary vascular remodeling and myocardial fibrosis in the rat with renovascular hypertension: response to captopril. Am J Hypertens 1991; 4: 51–55
2. Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345: 1689 – 1697
3. Epstein M. Aldosterone as a mediator of progressive renal dysfunction: evolving perspectives. Intern Med. 2001 Jul; 40(7):573-83
4. Рябов СИ, Каюков ИГ, Гадаев АГ и др. Морфофункциональные параллели при артериальной гипертензии у больных хроническим гломерулонефритом. Тер арх 1992; (6): 26-29
5. Котовой ЮО. Изменения ренин-ангиотензин-альдостероновой системы при гломерулонефрите. Урология и нефрология 1989; (5): 53-58
6. Каюков ИГ. Особенности нарушений водно-солевого гомеостаза у больных хроническим гломерулонефритом в доазотемическом периоде. Дисс. докт. мед. наук… СПб, 1994
7. Гадаев А. Механизмы артериальной гипертензии при хроническом гломерулонефрите. Дисс. докт. мед. наук…СПб, 1992
8. Котовой ЮО. Сосотояние ренин-ангиотензин-альлдостероновой системы при гломерулонефрите в динамике развития почечной недостаточности. Дисс. канд. мед. наук…СПб, 1986
9. Osmond DH, Loh EK, Loh AY et al. Kallikrein and plasmin as activators of inactive renin. Lancet 1978; 2, 8104-5: 1375-1376
10. Quan ZY, Walser M, Hill GS. Adrenalectomy ameliorates ablative nephropathy in the rat independently of corticosterone maintenance level. Kidney Int 1992; 41: 326–333
11. Nishiyama A, Yao L, Nagai Y et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 2004; 43: 841–848
12. Blasi ER, Rocha R, Rudolph AE et al. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int 2003; 63: 1791–1800
13. Peng H, Carretero OA, Raij L et al. Antifibrotic effects of N-acetyl-seryl-aspartyl-Lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension 2001; 37: 794–800
14. Nishiyama A, Abe Y. Molecular mechanisms and therapeutic strategies of chronic renal injury: renoprotective effects of aldosterone blockade. J Pharmacol Sci 2006 Jan; 100(1): 9-16. Epub 2006 Jan 6
15. Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98: 1063–1068
16. Rocha R, Chander PN, Khanna K et al. Mineralocorticoid blockade reduces vascular injury in stroke-prone hypertensive rats. Hypertension 1998; 31: 451–458
17. Conn JW, Knopf RF, Nesbit RM. Clinical characteristics of primary aldosteronism from an analysis of 145 cases. Am J Surg 1964; 107: 159-172
18. Nishimura M, Uzu T, Fujii T et al. Cardiovascular complications in patients with primary aldosteronism. Am J Kidney Dis 1999; 33: 261-266
19. Ribstein J, Du Cailar G, Fesler P, Mimran A. Relative glomerular hyperfiltration in primary aldosteronism. Am J Hypertens 2005 Jan; 18(1): 50-5
20. Epstein M. Aldosterone as a determinant of cardiovascular and renal dysfunction. J R Soc Med 2001 Aug; 94(8): 378-83
21. Connell JMC, Davies E. The new biology of aldosterone. J Endocrinology 2005; 186: 1-20
22. Epstein M. Aldosterone receptor blockade and the role of eplerenone: evolving perspectives. Nephrol Dial Transplant 2003 Oct; 18(10): 1984-1992
23. Мухин НА, Моисеев ВС, Кобалава ЖД, Моисеев СВ, Фомин ВВ. Кардиоренальные взаимодействия: клиническое значение и роль в патогенезе заболеваний сердечно-сосудистой системы и почек.Тер арх 2004; 76 (6):39-47
24. Карабаева АЖ. Альдостерон, сердечно-сосудистая система и почки. Нефрология 2006; 1(10): 25-34
25. Есаян АМ, Каюков ИГ, Карабаева АЖ. Минералкортикоидные рецепторы: строение, механизмы активации. Нефрология 2006; 2 (10): 28-32
26. Зверев ЯФ, Брюханов ВМ. Современные представления о механихмах почечного действия альдостерона. Нефрология 2001; 4(5): 9-16
27. Duprez D, de Buyzere M, Rietzchel ER, Clement DL. Aldosterone and vascular damage. Curr Hypertens Rep 2000; 2: 327-334
28. Epstein M. Aldosterone receptor blockade and the role of eplerenone: evolving perspectives. Nephrol Dial Transpl 2003; 18: 1984-1992
29. Fuller PJ, Young MJ. Mechanisms of mineralocorticoid action. Hypertension 2005; 46: 1227-1246
30. Weber KT, Anversa P, Armstrong PW et al. Remodeling and reparation of the cardiovascular system. J Am Coll Cardiol 1992; 20: 3-16
31. Nancy J. Brown, Kyung-Soo Kim, Yan-Qun Chen et al. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production 1. J Clinl Endocrinol Metabo 2000; 85(1): 336-344
32. Epstein M. Aldosterone as a mediator of progressive renal disease: Pathogenetic and clinical implications.Am J Kidney Dis 2001; 37: 677-688
33. Brown NJ, Vaughan DE, Fogo AB. Aldosterone and PAI – 1: implications for renal injury. J Nephrol 2002; 15 (3): 230-235
34. Huang Y, Wongamorntham S, Kasting J et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms.Kidney Int 2006; 69 (1): 105-113
ФАРМАТЕКА » Роль различных видов ренин-ангиотензиновой системы в патогенезе сердечно-сосудистых заболеваний. Фокус на валсартан
В обзоре представлены клинико-экспериментальные данные, касающиеся комплексной природы ренин-ангиотензин-альдостероновой системы (РААС), включая классическую эндокринную РААС, тканевые ренин-ангиотензиновые системы (РАС), расположенные на клеточной поверхности, и внутриклеточную РАС. Обсуждаются механизмы интракринной активации клеток сердечно-сосудистой системы, лежащие в основе гипертрофии сердца, его ремоделирования, фиброза и формирования сердечной недостаточности. Анализируются результаты действия валсартана – жирорастворимого антагониста АТ1-рецепторов, включая его влияние на три вида РАС, а также последствия его действия на АТ1-рецепторы разной локализации.
Артериальная гипертензия (АГ) остается одним из главных факторов риска ремоделирования сердца (гипертрофия миокарда) и сосудов (увеличение толщины внутреннего слоя с сужением их просвета, эндотелиальная дисфункция), развития сердечно-сосудистых заболеваний и осложнений, таких как атеросклероз, ишемическая болезнь сердца, инфаркт миокарда, сердечная недостаточность (СН) – часто с сохраненной фракцией выброса.
Полагают, что при подобных изменениях в сердечно-сосудистой системе задействованы многие механизмы, в частности активация ренин-ангиотензин-альдостероновой системы (РААС), активация симпато-адреналовой системы, увеличение объема циркулирующей жидкости и нарушения электролитного баланса. Следует сказать, что эти системы тесно взаимосвязаны и активация одного звена приводит к изменению функционирования других звеньев, участвующих в повышении артериального давления (АД) и структурно-функциональном ремоделировании органов-мишеней.
Для лечения повышенного АД используют восемь классов лекарственных препаратов, воздействующих на разные патогенетические звенья АГ, причем более трех четвертей пациентов с АГ требуют комбинированной терапии [1].
В соответствии с современными позициями по лечению пациентов с АГ в большинстве случаев рекомендуют использовать комбинации ингибиторов РААС с антагонистами кальция или с диуретиками. Препаратам, ингибирующим активность РААС, – ингибиторам ангиотензинпревращающего фермента (иАПФ) и сартанам, придают особое значение в связи с тем, что за последние годы наши представления о системах, регулирующих структурно-функциональное состояние сердца и сосудов, претерпели существенные изменения, были выявлены новые механизмы функционирования РААС и пути, по которым эти препараты оказывают влияние на РААС [2, 3].
В настоящее время описано три разновидности ренин-ангиотензиновой системы (РАС): это классическая гормональная РААС; тканевые РАС, расположенные на поверхности клеток, и внутриклеточные РАС.
Классическая РААС – одна из главных регуляторных систем организма, участвует в поддержании различных жизненно важных показателей в реакциях немедленного типа: контролирует АД, ионный гомеостаз, уровень ионов Na+, водный баланс, объем крови, а также функциональную активность почек. Ключевые ферменты РААС, продукты реакций, взаимодействие с калликреиновой системой и точки приложения лекарственных препаратов показаны на рис. 1.
Система тканевых, или локальных, РАС участвует в хроническом действии Ан II на клетки, которое не всегда связано с регуляцией АД, а вызывает разнообразные клеточные ответы, часто заканчивающиеся цитотоксическим эффектом.
Для этой системы характерны две особенности:
- Благодаря фиксации ренина и АПФ на поверхности клетки локально образуются высокие концентрации Ан II, изменяющие активность клеток-мишеней, в которых экспрессированы рецепторы для Ан II.
- Образовавшийся Ан II действует аутокринно, т.е. на клетку-продуцент, или паракринно – на окружающие клетки.
Система внутриклеточных РАС находится в цитоплазме некоторых видов клеток (кардиомиоцитах, адипоцитах, клетках почек), содержит все или, во всяком случае, ключевые компоненты РАС: ферменты, субстраты и продукты реакций, а также АТ1- и АТ2-подобные рецепторы, экспрессированные на лизосомах [4], митохондриях (МХ) [5], ядрах клеток [6, 7], и участвуют в фагоцитозе, энергетическом обмене и активации генов, продукты которых контролируют гипертрофию клеток, сократительную активность сердца и другие процессы (см. таблицу).
Интракринные эффекты внутриклеточного Ан II тесно связаны с изменением уровня одного из главных вторичных мессенджеров – ионов кальция (Са2+) в цитоплазме, который увеличивается в результате выхода Са2+ из внутриклеточных Са2+-депо (SR-депо) в цитоплазму (Е.И. Асташкин, Глезер, 2012).
Установлено, что выход Са2+ из внутриклеточных депо происходит по Са2+-каналам, формируемым рецепторами для инозитол трисфосфата (IP3). Об этом свидетельствует следующий факт: блокада IP3-рецепторов Са2+-депо подавляет действие интракринного Ан II: блокирует выход Са2+ из депо и тормозит Са2+-зависимую активацию генов, связанных с Са2+-зависимым фактором транскрипции генов NF-kB [6].
Активация АТ1- и АТ2-подобных рецепторов, локализующихся на внутренней мембране МХ [5], изменяет потребление МХ кислорода и тем самым влияет на синтез АТФ. Влияние Ан II на АТ1-рецепторы в МХ может прямо (в результате воздействия на ферментативные комплексы дыхательной цепи) или опосредованно (например, через поступление ионов Са2+ в матрикс МХ) изменять дыхание этих органелл. Жирорастворимые сартаны подавляют эффекты Ан II, связанные с активацией АТ1-рецепторов МХ [5], и восстанавливают картину экспрессии АТ1- и АТ2-рецепторов, наблюдаемую на молодых животных [4, 5, 7].
Показано, что в изолированных кардиомиоцитах экзогенный Ан II, введенный в клетку, связывался с ядром и активировал фактор транскрипции генов NF-kB. Этот процесс блокировался внутриклеточным введением валсартана, действующим на АТ1-подобные рецепторы ядра, но не на АТ2-рецепторы [6].
Следует отметить, что внутри группы как ингибиторы АПФ, так и сартаны существенным образом отличаются друг от друга. Они имеют разную химическую структуру, обладают различными физико-химическими свойствами, различаются по растворимости в воде и жирах. Это влияет на способность препаратов проникать в клетки и клеточные органеллы через липидный бислой мембран. Благодаря этому жирорастворимые иАПФ и сартаны могут не только блокировать активность АПФ и АТ1-рецепторов классической эндокринной РААС и тканевой поверхностной РАС, но и влиять на внутриклеточную РАС, изменяя интракринный характер действия Ан II [8].
Самое большое число исследований в этой области проведено с жирорастворимым препаратом валсартан.
У валсартана (Диован) из всех препаратов этой группы самый широкий спектр зарегистрированных показаний к применению: АГ, хроническая СН (II–IV функциональные классы по классификации NYHA – New York Heart Association) в составе комплексной терапии, повышение выживаемости пациентов с острым инфарктом миокарда, осложненным левожелудочковой недостаточностью и/или систолической дисфункцией левого желудочка, при наличии стабильных показателей гемодинамики.
Проведена масса исследований эффективности валсартана в отношении разных категорий пациентов с АГ. Его антигипертензивный эффект связан, с одной стороны, с блокадой АТ1-рецепторов, с другой – со стимуляцией АТ2-рецепторов под влиянием Ан II, а также опосредуется через образование Ан 1-7 и АПФ2, что сопровождается увеличением содержания брадикинина, который, как уже указывалось, стимулирует продукцию NO и тем самым увеличивает содержание циклического гуанозинмонофосфата (сGMP). Это приводит к улучшению эндотелиальных свойств сосудов, их расширению, снижению периферического сопротивления сосудов, снижению АД.
Валсартан входит в состав фиксированных комбинированных готовых лекарственных форм, содержащих амлодипин (препарат Эксфорж – амлодипин + валсартан) или амлодипин и гидрохлоротиазид (КО Эксфорж). Эти препараты широко и успешно используются в клинической практике [9, 10].
Помимо антигипертензивного эффекта валсартан обладает выраженными органопротективными свойствами: уменьшает выраженность гипертрофии миокарда, оказывает положительные эффекты, предупреждая постинфарктное ремоделирование сердца (исследование VALIANT) [11] и улучшая течение СН (исследование Val-He-FT) [12].
За последнее время получены важные данные о новых механизмах, по которым валсартан оказывает эти положительные эффекты. Многие из этих эффектов связаны с влиянием валсартана на интракринную РАС и обусловлены изменениями обмена в кардиомиоцитах или гладкомышечных клетках Ca2+. В связи с этим рассмотрим основные процессы обмена Cа2+ в кардиомиоцитах, схематично представленные на рис. 2.
Регуляция сократительной активности кардиомиоцитов в физиологических условиях
Как известно, сокращение миоцитов сердца в фазу систолы запускается транспортом небольших количеств ионов кальция (Са2+) через потенциал-регулируемые медленные Са2+-каналы сарколеммы (L-VOC- каналы) в цитоплазму при деполяризации мембраны [13]. Этот триггерный Са2+ служит сигналом для выхода больших количеств ионов Са2+ из внутриклеточного Са2+-депо саркоплазматического ретикулума (SR-депо) по кальциевым каналам, формируемым рианодиновыми рецепторами (RyR). Именно этот Са2+ используется актомиозиновым комплексом при мышечном сокращении.
Различные внеклеточные стимулы, например стимуляция β-адренорецепторов кардиомиоцитов норадреналином [14], увеличивают активность L-VOC-каналов вследствие стимуляции аденилатциклазной системы. Увеличение уровня циклического аденозинмонофосфата (сАМР) активируется сАМР-зависимой протеинкиназой А (PKA). Этот фермент фосфорилирует различные мишени, включая L-VOC-каналы, RyR Са2+-депо, ферменты, контролирующие Са2+-цикл и мышечное сокращение [15].
В фазу диастолы ионы Са2+ удаляются из цитоплазмы через плазматическую мембрану с помощью Са2+-АТФазы и Na+/Са2+-обменника, а также в результате активного транспорта ионов Са2+ в SR-депо, осуществляемого Са2+-АТФазой депо (Са2+-АТФаза сарко-эндоплазматического ретикулума – SERCA2а), что необходимо для последующего сокращения.
Активность SERCA2a подавляется фосфоламбаном (PLB). При фосфорилировании PLB активность SERCA2a возрастает. Уменьшение фосфорилирования PLB может быть обусловлено снижением активности и экспрессии PKA и стимуляцией фосфатазы РР1-α, которые фосфорилируют и дефосфорилируют PLB соответственно [16, 17].
Следует отметить, что в качестве резервного депо для ионов Са2+ выступают МХ, перегрузка которых ионами Са2+ приводит к гибели кардиомиоцитов.
Снижение выведения ионов Са2+ из цитоплазмы в фазу диастолы вызывает постепенное накопление ионов Са2+ в клетке, активирует процессы, лежащие в основе их гипертрофии и ремоделирования сердца, затрудняет расслабление кардиомиоцитов, что ведет к развитию диастолической СН.
Ионы Са2+ в клетке влияют на активность Са2+-связывающих белков, участвующих в регуляции разнообразных ферментативных каскадов. Регуляторные биохимические эффекты Са2+ в клетках реализуются в результате его связывания с кальмодулином (Ca2+/CaM) и воздействием этого комплекса на кальций-кальмодулинзависимую протеинкиназу (СаМРКII), которая фосфорилирует различные белки. Удаление фосфатных групп (дефосфорилирование) происходит под влиянием другого Са2+-зависимого фермента – кальцинеурина, который является серин-треониновой фосфатазой, активируется Ca2+/CaM и воздействует на ядерный фактор транскрипции генов – NF-kB. Другим важным Са2+-регулируемым белком является кальпаин – член большого семейства цитозольных Са2+-активируемых цистеиновых протеаз. При активации ионами Са2+ эта протеаза разрушает клеточные мембраны и разнообразные внутриклеточные белки и изменяет их активность. Таким образом, действие кальпаина необратимо связано с процессами дегенерации и апоптоза клеток.
Нарушение поступления и выведения Са2+ в цитоплазме (Са2+-цикл) может происходить на любом его этапе – при дефекте L-VOC-каналов, изменении структуры RyR, торможении активности Са2+-АТФаз и Nа+/Са2+-обменника, дисфункции кальмодулина, СаМРКII, кальцинеурина.
Патофизиологические условия, способствующие развитию гипертрофии миокарда и СН
Длительная активация эндокринной и тканевых РАС, сопровождающаяся пролонгированным образованием больших количеств Ан II, и стимуляция симпатической нервной системы с повышенным выделением из нервных окончаний норадреналина активируют внутриклеточные сигнальные процессы и гены, продукты которых контролируют рост клеток. Конечным итогом этих процессов служит развитие гипертрофии миокарда.
При гипертрофии миокарда меняются не только структура, размеры и форма камер сердца, но и состояние ферментов (метаболическое ремоделирование), контролирующих в т.ч. энергетический обмен, активный транспорт ионов в кардиомиоцитах, их электрические характеристики и процессы мышечного сокращения [13].
Установлено, что между нарушениями в кардиомиоцитах активности различных типов АТФаз, ответственных за потоки ионов через мембраны за счет энергии гидролиза АТФ (активный транспорт ионов), изменением ионного гомеостаза и сократительной активностью кардиомиоцитов существует тесная взаимосвязь [18].
Снижение активности Na+/K+-АТФазы, Са2+-АТФазы плазматической мембраны и SR-депо (SERCA2a), повышение активности Na+/Са2+-обменника наружной мембраны при гипертрофии миокарда тормозят активный транспорт ионов Na+, K+, Са2+, снижают ионные градиенты через наружную и внутриклеточные мембраны, прежде всего саркоплазматического ретикулума (SR-депо ионов Са2+). Это сопровождается увеличением уровней Na+ и Ca2+ в цитоплазме, а также приводит к поступлению ионов Са2+ в матрикс МХ [13, 19]. Нарушения ионного гомеостаза нарушают электромеханическое сопряжение в миоцитах сердца и вызывают формирование диастолической, а затем и систолической недостаточности [20–22].
Нарушению транспорта ионов способствует также снижение активности PKA, экспрессии образования фосфорилированного PLB (p-PLB) при возрастании активности протеин фосфатазы1-α (РР1-α).
Активация РАС и развитие гипертрофии сердца сопровождаются увеличением экспрессии Са2+ регулирующих белков – кальпаина I, экспрессии и активности кальцинеурина, а также двух изоформ СаМРКII – СаМРКII-δA и СаМРК II-δВ, при снижении уровней мРНК и белка изоформы СаМРК II-δС [16, 17, 23].
Действие валсартана на патологические изменения при гипертрофии и СН
Снижение выраженности гипертрофии под влиянием валсартана может быть обусловлено прямым блокирующим эффектом действия Ан II на АТ1-рецептор и уменьшением выраженности эффектов норадреналина [24].
Действие сартанов на АТ1-рецепторы приводит к коррекции активности кальциевого цикла как на уровне Са2+-каналов наружной мембраны клетки, так и в SR-депо, что сопровождается улучшением сократимости кардиомиоцитов. Блокада взаимодействия Ан II с АТ1-рецепторами подавляет снижение активности SERCA2 при СН и восстанавливает регуляцию внутриклеточного уровня ионов Са2+ в SR-депо.
Получены также данные, согласно которым валсартан может оказывать благоприятное действие на структурно-функциональное состояние сердца при СН, вызванное перегрузкой объемом и давлением. Валсартан в этих условиях ингибировал сигнальные пути, связанные с кальпаином I, кальцинеурином, экспрессией и активностью двух изоформ СаМРКII-δ: СаМРКII-δA и СаМРКII-δВ, уменьшал выраженность гипертрофии миокарда [26]. Сходные данные были получены в другом исследовании, где валсартан улучшал функциональную активность сердца у кроликов с СН в результате снижения экспрессии белка СаMPKII и активности этого фермента [27].
Валсартан повышает активность PKA и снижает активность протеинфосфатазы 1-α (фосфатазы РР1-α) (18), что сопровождается увеличением уровня фосфорилированного фосфоламбана ser16-р-PLB, а это увеличивает активность SERCA2a и транспорт Са2+ в SR-депо. При этом валсартан предупреждает развитие ремоделирования левого желудочка и улучшает систолическую дисфункцию, вызываемую длительной перегрузкой объемом и давлением.
При гипертрофии, индуцированной норадреналином, валсартан повышает активность исходно сниженных АТФазы миозина, Na+/K+-АТФазы и Са2+-АТФаз плазматической мембраны и SR-депо, что восстанавливает градиенты ионов Na+, K+ и Са2+, благоприятно влияет на электрическую активность сердца и усиливает сократительную активность миокарда.
При экспериментальной СН, вызванной перегрузкой объемом и увеличенной частотой сердечных сокращений, валсартан увеличивал сниженную активность SERCA2 [18] и, соответственно, активный транспорт Са2+ в SR-депо, снижал высокий уровень Са2+ в цитоплазме во время диастолы, который при СН был обусловлен неконтролируемым выходом Са2+ из SR-депо, восстанавливал стехиометрию и состав компонентов макромолекулярного комплекса RyR2 [16]. Все эти процессы улучшали Са2+-цикл и функциональную активность кардиомиоцитов.
Одно из объяснений такого защитного действия валсартана связано с его влиянием на АТ1-рецепторы в пресинаптической мембране симпатических нейронов, окончания которых иннервируют миокард, в результате чего через пресинаптическую мембрану симпатических нервов меньше секретируется норадреналина и возрастает его обратный захват в нейроны, что увеличивает внутриклеточный пул норадреналина. Это снижает поступление норадреналина к кардиомиоцитам, уменьшается величина адренергического стимула, влияющего на β-адренорецепторы кардиомиоцитов и, соответственно, активация β-адренорецепторов. Это тормозит аденилатциклазу, снижает синтез сАМР и уменьшает активность сАМР-зависимой PKA, фосфорилирующей RyR. Падение при действии валсартана фосфорилирования димерного комплекса RyR2 усиливает связывание ряда белков, включая белок FK BP16.6 с RyR2, что способствует стабилизации закрытого состояния Са2+-каналов этого рецептора и уменьшает выход Са2+ из SR-депо в цитоплазму во время диастолы. Это улучшает расслабление кардиомиоцитов, сопряжение между электрической активностью и сократимостью кардиомиоцитов.
Таким образом, валсартан в какой-то мере имитирует действие блокаторов β-адренорецепторов. Только в случае валсартана его защитный эффект обусловлен снижением уровня биогенных аминов, что ослабляет их действие на β-адренорецепторы, в то время как сами блокаторы β-адренорецепторов препятствуют взаимодействию биогенных аминов с рецепторами. Конечный эффект – образование сАМР: снижается при действии как валсартана, так и β-адреноблокаторов, что уменьшает гиперфосфорилирование RyR2 и восстанавливает их нормальную активность.
Совокупность этих результатов позволяет сделать следующее заключение: валсартан эффективно подавляет экспрессию генов, продукты которых играют центральную роль во внутриклеточной Са2+-регуляции разнообразных клеточных процессов, контролирующих не только гипертрофию сердца, но и его ремоделирование, электромеханическое сопряжение и мышечное сокращение.
Важным аспектом действия валсартана служит влияние на фиброз и апоптоз кардиомиоцитов. Длительное действие Ан II и норадреналина стимулирует также деление фибробластов – основных источников коллагена, отложение которого в соединительной ткани лежит в основе развития фиброза сердца и развития нарушения диастолического расслабления. Кроме того, происходит активация генетически запрограммированной клеточной гибели – апоптоза, и суммарное число кардиомиоцитов снижается, что нарушает сократительную функцию миокарда. Фиброз и апоптоз являются теми процессами, которые переводят компенсированную гипертрофию в некомпенсированную, т.е. ее патологическую форму.
Валсартан снижает содержание коллагена в миокарде и уменьшает фиброз сердца. Это очень важный вид активности валсартана, поскольку фиброз сердца не только связан с переходом от компенсированной гипертрофии к СН, но и вызывает аритмии, случаи внезапной сердечной смерти и различные серьезные осложнения [24, 25]. Интерстициальная ткань в сердце отвечает за функциональную интеграцию кардиомиоцитов и структурную целостность сердца как органа, что способствует скоординированному ответу клеток и контролирует силу суммарного мышечного сокращения. Способность валсартана нейтрализовывать систолическую и диастолическую дисфункции левого желудочка, а также снижать АД, обусловлена в т.ч. уменьшением содержания коллагена во внеклеточной ткани сердца.
Валсартан снижает развитие апоптоза в гипертрофированном сердце, о чем свидетельствует восстановление индекса мышечной массы сердца под влиянием валсартана.
Влияние валсартана на нарушения активности МХ
Глубокие изменения энергетического обмена в кардиомиоцитах являются одной из главных причин изменения функциональной активности миокарда при различных сердечно-сосудистых заболеваниях [28]. Основным источником синтеза АТФ в сердце остаются МХ, активность которых зависит от ряда факторов и процессов. Неблагоприятное влияние на клетки оказывает воздействие различных внеклеточных факторов, включая некоторые лекарственные препараты, которые при длительном применении подавляют активность МХ и приводят к развитию кардиомиопатий.
Описаны защитные эффекты валсартана при действии цитотоксических лекарственных препаратов, подавляющих активность МХ, в частности широко используемого для противоопухолевой терапии цитостатика доксирубицина [29, 30], который в определенные фазы клеточного цикла прямо взаимодействует, подавляет активность и синтез ДНК.
Одной из главных причин кардиомиопатии, вызываемой доксорубицином, является поражение МХ, в которых увеличивается образование радикалов кислорода; накапливаются ионы Са2+ в просвете МХ; развивается энергетический дисбаланс, связанный с торможением синтеза АТФ и возрастанием скорости его потребления. В экспериментальных моделях кардиомиопатии, вызванной доксирубицином, валсартан снижал набухание МХ и образование свободных радикалов кислорода, а также увеличивал мембранный потенциал на внутренней мембране МХ и синтез АТФ. В результате частичного восстановления структуры и функциональной активности сердца валсартан оказывал защитное действие при кардиомиопатии, индуцированной доксорубицином. Эти результаты показывают, что МХ выступают как одна из главных мишеней действия не только доксорубицина, но и валсартана [29].
Принимая во внимание, что на внутренней мембране МХ экспрессируются АТ1- и АТ2-подобные рецепторы [5], можно предположить, что защитные эффекты валсартана в той или иной степени связаны с его влиянием на МХ, в т.ч. при острой ишемии. При острой ишемии валсартан восстанавливает функциональную активность МХ в кардиомиоцитах в результате сохранения структуры этих органелл, увеличивает мембранный потенциал на внутренней мембране МХ, усиливает потребление кислорода и фосфорилирование АДФ, что приводит к увеличению синтеза АТФ и подавляет механизмы, вызывающие набухание и разрушение МХ, а затем и кардиомиоцитов [31].
Влияние валсартана на инсулиновую резистентность адипоцитов. Роль тканевых макрофагов
Известно, что ингибиторы АПФ и особенно сартаны снижают риск развития сахарного диабета у пациентов с АГ [32, 33]. Снижению инсулинорезистентности при действии этих препаратов придают большое значение в восстановлении нарушенной эндотелиальной функции при АГ, ожирении, сахарном диабете. Влияние на инсулинорезистентность пытались объяснить действием некоторых сартанов на систему РРАR. В настоящее время установлено, что валсартан, не взаимодействующий с РРАR, может уменьшать инсулинорезистентность жировых клеток. Было установлено, что валсартан увеличивал в присутствии макрофагов чувствительность адипоцитов к инсулину [34]. Подобный инсулин-сенсибилизирующий эффект валсартана воспроизводился не только на лабораторных моделях, но и в клинических условиях.
Валсартан вызывал полное восстановление процесса фосфорилирования субстрата-1 инсулинового рецептора и протеинкиназы В (Akt протеинкиназы) и чувствительности адипоцитов к действию инсулина. Более того, валсартан выраженным образом подавлял эндотоксин-индуцированную продукцию цитокинов воспаления, включая интерлейкин-1β (ИЛ-1β), ИЛ-6 и фактор некроза опухоли α, а также активацию ядерного фактора NF-kB и фосфорилирования терминальной киназы c-Jun. Защитный эффект валсартана также наблюдался в макрофагах с дефектом АТ1-рецепторов в наружной мембране и блокадой активности фактора транскрипции генов PPARγ. Эти результаты свидетельствуют о том, что валсартан подавляет воспалительный ответ макрофагов в обход АТ1-рецепторов плазматических мембран и активации внутриклеточного PPARγ. Очевидно, что мишень такого защитного действия валсартана находится внутри клеток [34].
Таким образом, функциональная активность РАС осуществляется на разных уровнях организации в рамках эндокринной РААС; тканевых РАС, экспрессированных на поверхности клеток-мишеней и тканевых внутриклеточных РАС. Валсартан не только корректирует эффекты, связанные с АТ1-рецепторами, экспрессированными на плазматических мембранах клеток-мишеней, но и оказывает влияние на АТ1-подобные рецепторы, расположенные на ядерной мембране; внутренней мембране МХ и лизосом. Участие валсартана в интракринной активности Ан II изменяет активацию генов и экспрессию их продуктов, контролирующих мышечное сокращение; перемещение из лизосом на ядро разнообразных регуляторов активности генов, включая экзогенный Ан II; регуляцию внутриклеточного Са2+, связанную с выходом Са2+ из Са2+-депо; образование радикалов кислорода, оксида азота и АТФ в МХ; активацию мегапоры МХ, индуцирующую апоптоз или некроз клеток.
- 2013 ESH/ESC Guidelines for the management of arterial hypertension J. Hypertens. 2013;31:1281–357.
- Асташкин Е.И., Глезер М.Г. Ренин-ангиотензиновая система – новые звенья в старой цепи. М., 2010. 20 с.
- Асташкин Е.И. Глезер М.Г. Новые данные о функционировании ренин-ангиотензиновой системы: роль внутриклеточной (итракринной) системы. Проблемы женского здоровья. 2012;7(4):47–54.
- Re R.N. Lysosomal action of intracrine angiotensin II. Focus on “Intracellular angiotensin II activates rat myometrium. Am. J. Physiol. Cell Physiol. 2011;301(3):553–54.
- Abadir P.M., Foster D.B., Crow M., et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. 2011;108(36):1484–54.
- Tadevosyan A., Maguy A, Villeneuve L.R., et al. Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J. Biol. Chem. 2010;285(29):22338–49.
- Kumar R., Yong Q.C., Thomas C.M., Baker K.M. Intracardiac intracellular angiotensin system in diabetes . Am. J. Physiol. Reg. Integ. Comp. Physiol. 2012;302(5):R510–17.
- Асташкин Е.И. Глезер М.Г. Жирорастворимые ингибиторы АПФ – особенности влияния на РАС. М., 2012. 12 с.
- Hu D., Liu L., Li W. Efficacy and safety of valsartan/amlodipine single-pill combination in 11,422 chinese patients with hypertension: an observational study. Adv Ther. 2014;31(7):762–75.
- Sison J., Assaad-Khalil S.H., Najem R., et al. Real-world clinical experience of amlodipine/valsartan and amlodipine/valsartan/hydrochlorothiazide in hypertension: the EXCITE study. Curr. Med. Res. Opin. 2014;17:1–9.
- Maggioni A.P., Fabbri G. VALIANT (VALsartan In Acute myocardial iNfarcTion) trial. Expert Opin. Pharmacother. 2005;6(3):507–12.
- Cohn J.N., Tognoni G. Valsartan Heart Failure Trial Investigators A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001;345(23):1667–75.
- Beers D.M. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205.
- Kurokawa J., Abriel H. Neurohormonal regulation of cardiac ion channels in chronic heart failure. J. Cardiovasc. Pharmacol. 2009;54(2):98–105.
- Benitah J.P., Alvarez J.L., Gomez A.M. L-type Ca(2+) current in ventricular cardiomyocytes. J. Mol. Cell Cardiol. 2010;48(1):26–36.
- Yano M., Ikeda Y., Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J. Clin. Investigation. 2005;115(3):556–64.
- Luo M., Anderson ME Mechanism of altered Ca2+ handling in heart failure Circ. Res. 2013;113(6):690–708.
- Hasenfuss G., Pieske B. Calcium cycling in congestive heart failure. J. Mol. Cell Cardiol. 2002;34(8):951–69.
- Dedkova E.N., Blatter L.A. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44(1):77–91.
- Hobai I.A., O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsiblefor defective excitation-contraction couplingin canine heart failure. Circulation. 2001;103(11):1577–84.
- Lou Q, Fedorov V.V., Glukhov A.V., Transmural heterogeneity and remodeling of ventricular excitation-contraction coupling in human heart failure. Circulation. 2011;123(17):1881–90.
- Liu T., Takimoto E., Dimaano V.L., et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014;115(1):44–54.
- Rokita A.G., Anderson M.E. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. 2012;126(17):2125–39.
- Yang X., Jun H. Effect of valsartan and fosinopril on cathecholamine-induced catdiac hypertrophy. Acta Pharmacol. Sin. 2000;21(9):850–54.
- Liu T., Brown D.A., O’Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J. Mol. Cell Cardiol. 2010;49(5):728–36.
- Lu J.X., Zhu J.H., Qin X.T., et al. Role of valsartan on myocardial Calpain I, calceunerin and Ca/calmodulin-dependent protein kinase II-d expression of renovascular hypertensive rats. Zhonghua xin guan bing za zhi. 2012;40(6):511–15.
- Qu F.Z., Lio Z.H., Jiang B., et al. Valsartan improved cardiac function in heart failure rabbits probably via down regulations myocardial CaMKII expression and activity. Zhonghua xin guan bing za zhi. 2009;37(6):501–05.
- Асташкин Е.И. Глезер М.Г. Роль L-карнитина в энергетическом обмене кардиомиоцитов и лечении заболеваний сердечно-сосудистой системы Кардиология и сердечно-сосудистая хирургия. 2012;6(2):58–65.
- Nakamae H., Tsumura K., Terada Y., et al. Notable effects of angiotensin II receptor blocker, valsartan, on acute cardiotoxic changes after standard chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisolone. Cancer. 2005;104(11):2492–98.
- Xu Li, Zhang Ying, Wang Jian, et al., Effect of valsartan on rats’ heart mitochondria of adriamycin-induced cardiomyopathy. J. Jiangsu University (Medicine Edition). 2010–01.
- Monteiro P., Duarte A.I., Gonzalves L.M., Providencia L.A. Valsartan improves mitochondrial function in hearts submitted to acute ischemia. Eur. J. Pharmacol. 2005;518(2–3):158–64.
- Rizos C.V., Elisaf M.S. Antihypertensive drugs and glucose metabolism. World J. Cardiol. 2014;6(7):517–30.
- Глезер М.Г. Антигипертензивная терапия и развитие новых случаев сахарного диабета. Можно ли снизить риск? Кардиоваскулярная терапия и профилактика. 2008;7(8):85–93.
- Iwashita M., Sakoda H., Kushiyama A., et al. Valsartan, independently of AT1 receptor or PPAR-gamma suppresses LPS-indused macrophage activation and improves insulin resistance in cocultured adipocytes. Am. J. Physiol. Endocrinol. Metab. 2012;302(3):E286–96.
Е.И. Асташкин –д.б.н., проф. кафедры патологии, зав.лабораторией экстремальных состояний НИЦ ГБОУ ВПО «Первый МГМУ им.И.М.Сеченова» Минздрава России;e-mail: [email protected].
М.Г. Глезер – д.м.н., проф. кафедры профилактической и неотложной кардиологии, зав.лабораторией функциональных методов исследования и рациональной фармакотерапии сердечно-сосудистых заболеваний НИЦ ГБОУ ВПО «Первый МГМУ им.И.М.Сеченова» Минздрава России
Ренин-ангиотензиновая система: новая возможная мишень для депрессии | BMC Medicine
Jakubovski E, Bloch MH. Подгруппы прогнозирования ответа на циталопрам в исследовании STAR * D. J Clin Psychiatry. 2014; 75: 738–47.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Мойлан С., Берк М., Дин О.М., Самуни Ю., Уильямс Л.Дж., О’Нил А. и др. Окислительный и нитрозативный стресс при депрессии: зачем столько стресса? Neurosci Biobehav Rev.2014; 45: 46–62.
CAS
PubMed
Статья
Google Scholar
Гар, ПР, Мэнди А., Сатклифф, Массачусетс. Доказательства возможной роли измененной функции ангиотензина в лечении депрессии, но не этиологии. Биол Психиатрия. 1999; 45: 1030–4.
CAS
PubMed
Статья
Google Scholar
Гар Пр. Роль ангиотензина II в познании и поведении.Eur J Pharmacol. 2002; 438: 1–14.
CAS
PubMed
Статья
Google Scholar
Гар пр. Ренин-ангиотензиновая система мозга: мишень для новых антидепрессантов и анксиолитиков. Drug Dev Res. 2005; 65: 270–7.
CAS
Статья
Google Scholar
Saab YB, Gard PR, Yeoman MS, Mfarrej B, El-Moalem H, Ingram MJ. Полиморфизм генов ренин-ангиотензиновой системы и депрессия.Prog Neuropsychopharmacol Biol Psychiatry. 2007; 31: 1113–8.
CAS
PubMed
Статья
Google Scholar
Saavedra JM, Pavel J. Антагонисты рецептора AT1 ангиотензина II ингибируют ось ангиотензин-CRF-AVP и потенциально полезны для лечения связанных со стрессом и расстройств настроения. Drug Dev Res. 2005. 65: 237–69.
CAS
Статья
Google Scholar
Филлипс М.И., Шмидт-Отт К.М. Открытие ренина 100 лет назад. Новости Physiol Sci. 1999; 14: 271–4.
CAS
PubMed
Google Scholar
Гантен Д., Бушер Р., Дженест Дж. Активность ренина в мозговой ткани щенков и взрослых собак. Brain Res. 1971; 33: 557–9.
CAS
PubMed
Статья
Google Scholar
Гантен Д., Маркес-Хулио А., Грейнджер П., Хайдук К., Карсунки К.П., Бушер Р. и др.Ренин в мозгу собаки. Am J Physiol. 1971; 221: 1733–7.
CAS
PubMed
Google Scholar
Bickerton RK, Buckley JP. Доказательства центрального механизма гипертонии, индуцированной ангиотензином. Exp Biol Med. 1961; 106: 834–6.
CAS
Статья
Google Scholar
Багги Дж., Джонсон А.К. Ангиотензин-индуцированная жажда: эффекты обструкции третьего желудочка и перивентрикулярной абляции.Brain Res. 1978; 149: 117–28.
CAS
PubMed
Статья
Google Scholar
Филлипс М.И., Феликс Д. Специфические рецептивные нейроны к ангиотензину II в субфорном органе кошки. Brain Res. 1976; 109: 531–40.
CAS
PubMed
Статья
Google Scholar
Джонсон А.К., Эпштейн А.Н. Желудочки головного мозга как канал дипсогенного действия внутричерепного ангиотензина.Brain Res. 1975. 86: 399–418.
CAS
PubMed
Статья
Google Scholar
Джонс Э.С., Винь А., Маккарти, Калифорния, Гаспари Т.А., Виддоп РЭ. Рецепторы AT2: функциональное значение при сердечно-сосудистых заболеваниях. Pharmacol Ther. 2008. 120: 292–316.
CAS
PubMed
Статья
Google Scholar
Оро С., Цянь Х., Томас В.Г. Фармакология рецепторов ангиотензина 1 типа: передача сигналов за пределы G-белков.Pharmacol Ther. 2007. 113: 210–26.
CAS
PubMed
Статья
Google Scholar
Сантос Р.А., Сильва А.С. Се, Марич С., Сильва Д.М., Мачадо Р.П., де Бур И. и др. Ангиотензин- (1-7) является эндогенным лигандом для рецептора, связанного с G-белком. Mas Proc Natl Acad Sci U S. A. 2003; 100: 8258–63.
CAS
PubMed
Статья
Google Scholar
Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T., Sraer JD.Ключевая роль рецептора ренина / проренина в продукции ангиотензина II и клеточных ответах на ренин. J Clin Invest. 2002; 109: 1417–27.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Harding JW, Cook VI, Miller-Wing AV, Hanesworth JM, Sardinia MF, Hall KL, et al. Идентификация сайта связывания AII (3-8) [AIV] в гиппокампе морской свинки. Brain Res. 1992; 583: 340–3.
CAS
PubMed
Статья
Google Scholar
Чаки С., Инагами Т. Идентификация и характеристика нового сайта связывания ангиотензина II в нейробластомных клетках мыши нейро-2А. Biochem Biophys Res Commun. 1992; 182: 388–94.
CAS
PubMed
Статья
Google Scholar
Беники Дж., Санчес-Лемус Э, Хонда М., Панг Т., Орекна М., Ван Дж. И др. Блокада рецептора AT1 ангиотензина II уменьшает воспаление головного мозга. Нейропсихофармакология. 2011; 36: 857–70.
CAS
PubMed
Статья
Google Scholar
Сааведра Дж. М., Санчес-Лемус Э., Беники Дж. Блокада рецепторов AT1 ангиотензина II головного мозга уменьшает стресс, беспокойство, воспаление мозга и ишемию: терапевтические последствия. Психонейроэндокринология. 2011; 36: 1–18.
CAS
PubMed
Статья
Google Scholar
Джоглар Б., Родригес-Палларес Дж., Родригес-Перес А.И., Рей П., Герра М.Дж., Лабандейра-Гарсиа Дж.Л. Воспалительный ответ в модели болезни Паркинсона МРТР опосредуется ангиотензином головного мозга: имеет отношение к прогрессированию заболевания.J Neurochem. 2009; 109: 656–69.
CAS
PubMed
Статья
Google Scholar
Rodriguez-Pallares J, Rey P, Parga JA, Muñoz A, Guerra MJ, Labandeira-Garcia JL. Ангиотензин головного мозга увеличивает гибель дофаминергических клеток за счет активации микроглии и АФК, производных НАДФН. Neurobiol Dis. 2008; 31: 58–73.
CAS
PubMed
Статья
Google Scholar
Завада В.М., Баннингер Г.П., Торнтон Дж., Марриотт Б., Канту Д., Рачубински А.Л. и др.Генерация активных форм кислорода в допаминергических нейронах, обработанных 1-метил-4-фенилпиридинием (MPP +), происходит в виде двухволнового каскада, зависимого от НАДФН-оксидазы. J Нейровоспаление. 2011; 8: 129.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Родригес-Перес А.И., Домингес-Мейджиде А, Лансьего JL, Герра М.Дж., Лабандейра-Гарсия JL. Ингибирование киназы Rho опосредует нейрозащитные эффекты эстрогена в модели МРТР болезни Паркинсона.Neurobiol Dis. 2013; 58: 209–19.
CAS
PubMed
Статья
Google Scholar
Тонгес Л., Франк Т., Татенхорст Л., Заал К.А., Кох Дж. К., Сего ЭМ и др. Ингибирование ро-киназы увеличивает выживаемость дофаминергических нейронов и снижает потерю аксонов на мышиной модели болезни Паркинсона. Мозг. 2012; 135: 3355–70.
PubMed
PubMed Central
Статья
Google Scholar
Villar-Cheda B, Valenzuela R, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Связанные со старением изменения в системе черного ангиотензина усиливают провоспалительные и прооксидантные маркеры и индуцированную 6-OHDA дофаминергическую дегенерацию. Neurobiol Aging. 2012; 33: e1 – e11.
Артикул
CAS
Google Scholar
Borrajo A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL. Микроглиальный TNF-α опосредует усиление дофаминергической дегенерации ангиотензином головного мозга.Глия. 2014; 62: 145–57.
PubMed
Статья
Google Scholar
de Souza Gomes JA, de Souza GC, Berk M, Cavalcante LM, de Sousa FC, Budni J, et al. Антиманиакальная активность кандесартана у мышей: возможное участие антиоксидантных, противовоспалительных и нейротрофических механизмов. Eur Neuropsychopharmacol. 2015; 25: 2086–97.
PubMed
Статья
CAS
Google Scholar
Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, et al. Повышенный уровень циркулирующего фактора некроза опухоли при болезни Альцгеймера. Neurosci Lett. 1991; 129: 318–20.
CAS
PubMed
Статья
Google Scholar
Hofman FM, Hinton DR, Johnson K, Merrill JE. Фактор некроза опухоли, выявленный при рассеянном склерозе головного мозга. J Exp Med. 1989; 170: 607–12.
CAS
PubMed
Статья
Google Scholar
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Фактор некроза опухоли альфа (TNF-альфа) увеличивается как в головном мозге, так и в спинномозговой жидкости у пациентов с паркинсонизмом. Neurosci Lett. 1994; 165: 208–10.
CAS
PubMed
Статья
Google Scholar
Агилера Г., Скотт Янг В., Кисс А., Батия А. Прямая регуляция нейронов гипоталамического кортикотропин-рилизинг-гормона ангиотензином II. Нейроэндокринология.1995; 61: 437–44.
CAS
PubMed
Статья
Google Scholar
Сумитомо Т., Суда Т., Накано И., Тозава Ф., Ямада М., Демура Х. Ангиотензин II увеличивает уровень рибонуклеиновой кислоты, посланника рибонуклеиновой кислоты, высвобождающего кортикотропин, в гипоталамусе крысы. Эндокринология. 1991; 128: 2248–52.
CAS
PubMed
Статья
Google Scholar
Aguilera G, Kiss A, Luo X.Повышенная экспрессия рецепторов ангиотензина II типа 1 в паравентрикулярном ядре гипоталамуса после стресса и введения глюкокортикоидов. J Neuroendocrinol. 1995; 7: 775–83.
CAS
PubMed
Статья
Google Scholar
Wincewicz D, Juchniewicz A, Waszkiewicz N, Braszko JJ. Блокада рецептора ангиотензина II типа 1 телмисартаном предотвращает вызванное стрессом ухудшение памяти за счет деактивации оси HPA и усиления экспрессии гена нейротрофического фактора мозга.Pharmacol Biochem Behav. 2016; 148: 108–18.
CAS
PubMed
Статья
Google Scholar
Balla T, Baukal AJ, Eng S, Catt KJ. Подтипы рецепторов ангиотензина II и биологические ответы в коре и мозговом веществе надпочечников. Mol Pharmacol. 1991; 40: 401–6.
CAS
PubMed
Google Scholar
Geerling JC, Loewy AD. Альдостерон в головном мозге. Am J Physiol Ren Physiol.2009; 297: F559–76.
CAS
Статья
Google Scholar
Мурк Х., Шюсслер П., Штайгер А. Ренин-ангиотензин-альдостероновая система: забытая гормональная система стресса: связь с депрессией и сном. Фармакопсихиатрия. 2012; 45: 83–95.
CAS
PubMed
Статья
Google Scholar
Gomez-Sanchez CE, Zhou MY, Cozza EN, Morita H, Foecking MF, Gomez-Sanchez EP.Биосинтез альдостерона в головном мозге крысы. Эндокринология. 1997. 138: 3369–73.
CAS
PubMed
Статья
Google Scholar
Райт Дж. У., Хардинг Дж. У. Ренин-ангиотензин мозга — новый взгляд на старую систему. Prog Neurobiol. 2011; 95: 49–67.
CAS
PubMed
Статья
Google Scholar
de Kloet AD, Wang L, Ludin JA, Smith JA, Pioquinto DJ, Hiller H, et al.Репортерная линия мышей позволяет по-новому взглянуть на распределение рецепторов ангиотензина типа 2 в центральной нервной системе. Функция структуры мозга. 2016; 221: 891–912.
PubMed
Статья
CAS
Google Scholar
Yu L, Shao C, Gao L. Паттерны экспрессии развития рецепторов ангиотензина в коже и головном мозге мышей. J Renin Angiotensin Aldosterone Syst. 2014; 15: 139–49.
PubMed
Статья
CAS
Google Scholar
Мао Ц., Ши Л., Сюй Ф., Чжан Л., Сюй З. Развитие ренин-ангиотензиновой системы мозга плода и гипертония, запрограммированная в зародышевом происхождении. Prog Neurobiol. 2009. 87: 252–63.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Meffert S, Stoll M, Steckelings UM, Bottari SP, Unger T. Рецептор AT2 ангиотензина II подавляет пролиферацию и способствует дифференцировке в клетках PC12W. Mol Cell Endocrinol. 1996. 122: 59–67.
CAS
PubMed
Статья
Google Scholar
Ли Дж. М., Моги М., Цукуда К., Томочика Х., Иванами Дж., Мин Л. Дж. И др. Индуцированная ангиотензином II нейральная дифференцировка через каскад рецептор ангиотензина II типа 2 (AT2)-MMS2, включающий взаимодействие между белком, взаимодействующим с рецептором AT2, и протеином-тирозинфосфатазой, содержащим домен Src-гомологии 2, 1. Мол эндокринол. 2007; 21: 499–511.
PubMed
Статья
CAS
Google Scholar
Stroth U, Meffert S, Gallinat S, Unger T. Ангиотензин II и NGF по-разному влияют на белки микротрубочек в клетках PC12W: роль рецептора AT2. Mol Brain Res. 1998. 53: 187–95.
CAS
PubMed
Статья
Google Scholar
Gendron L, Laflamme L, Rivard N, Asselin C, Payet MD, Gallo-Payet N. Сигналы от рецептора AT2 (ангиотензин типа 2) ангиотензина II ингибируют p21ras и активируют MAPK (митоген-активированная протеинкиназа ), чтобы вызвать морфологическую дифференцировку нейронов в клетках NG108-15.Мол Эндокринол. 1999; 13: 1615–26.
CAS
PubMed
Статья
Google Scholar
Cernes R, Mashavi M, Zimlichman R. Дифференциальный клинический профиль кандесартана по сравнению с другими блокаторами рецепторов ангиотензина. Vasc Health Risk Man. 2011; 7: 749–59.
CAS
Google Scholar
Алхусбан А., Фауда А.Ю., Бинду П., Ишрат Т., Солиман С., Фаган СК. Соединение 21 является проангиогеном для мозга и приводит к устойчивому выздоровлению после ишемического инсульта.J Hypertens. 2015; 33: 170–80.
CAS
PubMed
Статья
Google Scholar
Джозеф Дж. П., Мекка А. П., Регенхардт Р. В., Беннион Д. М., Родригес В., Десланд Ф. и др. Агонист рецептора ангиотензина типа 2 соединение 21 вызывает церебропротектор при ишемическом инсульте, вызванном эндотелином-1. Нейрофармакология. 2014; 81: 134–41.
CAS
PubMed
Статья
Google Scholar
McCarthy CA, Vinh A, Miller AA, Hallberg A, Alterman M, Callaway JK, et al. Прямая стимуляция рецептора ангиотензина AT2 с использованием нового агониста рецептора AT2, соединения 21, вызывает нейрозащиту у находящихся в сознании крыс с гипертензией. PLoS One. 2014; 9, e
.
PubMed
PubMed Central
Статья
CAS
Google Scholar
Мин Л.Дж., Моги М., Цукуда К., Цзин Ф., Охшима К., Накаока Х. и др. Прямая стимуляция рецептора ангиотензина II типа 2, инициированная после инсульта, улучшает ишемическое повреждение головного мозга.Am J Hypertens. 2014; 27: 1036–44.
PubMed
Статья
Google Scholar
Schwengel K, Namsolleck P, Lucht K, Clausen BH, Lambertsen KL, Valero-Esquitino V, et al. Стимуляция рецептора ангиотензина AT2 улучшает выживаемость и неврологический исход после экспериментального инсульта у мышей. J Mol Med (Берл). 2016; 94: 957–66.
CAS
Статья
Google Scholar
Fouda AY, Pillai B, Dhandapani KM, Ergul A, Fagan SC. Роль интерлейкина-10 в нейрозащитном эффекте агониста рецептора ангиотензина 2 типа, соединения 21, после ишемического / реперфузионного повреждения. Eur J Pharmacol. 2017; 799: 128–34.
CAS
PubMed
Статья
Google Scholar
Mascolo A, Sessa M, Scavone C, De Angelis A, Vitale C, Berrino L, et al. Новые и старые роли периферической и мозговой ренин-ангиотензин-альдостероновой системы (РААС): внимание к сердечно-сосудистым и неврологическим заболеваниям.Int J Cardiol. 2017; 227: 734–42.
CAS
PubMed
Статья
Google Scholar
Young D, Waitches G, Birchmeier C, Fasano O, Wigler M. Выделение и характеристика нового клеточного онкогена, кодирующего белок с множеством потенциальных трансмембранных доменов. Клетка. 1986; 45: 711–9.
CAS
PubMed
Статья
Google Scholar
Карник С.С., Унал Х., Кемп Дж.Р., Тирупула К.С., Эгучи С., Вандерхейден П.М. и др.Международный союз фундаментальной и клинической фармакологии. XCIX. Рецепторы ангиотензина: интерпретаторы патофизиологических ангиотензинергических стимулов. Pharmacol Rev.2015; 67: 754–819.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Сингх К.Д., Карник С.С. Рецепторы ангиотензина: структура, функция, передача сигналов и клиническое применение. J Cell Signal. 2016; 1: 1–8.
Google Scholar
Xu Q, Jensen DD, Peng H, Feng Y. Критическая роль (про) рецептора ренина центральной нервной системы в регулировании системного кровяного давления. Pharmacol Ther. 2016; 164: 126–34.
CAS
PubMed
Статья
Google Scholar
Куадра А.Е., Шан З., Самнерс К., Райзада МК. Современный взгляд на ренин-ангиотензиновую систему мозга: является ли (про) рениновый рецептор недостающим звеном? Pharmacol Ther. 2010; 125: 27–38.
CAS
PubMed
Статья
Google Scholar
Лью Р.А., Мустафа Т., Йе С., МакДауэлл С.Г., Чай С.И., Альбистон А.Л. Лиганды ангиотензина AT4 являются мощными конкурентными ингибиторами регулируемой инсулином аминопептидазы (IRAP). J Neurochem. 2003. 86: 344–50.
CAS
PubMed
Статья
Google Scholar
Loyens E, De Bundel D, Demaegdt H, Chai SY, Vanderheyden P, Michotte Y, et al. Подобные антидепрессантам эффекты окситоцина у мышей зависят от присутствия регулируемой инсулином аминопептидазы.Int J Neuropsychopharmacol. 2013; 16: 1153–63.
CAS
PubMed
Статья
Google Scholar
Альбистон А.Л., Фернандо Р.Н., Йитман Х.Р., Бернс П., Нг Л., Дасвани Д. и др. Нокаут гена инсулино-регулируемой аминопептидазы: потеря специфического сайта связывания ангиотензина IV и возрастной дефицит пространственной памяти. Neurobiol Learn Mem. 2010; 93: 19–30.
CAS
PubMed
Статья
Google Scholar
Tan PS, Killinger S, Horiuchi J, Dampney RA. Модуляция барорецепторного рефлекса циркулирующим ангиотензином II опосредуется рецепторами AT1 в солитарном ядре тракта. Am J Physiol Regul Integr Comp Physiol. 2007; 293: R2267–78.
CAS
PubMed
Статья
Google Scholar
Savoia C, Schiffrin EL. Воспаление сосудов при гипертонии и диабете: молекулярные механизмы и терапевтические вмешательства. Clin Sci (Лондон).2007; 112: 375–84.
CAS
Статья
Google Scholar
Ozacmak VH, Sayan H, Cetin A, Akyildiz-Igdem A. Блокатор рецепторов AT1 кандесартан — индуцированное ослабление повреждения головного мозга крыс, подвергшихся хронической гипоперфузии головного мозга. Neurochem Res. 2007. 32: 1314–21.
CAS
PubMed
Статья
Google Scholar
Андо Х., Чжоу Дж., Макова М., Имбоден Х., Сааведра Дж. М..Блокада рецептора AT1 ангиотензина II обращает вспять патологическую гипертрофию и воспаление в микрососудах головного мозга крыс со спонтанной гипертензией. Гладить. 2004; 35: 1726–31.
CAS
PubMed
Статья
Google Scholar
Nishimura Y, Ito T, Hoe KL, Saavedra JM. Хроническое периферическое введение антагониста рецепторов AT1 ангиотензина II кандесартана блокирует рецепторы AT1 мозга. Brain Res. 2000; 871: 29–38.
CAS
PubMed
Статья
Google Scholar
Ямакава Х., Джезова М., Андо Х., Сааведра Дж. М.. Нормализация экспрессии эндотелиальной и индуцибельной синтазы оксида азота в микрососудах головного мозга крыс со спонтанной гипертензией путем ингибирования рецептора AT1 ангиотензина II. J Cereb Blood Flow Metab. 2003; 23: 371–80.
CAS
PubMed
Статья
Google Scholar
Чжоу Дж., Андо Х., Макова М., Доу Дж., Сааведра Дж. М.. Блокада рецептора AT1 ангиотензина II устраняет воспаление микрососудов головного мозга и ответы белков теплового шока у гипертонических крыс.J Cereb Blood Flow Metab. 2005. 25: 878–86.
CAS
PubMed
Статья
Google Scholar
Бианкарди В.К., Стерн Дж. Э. Нарушение проницаемости гематоэнцефалического барьера: новый механизм, с помощью которого циркулирующий ангиотензин II передает сигналы симпато-возбуждающим центрам во время гипертонии. J Physiol. 2016; 594: 1591–600.
CAS
PubMed
Статья
Google Scholar
Saavedra JM. Ангиотензин II мозга: новые разработки, вопросы без ответов и терапевтические возможности. Cell Mol Neurobiol. 2005. 25: 485–512.
CAS
PubMed
Статья
Google Scholar
van Thiel BS, Góes Martini A, Te Riet L, Severs D, Uijl E, Garrelds IM, et al. Ренин-ангиотензиновая система мозга: существует ли? Гипертония. 2017; 69: 1136–44.
PubMed
Статья
CAS
Google Scholar
Grobe JL, Xu D, Sigmund CD. Внутриклеточная ренин-ангиотензиновая система в нейронах: факт, гипотеза или фантазия. Физиология (Bethesda). 2008; 23: 187–93.
CAS
Статья
Google Scholar
Охруи Т., Томита Н., Сато-Накагава Т., Мацуи Т., Маруяма М., Нива К. и др. Влияние проникающих в мозг ингибиторов АПФ на прогрессирование болезни Альцгеймера. Неврология. 2004; 63: 1324–5.
CAS
PubMed
Статья
Google Scholar
Раковина KM, Ленг X, Уильямсон Дж., Кричевский С.Б., Яффе К., Куллер Л. и др. Ингибиторы ангиотензин-превращающего фермента и снижение когнитивных функций у пожилых людей с гипертонией: результаты исследования сердечно-сосудистой системы. Arch Intern Med. 2009; 169: 1195–202.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Йонг Ф.В., Ривест С. Использование системной иммунной системы для лечения болезней мозга. Нейрон.2009; 64: 55–60.
CAS
PubMed
Статья
Google Scholar
Licinio J, Wong ML. Пути и механизмы передачи сигналов цитокинами центральной нервной системы. J Clin Invest. 1997; 100: 2941–7.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Brietzke E, Stertz L, Fernandes BS, Kauer-Sant’anna M, Mascarenhas M, Escosteguy Vargas A, et al.Сравнение уровней цитокинов у пациентов с депрессией, маниакальным синдромом и эутимией с биполярным расстройством. J влияет на Disord. 2009; 116: 214–7.
CAS
PubMed
Статья
Google Scholar
Fernandes BS, Molendijk ML, Köhler CA, Soares JC, Leite CM, Machado-Vieira R, et al. Нейротрофический фактор периферического мозга (BDNF) как биомаркер биполярного расстройства: метаанализ 52 исследований. BMC Med. 2015; 13: 289.
PubMed
PubMed Central
Статья
CAS
Google Scholar
de Oliveira GS, Ceresér KM, Fernandes BS, Kauer-Sant’Anna M, Fries GR, Stertz L, et al. Снижение нейротрофического фактора головного мозга у пациентов с биполярным расстройством, принимающих лекарства или не принимающих их. J Psychiatr Res. 2009; 43: 1171–4.
PubMed
Статья
Google Scholar
Fernandes BS, Gama CS, Ceresér KM, Yatham LN, Fries GR, Colpo G и др. Нейротрофический фактор головного мозга как индикатор состояния эпизодов настроения при биполярных расстройствах: систематический обзор и мета-регрессионный анализ.J Psychiatr Res. 2011; 45: 995–1004.
PubMed
Статья
Google Scholar
Фернандес Б.С., Берк М., Терк К.В., Штайнер Дж., Гонсалвес К. Снижение уровней нейротрофических факторов периферического мозга является биомаркером активности заболевания при основных психических расстройствах: сравнительный метаанализ. Мол Психиатрия. 2013; 19: 749–51.
Google Scholar
Фернандес Б.С., Штайнер Дж., Берк М., Молендийк М.Л., Гонсалес-Пинто А., Турк К.В. и др.Нейротрофический фактор периферического мозга при шизофрении и роль нейролептиков: метаанализ и последствия. Мол Психиатрия. 2015; 20: 1108–19.
CAS
PubMed
Статья
Google Scholar
Бэррон М., Гартлон Дж., Доусон Л.А., Аткинсон П.Дж., Pardon MC. Состояние делирия: расшифровка влияния воспаления на тау-патологию при болезни Альцгеймера. Exp Gerontol. 2017; 94: 103–7.
CAS
PubMed
Статья
Google Scholar
О’Донован А., Ахмадиан А.Дж., Нейлан Т.С., Пакульт М.А., Эдмондсон Д., Коэн Б.Е. Текущее посттравматическое стрессовое расстройство и повышенная чувствительность к угрозам, связанная с повышенным воспалением, в исследовании Mind Your Heart Study. Иммунное поведение мозга. 2017; 60: 198–205.
PubMed
Статья
Google Scholar
Киркпатрик Б., Миллер Б.Дж. Воспаление и шизофрения. Шизофр Бык. 2013; 39: 1174–9.
PubMed
PubMed Central
Статья
Google Scholar
Фернандес Б.С., Штайнер Дж., Бернштейн Х.Г., Додд С., Паско Дж. А., Дин О.М. и др. С-реактивный белок повышается при шизофрении, но не изменяется под действием нейролептиков: метаанализ и последствия. Мол Психиатрия. 2016; 21: 554–64.
CAS
PubMed
Статья
Google Scholar
Madore C, Leyrolle Q, Lacabanne C, Benmamar-Badel A, Joffre C, Nadjar A, et al. Нейровоспаление при аутизме: вероятная роль материнского воспаления, диетических омега-3 и микробиоты.Neural Plast. 2016; 2016: 3597209.
PubMed
PubMed Central
Статья
Google Scholar
Де Вирджилио А., Греко А., Фаббрини Г., Ингилери М., Риццо М.И., Галло А. и др. Болезнь Паркинсона: аутоиммунитет и нейровоспаление. Autoimmun Rev.2016; 15: 1005–11.
PubMed
Статья
CAS
Google Scholar
Hong S, Banks WA. Роль иммунной системы в ВИЧ-ассоциированном нейровоспалении и нейрокогнитивных последствиях.Иммунное поведение мозга. 2015; 45: 1–12.
CAS
PubMed
Статья
Google Scholar
McKee CA, Lukens JR. Новые роли иммунной системы при черепно-мозговой травме. Фронт Иммунол. 2016; 7: 556.
PubMed
PubMed Central
Статья
Google Scholar
Селми К., Барин Дж. Г., Роуз Н. Р.. Современные тенденции аутоиммунитета и нервной системы.J Autoimmun. 2016; 75: 20–9.
CAS
PubMed
Статья
Google Scholar
Ransohoff RM. Как нейровоспаление способствует нейродегенерации. Наука. 2016; 353: 777–83.
CAS
PubMed
Статья
Google Scholar
Берк М., Уильямс Л.Дж., Джека Ф.Н., О’Нил А., Паско Дж. А., Мойлан С. и др. Итак, депрессия — это воспалительное заболевание, но откуда взялось воспаление? BMC Med.2013; 11: 200.
PubMed
PubMed Central
Статья
CAS
Google Scholar
Леонард Б., Маес М. Механистические объяснения того, как клеточно-опосредованная активация иммунной системы, воспаление и пути окислительного и нитрозативного стресса, а также их последствия и сопутствующие факторы играют роль в патофизиологии униполярной депрессии. Neurosci Biobehav Rev.2012; 36: 764–85.
CAS
PubMed
Статья
Google Scholar
Fernandes BS, Steiner J, Molendijk ML, Dodd S, Nardin P, Gonçalves CA и др. Концентрации С-реактивного белка в спектре настроения при биполярном расстройстве: систематический обзор и метаанализ. Ланцетная психиатрия. 2016; 3: 1147–56.
PubMed
Статья
Google Scholar
Слепченко А., Маес М., Келер К.А., Андерсон Дж., Кеведо Дж., Алвес Г.С. и др. Клетки Т-хелперов 17 могут управлять нейропрогрессией при большом депрессивном расстройстве: предложение интегративной модели.Neurosci Biobehav Rev.2016; 64: 83–100.
CAS
PubMed
Статья
Google Scholar
Андреацца А.С., Кауэр-Сант’анна М., Фрей Б.Н., Бонд Д.Д., Капчински Ф., Янг Л.Т. и др. Маркеры окислительного стресса при биполярном расстройстве: метаанализ. J влияет на Disord. 2008; 111: 135–44.
CAS
PubMed
Статья
Google Scholar
Dowlati Y, Herrmann N, Swardfager W., Liu H, Sham L, Reim EK, et al.Мета-анализ цитокинов при большой депрессии. Биол Психиатрия. 2010. 67: 446–57.
CAS
PubMed
Статья
Google Scholar
Köhler CA, Freitas TH, Maes M, de Andrade NQ, Liu CS, Fernandes BS, et al. Изменения периферических цитокинов и хемокинов при депрессии: метаанализ 82 исследований. Acta Psychiatr Scand. 2017; 135: 373–87.
PubMed
Статья
CAS
Google Scholar
Hannestad J, DellaGioia N, Bloch M. Влияние лечения антидепрессантами на уровни воспалительных цитокинов в сыворотке: метаанализ. Нейропсихофармакология. 2011; 36: 2452–9.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Эллер Т., Вазар В., Шлик Дж., Марон Э. Провоспалительные цитокины и ответ на лечение эсциталопрсамом при большом депрессивном расстройстве. Prog Neuropsychopharmacology Biol Psychiatry.2008. 32: 445–50.
CAS
Статья
Google Scholar
Паско Дж. А., Паско Дж. А., Джека Ф. Н., Уильямс Л. Дж., Генри М. Дж., Николсон Г. К., Котович М. А. и др. Клинические последствия цитокиновой гипотезы депрессии: связь между использованием статинов и аспирина и риском большой депрессии. Psychother Psychosom. 2010; 79: 323–5.
PubMed
Статья
Google Scholar
Валканова В, Эбмайер К.П., Аллан КЛ. CRP, IL-6 и депрессия: систематический обзор и метаанализ продольных исследований. J влияет на Disord. 2013; 150: 736–44.
CAS
PubMed
Статья
Google Scholar
Виум-Андерсен М.К., Орстед Д.Д., Нордестгаард Б.Г. Повышенный уровень С-реактивного белка, депрессия, соматические заболевания и общая смертность: менделевское рандомизированное исследование. Биол Психиатрия. 2014; 76: 249–57.
CAS
PubMed
Статья
Google Scholar
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF и др. Рандомизированное контролируемое исследование инфликсимаба, антагониста фактора некроза опухолей, для лечения резистентной депрессии. JAMA Psychiat. 2013; 70: 31–41.
CAS
Статья
Google Scholar
Макино М., Китано Ю., Хирохаши М., Такасуна К. Повышение неподвижности в тесте принудительного плавания мышей путем обработки интерфероном человека. Eur J Pharmacol.1998; 356: 1–7.
CAS
PubMed
Статья
Google Scholar
Макино М., Китано Ю., Комияма К., Такасуна К. Человеческий интерферон-альфа увеличивает неподвижность в тесте принудительного плавания у крыс. Психофармакология (Берл). 2000; 148: 106–10.
CAS
Статья
Google Scholar
Ping F, Shang J, Zhou J, Zhang H, Zhang L. Рецептор 5-HT (1A) и апоптоз способствуют индуцированному интерфероном-α «депрессивному» поведению у мышей.Neurosci Lett. 2012; 514: 173–8.
CAS
PubMed
Статья
Google Scholar
Фишер К.В., Эскелунд А., Будак Д.П., Тиллманн С., Либенберг Н., Эльфвинг Б. и др. Лечение интерфероном-альфа вызывает депрессивное поведение, сопровождающееся повышенным уровнем хинолиновой кислоты в гиппокампе у крыс. Behav Brain Res. 2015; 293: 166–72.
CAS
PubMed
Статья
Google Scholar
Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Müller B, et al. Ингибитор циклооксигеназы-2 целекоксиб оказывает терапевтическое действие при большой депрессии: результаты двойного слепого рандомизированного плацебо-контролируемого дополнительного пилотного исследования к ребоксетину. Мол Психиатрия. 2006; 11: 680–4.
PubMed
Статья
CAS
Google Scholar
Келер О., Бенрос М.Э., Нордентофт М., Фарку М.Э., Айенгар Р.Л., Морс О. и др.Влияние противовоспалительного лечения на депрессию, депрессивные симптомы и побочные эффекты. JAMA Psychiat. 2014; 71: 1381–91.
Артикул
Google Scholar
Ухер Р., Карвер С., Пауэр Р.А., Морс О, Майер В., Ритчел М. и др. Нестероидные противовоспалительные препараты и эффективность антидепрессантов при большом депрессивном расстройстве. Psychol Med. 2012; 42: 2027–35.
CAS
PubMed
Статья
Google Scholar
Тайринг С., Готлиб А., Папп К., Гордон К., Леонарди С., Ван А. и др. Этанерцепт и клинические исходы, утомляемость и депрессия при псориазе: двойное слепое плацебо-контролируемое рандомизированное исследование III фазы. Ланцет. 2006; 367: 29–35.
CAS
PubMed
Статья
Google Scholar
Ментер А., Огюстин М., Синьорович Дж., Ю. П., Ву Э. К., Гупта С. Р. и др. Влияние адалимумаба на уменьшение симптомов депрессии у пациентов с псориазом средней и тяжелой степени тяжести: рандомизированное клиническое исследование.J Am Acad Dermatol. 2010; 62: 812–8.
CAS
PubMed
Статья
Google Scholar
Эртенли И., Озер С., Кираз С., Апрас С.Б., Акдоган А., Карадаг О. и др. Инфликсимаб, лечение антагонистом TNF-альфа у пациентов с анкилозирующим спондилитом: влияние на депрессию, тревожность и качество жизни. Rheumatol Int. 2012; 32: 323–30.
CAS
PubMed
Статья
Google Scholar
Лэнгли Р.Г., Фельдман С.Р., Хан Ч., Шенкель Б., Сапари П., Хсу М.К. и др. Устекинумаб значительно улучшает симптомы тревоги, депрессии и качество жизни, связанное с кожей, у пациентов с псориазом средней и тяжелой степени тяжести: результаты рандомизированного двойного слепого плацебо-контролируемого исследования III фазы. J Am Acad Dermatol. 2010; 63: 457–65.
CAS
PubMed
Статья
Google Scholar
Карсон А., Демирташ Т., Байрамгюрлер Д., Балчи Ф., Уткан Т.Хроническое введение инфликсимаба (ингибитора TNF-α) снижает депрессию и тревожное поведение на крысиной модели хронического легкого стресса. Basic Clin Pharmacol Toxicol. 2013; 112: 335–40.
CAS
PubMed
Статья
Google Scholar
Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N. Укороченное начало действия антидепрессантов при большой депрессии с помощью увеличения ацетилсалициловой кислоты: пилотное открытое исследование.Int Clin Psychopharmacol. 2006; 21: 227–31.
PubMed
Статья
Google Scholar
Fond G, Hamdani N, Kapczinski F, Boukouaci W., Drancourt N, Dargel A, et al. Эффективность и переносимость дополнительной терапии противовоспалительными препаратами при основных психических расстройствах: систематический качественный обзор. Acta Psychiatr Scand. 2014; 129: 163–79.
CAS
PubMed
Статья
Google Scholar
Берк М., Дин О., Дрексхэдж Х., Макнил Дж. Дж., Мойлан С., О’Нил А. и др. Аспирин: обзор его нейробиологических свойств и терапевтического потенциала при психических заболеваниях. BMC Med. 2013; 11: 74.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Алмейда О.П., Фликер Л., Йип ББ, Альфонсо Х., МакКол К., Хэнки Г.Дж. Аспирин снижает риск депрессии у пожилых мужчин с высоким уровнем гомоцистеина в плазме. Перевод Психиатрия.2012; 2, с151.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Берк М., Дин О., Коттон С.М., Гама С.С., Капчински Ф., Фернандес Б.С. и др. Эффективность N-ацетилцистеина в качестве дополнительного лечения биполярной депрессии: открытое испытание. J влияет на Disord. 2011; 135: 389–94.
CAS
PubMed
Статья
Google Scholar
Берк М., Дин О.М., Коттон С.М., Дживонс С., Таниус М., Кольманн К. и др. Эффективность дополнительного N-ацетилцистеина при большом депрессивном расстройстве: двойное слепое рандомизированное плацебо-контролируемое исследование. J Clin Psychiatry. 2014; 75: 628–36.
CAS
PubMed
Статья
Google Scholar
Фернандес Б.С., Дин О.М., Додд С., Малхи Г.С., Берк М. N-ацетилцистеин в депрессивных симптомах и функциональных возможностях: систематический обзор и метаанализ.J Clin Psychiatry. 2016; 77: e457–66.
PubMed
Статья
Google Scholar
Wood WG, Mΰller WE, Eckert GP. Статины и нейропротекция: необходимы основы фармакологии. Mol Neurobiol. 2014; 50: 214–20.
CAS
PubMed
Статья
Google Scholar
Li Q, Zhuang QK, Yang JN, Zhang YY. Статины обладают нейропротекцией при церебральной ишемии независимо от их гиполипидемического действия: потенциальные молекулярные механизмы.Eur Rev Med Pharmacol Sci. 2014; 18: 1113–26.
CAS
PubMed
Google Scholar
Malfitano AM, Marasco G, Proto MC, Laezza C, Gazzerro P, Bifulco M. Статины при неврологических расстройствах: обзор и обновление. Pharmacol Res. 2014; 88: 74–83.
CAS
PubMed
Статья
Google Scholar
О’Нил А., Санна Л., Редлих К., Сандерсон К., Джека Ф., Уильямс Л.Дж. и др.Влияние статинов на психологическое благополучие: систематический обзор и метаанализ. BMC Med. 2012; 10: 154.
PubMed
PubMed Central
Статья
CAS
Google Scholar
Янг-Сюй Ю., Чан К.А., Ляо Дж.К., Равид С., Блатт К.М. Длительное употребление статинов и психологическое благополучие. J Am Coll Cardiol. 2003. 42: 690–7.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Salagre E, Fernandes BS, Dodd S, Brownstein DJ, Berk M. Статины для лечения депрессии: метаанализ рандомизированных двойных слепых плацебо-контролируемых исследований. J влияет на Disord. 2016; 200: 235–42.
CAS
PubMed
Статья
Google Scholar
Appleton KM, Rogers PJ, Ness AR. Обновленный систематический обзор и метаанализ эффектов длинноцепочечных полиненасыщенных жирных кислот n-3 на депрессивное настроение. Am J Clin Nutr.2010; 91: 757–70.
CAS
PubMed
Статья
Google Scholar
Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. Полиморфизм вставки / делеции в гене фермента, превращающего ангиотензин I, составляет половину дисперсии уровней фермента в сыворотке. J Clin Invest. 1990; 86: 1343–6.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Wu Y, Wang X, Shen X, Tan Z, Yuan Y. I / D-полиморфизм гена ангиотензин-превращающего фермента при большом депрессивном расстройстве и терапевтический результат: исследование случай-контроль и метаанализ. J влияет на Disord. 2012; 136: 971–8.
CAS
PubMed
Статья
Google Scholar
López-León S, Janssens AC, González-Zuloeta Ladd AM, Del-Favero J, Claes SJ, Oostra BA, et al. Метаанализ генетических исследований большого депрессивного расстройства.Мол Психиатрия. 2008; 13: 772–85.
PubMed
Статья
CAS
Google Scholar
Багхай Т.С., Биндер Э.Б., Шуле С., Салякина Д., Эсер Д., Лука С. и др. Полиморфизм гена ангиотензинпревращающего фермента связан с униполярной депрессией, активностью АПФ и гиперкортизолизмом. Мол Психиатрия. 2006; 11: 1003–15.
CAS
PubMed
Статья
Google Scholar
Angunsri R, Sritharathikhun T, Suttirat S, Tencomnao T. Ассоциация однонуклеотидных полиморфизмов и гаплотипов промотора гена ангиотензинпревращающего фермента с большой депрессией в популяции северо-востока Таиланда. J Renin Angiotensin Aldosterone Syst. 2009. 10: 179–84.
CAS
PubMed
Статья
Google Scholar
Firouzabadi N, Shafiei M, Bahramali E, Ebrahimi SA, Bakhshandeh H, Таджик Н. Ассоциация полиморфизма гена ангиотензинпревращающего фермента (ACE) с повышенной активностью ACE в сыворотке и большой депрессией у иранского населения.Psychiatry Res. 2012; 200: 336–42.
CAS
PubMed
Статья
Google Scholar
Ancelin ML, Carrière I, Scali J, Ritchie K, Chaudieu I, Ryan J. Варианты генов ангиотензинпревращающего фермента связаны как с секрецией кортизола, так и с депрессией в позднем возрасте. Перевод Психиатрия. 2013; 3, с322.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Бонди Б., Багхай Т.С., Зилл П., Шуле С., Эзер Д., Деймл Т. и др. Генетические варианты гена ангиотензин I-конвертирующего фермента (ACE) и рецептора ангиотензина II (AT1) и клинические исходы при депрессии. Prog Neuropsychopharmacology Biol Psychiatry. 2005; 29: 1094–9.
CAS
Статья
Google Scholar
Кондо Д.Г., Спир М.К., Кришнан К.Р., Маккуойд Д.Р., Слифер С.Х., Пипер К.Ф. и др. Связь AGTR1 с 18-месячным результатом лечения депрессии позднего возраста.Am J Гериатр Психиатрия. 2007; 15: 564–72.
PubMed
Статья
Google Scholar
Zill P, Baghai TC, Schüle C, Born C, Früstück C, Büttner A, et al. Анализ метилирования ДНК гена ангиотензинпревращающего фермента (АПФ) при большой депрессии. PLoS One. 2012; 7, e40479.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Bahramali E, Firouzabadi N, Yavarian I., Shayesteh MR, Erfani N, Shoushtari AA, et al.Влияние гена АПФ на дифференциальный ответ на сертралин по сравнению с флуоксетином у пациентов с большой депрессией: рандомизированное контролируемое исследование. Eur J Clin Pharmacol. 2016; 72: 1059–64.
CAS
PubMed
Статья
Google Scholar
Baghai TC, Schule C, Zill P, Deiml T., Eser D, Zwanzger P, et al. Полиморфизм вставки / делеции фермента, превращающего ангиотензин I, влияет на терапевтический результат у женщин с тяжелой депрессией, но не у мужчин.Neurosci Lett. 2004; 363: 38–42.
CAS
PubMed
Статья
Google Scholar
Hou Z, Yuan Y, Zhang Z, Hou G, You J, Bai F, et al. Аллель D полиморфизма инсерции / делеции АПФ связан с региональными изменениями объема белого вещества и когнитивными нарушениями при ремитированной гериатрической депрессии. Neurosci Lett. 2010; 479: 262–6.
CAS
PubMed
Статья
Google Scholar
Wang Z, Yuan Y, Bai F, You J, Li L, Zhang Z. Аномальная сеть по умолчанию у носителей аллеля ангиотензинпревращающего фермента D с ремитирующейся гериатрической депрессией. Behav Brain Res. 2012; 230: 325–32.
CAS
PubMed
Статья
Google Scholar
Fudalej S, Fudalej M, Kostrzewa G, Ku Franniar P, Franaszczyk M, Wojnar M, et al. Полиморфизм ангиотензинпревращающего фермента и завершенное самоубийство: ассоциация у кавказцев и доказательства связи с методом самоповреждения.Нейропсихобиология. 2009; 59: 151–8.
CAS
PubMed
Статья
Google Scholar
Sparks DL, Hunsaker 3rd JC, Amouyel P, Malafosse A, Bellivier F, Leboyer M, et al. Полиморфизм I / D фермента, превращающего ангиотензин I, и суицидальное поведение. Am J Med Genet B Neuropsychiatr Genet. 2009; 150B: 290–4.
CAS
PubMed
Статья
Google Scholar
Sonino N, Tomba E, Genesia ML, Bertello C, Mulatero P, Veglio F и др. Психологическая оценка первичного альдостеронизма: контролируемое исследование. J Clin Endocrinol Metab. 2011; 96: E878–83.
CAS
PubMed
Статья
Google Scholar
Künzel HE. Психопатологические симптомы у пациентов с первичным гиперальдостеронизмом — возможные пути. Horm Metab Res. 2012; 44: 202–7.
PubMed
Статья
CAS
Google Scholar
Hlavacova N, Wes PD, Ondrejcakova M, Flynn ME, Poundstone PK, Babic S, et al. Субхроническое лечение альдостероном вызывает депрессивное поведение и изменения экспрессии генов, связанные с большим депрессивным расстройством. Int J Neuropsychopharmacol. 2012; 15: 247–65.
CAS
PubMed
Статья
Google Scholar
Главацова Н., Джезова Д. Хроническое лечение минералокортикоидным гормоном альдостероном приводит к усилению тревожного поведения.Horm Behav. 2008; 54: 90–7.
CAS
PubMed
Статья
Google Scholar
Моррис MJ, Na ES, Grippo AJ, Johnson AK. Влияние индуцированного дезоксикортикостероном аппетита натрия на гедонистическое поведение крыс. Behav Neurosci. 2006; 120: 571–9.
CAS
PubMed
Статья
Google Scholar
Главацова Н., Джезова Д. Влияние однократной терапии антигипертензивным препаратом эплереноном на уровень гормонов и тревожное поведение у крыс.Endocr Regul. 2008; 42: 147–53.
CAS
PubMed
Google Scholar
Бюттнер М., Джезова Д., Грин Б., Конрад С., Кирхер Т., Мурк Н. и др. Выбор целевого биомаркера — биомаркеры, связанные с минералокортикоидными рецепторами, и исход лечения большой депрессии. J Psychiatr Res. 2015; 66–7: 24–37.
Артикул
Google Scholar
Сегеда В., Изакова Л., Главацова Н., Беднарова А., Езова Д.Концентрация альдостерона в слюне отражает продолжительность и тяжесть депрессивного эпизода в зависимости от пола. J Psychiatr Res. 2017; 91: 164–8.
CAS
PubMed
Статья
Google Scholar
Холлберг Л., Вестрин А., Исакссон А., Джанелидзе С., Трэскман-Бендз Л., Брундин Л. Снижение альдостерона в плазме у лиц, пытающихся покончить жизнь самоубийством с большим депрессивным расстройством. Psychiatry Res. 2011; 187: 135–9.
CAS
PubMed
Статья
Google Scholar
Франклин М., Главацова Н., Бабич С., Покуса М., Бермудес И., Джезова Д. Альдостерон сигнализирует о начале депрессивного поведения в модели депрессии у самок крыс наряду с устойчивостью к лечению СИОЗС. Нейроэндокринология. 2015; 102: 274–87.
CAS
PubMed
Статья
Google Scholar
Берк М., Ниренберг А.А. Три пути к открытию лекарств в психиатрии. Am J Psychiatry. 2015; 172: 412–4.
PubMed
Статья
Google Scholar
Thöne-Reineke C, Steckelings UM, Unger T. Блокаторы рецепторов ангиотензина и церебральная защита при инсульте. J Hypertens. 2006; 24: S115–21.
Артикул
CAS
Google Scholar
Benigni A, Cassis P, Remuzzi G. Повторный визит к ангиотензину II: новые роли в воспалении, иммунологии и старении. EMBO Mol Med. 2010; 2: 247–57.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Куме К., Ханью Х., Сакураи Х., Такада Й., Онума Т., Ивамото Т. Влияние телмисартана на познание и региональный церебральный кровоток у гипертоников с болезнью Альцгеймера. Гериатр Геронтол Инт. 2012; 12: 207–14.
PubMed
Статья
Google Scholar
Li NC, Lee A., Whitmer RA, Kivipelto M, Lawler E, Kazis LE, et al. Использование блокаторов рецепторов ангиотензина и риск деменции у преимущественно мужского населения: проспективный когортный анализ.BMJ. 2010; 340: b5465.
PubMed
PubMed Central
Статья
Google Scholar
Окуяма С., Сакагава Т., Сугияма Ф., Фукамизу А., Мураками К. Снижение депрессивного поведения у мышей, лишенных ангиотензиногена. Neurosci Lett. 1999; 261: 167–70.
CAS
PubMed
Статья
Google Scholar
Giardina WJ, Ebert DM. Положительные эффекты каптоприла в тесте плавания на основе поведенческого отчаяния.Биол Психиатрия. 1989; 25: 697–702.
CAS
PubMed
Статья
Google Scholar
Martin P, Massol J, Puech AJ. Каптоприл как антидепрессант? Влияние на парадигму выученной беспомощности у крыс. Биол Психиатрия. 1990; 27: 968–74.
CAS
PubMed
Статья
Google Scholar
Айюб М., Наджми А.К., Ахтар М. Защитный эффект ирбесартана антагониста рецептора ангиотензина (АТ1) при непредсказуемой хронической депрессии, вызванной умеренным стрессом, у мышей.Drug Res (Штутг). 2017; 67: 59–64.
CAS
Google Scholar
Ping G, Qian W, Song G, Zhaochun S. Валсартан обращает вспять депрессивное / тревожное поведение и индуцирует нейрогенез в гиппокампе и экспрессию белка BDNF у мышей с непредсказуемым хроническим умеренным стрессом. Pharmacol Biochem Behav. 2014; 124: 5–12.
CAS
PubMed
Статья
Google Scholar
Асвар Ю., Чепурвар С., Шинтре С., Асвар М. Телмисартан ослабляет депрессию, вызванную диабетом, у крыс. Pharmacol Reports. 2017; 69: 358–64.
CAS
Статья
Google Scholar
Saavedra JM, Armando I, Bregonzio C, Juorio A, Macova M, Pavel J, et al. Анксиолитический анксиолитический антагонист рецептора AT1 ангиотензина II центрального действия предотвращает вызванное изоляционным стрессом снижение кортикального рецептора CRF1 и связывания бензодиазепина.Нейропсихофармакология. 2006; 31: 1123–34.
CAS
PubMed
Google Scholar
Llano López LH, Caif F, García S, Fraile M, Landa AI, Baiardi G, et al. Анксиолитический эффект лозартана, введенного в миндалину крыс, подвергшихся острому стрессу. Pharmacol Reports. 2012; 64: 54–63.
Артикул
Google Scholar
Шринивасан Дж., Суреш Б., Раманатан М.Дифференциальный анксиолитический эффект эналаприла и лозартана у крыс с нормотензивной и почечной гипертензией. Physiol Behav. 2003. 78: 585–91.
CAS
PubMed
Статья
Google Scholar
Косталл Б., Домени А.М., Джеррард П.А., Горовиц З.П., Келли М.Э., Нейлор Р.Дж. и др. Влияние каптоприла и SQ29,852 на поведение, связанное с тревогой, у грызунов и мартышек. Pharmacol Biochem Behav. 1990; 36: 13–20.
CAS
PubMed
Статья
Google Scholar
Golding BJ, General ADJ, Gard PR. Различия в штаммах и роль экспрессии рецептора AT1 при тревоге. Int J Mol Epidemiol Genet. 2011; 2: 51–5.
CAS
PubMed
Google Scholar
Кангуссу Л.М., Алмейда-Сантос А.Ф., Морейра Ф.А., Фонтес МАП, Сантос РАН, Агияр округ Колумбия и др. Снижение тревожного поведения у трансгенных крыс с хронической гиперпродукцией ангиотензина (1-7): роль рецептора Mas. Behav Brain Res.2017; 331: 193–8.
CAS
PubMed
Статья
Google Scholar
Ван Л., де Клоет А.Д., Пати Д., Хиллер Х., Смит Дж. А., Пиоквинто Д. Д. и др. Повышение активности ангиотензинпревращающего фермента 2 головного мозга снижает тревожное поведение самцов мышей за счет активации центральных рецепторов Mas. Нейрофармакология. 2016; 105: 114–23.
PubMed
PubMed Central
Статья
CAS
Google Scholar
Bild W, Ciobica A. Центральное введение ангиотензина (1-7) вызывает анксиолитические эффекты в приподнятом крестообразном лабиринте и снижает окислительный стресс в миндалине. J влияет на Disord. 2013; 145: 165–71.
CAS
PubMed
Статья
Google Scholar
Алмейда-Сантос AF, Kangussu LM, Moreira FA, Santos RA, Aguiar DC, Campagnole-Santos MJ. Анксиолитические и антидепрессантные эффекты ангиотензина (1-7) у трансгенных (mRen2) крыс с гипертензией 27.Clin Sci (Лондон). 2016; 130: 1247–55.
CAS
Статья
Google Scholar
Torika N, Asraf K, Roasso E, Danon A, Fleisher-Berkovich S. Ингибиторы ангиотензинпревращающего фермента уменьшают воспаление мозга, связанное с активацией микроглии: возможные последствия для болезни Альцгеймера. J Neuroimmune Pharmacol. 2016; 11: 774–85.
PubMed
Статья
Google Scholar
Torika N, Asraf K, Danon A, Apte RN, Fleisher-Berkovich S. Телмисартан модулирует активацию глии: исследования in vitro и in vivo. PLoS One. 2016; 11, e0155823.
PubMed
PubMed Central
Статья
CAS
Google Scholar
Торика Н., Асраф К., Коэн Х., Флейшер-Беркович С. Интраназальный телмисартан улучшает патологию головного мозга у пяти мышей с семейной болезнью Альцгеймера. Иммунное поведение мозга. 2017; 64: 80–90.
CAS
PubMed
Статья
Google Scholar
Санчес-Лемус Э., Мураками Ю., Ларрайоз-Ролдан И.М., Мугамян А.Дж., Павел Дж., Нисиоку Т. и др. Блокада рецептора AT1 ангиотензина II снижает вызванное липополисахаридом воспаление в надпочечниках крыс. Эндокринология. 2008; 149: 5177–88.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Санчес-Лемус Э., Беники Дж., Павел Дж., Ларрайоз И.М., Чжоу Дж., Балиова М. и др. Блокада ангиотензина II AT1 снижает индуцированный липополисахаридом врожденный иммунный ответ в селезенке крысы.Am J Physiol Regul Integr Comp Physiol. 2009; 296: R1376–84.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Амброзино С.В. Депрессивные реакции, связанные с резерпином. Штат Нью-Йорк J Med. 1974; 74: 860–4.
CAS
Google Scholar
Bevacqua BK, Fattouh M, Backonja M. Депрессия, ночные кошмары и бессонница, связанные с длительной интратекальной терапией клонидином.Pain Pract. 2007; 7: 36–8.
PubMed
Статья
Google Scholar
Ганизаде А. Бессонница, ночной ужас и депрессия, связанные с клонидином при синдроме дефицита внимания / гиперактивности. J Clin Psychopharmacol. 2008. 28: 725–6.
PubMed
Статья
Google Scholar
Голдштейн Б.И., Карнетон М.Р., Мэтьюз К.А., Макинтайр Р.С., Миллер Г.Э., Рагхувеер Г. и др.Большое депрессивное расстройство и биполярное расстройство предрасполагают молодежь к ускоренному атеросклерозу и ранним сердечно-сосудистым заболеваниям: научное заявление Американской кардиологической ассоциации. Тираж. 2015; 132: 965–86.
PubMed
Статья
Google Scholar
Scalco AZ, Scalco MZ, Azul JBS, Lotufo NF. Гипертония и депрессия. Клиники (Сан-Паулу). 2005; 60: 241–50.
Артикул
Google Scholar
Johansen A, Holmen J, Stewart R, Bjerkeset O. Симптомы тревоги и депрессии при артериальной гипертензии: влияние антигипертензивного лечения. Исследование HUNT, Норвегия. Eur J Epidemiol. 2012; 27: 63–72.
CAS
PubMed
Статья
Google Scholar
Хаффман Дж.С., Стерн Т.А. Психоневрологические последствия сердечно-сосудистых препаратов. Диалоги Clin Neurosci. 2007; 9: 29–45.
PubMed
PubMed Central
Google Scholar
Long J, Duan G, Tian W, Wang L, Su P, Zhang W и др. Гипертония и риск депрессии у пожилых людей: метаанализ проспективных когортных исследований. J Hum Hypertens. 2015; 29: 478–82.
CAS
PubMed
Статья
Google Scholar
Musselman DL, Evans DL, Nemeroff CB. Связь депрессии с сердечно-сосудистыми заболеваниями: эпидемиология, биология и лечение. Arch Gen Psychiatry. 1998; 55: 580–92.
CAS
PubMed
Статья
Google Scholar
Meng L, Chen D, Yang Y, Zheng Y, Hui R. Депрессия увеличивает риск возникновения гипертонии. J Hypertens. 2012; 30: 842–51.
CAS
PubMed
Статья
Google Scholar
Зубенко Г.С., Никсон Р.А. Эффект каптоприла для улучшения настроения у пациентов с депрессией. Am J Psychiatry. 1984; 141: 110–1.
CAS
PubMed
Статья
Google Scholar
Deicken РФ. Каптоприл лечение депрессии. Биол Психиатрия. 1986; 21: 1425–8.
CAS
PubMed
Статья
Google Scholar
Germain L, Chouinard G. Лечение рецидивирующей униполярной большой депрессии каптоприлом. Биол Психиатрия. 1988; 23: 637–41.
CAS
PubMed
Статья
Google Scholar
Герцман М., Адлер Л.В., Арлинг Б., Керн М.Лизиноприл может усиливать антидепрессивный ответ. J Clin Psychopharmacol. 2005; 25: 618–20.
PubMed
Статья
Google Scholar
Ратманн В., Хаастерт Б., Роземан Дж. М., Джани Г. Рецепты сердечно-сосудистых препаратов и риск депрессии у пациентов с диабетом. J Clin Epidemiol. 1999; 52: 1103–9.
CAS
PubMed
Статья
Google Scholar
Уильямс Л.Дж., Паско Дж.А., Кессинг Л.В., Куирк С.Е., Фернандес Б.С., Берк М.Ингибиторы ангиотензинпревращающего фермента и риск расстройств настроения. Psychother Psychosom. 2016; 85: 250–2.
PubMed
Статья
Google Scholar
Boal AH, Smith DJ, McCallum L, Muir S, Touyz RM, Dominiczak AF, et al. Монотерапия основными классами гипотензивных препаратов и риск госпитализации по поводу аффективных расстройств. Гипертония. 2016; 68: 1132–8.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Habra ME, Baker B, Frasure-Smith N, Swenson JR, Koszycki D, Butler G и др. Первый эпизод большого депрессивного расстройства и сосудистые факторы у пациентов с ишемической болезнью сердца: исходные характеристики и ответ на лечение антидепрессантами в исследовании CREATE. J Psychosom Res. 2010; 69: 133–41.
PubMed
Статья
Google Scholar
Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ, et al.Сравнение миртазапина и нортриптилина после двух последовательных неудачных курсов лечения амбулаторных пациентов с депрессией: отчет STAR * D. Am J Psychiatry. 2006; 163: 1161–72.
PubMed
Статья
Google Scholar
Хантер А.М., Кук И.А., Лойхтер А.Ф. Влияет ли предшествующее лечение большой депрессии антидепрессантами на функцию мозга во время текущего лечения? Eur Neuropsychopharmacol. 2012; 22: 711–20.
CAS
PubMed
Статья
Google Scholar
Коэн Л. М., Андерсон Г., Фирнхабер В. Р.. Эналаприл и гипертония. Am J Psychiatry. 1984; 141: 1012–3.
CAS
PubMed
Статья
Google Scholar
Croog SH, Levine S, Testa MA, Brown B, Bulpitt CJ, Jenkins CD, et al. Влияние гипотензивной терапии на качество жизни. N Engl J Med. 1986; 314: 1657–64.
CAS
PubMed
Статья
Google Scholar
Testa MA, Андерсон РБ, Nackley JF, Холленберг NK. Качество жизни и гипотензивная терапия у мужчин. Сравнение каптоприла и эналаприла. Группа исследования качества жизни гипертонии. N Engl J Med. 1993; 328: 907–13.
CAS
PubMed
Статья
Google Scholar
Павлатоу М.Г., Масторакос Г., Лекакис И., Лиатис С., Вамваку Г., Зумакис Е. и др. Хроническое введение антагониста рецепторов ангиотензина II сбрасывает ось гипоталамус-гипофиз-надпочечники (HPA) и улучшает влияние пациентов с сахарным диабетом 2 типа: предварительные результаты.Стресс. 2008; 11: 62–72.
CAS
PubMed
Статья
Google Scholar
Каллендер Дж. С., Ходсман Г. П., Хатчесон М. Дж., Левер А. Ф., Робертсон Дж. И.. Изменения настроения во время терапии каптоприлом при гипертонии. Двойное слепое пилотное исследование. Гипертония. 1983; 5: III90–3.
CAS
PubMed
Статья
Google Scholar
Уважаемый I, Capewell S, Hajducka C, Muir A.Эффекты каптоприла и атенолола на память, обработку информации и настроение: двойное слепое перекрестное исследование. Br J Clin Pharmacol. 1991; 32: 347–53.
CAS
PubMed
PubMed Central
Статья
Google Scholar
Омвик П., Таулов Е., Херланд О. Б., Эйде I, Мидха Р., Тернер Р. Р.. Двойное слепое параллельное сравнительное исследование качества жизни во время лечения амлодипином или эналаприлом у пациентов с легкой или умеренной артериальной гипертензией: многоцентровое исследование.J Hypertens. 1993; 11: 103–13.
CAS
PubMed
Статья
Google Scholar
Fletcher AE, Bulpitt CJ, Chase DM, Collins WC, Furberg CD, Goggin TK, et al. Качество жизни после трех антигипертензивных препаратов. Цилазаприл, атенолол, нифедипин. Гипертония. 1992; 19: 499–507.
CAS
PubMed
Статья
Google Scholar
Weir MR, Elkins M, Liss C, Vrecenak AJ, Barr E, Edelman JM.Эффективность, переносимость и качество жизни лозартана отдельно или с гидрохлоротиазидом по сравнению с нифедипином GITS у пациентов с гипертонической болезнью. Clin Ther. 1996; 18: 411–28.
CAS
PubMed
Статья
Google Scholar
Миниобзор: Обзор ренин-ангиотензиновой системы — эндокринной и паракринной системы | Эндокринология
Аннотация
С момента открытия ренина в качестве прессорного вещества в 1898 году система ренин-ангиотензин (РАС) широко изучалась, поскольку она остается главным кандидатом в качестве причинного фактора в развитии и поддержании гипертонии.Действительно, некоторые свойства физиологически активного компонента РАС, ангиотензина II, включают сужение сосудов, регулирование почечного поглощения натрия и воды и усиление жажды. Первоначально считалось, что его влияние на артериальное давление опосредуется в первую очередь классическим эндокринным путем; то есть образование передающегося с кровью ангиотензина с действием на ткани-мишени. Однако в последнее время стало понятно, что локальный аутокринный или паракринный РАС может существовать в ряде тканей и что они также могут играть важную роль в регулировании кровяного давления.Некоторые трудности в изучении тканевого РАС связаны с ограничениями фармакологии, связанными с неразличением между препаратами РАС, полученными системно, и продуктами, синтезируемыми на месте. Однако создание трансгенных животных с высокоспецифичными промоторами для нацеливания RAS на определенные ткани предоставило важные инструменты для анализа этих систем. Таким образом, в этом мини-обзоре будут обсуждаться последние достижения в понимании взаимосвязи между эндокринной и паракринной (тканевой) РАС с использованием трансгенных моделей.
РЕНИН-АНГИОТЕНСИНОВАЯ СИСТЕМА (РАС) хорошо известна своей регуляцией кровяного давления и гомеостаза жидкости. Ангиотензин II (Ang-II), конечный эффектор системы, вызывает сужение сосудов как прямо, так и косвенно, стимулируя рецепторы рецептора Ang-II типа 1 (AT-1), присутствующие в сосудистой сети, и увеличивая симпатический тонус и высвобождение аргинина вазопрессина. В хроническом порядке Ang-II регулирует кровяное давление, напрямую модулируя реабсорбцию натрия и воды в почках, стимулируя рецепторы AT-1 в почках или косвенно, стимулируя выработку и высвобождение альдостерона надпочечниками или стимулируя чувство жажды в почках. центральная нервная система (ЦНС).Ферментативный каскад, с помощью которого продуцируется Ang-II, состоит из ренина (REN), аспартил-протеазы, которая расщепляет ангиотензиноген (AGT) с образованием декапептида ангиотензина I (Ang-I; рис. 1). Затем Ang-I расщепляется ангиотензин-превращающим ферментом (ACE), дипептидилкарбоксипептидазой, с образованием октапептида Ang-II, физиологически активного компонента системы. Дальнейшая деградация (или процессинг) аминопептидазой A и N продуцирует ангиотензин III (Ang 2-8) и ангиотензин IV (Ang 3-8) соответственно.Действие Ang-II является результатом его связывания со специфическими рецепторами (AT-1 и AT-2), классифицируемыми по их дифференциальному сродству к различным непептидным антагонистам (1). Оба этих рецептора клеточной поверхности принадлежат к большому семейству рецепторов, связанных с G-белком, хотя используемые пути совершенно разные и сигнализируют явно противоположно. Например, рецепторы AT-1 опосредуют вазоконстрикторные реакции, тогда как рецепторы AT-2, как полагают, опосредуют вазодилататорные реакции. Подробные обзоры передачи сигналов AT-1 и AT-2 опубликованы (2–4).Эти рецепторы имеют широкое тканеспецифическое распределение и оба присутствуют в почках, головном мозге и надпочечниках. В целом рецепторы AT-1 присутствуют в сердечно-сосудистых тканях взрослых, тогда как AT-2 высоко экспрессируется во время внутриутробного развития (5).
Фармакологические исследования с использованием специфических антагонистов определили, что большинство физиологических действий Ang-II опосредовано рецептором AT-1 (1). Два подтипа этого рецептора, AT-1a и AT-1b, были идентифицированы у крысы (6), мыши (7), а рецептор AT-1b был зарегистрирован у людей (8), хотя общепринято считать, что люди экспрессируют только один тип рецептора AT-1.Эти подтипы рецепторов фармакологически неразличимы и, как считается, сигнализируют идентично, но являются продуктом разных генов ( Agtr1a и Agtr1b ), которые по-разному экспрессируются и регулируются (9, 10). Именно это дифференциальное выражение, скорее всего, отличает функцию двух подтипов рецепторов. AT-1a является преобладающим рецептором в большинстве органов, тогда как AT-1b более распространен в надпочечниках и гипофизе (11). Эксперименты по нацеливанию на гены были полезны для определения индивидуальной роли AT-1a и AT-1b на периферии (12, 13) и в ЦНС (14).Рецепторы AT-1a преимущественно участвуют в регуляции сосудистого тонуса и реабсорбции натрия на периферии, а также в прессорной реакции на Ang-II в ЦНС, тогда как рецепторы AT-1b необходимы для дипсогенного ответа на Ang-II в организме. ЦНС. С другой стороны, функция рецептора AT-2 еще полностью не определена. Недавние исследования показали, что он может противодействовать действию рецептора AT-1 в отношении артериального давления и клеточной пролиферации (15). Также было высказано предположение, что стимуляция рецептора AT-2 снижает реабсорбцию натрия в канальцах почек (16), а у мышей с нокаутом рецептора AT-2 наблюдаются изменения в поведении (17).
Другие рецепторы были описаны в отношении RAS. Например, был идентифицирован сайт связывания AT-4, который, в отличие от других рецепторов AT, по-видимому, не является рецептором, связанным с G-белком (18). Этот рецептор преимущественно связывает Ang 3-8, он локализован в различных тканях млекопитающих и, как предполагается, вызывает расширение сосудов (18, 19). Совсем недавно о наличии рецептора REN также сообщалось в сердце, головном мозге, плаценте, почках и печени (20). Сообщается, что этот рецептор связывает как REN, так и проренин, и это связывание увеличивает каталитическую активность REN по расщеплению AGT, делая проренин активным.
Рисунок 1.
Каскад REN-ангиотензина. Классический каскад РАС показан в блоке стрелок . Альтернативные средства для генерации Ang-II обозначены пунктирными стрелками . Ang 3-8, ангиотензин IV; Ang 2-8, ангиотензин III; CAGE, химостатин-чувствительный Ang-II-продуцирующий фермент; t-PA, тканевый активатор плазминогена.
Рисунок 1.
Каскад REN-ангиотензина. Классический каскад РАС показан в блоке стрелок .Альтернативные средства для генерации Ang-II обозначены пунктирными стрелками . Ang 3-8, ангиотензин IV; Ang 2-8, ангиотензин III; CAGE, химостатин-чувствительный Ang-II-продуцирующий фермент; t-PA, тканевый активатор плазминогена.
Локальный РАН
Тканевые RAS существуют в тканях, которые обладают способностью как к локальной генерации, так и к действию Ang-II (рис. 2). Все компоненты РАС можно найти в головном мозге (21, 22), сердце (21), сосудистой сети (21), жировой ткани (23), гонадах (24), поджелудочной железе (25), плаценте (26) и почках. (21) и другие.Предполагается, что внутрипочечный РАС регулирует системное артериальное давление и аспекты функции почек, такие как кровоток и реабсорбцию натрия (27), тогда как в головном мозге он может облегчать нейротрансмиссию и стимулировать высвобождение вазопрессина и симпатический отток (28, 29). Концепция тканевого RAS полностью поддерживается данными первичной экспрессии, показывающими все компоненты RAS в отдельных тканях. Однако важно отметить, что клинические наблюдения составляют основу концепции и потенциальной важности тканевых РАС.Эти наблюдения включают: 1) антигипертензивное действие ингибиторов АПФ лучше коррелирует с ингибированием тканевого АПФ, чем АПФ плазмы, и 2) пациентов с гипертонией с нормальным или даже низким уровнем системной активности РАС можно эффективно лечить ингибиторами РАС ( 30, 31).
Рисунок 2.
Выражение RAS. Показаны участки экспрессии различных компонентов РАС. Классические сайты синтеза эндокринного RAS выделены жирным шрифтом .
Рисунок 2.
Выражение RAS. Показаны участки экспрессии различных компонентов РАС. Классические сайты синтеза эндокринного RAS выделены жирным шрифтом .
В некоторых тканях могут быть обнаружены только некоторые компоненты RAS, что заставляет некоторых размышлять о существовании альтернативных путей для продукции Ang-II (Fig. 1). Например, сообщалось о превращении Ang-I в Ang-II с использованием таких ферментов, как катепсин G, химостатин-чувствительный фермент, генерирующий Ang-II, и химаза (32, 33), а также путь для REN- Также сообщалось о независимом производстве Ang-II из AGT (34, 35).Физиологическое значение этих путей остается неясным и не будет подробно рассматриваться в этом обзоре.
Модели тканей у трансгенных животных RAS
Нацелен на мозг
Был проявлен значительный интерес к УЗВ головного мозга на основе данных, свидетельствующих о его вкладе в состояние гипертонии во многих моделях животных, таких как крыса со спонтанной гипертензией, крыса с солевой гипертензией ацетата дезоксикортикостерона (DOCA) и крыса, чувствительная к соли Даля ( 36, 37).У крыс со спонтанной гипертензией острая и хроническая интрацеребровентрикулярная (ICV) инъекция ингибитора АПФ, блокатора рецепторов ангиотензина или антисмысловых олигонуклеотидов к рецепторам AT-1 или мРНК AGT ослабляет развитие гипертензии (38, 39). У мышей с двойной трансгенной гипертензией, системно экспрессирующих как человеческий REN (hREN), так и человеческий AGT (hAGT), мы сообщили о значительном снижении артериального давления после ICV-инъекции лозартана, блокатора рецепторов ангиотензина (40, 41).
Чтобы лучше оценить роль первичной продукции REN и AGT в головном мозге и различить эффекты глиальной и нейрональной продукции Ang-II, мы разработали трансгенных мышей, экспрессирующих hAGT и / или hREN, управляемые синапсином I (SYN I) промотор, нейрональный промотор или промотор глиального фибриллярного кислого белка (GFAP), глиальный промотор. Экспрессия трансгена у мышей GFAP-hAGT была очевидна в основном в астроцитах головного мозга, но hAGT также можно было обнаружить в клетках субфорного органа, который также содержал связанный с микротрубочками белок-2, нейрональный маркер (42).Когда мышей GFAP-hAGT скрещивали с мышами, системно экспрессирующими hREN, наблюдалось повышение артериального давления на 15 мм рт. Кроме того, эти двойные трансгенные мыши проявляли предпочтение физиологического раствора, когда им предоставлялся выбор между водопроводной водой и физиологическим раствором. Эти результаты согласуются с исследованиями, проведенными на трансгенных крысах, экспрессирующих антисмысловую РНК против мРНК AGT, управляемую промотором GFAP, TGR (ASrAOGEN), где наблюдалось значительное снижение артериального давления (43). Мы также получили трансгенных мышей, экспрессирующих hREN под контролем промотора GFAP (GFAP-hREN; Ref.44). Эти трансгенные мыши экспрессировали hREN в головном мозге, особенно в глии, с некоторой эктопической экспрессией в легких и жировой ткани, но не определяли плазменный hREN. Когда этих мышей скрещивали с мышами GFAP-hAGT, у двойных трансгенных животных наблюдалось повышение артериального давления, увеличение объема питья и увеличение потребления соли. Наблюдаемое повышение артериального давления было обращено внутривенной инъекцией лозартана, тогда как внутривенное введение той же дозы оказалось неэффективным. Это говорит о том, что наблюдаемое повышение артериального давления было связано с местной выработкой и действием Ang-II в головном мозге.Этот прессорный эффект может быть опосредован увеличением симпатической активности, поскольку гексаметоний, блокатор ганглиев, вызывал большее падение артериального давления у двойных трансгенных мышей, чем у отрицательных однопометников.
Мы также получили трансгенных мышей SYN I-hAGT, которые экспрессируют трансген только в нейронах головного мозга и на низких уровнях в почках и сердце, но не обнаруживают обнаруживаемого hAGT в плазме (45). У этих мышей наблюдали прессорный ответ после ICV, но не внутривенного введения очищенного рекомбинантного hREN, который можно было предотвратить путем предварительной обработки ICV лозартаном, что указывает на то, что прессорный ответ был зависимым от рецептора AT-1.Соответственно, когда мышей SYN I-hAGT скрещивали с мышами SYN I-hREN, которые также проявляли нейронально-специфический паттерн экспрессии в головном мозге, они были умеренно гипертоническими и демонстрировали повышенный объем питья и предпочтение соли (44). Обе модели GFAP и SYN ясно демонстрируют, что локальная продукция Ang-II в головном мозге имеет многочисленные физиологические эффекты, регулирующие кровяное давление, а также гомеостаз воды и электролитов.
Мы и другие (46, 47) сообщили об измененной форме мРНК REN, полученной в результате использования альтернативного сайта начала транскрипции в головном мозге.В случае трансляции эта мРНК будет кодировать внутриклеточную (несекретированную) и конститутивно активную форму белка, предполагая возможность аутокринного внутриклеточного пути продукции Ang-II в головном мозге. Поскольку физиологическое значение этого пути остается неизвестным, мы в настоящее время изучаем регуляцию кровяного давления и гомеостаза жидкости на новых трансгенных моделях, экспрессирующих эту внутриклеточную форму REN, управляемую промоторами GFAP или SYN I.
Направлено на почки
В почках REN экспрессируется в основном в юкстагломерулярных клетках, где он хранится в плотных секреторных гранулах ядра и высвобождается в интерстиций в ответ на различные физиологические сигналы.Оттуда REN попадает в системный кровоток, где он может действовать как часть эндокринного RAS. AGT экспрессируется в клетках проксимальных канальцев почек и проявляет поляризованную секрецию через апикальную мембрану в просвет канальцев (48, 49). В просвете REN либо фильтруется из кровотока, транспортируется из почечного интерстиция, либо вырабатывается в канальцах, непосредственно преобразуя AGT в Ang-I. В канальцевой жидкости Ang-I быстро превращается в Ang-II за счет высокой концентрации АПФ на мембране щеточной каймы проксимального канальца.
Чтобы проверить, может ли внутрипочечный РАС влиять на кровяное давление независимо от изменений циркулирующего Ang-II, мы создали модель, специфичную для почек, в результате экспрессии гена AGT, специфичной для проксимальных канальцев. Мы получили трансгенных мышей, экспрессирующих hAGT, управляемый андроген-регулируемым промотором белка почек, который специфически экспрессируется в проксимальных канальцах почек и очень чувствителен к андрогенам (50). Повышенные концентрации чАГТ наблюдались в моче, отражая его повышенную продукцию в клетках проксимальных канальцев и его высвобождение в просвет канальцев, но в системном кровотоке чАГТ обнаружено не было.Двойные трансгенные мыши-самцы, экспрессирующие почечный андроген-регулируемый белок-hAGT и системно экспрессируемый hREN (экспрессируемый в юкстагломерулярных клетках почек), имели повышенное кровяное давление, но нормальные уровни циркулирующего Ang-II (51). Повышение кровяного давления может быть вызвано у самок двойных трансгенных мышей обработкой тестостероном, причем повышение кровяного давления происходит параллельно индукции андроген-чувствительного трансгена. Учитывая высокую концентрацию hAGT в моче, мы предположили, что существует высокая концентрация Ang-II в жидкости проксимальных канальцев и, возможно, далее в жидкости вдоль дистальной части нефрона.Это согласуется с измерениями, показывающими, что уровень Ang-II в трубчатой жидкости нельзя объяснить фильтрацией циркулирующего Ang-II (27, 52). Ang-II оказывает прямое влияние на транспорт натрия в раннем нефроне, стимулируя натрий-водородный обмен в проксимальных канальцах, и косвенные эффекты в позднем нефроне, регулируя синтез эпителиальных натриевых каналов альдостероном, оба из которых могут происходить через связывание Ang-II с просветом. Рецепторы АТ-1 (53). Это подтверждает гипотезу о том, что гипертония у этих мышей может быть вызвана изменениями гомеостаза натрия или жидкости, возможно, из-за изменений в этих транспортных механизмах.Такие аффекты, по-видимому, являются общим основным механизмом, вызывающим высокое кровяное давление при ряде генетических синдромов человека (54).
Заключение
Недавняя демонстрация того, что локальные РАС существуют и физиологически активны во многих тканях, указывает на важность тканевого или паракринного пути генерации и действия Ang-II. Именно эта двойственность РАС, как тканевой, так и эндокринной систем, работающих одновременно, сделала систему чрезвычайно сложной, и поэтому после более чем 100-летнего изучения все еще остаются секреты, которые нужно открыть.По мере появления новых технологий и внедрения новых инструментов для решения этой проблемы мы, несомненно, узнаем больше о том, как эта сложная система работает in vivo . Например, наша лаборатория продемонстрировала, что отсутствие hAGT в печени, вызванное использованием системы рекомбиназы Cre-loxP для создания тканеспецифического нокаута AGT, вызывает потерю циркулирующего hAGT, что напрямую демонстрирует, что внепеченочные источники AGT не вносит значительного вклада в циркулирующий пул AGT (55).Более того, инфицирование двойных трансгенных мышей, содержащих hREN и флокированный трансген hAGT, аденовирусом, кодирующим cre-рекомбиназу, значительно снижает артериальное давление (56). Таким образом, в настоящее время мы используем cre-рекомбиназу в сочетании с клеточно-специфическими промоторами для изучения роли различных тканевых RAS.
Благодарности
Эта работа была поддержана постдокторской стипендией Американской кардиологической ассоциации Heartland Affiliate (J.L.L.). Мы с благодарностью отмечаем исследовательскую поддержку Carver Trust.
Сокращения
ACE
Ангиотензинпревращающий фермент;
AGT
Ang-I и II
AT-1
AT-2
CNS
GFAP фибриновый белок;
hAGT
hREN
ICV
RAS
ренин-ангиотензиновая система;
REN
Syn
1
Timmermans
PB
,
Wong
PC
,
Chiu
AT
,
000
Herblin
Carini
DJ
,
Lee
RJ
,
Wexler
RR
,
Saye
JA
,
Smith
RD
Ангиогенные рецепторы
9000 II3 рецепторы ангиогенов II3
Pharmacol Rev
45
:
205
—
251
2
Sayeski
PP
,
Bernstein
KE
2001
Механизмы передачи сигнала AT (1) ангиотензин II типа : выход за рамки парадигмы гетеротримерного G-белка.
J Ренин Ангиотензин-альдостерон Syst
2
:
4
—
10
3
Ishii
K
,
Takekoshi
K
,
Yamaha
000
000
000
Isobe
K
,
Nakai
T
2001
Рецептор подтипа 2 ангиотензина (AT2) отрицательно регулирует рецептор подтипа 1 (AT1) в путях передачи сигнала в культивируемых хромаффинных клетках мозгового вещества надпочечников свиней.
J Гипертензия
19
:
1991
—
1999
4
Eguchi
S
,
Inagami
T
2000
Передача сигнала рецептора 1 ангиотензина через рецептор к тирозина II типа
Regul Pept
91
:
13
—
20
5
Shanmugam
S
,
Corvol
P
,
Gasc
angyin
II
два типа
9000io4 1 подтипы рецепторов у крыс
.
Am J Physiol Endocrinol Metab
267
:
E828
—
E836
6
Iwai
N
,
Inagami
T
1992
рецепторов типа I .
FEBS Lett
298
:
257
—
260
7
Сасамура
H
,
Hein
L
,
Krieger
JE
000
000
000
000 RE
000
000 RE
000 BK
,
Dzau
VJ
1992
Клонирование, характеристика и экспрессия двух изоформ рецептора ангиотензина (AT-1) из генома мыши.
Biochem Biophys Res Commun
185
:
253
—
259
8
Konishi
H
,
Kuroda
S
,
Inada
Y
0003
Новый подтип рецептора человеческого ангиотензина II типа 1: клонирование и экспрессия кДНК.
Biochem Biophys Res Commun
199
:
467
—
474
9
Burson
JM
,
Aguilera
G
,
Gross
000
000
000
000
000
Дифференциальная экспрессия рецепторов ангиотензина 1A и 1B у мышей
.
Am J Physiol
267
:
E260
—
E267
10
Kakar
SS
,
Продавцы
JC
,
Devor
DC Musill
Neon
JD
1992
кДНК подтипа рецептора ангиотензина II типа 1: дифференциальная экспрессия в тканях и гормональная регуляция.
Biochem Biophys Res Commun
183
:
1090
—
1096
11
Gasc
JM
,
Shanmugam
S
,
Sibony
M
Тканевая экспрессия подтипов рецепторов ангиотензина II типа 1: исследование гибридизации in situ.
Гипертония
24
:
531
—
537
12
Ito
M
,
Oliverio
MI
,
Mannon
PJ
,
Best
,
Smithies
O
,
Coffman
T
1995
Регулирование артериального давления геном рецептора ангиотензина II типа 1A.
Proc Natl Acad Sci USA
92
:
3521
—
3525
13
Chen
XM
,
Li
WG
,
Yoshida
H
Nishimura
H
,
Takemoto
F
,
Okubo
S
,
Fogo
A
,
Matsusaka
T
,
Ichikawa 9000 9000 9000 9000 9000 9000 ang3 Ген рецептора 1B у мыши
.
Am J Physiol Renal Physiol
41
:
F299
—
F304
14
Davisson
RL
,
Oliverio
MI
,
Coffman
000
Coffman
TM
Дивергентные функции изоформ рецепторов ангиотензина II в головном мозге.
J Clin Invest
106
:
103
—
106
15
Carey
RM
,
Wang
ZQ
,
Siragy
HM
2000 ang. рецептор в регуляции артериального давления и функции почек.
Гипертония
35
:
155
—
163
16
Lo
M
,
Liu
KL
,
Lantelme
P
,
Subtype J 2 рецептора ангиотензина II контролируют давление-натрийурез у крыс.
J Clin Invest
95
:
1394
—
1397
17
Hein
L
,
Barsh
GS
,
Pratt
RE
000 Kz
,
000 KZ
000
BK
1995
Поведенческие и сердечно-сосудистые эффекты нарушения рецептора ангиотензина II типа 2 у мышей.
Nature
377
:
744
—
747
18
Swanson
GN
,
Hanesworth
JM
,
Sardinia
MF
000 JM
9000
,
Hall
KL
,
Miller-Wing
AV
,
Stobb
JW
,
Cook
VI
,
Harding
EC
1992
Открытие особого места для переплета ангиотензин II (3–8), предполагаемый рецептор ангиотензина IV.
Regul Pept
40
:
409
–
419
19
Coleman
JKM
,
Ong
B
,
Sardinia
M
9000 JR
JW
1992
Изменения почечного кровотока из-за инфузий ангиотензина-Ii (3–8) [Aiv] нормотензивным крысам
.
FASEB J
6
:
A981
20
Nguyen
G
,
Delarue
F
,
Burckle
C
,
Bouzhir
Liller
Liller
JD
2002
Ключевая роль рецептора ренина / проренина в продукции ангиотензина II и клеточных ответах на ренин.
J Clin Invest
109
:
1417
—
1427
21
Bader
M
,
Peters
J
,
Baltatu
O
9000 9000 9000
Muller
FC
,
Ganten
D
2001
Тканевые ренин-ангиотензиновые системы: новые идеи экспериментальных животных моделей в исследованиях гипертонии.
J Mol Med
79
:
76
—
102
22
Morimoto
S
,
Sigmund
CD
2002
Ангиотензин на мутантную ренин-систему мозга: a .
Нейропептиды
36
:
194
—
200
23
Энгели
S
,
Негрель
R
,
Шарма
AM
Ренология и патология тканей
9000 Ренология 9000 -ангиотензиновая система.
Гипертония
35
:
1270
—
1277
24
Speth
RC
,
Daubert
DL
,
Grove
KL
слишком репродуктивный?
Regul Pept
79
:
25
—
40
25
Sernia
C
2001
Критическая оценка внутренней системы генерации ангиотензина поджелудочной железы.
J Поджелудочная железа
2
:
50
—
55
26
Nielsen
AH
,
Schauser
KH
,
Poulsen
K
Система рен.
Плацента
21
:
468
—
477
27
Navar
LG
,
Imig
JD
,
Wang
CT
1997 9000ren
Производство Angratensal
Семин Нефрол
17
:
412
—
422
28
Коста
M
,
Majewski
H
1988
1988
Активация симпатических нервов через норадренатические рецепторы -адренорецепторы и рецепторы ангиотензина II.
Br J Pharmacol
95
:
993
—
1001
29
Steckelings
U
,
Lebrun
C
,
Qadri
0003 Velt
,
T
1992
Роль ангиотензина головного мозга в регуляции сердечно-сосудистой системы
.
J Cardiovasc Pharmacol
19
(
Suppl 6
):
S72
—
S79
30
Brunner
HR
,
Gavras
H
,
Waeber
Waeber
ингибитор ангиотензинпревращающего фермента при длительном лечении пациентов с артериальной гипертонией.
Ann Intern Med
2
:
1317
—
1325
31
Dzau
VJ
,
Bernstein
K
,
Celermajer
0003 9000 Cohen
B
,
Deanfield
J
,
Diez
J
,
Drexler
H
,
Ferrari
R
,
фургон Gilst
W Han
B
,
Husain
A
,
Johnston
C
,
Lazar
H
,
Lonn
E
,
Luscher
T
Mran A
,
Pepine
C
,
Rabelink
T
,
Remme
W
,
Ruilope
L
,
Ruzicka
M
,
Schunkert
H
,
Swedberg
K
,
Unger
T
,
Vaughan
D
000
Weber 9000
000
Weber 9000 Актуальность тканевого ангиотензин-превращающего фермента: проявления в механистических и конечных данных.
Am J Cardiol
88
:
1
L – 20L32
Liao
Y
,
Husain
A
1995
Химазно-ангиотензиновая система и потенциальная роль системы химазы-ангиотензина в организме человека сердечно-сосудистых заболеваний
.
Can J Cardiol
11
(
Suppl F
):
13F
—
19F
33
Urata
H
,
Ganten
D
Формирование тензина II:
Ангиотензия сердца
I превращающий фермент и химазу человека
.
Eur Heart J
14
(
Suppl I
):
177
—
182
34
Grise
C
,
Boucher
R
,
Thibault
GISS
1981
Образование ангиотензина II тонином из частично очищенного ангиотензиногена человека.
Can J Biochem
59
:
250
—
255
35
Boucher
R
,
Demassieux
S
,
Garcia
R
000
000 Genest
Тонин, система ангиотензина II.Обзор.
Circ Res
41
:
26
—
29
36
Bunnemann
B
,
Fuxe
K
,
Ganten
D
9000 ang3 9000 ang3
9000 ang3 система мозга локализация и общее значение
.
J Cardiovasc Pharmacol
19
:
S51
—
S62
37
Унгер
T
,
Badoer
E
,
Ganten
D
R
1988
Ангиотензин головного мозга: метаболические пути и фармакология
.
Обращение
77
:
I40
—
I54
38
Гюрко
R
,
Wielbo
D
,
Phillips
MI
мРНК ангиотензиногена в головном мозге крыс со спонтанной гипертензией снижает гипертензию нейрогенного происхождения.
Regul Pept
49
:
167
—
174
39
Филлипс
MI
,
Mann
JF
,
Haebara
H
000
000 WE
000
R
,
Schelling
P
,
Ganten
D
1977
Снижение артериальной гипертензии центральным саралазином при отсутствии ренина плазмы.
Nature
270
:
445
—
447
40
Дэвиссон
RL
,
Ян
G
,
Beltz
TG
,
Cassell
,
Sigmund
CD
1998
Ренин-ангиотензиновая система мозга способствует развитию гипертонии у мышей, содержащих как человеческий ренин, так и человеческий трансген ангиотензиногена.
Circ Res
83
:
1047
—
1058
41
Morimoto
S
,
Cassell
MD
,
Sigmund
CD
brain трансгенные мыши, несущие строго регулируемый трансген человеческого ренина.
Circ Res
90
:
80
—
86
42
Morimoto
S
,
Cassell
MD
,
Beltz
TG
,
Johnson
RL
,
Sigmund
CD
2001
Повышенное кровяное давление у трансгенных мышей со специфической для мозга экспрессией ангиотензиногена человека, управляемой промотором глиального фибриллярного кислого белка.
Circ Res
89
:
365
—
372
43
Schinke
M
,
Baltatu
O
,
Bohm
M
0003 Peters
0003 Peters
,
0003 Peters W
,
Bricca
G
,
Lippoldt
A
,
Ganten
D
,
Bader
M
1999
Снижение артериального давления и трансгенный несахарный диабет при трансгенном несахарном диабете.
Proc Natl Acad Sci USA
96
:
3975
—
3980
44
Morimoto
S
,
Cassell
MD
,
Sigmund
Gl
-специфическая экспрессия ренин-ангиотензиновой системы в головном мозге изменяет кровяное давление, потребление воды и предпочтение соли.
J Biol Chem
277
:
33235
—
33241
45
Morimoto
S
,
Cassell
MD
,
Sigmund
CD
специфическая экспрессия человека
ангиотензиноген в головном мозге вызывает повышенный солевой аппетит.
Physiol Genom
9
:
113
—
120
46
Sinn
PL
,
Sigmund
CD
2000
Идентификация трех альтернативных тканеспецифичных мРНК ренина человека инициация.
Physiol Genom
3
:
25
—
31
47
Lee-Kirsch
MA
,
Gaudet
F
,
Cardoso
MC
000
paint,
Отдельные изоформы ренина, генерируемые тканеспецифической инициацией транскрипции и альтернативным сплайсингом.
Circ Res
84
:
240
—
246
48
Rohrwasser
A
,
Morgan
T
,
Dillon
HFay
000 L
,
CW
,
Hillas
E
,
Zhang
S
,
Cheng
T
,
Inagami
T
,
Ward
K
,
Terreros 9000 L
Terreros
1999
Элементы паракринной трубчатой ренин-ангиотензиновой системы вдоль всего нефрона.
Гипертония
34
:
1265
—
1274
49
Loghman-Adham
M
,
Rohrwasser
A
,
Helin
C
D
,
Inoue
I
,
Lalouel
JM
1997
Условно иммортализованная линия клеток проксимального канальца мыши.
Kidney Int
52
:
229
—
239
50
Ding
Y
,
Davisson
RL
,
Hardy
DO
Zhu DC
,
Catterall
JF
,
Sigmund
CD
1997
Промотор почечного андроген-регулируемого белка (KAP) обеспечивает специфическую для клеток проксимальных канальцев почек и высокую андроген-чувствительную экспрессию гена ангиотензиногена человека. трансгенные мыши.
J Biol Chem
272
:
28142
—
28148
51
Davisson
RL
,
Ding
Y
,
Stec
DE
,
Stec
DE
CD
1999
Новый механизм гипертонии, выявленный клеточно-специфическим нацеливанием ангиотензиногена человека у трансгенных мышей.
Physiol Genom
1
:
3
—
9
52
Kobori
H
,
Harrison-Bernard
LM
,
Navar
LG
ангиоген, отражающий ангиогенез мочи
внутрипочечная продукция ангиотензиногена.
Kidney Int
61
:
579
—
585
53
Cogan
MG
1990
Ангиотензин II: мощный регулятор транспорта натрия в ранних проксимальных канальцах.
Гипертония
15
:
451
—
458
54
Шимкетс
RA
,
Warnock
DG
,
Bositis
DG
0003 Nelson
JH
,
Schambelan
M
,
Gill
JR
,
Ulick
S
,
Milora
RV
,
Findling
JW
Ross
a,
BC
,
Lifton
RP
1994
Синдром Лиддла: наследственная гипертензия человека, вызванная мутациями в субъединице β эпителиального натриевого канала.
Ячейка
79
:
407
—
414
55
Stec
DE
,
Davisson
RL
,
Haskell
RE
,
Davidson
1999
Эффективная печеночно-специфическая делеция флоксированного трансгена ангиотензиногена человека аденовирусной доставкой cre-рекомбиназы in vivo.
J Biol Chem
274
:
21285
—
21290
56
Stec
DE
,
Keen
HL
,
Sigmund
кровяное давление
CD
мышей после аденовирусной доставки cre-рекомбиназы.
Гипертония
39
:
629
—
633
Авторские права © 2003 Общество эндокринологов
Контррегуляторная ренин-ангиотензиновая система при сердечно-сосудистых заболеваниях
Феррарио, К. М. Роль ангиотензина II в терапевтических последствиях сердечно-сосудистых заболеваний — более чем столетние исследования. J. Renin Angiotensin Aldosterone Syst. 7 , 3–14 (2006).
CAS
PubMed
Google Scholar
Karnik, S. S. et al. Международный союз фундаментальной и клинической фармакологии. XCIX. Рецепторы ангиотензина: интерпретаторы патофизиологических ангиотензинергических стимулов [исправлено]. Pharmacol. Ред. 67 , 754–819 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Forrester, S.J. et al. Передача сигнала ангиотензина II: обновленная информация о механизмах физиологии и патофизиологии. Physiol. Ред. 98 , 1627–1738 (2018).
CAS
PubMed
PubMed Central
Google Scholar
Teixeira, L. B. et al. Ang- (1-7) представляет собой эндогенный агонист рецептора AT1, связанный с бета-аррестином, с защитным действием при гипертрофии сердца. Sci. Отчетность 7 , 11903 (2017).
PubMed
PubMed Central
Google Scholar
Иисус, И. К. Г. и др. Аламандин действует через MrgD, вызывая активацию AMPK / NO против гипертрофии ANG II в кардиомиоцитах. Am. J. Physiol. Клетка. Physiol. 314 , C702 – C711 (2018).
PubMed
Google Scholar
Mendoza-Torres, E. et al. Защита миокарда от ишемического / реперфузионного повреждения ангиотензин- (1-9) через AT2R и Akt-зависимый механизм. Pharmacol. Res. 135 , 112–121 (2018).
CAS
PubMed
Google Scholar
Li, T. et al. Критическая роль оси химаза / ангиотензин- (1-12) в модуляции сократимости кардиомиоцитов. Внутр. J. Cardiol. 264 , 137–144 (2018).
PubMed
PubMed Central
Google Scholar
Yu, L., Yuan, K., Phuong, HT, Park, BM & Kim, SH Ангиотензин- (1-5), активный медиатор ренин-ангиотензиновой системы, стимулирует секрецию ANP через рецептор Mas. . Пептиды 86 , 33–41 (2016).
CAS
PubMed
Google Scholar
Chang, L. et al. Bmal1 в периваскулярной жировой ткани регулирует артериальное давление в фазе покоя посредством регуляции транскрипции ангиотензиногена. Тираж 138 , 67–79 (2018).
CAS
PubMed
PubMed Central
Google Scholar
Tetzner, A. et al. Рецептор, связанный с G-белком, MrgD представляет собой рецептор ангиотензина (1-7) с участием аденилатциклазы, цАМФ и фосфокиназы A. Hypertension 68 , 185–194 (2016).
CAS
PubMed
Google Scholar
Bosnyak, S. et al. Относительное сродство пептидов ангиотензина и новых лигандов к рецепторам AT1 и AT2. Clin. Sci. 121 , 297–303 (2011).
CAS
PubMed
Google Scholar
Kostenis, E. et al. Рецептор, связанный с G-белком. Mas является физиологическим антагонистом рецептора ангиотензина II типа 1. Тираж 111 , 1806–1813 (2005).
CAS
PubMed
Google Scholar
Гайдаров И. и др. Ангиотензин (1-7) не взаимодействует напрямую с MAS1, но может сильно противодействовать передаче сигналов от рецептора AT1. Ячейка. Сигнал. 50 , 9–24 (2018).
CAS
PubMed
Google Scholar
Santos, R.A. et al. Ангиотензин- (1-7) является эндогенным лигандом для рецептора Mas, сопряженного с G-белком. Proc. Natl Acad. Sci. США 100 , 8258–8263 (2003). Это исследование показывает, что ангиотензин 1-7 связывается с рецептором Mas .
CAS
PubMed
Google Scholar
Мимс, Л.M. G. et al. Дизайн, синтез и действие инновационного биспецифического дизайнерского пептида. Гипертония 73 , 900–909 (2019). В этой статье описывается синтез пептида, который одновременно активирует рецептор Mas и рецептор гуанилилциклазы A в виде частиц с сильным антигипертензивным действием .
CAS
PubMed
Google Scholar
Leonhardt, J. et al. Доказательства гетеродимеризации и функционального взаимодействия рецептора ангиотензина 2 типа и рецептора MAS. Гипертония 69 , 1128–1135 (2017).
CAS
PubMed
Google Scholar
Zhang, H. et al. Структурная основа избирательности и разнообразия рецепторов ангиотензина II. Природа 544 , 327–332 (2017). В этой статье описывается кристаллическая структура человеческого AT .
2 R и доказательства, показывающие, что этот рецептор не связывается с G-белками или β-аррестинами .
CAS
PubMed
PubMed Central
Google Scholar
Лобо М. Д., Соботка П. А. и Патак А. Интервенционные процедуры и будущая лекарственная терапия гипертонии. евро. Харт Дж. 38 , 1101–1111 (2017).
PubMed
Google Scholar
Pan, X. et al. FGF21 Предотвращает индуцированную ангиотензином II гипертензию и сосудистую дисфункцию за счет активации оси ACE2 / ангиотензин- (1-7) у мышей. Cell Metab. 27 , 1323–1337.e5 (2018).
CAS
PubMed
Google Scholar
Лоусон, К., Виченсио, Дж. М., Йеллон, Д. М. и Дэвидсон, С. М. Микровезикулы и экзосомы: новые игроки в метаболических и сердечно-сосудистых заболеваниях. J. Endocrinol. 228 , R57 – R71 (2016).
PubMed
Google Scholar
Йеллон, Д.М. и Дэвидсон, С. М. Экзосомы: наночастицы, участвующие в кардиозащите? Circ. Res. 114 , 325–332 (2014).
CAS
PubMed
Google Scholar
Pironti, G. et al. Циркулирующие экзосомы, вызванные перегрузкой сердечным давлением, содержат функциональные рецепторы ангиотензина II типа 1. Тираж 131 , 2120–2130 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Лю, Л. и др. Критическая роль экзосом, происходящих из сердечных фибробластов, в активации ренин-ангиотензиновой системы в кардиомиоцитах. J. Mol. Клетка. Кардиол. 89 , 268–279 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Hamming, I. et al. Растущая роль ACE2 в физиологии и болезнях. J. Pathol. 212 , 1–11 (2007).
CAS
PubMed
Google Scholar
Yamazato, Y. et al. Профилактика легочной гипертензии путем переноса гена ангиотензинпревращающего фермента 2. Гипертония 54 , 365–371 (2009).
CAS
PubMed
PubMed Central
Google Scholar
Ferreira, A. J. et al. Доказательства использования ангиотензин-превращающего фермента 2 в качестве терапевтической мишени для профилактики легочной гипертензии. Am. J. Respir. Крит. Care Med. 179 , 1048–1054 (2009).
CAS
PubMed
PubMed Central
Google Scholar
Li, G. et al. Активация ангиотензин-превращающего фермента 2 улучшает легочную эндотелиальную дисфункцию у крыс с легочной артериальной гипертензией за счет фосфорилирования эндотелиальной синтазы оксида азота. J. Am. Soc. Гипертоническая болезнь. 11 , 842–852 (2017).
CAS
PubMed
Google Scholar
Sztuka, K., Orszulak-Michalak, D. & Jasinska-Stroschein, M. Систематический обзор и метаанализ вмешательств, протестированных на животных моделях легочной гипертензии. Vasc. Pharmacol. 110 , 55–63 (2018).
CAS
Google Scholar
Epelman, S. et al. Растворимый ангиотензин-превращающий фермент 2 при сердечной недостаточности человека: связь с функцией миокарда и клиническими исходами. J. Card. Провал. 15 , 565–571 (2009).
CAS
PubMed
PubMed Central
Google Scholar
Shao, Z. et al. Повышение активности растворимого ангиотензинпревращающего фермента 2 в сыворотке крови после интенсивной медикаментозной терапии связано с лучшим прогнозом острой декомпенсированной сердечной недостаточности. J. Card. Провал. 19 , 605–610 (2013).
CAS
PubMed
PubMed Central
Google Scholar
Джонсон, Дж. А., Уэст, Дж., Мейнард, К. Б. и Хемнес, А. Р. ACE2 улучшает функцию правого желудочка в модели перегрузки давлением. PLOS ONE 6 , e20828 (2011).
CAS
PubMed
PubMed Central
Google Scholar
Rathinasabapathy, A. et al. Терапия rhACE2 изменяет вызванную блеомицином легочную гипертензию за счет восстановления ремоделирования сосудов. Фронт. Physiol. 9 , 271 (2018).
PubMed
PubMed Central
Google Scholar
Hemnes, A. R. et al. Потенциальная терапевтическая роль ангиотензин-превращающего фермента 2 при легочной артериальной гипертензии человека. евро. Респир. J. 51 , 1702638 (2018).
PubMed
PubMed Central
Google Scholar
Khan, A. et al. Пилотное клиническое испытание рекомбинантного человеческого ангиотензин-превращающего фермента 2 при остром респираторном дистресс-синдроме. Crit. Уход 21 , 234 (2017).
PubMed
PubMed Central
Google Scholar
Hampl, V. et al. Внутрилегочная активация ангиотензин-превращающего фермента типа 2 / ангиотензин 1-7 / ось Mas-рецептора, связанного с G-белком, ослабляет легочную гипертензию у трансгенных крыс Ren-2, подвергшихся хронической гипоксии. Physiol. Res. 64 , 25–38 (2015).
CAS
PubMed
Google Scholar
Yamada, K., Iyer, S. N., Chappell, M.C., Ganten, D. & Ferrario, C.M. Конвертирующий фермент определяет плазменный клиренс ангиотензина- (1-7). Гипертония 32 , 496–502 (1998).
CAS
PubMed
Google Scholar
Кастин А. Дж. И Пан У. Концепции биологически активных пептидов. Curr. Pharm. Des. 16 , 3390–3400 (2010).
CAS
PubMed
PubMed Central
Google Scholar
Bennion, D. M. et al. Нейропротекция пероральным введением ангиотензина (1-7) после инсульта при ишемическом инсульте. Exp. Physiol. 103 , 916–923 (2018).
CAS
PubMed
Google Scholar
Becker, L. K. et al. Повреждение мышц при эксцентрической перегрузке ослабляется новым лечением ангиотензином (1-7). Внутр. J. Sports Med. 39 , 743–748 (2018).
CAS
PubMed
Google Scholar
Sabharwal, R. et al. Хроническое пероральное введение Ang- (1-7) улучшает фенотипы скелетных мышц, вегетативные и двигательные функции при мышечной дистрофии. Clin. Sci. 127 , 101–109 (2014).
CAS
PubMed
PubMed Central
Google Scholar
Bertagnolli, M. et al. Перорально активное соединение включения ангиотензина (1-7) и тренировка с физической нагрузкой вызывают аналогичные сердечно-сосудистые эффекты у крыс со спонтанной гипертензией. Пептиды 51 , 65–73 (2014).
CAS
PubMed
Google Scholar
Marques, F. D. et al. Благоприятные эффекты длительного приема перорального препарата ангиотензина (1-7) у крыс с инфарктом. Внутр. J. Hypertens. 2012 , 7
(2012).
PubMed
PubMed Central
Google Scholar
Marques, F.D. et al. Пероральный состав ангиотензина (1-7) оказывает кардиозащитное действие у крыс, перенесших инфаркт, и крыс, получавших изопротеренол. Гипертония 57 , 477–483 (2011). В этой статье описывается новый состав ангиотензина 1–7, который увеличивает период полураспада в плазме .
CAS
PubMed
Google Scholar
Lula, I. et al. Изучение ангиотензин- (1-7) вазоактивного пептида и его комплексов включения бета-циклодекстрина: полные назначения ЯМР для конкретных последовательностей и структурные исследования. пептидов 28 , 2199–2210 (2007).
CAS
PubMed
Google Scholar
Breitling, S. et al. Дозозависимый терапевтический потенциал ангиотензина (1-7) для лечения легочной артериальной гипертензии. Pulm. Circ. 5 , 649–657 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Малек В., Шарма Н., Санкритьаян Х. и Гайквад А. Б. Одновременное ингибирование неприлизина и модуляция ренин-ангиотензиновой системы предотвращали диабетическую нефропатию. Life Sci. 221 , 159–167 (2019).
CAS
PubMed
Google Scholar
Окаранза, М. П. и Джалил, Дж. Э. Защитная роль оси ACE2 / Ang- (1-9) в ремоделировании сердечно-сосудистой системы. Внутр. J. Hypertens. 2012 , 5 (2012).
PubMed
PubMed Central
Google Scholar
Ча, С. А., Парк, Б. М. и Ким, С. Х. Ангиотензин- (1-9) улучшает легочную артериальную гипертензию через рецептор ангиотензина типа II. Korean J. Physiol. Pharmacol. 22 , 447–456 (2018).
PubMed
PubMed Central
Google Scholar
Bruce, E. et al. Селективная активация рецепторов ангиотензина AT2 замедляет прогрессирование легочной гипертензии и подавляет сердечно-легочный фиброз. руб. J. Pharmacol. 172 , 2219–2231 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Wagenaar, G. T. et al. Агонисты онкогена MAS и рецепторов ангиотензина II типа 2 ослабляют сердечно-легочное заболевание у крыс с повреждением легких, вызванным неонатальной гипероксией. Am. J. Physiol. Легочная клетка. Мол. Physiol. 305 , L341 – L351 (2013).
CAS
PubMed
PubMed Central
Google Scholar
Эрнандес Прада, Дж. А. и др. Идентификация низкомолекулярных активаторов ангиотензинпревращающего фермента 2 на основе структуры как новых антигипертензивных средств. Гипертония 51 , 1312–1317 (2008).
CAS
PubMed
Google Scholar
De Maria, M. L. et al. Антигипертензивные эффекты диминазена ацетурата: активатора ангиотензинпревращающего фермента 2 у крыс. Protein Peptide Lett. 23 , 9–16 (2016).
Google Scholar
Wiemer, G., Dobrucki, L. W., Louka, F. R., Malinski, T. & Heitsch, H. AVE 0991, непептидный аналог эффектов ангиотензина (1-7) на эндотелий. Гипертония 40 , 847–852 (2002).
CAS
PubMed
Google Scholar
Savergnini, S.Q. et al. Расслабление сосудов, антигипертензивный эффект и кардиопротекция нового пептидного агониста рецептора MAS. Гипертония 56 , 112–120 (2010).
CAS
PubMed
Google Scholar
Liu, P. et al. Новое химерное слияние ACE2-Fc обеспечивает длительный контроль гипертензии и защиту органов на мышиных моделях системной активации ренин-ангиотензиновой системы. Kidney Int. 94 , 114–125 (2018).
CAS
PubMed
Google Scholar
Haber, P. K. et al. Независимое от ангиотензинпревращающего фермента 2 действие предполагаемых активаторов ангиотензинпревращающего фермента 2: исследования in vivo, ex vivo и in vitro. Гипертония 63 , 774–782 (2014).
CAS
PubMed
PubMed Central
Google Scholar
Шеной В. и др. Диминазен ослабляет легочную гипертензию и улучшает функции ангиогенных клеток-предшественников в экспериментальных моделях. Am. J. Respir. Крит. Care Med. 187 , 648–657 (2013).
CAS
PubMed
PubMed Central
Google Scholar
Сингх Ю., Сингх К. и Шарма П. Л. Эффект комбинации ингибитора ренина и агониста Mas-рецептора на гипертензию, индуцированную солью DOCA у крыс. Мол. Клетка. Biochem. 373 , 189–194 (2013).
CAS
PubMed
Google Scholar
Ma, Y. et al. AVE 0991 снижает гипертрофию сердца за счет снижения окислительного стресса. Biochem. Биофиз. Res. Commun. 474 , 621–625 (2016).
CAS
PubMed
Google Scholar
Кейдар, С., Стризевский, А., Раз, А., Гамлиель-Лазарович, А. Активность ACE2 повышена в макрофагах, полученных из моноцитов от предгипертензивных субъектов. Нефрол. Набирать номер. Пересадка. 22 , 597–601 (2007).
CAS
PubMed
Google Scholar
Ортис-Перес, Дж. Т. и др. Роль циркулирующего ангиотензинпревращающего фермента 2 в ремоделировании левого желудочка после инфаркта миокарда: проспективное контролируемое исследование. PLOS ONE 8 , e61695 (2013).
CAS
PubMed
PubMed Central
Google Scholar
Соро-Паавонен, А.и другие. Активность циркулирующего ACE2 повышена у пациентов с диабетом 1 типа и сосудистыми осложнениями. J. Hypertens. 30 , 375–383 (2012).
CAS
PubMed
Google Scholar
Робертс, М. А., Велкоска, Э., Иерино, Ф. Л. и Баррелл, Л. М. Активность ангиотензин-превращающего фермента 2 у пациентов с хронической болезнью почек. Нефрол. Набирать номер. Пересадка. 28 , 2287–2294 (2013).
CAS
PubMed
Google Scholar
Хлестова Г.В. и др. Динамика ренина, ангиотензина II и ангиотензина (1-7) во время беременности и предрасположенность к осложнениям, связанным с артериальной гипертензией. Бык. Exp. Биол. Med. 165 , 438–439 (2018).
CAS
PubMed
Google Scholar
Ferrario, C. M. et al. Характеристика ангиотензина- (1-7) в моче нормальных и эссенциальных гипертоников. Am. J. Hypertens 11 , 137–146 (1998).
CAS
PubMed
Google Scholar
Schinzari, F. et al. Благоприятное сосудистое действие ангиотензина (1-7) при ожирении у человека. Гипертония 71 , 185–191 (2018).
CAS
PubMed
Google Scholar
Lautner, R.Q. et al. Открытие и характеристика аламандина: нового компонента ренин-ангиотензиновой системы. Circ. Res. 112 , 1104–1111 (2013).
CAS
PubMed
Google Scholar
Soltani Hekmat, A., Javanmardi, K., Kouhpayeh, A., Baharamali, E. & Farjam, M. Различия в сердечно-сосудистых реакциях на аламандин у двупочечных, гипертензивных и нормотензивных крыс с одним зажимом. Circ. J. 81 , 405–412 (2017).
PubMed
Google Scholar
Соуза-Нето, Ф. П. и др. Аламандин ослабляет ремоделирование артерий, вызванное поперечным сужением аорты у мышей. Clin. Sci. 133 , 629–643 (2019).
PubMed
Google Scholar
Ichiki, T. et al. Влияние на кровяное давление и исследовательское поведение мышей, лишенных рецептора ангиотензина II типа 2. Природа 377 , 748–750 (1995).
CAS
PubMed
Google Scholar
Хайн, Л., Барш, Г. С., Пратт, Р. Э., Дзау, В. Дж. И Кобилка, Б. К. Поведенческие и сердечно-сосудистые эффекты нарушения рецептора ангиотензина II типа 2 у мышей. Природа 377 , 744–747 (1995).
CAS
PubMed
Google Scholar
Tsutsumi, Y. et al. Сверхэкспрессия рецептора ангиотензина II типа 2 активирует кининовую систему сосудов и вызывает расширение сосудов. J. Clin. Вкладывать деньги. 104 , 925–935 (1999).
CAS
PubMed
PubMed Central
Google Scholar
Ocaranza, M. P. et al. Ангиотензин- (1-9) обращает вспять экспериментальную гипертензию и сердечно-сосудистые повреждения путем ингибирования ангиотензин-превращающего фермента / оси Ang II. J. Hypertens. 32 , 771–783 (2014).
CAS
PubMed
Google Scholar
Али, К., Ву, Ю. и Хуссейн, Т.Хроническая активация рецептора AT2 увеличивает почечную активность ACE2, снижает функцию рецептора AT1 и артериальное давление у тучных крыс Zucker. Kidney Int. 84 , 931–939 (2013).
CAS
PubMed
PubMed Central
Google Scholar
Li, X. C. & Widdop, R. E. Опосредованная рецептором AT2 вазодилатация демаскируется блокадой рецептора AT1 у SHR в сознании. руб. J. Pharmacology 142 , 821–830 (2004).
CAS
Google Scholar
Benndorf, R., Boger, RH, Ergun, S., Steenpass, A. & Wieland, T. Рецептор ангиотензина II типа 2 ингибирует индуцированную фактором роста сосудистого эндотелия миграцию и образование эндотелия человека в трубках in vitro. клетки. Circ. Res. 93 , 438–447 (2003).
CAS
PubMed
Google Scholar
Окаранса, М.P. et al. Ингибирование Rho-киназы активирует ось гомологичного ангиотензинпревращающего фермента-ангиотензина (1-9) при экспериментальной гипертензии. J. Hypertens. 29 , 706–715 (2011).
CAS
PubMed
Google Scholar
Xu, X. et al. Связанная с RhoA-Rho передача сигналов киназы ведет к дисбалансу ренин-ангиотензиновой системы, а ангиотензин-превращающий фермент 2 играет защитную роль при острой легочной эмболии. Тромб. Res. 176 , 85–94 (2019).
PubMed
Google Scholar
Fattah, C. et al. Генная терапия ангиотензином (1-9) сохраняет систолическую функцию левого желудочка после инфаркта миокарда. J. Am. Coll. Кардиол. 68 , 2652–2666 (2016). Это исследование показало, что использование генной терапии для сверхэкспрессии ангиотензина 1–9 предотвращает сердечную дисфункцию и улучшает выживаемость после инфаркта миокарда у мышей .
CAS
PubMed
PubMed Central
Google Scholar
Flores-Munoz, M. et al. Ангиотензин- (1-9) ослабляет сердечный фиброз у склонных к инсульту крыс со спонтанной гипертензией через рецептор ангиотензина 2 типа. Гипертония 59 , 300–307 (2012). Ангиотензин 1–9 снижает гипертоническое ремоделирование сердечно-сосудистой системы .
CAS
PubMed
Google Scholar
Reddy, R. et al. Уровни циркулирующих пептидов ангиотензина при остром респираторном дистресс-синдроме коррелируют с клиническими результатами: пилотное исследование. PLOS ONE 14 , e0213096 (2019).
CAS
PubMed
PubMed Central
Google Scholar
Crackower, M. A. et al. Ангиотензин-превращающий фермент 2 является важным регулятором сердечной функции. Природа 417 , 822–828 (2002). Эта статья является первой публикацией о роли ACE2 в сердечной функции .
CAS
PubMed
PubMed Central
Google Scholar
Rentzsch, B. et al. Сверхэкспрессия трансгенного ангиотензин-превращающего фермента 2 в сосудах крыс SHRSP снижает кровяное давление и улучшает функцию эндотелия. Гипертония 52 , 967–973 (2008).
CAS
PubMed
Google Scholar
Xu, P., Sriramula, S. & Lazartigues, E. ACE2 / ANG- (1-7) / Путь Mas в мозге: ось добра. Am. J. Physiol. Regul. Интегр. Комп. Physiol. 300 , R804 – R817 (2011).
CAS
PubMed
Google Scholar
Oudit, G. Y. et al. Окислительный стресс и воспаление, опосредованные ангиотензином II, опосредуют возрастную кардиомиопатию у мышей с нулевым показателем ACE2. Cardiovasc. Res. 75 , 29–39 (2007).
CAS
PubMed
Google Scholar
Epelman, S. et al. Обнаружение растворимого ангиотензин-превращающего фермента 2 при сердечной недостаточности: понимание эндогенного контррегуляторного пути ренин-ангиотензин-альдостероновой системы. J. Am. Coll. Кардиол. 52 , 750–754 (2008).
CAS
PubMed
PubMed Central
Google Scholar
Редфилд М. М. Сердечная недостаточность с сохраненной фракцией выброса. N. Engl. J. Med. 375 , 1868–1877 (2016).
PubMed
Google Scholar
Zhong, J. et al. Ангиотензин-превращающий фермент 2 подавляет патологическую гипертрофию, фиброз миокарда и сердечную дисфункцию. Тираж 122 , 717–728 (2010).
CAS
PubMed
Google Scholar
Oliveira, A.C. et al. Генетическая делеция рецептора аламандина MRGD приводит к дилатационной кардиомиопатии у мышей. Am. J. Physiol. Heart Circ. Physiol. 316 , h223 – h233 (2019).
CAS
PubMed
Google Scholar
Гао, Дж., Цукер, И. Х. и Гао, Л. Активация центральных рецепторов ангиотензина 2 типа соединением 21 улучшает чувствительность артериального барорефлекса у крыс с сердечной недостаточностью. Am.J. Hypertens. 27 , 1248–1256 (2014).
CAS
PubMed
PubMed Central
Google Scholar
Burrell, L. M. et al. Инфаркт миокарда увеличивает экспрессию ACE2 у крыс и людей. евро. Heart J. 26 , 369–375; обсуждение 322–364 (2005).
CAS
PubMed
Google Scholar
Kassiri, Z. et al.Потеря ангиотензин-превращающего фермента 2 ускоряет дезадаптивное ремоделирование левого желудочка в ответ на инфаркт миокарда. Circ. Сердечная недостаточность. 2 , 446–455 (2009).
CAS
PubMed
Google Scholar
Der Sarkissian, S. et al. Сердечная сверхэкспрессия ангиотензинпревращающего фермента 2 защищает сердце от патофизиологии, вызванной ишемией. Гипертония 51 , 712–718 (2008).
Google Scholar
Santos, R.A. et al. Экспрессия слитого белка, продуцирующего ангиотензин (1-7), вызывает у крыс кардиозащитное действие. Physiol. Геномика 17 , 292–299 (2004).
CAS
PubMed
Google Scholar
Oishi, Y. et al. Кардиозащитная роль рецептора AT2 в постинфарктном ремоделировании левого желудочка. Гипертония 41 , 814–818 (2003).
CAS
PubMed
Google Scholar
Brede, M. et al. Гипертрофия сердца связана со снижением экспрессии eNOS у мышей с дефицитом рецептора ангиотензина AT2. Гипертония 42 , 1177–1182 (2003).
CAS
PubMed
Google Scholar
Adachi, Y. et al.Дефицит рецепторов ангиотензина II типа 2 обостряет сердечную недостаточность и снижает выживаемость после острого инфаркта миокарда у мышей. Тираж 107 , 2406–2408 (2003).
CAS
PubMed
Google Scholar
Yang, Z. et al. Сверхэкспрессия рецептора ангиотензина II типа 2 сохраняет функцию левого желудочка после инфаркта миокарда. Тираж 106 , 106–111 (2002).
CAS
PubMed
Google Scholar
Qi, Y. et al. Умеренная сердечноселективная сверхэкспрессия рецептора ангиотензина II типа 2 защищает сердечные функции от ишемического повреждения. Exp. Physiol. 97 , 89–101 (2012).
CAS
PubMed
Google Scholar
Lauer, D. et al. Стимуляция рецептора ангиотензина типа 2 улучшает фиброз и дисфункцию левого желудочка за счет регуляции тканевого ингибитора матричной металлопротеиназы 1 / матричной металлопротеиназы 9 и трансформирующего фактора роста бета1 в сердце крысы. Гипертония 63 , e60 – e67 (2014).
CAS
PubMed
Google Scholar
Рупарелия, Н., Чай, Дж. Т., Фишер, Э. А. и Чоудхури, Р. П. Воспалительные процессы при сердечно-сосудистых заболеваниях: путь к таргетной терапии. Нат. Rev. Cardiol. 14 , 133–144 (2017).
CAS
PubMed
Google Scholar
Ширази, Л. Ф., Биссет, Дж., Ромео, Ф. и Мехта, Дж. Л. Роль воспаления при сердечной недостаточности. Curr. Атеросклер. Отчетность 19 , 27 (2017).
PubMed
Google Scholar
Golia, E. et al. Воспаление и сердечно-сосудистые заболевания: от патогенеза к терапевтической цели. Curr. Атеросклер. Реп. 16 , 435 (2014).
PubMed
Google Scholar
Guzik, T. J. & Touyz, R.M. Окислительный стресс, воспаление и старение сосудов при гипертонии. Гипертония 70 , 660–667 (2017).
CAS
PubMed
Google Scholar
Макмастер, В. Г., Кирабо, А., Мадхур, М. С. и Харрисон, Д. Г. Воспаление, иммунитет и гипертоническое поражение органов-мишеней. Circ. Res. 116 , 1022–1033 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Hoch, N.E. et al. Регулирование функции Т-клеток эндогенно продуцируемым ангиотензином II. Am. J. Physiol. Regul. Интегр. Комп. Physiol. 296 , R208 – R216 (2009).
CAS
PubMed
Google Scholar
Jurewicz, M. et al. Человеческие Т и естественные киллерные клетки обладают функциональной ренин-ангиотензиновой системой: дополнительными механизмами воспаления, вызванного ангиотензином II. J. Am. Soc. Нефрол. 18 , 1093–1102 (2007).
CAS
PubMed
Google Scholar
Дин, К. Н., Драммонд, Г. Р., Соби, К. Г. и Криссоболис, С. Роли воспаления, окислительного стресса и сосудистой дисфункции при гипертонии. Biomed. Res. Int. 2014 , 406960 (2014).
PubMed
PubMed Central
Google Scholar
Rutkowska-Zapala, M. et al. Субпопуляции человеческих моноцитов проявляют дивергентную активность по преобразованию ангиотензина I. Clin. Exp. Иммунол. 181 , 126–132 (2015).
CAS
PubMed
PubMed Central
Google Scholar
da Silveira, K. D. et al. Противовоспалительные эффекты активации рецептора ангиотензина (1-7), MAS, в экспериментальных моделях артрита. J. Immunol. 185 , 5569–5576 (2010).
PubMed
Google Scholar
Oliveira-Lima, O.C. et al. Дефицит рецептора Mas усугубляет вызванное липополисахаридом церебральное и системное воспаление у мышей. Иммунобиология 220 , 1311–1321 (2015).
CAS
PubMed
Google Scholar
Пассос-Сильва, Д. Г., Верано-Брага, Т. и Сантос, Р. А. Ангиотензин- (1-7): за пределами сердечно-почечной деятельности. Clin. Sci. 124 , 443–456 (2013).
CAS
PubMed
Google Scholar
Magalhaes, G. S. et al. Ангиотензин- (1-7) ослабляет ремоделирование дыхательных путей и гиперреактивность в модели хронического аллергического воспаления легких. руб. J. Pharmacol. 172 , 2330–2342 (2015).
CAS
PubMed
PubMed Central
Google Scholar
Jawien, J. et al. Агонист Mas-рецептора ангиотензина (1-7) улучшает развитие атеросклероза у мышей с нокаутом по ароЕ. J. Physiol. Pharmacol. 63 , 77–85 (2012).
CAS
PubMed
Google Scholar
Tesanovic, S., Vinh, A., Gaspari, T. A., Casley, D. & Widdop, R.E. Вазопротекторные и атеропротекторные эффекты ангиотензина (1-7) у мышей с дефицитом аполипопротеина E. Артериосклер. Тромб. Васк. Биол. 30 , 1606–1613 (2010).
CAS
PubMed
Google Scholar
Magalhaes, G. S. et al. Ангиотензин- (1-7) способствует разрешению эозинофильного воспаления в экспериментальной модели астмы. Фронт. Иммунол. 9 , 58 (2018).
PubMed
PubMed Central
Google Scholar
Dhande, I., Ma, W. & Hussain, T. Стимуляция рецептора ангиотензина AT2 оказывает противовоспалительное действие в макрофагах THP-1, активируемых липополисахаридами, за счет увеличения продукции интерлейкина-10. Гипертоническая болезнь. Res. 38 , 21–29 (2015).
CAS
PubMed
Google Scholar
Dhande, I., Ali, Q. & Hussain, T. Рецепторы ангиотензина AT2 проксимальных канальцев опосредуют противовоспалительный ответ через интерлейкин-10: роль в ренопротекции у крыс с ожирением. Гипертония 61 , 1218–1226 (2013).
CAS
PubMed
PubMed Central
Google Scholar
Kaschina, E. et al. Стимуляция рецепторов ангиотензина II типа 2: новый вариант терапевтического вмешательства в систему ренин-ангиотензин при инфаркте миокарда? Тираж 118 , 2523–2532 (2008).
CAS
PubMed
Google Scholar
Gonzalez, L. et al. Ангиотензин (1-9) снижает сердечно-сосудистые и почечные воспаления при экспериментальной ренин-независимой гипертензии. Biochem.Pharmacol. 156 , 357–370 (2018). В этой статье описывается, что ангиотензин 1–9 является противовоспалительным средством .
CAS
PubMed
Google Scholar
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00886353 (2009).
Национальная медицинская библиотека США. ClinicalTrials.gov https: // Clinicaltrials.gov / ct2 / show / NCT00771810 (2017).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01884051 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03177603 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03000686 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03252093 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02245230 (2018).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT025
(2018).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02646475 (2018).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03159988 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03615196 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03240068 (2019).
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03604289 (2019).
Basu, R. et al. Роль пептидов ангиотензина и рекомбинантного человеческого ACE2 при сердечной недостаточности. J. Am. Coll. Кардиол. 69 , 805–819 (2017).
CAS
PubMed
Google Scholar
Ocaranza, M. P. et al. Ангиотензин- (1-9) регулирует гипертрофию сердца in vivo и in vitro. J. Hypertens. 28 , 1054–1064 (2010). В данной статье впервые описывается антигипертрофический эффект ангиотензина 1–9 .
CAS
PubMed
Google Scholar
Kluskens, L.D. et al. Ангиотензин- (1-7) с тиоэфирным мостиком: устойчивый к ангиотензинпревращающим ферментам мощный аналог ангиотензина (1-7). J. Pharmacol. Exp. Ther. 328 , 849–854 (2009).
CAS
PubMed
Google Scholar
Pandey, A. & Gaikwad, A. B. Соединение 21 агониста рецептора AT2: серебряная подкладка для диабетической нефропатии. евро. J. Pharmacol. 815 , 251–257 (2017).
CAS
PubMed
Google Scholar
Wan, Y. et al. Дизайн, синтез и биологическая оценка первого селективного непептидного агониста рецептора AT2. J. Med. Chem. 47 , 5995–6008 (2004).
CAS
PubMed
Google Scholar
Калра, С., Калра, Б. и Агравал, Н. Комбинированная терапия гипертонии: обновление. Диабетол. Метаб. Syndr. 2 , 44 (2010).
PubMed
PubMed Central
Google Scholar
Грэдман, А. Х., Базиль, Дж. Н., Картер, Б.Л., Бакрис, Г. Л. и писательская группа Американского общества гипертонии. Комбинированная терапия при артериальной гипертензии. J. Am. Soc. Гипертония 4 , 42–50 (2010).
CAS
Google Scholar
Гебрейоханнес, Э. А., Бхагаватула, А. С., Абебе, Т. Б., Тефера, Ю. Г. и Абегаз, Т. М. Побочные эффекты и несоблюдение антигипертензивных препаратов в комплексной специализированной больнице Гондарского университета. Clin. Гипертоническая болезнь. 25 , 1 (2019).
PubMed
PubMed Central
Google Scholar
Vrijens, B., Vincze, G., Kristanto, P., Urquhart, J. & Burnier, M. Приверженность назначенному антигипертензивному лечению: продольное изучение историй дозирования, составленных в электронном виде. BMJ 336 , 1114–1117 (2008).
PubMed
PubMed Central
Google Scholar
Burnier, M. & Egan, B.M. Соблюдение режима лечения артериальной гипертензии. Circ. Res. 124 , 1124–1140 (2019).
CAS
PubMed
Google Scholar
Grobe, J. L. et al. Предотвращение индуцированного ангиотензином II ремоделирования сердца с помощью ангиотензина (1-7). Am. J. Physiol. Heart Circ. Physiol. 292 , H736 – H742 (2007).
CAS
PubMed
Google Scholar
Machado-Silva, A., Passos-Silva, D., Santos, R.A. и Sinisterra, R.D. Терапевтическое применение ангиотензина- (1-7). Мнение эксперта. Ther. Пат. 26 , 669–678 (2016).
CAS
PubMed
Google Scholar
Yang, J. et al. Сравнение ангиотензин- (1-7), лозартана и их комбинации на образование атеросклеротических бляшек у мышей с нокаутом аполипопротеина E. Атеросклероз 240 , 544–549 (2015).
CAS
PubMed
Google Scholar
Янг Д., Уайтчес Г., Бирчмайер К., Фазано О. и Виглер М. Выделение и характеристика нового клеточного онкогена, кодирующего белок с множеством потенциальных трансмембранных доменов. Cell 45 , 711–719 (1986).
CAS
PubMed
Google Scholar
Джонсон, Х. и Драммер, О.H. Гидролиз ангиотензина I пептидазами в гомогенатах легкого и аорты крысы. Biochem. Pharmacol. 37 , 1131–1136 (1988).
CAS
PubMed
Google Scholar
Santos, R.A. et al. Преобразование активности ферментов и метаболизма ангиотензина в стволе мозга собаки. Гипертония 11 , I153 – I157 (1988).
CAS
PubMed
Google Scholar
Campagnole-Santos, M. J. et al. Сердечно-сосудистые эффекты ангиотензина (1-7), вводимого в спинной мозг крыс. Am. J. Physiol. 257 , h424 – h429 (1989).
CAS
PubMed
Google Scholar
Tipnis, S. R. et al. Человеческий гомолог ангиотензин-превращающего фермента. Клонирование и функциональная экспрессия нечувствительной к каптоприлу карбоксипептидазы. J. Biol. Chem. 275 , 33238–33243 (2000).
CAS
PubMed
Google Scholar
Donoghue, M. et al. Новая карбоксипептидаза, связанная с ангиотензинпревращающим ферментом (ACE2), превращает ангиотензин I в ангиотензин 1-9. Circulation Res. 87 , E1 – E9 (2000).
CAS
PubMed
Google Scholar
Феррейра А. Дж., Сантос Р. А. и Алмейда А. П. Ангиотензин- (1-7): кардиозащитный эффект при ишемии / реперфузии миокарда. Гипертония 38 , 665–668 (2001).
CAS
PubMed
Google Scholar
Национальная медицинская библиотека США. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00471562 (2017).
Liu, C. et al. Аламандин снижает артериальную гипертензию и гипертрофию сердца у гипертонических крыс. Аминокислоты 50 , 1071–1081 (2018).
CAS
PubMed
PubMed Central
Google Scholar
Парк, Б. М., Фуонг, Х. Т. А., Ю, Л. и Ким, С. Х. Аламандин защищает сердце от реперфузионного повреждения через рецептор MrgD. Circ. J. 82 , 2584–2593 (2018).
CAS
PubMed
Google Scholar
Danser, A.H. et al. Сердечный ренин и ангиотензины. Поглощение из плазмы по сравнению с синтезом in situ. Гипертония 24 , 37–48 (1994).
CAS
PubMed
Google Scholar
Лоуренс, А.С., Эвин, Г., Кладис, А. и Кэмпбелл, Д. Дж. Альтернативная стратегия радиоиммуноанализа пептидов ангиотензина с использованием антисыворотки, направленной на амино-конец: измерение восьми пептидов ангиотензина в плазме человека. J. Hypertens. 8 , 715–724 (1990).
CAS
PubMed
Google Scholar
Alexiou, T. et al. Число копий гена ангиотензиногена и ангиотензинпревращающего фермента и уровни пептидов ангиотензина и брадикинина у мышей. J. Hypertens. 23 , 945–954 (2005).
CAS
PubMed
Google Scholar
Ocaranza, M. P. et al. Эналаприл ослабляет регуляцию ангиотензин-превращающего фермента 2 в поздней фазе желудочковой дисфункции у крыс с инфарктом миокарда. Гипертония 48 , 572–578 (2006). Это исследование является первым, показывающим, что ангиотензин 1–9 противодействует патофизиологическим эффектам ангиотензина II .
CAS
PubMed
Google Scholar
Шарп С., Поглич М., Зилла П., Дэвис Н. Х. и Старрок Е. Д. Фармакодинамические эффекты С-домен-специфичных ингибиторов АПФ на ренин-ангиотензиновую систему у крыс с инфарктом миокарда. J. Renin Angiotensin Aldosterone Syst. 16 , 1149–1158 (2015).
CAS
PubMed
Google Scholar
Haschke, M. et al. Фармакокинетика и фармакодинамика рекомбинантного человеческого ангиотензин-превращающего фермента 2 у здоровых людей. Clin. Фармакокинет. 52 , 783–792 (2013).
CAS
PubMed
Google Scholar
Chappell, M. C. Биохимическая оценка ренин-ангиотензиновой системы: хорошее, плохое и абсолютное? Am. J. Physiol. Heart Circ. Physiol. 310 , h237 – h252 (2016).
PubMed
Google Scholar
Лорти, М., Барк, С., Бланц, Р. и Хук, В. Обнаружение вазоактивных пептидов с низким содержанием в плазме: прогресс в направлении абсолютного количественного определения с использованием наножидкостной хромато-масс-спектрометрии. Анал. Biochem. 394 , 164–170 (2009).
CAS
PubMed
PubMed Central
Google Scholar
Агилера, Г.Роль подтипов рецепторов ангиотензина II в регуляции секреции альдостерона в зоне клубочков надпочечников у крыс. Мол. Клетка. Эндокринол. 90 , 53–60 (1992).
CAS
PubMed
Google Scholar
Qadri, F. et al. Вызванное ангиотензином II высвобождение вазопрессина опосредуется через адренорецепторы альфа-1 и рецепторы AT1 ангиотензина II в супраоптическом ядре. J. Pharmacol. Exp.Ther. 267 , 567–574 (1993).
CAS
PubMed
Google Scholar
Хуанг, Б.С., Чен, А., Ахмад, М., Ван, Х. В. и Линен, Ф. Х. Минералокортикоидные рецепторы и рецепторы AT1 в паравентрикулярном ядре способствуют гиперактивности симпатической нервной системы и сердечной дисфункции у крыс после инфаркта миокарда. J. Physiol. 592 , 3273–3286 (2014).
CAS
PubMed
PubMed Central
Google Scholar
Айер, С. Н., Лу, Д., Катович, М. Дж. И Райзада, М. К. Хронический контроль высокого кровяного давления у крыс со спонтанной гипертензией путем доставки антисмысловых рецепторов ангиотензина типа 1. Proc. Natl Acad. Sci. США 93 , 9960–9965 (1996).
CAS
PubMed
Google Scholar
Li, Q., Feenstra, M., Pfaffendorf, M., Eijsman, L. & van Zwieten, P.A. Сравнительные сосудосуживающие эффекты ангиотензина II, III и IV в изолированной подкожной вене человека. J. Cardiovasc. Pharmacol. 29 , 451–456 (1997).
CAS
PubMed
Google Scholar
Садошима Дж. И Изумо С. Молекулярная характеристика индуцированной ангиотензином II гипертрофии сердечных миоцитов и гиперплазии сердечных фибробластов. Критическая роль подтипа рецептора AT1. Circ. Res. 73 , 413–423 (1993).
CAS
PubMed
Google Scholar
Schieffer, B. et al. Сравнительные эффекты хронического ингибирования ангиотензин-превращающего фермента и блокады рецепторов ангиотензина II типа 1 на ремоделирование сердца после инфаркта миокарда у крыс. Тираж 89 , 2273–2282 (1994).
CAS
PubMed
Google Scholar
Wolf, G. et al. Ангиотензин II активирует ядерный фактор транскрипции-каппаB через рецепторы AT1 и AT2. Kidney Int. 61 , 1986–1995 (2002).
CAS
PubMed
Google Scholar
Viswanathan, M., Stromberg, C., Seltzer, A. & Saavedra, J. M. Баллонная ангиопластика усиливает экспрессию рецепторов AT1 ангиотензина II в неоинтиме аорты крысы. J. Clin. Вкладывать деньги. 90 , 1707–1712 (1992).
CAS
PubMed
PubMed Central
Google Scholar
Jara, Z. P. et al. Сверхэкспрессия тонина у мышей снижает симпатическую вегетативную модуляцию и изменяет ответ рецептора ангиотензина 1 типа. Фронт. Med. 5 , 365 (2018).
Google Scholar
де Кейроз, Т. М., Монтейро, М. и Брага, В. А. Активные формы кислорода, полученные из ангиотензина-II, на чувствительность барорефлекса во время гипертонии: новые перспективы. Фронт. Physiol. 4 , 105 (2013).
PubMed
PubMed Central
Google Scholar
Kihara, M. et al. Ангиотензин II подавляет индуцированную интерлейкином-1 бета продукцию оксида азота в культивируемых мезангиальных клетках крыс. Kidney Int. 55 , 1277–1283 (1999).
CAS
PubMed
Google Scholar
van der Mark, J. & Kline, R. L. Натрийурез измененного давления при хронической гипертензии ангиотензина II у крыс. Am. J. Physiol. 266 , R739 – R748 (1994).
PubMed
Google Scholar
Парк, Б. М., Ча, С. А., Ли, С. Х. и Ким, С. Х. Ангиотензин IV защищает сердечное реперфузионное повреждение, подавляя апоптоз и воспаление через AT4R у крыс. Пептиды 79 , 66–74 (2016).
CAS
PubMed
Google Scholar
Ханда, Р. К., Кребс, Л. Т., Хардинг, Дж. У. и Ханда, С. Е. Ангиотензин IV АТ4-рецепторная система в почках крысы. Am. J. Physiol. 274 , F290 – F299 (1998).
CAS
PubMed
Google Scholar
Крамар, Э.А., Кришнан, Р., Хардинг, Дж. У. и Райт, Дж. У. Роль оксида азота в индуцированном ангиотензином IV увеличении церебрального кровотока. Регул. Pept. 74 , 185–192 (1998).
CAS
PubMed
Google Scholar
Coleman, J. K. et al. Авторадиографическая идентификация участков связывания ангиотензина IV в почках и индуцированных ангиотензином IV изменений коркового кровотока почек у крыс. пептидов 19 , 269–277 (1998).
CAS
PubMed
Google Scholar
Qiu, H. et al. Влияние берберина на сигнальный путь PPARalpha-NO при пролиферации гладкомышечных клеток сосудов, индуцированной ангиотензином IV. Pharm. Биол. 55 , 227–232 (2017).
CAS
PubMed
Google Scholar
Padia, S.H. et al. Превращение почечного ангиотензина II в ангиотензин III имеет решающее значение для опосредованного рецептором AT2 натрийуреза у крыс. Гипертония 51 , 460–465 (2008).
CAS
PubMed
Google Scholar
Фонтес, М.A. et al. Доказательства того, что ангиотензин- (1-7) играет роль в центральном контроле артериального давления в вентро-латеральном мозговом веществе, действуя через специфические рецепторы. Brain Res. 665 , 175–180 (1994).
CAS
PubMed
Google Scholar
Xia, H. & Lazartigues, E. Ангиотензин-превращающий фермент 2: центральный регулятор сердечно-сосудистой функции. Curr. Гипертоническая болезнь. Реп. 12 , 170–175 (2010).
CAS
PubMed
PubMed Central
Google Scholar
Li, P., Chappell, M. C., Ferrario, C. M. & Brosnihan, K. B. Ангиотензин (1-7) усиливает индуцированную брадикинином вазодилатацию, конкурируя с АПФ и высвобождая оксид азота. Гипертония 29 , 394–400 (1997).
CAS
PubMed
Google Scholar
DelliPizzi, A.М., Хилчи, С. Д., Белл-Квилли, С. П. Натрийуретическое действие ангиотензина (1-7). руб. J. Pharmacol. 111 , 1–3 (1994).
CAS
PubMed
PubMed Central
Google Scholar
Garcia-Espinosa, MA, Shaltout, HA, Gallagher, PE, Chappell, MC & Diz, D.I. Экспрессия ангиотензина (1-7) in vivo снижает артериальное давление и улучшает функцию барорефлекса у трансгенных (mRen2) 27 крыс. J. Cardiovasc. Pharmacol. 60 , 150–157 (2012).
CAS
PubMed
PubMed Central
Google Scholar
Sakima, A. et al. Регуляция барорецепторного рефлекса у анестезированных трансгенных крыс с низким уровнем ангиотензиногена, производного от глии. Am. J. Physiol. Heart Circ. Physiol. 292 , h2412 – h2419 (2007).
CAS
PubMed
Google Scholar
Соарес, Э. Р., Барбоза, К. М., Кампаньол-Сантос, М. Дж., Сантос, Р. А. С. и Альзамора, А. С. Гипотензивный эффект, вызванный микроинъекцией аламандина, производного ангиотензина (1-7), в хвостовой вентролатеральный мозг крыс с гипертензией 2K1C. Пептиды 96 , 67–75 (2017).
CAS
PubMed
PubMed Central
Google Scholar
Абдалла, С., Лотер, Х., Абдель-Таваб, А. М. и Квиттерер, У.Рецептор AT2 ангиотензина II является антагонистом рецептора AT1. J. Biol. Chem. 276 , 39721–39726 (2001).
CAS
PubMed
Google Scholar
Bedecs, K. et al. Рецепторы ангиотензина II типа 2 опосредуют ингибирование каскада митоген-активируемых протеинкиназ и функциональную активацию тирозинфосфатазы SHP-1. Biochem. J. 325 , 449–454 (1997).
CAS
PubMed
PubMed Central
Google Scholar
Хориучи М., Акисита М. и Дзау В. Дж. Молекулярный и клеточный механизм апоптоза, опосредованного ангиотензином II. Endocr. Res. 24 , 307–314 (1998).
CAS
PubMed
Google Scholar
Senbonmatsu, T. et al. Новый сигнальный путь рецептора ангиотензина II типа 2: возможная роль в сердечной гипертрофии. EMBO J. 22 , 6471–6482 (2003).
CAS
PubMed
PubMed Central
Google Scholar
Gao, S. et al. Агонист рецептора ангиотензина AT2 стимулирует секрецию ANP, индуцированную высоким растяжением, посредством пути PI3K / NO / sGC / PKG /. Пептиды 47 , 36–44 (2013).
CAS
PubMed
Google Scholar
Ча, С. А., Парк, Б. М., Гао, С. и Ким, С. Х. Стимуляция ПНП ангиотензином- (1-9) через рецептор ангиотензина 2 типа. Life Sci. 93 , 934–940 (2013).
CAS
PubMed
Google Scholar
Абадир П. М., Периасами А., Кэри Р. М. и Сираджи Х. М. Функциональная гетеродимеризация рецептора ангиотензина II типа 2-рецептора брадикинина B2. Гипертония 48 , 316–322 (2006).
CAS
PubMed
Google Scholar
Tao, X. et al. Ангиотензин- (1-7) ослабляет индуцированную ангиотензином II передачу сигналов, связанную с активацией тирозинфосфатазы в сердечных фибробластах крыс Sprague-Dawley. Biol. Клетка. 106 , 182–192 (2014).
CAS
PubMed
Google Scholar
McCollum, L.T., Gallagher, P.E. и Tallant, E.A. Ангиотензин- (1-7) отменяет стимулируемую митогенами пролиферацию сердечных фибробластов. пептидов 34 , 380–388 (2012).
CAS
PubMed
PubMed Central
Google Scholar
Shah, A. et al. Ангиотензин- (1-7) стимулирует высокую секрецию ANP, вызванную стимуляцией предсердий, через ось Mas / PI3-киназы / Akt и обменник Na + / H + . Am. J. Physiol. Heart Circ. Physiol. 298 , h2365 – h2374 (2010 г.).
CAS
PubMed
Google Scholar
Wang, L.P. et al. Защитная роль ACE2-Ang- (1-7) -Mas при фиброзе миокарда за счет подавления канала KCa3.1 через путь ERK1 / 2. Pflug. Arch. 468 , 2041–2051 (2016).
CAS
Google Scholar
Карвер, К. А., Смит, Т. Л., Галлахер, П. Э. и Таллант, Э. А. Ангиотензин- (1-7) предотвращает индуцированный ангиотензином II фиброз в микрососудах кремастера. Микроциркуляция 22 , 19–27 (2015).
CAS
PubMed
Google Scholar
Gomes, E.R. et al. Ангиотензин- (1-7) предотвращает патологическое ремоделирование кардиомиоцитов посредством 3 ‘, 5’-циклического монофосфат-зависимого пути оксида азота / гуанозина. Гипертония 55 , 153–160 (2010).
CAS
PubMed
Google Scholar
Обзор системы ренин-ангиотензин
- белок (в частности, гормон), секретируемый печенью, который служит исходным материалом для производства ангиотензина 1 и 2
- фермента, секретируемого и хранящегося в почках, который способствует выработке ангиотензина из ангиотензиногена
- мембраносвязанного фермента, который превращает ангиотензин 1 в активный сосудосуживающий ангиотензин 2
- мембраносвязанный фермент, который превращает ангиотензин 2 в сосудорасширяющий ангиотензин 1-7
- образование аномального количество фиброзной ткани в органе или другой части тела в результате воспаления, раздражения или заживления
- высокое кровяное давление
- сердечное лекарство, которое увеличивает количество крови, перекачиваемой вашим сердцем, и понижает кровяное давление путем расширения или расширения , ваши кровеносные сосуды
- вещество, которое расширяет (открывает) кровеносные сосуды, что позволяет крови течь больше легко
- вещество, которое сужает или закрывает кровеносный сосуд, что, в свою очередь, увеличивает кровяное давление
Вы знаете, когда вы идете к врачу, и они вставляют вашу руку в эту манжету, а затем сильно сжимают ее, чтобы проверить и сделать уверен, что ваше кровяное давление в пределах нормы? Что ж, оказывается, у нашего тела есть отличная система, позволяющая следить за тем, чтобы наше кровяное давление всегда оставалось под контролем, когда меняется диета, мы испытываем стресс или неизбежно забываем пить достаточно воды…
В наших телах наши хорошие друзья печень и почки усердно работают, производя ангиотензиноген — пептид (или короткую серию аминокислот, связанных вместе) — и фермент под названием ренин. Оба попадают в кровоток, где ренин отщепляет три аминокислотные субъединицы (или остатки) на конце ангиотензиногена с образованием нового пептида, называемого ангиотензин 1. И теперь, похоже, подходящее время упомянуть, что это действительно круто «сохранить кровь» система нормального давления »на самом деле называется ренин-ангиотензиновой системой (или иногда ренин-ангиотензин-альдостероновой системой).Итак, теперь, когда мы получили наш ангиотензин 1, настало время для этого пептида двигаться дальше, пока он не обнаружит фермент АПФ или ангиотензин-превращающий фермент, торчащий с поверхности соседней клетки. Когда ангиотензин 1 сталкивается с АПФ, фермент удаляет еще два остатка с конца ангиотензина 1, тем самым превращая этот пептид в его близкого члена семьи, ангиотензин 2.
А вот где история немного изменится. Итак, ангиотензин 1 был довольно ненавязчивым маленьким пептидом, который в основном существует только для того, чтобы превращаться в ангиотензин 2.С другой стороны, ангиотензин 2 играет довольно важную роль, помогая нам регулировать кровяное давление и объем жидкости, но, как и некоторые системы в нашем организме, слишком много хорошего может стать плохим. Если ангиотензина 2 слишком много, это заставляет наши кровеносные сосуды сужаться, что, в свою очередь, увеличивает кровяное давление. Ангиотензин 2 также увеличивает задержку жидкости, что также повышает кровяное давление. Этот избыток жидкости может вызвать рост сердца, что, как вы понимаете, может привести к довольно серьезным проблемам.Если этого было недостаточно, ангиотензин 2 также может вызвать воспаление и ремоделирование наших кровеносных сосудов, что может привести к фиброзу или утолщению ткани, а также может оказать пагубное воздействие на мозг. Итак, как мы можем убедиться, что мы не склоним чашу весов и не имеем слишком большого количества ангиотензина 2, которое разрастается и вызывает все плохие вещи, которые я только что перечислил?
Что ж, один из способов сделать это — принять лекарство, называемое ингибитором АПФ. Возможно, вы слышали об этом типе лекарств раньше, но теперь вы точно знаете, почему это такое важное лекарство для тех, кто борется с высоким кровяным давлением.Входят ингибиторы АПФ, и они блокируют преобразование АПФ ангиотензина 1 в ангиотензин 2. Я знаю, что это звучит плохо, потому что я сказал вам, что нам действительно нужен ангиотензин 2 для регулирования артериального давления, но не волнуйтесь, у нас много ферментов АПФ и препарат не блокирует их все, он просто помогает гарантировать, что мы не перегружаем нашу систему ангиотензином 2, который затем вызывает плохие вещи.
В нашем организме также есть то, что часто называют природным ингибитором АПФ, которым является не что иное, как ангиотензин-превращающий фермент 2 или АПФ2.Термин «ингибитор» подразумевает, что ACE2 выполняет то же действие, что и лекарство, а именно останавливает выработку ангиотензина 2. На самом деле это не так, поэтому вместо этого давайте назовем ACE2 отклонителем ангиотензина 2. Он в основном берет ангиотензин 2, который производит АПФ, и расщепляет его до пептида, который не повышает кровяное давление. Фактически, ACE2 превращает ангиотензин 2 в ангиотензин (1-7), который на самом деле является сосудорасширяющим средством, поэтому он прямо противодействует функции ангиотензина 2. Первоначально ACE2 был открыт в контексте сердечных заболеваний и сердечной недостаточности, и хотя это критически важно для предотвращения гипертонии (или высокого кровяного давления) и сохранения функции сердца этот фермент также экспрессируется в широком диапазоне тканей.ACE2 обнаружен в органах, таких как легкие, желудочно-кишечный тракт, кожа, мышцы и почки, что указывает на то, что он может играть более широкую роль в других биологических процессах, помимо гомеостаза артериального давления. Многие различные исследовательские группы изучают, какую еще роль ACE2 может играть при недоедании, заболеваниях почек, диабете, заболеваниях легких и даже вирусных инфекциях, поэтому этот маленький белок действительно может помочь нам лучше понять другие насущные вопросы о здоровье и болезнях человека.
Понимание оси ренин-ангиотензин-альдостерон-SARS-CoV: всесторонний обзор
Резюме
Важность: Коронавирусная болезнь 19 (COVID-19), заболевание, вызванное тяжелым острым респираторным синдромом коронавируса 2 (SARS). -CoV-2) была объявлена глобальной пандемией со значительной заболеваемостью и смертностью с момента ее первого появления в Ухане, Китай, в конце 2019 года. Поскольку многие страны борются с началом своих эпидемий, фармакотерапевтических средств по-прежнему не хватает.Окно возможностей для снижения заболеваемости и смертности в низовьях мало, но остается открытым. Ренин-ангиотензин-альдостероновая система (РААС) имеет решающее значение для гомеостаза сердечно-сосудистой и дыхательной систем. Важно отметить, что SARS-CoV-2 напрямую использует и прерывает этот путь, который можно описать как ось ренин-ангиотензин-альдостерон-SARS-CoV-2 (ось RAAS-SCoV). Существуют значительные разногласия и путаница относительно того, как гипотензивные средства могут действовать на этом пути.В этом обзоре исследуется текущее состояние знаний относительно оси RAAS-SCoV, основанное на предыдущих исследованиях SARS-CoV, как это связано с нашей в настоящее время развивающейся пандемией и как эти идеи могут направлять наши следующие шаги на основе фактических данных.
Наблюдения: В этом обзоре обсуждается роль оси RAAS-SCoV в остром повреждении легких, а также эффекты, риски и преимущества фармакологической модификации этой оси. Может появиться возможность использовать различные аспекты ингибиторов РААС для смягчения косвенного повреждения легких, вызванного вирусами.Высказывались опасения, что такая модуляция может усугубить болезнь. В то время как соответствующие доклинические экспериментальные модели на сегодняшний день подтверждают защитный эффект ингибирования оси RAAS-SCoV как на повреждение легких, так и на выживаемость, клинические данные, касающиеся роли модуляции RAAS в настройке SARS-CoV-2, остаются ограниченными.
Заключение: Предлагаемые вмешательства для SARS-CoV-2 в основном сосредоточены на вирусной микробиологии и направлены на подавление вирусного клеточного повреждения. Хотя эти методы лечения являются многообещающими, немедленное использование может оказаться невозможным, и временное окно их эффективности остается главным вопросом без ответа.Альтернативный подход — это модуляция специфических патофизиологических эффектов, вызываемых вирусом, которые приводят к заболеваемости и смертности. Мы предлагаем преобладание доказательств, подтверждающих клиническое равновесие в отношении эффективности вмешательств на основе RAAS, и неизбежную потребность в рандомизированном контролируемом клиническом исследовании с несколькими участками для оценки ингибирования оси RAAS-SCoV при остром повреждении легких при COVID-19.
Abstract
Взаимодействие SARS-CoV-2 с системой ренин-ангиотензин-альдостерон, вероятно, объясняет большую часть его уникальной патологии.Оценка степени и механизма этого взаимодействия подчеркивает потенциальные терапевтические возможности, включая блокаду (БРА).
Введение
COVID-19, инфекционное заболевание, вызванное тяжелым острым респираторным синдромом, коронавирусом 2 (SARS-CoV-2), покинуло более 180 стран и территорий, борющихся с разрушительной пандемией. В декабре 2019 года эпицентром этой вспышки был назван Ухань, Китай. На момент подачи этой заявки количество зарегистрированных случаев COVID-19 превысило 700 000, при этом более 30 000 умерли [1–3]. (на момент подачи статьи). Хотя ранние оценки различаются, а истинные значения остаются неопределенными, смертность оценивается в пределах от 0,4 до 3,4% [4, 5], при этом исходная заболеваемость и смертность непропорционально затрагивают пожилых пациентов [6]. Инфекционность ( R 0 ) оценивается от 2,24 до 3,58 [4, 7]. Госпитализации, связанные с COVID-19, в основном связаны с необходимостью респираторной поддержки и все более высокого уровня помощи, при этом дыхательная недостаточность является основной этиологией смертей, связанных с COVID-19 [8, 9].Поскольку многие страны борются с началом эпидемий, фармакотерапевтических средств по-прежнему не хватает [10, 11]. Информация о предыдущих пандемиях и связанных с ними вирусах может направить наши усилия на борьбу с распространением и лечение инфицированных [12].
Этот новый коронавирус тесно связан с коронавирусом тяжелого острого респираторного синдрома (SARS-CoV), который вызвал вспышку заболевания (SARS) в 2003 году. Генетические исследования показали, что SARS-CoV-2 разделяет почти 80% и 50% последовательности идентичность с SARS-CoV и ближневосточным респираторным вирусом коронавируса (MERS-CoV) соответственно [13].Генетические различия между SARS-CoV и SARS-CoV-2 приводят к различиям в инфекционности [14] и иммунном ответе [15]. Несмотря на эти различия, общий геном трансформируется в общие клинические, микробиологические и биохимические фенотипы. SARS-CoV-2 имитирует механизм заражения SARS-CoV, который использует ангиотензин-превращающий фермент 2 (ACE2), часть ренин-ангиотензин-альдостероновой системы (RAAS). После связывания активность ACE2 подавляется с помощью множества механизмов, не позволяя ему выполнять свою обычную функцию в состоянии здоровья, что более подробно описано ниже [13, 16–18].Связь между ACE2 и COVID-19 основана на двух концепциях: механизме заражения SARS-CoV-2 и регулирующей роли ACE2 во время сверхактивации RAAS. После вспышки атипичной пневмонии обширные исследования расширили наши знания о высокопатологических коронавирусных инфекциях [16–25]. Сходные взаимодействия ренин-ангиотензин-альдостерон-SARS-CoV-2-ось (RAAS-SCoV-ось), общие для двух коронавирусов, дают возможность углубить наше понимание этого уникального взаимодействия в нашем поиске вариантов лечения.
Ранние попытки разработать безопасные и эффективные вакцины против SARS или MERS не увенчались успехом. Предполагается, что текущие усилия потребуют времени для развития и могут оказаться эффективными или неэффективными в отношении этой или будущих пандемий коронавируса [10, 26–28]. Было предложено множество потенциальных методов лечения, часть из которых в настоящее время проходит исследования [29, 30]. Между тем, на сегодняшний день обширные усилия были должным образом сосредоточены на скрининге, идентификации и локализации посредством сотрудничества и координации между глобальными и местными органами здравоохранения и правительственными учреждениями [31].К сожалению, эти усилия в области общественного здравоохранения имели в лучшем случае неоднозначный успех в сдерживании распространения пандемии [32]. Учитывая быстрое распространение SARS-COV2 и значительную заболеваемость и смертность, связанные с инфекцией, медицинскому сообществу надлежит оценивать и использовать новые методы лечения для повышения эффективности, особенно если они уже доступны и имеют установленный профиль безопасности.
Блокада RAAS была предложена в качестве потенциального лечения SARS-CoV-2 [33–36]. Эта гипотеза была первоначально опубликована Sun et al. [35, 37] 4 февраля 2020 г. и усилены в публикации «Исследования в области разработки лекарств» 4 марта 2020 г. [33]. В других обзорах высказывалась озабоченность по поводу связи между COVID-19 и сердечно-сосудистыми заболеваниями [38], они заходили слишком далеко, чтобы постулировать, что продолжение блокады РААС может причинить вред, и рекомендовали рассмотреть возможность прекращения приема [39]. Предыдущий аргумент основан на наблюдении, что фармакологические блокаторы РААС могут активировать экспрессию ACE2, что может увеличить проникновение вируса в клетку [39].Доказательства, полученные от людей в поддержку такой гипотезы, скудны, и, как мы увидим в этом обзоре, доклинические и текущие наблюдательные данные Covid-19 подтверждают противоположную гипотезу — прекращение блокады РААС может оказаться вредным. Эти противоположные гипотезы подчеркивают острую необходимость оценки потенциальных механизмов, если таковые имеются, посредством которых модуляция РААС могла бы влиять на патофизиологию COVID-19 [35, 38, 40]. В этом обзоре мы намерены собрать существующие доказательства, чтобы обсудить, как мы можем восполнить пробелы в знаниях о взаимодействии между SARS-CoV-2, ACE2 и RAAS.
РААС в состоянии здоровья
Обзор
Ренин, ангиотензин и альдостерон представляют собой ядро сложной гормональной оси, называемой РААС, которая способствует контролю артериального давления, реабсорбции натрия, воспалению и фиброзу [41]. Дисбаланс или модификация РААС могут вызывать или лечить многие заболевания, включая сердечную недостаточность, гипотонию, диабет и атеросклероз, соответственно [42]. В этом обзоре основное внимание уделяется нескольким физиологическим и патологическим эффектам передачи сигналов клетками ангиотензина II (Ang II) (рис.1).
РИСУНОК 1
Ренин-ангиотензиновая система с COVID-19. Ингибитор ангиотензинпревращающего фермента ACEi, ангиотензинпревращающий фермент ACE1, ангиотензинпревращающий фермент 2 ACE2, блокатор рецептора ангиотензина ARB, AT 1 R рецептор ангиотензина II типа 1, AT 2 R рецептор ангиотензина II типа 2, рекомбинантный человеческий фермент rhACE2 2.
Ангиотензин II / AT
1 Взаимосвязь рецепторов
Ангиотензин II (Ang II), основной физиологический продукт системы РААС, является сильнодействующим вазоконстриктором.Как показано на рисунке 1, АПФ катализирует превращение ангиотензина I (Ang I) в Ang II. Ang II вызывает свои эффекты, активируя два рецептора: рецептор ангиотензина II типа 1 (AT 1 ) и рецептор ангиотензина II типа 2 (AT 2 ) [43]. Действие Ang II через рецептор AT 1 вызывает каскад с последующим воспалением, сужением сосудов и атерогенезом [44]. Эти эффекты также способствуют развитию инсулинорезистентности и тромбозов [45]. Напротив, стимуляция рецептора AT 2 вызывает расширение сосудов, снижение агрегации тромбоцитов и усиление действия инсулина.Однако у здоровых взрослых экспрессия рецептора AT 2 низкая [45]. Таким образом, эффекты Ang II у взрослых модулируются и уравновешиваются косвенно ферментом, превращающим ангиотензин II (ACE2), который превращает Ang II в защитный для легких ангиотензин- (1-7) (Ang- [1-7]), аналогично эффектам. видно из стимуляции рецептора AT 2 [41, 46]. Концептуальное объединение путей в рецептор Ang II / AT 1 и противоположный ему путь ACE2 / Ang- (1–7) помогает понять противодействующие силы и последствия при возникновении дисбаланса (рис.1).
Последствия чрезмерного Ang II включают вазоконстрикцию легких, воспаление и вызванное цитокинами повреждение органов [47], вторичное по отношению к повышенной проницаемости мембран [48], и усиление апоптоза эпителиальных клеток [49]. Эффекты преимущественно опосредованы безальтернативной активацией рецептора AT 1 , вторичной по отношению к снижению уровней ACE2 [50]. Кроме того, провоспалительный каскад [51] и повышенная проницаемость сосудов [48], вызванные чрезмерной активацией рецептора AT 1 в легких, непосредственно вызывают острое повреждение легких (ALI), острый респираторный дистресс-синдром (ARDS) и приводят к смерти [10, 33].
Ангиотензин-превращающий фермент 2
ACE2 экспрессируется во многих типах тканей, включая клетки респираторного эпителия [52]. ACE2 преимущественно экспрессируется на апикальной поверхности и превращает Ang II в защищающие легкие Ang- (1–7) и Ang- (1–9), катализируя путь, который предотвращает беспрепятственную активацию рецептора AT 1 . Следовательно, ACE2 действует как противовес своему структурно схожему аналогу ACE [53] посредством преобразования Ang II в Ang- (1-7), который в отличие от ACE способствует расширению сосудов, снижает пролиферацию и предотвращает апоптоз [53].Известно, что уровни ACE2 низкие у здоровых людей [54], а уровни ACE2 снижаются с возрастом [55]. Хронически повышенные уровни ACE2 являются независимым предиктором прогрессирования заболевания; но данные свидетельствуют о том, что эти изменения являются скорее компенсаторными, чем причинными [54].
ИАПФ и БРА
И ингибиторы ангиотензинпревращающего фермента (ИАПФ), и блокаторы рецепторов ангиотензина (БРА) повышают уровни ренина и Ang I в тканях и плазме крови. (50) ИАПФ ингибируют преобразование Ang I в Ang II, уменьшая сывороточный Ang II уровни до исходного уровня и практически не имеют сродства к ACE2 [56].В клинических дозах АПФ лишь частично влияют на это превращение, потому что 40% образования Ang II происходит вне пути АПФ [57]. Уровни АПФ, в свою очередь, снижаются за счет отрицательной обратной связи в состояниях высоких уровней Ang II [58]. Уровни Ang II повышаются в ответ на БРА на всех уровнях, кроме почек. Следствием этого увеличения является перенаправление активности Ang II через противовоспалительные и сосудорасширяющие пути рецепторов ACE2 и AT 2 [56]. В результате ACE2, по сравнению с ACE, в большей степени регулирует локальные уровни как Ang II, так и Ang- (1–7) [42].Тикеллис и др. . предполагают, что БРА, следовательно, обладают более высоким потенциалом благоприятного восстановления баланса пути РААС по сравнению с ИАПФ [42]. Кроме того, калликреин-кининовая система регулируется ACE и ACE2. Повышенная активность системы калликреин-кинин происходит при провоспалительных условиях [59], в частности, брадикинин и дез-Arg-BK 9 , что может привести к утечке из сосудов, если не контролируется ACE и ACE2, соответственно. Было высказано предположение, что этот механизм, по крайней мере частично, является причиной кашля [60] и отека легких [61], независимо от предполагаемого гидростатического давления, индуцированного Ang II.Роль дисрегулируемой системы калликреин-кинин остается теоретической при COVID-19; однако, если это так, блокада рецептора AT 1 может быть предпочтительнее (по сравнению с ингибированием АПФ) в попытке сохранить регуляторные механизмы этой системы [61]. Хотя ингибирование АПФ все еще может играть ключевую роль в контексте текущего заболевания, которое мы разъясним в следующих разделах, эти наблюдения подтверждают наше решение сузить фокус этого обзора до модификаторов RAAS, расположенных ниже по течению, БРА.
Ключевые моменты 1
Уровни ACE2 высоки в болезненных состояниях, вероятно, вторично по отношению к недостаточной компенсаторной реакции на сверхактивную активность RAAS
При COVID-19 частичная причина возникновения отека легких и кашля. для уменьшения распада брадикинина из-за снижения активности АПФ
БРА могут обеспечить решающую регуляцию сверхактивной оси RAAS-SCoV
RAAS при сердечно-сосудистых заболеваниях
Модуляция оси RAAS является центральной для лечения сердечно-сосудистых заболеваний [62].Сердечно-сосудистые заболевания, сердечная недостаточность, фибрилляция предсердий и заболевание почек связаны с повышенным уровнем ACE2 [60]. Крайне важно различать, являются ли эти повышения причиной или последствиями заболевания, чтобы отделить связь от причинно-следственной связи [63]. Более свежие мнения предполагают, что ACE2 является биомаркером, а не виновником сердечно-сосудистых заболеваний [64].
Рамчанд и др. . продемонстрировали, что повышенные уровни ACE2 являются независимым фактором риска серьезных неблагоприятных сердечных событий у пациентов с известным заболеванием коронарной артерии, хотя эти данные, как отмечают авторы, не позволяют дифференцировать связь и причинно-следственной связи [54].Чтобы начать анализ этого взаимодействия, мы обратимся к доклиническим моделям, в частности к мышам с нокаутом ACE2, которые характеризуются тяжелыми сердечными дефектами. Критически важно, что эти дефекты заметно отсутствуют в моделях с двойным ACE и ACE2 нокаутом, что подчеркивает высоко сбалансированное взаимодействие между ACE / Ang II / AT 1 и ACE2 / Ang- (1-7) [53, 65]. Подобные защитные эффекты для предотвращения сердечно-сосудистых событий и инсультов были продемонстрированы на моделях мышей со сверхэкспрессией ACE2 [66]. Рахманд и др. .пришли к выводу, что пациенты с повышенным уровнем ACE2 могут «отражать стойкий, хотя и недостаточный контррегуляторный процесс, чтобы сместить баланс в сторону от пагубных эффектов устойчивой активации Ang II». [54] Как таковая, избыточная экспрессия ACE2 может быть компенсаторным механизмом для смягчения эффекта беспрепятственной стимуляции Ang II за счет увеличения деградации в защитный Ang- (1-7) [67].
Важно отметить, что лечение лозартаном на доклинических моделях животных активизирует экспрессию мРНК ACE2 в сердце и увеличивает активность ACE2 [68, 69].Однако исследования на людях не обнаружили существенной разницы в уровнях ACE2 в плазме у пациентов, получавших ACEi, или ARB не подтвердили такой эффект, хотя локальные тканевые уровни ACE2 не измерялись [54]. Судя по неофициальным сообщениям, COVID-19 может быть связан с высокой частотой миокардита и сердечной недостаточности у тяжелобольных. Хотя это выходит за рамки этого предложения, отметим, что повышенный Ang II и пониженный ACE2 также связаны с нарушением сократительной способности сердца, которое можно исправить с помощью лозартана [70].Необходимы дополнительные данные наблюдений, чтобы оценить, предвещают ли другие признаки перегрузки РААС (гипокалиемия, гипертензия) плохие результаты и могут ли быть связаны с аритмией, отмеченной при заболеваниях [71]. Тем не менее, это может потенциально подчеркнуть потенциальные преимущества модуляции RAAS, которые выходят за рамки ослабления повреждения легких.
Ключевые моменты 2
ИАПФ и БРА прерывают дезадаптивные патофизиологические реакции на повышенную активацию РААС.
БРА и АПФ смягчают пагубные последствия активации пути рецептора AT1, что, в свою очередь, снижает воспаление, инсулинорезистентность и повреждение легких (ХОБЛ), пациенты, получавшие БРА (по сравнению с пациентами, принимавшими ИАПФ), имели менее тяжелые обострения, меньше обострений в целом, более низкую смертность, более низкие требования к ИВЛ и меньшее количество госпитализаций [72].Более того, у ветеранов старше 65 лет, которые принимали БРА до и во время госпитализации по поводу пневмонии, снизилась смертность по сравнению с пациентами без такого лечения [73].
Острое повреждение легких
Незамедлительная активация RAAS через рецептор Ang II / AT 1 вызывает воспаление [51], повышенную проницаемость сосудов [48] и тяжелое повреждение легких [10, 33], тогда как БРА значительно ослабляют эти изменения [ 48–51]. Важно отметить, что простое присутствие высоких концентраций Ang II может дополнительно регулировать экспрессию ACE2, что приводит к нарушению регуляции активности рецептора Ang II / AT 1 [74].У мышей лозартан снижал смертность за счет подавления связанного с Ang II увеличения содержания растворимой эпоксидгидролазы, промотора повреждения легких [75]. На животных моделях повреждения легких, связанного с аппаратом искусственной вентиляции легких, продемонстрирована польза от лозартана, снижающего активность Ang II и экспрессию рецептора AT 1 [76–78]. В то время как большинство исследований включает предварительно обработанные животные модели, спасательные модели также демонстрируют эффективность с восстановлением уровней ACE2, замедлением снижения PaO2 и ослаблением повреждения легких [50].
Генетические когортные исследования пациентов-людей позволяют лучше понять взаимосвязь между РААС и острым повреждением легких. Джернг и др. . обнаружили, что полиморфизмы в гене ACE связаны с исходами при ОРДС. Эти данные были подтверждены Адамзиком и соавторами , которые определили пациентов с генотипом ACE DD (связанным с повышенной активностью ACE) как имеющие самый высокий риск смерти, связанной с ОРДС (HR 5,7) [79]. Другие исследования на людях, оценивающие связь между ингибированием РААС и ОРДС, остаются наблюдательными.Ким и др. . обнаружили, что пациенты с ОРДС, которые принимали ИАПФ или БРА, имели лучшую выживаемость по сравнению с пациентами без ингибирования РААС [80]. Вторичный анализ рандомизированного контрольного исследования 2010 г. с участием пациентов с острой дыхательной недостаточностью показал, что лечение ИАПФ / БРА при выписке после эпизода острой дыхательной недостаточности было связано со снижением годовой смертности на 44% [81]. Совсем недавно Hsieh et al . наблюдали более низкие скорректированные шансы госпитальной смертности у пациентов с сепсисом (с шоком и без него), которые получали терапию БРА или ИАПФ [82].Мортенсон и др. . также было обнаружено снижение на 58% вероятности госпитальной летальности у пациентов, принимавших БРА перед госпитализацией [83]. На основании вышеизложенного были высказаны призывы к дальнейшему выяснению потенциальных преимуществ ACEi и ARB при ОРДС [84]. Однако рандомизированные контрольные испытания по этой теме не были обнаружены в рецензируемой литературе или на сайте Clinicaltrials.gov на момент подготовки этой рукописи.
Пневмония
Хотя грипп и другие типы пневмонии могут взаимодействовать с осью RAAS, исследования как на животных, так и на людях демонстрируют явное косвенное влияние на RAAS, в частности, в условиях определенных штаммов гриппа.Предыдущие исследования предполагают, что уровни Ang II позволяют прогнозировать смертность у недифференцированных пациентов с гриппом [85], а продолжение терапии ингибиторами РААС во время госпитализации связано со снижением госпитальной летальности и вероятностью интубации в случаях вирусной пневмонии [86]. РААС может иметь значение и для других вирусных пневмоний, как Gu et al . обнаружили, что дети с респираторно-синцитиальным вирусом, как правило, имеют более высокие уровни Ang II по сравнению со здоровыми детьми [87]. Основываясь на этом наблюдении, они продемонстрировали преимущество терапии рекомбинантным ACE2 в отношении инфекции RSV на доклинической модели мыши.
Критически важно, что как H7N9, так и H5N1 грипп вызывают повреждение легких за счет подавления ACE2, усиления Ang II и индуцированного рецептором AT 1 повреждения легких [88, 89]. Мышиные модели H5N1 и H7N9 продемонстрировали снижение уровня IL-6, отека легких, повреждения легких и смертности при лечении лозартаном [89, 90]. Однако механизм, с помощью которого лозартан предотвращает повреждение легких, может находиться не только в пути RAAS [51]. Лю и др. . предполагают, что лозартан подавляет активацию дендритных клеток легких [91].В исследовании крыс с пневмонией pseudomonas , блокаторы рецепторов AT 1 подавляли активацию нейтрофилов посредством механизма, который не вовлекает подавление регуляции рецептора AT 1 [51, 92]. Такие исследования вызвали опасения, что лечение лозартаном может снизить микробный клиренс. Напротив, результаты исследования фактически демонстрируют снижение вирусной нагрузки [90] и увеличение бактериального клиренса [51] в моделях повреждения легких, получавших лозартан. Учитывая сложность этих взаимодействий, необходимы дальнейшие исследования для дальнейшего выяснения этих взаимосвязей.
ACE2-связывающая вирусная инфекция SARS-CoV
В 2003 году ACE2 был идентифицирован как связывающий белок для SARS-CoV. В последующие годы исследования, демонстрирующие нисходящие последствия подавления ACE2 с результирующим увеличением концентрации Ang II, иллюстрируют порочный круг, вызванный нарушением RAAS, мы называем в этой рукописи RAAS-SARS-CoV-2 (RAAS -SCoV-ось) [18]. Имеет отношение к текущей пандемии, Wrapp et al . обнаружили, что у SARS-CoV-2 сродство к ACE2 в 10-20 раз выше, чем у SARS-CoV.Авторы предполагают, что это может вызвать повышенную инфекционность, что может помочь объяснить различия в развитии этих двух эпидемий.
Оба коронавируса SARS содержат четыре структурных белка, нуклеокапсид, мембрану, оболочку и спайковый белок [93]. Нуклеокапсид, мембрана и белки оболочки играют роль в репликации вируса, структурной целостности и ответе хозяина среди других механизмов [93]. Белок-шип, известный как S-белок, является неотъемлемой частью прикрепления и заражения клеток-хозяев [14, 93].Фан и др. . определили конкретные делеции, которые увеличивают способность спайка SARS-CoV-2 ускользать от иммунных ответов со временем. Эти изменения влияют как на начальную, так и на последующую тяжесть инфекции, иммунитет и вероятность разработки эффективной вакцины. Однако более актуальным для настоящего обзора является потенциальное влияние таких изменений на ось RAAS-SCoV, что может служить объяснением не только их склонности вызывать дыхательную недостаточность, но, возможно, других общих черт между этими инфекциями.
Инфекция SARS-CoV подавляет поверхностную экспрессию связывающего белка, ACE2, основного компонента для проникновения в хозяйскую клетку [17]. Примечательно, что низкая экспрессия ACE2 связана с повышенной серьезностью фенотипа в исследованиях in vitro эпителиальных клеток дыхательных путей человека [24]. Имаи и др. . подтвердили эти результаты на модели всего животного, продемонстрировав, что потеря ACE2 у мышей с нокаутом (KO) приводит к нарушениям оксигенации, усилению воспаления и ухудшению отека по сравнению с мышами WT после повреждения легких, вызванного кислотой [23].Предварительная обработка ингибиторами рецептора AT 1 до индуцированного кислотой ОРДС показала значительно меньшее повреждение легких, что подтверждает роль рецептора AT 1 в физиологической реакции [23]. Впоследствии, что наиболее важно, Куба и др. . продемонстрировали, что спайковый белок SARS-CoV усиливает увеличение Ang II и подавление ACE2, что приводит к повреждению легких [17]. В этой модели лозартан может уменьшить тяжесть острого повреждения легких из-за инфекции SARS-CoV как в модели до лечения, так и в более клинически значимой модели после инфекции (спасения) [17].
Последующие исследования подтвердили и основывались на этих выводах [19]. Блокада рецептора AT 1 активирует ACE2 посредством механизма отрицательной обратной связи и служит защитным механизмом легких за счет повышенного превращения Ang II в Ang- (1–7), эффективно притупляя повреждение легких вирусом [50]. Эти находки привели исследователей к выводу, что путь ACE2 / Ang- (1-7) может служить терапевтической мишенью для борьбы с патологическими эффектами Ang II [74]. С этой целью уже существует по крайней мере одно испытание терапии рекомбинантным человеческим ACE2 для лечения COVID-19, в котором предпринимается попытка усилить эту взаимосвязь и смягчить последующее повреждение легких [23, 35].
Механизм, с помощью которого SARS-CoV подавляет ACE2, также может иметь отношение к разработке новых терапевтических средств. Хага и др. . продемонстрировали, что связывание SARS-CoV с ACE2 индуцирует выделение ACE2 в виде растворимой формы в сыворотку [20], и дальнейшие исследования подтвердили эти результаты [19]. Кроме того, эндоцитоз белка ACE2 происходит после связывания с SARS-CoV, дополнительно снижая активность ACE2 [94], с потенциальными последствиями для баланса Ang II / Ang1-7, описанными в этом обзоре.Последний критический механизм, который нельзя упускать из виду, включает роль рецептора AT 1 в подавлении ACE2. Desholtz и др. . обнаружили, что подавление ACE2 вызывает стойкое повышение Ang II за счет локального взаимодействия с рецептором AT 1 , что запускает порочный цикл, когда Ang II подавляет ACE2, что приводит к дальнейшему увеличению локальных уровней Ang II в тканях [74]. Эти механизмы стимулируют беспрепятственную ось рецептора Ang II / AT -1 (рис.1).
Взаимодействие ACE2-SARS / COVID-19 привело к противоречиям с клиническими последствиями. Обнаружение того, что мыши ACE2 KO защищены от инфекции SARS-CoV [17], вызывает опасение, что повышенная регуляция ACE2 вредна в условиях инфекций SARS-CoV и COVID, поскольку вирус не может проникнуть в клетку. И наоборот, подавление ACE2 после инфекции связано с рассмотрением доклинических данных, обобщенных к этому моменту, которые вместо этого предполагают предполагаемый причинный механизм Ang II / Ang (1–7) в развитии острого повреждения легких, связанного с SARS-CoV-2.Даже если бы отмена ACE2 была возможной, Han et al . выявили альтернативные рецепторы DC / L-SIGN, которые опосредуют проникновение SARS-CoV независимо от ACE2 [21].
Ключевые моменты 3
SARS-CoV снижает поверхностную экспрессию ACE2 во время инфекции
Снижение активности ACE2 приводит к увеличению Ang II и дальнейшему подавлению ACE2 в порочном круге, вызывая острое повреждение легких.
Хотя основная точка входа включает ACE2, другие рецепторы могут независимо опосредовать инфекцию SARS-CoV
Споры относительно причинной роли ACE2 в COVID-19
В настоящее время существуют ограниченные механистические исследования COVID-19 [95 ].Ранние китайские данные демонстрируют прогрессивное повышение уровня Ang-II среди пациентов, госпитализированных в условиях подтвержденного COVID-19 [96], наряду с сопутствующим повышением уровней IL-6, с самыми высокими уровнями у тех, кто не выжил [97]. Аналогичным образом, анализ пациентов с H7N9 в 2014 г. показал прогрессивное повышение уровней Ang-II в течение до 4 недель после заражения, что связано с худшим прогнозом [89]. В недавнем исследовании пациентов с COVID-19 количественные уровни Ang-II были положительно коррелированы с вирусными титрами и тесно связаны с показателями PaO2 / FiO2, что предполагает связь между инфекцией, RAAS и повреждением легких.Соответственно, исследователи постулировали, что блокада рецепторов ангиотензина может представлять собой многообещающую терапевтическую мишень [96].
Как отмечалось ранее, уровни ACE2, как известно, низкие у здоровых людей [54], снижаются с возрастом и ниже у мужчин, чем у женщин [55, 98]. Возраст является независимым фактором риска смертности пациентов, госпитализированных с COVID-19 [97]. Данные также свидетельствуют о непропорциональной частоте возникновения, тяжести и худших исходах заболевания у мужчин по сравнению с женщинами во время этой пандемии [99].Эти ассоциации можно интерпретировать как указание на то, что низкий уровень ACE2 в периоды здоровья уместен, но способность регулировать сверхактивную ось RAAS или ось RAAS-SCoV во время болезни, по крайней мере частично, зависит от ACE2. Например, одно исследование выявило более высокую экспрессию защищающего легкие рецептора AT 2 у женщин, что могло бы объяснить непропорционально более низкую смертность у женщин с COVID-19 [100]. Интересно, что ACE2 расположен на X-хромосоме, что, как предполагалось, влияет на способность мужчин поддерживать адекватное производство ACE2 в ситуациях тяжелого заболевания ( i.е. , ARDS или COVID-19) [40]
Возможно, наиболее важным является то, что наиболее частыми сопутствующими заболеваниями у тяжелых пациентов были артериальная гипертензия, сахарный диабет и ишемическая болезнь сердца [9]. Эти сопутствующие заболевания связаны либо с чрезмерной активацией Ang II / AT 1 , либо с дефицитом ACE2 [42, 45], и все они показали пользу от блокады RAAS при хронических состояниях [101], включая ослабление AT 1 . экспрессия рецепторов лозартаном [102]. Кроме того, курение имеет тенденцию увеличивать экспрессию рецептора AT 1 [102] наряду с экспрессией ACE2 в эпителиальных клетках человека и мыши [103, 104].Активация РААС в состоянии стресса с высокими уровнями циркулирующего Ang II может быть особенно вредной для таких пациентов и может объяснить, по крайней мере, некоторые из наблюдаемых клинических исходов. В частности, данные из Китая показали, что вероятность прогрессирования заболевания у людей с COVID-19 у курильщиков в 14 раз выше [8]. Однако мы признаем, что неверное истолкование причинно-следственной связи может быть опасным и что патология, лежащая в основе острых и хронических состояний, может не быть эквивалентной [105]. Тем не менее манипуляции с РААС остаются привлекательной терапевтической целью не для лечения или предотвращения инфекции COVID-19, а для смягчения ее последующих последствий.
Побочные эффекты манипуляции с РААС по понятным причинам вызывают беспокойство, особенно гипотензия и острое повреждение почек на фоне острого заболевания. Наиболее серьезные риски БРА включают тератогенность на фоне беременности, гиперчувствительность, включая ангионевротический отек, симптоматическую гипотензию, ухудшение функции почек и электролитные нарушения; однако наиболее частые побочные эффекты включают усталость, слабость, диарею, боль в груди и анемию. Данные клинических испытаний и постмаркетингового наблюдения лозартана демонстрируют превосходный профиль безопасности.У более чем 4000 пациентов частота нежелательных явлений была низкой (2,1%), по сравнению с плацебо (3,7%) [106]. Эффекты, более частые для лозартана, чем плацебо, были клинически незначительными, включая головокружение (3% против 2%), инфекцию верхних дыхательных путей (8%, против 7%), заложенность носа (2% против 1%) и обратно. боль (2% против 1%). Хотя лозартан может вызывать симптоматическую гипотензию, четыре исследования показали, что лозартан оказывает незначительное влияние на артериальное давление в состоянии покоя и частоту сердечных сокращений у здоровых добровольцев [107–110].Эти обнадеживающие данные нельзя экстраполировать на пациентов с COVID-19; однако при оценке соотношения риск / польза для модуляции РААС исходный риск оказывается низким в здоровой популяции, тогда как повторное использование других лекарств может не иметь такого же благоприятного профиля. Также есть опасения, что усиление ACE2 с помощью ARB может ускорить проникновение SARS-CoV-2 в клетки-хозяева. Такие опасения становятся более актуальными, если учесть наблюдение, что пациенты с гипертонией, как правило, имеют худшие результаты, и многие пациенты с гипертонией принимают агенты, которые могут повышать экспрессию ACE2.Следовательно, некоторые клиницисты выступали за прекращение приема ИАПФ и БРА и перевод своих пациентов на разные классы антигипертензивных средств. Однако, насколько нам известно, нет данных, позволяющих предположить, что такое изменение полезно, равно как и не существует хорошо контролируемых анализов с поправкой на риск, показывающих, что эти лекарства вредны.
Напротив, данные, обобщенные в этом обзоре, фактически предполагают, что прекращение приема ACEi или ARB может представлять серьезный риск вреда из-за распространения чрезмерного опосредованного Ang II острого повреждения легких и подавления защитного ACE2.К этому моменту в недавнем препринте Liu et al. ретроспективно изучил 511 пациентов с COVID-19 (возраст> 65 лет) с артериальной гипертензией. Пациенты были разделены на категории в зависимости от режима приема антигипертензивных препаратов в домашних условиях. У пациентов, принимающих БРА, значительно снизились шансы развития тяжелой болезни COVID-19 при однофакторном анализе (OR 0,34, p = 0,025) и многомерном анализе (OR 0,25, p = 0,046), в то время как ни один из других препаратов не продемонстрировал улучшения результатов. Два последующих ретроспективных исследования продемонстрировали защитный эффект блокады РАС, включая значительное снижение вирусной нагрузки [111, 112].Наконец, в дополнение к ингибирующим эффектам на AT1R, предпечатная публикация из Ирана [113] идентифицировала лозартан как потенциальный кандидат на лечение, основанное на молекулярном стыковке и динамическом моделировании. Таким образом, помимо эффектов на гомеостаз ангиотензина, лозартан может напрямую ингибировать вирусный макродомен и клеточный цикл [113]. Еще до этих данных наблюдений, основанных на доклинических данных и исследованиях, не связанных с COVID-19, Международное общество гипертонии, Европейское общество кардиологов и другие международные комитеты настоятельно рекомендовали врачам и пациентам продолжать лечение их обычными антибиотиками. -гипертензивная терапия, поскольку нет клинических или научных данных, позволяющих предположить, что лечение ИАПФ или БРА следует прекратить в условиях пандемии SARS-CoV-2 в настоящее время [114–116].
Заключение
Текущих клинических данных недостаточно, чтобы рекомендовать лечение или отмену блокады РААС во время этой пандемии. Несмотря на то, что повышенные уровни ACE2 обнаруживаются при хронических болезненных состояниях, повышенная регуляция ACE2 кажется скорее адаптивной реакцией, чем виновником [67]. Значительные доклинические данные и ретроспективные данные на людях предполагают, что ингибирование РААС снижает повреждение легких и улучшает выживаемость, одновременно снижая вирусную нагрузку на животных моделях с вирусными инфекциями, которые используют рецептор ACE2.Клинические данные о влиянии ингибиторов РААС на исходы остаются скромными и являются областью активных исследований. Хотя связь между артериальной гипертензией и плохими исходами может быть опосредована активацией ACE2 и повышением чувствительности к вирусам, эти дифференциальные исходы, по крайней мере, с такой же вероятностью являются результатом дисрегулируемого Ang II-опосредованного повреждения легких. Мы считаем, что это состояние клинического равновесия, и предполагаем, что ингибирование РААС может улучшить исходы у пациентов, инфицированных COVID-19.Кроме того, широкий спектр тяжести во время пандемии подчеркивает неоднородность пациентов с COVID-19. По мере появления разных фенотипов определенные подгруппы (, т. Е. человека с гиперактивным РААС) могут различаться по своей реакции на терапию. Также нужно будет учитывать время. Модуляция RAAS может иметь значительно разные эффекты на ранней стадии заболевания по сравнению с более поздними, учитывая сложность оси RAAS-SCoV. Это подчеркивает необходимость получения высококачественных доказательств для исследования любой значительной связи между модуляцией РААС и патофизиологией легких с акцентом на ACE2-опосредованные вирусные инфекции, в частности, SARS-CoV-2.
ТАБЛИЦА 1 Резюме (в тексте).
ТАБЛИЦА 2 Резюме (в тексте)
ТАБЛИЦА 3 Резюме (в тексте)
Благодарности
Мы благодарны за редакционную помощь Майклу Лотти, старшему редактору Института инженерии в медицине Университета Миннесоты.
Сноски
Оттиски авторам не предоставляются. Все авторы внесли значительный вклад в разработку, написание и пересмотр этой рукописи.
Спонсор: Национальный институт сердца, легких и крови; DOI: http://dx.doi.org/10.13039/100000050; Грант: T32HL07741.
Конфликт интересов: доктору Ингрэхему нечего раскрывать.
Конфликт интересов: доктору Баракату нечего раскрывать.
Конфликт интересов: доктору Рейлкоффу нечего раскрывать.
Конфликт интересов: доктору Бездичеку нечего раскрывать.
Конфликт интересов: Dr.Шакеру нечего раскрывать.
Конфликт интересов: доктору Чипману нечего раскрывать.
Конфликт интересов: доктору Тиньянелли нечего раскрывать.
Конфликт интересов: доктору Пушкаричу нечего раскрывать.
- Получено 30 марта 2020 г.
- Принято 18 апреля 2020 г.
Система ренин-ангиотензин-альдостерон при воспалении и ремоделировании сосудов
РААС через свои физиологические эффекторы играет ключевую роль в продвижении и поддержании воспаление.Воспаление — важный механизм в развитии и прогрессировании сердечно-сосудистых заболеваний, таких как гипертония и атеросклероз. Помимо своей основной роли в регулировании артериального давления и роли при гипертонии, РААС оказывает провоспалительное и профибротическое действие на клеточном и молекулярном уровнях. Блокирование РААС оказывает благотворное влияние при лечении сердечно-сосудистых и почечных заболеваний. Данные показывают, что ингибирование РААС положительно влияет на ремоделирование сосудов, улучшая исходы ССЗ.Благоприятные сосудистые эффекты ингибирования РААС, вероятно, связаны с уменьшением сосудистого воспаления, окислительного стресса, эндотелиальной дисфункции и положительным влиянием на регенерацию эндотелиальных клеток-предшественников. Воспалительные факторы, такие как ICAM-1, VCAM-1, TNF, IL-6 и CRP, играют ключевую роль в опосредовании сосудистого воспаления, а блокирование RAAS отрицательно влияет на уровни этих воспалительных молекул. Некоторые из этих воспалительных маркеров клинически связаны с сердечно-сосудистыми заболеваниями. Требуются дополнительные исследования, чтобы установить долгосрочные эффекты ингибирования РААС на воспаление сосудов, регенерацию сосудистых клеток и клинические исходы сердечно-сосудистых заболеваний.В этом обзоре представлена важная информация о роли RAAS в сосудистом воспалении, ответах сосудистых клеток на RAAS и ингибировании передачи сигналов RAAS в контексте сосудистого воспаления, ремоделирования сосудов и ССЗ, связанных с сосудистым воспалением. Тем не менее, в обзоре также указывается на необходимость переосмыслить и заново открыть новые ингибиторы РААС.
1. Ренин-ангиотензин-альдостероновая система (РААС) и сердечно-сосудистые заболевания
Ренин-ангиотензин-альдостероновая система (РААС), одна из наиболее важных гормональных систем, контролирует функции сердечно-сосудистой системы, почек и надпочечников путем регулирования артериальное давление, объем жидкости и баланс натрия и калия [1].Классическая система RAAS была открыта более века назад, а в 1934 году Goldblatt et al. показали связь Ренина между функцией почек и артериальным давлением [2]. С тех пор были проведены обширные экспериментальные исследования для определения компонентов РААС и его роли в регуляции артериального давления. Аномальная активность РААС приводит к развитию ряда сердечно-сосудистых заболеваний (ССЗ; гипертония, атеросклероз и гипертрофия левого желудочка), сердечно-сосудистых событий (инфаркт миокарда, инсульт и застойная сердечная недостаточность) и почечной недостаточности [1].Еще в 1956 году Леональд Т. Скеггс предложил разработать препараты для регулирования ренин-ангиотензиновой системы (РАС), и с тех пор был разработан ряд ингибиторов. Из-за сложности сигнальных путей RAAS, чем считалось ранее, полвека спустя новые ингибиторы RAAS все еще разрабатываются [3]. Действительно, многочисленные экспериментальные и клинические данные показывают, что фармакологическое ингибирование РААС ингибиторами ангиотензинпревращающего фермента (ИАПФ), блокаторами рецепторов ангиотензина (БРА), прямыми ингибиторами реннина (ДРИ) и антагонистами минералокортикоидных рецепторов (МРА) эффективно при лечении артериальной гипертензии и диабетическое повреждение почек, и результаты показывают снижение частоты сердечно-сосудистых заболеваний и сердечно-сосудистых заболеваний во всем мире [1].В этом обзоре обсуждаются недавние открытия в нашем понимании роли компонентов РААС и их ингибирующего действия на воспаление сосудов, ремоделирование сосудов и сердечно-сосудистые заболевания.
1.1. RAAS
Ренин, активный протеолитический фермент, сначала синтезируется как неактивный препрогормон (проренин), подвергается последующим протеолитическим изменениям в афферентных артериолах почечных клубочков, а затем попадает в кровоток [4]. В кровотоке протеолитические и непротеолитические механизмы расщепляют проренин до активного ренина.Активный ренин действует на свой субстрат, ангиотензиноген, с образованием ангиотензина I (Ang I). Ang I расщепляется ангиотензин-превращающим ферментом (ACE), в результате чего образуется физиологически активный ангиотензин II (Ang II). Ang II, главный эффектор RAAS, опосредует его эффекты через рецептор Ang II типа 1 (AT1R). Однако немногие исследования предполагают существование дополнительных рецепторов проренина и ренина в сердце, почках, печени и плаценте [5]. Другие исследования предполагают наличие рецепторов ренина в висцеральной и подкожной жировой ткани, что указывает на местную продукцию Ang II.Активация проренина и рецепторов ренина стимулирует сигнальный путь, связанный с митоген-активируемой киназой (MAPK) / регулируемой внеклеточными сигналами киназой (ERK1 / 2) [6]. Поскольку лимитирующая стадия РААС находится под контролем ренина, идея ингибирования ренина для подавления РААС была предложена в середине 1950-х годов, но разработка ингибиторов ренина была долгим и трудным процессом [7]. Аналогичным образом, первый пероральный DRI, алискирен, был продан в 2007 году для лечения гипертонии [8]. Другой эффектор РААС, альдостерон, выполняет важные эндокринные функции, регулируя объем жидкости, гомеостаз натрия и калия, и прежде всего действует в почечных дистальных извитых канальцах.Альдостерон опосредует геномные и негеномные эффекты через минералокортикоидный рецептор (MR), AT1R, рецептор, связанный с G-белком, и рецепторы эпидермального фактора роста (EGFR). Последующие эффекторы этих рецепторов, такие как пути MAPK / ERK1 / 2 / p38, опосредуют биологию и физиологию сосудов, в частности ремоделирование сосудов, воспаление, фиброз и тонус сосудов. Кардиопатологические эффекты альдостерона включают фиброз и гипертрофию миокарда, а также ремоделирование и фиброз сосудов. Продукция альдостерона регулируется ангиотензином II, гиперкалиемией, адренокортикотропным гормоном (АКТГ) и уровнем натрия [9].Клинические испытания показали, что блокирование рецепторов альдостерона антагонистами рецепторов минералокортикоидов (MRA), спиронолактоном или эплереноном снижает артериальное давление, снижает альбуминурию и улучшает исходы у пациентов с сердечной недостаточностью, инфарктами миокарда или сердечно-сосудистыми осложнениями, связанными с сахарным диабетом [10]. Инфузия альдостерона на животной модели ишемии вызывает изменения сосудов через AT1R, поскольку блокирование AT1R ингибирует эффекты альдостерона, что указывает на перекрестную связь между компонентами RAAS.
Недавнее открытие и клонирование нового ангиотензинпревращающего фермента, ACE2, усложнили RAAS. ACE2 на 42% гомолог ACE1 и экспрессируется в сердце, почках, семенниках, эндотелии коронарных, внутрипочечных сосудов и эпителии почечных канальцев [11]. ACE2 представляет собой монопептидазу с ферментативным предпочтением гидрофобных / основных остатков C-конца Ang II, что приводит к образованию ангиотензина II (1-7). Экспериментальные исследования показывают, что Ang II (1–7) является конкурентом Ang II и действительно может иметь кардиоренальные защитные эффекты [12, 13].Ang II также продуцируется ферментами, не относящимися к АПФ, такими как химаза сериновой протеазы, которые были обнаружены в сердце, сосудистой сети и других тканях [14, 15].
2. Воспаление и сердечно-сосудистые заболевания
Воспаление играет ключевую роль в возникновении, прогрессировании и развитии ряда сердечно-сосудистых заболеваний, таких как гипертония, атеросклероз, рестеноз после баллонной ангиопластики, нефропатия и кардиомиопатия [16]. Типичным примером того, как воспаление лежит в основе развития сердечно-сосудистых заболеваний, является атеросклероз через активацию эндотелиальных клеток воспалительными цитокинами.Дисфункция эндотелия из-за повреждения воспалительным процессом была связана с сердечно-сосудистыми факторами риска, включая гипертензию, сахарный диабет или ожирение [17].
2.1. Маркеры воспаления
Фактор некроза опухоли альфа (TNF) является ключевым провоспалительным цитокином, регулирующим экспрессию многих генов воспаления, окислительного стресса и антиапоптотических сигнальных путей практически во всех типах клеток [18]. Аберрантная передача сигналов TNF приводит к развитию патологических состояний, включая сердечно-сосудистые заболевания, и терапевтическое блокирование передачи сигналов TNF было предложено для лечения нескольких воспалительных заболеваний, в частности ревматоидного артрита и заболеваний кишечника [18].TNF нарушает зависимую от эндотелия вазорелаксацию, опосредованную оксидом азота (NO), в коронарных артериях или сонных артериях за счет продукции супероксидных радикалов [19]. Пациенты с высоким уровнем циркулирующего TNF имеют больший риск развития сердечно-сосудистых заболеваний [20]. В эндотелиальных клетках TNF индуцирует экспрессию интерлейкина-6 (IL-6), моноцитарного хемоаттрактантного белка-1 (MCP-1) и молекул клеточной адгезии (CAM) [21]. Делеция TNF у мышей подавляет гиперплазию интимы после повреждения сонной артерии [22], тогда как повышенная экспрессия TNF усугубляет легочную гипертензию у мышей [23].Воспаление, опосредованное TNF, играет важную роль в ремоделировании сосудов. Гладкомышечные клетки сонной артерии человека реагируют на TNF повышенной пролиферацией клеток, тогда как ингибирование циркулирующего TNF предотвращает ремоделирование среды после повреждения сонной артерии и образование неоинтимы у крыс [24]. Было показано, что ингибирование TNF улучшает функцию эндотелия за счет стимуляции регенерации эндотелиальных клеток [25].
NF-B, провоспалительный фактор, расположенный ниже TNF, играет центральную роль в регуляции экспрессии сосудистых медиаторов воспаления интерлейкина-1 бета (IL-1), интерлейкина-6 (IL-6), TNF и MCP-1. в эндотелиальных клетках и других типах клеток [26].Активированный NF-B вызывает пролиферацию гладкомышечных клеток сосудов и опосредует гиперплазию неоинтимы после повреждения сосудов [27].
Еще одним маркером воспаления является С-реактивный белок (СРБ). CRP считается признаком острой фазы ответа и предиктором риска сердечно-сосудистых событий [28]. С-реактивный белок в основном продуцируется в печени [29], но другие типы клеток, такие как гладкие мышцы и эндотелиальные клетки атеросклеротических артерий, демонстрируют экспрессию CRP [30]. CRP играет роль в посредничестве сосудистых заболеваний. Исследования in vitro показывают, что CRP обладает провоспалительным и протромботическим действием [31], ингибирует дифференцировку и функцию эндотелиальных клеток-предшественников [32] и активирует AT1R [33]. CRP активирует классический сигнальный каскад комплемента, который играет ключевую роль в формировании неоинтимы в поврежденных сосудах [34]. Уровни циркулирующего CRP коррелируют с несколькими маркерами воспаления, включая воспалительные цитокины, молекулы клеточной адгезии, маркеры активированных тромбоцитов и лейкоциты [35].Все эти маркеры воспаления также позволяют прогнозировать события в коронарной артерии [36].
Интерлейкин-6, плейотропный цитокин, регулирует многие клеточные функции, включая пролиферацию и апоптоз. IL-6 играет важную роль в воспалении и модулирует развитие нескольких заболеваний, включая сердечно-сосудистые заболевания, такие как гипертония и другие связанные с ними заболевания. Высокие уровни циркулирующего IL-6 обнаруживаются у пациентов с гипертонией. Крысы с диабетом I типа имеют высокий уровень циркулирующего IL-6 и повышенную сократимость кровеносных сосудов [37].Сверхэкспрессия IL-6 у мышей вызывает ремоделирование легочных сосудов, подобное тому, которое наблюдается у пациентов с легочной гипертензией, и вызывает легочную гипертензию через пролиферативные и антиапоптотические механизмы [38]. IL-6 также модулирует реактивность сосудов. Обработка изолированных кровеносных сосудов человека из различных органов с помощью ИЛ-6 приводит к усилению сокращения [39]. IL-6 опосредует развитие окклюзионной болезни сосудов и является предиктором внезапной смерти от сердечно-сосудистых заболеваний [40]. Воздействие IL-6 на сосудистую систему опосредуется через передачу сигналов NF-B, которая играет ключевую роль в ремоделировании сосудов.Ингибирование NF-B посредством делеции IBNS, ядерного белка, регулирующего IB NF-B, снижает гиперплазию интимы после повреждения сосудов у мышей за счет продукции IL-6, опосредованной NF-B [41].
2.2. Молекулы межклеточной адгезии
Опосредованное воспалением повреждение эндотелия генерирует провоспалительный сигнальный каскад и экспрессию молекулы межклеточной адгезии-1 (ICAM-1) и молекулы адгезии сосудистых клеток-1 (VCAM-1), обе из которых привлекают моноциты крови для стенка сосудов, что способствует высвобождению большего количества цитокинов и хемокинов в месте повреждения, что приводит к развитию сосудистого заболевания, такого как атеросклероз.Уровни циркуляции ICAM-1 и VCAM-1 положительно коррелируют с соотношением интима-медиа сонной артерии [42]. Экспрессия VCAM-1 повышается с помощью Ang II в аорте крысы, тогда как спиронолактон, антагонист минералокортикоидных рецепторов, ингибирует экспрессию VCAM-1 и других воспалительных маркеров [43]. Обработка эндотелиальных клеток Ang II активирует VCAM-1 посредством окислительного стресса и активации NF-B [44]. Высокие циркулирующие уровни ICAM-1, VCAM-1 и других молекул адгезии воспалительных клеток связаны с гипертрофией левого желудочка (ГЛЖ) и диастолической дисфункцией у пожилого населения [45].
3. РААС и сосудистое воспаление
РААС играет решающую роль в инициировании и поддержании сосудистого воспаления и ремоделирования сосудов. Воспаление сосудов приводит к дисфункции эндотелия, а снижение функции эндотелия опосредует прогрессирование сердечно-сосудистых заболеваний. Дисфункциональный эндотелий является неплотным и способствует миграции воспалительных клеток в сосудистую стенку и стимулирует пролиферацию гладкомышечных клеток, процессы, которые снижают функцию сосудов и способствуют развитию сердечно-сосудистых заболеваний и повреждению тканей.Дисфункциональный эндотелий обеспечивает провоспалительную среду, так как способствует привлечению и прикреплению воспалительных клеток, которые, как хорошо известно, играют ключевую роль в развитии атеросклероза. Появляется все больше доказательств, указывающих на связь между гипертонией и атеросклерозом через воспаление, опосредованное Ang II. In vivo , острое лечение Ang II значительно увеличивает адгезию лейкоцитов в брыжеечных артериях крыс [46]. Исследования на животных и людях показывают, что Ang II имеет провоспалительные реакции в артериях, сердце и почках, регулируя экспрессию цитокинов и хемокинов.В клетках гладких мышц сосудов человека Ang II индуцирует активацию NF-B и экспрессию IL-6 [47], MCP-1 и TNF в моноцитах [48]. In vivo инфузия Ang II вызывает повышенную экспрессию VCAM-1 в аорте крысы через активацию транскрипции NF-B. Введение лозартана, антагониста AT1R, ингибирует Ang II-индуцированную активацию NF-B и накопление VCAM-1 [49]. In vitro Обработка клеток гладких мышц сосудов крыс с помощью Ang II активирует MCP-1, а блокада AT1R лозартаном предотвращает экспрессию MCP-1 и миграцию моноцитов в стенку сосудов и другие органы-мишени [50].Хотя Ang II является вазоконстриктором, он вызывает повреждение эндотелия, подавляя регенерацию эндотелиальных клеток. Ang II действует как второй посредник для активации внутриклеточных сигнальных путей, таких как митоген-активируемая протеинкиназа (MAPK) и AKT, путей, которые опосредуют пролиферацию и апоптоз клеток и, следовательно, сосудистую дисфункцию [51]. Ang II играет важную роль в инициации и прогрессировании атерогенеза, процесса, опосредованного воспалением. В поврежденных артериях Ang II обеспечивает петлю положительной обратной связи при воспалении сосудов за счет рекрутирования воспалительных клеток, которые затем продуцируют больше Ang II, тем самым сохраняя воспаление сосудов [1].Ang II — мощный прооксидант. Ang II индуцирует продукцию супероксид-анионов и активирует прооксидантную передачу сигналов NADH / NADPH [52]. Окислительный стресс, опосредованный Ang II, снижает уровень оксида азота (NO) и активирует чувствительные к окислению-восстановлению гены, особенно цитокины, молекулы адгезии и матриксные металлопротеиназы [53]. Ang II также является профибротическим фактором. Хроническое введение мышам Ang II приводит к повышению артериального давления, инфильтрации воспалительных клеток в миокард и сердечному фиброзу [54].В кардиомиоцитах крыс Ang II индуцирует кальциевые сигналы (Ca 2+ ) и окислительный стресс, которые совместно вызывают гипертрофию кардиомиоцитов [55]. Хроническое лечение гладкомышечных клеток аорты крысы с помощью Ang II вызывает гипертрофию клеток за счет увеличения синтеза белка [56]. Сердечные фибробласты крыс, обработанные Ang II, демонстрируют повышенную экспрессию киназ фокальной адгезии (FAK) и интегринов, тогда как сердечные миоциты экспрессируют высокие уровни c-fos, EGFR1, TGF и белков внеклеточного матрикса (109, 110).Воспаление опосредует эндотелиальное повреждение, которое изменяет архитектуру эндотелиальных клеток, так что эндотелий становится дисфункциональным. Было показано, что дисфункциональный эндотелий напрямую связан с гипертонией и атеросклерозом [17]. Функциональный эндотелий является ключевым регулятором высвобождения NO, а потеря биодоступности NO связана с высоким уровнем Ang II из-за окислительного стресса. Хотя развитие атеросклероза — это многофакторный сложный процесс, взаимодействие между эндотелиальной дисфункцией и окислительным стрессом играет важную роль в атеросклеротическом процессе.Повышенный окислительный стресс в сосудистой стенке является признаком сосудистых заболеваний, таких как атеросклероз, гипертония и диабет. Действительно, высокий уровень супероксида является важным фактором инициации атеросклероза за счет привлечения воспалительных клеток и эндотелиальной дисфункции. Полная генетическая делеция субъединицы НАДФН-оксидазы, Nox2, у мышей приводит к значительному снижению атеросклероза аорты [57]. Блокирование РААС валсартаном в комбинации с флувастатином (статином) в модели атеросклероза у мышей, нулевых по аполипопротеину Е (ApoE — / —), снижает уровень атеросклеротических поражений, супероксид-анион и уровень экспрессии MCP- 1 и ICAM-1, что указывает на то, что блокирование воспаления и окислительного стресса благотворно влияет на эндотелий [58].Действительно, клинические исследования показывают снижение сердечно-сосудистых событий помимо снижения артериального давления, например, положительное изменение структуры эндотелия / сосудистой стенки, что, в свою очередь, способствует уменьшению сердечно-сосудистых заболеваний. Некоторые ингибиторы РААС, такие как ИАПФ рамиприл и БРА лозартан, улучшают эндотелиальную активность и функцию сосудов за счет увеличения биодоступности NO [17]. NO оказывает защитное действие на сердечно-сосудистую и почечную системы. NO эффекты на сосудистую сеть многочисленны, от индукции расширения всех типов кровеносных сосудов до ингибирования агрегации и адгезии тромбоцитов или адгезии лейкоцитов к эндотелию.Кроме того, NO подавляет синтез ДНК, митогенез и пролиферацию гладкомышечных клеток сосудов, а также противодействует окислительному стрессу [59]. Биодоступность NO зависит от активности eNOS, а снижение активности eNOS связано с эссенциальной гипертензией [59].
Провоспалительные и профибротические эффекты РААС также опосредуются альдостероном. Альдостерон играет роль в фиброзе органов и ишемии тканей, и в сочетании с макрофагами он вызывает сердечный фиброз [60].Альдостерон способствует инсулинорезистентности и ремоделированию сосудов, а также влияет на развитие атеросклероза [61]. В гладкомышечных клетках сосудов альдостерон изменяет передачу сигналов инсулина за счет усиления экспрессии рецептора инсулиноподобного фактора роста-1 (IGF1R) и гибридного рецептора и модулирует структуру мембраны через рецепторы тирозинкиназы [62]. Хроническая инфузия альдостерона вызывает окислительный стресс в аорте крысы, а антагонист MR спиронолактон снижает образование активных форм кислорода [62].Исследования на животных также указывают на связь между альдостероном и снижением синтеза NO и эндотелиальными клетками-предшественниками (EPC) через окислительный стресс и низкие уровни VEFGR2 [63]. NO играет ключевую роль в гомеостазе сосудов, воздействуя на эндотелиальные и гладкомышечные клетки. В эндотелиальных клетках NO усиливает митогенные эффекты VEFG, тем самым стимулируя пролиферацию эндотелиальных клеток. В VSMC NO ограничивает их распространение и миграцию [64]. Помимо RAAS, присутствующего в системном кровотоке и его продуцирования в местных тканях, есть также сообщения об идентификации внутриклеточного RAAS в определенных типах клеток, таких как клетки гепатомы [65], клетки коры почек [66] или хромаффин мозгового вещества надпочечников и клетки гипофиза [67].Клетки коры надпочечников человека и крысы, стимулированные Ang II, продуцируют альдостерон за счет активации AT1R цитохрома P450 оксидазы B2 и повышения уровня перекиси водорода, тогда как предварительная обработка лозартаном и антиоксидантами отменяет эффекты Ang II [68]. Как показано на рисунке 1, Ang II, главный цитокин, TNF и альдостерон индуцируют экспрессию множества молекулярных эффекторов сигнального пути, связанных с воспалением и ремоделированием сосудов, фиброзом и окислительным стрессом. Некоторые молекулярные молекулы, такие как ERK1 / 2 и NADPH, также активируются Ang II и альдостероном и активируют NF-B-зависимую передачу сигналов в отсутствие TNF, перекрестный разговор, который указывает на сложность роли эффекторов RAAS в опосредовании сосудистого воспаления и ремоделирования ( Фигура 1).Более того, перекрестная связь между Ang II и альдостероном указывает на сложность системы RAAS в отношении патологии сердечно-сосудистой системы.
4. Блокаторы RAAS и сосудистое воспаление
Блокирование передачи сигналов RAAS с помощью ИАПФ, которые ингибируют образование ангиотензина, или БРА, блокирующих рецепторы ангиотензина, или ДРИ, которые ингибируют реакцию ренин-ангиотензиноген, или МРА, которые блокируют только альдостерон или в комбинации снижает смертность и заболеваемость при диабете, гипертонии, атеросклерозе, сердечной недостаточности и инсульте [69].Множественные биологические и физиологические эффекты ингибиторов РААС суммированы в таблице 1 и включают уменьшение воспаления, ремоделирования сосудов и фиброза, окислительного стресса, усиление функции эндотелия и оксида азота, а также поддержание брадикинина и факторов гиперполяризации эндотелия (EDHF). ), которые способствуют поддержанию сосудистого тонуса. Однако блокада РААС на одном уровне не очень эффективна для лечения гипертензии; поэтому блокирование РААС на нескольких уровнях, по-видимому, обеспечивает клиническую эффективность для лечения гипертензии и других форм сердечно-сосудистых заболеваний, включая атеросклероз [69].
DRI ACEI ARB MRB Ang 1–7 ASI (P) 9
9
(P) RRI 3
9
гипертрофия↑ Эндотелиальная
функция↑ Эндотелиальная
функция↓ Сердечный приступ Расширение сосудов ↓ Кровь
давление↓ Кровь
давление970 970
970 970 970 970 ↓ Воспаление
↓ Артериальное давление ↓ Пролиферация ↑ Эндотелиальная
Функция• Брадикинин ↑ 3 3 Оксидный стресс 3
0 ↑ Сердечный фиброз
↓ 933 71 Сердечное
ремоделирование↓ Сердечное
фиброз↓ Моноциты
Адгезия↓ Сосудистое
ремоделирование9516 ↓ 13 Сердечная болезнь 9516 9516 ↓ ↓ 133 Сердечная недостаточность
↓ Сердечная
гипертрофия↓ Воспаление сетчатки ↓ Воспаление • EDHF ↓ 8
8 ↓ Сосудистое
8 ремоделирование 97011
8 97011
18
↓ Гипертрофия интимы сосудов ↓ Окислительный стресс 812
ИАПФ: ингибиторы ангиотензинпревращающего фермента; БРА: блокаторы рецепторов Ang II типа 1; MRB: блокаторы минералокортикоидных рецепторов; DRI: прямые ингибиторы реннина; NO: оксид азота; EDHF: гиперполяризующий фактор эндотелия.Тонкая стрелка: увеличена или уменьшена; пуля: поддержание уровня. 4.1. Прямые ингибиторы ренина (DRI)
DRI блокируют RAAS, подавляя ферментативную активность ренина. Недавно одобренный DRI, алискирен, представляет собой прямой пероральный ингибитор ренина, который снижает артериальное давление, блокируя лимитирующую стадию РААС. В рандомизированном двойном слепом исследовании Andersen et al. [70] и другие [71] сообщили, что терапия на основе алискирена снижает артериальное давление (АД) и активность ренина плазмы (PRA), и эффекты сохраняются в течение четырех недель, что свидетельствует о долгосрочном влиянии алискирена на активность ренина.Однако активность ренина в плазме может быть увеличена с помощью ИАПФ или БРА; поэтому комбинация алискирена с ИАПФ или БРА считается предпочтительным вариантом лечения артериальной гипертензии, застойной сердечной недостаточности и хронического заболевания почек [72]. Алискирен благотворно влияет на эндотелий. У пациентов с диабетом I типа алискирен улучшал функцию эндотелия независимо от снижения артериального давления. На модели трансгенных мышей с атеросклерозом алискирен отдельно или в комбинации с аторвастатином подавлял развитие атеросклероза и прогрессирование бляшек за счет снижения адгезии моноцитов и уровней MCP-1 [73].У мышей с дефицитом eNOS алискирен предотвращал гипертрофию сердца, воспаление, ремоделирование коронарных артерий и гиперплазию интимы сосудов, а в комбинации с валсартаном были обнаружены даже более сильные эффекты. Механически комбинация алискирена и валсартана подавляет активность НАДФН-оксидазы и, следовательно, ослабляет окислительный стресс [59], который играет ключевую роль в инициировании развития сосудистого воспаления и сердечно-сосудистых заболеваний.
4.2. (Про) рецептор ренина и потенциальные ингибиторы
Открытие (Про) рецептора ренина (P) RR в 2002 г. пролило свет на тканевую ренин-ангиотензиновую систему и функцию циркулирующего проренина, неактивного предшественника ренина.Было показано, что проренин и ренин связывают (P) RR с высоким сродством; однако проренин проявляет более высокое сродство к (P) RR, что указывает на то, что действительно проренин может быть фактическим лигандом для (P) RR. Исследования показали, что после связывания проренина и активации (P) RR нижестоящая передача сигналов приводит к активации MAPK ERK1 / 2 / p38 и Akt во множестве клеток, включая VSMC [74]. Роль (P) RR в сердечно-сосудистой патологии оказалось более трудным для изучения, чем предполагалось, поскольку генетическая делеция (P) RR у мышей является летальной или продолжительность жизни очень короткая, что делает функциональный анализ этого рецептора практически невозможным для изучения. а также предполагая, что для гомеостаза необходим нормальный уровень (P) RR.Тем не менее, несколько исследований были сосредоточены на роли (P) RR в сердечно-сосудистых осложнениях, опосредованных диабетом. Диабетическая нефропатия значительно увеличивает уровень (P) RR в почках, а использование (R) блокатора RR, HRP, частично устраняет связывание ренина и проренина с (P) RR, нормализует уровни Ang II или ослабляет существующую нефропатию [75, 76]. В моделях на животных с гипертонией HRP улучшала функцию почек и левого желудочка и уменьшала фиброз. Однако HRP не удалось снизить артериальное давление или повреждение почек в более сложных моделях животных, особенно в моделях животных с избыточной экспрессией проренина и ангиотензиногена [77].В VSMC со сверхэкспрессией (P) RR человека HRP неспособна блокировать связывание проренина с (P) RR или активацию ERK1 / 2, тогда как в эндотелиальных клетках пупочной вены человека HRP неспособна связываться с (P) RR [77, 78]. Предполагается, что у пациентов с гемодиализом и повышенной смертностью от сердечно-сосудистых заболеваний ингибирование (P) RR в дополнение к ACEI или ARB может иметь потенциал для положительного снижения гипертрофии сердца или экскреции белка, по крайней мере, на основании исследований на животных; однако HRP обращает действие DRI на артериальное давление или коронарное кровообращение [79].Основываясь на информации, представленной выше, и сложности передачи сигналов RAS или петли отрицательной обратной связи ингибирования RAS, клиническое использование HRP должно сначала полностью понять исследования in vitro, и in vivo, , прежде чем последуют клинические исследования.
4.3. Ингибиторы ангиотензинпревращающего фермента (ИАПФ)
Использование ИАПФ является эффективным традиционным лечением артериальной гипертензии и снижает гипертрофию левого желудочка, поэтому ИАПФ улучшают сердечно-сосудистые исходы [80] Лечение пациентов с дисфункциональным эндотелием, вызванным различными патологическими состояниями, улучшает функции эндотелия, измеряемые с помощью вазодилатации, опосредованной плечевым потоком (FMD ) [81].Механически ИАПФ улучшают функцию эндотелия за счет повышения уровня NO за счет блокирования деградации брадикинина и ингибируют продукцию эндотелием эндотелина-1 (ЕТ-1) и Ang II [82]. Поддержание уровня брадикинина оказывает аддитивный эффект на эндотелий за счет увеличения уровня простациклина и EDHF, которые вызывают вазодилатацию и ингибируют пролиферацию гладкомышечных клеток сосудов и адгезию тромбоцитов [82–84]. Самые последние данные свидетельствуют о том, что окислительный стресс, воспаление и гипертония — это взаимосвязанные процессы, которые влияют друг на друга в патологии сердечно-сосудистых событий, связанных с гипертензией.Поэтому разрабатываются новые ингибиторы, нацеленные на АПФ и, в частности, обладающие антиоксидантной активностью; таким образом, вместе новые свойства одного ингибитора могут способствовать эндотелий-зависимым защитным эффектам против окислительного стресса и высокого кровяного давления. Было разработано несколько селеновых аналогов ингибитора АПФ, каптоприла, и было показано, что они обладают ингибирующим действием на АПФ и устраняют продукт окислительного стресса, пероксинитрит [85, 86]. Хотя эти новые ингибиторы АПФ демонстрируют многообещающие ингибирующие эффекты, необходимы расширенные исследования in vitro, и in vivo, , чтобы понять и определить их расширенный физиологический эффект.
4.4. Ang 1–7 и Mas Axis
В дополнение к Ang II, другие пептиды Ang III (Ang-2–8), Ang IV (Ang 3–8) и Ang 1–7 обладают биологическим действием в системе RAAS [87 ]. Уровень Ang 1-7 в тканях и циркулирующей крови аналогичен Ang II; однако, в отличие от Ang II, Ang 1–7 является сосудорасширяющим средством и опосредует антиангиогенные и антимитогенные эффекты [87]. Ang 1–7 ингибирует пролиферацию гладких мышц и ингибирует образование неоинтимы после повреждения сонной артерии или стентирования брюшной аорты [88, 89].Благоприятные сердечно-сосудистые эффекты Ang 1–7 опосредуются через рецептор Mas, связанный с G-белком (MasR). Было показано, что MasR присутствует в эндотелиальных клетках человека, афферентных артериолах, собирательных протоках почек и гломерулярных мезангиальных клетках [90]. Эффект Ang 1–7 на сосудистое русло обусловлен локальным синтезом Ang 1–7, особенно эндотелием, который опосредует ряд вазодилататоров, включая EDHF и NO. Однако в исследовании пациентов с сердечной недостаточностью, получавших ингибитор АПФ, местная инфузия Ang 1–7 не вызвала локального расширения сосудов [91].Действительно, считается, что действие лозартана на снижение артериального давления частично опосредуется через Ang 1–7, поскольку было обнаружено, что уровень Ang 1–7 в плазме повышается во время лечения ACEI или ARB [13]. Было показано, что регуляция ACE2 и MasR, по крайней мере, в сердце и почках с гипертонией нечувствительна к Ang II или передаче сигналов альдостерона [92]. Эти данные предполагают RAAS-независимую регуляцию ACE2 и MasR. Тем не менее, передача сигналов ACE2 / Ang 1-7 / MasR может рассматриваться как новый терапевтический подход к уравновешиванию оси ACE / Ang II / AT1R как новый подход, нацеленный на RAAS [87].
4.5. Блокаторы рецепторов ангиотензина (БРА)
Было показано, что блокада РААС БРА уменьшает воспаление и улучшает функцию эндотелия. In vitro и in vivo Исследования продемонстрировали, что противовоспалительный эффект кандесартана ARB проявляется через подавление воспалительных толл-подобных рецепторов 2 и 4 (TLR2 и TLR4) [17]. Действительно, TLR вовлечены в развитие и прогрессирование сердечно-сосудистых заболеваний. В моделях артериальной гипертензии на животных TLR4 способствует регуляции артериального давления и сужению сосудов малых резистентных артерий [93].Было показано, что у пациентов с артериальной гипертензией БРА ирбесартан улучшает функцию эндотелия и реактивность сосудов, а также снижает уровни CRP, ICAM-1, IL-6 и маркера окислительного стресса 8-изопростана [94]. Хорошо известно, что окислительный стресс играет важную роль в опосредовании дисфункции эндотелия. Было показано, что использование валсартана предотвращает образование активных форм кислорода (АФК) и подавляет активность NF-B, фактора транскрипции, который регулирует экспрессию воспалительных цитокинов и молекул клеточной адгезии, которые способствуют развитию сосудистой системы. воспаление и сосудистые события [95]. In vitro и in vivo Исследования показали, что БРА олмесартан ингибирует Ang II-индуцированную миграцию гладкомышечных клеток сосудов аорты и, следовательно, предотвращает ремоделирование сосудов [96]. Использование БРА у пациентов с сахарным диабетом 2 типа или со стабильной ишемической болезнью сердца увеличивает количество кардиозащитных и эндотелиальных клеток-предшественников [97, 98]. БРА оказывают положительное действие при лечении ишемической болезни сердца и атеросклероза. Лечение лозартаном улучшает вызванное кровотоком заболевание коронарной артерии у пациентов с атеросклерозом и нарушением функции эндотелия за счет биодоступности NO.
4.6. Антагонисты минералокортикоидных рецепторов (MRA)
Нарушение регуляции передачи сигналов минералокортикоидной системы влияет на артериальную гипертензию, атеросклероз и сердечную недостаточность независимо от воздействия MR почек на кровяное давление [99]. Аберрантно активированная МР отрицательно модулирует функцию эндотелия у пациентов с сердечно-сосудистыми факторами риска и заболеваниями. Было показано, что блокирование эффектов альдостерона на уровне MR с помощью доступных в настоящее время антагонистов, эплеренона и спиронолактона, является эффективным вариантом лечения гипертонии и сердечной недостаточности [100].Действие альдостерона на сердечно-сосудистую систему опосредуется MR, присутствующим в гладкомышечных клетках сосудов, эндотелиальных клетках и кардиомиоцитах [101]. Исследования показали, что активация MR вызывает множество сигнальных событий в сердечно-сосудистой системе, включая окислительный стресс [102], ингибирует расслабление сосудов и вызывает воспаление сосудов, фиброз и ремоделирование [62]. Активированный альдостероном MR в эндотелиальных клетках человека (EC) индуцирует экспрессию воспалительного фактора ICAM-1 и адгезию лейкоцитов к EC, а блокирование MR спиронолактоном ингибирует опосредованные альдостероном эффекты на EC [102].Альдостерон нарушает дифференцировку, миграцию и пролиферацию EPC, тогда как фармакологическое ингибирование или генетические манипуляции с альдостероном восстанавливают функции EPC [103, 104]. Ингибирование MR спиронолактоном улучшает эндотелий-зависимое расширение сосудов за счет ингибирования пути NAD (P) H оксидазы [104]. Более того, блокирование передачи сигналов альдостерона улучшает пролиферацию сердечных мышц и ремоделирование артериальной стенки, эндотелиальную функцию и синтез NO [60]. Многие клинические исследования показали, что фармакологическое ингибирование МР снижает частоту сердечных приступов, инсультов и смертность в дополнение к снижению артериального давления [101].Самые последние исследования также показывают, что дополнительные преимущества от блокады передачи сигналов альдостерона, в частности, уменьшают воспаление, уменьшают ремоделирование сердечно-сосудистой системы и уменьшают атеросклероз [60]. Однако было показано, что использование MRA действительно вызывает противодействие, такое как повышение уровня альдостерона, тем самым усиливая MR-независимые эффекты альдостерона, особенно негеномные эффекты, такие как стимуляция сократительной способности сердечно-сосудистой системы [105].
4.6.1. Разработка новых антагонистов минералокортикоидных рецепторов
В настоящее время ведутся исследования по разработке новых MRA из-за ограниченного количества клинически одобренных MRA, спиронолактона и эплеренона в США и канренона в Европе.Исследования дизайна лекарств на основе МРТ выявили несколько соединений; однако только ограниченное количество новых кандидатов MRA было продвинуто к клиническим исследованиям. Один из таких новых MRA, PF-3882845, значительно снижает артериальное давление, снижает уровень альбумина в моче и защищает почки на животной модели гипертонии, индуцированной солью Даля. Эти результаты продвинули эту новую MRA к текущим клиническим исследованиям [106]. Более поздние антагонисты MR, такие как дигидрофуран-1-он и дигидропиррол-2-он, показали очень многообещающую селективность связывания MR в исследованиях in vitro [107].Однако недавно была разработана другая селективная нестероидная молекула, BR-4628, которая демонстрирует высокую эффективность и селективность в отношении MR в исследованиях in vivo, и in vitro, [108]. У крыс с гипертонической болезнью DOCA-соли обработка BR-4628 значительно снизила уровень маркеров воспаления в сыворотке MCP-1, IL-1beta, CXCL-1 и маркеров повреждения почек MCP-1, тенасцина-1 и остеопонтина. Кроме того, обработка BR-4628 уменьшала индуцированный солью DOCA окислительный стресс и повреждение ДНК. Однако лечение BR-4628 вызывало умеренное снижение систолического артериального давления в группе с DOCA-солью, тогда как спиронолактон значительно снижал систолическое артериальное давление по сравнению с группой с DOCA-солью.Эти данные свидетельствуют о тканевой активации антагонизма MR и BR-4628 в тканях, а не о системном эффекте, таком как снижение артериального давления [109]. Для определения эффективности BR-4628 в снижении артериального давления необходимы дополнительные исследования.
4.7. Ингибиторы альдостерон-синтазы
Снижение синтеза альдостерона на стадии его фермента, альдостерон-синтазы (AS), CYP11B2, является новым альтернативным подходом к MRA для ограничения эффектов альдостерона [105]. Ингибиторы альдостерон-синтазы (ASI) представляют собой новейшую терапевтическую стратегию по снижению продукции альдостерона [110].Было показано, что фадрозол 286A (FAD 286A) дозозависимо ингибирует индуцированный Ang II синтез альдостерона в клетках адренокортикальной карциномы человека, тогда как на животных моделях он снижает активность ренина в плазме, гипертрофию сердца и ремоделирование сердца [111–113]. На основе FAD 286A был разработан новый ингибитор AS, LCI699, который представляет собой ASI первого поколения, прошедшее клинические испытания. В клинических испытаниях было показано, что введение LCI699 снижает уровень альдостерона в плазме и моче у участников с дефицитом натрия, снижает артериальное давление у пациентов с гипертонической болезнью и уравновешивает гипокалиемию у пациентов с альдостеронизмом [110, 114, 115].Более того, недавно было показано, что LCI699 незначительно снижает артериальное давление (АД) у пациентов с резистентной гипертензией [116]. Однако в исследовании Karns et al. [116] фармакологический эффект LCI699 был протестирован на первичный альдостеронизм, резистентную и неконтролируемую гипертензию и эссенциальную гипертензию [106]. В этом исследовании было обнаружено, что первое поколение ASI, LCI699, демонстрирует некоторые ограничения, то есть вмешивается в основные петли эндокринной обратной связи: RAAS и гипоталамо-гипофизарно-надпочечниковые (HPA).LCI699 потенцировал более высокий, чем принято, физиологический уровень 11-дезоксикортикостерона и адренокортикотропного гормона (АКТГ). Повышенные уровни 11-дезоксикортикостерона и АКТГ являются компонентами двух различных петель эндокринной обратной связи, которые действительно сходятся в одной точке: мощном рецепторе минералокортикоидов, данные, которые подтверждают умеренное влияние LCI699 на снижение артериального давления. Эти данные также предполагают необходимость проведения дополнительных исследований in vivo , чтобы полностью понять физиологический эффект LCI699.
5. РААС и ремоделирование сосудов
Ang II также вызывает ремоделирование сосудов, тромбоз и разрыв бляшек [117, 118]. Ремоделирование сосудов, опосредованное Ang II, происходит через экспрессию аутокринных факторов роста, основного фактора роста фибробластов (bFBS), трансформирующего фактора роста-1 (TGF1) и фактора роста инсулина (IGF) [119]. Ремоделирование сосудов, опосредованное Ang II, происходит из-за повышенной миграции сосудистых клеток и модификации состава внеклеточного матрикса [96, 120]. Изменения в структуре и функции кровеносных сосудов, особенно в кровеносных сосудах с малым сопротивлением, потенцируют осложнения гипертонии.Более того, ремоделирование мелких кровеносных сосудов происходит до гипертрофии левого желудочка и утолщения интима-медиа сонной артерии или повышения уровня микроальбуминурии. Действительно, у пациентов с артериальной гипертензией наблюдается меньший просвет и внешний диаметр мелких резистентных артерий [121]. Изменения функции мелких артерий связаны со снижением уровня вазодилататоров и повышенной чувствительностью к Ang II и связанным с ним путям передачи сигнала.
Помимо своей роли в гипертонии, ренин-ангиотензин-альдостероновая система также играет важную роль в опосредовании ремоделирования сосудов при неоинтимальной гиперплазии после ангиопластики и атеросклероза [122–124].Ang II также является фактором роста, регулирующим пролиферацию и дифференцировку клеток, гипертрофию и апоптоз. При ремоделировании сосудов эффекты ремоделирования, вызванные Ang II, обусловлены пролиферацией и гипертрофией гладкомышечных клеток сосудов. Эффекты роста Ang II, пролиферация против гипертрофии, зависят от типа клетки и генов, регулируемых клеточным циклом. Например, Ang II оказывает гипертрофический эффект на кардиомиоциты посредством передачи сигналов, опосредованной TGF1, а блокада рецептора TGF1 отменяет опосредованную Ang II гипертрофию кардиомиоцитов [125].В модели инфаркта миокарда (ИМ) БРА, телмисартан, подавляет ремоделирование сердца за счет уменьшения гипертрофии и фиброза кардиомиоцитов за счет противовоспалительного эффекта и активации рецептора гамма-активации, активируемого пролифераторами пероксисом (PPAR) [126]. Роль системной и местной ренин-ангиотензиновой системы в заболеваниях ремоделирования сосудов, таких как атеросклероз и гиперплазия неоинтимы после ангиопластики, хорошо известна [127]. Активация Ang II / AT1R в сосудистой ткани приводит к накоплению воспалительных клеток, фиброзу и миграции гладкомышечных клеток сосудов [128].Было показано, что блокирование передачи сигналов Ang II через ACEI или ARB ингибирует Ang II-опосредованную эндотелиальную дисфункцию и атеросклероз [129], а ARB эффективно ингибируют ремоделирование сосудов и неоинтимальную гиперплазию после сосудистого повреждения [130, 131]. Действительно, было показано, что БРА телмисартан подавляет гиперплазию неоинтимы после трансплантации сердца на модели мышей, что позволяет предположить, что телмисартан может оказывать положительный эффект в предотвращении отторжения трансплантата [132]. Было показано, что ARB лозартан снижает соотношение интима: среда в сонной артерии или в резистентных артериях у пациентов с гипертонией и предотвращает продукцию TGF, известного медиатора Ang II [133, 134].Альдостерон играет ключевую роль в сердечном фиброзе и ремоделировании за счет прямого воздействия на синтез и отложение коллагена в интерстиции сердца. Использование спиронолактона на крысиной модели фиброза миокарда предотвращает фиброз миокарда [135]. Следовательно, появляется все больше данных, указывающих на то, что фармакологическое блокирование передачи сигналов RAAS положительно регулирует сосудистое воспаление и ремоделирование, а также физиологию сердечно-сосудистой системы (краткое изложение приведенных выше результатов см. В Таблице 1).
6. Перспективы ингибирования РААС
Хотя система РААС изучалась более века, новые экспериментальные и клинические данные предполагают, что физиология РААС сложна и многофакторна, и новые или продолжающиеся исследования дадут лучшее понимание по физиологии РААС.Например, текущее понимание взаимодействия между Ang II и альдостероном для регуляции вредных клеточных процессов указывает на сложность передачи сигналов RAAS и обеспечивает основу для блокирования RAAS на нескольких уровнях. Хотя разрабатываются новые мишени и ингибиторы RAAS, необходимы дополнительные исследования для определения молекулярных механизмов и физиологических взаимодействий. Более того, идентификация новых мишеней / эффекторов системы RAAS, возможно, обеспечит основу для разработки новых терапевтических стратегий, направленных на предотвращение сосудистого воспаления и ремоделирования и, следовательно, улучшение исхода сердечно-сосудистых заболеваний.
Сокращения
AS: Альдостерон-синтаза ASI: Ингибиторы альдостерон-синтазы Ang: AC 8118118
Блокаторы рецепторов ангиотензина CRP: C-реактивный белок CVD: Сердечно-сосудистые заболевания DRI: Прямые ингибиторы реннина 6 EGH116
1
ERK: Киназа внеклеточного рецептора EGFR: Рецептор эпидермального фактора роста eNOS: Эндотелиальный фактор оксида азота 80H-фактор
80 Гиперолизирующий фактор H
80 9580 6 ET-1:
Эндотелин 1 IL-1: Интерлейкин 1 бета IL-6: Интерлейкин 6 Молекула ICAM-1: Внутриклеточная адгезия IGF: Фактор роста инсулина MCP-1: Моноциты, хемоаттрактантный белок 1 MIP-1: Макрофаги воспалительный белок 1 16 рецептор антагониста MRA11
NF-B: Ядерный фактор каппа B NO: Оксид азота PPAR: рецептор, активируемый пролифераторами пероксисом, гамма d1 RAAS-
-ангиостерон система
TGF: Трансформирующий фактор роста бета TNF: Некроз опухоли является фактором альфа VCAM-1: Молекула адгезии сосудистых клеток 1. Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов в отношении публикации данной статьи.
Выражение признательности
Это исследование финансировалось грантом Национальных институтов здравоохранения (грант № G12MD007581) через RCMI-Центр гигиены окружающей среды при Университете штата Джексон.
Ренин-ангиотензиновая система: комплексный взгляд на заболевание легких и коагулопатию при COVID-19 и терапевтические последствия | Журнал экспериментальной медицины
Ренин-ангиотензиновая система (РАС) долгое время считалась основным регулятором кровяного давления, но недавно была признана также как механизм модуляции воспаления.Несмотря на то, что пациенты с COVID-19 выражали обеспокоенность по поводу использования лекарств, нацеленных на эту систему, RAS не был полностью изучен как мишень, на которую можно воздействовать лекарствами. Сокращенное описание RAS предполагает, что его нарушение регуляции может быть в центре COVID-19.
Ангиотензин II является основным компонентом ренин-ангиотензиновой системы (РАС). Он вырабатывается из ангиотензина I ангиотензин-превращающим ферментом (ACE; Benigni et al., 2010). Примечательно, что в RAS существует положительная обратная связь, благодаря которой ангиотензин II может повышать уровни АПФ (Koka et al., 2008). Ангиотензин II также приводит к повышенной секреции альдостерона, что вызывает задержку натрия и выведение калия (Muñoz-Durango et al, 2016).
Существует два основных рецептора ангиотензина II, которые экспрессируются во многих клетках. Когда ангиотензин II связывается с первым из этих рецепторов, называемым рецептором ангиотензина 1 (AT1), экспрессирующимся на фибробластах, сердечных миоцитах, гладкомышечных клетках, клетках надпочечников, макрофагах, микроглии, гепатоцитах, эндотелиальных клетках и т. Д., Возникает вазоконстрикция и активация NF-κB и высвобождение провоспалительных цитокинов (Benigni et al., 2010; Дандона и др., 2007; Okamura et al., 1999). Когда ангиотензин II связывает AT1 на альвеолярных эпителиальных клетках, он вызывает апоптоз (Wang et al., 1999). Когда он задействует AT1 на эндотелиальных клетках, это приводит к повышенной экспрессии тканевого фактора (TF), необходимого компонента каскада свертывания (Dielis et al., 2005), а также к проницаемости сосудов и экстравазации нейтрофилов в ткани (Nabah et al. ., 2004). Вовлечение второго рецептора ангиотензина, AT2, также довольно повсеместно экспрессируемого ангиотензином II, приводит к расширению сосудов и подавлению воспаления (Crowley and Rudemiller, 2017).Как при острых, так и при хронических воспалительных состояниях уровень AT1 увеличивается, а уровень AT2 снижается (Crowley and Rudemiller, 2017; Koka et al., 2008; Tikellis and Thomas, 2012).
Второй ACE, ACE2, существует как мембраносвязанная протеаза на многих типах клеток (Clarke et al., 2012). ACE2 расщепляет ангиотензин II с образованием небольшого пептида ang1-7, который связывается с рецептором Mas, связанным с G-белком, чтобы вызвать противовоспалительную и антиапоптотическую программу и вазодилатацию (Simões e Silva et al., 2013). ACE2 является клеточным рецептором вируса SARS-CoV-2 (Lan et al., 2020), возбудителя COVID-19, и позволяет вирусу проникать в клетки, экспрессирующие ACE2. Однако, когда вирусный спайковый белок связывает ACE2, активируется другая протеаза, называемая TACE или ADAM 17; это вызывает отрыв ACE2 от клеточной мембраны, что приводит к снижению деградации ангиотензина II и снижению продукции ang1-7 (Shah and Catt, 2006). Интересно, что высокая концентрация альдостерона снижает экспрессию Mas, также нарушая это звено РАС (Stoll et al., 2019).
Так как это связано с COVID-19? Инфекция SARS-CoV-2 может вызвать особенно тяжелое заболевание у людей с гипертонией и ожирением (Kenyon, 2020; Richardson et al., 2020). Люди с гипертонией обычно имеют высокий уровень ангиотензина и высокий уровень TF, что приводит к состоянию гиперкоагуляции, которое наблюдается при гипертонии (Dielis et al., 2005). Важно отметить, что уровни ренина у афроамериканцев часто низкие, но на моделях грызунов есть доказательства того, что тканевые уровни ангиотензина могут быть высокими даже при низком уровне ренина (Williams et al., 2014). Ожирение связано с высоким уровнем АПФ и АТ1 (Barton et al., 2000). Таким образом, обе эти сопутствующие патологии усиливают провоспалительные, проапоптотические и прокоагулянтные звенья РАС.
Модель COVID-19 может заключаться в том, что SARS-CoV-2 инфицирует альвеолярные эпителиальные клетки легких, источник сурфактанта, вызывая цитопатический эффект. Кроме того, связывание ангиотензина II с альвеолярными эпителиальными клетками вызывает их гибель через апоптоз.Поскольку ACE2 выделяется при связывании с белком spike, мы предполагаем, что ang1-7 не генерируется. При уменьшении ang1-7 для связывания Mas может отсутствовать активация плеча пути RAS, защищающего от апоптоза. Поэтому, во-первых, мы предполагаем, что происходит коллапс альвеол с последующей гипоксией. Поскольку ангиотензин II увеличивает проницаемость сосудов, может происходить экстравазация нейтрофилов в альвеолы. Более того, ангиотензин II, действующий на эндотелиальные клетки, индуцирует IL-8, что приводит к рекрутированию нейтрофилов (Nabah et al., 2004). Эти нейтрофилы могут выделять АФК и образовывать внеклеточные ловушки нейтрофилов (Khan et al., 2017). Действительно, нейтрофилы в альвеолах были замечены при патологии легких у инфицированных пациентов (Barnes et al., 2020). Мы предполагаем, что это накопление нейтрофилов приведет к нетозу и гиперкапнии с неадекватной диффузией CO 2 через альвеолы, если наша модель верна. Поверхностно-активное вещество, которое обычно защищает от нетоза (Rodriguez et al., 2019), будет скудным из-за потери альвеолярных эпителиальных клеток.Это может привести к нарушению функции легких, которое похоже на то, что наблюдается при остром респираторном дистресс-синдроме, но при инфекции SARS-CoV-2 разрушение функции легких может прогрессировать по другому пути. При остром респираторном дистресс-синдроме потеря функции легких является следствием цитокинового шторма из-за активации врожденной и адаптивной иммунной систем. Мы предполагаем, что при COVID-19 легкое разрушается комбинацией цитопатического эффекта вируса и несбалансированного RAS со слишком большим количеством AT1 и недостаточным ACE2 (рис.1). Это суть проблемы, которая объясняет большую тяжесть заболевания у пожилых людей, а также у людей с гипертонией и ожирением, а не у молодых и здоровых людей. В этой модели ключевой проблемой является потеря ACE2 из-за вовлечения ACE2 в спайковый белок и активации TACE. Действительно, дефицит ACE2 усугубляет заболевание на мышиных моделях метициллин-устойчивого Staphylococcus aureus и двух штаммов гриппа (Khan et al., 2017; Yang et al., 2014; Zou et al., 2014). У пациентов, инфицированных птичьим гриппом H5N1, уровни ангиотензина II повышаются, причем высокие уровни связаны с большей тяжестью заболевания. Точно так же у мышей, инфицированных H5N1, уровни ангиотензина II предсказывают исход, а дефицит ACE2 приводит к большей смертности (Zou et al., 2014). У мышей, инфицированных H5N9, дефицит ACE2 также приводит к более тяжелому заболеванию. Ангиотензин, не расщепляемый ACE2, связывает рецептор AT1, способствуя патологии; ингибитор рецептора AT1, лозартан, улучшает исход заболевания (Yang et al., 2014). Интересно, что на мышиной модели инфекции Pseudomonas aeruginosa дефицит функции ACE2 приводит к увеличению накопления нейтрофилов в легких посредством IL-17-зависимого пути (Sodhi et al., 2019).
Повышенный TF в эндотелиальных клетках приводит к тромбозу, который присутствует у многих госпитализированных пациентов с COVID-19 (Bikdeli et al., 2020; Klok et al., 2020). Повышенный уровень альдостерона может быть причиной гипокалиемии, наблюдаемой у многих пациентов с COVID-19.Более предположительно, в то время как воспалительная ветвь RAS производит TNF, IL-6 и другие провоспалительные цитокины, не производя интерферон, ACE расщепляет брадикинин, известный супрессор интерферона, и поэтому может привести к увеличению интерферона у некоторых людей (Seliga et al. др., 2018). Некоторые люди с определенной генетической предрасположенностью могут продуцировать высокие уровни интерферона, и это могут быть люди, у которых развиваются «пальцы ног COVID» (Landa et al., 2020; Zhang et al., 2020). Эта предрасположенность может включать те гены с аллелями риска при системной красной волчанке (СКВ), которые имеют склонность к высокому производству интерферона (Ghodke-Puranik and Niewold, 2015).Интересно, что низкий уровень интерферона наблюдается у многих пациентов с COVID-19 и больше соответствует активации пути RAS, чем активации TLR (Hadjadj et al., 2020 Preprint ). Активация TLR индуцирует интерферон; активация AT1 ангиотензином II, напротив, приводит к провоспалительным цитокинам, но не к интерферону (Meng et al., 2017).
Есть последствия этой модели. Во-первых, у пациентов с COVID-19 будет высокий уровень ангиотензина II, возможно, даже у афроамериканцев, и низкий уровень ang1-7 из-за пониженной активности ACE2.Во-вторых, пациенты с COVID на пальцах ног могут иметь аллели риска в пути интерферона, аналогичные некоторым аллелям риска СКВ, поскольку аналогичная патология наблюдается при СКВ и интерферонопатиях (Kolivras et al., 2020). Высокий уровень интерферона может привести к эффективному противовирусному ответу, но также может иметь негативные последствия, приводящие к интерферон-индуцированной микроангиопатии (Massey and Jones, 2020). Ингибиторы АПФ могут быть скорее полезными, чем вредными. Блокаторы рецепторов ангиотензина, которые преимущественно нацелены на AT1, также могут быть полезны.