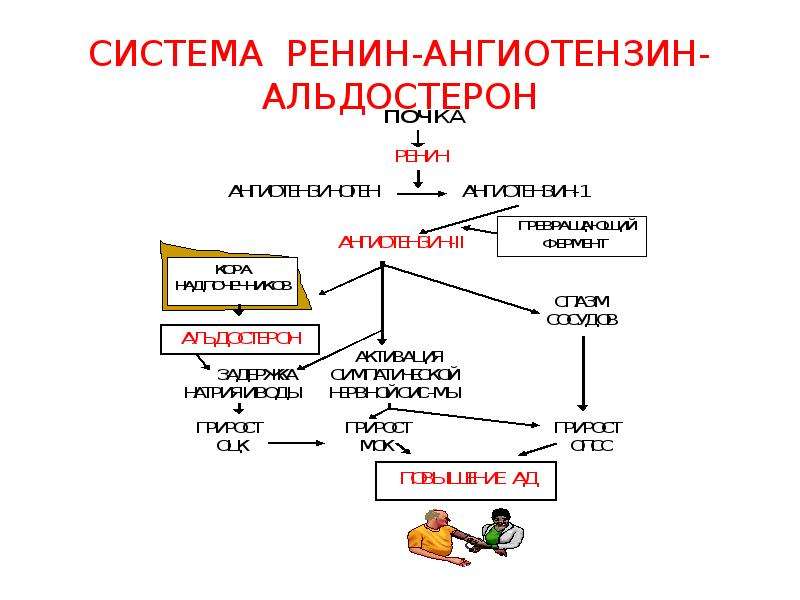
Характеристика ренин-ангиотензин-альдостероновой системы при полиморфных вариантах гена VDR у больных артериальной гипертензией и ожирением | Сенцова
1. Puzyrev V. I. Genetics of arterial hypertension (current research paradigms). Clinical Medicine 2003;1:12-18. Russiаn (Пузырев В. П. Генетика артериальной гипертензии (современные исследовательские парадигмы). Клиническая медицина 2003;1:12-18.)
2. Sun J., Zhao M., Miao S., Xi B. Polymorphisms of three genes (ACE, AGT and CYP11B2) in the renin-angiotensin-aldosterone system are not associated with blood pressure salt sensitivity: A systematic meta-analysis. Blood Press 2016; 25:117-122. DOI: 10.3109/08037051.2015.1110923
3. Kohli S., Kumar R., Gupta M. et al. Impact of interactions between risk alleles on clinical endpoints in hypertension. Heart Asia 2016;9:83-89. DOI: 10.1136/heartasia-2016-010723.
Heart Asia 2016;9:83-89. DOI: 10.1136/heartasia-2016-010723.
4. Ozaki K. Genetic background of heart failure: SNP association study for heart failure and the underlying diseases. Rinsho Byori 2013;2:167-175. PMID:23672095
5. Jhun M. A., Hu H., Schwartz J. et al. Effect modification by vitamin D. receptor genetic polymorphisms in the association beetwen cumulative lead exposure and puls pressure: a longitudinal study. Environ Health 2015;13:14-15. DOI: 10.1186/1476-069X-14-5
6. Wang L., Chu A., Buring J. E. et al. Common genetic variations in the vitamin D. pathway in relation to blood pressure. Am J. Hypertens 2014;11:1387-1395. DOI: 10.1093/ajh/hpu049
7. Basit S. Vitamin D.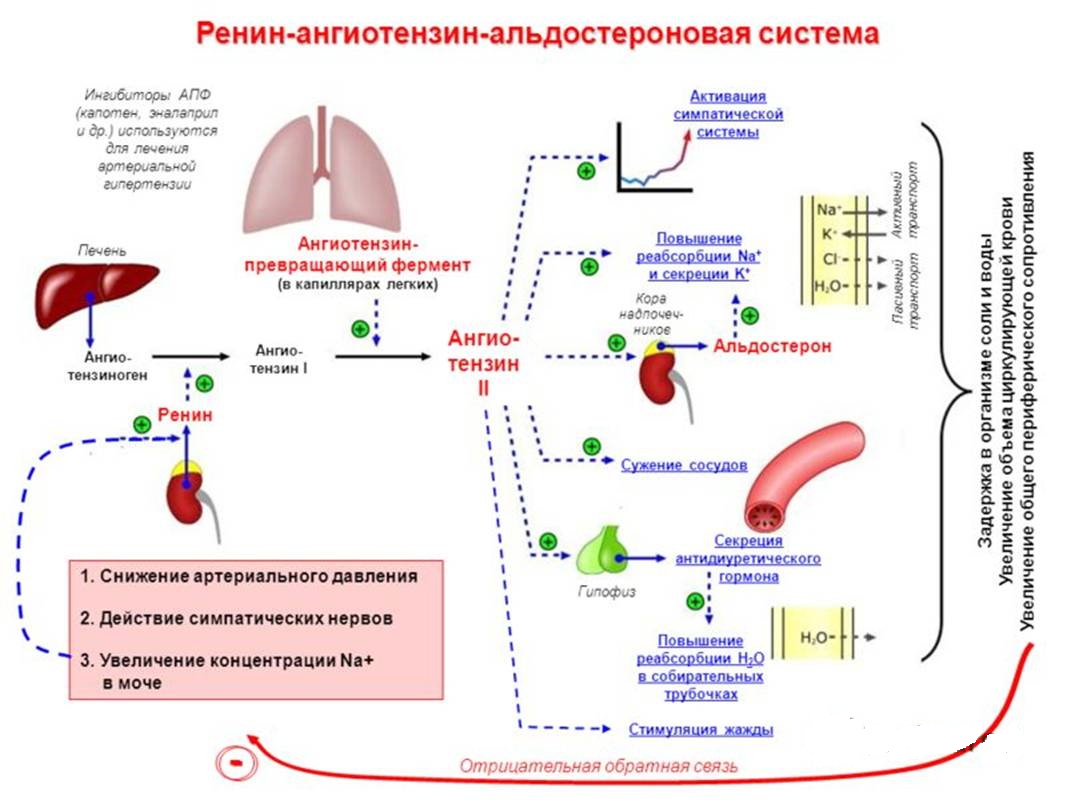 er Med 2016;4:2395-2399. DOI: 10.3892/etm. 2016.3667
er Med 2016;4:2395-2399. DOI: 10.3892/etm. 2016.3667
11. Cooke A. A., Connaughton R. M., Lyons C. L. et al. Fatty acids and chronic low grade inflammation associated with obesity and the metabolic syndrome. Eur J. Pharmacol 2016;5:207-214. DOI: 10.1016/j.ejphar. 2016.04.021
12. Favre G. A., Esnault V. L., Van Obberghen E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am J. Physiol Endocrinol Metab 2015;6:435-449. DOI: 10.1152/ajpendo. 00391.2014
13. Kang Y. S. Obesity associated hypertension: new insights into mechanism. Electrolyte Blood Press 2013;2:46-52. DOI: 10.5049/EBP. 2013.11.2.46
14. Дедов И. И., Тюльпаков А. Н., Чехонин В. П. и др. Персонализированная медицина: современное состояние и перспективы. Вестник Российской академии медицинских наук 2012; 12:4-12. DOI:10.15690/vramn. v67i12.474
Персонализированная медицина: современное состояние и перспективы. Вестник Российской академии медицинских наук 2012; 12:4-12. DOI:10.15690/vramn. v67i12.474
Ренин-ангиотензин-альдостероновая система как потенциальная мишень для терапии пациентов с кальцинирующим аортальным стенозом: обзор литературы | Костюнин
1. Lindman BR, Clavel M-A, Mathieu P, Iung B, Lancellotti P, Otto CM et al. Calcific aortic stenosis. Nature Reviews Disease Primers. 2016;2(1):16006. DOI: 10.1038/nrdp.2016.6
2. Authors/Task Force Members, Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Barón-Esquivias G et al. Guidelines on the management of valvular heart disease (version 2012). European Heart Journal. 2012;33(19):2451–96. DOI: 10.1093/eurheartj/ehs109
3. d’Arcy JL, Prendergast BD, Chambers JB, Ray SG, Bridgewater B. Valvular heart disease: the next cardiac epidemic. Heart. 2011;97(2):91–3. DOI: 10.1136/hrt.2010.205096
d’Arcy JL, Prendergast BD, Chambers JB, Ray SG, Bridgewater B. Valvular heart disease: the next cardiac epidemic. Heart. 2011;97(2):91–3. DOI: 10.1136/hrt.2010.205096
4. Iung B, Vahanian A. Degenerative calcific aortic stenosis: a natural history. Heart. 2012;98(Suppl 4):iv7–13. DOI: 10.1136/heartjnl-2012-302395
5. Osnabrugge RLJ, Mylotte D, Head SJ, Van Mieghem NM, Nkomo VT, LeReun CM et al. Aortic Stenosis in the Elderly: disease prevalence and number of candidates for transcatheter aortic valve replacement: a meta-analysis and modeling study. Journal of the American College of Cardiology. 2013;62(11):1002–12. DOI: 10.1016/j.jacc.2013.05.015
6. Thaden JJ, Nkomo VT, Enriquez-Sarano M. The Global Burden of Aortic Stenosis. Progress in Cardiovascular Diseases.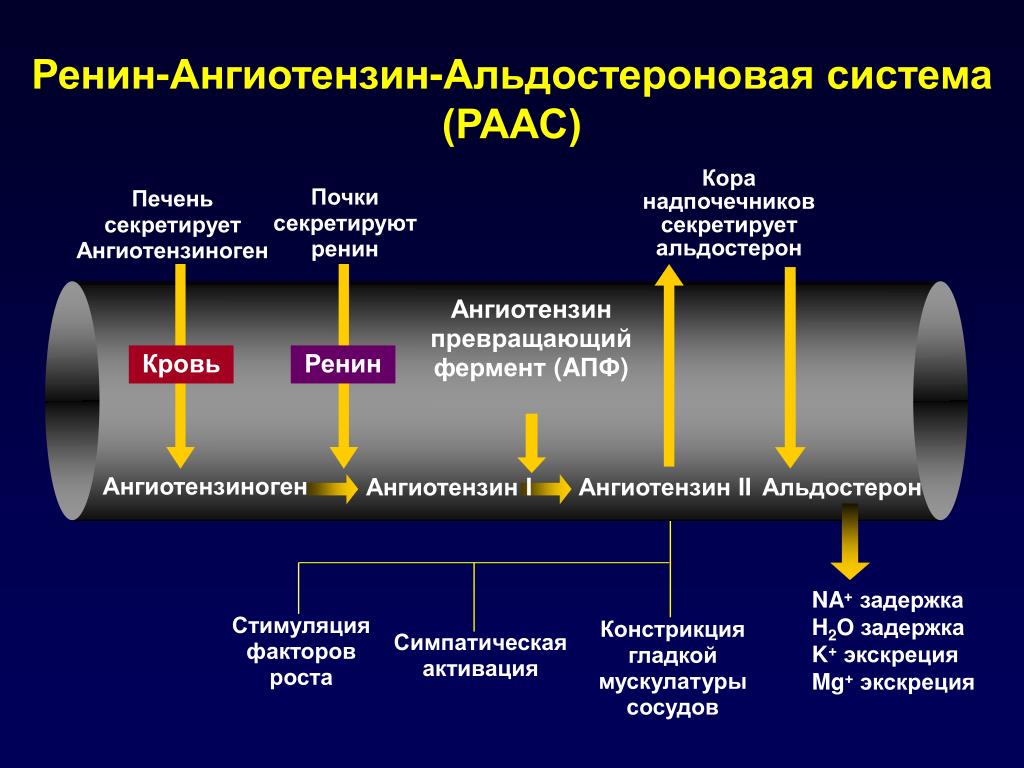 2014;56(6):565–71. DOI: 10.1016/j.pcad.2014.02.006
2014;56(6):565–71. DOI: 10.1016/j.pcad.2014.02.006
7. Marquis-Gravel G, Redfors B, Leon MB, Généreux P. Medical Treatment of Aortic Stenosis. Circulation. 2016;134(22):1766–84. DOI: 10.1161/CIRCULATIONAHA.116.023997
8. Salas MJ, Santana O, Escolar E, Lamas GA. Medical Therapy for Calcific Aortic Stenosis. Journal of Cardiovascular Pharmacology and Therapeutics. 2012;17(2):133–8. DOI: 10.1177/1074248411416504
9. Baumgartner H, Falk V, Bax JJ, De Bonis M, Hamm C, Holm PJ et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. European Heart Journal. 2017;38(36):2739–91. DOI: 10.1093/eurheartj/ehx391
10. Lindman BR, Bonow RO, Otto CM. Current Management of Calcific Aortic Stenosis. Circulation Research. 2013;113(2):223–37. DOI: 10.1161/CIRCRESAHA.111.300084
Circulation Research. 2013;113(2):223–37. DOI: 10.1161/CIRCRESAHA.111.300084
11. Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP, Fleisher LA et al. 2017 AHA/ACC Focused Update of the 2014 AHA/ACC Guideline for the Management of Patients with Valvular Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159–95. DOI: 10.1161/CIR.0000000000000503
12. Гуляев Н. И., Варавин Н. А., Коровин А. Е., Кузнецов В. В., Яковлев В. В., Гордиенко А. В. Современные аспекты патогенеза кальциноза аортальных полулуний (обзор литературы). Вестник СПбГУ. 2016;3:20-34. DOI: 10.21638/11701/spbu11.2016.302
13. Pacurari M, Kafoury R, Tchounwou PB, Ndebele K. The ReninAngiotensin-Aldosterone System in Vascular Inflammation and Remodeling.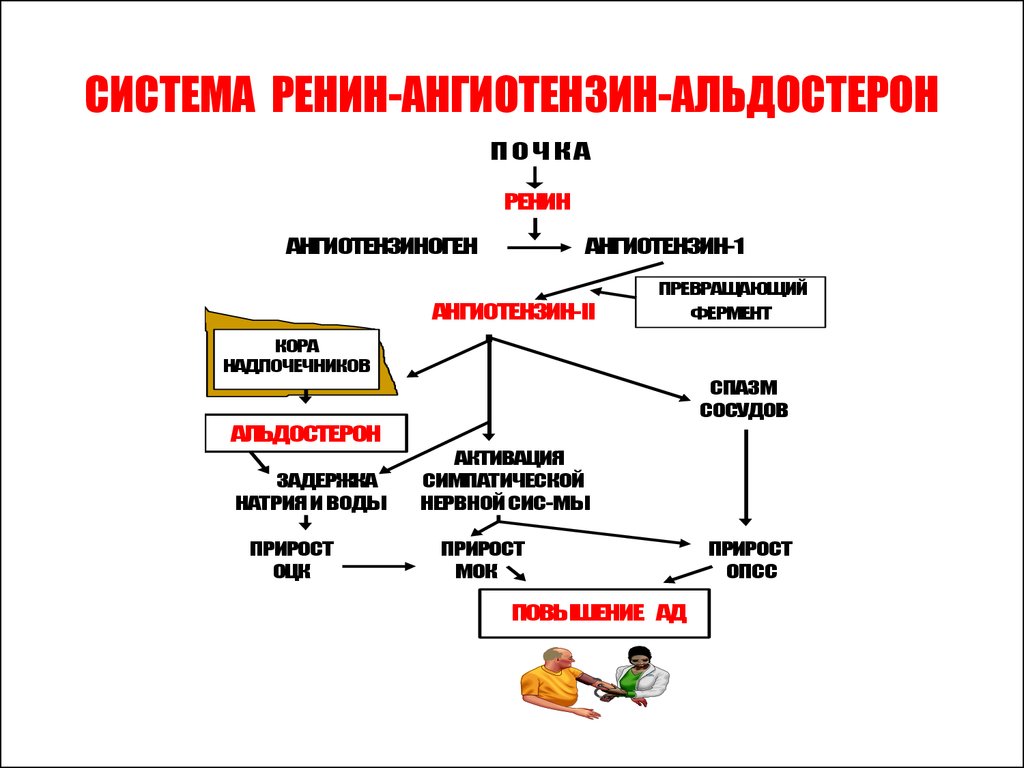 International Journal of Inflammation. 2014;2014:689360. DOI: 10.1155/2014/689360
International Journal of Inflammation. 2014;2014:689360. DOI: 10.1155/2014/689360
14. Cao W, Hu N, Yuan Y, Cheng J, Guo X, Wang Y et al. Effects of Tilianin on Proliferation, Migration and TGF-β/Smad Signaling in Rat Vascular Smooth Muscle Cells Induced with Angiotensin II: Pharmacological effect of Talinin on rat vascular smooth muscle cells. Phytotherapy Research. 2017;31(8):1240–8. DOI: 10.1002/ptr.5846
15. Chen T, Li M, Fan X, Cheng J, Wang L. Sodium Tanshinone IIA Sulfonate Prevents Angiotensin II-Induced Differentiation of Human Atrial Fibroblasts into Myofibroblasts. Oxidative Medicine and Cellular Longevity. 2018;2018:6712585. DOI: 10.1155/2018/6712585
16. Wu X, Liu Y, An J, Li J, Lv W, Geng S et al. Piperlongumine inhibits angiotensin II-induced extracellular matrix expression in cardiac fibroblasts. Journal of Cellular Biochemistry. 2018;119(12):10358–64. DOI: 10.1002/jcb.27379
Journal of Cellular Biochemistry. 2018;119(12):10358–64. DOI: 10.1002/jcb.27379
17. Barhoumi T, Fraulob-Aquino JC, Mian MOR, Ouerd S, IdrisKhodja N, Huo K-G et al. Matrix metalloproteinase-2 knockout prevents angiotensin II-induced vascular injury. Cardiovascular Research. 2017;113(14):1753–62. DOI: 10.1093/cvr/cvx115
18. Kong J, Zhang Y, Liu S, Li H, Liu S, Wang J et al. Melatonin attenuates angiotensin II-induced abdominal aortic aneurysm through the down-regulation of matrix metalloproteinases. Oncotarget. 2017;8(9):14283–93. DOI: 10.18632/oncotarget.15093
19. Guo F, Chen X-L, Wang F, Liang X, Sun Y-X, Wang Y-J. Role of Angiotensin II Type 1 Receptor in Angiotensin II-Induced Cytokine Production in Macrophages. Journal of Interferon & Cytokine Research.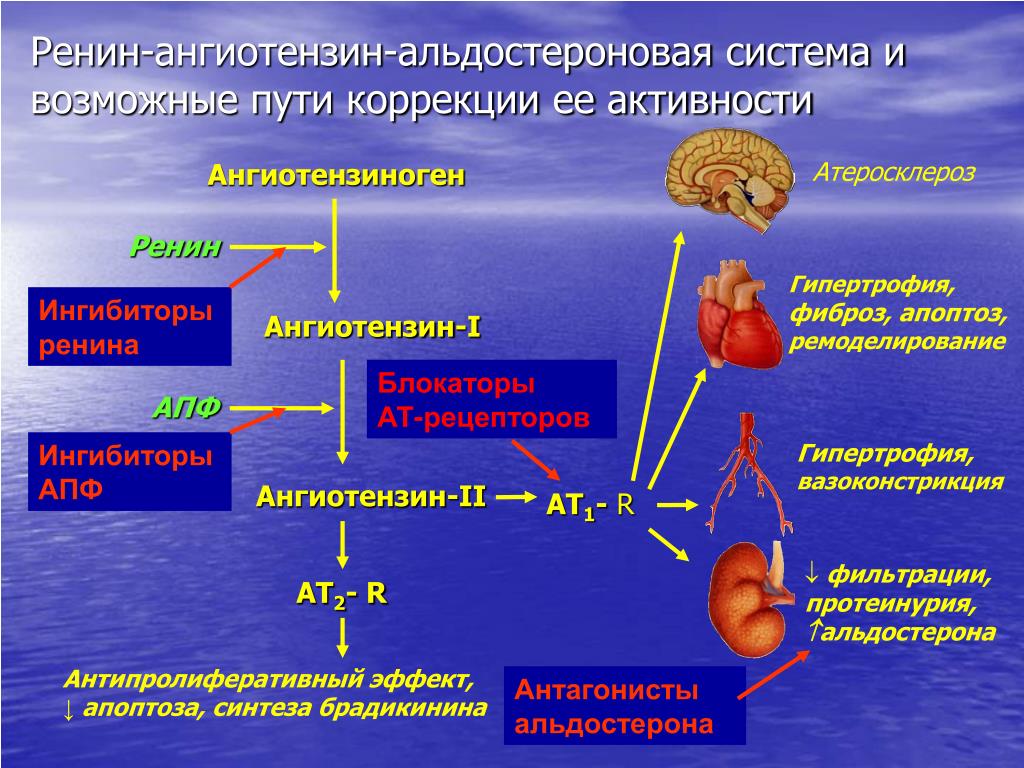 2011;31(4):351–61. DOI: 10.1089/jir.2010.0073
2011;31(4):351–61. DOI: 10.1089/jir.2010.0073
20. Manuneedhi Cholan P, Cartland SP, Dang L, Rayner BS, Patel S, Thomas SR et al. TRAIL protects against endothelial dysfunction in vivo and inhibits angiotensin-II-induced oxidative stress in vascular endothelial cells in vitro. Free Radical Biology and Medicine. 2018;126:341–9. DOI: 10.1016/j.freeradbiomed.2018.08.031
21. Tian H, Yu D, Hu Y, Zhang P, Yang Y, Hu Q et al. Angiotensin II upregulates cyclophilin A by enhancing ROS production in rat cardiomyocytes. Molecular Medicine Reports. 2018;18(5):4349–55. DOI: 10.3892/mmr.2018.9448
22. Han C, Liu J, Liu X, Li M. Angiotensin II induces C-reactive protein expression through ERK1/2 and JNK signaling in human aortic endothelial cells. Atherosclerosis. 2010;212(1):206–12. DOI: 10.1016/j.atherosclerosis.2010.05.020
DOI: 10.1016/j.atherosclerosis.2010.05.020
23. Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S et al. Angiotensin II regulates the synthesis of proinflam matory cytokines and chemokines in the kidney. Kidney International. 2002;82:S12–22. DOI: 10.1046/j.1523-1755.62.s82.4.x
24. Osako MK, Nakagami H, Shimamura M, Koriyama H, Nakagami F, Shimizu H et al. Cross-Talk of Receptor Activator of Nuclear Factor-κB Ligand Signaling With Renin–Angiotensin System in Vascular Calcification. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(6):1287–96. DOI: 10.1161/ATVBAHA.112.301099
25. Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(3):645–51. DOI: 10.1161/01.atv.20.3.645
Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(3):645–51. DOI: 10.1161/01.atv.20.3.645
26. Oparil S, Acelajado MC, Bakris GL, Berlowitz DR, Cífková R, Dominiczak AF et al. Hypertension. Nature Reviews Disease Primers. 2018;4(1):18014. DOI: 10.1038/nrdp.2018.14
27. Montecucco F, Pende A, Mach F. The Renin-Angiotensin System Modulates Inflammatory Processes in Atherosclerosis: Evidence from Basic Research and Clinical Studies. Mediators of Inflammation. 2009; 2009:752406. DOI: 10.1155/2009/752406
28. Sata M, Fukuda D. Crucial role of renin-angiotensin system in the pathogenesis of atherosclerosis. The Journal of Medical Investigation. 2010;57(1–2):12–25. DOI: 10.2152/jmi.57.12
29. George AJ, Thomas WG, Hannan RD. The renin–angiotensin system and cancer: old dog, new tricks. Nature Reviews Cancer. 2010;10(11):745–59. DOI: 10.1038/nrc2945
George AJ, Thomas WG, Hannan RD. The renin–angiotensin system and cancer: old dog, new tricks. Nature Reviews Cancer. 2010;10(11):745–59. DOI: 10.1038/nrc2945
30. Ishikane S, Takahashi-Yanaga F. The role of angiotensin II in cancer metastasis: Potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochemical Pharmacology. 2018; 151:96–103. DOI: 10.1016/j.bcp.2018.03.008
31. Chu KY, Leung PS. Angiotensin II in type 2 diabetes mellitus. Current Protein & Peptide Science. 2009;10(1):75–84. DOI: 10.2174/138920309787315176
32. Ribeiro-Oliveira AJr, Nogueira AI, Pereira RM, Boas WW, Dos Santos RA, Simões e Silva AC. The renin-angiotensin system and diabetes: An update. Vascular Health and Risk Management. 2008;4(4):787–803. DOI: 10.2147/VHRM.S1905
DOI: 10.2147/VHRM.S1905
33. Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of reninangiotensin-aldosterone system in the progression of chronic kidney disease. Kidney International. 2005;99:S57–65. DOI: 10.1111/j.1523-1755.2005.09911.x
34. Balakumar P, Jagadeesh G. A century old renin–angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cellular Signalling. 2014;26(10):2147–60. DOI: 10.1016/j.cellsig.2014.06.011
35. Peltonen T, Ohukainen P, Ruskoaho H, Rysä J. Targeting vasoactive peptides for managing calcific aortic valve disease. Annals of Medicine. 2017;49(1):63–74. DOI: 10.1080/07853890.2016.1231933
36. Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PML et al. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli. Pharmacological Reviews. 2015;67(4):754–819. DOI: 10.1124/pr.114.010454
Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PML et al. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli. Pharmacological Reviews. 2015;67(4):754–819. DOI: 10.1124/pr.114.010454
37. Helske S, Lindstedt KA, Laine M, Mäyränpää M, Werkkala K, Lommi J et al. Induction of local angiotensin II-producing systems in stenotic aortic valves. Journal of the American College of Cardiology. 2004;44(9):1859–66. DOI: 10.1016/j.jacc.2004.07.054
38. Peltonen T, Näpänkangas J, Vuolteenaho O, Ohtonen P, Soini Y, Juvonen T et al. Apelin and its receptor APJ in human aortic valve stenosis. The Journal of Heart Valve Disease. 2009;18(6):644–52. PMID: 20099713
39. O’Brien KD, Shavelle DM, Caulfield MT, McDonald TO, OlinLewis K, Otto CM et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106(17):2224–30. DOI: 10.1161/01.cir.0000035655.45453.d2
O’Brien KD, Shavelle DM, Caulfield MT, McDonald TO, OlinLewis K, Otto CM et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106(17):2224–30. DOI: 10.1161/01.cir.0000035655.45453.d2
40. Li XC, Zhuo JL. Nuclear factor-κB as a hormonal intracellular signaling molecule: focus on angiotensin II-induced cardiovascular and renal injury: Current Opinion in Nephrology and Hypertension. 2008;17(1):37–43. DOI: 10.1097/MNH.0b013e3282f2903c
41. Zablocki D, Sadoshima J. Angiotensin II and Oxidative Stress in the Failing Heart. Antioxidants & Redox Signaling. 2013;19(10):1095–109. DOI: 10.1089/ars.2012.4588
42. Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J, Pomerantzeff PMA et al. Oxidant Generation Predominates Around Calcifying Foci and Enhances Progression of Aortic Valve Calcification. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(3):463–70. DOI: 10.1161/ATVBAHA.107.156745
Oxidant Generation Predominates Around Calcifying Foci and Enhances Progression of Aortic Valve Calcification. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(3):463–70. DOI: 10.1161/ATVBAHA.107.156745
43. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña-Silva R, Heistad DD. Dysregulation of Antioxidant Mechanisms Contributes to Increased Oxidative Stress in Calcific Aortic Valvular Stenosis in Humans. Journal of the American College of Cardiology. 2008;52(10):843–50. DOI: 10.1016/j.jacc.2008.05.043
44. Rajamannan N. Role of Oxidative Stress in Calcific Aortic Valve Disease: From Bench to Bedside — The Role of a Stem Cell Niche. In: Oxidative Stress and Chronic Degenerative Diseases — A Role for Antioxidants Morales-Gonzalez JA, editor -Croatia: InTech;2013.
45. Morgan MJ, Liu Z. Reactive oxygen species in TNFα-induced signaling and cell death. Molecules and Cells. 2010;30(1):1–12. DOI: 10.1007/s10059-010-0105-0
Morgan MJ, Liu Z. Reactive oxygen species in TNFα-induced signaling and cell death. Molecules and Cells. 2010;30(1):1–12. DOI: 10.1007/s10059-010-0105-0
46. Belhadj Slimen I, Najar T, Ghram A, Dabbebi H, Ben Mrad M, Abdrabbah M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. International Journal of Hyperthermia. 2014;30(7):513–23. DOI: 10.3109/02656736.2014.971446
47. Xie C, Shen Y, Hu W, Chen Z, Li Y. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging Clinical and Experimental Research. 2016;28(2):181–7. DOI: 10.1007/s40520-015-0408-2
48. Fujisaka T, Hoshiga M, Hotchi J, Takeda Y, Jin D, Takai S et al. Angiotensin II promotes aortic valve thickening independent of elevated blood pressure in apolipoprotein-E deficient mice. Atherosclerosis. 2013;226(1):82–7. DOI: 10.1016/j.atherosclerosis.2012.10.055
Atherosclerosis. 2013;226(1):82–7. DOI: 10.1016/j.atherosclerosis.2012.10.055
49. Arishiro K, Hoshiga M, Negoro N, Jin D, Takai S, Miyazaki M et al. Angiotensin Receptor-1 Blocker Inhibits Atherosclerotic Changes and Endothelial Disruption of the Aortic Valve in Hypercholesterolemic Rabbits. Journal of the American College of Cardiology. 2007;49(13):1482–9. DOI: 10.1016/j.jacc.2006.11.043
50. Masuda C, Dohi K, Sakurai Y, Bessho Y, Fukuda H, Fujii S et al. Impact of Chronic Kidney Disease on the Presence and Severity of Aortic Stenosis in Patients at High Risk for Coronary Artery Disease. Cardiovascular Ultrasound. 2011;9(1):31. DOI: 10.1186/1476-7120-9-31
51. Rieck ÅE, Cramariuc D, Boman K, Gohlke-Bärwolf C, Staal EM, Lønnebakken MT et al. Hypertension in Aortic Stenosis: Implications for Left Ventricular Structure and Cardiovascular Events. Hypertension. 2012;60(1):90–7. DOI: 10.1161/HYPERTENSIONAHA.112.194878
Hypertension. 2012;60(1):90–7. DOI: 10.1161/HYPERTENSIONAHA.112.194878
52. Perkovic V, Hunt D, Griffin SV, du Plessis M, Becker GJ. Accelerated Progression of Calcific Aortic Stenosis in Dialysis Patients. Nephron Clinical Practice. 2004;94(2):c40–5. DOI: 10.1159/000071280
53. Tastet L, Capoulade R, Clavel M-A, Larose É, Shen M, Dahou A et al. Systolic hypertension and progression of aortic valve calcification in patients with aortic stenosis: results from the PROGRESSA study. European Heart Journal – Cardiovascular Imaging. 2017;18(1):70–8. DOI: 10.1093/ehjci/jew013
54. Liakos CI, Grassos CA, Papadopoulos DP, Dimitriadis KS, Tsioufis CP, Tousoulis D. Arterial hypertension and aortic valve stenosis: Shedding light on a common “liaison”. Hellenic Journal of Cardiology. 2017;58(4):261–6. DOI: 10.1016/j.hjc.2017.03.005
2017;58(4):261–6. DOI: 10.1016/j.hjc.2017.03.005
55. Dweck MR, Boon NA, Newby DE. Calcific Aortic Stenosis: a disease of the valve and the myocardium. Journal of the American College of Cardiology. 2012;60(19):1854–63. DOI: 10.1016/j.jacc.2012.02.093
56. Rattazzi M, Bertacco E, Del Vecchio A, Puato M, Faggin E, Pauletto P. Aortic valve calcification in chronic kidney disease. Nephrology Dialysis Transplantation. 2013;28(12):2968–76. DOI: 10.1093/ndt/gft310
57. Ahmad S, Varagic J, VonCannon JL, Groban L, Collawn JF, Dell’Italia LJ et al. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1-12) metabolizing enzyme. Biochemical and Biophysical Research Communications. 2016;478(2):559–64. DOI: 10.1016/j.bbrc.2016.07.100
58. Nagata S, Hatakeyama K, Asami M, Tokashiki M, Hibino H, Nishiuchi Y et al. Big angiotensin-25: A novel glycosylated angiotensin-related peptide isolated from human urine. Biochemical and Biophysical Research Communications. 2013;441(4):757–62. DOI: 10.1016/j.bbrc.2013.10.124
Nagata S, Hatakeyama K, Asami M, Tokashiki M, Hibino H, Nishiuchi Y et al. Big angiotensin-25: A novel glycosylated angiotensin-related peptide isolated from human urine. Biochemical and Biophysical Research Communications. 2013;441(4):757–62. DOI: 10.1016/j.bbrc.2013.10.124
59. Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin–angiotensin system. Biochemical and Biophysical Research Communications. 2006;350(4):1026–31. DOI: 10.1016/j.bbrc.2006.09.146
60. Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-Dependent Generation of Angiotensin II from Angiotensin-(1-12) in Human Atrial Tissue. PLoS ONE. 2011;6(12):e28501. DOI: 10.1371/journal.pone.0028501
61. Ahmad S, Wei C-C, Tallaj J, Dell’Italia LJ, Moniwa N, Varagic J et al. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. Journal of the American Society of Hypertension. 2013;7(2):128–36. DOI: 10.1016/j.jash.2012.12.003
Ahmad S, Wei C-C, Tallaj J, Dell’Italia LJ, Moniwa N, Varagic J et al. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. Journal of the American Society of Hypertension. 2013;7(2):128–36. DOI: 10.1016/j.jash.2012.12.003
62. Helske S, Syväranta S, Kupari M, Lappalainen J, Laine M, Lommi J et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. European Heart Journal. 2006;27(12):1495–504. DOI: 10.1093/eurheartj/ehi706
63. Coté N, Mahmut A, Bosse Y, Couture C, Pagé S, Trahan S et al. Inflammation Is Associated with the Remodeling of Calcific Aortic Valve Disease. Inflammation. 2013;36(3):573–81. DOI: 10.1007/s10753-012-9579-6
64. Šteiner I, Krbal L, Rozkoš T, Harrer J, Laco J. Calcific aortic valve stenosis: Immunohistochemical analysis of inflammatory infiltrate. Pathology — Research and Practice. 2012;208(4):231–4. DOI: 10.1016/j.prp.2012.02.009
Pathology — Research and Practice. 2012;208(4):231–4. DOI: 10.1016/j.prp.2012.02.009
65. Šteiner I, Stejskal V, Žáček P. Mast cells in calcific aortic stenosis. Pathology – Research and Practice. 2018;214(1):163–8. DOI: 10.1016/j.prp.2017.07.016
66. O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(4):523–32. DOI: 10.1161/01.atv.16.4.523
67. Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(5):1218–22. DOI: 10.1161/01.atv.19. 5.1218
5.1218
68. Parisi V, Leosco D, Ferro G, Bevilacqua A, Pagano G, de Lucia C et al. The lipid theory in the pathogenesis of calcific aortic stenosis. Nutrition, Metabolism and Cardiovascular Diseases. 2015;25(6):519–25. DOI: 10.1016/j.numecd.2015.02.001
69. Mohty D, Pibarot P, Després J-P, Côté C, Arsenault B, Cartier A et al. Association Between Plasma LDL Particle Size, Valvular Accumulation of Oxidized LDL, and Inflammation in Patients with Aortic Stenosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(1):187–93. DOI: 10.1161/ATVBAHA.107.154989
70. Tiede K, Stöter K, Petrik C, Chen WB, Ungefroren H, Kruse ML et al. Angiotensin II AT(1)-receptor induces biglycan in neonatal cardiac fibroblasts via autocrine release of TGFbeta in vitro. Cardiovascular Research. 2003;60(3):538–46. DOI: 10.1016/j.cardiores.2003.08.009
2003;60(3):538–46. DOI: 10.1016/j.cardiores.2003.08.009
71. Neufeld EB, Zadrozny LM, Phillips D, Aponte A, Yu Z-X, Balaban RS. Decorin and biglycan retain LDL in disease-prone valvular and aortic subendothelial intimal matrix. Atherosclerosis. 2014;233(1):113–21. DOI: 10.1016/j.atherosclerosis.2013.12.038
72. Osman N, Grande-Allen KJ, Ballinger ML, Getachew R, Marasco S, O’Brien KD et al. Smad2-dependent glycosaminoglycan elongation in aortic valve interstitial cells enhances binding of LDL to proteoglycans. Cardiovascular Pathology. 2013;22(2):146–55. DOI: 10.1016/j.carpath.2012.07.002
73. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circulation Research. 2000;87(5):E1-9. DOI: 10.1161/01.res.87.5.e1
Circulation Research. 2000;87(5):E1-9. DOI: 10.1161/01.res.87.5.e1
74. Ferreira AJ, Santos RAS, Bradford CN, Mecca AP, Sumners C, Katovich MJ et al. Therapeutic Implications of the Vasoprotective Axis of the Renin-Angiotensin System in Cardiovascular Diseases. Hypertension. 2010;55(2):207–13. DOI: 10.1161/HYPERTENSIONAHA.109.140145
75. Iwai M, Horiuchi M. Devil and angel in the renin–angiotensin system: ACE–angiotensin II–AT1 receptor axis vs. ACE2–angiotensin-(1–7)–Mas receptor axis. Hypertension Research. 2009;32(7):533–6. DOI: 10.1038/hr.2009.74
76. Alenina N, Xu P, Rentzsch B, Patkin EL, Bader M. Genetically altered animal models for Mas and angiotensin-(1-7): Transgenic animal models for Mas and angiotensin-(1-7). Experimental Physiology. 2008;93(5):528–37.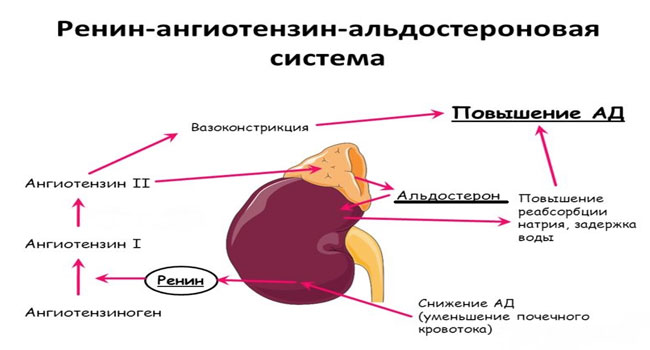 DOI: 10.1113/expphysiol.2007.040345
DOI: 10.1113/expphysiol.2007.040345
77. Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP et al. Loss of Angiotensin-Converting Enzyme 2 Accelerates Maladaptive Left Ventricular Remodeling in Response to Myocardial Infarction. Circulation: Heart Failure. 2009;2(5):446–55. DOI: 10.1161/CIRCHEARTFAILURE.108.840124
78. Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M et al. Angiotensin-Converting Enzyme 2 Suppresses Pathological Hypertrophy, Myocardial Fibrosis, and Cardiac Dysfunction. Circulation. 2010;122(7):717–28. DOI: 10.1161/CIRCULATIONAHA.110.955369
79. Trask AJ, Groban L, Westwood BM, Varagic J, Ganten D, Gallagher PE et al. Inhibition of Angiotensin-Converting Enzyme 2 Exacerbates Cardiac Hypertrophy and Fibrosis in Ren-2 Hypertensive Rats. American Journal of Hypertension. 2010;23(6):687–93. DOI: 10.1038/ajh.2010.51
American Journal of Hypertension. 2010;23(6):687–93. DOI: 10.1038/ajh.2010.51
80. Peltonen T, Näpänkangas J, Ohtonen P, Aro J, Peltonen J, Soini Y et al. (Pro)renin receptors and angiotensin converting enzyme 2/angiotensin-(1-7)/Mas receptor axis in human aortic valve stenosis. Atherosclerosis. 2011;216(1):35–43. DOI: 10.1016/j.atherosclerosis.2011.01.018
81. Collister JP, Nahey DB. Simultaneous administration of Ang(1-7) or A-779 does not affect the chronic hypertensive effects of angiotensin II in normal rats. Journal of the Renin-Angiotensin-Aldosterone System. 2010;11(2):99–102. DOI: 10.1177/1470320309359928
82. Velkoska E, Dean RG, Griggs K, Burchill L, Burrell LM. Angiotensin-(1–7) infusion is associated with increased blood pressure and adverse cardiac remodelling in rats with subtotal nephrectomy. Clinical Science. 2011;120(8):335–45. DOI: 10.1042/CS20100280
Clinical Science. 2011;120(8):335–45. DOI: 10.1042/CS20100280
83. Shao Y, He M, Zhou L, Yao T, Huang Y, Lu L. Chronic angiotensin (17) injection accelerates STZ-induced diabetic renal injury 1. Acta Pharmacologica Sinica. 2008;29(7):829–37. DOI: 10.1111/j.1745-7254.2008.00812.x
84. Mendoza-Torres E, Oyarzún A, Mondaca-Ruff D, Azocar A, Castro PF, Jalil JE et al. ACE2 and vasoactive peptides: novel players in cardiovascular/renal remodeling and hypertension. Therapeutic Advances in Cardiovascular Disease. 2015;9(4):217–37. DOI: 10.1177/1753944715597623
85. Westermeier F, Bustamante M, Pavez M, García L, Chiong M, Ocaranza MP et al. Novel players in cardioprotection: Insulin like growth factor-1, angiotensin-(1–7) and angiotensin-(1–9). Pharmacological Research.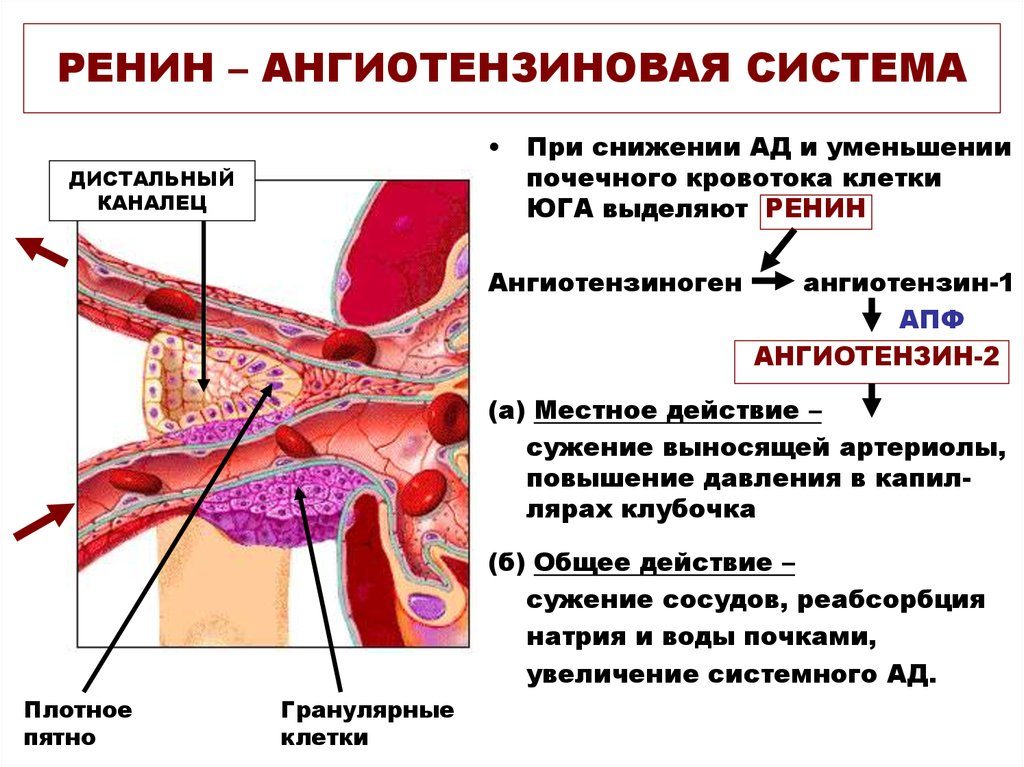 2015;101:41–55. DOI: 10.1016/j.phrs.2015.06.018
2015;101:41–55. DOI: 10.1016/j.phrs.2015.06.018
86. Ocaranza MP, Lavandero S, Jalil JE, Moya J, Pinto M, Novoa U et al. Angiotensin-(1–9) regulates cardiac hypertrophy in vivo and in vitro. Journal of Hypertension. 2010;28(5):1054–64. DOI: 10.1097/HJH.0b013e328335d291
87. Ocaranza MP, Moya J, Barrientos V, Alzamora R, Hevia D, Morales C et al. Angiotensin-(1–9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis: Journal of Hypertension. 2014;32(4):771–83. DOI: 10.1097/HJH.0000000000000094
88. Zheng H, Pu S-Y, Fan X-F, Li X-S, Zhang Y, Yuan J et al. Treatment with angiotensin-(1-9) alleviates the cardiomyopathy in streptozotocin-induced diabetic rats. Biochemical Pharmacology. 2015;95(1):38–45.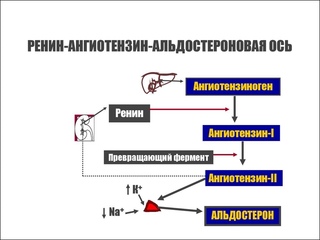 DOI: 10.1016/j.bcp.2015.03.009
DOI: 10.1016/j.bcp.2015.03.009
89. Cha SAh, Park BM, Gao S, Kim SH. Stimulation of ANP by angiotensin-(1-9) via the angiotensin type 2 receptor. Life Sciences. 2013;93(24):934–40. DOI: 10.1016/j.lfs.2013.10.020
90. Flores-Munoz M, Work LM, Douglas K, Denby L, Dominiczak AF, Graham D et al. Angiotensin-(1-9) Attenuates Cardiac Fibrosis in the Stroke-Prone Spontaneously Hypertensive Rat via the Angiotensin Type 2 Receptor. Hypertension. 2012;59(2):300–7. DOI: 10.1161/HYPERTENSIONAHA.111.177485
91. Yu L, Yuan K, Phuong HTA, Park BM, Kim SH. Angiotensin-(1-5), an active mediator of renin-angiotensin system, stimulates ANP secretion via Mas receptor. Peptides. 2016;86:33–41. DOI: 10.1016/j.peptides.2016.09.009
92. Hrenak J, Paulis L, Simko F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. International Journal of Molecular Sciences. 2016;17(7):1098. DOI: 10.3390/ijms17071098
Hrenak J, Paulis L, Simko F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. International Journal of Molecular Sciences. 2016;17(7):1098. DOI: 10.3390/ijms17071098
93. Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, CostaFraga F et al. Discovery and Characterization of Alamandine: A Novel Component of the Renin–Angiotensin System. Circulation Research. 2013;112(8):1104–11. DOI: 10.1161/CIRCRESAHA.113.301077
94. Villela DC, Passos-Silva DG, Santos RAS. Alamandine: a new member of the angiotensin family. Current Opinion in Nephrology and Hypertension. 2014;23(2):130–4. DOI: 10.1097/01.mnh.0000441052.44406.92
95. Habiyakare B, Alsaadon H, Mathai ML, Hayes A, Zulli A. Reduction of angiotensin A and alamandine vasoactivity in the rabbit model of atherogenesis: differential effects of alamandine and Ang(1-7).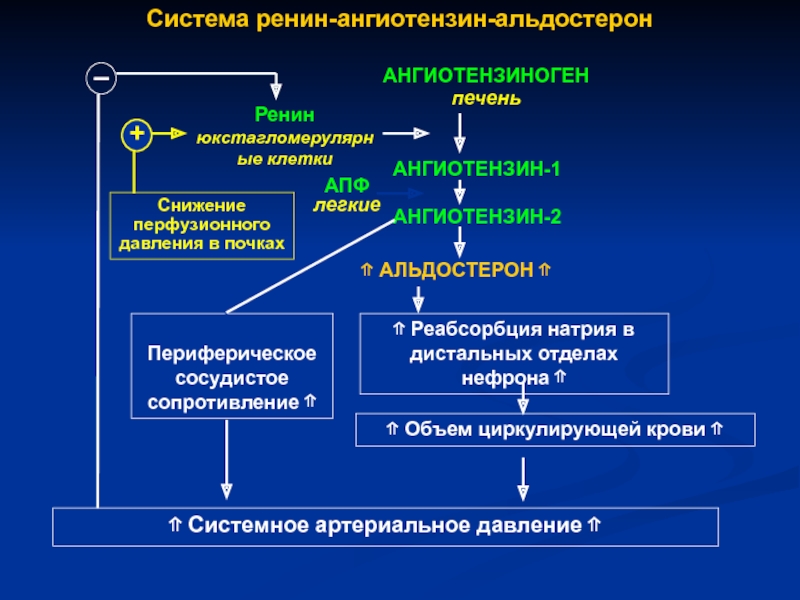 International Journal of Experimental Pathology. 2014;95(4):290–5. DOI: 10.1111/iep.12087
International Journal of Experimental Pathology. 2014;95(4):290–5. DOI: 10.1111/iep.12087
96. Jankowski V, Tölle M, Santos RAS, Günthner T, Krause E, Beyermann M et al. Angioprotectin: an angiotensin II-like peptide causing vasodilatory effects. The FASEB Journal. 2011;25(9):2987–95. DOI: 10.1096/fj.11-185470
97. Jankowski V, Vanholder R, van der Giet M, Tölle M, Karadogan S, Gobom J et al. Mass-Spectrometric Identification of a Novel Angiotensin Peptide in Human Plasma. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(2):297–302. DOI: 10.1161/01.ATV.0000253889.09765.5f
98. Yang R, Smolders I, Vanderheyden P, Demaegdt H, Van Eeckhaut A, Vauquelin G et al. Pressor and Renal Hemodynamic Effects of the Novel Angiotensin A Peptide Are Angiotensin II Type 1A Receptor Dependent. Hypertension. 2011;57(5):956–64. DOI: 10.1161/HYPERTENSIONAHA.110.161836
Hypertension. 2011;57(5):956–64. DOI: 10.1161/HYPERTENSIONAHA.110.161836
99. Coutinho DC, Foureaux G, Rodrigues KD, Salles RL, Moraes PL, Murça TM et al. Cardiovascular effects of angiotensin A: A novel peptide of the renin–angiotensin system. Journal of the ReninAngiotensin-Aldosterone System. 2014;15(4):480–6. DOI: 10.1177/1470320312474856
100. Ngo DT, Stafford I, Sverdlov AL, Qi W, Wuttke RD, Zhang Y et al. Ramipril retards development of aortic valve stenosis in a rabbit model: mechanistic considerations: Ramipril retards development of aortic valve stenosis. British Journal of Pharmacology. 2011;162(3):722–32. DOI: 10.1111/j.1476-5381.2010.01084.x
101. Simolin MA, Pedersen TX, Bro S, Mäyränpää MI, Helske S, Nielsen LB et al. ACE inhibition attenuates uremia-induced aortic valve thickening in a novel mouse model. BMC Cardiovascular Disorders. 2009;9(1):10. DOI: 10.1186/1471-2261-9-10
BMC Cardiovascular Disorders. 2009;9(1):10. DOI: 10.1186/1471-2261-9-10
102. Bull S, Loudon M, Francis JM, Joseph J, Gerry S, Karamitsos TD et al. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor Ramipril In Aortic Stenosis (RIAS trial). European Heart Journal – Cardiovascular Imaging. 2015;16(8):834–41. DOI: 10.1093/ehjci/jev043
103. Chockalingam A, Venkatesan S, Subramaniam T, Jagannathan V, Elangovan S, Alagesan R et al. Safety and efficacy of angiotensin-converting enzyme inhibitors in symptomatic severe aortic stenosis: symptomatic cardiac obstruction–pilot study of enalapril in aortic stenosis (SCOPE-AS). American Heart Journal. 2004;147(4):740. DOI: 10.1016/j.ahj.2003.10.017
104. Dalsgaard M, Iversen K, Kjaergaard J, Grande P, Goetze JP, Clemmensen P et al.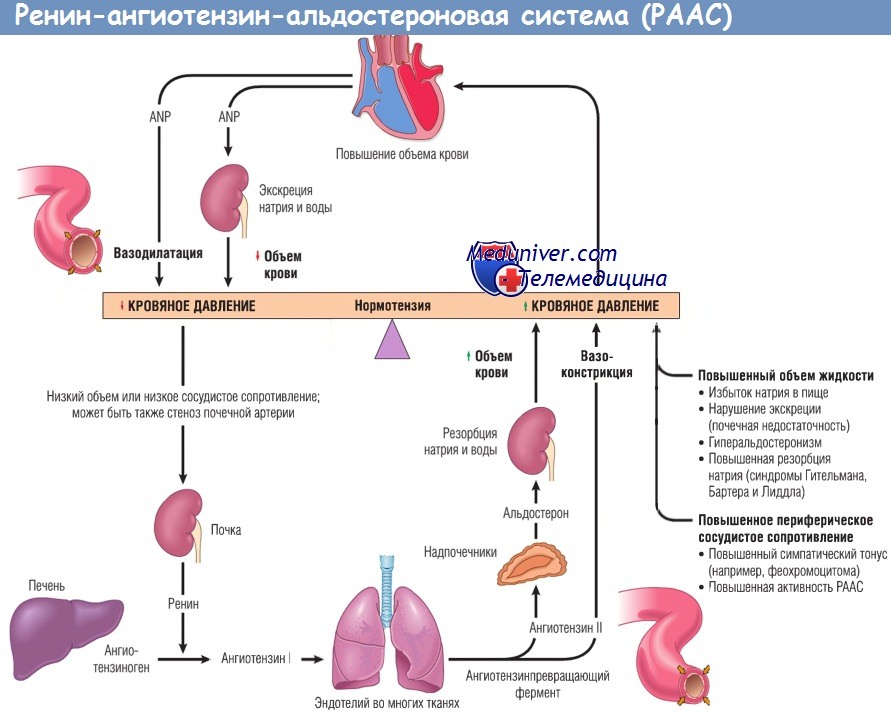 Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis. American Heart Journal. 2014;167(2):226–34. DOI: 10.1016/j.ahj.2013.11.002
Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis. American Heart Journal. 2014;167(2):226–34. DOI: 10.1016/j.ahj.2013.11.002
105. Capoulade R, Clavel M-A, Mathieu P, Côté N, Dumesnil JG, Arsenault M et al. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. European Journal of Clinical Investigation. 2013;43(12):1262–72. DOI: 10.1111/eci.12169
106. Côté N, Couture C, Pibarot P, Després J-P, Mathieu P. Angiotensin receptor blockers are associated with a lower remodelling score of stenotic aortic valves. European Journal of Clinical Investigation. 2011;41(11):1172–9. DOI: 10.1111/j.1365-2362.2011.02522.x
107. Yamamoto K, Yamamoto H, Yoshida K, Kisanuki A, Hirano Y, Ohte N et al. Prognostic factors for progression of early- and late-stage calcific aortic valve disease in Japanese: The Japanese Aortic Stenosis Study (JASS) Retrospective Analysis. Hypertension Research. 2010;33(3):269–74. DOI: 10.1038/hr.2009.225
Prognostic factors for progression of early- and late-stage calcific aortic valve disease in Japanese: The Japanese Aortic Stenosis Study (JASS) Retrospective Analysis. Hypertension Research. 2010;33(3):269–74. DOI: 10.1038/hr.2009.225
108. Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U et al. Statins but Not Angiotensin-Converting Enzyme Inhibitors Delay Progression of Aortic Stenosis. Circulation. 2004;110(10):1291–5. DOI: 10.1161/01.CIR.0000140723.15274.53
109. O’Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM et al. Angiotensin-Converting Enzyme Inhibitors and Change in Aortic Valve Calcium. Archives of Internal Medicine. 2005;165(8):858–62. DOI: 10.1001/archinte.165.8.858
110. Nadir MA, Wei L, Elder DHJ, Libianto R, Lim TK, Pauriah M et al. Impact of Renin-Angiotensin System Blockade Therapy on Outcome in Aortic Stenosis. Journal of the American College of Cardiology. 2011;58(6):570–6. DOI: 10.1016/j.jacc.2011.01.063
Impact of Renin-Angiotensin System Blockade Therapy on Outcome in Aortic Stenosis. Journal of the American College of Cardiology. 2011;58(6):570–6. DOI: 10.1016/j.jacc.2011.01.063
111. Chen J-H, Simmons CA. Cell–Matrix Interactions in the Pathobiology of Calcific Aortic Valve Disease: Critical Roles for Matricellular, Matricrine, and Matrix Mechanics Cues. Circulation Research. 2011;108(12):1510–24. DOI: 10.1161/CIRCRESAHA.110.234237
112. Yip CYY, Simmons CA. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovascular Pathology. 2011;20(3):177–82. DOI: 10.1016/j.carpath.2010.12.001
113. Balachandran K, Sucosky P, Jo H, Yoganathan AP. Elevated Cyclic Stretch Induces Aortic Valve Calcification in a Bone Morphogenic ProteinDependent Manner. The American Journal of Pathology. 2010;177(1):49–57. DOI: 10.2353/ajpath.2010.090631
The American Journal of Pathology. 2010;177(1):49–57. DOI: 10.2353/ajpath.2010.090631
114. Helske-Suihko S, Laine M, Lommi J, Kaartinen M, Werkkala K, Kovanen PT et al. Is Blockade of the Renin-angiotensin System Able to Reverse the Structural and Functional Remodeling of the Left Ventricle in Severe Aortic Stenosis? Journal of Cardiovascular Pharmacology. 2015;65(3):233–40. DOI: 10.1097/FJC.0000000000000182
115. Bang CN, Greve AM, Køber L, Rossebø AB, Ray S, Boman K et al. Renin–angiotensin system inhibition is not associated with increased sudden cardiac death, cardiovascular mortality or all-cause mortality in patients with aortic stenosis. International Journal of Cardiology. 2014;175(3):492–8. DOI: 10.1016/j.ijcard.2014.06.013
116. Dell’Italia LJ, Collawn JF, Ferrario CM. Multifunctional Role of Chymase in Acute and Chronic Tissue Injury and Remodeling. Circulation Research. 2018;122(2):319–36. DOI: 10.1161/CIRCRESAHA.117.310978
Multifunctional Role of Chymase in Acute and Chronic Tissue Injury and Remodeling. Circulation Research. 2018;122(2):319–36. DOI: 10.1161/CIRCRESAHA.117.310978
117. Ahmad S, Ferrario CM. Chymase inhibitors for the treatment of cardiac diseases: a patent review (2010–2018). Expert Opinion on Therapeutic Patents. 2018;28(11):755–64. DOI: 10.1080/13543776.2018.1531848
118. Nguyen G, Muller DN. The Biology of the (Pro)Renin Receptor. Journal of the American Society of Nephrology. 2010;21(1):18–23. DOI: 10.1681/ASN.2009030300
119. Cruciat C-M, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D et al. Requirement of Prorenin Receptor and Vacuolar H+-ATPase-Mediated Acidification for Wnt Signaling. Science. 2010;327(5964):459–63. DOI: 10.1126/science. 1179802
1179802
Ренин-ангиотензиновая система при новой коронавирусной инфекции COVID-2019 | Загидуллин
1. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H et al. Genomic characterization and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi:10.1016/S0140-6736(20)30251-8
2. Liu Y, Gayle AA, Wilder-Smith A, Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med. 2020;27(2): taaa021. doi:10.1093/jtm/taaa021
3. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020: e200994. doi:10.1001/jamainternmed.2020.0994
JAMA Intern Med. 2020: e200994. doi:10.1001/jamainternmed.2020.0994
4. Maksimov ML, Dralova OV, Starodubtsev AK. Angiotensin II type 1 receptor antagonists and ACE inhibitors in the regulation of hemodynamics and renin-angiotensin-aldosterone system activity: focus on the organ protection. Cardiovasc Ther Prev. 2010;9(2):115–124.
5. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE 2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87(5):1–9. doi:10.1161/01.res.87.5.e1
6. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captoprilin sensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–33243. doi:10.1074/jbc.M002615200
J Biol Chem. 2000;275(43):33238–33243. doi:10.1074/jbc.M002615200
7. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE 2, the putative receptor of Wuhan 2019-nCov. BioRxiv 919985 [Preprint]. 2020.
8. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J et al. Hydrolysis of biological peptides by human angiotensinconverting enzyme-related carboxypeptidase. J Biol Chem. 2002;277(17):14838–14843. doi:10.1074/jbc.M200581200
9. Raizada MK, Ferreira AJ. ACE 2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol. 2007;50(2):112–119. doi:10.1097/FJC.0b013e3180986219
10. Alenina N, Xu P, Rentzsch B, Patkin EL, Bader M.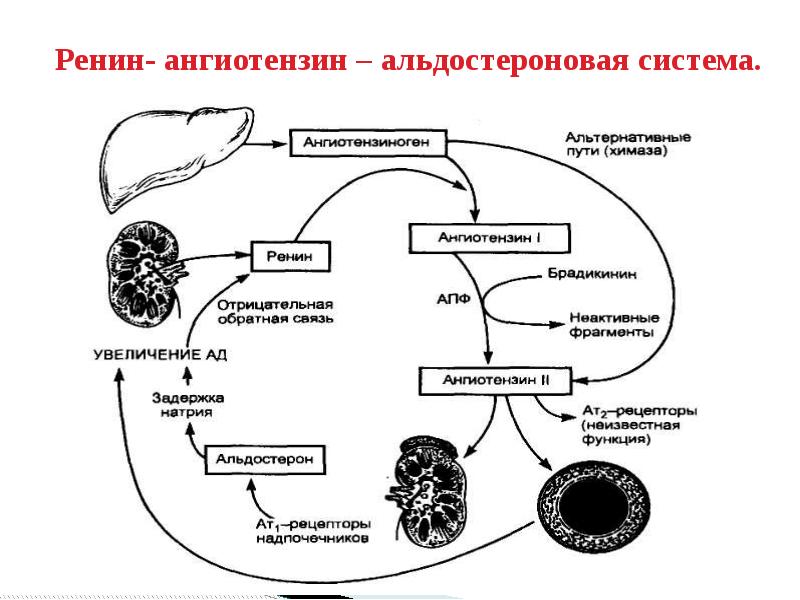 Genetically altered animal models for Mas and angiotensin-(1–7). Exp Physiol. 2008;93(5):528–537. doi:10.1113/expphysiol.2007.040345
Genetically altered animal models for Mas and angiotensin-(1–7). Exp Physiol. 2008;93(5):528–537. doi:10.1113/expphysiol.2007.040345
11. Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116(6):960–975. doi:10.1161/CIRCRESAHA.116.303587
12. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE 2/Angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res. 2016;118(8):1313–1326. doi:10.1161/CIRCRESAHA.116.307708
13. Santos R, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M et al. The ACE 2/Angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2018;98(1):505–553. doi:10.1152/physrev.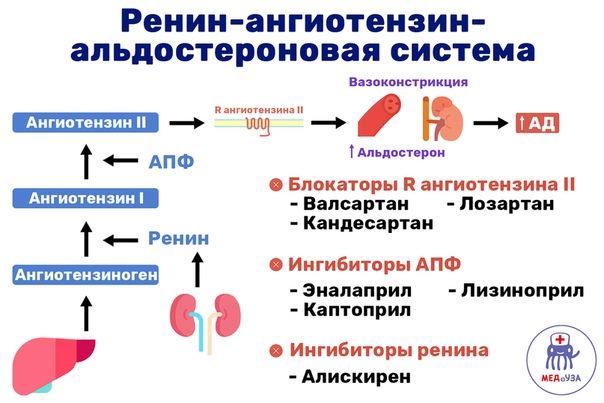 00023.2016
00023.2016
14. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi:10.1038/nature02145
15. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi:10.1038/nature00786
16. Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J et al. Characterization of renal angiotensinconverting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41(3):392–397. doi:10.1161/01.HYP.0000060689.38912.CB
17. Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow M et al. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensinconverting enzyme Homologue ACE 2. Circulation. 2003;108(14):1707–1712. doi:10.1161/01.CIR.0000094734.67990.99
Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensinconverting enzyme Homologue ACE 2. Circulation. 2003;108(14):1707–1712. doi:10.1161/01.CIR.0000094734.67990.99
18. Brake SJ, Barnsley K, Lu W, McAlinden KD, Eapen MS, Sohal SS. Smoking upregulates angiotensin-converting enzyme-2 receptor: a potential adhesion site for novel coronavirus SARS-CoV-2 (Covid-19). J Clin Med. 2020;9(3):841. doi:10.3390/jcm9030841
19. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):26052610. doi:10.1161/CIRCULATIONAHA.104.510461
20. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
21. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE 2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt1):45–51. doi:10.1042/BJ20040634
22. Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, Ma M et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1–7/mas receptor axis. Int J Biol Sci. 2011;7(8):1077–1092. doi:10.7150/ijbs.7.1077
23. Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT. Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1–7/ Mas receptor cascade. Biochem Pharmacol. 2017;144:90–99. doi:10.1016/j.bcp.2017.07.022
Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT. Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1–7/ Mas receptor cascade. Biochem Pharmacol. 2017;144:90–99. doi:10.1016/j.bcp.2017.07.022
24. Burchill LJ, Velkoska E, Dean RG, Griggs K, Patel SK, Burrell LM. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci (Lond). 2012;123(11):649–658. doi:10.1042/CS20120162
25. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374. doi:10.1007/s11427-020-1643-8
26. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al. A crucial role of angiotensin converting enzyme 2 (ACE 2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):8758787589. doi:10.1038/nm1267
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al. A crucial role of angiotensin converting enzyme 2 (ACE 2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):8758787589. doi:10.1038/nm1267
27. Henry С, Zaizafoun М, Stock E, Ghamande S, Arroliga AC, White HD. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Proc (Bayl Univ Med Cent). 2018;31(4):419–423. doi:10.1080/08998280.2018.1499293
28. Li J, Wang X, Chen J, Zhang H, Deng A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) Infection in Wuhan, China. J Am Med Assoc Cardiol. 2020; doi:10.1001/jamacardio.2020.1624
29. Guan W, Ni Z, Hu Y, Liang WH, Ou CQ, He JX et al.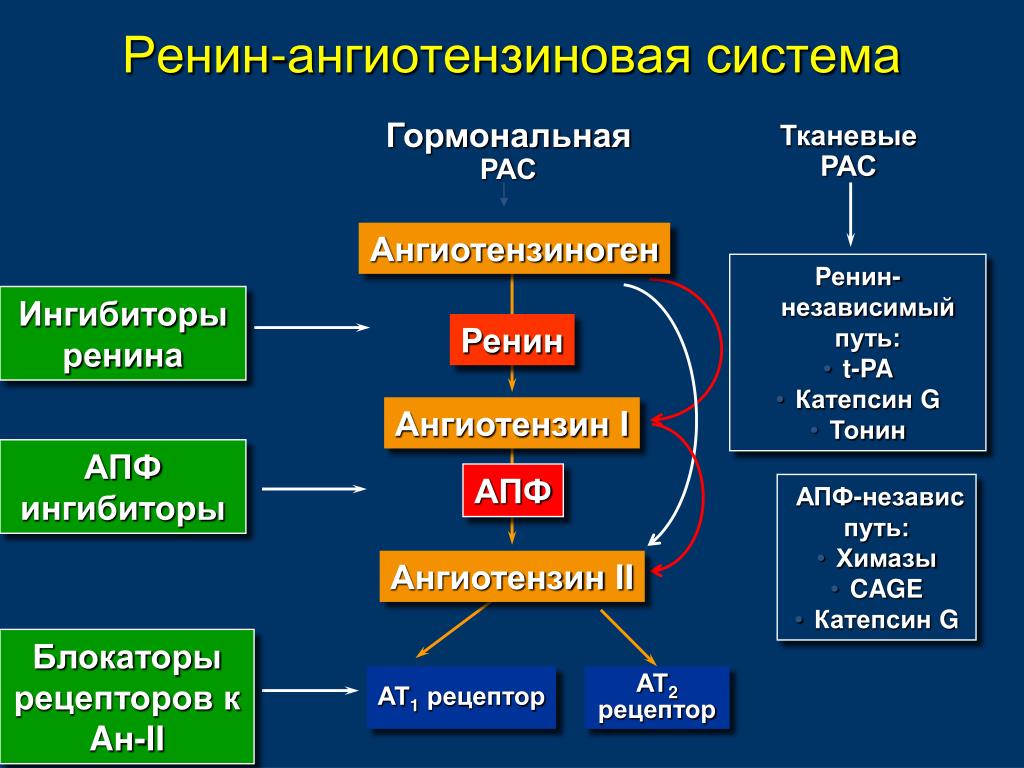 Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720. doi:10.1056/NEJMoa2002032
Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720. doi:10.1056/NEJMoa2002032
30. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Zh et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi:10.1016/S0140-6736(20)30566-3
31. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;1–3. doi:10.1007/s00134-020-05991-x
32. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J Am Med Assoc. 2020;323(11):1061–1069. doi:10.1001/jama.2020.1585
J Am Med Assoc. 2020;323(11):1061–1069. doi:10.1001/jama.2020.1585
33. Arentz M, Yim E, Klaff L, Lokhandwala Sh, Riedo FX, Chong M et al. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. J Am Med Assoc. 2020;323(16):1612–1614. doi:10.1001/jama.2020.4326
34. ACE Inhibitor Myocardial Infarction Collaborative Group. Indications for ACE inhibitors in the early treatment of acute myocardial infarction: systematic overview of individual data from 100,000 patients in randomized trials. Circulation. 1998;97(22):2202–2212. doi:10.1161/01.cir.97.22.2202
35. Шляхто Е. В., Конради А. О., Арутюнов Г. П., Арутюнов А. Г., Баутин А. Е., Бойцов С. А. и др. Руководство по диагностике и лечению болезней системы кровообращения в контексте пандемии COVID-19. Российский кардиологический журнал. 2020;25(3):3801. doi:10.15829/1560-4071-2020-3-3801.
Российский кардиологический журнал. 2020;25(3):3801. doi:10.15829/1560-4071-2020-3-3801.
36. Monteil V, Kwon H, Patricia P, Hagelkrüys A, Wimmer RA, Stahl M et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE 2. Cell. 2020; S 0092–8674(20):30399–8. doi:10.1016/j.cell.2020.04.004
37. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD et al. A pilot clinical trial of recombinant human angiotensinconverting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21(1):234. doi:10.1186/s13054-017-1823-x
38. Zhang H, Baker A. Recombinant human ACE 2: acing out angiotensin II in ARDS therapy. Crit Care. 2017;21(1):305. doi:10.1186/s13054-017-1882-z
Блокаторы РААС в клинической практике: новые возможности применения uMEDp
В последние годы в клинической практике все чаще назначаются блокаторы ренин-ангиотензин-альдостероновой системы (РААС). И это вполне естественно, поскольку РААС играет важную роль в развитии сердечно-сосудистых заболеваний. К блокаторам РААС, как известно, относят ингибиторы ангиотензинпревращающего фермента (АПФ) и блокаторы рецепторов ангиотензина II АТ1 (АРА).
И это вполне естественно, поскольку РААС играет важную роль в развитии сердечно-сосудистых заболеваний. К блокаторам РААС, как известно, относят ингибиторы ангиотензинпревращающего фермента (АПФ) и блокаторы рецепторов ангиотензина II АТ1 (АРА).
Таблица 1. Рекомендации по выбору лекарственных препаратов для лечения больных АГ (2008)
Рисунок 1. Механизм действия блокаторов рецепторов АТ1
Рисунок 2. HOPE: результаты исследования
Рисунок 3. План исследования ONTARGET
Рисунок 4. Достижение первичной конечной точки в исследовании ONTARGET
Таблица 2. Достижение первичной сердечно-сосудистой конечной точки
Ингибиторы АПФ давно применяются в клинической практике. На протяжении последнего десятилетия проведены многочисленные исследования, в которых изучалась клиническая эффективность ингибиторов АПФ при различных сердечно-сосудистых заболеваниях. Результаты этих исследований нашли отражение в отечественных рекомендациях, согласно которым ингибиторы АПФ рекомендовано применять у больных артериальной гипертонией, сердечной недостаточностью, острым и перенесенным инфарктом миокарда и диабетической нефропатией (1, 2).
На протяжении последнего десятилетия проведены многочисленные исследования, в которых изучалась клиническая эффективность ингибиторов АПФ при различных сердечно-сосудистых заболеваниях. Результаты этих исследований нашли отражение в отечественных рекомендациях, согласно которым ингибиторы АПФ рекомендовано применять у больных артериальной гипертонией, сердечной недостаточностью, острым и перенесенным инфарктом миокарда и диабетической нефропатией (1, 2).
Что касается АРА, то за последние годы этот класс препаратов сделал огромный скачок вперед в плане приобретения новых ниш в различных клинических ситуациях. Если раньше этот класс препаратов пребывал в тени ингибиторов АПФ (их назначение при артериальной гипертензии (АГ) ограничивалось в основном ситуациями, связанными с побочными эффектами приема ингибиторов АПФ), то в настоящее время ниша их применения достаточно обширная (таблица 1).
Обращают на себя внимание новые показания для применения АРА: профилактика мерцательной аритмии и хроническая сердечная недостаточность. Чтобы лучше понять возможности АРА, необходимо вспомнить их механизм действия. Он, как известно, заключается в блокировании АТ1-рецепторов ангиотензина II, через которые осуществляются основные негативные эффекты этого гормона. При этом, в отличие от ингибиторов АПФ, образование ангиотензина II не нарушается.
Чтобы лучше понять возможности АРА, необходимо вспомнить их механизм действия. Он, как известно, заключается в блокировании АТ1-рецепторов ангиотензина II, через которые осуществляются основные негативные эффекты этого гормона. При этом, в отличие от ингибиторов АПФ, образование ангиотензина II не нарушается.
К числу негативных эффектов ангиотензина II относятся: вазоконстрикция, увеличение секреции эндотелина, стимуляция образования перекисных радикалов, гипертрофия гладкомышечных клеток, увеличение активности ингибитора тканевого активатора плазминогена 1 типа. Многие из этих эффектов являются атерогенными. В то же время стимуляция ангиотензином II незаблокированных рецепторов 2 типа (АТ2) вызывает эффекты, противоположные вышеперечисленным, а именно вазодилатацию, увеличение продукции оксида азота, стимуляцию антипролиферативных процессов. Таким образом, AРА обладают двойным положительным механизмом действия, в котором заложен мощный антиатеротромбогенный потенциал (рисунок 1).
Иллюстрацией возможностей АРА является исследование LIFE (изучение эффективности лозартана в отношении снижения достижения конечных точек у лиц с артериальной гипертонией) (3). В этом исследовании не только впервые была доказана антигипертензивная эффективность АРА в плане влияния на конечные точки, но и продемонстрированы другие их возможности.
В двойном слепом рандомизированном контролируемом международном исследовании участвовали 9193 больных с АГ и гипертрофией левого желудочка в возрасте 55-80 лет. Участники исследования были рандомизированы на две группы для получения в качестве первоначального лечения либо лозартана, либо атенолола. Начальная доза препаратов составила соответственно 50 мг лозартана 1 раз в сутки и атенолола 50 мг 1 раз в сутки. Препараты можно было комбинировать с гидрохлоротиазидом – 12,5 мг/сут. и далее повышать их дозу до 100 мг/сут., чтобы достичь целевого снижения АД – менее 140/90 мм рт. ст. Наконец, если максимальные дозы исследуемых препаратов в комбинации с диуретиком не обеспечивали адекватного контроля АД, то разрешалось назначать дополнительные препараты (за исключением АРА, ингибиторов АПФ и b-адреноблокаторов).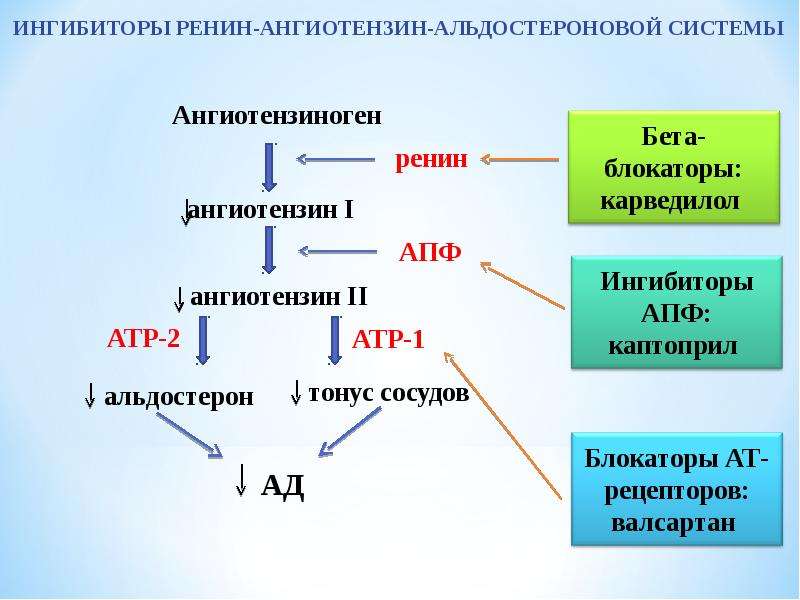 Длительность исследования составила в среднем 4,7 года.
Длительность исследования составила в среднем 4,7 года.
Основной целью исследования явилось изучение сравнительной эффективности лозартана и атенолола в плане снижения главной конечной точки, которая включала суммарно мозговой инсульт (МИ), инфаркт миокарда (ИМ) и смертность от сердечно-сосудистых причин. Другие конечные точки включали частоту возникновения новых случаев сахарного диабета (СД), смертность от всех причин, регресс гипертрофии миокарда левого желудочка, частоту госпитализаций по поводу стенокардии или сердечной недостаточности.
Частота главной конечной точки (смерть от сердечно-сосудистых причин, МИ и ИМ) на фоне лечения лозартаном оказалась 18%, а в группе пациентов, лечившихся атенололом, – 23% (р
Новой областью применения блокаторов РААС стало снижение сердечно-сосудистого риска у пациентов с различными клиническими проявлениями атеросклероза. Новая область применения этого класса препаратов не имеет ничего общего с традиционным их применением при АГ, СД или хронической сердечной недостаточности (ХСН). Долгосрочный эффект блокаторов РААС основан на ряде механизмов. Одним из важнейших механизмов является улучшение эндотелиальной функции (4, 5). Коррекция эндотелиальной функции, в основе которой лежит увеличение синтеза оксида азота, является залогом антиатеросклеротического эффекта, который может иметь долгосрочное значение в плане улучшения прогноза пациентов, имеющих высокий риск смертельных исходов.
Долгосрочный эффект блокаторов РААС основан на ряде механизмов. Одним из важнейших механизмов является улучшение эндотелиальной функции (4, 5). Коррекция эндотелиальной функции, в основе которой лежит увеличение синтеза оксида азота, является залогом антиатеросклеротического эффекта, который может иметь долгосрочное значение в плане улучшения прогноза пациентов, имеющих высокий риск смертельных исходов.
Впервые такая возможность была продемонстрирована для ингибиторов АПФ.
Это было показано в исследовании HOPE (6), в котором участвовали 9297 мужчин и женщин с подтвержденным атеросклерозом различной локализации (коронарная болезнь сердца, поражение периферических артерий, инсульт) или сахарным диабетом и по крайней мере еще одним фактором риска (артериальная гипертония, курение сигарет, микроальбуминурия или дислипидемия). 80% больных имели ишемическую болезнь сердца, 55% – стенокардию, 52% – инфаркт миокарда в анамнезе, 43% – атеросклероз периферических артерий, у 25% была нестабильная стенокардия в анамнезе, а у 26% – аортокоронарное шунтирование в анамнезе, у 18% – чрескожная реваскуляризация коронарных артерий, у 11% инсульт или транзиторная ишемическая атака.
Почти половина пациентов страдала АГ и около 40% – СД типа 2. Больным назначали плацебо или ингибитор АПФ рамиприл (с титрованием доз от 1,25 до 10 мг) и продолжали наблюдение в среднем 5 лет. Первичная конечная точка (сердечно-сосудистая смерть, ИМ или МИ) была зарегистрирована у 17,8% больных группы плацебо и 14,0% больных группы рамиприла – снижение риска на 22%, р
Терапия рамиприлом привела к снижению частоты важнейших компонентов конечной точки – ИМ, МИ. Кроме того, установлено снижение общей смертности (с 12,2 до 10,4% в течение 5 лет), необходимости реваскуляризации, диабетических осложнений, развития СД, остановки сердца, прогрессирования стенокардии или сердечной недостаточности. Представляет интерес тот факт, что снижение АД в группе рамиприла было сравнительно небольшим (АД –3/2 мм рт. ст.), поэтому результаты лечения нельзя объяснить только антигипертензивным действием препарата.
Таким образом, в этом исследовании была подтверждена протективная роль рамиприла в плане предупреждения развития осложнений у пациентов с клиническими проявлениями атеросклероза. Причем защитный эффект рамиприла никак не связан со снижением АД.
Причем защитный эффект рамиприла никак не связан со снижением АД.
Вместе с тем с момента окончания исследования НОРЕ возникал вопрос: а почему АРА не способны на такое же снижение риска? Ведь как указывалось выше, АРА обладают механизмом действия, который способен приостановить развитие атеросклероза и улучшить прогноз пациентов. Именно такая гипотеза проверялась в исследовании ONTARGET (The Ongoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial).
В данном исследовании изучали влияние телмисартана в сравнении с рамиприлом, а также комбинации этих двух препаратов на прогноз пациентов с различными проявлениями атеросклероза (поражения коронарных, периферических и церебральных артерий, СД типа 2, с органными поражениями) без признаков сердечной недостаточности (7). В двойном слепом рандомизированном исследовании в среднем в течение 56 месяцев 8576 больных получали рамиприл в дозе 10 мг; 8542 больных – телмисартан в дозе 80 мг; 8502 больных – оба препарата в указанных дозах дополнительно к ранее проводимой терапии (рисунок 3).
Препаратом сравнения для телмисартана был выбран рамиприл, ранее продемонстрировавший эффективность при лечении подобной категории пациентов. Проверялась гипотеза, что телмисартан будет не хуже, чем рамиприл, в профилактике осложнений у этой группы больных, а комбинация телмисартана с рамиприлом лучше, чем рамиприл.
В качестве первичной конечной точки была выбрана комбинация смерти от сердечно-сосудистых причин, ИМ, инсультов и госпитализация в связи с сердечной недостаточностью. В течение исследования частота первичной конечной точки составила 1412 больных в группе рамиприла (16,5%) и 1423 больных в группе телмисартана (16,7%). Различия между группами оказались недостоверны (рисунок 4). Вместе с тем частота побочных эффектов в группе телмисартана была ниже. К примеру, частота кашля в группе телмисартана составила 1,1%, а в группе рамиприла 4,2%, р
В группе комбинированной терапии при одинаковой частоте развития сердечно-сосудистых осложнений по сравнению с группой рамиприла (нет достоверных отличий) была хуже переносимость: повышенный риск развития гипотензивных проявлений (4,8% vs 1,7%, р
Таким образом, гипотеза, которая лежала в основе этого исследования, – «телмисартан будет не хуже в профилактике осложнений у больных с очень высоким риском их развития» – подтвердилась при лучшей переносимости лечения телмисартаном. А вот давно обсуждаемая потенциальная эффективность при сочетанном применении ингибиторов АПФ с АРА, особенно для усиления независимых от АД положительных механизмов, не только не подтвердилась, но и оказалась более опасной в плане развития нежелательных явлений.
А вот давно обсуждаемая потенциальная эффективность при сочетанном применении ингибиторов АПФ с АРА, особенно для усиления независимых от АД положительных механизмов, не только не подтвердилась, но и оказалась более опасной в плане развития нежелательных явлений.
Результаты исследования важны еще и потому, что, по данным завершившихся исследований, некоторыми учеными было сделано предположение о том, что АРА менее эффективны в профилактике ИМ, чем другие антигипертензивные препараты (8).
В исследовании ONTARGET частота развития ИМ на фоне лечения рамиприлом и телмисартаном достоверно не различались (таблица 2). В целом эта позиция не получила подтверждения в материалах опубликованного недавно всестороннего метаанализа, который показал одинаковую частоту ИМ в сравнении с другими препаратами.
В новом мета-регрессионном анализе Blood Pressure Lowering Treatment Trialist (BPLTT) оказалось, что АРА имеют ровно такие же связанные со снижением АД благоприятные эффекты на коронарные события, как и ингибиторы АПФ, однако последние могут иметь небольшое, не связанное с АД, благоприятное влияние.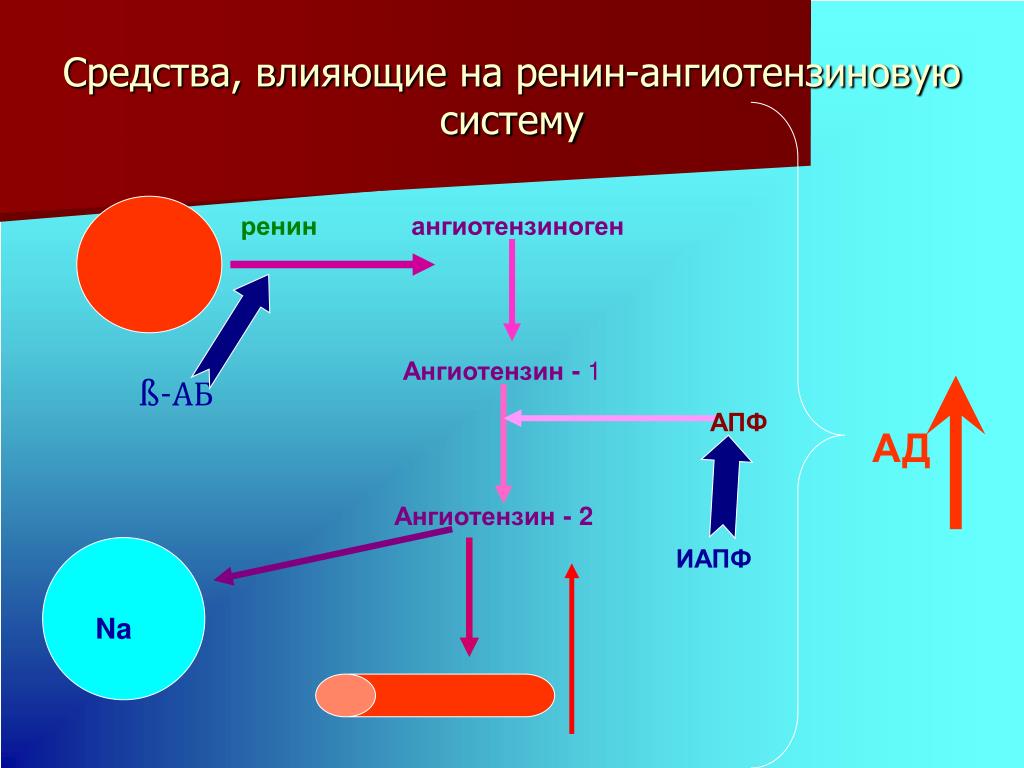 Прямое сравнение АРА (телмисартан) и ингибиторов АПФ (рамиприл) у больных с высоким риском развития осложнений в исследовании ONTARGET показало, что в обеих группах частота развития ИМ была одинаковой, т.е. кардиопротективное действие выражено одинаково.
Прямое сравнение АРА (телмисартан) и ингибиторов АПФ (рамиприл) у больных с высоким риском развития осложнений в исследовании ONTARGET показало, что в обеих группах частота развития ИМ была одинаковой, т.е. кардиопротективное действие выражено одинаково.
Исследование TRANSCEND (Telmisartan Randomised Assessment Study in ACE intolerant subjects with cardiovascular Disease), которое является частью программы ONTARGET, было организованно для изучения эффективности телмисартана у больных с сердечно-сосудистыми заболеваниями или СД с органными поражениями, не переносивших лечения ингибиторами АПФ (9).
В исследование было включено 5926 больных, которые были рандомизированы на две группы – телмисартана 80 мг (n = 2954) и плацебо (n = 2972). Первичной конечной точкой в исследовании была сумма смертей от сердечно-сосудистых причин, ИМ, инсультов и госпитализаций по поводу сердечной недостаточности. Средняя продолжительность исследования составила 56 месяцев.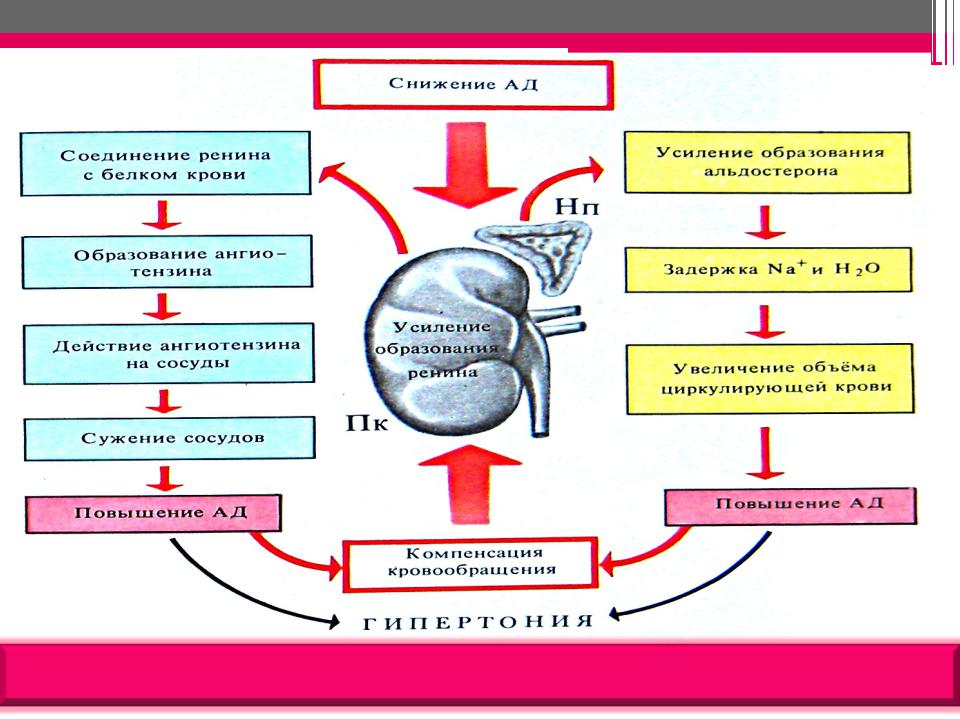 Артериальное давление было ниже в группе телмисартана по сравнению с плацебо на протяжении всего исследования (в среднем на 4,0/2,2 мм рт. ст.). В группе телмисартана было отмечено 465 (15,7%) событий первичной конечной точки в сравнении с 504 (17,0%) событиями в группе плацебо (отношение рисков 0,91; 95% доверительный интервал 0,81-1,05, р = 0,216).
Артериальное давление было ниже в группе телмисартана по сравнению с плацебо на протяжении всего исследования (в среднем на 4,0/2,2 мм рт. ст.). В группе телмисартана было отмечено 465 (15,7%) событий первичной конечной точки в сравнении с 504 (17,0%) событиями в группе плацебо (отношение рисков 0,91; 95% доверительный интервал 0,81-1,05, р = 0,216).
Следует отметить, что одна из вторичных конечных точек – сумма смертей от сердечно-сосудистых причин, ИМ и инсультов – была у 384 (13,0%) больных на телмисартане и у 440 (14,8%) больных на плацебо (отношение рисков 0,87; 95% доверительный интервал 0,76-1,00, р = 0,048), что оказалось достоверно меньше (см. рисунок 4). Именно по такому критерию была доказана эффективность рамиприла в сравнении с плацебо в исследовании HOPE (26). Больные, получавшие телмисартан, достоверно реже госпитализировались по сердечно-сосудистым причинам на 15% (р = 0,028). Телмисартан продемонстрировал хорошую толерантность у больных с непереносимостью ингибиторов АПФ.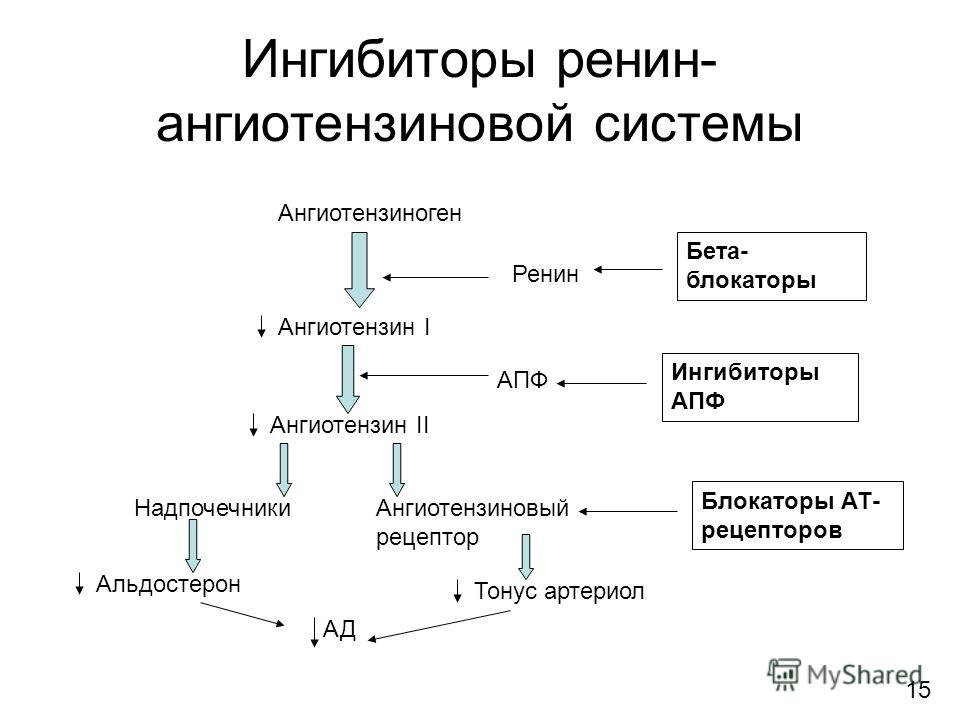
Результаты исследования ONTARGET/TRANSCEND послужили основанием для регистрации нового показания для телмисартана – снижение риска осложнений у пациентов с клиническими проявлениями атеросклероза. В октябре 2009 г. FDA (США) одобрила применение телмисартана для снижения риска ИМ (сердечной атаки), инсульта и смерти от сердечно-сосудистых причин у больных старше 55 лет с высоким сердечно-сосудистым риском, которые не переносят ингибиторы АПФ.
В ноябре 2009 г. Европейская комиссия (EMEA) зарегистрировала новое показание для телмисартана. Телмисартан рекомендуется для снижения сердечно-сосудистой заболеваемости у больных с клиническими проявлениями атеросклероза (ИБС, инсульт, поражение периферических артерий) и СД типа 2 с документированными органными поражениями. Таким образом, телмисартан стал первым препаратом из класса АРА, рекомендованным для назначения больным с высоким сердечно-сосудистым риском.
Заключение
Блокаторы ангиотензиновых рецепторов обладают выраженными органопротективными свойствами, что значительно расширяет возможности их применения у больных с АГ. Благодаря выигрышному сочетанию хорошей переносимости, органопротекции, благоприятного метаболического профиля и доказанного в клинических исследованиях снижения риска развития осложнений этот класс антигипертензивных препаратов следует рассматривать как средство первого выбора для многих больных с повышенным АД. Недавно было доказано, что сартаны не уступают ингибиторам АПФ в эффективности при лечении больных с высоким риском осложнений (исследование ONTARGET/TRANSCEND), а телмисартан – первый и единственный представитель класса АРА – получил официальное показание для применения у этих больных.
Благодаря выигрышному сочетанию хорошей переносимости, органопротекции, благоприятного метаболического профиля и доказанного в клинических исследованиях снижения риска развития осложнений этот класс антигипертензивных препаратов следует рассматривать как средство первого выбора для многих больных с повышенным АД. Недавно было доказано, что сартаны не уступают ингибиторам АПФ в эффективности при лечении больных с высоким риском осложнений (исследование ONTARGET/TRANSCEND), а телмисартан – первый и единственный представитель класса АРА – получил официальное показание для применения у этих больных.
РААС и фиброз. Реалии и перспективы
Стенограмма выступления профессора Драпкиной О.М. на II Международном Интернет Конгрессе специалистов по внутренним болезням (день 2).
Профессор Драпкина О.М.: – Мы продолжаем, и дальше я сделаю сообщение, которое будет касаться ренин-ангиотензин-альдостероновой системы и возможного ее влияния на процессы фиброза.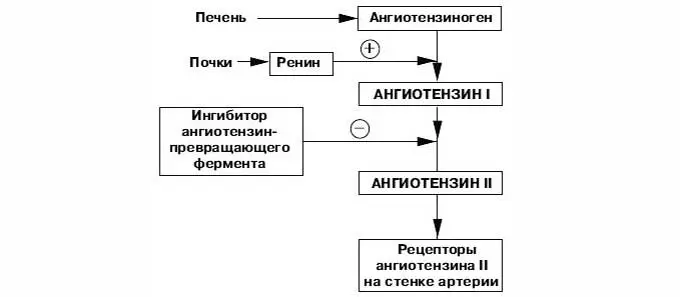 Я сразу приступлю…
Я сразу приступлю…
(00:15) Заставка: РААС и фиброз
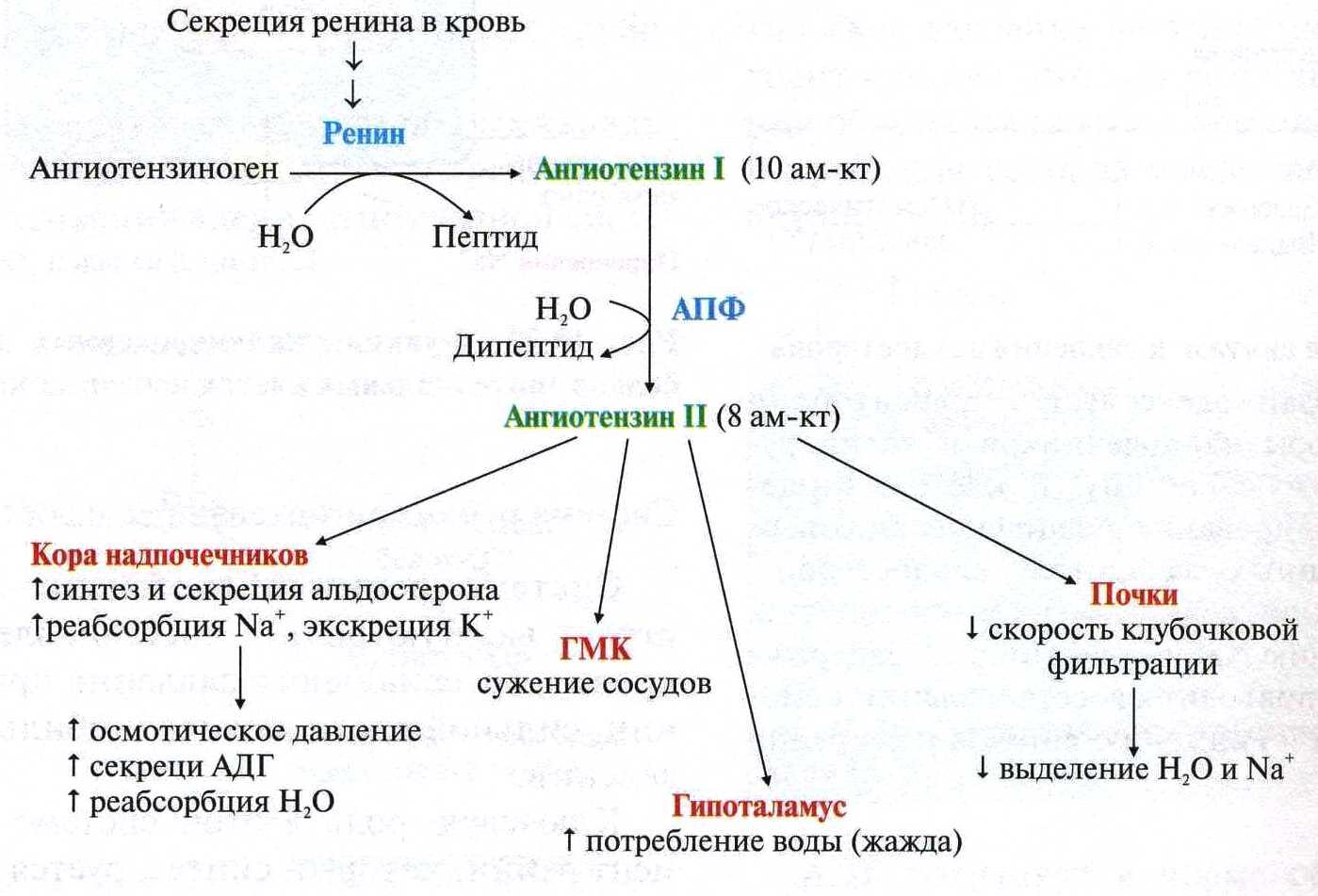
Профессор Драпкина О.М.: — …непосредственно к лекции. Итак, ренин-ангиотензин-альдостероновая система и фиброз. Ренин-ангиотензин-альдостероновая система известна очень давно, и в принципе мы, конечно, знаем, что эта система представляет собой систему очень нужную в организме. Нельзя ее блокировать просто для того, чтобы ее заблокировать. В минуты опасности, в минуты, предположим, чрезвычайно интенсивных ранений, когда надо поднять уровень артериального давления, когда нужно поднять сосудистое сопротивление, она мгновенно срабатывает. И в принципе, давно-давно, в далекие годы это, наверное, и защищало наших далеких предшественников от того, чтобы умереть от кровопотерь, от того, чтобы умереть от каких-то травм. Но, тем не менее, год за годом, столетие за столетием эта система стала превращаться в звено патогенеза очень большого количества заболеваний, и сегодня Анатолий Иванович Мартынов показал, как это происходит.
Ренин-ангиотензин-альдостероновая система – это часть того самого печального сердечно-сосудистого континуума, к которому мы привыкли. Хорошо известны действующие лица, это ангиотензиноген, ангиотензины I и II, альдостерон и два фермента, один из которых настоящим ферментом, в общем-то, и не является, я говорю о ренине, а второй настоящий фермент, ангиотензинпревращающий фермент, который служит мишенью для излюбленного класса препаратов кардиологов, терапевтов и других специалистов.
Конечно, путь к тому, чтобы открыть не только каждый из компонентов ренин-ангиотензин-альдостероновой системы, но и всю слаженную ее работу, был достаточно труден, но, тем не менее, неуклонно ученые шли к тому, чтобы разгадать эту загадку. Началось все в 1898 году, когда было обнаружено, что вытяжка из почек кролика способна повышать артериальное давление, это был как раз ренин. Затем наступили 1939 и 1940 годы, тогда был открыт ангиотонин, так его раньше называли, впоследствии он был известен и нам он известен как ангиотензин. Затем был выделен ангиотензин, и наконец, было выдвинуто предположение – это предположение затем было реализовано в соответствующий класс препаратов – о том, что существует некий фермент, блокирование которого приведет к тому, что ангиотензина II образуется меньше. Это как раз тот самый ангиотензинпревращающий фермент.
Затем был выделен ангиотензин, и наконец, было выдвинуто предположение – это предположение затем было реализовано в соответствующий класс препаратов – о том, что существует некий фермент, блокирование которого приведет к тому, что ангиотензина II образуется меньше. Это как раз тот самый ангиотензинпревращающий фермент.
И потом мысль человеческая привела к тому, что были созданы препараты, которые способны влиять на различные уровни ренин-ангиотензин-альдостероновой системы. Это и ингибиторы АПФ, и первый ингибитор АПФ был получен из яда бразильской змеи жарарака, маленький портрет которой представлен на этом слайде. Наконец, где-то через 10 лет был создан первый непептидный пероральный блокатор ангиотензиновых рецепторов.
Еще раз посмотрим на главных героев ренин-ангиотензин-альдостероновой системы, я кратко охарактеризую каждого из них. Про ангиотензиноген, наверное, говорить приходится меньше, здесь больше роль отводится ренину, который и стимулирует эту реакцию, это превращение ангиотензиногена в ангиотензин I. И затем нас интересует, конечно, ангиотензин II, который и реализует основные неблагоприятные эффекты активации ренин-ангиотензин-альдостероновой системы. И мы видим, что есть еще и альдостерон, и про него тоже не следует забывать практикующему врачу.
И затем нас интересует, конечно, ангиотензин II, который и реализует основные неблагоприятные эффекты активации ренин-ангиотензин-альдостероновой системы. И мы видим, что есть еще и альдостерон, и про него тоже не следует забывать практикующему врачу.
Давайте посмотрим на портреты главных героев. Это действительно портрет, в беленькой рамочке, так выглядит ренин. Происходит от латинского ren – «почка», и понятно, что вырабатывается он в почках, в юкстагломерулярном аппарате в стенках артериол почечных клубочков. Оттуда он поступает в кровь и лимфу и начинает уже свое действие. Как я уже сказала, ренин не является истинным гормоном, поскольку не имеет клеточной мишени, а воздействует на другой белок – это единственная его мишень, белок ангиотензиноген. Он присутствует в матке, в плаценте, в слюнных железах, в мозге, в стенках некоторых крупных артерий и представляет из себя 340 аминокислот с молекулярной массой 37 кДа.
Следующий очень важный агент – это ангиотензин II, тоже его портрет в уголке есть. Здесь их две структуры, потому что первая структура – это ангиотензин I, а вторая структура, от которой уже отщепилось несколько остатков – это ангиотензин II. Он известен под разными именами: он же ангиотонин, он же гипертензин – мы чаще называем эту группу ангиотензинами. Это пептид, который образуется в организме из ангиотензиногена, и он обуславливает эффекты, которые тоже представлены на слайде. Это и вазоконстрикция, и усиление реабсорбции натрия в проксимальных почечных канальцах, и секреция альдостерона – без него она как будто бы невозможна, но мы чуть ниже посмотрим, что возможна и без него. Секреция вазопрессина, эндотелина-1 – это главные враги, или антиподы релаксирующего фактора из эндотелия, оксида азота. Высвобождение ренина, усиление высвобождения норадреналина из симпатических нервных окончаний, и наконец, пролиферация гладкомышечных клеток сосудов, гиперплазия интимы и гипертрофия кардиомиоцитов.
Здесь их две структуры, потому что первая структура – это ангиотензин I, а вторая структура, от которой уже отщепилось несколько остатков – это ангиотензин II. Он известен под разными именами: он же ангиотонин, он же гипертензин – мы чаще называем эту группу ангиотензинами. Это пептид, который образуется в организме из ангиотензиногена, и он обуславливает эффекты, которые тоже представлены на слайде. Это и вазоконстрикция, и усиление реабсорбции натрия в проксимальных почечных канальцах, и секреция альдостерона – без него она как будто бы невозможна, но мы чуть ниже посмотрим, что возможна и без него. Секреция вазопрессина, эндотелина-1 – это главные враги, или антиподы релаксирующего фактора из эндотелия, оксида азота. Высвобождение ренина, усиление высвобождения норадреналина из симпатических нервных окончаний, и наконец, пролиферация гладкомышечных клеток сосудов, гиперплазия интимы и гипертрофия кардиомиоцитов.
Таким образом, мы можем сказать, что действительно практически на каждом этапе так называемого сердечно-сосудистого континуума ангиотензин вносит свою роль. Это начинается на самых первых этапах этой печальной дороги, когда мы говорим о факторах риска. А факторы риска для всех неинфекционных заболеваний одни и те же.
Это начинается на самых первых этапах этой печальной дороги, когда мы говорим о факторах риска. А факторы риска для всех неинфекционных заболеваний одни и те же.
Это высокий уровень артериального давления, это нарушение липидного обмена, это высокий уровень глюкозы и избыточная масса тела. Затем факторы риска реализуются в эндотелиальную дисфункцию, которой не видно, которая себя никаким образом не проявляет, но именно этот этап – самый лучший этап для того, чтобы внедриться в патологический процесс и повернуть его вспять. Если этого не происходит, то идет череда событий, таких как атеросклероз. Это заболевание, которое поражает сосуды крупного мышечного типа, и в первую очередь уязвимы сосуды коронарные. Ишемическая болезнь сердца, соответственно. Если вдруг происходит дестабилизация этой бляшки, которая была до поры до времени стабильной, то бригада скорой медицинской помощи доставляет такого пациента в отделение интенсивной терапии, поскольку бляшка изъязвляется, а это уже инфаркт миокарда.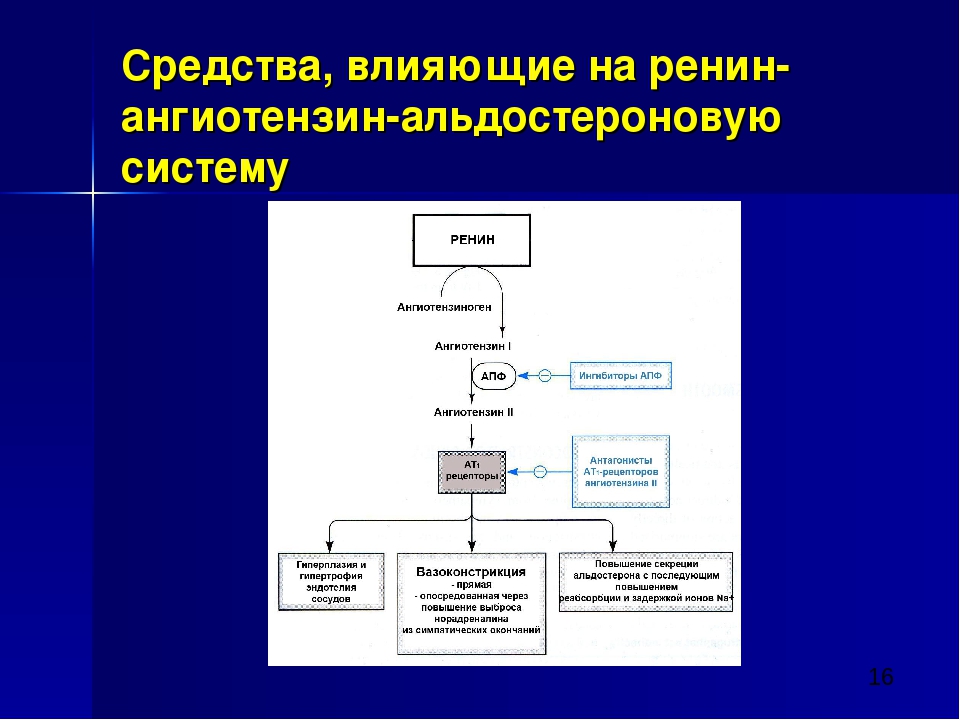 А дальше – как повезет. У кого-то возникнет миогенная дилатация, и пациент будет проявлять симптомы сердечной недостаточности. У кого-то рубец будет небольшим, и какое-то время ремоделирования миокарда не произойдет.
А дальше – как повезет. У кого-то возникнет миогенная дилатация, и пациент будет проявлять симптомы сердечной недостаточности. У кого-то рубец будет небольшим, и какое-то время ремоделирования миокарда не произойдет.
Поэтому для того, чтобы – возвращаемся к героям ренин-ангиотензин-альдостероновой системы – опосредовать свое действие, ангиотензин II должен связаться со специфическими рецепторами. И в основном таких рецепторов известно два типа, это ангиотензиновые рецепторы 1 и 2. Мы видим, что есть все основания – и это совершенно точно доказано – сделать так, чтобы заблокировать именно ангиотензиновые рецепторы 1 типа, потому что ангиотензиновые рецепторы 2 типа – это наши друзья, поскольку они существуют в «антагонистических» соотношениях с АТ1, и если АТ1 ответственны за вазоконстрикцию, то стимуляция АТ2 приведет к вазодилатации. Там задержка натрия – здесь выведение натрия. Здесь активация симпатоадреналовой системы и пролиферативные процессы – здесь обратный процесс, антипролиферативный эффект, противовоспалительный эффект. И надо сказать, что такие препараты тоже созданы, эти препараты влияют в какой-то степени избирательно – кто-то более избирательно, кто-то менее избирательно – на блокаду ангиотензиновых рецепторов 1 типа.
И надо сказать, что такие препараты тоже созданы, эти препараты влияют в какой-то степени избирательно – кто-то более избирательно, кто-то менее избирательно – на блокаду ангиотензиновых рецепторов 1 типа.
Есть еще один агент, это альдостерон. Альдостерон представляет из себя стероидную структуру, и физиологическая роль его известна давно, и не случайно мы практически в любой клинической ситуации, особенно когда пациент с сердечной недостаточностью, или он солечувствителен, или он солезависим, мы применяем блокаторы альдостерона, хорошо известные, которые есть в арсенале любого врача. Альдостерон представляет из себя гормон клубочковой зоны надпочечников, и он стимулируется в ответ на активацию ангиотензина II. Но обратите внимание, есть еще стимуляторы: это калий и, например, адренокортикотропный гормон. Он активирует минералокортикотропные рецепторы в цитоплазме и действует на уровне дистальных канальцев нефрона, вызывая те эффекты, за которые он отвечает, а именно, задержка натрия и воды, соответственно, объемозависимая гипертензия, экскреция калия, вывод его, и, как я уже сказала, задержка жидкости. Надо помнить, как я уже сказала, что помимо классической концепции, когда и ренин осуществляет свою работу, и ангиотензинпревращающий фермент осуществляет свою работу, и ангиотензина II образовалось много, и он стимулирует секрецию альдостерона (хотя лучше бы этого не было), если мы блокируем эту цепь, которая представлена на слайде слева от меня – у нас есть способы это заблокировать – то альдостерон все равно может вырабатываться под воздействием других факторов.
Надо помнить, как я уже сказала, что помимо классической концепции, когда и ренин осуществляет свою работу, и ангиотензинпревращающий фермент осуществляет свою работу, и ангиотензина II образовалось много, и он стимулирует секрецию альдостерона (хотя лучше бы этого не было), если мы блокируем эту цепь, которая представлена на слайде слева от меня – у нас есть способы это заблокировать – то альдостерон все равно может вырабатываться под воздействием других факторов.
Как я уже сказала, высокий уровень калия, высокий уровень эндотелина, кортикотропина может приводить к тому, что альдостерона тоже будет много. Поэтому синтез альдостерона – это тоже тот факт, который надо учитывать, когда мы обдумываем схему ведения пациента с артериальной гипертензией. Рецепторы, которые чувствительны к альдостерону, найдены практически везде, поэтому мы тоже можем с определенной долей допущения сказать, что блокада альдостерона приведет к тому, что мы получим благоприятные эффекты активации ренин-ангиотензин-альдостероновой системы на так называемом тканевом уровне, не только плазменную, но и тканевую, которая и опосредует большие неприятности в плане, например, ремоделирования. Фиброз миокарда в основном связан, как мы немножко ниже увидим, именно с активацией ангиотензина II в большей степени и альдостерона.
Фиброз миокарда в основном связан, как мы немножко ниже увидим, именно с активацией ангиотензина II в большей степени и альдостерона.
Раннее ремоделирование левого желудочка после инфаркта миокарда, давайте представим себе эту ситуацию или вспомним эту ситуацию из нашей клинической практики. Первые часы, зона инфаркта есть, но нет пока клинически значимых изменений общей геометрии полости левого желудочка. А дальше все зависит от того, насколько быстро больной будет доставлен в специализированное отделение интенсивной терапии, скорее всего, пройдут часы. Мы видим, что зона пораженного миокарда расширяется, и та самая сферизация полости левого желудочка, конечно, требует не только месяцев, но начинается она через дни, поэтому здесь очень важно как можно раньше заблокировать ренин-ангиотензин-альдостероновую систему, например, в первые часы после инфаркта миокарда, и такие работы тоже есть и выполняются. Существенным моментом служит и то, что этот каскад – ангиотензиноген, ангиотензин II и связывание его с ангиотензиновыми рецепторами – имеет и некие альтернативные пути.
Например, если мы даже и блокируем ангиотензинпревращающий фермент, то возможно формирование или синтез ангиотензина II за счет АПФ независимого образования, и здесь самую большую роль играют химазы. Обратите внимание, образуется брадикинин. Мы не любим этот агент, поскольку он вызывает кашель у нашего пациента. Но если повнимательнее присмотреться уже к этому герою, мы увидим, как много положительных черт он в себе таит. Это вазодилатация, в том числе, коронарных артерий, это усиление натрийуреза, это кардиопротективное действие. Это увеличение фибринолитической активности плазмы крови, и это еще далеко не полный перечень. Брадикинин участвует в процессах цитопротекции в условиях гипоксии, стимулирует рост капилляров, а самое главное, является мощным индуктором для того, чтобы освободился оксид азота. Он тормозит высвобождение норадреналина из нервных окончаний симпатоадреналовой системы и таит в себе очень большой антифибротический потенциал, который тоже активно изучается.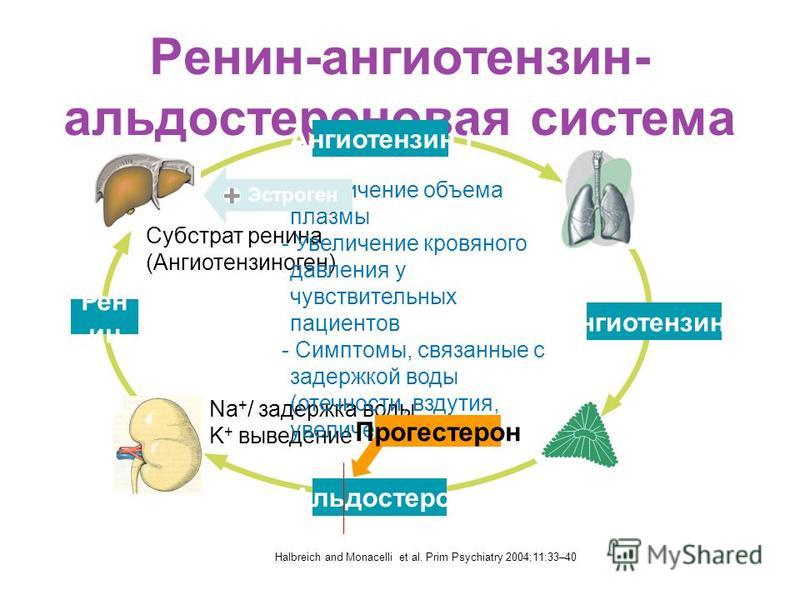
Вот она, наша ренин-ангиотензин-альдостероновая система, если ее представить «в полной красе», и у нас появляются и есть огромные возможности ее заблокировать везде: и на уровне фермента ангиотензинпревращающего, и на уровне рецепторов, и даже появились ингибиторы вазопептидаз и прямые ингибиторы ренина, о чем сегодня тоже говорил Анатолий Иванович.
Нужно ли блокировать на разных этапах ее? Исследования последние говорят, что не стоит этого делать, поскольку концепция соединить, например, блокаторы ангиотензиновых рецепторов с ингибиторами АПФ себя не оправдала. Тому есть, например, подтверждение в исследовании ONTARGET, когда никаких благоприятных эффектов от этой комбинации не было получено, а когда попытались ингибиторы АПФ соединить с алискиреном (исследование ALTITUDE), тоже ничего хорошего не получилось. По-видимому, этого делать не стоит.
Но всегда возникают вопросы, и сейчас нам уважаемая Интернет-аудитория задала вопросы по поводу того, что же выбрать, выбрать ли блокаторы ангиотензиновых рецепторов или же ингибиторы АПФ? Для того чтобы ответить на этот вопрос, давайте мы еще раз вспомним, казалось бы, хорошо знакомую нам схему. Вы видите, сигнализируют нам пути образования ангиотензина II, и если мы применяем блокаторы ангиотензиновых рецепторов, то ангиотензин II увеличенный начинает связываться с АТ2 рецепторами, и действительно мы получаем, что гипертрофия миокарда снижается, ремоделирование сердца и сосудов снижается. Но появляются новые данные. Оказывается, немного повышаются провоспалительные хемокины, как будто бы несколько активируется процесс апоптоза кардиомиоцитов, и возникает вопрос: что еще? Может быть, мы еще не все знаем. Если происходит или если мы блокируем ангиотензинпревращающий фермент, то ангиотензин II действительно уменьшается, а его «остатки» будут связываться с тем, что есть, поровну.
Вы видите, сигнализируют нам пути образования ангиотензина II, и если мы применяем блокаторы ангиотензиновых рецепторов, то ангиотензин II увеличенный начинает связываться с АТ2 рецепторами, и действительно мы получаем, что гипертрофия миокарда снижается, ремоделирование сердца и сосудов снижается. Но появляются новые данные. Оказывается, немного повышаются провоспалительные хемокины, как будто бы несколько активируется процесс апоптоза кардиомиоцитов, и возникает вопрос: что еще? Может быть, мы еще не все знаем. Если происходит или если мы блокируем ангиотензинпревращающий фермент, то ангиотензин II действительно уменьшается, а его «остатки» будут связываться с тем, что есть, поровну.
На самом деле ангиотензиновых рецепторов намного больше, мы только лучше знаем первые два типа, АТ1 и АТ2 рецепторы, но их уже около семи. Если про АТ1 и АТ2, как я уже сказала, мы много знаем, то, например, функция АТ3 на 2011 год была не совсем известна, они мало изучены, и что будет, если ангиотензин II начнет связываться и с ними, пока не совсем ясно.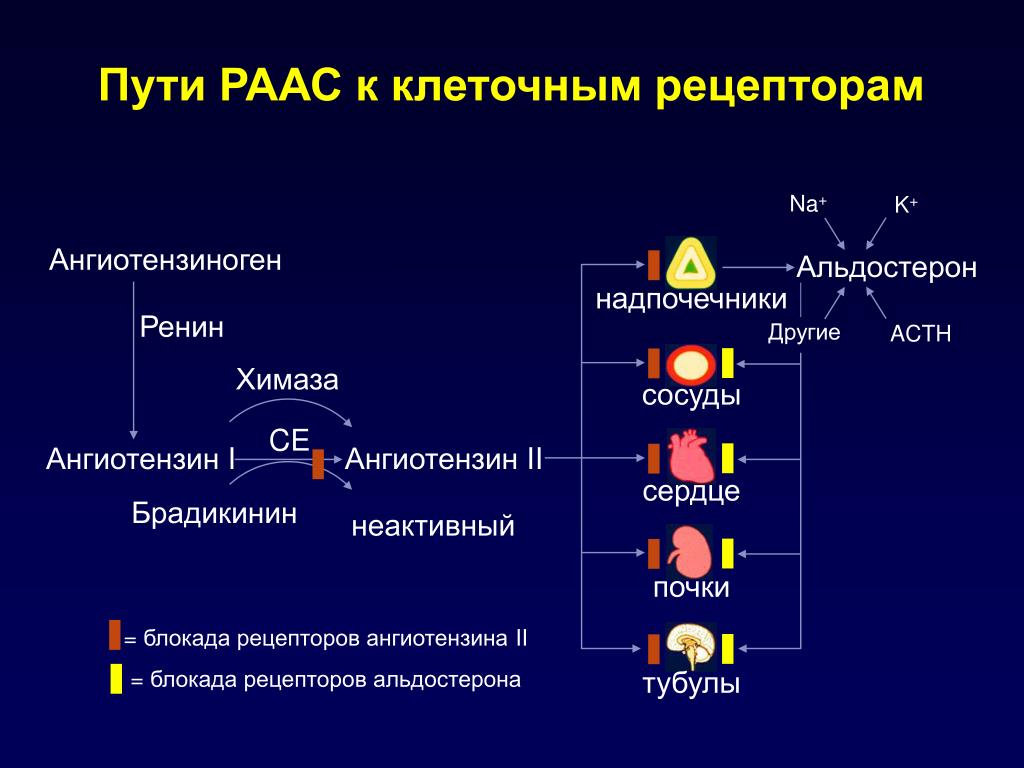 АТ4 локализованы в головном мозге и участвуют в местной регуляции кровотока и в формировании памяти.
АТ4 локализованы в головном мозге и участвуют в местной регуляции кровотока и в формировании памяти.
Поэтому, наверное, мы хотим или мы должны сделать так, чтобы этих знаний, немножко пока недостающих, было больше. Было несколько мета-анализов, которые говорили о том, что все-таки блокаторы ангиотензиновых рецепторов несколько хуже защищают от инфаркта миокарда, чем другие препараты, хотя надо сказать, что следующие мета-анализы показали, что нет оснований беспокоиться, тем не менее, об этом мы иногда помним.
В чем же, на мой взгляд, основное отличие? Это основное отличие как раз в том кашле, который мы очень не любим у наших пациентов. И действительно, когда он мучительный, сухой кашель, то мы вынуждены менять ингибиторы ангиотензинпревращающего фермента на блокаторы ангиотензиновых рецепторов, но здесь мы лишаемся тогда еще одного эффекта – хорошего действия оксида азота.
Давайте рассмотрим эту схему. Это клетка эндотелия, BK2-R – это брадикининовый рецептор, NO-синтаза эндотелиальная – это как раз та NO-синтаза, которая ответственна за выработку оксида азота в эндотелиальной клетке, и мы тоже очень хорошо знаем, за это в 1992 году была присуждена Нобелевская премия, за открытие роли оксида азота как сигнальной молекулы. При наличии L-аргинина и при активности эндотелиальной NO-синтазы происходит цепь реакций, которая приводит к тому, что образуется L-цитруллин и оксид азота.
При наличии L-аргинина и при активности эндотелиальной NO-синтазы происходит цепь реакций, которая приводит к тому, что образуется L-цитруллин и оксид азота.
Но если мы блокируем ангиотензинпревращающий фермент, даем ингибиторы АПФ, то количество кининов, брадикинина в частности, возрастает, это приводит к большей активации эндотелиальной NO-синтазы, и мы видим, что оксида азота образуется еще больше. Это дополнительный и кардиопротективный, и сосудистопротективный эффект. Безусловно, когда человек кашляет мучительно, я повторюсь, мы вынуждены отменять ингибиторы АПФ, но подумайте несколько раз перед тем, как это сделать, и если это легкое покашливание, то, на мой взгляд, нет убедительных мотивов для того, чтобы это сделать.
Как же связать ренин-ангиотензин-альдостероновую систему с фиброзом? Уже частично я коснулась этого вопроса, когда характеризовала каждый из агентов этого каскада. Для того чтобы сегодня не охватить – это будет невозможно, охватить все, куда приложил свою руку фиброз – я хочу сказать, что в основном я коснусь интерстициального фиброза.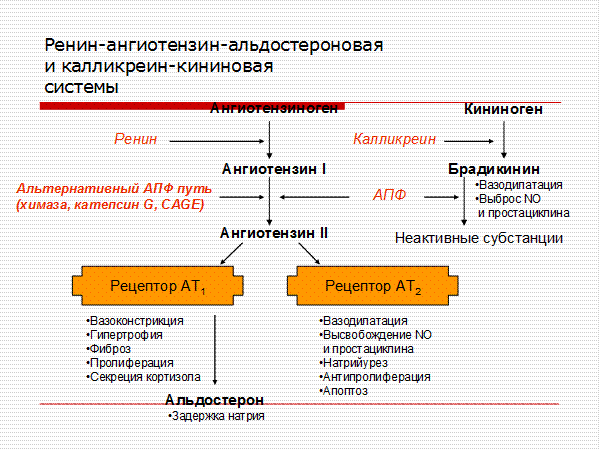 Вообще, его можно подразделить на интерстициальный и периваскулярный.
Вообще, его можно подразделить на интерстициальный и периваскулярный.
Под фиброзом понимают увеличение фракции коллагена в 2-3 раза, и совершенно логично предположить, что чем больше коллагена, тем больше жесткости не только, например, в сосудистой стенке, но и в стенке миокарда, и сейчас есть методы для того, чтобы определить, насколько больше этой жесткости стало. Мы видим, что последствия фиброза, так же как и последствия активации ренин-ангиотензин-альдостероновой системы, идентичны. Это сердечная недостаточность, это ишемическая болезнь сердца и это внезапная сердечная смерть.
Что же такое фиброз? Фиброз – это преобладание синтеза коллагена над его распадом. Нас интересует, конечно, только структурный коллаген, нормальный коллаген. И для того, чтобы на это соотношение – либо преобладание синтеза, либо распад – на него влиять, необходимо знать, какова активность металлопротеиназ или их ингибиторов. История изучения этого вопроса берет свое начало еще с 1872 года, когда впервые был охарактеризован нежный фиброз, так называемый «артерио-капиллярный фиброз», который развивается при болезнях почек.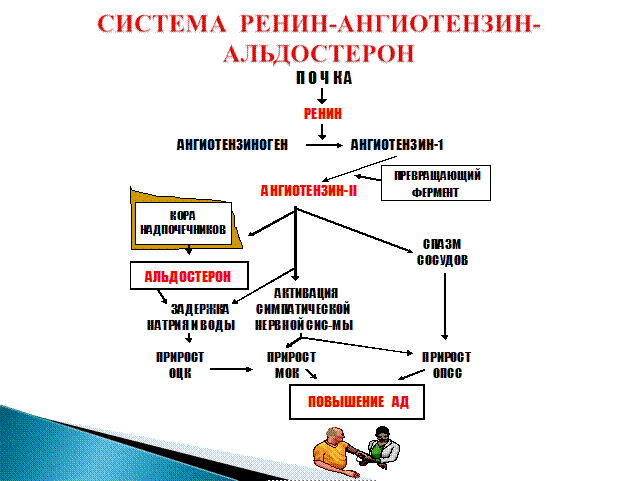 В 60-е годы идет интенсивное изучение этого процесса, и Браунвальд уже изучает влияние ангиотензина на функцию сердца, появляются первые данные о взаимосвязи сахарного диабета, гипертензии и фиброза. И в XXI веке становится возможной количественная и, самое главное, неинвазивная оценка фиброза.
В 60-е годы идет интенсивное изучение этого процесса, и Браунвальд уже изучает влияние ангиотензина на функцию сердца, появляются первые данные о взаимосвязи сахарного диабета, гипертензии и фиброза. И в XXI веке становится возможной количественная и, самое главное, неинвазивная оценка фиброза.
Теперь у нас портреты тех агентов, которые действуют на фиброз, и как будто бы мы здесь не замечаем того, что видели при активности ренин-ангиотензин-альдостероновой системы, за исключением последнего портрета – профибротические цитокины, и это как раз TGF beta, трансформирующий фактор роста бета-типа. Коллаген – чрезвычайно твердая субстанция, он представляет из себя множество сшитых вместе стержнеобразных молекул. Они образуют столь прочные соединения, которые сравнимы приблизительно с прочностью стали. И понятно, если он образовался, то очень трудно повернуть вспять этот процесс.
Тем не менее, основной белок волокнистой ткани – это как раз коллаген, и он составляет до 30% всех белков тела человека.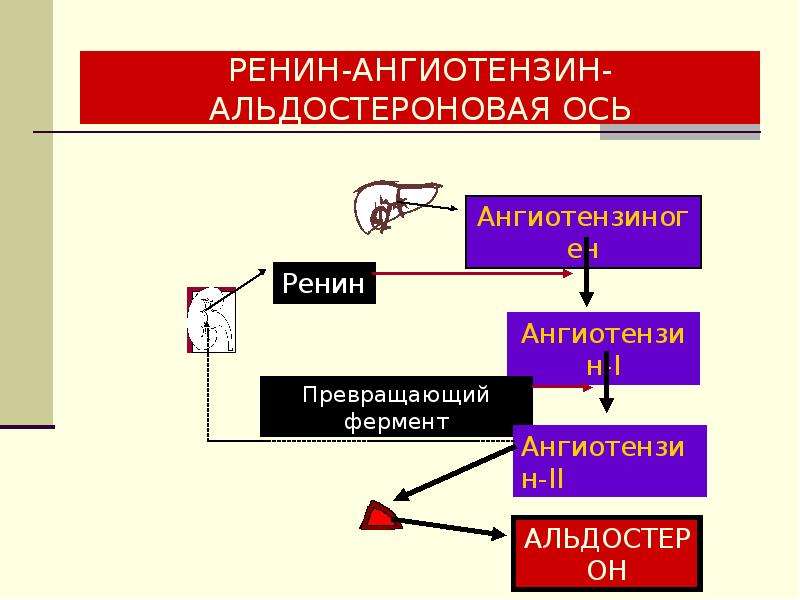 Синтез коллагена подразумевает влияние и на уровне саркоплазматического ретикулума, и на уровне самой клетки. Мы видим, что здесь необходима нормальная работа комплекса Гольджи, а также процессы гидроксилирования и гликозилирования, которые происходят после того, как произошел экцоцитоз.
Синтез коллагена подразумевает влияние и на уровне саркоплазматического ретикулума, и на уровне самой клетки. Мы видим, что здесь необходима нормальная работа комплекса Гольджи, а также процессы гидроксилирования и гликозилирования, которые происходят после того, как произошел экцоцитоз.
На данном слайде представлены ультраструктурные микрофотографии фрагментов миокарда и левого предсердия и левого желудочка. Мы видим, что коллагеновые волокна образуются в пространстве между отростком фибробласта и капилляром. То есть, процессу фиброза подвержен весь миокард, и миокард предсердий, и миокард желудочков. Различные типы фиброза есть, и сегодня, если мы говорим о ренин-ангиотензин-альдостероновой системе, нас больше интересует именно реактивный фиброз. Верхняя часть, которая чередует участки кардиомиоцитов, изображенных красным цветом, и участки фиброза бледным голубым цветом. Конечно, реактивный фиброз – это, я бы сказала, закономерное осложнение гипертоника, который не лечился и у которого происходила хроническая активация ренин-ангиотензин-альдостероновой системы.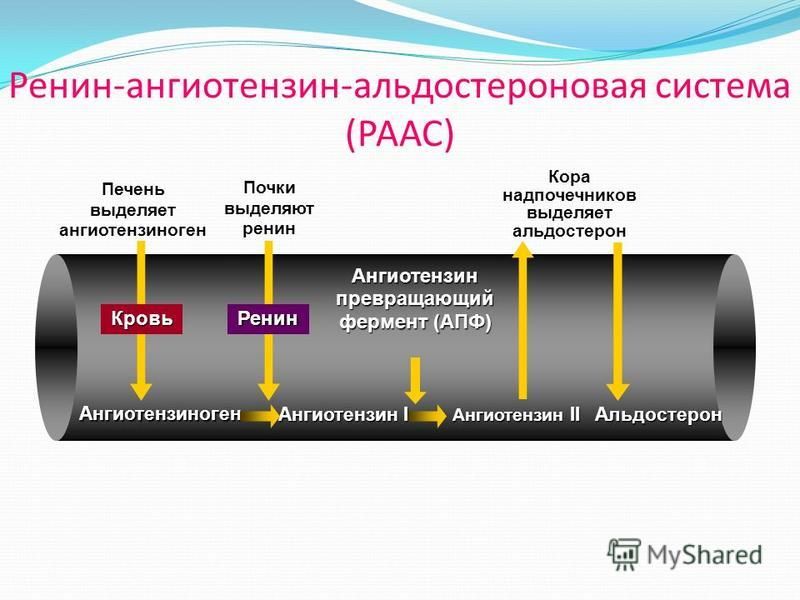
Фиброз, и именно фиброз, повинен в том, что такие пациенты часто демонстрируют суправентрикулярные нарушения ритма, и мы видим на срезе легочной вены – как раз там, в основном, происходит операция радиочастотной абляции – голубым цветом показан фиброз, пучки коллагена, которые окутывают легочную вену. Конечно, у такого пациента будет склонность к фибрилляции предсердия, и конечно, если он гипертоник, то артериальная гипертензия будет служить причиной его фибрилляции. У пациентов с артериальной гипертензией и с сахарным диабетом еще больше возможности иметь фиброз. При этом даже могут отмечаться так называемые «чистые» коронарные артерии, но ангинозные боли могут быть связаны с экстравазальной компрессией коронарных артерий, и здесь большая роль придается высокому уровню глюкозы, потому что этот высокий уровень приводит к тому, что образуются так называемые «сшивки» коллагена, и этот агент становится очень прочным.
Есть некоторые интересные факты, которые требуют переосмысления, я хочу их представить.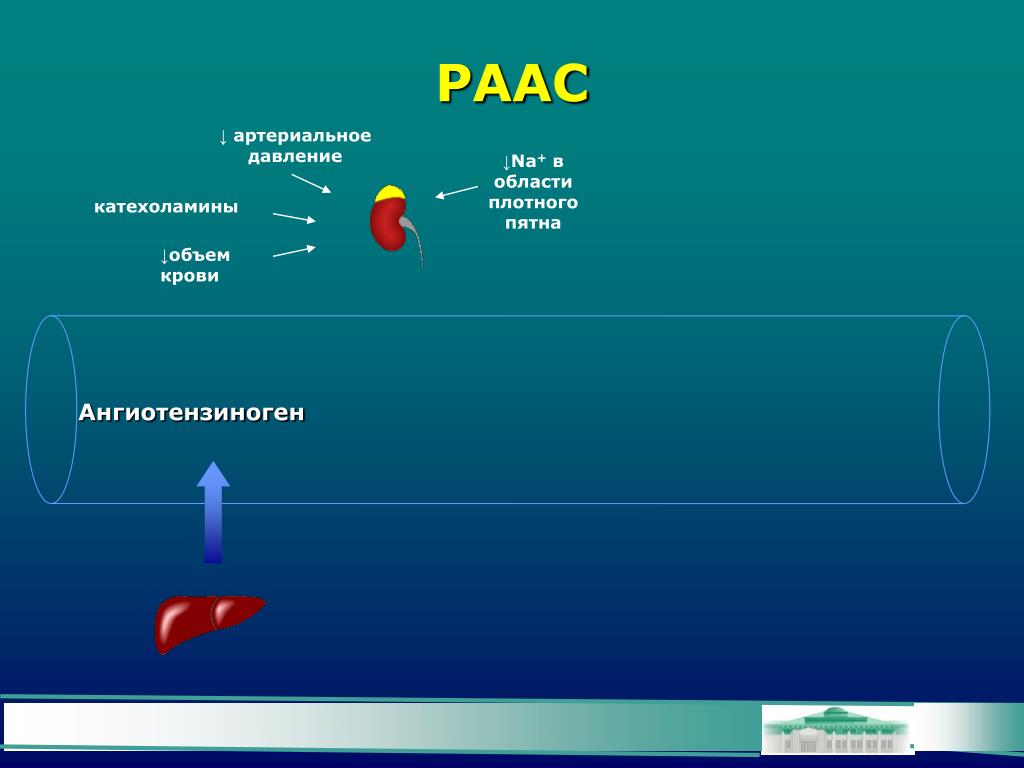 На самом деле, ответа на них я не знаю. Может быть, наша уважаемая аудитория, которая сегодня нас смотрит, пришлет свои предположения по поводу того, что повышение активности матричной металлопротеиназы может приводить и к росту фиброза миокарда, потому что матричные металлопротеиназы могут перестраивать внеклеточный матрикс. Кроме того, есть убедительные данные того, что при хронической сердечной недостаточности фиброз может вызываться клетками костного мозга, которые по каким-то причинам мигрируют в миокард и дифференцируются в фибробласты, а не в кардиомиоциты. Вот факторы, которые влияют на фиброз.
На самом деле, ответа на них я не знаю. Может быть, наша уважаемая аудитория, которая сегодня нас смотрит, пришлет свои предположения по поводу того, что повышение активности матричной металлопротеиназы может приводить и к росту фиброза миокарда, потому что матричные металлопротеиназы могут перестраивать внеклеточный матрикс. Кроме того, есть убедительные данные того, что при хронической сердечной недостаточности фиброз может вызываться клетками костного мозга, которые по каким-то причинам мигрируют в миокард и дифференцируются в фибробласты, а не в кардиомиоциты. Вот факторы, которые влияют на фиброз.
Мы смотрим на стимуляторы и на ингибиторы и пытаемся найти те агенты, о которых сегодня уже говорили. Ангиотензин II есть, альдостерон есть в стимуляторах. И обратите внимание, в ингибиторах есть брадикинин и оксид азота. Если проанализировать влияние гормонов на функцию сердечных фибробластов, опять я обращаю внимание на ангиотензин II и альдостерон. Мы видим, что ангиотензин II больше повинен в аккумуляции коллагена, поскольку он действует положительно на синтез коллагена, стимулируя его, снижает при этом деградацию коллагена, и аккумуляция поэтому увеличивается.![]() Альдостерон тоже действует на синтез коллагена, но практически не влияет на деградацию, и это приводит к тому, что вместо двух плюсиков (это весьма схематично) аккумуляция коллагена будет представлять из себя один плюсик. Если мы вспомним тех агентов, которые в основном ответственны за фиброз, там как раз был TGF beta 1 типа, и так все происходит в кардиомиоците. Здесь показан кардиомиоцит розовым цветом, а фибробласты изображены полигональными бледно-голубыми клетками. Если происходит активация ренин-ангиотензин-альдостероновой системы и ангиотензина II много, это приводит к тому, что активируется TGF beta, и его активация приводит к тому, что стимулы идут, активно вырабатываются фибробласты, которые в свою очередь приводят к синтезу межклеточного матрикса и к фиброзу.
Альдостерон тоже действует на синтез коллагена, но практически не влияет на деградацию, и это приводит к тому, что вместо двух плюсиков (это весьма схематично) аккумуляция коллагена будет представлять из себя один плюсик. Если мы вспомним тех агентов, которые в основном ответственны за фиброз, там как раз был TGF beta 1 типа, и так все происходит в кардиомиоците. Здесь показан кардиомиоцит розовым цветом, а фибробласты изображены полигональными бледно-голубыми клетками. Если происходит активация ренин-ангиотензин-альдостероновой системы и ангиотензина II много, это приводит к тому, что активируется TGF beta, и его активация приводит к тому, что стимулы идут, активно вырабатываются фибробласты, которые в свою очередь приводят к синтезу межклеточного матрикса и к фиброзу.
Обратите внимание на другой подход к этой гипертрофии и к фиброзу. Оказывается, само по себе механическое растяжение тоже будет приводить к тому, что активируется TGF beta. Значит, если уже камера большая, она растянулась, или там есть высокое давление, сам по себе этот факт будет приводить к тому, что фиброз будет усиливаться, и соответственно, ремоделирование как минимум не будет уменьшаться. Поэтому нам, конечно, для того, чтобы быть эффективными, надо снизить и активность ренин-ангиотензин-альдостероновой системы, уменьшить концентрацию ангиотензина II, и каким-то образом воздействовать на ремоделирование миокарда, то есть уменьшать размеры этой большой камеры, снижать механическое растяжение.
Поэтому нам, конечно, для того, чтобы быть эффективными, надо снизить и активность ренин-ангиотензин-альдостероновой системы, уменьшить концентрацию ангиотензина II, и каким-то образом воздействовать на ремоделирование миокарда, то есть уменьшать размеры этой большой камеры, снижать механическое растяжение.
Таким образом, совершенно точно доказана дружба между ангиотензином II и TGF beta – это так называемая профибротическая связка. Мало того, что ангиотензин II стимулирует TGF beta, он также может резко снижать активность матричной металлопротеиназы и влиять еще на некоторые процессы. Например, он может влиять на превращение фибробластов в миофибробласты и активировать фактор роста соединительной ткани.
На мой взгляд, удивительные данные на этом слайде представлены. Давайте вспомним пациента с гипертрофией, например, левого желудочка. Казалось бы, у них кардиомиоциты должны быть большие, либо в стадии гипертрофии, в стадии дистрофии, в стадии, может быть, воспаления. Но оказалось, что при гипертрофии левого желудочка размер кардиомиоцита даже уменьшается. Почему? Потому что все больший процент идет на внеклеточный матрикс, как раз на тот внеклеточный матрикс, который и ответственен за фиброз миокарда. Значит, гипертрофия, судя по этим данным, это в большей степени фиброз, нежели, например, перегрузка давлением, как мы тоже часто представляем.
Но оказалось, что при гипертрофии левого желудочка размер кардиомиоцита даже уменьшается. Почему? Потому что все больший процент идет на внеклеточный матрикс, как раз на тот внеклеточный матрикс, который и ответственен за фиброз миокарда. Значит, гипертрофия, судя по этим данным, это в большей степени фиброз, нежели, например, перегрузка давлением, как мы тоже часто представляем.
Что происходит после инфаркта миокарда? Мы видели, что происходит с ренин-ангиотензин-альдостероновой системой, с ангиотензином II, с альдостероном. Оказывается, что рост плотности ангиотензинпревращающего фермента и рецепторов к ангиотензину после инфаркта миокарда резко увеличивается. Это тоже доказывает, что влияние раннее на активность, на блокирование этих рецепторов или ангиотензинпревращающего фермента должно быть полезным. Не может быть жестким сердце без жестких сосудов, поэтому фиброз сосудов – это тоже та область, которая чрезвычайно увлекательна, и есть причины повышения их жесткости при артериальной гипертензии.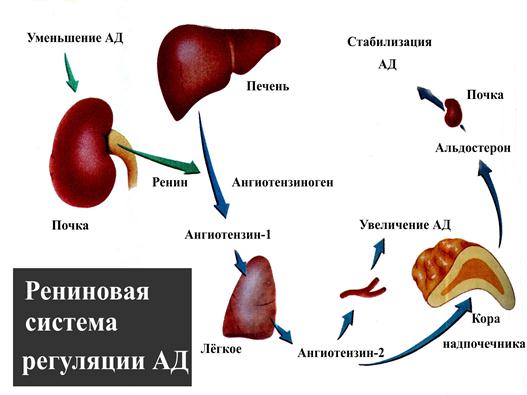 Но что сначала становится более жестким, периферия или центр – я имею в виде сердце, – конечно, на этот вопрос пока ответа нет.
Но что сначала становится более жестким, периферия или центр – я имею в виде сердце, – конечно, на этот вопрос пока ответа нет.
Возникает такой вопрос: может ли фиброз иметь протективное значение? Да, может, если мы говорим о фиброзе в бляшке, потому что нам нужна, конечно, стабильная бляшка, а активация металлопротеиназ приводит к тому, что бляшка становится нестабильной. Коротко остановлюсь на том, что процессы фиброза одни и те же, во многом схожи и в сердце, и в печени. И обратите внимание, фиброз в печени тоже начинается с сосудов, потому что синусоиды теряют фенестрации, и именно с этого участка происходит фиброз печени. И сейчас все больше доказательств о том, что статины, в частности, розувастатин может влиять на фиброз печени и может быть полезным. Кроме того, блокирование эффектов ренин-ангиотензин-альдостероновой системы – это хорошо теперь не только для сердца, но это хорошо и для печени.
Ингибиторов много, можно выбрать любой. Анатолий Иванович сказал о том, что преимущества в данном случае, например, лизиноприла есть у действительно тучного пациента, у пациента с какими-то диффузными заболеваниями печени. И мы это подтвердили, проделав свое исследование, в которое вошли пациенты от 18 до 65 лет с артериальной гипертензией, и они имели патологию печени.
И мы это подтвердили, проделав свое исследование, в которое вошли пациенты от 18 до 65 лет с артериальной гипертензией, и они имели патологию печени.
Что оказалось? Оказалось, что прием диротона, лизиноприла, с титрованием дозы от 5 до 20 мг в сутки был чрезвычайно благоприятен для них. Почему? Потому что, с одной стороны, его высокая эффективность приводила к тому, что мы обеспечили надежный контроль гемодинамики, а с другой стороны, все это привело к тому, что даже у пациентов с повышенным уровнем трансаминаз это не привело к дальнейшему ухудшению. Большое спасибо за внимание.
| Citation: | Солонская А. В. Роль ренин-ангиотензин-альдостероновой системы в развитии патологии сердечно-сосудистой системы / А. В. Солонская, Т. Н. Амбросова // Імплементація принципів біоетики у клінічну практику : наукова конференція молодих вчених і студентів, присвячена 210-річчю ХНМУ, Харків, 10 квітня 2015 р.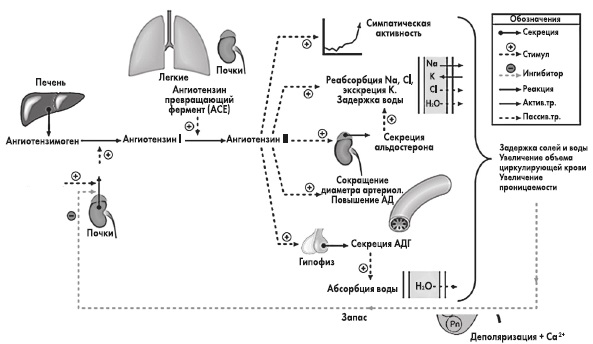 : матеріали конференції. – Харків, 2015. – С. 74. : матеріали конференції. – Харків, 2015. – С. 74. | Abstract: | Компоненты ренин-ангиотензин-альдостероновой системы (РААС) относятся к основным регуляторам сосудистого тонуса, артериального давления, функционального состояния сердца, электролитного и кислотно-основного равновесия, баланса натрия. Но также они принимают непосредственное участие в развитии заболеваний сердечно-сосудистой системы и оказывают влияние на их клиническое течение. По данным ВООЗ патология сердечно-сосудистой системы занимает первое место в структуре заболеваемости и смертности населения. Поэтому изучение роли РААС имеет важное значение для диагностики и лечения таких пациентов.
Компонентами РААС является: Ренин – гормон, который вырабатывается юксто-гломерулярным аппаратом (ЮГА) почек и играет ключевую роль в регуляции артериального давления, ренальной гемодинамики, водного и электролитного гомеостаза. Ангиотензин – гормон, который вырабатывается в печени из альфа-глобулина под действием ренина, откуда попадает в кровь.  Когда кровь проходит через легкие, под действием ангиотензин превращающего фермента (АПФ) ангиотензин I превращается в ангиотензин II. Этот пептид вызывает сужение кровеносных сосудов, а также стимулирует выработку таких гормонов, как вазопрессин и альдостерон. Альдостерон — основной минералокортикостероидный гормон коры надпочечников. Регулирует водно-солевой обмен, способствует задержке натрия и выведению калия. Когда кровь проходит через легкие, под действием ангиотензин превращающего фермента (АПФ) ангиотензин I превращается в ангиотензин II. Этот пептид вызывает сужение кровеносных сосудов, а также стимулирует выработку таких гормонов, как вазопрессин и альдостерон. Альдостерон — основной минералокортикостероидный гормон коры надпочечников. Регулирует водно-солевой обмен, способствует задержке натрия и выведению калия.
Основной целью активации РААС является поддержание системного артериального давления и достаточного кровотока в таких жизненно важных органах, как головной мозг, сердце, почки и печень. РААС играет роль «скорой помощи» при кровотечении, падении артериального давления, инфаркте миокарда и других острых состояниях. Она также оказывает регулирующее влияние на сердечно — сосудистый и почечный гомеостаз, способствует развитию компенсаторных процессов. Причиной многих заболеваний сердечно-сосудистой системы является хроническая гиперактивация РААС вследствии гипоперфузии почек и гипоксии. 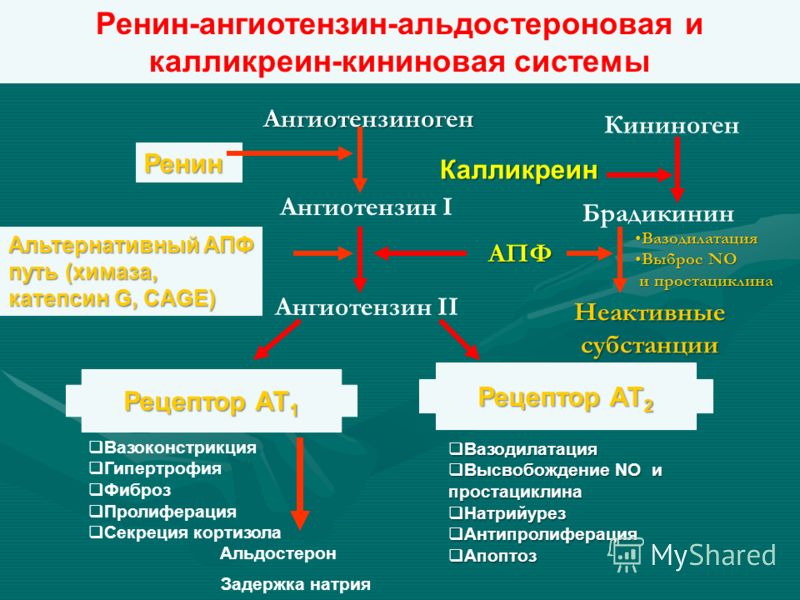 Это ведёт к стимуляции образования ренина. Он в свою очередь приводит к дополнительному образованию ангиотензина II . Этот гормон стимулирует сокращение миоцитов артериол, повышает чувствительность стенок сосудов к вазоконстрикторам, активирует высвобождение катехоламинов, стимулирует продукцию альдостерона, который увеличивает реабсорбцию натрия и выведение калия. Также ангиотензин II вызывает сужение артериальных сосудов, повышает сократимость миокарда, активирует симпатическую нервную систему (как на уровне центров, так и на уровне синапсов способствуя синтезу и освобождению норадреналина). Весь этот механизм приводит к спазму и увеличению переферического сопротивления сосудов, задержке жидкости, увеличению ОЦК, сердечного выброса, гипоперфузии органов и тканей, ремоделированию миокарда и сосудов. Эти изменения позже становятся причиной таких заболеваний как гипертоническая болезнь, атеросклероз, ишемическая болезнь сердца, сердечная недостаточность, нарушения ритма. Это ведёт к стимуляции образования ренина. Он в свою очередь приводит к дополнительному образованию ангиотензина II . Этот гормон стимулирует сокращение миоцитов артериол, повышает чувствительность стенок сосудов к вазоконстрикторам, активирует высвобождение катехоламинов, стимулирует продукцию альдостерона, который увеличивает реабсорбцию натрия и выведение калия. Также ангиотензин II вызывает сужение артериальных сосудов, повышает сократимость миокарда, активирует симпатическую нервную систему (как на уровне центров, так и на уровне синапсов способствуя синтезу и освобождению норадреналина). Весь этот механизм приводит к спазму и увеличению переферического сопротивления сосудов, задержке жидкости, увеличению ОЦК, сердечного выброса, гипоперфузии органов и тканей, ремоделированию миокарда и сосудов. Эти изменения позже становятся причиной таких заболеваний как гипертоническая болезнь, атеросклероз, ишемическая болезнь сердца, сердечная недостаточность, нарушения ритма.
Ингибирование гиперактивации РААС оказывает прямое кардиопротективное и вазопротективное действие, обеспечивает антиатеросклеротические эффекты, улучшает эндотелиальную функцию.  Блокада РААС фармакологически может быть осуществлена при применении 3 основных классов антагонистов РААС, к числу которых относят ингибиторы АПФ, блокаторы рецепторов антитензина, бета-адреноблокаторы. Данные препараты успешно применяют при лечении артериальной гипертензии, ишемической болезни сердца, сердечной недостаточности. Блокада РААС фармакологически может быть осуществлена при применении 3 основных классов антагонистов РААС, к числу которых относят ингибиторы АПФ, блокаторы рецепторов антитензина, бета-адреноблокаторы. Данные препараты успешно применяют при лечении артериальной гипертензии, ишемической болезни сердца, сердечной недостаточности. |
Значение нефропротекции при выборе антигипертензивной терапии
у пациентов с сахарным диабетом второго типа на основе изучения
реактивности ренин»ангиотензин»альдостероновой системы | Соколов
1. Арутюнов Г.П., Чернявская Т.К., Лукичева Т.И. и др. Микроальбуминурия: клиничес кие аспекты и пути медикаментозной кор рекции. Клин. фарм. и тер. 1999. № 8, с. 23;28.
2. Дедов И.И., Шестакова М.В., МиленькаяТ.М. Сахарный диабет: ретинопатия, нефропа тия. М.: Медицина. 2001.
3. Добронравов В.А. Современные подходы к диагностике и лечению диабетической не фропатии (пособие для врачей). Нефроло; гия. 2003. №2, с. 93;100.
Добронравов В.А. Современные подходы к диагностике и лечению диабетической не фропатии (пособие для врачей). Нефроло; гия. 2003. №2, с. 93;100.
4. Зонис Б.Я., Волкова Н.И., Соколов О.Ю. Влияние эднита на активность pенинан гиотензинальдостеpоновой системы и функцию почек у больных аpтеpиальными гипеpтониями в сочетании с сахаpным диабетом. Терапевтический архив. 2000. №2, с. 53;55.
5. Мухин Н.А. Современная нефропротектив ная стратегия лечения хронических про грессирующих заболеваний почек. Клин. фармакол. тер. 2001. № 11. С. 58;62.
6. Постнов Ю.В. О роли недостаточности митохондриального энергообразования в развитии первичной гипертензии: нейро генная составляющая патогенеза гипертен зиии. Кардиология. 2004. №9, с. 18;22.
7. Шестакова М.В., Кутырина И.М. Ингиби торы ангиотензинпревращающего фермен 88 та при заболеваниях почек. Кардиология. 2002. №9, с. 27;29.
Шестакова М.В., Кутырина И.М. Ингиби торы ангиотензинпревращающего фермен 88 та при заболеваниях почек. Кардиология. 2002. №9, с. 27;29.
8. Шулутко Б.И. Артериальная гипертензия 2000. СПб., 2001. — 304 с.
9. Bakris G., Copley J., Vicknair N. et al. Calcium channel blockers versus other antihypertensive therapies on progression of NIDDM associated nephropathy. Kidney Int 1996. Vol. 50. P. 1641; 1650.
10. Bakris G., Williams M., Dworkin L. et al. Pre serving renal function in adults with hyper tension and diabetes: a consensus approach. Am J Kid Dis. 2000. Vol.36. P. 646-661.
11. Brenner B., Mackenzie H. Nefron mass as a risk factor for progression of renal disease. Kidney Int.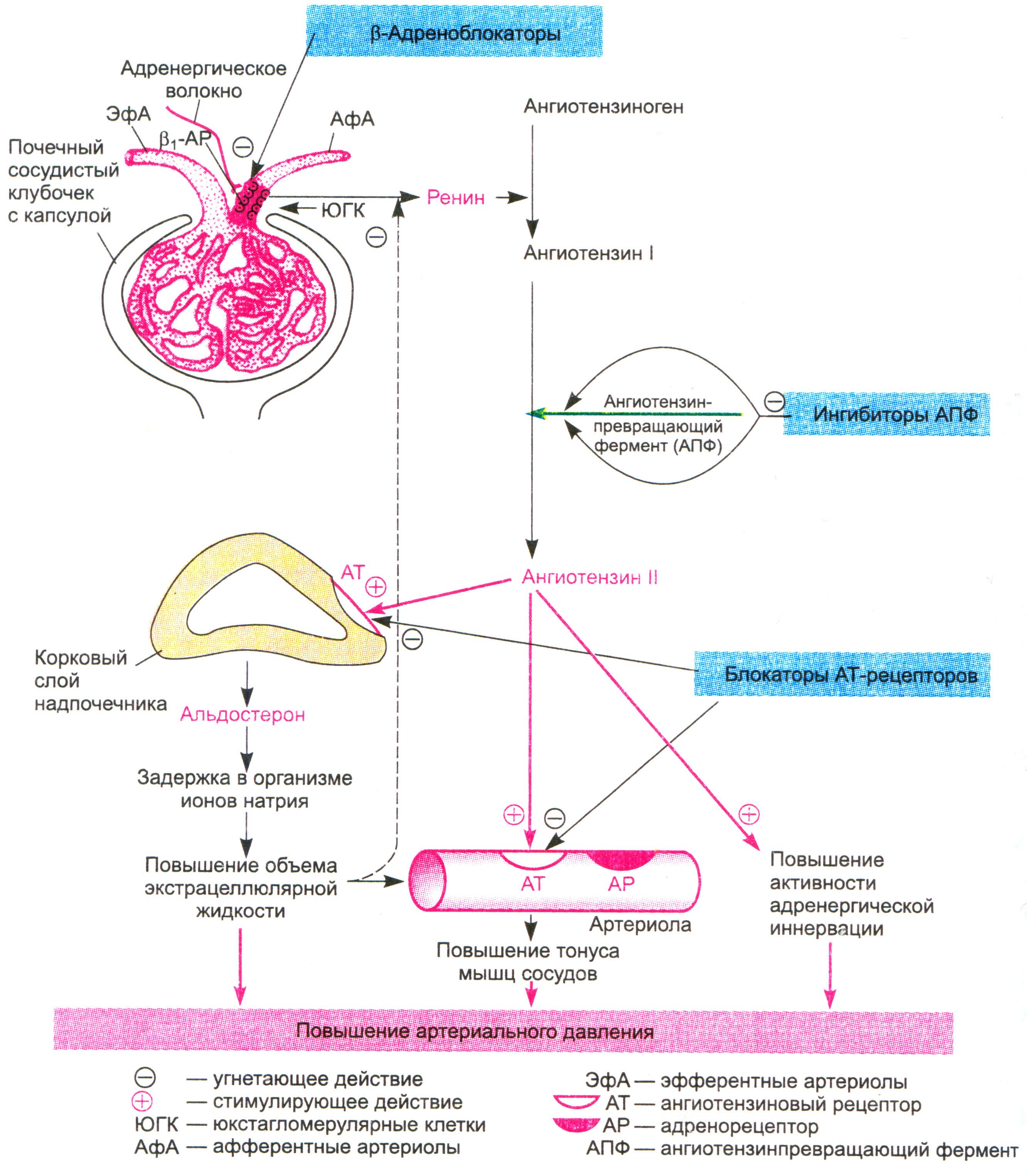 1997. Vol. 52 (Suppl 63). P. 124;127.
1997. Vol. 52 (Suppl 63). P. 124;127.
12. Brenner B.M. The history and future of renoprotection. Kidney Int. 2003. Vol. 64(4). P. 1163;1167.
13. Brown M.J., Castaigne A., de Leeuw P.W., et al. Influence of diabetes and type of hypertension on response to antihypertensive treatment. Hypertension. 2000. V. 35. P. 1038-1042.
14. Carmines P.K., Ohishi K., Ikenaga H. Func tional impairment of renal afferent arteriolar voltagegated calcium channels in rats with diabetes mellitus. J Clin Invest. 1996. Vol. 98. P. 2564;2571.
15. Kim S., Iwao H. Molecular and cellular mecha nisms of angiotensin IImediated cardio vascular and renal diseases. Pharmacol Rev. 2000. Vol. 52(3). P. 11;34.
16. Kvam F., Ofstad J., Iverson B.M. Effects of antihypertensive drugs on autoregulation of RBF and glomerular capillary pressure in SHR. Am J Physiol. 1998. Vol. 275. P. 576;584.
17. Navar L.G. Integrating multiple paracrine re gulators of renal microvascular dynamics. Am J Physiol Renal Physiol. 1998. Vol. 274. P.433;444.
18. Weidmann P., Schneider M., Bohlen L. Thera peutic efficacy of different antihypertensive drugs in human diabetic nephropathy: An updated metanalysis. Nephrol Dial Transplant. 1995. Vol. 10 (Suppl9). P. 39;45.
19. Winer N., Sowers J. Vascular compliсance in dia betes. Curr Diab Rep. 2003. Vol. 3. P. 230;234.
Что такое система ренин-ангиотензин-альдостерон?
Ренин-ангиотензин-альдостероновая система (РААС) или ренин-ангиотензиновая система (РАС) является регулятором кровяного давления и сердечно-сосудистой функции. Нарушение регуляции РААС вызывает высокое кровяное давление, сердечно-сосудистые заболевания и заболевания почек, и лекарства, нацеленные на РААС, могут улучшить эти состояния.
Нарушение регуляции РААС вызывает высокое кровяное давление, сердечно-сосудистые заболевания и заболевания почек, и лекарства, нацеленные на РААС, могут улучшить эти состояния.
Кредит изображения: kurhan / Shutterstock.com
Дисфункциональный РААС также считается причиной серьезности COVID-19, и нацеливание на РААС может быть потенциальной терапевтической стратегией при осложнениях COVID-19.
Что такое система ренин-ангиотензин-альдостерон (РААС)?
РААС — это сложная многоорганная эндокринная (гормональная) система, участвующая в регуляции артериального давления путем уравновешивания уровней жидкости и электролитов, а также регулирования сопротивления и тонуса сосудов. РААС регулирует всасывание натрия и воды в почках, напрямую влияя на системное артериальное давление.
Обычно РААС активируется при падении артериального давления (уменьшении объема крови) для увеличения реабсорбции воды и электролитов в почках; который компенсирует падение объема крови, повышая кровяное давление.
Ренин
Юкстагломерулярные клетки в почках изобилуют неактивным белком-предшественником, называемым проренином, который постоянно секретируется. Снижение артериального давления активирует юкстагломерулярные клетки, что приводит к расщеплению проренина до его активной формы — ренина, который затем секретируется в кровоток.
Ангиотензин
Ренин действует на ангиотензин (непрерывно продуцируемый печенью), отщепляя 10-аминокислотный пептид от N-конца с образованием ангиотензина I (неактивного).Ангиотензин-превращающий фермент (АПФ) дополнительно расщепляет ангиотензин I с образованием ангиотензина II — основного активного пептида РААС. АПФ в основном обнаруживается в эндотелии сосудов легких и почек.
Ангиотензин II связывается с рецепторами ангиотензина I типа A (AT 1A R) или B (AT 1B R), а также с рецепторами типа II (AT 2 R). Ангиотензин II является сильнодействующим вазоконстриктором, который приводит к повышению артериального давления. Обычно, если артериальное давление слишком высокое, предсердный натрийуретический пептид (ПНП), секретируемый клетками сердечной мышцы, приводит к уменьшению объема крови, что приводит к снижению артериального давления.
Обычно, если артериальное давление слишком высокое, предсердный натрийуретический пептид (ПНП), секретируемый клетками сердечной мышцы, приводит к уменьшению объема крови, что приводит к снижению артериального давления.
Связывание
ангиотензина II с AT 1 R может приводить к каскаду воспаления, сужения и развития атеросклероза. Кроме того, эти события могут также привести к инсулинорезистентности и тромбозу, тогда как связывание с AT 2 R имеет противоположные эффекты: вазодилатация снижает агрегацию тромбоцитов и повышает активность инсулина.
Ангиотензин II также действует на мозг, где он может связываться с гипоталамусом, чтобы стимулировать жажду и увеличить потребление воды.Действуя на гипофиз, ангиотензин II также стимулирует секрецию вазопрессина; также известен как антидиуретический гормон, который увеличивает задержку воды в почках за счет добавления водных каналов (аквапорин) в собирательный проток.
Кредит изображения: StudioMolekuul / Shutterstock.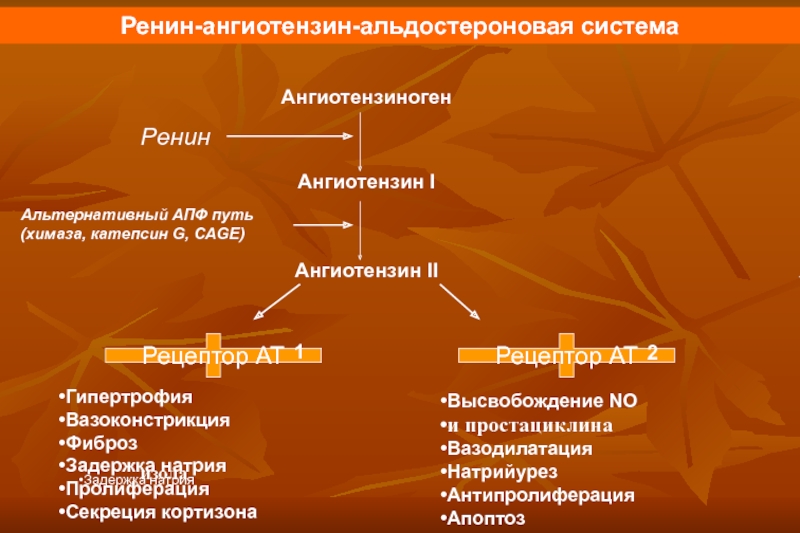 com
com
Альдостерон
Ангиотензин II стимулирует секрецию альдостерона, который вырабатывается клубочковой оболочкой коры надпочечников (надпочечников) и участвует в задержке натрия в почках и других железах.Удержание воды и натрия приводит к увеличению объема крови и, следовательно, артериального давления.
Неисправная РААС
Первичные патологии нарушения РААС включают хроническую гипертензию, почечную и сердечную недостаточность. Аномально активный РААС приводит к хронической гипертензии, и поэтому необходимо уравновешивать высокое кровяное давление.
Ингибиторы АПФ — это обычно используемые препараты для лечения гипертонии и сердечной недостаточности. Ингибирование АПФ предотвращает превращение ангиотензина I в ангиотензин II, таким образом удерживая ангиотензин в неактивном состоянии.
Связывание ангиотензина II с AT 1 R может также вызывать сердечную дисфункцию, включая гипертрофию, аритмию и нарушение функции желудочков.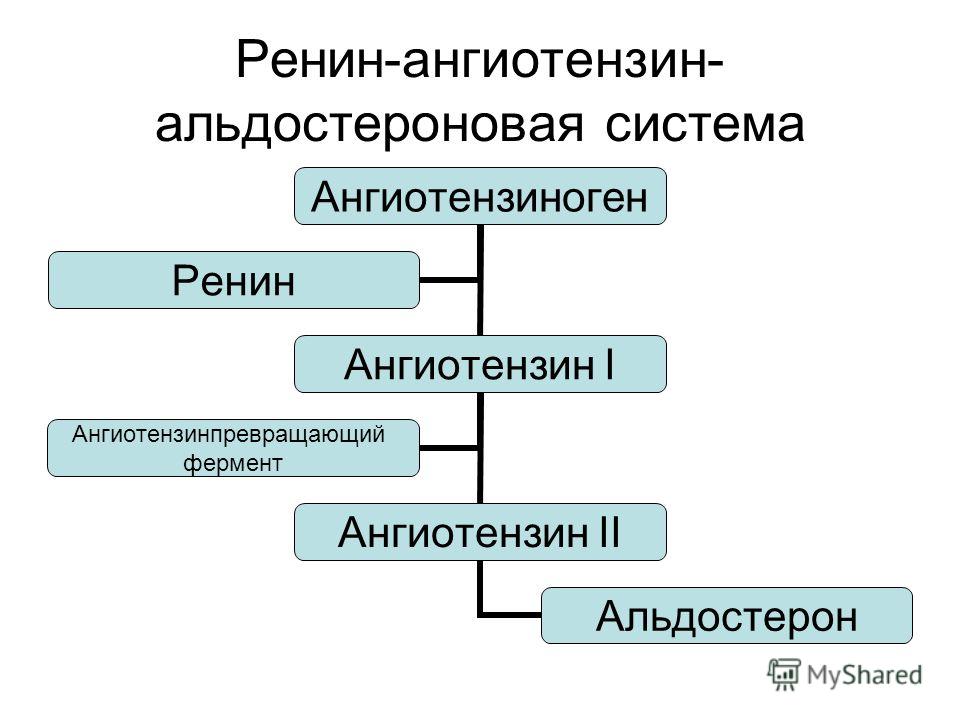 Избыточный ангиотензин II может также приводить к большим воспалительным изменениям, таким как цитокин-индуцированное повреждение органов, в дополнение к повышенной проницаемости мембран и апоптозу эпителиальных клеток.
Избыточный ангиотензин II может также приводить к большим воспалительным изменениям, таким как цитокин-индуцированное повреждение органов, в дополнение к повышенной проницаемости мембран и апоптозу эпителиальных клеток.
Поскольку ангиотензин II является вазоконстриктором, его облегчение вызывает вазодилатацию, тем самым снижая уровень системного артериального давления. Ингибиторы АПФ также приводят к увеличению выработки брадикинина — сосудорасширяющего средства.Другие препараты включают блокаторы рецепторов ангиотензина (БРА), которые блокируют связывание ангиотензина II с ATR. Ингибиторы АПФ и БРА также являются эффективными лекарствами при сердечной недостаточности и осложнениях диабета.
В почках стеноз почечной артерии приводит к уменьшению объема крови, поступающей в почки, следовательно, юкстагломерулярные клетки воспринимают уменьшенный объем крови, таким образом, активируя РААС. Это отрицательно увеличивает кровяное давление, что не соответствует системному кровообращению, а также повышает артериальный тонус. Таким образом, ингибиторы АПФ и БРА могут также использоваться у пациентов со стенозом почечной артерии для ограничения несоответствующей активации РААС.
Таким образом, ингибиторы АПФ и БРА могут также использоваться у пациентов со стенозом почечной артерии для ограничения несоответствующей активации РААС.
РААС и COVID-19
Вирус, вызывающий COVID-19; SARS-CoV-2 использует ACE2 для проникновения в клетки респираторного эпителия (вместе с TMPRSS2 ). Когда SARS-CoV-2 связывается с ACE2, активность ACE2 снижается, что препятствует его нормальной функции, что приводит к респираторным симптомам кашля и отека, а также к повышению уровня ангиотензина II.
Кредит изображения: Катерина Кон / Shutterstock.com
Кроме того, также была предложена связь между дисфункцией РААС при артериальной гипертензии и сердечно-сосудистыми заболеваниями и усилением осложнений COVID-19 и летального исхода (ось RAAS-SCoV).
Осложнения COVID-19 и показатели смертности выше у пациентов с артериальной гипертензией и сердечными / сердечно-сосудистыми заболеваниями и могут отражать аберрантный РААС (повышенный уровень ангиотензина II и пониженный уровень ACE2).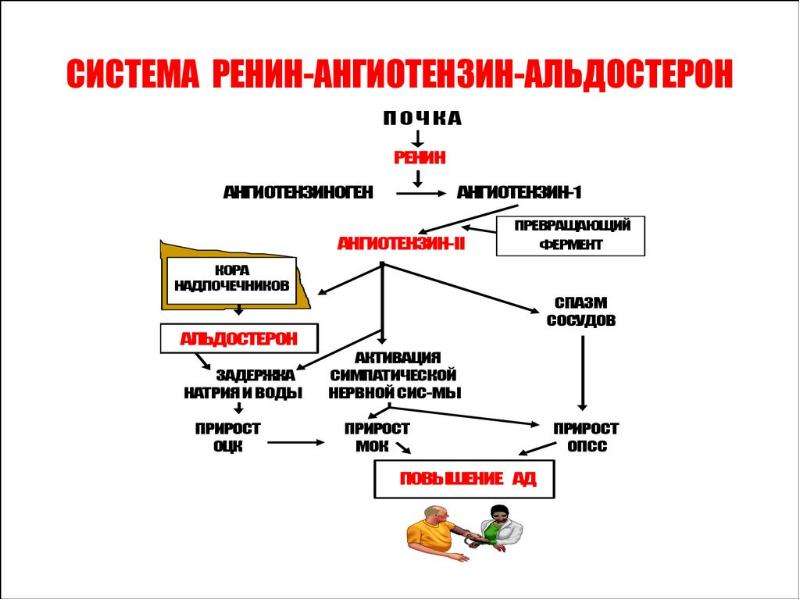
Как упоминалось ранее, связывание ангиотензина II с AT 1 R может приводить к воспалению, ведущему к повреждению тканей эпителиальными клетками легких, что может привести к острому респираторному дистресс-синдрому (ARDS).Связывание ангиотензина II с AT 2 R может обратить негативные эффекты связывания AT 1 R (обсуждалось выше).
Однако важно отметить, что экспрессия AT 2 R относительно низкая у здоровых взрослых людей, таким образом, первичные модулирующие эффекты ангиотензина II опосредуются ACE2, превращая ангиотензин II в защитный для легких ангиотензин 1-7 / 1-9 (против ACE).
Повышенная экспрессия ACE2 может быть компенсатором у пациентов с сердечно-сосудистыми заболеваниями и сердечной недостаточностью.Действительно, грипп и коронавирусы (наряду с вирусными формами пневмонии), как правило, демонстрируют снижение экспрессии ACE2 и повышение уровня ангиотензина II.
Таким образом, люди с гиперактивным RAAS (повышенная экспрессия ангиотензина II и ACE2) могут иметь более высокий риск развития тяжелой патологии в результате инфекции SARS-CoV-2. У здоровых взрослых экспрессия ACE2 низкая, и экспрессия ACE2 может быть независимым предиктором риска и тяжести COVID-19, а вирус SARS-CoV-2 использует ACE2.
Кроме того, женщины имеют более высокую экспрессию AT 2 R по сравнению с мужчинами (поскольку ACE2 находится на X-хромосоме), и это может отражать, что инфекции и осложнения COVID-19, как правило, выше у мужчин.
Ингибиторы
АПФ и БРА приводят к увеличению уровней ренина и ангиотензина I (неактивны) и ингибируют преобразование ангиотензина I в II, таким образом защищая организм от негативных эффектов связывания ангиотензина II с AT 1 R (как обсуждалось).
Однако
БРА приводят к повышению уровня ангиотензина II — однако это затем направлено на модуляцию ACE2 и связывание AT 2 R, чтобы иметь защитные эффекты, таким образом, БРА могут быть лучше ингибиторов АПФ для восстановления баланса пути РААС.Таким образом, БРА считаются средством лечения COVID-19.
Лечение пациентов с ХОБЛ БРА может привести к снижению смертности и снижению потребности в искусственной вентиляции легких. Кроме того, у тех, кто принимал БРА до госпитализации по поводу пневмонии, смертность ниже, чем у тех, кто не принимал БРА.
Существует множество ассоциаций между уровнями ангиотензина II и прогнозом смертности от гриппа, и тем, кто использует ингибиторы РААС (БРА), часто не требуется интубация пациентам с вирусной пневмонией.Похоже, то же самое и с COVID-19, и препараты RAAS могут быть защитными, уменьшая осложнения и необходимость госпитализации.
Подводя итог связи между COVID-19 и RAAS, SARS-CoV-2 связывается с ACE2, чтобы проникнуть в респираторные эпителиальные клетки. Обычно связывание ангиотензина II с AT 1 R вызывает отрицательные эффекты, и эти эффекты могут модулироваться связыванием ACE2 и AT 2 R.
Однако SARS-CoV-2 снижает экспрессию ACE2, что приводит к повреждению легких в результате каскада воспаления.Ингибиторы АПФ и БРА могут изменить баланс пути РААС, чтобы способствовать функции ACE2, чтобы противостоять негативным эффектам ангиотензина II и превращать его в защитный для легких ангиотензин 1-7.
Многие пациенты с вирусной пневмонией и ХОБЛ, принимающие БРА, обычно имеют лучший прогноз, чем пациенты без них, особенно в отношении госпитализации и смертности. Излишне говорить, что связь между взаимодействием SARS-CoV-2 и ACE2 в рамках RAAS (ось RAAS-SCoV) все еще плохо изучена и требует дальнейшего изучения на данном этапе.
Таким образом, ренин-ангиотензин-альдостероновая система (РААС) является важным регулятором артериального давления (объема крови и баланса электролитов), а также тонуса и сопротивления сосудов. Обычно ренин секретируется, если артериальное давление слишком низкое, таким образом активируя ангиотензин II, повышая артериальное давление и сопротивление сосудов.
Аномальная активация РААС приводит к хронической гипертензии, сердечной недостаточности и заболеваниям почек и, возможно, является предиктором риска осложнений COVID-19.Лекарства, ингибирующие путь РААС, такие как ингибиторы АПФ и БРА, являются эффективным средством лечения гипертонии и сердечной недостаточности и, возможно, важным вариантом для минимизации осложнений COVID-19.
Дополнительная литература
Ренин-ангиотензин-альдостероновая система и прогрессирование почечной недостаточности
Реферат
Подавление ренин-ангиотензин-альдостероновой системы (РААС) — один из самых действенных способов замедлить прогрессирование почечной недостаточности. Ангиотензин II (AngII) появился в последнее десятилетие как многофункциональный цитокин, который проявляет множество негемодинамических свойств, таких как действие как фактор роста и профиброгенный цитокин, и даже обладающий провоспалительными свойствами.Многие из этих вредных функций опосредуются другими факторами, такими как TGF-β и хемоаттрактанты, которые индуцируются в почках с помощью AngII. Более того, понимание РААС стало намного более сложным в последние годы с идентификацией новых пептидов (, например, , AngIV), которые могут связываться со специфическими рецепторами, выясняя вредные эффекты и опосредованные неангиотензинпревращающим ферментом (АПФ). поколение AngII. Способность почечных клеток продуцировать AngII в концентрации, намного превышающей концентрацию в системном кровотоке, и наблюдение, что альдостерон может непосредственно участвовать в профиброгенных процессах, независимо от гипертензии, усложнили RAAS.В настоящее время установлено, что даже ренин имеет «самостоятельную жизнь» и опосредует профибротические эффекты посредством связывания со специфическими рецепторами. Наконец, препараты, которые используются для блокирования РААС, такие как ингибиторы АПФ или некоторые антагонисты рецепторов AngII типа 1, могут обладать свойствами на клетки, независимыми от AngII (стимулирующие эффекты рецептора, опосредованные ингибитором АПФ и активированным пролифератором пероксисом. некоторых сартанов). Хотя блокада РААС ингибиторами АПФ, антагонистами рецепторов AngII типа 1 или их комбинацией должна быть частью каждой стратегии замедления прогрессирования почечной недостаточности, лучшее понимание новых аспектов РААС должно способствовать развитию инновационных стратегии не только для полной остановки прогрессирования, но и для индукции регресса почечной недостаточности у человека.
Одной из основных проблем современной нефрологии является рост числа пациентов с ТПН. Более того, в последнее время стало понятно, что пациенты с уже относительно незначительным нарушением функции почек имеют повышенный риск сердечно-сосудистых заболеваний задолго до того, как будет достигнута ТПН. Следовательно, лучшее понимание патофизиологии хронической болезни почек является обязательным для разработки стратегий предотвращения прогрессирования почечной недостаточности или даже восстановления функции почек путем индукции регрессии (1).Независимо от первичной формы прогрессирование почечной недостаточности характеризуется патоморфологическими изменениями, которые включают раннее воспаление почек с последующим тубулоинтерстициальным фиброзом, тубулярной атрофией и гломерулосклерозом (1). Ренин-ангиотензин-альдостероновая система (РААС) играет ключевую роль во многих патофизиологических изменениях, ведущих к прогрессированию почечной недостаточности. В этой статье освещаются некоторые из недавних молекулярных исследований сложности РААС и ее роли в хронической почечной недостаточности.Из-за нехватки места многие клинические исследования, посвященные RAAS, не охвачены, и читатель может быть отослан к превосходным обзорам (2, 3).
Как видели РАСН в прошлом
Традиционно РААС рассматривали как эндокринную систему с ангиотензиногеном, продуцируемым в печени, который расщепляется ренином, высвобождаемым юкстагломерулярными клетками почек (4). Таким образом образуется ангиотензин I (AngI), который, в свою очередь, дополнительно расщепляется под действием ангиотензин-превращающего фермента (ACE) легких в активную форму AngII.Затем AngII связывается со специфическими рецепторами коры надпочечников, что приводит к высвобождению альдостерона. С этой классической точки зрения основной функцией РААС является поддержание АД за счет сужения сосудов, вызванного AngII, и опосредованной альдостероном задержки натрия в собирательном канале (4). Однако в последние годы RAAS стала сложной, и были выявлены новые компоненты этой сети. На рисунке 1 представлен обзор нашего текущего понимания.
Рисунок 1.
Обзор ренин-ангиотензин-альдостероновой системы (РААС).Система становится все более сложной с альтернативными способами образования ангиотензина II (AngII) помимо ангиотензин-превращающего фермента (ACE; (химаза, химостатин-чувствительный AngII-генерирующий фермент [CAGE]), второй формы ACE (ACE2) и нового пептиды, такие как AngIV, ангиотензин 1-9 и ангиотензин 1-7. Клинически важным может быть то, что AngII может связываться с рецепторами AngII типа 2 (AT 2 ), а AngIV — с рецепторами AT 4 , которые не антагонизируются сартанами, вызывая провоспалительные и профибротические эффекты ( e.грамм. , индукция хемокинов, стимуляция ингибитора активатора плазминогена-1).
Получение AngII и других пептидов
Наиболее широко известным ферментом, способным к образованию AngII, является АПФ, но он не единственный. Другие ферменты, генерирующие AngII, включают химазу сериновой протеазы, которая, как предполагается, опосредует> 80% образования AngII в сердце и> 60% в сосудах (5). Ингибиторы АПФ не снижают активность химазы. Повышенная регуляция химазы, в основном в канальцах, наблюдается при биопсии почек у пациентов с диабетической нефропатией (5).Эти данные показывают, что при патологических состояниях происходит усиление активности химазы, и повышенное локальное образование AngII не может быть ослаблено ингибиторами АПФ. Механический стресс подоцитов стимулирует локальный синтез AngII не-АПФ путями, которые предположительно включают химазу (6), но точная роль опосредованного химазой образования AngII для почечной болезни у людей неясна, а ингибиторы АПФ явно замедляют прогрессирование заболевания.
Был идентифицирован новый фермент, похожий на АПФ, называемый ангиотензинпревращающим ферментом 2 (АПФ2) (7).ACE2 преимущественно экспрессируется в эндотелиальных клетках сосудов, включая клетки почек (8). В отличие от «классического» ACE, который преобразует AngI в октапептид AngII, ACE2 отщепляет от AngI на одну аминокислоту меньше, так что на первом этапе образуется ангиотензин 1-9 (7). Считается, что ангиотензин 1-9 усиливает опосредованную AngII вазоконстрикцию изолированных колец аорты крысы и оказывает вазодепрессорное действие у крыс, находящихся в сознании. Также было обнаружено, что ангиотензин 1-9 усиливает действие брадикинина на его рецептор B2, вероятно, путем индукции конформационных изменений (9).На втором этапе ангиотензин 1-9 может быть преобразован в ангиотензин 1-7 с помощью «классического» АПФ. Основной путь деградации ангиотензина 1-7, превращающий пептид в неактивные фрагменты, опосредуется самим АПФ. Известно, что ангиотензин 1-7 действует как вазодепрессор и участвует в апоптозе и остановке роста. Белковый продукт гена c-mas является рецептором ангиотензина 1-7. Дальнейшие экспериментальные исследования показывают противовоспалительное и антифибротическое действие ангиотензина 1-7. Локальная экспрессия ACE2 тесно коррелирует с концентрацией ангиотензина 1-7 и приводит к, по крайней мере, частичному антагонизму AngII.Таким образом, ингибирование АПФ может приводить к повышению уровня ангиотензина 1-7 при одновременном снижении AngII (9). Однако следует знать, что большая часть данных по ангиотензину 1-7 поступает из сердечно-сосудистой системы, и их значение для почечной недостаточности неясно.
AngII метаболизируется пептидазами, такими как аминопептидаза A, в AngIII и далее в AngIV (10). AngIII взаимодействует с рецепторами AngII типа 1 (AT 1 ) и AT 2 , хотя и с более низким сродством, и проявляет принципиально подобные эффекты, как AngII.AngIV связывается со специфическим рецептором под названием AT 4 , который широко экспрессируется в почках, включая эндотелиальные клетки и проксимальные, а также извитые канальцы. Рецептор AT 4 путем очистки белка и секвенирования пептидов был идентифицирован как регулируемая инсулином аминопептидаза (11).
Рецепторы AngII
Множественные эффекты AngII опосредуются разными рецепторами. Два основных рецептора AngII, AT 1 и AT 2 , по-разному экспрессируются в почках (12).Оба характеризуются конфигурацией семистрансмембранного рецептора, но обладают лишь приблизительно 30% гомологией на уровне белка. Рецепторы AT 1 связаны с гетеротримерными G-белками и опосредуют различные пути передачи сигналов, в основном вторичные мессенджеры, такие как активация фосфолипаз, ингибирование аденилатциклазы, стимуляция фосфорилирования тирозина, киназы 1 и 2, регулируемые внеклеточным сигналом, фосфатидилинозитол 3-киназа-зависимая киназа Akt и мишень рапамицин / S6-киназного пути у млекопитающих (12).Активация рецептора AT 1 также стимулирует высвобождение активных форм кислорода по механизму, который включает активацию мембраносвязанной NAD (P) H оксидазы.
Lautrette et al. (13) недавно описал новые механизмы, с помощью которых AngII трансактивирует рецептор EGF во время почечного повреждения на модели хронической инфузии AngII в течение 2 месяцев. Они обнаружили, что AngII индуцирует секрецию TGF-α, который связывается с рецептором EGF и активирует его, объясняя, как AngII посредством трансактивации рецептора EGF может проявлять тирозинкиназную активность (13).
AT 2 Рецепторы участвуют в повышении активности внутриклеточной протеинфосфатазы (14). Количество рецепторов AT 1 и AT 2 регулируется в процессе развития, и во время созревания почек экспрессия рецептора AT 1 становится более обильной.
AT 1 Экспрессия рецептора активируется различными стимулами, такими как гиперхолестеринемия или изменение осмолярности, но подавляется высокими концентрациями AngII или глитазонами (12).N-ацетилцистеин, антиоксидант, восстанавливающий дисульфидные связи, снижает связывание AngII с рецепторами AT 1 (15). Рецепторы AT 2 не подавляются AngII, но, что интересно, они активируются в поврежденной ткани и во время воспаления (14). В частности, рецепторы AT 2 повторно экспрессируются в почках во время почечного повреждения и ремоделирования нефронов. Способность рецепторов AT 1 образовывать гомодимеры, а также гетеродимеры с другими рецепторами, такими как рецептор брадикинина, приводит к значительному ускорению активности передачи сигнала после стимуляции AngII (16).
Почти все индуцированные AngII физиологические и патофизиологические функции, такие как сужение сосудов, высвобождение альдостерона, стимуляция канальцевого транспорта, провоспалительные эффекты, профиброгенное и стимулирующее рост действия, опосредуются рецепторами AT 1 (12). Роль рецепторов AT 2 кажется менее ясной. Активация рецепторов AT 2 приводит к снижению АД за счет высвобождения оксида азота, подавляет рост и индуцирует дифференцировку, а также участвует в опосредовании апоптоза (14).Недавние данные свидетельствуют о том, что активация NF-κB, важного провоспалительного фактора транскрипции, опосредуется рецепторами AT 1 и AT 2 . Вопрос о том, какие патофизиологические эффекты опосредуются через рецептор AT 2 , имеет клиническое значение, поскольку не все важные патофизиологические функции AngII могут быть антагонизированы блокаторами рецепторов AT 1 .
Агонистические антитела против рецепторов AT 1 были идентифицированы у беременных с преэклампсией и у пациентов со вторичной злокачественной гипертензией (17).Эти аутоантитела против рецепторов AT 1 приводят к стимуляции рецептора (18). Некоторые пациенты с трансплантатом почки с хронической недостаточностью аллотрансплантата без классических HLA-антител имеют такие агонистические антитела против рецептора AT 1 , и они были вовлечены в васкулит с разрушением почечного аллотрансплантата (18).
Полиморфизмы для различных компонентов РААС, таких как АПФ, ангиотензиноген или рецепторы AT 1 , были описаны с противоречивыми результатами (19), в основном объясняемыми различным этническим происхождением исследуемых популяций.Хуанг и др. (20) индуцировал диабет у мышей, которые имели одну, две или три копии гена ACE. Двенадцать недель спустя у трехкопийных мышей с диабетом было повышенное АД и явная протеинурия. Протеинурия коррелировала с уровнем АПФ в плазме у мышей с трехкопийным диабетом. Таким образом, умеренное генетическое повышение уровня АПФ приводит к обострению диабетической нефропатии у мышей по сравнению со сниженной экспрессией гена АПФ (20). Эти данные указывают на то, что, вероятно, существует некоторое генетическое влияние на активность РААС, но остается неясным, как это влияет на индивидуальный риск предрасположенности или прогрессирования почечной недостаточности.
Местный RAAS
В течение последних двух десятилетий было описано, что местные RAAS действуют независимо от системного аналога (21). Местный РААС, включая все его компоненты, мог быть показан в клетках проксимальных канальцев почек (рис. 2). Клетки проксимальных канальцев активно продуцируют AngII, а также секретируют ангиотензиноген с мочой (21). Внутрипросветный ангиотензиноген может превращаться в дистальных канальцах в AngII, и недавние наблюдения показывают, что это приводит к индукции натриевых каналов независимо от альдостерона (22).Гипергликемия и протеинурия могут стимулировать локальный синтез AngII, в основном за счет форм кислорода в качестве передатчиков сигнала (12). Повреждение почек прямо или косвенно активирует местный РААС. Например, снижение уровня кальцитриола стимулирует транскрипцию ренина, сопровождаемую локальным увеличением AngII, что свидетельствует о непрямой активации (12).
Рисунок 2.
Проксимальные тубулярные клетки как пример местного РААС. Трубчатые клетки могут генерировать AngII в наномолярных концентрациях и секретироваться в трубчатую жидкость, а также в интерстициальное пространство.Кроме того, канальцевые клетки секретируют интактный ангиотензиноген в канальцевую жидкость. Клетки также могут забирать ренин и ангиотензиноген из кровотока, что указывает на тесное взаимодействие с системным РААС. Хотя клетки проксимальных канальцев имеют ACE в мембранах щеточной каймы, остается спорным вопрос, вносит ли внутриклеточный ACE вклад в формирование AngII.
Клиническое значение имеет наблюдение, что полное системное ингибирование образования AngII ингибитором АПФ не сопровождается значительным снижением внутрипочечной продукции AngII (23).Внутрипочечный AngII обнаруживается регионально компартментализированным (24). Неповрежденный AngII обнаруживается в эндосомах, происходящих из рецептор-опосредованного эндоцитоза. Это может быть важным механизмом, поскольку наблюдения в некоторых клетках продемонстрировали, что AngII может перемещаться в ядро, где он непосредственно регулирует транскрипцию гена (12). AngII оказывает множество разнообразных эффектов на почечные клетки, некоторые из которых изображены на рисунке 3.
Рисунок 3.
AngII — это цитокин, оказывающий множество эффектов на почки, явно выходящие за рамки классической функции гемодинамического медиатора.
Протеинурия
Помимо гипертонии, протеинурия является одним из важнейших факторов риска прогрессирования почечных заболеваний. Как подробно описано в сопроводительной статье Remuzzi et al. , повышенная абсорбция фильтрованных белков канальцами вызывает тубулоинтерстициальное воспаление, что в конечном итоге приводит к атрофии канальцев, интерстициальному фиброзу и потере функции почек. РААС играет важную роль во многих патофизиологических процессах, связанных с протеинурией.Во-первых, AngII является медиатором протеинурии. Он преимущественно повышает эфферентное сопротивление артериол клубочка. AngII индуцирует TGF-β1 в различных почечных клетках (25). Sharma et al. (26) недавно показали, что TGF-β1 нарушает ауторегуляцию афферентных артериол. Поскольку афферентные артериолы реагируют на повышение артериального давления сужением сосудов, нарушение ауторегуляции в присутствии TGF-β1 приводит к повышению транскапиллярного давления, особенно при системной гипертензии.Таким образом, AngII прямо (эфферентная вазоконстрикция) и косвенно (TGF-β1-опосредованное нарушение ауторегуляции афферентных артериол) усиливает давление капиллярной фильтрации.
Более того, AngII оказывает прямое влияние на целостность ультрафильтрационного барьера. Было показано, что AngII снижает синтез отрицательно заряженных протеогликанов и дополнительно подавляет транскрипцию нефрина (12,27). Поскольку интактная передача сигналов нефрин-нефрин важна для выживания подоцитов, AngII-опосредованное подавление нефрина приводит к апоптозу подоцитов.Фактор роста эндотелия сосудов (VEGF) также может играть важную роль в повышении проницаемости ультрафильтрационного барьера. Нейтрализация VEGF с помощью антител снижает протеинурию наполовину в модели диабетической нефропатии (28). AngII стимулирует экспрессию VEGF через рецепторы AT 1 и AT 2 . Увеличение экспрессии VEGF через рецепторы AT 2 предположительно опосредовано увеличением индуцируемого гипоксией фактора 1α, поскольку активация рецептора AT 2 привела к подавлению активности пролилгидроксилазы 3, фермента, который важен для инициирования деградации гипоксии. -индуцируемый фактор 1α (29).AngII-индуцированный синтез α3-цепи коллагена типа IV, основного ингредиента базальной мембраны клубочков, опосредуется VEGF и TGF-β1 (30). Следовательно, AngII через гемодинамические и негемодинамические механизмы увеличивает протеинурию.
В проксимальном канальце альбумин и другие ультрафильтрованные белки реабсорбируются за счет эндоцитоза с участием мегалина и кубулина (31). AngII стимулирует эндоцитоз альбумина в клетках проксимальных канальцев посредством активации протеинкиназы B, опосредованной рецептором AT 2 .Однако увеличение реабсорбции канальцевого альбумина активирует канальцевую РААС, что приводит к порочному кругу (32). Поглощение альбумина вызывает поток провоспалительных и профиброгенных цитокинов, таких как RANTES, моноцитарный хемоаттрактантный белок-1, IL-8, эндотелин и TGF-β1 (33). Это стимулирует миграцию иммунокомпетентных клеток в интерстиций.
Воспаление
AngII активирует через AT 1 и AT 2 провоспалительный фактор транскрипции NF-κB (34).Кроме того, AngIII и AngIV могут стимулировать NF-κB (33). Следовательно, очевидно, что сартаны могут блокировать только некоторые провоспалительные эффекты РААС. Путь Rho-киназы участвует в AngII-опосредованной активации NF-κB. Кроме того, AngII стимулирует фактор транскрипции Ets, и этот фактор является критическим регулятором сосудистого воспаления с привлечением Т-клеток и макрофагов / моноцитов к стенке сосуда (35). Недавно мы продемонстрировали другой механизм того, как AngII может способствовать воспалению почек.AngII активирует Toll-подобные 4 рецепторы мезангиальных клеток, которые связывают LPS (36). Эта AngII-опосредованная активация рецептора Toll-like 4 привела к усиленной активации NF-κB (36). Вовлечение воспалительных клеток в клубочки, а также в тубулоинтерстиций играет ключевую роль в прогрессировании хронического заболевания почек. AngII стимулирует повышающую регуляцию молекул адгезии, таких как молекула клеточной адгезии сосудов-1, молекула внутриклеточной адгезии-1 и интегрины, позволяя циркулирующим иммунным клеткам прилипать к капиллярам.NF-κB-опосредованная транскрипция хемокинов, включая хемоаттрактантный белок-1 моноцитов, RANTES и другие хемокины, затем отвечает за инфильтрацию почечной ткани лейкоцитами. Кроме того, AngII может напрямую стимулировать пролиферацию лимфоцитов (33). Интересно, что лимфоциты являются активным источником AngII, дополнительно усиливая провоспалительные эффекты (37). Очевидно, что провоспалительные эффекты, опосредованные AngII, усиливают патофизиологические изменения, вызываемые протеинурией.
Эффекты роста и апоптоз
AngII стимулирует пролиферацию мезангиальных клеток, эндотелиальных клеток клубочков и фибробластов. Напротив, пептид вызывает гипертрофию клеток проксимальных канальцев за счет остановки клеточного цикла, опосредованной p27 Kip1 (38,39). Пролиферация клубочковых клеток и фибробластов может усилить структурное повреждение почек и фиброз. AngII-индуцированная гипертрофия канальцев, хотя изначально является адаптивной реакцией на потерю функциональных нефронов, в долгосрочной перспективе является дезадаптивной и, вероятно, способствует развитию атрофии канальцев и интерстициального фиброза.Более того, AngII индуцирует апоптоз при определенных условиях in vivo и in vitro (39). Экспериментальные данные свидетельствуют о том, что рецепторы AT 1 и AT 2 участвуют в этом эффекте. Решение о том, подвергаются ли клетки эффекту стимуляции роста (пролиферация, гипертрофия) или, скорее, апоптозу, может зависеть от присутствия дополнительных факторов роста и цитокинов, а также от состояния активации клетки. Более того, концентрация AngII может играть роль, но почему пептид опосредует эффекты стимуляции роста в определенных экспериментальных условиях и индуцирует апоптоз в других условиях, не совсем понятно.
Фиброз
Вероятно, наиболее прямое доказательство того, что AngII участвует в рубцевании почек, связано с направленной сверхэкспрессией ренина и ангиотензиногена в клубочках крыс (25). Через семь дней после трансфекции внеклеточный матрикс (ЕСМ) увеличивался у крыс с избыточной экспрессией гломерулярного ренина и ангиотензиногена без системной гипертензии. AngII индуцирует мРНК, кодирующую белки ЕСМ проколлаген I типа и фибронектин в культивируемых мезангиальных клетках, а также стимулирует транскрипцию и синтез коллагена типа α1 (IV) и α3 (IV), но не типа I, в культивируемых клетках проксимальных канальцев (25).Стимулирующее действие AngII на экспрессию коллагена зависит от экспрессии TGF-β1. AngII стимулирует пролиферацию культивируемых фибробластов почек и увеличивает экспрессию мРНК TGF-β, фибронектина и коллагена I типа. Новым поворотом в этой истории является недавнее наблюдение, что ренин сам по себе через определенный рецептор стимулирует TGF-β1 в мезангиальных клетках (40). Эти данные поднимают интригующую возможность того, что повышенный уровень ренина, как следствие лечения ингибитором АПФ или рецептором AT 1 , может непосредственно способствовать почечному фиброзу через TGF-β1, несмотря на блокаду AngII.AngII увеличивает фактор роста соединительной ткани (CTGF) в почках (41). CTGF — это новый медиатор фиброза, который стимулируется TGF-β. Однако индуцированная AngII экспрессия CTGF также происходит независимо от TGF-β (41).
Тонкий баланс между синтезом и деградацией ECM в физиологических условиях предотвращает фиброз. AngII индуцирует через AT 1 рецепторов, ингибитор активатора плазминогена-1 (PAI-1) и тканевой ингибитор матриксных металлопротеиназ-1 (TIMP-1).PAI-1 и TIMP-1 ингибируют металлопротеиназы и, таким образом, метаболизм матрикса, что приводит к накоплению ECM. Через стимуляцию экспрессии PAI-1 в проксимальных канальцах через рецептор AT 4 , AngIV может играть роль в развитии почечного фиброза независимо от активации рецепторов AT 1 и AT 2 (42 ). Из-за повышенной регуляции AngIV, который генерирует ферменты в условиях с высокими локальными концентрациями AngII и при диабетической нефропатии (10), ускоренная деградация AngII в AngIV может активировать рецепторы AT 4 , индуцируя PAI-1.
Изобретательные эксперименты продемонстрировали, что более одной трети локальных фибробластов при почечном интерстициальном фиброзе происходит из канальцевых эпителиальных клеток в результате процесса, называемого переходом от эпителия к мезенхиме (EMT). Молекулярный механизм ЭМП был подробно рассмотрен ранее (43). EMT может иметь важное значение на более поздних стадиях прогрессирования почечной недостаточности, приводя к интерстициальному фиброзу и атрофии канальцев из-за исчезновения эпителиальных клеток. Одним из важных посредников EMT является TGF-β1, и AngII может вносить вклад в EMT за счет индукции этого профибротического фактора (43).EMT противодействует фактору роста гепатоцитов. Поскольку AngII подавляет синтез фактора роста гепатоцитов, AngII может дополнительно стимулировать ЕМТ за счет снижения на его антагониста (44).
Эффекты ингибитора АПФ
и рецептора АТ
1 , не зависящие от RAAS
Недавние данные свидетельствуют о том, что ингибиторы АПФ, а также блокаторы рецепторов AT 1 могут влиять на клеточные функции независимо от ингибирования РААС. Например, ингибиторы АПФ блокируют гидролиз N-ацетилсерил-аспартил-лизил-пролина (Ac-SDKP).Некоторые защитные эффекты ингибитора АПФ (, например, , ингибирование фиброза, уменьшение инфильтрации воспалительных клеток) являются результатом ингибирования гидролиза Ac-SDKP, а не ингибирования образования AngII в модели AngII-индуцированного сердечного фиброза (45). . Остается неясным, действуют ли аналогичные механизмы в почках.
Используя эндотелиальные клетки, было продемонстрировано, что ингибиторы АПФ рамиприлат и периндоприлат увеличивают CK2-опосредованное фосфорилирование серина 1270 (46).Кроме того, лечение ингибитором АПФ увеличивало активность N-концевой киназы в эндотелиальных клетках. Эти провокационные данные указывают на то, что ингибиторы АПФ могут опосредовать клеточную функцию посредством передачи сигналов «извне-внутрь» непосредственно через АПФ, эффект, который полностью не зависит от образования AngII (46).
Накапливаются данные о том, что некоторые антагонисты рецептора AT 1 , такие как телмисартан, активируют рецептор-γ, активируемый пролифератором пероксисом (PPAR-γ), широко известную мишень для лечения метаболического синдрома и диабета (47).Недавние исследования показали, что в дополнение к противодиабетическим свойствам активаторы PPAR-γ могут улучшить состояние почек, нормализовать гиперфильтрацию и снизить протеинурию (47). PPAR-γ-активирующие свойства некоторых сартанов не требуют присутствия рецепторов AT 1 и обусловлены молекулярной структурой специфических сартанов (47). Следовательно, возможно, что некоторые из защитных эффектов блокаторов рецепторов AT 1 в замедлении прогрессирования хронического заболевания почек обусловлены действиями, которые не зависят от действия РААС.
А как насчет альдостерона?
Классическое понимание альдостерона как гормона, вырабатываемого в коре надпочечников и участвующего в реабсорбции натрия и секреции калия и протонов в собирательном канале, требует расширения. Эти эффекты альдостерона были объяснены как геномные эффекты, которые вызваны повышенной транскрипцией различных генов-мишеней после связывания альдостерона с цитоплазматическими рецепторами. Новые данные свидетельствуют о том, что негеномные эффекты альдостерона, такие как активация определенных путей передачи сигнала, происходят в нескольких органах, включая почки (48).Альдостерон также вырабатывается во многих других тканях, помимо коры надпочечников. В различных моделях почечных заболеваний на животных альдостерон участвует в эндотелиальной дисфункции, воспалении, протеинурии и фиброзе (48). Альдостерон усиливает действие AngII, индуцирует образование активных форм кислорода и приводит к ускорению вызванной AngII активации митоген-активируемых протеинкиназ (49). Эти данные показывают, что блокада минералокортикоидных рецепторов предположительно полезна даже в ситуациях с высоким AngII, потому что общие пути передачи сигнала между двумя системами прерваны.Первые клинические исследования, по-видимому, показывают, что блокада рецепторов альдостерона обеспечивает дополнительную защиту почек даже при наличии ингибирования АПФ или антагонизма рецепторов AT 1 . Однако потенциальная угроза гиперкалиемии при таком подходе требует дальнейших клинических исследований для проверки безопасности этого лечения.
Заключение
РААС прошла долгий путь с тех пор, как мы описали более 15 лет назад структурные и профибротические эффекты AngII на почечные клетки (38).Эта увлекательная система становится все более сложной, а поиск новых членов продолжается. AngII возник из вазоконстриктора основного многофакторного пептида, участвующего в прогрессировании почечной недостаточности. Исчерпывающее понимание РААС с новыми фармакологическими подходами к вмешательству в его члены (, например, , ингибитор ренина аскирен), мы надеемся, предоставит инструменты, чтобы полностью остановить прогрессирование и вызвать регресс почечной недостаточности.
Благодарности
Оригинальные исследования в лаборатории авторов поддерживаются Deutsche Forschungsgemeinschaft.
Мы приносим свои извинения всем участникам в этой быстрорастущей области, чьи публикации не могли быть признаны из-за ограничений на количество ссылок.
G.W. благодарит Эрика Г. Нейлсона за поддержку, ободрение и невероятное предвидение в конце 1980-х, когда никто не верил, что AngII — это нечто большее, чем сосудосуживающее средство.
- © 2006 Американское общество нефрологов
Ссылки
- ↵
Remuzzi G, Benigni A, Remuzzi A: Механизмы прогрессирования и регресса почечных поражений при хронических нефропатиях и диабете.J Clin Invest 116: 288–296, 2006
- ↵
Campbell RC, Ruggeneti P, Remuzzi G: Остановить прогрессирование хронической нефропатии. J Am Soc Nephrol 13: 190–195, 2002
- ↵
Wolf G, Ritz E: Комбинированная терапия с ингибиторами АПФ и блокаторами рецепторов ангиотензина II для остановки прогрессирования хронического заболевания почек: патофизиология и показания. Почки Инт 67: 799–812, 2005
- ↵
Wolf G, Neilson EG: От ингибирования превращения фермента до блокады рецептора ангиотензина II: новые взгляды на подтипы рецепторов ангиотензина II в почках.Опыт Нефрол 4 [Дополнение 1]: 8–19, 1996
- ↵
Huang XR, Chen WY, Truong LD, Lan HY: Химаза активируется при диабетической нефропатии: последствия для альтернативного пути развития диабетических заболеваний почек и сосудов, опосредованных ангиотензином II. J Am Soc Nephrol 14: 1738 –1747, 2003
- ↵
Дурвасула Р.В., Петерманн А.Т., Хиромура К., Блонски М., Пиппин Дж., Мундель П., Пихлер Р., Гриффин С., Кузер В.Г., Шенкланд С.Дж.: Активация местной тканевой ангиотензиновой системы в подоцитах механической нагрузкой.Почки Инт 65: 30–39, 2004
- ↵
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM: Ангиотензин-превращающий фермент 2 является важным регулятором сердечной функции. Nature 417: 822–828, 2002
- ↵
Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, Cooper ME: Характеристика почечного ангиотензин-превращающего фермента 2 при диабетической нефропатии.Гипертония 41: 392 –397, 2003
- ↵
Данилчик У., Пеннингер Дж. М.: Ангиотензин-превращающий фермент II в сердце и почках. Circ Res 98: 463–471, 2006
- ↵
Wolf G, Mentzel S, Assmann KJ: Аминопептидаза A: ключевой фермент во внутрипочечной деградации ангиотензина II. Опыт Нефрола 5: 364–369, 1997
- ↵
Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, Lee J, Mendelsohn FA, Simpson RJ, Connolly LM, Chai SY: доказательства того, что рецептор ангиотензина IV (AT (4)) является ферментом регулируемая инсулином аминопептидаза.J Biol Chem 276: 48623–48626, 2001
- ↵
Wolf G, Butzmann U, Wenzel UO: Система ренин-ангиотензин и прогрессирование или почечная болезнь: от гемодинамики до клеточной биологии. Нефрон Физиол 93: 3–13, 2003
- ↵
Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F: Перекрестный разговор рецепторов ангиотензина II и EGF при хронических заболеваниях почек: новый терапевтический подход.Nat Med 8: 867–874, 2005
- ↵
Кэри Р.М.: Обновленная информация о роли рецептора AT2. Curr Opin Nephrol Hypertens 14: 67–71, 2005
- ↵
Ullian ME, Gelasco AK, Fitzgibbon WR, Beck CN, Morinelli TA: N-ацетилцистеин снижает связывание рецептора ангиотензина II в гладкомышечных клетках сосудов. J Am Soc Nephrol 16: 2346–2353, 2005
- ↵
Abdalla S, Lother H, Langer A, el Faramawy Y, Quitterer U: трансглутаминаза фактора XIIIA сшивает димеры рецептора AT1 моноцитов в начале атеросклероза.Ячейка 119: 343–354, 2004 г.
- ↵
Thway TM, Шлыков С.Г., Day MC, Sanborn BM, Gilstrap LC 3rd, Xia Y, Kellems RE: Антитела от пациентов с преэклампсией стимулируют повышенную мобилизацию внутриклеточного Ca2 + за счет активации рецептора ангиотензина. Тираж 110: 1612–1619, 2004 г.
- ↵
Драгун Д., Мюллер Д. Н., Бразен Дж. Х., Фриче Л., Ниеминен-Келха М., Дехенд Р., Кинтчер Ю., Рудольф Б., Хёбеке Дж., Эккерт Д., Мазак I, Плем Р., Шонеман К., Унгер Т., Бадде К., Ноймайер HH, Luft FC, Wallukat G: антитела, активирующие рецептор ангиотензина II типа 1 при отторжении почечного аллотрансплантата.N Engl J Med 352: 558 –569, 2005
- ↵
Jardine AG, Padmanabhan N, Connell JMC: Полиморфизм генов ангиотензинпревращающего фермента и заболевание почек. Curr Opin Nephrol Hypertens 7: 259–264, 1998
- ↵
Huang W, Gallois Y, Bouby N, Bruneval P, Heudes D, Belair MF, Krege JH, Meneton P, Marre M, Smithies O, Alhenc-Gelas F: генетически повышенный уровень фермента, преобразующего ангиотензин I, и почечные осложнения в диабетическая мышь.Proc Natl Acad Sci U S A 98: 13330–13334, 2001
- ↵
Nishiyama A, Seth DM, Navar LG: Концентрации ангиотензинов I и II в почечной интерстициальной жидкости у анестезированных крыс. Гипертония 39: 129–134, 2002
- ↵
Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA: Долгосрочная регуляция экспрессии ENaC в почках ангиотензином II.Гипертония 41: 1143–1150, 2003
- ↵
Nishiyama A, Seth DM, Navar LG: Концентрации почечной интерстициальной жидкости I и ангиотензина II во время местного ингибирования ангиотензинпревращающего фермента. J Am Soc Nephrol 13: 2207 –2212, 2002
- ↵
Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG: Накопление Ang II в почечных эндосомах крыс во время гипертензии, индуцированной Ang II. Роль рецептора AT1.Гипертония 39: 116 –121, 2002
- ↵
Wolf G: Связь между ангиотензином II и TGF-бета в почках. Miner Electrolyte Metab 24: 174–180, 1998
- ↵
Sharma K, Cook A, Smith M, Valancius C, Inscho EW: TGF-бета нарушает ауторегуляцию почек посредством генерации ROS. Am J Physiol Renal Physiol 288: F1069 –F1077, 2005
- ↵
Brinkkoetter PT, Holtgrefe S, van der Woude FJ, Yard BA: Опосредованные рецептором ангиотензина II типа 1 изменения протеогликанов гепарансульфата в подоцитах человека, трансформированных SV40.J Am Soc Nephrol 15: 33–40, 2004
- ↵
Вольф Г., Чен С., Зияде Ф. Н.: От периферии стенки капилляров клубочка к центру заболевания. Повреждение подоцитов наступает в зрелом возрасте при диабетической нефропатии. Диабет 54: 1626–1634, 2005
- ↵
Вольф Г., Шредер Р., Шталь RAK: Ангиотензин II индуцирует индуцируемый гипоксией фактор-1альфа в клетках PC 12 через посттранскрипционный механизм: роль рецепторов AT2.Am J Nephrol 24: 415–421, 2004
- ↵
Chen S, Lee JS, Iglesias-de la Cruz MC, Kasama Y, Izquierdo-Lahuerta A, Wolf G, Ziyadeh FN: Ангиотензин II стимулирует выработку альфа-3 (IV) коллагена в подоцитах мышей через передачу сигналов TGF-бета и VEGF: последствия при диабетической нефропатии. Трансплантат Nephrol Dial 20: 1320–1328, 2005
- ↵
Birn H, Christensen EI: Поглощение альбумина почками в физиологии и патологии.Почки Инт 69: 440–449, 2006
- ↵
Caruso-Neves C, Kwon SH, Guggino WB: Эндоцитоз альбумина в клетках проксимальных канальцев модулируется ангиотензином II посредством активации протеинкиназы B, опосредованной рецептором AT2. Proc Nat Acad Sci U S A 102: 17513 –17518, 2005
- ↵
Руис-Ортега М., Эстебан В., Руперес М., Санчес-Лопес Е., Родригес-Вита Дж., Карвахаль Г., Эджидо Дж. Воспаление, вызванное почечной и сосудистой гипертензией: роль ангиотензина II.Curr Opin Nephrol Hypertens 15: 159–166, 2006
- ↵
Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F: Ангиотензин II активирует ядерный фактор транскрипции-kappaB через рецепторы AT1 и AT2. Почки Int 61: 1986–1995, 2002
- ↵
Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC, Oettgen P: Ets-1 является критическим регулятором Ang II-опосредованного воспаления и ремоделирования сосудов.J Clin Invest 115: 2508 –2516, 2005
- ↵
Wolf G, Bondeva T, Bohlender J, Roger T, Thaiss F, Wenzel U: ангиотензин II активирует Toll-подобный рецептор 4 на мезангиальных клетках. J Am Soc Nephrol 17: 1585 –1593, 2006
- ↵
Янковский В., Ванхолдер Р., ван дер Грит М., Хеннинг Л., Толле М., Шонфельдер Г., Краков А., Карадоган С., Густавссон Н., Гобом Дж., Уэбб Дж., Лехрах Н., Гибинг Г., Шлютер Н., Хильгерс К. Ф., Зидек W, Jankowski J: Обнаружение ангиотензина II в супернатантах стимулированных мононуклеарных лейкоцитов с помощью матричного масс-анализа с лазерной десорбцией и ионизацией по времени пролета / времени пролета.Гипертония 46: 591 –597, 2005
- ↵
Wolf G, Neilson EG: Ангиотензин II вызывает клеточную гипертрофию в культивируемых клетках проксимальных канальцев мышей. Am J Physiol 259: F768 –F777, 1990
- ↵
Wolf G, Wenzel U: Ангиотензин II и регуляция клеточного цикла. Гипертония 43: 693–698, 2004
- ↵
Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L, Noble NA, Border W. Ренин увеличивает мезангиальные клетки, трансформирующие фактор роста-бета1 и матричные белки посредством рецепторно-опосредованных, независимых от ангиотензина II механизмов.Почки Инт 69: 105–113, 2006
- ↵
Родригес-Вита Дж., Сабчес-Лопес Э., Эстебан В., Руперез М., Эгидо Дж., Руис-Ортега М.: Ангиотензин II активирует путь smad в гладкомышечных клетках сосудов с помощью механизма, независимого от трансформирующего фактора роста бета. Тираж 111: 2509–2517, 2005
- ↵
Abrahamsen CT, Pullen MA, Schnackenberg CG, Grygielko ET, Edwards RM, Laping NJ, Brooks DP: Влияние ангиотензинов II и IV на кровяное давление, функцию почек и экспрессию PAI-1 в сердце и почках крысы.Фармакология 66: 26–30, 2002
- ↵
Каллури Р., Нейлсон Э.Г .: Эпителиально-мезенхимальный переход и его последствия для фиброза. J Clin Invest 112: 1776 –1784, 2003
- ↵
Мацумото К., Моришита Р., Томита Н., Моригути А., Комай Н., Аоки М., Мацумото К., Накамура Т., Хигаки Дж., Огихара Т.: улучшение эндотелиальной дисфункции за счет блокады ангиотензина II, сопровождающейся индукцией системы факторов роста сосудистых гепатоцитов в диабетические спонтанно гипертонические крысы.Сосуды сердца 18: 18–25, 2003
- ↵
Peng H, Carretero OA, Vuljai N, Liao TD, Motivala A, Peterson EL, Rhaleb NE: ингибиторы ангиотензинпревращающего фермента. Новый механизм действия. Тираж 112: 2436–2445, 2005
- ↵
Fleming I, Kohlstedt K, Busse R: Тканевая система ренин-ангиотензин и внутриклеточная передача сигналов. Curr Opin Nephrol Hypertens 15: 8–13, 2006
- ↵
Schupp M, Clemenz M, Gineste R, Witt H, Janke J, Helleboid S, Hennuyer N, Ruiz P, Unger T., Staels B, Kintscher U: Молекулярная характеристика новых селективных гамма-модуляторов рецепторов, активируемых пролифератором пероксисом, с рецептором ангиотензина блокировка активности.Диабет 54: 3442–3452, 2005
- ↵
Nishiyama A, Abe Y: Молекулярные механизмы и терапевтические стратегии хронического почечного повреждения: Renoprotective эффекты блокады альдостерона. J Pharmacol Sci 100: 9–16, 2006
- ↵
Mazak I, Fiebeler A, Muller DN, Park JK, Shagdarsuren E, Lindschau C, Dechend R, Viedt C, Pilz B, Haller H, Luft FC: Альдостерон усиливает индуцированную ангиотензином II передачу сигналов в гладкомышечных клетках сосудов.Тираж 109: 2792–2800, 2004
Ингибиторы ренин-ангиотензин-альдостероновой системы — AMBOSS
Последнее обновление: 1 февраля 2021 г.
Резюме
Ингибиторы ренин-ангиотензин-альдостероновой системы (РААС) — это группа препаратов, которые действуют путем ингибирования ренин-ангиотензиновой системы. альдостероновой системы (РААС) и включают ингибиторы ангиотензинпревращающего фермента (ингибиторы АПФ), блокаторы рецепторов ангиотензина (БРА) и прямые ингибиторы ренина. Ингибиторы АПФ и БРА обычно используются для лечения пациентов с артериальной гипертензией, сердечной недостаточностью с пониженной фракцией выброса и некоторыми типами хронических заболеваний почек, а также пациентов, перенесших инфаркт миокарда.Они особенно важны при лечении пациентов с гипертоническим диабетом, поскольку предотвращают развитие диабетической нефропатии. Распространенным побочным эффектом ингибиторов АПФ является кашель, вызванный брадикинином, который может потребовать перехода на альтернативную терапию (например, БРА), в то время как ангионевротический отек и гиперкалиемия могут возникать при применении как БРА, так и ингибиторов АПФ. Прямые ингибиторы ренина могут быть рассмотрены у пациентов с артериальной гипертензией, если ингибиторы АПФ или БРА плохо переносятся; однако их никогда не следует использовать в сочетании с другими ингибиторами РААС.
Обзор
Типы ингибиторов РААС
Ингибиторы ангиотензинпревращающего фермента (ингибиторы АПФ)
- Названия лекарств: эналаприл, лизиноприл, рамиприл, каптоприл, беназеприл.
- Показания
Блокатор ангиотензиновых рецепторов (БРА, сартаны)
- Названия лекарств: валсартан, кандесартан, лозартан, ирбесартан.
- Показания: такие же, как и ингибиторы АПФ, в основном используются в качестве терапии второй линии, если ингибиторы АПФ не переносятся
- Ангионевротический отек: можно попробовать под тщательным наблюдением, если нет адекватной альтернативы [5]
- Неопасные для жизни побочные эффекты (например,г., сухой кашель; ): обычно используется
Прямые ингибиторы ренина
Фармакодинамика
БРА
- Механизм действия: угнетение рецептора ангиотензина II типа 1 (рецептор АТ 1 ).
- Основные эффекты
- Другие эффекты
Прямые ингибиторы ренина
aLESkiREN: МЕНЬШЕ RENin с алискиреном.
Побочные эффекты
Побочные эффекты каптоприла: кашель, ангионевротический отек, вульгарная пузырчатка, тератогенность, гипотензия, высокий уровень калия, почечная недостаточность, повышение креатинина, низкая СКФ.
Острое повреждение почек является потенциальным побочным эффектом всех типов ингибиторов РААС, особенно у пациентов с уже существующим заболеванием почек или в комбинации с НПВП
Мы перечисляем наиболее важные побочные эффекты. Выбор не исчерпывающий.
Противопоказания
Противопоказания для ингибиторов АПФ и БРА
- Абсолютные противопоказания
- Относительные противопоказания
В норме ангиотензин II сужает эфферентные сосуды, увеличивая СКФ.Ингибиторы АПФ препятствуют превращению ангиотензина I в ангиотензин II, снижая СКФ.
Противопоказания для прямых ингибиторов ренина
Перечислим наиболее важные противопоказания. Выбор не исчерпывающий.
Взаимодействия
Не комбинируйте прямые ингибиторы ренина с ингибиторами АПФ или БРА, особенно у пациентов с диабетом или ранее существовавшим заболеванием почек.
Дополнительные сведения
Ссылки
- Стандарты медицинской помощи при диабете 2016 г. http://care.diabetesjournals.org/content/suppl/2015/12/21/39.Supplement_1.DC2/2016-Standards-of-Care.pdf .
Обновлено: 1 января 2016 г.
Доступ: 22 февраля 2017 г. Роэтт М.А., Лигл С., Джаббарпур Ю. Диабетическая нефропатия — роль семейного врача. Ам Фам Врач . 2012; 85
(9): с.883-889.Юсуф С., Питт Б., Дэвис К.Э., Худ В.Б., Кон Дж. Влияние эналаприла на выживаемость у пациентов со сниженной фракцией выброса левого желудочка и застойной сердечной недостаточностью. N Engl J Med . 1991; 325
(5): с.293-302.
DOI: 10,1056 / nejm19- 13250501. | Открыть в режиме чтения QxMD
Франзози М.Г., Санторо Э., Зуанетти Г. и др. Показания к применению ингибиторов АПФ в раннем лечении острого инфаркта миокарда: систематический обзор индивидуальных данных от 100 000 пациентов в рандомизированных исследованиях. Тираж . 1998; 97
(22): p.2202-2212.
DOI: 10.1161 / 01.cir.97.22.2202. | Открыть в режиме чтения QxMDХеймор Б.Р., Юн Дж., Никита С.П., Клот ММ, Дези К.Дж.Риск ангионевротического отека с блокаторами рецепторов ангиотензина у пациентов с предшествующим ангионевротическим отеком, связанным с ингибиторами ангиотензинпревращающего фермента: метаанализ. Ann Allergy Asthma Immunol. . 2008; 101
(5): с.495-499.
DOI: 10,1016 / s1081-1206 (10) 60288-8. | Открыть в режиме чтения QxMDКарлберг БЭ. Кашель и угнетение ренин-ангиотензиновой системы .. J Hypertens Suppl . 1993; 11
(3): С. S49-52.Yılmaz İ.Ингибиторы ангиотензинпревращающего фермента вызывают кашель .. Турецкий торакальный журнал . 2019; 20
(1): с.36-42.
DOI: 10.5152 / TurkThoracJ.2018.18014. | Открыть в режиме чтения QxMDNavis G, Faber HJ, de Zeeuw D, de Jong PE. Ингибиторы АПФ и почки. Сейф с наркотиками . 1996; 15
(3): с.200-211.
DOI: 10.2165 / 00002018-199615030-00005. | Открыть в режиме чтения QxMDАятоллахи А., Тусси П., Юнеспур С., Робати Р.Сывороточный ангиотензинпревращающий фермент при вульгарной пузырчатке. Индийский J Dermatol . 2014; 59
(4): 348 с.
DOI: 10.4103 / 0019-5154.135478. | Открыть в режиме чтения QxMD- Использование ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов ангиотензина при ХБП.
https://www2.kidney.org/professionals/kdoqi/guidelines_bp/guide_11.htm .
Обновлено: 1 января 2004 г.
Доступ: 11 апреля 2018 г. Бикет ДП.Правильное использование ингибиторов АПФ. Ам Фам Врач . 2002; 66
(3): с.461-469.Shionoiri H. Фармакокинетические лекарственные взаимодействия с ингибиторами АПФ. Клин Фармакокинет . 1993; 25
(1): с.20-58.
DOI: 10.2165 / 00003088-199325010-00003. | Открыть в режиме чтения QxMDPolónia J. Взаимодействие гипотензивных препаратов с противовоспалительными средствами. Кардиология . 1997; 88
(3): стр.47-51.
DOI: 10,1159 / 000177507. | Открыть в режиме чтения QxMDШтамп LK, Chapman PT. Подагра и сопутствующие ей заболевания: значение для терапии. Ревматология . 2012; 52
(1): с.34-44.
DOI: 10.1093 / ревматология / kes211. | Открыть в режиме чтения QxMDВайдьянатан С., Яругула В., Дитрих Х.А., Ховард Д., Доул В.П. Клиническая фармакокинетика и фармакодинамика Алискирена. Клин Фармакокинет . 2008; 47
(8): с.515-531.
DOI: 10.2165 / 00003088-200847080-00002. | Открыть в режиме чтения QxMDХарел З., Гилберт С., Вальд Р. и др. Влияние комбинированного лечения алискиреном и блокаторами ренин-ангиотензиновой системы на гиперкалиемию и острое повреждение почек: систематический обзор и метаанализ. BMJ . 2012; 344
: p.e42.
DOI: 10.1136 / bmj.e42. | Открыть в режиме чтения QxMD
Какова роль ренин-ангиотензин-альдостероновой системы (РААС) в патофизиологии сердечной недостаточности?
Хо К.К., Пинский Ю.Л., Каннел В.Б., Леви Д.Эпидемиология сердечной недостаточности: исследование Framingham. Джам Колл Кардиол . 1993, 22 октября (4 приложения A): 6A-13A. [Медлайн]. [Полный текст].
Американская кардиологическая ассоциация. Классы сердечной недостаточности. Доступно по адресу http://www.heart.org/HEARTORG/Conditions/HeartFailure/AboutHeartFailure/Classes-of-Heart-Failure_UCM_306328_Article.jsp#.WUcGf-vyuHs. Обновлено: 8 мая 2017 г .; Дата обращения: 18 июня 2017 г.
[Рекомендации] Янси К.В., Джессап М., Бозкурт Б. и др., Фонд Американского колледжа кардиологов / Целевая группа Американской кардиологической ассоциации по практическим рекомендациям.Руководство ACCF / AHA по лечению сердечной недостаточности, 2013 г.: отчет Фонда Американского колледжа кардиологов / Целевой группы Американской кардиологической ассоциации о практических рекомендациях. Тираж . 2013 15 октября 128 (16): e240-327. [Медлайн]. [Полный текст].
[Рекомендации] Пониковски П., Вурс А.А., Анкер С.Д. и др. Для авторов / членов рабочей группы. Рекомендации ESC по диагностике и лечению острой и хронической сердечной недостаточности, 2016: рабочая группа Европейского общества кардиологов (ESC) по диагностике и лечению острой и хронической сердечной недостаточности.Разработано при особом участии Ассоциации сердечной недостаточности (HFA) ESC. Евро Сердце J . 2016 14 июля. 37 (27): 2129-200. [Медлайн]. [Полный текст].
[Рекомендации] Линденфельд Дж., Альберт Н.М., Бёмер Дж. П. и др. Для Американского общества сердечной недостаточности. Комплексное практическое руководство HFSA 2010 по сердечной недостаточности. J Card Fail . 2010 июн.16 (6): e1-194. [Медлайн]. [Полный текст].
Stiles S. FDA одобрило средство визуализации сердечной симпатической активности для оценки сердечной недостаточности.Новости Medscape от WebMD. 22 марта 2013 г. Доступно по адресу http://www.medscape.com/viewarticle/781309. Доступ: 5 апреля 2013 г.
Браунвальд Э. Патогенез застойной сердечной недостаточности: тогда и сейчас. Медицина . 1991, январь 70 (1): 68-79. [Полный текст].
Браунвальд Э., Росс-младший, Зонненблик Э. Механизмы сокращения нормального и больного сердца . 2-е изд. Бостон: Little Brown & Co; 1976.
Грейсон CR.Патофизиология правожелудочковой недостаточности. Crit Care Med . 2008 г., 36 января (1 доп.): С57-65. [Медлайн].
Хаддад Ф, Дойл Р., Мерфи Ди-джей, Хант С.А. Функция правого желудочка при сердечно-сосудистых заболеваниях, часть II: патофизиология, клиническое значение и лечение правожелудочковой недостаточности. Тираж . 2008 г. 1. 117 (13): 1717-31. [Медлайн].
Onwuanyi A, Taylor M. Острая декомпенсированная сердечная недостаточность: патофизиология и лечение. Ам Дж. Кардиол . 2007 26 марта, 99 (6B): 25D-30D. [Медлайн].
Росс-младший, Браунвальд Э. Исследования закона сердца Старлинга. IX. Влияние замедления венозного возврата на работу нормального и поврежденного левого желудочка человека. Тираж . 1964, 30 ноября: 719-27. [Медлайн]. [Полный текст].
Gheorghiade M, Pang PS. Синдромы острой сердечной недостаточности. Джам Колл Кардиол . 2009 17 февраля. 53 (7): 557-73. [Медлайн].
Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Разрастание миоцитов при терминальной стадии сердечной недостаточности у людей. Proc Natl Acad Sci U S A . 1998 21 июля. 95 (15): 8801-5. [Медлайн]. [Полный текст].
Cohn JN. Структурная основа сердечной недостаточности. Ремоделирование желудочков и его фармакологическое торможение. Тираж . 1995 15 мая. 91 (10): 2504-7. [Медлайн].
Коди Р.Дж.Гормональные изменения при сердечной недостаточности. В: Hosenpud JB, Greenberg BH, eds. Застойная сердечная недостаточность: патофизиология, диагностика и комплексный подход к лечению . Филадельфия, Пенсильвания: Липпинкотт Уильямс и Уилкинс; 2000. 199-212.
Anversa P, Nadal-Ginard B. Обновление миоцитов и ремоделирование желудочков. Природа . 2002, 10 января. 415 (6868): 240-3. [Медлайн].
Лери А., Клаудио П.П., Ли Кью и др. Высвобождение ангиотензина II, опосредованное растяжением, вызывает апоптоз миоцитов за счет активации р53, который усиливает локальную систему ренин-ангиотензин и снижает соотношение белков Bcl-2 и Bax в клетке. Дж. Клин Инвест . 1998 г. 1. 101 (7): 1326-42. [Медлайн]. [Полный текст].
Кайстура Дж., Лери А., Кастальдо С., Надаль-Жинард Б., Анверса П. Рост миоцитов в больном сердце. Surg Clin North Am . 2004 Февраль 84 (1): 161-77. [Медлайн].
Хенес Дж, Розенбергер П. Систолическая сердечная недостаточность: диагностика и терапия. Curr Opin Anaesthesiol . 2016 29 февраля (1): 55-60. [Медлайн].
Никоара А., Джонс-Хейвуд М.Диастолическая сердечная недостаточность: диагностика и терапия. Curr Opin Anaesthesiol . 2016 29 февраля (1): 61-7. [Медлайн].
Feldman AM, Combes A, Wagner D, et al. Роль фактора некроза опухолей в патофизиологии сердечной недостаточности. Джам Колл Кардиол . 2000 г. 1. 35 (3): 537-44. [Медлайн].
Гэри Р., Дэвис Л. Диастолическая сердечная недостаточность. Сердце легкое . 2008 ноябрь-декабрь. 37 (6): 405-16. [Медлайн].
Моррис Д.А., Гайлани М., Ваз Перес А. и др.Систолическая и диастолическая дисфункция миокарда правого желудочка при сердечной недостаточности с нормальной фракцией выброса левого желудочка. J Am Soc Echocardiogr . 2011 24 августа (8): 886-97. [Медлайн].
Джусилахти П., Харальд К., Юла А. и др., Национальный институт здравоохранения и социального обеспечения-THL — Хельсинки — Финляндия. Потребление соли и риск сердечной недостаточности [аннотация 1192]. Представлено на: Конгрессе Европейского общества кардиологов 2017; 27 августа 2017 г .; Барселона, Испания. Евро Сердце J . Август 2017. 38 (приложение 1): 240. [Полный текст].
Давенпорт Л. Высокое потребление соли связано с повышенным риском сердечной недостаточности. Новости Medscape от WebMD. 28 августа 2017 г. Доступно по адресу http://www.medscape.com/viewarticle/884824. Доступ: 1 сентября 2017 г.
Лам С.С., Лясс А., Крейгер-Крайнер Э. и др. Сердечная дисфункция и несердечная дисфункция как предвестники сердечной недостаточности со сниженной и сохраненной фракцией выброса в сообществе. Тираж . 2011 июл 5. 124 (1): 24-30. [Медлайн].
Хо К.К., Андерсон К.М., Каннель В.Б., Гроссман В., Леви Д. Выживание после начала застойной сердечной недостаточности у субъектов Фрамингемского исследования сердца. Тираж . 1993 июл.88 (1): 107-15. [Медлайн].
Halley CM, Houghtaling PL, Khalil MK, Thomas JD, Jaber WA. Смертность у пациентов с диастолической дисфункцией и нормальной систолической функцией. Arch Intern Med .2011, 27 июня. 171 (12): 1082-7. [Медлайн].
Мерфи RT, Starling RC. Генетика и кардиомиопатия: где мы сейчас ?. Клив Клин Дж. Мед . 2005 июн.72 (6): 465-6, 469-70, 472-3 passim. [Медлайн].
Benjamin EJ, Blaha MJ, Chiuve SE и др., Для Статистического комитета Американской кардиологической ассоциации и Подкомитета по статистике инсультов. Обновление статистики сердечных заболеваний и инсульта за 2017 г .: отчет Американской кардиологической ассоциации. Тираж . 2017 7 марта. 135 (10): e146-e603. [Медлайн]. [Полный текст].
Ni H, Xu J. Последние тенденции смертности от сердечной недостаточности: США, 2000–2014 гг. Центры по контролю и профилактике заболеваний. 31 декабря 2015 г. Доступно по адресу http://www.cdc.gov/nchs/data/databriefs/db231.htm. Доступ: 5 января 2016 г.
Brauser D. CDC: Уровень смертности от сердечной недостаточности растет после десятилетнего снижения. Heartwire от Medscape.4 января 2016 г. Доступно по адресу http://www.medscape.com/viewarticle/856704. Доступ: 5 января 2016 г.
Chen J, Normand SL, Wang Y, Krumholz HM. Национальные и региональные тенденции в отношении госпитализаций и смертности от сердечной недостаточности для получателей медицинской помощи, 1998-2008 гг. ЯМА . 2011 Октябрь 19, 306 (15): 1669-78. [Медлайн].
Kolte D, Abbott JD, Aronow HD. Интервенционные методы лечения сердечной недостаточности у пожилых людей. Клиника сердечной недостаточности .2017 июл.13 (3): 535-70. [Медлайн].
Dharmarajan K, Rich MW. Эпидемиология, патофизиология и прогноз сердечной недостаточности у пожилых людей. Клиника сердечной недостаточности . 2017 июл.13 (3): 417-26. [Медлайн].
Дженкс С.Ф., Уильямс М.В., Коулман Е.А. Повторные госпитализации среди пациентов по программе оплаты медицинских услуг Medicare. N Engl J Med . 2009 апр. 2. 360 (14): 1418-28. [Медлайн].
Стюарт С., Уилкинсон Д., Хансен С. и др.Преобладание сердечной недостаточности в когорте Heart of Soweto Study: новые проблемы для городских африканских сообществ. Тираж . 2008 декабрь 2. 118 (23): 2360-7. [Медлайн].
Damasceno A, Cotter G, Dzudie A, Sliwa K, Mayosi BM. Сердечная недостаточность в Африке к югу от Сахары: время действовать. Джам Колл Кардиол . 2007 октября 23:50 (17): 1688-93. [Медлайн].
Mbewu A, Mbanya JC. Сердечно-сосудистые заболевания. В: Джеймисон Д.Т., Фичем Р.Г., Макгоба М.В. и др., Ред. Болезни и смертность в Африке к югу от Сахары . 2-е изд. Вашингтон, округ Колумбия: публикации Всемирного банка; 2006. 305-28.
Дрис Д.Л., Экснер Д.В., Домански М.Дж., Гринберг Б., Стивенсон Л.В. Прогностические последствия почечной недостаточности у бессимптомных и симптоматических пациентов с систолической дисфункцией левого желудочка. Джам Колл Кардиол . 2000 г. 1. 35 (3): 681-9. [Медлайн].
Фонаров Г.К., Адамс К.Ф. мл., Абрахам В.Т., Янси К.В., Боскардин В.Дж., Научно-консультативный комитет ADHERE и др.Стратификация риска госпитальной смертности при острой декомпенсированной сердечной недостаточности: классификация и анализ дерева регрессии. ЯМА . 2005 2 февраля. 293 (5): 572-80. [Медлайн].
Леви Д., Кенчайа С., Ларсон М.Г. и др. Долгосрочные тенденции частоты сердечной недостаточности и выживаемости с ней. N Engl J Med . 2002 31 октября. 347 (18): 1397-402. [Медлайн].
Лукас С., Джонсон В., Гамильтон М.А. и др. Отсутствие заложенности носа обеспечивает хорошую выживаемость, несмотря на предыдущие симптомы сердечной недостаточности IV класса. Am Heart J . 2000 декабрь 140 (6): 840-7. [Медлайн].
Макинтайр К., Кейпвелл С., Стюарт С. и др. Доказательства улучшения прогноза при сердечной недостаточности: тенденции летальности у 66 547 пациентов, госпитализированных в период с 1986 по 1995 год. Circulation . 2000 Сен 5. 102 (10): 1126-31. [Медлайн].
Ketchum ES, Леви WC. Установление прогноза при сердечной недостаточности: многомаркерный подход. Программа Cardiovasc Dis . 2011 сентябрь-октябрь.54 (2): 86-96. [Медлайн].
van Diepen S, Bakal JA, McAlister FA, Ezekowitz JA. Смертность и повторная госпитализация пациентов с сердечной недостаточностью, фибрилляцией предсердий или ишемической болезнью сердца, перенесших некардиальные операции: анализ 38 047 пациентов. Тираж . 2011 июля 19, 124 (3): 289-96. [Медлайн].
Бурси Ф., МакНаллан С.М., Редфилд М.М. и др. Легочное давление и смерть при сердечной недостаточности: исследование сообщества. Джам Колл Кардиол .2012 17 января. 59 (3): 222-31. [Медлайн]. [Полный текст].
Ho JE, Liu C, Lyass A, et al. Галектин-3, маркер сердечного фиброза, позволяет прогнозировать сердечную недостаточность у населения. Джам Колл Кардиол . 2012 октября 2. 60 (14): 1249-56. [Медлайн].
Nayar P, Yu F, Chandak A, Kan GL, Lowes B, Apenteng BA. Факторы риска госпитальной смертности у пациентов с сердечной недостаточностью: имеет ли значение местность, плательщик или источник госпитализации? J Здоровье в сельской местности .2018 декабрь 34 (1): 103-8. [Медлайн].
Dunlay SM, Eveleth JM, Shah ND, McNallan SM, Roger VL. Приверженность к лечению среди местных пациентов с сердечной недостаточностью. Mayo Clin Proc . 2011 Апрель, 86 (4): 273-81. [Медлайн]. [Полный текст].
DeWalt DA, Schillinger D, Ruo B и др. Многосайтовое рандомизированное исследование одноразового и многосессионного вмешательства по самопомощи с учетом грамотности пациентов с сердечной недостаточностью. Тираж .2012, 12 июня. 125 (23): 2854-62. [Медлайн].
Lainscak M, Cleland JG, Lenzen MJ, Follath F, Komajda M, Swedberg K. Международные вариации лечения и сопутствующие заболевания систолической дисфункции левого желудочка: данные EuroHeart Failure Survey. Eur J Heart Fail . 2007 марта 9 (3): 292-9. [Медлайн].
Panjrath G, Ahmed A. Диагностика и лечение сердечной недостаточности у пожилых людей. Клиника сердечной недостаточности .2017 июл.13 (3): 427-44. [Медлайн].
Стивенсон Л.В., Перлофф Дж. Ограниченная надежность физических признаков для оценки гемодинамики при хронической сердечной недостаточности. ЯМА . 1989, 10 февраля. 261 (6): 884-8. [Медлайн].
Steinhart B, Thorpe KE, Bayoumi AM, Moe G, Januzzi JL Jr, Mazer CD. Улучшение диагностики острой сердечной недостаточности с помощью проверенной модели прогнозирования. Джам Колл Кардиол . 2009 Октябрь 13, 54 (16): 1515-21.[Медлайн].
[Рекомендации] Янси К.В., Джессап М., Бозкурт Б. и др. Обновление рекомендаций ACC / AHA / HFSA по лечению сердечной недостаточности от 2013 г., посвященное ACC / AHA / HFSA: Отчет Американского колледжа кардиологии / Американской кардиологической ассоциации по клиническим практическим рекомендациям и Американского общества сердечной недостаточности. Тираж . 2017 8 августа. 136 (6): e137-e161. [Медлайн]. [Полный текст].
Рич М.В., МакШерри Ф., Уиллифорд В.О., Юсуф С., для исследовательской группы Digitalis.Влияние возраста на смертность, госпитализацию и реакцию на дигоксин у пациентов с сердечной недостаточностью: исследование DIG. Джам Колл Кардиол . 2001 Сентябрь 38 (3): 806-13. [Медлайн].
[Рекомендации] Янси К.В., Джессап М., Бозкурт Б. и др. Для членов комитета по написанию. ACC / AHA / HFSA 2016 сосредоточили внимание на обновленной информации о новой фармакологической терапии сердечной недостаточности: обновленное руководство ACCF / AHA по лечению сердечной недостаточности от 2013 года: отчет Целевой группы Американского колледжа кардиологов / Американской кардиологической ассоциации по клиническим практическим рекомендациям и Американское общество сердечной недостаточности. Тираж . 2016 27 сентября. 134 (13): e282-93. [Медлайн]. [Полный текст].
Бадве С.В., Робертс М.А., Хоули С.М. и др. Эффекты антагонистов бета-адренорецепторов у пациентов с хронической болезнью почек: систематический обзор и метаанализ. Джам Колл Кардиол . 2011 6 сентября. 58 (11): 1152-61. [Медлайн].
Майзел А.С., Кришнасвами П., Новак Р.М. и др., Для исследователей многонационального исследования «Неправильное дыхание». Быстрое определение натрийуретического пептида B-типа в экстренной диагностике сердечной недостаточности. N Engl J Med . 2002 18 июля. 347 (3): 161-7. [Медлайн].
Януцци JL мл., Камарго Калифорния, Анваруддин С. и др. N-терминальное исследование одышки с про-BNP в исследовании отделения неотложной помощи (PRIDE). Ам Дж. Кардиол . 2005, 15 апреля. 95 (8): 948-54. [Медлайн].
Ван К.С., Фитцджеральд Дж. М., Шульцер М., Мак Э., Аяс Н. Т.. У этого пациента с одышкой в отделении неотложной помощи застойная сердечная недостаточность? ЯМА .2005 Октябрь 19, 294 (15): 1944-56. [Медлайн].
Groenveld HF, Januzzi JL, Damman K и др. Анемия и смертность у пациентов с сердечной недостаточностью: систематический обзор и метаанализ. Джам Колл Кардиол . 2008 сентябрь 2. 52 (10): 818-27. [Медлайн].
Grote Beverborg N, van Veldhuisen DJ, van der Meer P. Анемия при сердечной недостаточности: все еще актуально ?. JACC Heart Fail . 8 ноября 2017 г. [Medline].
Бидкар А, Парих Р., Дешмук П.Сердечная недостаточность и дефицит железа. Дж. Ассошиэйтед врачей Индия . 2017 Ноябрь 65 (11): 79-80. [Медлайн].
Hoes MF, Grote Beverborg N, Kijlstra JD, et al. Дефицит железа снижает сократительную способность кардиомиоцитов человека из-за снижения функции митохондрий. Eur J Heart Fail . 2018 27 февраля. [Medline].
Rocha BML, Cunha GJL, Menezes Falcao LF. Бремя дефицита железа при сердечной недостаточности: терапевтический подход. Джам Колл Кардиол . 2018 20 февраля. 71 (7): 782-93. [Медлайн].
Майзел А.С., МакКорд Дж., Новак Р.М. и др., Для исследователей многонационального исследования «Неправильное дыхание». Прикроватный натрийуретический пептид B-типа в неотложной диагностике сердечной недостаточности со сниженной или сохраненной фракцией выброса. Результаты многонационального исследования «Неправильное дыхание». Джам Колл Кардиол . 4 июня 2003 г. 41 (11): 2010-7. [Медлайн].
Januzzi JL, van Kimmenade R, Lainchbury J, et al.Тестирование NT-proBNP для диагностики и краткосрочного прогноза острой дестабилизированной сердечной недостаточности: международный объединенный анализ 1256 пациентов: Международное совместное исследование NT-proBNP. Евро Сердце J . 2006 27 февраля (3): 330-7. [Медлайн].
Маэда К., Цутамото Т., Вада А., Хисанага Т., Киношита М. Натрийуретический пептид плазмы мозга как биохимический маркер высокого конечного диастолического давления левого желудочка у пациентов с симптоматической дисфункцией левого желудочка. Am Heart J . 1998 Май. 135 (5 Пет 1): 825-32. [Медлайн].
Fisher C, Berry C, Blue L, Morton JJ, McMurray J. N-концевой натрийуретический пептид про B типа, но не новый предполагаемый сердечный гормон релаксин, предсказывает прогноз у пациентов с хронической сердечной недостаточностью. Сердце . 2003 августа 89 (8): 879-81. [Медлайн]. [Полный текст].
Hall C, Rouleau JL, Moye L, et al. N-концевой предсердный натрийуретический фактор. Независимый предиктор отдаленного прогноза инфаркта миокарда. Тираж . 1994 Май. 89 (5): 1934-42. [Медлайн].
Андерссон Б., Холл С. N-концевой предсердный натрийуретический пептид и прогноз у пациентов с сердечной недостаточностью и сохраненной систолической функцией. J Card Fail . 2000 Сентябрь 6 (3): 208-13. [Медлайн].
Чен Х. Х., Бернетт Дж. Натрийуретические пептиды в патофизиологии застойной сердечной недостаточности. Curr Cardiol Rep . 2000 Май. 2 (3): 198-205. [Медлайн].
Ченг В., Казанагра Р., Гарсия А. и др.Быстрый прикроватный тест на пептид B-типа позволяет прогнозировать результаты лечения пациентов, госпитализированных по поводу декомпенсированной сердечной недостаточности: пилотное исследование. Джам Колл Кардиол . 2001 г., 37 (2): 386-91. [Медлайн].
Cowie MR, Struthers AD, Wood DA, et al. Значение натрийуретических пептидов в оценке пациентов с возможной новой сердечной недостаточностью в первичной медико-санитарной помощи. Ланцет . 1997, 8 ноября. 350 (9088): 1349-53. [Медлайн].
Дао К., Кришнасвами П., Казанегра Р. и др.Применение натрийуретического пептида B-типа в диагностике застойной сердечной недостаточности в условиях неотложной помощи. Джам Колл Кардиол . 2001 г., 37 (2): 379-85. [Медлайн].
Маэда К., Цутамото Т., Вада А., Хисанага Т., Киношита М. Натрийуретический пептид плазмы мозга как биохимический маркер высокого конечного диастолического давления левого желудочка у пациентов с симптоматической дисфункцией левого желудочка. Am Heart J . 1998 Май. 135 (5 пт 1): 825-32. [Медлайн].
Майзель А.С., Кун Дж., Хоуп Дж. И др. Быстрый прикроватный тест на натрийуретический пептид головного мозга точно предсказывает сердечную функцию у пациентов, направленных на эхокардиографию. Am Heart J . 2001. 141: 374-9.
Masson S, Vago T, Baldi G и др. Сравнительное измерение N-концевого натрийуретического пептида головного мозга и натрийуретического пептида головного мозга у амбулаторных пациентов с сердечной недостаточностью. Clin Chem Lab Med . 2002 г., 40 (8): 761-3.[Медлайн].
Song BG, Jeon ES, Kim YH и др. Корреляция между уровнями N-концевого натрийуретического пептида про-B-типа и степенью сердечной недостаточности. Korean J Intern Med . 2005 20 марта (1): 26-32. [Медлайн].
Хоббс Ф. Д., Дэвис Р. К., Роальф А. К., Харе Р., Дэвис М. К., Кенкре Дж. Э.. Надежность анализа N-терминального про-мозгового натрийуретического пептида в диагностике сердечной недостаточности: когортное исследование в репрезентативных популяциях и группах высокого риска. BMJ . 2002, 22 июня. 324 (7352): 1498. [Медлайн]. [Полный текст].
Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC Jr. Концентрация натрийуретического пептида в плазме мозга: влияние возраста и пола. Джам Колл Кардиол . 2002 Сентябрь 4. 40 (5): 976-82. [Медлайн].
Сент-Питер СП, Хартли Г.Г., Мураками М.М., Apple FS. Натрийуретический пептид B-типа (BNP) и N-концевой про-BNP у пациентов с ожирением без сердечной недостаточности: связь с индексом массы тела и операцией обходного желудочного анастомоза. Clin Chem . 2006 апр. 52 (4): 680-5. [Медлайн].
Ривера М., Кортес Р., Сальвадор А. и др. Субъекты с ожирением и сердечной недостаточностью имеют более низкие уровни N-концевого про-мозгового натрийуретического пептида в плазме независимо от этиологии. Eur J Heart Fail . 2005 Декабрь 7 (7): 1168-70. [Медлайн].
Hermann-Arnhof KM, Hanusch-Enserer U, Kaestenbauer T, et al. N-терминальный натрийуретический пептид про-B-типа как индикатор возможного сердечно-сосудистого заболевания у лиц с тяжелым ожирением: сравнение с пациентами на разных стадиях сердечной недостаточности. Clin Chem . 2005, январь, 51 (1): 138-43. [Медлайн].
Seino Y, Ogawa A, Yamashita T, et al. Применение измерений NT-proBNP и BNP в кардиологической помощи: более точный маркер для обнаружения и оценки сердечной недостаточности. Eur J Heart Fail . 2004 15 марта. 6 (3): 295-300. [Медлайн].
Colucci WS, Elkayam U, Horton DP, et al. Внутривенное введение натрийуретического пептида несиритида при лечении декомпенсированной застойной сердечной недостаточности.Группа изучения несиритида. N Engl J Med . 2000 27 июля. 343 (4): 246-53. [Медлайн].
Ezekowitz JA, Hernandez AF, Starling RC, et al. Стандартизация помощи при острой декомпенсированной сердечной недостаточности в большом мегатриале: подход к острым исследованиям клинической эффективности несиритида у субъектов с декомпенсированной сердечной недостаточностью (ASCEND-HF). Am Heart J . 2009 Февраль 157 (2): 219-28. [Медлайн].
Миллс Р.М., ЛеДжемтель Т.Х., Хортон Д.П. и др.Устойчивые гемодинамические эффекты инфузии несиритида (человеческого натрийуретического пептида b-типа) при сердечной недостаточности: рандомизированное двойное слепое плацебо-контролируемое клиническое исследование. Исследовательская группа Natrecor. Джам Колл Кардиол . 1999 июл. 34 (1): 155-62. [Медлайн].
Silver MA, Horton DP, Ghali JK, Elkayam U. Влияние несиритида по сравнению с добутамином на краткосрочные результаты лечения пациентов с острой декомпенсированной сердечной недостаточностью. Джам Колл Кардиол .2002 6 марта. 39 (5): 798-803. [Медлайн].
Миллер В.Л., Хартман К.А., Бурритт М.Ф., Борхесон Д.Д., Бернетт Дж.С. мл., Джаффе А.С. Биомаркеры во время и после лечения инфузией несиритида у пациентов с декомпенсированной хронической сердечной недостаточностью. Clin Chem . 2005 Март 51 (3): 569-77. [Медлайн].
Фитцджеральд Р.Л., Кремо Р., Гардетто Н. и др. Влияние несиритида в сочетании со стандартной терапией на сывороточные концентрации натрийуретических пептидов у пациентов, госпитализированных по поводу декомпенсированной застойной сердечной недостаточности. Am Heart J . 2005 Сентябрь 150 (3): 471-7. [Медлайн].
Михельс В.В., Молл П.П., Миллер Ф.А. и др. Частота семейной дилатационной кардиомиопатии у ряда пациентов с идиопатической дилатационной кардиомиопатией. N Engl J Med . 1992, 9 января. 326 (2): 77-82. [Медлайн].
Бейг М.К., Голдман Дж. Х., Кафорио А. Л., Кунар А. С., Килинг П. Дж., Маккенна В. Дж.. Семейная дилатационная кардиомиопатия: сердечные аномалии часто встречаются у бессимптомных родственников и могут указывать на раннее заболевание. Джам Колл Кардиол . 1998 31 января (1): 195-201. [Медлайн].
Grunig E, Tasman JA, Kucherer H, Franz W., Kubler W, Katus HA. Частота и фенотипы семейной дилатационной кардиомиопатии. Джам Колл Кардиол . 1998 31 января (1): 186-94. [Медлайн].
McNally E, MacLeod H, Dellefave-Castillo L, et al. Аритмогенная кардиомиопатия правого желудочка. Джин Ревьюз . 1993. [Medline]. [Полный текст].
Cheitlin MD, Alpert JS, Armstrong WF и др.Рекомендации ACC / AHA по клиническому применению эхокардиографии. Отчет рабочей группы Американского колледжа кардиологов / Американской кардиологической ассоциации по практическим рекомендациям (Комитет по клиническому применению эхокардиографии). Разработан в сотрудничестве с Американским обществом эхокардиографии. Тираж . 1997 18 марта. 95 (6): 1686-744. [Медлайн].
Patel AR, Alsheikh-Ali AA, Mukherjee J, et al. 3D-эхокардиография для оценки корреляции давления в правом предсердии при острой декомпенсированной сердечной недостаточности с инвазивной гемодинамикой. JACC Cardiovasc Imaging . 2011 Сентябрь 4 (9): 938-45. [Медлайн].
Prior D, Coller J. Эхокардиография при сердечной недостаточности — руководство для общей практики. Врач Aust Fam . 2010 декабрь 39 (12): 904-9. [Медлайн].
Киркпатрик JN, Wiegers SE, Lang RM. Вспомогательные устройства для левого желудочка и другие устройства для терминальной стадии сердечной недостаточности: полезность эхокардиографии. Curr Cardiol Rep . 2010 май. 12 (3): 257-64.[Медлайн].
Abraham J, Abraham TP. Роль эхокардиографии в оценке гемодинамики при сердечной недостаточности. Клиника сердечной недостаточности . 2009 Апрель 5 (2): 191-208. [Медлайн].
Гупта В.А., Нанда Северная Каролина, Соррелл В.Л. Роль эхокардиографии в диагностической оценке и этиологии сердечной недостаточности у пожилых людей: затемнение, количественная оценка и исправление. Клиника сердечной недостаточности . 2017 июл.13 (3): 445-66. [Медлайн].
Meersch M, Schmidt C, Zarbock A.Эхофизиология: чреспищеводный эхо-зонд в качестве неинвазивного катетера Свана-Ганца. Curr Opin Anaesthesiol . 2016 29 февраля (1): 36-45. [Медлайн].
Ким Р.Дж., Ву Э., Рафаэль А. и др. Использование магнитно-резонансной томографии с контрастным усилением для выявления обратимой дисфункции миокарда. N Engl J Med . 2000 16 ноября. 343 (20): 1445-53. [Медлайн].
[Рекомендации] Ричи Дж. Л., Бейтман Т. М., Боноу Р. О. и др. Руководство по клиническому использованию радионуклидной визуализации сердца.Отчет Американского колледжа кардиологии / Американской кардиологической ассоциации по оценке диагностических и терапевтических сердечно-сосудистых процедур (Комитет по радионуклидной визуализации), разработанный в сотрудничестве с Американским обществом ядерной кардиологии. Джам Колл Кардиол . 1995 25 февраля (2): 521-47. [Медлайн].
Taillefer R, DePuey EG, Udelson JE, Beller GA, Latour Y, Reeves F. Сравнительная диагностическая точность изображений Tl-201 и Tc-99m sestamibi SPECT (перфузия и ECG-gated SPECT) при обнаружении ишемической болезни сердца у женщин. Джам Колл Кардиол . 1997, 29 января (1): 69-77. [Медлайн].
Боноу Р.О., Маурер Г., Ли К.Л. и др., Для исследователей исследования STICH. Жизнеспособность миокарда и выживаемость при ишемической дисфункции левого желудочка. N Engl J Med . 2011 28 апреля. 364 (17): 1617-25. [Медлайн].
Бинанай К., Калифф Р.М., Хассельблад В. и др., Для исследователей ESCAPE и координаторов исследований ESCAPE. Оценка исследования застойной сердечной недостаточности и эффективности катетеризации легочной артерии: исследование ESCAPE. ЯМА . 2005 Октябрь 5. 294 (13): 1625-33. [Медлайн].
[Рекомендации] Дикштейн К., Вардас П.Е., Ауриккио А. и др. Для Целевой группы по острой сердечной недостаточности Европейского общества кардиологов. Специальное обновление рекомендаций ESC по аппаратной терапии при сердечной недостаточности, 2010 г.: обновление рекомендаций ESC 2008 г. по диагностике и лечению острой и хронической сердечной недостаточности, а также рекомендаций ESC 2007 г. по сердечной и ресинхронизирующей терапии. Разработано при особом участии Ассоциации сердечной недостаточности и Европейской ассоциации сердечного ритма. Евро Сердце J . 2010 31 ноября (21): 2677-87. [Медлайн]. [Полный текст].
Горький Т., Вестерхайд Н, Принц С. и др. Дыхание Чейна-Стокса и обструктивное апноэ во сне являются независимыми факторами риска злокачественных желудочковых аритмий, требующих соответствующей терапии кардиовертер-дефибриллятор у пациентов с застойной сердечной недостаточностью. Евро Сердце J . 2011 января, 32 (1): 61-74. [Медлайн].
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R.Кардиоренальный синдром. Джам Колл Кардиол . 2008, 4 ноября. 52 (19): 1527-39. [Медлайн].
House AA, Haapio M, Lassus J, Bellomo R, Ronco C. Терапевтические стратегии сердечной недостаточности при кардиоренальных синдромах. Ам Дж. Почек . 2010 Октябрь 56 (4): 759-73. [Медлайн].
Джамузис Дж., Батлер Дж., Старлинг Р.К. и др. Влияние инфузии дофамина на функцию почек у госпитализированных пациентов с сердечной недостаточностью: результаты исследования допамина при острой декомпенсированной сердечной недостаточности (DAD-HF). J Card Fail . 2010 16 (12): 922-30. [Медлайн].
Pleister AP, Baliga RR, Haas GJ. Острое исследование клинической эффективности несиритида при декомпенсированной сердечной недостаточности: несиритид редукс. Curr Heart Fail Rep . 2011 Сентябрь 8 (3): 226-32. [Медлайн].
О’Коннор С.М., Старлинг Р.С., Эрнандес А.Ф. и др. Эффект несиритида у пациентов с острой декомпенсированной сердечной недостаточностью. N Engl J Med . 2011 июл 7. 365 (1): 32-43.[Медлайн].
Konstam MA, Gheorghiade M, Burnett JC Jr и др., За «Эффективность антагонизма вазопрессина в исследовании исходов сердечной недостаточности с участием исследователей толваптана (ЭВЕРЕСТ)». Эффекты перорального толваптана у пациентов, госпитализированных по поводу обострения сердечной недостаточности: испытание результатов ЭВЕРЕСТ. ЯМА . 2007 28 марта, 297 (12): 1319-31. [Медлайн].
Мэсси Б.М., О’Коннор К.М., Метра М. и др., Для исследователей и комитетов PROTECT.Ролофиллин, антагонист аденозиновых A1-рецепторов, при острой сердечной недостаточности. N Engl J Med . 2010 октябрь 7. 363 (15): 1419-28. [Медлайн].
Бадве С.В., Робертс М.А., Хоули С.М. и др. Эффекты антагонистов бета-адренорецепторов у пациентов с хронической болезнью почек: систематический обзор и метаанализ. Джам Колл Кардиол . 2011 6 сентября. 58 (11): 1152-61. [Медлайн].
Рой Д., Таладжич М., Наттель С. и др., Исследователи фибрилляции предсердий и застойной сердечной недостаточности.Контроль ритма по сравнению с контролем частоты при фибрилляции предсердий и сердечной недостаточности. N Engl J Med . 19 июня 2008 г. 358 (25): 2667-77. [Медлайн].
Уилтон С.Б., Фундитус А., Гали В.А. и др. Метаанализ эффективности и безопасности катетерной аблации фибрилляции предсердий у пациентов с систолической дисфункцией левого желудочка и без нее. Ам Дж. Кардиол . 2010 г. 1. 106 (9): 1284-91. [Медлайн].
Макдональд М.Р., Коннелли Д.Т., Хокинс Н.М. и др.Радиочастотная абляция для стойкой фибрилляции предсердий у пациентов с тяжелой сердечной недостаточностью и тяжелой систолической дисфункцией левого желудочка: рандомизированное контролируемое исследование. Сердце . 2011 Май. 97 (9): 740-7. [Медлайн].
Чен Ю.М., Ли З.Б., Чжу М., Цао Ю.М. Влияние тренировок на ремоделирование левого желудочка у пациентов с сердечной недостаточностью: обновленный метаанализ рандомизированных контролируемых исследований. Int J Clin Pract . 2012 августа 66 (8): 782-91.[Медлайн].
Mozaffarian D, Lemaitre RN, King IB, et al. Циркулирующие длинноцепочечные жирные кислоты омега-3 и частота застойной сердечной недостаточности у пожилых людей: исследование сердечно-сосудистой системы: когортное исследование. Энн Интерн Мед. . 2011, 2 августа. 155 (3): 160-70. [Медлайн].
Маркиоли Р., Левантеси Дж., Силлетта М.Г. и др. Для исследователей GISSI-HF. Эффект n-3 полиненасыщенных жирных кислот и розувастатина у пациентов с сердечной недостаточностью: результаты исследования GISSI-HF. Эксперт Rev Cardiovasc Ther . 2009 июл.7 (7): 735-48. [Медлайн].
Армстронг П. У., Пиеске Б., Анстром К. Дж. И др. Для исследовательской группы VICTORIA. Верицигуат у пациентов с сердечной недостаточностью и сниженной фракцией выброса. N Engl J Med . 2020 14 мая. 382 (20): 1883-93. [Медлайн]. [Полный текст].
Сведберг К., Комайда М., Бём М. и др., Для исследователей SHIFT. Ивабрадин и исходы при хронической сердечной недостаточности (SHIFT): рандомизированное плацебо-контролируемое исследование. Ланцет . 11 сентября 2010 г. 376 (9744): 875-85. [Медлайн]. [Полный текст].
Borer JS, Bohm M, Ford I, et al, для исследователей SHIFT. Влияние ивабрадина на повторную госпитализацию по поводу обострения сердечной недостаточности у пациентов с хронической систолической сердечной недостаточностью: исследование SHIFT. Евро Сердце J . 2012 ноябрь 33 (22): 2813-20. [Медлайн]. [Полный текст].
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США. FDA одобрило новый препарат для лечения сердечной недостаточности.7 июля 2015 г. Доступно по адресу http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm453845.htm. Доступ: 8 июля 2015 г.
МакМюррей Дж. Дж., Пакер М., Десаи А. С. и др., Исследователи и комитеты PARADIGM-HF. Ингибирование ангиотензин-неприлизина по сравнению с эналаприлом при сердечной недостаточности. N Engl J Med . 2014 11 сентября. 371 (11): 993-1004. [Медлайн]. [Полный текст].
Solomon SD, Zile M, Pieske B и др., За проспективное сравнение ARNI с ARB при лечении сердечной недостаточности с сохраненной фракцией выброса (PARAMOUNT).Ингибитор рецептора ангиотензина неприлизина LCZ696 при сердечной недостаточности с сохраненной фракцией выброса: двойное слепое рандомизированное контролируемое исследование фазы 2. Ланцет . 20 октября 2012 г. 380 (9851): 1387-95. [Медлайн].
Entresto (сакубитрил / валсартан) [вкладыш в упаковке]. Восточный Ганновер, Нью-Джерси: Novartis Pharmaceuticals Corp., октябрь 2019 г. Доступно на [Полный текст].
Мэсси Б.М., Коллинз Дж.Ф., Аммон С.Е. и др., Для исследователей испытаний WATCH.Рандомизированное исследование варфарина, аспирина и клопидогреля у пациентов с хронической сердечной недостаточностью: исследование варфарина и антитромбоцитарной терапии при хронической сердечной недостаточности (WATCH). Тираж . 2009 31 марта, 119 (12): 1616-24. [Медлайн].
Freeman JV, Ян Дж., Сунг Ш., Хлатки М. А., Go AS. Эффективность и безопасность дигоксина у современных взрослых с систолической сердечной недостаточностью. Результаты Circ Cardiovasc Qual . 2013 г. 1. 6 (5): 525-33. [Медлайн].
Konstantinou DM, Karvounis H, Giannakoulas G. Дигоксин при сердечной недостаточности с пониженной фракцией выброса: фактор риска или маркер риска. Кардиология . 2016. 134 (3): 311-9. [Медлайн].
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Тенденции распространенности и исходов сердечной недостаточности с сохраненной фракцией выброса. N Engl J Med . 2006 20 июля. 355 (3): 251-9. [Медлайн].
Хогг К., Сведберг К., МакМюррей Дж.Сердечная недостаточность с сохраненной систолической функцией левого желудочка; эпидемиология, клинические характеристики и прогноз. Джам Колл Кардиол . 2004 г., 4 февраля. 43 (3): 317-27. [Медлайн].
Георгиад М., Абрахам В.Т., Альберт Н.М. и др. Для исследователей и координаторов OPTIMIZE-HF. Систолическое артериальное давление при поступлении, клинические характеристики и исходы у пациентов, госпитализированных с острой сердечной недостаточностью. ЯМА . 2006 г. 8 ноября. 296 (18): 2217-26.[Медлайн].
Масип Дж., Роке М., Санчес Б., Фернандес Р., Субирана М., Экспозито Дж. А. Неинвазивная вентиляция при остром кардиогенном отеке легких: систематический обзор и метаанализ. ЯМА . 2005 28 декабря. 294 (24): 3124-30. [Медлайн].
Питер Дж. В., Моран Дж. Л., Филлипс-Хьюз Дж., Грэм П., Берстен А. Д.. Влияние неинвазивной вентиляции с положительным давлением (NIPPV) на смертность пациентов с острым кардиогенным отеком легких: метаанализ. Ланцет . 2006 г., 8 апреля. 367 (9517): 1155-63. [Медлайн].
Винк Дж. С., Азеведо Л. Ф., Коста-Перейра А, Антонелли М., Вятт Дж. Эффективность и безопасность неинвазивной вентиляции в лечении острого кардиогенного отека легких — систематический обзор и метаанализ. Crit Care . 2006. 10 (2): R69. [Медлайн]. [Полный текст].
Vital FM, Saconato H, Ladeira MT, et al. Неинвазивная вентиляция с положительным давлением (CPAP или двухуровневая NPPV) при кардиогенном отеке легких. Кокрановская база данных Syst Rev . 2008 16 июля. CD005351. [Медлайн].
Maeder MT, Kaye DM. Сердечная недостаточность с нормальной фракцией выброса левого желудочка. Джам Колл Кардиол . 2009 17 марта. 53 (11): 905-18. [Медлайн].
Gray A, Goodacre S, Newby DE и др., Для исследователей 3CPO. Неинвазивная вентиляция при остром кардиогенном отеке легких. N Engl J Med . 2008 г. 10 июля. 359 (2): 142-51. [Медлайн].
CONSENSUS Trial Study Group.Влияние эналаприла на смертность при тяжелой застойной сердечной недостаточности. Результаты совместного исследования выживания эналаприла в Северной Скандинавии (CONSENSUS). N Engl J Med . 4 июня 1987 г.. 316 (23): 1429-35. [Медлайн].
Юсуф С., Питт Б., Дэвис К.Э., Худ В.Б., Кон Дж. Н., для следователей СОЛВД. Влияние эналаприла на выживаемость у пациентов со сниженной фракцией выброса левого желудочка и застойной сердечной недостаточностью. N Engl J Med . 1 августа 1991 г. 325 (5): 293-302.[Медлайн].
Zannad F, Alla F, Dousset B, Perez A, Pitt B. Ограничение чрезмерного оборота внеклеточного матрикса может способствовать повышению выживаемости от терапии спиронолактоном у пациентов с застойной сердечной недостаточностью: выводы из рандомизированного исследования по оценке альдактона (RALES). Рейлс следователи. Тираж . 2000, 28 ноября. 102 (22): 2700-6. [Медлайн].
Sodhi N, Lasala JM. Механическая поддержка кровообращения при острой декомпенсированной сердечной недостаточности и шоке. Интервал Кардиол Клин . 2017 Июль 6 (3): 387-405. [Медлайн].
Веласкес Э.Дж., Ли К.Л., Джонс Р.Х. и др. Для исследователей STICHES. Аортокоронарное шунтирование у пациентов с ишемической кардиомиопатией. N Engl J Med . 2016 21 апреля. 374 (16): 1511-20. [Медлайн].
Буско М. Клевидипин перспективен при острой сердечной недостаточности с высоким АД. Медицинские новости Medscape от WebMD. 10 февраля 2014 г. Доступно по адресу http: //www.medscape.com / viewarticle / 820377. Доступ: 18 февраля 2014 г.
Felker GM, Lee KL, Bull DA и др., Для сети клинических исследований сердечной недостаточности NHLBI. Диуретические стратегии у пациентов с острой декомпенсированной сердечной недостаточностью. N Engl J Med . 2011 г., 3 марта. 364 (9): 797-805. [Медлайн].
Лю ПП. Кардиоренальный синдром при сердечной недостаточности: взгляд кардиолога. Банка J Cardiol . 24 июля 2008 г., приложение B: 25B-9B. [Медлайн]. [Полный текст].
Kramer BK, Schweda F, Riegger GA. Лечение диуретиками и резистентность к диуретикам при сердечной недостаточности. Ам Дж. Мед . 1999 Январь 106 (1): 90-6. [Медлайн].
Neuberg GW, Miller AB, O’Connor CM, et al, для PRAISE Investigators. Проспективная рандомизированная оценка выживаемости с применением амлодипина. Устойчивость к диуретикам позволяет прогнозировать смертность у пациентов с тяжелой сердечной недостаточностью. Am Heart J . 2002 Июль 144 (1): 31-8. [Медлайн].
Costanzo MR, Saltzberg M, O’Sullivan J, Sobotka P.Ранняя ультрафильтрация у пациентов с декомпенсированной сердечной недостаточностью и резистентностью к диуретикам. Джам Колл Кардиол . 2005 декабрь 6. 46 (11): 2047-51. [Медлайн].
Янг Дж. Б., Абрахам В. Т., Стивенсон Л. В. и др. Результаты исследования VMAC: вазодилатация в лечении острой застойной сердечной недостаточности. Цирк . 2000 28 ноября. 102 (22): a2794. [Полный текст].
Комитет по публикациям исследователей VMAC (Расширение сосудов в лечении острой ЗСН).Внутривенное введение несиритида в сравнении с нитроглицерином для лечения декомпенсированной застойной сердечной недостаточности: рандомизированное контролируемое исследование. ЯМА . 2002, 27 марта. 287 (12): 1531-40. [Медлайн].
Костанцо М.Р., Джессап М. Лечение застойных явлений при сердечной недостаточности с помощью диуретиков и экстракорпоральной терапии: влияние на симптомы, функцию почек и прогноз. Heart Fail Ред. . 2012 марта 17 (2): 313-24. [Медлайн].
Костанцо М.Р., Гуглин М.Э., Зальцберг М.Т. и др. Для исследователей UNLOAD Trial.Ультрафильтрация по сравнению с внутривенными диуретиками у пациентов, госпитализированных по поводу острой декомпенсированной сердечной недостаточности. Джам Колл Кардиол . 2007 13 февраля. 49 (6): 675-83. [Медлайн].
Следователи и комитеты CIBIS. Рандомизированное исследование бета-блокады при сердечной недостаточности. Исследование сердечной недостаточности бисопролола (CIBIS). Исследователи и комитеты CIBIS. Тираж . 1994 Октябрь 90 (4): 1765-73. [Медлайн].
Чаудри С.И., Маттера Дж. А., Кертис Дж. П. и др.Телемониторинг у больных с сердечной недостаточностью. N Engl J Med . 2010 декабрь 9. 363 (24): 2301-9. [Медлайн].
Келер Ф., Винклер С., Шибер М. и др., Телемедицинский интервенционный мониторинг у исследователей сердечной недостаточности. Влияние удаленного телемедицинского управления на смертность и госпитализацию амбулаторных пациентов с хронической сердечной недостаточностью: телемедицинский интервенционный мониторинг в исследовании сердечной недостаточности. Тираж . 2011 3 мая. 123 (17): 1873-80.[Медлайн].
Сообщество P&T. FDA одобрило первое имплантируемое беспроводное устройство с дистанционным мониторингом для измерения давления в легочной артерии у некоторых пациентов с сердечной недостаточностью (пресс-релиз). Доступно по адресу https://www.ptcommunity.com/news/20170422/fda-approves-first-implantable-device-remote-monitoring-measure-pa-pressure-heart. 28 мая 2014 г .; Доступ: 2 июня 2014 г.
O’Riordan M. FDA одобрило первое имплантируемое устройство для дистанционного наблюдения за пациентами с сердечной недостаточностью.Heartwire. Доступно на http://www.medscape.com/viewarticle/825805. 28 мая 2014 г .; Доступ: 2 июня 2014 г.
Abraham W.T., Adamson PB, Bourge RC и др. Для группы экспериментальных исследований CHAMPION. Беспроводной мониторинг гемодинамики легочной артерии при хронической сердечной недостаточности: рандомизированное контролируемое исследование. Ланцет . 19 февраля 2011 г. 377 (9766): 658-66. [Медлайн].
Eisen HJ, Kobashigawa J, Keogh A, et al, для исследователей кардиологического исследования микофенолата мофетила.Трехлетние результаты рандомизированного двойного слепого контролируемого исследования микофенолата мофетила по сравнению с азатиоприном у реципиентов сердечного трансплантата. J Пересадка легкого сердца . 2005 Май. 24 (5): 517-25. [Медлайн].
Янси К.В., Лопатин М., Стивенсон Л.В., Де Марко Т., Фонаров Г.К., для Научного консультативного комитета и исследователей ADHERE. Клиническая картина, ведение и результаты госпитализации пациентов, поступивших с острой декомпенсированной сердечной недостаточностью с сохраненной систолической функцией: отчет из базы данных Национального реестра острой декомпенсированной сердечной недостаточности (ADHERE). Джам Колл Кардиол . 2006 г. 3 января, 47 (1): 76-84. [Медлайн].
Новости сердечного ритма. FDA одобряет расширенные показания для определенных кардиостимуляторов и дефибрилляторов, используемых для лечения сердечной недостаточности. Доступно по адресу https://cardiacrhythmnews.com/fda-approves-expanded-indication-for- specific-pacemakers-and-defibrillators-used-to-treat-heart-failure/. 15 апреля 2014 г .; Доступ: 15 апреля 2014 г.
Стайлз С. FDA одобряет услуги Medtronic для ЭЛТ легкой формы HF с AV-блокадой.Медицинские новости Medscape. Доступно на http://www.medscape.com/viewarticle/823485. 10 апреля 2014 г .; Доступ: 15 апреля 2014 г.
Леви В.К., Ли К.Л., Хеллкамп А.С. и др. Увеличение выживаемости с помощью имплантируемого кардиовертера-дефибриллятора для первичной профилактики у пациентов с сердечной недостаточностью. Тираж . 2009 8 сентября. 120 (10): 835-42. [Медлайн].
Santangelo G, Bursi F, Negroni MS, et al. Прогнозирование аритмических событий у пациентов с сердечной недостаточностью и сниженной фракцией выброса. Дж. Кардиоваск Мед (Хагерстаун) . 2021, 1 февраля. 22 (2): 110-7. [Медлайн].
Оджо А., Тарик С., Харикришнан П., Иваи С., Джейкобсон Дж. Т.. Сердечная ресинхронизирующая терапия при сердечной недостаточности. Интервал Кардиол Клин . 2017 июл.6 (3): 417-26. [Медлайн].
Rosanio S, Schwarz ER, Ahmad M, et al. Преимущества, нерешенные вопросы и технические вопросы сердечной ресинхронизирующей терапии при сердечной недостаточности. Ам Дж. Кардиол .2005 Сентябрь 1. 96 (5): 710-7. [Медлайн].
Tang AS, Wells GA, Talajic M и др., За повторную синхронизацию-дефибрилляцию для исследователей амбулаторных исследований сердечной недостаточности. Сердечная ресинхронизирующая терапия при сердечной недостаточности легкой и средней степени тяжести. N Engl J Med . 2010 16 декабря. 363 (25): 2385-95. [Медлайн].
Giraldi F, Cattadori G, Roberto M, et al. Долгосрочная эффективность сердечной ресинхронизирующей терапии у пациентов с сердечной недостаточностью с неблагоприятной анатомией сердечных вен, сравнение хирургической и гемодинамической процедуры. Джам Колл Кардиол . 2011 26 июля. 58 (5): 483-90. [Медлайн].
Абрахам В.Т., Фишер В.Г., Смит А.Л. и др. Для исследовательской группы MIRACLE. Многоцентровая рандомизированная клиническая оценка InSync. Ресинхронизация сердца при хронической сердечной недостаточности. N Engl J Med . 2002, 13 июня. 346 (24): 1845-53. [Медлайн].
Мосс А.Дж., Холл В.Дж., Кэнном Д.С. и др. Для исследователей испытаний MADIT-CRT. Сердечная ресинхронизирующая терапия для профилактики сердечной недостаточности. N Engl J Med . 2009 Октябрь 1. 361 (14): 1329-38. [Медлайн].
Аршад А., Мосс А.Дж., Фостер Е. и др. От Исполнительного комитета MADIT-CRT. Сердечная ресинхронизирующая терапия более эффективна у женщин, чем у мужчин: исследование MADIT-CRT (Испытание по имплантации многоцентрового автоматического дефибриллятора с сердечной ресинхронизирующей терапией). Джам Колл Кардиол . 2011 15 февраля. 57 (7): 813-20. [Медлайн].
Ruwald MH, Ruwald AC, Jons C и др.Влияние метопролола по сравнению с карведилолом на результаты в MADIT-CRT (испытание многоцентровой автоматической имплантации дефибриллятора с сердечной ресинхронизирующей терапией). Джам Колл Кардиол . 2013, 9 апреля. 61 (14): 1518-26. [Медлайн].
Cleland JG, Daubert JC, Erdmann E, et al, для исследователей исследования сердечной ресинхронизации-сердечной недостаточности (CARE-HF). Влияние сердечной ресинхронизации на заболеваемость и смертность при сердечной недостаточности. N Engl J Med .2005, 14 апреля. 352 (15): 1539-49. [Медлайн].
Bristow MR, Saxon LA, Boehmer J, et al., Для сравнения медицинской терапии, Pacing, et al. Сердечная ресинхронизирующая терапия с имплантируемым дефибриллятором или без него при запущенной хронической сердечной недостаточности. N Engl J Med . 2004 20 мая. 350 (21): 2140-50. [Медлайн].
Curtis AB, Worley SJ, Adamson PB и др., Для исследователей испытаний бивентрикулярной стимуляции и стимуляции правого желудочка у пациентов с сердечной недостаточностью и атриовентрикулярной блокадой (BLOCK HF).Бивентрикулярная стимуляция при атриовентрикулярной блокаде и систолической дисфункции. N Engl J Med . 2013 25 апреля. 368 (17): 1585-93. [Медлайн].
Кооперативная исследовательская группа по хирургии коронарного шунтирования администрации ветеранов. Одиннадцатилетняя выживаемость в рандомизированном исследовании Администрации ветеранов хирургического коронарного шунтирования стабильной стенокардии. N Engl J Med . 1984, 22 ноября. 311 (21): 1333-9. [Медлайн].
VA Совместная исследовательская группа по хирургии коронарного шунтирования.Восемнадцатилетнее наблюдение в Совместном исследовании по делам ветеранов хирургического шунтирования коронарной артерии при стабильной стенокардии. Тираж . 1992 июл. 86 (1): 121-30. [Медлайн].
Караччоло Э.А., Дэвис К.Б., Сопко Г. и др. Сравнение выживаемости хирургической и медицинской групп у пациентов с эквивалентной болезнью левой основной коронарной артерии. Многолетний опыт работы в CASS. Тираж . 1995 May 1. 91 (9): 2335-44. [Медлайн].
Робинсон Т.Н., Моррелл Т.Д., Померанц Б.Дж., Хаймбах Дж.К., Кэрнс CB, Харкен А.Х.Терапевтически доступные клинические сердечные состояния. Джей Ам Колл Сург . 2000 Октябрь 191 (4): 452-63. [Медлайн].
Senior R, Lahiri A, Kaul S. Влияние реваскуляризации на ремоделирование левого желудочка у пациентов с сердечной недостаточностью от тяжелой хронической ишемической дисфункции левого желудочка. Ам Дж. Кардиол . 2001 15 сентября. 88 (6): 624-9. [Медлайн].
Веласкес Э.Дж., Ли К.Л., Дежа М.А. и др. Для исследователей STICH.Аортокоронарное шунтирование у пациентов с дисфункцией левого желудочка. N Engl J Med . 2011 28 апреля. 364 (17): 1607-16. [Медлайн]. [Полный текст].
Elefteriades JA, Morales DL, Gradel C, Tollis G Jr, Levi E, Zaret BL. Результаты коронарного шунтирования одним хирургом у пациентов с фракцией выброса левого желудочка <или = 30%. Ам Дж. Кардиол . 1997 15 июня. 79 (12): 1573-8. [Медлайн].
Крон Иллинойс, Фланаган ТЛ, Блэкборн ЛХ, Шредер Р.А., Нолан СП.Коронарная реваскуляризация, а не трансплантация сердца при хронической ишемической кардиомиопатии. Энн Сург . 1989, сентябрь 210 (3): 348-52; обсуждение 352-4. [Медлайн]. [Полный текст].
Доенст Т., Веласкес Э.Дж., Бейерсдорф Ф. и др. Для исследователей STICH. STICH или нет: мы знаем ответ, но понимаем ли мы вопрос? J Thorac Cardiovasc Surg . 2005 Февраль 129 (2): 246-9. [Медлайн].
Джойс Д., Лёбе М., Нун Г.П. и др.Реваскуляризация и восстановление желудочков у пациентов с ишемической сердечной недостаточностью: исследование STICH. Curr Opin Cardiol . 2003 ноября 18 (6): 454-7. [Медлайн].
Sharoni E, Song HK, Peterson RJ, Guyton RA, Puskas JD. Операция по аортокоронарному шунтированию без помпы при значительной дисфункции левого желудочка: безопасность, осуществимость и тенденции в методологии с течением времени — первый опыт. Сердце . 2006 апр. 92 (4): 499-502. [Медлайн]. [Полный текст].
Калафиоре А.М., Ди Джиаммарко Дж., Теодори Дж. И др.Поздние результаты первой реваскуляризации миокарда при множественном поражении сосудов: одиночная или двусторонняя внутренняя артерия молочной железы с трансплантатами подкожной вены или без них. евро J Cardiothorac Surg . 2004 Сентябрь 26 (3): 542-8. [Медлайн].
Nishimura RA, Grantham JA, Connolly HM, Schaff HV, Higano ST, Holmes DR Jr. Низкопроизводительный и низкоградиентный стеноз аорты у пациентов со сниженной систолической функцией левого желудочка: клиническая полезность провокации добутамином в лаборатории катетеризации . Тираж . 2002 13 августа. 106 (7): 809-13. [Медлайн].
Carabello BA. Клиническая практика. Стеноз аорты. N Engl J Med . 2002 28 февраля. 346 (9): 677-82. [Медлайн].
Lindblom D, Lindblom U, Qvist J, Lundström H. Долгосрочные относительные показатели выживаемости после замены сердечного клапана. Джам Колл Кардиол . 1990, 1. 15 (3): 566-73. [Медлайн].
Connolly HM, Oh JK, Schaff HV, et al.Тяжелый стеноз аорты с низким трансклапанным градиентом и тяжелой дисфункцией левого желудочка: результат протезирования аортального клапана у 52 пациентов. Тираж . 2000 25 апреля. 101 (16): 1940-6. [Медлайн].
deFilippi CR, Willett DL, Brickner ME, et al. Полезность добутаминовой эхокардиографии для отличия тяжелого клапанного стеноза аорты от нетяжелого у пациентов с угнетением функции левого желудочка и низкими трансклапанными градиентами. Ам Дж. Кардиол .1995 15 января. 75 (2): 191-4. [Медлайн].
Vaquette B, Corbineau H, Laurent M, et al. Замена клапана у пациентов с критическим стенозом аорты и сниженной функцией левого желудочка: предикторы операционного риска, восстановления функции левого желудочка и отдаленные результаты. Сердце . 2005 октябрь 91 (10): 1324-9. [Медлайн]. [Полный текст].
Dujardin KS, Enriquez-Sarano M, Schaff HV, Bailey KR, Seward JB, Таджикский AJ. Смертность и заболеваемость аортальной регургитацией в клинической практике.Долгосрочное последующее исследование. Тираж . 1999 г., 13 апреля 99 (14): 1851-7. [Медлайн].
Chaliki HP, Mohty D, Avierinos JF и др. Результаты после протезирования аортального клапана у пациентов с тяжелой аортальной регургитацией и значительным снижением функции левого желудочка. Тираж . 2002 19 ноября. 106 (21): 2687-93. [Медлайн].
Энрикес-Сарано М., Таджик А.Дж. Клиническая практика. Аортальная регургитация. N Engl J Med .2004, 7 октября. 351 (15): 1539-46. [Медлайн].
Ланселотти П., Джерард П.Л., Пиерард Л.А. Отдаленные результаты пациентов с сердечной недостаточностью и динамической функциональной митральной регургитацией. Евро Сердце J . 2005 26 августа (15): 1528-32. [Медлайн].
Патель Дж. Б., Борхесон Д. Д., Барнс М. Э., Рихал С. С., Дейли Р. К., Редфилд ММ. Митральная регургитация у пациентов с тяжелой систолической сердечной недостаточностью. J Card Fail . 2004 10 августа (4): 285-91. [Медлайн].
Боллинг С.Ф., Пагани Ф.Д., Диб Г.М., Бах Д.С. Промежуточный результат митральной реконструкции при кардиомиопатии. J Thorac Cardiovasc Surg . 1998, февраль, 115 (2): 381-6; обсуждение 387-8. [Медлайн].
Акасака Т., Ёсида К., Ходзуми Т. и др. Ограниченный резерв коронарного кровотока у пациентов с митральной регургитацией улучшается после митральной реконструктивной хирургии. Джам Колл Кардиол . 1998 Декабрь 32 (7): 1923-30. [Медлайн].
Wu AH, Aaronson KD, Bolling SF, Pagani FD, Welch K, Koelling TM. Влияние аннулопластики митрального клапана на риск смерти у пациентов с митральной регургитацией и систолической дисфункцией левого желудочка. Джам Колл Кардиол . 2005 г. 1. 45 (3): 381-7. [Медлайн].
McGee EC, Gillinov AM, Blackstone EH, et al. Рецидивирующая митральная регургитация после аннулопластики по поводу функциональной ишемической митральной регургитации. J Thorac Cardiovasc Surg .2004 декабрь 128 (6): 916-24. [Медлайн].
Srichai MB, Grimm RA, Stillman AE, et al. Ишемическая митральная регургитация: влияние левого желудочка и митрального клапана у пациентов с систолической дисфункцией левого желудочка. Энн Торак Хирургия . 2005 июл. 80 (1): 170-8. [Медлайн].
Моришита А., Шимакура Т., Нонояма М., Такасаки Т. Замена митрального клапана при ишемической митральной регургитации. Сохранение как передних, так и задних митральных створок. J Cardiovasc Surg (Турин) . 2002 апр. 43 (2): 147-52. [Медлайн].
Юн К.Л., Синтек С.Ф., Миллер Д.К. и др. Рандомизированное исследование, сравнивающее частичную и полную замену митрального клапана с сохранением хорды: влияние на объем и функцию левого желудочка. J Thorac Cardiovasc Surg . 2002 Апрель 123 (4): 707-14. [Медлайн].
Гиллинов AM, Wierup PN, Blackstone EH, et al. Предпочтительнее ли восстановление или замена ишемической митральной регургитации? J Thorac Cardiovasc Surg . 2001 декабрь 122 (6): 1125-41. [Медлайн].
Миллер округ Колумбия. Редукция ишемической митральной регургитации — исправить или заменить ?. J Thorac Cardiovasc Surg . 2001 декабрь 122 (6): 1059-62. [Медлайн].
Орбан М., Хауслейтер Дж. Ремонт митрального клапана от края до края: надежные данные и благополучное будущее. Сердце . 2017 5 августа [Medline].
Vahanian A, Urena M, Ince H, Nickenig G.Митральный клапан: ремонт / зажимы / защемление / хорды. ЕвроВмешательство . 2017 24 сентября. 13 (AA): AA22-30. [Медлайн].
Фельдман Т., Кар С., Ринальди М. и др., Для исследователей ЭВЕРЕСТ. Чрескожное восстановление митрального клапана с помощью системы MitraClip: безопасность и долговечность в исходной когорте EVEREST (исследование Endovascular Valve Edge-to-Edge REpair). Джам Колл Кардиол . 2009 18 августа. 54 (8): 686-94. [Медлайн].
Chiarito M, Pagnesi M, Martino EA и др.Результат после чрескожной пластики митрального клапана от края до края при функциональной и дегенеративной митральной регургитации: систематический обзор и метаанализ. Сердце . 29 июня 2017 г. [Medline].
Geis NA, Puls M, Lubos E, et al. Безопасность и эффективность терапии MitraClip у пациентов с тяжелыми нарушениями фракции выброса левого желудочка: результаты немецкого реестра транскатетерных вмешательств на митральном клапане (TRAMI). Eur J Heart Fail . 2017 18 августа.[Медлайн].
Buckberg GD. Восстановление желудочков — хирургический подход к обратному ремоделированию желудочков. Heart Fail Ред. . 2004 октября, 9 (4): 233-9; обсуждение 347-51. [Медлайн].
Ямагути А., Ино Т., Адачи Х. и др. Объем левого желудочка позволяет прогнозировать послеоперационное течение у пациентов с ишемической кардиомиопатией. Энн Торак Хирургия . 1998 Февраль 65 (2): 434-8. [Медлайн].
Миклборо Л.Л., Карсон С., Иванов Дж.Восстановление дискинетической или акинетической аневризмы левого желудочка: результаты, полученные с помощью модифицированного линейного закрытия. J Thorac Cardiovasc Surg . 2001 апр. 121 (4): 675-82. [Медлайн].
Отт Д.А., Парравачини Р., Кули Д.А. и др. Улучшение сердечной функции после резекции аневризмы левого желудочка: пред- и послеоперационные исследования эффективности у 150 пациентов. Tex Heart Inst J . 1982 сентября, 9 (3): 267-73. [Медлайн]. [Полный текст].
Athanasuleas CL, Buckberg GD, Stanley AW и др. Для группы RESTORE.Хирургическое восстановление желудочков в лечении застойной сердечной недостаточности вследствие расширения желудочков после инфаркта миокарда. Джам Колл Кардиол . 2004 Октябрь 6. 44 (7): 1439-45. [Медлайн].
Джонс Р.Х., Веласкес Э.Дж., Михлер Р.Э. и др., Для исследователей гипотезы STICH 2. Коронарное шунтирование с хирургической реконструкцией желудочка или без нее. N Engl J Med . 2009, 23 апреля. 360 (17): 1705-17. [Медлайн]. [Полный текст].
Имамура Т., Чунг Б., Нгуен А., Сайер Г., Уриэль Н.Клинические последствия гемодинамической оценки во время терапии вспомогательным устройством левого желудочка. Дж Кардиол . 2018 Апрель 71 (4): 352-8. [Медлайн].
Тиммс Д. Обзор клинических желудочковых вспомогательных устройств. Med Eng Phys . 2011 ноябрь 33 (9): 1041-7. [Медлайн].
Pamboukian SV, Tallaj JA, Brown RN, et al. Повышение 2-летней выживаемости пациентов с вспомогательными устройствами для желудочков после внедрения протокола интенсивного наблюдения. J Пересадка легкого сердца . 2011 30 августа (8): 879-87. [Медлайн].
Lietz K, Long JW, Kfoury AG, et al. Результаты имплантации вспомогательного устройства левого желудочка в качестве целевой терапии в эпоху после REMATCH: значение для отбора пациентов. Тираж . 31 июля 2007 г., 116 (5): 497-505. [Медлайн].
Данешманд М.А., Раджагопал К., Лима Б. и др. Целевое назначение вспомогательного устройства левого желудочка в сравнении с трансплантацией сердца с расширенными критериями. Энн Торак Хирургия . 2010 апр. 89 (4): 1205-9; обсуждение 1210. [Medline].
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США. Недавно одобренные устройства: HeartWare HVAD — P100047 / S090. Доступно по адресу https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm581473.htm. 27 сентября 2017 г. [Обновлено 31 октября 2017 г.]; Дата обращения: 31 октября 2017 г.
Национальная медицинская библиотека США. Оценка системы помощи левого желудочка Jarvik 2000 с постурикулярным соединителем — исследование целевой терапии.ClinicalTrials.gov. Доступно на https://clinicaltrials.gov/ct2/show/NCT01627821. Обновлено: 16 октября 2017 г .; Дата обращения: 31 октября 2017 г.
Rose EA, Gelijns AC, Moskowitz AJ, et al, за рандомизированную оценку механической помощи при лечении застойной сердечной недостаточности (REMATCH). Длительное использование вспомогательного аппарата левого желудочка при терминальной стадии сердечной недостаточности. N Engl J Med . 2001 15 ноября. 345 (20): 1435-43. [Медлайн].
Park SJ, Tector A, Piccioni W и др.Вспомогательные устройства для левого желудочка в качестве целевой терапии: новый взгляд на выживание. J Thorac Cardiovasc Surg . 2005 Январь 129 (1): 9-17. [Медлайн].
Starling RC, Naka Y, Boyle AJ и др. Результаты пост-США. Одобренное Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов исследование с использованием вспомогательного устройства для левого желудочка с непрерывным потоком в качестве моста к трансплантации сердца: проспективное исследование с использованием INTERMACS (Межведомственного реестра для механической поддержки кровообращения). Джам Колл Кардиол .2011 10 мая. 57 (19): 1890-8. [Медлайн].
Ventura PA, Alharethi R, Budge D, et al. Различное влияние на исходы после трансплантации вспомогательных устройств для левого желудочка с пульсирующим и непрерывным потоком. Клиническая трансплантация . 2011 июл-авг. 25 (4): E390-5. [Медлайн].
Slaughter MS, Pagani FD, Rogers JG и др., Для клинических исследователей HeartMate II. Клиническое управление вспомогательными устройствами для левого желудочка с непрерывным потоком на поздней стадии сердечной недостаточности. J Пересадка легкого сердца . 2010 г., 29 апреля (4 доп.): S1-39. [Медлайн].
Кирклин Дж. К., Нафтел, округ Колумбия, Кормос Р. Л. и др. Третий годовой отчет INTERMACS: эволюция целевой терапии в США. J Пересадка легкого сердца . 2011, 30 февраля (2): 115-23. [Медлайн].
Тейлор Д.О., Эдвардс Л.Б., Боучек М.М. и др. Реестр Международного общества трансплантации сердца и легких: двадцать второй официальный отчет о трансплантации сердца взрослым — 2005 г. J Пересадка легкого сердца . 2005 24 августа (8): 945-55. [Медлайн].
Boucek MM, Edwards LB, Keck BM, Trulock EP, Taylor DO, Hertz MI. Реестр Международного общества трансплантации сердца и легких: восьмой официальный педиатрический отчет — 2005 г. J Пересадка легкого сердца . 2005 24 августа (8): 968-82. [Медлайн].
Объединенная сеть обмена органами (США). Национальные данные. UNOS. Доступно на https://unos.org/data/. 24 октября 2017 г .; Доступ: 1 ноября 2017 г.
Стивенсон Л.В., Кормос Р.Л., Бурж Р.К. и др. Механическая поддержка сердца 2000: текущие применения и дизайн будущих исследований. 15-16 июня 2000 г., Бетесда, штат Мэриленд. Джам Колл Кардиол . 2001, январь, 37 (1): 340-70. [Медлайн].
Gray NA Jr, Selzman CH. Текущее состояние тотального искусственного сердца. Am Heart J . 2006 июл.152 (1): 4-10. [Медлайн].
Кули Д.А., Лиотта Д., Холлман Г.Л., Бладуэлл Р.Д., Личман Р.Д., Милам Д.Д.Ортотопический протез сердца для двухэтапной замены сердца. Ам Дж. Кардиол . 1969, 24 ноября (5): 723-30. [Медлайн].
Платис А., Ларсон Д.Ф. Временное тотальное искусственное сердце CardioWest. Перфузия . 2009 Сентябрь 24 (5): 341-6. [Медлайн].
Roussel JC, Senage T, Baron O, et al. Тотальное искусственное сердце CardioWest (Jarvik): опыт одного центра с 42 пациентами. Энн Торак Хирургия . 2009 г., 87 (1): 124–9; Обсуждение 130.[Медлайн].
Anderson E, Jaroszewski D, Pierce C, DeValeria P, Arabia F. Параллельное применение экстракорпоральной мембранной оксигенации и тотального искусственного сердца CardioWest в качестве моста для трансплантации. Энн Торак Хирургия . 2009 ноябрь 88 (5): 1676-8. [Медлайн].
Моррис Р.Дж. Тотальное искусственное сердце — концепции и клиническое применение. Semin Thorac Cardiovasc Surg . Осень 2008 г. 20 (3): 247-54. [Медлайн].
Мейер А., Слотер М.Тотальное искусственное сердце. Panminerva Med . 2011 Сентябрь 53 (3): 141-54. [Медлайн].
Национальная медицинская библиотека США. Европейская клиническая оценка общего искусственного сердца Carmat (предварительная HF). ClinicalTrials.gov. Доступно на https://clinicaltrials.gov/ct2/show/NCT02962973. Обновлено: 25 ноября 2016 г .; Доступ: 1 ноября 2017 г.
[Рекомендации] Акерман М.Дж., Приори С.Г., Виллемс С. и др., Общество сердечного ритма (HRS), Европейская ассоциация сердечного ритма (EHRA).Консенсусное заявление экспертов HRS / EHRA о состоянии генетического тестирования каннелопатий и кардиомиопатий: этот документ был разработан в рамках партнерства между Обществом сердечного ритма (HRS) и Европейской ассоциацией сердечного ритма (EHRA). Europace . 2011 августа 13 (8): 1077-109. [Медлайн]. [Полный текст].
Маркус Ф.И., Маккенна В.Дж., Шерилл Д. и др. Диагностика аритмогенной кардиомиопатии / дисплазии правого желудочка: предлагаемая модификация критериев рабочей группы. Евро Сердце J . 2010 г. 31 (7): 806-14. [Медлайн]. [Полный текст].
[Рекомендации] О’Мира Э, Макдональд М., Чан М. и др. Рекомендации CCS / CHFS по сердечной недостаточности: обновленные данные о клинических испытаниях функциональной митральной регургитации, ингибиторов SGLT2, ARNI при HFpEF и тафамидисе при амилоидозе. Банка J Cardiol . 2020 Февраль 36 (2): 159-69. [Медлайн]. [Полный текст].
[Рекомендации] Трейси С.М., Эпштейн А.Е., Дарбар Д. и др. 2012 г. ACCF / AHA / HRS, посвященное обновлению рекомендаций 2008 г. по аппаратной терапии нарушений сердечного ритма: отчет Фонда Американского колледжа кардиологов / Целевой группы Американской кардиологической ассоциации по практическим рекомендациям и Общества сердечного ритма.[исправлено]. Тираж . 2 октября 2012 г. 126 (14): 1784-800. [Медлайн]. [Полный текст].
[Рекомендации] Priori SG, Blomstrom-Lundqvist C, Mazzanti A, et al. Руководство ESC 2015 г. по ведению пациентов с желудочковыми аритмиями и предотвращению внезапной сердечной смерти: Целевая группа по ведению пациентов с желудочковыми аритмиями и профилактике внезапной сердечной смерти Европейского общества кардиологов (ESC). Принято: Ассоциация европейской детской и врожденной кардиологии (AEPC). Евро Сердце J . 2015 г., 1. 36 (41): 2793-867. [Медлайн]. [Полный текст].
[Рекомендации] Рихал С.С., Найду С.С., Живертц М.М. и др., Общество сердечно-сосудистой ангиографии и вмешательств (SCAI), Американское общество сердечной недостаточности (HFSA) и др. Консенсусное заявление клинических экспертов SCAI / ACC / HFSA / STS от 2015 года об использовании устройств для чрескожной механической поддержки кровообращения при лечении сердечно-сосудистых заболеваний: одобрено Американской кардиологической ассоциацией, Кардиологическим обществом Индии и Sociedad Latino Americana de Cardiologia Intervencion; подтверждение ценности Канадской ассоциацией интервенционной кардиологии-Association Canadienne de Cardiologie d’intervention. Джам Колл Кардиол . 2015 19 мая. 65 (19): e7-e26. [Медлайн]. [Полный текст].
[Рекомендации] Фельдман Д., Памбукян С.В., Тойтеберг Дж. Дж. И др., Международное общество трансплантации сердца и легких. Рекомендации Международного общества по трансплантации сердца и легких по механической поддержке кровообращения, 2013: краткое содержание. J Пересадка легкого сердца . 2013 Февраль 32 (2): 157-87. [Медлайн]. [Полный текст].
[Рекомендации] Пеура Дж. Л., Колвин-Адамс М., Фрэнсис Г. С. и др.Рекомендации по использованию механической поддержки кровообращения: стратегии устройств и выбор пациентов: научное заявление Американской кардиологической ассоциации. Тираж . 2012 27 ноября, 126 (22): 2648-67. [Медлайн]. [Полный текст].
Masini M, Elia E, Vianello PF и др. Частота, предикторы и прогностическое влияние разрядов имплантируемого кардиовертера-дефибриллятора в популяции первичной профилактики сердечной недостаточности и сниженной фракции выброса. Дж. Кардиоваск Мед (Хагерстаун) .2021, 1 февраля. 22 (2): 118-25. [Медлайн].
Новартис. Novartis объявляет о положительной рекомендации Консультативного комитета FDA по использованию Entresto для лечения пациентов с HFpEF [пресс-релиз]. Доступно по адресу https://www.novartis.com/news/media-releases/novartis-announces-positive-fda-advisory-committee-recommendation-use-entresto-treat-patients-hfpef. 16 декабря 2020 г .; Дата обращения: 26 января 2021 г.
Информационный документ Консультативного комитета FDA: Entresto (сакубитрил / валсартан) для лечения хронической сердечной недостаточности и сохраненной фракции выброса.15 декабря 2020 г. Доступно по адресу http://fda.gov/media/144379/download.
Ллойд-Джонс Д., Адамс Р.Дж., Браун TM и др. Для Статистического комитета Американской кардиологической ассоциации и Подкомитета по статистике инсультов. Резюме: статистика сердечных заболеваний и инсульта — обновление 2010 г .: отчет Американской кардиологической ассоциации. Тираж . 2010 г. 23 февраля. 121 (7): 948-54. [Медлайн].
Temporelli PL, Scapellato F, Eleuteri E, Imparato A, Giannuzzi P.Допплер-эхокардиография при тяжелой систолической сердечной недостаточности: неинвазивная альтернатива катетеру Свана-Ганца. Циркулярная сердечная недостаточность . 2010 май. 3 (3): 387-94. [Медлайн].
Miller R. AHA издает рекомендации по механической поддержке кровообращения для справочных документов. Медицинские новости Medscape. Доступно на http://www.medscape.com/viewarticle/773806. 1 ноября 2012 г .; Дата обращения: 7 ноября 2012 г.
[Рекомендации] Бангалор С., Кумар С., Мессерли Ф. Х.Когда обычной терапии сердечной недостаточности недостаточно: блокатор рецепторов ангиотензина, прямой ингибитор ренина или антагонист альдостерона? Застойная сердечная недостаточность . 2013 май-июнь. 19 (3): 107-15. [Медлайн].
Cleland JG, Teerlink JR, Senior R и др. Эффекты сердечного активатора миозина, омекамтива мекарбила, на сердечную функцию при систолической сердечной недостаточности: двойное слепое плацебо-контролируемое перекрестное исследование фазы 2 с ранжированием доз. Ланцет . 2011 20 августа.378 (9792): 676-83. [Медлайн].
Эклинд-Червенка М., Бенсон Л., Дальстрем Ю., Эднер М., Розенквист М., Лунд Л. Связь кандесартана и лозартана со смертностью от всех причин у пациентов с сердечной недостаточностью. ЯМА . 2011 12 января 305 (2): 175-82. [Медлайн].
Фумото Х., Хорват Д. Д., Рао С. и др. Саморегулирующееся искусственное сердце с непрерывным потоком in vivo в клинике Кливленда. J Пересадка легкого сердца .2010 29 января (1): 21-6. [Медлайн]. [Полный текст].
Дженнингс Д.Л., Чемберс Р.М., Шиллиг Дж. М.. Фармакотерапия HeartMate II, вспомогательного устройства для левого желудочка с непрерывным потоком, у пациентов с тяжелой сердечной недостаточностью: интеграция заболевания, устройства и лекарства. Энн Фармакотер . 2010 Октябрь 44 (10): 1647-50. [Медлайн].
Opie LH. Digitalis вчера и сегодня, но не навсегда. Результаты Circ Cardiovasc Qual .2013 г. 1. 6 (5): 511-3. [Медлайн].
Plank B, Kutyifa V, Moss AJ, et al. Курение связано с повышенным риском возникновения первых и рецидивов желудочковых тахиаритмий у ишемических и неишемических пациентов с легкой сердечной недостаточностью: субисследование MADIT-CRT. Ритм сердца . 2014 май. 11 (5): 822-7. [Медлайн].
Стайлз С. Удвоение блокады РААС при ВЧ? Согласно метаанализу, антагонисты альдостерона, а не БРА. Медицинские новости Medscape.Доступно на http://www.medscape.com/viewarticle/776725. 26 декабря 2012 г .; Доступ: 31 декабря 2012 г.
Топильский Ю., О Дж. К., Атчисон Ф. В. и др. Результаты эхокардиографии у стабильных амбулаторных пациентов с правильно функционирующими вспомогательными устройствами для левого желудочка HeartMate II. J Am Soc Echocardiogr . 2011 24 февраля (2): 157-69. [Медлайн].
Haddad F, Elmi-Sarabi M, Fadel E, Mercier O, Denault AY. Жемчужины и подводные камни в лечении правожелудочковой недостаточности в кардиохирургии. Curr Opin Anaesthesiol . 2016 29 февраля (1): 68-79. [Медлайн].
Bois JP, Chareonthaitawee P. Радионуклидная визуализация при застойной сердечной недостаточности: оценка жизнеспособности, саркоидоз и амилоидоз. Кардиол Клин . 2016 Февраль 34 (1): 119-32. [Медлайн].
Hanzel GS, Dixon S, Goldstein JA. Определение приоритетов и сочетание методов лечения сердечной недостаточности в эпоху механических поддерживающих устройств. Интервал Кардиол Клин .2017 Июль 6 (3): 465-80. [Медлайн].
Geis N, Raake P, Mereles D, et al. Чрескожное лечение тяжелой регургитации митрального клапана, вторичной по отношению к разрыву хорд, у восьмидесятилетних детей с использованием MitraClip. J Интервал Кардиол . 12 октября 2017 г. [Medline].
Faletra FF, Berrebi A, Pedrazzini G, et al. Трехмерная чреспищеводная эхокардиография: новый инструмент визуализации для оценки митральной регургитации и управления чрескожной пластикой митрального клапана от края до края. Программа Cardiovasc Dis . 19 октября 2017 г. [Medline].
Ренин-ангиотензин-альдостероновая система при сердечной недостаточности для неспециалистов: прошлое, настоящее и будущее
Ингибиторы рецепторов ангиотензина и неприлизина
До сих пор основное внимание уделялось ингибированию РААС. Однако патология сердечной недостаточности — это дисбаланс между регуляторными и противорегулирующими механизмами (рисунок 2). Лучше всего охарактеризованными медиаторами, противостоящими пагубным эффектам активации РААС при сердечной недостаточности, являются НЧ.Следовательно, ингибиторы неприлизина, фермента, разрушающего НЧ, были использованы в новых методах лечения сердечной недостаточности.
Первоначальные испытания ингибитора АПФ и комбинации ингибитора неприлизина, омапатрилата, в когортах пациентов с сердечной недостаточностью и гипертонией не увенчались успехом из-за неприемлемо высокой частоты ангиоэдемы. Это связано с избыточным накоплением брадикинина, поскольку ингибирование как АПФ, так и неприлизина блокирует путь деградации брадикинина.31–34 Однако, поскольку БРА не повышают уровень брадикининов, комбинация БРА недавно изучалась в исследовании PARDIGM-HF.PARDIGM-HF набирал пациентов с ФВ <40% и сердечной недостаточностью II – IV классов по NYHA и повышенным BNP, несмотря на оптимальную терапию против сердечной недостаточности, и рандомизировал их на группы эналаприла или ARNI, сакубитрил-валсартан. Исследование показало новый «вводный» период, когда пациенты получали оба препарата по очереди в течение короткого периода, чтобы убедиться, что пациенты могут переносить оба препарата в необходимых дозах до рандомизации. Девяносто три процента пациентов были установлены на терапию BB, 55,6% на MRA и 7% получали сердечную ресинхронизирующую терапию.
Все первичные исходы исследования были значительно снижены в группе ARNI со снижением смертности от сердечно-сосудистых причин или первой госпитализации с сердечной недостаточностью с 26,5% до 21,8% (HR 0,8, 95% ДИ от 0,73 до 0,87, p> 0,001). смерть от сердечно-сосудистых причин от 16,5% до 13,3% (отношение рисков 0,8, 95% доверительный интервал от 0,71 до 0,89, p <0,001) и, что особенно важно, смертность от всех причин от 19,8% до 17% (отношение рисков 0,84, 95% доверительный интервал от 0,76 до 0,93, p < 0,001). Важно отметить, что побочные эффекты также были тщательно изучены. Следует отметить, что в течение «вводного» периода 12% группы пациентов с ОРНИ прекратили лечение из-за побочных эффектов, в частности кашля, гиперкалиемии, почечной дисфункции или симптоматической гипотензии, хотя в целом более частые отмены были отмечены в группе эналаприла.В основном исследовании сакубитрил-валсартан вызывал значительно более выраженную симптоматическую гипотензию, но значительно меньшую гиперкалиемию, кашель и почечную недостаточность, чем эналаприл. Несмотря на то, что в группе ARNI наблюдалось незначительное увеличение частоты возникновения отека Квинке (19 случаев против 10 случаев, p = 0,13), в исследовании в целом не было сообщений о нарушении дыхательных путей7
Таким образом, Появление ОРНИ является ключевым событием в лечении систолической сердечной недостаточности. В прямом сравнении с ингибиторами АПФ, ARNI демонстрируют значительное улучшение показателей смертности, заболеваемости и госпитализации.Это впечатляет в контексте пациентов, уже оптимизированных с помощью BB, MRA и аппаратной терапии, которые сами по себе дают значительные преимущества. Неоспоримым фактом является то, что перевод пациентов с установленного золотого стандарта ингибиторов АПФ на АРНИ значительно улучшит заболеваемость и смертность у пациентов с систолической сердечной недостаточностью; это первый новый класс агентов в терапии сердечной недостаточности с момента появления ББ 15 лет назад. Следовательно, ARNI могут ускорить изменение парадигмы в лечении сердечной недостаточности.
ARNI недавно были включены в руководящие документы. В Великобритании Национальный институт здравоохранения и передового опыта рекомендует сакубитрил-валсартан пациентам с симптоматической хронической сердечной недостаточностью с ФВ <35%, которые уже принимают стабильные дозы ингибиторов АПФ или БРА. Рекомендуется, чтобы новую терапию начали только специалисты по сердечной недостаточности, имеющие доступ к многопрофильной бригаде по сердечной недостаточности. Между тем, в недавно обновленных рекомендациях Европейского общества кардиологов по лечению сердечной недостаточности сакубитрил-валсартан получил рекомендацию класса IB у пациентов с ФВ <35% и повышенными НЧ (BNP ≥150 пг / мл или N-концевой про Натрийуретический пептид B-типа (NT-proBNP) ≥600 пг / мл, за исключением случаев госпитализации по поводу сердечной недостаточности в предыдущие 12 месяцев, когда пороговые значения составляют BNP ≥100 пг / мл или NT-proBNP ≥400 пг / мл), которые остаются симптоматичными, несмотря на ингибитор АПФ, ВВ и МРА в наивысшей переносимой дозе.35, 36
Сакубитрил-валсартан выпускается в трех дозах: 26/24 мг, 51/49 мг или 103/97 мг два раза в сутки. Начальная доза 26/24 мг два раза в день подходит, когда ОРНИ заменяет низкие дозы ингибитора АПФ или БРА или когда начальное систолическое артериальное давление <110 мм рт. Ст., Ее следует увеличивать каждые 3-4 недели по мере переносимости; Тем, кто уже стабилизировался на ингибиторе АПФ или БРА, рекомендуется 51/49 мг два раза в день, и эту дозу следует увеличить через 2–4 недели по мере переносимости. Более низкая начальная доза 26/24 мг необходима при умеренной печеночной недостаточности (классификация B по классификации Чайлд-Пью) или тяжелой почечной недостаточности (расчетная скорость клубочковой фильтрации <30 мл / мин / л.73 м 2 ). У всех пациентов доза должна быть увеличена по мере переносимости через интервалы, указанные выше, до максимальной дозы 103/97 мг два раза в день. Сакубитрил-валсартан противопоказан при тяжелой печеночной недостаточности, предшествующем ангиоэдеме с применением ингибитора АПФ / БРА, беременности, кормлении грудью, систолическом артериальном давлении <100 мм рт.ст. и одновременном применении алискирена пациентам с диабетом.
Критически важно, что сакубитрил-валсартан не следует назначать вместе с ингибитором АПФ или БРА.Чтобы свести к минимуму риск ангиоэдемы, обусловленного одновременным ингибированием АПФ и неприлизина, прием сакубитрил-валсартана следует начинать только после 36-часового периода без введения ингибиторов АПФ. Пациентов следует предупреждать о часто встречающихся побочных эффектах, включая тошноту, диахоррею, гастрит, анемию, гипогликемию, гипокалиемию и головокружение.18, 35
Сахарный диабет, система ренин-ангиотензин-альдостерон и сердце | Кардиология | JAMA Internal Medicine
Поскольку сахарный диабет достигает масштабов эпидемии, в основном вторичный по отношению к ожирению, влияние сердечно-сосудистых заболеваний из-за этого сочетания делает его доминирующей проблемой общественного здравоохранения в первой четверти 21 века.Сложное взаимодействие, которое приводит к диабетической болезни сердца, создается перекрывающимися механизмами. Существует склонность к развитию преждевременной диффузной атеросклеротической коронарной болезни, которая связана с неблагоприятными краткосрочными и долгосрочными заболеваниями и смертностью. Имеются структурные и функциональные аномалии микрососудов, вегетативная дисфункция и внутренняя недостаточность сокращения миокарда (так называемая диабетическая кардиомиопатия). Эти изменения усугубляются артериальной гипертензией и заболеванием почек.В этом обзоре мы рассматриваем роль ренин-ангиотензин-альдостероновой системы и то, как она является решающим фактором большинства патофизиологических механизмов, лежащих в основе диабетической болезни сердца, и почему за последние 5 лет блокирование этой системы у пациентов с диабетом стало решающим фактором. критическое терапевтическое вмешательство.
Распространенность сахарного диабета (СД) переживает беспрецедентный рост. Согласно прогнозам, нынешняя распространенность заболевания среди 150 миллионов человек во всем мире увеличится до 220 миллионов к 2010 году и до 300 миллионов к 2025 году. 1 Этот драматический эффект в значительной степени обусловлен связанной эпидемией ожирения. 2 Сахарный диабет тесно связан с возникновением окклюзии коронарной артерии и другими сосудистыми событиями по данным популяционных исследований. Фрамингемское исследование 3 показало повышенный риск ишемической болезни сердца на 66% у мужчин и 203% у женщин с СД после контроля других сердечно-сосудистых факторов риска, тогда как исследование множественных факторов риска (MRFIT) 4 продемонстрировало усиленный риск. приписывается DM.Фактически, исследования 5 -8 подтвердили, что пациенты с СД, но без ишемической болезни сердца, испытывают такую же частоту коронарных событий, как и недиабетические пациенты с инфарктом миокарда (ИМ) в анамнезе. Неясно, является ли это открытие просто результатом сочетания обычных факторов или, более конкретно, сложными метаболическими эффектами панкреатической недостаточности. Ясно то, что потребность в специфическом, проактивном управлении сердечным и сосудистым риском остро стоит у пациентов с СД, независимо от того, есть ли у них явная коронарная болезнь или какое-либо сердечное поражение.Следует предположить, что это доминирует в их мировоззрении. В этом обзоре исследуется роль ренин-ангиотензин-альдостероновой системы (РААС) при диабетической болезни сердца и доказательная база для блокирования этой системы в качестве ключевого медиатора нежелательных явлений у пациентов с диабетом.
Основные физиологические и фармакологические процессы
Ренин секретируется в виде прогормона, активируемого для высвобождения активного ренина, который является лимитирующим ферментом, контролирующим гомеостаз почек, натрия и объема.Проренин продуцируется юкстагломерулярными клетками в почках в ответ на различные стимулы, включая снижение перфузионного давления почек, активацию симпатической нервной системы и снижение доставки натрия по канальцам в плотное пятно. Ферментный каскад инициирует выработку относительно неактивного декапептида ангиотензина I (Ang I) из циркулирующего, преимущественно печеночного, ангиотензиногена. Затем неспецифический фермент ангиотензин-превращающий фермент (АПФ) отщепляет ангиотензин II (Ang II) от Ang I. АПФ (или кининаза II) играет важную роль в системе калликреин-кинин (где он имеет более высокое ферментное сродство к брадикинину и , в меньшей степени каллидин).Помимо образования Ang II, АПФ отдельно катализирует деградацию брадикинина. Следовательно, терапевтическое лечение препаратами ингибитора АПФ (ИАПФ) приводит к накоплению брадикинина и подавлению Ang II. 9 Было высказано предположение, что брадикинин частично противодействует сосудистым и тканевым эффектам Ang II, поскольку первый улучшает коронарную перфузию и работу желудочков, одновременно защищая от гипертрофии левого желудочка. Существуют некоторые разногласия относительно того, могут ли некоторые из положительных эффектов блокирования РААС посредством ингибирования АПФ быть связаны с потенцированием кининов в такой же степени, как с подавлением Ang II или альдостерона. 10 , 11 Эта довольно простая, давняя гипотеза еще не прояснена и не более и не менее актуальна для СД, чем для общего воздействия лечения препаратами ИАПФ.
Помимо системы циркулирующих гормонов, имеется также местный тканевый РААС. Связанный с тканями АПФ может составлять до 90% от того, что содержится в организме. 12 Активность широко распространена, эндотелиальные клетки являются одним из важных участков в стенке сосуда.Стенка сосуда способна продуцировать Ang II из циркулирующего и локально продуцируемого Ang I и, следовательно, действовать как локальная «тканевая ренин-ангиотензиновая система (РАС)». 13 , 14
Хотя циркулирующий RAS важен для системной регуляции сердечно-сосудистой системы, активация этого тканевого RAS (с выработкой Ang II) изменяет локальную функцию и, следовательно, может оказывать прямое аутокринное и паракринное воздействие на эндотелий и гладкомышечные клетки независимо от его эндокринных эффектов. по системной гемодинамике.Ангиотензин II, действуя преимущественно через рецепторы ангиотензина 1 типа, вызывает широкий спектр эффектов, включая воспаление сосудов и окислительное повреждение (Таблица 1). 9 , 15 -18 Повышенный окислительный стресс, в свою очередь, активирует пути гибели клеток (апоптоз), 19 , тогда как воспаление сосудов непосредственно участвует в атеросклеротическом процессе. 20 Эти процессы в настоящее время вовлечены в широкий спектр сердечно-сосудистых заболеваний, таких как острые коронарные синдромы и сердечная недостаточность, а также в хроническую почечную недостаточность.
Клеточный окислительный метаболический стресс и эндотелиальная дисфункция являются ключевыми в генерализованном патогенезе сердечно-сосудистых заболеваний и тесно связаны с циркулирующими и тканевыми уровнями Ang II (рис. 1). 13 Это открытие основано на экспериментальных исследованиях и клинических наблюдениях, демонстрирующих стимуляцию РАС и одновременную активацию никотинамидадениндинуклеотид / никотинамидадениндинуклеотидфосфатоксидазы в стенке артерии. 21 -23 Однако более конкретное взаимодействие может связывать DM и RAAS.
Во-первых, краткосрочная умеренная гипергликемия без глюкозурии на ранних стадиях СД была связана с увеличением активности ренина плазмы, среднего артериального давления и сопротивления сосудов почек, 24 с активацией циркулирующей и локальной (внутрипочечной) РАС. Например, в исследовании Miller, 25 , среднее артериальное давление было значительно выше во время гипергликемии, чем при эугликемии, и артериальное давление хорошо реагировало на терапию лозартаном калия, тогда как реакция на терапию лозартаном во время эугликемии была минимальной.Osei et al., , 26, , также продемонстрировали усиленный почечный вазодилататорный ответ на применение каптоприла и эпросартана во время гипергликемии, предполагая, что гипергликемия приводит к повышению тонуса сосудов почек, опосредованного Ang II. Это может происходить из-за повышенной реактивности, которая может быть прямой или косвенной в зависимости от воздействия других систем, таких как вегетативная нервная система и дуга барорефлекса, которые также являются аномальными при СД.
Во-вторых, гипергликемия приводит к гликозилированию p53, что связано с транскрипцией ангиотензиногена и последующим продуцированием Ang II из местного RAS. 27 , 28 Этот вывод подтверждается экспериментом Fiordaliso и его коллегами, 29 , которые продемонстрировали прямую корреляцию между уровнями глюкозы, экспрессией p53 и количеством Ang II. Синтез ангиотензина II увеличивался с увеличением степени гликемии, и это ослаблялось ингибированием гликозилирования p53. Признано, что ангиотензин II обладает проапоптотическими свойствами 30 и, таким образом, представляет вероятную роль RAS в патогенезе диабетической болезни сердца.Специфическая блокада РАС с помощью ИАПФ и блокаторов рецепторов ангиотензина (БРА), например, может ослабить некоторые из этих эффектов.
В-третьих, недавние исследования клинических исходов, такие как исследование Heart Outcomes Prevention Evaluation (HOPE), проект Captopril Prevention Project (CAPPP) и недавнее исследование лозартана для снижения конечной точки при гипертонии (LIFE), неизменно и неожиданно предлагали снижение заболеваемости впервые возникшим СД. Это не было заранее определенной первичной конечной точкой ни в одном из этих исследований.В исследовании HOPE, , 31, , , 32, , диагноз СД был поставлен участниками испытания самостоятельно и не был подтвержден измерениями глюкозы, что могло подорвать любые твердые выводы. В CAPPP 33 появился новый диагноз СД в качестве вторичной конечной точки. В этом исследовании диагноз был подтвержден в соответствии с критериями Всемирной организации здравоохранения 1985 года. В этом исследовании 33 сообщалось о снижении риска развития СД в группе, получавшей каптоприл, на 11% (337 из 5183 пациентов в группе каптоприла по сравнению с 380 из 5230 пациентов в группе обычного лечения).Наконец, исследование LIFE 34 продемонстрировало значительное снижение на 25% впервые возникшего СД ( P = 0,001), что, опять же, было предварительно определенной вторичной конечной точкой. Однако в этом последнем исследовании в качестве средства сравнения использовался атенолол (с известным, хотя и умеренным неблагоприятным метаболическим профилем). Согласованность результатов этих крупных испытаний требует дальнейшего первичного расследования. Гипотеза о том, что специфическое ингибирование РААС, особенно с помощью ИАПФ, уменьшит развитие нового СД, формально проверяется в 2 крупных рандомизированных контролируемых исследованиях: в исследовании «Оценка снижения диабета с помощью рамиприла и розиглитазона» (DREAM) и натеглинида и валсартана в исследовании. Исследование исходов нарушенной толерантности к глюкозе (НАВИГАТОР).
Крупномасштабные клинические испытания в течение последних двух десятилетий, в частности, привели к клинически значимым улучшениям в выживаемости от ишемической болезни сердца в целом. Однако данные реестра Всемирной организации здравоохранения по мониторингу тенденций и детерминантов сердечно-сосудистых заболеваний (MONICA) — Аугсбургский регистр показывают небольшое улучшение выживаемости пациентов с СД после ИМ, в отличие от пациентов без диабета, 35 , несмотря на значительные шаги вперед в лечении, такие, как четко определено в исследовании «Диабет и инфузия инсулина и глюкозы при остром инфаркте миокарда» (DIGAMI). 36
Присутствие СД вдвое увеличивает летальность по сравнению с пациентами без диабета. 37 Частично предполагалось, что это открытие связано с учащением симптомов сердечной недостаточности и нарушением функции левого желудочка у пациентов с диабетом. 38 Систолическая функция левого желудочка — хорошо известный независимый предиктор сердечно-сосудистой заболеваемости и смертности. 39 Основным процессом может быть обычная ишемическая болезнь сердца, которая может быть симптоматической, атипичной по симптоматике или даже клинически бессимптомной, хотя вся эта область плохо определена. 40 -42 Также возможно, что частота систолической сократительной недостаточности может быть связана с большим размером инфаркта у пациентов с установленной коронарной болезнью, но клинические наблюдения говорят об обратном. 43 , 44
Воздействие DM на сердце сложное и многофакторное. Существуют элементы проявления и времени появления симптомов у пациента с диабетом, тяжесть основных установленных факторов риска атеросклероза (которые обычно объединяются с СД) и, возможно, специфические особенности, связанные с самим СД.Несмотря на то, что они описаны отдельно, среди предлагаемых патофизиологических процессов, описанных в следующих подразделах, существует значительное совпадение. В большинстве анализов оказалось невозможным выделить компоненты, способствующие диабетической болезни сердца.
Гипертония и гипертрофия желудочков
Увеличивается распространенность артериальной гипертензии у пациентов с СД (особенно СД 2 типа). 45 До 70% пациентов с диабетом 2 типа с ожирением имеют гипертонию 46 (определяется как артериальное давление [АД]> 140/90 мм рт. соотношение или, более конкретно, во время клэмп-исследований эугликемии-гиперинсулинемии), по оценкам, присутствует примерно у 50% пациентов с гипертензией. 47 Эта общая ассоциация действует синергетически, приводя к увеличению частоты сердечно-сосудистых событий и, следовательно, к существенному абсолютному преимуществу при строгом снижении АД.
Проспективное исследование диабета в Соединенном Королевстве (UKPDS) показало, что у включенных в исследование пациентов с диабетом каждое снижение среднего систолического АД на 10 мм рт. и микрососудистых осложнений на 13%. 48 , 49 Подобные данные из специального анализа подгрупп пациентов с диабетом из исследования оптимального лечения гипертонии (HOT) 50 предполагают пользу в снижении АД даже до референсного диапазона (АД <140/85 мм рт.Кроме того, исследование «Систолическая гипертензия в Европе» (Syst-Eur) 51 показало, что пациенты с СД получали больше преимуществ с точки зрения сердечно-сосудистой заболеваемости и смертности от снижения АД, чем пациенты без диабета, хотя и за счет большей антигипертензивной терапии (таблица 2). . Эти и другие исследования привели к консенсусу во многих национальных и международных рекомендациях, направленных на снижение целевого АД у пациентов с СД. Это подтверждается недавним заявлением о позиции Американской диабетической ассоциации 48 и отчетом Рабочей группы 52 Национального фонда почек по гипертонии и диабету, в котором рекомендуется целевое АД ниже 130/80 мм рт.
Природа взаимодействия между повышением артериального давления, сосудистым сопротивлением, сосудистым повреждением и окклюзионными сосудистыми событиями у пациента с гипертоническим диабетом сложна и вряд ли напрямую связана только с давлением. Несколько небольших исследований и 2 недавних крупномасштабных терапевтических клинических испытания — HOPE и LIFE — с участием разумного числа пациентов с диабетом и изучением эффектов терапии ИАПФ и БРА предполагают важную роль РААС независимо от эффектов снижения давления.В исследовании HOPE изучались эффекты терапии рамиприлом у «пациентов высокого риска» с диабетической подгруппой из 3577 пациентов (проанализированы отдельно в исследовании MICRO-HOPE 31 ). Хотя неконтролируемая гипертензия была критерием исключения, участники могли лечить гипертонию. Входные значения АД 141,7 / 80 мм рт. Ст. И 142,3 / 79,3 мм рт. Ст. В группах рамиприла и плацебо соответственно 31 были связаны со скромным эффектом лечения АД (3/2 мм рт. Ст.). Существенная польза (число, необходимое для лечения для предотвращения 1 инфаркта миокарда, инсульта, сердечно-сосудистой смерти, госпитализации по поводу сердечной недостаточности, процедуры реваскуляризации, развития явной нефропатии, лазерной терапии ретинопатии и почечного диализа, составила 15) превзошла то, что большинство наблюдателей могло бы отнести к этот небольшой эффект снижения АД.Однако многие исследователи полагают, что влияние АД могло быть неоптимально определено и систематически недооцениваться. 53
Аналогичным образом, исследование LIFE, 34 с диабетической когортой из 1195 пациентов, продемонстрировало более высокую эффективность лозартана по сравнению с атенололом в снижении сердечно-сосудистой заболеваемости и смертности (преимущественно инсульта) у пациентов с СД, артериальной гипертензией и гипертрофией левого желудочка (первичная гипертрофия). составная конечная точка: отношение рисков, 0.76; P = 0,03). Среднее АД в конце исследования составило 146/79 и 148/79 мм рт. Ст. В группах лозартана и атенолола соответственно. Поскольку систолическое АД не было связано с какими-либо изменениями в основных составных конечных точках, большее преимущество было приписано специфической блокаде Ang II. В обоих этих исследованиях положительный эффект был получен в группах пациентов, включающих большое количество людей с диабетом, хотя испытания не проводились только на пациентах с диабетом. Хотя неразумно выводить дополнительную пользу от специфического ингибирования РААС при СД, общая гипотеза изучалась в течение нескольких лет и становится все более обоснованной.
Одна из проблем диабетической болезни сердца, заслуживающая особого внимания, — это распространенность гипертрофии левого желудочка. Артериальное давление имеет прямую дифференцированную связь с распространенностью и тяжестью гипертрофии левого желудочка у отдельных пациентов, что, в свою очередь, является независимым популяционным предиктором будущей сердечно-сосудистой заболеваемости и смертности. 54 Хотя любое лечение, снижающее АД, вызывает некоторый регресс гипертрофии левого желудочка, лечение, блокирующее РААС, может быть более эффективным в этом отношении.Однако изменения незначительны, происходят в течение длительных периодов лечения и зависят от тщательно контролируемых серийных эхокардиографических измерений. Большая часть доказательной базы этого эффекта получена в результате метаанализа небольших исследований. Насколько нам известно, нет конкретных исследований, предполагающих, что диабетическая гипертрофия левого желудочка более или менее подвержена этому эффекту.
Опять же, в более общем плане было показано, что использование лизиноприла ИАПФ 55 -57 улучшает резерв перфузии миокарда и максимальный коронарный кровоток у пациентов с гипертрофией левого желудочка, вызванной гипертензией, возможно, за счет увеличения плотности капилляров миокарда.Этот эффект не наблюдался при терапии лозартаном, и это заставило некоторых исследователей предположить, что это один из примеров механизма, опосредованного брадикинином. 57 Использование ИАПФ может также обратить вспять эндотелиальную дисфункцию у пациентов с ишемической болезнью сердца, гипертонией и СД и может благоприятно влиять на фибринолитический баланс, возможно, за счет ослабления эффектов Ang II и усиления брадикинин-зависимых сосудистых эффектов. 58 , 59 Продолжающееся европейское испытание по уменьшению сердечных событий с помощью периндоприла при стабильной коронарной болезни сердца (EUROPA) и предотвращению событий с помощью ингибирования ангиотензин-превращающего фермента (PEACE), хотя опять же не первичные исследования диабетического сердца болезнь, будет содержать анализ заранее определенных подгрупп и должен пролить больше света на этот вопрос.
Эффект острой коронарной окклюзии и, следовательно, некроза миокарда на сердце зависит от размера зоны инфаркта и от реакции выжившего миокарда на поддержание общей сердечной функции. Неинфарктный миокард при наличии адекватной перфузии обычно демонстрирует компенсаторный гиперкинез для поддержания нормальной фракции выброса. 60 Этот компенсаторный ответ нарушен у пациентов с СД, 37 , 61 , что может просто отражать более диффузное многососудистое заболевание при СД.Одно только это открытие может независимо предсказывать неблагоприятный краткосрочный результат, но, кроме того, снижение резерва кровотока и нарушение коронарной вазодилатации могут усилить ишемическое повреждение в неинфарктных сегментах за счет водораздела.
Коронарное расширение сосудов зависит от структурной и функциональной целостности эндотелия. Изменения эндотелия хорошо распознаются при СД 2-го типа и предшествуют развитию микроангиопатии. 62 Широко распространено мнение, что гипергликемия является важной причиной эндотелиальной дисфункции.Гипергликемия вызывает внутриклеточные изменения окислительно-восстановительного состояния с активацией диацилглицерина и фосфокиназы C и последующим истощением пула никотинамидадениндинуклеотид / никотинамидадениндинуклеотидфосфат. Никотинамидадениндинуклеотид / никотинамидадениндинуклеотидфосфат необходим для образования оксида азота, 63 , важного вазодилататора, производного от эндотелия. Кроме того, локально продуцируемый эндотелиальный оксид азота может быть инактивирован взаимодействиями с продвинутыми гликозилированными конечными продуктами, образованными посредством неферментативных взаимодействий между глюкозой и аминогруппами белков, липидов и нуклеиновых кислот.Эта реакция усиливается при гипергликемии и при наличии СД. 64 Кроме того, эти гликозилированные продукты обычно обнаруживаются в атеросклеротических поражениях 65 и могут приводить к активации ядерного фактора фактора транскрипции κB, 66 , модулирующего транскрипцию гена для эндотелина-1, молекул адгезии, тканевого фактора и тромбомодулина. . Эти процессы приводят к дальнейшему окислительному стрессу, сужению сосудов и протромбическому состоянию, 64 , способствуя окклюзии сосудов при СД.
Ангиотензин II может непосредственно способствовать окислительному стрессу за счет увеличения производства в сосудах супероксидных радикалов (Таблица 1), 67 , который, реагируя с оксидом азота (образуя пероксинитрит), снижает уровни свободного оксида азота. Сами свободные радикалы увеличивают адгезию лейкоцитов к эндотелию, агрегацию тромбоцитов и экспрессию цитокинов, что приводит к инфильтрации макрофагами в атеросклеротическом участке и, следовательно, к увеличению вероятности нестабильности бляшек.Ангиотензин II может циркулировать или вырабатываться с использованием местного или циркулирующего Ang I в качестве субстрата. Доказательства этого исходят из демонстрации накопления тканевого АПФ в физической области атеросклеротических бляшек. 68
Следовательно, во время или после ишемии миокарда, диффузное атеросклеротическое заболевание, структурные аномалии микрососудов коронарных артерий, ограниченный резерв коронарного кровотока и нарушение коронарной вазодилатации — все это в совокупности снижает перфузию неинфарктного миокарда.Это приводит к ограничению компенсаторной гиперкинезии, тогда как продолжающийся оксидативный стресс может еще больше усугубить повреждение миокарда при СД. 69
Ожирение достигает масштабов эпидемии и тесно связано с СД 2 типа. Почти 65% взрослых в Соединенных Штатах имеют избыточный вес (индекс массы тела [ИМТ] [рассчитывается как вес в килограммах, разделенный на квадрат роста в метрах] ≥25) или страдают ожирением, причем 31% имеют ИМТ 30 или выше. . 70 Kenchaiah и его коллеги 71 исследовали связь между ИМТ и частотой сердечной недостаточности в когорте Framingham Heart Study и продемонстрировали градуированный риск от нормального ИМТ до диапазона ожирения без каких-либо доказательств порогового значения, что предполагает возможную причинную роль . Повышенная гемодинамическая нагрузка и активация нейроэндокринной системы были предложены в качестве двух возможных объяснений. 72 , 73
Адипоциты могут предлагать альтернативный или, возможно, дополнительный механизм.Экспрессия ангиотензиногена, хотя и обнаруживается преимущественно в печени, была продемонстрирована в жировой ткани человека. Имеются также сообщения об экспрессии АПФ и присутствии рецепторов AT в висцеральных адипоцитах человека, что подтверждает существование местного RAS в жировой ткани. 74 Поскольку продукция печенкой ангиотензиногена остается чувствительной к инсулину перед лицом инсулинорезистентности, состояние компенсаторной гиперинсулинемии, такое как обнаруживаемая при СД 2 типа, будет увеличивать ангиотензиноген субстрата ренина, что приводит к увеличению местной и, возможно, системной продукции Ang II.Исследование Price et al. 75 подтверждает эту гипотезу, демонстрируя корреляцию между повышенным почечным плазмотоком в ответ на терапию ирбесартаном и повышением ИМТ у 12 пациентов с ожирением и СД 2 типа.
Вегетативная невропатия — это хорошо известное осложнение СД и обычно явно проявляется на поздних стадиях плохо контролируемого заболевания. Тем не менее, это может быть продемонстрировано до 40% пациентов с диабетом при надлежащем клиническом тестировании 76 или позитронно-эмиссионных томографических исследованиях для изучения сердечной симпатической денервации. 77
Вегетативная дисфункция влечет за собой неблагоприятный прогноз со стороны сердечно-сосудистой системы с повышенной смертностью, дисфункцией левого желудочка и риском внезапной смерти у пациентов с диабетом с ишемией миокарда или без нее. 78 -83 Такой вывод может быть вызван несколькими причинами. Во-первых, пациенты с диабетом с вегетативной дисфункцией имеют более высокую частоту сердечных сокращений в состоянии покоя, чем пациенты без диабета, 84 , что может логически увеличить потребность миокарда в кислороде, уменьшить коронарный кровоток (укороченную диастолу) и усугубить ишемию миокарда.Во-вторых, сенсорная вегетативная дисфункция может привести к нарушению или изменению восприятия ишемической боли в области сердца в груди, что приведет к «тихим» инфарктам или атипичным проявлениям, потенциально задерживая доступ к неотложной помощи и увеличивая риск внезапной смерти и осложнений ИМ. Этот пункт, хотя и общепринятый, 85 еще предстоит окончательно продемонстрировать. 86 Кроме того, вегетативная нейропатия первоначально проявляется как парасимпатическая дисфункция, которая приводит к вегетативному дисбалансу с преобладанием (особенно ночной) симпатической активности.Известно, что повышенная симпатическая активность связана с активацией РААС, повышенным артериальным давлением, сердечной недостаточностью и аритмогенезом (и, следовательно, внезапной сердечной смертью). Результаты экспериментальных исследований 54 также предполагают прямые кардиотрофические эффекты, способствующие гипертрофии миокарда. Хотя это может быть прямым эффектом катехоламинов, сопутствующая активация RAAS и, следовательно, высвобождение Ang II, вероятно, будет играть определенную роль.
Существование специфической диабетической кардиомиопатии (специфическая недостаточность систолического сокращения сердечной мышцы, отличная от эффектов коронарного кровотока или инфаркта) было областью интенсивных исследований и споров с 1970-х годов.Ранние эпидемиологические исследования 87 , 88 показали более высокий риск сердечной недостаточности у пациентов с СД. Это подтверждается различными патологическими данными, такими как фиброз миокарда, утолщение базальной мембраны и капиллярные микроаневризмы. 89 , 90 Эти изменения не являются уникальными для СД и напоминают патологические особенности любой другой идиопатической кардиомиопатии. Кроме того, эти изменения не были описаны повсеместно и, если они присутствовали, не обязательно коррелировали с наличием классических симптомов сердечной недостаточности. 89
Последующие экспериментальные исследования СД на животных моделях продемонстрировали нарушение систолической сократимости и диастолической релаксации. Ранняя работа Regan et al. 91 на собаках с аллоксан-индуцированным диабетом продемонстрировала снижение податливости желудочков за счет накопления интерстициального гликопротеина и коллагена. Jackson et al., , 92, , например, на крысах с индуцированным стрептозотоцином диабетом показали более низкое давление в левом желудочке и сократительную способность с прогрессирующим увеличением отложения липидов и ухудшением целостности кардиомиоцитов до 24 недель.Однако химически индуцированный СД у животных мало похож на клинический СД, поскольку эти гипоинсулинемические (по сравнению с гиперинсулинемией в СД 2-го типа) модели не требуют инсулина для выживания (в отличие от СД 1-го типа). Это открытие привело к использованию крыс со спонтанным диабетом, таких как модели крыс BioBreed 93 и Otsuka Long-Evans Tokushima Fatty 94 , имитирующие СД 1 и 2 типа соответственно. Mizushige и его коллеги 95 исследовали серийные изменения в динамике наполнения левого желудочка с помощью допплеровской эхокардиографии и гистопатологические изменения в сердце спонтанно диабетических крыс Otsuka Long-Evans Tokushima Fatty по сравнению с недиабетическими крысами.Некоторые исследователи продемонстрировали диастолические нарушения на преддиабетической стадии (или стадии нарушения толерантности к глюкозе), которые сопровождались серологическими и патологическими изменениями кардиомиоцитов или интерстициальным повреждением (накопление коллагена без атеросклеротических изменений и повышенная экспрессия рецептора 2 трансформирующего фактора роста β1 в миоцитах и эндотелии. ). Несмотря на полезность, присущие экспериментам на животных ограничения запрещают прямую экстраполяцию этих результатов на людей.
Многочисленные инвазивные и неинвазивные исследования гемодинамики были выполнены в попытке охарактеризовать эту потенциальную форму кардиомиопатии у людей.Однако во многих из этих исследований есть недостатки из-за набора пациентов с легкой гипертензией (по сегодняшним стандартам), 96 -100 отсутствия различия между СД 1 и 2 типа, 96 -98,101 и, самое главное, как правило, неадекватное исключение возможной клинически «тихой» коронарной болезни. 101 Некоторые исследования 102 не обнаружили никаких доказательств такой кардиомиопатии у молодых людей с нормотензивным диабетом, не страдающих коронарной болезнью.
В целом, похоже, существует общее мнение о существовании отдельного миопатического состояния, связанного с СД, которое, вероятно, первоначально проявляется как субклиническая диастолическая дисфункция. 89 , 103 -106 Однако здесь имеется явный пробел в знаниях, помимо согласия, что он, вероятно, действительно существует. 103 Его клиническое течение неизвестно, реакция на гликемический контроль непоследовательно демонстрируется в клинических исследованиях, 107 -109 , и лечение предполагаемых нарушений диастолической функции еще требует адекватного определения. 103 Во многих отношениях это может частично совпадать с дебатами о существовании симптомов сердечной недостаточности из-за нарушений диастолического наполнения у недиабетической популяции. Они тоже не очень хорошо определены.
Причины специфической диабетической кардиомиопатии также неуловимы, хотя несколько механизмов легко связаны с сократительной недостаточностью. Во-первых, абсолютный или относительный дефицит инсулина приводит к аномальному метаболизму субстрата и, следовательно, сокращению.Поглощение глюкозы переносчиками глюкозы и гликолиз снижены в сердце диабетика, тогда как использование жирных кислот увеличивается. 110 Это приводит к аномалиям, среди прочего, сарколеммы и саркоплазматического ретикулума, важных для обработки кальция и, следовательно, сократительной функции сердца. 110 Во-вторых, гипергликемия через гликирование белков, как описано ранее, или через активацию диацилглицерина и протеинкиназы C, вызывает окислительный стресс, который широко вовлечен в патогенез диабетических осложнений. 111 В эксперименте Wakasaki et al., 112 сверхэкспрессия изоформы β2 протеинкиназы C в миокарде крысы приводила к гипертрофии сердца, повреждению кардиомиоцитов и фиброзу, тогда как лечение LY333531, ингибитором изоформы β2 протеинкиназы C, предотвращало большинство этих изменений.
Наконец, как описано ранее в данном документе, текущие данные свидетельствуют о том, что DM ассоциирован с активированной системной и локальной RAS и продукцией Ang II. Это приводит к образованию супероксида за счет стимуляции никотинамидадениндинуклеотид / никотинамидадениндинуклеотидфосфатоксидазы, что приводит к дальнейшему окислительному стрессу. 67 Это может напрямую нарушать систолическое сокращение и, таким образом, в целом может иметь решающее значение в патогенезе сердечной дисфункции при СД (рис. 2). 110
Сахарный диабет — наиболее частая причина терминальной стадии почечной недостаточности в развитых странах. В целом диабетическая нефропатия составляет 40% новых случаев терминальной почечной недостаточности в США. С увеличением распространенности во всем мире и увеличением числа пациентов с диабетом, принимаемых в программы лечения терминальной почечной недостаточности, эта тенденция будет продолжаться. 113
Диабетическая нефропатия, однако, имеет более далеко идущие последствия. Изолированный сахарный диабет уже создает неблагоприятный профиль сердечно-сосудистого риска, но этот риск усиливается в сочетании с нарушением функции почек, что приводит к 9-кратному увеличению относительной сердечно-сосудистой смертности. 114 Фактически, сердечно-сосудистый риск имеет прямую связь с нарушением функции почек, увеличиваясь до 20 раз у пациентов, находящихся на поддерживающем гемодиализе. 115
Какова роль РААС при диабетической нефропатии? Артериальное давление и почки тесно и неразрывно связаны. Заболевания почек являются важными причинами гипертонии, а гипертония вызывает и ускоряет прогрессирование почечной дисфункции. Антигипертензивная терапия (и контроль гликемии) замедляет прогрессирование почечной дисфункции при СД 1 и 2 типа. 116 , 117
Уже несколько лет установлено, 118 и продолжает подтверждаться новыми результатами исследований, 119 , что ишемическая и неишемическая почечная недостаточность специфически чувствительны к блокаде РААС.Важное исследование, проведенное Lewis et al. –120 в 1993 г., показало дополнительный независимый от АД ренопротекторный эффект каптоприла короткого действия АПФ при СД 1 типа (с явной нефропатией). Исследовательская группа EUCLID 121 расширила эти результаты с помощью лизиноприла, когда они продемонстрировали снижение прогрессирования почечной недостаточности у пациентов с «нормотензивным» (целевое систолическое АД <155 мм рт. Ст.) Диабетом 1 типа с микроальбуминурией. Аналогичная работа с ИАПФ при СД 2-го типа была недавно проведена с UKPDS (отчет 39) и анализом подгруппы MICRO-HOPE (см. Подраздел «Гипертония и гипертрофия желудочков»).Однако эти исследования, похоже, дают несколько противоречивые результаты. В UKPDS снижение АД на 117 с помощью атенолола или каптоприла ACEI короткого действия снижало скорость прогрессирования микроальбуминурии без какой-либо значительной разницы между ними. MICRO-HOPE, 31 с использованием ИАПФ рамиприла длительного действия, принимаемого дважды в день, предположил наличие специфического для ИАПФ и независимого от АД эффекта. Роль РААС стала немного яснее с дальнейшими результатами клинических испытаний с использованием препаратов, блокирующих рецепторы (таблица 3). 122 -126 Это множество доказательств в поддержку БРА привело к рекомендации Американской диабетической ассоциации 113 использовать БРА в качестве первой линии лечения диабетической нефропатии, хотя мало что указывает на то, что результаты будут специфичными для класса ARB, а не для любого блокировщика RAAS. Действительно, в сердечной сфере данные свидетельствуют о том, что верно обратное и что ИАПФ лучше. Эти исследования показывают, что ингибирование РААС с помощью БРА снижает прогрессирование диабетической нефропатии, эффект, вероятно, частично зависит и частично не зависит от АД и подтверждает роль РААС в прогрессировании диабетической нефропатии.
Существует сложная взаимосвязь между генерализованным атеросклерозом и отдельным процессом недостаточности поджелудочной железы, будь то абсолютная, как при СД 1 типа, или относительная, как при СД 2 типа. Ожирение и его эпидемические последствия для западных культур и диетических привычек играют ключевую роль в артериальном давлении и его влиянии на сосудистое дерево и сердце. Диабетические метаболические изменения могут напрямую влиять на сердце с точки зрения структуры и функции и могут сливаться с усиленными эффектами повышенного артериального давления.Различить относительную важность каждого из этих элементов оказалось сложно, но совершенно очевидно, что индивидуальный сосудистый и сердечный риск пациента с диабетом велик независимо от того, есть у него симптомы или нет. Специфические вмешательства, направленные на РААС, по-видимому, имеют особую эффективность в этой группе пациентов, что подразумевает ключевую роль этой системы в патогенезе диабетических заболеваний сердца и сосудов. Откуда появятся новые расширения этого принципа?
Интересная перспектива возникла с открытием тиазолидиндионов — активатора рецептора, активируемого пролифератором пероксисом (PPAR).PPAR представляет собой рецептор ядерного гормона, который после связывания с лигандом активируется и образует активированный комплекс с рецептором 9- цис- -ретиноевой кислоты. Этот комплекс, в свою очередь, связывается со специфическими элементами ответа пролифератора пероксисом, что в конечном итоге приводит к усилению транскрипции и синтезу белка. В дополнение к своим антигипергликемическим эффектам агонисты PPAR (PPAR-γ и, в определенной степени, PPAR-α), такие как гидрохлорид пиоглитазона и малеат розиглитазона, изменяют другие «нетрадиционные» маркеры сердечно-сосудистых заболеваний.В частности, было показано, что они снижают сывороточные уровни матриксной металлопротеиназы 9 (вовлеченной в разрыв бляшек), 127 С-реактивного белка, 127 фактора некроза опухоли α, 128 молекулы адгезии клеток сосудов 1 (рекрутирование лейкоцитов в атеросклеротические клетки). поражения), 129 и тканевой фактор (тромбогенность, связанная с разрывом бляшки). 130 Более важно то, что Ang II подавляет мессенджерную РНК и белок PPAR, 131 , тем самым способствуя воспалению сосудов и атеросклерозу.Является ли это потенциальной связью между этими двумя, казалось бы, разрозненными системами? Проспективное клиническое испытание пиоглитазона при макрососудистых событиях (PROactive), которое в настоящее время исследует влияние пиоглитазона на основные конечные точки смертности от всех причин и совокупность сердечно-сосудистых исходов, предоставит дополнительную информацию о роли PPAR и его агонистов.
Во-вторых, существуют явные различия между ИАПФ и БРА, хотя оба являются ингибиторами РААС и полезны для пациентов с диабетом.Некоторые из этих различий могут быть связаны с накоплением брадикинина при использовании ИАПФ (обсуждается в подразделе «Основные физиологические и фармакологические процессы»), но есть много других систем, на которые воздействуют ИАПФ, которые в равной степени можно рассматривать и заслуживают оценки. Эти альтернативные пути потенциально клинически значимы с учетом следующего: (1) хроническое лечение полными дозами ИАПФ было связано с реактивацией Ang II 132 и повышенными уровнями альдостерона 133 ; (2) нейроэндокринная активация связана с плохим прогнозом при таких состояниях, как симптоматическая сердечная недостаточность 134 ; (3) специфическое ингибирование альдостерона спиронолактоном значительно улучшает заболеваемость и смертность при тяжелой сердечной недостаточности 135 ; (4) почти 40% Ang I может быть преобразовано в Ang II не-АПФ путями, и это может быть увеличено в DM 136 ; и (5) хотя ИАПФ доказали свою пользу у пациентов с высоким сердечно-сосудистым риском (исследование HOPE) и сердечной недостаточностью (CONSENSUS, 137 Профилактика и лечение SOLVD, 138 , 139 AIRE, 140 и SAVE, 141 исследований), доказательство эквивалентной пользы от использования БРА оказалось трудно продемонстрировать (см. Оптимальное исследование инфаркта миокарда с антагонистом ангиотензина II лозартаном [OPTIMAAL] в следующем абзаце). 142
Исследование лозартана в пожилом возрасте (ELITE), 143 , которое было разработано для проверки превосходства БРА (лозартан) над ИАПФ (каптоприл), а также исследование сердечной недостаточности валсартана (Val-HeFT), 144 , которое был разработан для проверки превосходства БРА и ИАПФ над одними ИАПФ, только БРА или БРА в сочетании с ИАПФ не показали какого-либо дополнительного преимущества в отношении смертности, хотя группа ИАПФ показала снижение комбинированных конечных точек смертности и заболеваемости.Действительно, последнее исследование БРА, OPTIMAAL, , 142, , сравнивающее лозартан с каптоприлом у пациентов с ИМ и сердечной недостаточностью, новым ИМ с зубцом Q или повторным инфарктом, показало незначительную тенденцию в пользу каптоприла (хотя лозартан переносился лучше). Отсутствие превосходства над ИАПФ поднимает вопросы о точной роли РААС и БРА, по крайней мере, у пациентов с сердечной недостаточностью. С нетерпением ожидаются продолжающиеся исследования БРА, которые будут включать заранее определенные диабетические подгруппы (таблица 4).
Более того, до сих пор отсутствуют доказательства специфической эффективности блокирования РААС при СД среди небелых этнических групп, несмотря на обсервационные исследования, предполагающие более высокую заболеваемость СД 2 типа 145 и риск сердечно-сосудистой заболеваемости и смертности. 146 В этом отношении, 2 недавних исследования, Афро-американское исследование болезней почек и гипертонии (AASK) 119 и исследование гипотензивных и гиполипидемических средств для предотвращения сердечного приступа (ALLHAT), 147 , оба со значительными числами пациентов из числа афроамериканцев, позволяют лучше понять возможные различия и сходства между этническими группами.AASK, 119 , который исключал пациентов с диабетом, продемонстрировал более сильные ренопротекторные эффекты при использовании рамиприла по сравнению с применением амлодипина при гипертонической болезни почек. Тем не менее, ALLHAT, 147 , в котором примерно 36% всех пациентов были афроамериканцами, предполагал меньший эффект снижения АД при монотерапии ИАПФ. Принимая во внимание эти и другие исследования, Рабочая группа по гипертонии у афроамериканцев 148 (1) признала, что для достижения целевого уровня АД часто может потребоваться комбинированная терапия, (2) рекомендовала аналогичное целевое значение АД менее 130/80 мм рт. у пациентов с высоким сердечно-сосудистым риском (особенно с СД 2 типа) и (3) заявили, что любые показания для терапии ИАПФ должны в равной степени применяться к пациентам афроамериканского происхождения.Это важные шаги вперед, но в наших знаниях остаются значительные пробелы. Хотя может показаться разумным предположение об эффективности в отношении других этнических меньшинств, такие предположения, тем не менее, потребуют аналогичного подтверждения. Эта потребность в настоящее время становится более насущной, поскольку DM в настоящее время переживает всемирную эпидемию (и связанное с ней сердечно-сосудистое бремя следует той же тенденции), особенно в Китае и на Индийском субконтиненте. 1
Наконец, существует реальная потребность в оценке существующих стратегий лечения с помощью исследований результатов, особенно при СД.Во многих исследованиях исходы по диабету оставляют в виде анализа подгрупп или используют суррогатные конечные точки, такие как гликемический контроль, контроль АД и альбуминурия. Хотя разумно предположить, что эти суррогатные конечные точки приведут к клинически значимым улучшениям в исходе, включая смертность, парадоксально, что данные о смертности не собираются, особенно там, где уровень смертности настолько высок. Кроме того, многие прошлые клинические испытания показали, что такая экстраполяция не всегда может быть обоснованной, например, Испытание подавления сердечной аритмии (CAST). 149 Аналогичным образом, исследования диабетической нефропатии с применением БРА также показали, что снижение альбуминурии не может автоматически приводить к улучшению общей выживаемости, несмотря на тот факт, что ухудшение функции почек связано с прогрессивным повышением риска сердечно-сосудистых заболеваний.
Диабетическая болезнь сердца представляет собой сложную взаимосвязь между гипертонией, сосудистым повреждением, общепринятыми факторами риска атеросклероза, вегетативной нейропатией и нефропатией, которые объединяются в СД.Тромбоз, эндотелиальная дисфункция, повреждение сосудов и воспаление являются важными эффекторными механизмами и сильно зависят от метаболических изменений явного и субклинического СД (рис. 3).
Заболеваемость и смертность от воздействия DM на сердце остаются значительными, несмотря на недавний прогресс в лечении сердечно-сосудистых заболеваний. Хотя диабетическая болезнь сердца не является исключительно опосредованной RAAS, улучшенное понимание патогенеза и результаты очень крупных общих клинических испытаний, в которых пациенты с диабетом составляют значительную подгруппу, помогли указать путь вперед.Дополнительные исследования конкретных исходов при СД были бы полезны этой подгруппе для рассмотрения более общих аспектов сосудистой патологии, особенно в более восприимчивых этнических группах, и для развеивания мифов о ИАПФ, которые, по-видимому, хронически ограничивают их широкое использование у этих пациентов ( Таблица 5). 150
Для корреспонденции: Грегори И. Х. Лип, доктор медицины, FRCP, Медицинский факультет Университета, Городская больница, Бирмингем, B18 7QH, Англия (электронная почта: [email protected]).
Принята к публикации 30 июня 2003 г.
2.Wannamethee
SGShaper
AG Изменение веса и продолжительность избыточной массы тела и ожирения в частоте диабета 2 типа. Уход за диабетом. 1999; 221266-1272PubMedGoogle ScholarCrossref 4.Stamler
JVaccaro
ONeaton
J Диабет, другие факторы риска и 12-летняя смертность от сердечно-сосудистых заболеваний среди мужчин в испытании множественных факторов риска. Уход за диабетом. 1993; 16434-444PubMedGoogle ScholarCrossref 5.Haffner
С.М.Лехто
SRonnemaa
Т
и другие. Смертность от ишемической болезни сердца у субъектов с диабетом 2 типа и у субъектов без диабета с инфарктом миокарда и без него. N Engl J Med. 1998; 339229-234PubMedGoogle ScholarCrossref 6.Malmberg
КЮсуф
SGerstein
HC
и другие. Влияние диабета на долгосрочный прогноз у пациентов с нестабильной стенокардией и инфарктом миокарда без зубца Q: результаты реестра OASIS. Тираж. 2000; 1021014-1019PubMedGoogle ScholarCrossref 7.Cho
ERimm
EBStampfer
MJ
и другие. Влияние диабета и перенесенного инфаркта миокарда на смертность от всех причин и от ишемической болезни сердца у мужчин. J Am Coll Cardiol. 2002; 40954-960PubMedGoogle ScholarCrossref 8.Hu
FBStampfer
MJSolomon
CG
и другие. Влияние сахарного диабета на смертность от всех причин и ишемическую болезнь сердца у женщин. Arch Intern Med. 2001; 1611717-1723PubMedGoogle ScholarCrossref 13.Дзау
VJ Тканевый ангиотензин и патобиология сосудистых заболеваний: объединяющая гипотеза. Гипертония. 2001; 371047-1052PubMedGoogle ScholarCrossref 21.Laursen
JBRajagopalan
SGalis
ZS
и другие. Роль супероксида в индуцированной ангиотензином II, но не индуцированной катехоламином гипертензии. Тираж. 1997; 95588-593PubMedGoogle ScholarCrossref 22.Warnholtz
ANickenig
GSchulz
E
и другие.Повышенная продукция супероксида, опосредованная НАДН-оксидазой, на ранних стадиях атеросклероза. Тираж. 1999; 9
-2033PubMedGoogle ScholarCrossref 24.Miller
JAFloras
JSZinman
B
и другие. Влияние гипергликемии на артериальное давление, активность ренина плазмы и функцию почек при раннем диабете. Clin Sci. 1996;
-195PubMedGoogle Scholar25.Miller
JA Влияние гипергликемии на ренин-ангиотензиновую систему при раннем сахарном диабете 1 типа у человека. J Am Soc Nephrol. 1999; 101778-1785PubMedGoogle Scholar26.Osei
SYPrice
DALaffel
ЛКМ
и другие. Влияние антагониста ангиотензина II эпросартана на индуцированную гипергликемией активацию внутрипочечной ренин-ангиотензиновой системы у здоровых людей. Гипертония. 2000; 36122-126PubMedGoogle ScholarCrossref 27.Leri
AClaudio
PPWang
XX
и другие. Высвобождение ангиотензина II, опосредованное растяжением, индуцирует апоптоз миоцитов за счет активации р53, который усиливает локальную ренин-ангиотензиновую систему и снижает соотношение белков Bel-2 и Bax в клетке. J Clin Invest. 1998; 1011326-1342PubMedGoogle ScholarCrossref 28.Leri
AFiordaliso
FSetoguchi
M
и другие. Ингибирование функции р53 предотвращает активацию ренин-ангиотензиновой системы и апоптоз миоцитов, опосредованный растяжением. Am J Pathol. 2000; 157843-857PubMedGoogle ScholarCrossref 29.Fiordaliso
FLeri
ACesselli
D
и другие. Гипергликемия активирует регулируемые p53 и p53 гены, что приводит к гибели клеток миоцитов. Диабет. 2001; 502363-2375PubMedGoogle ScholarCrossref 30.Horiuchi
Макишита
MDzau
VJ Недавний прогресс в исследованиях рецепторов ангиотензина II типа 2 в сердечно-сосудистой системе. Гипертония. 1999; 33613-621PubMedGoogle ScholarCrossref 31. Исследователи исследования по оценке профилактики сердечных заболеваний (HOPE), влияние рамиприла на сердечно-сосудистые и микрососудистые исходы у людей с сахарным диабетом. Ланцет. 2000; 355253-259PubMedGoogle ScholarCrossref 33.Hansson
LLindholm
LHNiskanen
L
и другие. Влияние ингибирования ангиотензинпревращающего фермента по сравнению с традиционной терапией на сердечно-сосудистую заболеваемость и смертность при гипертонии: рандомизированное исследование Captopril Prevention Project (CAPPP). Ланцет. 1999; 353611-616PubMedGoogle ScholarCrossref 34.Lindholm
Л.Х.Ибсен
HDahlof
B
и другие. LIFE Study Group, Сердечно-сосудистые заболевания и смертность у пациентов с диабетом в исследовании «Вмешательство лозартана для снижения конечной точки в исследовании гипертонии» (LIFE): рандомизированное исследование атенолола. Ланцет. 2002; 35
-1010PubMedGoogle ScholarCrossref 35.Bloomgarden
ZT Ежегодное собрание Европейской ассоциации по изучению диабета в 1999 г. — осложнения диабета. Уход за диабетом. 2000; 231423-1428PubMedGoogle ScholarCrossref 36.Malmberg
KRyden
LEfendic
S
и другие. Рандомизированное исследование инфузии инсулина и глюкозы с последующим подкожным лечением инсулином у диабетических пациентов с острым инфарктом миокарда (исследование DIGAMI). J Am Coll Cardiol. 1995; 2657-65PubMedGoogle ScholarCrossref 37.Woodfield
SLundergan
RReiner
J
и другие. Ангиографические данные и исходы у больных сахарным диабетом, получавших тромболитическую терапию при остром инфаркте миокарда: опыт GUSTO-1. J Am Coll Cardiol. 1996; 281661-1669PubMedGoogle ScholarCrossref 38.Fava
Саццопарди
JMuscat
HA
и другие. Факторы, влияющие на исход у диабетиков с инфарктом миокарда. Уход за диабетом. 1993; 161615-1618PubMedGoogle ScholarCrossref 39. Исследователи GUSTO-1, Предикторы 30-дневной смертности в эпоху реперфузии при остром инфаркте миокарда. Тираж. 1995;
9-1668PubMedGoogle ScholarCrossref 40.Юдкин
JSOswald
GA Детерминанты госпитализации и летальности у больных сахарным диабетом с инфарктом миокарда. Уход за диабетом. 1988; 11351-358PubMedGoogle ScholarCrossref 41.Jaffe
АССпадаро
JJSchechtman
K
и другие.Усиление застойной сердечной недостаточности после инфаркта миокарда умеренной степени у больных сахарным диабетом. Am Heart J. 1984; 10831-37PubMedGoogle ScholarCrossref 42.Aronson
DRayfield
EJChesebro
JH Механизмы, определяющие течение и исходы больных сахарным диабетом, перенесших острый инфаркт миокарда. Ann Intern Med. 1997; 126296-306PubMedGoogle ScholarCrossref 43. Gwilt
DJPetri
MLewis
PW
и другие. Размер инфаркта миокарда и смертность у больных диабетом. Br Heart J. 1985; 54466-472PubMedGoogle ScholarCrossref 44. Исследовательская группа МИЛИС, Влияние сахарного диабета на прогноз и серийную функцию левого желудочка после инфаркта миокарда. J Am Coll Cardiol. 1989; 1449–57PubMedGoogle ScholarCrossref 47.Reaven
GM Интенсивный контроль артериального давления / глюкозы при диабете 2 типа: почему так трудно уменьшить ишемическую болезнь сердца? J Hum Hypertens. 1999; 13S19- S23PubMedGoogle ScholarCrossref 48.Американская диабетическая ассоциация, Лечение гипертонии у взрослых с диабетом. Уход за диабетом. 2002; 25S71- S73Google ScholarCrossref 49.UK Prospective Diabetes Study Group, Жесткий контроль артериального давления и риск макрососудистых и микрососудистых осложнений при диабете 2 типа: UKPDS 38. BMJ. 1998; 317703-713PubMedGoogle ScholarCrossref 50. Группа исследования HOT, Эффекты интенсивного снижения артериального давления и низких доз аспирина у пациентов с гипертонией: основные результаты рандомизированного исследования оптимального лечения гипертонии (HOT). Ланцет. 1998; 3511755-1762PubMedGoogle ScholarCrossref 51. Систолическая гипертензия в испытаниях европейских исследователей, Эффекты блокады кальциевых каналов у пожилых пациентов с диабетом и систолической гипертензией. N Engl J Med. 1999; 340677-684PubMedGoogle ScholarCrossref 52.Bakris
GLWilliams
MDworkin
L
и другие. Рабочая группа исполнительных комитетов Национального фонда почек по гипертензии и диабету, Сохранение функции почек у взрослых с гипертонией и диабетом. Am J Kidney Dis. 2000; 36646-661PubMedGoogle ScholarCrossref 53.Svensson
Pde Faire
USleight
п
и другие. Сравнительные эффекты рамиприла на амбулаторное и офисное артериальное давление: подисследование НАДЕЖДА. Гипертония. 2001; 38E28- E32PubMedGoogle ScholarCrossref 54.Lip
GYHFelmeden
DCLi-Saw-Hee
FLBeevers
Д.Г. Гипертоническая болезнь сердца: комплексный синдром или гипертоническая «кардиомиопатия»? Eur Heart J. 2000; 211653–1665PubMedGoogle ScholarCrossref 55.Брилла
CGJanicki
JSWeber
KT Кардиорепаративные эффекты лизиноприла у крыс с генетической гипертензией и гипертрофией левого желудочка. Тираж. 1991; 831771-1779PubMedGoogle ScholarCrossref 56.Motz
WStrauer
BE Улучшение резерва коронарного кровотока после длительной терапии эналаприлом. Гипертония. 1996; 271031-1038PubMedGoogle ScholarCrossref 57.Akinboboye
OOChou
Р.Л.Бергманн
SR Увеличение кровотока в миокарде при гипертонической болезни сердца антагонистами ангиотензина. J Am Coll Cardiol. 2002; 40703-709PubMedGoogle ScholarCrossref 58.Mancini
GBHenry
GCMacaya
C
и другие. Ингибирование ангиотензин-превращающего фермента с помощью квинаприла улучшает эндотелиальную вазомоторную дисфункцию у пациентов с ишемической болезнью сердца: исследование TREND (Испытание по обращению эндотелиальной дисфункции). Тираж. 1996; 94258-265PubMedGoogle ScholarCrossref 59. Hornig
BKohler
CDrexler
H Роль брадикинина в опосредовании сосудистых эффектов ингибиторов ангиотензинпревращающего фермента у людей. Тираж. 1997; 951115-1118PubMedGoogle ScholarCrossref 60. Крупы
CLTopol
EJCaliff
RM
и другие. Прогностические последствия и предикторы усиления регионарного движения стенки в зоне без инфаркта миокарда после тромболизиса и ангиопластической терапии острого инфаркта миокарда. Тираж. 1989; 80245-253PubMedGoogle ScholarCrossref 61.Iwasaka
TTakahashi
Nakamura
S
и другие. Остаточная насосная функция левого желудочка после острого инфаркта миокарда у пациентов с NIDDM. Уход за диабетом. 1992; 151522-1526PubMedGoogle ScholarCrossref 62.Balletshofer
BMRittig
KEnderle
MD
и другие. Эндотелиальная дисфункция выявляется у молодых нормотензивных родственников первой степени родства пациентов с диабетом 2 типа в сочетании с инсулинорезистентностью. Тираж. 2000; 1011780-1784PubMedGoogle ScholarCrossref 65.Miyata
SMonnier
В.М. Иммуногистохимическое определение AGE в диабетических тканях с использованием моноклональных антител к пирралину. J Clin Invest. 1992; 8
-1122PubMedGoogle ScholarCrossref 66. Палински
В.Кощинский
TButler
S
и другие. Иммунологические доказательства наличия AGE в атеросклеротических поражениях эугликемических кроликов. Arterioscler Thromb Vasc Biol. 1995; 15571-582PubMedGoogle ScholarCrossref 67.Berry
Чамильтон
CABrosnan
J
и другие. Исследование источников супероксида в кровеносных сосудах человека: ангиотензин II увеличивает выработку супероксида во внутренних артериях молочной железы человека. Тираж. 2000; 1012206-2212PubMedGoogle ScholarCrossref 68. Диета
FPratt
REBerry
ГДж
и другие. Повышенное накопление тканевого АПФ при атеросклеротической болезни коронарных артерий человека. Тираж. 1996; 942756-2767PubMedGoogle ScholarCrossref 69.van Harsdorf
RLi
П.Ф.Дитц
R Сигнальные пути в апоптозе кардиомиоцитов, вызванном активными формами кислорода. Тираж. 1999; 9-2941PubMedGoogle ScholarCrossref 70.Должен
Аспадано
Дж. Коукли
EH
и другие. Бремя болезней, связанных с избыточным весом и ожирением. JAMA. 1999; 2821523-1529PubMedGoogle ScholarCrossref 72.Messerli
FHSungaard-Riise
KReisin
E
и другие. Несопоставимые сердечно-сосудистые эффекты ожирения и артериальной гипертензии. Am J Med. 1983; 74808-812PubMedGoogle ScholarCrossref 73.Engeli
SSharma
AM Ренин-ангиотензиновая система и натрийуретические пептиды при гипертонии, связанной с ожирением. J Mol Med. 2001; 7921-29PubMedGoogle ScholarCrossref 74.Engeli
СНегрель
RSharma
AM Физиология и патофизиология ренин-ангиотензиновой системы жировой ткани. Гипертония. 2000; 351270-1277PubMedGoogle ScholarCrossref 75.Цена
DALansang
MCOsei
SY
и другие. Диабет 2 типа, ожирение и реакция почек на блокирование рениновой системы ирбесартаном. Diabet Med. 2002; 19858-862PubMedGoogle ScholarCrossref 77.Стивенс
MSRaffel
DMAllman
KC
и другие. Симпатическая дисиннервация сердца при диабете: последствия для повышенного сердечно-сосудистого риска. Тираж. 1998; 98961-968PubMedGoogle ScholarCrossref 78.Zola
BKahn
JKJuni
JE
и другие. Нарушение сердечной функции у больных сахарным диабетом с вегетативной нейропатией при отсутствии ишемической болезни сердца. J Clin Endocrinol Metab. 1986; 63208-214PubMedGoogle ScholarCrossref 79.О’Брайен
IAMcFadden
JPorral
RJM Влияние вегетативной нейропатии на смертность при инсулинозависимом диабете. Q J Med. 1991; 79495-502Google Scholar 80.Rathman
WZiegler
DJahnke
M
и другие. Смертность у больных сахарным диабетом с сердечно-сосудистой вегетативной нейропатией. Diabet Med. 1993; 10820-824PubMedGoogle ScholarCrossref 81.Monteagudo
Шумы
В.А.Кольманн
О
и другие. Влияние вегетативной нейропатии на дисфункцию левого желудочка у пациентов с инсулинозависимым диабетом. Clin Cardiol. 2000; 23371-375PubMedGoogle ScholarCrossref 82.Scognamiglio
Равогаро
ACasara
D
и другие. Дисфункция миокарда и адренергическая иннервация сердца у пациентов с инсулинозависимым сахарным диабетом. J Am Coll Cardiol. 1998; 31404-412PubMedGoogle ScholarCrossref 83.Toyry
JPNiskanen
LKMantysaari
MJ
и другие. Возникновение, предикторы и клиническое значение вегетативной нейропатии при NIDDM. Диабет. 1996; 45308-315PubMedGoogle ScholarCrossref 84.Ewing
DJCampbell
И. В. Кларк
BF Оценка сердечно-сосудистых эффектов при диабетической вегетативной нейропатии и прогностические последствия. Ann Intern Med. 1980; -311PubMedGoogle ScholarCrossref 86.Airaksinen
KEJ Тихая ишемическая болезнь сердца при диабете: признак вегетативной нейропатии или ускоренный атеросклероз? Diabetologia. 2001; 44259-266PubMedGoogle ScholarCrossref 88.Kannel
WBHjortland
MCastelli
WP Роль диабета в застойной сердечной недостаточности: исследование Framingham. Am J Cardiol. 1974; 3429-34PubMedGoogle ScholarCrossref 91.Regan
TJEttinger
POKahn
MI
и другие. Нарушение функции миокарда и метаболизма при хроническом сахарном диабете без ишемии у собак. Circ Res. 1974; 35222-237Google ScholarCrossref 92.Jackson
CVMcGrath
GMTahiliani
AG
и другие. Функциональный и ультраструктурный анализ экспериментального диабетического миокарда крысы. Диабет. 1985; 34876-883PubMedGoogle ScholarCrossref 93.Ren
JBode
AM Измененная связь возбуждения и сокращения сердца в миоцитах желудочков у крыс BB со спонтанным диабетом. Am J Physiol Heart Circ Physiol. 2000; 279h338- h344PubMedGoogle Scholar94.Kawano
Хирасима
TMori
S
и другие. Крысы со спонтанной длительной гипергликемией с диабетическими осложнениями: линия Оцука Лонг-Эванс Токусима Фатти (OLETF). Диабет. 1993; 411422-1428Google ScholarCrossref 95.Mizushige
Кяо
LNoma
Т
и другие.Изменение диастолического наполнения левого желудочка и накопление миокардиального коллагена на инсулинорезистентной предиабетической стадии на модели крыс с диабетом II типа. Тираж. 2000; 101899-907PubMedGoogle ScholarCrossref 96.Shapiro
LMHowat
APCalter
М. М. Функция левого желудочка при сахарном диабете I: методология, распространенность и спектр отклонений. Br Heart J. 1981; 45122-128PubMedGoogle ScholarCrossref 97.Shapiro
LMЛезердейл
BAMackinnon
J
и другие.Функция левого желудочка при сахарном диабете II: связь между клиническими особенностями и функцией левого желудочка. Br Heart J. 1981; 45129-132PubMedGoogle ScholarCrossref 98.Shapiro
Л.М. Эхокардиографические особенности нарушения функции желудочков при сахарном диабете. Br Heart J. 1982; 47439-444PubMedGoogle ScholarCrossref 101.Galderisi
MAnderson
KMWilson
PWLevy
D Эхокардиографические доказательства существования отчетливой диабетической кардиомиопатии (Framingham Heart Study). Am J Cardiol. 1991; 6885-89PubMedGoogle ScholarCrossref 102.Borow
KMJaspan
JBWilliam
KA
и другие. Механика миокарда у молодых взрослых пациентов с сахарным диабетом. J Am Coll Cardiol. 1990; 151508-1517PubMedGoogle ScholarCrossref 105.Bell
Д.С. Диабетическая кардиомиопатия: уникальное явление или осложнение ишемической болезни сердца? Уход за диабетом. 1995; 18708-714PubMedGoogle ScholarCrossref 106.Raev
DC Какая функция левого желудочка нарушена на более раннем этапе развития диабетической кардиомиопатии? Уход за диабетом. 1994; 17633-639PubMedGoogle ScholarCrossref 107.Poirier
ПБогаты
PGarneau
C
и другие. Диастолическая дисфункция у мужчин с нормальным давлением и хорошо контролируемым диабетом 2 типа. Уход за диабетом. 2001; 15-10Google ScholarCrossref 108.Hirai
Джуеда
К.Такегоши
Т
и другие. Влияние метаболического контроля на функцию желудочков у пациентов с диабетом 2 типа. Intern Med. 1992; 31725-730PubMedGoogle ScholarCrossref 109.Uusitupa
MSiitonen
ОАро
А
и другие.Влияние коррекции гипергликемии на функцию левого желудочка у инсулиннезависимых (тип 2) диабетиков. Acta Med Scand. 1983; 213363-368PubMedGoogle ScholarCrossref 110.Dhalla
NSLiu
XPanagia
V
и другие. Субклеточное ремоделирование и дисфункция сердца при хроническом диабете. Cardiovasc Res. 1998; 40239-247PubMedGoogle ScholarCrossref 112.Wakasaki
HKoya
DSchuen
FJ
и другие. Нацеленная сверхэкспрессия изоформы протеинкиназы C в миокарде вызывает кардиомиопатию. Proc Natl Acad Sci U S A. 1997; 949320-9325PubMedGoogle ScholarCrossref 114.Opie
LHParving
HH Диабетическая нефропатия: можно ли экстраполировать ренопротекцию на защиту сердечно-сосудистой системы? Тираж. 2002; 106643-645PubMedGoogle ScholarCrossref 115.Ruilope
LMvan Veldhuisen
DJRitz
E
и другие. Функция почек: Золушка профиля риска сердечно-сосудистых заболеваний. J Am Coll Cardiol. 2001; 381782-1787PubMedGoogle ScholarCrossref 116.Группа исследований по контролю и осложнениям диабета, Влияние интенсивного лечения диабета на развитие и прогрессирование отдаленных осложнений инсулинозависимого сахарного диабета. N Engl J Med. 1993; 329977-986PubMedGoogle ScholarCrossref 117. UK Prospective Diabetes Study Group, Эффективность атенолола и каптоприла в снижении риска макрососудистых осложнений при диабете 2 типа: UKPDS 39. BMJ. 1998; 317713-720PubMedGoogle ScholarCrossref 118.Кокс
MJde Zeeuw
DNavis
GJ Оптимальный контроль артериального давления и антигипертензивные режимы при гипертонической болезни почек. Curr Opin Nephrol Hypertens. 2002; 11135-140PubMedGoogle ScholarCrossref 119.Wright
JTBakris
GLGreene
Т
и другие. Афро-американская группа по изучению заболеваний почек и гипертонии, Эффект снижения артериального давления и класса антигипертензивных препаратов на прогрессирование гипертонической болезни почек. JAMA. 2002; 2882421-2431PubMedGoogle ScholarCrossref 120.Lewis
EJHunsicker
LGBain
RP
и другие. Влияние ингибитора ангиотензин-превращающего фермента на диабетическую нефропатию. N Engl J Med. 1993; 32
-1462PubMedGoogle ScholarCrossref 121.EUCLID Study Group, Рандомизированное плацебо-контролируемое исследование лизиноприла у пациентов с нормальным давлением, диабетом и нормоальбуминурией или микроальбуминурией. Ланцет. 1997; 34
-1792PubMedGoogle ScholarCrossref 122.Могенсен
CENeldam
STikkanen
я
и другие. Рандомизированное контролируемое исследование двойной блокады ренин-ангиотензиновой системы у пациентов с артериальной гипертензией, микроальбуминурией и инсулинозависимым диабетом: исследование кандесартана и лизиноприла микроальбуминурии (CALM). BMJ. 2000; 3211440-1444PubMedGoogle ScholarCrossref 123. Исследователи исследования RENAAL, Эффекты лозартана на почечные и сердечно-сосудистые исходы у пациентов с диабетом 2 типа и нефропатией. N Engl J Med. 2001; 345861-869PubMedGoogle ScholarCrossref 124.Lewis
EJHunsicker
LGClarke
WR
и другие. Совместная исследовательская группа, Ренопротективные эффекты блокатора рецепторов ангиотензина ирбесартана у пациентов с нефропатией, вызванной диабетом 2 типа. N Engl J Med. 2001; 345851-860PubMedGoogle ScholarCrossref 125. Ирбесартан у пациентов с диабетом 2 типа и микроальбуминурией. Группа исследования, Влияние ирбесартана на развитие диабетической нефропатии у пациентов с диабетом 2 типа. N Engl J Med. 2001; 345870-878PubMedGoogle ScholarCrossref 126. Исследователи исследования MARVAL, Снижение микроальбуминурии с помощью валсартана у пациентов с сахарным диабетом 2 типа. Тираж. 2002; 106672-678PubMedGoogle ScholarCrossref 127.Haffner
С.М.Гринберг
ASWeston
WM
и другие. Влияние лечения розиглитазоном на нетрадиционные маркеры сердечно-сосудистых заболеваний у пациентов с сахарным диабетом 2 типа. Тираж. 2002; 106679-684PubMedGoogle ScholarCrossref 128.Мурасе
Кодака
HSuzuki
N
и другие. Пиоглитазон со временем снижает уровень фактора некроза опухоли α в мышцах и улучшает метаболические нарушения у жирных крыс линии Вистар. Diabetologia. 1998; 41257-264PubMedGoogle ScholarCrossref 129.Pasceri
VWu
HDWillerson
JT
и другие. Модуляция сосудистого воспаления in vitro и in vivo с помощью активаторов рецептора-γ, активируемых пролифератором пероксисом. Тираж. 2000; 101235-238PubMedGoogle ScholarCrossref 130.Неве
BPCorseaux
DChinetti
грамм
и другие. Агонисты PPAR-α подавляют экспрессию тканевого фактора в моноцитах и макрофагах человека. Тираж. 2001; 103207-212PubMedGoogle ScholarCrossref 131.Tham
DMMartin-McNulty
BWang
Y
и другие. Ангиотензин II связан с активацией генов, опосредованных NF-κB, и подавлением PPAR. Physiol Genomics. 2002; 1121-30PubMedGoogle Scholar132.MacFadyen
RJLee
AFCMorton
JJ
и другие.Как часто повышаются концентрации ангиотензина II и альдостерона при хронической терапии ингибиторами АПФ при сердечной недостаточности? Сердце. 1999; 8257-61PubMedGoogle Scholar133.Jorde
UPVittorio
TKatz
SD
и другие. Повышенный уровень альдостерона в плазме, несмотря на полное ингибирование сосудистого ангиотензин-превращающего фермента при хронической сердечной недостаточности. Тираж. 2002; 1061055-1057PubMedGoogle ScholarCrossref 134. Исследовательская группа CONSENSUS, Гормоны, регулирующие сердечно-сосудистую функцию у пациентов с тяжелой застойной сердечной недостаточностью, и их связь со смертностью. Тираж. 1990; 821730-1736PubMedGoogle ScholarCrossref 135.Pitt
BZannad
FRemme
WJ
и другие. Влияние спиронолактона на заболеваемость и смертность у пациентов с тяжелой застойной сердечной недостаточностью. N Engl J Med. 1999; 341709-717PubMedGoogle ScholarCrossref 136.Rincon-Choles
HKasinath
Б.С.Горин
Y
и другие. Ангиотензин II и факторы роста в патогенезе диабетической нефропатии. Kidney Int Suppl. 2002; 828-11PubMedGoogle ScholarCrossref 137. Группа экспериментального исследования КОНСЕНСУС, Влияние эналаприла на смертность при тяжелой застойной сердечной недостаточности: результаты Совместного североскандинавского исследования выживаемости с применением эналаприла (КОНСЕНСУС). N Engl J Med. 1987; 3161429-1435PubMedGoogle ScholarCrossref 138.SOLVD Investigators, Влияние эналаприла на выживаемость у пациентов со сниженной фракцией выброса левого желудочка и застойной сердечной недостаточностью. N Engl J Med. 1991; 325293-302PubMedGoogle ScholarCrossref 139.Исследователи СОЛВД, Влияние эналаприла на смертность и развитие сердечной недостаточности у бессимптомных пациентов со сниженной фракцией выброса левого желудочка. N Engl J Med. 1992; 327685-691PubMedGoogle ScholarCrossref 140. Исследовательская группа AIRE, Влияние рамиприла на смертность и заболеваемость выживших после острого инфаркта миокарда с клиническими доказательствами сердечной недостаточности. Ланцет. 1993; 342821-828PubMedGoogle Scholar141.SAVE Investigators, Влияние каптоприла на смертность и заболеваемость у пациентов с дисфункцией левого желудочка после инфаркта миокарда. N Engl J Med. 1992; 327669-677PubMedGoogle ScholarCrossref 142. Руководящий комитет OPTIMAAL Исследовательской группы OPTIMAAL, Влияние лозартана и каптоприла на смертность и заболеваемость у пациентов из группы высокого риска после острого инфаркта миокарда: рандомизированное исследование OPTIMAAL. Ланцет. 2002; 360752-760PubMedGoogle ScholarCrossref 143.Pitt
Б.П.ул-Уилсон
ПАСегал
р
и другие. Влияние лозартана по сравнению с каптоприлом на смертность у пациентов с симптоматической сердечной недостаточностью: рандомизированное исследование — исследование выживания при сердечной недостаточности с лозартаном ELITE II. Ланцет. 2000; 3551582-1587PubMedGoogle ScholarCrossref 144.Valsartan Heart Failure Trial Investigators, рандомизированное исследование валсартана, блокатора рецепторов ангиотензина, при хронической сердечной недостаточности. N Engl J Med. 2001; 3451667-1675PubMedGoogle ScholarCrossref 145.Cappuccio
FPCook
Д. Г. Аткинсон
RW
и другие. Распространенность, выявление и лечение факторов риска сердечно-сосудистых заболеваний в различных этнических группах на юге Лондона. Сердце. 1997; 78555-563PubMedGoogle Scholar146.Балараджан
R Этнические различия в смертности от ишемической болезни сердца и цереброваскулярных заболеваний в Англии и Уэльсе. BMJ. 1991; 302560-564PubMedGoogle ScholarCrossref 147. ВСЕ должностные лица и координаторы; Группа совместных исследований ALLHAT, Основные результаты у пациентов с высоким риском гипертонии, рандомизированных для приема ингибитора ангиотензинпревращающего фермента или блокатора кальциевых каналов по сравнению с диуретиком: гипотензивное и гиполипидемическое лечение для предотвращения сердечного приступа (ALLHAT). JAMA. 2002; 2882981-2997PubMedGoogle ScholarCrossref 148.Douglas
JGBakris
Гепштейн
M
и другие. Гипертония в рабочей группе афроамериканцев, Управление высоким кровяным давлением у афроамериканцев: консенсусное заявление Рабочей группы по гипертонии у афроамериканцев Международного общества по гипертонии у чернокожих. Arch Intern Med. 2003; 163525-541PubMedGoogle ScholarCrossref 149.Epstein
AEBigger
JTWyse
DG
и другие.События в испытании подавления сердечной аритмии (CAST): смертность во всем включенном населении. J Am Coll Cardiol. 1991; 1814-1919PubMedGoogle ScholarCrossref
Система ренин-ангиотензин (РАА) и артериальное давление
Система ренин-ангиотензин (РАА) — это группа родственных гормонов, которые действуют вместе, регулируя кровяное давление. Это называется системой, потому что каждая часть влияет на другие части, и все они необходимы для правильного функционирования целого. Ренин-ангиотензиновая система, работающая вместе с почками, является жизненно важной частью системы регуляции кровяного давления в организме.Взаимодействие с другими людьми
Хотя краткосрочные изменения артериального давления вызваны множеством факторов, почти все долгосрочные изменения артериального давления являются обязанностью почек и ренин-ангиотензиновой системы.
Изображения героев / Getty Images
Как работает ренин-ангиотензиновая система
Важными членами ренин-ангиотензиновой системы являются:
- Ренин
- Ангиотензин I
- Ангиотензин II
- Ангиотензинпревращающий фермент (АПФ)
Когда по какой-либо причине артериальное давление падает, специальные клетки в почках обнаруживают это изменение и выпускают ренин в кровоток.Сам по себе ренин не влияет на артериальное давление.
Вместо этого он плавает вокруг и превращает неактивные формы ангиотензина в ангиотензин I. Эти неактивные формы ангиотензина, которые вырабатываются печенью, не способны изменять кровяное давление до тех пор, пока ренин не превратит их в ангиотензин I.
Ангиотензин I способен до некоторой степени изменять кровяное давление, но он недостаточно силен, чтобы вызывать большие изменения. Вместо этого большая часть ангиотензина I превращается в ангиотензин II, гораздо более мощный гормон, который действительно вызывает большие изменения артериального давления.
Это второе преобразование происходит в основном в легких под действием другой молекулы, называемой ангиотензин-превращающим ферментом (АПФ). (Это преобразование может быть остановлено лекарствами, называемыми ингибиторами АПФ, важным типом лекарств от высокого кровяного давления).
Ангиотензин II — сильный гормон, который может воздействовать непосредственно на кровеносные сосуды, повышая кровяное давление. У него есть еще одна важная функция — стимуляция высвобождения альдостерона.
Альдостерон является очень сильным сосудосуживающим средством, которое вызывает значительное повышение артериального давления, но более важно, потому что он может фактически изменить базовую фильтрующую активность почек.Альдостерон заставляет почки удерживать как соль, так и воду, что со временем увеличивает количество жидкости в организме. Это увеличение, в свою очередь, повышает кровяное давление.
Через некоторое время ангиотензин I, ангиотензин II и альдостерон расщепляются на другие молекулы. Ренин-ангиотензиновая система в целом реагирует как на краткосрочные, так и на долгосрочные изменения артериального давления. Он активируется при резких падениях артериального давления, например, после кровопотери, но также стимулируется меньшими, менее резкими колебаниями артериального давления.
Ренин-ангиотензиновая система как долговременный регулятор артериального давления имеет постоянный базовый уровень активности и фактически работает так же, как педаль газа в автомобиле. Постоянное нажатие на педаль газа необходимо, чтобы автомобиль двигался вперед, даже если вы просто хотите ехать с той же скоростью.
Однако, если вам нужно, вы можете резко нажать на педаль, чтобы быстро ускориться. Точно так же постоянная активность ренин-ангиотензиновой системы поддерживает стабильное кровяное давление в течение длительного времени, но возможны внезапные всплески активности, когда требуется быстрый ответ.
Почему система RAA важна при высоком кровяном давлении
О важности ренин-ангиотензиновой системы в регуляции артериального давления написаны научные статьи, презентации на конференциях и даже целые учебники. Эта область исследований все еще продолжается более чем через 50 лет после открытия системы.
Ренин-ангиотензиновая система привлекает столько внимания, потому что, как известно, она является важным фактором, который может помочь нам понять:
- Почему у людей вообще развивается высокое кровяное давление
- Почему некоторые люди плохо реагируют на обычное лечение высокого кровяного давления
- Почему у некоторых людей с высоким артериальным давлением развивается больше осложнений, чем у других
Например, афроамериканские пациенты с высоким кровяным давлением часто не реагируют на ингибиторы АПФ так же хорошо, как на другие лекарства.Вероятно, это связано с тем, что у афроамериканцев другой уровень активности ренин-ангиотензиновой системы, что делает их менее чувствительными к лекарствам, которые действуют, блокируя систему.
Ряд эффективных методов лечения высокого кровяного давления был разработан как прямой результат нашего понимания ренин-ангиотензиновой системы. Наряду с ингибиторами АПФ, которые останавливают превращение ангиотензина I в ангиотензин II, другие препараты действуют, воздействуя на различные части системы. Блокаторы рецепторов ангиотензина (БРА), например, предотвращают связывание ангиотензина I и ангиотензина II с кровеносными сосудами. и вызывая сужение сосудов.
Хотя тонкие детали системы ренин-ангиотензин все еще открываются, наше понимание этого важного регулирующего механизма уже привело к разработке нескольких методов лечения высокого кровяного давления и лучшему пониманию того, как управлять высоким кровяным давлением в долгосрочной перспективе.
.