Бета-талассемия — Beta thalassemia — qaz.wiki
Талассемия, характеризующаяся сниженным или отсутствующим синтезом бета-глобиновых цепей гемоглобина
| Бета-талассемия | |
|---|---|
| Другие имена | Микроцитемия, бета-тип |
| Генетика бета-талассемии, изображение показывает один пример того, как бета-талассемия передается по наследству. Ген бета-глобина расположен на хромосоме 11. Ребенок наследует два гена бета-глобина (по одному от каждого родителя). | |
| Специальность | Гематология |
| Типы | Малая, средняя и большая талассемия |
| Причины | Мутации в гене HBB |
| Диагностический метод | Анализ ДНК |
| лечение | Зависит от типа (см. Типы) |
Бета-талассемии ( β-талассемии ) — это группа наследственных заболеваний крови . Они являются одной из форм талассемии , вызванные снижением или полным отсутствием синтеза бета — цепей из гемоглобина , которые приводят к переменным результатам , начиная от тяжелой анемии клинически бессимптомных лиц. Ежегодная заболеваемость во всем мире оценивается в 1 случай на 100 000. Бета-талассемия возникает из-за нарушения функции бета-субъединицы гемоглобина или HBB. Степень тяжести заболевания зависит от характера мутации.
Они являются одной из форм талассемии , вызванные снижением или полным отсутствием синтеза бета — цепей из гемоглобина , которые приводят к переменным результатам , начиная от тяжелой анемии клинически бессимптомных лиц. Ежегодная заболеваемость во всем мире оценивается в 1 случай на 100 000. Бета-талассемия возникает из-за нарушения функции бета-субъединицы гемоглобина или HBB. Степень тяжести заболевания зависит от характера мутации.
Блокада HBB со временем приводит к снижению синтеза бета-цепи. Неспособность организма строить новые бета-цепи приводит к недостаточной продукции HbA . Снижение уровня HbA, доступного для наполнения красных кровяных телец, в свою очередь, приводит к микроцитарной анемии . Микроцитарная анемия в конечном итоге развивается из-за недостаточного количества белка HBB для достаточного функционирования красных кровяных телец. Из-за этого фактора пациенту может потребоваться переливание крови, чтобы восполнить блокировку бета-цепей. Повторные переливания крови вызывают серьезные проблемы, связанные с перегрузкой железом .
Признаки и симптомы
Рука человека с тяжелой анемией (слева, в кольце) по сравнению с рукой без
Описаны три основные формы: большая талассемия, промежуточная талассемия и малая талассемия. Все люди с талассемией подвержены осложнениям со здоровьем, связанным с селезенкой (которая часто увеличивается и часто удаляется) и желчными камнями . Эти осложнения чаще всего встречаются у пациентов с большой и средней талассемией. Люди с большой бета-талассемией обычно в течение первых двух лет жизни проявляют тяжелую анемию, замедленный рост и аномалии скелета в младенчестве. Отсутствие лечения большой талассемии в конечном итоге приводит к смерти, обычно от сердечной недостаточности ; поэтому скрининг при рождении очень важен.
Избыток железа вызывает серьезные осложнения со стороны печени, сердца и эндокринных желез . Тяжелые симптомы включают цирроз печени , фиброз печени , и в крайних случаях, рак печени . Сердечная недостаточность, нарушение роста, диабет и остеопороз — опасные для жизни состояния, которые могут быть вызваны ТМ. Основные сердечные аномалии, наблюдаемые в результате талассемии и перегрузки железом, включают систолическую и диастолическую дисфункцию левого желудочка, легочную гипертензию, вальвулопатию, аритмии и перикардит. Повышенное всасывание железа в желудочно-кишечном тракте наблюдается при всех степенях бета-талассемии, а повышенное разрушение эритроцитов селезенкой из-за неэффективного эритропоэза дополнительно высвобождает дополнительное количество железа в кровоток .
Основные сердечные аномалии, наблюдаемые в результате талассемии и перегрузки железом, включают систолическую и диастолическую дисфункцию левого желудочка, легочную гипертензию, вальвулопатию, аритмии и перикардит. Повышенное всасывание железа в желудочно-кишечном тракте наблюдается при всех степенях бета-талассемии, а повышенное разрушение эритроцитов селезенкой из-за неэффективного эритропоэза дополнительно высвобождает дополнительное количество железа в кровоток .
Причина
Мутации
Можно выделить две основные группы мутаций:
- Неделеционные формы: эти дефекты, как правило, связаны с заменой одного основания или небольшими вставками рядом или выше гена β-глобина. Чаще всего мутации происходят в промоторных областях, предшествующих генам бета-глобина. Реже считается, что заболеванию способствуют аномальные варианты сплайсинга.
- Формы делеции: Делеции разного размера с участием гена β-глобина вызывают различные синдромы, такие как (β o ) или наследственная персистенция синдромов гемоглобина плода .


Аллели без мутации, снижающей функцию, характеризуются как (β). Мутации обозначаются (βo), если они предотвращают образование β-цепей, мутации обозначаются как (β +), если они допускают образование некоторых β-цепей.
| имя | Старые синонимы | Описание | Аллели |
|---|---|---|---|
| Малая талассемия | Гетерозиготная форма: только один из аллелей β-глобина несет мутацию. Люди будут страдать от микроцитарной анемии . Обнаружение обычно предполагает более низкое, чем обычно, значение среднего корпускулярного объема (<80 мкл). | β + / β β о / β | |
| Промежуточная талассемия | Больные часто могут вести нормальный образ жизни, но могут нуждаться в периодических переливаниях, например, во время болезни или беременности, в зависимости от степени тяжести анемии. | β + / β + β o / β + | |
| Большая талассемия | Средиземноморская анемия; Анемия Кули | Гомозиготная форма : возникает, когда оба аллеля имеют мутации талассемии. Это тяжелая микроцитарная гипохромная анемия. Без лечения он вызывает анемию, спленомегалию и серьезные деформации костей. До 20 лет заболевание прогрессирует до смерти. Лечение заключается в периодическом переливании крови ; спленэктомия при спленомегалии и хелатирование перегрузки железом, связанной с переливанием крови. Это тяжелая микроцитарная гипохромная анемия. Без лечения он вызывает анемию, спленомегалию и серьезные деформации костей. До 20 лет заболевание прогрессирует до смерти. Лечение заключается в периодическом переливании крови ; спленэктомия при спленомегалии и хелатирование перегрузки железом, связанной с переливанием крови. | β о / β о |
сборка мРНК
Протеин HBB PDB 1a00: это здоровый белок бета-глобина
Бета-талассемия — это наследственное заболевание, влияющее на гемоглобин. Как и в случае примерно половины всех наследственных заболеваний, наследственная мутация повреждает сборку РНК-мессенджера (мРНК), которая транскрибируется с хромосомы . ДНК содержит как инструкции ( гены ) для связывания аминокислот в белки , так и участки ДНК, которые играют важную роль в регулировании уровней продуцируемого белка .
При талассемии в мРНК включается дополнительная непрерывная длина или прерывистый фрагмент некодирующих инструкций.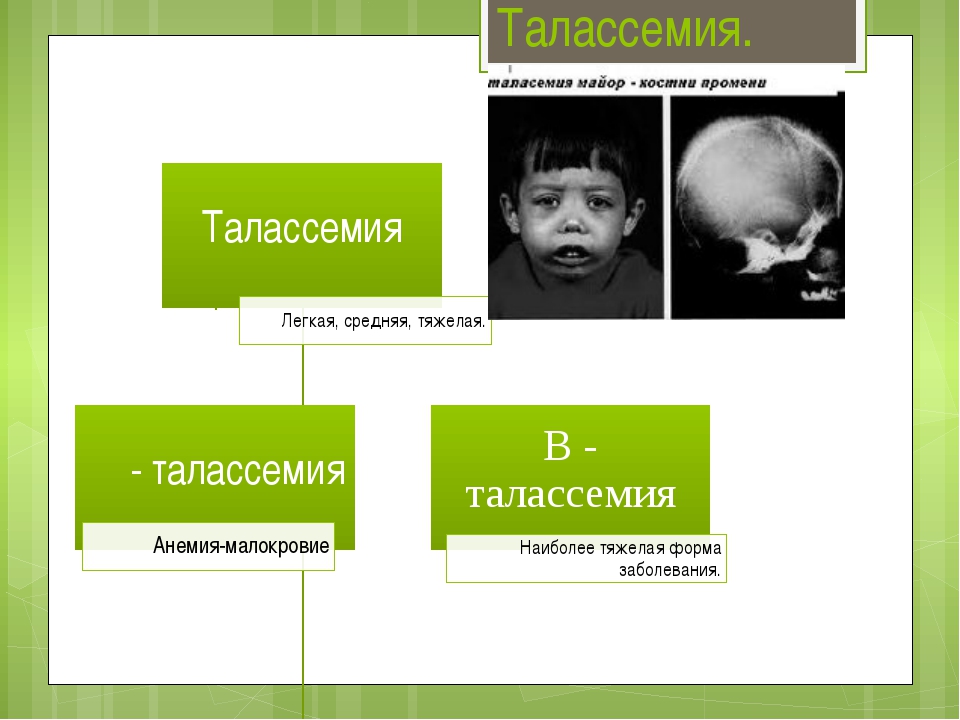 Это происходит потому, что мутация стирает границу между интронной и экзонной частями матрицы ДНК. Поскольку все кодирующие участки могут все еще присутствовать, может образовываться нормальный гемоглобин, а добавленный генетический материал, если он вызывает патологию, вместо этого нарушает регуляторные функции в достаточной степени, чтобы вызвать анемию. Каждая нормальная альфа- и бета-субъединица гемоблогина имеют железосодержащую центральную часть (гем), которая позволяет белковой цепи субъединицы складываться вокруг нее. Нормальный взрослый гемоглобин содержит 2 альфа и 2 бета субъединицы. Талассемии обычно влияют только на мРНК для производства бета-цепей (отсюда и название). Поскольку мутация может быть изменением только одного основания ( однонуклеотидный полиморфизм ), текущие усилия направлены на поиск генных терапий для внесения этой единственной коррекции.
Это происходит потому, что мутация стирает границу между интронной и экзонной частями матрицы ДНК. Поскольку все кодирующие участки могут все еще присутствовать, может образовываться нормальный гемоглобин, а добавленный генетический материал, если он вызывает патологию, вместо этого нарушает регуляторные функции в достаточной степени, чтобы вызвать анемию. Каждая нормальная альфа- и бета-субъединица гемоблогина имеют железосодержащую центральную часть (гем), которая позволяет белковой цепи субъединицы складываться вокруг нее. Нормальный взрослый гемоглобин содержит 2 альфа и 2 бета субъединицы. Талассемии обычно влияют только на мРНК для производства бета-цепей (отсюда и название). Поскольку мутация может быть изменением только одного основания ( однонуклеотидный полиморфизм ), текущие усилия направлены на поиск генных терапий для внесения этой единственной коррекции.
Факторы риска
Семейный анамнез и происхождение являются факторами, повышающими риск бета-талассемии. В зависимости от семейного анамнеза, если у родителей или бабушек и дедушек была большая или промежуточная бета-талассемия, существует вероятность 75% (3 из 4) (см. Диаграмму наследования вверху страницы) того, что мутировавший ген унаследуется потомством. Даже если у ребенка нет большой или промежуточной бета-талассемии, он все равно может быть носителем, что может привести к тому, что будущие поколения их потомков будут иметь бета-талассемию.
Диаграмму наследования вверху страницы) того, что мутировавший ген унаследуется потомством. Даже если у ребенка нет большой или промежуточной бета-талассемии, он все равно может быть носителем, что может привести к тому, что будущие поколения их потомков будут иметь бета-талассемию.
Еще один фактор риска — это происхождение. Бета-талассемия чаще всего встречается у людей итальянского, греческого, ближневосточного, южноазиатского и африканского происхождения.
Диагностика
Боль в животе из-за гиперспленизма , инфаркта селезенки и боль в правом подреберье, вызванная желчными камнями, являются основными клиническими проявлениями. Однако диагностировать талассемию только на основании симптомов недостаточно. Врачи считают эти признаки ассоциативными из-за сложности заболевания. Следующие ассоциативные признаки могут свидетельствовать о тяжести фенотипа : бледность, плохой рост, недостаточное потребление пищи, спленомегалия , желтуха , гиперплазия верхней челюсти, неправильный прикус зубов , желчнокаменная болезнь , шум систолического выброса при тяжелой анемии и патологических переломах.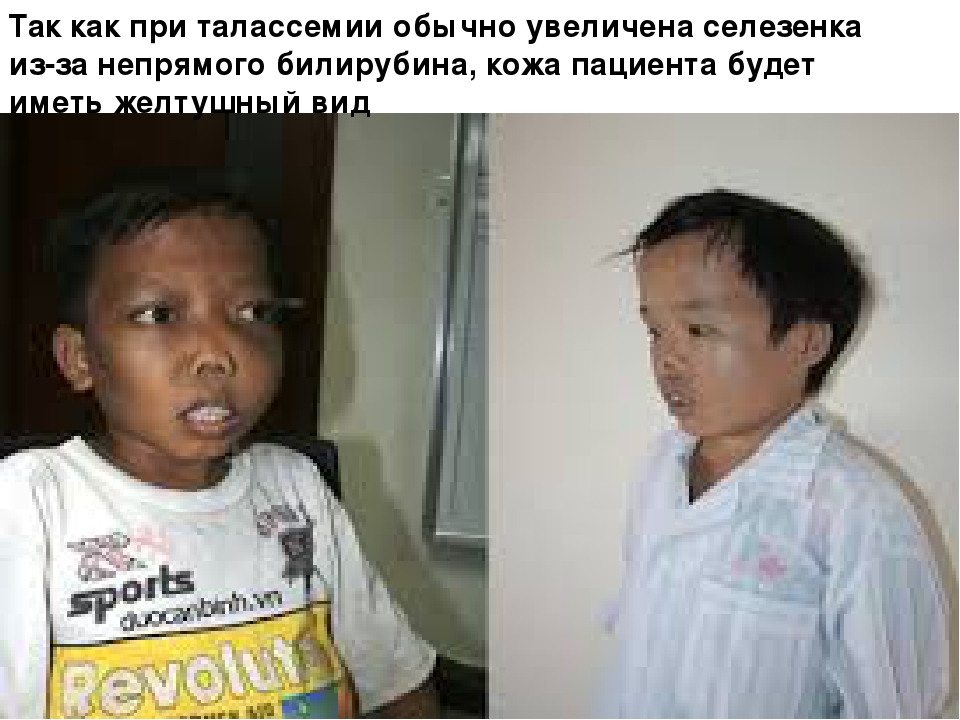 На основании симптомов назначаются анализы для дифференциальной диагностики . Эти анализы включают полный анализ крови ; электрофорез гемоглобина ; трансферрин сыворотки , ферритин , общая железосвязывающая способность ; мочи уробилина и urobilogen; мазок периферической крови , который может показать кодоциты или клетки-мишени; гематокрит ; и билирубин сыворотки. Ожидаемый рисунок на электрофорезе гемоглобина у людей с бета-талассемией является повышенным уровнем гемоглобина А2 и незначительно увеличился гемоглобин F .
На основании симптомов назначаются анализы для дифференциальной диагностики . Эти анализы включают полный анализ крови ; электрофорез гемоглобина ; трансферрин сыворотки , ферритин , общая железосвязывающая способность ; мочи уробилина и urobilogen; мазок периферической крови , который может показать кодоциты или клетки-мишени; гематокрит ; и билирубин сыворотки. Ожидаемый рисунок на электрофорезе гемоглобина у людей с бета-талассемией является повышенным уровнем гемоглобина А2 и незначительно увеличился гемоглобин F .
Изменения скелета, связанные с расширением костного мозга:
- лицо бурундука : выпуклость черепа, выдающееся скуловое возвышение, депрессия переносицы, склонность к монголоидному наклону глаза и обнажение верхних зубов из-за гипертрофии верхних челюстей.
- волосы на конце (или «ежик») на рентгеновском снимке черепа: образование новой кости из-за внутреннего стола.
Анализ ДНК
При всех бета-талассемии могут быть выявлены аномальные эритроциты, семейный анамнез сопровождается анализом ДНК. Этот тест используется для исследования делеций и мутаций в генах, продуцирующих альфа- и бета-глобин. Семейные исследования могут быть проведены для оценки статуса носителя и типов мутаций, присутствующих у других членов семьи. ДНК-тестирование не является рутинным, но может помочь диагностировать талассемию и определить статус носителя. В большинстве случаев лечащий врач использует предварительный клинический диагноз для оценки симптомов анемии: утомляемости, одышки и плохой переносимости физических нагрузок. Дальнейший генетический анализ может включать ВЭЖХ, если рутинный электрофорез окажется затруднительным.
Этот тест используется для исследования делеций и мутаций в генах, продуцирующих альфа- и бета-глобин. Семейные исследования могут быть проведены для оценки статуса носителя и типов мутаций, присутствующих у других членов семьи. ДНК-тестирование не является рутинным, но может помочь диагностировать талассемию и определить статус носителя. В большинстве случаев лечащий врач использует предварительный клинический диагноз для оценки симптомов анемии: утомляемости, одышки и плохой переносимости физических нагрузок. Дальнейший генетический анализ может включать ВЭЖХ, если рутинный электрофорез окажется затруднительным.
Профилактика
Бета-талассемия — это наследственное заболевание, позволяющее проводить профилактическое лечение путем скрининга на носительство и пренатальной диагностики. Это можно предотвратить, если у одного из родителей есть нормальные гены, что дает основания для скрининга, позволяющего носителям выбирать партнеров с нормальным гемоглобином. Исследование направлено на выявление генов, которые могут дать потомство с серповидно-клеточной анемией. Пациенты с диагнозом бета-талассемия имеют MCH ≤ 26 пг и RDW <19. Из 10 148 пациентов у 1739 пациентов был фенотип гемоглобина и RDW, соответствующие бета-талассемии. После сужения числа пациентов уровень HbA2 был протестирован на 77 пациентах с бета-талассемией. Эта процедура скрининга оказалась нечувствительной в популяциях западноафриканского происхождения из-за высокой распространенности показателей альфа-талассемии. В странах есть программы распространения информации о репродуктивных рисках, связанных с носителями гемоглобинопатий. Программы скрининга носителей талассемии включают образовательные программы в школах, вооруженных силах и через средства массовой информации, а также консультирование носителей и пары носителей. Скрининг показал снижение заболеваемости; к 1995 г. распространенность в Италии снизилась с 1: 250 до 1: 4000, а в этом регионе — на 95%. Снижение заболеваемости принесло пользу больным талассемией, поскольку потребность в крови снизилась, что улучшило предложение лечения.
Пациенты с диагнозом бета-талассемия имеют MCH ≤ 26 пг и RDW <19. Из 10 148 пациентов у 1739 пациентов был фенотип гемоглобина и RDW, соответствующие бета-талассемии. После сужения числа пациентов уровень HbA2 был протестирован на 77 пациентах с бета-талассемией. Эта процедура скрининга оказалась нечувствительной в популяциях западноафриканского происхождения из-за высокой распространенности показателей альфа-талассемии. В странах есть программы распространения информации о репродуктивных рисках, связанных с носителями гемоглобинопатий. Программы скрининга носителей талассемии включают образовательные программы в школах, вооруженных силах и через средства массовой информации, а также консультирование носителей и пары носителей. Скрининг показал снижение заболеваемости; к 1995 г. распространенность в Италии снизилась с 1: 250 до 1: 4000, а в этом регионе — на 95%. Снижение заболеваемости принесло пользу больным талассемией, поскольку потребность в крови снизилась, что улучшило предложение лечения.
лечение
Бета-талассемия
Хирургически удалена селезенка у ребенка с талассемией. Это примерно в 15 раз больше обычного.
Больным детям требуется регулярное переливание крови на протяжении всей жизни, и у них могут быть осложнения, в том числе со стороны селезенки. Трансплантация костного мозга может быть излечивающей для некоторых детей. Пациентам часто делают переливания крови, что приводит к перегрузке железом или усиливает ее . Лечение хелатированием железа необходимо для предотвращения повреждения внутренних органов. Достижения в области хелатирования железа позволяют пациентам с большой талассемией прожить долгую жизнь с доступом к надлежащему лечению. Популярные хелаторы включают дефероксамин и деферипрон .
Наиболее частые жалобы пациентов на дефероксамин — это болезненность и неудобства. Пероральный хелатор деферасирокс был одобрен для использования в 2005 году в некоторых странах, он дает некоторую надежду на соблюдение режима лечения по более высокой цене. Трансплантация костного мозга — единственное лекарство, которое показано пациентам с тяжелой формой большой талассемии. Трансплантация может устранить зависимость пациента от переливаний. При отсутствии подходящего донора с помощью преимплантационной генетической диагностики (ПГД) можно зачать брата-спасителя, свободного от заболевания, а также соответствующего лейкоцитарному антигену человека (HLA) реципиента .
Трансплантация костного мозга — единственное лекарство, которое показано пациентам с тяжелой формой большой талассемии. Трансплантация может устранить зависимость пациента от переливаний. При отсутствии подходящего донора с помощью преимплантационной генетической диагностики (ПГД) можно зачать брата-спасителя, свободного от заболевания, а также соответствующего лейкоцитарному антигену человека (HLA) реципиента .
Ученые из Медицинского колледжа Вейлла Корнелла разработали стратегию генной терапии, с помощью которой можно эффективно лечить как бета-талассемию, так и серповидно-клеточную анемию. Технология основана на доставке лентивирусного вектора, несущего как ген β-глобина человека, так и инсулятор анкирина, для улучшения транскрипции и трансляции гена, а также повышения уровня продукции β-глобина.
Хирургический
Пациенты с большой талассемией более склонны к спленэктомии. В последние годы число медицинских случаев спленэктомии сокращается из-за снижения распространенности гиперспленизма у пациентов, которым адекватно переливается кровь. Пациенты с гиперспленизмом склонны иметь меньшее количество здоровых клеток крови в своем теле, чем обычно, и у них обнаруживаются симптомы анемии. Пациентам, богатым железом, необходима спленэктомия, чтобы снизить вероятность перегрузки железом. Различные хирургические методы — это открытый и лапароскопический метод. Лапароскопический метод требует более длительного времени операции, но более короткого периода восстановления без хирургического рубца. Если нет необходимости удалять всю селезенку, может потребоваться частичная спленэктомия; этот метод сохраняет часть иммунной функции, снижая при этом вероятность гиперспленизма. Хирурги, выбравшие лапароскопическую спленэктомию, должны провести соответствующую иммунизацию как минимум за две недели до операции. На операционном столе пациент должен находиться в положении от 30 ° до 40 °, при этом его левая рука должна быть поднята над головой, чтобы правильно сделать разрез. Камера вводится вместе с четырьмя другими троакарами: один размещается в левой подреберной области, один вставляется в средней точке между первым и третьим, один на 4 см справа от средней линии, а четвертый расположен по средней линии для втягивания селезенки.
Пациенты с гиперспленизмом склонны иметь меньшее количество здоровых клеток крови в своем теле, чем обычно, и у них обнаруживаются симптомы анемии. Пациентам, богатым железом, необходима спленэктомия, чтобы снизить вероятность перегрузки железом. Различные хирургические методы — это открытый и лапароскопический метод. Лапароскопический метод требует более длительного времени операции, но более короткого периода восстановления без хирургического рубца. Если нет необходимости удалять всю селезенку, может потребоваться частичная спленэктомия; этот метод сохраняет часть иммунной функции, снижая при этом вероятность гиперспленизма. Хирурги, выбравшие лапароскопическую спленэктомию, должны провести соответствующую иммунизацию как минимум за две недели до операции. На операционном столе пациент должен находиться в положении от 30 ° до 40 °, при этом его левая рука должна быть поднята над головой, чтобы правильно сделать разрез. Камера вводится вместе с четырьмя другими троакарами: один размещается в левой подреберной области, один вставляется в средней точке между первым и третьим, один на 4 см справа от средней линии, а четвертый расположен по средней линии для втягивания селезенки.
Лечебный
Длительная трансфузионная терапия для поддержания уровня гемоглобина пациента выше 9-10 г / дл (нормальный уровень составляет 13,8 для мужчин и 12,1 для женщин). Переливание крови происходит при соблюдении строгих критериев, обеспечивающих их безопасность. У них должны быть: подтвержденный лабораторный диагноз большой талассемии и уровень гемоглобина менее 7 г / дл, чтобы иметь право на переливание. Чтобы обеспечить качественное переливание крови, упакованные эритроциты должны быть выделены лейкоцитами с содержанием гемоглобина не менее 40 г. Имея пакеты с кровью с пониженным содержанием лейкоцитов, пациент подвергается меньшему риску развития побочных реакций со стороны зараженных лейкоцитов и предотвращения аллоиммунизации тромбоцитов. Фильтрация цельной крови перед хранением обеспечивает высокую эффективность удаления и низкий уровень остаточных лейкоцитов; Это предпочтительный метод уменьшения лейкоцитов по сравнению с пре-переливанием и фильтрацией у постели больного. Пациенты с аллергическими реакциями на переливание крови или необычными антителами к эритроцитам должны получать «промытые эритроциты» или «криоконсервированные эритроциты». Из промытых эритроцитов были удалены белки плазмы, которые могли стать мишенью для антител пациента, что позволило безопасно провести переливание крови. Криоконсервированные эритроциты используются для поддержания запаса редких донорских единиц для пациентов с необычными антителами к эритроцитам или отсутствием общих антигенов эритроцитов. Доступные программы переливания включают регулярное переливание крови на протяжении всей жизни, чтобы поддерживать уровень гемоглобина перед переливанием выше 9-10 г / гл. Ежемесячные переливания крови способствуют нормальному росту, физической активности, подавляют активность костного мозга и сводят к минимуму накопление железа. Было объявлено о начале первого клинического испытания CRISPR / Cas9 в Европе в 2018 году.
Пациенты с аллергическими реакциями на переливание крови или необычными антителами к эритроцитам должны получать «промытые эритроциты» или «криоконсервированные эритроциты». Из промытых эритроцитов были удалены белки плазмы, которые могли стать мишенью для антител пациента, что позволило безопасно провести переливание крови. Криоконсервированные эритроциты используются для поддержания запаса редких донорских единиц для пациентов с необычными антителами к эритроцитам или отсутствием общих антигенов эритроцитов. Доступные программы переливания включают регулярное переливание крови на протяжении всей жизни, чтобы поддерживать уровень гемоглобина перед переливанием выше 9-10 г / гл. Ежемесячные переливания крови способствуют нормальному росту, физической активности, подавляют активность костного мозга и сводят к минимуму накопление железа. Было объявлено о начале первого клинического испытания CRISPR / Cas9 в Европе в 2018 году.
Фармацевтическая
Перегрузка железом — неизбежное последствие хронической трансфузионной терапии, необходимой пациентам с бета-талассемией. Хелатирование железа — это лекарственная терапия, позволяющая избежать осложнений, связанных с перегрузкой железом. Перегрузку железом можно устранить с помощью деферазирокса, перорального хелатора железа, который оказывает дозозависимое влияние на содержание железа. Каждая единица перелитой крови содержит 200–250 мг железа, и в организме нет естественного механизма для удаления лишнего железа. Деферасирокс — жизненно важный компонент для здоровья пациентов после переливания крови. Во время нормального гомеостаза железа циркулирующее железо связано с трансферрином, но при перегрузке железом способность трансферрина связывать железо превышается и образуется железо, не связанное с трансферрином. Он представляет собой потенциально токсичную форму железа из-за его высокой склонности индуцировать формы кислорода и отвечает за повреждение клеток. Профилактика перегрузки железом защищает пациентов от заболеваемости и смертности. Основная цель заключается в связывании и удалении железа из организма со скоростью, равной скорости поступления железа при переливании или большей, чем поступление железа.
Хелатирование железа — это лекарственная терапия, позволяющая избежать осложнений, связанных с перегрузкой железом. Перегрузку железом можно устранить с помощью деферазирокса, перорального хелатора железа, который оказывает дозозависимое влияние на содержание железа. Каждая единица перелитой крови содержит 200–250 мг железа, и в организме нет естественного механизма для удаления лишнего железа. Деферасирокс — жизненно важный компонент для здоровья пациентов после переливания крови. Во время нормального гомеостаза железа циркулирующее железо связано с трансферрином, но при перегрузке железом способность трансферрина связывать железо превышается и образуется железо, не связанное с трансферрином. Он представляет собой потенциально токсичную форму железа из-за его высокой склонности индуцировать формы кислорода и отвечает за повреждение клеток. Профилактика перегрузки железом защищает пациентов от заболеваемости и смертности. Основная цель заключается в связывании и удалении железа из организма со скоростью, равной скорости поступления железа при переливании или большей, чем поступление железа.
Бета-талассемия промежуточная
Пациентам могут потребоваться эпизодические переливания крови. У пациентов, зависимых от переливания крови, развивается перегрузка железом, и им требуется хелатная терапия для удаления избытка железа. Передача аутосомно-рецессивная ; однако сообщалось о доминантных мутациях и сложных гетерозиготах . Рекомендуется генетическое консультирование и может быть предложена пренатальная диагностика . Аллели без мутации, снижающей функцию, характеризуются как (β). Мутации обозначаются (βo), если они предотвращают образование β-цепей, мутации обозначаются как (β +), если они допускают образование некоторых β-цепей.
Малая бета-талассемия
Пациенты часто наблюдаются без лечения. Хотя многие из лиц с незначительным статусом не нуждаются в переливании крови, они все же рискуют перегрузкой железом, особенно в печени . Анализ сывороточного ферритина проверяет уровень железа и может указать на необходимость дальнейшего лечения. Хотя это не опасно для жизни само по себе, оно может повлиять на качество жизни из-за анемии. Незначительное часто сосуществует с другими состояниями, такими как астма, и может вызывать перегрузку печени железом, а у пациентов с неалкогольной жировой болезнью печени — к более тяжелым исходам.
Незначительное часто сосуществует с другими состояниями, такими как астма, и может вызывать перегрузку печени железом, а у пациентов с неалкогольной жировой болезнью печени — к более тяжелым исходам.
Эпидемиология
Бета-форма талассемии особенно распространена среди средиземноморских народов, и эта географическая ассоциация отвечает за ее название: таласса ( θάλασσα ) — греческое слово, обозначающее море, а хайма ( αἷμα ) — греческое слово, обозначающее кровь. В Европе самые высокие концентрации заболевания обнаружены в Греции и прибрежных районах Турции . Особенно сильно пострадали основные средиземноморские острова (кроме Балеарских островов ), такие как Сицилия , Сардиния , Корсика , Кипр , Мальта и Крит . Другие средиземноморские народы , а также народы , проживающие вблизи Средиземного моря, также имеют высокие показатели заболеваемости, включая выходцев из Западной Азии и Северной Африки . Данные показывают, что 15% греческих и турецких киприотов являются носителями генов бета-талассемии, а 10% населения являются носителями генов альфа-талассемии .
Эволюционная адаптация
Признак талассемии может обеспечивать определенную степень защиты от малярии , которая преобладала или преобладала в регионах, где этот признак является обычным, что дает носителям селективное преимущество выживания (известное как преимущество гетерозиготности ), таким образом закрепляя мутацию. В этом отношении различные талассемии напоминают другое генетическое заболевание, влияющее на гемоглобин, — серповидно-клеточную анемию .
Заболеваемость
Расстройство поражает всех полов, но чаще встречается в определенных этнических и возрастных группах. 20 человек умирают в год, в результате чего талассемия причисляется к «редким заболеваниям». В Соединенных Штатах распространенность талассемии составляет примерно 1 на 272 000 или 1 000 человек. В 2002 году в Англии было 4 000 госпитализированных больных и 9 233 случая талассемии. На мужчин приходилось 53% обращений к консультантам в больницах, а на женщин — 47%. Средний возраст пациентов составляет 23 года, только 1% консультантов старше 75 лет, а 69% — в возрасте от 15 до 59 лет. Детская больница Окленда сформировала международную сеть по борьбе с талассемией. «Это наиболее распространенное генетическое заболевание крови в мире, и его число быстро растет». 7% населения мира являются носителями, и ежегодно 400 000 детей рождаются с этим признаком. Если сразу не начать переливание крови, это обычно приводит к летальному исходу в младенчестве.
Детская больница Окленда сформировала международную сеть по борьбе с талассемией. «Это наиболее распространенное генетическое заболевание крови в мире, и его число быстро растет». 7% населения мира являются носителями, и ежегодно 400 000 детей рождаются с этим признаком. Если сразу не начать переливание крови, это обычно приводит к летальному исходу в младенчестве.
Смотрите также
Ссылки
дальнейшее чтение
- Цао, Антонио; Галанелло, Ренцо (2010). «Бета-талассемия» . В Пагоне, Роберта А; Bird, Thomas D; Долан, Синтия Р.; Стивенс, Карен; Адам, Маргарет П. (ред.). GeneReviews . Вашингтонский университет, Сиэтл. PMID 20301599 .
- Бахал, Раман; Макнер, Николь Али; Кихано, Элиас; Лю, Яньфэн; Сулковски, Паркер; Турчик, Одри; Лу, И-Цзянь; Bhunia, Dinesh C .; Манна, Арунава; Greiner, Dale L .; Brehm, Michael A .; Ченг, Кристофер Дж .; Лопес-Хиральдес, Франсеск; Риккарди, Адель; Белоор, Джагадиш; Краузе, Дайан С .; Кумар, Прити; Галлахер, Патрик Дж . ; Брэддок, Деметриос Т .; Зальцман, В. Марк; Ly, Danith H .; Глейзер, Питер М. (26 октября 2016 г.). «Коррекция in vivo анемии у β-талассемических мышей с помощью γPNA-опосредованного редактирования генов с доставкой наночастиц» . Nature Communications . 7 : 13304. Bibcode : 2016NatCo … 713304B . DOI : 10.1038 / ncomms13304 . ISSN 2041-1723 . PMC 5095181 . PMID 27782131 .
 ; Брэддок, Деметриос Т .; Зальцман, В. Марк; Ly, Danith H .; Глейзер, Питер М. (26 октября 2016 г.). «Коррекция in vivo анемии у β-талассемических мышей с помощью γPNA-опосредованного редактирования генов с доставкой наночастиц» . Nature Communications . 7 : 13304. Bibcode : 2016NatCo … 713304B . DOI : 10.1038 / ncomms13304 . ISSN 2041-1723 . PMC 5095181 . PMID 27782131 .
; Брэддок, Деметриос Т .; Зальцман, В. Марк; Ly, Danith H .; Глейзер, Питер М. (26 октября 2016 г.). «Коррекция in vivo анемии у β-талассемических мышей с помощью γPNA-опосредованного редактирования генов с доставкой наночастиц» . Nature Communications . 7 : 13304. Bibcode : 2016NatCo … 713304B . DOI : 10.1038 / ncomms13304 . ISSN 2041-1723 . PMC 5095181 . PMID 27782131 .внешняя ссылка
причины, симптомы, диагностика и лечение
Талассемии — это группа нарушений крови, которые передаются по наследству. Данные нарушения имеют общее основание, которое проявляется в недостаточном образовании гемоглобина, благодаря которому эритроциты переносят кислород и углекислый газ.
Говоря более научным языком, талассемии — это гетерогенная группа гемоглобинопатии, которая основана на снижении синтеза полипептидных цепей, которые входят в структуру правильного гемоглобина А. Данная форма патологии характеризуется гипохромной анемией при повышенном или нормальном содержании железа сыворотки.
Первый раз данное заболевание описали в 1925 году американские педиатры, которые наблюдали пятерых детей, принадлежащих к семьям итальянцев-эмигрантов. У них были симптомы гипохромной анемии, которая имела тяжелое течение. После того, как об этом опубликовали, появилось сообщение итальянских авторов, которые описали сходную форму талассемии, имеющей более легкое протекание.
до змісту↑
Причины
При данном заболевании одна цепь глобина синтезируется не в сниженном количестве или же синтеза не происходит вообще. В нормальном состоянии синтез сбалансирован, количество свободных цепей глобина нет, так как число а- и не а-цепей одно и тоже. Из-за нарушения синтеза происходит нарушение баланса. Цепь, производимая в слишком большом количестве, откладывается в эритрокариоцитах, с чем связана основная часть клинических проявлений заболевания.
Также талассемии можно описать как мишеневидноклеточную анемию, которая имеет по биохимическим показателям нарушенное соотношение HbA и HbF. То есть может наблюдаться полное отсутствие определенной цепи в случае преобладания другой или же частичная недостаточность этой цепи. Например, если нарушен синтез в-цепи, то преобладать будут а-цепи. При бета-талассемии снижена продукция в-цепей гемоглобина.
Есть различные виды болезни. В этом разделе упомянем о нескольких из них.
- Гомозиготная бета-талассемия. Это результат наследования от обоих родителей ребенком гена с данной патологией.
- Гетерозиготная форма. При этом происходит наследование одного мутантного аллеля, который обуславливает снижение скорости синтеза бета-цепей.
Есть еще гетерозиготная дельтабета, являющаяся результатом мутации гена, который вызывает одновременное угнетение скорости синтеза бета- и дельта-цепей. Основу а-талассемии составляет нарушение синтеза а-цепей. Эта цепь является составляющей всех фракций гемоглобина, поэтому при обсуждаемом нами заболевании они равномерно снижены.
до змісту↑
Симптомы
Клинические проявления наблюдаются уже в детском возрасте. Больные дети имеют своеобразный башенный череп и лицо монголоидного характера с большой верхней челюстью. Бета-талассемия имеет две формы: малая форма и анемия Кули, которую можно назвать большой формой. Ранним признаком болезни Кули является сплено- и гепатомегалия. Они развиваются из-за гемосидероза и экстрамедуллярного кроветворения. Впоследствии у таких пациентов развиваются:
- сахарный диабет;
- цирроз печени;
- застойная сердечная недостаточность.
Бета-талассемия гомозиготного характера, то есть анемия Кули, имеет следующие отличительные черты:
- увеличение содержания HbF;
- резкое снижение образования HbA1;
- повышенное, нормальное или низкое содержание HbA2.
Содержание HbF способно колебаться от тридцати до девяноста процентов, а может быть и ниже десяти процентов. Заболевание протекает в виде тяжелой гемолитической анемии, которая проявляется в конце первого года жизни. Лицо монголоидное, череп башенный, также наблюдается гепато- и спленомегалия, бледность кожи, желтушность и отставание в развитии физического характера, в области голеней могут развиться язвы.
Гетерозиготные бета-талассемии могут протекать как бессимптомная форма, так и в виде манифестной формы, при которой наблюдается незначительное увеличение селезенки и специфические костные изменения. Также есть другие признаки, которые выявляются при обследовании больного.
Среди тех, у кого обнаруживается заболевание, есть лица с гомозиготными и гетерозиготными формами A2F-талассемии. Признаки, по которым они определяются, не особо отличаются от бета формы. В группе пациентов, у которых обнаружена бета форма заболевания, случаи большой талассемии, которая имеет выраженные проявления, встречаются не так часто, как малая форма недуга или промежуточная форма.
А-талассемии также бывают нескольких видов.
- Водянка плода. Гемоглобин Barts. Это гомозиготное состояние, которое с жизнью несовместимо. В таких случаях беременность прерывается непроизвольно, а у плода выявляется гепатомегалия и водянка мозга.
- Гемоглобинопатия H. Она проявляется как гемолитическая анемия. Наблюдаются тяжелые костные изменения и увеличение селезенки.
- А-талассемия-1. Данный вид характеризуется небольшой анемией. При этом нормальный ген синтеза а-цепи сочетается с геном a-th-1, из-за чего и возникает эта форма.
- А-талассемия-2. Считается минимальной формой заболевания. Происходит из-за сочетания нормального гена синтеза а-цепи с геном a-th-2. Клинических проявлений не наблюдается.
до змісту↑
Диагностика
В первую очередь сдается анализ крови, который помогает определить степень тяжести гипохромной гиперрегенераторной анемии. Мазок крови дает информацию о гипохромных эритроцитах малых размеров, которые имеют различную форму. Также в мазке крови обнаруживается много нормоцитов.
Биохимический анализ крови позволяет выявить гипербилирубинемию из-за свободной фракции. Также выявляется повышение активности ЛДГ, снижение ОЖСС и гиперсидеремия. Уровень фетального гемоглобина в эритроцитах повышен.
Альфа-талассемии распространены в таких странах, как Китай, Юго-Восточная Азия, Африка, а также в Средиземноморье. При синтезе а-цепей происходит кодирование четырех генов, в связи с чем их синтез нарушен меньше, чем при бета форме. Дисбаланс наиболее выражен лишь в случае поражения всех четырех генов. При альфа форме гемолиз выражен не так сильно, как при бета форме, но более эффективен эритропоэз. Это связано с тем, что агрегаты из бета цепей (при альфа форме их больше, чем необходимо) растворимы больше, чем агрегаты из альфа цепей. Это дает основание полагать, что лабораторные и клинические данные при альфа форме заболевания не такие отчетливые, как при бета форме. Главное отличие проявляется в биохимическом составе гемоглобина.
При гомозиготной бета форме талассемии важные данные предоставляет рентгенологическое исследование. С помощью его проведения обнаруживается так называемый симптом «щетки» или «ежика». При большом содержании HbF он положителен, при недостаточном содержании HbF2 отрицателен. В возрастном детском промежутке от полугода до года истончение коркового слоя наблюдается в мелких костях кистей и стоп. Происходит вздутие кости, и образуется грубосетчатая структура костного мозга.
После первого года жизни нарушается развитие костей, что прогрессирует вплоть до начала полового созревания. В желчных путях часто образуются билирубиновые камни. Уровень гемоглобина от тридцати до пятидесяти. В мазках крови есть мишеневидные эритроциты, которые отличаются небольшим содержанием гемоглобина.
до змісту↑
Лечение
Решение о лечении тяжелых форм талассемии принимают совместно родители с врачом. Конечно же, перед этим проводится полное обследование ребенка и делается прогноз его состояния.
Гетерозиготная бета форма талассемии чаще всего не требует лечения. Состояние больных удовлетворительное. Уровень гемоглобина обычно находится в пределах от 90 до 100. В редких случаях удаляют селезенку, если она достигает слишком больших размеров. Врач может назначить прием десферала в том случае, если после введения 500 мг этого препарата в моче наблюдается большое количество железа или сывороточного железа. Но этот препарат только предотвращает сидероз и не повышает уровень гемоглобина.
Если уровень гемоглобина снижен из-за инфекционного заболевания, то в период беременности врач назначит прием фолиевой кислоты. Это обусловлено тем, что при неэффективном кроветворении потребляется большое количество этой кислоты. При гетерозиготной талассемии нельзя принимать препараты железа. Самочувствие больных, которые все же делают это, значительно ухудшается.
А-талассемии лечатся практически так же, как и гетерозиготная форма, Исключение составляет гемоглобинопатия H. Этот гемоглобин нестабилен и выпадает в осадок, в связи с чем наблюдается неэффективное кроветворение и яркое разрушение эритроцитов периферической крови. Этот процесс главным образом происходит в селезенке, из-за чего она увеличивается. Зачастую это приводит к ее удалению, что становится главным методом лечения данного вида талассемии.
до змісту↑
Последствия
Прогноз талассемии зависит от того, когда начинают проявляться клинические симптомы. Чем раньше это происходит, тем хуже последствия. В зависимости от того, насколько интенсивны признаки заболевания, выделяют три степени анемии средиземноморской.
- Тяжелая форма. Она проявляется у новорожденных. Часто они умирают уже на первом году.
- Менее тяжелая форма талассемии. Обычно она проявляется к концу первого года жизни. Дети часто доживают до школы.
- Легкая форма талассемии. Клинические проявления наблюдаются на втором году после рождения. Пациенты могут дожить до зрелого возраста.
до змісту↑
Профилактика
Профилактика заболевания главным образом направлена на то, чтобы выявить лица, которые подвержены риску. Это можно сделать посредством скрининга носителей, а также с помощью изучения истории семьи.
Бета-талассемии имеют интересное свойство: здоровых носителей можно выявить точным, простым и недорогим анализом крови. То есть можно выявить пару носителей, что позволит информировать их о генетическом риске. Недорогим и доступным способом является скрининг.
Если человек предупрежден, он сам решает, что делать дальше. В любом случае, на него не нужно оказывать давление. Талассемии являются наследственным опасным заболеванием, однако, как мы увидели, не все его формы несут сильную угрозу жизни человека. Очень важно вовремя обследоваться не только уже существующим родителям, но и будущим семьям, чтобы быть готовыми к самым непредвиденным ситуациям.
причины болезни, симптомы, признаки, формы
Талассемия это наследуемое заболевание крови, то есть талассемия передается от родителей к детям через гены.
Талассемия «заставляет» организм производить меньше, чем обычно здоровых красных кровяных клеток и гемоглобина. Гемоглобин является богатым железом белком в красных кровяных клетках. Он переносит кислород ко всем частям тела. Гемоглобин также осуществляет вывод из организма двуокиси углерода (газа) — отходов жизнедеятельности легких.
Болезнь талассемия может принести с собой другую известную проблему — анемию — в мягкой или тяжелой форме. Анемия обусловлена более низким чем обычно количеством красных кровяных клеток или недостаточным уровнем гемоглобина в красных кровяных клетках.
Причины талассемии
Ваше тело производит три типа клеток крови: эритроциты, лейкоциты и тромбоциты. Красные кровяные клетки содержат гемоглобин, богатый железом белок, переносящий кислород от легких ко всем частям тела.
Гемоглобин имеет два вида белковых цепей: альфа и бета-глобины. Если ваше тело не производит достаточное количество этих белковых цепей, или они ненормальные, красные кровяные клетки не образуются правильно или не переносят достаточно кислорода. Ваше тело не будет работать хорошо, если красные кровяные клетки не производят нужное количество здорового гемоглобина.
Гены определяют то как тело производит цепи гемоглобина, когда эти гены отсутствуют или изменены, происходит болезнь талассемия.
Талассемия это расстройство, которое передается от родителей к детям через гены. Люди, которые наследуют дефектные гены гемоглобина от одного родителя, но нормальные гены от другого, называются носителями. Носители часто не имеют никаких признаков талассемии, кроме мягкой анемии. Тем не менее, они могут передать дефектные гены своим детям.
Люди с умеренной до тяжелой формой талассемии, унаследовали дефектные гены от обоих родителей.
Формы талассемии
Альфа талассемия
Вы должны иметь четыре гена (два от каждого родителя), чтобы производить достаточно белков цепи альфа-глобина. Если не хватает одного или более генов, форма болезни будет альфа талассемия. Это означает, что ваше тело не производит достаточно белка альфа-глобина.
- Если у вас отсутствует только один ген, вы «молчаливый» носитель. Это означает, что вы не будете иметь никаких признаков болезни.
- Если вам не хватает двух генов, у вас альфа-талассемия (также называется альфа-талассемия минор). Вы можете иметь умеренную анемию.
- Если вам не хватает трех генов, у вас, вероятно, болезнь Гемоглобинопатия H (может обнаружить анализ крови ). Эта форма талассемии вызывает от умеренной до тяжелой анемии.
Очень редко, у ребенка отсутствуют все четыре гена. Это состояние называется гомозиготная форма альфа талассемиии или водянка плода. Дети, которые имеют водянку плода, как правило, умирают до или вскоре после рождения.
Бета талассемия
Вы нуждаетесь в двух генах (по одному от каждого родителя), чтобы произвести достаточно цепей бета-глобина. Если один или оба из этих генов изменяются, вы будете иметь бета-талассемию. Это означает, что ваше тело не будет делать достаточно производить белка бета-глобина.
- Если у вас есть один измененный ген, вы носитель. Это состояние называется незначительная бета-талассемия и вызывает умеренную анемию.
- Если оба гена изменены, вы будете иметь бета-талассемию интермедия (также называемая анемия Кули). Промежуточный форма расстройства вызывает умеренную анемию. Основная форма вызывает тяжелую анемию.
Риск развития талассемии
Семейная история и происхождение являются двумя факторам риска талассемии.
История семьи
Талассемия наследуются, то есть дефектные гены, которые с передаются от родителей к детям. Если ваши родители имеют отсутствующие или измененные гены, производящие гемоглобин, у вас может быть талассемии.
Происхождение
Талассемия чаще всего возникает у жителей Средиземноморья, Ближнего Востока, Южной Азии, Африки.
Признаки и симптомы талассемии
Недостаток кислорода в крови вызывает признаки и симптомы талассемии. Недостаток кислорода происходит из-за того, что организм не вырабатывает достаточного количества здоровых красных кровяных клеток и гемоглобина. Тяжесть симптомов зависит от тяжести заболевания.
Талассемия без симптомов
Молчаливая альфа талассемия, как правило, не имеет серьезных признаков или симптомов расстройства. Отсутствие белка альфа-глобина настолько незначительно, что гемоглобин в организме работает нормально.
Умеренная талассемия
Люди, с альфа или бета-талассемией могут иметь мягкую анемию, которая способна заставить вас чувствовать себя усталым. Мягкая анемия, вызванная альфа талассемией, бывает ошибочно принята за железодефицитную анемию.
Люди, у которых бета-талассемия интермедия имеют от легкой до умеренной анемии. Они также могут иметь и другие проблемы со здоровьем, такие как:
- Замедление роста и задержка полового созревания. Анемия может замедлить рост и развитие ребенка.
- Проблемы костей. Талассемии может привести к расширению костного мозга. Костный мозг это губчатое вещество внутри кости, что производит клетки крови. Когда расширяется костный мозг, кости становятся шире, чем обычно. Они могут стать хрупкими и легко ломаются.
- Увеличение селезенки. Селезенка является органом, который помогает вашему организму бороться с инфекциями и удаляет нежелательные агенты. Когда человек имеет талассемию, селезенки приходится работать очень трудно. В результате селезенка становится больше, чем обычно, что ухудшает анемию. Если селезенка становится слишком большой, она должна быть удалена.
Тяжелая анемия и другие признаки и симптомы талассемии
Люди, у которых заболевание Гемоглобинопатия H или бета-талассемия (также называемая анемия Кули), имеют серьезные симптомы талассемии. Признаки и симптомы обычно происходят в течение первых 2 лет жизни. Они могут включать в себя тяжелую анемию и другие проблемы со здоровьем, такие как:
Подпишитесь на наш Ютуб-канал!
- Бледность и апатичность
- Плохой аппетит
- Темная моча (признак того, что красные кровяные клетки разрушаются)
- Замедление роста и задержка полового созревания
- Желтуха (желтоватый цвет кожи или белков глаз)
- Увеличение селезенки, печени, сердца
- Проблемы костей (особенно с костями лица)
Осложнения талассемии
Лучшие методы лечения талассемии в настоящее время позволяют людям, которые имеют умеренные и тяжелые формы заболевания, жить гораздо дольше, что снижает осложнения этих заболеваний, которые возникают с течением времени.
Болезни сердца и печени при талассемии
Регулярные переливания крови являются стандартом лечения талассемии. Переливание может привести к накоплению железа в крови (перегрузка железом). Это может привести к повреждению органов и тканей, особенно сердца и печени.
Сердечные заболевания, вызванные перегрузкой железом, являются основной причиной смерти у людей, которые болеют талассемией. Болезни сердца при талассемии включает в себя сердечную недостаточность, аритмию (нерегулярное сердцебиение), и сердечный приступ.
Инфекции при талассемии
Заболевание талассемия связано с частными инфекциями, которые являются второй наиболее распространенной причиной смерти. Люди после удаления селезенки находятся в еще более высокой зоне риска, потому у них больше нет органа, который организует борьбу с инфекциями.
Остеопороз и талассемия
Многие люди с талассемией имеют проблемы костей, в том числе остеопороз. Это состояние, при котором кости становятся слабыми и ломкими.
Жизнь с талассемией
Выживание и качество жизни больных с умеренной и даже тяжелой талассемией улучшились, потому что:
- Больше людей имеют возможность получить переливания крови в настоящее время.
- Скрининг крови снизил число инфекций при переливании крови. Кроме того, улучшились методы лечения других видов инфекций.
- Некоторые люди получают лечение с помощью переливания крови и трансплантации костного мозга и стволовых клеток
Жизнь с талассемией может быть сложной, но варианты ее улучшения есть.
Следуйте плану лечения талассемии
Это очень важно. Например, обязательно пройдите процедуру переливания крови, если врач рекомендует ее, и принимайте как предписано все лекарства.
Лечение талассемии может быть долгим и даже слегка болезненным. Тем не менее, не переставайте принимать лекарства. Ведущей причиной смерти людей, которые имеют талассемию, являются болезни сердца, вызванные перегрузкой железом. Накопление железа может привести к повреждению сердца, печени и других органов.
Принимайте фолиевую кислоту, если ваш врач прописывает ее. Фолиевая кислота это витамин В, который помогает строить здоровые красные кровяные клетки. Кроме того, поговорите с вашим врачом о том, нужны ли Вы другие витаминные и минеральные добавки, такие как витамины А, С, D или или селен.
Исполняйте все запланированные медицинские назначения, и сдавайте все анализы и проходите все тесты, которые назначает врач. Эти анализы на талассемию могут включать:
- Ежемесячные полный анализ крови и уровня железа каждые 3 месяца
- Ежегодные тесты функции сердца, печени и вирусных инфекций (например, гепатит В и С и ВИЧ)
- Ежегодные анализы для проверки наращивания железа в печени
- Ежегодная проверка зрения и слуха
- Регулярные осмотры, чтобы убедиться, что переливание крови работают
- Другие тесты (например, оценка легочных функций, генетические тесты, и тесты совместимости тканей с возможным донором, если рассматривается пересадка стволовых клеток )
Дети с талассемией должны проходить ежегодные осмотры по поводу контроля их роста и развития, которые включают физический осмотр, в том оценка их роста и веса и любые другие необходимые тесты.
Автор статьи: Ирина Суркова, «Московская медицина»©
Отказ от ответственности: Информация, представленная в этой статье про причины и симптомы талассемии, предназначена только для информирования читателя. Она не может быть заменой для консультации профессиональным медицинским работником.
Талассемия — Диагностика и лечение
Диагноз
У большинства детей с умеренной и тяжелой талассемией признаки и симптомы появляются в течение первых двух лет жизни. Если ваш врач подозревает, что у вашего ребенка талассемия, он может подтвердить диагноз с помощью анализов крови.
Анализы крови могут выявить количество красных кровяных телец и отклонения от нормы по размеру, форме или цвету. Анализы крови также можно использовать для анализа ДНК для поиска мутировавших генов.
Пренатальное тестирование
Тестирование можно провести до рождения ребенка, чтобы выяснить, есть ли у него талассемия, и определить, насколько серьезной она может быть. Тесты, используемые для диагностики талассемии у плода, включают:
- Взятие пробы ворсинок хориона. Обычно этот тест проводится примерно на 11-й неделе беременности и включает в себя удаление крошечного кусочка плаценты для оценки.
- Амниоцентез. Обычно этот тест проводится примерно на 16-й неделе беременности. Этот тест включает исследование образца жидкости, окружающей плод.
Дополнительная информация
Показать дополнительную информацию
Лечение
Легкие формы талассемии в лечении не нуждаются.
Лечение талассемии от умеренной до тяжелой может включать:
- Частые переливания крови. Более тяжелые формы талассемии часто требуют частых переливаний крови, возможно, каждые несколько недель. Со временем переливание крови вызывает накопление железа в крови, что может повредить сердце, печень и другие органы.
Хелатотерапия. Это лечение для удаления излишка железа из крови. Железо может накапливаться в результате регулярных переливаний. У некоторых людей с талассемией, которым не делают регулярных переливаний, также может развиться избыток железа. Удаление лишнего железа жизненно важно для вашего здоровья.
Чтобы избавить ваш организм от лишнего железа, вам может потребоваться пероральный прием лекарств, таких как деферазирокс (Exjade, Jadenu) или деферипрон (Ferriprox). Другой препарат, дефероксамин (десферал), вводится через иглу.
Пересадка стволовых клеток. Трансплантация стволовых клеток, также называемая трансплантатом костного мозга, в некоторых случаях может быть вариантом. Для детей с тяжелой формой талассемии это может устранить необходимость в пожизненных переливаниях крови и лекарствах для контроля перегрузки железом.
Эта процедура включает введение стволовых клеток от совместимого донора, обычно брата или сестры.
Образ жизни и домашние средства
Вы можете помочь справиться с талассемией, следуя своему плану лечения и придерживаясь здорового образа жизни.
- Избегайте лишнего железа. Если ваш врач не рекомендует это, не принимайте витамины или другие добавки, содержащие железо.
Придерживайтесь здоровой диеты. Здоровое питание поможет вам почувствовать себя лучше и зарядиться энергией. Ваш врач может также порекомендовать добавку фолиевой кислоты, чтобы помочь вашему организму вырабатывать новые эритроциты.
Чтобы ваши кости оставались здоровыми, убедитесь, что в вашем рационе содержится достаточно кальция и витамина D. Спросите своего врача, какое количество вам подходит и нужны ли вам добавки.
Спросите своего врача о приеме других добавок, например, фолиевой кислоты. Это витамин B, который помогает создавать красные кровяные тельца.
Избегайте инфекций. Часто мойте руки и избегайте больных. Это особенно важно, если вам удалили селезенку.
Вам также понадобится ежегодная прививка от гриппа, а также вакцины для предотвращения менингита, пневмонии и гепатита B. Если у вас поднимется температура или появятся другие признаки и симптомы инфекции, обратитесь к врачу для лечения.
Как справиться и поддержать
Справиться с талассемией, вашей собственной или вашего ребенка, может быть непросто. Не стесняйтесь обращаться за помощью. Если у вас есть вопросы или вам нужен совет, поговорите с членом вашей медицинской бригады.
Вам также может быть полезно присоединиться к группе поддержки, которая может предоставить как отзывчивое внимание, так и полезную информацию. Спросите члена вашей медицинской бригады о группах в вашем районе.
Подготовка к приему
У людей со средними и тяжелыми формами талассемии обычно диагностируют в течение первых двух лет жизни.Если вы заметили некоторые признаки и симптомы талассемии у своего младенца или ребенка, обратитесь к семейному врачу или педиатру. Затем вас могут направить к врачу, специализирующемуся на заболеваниях крови (гематологу).
Вот некоторая информация, которая поможет вам подготовиться к встрече.
Что вы можете сделать
Составьте список:
- Симптомы вашего ребенка, включая любые, которые могут показаться не связанными с причиной, по которой вы записались на прием, и когда они начались
- Члены семьи , перенесшие талассемию
- Все лекарства, витамины и другие добавки , которые принимает ваш ребенок, включая дозы
- Вопросы, которые следует задать своему врачу
В отношении талассемии вам следует задать врачу следующие вопросы:
- Какая наиболее вероятная причина симптомов у моего ребенка?
- Есть ли другие возможные причины?
- Какие тесты необходимы?
- Какие методы лечения доступны?
- Какие процедуры вы рекомендуете?
- Каковы наиболее частые побочные эффекты от каждого лечения?
- Как лучше всего справиться с этим при других заболеваниях?
- Есть ли диетические ограничения, которым нужно следовать? Вы рекомендуете пищевые добавки?
- Вы можете дать мне печатные материалы? Какие сайты вы рекомендуете?
Не стесняйтесь задавать другие вопросы.
Чего ожидать от врача
Ваш врач, вероятно, задаст вам ряд вопросов, в том числе:
- Симптомы возникают постоянно или приходят и уходят?
- Насколько серьезны симптомы?
- Кажется, что-нибудь улучшает симптомы?
- Что может ухудшить симптомы?
22 ноября 2019 г.
Что такое талассемия? | CDC
Талассемия передается по наследству (т.е. передается от родителей к детям через гены) заболевание крови, вызванное тем, что организм не производит достаточного количества белка, называемого гемоглобином, который является важной частью красных кровяных телец. Когда гемоглобина не хватает, красные кровяные тельца в организме не функционируют должным образом, и они существуют в течение более коротких периодов времени, поэтому в кровотоке перемещается меньше здоровых эритроцитов.
Красные кровяные тельца переносят кислород ко всем клеткам тела. Кислород — это пища, которую клетки используют для функционирования.Когда не хватает здоровых красных кровяных телец, также не хватает кислорода, доставляемого всем другим клеткам тела, что может вызвать у человека чувство усталости, слабости или одышки. Это состояние называется анемией. У людей с талассемией может быть легкая или тяжелая анемия. Тяжелая анемия может повредить органы и привести к смерти.
Какие бывают типы талассемии?
Когда мы говорим о различных «типах» талассемии, мы можем говорить об одном из двух: о конкретной части гемоглобина, которая поражена (обычно «альфа» или «бета»), или о степени тяжести талассемии, которая отмечается такими словами, как черта, носитель, промежуточное звено или мажор.
Гемоглобин, доставляющий кислород ко всем клеткам тела, состоит из двух разных частей, называемых альфа и бета. Когда талассемия называется «альфа» или «бета», это относится к той части гемоглобина, которая не вырабатывается. Если альфа или бета часть не производится, то не хватает строительных блоков для производства нормального количества гемоглобина. Низкая альфа называется альфа-талассемией. Низкий бета-тест называется бета-талассемией.
Когда используются слова «черта», «незначительная», «промежуточная» или «большая», эти слова описывают, насколько серьезна талассемия.У человека с признаками талассемии могут отсутствовать какие-либо симптомы или у него может быть только легкая анемия, тогда как у человека с большой талассемией могут быть тяжелые симптомы и может потребоваться регулярное переливание крови.
Точно так же, как особенности цвета волос и строения тела передаются от родителей к детям, черты талассемии передаются от родителей к детям. Тип талассемии, который есть у человека, зависит от того, сколько и какой тип признаков талассемии у человека в
Талассемия Awareness | CDC
Знаете ли вы, что большая бета-талассемия, самая тяжелая форма талассемии? Сохраняя приверженность долгосрочному лечению, люди с талассемией могут жить полноценной жизнью.
Талассемия — это группа заболеваний крови, передающихся от родителей к детям по наследству. У человека с талассемией вырабатывается меньше здоровых эритроцитов. Их красные кровяные тельца не производят достаточного количества гемоглобина — белка, переносящего кислород по всему телу. Люди с тяжелой формой талассемии могут иметь различные медицинские осложнения. Им также может потребоваться пожизненное переливание крови для лечения.
Жизнь с талассемией
Специализированный уход на протяжении всей жизни может помочь людям, больным талассемией, жить как можно более здоровыми.Талассемия — это излечимое заболевание, которое можно хорошо контролировать с помощью переливания крови и хелатной терапии. Больному талассемией необходимо регулярно получать медицинскую помощь у гематолога (медицинского специалиста, занимающегося лечением заболеваний или нарушений со стороны крови). Если врач назначил переливание крови или хелатную терапию, самое важное, что может сделать человек с талассемией, — это придерживаться своего графика лечения, чтобы предотвратить тяжелую анемию (низкое количество красных кровяных телец) и возможное повреждение органов от перегрузки железом.
Нур Альтахафи
Личная история
В возрасте 5 лет Нур Альтахафи начал получать переливание эритроцитов — средство от талассемии, несмотря на то, что он жил в очень сложных условиях. Талассемия сильно повлияла на ее жизнь. «Талассемия и сопутствующие ей проблемы помогли мне стать сильнее и преодолеть множество трудностей», — говорит Нур. Узнайте больше о том, как семья Нур поддерживала ее постоянное лечение талассемии, когда она жила в зоне боевых действий.
Подробнее об истории Нура можно узнать здесь .
Работа CDC
Национальный центр CDC по врожденным дефектам и порокам развития проводит следующие мероприятия:
Мониторинг безопасности крови для людей с заболеваниями крови
CDC финансирует один проект по мониторингу безопасности крови у людей с заболеваниями крови. Этот проект называется «Характеристика осложнений, связанных с терапевтическими переливаниями крови при гемоглобинопатиях.Государственный университет Джорджии, Медицинский колледж Джоан и Сэмфорд И. Вейл Корнельского университета и Калифорнийский университет в Сан-Франциско получили финансирование для изучения связанных с переливанием крови осложнений у людей с нарушениями гемоглобина (серповидноклеточная анемия и талассемия) и разработать подходы для уменьшения этих осложнений. Кроме того, CDC финансирует Ассоциацию лабораторий общественного здравоохранения для оказания технической помощи общественному здравоохранению в проведении скрининговых мероприятий (тест для выявления заболевания до того, как оно станет заметным), включая оценку потребностей лабораторий, а также обучение пациентов, лиц, осуществляющих уход, и медицинские работники, участвующие в программах скрининга гемоглобинопатии.
Укрепление здоровья
Чтобы лучше понять проблемы, связанные с лечением талассемии, CDC финансирует Фонд Кули по борьбе с анемией (CAF), чтобы продолжать оказывать помощь людям, больным талассемией. CAF обращается к людям, страдающим этим расстройством, чтобы предоставить им информацию и услуги, которые помогут в лечении талассемии. CAF также предоставляет техническую помощь общественному здравоохранению в рамках серии веб-семинаров CDC по вопросам общественного здравоохранения по заболеваниям крови.
Причины, симптомы, диагностика и лечение
Что такое талассемия?
Талассемия — это наследственное заболевание крови, при котором организм вырабатывает аномальную форму гемоглобина.Гемоглобин — это белковая молекула красных кровяных телец, переносящая кислород.
Заболевание приводит к чрезмерному разрушению эритроцитов, что приводит к анемии. Анемия — это состояние, при котором в вашем организме не хватает нормальных здоровых эритроцитов.
Талассемия передается по наследству, что означает, что хотя бы один из ваших родителей должен быть носителем заболевания. Это вызвано генетической мутацией или удалением определенных ключевых фрагментов гена.
Малая талассемия — менее серьезная форма заболевания.Существуют две более серьезные формы талассемии. При альфа-талассемии по крайней мере один из генов альфа-глобина имеет мутацию или аномалию. При бета-талассемии затрагиваются гены бета-глобина.
Каждая из этих форм талассемии имеет разные подтипы. Точная форма, которая у вас есть, повлияет на тяжесть ваших симптомов и ваше мировоззрение.
Симптомы талассемии могут быть разными. Вот некоторые из наиболее распространенных:
Не у всех есть видимые симптомы талассемии.Признаки расстройства также проявляются позже, в детстве или подростковом возрасте.
Талассемия возникает, когда есть аномалия или мутация в одном из генов, участвующих в производстве гемоглобина. Вы унаследовали эту генетическую аномалию от своих родителей.
Если только один из ваших родителей является носителем талассемии, у вас может развиться форма заболевания, известная как малая талассемия. Если это произойдет, у вас, вероятно, не будет симптомов, но вы будете носителем. У некоторых людей с малой талассемией появляются незначительные симптомы.
Если оба ваших родителя являются носителями талассемии, у вас больше шансов унаследовать более серьезную форму заболевания.
Талассемия чаще всего встречается у людей из Азии, Ближнего Востока, Африки и стран Средиземноморья, таких как Греция и Турция.
Существует три основных типа талассемии (и четыре подтипа):
- бета-талассемия, которая включает подтипы большой и промежуточный
- альфа-талассемия, которые включают подтипы гемоглобина H и водянки плода
- малая талассемия
Все этих типов и подтипов различаются симптомы и тяжесть.Начало также может незначительно отличаться.
Если ваш врач пытается диагностировать талассемию, он, скорее всего, возьмет образец крови. Они отправят этот образец в лабораторию для проверки на анемию и аномальный гемоглобин. Лаборант также посмотрит на кровь под микроскопом, чтобы увидеть, не имеют ли красные кровяные тельца странной формы.
Эритроциты неправильной формы являются признаком талассемии. Лаборант может также провести тест, известный как электрофорез гемоглобина. Этот тест отделяет различные молекулы в красных кровяных тельцах, позволяя им идентифицировать аномальный тип.
В зависимости от типа и степени тяжести талассемии медицинский осмотр также может помочь вашему врачу поставить диагноз. Например, сильно увеличенная селезенка может указывать на то, что у вас болезнь гемоглобина Н.
Лечение талассемии зависит от типа и тяжести заболевания. Ваш врач назначит вам курс лечения, который лучше всего подойдет для вашего конкретного случая.
Некоторые виды лечения включают:
Ваш врач может посоветовать вам не принимать витамины или добавки, содержащие железо.Это особенно актуально, если вам необходимо переливание крови, потому что люди, которые его получают, накапливают лишнее железо, от которого организм не может легко избавиться. Железо может накапливаться в тканях, что может быть смертельным.
Если вам делают переливание крови, вам может потребоваться хелатная терапия. Обычно это включает инъекцию химического вещества, которое связывается с железом и другими тяжелыми металлами. Это помогает удалить лишнее железо из вашего тела.
Бета-талассемия возникает, когда организм не может производить бета-глобин.Два гена, по одному от каждого родителя, передаются по наследству для образования бета-глобина. Этот тип талассемии бывает двух серьезных подтипов: большая талассемия (анемия Кули) и промежуточная талассемия.
Большая талассемия
Большая талассемия — самая тяжелая форма бета-талассемии. Он развивается, когда отсутствуют гены бета-глобина.
Симптомы большой талассемии обычно проявляются до второго дня рождения ребенка. Связанная с этим заболеванием тяжелая анемия может быть опасной для жизни.Другие признаки и симптомы включают:
Эта форма талассемии обычно настолько серьезна, что требует регулярных переливаний крови.
Промежуточная талассемия
Промежуточная талассемия — менее тяжелая форма. Он развивается из-за изменений в обоих генах бета-глобина. Людям с промежуточной талассемией переливание крови не требуется.
Альфа-талассемия возникает, когда организм не может производить альфа-глобин. Чтобы произвести альфа-глобин, вам нужно иметь четыре гена, по два от каждого родителя.
Этот тип талассемии также имеет два серьезных типа: болезнь гемоглобина H и водянка плода.
Гемоглобин H
Гемоглобин H развивается, когда у человека отсутствуют три гена альфа-глобина или происходят изменения в этих генах. Это заболевание может привести к проблемам с костями. Щеки, лоб и челюсть могут разрастаться. Кроме того, болезнь гемоглобина H может вызывать:
Hydrops fetalis
Hydrops fetalis — чрезвычайно тяжелую форму талассемии, которая возникает до рождения.Большинство детей с этим заболеванием либо рождаются мертвыми, либо умирают вскоре после рождения. Это состояние развивается, когда все четыре гена альфа-глобина изменены или отсутствуют.
Талассемия может быстро привести к анемии. Это состояние характеризуется недостатком кислорода, транспортируемого к тканям и органам. Поскольку за доставку кислорода отвечают эритроциты, уменьшенное количество этих клеток означает, что в организме не хватает кислорода.
Ваша анемия может быть легкой или тяжелой.Симптомы анемии включают:
Анемия также может вызвать потерю сознания. Тяжелые случаи могут привести к обширному повреждению органов, которое может быть фатальным.
Талассемия имеет генетическую природу. Для развития полной талассемии и ваших родителей должны быть носителями этой болезни. В результате у вас будет два мутировавших гена.
Также возможно стать носителем талассемии, если у вас есть только один мутировавший ген, а не два от обоих родителей. Либо один из ваших родителей, либо оба должны иметь это условие или быть носителем или .Это означает, что вы наследуете один мутировавший ген от одного из своих родителей.
Важно пройти обследование, если у одного из ваших родителей или родственников есть какая-либо форма заболевания.
Малая талассемия
В случаях альфа-минор отсутствуют два гена. В бета-минор отсутствует один ген. У людей с малой талассемией обычно нет никаких симптомов. Если это так, скорее всего, это небольшая анемия. Состояние классифицируется как альфа- или бета-талассемия.
Даже если малая талассемия не вызывает каких-либо заметных симптомов, вы все равно можете быть переносчиком болезни.Это означает, что если у вас есть дети, у них может развиться какая-то форма генной мутации.
По оценкам, из всех детей, ежегодно рождающихся с талассемией, во всем мире 100 000 рождаются с тяжелыми формами.
Дети могут проявлять симптомы талассемии в течение первых двух лет жизни. Вот некоторые из наиболее заметных признаков:
- усталость
- желтуха
- бледная кожа
- плохой аппетит
- медленный рост
Очень важно быстро диагностировать талассемию у детей.Если вы или другой родитель вашего ребенка являетесь носителями, вам следует пройти тестирование заранее.
Если не лечить, это состояние может привести к проблемам с печенью, сердцем и селезенкой. Инфекции и сердечная недостаточность — самые частые опасные для жизни осложнения талассемии у детей.
Как и взрослые, дети с тяжелой формой талассемии нуждаются в частых переливаниях крови, чтобы избавиться от избытка железа в организме.
Обезжиренная растительная диета — лучший выбор для большинства людей, включая больных талассемией.Однако вам может потребоваться ограничить употребление продуктов, богатых железом, если у вас уже высокий уровень железа в крови. Рыба и мясо богаты железом, поэтому вам, возможно, придется ограничить их в своем рационе.
Вы также можете отказаться от обогащенных злаков, хлеба и соков. Они также содержат высокий уровень железа.
Талассемия может вызвать дефицит фолиевой кислоты (фолиевой кислоты). Этот витамин B, естественным образом содержащийся в таких продуктах, как темная зелень и бобовые, необходим для предотвращения воздействия высокого уровня железа и защиты красных кровяных телец.Если вы не получаете достаточного количества фолиевой кислоты с пищей, ваш врач может порекомендовать ежедневный прием добавок 1 мг.
Не существует единой диеты, которая может вылечить талассемию, но правильная пища может помочь. Обязательно заранее обсудите с врачом любые диетические изменения.
Поскольку талассемия — это генетическое заболевание, предотвратить ее невозможно. Однако есть способы справиться с болезнью, чтобы предотвратить осложнения.
В дополнение к постоянному медицинскому обслуживанию CDC рекомендует всем людям с расстройством защищать себя от инфекций, не отставая от следующих вакцин:
В дополнение к здоровому питанию, регулярные упражнения могут помочь справиться с симптомами и привести к более сильному развитию болезни. прогноз положительный.Обычно рекомендуются тренировки средней интенсивности, поскольку тяжелые упражнения могут ухудшить ваши симптомы.
Ходьба и езда на велосипеде являются примерами тренировок средней интенсивности. Плавание и йога — другие варианты, которые также полезны для суставов. Главное — найти то, что вам нравится, и продолжать двигаться.
Талассемия — серьезное заболевание, которое при отсутствии лечения или недостаточном лечении может привести к опасным для жизни осложнениям. Хотя трудно определить точную продолжительность жизни, общее правило состоит в том, что чем тяжелее состояние, тем быстрее талассемия может стать фатальной.
По некоторым оценкам, люди с бета-талассемией — наиболее тяжелой формой — обычно умирают к 30 годам. Сокращение продолжительности жизни связано с перегрузкой железом, которая в конечном итоге может повлиять на ваши органы.
Исследователи продолжают изучать генетическое тестирование, а также возможности генной терапии. Чем раньше будет выявлена талассемия, тем раньше можно будет получить лечение. В будущем генная терапия, возможно, сможет реактивировать гемоглобин и деактивировать аномальные генные мутации в организме.
Талассемия также вызывает различные опасения, связанные с беременностью. Заболевание влияет на развитие репродуктивных органов. Из-за этого женщины с талассемией могут столкнуться с проблемами фертильности.
Чтобы обеспечить здоровье и вам, и вашему ребенку, важно планировать как можно больше заранее. Если вы хотите завести ребенка, обсудите это со своим врачом, чтобы убедиться, что у вас самое лучшее здоровье.
Необходимо тщательно следить за уровнем железа.Также рассматриваются ранее существовавшие проблемы с основными органами.
Пренатальное тестирование на талассемию можно проводить на 11 и 16 неделях. Это делается путем взятия образцов жидкости из плаценты или плода соответственно.
Беременность связана со следующими факторами риска у женщин с талассемией:
Если у вас талассемия, ваш прогноз зависит от типа заболевания. Люди, у которых есть легкие или легкие формы талассемии, обычно могут вести нормальный образ жизни.
В тяжелых случаях возможна сердечная недостаточность.Другие осложнения включают заболевание печени, аномальный рост скелета и эндокринные проблемы.
Ваш врач может дать вам более подробную информацию о вашем прогнозе. Они также объяснят, как ваше лечение может помочь улучшить качество вашей жизни или увеличить ее продолжительность.
Что такое талассемия? — Thalassemia.com
Талассемия — это генетическое заболевание крови.
Люди с талассемией не могут вырабатывать достаточное количество гемоглобина, что вызывает тяжелую анемию.Гемоглобин содержится в красных кровяных тельцах и переносит кислород во все части тела.
Когда в красных кровяных тельцах недостаточно гемоглобина, кислород не может добраться до всех
части тела. Затем органы испытывают недостаток кислорода и
не могут нормально функционировать.
Существует два основных типа талассемии: болезнь альфа-талассемия и
Бета-талассемия. Большая бета-талассемия (также называемая анемией Кули) — это
серьезная болезнь.Симптомы появляются в первые два года жизни и включают бледность
кожа, плохой аппетит, раздражительность и задержка роста. Правильное лечение включает рутинный
переливание крови и другие методы лечения.
Существует два основных типа болезни альфа-талассемии. Большая альфа-талассемия
— очень серьезное заболевание, при котором тяжелая анемия начинается еще до рождения.
Беременные женщины, вынашивающие пораженный плод, сами подвергаются риску серьезной беременности и
осложнения доставки.Другой тип альфа-талассемии — болезнь гемоглобина Н.
Существуют различные степени болезни гемоглобина Н.
Талассемия — сложная группа заболеваний, которые относительно редко встречаются в
США, но распространен в регионах Средиземноморья, Южной и Юго-Восточной Азии.
Ежегодно во всем мире 350000 новорожденных с серьезными гемоглобинопатиями.
(заболевания крови). В Соединенных Штатах, в результате иммиграции,
увеличивается количество случаев талассемии.
Талассемии — это разнообразная группа генетических заболеваний крови, характеризующихся
отсутствием или снижением выработки нормального гемоглобина, что приводит к
микроцитарный
анемия разной степени. Талассемии имеют сопутствующее распространение
с местами, где распространена малярия, вызванная P. falciparum. Альфа-талассемия
сосредоточены в Юго-Восточной Азии, Малайзии и южном Китае. В
бета-талассемии встречаются в основном в районах, прилегающих к Средиземному морю.
Море, Африка и Юго-Восточная Азия.Из-за глобальных схем миграции там
наблюдается рост заболеваемости талассемией в Северной Америке.
в последние десять лет, в первую очередь за счет иммиграции из Юго-Восточной Азии.
У нормального взрослого человека гемоглобин A состоит из двух альфа и
два бета-глобина (A 2 Β 2 ),
является наиболее распространенным, составляя около 95% всего гемоглобина. Два второстепенных
также встречаются гемоглобины: гемоглобин А 2 , состоящий из двух альфа
и два дельта-глобина
(α 2 δ 2 )
состоит из 2-3.5% гемоглобина, а гемоглобин F состоит из двух альфа
и два гамма-глобина
(α 2 γ 2 )
содержит менее 2% гемоглобина.
Гемоглобин F, или гемоглобин плода, вырабатывается плодом in utereo.
и примерно до 48 недель после рождения. Гемоглобин F имеет высокое сродство к кислороду
чтобы привлечь кислород из материнской крови и доставить его к плоду.
После рождения производство взрослого гемоглобина быстро увеличивается и
продукция гемоглобина плода падает.
Гены, контролирующие выработку глобина, находятся на хромосоме 16.
(гены альфа-глобина: «α»),
и хромосома 11
(бета: «β», гамма: «γ» и дельта: «δ» гены).
Как видно на диаграмме, концентрация молекулы альфа-глобина
достаточно стабилен во внутриутробной и взрослой жизни, так как необходим как
производство гемоглобина плода и взрослого. Бета-глобин появляется рано
продолжительность жизни плода на низком уровне и начинает быстро увеличиваться через 30 недель
гестационный возраст, достигающий максимума около 30 недель послеродового периода.Гамма
молекула глобина достигает высокого уровня в начале жизни плода примерно в 6 недель
и начинает снижаться около 30 недель гестации, достигая низкого уровня
около 48 недель постгестационного возраста. Дельта-глобин оказывается на низком уровне
уровень примерно на 30 неделе беременности и сохраняет низкий уровень на протяжении всего
жизнь.
У пациента с талассемией мутация или делеция генов, контролирующих
происходит производство глобина.Это приводит к снижению производства
соответствующие цепи глобина и ненормальное соотношение гемоглобина
(α: не α).
Это ненормальное соотношение приводит к снижению синтеза гемоглобина и
проявление талассемии. Глобин, который вырабатывается в нормальных количествах
сворачивается в избытке и образует агрегаты или включения красных кровяных телец.
Эти агрегаты окисляются и повреждают клеточную мембрану, что приводит к
либо на гемолиз,
неэффективный эритропоэз,
или оба.Количество и свойства этих агрегатов цепей глобина
определить характер и степень тяжести талассемии.
Бета-талассемия приводит к избытку
альфа-глобинов, что приводит к образованию тетрамеров альфа-глобина (α 4 )
накапливаются в эритробласте (незрелые эритроциты). Эти агрегаты очень нерастворимы.
и осаждение мешает эритропоэзу, созреванию клеток и
функция клеточных мембран, приводящая к неэффективному эритропоэзу и анемии.
Альфа-талассемия приводит к избытку
бета-глобинов, что приводит к образованию тетрамеров бета-глобина (β 4 )
называется гемоглобином H. Эти тетрамеры более стабильны и растворимы,
но при особых обстоятельствах может привести к гемолизу, как правило, к сокращению
продолжительность жизни красной клетки.
Условия оксидантного стресса вызывают осаждение Hgb H, препятствуя
с функцией мембраны и приводит к разрушению эритроцитов.H-постоянная гемоглобина
Весенняя болезнь — более тяжелая форма этого гемолитического расстройства. Большинство
тяжелая талассемия — это большая альфа-талассемия, при которой плод производит
нет альфа-глобинов, что вообще несовместимо с жизнью.
747 52nd Street, Окленд, Калифорния 94609 •
Телефон: (510) 428-3347 • Факс: (510) 450-5647
© 2003-2012 Детская больница и исследовательский центр Окленда
Талассемия: симптомы и диагностика — Талассемия.com
СИМПТОМЫ
В большинстве штатов талассемия выявляется при обследовании новорожденных. Таким образом, пациенты проходят обследование до появления симптомов.
Если пациенту не поставлен диагноз при скрининге новорожденных, симптомы могут включать:
- Бледная или желтушная бледность
- Усталость
- Одышка
- Легочная гипертензия
- Слабый рост
- Изменения костей
Если не лечить талассемию, может возникнуть следующее:
- Увеличенные печень, селезенка и сердце.
- Кости тонкие и хрупкие.
- Опухолевые образования экстрамедуллярной эритропоэтической ткани являются частым осложнением у пациента с непереносимой талассемией.
- Тяжелая анемия.
- Застойная сердечная недостаточность.
- Преждевременная смерть.
ДИАГНОСТИКА
Перед рассмотрением вопроса о трансфузионной терапии очень важно:
подтвердить диагноз пациента. Помимо полной крови
подсчет (CBC), электрофорез гемоглобина является первым диагностическим
контрольная работа.Фракции гемоглобина A, A2, F, H, E и другие варианты
измеряются. Анализ гемоглобина электрофорезом гемоглобина
или используется высокоэффективная жидкостная хроматография. Мутации
может совпадать со скрининговым тестом, что приводит к неверному диагнозу
или ложноотрицательный. Таким образом, генетический анализ как бетаталассемии
и мутации альфа-талассемии необходимы. В
Кроме того, необходимо обследовать родителей, братьев и сестер. Иногда
(до 20 процентов времени) только одна мутация будет
обнаружено, что указывает на признак талассемии.Некоторые такие случаи приводят
от аутосомно-доминантной формы талассемии и других от
наследование мутации, которая не обнаруживается зондами, используемыми в
ДНК-тестирование. Трипликация альфа-генов является распространенным сопутствующим фактором
которые могут превратить признак талассемии в болезнь или усугубить доброкачественные
мутация. Тестирование на ко-мутации необходимо запрашивать у
Лаборатория ДНК — иначе не будет.
Пациенты с промежуточной талассемией могли преувеличивать
анемия из-за временной недостаточности питания или инфекционного
осложнения.Важно заполнить подробную медицинскую
история факторов, которые могут временно снизить
гемоглобин, включая вирусные заболевания, подавляющие костный мозг
лекарства или воздействие факторов окружающей среды, таких как свинец.
Недостаток фолиевой кислоты или железа в питании может преувеличивать
анемия. Исправление этих недостатков может повысить гемоглобин.
уровень, достаточный, чтобы избежать необходимости переливания крови. * Следовательно,
лабораторный скрининг пациентов необходим для исключения других
причины анемии.
* Следует измерить уровень G6PD в сыворотке
ферритин, общая железосвязывающая способность, сывороточное железо и эритроциты
фолиевая кислота. Краткое терапевтическое испытание железа (6 мг / кг / день от четырех до
восемь недель) и фолиевая кислота (1 мг / день) показаны при значительном
обнаружены лабораторные недостатки.