Случай ольфактогенитальной дисплазии (синдром Каллмана) у женщин | Бабарина
В 1944 г. F. J. Kaliman в работе «Генетические аспекты первичного евнухоидизма» впервые описал синдром, характеризующийся задержкой или отсутствием полового развития и аносмией, впоследствии получивший название синдрома Калл- мана (ольфактогенитальная дисплазия) [3, 4].
Данный синдром развивается вследствие нарушения миграции нейрональных ГнРГ (гонадотропин-рилизинг-гормон)-продуцирующих клеток из медиальной ольфакторной зоны головного мозга в преоптические ядра гипоталамуса. В настоящее время доказана связь развития ольфактогенитальной дисплазии с мутацией гена, локализующегося в регионе р.22.3 Х-хромосомы. Обнаружено, что указанный регион длиной 67 т. п. о. существенно уменьшен у лиц с синдромом Каллмана и впоследствии он был назван ADMLX (adhesion molecule-like from X- chromosome) [1]. В дальнейшем была выделена комплементарная ДНК для этой области и ген идентифицирован как KALIG-1 (Kaliman syndrome interval gene 1) [2].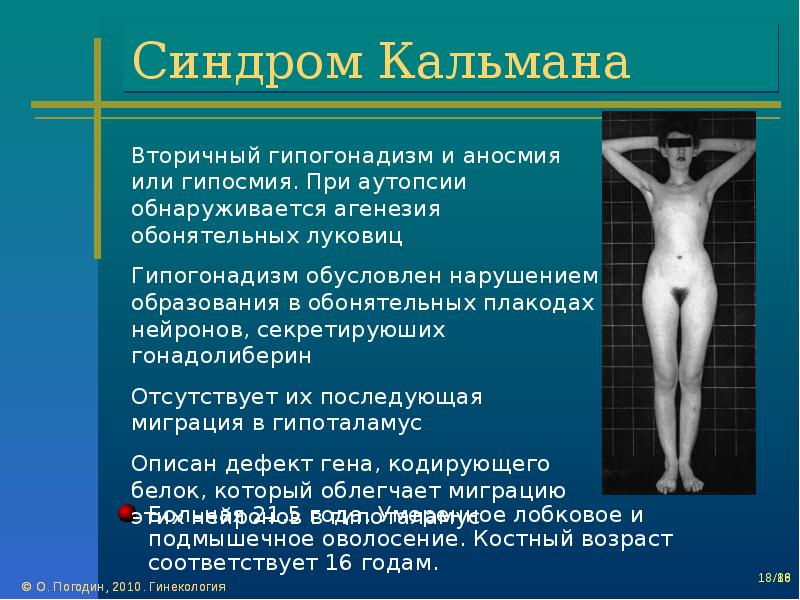 Анализ предполагаемой аминокислотной последовательности белка выявил гомологичность с молекулами адгезии нервных клеток, которые играют важную роль в регуляции развития и морфогенеза нервной ткани. Предполагается, что KALIG-1 может кодировать новый тип нейрогенного миграционного фактора. Заболевание имеет три варианта наследования: Х-сцепленный, аутосомно-доминантный, аутосомно-рецессивный [5, 6].
Анализ предполагаемой аминокислотной последовательности белка выявил гомологичность с молекулами адгезии нервных клеток, которые играют важную роль в регуляции развития и морфогенеза нервной ткани. Предполагается, что KALIG-1 может кодировать новый тип нейрогенного миграционного фактора. Заболевание имеет три варианта наследования: Х-сцепленный, аутосомно-доминантный, аутосомно-рецессивный [5, 6].
Клинически ольфактогенитальная дисплазия у женщин проявляется первичной аменореей и как следствие — первичным бесплодием. При осмотре может быть выявлено телосложение евнухоидного типа, редко наблюдается умеренное развитие молочных желез. У мужчин — гипоплазированные яички, к подростковому возрасту формируется евнухоидная внешность (высокий рост, яички препубертатного размера, инфантильный половой член, полное отсутствие вторичных половых признаков).
Гипоосмия или аносмия — сопутствующий симптом заболевания, вследствие частичной или полной агенезии обонятельных луковиц и ольфакторного тракта. Нейроны, секретирующие ГнРГ, так же как и ольфакторные нейроны, в период эмбриогенеза формируются в зоне ольфакторной пластины, а затем совместно мигрируют, пересекая этмоидальную пластину, в различные отделы мозга. Контакт ольфакторных нейронов с передними отделами мозга необходим для нормального развития обонятельных луковиц. ГнРГ-нейроны мигрируют, проходя более значительное расстояние, достигая преоптических ядер гипоталамуса [И, 13]. Исследования, проводимые у эмбрионов с синдромом Каллмана, показали, что и ГнРГ-секретирующие нейроны, и аксоны ольфакторного тракта формируются нормально, но миграция их заканчивается преждевременно, в пределах оболочек мозга, и нейроны не достигают их нормальной конечной локализации [14].
Нейроны, секретирующие ГнРГ, так же как и ольфакторные нейроны, в период эмбриогенеза формируются в зоне ольфакторной пластины, а затем совместно мигрируют, пересекая этмоидальную пластину, в различные отделы мозга. Контакт ольфакторных нейронов с передними отделами мозга необходим для нормального развития обонятельных луковиц. ГнРГ-нейроны мигрируют, проходя более значительное расстояние, достигая преоптических ядер гипоталамуса [И, 13]. Исследования, проводимые у эмбрионов с синдромом Каллмана, показали, что и ГнРГ-секретирующие нейроны, и аксоны ольфакторного тракта формируются нормально, но миграция их заканчивается преждевременно, в пределах оболочек мозга, и нейроны не достигают их нормальной конечной локализации [14].
Клинические проявления синдрома Каллмана могут быть достаточно вариабельными. Даже больные в одной семье могут иметь разные клинические проявления: от легкой аносмии, выявляемой только с помощью специальных тестов, и нормального полового развития до выраженной аносмии и глубокого гипогонадизма. В редких случаях гипогонадизм и аносмия могут сочетаться с другими генетическими аномалиями: спастические параплегии, глухота, горизонтальный нистагм, нарушения цветового зрения, незаращение неба и верхней губы, задержка умственного развития. Многие из этих аномалий связаны с пороком развития миндалевидного тракта, что предполагает связь заболевания с пороком развития этой области. Возможны также симптомы, связанные с пороком развития мочеполовой системы: агенезия почек, подковообразная почка, крипторхизм у мужчин [10, 15].
В редких случаях гипогонадизм и аносмия могут сочетаться с другими генетическими аномалиями: спастические параплегии, глухота, горизонтальный нистагм, нарушения цветового зрения, незаращение неба и верхней губы, задержка умственного развития. Многие из этих аномалий связаны с пороком развития миндалевидного тракта, что предполагает связь заболевания с пороком развития этой области. Возможны также симптомы, связанные с пороком развития мочеполовой системы: агенезия почек, подковообразная почка, крипторхизм у мужчин [10, 15].
При гормональном исследовании выявляют низкое содержание лютеинизирующего гормона (ЛГ), фолликулостимулирующего гормона (ФСГ) и эстрадиола у женщин и тестостерона у мужчин, нормальный уровень пролактина в крови.
По данным различных авторов, заболевание встречается преимущественно у мужчин, частота встречаемости в популяции, по данным разных авторов: у мужчин 1:10 000, у женщин от 1:50 000 до 1:80 000. В связи с редкой встречаемостью этой патологии у женщин приводим описание клинического случая.
В отделение нейроэндокринологии Эндокринологического научного центра РАМН 14 марта 2005 г. госпитализирована пациентка А., 33 лет, жительница Санкт-Петербурга, с диагнозом: «первичная аменорея». При поступлении предъявляла жалобы на отсутствие менструаций, бесплодие.
Из анамнеза установлено, что самостоятельных менструаций никогда не было, по поводу чего неоднократно обращалась к гинекологу. С 21 года нерегулярно получала заместительную гормональную терапию мерсилоном с менструальноподобной реакцией. Необходимо отметить, что активно пациентка на аносмию не жаловалась. При целенаправленном расспросе, когда по данным предварительного обследования было очевидно, что у пациентки имеет место гипогонадотропный гипогонадизм, нами выявлена аносмия (например, пациентка не отличает запах жареной картошки от бензина). Также при активном расспросе выявлено, что имеется аносмия у бабушки по материнской линии, первичная аменорея у сестры. При обследовании в 2003 г. по месту жительства на 5-й день индуцированного менструального цикла: ФСГ 1,84 ЕД/л (1,9—11,6), ЛГ 0,12 ЕД/л (2,5—12), пролактин 48,2 мЕ/л (90—540), ультразвуковое исследование (УЗИ) органов малого таза — признаки гипоплазии матки. При магнитно-резонансной томографии (МРТ) головного мозга данных о наличии аденомы не получено. Кариотипирование: кариотип 46 XX.
При магнитно-резонансной томографии (МРТ) головного мозга данных о наличии аденомы не получено. Кариотипирование: кариотип 46 XX.
Объективно: Состояние пациентки при поступлении удовлетворительное. Рост 169 см, масса 67 кг, индекс массы тела (ИМТ) 23 кг/м2. Особенности объективного статуса: аносмия, телосложение нормостеническое, определяется высокая талия, слабая пигментация ореола молочных желез, недостаточное оволосение подмышечной и лобковой областей. Артериальное давление 115/70 мм. рт. ст., ЧСС 70 в 1 мин. Щитовидная железа при пальпации не увеличена, плотноэластической консистенции, однородной структуры, безболезненная.
Клинический анализ крови и мочи, биохимическое исследование крови — без патологии.
Гормональное исследование на 5-й день индуцированного менструального цикла: ЛГ <0,1 (норма 1,1-11,6), ФСГ <0,1 (норма 2,8—11,3) мМЕД/мл, эстрадиол < 0,07 (норма 0,11—0,73) нмоль/л, пролактин — 110 (норма 40—530) мМЕД/л.
УЗИ органов малого таза: матка в anteflexio 3,9 х 2,7 х 3,7 см. Длина шейки матки 2,8 см. Эндометрий 0,1—0,2 см, «линейный». Яичники не визуализируются. Заключение: гипоплазия матки и яичников.
Длина шейки матки 2,8 см. Эндометрий 0,1—0,2 см, «линейный». Яичники не визуализируются. Заключение: гипоплазия матки и яичников.
Осмотр гинеколога: оволосение по женскому типу. Молочные железы мягкие, выделений нет. Status genitalis: наружные половые органы развиты правильно. Клитор нормальных размеров. «Симптома зрачка» нет. Выделения слизистые. Шейка матки обычной консистенции. Тело матки в anteflexio. Матка небольшая. Придатки отдельно не пальпируются.
МРГ головного мозга: неоднородность структуры аденогипофиза. Данных о наличии аденомы гипофиза нет. Прицельное исследование состояния обонятельных луковиц не проводили ввиду значительной продолжительности МРТ (несколько часов) и низкой информативности, что связано с малыми размерами bulbus olphactorius.
По данным двухэнергетической рентгеновской денситометрии на аппарате «Prodigy» костной денситометрии, умеренный остеопороз поясничного отдела позвоночника в целом (Т-критерий max = -3,1). Остеопения проксимального отдела бедренной кости (Т-критерий в шейке бедренной кости равен -2,4).
На основании данных анамнеза, наследственных факторов и гормональных исследований установлен диагноз: синдром Каллмана; гипогонадотропный гипогонадизм; аменорея I; аносмия; гипо гонадальный остеопороз с преимущественным поражением поясничного отдела позвоночника.
Пациентке рекомендованы препараты кальция и витамина D3, заместительная терапия эстрогенами. Ввиду того что основной целью обращения пациентки было выяснение причин бесплодия, дальнейшее обследование с целью определения генетических маркеров, ответной реакции яичников на экзогенную гонадотропную стимуляцию пациентке будут проводить по месту жительства с последующим наблюдением гинеколога-эндокринолога.
В схеме лечения бесплодия таким пациентам показаны гонадотропные гормоны: хорионический гонадотропин и менотропин или гонадолиберин в импульсном режиме [7, 8]. Человеческий хорионический гонадотропин вначале назначают по 1000 ЕД внутримышечно 2 раза в неделю, затем дозу постепенно увеличивают до 2000—3000 ЕД 2 раза в неделю и вводят на протяжении 2—3 лет. В течение 2-го и 3-го года лечения хорионическим гонадотропином подключают менотропин (менопаузальный гонадотропин; экстракт из мочи женщин в постменопаузе, содержащий ЛГ и ФСГ) в дозе 75 ЕД внутримышечно 3 раза в неделю. Комбинированное лечение продолжают 12—15 мес. Стимуляцию овуляции проводят гонадолиберином (гонадорели- на ацетат) в импульсном режиме: каждые 90 мин подкожно вводят 5 мкг препарата внутривенно или с помощью программируемого носимого дозатора на протяжении 7 сут. Интервал между курсами 3 нед [9, 12]. Благоприятный прогноз наступления беременности возможен в 70% случаев. В клинической практике есть наблюдения рождения двух детей в одной семье при адекватной заместительной гормональной терапии [16].
В течение 2-го и 3-го года лечения хорионическим гонадотропином подключают менотропин (менопаузальный гонадотропин; экстракт из мочи женщин в постменопаузе, содержащий ЛГ и ФСГ) в дозе 75 ЕД внутримышечно 3 раза в неделю. Комбинированное лечение продолжают 12—15 мес. Стимуляцию овуляции проводят гонадолиберином (гонадорели- на ацетат) в импульсном режиме: каждые 90 мин подкожно вводят 5 мкг препарата внутривенно или с помощью программируемого носимого дозатора на протяжении 7 сут. Интервал между курсами 3 нед [9, 12]. Благоприятный прогноз наступления беременности возможен в 70% случаев. В клинической практике есть наблюдения рождения двух детей в одной семье при адекватной заместительной гормональной терапии [16].
Особенностью данного случая является развитие остеопороза поясничного отдела позвоночника, подлежащего соответствующей коррекции.
1. Семичева Т. В., Баканова Т.Д. // Пробл. эндокринол. — 2004. — № 3. — С. 21-24.
В., Баканова Т.Д. // Пробл. эндокринол. — 2004. — № 3. — С. 21-24.
2. Baird D. Т. // Lancet. — 1997. — Vol. 350. — P. 275-279.
3. Bick D., Franco В., Sherins R. J. et al. // N. Engl. J. Med. — 1992. — Vol. 326. — P. 1752-1755.
4. Roux N., Young J., Misrahi M. et al. // N. Engl. J. Med. — 1997.- Vol. 337. — P. 1597-1602.
5. Dissaneevate P., Warne G. L., Zacharin M. R. // J. Pediatr. Endocrinol. Metab. — 1998. — Vol. 11, N 5. — P. 631-638.
6. Fox K. M., Swan L. // J. Clin. Endocrinol. Metab. — 1999.- Vol. 72. — P. 808-813.
7. Fuerxer F., Carlier R., Iffenecker C. et al. // J. Neuroradiol. — 1996. — Vol. 23, N 4. — P. 223-230.
Fuerxer F., Carlier R., Iffenecker C. et al. // J. Neuroradiol. — 1996. — Vol. 23, N 4. — P. 223-230.
8. Ghai K., Cara J. F., Rosenfield R. L. // J. Clin. Endocrinol. Metab. — 1995. — Vol. 80, N 10. — P. 2980-2986.
9. Handelin J. P., Levilliers J., del Castillo I. et al. // Proc. Natl. Acad. Sci. USA. — 1992. — Vol. 89. — P. 8190-8194.
10. Handelin J. P., Levilliers J., Young J. et al. // J. Clin. Endocrinol. Metab. — 1993. — Vol. 76. — P. 827-831.
11. Layman L. C, Cohen D. P., Jin M. et al. // Nature Genet. — 1998.- Vol. 18. — P. 14-15.
12. Lieblich J. M., Rogol A. D., White B. J., Rosen S. W. // Am. J. Med. — 1982. — Vol. 73. — P. 506-519.
M., Rogol A. D., White B. J., Rosen S. W. // Am. J. Med. — 1982. — Vol. 73. — P. 506-519.
13. Meitinger Т., Неуе В., Petit С. et al. // Am. J. Hum. Genet. — 1990. — Vol. 47. — P. 664-669.
14. Nakayama Y., Wondisford F. E., Lash R. W. et al. // J. Clin. Endocrinol. Metab. — 1990. — Vol. 70. — P. 1233-1238.
15. Sitvera L., Tanriverdi F., Maccoll G. et al. // 12-th International Congress of Endocrinology. — Lisbon, Aug.-Sep., 2004.
16. Weiss J., Crowley W. F, Jameson J. L. // J. Clin. Endocr. Metab. — 1989. — Vol. 69. — P.299-303.
Синдром Каллмана — Kallmann syndrome
Синдром Каллмана ( СК ) — это генетическое заболевание, которое не позволяет человеку начать или полностью завершить половое созревание .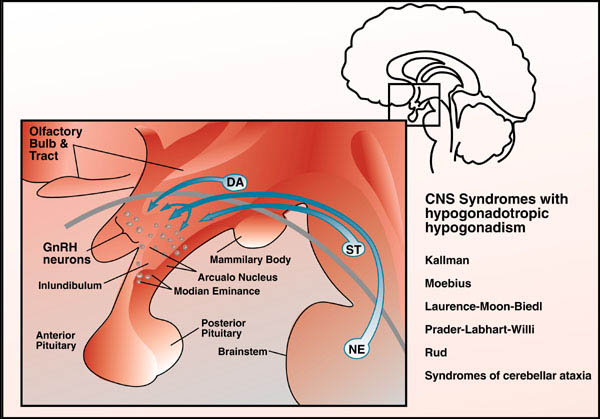 Синдром Каллмана — это форма группы состояний, называемых гипогонадотропным гипогонадизмом . Чтобы отличить его от других форм гипогонадотропного гипогонадизма, синдром Каллмана имеет дополнительный симптом в виде полного отсутствия обоняния (аносмия) или пониженного обоняния . Если не лечить, у людей будут плохо определяемые вторичные половые признаки , проявятся признаки гипогонадизма , они почти всегда будут бесплодны и подвержены повышенному риску развития остеопороза . Также может возникнуть ряд других физических симптомов, влияющих на лицо, руки и скелетную систему.
Синдром Каллмана — это форма группы состояний, называемых гипогонадотропным гипогонадизмом . Чтобы отличить его от других форм гипогонадотропного гипогонадизма, синдром Каллмана имеет дополнительный симптом в виде полного отсутствия обоняния (аносмия) или пониженного обоняния . Если не лечить, у людей будут плохо определяемые вторичные половые признаки , проявятся признаки гипогонадизма , они почти всегда будут бесплодны и подвержены повышенному риску развития остеопороза . Также может возникнуть ряд других физических симптомов, влияющих на лицо, руки и скелетную систему.
Основной причиной является сбой в правильном производстве или активности гонадотропин-рилизинг-гормона по гипоталамусе . Это приводит к низкому уровню половых гормонов тестостерона у мужчин или эстрогена и прогестерона у женщин. Диагноз обычно ставится в подростковом возрасте, когда половое созревание не начинается.
Обычно требуется пожизненное лечение для всех полов. Заместительная гормональная терапия (ЗГТ) является основной формой лечения, направленной на восполнение недостающего тестостерона или эстрогена и прогестерона. Также доступны специализированные процедуры для лечения бесплодия.
Заместительная гормональная терапия (ЗГТ) является основной формой лечения, направленной на восполнение недостающего тестостерона или эстрогена и прогестерона. Также доступны специализированные процедуры для лечения бесплодия.
Заболевание чаще диагностируется у мужчин, чем у женщин. В 2011 году исследование финского населения показало, что заболеваемость составляет 1 случай из 48 000 человек, из которых 1 из 30 000 среди мужчин и 1 из 125 000 среди женщин. Синдром Кальмана был впервые описан по имени в статье , опубликованной в 1944 году Франц Йозеф Каллманна , в немецком — американский генетик . Связь между аносмией и гипогонадизмом уже была отмечена испанским врачом Аурелиано Маэстре де Сан-Хуан в 1856 году.
Признаки и симптомы
19 лет с синдромом Каллмана до диагностики и лечения
Певец Джимми Скотт (справа), чей необычный голос был вызван синдромом Каллмана
Обычно трудно отличить случай синдрома Каллмана (СК) / гипогонадотропного гипогонадизма (ГГ) от прямой конституциональной задержки полового созревания . Однако, если половое созревание не наступило ни к 14 годам (девочкам), ни к 15 годам (мальчикам) и присутствует один или несколько не репродуктивных признаков, упомянутых ниже, то может быть целесообразно направление к репродуктивному эндокринологу .
Однако, если половое созревание не наступило ни к 14 годам (девочкам), ни к 15 годам (мальчикам) и присутствует один или несколько не репродуктивных признаков, упомянутых ниже, то может быть целесообразно направление к репродуктивному эндокринологу .
Характеристики KS и других форм HH можно разделить на две разные категории; «репродуктивный» и «непродуктивный».
Репродуктивные особенности
Не репродуктивные особенности
- Полное отсутствие обоняния ( аносмия ) или заметно сниженное обоняние (гипосмия). Это определяющая черта синдрома Каллмана; это не наблюдается в других случаях HH. Примерно 50% случаев ГГ происходят с аносмией и могут быть названы синдромом Каллмана.
- Расщелина неба , губы или другие дефекты черепа и лица средней линии.
- Нервное нарушение слуха
- Отсутствие одной из почек (односторонняя агенезия почек)
- Скелетные дефекты, включая расщепление кисти и стопы ( эктродактилия ), укороченный средний палец (пястные кости) или сколиоз
- Мануальные синкинезии (зеркальные движения рук)
- Отсутствие зубов (гиподонтия)
- Плохое равновесие или координация из-за церебральной атаксии .

- Дефекты глаз, такие как колобома или птоз .
- Увеличение числа случаев дальтонизма

Точная генетическая природа каждого конкретного случая KS / HH будет определять, какие из не репродуктивных особенностей, если таковые имеются, будут иметь место. Выраженность симптомов также будет варьироваться от случая к случаю. Даже у членов семьи не будет такого же диапазона или серьезности симптомов.
KS / HH чаще всего присутствует с рождения, но версии с началом у взрослых встречаются как у мужчин, так и у женщин. Гипоталамо-гипофизарно-гонадной оси (ось HPG) функционирует нормально при рождении и хорошо во взрослую жизнь, давая нормальное половое созревание и нормальную репродуктивную функцию. Затем ось HPG либо полностью выходит из строя, либо снижается до очень низкого уровня высвобождения GnRH во взрослой жизни без очевидной причины (например, опухоль гипофиза). Это приведет к падению уровня тестостерона или эстрогена и бесплодию.
Функциональная гипоталамическая аменорея наблюдается у женщин, где ось HPG подавляется в ответ на физический или психологический стресс или недоедание, но обратима при удалении фактора стресса.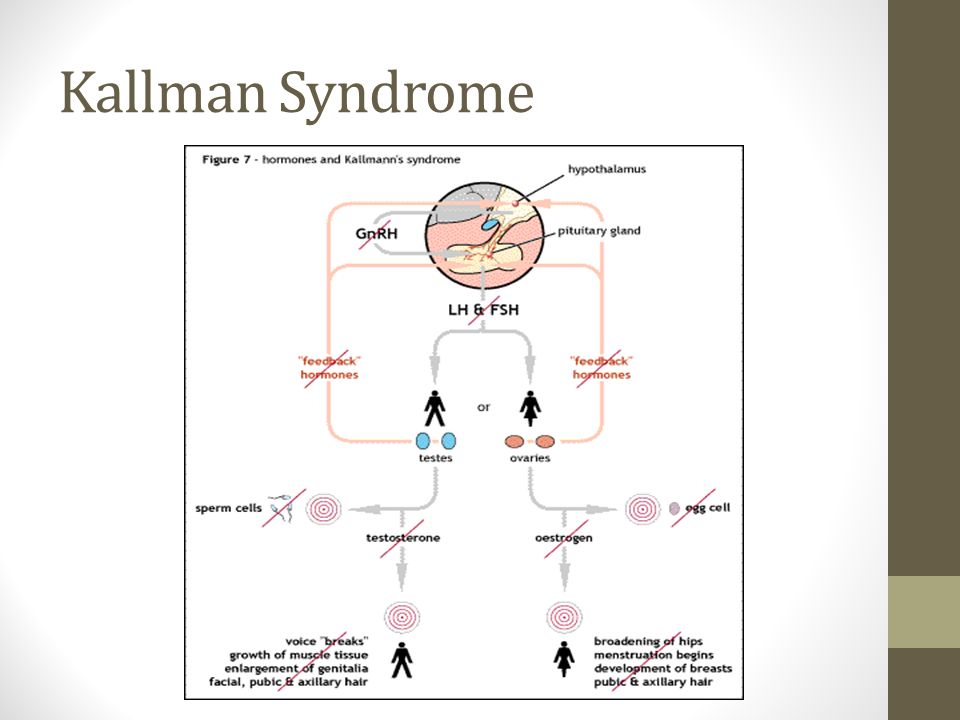
Некоторые случаи KS / HH, по-видимому, меняются в течение взрослой жизни, когда ось HPG восстанавливает свою нормальную функцию, а уровни GnRH, LH и FSH возвращаются к нормальным уровням. Это происходит примерно у 10–22% людей, в первую очередь у нормосмических случаев ЗГГ, а не у пациентов с СК, и обнаруживается только у людей, которые прошли какую-либо форму заместительной терапии тестостероном. Обычно это обнаруживается только тогда, когда объем яичек увеличивается во время лечения только тестостероном, а уровень тестостерона возвращается к норме после прекращения лечения. Этот тип KS / HH редко встречается в тех случаях, когда у мужчин в анамнезе не опускались яички.
Люди, страдающие СК и другими формами ГГ, почти всегда рождаются с нормальной половой дифференциацией; т.е. физически они мужчины или женщины. Это происходит из-за человеческого хорионического гонадотропина (ХГЧ), вырабатываемого плацентой примерно на сроке от 12 до 20 недель беременности (беременность), на который обычно не влияет СК или ХГГ.
У людей с KS / HH отсутствует всплеск GnRH, LH и FSH, который обычно возникает между рождением и шестимесячным возрастом. Этот всплеск особенно важен для мальчиков, так как он помогает при опускании яичек в мошонку. Всплеск GnRH / LH / FSH у детей без KS / HH дает определяемые уровни тестостерона у мальчиков и эстрогена и прогестерона у девочек. Отсутствие этого всплеска иногда может быть использовано в качестве диагностического инструмента, если подозревается саркома-шевелюра / ГГ у новорожденного мальчика, но, как правило, оно недостаточно отчетливо для диагностики у девочек.
Остеопороз
Одним из возможных побочных эффектов KS / CHH является повышенный риск развития вторичного остеопороза или остеопении . Эстроген (женщины) или тестостерон (мужчины) необходимы для поддержания плотности костей . Дефицит тестостерона или эстрогена может увеличить скорость резорбции кости , в то же время замедляя скорость образования кости . В целом это может привести к ослаблению и хрупкости костей, которые более склонны к переломам.
Даже непродолжительное время с низким уровнем эстрогена или тестостерона, поскольку в случаях поздней диагностики KS / CHH может привести к повышенному риску развития остеопороза, но при этом присутствуют и другие факторы риска, такие как курение, поэтому риск его развития будет варьироваться от человека к человеку. человек. Для контроля минеральной плотности кости рекомендуется сканирование плотности костей.
Сканирование плотности кости известно как двухэнергетическое рентгеновское абсорбциометрическое сканирование (DEXA или DXA сканирование). Это простой тест, на выполнение которого требуется менее 15 минут. Он включает в себя получение специального рентгеновского снимка позвоночника и бедер, измерение минеральной плотности костной ткани и сравнение результата со средним значением для молодого здорового взрослого в общей популяции.
Адекватный уровень кальция и, возможно, что более важно, уровень витамина D необходимы для здоровой плотности костей. Некоторым людям с KS / CHH будут проверяться их уровни, и им могут прописать дополнительные таблетки или инъекции витамина D, чтобы попытаться предотвратить ухудшение состояния. Роль витамина D для общего состояния здоровья в настоящее время находится под пристальным вниманием, и некоторые исследователи утверждают, что дефицит витамина D распространен во многих группах населения и может быть связан с другими заболеваниями.
Некоторым людям с KS / CHH будут проверяться их уровни, и им могут прописать дополнительные таблетки или инъекции витамина D, чтобы попытаться предотвратить ухудшение состояния. Роль витамина D для общего состояния здоровья в настоящее время находится под пристальным вниманием, и некоторые исследователи утверждают, что дефицит витамина D распространен во многих группах населения и может быть связан с другими заболеваниями.
Некоторым людям с тяжелым остеопорозом могут быть назначены бисфосфонаты для сохранения костной массы в дополнение к заместительной гормональной терапии.
Генетика
Генетические и молекулярные основы идиопатического гипогонадотропного гипогонадизма
На сегодняшний день по меньшей мере 25 различных генов вовлечены в возникновение синдрома Каллмана или других форм гипогонадотропного гипогонадизма через нарушение производства или активности гонадолиберина (37). Эти вовлеченные гены охватывают все формы наследования, и ни один дефект гена не является общим для всех случаев, что затрудняет генетическое тестирование и прогнозирование наследования.
Число известных генов, вызывающих случаи KS / CHH, все еще увеличивается. Кроме того, считается, что некоторые случаи KS / CHH вызваны двумя отдельными генными дефектами, возникающими одновременно.
Дефекты отдельных генов могут быть связаны со специфическими симптомами, которые могут помочь в определении того, какие гены нужно проверить. От 35 до 45% случаев KS / CHH имеют неизвестную генетическую причину.
ANOS1 дефект гена (ранее известный как Кал-1) был первым , кто открыл и одним из наиболее часто испытывали на. Он вызывает Х-сцепленную форму синдрома Каллмана и связан с дополнительными симптомами аносмии , бимануального синкинезии и агенеза почек . Считается, что этот дефект является причиной от 5 до 10% всех случаев синдрома Каллмана / ХГГ.
Патофизиология
Показывает нормальный гормональный контроль полового созревания от гипоталамуса до семенников или яичников и их механизмы отрицательной обратной связи. Контроль отрицательной обратной связи позволяет высвобождать только нужное количество гормона в соответствии с потребностями организма в данный момент.
Показывает эффект прерывания высвобождения гормона GnRH из гипоталамуса и последующую неспособность семенников и яичников правильно функционировать в период полового созревания, как это видно в случаях KS / HH. В большинстве случаев KS / HH яички и яичники могут функционировать правильно, но не могут этого делать, потому что у них нет правильных гормональных сигналов.
Структура GNRh2
(из PDB : 1YY1 )
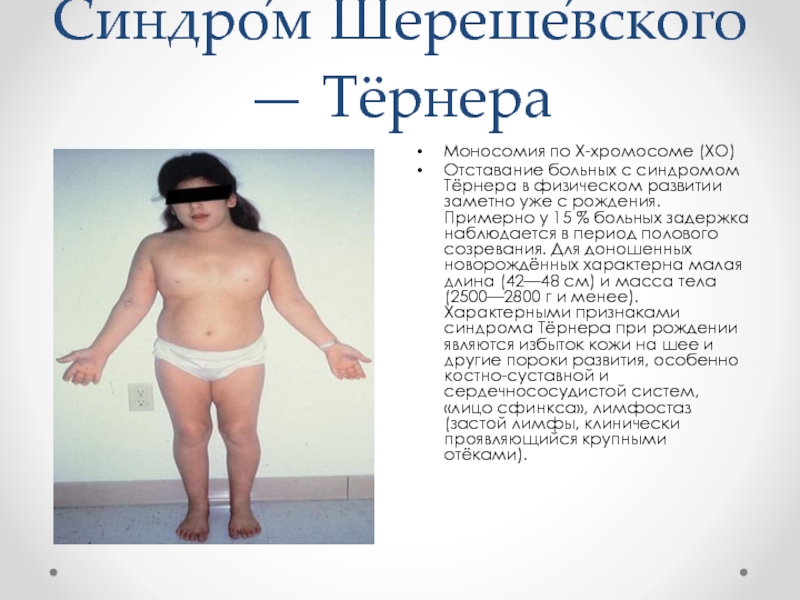
Основная причина синдрома Каллмана или других форм гипогонадотропного гипогонадизма — нарушение правильного действия гипоталамического гормона ГнРГ . Термин изолированный дефицит гонадолиберина (IGD) все чаще используется для описания этой группы состояний, поскольку он подчеркивает основную причину этих состояний и отличает их от других состояний, таких как синдром Клайнфельтера или синдром Тернера, которые имеют некоторые сходные симптомы, но имеют другую этиологию. . Термин гипогонадизм описывает низкий уровень циркулирующих половых гормонов ; тестостерон у мужчин и эстроген и прогестерон у женщин. Гипогонадизм может возникать по разным причинам. Использование термина гипогонадотропный связано с тем фактом, что гипогонадизм, обнаруживаемый при ГГ, вызван нарушением выработки гормонов гонадотропина, которые обычно выделяются передней долей гипофиза, известных как лютеинизирующий гормон (ЛГ) и фолликулостимулирующий гормон (ФСГ). В противном случае нарушение активности GnRH может быть связано с отсутствием нейронов, высвобождающих GnRH, внутри гипоталамуса. ГГ может возникать как изолированное состояние, при котором затрагивается только выработка ЛГ и ФСГ, или он может возникать в условиях комбинированного дефицита гипофиза.
Гипогонадизм может возникать по разным причинам. Использование термина гипогонадотропный связано с тем фактом, что гипогонадизм, обнаруживаемый при ГГ, вызван нарушением выработки гормонов гонадотропина, которые обычно выделяются передней долей гипофиза, известных как лютеинизирующий гормон (ЛГ) и фолликулостимулирующий гормон (ФСГ). В противном случае нарушение активности GnRH может быть связано с отсутствием нейронов, высвобождающих GnRH, внутри гипоталамуса. ГГ может возникать как изолированное состояние, при котором затрагивается только выработка ЛГ и ФСГ, или он может возникать в условиях комбинированного дефицита гипофиза.
В первые 10 недель нормального эмбрионального развития нейроны, высвобождающие гонадолиберин, мигрируют из своего первоначального источника в носовую область и в конечном итоге попадают в гипоталамус. Эти нейроны происходят из области развивающейся головы, обонятельной плакоды , которая дает начало обонятельному эпителию; Затем они проходят через решетчатую пластинку вместе с волокнами обонятельных нервов в ростральный передний мозг . Оттуда они мигрируют в то, что станет гипоталамусом. Любые проблемы с развитием волокон обонятельного нерва будут препятствовать продвижению нейронов, высвобождающих гонадолиберин, к мозгу.
Оттуда они мигрируют в то, что станет гипоталамусом. Любые проблемы с развитием волокон обонятельного нерва будут препятствовать продвижению нейронов, высвобождающих гонадолиберин, к мозгу.
Диагностика
Диагностика KS и других форм CHH осложняется тем, что трудно различить нормальную конституциональную задержку полового созревания и случай KS / CHH. Диагноз часто ставится при диагностике задержки полового созревания .
У мужчин использование соответствующих возрасту уровней тестостерона может помочь отличить случай KS / CHH от случая задержки полового созревания. Если полового созревания не наблюдается, особенно при отсутствии развития яичек, может потребоваться проверка эндокринолога-репродуктолога. Если половая зрелость не проявляется к 16 годам, человека следует направить на эндокринологическое обследование. Постнатальная диагностика KS / CHH в возрасте до 6 месяцев иногда возможна, поскольку нормальный послеродовой гормональный всплеск гонадотропинов вместе с тестостероном или эстрогеном отсутствует у детей с KS / CHH. Этот недостаток обнаруживаемых гормонов в крови может использоваться как диагностический индикатор, особенно у младенцев мужского пола.
Этот недостаток обнаруживаемых гормонов в крови может использоваться как диагностический индикатор, особенно у младенцев мужского пола.
У женщин постановка диагноза иногда откладывается, так как другие причины аменореи обычно необходимо исследовать, прежде чем рассматривать случай KS / CHH.
Чешуйчатая кожевница
Диагностика нормального KS / CHH включает ряд клинических, биохимических и радиологических тестов, чтобы исключить другие состояния, которые могут вызывать аналогичные симптомы.
Клинические испытания
- Сравнение роста со стандартными диаграммами роста.
- Определение стадии полового развития по Таннеру . (Мужчины с KS / CHH обычно находятся на стадии I или II с развитием гениталий, женщины на стадии I с развитием груди, а мужчины и женщины — на стадии III с развитием лобковых волос).
- Проверка на микропенис и неопущение яичек ( крипторхизм ) у мужчин.
- Измерение объема яичка.
- Проверка развития груди и возраста менархе у женщин.
- Проверка обоняния с помощью панели одорантов или теста идентификации запаха Университета Пенсильвании (UPSIT)
- Проверка на нарушение слуха.
- Проверка на отсутствие зубов или расщелины губы и / или неба .
- Проверка на пигментацию кожи и волос.
- Проверка зеркальных движений рук или признаков задержки нервного развития .
Лабораторные тесты
Медицинская визуализация
лечение
Саше с гелем тестостерона, инъекции ундеканоата тестостерона (Небидо), инъекции хорионического гонадотропина человека (ХГЧ), инъекции менотропина (ЧМГ).
И у мужчин, и у женщин первоначальной целью лечения является развитие вторичных половых признаков, обычно наблюдаемых в период полового созревания. Как только это будет достигнуто, как мужчинам, так и женщинам потребуется продолжение заместительной гормональной терапии для поддержания половой функции, здоровья костей, либидо и общего благополучия. У мужчин заместительная терапия тестостероном необходима для поддержания нормальной мышечной массы.
У мужчин заместительная терапия тестостероном необходима для поддержания нормальной мышечной массы.
Раннее лечение иногда требуется для младенцев мужского пола с подозрением на KS / CHH, чтобы исправить непопущенные семенники и микропенис, если они присутствуют при использовании или хирургическом вмешательстве или лечении гонадотропином или DHT . Женщины с KS / CHH обычно не нуждаются в лечении до подросткового возраста. В настоящее время не существует лечения отсутствия обоняния, зеркального движения рук или отсутствия одной почки.
Лечение как мужчин, так и женщин с KS / CHH обычно состоит из одного из трех вариантов, которые можно использовать как для заместительной гормональной терапии, так и для лечения бесплодия.
- Замещение половых гормонов (тестостерон или эстроген и прогестерон).
- Гонадотропная терапия (препараты, которые воспроизводят активность ФСГ и ЛГ).
- Пульсирующая терапия ГнРГ.
Заместительная гормональная терапия
Метод и доза лечения будут варьироваться в зависимости от пациента, которого лечат. Первоначальное лечение обычно проводится более низкими дозами у более молодых пациентов, чтобы развить вторичные половые признаки до того, как будут достигнуты дозы для взрослых.
Первоначальное лечение обычно проводится более низкими дозами у более молодых пациентов, чтобы развить вторичные половые признаки до того, как будут достигнуты дозы для взрослых.
Для мужчин с KS / CHH типы доставки тестостерона включают ежедневные пластыри, ежедневное использование геля, ежедневные капсулы, подкожные или внутримышечные инъекции или шестимесячные имплантаты. Используются различные составы тестостерона для обеспечения как анаболических, так и андрогенных эффектов тестостерона. Способы назальной доставки тестостерона были разработаны, но их использование в лечении KS / CHH официально не оценивалось.
Терапия гонадотропинами в виде инъекций хорионического гонадотропина человека (ХГЧ) с использованием или без использования ФСГ также может применяться у пациентов мужского пола для индукции развития вторичных половых признаков наряду с возможным индукцией фертильности.
Для женщин заместительная гормональная терапия предполагает использование эстрогена и прогестерона. Сначала эстроген используется в форме таблеток или геля для максимального развития груди, затем используется комбинация эстрогена и прогестерона. Циклический прогестерон обычно необходим для поддержания здоровья эндометрия (слизистой оболочки матки ).
Сначала эстроген используется в форме таблеток или геля для максимального развития груди, затем используется комбинация эстрогена и прогестерона. Циклический прогестерон обычно необходим для поддержания здоровья эндометрия (слизистой оболочки матки ).
У мужчин для мониторинга лечения обычно требуется измерение сывороточного тестостерона, ингибина B , гематокрита и простатоспецифического антигена (PSA). Если используются инъекции, измеряются минимальные уровни, чтобы обеспечить достижение адекватного уровня тестостерона на протяжении всего цикла инъекции.
У женщин мониторинг обычно состоит из измерения эстрогена, ФСГ, ЛГ, ингибина В и антимюллерова гормона (АМГ).
Стандартная заместительная гормональная терапия обычно не способствует фертильности ни у мужчин, ни у женщин, при этом рост яичек у мужчин отсутствует. Раннее лечение в подростковом возрасте может помочь в психологическом благополучии людей с KS / CHH.
Лечение бесплодия
Терапия гонадотропинами может использоваться как для мужчин, так и для женщин, чтобы добиться фертильности у некоторых людей.
Пульсирующая терапия гонадолиберином также может использоваться для стимуляции фертильности, особенно у женщин, но ее применение ограничено несколькими специализированными лечебными центрами.
У мужчин с KS / CHH бесплодие в первую очередь связано с отсутствием выработки спермы в семенниках . Производство спермы может быть достигнуто либо за счет использования ГнРГ , вводимого с помощью микроинфузионного насоса, либо за счет инъекций гонадотропина ( ХГЧ , ФСГ, ЧМГ ). Время, необходимое для достижения адекватного производства спермы для естественного зачатия, будет варьироваться от человека к человеку. Если до лечения яички очень маленькие и в анамнезе не опускались яички, может потребоваться больше времени для получения спермы. В этих случаях могут потребоваться вспомогательные репродуктивные технологии , такие как извлечение сперматозоидов с использованием экстракции сперматозоидов из яичек (TESE) и / или интрацитоплазматическая инъекция сперматозоидов (ICSI) .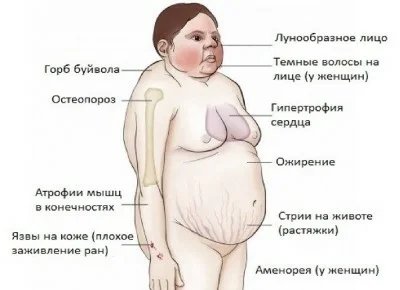
У самок с KS / CHH бесплодие в первую очередь связано с отсутствием созревания яиц, расположенных в яичниках . Стимуляция овуляции может быть достигнута либо с помощью пульсирующей терапии гонадолиберином, либо с помощью инъекций гонадотропина (ХГЧ, ФСГ, ЧМГ), вводимых через определенные промежутки времени, чтобы вызвать созревание и высвобождение яйца для естественного зачатия.
Прогноз
Об исчезновении симптомов сообщалось от 10% до 22% случаев.
Случаи обратного развития наблюдались как при KS, так и при нормосмическом CHH, но, по-видимому, менее распространены в случаях KS (где также нарушается обоняние). Обратное изменение не всегда является постоянным, и точные генетические причины еще не полностью поняты.
Эпидемиология
Эпидемиология синдрома Каллмана до конца не изучена. Отдельные исследования включают отчет 1986 года с обзором медицинских карт в сардинской армии, который выявил распространенность 1 из 86 000 мужчин и отчет 2011 года из Финляндии, в котором обнаружена распространенность 1: 30 000 для мужчин и 1: 125 000 для женщин.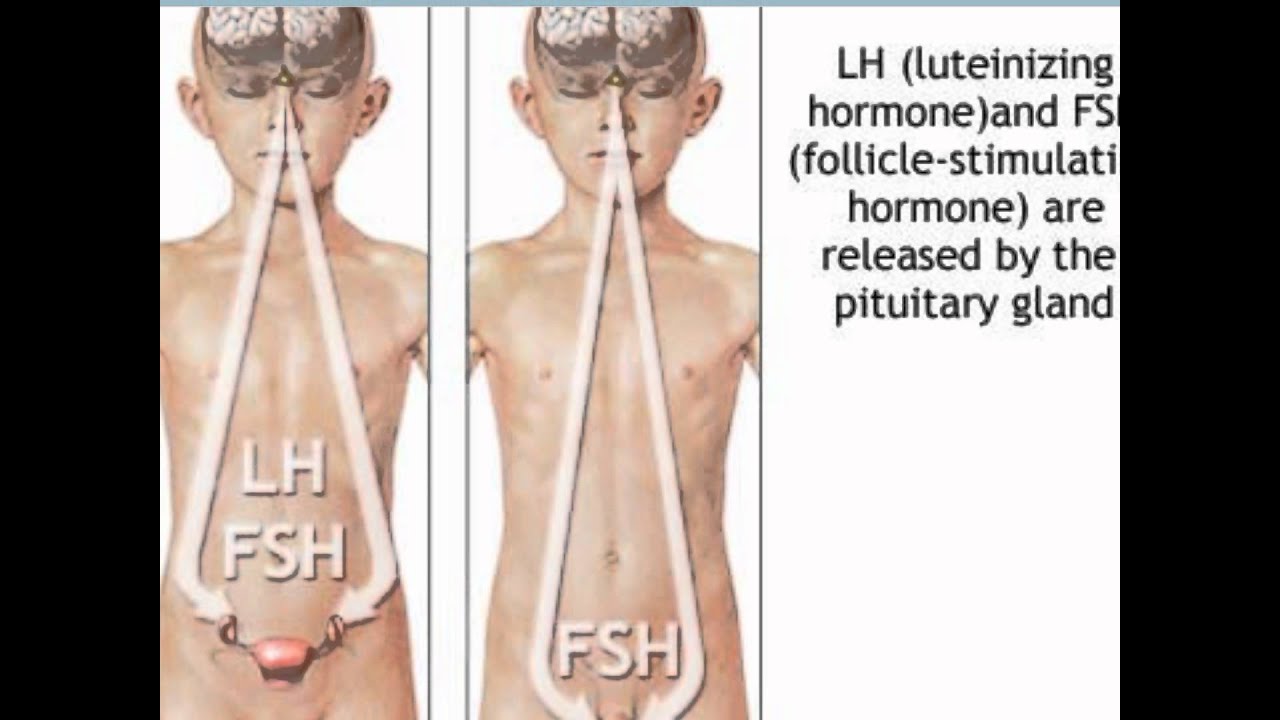
Синдром Каллмана встречается примерно в 4 раза чаще у мужчин, чем у женщин, но только в 2,5 раза чаще встречается у мужчин в семейных случаях.
История
Синдром Кальмана был впервые описан по имени в статье , опубликованной в 1944 году Франц Йозеф Каллманна , в немецком — американский генетик . Связь между аносмией и гипогонадизмом уже была отмечена испанским врачом Аурелиано Маэстре де Сан-Хуаном в 1856 году. В 1950-х годах де Морсье и Готье сообщили о частичном или полном отсутствии обонятельной луковицы в мозгу мужчин с гипогонадизмом.
Терминология
Терминология, используемая при описании случаев ДГ, различается и может включать:
Исследование
Кисспептин — это белок, который регулирует высвобождение гонадолиберина из гипоталамуса, который, в свою очередь, регулирует высвобождение ЛГ и, в меньшей степени, ФСГ из передней доли гипофиза. Известно, что кисспептин и связанный с ним рецептор KISS1R участвуют в регуляции полового созревания. Исследования показали, что кисспептин может использоваться в диагностике и лечении некоторых случаев синдрома Каллмана и хронического гепатита.
Исследования показали, что кисспептин может использоваться в диагностике и лечении некоторых случаев синдрома Каллмана и хронического гепатита.
Рекомендации
внешние ссылки
Бесплодие — Infertility — qaz.wiki
Невозможность воспроизводства естественным путем
Бесплодие — это неспособность человека, животного или растения к воспроизводству естественным путем. Обычно это не естественное состояние здорового взрослого человека, за исключением некоторых эусоциальных видов (в основном гаплодиплоидных насекомых).
У людей бесплодие — это невозможность забеременеть после одного года полового акта без контрацепции с участием партнера мужского и женского пола. Существует множество причин бесплодия, в том числе те, которые можно лечить с помощью медицинского вмешательства . По оценкам 1997 года, около пяти процентов всех гетеросексуальных пар во всем мире имеют нерешенную проблему бесплодия. Однако гораздо больше пар страдают от недобровольной бездетности по крайней мере в течение одного года: оценки варьируются от 12% до 28%. На мужское бесплодие приходится 20–30% случаев бесплодия, 20–35% — из-за женского бесплодия , а 25–40% — из-за сочетанных проблем в обеих частях. В 10–20% случаев причина не обнаруживается. Наиболее частой причиной женского бесплодия являются проблемы с овуляцией, которые обычно проявляются редкими или отсутствующими менструациями. Наиболее частой причиной бесплодия среди западного населения сегодня является задержка родов, потому что качество ооцитов резко снижается с возрастом, особенно после 35 лет. Мужское бесплодие чаще всего возникает из-за недостатка спермы , и качество спермы используется как суррогатный показатель мужской плодовитости .
На мужское бесплодие приходится 20–30% случаев бесплодия, 20–35% — из-за женского бесплодия , а 25–40% — из-за сочетанных проблем в обеих частях. В 10–20% случаев причина не обнаруживается. Наиболее частой причиной женского бесплодия являются проблемы с овуляцией, которые обычно проявляются редкими или отсутствующими менструациями. Наиболее частой причиной бесплодия среди западного населения сегодня является задержка родов, потому что качество ооцитов резко снижается с возрастом, особенно после 35 лет. Мужское бесплодие чаще всего возникает из-за недостатка спермы , и качество спермы используется как суррогатный показатель мужской плодовитости .
У фертильных женщин наступает естественный период фертильности до и во время овуляции , и они естественно бесплодны в течение остальной части менструального цикла . Методы определения фертильности используются, чтобы определить, когда происходят эти изменения, путем отслеживания изменений цервикальной слизи или базальной температуры тела .
Определение
«Демографы склонны определять бесплодие как бездетность в популяции женщин репродуктивного возраста», тогда как «эпидемиологическое определение относится к« попыткам »или« времени до »беременности, обычно в группе женщин, подверженных« вероятности зачатия ». . В настоящее время максимальная фертильность женщин достигает пика в возрасте 24 лет и снижается после 30 лет, при этом беременность редко наступает после 50 лет. Самка наиболее фертильна в течение 24 часов после овуляции. Пик мужской фертильности обычно достигается в возрасте 25 лет и снижается после 40 лет. Время, необходимое для того, чтобы пара пыталась забеременеть, чтобы у этой пары диагностировали бесплодие, различается в разных юрисдикциях. Существующим определениям бесплодия не хватает единообразия, что затрудняет сравнение распространенности между странами или во времени. Таким образом, данные по оценке распространенности бесплодия из разных источников существенно различаются. Пара, которая безуспешно пытается завести ребенка через определенный период времени (часто короткий период, но определения различаются), иногда считается субфертильной , то есть менее плодородной, чем типичная пара.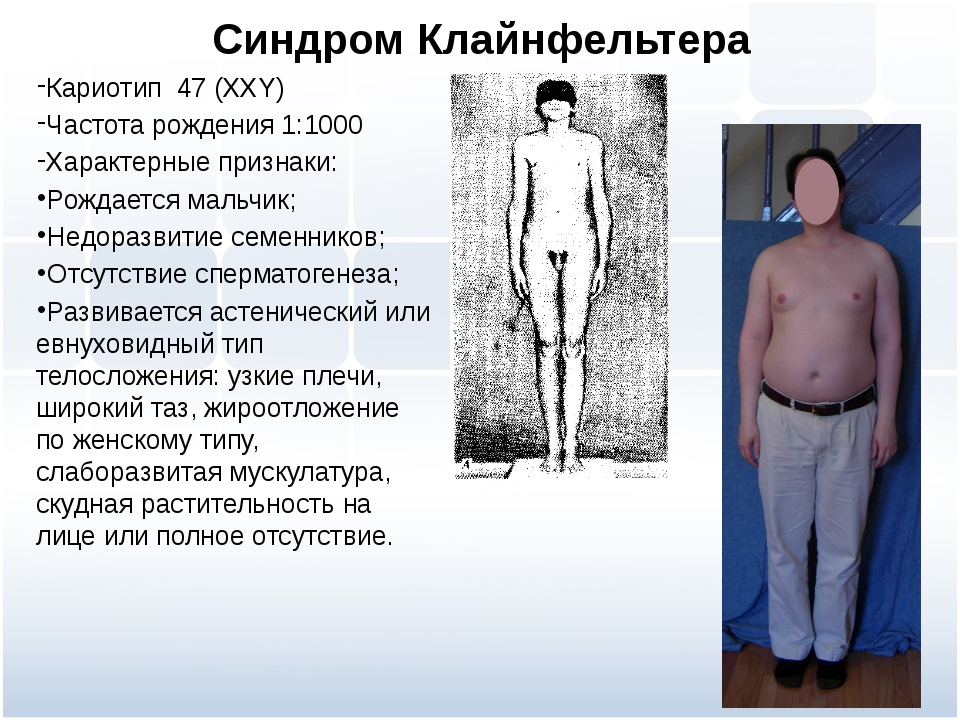 И бесплодие, и субфертильность определяются как неспособность зачать ребенка по прошествии определенного периода времени (продолжительность которого варьируется), поэтому часто эти два термина частично совпадают.
И бесплодие, и субфертильность определяются как неспособность зачать ребенка по прошествии определенного периода времени (продолжительность которого варьируется), поэтому часто эти два термина частично совпадают.
Всемирная организация здоровья
Всемирная организация здравоохранения определяет бесплодию следующим образом :
Бесплодие — это «заболевание репродуктивной системы, определяемое невозможностью наступления клинической беременности после 12 месяцев или более регулярного незащищенного полового акта (и нет другой причины, такой как кормление грудью или послеродовая аменорея ). Первичное бесплодие — это бесплодие у пары, у которой никогда не было ребенка. Вторичное бесплодие — это невозможность зачать ребенка после предыдущей беременности. Бесплодие может быть вызвано инфекцией у мужчины или женщины, но часто очевидной первопричины нет.
Соединенные Штаты
Одно из определений бесплодия, которое часто используется в Соединенных Штатах эндокринологами-репродуктологами , врачами, специализирующимися на бесплодии, чтобы считать пару подходящими для лечения:
- женщина до 35 лет не забеременела после 12 месяцев полового акта без контрацептивов. Двенадцать месяцев это нижний опорный предел для времени до беременности (ТТП) Всемирной организации здравоохранения.
- женщина старше 35 лет не забеременела после шести месяцев полового акта без противозачаточных средств .
 Двенадцать месяцев это нижний опорный предел для времени до беременности (ТТП) Всемирной организации здравоохранения.
Двенадцать месяцев это нижний опорный предел для времени до беременности (ТТП) Всемирной организации здравоохранения.Казалось бы, эти временные интервалы перевернуты; Это область, в которой государственная политика важнее науки. Идея состоит в том, что для женщин старше 35 лет на счету каждый месяц, и если заставить подождать еще шесть месяцев, чтобы доказать необходимость медицинского вмешательства, проблема может усугубиться. Следствием этого является то, что по определению неспособность зачать ребенка у женщин до 35 лет не рассматривается с такой же остротой, как у женщин старше 35 лет.
объединенное Королевство
В Великобритании в предыдущих рекомендациях NICE бесплодие определялось как неспособность зачать ребенка после регулярных незащищенных половых контактов в течение двух лет при отсутствии известной репродуктивной патологии. Обновленные руководства NICE не содержат конкретного определения, но рекомендуют, чтобы «женщине репродуктивного возраста, которая не забеременела после 1 года незащищенного вагинального полового акта, при отсутствии какой-либо известной причины бесплодия, должна быть предложена дальнейшая клиническая оценка и исследование. вместе со своим партнером, с более ранним обращением к специалисту, если женщина старше 36 лет ».
Обновленные руководства NICE не содержат конкретного определения, но рекомендуют, чтобы «женщине репродуктивного возраста, которая не забеременела после 1 года незащищенного вагинального полового акта, при отсутствии какой-либо известной причины бесплодия, должна быть предложена дальнейшая клиническая оценка и исследование. вместе со своим партнером, с более ранним обращением к специалисту, если женщина старше 36 лет ».
Другие определения
Исследователи обычно основывают демографические исследования распространенности бесплодия на пятилетнем периоде. Однако практические проблемы измерения существуют для любого определения, потому что трудно измерить постоянную подверженность риску беременности в течение многих лет.
Первичное бесплодие и вторичное бесплодие
Первичное бесплодие определяется как отсутствие живорождения у женщин, желающих иметь ребенка и состоящих в браке не менее 12 месяцев, в течение которых они не использовали никаких противозачаточных средств. Всемирная организация здравоохранения также добавляет, что «женщины, у которых беременность вызывает самопроизвольный выкидыш или у которых рождается мертворожденный ребенок, но никогда не родивший живого ребенка, в первую очередь страдают бесплодием».
Всемирная организация здравоохранения также добавляет, что «женщины, у которых беременность вызывает самопроизвольный выкидыш или у которых рождается мертворожденный ребенок, но никогда не родивший живого ребенка, в первую очередь страдают бесплодием».
Вторичное бесплодие определяется как отсутствие живорождения у женщин, которые хотят иметь ребенка и состоят в браке не менее 12 месяцев с момента их последнего живорождения, в течение которого они не использовали никаких противозачаточных средств.
Таким образом, отличительным признаком является то, была ли у пары когда-либо беременность, которая привела к рождению живого ребенка.
Последствия
Психологические
Последствия бесплодия разнообразны и могут включать в себя социальные последствия и личные страдания. Достижения в области вспомогательных репродуктивных технологий, таких как ЭКО , могут дать надежду многим парам, где лечение доступно, хотя существуют препятствия с точки зрения медицинского страхования и доступности. Медикаментозность бесплодия невольно привела к пренебрежению эмоциональных реакций , которые испытывают пары, которые включают в себя страдание, потерю контроля, стигматизации и нарушение в траектории развития взрослой жизни. Одной из основных проблем при оценке уровня дистресса у женщин с бесплодием является точность самоотчетов. Возможно, женщины «притворяются хорошими», чтобы казаться психически здоровее, чем они есть. Также возможно, что женщины испытывают чувство надежды / повышенного оптимизма до начала лечения бесплодия, когда собирается большинство оценок дистресса. Некоторые ранние исследования пришли к выводу, что бесплодные женщины не сообщают о каких-либо существенных различиях в симптомах тревоги и депрессии, чем фертильные женщины. Чем дальше проходит лечение, тем чаще у него проявляются симптомы депрессии и тревоги. Пациенты с одной неудачей лечения имели значительно более высокий уровень тревоги, а пациенты с двумя неудачами испытывали большую депрессию по сравнению с пациентами, не лечившимися в анамнезе.
Медикаментозность бесплодия невольно привела к пренебрежению эмоциональных реакций , которые испытывают пары, которые включают в себя страдание, потерю контроля, стигматизации и нарушение в траектории развития взрослой жизни. Одной из основных проблем при оценке уровня дистресса у женщин с бесплодием является точность самоотчетов. Возможно, женщины «притворяются хорошими», чтобы казаться психически здоровее, чем они есть. Также возможно, что женщины испытывают чувство надежды / повышенного оптимизма до начала лечения бесплодия, когда собирается большинство оценок дистресса. Некоторые ранние исследования пришли к выводу, что бесплодные женщины не сообщают о каких-либо существенных различиях в симптомах тревоги и депрессии, чем фертильные женщины. Чем дальше проходит лечение, тем чаще у него проявляются симптомы депрессии и тревоги. Пациенты с одной неудачей лечения имели значительно более высокий уровень тревоги, а пациенты с двумя неудачами испытывали большую депрессию по сравнению с пациентами, не лечившимися в анамнезе. Однако также было показано, что чем больше депрессия у бесплодной женщины, тем меньше у нее шансов начать лечение бесплодия и тем выше вероятность того, что она бросит лечение только после одного цикла. Исследователи также показали, что, несмотря на хороший прогноз и наличие финансовых средств для оплаты лечения, прекращение лечения чаще всего происходит по психологическим причинам. Фертильность не увеличивается, когда женщины принимают антиоксиданты для снижения окислительного стресса, вызванного ситуацией.
Однако также было показано, что чем больше депрессия у бесплодной женщины, тем меньше у нее шансов начать лечение бесплодия и тем выше вероятность того, что она бросит лечение только после одного цикла. Исследователи также показали, что, несмотря на хороший прогноз и наличие финансовых средств для оплаты лечения, прекращение лечения чаще всего происходит по психологическим причинам. Фертильность не увеличивается, когда женщины принимают антиоксиданты для снижения окислительного стресса, вызванного ситуацией.
Бесплодие может иметь психологические последствия. Партнеры могут стать более озабоченными зачатием, что увеличивает сексуальную дисфункцию . Часто возникают разногласия в браке, особенно когда они вынуждены принимать медицинские решения. Женщины, пытающиеся зачать ребенка, часто страдают депрессией, как и женщины с сердечными заболеваниями или раком. Эмоциональный стресс и супружеские трудности больше в тех парах, где бесплодие связано с мужчиной.
Пожилые люди со взрослыми детьми живут дольше. Почему это так, неясно и может частично зависеть от тех, у кого есть дети, которые ведут более здоровый образ жизни, поддержки со стороны детей или обстоятельств, которые привели к тому, что детей не было.
Почему это так, неясно и может частично зависеть от тех, у кого есть дети, которые ведут более здоровый образ жизни, поддержки со стороны детей или обстоятельств, которые привели к тому, что детей не было.
Социальное
Во многих культурах неспособность зачать ребенка является клеймом. В закрытых социальных группах степень неприятия (или чувство, что пара отвергнута) может вызвать значительное беспокойство и разочарование. Некоторые в ответ активно избегают проблемы; мужчины из среднего класса, скорее всего, отреагируют подобным образом.
В Соединенных Штатах некоторые виды лечения бесплодия, включая диагностические тесты, хирургическое вмешательство и терапию депрессии, могут подпадать под действие отпуска по семейным обстоятельствам и отпуска по болезни . Было предложено классифицировать бесплодие как форму инвалидности.
Причины
Иммунное бесплодие
Антиспермальные антитела (ASA) считаются причиной бесплодия примерно у 10–30% бесплодных пар. Как у мужчин, так и у женщин продукция ASA направлена против поверхностных антигенов в сперматозоидах, которые могут мешать подвижности и транспортировке сперматозоидов по женским репродуктивным трактам, подавляя емкостную и акросомную реакцию , нарушая оплодотворение , влияя на процесс имплантации и нарушая рост и развитие. из эмбриона . Антитела делятся на разные группы: есть антитела IgA, IgG и IgM. Они также различаются расположением сперматозоидов, с которыми они связываются (голова, средняя часть, хвост). Факторами, способствующими образованию антиспермальных антител у женщин, являются нарушение нормальных иммунорегуляторных механизмов, инфекция, нарушение целостности слизистых оболочек, изнасилование и незащищенный оральный или анальный секс. Факторы риска образования антиспермальных антител у мужчин включают нарушение гематоэнцефалического барьера, травмы и хирургические вмешательства, орхит, варикоцеле , инфекции, простатит , рак яичек , отсутствие иммуносупрессии и незащищенный рецептивный анальный или оральный секс с мужчинами.
Как у мужчин, так и у женщин продукция ASA направлена против поверхностных антигенов в сперматозоидах, которые могут мешать подвижности и транспортировке сперматозоидов по женским репродуктивным трактам, подавляя емкостную и акросомную реакцию , нарушая оплодотворение , влияя на процесс имплантации и нарушая рост и развитие. из эмбриона . Антитела делятся на разные группы: есть антитела IgA, IgG и IgM. Они также различаются расположением сперматозоидов, с которыми они связываются (голова, средняя часть, хвост). Факторами, способствующими образованию антиспермальных антител у женщин, являются нарушение нормальных иммунорегуляторных механизмов, инфекция, нарушение целостности слизистых оболочек, изнасилование и незащищенный оральный или анальный секс. Факторы риска образования антиспермальных антител у мужчин включают нарушение гематоэнцефалического барьера, травмы и хирургические вмешательства, орхит, варикоцеле , инфекции, простатит , рак яичек , отсутствие иммуносупрессии и незащищенный рецептивный анальный или оральный секс с мужчинами.
Инфекции, передающиеся половым путем
Инфекции, вызываемые следующими возбудителями, передающимися половым путем, отрицательно влияют на фертильность: Chlamydia trachomatis и Neisseria gonorrhoeae . Существует последовательная ассоциация инфекции Mycoplasma genitalium и синдромов женских половых путей. Инфекция M. genitalium связана с повышенным риском бесплодия.
Генетический
Мутации гена NR5A1, кодирующего стероидогенный фактор-1 (SF-1), были обнаружены у небольшой подгруппы мужчин с бесплодием с необструктивным мужским фактором, причина которого неизвестна. Результаты одного исследования, в котором изучалась когорта из 315 мужчин, выявили изменения в шарнирной области SF-1 и отсутствие редких аллельных вариантов у фертильных контрольных мужчин. У больных наблюдались более тяжелые формы бесплодия, такие как азооспермия и тяжелая олигозооспермия .
Другие причины
Факторы, которые могут вызвать мужское, а также женское бесплодие:
- Повреждение ДНК
- Повреждение ДНК снижает фертильность в женских овоцитах, что вызвано курением, другими агентами, повреждающими ДНК ксенобиотиков (такими как радиация или химиотерапия) или накоплением окислительного повреждения ДНК 8-гидроксидезоксигуанозина
- Повреждение ДНК снижает фертильность мужской спермы, что вызвано окислительным повреждением ДНК, курением, другими агентами, повреждающими ДНК ксенобиотиков (такими как лекарства или химиотерапия) или другими агентами, повреждающими ДНК, включая активные формы кислорода, лихорадку или высокую температуру яичек. Поврежденная ДНК, связанная с бесплодием, проявляется повышенной восприимчивостью к денатурации, вызванной нагреванием или кислотой, или наличием двухцепочечных разрывов, которые можно обнаружить с помощью анализа TUNEL .
- Общие факторы
- Гипоталамо-гипофизарные факторы
- Факторы окружающей среды
 Поврежденная ДНК, связанная с бесплодием, проявляется повышенной восприимчивостью к денатурации, вызванной нагреванием или кислотой, или наличием двухцепочечных разрывов, которые можно обнаружить с помощью анализа TUNEL .
Поврежденная ДНК, связанная с бесплодием, проявляется повышенной восприимчивостью к денатурации, вызванной нагреванием или кислотой, или наличием двухцепочечных разрывов, которые можно обнаружить с помощью анализа TUNEL .Немецкие ученые сообщили, что вирус, называемый аденоассоциированным вирусом, может играть роль в мужском бесплодии, хотя в остальном он не опасен. Другие заболевания, такие как хламидиоз и гонорея, также могут вызывать бесплодие из-за внутренних рубцов ( непроходимость маточных труб ).
- Пищевые привычки
- Ожирение : ожирение может иметь значительное влияние на мужскую и женскую фертильность. ИМТ (индекс массы тела) может быть значительным фактором фертильности, поскольку увеличение ИМТ у мужчин всего на три единицы может быть связано с бесплодием. Несколько исследований показали, что увеличение ИМТ коррелирует с уменьшением концентрации сперматозоидов, снижением подвижности и увеличением повреждения ДНК в сперматозоидах. Также существует связь между ожирением и эректильной дисфункцией (ЭД). ЭД может быть следствием превращения андрогенов в эстрадиол. За это преобразование отвечает фермент ароматаза, который содержится в основном в жировой ткани. По мере увеличения количества жировой ткани становится больше доступной ароматазы для преобразования андрогенов, и уровни эстрадиола в сыворотке повышаются. На другие гормоны, включая ингибин B и лептин, также может влиять ожирение. Сообщалось, что уровни ингибина B снижаются с увеличением веса, что приводит к уменьшению количества клеток Сертоли и выработки спермы. Согласно многим исследованиям, лептин — это гормон, связанный с многочисленными эффектами, включая контроль аппетита, воспаление и снижение секреции инсулина. У женщин с ожирением выше частота повторных выкидышей на ранних сроках по сравнению с женщинами, не страдающими ожирением.
- Низкий вес: ожирение — не единственный способ воздействия веса на фертильность. У мужчин с пониженным весом концентрация сперматозоидов обычно ниже, чем у мужчин с нормальным ИМТ. У женщин недостаточный вес и крайне низкое количество жира в организме связаны с дисфункцией яичников и бесплодием, и у них выше риск преждевременных родов. Расстройства пищевого поведения, такие как нервная анорексия, также связаны с чрезвычайно низким ИМТ. Несмотря на то, что расстройства пищевого поведения относительно редки, они могут отрицательно влиять на менструацию, фертильность, а также на благополучие матери и плода.
- Ожирение : ожирение может иметь значительное влияние на мужскую и женскую фертильность. ИМТ (индекс массы тела) может быть значительным фактором фертильности, поскольку увеличение ИМТ у мужчин всего на три единицы может быть связано с бесплодием. Несколько исследований показали, что увеличение ИМТ коррелирует с уменьшением концентрации сперматозоидов, снижением подвижности и увеличением повреждения ДНК в сперматозоидах.
 Также существует связь между ожирением и эректильной дисфункцией (ЭД). ЭД может быть следствием превращения андрогенов в эстрадиол. За это преобразование отвечает фермент ароматаза, который содержится в основном в жировой ткани. По мере увеличения количества жировой ткани становится больше доступной ароматазы для преобразования андрогенов, и уровни эстрадиола в сыворотке повышаются. На другие гормоны, включая ингибин B и лептин, также может влиять ожирение. Сообщалось, что уровни ингибина B снижаются с увеличением веса, что приводит к уменьшению количества клеток Сертоли и выработки спермы. Согласно многим исследованиям, лептин — это гормон, связанный с многочисленными эффектами, включая контроль аппетита, воспаление и снижение секреции инсулина. У женщин с ожирением выше частота повторных выкидышей на ранних сроках по сравнению с женщинами, не страдающими ожирением.
Также существует связь между ожирением и эректильной дисфункцией (ЭД). ЭД может быть следствием превращения андрогенов в эстрадиол. За это преобразование отвечает фермент ароматаза, который содержится в основном в жировой ткани. По мере увеличения количества жировой ткани становится больше доступной ароматазы для преобразования андрогенов, и уровни эстрадиола в сыворотке повышаются. На другие гормоны, включая ингибин B и лептин, также может влиять ожирение. Сообщалось, что уровни ингибина B снижаются с увеличением веса, что приводит к уменьшению количества клеток Сертоли и выработки спермы. Согласно многим исследованиям, лептин — это гормон, связанный с многочисленными эффектами, включая контроль аппетита, воспаление и снижение секреции инсулина. У женщин с ожирением выше частота повторных выкидышей на ранних сроках по сравнению с женщинами, не страдающими ожирением. У женщин недостаточный вес и крайне низкое количество жира в организме связаны с дисфункцией яичников и бесплодием, и у них выше риск преждевременных родов. Расстройства пищевого поведения, такие как нервная анорексия, также связаны с чрезвычайно низким ИМТ. Несмотря на то, что расстройства пищевого поведения относительно редки, они могут отрицательно влиять на менструацию, фертильность, а также на благополучие матери и плода.
У женщин недостаточный вес и крайне низкое количество жира в организме связаны с дисфункцией яичников и бесплодием, и у них выше риск преждевременных родов. Расстройства пищевого поведения, такие как нервная анорексия, также связаны с чрезвычайно низким ИМТ. Несмотря на то, что расстройства пищевого поведения относительно редки, они могут отрицательно влиять на менструацию, фертильность, а также на благополучие матери и плода.Самки
Следующие причины бесплодия могут быть обнаружены только у женщин. Чтобы женщина могла зачать ребенка, должны произойти определенные вещи: вагинальный половой акт должен иметь место примерно в то время, когда яйцеклетка выходит из ее яичника; система, производящая яйца, должна работать на оптимальном уровне; и ее гормоны должны быть сбалансированы.
У женщин проблемы с оплодотворением возникают в основном либо из-за структурных проблем в фаллопиевых трубах или матке, либо из-за проблем с выделением яиц. Бесплодие может быть вызвано закупоркой фаллопиевых труб из-за пороков развития, инфекций, таких как хламидиоз или рубцовая ткань. Например, эндометриоз может вызвать бесплодие из-за разрастания ткани эндометрия в фаллопиевых трубах или вокруг яичников. Эндометриоз обычно чаще встречается у женщин в возрасте от двадцати пяти лет и старше, особенно если роды были отложены.
Бесплодие может быть вызвано закупоркой фаллопиевых труб из-за пороков развития, инфекций, таких как хламидиоз или рубцовая ткань. Например, эндометриоз может вызвать бесплодие из-за разрастания ткани эндометрия в фаллопиевых трубах или вокруг яичников. Эндометриоз обычно чаще встречается у женщин в возрасте от двадцати пяти лет и старше, особенно если роды были отложены.
Другой важной причиной бесплодия у женщин может быть невозможность овуляции . Нарушения овуляции составляют 25% известных причин женского бесплодия. Олигоовуляция или ановуляция приводит к бесплодию, потому что ежемесячно не выделяются ооциты. При отсутствии ооцита нет возможности для оплодотворения и беременности. Всемирная организация здравоохранения подразделяет нарушения овуляции на четыре класса:
- Гипогонадотропная гипогонадальная ановуляция: то есть гипоталамическая аменорея.
- Нормогонадотропная нормоэстрогенная ановуляция: синдром поликистозных яичников (СПКЯ)
- Гипергонадотропная гипоэстрогенная ановуляция: преждевременная недостаточность яичников.
- Гиперпролактинемическая ановуляция: аденома гипофиза.

Malformation of the eggs themselves may complicate conception. For example, polycystic ovarian syndrome is when the eggs only partially develop within the ovary and there is an excess of male hormones. Some women are infertile because their ovaries do not mature and release eggs. In this case synthetic FSH by injection or Clomid (Clomiphene citrate) via a pill can be given to stimulate follicles to mature in the ovaries.
Другие факторы, которые могут повлиять на шансы женщины на зачатие, включают избыточный или недостаточный вес, или ее возраст, поскольку женская фертильность снижается после 30 лет.
Иногда это может быть комбинация факторов, а иногда явная причина никогда не устанавливается.
К частым причинам бесплодия у женщин относятся:
Самцы
Мужское бесплодие определяется как неспособность мужчины забеременеть от фертильной женщины в течение как минимум одного года незащищенного полового акта.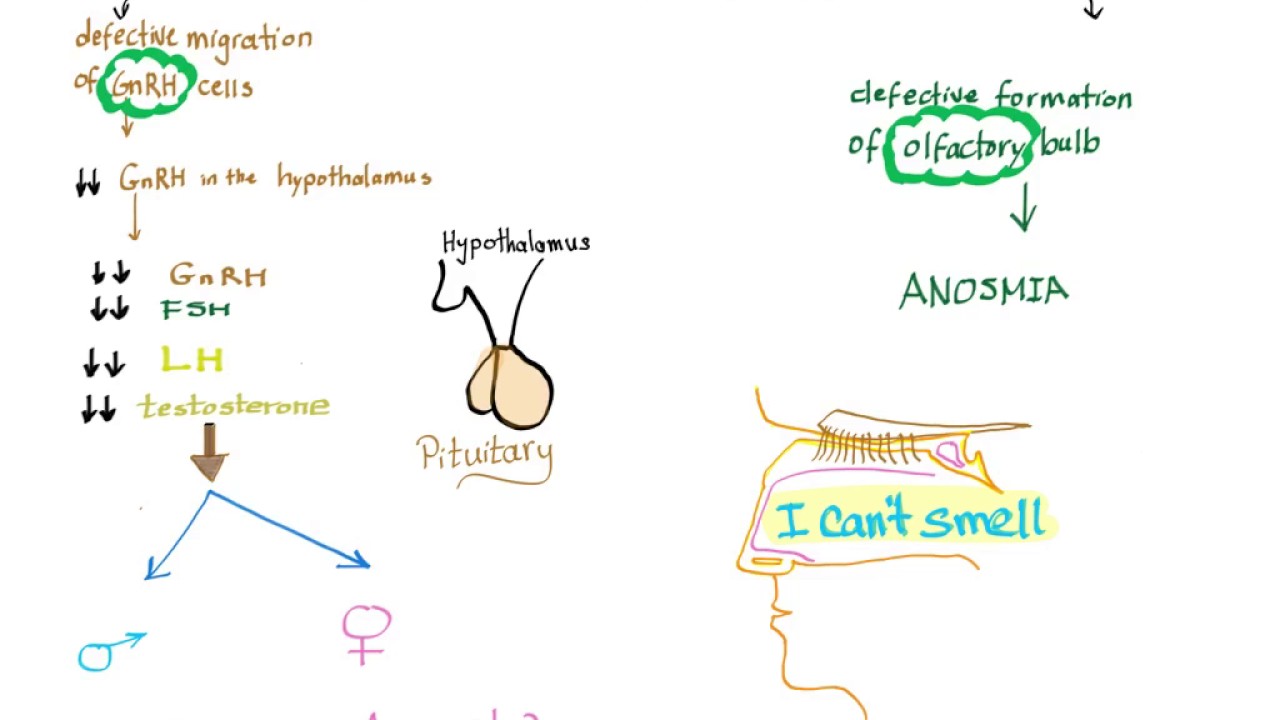 Есть несколько причин мужского бесплодия. К ним относятся эндокринные нарушения (обычно из-за гипогонадизма) примерно от 2% до 5%), нарушения транспорта сперматозоидов (например, вазэктомия) у 5%, первичные дефекты яичек (которые включают аномальные параметры сперматозоидов без какой-либо идентифицируемой причины) от 65% до 80% и идиопатический (когда бесплодный мужчина имеет нормальные параметры спермы и спермы) от 10% до 20%.
Есть несколько причин мужского бесплодия. К ним относятся эндокринные нарушения (обычно из-за гипогонадизма) примерно от 2% до 5%), нарушения транспорта сперматозоидов (например, вазэктомия) у 5%, первичные дефекты яичек (которые включают аномальные параметры сперматозоидов без какой-либо идентифицируемой причины) от 65% до 80% и идиопатический (когда бесплодный мужчина имеет нормальные параметры спермы и спермы) от 10% до 20%.
Основная причина мужского бесплодия — низкое качество спермы. У мужчин, у которых есть репродуктивные органы, необходимые для продолжения рода, бесплодие может быть вызвано низким количеством сперматозоидов из-за эндокринных проблем, лекарств, радиации или инфекции. Могут быть пороки развития яичек, гормональный дисбаланс или закупорка протоковой системы мужчины. Хотя многие из них можно вылечить с помощью хирургического вмешательства или гормональной замены, некоторые из них могут быть неопределенными. Бесплодие, связанное с жизнеспособными, но неподвижными сперматозоидами, может быть вызвано первичной цилиарной дискинезией . Сперма должна обеспечить зиготу ДНК , центриолями и фактором активации для развития эмбриона. Дефект любой из этих структур сперматозоидов может привести к бесплодию, которое не будет обнаружено анализом спермы. Антиспермальные антитела вызывают иммунное бесплодие. Муковисцидоз может привести к бесплодию у мужчин.
Сперма должна обеспечить зиготу ДНК , центриолями и фактором активации для развития эмбриона. Дефект любой из этих структур сперматозоидов может привести к бесплодию, которое не будет обнаружено анализом спермы. Антиспермальные антитела вызывают иммунное бесплодие. Муковисцидоз может привести к бесплодию у мужчин.
Комбинированное бесплодие
В некоторых случаях и мужчина, и женщина могут быть бесплодными или субфертильными, а бесплодие пары возникает в результате сочетания этих состояний. В других случаях предполагается, что причина является иммунологической или генетической; может случиться так, что каждый партнер может самостоятельно зачать ребенка, но пара не может зачать ребенка вместе без посторонней помощи.
Необъяснимое бесплодие
В США до 20% бесплодных пар страдают необъяснимым бесплодием. В этих случаях аномалии могут присутствовать, но не обнаруживаются существующими методами. Возможные проблемы могут заключаться в том, что яйцеклетка не выходит в оптимальное время для оплодотворения, что она не может попасть в маточную трубу, сперма не может достичь яйцеклетки, может не произойти оплодотворение, может быть нарушен транспорт зиготы, или имплантация не удалась. Все больше признается, что качество яиц имеет решающее значение, и у женщин пожилого возраста яйцеклетки снижены для нормального и успешного оплодотворения. Кроме того, полиморфизм в генах фолатного пути может быть одной из причин осложнений фертильности у некоторых женщин с необъяснимым бесплодием. Тем не менее, все больше данных свидетельствует о том, что отчасти это могут быть эпигенетические модификации сперматозоидов.
Все больше признается, что качество яиц имеет решающее значение, и у женщин пожилого возраста яйцеклетки снижены для нормального и успешного оплодотворения. Кроме того, полиморфизм в генах фолатного пути может быть одной из причин осложнений фертильности у некоторых женщин с необъяснимым бесплодием. Тем не менее, все больше данных свидетельствует о том, что отчасти это могут быть эпигенетические модификации сперматозоидов.
Диагностика
Если оба партнера молоды и здоровы и безуспешно пытались забеременеть в течение одного года, посещение врача или практикующей медсестры (WHNP) может помочь выявить потенциальные медицинские проблемы раньше, чем позже. Врач или WHNP также могут предложить изменить образ жизни, чтобы повысить шансы на зачатие.
Женщинам старше 35 лет следует обратиться к своему врачу или WHNP через шесть месяцев, поскольку для завершения тестов на фертильность может потребоваться некоторое время, а возраст может повлиять на варианты лечения, доступные в этом случае.
Врач или WHNP изучают историю болезни и проводят медицинский осмотр. Они также могут провести некоторые базовые тесты на обоих партнерах, чтобы узнать, есть ли явная причина недобестения. При необходимости они направляют пациентов в клинику репродуктивной медицины или местную больницу для более специализированных тестов. Результаты этих тестов помогают определить лучшее лечение бесплодия.
Уход
Лечение зависит от причины бесплодия, но может включать консультирование, лечение бесплодия, в том числе экстракорпоральное оплодотворение. Согласно рекомендациям ESHRE , парам, у которых показатель живорождений составляет 40% или выше в год, рекомендуется продолжать стремиться к спонтанной беременности. Методы лечения бесплодия можно разделить на медицинские или дополнительные и альтернативные. Некоторые методы могут использоваться вместе с другими методами. Лекарства, используемые как для женщин, так и для мужчин, включают цитрат кломифена , менопаузальный гонадотропин человека (чМГ), фолликулостимулирующий гормон (ФСГ), хорионический гонадотропин человека (ХГЧ), аналоги гонадотропин-рилизинг-гормона (ГнРГ) , ингибиторы ароматазы и метформин .
Лечебные процедуры
Медикаментозное лечение бесплодия обычно включает использование лекарств от бесплодия , медицинских устройств, хирургического вмешательства или комбинации следующих средств. Если сперма хорошего качества и репродуктивные структуры женщины в порядке (открытые маточные трубы, отсутствие спаек или рубцов), можно использовать курс индукции овуляции . Врач или WHNP могут также предложить использовать шейный колпачок для зачатия , который пациент использует дома, помещая сперму внутрь колпачка и помещая устройство для зачатия на шейку матки, или внутриматочную инсеминацию (IUI), при которой врач или WHNP вводят сперма в матку во время овуляции через катетер. В этих методах оплодотворение происходит внутри тела.
Если консервативными методами лечения не удается достичь доношенной беременности, врач или WHNP могут предложить пациентке пройти экстракорпоральное оплодотворение (ЭКО). ЭКО и связанные с ним методы ( ИКСИ , ZIFT , GIFT ) называются методами вспомогательных репродуктивных технологий (ВРТ).
Методы ВРТ обычно начинаются со стимуляции яичников для увеличения яйценоскости. После стимуляции врач хирургическим путем извлекает одну или несколько яйцеклеток из яичника и объединяет их со спермой в лабораторных условиях с целью получения одного или нескольких эмбрионов. Оплодотворение происходит вне тела, и оплодотворенная яйцеклетка повторно вводится в репродуктивный тракт женщины в ходе процедуры, называемой переносом эмбриона .
К другим медицинским методам относятся, например, тубопластика , вспомогательное вылупление и преимплантационная генетическая диагностика .
Экстракорпоральное оплодотворение
Изображение процедуры экстракорпорального оплодотворения.
ЭКО — наиболее часто применяемое ВРТ. Было доказано, что он полезен в преодолении состояний бесплодия, таких как закупорка или повреждение трубок, эндометриоз, повторная неудача ВМИ, необъяснимое бесплодие, плохой овариальный резерв, плохое или даже нулевое количество сперматозоидов.
Интрацитоплазматическая инъекция спермы
Техника ИКСИ используется в случае плохого качества спермы, низкого количества сперматозоидов или неудачных попыток оплодотворения во время предыдущих циклов ЭКО. Этот метод включает инъекцию одной здоровой спермы непосредственно в зрелую яйцеклетку. Затем оплодотворенный эмбрион переносится в матку.
Туризм
Туризм для лечения бесплодия — это практика поездок в другую страну для лечения бесплодия. Его можно рассматривать как вид медицинского туризма . Основными причинами развития плодородного туризма являются правовое регулирование искомой процедуры в стране проживания или более низкая цена. Экстракорпоральное оплодотворение и донорское оплодотворение являются основными процедурами.
Терапия стволовыми клетками
В настоящее время существует несколько методов лечения (все еще в стадии экспериментов), связанных с терапией стволовыми клетками . Это новая возможность не только для партнеров с недостатком гамет, но также для гомосексуалистов и одиноких людей, которые хотят иметь потомство. Теоретически с помощью этой терапии мы можем получить искусственные гаметы in vitro . Есть разные исследования как для женщин, так и для мужчин.
Теоретически с помощью этой терапии мы можем получить искусственные гаметы in vitro . Есть разные исследования как для женщин, так и для мужчин.
- Трансплантация сперматогониальных стволовых клеток: проходит в семенных канальцах. Благодаря этому лечению пациент испытывает сперматогенез, и, следовательно, у него есть шанс иметь потомство, если он этого хочет. Он специально предназначен для онкологических пациентов, сперма которых разрушается из-за гонадотоксического лечения, которому они подвергаются.
- Стволовые клетки яичников: считается, что у женщин с самого начала есть конечное число фолликулов. Тем не менее, ученые обнаружили эти стволовые клетки, которые могут генерировать новые ооциты в постнатальных условиях. По всей видимости, их всего 0,014% (это может быть объяснением того, почему они не были обнаружены до сих пор). По поводу их существования до сих пор ведутся споры, но если открытия верны, это может быть новым методом лечения бесплодия.

Терапия стволовыми клетками действительно нова, и все еще исследуются. Кроме того, это может быть будущее для лечения множества заболеваний, включая бесплодие. Пройдет время, прежде чем эти исследования станут доступны для клиник и пациентов.
Эпидемиология
Распространенность бесплодия варьируется в зависимости от определения, то есть от периода времени, связанного с невозможностью зачать ребенка.
- Уровень бесплодия увеличился на 4% с 1980-х годов, в основном из-за проблем с плодовитостью из-за увеличения возраста.
- Проблемы с фертильностью затрагивают каждую седьмую пару в Великобритании. Большинство пар (около 84%), которые имеют регулярные половые контакты (то есть каждые два-три дня) и не используют противозачаточные средства, беременеют в течение года. Примерно 92 из 100 пар, пытающихся забеременеть, делают это в течение двух лет.
- С возрастом женщины становятся менее плодовитыми. Около 94% женщин в возрасте 35 лет, которые имеют регулярные незащищенные половые отношения, забеременеют после трех лет попыток. Однако для женщин в возрасте 38 лет только около 77%. Влияние возраста на фертильность мужчин менее очевидно.
- У людей, планирующих ЭКО в Великобритании, примерно половина проблем с фертильностью с установленной причиной возникает из-за проблем с мужчиной, а примерно половина — с проблемами с женщиной. Однако примерно каждый пятый случай бесплодия не имеет четко установленной причины.
- В Великобритании мужское бесплодие составляет 25% бесплодных пар, а 25% остаются невыясненными. 50% — женские причины, 25% — из-за ановуляции и 25% трубных / других проблем.
- В Швеции около 10% пар, желающих иметь детей, бесплодны. Примерно в одной трети этих случаев фактором является мужчина, в одной трети — женщина, а в оставшейся трети бесплодие является результатом действия факторов с обеих сторон.
 Однако для женщин в возрасте 38 лет только около 77%. Влияние возраста на фертильность мужчин менее очевидно.
Однако для женщин в возрасте 38 лет только около 77%. Влияние возраста на фертильность мужчин менее очевидно.Общество и культура
Возможно, за исключением бесплодия в научной фантастике , фильмы и другая фантастика, изображающие эмоциональную борьбу вспомогательных репродуктивных технологий, пережили подъем сначала в конце 2000-х годов, хотя методы были доступны уже несколько десятилетий. Тем не менее, количество людей, которые так или иначе могут относиться к этому на собственном опыте, постоянно растет, а разнообразие испытаний и борьбы огромно.
Тем не менее, количество людей, которые так или иначе могут относиться к этому на собственном опыте, постоянно растет, а разнообразие испытаний и борьбы огромно.
Pixar ‘s Up содержит изображение бесплодия в продолжительной жизни, которая длится первые несколько минут фильма.
Другие отдельные примеры относятся к отдельным разделам вспомогательных репродуктивных технологий.
Этика
Есть несколько этических проблем, связанных с бесплодием и его лечением.
- Для некоторых пар дорогостоящие процедуры недоступны.
- Споры о том, должны ли медицинские страховые компании (например, в США) обязать покрывать лечение бесплодия.
- Выделение медицинских ресурсов, которые можно было бы использовать в другом месте
- Правовой статус эмбрионов, оплодотворенных in vitro и не перенесенных in vivo . (См. Также начало спора о беременности ).
- Противодействие жизни против уничтожения эмбрионов, не перенесенных in vivo.
- ЭКО и другие методы лечения бесплодия привели к увеличению числа многоплодных родов , что повлекло за собой этический анализ из-за связи между многоплодной беременностью, преждевременными родами и множеством проблем со здоровьем.
- Мнения религиозных лидеров о методах лечения бесплодия; Например, Римско-католическая церковь рассматривает бесплодие как призыв принять или использовать естественные методы лечения (лекарства, хирургические операции или составление графика цикла), и ее члены должны отказаться от вспомогательных репродуктивных технологий.
- Бесплодие, вызванное дефектами ДНК на Y-хромосоме, передается от отца к сыну. Если естественный отбор является основным механизмом исправления ошибок, который предотвращает случайные мутации в Y-хромосоме, то лечение бесплодия мужчин с аномальной спермой (в частности, ИКСИ ) только переносит основную проблему на следующее мужское поколение.

Во многих странах существуют специальные механизмы для решения этических и социальных вопросов, связанных с лечением бесплодия.
- Одним из наиболее известных является HFEA — британский регулятор по лечению бесплодия и исследованиям эмбрионов. Он был создан 1 августа 1991 года после подробного расследования комиссии, возглавляемой Мэри Уорнок в 1980-х годах.
- Модель, аналогичная HFEA, была принята в остальных странах Европейского Союза. В каждой стране есть свой орган или органы, ответственные за инспекцию и лицензирование лечения бесплодия в соответствии с Директивой ЕС по тканям и клеткам.
- Регулирующие органы также находятся в Канаде и в штате Виктория в Австралии.
Смотрите также
Рекомендации
- Инхорн MC (2003). «Глобальное бесплодие и глобализация новых репродуктивных технологий: иллюстрации из Египта». Социальные науки и медицина . 56 (9): 1837–1851. DOI : 10.1016 / s0277-9536 (02) 00208-3 . PMID 12650724 .
- Лок, Маргарет и Винь-Ким Нгуен. 2011. Антропология биомедицины: Wiley-Blackwell.
- Герритс Т., Шоу М. (2010). «Биомедицинское лечение бесплодия в Африке к югу от Сахары: обзор современной практики, опыта и точек зрения в области социальных наук». Факты, взгляды и видение в ObGyn . 2 (3): 194–207. PMID 25013712 .

дальнейшее чтение
внешняя ссылка
Аносмия — Anosmia — qaz.wiki
Неспособность обонять
| Аносмия | |
|---|---|
| Другие имена | Потеря обоняния, обонятельная слепота, обонятельная слепота |
| Воспаление слизистой оболочки носа, вызывающее аносмию | |
| Произношение | |
| Специальность | Оториноларингология |
| Типы | Частично, полностью |
Аносмия , также известная как обонятельная слепота , — это потеря способности обнаруживать один или несколько запахов . Аносмия может быть временной или постоянной. Он отличается от гипосмии — пониженной чувствительностью к некоторым или всем запахам.
Аносмия может быть временной или постоянной. Он отличается от гипосмии — пониженной чувствительностью к некоторым или всем запахам.
Аносмия может быть из — за целый ряд факторов, в том числе воспаления в слизистой оболочке носа , закупоркой носовых ходов или уничтожения одной височной доли . Воспаление возникает из-за хронических изменений слизистой оболочки придаточных пазух носа, а также средних и верхних носовых раковин .
Когда аносмия вызвана воспалительными изменениями носовых ходов, ее лечат просто за счет уменьшения воспаления. Это может быть вызвано хроническим менингитом и нейросифилисом , которые увеличивают внутричерепное давление в течение длительного периода времени, а в некоторых случаях — цилиопатией , в том числе цилиопатией, вызванной первичной цилиарной дискинезией .
Термин происходит от нового латинского обоняния , основанные на древних греческих ἀν- (ап-) + ὀσμή ( osmḗ , «запах», другой соответствующий термин, гиперосмии , относится к повышенной способности к запаху). Некоторые люди могут страдать аносмией из-за одного конкретного запаха — состояния, известного как «специфическая аносмия». Отсутствие обоняния с рождения известно как врожденная аносмия.
Некоторые люди могут страдать аносмией из-за одного конкретного запаха — состояния, известного как «специфическая аносмия». Отсутствие обоняния с рождения известно как врожденная аносмия.
В Соединенных Штатах аносмией страдают 3% людей старше 40 лет.
Определение
Аносмия — это неспособность обонять . Он может быть частичным или полным и может быть специфическим для определенных запахов. Пониженная чувствительность к некоторым или всем запахам — это гипосмия .
Признаки и симптомы
Аносмия может иметь ряд вредных последствий. Люди с внезапной аносмией могут посчитать пищу менее аппетитной, хотя врожденные аносмики редко жалуются на это, и никто не сообщает о потере веса. Потеря запаха также может быть опасной, поскольку препятствует обнаружению утечек газа , огня и испорченных продуктов. Распространенное мнение об аносмии как об банальности может затруднить получение пациентом той же медицинской помощи, что и человеку, потерявшему другие чувства, такие как слух или зрение.
Многие испытывают одностороннюю потерю обоняния, часто в результате незначительной травмы головы. Этот тип аносмии обычно выявляется только в том случае, если обе ноздри исследуются отдельно. Использование этого метода тестирования каждой ноздри в отдельности часто показывает ослабленное или даже полное отсутствие обоняния либо в одной, либо в обеих ноздрях, что часто не обнаруживается при одновременном тестировании обеих ноздрей.
Известно, что потеря устоявшихся и сентиментальных воспоминаний об запахах (например, запах травы, чердака бабушек и дедушек, конкретной книги, близких или самого себя) вызывает чувство депрессии .
Утрата обоняния может привести к потере либидо , хотя обычно это не относится к потере обоняния, присутствующей при рождении.
Часто люди, потерявшие обоняние при рождении, сообщают, что в детстве они притворялись способными обонять, потому что считали, что обоняние — это то, что могут делать пожилые / зрелые люди, или не понимали концепцию обоняния, но не хотели выглядеть иначе. от других. Когда дети становятся старше, они часто понимают и сообщают своим родителям, что на самом деле у них нет обоняния, часто к удивлению родителей.
от других. Когда дети становятся старше, они часто понимают и сообщают своим родителям, что на самом деле у них нет обоняния, часто к удивлению родителей.
Исследование, проведенное на пациентах, страдающих аносмией, показало, что при тестировании обеих ноздрей аносмия не выявлялась; однако при тестировании каждой ноздри в отдельности тесты показали, что обоняние обычно затрагивается только в одной из ноздрей, а не в обеих. Это продемонстрировало, что односторонняя аносмия не редкость у пациентов с аносмией.
Причины
Временная потеря обоняния может быть вызвана заложенным носом или инфекцией. Напротив, необратимая потеря обоняния может быть вызвана гибелью нейронов обонятельных рецепторов в носу или травмой головного мозга, при которой имеется повреждение обонятельного нерва или повреждение областей мозга, которые обрабатывают запах (см. Обонятельная система ). Отсутствие обоняния при рождении, обычно из-за генетических факторов, называется врожденной аносмией. Члены семьи пациента, страдающего врожденной аносмией, часто имеют похожие истории болезни; это говорит о том, что аносмия может происходить по аутосомно-доминантному типу. Иногда аносмия может быть ранним признаком дегенеративного заболевания головного мозга, такого как болезнь Паркинсона и болезнь Альцгеймера .
Члены семьи пациента, страдающего врожденной аносмией, часто имеют похожие истории болезни; это говорит о том, что аносмия может происходить по аутосомно-доминантному типу. Иногда аносмия может быть ранним признаком дегенеративного заболевания головного мозга, такого как болезнь Паркинсона и болезнь Альцгеймера .
Другой специфической причиной необратимой потери может быть повреждение нейронов обонятельных рецепторов из-за использования определенных типов назального спрея ; то есть те, которые вызывают вазоконстрикцию носовой микроциркуляции. Чтобы избежать таких повреждений и последующего риска потери обоняния, сосудосуживающие назальные спреи следует использовать только в случае крайней необходимости и только в течение короткого периода времени. Не сужающие сосуды спреи, например, те, которые используются для лечения заложенности носа, связанной с аллергией, безопасны для использования в течение предписанных периодов времени. Аносмия также может быть вызвана полипами в носу. Эти полипы обнаруживаются у людей с аллергией, синуситом и семейным анамнезом. У людей с муковисцидозом часто возникают полипы носа.
Эти полипы обнаруживаются у людей с аллергией, синуситом и семейным анамнезом. У людей с муковисцидозом часто возникают полипы носа.
Амиодарон — препарат, применяемый при лечении аритмий сердца. Клиническое исследование показало, что у некоторых пациентов применение этого препарата вызывало аносмию. В редких случаях был случай, когда 66-летний мужчина лечился амиодароном от желудочковой тахикардии . После применения препарата у него начались обонятельные расстройства, однако после уменьшения дозировки амиодарона тяжесть аносмии соответственно уменьшилась, что коррелировало использование амиодарона с развитием аносмии.
Аносмия, связанная с COVID-19
Хемосенсорные нарушения, в том числе потеря обоняния или вкуса, являются преобладающим неврологическим симптомом COVID-19 . У 80% пациентов с COVID-19 наблюдаются некоторые изменения в хеместезе , включая запах. Согласно результатам опроса 2 миллионов участников в Великобритании и США, потеря обоняния более предсказуема для COVID-19, чем все другие симптомы, включая жар, кашель или усталость. Поисковые запросы Google по словам «запах», «потеря обоняния», «аносмия» и другие подобные термины увеличились с первых месяцев пандемии и сильно коррелировали с увеличением ежедневных случаев заболевания и смертей. В настоящее время продолжаются исследования механизмов, лежащих в основе этих симптомов.
Поисковые запросы Google по словам «запах», «потеря обоняния», «аносмия» и другие подобные термины увеличились с первых месяцев пандемии и сильно коррелировали с увеличением ежедневных случаев заболевания и смертей. В настоящее время продолжаются исследования механизмов, лежащих в основе этих симптомов.
Многие страны причисляют аносмию к официальным симптомам COVID-19, а некоторые разработали «тесты на запах» в качестве потенциальных инструментов скрининга.
В 2020 году Глобальный консорциум хемосенсорных исследований, совместная исследовательская организация международных исследователей запаха и вкуса, был сформирован для исследования потери обоняния и связанных с ней хемосенсорных симптомов.
Список причин
- Инфекция верхних дыхательных путей (например, синусит , простуда )
- COVID-19
- Носовые полипы
- Идиопатический гипогонадотропный гипогонадизм
- Гипотиреоз
- Травма головы, повреждение решетчатой кости
- Деменция с тельцами Леви
- Опухоли по лобной доли
- Антибиотики
- Фибромиалгия
- Рассеянный склероз
- Воздействие сероводорода ( H
2 S ) - Гипогликемия
- Сахарный диабет
- Астма или аллергия
- Сенная лихорадка
- Хроническая обструктивная болезнь легких (ХОБЛ)
- Длительный алкоголизм
- синдром Кушинга
- Воздействие химического вещества, вызывающего ожоги носа
- Гладить
- Эпилепсия
- Лучевая терапия головы и шеи
- Болезнь печени или почек
- болезнь Паркинсона
- Болезнь Альцгеймера
- Токсины (особенно акрилаты , метакрилаты и кадмий )
- Старость
- Синдром Каллмана
- Первичная цилиарная дискинезия
- Постперфузионный синдром
- Ларингэктомия с постоянной трахеостомией
- Эстезионейробластома — чрезвычайно редкая раковая опухоль, которая возникает в обонятельном нерве или рядом с ним . Симптомы — аносмия (потеря обоняния), часто сопровождающаяся хроническим синуситом .
- Интраназальное употребление наркотиков
- Триада Самтера, также известная как AERD (аспирин, обостряющий респираторное заболевание)
- Синдром Фостера Кеннеди
- Отравление кадмием
- Курение
- Нейротропный вирус
- Шизофрения
- Злокачественная анемия
- Дефицит цинка
- Паралич Белла или паралич и повреждение нерва
- Идиопатическая внутричерепная гипертензия
- Супраселлярная менингиома
- Болезнь Рефсума
- Адренергические агонисты или отказ от альфа-адреноблокаторов (сужение сосудов)
- Саркоидоз
- Продукты от простуды для интраназального введения на основе цинка, включая средства с пометкой «гомеопатические».
- Хронический атрофический ринит
- Костная болезнь Педжета
- Церебральная аневризма
- Гранулематоз Вегенера
- Первичный амебный менингоэнцефалит
- Миастения
- Укус змеи
- Идиопатическая аносмия (причину установить невозможно)
 Симптомы — аносмия (потеря обоняния), часто сопровождающаяся хроническим синуситом .
Симптомы — аносмия (потеря обоняния), часто сопровождающаяся хроническим синуситом .Диагностика
Врачи могут диагностировать аносмию с помощью тестов на ацетилцистеин .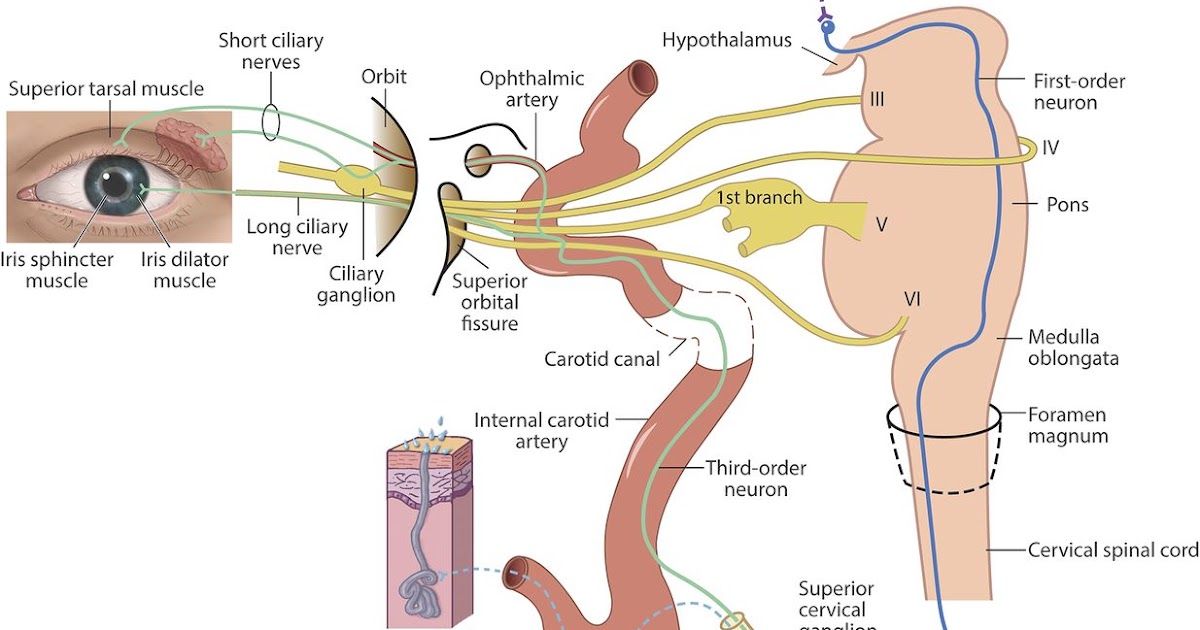 Врачи начнут с подробного изучения истории болезни. Затем врач спросит о любых травмах, связанных с аносмией, которые могут включать инфекции верхних дыхательных путей или травму головы. Психофизическая оценка порядка и идентификации вкуса может использоваться для выявления аносмии. Проводится обследование нервной системы, чтобы увидеть, не повреждены ли черепные нервы. Диагноз, а также степень нарушения могут быть теперь проверены гораздо эффективнее и действеннее, чем когда-либо прежде, благодаря появившимся «комплектам для проверки запаха», а также скрининговым тестам, в которых используются материалы, которые легко доступны в большинстве клиник. Иногда после несчастных случаев у пациента меняется обоняние. Особых запахов, которые были раньше, больше нет. Иногда после травм головы встречаются пациенты с односторонней аносмией. Обоняние следует проверять индивидуально в каждой ноздре.
Врачи начнут с подробного изучения истории болезни. Затем врач спросит о любых травмах, связанных с аносмией, которые могут включать инфекции верхних дыхательных путей или травму головы. Психофизическая оценка порядка и идентификации вкуса может использоваться для выявления аносмии. Проводится обследование нервной системы, чтобы увидеть, не повреждены ли черепные нервы. Диагноз, а также степень нарушения могут быть теперь проверены гораздо эффективнее и действеннее, чем когда-либо прежде, благодаря появившимся «комплектам для проверки запаха», а также скрининговым тестам, в которых используются материалы, которые легко доступны в большинстве клиник. Иногда после несчастных случаев у пациента меняется обоняние. Особых запахов, которые были раньше, больше нет. Иногда после травм головы встречаются пациенты с односторонней аносмией. Обоняние следует проверять индивидуально в каждой ноздре.
Многие случаи врожденной аносмии остаются незарегистрированными и невыявленными. Поскольку расстройство присутствует с рождения, человек может плохо понимать обоняние или не понимать его, следовательно, не осознает этого дефицита.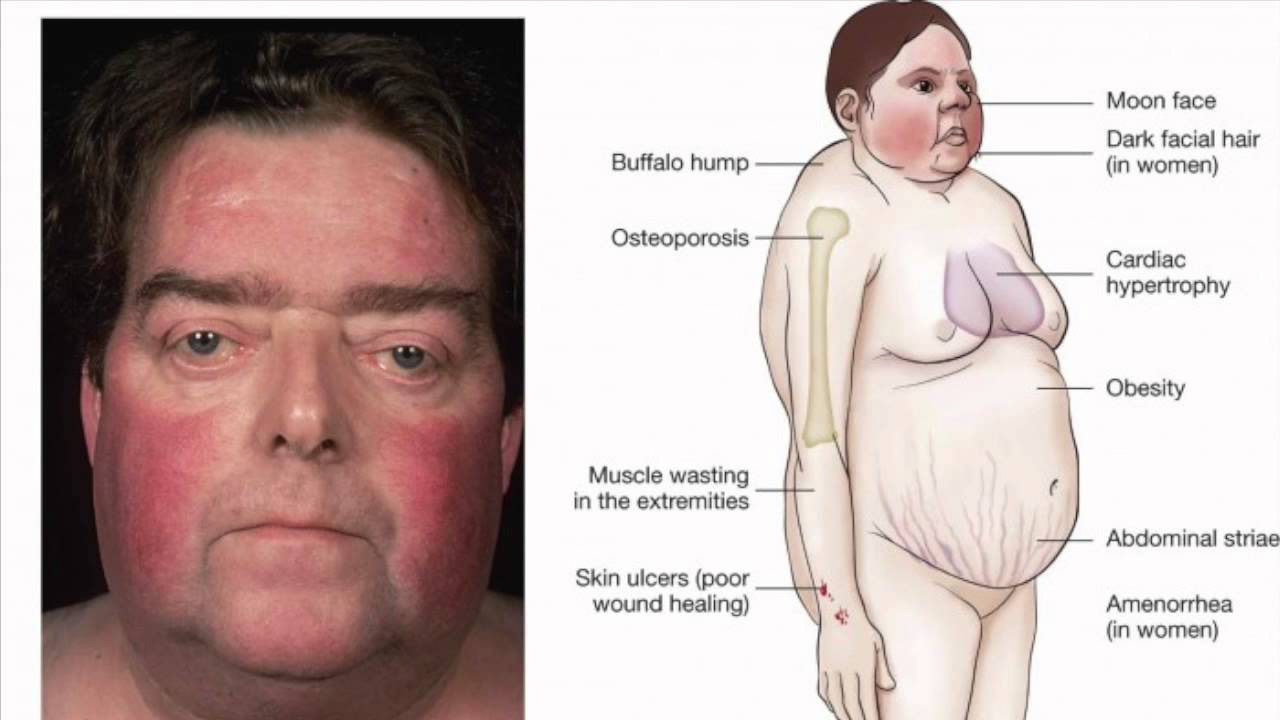 Это также может привести к снижению аппетита.
Это также может привести к снижению аппетита.
Уход
Хотя аносмию, вызванную повреждением головного мозга, нельзя лечить, аносмию, вызванную воспалительными изменениями слизистой оболочки, можно лечить глюкокортикоидами . Уменьшение воспаления за счет пероральных глюкокортикоидов, таких как преднизон, с последующим применением назального спрея с глюкокортикоидами для длительного применения, легко и безопасно лечит аносмию. Режим преднизона корректируется в зависимости от толщины слизистой оболочки, наличия отека и наличия или отсутствия полипов носа. Однако лечение непостоянно и, возможно, через короткое время его придется повторить. Вместе с лекарствами необходимо уменьшить давление в верхней части носа с помощью аэрации и дренажа.
Аносмию, вызванную полипом в носу, можно лечить стероидными препаратами или удалением полипа.
Несмотря на то, что генная терапия находилась на очень раннем этапе развития, она восстановила обоняние у мышей с врожденной аносмией, вызванной цилиопатией . В этом случае генетическое заболевание повлияло на реснички в их организме, что обычно позволяло им обнаруживать переносимые по воздуху химические вещества, и аденовирус был использован для имплантации рабочей версии гена IFT88 в дефектные клетки носа, что восстановило реснички и позволил обоняние.
В этом случае генетическое заболевание повлияло на реснички в их организме, что обычно позволяло им обнаруживать переносимые по воздуху химические вещества, и аденовирус был использован для имплантации рабочей версии гена IFT88 в дефектные клетки носа, что восстановило реснички и позволил обоняние.
Эпидемиология
В Соединенных Штатах 3% людей старше 40 лет страдают аносмией.
В 2012 году обоняние оценивалось у лиц в возрасте 40 лет и старше, при этом показатели аносмии / тяжелой гипосмии составляли 0,3% в возрасте 40–49 лет, а в возрасте 80+ — 14,1%. Частота гипосмии была намного выше: 3,7% в возрасте 40–49 лет и 25,9% в возрасте 80+.
Смотрите также
Рекомендации
дальнейшее чтение
внешняя ссылка
Лечение синдрома Каллмана в Москве: диагностика, услуги, врачи
Эндокринологи Москвы — последние отзывы
Очень внимательный и понимающий врач. Она выслушала меня, тщательно осмотрела, дала рекомендации и заменила все препараты. Приём прошёл на высшем уровне! Я очень довольна, что попала к Софье Владиславовне и выбирала её по отзывам!
Она выслушала меня, тщательно осмотрела, дала рекомендации и заменила все препараты. Приём прошёл на высшем уровне! Я очень довольна, что попала к Софье Владиславовне и выбирала её по отзывам!
На модерации,
21 февраля 2021
Очень добродушный, профессиональный и вежливый врач. Она взяла анализы и провела УЗИ щитовидной железы у ребёнка. Я пришла к ней по отзывам.
На модерации,
20 февраля 2021
Доктор внимательный, тактичный, профессиональный и заинтересованный. Она расспросила меня о жалобах, задала вопросы, поговорила на разные темы, обсудила некоторые лекарства, собрала полный анамнез, назначила сдачу анализов, прописала витамины и предложила пройти других специалистов, для более точного анализа.
Она расспросила меня о жалобах, задала вопросы, поговорила на разные темы, обсудила некоторые лекарства, собрала полный анамнез, назначила сдачу анализов, прописала витамины и предложила пройти других специалистов, для более точного анализа.
На модерации,
20 февраля 2021
Замечательный, внимательный и вдумчивый доктор. На приёме он мне все разъяснил и помог.
Тамара,
04 февраля 2021
Хороший и профессиональный врач. Она чётко ответила на все мои вопросы, расшифровала результаты исследований и назначила сдачу анализов.
Она чётко ответила на все мои вопросы, расшифровала результаты исследований и назначила сдачу анализов.
Гуландом,
21 января 2021
Компетентный врач. Она дала мне рекомендации. Я довольна и была не в первый раз у данного специалиста!
Ирина,
04 декабря 2020
Врач потрясающий и очень грамотный. Мы пошли к ней по рекомендации знакомых. Она нашла подход к мужу, ответила на все вопросы, дала много дельных советов и назначила обследование. Огромное спасибо этом доктору!
Мы пошли к ней по рекомендации знакомых. Она нашла подход к мужу, ответила на все вопросы, дала много дельных советов и назначила обследование. Огромное спасибо этом доктору!
Наталья,
25 августа 2020
Анна Владимировна профессиональный, знающий и квалифицированный специалист. Приём прошёл вовремя. Доктор выслушала меня, провела УЗИ, наметила программу действий и направила к ЛОРу.
Михаил,
30 апреля 2020
Очень внимательный врач. Она полностью помогла решить мой вопрос. Я доволен!
Она полностью помогла решить мой вопрос. Я доволен!
Аноним,
26 февраля 2020
Понравилась нам доктор, внимательная, вежливая, с детьми хорошо общается, грамотная, как специалист, не суетливая. Персонал очень вежливо общался. Нам не удобно было ехать, но ради такого врача, можно было съездить.
Святослав,
15 августа 2016
Показать 10 отзывов из 4176
Синдром Каллмана — европейский журнал генетики человека
Аннотация
Синдром Каллмана (KS) сочетает гипогонадотропный гипогонадизм (ДГ) с аносмией. Это клинически и генетически гетерогенное заболевание. KAL1, кодирующий внеклеточный гликопротеин аносмин-1, ответственен за рецессивную форму заболевания, связанную с Х-хромосомой. Мутации в FGFR1 или FGF8, кодирующие рецептор-1 фактора роста фибробластов и фактор-8 роста фибробластов, соответственно, лежат в основе аутосомно-доминантной формы с неполной пенетрантностью. Наконец, мутации в PROKR2 и PROK2, кодирующих прокинетиновый рецептор-2 и прокинетин-2, были обнаружены в гетерозиготном, гомозиготном и сложном гетерозиготных состояниях. Эти два гена могут быть вовлечены как в моногенный рецессивный, так и в дигенный / олигогенный режимы передачи KS. Примечательно, что мутации в любом из вышеупомянутых генов KS были обнаружены менее чем у 30% пациентов с KS, что указывает на то, что другие гены, вовлеченные в заболевание, еще предстоит обнаружить.
Это клинически и генетически гетерогенное заболевание. KAL1, кодирующий внеклеточный гликопротеин аносмин-1, ответственен за рецессивную форму заболевания, связанную с Х-хромосомой. Мутации в FGFR1 или FGF8, кодирующие рецептор-1 фактора роста фибробластов и фактор-8 роста фибробластов, соответственно, лежат в основе аутосомно-доминантной формы с неполной пенетрантностью. Наконец, мутации в PROKR2 и PROK2, кодирующих прокинетиновый рецептор-2 и прокинетин-2, были обнаружены в гетерозиготном, гомозиготном и сложном гетерозиготных состояниях. Эти два гена могут быть вовлечены как в моногенный рецессивный, так и в дигенный / олигогенный режимы передачи KS. Примечательно, что мутации в любом из вышеупомянутых генов KS были обнаружены менее чем у 30% пациентов с KS, что указывает на то, что другие гены, вовлеченные в заболевание, еще предстоит обнаружить.
Вкратце
KS является генетически гетерогенным заболеванием развития, которое чаще всего проявляется в отсутствии спонтанного полового созревания в сочетании с дефектом обоняния (гипосмия или аносмия).
Могут также присутствовать некоторые непродуктивные не обонятельные аномалии, в зависимости от генетической формы заболевания.
Распространенность заболевания была приблизительно оценена в 1: 8000 мужчин и 1: 40000 женщин, но может быть недооценена, особенно среди женщин.
Основными дифференциальными диагнозами являются нормосмический идиопатический гипогонадотропный гипогонадизм и синдром CHARGE.
Различные способы передачи KS включают связанный с Х-хромосомой рецессивный, аутосомно-рецессивный, аутосомно-доминантный с неполной пенетрантностью и, наиболее вероятно, дигеническое / олигогенное наследование.
Мутации в любом из пяти известных генов заболевания ( KAL1, FGFR1, FGF8, PROKR2, PROK2 ) были выявлены у относительно небольшой части (менее 30%) пациентов.
Целых 30% мутаций, обнаруженных в FGFR1, могут быть мутациями de novo, что, безусловно, должно быть рассмотрено до оценки риска рецидива этой генетической формы в семье.
Стратегия генетического тестирования (рис. 1) основана на поле пациента, семейном анамнезе (если есть) и предполагаемом типе наследования заболевания, а также наличии дополнительных клинических аномалий, которые могут направить генетика к определенному гену заболевания или иногда к синдрому смежного гена.
Лечение КС — это лечение гипогонадизма. В настоящее время нет лечения обонятельного дефицита. У обоих полов заместительная гормональная терапия используется для стимулирования развития вторичных половых признаков во время полового созревания, а затем для стимулирования фертильности.

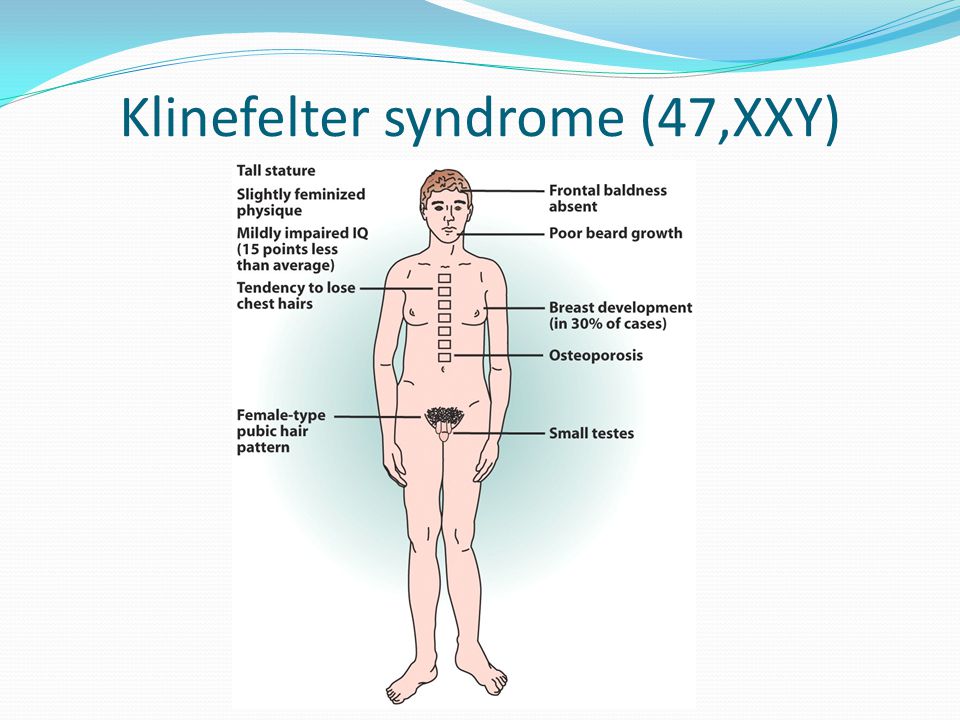
Вступление
Maestre de San Juan, вероятно, был первым, кто сообщил в 1856 году об отсутствии обонятельных структур в головном мозге и о наличии маленьких яичек у человека. 1 Синдром был идентифицирован как клиническая единица в 1944 году американским медицинским генетиком Каллманном, который провел исследование о распространении гипогонадизма, сопровождающегося аносмией, в трех затронутых семьях. 2 Он показал совокупную аносмию и гипогонадизм у всех пораженных людей и поэтому установил, что этот синдром может быть наследственным. В 1950-х годах швейцарский анатом де Морсье задокументировал заболевание, описав недоразвитие или отсутствие обонятельных луковиц и трактов у нескольких пациентов мужского пола с гипогонадизмом. 3 Несколько лет спустя гипогонадизм был приписан дефициту гонадотропин-рилизинг-гормона (ГнРГ). 4
2 Он показал совокупную аносмию и гипогонадизм у всех пораженных людей и поэтому установил, что этот синдром может быть наследственным. В 1950-х годах швейцарский анатом де Морсье задокументировал заболевание, описав недоразвитие или отсутствие обонятельных луковиц и трактов у нескольких пациентов мужского пола с гипогонадизмом. 3 Несколько лет спустя гипогонадизм был приписан дефициту гонадотропин-рилизинг-гормона (ГнРГ). 4
Распространенность КС до сих пор неизвестна. Он был приблизительно оценен в один из 8000 у мальчиков. Предполагается, что у девочек распространенность в пять раз ниже, но, вероятно, ее недооценивают, поскольку у некоторых пораженных женщин наблюдается лишь умеренный гипогонадизм (см. Ниже). Кроме того, первичная аменорея у женщин часто остается неизученной.
Клинический обзор
Синдром Каллмана, как правило, сочетает тяжелую ДГ с полным отсутствием обоняния (аносмия). Степень гипогонадизма и дефицита запаха может, однако, значительно различаться не только между неродственными пациентами, но и в пределах затронутых семей 5, 6 (и см. Родословные в ссылках 7, 8, 9, 10, 11, 12, 13, 14 ) даже между монозиготными близнецами. 15, 16 В некоторых семьях описаны как типичные фенотипы KS, так и диссоциированные фенотипы с гипогонадизмом или аносмией. 7, 10, 13, 14, 17 Кроме того, у нескольких пациентов с КС отмечалось явное изменение гипогонадизма после прекращения гормонального лечения. 9, 18, 19 Наконец, различные непроизводительные не обонятельные дополнительные аномалии присутствуют только у части пациентов с KS. Эти расстройства включают непроизвольные движения зеркал верхней конечности (бимануальный синкинез), 17, 20, 21, 22 аномальные движения глаз, 21, 23 врожденный птоз, 24, 25 аномальное зрительное пространственное внимание, 26 ухудшение слуха, 5, 6, 8, 27, 28, 29 агенезия мозолистого тела, 7, 13 односторонние (иногда двусторонние) почечные агенезы, 30, 31, 32 расщелины губы или неба, 5, 6, 7, 33 агенез одного или нескольких зубов (гиподонтия), 7, 24, 33, 34 ожирение 6, 10 и другие менее документированные аномалии (см.
Родословные в ссылках 7, 8, 9, 10, 11, 12, 13, 14 ) даже между монозиготными близнецами. 15, 16 В некоторых семьях описаны как типичные фенотипы KS, так и диссоциированные фенотипы с гипогонадизмом или аносмией. 7, 10, 13, 14, 17 Кроме того, у нескольких пациентов с КС отмечалось явное изменение гипогонадизма после прекращения гормонального лечения. 9, 18, 19 Наконец, различные непроизводительные не обонятельные дополнительные аномалии присутствуют только у части пациентов с KS. Эти расстройства включают непроизвольные движения зеркал верхней конечности (бимануальный синкинез), 17, 20, 21, 22 аномальные движения глаз, 21, 23 врожденный птоз, 24, 25 аномальное зрительное пространственное внимание, 26 ухудшение слуха, 5, 6, 8, 27, 28, 29 агенезия мозолистого тела, 7, 13 односторонние (иногда двусторонние) почечные агенезы, 30, 31, 32 расщелины губы или неба, 5, 6, 7, 33 агенез одного или нескольких зубов (гиподонтия), 7, 24, 33, 34 ожирение 6, 10 и другие менее документированные аномалии (см.![]() Ссылку 35 для обзора).
Ссылку 35 для обзора).
Дифференциальный диагноз: нормосмический идиопатический синдром HH и CHARGE
Трудности встречаются на обоих концах фенотипического спектра KS, который заключается либо в отсутствии заметного дефицита запаха, либо в случае наличия непродуктивных не обонятельных дополнительных аномалий в верхней части типичного KS (см. Рисунок 1).
Стратегия генетического тестирования синдрома Каллмана. Стратегия основана на поле пациента, семейном анамнезе (если есть) и предполагаемом типе наследования болезни, а также наличии дополнительных клинических аномалий, которые могут направить генетика к определенному гену заболевания или, иногда, к синдрому смежного гена в Xp22.3. 36 или 8p11.2 p12. 7 Поиск мутаций KAL1 ограничен больными мужчинами, либо единичными случаями, либо пациентами с семейным анамнезом, совместимым с Х-сцепленным рецессивным типом наследования. Скрининг мутаций известных генов KS ( KAL1, FGFR1, FGF8, PROKR2, PROK2 ) приводит к выявлению мутации менее чем у трети пациентов. Примечательно, что до 30% мутаций, обнаруженных в FGFR1, могут быть мутациями de novo, что, безусловно, должно быть рассмотрено до оценки риска рецидива этой генетической формы в семье. Основными дифференциальными диагнозами КС являются нормосмический идиопатический гипогонадотропный гипогонадизм и синдром CHARGE.
Примечательно, что до 30% мутаций, обнаруженных в FGFR1, могут быть мутациями de novo, что, безусловно, должно быть рассмотрено до оценки риска рецидива этой генетической формы в семье. Основными дифференциальными диагнозами КС являются нормосмический идиопатический гипогонадотропный гипогонадизм и синдром CHARGE.
Изображение в полном размере
Учитывая переменную степень гипосмии в KS, различие между KS и нормосмическим идиопатическим HH (nIHH) в настоящее время неясно, особенно поскольку пациенты HH не всегда проходят детальное обонятельное тестирование. Однако существует генетическое свидетельство того, что nIHH и KS представляют различные нозологические объекты. Действительно, гены, кодирующие рецепторы GnRH и целепептина, которые участвуют в nIHH 37, 38, 39, по-видимому, не требуются для эмбриональной миграции нейроэндокринных клеток GnRH, процесс, вероятно, является дефектным у пациентов с KS (см. Ниже). Крупномасштабное генетическое тестирование подлинных случаев nIHH на наличие мутаций в генах KS должно помочь прояснить эту проблему. Недавнее сообщение о семье, в которой вредные мутации GNRHR и FGFR1, совместно сгруппированные у особей nIHH, указывают, однако, что ситуация может быть более сложной, чем предполагалось. 40
Недавнее сообщение о семье, в которой вредные мутации GNRHR и FGFR1, совместно сгруппированные у особей nIHH, указывают, однако, что ситуация может быть более сложной, чем предполагалось. 40
Синдром CHARGE имеет предполагаемую рождаемость 1 на 8500–12 000 человек. Определяющими признаками, которые делают акроним, являются колобома, аномалии сердца, атрезия хоан, задержка роста и / или развития, аномалии половых органов и ушей. Тем не менее, ни один признак не является универсальным или достаточным для диагностики синдрома CHARGE. Другие часто встречающиеся признаки включают характерную дисморфию лица и руки, гипотонию, архинэнцефалию, агенез или гипоплазию полукруглого канала, нарушение слуха, аномалии мочевыводящих путей, расщелину лицевого отдела, дисфагию и трахео-эзофагальные аномалии. В последние несколько лет были предложены новые диагностические критерии (см. Ссылку 41 ). Более того, сообщалось, что у большинства, если не у всех пациентов с CHARGE, есть и аплазия обонятельной луковицы или гипоплазия, и HH, 42, 43, то есть две определяющие особенности KS. Следовательно, ранее сообщенные случаи KS, связанные с врожденным пороком сердца 44 или атрезией хоан 45, могут фактически представлять собой нераспознанные легкие случаи CHARGE. 46 Синдром CHARGE имеет общие черты с генетической формой KS2 KS (см. Ниже), включая расщелину губы или неба, присутствующую у 20–35% пациентов с KAL2 7, 11, 12, 13 и CHARGE 47, уродство наружного уха, отмеченное у практически все пациенты CHARGE 47 и несколько пациентов KAL2, 48 агенеза мозолистого тела, сообщили о нескольких пациентах CHARGE 47 и KAL2, 7, 13 и колобома, которая широко распространена у пациентов CHARGE 47 и была отмечена по крайней мере у одного пациента KAL2 тоже. 7 Большинство людей с синдромом CHARGE гетерозиготны по мутациям с потерей функции в CHD7, который кодирует ДНК-связывающий белок геликазы хромодомена (домен модификатора организации хроматина). 49, 50 Из-за сходства между фенотипами CHARGE и KAL2 заманчиво предположить, что существуют функциональные взаимодействия между CHD7 и FGFR1-сигнальным путем.
Следовательно, ранее сообщенные случаи KS, связанные с врожденным пороком сердца 44 или атрезией хоан 45, могут фактически представлять собой нераспознанные легкие случаи CHARGE. 46 Синдром CHARGE имеет общие черты с генетической формой KS2 KS (см. Ниже), включая расщелину губы или неба, присутствующую у 20–35% пациентов с KAL2 7, 11, 12, 13 и CHARGE 47, уродство наружного уха, отмеченное у практически все пациенты CHARGE 47 и несколько пациентов KAL2, 48 агенеза мозолистого тела, сообщили о нескольких пациентах CHARGE 47 и KAL2, 7, 13 и колобома, которая широко распространена у пациентов CHARGE 47 и была отмечена по крайней мере у одного пациента KAL2 тоже. 7 Большинство людей с синдромом CHARGE гетерозиготны по мутациям с потерей функции в CHD7, который кодирует ДНК-связывающий белок геликазы хромодомена (домен модификатора организации хроматина). 49, 50 Из-за сходства между фенотипами CHARGE и KAL2 заманчиво предположить, что существуют функциональные взаимодействия между CHD7 и FGFR1-сигнальным путем.
Диагностические подходы
Большинство случаев диагностируется во время полового созревания из-за недостаточного полового развития, определяемого маленькими яичками и отсутствующей вирилизации у мужчин или отсутствия развития груди и первичной аменореи у женщин. KS диагностируется, когда гонадотропины и гонадные стероиды с низким содержанием сыворотки сочетаются с нарушенным обонянием. Последнее должно быть установлено с помощью подробного опроса и обонятельного скрининга 51, 52, 53, 54, потому что это редко упоминается спонтанно. Магнитно-резонансная томография (МРТ) переднего мозга может быть выполнена, чтобы показать гипоплазию или аплазию обонятельных луковиц и трактов 55 (рис. 2). МРТ также полезно для исключения поражений гипоталамуса или гипофиза в качестве причины возникновения ГГ. 56 Дефицит ГнРГ может быть косвенно оценен с помощью эндокринологических тестов (см. Ссылку 57 ).
Магнитно-резонансная томография (МРТ) области обонятельной луковицы у контрольного человека ( a и b ) и человека, страдающего синдромом Каллмана ( c и d ). ( a и b ) МРТ T2-взвешенная последовательность в коронарной плоскости показывает нормальные обонятельные луковицы у контрольной особи ( а, белые стрелки), а позади обонятельных луковиц хорошую дифференцировку бороздчатой борозды ( b, белые стрелки) и обонятельных путей ( б, белые стрелки). ( c ) МРТ T2-взвешенная последовательность в коронарной плоскости показывает очень маленькие обонятельные луковицы у пациента с KS (белые стрелки). На правой стороне видна борозда носа (белая стрелка), с хорошей дифференциацией между правой извилиной и орбитальной извилиной. С левой стороны нет борозды на носу (черная стрелка) и различий между правой и орбитальной извилинами. ( d ) МРТ T1-взвешенная последовательность в коронарной плоскости, позади обонятельных луковиц, подтверждает наличие бороздки носа с правой стороны (белая стрелка), с относительно хорошим различием между правой (белая стрелка) и орбитальной (черная стрелка) ) извилины и отсутствие носовой борозды на левой стороне (черная стрелка).
( a и b ) МРТ T2-взвешенная последовательность в коронарной плоскости показывает нормальные обонятельные луковицы у контрольной особи ( а, белые стрелки), а позади обонятельных луковиц хорошую дифференцировку бороздчатой борозды ( b, белые стрелки) и обонятельных путей ( б, белые стрелки). ( c ) МРТ T2-взвешенная последовательность в коронарной плоскости показывает очень маленькие обонятельные луковицы у пациента с KS (белые стрелки). На правой стороне видна борозда носа (белая стрелка), с хорошей дифференциацией между правой извилиной и орбитальной извилиной. С левой стороны нет борозды на носу (черная стрелка) и различий между правой и орбитальной извилинами. ( d ) МРТ T1-взвешенная последовательность в коронарной плоскости, позади обонятельных луковиц, подтверждает наличие бороздки носа с правой стороны (белая стрелка), с относительно хорошим различием между правой (белая стрелка) и орбитальной (черная стрелка) ) извилины и отсутствие носовой борозды на левой стороне (черная стрелка).
Изображение в полном размере
Примечательно, что KS может также подозреваться у мальчиков уже в младенчестве при наличии крипторхизма или микропениса в сочетании с субнормальными концентрациями ЛГ и ФСГ. Действительно, послеродовый всплеск FSH, LH и тестостерона у младенца мужского пола как следствие продолжающейся функции генератора импульсов GnRH плода предоставляет 6-месячный интервал для установления диагноза HH, 58 и оповещения клинициста возможность его ассоциации с обонятельным нарушением. В связи с этим следует подчеркнуть полезность МРТ переднего мозга для диагностики заболевания у детей, слишком маленьких, чтобы пройти осмысленное тестирование обоняния или гипоталамо-гипофизарно-гонадной оси, 59, 60, даже несмотря на то, что нормальные изображения обонятельных луковиц были описаны в несколько пациентов с KS. 22, 29
Наконец, наличие непроизводительных без обонятельных дополнительных расстройств, включая движения зеркала, аномалии неба, почечную агенезию (ультрасонография), нарушение слуха (аудиометрическое тестирование) и агенез зубов, следует тщательно исследовать у пациентов и, по возможности, их родственники первой степени, потому что такие аномалии могут направить генетика к определенным генетическим формам заболевания (см. ниже и рисунок 1). В семьях, пораженных KS, расщелина нёба или почечный агенез, диагностированные с помощью УЗИ плода, могут иногда выявить заболевание до рождения.
ниже и рисунок 1). В семьях, пораженных KS, расщелина нёба или почечный агенез, диагностированные с помощью УЗИ плода, могут иногда выявить заболевание до рождения.
Комплексная генетика К.С.
Несмотря на то, что большинство пациентов с КС встречаются в виде спорадических случаев, многие случаи являются явно семейными, при этом сообщается о трех типах наследования: X-хромосомно-рецессивный (OMIM № 308700), аутосомно-доминантный (OMIM № 147950) и аутосомно-рецессивный (OMIM № 244200) (//www.ncbi.nlm.nih.gov/omim/). В аутосомно-доминантной форме была подчеркнута неполная пенетрантность. 5, 61
Пять причинных генов были идентифицированы на сегодняшний день, а именно, в хронологическом порядке обнаружения, KAL1, 62, 63, 64 FGFR1, 7 PROKR2 и PROK2, 10 и FGF8. 65 Различные мутации потери функции в KAL1, кодирующие гликопротеин внеклеточного матрикса аносмин-1, и в FGFR1 или FGF8, кодирующие рецептор-1 фактора роста фибробластов и фактор-8 роста фибробластов, лежат в основе Х-хромосомно-связанной формы (KAL1) и аутосомно-доминантная форма (KAL2) KS соответственно (список дополнительных мутаций см. в дополнительных таблицах S1, S2 и S3). Генетические формы KAL1 и KAL2 составляют примерно 8% и 10% всех случаев KS, соответственно. Мутации в KAL1 представляют собой в основном нонсенс-мутации, мутации со сдвигом рамки или большие делеции генов, тогда как большинство мутаций в FGFR1 (т.е. приблизительно 70%) или FGF8 (все шесть мутаций, о которых сообщалось до сих пор) являются ошибочными мутациями. Примечательно, что до 30% мутаций FGFR1, обнаруженных у пациентов, могут быть мутациями de novo 13, 48, 66 (C Dodé, неопубликованные результаты). Предполагаемые мутации потери функции в PROKR2 или PROK2, кодирующих прокинетиновый рецептор-2 и прокинетин-2, соответственно, были обнаружены примерно у 9% пациентов с KS (список дополнительных мутаций см. В дополнительной таблице S4). Большинство из этих мутаций являются миссенс-мутациями, и многие из них также были обнаружены у лиц, по-видимому, не затронутых, что ставит под сомнение их патогенную роль в заболевании. Однако вредное воздействие на передачу сигналов прокинетина было показано in vitro почти для всех миссенс-мутаций.
в дополнительных таблицах S1, S2 и S3). Генетические формы KAL1 и KAL2 составляют примерно 8% и 10% всех случаев KS, соответственно. Мутации в KAL1 представляют собой в основном нонсенс-мутации, мутации со сдвигом рамки или большие делеции генов, тогда как большинство мутаций в FGFR1 (т.е. приблизительно 70%) или FGF8 (все шесть мутаций, о которых сообщалось до сих пор) являются ошибочными мутациями. Примечательно, что до 30% мутаций FGFR1, обнаруженных у пациентов, могут быть мутациями de novo 13, 48, 66 (C Dodé, неопубликованные результаты). Предполагаемые мутации потери функции в PROKR2 или PROK2, кодирующих прокинетиновый рецептор-2 и прокинетин-2, соответственно, были обнаружены примерно у 9% пациентов с KS (список дополнительных мутаций см. В дополнительной таблице S4). Большинство из этих мутаций являются миссенс-мутациями, и многие из них также были обнаружены у лиц, по-видимому, не затронутых, что ставит под сомнение их патогенную роль в заболевании. Однако вредное воздействие на передачу сигналов прокинетина было показано in vitro почти для всех миссенс-мутаций. 67, 68 Обнаружение для данных мутаций PROKR2 и PROK2 как гетерозиготных, так и гомозиготных (или сложных гетерозиготных) неродственных пациентов 10, 69 весьма примечательно и свидетельствует в пользу дигенного или олигогенного способа наследования у гетерозиготных пациентов. На сегодняшний день наследственное наследование KS было показано у трех таких пациентов, у которых были моноаллельные миссенс-мутации как в PROKR2, так и в PROK2, 67 FGFR1 (C Dodé, неопубликованный) или KAL1 . 10 Заманчиво предположить, что у последнего пациента имеется гипоморфный аллель KAL1, кодирующий вариант белка, который все еще сохраняет некоторую биологическую активность, в то время как подавляющее большинство описанных к настоящему времени мутаций KAL1 приводят к нулевым аллелям, которые, по-видимому, достаточны для образования аномального фенотип у мужчин. Ожидается, что другие пациенты, несущие гетерозиготные мутации в PROKR2, PROK2 или гипоморфные мутации в KAL1, несут дополнительные мутации в других, пока неизвестных, генах KS.
67, 68 Обнаружение для данных мутаций PROKR2 и PROK2 как гетерозиготных, так и гомозиготных (или сложных гетерозиготных) неродственных пациентов 10, 69 весьма примечательно и свидетельствует в пользу дигенного или олигогенного способа наследования у гетерозиготных пациентов. На сегодняшний день наследственное наследование KS было показано у трех таких пациентов, у которых были моноаллельные миссенс-мутации как в PROKR2, так и в PROK2, 67 FGFR1 (C Dodé, неопубликованный) или KAL1 . 10 Заманчиво предположить, что у последнего пациента имеется гипоморфный аллель KAL1, кодирующий вариант белка, который все еще сохраняет некоторую биологическую активность, в то время как подавляющее большинство описанных к настоящему времени мутаций KAL1 приводят к нулевым аллелям, которые, по-видимому, достаточны для образования аномального фенотип у мужчин. Ожидается, что другие пациенты, несущие гетерозиготные мутации в PROKR2, PROK2 или гипоморфные мутации в KAL1, несут дополнительные мутации в других, пока неизвестных, генах KS. В самом деле, мутации в пяти известных генах вместе составляют менее 30% случаев KS, что указывает на то, что другие гены, ответственные за заболевание, еще предстоит обнаружить, некоторые из которых также могут быть вовлечены в передачу сигналов FGF или передачу прокинетина. Примечательно, что последние данные указывают на то, что олигогенный тип наследования может также применяться к пациентам с мутациями в FGFR1 или FGF8 . У трех пациентов, несущих миссенс-мутации в FGFR1, действительно было обнаружено, что они также имеют моноаллельную или двуаллельную мутацию в FGF8, 64 или моноаллельную мутацию в PROKR2 (см. Выше).
В самом деле, мутации в пяти известных генах вместе составляют менее 30% случаев KS, что указывает на то, что другие гены, ответственные за заболевание, еще предстоит обнаружить, некоторые из которых также могут быть вовлечены в передачу сигналов FGF или передачу прокинетина. Примечательно, что последние данные указывают на то, что олигогенный тип наследования может также применяться к пациентам с мутациями в FGFR1 или FGF8 . У трех пациентов, несущих миссенс-мутации в FGFR1, действительно было обнаружено, что они также имеют моноаллельную или двуаллельную мутацию в FGF8, 64 или моноаллельную мутацию в PROKR2 (см. Выше).
Генотип-фенотипическая корреляция
Для каждой генетической формы KS, выявленной до настоящего времени, клиническая гетерогенность заболевания в затронутых семьях ясно указывает на то, что проявление фенотипов KS зависит от факторов, отличных от самого мутированного гена. Эти факторы, вероятно, включают эпигенетические факторы и гены-модификаторы, которые еще не были идентифицированы. Кроме того, наследственное или олигогенное наследование, по-видимому, частично объясняет давно признанную неполную пенетрантность заболевания. Тем не менее, некоторые общие черты появились из клинических наблюдений у пациентов, пораженных различными генетическими формами KS. Например, у пациентов с мутациями у FGFR1, FGF8, PROKR2 или PROK2 наблюдалась большая вариабельность степени гипогонадизма, чем у пациентов с KAL1. 7, 10, 65, 70, 71, 72 В частности, спонтанно плодовитые особи, несущие мутации в любом из четырех аутосомных генов KS, ответственны за передачу заболевания в течение нескольких поколений, тогда как Х-сцепленная форма KS обычно передается женщины-носители мутаций KAL1, которые клинически не затронуты. Среди множества непродуктивных и не обонятельных нарушений, которые затрагивают часть пациентов с KS, некоторые были зарегистрированы для определенных генетических форм болезни. Например, односторонний почечный агенез происходит примерно у 30% пациентов с KAL1, 31, 32, но до сих пор не сообщалось о пациентах с мутациями FGFR1, FGF8, PROKR2 или PROK2 .
Кроме того, наследственное или олигогенное наследование, по-видимому, частично объясняет давно признанную неполную пенетрантность заболевания. Тем не менее, некоторые общие черты появились из клинических наблюдений у пациентов, пораженных различными генетическими формами KS. Например, у пациентов с мутациями у FGFR1, FGF8, PROKR2 или PROK2 наблюдалась большая вариабельность степени гипогонадизма, чем у пациентов с KAL1. 7, 10, 65, 70, 71, 72 В частности, спонтанно плодовитые особи, несущие мутации в любом из четырех аутосомных генов KS, ответственны за передачу заболевания в течение нескольких поколений, тогда как Х-сцепленная форма KS обычно передается женщины-носители мутаций KAL1, которые клинически не затронуты. Среди множества непродуктивных и не обонятельных нарушений, которые затрагивают часть пациентов с KS, некоторые были зарегистрированы для определенных генетических форм болезни. Например, односторонний почечный агенез происходит примерно у 30% пациентов с KAL1, 31, 32, но до сих пор не сообщалось о пациентах с мутациями FGFR1, FGF8, PROKR2 или PROK2 . С другой стороны, потеря носового хряща, гипоплазия наружного уха и скелетные аномалии рук или ног были зарегистрированы только у пациентов с KAL2. 7, 13, 48 Напротив, нарушение слуха является общим для нескольких генетических форм KS, 7, 8, 13, 23, 24, 29, 65, хотя следует отметить, что основной дефект (проводящий, восприимчивый или смешанный) может варьироваться между различными генетическими формами. Дефекты неба также следует рассматривать как одну из этих общих черт, хотя степень тяжести различается между KAL1 (с высоким сводчатым небом) и KAL2 (с расщелиной неба). Расщелина губы и / или неба может возникать в 25–30% случаев KAL2. 7, 11, 12, 13, 65, 73 Наконец, бимануальный синкинез широко распространен в KAL1 (возможно, > 75% случаев), 17, 22, но, по-видимому, гораздо реже в KAL2. 7, 12 В таблице 1 показано сравнение клинических признаков KAL1 и KAL2. До сих пор не сообщалось о дополнительных аномалиях у пациентов с KS, несущих мутации в PROKR2 или PROK2, с заметным исключением тяжелого нарушения сна и выраженного ожирения у одного пациента 10, что может быть связано с известной функцией передачи сигналов прокинетин-2 в поведенческом поведении.
С другой стороны, потеря носового хряща, гипоплазия наружного уха и скелетные аномалии рук или ног были зарегистрированы только у пациентов с KAL2. 7, 13, 48 Напротив, нарушение слуха является общим для нескольких генетических форм KS, 7, 8, 13, 23, 24, 29, 65, хотя следует отметить, что основной дефект (проводящий, восприимчивый или смешанный) может варьироваться между различными генетическими формами. Дефекты неба также следует рассматривать как одну из этих общих черт, хотя степень тяжести различается между KAL1 (с высоким сводчатым небом) и KAL2 (с расщелиной неба). Расщелина губы и / или неба может возникать в 25–30% случаев KAL2. 7, 11, 12, 13, 65, 73 Наконец, бимануальный синкинез широко распространен в KAL1 (возможно, > 75% случаев), 17, 22, но, по-видимому, гораздо реже в KAL2. 7, 12 В таблице 1 показано сравнение клинических признаков KAL1 и KAL2. До сих пор не сообщалось о дополнительных аномалиях у пациентов с KS, несущих мутации в PROKR2 или PROK2, с заметным исключением тяжелого нарушения сна и выраженного ожирения у одного пациента 10, что может быть связано с известной функцией передачи сигналов прокинетин-2 в поведенческом поведении. циркадные ритмы, включая сон-бодрствование и пищеварительное поведение. 74 Распространенность нарушений сна и питания у пациентов с КС, однако, еще предстоит определить.
циркадные ритмы, включая сон-бодрствование и пищеварительное поведение. 74 Распространенность нарушений сна и питания у пациентов с КС, однако, еще предстоит определить.
Таблица в натуральную величину
Лечение гипогонадизма
Лечение гипогонадизма при КС направлено, во-первых, на начало вирилизации или развития молочной железы, а во-вторых, на развитие фертильности. Заместительная гормональная терапия, обычно с тестостероном для мужчин и комбинированным эстрогеном и прогестероном для женщин, является терапией, стимулирующей развитие вторичных половых признаков. Для тех, кто желает иметь фертильность, гонадотропины или пульсирующий ГнРГ могут быть использованы для получения роста яичка и выработки спермы у мужчин или овуляции у женщин. Оба метода лечения восстанавливают фертильность у подавляющего большинства больных. 75 До сих пор неизвестно, может ли транзиторная заместительная гормональная терапия у затронутых младенцев мужского пола для симуляции послеродового выброса гонадотропинов позже повлиять на их половую жизнь и репродуктивный прогноз (см. Ссылку 58 ).
Ссылку 58 ).
патофизиология
Большинство фенотипических аномалий, о которых сообщается при KS, могут быть результатом нарушений развития в течение периода органогенеза, между 4 и 10 эмбриональными неделями (см. Ссылку 76 для обзора). Нарушение развития, приводящее к отсутствию (или гипоплазии) обонятельных луковиц и трактов и к аносмии в KS, полностью не изучено. Это может быть связано с нарушением терминального удлинения или нацеливания обонятельных аксонов, первичным морфогенетическим дефектом обонятельных луковиц (в конце 6-й эмбриональной недели) и более поздним дефектом разветвления аксонов выходных нейронов обонятельной луковицы. В конце 1980-х годов был пролен новый свет на механизм дефицита GnRH, лежащего в основе гипогонадизма KS, с открытием тесной топографической связи между периферической обонятельной системой и нейроэндокринными клетками GnRH в течение эмбриональной жизни. Эти клетки подвергаются миграции, начиная с 6-й эмбриональной недели, из обонятельного эпителия в передний мозг по пути обонятельного нерва. 77 На человеческом плоде, несущем хромосомную делецию на Xp22.3, которая включала KAL1, было показано, что клетки GnRH не мигрировали нормально и накапливались в верхней части носа. 78 В настоящее время нет патогистологических данных, подтверждающих, что эмбриональная миграция клеток GnRH задерживается у людей, пораженных другими генетическими формами KS. Однако у гомозиготных нокаутных мышей Prokr2 или Prok2 и мышей, несущих гипоморфные мутации Fgfr1 или Fgf8 в гомозиготном состоянии, миграция этих клеток также нарушается. 14, 65, 79, 80 Механизм предполагаемого дефекта миграции клеток GnRH в KS все еще остается предположительным. Это может быть либо следствием ранней дегенерации обонятельных нервов и аксонов терминальных нервов, которые действуют как направляющие сигналы, либо процессом, непосредственно затрагивающим сами клетки GnRH. Кроме того, дефекты спецификации судьбы клеток GnRH, дифференцировки, удлинения аксонов или нацеливания аксонов на срединное возвышение гипоталамуса также могут способствовать дефициту GnRH, по крайней мере, в генетической форме заболевания KAL2 (см.
77 На человеческом плоде, несущем хромосомную делецию на Xp22.3, которая включала KAL1, было показано, что клетки GnRH не мигрировали нормально и накапливались в верхней части носа. 78 В настоящее время нет патогистологических данных, подтверждающих, что эмбриональная миграция клеток GnRH задерживается у людей, пораженных другими генетическими формами KS. Однако у гомозиготных нокаутных мышей Prokr2 или Prok2 и мышей, несущих гипоморфные мутации Fgfr1 или Fgf8 в гомозиготном состоянии, миграция этих клеток также нарушается. 14, 65, 79, 80 Механизм предполагаемого дефекта миграции клеток GnRH в KS все еще остается предположительным. Это может быть либо следствием ранней дегенерации обонятельных нервов и аксонов терминальных нервов, которые действуют как направляющие сигналы, либо процессом, непосредственно затрагивающим сами клетки GnRH. Кроме того, дефекты спецификации судьбы клеток GnRH, дифференцировки, удлинения аксонов или нацеливания аксонов на срединное возвышение гипоталамуса также могут способствовать дефициту GnRH, по крайней мере, в генетической форме заболевания KAL2 (см. Ссылку 35, 80 ).
Ссылку 35, 80 ).
Нерешенные вопросы
Хотя KS был идентифицирован как наследственное заболевание более 60 лет назад, его генетика до сих пор не до конца изучена, включая его гораздо более высокую распространенность у мужчин, чем у женщин. Среди многих нерешенных генетических и клинических вопросов можно выделить следующие:
Какова фактическая распространенность KS, особенно у женщин?
Каков фенотипический спектр KS, особенно в отношении непродуктивных, не обонятельных дополнительных нарушений?
Является ли наследственная аносмия без явного гипогонадизма клинической формой КС?
Где находится нозологическая граница между KS и нормосмическим HH?
Сколько различных генов заболевания вовлечено в KS и как они функционально взаимодействуют в развитии обонятельной и GnRH нейроэндокринной систем (см. Ссылку 81 для текущих гипотез)?
Какова распространенность дигенического / олигогенного типа наследования среди пациентов с КС? Действительно, ответ на этот вопрос является необходимым условием для оценки риска рецидива заболевания в затронутых семьях.
Почему КС чаще встречается у мужчин, чем у женщин?
Учитывая, что рецессивная форма, связанная с Х-хромосомой, не учитывает более высокую распространенность заболеваний у мужчин, возможно, женщины в какой-то степени защищены от возникновения заболеваний физиологически более высокими уровнями экспрессии KAL1 в течение эмбриональной жизни по сравнению с мужчинами (поскольку KAL1 частично ускользает от процесса инактивации Х-хромосомы у людей?
Дополнительная информация
Word документы
- 1.
Дополнительные таблицы S1
- 2.
Дополнительные таблицы S2
- 3.
Дополнительные таблицы S3
- 4.
Дополнительные таблицы S4
Дополнительная информация прилагается к статье на веб-сайте Европейского журнала генетики человека (//www.nature.com/ejhg)
Синдром Кальмана [LifeBio.wiki]
Синдром Кальмана представляет собой генетическое заболевание, первичным симптомом которого является отсутствие полового созревания или частичное половое созревание. Оно встречается и у мужчин, и у женщин и имеет такие дополнительные симптомы как гипогонадизм и почти всегда бесплодие. Синдром Кальмана также имеет такие симптомы как изменение обоняния либо полное его отсутствие (аносмия) или снижение обоняния в значительной степени (гипосмия)1). Синдром Кальмана возникает, когда нейроны гипоталамуса, которые отвечают за высвобождение гонадотропин-рилзинг гормона (нейроны, секретирующие ГнРГ), неспособны мигрировать в гипоталамус в ходе эмбрионального развития2).
Оно встречается и у мужчин, и у женщин и имеет такие дополнительные симптомы как гипогонадизм и почти всегда бесплодие. Синдром Кальмана также имеет такие симптомы как изменение обоняния либо полное его отсутствие (аносмия) или снижение обоняния в значительной степени (гипосмия)1). Синдром Кальмана возникает, когда нейроны гипоталамуса, которые отвечают за высвобождение гонадотропин-рилзинг гормона (нейроны, секретирующие ГнРГ), неспособны мигрировать в гипоталамус в ходе эмбрионального развития2).
Обзор
Синдром Кальмана является частью группы заболеваний, которые объединены термином «гипогонадотропный гипогонадизм». Влияние на обоняние наблюдается только примерно в 50% случаев гипогонадотропного гипогонадизма, эти случаи получили название синдрома Кальмана. Кроме обоняния, гипогонадотропного гипогонадизма и синдрома Кальмана не имеют отличий в диагностировании или лечении3).
Терминология, используемая при описании случаев гипогонадотропного гипогонадизма, может отличаться. Термин «врожденный гипогонадотропный гипогонадизм» в настоящее время используется наиболее часто. Другие используемые термины включают идиопатический/изолированный гипогонадотропный гипогонадизм, нормосмический гипогонадотропный гипогонадизм или гипоталамический гипогонадизм. Термин гипогонадотропный гипогонадизм может использоваться для обозначения всех случаев, в том числе синдрома Кальмана.
Термин «врожденный гипогонадотропный гипогонадизм» в настоящее время используется наиболее часто. Другие используемые термины включают идиопатический/изолированный гипогонадотропный гипогонадизм, нормосмический гипогонадотропный гипогонадизм или гипоталамический гипогонадизм. Термин гипогонадотропный гипогонадизм может использоваться для обозначения всех случаев, в том числе синдрома Кальмана.
Термин гипогонадизм описывает низкий уровень в крови половых гормонов, тестостерона у мужчин и эстрогена и прогестерона у женщин. Гипогонадизм может развиваться посредством нескольких механизмов. Использование термина «гипогонадотропный» относится к тому факту, что гипогонадизм при гипогонадотропном гипогонадизме вызван нарушением выработки гонадотропных гормонов, в норме секретируемых аденогипофизом, а именно лютеинизирующего гормона (ЛГ) и фолликулостимулирующего гормона (ФСГ).
ЛГ и ФСГ оказывают прямое влияние на яичники у женщин и семенники у мужчин. Отсутствие ЛГ и ФСГ означает, что первоначально пубертатный период не наступает в соответствующее время и впоследствии яичники и семенники не выполняют свою репродуктивную функцию при созревании, а именно, высвобождение яйцеклеток у женщин и выработку сперматозоидов у мужчин совместно с их ролью в выработке половых гормонов.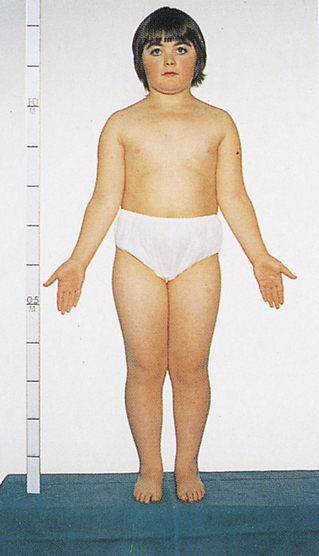
Первопричина неспособности вырабатывать ЛГ и ФСГ состоит в нарушении функции гипоталамуса в высвобождении гормона ГнРГ, который при нормальном состоянии стимулирует выработку ЛГ и ФСГ. Без корректного высвобождения ГнРГ, гипофиз неспособен высвобождать ЛГ и ФСГ, что, в свою очередь, приводит к невозможности корректного функционирования яичников и семенников. Такая неспособность вырабатывать ГнРГ может быть обусловлена либо отсутствием нейронов, секретирующих ГнРГ, внутри гипоталамуса, либо неспособностью гипоталамуса высвобождать ГнРГ корректным пульсативным способом для обеспечения высвобождения ЛГ и ФСГ из гипофиза.
Гипогонадотропный гипогонадизм может возникнуть в качестве отдельного заболевания с воздействием только на выработку ЛГ и ФСГ или совместно с гипофизарной недостаточностью, например, синдромом CHARGE.
По последним данным, как минимум, шестнадцать различных генов вовлечены в развитие синдрома Кальмана или иные формы гипогонадотропного гипогонадизма посредством нарушения выработки или активности ГнРГ. Вовлеченные гены представляют все формы наследуемости, и ни один генный дефект не был отмечен общим для всех случаев, что делает генетический анализ и прогнозирование наследования весьма проблематичным.
Вовлеченные гены представляют все формы наследуемости, и ни один генный дефект не был отмечен общим для всех случаев, что делает генетический анализ и прогнозирование наследования весьма проблематичным.
Синдром Кальмана был описан в работе, опубликованной в 1944 году Францем Иосифом Кальманом, немецко-американским генетиком4). Однако другие ученые, такие как доктор Аурелиано Маэстро де Сан Хуан в 1856 году, отмечали корреляцию между аносмией и гипогонадизмом5).
Заболевание имеет низкую распространенность, оцененную всего 1 случай на 4000 человек гипогонадотропного гипогонадизма для мужчин и 1 случай на 50 000 человек для синдрома Кальмана. Он встречается в 3-5 раз чаще у мужчин, чем у женщин.
Известной персоной с синдромом Кальмана был джазовый вокалист Джимми Скотт; данный синдром являлся сопутствующим фактором необычно высокого и характерного певческого голоса Скотта. В 2004 году канадский писатель Брайан Бретт опубликовал мемуары Uproar’s Your Only Music о взрослении человека с синдромом Кальмана6).
Клинические признаки
Симптомы синдрома Кальмана и гипогонадотропного гипогонадизма могут быть поделены на две разные группы: связанные и не связанные с репродуктивностью. Не все симптомы обязательно проявляются при синдроме Кальмана/гипогонадотропном гипогонадизме, даже среди членов одной семьи. Некоторые из этих симптомов связаны с дефектами генов, которые, как известно, отвечают за развитие синдрома Кальмана и гипогонадотропного гипогонадизма, но в некоторых случаях наличие этих симптомов не подлежит объяснению. По оценкам, в 60% случаев синдрома Кальмана/гипогонадотропного гипогонадизма не наблюдается симптомов, не связанных с репродуктивной функцией.
Обычно сложно отличить синдром Кальмана/гипогонадотропный гипогонадизм от простой системной задержки полового созревания. Однако если пубертатный период не наступает к 14 (девочки) или 15 (мальчики) годам и при наличии одного из данных симптомов, не связанных с репродуктивной функцией, рекомендуется получить консультацию эндокринолога.
Симптомы, связанные с репродуктивной функцией
Задержка или полное отсутствие полового созревания у мужчин и женщин.
Отсутствие развития семенников у мужчин; размер менее 3 см3. Нормальный диапазон составляет от 12 до 30 см3.
Первичная аменорея или нарушение начала менструации у женщин.
Слабо выраженные вторичные половые признаки у мужчин и женщин.
Бесплодие.
Симптомы, не связанные с репродуктивной функцией
Гипогонадотропный гипогонадизм (отсутствие гормонов гипофиза – лютеинизирующего гормона и фолликулостимулирующего гормона).
Врожденное заболевание (присутствует с рождения).
Полное отсутствие обоняния (аносмия) или значительно сниженное обоняние (гипосмия). Это отличительный симптом синдрома Кальмана, он не наблюдается при других случаях гипогонадотропного гипогонадизма. Примерно в 50% случаев гипогонадотропного гипогонадизма наблюдается аносмия, что может быть определено как синдром Кальмана.
Волчья пасть или иные краниофациальные дефекты.
Односторонний почечный агенез или аплазия; отсутствие или недостаточное функционирование одной почки.
Крипторхизм, неопущение яичек при рождении, возникает в 30% случаев синдрома Кальмана/гипогонадотропного гипогонадизма.
Микропенис, наблюдается менее чем в 5-10% случаев синдрома Кальмана/гипогонадотропного гипогонадизма.
Невральные дефекты слуха.
Синкинезия или зеркальные движения рук.
Дентальные нарушения.
- Нормальное телосложение, но может наблюдаться увеличение роста при задержке лечения из-за отсутствия тестостерона или эстрогена, что приводит к избыточному росту костей рук и ног.
В одно время дальтонизм связывали с синдромом Кальмана/гипогонадотропным гипогонадизмом, но данная теория была опровергнута.
В ряде случаев начало заболевания может наблюдаться после пубертатного периода, что приводит к развитию фенотипически нормального пениса у мужчин с последующей атрофией яичек и потерей некоторых вторичных половых признаков. У таких мужчин обычно наблюдается ухудшение половой функции и понижение либидо. У женщин позднее начало синдрома Кальмана приводит ко вторичной аменорее. Аносмия у таких пациентов может присутствовать или отсутствовать.
У таких мужчин обычно наблюдается ухудшение половой функции и понижение либидо. У женщин позднее начало синдрома Кальмана приводит ко вторичной аменорее. Аносмия у таких пациентов может присутствовать или отсутствовать.
Пациенты с синдромом Кальмана и другими формами гипогонадотропного гипогонадизма почти всегда рождаются с нормальной половой дифференциацией, т.е. физически они являются мужчинами или женщинами. Это происходит за счет действия хорионического гонадотропина человека (ХГЧ), вырабатываемого плацентой примерно на 12-20 неделе гестации (беременности), на активность которого в норме не влияет развитие синдрома Кальмана или гипогонадотропного гипогонадизма.
У пациентов с синдромом Кальмана/гипогонадотропным гипогонадизмом наблюдается недостаток выброса ГнРГ, ЛГ и ФСГ, который происходит между рождением и шестью месяцами жизни7). Этот выброс, в частности, важен для новорожденных мальчиков, поскольку он способствует опущению яичек в мошонку. Небольшой процент мальчиков с синдромом Кальмана/гипогонадотропным гипогонадизмом рождается с микропенисом и/или крипторхизмом, оба состояния могут лечиться хирургически в первый год жизни. Выброс ГнРГ/ЛГ/ФСГ у здоровых детей приводит к заметному увеличению уровня тестостерона у мальчиков и эстрогена у девочек. Отсутствие такого выброса может иногда использоваться для диагностики при подозрении на синдром Кальмана/гипогонадотропный гипогонадизм у новорожденных мальчиков, но недостаточен для диагностики данных заболеваний у девочек.
Выброс ГнРГ/ЛГ/ФСГ у здоровых детей приводит к заметному увеличению уровня тестостерона у мальчиков и эстрогена у девочек. Отсутствие такого выброса может иногда использоваться для диагностики при подозрении на синдром Кальмана/гипогонадотропный гипогонадизм у новорожденных мальчиков, но недостаточен для диагностики данных заболеваний у девочек.
Диагностика
Диагностика часто представляет собой метод исключения при обследовании задержки полового созревания8).
В работе, опубликованной в 2012 году профессором Яскесом Янгом9), представлен типичный пример диагностического обследования при подозрении на синдром Кальмана/гипогонадотропный гипогонадизм.
Одной из важнейших проблем в диагностике синдрома Кальмана и иных форм гипогонадотропного гипогонадизма является способность различить нормальную конституциональную задержку в половом созревании и синдром Кальмана или гипогонадотропный гипогонадизм.
Основными биохимическими параметрами у мужчин является низкий уровень тестостерона в сыворотке крови и низкий уровень гонадотропинов ЛГ и ФСГ, а у женщин — низкий уровень эстрогена в сыворотке крови и низкий уровень ЛГ и ФСГ.
Для мужчин и женщин с конституциональной задержкой полового созревания, эндогенная половая зрелость в итоге наступает без лечения. Однако задержка в лечении при синдроме Кальмана/гипогонадотропном гипогонадизме приводит к задержке физического развития пациента и может вызвать серьезные физиологические нарушения. Выжидательная тактика, применяемая к пациентам-«переросткам», вероятно, неэффективна, тогда как в качестве инструмента диагностики может использоваться поэтапный подход с применением гормонозаместительной терапии.
У женщин иногда диагностика является более отсроченной, поскольку в норме до рассмотрения возможности постановки диагноза синдром Кальмана/гипогонадотропный гипогонадизм должны быть исследованы другие причины аменореи.
У мужчин для отличия синдрома Кальмана/гипогонадотропного гипогонадизма от задержки полового созревания может использоваться лечение при уровнях тестостерона, соответствующих возрасту. При простой задержке, тестостерон может дать толчок эндогенному половому созреванию, что видно по увеличению семенников, тогда как при синдроме Кальмана/гипогонадотропном гипогонадизме не будет наблюдаться увеличения семенников только при проведении терапии тестостероном. При очевидном отсутствии полового созревания, особенно при отсутствии развития семенников, может потребоваться консультация эндокринолога. Доктор Ричард Куинтон, ведущий британский эксперт в области синдрома Кальмана/гипогонадотропного гипогонадизма, предполагает, что, если половое созревание отсутствует к возрасту 16 лет, пациент должен быть направлен к эндокринологу10).
При очевидном отсутствии полового созревания, особенно при отсутствии развития семенников, может потребоваться консультация эндокринолога. Доктор Ричард Куинтон, ведущий британский эксперт в области синдрома Кальмана/гипогонадотропного гипогонадизма, предполагает, что, если половое созревание отсутствует к возрасту 16 лет, пациент должен быть направлен к эндокринологу10).
Полное обследование эндокринной функции потребуется для измерения уровней других гормонов гипофиза, особенно пролактина, для проверки корректной работы гипофиза. Могут наблюдаться другие проблемы общего состояния здоровья, такие как избыточный вес или первопричинные хронические или острые заболевания, которые могли вызвать задержку полового созревания. Это является существенным для получения полного обзора эндокринной функции пациентов для различения синдрома Кальмана/гипогонадотропного гипогонадизма и иных причин задержки полового созревания.
Может быть оценен костный возраст с помощью рентгенологического исследования костей рук и запястий. Если костный возраст значительно ниже, чем биологический возраст пациента, можно предположить задержку полового созревания, если нет иных первопричин такого несоответствия.
Если костный возраст значительно ниже, чем биологический возраст пациента, можно предположить задержку полового созревания, если нет иных первопричин такого несоответствия.
Для исключения синдрома Клайнфельтера и синдрома Тернера может быть определен кариотип, хотя уровень гормонов будет также исключать обе этих относительно общих причины гипогонадизма.
Магнитно-резонансная томография (МРТ) может использоваться для определения наличия обонятельной луковицы и для проверки физических нарушений гипофиза или гипоталамуса.
Для проверки наличия аносмии может использоваться стандартный тест на обоняние, но следует помнить, что даже при полной аносмии различные вещества (такие как ментол и спирт) могут также выявляться посредством прямой стимуляции тройничного нерва.
Может быть выполнен генетический скрининг, особенно для определения мутации гена KAL-1, но с учетом неопределенного генетического происхождения большинства случаев синдрома Кальмана и гипогонадотропного гипогонадизма, отрицательный результат не будет исключать возможный диагноз.
В обзорной статье, опубликованной в 2014 году11), отмечается необходимость принятия к сведению врачом возможного диагноза синдрома Кальмана/гипогонадотропного гипогонадизма, если задержка полового созревания обнаружена вместе со связанными настораживающими симптомами. Симптомы, перечисленные в работе, были поделены на две группы: симптомы, относящиеся к репродуктивной функции, связанные с отсутствием минимального полового созревания от момента рождения до шестимесячного возраста, и симптомы, не относящиеся к репродуктивной функции, связанные со специфическими формами гипогонадотропного гипогонадизма. Как и в других обзорных статьях, авторы также отмечают нежелательность применения выжидательной тактики при задержке полового созревания.
Симптомы, связанные с репродуктивной функцией:
микропенис,
крипторхизм.
Симптомы, не связанные с репродуктивной функцией:
аносмия,
понижение слуха (обычно нервная глухота),
синкинезия рук (зеркальные движения),
почечный агенез (отсутствие одной почки),
дентальные нарушения,
синдактилия или иные аномалии рук,
заячья губа или волчья пасть,
колобома.
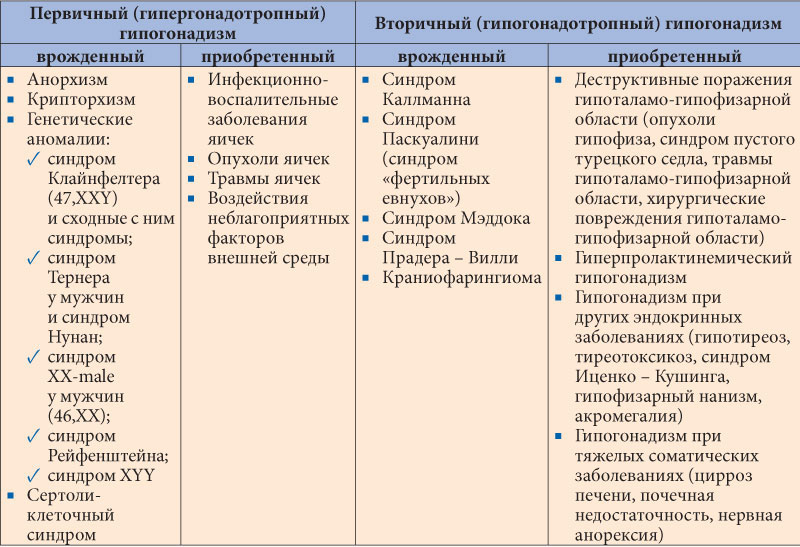
Патофизиология
Синдром Кальмана и иные формы гипогонадотропного гипогонадизма классифицируются как нарушения работы гипофиза или эндокринные нарушения. Если конечным результатом является нарушение функции гипофиза и развитие вторичных половых признаков, первопричина нарушения локализуется между двумя эндокринными железами головного мозга.
Гипоталамус и гипофиз могут рассматриваться как контрольные станции для всей гормональной активности организма. Эти железы секретируют различные гормоны с различным действием на организм. Синдром Кальмана/гипогонадотропный гипогонадизм развивается в результате нарушения коммуникации между гипоталамусом и гипофизом только относительно одного набора гормонов. Все иные эффекты гипоталамуса и гипофиза остаются незатронутыми.
В норме гипоталамус высвобождает гормон, который называется гонадотропин-рилизинг гормон (ГнРГ). ГнРГ высвобождается из гипоталамуса пульсативным способом через определенные промежутки времени в течение суток с помощью гипофизарной портальной системы и действует на аденогипофиз, вызывая высвобождение им двух гонадотропных гормонов. Эти гормоны представляют собой лютеинизирующий гормон (ЛГ) и фолликулостимулирующий гормон (ФСГ), которые оказывают прямое влияние на семенники у мужчин и яичники у женщин. ЛГ и ФСГ являются существенными факторами стимулирования развития вторичных половых признаков, наблюдаемых в пубертатный период, а также обеспечения нормальной половой функции у мужчин и женщин, включая поддержание корректного уровня половых стероидов: тестостерона у мужчин и эстрогена у женщин.
Эти гормоны представляют собой лютеинизирующий гормон (ЛГ) и фолликулостимулирующий гормон (ФСГ), которые оказывают прямое влияние на семенники у мужчин и яичники у женщин. ЛГ и ФСГ являются существенными факторами стимулирования развития вторичных половых признаков, наблюдаемых в пубертатный период, а также обеспечения нормальной половой функции у мужчин и женщин, включая поддержание корректного уровня половых стероидов: тестостерона у мужчин и эстрогена у женщин.
При синдроме Кальмана/гипогонадотропном гипогонадизме высвобождение ГнРГ либо полностью блокируется, либо значительно снижается. ГнРГ высвобождается гипоталамусом посредством специализированных нервных клеток или нейронов. В ходе развития головного мозга в первые 10 недель нейроны, секретирующие ГнРГ, мигрируют из своего первоначального источника и завершают развитие внутри гипоталамуса.
Нейроны, секретирующие ГнРГ, берут начало в области развивающегося головного мозга, называемого обонятельная плакода, затем они проходят через ситовидную пластинку и попадают в структуру, называемую обонятельная луковица, где генерируется ощущение обоняния. Оттуда они мигрируют в структуру, которая затем становится гипоталамусом. Любые проблемы в развитии обонятельной луковицы препятствуют прохождению нейронов, секретирующих ГнРГ, через нее. Если нейроны, секретирующие ГнРГ, не достигают гипоталамуса, ГнРГ не высвобождается, и, в свою очередь, не высвобождаются ФСГ или ЛГ, что приводит к нарушению полового созревания и недостаточной выработке тестостерона у мужчин и эстрогена у женщин.
Оттуда они мигрируют в структуру, которая затем становится гипоталамусом. Любые проблемы в развитии обонятельной луковицы препятствуют прохождению нейронов, секретирующих ГнРГ, через нее. Если нейроны, секретирующие ГнРГ, не достигают гипоталамуса, ГнРГ не высвобождается, и, в свою очередь, не высвобождаются ФСГ или ЛГ, что приводит к нарушению полового созревания и недостаточной выработке тестостерона у мужчин и эстрогена у женщин.
При синдроме Кальмана обонятельная луковица отсутствует или не развита полностью, что дополнительно выражается в отсутствии обоняния (аносмия) или значительном снижении обонянии (гипосмия). При иных формах гипогонадотропного гипогонадизма, обонятельная луковица развивается корректно, так что обоняние является нормальным, но миграция нейронов, секретирующих ГнРГ, прерывается в другом месте и также прпятствует своевременному высвобождению ГнРГ.
Однако данная связь между обонятельной луковицей и нейронами, секретирующими ГнРГ, не так прочна, как считалось ранее.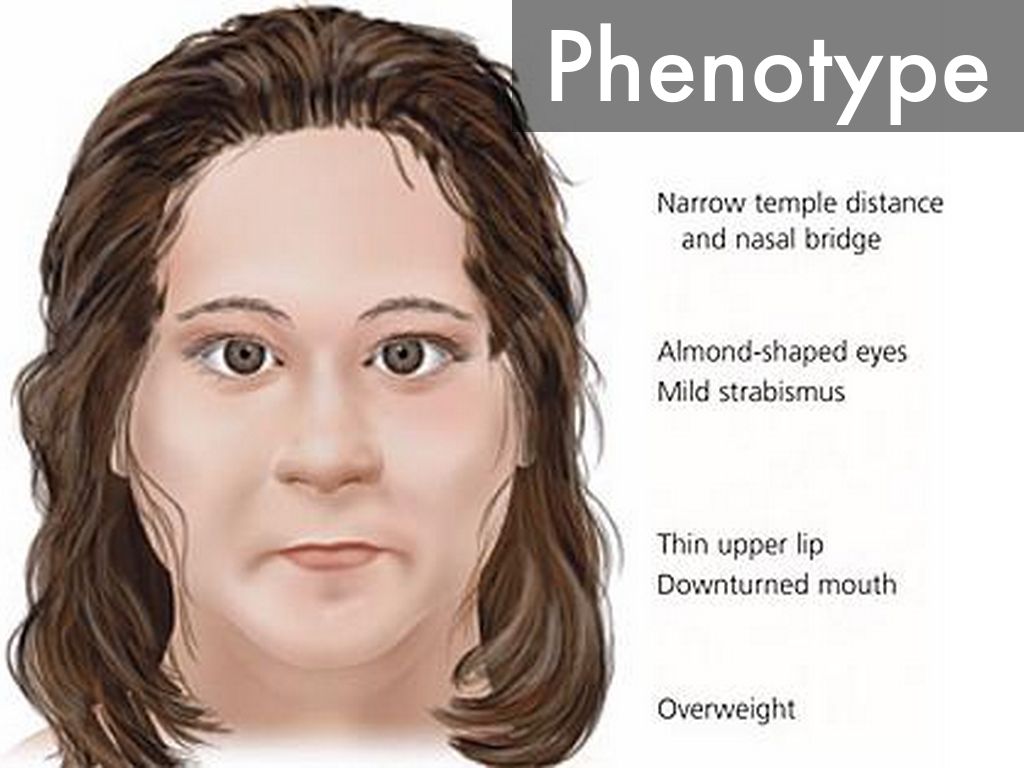 Большинство пациентов с врожденной аносмией с отсутствием обонятельной луковицы не имеют дефицита репродуктивных гормонов12). В одной и той же семье с теми же генами, отвечающими за развитие синдрома Кальмана/гипогонадотропного гипогонадизма, некоторые члены семьи могут иметь синдром Кальмана с отсутствием обоняния, другие могут иметь идиопатический гипогонадотропный гипогонадизм с наличием чувства обоняния, а другие могут иметь изолированную аносмию без дефицита гормонов.
Большинство пациентов с врожденной аносмией с отсутствием обонятельной луковицы не имеют дефицита репродуктивных гормонов12). В одной и той же семье с теми же генами, отвечающими за развитие синдрома Кальмана/гипогонадотропного гипогонадизма, некоторые члены семьи могут иметь синдром Кальмана с отсутствием обоняния, другие могут иметь идиопатический гипогонадотропный гипогонадизм с наличием чувства обоняния, а другие могут иметь изолированную аносмию без дефицита гормонов.
Большинство генов, которые были связаны с развитием синдрома Кальмана или гипогонадотропного гипогонадизма, играют роль либо в образовании, либо в миграции, либо в активности этих нейронов, секретирующих ГнРГ, и их способности стимулировать выработку ФСГ и ЛГ.
В работе, опубликованной в 2007 году Питтлаудом и др., предлагается возможная дигенная модель синдрома Кальмана и иных форм гипогонадотропного гипогонадизма. Возможность наличия дефектов двух отдельных генов, действующих совместно, может объяснять некоторую вариацию симптомов, наблюдаемых при синдроме Кальмана, даже у членов одной семьи.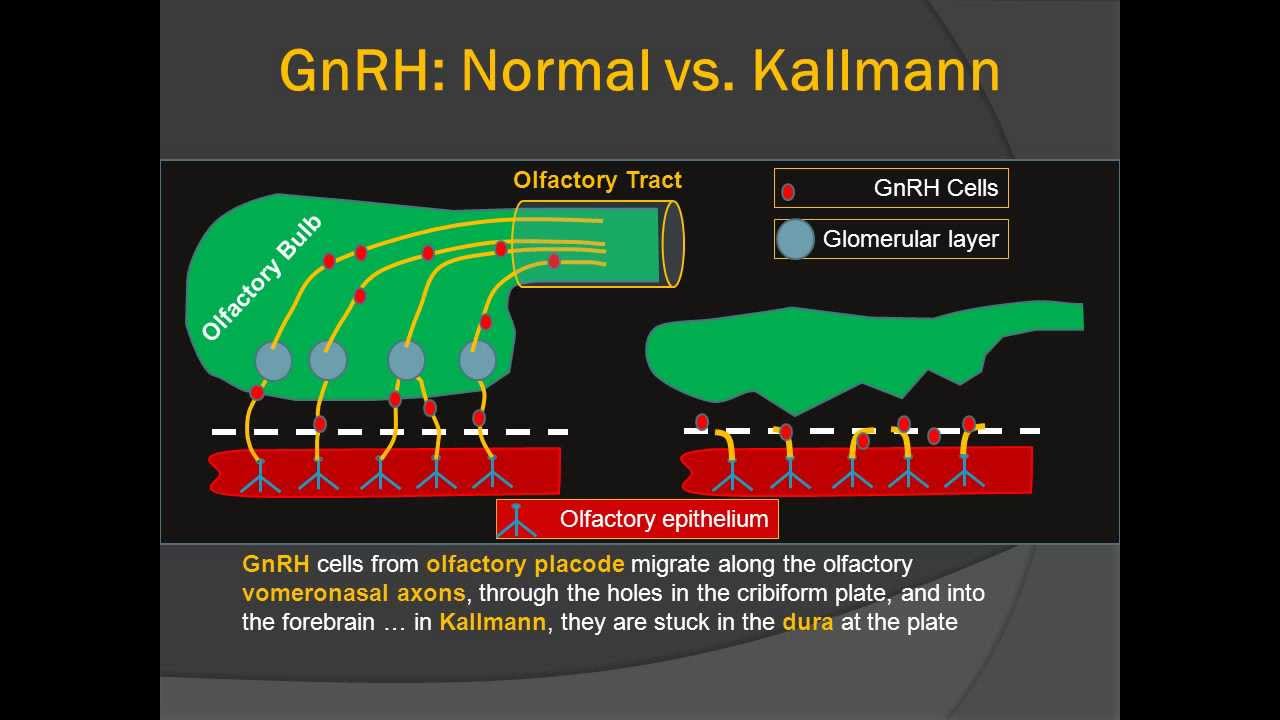
Генетика синдрома Кальмана и иных форм гипогонадотропного гипогонадизма все еще не достаточно изучена, и примерно 70% случаев имеют неизвестное генетическое происхождение.
В дальнейшем исследовании, опубликованном Анной Митчел и др., подчеркивается факт, что число генетических локусов, которые, как известно, вызывают развитие синдрома Кальмана и гипогонадотропного гипогонадизма, было увеличено до десяти. В работе освещен широкий спектр физических симптомов – связанных и не связанных с репродуктивной функцией – которые могут наблюдаться при синдроме Кальмана и гипогонадотропном гипогонадизме, даже внутри одной семьи.
Фенотипический спектр при синдроме Кальмана/гипогонадотропном гипогонадизме
Синдром Кальмана и гипогонадотропный гипогонадизм не являются четко выраженными заболеваниями. В каждом случае может наблюдаться различный диапазон симптомов с различной тяжестью. У каждого члена семьи не всегда наблюдается одинаковая степень проявления симптомов. Случаи синдрома Кальмана/гипогонадотропного гипогонадизма могут быть поделены на две разные группы в зависимости от мутаций вовлеченных генов. Серьезность может варьировать от полного отсутствия полового созревания с аносмией до небольшой задержки полового созревания с аносмией или без таковой.
Серьезность может варьировать от полного отсутствия полового созревания с аносмией до небольшой задержки полового созревания с аносмией или без таковой.
Классический гипогонадотропный гипогонадизм
Этот тип гипогонадотропного гипогонадизма присутствует с рождения и наблюдается в течение всей жизни. Примерно в двух третях случаев классического гипогонадотропного гипогонадизма присутствует низкий уровень пульсативного высвобождения ГнРГ из гипоталамуса, что приводит к частичному половому созреванию, тогда как примерно у одной трети пациентов наблюдается отсутствие выброса ГнРГ и полового созревания.
Симптомы, не связанные с репродуктивной функцией, упомянутые ранее в данной статье, присутствуют примерно в половине случаев. Наиболее распространенными из них являются аносмия, которая позволяет отличить синдром Кальмана от иных форм гипогонадотропного гипогонадизма. У мужчин с классическим гипогонадотропным гипогонадизмом в анамнезе может наблюдаться неопущение яичек и/или микропенис.
Было доказано, что данный тип гипогонадотропного гипогонадизма вызван мутациями полиаллельных генов у мужчин и женщин и аутосомно-доминантными мутациями и сцепленными с Х-хромосомой рецессивными мутациями у мужчин, упомянутыми ранее в данной статье.
Гипогонадотропный гипогонадизм с началом во взрослом возрасте
Данный тип гипогонадотропного гипогонадизма наблюдается только у мужчин. Гипоталамно-гипофизарно-надпочечниковая ось функционирует нормально при рождении и в течение взрослой жизни с нормальным половым созреванием.
Гипоталамно-гипофизарно-надпочечниковая ось затем либо полностью перестает функционировать, либо высвобождение ГнРГ снижается до очень низкого уровня, без очевидных причин во взрослом возрасте, таких как опухоль гипофиза. Это приводит к снижению уровня тестостерона и бесплодию. Данный тип гипогонадотропного гипогонадизма не связан с симптомами, не относящимися к репродуктивной функции, и было показано, что он вызван мутациями моноаллельных генов.
Обратимый синдром Кальмана/гипогонадотропный гипогонадизм
Данный тип синдрома Кальмана/гипогонадотропного гипогонадизма является классической формой и вначале проявляется в течение всей жизни, но в некоторый момент взрослой жизни гипоталамно-гипофизарно-надпочечниковая ось восстанавливает свою нормальную функцию, и уровни ГнРГ, ЛГ и ФСГ возвращаются к нормальному значению. Это наблюдается в 10% случаев, преимущественно у пациентов с синдромом Кальмана, чем с гипогонадотропным гипогонадизмом, и только у пациентов, которые подвергались некоторой форме замещающей терапии тестостероном. Такой результат в норме обнаруживается только при увеличении объема семенников при отдельной терапии тестостероном, и уровень тестостерона возвращается к нормальному значению при прекращении лечения.
Такой тип синдрома Кальмана/гипогонадотропного гипогонадизма редко встречается у мужчин, которые имеют в анамнезе неопущение яичек и/или микропенис, и было доказано, что он вызван мутациями моноаллельных генов.
Гипоталамическая аменорея
Данный тип гипогонадотропного гипогонадизма наблюдается у женщин, когда функция гипоталамно-гипофизарно-надпочечниковой оси подавляется в ответ на физический или психологический стресс или недостаток питания. Он является обратимым при устранении фактора стресса.
Данный тип гипогонадотропного гипогонадизма не связан с симптомами, не относящимися к репродуктивной функции, и было показано, что он вызван мутациями моноаллельных генов. Исследования предполагают, что, возможно, на какой-то ранней стадии развития организма мужчины присутствовало эволюционное преимущество для этих генов, когда отсутствие репродуктивности для женщин было преимущественным при недостаточности пищи в сообществе.
Было доказано, что данный тип гипогонадотропного гипогонадизма был вызван мутациями моноаллельных генов.
Нормальное половое созревание без симптомов, не связанных с репродуктивностью
При данном типе гипогонадотропного гипогонадизма половое созревание наступает нормально или слега задерживается, также возможно наличие симптомов, не связанных с репродуктивной функцией, таких как аносмия. Было доказано, что данный тип гипогонадотропного гипогонадизма был вызван мутациями моноаллельных генов.
Лечение
Лечение синдрома Кальмана и иных форм гипогонадотропного гипогонадизма можно разделить на две разные группы13):
Гормонозаместительная терапия
Целью гормонозаместительной терапии для мужчин и женщин является обеспечение нормального физиологического уровня гормонов в крови (тестостерона для мужчин и эстрогена/прогестерона для женщин) согласно возрасту пациента. Сначала лечение вызывает большинство физических и психологических изменений в пубертатном периоде, с основным исключением отсутствия развития семенников у мужчин и овуляции у женщин.
После достижения оптимального физического развития гормонозаместительная терапия для мужчин продолжается для поддержания нормальной андрогенной функции, такой как либидо, развития мышц, уровня энергии, роста волос и половой функции. У женщин различные типы гормонозаместительной терапии либо приводят к началу менструального цикла, либо нет, в зависимости от предпочтений пациентки. Гормонозаместительная терапия очень важна и для мужчин, и для женщин для поддержания плотности костной ткани и снижения риска раннего начала остеопороза.
Лечение бесплодия для мужчин и женщин также включает гормонозаместительную терапию.
Существует ряд различных препаратов, доступных для гормонозаместительной терапии для мужчин и женщин, большинство их них используется для стандартных протоколов гормонозаместительной терапии, когда уровень гормонов падает позднее в течение жизни или после наступления менопаузы.
Для мужчин заместительная терапия тестостероном включает либо ежедневный прием капсул, либо использование геля или пластырей, инъекций раз в две недели, инъекций раз в три месяца или имплантатов раз в шесть месяцев. Гормонозаместительная терапия в виде таблеток/капсул редко приводит к достижению достаточного уровня тестостерона для мужчин с синдромом Кальмана/гипогонадотропным гипогонадизмом.
Инъекции тестостерона ундеканоата раз в три месяца стали весьма популярны в течение последних десяти лет. Впервые произведены компанией «Bayer pharmaceutical» под наименованием Небидо, Риндрон или Авид. В начале 2014 года Авид получил лицензию на использование в США Управлением по продовольствию и лекарствам, с формой выпуска в виде ампул 3 мл в отличие от стандартных ампул 4 мл, используемых по всему миру.
После первых двух инъекций с интервалом шесть месяцев, инъекции проводятся каждые три месяца и приводят к оптимальному уровню тестостерона в течение периода три месяца без значительного снижения уровня в конце цикла инъекций. Некоторым пациентам требуются инъекции только каждые шесть месяцев. Эти интервалы между инъекциями могут регулироваться в зависимости от ответа конкретного пациента.
Некоторое лечение может иметь более значительные результаты у некоторых пациентов, так что врач индивидуально назначает лечение для каждого пациента.
Не существует отдельно специалистов по гормонозаместительной терапии только для женщин с синдромом Кальмана/гипогонадотропным гипогонадизмом, но на рынке предлагается множество различных препаратов гормонозаместительной терапии, в том числе пероральные контрацептивы и стандартные препараты для лечения постменопаузы. Наиболее популярны таблетки, но также доступны пластыри. Поиск соответствующей гормонозаместительной терапии может потребовать применения метода некоторых проб и ошибок в зависимости от реакции организма на определенные препараты. Необходимо проконсультироваться со специалистом для обеспечения поддержания корректного уровня эстрогена и прогестерона каждый месяц в зависимости от того, требуется ли пациентке непрерывная гормонозаместительная терапия (без вызывания менструации) или опция отмены для вызывания кровотечения «менструального» типа. Такое кровотечение при отмене может быть ежемесячным или через более длительные промежутки времени в зависимости от типа используемых препаратов.
Лечение бесплодия
Лечение бесплодия у пациентов с синдромом Кальмана/гипогонадотропным гипогонадизмом требует консультации с квалифицированным специалистом в области репродуктивной эндокринологии. Существует хороший процент успешных результатов в достижении репродуктивной функции для пациентов с синдромом Кальмана/гипогонадотропным гипогонадизмом, некоторые эксперты называют значение до 70% при совместном использовании техник ЭКО. Однако существуют факторы, которые могут оказывать негативное влияние на репродуктивную функцию, и для определения успешности лечения необходима консультация со специалистом.
Лечение бесплодия включает введение гонадотропинов ЛГ и ФСГ для стимулирования выработки и высвобождения яйцеклеток и сперматозоидов. Женщины с синдромом Кальмана или гипогонадотропным гипогонадизмом имеют преимущество над мужчинами, поскольку их яичники в норме содержат нормальное количество яйцеклеток и иногда необходимо только пару недель лечения для достижения нормальной репродуктивной функции, тогда как у мужчин лечение бесплодия может занять до двух лет.
Потенциально новая форма лечения бесплодия прошла клинические испытания в 2013 и 2014 году компанией Merck Sharp & Dohme. В исследовании проводилась оценка формы ФСГ более длительного действия в форме корифоллитропина альфа. Инъекции вводились раз в две недели вместо стандартного введения два раза в неделю, предполагая, что это вызовет выработку сперматозоидов в течение нескольких месяцев, а не двух лет, что достигается при применении лекарственных препаратов, используемых в настоящее время. Конец исследования был запланирован на май 2015 года.
Хорионический гонадотропин человека (ХГЧ) иногда используется для стимуляции выработки тестостерона у мужчин и овуляции у женщин. У мужчин он действует аналогично ЛГ; для выработки тестостерона применяется стимуляция клеток Лейдига в семенниках. Распространенные торговые наименования препаратов содержанием ХГЧ включают Прегнил, Фоллютеин, Профази и Хорагон. Некоторым мужчинам с синдромом Кальмана или гипогонадотропным гипогонадизмом назначают ХГЧ исключительно для выработки тестостерона.
Менопаузальный гонадотропин человека (ЧМГ) используется для стимуляции выработки спермы у мужчин и для выработки нескольких яйцеклеток и овуляции у женщин. Он содержит смесь ЛГ и ФСГ. У мужчин ФСГ действует на сперму, стимулируя выработку клеток Сертоли в семенниках. Это может привести к увеличению семенников, но для достижения оптимального уровня выработки спермы может потребоваться от 6 месяцев до 2 лет. Распространенные торговые наименования препаратов ЧМГ включают Менопур, Меногон, Репронекс и Пергонал.
Также доступны очищенные формы ФСГ и иногда они используются вместе с ХГЧ вместо ЧМГ.
Инъекции могут вводиться внутримышечно, но в норме вводятся подкожно два или три раза в неделю.
И для мужчин, и для женщин альтернативным методом (но не широко доступным) является использование инфузионного дозатора для доставки ГнРГ (или ЛГРГ) пульсирующими дозами в течение суток. Это стимулирует высвобождение гипофизом естественного ЛГ и ФСГ для активирования функции семенников или яичников.
Купирование симптомов
Купирование симптомов было отмечено в 15-22% случаев14). Причины все еще исследуются, но они были отмечены и у мужчин, и у женщин.
Купирование симптомов, вероятно, обусловлено 14 известными дефектами генов, связанными с синдромом Кальмана/врожденным гипогонадотропным гипогонадизмом. Исследование предполагает отсутствие очевидного генного дефекта, показывая тенденцию к возможному исчезновению симптомов. Существует предположение, что при мутации TAC3 и TACR3 может наблюдаться немного больший шанс купирования симптомов, но для подтверждения этому недостаточно результатов. Вероятно, мутация KAL-1 как минимум, позволяет купировать симптомы, в настоящее время отмечен только единственный случай в медицинской литературе.
Купирование симптомов может не быть постоянным, и возможно развитие рецидива на любом этапе; в работе предполагается, что это может быть связано с уровнем стресса. В работе отмечается случай купирования симптомов, который привел к ремиссии, но впоследствии снова было достигнуто купирование симптомов, что явно предполагает связь с окружающей средой.
Очевидное купирование симптомов будет наблюдаться в том случае, если у мальчиков наблюдается развитие семенников, тогда как при отдельной терапии тестостероном или у женщин, которые достигли менструации или беременности при отсутствии терапии.
Кисс-пептин
Кисс-пептин представляет собой протеин, которые регулирует высвобождение ГнРГ из гипоталамуса, который, в свою очередь, регулирует высвобождение ЛГ и в меньшей степени ФСГ из аденогипофиза. Известно, что кисс-пептин и его связанные лиганды рецептора GPR54 вовлечены в регуляцию полового созревания.
Исследования показали, что кисс-пептин имеет потенциал в диагностике и лечении таких заболеваний как синдром Кальмана и врожденный гипогонадотропный гипогонадизм в некоторых случаях15).
В настоящее время существуют ограничения потенциальной терапевтической роли кисс-пептина при лечении синдрома Кальмана и врожденного гипогонадотропного гипогонадизма. Исследования показали, что высвобождение ГнРГ и, следовательно, ответ ЛГ и ФСГ действительно снижается со временем при продолжительном введении кисс-пептина, и должно быть достаточное количество нейронов, секретирующих ГнРГ, в гипоталамусе для эффективного действия кисс-пептина. Однако это текущая область исследований синдрома Кальмана и иных клинических состояний, связанных с дисрегуляцией гипоталамно-гипофизарно-надпочечниковой оси.
Неспособность выработки ЛГ и ФСГ при введении кисс-пептина через 24 часа может использоваться как диагностический маркер синдрома Кальмана и врожденного гипогонадотропного гипогонадизма, при которых наблюдается отсутствие нейронов, секретирующих ГнРГ.
Вероятно, существует роль в лечении кисс-пептином приобретенной аменореи у женщин и в некоторых случаях бесплодия.
Остеопороз
Одним из возможных побочных явлений синдрома Кальмана/гипогонадотропного гипогонадизма является увеличенный риск развития вторичного остеопороза или остеопении. Эстроген (у женщин) или тестостерон (у мужчин) являются существенными факторами поддержания плотности костной ткани. Дефицит тестостерона или эстрогена может повышать степень резорбции кости, в то же время снижая скорость формирования костной ткани. В целом это может привести к ослабленным хрупким костям, которые имеют большую склонность к переломам.
Даже при кратковременном дефиците эстрогена или тестостерона, например, при задержке в диагностике синдрома Кальмана/гипогонадотропного гипогонадизма, может наблюдаться повышенный риск развития остеопороза, но другие факторы риска также вовлечены, так что риск развития отличается для каждого пациента.
Пациенты с синдромом Кальмана/гипогонадотропным гипогонадизмом должны проходить сканирование плотности костной ткани не менее одного раза в пять лет, даже если они находятся на постоянной гормонозаместительной терапии. Этот интервал может быть сокращен до трех лет, если пациент уже находится в зоне риска (остеопения), или до одного года при наличии остеопороза.
Исследование плотности костной ткани известно как двухэнергетическая рентгеновская абсорбциометрия (ДЭРА). Это очень простое эффективное исследование, проведение которого занимает менее 15 минут. Оно включает проведение специализированного рентгенологического исследования позвоночника и бедренных костей, а также измерение минеральной плотности костной ткани и сравнение результатов со средними значениями молодого взрослого человека из общей популяции16).
Оптимальный уровень кальция и, вероятно, более важный уровень витамина D являются существенными для нормальной плотности костной ткани. Некоторым пациентам с синдромом Кальмана/гипогонадотропным гипогонадизмом после проверки уровня данных веществ могут быть прописаны таблетки с витамином D или инъекции для предотвращения ухудшения состояния. Роль витамина D для общего состояния здоровья находится под пристальным рассмотрением в настоящее время некоторыми исследователями, утверждающими, что дефицит витамина D преобладает во многих популяциях и может быть связан с иными заболеваниями.
Некоторым пациентам с тяжелой степенью остеопороза могут прописываться бисфосфонаты. Упражнения, особенно весовые нагрузки и упражнения с сопротивлением, снижают риск развития остеопороза.
Прогноз
Синдром Кальмана может оказывать значительное действие на жизнь пациента, однако влияние на каждого пациента различно. Возраст постановки диагноза и начала лечения играют ключевую роль в том, насколько хорошо отдельный пациент справляется с заболеванием. Для некоторых пациентов способность диагностировать заболевание и осознание, что они не единственные в мире с таким заболеванием, очень обнадеживает. С ключевым симптомом недостижения половозрелости в нормальном возрасте это может вызывать значительное влияние на социальное, а также физическое развитие человека.
Симптомы различаются для каждого пациента, но в целом мужчины с синдромом Кальмана имеют меньший размер пениса, чем среднее значение в популяции, что в дополнение к отсутствию развития семенников может влиять на уверенность в себе до такой степени, что половая активность даже не рассматривается такими пациентами. Большинство мужчин с синдромом Кальмана могут иметь нормальную активную половую жизнь, но уверенность, требуемая для достижения этого, иногда снижена у таких пациентов, и они имеют меньшую половую активность, чем другие люди их возраста.
Другим аспектом синдрома Кальмана является социальная изоляция. Поскольку заболевание является довольно редким, большинство пациентов с синдромом Кальмана никогда не встречаются и не общаются с пациентами с таким же диагнозом. Возможности встречаться и общаться с другими пациентами достаточно для помощи таким людям примириться со своим заболеванием17).
Лечение пациентов с синдромом Кальмана достаточно простое после постановки диагноза с использованием гормонозаместительной терапии и инъекций для лечения репродуктивной функции в некоторых случаях. Более значительной проблемой является постановка первоначального диагноза, особенно в решающем подростковом возрасте. Пациенты, которые в большей степени примиряются с синдромом Кальмана, это те, которым заболевание было диагностировано в возрасте до 16 лет, и было предпринято немедленное лечение. Проблемы могут возникать, если к пациентам слишком долго применяется выжидательная тактика в подростковом возрасте вместо консультирования эндокринолога, когда задержка полового созревания может быть дифференцирована от синдрома Кальмана.
Синдром Кальмана не является заболеванием с внешними видимыми проявлениями. После начала лечения и восстановления нормального уровня гормонов не наблюдается побочных явлений или пролбем продолжительности жизни, связанных с синдромом Кальмана.
В недавнем исследовании Эндрю Двайера18), выполненном при общении онлайн и при личном интервью с пациентами с синдромом Кальмана/гипогонадотропным гипогонадизмом, отмечаются некоторые вопросы и проблемы, которые испытывают пациенты в отношении своего диагноза и лечения. Одним из ключевых результатов данного исследования было мнение, что большинство пациентов получили пользу от возможности общаться непосредственно с пациентами с аналогичным диагнозом, лично и онлайн.
Эпидемиология
Распространенность идиопатического гипогонадотропного гипогонадизма и синдрома Кальмана была оценена как 1 случай на 10 000 рожденных мальчиков. Эта цифра взята из исследования 1973 года призывников французского иностранного легиона.
Поскольку отсутствует согласованное мнение относительно генетики синдрома Кальмана и гипогонадотропного гипогонадизма, сложно определить точную цифру встречаемости заболевания. Существует определенный уровень достоверности в цифрах, полученных из исследования 1973 года, поскольку цифры для синдрома Клайнфельтера тесно связаны с текущим принятым процентным соотношением.
Считается, что заболевание в пять раз чаще поражает мужчин, чем женщин, но для этого нет очевидных генетических причин, даже хотя два связанных дефекта гена сцеплены с Х-хромосомой.
При синдроме Кальмана и гипогонадотропном гипогонадизме наблюдаются все варианты генетического наследования, сцепленного с Х-хромосомой и аутосомного и доминантного или рецессивного типа наследования. Мутации гена KAL-1 в Х-хромосоме могут вызывать сцепленный с Х-хромосомой синдром Кальмана отдельно, но другие случаи синдрома Кальмана и идиопатического гипогонадотропного гипогонадизма показывают вероятные дигенные свойства с комбинацией дефектов двух генов.
Тогда как синдром Кальмана и идиопатический гипогонадотропный гипогонадизм в норме считаются наследственными заболеваниями, были отмечены другие формы, в том числе идиопатический гипогонадотропный гипогонадизм с началом во взрослом возрасте и потенциально обратимый идиопатический гипогонадотропный гипогонадизм. Случаи в рамках одной семьи не показывают одинаковый диапазон симптомов, вероятно, предполагая обширную генетическую природу заболеваний.
Также, вероятно, может отсутствовать очевидный семейный анамнез наследуемости (единичные или изолированные случаи), но любые случаи синдрома Кальмана или идиопатического гипогонадотропного гипогонадизма имеют потенциал наследования будущими поколениями.
Если отсутствуют сопутствующие заболевания, такие как нарушения сердечной и нервной деятельности, в норме нет влияния на ожидаемую продолжительность жизни.
Ранее начало остеопороза из-за низкого уровня тестостерона или эстрогена может вызвать проблемы, но в других случаях синдром Кальмана и идиопатический гипогонадотропный гипогонадизм при правильном лечении не связаны с увеличением уровня смертности.
Европейское сообщество
В 2011 году группа под руководством профессора Нелли Питтлауд и Эндрю Двайера Университетского госпиталя Лозанны в Швейцарии предложили создание Европейского обширного исследовательского сообщества, основанного Европейской организацией по сотрудничеству по проблемам науки, техники и технологиям, которое будет представлять платформу для практикующих врачей и исследователей для сотрудничества в области исследования заболеваний дефицита ГнРГ, в том числе синдрома Кальмана и иных форм гипогонадотропного гипогонадизма. Первая встреча по работе организации по сотрудничеству по проблемам науки, техники и технологиям BM1105 была проведена в Брюсселе в феврале 2013 года. Веб-сайт сообщества (www.gnrhnetwork.eu) был запущен в марте 2013 года и содержит данные для пациентов с заболеваниями дефицита ГнРГ и современные клинические руководства для диагностики и лечения таких заболеваний.
:Tags
Читать еще: Валацикловир , Гамма-оксимасляная кислота (ГОМК) , Глицерол (глицерин) , Мускатный орех , Рамиприл ,
Список использованной литературы:
1)
MacColl G, Bouloux P, Quinton R (2002). «Kallmann syndrome: adhesion, afferents, and anosmia». Neuron 34 (5): 675–8. doi:10.1016/S0896-6273(02)00720-1. PMID 12062015.
2)
Valdes-Socin, H (2014). «Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes». Frontiers in Endocrinology 5: 109. doi:10.3389/fendo.2014.00109.
3)
Guo CY, Jones TH, Eastell R (1997). «Treatment of isolated hypogonadotropic hypogonadism effect on bone mineral density and bone turnover.». J Clin Endocrinol Metab. 82 (2): 658–65. doi:10.1210/jcem.82.2.3758. PMID 9024272.
4)
synd/2549 at Who Named It?
5)
Maestre de San Juan, Aureliano (1856). «Teratolagia: falta total de los nervios olfactorios con anosmia en un individuo en quien existia una atrofia congenita de los testiculos y miembro viril.». El Siglo Médico 3: 211–221.
6)
Brian Brett at abcbookworld
7)
Bouvattier C et al. (2011). «Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism.». Nature Reviews Endocrinology 18;8 (3): 172–82. doi:10.1038/nrendo.2011.164. PMID 22009162.
8)
Male Hypogonadism. Friedrich Jockenhovel. Uni-Med Science. 2004. ISBN 3-89599-748-X. Chapter 3. Diagnostic work up of hypogonadism.
9)
Young J (2012). «Approach to the Male Patient with Congenital Hypogonadotropic Hypogonadism». J Clin Endocrinol Metab. 97 (3): 707–718. doi:10.1210/jc.2011-1664. PMID 22392951.
10)
Quinton R. (2005). «Adolescent development: advice in ABC of adolescence is potentially misleading.». BMJ. 330 (7494) (Apr 2): 789. doi:10.1136/bmj.330.7494.789. PMC 555895. PMID 15802728.
11)
Dunkel L, Quinton R. (2014). «Transition in endocrinology: induction of puberty.». Eur J Endocrinol. 170 (6): R229–39. doi:10.1530/EJE-13-0894. PMID 24836550.
12)
Yousem DM, Geckle RJ, Bilker W, McKeown DA, Doty RL. (1996). «MR evaluation of patients with congenital hyposmia or anosmia». AJR Am J Roentgenol. 166 (2): 439–43. doi:10.2214/ajr.166.2.8553963. PMID 8553963.
13)
Han TS, Bouloux PM (2010). «What is the optimal therapy for young males with hypogonadotropic hypogonadism?». Clin Endocrinol (Oxf) 72 (6): 731–7. doi:10.1111/j.1365-2265.2009.03746.x. PMID 19912242.
14)
Sidhoum VF, Chan YM, Lippincott MF et al. (2013). «Reversal and Relapse of Hypogonadotropic Hypogonadism: Resilience and Fragility of the Reproductive Neuroendocrine System.». J Clin Endocrinol Metab. Jan.
15)
Jyothis T. George and Stephanie B. Seminara. «Kisspeptin and the Hypothalamic Control of Reproduction: Lessons from the Human». Endocrinology 153 (11): 5130–5136. doi:10.1210/en.2012-1429. PMC 3473216.
16)
Laitinen EM, Hero M, Vaaralahti K, Tommiska J, Raivio T (2012). «Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism.». Int J Androl. 35 (4): 534–40. doi:10.1111/j.1365-2605.2011.01237.x. PMID 22248317.
17)
Caronia LM, Martin C, Welt CK, Sykiotis GP et al. (2011). «A genetic basis for functional hypothalamic amenorrhea.». N Engl J Med. 20;364 (3): 215–25. doi:10.1056/NEJMoa0911064. PMC 3045842. PMID 21247312.
18)
Dwyer AA, Quinton R, Morin D, Pitteloud N. (2014). «Identifying the unmet health needs of patients with congenital hypogonadotropic hypogonadism using a web-based needs assessment: implications for online interventions and peer-to-peer support.». Orphanet J Rare Dis. 83 (9): 1750–1172. doi:10.1186/1750-1172-9-83. PMID 24915927.
синдром_кальмана.txt · Последние изменения: 2019/08/06 13:03 — nataly
Синдром Каллмана: MedlinePlus Genetics
С синдромом Каллмана связаны изменения более чем 20 генов. Среди наиболее частых причин этого состояния — мутации в гене ANOS1 , CHD7 , FGF8 , FGFR1 , PROK2 или PROKR2 . В некоторых случаях у пораженных людей есть мутации более чем в одном из этих генов. Кроме того, исследователи выявили мутации в других генах, которые могут способствовать развитию и особенностям синдрома Каллмана, но вряд ли сами по себе могут вызвать болезнь.
Гены, связанные с синдромом Каллмана, играют роль в развитии определенных областей мозга до рождения. Хотя некоторые из их конкретных функций неясны, эти гены, по-видимому, участвуют в формировании и движении (миграции) группы нервных клеток, которые специализируются на обработке обоняния (обонятельные нейроны). Эти нервные клетки берут начало в развивающемся носу, а затем мигрируют вместе в структуру в передней части мозга, называемую обонятельной луковицей, которая имеет решающее значение для восприятия запахов.Исследования показывают, что гены, связанные с синдромом Каллмана, также участвуют в миграции нейронов, вырабатывающих гормон, называемый гонадотропин-рилизинг-гормоном (ГнРГ). Подобно обонятельным нейронам, нейроны, продуцирующие ГнРГ, мигрируют из развивающегося носа в переднюю часть мозга. ГнРГ контролирует выработку нескольких гормонов, управляющих половым развитием до рождения и в период полового созревания. Эти гормоны важны для нормальной функции яичников у женщин и яичек у мужчин.
Исследования показывают, что мутации в генах, связанных с синдромом Каллмана, нарушают миграцию обонятельных нервных клеток и нервных клеток, продуцирующих GnRH, в развивающемся головном мозге.Если обонятельные нервные клетки не распространяются на обонятельную луковицу, обоняние человека будет нарушено или отсутствует. Неправильное размещение нейронов, продуцирующих гонадолиберин, в головном мозге предотвращает выработку других половых гормонов, что мешает нормальному половому развитию и вызывает характерные черты гипогонадотропного гипогонадизма. Неясно, как мутации генов приводят к другим признакам и симптомам, которые могут возникать при синдроме Каллмана. Поскольку особенности этого состояния различаются у разных людей, этому заболеванию, вероятно, способствуют дополнительные генетические факторы и факторы окружающей среды.
Вместе мутации в известных генах составляют около 30 процентов всех случаев синдрома Каллмана. В случаях без мутации в одном из идентифицированных генов причина состояния неизвестна. Исследователи ищут дополнительные генетические изменения, которые могут вызвать это заболевание.
Синдром Каллмана — NORD (Национальная организация по редким заболеваниям)
Topaloglu AK, Tello JA, Kotan LD, et al. Инактивация мутации KISS1 и гипогонадотропного гипогонадизма. Медицинский журнал Новой Англии.2012; 366 (7): 629-35.
Джордж JT, Seminara SB. Кисспептин и гипоталамический контроль репродукции: уроки человека. Эндокринология. 2012; 153 (11): 5130-6.
Gianetti E, Hall JE, Au MG, et al. Когда генетическая нагрузка не коррелирует с фенотипическим спектром: уроки рецептора GnRH (GNRHR). Журнал клинической эндокринологии и метаболизма. 2012; 97 (9): E1798-807.
Hanchate NK, Giacobini P, Lhuillier P, et al. SEMA3A, ген, участвующий в поиске аксонального пути, мутирует у пациентов с синдромом Каллмана.PLoS генетика. 2012; 8 (8): e1002896.
Янг Дж., Метэй С., Булиганд Дж. И др. Делеция SEMA3A в семье с синдромом Каллмана подтверждает роль семафорина 3A в половом созревании человека и развитии обонятельной системы. Hum Reprod. 2012; 27 (5): 1460-5.
Hanchate NK, Giacobini P, Lhuillier P, et al. SEMA3A, ген, участвующий в поиске аксонального пути, мутирован у пациентов с синдромом Каллмана. PLoS Genet. 2012; 8 (8): e1002896.
Forsythe E, Beales PL. Синдром Барде-Бидля. Eur J Hum Genet.2012. DOI: 10.1038 / ejhg.2012.115. [Epub перед печатью]
Лайтинен Е.М., Вааралахти К., Томмиска Дж. И др. Заболеваемость, фенотипические особенности и молекулярная генетика синдрома Каллмана в Финляндии. Orphanet J Rare Dis. 2011; 6: 41.
Карибони А., Дэвидсон К., Ракич С., Магги Р., Парнавелас Дж. Г., Рурберг С. Дефектная миграция нейронов гонадотропин-рилизинг-гормона у мышей, лишенных передачи сигналов SEMA3A через NRP1 и NRP2: последствия для этиологии гипогонадотропного гипогонадизма. Hum Mol Genet.2011; 20 (2): 336-44.
Шоу Н.Д., Семинара С.Б., Велт СК и др. Расширение фенотипа и генотипа дефицита женского гонадолиберин. Журнал клинической эндокринологии и метаболизма. 2011; 96 (3): E566-76.
Balasubramanian R, Crowley WF Jr. Изолированный дефицит GnRH: модель заболевания, служащая уникальной призмой в системной биологии нейронной сети GnRH. Mol Cell Endocrinol. 2011; 346 (1-2): 4-12.
Чан Ю.М., Бродер-Фингерт С., Парасхос С. и др. Фенотипы с дефицитом ГнРГ у людей и мышей с гетерозиготными вариантами в KISS1 / Kiss1.Журнал клинической эндокринологии и метаболизма. 2011; 96 (11): E1771-81.
Balasubramanian R, Plummer L, Sidis Y, et al. Загадки пути прокинетицина 2 в репродукции человека. Mol Cell Endocrinol. 2011; 346 (1-2): 44-50.
Quaynor SD, Kim HG, Cappello EM, et al. Распространенность дигенных мутаций у пациентов с нормосмическим гипогонадотропным гипогонадизмом и синдромом Каллмана. Плодовитость и бесплодие. 2011; 96 (6): 1424-30 e6.
Сюй Н., Ким Х.Г., Бхагават Б. и др.Мутации носового эмбрионального фактора LHRH (NELF) у пациентов с нормосмическим гипогонадотропным гипогонадизмом и синдромом Каллмана. Плодовитость и бесплодие. 2011; 95 (5): 1613-20 e1-7.
Торнберг Дж., Сикиотис Г.П., Киф К. и др. Гепарансульфат-6-O-сульфотрансфераза 1, ген, участвующий во внеклеточных модификациях сахара, мутирует у пациентов с идиопатическим гипогонадотропным гипогонадизмом. Труды Национальной академии наук Соединенных Штатов Америки. 2011; 108 (28): 11524-9.
Caronia LM, Martin C, Welt CK и др.Генетическая основа функциональной гипоталамической аменореи. N Engl J Med. 2011; 364 (3): 215-25.
Ким Х.Г., Ан Дж.В., Курт И., Ульманн Р., Ким Х.Т., Кулхарья А. и др. WDR11, белок WD, который взаимодействует с фактором транскрипции EMX1, мутирован при идиопатическом гипогонадотропном гипогонадизме и синдроме Каллмана. Американский журнал генетики человека. 2010; 87 (4): 465-79.
Sykiotis GP, Plummer L, Hughes VA, et al. Олигогенная основа изолированного дефицита гонадотропин-рилизинг-гормона. Труды Национальной академии наук Соединенных Штатов Америки.2010; 107 (34): 15140-4.
Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB, Crowley WF, Jr. Дефицит человеческого GnRH: уникальная модель заболевания, позволяющая раскрыть онтогенез нейронов GnRH. Нейроэндокринология. 2010; 92 (2): 81-99.
Sykiotis GP, Plummer L, Hughes VA, et al. Олигогенная основа изолированного дефицита гонадотропин-рилизинг-гормона. Proc Natl Acad Sci U S. A. 2010; 107 (34): 15140-4.
Bailleul-Forestier I, Gros C, Zenaty D, Bennaceur S, Leger J, de Roux N.Агенезия зубов у людей с синдромом Каллмана с мутациями FGFR1. Int J Paediatr Dent. 2010; 20 (4): 305-12.
Джанетти Э., Тассет С., Ноэль С.Д. и др. Мутации TAC3 / TACR3 демонстрируют преимущественную активацию высвобождения гонадотропин-рилизинг гормона нейрокинином B в неонатальной жизни с последующим обращением в зрелом возрасте. Журнал клинической эндокринологии и метаболизма. 2010; 95 (6): 2857-67.
Dode C, Hardelin JP. Синдром Каллмана. Eur J Hum Genet. 2009; 17 (2): 139-46.
Чан Ю.М., Бродер-Фингерт С, Семинара СБ.Репродуктивные функции кисспептина и Gpr54 в течение жизненного цикла мышей и людей. Пептиды. 2009; 30 (1): 42-8.
Bouligand J, Ghervan C, Tello JA и др. Изолированный семейный гипогонадотропный гипогонадизм и мутация GNRh2. N Engl J Med. 2009; 360 (26): 2742-8.
Чан Ю.М., де Гильебон А., Ланг-Муритано М., Пламмер Л., Черрато Ф., Циарас С. и др. Мутации GNRh2 у пациентов с идиопатическим гипогонадотропным гипогонадизмом. Proc Natl Acad Sci U S. A. 2009; 106 (28): 11703-8.
Топалоглу А.К., Рейманн Ф., Гуклу М. и др.Мутации TAC3 и TACR3 при семейном гипогонадотропном гипогонадизме раскрывают ключевую роль нейрокинина B в центральном контроле воспроизводства. Нат Жене. 2009; 41 (3): 354-8.
Falardeau J, Chung WC, Beenken A, et al. Снижение передачи сигналов FGF8 вызывает дефицит гонадотропин-рилизинг-гормона у людей и мышей. J Clin Invest. 2008; 118 (8): 2822-31.
Коул Л.В., Сидис Ю., Чжан С. и др. Мутации в генах прокинетицина 2 и рецептора прокинетицина 2 при дефиците гонадотропин-рилизинг гормона человека: молекулярная генетика и клинический спектр.Журнал клинической эндокринологии и метаболизма. 2008; 93 (9): 3551-9.
Ким Х.Г., Курт И., Лан Ф. и др. Мутации в CHD7, кодирующем белок-ремоделирующий хроматин, вызывают идиопатический гипогонадотропный гипогонадизм и синдром Каллмана. Am J Hum Genet. 2008; 83 (4): 511-9.
Питтелуд Н., Куинтон Р., Пирс С. и др. Дигенные мутации являются причиной вариабельных фенотипов при идиопатическом гипогонадотропном гипогонадизме. J Clin Invest. 2007; 117 (2): 457-63.
Георгопулос Н.А., Койка В., Галли-Цинопулу А. и др.Дисгенезия почек и дефекты гена KAL1 у пациентов со спорадическим синдромом Каллмана. Fertil Steril. 2007; 88 (5): 1311-7.
Dode C, Fouveaut C, Mortier G и др. Новые варианты последовательности FGFR1 при синдроме Каллмана и генетические доказательства того, что изоформа FGFR1c необходима для морфогенеза обонятельной луковицы и неба. Хум Мутат. 2007; 28 (1): 97-8.
Pitteloud N, Zhang C, Pignatelli D, et al. Мутация потери функции в гене прокинетицина 2 вызывает синдром Каллмана и нормосмический идиопатический гипогонадотропный гипогонадизм.Труды Национальной академии наук Соединенных Штатов Америки. 2007; 104 (44): 17447-52.
Doty RL. Офисные процедуры для количественной оценки обонятельной функции. Am J Rhinol. 2007; 21 (4): 460-73.
Райвио Т., Фалардо Дж., Дуайер А. и др. Лечение идиопатического гипогонадотропного гипогонадизма. N Engl J Med. 2007; 357 (9): 863-73.
Badano JL, Leitch CC, Ansley SJ и др. Рассечение эпистаза при олигогенном синдроме Барде-Бидля. Природа. 2006; 439 (7074): 326-30.
Pitteloud N, Acierno JS Jr, Meysing A и др. Мутации рецептора 1 фактора роста фибробластов вызывают как синдром Каллмана, так и нормосмический идиопатический гипогонадотропный гипогонадизм. Труды Национальной академии наук Соединенных Штатов Америки. 2006; 103 (16): 6281-6.
Dode C, Teixeira L, Levilliers J, et al. Синдром Каллмана: мутации в генах, кодирующих прокинетицин-2 и рецептор прокинетицина-2. PLoS Genet. 2006; 2 (10): e175.
Фаруки С., О’Рахилли С.Генетика ожирения у человека. Endocr Rev.2006; 27 (7): 710-18.
Grumbach MM. Окно возможностей: диагностика дефицита гонадотропинов у младенцев мужского пола. Журнал клинической эндокринологии и метаболизма. 2005; 90 (5): 3122-7.
Ким Х.Г., Херрик С.Р., Лемир Э. и др. Гипогонадотропный гипогонадизм и расщелина губы и неба, вызванные сбалансированной транслокацией, вызывающей гаплонедостаточность для FGFR1. J Med Genet. 2005; 42 (8): 666-72.
Пинто Г., Абади В., Меснаж Р. и др.Синдром CHARGE включает гипогонадотропный гипогонадизм и аномальное развитие обонятельных луковиц. Журнал клинической эндокринологии и метаболизма. 2005; 90 (10): 5621-6.
Кацанис Н. Олигогенные свойства синдрома Барде-Бидля. Hum Mol Genet. 2004; 13 ТУ № 1: Р65-71.
Eichers ER, Льюис Р.А., Катсанис Н., Лупски-младший. Триаллельное наследование: мост между менделевскими и многофакторными признаками. Ann Med. 2004; 36 (4): 262-72.
Сато Н., Кацумата Н., Кагами М. и др.Клиническая оценка и анализ мутаций синдрома Каллмана 1 (KAL1) и рецептора 1 фактора роста фибробластов (FGFR1 или KAL2) в пяти семьях и 18 спорадических пациентах. Журнал клинической эндокринологии и метаболизма. 2004; 89 (3): 1079-88.
Miura K, Acierno JS, Jr., Seminara SB. Характеристика гена фактора носового эмбрионального LHRH человека, NELF, и скрининг мутаций среди 65 пациентов с идиопатическим гипогонадотропным гипогонадизмом (IHH). J Hum Genet. 2004; 49 (5): 265-8.
Gonzalez-Martinez D, Kim SH, Hu Y, et al.Аносмин-1 модулирует передачу сигналов рецептора 1 фактора роста фибробластов в обонятельных нейробластах гонадотропин-рилизинг-гормона человека посредством гепарансульфат-зависимого механизма. J Neurosci. 2004; 24 (46): 10384-92.
de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Гипогонадотропный гипогонадизм из-за потери функции производного KiSS1 пептидного рецептора GPR54. Труды Национальной академии наук Соединенных Штатов Америки. 2003; 100 (19): 10972-6.
Seminara SB, Messager S, Chatzidaki EE, et al.Ген GPR54 как регулятор полового созревания. Медицинский журнал Новой Англии. 2003; 349 (17): 1614-27.
Dode C, Levilliers J, Dupont JM, et al. Мутации с потерей функции в FGFR1 вызывают аутосомно-доминантный синдром Каллмана. Генетика природы. 2003; 33 (4): 463-5.
Hebert JM, Lin M, Partanen J, Rossant J, McConnell SK. Передача сигналов FGF через FGFR1 необходима для морфогенеза обонятельной луковицы. Разработка. 2003; 130 (6): 1101-11.
Дженнингс Дж. Э., Костиган С., Рирдон В. Последовательность Мебиуса и гипогонадотропный гипогонадизм.Am J Med Genet A. 2003; 123A (1): 107-10.
Soussi-Yanicostas N, de Castro F, Julliard AK, Perfettini I, Chedotal A, Petit C. Аносмин-1, дефектный по Х-сцепленной форме синдрома Каллмана, способствует формированию аксональных ветвей из выходных нейронов обонятельной луковицы. Клетка. 2002; 109 (2): 217-28.
Salvi R, Gomez F, Fiaux M, et al. Прогрессирующее начало надпочечниковой недостаточности и гипогонадизма гипофизарного происхождения, вызванное сложной генетической перестройкой в DAX-1. Журнал клинической эндокринологии и метаболизма.2002; 87 (9): 4094-100.
Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Предикторы исхода долгосрочной терапии гонадолиберином у мужчин с идиопатическим гипогонадотропным гипогонадизмом. Журнал клинической эндокринологии и метаболизма. 2002; 87 (9): 4128-36.
Куинтон Р., Дюк В.М., Робертсон А. и др. Идиопатический дефицит гонадотропина: генетические вопросы, решаемые посредством фенотипической характеристики. Клин Эндокринол (Oxf). 2001; 55 (2): 163-74.
Kramer PR, Wray S.Новый ген, экспрессируемый в области носа, влияет на рост обонятельных аксонов и миграцию нейронов рилизинг-гормона лютеинизирующего гормона (LHRH). Genes Dev. 2000; 14 (14): 1824-34.
Hardelin JP, Julliard AK, Moniot B и др. Аносмин-1 является регионально ограниченным компонентом базальных мембран и интерстициальных матриц во время органогенеза: последствия для аномалий развития синдрома Каллмана, сцепленного с Х-хромосомой. Dev Dyn. 1999; 215 (1): 26-44.
Крамс М., Куинтон Р., Эшбернер Дж. И др.Синдром Каллмана: зеркальные движения, связанные с двусторонней гипертрофией кортикоспинального тракта. Неврология. 1999; 52 (4): 816-22.
Seminara SB, Hayes FJ, Crowley WF Jr. Дефицит гонадотропин-рилизинг-гормона у человека (идиопатический гипогонадотропный гипогонадизм и синдром Каллмана): патофизиологические и генетические соображения. Эндокринные обзоры. 1998; 19 (5): 521-39.
Layman LC, Коэн Д.П., Джин М. и др. Мутации в гене рецептора гонадотропин-рилизинг-гормона вызывают гипогонадотропный гипогонадизм.Генетика природы. 1998; 18 (1): 14-5.
Seminara SB FJH, Crowley WF Jr. Дефицит гонадотропин-высвобождающего гормона у человека (идиопатический гипогонадотропный гипогонадизм и синдром Каллмана): соображения патофизиологии и генетики. Эндокринные обзоры. 1998: 19 (5): 521-39.
Лафлин Г.А., Домингес СЕ, Йен СС. Нутриционные и эндокринно-метаболические аберрации у женщин с функциональной гипоталамической аменореей. J Clin Endocrinol Metab. 1998; 83 (1): 25-32.
Крамс М., Куинтон Р., Мейстон М.Дж. и др.Зеркальные движения при Х-сцепленном синдроме Каллмана. II. ПЭТ-исследование. Мозг. 1997; 120 (Pt 7): 1217-28.
де Ру Н., Янг Дж., Мисрахи М. и др. Семья с гипогонадотропным гипогонадизмом и мутациями рецептора гонадотропин-рилизинг-гормона. Медицинский журнал Новой Англии. 1997; 337 (22): 1597-602.
Nachtigall LB, Boepple PA, Pralong FP, Crowley WF Jr. Идиопатический гипогонадотропный гипогонадизм с началом у взрослых — излечимая форма мужского бесплодия. N Engl J Med. 1997; 336 (6): 410-5.
Waldstreicher J, Seminara SB, Jameson JL, Geyer A, et al. Генетическая и клиническая гетерогенность дефицита гонадотропин-рилизинг гормона у человека. J Clin Endocrinol Metab. 1996; 81 (12): 4388-95.
Барайцер М., Радж П. Синдром Мебиуса, аксональная нейропатия и гипогонадизм. Clin Dysmorphol. 1996; 5 (4): 351-5.
Кирк Дж. М., Грант Д. Б., Бессер Г. М. и др. Односторонняя аплазия почек при Х-сцепленном синдроме Каллмана. Clin Genet. 1994; 46 (3): 260-2.
Ругарли Э.И., Лутц Б., Куратани С.К. и др.Паттерн экспрессии гена синдрома Каллмана в обонятельной системе предполагает его роль в нацеливании на нейроны. Генетика природы. 1993; 4 (1): 19-26.
Whitcomb RW, Crowley WF Jr. Мужской гипогонадотропный гипогонадизм. Endocrinol Metab Clin North Am. 1993; 22 (1): 125-43.
Schwanzel-Fukuda M, Jorgenson KL, Bergen HT, Weesner GD, Pfaff DW. Биология нормальных нейронов рилизинг-гормона лютеинизирующего гормона во время и после их миграции из обонятельной плакоды. Эндокринные обзоры. 1992; 13 (4): 623-34.
del Castillo I, Cohen-Salmon M, Blanchard S, Lutfalla G, Petit C. Структура гена X-сцепленного синдрома Каллмана и его гомологичного псевдогена на Y-хромосоме. Генетика природы. 1992; 2 (4): 305-10.
Franco B, Guioli S, Pragliola A, et al. Ген, удаленный при синдроме Каллмана, имеет гомологию с адгезией нервных клеток и молекулами, определяющими путь аксонов. Природа. 1991; 353 (6344): 529-36.
Legouis R, Hardelin JP, Levilliers J, et al. Ген-кандидат для Х-сцепленного синдрома Каллмана кодирует белок, связанный с молекулами адгезии.Клетка. 1991; 67 (2): 423-35.
Мартин К., Санторо Н., Холл Дж., Филикори М., Вирман М., Кроули В. Ф. младший. Клинический обзор 15: Управление овуляторными расстройствами с помощью пульсирующего гонадотропин-рилизинг-гормона. J Clin Endocrinol Metab. 1990; 71 (5): 1081A-G.
Бик Д., Карри С.Дж., Макгилл Дж. Р., Шордерет Д. Ф., Букс Р. К., Мур К. М.. Младенец мужского пола с ихтиозом, синдромом Каллмана, точечной хондродисплазией и делецией хромосомы Xp. Американский журнал медицинской генетики. 1989; 33 (1): 100-7.
Баллабио А., Паренти Дж., Типпет П. и др.Х-связанный ихтиоз из-за дефицита стероид-сульфатазы, связанный с синдромом Каллмана (гипогонадотропный гипогонадизм и аносмия): взаимосвязь с Xg и клонированными последовательностями ДНК из дистального короткого плеча Х-хромосомы. Генетика человека. 1986; 72 (3): 237-40.
Conn PM, Marian J, McMillian M, et al. Действие гонадотропин-рилизинг-гормона в гипофизе: трехступенчатый механизм. Эндокринные обзоры. 1981; 2 (2): 174-85.
Уоррен МП. Влияние упражнений на прогрессирование полового созревания и репродуктивную функцию у девочек.J Clin Endocrinol Metab. 1980; 51 (5): 1150-7.
Боярский РМ. Контроль наступления полового созревания. Annu Rev Med. 1978; 29: 509-20.
Абид Ф, Холл Р., Хаджсон П., Вейзер Р. Синдром Мебиуса, периферическая невропатия и гипогонадотропный гипогонадизм. J Neurol Sci. 1978; 35 (2-3): 309-15.
Маршалл, Вашингтон, Таннер Дж. М.. Вариации характера пубертатных изменений у мальчиков. Arch Dis Child. 1970; 45 (239): 13-23.
Pasqualini RQ BE. Синдром гипоандрогенического происхождения при консервативном гаметогенезе: классификация инсуфицинского яичка.Rev Assoc Med. 1950 (46: 6).
Kallmann FJ, Schoenfeld W, Barrera S. Генетические аспекты первичного евнухоидизма. Am J Ment Defic. 1944; 48: 203-36.
Маэстре де Сан-Хуан А. Тератология: общая совокупная нервно-лучевая терапия, содержащаяся в индивидуальном анонимном существе, врожденная атрофия тестикулос и мимобро вириль. El Singlo Med. 1856; 3: 211-21.
ИНТЕРНЕТ
Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF. (Обновлено 18 августа 2011 г.). Синдром Каллмана.В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторское право, Вашингтонский университет, Сиэтл. 1997-2012 гг. Доступно на http://www.genetests.org. По состоянию на 2 ноября 2012 г.
Признаки и симптомы синдрома Каллмана
Синдром Каллмана, также известный как идиопатический гипогонадотропный гипогонадизм с аносмией, является врожденным заболеванием, которое проявляется аномально низкой выработкой гормонов, участвующих в половом развитии. Отличительные симптомы этого состояния включают задержку или отсутствие полового созревания, а также нарушение или отсутствие обоняния.
Поскольку нормальное течение полового созревания существенно различается у мужчин и женщин, по этой причине симптомы этого состояния сильно различаются между полами. Хотя нарушение обоняния очевидно у большинства пациентов, оно обычно не регистрируется как симптом и выявляется при диагностическом тестировании.
Отсроченное половое созревание
Отсутствие полового созревания — это клинический признак, который встречается как у мужчин, так и у женщин с этим заболеванием. Задержка или отсутствие полового созревания известна как гипогонадизм.Он включает снижение уровня половых гормонов (тестостерона у мужчин и эстрогена у женщин) и гонадотропинов (лютеинизирующий гормон и фолликулостимулирующий гормон). Признаки и симптомы задержки полового созревания зависят от пола человека.
Некоторые признаки, характерные для мужчин с синдромом Каллмана, присутствуют до полового созревания, такие как микропенис (аномально маленький половой член) и крипторхизм (неопущение яичек). Однако симптомы этого состояния становятся более очевидными в возрасте полового созревания, когда вторичные половые признаки, такие как снижение голоса, увеличение гениталий, рост волос на лобке и лице, не развиваются.Больные мужчины также могут сообщать о снижении либидо и иметь проблемы с эректильной дисфункцией.
Женщины, страдающие синдромом Каллманна, обычно начинают замечать признаки заболевания примерно в возрасте полового созревания. У них обычно не начинается менструация в обычном возрасте, а у некоторых может вообще не наступить половая зрелость. Также наблюдается незначительное развитие груди или его отсутствие.
Степень полового созревания в этом состоянии сильно варьируется как у мужчин, так и у женщин. Некоторые больные люди могут не проявлять никаких признаков вторичного полового развития.Другие демонстрируют частичные признаки полового созревания, которые не развиваются, как ожидалось, у здоровых людей.
Обоняние
Характерным признаком синдрома Каллмана является ослабление или отсутствие обоняния, известное как гипосмия или аносмия соответственно. Это отличает синдром от других типов гипогонадотропного гипогонадизма, которые не связаны с изменениями обоняния.
У многих пациентов неспособность чувствовать запахи не рассматривается как симптом, потому что они не осознают своего нарушения.По этой причине часто требуются диагностические тесты, чтобы идентифицировать аномалию и отличить синдром Каллмана от других подобных состояний.
Другие симптомы
Клинические признаки и симптомы синдрома Каллмана могут значительно различаться у разных пациентов. В дополнение к уже описанным основным симптомам, другие признаки и симптомы состояния могут включать:
- Односторонняя агенезия почек (недостаточность развития одной почки)
- Расщелина губы или неба
- Аномальные движения глаз
- Аномальное развитие зубов
- Потеря слуха
У некоторых пациентов может также наблюдаться бимануальная синкинезия, которая заключается в отражении движений рук на другой стороне тела.Это может усложнить выполнение задач, требующих точных движений рук, таких как игра на музыкальном инструменте.
Список литературы
Дополнительная литература
Синдром Каллмана | Детская больница Филадельфии
Синдром Каллмана сочетает в себе нарушение обоняния с гормональным расстройством, которое задерживает или предотвращает половое созревание.
Гормональное расстройство возникает из-за недоразвития определенных нейронов или нервов в головном мозге, которые сигнализируют о гипоталамусе.Без этих нейронов гипоталамус не может должным образом стимулировать выработку и высвобождение определенных гормонов гипофизом. При нормальном развитии гипоталамус выделяет выбросы гонадотропин-рилизинг-гормона (ГнРГ) в период полового созревания. Эти всплески гонадолиберина заставляют гипофиз вырабатывать гормоны, которые, в свою очередь, вызывают выброс мужских и женских половых гормонов гонадами (яичками и яичниками) и развитие сперматозоидов и яйцеклеток. При синдроме Каллмана гипоталамус не может секретировать эти выбросы гонадолиберина в утробе матери, в младенчестве и в период полового созревания.
Синдром Каллмана влияет на раннее развитие гипоталамуса и обоняния у человеческого эмбриона. Часть гипоталамуса, производящая гонадолиберин, изначально формируется как часть носа и мигрирует на ранних стадиях эмбрионального развития, чтобы присоединиться к остальной части гипоталамуса. При синдроме Каллмана и эта часть гипоталамуса, и нейроны головного мозга, обнаруживающие запах (обонятельные), не развиваются полностью.
Когда у человека характерен гормональный дефицит, характерный для синдрома Каллмана, но при этом он имеет нормальное обоняние, это состояние известно как нормосмический идиопатический гипогонадотропный гипогонадизм (НИЗГ).
Синдром Каллмана и нИЗГ являются генетическими заболеваниями. Они вызваны мутациями в любом из нескольких различных генов. Некоторые, но не все, из них были идентифицированы, и шаблоны наследования отображены.
Различные мутации генов, вызывающие синдром Каллмана и нИГГ, имеют разные паттерны наследования.
- Некоторые должны быть унаследованы от обоих родителей (аутосомно-рецессивное наследование). У этих типов носителями могут быть оба родителя, а это значит, что у них нет симптомов заболевания.
- Другие могут быть унаследованы от матери или отца (аутосомно-доминантное наследование).
- У других типов женщины являются носителями, а мужчины имеют (выражают) это заболевание (Х-сцепленное наследование). У этих типов заболевание может передаваться от матери к сыновьям, но не от отцов к сыновьям. Любой из родителей может передать заболевание дочери как носителю.
- Недавно исследователи определили четвертый образец наследования, при котором мутации более чем в одном гене могут объединяться, чтобы вызвать состояние (олигогенное наследование).
Исследователи работают над выявлением всех генетических мутаций, связанных с синдромом Каллмана и НИЗГ.
Основным симптомом синдрома Каллмана или нИГГ является задержка или неполное половое созревание. При синдроме Каллмана это сочетается с нарушением обоняния, состоянием, присутствующим с рождения, но часто не доводящимся до сведения врача, пока его не спросят об этом в ходе диагностики причины задержки полового созревания.
Другие характеристики также могут присутствовать у детей и подростков с синдромом Каллмана или нИЗГ.Сюда могут входить:
- Неопущенные или частично опущенные яички
- Маленький размер полового члена
- Дефекты лица, например заячья губа или нёбо
- Короткие пальцы рук или ног, особенно безымянный палец
- Развитие только одной почки
- Потеря слуха
- Дальтонизм
- Аномальные движения глаз
- Аномальное развитие зубов
- Зеркальные движения рук (бимануальная синкинезия), при которых движения одной руки отражаются движениями другой
Ваш врач обычно начинает с медицинского осмотра и с вопросов о любых симптомах, которые вы, возможно, заметили, особенно о тех, которые связаны с задержкой полового созревания и нарушением обоняния.Поскольку это наследственное заболевание, врач может также спросить о родственниках, у которых наблюдается задержка полового созревания или проблемы с фертильностью.
Если признаки указывают на возможность синдрома Каллмана или НИЗГ, дополнительные тесты могут включать:
- Анализы крови, специально предназначенные для определения уровня гормонов в периферических венах, происходящих из гипофиза
- Магнитно-резонансная томография (МРТ) гипоталамуса, гипофиза и носа для поиска анатомических аномалий
- Молекулярно-генетическое тестирование для поиска специфических генных мутаций
Синдром Каллмана и нИЗГ лечат заместительной гормональной терапией с применением специальных лекарств и доз, адаптированных к потребностям пациента.В первую очередь это лечение направлено на стимулирование полового созревания и поддержание нормального уровня гормонов. Позже лечение может быть изменено, чтобы вызвать фертильность.
Также могут потребоваться лекарства для восстановления здоровья костей, так как отсутствие гормонов, которое приводит к задержке полового созревания, также может привести к ослаблению плотности и прочности костей.
В долгосрочной перспективе заместительная гормональная терапия для мужчин может быть сокращена или периодически отменена, чтобы увидеть, не изменило ли их состояние и вырабатывает ли их нормальный уровень гормонов.Это изменение происходило у 10-15 процентов пациентов мужского пола в исследовании в одном центре.
Нейроэндокринный центр и Центр надпочечников и полового созревания детской больницы Филадельфии (CHOP) являются партнерами Программы сохранения фертильности больницы Пенсильванского университета. Эта команда репродуктивных эндокринологов специализируется на всех аспектах репродуктивной эндокринологии для взрослых мужчин и женщин с такими состояниями, как синдром Каллмана, включая варианты консультирования по вопросам бесплодия и индукции.Подробности направления к этой конкретной группе можно обсудить с эндокринным поставщиком в нейроэндокринном центре CHOP.
Когда лекарства необходимы для увеличения выработки гормонов, необходимы периодические контрольные тесты, чтобы гарантировать, что лечение продолжает работать эффективно. Уровни дозировки и комбинацию лекарств могут со временем корректироваться.
Детям с Каллманном часто требуется помощь многих педиатров.
Нейроэндокринный центр и Центр надпочечников и полового созревания при детской больнице Филадельфии предлагают семьям скоординированный и междисциплинарный подход к лечению синдрома Каллмана.Наша команда объединяет опыт детских эндокринологов, нейроонкологов, нейрохирургов, нейроофтальмологов, нейрорадиологов и патологов.
Все члены нашей команды имеют обширный опыт лечения сложных нейроэндокринных состояний, таких как синдром Каллмана.
синдром Каллмана | European Journal of Human Genetics
Maestre de San Juan A: Falta total de los nervios olfactorios con anosmía en un Individual en quien existia una atrofía congénita de los testículos y miembro viril. Siglo Medico 1856; 131 : 211.
Google ученый
Каллманн Ф.Дж., Шенфельд В.А., Баррера С.Е.: Генетические аспекты первичного евнухоидизма. Am J Умственная отсталость 1944; XLVIII : 203–236.
Google ученый
de Morsier G, Gauthier G: La dysplasie olfacto-génitale. Патол Биол 1963; 11 : 1267–1272.
CAS
PubMed
Google ученый
Нафтолин Ф., Харрис Г.В., Бобров М.: Влияние очищенного рилизинг-фактора лютеинизирующего гормона на нормальных и гипогонадотропных аносмических мужчин. Nature 1971; 232 : 496–497.
CAS
Статья
Google ученый
Сантен Р. Дж., Полсен, Калифорния: гипогонадотропный евнухоидизм. I. Клиническое исследование способа наследования. J Clin Endocrinol Metab 1972; 36 : 47–54.
Артикул
Google ученый
Либлих Дж. М., Роголь А. Д., Уайт Б. Дж., Розен С. В.: Синдром аносмии с гипогонадотропным гипогонадизмом (синдром Каллмана). Am J Med 1982; 73 : 506–519.
CAS
Статья
Google ученый
Доде С., Левилье Дж., Дюпон Дж. М. и др. : Мутации с потерей функции в FGFR1 вызывают аутосомно-доминантный синдром Каллмана. Nat Genet 2003; 33 : 463–465.
Артикул
Google ученый
Massin N, Pêcheux C, Eloit C et al. : Х-хромосомный синдром Каллмана: клиническая гетерогенность у трех братьев и сестер, несущих внутригенную делецию гена KAL-1. J Clin Endocrinol Metab 2003; 88 : 2003–2008.
CAS
Статья
Google ученый
Питтелуд Н., Асьерно Дж., Мейсинг А., Дуайер А., Хейс Ф., Кроули В.: обратимый синдром Каллмана, задержка полового созревания и изолированная аносмия, встречающиеся в одной семье с мутацией в гене FGFR1 . J Clin Endocrinol Metab 2005; 90 : 1317–1322.
CAS
Статья
Google ученый
Dodé C, Teixeira L, Levilliers J et al : синдром Каллмана: мутации в генах, кодирующих прокинетицин-2 и рецептор прокинетицина-2. PLoS Genetics 2006; 2 : 1648–1652.
Артикул
Google ученый
Pitteloud N, Acierno Jr JS, Meysing A et al : Мутации рецептора 1 фактора роста фибробластов вызывают как синдром Каллмана, так и нормосмический идиопатический гипогонадотропный гипогонадизм. Proc Natl Acad Sci USA 2006; 103 : 6281–6286.
CAS
Статья
Google ученый
Trarbach EB, Costa EM, Versiani B et al : Новые мутации рецептора 1 фактора роста фибробластов у пациентов с врожденным гипогонадотропным гипогонадизмом с аносмией и без нее. J Clin Endocrinol Metab 2006; 91 : 4006–4012.
CAS
Статья
Google ученый
Dodé C, Fouveaut C, Mortier G et al : Новые варианты последовательности FGFR1 при синдроме Каллмана и генетические доказательства того, что изоформа FGFR1c необходима для морфогенеза обонятельной луковицы и неба. Hum Mutat 2007; 28 : 97–98.
Артикул
Google ученый
Питтелуд Н., Чжан С., Пигнателли Д. и др. : Мутация потери функции в гене прокинетицина 2 вызывает синдром Каллмана и нормосмический идиопатический гипогонадотропный гипогонадизм. Proc Natl Acad Sci USA 2007; 104 : 17447–17452.
CAS
Статья
Google ученый
Hermanussen M, Sippell WG: Гетерогенность синдрома Каллмана. Clin Genet 1985; 28 : 106–111.
CAS
Статья
Google ученый
Хипкин Л., Кассон И., Дэвис Дж. Однояйцевые близнецы, не согласующиеся с синдромом Каллмана. J Med Genet 1990; 27 : 198–199.
CAS
Статья
Google ученый
Куинтон Р., Дюк В.М., Робертсон А и др. : Идиопатический дефицит гонадотропина: генетические вопросы, решаемые с помощью фенотипической характеристики. Clin Endocrinol (Oxf) 2001; 55 : 163–174.
CAS
Статья
Google ученый
Ribeiro RS, Vieira TC, Abucham J: Обратимый синдром Каллмана: отчет о первом случае с мутацией KAL1 и обзор литературы. Eur J Endocrinol 2007; 156 : 285–290.
CAS
Статья
Google ученый
Райвио Т., Фалардо Дж., Дуайер А. и др. : Обращение идиопатического гипогонадотропного гипогонадизма. N Engl J Med 2007; 357 : 863–873.
CAS
Статья
Google ученый
Конрад Б., Крибель Дж., Хетцель В.Д .: Наследственный бимануальный синкинез в сочетании с гипогонадотропным гипогонадизмом и аносмией у четырех братьев. J Neurol 1978; 218 : 263–274.
CAS
Статья
Google ученый
Schwankhaus JD, Currie J, Jaffe MJ, Rose SR, Sherins RJ: Неврологические находки у мужчин с изолированным гипогонадотропным гипогонадизмом. Неврология 1989; 39 : 223–226.
CAS
Статья
Google ученый
Quinton R, Duke VM, de Zoysa PA et al : нейрорадиология синдрома Каллмана: генотипический и фенотипический анализ. J Clin Endocrinol Metab 1996; 81 : 3010–3017.
CAS
PubMed
Google ученый
Söderlund D, Canto P, Méndez J: Идентификация трех новых мутаций в гене KAL1 у пациентов с синдромом Каллмана. J Clin Endocrinol Metab 2002; 87 : 2589–2592.
Артикул
Google ученый
Hardelin JP, Levilliers J, Young J et al. : делеции Xp22.3 при изолированном семейном синдроме Каллмана. J Clin Endocrinol Metab 1993; 76 : 827–831.
CAS
PubMed
Google ученый
Рирдон В. Синдром Каллмана, проявляющийся врожденным птозом у братьев. Clin Dysmorphol 2007; 16 : 207–208.
Артикул
Google ученый
Керцман К., Робинсон Д.Л., Шеринс Р.Дж., Шванкхаус Дж. Д., Мак-Клеркин Дж. В.: Нарушения визуального пространственного внимания у мужчин с зеркальными движениями, связанные с изолированным гипогонадотропным гипогонадизмом. Неврология 1990; 40 : 1057–1063.
CAS
Статья
Google ученый
Hill J, Elliott C, Colquhoun I. Аудиологические, вестибулярные и радиологические аномалии при синдроме Каллмана. Дж Ларингол Отол 1992; 106 : 530–534.
CAS
Статья
Google ученый
Коутсворт А.П., Вудхед К.Дж.: Кондуктивная потеря слуха, связанная с синдромом Каллмана. Дж Ларингол Отол 2002; 116 : 125–126.
CAS
PubMed
Google ученый
Сато Н., Кацумата Н., Кагами М. и др. : Клиническая оценка и анализ мутаций синдрома Каллмана 1 ( KAL1 ) и рецептора фактора роста фибробластов 1 ( FGFR1 или KAL2 ) в пяти семьи и 18 спорадических пациентов. J Clin Endocrinol Metab 2004; 89 : 1079–1088.
CAS
Статья
Google ученый
Wegenke JD, Uehling DT, Wear Jr JB et al : Семейный синдром Каллмана с односторонней аплазией почек. Clin Genet 1975; 7 : 368–381.
CAS
Статья
Google ученый
Hardelin J-P, Levilliers J, Blanchard S et al : Гетерогенность мутаций, ответственных за синдром Каллмана, связанный с X-хромосомой. Hum Mol Genet 1993; 2 : 373–377.
CAS
Статья
Google ученый
Кирк Дж., Грант Д., Бессер Г. и др. : Односторонняя почечная аплазия при Х-сцепленном синдроме Каллмана. Clin Genet 1994; 46 : 260–262.
CAS
Статья
Google ученый
Molsted K, Kjaer I, Giwercman A, Versterhauge S, Skakkebaek N: черепно-лицевая морфология у пациентов с синдромом Каллмана с расщелиной губы и неба и без нее. Расщелина неба и черепа J 1997; 34 : 417–424.
CAS
Статья
Google ученый
de Zegher F, Lagae L, Declerck D., Vinckier F: синдром Каллмана и задержка полового созревания, связанная с агенезом боковых резцов верхней челюсти. J Craniofac Genet Dev Biol 1995; 15 : 87–89.
CAS
PubMed
Google ученый
Tsai P-S, Gill J: Механизмы болезни: понимание Х-сцепленного и аутосомно-доминантного синдрома Каллмана. Нат Клин Практик Эндокринол Метаб 2006; 2 : 160–171.
CAS
Статья
Google ученый
Ballabio A, Andria G: Делеции и транслокации, вовлекающие дистальное короткое плечо X-хромосомы человека: обзор и гипотезы. Hum Mol Genet 1992; 1 : 221–227.
CAS
Статья
Google ученый
де Ру Н., Янг Дж., Мисрахи М. и др. : Семья с гипогонадотропным гипогонадизмом и мутациями в рецепторе гонадотропин-рилизинг-гормона. N Engl J Med 1997; 337 : 1597–15602.
CAS
Статья
Google ученый
de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E: Гипогонадотропный гипогонадизм из-за потери функции производного KiSS1 пептидного рецептора GPR54. Proc Natl Acad Sci USA 2003; 100 : 10972–10976.
CAS
Статья
Google ученый
Seminara SB, Messager S, Chatzidaki EE et al : Ген GPR54 как регулятор полового созревания. N Engl J Med 2003; 349 : 1614–1627.
CAS
Статья
Google ученый
Питтелуд Н., Куинтон Р., Пирс С. и др. : Дигенные мутации определяют вариабельные фенотипы при идиопатическом гипогонадотропном гипогонадизме. J Clin Invest 2007; 117 : 457–463.
CAS
Статья
Google ученый
Sanlaville D, Verloes A: Синдром ЗАРЯДА: обновление. евро J Hum Genet 2007; 15 : 389–399.
CAS
Статья
Google ученый
Пинто Г., Абади В., Меснаж Р и др. : Синдром ЗАРЯДА включает гипогонадотропный гипогонадизм и аномальное развитие обонятельных луковиц. J Clin Endocrinol Metab 2005; 90 : 5621–5626.
CAS
Статья
Google ученый
Blustajn J, Kirsch CF, Panigrahy A, Netchine I: Обонятельные аномалии при синдроме CHARGE: результаты визуализации — потенциальный главный диагностический критерий. AJNR Am J Neuroradiol 2008; 29 : 1266–1269.
CAS
Статья
Google ученый
Cortez AB, Galindo A, Arensman FW, Van Dop C: Врожденный порок сердца, связанный со спорадическим синдромом Каллмана. Am J Med Genet 1993; 46 : 551–554.
CAS
Статья
Google ученый
Кляйн В.Р., Фридман Дж.М., Брукшир Г.С., Браун О.Е., Эдман К.Д.: Синдром Каллмана, связанный с атрезией хоан. Clin Genet 1987; 31 : 224–227.
CAS
Статья
Google ученый
Огата Т., Фудзивара И., Огава Э., Сато Н., Удака Т., Косаки К. Фенотип синдрома Каллмана у пациентки с синдромом CHARGE и мутацией CHD7 . Endocr J 2006; 53 : 741–743.
CAS
Статья
Google ученый
Теллье А.Л., Кормье-Дайр В., Абади В. и др. : Синдром ЗАРЯДА: отчет о 47 случаях и обзор. Am J Med Genet 1998; 76 : 402–409.
CAS
Статья
Google ученый
Зенати Д., Бретонес П., Ламбе С. и др. : детский фенотип синдрома Каллмана из-за мутаций рецептора 1 фактора роста фибробластов (FGFR1). Mol Cell Endocrinol 2006; 254–255 : 78–83.
Артикул
Google ученый
Vissers LE, van Ravenswaaij CM, Admiraal R et al : Мутации в новом члене семейства генов хромодомена вызывают синдром CHARGE. Nat Genet 2004; 36 : 955–957.
CAS
Статья
Google ученый
Лалани С.Р., Сафиуллах А.М., Фернбах С.Д. и др. : спектр мутаций CHD7 у 110 человек с синдромом CHARGE и генотип-фенотипическая корреляция. Am J Hum Genet 2006; 78 : 303–314.
CAS
Статья
Google ученый
Rosen S, Gann P, Rogol A: Врожденная аносмия: пороги обнаружения семи классов одорантов у пациентов с гипогонадизмом и эугонадизмом. Ann Otol Rhinol Laryngol 1979; 88 : 288.
CAS
Статья
Google ученый
Доти Р., Шаман П., Данн М.: Разработка теста идентификации запаха Университета Пенсильвании, стандартизированного теста обонятельной функции с микрокапсулами. Physiol Behav 1984; 32 : 489–502.
CAS
Статья
Google ученый
Дэвидсон TM, Мерфи C: Быстрая клиническая оценка аносмии.Тест на запах алкоголя. Arch Otolaryngol Head Neck Surg 1997; 123 : 591–594.
CAS
Статья
Google ученый
Chalouhi C, Faulcon P, Le Bihan C, Hertz-Pannier L, Bonfils P, Abadie V: Обонятельная оценка у детей: приложение к синдрому CHARGE. Педиатрия 2005; 116 : e81 – e88.
Артикул
Google ученый
Klingmüller D, Duwes W, Krahe T., Brecht G, Schweikert H-U: Магнитно-резонансная томография головного мозга у пациентов с аносмией и гипоталамическим гипогонадизмом (синдром Каллмана). J Clin Endocrinol Metab 1987; 65 : 581–584.
Артикул
Google ученый
Whitcomb R, Crowley WJ: Мужской гипогонадотропный гипогонадизм. Endocrinol Metab Clin North Am 1993; 22 : 125–143.
CAS
Статья
Google ученый
Seminara SB, Hayes FJ, Crowley Jr WF: Дефицит гонадотропин-рилизинг-гормона у человека (идиопатический гипогонадотропный гипогонадизм и синдром Каллмана): патофизиологические и генетические соображения. Endocr Rev 1998; 19 : 521–539.
CAS
PubMed
Google ученый
Grumbach MM: Окно возможностей: диагностика дефицита гонадотропинов у младенцев мужского пола. J Clin Endocrinol Metab 2005; 90 : 3122–3127.
CAS
Статья
Google ученый
Truwit CL, Barkovich AJ, Grumbach MM, Martini JJ: МРТ синдрома Каллмана, генетического нарушения миграции нейронов, влияющего на обонятельную и генитальную системы. AJNR Am J Neuroradiol 1993; 14 : 827–838.
CAS
PubMed
Google ученый
Birnbacher R, Wandl-Vergesslich K, Frisch H: Диагностика X-рецессивного синдрома Каллмана в раннем младенчестве. Признаки гипоплазии носового мозга. Eur J Pediatr 1994; 153 : 245–247.
CAS
Статья
Google ученый
Белый Б.Дж., Рогол А.Д., Браун К.С., Либлих Дж.М., Розен С.В.: Синдром аносмии с гипогонадотропным гипогонадизмом: генетическое исследование 18 новых семей и обзор. Am J Med Genet 1983; 15 : 417–435.
CAS
Статья
Google ученый
Franco B, Guioli S, Pragliola A et al : Ген, удаленный при синдроме Каллманна, имеет гомологию с адгезией нервных клеток и молекулами, определяющими путь аксонов. Nature 1991; 353 : 529–536.
CAS
Статья
Google ученый
Legouis R, Hardelin J-P, Levilliers J et al : Ген-кандидат для X-сцепленного синдрома Каллмана кодирует белок, связанный с молекулами адгезии. Cell 1991; 67 : 423–435.
CAS
Статья
Google ученый
Hardelin J-P, Levilliers J, del Castillo I et al : Х-хромосомно-связанный синдром Каллмана: остановка мутаций подтверждает достоверность гена-кандидата. Proc Natl Acad Sci USA 1992; 89 : 8190–8194.
CAS
Статья
Google ученый
Falardeau J, Chung WC, Beenken A et al : Снижение передачи сигналов FGF8 вызывает дефицит гонадотропин-рилизинг-гормона у людей и мышей. J Clin Invest 2008; 118 : 2822–2831.
CAS
Статья
Google ученый
Сато Н., Охьяма К., Фуками М., Окада М., Огата Т.: Синдром Каллмана: соматические и зародышевые мутации гена рецептора 1 фактора роста фибробластов у матери и сына. J Clin Endocrinol Metab 2006; 91 : 1415–1418.
CAS
Статья
Google ученый
Коул Л.В., Сидис Ю., Чжан С и др. : Мутации в прокинетицине 2 (PROK2) и рецепторе 2 PROK2 (PROKR2) при дефиците гонадотропин-высвобождающего гормона человека: молекулярная генетика и клинический спектр. J Clin Endocrinol Metab 2008; 93 : 3551–3559.
CAS
Статья
Google ученый
Monnier C, Dodé C, Fabre L et al : PROKR2 миссенс-мутации, связанные с синдромом Каллмана, нарушают сигнальную активность рецептора. Hum Mol Genet 2008 [Epub перед печатью]. PMID 18826963.
Leroy C, Fouveaut C, Leclercq S et al : двуаллельные мутации в гене прокинетицина-2 в двух спорадических случаях синдрома Каллмана. евро J Hum Genet 2008; 16 : 865–868.
CAS
Статья
Google ученый
Оливейра Л.М., Семинара С.Б., Беранова М. и др. : Важность аутосомных генов в синдроме Каллмана: корреляции генотип-фенотип и нейроэндокринные характеристики. J Clin Endocrinol Metab 2001; 86 : 1532–1538.
CAS
PubMed
Google ученый
Питтелуд Н., Мейсинг А., Куинтон Р. и др. : Мутации в рецепторе 1 фактора роста фибробластов вызывают синдром Каллмана с широким спектром репродуктивных фенотипов. Mol Cell Endocrinol 2006; 254–255 : 60–69.
Артикул
Google ученый
Salenave S, Chanson P, Bry H et al : Синдром Каллмана: сравнение репродуктивных фенотипов у мужчин, несущих мутации KAL1 и FGFR1 / KAL2 . J Clin Endocrinol Metab 2008; 93 : 758–763.
CAS
Статья
Google ученый
Альбуиссон Дж., Пешо С., Карел Дж. С. и др. : Синдром Каллмана: 14 новых мутаций в KAL1 и FGFR1 ( KAL2 ). Hum Mutat 2005; 25 : 98–99.
Артикул
Google ученый
Zhou QY, Cheng MY: Prokineticin 2 и выход циркадных часов. FEBS J 2005; 272 : 5703–5709.
CAS
Статья
Google ученый
Buchter D, Behre HM, Kliesch S, Nieschlag E: Пульсирующий ГнРГ или хорионический гонадотропин человека / менопаузальный гонадотропин человека как эффективное лечение для мужчин с гипогонадотропным гипогонадизмом: обзор 42 случаев. Eur J Endocrinol 1998; 139 : 298–303.
CAS
Статья
Google ученый
Hardelin J-P, Dodé C: KAL1, FGFR1, PROKR2, PROK2 и синдром Каллмана; in: Epstein C, Erickson R, Wynshaw-Boris A (eds): Врожденные ошибки развития . Нью-Йорк: Oxford University Press, 2008, глава 47, стр. 482–490.
Google ученый
Schwanzel-Fukuda M, Crossin KL, Pfaff DW, Bouloux PMG, Hardelin J-P, Petit C: Миграция нейронов высвобождающего гормона лютеинизирующего гормона (LHRH) нейронов в ранних эмбрионах человека. J Comp Neurol 1996; 366 : 547–557.
CAS
Статья
Google ученый
Schwanzel-Fukuda M, Bick D, Pfaff DW: Клетки, экспрессирующие лютеинизирующий гормон-рилизинг-гормон (LHRH), не мигрируют нормально при наследственном гипогонадальном синдроме (Kallmann). Mol Brain Res 1989; 6 : 311–326.
CAS
Статья
Google ученый
Мацумото С., Ямадзаки С., Масумото К.Х. и др. : Аномальное развитие обонятельной луковицы и репродуктивной системы у мышей, лишенных рецептора прокинетицина PKR2. Proc Natl Acad Sci USA 2006; 103 : 4140–4145.
CAS
Статья
Google ученый
Chung WC, Moyle SS, Tsai PS: Передача сигналов фактора роста фибробластов 8 через рецептор 1 Fgf необходима для появления нейронов гонадотропин-рилизинг-гормона. Эндокринология 2008; 149 : 4997–5003.
CAS
Статья
Google ученый
Hardelin J-P, Dodé C: Сложная генетика синдрома Каллмана: KAL1 , FGFR1 , FGF8 , PROKR2 , PROK2 , и др. и др. Sex Dev 2008; 2 : 181–193.
CAS
Статья
Google ученый
Исследование уникального мужского и женского родства с синдромом Каллмана и дисгенезией гонад 46, XX при низком росте | Репродукция человека
Аннотация
Описано родство, где брат и сестра оба страдают синдромом Каллмана (аносмия и дефицит гонадотропин-рилизинг-гормона), а у женщины также есть полоса на яичниках.Хотя есть несколько состояний, которые могут возникать при синдроме Каллманна, нет известных сообщений о дисгенезии яичников, связанной с этим расстройством. Цитогенетический анализ не показал перестройки или серьезных делеций хромосом. Анализ сцепления с использованием информативных микросателлитных маркеров предсказывает, что ген, отличный от KAL1 (на Xp22.3), вовлечен в синдром Каллмана, проявляющийся одновременно с дисгенезом яичников, обнаруженным в этом семействе.
Введение
Было идентифицировано уникальное родство мужского и женского пола, у обоих братьев и сестер проявлялся гипогонадотропный гипогонадизм с аносмией, что согласуется с диагнозом синдрома Каллмана.У брата или сестры также есть дисгенезия яичников и низкий рост. О женщинах с синдромом Каллмана и полосатыми яичниками ранее не сообщалось. Синдром Каллманна встречается менее чем у 1 из 10 000 новорожденных, при этом заболевание мужчин и женщин в 5 раз больше (Jones and Kemmann, 1976). Наблюдались следующие типы наследования: Х-сцепленный, аутосомно-доминантный с переменной пенетрантностью и аутосомно-рецессивный (McKusick, 1994). Ген ( KAL1 ) был выделен из Xp22.3 (Franco et al ., 1991; Legouis et al ., 1991), и мутации в этом гене, как было установлено, ответственны за Х-сцепленную форму синдрома Каллмана (Meitinger et al ., 1990 ; Bick et al. ., 1992; Hardelin et al. ., 1992). Считается, что локус KAL1 подвергается X-инактивации (Franco et al. ., 1991). Предполагается, что гликопротеин внеклеточного матрикса, полученный из этого гена, участвует в адгезии нервных клеток и поиске пути аксонов, и его отсутствие, вероятно, является причиной неспособности нейронов, секретирующих гонадотропин-рилизинг-гормон (GnRH), развиваться и мигрировать в эмбриональной жизни (Rugarli et al. ., 1993, 1996). Обсуждается исключение участия локуса KAL1 в Xp22.3 в этой родословной.
Клинико-патологическое заключение
Женщина-пробанд поступила в возрасте 16 лет и 11 месяцев, у нее никогда не было менструации и не было признаков развития груди или каких-либо других признаков спонтанного полового созревания. Вес при рождении 3 кг. На презентации ее вес составлял 41,2 кг, а рост — 145 см. Она также была полностью аносмической. Базальные концентрации лютеинизирующего гормона (ЛГ) были менее 1 МЕ / л и не изменились с i.v. Стимуляция гонадолиберина (Wyeth-Ayerst HRF ® ; Ayerst, Rouses Point, США) и концентрация фолликулостимулирующего гормона (ФСГ) началась во время стимуляции с 0,8 МЕ / л и повысилась до 3,1 МЕ / л. В остальном передний гипофизарный резерв при динамическом тестировании был нормальным. Эти данные соответствовали диагнозу синдрома Каллмана (обонятельная дисплазия). Была начата терапия эстрогенами, доза увеличилась через 3 месяца и добавлен циклический прогестин. Была достигнута конечная высота 156 см.Пациентке сообщили, что для наступления беременности потребуется поддержка гонадотропинами.
В возрасте 30 лет пробанд вышла замуж и пожелала беременности. Однако она была полностью невосприимчива к повторным и продолжительным попыткам индукции овуляции сначала пульсирующим ГнРГ (Wyeth-Ayerst HRF ® ) (Jansen, 1993), а затем высокими дозами внутримышечно. гонадотропины (Pergonal ® ; Laboratories Serono SA, Обон, Швейцария). При лапароскопическом исследовании были отмечены двусторонние «полоски яичников» и выполнена биопсия яичников.Матка и маточные трубы выглядели нормально. Гистероскопическое исследование: полость матки 7 см в норме. Яичников, антинуклеарных, митохондриальных, париетальных клеток, островков клеток Лангерганса, надпочечников, ревматоидного фактора, ацетилхолинэстеразы, аутоантител к микросомам щитовидной железы и тиреоглобулину сыворотки не обнаружено. При УЗИ почечные пути в норме.
Пробанд зачат в 34 года после переноса двух замороженных-размороженных эмбрионов, помещенных на карантин в течение предыдущих 6 месяцев после ЭКО донорских ооцитов.Для подготовки и поддержки эндометрия использовались таблетки этинилэстрадиола (Progynon-C; Schering, Берлин, Германия) до 5 недель после переноса эмбриона и непатентованные прогестероновые пессарии (порошок прогестерона; Sigma, Сент-Луис, Миссури, США) до 7 недель после перенос эмбрионов. Здоровая женщина весом 3395 г родилась через 36 недель после переноса эмбриона путем планового кесарева сечения. Вагинальные роды не предпринимались из-за клинически суженного таза. Приливы были отмечены через неделю после послеродового периода, и заместительная гормональная терапия была возобновлена.
При обращении было отмечено, что ее брат, возраст 24 года, не достиг половой зрелости и страдает аносмией. Диагноз синдрома Каллмана у брата или сестры мужского пола подтвержден эндокринными исследованиями: ФСГ 0,6 МЕ / л; ЛГ <1,0 МЕ / л; пролактин 19 нмоль / л; общий тестостерон 1,2 нмоль / л и свободный тестостерон 19 пмоль / л. Скелетный фенотип был гипогонадальным; рост 172 см, размах рук 184 см, сегментарная диспропорция (верхний сегмент 73 см, нижний сегмент 79 см). Распределение волос хорошее, наблюдалась двусторонняя гинекомастия, объем яичек 8 мл.Анализ семенной жидкости выявил азооспермию. Затем дважды в неделю вводили инъекции хорионического гонадотропина человека (ХГЧ) в течение ряда лет. Обработка Pergonal ® инициировала сперматогенез, и анализ спермы зафиксировал объем 4,6 мл, концентрацию 4,6 × 10 6 сперматозоидов на мл и подвижность 50%. Морфология сперматозоидов не исследовалась. Заместительная андрогенная терапия проводилась подкожно. гранулы тестостерона (Conway и др. , 1988). Дальнейшая попытка получения сперматогенеза была предпринята в возрасте 41 года для целей ЭКО.Использование инъекций ХГЧ в дозе 2000 ЕД п / к. трижды в неделю (Profasi; Laboratories Serono SA, Обон, Швейцария), и когда через 6 недель была зарегистрирована концентрация сывороточного тестостерона 10 нмоль / л, ФСГ вводили в дозах 150 единиц трижды в неделю (Puregon; Organon, Oss, Нидерланды). Через четыре месяца после введения ФСГ анализ спермы показал 1,5 × 10 6 сперматозоидов на мл, 64% сперматозоидов показали быструю подвижность и 6% нормальную морфологию. Дальнейшие анализы в течение нескольких месяцев зафиксировали аналогичные параметры спермы.Сперматозоиды также были заморожены для длительного хранения. Оплодотворение было достигнуто с помощью свежей эякулированной спермы и интрацитоплазматической инъекции сперматозоидов всех пяти ооцитов, полученных после подавления активности гипофиза и стимуляции яичников. Анализ спермы проводился в соответствии с методологией, рекомендованной Всемирной организацией здравоохранения (1992). Беременность наступила не в результате переноса двух эмбрионов. Три лишних эмбриона подвергаются криоконсервации.
У братьев и сестер нормальный интеллект и слух.Офтальмологическое обследование сестры или сестры не выявило признаков глазного альбинизма 1 типа ( OA1 ). Кожных высолов, свидетельствующих об ихтиозе, не было. У братьев и сестер не вызывали зеркальных движений. У матери не было преждевременной менопаузы. Гипосмия у матери выявлена в анамнезе и клинически. Ни у одного из родителей не было обнаружено зеркальных движений. Родители эмигрировали в Австралию из разных районов Сицилии. В семейном анамнезе не было отложенного полового созревания, бесплодия, кровного родства или аносмии.
Магнитно-резонансная томография
Было получено
сагиттальных изображений T1, коронарных T1 и FSE и аксиальных двойных эхо-изображений T2 гипофиза, гипоталамуса, передней комиссуры, обонятельных луковиц и борозд. Не было обнаружено никаких нарушений со стороны гипофиза, верхнего таламуса или обонятельного пути.
Гистопатология полосковой биопсии яичника
Серый фрагмент биопсии размером 13 × 5 × 2 мм, полученный из яичника с правой полоской, был полностью залит парафином.Были приготовлены шестнадцать серийных срезов и окрашены гематоксилином-эозином для микроскопического исследования. Аттенуированная полоса яичника характеризовалась наличием тонкой, плотно гипоцеллюлярной коры, покрывающей очевидно нормальную вагинальную оболочку и мезотелиальную поверхность (рис. 1). Внутри тонкой коры был замечен случайный примордиальный фолликул (рис. 2), содержащий морфологически жизнеспособный ооцит, который был окружен кубовидными клетками прегранулезы (~ 1/200 мм 2 ). При серийной биопсии яичников не было других доказательств фолликулярной активности, ни в настоящем, ни в прошлом.В коре головного мозга были отмечены редкие, мелкие, незаметные рубцы, ни один из которых не свидетельствовал о наличии остатков фолликулов, а в одном из них была обнаружена гигантская клетка инородного тела, содержащая инородный материал. Серозных включений (Мюллерова) или инфильтратов воспалительных клеток в коре яичников не наблюдалось. Точно так же гипотрофический мозг полностью лишен фолликулярных структур или остатков (рис. 1). В отличие от многих полосковых гонад, клетки ворот (клетки Лейдига) не наблюдались в глубоком мозговом веществе или прикорневой области яичника.
Цитогенетические исследования
Бэндинг
G с исследованием 20 метафаз периферических лимфоцитов обоих братьев и сестер подтвердил нормальный кариотип. Микроскопия с высоким разрешением была выполнена для Х-хромосом, дистальных областей 1q и 10q обоих братьев и сестер, и не выявила аномалий.
Молекулярно-генетические исследования
Было собрано
образцов периферической крови у родителей и двух братьев и сестер. Геномная ДНК была выделена с использованием модификации метода высаливания (Miller et al ., 1988). Олигонуклеотидные праймеры для экзонов KAL1 были получены от Pierre Bouloux (Royal Free Hospital, Лондон, Великобритания). Амплификацию экзона 12 KAL1 с помощью полимеразной цепной реакции (ПЦР) проводили, как описано ранее (Hardelin и др. ., 1993; Quinton и др. ., 1996b). Олигонуклеотидные праймеры для полиморфизма динуклеотидных повторов в локусе KAL1 (Bouloux et al ., 1991) и олигонуклеотидные праймеры для полиморфизмов DXS996, DXS7103 были получены от Research Genetics, Inc.(Хантсвилл, Алабама, США). Реакции ПЦР для полиморфных локусов DXS996 и DXS7103 проводили в соответствии с условиями, впервые описанными Джин Вайссенбах и полученными из базы данных генома (доступной в Интернете http://gdbwww.gdb.org). Продукты ПЦР анализировали на 7% (19: 1) полиакриламидных гелях и визуализировали с использованием метода окрашивания серебром (Budowle et al ., 1991).
ПЦР с использованием внутригенных праймеров KAL1 , фланкирующих экзон 12, приводили к продуктам для обоих братья и сестры.Для исследований сцепления внутригенный полиморфный локус KAL1 (Bouloux et al. ., 1991) оказался неинформативным в родословной; однако использование полиморфных локусов DXS996 и DXS7103, фланкирующих локус KAL1 , показало, что братья и сестры унаследовали разные материнские аллели для этих локусов (рис. 3). Вероятность событий рекомбинации между полиморфными локусами, DXS996 и DXS7103, была рассчитана с использованием данных сантиморганов, полученных от Cedar Genetics (доступно в Интернете http: // cedar.генетика). DXS996, как утверждается, составляет 2,69 сантиморгана по отношению к KAL1 , тогда как DXS7103 составляет 3,79 сантиморгана по отношению к KAL1 . Вероятность двойной рекомбинации между этими локусами составила 1/981 (или 0,1%).
Обсуждение
Синдром Каллмана демонстрирует генетическую гетерогенность (McKusick, 1994). После выделения KAL1 , гена-кандидата для Х-связанной формы заболевания, было проведено несколько исследований по поиску мутаций в этом гене.Родословные, использованные в этих исследованиях, обычно имели по крайней мере одного затронутого мужчины либо в материнской семье пробанда, либо среди братьев и сестер мужского пола. Пораженные самки отсутствуют (Hardelin et al ., 1993). Самки-носители часто страдают аносмией или гипосмией, но очень мало сообщений о гипогонадотропных родственниках пробанда-самца, если таковые вообще имеются. Центральный дефект гипоталамуса у пробанда демонстрируется результатами динамического тестирования и данными биопсии яичников в глубоком мозговом / прикорневом отделе.Поскольку стимуляция гонадотропинами отсутствовала, в отличие от многих полосковых гонад, не наблюдалось развития клеток ворот (клетки Лейдига).
Женщины с синдромом Каллмана обычно встречаются спорадически среди населения, у которого нет других затронутых родственников (П. М. Булу, личное сообщение). Другие состояния были связаны с синдромом Каллмана, такие как зеркальные движения (бимануальная синкинезия), аномальное пространственное внимание, окулярная моторная апраксия, односторонняя почечная дисплазия и, реже, односторонняя нейросенсорная глухота, короткие ключицы, хориоретинальная колобома, гидроцефалия новорожденных, расщелина губы / неба, деформация кавус (Hardelin и др. ., 1993; Quinton и др. ., 1996a). Дисплазия почек, в частности, часто возникает у пациентов с Х-сцепленным синдромом Каллмана (Kirk и др. ., 1994), а также у этих пациентов часто встречается одна почка (Hardelin и др. ., 1993). Ультразвуковое исследование почек у братьев и сестер было нормальным. Этот вывод не может быть использован для исключения локуса KAL1 . Обнаружение того, что братья и сестры не вызывали зеркальных движений, является надежным нейрофизиологическим доказательством того, что родственные братья не обладают мутацией локуса KAL1 (Quinton et al ., 1996а).
Возможно, что аберрантные клеточные взаимодействия могут лежать в основе феноменов синдрома Каллмана и агенеза почек. Агенезия почек, по-видимому, не происходит при синдроме Каллмана, не сцепленном с Х-хромосомой (Quinton et al. ., 1996b). Почечные аномалии наблюдаются при дисгенезии яичников 45 X, но без обонятельных аномалий (Plouffe and McDonough, 1996). Метанефрос и гонадный гребень развиваются рядом, и дефект поля может указывать на аберрантные клеточные взаимодействия.Тем не менее, почечное развитие у братьев кажется нормальным. Во время гаструляции примордиальные половые клетки начинают миграцию из каудальной примитивной полоски / эпибласта (Lawson and Hage, 1994). Эпибласт также дифференцируется, чтобы стать нейроэктодермой (Larsen, 1997). Следовательно, участие половых клеток и обонятельное / гипоталамическое расстройство предполагает возможную связь на уровне развития эпибласта.
При обследовании братьев и сестер не было обнаружено доказательств наличия известных синдромов смежных генов.Были описаны синдромы смежных генов, при которых делеции, затрагивающие дистальное короткое плечо Х-хромосомы, приводили к пациентам с сочетанием низкого роста, точечной хондродисплазии, умственной отсталости, дефицита стероид-сульфатазы (STS) и синдрома Каллманна (Ballabio et al . , 1989). Клинический анамнез и обследование исключили синдромы ближайших смежных смежных генов, STS (ихтиоз) и OA1, фланкирующие локус KAL1 . ПЦР-амплификации экзона 12 KAL1 и внутригенного полиморфизма KAL1 привели к обнаружению соответствующих продуктов для обоих братьев и сестер, и поэтому микроделеции всего локуса KAL1 могут быть исключены.
Хотя Х-связанная передача расстройства не была указана в этом семействе, для подтверждения на молекулярном уровне был проведен сегрегационный анализ нарушения с областью KAL1 Х-хромосомы (рис. 3). Ввиду результатов сегрегации аллелей KAL1 вовлечение локуса потребует двойной рекомбинации материнского аллеля. Вероятность события двойной рекомбинации была рассчитана как 1/981. Однако знаменатель, вероятно, недооценен, поскольку физическая « интерференция » снижает близкую частоту двойной рекомбинации (Schmitt et al ., 1994; Strachen and Read, 1996). Вероятность вовлечения локуса KAL1 , таким образом, вероятно, меньше, чем вычисленное сантиморганом значение 0,1%.
Информация о родословной и три отчета о транслокации позволяют предположить, что существуют аутосомные формы синдрома Каллмана (Best et al ., 1990; Casamassima et al ., 1993; McKusick, 1994; Schinzel et al ., 1995). Диагноз синдрома Каллмана в двух отчетах о транслокации спорен.Бест и др. . (1990), возможно, ошиблись, утверждая, что случай реципрокной транслокации 7q; 12q имел синдром Каллмана, поскольку концентрации гонадотропина фактически были в пределах нормы; молекулярное исключение гена KAL1 не проводили. Дело, представленное Casamassima et al . (1993) не исключили участия гена KAL1 в молекулярных исследованиях; также концентрация ФСГ 3 МЕ / л не является особенно низкой и несовместимой с синдромом Каллмана.Шинцель и др. . (1995) представили случай, когда скрининг саузерн-блоттингом не обнаружил сдвигов полос в гене KAL1 . Могут присутствовать необнаруженные мутации гена KAL1 . Базальные концентрации гонадотропинов были низкими, что соответствует синдрому Каллмана. Принимая во внимание случай транслокации, описанный Schinzel et al. . (1995) была проведена микроскопия с высоким разрешением у братьев и сестер для дистальных областей 1q и 10q, и отклонений не было обнаружено.Гипогонадотропный гипогонадизм также наблюдается при аутосомно-рецессивном синдроме Буше – Нойхаузера. Однако эти сибсы не обладают типичными характеристиками хориоретинальной дистрофии и мозжечковой атаксии (Rump et al ., 1997).
Предполагаемый ген рецептора KALP , который не был клонирован, был предложен в качестве кандидата на аутосомную форму синдрома Каллмана (Quinton et al. ., 1996a). Недавнее исследование рассматривало идиопатический гипогонадотропный гипогонадизм как альтернативное проявление синдрома Каллманна и показало, что Х-сцепленный тип наследования составлял только 36% из 106 случаев (Waldstreicher et al ., 1996). Исследователи также обнаружили низкую распространенность мутации гена KAL1 при изолированном дефиците GnRH (Georgopoulos et al ., 1997). Было высказано предположение, однако, что синдром Каллмана не следует рассматривать вместе с идиопатическим гипогонадотропным гипогонадизмом, поскольку последний обычно представляет собой отчетливый дефект поля развития (Dean et al ., 1990; Quinton et al ., 1996b). Если так, то доля аутосомных форм синдрома Каллмана значительно меньше.
При синдроме врожденной гипоплазии надпочечников и гипогонадотропного гипогонадизма ген DAX1 в Xp21.3 мутирует (Zanaria et al. ., 1994). Основываясь на исследовании гена DAX1 мыши, предполагается, что экспрессия влияет на дифференцировку гонад (Swain et al ., 1996). Хотя совпадение гонадных и гипоталамических эффектов впечатляет, мутация гена DAX1 , тем не менее, не вызывает аносмию. Гипоплазии надпочечников у братьев и сестер не наблюдалось, и Х-сцепленное заболевание маловероятно.Следовательно, мутация гена DAX-1 у братьев и сестер маловероятна.
Было замечено, что делеции Х-хромосомы, связанные с полным нарушением функции яичников, а не вторичной аменореей, неизменно затрагивают проксимальные области Х-хромосомы (Simpson, 1986). Эти области более чем вероятно X-инактивированы. Если кто-то постулирует, что причинный локус находится в этой области, то следует предположить, что родной брат или сестра является проявленным носителем. Это предположение требует возникновения еще одного случайного события, такого как искаженная X-инактивация или монородительская изодисомия.Перекошенная X-инактивация нормального аллеля была зарегистрирована при мышечной дистрофии Дюшенна, но нечасто (Azofeifa et al ., 1995). Гомозиготный мутировавший статус KAL1 у женщины крайне маловероятен. Однородительская изодисомия у женщины с мутированным геном KAL1 может быть отвергнута из-за гетерозиготности аллелей, обнаруженной в трех полиморфных сайтах: KAL1 , DXS996 и DXS7103.
Делеции, обнаруженные в длинном плече Х-хромосомы (Xq), хорошо известны как причина преждевременной недостаточности яичников (Krauss et al ., 1987). Сообщалось о вероятности делеций смежных генов у женщин с недостаточностью функции яичников, сопровождающей синдром блефарофимоза (Smith et al. ., 1989). Такие удаления кажутся аутосомными. Исследования сцепления показывают, что синдром блефарофимоза-птоза-эпикантуса-обратного типа 1 (BPES1), связанный с дисгенезией яичников, локализован в 3q22-q23 (Small et al ., 1995; Amati et al ., 1996). Синдром BPES1 не был связан с синдромом Каллмана.
Семьдесят пять семей с 46, XX дисгенезией яичников (ODG) были идентифицированы в Финляндии, и некоторые родословные предполагают существование аутосомно-рецессивного гена (Aittomaki, 1994). Посредством систематического поиска сцепления в мультиплексно пораженных семьях локус ODG был картирован на хромосоме 2р. Ген рецептора фолликулостимулирующего гормона ( FSHR ) ранее был отнесен к 2p. Поиск мутаций выявил переход C566T в экзоне 7 FSHR , который сегрегирован с фенотипом заболевания (Aittomaki et al ., 1995). Дальнейшее исследование 22 пациенток с дисгенезом яичников и мутацией 566C >> T в гене рецептора ФСГ позволило предположить подмножество патогенетически отличных пациентов с дисгенезом яичников. Возможно, из-за остаточной рецепторной активности этих пациентов можно идентифицировать, продемонстрировав наличие фолликулов яичников и подтвердив анализ мутаций (Aittomaki et al ., 1996). Гипогонадотрофизм и развитие сперматогенеза после терапии гонадотропинами позволяют предположить, что дефицит рецептора ФСГ у родства маловероятен.
Аутосомно-рецессивные расстройства, связанные с дефицитом одного гонадотропина, либо ФСГ, либо ЛГ, могут препятствовать нормальному развитию полового созревания и фертильности (Weiss et al ., 1992; Layman et al ., 1993, 1997). Имеются данные о том, что ФСГ не требуется для сперматогенеза (Kumar et al ., 1997). Однако при изолированном дефиците ФСГ не наблюдается одновременного дефицита высвобождения ЛГ, как это наблюдалось у братьев и сестер. Самцы подвергаются нормальной вирилизации, чего не наблюдалось у пробандов мужского пола (Rabin et al ., 1972; Рабин, 1975). Изолированный дефицит гонадотропина также не объясняет наблюдаемую дисгенезию яичников. Имеется сообщение о дисгенезии гонад 47, XXY с синдромом Каллмана (Hazard et al ., 1986). Возможно, это указывает на наличие гоносомного гена, влияющего на синдром Каллмана. Трудно предположить наличие X-связанной рецессивной причины, такой как мутация локуса KAL1 , если только X-хромосома не была дублирована. Ошибка дублирования могла привести к неразъединению.
Причины нарушения персистенции половых клеток при дисгенезии гонад 46, XX неизвестны.У «нокаутных» мышей, дефектных по гену репарации ошибочного спаривания ДНК Mlh2 , были отмечены нарушения механизмов контроля мейоза и маленькие яичники с очень небольшим количеством фолликулов (Baker et al ., 1996). Не сообщалось об эндокринных данных, и синдром Каллмана не был связан с нарушениями генов восстановления несоответствия ДНК. Эти различные отчеты предполагают, что более одного гена кодирует факторы, необходимые для выживания ооцитов в течение репродуктивной жизни. Вполне могут быть другие женщины с синдромом Каллмана, а также с дисгенезией гонад 46, XX.Такие женщины будут идентифицированы по невосприимчивым попыткам овуляции. Сообщений об индукции овуляции и результатах беременности немного (Сунгуртекин и др. , 1995).
Это первое сообщение о братьях и сестрах мужского и женского пола с синдромом Каллмана, проявляющимся одновременно с дисгенезией яичников. Исключение вовлечения с помощью анализа сцепления локуса KAL1 и представление мужских и женских фенотипов предполагает, что весьма вероятно, что задействован аутосомный ген.Когда будет получено достаточное количество родословных самок, страдающих синдромом Каллмана, станет возможным анализ молекулярных связей. Затем исследования могут выявить ген, участвующий, хотя и непрерывно, в выживании ооцитов.
Рисунок 1.
Обзор полоски яичника. Оригинальное увеличение × 25.
Рисунок 1.
Обзор полосатого яичника. Оригинальное увеличение × 25.
Рисунок 2.
Вид тонкой коры яичника. Оригинальное увеличение × 200.
Рис. 2.
Вид тонкой коры яичника. Оригинальное увеличение × 200.
Рисунок 3.
Схема, представляющая фланкирующие и внутригенные фрагменты полимеразной цепной реакции (ПЦР), действующие как маркеры сегрегации для аллелей KAL1 в родословной. Цифры 1, 2 и 3 различают длины трех аллельных фрагментов ПЦР.
Рисунок 3.
Схема, представляющая фланкирующие и внутригенные фрагменты полимеразной цепной реакции (ПЦР), действующие как маркеры сегрегации для аллелей KAL1 в родословной. Цифры 1, 2 и 3 различают длины трех аллельных фрагментов ПЦР.
Авторы хотели бы поблагодарить следующих: Pierre M.G.Bouloux, Veronique M.Duke и Richard Quinton из отделения эндокринологии Royal Free Hospital, Лондон, Великобритания, которые предоставили учебники и советы; Стюарт Первис-Смит и Мо-Ин Йип из отдела цитогенетики и клеточной биологии Королевской больницы Южного Сиднея, Зетланд, Новый Южный Уэльс; Энн Конвей, андролог; Джефф Уотсон и Питер Рассел, патологи, Джуди Сопер, радиолог, госпиталь Королевского принца Альфреда, Кампердаун, Новый Южный Уэльс; Дэвид Маккей из офтальмологической генетической службы, больница Конкорд, Конкорд, Новый Южный Уэльс; и Майкл Хоуанг из Сиднея КТ и МРТ.Мы также благодарны за помощь Des Cooper и Gavan Harrison из Школы биологических наук Университета Маккуори, Норт-Райд, Новый Южный Уэльс.
Список литературы
Айттомаки К. (
1994
) Генетика дисгенезии XX гонад.
Am . Дж . Хум . Генет .
,
54
,
844
–851.
Айттомаки К., Лусена Дж. Л., Пакаринен П. и др. . (
1995
) Мутация в гене рецептора фолликулостимулирующего гормона вызывает наследственную гипергонадотропную недостаточность яичников.
Ячейка
,
82
,
959
–968.
Aittomaki, K., Herva, R., Stenman, U.H. и др. . (
1996
) Клинические особенности первичной недостаточности яичников, вызванной точечной мутацией в гене рецептора фолликулостимулирующего гормона.
Дж . Клин . Endocr . Метаб .
,
81
,
3722
–3726.
Амати П., Гаспарини П., Злотогора Дж. и др. . (
1996
) Ген преждевременной недостаточности яичников, связанной с пороком развития век, отображается на хромосоме 3q22-q23.
Am . Дж . Хум . Генет .
,
58
,
1089
–1092.
Azofeifa, J., Voit, T., Hubner, C. и Cremer, M. (
1995
) Метилирование Х-хромосомы у проявляющихся и здоровых носителей дистрофинопатий: соответствие коэффициентов активации среди родственниц первой степени родства и искаженная инактивация как причина пораженных фенотипов.
Гм . Генет .
,
96
,
167
–176.
Бейкер, С.М., Плаг, А.В., Пролла, Т.А. и др. . (
1996
) Вовлечение мышиного Mlh2 в репарацию ошибочного спаривания ДНК и мейотический кроссинговер.
Nature Genet .
,
13
,
336
–342.
Баллабио, А., Бардони, Б., Карроццо, Р. и др. . (
1989
) Синдромы смежных генов из-за делеций в дистальном коротком плече Х-хромосомы человека.
Proc . Нац. . Акад. . Sci . США
,
86
,
10001
–10005.
Бест, Л.Г., Васдал, В.А., Ларсон, Л.М. и Стурлаугсон, Дж. (
1990
) Хромосомная аномалия при синдроме Каллмана.
Am . Дж . Мед . Генет .
,
35
,
306
–309.
Бик, Д., Франко, Б., Шеринс, Р.Дж. и др. . (
1992
) Краткое сообщение: внутригенная делеция гена KALIG-1 при синдроме Каллмана.
№ . Англ. . Дж . Мед .
,
326
,
1752
–1755.
Bouloux, P.M., Hardelin, J.P., Munroe, P. et al. . (
1991
) Полиморфизм динуклеотидных повторов в локусе Каллмана (Xp22.3).
Nucleic Acids Res .
,
19
,
5453
Budowle, B., Chakraborty, R., Giusti, A.M. и др. . (
1991
) Анализ локуса VNTR D1S80 с помощью ПЦР с последующим ПААГ высокого разрешения.
Am . Дж . Хум . Генет .
,
48
,
137
–144.
Casamassima, A.C., Wilmot, P.L., Vibert, B.K. и Шапиро, Л. (
1993
) Синдром Каллмана, связанный со сложной перестройкой хромосом.
Am . Дж . Мед . Генет .
,
45
,
539
–541.
Конвей, А.Дж., Бойлан, Л.М., Хау, К. и др. . (
1988
) Рандомизированное клиническое испытание заместительной терапии тестостероном у мужчин с гипогонадизмом.
Внутр. . Дж . Андрол .
,
11
,
247
–264.
Dean, J.C., Johnston, A.W. и Клоппер, А. (
1990
) Изолированный гипогонадотропный гипогонадизм: семья с аутосомно-доминантным наследованием.
Клин . Endocr .
,
32
,
341
–347.
Франко Б., Гуйоли С., Праглиола А. и др. . (
1991
) Ген, удаленный при синдроме Каллмана, имеет гомологию с адгезией нервных клеток и молекулами, определяющими путь аксонов.
Природа
,
353
,
529
–536.
Georgopoulos, N.A., Pralong, F.P., Seidman, C.E. et al. . (
1997
) Генетическая гетерогенность, о чем свидетельствует низкая частота мутаций гена KAL-1 в спорадических случаях дефицита гонадотропин-рилизинг-гормона.
Дж . Клин . Endocr . Метаб .
,
82
,
213
–217.
Hardelin, J.P., Levilliers, J., delCastillo, I. и др. . (
1992
) Синдром Каллмана, сцепленный с Х-хромосомой: остановка мутаций подтверждает достоверность гена-кандидата.
Proc . Нац. . Акад. . Sci . США
,
89
,
8190
–8194.
Hardelin, J.P., Levilliers, J., Blanchard, S. et al. . (
1993
) Неоднородность мутаций, ответственных за синдром Каллмана, связанный с Х-хромосомой.
Гм . Мол . Генет .
,
2
,
373
–377.
Хазард, Дж., Розенберг, И., Перлемутер, Л. и др. . (
1986
) Реакции гонадотропинов на пульсирующее введение низких доз ГнРГ в случае аносмии с гипогонадотропным гипогонадизмом, связанным с дисгенезией гонад 47 XXY.
Acta Endocrinol .
,
113
,
593
–597.
Янсен, Р.П.С. (
1993
) Пульсирующее внутривенное введение гонадотропин-рилизинг-гормона для индукции овуляции: детерминанты реакций фолликулярной и лютеиновой фаз.
Гм . Репродукция .
,
8 (Дополнение 2)
,
193
–196.
Джонс, Дж. Р. и Кемманн, Э. (
1976
) Олфакто-генитальная дисплазия у женщин.
Акушерский . Гинеколь . Энн .
,
5
,
443
–466.
Кирк, Дж. М., Грант, Д. Б., Бессер, Г. М. и др. . (
1994
) Односторонняя аплазия почек при Х-сцепленном синдроме Каллмана.
Клин . Генет .
,
46
,
260
–262.
Краусс К.М., Тюрксой Р.Н., Аткинс Л. и др. . (
1987
) Семейная преждевременная недостаточность яичников из-за интерстициальной делеции длинного плеча Х-хромосомы.
№ . Англ. . Дж . Мед .
,
317
,
125
–131.
Кумар, Т.Р., Ван, Ю., Лу, Н. и Мацук, М.М. (
1997
) Фолликулостимулирующий гормон необходим для созревания фолликула яичника, но не для мужской фертильности.
Nature Genet .
,
15
,
201
–204.
Larsen, W.J. (1997) Human Embryology , 2nd edn. Черчилль Ливингстон, Нью-Йорк.
Лоусон, К.А. и Hage, W.J. (
1994
) Клональный анализ происхождения первичных зародышевых клеток мыши.
Ciba Foundn Symp .
,
182
,
68
–84.
Лейман, Л.С., Ли, Э.-Дж., Пик, Д. и др. . (
1997
) Краткий отчет: задержка полового созревания и гипогонадизм, вызванные мутациями в гене β-субъединицы фолликулостимулирующего гормона.
№ . Англ. . Дж . Мед .
,
337
,
607
–611.
Layman, L.C., Shelley, M.E., Huey, L.O. и др. . (
1993
) Структура гена фолликулостимулирующего гормона бета при преждевременной недостаточности яичников.
Фертиль . Стерил .
,
60
,
852
–857.
Legouis, R., Hardelin, J.P., Levilliers, J. et al. . (
1991
) Ген-кандидат для Х-сцепленного синдрома Каллмана кодирует белок, связанный с молекулами адгезии.
Ячейка
,
67
,
423
–435.
МакКусик, В.А. (1994) 308700 Синдром Каллмана. В McKusick, В.А. (ред.), Менделирующее наследование в человеке , 11-е изд. Издательство Университета Джона Хопкинса, Балтимор.
Meitinger, T., Heye, B., Petit, C. et al. . (1990) Окончательная локализация Х-сцепленного синдрома Каллмана (гипогонадотропный гипогонадизм и аносмия) в Xp22.3: тесная связь с гипервариабельной повторяющейся последовательностью CRI-S232 Am . Дж . Хум . Genet ., 47 , 664–669. [Опубликованная ошибка появляется в Am . Дж . Хум . Genet ., 47 , 883.]
Miller, S.A., Dykes, D.D. and Polesky, H.F. (
1988
) Простая процедура высаливания для извлечения ДНК из ядерных клеток человека.
Nucleic Acids Res .
,
16
,
1215
Plouffe Jr, L. и McDonough, P.Г. (1996) Агенезия и дисгенезия яичников. In Adashi, E.Y., Rock, J.A. and Rosenwaks, Z. (eds), Reproductive Endocrinology , Surgery , and Technology . Липпинкотт-Рэйвен, Филадельфия, стр. 1366–1384.
Quinton, R., Duke, V.M., De Zoysa, P.A. и Bouloux, P.M.G. (
1996a
) Нейробиология синдрома Каллмана.
Дж . Br . Фертиль . Соц .
,
1
,
121
–127.
Quinton, R., Duke, V.M., de, Z.P., Platts, A.D. и др. . (1996b) Нейрорадиология синдрома Каллмана: генотипический и фенотипический анализ J . Клин . Endocr . Метаб ., 81 , 3010–3017. [Опубликованная ошибка появляется в J . Клин . Эндокринол . Metab ., 81, 3614].
Рабин Д. (
1975
) Синдромы изолированной недостаточности гонадотропина.
Врожденные дефекты: серия оригинала
,
11
,
73
–80.
Рабин Д., Шпиц И., Берковичи Б. и др. . (
1972
) Изолированный дефицит фолликулостимулирующего гормона. Клинико-лабораторные особенности.
№ . Англ. . Дж . Мед .
,
287
,
1313
–1317.
Rugarli, E.I., Ghezzi, C., Valsecchi, V. и Ballabio, A. (
1996
) Продукт гена синдрома Каллмана, экспрессируемый в клетках COS, расщепляется на поверхности клетки с образованием диффундирующего компонента.
Гм . Мол . Генет .
,
5
,
1109
–1115.
Ругарли, Э.И., Лутц, Б., Куратани, С.С. и др. . (
1993
) Паттерн экспрессии гена синдрома Каллмана в обонятельной системе предполагает его роль в нацеливании на нейроны.
Nature Genet .
,
4
,
19
–26.
Рамп, П., Хамель, Б.С.Дж., Пинкерс, А.Дж.Л.Г. и ванДоп П.А. (
1997
) Два родных брата с хориоретинальной дистрофией, гипогонадотропным гипогонадизмом и мозжечковой атаксией: синдром Буше – Нойхаузера.
Дж . Мед . Генет .
,
34
,
767
–771.
Шинцель А., Лорда-Санчес И., Бинкерт Ф. и др. . (
1995
) Синдром Каллмана у мальчика с транслокацией t (1; 10), обнаруженной с помощью обратной окраски хромосом.
Дж . Мед . Генет .
,
32
,
957
–961.
Schmitt, K., Lazzeroni, L.C., Foote, S. et al. . (
1994
) Многоточечная карта сцепления псевдоавтосомальной области человека, основанная на типировании одного сперматозоида: происходят ли двойные кроссоверы во время мужского мейоза?
Am . Дж . Хум . Генет .
,
55
,
423
–430.
Симпсон, Дж. Л. (1986) Фенотипико-кариотипические корреляции гонадных детерминант: текущий статус и связь с молекулярными исследованиями. В Sperling, K. и Vogel, F. (eds), Proceedings 7th International congress on Human Genetics Berlin , 1986 . Springer-Verlag, Heidelberg, стр. 224–232.
Смолл, К.В., Сталвей, М., Фишер, Л. и др. . (
1995
) Синдром блефарофимоза связан с хромосомой 3q.
Гм . Мол . Генет .
,
4
,
443
–448.
Smith, A., Fraser, I.S., Shearman, R.P., Russell, P. (
1989
) Блефарофимоз плюс недостаточность яичников: вероятный кандидат на синдром смежных генов.
Дж . Мед . Генет .
,
26
,
434
–438.
Soussi-Yanicostas, N., Hardelin, J.P., Arroyo-Jiminez, M.M. и др. . (
1996
) Первоначальная характеристика аносмина-1, предполагаемого белка внеклеточного матрикса, синтезируемого определенными популяциями нейрональных клеток в центральной нервной системе.
Дж . Cell Sci .
,
109
,
1749
–1757.
Strachen, T. и Read, A.P. (1996) Human Molecular Genetics . Биос, Оксфорд, стр. 329.
Сунгуртекин У., Фрейзер И.С. and Shearman, R.P. (
1995
) Беременность у женщин с синдромом Каллмана.
Фертиль . Стерил .
,
63
,
494
–499.
Суэйн, А., Занария, Э., Хакер, А. и др. . (
1996
) Экспрессия Dax1 мыши согласуется с ролью в определении пола, а также в функции надпочечников и гипоталамуса.
Nature Genet .
,
12
,
404
–409.
Waldstreicher, J., Seminara, S.B., Jameson, J.L. и др. . (
1996
) Генетическая и клиническая гетерогенность дефицита гонадотропин-рилизинг-гормона у человека.
Дж . Клин . Endocr . Метаб .
,
81
,
4388
–4395.
Weiss, J., Axelrod, L., Whitcomb, R.W. et al. . (
1992
) Гипогонадизм, вызванный заменой одной аминокислоты в бета-субъединице лютеинизирующего гормона.
№ . Англ. . Дж . Мед .
,
326
,
179
–183.
Занария, Э., Мускателли, Ф., Бардони, Б. и др. . (
1994
) Необычный член суперсемейства ядерных гормональных рецепторов, ответственный за врожденную Х-сцепленную гипоплазию надпочечников.
Природа
,
372
,
635
–641.
© Европейское общество репродукции человека и эмбриологии
Синдром Каллмана, статья
[1]
Laitinen EM, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, Raivio T., Заболеваемость, фенотипические особенности и молекулярная генетика синдрома Каллмана в Финляндии.Орфанский журнал редких болезней. 2011, 17 июня; [PubMed PMID: 21682876]
[2]
Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton Р., Янг Дж., Экспертный консенсусный документ: Европейское консенсусное заявление о врожденном гипогонадотропном гипогонадизме — патогенез, диагностика и лечение.Обзоры природы. Эндокринология. 2015 сен; [PubMed PMID: 26194704]
[3]
Iolascon G, Frizzi L, Bianco M, Gimigliano F, Palumbo V, Sinisi AM, Sinisi AA, Поражение костей у мужчин с болезнью Каллмана. Клинические и экспериментальные исследования старения. 2015 окт; [PubMed PMID: 26201943]
[4]
Balasubramanian R, Crowley WF Jr, Дефицит изолированного гонадотропин-высвобождающего гормона (GnRH) 1993; [PubMed PMID: 20301509]
[5]
Ким Ш., Врожденный гипогонадотропный гипогонадизм и синдром Каллмана: прошлое, настоящее и будущее.Эндокринология и метаболизм (Сеул, Корея). 2015 Dec; [PubMed PMID: 267
]
[6]
Лима Амато Л.Г., Латронико А.С., Гонтихо Сильвейра Л.Ф., Молекулярные и генетические аспекты врожденного изолированного гипогонадотропного гипогонадизма. Клиники эндокринологии и обмена веществ Северной Америки. 2017 июн; [PubMed PMID: 28476224]
[7]
Vezzoli V, Duminuco P, Bassi I, Guizzardi F, Persani L, Bonomi M, Комплексная генетическая основа врожденного гипогонадотропного гипогонадизма.Minerva endocrinologica. 2016 июн; [PubMed PMID: 26934720]
[8]
Rhie YJ, Система рецепторов-54, связанных с кисспептином / G-белком, как важный привратник полового развития. Анналы детской эндокринологии [PubMed PMID: 24
2]
[9]
Cioppi F, Riera-Escamilla A, Manilall A, Guarducci E, Todisco T, Corona G, Colombo F, Bonomi M, Flanagan CA, Krausz C, Genetics of ncHH: от своеобразного наследования новой мутации GNRHR до всестороннего обзора литературы.Андрология. 2019 янв. [PubMed PMID: 30575316]
[10]
Комнинос А.Н., Дилло В.С., Новые роли кисспептина в сексуальной и эмоциональной обработке мозга. Нейроэндокринология. 2018 [PubMed PMID: 28866668]
[11]
Mills EGA, Dhillo WS, Comninos AN, Kisspeptin и контроль эмоций, настроения и репродуктивного поведения Журнал эндокринологии.1 октября 2018 г. [PubMed PMID: 30306845]
[13]
Mikkelsen JD, Simonneaux V, Нейроанатомия системы кисспептина в мозге млекопитающих. Пептиды. 2009, янв [PubMed PMID: 18840491]
[14]
Lehman MN, Hileman SM, Goodman RL, Нейроанатомия сигнальной системы кисспептина у млекопитающих: сравнительные аспекты и аспекты развития.Успехи экспериментальной медицины и биологии. 2013 [PubMed PMID: 23550001]
[17]
AminiLari M, Manjoo P, Craigie S, Couban R, Wang L, Busse JW, Заместительная гормональная терапия и снижение дозы опиоидов для индуцированного опиоидами гипогонадизма среди пациентов с хронической нераковой болью: систематический обзор. Лекарство от боли (Мальден, Массачусетс). 2 мая 2018 г .; [PubMed PMID: 29727002]
[18]
Pierzchlewska MM, Robaczyk MG, Vogel I, Индукция полового созревания с помощью человеческого хорионического гонадотропина (ХГЧ) с последующим обращением гипогонадотропного гипогонадизма при синдроме Каллмана.Endokrynologia Polska. 2017; [PubMed PMID: 2
42]
[19]
McCabe MJ, Bancalari RE, Dattani MT, Диагностика и оценка гипогонадизма.