Врожденные мышечные дистрофии: классификация и диагностика | Rivier
1. Sparks S., Quijano-Roy S., Harper A. et al. Congenital muscular dystrophy overview in: Pagon RA, Bird TD, Dolan CR, et al. Gene reviews. 1993–2001.
2. Muntoni F., Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14(10):635–49.
3. Sparks S.E., Escolar D.M. Congenital muscular dystrophies. Handb Clin Neurol 2011;101:47–9.
4. Mercuri E., Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann Neurol 2012;72(1):9–17.
5. Godfrey C., Foley A. R., Clement E. et al. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011;21(3):278–85.
R., Clement E. et al. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011;21(3):278–85.
6. Mathews K.D., Stephan C.M., Laubenthal K. et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 2011;76(2):194–5.
7. Clement E.M., Feng L., Mein R. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord 2012;22(6):522–7.
8. Hayashi Y.K., Chou F.L., Engvall E. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 1998;19(1):94–7.
9. Hara Y., Balci-Hayta B., Yoshida-Moriguchi T. A dystroglycan mutation associated with limbgirdle muscular dystrophy.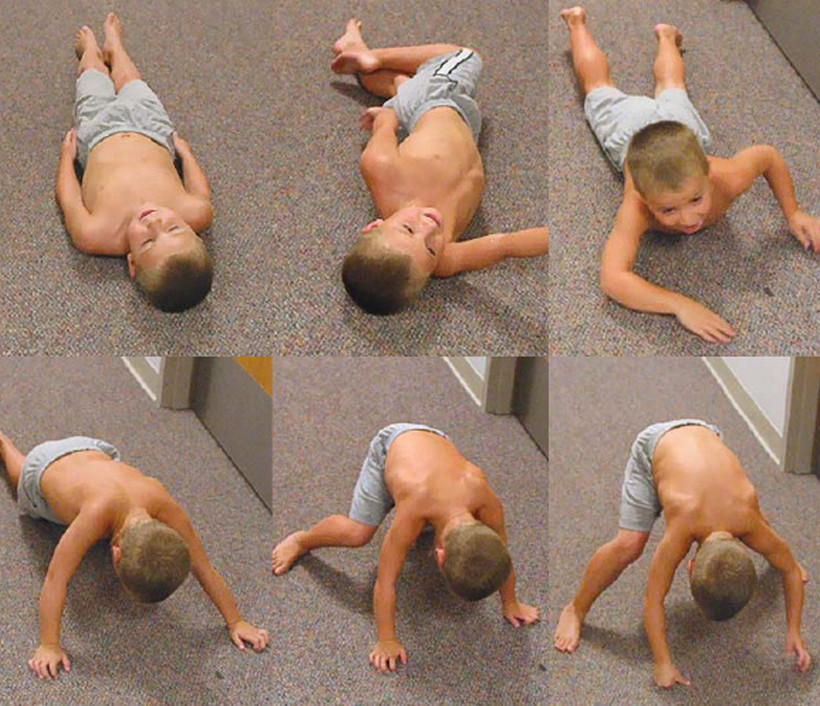 N Engl J Med 2011;364(10):939–46.
N Engl J Med 2011;364(10):939–46.
10. Godfrey C., Clement E., Mein R. et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130(10):2725–35.
11. Mercuri E., Messina S., Bruno C. et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology 2009;72(21):1802–9.
12. Ferreiro A., Quijano-Roy S., Pichereau C. et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002;71(4):739–49.
13. Mitsuhashi S., Ohkuma A., Talim B. et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am J Hum Genet 2011;88(6):845–51.
Mitsuhashi S., Ohkuma A., Talim B. et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am J Hum Genet 2011;88(6):845–51.
14. Tomé F.M., Evangelista T., Leclerc A. et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III 1994;317(4):351–7.
15. Helbling-Leclerc A., Zhan X., Topaloglu H. et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet 1995;11(2):216–8.
16. Geranmayeh F., Clement E., Feng L.H. et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20(4):241–50.
17. Lamer S., Carlier R.Y., Pinard J.M. et al. Congenital muscular dystrophy: use of brain MR imaging findings to predict merosin deficiency. Radiology 1998;206(3):811–6.
Lamer S., Carlier R.Y., Pinard J.M. et al. Congenital muscular dystrophy: use of brain MR imaging findings to predict merosin deficiency. Radiology 1998;206(3):811–6.
18. Okada M., Kawahara G., Noguchi S. et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69(10):1035–42.
19. Allamand V., Briñas L., Richard P. et al. ColVI myopathies: where do we stand, where do we go? Skelet Muscle 2011;1:30.
20. Briñas L., Richard P., Quijano-Roy S. et al. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol 2010;68(4):511–20.
21. Nadeau A., Kinali M., Main M. et al. Natural history of Ullrich congenital muscular dystrophy.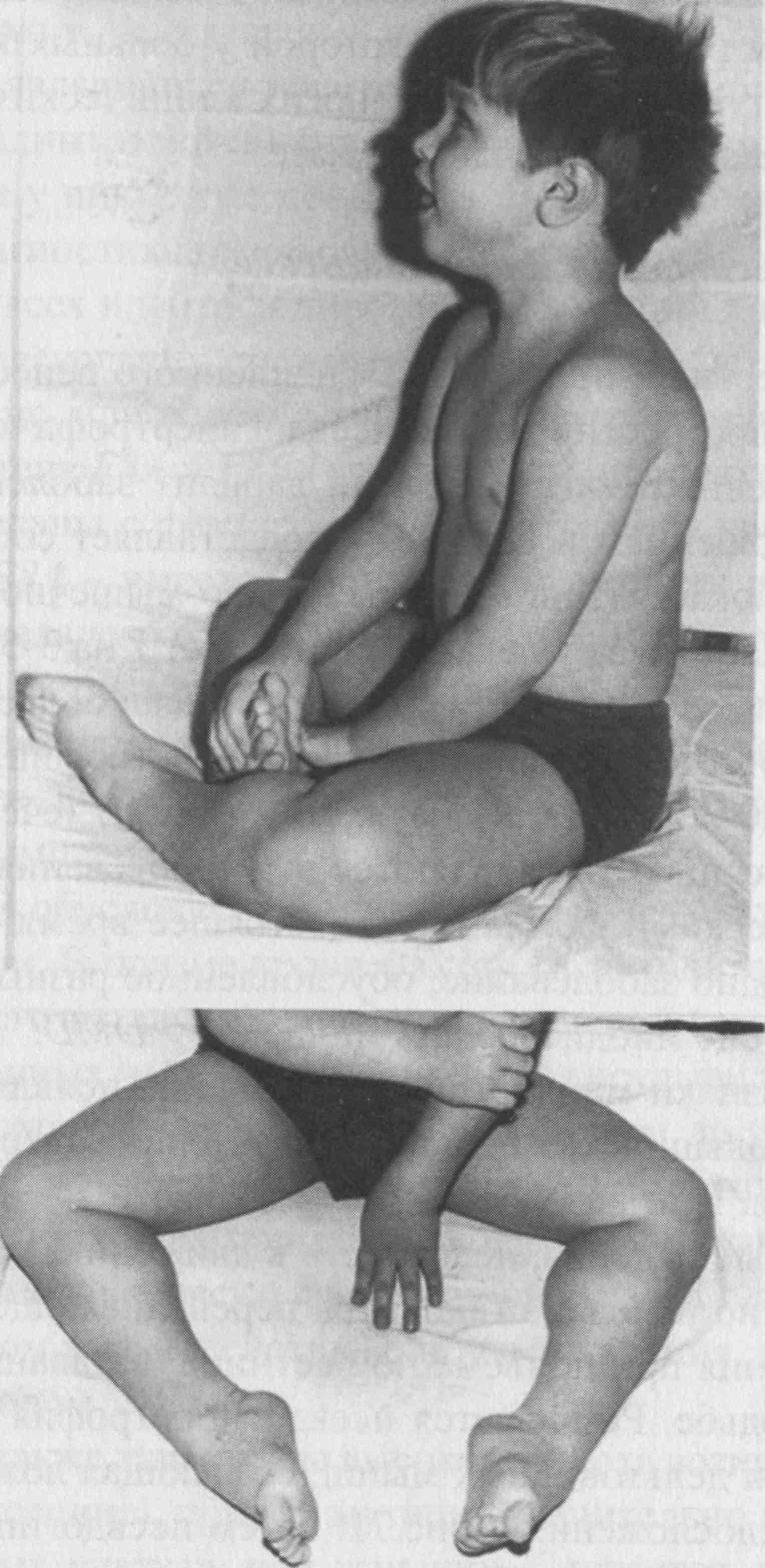 Neurology 2009;73(1):25–31.
Neurology 2009;73(1):25–31.
22. Mercuri E., Lampe A., Allsop J. et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303–10.
23. Quijano-Roy S., Avila-Smirnow D., Carlier R.Y. et al. Whole body muscle MRI protocol: pattern recognition in early onset NM disorders. Neuromuscul Disord 2012;22.
24. Hicks D., Lampe A.K., Barresi R. et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology 2008;70(14):1192–9.
25. Moore C.J., Winder S.J. The inside and out of dystroglycan post-translational modification. Neuromuscul Disord 2012;22(11):959–65.
26. Wells L. The o-mannosylation pathway: glycosyltransferases and proteins implicated in congenital muscular dystrophy. J Biol Chem 2013;288(10):6930–5.
Wells L. The o-mannosylation pathway: glycosyltransferases and proteins implicated in congenital muscular dystrophy. J Biol Chem 2013;288(10):6930–5.
27. Kobayashi K., Nakahori Y., Miyake M. et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature, 1998;394(6691):388–92.
28. Yoshida A., Kobayashi K., Manya H. et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001;1(5):717–24.
29. Brockington M., Blake D.J., Prandini P. et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001;69(6):1198–209.
30. Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A. et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71(5):1033–43.
31. van Reeuwijk J., Janssen M., van den Elzen C. et al. POMT2 mutations cause alphadystroglycan hypoglycosylation and Walker- Warburg syndrome. J Med Genet.2005 Dec;42(12):907–12.
32. Longman C., Brockington M., Torelli S et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 2003;12(21):2853–61.
33. Cirak S. , Foley A.R., Herrmann R. et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain 2013;136(Pt1):269–81.
, Foley A.R., Herrmann R. et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain 2013;136(Pt1):269–81.
34. Barone R., Aiello C., Race V. et al. DPM2-CDG: a muscular dystrophydystroglycanopathy syndrome with severe epilepsy. Ann Neurol 2012;72(4):550–8.
35. Lefeber D.J., de Brouwer A.P., Morava E. et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PloS Genet 2011;7(12).
36. Lefeber D.J., Schönberger J., Morava E. et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 2009;85(1):76–86.
37. Willer T., Lee H., Lommel M. et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 2012;44(5):575–80.
Willer T., Lee H., Lommel M. et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 2012;44(5):575–80.
38. Roscioli T., Kamsteeg E.J., Buysse K. et al.Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat Genet. 2012;44(5):581–5.
39. Manzini M.C., Tambunan D.E., Hill R.S. et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet 2012;91(3):541–7.
40. Vuillaumier-Barrot S., Bouchet-Séraphin C., Chelbi M. et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am J Hum Genet 2012;91(6):1135–43.
41.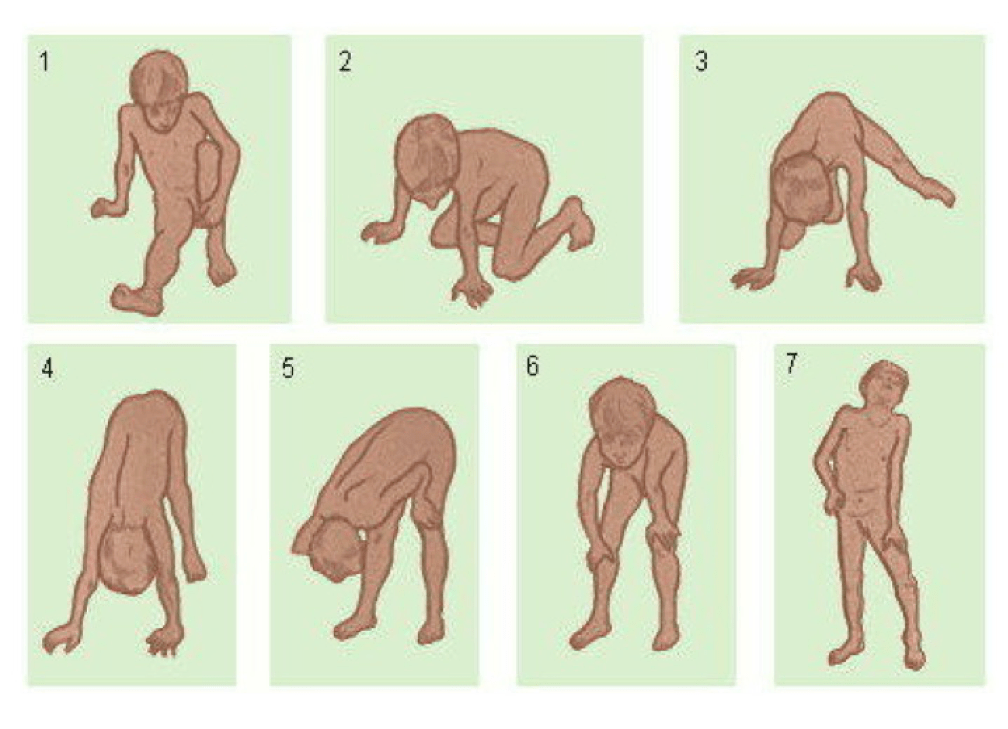 Stevens E., Carss K.J., Cirak S. et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;92(3):354–65.
Stevens E., Carss K.J., Cirak S. et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;92(3):354–65.
42. Buysse K., Riemersma M., Powell G. et al. Missense mutations in β-1,3-Nacetylglucosaminyltransferase 1 (B3GnT1) cause Walker-Warburg syndrome. Hum Mol Genet 2013;22(9):1746–54.
43. Carss K.J., Stevens E., Foley A.R. et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;93(1):29–41.
44. Yang A.C., Ng B.G., Moore S.A. et al. Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy. Mol Genet Metab 2013; 110(3):345–51.
45. Vuillaumier-Barrot S., Quijano-Roy S., Bouchet-Seraphin C. et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul Disord 2009;19(3):182–8.
46. Schara U., Kress W., Bönnemann C.G. et al. The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol 2008;12(3):224–30.
47. Scoto M., Cirak S., Mein R. et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76(24):2973–8.
48. Mercuri E., Pichiecchio A., Allsop J. et al. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging. 2007;25(2):433–40.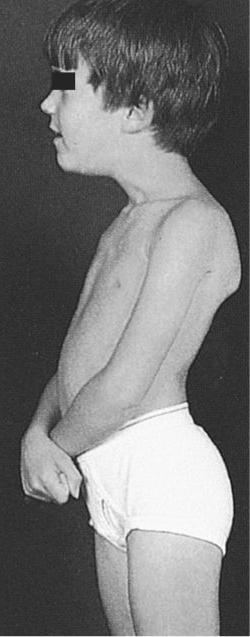
49. Quijano-Roy S., Mbieleu B., Bönnemann C.G. et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008;64(2):177–86.
50. Ben Yaou et al. Les Cahiers de myologie. 2010(3) :24–33.
51. Bonne G., Quijano-Roy S. Emery–Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol 2013;113:1367–76.
52. Hattori A., Komaki H., Kawatani M. et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul Disord 2012;22(2):149–51.
53. Mercuri E., Clements E., Offiah A. et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010;67(2):201–8.
et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010;67(2):201–8.
54. Makri S., Clarke N.F., Richard P. et al. Germinal mosaicism for LMNA mimics autosomal recessive congenital muscular dystrophy. Neuromuscul Disord 2009;19(1):26–8.
МЕРОЗИНДЕФИЦИТНАЯ ВРОЖДЕННАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ВМД1А): КЛИНИЧЕСКИЙ ПРИМЕР ВРОЖДЕННОЙ МЫШЕЧНОЙ ДИСТРОФИИ С ВОВЛЕЧЕНИЕМ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | Клочкова
1. Rivier F., Meyer P., Walther-Louvie U., Mercier M., Echenne B., Quijano-Roy S. Врожденные мышечные дистрофии: классификация и диагностика. Нервно-мышечные болезни. 2014; 1: 6–20.
2. Tome F. M., Evangelista T., Leclerc A.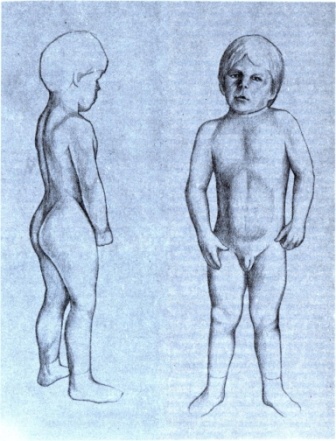 , Sunada Y., Manole E., Estournet B., Barois A., Campbell K. P., Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994; 317 (4): 351–7.
, Sunada Y., Manole E., Estournet B., Barois A., Campbell K. P., Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994; 317 (4): 351–7.
3. Hillaire D., Leclerc A., Faure S., Topaloglu H., Chiannilkulchai N., Guicheney P., Grinas L., Legos P., Philpot J., Evangelista T. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum Mol Genet. 1994; 3 (9): 1657–61.
4. Clement E. M., Feng L., Mein R., Sewry C. A., Robb S. A., Manzur A. Y., Mercuri E., Godfrey C., Cullup T., Abbs S., Muntoni F. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord. 2012; 22 (6): 522–7.
5. Darin N., Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord. 2000; 10 (1): 1–9.
Neuromuscul Disord. 2000; 10 (1): 1–9.
6. Norwood F. L., Harling C., Chinnery P. F., Eagle M., Bushby K., Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009; 132 (Pt. 11): 3175–86.
7. Peat R. A., Smith J. M., Compton A. G., Baker N. L., Pace R. A., Bur-kin D. J., Kaufman S. J., Lamande S. R., North K. N. Diagnosis and etiology of congenital muscular dystrophy. Neurology. 2008; 71 (5): 312–21.
8. Аверьянов Ю. Н. Врожденная мышечная дистрофия с лейкоэнцефалопатией. Журнал неврологии и психиатрии им. C. C. Корсакова. 1993; 5: 27–29.
9. Руденская Г. Е., Галкина В. А., Дунаевская Г. Н. Редкие формы наследственных прогрессирующих мышечных дистрофий с контрактурами.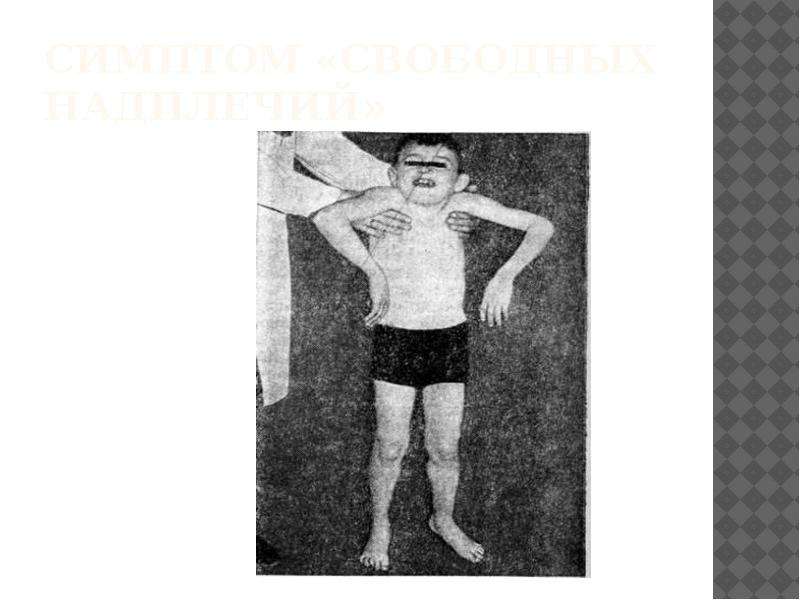 Теоретические и прикладные проблемы мед. генетики. 1993. С. 105–119.
Теоретические и прикладные проблемы мед. генетики. 1993. С. 105–119.
10. Руденская Г. Е., Дадали Е. Л., Ситников В. Ф. Наследственная сочетанная церебромышечная патология в детском возрасте. Организационные и клинические проблемы детской неврологии и психиатрии. 1993. С. 253–255.
11. Дадали Е. Л., Руденская Г. Е., Щагина О. А., Тибуркова Т. Б., Сухоруков В. С., Харламов Д. А., Поляков А. В. Мерозин-дефицитная врожденная мышечная дистрофия (ВМД1А). Журнал неврологии и психиатрии им. C. C. Корсакова. 2010; 110 (3): 83–89.
12. Комарова Н. В., Тибуркова Т. Б., Щагина О. А., Дадали Е. Л., Руденская Г. Е., Поляков А. В. Врожденная мышечная дистрофия, мерозин-негативная (ВМД1А) у российских больных. Материалы VI съезда Российского общества медицинских генетиков. Медицинская генетика (прил. к № 5). 2010. 88 с.
Медицинская генетика (прил. к № 5). 2010. 88 с.
13. Mendell J. R., Boue D. R., Martin P. T. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol. 2006; 9 (6): 427–43.
14. Zhang X., Vuolteenaho R., Tryggvason K. Structure of the human laminin alpha2-chain gene (LAMA2), which is affected in congenital muscular dystrophy. J Biol Chem. 1996; 271 (44): 27664–9.
15. Gawlik K. I., Durbeej M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle. 2011; 1 (1): 9.
16. Geranmayeh F., Clement E., Feng L. H., Sewry C., Pagan J., Mein R., Abbs S., Brueton L., Childs A. M., Jungbluth H., De Goede C. G., Lynch B.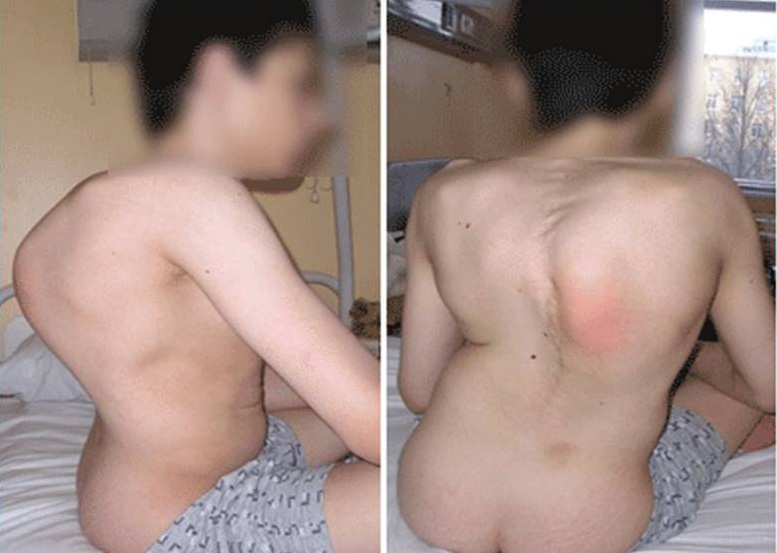 , Lin J. P., Chow G., Sousa Cd, O’Mahony O., Majumdar A., Straub V., Bushby K., Muntoni F. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010; 20 (4): 241–50.
, Lin J. P., Chow G., Sousa Cd, O’Mahony O., Majumdar A., Straub V., Bushby K., Muntoni F. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010; 20 (4): 241–50.
17. Philpot J., Bagnall A., King C., Dubowitz V., Muntoni F. Feeding problems in merosin deficient congenital muscular dystrophy. Arch Dis Child. 1999; 80 (6): 542–7.
18. Tezak Z., Prandini P., Boscaro M., Marin A., Devaney J., Marino M., Fanin M., Trevisan C. P., Park J., Tyson W., Finkel R., Garcia C., Angelini C., Hoffman E. P., Pegoraro E. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat. 2003; 21 (2): 103–11.
19. Jones K. J., Morgan G., Johnston H. , Tobias V., Ouvrier R. A., Wilkinson I., North K. N. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001; 38 (10): 649–57.
, Tobias V., Ouvrier R. A., Wilkinson I., North K. N. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001; 38 (10): 649–57.
20. Leite C. C., Lucato L. T., Martin M. G., Ferreira L. G., Resende M. B., Carvalho M. S., Marie S. K., Jinkins J. R., Reed U. C. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol. 2005; 35 (6): 572–9.
21. Philpot J., Pennock J., Cowan F., Sewry C. A., Dubowitz V., Bydder G., Muntoni F. Brain magnetic resonance imaging abnormalities in merosin-positive congenital muscular dystrophy. Eur J Paediatr Neurol. 2000; 4 (3): 109–14.
22. Bonnemann C. G., Wang C. H., Quijano-Roy S., Deconinck N., Bertini E., Ferreiro A. , Muntoni F., Sewry C., Beroud C., Mathews K. D., Moore S. A., Bellini J., Rutkowski A., North K. N. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014; 24 (4): 289–311.
, Muntoni F., Sewry C., Beroud C., Mathews K. D., Moore S. A., Bellini J., Rutkowski A., North K. N. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014; 24 (4): 289–311.
23. Oliveira J., Santos R., Soares-Silva I., Jorge P., Vieira E., Oliveira M. E., Moreira A., Coelho T., Ferreira J. C., Fonseca M. J., Barbosa C., Prats J., Ariztegui M. L., Martins M. L., Moreno T., Heinimann K., Barbot C., Pascual-Pascual S. I., Cabral A., Fineza I., Santos M., Bronze-da-Rocha E. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. 2008; 74 (6): 502–12.
24. Vainzof M., Richard P., Herrmann R., Jimenez-Mallebrera C., Talim B., Yamamoto L. U., Ledeuil C., Mein R., Abbs S., Brockington M., Romero N. B., Zatz M., Topaloglu H., Voit T., Sewry C., Muntoni F., Guicheney P., Tome F. M. Prenatal diagnosis in laminin alpha2 chain (merosin)-deficient congenital muscular dystrophy: a collective experience of five international centers. Neuromuscul Disord. 2005; 15 (9–10): 588–94.
B., Zatz M., Topaloglu H., Voit T., Sewry C., Muntoni F., Guicheney P., Tome F. M. Prenatal diagnosis in laminin alpha2 chain (merosin)-deficient congenital muscular dystrophy: a collective experience of five international centers. Neuromuscul Disord. 2005; 15 (9–10): 588–94.
25. Matsumura K., Yamada H., Saito F., Sunada Y., Shimizu T. Peripheral nerve involvement in merosin-deficient congenital muscular dystrophy and dy mouse. Neuromuscul Disord. 1997; 7 (1): 7–12.
26. Vigliano P., Dassi P., Di Blasi C., Mora M., Jarre L. LAMA2 stop-codon mutation: merosin-deficient congenital muscular dystrophy with occipital polymicrogyria, epilepsy and psychomotor regression. Eur J Paediatr Neurol. 2009; 13 (1): 72–6.
Врождённые мышечные дистрофии. Врождённые миопатии
Содержание страницы:
- Общее описание врождённых мышечных дистрофий
- Диагностика врождённых мышечных дистрофий / миопатий
- Что важно знать при врождённых мышечных дистрофиях / миопатиях?
- Регистры пациентов с врождёнными мышечными дистрофиями / миопатиями
- Организации и сообщества, посвящённые врождённым мышечным дистрофиям / миопатиям
- Статьи о ВМД на нашем сайте
Общее описание врождённых мышечных дистрофий
Перевод материалов Muscular Dystrophy Association. Ссылка на источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Ссылка на источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Что такое врождённая мышечная дистрофия (ВМД, англ. congenital muscular dystrophy (CMD)?
Врождённая мышечная дистрофия относится к группе мышечных дистрофий, которые становятся заметными при рождении или сразу после рождения.
Мышечные дистрофии являются, в основном, генетическими дегенеративными заболеваниями, поражающие большей частью произвольно сокращающиеся мышцы.
Дети с врождённой мышечной дистрофией — слабые уже при рождении и могут испытывать трудности с дыханием и глотанием.
В настоящее время методы поддержки больных с ВМД значительно улучшились и повысили выживаемость пациентов, а клинические испытания препаратов против этой болезни — уже не в таком далёком будущем, как это было раньше.
Более подробно см. на mda.org: Types of CMD.
Симптомы ВМД
ВМД приводит к общей мышечной слабости с возможным развитием суставных контрактур, также возможно ослабление суставов.
В зависимости от типа при ВМД может появиться искривление позвоночника, дыхательная недостаточность, ментальные нарушения, сложности с обучением, дефекты глаз или судороги.
Более подробно см. на mda.org: Types of CMD and Signs and Symptoms.
Причины появления ВМД
ВМД возникает из-за мутаций в генах, отвечающих за определённые белки, необходимые для мышц, а в некоторых случаях — для глаз или мозга.
Более подробно см. на mda.org: Causes/Inheritance.
Прогрессирование ВМД
ВМД проявляется при рождении или через короткое время после него. Прогрессирование заболевание зависит от типа. Многие типы прогрессируют медленно, но некоторые приводят к сокращению продолжительности жизни.
Статус исследований по ВМД
Исследователи определили многие из генов, дефекты в которых приводят к появлению ВМД. Эти открытия ведут нас к лучшему пониманию природы болезни и улучшают диагностику, а также стратегии лечения.
Более подробно см. на mda.org: Research.
Источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Диагностика врождённых мышечных дистрофий / миопатий
Общий список клиник и врачей, занимающихся нервно-мышечными заболеваниями, — по ссылке.
- Статья из журнала «Нервно-мышечные болезни» — «Врождённые мышечные дистрофии: классификация и диагностика» (2014 год, выпуск №1). Авторы: François Rivier, Pierre Meyer, Ulrike Walther-Louvie, Moïse Mercier, Bernard Echenne, Susana Quijano-Roy. Если статья не видна во врезке ниже, вы можете ознакомиться с текстом на сайте журнала: http://nmb.abvpress.ru/jour/article/view/7.
[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-RnIzMDVLNTFCZ2M/preview?usp=drivesdk» title=»2014_Диагностика врожденных МД_Журнал НМБ.pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
[google-drive-embed url=»https://drive. google.com/file/d/0B0m38yuXU_b-cUhobHF4LUNRMmc/preview?usp=drivesdk» title=»2011_Диагностика врожденных миопатий_Журнал НМБ.pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
google.com/file/d/0B0m38yuXU_b-cUhobHF4LUNRMmc/preview?usp=drivesdk» title=»2011_Диагностика врожденных миопатий_Журнал НМБ.pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
Что важно знать при врождённых мышечных дистрофиях / миопатиях?
- Семейное руководство по медицинскому уходу при врожденной миопатии (если руководство не отображается во врезке ниже, то вы можете его просмотреть по ссылке https://goo.gl/woCsw6):
[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-dTNOVlF1N1pmN0U/preview?usp=drivesdk» title=»2017-03-31_Семейное-руководство-врождённые миопатии.pdf» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/pdf» width=»100%» height=»400″ style=»embed»]
- Семейное руководство по медицинскому уходу при врожденной мышечной дистрофии (если руководство не отображается во врезке ниже, то вы можете его просмотреть по ссылке https://goo.
 gl/YKGySX
gl/YKGySX
 gl/YKGySX
gl/YKGySX[embeddoc url=»https://drive.google.com/file/d/0B0m38yuXU_b-TmlyT3diRXotSDA/preview?usp=drive_web» viewer=»drive» ]
- Для врачей «Заявление на основе консенсуса касательно стандартного лечения врожденных миопатий» (если руководство не отображается во врезке ниже, то вы можете его просмотреть по ссылке https://goo.gl/sV4B1B):
[google-drive-embed url=»https://drive.google.com/file/d/0B0m38yuXU_b-b2J4ZkQxZUgwdDg/preview?usp=drivesdk» title=»2017-03-31_Для врачей_Стандарты лечения врождённых миопатий.docx» icon=»https://drive-thirdparty.googleusercontent.com/16/type/application/vnd.openxmlformats-officedocument.wordprocessingml.document» width=»100%» height=»400″ style=»embed»]
Важно! Регистры (реестры) пациентов с врождённой мышечной дистрофией / миопатией
Регистры пациентов в разных странах вы можете узнать по ссылке: http://www.treat-nmd.eu/resources/patient-registries/list/CMD/.
Международный регистр по врождённым мышечным заболеваниям (CMDIR — Congenital muscle disease international register).
Почему нужно регистрироваться в реестре?
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку генетические нервно-мышечные заболевания являются редкими (орфанными) заболеваниями.
Для этого в разных странах ведутся реестры пациентов — базы данных по генетической и клинической информации о людях, страдающих от генетических нервно-мышечных заболеваний и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Организации и сообщества, посвящённые врождённым мышечным дистрофиям / миопатиям
Статьи о ВМД на нашем сайте
Статьи о ВМД типа 1А на нашем сайте
Мышечная дистрофия | Неврология | Заболевания
Мышечная дистрофия – это патологическое заболевание, которое характерно для людей, ведущих лежачий образ жизни. Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Виды
Мышечная дистрофия очень распространенное патологическое заболевание. Бывает детская и взрослая дистрофия мышц. Также мышечная дистрофия имеет наследственный характер (генетическая и наследственная дистрофия). По характеру и месту локализации различают:
-
инфекционную и неинфекционную; -
миотоническую; -
тазово-плечевую; -
врожденную; -
плечелопаточную;
.
Симптомы
Мышечная дистрофия прогрессирует заболевание мышечной слабости и потери трудоспособности Мышечная дистрофия прогрессирующее заболевание. Поэтому не следует откладывать визит к доктору.
Мышечная дистрофия прогрессирующее заболевание. Поэтому не следует откладывать визит к доктору.
Диагностика
Обратиться к доктору следует немедленно, как только вы заметили мышечную слабость.. К методам диагностики на данном этапе развития медицины относят МРТ. Оно покажет анатомические и физиологические изменения в организме. При этом заболевании также стоит сдать общий анализ крови, мочи и кала. После проведенных диагностик доктор поставит диагноз и направит на лечение.
Лечение
Лечение проводиться с помощью комплексной терапии: консервативное лечение и физиотерапия. Еще не разработано лечение, которые бы полностью устранило это заболевание. Консервативное лечение составляет прием кортикостероидов и препаратов для улучшения мышечной массы. Физиотерапия очень распространенный метод.
Профилактика
Лечение мышечной дистрофии продолжается его профилактикой. Очень важно после выписки с госпиталя не забыть о приписках врача. Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
Клинико-генетические характеристики больных с врожденной мышечной дистрофией, обусловленной мутациями в гене LMNA
Врожденные мышечные дистрофии (ВМД) — группа генетически гетерогенных заболеваний, характеризующихся диффузной мышечной гипотонией, гипорефлексией, снижением объема активных движений, возникающих с рождения или в период новорожденности. К настоящему времени идентифицировано более десяти генов, ответственных за возникновение изолированных форм ВМД, и охарактеризованы белковые продукты их экспрессии [1]. Один из самых редких вариантов этой группы заболеваний обусловлен мутациями в гене LMNA, картированном на хромосоме 1q11—q23 [2] и состоящем из 12 экзонов [3]. Кодируемый им белок преламин, А является предшественником зрелых форм ламинов, А и С, которые формируют структуру внутренней мембраны ядра клетки.
Описано одиннадцать основных фенотипов, обусловленных мутациями в гене LMNA [4—7].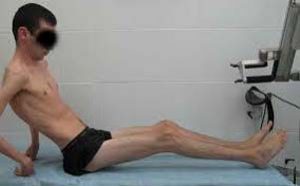 Наиболее часто они приводят к формированию прогрессирующих мышечных дистрофий (ПМД) с аутосомно-доминантным типом наследования, которые имеют различный возраст манифестации и особенности клинических проявлений: ПМД Эмери—Дрейфуса 2-го типа (OMIM:181350), поясно-конечностная ПМД 1 В типа (OMIM:159001) и ВМД (OMIM:613205). В ряде работ были сделаны попытки объяснения особенностей течения ПМД на основании различий в характере мутаций в гене LMNA. Так, некоторые авторы показали, что к возникновению более тяжелых фенотипов приводят мутации, обладающие доминантно-негативным эффектом, в то время как у больных с наличием миссенс-мутаций в этом гене, приводящих только к гаплонедостаточности, отмечается более мягкое течение заболевания [8]. Однако E. Mercuri и соавт. [9] описали 5 больных с различными вариантами ПМД, возникающими с рождения до взрослого возраста, у которых была обнаружена одна и та же миссенс-мутация Gln358Lys.
Наиболее часто они приводят к формированию прогрессирующих мышечных дистрофий (ПМД) с аутосомно-доминантным типом наследования, которые имеют различный возраст манифестации и особенности клинических проявлений: ПМД Эмери—Дрейфуса 2-го типа (OMIM:181350), поясно-конечностная ПМД 1 В типа (OMIM:159001) и ВМД (OMIM:613205). В ряде работ были сделаны попытки объяснения особенностей течения ПМД на основании различий в характере мутаций в гене LMNA. Так, некоторые авторы показали, что к возникновению более тяжелых фенотипов приводят мутации, обладающие доминантно-негативным эффектом, в то время как у больных с наличием миссенс-мутаций в этом гене, приводящих только к гаплонедостаточности, отмечается более мягкое течение заболевания [8]. Однако E. Mercuri и соавт. [9] описали 5 больных с различными вариантами ПМД, возникающими с рождения до взрослого возраста, у которых была обнаружена одна и та же миссенс-мутация Gln358Lys.
Первые сведения о единичных случаях этой формы ПМД были опубликованы в 2004 г. E. Mercuri и соавт. [9] и 2005 г. A. D`Amico и соавт. [10]. Однако выделение врожденной ламинопатии в отдельный вариант ВМД было предложено в 2008 г. S. Quijano-Roy и соавт. [11], которые суммировали клинико-генетические характеристики 15 больных с ВМД, обусловленными мутациями в гене LMNA. К настоящему времени описано более 40 больных с этим вариантом ВМД из популяций Италии, Франции, Бразилии, Германии и др. [6, 11—15].
E. Mercuri и соавт. [9] и 2005 г. A. D`Amico и соавт. [10]. Однако выделение врожденной ламинопатии в отдельный вариант ВМД было предложено в 2008 г. S. Quijano-Roy и соавт. [11], которые суммировали клинико-генетические характеристики 15 больных с ВМД, обусловленными мутациями в гене LMNA. К настоящему времени описано более 40 больных с этим вариантом ВМД из популяций Италии, Франции, Бразилии, Германии и др. [6, 11—15].
Цель настоящей работы — представление клинико-генетических характеристики 5 больных с ВМД, обусловленной мутациями в гене LMNA, впервые выявленных в России.
Материал и методы
Были исследованы образцы ДНК 42 пробандов, наблюдающихся в ФБГНУ «Медико-генетический научный центр», с характерными признаками ВМД в возрасте от 2 мес до 9 лет из неродственных семей, проживающих на территории Р.Ф. Диагноз устанавливали на основании данных генеалогического анализа, неврологического осмотра, показателей активности креатинфосфокиназы (КФК) в сыворотке крови, данных электронейромиографии (ЭНМГ). Большинство больных имели результаты проведенной ранее магнитно-резонансной томографии (МРТ) головного мозга и исследования мышечного биоптата.
Большинство больных имели результаты проведенной ранее магнитно-резонансной томографии (МРТ) головного мозга и исследования мышечного биоптата.
Выделение геномной ДНК проводили из лимфоцитов периферической крови с помощью набора реактивов DLAtomTM DNA Prep100 («Isogene Lab.ltd.», Россия) по протоколу производителя. Исследуемые участки ДНК амплифицировали методом ПЦР с использованием уникальных праймеров, дизайн которых выполнен в лаборатории ДНК-диагностики, синтез — в ЗАО «Евроген». Качественную визуализацию продуктов ПЦР-реакции осуществляли при помощи электрофореза в 7% полиакриламидном геле (ПААГ) с соотношением акриамида и бисакриламида 29:1. Определение нуклеотидной последовательности всей кодирующей области и участков экзон-интронных соединений гена LMNA проводили при помощи метода прямого автоматического секвенирования на приборе ABI Prism 3100 («Applied Biosystems», США) с использованием протокола фирмы-производителя. Полученные данные анализировали при помощи программы Chromas 2. 4.1.
4.1.
Результаты и обсуждение
В результате проведения ДНК-диагностики, направленной на поиск мутаций в гене LMNA, выявлено 5 больных (4 мальчика и 1 девочка) в возрасте от 4 мес до 7 лет. Клинико-генетические характеристики больных с ВМД, обусловленной мутациями в гене LMNA, представлены в таблице.
Клинико-генетические характеристики больных ВМД, обусловленной мутациями в гене LMNA
Как видно из таблицы, все больные имели классические проявления врожденной ламинопатии. Однако у 1 из них имелись отличия в возрасте манифестации, тяжести течения заболевания и типе мутации, что позволило разделить наблюдаемых нами больных на 2 группы. Это коррелирует с данными других исследователей, поскольку, по мнению ряда авторов [5, 7, 11, 13], существуют два клинических варианта ВМД, обусловленных мутациями в гене LMNA, отличающихся по возрасту начала и тяжести течения заболевания.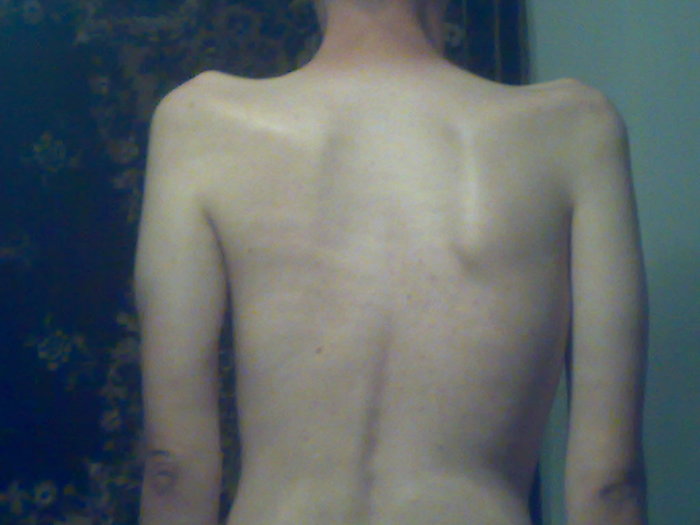
Первую группу составили 4 больных (3 мальчика и 1 девочка) с тяжелой формой болезни имели отягощенный акушерский анамнез и страдали с рождения. На момент осмотра у всех отмечались выраженная гипотрофия, гипотония, мышечная слабость с преимущественным поражением аксиальных мышц и мышц лопаточно-плечевого региона, что приводило к возникновению симптома «свисающей головы». В основном их моторное развитие было минимальным. Лишь 1 мальчик мог передвигаться с поддержкой с 18 мес до 4 лет. Достаточно рано у всех больных этой группы появлялась тугоподвижность в голеностопных суставах, что в последующем приводило к возникновению фиксированной сгибательной контрактуры, а по мере течения заболевания контрактуры формировались и в других крупных суставах (в том числе в локтевых — у 3 больных). Нарушение сердечного ритма имели 2 больных. Дыхательные нарушения, требующие специальной поддержки, наблюдались только у 5-летней больной, которой установили трахеостому в 4,5 года. Однако некоторые авторы [13] считают, что возникновение дыхательной недостаточности с необходимостью использования вспомогательных средств характерно для всех больных с этим вариантом ВМД. У 2 больных на МРТ головного мозга были выявлены признаки перивентрикулярной лейкоэнцефалопатии. Хотя в литературе есть описание [16] 1 больного с ВМД, обусловленной мутацией в гене LMNA, также имевшего локально повышенный, неспецифический МР-сигнал от белого вещества мозга, мы склонны считать, что в нашем случае выявленные признаки перивентрикулярной лейкоэнцефалопатии, вероятнее всего, были обусловлены присоединившимся перинатальным гипоксически-имшемическим поражением ЦНС. У одного пробанда в ранее проведенном исследовании мышечного биоптата были выявлены признаки митохондриальной миопатии, а у другого — неспецифического воспалительного процесса в исследуемой мышце. В литературе [11, 17, 18] также имеются сведения о сходном характере изменений в мышечных биоптатах у некоторых больных с этой формой ВМД. 2 мальчика из этой группы внезапно скончались в возрасте 10 мес и 9 лет. У всех больных в этой группе при проведении ДНК-диагностики была обнаружена мутация c.94_96delAAG в гетерозиготном состоянии в 1-м экзоне гена LMNA, приводящая к делеции аминокислоты лизин (Lys) в 32-м положении полипептидной цепи.
У 2 больных на МРТ головного мозга были выявлены признаки перивентрикулярной лейкоэнцефалопатии. Хотя в литературе есть описание [16] 1 больного с ВМД, обусловленной мутацией в гене LMNA, также имевшего локально повышенный, неспецифический МР-сигнал от белого вещества мозга, мы склонны считать, что в нашем случае выявленные признаки перивентрикулярной лейкоэнцефалопатии, вероятнее всего, были обусловлены присоединившимся перинатальным гипоксически-имшемическим поражением ЦНС. У одного пробанда в ранее проведенном исследовании мышечного биоптата были выявлены признаки митохондриальной миопатии, а у другого — неспецифического воспалительного процесса в исследуемой мышце. В литературе [11, 17, 18] также имеются сведения о сходном характере изменений в мышечных биоптатах у некоторых больных с этой формой ВМД. 2 мальчика из этой группы внезапно скончались в возрасте 10 мес и 9 лет. У всех больных в этой группе при проведении ДНК-диагностики была обнаружена мутация c.94_96delAAG в гетерозиготном состоянии в 1-м экзоне гена LMNA, приводящая к делеции аминокислоты лизин (Lys) в 32-м положении полипептидной цепи. Впервые эту мутацию обнаружили M. Vytopil и соавт. [19] в 2002 г. у больной с тяжелой клинической картиной ПМД Эмери—Дрейфуса. Авторы показали, что женщина унаследовала ее от отца, который в возрасте 52 лет был здоров (на основании чего авторы предположили наличие неполной пенетрантности гена при наличии данной мутации). В литературе также имеются описания больных с другими фенотипами ПМД, манифестирующих с периода новорожденности [10] или в юношеском и взрослом возрасте [20, 21], обусловленными мутацией c.94_96delAAG в гене LMNA.
Впервые эту мутацию обнаружили M. Vytopil и соавт. [19] в 2002 г. у больной с тяжелой клинической картиной ПМД Эмери—Дрейфуса. Авторы показали, что женщина унаследовала ее от отца, который в возрасте 52 лет был здоров (на основании чего авторы предположили наличие неполной пенетрантности гена при наличии данной мутации). В литературе также имеются описания больных с другими фенотипами ПМД, манифестирующих с периода новорожденности [10] или в юношеском и взрослом возрасте [20, 21], обусловленными мутацией c.94_96delAAG в гене LMNA.
Представляем описание 2 клинических случаев из этой группы больных ВМД.
Девочка Б-на, впервые осмотрена в 13 мес по поводу жалоб на выраженную мышечную гипотонию с рождения и задержку темпов моторного развития. Из анамнеза известно, что ребенок от первой беременности, протекавшей в I триместре на фоне токсикоза и фетоплацентарной недостаточности, с формированием внутриутробной гипотрофии плода в III триместре. Роды срочные, самостоятельные на 39-й неделе, с использованием медикаментозной стимуляции и приемов выдавливания плода. При рождении — вес 2800 г, длина 49 см, по шкале Апгар 5/7 баллов за счет аспирации мекониальными водами. Получала ИВЛ в течение 5 сут. Домой выписана на 22-е сутки. С рождения отмечался симптомокомплекс вялого ребенка. На МРТ головного мозга, проведенной в периоде новорожденности, выявлены диффузная субкортикальная субатрофия головного мозга и перивентрикулярная лейкоэнцефалопатия. На ЭНМГ: первично-мышечный уровень поражения. При объективном осмотре в 13 мес: выраженная диффузная мышечная гипотрофия, гипотония с минимальной двигательной активностью в верхних и нижних конечностях, сухожильная арефлексия. Выраженная слабость аксиальных мышц. Симптом «свисающей головы». Контрактур нет. Психоэмоциональная сфера по возрасту. Уровень КФК в сыворотке крови 2000 ед/л. В 15 мес при исследовании мышечного биоптата выявлены признаки неспецифического воспалительного процесса, по поводу чего была предпринята безуспешная попытка терапии гормональными препаратами. При осмотре в 20 мес — голова свисает, удерживать ее может только при подъеме и фиксации плеч, не может поднять руки выше горизонтального уровня и подтянуть ноги к животу, сухожильные рефлексы не вызываются.
При рождении — вес 2800 г, длина 49 см, по шкале Апгар 5/7 баллов за счет аспирации мекониальными водами. Получала ИВЛ в течение 5 сут. Домой выписана на 22-е сутки. С рождения отмечался симптомокомплекс вялого ребенка. На МРТ головного мозга, проведенной в периоде новорожденности, выявлены диффузная субкортикальная субатрофия головного мозга и перивентрикулярная лейкоэнцефалопатия. На ЭНМГ: первично-мышечный уровень поражения. При объективном осмотре в 13 мес: выраженная диффузная мышечная гипотрофия, гипотония с минимальной двигательной активностью в верхних и нижних конечностях, сухожильная арефлексия. Выраженная слабость аксиальных мышц. Симптом «свисающей головы». Контрактур нет. Психоэмоциональная сфера по возрасту. Уровень КФК в сыворотке крови 2000 ед/л. В 15 мес при исследовании мышечного биоптата выявлены признаки неспецифического воспалительного процесса, по поводу чего была предпринята безуспешная попытка терапии гормональными препаратами. При осмотре в 20 мес — голова свисает, удерживать ее может только при подъеме и фиксации плеч, не может поднять руки выше горизонтального уровня и подтянуть ноги к животу, сухожильные рефлексы не вызываются. Контрактур нет. Говорит несколько слов. Уровень КФК в сыворотке крови снизился до 700 ед/л. В возрасте 29 мес родители отметили появление контрактур в голеностопных суставах. При объективном осмотре в 3 года 8 мес — не садится, не стоит, не ходит, сидит с опорой, лицо «миопата», выраженная диффузная гипотрофия и гипотония, голову не удерживает, симптом «свисающей головы», повороты головы и поднятие плеч ограничены, «крыловидные лопатки», выраженный гиперлордоз и сколиоз поясничного отдела позвоночника. Контрактуры в голеностопных, коленных, тазобедренных, локтевых, межфаланговых суставах. Уровень КФК сыворотки крови составляет 441 ед/л. На ЭКГ: незначительная синусовая тахикардия, аритмия с частотой сердечных сокращений 103—130 ударов в 1 мин. Электрическая ось сердца отклонена вправо. Неполная блокада правой ножки пучка Гиса. При проведении ЭХО-кардиографии пороков сердца не выявлено. Полости сердца не расширены. Клапаны интактны, сократительная способность удовлетворительная. В 4,5 года появились дыхательные нарушения, требующие специальной поддержки — установлена трахеостома.
Контрактур нет. Говорит несколько слов. Уровень КФК в сыворотке крови снизился до 700 ед/л. В возрасте 29 мес родители отметили появление контрактур в голеностопных суставах. При объективном осмотре в 3 года 8 мес — не садится, не стоит, не ходит, сидит с опорой, лицо «миопата», выраженная диффузная гипотрофия и гипотония, голову не удерживает, симптом «свисающей головы», повороты головы и поднятие плеч ограничены, «крыловидные лопатки», выраженный гиперлордоз и сколиоз поясничного отдела позвоночника. Контрактуры в голеностопных, коленных, тазобедренных, локтевых, межфаланговых суставах. Уровень КФК сыворотки крови составляет 441 ед/л. На ЭКГ: незначительная синусовая тахикардия, аритмия с частотой сердечных сокращений 103—130 ударов в 1 мин. Электрическая ось сердца отклонена вправо. Неполная блокада правой ножки пучка Гиса. При проведении ЭХО-кардиографии пороков сердца не выявлено. Полости сердца не расширены. Клапаны интактны, сократительная способность удовлетворительная. В 4,5 года появились дыхательные нарушения, требующие специальной поддержки — установлена трахеостома. При осмотре в 5 лет у девочки на фоне выраженной диффузной гипотрофии, гипотонии, сухожильной арефлексии имеет место ригидный поясничный лордосколиоз, сохраняющийся даже в положении лежа на спине, контрактуры во всех крупных и межфаланговых суставах. Трудности при глотании и жевании твердой пищи. Интеллект соответствует возрасту.
При осмотре в 5 лет у девочки на фоне выраженной диффузной гипотрофии, гипотонии, сухожильной арефлексии имеет место ригидный поясничный лордосколиоз, сохраняющийся даже в положении лежа на спине, контрактуры во всех крупных и межфаланговых суставах. Трудности при глотании и жевании твердой пищи. Интеллект соответствует возрасту.
Мальчик К-в, осмотрен впервые в 7 лет. Из анамнеза известно, что ребенок от четвертой беременности, протекавшей с токсикозом. Роды вторые, срочные, самостоятельные. Вес при рождении 3200 г, длина 52 см, по шкале Апгар 7/8 баллов, закричал не сразу. На 3-и сутки установлен диагноз «врожденный порок сердца: дефект межжелудочковой перегородки», по поводу чего перенес успешное оперативное лечение в возрасте 5 мес. С рождения отмечался симтомокомплекс вялого ребенка. Голову держит с 8 мес, сам никогда не садился, ходил с поддержкой с 18 мес до 4 лет. С 6 мес развился эпилептический синдром в виде левосторонних гемиклоний и кивков, купированных по настоящее время приемом противоэпилептических препаратов (конвулекс до 30 мг/кг). При проведении биопсии мышц выявлены признаки митохондриальной миопатии. Уровень КФК в сыворотке крови больного варьировал от 2300 ед/л в начале заболевания до 489 ед/л в возрасте 5 лет. Объективно: диффузная гипотрофия, гипотония, больше выраженная в аксиальных мышцах, симптом «свисающей головы», «лицо миопата», сухожильная арефлексия, выраженный гиперлордоз в поясничном отделе позвоночника, сохраняющийся даже в положении лежа на спине, фиксированный сколиоз грудного отдела позвоночника. Фиксированные контрактуры в голеностопных, коленных, тазобедренных суставах (рис. 1). Трудности при глотании и жевании твердой пищи, носовой оттенок голоса, псевдобульбарный парез. Интеллект соответствует возрасту.
При проведении биопсии мышц выявлены признаки митохондриальной миопатии. Уровень КФК в сыворотке крови больного варьировал от 2300 ед/л в начале заболевания до 489 ед/л в возрасте 5 лет. Объективно: диффузная гипотрофия, гипотония, больше выраженная в аксиальных мышцах, симптом «свисающей головы», «лицо миопата», сухожильная арефлексия, выраженный гиперлордоз в поясничном отделе позвоночника, сохраняющийся даже в положении лежа на спине, фиксированный сколиоз грудного отдела позвоночника. Фиксированные контрактуры в голеностопных, коленных, тазобедренных суставах (рис. 1). Трудности при глотании и жевании твердой пищи, носовой оттенок голоса, псевдобульбарный парез. Интеллект соответствует возрасту.
Рис. 1. Больной К-в, 7 лет. Вид лежа на спине (а) и сидя (б).
Клинические проявления второго варианта BMD, обусловленной мутациями в гене LMNA (более легкая форма) по данным некоторых авторов [5, 7, 11, 13] появляются на 1-м году жизни после периода нормального моторного развития и, хотя также характеризуются прогрессирующей слабостью мышц аксиальных и поясов конечностей, больные более длительно могут передвигаться самостоятельно.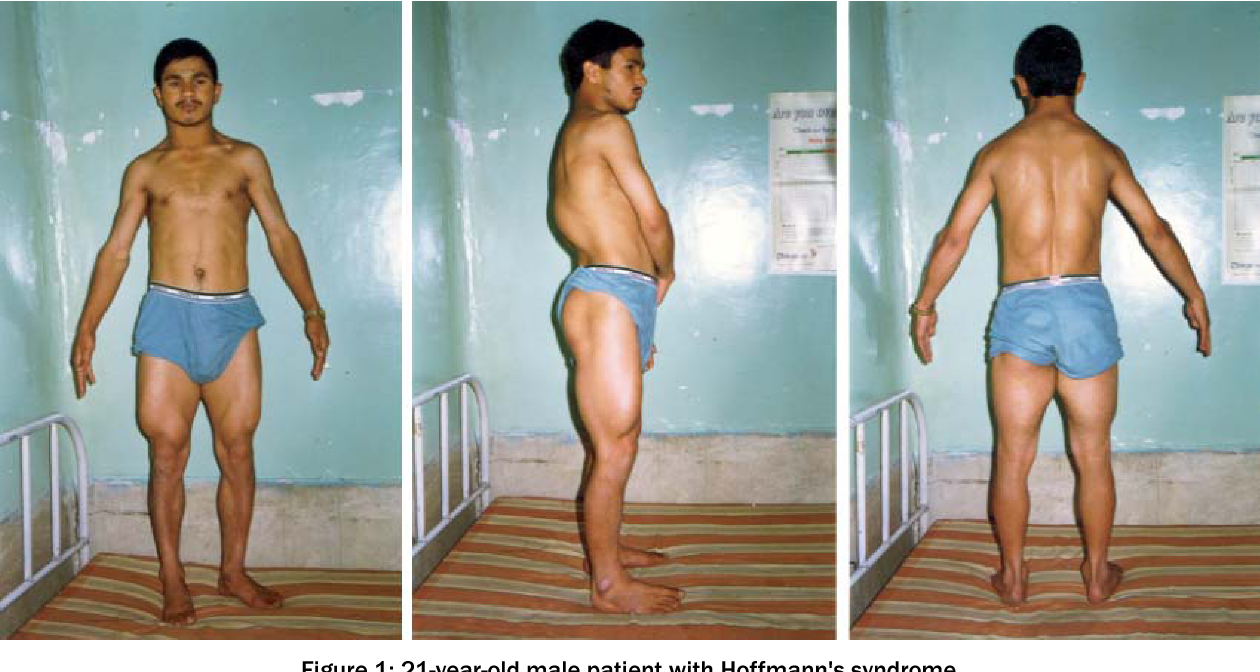
В одном случае и мы наблюдали такой вариант развития ВМД, обусловленный мутацией в гене LMNA. Мальчик Б-н, осмотрен в 5 лет по поводу жалоб на нарушение походки, общую слабость, трудности подъема с пола. Из анамнеза известно, что ребенок от девятой беременности, протекавшей на фоне угрозы прерывания. Роды третьи, самостоятельные, в срок. При рождении вес 4000 г, длина 53 см, закричал сразу. Домой выписан на 5-е сутки. Голову держит с 1,5 мес, сел самостоятельно в 15 мес, пошел в 18 мес. Первые признаки заболевания появились в 3 мес, когда родители обратили внимание на то, что ребенок стал хуже удерживать голову. При проведении ЭНМГ выявлен первично мышечный уровень поражения. На ЭКГ в 4 года выявлена выраженная синусовая тахикардия, частота сердечных сокращений до 112 ударов в 1 мин. Снижен вольтаж QRS в стандартных и усиленных отведениях. При проведении ЭХО-кардиографии обнаружен врожденный порок сердца — открытый аортальный проток (диаметр протока по цветному допплеровскому картированию примерно 2,0 мм), открытое овальное окно (диаметр окна до 3 мм с лево-правым сбросом крови), умеренно расширена легочная артерия. При объективном осмотре в 5 лет: диффузная мышечная гипотония и гипотрофия, сухожильная арефлексия. Слабость аксиальных мышц, голову удерживает плохо, отмечается привычная установка головы с наклоном влево и свисанием вперед, «крыловидные лопатки», слабость лицевой мускулатуры. Небный и глоточный рефлексы несколько снижены. Отмечается выраженная ригидность спины и шеи, выраженный гиперлордоз поясничного отдела позвоночника, фиксированные контрактуры в тазобедренных и локтевых, начинающиеся — в коленных и голеностопных (больше слева) суставах (рис. 2). В настоящее время может стоять с опорой и пройти с поддержкой до 1 м. В 2014 г. по желанию родителей мальчик обследован во Франции, где обнаружена мутация c.1361Т>А в гене LMNA в гетерозиготном состоянии, приводящая к аминокислотной замене лейцина (Leu) на глутаминовую кислоту (Gln) в 454 положении полипептидной цепи. Эта мутация выявлена впервые.
При объективном осмотре в 5 лет: диффузная мышечная гипотония и гипотрофия, сухожильная арефлексия. Слабость аксиальных мышц, голову удерживает плохо, отмечается привычная установка головы с наклоном влево и свисанием вперед, «крыловидные лопатки», слабость лицевой мускулатуры. Небный и глоточный рефлексы несколько снижены. Отмечается выраженная ригидность спины и шеи, выраженный гиперлордоз поясничного отдела позвоночника, фиксированные контрактуры в тазобедренных и локтевых, начинающиеся — в коленных и голеностопных (больше слева) суставах (рис. 2). В настоящее время может стоять с опорой и пройти с поддержкой до 1 м. В 2014 г. по желанию родителей мальчик обследован во Франции, где обнаружена мутация c.1361Т>А в гене LMNA в гетерозиготном состоянии, приводящая к аминокислотной замене лейцина (Leu) на глутаминовую кислоту (Gln) в 454 положении полипептидной цепи. Эта мутация выявлена впервые.
Рис. 2. Больной Б-н, 5 лет. Вид сидя (а) и стоя (б).
Таким образом, нами показано, что на долю ВМД, обусловленной мутациями в гене LMNA, в российской популяции приходится не менее 12% всех случаев этой группы заболеваний. Также полученные результаты свидетельствуют в пользу наличия мажорной мутации в гене LMNA c.94_96delAAG, которая выявлена у 4 из 5 диагностированных больных.
Также полученные результаты свидетельствуют в пользу наличия мажорной мутации в гене LMNA c.94_96delAAG, которая выявлена у 4 из 5 диагностированных больных.
Фенотипические проявления этой формы ВМД имеют специфические особенности и характеризуются симптомами прогрессирующего вялого пареза с преимущественным поражением мышц аксиальных и ответственных за подошвенное сгибание стопы. Заболевание манифестирует в интервале от периода новорожденности до грудного возраста. В клинической картине на фоне диффузной гипотрофии и гипотонии отмечается раннее формирование симптома «свисающей головы», контрактур в дистальных отделах нижних конечностей. По мере прогрессирования заболевания у больных формируются контрактуры в других крупных суставах и выраженный гиперлордоз поясничного отдела позвоночника, сохраняющийся даже в положении лежа на спине. В большинстве случаев отмечается заинтересованность лицевой и дыхательной мускулатуры.
Ограниченность выборки не позволяет сделать однозначное заключение, но мы можем согласиться с мнением других авторов, которые указывают на существование двух фенотипов в этой группе ВМД.
| Подробное описание | Исследование пациентов с врожденными мышечными заболеваниями и результатов, сообщаемых их доверенными лицами (CMDPROS), является продольное 10-летнее обсервационное исследование для определения ключевых параметров ухода и тенденций нежелательные явления при врожденных заболеваниях мышц с использованием врожденного заболевания мышц Международный регистр (CMDIR). CMDIR регистрирует людей с генетическими подтверждение, кому был поставлен клинический диагноз врожденной мышечной дистрофии, врожденная миопатия и врожденный миастенический синдром или миофибриллярная миопатия через пояс конечностей / спектр позднего начала. Выявление параметров лечения и побочных эффектов при редких генетических нервно-мышечных заболеваниях может быть трудным. Уход фрагментирован, генетическое подтверждение не может быть приоритетным для медицинских сообщества или покрытые медицинской страховкой, и пациенты рассеяны по всему миру с потенциальными проблемы с агрегацией данных по центрам. Гипотеза исследования: 1. Использовать ответы на опрос пациентов и доверенных лиц и медицинские отчеты для построения база данных лонгитюдной помощи и результатов по врожденным заболеваниям мышц. 2. Определить частоту нежелательных явлений, специфичных для подтипа врожденных мышечных заболеваний, и сопоставить с ключевыми параметрами ухода. Первичный результат — выживаемость, измеряемая от даты рождения до даты смерти. Первичный результат будет проанализирован по подтипу врожденного мышечного заболевания и достигнутому максимальному амбулаторному статусу. Вторичные исходы включают частоту нежелательных явлений, связанных с конкретным заболеванием, включая частоту госпитализация, частота использования антибиотиков, частота легочных инфекций, пневмоторакса, ателектаз, аспирация и неблагоприятные жалобы, включая вздутие живота, запор, боль в груди, одышка, оцененная с помощью проверенной оценки дыхания, рвота, тошнота и затрудненное состояние принимать пищу. Отчеты о госпитализации пациентов и доверенных лиц, пневмотораксе и ателектазах будут подтверждается получением выписки из больницы. Дополнительные вторичные результаты включают: фракция выброса (зависит от подтипа), форсированная жизненная емкость легких в литрах, вес, Индекс гипопноэ апноэ во сне с быстрым движением глаз (REM) и средняя сатурация кислорода во время REM и исследование общего сна, возраст, пол, тип расположения лечебного центра (национальный справочный центр, больница третичного уровня, общественная больница), гастростомическая трубка, общее количество переломов и Tscore / Zscore бедра и позвоночника на сканировании DEXA. Предварительные исследования могут быть сосредоточены на конкретных подтипах врожденных мышечных заболеваний и их использовании. ретроспективный сбор данных через регистрацию, обследование обезьян и телефонные интервью с оценить частоту нежелательных явлений за последний месяц и за последний год, чтобы ограничить систематическую ошибку воспоминаний. Перспективный набор тех же участников исследования в течение 12 месяцев будет оценивать ежемесячные показатели неблагоприятных события и жалобы. Предварительное исследование CMD PROADE (Пациенты и доверенные лица сообщают о негативных последствиях). Частота событий) планируется для двух подтипов врожденной мышечной дистрофии: коллагеновой 6 миопатии и LAMA 2 Связанный CMD. Деидентифицированные данные из CMDIR будут доступны для исследований естественной истории, утвержденных IRB. при врожденных заболеваниях мышц. |
|---|
 Исследования естествознания в настоящее время запущен. Однако потенциальные предубеждения в отношении участия включают вербовку менее серьезных пострадавшие пациенты, столкнувшиеся с трудностями при путешествии, вызванными болезнью с медицинской точки зрения. В настоящее время не существует лечения этих состояний; через оптимизацию и стандартизацию уход и оказание помощи могут способствовать значительному повышению качества жизни и выживаемости. Выявление параметров лечения, характерных для конкретного заболевания, и сопоставление этих параметров с неблагоприятными частота событий будет способствовать не только разработке руководств, основанных на фактических данных, но и информирует о клинически значимых результатах для будущих клинических испытаний.
Исследования естествознания в настоящее время запущен. Однако потенциальные предубеждения в отношении участия включают вербовку менее серьезных пострадавшие пациенты, столкнувшиеся с трудностями при путешествии, вызванными болезнью с медицинской точки зрения. В настоящее время не существует лечения этих состояний; через оптимизацию и стандартизацию уход и оказание помощи могут способствовать значительному повышению качества жизни и выживаемости. Выявление параметров лечения, характерных для конкретного заболевания, и сопоставление этих параметров с неблагоприятными частота событий будет способствовать не только разработке руководств, основанных на фактических данных, но и информирует о клинически значимых результатах для будущих клинических испытаний.

Мерозин-дефицитная мышечная дистрофия
Генетическое исследование является конечной точкой диагностики . Проводится секвенирование гена LAMA2. Для подтверждения диагноза необходимо выявление гомозиготной или двух компаунд-гетерози готных мутаций. В случае обнаружения только одной точечной мутации в гетерозиготном состоянии при наличии характерной клинической картины или значимого повышения уровня КФК в крови и гиперсигнала от белого вещества на Т2 по данным МРТ головного мозга целесообразно проведение хромосомного микроматричного анализа с целью поиска крупных делеций и дупликаций, которые встречаются с высокой частотой — по разным данным, от 18% до 30–40% мутаций в гене LAMA2. Корреляции генотипа и фенотипа Прогноз тяжести заболевания зависит от возраста дебюта первых симптомов, наличия или отсутствия экспресcии ламинина-α2 при проведении иммуногистохимического тестирования мышечного биоптата, типа мутации и ее влияния на функцию белка. Для МД-МД приобретение навыка самостоятельной ходьбы является критически важной клинической характеристикой тяжести заболевания. Фенотип ВМД 1А типа ассоциирован с мутациями, приводящими к прекращению синтеза белка.
Проводится секвенирование гена LAMA2. Для подтверждения диагноза необходимо выявление гомозиготной или двух компаунд-гетерози готных мутаций. В случае обнаружения только одной точечной мутации в гетерозиготном состоянии при наличии характерной клинической картины или значимого повышения уровня КФК в крови и гиперсигнала от белого вещества на Т2 по данным МРТ головного мозга целесообразно проведение хромосомного микроматричного анализа с целью поиска крупных делеций и дупликаций, которые встречаются с высокой частотой — по разным данным, от 18% до 30–40% мутаций в гене LAMA2. Корреляции генотипа и фенотипа Прогноз тяжести заболевания зависит от возраста дебюта первых симптомов, наличия или отсутствия экспресcии ламинина-α2 при проведении иммуногистохимического тестирования мышечного биоптата, типа мутации и ее влияния на функцию белка. Для МД-МД приобретение навыка самостоятельной ходьбы является критически важной клинической характеристикой тяжести заболевания. Фенотип ВМД 1А типа ассоциирован с мутациями, приводящими к прекращению синтеза белка. В основном, это нонсенс-варианты, при которых формируется стоп-кодон. Для таких пациентов характерно тяжелое течение заболевания с ранним началом, дыхательными нарушениями, часто с необходимостью респираторной поддержки, отсутствием самостоятельной ходьбы. По данным биопсии определяется полное отсутствие экспрессии ламинина-α2 в мышцах и коже.
В основном, это нонсенс-варианты, при которых формируется стоп-кодон. Для таких пациентов характерно тяжелое течение заболевания с ранним началом, дыхательными нарушениями, часто с необходимостью респираторной поддержки, отсутствием самостоятельной ходьбы. По данным биопсии определяется полное отсутствие экспрессии ламинина-α2 в мышцах и коже.
Формы заболевания с началом в детском или взрослом возрасте, более мягким течением, достижением навыка самостоятельной ходьбы вызваны гомозиготными или компаунд-гетерозиготными миссенс-заменами, а также крупными делециями с восстановлением рамки считывания. Эти мутации приводят к снижению или почти нормальной экспрессии ламинина-α2 и влияют на полимеризацию мерозина. Большинство исследований подтверждают прямую корреляцию генотипа и фенотипа. Несмотря на это F. Geranmayeh и соавт. сообщили о семье, члены которой имели гомозиготные миссенсмутации в гене LAMA2, при этом не все приобрели способность самостоятельной ходьбы.
Дифференциальный диагноз Мерозин-негативную ВМД следует дифференцировать с заболеваниями, сопровождающимися мышечной слабостью и гипотонией с первых месяцев жизни.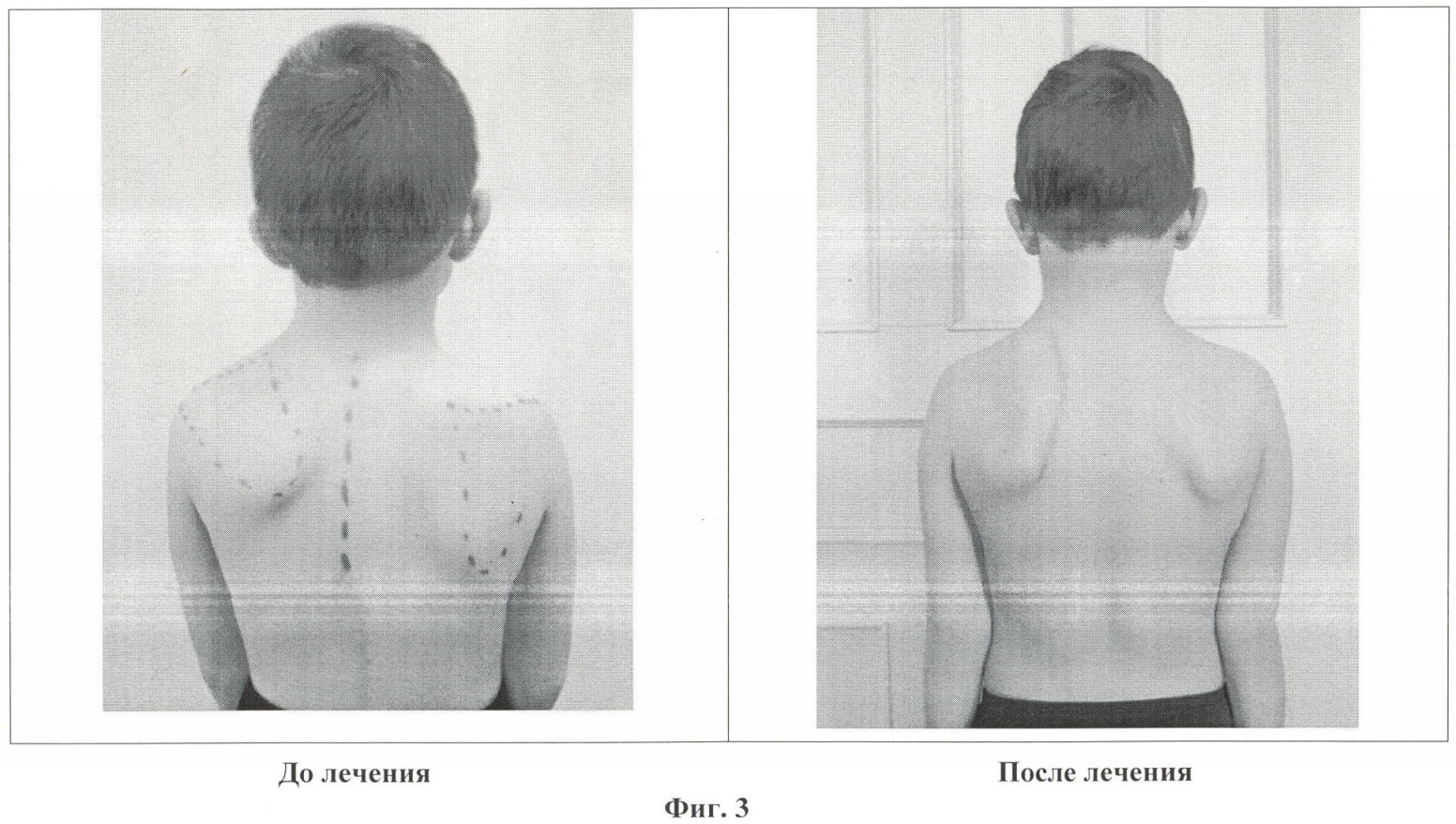 В первую очередь, это другие формы врожденных миопатий (ВМД Ульриха, синдром ригидного позвоночника, болезнь центрального стержня, немалиновая, центронуклеарная миопатии, дистрогликанопатии, ВМД Фукуямы), врожденные миастенические синдромы, спинальная мышечная атрофия. Отличительными чертами ВМД 1А типа являются наиболее высокие среди всех врожденных миопатий показатели КФК, превышающие норму в 2–17 раз, и изменения на МРТ головного мозга (гиперинтенсивный сигнал от белого вещества в режиме Т2, структурные аномалии). При ВМД Фукуямы и дистрогликанопатиях может отмечаться частичный дефицит мерозина, выявляемый при биопсии мышечной ткани; антитела к длинному фрагменту α2-цепи ламинина помогают отличить первичный дефицит мерозина от вторичного. Структурные изменения головного мозга, встречающиеся при ВМД с дефектами гликозилирования (мышечно-глазо-мозговой синдром, ВМД Фукуямы, синдром Уокера–Варбурга), обычно более значительны и сопровождаются грубой задержкой психического развития и высокой частотой развития эпилепсии.
В первую очередь, это другие формы врожденных миопатий (ВМД Ульриха, синдром ригидного позвоночника, болезнь центрального стержня, немалиновая, центронуклеарная миопатии, дистрогликанопатии, ВМД Фукуямы), врожденные миастенические синдромы, спинальная мышечная атрофия. Отличительными чертами ВМД 1А типа являются наиболее высокие среди всех врожденных миопатий показатели КФК, превышающие норму в 2–17 раз, и изменения на МРТ головного мозга (гиперинтенсивный сигнал от белого вещества в режиме Т2, структурные аномалии). При ВМД Фукуямы и дистрогликанопатиях может отмечаться частичный дефицит мерозина, выявляемый при биопсии мышечной ткани; антитела к длинному фрагменту α2-цепи ламинина помогают отличить первичный дефицит мерозина от вторичного. Структурные изменения головного мозга, встречающиеся при ВМД с дефектами гликозилирования (мышечно-глазо-мозговой синдром, ВМД Фукуямы, синдром Уокера–Варбурга), обычно более значительны и сопровождаются грубой задержкой психического развития и высокой частотой развития эпилепсии. Сочетание задержки развития и изменений на МРТ нередко заставляет дифференцировать ВМД 1А типа с лейкодистрофиями. Высокий уровень КФК, медленное прогрессирование со стабилизацией состояния после 1-го года жизни при мерозин-негативной ВМД и, напротив, быстрое неуклонно прогрессирующее течение, характерное для лейкодистрофий, помогают различить эти два состояния. Дифференциальный диагноз поздних форм LAMA2-связанной МД более затруднителен. I. Nelson и соавт. сообщили о 4 пациентах, двое из которых наблюдались с диагнозом миопатии Бетлема, двое с миодистрофией Эмери–Дрейфуса. У пациентов с диагнозом миопатии Бетлема определялся типичный фенотип коллагенопатии (кожные изменения, контрактуры крупных суставов). Пациенты с миодистрофией Эмери–Дрейфуса имели контрактуры в локтевых суставах, дилатационную кардиомиопатию и тяжелые нарушения сердечного ритма в виде желудочковой тахикардии и фибрилляции. Картина МРТ мышц, повышение уровня КФК в крови также соответствовали предполагаемым диагнозам.
Сочетание задержки развития и изменений на МРТ нередко заставляет дифференцировать ВМД 1А типа с лейкодистрофиями. Высокий уровень КФК, медленное прогрессирование со стабилизацией состояния после 1-го года жизни при мерозин-негативной ВМД и, напротив, быстрое неуклонно прогрессирующее течение, характерное для лейкодистрофий, помогают различить эти два состояния. Дифференциальный диагноз поздних форм LAMA2-связанной МД более затруднителен. I. Nelson и соавт. сообщили о 4 пациентах, двое из которых наблюдались с диагнозом миопатии Бетлема, двое с миодистрофией Эмери–Дрейфуса. У пациентов с диагнозом миопатии Бетлема определялся типичный фенотип коллагенопатии (кожные изменения, контрактуры крупных суставов). Пациенты с миодистрофией Эмери–Дрейфуса имели контрактуры в локтевых суставах, дилатационную кардиомиопатию и тяжелые нарушения сердечного ритма в виде желудочковой тахикардии и фибрилляции. Картина МРТ мышц, повышение уровня КФК в крови также соответствовали предполагаемым диагнозам. Двое наблюдаемых страдали фармакорезистентной эпилепсией. При проведении МРТ головного мозга патологические изменения выявлены у 3 человек. Всем пациентам было проведено генетическое тестирование, при котором обнаружены гомозиготные (2 случая) и компаунд-гетерозиготные (2 случая) мутации в гене LAMA2. На основании этого наблюдения авторами было предложено всем больным миопатией, в том числе взрослым, имеющим контрактуры в крупных суставах, изменения на МРТ головного мозга и даже изолированную кардиомиопатию, включать в диагностический поиск ген LAMA2. Также мягкие формы иногда приходится дифференцировать с невропатиями, при которых поражение нервов выходит на передний план в клинической картине, а легкая проксимальная мышечная слабость мало беспокоит пациентов и выявляется лишь при неврологическом тестировании.
Двое наблюдаемых страдали фармакорезистентной эпилепсией. При проведении МРТ головного мозга патологические изменения выявлены у 3 человек. Всем пациентам было проведено генетическое тестирование, при котором обнаружены гомозиготные (2 случая) и компаунд-гетерозиготные (2 случая) мутации в гене LAMA2. На основании этого наблюдения авторами было предложено всем больным миопатией, в том числе взрослым, имеющим контрактуры в крупных суставах, изменения на МРТ головного мозга и даже изолированную кардиомиопатию, включать в диагностический поиск ген LAMA2. Также мягкие формы иногда приходится дифференцировать с невропатиями, при которых поражение нервов выходит на передний план в клинической картине, а легкая проксимальная мышечная слабость мало беспокоит пациентов и выявляется лишь при неврологическом тестировании.
Тактика ведения пациентов
В 2010 г. был опубликован Международный консенсус по стандартам лечения пациентов с ВМД, в котором подробно описаны объем и порядок оказания необходимой им помощи. Дети должны наблюдаться мультидисциплинарной командой специалистов, включающей педиатра, невролога, ортопеда, пульмонолога, диетолога, кардиолога, физического терапевта. Пациенты до 1 года и старше с тяжелыми или прогрессирующими формами (рефрактерные эпилептические приступы, тяжелая гипотония, нутритивные проблемы) должны осматриваться специалистами каждые 3–4 мес. Дети старше 1 года в стабильном состоянии нуждаются в рутинном обследовании каждые 4–6 мес. Обследование включает в себя оценку статуса питания, сердечной функции и ортопедических осложнений, выявление гастроэзофагального рефлюкса, проведение функциональных легочных тестов, ночной пульсоксиметрии или полисомнографии ослабленным детям с рецидивирующими респираторными инфекциями. Поскольку подавляющее большинство пациентов с ВМД не приобретают навык самостоятельной ходьбы и имеют прогрессирующее снижение мышечной массы, часто такие дети имеют низкий вес. Консультации гастроэнтеролога, диетолога рекомендованы 2 раза в год. Целью не является нормализация массы тела до стандартных значений, достаточна положительная динамика в ежегодной прибавке веса.
Дети должны наблюдаться мультидисциплинарной командой специалистов, включающей педиатра, невролога, ортопеда, пульмонолога, диетолога, кардиолога, физического терапевта. Пациенты до 1 года и старше с тяжелыми или прогрессирующими формами (рефрактерные эпилептические приступы, тяжелая гипотония, нутритивные проблемы) должны осматриваться специалистами каждые 3–4 мес. Дети старше 1 года в стабильном состоянии нуждаются в рутинном обследовании каждые 4–6 мес. Обследование включает в себя оценку статуса питания, сердечной функции и ортопедических осложнений, выявление гастроэзофагального рефлюкса, проведение функциональных легочных тестов, ночной пульсоксиметрии или полисомнографии ослабленным детям с рецидивирующими респираторными инфекциями. Поскольку подавляющее большинство пациентов с ВМД не приобретают навык самостоятельной ходьбы и имеют прогрессирующее снижение мышечной массы, часто такие дети имеют низкий вес. Консультации гастроэнтеролога, диетолога рекомендованы 2 раза в год. Целью не является нормализация массы тела до стандартных значений, достаточна положительная динамика в ежегодной прибавке веса. Адекватное увеличение веса у таких детей может быть достигнуто дополнительным приемом гиперкалорийных смесей. Пациентам с нарушениями глотания, гастроэзофагеальным рефлюксом, аспирационными пневмониями могут потребоваться установка назогастрального зонда или чрескожной гастростомы. Посещение пульмонолога для оценки дыхательной функции должно быть как минимум ежегодным. Респираторные тесты включают в себя спирометрию и оценку эффективности кашля, проводятся с 4–6 лет. Показатели форсированной жизненной емкости легких (ФЖЕЛ) <60% от прогнозируемого значения связаны с нарушениями дыхания во сне, ФЖЕЛ <40% от возрастной нормы связано с высоким риском ночной гиповентиляции. Таким пациентам обязательно измерение уровня сатурации и гиперкапнии во время ночного сна, при патологических показателях — проведение неинвазивной вентиляции легких. Больным с выраженными деформациями позвоночника, рецидивирующими дыхательными инфекциями рекомендовано проведение КТ грудной клетки для оценки наличия хронических ателектазов и компрессии дыхательных путей телами позвонков.
Адекватное увеличение веса у таких детей может быть достигнуто дополнительным приемом гиперкалорийных смесей. Пациентам с нарушениями глотания, гастроэзофагеальным рефлюксом, аспирационными пневмониями могут потребоваться установка назогастрального зонда или чрескожной гастростомы. Посещение пульмонолога для оценки дыхательной функции должно быть как минимум ежегодным. Респираторные тесты включают в себя спирометрию и оценку эффективности кашля, проводятся с 4–6 лет. Показатели форсированной жизненной емкости легких (ФЖЕЛ) <60% от прогнозируемого значения связаны с нарушениями дыхания во сне, ФЖЕЛ <40% от возрастной нормы связано с высоким риском ночной гиповентиляции. Таким пациентам обязательно измерение уровня сатурации и гиперкапнии во время ночного сна, при патологических показателях — проведение неинвазивной вентиляции легких. Больным с выраженными деформациями позвоночника, рецидивирующими дыхательными инфекциями рекомендовано проведение КТ грудной клетки для оценки наличия хронических ателектазов и компрессии дыхательных путей телами позвонков. Занятия с физическим терапевтом направлены на уменьшение мышечной гипотонии, увеличение двигательных возможностей пациента, предотвращение контрактур и респираторной дисфункции. Они включают ежедневную вертикализацию в ортопедических аппаратах, упражнения на растяжку и плавание в бассейне. Осмотр ортопедом и проведение рентгенографии позвоночника должны осуществляться, как минимум, раз в год. Более частая оценка оправдана в периоды быстрого роста и прогрессировании деформаций позвоночного столба. Ортезы и туторы используются для предотвращения прогрессирования деформаций суставов, обязательно использование корсета для поддержания осанки.
Занятия с физическим терапевтом направлены на уменьшение мышечной гипотонии, увеличение двигательных возможностей пациента, предотвращение контрактур и респираторной дисфункции. Они включают ежедневную вертикализацию в ортопедических аппаратах, упражнения на растяжку и плавание в бассейне. Осмотр ортопедом и проведение рентгенографии позвоночника должны осуществляться, как минимум, раз в год. Более частая оценка оправдана в периоды быстрого роста и прогрессировании деформаций позвоночного столба. Ортезы и туторы используются для предотвращения прогрессирования деформаций суставов, обязательно использование корсета для поддержания осанки.
Кардиологическое обследование в отсутствие жалоб проводится в 5 и 10 лет, затем каждые 2 года и включает в себя электрокардиографию и эхокардиографию. Пациентам с тяжелой дыхательной недостаточностью, находящимся на аппаратной вентиляционной поддержке, обязательно ежегодное проведение эхокардиографии. Пациентам с жалобами на учащенное сердцебиение, повышенную утомляемость дополнительно проводится холтеровское мониторирование сердечного ритма.
Стратегии терапии
В настоящий момент исследуются несколько вариантов восстановления структуры базальной мембраны при МД-МД: Трансгенная экспрессия кДНК, кодирующей ламинин-α1, с использованием промотера куриного β-актина (CAG). Проводилась на мышиной модели dy3K/dy3K, у которой отмечено существенное восстановление мышц и периферических нервов с повышением силы. Эти исследования доказали, что ламинин-α1 может полностью заменить ламинин-α2. Однако этот подход не может быть применен в качестве генной терапии для доставки вирусными векторами, поскольку к ДНК белка ламинина слишком велика. Парентеральное введение рекомбинантного ламинина-111. Трудности применения у человека также заключаются в большом размере белка. Использование линкерных белков. У пациентов с МД-МД увеличена экспрессия ламинина-411, однако этот белок имеет низкое сродство с α-дистрогликаном и α7β1-интегрином. Была разработана уменьшенная версия белка агрина (мини-агрин, или маг), который значительно улучшает связывание с α-дистрогликаном.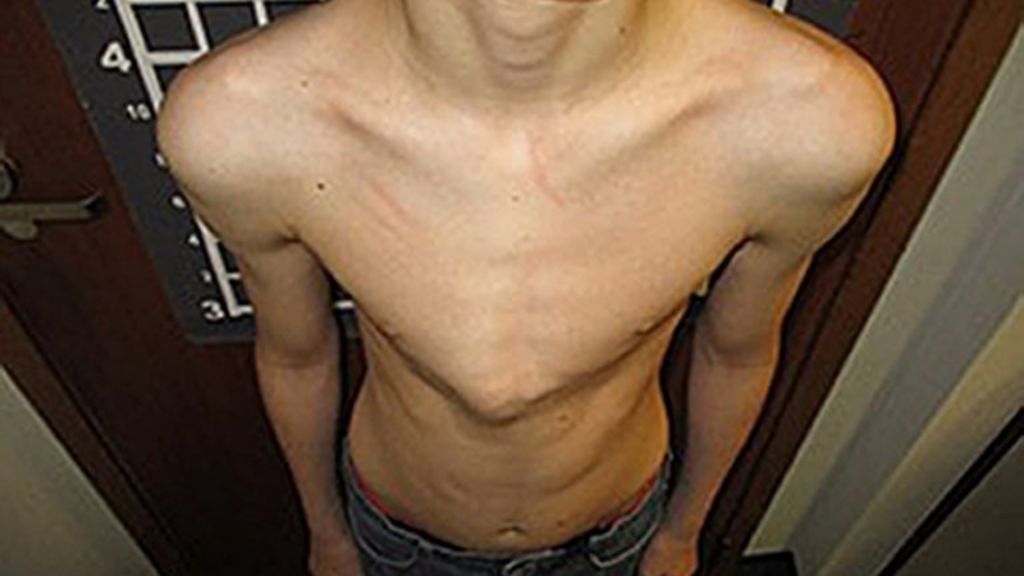 Инициация полимеризации ламинина. Для восстановления полимеризации ламинина был созданлинкерный белок, состоящий из фрагментов ламинина и нидогена αLNNd). Отмечено восстановление силы у мышей dy2J/dy2J, редукция фиброза по данным гистологии. Данный вариант лечения подходит лишь для малой группы пациентов, у которых экспрессия ламинина-α2 снижена незначительно.
Инициация полимеризации ламинина. Для восстановления полимеризации ламинина был созданлинкерный белок, состоящий из фрагментов ламинина и нидогена αLNNd). Отмечено восстановление силы у мышей dy2J/dy2J, редукция фиброза по данным гистологии. Данный вариант лечения подходит лишь для малой группы пациентов, у которых экспрессия ламинина-α2 снижена незначительно.
Использование технологий CRISPR/Cas9. Отмечено восстановление синтеза полноразмерного ламинина-α2 в мышиной модели dy2J/dy2J, повышение силы мышц, уменьшение фиброза. Учитывая многообещающие результаты, финансирование и дальнейшая разработка технологий LAMA2 CRISPR является одной из приоритетных задач исследований. Помимо лечения, направленного на восстановление структуры базальной мембраны, разрабатывается терапия по предотвращению последствий повреждения мышц. Завершена I фаза открытого клинического исследования препарата омигаприла для пациентов с LAMA2- и COL6-связанными мышечными дистрофиями (CALLISTO). Омигаприл является ингибитором апоптоза, блокируя глицеральдегид-3-фосфатдегидрогеназу. Он уменьшает потерю массы, дегенерацию мышечных волокон, особенно в дыхательной мускулатуре, что способствует улучшению респираторных функций и профилактике осложнений. Другой препарат — лозартан — является блокатором рецепторов ангиотензина II типа I. Он влияет на активность трансформирующего фактора роста, уменьшая фиброз и улучшая клинические проявления. Другие препараты (бортезомид, преднизолон) не показали свою эффективность или имели значительные побочные эффекты при ВМД 1А типа. Несмотря на то что методы терапии, направленные на восстановление структурного дефекта, показывают более значимую степень клинического улучшения у пациентов с МД-МД, чем препараты патогенетической терапии, нельзя быть уверенными, что один метод лечения окажется достаточным для пациентов. Вероятно, наиболее успешными могут оказаться протоколы с комбинацией этих двух групп терапии.
Он уменьшает потерю массы, дегенерацию мышечных волокон, особенно в дыхательной мускулатуре, что способствует улучшению респираторных функций и профилактике осложнений. Другой препарат — лозартан — является блокатором рецепторов ангиотензина II типа I. Он влияет на активность трансформирующего фактора роста, уменьшая фиброз и улучшая клинические проявления. Другие препараты (бортезомид, преднизолон) не показали свою эффективность или имели значительные побочные эффекты при ВМД 1А типа. Несмотря на то что методы терапии, направленные на восстановление структурного дефекта, показывают более значимую степень клинического улучшения у пациентов с МД-МД, чем препараты патогенетической терапии, нельзя быть уверенными, что один метод лечения окажется достаточным для пациентов. Вероятно, наиболее успешными могут оказаться протоколы с комбинацией этих двух групп терапии.
Заключение
МД-ВМД является самой распространенной формой среди ВМД. Она крайне гетерогенна генетически и клинически, проявляясь, в подавляющем большинстве случаев, тяжелыми неамбулаторными фенотипами с полным отсутствием экспрессии мерозина в тканях, но также встречаются и легкие КП-формы с поздним дебютом заболевания, связанные со сниженным содержанием ламинина-α2 в мышцах и периферических нервах.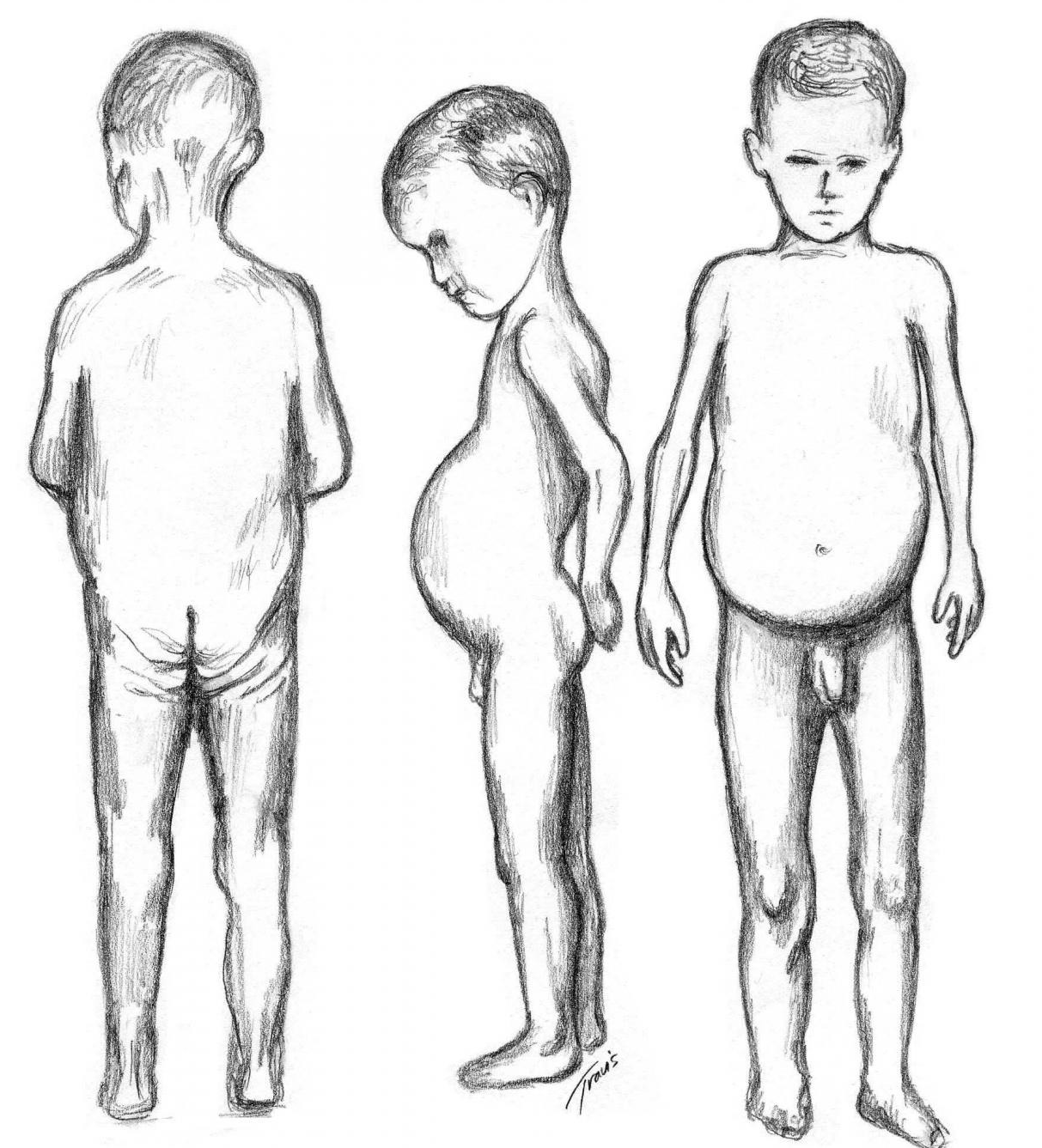 Такая гетерогенность ведет к трудностям диагностики, особенно нетяжелых форм болезни. Знание особенностей клинического течения и патогенеза LAMA2-связанных мышечных дистрофий особенно актуально в условиях активно разрабатываемых специфических методов их терапии.
Такая гетерогенность ведет к трудностям диагностики, особенно нетяжелых форм болезни. Знание особенностей клинического течения и патогенеза LAMA2-связанных мышечных дистрофий особенно актуально в условиях активно разрабатываемых специфических методов их терапии.
Врожденная мышечная дистрофия — NORD (Национальная организация по редким заболеваниям)
Начало, специфические симптомы и тяжесть ВМД значительно различаются даже среди пораженных членов одной семьи. Было предложено несколько различных методов классификации CMD. Одна классификация разделяет эти расстройства на основе первичного генетического дефекта. Эта классификация состоит из трех основных категорий: CMD, вызванные дефектными генами, которые продуцируют структурные белки базальной мембраны или внеклеточного матрикса, сложной структуры, которая окружает и поддерживает клетки; CMD, вызванные дефектными генами, которые продуцируют белки, необходимые для нормального прикрепления или связывания (гликозилирования) молекул сахара с дистрогликаном, белком, обнаруженным на мембране (сарколемме) мышечных клеток; или CMD, вызванные дефектным геном селенопротеина 1 (SEPN1), который продуцирует белок без известной в настоящее время функции. Недавно дефектные гены, которые производят белки ядерной оболочки, двухслойной мембраны, покрывающей ядра некоторых клеток, также были связаны с CMD.
Недавно дефектные гены, которые производят белки ядерной оболочки, двухслойной мембраны, покрывающей ядра некоторых клеток, также были связаны с CMD.
CMDS, ВЫЗВАННЫЕ ДЕФЕКТНЫМИ СТРУКТУРНЫМИ БЕЛКАМИ БАЗАЛЬНОЙ МЕМБРАНЫ ИЛИ ВНЕКЛЕТОЧНОЙ МАТРИЦЫ МЫШЕЧНЫХ ВОЛОКОН.
* Врожденная мышечная дистрофия типа 1A (MDC1A; CMD с дефицитом мерозина; CMD с дефицитом ламинина альфа 2)
Эта форма CMD может быть связана с полным дефицитом белка мерозина в мышцах, белка, обнаруженного в ткани, которая окружает мышечные волокна.Младенцы обычно демонстрируют пониженный мышечный тонус (гипотония) и мышечную слабость при рождении. Некоторые младенцы могут испытывать затруднения с дыханием и кормлением вскоре после рождения (неонатальный период). Проблемы с кормлением могут привести к тому, что пораженные дети не смогут набрать вес и расти с ожидаемой скоростью (неспособность развиваться). Мышечная слабость является серьезной, и у большинства детей с ней наблюдается задержка в достижении или неспособность достичь многих моторных вех. Большинство пострадавших младенцев могут сидеть без опоры, а некоторые могут стоять без посторонней помощи.Лишь немногие дети с тяжелой формой MDC1A в конечном итоге могут ходить без посторонней помощи.
Большинство пострадавших младенцев могут сидеть без опоры, а некоторые могут стоять без посторонней помощи.Лишь немногие дети с тяжелой формой MDC1A в конечном итоге могут ходить без посторонней помощи.
Могут возникать дополнительные симптомы, включая контрактуры суставов, врожденный вывих бедра, прогрессирующее искривление позвоночника (сколиоз) и слабость мышц вокруг глаз, которая постепенно ограничивает движения глаз (офтальмоплегия). Некоторые люди с MDC1A испытывают судороги. По мере взросления у детей проблемы с дыханием могут прогрессировать и вызывать такие осложнения, как нарушение дыхания ночью (ночная гиповентиляция).Судорожные припадки были зарегистрированы примерно у 20-30% пострадавших.
Хотя встречается реже, у некоторых больных детей наблюдается лишь частичный дефицит мерозина. Тяжесть частичной недостаточности мерозина сильно варьируется и может быть намного слабее. Слабость мышц может отсутствовать. Возраст начала может быть в младенчестве, подростковом или взрослом возрасте. В младенчестве у пораженных людей может наблюдаться снижение мышечного тонуса (гипотония), контрактуры и задержки в достижении моторных вех. Хотя в редких случаях частичный дефицит мерозина может напоминать классический MDC1A, симптомы обычно не проявляются до второго десятилетия жизни, а кормление или искусственная вентиляция легких обычно не требуется или не требуется до гораздо более позднего возраста.Начало в подростковом или взрослом возрасте обычно напоминает аналогичное мышечное заболевание, известное как мышечная дистрофия конечностей. (Для получения дополнительной информации об этом заболевании см. Раздел, посвященный связанным расстройствам в этом отчете.)
В младенчестве у пораженных людей может наблюдаться снижение мышечного тонуса (гипотония), контрактуры и задержки в достижении моторных вех. Хотя в редких случаях частичный дефицит мерозина может напоминать классический MDC1A, симптомы обычно не проявляются до второго десятилетия жизни, а кормление или искусственная вентиляция легких обычно не требуется или не требуется до гораздо более позднего возраста.Начало в подростковом или взрослом возрасте обычно напоминает аналогичное мышечное заболевание, известное как мышечная дистрофия конечностей. (Для получения дополнительной информации об этом заболевании см. Раздел, посвященный связанным расстройствам в этом отчете.)
* Расстройства, связанные с коллагеном VI типа
Расстройства, связанные с коллагеном VI типа, представляют собой спектр заболеваний, включающий два расстройства, которые ранее считались отдельными состояниями, Миопатия Бетлема и врожденная мышечная дистрофия Ульриха. Миопатия Бетлема представляет собой умеренную часть этого спектра; Врожденная мышечная дистрофия Ульриха представляет собой тяжелую часть этого спектра. Обычны промежуточные формы. (Для получения дополнительной информации см. Отдельную запись NORD о «расстройствах, связанных с коллагеном VI типа» в базе данных редких заболеваний NORD.)
Обычны промежуточные формы. (Для получения дополнительной информации см. Отдельную запись NORD о «расстройствах, связанных с коллагеном VI типа» в базе данных редких заболеваний NORD.)
Характерными симптомами этой формы CMD являются снижение мышечного тонуса (гипотония), мышечная слабость, аномалии передних конечностей. искривление позвоночника из стороны в сторону и назад (кифосколиоз) и аномально гибкие (гиперэластичные) суставы запястий и лодыжек, а также пальцев рук и ног. Контрактуры могут присутствовать при рождении или возникать позже.Могут присутствовать дополнительные симптомы, а степень тяжести сильно варьируется. В некоторых случаях может развиться врожденный вывих бедра, мышечные спазмы шеи (кривошея) или снижение плотности костей. Пострадавшие люди также могут испытывать затруднения с дыханием (респираторными) и неспособность к развитию.
Интеллект в большинстве случаев нормальный. Степень моторного развития варьируется от случая к случаю. Некоторые дети могут ходить самостоятельно; другим требуется помощь при ходьбе. В некоторых случаях пораженные дети могут никогда не ходить.Кроме того, некоторые дети, у которых развивается способность самостоятельно ходить, теряют эту способность из-за прогрессирования заболевания и обострения контрактур. Другие сохраняют способность ходить в зрелом возрасте.
В некоторых случаях пораженные дети могут никогда не ходить.Кроме того, некоторые дети, у которых развивается способность самостоятельно ходить, теряют эту способность из-за прогрессирования заболевания и обострения контрактур. Другие сохраняют способность ходить в зрелом возрасте.
Могут возникать дополнительные симптомы, включая недостаточность дыхания и состояние кожи, характеризующееся утолщением и затвердеванием (гиперкератоз) волосяных фолликулов, что приводит к развитию грубых возвышенных наростов (папул) на коже. Однако кожа на ладонях рук и подошвах ног бархатистая и очень мягкая.
У лиц с этой формой ВМД развивается ригидность спины, часто со сколиозом.
* Врожденная мышечная дистрофия с дефицитом интегрина 7 альфа
Это чрезвычайно редкая форма ВМД, описанная лишь у нескольких человек. Симптомы включают мышечную слабость и задержку в достижении моторных вех.
* Врожденная мышечная дистрофия с дефицитом интегрина 9 альфа
Эта чрезвычайно редкая форма ВМД была описана лишь у небольшого числа людей.У больных может наблюдаться гипотония, контрактуры, гиперсслабление суставов и сколиоз. При этой форме CMD интеллект не пострадал, и люди сохраняют способность ходить во взрослую жизнь. У некоторых больных может развиться снижение респираторной функции, но, как правило, поддержка аппарата искусственной вентиляции легких не требуется.
CMDS, ВЫЗВАННЫЕ ДЕФЕКТНЫМИ БЕЛКАМИ, НЕОБХОДИМЫМИ ДЛЯ СВЯЗИ МОЛЕКУЛ САХАРА С ДИСТРОГЛИКАНОМ, КОЛЛЕКТИВНО ИЗВЕСТНЫМ КАК ДИСТРОГЛИКАНОПАТИИ.
Несколько различных генов были связаны с дистрогликанопатиями, и исследователи определили, что эти отдельные гены потенциально могут быть связаны с более чем одним расстройством, описанным ниже.Хотя эти расстройства когда-то считались отдельными состояниями, эта подгруппа CMD теперь считается спектром заболеваний, которые варьируются от легких проявлений (фенотипов) до тяжелых. Мутации в некоторых генах, вызывающие эти дистрогликанопатии, также могут вызывать легкие формы мышечной дистрофии конечностей и поясов.
* Врожденная мышечная дистрофия типа 1C (MDC1C; ВМД с вторичной недостаточностью мерозина типа 2)
MDC1C — потенциально тяжелая форма ВМД, которая характеризуется пониженным мышечным тонусом (гипотонией) и мышечной слабостью при рождении.У заболевших младенцев также могут развиться затруднения дыхания и кормления. Проблемы с дыханием прогрессируют и часто вызывают дыхательную недостаточность (дыхательную недостаточность). Также случаются задержки в достижении моторных вех. Больные младенцы обычно могут сидеть без посторонней помощи, но лишь некоторые из них могут ходить без посторонней помощи.
Могут возникнуть дополнительные симптомы, включая чрезмерный рост (гипертрофию) мышц ног, аномально увеличенный язык (макроглоссия), слабость и истощение (атрофия) мышц рук и контрактуры, особенно ахиллова сухожилия, сгибателя бедра. , и пальцы.Слабость и истощение также могут влиять на мышцы лица и плеч. У некоторых людей в конечном итоге развивается заболевание сердечной мышцы, в частности дилатационная кардиомиопатия. Это состояние характеризуется аномальным увеличением или расширением (дилатацией) одной или нескольких камер сердца, что приводит к ослаблению насосной активности сердца, вызывая ограниченную способность циркулировать кровь к легким и остальным частям тела и приводя к накопление жидкости в сердце, легких и различных тканях организма (застойная сердечная недостаточность).
Большинство случаев MDC1C не связаны со структурными аномалиями мозга, и интеллект в норме. Однако в некоторых редких случаях у пораженных людей действительно есть такие аномалии и часто наблюдается легкая умственная отсталость. В некоторых случаях также сообщалось об изъятиях.
* Спектр заболевания мышцы-глаз-мозг (MEB)
Симптомы и тяжесть заболевания MEB могут различаться. У пораженных младенцев обычно резко снижается мышечный тонус (гипотония) и мышечная слабость при рождении.Слабость мышц часто поражает руки, ноги и туловище. Больные младенцы могут не набирать вес и не расти с ожидаемой скоростью (неспособность развиваться). Задержка развития и глазные (глазные) аномалии также являются частыми находками. При легких формах MEB у пораженных людей наблюдается задержка в достижении основных этапов развития, но в конечном итоге они могут ходить самостоятельно. В тяжелых случаях достигается несколько этапов развития моторики (например, отсутствие контроля над головой или способность держать голову вверх). MEB обычно прогрессирует, и некоторые люди теряют ранее приобретенные навыки, такие как самостоятельная ходьба.Снижение моторики также может произойти в результате увеличения спастичности.
Пораженные младенцы могут иметь большую голову с выступающим лбом и широким «мягким пятном» (родничок) и отличительными чертами лица, включая аномально маленькую челюсть (микрогнатия), недоразвитие средней части лица (гипоплазия средней части лица), короткий нос. , и аномально маленькая бороздка между верхней губой и кончиком носа (желобок). Глазные аномалии, связанные с заболеванием MEB, включают повышенное давление в глазах (инфантильная глаукома), помутнение хрусталика глаз (катаракта), быстрые непроизвольные движения глаз (нистагм), недоразвитие богатой нервом мембраны, выстилающей глаза (гипоплазия сетчатки) , и тяжелая близорукость (врожденная миопия).
Также может развиваться поражение центральной нервной системы, такое как усиление рефлексов или непроизвольные мышечные спазмы (спастичность), которые приводят к медленным, жестким движениям ног. Умственная отсталость и судороги также могут возникать у людей с заболеванием MEB. У всех людей с MEB есть небольшой ствол мозга и мозжечок, а у некоторых есть аномалии коры головного мозга, такие как пахигирия или лиссэнцефалия, структурный дефект мозга, при котором мозг гладкий, без нормальных складок.
* Врожденная мышечная дистрофия типа Фукуямы и синдром Уокера-Варбурга
Как и болезнь MEB, врожденная мышечная дистрофия Фукуямы (FCMD) и синдром Уокера-Варбурга (WSS) являются мультисистемными расстройствами, характеризующимися мышечной слабостью, структурными дефектами мозга и аномалиями глаз.
Младенцы с FCMD имеют общую мышечную слабость, снижение мышечного тонуса (гипотония), плохую сосательную способность и слабый плач. Контрактуры бедра, колена, лодыжек и локтей — обычное явление в течение первого года жизни. У людей с FCMD также есть глазные (глазные) аномалии, такие как косоглазие (косоглазие), катаракта, близорукость (миопия), аномальные движения глаз и, в тяжелых случаях, отслоение сетчатки и аномально маленькие глаза (микрофтальм). FCMD также характеризуется судорогами, умственной отсталостью и проблемами речи.В конце концов, у пораженных людей развиваются респираторные проблемы и заболевание сердечной мышцы (дилатационная кардиомиопатия).
Синдром Уокера-Варбурга характеризуется мышечной слабостью, лиссэнцефалией II типа и аномальным развитием богатой нервом мембраны в задней части глаза (дисплазия сетчатки). У пораженных людей также есть обструктивная гидроцефалия — состояние, при котором нарушение нормальной циркуляции спинномозговой жидкости приводит к давлению на мозг. Также может возникнуть затылочное энцефалоцеле.У пораженных младенцев также обычно наблюдается серьезная задержка роста; необычно маленькая голова; судороги; и дополнительные аномалии глаз, включая отслоение сетчатки, аномально маленькие глаза (микрофтальм) и помутнение хрусталика (катаракта) и роговицы.
(Для получения дополнительной информации см. Отдельные записи для FCMD и WSS в базе данных редких заболеваний NORD.)
* Врожденная мышечная дистрофия типа 1D (MDC1D)
По состоянию на 2012 год, MDC1D был зарегистрирован у нескольких человек из четырех разных семей. (родичи).У больных развилась тяжелая умственная отсталость, гипотония, задержка развития, легкие контрактуры, мышечная слабость и дегенерация (атрофия). Сообщалось также о структурных дефектах мозга.
CMDS, ВЫЗВАННЫЕ ДЕФЕКТАМИ ГЕНА СЕЛЕНОПРОТЕИН1 (SEPN1).
* Миопатия, связанная с SEPN1
Лица с этой формой CMD первоначально назывались мышечной дистрофией ригидного позвоночника (RSMD1) или CMD с ранней ригидностью позвоночника.Однако наблюдались люди с ригидным позвоночником и мутацией гена селенопротеина N1 (SEPN1), которые соответствовали критериям для четырех различных мышечных состояний, включая мышечную дистрофию ригидного позвоночника (синдром ригидного позвоночника), врожденную диспропорцию типа волокон, миопатию, связанную с десмином. , и миопатия с множественными ядрами. В настоящее время исследователи вместе называют этих людей страдающими миопатией, связанной с SEPN1.
Отличительной чертой этой формы ВМД является ранняя ригидность позвоночника, которая часто развивается в течение первого года жизни, и медленно прогрессирующее искривление позвоночника (сколиоз), которое развивается с 3 до 12 лет.Дополнительные симптомы включают прогрессирующую мышечную слабость, снижение мышечного тонуса (гипотония), затрудненное дыхание (дыхание) в раннем возрасте, контрактуры ахилловых сухожилий и слабость мышц шеи, приводящую к плохому контролю над головой. Мышечная слабость незначительна по сравнению с другими формами CMD.
Проблемы с дыханием прогрессируют, и к подростковому возрасту могут быть затронуты мышцы легких. В конце концов, больные люди испытывают неадекватное дыхание ночью (ночная гиповентиляция) и, возможно, дыхательная недостаточность.Начало затрудненного дыхания сильно варьируется, обычно происходит в подростковом возрасте, но также встречается в младенчестве и уже в четвертом десятилетии жизни.
Люди с миопатией, связанной с SEPN1, обычно не задерживаются в достижении двигательных вех. Большинство людей могут ходить самостоятельно, хотя из-за прогрессирующей ригидности позвоночника и сколиоза они могут испытывать трудности при ходьбе в более позднем возрасте.
CMDS, ОБРАЗУЮЩИЕСЯ ИЗ-ЗА ДЕФЕКТНЫХ БЕЛКОВ ЯДЕРНОЙ ОБОЛОЧКИ.
* CMD, связанная с LMNA
Эта форма CMD часто связана с тяжелым предлежанием, которое происходит в течение первых 6 месяцев жизни. У заболевших младенцев может наблюдаться гипотония, отсутствие опоры для головы / туловища (синдром опущенной головы), ригидность позвоночника и общее истощение мышц и слабость, которая проявляется в области шеи, рук и ног. Также могут развиваться сколиоз и контрактуры нижних конечностей. По мере прогрессирования мышечной слабости может развиться заболевание легких, приводящее к затруднению дыхания.Людям с тяжелыми заболеваниями может потребоваться помощь при дыхании (искусственная вентиляция легких).
Эта форма CMD вызывается мутациями гена LMNA. Также было показано, что мутации этого гена вызывают широкий спектр других расстройств (аллельные расстройства), включая семейную частичную липодистрофию 2 типа (липодистрофию Даннигана), мандибулоакральную дисплазию, пару форм мышечной дистрофии Эмери-Дрейфуса, форму поясничной конечности мышечная дистрофия, форма наследственной спастической параплегии, форма болезни Шарко-Мари-Тута, форма дилатационной кардиомиопатии, синдром Малуфа и некоторые случаи синдрома прогерии Хатчинсона-Гилфорда.О лицах, симптомы которых частично совпадают с этими расстройствами, сообщалось в медицинской литературе. (Для получения дополнительной информации об этих расстройствах выберите конкретное заболевание в качестве поискового запроса в базе данных редких заболеваний.)
* CMD, связанный с SYNE1
Об этой чрезвычайно редкой форме CMD сообщалось только у двух братьев и сестер, у которых были сведены большие пальцы. , умственная отсталость, катаракта и офтальмоплегия. У этих братьев и сестер также было недоразвитие мозжечка (гипоплазия мозжечка), что могло вызвать проблемы с равновесием и координацией.Эта форма CMD вызвана мутациями гена SYNE1.
ДОПОЛНИТЕЛЬНЫЕ РЕДКИЕ ФОРМЫ CMDS С ИЗВЕСТНЫМ ГЕНЕТИЧЕСКИМ ДЕФЕКТОМ ИЛИ БЕЗ ИЗВЕСТНОГО ДЕФЕКТА.
Существует несколько дополнительных форм CMD, которые не могут быть связаны с конкретным генетическим дефектом. Эти формы включают случаи WSS, не связанные с мутациями в каком-либо известном гене, связанном с ферментами гликозилтрансферазы, и случаи синдрома ригидного позвоночника, не связанные с мутациями в гене SEPN1. Сообщалось также о случаях, связанных с поражением мозжечка.
* Врожденная мышечная дистрофия типа 1B (MDC1B; CMD с вторичной недостаточностью мерозина 1 типа)
MDC1B характеризуется пониженным мышечным тонусом (гипотония), мышечной слабостью мышц ближе к центру тела (проксимальные мышцы), генерализованной разрастание некоторых мышц (гипертрофия), ригидность позвоночника и контрактуры, особенно ахиллова сухожилия. Эта форма CMD была связана с еще не идентифицированным геном на хромосоме 1 и классифицируется как подтип дистрогликанопатий.
* Заболевание мышц, связанное с CHKB
Об этой чрезвычайно редкой форме CMD сообщили менее чем у двадцати человек. У больных наблюдается умственная отсталость, гипотония и общая мышечная слабость. Пострадавшие могли ходить без посторонней помощи. Дополнительные симптомы, которые могут возникнуть, включают дилатационную кардиомиопатию и структурные аномалии митохондрий — частей клетки, выделяющих энергию. Это расстройство также известно как CMD мегакониального типа или CMD с митохондриальными структурными аномалиями (CMDmt).
Врожденная мышечная дистрофия: история болезни, патофизиология, эпидемиология
Фукуяма Й., Квазура М., Харуна Х. Своеобразная форма врожденной мышечной дистрофии. Педиатрический университет Токио . 1960. 4: 5-8:
Sparks SE, Escolar DM. Врожденные мышечные дистрофии. Handb Clin Neurol . 2011. 101: 47-79. [Медлайн].
Mercuri E, Muntoni F. Постоянно расширяющийся спектр врожденных мышечных дистрофий. Энн Нейрол . 2012 июл.72 (1): 9-17. [Медлайн].
Грациано А., Бьянко Ф, Д’Амико А., Морони I, Мессина С. и др. Распространенность врожденной мышечной дистрофии в Италии: популяционное исследование. Неврология . 2015 3 марта. 84 (9): 904-11. [Медлайн].
Clement EM, Feng L, Mein R, Sewry CA, Robb SA, Manzur AY, et al. Относительная частота подтипов врожденных мышечных дистрофий: анализ диагностической службы Великобритании 2001-2008 гг. Нервно-мышечное расстройство . 2012 июн. 22 (6): 522-7. [Медлайн].
Peat RA, Smith JM, Compton AG, Baker NL, Pace RA, Burkin DJ и др. Диагностика и этиология врожденной мышечной дистрофии. Неврология . 29 июля 2008 г. 71 (5): 312-21. [Медлайн].
Окада М., Кавахара Г., Ногучи С., Суги К., Мураяма К., Нонака И. и др. Первичный дефицит коллагена VI — вторая по распространенности врожденная мышечная дистрофия в Японии. Неврология .2007 сентября 4. 69 (10): 1035-42. [Медлайн].
Geranmayeh F, Clement E, Feng LH, et al. Корреляция генотип-фенотип в большой популяции пациентов с мышечной дистрофией с мутациями LAMA2. Нервно-мышечное расстройство . 2010 Апрель 20 (4): 241-50. [Медлайн].
Bonnemann CG. Миопатии, связанные с коллагеном VI: мышца встречает свою матрицу. Нат Рев Нейрол . 2011, 21 июня. 7 (7): 379-90. [Медлайн].
Scacheri PC, Gillanders EM, Subramony SH, et al.Новые мутации в генах коллагена VI: расширение фенотипа миопатии Бетлема. Неврология . 2002 26 февраля. 58 (4): 593-602. [Медлайн].
Merlini L, Martoni E, Grumati P, et al. Аутосомно-рецессивная миосклерозная миопатия — это нарушение коллагена VI. Неврология . 14 октября 2008 г. 71 (16): 1245-53. [Медлайн].
Накашима Х., Кибе Т., Йокочи К. Врожденная мышечная дистрофия, вызванная дефицитом интегрина альфа7 ». Дев Мед Детский Нейрол .2009 Март 51 (3): 245. [Медлайн].
Форрест К., Меллерио Дж. Э., Робб С. и др. Врожденная мышечная дистрофия, миастенические симптомы и простой буллезный эпидермолиз (EBS), связанные с мутациями в гене PLEC1, кодирующем плектин. Нервно-мышечное расстройство . 2010 20 ноября (11): 709-11. [Медлайн].
Yiu EM, Klausegger A, Waddell LB, et al. Буллезный эпидермолиз с поздней мышечной дистрофией и недостаточностью плектина. Мышечный нерв .2011 Июль 44 (1): 135-41. [Медлайн].
Gundesli H, Talim B, Korkusuz P и др. Мутация в экзоне 1f PLEC, приводящая к разрушению изоформы 1f плектина, вызывает аутосомно-рецессивную мышечную дистрофию конечностей и поясов. Ам Дж. Хам Генет . 2010 декабрь 10. 87 (6): 834-41. [Медлайн].
Schara U, Kress W, Bonnemann CG, et al. Фенотип и отдаленное наблюдение у 11 пациентов с ювенильной селенопротеидной N1-зависимой миопатией. Eur J Paediatr Neurol .2008 май. 12 (3): 224-30. [Медлайн].
Schoenmakers E, Agostini M, Mitchell C, et al. Мутации в гене белка 2, связывающего последовательность вставки селеноцистеина, приводят к мультисистемному нарушению селенопротеиновой недостаточности у людей. Дж. Клин Инвест . 2010 декабрь 1. 120 (12): 4220-35. [Медлайн]. [Полный текст].
Mitsuhashi S, Ohkuma A, Talim B и др. Врожденная мышечная дистрофия со структурными аномалиями митохондрий, вызванная нарушением биосинтеза фосфатидилхолина de novo. Ам Дж. Хам Генет . 2011, 10 июня. 88 (6): 845-51. [Медлайн]. [Полный текст].
Акияма Т., Оцука Ю., Таката Т., Хаттори Дж., Кавакита Ю., Сайто К. Самый легкий известный случай врожденной мышечной дистрофии типа Фукуяма. Brain Dev . 2006 Сентябрь 28 (8): 537-40. [Медлайн].
Годфри С., Эсколар Д., Брокингтон М. и др. Мутации гена фукутина при стероид-зависимой мышечной дистрофии пояса конечностей. Энн Нейрол . 2006 ноя.60 (5): 603-10. [Медлайн].
Мураками Т., Хаяси Ю.К., Ногучи С., Огава М., Нонака И., Танабэ Ю. Мутации гена Фукутина вызывают дилатационную кардиомиопатию с минимальной мышечной слабостью. Энн Нейрол . 2006 ноябрь 60 (5): 597-602. [Медлайн].
Годфри С., Клемент Е., Мейн Р., Брокингтон М., Смит Дж., Талим Б. Уточнение корреляций фенотипа генотипа при мышечных дистрофиях с дефектным гликозилированием дистрогликана. Мозг . 2007 окт.130 (Pt 10): 2725-35. [Медлайн].
Messina S, Tortorella G, Concolino D и др. Врожденная мышечная дистрофия с дефектным альфа-дистрогликаном, гипоплазия мозжечка и эпилепсия. Неврология . 2009 10 ноября. 73 (19): 1599-601. [Медлайн].
Mercuri E, Messina S, Bruno C и др. Врожденные мышечные дистрофии с дефектным гликозилированием дистрогликана: популяционное исследование. Неврология . 2009 26 мая. 72 (21): 1802-9.[Медлайн].
Balci B, Uyanik G, Dincer P, Gross C, Willer T., Talim B. Аутосомно-рецессивная мышечная дистрофия пояса конечностей (LGMD2) с легкой умственной отсталостью является аллельной к синдрому Уокера-Варбурга (WWS), вызванному мутацией в Ген POMT1. Нервно-мышечное расстройство . 2005 апр. 15 (4): 271-5. [Медлайн].
Бианчери Р., Фалас А., Тесса А. и др. Мутация гена POMT2 при мышечной дистрофии конечностей с воспалительными изменениями. Биохимия Биофиз Рес Коммуна . 2007 30 ноября. 363 (4): 1033-7. [Медлайн].
Clarke NF, Maugenre S, Vandebrouck A, et al. Врожденная мышечная дистрофия 1D (MDC1D) из-за большой внутригенной вставки / делеции с участием интрона 10 гена LARGE. евро J Hum Genet . 2011 апр.19 (4): 452-7. [Медлайн].
van Reeuwijk J, Grewal PK, Salih MA, et al. Внутригенная делеция в гене LARGE вызывает синдром Уокера-Варбурга. Хум Генет . 2007 июль 121 (6): 685-90. [Медлайн].
Zou Y, Zhang RZ, Sabatelli P, Chu ML, Bonnemann CG. Мышечные интерстициальные фибробласты являются основным источником синтеза коллагена VI в скелетных мышцах: последствия для врожденных мышечных дистрофий типов Ульриха и Бетлема. J Neuropathol Exp Neurol . 2008 Февраль 67 (2): 144-54. [Медлайн].
Бринас Л., Ричард П., Кихано-Рой С. и др. Миопатии с ранним началом коллагена VI: генетические и клинические корреляции. Энн Нейрол . 2010 Октябрь 68 (4): 511-20. [Медлайн].
Лампе А.К., Зоу Й., Судано Д. и др. Мутации с пропуском экзона в коллагене VI являются распространенными и позволяют прогнозировать тяжесть и наследственность. Хум Мутат . 29 июня 2008 г. (6): 809-22. [Медлайн].
Кон РД. Дистрогликан: важный игрок в скелетных мышцах и не только. Нервно-мышечное расстройство . 2005 15 марта (3): 207-17. [Медлайн].
Chavanas S, Pulkkinen L, Gache Y, et al.Гомозиготная нонсенс-мутация в гене PLEC1 у пациентов с простым буллезным эпидермолизом с мышечной дистрофией. Дж. Клин Инвест . 1996 15 ноября. 98 (10): 2196-200. [Медлайн]. [Полный текст].
Pulkkinen L, Smith FJ, Shimizu H, et al. Гомозиготные делеционные мутации в гене плектина (PLEC1) у пациентов с простым буллезным эпидермолизом, ассоциированным с поздней мышечной дистрофией. Хум Мол Генет . 1996 5 октября (10): 1539-46. [Медлайн].
Резничек Г.А., Уолко Г., Виче Г.Дефекты гена плектина приводят к различным формам простого буллезного эпидермолиза. Дерматол Клин . 2010, 28 января (1): 33-41. [Медлайн].
Хименес-Маллебрера С., Торелли С., Фенг Л. и др. Сравнительное исследование гликозилирования альфа-дистрогликана при дистрогликанопатиях предполагает, что гипогликозилирование альфа-дистрогликана не всегда коррелирует с клинической тяжестью. Brain Pathol . 2009 октября 19 (4): 596-611. [Медлайн]. [Полный текст].
Clement EM, Godfrey C, Tan J, et al.Легкие мутации POMGnT1 лежат в основе нового варианта мышечной дистрофии конечностей и поясов. Arch Neurol . 2008, январь, 65 (1): 137-41. [Медлайн].
Keramaris-Vrantsis E, Lu PJ, Doran T, Zillmer A, Ashar J, Esapa CT. Связанный с фукутином белок локализуется в аппарате Гольджи, и мутации приводят к неправильной локализации в мышцах in vivo. Мышечный нерв . 2007 Октябрь, 36 (4): 455-65. [Медлайн].
Roscioli T, Kamsteeg EJ, Buysse K, et al.Мутации в ISPD вызывают синдром Уокера-Варбурга и дефектное гликозилирование α-дистрогликана. Нат Генет . 2012 май. 44 (5): 581-5. [Медлайн]. [Полный текст].
Willer T, Lee H, Lommel M, et al. Мутации с потерей функции ISPD нарушают O-маннозилирование дистрогликана и вызывают синдром Уокера-Варбурга. Нат Генет . 2012 май. 44 (5): 575-80. [Медлайн]. [Полный текст].
Vuillaumier-Barrot S, Bouchet-Séraphin C, Chelbi M, et al.Выявление мутаций TMEM5 и ISPD как причины тяжелой лиссэнцефалии булыжника. Ам Дж. Хам Генет . 2012 декабрь 7. 91 (6): 1135-43. [Медлайн]. [Полный текст].
Cirak S, Foley AR, Herrmann R, et al. Мутации гена ISPD — частая причина врожденных и конечностно-поясных мышечных дистрофий. Мозг . 2013, январь, 136: 269–81. [Медлайн]. [Полный текст].
Manzini MC, Tambunan DE, Hill RS и др. Секвенирование экзома и функциональная проверка у рыбок данио идентифицируют мутации GTDC2 как причину синдрома Уокера-Варбурга. Ам Дж. Хам Генет . 2012 сентябрь 7. 91 (3): 541-7. [Медлайн]. [Полный текст].
Jae LT, Raaben M, Riemersma M, et al. Расшифровка гликозилома при дистрогликанопатиях с использованием гаплоидных экранов на проникновение вируса Ласса. Наука . 2013 26 апреля. 340 (6131): 479-83. [Медлайн].
Стивенс Э., Карсс К.Дж., Цирак С. и др. Мутации в B3GALNT2 вызывают врожденную мышечную дистрофию и гипогликозилирование α-дистрогликана. Ам Дж. Хам Генет .2013 7 марта. 92 (3): 354-65. [Медлайн]. [Полный текст].
Jae LT, Raaben M, Riemersma M, et al. Расшифровка гликозилома при дистрогликанопатиях с использованием гаплоидных экранов на проникновение вируса Ласса. Наука . 2013 26 апреля. 340 (6131): 479-83. [Медлайн].
Buysse K, Riemersma M, Powell G, et al. Миссенс-мутации в β-1,3-N-ацетилглюкозаминилтрансферазе 1 (B3GNT1) вызывают синдром Уокера-Варбурга. Хум Мол Генет . 2013 1 мая.22 (9): 1746-54. [Медлайн]. [Полный текст].
Карсс К.Дж., Стивенс Э., Фоли А.Р. и др. Мутации GDP-маннозопирофосфорилазы B Причина врожденных и конечностей-поясных мышечных дистрофий, связанных с гипогликозилированием a-дистрогликана. Американский журнал генетики человека . 2013/07. 93: 1-13. [Полный текст].
Кранц С., Юнгеблут С., Денеке Дж. И др. Нарушение биосинтеза долихолфосфата вызывает новое наследственное заболевание со смертью в раннем младенчестве. Ам Дж. Хам Генет . 2007 Март 80 (3): 433-40. [Медлайн]. [Полный текст].
Капуста Л., Цукер Н., Френкель Г. и др. От дискретной дилатационной кардиомиопатии до успешной трансплантации сердца при врожденных нарушениях гликозилирования из-за дефицита долихолкиназы (DK1-CDG). Heart Fail Ред. . 2013 18 марта (2): 187-96. [Медлайн]. [Полный текст].
Barone R, Aiello C, Race V и др. DPM2-CDG: синдром мышечной дистрофии-дистрогликанопатии с тяжелой эпилепсией. Энн Нейрол . 2012 Октябрь 72 (4): 550-8. [Медлайн].
Lefeber DJ, Schonberger J, Morava E, et al. Дефицит субъединицы DPM3 синтазы Dol-P-Man связывает врожденные нарушения гликозилирования с дистрогликанопатиями. Ам Дж. Хам Генет . 2009 Июль 85 (1): 76-86. [Медлайн]. [Полный текст].
Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R, et al. Корреляция генотип-фенотип в большой популяции пациентов с мышечной дистрофией с мутациями LAMA2. Нервно-мышечное расстройство . 2010 апр.20 (4): 241-50. [Медлайн].
Pane M, Messina S, Vasco G, Foley AR, Morandi L, Pegoraro E, et al. Дыхательная и сердечная функция при врожденных мышечных дистрофиях с дефицитом альфа-дистрогликана. Нервно-мышечное расстройство . 2012 22 августа (8): 685-9. [Медлайн].
Scoto M, Cirak S, Mein R, Feng L, Manzur AY, Robb S и др. Миопатии, связанные с SEPN1: клиническое течение у большой группы пациентов. Неврология . 2011, 14 июня. 76 (24): 2073-8. [Медлайн].
Mercuri E, Clements E, Offiah A, et al. Магнитно-резонансная томография мышц при мышечных дистрофиях с ригидностью позвоночника. Энн Нейрол . 2010 Февраль 67 (2): 201-8. [Медлайн].
Канг П.Б., Моррисон Л., Ианнакконе С.Т., Грэм Р.Дж., Беннеманн К.Г. и др. Резюме рекомендаций, основанных на фактах: оценка, диагностика и лечение врожденной мышечной дистрофии: отчет Подкомитета по разработке рекомендаций Американской академии неврологии и Группы по анализу практических вопросов Американской ассоциации нервно-мышечной и электродиагностической медицины. Неврология . 2015 31 марта. 84 (13): 1369-78. [Медлайн].
Бейкер Н.Л., Моргелин М., Торф Р. и др. Доминирующие мутации коллагена VI являются частой причиной врожденной мышечной дистрофии Ульриха. Хум Мол Генет . 2005 15 января. 14 (2): 279-93. [Медлайн].
Barresi R, Michele DE, Kanagawa M. LARGE может функционально обходить дефекты гликозилирования альфа-дистрогликана при различных врожденных мышечных дистрофиях. Нат Мед .2004 июл.10 (7): 696-703. [Медлайн].
Рейка FE. Три случая миопатии инфантильного типа. Мозг . 1903. 26: 147-8.
Бельтран-Валеро де Бернабе Д., Карриер С., Штайнбрехер А. и др. Мутации в гене POMT1 O-маннозилтрансферазы вызывают тяжелое нарушение миграции нейронов, синдром Уокера-Варбурга. Ам Дж. Хам Генет . 2002 ноябрь 71 (5): 1033-43. [Медлайн]. [Полный текст].
Брокингтон М., Торелли С., Прандини П. и др.Локализация и функциональный анализ БОЛЬШОГО семейства гликозилтрансфераз: значение для мышечной дистрофии. Хум Мол Генет . 2005 г., 1. 14 (5): 657-65. [Медлайн].
Камачо Ванегас О., Бертини Э., Чжан Р.З. и др. Склероатоническая мышечная дистрофия Ульриха вызывается рецессивными мутациями коллагена VI типа. Proc Natl Acad Sci U S A . 2001, 19 июня. 98 (13): 7516-21. [Медлайн].
Центр человеческой и клинической генетики.Медицинский центр Лейденского университета. Лейденская мышечная дистрофия. Страницы: мышечные дистрофии Дюшенна и Дюшенна. Доступно на: http://www.dmd.nl. [Полный текст].
Currier SC, Lee CK, Chang BS и др. Мутации в POMT1 обнаруживаются у меньшинства пациентов с синдромом Уокера-Варбурга. Ам Дж. Мед Генет А . 2005 15 февраля. 133A (1): 53-7. [Медлайн].
Д’Амико А., Халилоглу Г., Ричард П. и др. Два пациента с синдромом опущенной головы из-за мутаций в генах LMNA или SEPN1. Нервно-мышечное расстройство . 2005 15 августа (8): 521-4. [Медлайн].
Д’Амико А., Тесса А., Бруно С. и др. Расширение клинического спектра фенотипа POMT1. Неврология . 2006 г. 23 мая. 66 (10): 1564-7; обсуждение 1461. [Medline].
Di Blasi C, Piga D, Brioschi P, et al. Анализ гена LAMA2 при врожденной мышечной дистрофии: новые мутации, пренатальная диагностика и эффект основателя. Arch Neurol . 2005 Октябрь 62 (10): 1582-6.[Медлайн].
Дубовиц В. Синдром жесткого позвоночника: мышечный синдром в поисках имени. Proc R Soc Med . 1973 Mar.66 (3): 219-20. [Медлайн].
Esapa CT, McIlhinney RA, Blake DJ. Связанные с фукутином белковые мутации, вызывающие врожденную мышечную дистрофию, приводят к задержке мутантного белка в ER в культивируемых клетках. Хум Мол Генет . 2005 15 января. 14 (2): 295-305. [Медлайн].
Джусти Б., Лукарини Л., Пьетрони В. и др.Доминантные и рецессивные мутации COL6A1 при склероатонической мышечной дистрофии Ульриха. Энн Нейрол . 2005 Сентябрь 58 (3): 400-10. [Медлайн].
Grewal PK, Holzfeind PJ, Bittner RE, Hewitt J.E. Мутантная гликозилтрансфераза и измененное гликозилирование альфа-дистрогликана у мышей с миодистрофией. Нат Генет . 2001 июн. 28 (2): 151-4. [Медлайн].
Grewal PK, McLaughlan JM, Moore CJ, Browning CA, Hewitt JE. Характеристика БОЛЬШОГО семейства предполагаемых гликозилтрансфераз, связанных с дистрогликанопатиями. Гликобиология . 2005 октября, 15 (10): 912-23. [Медлайн].
Guglieri M, Magri F, Comi GP. Молекулярный этиопатогенез мышечных и врожденных мышечных дистрофий пояса конечностей: границы и соприкосновения. Клин Чим Акта . 2005 ноябрь 361 (1-2): 54-79. [Медлайн].
Haliloglu G, Gross C, Senbil N, Talim B, Hehr U, Uyanik G. Клинический спектр заболеваний мышц, глаз и головного мозга: от типичных проявлений до тяжелых аутистических особенностей. Acta Myol . 2004 г., 23 (3): 137-9. [Медлайн].
Hayashi YK, Chou FL, Engvall E, et al. Мутации в гене интегрина альфа7 вызывают врожденную миопатию. Нат Генет . 1998 Май. 19 (1): 94-7. [Медлайн].
Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Мутации в гене 2-цепи ламинина альфа (LAMA2) вызывают врожденную мышечную дистрофию с дефицитом мерозина. Нат Генет . 1995 г., 11 (2): 216-8. [Медлайн].
Henion TR, Qu Q, Smith FI. Экспрессия дистрогликана, фукутина и POMGnT1 во время развития мозжечка у мышей. Brain Res Mol Brain Res . 2003 10 апреля. 112 (1-2): 177-81. [Медлайн].
Говард РА. Случай врожденного порока мышечной системы (dystrophia muscularis congenita) и его связь с врожденной эквиноварусной косолапостью. Proc R Soc Med . 1908. 1: 157-66.
Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F.Врожденная мышечная дистрофия: молекулярные и клеточные аспекты. Cell Mol Life Sci . 2005 апр. 62 (7-8): 809-23. [Медлайн].
Кобаяши К., Накахори Ю., Мияке М. и др. Древнее ретротранспозирование вызывает врожденную мышечную дистрофию типа Фукуямы. Природа . 1998, 23 июля. 394 (6691): 388-92. [Медлайн].
Лампе АК, Бушби КМ. Связанные с коллагеном VI мышечные расстройства. Дж. Мед Генет . 2005 сентябрь 42 (9): 673-85.[Медлайн]. [Полный текст].
Лю Дж., Болл С.Л., Ян Й. и др. Генетическая модель заболевания мышцы-глаза-мозг у мышей, лишенных протеин-O-маннозы 1,2-N-ацетилглюкозаминилтрансферазы (POMGnT1). Механический разработчик . 2006 Март 123 (3): 228-40. [Медлайн].
Longman C, Brockington M, Torelli S и др. Мутации в гене LARGE человека вызывают MDC1D, новую форму врожденной мышечной дистрофии с тяжелой умственной отсталостью и аномальным гликозилированием альфа-дистрогликана. Хум Мол Генет . 1 ноября 2003 г. 12 (21): 2853-61. [Медлайн].
Мартин П.Т. Дистрогликанопатии: новые нарушения О-связанного гликозилирования. Семин Педиатр Нейрол . 2005 Сентябрь 12 (3): 152-8. [Медлайн]. [Полный текст].
Мацумото Х., Хаяси Ю.К., Ким Д.С. и др. Врожденная мышечная дистрофия с дефектами гликозилирования альфа-дистрогликана в Японии. Нервно-мышечное расстройство . 2005 г., май. 15 (5): 342-8. [Медлайн].
Mayer U, Saher G, Fassler R, et al. Отсутствие интегрина альфа 7 вызывает новую форму мышечной дистрофии. Нат Генет . 1997 17 ноября (3): 318-23. [Медлайн].
Mercuri E, Topaloglu H, Brockington M и др. Спектр изменений мозга у пациентов с врожденной мышечной дистрофией и мутациями гена FKRP. Arch Neurol . 2006 Февраль 63 (2): 251-7. [Медлайн].
Moghadaszadeh B, Petit N, Jaillard C, et al.Мутации в SEPN1 вызывают врожденную мышечную дистрофию с ригидностью позвоночника и рестриктивным респираторным синдромом. Нат Генет . 2001 Сентябрь 29 (1): 17-8. [Медлайн].
Мур С.А., Сайто Ф., Чен Дж. И др. Удаление дистрогликана головного мозга повторяет аспекты врожденной мышечной дистрофии. Природа . 2002 25 июля. 418 (6896): 422-5. [Медлайн].
Пестронк А. Веб-страница Центра нервно-мышечных заболеваний Вашингтонского университета.1999. Доступно по адресу: http://www.neuro.wustl.edu/neuromuscular. [Полный текст].
Raitta C, Lamminen M, Santavuori P, Leisti J. Офтальмологические находки при новом синдроме с вовлечением мышц, глаз и мозга. Acta Ophthalmol (Копен) . 1978 июн 56 (3): 465-72. [Медлайн].
Rederstorff M, Krol A, Lescure A. Понимание важности селена и селенопротеидов для работы мышц. Cell Mol Life Sci . 2006 Янв.63 (1): 52-9. [Медлайн]. [Полный текст].
Сантавуори П., Лейсти Дж., Круус С. Заболевание мышц, глаз и головного мозга: новый синдром. Neuropadiatrie . 1977. 8 (доп.): 550.
Танигучи К., Кобаяши К., Сайто К. и др. Распространение по всему миру и более широкий клинический спектр заболеваний мышц глаз и головного мозга. Хум Мол Генет . 1 марта 2003 г. 12 (5): 527-34. [Медлайн].
Том FM, Евангелиста Т., Леклерк А. и др. Врожденная мышечная дистрофия с дефицитом мерозина. C R Acad Sci III . 1994 Апрель, 317 (4): 351-7. [Медлайн].
Tsao CY, Mendell JR. Детские мышечные дистрофии: наведение порядка из хаоса. Семин Нейрол . 1999. 19 (1): 9-23. [Медлайн].
Вайнзоф М., Ричард П., Херрманн Р. и др. Пренатальная диагностика врожденной мышечной дистрофии с недостаточностью цепи ламинина альфа2 (мерозин): коллективный опыт пяти международных центров. Нервно-мышечное расстройство .2005 15 октября (9-10): 588-94. [Медлайн].
van Reeuwijk J, Janssen M, van den Elzen C, et al. Мутации POMT2 вызывают гипогликозилирование альфа-дистрогликана и синдром Уокера-Варбурга. Дж. Мед Генет . 2005 декабрь 42 (12): 907-12. [Медлайн]. [Полный текст].
van Reeuwijk J, Maugenre S, van den Elzen C, et al. Расширяющийся фенотип мутаций POMT1: от синдрома Уокера-Варбурга до врожденной мышечной дистрофии, микроцефалии и умственной отсталости. Хум Мутат . 2006 май. 27 (5): 453-9. [Медлайн].
Войт Т., Том Ф.С. Врожденные мышечные дистрофии. В: Engel AG, Franzini-Armstrong C, eds. Миология. Нью-Йорк: МакГроу-Хилл . 2004: 1203-38:
Уокер AE. Лиссэнцефалия. Arch Neurol Psychiat . 1942. 48: 13-29.
Варбург М. Неоднородность врожденного неприсоединения сетчатки, серповидные складки и дисплазия сетчатки.Руководство по генетическому консультированию. Наследие . 1976. 26 (2): 137-48. [Медлайн].
Willer T, Prados B, Falcon-Perez JM, et al. Целенаправленное нарушение гена Pomt1 синдрома Уокера-Варбурга у мышей приводит к эмбриональной летальности. Proc Natl Acad Sci U S A . 2004, 28 сентября. 101 (39): 14126-31. [Медлайн]. [Полный текст].
Йошида А., Кобаяши К., Манья Х. и др. Мышечная дистрофия и нарушение миграции нейронов, вызванные мутациями гликозилтрансферазы, POMGnT1. Dev Cell . 2001 ноября 1 (5): 717-24. [Медлайн].
Врожденная мышечная дистрофия (ВМД) | Мышечная дистрофия UK
Врожденная мышечная дистрофия (ВМД) — это термин, используемый для группы генетических состояний истощения мышц, симптомы которых проявляются в раннем возрасте (врожденное означает «с рождения»).
Они вызывают со временем ослабление и истощение мышц, что приводит к увеличению инвалидности. Они также могут вызвать трудности в обучении.
Считается, что из 10 000 жителей Великобритании, страдающих мышечной дистрофией, около 400 имеют ВМД.Двумя наиболее распространенными формами этого состояния являются врожденная мышечная дистрофия Ульриха, которая поражает около 50 процентов людей с ВМД, и врожденная мышечная дистрофия с дефицитом мерозина, которая поражает 25 процентов.
CMD вызывается генетическими мутациями, которые приводят к нехватке различных белков, жизненно важных для здоровой структуры или функции мышц. Мутации в разных генах вызывают разные типы CMD.
Тяжесть CMD сильно различается между разными типами и людьми.Некоторые затронутые дети смогут ходить, хотя они могут ходить позже, чем другие дети. Некоторые утратят эту способность по мере того, как они станут тяжелее и нагрузка на их мышцы увеличится.
Некоторые люди могут испытывать проблемы с дыханием, вызванные ослаблением грудных мышц, что может быть опасным для жизни. Поэтому жизненно важно, чтобы они получали правильную специализированную помощь.
В настоящее время не существует лечения для устранения основной генетической причины CMD. Muscular Dystrophy UK в настоящее время финансирует исследования, направленные на выяснение первопричины генетической причины CMD.Это поможет поставить точный генетический диагноз и спрогнозировать, насколько быстро симптомы будут прогрессировать, что позволит семьям сделать осознанный выбор на будущее.
Узнайте больше о миопатии Бетлема , врожденной мышечной дистрофии , SEPN1-связанной миопатии .
Посмотрите наш информационный видеоролик о том, на какое медицинское обслуживание и поддержку вы имеете право:
Врожденная мышечная дистрофия Фукуямы: MedlinePlus Genetics
Врожденная мышечная дистрофия Фукуямы — это наследственное заболевание, которое преимущественно поражает мышцы, мозг и глаза.Врожденные мышечные дистрофии — это группа генетических состояний, которые вызывают мышечную слабость и истощение (атрофию), которое начинается в очень раннем возрасте.
Врожденная мышечная дистрофия Фукуямы поражает скелетные мышцы — мышцы, которые тело использует для движения. Первые признаки расстройства появляются в раннем младенчестве и включают слабый плач, плохое питание и слабый мышечный тонус (гипотония). Слабость лицевых мышц часто приводит к характерному внешнему виду лица, включая опущенные веки (птоз) и открытый рот.В детстве мышечная слабость и деформации суставов (контрактуры) ограничивают движения и мешают развитию двигательных навыков, таких как сидение, стояние и ходьба.
Врожденная мышечная дистрофия Фукуямы также ухудшает развитие мозга. Люди с этим заболеванием имеют аномалию мозга, называемую лиссэнцефалией булыжника, при которой поверхность мозга приобретает неровный, неровный вид (как у булыжника). Эти изменения в структуре мозга приводят к значительной задержке развития речи и моторики, а также к умственной отсталости от умеренной до тяжелой.Социальные навыки нарушены в меньшей степени. Большинство детей с врожденной мышечной дистрофией Фукуямы никогда не могут стоять или ходить, хотя некоторые могут сидеть без поддержки и скользить по полу в сидячем положении. Более половины всех пострадавших детей также испытывают судороги.
Другие признаки и симптомы врожденной мышечной дистрофии Фукуямы включают нарушение зрения, другие глазные аномалии и медленно прогрессирующие проблемы с сердцем после 10 лет. По мере прогрессирования болезни у больных могут развиваться трудности с глотанием, что может привести к бактериальной инфекции легких, называемой аспирационной пневмонией .Из-за серьезных медицинских проблем, связанных с врожденной мышечной дистрофией Фукуямы, большинство людей с этим заболеванием доживают до позднего детства или подросткового возраста.
Врожденные мышечные дистрофии — TREAT-NMD
ВМД или врожденная мышечная дистрофия на самом деле представляет собой не одно отдельное заболевание, а ряд различных наследственных нервно-мышечных состояний, которые сгруппированы под ярлыком «врожденные», потому что они обычно проявляются симптомами при рождении или от очень ранний возраст. Подсчитано, что 1 ребенок из 20 000–50 000 рождается с врожденной мышечной дистрофией.Различные типы CMD вызываются мутациями в разных генах и могут иметь разные симптомы.
Все врожденные мышечные дистрофии поражают мышцы. Некоторые также влияют на мозг. Это означает, что у некоторых детей мышечная слабость, охватывающая все мышцы, но при этом нормальный интеллект, в то время как у других может быть мышечная слабость и трудности в обучении. В некоторых случаях поражение головного мозга также может вызывать судороги. Проблемы с обучением могут быть легкими, средними или серьезными.
Младенцы с врожденной мышечной дистрофией часто выглядят «вялыми» (врачи также могут описать это как низкий мышечный тонус или гипотонию).Также часто встречаются контрактуры (стеснение) в суставах — лодыжках, бедрах, коленях и локтях. Иногда контрактуры могут быть серьезными и поражать несколько суставов. Это называется артрогрипозом. У некоторых младенцев могут быть проблемы с дыханием из-за слабости дыхательных мышц. У некоторых детей, у которых нет контрактур, первые проблемы могут быть замечены только через несколько месяцев, когда ребенок не достигает своих моторных вех в том же возрасте, что и другие дети, и трудности с поднятой головой или задержка в обучении сидению. без посторонней помощи замечаются стоять или ходить.
Большинство форм CMD наследуются по аутосомно-рецессивному типу, что означает, что для того, чтобы ребенок был поражен, оба родителя должны быть носителями аномального гена, и оба должны передать этот ген своему ребенку. Родители-носители имеют 1 из 4 шансов иметь больного ребенка, 2 из 4 шансов иметь ребенка-носителя и 1 из 4 шансов иметь ребенка, который полностью свободен от пораженного гена.
Здесь представлен обзор распространенных типов CMD. Более подробная информация о различных типах CMD и их генетическом происхождении также доступна на веб-сайте правозащитной организации Cure CMD.
Врожденный MD | MD Австралия
Врожденный означает «от рождения», и поэтому у лиц с врожденной мышечной дистрофией (ВМД) симптомы обычно проявляются при рождении или в первые несколько месяцев. Существует как минимум 30 различных типов ВМК, каждый из которых имеет свою генетическую причину и свой набор симптомов, но все они в первую очередь влияют на мышцы, используемые для движения. Тяжесть CMD сильно варьируется от человека к человеку.
Младенцы с врожденной мышечной дистрофией часто выглядят «вялыми» (врачи также могут описать это как низкий мышечный тонус или гипотонию).Также часто встречаются контрактуры (стеснение) в суставах — лодыжках, бедрах, коленях и локтях. Контрактуры иногда бывают тяжелыми и поражают несколько суставов. Это называется артрогрипозом.
Некоторые типы CMD также влияют на мозг, вызывая трудности в обучении, а иногда и судороги. У некоторых младенцев также могут быть проблемы с дыханием и кормлением. У некоторых детей, у которых нет контрактур, первые проблемы могут быть замечены только через несколько месяцев, когда ребенок не достигает своих моторных вех в том же возрасте, что и другие дети.
Трудно сделать общие выводы о прогрессировании CMD, поскольку разные подтипы прогрессируют по-разному, а также прогрессирование варьируется от человека к человеку. Однако это состояние обычно считается довольно стабильным с точки зрения силы мышц рук и ног, и часто кажется, что ребенок набирает силу в первое десятилетие жизни. Некоторые дети будут ходить, но иногда это может быть отложено до пяти лет и старше, а иногда требуются шины для ног. Некоторые дети, достигшие самостоятельной ходьбы, могут потерять эту способность позже, потому что, когда они становятся старше и тяжелее, мышцы не могут справиться с большим напряжением.
Дети со всеми типами ВМД имеют повышенный риск развития респираторных заболеваний из-за слабых дыхательных мышц. Возраст, когда могут возникнуть проблемы с дыханием, и степень тяжести варьируется от человека к человеку. Следовательно, необходим тщательный мониторинг и контроль дыхания. При некоторых типах CMD могут развиваться проблемы с сердцем, особенно CMD, связанные с LMNA, и дистрогликанопатии (см. Таблицу ниже), поэтому людям с диагнозом этих состояний требуется частый мониторинг сердечной функции и соответствующее лечение.
По оценкам, примерно 1 ребенок из 20 000 рождается с врожденной мышечной дистрофией.
Какие бывают типы CMD?
Наиболее распространенным типом CMD является CMD Ульриха, за которым следует CMD с дефицитом мерозина (MDC1A). Некоторые из наиболее распространенных типов CMD перечислены в таблице ниже.
Имя | Участвующий ген / белок | Прочие наименования |
| ЦМД Ульриха и миопатия Бетлема | Коллаген VI | Миопатии, связанные с коллагеном 6 |
| ЦМД с дефицитом мерозина (MDC1A) | Ламинин альфа 2 | Мышечная дистрофия, связанная с LAMA2 |
| Врожденная мышечная дистрофия 1С (MDC1C) | Фукутин-родственный белок (FKRP) | Дистрогликанопатия |
| Врожденная мышечная дистрофия 1D типа (MDC1D) | Ацетилглюкозаминилтрансферазоподобный белок (большой) | Дистрогликанопатия |
| Врожденная мышечная дистрофия Фукуямы | Фукутин (ФКМД) | Дистрогликанопатия |
| Заболевание мышц, глаз и головного мозга | ПОМГнТ1, ФКРП, ФКМД | Дистрогликанопатия |
| Синдром Уокера-Варбурга | ПОМТ1, ПОМТ2, ФКРП, FCMD | Дистрогликанопатия |
| CMD, связанный с SEPN1 | Селенопротеин N | Мышечная дистрофия ригидного позвоночника, миопатия миопатия |
| CMD, относящаяся к LMNA | Ламин A / C | L-CMD, синдром опущенной головы |
Что вызывает CMD и как передается по наследству?
CMD вызывается изменением гена, который содержит инструкции по построению белка, необходимого для здоровья мышц.В некоторых условиях затронутый белок играет структурную роль, например коллаген и ламинин, которые обеспечивают структурную поддержку мышечным клеткам.
Некоторые типы CMD получили общее название «дистрогликанопатии» (см. Таблицу выше), поскольку все они вызваны дефектами процесса добавления молекул сахара к белку, называемому «альфа-дистрогликан». Этот белок образует важное звено между мышечной клеткой и окружающей средой. Добавление молекул сахара необходимо для эффективного функционирования альфа-дистрогликана.
Большинство форм CMD наследуются по аутосомно-рецессивному типу, что означает, что для того, чтобы ребенок был поражен, оба родителя должны быть носителями аномального гена, и оба должны передать этот ген своему ребенку. Родители-носители имеют 1 из 4 шансов иметь больного ребенка.
LMNA-связанные CMD, миопатия Бетлема и иногда CMD Ульриха наследуются по аутосомно-доминантному типу. Это означает, что одной копии измененного гена, унаследованной от любого из родителей, достаточно, чтобы вызвать заболевание.Существует 50% (каждый второй) шанс того, что ребенок пострадавшего унаследует это заболевание.
Вскоре после постановки диагноза ВМД необходимо организовать генетическое консультирование. Генетическое консультирование предоставляет информацию о типе наследования, рисках для других членов семьи и «прогнозе» (вероятном исходе заболевания). Генетические консультанты стремятся помочь отдельным лицам, парам и семьям понять и адаптироваться к медицинским и психологическим последствиям диагностики генетического заболевания в их семье.Консультант-генетик также может объяснить варианты планирования семьи, чтобы снизить риск передачи заболевания будущим детям. Для получения дополнительной информации о генетическом консультировании посетите нашу информационную страницу по генетике.
Как диагностируется CMD?
Когда врачи впервые замечают, что ребенок или младенец болеет, может быть проведена серия тестов, чтобы попытаться поставить точный диагноз. Во-первых, можно провести анализ крови, чтобы измерить уровень мышечного фермента, называемого креатинфосфокиназой (КФК).СК протекает из поврежденной мышцы и может быть признаком нескольких различных типов мышечной дистрофии. Примерно у 40 процентов детей с ВМД уровень КК в 5-20 раз выше нормы.
Также может быть проведен тест электромиографии (ЭМГ), при котором небольшая игла вводится в мышцу и регистрируется электрическая активность. Этот тест может предоставить доказательства аномального характера электрической активности в мышце. Этот тест обычно не требуется детям с повышенным уровнем КФК в крови.Для сбора дополнительной информации также может быть выполнено сканирование мозга и мышц.
Биопсия мышцы почти всегда необходима, чтобы сузить тип CMD. Это хирургическая процедура, при которой удаляется и исследуется небольшой образец мышцы. Это считается незначительной операцией и обычно проводится в дневном режиме под местной или общей анестезией. Диагноз CMD ставится на основании появления мышечных клеток под микроскопом. Определенные мышечные белки также можно изучить под микроскопом, чтобы попытаться определить конкретную причину.
После биопсии мышцы можно провести генетическое тестирование, чтобы определить ген, вызывающий заболевание, и поставить окончательный диагноз. Это включает анализ крови. Генетическое тестирование — это процесс исключения, и может потребоваться некоторое время, чтобы определить ген, вызывающий заболевание. Также важно знать, что иногда невозможно установить генетический диагноз. Однако исследователи постоянно узнают больше об этих состояниях и генетических причинах, поэтому со временем все больше и больше людей смогут получить генетический диагноз.Есть лечение?
В настоящее время нет специального лечения CMD, но есть много вещей, которые можно сделать, чтобы управлять симптомами. В идеале, дети и взрослые с ВМК должны регулярно посещаться в специализированной нервно-мышечной клинике, имея при необходимости доступ к физиотерапевтам, ортопедам, респираторным, ортопедическим, спинальным и генетическим специалистам.
Руководство для семей под названием «Ведение врожденной мышечной дистрофии (ВМД)» доступно для загрузки, в котором описывается медицинское лечение, рекомендованное группой из 82 международных экспертов.В этом руководстве вы найдете информацию о физиотерапии, респираторной и кардиологической помощи, питании и кормлении, ортопедии и неврологическом лечении (включая трудности с обучением и судороги).
Какие исследования проводятся?
Большая часть исследований врожденной мышечной дистрофии сосредоточена на понимании того, какие генетические изменения вызывают эти состояния, а также молекулярные процессы, которые происходят внутри тела и приводят к появлению симптомов. Достижения в этих областях уже привели к открытию некоторых потенциальных методов лечения, которые сейчас находятся на ранних этапах тестирования.
Швейцарская фармацевтическая компания Santhera в 2014 году объявила о начале клинических испытаний препарата омигапил у детей с врожденной мышечной дистрофией (ВМД). Считается, что омигапил предотвращает гибель клеток, и тестирование на мышиной модели MDC1A показало, что он может уменьшить тяжесть симптомов. Исследование (названное CALLISTO) с участием детей с CMD Ульриха и мерозин-дефицитным CMD (MDC1A) проводится в США. Дополнительная информация об испытании доступна в клинических испытаниях.gov веб-сайт.
Другой возможный путь, рассматриваемый для разработки методов лечения, включает лекарства, которые уменьшают воспаление и «фиброз» или рубцевание в мышцах, которые, как считается, являются основным фактором мышечной слабости при CMD. Лозартан, обычно назначаемое лекарство от высокого кровяного давления, является одним из возможных кандидатов для тестирования в клинических испытаниях, поскольку было показано, что он снижает фиброз на моделях CMD у мышей. Кортикостероидные препараты, такие как преднизон, обладающие противовоспалительными свойствами, иногда давали пациентам с некоторыми типами CMD, и сейчас рассматривается возможность проведения клинических испытаний для тестирования преднизона.
Эти подходы не устраняют основную причину состояния, поэтому могут только замедлить прогрессирование некоторых симптомов. В идеале лечение должно привести к исправлению генетических изменений. Генная терапия для исправления генетических мутаций исследуется для других генетических состояний, включая мышечную дистрофию, и, если они окажутся успешными, возможно, можно будет применить эту технологию для разработки методов лечения CMD. Исследования генной терапии с использованием некоторых генов CMD, таких как ген белка, связанного с фукутином, и ген LARGE, продолжаются на лабораторных мышах.
Регистр пациентов
Вы можете быть заинтересованы в регистрации в Международном регистре врожденных мышечных заболеваний (CMDIR). Это реестр пациентов: база данных, содержащая информацию о пациентах с определенным заболеванием. Организаторы клинических испытаний и другие исследователи используют эту (анонимную) информацию, чтобы узнать больше об условиях и спланировать клинические испытания.
Если бы клиническое испытание началось, регистр использовался бы для связи с подходящими потенциальными участниками и приглашения их принять участие.Регистры пациентов также являются полезным источником информации для пациентов и их семей, поскольку рассылаются регулярные информационные бюллетени. Вы можете узнать больше о реестрах пациентов на нашем веб-сайте.
Дополнительная информация
• Отдельные информационные бюллетени доступны для следующих типов CMD:
- Ульрих CMD
- Миопатия Бетлема
- Фукуяма CMD
• Клинические испытания — ответы на ваши вопросы
• Определения любых терминов, которые вам не знакомы, можно найти в нашем глоссарии
• Американская организация CureCMD является полезным источником информации, касающейся всех типов CMD
• Вы можете получать регулярные обновления, став другом страницы MDA на Facebook
Для получения дополнительной информации по любому из вопросов, рассмотренных выше, свяжитесь с MDA:
Телефон: (03) 9320 9555
Эл. Почта: info @ mda.org.au
Пересмотрено 28 июня 2018 г.
Список литературы
Ведение врожденной мышечной дистрофии (ВМД): руководство для семей, опубликовано в 2011 году.
Мышечная дистрофия, Великобритания. Информационный бюллетень по врожденной мышечной дистрофии: обзор, октябрь 2013 года.
Susan Sparks et al. Обзор генов: обзор врожденной мышечной дистрофии. Последняя редакция: 23 августа 2012 г.
Врожденная мышечная дистрофия — обзор
Врожденная мышечная дистрофия
Врожденные мышечные дистрофии (ВМД) — это группа клинически и генетически разнообразных заболеваний, характеризующихся мышечной слабостью, начинающейся в раннем детстве и часто проявляющейся при рождении [59].Многие из этих нарушений вызваны мутациями белков, связанных с внеклеточным матриксом, или посттрансляционной модификацией дистрогликана, основного связывающего дистрофин белка. Мутации в других генах, включая те, которые кодируют белки, участвующие в хранении гликогена, синтез базальной пластинки и белки ядерных пор, также могут приводить к подобному фенотипу. Гистологически эти расстройства имеют сходные характеристики на ранних стадиях заболевания, и они включают изменение размера волокон, увеличение количества ядер на волокно и мононуклеарный воспалительный инфильтрат.Тяжелые формы CMD будут развиваться с развитым фибро-жировым инфильтратом, некрозом и воспалением, которые могут затрагивать или не затрагивать сердце. Слабость дыхательных мышц, приводящая к гиповентиляции, является серьезной проблемой при CMD [60]. Сердечно-легочное поражение варьирует в зависимости от CMD. Однако слабость дыхательных мышц более универсальна для ВМД, чем для кардиомиопатии, и снижение показателей легочной функции на протяжении второго десятилетия жизни является основным признаком ВМД. Некоторые подмножества CMD преимущественно влияют на сердце по причинам, которые не совсем понятны.В частности, CMD, связанный с FKTN-, фукутин- ( FKRP ) и протеин-O-маннозилтрансферазой 1, ( POMT1 ), оба характеризуются тяжелой и ранней кардиомиопатией с систолическая дисфункция левого желудочка, развивающаяся к 12 годам [61–63].
Дефицит мерозина или LAMA2 (MDC1A) вызван мутациями в гене LAMA2 , который кодирует тяжелую α2-изоформу ламинина, структурного белка внутренней ядерной створки [64].MDC1A примечателен тем, что он может проявляться очень ранней кардиомиопатией, а также слабостью, гипотонией и контрактурами суставов уже при рождении. Уровни КФК в сыворотке также обычно повышены при рождении, и некоторые сообщения предполагают наличие сердечной недостаточности вскоре после этого. К сожалению, фракция выброса левого желудочка ухудшается до стабилизации, и ее снижение можно отследить с помощью эхокардиографии [65,66].
Коллаген VI является основным компонентом матрикса вокруг миофибрилл.Как и все коллагены, зрелый коллаген VI состоит из тримерного массива трех мономеров α-коллагена, переплетенных для обеспечения прочности на разрыв. Каждая субъединица кодируется отдельным геном: COL6A1 , COL6A2 и COL6A3 , все из которых экспрессируются вместе с одинаковыми промоторами. Хотя коллаген VI экспрессируется во многих типах тканей, он сильно экспрессируется в мышцах, где, по-видимому, важен для стабилизации внеклеточного матрикса от повреждений, вызванных работой.