Дефицит альфа-1-антитрипсина у детей: описание серии случаев | Мельник
1. Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397–415.
2. Основы клинической гепатологии. Заболевания печени и билиарной системы. Учебное пособие для системы последипломного образования врачей / Под ред. В.Г. Радченко, А.В. Шаброва, Е.Н. Зиновьева. — СПб.: Диалект; М.: БИНОМ; 2005. — 862 с. [Osnovy klinicheskoi gepatologii. Zabolevaniya pecheni i biliarnoi sistemy. Uchebnoe posobie dlya sistemy poslediplomnogo obrazovaniya vrachei. Ed by Radchenko V.G., Shabrov A.V., Zinov’ev E.N. St. Petersburg: Dialekt; Moscow: BINOM; 2005. 862 p. (In Russ).]
3. Жигальцова-Кучинская А., Сивицкая Л.Н., Даниленко Н.Г. и др. Дефицит альфа-1- антитрипсина: генетические основы, эпидемиология, значение в развитии бронхо- легочной патологии // Вестник Витебского государственного медицинского университета. — 2015. — Т. 14. — № 6 — С. 39–52. [Zhigaltsova-Kuchinskaya OA, Sivitskaya LN, Danilenko NG, et al. Alpha-1-antitrypsin deficiency: genetic fundamentals, epidemiology, role in the development of bronchopulmonary pathology. Vestnik Vitebskogo gosudarstvennogo meditsinskogo universiteta. 2015;14(6):39–52. (In Russ).]
4. Колесникова Е.В. Альфа-1-антитрипсиновая недостаточность: Современный взгляд на проблему // Сучасна гастроентерологія. — 2008. — № 2 — С. 93–98. [Kolesnikova EV. Al’fa-1-antitripsinovaya nedostatochnost’: Sovremennyi vzglyad na problemu. Contemporary gastroenterology. 2008;(2):93–98. (In Russ).]
5. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi: 10.1164/rccm.168.7.818.
Am J Respir Crit Care Med. 2003;168(7):818–900. doi: 10.1164/rccm.168.7.818.
6. Churg A, Wang X, Wang RD, et al. 1-Antitrypsin suppresses TNF- and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144– 151. doi: 10.1165/rcmb.2006-0345OC.
7. Sharp HL. Alpha-1-antitrypsin deficiency. Hosp Pract. 1971; 6(5):83–96. doi: 10.1080/21548331.1971.11706032.
8. who.int [Internet]. International statistical classification of dise ases and related health problems 10th revision [cited 2016 Nov 9]. Available from: http://apps.who.int/classifications/icd10/browse/2016/en.
9. europeanlung.org [интернет]. Европейский пульмонологический фонд. Дефицит альфа1-антитрипсина [доступ от 21.10.2016]. Доступ по ссылке http://www.europeanlung.org/assets/files/ru/publications/alpha1-anti-trypsin-ru.pdf.
10. Hutchinson DCS. Аlpha-1-Antitrypsin deficiency in Europe: geographical distribution of Pi types S and Z. Respir Med. 1998; 92(3):367-377. doi: 10.1016/S0954-6111(98)90278-5.
11. Laurell CB, Eriksson S. The electrophoretic pattern alpha-1-globulin pattern of serum in alpha-l-antitrypsin deficiency. Scan J Clin Lab Invest. 1963;15(2):132–140. doi: 10.3109/00365516309051324.
12. Pariente EA, Degott C, Martin JP, et al. Hepatocytic PAS-positive diastase-resistance inclusions in the absence of alpha-1-antitrypsin deficiency — high prevalence in alcoholic cirrhosis. Am J Clin Pathol. 1981;76(3):299-302.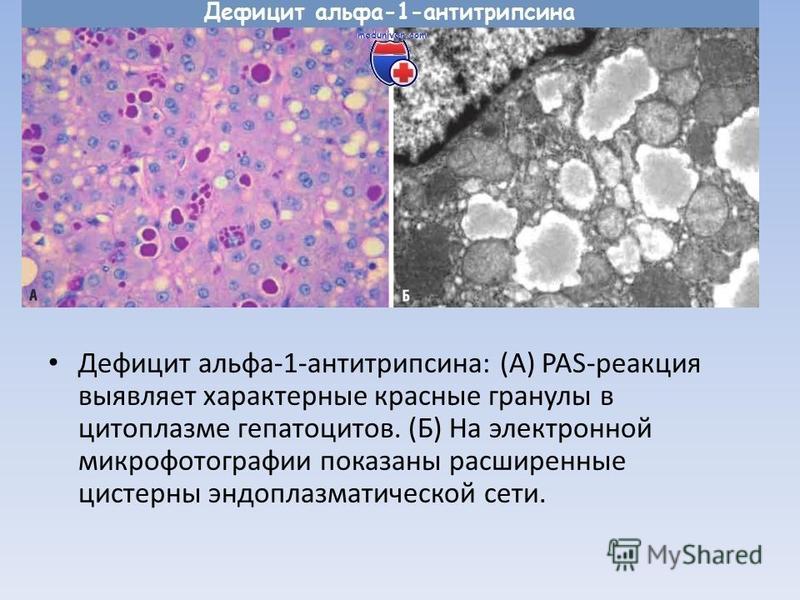 doi: 10.1093/ajcp/76.3.299.
doi: 10.1093/ajcp/76.3.299.
13. Протеолиз в норме и при патологии / Под ред. К.Н. Веремеенко, О.П. Голобородько, А.И. Кизим. — Киев: Здоров’я; 1988. — 199 с. [Proteoliz v norme i pri patologii. Ed by Veremeenko K.N., Goloborod’ko O.P., Kizim A.I. Kiev: Zdorov’ya; 1988. 199 p. (In Russ).]
14. Дидковский Н.А., Жарова М.А. Значение наследственных факторов в развитии эмфиземы легких // Терапевтический архив. — 2006. — Т. 78. — № 3 — С. 70–74. [Didkovsky NA, Zharova MA. The role of hereditary factors in development of pulmonary emphysema. Ter Arkh. 2006;78(3):70–74. (In Russ).]
15. Веремеенко К.Н. Ферменты протеолиза и их ингибиторы в медицинской практике. — Киев; 1971. — 216 с. [Veremeenko KN. Fermenty proteoliza i ikh ingibitory v meditsinskoi praktike. Kiev; 1971. 216 p. (In Russ).]
16. Hubbard RC, Crystal RG. Strategies for aerosol therapy of alpha 1-antitrypsin deficiency by the aerosol route. Lung. 1990;168 Suppl:565–578. doi: 10.1007/bf02718179.
17. Crowther DC, Belorgey D, Miranda E, et al. Practical genetics: alpha-1-antitrypsin deficiency and the serpinopathies. Eur J Hum Genet. 2004;12(3):167–172. doi: 10.1038/sj.ejhg.5201127.
18. Шапошникова Н.А., Шулятьев И.С., Варванина Г.Г., Дроздов В.Н. Клиническое значение наследственного и приобретенного дефицита альфа-1-антитрипсина у больных циррозом печени и болезнью Вильсона-Коновалова // Лабораторная служба. — 2010. — № 10 — С. 12–16. [Shaposhnikova NA, Shulyat’ev IS, Varvanina GG, Drozdov VN. Klinicheskoe znachenie nasledstvennogo i priobretennogo defitsita al’fa-1-antitripsina u bol’nykh tsirrozom pecheni i bolezn’yu Vil’sona-Konovalova. Laboratornaya sluzhba. 2010; (10):12–16. (In Russ).]
Laboratornaya sluzhba. 2010; (10):12–16. (In Russ).]
19. Бродская О.Н. Наследственная недостаточность 1-антитрипсина // Практическая пульмонология. — 2008. — № 4 — С. 58–59. [Brodskaya ON. Nasledstvennaya nedostatochnost’ 1-antitripsina. Prakticheskaya pul’monologiya. 2008;(4):58–59. (In Russ).]
20. Аверьянов А.В., Поливанова А.Э. Дефицит 1-антитрипсина и хроническая обструктивная болезнь легких // Пульмонология. — 2007. — № 3 — С. 103–109. [Averyanov AV, Polivanova AE. Alfa-1-antitripsin deficiency and chronic obstrictivc pulmonary disease. Pul’monologiya. 2007;(3):103–109. (In Russ).]
21. Castaldi PJ, DeMeo DL, Kent DM, et al. Development of predictive models for airflow obstruction in alpha-1-antitrypsin deficiency. Am J Epidemiol. 2009;170(8):1005–1013. doi: 10.1093/aje/kwp216.
22. Davis ID, Burke B, Freese D, et al. The pathologic spectrum of the nephropathy associated with 1-antitrypsin deficiency. Hum Pathol. 1992;23(1):57–62. doi: 10.1016/0046- 8177(92)90012-r.
23. Churg A, Wang X, Wang RD, et al. Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144–151. doi: 10.1165/rcmb.2006-0345OC.
24. Видаль Р., Бланко И., Касас Ф. и др. Рекомендации по диагностике и ведению больных с дефицитом 1-антитрипсина Испанского общества пульмонологии и торакальной хирургии (SEPAR) // Пульмонология. — 2008. — № 1 — С. 14–28. [Vidal R, Blanco I, Casas F, et al. The National Alpha-1 Antitrypsin Registry Committee Guidelines for the diagnosis and management of alpha-1-antitrypsin deficiency of SEPAR. Pul’monologiya. 2008;(1):14–28. (In Russ).]
Pul’monologiya. 2008;(1):14–28. (In Russ).]
25. Назаров П.Г. Реактанты острой фазы воспаления.— СПб.: Наука; 2001. — 423 с. [Nazarov PG. Reaktanty ostroi fazy vospaleniya. St. Petersburg: Nauka; 2001. 423 p. (In Russ).]
26. Tanash HA, Nilsson PM, Nilsson JA, et al. Survival in severe alpha-1-antitrypsin deficiency (PiZZ). Respir Res. 2010;11:44. doi: 10.1186/1465-9921-11-44.
27. Bornhorst J, Calderon F, Procter M, et al. Genotypes and serum concentrations of human alpha-1-antitrypsin “P” protein variants in a clinical population. J Clin Pathol. 2007;60(10):1124–1128. doi: 10.1136/jcp.2006.042762.
28. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi: 10.1164/rccm.168.7.818.
29. Elzouki AN, Segelmark M, Wieslander J, Eriksson S. Strong link between the alpha 1- antitrypsin PiZ allele and Wegener’s granulomatosis. J Intern Med. 1994;236(5):543–548. doi: 10.1111/j.1365-2796.1994.tb00842.x.
30. Askari FK. Molecular mechanism of hepatocellular injury in alpha 1 antitrypsin deficiency. Hepatology. 1995;21(6):1745–1747. doi: 10.1002/hep.1840210638.
31. Strange C, Dickson R, Carter C, et al. Genetic testing for alpha1-antitrypsin deficiency. Genet Med. 2004;6(4):204–210. doi: 10.109701.GIM.0000132669.09819.79.
32. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. doi: 10.1164/ajrccm.153.2.8564092.
Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. doi: 10.1164/ajrccm.153.2.8564092.
33. Fregonese L, Stolk J. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis. 2008;3:16. doi: 10.1186/1750-1172-3-16.
34. McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1- Antitrypsin Deficiency Registry Study Group. Chest. 1997;111(2):394–403. doi: 10.1378/chest.111.2.394.
35. Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: Clinical manifestations and natural history. Thorax. 2004;59(5): 441–445. doi: 10.1136/thx.2003.006510.
36. Katz RM, Lieberman J, Siegel SC. Alpha-1 antitrypsin levels and prevalence of Pi variant phenotypes in asthmatic children. J Allergy Clin Immunol. 1976;57(1):41–45. doi: 10.1016/0091-6749(76)90077-4.
37. Bruttmann G. [Reagin asthma and familial alpha-1antitrypsin deficiency. (In French).] Nouv Presse Med. 1974;3(10):589–591.
38. Bruttmann G. [Asthma associated with a familial deficiency in alpha-1-antitrypsin. (In French).] Revue francaise d’allergologie. 1973;13(4):411–418 doi: 10.1016/S0035- 2845(73)80062-9.
39. Browne RJ, Mannino DM, Khoury MJ. Alpha 1-antitrypsin deficiency deaths in the United States from 1979–1991. An analysis using multiple-cause mortality data. Chest. 1996;110(1):78–83. doi: 10.1378/chest.110.1.78.
Alpha 1-antitrypsin deficiency deaths in the United States from 1979–1991. An analysis using multiple-cause mortality data. Chest. 1996;110(1):78–83. doi: 10.1378/chest.110.1.78.
40. Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294(24): 1316–1321. doi: 10.1056/NEJM197606102942404.
Альфа-1-антитрипсин, концентрация (А1АТ, Alpha-1-Antitrypsin сoncentration, A1-Antitrypsin, A1A, AAT)
Метод определения
Иммунотурбидиметрия
Исследуемый материал
Сыворотка крови
Синонимы: α1-антитрипсин концентрация; А1АТ концентрация; ААТ.
Alpha1-antitrypsin; α1-antitrypsin.
Краткое описание исследования Альфа-1-антитрипсин, концентрация
Скрининговый тест для выявления альфа-1-антитрипсиновой недостаточности.
Альфа-1-антитрипсин – гликопротеин с молекулярной массой 50 кДа, основной компонент альфа-1-фракции при электрофорезе белков сыворотки крови. Подавляющая часть альфа-1-антитрипсина сыворотки образуется в печени. Молекулы этого белка имеют небольшой размер и легко диффундируют из плазмы в ткани. Уровень А1АТ повышается при воспалении. Этот белок обладает способностью блокировать активность протеолитических ферментов: систем кинина, комплемента, фибринолитической системы, протеаз, выделяемых нейтрофилами. При воспалительном процессе в легочной ткани альфа-1-антитрипсин эффективно подавляет функцию эластазы, выделяющейся из нейтрофилов, предотвращая деградацию белка соединительной ткани − эластина − в стенках альвеол и развитие эмфиземы легких. Он модулирует локальный иммунный ответ, обладает антиоксидантным и антимикробным действием, ингибирует протеолитические ферменты апоптоза.
При каких состояниях может повышаться сывороточная концентрация Альфа-1-антитрипсина
Концентрация А1АТ значительно повышается при остром воспалении, инфекционных заболеваниях, ревматических заболеваниях, повреждении или некрозе тканей, некоторых злокачественных процессах, действии эстрогенов (при заместительной терапии эстрогенами, приеме пероральных контрацептивов, повышении уровня эстрогенов при беременности – в третьем триместре до двукратного повышения), воспалительном процессе в печени.
При каких патологиях развивается дефицит альфа-1-антитрипсина в организме и как он проявляется
Генетически детерминированный дефицит А1АТ коррелирует с высоким риском развития патологии легких. Связь дефицита альфа-1-антитрипсина с повышенным риском развития эмфиземы легких была открыта Laurell и Eriksson, которые в 1963 году впервые описали отсутствие альфа-1-фракции у больных с панацинарной эмфиземой. Позднее была описана связь врожденного дефицита данного белка с заболеваниями печени.
Дефицит А1АТ (альфа-1-антитрипсиновая недостаточность) представляет собой наследственное заболевание, обусловленное сниженной сывороточной концентрацией альфа-1-антитрипсина, с общей частотой встречаемости 1:3000-5000. Среди лиц с эмфиземой, астмой, хронической обструктивной болезнью легких распространенность недостаточности альфа-1-антитрипсина существенно выше, чем в общей популяции, и достигает 1:100-1:10. Риск развития патологии легких на фоне дефицита А1АТ значительно увеличивается у курильщиков: эмфизема и тяжелая хроническая обструктивная болезнь легких у курящих лиц с дефицитом альфа-1-антитрипсина развивается, в среднем, к 32-41 году. Количественное определение А1АТ входит в современные рекомендации по обследованию пациентов с хроническими обструктивными заболеваниями легких и семейной историей подобных заболеваний, а также при возникновении такой патологии в возрасте до 45 лет.
Дефицит альфа-1-антитрипсина уже в детском возрасте может вызывать поражение печени, его можно подозревать при синдроме тяжелого гепатита у новорожденных, холестатическом синдроме в детском и молодом возрасте. У лиц старше 50 лет повышен риск развития цирроза печени и гепатоцеллюлярной карциномы. Более редкими клиническими проявлениями недостаточности А1АТ являются некротический панникулит, системный гранулематозный васкулит.
У лиц старше 50 лет повышен риск развития цирроза печени и гепатоцеллюлярной карциномы. Более редкими клиническими проявлениями недостаточности А1АТ являются некротический панникулит, системный гранулематозный васкулит.
Помимо генетически обусловленной недостаточности альфа-1-антитрипсина, снижение сывороточной концентрации А1АТ может наблюдаться и при увеличенном его потреблении – при идиопатическом респираторном дистресс-синдроме у детей, тяжелом гепатите у новорожденных, тяжелом поражении поджелудочной железы. Уровень альфа-1-антитрипсина низок при нефротическом синдроме вследствие потерь белка (если отсутствует воспаление), энтеропатиях с потерей белка, при голодании.
Для подтверждения генетически детерминированного дефицита необходимо исследование генотипа или фенотипа альфа-1-антитрипсина у пациента (см. тест №832A1A «Альфа-1-антитрипсин, фенотипирование»).
С какой целью определяют сывороточную концентрацию альфа-1-антитрипсина
Скрининговый тест для выявления альфа-1-антитрипсиновой недостаточности.
Литература
Основная литература:
- Алан Г.Б., Ву А. Клиническое руководство Тица по лабораторным тестам. Пер. с англ. В.В. Меньшикова, 4-е изд. — М., Лабора. 2013:1280.
- Дидковский Н.А. Дефицит альфа-1-антитрипсина. Монография/ Хронические заболевания легких у детей./Под ред. Н.Н. Розиновой и Ю.Л. Мизерницкого. — М., Практика. 2011:124-129.
- Инструкции к набору реагентов.
- American Thoracic Society; European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. American Journal of Respiratory and Critical Care Medicine. 2003;168(7):818-900.
- Stoller J.K., Aboussouan L.S. A review of a1-antitrypsin deficiency. American Journal of Respiratory and Critical Care Medicine. 2012;185(3):246-259.
Поражение печени у детей при недостаточности альфа-1-антитрипсина
Поражение печени у детей при недостаточности альфа-1-антитрипсина
Специалисты ОСП «Научно-исследовательский клинический институт педиатрии им. академика Ю.Е. Вельтищева» и ФГАОУ ВО РНИМУ им. Н.И. Пирогова МЗ РФ представили обзор патофизиологии поражения печени при дефиците альфа-1-антитрипсина у детей. Также авторами представлены рекомендации по ведению детей с подозрением и подтвержденным дефицитом альфа-1-антитрипсина.
академика Ю.Е. Вельтищева» и ФГАОУ ВО РНИМУ им. Н.И. Пирогова МЗ РФ представили обзор патофизиологии поражения печени при дефиците альфа-1-антитрипсина у детей. Также авторами представлены рекомендации по ведению детей с подозрением и подтвержденным дефицитом альфа-1-антитрипсина.
Основное место синтеза альфа-1-антитрипсина – печень, хотя этот белок также вырабатывается в энтероцитах и некоторых мононуклеарных лейкоцитах. Физиологическая роль альфа-1-антитрипсина заключается в ингибировании нейтрофильных протеаз во время воспалительного ответа и фагоцитоза, направленного против микроорганизмов. Однако показано, что альфа-1-антитрипсин, вероятно, имеет и другие функции в иммунном ответе, в частности ингибирует эластазу и защищает таким образом ткани от протеолитического действия указанного фермента.
Дефицит альфа-1-антитрипсина – наиболее частое из генетически детерминированных заболеваний печени у детей. Спектр клинических проявлений варьирует от длительной желтухи и повышения уровня цитолитической активности до хронического гепатита и цирроза. Признаки поражения печени чаще возникают у новорожденных, поскольку у них клетки печени менее способны расщеплять мутантный альфа-1-антитрипсин.
Ген PI высоко полиморфен: известно более 500 его аллельных вариантов, из которых около 30 имеют клиническое значение. Наиболее часто встречаемые генотипы образованы комбинациями PIM-, PIS- и PIZ- аллелей: PIMM, PIMS, PISS, PIMZ, PISZ и PIZZ. Аллели гена PI, которые обусловливают снижение уровня альфа-1-антитрипсина в сыворотке крови, называют дефицитными, из них наиболее часто встречаются варианты S и Z. Следствием полиморфизма гена PI являются разнообразные механизмы развития недостаточности альфа-1-антитрипсина в крови и различия его функциональной активности. Нормальный белок альфа-1-антитрипсин обозначается буквой «М», аномальный белок – буквой «Z».
Лица, у которых уровень альфа-1-антитрипсина в крови соответствует норме, гомозиготны (PI*MM) по нормальному («дикому») аллелю М, пациенты с тяжелым дефицитом альфа-1-антитрипсина гомозиготны по аллелю Z (PI*ZZ). Кроме того, к более низкому содержанию этого белка в крови приводит наличие аллеля S. В то время как PI*ZZ служит основным генотипом, ведущим к заболеванию печени, аллель S не связан с заболеванием печени, за исключением случаев, когда он сочетается с аллелем Z. Болезнь печени должна быть исключена даже у пациентов с гетерозиготными мутациями гена PI.
Кроме того, к более низкому содержанию этого белка в крови приводит наличие аллеля S. В то время как PI*ZZ служит основным генотипом, ведущим к заболеванию печени, аллель S не связан с заболеванием печени, за исключением случаев, когда он сочетается с аллелем Z. Болезнь печени должна быть исключена даже у пациентов с гетерозиготными мутациями гена PI.
За последнее десятилетие было продемонстрировано, что альфа-1-антитрипсин обладает широким спектром действия: противовоспалительное, иммуномодулирующеее, противоинфекционное и репаративное.
Альфа-1-антитрипсин в печени вырабатывается постоянно, по количеству уступая первенство только альбумину. В эндоплазматическом ретикулуме гепатоцитов синтезируется неактивный предшественник этого белка, путем отщепления N-концевых пептидов образуется активная форма, которая секретируется в кровь.
При дефиците альфа-1-антитрипсина в большинстве случаев заболевание печени связано с гомозиготной мутацией по аллелю Z – генотипом PI*ZZ. Считается, что для противодействия накоплению полимеров альфа-1-антитрипсина в эндоплазматическом ретикулуме гепатоцитов имеются два пути:
1) связывание неполимеризованного Z-белка с трансмембранным калнексином эндоплазматического ретикулума с последующим связыванием с убиквитином и дальнейшей деградацией этого комплекса;
2) аутофагическая деградация, апоптоз и гибель гепатоцитов с наибольшим накоплением мутантного белкового полимера; гепатоциты с более низким уровнем накопления полимера Z-белка пролиферируют, чтобы поддерживать функциональную массу клеток печени.
Считается, что эффективность механизма деградации Z-белка с участием кальнексина служит фактором, определяющим восприимчивость к повреждению печени у человека с генотипом PI*ZZ.
Отдельные «мономерные» мутантные Z-молекулы альфа-1-антитрипсина содержатся в эндоплазматическом ретикулуме гепатоцитов, а затем подвергаются протеолизу . Однако некоторые из них агрегируют, образуя большие массы. Часто эти включения, называемые глобулами, достаточно велики, и их можно увидеть с помощью световой микроскопии. Считается, что именно эти скопления мутантного Z-белка в гепатоцитах запускают внутриклеточный каскад патологических процессов, повреждающий гепатоцит, и это приводит к таким заболеваниям печени, как хронический гепатит, цирроз и гепатоцеллюлярная карцинома. Однако одного только накопления Z-белка для поражения органа недостаточно, поскольку, по клиническим наблюдениям, не у всех лиц с генотипом PI*ZZ развивается заболевание печени. Хотя накопление мутантного Z-белка в гепатоцитах служит стимулирующим фактором повреждения печени, большинство удерживаемых молекул мутантного альфа-1-антитрипсина подвергается внутриклеточному протеолизу и распадается на составляющие их аминокислоты.
Часто эти включения, называемые глобулами, достаточно велики, и их можно увидеть с помощью световой микроскопии. Считается, что именно эти скопления мутантного Z-белка в гепатоцитах запускают внутриклеточный каскад патологических процессов, повреждающий гепатоцит, и это приводит к таким заболеваниям печени, как хронический гепатит, цирроз и гепатоцеллюлярная карцинома. Однако одного только накопления Z-белка для поражения органа недостаточно, поскольку, по клиническим наблюдениям, не у всех лиц с генотипом PI*ZZ развивается заболевание печени. Хотя накопление мутантного Z-белка в гепатоцитах служит стимулирующим фактором повреждения печени, большинство удерживаемых молекул мутантного альфа-1-антитрипсина подвергается внутриклеточному протеолизу и распадается на составляющие их аминокислоты.
Клетка использует различные протеолитические процессы в попытке уменьшить повреждение мутантным Z-белком. К ним относятся убиквитин-зависимые и убиквитин-независимые протеасомные пути протеолиза, а также другие механизмы, иногда называемые «ЭР-ассоциированным протеолизом» (ЭРАП). Эти протеосомные пути как часть ЭРАП служат основным путем деградации мутантных мономерных Z-молекул альфа-1-антитрипсина в неполимеризованной конформации.
Другой важный протеолитический путь – аутофагия, представляющая собой высоко консервативную систему деградации, в которой специализированные вакуоли разлагают аномальные белки и более крупные структуры, такие как стареющие органеллы. Исследования показывают, что накопление полимеризованного мутантного Z-белка альфа-1-антитрипсина внутри клеток индуцирует аутофагический ответ. В экспериментальных системах повреждение печени было уменьшено за счет усиления аутофагической деградации мутантного альфа-1-антитрипсина.
Клинически поражение печени у людей с PI*ZZ генотипом обычно представляет собой медленный процесс, который длится на протяжении многих лет или десятилетий; анализ ткани печени человека показал, что накопление в гепатоцитах мутантного альфа-1-антитрипсина очень неоднородно.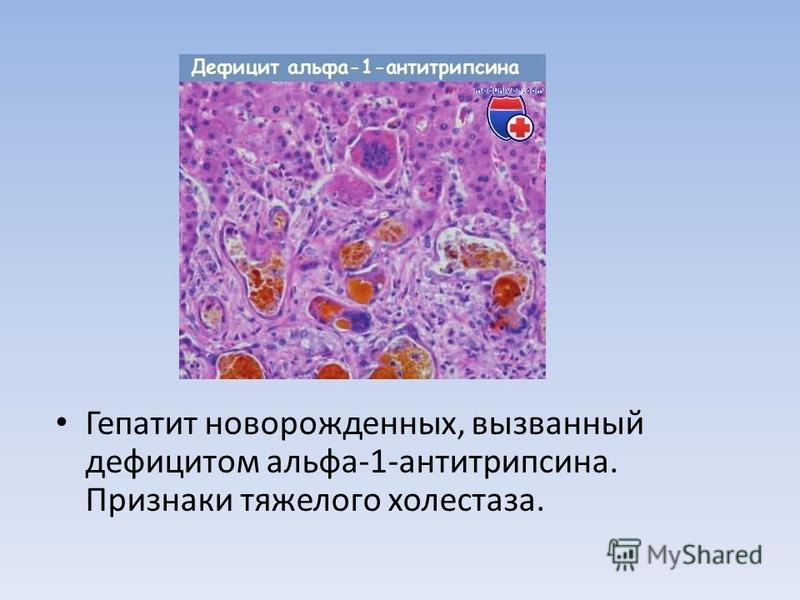 Результаты недавних исследований показывают, что каскад повреждения клеток запускается в небольшой популяции гепатоцитов, имеющих наибольшее накопление мутантного полимеризованного альфа-1-антитрипсина. Указанные гепатоциты, а их только несколько процентов от общего числа клеток, возможно, имеют повышенную активацию каспазы и повышенную восприимчивость к апоптозу.
Результаты недавних исследований показывают, что каскад повреждения клеток запускается в небольшой популяции гепатоцитов, имеющих наибольшее накопление мутантного полимеризованного альфа-1-антитрипсина. Указанные гепатоциты, а их только несколько процентов от общего числа клеток, возможно, имеют повышенную активацию каспазы и повышенную восприимчивость к апоптозу.
Существует также недавно признанный компонент окислительного повреждения клеток. Эти процессы обусловливают низкий (но более высокий, чем обычно) исходный уровень гибели гепатоцитов с Z-белком альфа-1-антитрипсина. Клетки с низким накоплением мутантного Z-белка пролиферируют для поддержания функциональной массы печени. Со временем продолжающийся стресс, гибель и восстановление клеток приводят к фиброзу, циррозу печени, гепатоцеллюлярной карциноме. Предполагается, что экологические и генетические модификаторы секреции, деградации, апоптоза или регенерации белка влияют на прогрессирование заболевания печени у пациентов.
Проявления недостаточности альфа-1-антитрипсина у детей.
Прогрессирующее нарушение функции печени встречается нечасто. Большинство детей клинически выздоравливают, однако при наличии спленомегалии может развиться цирроз печени и 5% детей с поражением печени нуждаются в трансплантации органа в течение первых 4 лет жизни. В некоторых случаях у пациентов детского возраста развивается гепатоцеллюлярная карцинома, иногда фульминантная печеночная недостаточность.
К клиническим признакам, свидетельствующим о дефиците альфа-1-антитрипсина, обусловленном генотипом PI*ZZ в детском возрасте, относятся следующие: повышенный уровень трансаминаз и/или билирубина; синдром неонатального гепатита у ребенка; гепатомегалия или гепатоспленомегалия у детей и подростков; витамин К-дефицитная коагулопатия у ребенка; симптомы хронического заболевания печени у детей и подростков; генотип PI*ZZ у родственника первой линии.
К факторам, которые указывают на потенциально более тяжелый прогноз при поражении печени у детей с генотипом PI*ZZ, относятся: неонатальный холестаз; мужской пол; длительная гипербилирубинемия; значительная гепатомегалия; ранняя спленомегалия; удлиненное протромбиновое время; постоянно повышенный уровень гамма-глютамилтранспептидазы.
В неонатальном периоде заболевание обычно носит холестатический характер и сопровождается длительной холестатической желтухой, кожным зудом, определить который объективно можно лишь в возрасте 6 мес, снижением аппетита и отставанием в прибавке массы тела, гепато- и спленомегалией.
У детей старшего возраста дефицит альфа-1-антитрипсина может проявляться бессимптомным хроническим гепатитом. Прогрессирующее заболевание печени у молодых или людей среднего возраста, по-видимому, встречается редко, но риск его возникновения увеличивается с возрастом.
Приблизительно 50% взрослых пациентов с генотипом PI*ZZ умирают от тяжелой болезни легких в среднем в возрасте 52 лет и имеют незначительные признаки поражения печени или не имеют их вовсе, тогда как у лиц, умерших в возрасте 62 лет, обнаруживаются проявления хронического прогрессирующего заболевания печени.
Цирроз печени, вызванный дефицитом альфа-1-антитрипсина, служит установленным фактором риска развития гепатоцеллюлярной карциномы; выживаемость после установления диагноза цирроза печени снижается и у 30% пациентов при вскрытии обнаруживается первичный рак печени. Люди с дефицитом альфа-1-антитрипсина должны быть обследованы для выявления гепатоцеллюлярной карциномы с использованием биомаркеров или методов визуализации.
Клиническое течение и тяжесть заболевания печени, связанного с дефицитом альфа-1-антитрипсина, могут зависеть от генетических факторов и факторов окружающей среды. В частности, факторами риска развития заболеваний печени в зрелом возрасте являются мужской пол, болезни печени и ожирение в детском возрасте.
Поскольку на ранних стадиях заболевание печени вследствие недостаточности альфа-1-антитрипсина может проявляться у новорожденных и детей, протекая относительно доброкачественно, тестирование на дефицит альфа-1-антитрипсина должно быть частью дифференциальной диагностики у детей с нарушениями функции печени.
Ключом к успешной диагностике служит определение уровня альфа-1-антитрипсина с последующим клиническим исследованием фенотипических проявлений и анализом генотипа при обнаружении низкой концентрации его в сыворотке. Установление диагноза позволяет осуществлять генетическое консультирование и в отдельных случаях применять дополнительную терапию.
Установление диагноза позволяет осуществлять генетическое консультирование и в отдельных случаях применять дополнительную терапию.
Биопсия печени для установления диагноза дефицита альфа-1-антитрипсина не требуется, хотя это исследование может быть полезно в целях исключения других причин поражения печени и использовано для определения степени фиброза, изменений паренхимы и стадии заболевания.
Показания к трансплантации печени включают постоянный и рецидивирующий холестаз, ухудшение показателей гемокоагуляции, высокий уровень аланинаминотрансферазы (АлАТ) и аспартатаминотрансферазы (АсАТ), тяжелый гломерулонефрит, связанный с дефицитом альфа-1-антитрипсина, и асцит в результате портальной гипертензии.
При наблюдении за изменениями легочной функции пациентов было показано, что трансплантация печени стабилизирует состояние легких.
Рекомендации по ведению пациентов с недостаточностью альфа-1-апантитрипсина.
Ключевая особенность диагностики дефицита альфа-1-антитрипсина как при заболеваниях печени, так и при заболеваниях легких, заключается в том, что однозначные доказательства могут быть получены только по результатам генетического тестирования. Ранняя диагностика необходима, чтобы: лица с дефицитом альфа-1-антитрипсина могли принять меры для сохранения легочной функции; члены семьи могли быть обследованы для выявления гетерозиготного носительства; могло быть предложено эффективное лечение.
Если заболевание печени вызвано дефицитом альфа-1-антитрипсина, то последующее наблюдение должно включать базовую оценку функции легких. Если таковая нарушена, пациентов следует проконсультировать у пульмонолога и ежегодно проводить обследование.
Таким образом, правильный диагноз важен для эффективного клинического наблюдения и генетического консультирования. Обнаружение заболеваний печени из-за дефицита альфа-1-антитрипсина у детей может помочь предотвратить развитие заболеваний легких в зрелом возрасте.
Источник: Патофизиологические аспекты поражения печени у детей при недостаточности альфа-1-антитрипсина
Г. В. Волынец, А.В. Никитин
В. Волынец, А.В. Никитин
Метки: научные исследования
12.08.2020
ХАРАКТЕРИСТИКА ПРОФИЛЯ ПРОВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ У БОЛЬНЫХ С РАЗЛИЧНЫМИ ФЕНОТИПАМИ АЛЬФА-1-АНТИТРИПСИНА | Первакова
1. Шевченко О.П. Белки острой фазы воспаления. Лаборатория, 1996, № 1. С. 10-17. [Shevchenko O.P. Acute phase proteins. Laboratoriya = Laboratory, 1996, no. 1, pp. 10-17. (In Russ.])
2. Stoller J.K., Lacbawan F.L., Aboussouan L.S. Alpha-1 antitrypsin deficiency. Gene reviews, 2006.
3. Lockett A.D., van Demark M., Gu Y., Schweitzer K.S., Sigua N., Kamocki K., Fijalkowska I., Garrison J., Fisher A.J., Serban K., Wise R.A., Flotte T.R., Mueller C., Presson R.G.Jr., Petrache H.I., Tuder R.M., Petrache I. Effect of cigarette smoke exposure and structural modifications on the alpha-1 Antitrypsin interaction with caspases. Mol. Med., 2012, Vol. 18, pp. 445-454.
4. Feng Y., Xu J., Zhou Q., Wang R., Liu N., Wu Y., Yuan H., Che H. Alpha-1 Antitrypsin prevents the development of preeclampsia through suppression of oxidative stress. Front Physiol., 2016, Vol. 7, p. 176.
5. Stockley R.A. The multiple facets of alpha-1-antitrypsin. Ann. Transl. Med., 2015, Vol. 3, no. 10, p. 130.
6. Zampronio A.R., Soares D.M., Souza G.E. Central mediators involved in the febrile response: effects of antipyretic drugs. Temperature (Austin), 2015, Vol. 2, no. 4, pp. 506-521.
7. Koo J.B., Han J.S. Cigarette smoke extract-induced interleukin-6 expression is regulated by phospholipase D1 in human bronchial epithelial cells. J. Toxicol. Sci., 2016, Vol. 41, no. 1, pp. 77-89.
J. Toxicol. Sci., 2016, Vol. 41, no. 1, pp. 77-89.
8. de Carvalho F.O., Felipe F.A., de Melo Costa A.C., Teixeira L.G., Silva É.R., Nunes P.S., Shanmugam S., de Lucca Junior W., Quintans J.S., de Souza Araújo A.A. Inflammatory mediators and oxidative stress in animals subjected to smoke inhalation: a systematic review. Lung, 2016.
9. Ansarin K., Rashidi F., Namdar H., Ghaffari M., Sharifi A. Echocardiographic evaluation of the relationship between inflammatory factors (IL6, TNFalpha, hs-CRP) and secondary pulmonary hypertension in patients with COPD. A Cross Sectional Study. Pneumologia, 2015, Vol. 64, no. 3, pp. 31-35.
10. Chen Z., Shao X., Dou X., Zhang X., Wang Y., Zhu C., Hao C., Fan M., Ji W., Yan Y. Role of the mycoplasma pneumoniae/interleukin-8/neutrophil axis in the pathogenesis of pneumonia. PLoS One, 2016, Vol. 11, no. 1, p. e0146377.
11. Majak P. Tumor necrosis factor alpha as an asthma biomarker in early childhood. Pneumonol. Alergol. Pol., 2016, Vol. 84, no. 3, pp. 143-144.
12. Cosmi L., Liotta F., Annunziato F. Th27 regulating lower airway disease. Curr Opin Allergy Clin. Immunol., 2016, Vol. 16, no. 1, pp. 1-6.
13. Miossec P., Korn T., Kuchroo V.K. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med., 2009, Vol. 361, no. 9, pp. 888-898.
14. Ortiz G, Salica J.P., Chuluyan E.H., Gallo J.E. Diabetic retinopathy: could the alpha-1 antitrypsin be a therapeutic option? Biol. Res., 2014, Vol. 47, p. 58.
15. Lewis E.C., Shapiro L., Bowers O.J., Dinarello C.A. Alpha1-antitrypsin monotherapy prolongs islet allograſt survival in mice. Proc. Natl. Acad. Sci. USA, 2005, Vol. 102, no. 34, pp. 12153-12158.
Lewis E.C., Shapiro L., Bowers O.J., Dinarello C.A. Alpha1-antitrypsin monotherapy prolongs islet allograſt survival in mice. Proc. Natl. Acad. Sci. USA, 2005, Vol. 102, no. 34, pp. 12153-12158.
16. Subramaniyam D., Virtala R., Pawłowski K., Clausen I.G., Warkentin S., Stevens T., Janciauskiene S. TNF-alpha-induced self expression in human lung endothelial cells is inhibited by native and oxidized alpha1-antitrypsin. Int. J. Biochem. Cell Biol., 2008, Vol. 40, no. 2, pp. 258-271.
17. Bergin D.A., Reeves E.P., Hurley K., Wolfe R., Jameel R., Fitzgerald S., McElvaney N.G. The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci. Transl. Med., 2014, Vol. 6, no. 217, p. 217ra1.
18. Pott G.B., Chan E.D., Dinarello C.A., Shapiro L. Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J. Leukoc. Biol., 2009, Vol. 85, no. 5, pp. 886-895.
19. Subramanian S., Shahaf G., Ozeri E., Miller L.M., Vandenbark A.A., Lewis E.C., Offner H. Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab. Brain Dis., 2011, Vol. 26, no. 2, pp. 107-113.
20. Stone H., McNab G., Wood A.M., Stockley R.A., Sapey E. Variability of sputum inflammatory mediators in COPD and alpha1-antitrypsin deficiency. Eur. Respir. J., 2012, Vol. 40, no. 3, pp. 561-569.
21. Seixas S., Garcia O., Trovoada M. J., Santos M.T., Amorim A., Rocha J. Patterns of haplotype diversity within the serpin gene cluster at 14q32.1: insights into the natural history of the alpha1-antitrypsin polymorphism. Hum. Genet., 2001, Vol. 108, no. 1, pp. 20-30.
J., Santos M.T., Amorim A., Rocha J. Patterns of haplotype diversity within the serpin gene cluster at 14q32.1: insights into the natural history of the alpha1-antitrypsin polymorphism. Hum. Genet., 2001, Vol. 108, no. 1, pp. 20-30.
22. Keren D.F. Protein electrophoresis in clinical diagnosis. Ed. A. London, 2003, pp. 71-77.
23. Salahuddin P. Genetic variants of alpha1-antitrypsin. Curr. Protein Pept. Sci., 2010, Vol. 11, no. 2, pp. 101-117.
24. Olfert I.M., Malek M.H., Eagan T.M., Wagner H., Wagner P.D. Inflammatory cytokine response to exercise in alpha-1-antitrypsin deficient COPD patients ‘on’ or ‘off’ augmentation therapy. BMC Pulm. Med., 2014, Vol. 14, p. 106.
25. Pervakova M.Yu., Emanuel V.L., Titova O.N., Lapin S.V., Mazurov V.I., Belyaeva I.B., Chudinov A.L., Blinova T.V., Surkova E.A. The diagnostic value of alpha-1-antitrypsin phenotype in Patients with granulomatosis with polyangiitis. International Journal of Rheumatology, 2016, pp. 1-5.
26. Pervakova M.Y., Emanuel V.L., Surkova E.A., Mazing A.V., Lapin S.V., Kovaleva I.S., Sysoeva S.N. The comparison of techniques of electrophoresis, immune turbidynamic measurement and phenotyping of alpha-1-antitrypsin for diagnostic of alpha-1-antitrypsin insufficiency. Clin. Lab. Diagn., 2015, Vol. 10, pp. 28-32.
27. Mahr A.D., Edberg J.C., Stone J.H., Hoffman G.S., St Clair E.W., Specks U., Dellaripa P.F., Seo P., Spiera R.F., Rouhani F.N., Brantly M.L., Merkel P.A. Alpha(1)-antitrypsin deficiency-related alleles Z and S and the risk of Wegener’s granulomatosis.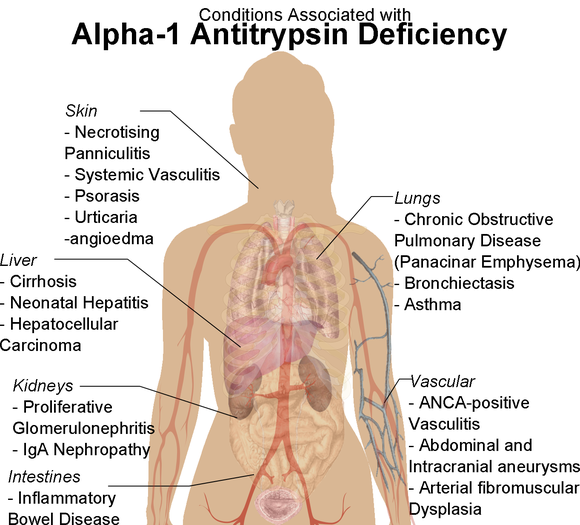 Arthritis Rheum., 2010, Vol. 62, no. 12, pp. 3760-3767.
Arthritis Rheum., 2010, Vol. 62, no. 12, pp. 3760-3767.
28. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med., 2003, Vol. 168, no. 7, pp. 818-900.
29. Duan M.C., Zhang J.Q., Liang Y., Liu G.N., Xiao J., Tang H.J., Liang Y. Infiltration of IL-17-producing T cells and Treg cells in a mouse model of smoke-induced emphysema. Inflammation, 2016.
30. Singh D. Chronic Obstructive Pulmonary Disease, Neutrophils and Bacterial Infection: A complex web involving IL-17 and IL-22 unravels. EBioMedicine, 2015, Vol. 2, no. 11, pp. 1580-1581.
31. Chen Q.R., Wang L.F., Xia S.S., Zhang Y.M., Xu J.N., Li H., Ding Y.Z. Role of interleukin-17A in early graſt rejection aſter orthotopic lung transplantation in mice. J. Thorac. Dis., 2016, Vol. 8, no. 6, pp. 1069-1079.
32. Bergin D.A., Reeves E.P., Meleady P., Henry M., McElvaney O.J., Carroll T.P., Condron C., Chotirmall S.H., Clynes M., O’Neill S.J., McElvaney N.G. Alpha-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin. Invest., 2010, Vol. 120, no. 12, pp. 4236-4250.
33. Aldonyte R., Eriksson S., Piitulainen E., Wallmark A., Janciauskiene S. Analysis of systemic biomarkers in COPD patients. COPD, 2004, Vol. 1, no. 2, pp. 155-164.
34. Honda K., Wada H., Nakamura M., Nakamoto K., Inui T., Sada M., Koide T. , Takata S., Yokoyama T., Saraya T., Kurai D., Ishii H., Goto H., Takizawa H. IL-17A synergistically stimulates TNF-alpha-induced IL-8 production in human airway epithelial cells: A potential role in amplifying airway inflammation. Exp. Lung. Res., 2016, Vol. 42, no. 4, pp. 205-216.
, Takata S., Yokoyama T., Saraya T., Kurai D., Ishii H., Goto H., Takizawa H. IL-17A synergistically stimulates TNF-alpha-induced IL-8 production in human airway epithelial cells: A potential role in amplifying airway inflammation. Exp. Lung. Res., 2016, Vol. 42, no. 4, pp. 205-216.
Недостаточность альфа-1-антитрипсина: диагностика и лечение (обзор литературы) | Ларшина
1. Torres-Durán M., Lopez-Campos J.L., Barrecheguren M., et al. Alpha-1 antitrypsin deficiency: outstanding questions and future directions. Orphanet J Rare Dis. 2018;13(1):114.
2. Crossley D., Stockley R., Sapey E. Alpha-1 Antitrypsin Deficiency and Accelerated Aging: A New Model for an Old Disease? Drugs Aging. 2019;36(9):823-840.
3. Cox D.W., Billingsley G.D., Mansfield T. DNA restriction-site polymorphisms associated with the alpha 1-antitrypsin gene. Am J Hum Genet. 1987;41(5):891-906.
4. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818-900.
5. de Serres F., Blanco I. Role of alpha-1 antitrypsin in human health and disease. J Intern Med. 2014;276(4):311-35.
6. Kalfopoulos M., Wetmore K., ElMallah M.K. Pathophysiology of Alpha-1 Antitrypsin Lung Disease. Methods Mol Biol. 2017;1639:9-19.
7. de Serres F.J., Blanco I. Prevalence of α1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review.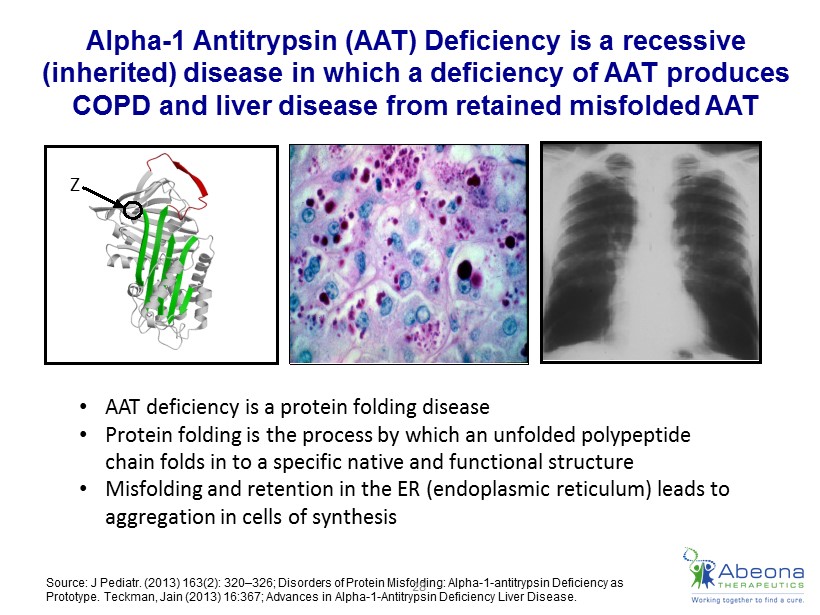 Ther Adv Respir Dis. 2012;6(5):277-95.
Ther Adv Respir Dis. 2012;6(5):277-95.
8. Brantly M., Campos M., Davis A.M., et al. Detection of alpha-1 antitrypsin deficiency: the past, present and future. Orphanet J Rare Dis. 2020;15(1):96.
9. Stoller J.K., Aboussouan L.S. A review of α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2012;185(3):246-59.
10. Мельник С.И., Власов Н.Н., Пиневская М.В, и др. Дефицит альфа-1-антитрипсина у детей: описание серии случаев. Вопросы современной педиатрии. 2016;15(6):619-624.
11. Teckman J.H., Blomenkamp K.S. Pathophysiology of Alpha-1 Antitrypsin Deficiency Liver Disease. Methods Mol Biol. 2017;1639:1-8.
12. Al-Jameil N., Hassan A.A., Hassanato R., et al. The prevalence of PI*S and PI*Z SERPINA1 alleles in healthy individuals and COPD patients in Saudi Arabia: A case-control study. Medicine (Baltimore). 2017;96(42):e8320.
13. Craig T.J., Henao M.P. Advances in managing COPD related to α(1) -antitrypsin deficiency: An under-recognized genetic disorder. Allergy. 2018;73(11):2110-2121.
14. Silva D., Oliveira M.J., Guimarães M., et al. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: Three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med. 2016;116:8-18.
15. Mitchell E.L., Khan Z. Liver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future Directions. Curr Pathobiol Rep.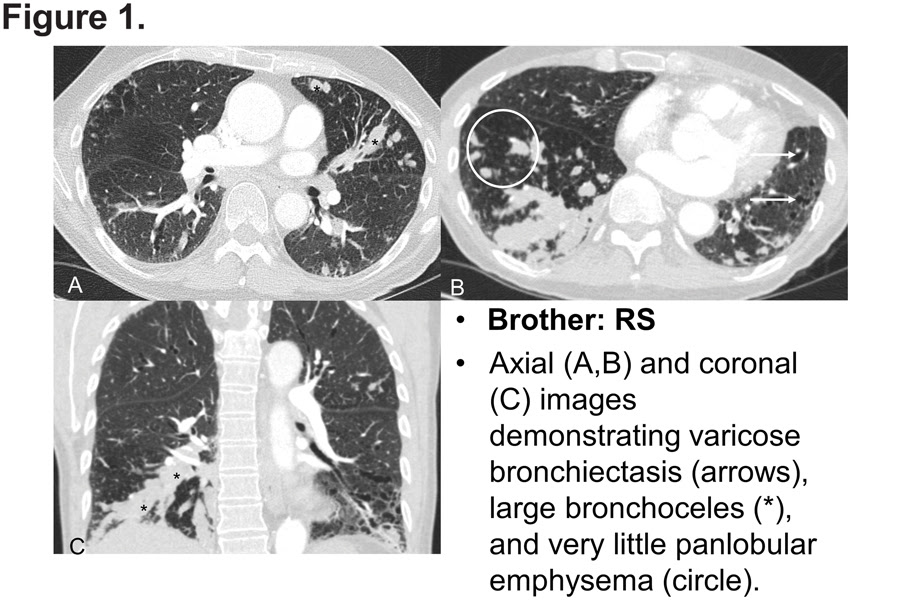 2017;5(3):243-252.
2017;5(3):243-252.
16. McGee D., Schwarz L., McClure R., et al. Is PiSS Alpha-1 Antitrypsin Deficiency Associated with Disease? Pulm Med. 2010;2010:570679.
17. Matamala N., Martínez M.T., Lara B., et al. Alternative transcripts of the SERPINA1 gene in alpha-1 antitrypsin deficiency. J Transl Med. 2015;13:211.
18. Crowther D.C., Belorgey D. Miranda E., et al. Practical genetics: alpha-1-antitrypsin deficiency and the serpinopathies. Eur J Hum Genet. 2004;12(3):167-72.
19. Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397-415.
20. Lace B., Sveger T., Krams A., et al. Age of SERPINA1 gene PI Z mutation: Swedish and Latvian population analysis. Ann Hum Genet. 2008;72(Pt 3):300-4.
21. Ferrarotti I., Ottaviani S., De Silvestri A., et al. Update on α(1)-antitrypsin deficiency. Breathe (Sheff). 2018;14(2):e17-e24.
22. Miskoff J.A., Khan B., Chaudhri M., et al. Identifying Alpha-1 Antitrypsin Deficiency Based on Computed Tomography Evidence of Emphysema. Cureus. 2019;11(1):e3971.
23. DeMeo D.L., Silverman E.K. Alpha1-antitrypsin deficiency. 2: genetic aspects of alpha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax. 2004;59(3):259-64.
24. Chiuchiolo M.J., Crystal R.G. Gene Therapy for Alpha-1 Antitrypsin Deficiency Lung Disease.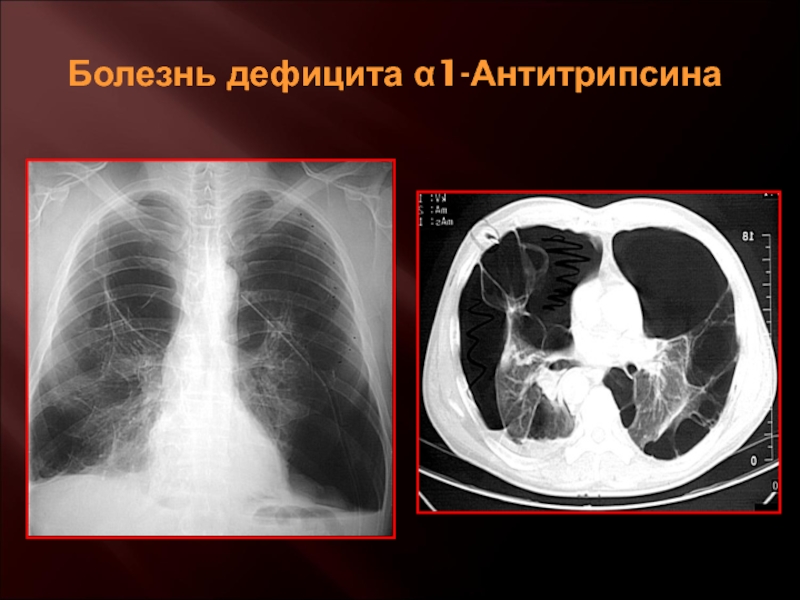 Ann Am Thorac Soc. 2016;13 Suppl 4(Suppl 4):S352-69.
Ann Am Thorac Soc. 2016;13 Suppl 4(Suppl 4):S352-69.
25. Mulgrew A.T., Taggart C.C., McElvaney N.G. Alpha-1-antitrypsin deficiency: current concepts. Lung. 2007;185(4):191-201.
26. Luisetti M., Seersholm N. Alpha1-antitrypsin deficiency. 1: epidemiology of alpha1-antitrypsin deficiency. Thorax. 2004;59(2):164-9.
27. Жигальцова-Кучинская О.А., Сивицкая Л.Н., Даниленко Н.Г., и др. Дефицит альфа-1-антитрипсина: генетические основы, эпидемилогия, значение в развитии бронхо-легочной патологии. Вестник ВГМУ. 2015;14(6):39-52.
28. Blanco I., Bueno P., Diego I., et al. Alpha-1 antitrypsin Pi*SZ genotype: estimated prevalence and number of SZ subjects worldwide. Int J Chron Obstruct Pulmon Dis. 2017;12:1683-1694.
29. Greulich T., Ottaviani S., Bals R., et al. Alpha1-antitrypsin deficiency — diagnostic testing and disease awareness in Germany and Italy. Respir Med. 2013;107(9):1400-8.
30. Miravitlles M., Dirksen A., Ferrarotti I., et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. Eur Respir J. 2017;50(5).
31. Rigobello C., Baraldo S., Tinè M., et al. Exome Sequencing Reveals Immune Genes as Susceptibility Modifiers in Individuals with α(1)-Antitrypsin Deficiency. Sci Rep. 2019;9(1):13088.
32. Brantly M.L., Lascano J.E., Shahmohammadi A. Intravenous Alpha-1 Antitrypsin Therapy for Alpha-1 Antitrypsin Deficiency: The Current State of the Evidence. Chronic Obstr Pulm Dis. 2018;6(1): 100-114.
Chronic Obstr Pulm Dis. 2018;6(1): 100-114.
33. Stiles K.M., Sondhi D., Kaminsky S.M., et al. Intrapleural Gene Therapy for Alpha-1 Antitrypsin Deficiency-Related Lung Disease. Chronic Obstr Pulm Dis. 2018;5(4):244-257.
34. Kaushal S., Annamali M., Blomenkamp K., et al. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med (Maywood). 2010;235(6):700-9.
35. Hidvegi T., Ewing M., Hale P., et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229-32.
36. Wang Y., Cobanoglu M.C., Li J., et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded α1-antitrypsin Z. PLoS One. 2019;14(1):e0209748.
37. Flotte T.R., Mueller C. Gene therapy for alpha-1 antitrypsin deficiency. Hum Mol Genet. 2011;20(R1):R87-92.
ИДЦ — Иркутский диагностический центр
Альфа-1-антитрипсин (сыворотка крови) (количественный)
Описание услуги
Код услуги:
2Ж1010
Готовность результатов:
по графику, ближайшие даты: 23. 03.21, 26.03.21, 30.03.21, 02.04.21, результат на следующий рабочий день
03.21, 26.03.21, 30.03.21, 02.04.21, результат на следующий рабочий день
Альфа-1-антитрипсин – положительный реактант острой фазы. Является
основным компонентом α-1- полосы при электрофорезе белков сыворотки крови
и наибольшим по концентрации ингибитором протеаз в крови. Важнейшая
физиологическая роль α-1-Антитрипсина состоит в торможении протеаз,имеющих
в своём составе аминокислоту серин, выделяющихся из лейкоцитов при
фагоцитозе. Норма: 83 – 199 мг/дл в сыворотке крови.
Клинико-диагностическое значение: Повышение концентрации: Воспаление
(инфекции, ревматические заболевания), некроз ткани, злокачественный рост,
травма (включая хирургическую), эстрогены, беременность. Воспаление
паренхиматозных клеток печени часто сочетается с повышением
α-1-Антитрипсина при отсутствии других элементов реакции острой фазы.
Снижение концентрации: Респираторный дистресс-синдром у детей, синдром
тяжёлого гепатита у новорождённых, тяжёлое поражение поджелудочной железы
и печени, нефротический синдром. Врождённый дефицит альфа-1-антитрипсин
часто сочетается с заболеваниями печени, особенно в детском возрасте
(неонатальный гепатит, инфантильный цирроз), хронические заболевания
лёгких у взрослых, криптогенный цирроз, гепатомы.
Для
сдачи биоматериала (кровь из вены) обратиться в регистратуру
клинико-диагностической лаборатории на 2 этаже. Перед сдачей анализов
рекомендуется воздержаться от приема пищи в течение не менее 3 часов. Можно
пить воду без газа.
Респикам инструкция по применению: показания, противопоказания, побочное действие – описание Respikam Раствор для инфузий (39356)
Респикам представляет собой стерильный, готовый к употреблению раствор, содержащий очищенный ингибитор альфа1-протеиназы (другое название — альфа1-антитрипсин (ААТ)).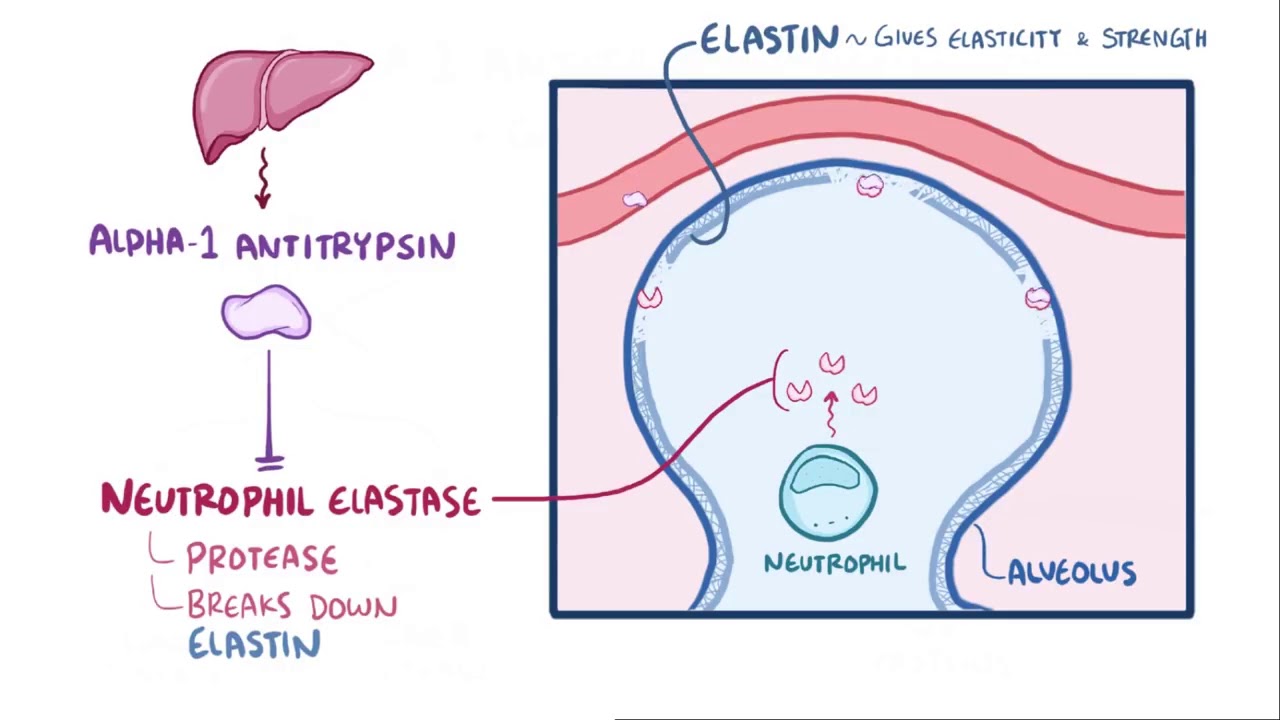
Респикам изготавливается из плазмы крови здоровых людей с помощью ионообменной хроматографии. С целью предотвращения риска переноса возбудителей инфекционных заболеваний, каждая порция плазмы человека, используемая для производства препарата, проверяется на содержание вируса иммунодефицита человека (ВИЧ), антител к гепатиту С (анти-ВСГ), поверхностного антигена гепатита В (HBsAg), а также проводится серологическая реакция на определение возбудителя сифилиса.
Респикам оказывает терапевтический эффект при дефиците ААТ — хроническом наследственном, аутосомно-рецессивном заболевании, характеризующимся развитием тяжелой, медленно прогрессирующей панацинозной эмфиземы лёгких, чаще всего наблюдающейся на третьем-четвертом десятилетии жизни. Предполагается, что развитие этого патологического состояния обусловлено дисбалансом между эластазой (ферментом, способным разрушать эластин тканей и продуцируемым преимущественно нейтрофилами, в нижних отделах дыхательных путей) и ААТ — главным ингибитором эластазы. В результате длительного воздействия эластазы разрушается эластин тканей и развивается эмфизема легких. Примерно у 10 % новорожденных с ААТ-дефицитом наблюдается гигантоклеточный гепатит с холестатической желтухой. У взрослых дефицит ААТ может сопровождаться циррозом печени.
Дефицит ААТ является генетической патологией в локусе гена PI, который был найден в хромосоме 14q32.1. Концентрация ААТ в плазме крови, связанная с PI — типом гомозиготного наследования, обычно находится в диапазоне 12-18 % от нормы, а гетерозиготного наследования — составляет примерно 35 % от нормы. Множество исследований эффективности заместительной терапии основывались на поддержании концентрации AAT в плазме крови человека >80 мг/дл. Такая концентрация оказалась достаточной для предотвращения развития эмфиземы легких у таких больных.
Детский дефицит антитрипсина альфа-1 | Детский Питтсбург
Что такое дефицит антитрипсина альфа-1?
Дефицит антитрипсина альфа-1 — распространенное наследственное заболевание, характеризующееся пониженным уровнем антитрипсина альфа-1.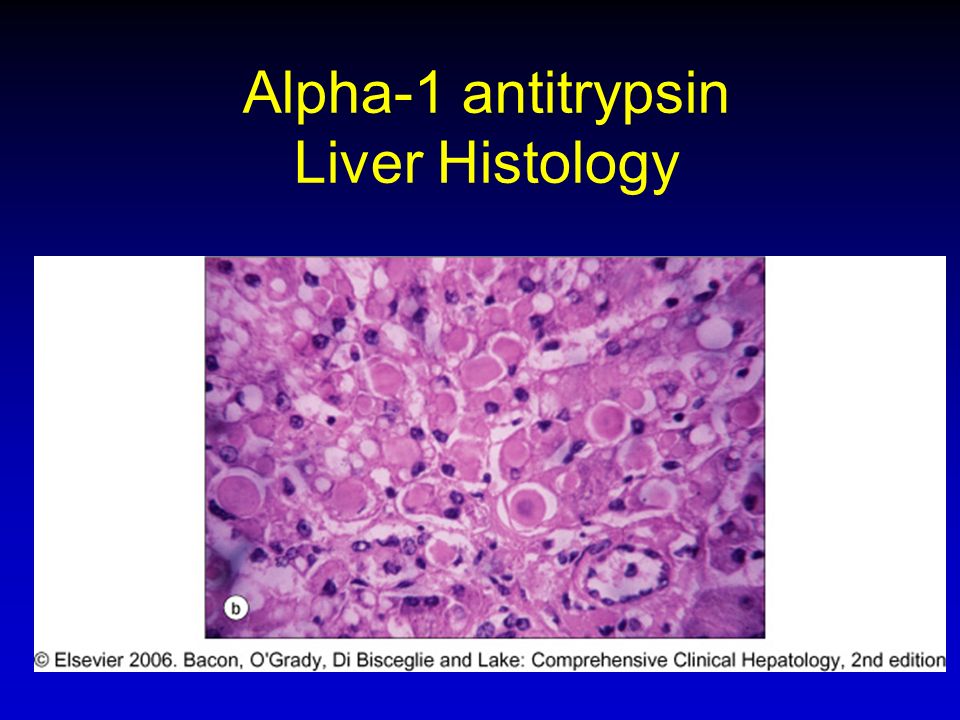 Альфа-1-антитрипсин — это белок крови, который вырабатывается в печени; его основная функция — защищать легкие, чтобы они могли нормально работать.
Альфа-1-антитрипсин — это белок крови, который вырабатывается в печени; его основная функция — защищать легкие, чтобы они могли нормально работать.
После того, как печень попадает в кровоток, альфа-1 диффундирует в ткани и защищает ткани от переваривания ферментами, выделяемыми воспалительными клетками, такими как белые кровяные тельца.Белые кровяные тельца в организме защищают от инфекции; они также выделяют фермент, называемый эластазой нейтрофилов. Эластаза нейтрофилов служит полезной цели в легочной ткани: она переваривает поврежденные или стареющие клетки и бактерии. Это способствует заживлению или росту новых, более здоровых клеток. Однако эластаза нейтрофилов не знает, когда прекратить переваривание; оставленный сам по себе, он скоро перейдет к атаке на здоровые ткани. В нормальных легких альфа-1-антитрипсин защищает легочную ткань, улавливая и разрушая эластазу нейтрофилов до того, как она переживет свою полезность и вызовет повреждение.
Определенные генные мутации могут вызвать аномальную форму антитрипсина альфа-1, которая застревает в печени и не может попасть в кровоток. Фактическое количество продуцируемого альфа-1 может быть близко к норме, но печень не выделяет достаточного количества в кровоток. Кроме того, аномальный альфа-1 часто является дефектным, так что высвободившееся небольшое количество не может вовремя эффективно «улавливать» эластазу нейтрофилов. Различные мутации вызывают разные разновидности этого расстройства, некоторые из которых более серьезны, чем другие.
Когда в легких недостаточно альфа-1-антитрипсина, эластаза нейтрофилов может разрушить ткань. Ткань легких особенно уязвима, поскольку легкие постоянно подвергаются воздействию токсинов из окружающей среды. Это приводит к заболеванию легких (чаще всего эмфиземе), а иногда и к заболеванию печени.
Детская болезнь печени при дефиците антитрипсина альфа-1
Патофизиология (функциональные изменения, связанные с заболеванием или травмой или в результате) заболевания печени при дефиците альфа-1-антитрипсина менее изучена, чем патофизиология связанного с ней заболевания легких.
Обычно считается, что заболевание печени, связанное с дефицитом альфа-1-антитрипсина, вызвано не дефицитом альфа-1 в кровотоке, а чрезмерным количеством альфа-1, застрявшим в клетках печени.
Повреждение печени встречается примерно у 10% младенцев, рожденных с тяжелой формой дефицита альфа-1-антитрипсина. У некоторых пациентов с дефицитом альфа-1-антитрипсина наблюдается цирроз печени. Это влияет на очень маленьких детей с дефицитом альфа-1-антитрипсина, а также у 12-15 процентов взрослых пациентов с дефицитом альфа-1-антитрипсина.
Симптомы дефицита антитрипсина альфа-1
Дефицит антитрипсина альфа-1 у детей или младенцев влияет на их печень, могут наблюдаться некоторые или все следующие симптомы:
- Желтуха, пожелтение кожи и склер (белков глаз) при рождении
- Стул с неприятным запахом или бледный, почти белый
- Цирроз
- Плохая прибавка в весе
- Медленное питание
- Рвота, тошнота или рефлюкс
- Проблемы с уходом
- Зуд
- Увеличенная селезенка, брюшная полость
- Потеря аппетита
- Недостаток энергии
Диагностика дефицита антитрипсина альфа-1
Диагноз болезни печени, вызванной недостаточностью альфа-1-антитрипсина, может быть установлен с помощью простого анализа крови.Если симптомы пациента указывают на более серьезную дисфункцию печени, может быть рекомендована биопсия печени.
Лечение дефицита антитрипсина альфа-1
Не существует специального лечения заболеваний печени, связанных с дефицитом антитрипсина альфа-1; Клиническая помощь в первую очередь лечит симптомы по мере их появления и направлена на предотвращение возможных осложнений. Избегание веществ, о которых известно, что они способны вызвать повреждение печени, а также правильное питание — важная часть лечения.
Для тех, кто страдает тяжелым прогрессирующим поражением печени, трансплантация печени в настоящее время является единственным вариантом, позволяющим выжить.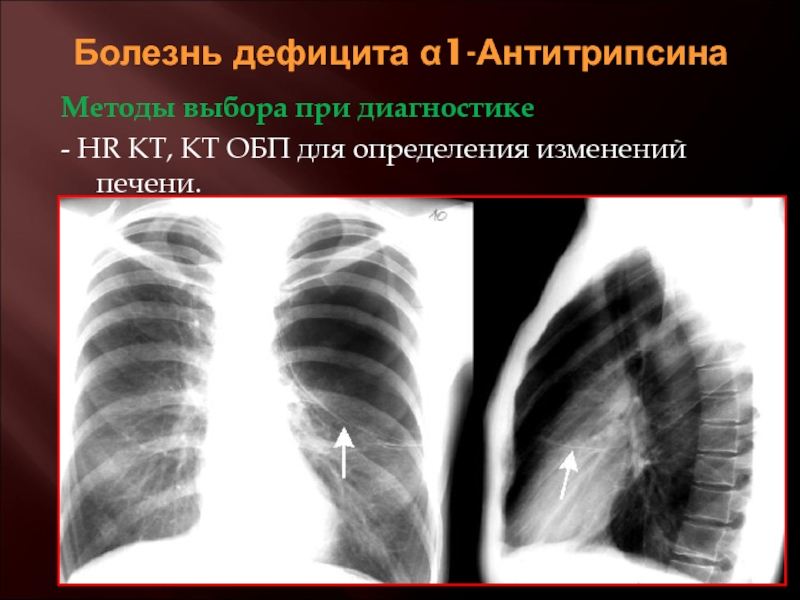 К счастью, исследования показывают, что только около 10% людей с дефицитом альфа-1-антитрипсина имеют настолько серьезное повреждение печени, что требуется трансплантация печени. Обычно трансплантация печени проводится детям с инфантильным гепатитом, связанным с тяжелым дефицитом альфа-1-антитрипсина. У этих пациентов трансплантат полностью заменяет клетки печени, которые продуцируют мутировавший альфа-1, тем самым корректируя белковую аномалию.
К счастью, исследования показывают, что только около 10% людей с дефицитом альфа-1-антитрипсина имеют настолько серьезное повреждение печени, что требуется трансплантация печени. Обычно трансплантация печени проводится детям с инфантильным гепатитом, связанным с тяжелым дефицитом альфа-1-антитрипсина. У этих пациентов трансплантат полностью заменяет клетки печени, которые продуцируют мутировавший альфа-1, тем самым корректируя белковую аномалию.
Трансплантация печени, связанная с живыми, становится все более популярной по мере совершенствования медицинских знаний и методов. Обычный донор — родитель; однако могут быть рассмотрены родные братья или сестры 18 лет и старше, дедушка или бабушка или другой член семьи.
Для сохранения функции легких каждые одну или две недели вводят внутривенное замещение человеческого альфа-1, полученного из крови человека (Prolastin®). Inhaled Alpha-1 в настоящее время проходит испытания.
Генная терапия в настоящее время также изучается.При генной терапии нормальный ген альфа-1 помещается в неинфекционный вирус. Когда вирус вводится пациенту (путем ингаляции или другими способами), он инфицирует клетки ткани, вызывая выработку нормального альфа-1 содержащимся в нем нормальным геном. Пока что этим методом достигнут лишь минимальный успех, но он очень многообещающий. Однако эта терапия будет полезна только при лечении легочного заболевания, связанного с дефицитом альфа-1-антитрипсина, поскольку заболевание печени, связанное с дефицитом альфа-1-антитрипсина, не связано со сниженным уровнем циркулирующего альфа-1.
Узнайте о других заболеваниях печени.
Дефицит антитрипсина альфа-1 | Детская больница Филадельфии
Дефицит антитрипсина альфа-1 — это наследственное генетическое заболевание, при котором ткани печени или легких могут быть повреждены, что мешает им работать должным образом. Первые симптомы дефицита альфа-1-антитрипсина обычно возникают в возрасте от 20 до 50 лет, но некоторые младенцы или дети также могут быть поражены этим заболеванием. Примерно один из 3000–5000 человек страдает дефицитом альфа-1-антитрипсина.
Примерно один из 3000–5000 человек страдает дефицитом альфа-1-антитрипсина.
Дефицит антитрипсина альфа-1 является результатом генетической мутации, которая заставляет организм вырабатывать пониженные уровни или аномальную форму белка, называемого антитрипсином альфа-1. Альфа-1-антитрипсин обычно защищает организм от мощного фермента, вырабатываемого лейкоцитами. У пациентов с дефицитом альфа-1-антитрипсина этот фермент не контролируется должным образом, и он может повредить легкие.
Лица с дефицитом альфа-1-антитрипсина обычно испытывают респираторные симптомы, включая одышку после небольшой активности, снижение способности выполнять упражнения, хрипы, кашель, повторяющиеся респираторные инфекции, утомляемость и учащенное сердцебиение при стоянии.В конце концов, у пациентов может развиться эмфизема — состояние, при котором маленькие воздушные мешочки в легких (так называемые альвеолы) повреждаются. У пациентов с эмфиземой возникает затрудненное дыхание, отрывистый кашель и может развиться грудная клетка в форме бочонка. Респираторные симптомы развиваются у пациентов в результате повреждения тканей легких.
У некоторых пациентов с дефицитом альфа-1-антитрипсина может развиться заболевание печени и появиться такие симптомы, как вздутие живота, опухшие ступни или ноги, а также пожелтение кожи и белков глаз.
В настоящее время не существует известного способа предотвратить дефицит альфа-1-антитрипсина. У пациентов с дефицитом альфа-1-антитрипсина может быть сокращена продолжительность жизни в зависимости от тяжести симптомов.
Известно, что дефицит антитрипсина альфа-1 является результатом наследования мутированной формы гена SERPINA1. SERPINA1 производит белок, который обычно защищает организм от мощного фермента, вырабатываемого лейкоцитами, под названием эластаза нейтрофилов. У людей обычно есть две копии гена SERPINA1.Человек с одной мутированной копией SERPINA1 имеет низкую вероятность развития дефицита альфа-1-антитрипсина, но человек, несущий две мутированные копии SERPINA1, скорее всего, разовьется заболеванием. Следовательно, пары, в которых каждый родитель имеет мутировавший ген SERPINA1, подвергаются большему риску рождения ребенка с дефицитом антитрипсина альфа-1, поскольку каждый родитель может передать ребенку мутированную копию SERPINA1.
Следовательно, пары, в которых каждый родитель имеет мутировавший ген SERPINA1, подвергаются большему риску рождения ребенка с дефицитом антитрипсина альфа-1, поскольку каждый родитель может передать ребенку мутированную копию SERPINA1.
Некоторые мутации в гене SERPINA1 не так серьезны, как другие, и лишь незначительно снижают способность гена функционировать должным образом.Лица, несущие эти менее тяжелые мутировавшие формы SERPINA1, имеют более низкий риск передачи болезни детям.
Дефицит антитрипсина альфа-1 является результатом мутации в гене SERPINA1. Эта мутация заставляет организм вырабатывать пониженные уровни или аномальную форму белка, называемого альфа-1-антритрипсином.
Альфа-1-антитрипсин обычно защищает организм от мощного фермента, вырабатываемого лейкоцитами, называемого эластазой нейтрофилов. Нормальная функция эластазы нейтрофилов — бороться с инфекциями.Однако у пациентов с дефицитом альфа-1-антитрипсина эластаза нейтрофилов не контролируется должным образом и может повредить легкие.
Если в организме в результате мутации вырабатывается аномальный белок альфа-1-антитрипсина, он может накапливаться в печени, вызывая заболевания печени и желудочно-кишечные (GI) проблемы.
Респираторные проблемы: Лица с дефицитом альфа-1-антитрипсина обычно испытывают респираторные симптомы, включая одышку после легкой активности, снижение способности выполнять упражнения, кашель, хрипы, повторяющиеся респираторные инфекции, утомляемость и учащенное сердцебиение при стоянии.Пациенты также могут срыгивать слизь из дыхательных путей.
Проблемы с печенью: Примерно у 10-15 процентов пациентов с дефицитом альфа-1-антитрипсина развивается заболевание печени с такими симптомами, как вздутие живота, опухшие ступни или ноги, а также пожелтение кожи и белков глаз. Также может возникнуть рубцевание печени, называемое циррозом.
Дефицит антитрипсина альфа-1 может быть трудно диагностировать, потому что нельзя использовать один физический признак или симптом для подтверждения диагноза.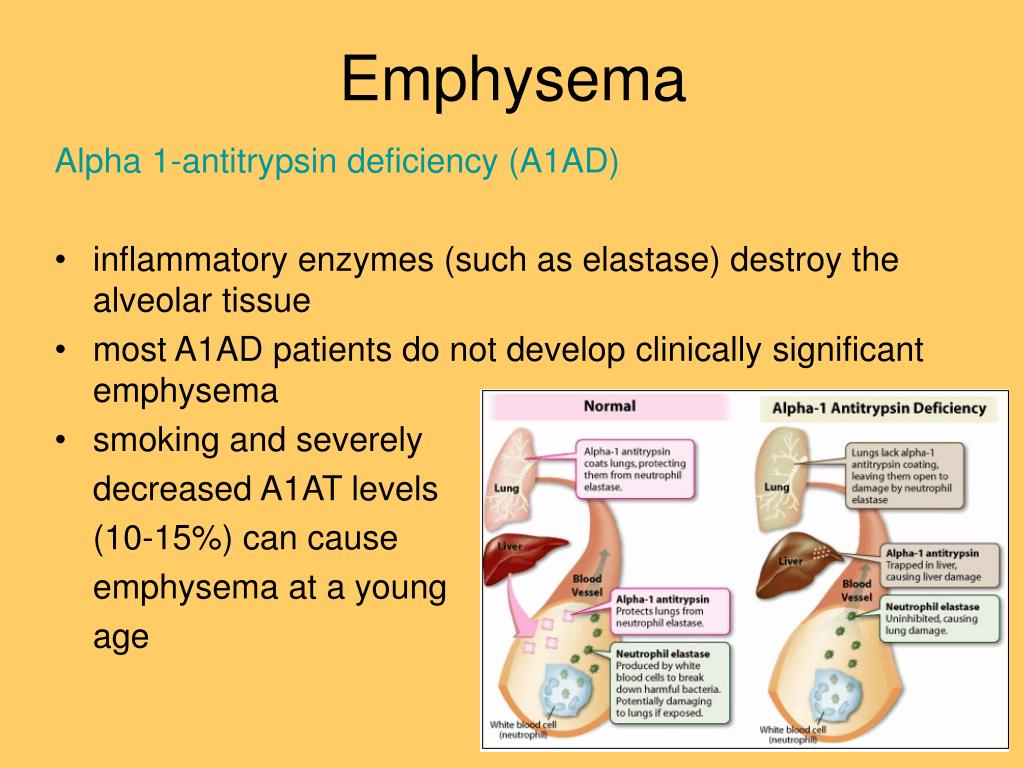 Генетическое тестирование и анализы крови могут дать более убедительный диагноз.
Генетическое тестирование и анализы крови могут дать более убедительный диагноз.
- Клинический осмотр: Клиническое обследование дыхательной системы и грудной клетки с помощью рентгеновских лучей или путем прослушивания аномальных звуков с помощью стетоскопа может помочь выявить респираторные нарушения у пациентов. Проверка различных респираторных симптомов, связанных с заболеванием, может помочь в постановке диагноза. Врачи также могут искать признаки повышенной респираторной работы или обструкции дыхательных путей.
- Генетические тесты: Известно, что мутации в гене SERPINA1 вызывают дефицит антитрипсина альфа-1. Генетические тесты могут использоваться для проверки этих мутаций и диагностики дефицита антитрипсина альфа-1.
- Анализы крови: Можно провести тесты для измерения уровня альфа-1-антитрипсина в крови пациента. Низкий уровень может указывать на то, что у пациента наблюдается дефицит альфа-1-антитрипсина.
- Панникулит: У небольшого числа пациентов с дефицитом альфа-1-антитрипсина может развиться панникулит — состояние, при котором кожа становится твердой и появляются болезненные уплотнения и пятна.
- Эмфизема: у пациентов может развиться эмфизема, состояние, при котором маленькие воздушные мешочки в легких (так называемые альвеолы) повреждаются. Пациенты с эмфиземой испытывают затрудненное дыхание, отрывистый кашель и бочкообразную грудь.
- Проблемы с дыхательными путями: Дефицит антитрипсина альфа-1 может привести к ряду других респираторных проблем, включая хронический бронхит (воспаление дыхательных путей легких) и бронхоэктазы (необратимое расширение некоторых дыхательных путей в легких).У некоторых пациентов может наблюдаться инфекция верхних дыхательных путей.
- Эмфизема и цирроз печени представляют собой опасные для жизни состояния и могут способствовать смерти пациентов с дефицитом альфа-1-антитрипсина. Однако не у всех пациентов с дефицитом альфа-1-антитрипсина развиваются эти тяжелые осложнения.
- Альфа-1-антитрипсин обычно защищает организм от мощного фермента, вырабатываемого лейкоцитами, называемого эластазой нейтрофилов. Нормальная функция эластазы нейтрофилов — бороться с инфекциями.Однако у пациентов с дефицитом альфа-1-антитрипсина эластаза нейтрофилов не контролируется должным образом и может повредить легкие.
- Заболевание печени: Если в результате мутации организм производит аномальный белок альфа-1-антитрипсина, он может накапливаться в печени, вызывая заболевание печени.
В настоящее время не существует известного лекарства от дефицита альфа-1-антитрипсина. Существует ряд методов лечения, помогающих пациентам управлять симптомами этого расстройства.
- Антитрипсиновый белок альфа-1: Дефицит альфа-1-антитрипсина можно лечить с помощью альфа-1-антитрипсинового белка, полученного из крови здоровых людей.Если нормальный белок альфа-1-антитрипсина вводится пациентам внутривенно, это может замедлить течение болезни и предотвратить дальнейшее повреждение легких.
- Трансплантация: Если заболевание печени становится тяжелым, может потребоваться трансплантация печени. Трансплантация легких может потребоваться в тех случаях, когда легкие серьезно повреждены.
- Избегать раздражителей: Пациентам с дефицитом альфа-1-антитрипсина следует избегать курения. Курение может ускорить повреждение легких пациента.Также рекомендуется избегать попадания пыли, паров и других раздражителей окружающей среды.
- Антибиотики: Существуют антибиотики, которые можно использовать для лечения инфекций дыхательных путей.
- Бронходилататоры: Бронходилататоры, которые представляют собой лекарства, которые можно использовать для расширения дыхательных путей, могут помочь пациентам с затрудненным дыханием. Семейство бета-агонистов бронходилататоров, которые вызывают расслабление мышц бронхов, можно использовать для лечения дефицита альфа-1-антитрипсина.Побочные эффекты бета-агонистов бронходилататоров включают нервозность, учащенное сердцебиение и нервное чувство.
Дефицит антитрипсина альфа-1 (AATD) | Симптомы и лечение
Причины дефицита антитрипсина альфа-1
Антитрипсин альфа-1 — это белок, который вырабатывается в печени и затем попадает в кровоток. При AATD организм не вырабатывает этот белок в правильной форме. Нашему организму нужен альфа-1-антитрипсин для защиты тканей организма, особенно легких.Недостаток этого белка в легких приводит к заболеванию легких во взрослом возрасте. В то же время накопление аномального белка в печени приводит к заболеванию печени. Около 10 процентов пациентов с тяжелой формой AATD страдают заболеванием печени, которое в конечном итоге требует трансплантации печени.
AATD является наследственным заболеванием и не возникает, если человек не получает один и тот же дефектный ген от обоих родителей. Если оба родителя несут аномальный ген AATD, это:
- Вероятность того, что их ребенок заболеет, составляет 25 процентов
- 50-процентная вероятность того, что их ребенок получит один аномальный ген от одного из родителей, что означает, что у ребенка не будут проявляться симптомы заболевания, но он является «носителем».
- Вероятность 25%, что их ребенок получит оба нормальных гена, по одному от каждого родителя, не будет затронут
Важно понимать, что тяжесть заболеваний печени и легких, связанных с ДАТД, может сильно различаться.Многие люди с AATD могут не иметь поражения печени или иметь минимальное поражение.
Признаки и симптомы
- Желтуха (или пожелтение кожи и глаз), которая не проходит
- Темная моча или бледный стул
- Кожный зуд
- Увеличенная печень
- Кровотечение
- Накопление жидкости в животе (асцит)
- Проблемы с едой
- Плохой рост или неспособность к развитию
- Повышенный уровень ферментов печени
Другие дети могут не проявлять признаков этого состояния до раннего детства.Первые признаки в этом возрасте могут включать:
- Легко устает
- Потеря аппетита
- Желтуха
- Накопление жидкости в животе (асцит)
- Сильный кожный зуд
- Панникулит (редкая форма кожного заболевания, при котором кожа затвердевает и образуются комочки)
- Повышенный уровень ферментов печени
- Увеличение печени или селезенки
Диагностика дефицита альфа-1-антитрипсина
AATD использует простой анализ крови, который определяет тип альфа-1-антитрипсина, обнаруженного в крови.Этот тест может определить, есть ли у человека AATD или он является носителем. Анализ крови можно сделать вскоре после рождения ребенка, если в семейном анамнезе имеется дефицит. Также доступен тест, чтобы проверить, есть ли это заболевание у ребенка в утробе матери.
Заболевание печени от AATD диагностируется при физикальном обследовании и аномальных изменениях в крови. Во время медицинского осмотра врач может на ощупь определить, что что-то не в порядке. Такие тесты, как УЗИ печени и селезенки и биопсия печени, могут помочь подтвердить диагноз.
Если уровень билирубина (жидкость, вырабатываемая в печени, которая выводит токсины из организма и помогает расщеплять жиры в пище) выше нормы, это может быть признаком AATD. Повышенные уровни определенных ферментов и аномальные соотношения определенных белков также могут указывать на заболевание печени.
Лечение дефицита антитрипсина альфа-1
Дефицит антитрипсина альфа-1 по-разному влияет на детей. Один ребенок может не показывать никаких признаков заболевания печени, а другой ребенок может серьезно пострадать.В целом, лишь у небольшого процента детей развивается заболевание печени, связанное с ДААТ.
Нет лекарства от AATD. Если развивается тяжелое заболевание печени, трансплантация печени в настоящее время является единственным вариантом, доступным для выживания. Цель лечения — облегчить симптомы.
- При сильном зуде можно дать лекарство.
- Мочегонные средства (лекарства, которые помогают удалить излишки жидкости из организма) могут использоваться для уменьшения скопления жидкости в животе.
- Здоровое питание и витаминные добавки могут обеспечить необходимые питательные вещества и улучшить общее состояние здоровья.
- Витаминные / пищевые добавки могут повысить эффективность процесса пищеварения и повысить уровень энергии.
Трансплантация приводит к излечению от AATD. Если трансплантат — лучший вариант лечения, врач и другие члены бригады по уходу за пациентом сосредотачиваются на предотвращении осложнений. Они будут лечить симптомы, пока ваш ребенок ждет пожертвования печени. Пересадка печени полностью заменяет аномальные клетки печени, которые вызывают аномальный дефицит, и исправляет белковые аномалии.
Для ребенка с AATD крайне важно избегать курения или воздействия вторичного табачного дыма.
Долгосрочная перспектива
Есть хорошие шансы избежать заболевания печени, поскольку только около 10 процентов детей с дефицитом альфа-1-антитрипсина заболевают серьезным заболеванием печени. Трансплантация печени оказалась эффективной в обращении симптомов печеночной недостаточности из-за дефицита альфа-1-антитрипсина.
Сопутствующие ресурсы
Дефицит антитрипсина альфа-1 | Архив детских болезней
α-1-антитрипсин синтезируется в печени и защищает альвеолярные ткани легких от разрушения эластазой нейтрофилов.Дефицит антитрипсина 1α-1 является распространенным аутосомно-рецессивным заболеванием (от 1: 1600 до 1: 1800), при котором заболевание печени возникает в результате удержания аномального полимеризованного антитрипсина α-1 в эндоплазматическом ретикулуме гепатоцитов, а эмфизема возникает в результате повреждения стенки альвеол. Клиническими последствиями дефицита α-1-антитрипсина в детстве являются геморрагическая болезнь в младенчестве, холестаз в младенчестве или хроническое заболевание печени. Заболевания легких, связанные с дефицитом α-1-антитрипсина, не возникают в детстве, но у взрослых тесно связаны с курением.Мембранопролиферативный гломерулонефрит, панникулит и некротизирующий васкулит связаны с дефицитом α-1-антитрипсина во взрослом возрасте.2–4 Диагностические методы сведены в таблицу 1.
Таблица 1
Методы диагностики дефицита α-1-антитрипсина
Фенотипы
α-1-антитрипсин является ингибитором протеазы, и обычные варианты Pi были названы по их электрофоретической подвижности. PiM, у которого есть несколько второстепенных вариантов, является нормальным белком.PiZ, мутант, ответственный за более чем 95% случаев заболеваний легких и печени, связанных с дефицитом α-1-антитрипсина, наиболее часто встречается в Скандинавии и все реже встречается по мере путешествия на юг в Европу. PiS, напротив, наиболее распространен на Пиренейском полуострове.
Эмфизема связана как с нулевыми мутациями (белок не продуцируется), так и с мутациями, продуцирующими дефектный или неэкспортируемый белок. Заболевание печени связано только с теми мутациями, которые продуцируют пептид, который образует полимеры петлевых листов, которые удерживаются в печени, а именно с гомозиготами по PiZ, PiM (Мальтон) и сложным гетерозиготам PiZ-, PiSZ и PiZI.6 Номенклатура генетических вариантов немного сбивает с толку: предполагаемые гомозиготные аномалии, такие как PiZZ, обычно называют PiZ, если только нулевой ген не был исключен из фенотипа; таким образом, PiZ может фактически быть PiZ− (Z плюс ноль) или PiZZ. Младенец с дефицитом α-1-антитрипсина считается PiZZ, если оба родителя несут аллель Z; это может быть подтверждено генотипом.
Молекулярная патология
Есть несколько недавних обзоров молекулярной патологии дефицита α-1-антитрипсина.7-9 Новообразованный пептид-1-антитрипсин попадает в эндоплазматический ретикулум, где ряд молекулярных шаперонов контролирует упорядоченный процесс гликозилирования, образования дисульфидных мостиков и фолдинга. Достигнув своей третичной структуры, α-1-антитрипсин покидает эндоплазматический ретикулум и перемещается по Гольджи и секреторным пузырькам к плазматической мембране, где он секретируется в виде гликопротеина 55 кДа со скоростью приблизительно 34 мг / кг / день. Недавно синтезированный α-1-антитрипсин подвергается процессу контроля качества — белок, которому не удается достичь правильной третичной структуры, вступает в деградационный путь, в котором неправильно свернутый α-1-антитрипсин остается связанным с калнексином в эндоплазматическом ретикулуме, индуцирует конъюгацию с убиквитином и оставляет эндоплазматический ретикулум разрушается в протеасомах.
Мутантный пептид, обнаруженный при дефиците α-1-антитрипсина общего фенотипа PiZZ (α-1-антитрипсин Z, AAT-Z), отличается от α-1-антитрипсина M (AAT-M) только заменой одной аминокислоты, gly 342 → лиз. Эффектом этого является серьезное снижение скорости сворачивания пептида.10 Медленное сворачивание позволяет пептидным мономерам объединяться с помощью механизма вставки петлевых листов с образованием полимера AAT-Z, который удерживается в эндоплазматическом ретикулуме.12 В печени В биоптатах пациентов с дефицитом α-1-антитрипсина полимеризованный AAT-Z может быть продемонстрирован в эндоплазматическом ретикулуме с помощью электронной микроскопии и гистохимически обнаружен как характерные PAS-положительные, устойчивые к диастазе глобулы.
Существует множество генетических нарушений, при которых мутировавший пептид не может достичь правильной конформации и остается в эндоплазматическом ретикулуме. Как и в случае с AAT-Z, мутировавший белок может быть функционально активным, но болезнь возникает из-за невозможности доставки в его правильное местоположение, будь то плазматическая мембрана (например, кистозный фиброз или дефицит сахаразы-изомальтазы), мембраны других органелл (болезнь Вильсона). ) или внеклеточной жидкости (дефицит фибриногена, протеина С или тиреоглобулина).7
8
Почему дефицит α-1-антитрипсина отличается от всех этих нарушений тем, что сохраненный мутантный пептид повреждает клетку печени? Возможно, просто α-1-антитрипсин синтезируется в больших количествах, но, вероятно, более важным является тот факт, что полимеризованный AAT-Z сопротивляется разложению. Можно фантастически представить, что он делает с эндоплазматическим ретикулумом то же самое, что гемоглобин S делает с эритроцитом при серповидно-клеточной анемии.
Почему тогда только у 10% детей PiZZ развивается заболевание печени? Среди предлагаемых приобретенных сопутствующих факторов можно выделить следующие:
внутриутробная инфекция , потому что дети PiZ с заболеваниями печени, как правило, были маленькими для гестационного возраста;
протеазы кишечника , предполагая, что грудное вскармливание может быть защитным;
аутоиммунитет , но антитела, направленные на печень, скорее всего, будут вторичными по отношению к повреждению печени, а не первичными;
лихорадка , потому что повышенная температура увеличивает как продукцию, так и скорость полимеризации α-1-антитрипсина.Следовательно, временное повышение уровня трансаминаз в плазме действительно происходит во время инфекций у детей с дефицитом α-1-антитрипсина14;
инфекция криптического гепатита B или C , 12 но это не было подтверждено.13
Ни один из этих приобретенных факторов не кажется достаточным, чтобы полностью объяснить огромные вариации фенотипа печени. Более привлекательная гипотеза касается возможных генетических различий в деградации AAT-Z. В трансфицированных фибробластах кожи пациентов PiZZ, у которых развилось заболевание печени, AAT-Z деградировал менее быстро, чем в фибробластах пациентов PiZZ, у которых не развилось заболевание печени.15 Это говорит о том, что пациенты PiZZ, у которых развивается заболевание печени, имеют одну из ряда возможных врожденных ошибок клиренса AAT-Z. Заманчиво экстраполировать эту концепцию на другие нарушения, связанные с торговлей людьми.
Эпидемиология болезни печени, вызванной недостаточностью α-1-антитрипсина, в детском возрасте
Классический пример проспективной эпидемиологии, 14
16–20 Свегер изучил 200 000 младенцев в 1972–74 годах, выявив среди них 127 детей PiZ и проследив их до возраста 18 лет. Только у 14 из них в младенчестве развилась холестатическая желтуха.Еще у восьми была гепатоспленомегалия и легкие нарушения функциональных тестов печени.
ПРОГНОЗ РЕБЕНКА С ДЕФИЦИТОМ α-1 АНТИТРИПСИНА, НЕ РАЗВИВАЮЩИМ ДЕТСКИЙ ХОЛЕСТАЗ
Частота аномальных тестов функции печени у лиц с PiZZ с дефицитом α-1-антитрипсина, у которых не развивается желтуха или гепатомегалия в младенчестве, снижается с 60% в 6 месяцев до плато примерно 15% в 12–18 лет, без клинических доказательств. заболеваний печени (таблица 2). Аномальные функциональные пробы печени могут быть временными — большинство из тех, у кого были аномальные функциональные пробы в 16 лет, были нормальными в 18 и наоборот.Время от времени появляются сообщения о детях PiZZ, у которых в детстве развилось новое или ухудшающееся нарушение функции печени, часто в связи с другими тяжелыми заболеваниями: по одному случаю аппендицита и пневмонии в серии исследований Свегера и одного случая панкреатита в другом сообщении. Эти случаи кажутся редкими и оправдывают исключение дефицита α-1-антитрипсина при исследовании недиагностированного заболевания печени в детстве.
Таблица 2
Частота (%) аномалий аланинтрансаминазы и / или γ-глутаматтрансферазы у проспективно идентифицированных шведских детей с дефицитом α-1-антитрипсина, у которых не развился детский холестаз или гепатомегалия
Прогноз для младенцев PiSZ еще лучше.Ни у одного из участников серии исследований Свегера желтуха в младенчестве не развивалась, а частота аномальных функциональных тестов печени была ниже в детстве (таблица 2).
Риск цирроза у взрослых гораздо труднее определить количественно, потому что доступные данные получены либо от пациентов, у которых известно о заболевании легких с дефицитом α-1-антитрипсина, либо из исследований дефицита α-1-антитрипсина в случаях цирроза. Таким образом, у 12% взрослых шведов, выявленных при госпитализации, был цирроз печени.21 Неизвестно, имеет ли алкоголь какое-либо отношение к заболеванию печени при дефиците α-1 антитрипсина; поэтому у нас нет оснований настаивать на воздержании, но мы советуем умеренность.
В то время как гетерозиготное состояние PiMZ не несет риска в детстве, оно может быть связано с заболеванием печени у взрослых. Фенотип PiMZ был обнаружен у 8% взрослых кандидатов на трансплантацию с хронической печеночной недостаточностью, в том числе у 27% пациентов с криптогенным циррозом, по сравнению с 2–4% в общей популяции.22 Однако инвертирование этой статистики показывает, что риск заболевания печени у конкретного индивидуума PiMZ должен быть небольшим.
ПРОГНОЗ РЕБЕНКА С дефицитом α-1-антитрипсина, у которого развивается детский холестаз
Из 22 детей в Швеции с клинически поражением и дефицитом α-1-антитрипсина двое умерли от цирроза печени примерно в 7 лет, а один умер от апластической анемии и имел цирроз печени при вскрытии.
16–20 Из 74 детей, поступивших в больницу Кингз-Колледжа и проследивших до 17 лет, 20 умерли, 20 страдали циррозом печени, 19 имели постоянно отклоняющиеся от нормы показатели функции печени и только 15 полностью выздоровели.23 Однако это были пациенты, у которых детский холестаз был достаточно тяжелым для направления к специалистам.
Более поздний обзор24 показал, что, возможно, неудивительно, что у детей, у которых болезнь печени прогрессировала до конечной стадии, в младенчестве наблюдались более серьезные аномалии. У них с большей вероятностью оставалась желтуха более 6 недель, у них был более высокий уровень аспартатаминотрансферазы при поступлении и имелись более серьезные изменения при первоначальной биопсии (включая тяжелую редупликацию желчных протоков, тяжелый фиброз с перегородками и установленный цирроз. ).Тем не менее, следует с осторожностью относиться к перспективам отдельного ребенка с желтухой и дефицитом α-1-антитрипсина. Volpert и др. отмечают, что среди пораженных детей с установленным заболеванием печени, которым сразу не была показана трансплантация, есть группа, которая остается клинически стабильной в течение длительного периода после установления наличия цирроза или портальной гипертензии.13 Они изначально не различимы. от тех детей, у которых функция печени ухудшается быстрее.
Три диагностических пункта заслуживают внимания при холестатическом младенчестве. Во-первых, первые признаки биопсии печени с заметными изменениями портального тракта и еще не обнаруженными гранулами, положительными по PAS и отрицательной диастазой, могут имитировать атрезию желчевыводящих путей. Трансформация гигантских клеток гепатоцитов при дефиците α-1-антитрипсина нехарактерна. Во-вторых, концентрация α-1-антитрипсина в плазме у пациента с дефицитом может быть повышена как реагент острой фазы, поэтому необходимо получить фенотип α-1-антитрипсина.В-третьих, определение фенотипа α-1-антитрипсина может быть наиболее трудоемким лабораторным исследованием, поэтому его необходимо (как и скрининг коагуляции) запрашивать незамедлительно.
Показания к трансплантации печени такие же, как и при других заболеваниях печени. После атрезии желчных путей дефицит α-1-антитрипсина является наиболее частой причиной трансплантации печени в детском возрасте. Легкие не представляют особых проблем, а периоперационный и послеоперационный уход не отличается от обычных процедур.Результаты хорошие.25
26 Ортотопическая трансплантация печени от родителя-донора была успешно проведена.27 После трансплантации реципиент проявляет фенотип донора и, как ожидается, не будет подвергаться риску эмфиземы.
Каков риск тяжелого заболевания печени у последующего брата или сестры по PiZ серьезно пораженного пробанда? Псахаропулос и др. сообщили, что риск составлял 78%, 23 в то время как другие сообщили менее пессимистические цифры. Родители, у которых был ребенок с тяжелым заболеванием, с большей вероятностью выберут прерывание последующего плода PiZ, поэтому может быть трудно получить дополнительные данные для уточнения этого риска.
Поздняя геморрагическая болезнь в младенчестве
В то время как ранняя неонатальная геморрагическая болезнь у младенцев на грудном вскармливании предотвращается пероральным приемом витамина К при рождении, случаи поздних зависимых от витамина К кровотечений продолжают иметь место, обычно у младенцев с недиагностированным холестазом, из которых наиболее распространены недостаточность антитрипсина α-1 и атрезия желчных путей. общий. Из 182 000 младенцев в Северо-Восточном регионе Соединенного Королевства, которым при рождении давали 1 мг витамина К перорально, а детям, находящимся на грудном вскармливании, рекомендовалось получать еще три дозы по 1 мг с двухнедельными интервалами, у четверых развилась поздняя геморрагическая болезнь.28 Двое из них не получали витамин К, а двое имели дефицит α-1-антитрипсина. Учитывая, что частота грудного вскармливания составляла примерно 30% (Варияр Ю., личное сообщение), а частота дефицита α-1-антитрипсина составляет примерно 1: 1800, похоже, что геморрагическая болезнь возникла у 2/30 грудных детей, вскармливаемых α-1-антитрипсином. дефицит.
Из 332 686 шведских младенцев, около 80% которых получали 1 или 2 мг перорального витамина К при рождении, и среди которых частота грудного вскармливания, как сообщается, была высокой, у 17 развилась поздняя геморрагическая болезнь.29 Пятнадцать из них имели холестатическое заболевание печени, в том числе трое с дефицитом α-1-антитрипсина, пять с атрезией желчных путей и семь с другими состояниями. Таким образом, у 1/35 младенцев с дефицитом α-1-антитрипсина было кровотечение. В других сообщениях дефицит α-1-антитрипсина также был причиной поздних геморрагических заболеваний.30
31 Это трагическое последствие дефицита α-1-антитрипсина и других инфантильных холестазов предотвращается более физиологичным голландским протоколом, согласно которому грудным детям вводят 1 мг витамина К при рождении и 25 мкг ежедневно с 2–13 недель жизни, хотя на поздней стадии геморрагическая болезнь. все еще может произойти из-за несоблюдения требований.32
Несмотря на заболевание печени, значительно увеличенное протромбиновое время резко улучшается в течение нескольких часов после парентерального введения витамина К.
Последующие зараженные братья и сестры пробанда с дефицитом α-1-антитрипсина всегда должны получать внутримышечно витамин К при рождении.
Заболевания легких при дефиците α-1-антитрипсина
Повреждение, вызванное неингибированной эластазой нейтрофилов в легких, проявляется клинически через много лет.33 Характерной патологией, наблюдаемой при дефиците α-1-антитрипсина, является эмфизема, вызванная потерей упругой отдачи.У детей и подростков с дефицитом α-1-антитрипсина не было обнаружено значительных нарушений функции легких.
35 Хотя исследование пораженных детей с заболеванием печени показало склонность к гиперинфляции, 36 этого не было обнаружено в последующем исследовании Свегера с участием 150 подростков34. В возрасте 30–35 лет наблюдается ускоренное снижение объема форсированного выдоха за одну секунду. (FEV 1 ), которая значительно ухудшается при курении сигарет. У некурящих симптомы обычно проявляются примерно к 50 годам, тогда как у курильщиков симптомы проявляются к 30-40 годам.Хотя продолжительность жизни труднее оценить с точностью, комбинация трех исследований дает средний возраст смерти 50 лет у курильщиков по сравнению с 66 годами у некурящих.37-39 Интересно, что только 3% подростков с α- 1 дефицит антитрипсина отмечен в исследовании Свегера 34, что свидетельствует о том, что санитарное просвещение может быть эффективным в этой группе детей.
Педиатры, как правило, включают фенотип антитрипсина α-1 в набор тестов для выявления необъяснимых легочных симптомов.В поддержку этого мало свидетельств, хотя теоретически возможно, что сопутствующее воспалительное заболевание может усугубляться дефицитом α-1-антитрипсина даже в детстве. В исследовании взрослых с бронхоэктазами не было отмечено увеличения распространенности аллелей дефицита α-1-антитрипсина, но увеличилось количество эмфиземы, если оба заболевания сосуществовали вместе.40 Если дефицит α-1-антитрипсина обнаружен у ребенка с легочными симптомами, этого не должно быть. принимается как основная причина проблемы, но может быть фактором, усугубляющим прогрессирование заболевания.
Перспективы фармакологического лечения дефицита α-1-антитрипсина
Логическая терапевтическая амбиция состоит в том, чтобы разработать способ перемещения AAT-Z из эндоплазматического ретикулума печени, где он вызывает повреждение, в плазму, где его антипротеазная активность — хотя и ниже, чем у AAT-M дикого типа — принесет пользу пациентам. легкое. Это может быть достигнуто либо путем ингибирования полимеризации AAT-Z41, либо путем сопровождения неправильно свернутого AAT-Z из эндоплазматического ретикулума в секреторный путь.2 Последнее понятие является общим для всех заболеваний ретикулума эндоплазматического ретикулума. В отличие от муковисцидоза ΔF508, инкубация клеток при более низкой температуре не улучшает секреторный дефект при дефиците α-1-антитрипсина, но есть многообещающие результаты от химических шаперонов. В культивируемых гепатоцитах мыши или трансфицированных фибробластах кожи глицерин и 4-фенилмасляная кислота приводили к увеличению секреции AAT-Z с 3% в контроле до 25% и 17% соответственно. 4-фенилмасляная кислота также повысила уровни в плазме, достигнув 20–50% от уровней, присутствующих у мышей PiM с дефицитом трансгенного α-1-антитрипсина.42 Фенилбутират натрия уже используется в клинической практике при нарушениях цикла мочевины и является потенциальной формой лечения. Однако способ его действия еще предстоит определить. Как ингибитор трансляции белка циклогексимид, так и специфический ингибитор функции протеасом, лактацистин, предотвращали внутриклеточную деградацию AAT-Z и частично восстанавливали его везикулярный транспорт в трансфицированных клетках СНО и альвеолярных макрофагах человека.43 В других исследованиях клеточных культур также используются ингибиторы глюкозидазы и маннозидазы. достигнута повышенная секреция.44 год
Было показано, что усиливающая терапия α-1-антитрипсином внутривенно восстанавливает уровни антипротеаз в сыворотке и мокроте. Очевидно, что это не принесет пользы младенцам с заболеваниями печени, но дает возможность профилактики или лечения заболеваний легких. В нерандомизированных обсервационных исследованиях внутривенная увеличивающая терапия ассоциировалась с более медленным снижением ОФВ 1 4 и улучшением выживаемости, но пока нет доказательств долгосрочной пользы от рандомизированного контролируемого исследования.Если видны преимущества, они, по-видимому, ограничиваются подгруппой пациентов с ОФВ 1 ниже 65% от прогнозируемого. Одно небольшое рандомизированное испытание, в котором сравнивали 28 пациентов, получавших лечение, и 28 пациентов контрольной группы, показало незначительные преимущества по внешнему виду на компьютерной томографии, но отсутствие значительных различий в функции легких после трех лет лечения. 46 В настоящее время лечение не лицензировано в Соединенном Королевстве. В настоящее время становится доступным рекомбинантный α-1-антитрипсин, и планируются испытания его введения с помощью небулайзера.Однако у ребенка с дефицитом α-1-антитрипсина может никогда не развиться серьезное заболевание легких, если избегать других повреждающих факторов, и заместительная терапия (при ее наличии) в настоящее время рекомендуется только при значительной или быстро прогрессирующей эмфиземе, а не в качестве упреждающего лечения.
Есть многообещающие первые результаты генной терапии. Нормальный ген антитрипсина α-1 в плазмидно-катионном липосомном комплексе был доставлен в одну ноздрю каждого из пяти пораженных пациентов. Белок α-1-антитрипсина увеличился, а интерлейкин 8 (как маркер воспаления) уменьшился в жидкости для промывания носа, с максимальным эффектом на 5 день.47 Хотя технически возможно вставить ген AAT-M в печень мышей AAT-Z, 48 это явно не исправит клеточный дефект.
Выводы
Поздняя геморрагическая болезнь остается опасностью для младенцев с дефицитом α-1-антитрипсина на грудном вскармливании с текущим протоколом введения витамина К. Для своевременного выявления холестаза в младенчестве необходима постоянная бдительность. У нас есть убедительные доказательства прогноза недостаточности α-1-антитрипсина для печени, и недавние знания о молекулярной патологии дают надежду на новые терапевтические подходы к этому и другим нарушениям ретикулума эндоплазматического ретикулума.Обозначены показания и результаты трансплантации печени.
Какой совет мы должны дать молодым людям с дефицитом α-1-антитрипсина? Основная идея не изменилась с тех пор, как в 1985 году Янус и др. открыто заявили: «Если они курят, к среднему возрасту у них разовьется парализующая эмфизема; если они не курят, у них есть разумная вероятность прожить полную жизнь ». Пока не будет доступно простое и безопасное лечение, это остается единственным способом обеспечить здоровье дыхательных путей при дефиците α-1-антитрипсина.
детей с альфа-1: осведомленность об альфа-1
Мой папа умер, когда мне было 3 года. Я очень хорошо его помню по нашему дому в Эшби-де-ла-Зауч. Я помню, как играл с ним, и он рассказывал мне действительно интересные вещи. Мне грустно, что его больше нет здесь, чтобы развлекаться со мной, мамой и Тедом. Он не видел меня в форме, когда я пошел в школу, и не знал, что Тед (это мой младший брат) любит играть в футбол и что он тоже сейчас в школе!
Моя мама сказала мне, что когда папа родился очень-очень давно (задолго до моего рождения), он действительно был болен.Он был весь желтый и очень крошечный. Он провел три месяца в больнице (это много сна), прежде чем он смог вернуться домой к маме и папе (то есть бабушке Кристине и дедушке Джону). Мама говорит, что врачи никогда не знали, почему папа так плохо себя чувствовал, и ему только что стало лучше. Ну, когда я родился, мама говорит, что папа снова стал болеть.
Когда мне было 9 месяцев (то есть мне было всего 0!), Врачи сказали папе, что у него болезнь под названием дефицит антитрипсина альфа-1. Это очень громкие слова, и я не понимаю, что это за болезнь, но она как-то связана с вашей печенью и легкими.Я знаю, что они являются важными частями вашего тела, потому что учитель сказал мне об этом в школе.
Я не помню, чтобы папа болел, но знаю, что он много времени проводил в постели. Мама говорила мне молчать и дать папе поспать, и мы (это я и Тед) забирались в двухместную коляску, и мама толкала нас в парк. Иногда мы ссорились … а однажды Тед выпадал из-за того, что мама забыла его пристегнуть!
Мама сказала, что после того, как родился Тед, папа стал очень, очень плохо болеть и у него начались судороги.Я тоже этого не понимаю, но знаю, что это означало, что папа не мог контролировать свое тело, и ему пришлось лечь в больницу. Однажды мы ходили по магазинам, и у папы была одна из этих вещей. Скорая приехала долго, потому что мама не могла найти никого, чтобы рассказать, потому что ей приходилось заботиться обо мне и Теде. Потом я захотел крошки, поэтому ей пришлось попросить кого-нибудь присмотреть за Тедом и папой, пока она забирала меня! Я был в Макдональдсе. Когда я вернулся из туалета, хороший человек играл в игру с Тедом, я присоединился к нему, а затем приехала скорая помощь, положила папу на кровать с колесами и в машину скорой помощи.Я помню другой раз, когда скорая помощь была у нас дома, когда я вернулся из детской, и у папы была маска на рту, чтобы помочь ему дышать, и он был весь в синяках. Мама сказала, что у него в саду случился приступ, и он поранился.
Я помню, как собирался остановиться в фургоне с мамой, папой и Тедом. Это было в Чармуте у моря. Это был последний отпуск, который у меня был с папой. Это было действительно весело. Однажды ночью мама вытащила меня из постели, потому что была фантастическая радуга. В этот праздник папа не мог далеко ходить, поэтому мы много обнимались и много садились.
Когда мы снова вернулись домой, я помню, что папа много спал. Потом бабушка и бабушка приехали, а мама продолжала уходить. Папы не было. Он был в больнице, и мама каждый день ходила к нему в гости. Мама много плакала, и я не понимала почему. Однажды мы с Тедом пошли навестить папу. Когда я его увидел, я был очень счастлив, но потом немного испугался. Было много машин и проводов, и я не хотел подходить к папе. Тед сделал. Он лег рядом с папой, и папа прижал его к себе.Папа держал меня за руку, но я не хотел. Затем мы пошли домой с бабушкой.
Папа не пришел домой. Мама сказала, что он очень плохо себя чувствует и его тело перестало работать. Я так по нему соскучилась. Я люблю пить лимонный сок, потому что знаю, что он был одним из любимых папин. Люди говорят, что я похож на папу, и я такой же глупый, как он — мне нравится, когда люди говорят это, потому что это заставляет меня гордиться. Мне жаль Теда, потому что он был еще младенцем, когда умер папа, поэтому он не может его вспомнить — он говорит, что помнит, но я ему не верю.Он много просит нового папу. Однажды я спросил маму, могу ли я отправиться на небеса, чтобы увидеться с папой, но я не думаю, что ты сможешь это сделать. У нас есть много фотографий, на которые мы часто смотрим с мамой, и все мы любим это делать.
Мама сказала мне, что не многие знают о болезни папы. Она говорит, что до сих пор рождаются тяжелобольные дети, которые, возможно, болеют папиной болезнью, но мало кто знает об этом. Мама также рассказывала мне, что, когда папа плохо себя чувствовал, за ним ухаживали множество разных врачей, которые все были очень сбиты с толку и не знали, какое лекарство ему дать.Если бы о болезни папы знали больше людей, то он, возможно, получил бы правильное лекарство. Тогда он мог бы увидеть, как я пошел в школу.
Мама говорит, что папа был потрясающим. Она сказала мне, что даже при том, что он был в таком плохом состоянии, он никогда не стонал и был хорошим папой и другом моей мамы. Мама до сих пор много плачет. Я стараюсь делать то, что сделал бы мой папа, и заставлять ее снова чувствовать себя счастливой и помогать с работой по дому.
Мы все скучаем по папе. Я не хочу, чтобы младенцы больше страдали, а мальчики и девочки грустили, если их папа умрет.Я был бы счастлив, если бы все мы узнали больше о болезни папы.
Бобби Гуд, 6 ¾ лет
Основные и клинические аспекты альфа1-антитрипсина
Реферат
Дефицит основного сывороточного α 1 -глобулина, α 1 -антитрипсина, был впервые описан Лауреллом и Эриксоном в Швеции в 1963 году у пяти пациентов. Вскоре стало очевидно, что тяжелый дефицит α 1 -антитрипсина был семейным и сильно ассоциировался с хроническим заболеванием легких, начавшимся в третьем или четвертом десятилетии жизни.Со времени первых описаний этого общего состояния дефицита оно стало четко ассоциироваться с семейной эмфиземой в одних семьях, семейным инфантильным циррозом в других и иногда с комбинацией детских болезней легких и печени у братьев и сестер. Для педиатра тяжелый дефицит α 1 -антитрипсина теперь входит в дифференциальный диагноз как хронической болезни легких в детстве, так и механической желтухи в период новорожденности. Кроме того, низкие уровни α 1 -анриртизина в сыворотке крови характерны для респираторного дистресс-синдрома, и повышение этого белка может быть обнаружено в различных клинических ситуациях.
α 1 -антитрипсин, вероятно, функционирует как главный контрольный белок против повреждающего воздействия на ткани как эндогенных, так и экзогенных ферментов. В этом обзоре будут рассмотрены несколько основных и клинических характеристик этого белка с учетом его важности в педиатрии.
ХИМИЯ И МЕТАБОЛИЗМ
Гликопротеин с молекулярной массой 50 000, α 1 -антитрипсин синтезируется в паренхимных клетках печени и секретируется в сыворотку как основной α 1 -глобулин, составляющий примерно 4% от общего уровня сывороточного белка (нормальный концентрации составляют примерно 2.2 мг / мл). Альфа 1 -антитрипсин присутствует во многих жидкостях организма, в количествах микрограммов на миллилитр в выделениях из носа, слезах, слюне, легких, двенадцатиперстной кишке, спинномозговой жидкости, кобстру и материнском молоке.
- Получено 18 ноября 1974 г.
- Принято 9 декабря 1974 г.
- Авторские права © 1975 Американской академии педиатрии
Генетическое тестирование на дефицит антитрипсина альфа-1 | Детская больница CS Mott
Что такое дефицит антитрипсина альфа-1?
Альфа-1-антитрипсин (ААТ) — это белок, обычно обнаруживаемый в легких и кровотоке.Он помогает защитить легкие от таких заболеваний, как эмфизема легких и хроническая обструктивная болезнь легких (ХОБЛ). Некоторые люди вырабатывают недостаточно этого белка или вырабатывают аномальный тип ААТ, любой из которых может вызвать дефицит ААТ. Эти люди чаще болеют легочными заболеваниями и заболевают ими в более молодом, чем обычно, возрасте (от 30 до 40 лет). Некоторые типы аномальных ААТ также могут повредить печень. Дефицит ААТ — редкое заболевание и единственный известный генетический (наследственный) фактор, который увеличивает ваши шансы на развитие эмфиземы.
Дефицит антитрипсина альфа-1 вызван изменением или мутацией гена, который сообщает организму, как вырабатывать антитрипсин альфа-1. В этом гене есть много возможных изменений, но только некоторые из них вызывают проблемы. Чтобы иметь это состояние, вы должны получить измененный ген от обоих родителей.
Если вы получили только один измененный ген, вы не больны, но являетесь носителем. Хорошей копии гена, полученной вами от другого родителя, достаточно, чтобы сообщить вашему организму, как правильно вырабатывать альфа-1-антитрипсин.Некоторые люди, несущие измененный ген, могут иметь очень легкие симптомы дефицита.
Лечение дефицита альфа-1-антитрипсина заключается в отказе от веществ, особенно сигаретного дыма, которые могут нанести вред вашим легким. Также старайтесь избегать пыли и химикатов на рабочем месте. Вы также можете отказаться от алкоголя из-за риска повреждения печени. Физические упражнения могут улучшить вашу выносливость и общее состояние здоровья. Вам также могут потребоваться лекарства и другие методы лечения, чтобы облегчить дыхание и оставаться максимально здоровым и сильным.
Единственное средство для лечения недостатка белка — это плазма, содержащая альфа-1-антитрипсин. Обычно это назначают только людям с очень низким уровнем ААТ в крови. Неясно, лучше ли это лечение, чем отказ от дыма и других химикатов, повреждающих легкие. Плазма производится из крови многих доноров и обрабатывается, чтобы снизить вероятность распространения инфекционного заболевания. Вы получаете плазму через капельницу, обычно каждые 3-4 недели на всю жизнь.
Что такое тест на дефицит AAT?
Анализ крови может определить количество альфа-1-антитрипсина (ААТ) в вашей крови.У вас может быть дефицит ААТ, если у вас низкий уровень или если анализ крови не может найти ААТ в вашей крови. Если ваш уровень ААТ ниже нормы, можно провести анализ крови на предмет аномальных типов антитрипсина альфа-1. Люди, несущие измененный ген, могут подвергаться большему риску развития симптомов, если у них есть определенные типы альфа-1-антитрипсина.
Точность тестирования?
Хотя этот анализ крови очень надежен, ни один тест не является точным на 100%.Этот тест не может предсказать, когда или появятся ли у вас симптомы, или насколько они серьезны.
Следует ли мне проходить тестирование?
Решение о прохождении теста — личное. У вас могут быть эмоциональные, финансовые или семейные причины для прохождения или отказа от прохождения теста.
Вы можете пройти тест, потому что:
- У вас необъяснимые проблемы с легкими и вы хотите узнать, есть ли у вас это заболевание.
- У других членов вашей семьи дефицит ААТ.
- Другие члены вашей семьи страдают необъяснимым заболеванием легких или печени.
- Вы хотите принять меры для защиты своего здоровья, если обнаружите, что у вас есть это заболевание.
- Вы с облегчением узнаете, что у вас нет измененного гена.
- Вы измените свое решение иметь детей из-за результатов тестирования.
- Тест оплачивает ваша страховка.
Почему бы мне не пройти тестирование?
Вы можете отказаться от тестирования, потому что:
- У вас нет симптомов заболевания легких.
- Никто в вашей семье не страдает дефицитом ААТ, заболеваниями легких или печени.
- Вы бы забеспокоились, если бы узнали, что когда-нибудь можете получить заболевание легких или печени.
- Вы не курите и уже хорошо о себе заботитесь. Вы думаете, что больше ничего не можете сделать, чтобы предотвратить или отсрочить болезнь.
- Тест не может предсказать, появятся ли у вас симптомы этого состояния.
- У вас нет страховки, иначе ваша страховка не оплатит тест.
- Вы обеспокоены тем, что результаты теста могут вызвать у вас проблемы на работе или затруднить получение медицинской страховки. Важно знать, что закон в Соединенных Штатах, называемый Законом о запрещении генетической информации от 2008 года (GINA), помогает защитить людей с генетическими различиями, которые могут повлиять на их здоровье. GINA не позволяет работодателям и медицинским страховым компаниям использовать генетическую информацию о людях, чтобы влиять на решения. Но у этого есть ограничения. Например, этот закон не распространяется на страхование жизни, страхование на случай инвалидности или страхование на случай длительного ухода.И это не защищает людей, которые работают в компаниях с менее чем 15 сотрудниками.
Что такое генетическое консультирование?
Информация, полученная в результате генетического тестирования, может иметь большое влияние на вашу жизнь. Попросите пройти генетическую консультацию, прежде чем принимать решение о тестировании. Консультанты-генетики обучены объяснять тест и его результаты, но решение о том, проходить ли тест, принимаете вы. Консультант по генетическим вопросам может помочь вам принять обоснованное решение. Генетическое консультирование может помочь вам и вашей семье:
- Разберитесь в медицинских фактах, в том числе в том, что вызывает заболевания, как ставится диагноз и что вы можете сделать, чтобы помочь вам справиться с болезнью.
- Узнайте, как ваша семейная история способствует развитию болезни.
- Помогите вам узнать об уходе за членом семьи с генетическим заболеванием, включая направление к специалистам или присоединение к группам поддержки.
Консультанты-генетики обучены помогать вам и вашей семье в принятии осознанных решений. Они чувствительны к физическим и эмоциональным аспектам этих решений. Ваша конфиденциальность и конфиденциальность тщательно защищены.
Список литературы
Цитаты
- Fischbach FT, Dunning MB III, ред.(2009). Руководство по лабораторным и диагностическим исследованиям , 8 изд. Филадельфия: Липпинкотт Уильямс и Уилкинс.
Консультации по другим работам
- Pagana KD, Pagana TJ (2010). Руководство Мосби по диагностическим и лабораторным исследованиям, 4-е изд. Сент-Луис: Мосби.
Кредиты
Текущий по состоянию на:
26 октября 2020 г.
Автор: Healthwise Staff
Медицинский обзор:
E.Грегори Томпсон, врач-терапевт
Адам Хусни, доктор медицины, семейная медицина
Элизабет Т. Руссо, врач-терапевт
Кен Йонеда, доктор медицины, пульмонология
По состоянию на: 26 октября 2020 г.
Автор:
Здоровый персонал
Медицинское обозрение: E. Грегори Томпсон, врач-терапевт и Адам Хусни, доктор медицины, семейная медицина и Элизабет Т.Руссо, врач-терапевт и Кен Йонеда, доктор медицины, пульмонология,
.