Болезнь Гентингтона: симптомы, лечение, фото
Содержание статьи:
Болезнь Гентингтона – это тяжелое нейродегенеративное заболевание головного мозга с прогрессирующим течением, которое характеризуется нарастающей деменцией и хореическим гиперкинезом. Встречается с частотой 10 случаев на 100 000 населения, у мужчин и женщин в равной степени. Первые симптомы могут проявиться в любом возрасте, но обычно дебют заболевания приходится на 30–50 лет.
Синонимы: хорея Гентингтона, хореическая деменция, хорея наследственная, прогрессирующая хроническая хорея, дегенеративная хорея.
 Участки мозга, наиболее страдающие при болезни Гентингтона
Участки мозга, наиболее страдающие при болезни Гентингтона
Причины возникновения и факторы риска
Болезнь Гентингтона – это генетическое заболевание, имеющее аутосомно-доминантный тип наследования. Вероятность рождения больного ребенка в семейной паре, где один из родителей страдает этой патологией, составляет 50%.
Мутированный ген HD локализован в хромосоме 4р16.3. Он отвечает за синтез особого белка – гентингтина. Названный белок токсичен для некоторых типов клеток, особенно для нервных клеток полосатого тела головного мозга, основными функциями которого являются:
- уменьшение тонуса мышц;
- регуляция работы внутренних органов;
- участие в формовании условных рефлексов;
- участие в осуществлении некоторых поведенческих реакций.
 Наследование болезни Гентингтона обусловлено генетически
Наследование болезни Гентингтона обусловлено генетически
Летальный исход при болезни Гентингтона наступает через 10–15 лет от появления первых симптомов заболевания. Средняя продолжительность жизни пациентов с данным диагнозом – 45–55 лет.
Формы заболевания
В зависимости от возраста, в котором возникло заболевание, выделяются две формы болезни Гентингтона:
- Типичная. Наблюдается в 90% всех случаев. Развивается в 35–40 лет. Первыми симптомами обычно являются избыточные движения, происходящие на фоне пониженного тонуса мышц.
- Ювенильная (вариант Вестфаля). Возникает до 20 лет. Характеризуется повышенным мышечным тонусом и меньшей выраженностью непроизвольных движений.
Симптомы
У взрослых болезнь Гентингтона обычно стартует с развития хореического синдрома. Первые симптомы заболевания – суетливость и неусидчивость. В дальнейшем двигательные нарушения нарастают. Наиболее характерные признаки хореического синдрома при болезни Гентингтона:
- судорожные неритмичные движения туловища и (или) конечностей;
- нарушения артикуляции;
- всхлипывания;
- подергивания щек;
- поочередное поднятие и (или) нахмуривание бровей;
- высовывание языка;
- «танцующая» походка.
 Прогрессирование болезни Гентингтона приводит к инвалидности
Прогрессирование болезни Гентингтона приводит к инвалидности
У пациентов уже на самых ранних стадиях развития болезни Гентингтона отмечается нарушение исполнительных функций, мышления, снижается концентрация внимания. Развиваются нарушения психики:
- периодическая расторможенность;
- раздражительность;
- отчужденность;
- апатия;
- депрессия;
- навязчивые состояния;
- бред.
Болезнь Гентингтона встречается с частотой 10 случаев на 100 000 населения, у мужчин и женщин в равной степени. Первые симптомы могут проявиться в любом возрасте, но обычно дебют заболевания приходится на 30–50 лет.
Особенности протекания заболевания у детей
Случаи ювенильной формы болезни Гентингтона довольно редки. Заболевание у детей и подростков обычно начинается с замедления активных и содружественных движений (брадикинезии), а также со значительного повышения мышечного тонуса (ригидности). В отличие от взрослых пациентов, у детей, страдающих болезнью Гентингтона, зачастую наблюдаются судороги.
 У детей с болезнью Гентингтона, часто наблюдаются судороги
У детей с болезнью Гентингтона, часто наблюдаются судороги
По мере прогрессирования патологии нарушается речевая функция. Уже на ранних стадиях наблюдаются проблемы с произношением определенных звуков. В то же время синтаксическая и семантическая структура речи сохраняется довольно долго.
Читайте также:
9 советов мужчине, мечтающему о здоровом потомстве
5 опасений будущих рожениц
Прогнозы ученых: 8 изменений человека будущего
Диагностика
При подозрении на болезнь Гентингтона пациентам рекомендуется проведение магнитно-резонансной или компьютерной томографии головного мозга. На томограммах определяют признаки атрофии хвостатых ядер, выраженность которых нарастает по мере прогрессирования заболевания.
 Для диагностики болезни Гентингтона проводят МРТ головного мозга
Для диагностики болезни Гентингтона проводят МРТ головного мозга
Верифицируют диагноз по результатам молекулярно-генетического исследования. При помощи полимеразной цепной реакции (ПЦР) устанавливают количество поворотов триплета цитозин – аденин – гуанин в гене HD. Диагноз считается подтвержденным, если у подростков число повторов составляет 50 и более, а у взрослых превышает 36.
Вероятность рождения больного ребенка в семейной паре, где один из родителей страдает болезнью Гентингтона, составляет 50%.
Лечение
Специфическая терапия болезни Гентингтона не разработана. Лечение направлено на борьбу с гиперкинезами. Для этого пациентам назначают препараты фенотиазинового ряда, являющиеся антагонистами допамина, и транквилизаторы.
Хирургическое лечение болезни Гентингтона (стереотаксические операции) неэффективно.
 Основное направление в лечении болезни Гентингтона – борьба с гиперкинезами с помощью лекарственных препаратов
Основное направление в лечении болезни Гентингтона – борьба с гиперкинезами с помощью лекарственных препаратов
Возможные осложнения и последствия
Болезнь Гентингтона может осложняться:
- пневмонией;
- хронической сердечной недостаточностью.
Прогноз
Прогноз неблагоприятный. Летальный исход наступает через 10–15 лет от появления первых симптомов заболевания. Средняя продолжительность жизни пациентов с данным диагнозом – 45–55 лет.
Профилактика
Не существует методов, позволяющих выявить носителей мутировавшего гена до развития у них клинических признаков болезни Гентингтона, в связи с чем при проведении медико-генетического консультирования возникают определенные сложности. Если в семье есть ребенок, страдающий данным заболеванием, или оно развилось у одного из родителей, рекомендуется отказаться от дальнейшей возможности иметь детей.
Болезнь Гентингтона (код по МКБ-10 – G10) – это очень редкое наследственное нейродегенеративное заболевание, вызываемое нарушениями клеток в мозге (в стриатуме) с их последующим отмиранием и поражением ЦНС.
Описание
Хорея Гентингтона – что за болезнь, в чем ее суть? Это серьезное наследственное заболевание с типичными психическими и физическими симптомами, которые обычно возникают между 30 и 45 годами. Но признаки могут появиться раньше. Если они наблюдаются в возрасте около 20 лет, патология упоминается как ювенильная болезнь Гентингтона (вариант Вестфаля). Как правило, она характеризуется неконтролируемым подергиванием конечностей, туловища, головокружением. Заболевание приводит к психологическому расстройству личности, слабоумию.
Синдром Гентингтона затрагивает около 5-10 человек на 100000. К сожалению, ее невозможно предотвратить, замедлить или вылечить. Ожидаемая продолжительность жизни уменьшается не самой болезнью, а ослабленным иммунитетом.
Патология получила свое название в честь американского врача Джорджа Гентингтона, впервые описавшего ее в 1872 г.
Причины и факторы риска
Причина заболевания – изменение (мутация) гена IT15 на 4-й хромосоме.
Основой мутации при болезни Гентингтона является размножение триплета CAG (цитозин-аденин-гуанин). Нормальное состояние – до 35 триплетов CAG, при количестве 36-39 прогноз неясен, при более 40 триплетах человек наверняка заболеет этой коварной болезнью. Правило «чем больше триплетов CAG, тем быстрее вспышка заболевания» особенно верно для людей с экстремальным количеством триплетов CAG.
Ген, содержащий менее 36 триплетов, продуцирует белок хантингтин, необходимый для правильного развития мозга. Болезнь Гентингтона – это заболевание с аутосомно-доминантным типом наследования. Это означает, что она встречается в основном в каждом поколении. Если один из родителей страдает этим расстройством, существует 50% риск того, что ребенок также заболеет.

При диагностике болезни Гентингтона необходимо учитывать, что не всегда известно присутствие в семье носителя мутаций. Причины – преждевременная смерть от другой патологии, развод, неизвестность отца. Новые мутации встречаются очень редко.
Пациентам, страдающим от заболевания, рекомендуется не иметь детей. Но проблема заключается в том, что в большинстве случаев оно диагностируется только после рождения ребенка. Генетическая мутация присутствует с рождения, но обычно начинает проявляться во взрослом возрасте.
Симптомы
Человек обычно приходит к врачу из-за проблем с движением, сопровождающихся изменением психики. Симптомы и признаки болезни Гентингтона чаще всего появляются около 40 лет. Клиническая картина постепенно ухудшается в результате повреждения головного мозга.
Вначале наблюдаются мелкие мышечные подергивания и постоянные движения конечностей. Позже развивается полная хорея. Она сначала поражает руки и голову, человек делает различные гримасы, открывает рот. Затем затрагиваются нижние конечности. Эти непроизвольные движения значительно усиливаются при стрессе, в то же время, вообще не появляются во время сна. Движения прогрессируют, человек становится неспособным к самостоятельности. Признаки проявляются на обеих сторонах тела.
Типичный симптом болезни хорея Гентингтона – нарушение походки, которая иногда напоминает танец. На более поздней стадии происходят падения, неуверенные движения. Непроизвольные движения могут больше не проявляться, развивается ригидность, человеку трудно двигаться.
Заболевание часто сопровождается нарушением речи (она становится непонятной). Кроме того, наблюдается расстройство глотания, приводящее к серьезным проблемам с приемом пищи. Пищевые расстройства вызывают потерю веса.
В большинстве случаев при болезни Гентингтона возникает недержание мочи.
Значимые проявления генетического заболевания хорея Гентингтона – психические проблемы. Первоначально возникает незначительная раздражительность, затем появляются другие признаки, такие как:
- агрессивность;
- апатия;
- безжалостное поведение;
- склонность к воровству;
- сниженный интерес к внешности;
- общие изменения личности.
Хотя окружающие замечают эти проблемы, немногие приписывают их болезни.
Депрессия возникает уже на ранних стадиях заболевания. Появляется страх перед новым днем, опасения потери производительности, неудач, нехватка энергии, воли к действию. Человек становится безразличным к окружающему миру, раздражается, имеет трудности с чувством радости, не концентрируется, страдает нарушением памяти. В результате психических проблем возникает снижение аппетита, потеря веса, нарушения сна. Больного могут даже посещать мысли о самоубийстве. Частью депрессии бывает тревожное расстройство.
Депрессия может чередоваться с периодом гипомании, которому присуще чувство полноты энергии, активности.

Больному не хватает проницательности и самокритики, поэтому ему трудно понять, что он больше не может продолжать принимать решения о содержании семьи или управлять автомобилем.
Серьезные проблемы включают галлюцинации и бред. Галлюцинация – это расстройство восприятия, при котором пациент не может провести различие между реальностью и картинами, которые рисует его воображение. Он воспринимает звуковые и визуальные галлюцинации как истинные, попытки переубеждения бессмысленны. Бред – это расстройство мышления, при котором больные страдают манией преследования, считают, что имеют особое происхождение и способности, бессмертность, безнаказанность и т.д.
Основные клинические симптомы болезни Гентингтона включают слабоумие, встречающееся у всех пациентов. Человек перестает выполнять обычную работу, не может сосредоточиться, постоянно забывает.
Ограниченная подвижность и слабоумие – одни из самых печальных проявлений, потому что они лишают человека самостоятельности на всю жизнь.
Выделяются 3 основных типа болезни Гентингтона:
- Ювенильная форма. Появляется к 20 годам. При этом типе непроизвольные движения обычно отсутствуют, но возникает мышечная скованность. Наблюдается быстрое развитие деменции, расстройств личности, речевых нарушений.
- Классическая форма. Проявляется в возрасте 35-50 лет и встречается в 90% случаев. Характеризуется типичным курсом, описанным выше.
- Форма позднего старта. Появляется после 60 лет. Из всех трех типов этот является одним из самых легких – его течение медленное, значительное слабоумие отсутствует, человек сохраняет самостоятельность.
Диагностика
Фундаментальное обследование – генетическое, доказывающее умножение триплетов CAG. В соответствии с уровнем мутаций оно определяет приблизительный возраст, в котором может проявиться расстройство. Такое тестирование предполагает соблюдение очень строгих этических правил. Только пациенты, считающие это целесообразным, полностью согласные с тестом, могут сдавать анализы. Совершенно недопустимо принуждать человека к тестированию, переубеждать его. Из-за невозможности лечения необходимо связать эти тесты с поддержкой психолога и, при необходимости, социальных работников.

Из вспомогательных исследований (а также в рамках дифференциальной диагностики) можно выполнить КТ или МРТ. Цель этих обследований – определение нарушения базальных ганглиев, особенно хвостатого ядра.
У пациентов с подозрением на болезнь Гентингтона проводится специальный тест. Он подтверждает или исключает подозрение со 100% достоверностью. Всегда необходима подпись пациента, фиксирующая ознакомление с правилами диагностики.
Предиктивный (предсимптомный и пренатальный) тест проводится у бессимптомных людей с заболеванием в семейной истории, желающих узнать, поражены ли они болезнью. Но генетический тест только подтверждает наличие мутации и предпосылок к болезни, а не сам диагноз.
При беременности у женщин, у которых развилась хорея Гентингтона, диагностика может заключаться в проведении теста, направленного на определение генетического статуса их будущего ребенка. Этот пренатальный тест выполняется путем отбора амниотической жидкости (амниоцентез).
Лица, желающие пройти предсимптомное тестирование, должны тщательно рассмотреть последствия. Они имеют право не знать свой генетический статус.
В связи с ответственностью, предсимптомное тестирование не проводится для лиц младше 18 лет.
Лечение
Болезнь Гентингтона в настоящее время неизлечима. Врачи пытаются противодействовать многим ее проявлениям. Заболевание требует сотрудничества специалистов из области неврологии, психологии, психиатрии, физиотерапии, трудотерапии, логопедии и др.
В очень серьезных случаях непроизвольных движений используются антипсихотические, успокоительные средства или нейролептики. Они также применяются в случаях бреда, галлюцинаций, агрессии, беспокойства. Однако такие лекарства имеют много побочных эффектов, поэтому находят применение только в действительно тяжелых случаях, после тщательной оценки возможных рисков.

В процессе лечения хореи Гентингтона важно бороться с потерей веса, принимая высококалорийную пищу (более 5000 ккал/день). Желательно обсудить питание с диетологом. Рекомендуется употреблять жирное мясо, цельные молочные продукты, жирную рыбу, сладкие соки, картофель, шоколад.
Против депрессии эффективны антидепрессанты. При нарушениях сна, вызванных беспокойством, по рекомендации врача принимаются снотворные препараты. Возможно также применение народных методов, например, успокаивающих травяных чаев.
К сожалению, самое серьезное проявление заболевания – слабоумие – совершенно неизлечимо. Его даже нельзя замедлить.
Осложнения
Одно из осложнений – инфекции. Наиболее распространенная инфекция – пневмония, способная быть смертельной для пациента из-за ослабленного иммунитета.
Сосудистые нарушения, закупорка артерий могут привести к сердечной недостаточности. Серьезное осложнение – расстройство глотания и приема пищи. Пациент сильно худеет, истощается.

Все остальные осложнения связаны с симптомами, относящимися к самой болезни. Особенно разрушительным является изменение в поведении. Часто человек становится эгоцентричным, агрессивным, навязчивым. Осложнение этого поведения – более высокая склонность к зависимости. Опасность представляют тенденции к самоубийству, которые при болезни Гентингтона намного выше, чем в среднем по населению.
Важно знать
Болезнь Гентингтона связана не только с самим пациентом, но и с его семьей, окружением. Самые большие опасения относятся к здоровью потомства – генетический риск очень высок. Также нелегко объяснить детям, что на самом деле происходит с их родителями. Почему мама ведет себя так странно, что делает со своими руками и т.д. Часто тема заболевания становится запретной в широкой семье или в обществе, что усугубляет психическое состояние пациента.
Жизнь партнера больного человека выворачивается наизнанку. Он постепенно теряет свое свободное время, которое должен посвятить, заботе о нем. У большинства людей, живущих в семье с больным, развивается депрессия, негативное отношение к миру, вспышки гнева, постоянная грусть и т.д. Нередки случаи пристрастия к алкоголю.
К сожалению, в нашей стране нет специализированного учреждения долгосрочного или краткосрочного лечения этого заболевания. В то же время социальная поддержка играет жизненно важную роль в оказании помощи пациентам и их семьям.
Патоморфология болезни Гентингтона
Болезнь Гентингтона характеризуется гибелью нейронов преимущественно в хвостатом ядре и скорлупе, в некоторой степени также в коре и других структурах головного мозга. Общий вес мозга при болезни Гентингтона снижается не только за счет снижения численности нейронов, но вследствие утраты белого вещества. В коре больших полушарий в наибольшей степени поражаются клетки в слоях V и VI. Выраженность микро- и макроскопических дегенеративных изменений (с коррекцией на возраст к моменту смерти) коррелирует с числом повторов ЦАГ. Детальный патоморфологический анализ изменений нескольких сотен случаев болезни Гентингтона показал, что дегенерация стриатума начинается с дорсомедиальной части хвостатого ядра и дорсолатеральной части скорлупы, а затем распространяется в вентральном направлении. Различные группы нейронов хвостатого ядра и скорлупы страдают не в одинаковой степени. Вставочные нейроны в стриатуме остаются относительно сохранными, но избирательно поражаются некоторые проекционные нейроны. При ювенильной форме болезни Гентингтона патоморфологические изменения в стриатуме более выражены и имеют более распространенный характер, вовлекая кору больших полушарий, мозжечок, таламус, бледный шар.
 [3], [4], [5], [6], [7], [8], [9], [10], [11], [12]
[3], [4], [5], [6], [7], [8], [9], [10], [11], [12]
Нейрохимические изменения при болезни Гентингтона
ГАМК. При нейрохимическом исследовании мозга у больных с болезнью Гентингтона выявлено значительное снижение концентрации ГАМК в стриатуме. Последующие исследования подтвердили, что при болезни Гентингтона снижается численность ГАМКергических нейронов, и показали, что концентрация ГАМК снижена не только в стриатуме, но и в его проекционных зонах — наружном и внутреннем сегментах бледного шара, а также черной субстанции. В мозге при болезни Гентингтона обнаружено также изменение ГАМК-рецепторов с помощью исследований связывания рецепторов и гибридизации in situ мРНК Число ГАМК-рецепторов оказалось умеренно сниженным в хвостатом ядре и скорлупе, но повышено в ретикулярной части черной субстанции и наружном сегменте бледного шара, что, вероятно, объясняется денервационной гиперчувствительностью.
Ацетилхолин. Ацетилхолин используют в качестве нейромедиатора крупные нешипо-видные вставочные нейроны в полосатом теле. В ранних посмертных исследованиях у больных с болезнью Гентингтона было выявлено снижение активности холинацетылтрансферазы (ХАТ) в стриатуме, что могло свидетельствовать об утрате холинергических нейронов. Однако в сравнении со значительным снижением численности ГАМКергических нейронов, холинергические вставочные нейроны остаются относительно сохранными. Следовательно, плотность ацетилхолинэстераза-позитивных нейронов и активность ХАТ в полосатом теле на самом деле относительно повышены, в сравнении с контролем, уравненным по возрасту.
Субстанция Р. Субстанция Р содержится во многих средних шиловидных нейронах полосатого тела, которые преимущественно проецируются на внутренний сегмент бледного шара и черную субстанцию и обычно содержат также динорфин и ГАМК. Уровень субстанции Р в стриатуме и ретикулярной части черной субстанции при болезни Гентингтона снижен. На терминальной стадии заболевания с помощью иммуногистохимических исследований выявлено значительное снижение численности нейронов, содержащих субстанцию Р. На более ранних стадиях нейроны, содержащие субстанцию Р и проецирующиеся на внутренний сегмент бледного шара, относительно сохранны, по сравнению с нейронами, проецирующимися на ретикулярную часть черной субстанции.
Опиоидные пептиды. Энкефалин содержится в средних шиловидных проекционных ГАМКергических нейронах непрямого пути, проецирующихся на наружный сегмент бледного шара и несущих на себе D2-рецепторы. С помощью иммуногистохимических исследований было показано, что на ранней стадии болезни Гентингтона происходит утрата энкефалин-содержащих нейронов, проецирующихся на наружный сегмент бледного шара. Эти клетки, по-видимому, гибнут раньше, чем клетки, содержащие субстанцию Р и проецирующиеся на внутренний сегмент бледного шара.
Катехоламины. Нейроны, содержащие биогенные амины (дофамин, серотонин) и проецирующиеся на полосатое тело, расположены в компактной части черной субстанции, вентральной покрышке и ядрах шва. В то время как норадренергические проекции в полосатое тело человека минимальны, уровни серотонина и дофамина (в пересчете на грамм ткани) в стриатуме оказываются повышенными, что свидетельствует о сохранности этих афферентных проекций на фоне выраженной утраты собственных нейронов стриатума. Дофаминергические нейроны черной субстанции остаются сохранными как при классической, так и при ювенильной формах болезни Гентингтона.
Соматостатин/нейропептид Y и синтетаза оксида азота. Измерение уровня соматостатина и нейропептида Y в стриатуме при болезни Гентингтона выявило их 4-5-кратное увеличение, по сравнению с нормальными тканями. С помощью иммуногистохимических исследований констатирована абсолютная сохранность вставочных нейронов стриатума, содержащих нейропептид Y, соматостатин и синтетазу оксида азота. Таким образом, эти нейроны резистентны к патологическому процессу.
Возбуждающие аминокислоты. Высказывалось предположение, что селективная гибель клеток при болезни Гентингтона связана с индуцированным глутаматом нейротоксическим эффектом. Уровни глутамата и хинолиновой кислоты (эндогенный нейротоксин, представляющий собой побочный продукт метаболизма серотонина и являющийся агонистом глугаматных рецпторов) в стриатуме при болезни Гентингтона изменены незначительно, однако недавнее исследование с помощью МР—спектроскопии выявило in vivo повышение уровня глутамата. Уровень глиального фермента, ответственного за синтез хинолиновой кислоты, в стриатуме при болезни Гентингтона увеличен по сравнению с нормой примерно в 5 раз, в то время как активность фермента, обеспечивающего деградацию хинолиновой кислоты, повышена при болезни Гентингтона только на 20-50%. Таким образом, синтез хинолиновой кислоты при болезни Гентингтона может быть повышен.
Исследования рецепторов возбуждающих аминокислот (ВАК) при болезни Гентингтона выявили значительное снижение численности NMDA-, АМРА-, каинатных и метаботропных глугаматных рецепторов в стриатуме, а также АМРА- и каинатных рецепторов в коре больших полушарий. На поздней стадии болезни Гентингтона NMDA-рецепторы практически отсутствовали, на предклинической и ранней стадии отмечалось значительное снижение численности этих рецепторов.
Избирательная чувствительность. При болезни Гентингтона избирательно гибнут определенные типы стриарных клеток. Средние шиловидные нейроны, проецирующиеся на наружный сегмент бледного шара и содержащие ГАМК и энкефалин, гибнут уже на очень ранней стадии заболевания, так же, как и нейроны, содержащие ГАМК и субстанцию Р и проецирующиеся на ретикулярную часть черной субстанции. Утрата нейронов, содержащих ГАМК и энкефалин и проецирующихся на наружный сегмент бледного шара, растормаживает эту структуру, что, в свою очередь, ведет к активному торможению субталамического ядра. Снижением активности субталамического ядра, по-видимому, можно объяснить хореиформные движения, возникающие при болезни Гентингтона. Давно известно, что очаговые поражения субталамического ядра могут быть причиной хореи. Утрата нейронов, содержащих ГАМК и субстанцию Р и проецирующихся на ретикулярную часть черной субстанции, вероятно, может быть причиной глазодвигательных нарушений, наблюдаемых при болезни Гентингтона. Этот путь в норме тормозит нейроны ретикулярной части черной субстанции, проецирующиеся на верхние бугорки четверохолмия, которые, в свою очередь, регулируют саккады. При ювенильной форме болезни Гентингтона пути, указанные выше, страдают более тяжело и, кроме того, рано утрачиваются стриарные проекции во внутренний сегмент бледного шара.
Белок гентингтин, кодируемый геном, мутация которого вызывает болезнь Гентингтона, выявляется в различных структурах головного мозга и других тканях. В норме гентингтин преимущественно обнаруживается в цитоплазме нейронов. Белок выявляется в большинстве нейронов мозга, но, как показывают последние данные, его содержание выше в матриксных, чем в стриосомных нейронах, а в проекционных нейронах выше, чем во вставочных нейронах. Таким образом, избирательная чувствительность нейронов коррелирует с содержанием в них гентингтина, который в норме представлен в определенных популяциях нейронов.
Как и в мозге больных с болезнью Гентингтона, у мышей, трансгенных по N-терминальному фрагменту гена болезни Гентингтона с увеличенным числом повторов, гентингтин образует плотные агрегаты в ядрах нейронов. Эти внутриядерные включения формируются в стриарных проекционных (но не во вставочных) нейронах. У трансгенных мышей включения образуются за несколько недель до появления симптомов. Эти данные свидетельствуют, что белок гентингтин, содержащий увеличенное число глутаминовых остатков, включение которых кодируют тринуклетидные повторы, или его фрагмент накапливается в ядре, в результате может страдать осуществляемый им контроль клеточных функций.
[13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23]
что это за болезнь, симптомы, диагностика, лечение
Хорея Гентингтона – прогрессирующая, нейродегенеративная болезнь, которая характеризуется двигательными, психическими нарушениями, ухудшением когнитивных функций, неизбежно приводит к летальному исходу. Хореические гиперкинезы (неконтролируемые, беспорядочные движения, возникающие в разных частях тела) составляют основу клинической картины при классической форме течения. Распространенность патологии составляет меньше 5-7 случаев на 100 тысяч населения.

Характеристика
В Википедии хорея описывается как гиперкинез (неконтролируемая, патологическая двигательная активность), который проявляется в виде беспорядочных, отрывистых движений, похожих на нормальные, но отличающихся от вариантов нормы интенсивностью и амплитудой. В неврологии хорея Гентингтона – это болезнь, течение которой можно разделить на бессимптомный период и этап выраженных клинических проявлений, что предполагает особый подход к диагностике на ранней стадии.
Болезнь Гентингтона протекает в три стадии – начальная, умеренных проявлений и развернутой клинической картины. Для патологии типично постепенное, затяжное начало и медленное прогрессирование. Если симптоматика проявляется остро, стремительно, проводится дифференциальная диагностика и поиск вероятных других причин.
Против диагноза БГ свидетельствуют периоды ослабления симптомов или полной ремиссии (отсутствие симптомов). Распространенные ошибочные названия хореи Гентингтона – хорея Лемпингтона или Кенсингтона. Выделяют формы заболевания с учетом доминирующих симптомов:
- Гиперкинетическая (клиническая картина представлена преимущественно гиперкинезами).
- Ригидная (в клинике преобладают расстройства, связанные с повышением тонуса скелетной мускулатуры). В рамках ригидной различают ювенильную форму, которая дебютирует в возрасте 20 лет и более раннем возрасте, и позднюю форму.
- Психическая (в клинике преобладают психические расстройства).
В 10% случаев при поздней ригидной форме хореи Гентингтона, известной так же как болезнь Хантингтона, начальные проявления представлены в виде акинетико-ригидного синдрома (ограничение объема, скорости, амплитуды произвольных движений на фоне повышения мышечного тонуса) с минимальными признаками гиперкинезов.
Нередко поздняя ригидная форма развивается как трансформация гиперкинетической. Тогда заболевание дебютирует гиперкинетическим синдромом (патологическая, повышенная двигательная активность), позже в клинической картине преобладают признаки – брадикинезия (замедление двигательной активности) и ригидность мышц экстрапирамидного типа (изменение мышечного тонуса проявляется как сопротивление пассивному растяжению). В МКБ-10 патология значится под кодом «G10».
Причины возникновения
Болезнь Гентингтона – это хорея первичной, наследственной формы, такое заболевание, которое развивается на фоне мутации генов, что предопределяет вероятность наследования потомками. Заболевание развивается вследствие мутации гена HTT, наследуется потомками обоих полов в равной степени. Чтобы понять, как передается хорея, следует учитывать характер наследования.

Тип наследования хореи Гентингтона – аутосомно-доминантный. Болезнь передается по наследству, если у человека имеется хотя бы один дефектный ген, который находится в аутосомах, вне половых клеток. Если родитель является гетерозиготным носителем мутировавшего гена, его дети с вероятностью 50% унаследуют мутацию. В случаях, когда родитель является гомозиготным носителем дефектного гена, мутация наследуется потомками с вероятностью 100%.
Гомозиготное носительство встречается реже, чем гетерозиготное. Гомозиготное носительство не влияет на особенности клинической картины или на период, когда происходит дебют заболевания. Мутация проявляется увеличением количества повторов CAG – азотистого основания, состоящего из комплекса цитозин-аденин-гуанин.
Мутация приводит к синтезу белка гентингтина, который отличается измененной структурой (избыточная концентрация глутамина). Белок гентингтин оказывает токсическое воздействие на нервную ткань, провоцируя массовую гибель нервных клеток. Чаще атрофические процессы дебютируют в области полосатого тела, затрагивая такие отделы, как скорлупа, бледный шар и хвостатое ядро.
Затем патологический процесс распространяется на близлежащие ткани мозга, охватывая в том числе корковый слой полушарий. Возраст, когда происходит дебют болезни, зависит от числа повторов CAG. Чем больше число повторов, тем раньше происходит развитие болезни. К примеру, число CAG-повторов в границах 36-39 приводит к появлению выраженных симптомов в возрасте старше 65 лет.
Если число повторов превышает 60, дебют болезни приходится на возраст около 20 лет. Влияние числа CAG-повторов на дебют выявляется в 56% случаев. У других пациентов этот период определяется другими влияющими факторами (воздействие внешней среды, мозаицизм – наличие в однотипной ткани генетически различающихся клеток, нестабильность CAG-повторов).
Хорея – это такое заболевание, которое может начаться в любом возрасте, что обусловлено в первую очередь числом CAG-повторов, а также другими влияющими факторами. Течение патологии предполагает наличие периода бессимптомного носительства мутировавшего гена.
Число CAG-повторов чаще увеличивается у представителей следующих поколений. В 5-10% у пациентов не выявляется отягощенный семейный анамнез – наличие родственников, которые являются носителями дефектного гена. В этих случаях речь идет о спорадической форме болезни, о новой мутации.
Симптоматика
Первые, слабо выраженные симптомы хореи Гентингтона в виде незначительных двигательных, психических, интеллектуально-мнестических расстройств могут появиться за 10 лет до развития развернутой формы заболевания с выраженной клинической картиной. За 10-15 лет до развернутой формы болезни локальные, сегментарные атрофические изменения в нервной ткани обнаруживаются в ходе исследования головного мозга в формате МРТ, КТ.
В продромальный (предшествующий появлению симптоматики) период невозможно поставить диагноз на основании клинических наблюдений или по результатам инструментального исследования. Клиническая картина при БГ характеризуется многообразием вариантов. Симптомы болезни Гентингтона отражают двигательные нарушения, которые проявляются в форме:
- Хореи (неконтролируемые патологические движения).
- Атетоза (гиперкинез, проявляющийся медленной судорогой тонического типа в зоне лица, конечностей, туловища).
- Дистонии (медленные, неритмичные сокращения мышц, часто провоцирующие возникновение патологической позы).
- Постуральной неустойчивости (нарушение равновесия вследствие ослабления стабилизирующих рефлексов).
- Миоклонии (кратковременное, быстрое сокращение мышц).
- Баллизма (резкие движения крупной амплитуды размашистого характера с вращательными компонентами, затрагивающие проксимальные отделы конечностей).
- Брадикинезии (замедление произвольных содружественных движений на фоне повышения мышечного тонуса).
- Ригидности (твердости, неподатливости) мышц.
- Окуломоторных (глазодвигательных) расстройств.
Психические нарушения при хорее Гентингтона (Хантингтона) представлены повышенной раздражительностью, апатией, отсутствием интереса к окружающей действительности, усиленной тревожностью. У больных нередко выявляется депрессивное состояние, расстройства обсессивно-компульсивного типа, психопатии, гиперсексуальность.
Признаки нарушения двигательной и психической деятельности при болезни Гентингтона (Хантингтона) дополняются симптомами расстройства интеллектуально-мнестических функций. Нередко выявляются такие признаки, как расстройство сна, снижение массы тела, иногда кахексия (крайнее истощение, проявляющееся сокращением мышечной массы, общей слабостью, замедлением физиологических процессов в организме).

Хорея Гентингтона, известная так же как синдром Хантингтона, проявляется неусидчивостью, моторным возбуждением, сложностями при совершении движений, основанных на тонкой координации. Пациенты жалуются на неловкость, трудности, возникающие при двигательной активности. Типичное проявление при хорее Гентингтона – дисфагия (нарушение функции глотания), которая нередко становится причиной аспирационного синдрома (попадание пищи в дыхательные пути) и смерти пациента.
Пациенты жалуются на беспокойство в верхних и нижних конечностях, которое усиливается под воздействием провоцирующих факторов – стресс, физическая активность (ходьба, работа руками). Клиническая картина может изменяться по мере прогрессирования патологии. Нередко выраженная на начальных этапах хорея сменяется другими неврологическими нарушениями. В некоторых случаях ухудшение когнитивных способностей и психические расстройства предваряют моторную дисфункцию.
Болезнь Хантингтона обязательно сопровождается глазодвигательными расстройствами, что представлено, к примеру, замедлением саккад – содружественных движений, совершаемых глазными яблоками, такие нарушения нередко появляются на ранних этапах. Прогрессирование БГ приводит к ухудшению глазодвигательных функций. Пациент не может сфокусировать взгляд на предмете.
Для поздних стадий течения типичны экстрапирамидные расстройства, проявляющиеся паркинсонизмом и дистонией. Пациенту сложно удерживать позу, выполнять движения, обусловленные тонкой моторикой и точной координацией. Темп движений существенно замедляется, нередки случаи потери равновесия и падения. В клинической картине появляется дизартрия (нарушение функции речи вследствие расстройства иннервации элементов речевого аппарата).
При ювенильной (юношеской) форме нередки эпилептические приступы, которые практически не встречаются, когда болезнь дебютировала у взрослых пациентов. Для ювенильной формы характерно тяжелое течение и малая продолжительность жизни. Когнитивные нарушения – универсальный диагностический критерий.
Они появляются на ранних этапах течения и затрагивают преимущественно исполнительные функции, которые управляют когнитивными процессами. Пациент не способен планировать будущее, адекватно оценивать последствия поступков, регулировать поведение. Для больных типичны импульсивные действия, отсутствие инициативы, апатия. Распространенные психические нарушения:
- Раздражительность, агрессия.
- Персеверации (устойчивое повторение фразы, эмоции, деятельности).
- Психозы, обсессивно-компульсивные расстройства.

Среди больных частота реализованных суицидов в 4 раз превышает общие показатели в популяции. Для БГ характерно снижение показателей массы тела. Причем снижение веса не связано с анорексией, часто происходит на фоне повышения аппетита. По данным научных исследований более высокий индекс массы тела ассоциируется с замедлением темпов прогрессирования патологии.
Диагностика
С целью выявления заболевания проводится пресимптоматическое (до появления клинических проявлений) обследование лиц, чьи близкие родственники болели БГ, на наличие генов, ответственных за развитие патологии. Диагностика хореи Гентингтона предполагает сбор анамнеза. Наличие в клинической картине когнитивных нарушений ассоциируется с необъективной оценкой пациентом своего состояния.
В этом случае рекомендовано провести опрос родственников больного. Большое значение имеет скорость прогрессирования неврологической симптоматики. Дифференциальная диагностика проводится в отношении инфекционных поражений, метаболических нарушений, интоксикаций, которые провоцируют появление гиперкинезов.
При постановке диагноза учитывают, что гиперкинетический синдром и дискинезии нередко являются следствием приема фармацевтических препаратов (нейролептики, препараты Леводопы, блокаторы рецепторов дофамина, бензодиазепины, антиконвульсанты, холинолитики). К примеру, хорея беременных развивается обычно в I триместре гестации, не сопровождается другими неврологическими или психическими нарушениями. Спровоцировать хорею у беременных могут такие заболевания и состояния в анамнезе, как ревматическая лихорадка, антифосфолипидный синдром.
Хорея может появляться как следствие тиреотоксикоза (гиперфункция щитовидной железы) или красной волчанки системного типа. Повреждение церебральных сосудов, пролегающих в зоне базальных ядер, нарушения мозгового кровотока, могут стать причиной возникновения хореи. Оценка когнитивных функций осуществляется по шкале ММSE (Mini-Mental State Examination). Для выявления мутантного гена проводится генетическое тестирование.
Инструментальные методы диагностики включают МРТ мозгового вещества, электроэнцефалографию (при наличии эпилептических приступов в анамнезе). Исследования в формате КТ и МРТ не имеют решающего диагностического значения при обнаружении дефектного гена в ходе генетического тестирования. Врач назначает лечение, опираясь на результаты анализов и инструментальной диагностики.

Лечение
Болезнь Гентингтона неизлечима, лечение направлено на устранение симптомов. Современные методы терапии не способны остановить прогрессирование заболевания, однако улучшают качество жизни пациента, облегчают уход за ним, осуществляемый родственниками. Фармацевтические препараты не назначают носителям дефектного гена с целью профилактики заболевания.
Медикаментозное лечение хореи направлено на коррекцию психических и двигательных нарушений, стимуляцию когнитивных функций. Устранение хореи осуществляется препаратом Тетрабеназин, если у пациента не проявляются такие нарушения, как раздражительность, депрессия, суицидальные настроения. Дозировка подбирается индивидуально.
На фоне длительного приема эффективность Тетрабеназина понижается независимо от увеличения дозы. При наличии депрессии целесообразно сочетать Тетрабеназин с антидепрессантом. При наличии противопоказаний к назначению Тетрабеназина показаны нейролептики (Тиаприд, Арипипразол, Галоперидол).
Лечение хореи Гентингтона предполагает применение противоэпилептического препарата Клоназепам, который эффективен в отношении миоклоний и дистонии. Препараты с противоэпилептическим, противосудорожным действием (Вальпроевая кислота, Леветирацетам) способствуют уменьшению проявлений миоклонии и увеличению массы тела.
В ряде случаев коррекция дистонии осуществляется инъекциями ботулотоксина. Иногда для уменьшения проявлений дистонии назначают лечебную гимнастику. Препарат Ривастигмин показан для стимуляции когнитивных функций. Для лечения депрессии назначают антидепрессанты (Венлафаксин, Флуоксетин, Сертралин). Занятия с логопедом на ранних стадиях показаны при дизартрии.

Последствия и профилактика
Прогноз относительно неблагоприятный. Эффективного лечения хореи Гентингтона не найдено – летальный исход неизбежен. Продолжительность жизни обычно варьируется в пределах 15-20 лет от момента появления начальных признаков. Причины смерти связаны с осложнениями, в числе которых нарушение сердечной деятельности, травмы. В среднем в 5% случаев летальный исход наступает в результате реализованной попытки суицида.
Частое осложнение дисфагии при БГ – аспирационная пневмония (воспаление легких, развивающееся на фоне попадания в органы дыхания частиц пищи), которая часто является причиной смерти больных. В качестве профилактической меры при беременности носителя дефектного гена рекомендовано пренатальное (дородовое) генетическое обследование.
Хорея Гентингтона – генетическое заболевание, которое наследуется по аутосомно-доминантному типу, сопровождается двигательными и психическими нарушениями, ухудшением когнитивных функций.
ПОЖАЛУЙСТА, ОЦЕНИТЕ СТАТЬЮ!
Просмотров: 94
Болезнь Гентингтона — Википедия
Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона или Хантингтона) — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Эпидемиология
В настоящее время от хореи Хантингтона в США страдает около 7000 человек. Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000, и 1:1000000 среди остальных рас[1]. Название болезни дано в честь трёх поколений врачей, изучавших её в штате Коннектикут. В частности, считается, что заболевание названо в честь американского врача Джорджа Хантингтона, первым давшего его классическое описание[2][3].
Генетика
Ген HTT, присутствующий у всех людей, кодирует белок хантингтин (Htt). Ген HTT расположен на коротком плече 4-й хромосомы (4p16.3)[4]. Этот ген содержит в себе участок с повторяющейся последовательностью трёх азотистых оснований — цитозин-аденин-гуанин (то есть, ЦАГЦАГЦАГ…). Триплет ЦАГ кодирует аминокислоту глутамин, поэтому синтезируемый белок хантингтин содержит последовательность глутаминовых аминокислот, называемую полиглутаминовый тракт[5].
Количество ЦАГ триплетов различно у отдельных лиц и может изменяться с последующими поколениями. Если их становится больше 36, то синтезируется удлинённый полиглутаминовый тракт и происходит образование мутантного белка хантингтина (mHtt)[6], который оказывает токсичное действие на клетки и вызывает болезнь Хантингтона. Как правило, от числа ЦАГ повторов зависит степень повреждений, наличие около 60 % повторов сверх нормы вызывает появление симптомов в различном возрасте[4]. 36—40 повторов приводят к редуцированной пенетрантности формы этого заболевания, которая намного позже проявляется и медленнее прогрессирует. В некоторых случаях начало болезни может быть настолько поздним, что симптомы никогда не обнаруживаются[7]. При очень большом количестве повторов болезнь Хантингтона имеет полную пенетрантность и может проявиться до 20 лет, тогда болезнь классифицируется как ювенильная, акинетически-ригидная или Вестфаль варианты. Составляет приблизительно 7 % случаев болезни Хантингтона[8].
Мутантный ген был предположительно завезён в США в 1630 году двумя братьями, эмигрировавшими из Эссекса в Бостон[9][10].
Ребенок, не унаследовавший заболевание, не может передать его своим детям[11].
Патогенез
Htt-белок взаимодействует с сотней других белков и, вероятно, выполняет множество биологических функций[12]. Механизм действия mHtt до конца не ясен, но известно, что он токсичен для некоторых типов клеток, особенно в головном мозге. В основном происходит поражение полосатого тела (стриатума), но при прогрессировании заболевания и другие области головного мозга значительно повреждаются[6]. Планирование и коррекция движений — основная функция полосатого тела, и нарушения в этой области провоцируют симптомы[6].
Функция Htt
Htt образуется во всех клетках млекопитающих. Наибольшая его концентрация — в головном мозге и яичках, а также в умеренных количествах в печени, сердце и лёгких[6]. Функция Htt у человека не ясна. Он взаимодействует с белками, участвующими в транскрипции, передаче сигнала в клетке и внутриклеточном транспорте[6][13]. Некоторые функции Htt обнаружены в экспериментальных моделях животных: играет важную роль в развитии эмбриона и связан с гибелью эмбриона при отсутствии белка[14]. Он также выступает в качестве анти-апоптозного агента, предотвращая запрограммированную гибель клеток, и контролирует образование нейротрофического фактора мозга (белок, защищающий нейроны и регулирующий их образование во время нейрогенеза). Если экспрессия Htt возрастает, выживаемость нервных клеток увеличивается и эффект mHtt уменьшается, наоборот, понижение экспрессии Htt даёт картину более типичную присутствию mHtt[14]. У людей разрушение нормального гена не приводит к болезни. В настоящее время считается, что болезнь вызывает не недостаточное образование Htt, а усиление токсического эффекта mHtt[6].
Клеточные изменения под действием mHtt
Под действием образовавшегося mHtt происходит множество изменений в клетке, что вызывает болезнь Хантингтона. Удлинение полиглутаминовой последовательности изменяет конформацию белка хантингтина и прочно соединяет его с другими белками[15]. Это приводит к агрегации хантингтина, при этом образуются так называемые внутриклеточные тельца включения[16]. Эти включения механически препятствуют движению везикул, содержащих нейромедиаторы, через цитоскелет, что нарушает передачу сигналов в нейронах[16]. Тельца включения обнаруживаются как в ядрах клеток, так и в цитоплазме. Некоторые эксперименты показали, что они могут быть токсичны для клеток, а другие — что тельца, наоборот, защищают нейрон от смерти, аккумулируя мутантный хантингтин, и именно неагрегированный белок токсичен[17].
Существует несколько путей, при которых mHtt вызывает гибель клеток. К ним относят: влияние на белки-шапероны; взаимодействие с каспазами, которые участвуют в апоптозе; токсическое действие глутамина на нервные клетки; нарушение выработки энергии в клетках и влияние на экспрессию генов. Токсическое действие mHtt значительно усиливается при взаимодействии с белком RASD2 (Rhes), который образуется преимущественно в стриатуме. RASD2 вызывает сумоляцию (SUMOylation) mHtt к образованию белковых сгустков и дезагрегации — исследования в культуре клеток показали, что сгустки менее токсичны, чем дезагрегированная форма[18].
Макроскопические изменения под действием mHtt
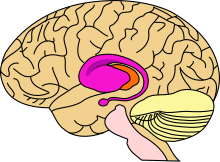 Область головного мозга, поражающаяся при болезни Хантингтона, — стриатум (розовым цветом)
Область головного мозга, поражающаяся при болезни Хантингтона, — стриатум (розовым цветом)
Болезнь Хантингтона поражает специфические области мозга. Наиболее заметные ранние изменения затрагивают область базальных ганглиев, называемую полосатым телом, которое состоит из хвостатого ядра и скорлупы[6]. Другие повреждаемые области включают чёрную субстанцию, 3, 5 и 6 слои коры головного мозга, гиппокамп, клетки Пуркинье в мозжечке, боковые туберальные ядра гипоталамуса и часть таламуса[4]. Эти области получают повреждения в соответствии с их структурой и типами содержащихся в них нейронов, уменьшаясь в размерах в связи с гибелью клеток[4]. Звёздчатые нейроны полосатого тела наиболее уязвимы, особенно проецирующиеся в направлении поверхности бледного шара, вставочные и звёздчатые нейроны, проецирующиеся к центру бледного шара, получают меньше повреждений[4][19]. Болезнь Хантингтона также вызывает аномальное увеличение астроцитов[20].
Базальные ганглии — часть головного мозга, наиболее заметно повреждающиеся при болезни Хантингтона — играют ключевую роль в контроле движений и поведения. Их функция полностью не ясна, но современные теории предполагают, что они являются частью когнитивной исполнительной системы.
Базальные ганглии в норме ингибируют большое число контуров (circuit), генерирующих специфические движения. Для инициации специфических движений кора посылает сигналы базальным ганглиям для снятия ингибирования. Повреждение базальных ганглиев может привести к снятию ингибирования или его постоянным неконтролируемым изменениям, что служит причиной затруднения начала движения или к их непроизвольной инициации, или движение может быть прервано до или после достижения желаемого результата. Накапливающиеся повреждения в этой области приводят к беспорядочным движениям, характерным для болезни Хантингтона[21].
Симптомы
Симптомы болезни Хантингтона могут проявиться в любом возрасте, но чаще это происходит в 35-44 года[22][23]. На ранних стадиях происходят небольшие изменения личности, когнитивных способностей и физических навыков[22]. Обычно первыми обнаруживают физические симптомы, так как когнитивные и психические расстройства не столь выражены в ранних стадиях[22]. Почти у всех пациентов болезнь Хантингтона в итоге проявляется схожими физическими симптомами, но начало заболевания, прогрессирование и степень когнитивных и психических нарушений различаются у отдельных лиц[24][25].
Для начала заболевания наиболее характерна хорея — беспорядочные, неконтролируемые движения. Хорея в начале может проявляться в беспокойстве, небольших непроизвольных или незавершённых движениях, нарушении координации и замедлении скачкообразных движений глаз[22].
В самом начале обычно возникают проблемы из-за физических симптомов, которые выражаются в резких, внезапных и не поддающихся контролю движениях. В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Чаще всего происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, может возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм, агрессия, навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания.
Диагностика
Клинические методы
Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить область распространения болезни[22]. Медицинская визуализация (компьютерная томография (КТ), магнитно-резонансная томография (МРТ)) показывает только видимую атрофию мозга на прогрессирующей стадии заболевания. Методы функциональной нейровизуализации (фМРТ и позитронно-эмиссионная томография (ПЭТ)) могут показать изменения в активности мозга до появления клинических симптомов[4].
Генетические методы
Для проведения генетической диагностики болезни Хантингтона необходим забор крови с последующим определением количества повторов ЦАГ в каждом НТТ-аллеле[26]. Положительный результат не подтверждает диагноз, поскольку может быть получен за несколько лет до появления первых симптомов. Однако отрицательный результат однозначно свидетельствует об отсутствии вероятности развития болезни Хантингтона[4].
Эмбрионы
Эмбрионы, полученные в результате экстракорпорального оплодотворения, могут быть подвержены генетической диагностике болезни Хантингтона с применением преимплантационной генетической диагностики. При этом методе забирается одна клетка из 4-8-клеточного эмбриона и затем проверяется на генетическую патологию. Полученная информация может впоследствии быть использована при выборе здорового эмбриона для имплантации. Кроме того, возможна пренатальная диагностика для эмбриона или плода в утробе матери[27].
Дифференциальная диагностика
Около 90 % диагнозов болезни Хантингтона, основанных на обнаружении типичных симптомов и семейном анамнезе, подтверждаются генетическим тестированием. Большинство других расстройств с аналогичными симптомами называют ХГ-подобными расстройствами (англ. HD-like disorders, HDL)[28]. Причины большинства HDL-заболеваний неизвестны. Известно лишь, что некоторые из них возникают в результате мутаций генов PRNP (HDL1), junctophilin 3 (HDL2), рецессивно наследуемого HTT гена (HDL3 — обнаружен у одной семьи и мало изучен) и гена, кодирующего ТАТА-связывающий белок (HDL4/SCA17)[28]. К другим заболеваниям с аутосомно-доминантным наследованием, которые схожи с болезнью Хантингтона, относят дентаторубро-паллидолюисовую атрофию и нейроферритинопатию[28].
Лечение
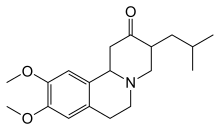 Химическая структура тетрабеназина, разрешённого для лечения болезни Хантингтона
Химическая структура тетрабеназина, разрешённого для лечения болезни Хантингтона
Болезнь Хантингтона неизлечима, но существует лечение, способное облегчить некоторые симптомы.[29]
Тетрабеназин был разработан специально для уменьшения тяжести симптомов болезни Хантингтона[30], был утвержден в 2008 году в США[31]. Нейролептики и бензодиазепины помогают уменьшить проявления хореи[23]. Амантадин и ремацемид находятся в стадии исследования, но показали положительные результаты[32]. Для облегчения гипокинезии и ригидности мышц назначают противопаркинсонические лекарства, для облегчения миоклонической гиперкинезии — вальпроевую кислоту[23].В России препарат продается под торговым названием Нормокинезтин.С 1 января 2018 года Нормокинезтин включен в обновленный перечень жизненно необходимых лекарственных препаратов.
Для устранения депрессии применяют селективные ингибиторы обратного захвата серотонина и миртазапин, а при психозах и нарушениях поведения назначают атипичные антипсихотики[33].
В настоящий момент ведутся активные исследования по разработке способа лечения, исследуются потенциальные направления для лечения болезни Хантингтона[34]. Так компания Teva исследовала препарат-иммуномодулятор лахинимод, обладающий протективным действием по отношению к ЦНС. Испытания препарата дошли до II фазы, но в ходе КИ препарату не удалось достигнуть конечной точки оценки эффективности. Однако испытующие определили, что на фоне терапии лахинимодом происходит снижение скорости атрофии головного мозга. Исходя из опыта неудавшегося исследования, компания приняла решение об отказе от дальнейшего изучения лекарственного препарата[35][36].
Прогноз
С момента появления первых симптомов продолжительность жизни составляет около 15—20 лет.
Смерть обычно происходит не из-за болезни Хантингтона, а из-за сопутствующих ей осложнений, включая пневмонию, заболевания сердца и травмы.
Частой причиной смерти является суицид.
Информационные ресурсы
АНО «Редкие Люди»- единственная благотворительная организация в РФ помогающая пациентам с диагнозом болезнь Гентингтона.
Информационный портал о болезни Гентингтона www.Гентингтона.рф
Примечания
- ↑ NCBI OMIM. Huntington’s Disease. Проверено 22 мая 2008.
- ↑ George Huntington (1850—1916) and Hereditary Chorea
- ↑ George Huntington (1850—1916) and hereditary chorea
- ↑ 1 2 3 4 5 6 7 Walker FO (2007). «Huntington’s disease». Lancet 369 (9557). DOI:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ Katsuno M, Banno H, Suzuki K, et al. (2008). «Molecular genetics and biomarkers of polyglutamine diseases». Curr. Mol. Med. 8 (3): 221–34. DOI:10.2174/156652408784221298. PMID 18473821. Проверено 2009-04-01.
- ↑ 1 2 3 4 5 6 7 Walker FO (2007). «Huntington’s disease». Lancet 369 (9557). DOI:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ Walker FO (2007). «Huntington’s disease». Lancet 369 (9557). DOI:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ Nance MA, Myers RH (2001). «Juvenile onset Huntington’s disease—clinical and research perspectives». Ment Retard Dev Disabil Res Rev 7 (3): 153–7. DOI:10.1002/mrdd.1022. PMID 11553930.
- ↑ Vessie, P. R. (1932) «On the transmission of Huntington’s chorea for 300 years—the Bures family group». Journal of Nervous and Mental Disease, Baltimore 76: 553—573.
- ↑ Wexler, A. (2008) «The Woman Who Walked into the Sea: Huntington’s and the Making of a Genetic Disease», Yale University Press; 1 edition
- ↑ Медицинская энциклопедия. — Астрель, 2009. — С. 186.
- ↑ Goehler H, Lalowski M, Stelzl U, et al. (2004). «A protein interaction network links GIT1, an enhancer of Huntingtin aggregation, to Huntington’s disease». Mol. Cell 15 (6): 853–65. DOI:10.1016/j.molcel.2004.09.016. PMID 15383276. Проверено 2009-04-27.
- ↑ Harjes P, Wanker EE (2003). «The hunt for huntingtin function: interaction partners tell many different stories». Trends Biochem. Sci. 28 (8): 425–33. DOI:10.1016/S0968-0004(03)00168-3. PMID 12932731. Проверено 2009-04-27.
- ↑ 1 2 Cattaneo E, Zuccato C, Tartari M (2005). «Normal huntingtin function: an alternative approach to Huntington’s disease». Nat. Rev. Neurosci. 6 (12): 919–30. DOI:10.1038/nrn1806. PMID 16288298.
- ↑ Rubinsztein DC, Carmichael J (2003). «Huntington’s disease: Molecular basis of neurodegeneration». Expert Rev Mol Med 5 (20): 1–21. DOI:10.1017/S1462399403006549. PMID 14585171.
- ↑ 1 2 Huntingtin Protein and Protein Aggregation | HOPES — A guide to the science of Huntington’s disease (недоступная ссылка — история). Архивировано 31 августа 2010 года.
- ↑ Arrasate, M. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431(7010):805-10.
- ↑ Subramaniam S, Sixt KM, Barrow R, Snyder SH (2009). «Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity». Science 324 (5932): 1327–30. DOI:10.1126/science.1172871. PMID 19498170.
- ↑ Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia A-S, McNamara JO, Williams SM. Modulation of Movement by the Basal Ganglia – Circuits within the Basal Ganglia System // Neuroscience / Purves D. — 2nd. — Sunderland, MA : Sinauer Associates, 2001. — ISBN 0-87893-742-0.
- ↑ Lobsiger CS, Cleveland DW (2007). «Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease». Nat. Neurosci. 10 (11): 1355–60. DOI:10.1038/nn1988. PMID 17965655.
- ↑ Crossman AR (2000). «Functional anatomy of movement disorders» (PDF). J. Anat. 196 (Pt 4): 519–25. DOI:10.1046/j.1469-7580.2000.19640519.x. PMID 10923984.
- ↑ 1 2 3 4 5 Walker FO (2007). «Huntington’s disease». Lancet 369 (9557). DOI:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ 1 2 3 Huntington Disease. genereviews bookshelf. University of Washington (19 июля 2007). Проверено 12 марта 2009.
- ↑ Kremer B. Clinical neurology of Huntington’s disease // Huntington’s Disease – Third Edition / Bates G, Harper P, and Jones L. — Oxford : Oxford University Press, 2002. — P. 28–53. — ISBN 0-19-851060-8.
- ↑ Wagle, A C; Wagle SA, Marková IS, Berrios GE (2000). «Psychiatric Morbidity in Huntington’s disease.». Neurology, Psychiatry and Brain Research (8): 5–16.
- ↑ Myers RH (2004). «Huntington’s disease genetics». NeuroRx 1 (2): 255–62. DOI:10.1602/neurorx.1.2.255. PMID 15717026.
- ↑ Kuliev A, Verlinsky Y (2005). «Preimplantation diagnosis: A realistic option for assisted reproduction and genetic practice». Curr. Opin. Obstet. Gynecol. 17 (2): 179–83. DOI:10.1097/01.gco.0000162189.76349.c5. PMID 15758612. Проверено 2009-04-01.
- ↑ 1 2 3 Schneider SA, Walker RH, Bhatia KP (2007). «The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test». Nat Clin Pract Neurol 3 (9): 517–25. DOI:10.1038/ncpneuro0606. PMID 17805246. Проверено 2009-03-18.
- ↑ Frank S, Jankovic J. (2010). «Advances in the Pharmacological Management of Huntington’s Disease». Drugs 70 (5): 561–71. DOI:10.2165/11534430-000000000-00000. PMID 20329804. Проверено 2011-04-25.
- ↑ Walker FO (2007). «Huntington’s disease». Lancet 369 (9557). DOI:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ FDA Approves First Drug for Treatment of Chorea in Huntington’s Disease. FDA Approves First Drug for Treatment of Chorea in Huntington’s Disease. U.S. Food and Drug Administration (August 15, 2008). Проверено 10 августа 2008. Архивировано 1 июня 2012 года.
- ↑ Walker FO (2007). «Huntington’s disease». Lancet 369 (9557). DOI:10.1016/S0140-6736(07)60111-1. PMID 17240289.
- ↑ Bonelli RM, Wenning GK, Kapfhammer HP (2004). «Huntington’s disease: present treatments and future therapeutic modalities». Int Clin Psychopharmacol 19 (2): 51–62. DOI:10.1097/00004850-200403000-00001. PMID 15076012. Проверено 2009-04-01.
- ↑ Обнаружено потенциальное направление для лечения болезни Гентингтона Архивная копия от 17 мая 2012 на Wayback Machine
- ↑ Лахинимод оказался неэффективен против болезни Хантингтона remedium.ru, 06.09.2018
- ↑ Teva drops development of laquinimod PharmaTimes Media, 06.09.2018
Болезнь Гентингтона – это патология нервной системы, которая имеет и другие названия – хорея Гентингтона, хорея дегенеративная, хорея прогрессирующая, хорея пляска Витта. Обычно недуг проявляется в самом продуктивном возрасте от 25 до 50 лет в виде «двигательного хаоса». У болезни неблагоприятный исход, летальность наступает обычно через 10-13 лет после дебюта. В мире не научились лечить эту тяжелую патологию, но в Германии благодаря активному внедрению новейших технологий добиваются замедления ее проявлений и улучшения качества жизни больного человека.
Хорея Гентингтона – что это такое?
Хорея Гентингтона – что это такое, что это за болезнь? Патология была описана американским врачом в 19 веке и была названа его именем. Болезнь вызывает дегенеративное поражение нервной системы в связи с синтезом патологического белка гентингтина и характеризуется сочетанием снижения интеллекта и прогрессирующего гиперкинеза. Непроизвольные быстрые движения разной силы («пляска Витта») делают жизнь пациента невыносимой, а его слабоумие отражается на жизни окружающих людей.
Болезнь практически не встречается в детском возрасте. Симптомы заболевания проявляются по мере взросления человека и достигают максимального эффекта в возрасте 30-35 лет. Но болезнь может манифестировать и к 80 годам. Все носители патологического гена в итоге заболевают. Но в преклонном возрасте чаще останавливаются на диагнозе старческой деменции, не рассматривая вариант проявления хореи. Женщины болеют несколько реже мужчин, то есть шансов передачи дефектного гена мальчику в несколько раз больше, чем девочке.
Прогноз болезни неблагоприятный. Болезнь заканчивается распадом личности и гибелью больного от осложнений.
Причины хореи Гентингтона
Причины хореи Гентингтона тщательно изучаются. Считается, что патология вызвана генетическим дефектом, который под влиянием некоторых внешних факторов дает о себе знать. Пораженный ген передается по наследству, нарушает биохимические связи и приводит к изменению нормальной функции нейронов головного мозга из-за образования патологического белка. Нервные клетки изменяют свою структуру, участки коры и некоторые отделы головного мозга атрофируются и перестают контролировать двигательную активность. Кроме того, масса мозга больных неотвратимо уменьшается, что влечет прогрессирование слабоумия.
Признаки хореи Гентингтона
Болезнь начинается как бы исподволь и прогрессирует очень медленно. Поэтому от первых признаков до серьезного ухудшения состояния может пройти 10-15 лет. Очень редко дебют болезни возникает в детском и подростковом возрасте (вариант Вестфаля). В этом случае болезнь протекает более злокачественно.
Наиболее характерные для прогрессирующей хореи следующие признаки:
- Гиперкинезы.
В связи с снижением тормозного влияния коры головного мозга на двигательную активность, появляются непроизвольные хаотические движения. Сначала усиливается мимика лица и жестикуляция. Окружающие не воспринимают эти проявления как начало болезни. В дальнейшем лицевая мимика становится выраженной и напоминает гримасы. Пациент может высовывать язык, поочередно хмурить брови, надувать щеки и активно жестикулировать. В этом периоде близкие могут обратиться к врачу.
Помимо гипертрофированной мимики появляются отрывистые движения в конечностях. Пальцы быстро сгибаются и разгибаются. Ноги скрещиваются или поочередно разводятся в сторону. Гиперкинез распространяется и на походку. Пациент как будто танцует, передвигаясь, или шатается, как пьяный. В дальнейшем гиперкинез усиливается и приобретает характер атетоза. Пациент самостоятельно не может передвигаться. Он не может сам себя обслуживать. Появляются проблемы с глотанием. Если заболевание возникает в молодом возрасте, болезнь могут сопровождать эпилептические припадки.
- Расстройство интеллекта вплоть до слабоумия.
В первое время человек жалуется на трудность концентрирования, неусидчивость, повышенную суетливость. Память сохранена долгое время, но мышление меняется уже на начальных этапах болезни. Пациент не может решить элементарные задачки, его подводит логика, а в дальнейшем больной не может критически оценить свои поступки и действия. Он перестает узнавать знакомые предметы и лица, усиливаются вредные привычки, возникают фобии, дезориентация в пространстве и времени. Иногда появляется бред, навязчивые мысли.
- Перепады настроения.
Больные страдают эмоциональной лабильностью. Для них характерны приступы ярости, раздражительности и паники без особых на то причин. У некоторых пациентов появляется повышенное либидо, гиперсексуальность. Перепады настроения могут заканчиваться тяжелой депрессией и попытками суицида. Психика человека сильно изменяется.
- Глазодвигательное расстройство.
Изменения будут видны уже на ранних стадиях болезни. Снижается скорость перевода взгляда и точность слежения. По мере прогрессирования возникает нистагм. Быстрое движение глаз нарушает сон.
- Нарушение речевой функции.
Речь прерывается внезапными причмокиванием, всхлипыванием, больной шмыгает носом. Нарушается артикуляция. В дальнейшем начинаются проблемы со звукоизвлечением, меняется скорость и ритм речи. На поздних стадиях пациент не может внятно произносить слова, его трудно понять.
Больной умирает из-за осложнений (сердечная недостаточность, пневмония). Через 15-20 лет после начала проявлений болезни организм его полностью истощен, как в физическом, так и в нервном отношении.
Диагностика хореи Гентингтона
Диагностика болезни основывается на сборе анамнеза (желательно присутствие при этом близких людей), общении, осмотре пациента и молекулярно-генетических исследованиях. Инструментальные виды обследования позволяют исключить другие болезни головного мозга, но они не являются специфическими, поэтому их назначение не обязательно.
- Осмотр и сбор анамнеза пациента
На этом этапе врач выясняет, когда появились первые симптомы болезни, есть ли родственники с похожими симптомами. При осмотре на ранней стадии специалист обращает внимание на поведение пациента, его осознанность, самокритичность и адекватность.
- Генетические и молекулярные исследования
Исследование крови в генетической лаборатории помогает выявить дефект в образцах ДНК. Методика полимеразной цепной реакции одна из самых последних разработок, которая дает основание поставить диагноз хореи Гентингтона.
Лечение хореи Гентингтона
Терапия носит комплексный характер. Специфического лечения нет, поэтому терапия не приносит выздоровление, но улучшить качество жизни, облегчить состояние больного и отсрочить появление осложнений современная медицина вполне может.
Лечение будет симптоматическим и поддерживающим. Оно зависит от тяжести протекания синдрома, от возраста и индивидуальных особенностей организма. С пациентом общается психоневролог, психиатр, при необходимости специалисты смежных специальностей. Для сохранения сознания необходима грамотная реабилитация и длительный качественный присмотр специалистами.
Преимущества лечения хореи Гентингтона в Германии
- Современные лаборатории, в которых возможно проведение генетического исследования на должном уровне и на новейшем оборудовании.
- Специализированные центры по лечению хореи, в которых работают опытные специалисты.
- Качественные лекарственные препараты, которые доказали свою эффективность, лицензированы и допущены к применению при лечении хореи.
- Дружелюбная атмосфера и чуткое отношение к пациентам всего медперсонала.
- Специалисты работают не только с пациентом, но и с его родственниками, обучают их правильному общению и грамотному уходу за больным, оказывают моральную психологическую поддержку.
- Оптимальное соотношение цены и качества лечения.
- Конфиденциальность.
- Продуманная индивидуальная поддерживающая терапия.
Хорея Гентингтона – тяжелое испытание для родных. Чем раньше выявлено заболевание, тем больше возможностей продлить осознанную жизнь пациента, облегчить состояние больного и жизнь близких людей. Поэтому при подозрении на болезнь надо срочно обращаться к квалифицированным и знающим специалистам.
Недавно подтверждена безопасность для человека одного из перспективных препаратов для лечения болезни Гентингтона – HTTRx, представляющего собой короткий синтетический нуклеотид. Препарат показал свою эффективность на мышах, у которых снижал концентрацию мутантного белка гентингтина, вызывающего болезнь, в спинномозговой жидкости. Также он смягчил моторные и когнитивные симптомы заболевания.Обнадеживающее сообщение появилось на страницах журнала The New England Journal of Medicine.
Структура гентингтина
Болезнь Гентингтона, также известная как хорея Гентингтона из-за характерных патологических движений больных – это фатальное неизлечимое неврологическое заболевание, проявляющееся обычно в возрасте 30-50 лет. Симптомы болезни прогрессируют постепенно и похожи на сущий кошмар: расстройства движения, психические нарушения, потеря памяти и речи и, наконец, смерть.
Заболевание обусловлено большим количеством повторов CAG в гене, кодирующем белок гентингтин, функции которого в настоящее время не ясны. При болезни Гентингтона происходит постепенная атрофия подкорковых структур мозга – полосатых тел, а на поздних стадиях – и коры головного мозга. Заболевание наследуется аутосомно-доминантно, то есть проявляется всегда, даже если в геноме есть хоть одна копия мутантного гена. Оно неизлечимо, поэтому тем людям, один из родителей которых скончался от этой болезни, пока остается лишь ждать наступления роковых симптомов (которые, впрочем, могут так и не проявиться, если хромосома с мутантной версией гена не была унаследована).
Тем не менее поиски лечения ведутся постоянно. Полтора года назад впервые появилась информация об успешности генной терапии для лечения этой болезни, правда, пока лишь у мышей и без сведений о возможных побочных эффектах. Еще через год вышла новая работа, где проверялась эффективность четырех различных вариантов молекул, которые бы могли взаимодействовать с мутантным геном в нужную для пациента сторону.
Теперь известно, что нынешний экспериментальный препарат против болезни Гентингтона, известный как HTTRx и подавляющий образование белка гентингтина, безопасен для людей. HTTRx представляет собой короткую синтетическую молекулу ДНК. При введении этого вещества мышам с мутантным гентингтином, обладающим характерной для болезни моторной и когнитивной симптоматикой, уровень мутантного белка в спинномозговой жидкости снижался, а симптомы становились существенно менее выражены. Эффект препарата оказался дозозависимым: чем больше была его доза, тем сильнее падала концентрация мутантного гентингтина.
Однако, начальные испытания на пяти группах пациентов (46 человек), по сообщениям авторов публикации, пока не показали какой-либо разницы между теми, кто получал HTTRx в различных дозах, и теми, кто получал плацебо.
Тем не менее, уже запущено клиническое испытание препарата с участием 660 пациентов с болезнью Гентингтона, и в январе этого года первый пациент уже начал получать препарат. Завершить сбор данных планируется к марту 2022 года. Будем надеяться, что новый препарат подарит надежду больным и их детям, над которыми висит рок тяжелого наследственного заболевания.
Текст: Елизавета Минина
Sarah J. Tabrizi et. al. (2019) Targeting Huntingtin Expression in Patients with Huntington’s Disease. The New England Journal of Medicine.
doi:10.1056/NEJMoa1900907
Читайте материалы нашего сайта в Facebook, ВКонтакте, Яндекс-Дзен, Одноклассниках и канале в Telegram, а также следите за новыми картинками дня в Instagram.
болезнь Хантингтона — диагностика и лечение
Диагноз
Предварительный диагноз болезни Хантингтона основывается прежде всего на ваших ответах на вопросы, общем физическом осмотре, обзоре истории болезни вашей семьи, а также неврологических и психиатрических осмотрах.
Неврологическое обследование
Невролог задаст вам вопросы и проведет относительно простые тесты вашего:
- Моторные симптомы, , такие как рефлексы, мышечная сила и баланс
- Сенсорные симптомы, , включая осязание, зрение и слух
- Психиатрические симптомы, такие как настроение и психическое состояние
Нейропсихологическое тестирование
Невролог может также выполнить стандартизированные тесты для проверки вашего:
- Память
- Аргументация
- Умственная ловкость
- Знание языков
- Пространственные рассуждения
Психиатрическое обследование
Скорее всего, вас направят к психиатру для обследования, чтобы выяснить ряд факторов, которые могут повлиять на ваш диагноз, в том числе:
- Эмоциональное состояние
- Модели поведения
- Качество суждения
- Навыки преодоления трудностей
- Признаки беспорядка мышления
- Доказательство токсикомании
Визуализация и функционирование мозга
Ваш врач может заказать тесты для визуализации мозга для оценки структуры или функции мозга.Технологии визуализации могут включать в себя сканирование MRI или CT , которые показывают детальные изображения головного мозга.
Эти изображения могут показать изменения в мозге в областях, пораженных болезнью Хантингтона. Эти изменения могут не проявляться на ранних стадиях заболевания. Эти тесты также можно использовать для исключения других состояний, которые могут вызывать симптомы.
Генетическое консультирование и тестирование
Если симптомы явно указывают на болезнь Хантингтона, ваш врач может порекомендовать генетический тест на дефектный ген.
Этот тест может подтвердить диагноз. Это также может быть полезно, если неизвестна семейная история болезни Хантингтона или если диагноз другого члена семьи не подтвержден генетическим тестом. Но тест не предоставит информацию, которая может помочь определить план лечения.
Прежде чем сдать такой тест, генетический консультант объяснит преимущества и недостатки результатов обучения. Генетический консультант также может ответить на вопросы о типах наследования болезни Хантингтона.
Прогностический генетический тест
Генетический тест может быть проведен, если у вас есть семейная история болезни, но у вас нет симптомов. Это называется прогностическим тестированием. Тест не может сказать вам, когда болезнь начнется или какие симптомы появятся первыми.
Некоторые люди могут пройти тест, потому что они не знают, что они более стрессовые. Другие могут захотеть пройти тест до рождения детей.
Риски могут включать в себя проблемы со страховкой или будущей работой, а также стрессы, возникающие при смертельной болезни.В принципе, существуют федеральные законы, запрещающие использование информации о генетическом тестировании для дискриминации людей с генетическими заболеваниями.
Эти тесты проводятся только после консультации с генетическим консультантом.
Лечение
Никакое лечение не может изменить течение болезни Хантингтона. Но лекарства могут уменьшить некоторые симптомы движения и психические расстройства. А многочисленные вмешательства могут помочь человеку адаптироваться к изменениям своих способностей в течение определенного периода времени.
Лекарства, вероятно, будут развиваться в течение болезни, в зависимости от общих целей лечения. Кроме того, лекарства, которые лечат некоторые симптомы, могут привести к побочным эффектам, которые ухудшают другие симптомы. Цели лечения будут регулярно пересматриваться и обновляться.
Препараты для лечения двигательных расстройств
Препараты для лечения двигательных расстройств включают в себя следующее:
- Препараты для контроля движения включают тетрабеназин (ксеназин) и дейтетрабеназин (Аустедо), которые были специально одобрены Управлением по контролю за продуктами и лекарствами для подавления непроизвольных подергиваний и корчей (хорея), связанных с болезнью Хантингтона.Однако эти препараты не влияют на прогрессирование заболевания. Возможные побочные эффекты включают сонливость, беспокойство и риск ухудшения или спровоцирования депрессии или других психических состояний.
Антипсихотические препараты , такие как галоперидол (Haldol) и флуфеназин, имеют побочный эффект подавления движений. Следовательно, они могут быть полезны при лечении хореи. Однако эти препараты могут усугубить непроизвольные сокращения (дистония), беспокойство и сонливость.
Другие лекарства, такие как рисперидон (Risperdal), оланзапин (Zyprexa) и кветиапин (Seroquel), могут иметь меньше побочных эффектов, но все же должны использоваться с осторожностью, поскольку они могут также ухудшить симптомы.
- Другие лекарства , которые могут помочь подавить хорею, включают амантадин (Gocovri ER, Osmolex ER), леветирацетам (Keppra, Elepsia XR, Spritam) и клоназепам (Klonopin). Однако побочные эффекты могут ограничивать их использование.
Лекарства для лечения психических расстройств
Лекарства для лечения психических расстройств будут различаться в зависимости от расстройств и симптомов.Возможные методы лечения включают следующее:
- Антидепрессанты включают такие препараты, как циталопрам (Celexa), эсциталопрам (Lexapro), флуоксетин (Prozac, Sarafem) и сертралин (Zoloft). Эти препараты могут также оказывать некоторое влияние на лечение обсессивно-компульсивного расстройства. Побочные эффекты могут включать тошноту, диарею, сонливость и низкое кровяное давление.
- Антипсихотические препараты , такие как кветиапин (Seroquel), рисперидон (Risperdal) и оланзапин (Zyprexa), могут подавлять вспышки насилия, возбуждение и другие симптомы расстройств настроения или психоза.Однако эти препараты сами могут вызывать различные двигательные расстройства.
- Препараты для стабилизации настроения , которые могут помочь предотвратить взлеты и падения, связанные с биполярным расстройством, включают противосудорожные средства, такие как дивальпроэкс (Депакот), карбамазепин (Карбатрол, Эпитол и др.) И ламотригин (Ламиктал).
Психотерапия
Психотерапевт — психиатр, психолог или медицинский социальный работник — может предоставить психотерапию, чтобы помочь с поведенческими проблемами, разработать стратегии выживания, управлять ожиданиями во время прогрессирования заболевания и способствовать эффективному общению между членами семьи.
Логопедия
Болезнь Гентингтона может значительно ослабить контроль над мышцами рта и горла, которые необходимы для речи, еды и глотания. Логопед может помочь улучшить вашу способность говорить ясно или научить вас пользоваться устройствами связи, такими как доска, на которой изображены предметы повседневного обихода и занятия. Логопеды могут также решать проблемы с мышцами, используемыми при еде и глотании.
Физическая терапия
Физиотерапевт может научить вас подходящим и безопасным упражнениям, которые повышают силу, гибкость, равновесие и координацию.Эти упражнения могут помочь сохранить подвижность как можно дольше и могут снизить риск падений.
Инструкция по правильной осанке и использованию опор для улучшения осанки может помочь уменьшить тяжесть некоторых проблем с движением.
Когда требуется использование ходунков или инвалидных колясок, физиотерапевт может предоставить инструкции по правильному использованию устройства и осанки. Кроме того, режимы тренировок могут быть адаптированы к новому уровню мобильности.
Профессиональная терапия
Профессиональный терапевт может помочь человеку с болезнью Хантингтона, членам его семьи и лицам, осуществляющим уход, использовать вспомогательные устройства, улучшающие функциональные способности.Эти стратегии могут включать в себя:
- Поручни на дому
- Вспомогательные устройства для таких мероприятий, как купание и одевание
- Посуда для еды и питья, приспособленная для людей с ограниченными возможностями мелкой моторики
Образ жизни и домашние средства
Лечение болезни Хантингтона требует от человека, страдающего расстройством, членов семьи и других лиц, осуществляющих уход на дому.По мере прогрессирования заболевания человек становится все более зависимым от лиц, обеспечивающих уход. Необходимо будет решить ряд вопросов, и стратегии для их решения будут развиваться.
Питание и питание
Факторы, касающиеся питания и питания, включают следующее:
- Сложность поддержания здоровой массы тела. Причиной могут быть трудности с питанием, повышенная потребность в калориях из-за физических нагрузок или неизвестные метаболические проблемы.Чтобы получить адекватное питание, вам может потребоваться более трех раз в день или использовать пищевые добавки.
- Трудности с жеванием, глотанием и мелкой моторикой. Эти проблемы могут ограничить количество пищи, которую вы едите, и увеличить риск удушья. Проблемы могут быть минимизированы путем устранения отвлекающих факторов во время еды и выбора продуктов, которые легче есть. Посуда, предназначенная для людей с ограниченными мелкими моторными навыками и накрытая чашками с соломкой или носиком, также может помочь.
В конце концов, человеку с болезнью Хантингтона понадобится помощь в еде и питье.
Управление когнитивными и психическими расстройствами
Семья и лица, обеспечивающие уход, могут помочь создать среду, которая может помочь человеку с болезнью Хантингтона избежать стрессоров и справиться с когнитивными и поведенческими проблемами. Эти стратегии включают в себя:
- Использование календарей и расписаний для поддержания регулярности рутины
- Инициирование задач с напоминаниями или помощью
- Приоритезация или организация работы или деятельности
- Разбиение задач на управляемые шаги
- Создание максимально спокойной, простой и структурированной среды
- Выявление и устранение стрессоров, которые могут вызвать вспышки, раздражительность, депрессию или другие проблемы
- Для детей школьного возраста или подростков, консультации с персоналом школы для разработки соответствующего индивидуального плана обучения
- Предоставление человеку возможности поддерживать социальные контакты и дружбу в максимально возможной степени
Решение проблем и поддержка
Ряд стратегий может помочь людям с болезнью Хантингтона и их семьям справиться с проблемами болезни.
Вспомогательные услуги
Вспомогательные услуги для людей с болезнью Хантингтона и их семей включают следующее:
- Некоммерческие агентства, , такие как Американское общество по болезням Хантингтона, предоставляют образование по уходу, направления в сторонние службы и группы поддержки для людей с заболеванием и попечителей.
- Местные и государственные агентства здравоохранения или социального обслуживания могут предоставлять дневной уход за людьми с заболеванием, программы помощи в приеме пищи или временную помощь лицам, осуществляющим уход.
Планирование стационарного лечения и ухода в конце жизни
Поскольку болезнь Хантингтона вызывает постепенную потерю функции и смерть, важно предвидеть уход, который будет необходим на поздних стадиях заболевания и ближе к концу жизни. Ранние дискуссии об этом типе ухода позволяют человеку с болезнью Хантингтона участвовать в этих решениях и сообщать о своих предпочтениях по уходу.
Создание юридических документов, определяющих уход в конце жизни, может быть полезным для всех.Они расширяют возможности человека, страдающего этой болезнью, и могут помочь членам семьи избежать конфликтов на поздних стадиях развития болезни. Ваш врач может дать совет о преимуществах и недостатках вариантов ухода в то время, когда все варианты могут быть тщательно рассмотрены.
Вопросы, которые могут потребоваться решить, включают:
- Уход. Уход на поздних стадиях заболевания, скорее всего, потребует ухода на дому или ухода в учреждении, где имеется помощь, или в доме престарелых.
- Уход за хосписом. Услуги хосписа обеспечивают уход в конце жизни, который помогает человеку приблизиться к смерти с минимальным дискомфортом, насколько это возможно. Этот уход также обеспечивает поддержку и образование для членов семьи, чтобы помочь им понять процесс смерти.
- Живые завещания. Живые завещания — это юридические документы, которые позволяют человеку изложить предпочтения по уходу, когда он или она не в состоянии принимать решения. Например, эти указания могут указывать, хочет ли человек поддерживать жизнедеятельность или проводить агрессивное лечение инфекции.
- Предварительные указания. Эти юридические документы позволяют вам идентифицировать одного или нескольких людей для принятия решений от вашего имени. Вы можете создать предварительное распоряжение для медицинских решений или финансовых вопросов.
Подготовка к вашему приёму
Если у вас есть какие-либо признаки или симптомы, связанные с болезнью Хантингтона, вы, вероятно, будете направлены к неврологу после первоначального посещения вашего семейного врача.
Обзор ваших симптомов, психического состояния, истории болезни и истории болезни семьи может быть важен при клинической оценке потенциального неврологического расстройства.
Что вы можете сделать
Перед назначением составьте список, который включает следующее:
- Признаки или симптомы — или любые изменения по сравнению с тем, что является нормальным для вас — это может вызывать беспокойство
- Последние изменения или стрессы в вашей жизни
- Все лекарства, включая безрецептурные препараты и пищевые добавки, и дозы, которые вы принимаете
- Семейная история болезни Хантингтона или других расстройств, которые могут вызвать двигательные расстройства или психические расстройства
Вы можете попросить члена семьи или друга сопровождать вас на приеме.Этот человек может оказать поддержку и предложить иной взгляд на влияние симптомов на ваши функциональные способности.
Чего ожидать от вашего доктора
Ваш доктор, вероятно, задаст вам ряд вопросов, включая следующие:
- Когда вы начали испытывать симптомы?
- Ваши симптомы были постоянными или прерывистыми?
- Кто-нибудь в вашей семье когда-либо был диагностирован с болезнью Хантингтона?
- Кто-нибудь в вашей семье был диагностирован с другим двигательным расстройством или психическим расстройством?
- У вас проблемы с выполнением работы, учебы или ежедневных заданий?
- Кто-нибудь в вашей семье умер молодым?
- Кто-нибудь в вашей семье в доме престарелых?
- Кто-нибудь в вашей семье нервничает или постоянно движется?
- Вы заметили изменение вашего общего настроения?
- Тебе все время грустно?
- Ты когда-нибудь думал о самоубийстве?
,
Болезнь Хантингтона — Genetics Home Reference
Bates GP. История генетической болезни: молекулярная генетика болезни Гентингтона — история. Nat Rev Genet. 2005 окт; 6 (10): 766-73.
Гонсалес-Алегри П, Афифи АК. Клиническая характеристика ранней (ювенильной) болезни Гентингтона у детей: отчет о 12 пациентах и обзор литературы. J Child Neurol. 2006 март; 21 (3): 223-9.
Имарисио С., Кармайкл Дж., Корольчук В., Чен С. В., Сайки С., Роуз С., Кришна Г., Дэвис Д. Е., Ттофи Е., Андервуд Б. Р., Рубинштейн Д. К.Болезнь Хантингтона: от патологии и генетики к потенциальной терапии. Biochem J. 2008 Jun 1; 412 (2): 191-209. doi: 10.1042 / BJ20071619. Обзор.
Джонс Л., Хьюз А. Патогенные механизмы при болезни Хантингтона. Int Rev Neurobiol. 2011; 98: 373-418. doi: 10.1016 / B978-0-12-381328-2.00015-8. Обзор.
Кент А. Болезнь Хантингтона. Нурс Стенд. 21 апреля 2004 года; 18 (32): 45-51; викторина 52-3. Обзор.
Maiuri T, Мокл AJ, Хунг CL, Ся J, Ван Рун-Мом WM, Truant R.Хантингтин представляет собой каркасный белок в комплексе ответа на окислительное повреждение ДНК АТМ. Хум Мол Генет. 2017 янв. 15; 26 (2): 395-406. дои: 10.1093 / мг / ддв395.
Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WM, Truant R. Huntingtin — это каркасный белок в комплексе ответа на окислительное повреждение ДНК АТМ. Хум Мол Генет. 2017 янв. 15; 26 (2): 395-406. дои: 10.1093 / мг / ддв395.
Sturrock A, Leavitt BR. Клинико-генетические особенности болезни Хантингтона.J Geriatr Психиатрия Neurol. 2010 дек; 23 (4): 243-59. doi: 10.1177 / 0891988710383573. Epub 2010 5 октября. Обзор.
Tost H, Wendt CS, Schmitt A, Heinz A, Braus DF. Болезнь Хантингтона: феноменологическое разнообразие психоневрологического состояния, которое бросает вызов традиционным представлениям в неврологии и психиатрии. Я J Психиатрия. 2004 янв; 161 (1): 28-34.
Walker FO. Болезнь Хантингтона. Lancet. 2007 янв 20; 369 (9557): 218-28. Обзор.
Warby SC, Graham RK, Hayden MR.Болезнь Хантингтона. 1998 окт 23 [обновлено 2014 дек 11]. В: Пагон Р.А., Адам М.П., Ардингер Х.Х., Уоллес С.Е., Амемия А., Боб Л.Дж., Берд Т.Д., Ледбеттер Н., Меффорд Х.К., Смит Р.Дж., Стивенс К., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017. Доступно по адресу: http://www.ncbi.nlm.nih.gov/books/NBK1305/
Young AB. Охота на здоровье и болезни. J Clin Invest. 2003 Фев; 111 (3): 299-302. Обзор.
.
Болезнь Гентингтона — это наследственное заболевание, при котором нервные клетки вашего мозга постепенно разрушаются. Это влияет на ваши физические движения, эмоции и когнитивные способности. Нет лекарства, но есть способы справиться с этим заболеванием и его симптомами.
Болезнь Хантингтона гораздо чаще встречается у людей с европейским происхождением, поражая примерно три-семь из каждых 100 000 человек европейского происхождения.
Существует два типа болезни Хантингтона: взрослое начало и раннее начало.
Взрослое начало
Взрослое начало является наиболее распространенным типом болезни Хантингтона. Симптомы обычно начинаются, когда людям за 30 или 40 лет. Начальные признаки часто включают в себя:
- депрессия
- раздражительность
- галлюцинации
- психоз
- незначительные непроизвольные движения
- плохая координация
- трудности с пониманием новой информации
- проблемы с принятием решений
Симптомы, которые могут возникнуть по мере прогрессирования заболевания, включают :
- неконтролируемые подергивания, называемые хореей
- трудности при ходьбе
- проблемы с глотанием и речью
- путаница
- потеря памяти
- изменения личности
- изменения речи
- снижение когнитивных способностей
раннее начало
Этот тип Болезнь Хантингтона встречается реже.Симптомы обычно начинают появляться в детстве или в подростковом возрасте. Раннее начало болезни Хантингтона вызывает психические, эмоциональные и физические изменения, такие как:
- слюнотечение
- неуклюжесть
- невнятная речь
- медленные движения
- частые падения
- жесткие мышцы
- судороги
- судороги
- внезапное снижение успеваемости в школе
Дефект в одном гене вызывает болезнь Хантингтона. Это считается аутосомно-доминантным расстройством.Это означает, что одной копии ненормального гена достаточно, чтобы вызвать заболевание. Если у одного из ваших родителей есть этот генетический дефект, у вас есть 50-процентный шанс унаследовать его. Вы также можете передать его своим детям.
Генетическая мутация, ответственная за болезнь Хантингтона, отличается от многих других мутаций. В гене нет замены или отсутствующего участка. Вместо этого происходит ошибка копирования. Область внутри гена копируется слишком много раз. Количество повторных копий имеет тенденцию увеличиваться с каждым поколением.
В общем, симптомы болезни Хантингтона проявляются раньше у людей с большим количеством повторений. Болезнь также прогрессирует быстрее, так как увеличивается количество повторений.
Семейный анамнез играет важную роль в диагностике болезни Хантингтона. Однако для диагностики проблемы могут быть проведены различные клинические и лабораторные исследования.
Неврологические тесты
Невролог проведет тесты, чтобы проверить ваши:
- рефлексов
- координация
- баланс
- мышечный тонус
- сила
- чувство осязания
- слух
- зрение
Функция мозга и визуализация Тесты
Если у вас были судороги, вам может потребоваться электроэнцефалограмма (ЭЭГ).Этот тест измеряет электрическую активность в вашем мозгу.
Тесты для визуализации мозга также можно использовать для обнаружения физических изменений в вашем мозге.
- При сканировании магнитно-резонансной томографии (МРТ) используются магнитные поля для записи изображений головного мозга с высоким уровнем детализации.
- Компьютерная томография (КТ) объединяет несколько рентгеновских снимков для получения поперечного сечения вашего мозга.
Психиатрические тесты
Ваш врач может попросить вас пройти психиатрическое обследование.Эта оценка проверяет ваши навыки совладания, эмоциональное состояние и поведенческие паттерны. Психиатр также будет искать признаки нарушения мышления.
Вас могут обследовать на злоупотребление психоактивными веществами, чтобы узнать, могут ли наркотики объяснить ваши симптомы.
Генетическое тестирование
Если у вас есть несколько симптомов, связанных с болезнью Хантингтона, ваш врач может порекомендовать вам генетическое тестирование. Генетический тест может окончательно диагностировать это состояние.
Генетическое тестирование также может помочь вам решить, стоит ли иметь детей.Некоторые люди с Хантингтоном не хотят рисковать передачей дефектного гена следующему поколению.
Медикаменты
Медикаменты могут помочь вам избавиться от некоторых физических и психических симптомов. Типы и количество необходимых лекарств будут меняться в зависимости от вашего состояния.
- Непроизвольные движения можно лечить с помощью тетрабеназина и антипсихотических препаратов.
- Диазепамом можно лечить ригидность мышц и непроизвольные сокращения мышц.
- Депрессию и другие психиатрические симптомы можно лечить с помощью антидепрессантов и стабилизирующих настроение лекарств.
Терапия
Физическая терапия может помочь улучшить вашу координацию, баланс и гибкость. Благодаря этой тренировке ваша мобильность улучшается, и падения могут быть предотвращены.
Трудовая терапия может использоваться для оценки вашей повседневной деятельности и рекомендовать устройства, которые помогают:
- движение
- еда и питье
- купание
- одевание
Логопедия может помочь вам четко говорить. Если вы не можете говорить, вас научат другим типам общения.Логопеды также могут помочь с глотанием и проблемами с питанием.
Психотерапия может помочь вам справиться с эмоциональными и психическими проблемами. Это также может помочь вам развить навыки преодоления трудностей.
Нет способа остановить развитие этой болезни. Скорость прогрессирования отличается для каждого человека и зависит от количества генетических повторов, присутствующих в ваших генах. Меньшее число обычно означает, что болезнь будет прогрессировать медленнее.
Люди с начальной формой болезни Хантингтона обычно живут от 15 до 20 лет после появления симптомов.Форма с ранним началом обычно прогрессирует быстрее. Люди могут жить только 10-15 лет после появления симптомов.
Причины смерти среди людей с болезнью Хантингтона включают:
- инфекций, таких как пневмония
- самоубийств
- травм в результате падения
- осложнений из-за невозможности глотать
Если у вас возникли проблемы с преодолением вашего состояния, рассмотреть возможность присоединения к группе поддержки. Это может помочь встретить других людей с болезнью Хантингтона и поделиться своими проблемами.
Если вам нужна помощь в выполнении повседневных задач или передвижения, обратитесь в агентства здравоохранения и социальных служб в вашем районе. Возможно, они смогут организовать дневной уход.
Обратитесь к своему врачу за советом о типе ухода, который может вам понадобиться по мере прогрессирования вашего состояния. Возможно, вам придется переехать в приют для престарелых или организовать медицинский уход на дому.
Болезнь Гентингтона: симптомы, причины и лечение
Болезнь Хантингтона — это неизлечимое наследственное заболевание головного мозга. Это разрушительное заболевание, которое вызывает повреждение клеток головного мозга или нейронов.
Это происходит, когда неисправный ген вызывает накопление токсичных белков в мозге.
Болезнь Хантингтона (HD) поражает одного человека из каждых 10 000, или около 30 000 человек в Соединенных Штатах. Еще 150 000 или более человек подвержены риску развития этого заболевания.
Первые признаки обычно появляются в возрасте от 30 до 50 лет.
Быстрые факты о болезни Хантингтона
- Болезнь Хантингтона (HD) — это наследственное заболевание, которое постепенно поражает нервные клетки.
- Заболевание возникает, когда дефектный ген образует аномальную версию белка хантингтина.
- Ранние симптомы могут включать перепады настроения, неуклюжесть и необычное поведение.
- На поздних стадиях заболевания удушье становится серьезной проблемой.
- В настоящее время нет лекарства, но лекарства могут помочь облегчить симптомы.
Болезнь Гентингтона (HD) является неврологическим состоянием. Это наследственное заболевание, которое происходит из-за дефектных генов. Токсичные белки накапливаются в мозге и вызывают повреждения, что приводит к неврологическим симптомам.
По мере разрушения частей мозга это влияет на движение, поведение и познание. Становится все труднее ходить, думать, рассуждать, глотать и говорить. В конце концов, человеку понадобится полный рабочий день. Осложнения обычно смертельны.
В настоящее время лечения нет, но лечение может помочь с симптомами.
Поделиться в PinterestГантингтон обычно начинается в возрасте от 30 до 50 лет. Изменения в настроении и мышлении могут быть ранним признаком.
Признаки и симптомы чаще всего появляются в возрасте от 30 до 50 лет, но они могут возникнуть в любом возрасте. Они имеют тенденцию ухудшаться в течение 10-20 лет.
В конечном счете болезнь Хантингтона или ее осложнения заканчиваются смертельным исходом.
По данным Американского общества по болезням Хантингтона (HDSA), симптомы HD могут ощущаться как боковой амиотрофический склероз (ALS), болезни Паркинсона и Альцгеймера — все в одном.
Ключевые симптомы включают в себя:
- изменения личности, перепады настроения и депрессию
- проблемы с памятью и суждением
- неустойчивая ходьба и неконтролируемые движения
- трудности с речью и глотанием и потеря веса
Как развиваются признаки и симптомы может варьироваться между людьми. У некоторых людей депрессия возникает до того, как будут затронуты моторные навыки. Перепады настроения и необычное поведение — обычные ранние признаки.
Ранние признаки и симптомы
Ранние симптомы могут быть не распознаны, если HD ранее не встречался в семье.Для установления диагноза может потребоваться много времени.
Первоначальные признаки и симптомы включают в себя:
- легкие неконтролируемые движения
- небольшие изменения в координации и неловкости
- спотыкание
- легкие признаки настроения и эмоциональных изменений
- отсутствие внимания, проблемы с небольшой концентрацией и трудности функционирования, например , на работе
- провалов в кратковременной памяти
- депрессия
- раздражительность
Человек может потерять мотивацию и сосредоточенность, выглядеть вялым и не иметь инициативы.
Другие возможные признаки HD могут включать в себя спотыкаться, бросать вещи и забывать имена людей. Тем не менее, большинство людей делают это время от времени.
Средняя и поздняя стадии
Со временем симптомы становятся более серьезными.
К ним относятся физические изменения, потеря контроля над движением, а также эмоциональные и когнитивные изменения.
Физические изменения
Человек может испытывать:
- трудности с речью, в том числе поиск слов и намазывание
- потеря веса, что приводит к слабости
- трудности с едой и глотанием, так как мышцы рта и диафрагмы могут не работать должным образом
- риск удушья, особенно на более поздних стадиях
- неконтролируемые движения
Возможны неконтролируемые движения тела, в том числе:
- неконтролируемые движения лица
- подергивания частей лица и головы
- взмахами или беспокойные движения рук, ног и тела
- кренившись и спотыкаясь
По мере развития HD неконтролируемые движения происходят чаще и с большей интенсивностью.В конце концов они могут стать медленнее, так как мышцы становятся более жесткими.
Эмоциональные изменения
Они могут чередоваться, а не происходить последовательно.
Они включают в себя:
- агрессия
- гнев
- антисоциальное поведение
- апатия
- депрессия
- возбуждение
- разочарование
- отсутствие эмоций становится все более очевидным
- капризность
- упрямство
- когнитивные изменения
может быть:
- потеря инициативы
- потеря организационных навыков
- дезориентация
- трудности с фокусировкой
- проблемы с многозадачностью
Поздняя стадия
В конце концов, человек больше не сможет ходить или говорить , и они будут нуждаться в полном уходе.
Тем не менее, они, как правило, понимают большинство сказанного и знают друзей и членов семьи.
Невозможность делать вещи, которые раньше были легкими, может привести к разочарованию и депрессии.
Потеря веса может усугубить симптомы и ослабить иммунную систему пациента, делая их более уязвимыми для инфекций и других осложнений.
HD само по себе обычно не смертельно, но может быть удушье, пневмония или другая инфекция.
На всех этапах важно скорректировать диету пациента, чтобы обеспечить адекватное потребление пищи.
HD вызван дефектным геном (mhTT) в хромосоме номер 4.
Нормальная копия гена продуцирует хинтингтин, белок. Неисправный ген больше, чем должен быть. Это приводит к чрезмерной выработке цитозина, аденина и гуанина (CAG), строительных блоков ДНК. Обычно CAG повторяется от 10 до 35 раз, но в HD повторяется от 36 до 120 раз. Если это повторяется 40 или более раз, симптомы вероятны.
Это изменение приводит к большей форме охоты.Это токсично, и, поскольку оно накапливается в мозге, оно вызывает повреждение клеток мозга.
Некоторые клетки мозга чувствительны к большей форме охоты, особенно те, которые связаны с движением, мышлением и памятью. Это подрывает их функцию и в конечном итоге разрушает их. Ученые не уверены, как именно это происходит.
Как это передается?
HD известен как аутосомно-доминантное заболевание. Это означает, что только одна копия дефектного гена, унаследованная от матери или отца, необходима для возникновения заболевания.
Человек с геном имеет одну хорошую копию гена и одну ошибочную копию гена. Любое потомство унаследует либо хорошую копию, либо неисправную. У ребенка, который унаследует хорошую копию, не будет развиваться HD. Ребенок, который наследует ошибочную копию, будет.
Каждый ребенок имеет 50-процентный шанс унаследовать неисправный ген. Если они унаследуют дефектный ген, у каждого из их детей будет 50-процентный шанс его унаследовать. HD может повлиять на несколько поколений.
Человек, который не наследует дефектный ген, не разовьет HD и не сможет передать его своим детям.У ребенка, который наследует дефектный ген, разовьется HD, если он достигнет возраста, когда появятся симптомы.
Около 10 процентов случаев ГБ начинаются в возрасте до 20 лет. Это известно как ювенильный HD (JHD).
Симптомы разные и могут включать в себя скованность ног, дрожь и регрессию в обучении.
От 30 до 50 процентов людей с JHD имеют судороги.
HD в настоящее время неизлечим. Не существует лечения, которое могло бы обратить вспять его прогрессирование или замедлить его.
Однако некоторые симптомы можно устранить с помощью лекарств и методов лечения.
Медикаменты
Тетрабеназин (ксеназин) одобрен Управлением по контролю за продуктами и лекарствами США (FDA) для лечения резких, непроизвольных движений или хореи, связанных с ГБ.
Побочные эффекты включают депрессию и суицидальные мысли или действия.
Симптомы включают в себя:
- чувство грусти и плачущие заклинания
- потерю интереса к друзьям и ранее приятным занятиям
- сон более или менее привычный и чувство усталости
- чувство вины или неважности
- чувство раздражительности, злости или чем раньше, чем
- едят меньше, чем обычно, возможно, с потерей веса
- с трудностями сосредоточения
- думая о том, чтобы навредить себе или покончить с жизнью
О любом из этих симптомов или других изменениях настроения следует немедленно сообщать врачу.
Тот, кто имеет диагноз депрессии, особенно с суицидальными мыслями, не должен использовать третрабеназин.
Препараты для контроля движений, вспышек и галлюцинаций могут включать в себя:
- клоназепам (клонопин)
- галоперидол
- клозапин (клоразил)
Эти препараты могут вызывать седативный эффект, а также жесткость и ригидность.
Для депрессии и некоторых обсессивно-компульсивных особенностей, которые могут появиться при HD, врач может назначить:
- флуоксетин (Prozac, Sarafem)
- сертралин (Zoloft)
- нортриптилин (Pamelor)
Литий может помочь при экстремальных состояниях эмоции и перепады настроения.
Логопедия
Логопедия может помочь пациентам найти способы выразить слова и фразы и более эффективно общаться.
Физическая и профессиональная терапия
Физиотерапевт может помочь улучшить мышечную силу и гибкость, приводя к лучшему балансу и уменьшая риск падения.
Психотерапевт может помочь пациенту разработать стратегии, позволяющие справиться с проблемами концентрации и памяти, а также повысить безопасность дома.
Врач осмотрит пациента и расспросит о семье и истории болезни, а также симптомах, таких как недавние эмоциональные изменения.
Если они подозревают HD, они направят пациента к неврологу.
Визуальные тесты, такие как компьютерная томография или МРТ, иногда используются для выявления изменений в структуре мозга пациента и для исключения других нарушений.
Генетическое тестирование может быть рекомендовано для подтверждения диагноза.
HD оказывает серьезное эмоциональное, психическое, социальное и экономическое влияние на жизнь человека и его семьи.После постановки диагноза человек обычно проживет от 15 до 20 лет, но продолжительность колеблется от 10 до 30 лет.
Человек с JHD, вероятно, будет жить около 10 лет. Эта форма прогрессирует быстрее.
Причиной смерти часто является осложнение, такое как пневмония или удушье.
Хотя в настоящее время лечения нет, некоторые методы лечения могут помочь людям справиться с этим заболеванием и улучшить качество жизни.
Надежда на будущее?
В будущем ученые надеются, что генная терапия найдет решение этой болезни.Исследователи искали способы использования генной терапии для лечения, замедления или предотвращения HD.
Одной из обнадеживающих стратегий является использование молекул, известных как синтетические малые интерферирующие РНК (миРНК), для подавления производства белка из дефектного гена. Это остановит сбор токсичного белка Хантингтина и вызовет симптомы.
Однако остающаяся проблема заключается в том, как доставить миРНК в соответствующие клетки мозга, чтобы они могли быть эффективными.
В 2017 году ученые из Университета Эмори предположили, что методы CRISPR / Cas9, которые включают «вырезание и вставку» ДНК, могут помочь предотвратить HD в будущем.
Когда исследователи сконструировали дефектный ген у мышей, они обнаружили «значительные улучшения» через 3 недели. Большая часть следов разрушающего белка исчезла, и нервные клетки показали признаки своего заживления.
Требуется гораздо больше исследований, прежде чем это можно будет применить к людям, однако.
Такие организации, как HDSA, предлагают поддержку людям с HD и их семьям.
Генетическое тестирование на HD стало возможным в 1993 году. Любой человек с семейным анамнезом HD может спросить своего врача о генетическом тестировании, чтобы выяснить, несут ли они дефектный ген.
Некоторые люди предпочитают выяснять, есть ли у них ген, и могут ли они развить симптомы, тогда как другие предпочли бы этого не знать. Генетический консультант может помочь с принятием решения.
HD, генетика и беременность
Если пара хочет иметь ребенка, а один из родителей имеет дефектный ген, можно пройти оплодотворение in vitro (ЭКО). Затем эмбрион проходит генетическое тестирование в лаборатории и имплантируется женщине, только если у него нет дефектного гена.
Генетическое тестирование также может проводиться во время беременности, если есть семейный анамнез HD. Это можно сделать с помощью образца ворсин хориона (CVS) через 10–11 недель или через амниоцентез через 14–18 недель.