
Прогерия – это редкое генетическое заболевание, впервые описанное Гилфордом, которое проявляется преждевременным старением организма, связанное с его недоразвитием. Прогерия классифицируется на детскую, получившую название синдрома Гетчинсона (Хатчинсона)-Гилфорда и взрослую – синдром Вернера.
При этом заболевании отмечается сильное отставание в росте с самого детства, изменение структуры кожи, кахексия, отсутствие вторичных половых признаков и волос, недоразвитие внутренних органов и вид старого человека. При этом психическое состояние больного соответствует возрасту, эпифизарная хрящевая пластина закрывается рано, а тело имеет детские пропорции.
Прогерия относится к неизлечимым заболеваниям и является причиной появления серьёзного атеросклероза, что в результате развивает инсульты и различные болезни сердца. А в итоге эта генетическая патология приводит к летальному исходу, т.е. она фатальна. Как правило, ребёнок может прожить, в среднем, тринадцать лет, хотя встречаются случаи с продолжительностью жизни более двадцати лет.
Детская прогерия Хатчинсона-Гилфорда
Это заболевание встречается крайне редко в соотношении 1:4000000 новорождённых в Нидерландах и 1:8000000 в США. Причём заболевание поражает больше мальчиков, чем девочек (1,2:1).
Рассматривают две формы прогерии Хатчинсона-Гилфорда: классическую и неклассическую.
В настоящее время описано более ста случаев детской прогерии. Причём в основном это заболевание поражает детей белой расы. Для прогерии Хатчинсона-Гилфорда свойственно полиморфное поражение. Дети, имеющие такой синдром, выглядят вполне нормальными при рождении. Но уже к году или двум наблюдается серьёзное отставание в росте. Обычно такие дети отличаются слишком маленьким ростом и ещё более низкой массой тела в соответствии с его длиной.
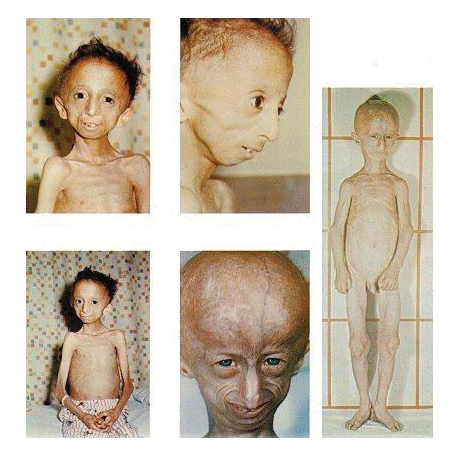
Для детей с прогерией характерно полное облысение не только волосистой части головы, но и отсутствие ресниц, бровей ещё с раннего возраста. Кожа выглядит слабой и морщинистой в результате абсолютной потери подкожного жира, присутствует цианоз кожных покровов. Для головы характерна непропорциональность черепно-лицевых костей, которые напоминают лицо птицы с крючковатым носом, аномально небольшой нижней челюстью, выпученными глазными яблоками и оттопыренными ушами. Именно эти черты, большая лысина и маленькая челюсть, придают внешности ребёнка вид старого человека.
Другие клинические проявления прогерии включают: неправильное и позднее прорезывание зубов, тонкий и высокий голос, грушевидная грудная клетка и уменьшенные в размерах ключицы. Конечности обычно тонкие, а изменённые локтевые и коленные суставы придают больному ребёнку «позу наездника».
У детей ещё до года отмечаются склероподобные уплотнения, врождённого или приобретённого характера, на ягодицах, бёдрах и внизу живота. Детям с прогерией характерна гиперпигментация кожи, которая только усиливается с годами и гипоплазия ногтей, при которой они становятся жёлтыми, тонкими и выпуклыми, напоминающие часовые стёкла. Однако, начиная с пятилетнего возраста, развивается распространённая форма атеросклероза с большим поражением аорты и артерий, особенно брыжеечных и коронарных. А уже гораздо позже появляются сердечные шумы и гипертрофия сердца, в левом желудочке. Раннее возникновение у детей атеросклероза, становится причиной непродолжительности их жизни. А вот основной причиной смерти считается инфаркт миокарда.
При прогерии известны случаи ишемического инсульта. Такие дети в умственном развитии абсолютно ничем не отличаются от здоровых детей, иногда даже опережают их. Дети с таким диагнозом в среднем живут около четырнадцати лет.
При детской прогерии неклассической формы длина тела от массы отстаёт незначительно, на протяжении длительного времени волосы сохраняются, а липодистрофия прогрессирует гораздо медленнее; возможен рецессивный тип наследования.
Прогерия причины
До сих пор точные причины возникновения прогерии не выяснены. Предположительной этиологией развития этого заболевания является нарушение обмена в соединительной ткани, в результате разрастания фибробластов путём клеточного деления и увеличения образования коллагена при сниженном синтезе гликозаминогликанов. Медленное формирование фибробластов объясняется нарушениями в межклеточном веществе.
В причинах детского синдрома прогерии считаются мутации в гене LMNA, который отвечает за кодирование ламина А. Это такой белок, из которого состоит один из слоёв ядра клеточной оболочки.
Во многих случаях прогерия проявляется спорадически, а в некоторых семьях встречается у сибсов, особенно при кровнородственных браках, а это говорит о возможном аутосомно-рецессивном типе наследования. При исследованиях кожи больных были обнаружены клетки, в которых нарушена способность исправлять разрывы и повреждения в ДНК, а также воспроизводить генетически однородные фибробласты, изменять атрофическую дерму и эпидермис, способствующие исчезновению подкожной клетчатки.
Для прогерии взрослых характерно аутосомно-рецессивное наследование с дефектным геном АТФ-зависимой хеликазой или WRN. Существует предположение в связывающей цепочки нарушений между репарацией ДНК и обменом соединительной ткани.
Также установлено, что прогерия Хатчинсона-Гилфорда имеет нарушения в клетках-носителях, которые не могут полноценно избавляться от сшивок ДНК, вызываемые химическими агентами. При диагностировании этих клеток с таким синдромом выяснили, что они не способны полноценно пройти процесс деления.
В 1971 году Оловниковым было высказано предположение об укороченных размерах теломер в процессе образования клеток. А в 1992 году это было уже доказано на пациентах с синдромом прогерии для взрослых. Анализ, который связывает лимит Хейфлика, длину теломера и активность фермента теломеразы, позволяет объединить естественный процесс старения с формированием клинической симптоматики детской прогерии Хатчинсона-Гилфорда. Так как эта форма прогерии встречается крайне редко, то можно только высказывать гипотезу о типе наследования, который имеет сходства с синдромом Коккейна и проявляется отдельными чертами преждевременного старения.
Имеются также высказывания о принадлежности прогерии Хатчинсона-Гилфорда к мутации, аутосомно-доминантной, которая возникла de novo, т.е. без наследования. Она стала косвенным подтверждением синдрома, в основу которого вошли измерения теломеров у носителей заболевания, их родителей и доноров.
Прогерия симптомы
Клиническая картина детской прогерии отличается характерным преждевременным атеросклерозом, фиброзом миокарда, нарушениями мозгового кровообращения, повышением липопротеидов и уровнем холестерина, протромбиновым временем в анализах, ранними инфарктами, скелетными аномалиями. В данном случае имеются выраженные диспропорции лица и черепа, недоразвитие челюсти и зубов, смещение бёдер. Длинные кости при нормальной корковой структуре и прогрессировании периферической деминерализации подвергаются рецидивирующим патологическим переломам.
Суставам характерна тугая подвижность, особенно коленным с возможными контрактурами тазобедренных, голеностопных, локтевых и лучезапястных суставов. При рентгенологических исследованиях обнаруживается деминерализация около суставов с остеопорозом, варусными и вальгусными деформациями нижних конечностей. Также очень часто развиваются опухоли и утолщение коллагеновых волокон.
Синдром Вернера или взрослая прогерия проявляется от 14 до 18 лет и характеризуется отставанием в росте, универсальным поседением с параллельным прогрессированием алопеции.
Как правило, синдром прогерия развивается после двадцати лет и отличается ранним облысением, истончением кожных покровов на лице и конечностях, характерной бледностью. Под слишком натянутой кожей просматриваются поверхностные кровеносные сосуды, а подкожная жировая клетчатка и мышцы, расположенные под ней, полностью атрофируются, поэтому конечности выглядят непропорционально тонко.
Затем кожа над выступами костей постепенно становится толще и изъязвляется. После тридцати лет у больных прогерией развивается катаракта обоих глаз, голос становится слабым, высоким и хриплым, заметно поражаются кожные покровы. Это проявляется в виде склероцермоподобных изменений конечностей и лица, сухости кожи, язв на ногах, мозолей на стопах и телеангиэктазией. Такие больные, как правило, низкого роста, с лунообразным лицом, клювоподобным носом, как у птицы, суженным ротовым отверстием и выступающим резко подбородком, полным туловищем и тонкими конечностями.
У больных прогерией нарушаются функции потовых и сальных желёз. На выступах костей образовывается гиперкератоз, проявляется общая гиперпигментация, изменяется форма ногтевых пластин. А после различных травм на голенях и стопах появляются трофические язвы. Кроме атрофии и истончений, у больных отмечаются значительные изменения в мышцах и костях, кальцификация, остеопороз генерализованного характера, остеоартриты с эрозиями. Такие больные ограничены в движениях пальцев и сгибательной контрактуре. Для больных прогерией свойственна деформация костей, как при ревматоидном артрите, боль в конечностях, плоскостопие и остеомиелит.
Во время обследований на рентенографии выявляются остеопороз костей, гетеротопические кальцинаты кожи и подкожной клетчатки, связок и сухожилий. Также, медленно прогрессирует катаракта, развивается атеросклероз, нарушающий деятельность сердечно-сосудистой системы. У большинства больных снижается интеллект.
После сорока лет к прогерии на фоне сахарного диабета, дисфункций паращитовидных желёз и других заболеваний почти у 10% пациентов развиваются опухолевые патологии в виде остеогенной саркомы, астроцитомы, тиреоидной аденокарциномы, рака молочной железы и кожи.
Летальный исход обычно является следствием сердечно-сосудистых патологий и злокачественных опухолей.
При гистологическом анализе синдрома прогерии устанавливают атрофию придатков кожи, где сохраняются эккринные железы; дерма при этом имеет утолщение, гиалинизируются волокна из коллагена, а нервные волокна разрушаются.
У больных полностью атрофируются мышцы, отсутствует подкожный жир.
Заболевание диагностируется на основании клинической симптоматики прогерии. При сомнениях в диагнозе определяют способность фибробластов размножаться в культуре (сниженный показатель для синдрома Вернера). Для дифференциального диагноза прогерии учитывают синдромы Хатчинсона-Гилфорда, Ротмунда-Томсона и системную склеродермию.
Прогерия лечение
До настоящего времени не существует конкретного лечения прогерии, его ещё не разработали. В основном терапия носит симптоматический характер с профилактикой осложнений после атеросклероза и в излечении трофических язв, сахарного диабета.
Для анаболического эффекта назначается СТГ, который у некоторых больных увеличивает массу тела и длину. Весь терапевтический процесс проводится рядом специалистов, таких как эндокринолог, терапевт, кардиолог, онколог и других, в зависимости от превалирующей симптоматики.
Но в 2006 году исследователями США было отмечено прогрессирование в лечении прогерии, как неизлечимого заболевания. Они внесли в культуру нарушенных фибробластов ингибитор фарнезилтрансферазы, который ранее проходил испытания на онкологических больных. И этот процесс вернул стареющим клеткам нормальную форму. Такой препарат был хорошо перенесён, поэтому сейчас существует надежда, что в будущем появится возможность в его применении, чтобы предотвратить прогерию ещё в детском возрасте.
Эффективность Лонафарниба (ингибитора фарнезилтрансферазы) заключается в увеличении количества жира под кожей, в массе тела, минерализации костей, что в итоге сократит переломы.
Но, тем не менее, пока это заболевание характеризуется неблагоприятными прогнозами. В среднем больные прогерией доживают до тринадцатилетнего возраста, умирая от кровоизлияний и инфарктов.
Детская прогерия Хатчинсона-Гилфорда
Это болезнь встречается очень изредка в соотношении 1:4000000 новорождённых в Нидерландах и 1:8000000 в США. Причём болезнь поражает больше мальчишек, чем девченок (1,2:1).
Рассматривают две формы прогерии Хатчинсона-Гилфорда: традиционную и неклассическую.
В истинное время описано более 100 случаев детской прогерии. Причём в главном это болезнь поражает малышей белоснежной расы. Для прогерии Хатчинсона-Гилфорда характерно полиморфное поражение. Детки, имеющие таковой синдром, смотрятся полностью нормальными при рождении. Но уже к году либо двум наблюдается серьёзное отставание в росте. Как правило такие детки отличаются очень небольшим ростом и ещё более низкой массой тела в согласовании с его длиной.
Для малышей с прогерией типично полное облысение не только лишь волосистой части головы, да и отсутствие ресниц, бровей ещё с ранешнего возраста. Кожа смотрится слабенькой и морщинистой в итоге абсолютной утраты подкожного жира, находится цианоз кожных покровов. Для головы свойственна несимметричность черепно-лицевых костей, которые напоминают лицо птицы с крючковатым носом, аномально маленький нижней челюстью, выпученными глазными яблоками и оттопыренными ушами. Конкретно эти черты, большая плешина и малая челюсть, присваивают наружности ребёнка вид старенького человека.
Другие клинические проявления прогерии включают: неверное и позже прорезывание зубов, узкий и высочайший глас, грушевидная грудная клеточка и уменьшенные в размерах ключицы. Конечности как правило тонкие, а изменённые локтевые и коленные суставы присваивают нездоровому ребёнку «позу наездника».
У малышей ещё до года отмечаются склероподобные уплотнения, врождённого либо приобретённого нрава, на ягодицах, бёдрах и понизу животика. Детям с прогерией свойственна гиперпигментация кожи, которая только усиливается с возрастом и гипоплазия ногтей, при которой они становятся жёлтыми, тонкими и выпуклыми, напоминающие часовые стёкла. Но, начиная с пятилетнего возраста, развивается распространённая форма атеросклероза с огромным поражением аорты и артерий, в особенности брыжеечных и коронарных. А уже еще позднее возникают сердечные шумы и гипертрофия сердца, в левом желудочке. Преждевременное появление у малышей атеросклероза, становится предпосылкой непродолжительности их жизни. А вот основной предпосылкой погибели считается инфаркт миокарда.
При прогерии известны случаи ишемического инфаркта. Такие детки в интеллектуальном развитии полностью ничем не отличаются от здоровых малышей, время от времени даже опережают их. Детки с таким диагнозом в среднем живут около 14-ти лет.
При детской прогерии неклассической формы длина тела от массы отстаёт некординально, в протяжении долгого времени волосы сохраняются, а липодистрофия прогрессирует еще медлительнее; вероятен рецессивный тип наследования.

детская прогерия фото
Прогерия причины
До этого времени четкие предпосылки появления прогерии не выяснены. Предположительной этиологией развития этого заболевания является нарушение обмена в соединительной ткани, в итоге разрастания фибробластов путём клеточного деления и роста образования коллагена при сниженном синтезе гликозаминогликанов. Неспешное формирование фибробластов разъясняется нарушениями в межклеточном веществе.
В причинах детского синдрома прогерии числятся мутации в гене LMNA, который отвечает за кодирование ламина А. Это таковой белок, из которого состоит один из слоёв ядра клеточной оболочки.
Во многих случаях прогерия проявляется спорадически, а в неких семьях встречается у сибсов, в особенности при кровнородственных браках, а это гласит о вероятном аутосомно-рецессивном типе наследования. При исследовательских работах кожи нездоровых были обнаружены клеточки, в каких нарушена способность исправлять разрывы и повреждения в ДНК, также воспроизводить на генном уровне однородные фибробласты, изменять атрофическую дерму и эпидермис, содействующие исчезновению подкожной клетчатки.
Для прогерии взрослых типично аутосомно-рецессивное наследование с дефектным геном АТФ-зависимой хеликазой либо WRN. Существует предположение в связывающей цепочки нарушений меж репарацией ДНК и обменом соединительной ткани.
Также установлено, что прогерия Хатчинсона-Гилфорда имеет нарушения в клеточках-носителях, которые не могут всеполноценно избавляться от сшивок ДНК, вызываемые хим агентами. При диагностировании этих клеток с таким синдромом узнали, что они не способны всеполноценно пройти процесс деления.
В 1971 году Оловниковым было высказано предположение об укороченных размерах теломер в процессе образования клеток. А в 1992 году это было уже подтверждено на пациентах с синдромом прогерии для взрослых. Анализ, который связывает предел Хейфлика, длину теломера и активность фермента теломеразы, позволяет соединить естественный процесс старения с формированием медицинской симптоматики детской прогерии Хатчинсона-Гилфорда. Потому что эта форма прогерии встречается очень изредка, то можно только высказывать догадку о типе наследования, который имеет сходства с синдромом Коккейна и проявляется отдельными чертами раннего старения.
Имеются также выражения о принадлежности прогерии Хатчинсона-Гилфорда к мутации, аутосомно-доминантной, которая появилась de novo, т. е. без наследования. Она стала косвенным доказательством синдрома, в базу которого вошли измерения теломеров у носителей заболевания, их папы и мамы и доноров.
Прогерия симптомы
Клиническая картина детской прогерии отличается соответствующим ранним атеросклерозом, фиброзом миокарда, нарушениями мозгового кровообращения, увеличением липопротеидов и уровнем холестерина, протромбиновым временем в анализах, ранешними инфарктами, скелетными аномалиями. В этом случае имеются выраженные диспропорции лица и черепа, недоразвитие челюсти и зубов, смещение бёдер. Длинноватые кости при обычной корковой структуре и прогрессировании периферической деминерализации подвергаются рецидивирующим патологическим переломам.
Суставам свойственна тугая подвижность, в особенности коленным с вероятными контрактурами тазобедренных, голеностопных, локтевых и лучезапястных суставов. При рентгенологических исследовательских работах находится деминерализация около суставов с остеопорозом, варусными и вальгусными деформациями нижних конечностей. Также очень нередко развиваются опухоли и утолщение коллагеновых волокон.
Синдром Вернера либо взрослая прогерия проявляется от 14 до 18 лет и характеризуется отставанием в росте, универсальным седением с параллельным прогрессированием алопеции.
Как правило, синдром прогерия развивается после 20 лет и отличается ранешным облысением, истончением кожных покровов на лице и конечностях, соответствующей бледностью. Под очень натянутой кожей просматриваются поверхностные кровяные сосуды, а подкожная жировая клетчатка и мускулы, расположенные под ней, на сто процентов атрофируются, потому конечности смотрятся диспропорционально тонко.
Затем кожа над выступами костей равномерно становится толще и изъязвляется. После 30 лет у нездоровых прогерией развивается катаракта обоих глаз, глас становится слабеньким, высочайшим и осиплым, приметно поражаются кожные покровы. Это проявляется в виде склероцермоподобных конфигураций конечностей и лица, сухости кожи, язв на ногах, мозолей на стопах и телеангиэктазией. Такие нездоровые, обычно, низкого роста, с лунообразным лицом, клювоподобным носом, насколько у птицы, суженным ротовым отверстием и выступающим резко подбородком, полным туловищем и тонкими конечностями.
У нездоровых прогерией нарушаются функции потовых и сальных желёз. На выступах костей создается гиперкератоз, проявляется общая гиперпигментация, меняется форма ногтевых пластинок. А после разных травм на голенях и стопах возникают трофические язвы. Не считая атрофии и истончений, у нездоровых отмечаются значимые конфигурации в мышцах и костях, кальцификация, остеопороз генерализованного нрава, остеоартриты с эрозиями. Такие нездоровые ограничены в движениях пальцев и сгибательной контрактуре. Для нездоровых прогерией характерна деформация костей, насколько при ревматоидном артрите, боль в конечностях, плоскостопие и остеомиелит.
Во время обследований на рентенографии выявляются остеопороз костей, гетеротопические кальцинаты кожи и подкожной клетчатки, связок и сухожилий. Также, медлительно прогрессирует катаракта, развивается склероз, нарушающий деятельность сердечно-сосудистой системы. Практически у всех нездоровых понижается ум.
После сорока лет к прогерии на фоне сладкого диабета, дисфункций паращитовидных желёз и других болезней практически у 10% пациентов развиваются опухолевые патологии в виде остеогенной саркомы, астроцитомы, тиреоидной аденокарциномы, рака молочной железы и кожи.
Летальный финал как правило является следствием сердечно-сосудистых патологий и злокачественных опухолей.
При гистологическом анализе синдрома прогерии устанавливают атрофию придатков кожи, где сохраняются эккринные железы; дерма при всем этом имеет утолщение, гиалинизируются волокна из коллагена, а нервные волокна разрушаются.
У нездоровых стопроцентно атрофируются мускулы, отсутствует подкожный жир.
Заболевание диагностируется на основании медицинской симптоматики прогерии. При колебаниях в диагнозе определяют способность фибробластов плодиться в культуре (сниженный показатель для синдрома Вернера). Для дифференциального диагноза прогерии учитывают синдромы Хатчинсона-Гилфорда, Ротмунда-Томсона и системную склеродермию.
Прогерия лечение
До реального времени не существует определенного исцеления прогерии, его ещё не разработали. В главном терапия носит симптоматический нрав с профилактикой осложнений после атеросклероза и в исцелении трофических язв, сладкого диабета.
Для анаболического эффекта назначается СТГ, который у неких нездоровых наращивает массу тела и длину. Весь терапевтический процесс проводится рядом профессионалов, таких насколько эндокринолог, терапевт, кардиолог, онколог и других, зависимо от превалирующей симптоматики.
Но в 2006 году исследователями США было отмечено прогрессирование в лечении прогерии, насколько неизлечимого заболевания. Они занесли в культуру нарушенных фибробластов ингибитор фарнезилтрансферазы, который ранее проходил тесты на онкологических нездоровых. И этот процесс возвратил стареющим клеточкам нормальную форму. Таковой продукт был отлично перенесён, потому на данный момент существует надежда, что в дальнейшем появится возможность в его применении, чтоб предупредить прогерию ещё в детском возрасте.
Эффективность Лонафарниба (ингибитора фарнезилтрансферазы) заключается в увеличении количества жира под кожей, в массе тела, минерализации костей, что в конечном итоге уменьшит переломы.
Но, все же, пока это болезнь характеризуется неблагоприятными прогнозами. В среднем нездоровые прогерией доживают до тринадцатилетнего возраста, умирая от кровоизлияний и инфарктов.
Warning: date() [function.date
]: It is not safe to rely on the system’s timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected ‘Europe/Moscow’ for ‘MSK/3.0/no DST’ instead in /home/k45201/public_html/angina03.ru/mycode/main.php on line 3
15.04.2019
Синдром Готтрона. Эпидемиология
Считается, что синдром Готрона развивается у лиц женского пола чаще, чем у лиц мужского пола. В медицинской литературе было описано примерно 40 случаев.
Синдром Готтрона. Причины
Синдром Готтрона является редким аутосомно-рецессивным расстройством. Точная распространенность этого синдрома неизвестна.
Синдром Готтрона. Похожие расстройства
- Синдром Хатчинсона-Гилфорда является тяжелой формой прогерии (преждевременное старение). Главные особенности этого синдрома включают в себя быстрое старение и очень низкий рост. Дети с этим синдромом имеют характерные черты лица: большая голова, маленькое лицо, нос в форме клюва и аномальный подбородок. Глаза могут выступать из орбит, а склеры могут иметь голубоватый оттенок. Другие клинические особенности этого синдрома могут включать в себя сухую, тонкую и морщинистую кожу. Дети с синдромом Хатчинсона-Гилфорда, как правило, имеют нормальный интеллект, но продолжительность их жизни сокращается.
- Синдром Вернера – редкое прогрессирующее заболевание, оно характеризуется необычно ускоренным старением (прогерией). Хотя это расстройство, как правило, признается только в возрасте 30-40 лет, некоторые характерные особенности выявляются в детстве, юности и в начале взрослой жизни. У детей с синдромом Вернера фиксируется аномально медленная скорость роста, в конце концов, набор роста заканчивается в период полового созревания. В результате, лица с этим синдромом имеют необычно низкий рост и малый вес. К 25 годам, большинство лиц начинают проявлять раннее поседение и преждевременную потерю волос на голове (алопеция). По мере прогрессирования болезни, у пациентов появляются дополнительные нарушения, они включают: потеря жирового слоя под кожей, тяжелое истощение (атрофия) мышечной ткани в определенных областях тела и дегенеративные изменения кожи, особенно в области лица, плеч и рук, в нижней части ног и на стопах. Из-за дегенеративных изменений, затрагивающих область лица, у человека могут быть необычно выпуклые глаза, нос в форме клюва и / или другие характерные аномалии лица.
Синдром Готтрона. Симптомы и проявления
Уже в младенчестве, дети с синдромом Готтрона выглядят старше чем их сверстники. У детей кожа может быть необычайно тонкой и элластичной, у большинства лиц с этим синдромом, поверхность кожи может чем-то напоминать пергамент. В абсолютном большинстве случаев, такие кожные изменения происходят на руках и ногах. Более того, руки и ноги могут быть аномально маленькими до тех пор, пока пациенты не достигнут взрослого возраста. Вены на груди часто очень заметны, из-за уменьшенного количества подкожного жира.
Синдром Готтрона. Лечение
Лечение синдрома Готтрона только симптоматическое и поддерживающее. Генетическое консультирование может быть полезным для пациентов и их семей.
Прогерия (синдром Хатчинсона-Гилфорда) – редкая патология, вызванная мутацией гена, ответственного за синтез белка. При такой патологии появляются изменения кожи и внутренних органов, что вызваны преждевременным старением.
Детская прогерия, симптомы которой появляются с возраста 2 лет, вызывает преждевременное старение: больные доживают в среднем до 13 лет и умирают от атеросклероза и связанных с ним заболеваний — инсульта, инфаркта миокарда. Несмотря на генетический характер болезни, по наследству не передается.
Взрослая форма – синдром Вернера – генетическая патология, передается по наследству, начинается после 18 лет, характеризуется ранним старением, развитием болезней пожилого возраста: атеросклероза, остеопороза, катаракты. Приводит к летальному исходу.
Синдром Хатчинсона-Гилфорда – это следствие мутации, изменения структуры гена, которое происходит самопроизвольно или под влиянием внешних факторов. Носителем наследственности человека является молекула ДНК. Ген состоит из аминокислот, соединенных между собой в строгой последовательности. Изменение состава полипептидной цепочки приводит к генетическим заболеваниям.
При прогерии происходят структурные изменения гена, ответственного за синтез белка ламина. Происходит замена аминокислоты цитизин на тимин. Патологический ламин называют прогерином, накопление которого приводит к преждевременной гибели клеток. Молекулярные изменения ведут к процессам, подобным естественному старению.
Прогерия взрослых также является следствием мутации гена. Нарушается синтез фермента, отвечающего за работу ДНК. Возникающие повреждения генетического аппарата вызывают преждевременное старение соматических клеток.
Детская прогерия симптомы имеет следующие:
- маленький рост;
- отсутствие подкожной клетчатки;
- расширенная вена под кожей;
- несоразмерно большой череп;
- отсутствие волос на голове;
- плохое физическое развитие;
- большие глаза;
- дефекты зубов;
- «килевидная грудь»;
- высокий голос.
Несмотря на отставание в физическом развитии, дети с синдромом Хатчинсона-Гилфорда интеллектуально развиты, не отстают от сверстников в психическом развитии. Детская прогерия сопровождается прогрессированием атеросклероза уже с 5 лет и нарастанием сердечной патологии — появляются шумы при аускультации, симптомах гипертрофии миокарда. Кардиологические заболевания – самая частая причина смерти.
Случаи прогерии у взрослых, то есть синдром Вернера, характеризуются следующими состояниями:
- ранняя седина и облысение;
- появление старческих морщин в молодом возрасте;
- пигментация, сухость кожи;
- фиброзные уплотнения в подкожной клетчатке;
- голос становится глухим.
Прогерия – причина бесплодия мужчин и женщин. На поздних стадиях заболевания появляются трофические язвы на голенях. Из-за мышечной атрофии истончаются конечности, развиваются контрактуры суставов, плоскостопие. Характерна «поза всадника» из-за полусогнутых рук. Деформируются кисти, ногти желтеют, приобретают вид «часовых стекол».
При рентгенографии наблюдается остеопороз и отложение извести в околосуставных тканях, связочном аппарате суставов. Прогерию взрослых часто сопровождают доброкачественные опухоли различной локализации, эндокринные заболевания, сахарный диабет. В 8-12% возникают злокачественные опухоли. Поэтому прогерия симптомы имеет часто смазанные.

Фото детей, страдающих прогерией
Синдром Хатчинсона-Гилфорда – фатальное заболевание, всегда заканчивается смертью. Этиотропного лечения, устраняющего причину патологии, не существует. К смерти приводит атеросклероз, при котором на внутренней стенке сосудов откладывается холестерин, сужающий просвет артерий, нарушается кровоток. Развивается ишемическая болезнь, инфаркт миокарда. Атеросклеротические бляшки вызывают формирование тромба, который может оторваться от стенки сосуда и стать причиной нарушений мозгового кровообращения, инсульта.
Лечение прогерии направлено на уменьшение проявлений атеросклероза, предусматривает диету с низким содержанием животных жиров, богатую белковыми продуктами: нежирное мясо, рыба, творог. Медикаментозная терапия предусматривает применение статинов – препаратов, снижающих уровень холестерина в крови:
- «Аторвастатин Пфайзер»;
- «Липофен»;
- «Розувастатин Сандоз»;
- «Симвастатин»;
- «Эпадол-нео».
Препараты этой группы снижают концентрацию холестерина, влияют на содержание липидов в крови.
При прогерии необходим постоянный мониторинг состояния сердечно-сосудистой системы. Для предупреждения и лечения заболеваний сердца применяют медикаменты, снижающие свертывающую способность крови, обладающие антиагрегантными свойствами:
- «Кардиомагнил»;
- «Варфарин орион»;
- «Гепарин»;
- «Ипатон».
Применяют гормон роста, физиотерапевтические процедуры для восстановления функции суставов. Молочные зубы удаляют, так как прогерия у детей приводит к нарушению их роста.
Появились препараты, продлевающие жизнь больных прогерией, а вместе с ними и надежда, что с развитием генетических исследований, появится возможность излечивать заболевание, считавшееся фатальным.
Интенсивное изучение генетической патологии в России и во всем мире началось в 21 веке. Исследователи установили, что прогерин в небольших количествах накапливается в здоровом организме, и содержание его в клетках увеличивается с возрастом. Синдром Хатчинсона-Гилфорда и естественное старение имеют общие причины. С развитием медицинской науки станет возможным не только излечивать тяжелое заболевание, но и бороться со старостью.
Все ли корректно в статье с медицинской точки зрения?
Ответьте только в том случае, если у вас есть подтвержденные медицинские знания
Поделиться статьей:
Читать нас на Яндекс.Дзен
Заболевания со схожими симптомами:
Гиперплазия коры надпочечников – патологическое состояние, при котором наблюдается стремительное умножение тканей, из которых состоят данные железы. В результате этого орган увеличивается в размерах и его функционирование нарушается. Недуг диагностируется как у взрослых мужчин и женщин, так и у маленьких детей. Стоит отметить, что чаще встречается такая форма патологии, как врождённая гиперплазия коры надпочечников. В любом случае болезнь является достаточно опасной, поэтому при появлении её первых симптомов следует незамедлительно обратиться в медицинское учреждение для проведения всестороннего обследования и назначения эффективного метода терапии.
…
Амиотрофия – это патологический процесс врожденного характера, который характеризуется дегенеративно-дистрофическими изменениями мышц с их последующей атрофией. В большинстве случаев такое заболевание носит характер необратимого, что делает прогноз крайне неблагоприятным.
…
Анорексия подразумевает особый синдром в различных вариантах его проявления, возникающий под воздействием определенного ряда причин и проявляющийся в абсолютном отсутствии у больных аппетита, вне зависимости от того, что существует объективная необходимость в питании для самого организма. Анорексия, симптомы которой проявляются при актуальных метаболических заболеваниях, заболеваниях ЖКТ, паразитарных и инфекционных заболеваниях, а также при определенных психических расстройствах, может привести к белково-энергетической недостаточности.
…
Болезнь Аддисона или бронзовая болезнь – это патологическое поражение коры надпочечников. Как следствие этого уменьшается секреция гормонов надпочечников. Болезнью Аддисона могут болеть как мужчины, так и женщины. В основной группе риска люди возрастной группы 20–40 лет. Аддисонова болезнь характеризуется как прогрессивное заболевание с тяжёлой клинической картиной.
…
Болезнь Тея – Сакса — это генетическая патология, при которой происходит поражение головного мозга и нервной системы. Сюда входит спинной мозг и оболочки мозга. В первые шесть месяцев жизни ребенка его развитие проходит нормально. Затем начинаются сбои в работе мозговой деятельности. Новорожденным детям диагностируют ганглиозидоз gm1. Такие больные умирают через 3 ‒ 4 года. Эта патология имеет второе название – ганглиозидоз gm2.
…
Прогерия — Википедия
Прогери́я (др.-греч. προσ- — сверх, γέρων — старик) — один из редчайших генетических дефектов. При прогерии возникают изменения кожи и внутренних органов, которые обусловлены преждевременным старением организма. Классифицируют детскую прогерию (синдром Гетчинсона (Хатчинсона)-Гилфорда) и прогерию взрослых (синдром Вернера).
В мире зафиксировано не более 350 случаев прогерии, в числе которых 11-летняя Адалия Роуз (Adalia Rose), 17-летняя девочка Хэйли Окинс, 14-летняя Ашанти Элиотт-Смит, а также 18-летняя Онталаметсе Фалатсе (Ontlametse Phalatse) из городка Хеброн неподалеку от Йоханнесбурга, являющаяся единственной больной прогерией представительницей негроидной расы. Широкую известность в мире получили скончавшийся 5 июня 2011 года Леон Бота, диджей и исполнитель xип-хопa, ведущий видеоблог на YouTube и страдавший прогерией[1], а также американский мотивационный спикер Сэм Бернс, умерший 10 января 2014 в возрасте 17 лет.
У детей
Причина детской прогерии — мутации гена LMNA, кодирующего ламин А. Ламины — белки, из которых выстроен особый слой оболочки клеточного ядра. В большинстве случаев прогерия встречается спорадически, в нескольких семьях зарегистрирована у сибсов, в том числе от кровнородственных браков, что свидетельствует о возможности аутосомно-рецессивного типа наследования. В клетках кожи больных обнаружены нарушения репарации ДНК и клонирования фибробластов, а также атрофические изменения эпидермиса и дермы, исчезновение подкожной клетчатки.
Хотя детская прогерия может быть врождённой, у большинства больных клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется рост ребёнка, отмечаются атрофические изменения дермы, подкожной клетчатки, особенно на лице, конечностях. Кожа истончается, становится сухой, морщинистой, на туловище могут быть склеродермоподобные очаги, участки гиперпигментации. Сквозь истонченную кожу просвечивают вены. Внешний вид больного: большая голова, лобные бугры выступают над маленьким заостренным («птичьим») лицом с клювовидным носом, нижняя челюсть недоразвита. Наблюдаются также атрофия мышц, дистрофические процессы в зубах, волосах и ногтях; отмечаются изменения костно-суставного аппарата, миокарда, гипоплазия половых органов, нарушение жирового обмена, помутнение хрусталика, атеросклероз.
Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство источников указывают возраст смерти от 7 до 27 лет, при этом случаи достижения совершеннолетия очень редки. Известен только один случай пациента, пережившего 27-летний рубеж — японец, описанный Огихарой и другими в 1986 году и проживший 45 лет[2].
У взрослых
Прогерия взрослых имеет аутосомно-рецессивный тип наследования. Дефектный ген — WRN (ген АТФ-зависимой хеликазы). Предполагается связь процесса с нарушением репарации ДНК, обмена соединительной ткани.
Гистологическая картина: уплощение эпидермиса, гомогенизация и склероз соединительной ткани, атрофия подкожной клетчатки с замещением её соединительнотканными волокнами. Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост, симптомы гипогонадизма. Обычно на третьем десятилетии жизни у больного седеют и выпадают волосы, развивается катаракта, постепенно истончается кожа и атрофируется подкожная клетчатка на лице и конечностях, вследствие чего руки и особенно ноги становятся тонкими. Появляются очаги склеродермоподобного уплотнения, дисхромии, наиболее выраженные в дистальных отделах конечностей и на лице, что наряду с тонким клювовидным носом, суженным ротовым отверстием придает ему маскообразность. На местах, подвергающихся давлению, развиваются гиперкератоз, хронические плохо заживающие трофические язвы. Обнаруживаются остеопороз, метастатическая кальцификация мягких тканей, реже остеомиелит. Часто наблюдается сахарный диабет, признаки которого, как и симптомы раннего генерализованного атеросклероза, обычно выявляются у больных в возрасте 30—40 лет; возможны злокачественные новообразования (например, рак кожи, саркома, аденокарцинома).
Диагноз устанавливают на основании клинической картины. Дифференциальный диагноз проводят с врожденной пойкилодермией, склеродермией. Лечение симптоматическое, в основном направлено на профилактику атеросклеротических осложнений, устранение сахарного диабета, трофических язв. Оно проводится терапевтом, эндокринологом или другим специалистом в зависимости от превалирующих клинических симптомов. Прогноз для выздоровления неблагоприятный; большинство больных погибает от атеросклеротических осложнений и злокачественных новообразований. Профилактика не разработана.
Старение
Установлено, что тяжёлая форма прогерии человека — синдром Хатчинсона-Гилфорда — связана с молекулярными изменениями, которые характерны для нормального старения, такими как геномная нестабильность, уменьшение длины теломер и нарушение гомеостаза стволовых клеток. Эти данные вместе с генетическими исследованиями продолжительности жизни привели к гипотезе, согласно которой при синдромах прогерии ускоряется ряд патологических изменений, которые, как правило, управляют обычным процессом старения[3].
Примечания
Литература
- Фёдорова Е. В. О врождённой прогерии. — 1980. — Т. 4. — С. 66. — (Педиатрия).
Синдром Хатчинсона-Гилфорда, или прогерия детей, — крайне редкое заболевание. Его частота составляет 1 на 1 000 000 человек. Именно этот синдром занесен под названием прогерии в OMIM (Online Mendeltan inheritance in Man).
Фенотип пациентов чрезвычайно характерный: маленький рост, «птичье лицо» с клювообразным профилем, преобладание размеров мозговой части черепа над лицевой, выступающая венозная сеть на коже мозговой части, как правило, обнаженной вследствие аллопеции, часто тотальной, с выпадением бровей и ресниц. Наблюдается резкая гипоплазия ключиц, дефекты формы и числа зубов, сухая истонченная кожа, практически полное отсутствие подкожной жировой клетчатки, отставание в развитии, особенно физическом. Больные бесплодны, хотя в литературе описан случай рождения ребенка у пацентки с синдромом Хатчинсона-Гилфорда. В крови повышен уровень Средняя продолжительность жизни описанных носителей синдрома- 13,4 лет (как редкое наблюдение описан единственный 45-летний пациент). Причиной смерти, как правило, служит инфаркт миокарда, с выявлением на аутопсии генерализованного атеросклероза и фиброза миокарда, а также отложения жироподобного вещества в тканях мозга и паренхиматозных органов.
Репарация ДНК при синдроме Хатчинсона-Гилфорда нарушена: установлено, что клетки его носителей не способны избавляться от вызываемых химическими агентами сшивок ДНК-белок. Но главная диагностическая особенность клеток больных с данным синдромом состоит в резко сниженном, по сравнению с нормой, количестве делений, которое способны пройти клетки в культуре (так называемый лимит, или число Хейфлика). В 1971 г. А.М. Оловников высказал предположение об укорочении хромосомных теломер в процессе развития клеток. А в 1992 г. было показано, что для клеток пациентов с синдромом Хатчинсона-Гилфорда характерно врожденное укорочение теломер. Анализ взаимосвязи между лимитом Хейфлика, длиной теломер и активностью теломеразы (фермента, способного наращивать конец теломерной ДНК) дает возможность соотнести естественное старение и процесс формирования клинической картины при синдроме Хатчинсона-Гилфорда.
Крайне низкая частота встречаемости данной формы прогерии позволяет лишь высказывать гипотезы о типе наследования. Аутосомно-рецессивный тип предполагается по аналогии с синдромом Коккейна, имеющим отдельные черты преждевременного старения. Но есть и предположение о развитии синдрома Хатчинсона-Гилфорда вследствие доминантной аутосомной мутации, возникшей de novo. Оно получило косвенное подтверждение на основе измерения теломер у носителей синдрома, их родителей и здоровых доноров. Было показано, существенное укорочение теломер у больных по сравнению с другими двумя испытуемыми группами.
|
|
Что такое прогерия (синдром Хатчинсона-Гилфорда)
Детская прогерия – редчайшее в современном мире генетическое заболевание, вызывающее сверхбыстрое старение детского, как правило, организма. Хотя и были зафиксированы случаи, при которых прогерия развивалась у взрослых, вследствие чего прогерию и принято разделять на детскую и взрослую. Эта болезнь, получившая также название синдрома Хатчинсона-Гилфорда, вызывается мутацией генов, в силу чего обнаружить ее можно лишь на 2-ом – 3-ем годах жизни больного прогерией ребенка.
прогерия у мальчика
В клетках больных, у которых обнаружена детская прогерия, зафиксировано нарушение репарации в ДНК и уменьшение до полного исчезновения подкожной клетчатки, атрофическое изменение эпидермиса и дермы. Прогерия Хатчинсона-Гилфорда у взрослых характеризуется аутосомно-рецессивным типом наследования, что предположительно связанно с нарушением обмена соединительных тканей и нарушением репараций ДНК. Увы, но данное заболевание хоть и дает некоторые подсказки, не позволяет нормализовать процессы старения у тех, кто болен им.
В тяжелой форме прогерия связана с ускоренным молекулярным изменением, характерным для естественного возрастного старения, с нарушением гемостаза стволовых клеток, с уменьшением, порой критическим, длины теломер. В клетках больного тела мутируют белки Lamin A, и увеличивается содержание progerin. Ученые довольно долго сомневались, существует ли progerin в клетках людей, проходящих через естественно старение. Но выяснилось: progerin можно найти в организме каждого человека, точнее – кожных и артериальных клетках. Однако не было ответа, что именно способствует накоплению организмом progerin и Lamin A.
Исследователи установили, что progerin активируется в здоровых людских клетках постепенно, со временем: истончаются, стираясь со временем теломеры, и их повреждение и служит катализатором для выработки progerin.
Увы, но ученые пока еще не понимают, каким именно образом теломеры оказывают стимулирующее воздействие на выработку progerin. А также, существует ли способ корректировать данный процесс. Если бы подобная возможность имелась, то синдром Хатчинсона-Гилфорда не перестал бы оставаться фатальным, ведь неумирающие клетки способны вызывать такую болезнь, как рак.
Однако медики справедливо полагают, что выявление связей между влиянием теломер на progerin выработку – огромный скачок вперед. Ранние исследования оказывались не столь информативными из-за небольшого числа пациентов, страдавших данным заболеванием, и в силу отсутствие современной научной базы, которая бы позволяла изучать эту проблему. А ведь выяснение связи между двумя этими факторами – невероятный прорыв.
Детская прогерия – психологический взгляд
Прогерия Хатчинсона-Гилфорда, наблюдаемая у детей, или детская прогерия, является и довольно уникальным заболеванием с позиций прикладной психологии. Ведь личность детей, у которых данная болезнь диагностирована, как бы подвергается расщеплению, в силу чего в них живет два человека:
— один — маленький ребенок, не собирающийся мириться, что он очень скоро умрет;
— второй — умудренный мыслью о скорой смерти взрослый, не обладающий достаточным жизненным опытом, чтобы действительно считаться по-настоящему взрослым человеком.
У ребенка, у которого была обнаружена синдром Хатчинсона-Гилфорда, развивается невероятной силы самостоятельная личность. Семи-девятилетний, страдающий прогерией малыш, способен принимать решения на уровне взрослого человека. Особенно, если подобные решения касаются его судьбы, здоровья, назначенных ему лечений и операций. Он достаточно психологически взросл, чтобы нести осознанную ответственность за себя, и он ее с почетом несет.
А подобные дети болеют невероятно часто. Хоть большинство болезней, из тех, что наблюдаются у них, детям вообще не свойственны. Рак, инсульты, инфаркты, закупорка сосудов, артриты, облысение и прочие визуальные признаки старения – это приводит, увы, к тому, что редкий ребенок с подобным заболеванием, доживает до 25-ти лет, умирая от какой-либо из возрастных болезней.
К сожалению, из-за упомянутой чуть ранее редкости (порядка 80-90-то зафиксированных случаев на всей Земле) детская прогерия все еще считается неизлечимым заболеванием. Ведь по-настоящему его начали изучать всего в 1999-том году, сделав первый прорыв лишь в начале этого века.
Правда есть предположения, что прогерия возникает гораздо чаще. Просто в мировой статистике не учтены больные дети и взрослые из латиноамериканских и африканских стран, а также из Китая и Индии. Но и при подобной поправке число больных, у которых может быть диагностирован синдром Хатчинсона-Гилфорда, едва ли превысит 150-280 человек.
Если прогерия Хатчинсона-Гилфорда у вашего ребенка
Как понятно из опубликованной нами информация, вероятность того, что детская прогерия будет диагностирована у вашего ребенка, ничтожно мала. Но если подобная беда произошла с вами, не стоит относиться к ней, как к беде. Помните, что прогерия – не приговор. Он просто факт того, что судьба решила позволить вашему малышу жить пусть короче, но гораздо ярче своих сверстников.
И жизнь уже ставших легендами людей, больных прогерией, тому прекрасное доказательство. Многие из больных детей стали музыкантами, художниками, поэтами, известными блоггерами. И мир не отвернулся от них. Как раз напротив, видя, с какой решимостью ими преодолевается эта нелегкая болезнь, заставила людей относиться к таким больным, как к настоящим героям. Человечество давно стало умней и добрей, больше не отталкивая не похожих на него.
Именно как к герою, вы и должны отслоиться к своему малышу, сделав все, чтобы он успел реализоваться и сделать все задуманное, несмотря на ту короткую жизнь, что отведена ему прогерией.
Синдром Хатчинсона-Гилфорда — это генетическое заболевание, которое характеризуется преждевременным и очень быстрым старением, которое начинается с момента рождения ребенка. Следует отметить, что во всем мире таких больных насчитывается очень мало. Патология имеет и другое название — прогерия.

Симптоматика заболевания
Синдром Хатчинсона-Гилфорда имеет очевидные признаки:
- небольшой рост;
- череп имеет ненормально увеличенные габариты;
- отсутствие волос, бровей и ресниц;
- «птичье лицо»;
- костная и другие системы организма являются деформированными;
- отсутствие подкожной жировой прослойки;
- сильное отставание в физическом развитии, при этом мышление и психика являются нормальными.
Следует сказать, что люди имеют определенную среднюю продолжительность жизни: всего 14 лет, хотя известен уникальный случай, когда человек с таким диагнозом прожил до 45 лет. Смерть чаще всего наступает вследствие сердечной недостаточности, а также чрезмерного отложения жира в мозговых тканях.
Причины появления заболевания и особенности лечения
Синдром Хатчинсона-Гилфорда — это патология, спровоцированная нарушениями в хромосомах человека, то есть полному излечению на данный момент она не поддается. Смысл дефекта заключается в том, что количество делений клеток существенно снижается, по сравнению с нормой.
Следует отметить, что синдромом Хатчинсона-Гилфорда (детская форма) может заболеть любой ребенок (независимо от возраста и образа жизни родителей). Он становится заметным в возрасте двух лет. Существует также болезнь Вернера, которая поражает уже взрослых людей. Рост организма останавливается в возрасте трех лет.
Специальной диагностики данной патологии нет, так как она тщательно не изучена. Кроме того, она явно видна по перечисленным симптомам. Что касается лечения заболевания, то эффективных препаратов, которые могли бы останосить его и вернуть человека к нормальной жизни, нет. Однако дети, которые имеют синдром Хатчинсона-Гилфорда должны находиться на учете у врача и постоянно обследоваться. Определенные процедуры могут помочь замедлить прогрессирование патологии.
План лечения синдрома составляется отдельно для каждого пациента. Оно включает повышение двигательной активности ребенка, физиотерапевтические процедуры. Благодаря употребелнию небольших доз аспирина у малыша снижается риск возникновения инсульта. Иногда больному проводятся хирургические вмешательства, которые направлены на шунтирование сосудов, удаление молочных зубов.
 Если вы не представляете, как выглядит ребенок с синдромом Хатчинсона-Гилфорда, фото, размещенные в статье, покажут вам это.
Если вы не представляете, как выглядит ребенок с синдромом Хатчинсона-Гилфорда, фото, размещенные в статье, покажут вам это.
Особенности развития детей, страдающих представленным заболеванием
Если физически ребенок расти не может, то мышление и психика его не страдает. Малыш может научиться читать, может получать знания. Естественно, процесс обучения будет проходить в домашних условиях.
Что касается последующего прогноза, то он неутешителен. Долго такие дети не живут. Дело в том, что за один год у них проходит 6-8 лет. Естественно, ученые работают над тем, чтобы раскрыть механизм появления заболевания и способы его остановки. Однако современный уровень развития медицины может только немного улучшить качество жизни больного, а также замедлить процесс старения.

В октябре 2005 года в московской клинике врачи сделали первую операцию пациентке, страдающей синдромом преждевременного старения. Прогерия – весьма редкая болезнь. Медицинские светила всего мира утверждают, что с момента «пробуждения» в организме этого заболевания люди в среднем живут всего 13 лет.
По статистике, с подобным генетическим дефектом рождается примерно 1 человек на 4 миллиона. Прогерию подразделяют на детскую, называемую синдромом Хатчинсона-Гилфорда, и прогерию у взрослых – синдром Вернера. В обоих случаях происходит поломка генного механизма и начинается противоестественное истощение всех систем жизнеобеспечения. При синдроме Гетчинсона-Гилфорда задерживается физическое развитие детей при одновременном появлении у них в первые же месяцы жизни признаков старческого поседения, облысения, морщин.
К пяти годам такой ребенок страдает всеми старческими недугами: снижением слуха, артритом, атеросклерозом, и не доживает даже до 13 лет. При синдроме Вернера молодые люди начинают быстро стареть в возрасте 16-20 лет, и уже к 30-40 годам такие больные умирают при всех симптомах глубокой старости.
Лекарства от прогерии нет – используя все научные достижения, можно лишь замедлить необратимый процесс.
Похищенная молодость
Случаи внезапного старения весьма прозаичны: живущий в нормальных условиях ребенок поначалу удивляет окружающих своим быстрым развитием. В малолетнем возрасте он выглядит как совершеннолетний, а затем у него начинают проявляться все признаки… приближающейся старости.

В 1716 году в английском городе Ноттингеме умер восемнадцатилетний сын графа Уильяма Шеффилда, начавший стареть в тринадцатилетнем возрасте. Молодой Шеффилд выглядел намного старше своего отца: cедые волосы, наполовину выпавшие зубы, морщинистая кожа. У злосчастного юноши был вид потрепанного жизнью мужчины, он очень от этого страдал и принял смерть как избавление от мук.
Есть случаи подобного рода и среди представителей королевских родов. Венгерский король Людвиг II в девятилетнем возрасте уже достиг полового созревания и с удовольствием развлекался с придворными девицами. В четырнадцать он обзавелся густой окладистой бородой и стал выглядеть минимум на 35 лет. Год спустя он женился, а к шестнадцатилетию супруга подарила ему сына. Но в восемнадцать лет Людвиг полностью поседел, а еще два года спустя скончался со всеми признаками старческого одряхления.
Любопытно, что ни сын короля, ни дальнейшие его потомки подобной болезни не унаследовали. Из примеров ХIХ века можно выделить историю простой деревенской девушки, француженки Луизы Равальяк. В восьмилетнем возрасте Луиза, полностью сформировавшаяся как женщина, забеременела от местного пастуха и родила вполне здорового ребенка. К шестнадцатилетию у нее уже было трое детей и она выглядела старше своей матери, в 25 она превратилась в дряхлую старуху и, не дожив до 26, умерла от старости.
Не меньший интерес вызывают судьбы тех, кто жил в XX веке. Кое-кому из них повезло несколько больше, чем другим. Например, родившийся в 1905 году житель американского города Сан-Бернардино Майкл Соммерс, рано созревший и постаревший, смог дожить до 31 года. Поначалу сверхбыстрое вступление во взрослую жизнь его даже радовало. Но когда в семнадцать Майкл с ужасом понял, что начал стареть, он стал предпринимать отчаянные попытки остановить этот губительный процесс.
Но врачи только разводили руками, не в силах чем-либо помочь. Немного замедлить дряхление Соммерсу удалось после того, как он, перебравшись на постоянное жительство в деревню, стал проводить много времени на свежем воздухе. Но все же к 30 годам он превратился в старика, а через год его доконал обыкновенный грипп. Среди прочих подобных феноменов можно выделить англичанку Барбару Дэлин, которая умерла в 1982 году в возрасте 26 лет.
К 20 годам успевшая побывать замужем и родить двоих детей, Барбара быстро и необратимо состарилась. Именно поэтому ее бросил молодой муж, не пожелавший жить со «старой развалиной». В 22 года от ухудшения здоровья и перенесенных потрясений «старушка» ослепла и до самой смерти передвигалась на ощупь или в сопровождении собаки-поводыря, подаренной ей властями ее родного Бирмингема.
Полю Демонжо из французского города Марселя двадцать три года. При этом выглядит он на все 60 и ощущает себя человеком преклонного возраста. Однако пока не теряет надежды, что свершится чудо и будет найдено средство, которое прекратит его стремительное одряхление. Его собрату по несчастью, сицилийцу из города Сиракузы Марио Термини нет и 20 лет лет, однако на вид ему намного больше 30. Сын богатых родителей, Термини ни в чем себе не отказывает, встречается с местными красотками и ведет разгульный образ жизни.
А что у нас?
«Скороспелые» люди жили и в нашей стране. Еще во времена Ивана Грозного сын бояр Михайловых Василий умер в 19-лет дряхлым стариком. В 1968 году в возрасте 22 лет в Свердловске скончался рабочий одного из заводов Николай Шориков. Стареть он начал еще в шестнадцатилетнем возрасте, чем крайне озадачил врачей. Светила медицины только разводили руками: «Такого не может быть!»
Став стариком в том возрасте, когда все только начинается, Николай потерял всякий интерес к жизни и покончил с собой, наглотавшись таблеток… А тринадцать лет спустя в Ленинграде умер 28-летний «старец» Сергей Ефимов. Юношеский период у него закончился к одиннадцати годам, а заметно стареть он начал после двадцати и умер дряхлым стариком, за год до смерти почти полностью потеряв способность здраво мыслить.
Во всем виноваты гены
Многие ученые считают, что основная причина этого заболевания – генетическая мутация, ведущая к накоплению большого количества протеина в клетках. Экстрасенсы и маги утверждают, что есть специальные методики насылания «порчи» с целью состарить человека.

Кстати, эта болезнь встречается не только у людей, но и у животных. У них также жизненные циклы и периоды порой идут по сценарию год за три, а то и за десять лет. Возможно, решение проблемы будет найдено именно после многолетних экспериментов на братьях наших меньших.
Как удалось установить исследователям из Калифорнийского Университета, препарат под названием «ингибитор фарнезилтрансферазы» существенно снижает скорость проявления симптомов преждевременного старения у лабораторных мышей. Возможно, это лекарство окажется пригодным и для лечения людей.
Вот как характеризует симптомы недуга у детей кандидат биологических наук Игорь Быков: «Прогерия возникает внезапно с появления крупных пигментных пятен на теле. Затем людей начинают одолевать самые настоящие старческие хвори. У них развиваются болезни сердца, сосудов, диабет, выпадают волосы и зубы, исчезает подкожный жир. Кости делаются ломкими, кожа морщинистой, а тела – сгорбленными. Процесс старения у таких больных протекает примерно в десять раз быстрее, чем у здорового человека. Зло коренится, скорее всего, в генах. Есть гипотеза, что они вдруг перестают отдавать клеткам команду делиться. И те быстро приходят в негодность».
Гены перестают отдавать клеткам команду делится вроде бы от того, что укорачиваются кончики ДНК в хромосомах, – так называемые теломеры, длиной которых предположительно и отмерен срок человеческой жизни. Подобные процессы идут и у нормальных людей, но гораздо медленнее. Но совершенно непонятно, в результате какого именно нарушения укорачиваются теломеры и начинается ускорение старения как минимум в 10 раз. Сейчас ученые с помощью ферментов пытаются удлинить теломеры. Появились даже сообщения, что американским генетикам удалось таким образом продлить жизнь мухам. Но до результатов, применимых на практике, пока далеко. Людям не удается помочь даже на уровне экспериментов. К счастью, по наследству недуг не передается.
Предполагается, что сбой в геноме происходит еще в период внутриутробного развития. Пока наука не может отслеживать и управлять этим сбоем: она может только констатировать факт, но, возможно в недалеком будущем геронтология ответит миру на этот вопрос.
Де Сандре-Джованноли А, Бернард Р, Кау П, Наварро С, Амиэль Дж, Боккаччо I, Лионнет С, Стюарт CL, Мюнхен А, Ле Меррер М., Леви Н. Ламина усечение в прогерии Хатчинсона-Гилфорда. Наука. 2003 июнь 27; 300 (5628): 2055. Epub 2003 17 апреля.
Эрикссон М., Браун В.Т., Гордон Л.Б., Глинн М.В., Сингер Дж., Скотт Л., Эрдос М.Р., Роббинс С.М., Моисей Т.Ю., Берглунд П., Дутра А, Пак Э, Дуркин С., Чока А.Б. Боэнке М, Гловер Т.В., Коллинз Ф.С.Рецидивирующие точечные мутации de novo в ламине А вызывают синдром Хатчинсона-Гилфорда. Природа. 2003 15 мая; 423 (6937): 293-8. Epub 2003 25 апреля.
Гош С., Чжоу З. Генетика старения, прогерии и расстройств ламинации. Curr Opin Genet Dev. 2014 июнь; 26: 41-6. doi: 10.1016 / j.gde.2014.05.003. Epub 2014 6 июля. Обзор.
Гольдман Р.Д., Шумейкер Д.К., Эрдос М.Р., Эрикссон М., Гольдман А.Е., Гордон Л.Б., Грюнбаум Й., Хуон С., Мендез М., Варга Р., Коллинз Ф.С. Накопление мутантного ламина А вызывает прогрессивные изменения в ядерной архитектуре при синдроме прогерии Хатчинсона-Гилфорда.Proc Natl Acad Sci U S A. 2004 Jun 15; 101 (24): 8963-8. Epub 2004 Jun 7.
Gonzalez JM, Pla D, Perez-Sala D, Andres V. Ламины А-типа и синдром Хатчинсона-Гилфорда прогерии: патогенез и терапия. Фронт Биоски (Schol Ed). 2011 июня 1; 3: 1133-46. Обзор.
Гордон Л.Б., Браун В.Т., Коллинз Ф.С. Синдром Хатчинсона-Гилфорда Прогерия. 2003 12 декабря [обновлено 2015 8 января]. В: Пагон Р.А., Адам М.П., Ардингер Х.Х., Уоллес С.Е., Амемия А., Боб Л.Дж., Берд Т.Д., Ледбеттер Н., Меффорд Х.К., Смит Р.Дж.Х., Стивенс К., редакторы.GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017. Доступно по адресу: http://www.ncbi.nlm.nih.gov/books/NBK1121/
Halaschek-Wiener J, Brooks-Wilson A. Прогерия стволовых клеток: истощение стволовых клеток при синдроме прогерии Хатчинсона-Гилфорда. J Gerontol A Biol Sci Med Sci. 2007 янв; 62 (1): 3-8. Обзор.
Хеннекам RC. Прогрессирующий синдром Хатчинсона-Гилфорда: обзор фенотипа. Am J Med Genet A. 2006 Dec 1; 140 (23): 2603-24.Обзор.
Pollex RL, Hegele RA. Синдром Хатчинсона-Гилфорда. Clin Genet. 2004 ноябрь; 66 (5): 375-81. Обзор.
.
Синдром Хатчинсона-Гилфорда Прогерия | IntechOpen
1. Введение
Синдром Хатчинсона-Гилфорда (HGPS) представляет собой летальное врожденное заболевание, характеризующееся преждевременным появлением ускоренного старения у детей. Хотя HGPS был впервые описан Джонатаном Хатчинсоном [1], а затем Гастингсом Гилфордом [2] более века назад, только в 2003 году была раскрыта генетическая основа HGPS [3, 4]. Приблизительно 90% пациентов с HGPS имеют идентичную мутацию в отцовском аллеле гена LMNA — замена цитозина на тимин в нуклеотиде 1824, c.1824C> Т. Хотя, по-видимому, молчащая мутация (то есть без изменений в аминокислоте, G608G), она вызывает аберрантный сплайсинг мРНК, что приводит к образованию усеченного и частично обработанного белка пре-ламина А, называемого «прогерин» [3, 4] , Считается, что накопление прогерина лежит в основе патофизиологии HGPS. Лица с HGPS, по-видимому, проявляют связанные со старением фенотипы гораздо быстрее, чем обычно, что приводит к тому, что у маленьких детей появляется внешний вид и состояние здоровья пожилого человека.Сообщаемая частота HGPS составляет от 1 до 4-8 миллионов новорожденных, и в настоящее время известно, что 89 пациентов во всем мире живут с HGPS [5]. Наблюдаемое соотношение заболеваемости HGPS между мужчинами и женщинами составляет 1,2: 1, и не было сообщений об этническом рецидиве. HGPS влияет на различные системы организма, включая рост, скелет, жировые отложения, кожу, волосы и сердечно-сосудистую систему. Однако пациенты не обнаруживают дефектов в своих умственных и интеллектуальных способностях [6-8]. Удивительно, но прогерин также был обнаружен у нормальных незатронутых людей, и его уровень увеличивается с возрастом, что предполагает сходный генетический механизм в прогерии, как и при нормальном физиологическом старении.Таким образом, были разработаны многочисленные модели на животных, чтобы лучше понять механизм (ы) HGPS и разработать лекарство от этой разрушительной болезни.
В этой главе будут обсуждаться основные аспекты HGPS, такие как признаки и симптомы, генетическая основа, модели на животных и методы лечения.
2. Признаки и симптомы
Средний возраст при постановке диагноза HGPS составляет 2,9 года [6]. Диагноз, как правило, прост, поскольку больные пациенты демонстрируют классические симптомы и сильно напоминают друг друга.У пострадавших людей нет никаких признаков заболевания при рождении, но в течение первых лет жизни у них постепенно развивается вид, часто называемый пожилым [9, 10]. Некоторые из типичных физических характеристик HGPS включают алопецию (выпадение волос, включая кожу головы и брови), выраженные вены кожи головы и лба, классические черты лица, включая лобное выпячивание, выступающие уши с отсутствующими долями, глифический (широкий, слегка вогнутый носовой выступ) нос видные глаза, тонкие губы и микрогнатия (маленькая челюсть) с вертикальной срединной бороздкой на подбородке [7, 11, 12] (рис. 1).Ненормальные и отсроченные зубные ряды также распространены, а тонкая и часто стянутая кожа является следствием значительной потери подкожного жира [7, 10] (рис. 1). Пациенты с HGPS имеют высокие голоса, позицию верховой езды, ограниченную подвижность суставов и имеют низкий рост (средняя конечная высота 100-110 см; средняя конечная масса 10-15 кг). По мере взросления у них развивается остеолиз, особенно с участием дистальных фаланг и ключиц [6-8, 11, 13]. В среднем смерть наступает в возрасте 13 лет, при этом по меньшей мере 90% субъектов HGPS умирают от прогрессирующего атеросклероза коронарных и цереброваскулярных артерий [7].
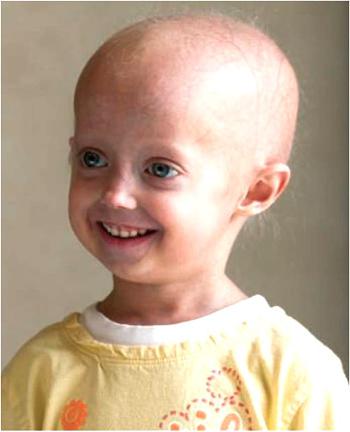
Рисунок 1.
Фотографии 7-летней девочки с HGPS (LMNA c.1824C> T, p.G608G). Этот пациент имеет типичные фенотипы, включая алопецию, тонкую и плотную кожу, потерю подкожного жира, заметные вены кожи головы и лба, выдающиеся глаза, выпученные уши, тонкие губы и маленькую челюсть. Фотографии предоставлены Фондом исследований Progeria.
Недавно, Olive и др. . сообщили о сходстве между многими аспектами сердечно-сосудистых заболеваний у пациентов с HGPS и нормальными взрослыми людьми с атеросклерозом и предположили, что прогерин может быть фактором риска атеросклероза в общей популяции [14].У пациентов с HGPS обнаружены признаки, которые классически связаны с атеросклерозом старения, в том числе наличие бляшек в коронарных артериях, поражение артерий, показывающее кальцификацию, воспаление и признаки эрозии или разрыва бляшки. Авторы предполагают, что накопление прогерина в сосудистых клетках вызывает ядерные дефекты и увеличивает восприимчивость к механическим нагрузкам, что, в свою очередь, вызывает гибель клеток и воспалительный ответ, что приводит к атеросклерозу [14].
Интересно, что, несмотря на наличие множества симптомов преждевременного старения, многие другие органы, такие как печень, почка, легкое, мозг, желудочно-кишечный тракт и костный мозг, по-видимому, не затронуты.Кроме того, не все процессы старения развиваются у пораженных детей. Например, распространенность умственных расстройств, рака и катаракты не выше у пациентов с HGPS [7]. На сегодняшний день существует мало объяснений того, почему в HGPS поражены только определенные органы. Тем не менее, исследователи пытаются уточнить некоторые из этих удивительных наблюдений. Недавно Юнг и его коллеги предположили, что отсутствие когнитивных нарушений у пациентов с HGPS можно объяснить понижающей регуляцией экспрессии пре-ламина А в мозге [15].Кроме того, авторы предположили, что низкий уровень пре-ламина А в мозге может регулироваться мозговой специфической микроРНК (miRNA), miRNA-9. В поддержку результатов этого исследования Nissan и др. . Недавно был опубликован многообещающий результат, показывающий, что miRNA-9 обратно регулирует экспрессию ламина А и прогерина в нервных клетках, и предположил, что защита нервных клеток от токсического накопления прогерина в HGPS может быть обусловлена экспрессией miRNA-9 [16]. Для изучения изменений экспрессии miRNA-9 и его влияния на уровень прогерина в мозге необходимы дальнейшие исследования, возможно, с использованием моделей на животных.
Клинические признаки, наблюдаемые в HGPS, сильно напоминают несколько аспектов естественного старения. По этой причине HGPS послужил полезной моделью для расшифровки некоторых механизмов, лежащих в основе физиологического старения. Первое свидетельство изменений ядерной архитектуры во время нормального процесса старения было получено в работе в C. elegans [17]. В этом исследовании авторы продемонстрировали, что ядерные дефекты накапливаются при старении, и предположили, что HGPS может быть результатом увеличения скорости нормального процесса старения [17].Scaffidi и Misteli показали, что клетки пациентов с HGPS и людей в возрасте обычно имеют несколько общих ядерных дефектов [18]. Кроме того, небольшое количество белка прогерина было обнаружено в белковых экстрактах, полученных от пожилых людей, которое отсутствовало в молодых образцах [19]. Родригес и др. . количественно оценили уровни транскриптов прогерина с использованием количественной ОТ-ПЦР в реальном времени и показали, что транскрипт прогерина присутствует у незатронутых пожилых людей, хотя на очень низком уровне по сравнению с пациентами HGPS, и этот уровень увеличился с в условиях старения in vitro, аналогично Клетки HGPS [20].Недавно, Olive и другие также сообщили, что, хотя уровень прогерина значительно выше у пациентов с HGPS, прогерин также присутствует в коронарных артериях людей, не страдающих HGPS, и значительно увеличивается с возрастом [14]. В целом, накопление прогерина, который образуется редко с течением времени в результате процесса старения, представляется возможным кандидатом и частично отвечает за клеточное старение и нестабильность генома, которая наблюдается в стареющих клетках.В HGPS это происходит значительно быстрее по сравнению с клетками нормального возраста из-за более активного использования донорного сайта с загадочным сплайсингом, продуцируя более высокий уровень прогерина. Связь между этой болезнью ускорения старения и появлением аналогичных симптомов в течение жизни нормального человека неясна. Тем не менее, идея о том, что прогерин может играть роль в общем старении человека, подтверждается многочисленными исследованиями, упомянутыми выше.
3. Генетическая основа
Ген LMNA , как известно, является горячей точкой для вызывающих заболевание мутаций, и ему уделяется большое внимание благодаря его связи с различными заболеваниями человека.На сегодняшний день было обнаружено более 400 мутаций, распространяющихся по области, кодирующей белок гена LMNA (см. Обзор [21]). Ген LMNA обнаружен в хромосоме 1q21.2-q21.3 и состоит из 12 экзонов. Посредством альтернативного сплайсинга ген LMNA кодирует ламины A-типа, ламины A и C (ламин A, AΔ10, C и C2), из которых ламин A (кодируется экзонами 1-12) и ламин C (кодируется экзоны 1-10) являются основными изоформами, экспрессируемыми во всех дифференцированных клетках позвоночных [22, 23].Ламины типа В, ламины В1 и В2, представляют собой еще один тип ламинов, которые кодируются генами LMNB1 и LMNB2 соответственно. Ламины типа В обнаружены во всех клетках и экспрессируются в процессе развития. Ламины A, C, B1 и B2 являются ключевыми структурными компонентами ядерной пластинки, промежуточной филаментной структуры, которая лежит на внутренней поверхности внутренней ядерной мембраны и отвечает за поддержание структурной стабильности и организацию хроматина (см. Обзор [24]).Ядерная пластинка определяет форму и размер ядра клетки и участвует в репликации и транскрипции ДНК. Кроме того, было показано, что ядерная пластинка взаимодействует с несколькими белками, ассоциированными с ядерной мембраной, факторами транскрипции, а также с самим гетерохроматином. Ядерная пластинка необходима для большинства ядерных активностей, таких как организация хроматина, репликация ДНК, регуляция клеточного цикла, позиционирование ядра внутри клетки, сборка / разборка ядра во время деления клетки, а также для модуляции основных регуляторных генов и сигнальных путей [ 25-27].Существует более 10 различных нарушений, которые вызваны мутациями в гене LMNA , и эти нарушения в совокупности называются ламинопатиями и включают невропатии, мышечные дистрофии, кардиомиопатии, липодистрофии, в дополнение к прогероидным синдромам (см. Главу о ламинопатиях).
Генетическая основа для HGPS была неизвестна до тех пор, пока не было обнаружено, что она является единственной нуклеотидной мутацией в отцовском аллеле с аутосомно-доминантной экспрессией [3, 4]. Несмотря на то, что многочисленные мутации, как сообщалось, вызывают HGPS [4, 28-33], примерно 90% случаев вызваны рецидивирующей, доминантной, de novo гетерозиготной молчащей аминокислотной заменой на c.1824C> T, G608G (переход от глицин-GGC к глицину-GGT, называемый G608G) гена LMNA [4] (рис. 2). Эта мутация локализована в 11 экзоне гена LMNA и приводит к повышенной активации донорного сайта криптического сплайсинга, сплайсируя ген LMNA на 5 нуклеотидах выше по течению от мутации, что приводит к накоплению аберрантной транскрипции мРНК, при этом отсутствует 150 нуклеотидов из нормальный пре-ламин А. Эта мутированная мРНК затем транслируется в белок, называемый «прогерин», в котором отсутствуют 50 аминокислотных остатков из его С-концевой области.Было высказано предположение, что различные мутации вызывают активацию одного и того же криптического сайта сплайсинга в экзоне 11 гена LMNA , и тяжесть заболевания коррелирует с использованием этого сайта сплайсинга (рис. 2). Например, Moulson и другие описали двух пациентов с особенно тяжелыми прогероидными симптомами, явно более серьезными, чем типичный случай HGPS [30]. В обоих случаях количество прогерина относительно правильно обработанного пре-ламина А было значительно больше, чем у типичного HGPS, что позволяет предположить, что тяжесть заболевания, по-видимому, зависит от количества прогерина в клетках [30].Совсем недавно Reunert и соавторы сообщили о еще одном более серьезном случае [31]. Этот пациент имел гетерозиготную мутацию LMNA c.1821G> A, которая приводит к прогерии новорожденных со смертью на первом году жизни [31]. Авторы показали, что отношение белка прогерина к зрелому ламину A было выше у этого пациента по сравнению с классическим HGPS, а также предположили, что это соотношение определяет тяжесть заболевания при прогерии [31]. Противоположные случаи были также показаны Хисамой и коллегами.В этом исследовании мутации в соединении экзона 11 и интрона 11 гена LMNA привели к значительно более низкому уровню прогерина по сравнению с HGPS, что привело к появлению прогероидного синдрома у взрослых, очень напоминающего синдром Вернера [33].
Рисунок 2.
Схематическая диаграмма, показывающая точечные мутации, приводящие к усиленной активации сайта криптического сплайсинга в экзоне 11 гена LMNA [4, 30, 31, 33]. Все эти мутации приводят к внутренней делеции 150 нуклеотидов экзона 11, что в конечном итоге приводит к выработке аномально обработанного белка, называемого «прогерин».Интересно отметить, что нормальная последовательность LMNA также может быть аномально сплайсирована, удаляя 150 нуклеотидов экзона 11 у здоровых людей, и эта частота может возрастать с возрастом, приводя к клеточному старению [18, 20].
В нормальных условиях зрелый белок ламина А получают из предшественника, предварительно ламина А, посредством серии посттрансляционных стадий обработки, которые начинаются на С-терминальном конце. CaaX мотив в C-терминальном хвосте (где C представляет собой цистеин, a являются алифатическими аминокислотами, а X могут быть любыми аминокислотами) сигналы для 4 последовательных модификаций (рис. 3А) ).Сначала цистеин из мотива CaaX фарнезилируется фарнезилтрансферазой (FTase), затем последние три аминокислоты ( aaX ) расщепляются с помощью металлопротеазы цинка, ZMPSTE24 (мышь) или FACE-1 (человек). После этого расщепления фарнезилированный C-концевой цистеин метилируется изопренилцистеинкарбоксиметилтрансферазой (ICMT). Наконец, последние 15 аминокислот белка снова расщепляются ZMPSTE24, в результате чего получается зрелый ламин A.
Рис. 3.
Посттрансляционная обработка пре-ламина A в нормальном состоянии (A) и усеченного пре-ламина A («Пре-прогерин») в (B) HGPS.Сайт протеолитического расщепления (мотив RSYLLG) находится в пределах области из 50 аминокислот, которая теряется из-за мутации HGPS, и в результате эндопротеаза ZMPSTE24 не может распознавать и выполнять последующее расщепление в восходящем направлении. Следовательно, усеченный белок ламина А (то есть прогерин) остается фарнезилированным, что, как полагают, оказывает доминирующее негативное влияние на HGPS.
В HGPS могут быть выполнены первые 3 этапа посттрансляционного созревания (то есть фарнезилирование, расщепление и метилирование), в то время как четвертый этап обработки не может быть завершен, поскольку мутация G608G исключает второй сайт расщепления, распознаваемый ZMPSTE24 из пре-ламин А, образующий постоянно фарнезилированную форму прогерина (рис. 3В) [34].Считается, что этот неправильно обработанный белок в HGPS лежит в основе прогрессирования фенотипа заболевания [35]. Поскольку прогерин в отличие от зрелого ламина А остается фарнезилированным, он приобретает высокое сродство к ядерной мембране, что приводит к нарушению целостности ядерной пластинки. Действительно, клетки пациента HGPS обнаруживают ряд нарушений в структуре и функции ядра. При непрямой иммунофлуоресцентной маркировке антителами, направленными против ламинов A / C, фибробласты от индивидуумов с HGPS характеризовались наличием дисморфных ядер с измененным размером и формой, наличием долек, морщин, грыж ядерной оболочки, утолщением ядерной пластинки, потеря периферического гетерохроматина и кластеризация ядерных пор [4, 36, 37].Эти особенности ухудшаются с пассажами в культуре клеток и коррелируют с очевидным внутриядерным накоплением прогерина (Figure 4) [36, 38]. В дополнение к постоянному фарнезилированию прогерина было выдвинуто предположение, что делеция сайта фосфорилирования (Ser 625), обнаруженная в области, удаленной из 50 аминокислот, может также учитывать некоторые из фенотипов HGPS в качестве зависимого от клеточного цикла фосфорилирования ламина A важен для своей нормальной функции [4, 39].
Рисунок 4.
Иммуноокрашивание фибробластов кожи, взятых у нормального человека (слева) и пациента HGPS (справа), демонстрирующего ядерный блеббинг.Ламин A / C обозначен красным, а ламинат B1 — зеленым. Обратите внимание, что экспрессия ламина B1 теряется в области blebbed. Фигура была адаптирована из Shimi et al. (2012) [40], с разрешения Elsevier.
Многочисленные исследования касались возрастных характеристик клеток HGPS, которые интригующе параллельны свойствам фибробластов у пожилых людей. Клеточное старение является отличительной чертой процесса старения, и ядра клеток у старых людей имеют дефекты, сходные с дефектами клеток пациента с HGPS, включая повышенное повреждение ДНК [18, 41, 42], подавление регуляции некоторых ядерных белков, таких как гетерохроматиновый белок HP1 и группа LAP2 белков, ассоциированных с ламином А [18, 37], и изменения в модификациях гистонов [18].Гетерохроматин становится более дезорганизованным с увеличением старения у пациентов [43], а нарушение регуляции организации хроматина является распространенным явлением в HGPS, где прогерин, как известно, изменяет метилирование гистонов [44, 45]. Интересно, что загадочный сайт сплайсинга, который постоянно активируется в HGPS, редко используется при «нормальной» обработке перед ламинацией A у здоровых
.
Синдром Шетре-Чотзена — Genetics Home Reference
Cai J, Goodman BK, Patel AS, Mulliken JB, Ван Малдергем L, Хогансон GE, Paznekas WA, Бен-Нериа Z, Шеффер R, Каннингем ML, Daentl DL, Jabs EW , Повышенный риск задержки развития при синдроме Saethre-Chotzen связан с делециями TWIST: улучшенная стратегия скрининга мутаций TWIST. Hum Genet. 2003 Dec; 114 (1): 68-76. Epub 2003 Sep 25.
Cai J, Shoo BA, Sorauf T, Jabs EW. Новая мутация в гене TWIST, вовлеченная в синдром Saethre-Chotzen, обнаружена в оригинальном случае синдрома Robinow-Sorauf.Clin Genet. 2003 июл; 64 (1): 79-82.
Чун К., Тиби А.С., Юнг Дж. Х., Кеннеди С., Лафрамбуаз Р., Мешино В. С., Накабаяши К., Шерер С. В., Рэй П. Н., Тешима И. Генетический анализ пациентов с фенотипом Сэтре-Чотзена. Am J Med Genet. 2002 15 июня, 110 (2): 136-43.
Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Синдромный краниосиностоз: от истории к водородным связям. Ортод Краниофак Рез. 2007 май; 10 (2): 67-81. Обзор.
Gallagher ER, Ratisoontorn C, Cunningham ML.Синдром Саэтре-Чотцена. 2003 16 мая [обновлено 2012 14 июня]. В: Пагон Р.А., Адам М.П., Ардингер Х.Х., Уоллес С.Е., Амемия А., Боб Л.Дж., Берд Т.Д., Ледбеттер Н., Меффорд Х.К., Смит Р.Дж.Х., Стивенс К., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017. Доступно по адресу: http://www.ncbi.nlm.nih.gov/books/NBK1189/
Gripp KW, Zackai EH, Stolle CA. Мутации в гене TWIST человека. Хум Мутат. 2000; 15 (2): 150-5. Обзор. Ошибка в: Hum Mutat 2000; 15 (5): 479.
Kress W, Schropp C, Lieb G, Петерсен B, Büsse-Ratzka M, Kunz J, Рейнхарт E, Schäfer WD, продано J, Hoppe F, Панке J, Трузен A, Соренсен N, Краусс J, Колльманн H Синдром Saethre-Chotzen, вызванный мутациями гена TWIST 1: функциональная дифференциация от синдрома коронарного синостоза Muenke. Eur J Hum Genet. 2006 янв; 14 (1): 39-48.
де Хеер И.М., де Кляйн А., ван ден Уувеланд А.М., Вермей-Кирс С., Ваутерс К., Ваандрагер Ю.М., Ховиус С.Е. Клинико-генетический анализ пациентов с синдромом Saethre-Chotzen.Plast Reconstr Surg. 2005 июнь; 115 (7): 1894-902; обсуждение 1903-5.
.
Синдром Ли — Genetics Home Reference
Синдром Ли может быть вызван мутациями одного из более чем 75 различных генов. У людей большинство генов содержится в ДНК в клетке, называемой ядерной ДНК. Тем не менее, некоторые гены находятся в ДНК в специализированных структурах в клетке под названием. Этот тип ДНК известен как митохондриальная ДНК (мтДНК). Хотя большинство людей с синдромом Ли имеют мутацию в ядерной ДНК, около 20 процентов имеют мутацию в мтДНК.
Большинство генов, связанных с синдромом Ли, участвуют в процессе производства энергии в митохондриях.Митохондрии используют кислород для преобразования энергии из пищи в форму, которую клетки могут использовать в процессе, называемом. Пять белковых комплексов, состоящих из нескольких белков каждый, участвуют в этом процессе. Комплексы названы комплексом I, комплексом II, комплексом III, комплексом IV и комплексом V. Во время окислительного фосфорилирования белковые комплексы шаг за шагом способствуют выработке аденозинтрифосфата (АТФ), основного источника энергии в клетке. Перенос отрицательно заряженных частиц называется электронами.Многие генные мутации, связанные с синдромом Ли, влияют на белки в этих комплексах или нарушают их сборку. Эти мутации снижают или устраняют активность одного или нескольких из этих комплексов, что может привести к синдрому Ли.
Разрушение комплекса I, также называемого NADH: убихиноноксидоредуктазой, является наиболее частой причиной синдрома Ли, на которую приходится почти треть случаев заболевания. По меньшей мере 25 генов, участвующих в образовании комплекса I, обнаруженного в ядерной или митохондриальной ДНК, были связаны с синдромом Ли.
Разрушение комплекса IV, также называемого цитохром с оксидазой или ЦОГ, также является частой причиной синдрома Ли, на который приходится примерно 15 процентов случаев. Одним из наиболее часто мутирующих генов при синдроме Ли является SURF1. Этот ген, который находится в ядерной ДНК, предоставляет инструкции для создания белка, который помогает собирать белковый комплекс ЦОГ (комплекс IV). Этот комплекс, который участвует в последнем этапе переноса электрона в окислительном фосфорилировании, обеспечивает энергию, которая будет использоваться на следующем этапе процесса для генерации АТФ.Мутации в гене SURF1 обычно приводят к аномально короткому белку SURF1, который расщепляется в клетках, что приводит к отсутствию функционального белка SURF1. Потеря этого белка уменьшает образование нормальных комплексов ЦОГ, что ухудшает выработку митохондриальной энергии.
Наиболее распространенная мутация мтДНК при синдроме Ли затрагивает ген MT-ATP6, который предоставляет инструкции для создания фрагмента комплекса V, также известного как. Используя энергию, обеспечиваемую другими белковыми комплексами, комплекс АТФ-синтазы генерирует АТФ.Мутации гена MT-ATP6, обнаруженные примерно у 10 процентов людей с синдромом Ли, блокируют генерацию АТФ. Другие мутации мтДНК, связанные с синдромом Ли, снижают активность других комплексов белков окислительного фосфорилирования или приводят к уменьшению образования митохондриальных белков, которые ухудшают выработку митохондриальной энергии.
Другие генные мутации, связанные с синдромом Ли, снижают активность одного или нескольких белковых комплексов окислительного фосфорилирования или влияют на дополнительные стадии, связанные с выработкой энергии.Например, синдром Ли может быть вызван мутациями в генах, которые образуют комплекс пируватдегидрогеназы или коэнзим Q10, оба из которых участвуют в выработке митохондриальной энергии. Мутации в генах, которые управляют репликацией мтДНК или продукцией митохондриальных белков, также могут нарушать выработку митохондриальной энергии.
Хотя точный механизм неясен, исследователи считают, что нарушение окислительного фосфорилирования может привести к гибели клеток из-за уменьшения энергии, доступной в клетке.Некоторые ткани, которые требуют большого количества энергии, такие как мозг, мышцы и сердце, кажутся особенно чувствительными к снижению клеточной энергии. Гибель клеток в головном мозге, вероятно, вызывает характерные поражения, наблюдаемые при синдроме Ли, которые способствуют появлению признаков и симптомов состояния. Гибель клеток в других чувствительных тканях также может способствовать развитию синдрома Ли.