Пороки развития
Пороки и дефекты развития конечностей
Пороки развития — стойкие морфологические изменения органа, системы или организма, которые выходят за пределы вариаций их строения и возникают внутриутробно в результате нарушений развития зародыша или после рождения ребенка, как следствие нарушения дальнейшего формирования органов. Пороки развития возникают в результате генных мутаций; хромосомных и геномных мутаций; комбинированного воздействия генных мутаций и факторов внешней по отношению к зародышу среды; тератогенных факторов.
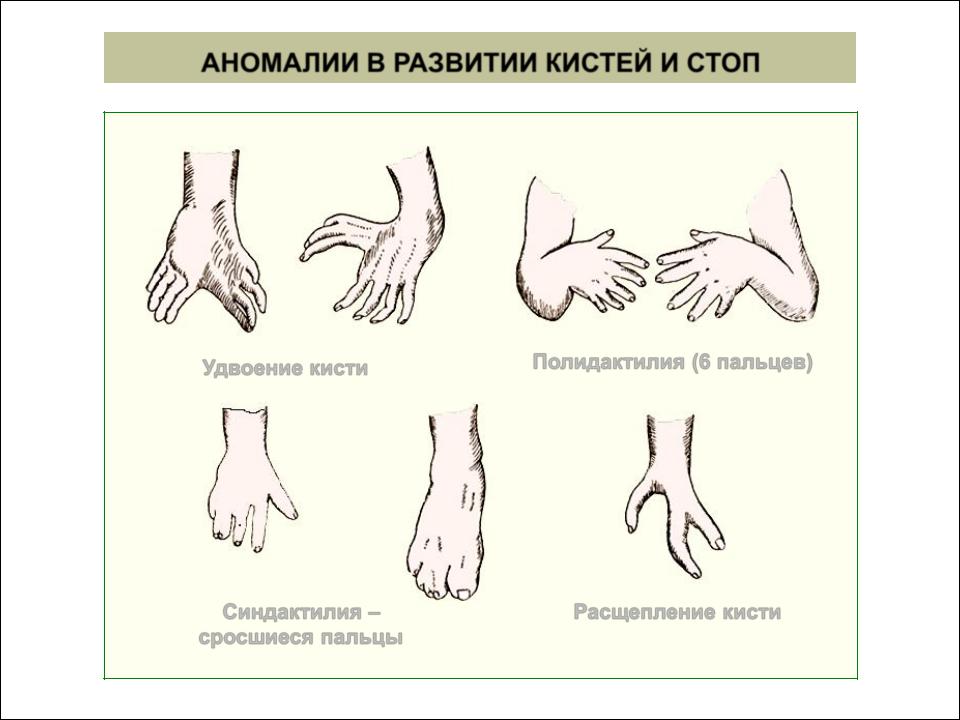
Аномалии, возникающие в результате недостаточности формирования частей конечностей.
Амелия — полное отсутствие конечности (исключая плечевой пояс и таз). Различают верхнюю и нижнюю амелию, в частности отсутствие двух верхних конечностей (абрахия), одной верхней конечности (монобрахия), двух нижних конечностей (апус), одной нижней конечности (монопус).
Фокомелия (тюленеобразные конечности) — отсутствие проксимальных и (или) средних частей конечности и соответствующих суставов (плечевого, тазобедренного).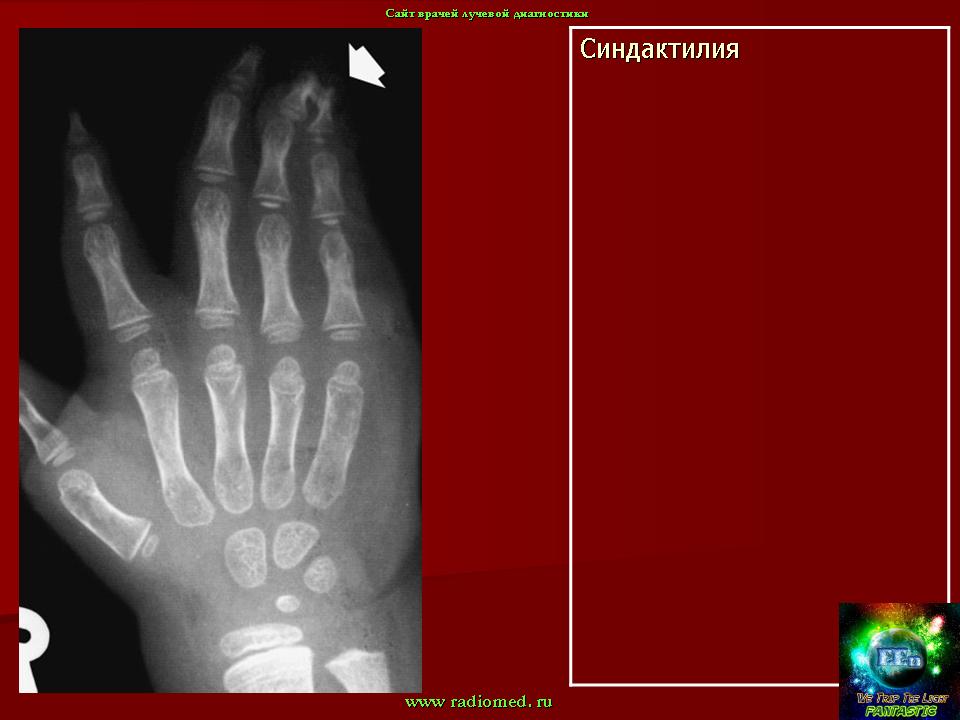 Различают проксимальную, дистальную и полную фокомелию. Проксимальная фокомелия — отсутствие плеча или бедра, дистальная — отсутствие предплечья (радиоульнарная форма) или голени (тибиофибулярная форма), полная фокомелия — отсутствие плеча и предплечья или бедра и голени. Соответственно вполне сформированная кисть или стопа может отходить непосредственно от туловища (полная фокомелия), соединяться с ним посредством сохранившихся костей предплечья, голени (проксимальная форма) или прикрепляться к плечу, бедру (дистальная форма). Фокомелия бывает одно- и двусторонней, иногда в процесс вовлекаются все четыре конечности.
Различают проксимальную, дистальную и полную фокомелию. Проксимальная фокомелия — отсутствие плеча или бедра, дистальная — отсутствие предплечья (радиоульнарная форма) или голени (тибиофибулярная форма), полная фокомелия — отсутствие плеча и предплечья или бедра и голени. Соответственно вполне сформированная кисть или стопа может отходить непосредственно от туловища (полная фокомелия), соединяться с ним посредством сохранившихся костей предплечья, голени (проксимальная форма) или прикрепляться к плечу, бедру (дистальная форма). Фокомелия бывает одно- и двусторонней, иногда в процесс вовлекаются все четыре конечности.
Перомелия — вариант фокомелии, сочетающейся с недоразвитием кистей или стоп. Различают полную (рука или нога отсутствует, соответствующий отдел туловища заканчивается одним рудиментарным пальцем или кожным выступом) и неполную (плечо или бедро недоразвито, заканчивается также одним рудиментарным пальцем или кожным выступом) формы.
Встречаются также лучевая и локтевая косорукость, аплазия большеберцовой кости, аплазия малоберцовой кости, адактилия — отсутствие пальцев, афалангия — отсутствие фаланг, монодактилия — наличие одного пальца на кисти или стопе, ахейрия — отсутствие кисти.
Расщепление кисти (эктродактилия, клешнеобразная кисть, «кисть омара») — аплазия центральных компонентов кисти (пальцев и нередко пястных костей) с бороздой (расщелиной) на месте отсутствующих костей. Выделяют типичные и атипичные формы. Типичная форма характеризуется аплазией III пальца и (нередко) соответствующей пястной кости, а также (иногда) дистального ряда костей запястья, что обусловливает наличие глубокой расщелины. Атипичная расщелина проявляется недоразвитием (реже отсутствием) средних пальцевых компонентов кисти или стопы. При атипичной форме расщелина неглубокая, но широкая; иногда она имеет вид чрезмерно широкого межпальцевого промежутка. Чаще встречается расщепление правой кисти.
Аномалии, возникающие в результате недостаточной дифференцировки частей конечности.
К ним относятся пороки развития лопатки (ладьевидная лопатка, поднятая лопатка), синостозы, синдактилии, брахидактилия, врожденная косолапость, врожденный вывих бедра, артрогрипоз, клинодактилия — укорочение средней фаланги пальцев кистей (чаще V пальца), обычно являющиеся составным компонентом синдромов.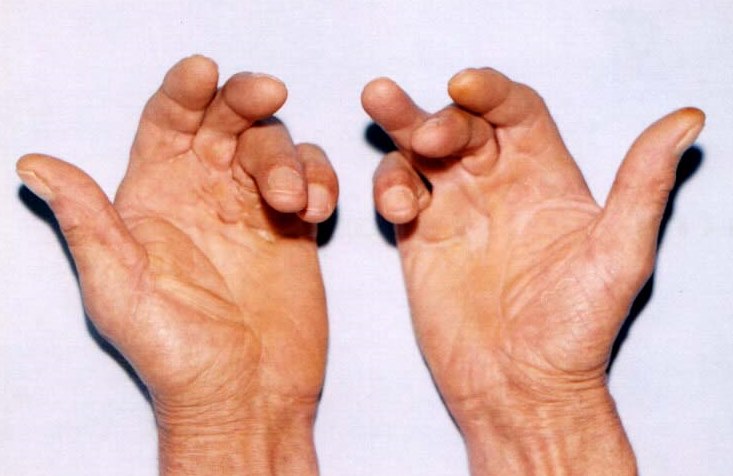
Камптодактилия (кампилодактилия) — сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти. В процесс может вовлекаться любой палец, кроме I. Встречается редко.
Сиреномелия (симподия, симмелия, синдром каудальной регрессии) — слияние нижних конечностей. Слияние может касаться мягких тканей и (чаще) некоторых длинных трубчатых костей, а также сопровождаться гипо- и (или) аплазией отдельных костей конечностей и таза. Стопы могут отсутствовать (sympus apus), бывают сформированы две (sympus dipus) или одна стопа (sympus monopus). Иногда имеется одна рудиментарная стопа с единственным пальцем. Сиреномелия сопровождается аплазией наружных и внутренних половых органов, аплазией мочевой системы, атрезией заднепроходного отверстия и прямой кишки, аплазией одной пупочной артерии.
Аномалии, обусловленные удвоением: полидактилия, диплоподия — удвоение стопы, полимелия — увеличение числа нижних конечностей. Полимелия может быть симметричной и асимметричной, обычно сочетается с пороками, несовместимыми с жизнью.
Аномалии, обусловленные чрезмерным ростом, включают макотородактилию и гигантизм конечности (парциальный гигантизм, односторонняя макросомия, гемигипертрофия), проявляющийся односторонним увеличением относительно пропорционально развитой конечности.
Аномалии, обусловленные недостаточным ростом. В их число входят аномалии, проявляющиеся гипоплазией различных отделов костей конечностей.
Врожденные перетяжки — порок развития амниона в виде тканевых тяжей, проходящих внутри плодовместилища и связывающих между собой плодовую поверхность последа с поверхностью плода, разные точки плодовой поверхности последа и несколько точек поверхности плода.
Генерализованные (системные) скелетные деформации. В их основе лежит нарушение эмбриогенеза соединительной ткани, включая костную ткань. К этой группе относятся хондродисплазии, остеодисплазии.
Клинические проявления пороков и прогноз во многом зависят от того, насколько жизненно важным является пораженный орган, от степени нарушения его функций, а также от присоединившихся осложнений.
Основная часть пороков опорно-двигательного аппарата корригируются хирургическим путем. Применяются различные пластические операции на коже и мягких тканях, так же костно-пластические операции при выраженных деформациях конечностей и позвоночника с применением различных корригирующих систем и аппаратов внешней фиксации, а так же костной пластики различными трансплантатами.
Детская ортопедия: аномалии развития кисти | 161.ru
Довольно часто к ортопеду сегодня обращаются пациенты с различными врожденными пороками развития опорно-двигательного аппарата. Чаще всего это – аномалии развития верхних конечностей, а именно – кисти.
Врожденные пороки развития кисти относительно часты и весьма разнообразны, на их долю приходится 65-70% всех аномалий верхних конечностей. Наиболее распространены такие врожденные пороки развития кисти, как полидактилия и синдактилия.
Синдактилия
Синдактилия – это сращивание одного или нескольких пальцев с нарушением функции или косметического состояния пораженного сегмента конечности. Ее причинами могут быть генетический фактор, воздействие токсических веществ во время беременности, а также инфекции.
Ее причинами могут быть генетический фактор, воздействие токсических веществ во время беременности, а также инфекции.
Существует несколько вариантов синдактилии. По протяженности она может быть полной (сращение всех фаланг) и неполной (сращение основных или основных и части средних фаланг). По виду сращения синдактилия бывает мягкотканая (то есть перепончатая с присутствием тонкой перепонки; кожная с присутствием толстой перемычки состоящей из кожи и мягких тканей) и костная (с наличием костной спайки пястных костей или фаланг на различном уровне). Также по состоянию поражения пальцев существуют простая (сращение нормально развитых пальцев имеющих полный объем движений), сложная (сращение пальцев со стабильной контрактурой, клинодактилией и недоразвитием) виды этого порока кисти.
Кроме того, выделяют и генетические виды синдактилий:
- тип первый (зигодактилия) – полное или частичное сращение третьего и четвертого пальцев кисти;
- тип второй (синполидактилия) – сращение третьего и четвертого пальцев кисти с одновременным удвоением четвертого пальца;
- тип третий – полная двусторонняя синдактилия четвертого и пятого пальцев кисти, мизинец укорочен за счет рудиментарной или отсутствующей средней фаланги;
- тип четвертый (синдактилия Газа) – полная двусторонняя кожная синдактилия, полидактилия, из-за чего форма кисти становится «ложкообразной»;
- тип пятый – синдактилия со слиянием пястных костей.
 На кистях чаще встречается кожное сращение третьего-четвертого пальцев.
На кистях чаще встречается кожное сращение третьего-четвертого пальцев.
 На кистях чаще встречается кожное сращение третьего-четвертого пальцев.
На кистях чаще встречается кожное сращение третьего-четвертого пальцев.=
Полидактилия
Полидактилия – анатомическая аномалия, проявляющаяся в увеличении количества пальцев на руках или ногах. Количество добавочных пальцев может достигать десяти. Разновидностью полидактилии также является раздвоение пальца – pollex duplex.
Полидактилия может быть единственной аномалией развития или же сочетаться с другими пороками развития опорно-двигательного аппарата. Чаще всего это синдактилия, брахидактилия, дисплазия суставов.
Основной причиной полидактилии считается наследственный фактор, встречаются семейные формы полидактилий. Также повлиять на возникновения этого порока может воздействие каких-либо токсических веществ на плод во время беременности. Еще одна причина – это спайки, находящиеся внутри оболочек, окружающих плод.
Во врачебной практике выделяют следующие виды и типы полидактилий:
По локализации:
- преаксиальная – дублирование сегментов большого пальца;
- центральная – аномалия развития второго-четвертого пальцев;
- ульнарная – удвоение (дублирование) пятого пальца.

По типу удвоения:
- первый тип – добавочный палец представлен рудиментом состоящим из кожи и мягких тканей;
- второй тип – добавочный палец образован в результате раздвоения основного;
- третий тип – добавочный палец является полноценным, имеет нормальную форму и размеры.
Синдактилия и полидактилия выявляется у новорожденного сразу после рождения врачом-неонатологом. Дальнейшее обследование и наблюдение ребенка проводится детским ортопедом.
Лечение
Лечение врожденных пороков развития кисти осуществляется только оперативным путем. Сроки и оптимальный способ хирургического вмешательства определяются с учетом формы и характера порока, а также дополнительного инструментального исследования – рентгенография кистей.
При полидактилии, если добавочный палец соединяется с основным только с помощью кожной мембраны, его удаление проводится в первые месяцы жизни ребенка.
Варианты хирургической коррекции полидактилии в зависимости от типа деформации могут быть различны:
- удаление добавочного сегмента (пальца) без операции на основном пальце;
- удаление добавочного сегмента (пальца) с коррегирующей остеотомией основного пальца;
- удаление добавочного сегмента (пальца) с выполнением кожной, сухожильной или костной пластики.
Хирургическое лечение синдактилии можно разделить на пять групп:
- Разделение сросшихся пальцев без кожной пластики.
- Разделение сросшихся пальцев с кожной пластикой местными тканями.
- Разделение сросшихся пальцев, дополненное свободной кожной пластикой расщепленным или полнослойным (нерасщепленным) кожным лоскутом.
- Разделение сросшихся пальцев, дополненное комбинированной кожной пластикой с использованием местных тканей и свободных аутотрансплантатов.
- Многоэтапные операции с кожной, сухожильно-мышечной и костной пластикой.
В послеоперационном периоде с целью иммобилизации пальцев на конечность накладывается гипсовая лонгета. После снятия гипсовой лонгеты приступают к восстановительному лечению, включающему физико-функциональное лечение и ЛФК.
Консультативный прием завотделения в отделении детской ортопедии МБУЗ ГБ№20.
Руководитель отделения детской травматологии и ортопедии для детей – главный детский ортопед Ростова-на-Дону, к.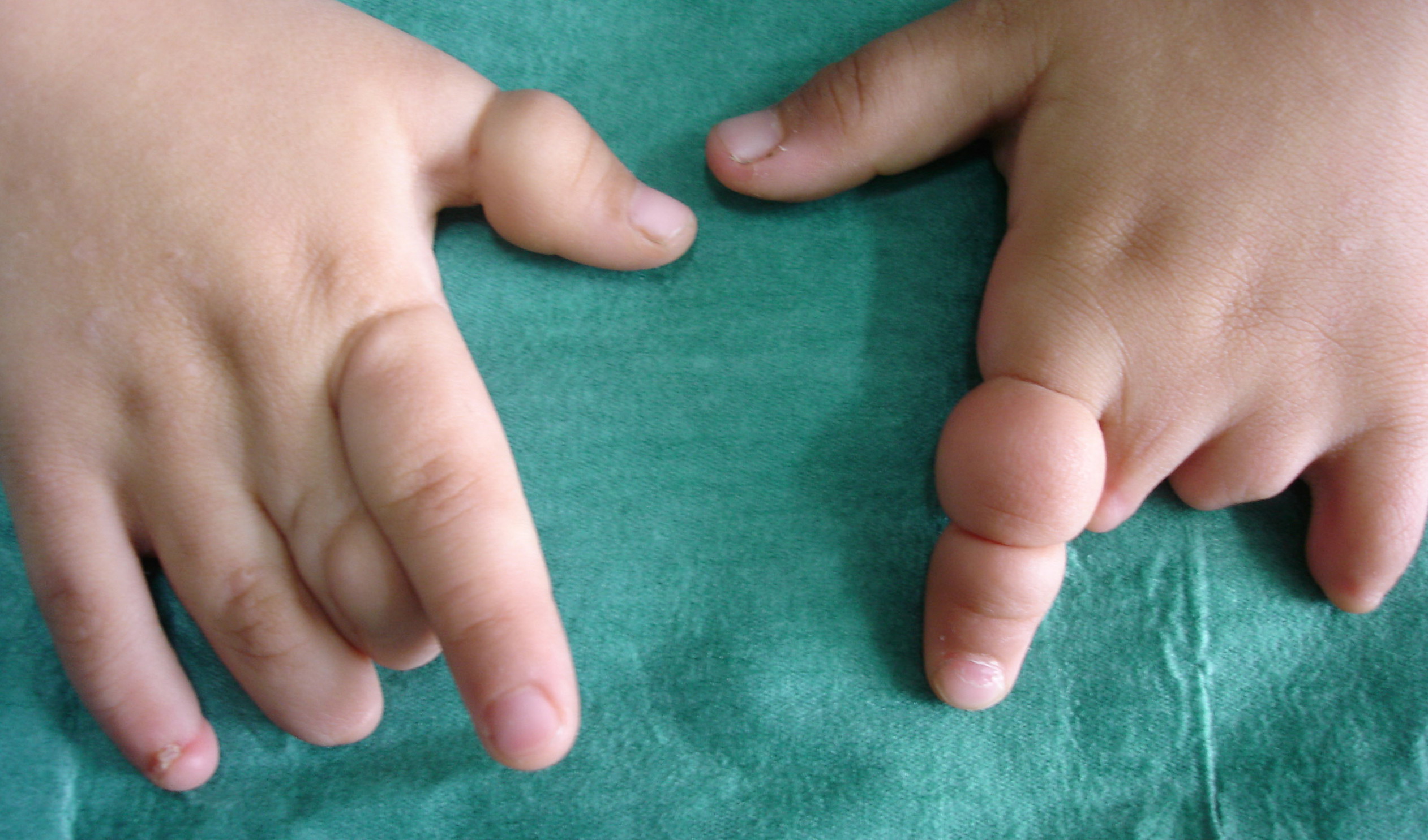 м.н. Мурадьян Владимир Юрьевич.
м.н. Мурадьян Владимир Юрьевич.
Адрес: Коммунистический, 39, 4 этаж;
часы приема: по субботам, с 10:00 до 13:00;
контактный телефон: 271-97-20.
ОБЩИЕ ПРИНЦИПЫ ЛЕЧЕНИЯ ВРОЖДЕННЫХ АНОМАЛИЙ РАЗВИТИЯ ВЕРХНЕЙ КОНЕЧНОСТИ. ЧАСТЬ I
Данная статья представляет собой краткий экскурс по основным принципам лечения врожденных аномалий развития верхней конечности. В ней представлена последняя, актуальная на данный момент классификация врожденных пороков верхней конечности, освещены вопросы их диагностики и общие принципы оператив- ного лечения. В первой части подробно рассмотрены часто встречающиеся врожденные деформации, такие как полидактилия, синдактилия, лучевая недостаточность и синдром врожденных амниотических перетяжек. В статье автор опирается как на данные современной мировой литературы, так и на личный опыт, приводя в качестве иллюстративного материала случаи из своей практики.
THE MAIN PRINCIPLES OF MANAGEMENT OF CONGENITAL ANOMALIES OF THE UPPER LIMB. PART I.pdf ВВЕДЕНИЕ Какой бы узкой специальностью ни считалась хирургия пороков развития верхней конечности, едва ли найдется детский ортопед, который за годы своей практики ни разу бы ни столкнулся ни с одной из врожденных аномалий рук у ребенка. К сожалению, количество литературы на русском языке в данной области очень ограничено, и систематизация знаний, полученных из различных литературных источников, требует значительного количества времени, которое достаточно тяжело найти практикующему врачу. Мы постарались отразить в этой статье самые важные аспекты практической информации, необходимой врачу, столкнувшемуся в своей практике с ребенком с врожденной аномалией развития верхней конечности. Классификация, терминология, ключевые моменты диагностики, возраст ребенка, в котором его следует направлять на оперативное лечение, и описание наиболее часто встречающихся пороков развития верхней конечности у детей являются теми практическими вопросами, ответы на которые предлагаются читателю в этой статье.
PART I.pdf ВВЕДЕНИЕ Какой бы узкой специальностью ни считалась хирургия пороков развития верхней конечности, едва ли найдется детский ортопед, который за годы своей практики ни разу бы ни столкнулся ни с одной из врожденных аномалий рук у ребенка. К сожалению, количество литературы на русском языке в данной области очень ограничено, и систематизация знаний, полученных из различных литературных источников, требует значительного количества времени, которое достаточно тяжело найти практикующему врачу. Мы постарались отразить в этой статье самые важные аспекты практической информации, необходимой врачу, столкнувшемуся в своей практике с ребенком с врожденной аномалией развития верхней конечности. Классификация, терминология, ключевые моменты диагностики, возраст ребенка, в котором его следует направлять на оперативное лечение, и описание наиболее часто встречающихся пороков развития верхней конечности у детей являются теми практическими вопросами, ответы на которые предлагаются читателю в этой статье.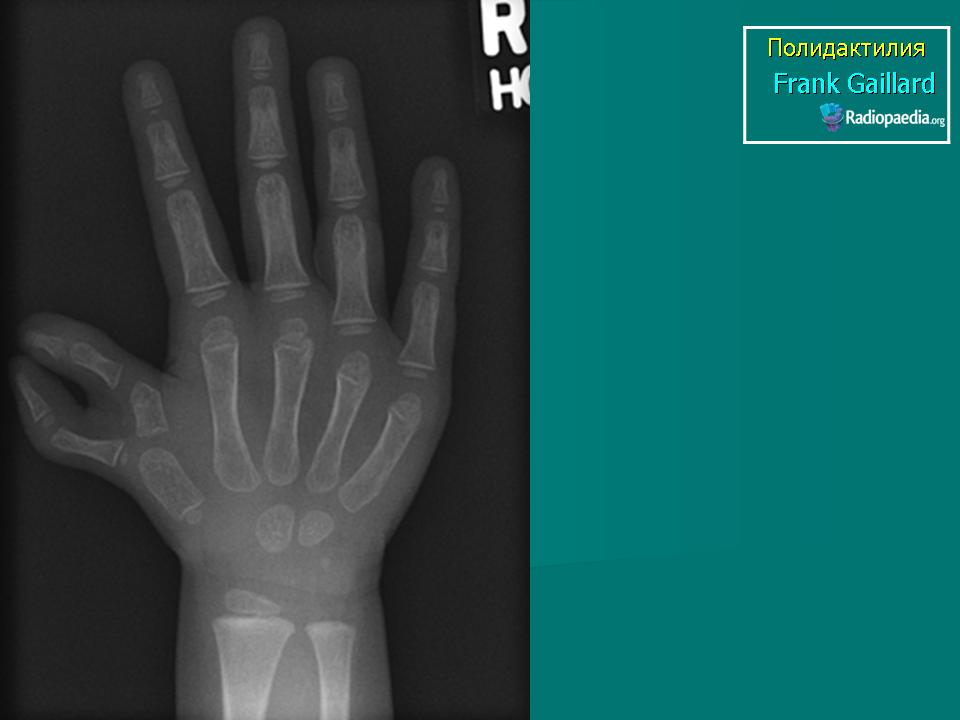 КЛАССИФИКАЦИЯ Пороки развития кисти и верхней конечности столь разнообразны, что систематизация их в классификации является непростой задачей. Одной из первых классификаций, которая была основана на уровне понимания для своего времени процессов развития верхней конечности, была классификация A. Swanson [1], одобренная в 1975 г. комитетом Американского общества Реконструктивная хирургия 41 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 хирургии кисти и Международной федерацией обществ хирургии кисти (IFSSH), после чего на втором съезде IFSSH эта классификация была рекомендована для всеобщего применения. В России с 2005 г. наиболее часто использовалась классификация И.В. Шведовченко [2]. Успехи развития молекулярной биологии и генетики последних десятилетий позволили более подробно изучить механизмы формирования верхней конечности в эмбриогенезе, что явилось толчком к появлению новой классификации ОМТ [3]. Рекомендованная к всеобщему применению комитетом IFSSH в 2014 г.
КЛАССИФИКАЦИЯ Пороки развития кисти и верхней конечности столь разнообразны, что систематизация их в классификации является непростой задачей. Одной из первых классификаций, которая была основана на уровне понимания для своего времени процессов развития верхней конечности, была классификация A. Swanson [1], одобренная в 1975 г. комитетом Американского общества Реконструктивная хирургия 41 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 хирургии кисти и Международной федерацией обществ хирургии кисти (IFSSH), после чего на втором съезде IFSSH эта классификация была рекомендована для всеобщего применения. В России с 2005 г. наиболее часто использовалась классификация И.В. Шведовченко [2]. Успехи развития молекулярной биологии и генетики последних десятилетий позволили более подробно изучить механизмы формирования верхней конечности в эмбриогенезе, что явилось толчком к появлению новой классификации ОМТ [3]. Рекомендованная к всеобщему применению комитетом IFSSH в 2014 г.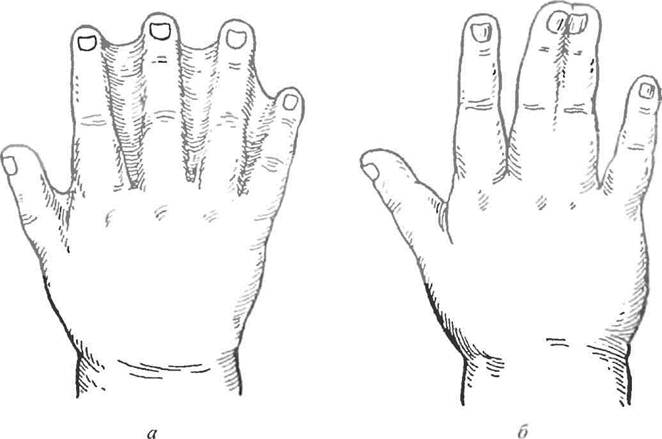 , данная классификация основана, прежде всего, на понимании того, нарушения в работе какого механизма приводят к определенному пороку развития верхней конечности (табл. 1). Таблица 1 Классификация врожденных аномалий развития верхней конечности OMT (Oberg, Manske, Tonkin) Классификация ОМТ I. Нарушения формирования A. Нарушения осевого формирования / дифференциации — вся верхняя конечность 1. Проксимально-дистальное направление (ось) i. Брахимелия с брахидактилией ii. Симбрахидактилия (a) Синдром Poland (b) Вся конечность, исключая синдром Poland iii. Поперечная недостаточность (a) Амелия (b) На уровне ключицы/лопатки (c) Плечевая (выше локтевого сустава) (d) На уровне предплечья (ниже локтевого сустава) (e) На уровне запястья (отсутствие костей запястья / на уровне проксимального ряда костей запястья / на уровне дистального ряда костей запястья) (с вовлечением предплечья/верхней конечности) (f) На уровне пястных костей (с вовлечением предплечья / верхней конечности) (g) На уровне фаланг (проксимальных / средних / дистальных) (с вовлечением предплечья/верхней конечности) iv.
, данная классификация основана, прежде всего, на понимании того, нарушения в работе какого механизма приводят к определенному пороку развития верхней конечности (табл. 1). Таблица 1 Классификация врожденных аномалий развития верхней конечности OMT (Oberg, Manske, Tonkin) Классификация ОМТ I. Нарушения формирования A. Нарушения осевого формирования / дифференциации — вся верхняя конечность 1. Проксимально-дистальное направление (ось) i. Брахимелия с брахидактилией ii. Симбрахидактилия (a) Синдром Poland (b) Вся конечность, исключая синдром Poland iii. Поперечная недостаточность (a) Амелия (b) На уровне ключицы/лопатки (c) Плечевая (выше локтевого сустава) (d) На уровне предплечья (ниже локтевого сустава) (e) На уровне запястья (отсутствие костей запястья / на уровне проксимального ряда костей запястья / на уровне дистального ряда костей запястья) (с вовлечением предплечья/верхней конечности) (f) На уровне пястных костей (с вовлечением предплечья / верхней конечности) (g) На уровне фаланг (проксимальных / средних / дистальных) (с вовлечением предплечья/верхней конечности) iv.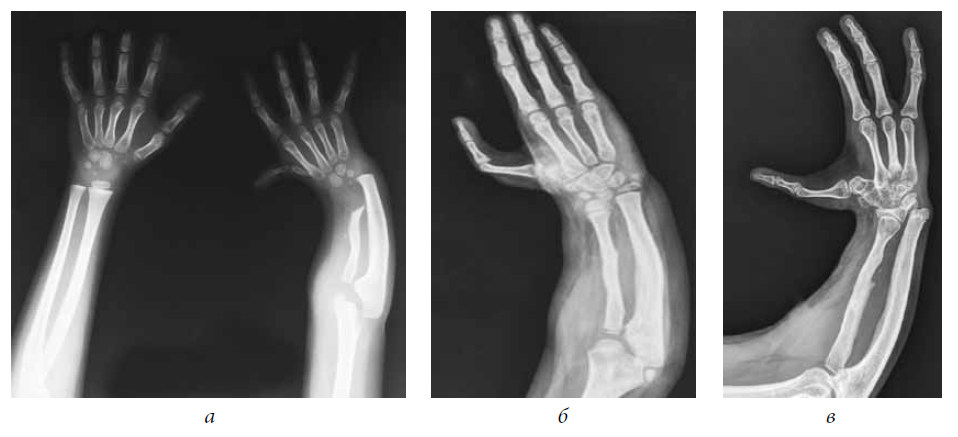 Межсегментарная недостаточность (a) Проксимальная (плечевая — ризомелия) (b) Дистальная (предплечье — мезомелия) (c) Тотальная (фокомелия) v. Полное удвоение/трипликация верхней конечности 2. Радиально-ульнарное (переднее-заднее) направление (ось) i. Продольная лучевая недостаточность — гипоплазия первого пальца (с вовлечением проксимального отдела конечности) ii. Продольная локтевая недостаточность iii. Ульнарная димелия iv. Радиоульнарный синостоз v. Врожденный вывих головки лучевой кости vi. Плече-лучевой синостоз — анкилозы локтевого сустава vii. Деформация Madelung viii. Вентральная димел 3. Дорсально-вентральное направление (ось) i. Вентральная димелия (a) Синдромы Furmann / Al-Awadi / Raas-Rotshild (b) Nail-patella синдром ii. Аплазия / гипоплазия мышц сгибателей /разгибателей 4. Неспецифичное направление (ось) i. Плечевой пояс (a) Врожденное высокое стояние лопатки (деформация Шпренгеля) (b) Аномалии развития мышц плечевого пояса ii. Артрогрипоз (Врожденный множественный артрогрипоз) B.
Межсегментарная недостаточность (a) Проксимальная (плечевая — ризомелия) (b) Дистальная (предплечье — мезомелия) (c) Тотальная (фокомелия) v. Полное удвоение/трипликация верхней конечности 2. Радиально-ульнарное (переднее-заднее) направление (ось) i. Продольная лучевая недостаточность — гипоплазия первого пальца (с вовлечением проксимального отдела конечности) ii. Продольная локтевая недостаточность iii. Ульнарная димелия iv. Радиоульнарный синостоз v. Врожденный вывих головки лучевой кости vi. Плече-лучевой синостоз — анкилозы локтевого сустава vii. Деформация Madelung viii. Вентральная димел 3. Дорсально-вентральное направление (ось) i. Вентральная димелия (a) Синдромы Furmann / Al-Awadi / Raas-Rotshild (b) Nail-patella синдром ii. Аплазия / гипоплазия мышц сгибателей /разгибателей 4. Неспецифичное направление (ось) i. Плечевой пояс (a) Врожденное высокое стояние лопатки (деформация Шпренгеля) (b) Аномалии развития мышц плечевого пояса ii. Артрогрипоз (Врожденный множественный артрогрипоз) B. Нарушения осевого формирования/ дифференциации — кисть 1. Проксимально — дистальное направление (ось) i. Брахидактилия (без поражения плеча/ предплечья) ii. Симбрахидактилия (без поражения плеча/ предплечья) iii. Поперечная недостаточность(без поражения плеча/ предплечья) (a) На уровне запястья (отсутствие костей запястья/на уровне проксимального ряда костей запястья/ на уровне дистального ряда костей запястья) (с вовлечением предплечья/верхней конечности) (b) На уровне пястных костей (с вовлечением предплечья/верхней конечности) (c) На уровне фаланг (проксимальных/ средних / дистальных) (с вовлечением предплечья/верхней конечности) 2. Радиально-ульнарное (переднее-заднее) направление (ось) i. Лучевая недостаточность (первый палец — без поражения плеча/ предплечья) ii. Локтевая недостаточность (без поражения плеча / предплечья) iii. Радиальная полидактилия iv. Трехфалангизм v. Ульнарная димелия (Зеркальная кисть, без поражения плеча / предплечья) vi. Ульнарная полидактилия 3. Дорсально-вентральное направление (ось) i.
Нарушения осевого формирования/ дифференциации — кисть 1. Проксимально — дистальное направление (ось) i. Брахидактилия (без поражения плеча/ предплечья) ii. Симбрахидактилия (без поражения плеча/ предплечья) iii. Поперечная недостаточность(без поражения плеча/ предплечья) (a) На уровне запястья (отсутствие костей запястья/на уровне проксимального ряда костей запястья/ на уровне дистального ряда костей запястья) (с вовлечением предплечья/верхней конечности) (b) На уровне пястных костей (с вовлечением предплечья/верхней конечности) (c) На уровне фаланг (проксимальных/ средних / дистальных) (с вовлечением предплечья/верхней конечности) 2. Радиально-ульнарное (переднее-заднее) направление (ось) i. Лучевая недостаточность (первый палец — без поражения плеча/ предплечья) ii. Локтевая недостаточность (без поражения плеча / предплечья) iii. Радиальная полидактилия iv. Трехфалангизм v. Ульнарная димелия (Зеркальная кисть, без поражения плеча / предплечья) vi. Ульнарная полидактилия 3. Дорсально-вентральное направление (ось) i.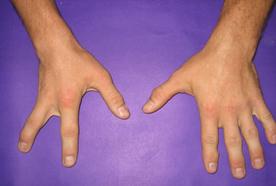 Дорсальная димелия ii. Вентральная димелия (включая гипоплазию/ аплазию ногтевых пластинок) 4. Неспецифичное направление (ось) i. Мягкие ткани 42 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии (a) Синдактилия (b) Камптодактилия (c) Сгибательно-приводящая контрактура первого пальца (d) Дистальные формы артрогрипоза ii. Скелет (a) Клинодактилия (b) Деформация Kirner (c) Синостоз / симфалангизм (костей запястья / пястных костей / фаланг) iii. Смешанные (a) Сложная форма синдактилии (b) Синполидактилия — центральная (c) Расщепление кисти (d) Синостоз / симфалангизм (костей запястья / пястных костей / фаланг) (e) Синдактилия в составе синдрома (f) Другие II. Деформации [1] Синдром врожденных (амниотических) перетяжек Стенозирующий лигаментит Другие III. Дисплазии A. Гипертрофии 1. Вся конечность i. Гемигипертрофия ii. Аберрантные сгибатели / разгибатели / собственные мышцы кисти 2. Парциальные гипертрофии i. Макродактилия ii. Аберрантные собственные мышцы кисти B.
Дорсальная димелия ii. Вентральная димелия (включая гипоплазию/ аплазию ногтевых пластинок) 4. Неспецифичное направление (ось) i. Мягкие ткани 42 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии (a) Синдактилия (b) Камптодактилия (c) Сгибательно-приводящая контрактура первого пальца (d) Дистальные формы артрогрипоза ii. Скелет (a) Клинодактилия (b) Деформация Kirner (c) Синостоз / симфалангизм (костей запястья / пястных костей / фаланг) iii. Смешанные (a) Сложная форма синдактилии (b) Синполидактилия — центральная (c) Расщепление кисти (d) Синостоз / симфалангизм (костей запястья / пястных костей / фаланг) (e) Синдактилия в составе синдрома (f) Другие II. Деформации [1] Синдром врожденных (амниотических) перетяжек Стенозирующий лигаментит Другие III. Дисплазии A. Гипертрофии 1. Вся конечность i. Гемигипертрофия ii. Аберрантные сгибатели / разгибатели / собственные мышцы кисти 2. Парциальные гипертрофии i. Макродактилия ii. Аберрантные собственные мышцы кисти B. Опухолеподобные состояния 1. Сосудистые i. Гемангиома ii. Сосудистые мальформации iii. Другие 2. Нейрогенные i. Нейрофиброматоз ii. Другие 3. Соединительнотканные i. Ювенильная апоневротическая фиброма ii. Инфантильная фиброма пальцев iii. Другие 4. Скелетные i. Остеохондроматоз ii. Энхондроматоз iii. Фиброзная дисплазия iv. Эпифизарные дисплазии IV. Синдромы A. Специфичные 1. Акрофациальный дизостоз 1 (синдром Nager) 2. Apert 3. Al-Awadi / Raas-Rothschild / фокомелия Schinzel 4. Baller-Gerold 5. Bardet-Biedl Carpenter 6. Beales 7. Catel-Manzke 8. Врожденных перетяжек (Амниотических перетяжек) 9. Cornelia de Lange (типы 1-5) 10. Crouzon 11. Down 12. EEC 13. Панцитопения Fanconi 14. Fuhrmann 15. Goltz 16. Gorlin 17. Цефалополисиндактилия Greig 18. Hajdu-Cheney 19. Гемифациальная микросомия (синдром Goldenhar) 20. Holt-Oram 21. Ушно-слезо-зубно-пальцевой (Levy-Hollister) 22. Larsen 23. Дисхондростеоз Leri-Weill 24. Moebius 25. Множественных синостозов 26. Nail-Patella 27. Noonan 28. Глазо-зубо-пальцевая дисплазия 29.
Опухолеподобные состояния 1. Сосудистые i. Гемангиома ii. Сосудистые мальформации iii. Другие 2. Нейрогенные i. Нейрофиброматоз ii. Другие 3. Соединительнотканные i. Ювенильная апоневротическая фиброма ii. Инфантильная фиброма пальцев iii. Другие 4. Скелетные i. Остеохондроматоз ii. Энхондроматоз iii. Фиброзная дисплазия iv. Эпифизарные дисплазии IV. Синдромы A. Специфичные 1. Акрофациальный дизостоз 1 (синдром Nager) 2. Apert 3. Al-Awadi / Raas-Rothschild / фокомелия Schinzel 4. Baller-Gerold 5. Bardet-Biedl Carpenter 6. Beales 7. Catel-Manzke 8. Врожденных перетяжек (Амниотических перетяжек) 9. Cornelia de Lange (типы 1-5) 10. Crouzon 11. Down 12. EEC 13. Панцитопения Fanconi 14. Fuhrmann 15. Goltz 16. Gorlin 17. Цефалополисиндактилия Greig 18. Hajdu-Cheney 19. Гемифациальная микросомия (синдром Goldenhar) 20. Holt-Oram 21. Ушно-слезо-зубно-пальцевой (Levy-Hollister) 22. Larsen 23. Дисхондростеоз Leri-Weill 24. Moebius 25. Множественных синостозов 26. Nail-Patella 27. Noonan 28. Глазо-зубо-пальцевая дисплазия 29. Рото-лице-пальцевой 30. Ушно-небно-пальцевой 31. Pallister-Hall 32. Pfeiffer 33. Pierre Robin 34. Poland 35. Proteus 36. Фокомелия Roberts-SC 37. Rothmund-Thomson 38. Rubinstein-Taybi 39. Saethre-Chotzen 40. TAR (Thrombocytopenia Absent Radius) 41. Townes-Brock 42. Трихо-рино-фалангеальный (типы 1-3) 43. Ульнарно-маммарный 44. VACTERLS ассоциация Классификация ОМТ представлена четырьмя крупными группами (Нарушения формирования; Деформации; Дисплазии; Синдромы). I. Нарушение формирования. Основываясь на современных знаниях о развитии конечности в эмбриогенезе в трех направлениях (по трем осям: проксимально-дистальной, передне-задней и дорсально-вентральной), нарушение формирования описывается отдельно для каждой из этих осей. В зависимости от уровня поражения группа делится на две части — вся верхняя конечность или только кисть. Внутри каждая из подгрупп имеет одну и ту же структуру — в зависимости от нару- Реконструктивная хирургия 43 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 шения формирования по одной из осей делится на проксимально-дистальную ось, переднезаднюю и дорсально-вентральную.
Рото-лице-пальцевой 30. Ушно-небно-пальцевой 31. Pallister-Hall 32. Pfeiffer 33. Pierre Robin 34. Poland 35. Proteus 36. Фокомелия Roberts-SC 37. Rothmund-Thomson 38. Rubinstein-Taybi 39. Saethre-Chotzen 40. TAR (Thrombocytopenia Absent Radius) 41. Townes-Brock 42. Трихо-рино-фалангеальный (типы 1-3) 43. Ульнарно-маммарный 44. VACTERLS ассоциация Классификация ОМТ представлена четырьмя крупными группами (Нарушения формирования; Деформации; Дисплазии; Синдромы). I. Нарушение формирования. Основываясь на современных знаниях о развитии конечности в эмбриогенезе в трех направлениях (по трем осям: проксимально-дистальной, передне-задней и дорсально-вентральной), нарушение формирования описывается отдельно для каждой из этих осей. В зависимости от уровня поражения группа делится на две части — вся верхняя конечность или только кисть. Внутри каждая из подгрупп имеет одну и ту же структуру — в зависимости от нару- Реконструктивная хирургия 43 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 шения формирования по одной из осей делится на проксимально-дистальную ось, переднезаднюю и дорсально-вентральную. Для смешанных пороков, имеющих признаки нарушения формирования по нескольким осям, предусмотрен раздел «неспецифичная ось». II. Деформации. К ним относятся синдром врожденных перетяжек, стенозирующий лигаментит и др. III. Дисплазии, к которым отнесены гипертрофии и опухолевые состояния. IV. Синдромы. К этой группе отнесен на данный момент список из 44 заболеваний, который, является далеко не полным перечнем синдромов, куда могут входить аномалии развития верхней конечности. ВСТРЕЧАЕМОСТЬ Встречаемость врожденных аномалий развития верхних конечностей варьирует, по данным разных авторов, и зависит в первую очередь от страны, где проводилось исследование. Одни из наиболее усредненных цифр были представлены Комитетом по врожденным деформациям Международной федерации обществ кистевых хирургов в 1982 г. [4]. Общая встречаемость по этим данным составила 22,91 случая на 10 тыс. новорожденных. Из них 9,5 случая приходилось на различные виды полидактилии, 5,8 — на группу поперечных недоразвитий на любом уровне конечности, 1,5 — на долю синдактилии, 1,3 — на гипоплазию, 0,8 — на лучевую косорукость и 0,6 случая — на синдром врожденных перетяжек.
Для смешанных пороков, имеющих признаки нарушения формирования по нескольким осям, предусмотрен раздел «неспецифичная ось». II. Деформации. К ним относятся синдром врожденных перетяжек, стенозирующий лигаментит и др. III. Дисплазии, к которым отнесены гипертрофии и опухолевые состояния. IV. Синдромы. К этой группе отнесен на данный момент список из 44 заболеваний, который, является далеко не полным перечнем синдромов, куда могут входить аномалии развития верхней конечности. ВСТРЕЧАЕМОСТЬ Встречаемость врожденных аномалий развития верхних конечностей варьирует, по данным разных авторов, и зависит в первую очередь от страны, где проводилось исследование. Одни из наиболее усредненных цифр были представлены Комитетом по врожденным деформациям Международной федерации обществ кистевых хирургов в 1982 г. [4]. Общая встречаемость по этим данным составила 22,91 случая на 10 тыс. новорожденных. Из них 9,5 случая приходилось на различные виды полидактилии, 5,8 — на группу поперечных недоразвитий на любом уровне конечности, 1,5 — на долю синдактилии, 1,3 — на гипоплазию, 0,8 — на лучевую косорукость и 0,6 случая — на синдром врожденных перетяжек.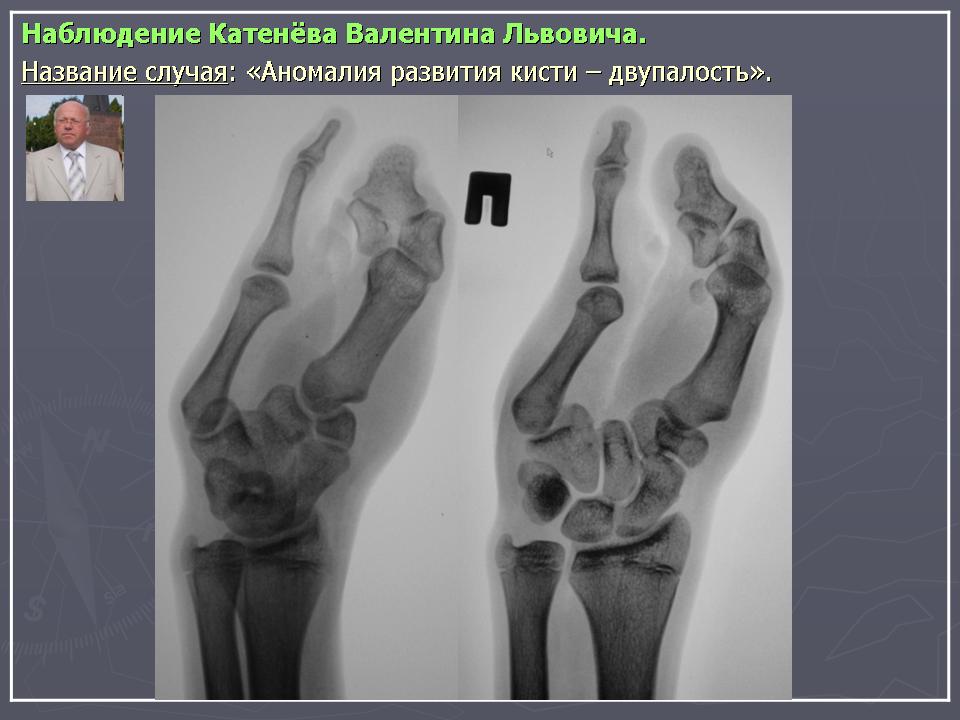 ДИАГНОСТИКА Возможности антенатальной ультразвуковой диагностики (рис. 1) становятся шире с каждым годом. Важность ее для своевременной подготовки родителей к рождению ребенка с особенностями не вызывает сомнений [5]. Визуализация верхней конечности на ультразвуковом исследовании (УЗИ) становится возможной с момента начала оссификации ее костей. Кости предплечья и плеча оссифицируются на 11-й нед от последнего менструального цикла, а пястные кости — с 12-й нед [6]. Рентгенологическое исследование является необходимым при большинстве аномалий развития верхней конечности. Ключевым вопросом считается возраст, в котором оно должно проводиться. Выполненные сразу после рождения рентгеновские снимки могут дать некоторую общую картину, однако снимки, выполненные накануне оперативного вмешательства, как правило, оказываются намного более информативными [5]. Это связано с постепенным замещением хрящевой модели костей верхней конечности костной, которая лучше позволяет оценить истинные пропорции и соотношения частей скелета.
ДИАГНОСТИКА Возможности антенатальной ультразвуковой диагностики (рис. 1) становятся шире с каждым годом. Важность ее для своевременной подготовки родителей к рождению ребенка с особенностями не вызывает сомнений [5]. Визуализация верхней конечности на ультразвуковом исследовании (УЗИ) становится возможной с момента начала оссификации ее костей. Кости предплечья и плеча оссифицируются на 11-й нед от последнего менструального цикла, а пястные кости — с 12-й нед [6]. Рентгенологическое исследование является необходимым при большинстве аномалий развития верхней конечности. Ключевым вопросом считается возраст, в котором оно должно проводиться. Выполненные сразу после рождения рентгеновские снимки могут дать некоторую общую картину, однако снимки, выполненные накануне оперативного вмешательства, как правило, оказываются намного более информативными [5]. Это связано с постепенным замещением хрящевой модели костей верхней конечности костной, которая лучше позволяет оценить истинные пропорции и соотношения частей скелета.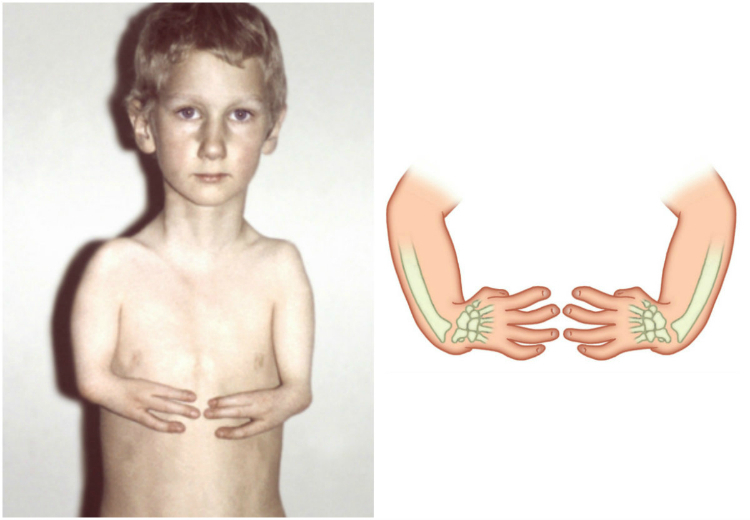 Кроме того, выполнение рентгенологического исследования при подавляющем большинстве пороков развития верхней конечности не является критерием для определения показаний к оперативному лечению, а только помогает определиться непосредственно с техническими аспектами выбора метода вмешательства, поэтому мы не считаем выполнение данного вида исследования необходимым в раннем возрасте, что позволяет снизить лучевую нагрузку на ребенка, которому в дальнейшем предстоит одна или целая серия операций с возможным рентгенологическим контролем на этапах лечения. а б Рис. 1. Сравнительное изображение левой кисти с брахидактилией, полученное при ультазвуковой диагностике у плода (а) и рентгенограмма этой же кисти, выполненная через 6 мес после рождения ребенка (б) 44 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии ВОЗРАСТ ОПЕРАЦИИ На сегодняшний день большинство специалистов считают, что лечение врожденных аномалий верхней конечности следует начинать как можно раньше, поскольку, с одной стороны, деформации при большинстве аномалий прогрессируют, с другой стороны, способности организма к значимому ремоделированию скелета сохраняются до возраста двух — максимум трех лет [7].
Кроме того, выполнение рентгенологического исследования при подавляющем большинстве пороков развития верхней конечности не является критерием для определения показаний к оперативному лечению, а только помогает определиться непосредственно с техническими аспектами выбора метода вмешательства, поэтому мы не считаем выполнение данного вида исследования необходимым в раннем возрасте, что позволяет снизить лучевую нагрузку на ребенка, которому в дальнейшем предстоит одна или целая серия операций с возможным рентгенологическим контролем на этапах лечения. а б Рис. 1. Сравнительное изображение левой кисти с брахидактилией, полученное при ультазвуковой диагностике у плода (а) и рентгенограмма этой же кисти, выполненная через 6 мес после рождения ребенка (б) 44 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии ВОЗРАСТ ОПЕРАЦИИ На сегодняшний день большинство специалистов считают, что лечение врожденных аномалий верхней конечности следует начинать как можно раньше, поскольку, с одной стороны, деформации при большинстве аномалий прогрессируют, с другой стороны, способности организма к значимому ремоделированию скелета сохраняются до возраста двух — максимум трех лет [7]. При этом недостатками слишком раннего начала оперативного лечения являются сложности на подготовительном этапе к вмешательству при заборе крови у ребенка для анализов, большие сложности и риски при проведении анестезии и более высокие риски развития рубцовых контрактур [8]. Поэтому при подавляющим большинстве пороков развития, оперативное лечение в нашей клинике мы начинаем в возрасте пациентов от 10 мес до двух лет. Исключения составляют такие пороки, как синдром амниотических перетяжек при наличии выраженного лимфостаза и (или) скомпроментированного кровообращения в сегменте, расположенного дистальнее перетяжки, когда операция может быть проведена в первые часы жизни, а также при формах синдактилии с костным сращением ногтевых фаланг I-II или IV-V пальцев, когда непропорциональный рост пальцев приводит к прогрессирующей костной деформации. Такие формы синдактилии мы стараемся оперировать в возрасте 6- 10 мес. НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЕМЫЕ ПОРОКИ РАЗВИТИЯ ВЕРХНЕЙ КОНЕЧНОСТИ Синдактилия — порок, который часто называют сросшимися пальцами, на самом деле является следствием неразделения пальцев на 8-й нед эмбриогенеза по причине нарушения процессов апоптоза (рис.
При этом недостатками слишком раннего начала оперативного лечения являются сложности на подготовительном этапе к вмешательству при заборе крови у ребенка для анализов, большие сложности и риски при проведении анестезии и более высокие риски развития рубцовых контрактур [8]. Поэтому при подавляющим большинстве пороков развития, оперативное лечение в нашей клинике мы начинаем в возрасте пациентов от 10 мес до двух лет. Исключения составляют такие пороки, как синдром амниотических перетяжек при наличии выраженного лимфостаза и (или) скомпроментированного кровообращения в сегменте, расположенного дистальнее перетяжки, когда операция может быть проведена в первые часы жизни, а также при формах синдактилии с костным сращением ногтевых фаланг I-II или IV-V пальцев, когда непропорциональный рост пальцев приводит к прогрессирующей костной деформации. Такие формы синдактилии мы стараемся оперировать в возрасте 6- 10 мес. НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЕМЫЕ ПОРОКИ РАЗВИТИЯ ВЕРХНЕЙ КОНЕЧНОСТИ Синдактилия — порок, который часто называют сросшимися пальцами, на самом деле является следствием неразделения пальцев на 8-й нед эмбриогенеза по причине нарушения процессов апоптоза (рис. 2). Синдактилию разделяют на простую (сращение только за счет мягких тканей) и сложную (имеется сращение костных структур) формы. По уровню сращения она может быть полной, или тотальной (на протяжении всей длины пальцев), или неполной. Кроме того, выделяют вариант базальной (сращение только у основания) синдактилии [9]. Встречаемость синдактилии большинство авторов оценивают в 2-3 случая на 10 тыс. новорожденных [9, 10]. Кроме того, что синдактилия является отдельным пороком развития, она может быть также составной частью других аномалий и синдромов. Синдактилия часто сопутствует такому недоразвитию кисти, как брахидактилия, в этом случае аномалия называется симбрахидактилией. Рис. 2. Простая форма синдактилии III-IV пальцев Одним из частых синдромов, включающих симбрахидактилию, является синдром Poland, при котором на стороне поражения имеется недоразвитие большой грудной мышцы, которое, в зависимости от выраженности поражения, может захватывать малую грудную мышцу и даже ребра [11]. Другим синдромом, в котором синдактилия является постоянным признаком, является синдром Apert, относящийся к группе акроцефалосиндактилий [12, 13].
2). Синдактилию разделяют на простую (сращение только за счет мягких тканей) и сложную (имеется сращение костных структур) формы. По уровню сращения она может быть полной, или тотальной (на протяжении всей длины пальцев), или неполной. Кроме того, выделяют вариант базальной (сращение только у основания) синдактилии [9]. Встречаемость синдактилии большинство авторов оценивают в 2-3 случая на 10 тыс. новорожденных [9, 10]. Кроме того, что синдактилия является отдельным пороком развития, она может быть также составной частью других аномалий и синдромов. Синдактилия часто сопутствует такому недоразвитию кисти, как брахидактилия, в этом случае аномалия называется симбрахидактилией. Рис. 2. Простая форма синдактилии III-IV пальцев Одним из частых синдромов, включающих симбрахидактилию, является синдром Poland, при котором на стороне поражения имеется недоразвитие большой грудной мышцы, которое, в зависимости от выраженности поражения, может захватывать малую грудную мышцу и даже ребра [11]. Другим синдромом, в котором синдактилия является постоянным признаком, является синдром Apert, относящийся к группе акроцефалосиндактилий [12, 13].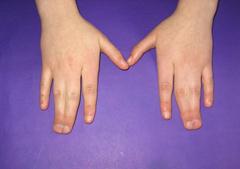 Синдактилия при синдроме врожденных перетяжек отличается от всех ее других врожденных вариантов механизмом образования и некоторыми авторами называется псевдосиндактилией [14]. Пальцы, которые разделились в положенный срок эмбриогенеза, оказываются вновь сведенными амниотической перетяжкой, и в этой ситуации синдактилия действительно является «сросшимися пальцами», что обусловливает некоторые ее анатомические особенности. Например, наличие эпителиальных ходов, дермоидных кист и анатомически нормальное расположение уровня развилки общей межпальцевой артерии на собственные пальцевые. Для успешного оперативного лечения синдактилии необходимо следовать следующим принципам: 1. Формирование межпальцевого промежутка. Вне зависимости от дизайна и применяемой методики, для формирования межпальцевого промежутка должны использоваться кровоснабжаемые лоскуты. Наиболее часто применяемыми для этой цели являются прямоугольный тыльный лоскут, прямоугольный ладонный лоскут или два треугольных лоскута с ладонной и тыльной поверхности.
Синдактилия при синдроме врожденных перетяжек отличается от всех ее других врожденных вариантов механизмом образования и некоторыми авторами называется псевдосиндактилией [14]. Пальцы, которые разделились в положенный срок эмбриогенеза, оказываются вновь сведенными амниотической перетяжкой, и в этой ситуации синдактилия действительно является «сросшимися пальцами», что обусловливает некоторые ее анатомические особенности. Например, наличие эпителиальных ходов, дермоидных кист и анатомически нормальное расположение уровня развилки общей межпальцевой артерии на собственные пальцевые. Для успешного оперативного лечения синдактилии необходимо следовать следующим принципам: 1. Формирование межпальцевого промежутка. Вне зависимости от дизайна и применяемой методики, для формирования межпальцевого промежутка должны использоваться кровоснабжаемые лоскуты. Наиболее часто применяемыми для этой цели являются прямоугольный тыльный лоскут, прямоугольный ладонный лоскут или два треугольных лоскута с ладонной и тыльной поверхности. При формировании промежутка необходимо учитывать, что в норме его ладонный край расположен значительно дистальнее тыльного, образуя наклон от 40 до 45° по отношению к саггитальной оси. Второй и третий межпальцевые промежутки лежат на одном уровне или индивидуально Реконструктивная хирургия 45 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 могут отличаться незначительно. Четвертый межпальцевой промежуток является самым глубоким, что необходимо учитывать при планировании операции. Однако, если при наиболее часто встречаемой синдактилии III-IV пальцев можно ориентироваться на глубину смежных промежутков, то гораздо сложнее дело обстоит при синдактилии всех трехфаланговых пальцев. Ориентиром в этой ситуации может являться правило, согласно которому межпальцевой промежуток при нормально развитых пальцах располагается ровно посредине линии, соединяющей складку проксимального межфалангового сустава (МФС) с дистальной ладонной складкой [15, 16]. 2. Закрытие боковых поверхностей пальцев.
При формировании промежутка необходимо учитывать, что в норме его ладонный край расположен значительно дистальнее тыльного, образуя наклон от 40 до 45° по отношению к саггитальной оси. Второй и третий межпальцевые промежутки лежат на одном уровне или индивидуально Реконструктивная хирургия 45 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 могут отличаться незначительно. Четвертый межпальцевой промежуток является самым глубоким, что необходимо учитывать при планировании операции. Однако, если при наиболее часто встречаемой синдактилии III-IV пальцев можно ориентироваться на глубину смежных промежутков, то гораздо сложнее дело обстоит при синдактилии всех трехфаланговых пальцев. Ориентиром в этой ситуации может являться правило, согласно которому межпальцевой промежуток при нормально развитых пальцах располагается ровно посредине линии, соединяющей складку проксимального межфалангового сустава (МФС) с дистальной ладонной складкой [15, 16]. 2. Закрытие боковых поверхностей пальцев. Формирование треугольных пальцевых лоскутов позволяет избежать линейного рубца и является по сути дела «пружиной», которая, растягиваясь, позволяет в ходе роста ребенка избежать осложнений в виде рубцовых контрактур пальцев (рис. 3, 4). Рис. 3. Формирование зигзагообразного рубца при устранении синдактилии для профилактики образования рубцовых контрактур; созданный из тыльного лоскута межпальцевой промежуток достаточной ширины и правильной формы Рис. 4. Рубцовая контрактура III пальца, развившаяся вследствие нарушения оперативной техники при устранении простой формы синдактилии и создания линейного рубца по боковой поверхности пальца в условиях избыточного натяжения кожи При формировании треугольных лоскутов необходимо придерживаться «золотой середины» между слишком остроугольными лоскутами, имеющими более высокий риск некроза, и тупоугольными лоскутами, которые с ростом ребенка быстрее могут распрямиться в линейный тянущий рубец. Пересадка некровоснабжаемых кожных аутотрансплантатов является при большинстве форм синдактилии непременным элементом операции, поскольку закрытие боковых поверхностей пальцев только за счет треугольных лоскутов возможно только при наличии достаточного его запаса в виде кожной «перепонки».
Формирование треугольных пальцевых лоскутов позволяет избежать линейного рубца и является по сути дела «пружиной», которая, растягиваясь, позволяет в ходе роста ребенка избежать осложнений в виде рубцовых контрактур пальцев (рис. 3, 4). Рис. 3. Формирование зигзагообразного рубца при устранении синдактилии для профилактики образования рубцовых контрактур; созданный из тыльного лоскута межпальцевой промежуток достаточной ширины и правильной формы Рис. 4. Рубцовая контрактура III пальца, развившаяся вследствие нарушения оперативной техники при устранении простой формы синдактилии и создания линейного рубца по боковой поверхности пальца в условиях избыточного натяжения кожи При формировании треугольных лоскутов необходимо придерживаться «золотой середины» между слишком остроугольными лоскутами, имеющими более высокий риск некроза, и тупоугольными лоскутами, которые с ростом ребенка быстрее могут распрямиться в линейный тянущий рубец. Пересадка некровоснабжаемых кожных аутотрансплантатов является при большинстве форм синдактилии непременным элементом операции, поскольку закрытие боковых поверхностей пальцев только за счет треугольных лоскутов возможно только при наличии достаточного его запаса в виде кожной «перепонки». Попытки закрыть боковые поверхности пальцев без пересадки кожных аутотрансплантатов за счет сильного натяжения треугольных лоскутов могут привести к развитию некрозов последних и формированию грубых тянущих рубцов, образованию контрактур, рубцовому рецидиву синдактилии и даже потере части пальца. При наличии костного сращения фаланг, треугольные лоскуты необходимо планировать с учетом закрытия ими опила кости, а в случае невозможности выполнения этого закрывать опил лоскутами из подкожно-жировой и межпальцевой клетчатки перед пересадкой на нее кожных аутотрансплантатов или предусматривать двухэтапную методику устранения синдактилии. 3. Аккуратное выделение собственных пальцевых сосудисто-нервных пучков во избежание их повреждения и определения уровня бифуркации. В случае, когда последняя расположена дистальнее планируемого межпальцевого промежутка, «натягивание» лоскутов поверх нее будет приводить к формированию базальной синдактилии. Методом выбора в данной ситуации должна являться аккуратная интрафасцикулярная препаровка общего пальцевого нерва с разделением его на два собственных и коагуляция с последующим пересечением одной из собственных пальцевых артерий.
Попытки закрыть боковые поверхности пальцев без пересадки кожных аутотрансплантатов за счет сильного натяжения треугольных лоскутов могут привести к развитию некрозов последних и формированию грубых тянущих рубцов, образованию контрактур, рубцовому рецидиву синдактилии и даже потере части пальца. При наличии костного сращения фаланг, треугольные лоскуты необходимо планировать с учетом закрытия ими опила кости, а в случае невозможности выполнения этого закрывать опил лоскутами из подкожно-жировой и межпальцевой клетчатки перед пересадкой на нее кожных аутотрансплантатов или предусматривать двухэтапную методику устранения синдактилии. 3. Аккуратное выделение собственных пальцевых сосудисто-нервных пучков во избежание их повреждения и определения уровня бифуркации. В случае, когда последняя расположена дистальнее планируемого межпальцевого промежутка, «натягивание» лоскутов поверх нее будет приводить к формированию базальной синдактилии. Методом выбора в данной ситуации должна являться аккуратная интрафасцикулярная препаровка общего пальцевого нерва с разделением его на два собственных и коагуляция с последующим пересечением одной из собственных пальцевых артерий. При наличии синдактилий нескольких смежных пальцев необходимо заранее планировать выбор коагуляции собственных пальцевых артерий таким образом, чтобы каждый из пальцев сохранял, как минимум, один источник кровоснабжения в виде ладонной собственной пальцевой артерии. 4. Формирование ногтевого ложа является непростой задачей при наличии тотальной синдактилии со сращением ногтевых пластинок и отсутствием ногтевых валиков со стороны сращения. Предложенные D. Buck-Gramcko лоскуты с подушечек пальцев [17] помогают в формировании бокового ногтевого валика и закрытии опила сращенных ногтевых фаланг (рис. 5). 46 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии Рис. 5. Формирование лоскутов по D. Buck-Gramcko с подушечек пальцев для закрытия боковых поверхностей ногтевых фаланг при тотальной синдактилии Полидактилия является самым часто встречающимся пороком развития кисти, она подразделяется на преаксиальную (полидактилия I пальца, радиальная полидактилия), постаксиальную (полидактилия V пальца, ульнарная полидактилия) и центральную, проявляющуюся чаще всего синполидактилией.
При наличии синдактилий нескольких смежных пальцев необходимо заранее планировать выбор коагуляции собственных пальцевых артерий таким образом, чтобы каждый из пальцев сохранял, как минимум, один источник кровоснабжения в виде ладонной собственной пальцевой артерии. 4. Формирование ногтевого ложа является непростой задачей при наличии тотальной синдактилии со сращением ногтевых пластинок и отсутствием ногтевых валиков со стороны сращения. Предложенные D. Buck-Gramcko лоскуты с подушечек пальцев [17] помогают в формировании бокового ногтевого валика и закрытии опила сращенных ногтевых фаланг (рис. 5). 46 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии Рис. 5. Формирование лоскутов по D. Buck-Gramcko с подушечек пальцев для закрытия боковых поверхностей ногтевых фаланг при тотальной синдактилии Полидактилия является самым часто встречающимся пороком развития кисти, она подразделяется на преаксиальную (полидактилия I пальца, радиальная полидактилия), постаксиальную (полидактилия V пальца, ульнарная полидактилия) и центральную, проявляющуюся чаще всего синполидактилией. При постаксиальной полидактилии добавочный палец может быть представлен как полноценным лучом (тип А), так и рудиментом на тонкой кожной ножке (тип В) [18]. При удалении хорошо развитого добавочного V пальца необходимо переносить на основной V палец такие важные анатомические структуры, как сухожилие мышцы, отводящей мизинец, и коллатеральную ульнарную связку при наличии общего пястно-фалангового сустава (ПФС) у основного и добавочного V пальца [14]. Одним из наиболее распространенных способов удаления рудиментарного V пальца, соединяющегося с кистью тонкой кожной ножкой, является его перевязка нитью в роддоме, после чего постепенно происходит сухая гангрена и «отпадение» этого пальца. Результатом такого лечения часто является наличие выпуклого бугорка на месте удаленного пальца и (или) болезненного рубца, связанного с запаиванием в него собственного пальцевого нерва, проходящего в кожной ножке к добавочному пальцу. Мы не можем адекватно оценить, насколько может быть болезненна для новорожденного ребенка эта процедура.
При постаксиальной полидактилии добавочный палец может быть представлен как полноценным лучом (тип А), так и рудиментом на тонкой кожной ножке (тип В) [18]. При удалении хорошо развитого добавочного V пальца необходимо переносить на основной V палец такие важные анатомические структуры, как сухожилие мышцы, отводящей мизинец, и коллатеральную ульнарную связку при наличии общего пястно-фалангового сустава (ПФС) у основного и добавочного V пальца [14]. Одним из наиболее распространенных способов удаления рудиментарного V пальца, соединяющегося с кистью тонкой кожной ножкой, является его перевязка нитью в роддоме, после чего постепенно происходит сухая гангрена и «отпадение» этого пальца. Результатом такого лечения часто является наличие выпуклого бугорка на месте удаленного пальца и (или) болезненного рубца, связанного с запаиванием в него собственного пальцевого нерва, проходящего в кожной ножке к добавочному пальцу. Мы не можем адекватно оценить, насколько может быть болезненна для новорожденного ребенка эта процедура. Избежать подобных осложнений поможет выполнение хирургического удаления пальца с предварительным выделением и коагуляцией пальцевой артерии и отсечением пальцевого нерва, выделенного максимально в проксимальном направлении под кратковременной общей анестезий или седацией в сочетании с местной анестезией. Преаксиальная полидактилия может быть представлена более разнообразными формами, в основе классификации которых лежит система H.D. Wassel (рис. 6). Согласно этой классификации, четные типы II, IV, VI имеют общие суставы, и операция по удалению добавочного пальца при данных типах всегда будет сопровождаться внутрисуставным вмешательством. I, III и V типы удвоения первого луча характеризуются синостозом на уровне фаланг или пястной кости с наличием раздельных суставов, что позволяет давать при данных типах более хороший функциональный прогноз оперативного лечения. При этом самым часто встречаемым (более 50% случаев) является IV тип полидактилии, при котором наблюдаются наиболее серьезные деформации, как ульнарного, так и радиального первых пальцев [20].
Избежать подобных осложнений поможет выполнение хирургического удаления пальца с предварительным выделением и коагуляцией пальцевой артерии и отсечением пальцевого нерва, выделенного максимально в проксимальном направлении под кратковременной общей анестезий или седацией в сочетании с местной анестезией. Преаксиальная полидактилия может быть представлена более разнообразными формами, в основе классификации которых лежит система H.D. Wassel (рис. 6). Согласно этой классификации, четные типы II, IV, VI имеют общие суставы, и операция по удалению добавочного пальца при данных типах всегда будет сопровождаться внутрисуставным вмешательством. I, III и V типы удвоения первого луча характеризуются синостозом на уровне фаланг или пястной кости с наличием раздельных суставов, что позволяет давать при данных типах более хороший функциональный прогноз оперативного лечения. При этом самым часто встречаемым (более 50% случаев) является IV тип полидактилии, при котором наблюдаются наиболее серьезные деформации, как ульнарного, так и радиального первых пальцев [20]. Рис. 6. Классификация преаксиальной полидактилии по H.D. Wassel (1969) [20] Основной целью оперативного вмешательства при преаксиальной полидактилии должно являться не удаление добавочного пальца, а реконструкция и создание прямого, стабильного, подвижного и функционального I пальца. При I и II типе по классификации Wassel имеются два основных варианта оперативного лечения, у каждого из которых есть свои достоинства и недостатки, и как следствие, сторонники и противники. Один из вариантов — операция Bilhaut-Cloquet, основная идея которой заключается в создании одного пальца нормального размера из двух недоразвитых. При этом производится резекция меньших боковых, обращенных друг к другу частей основного и добавочного пальцев (рис. 7), а большие порции, соединяясь, создают более широкий палец, имеющий стабильные коллатеральные связки МФС и широкую ногтевую пластинку. Рис. 7. Схема операции Bilhaut-Cloquet Реконструктивная хирургия 47 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 Одним из недостатков данной методики является непредсказуемость эстетического результата в связи с тем, что на ногтевой пластинке, соединенной из двух частей, в большинстве случаев место стыка очень заметно и выступает в виде гребня (рис.
Рис. 6. Классификация преаксиальной полидактилии по H.D. Wassel (1969) [20] Основной целью оперативного вмешательства при преаксиальной полидактилии должно являться не удаление добавочного пальца, а реконструкция и создание прямого, стабильного, подвижного и функционального I пальца. При I и II типе по классификации Wassel имеются два основных варианта оперативного лечения, у каждого из которых есть свои достоинства и недостатки, и как следствие, сторонники и противники. Один из вариантов — операция Bilhaut-Cloquet, основная идея которой заключается в создании одного пальца нормального размера из двух недоразвитых. При этом производится резекция меньших боковых, обращенных друг к другу частей основного и добавочного пальцев (рис. 7), а большие порции, соединяясь, создают более широкий палец, имеющий стабильные коллатеральные связки МФС и широкую ногтевую пластинку. Рис. 7. Схема операции Bilhaut-Cloquet Реконструктивная хирургия 47 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 Одним из недостатков данной методики является непредсказуемость эстетического результата в связи с тем, что на ногтевой пластинке, соединенной из двух частей, в большинстве случаев место стыка очень заметно и выступает в виде гребня (рис. 8) или, наоборот, пластинка состоит из двух раздельных частей [21]. Вторым отрицательным моментом данной методики является объем внутрисуставного вмешательства, который приводит к существенному снижению амплитуды движений в МФС, что заставило многих хирургов отказаться от этого варианта вмешательства. а б Рис. 8. Отдаленный результат (более 15 лет) операции Bilhaut-Cloquet. Деформация ногтевой пластинки (а) и двойной контур подушечки I пальца по ладонной и торцевой поверхности (б) Применяемый нами вариант заключается в формировании латерального кожного лоскута с удаляемого пальца, удалении добавочной фаланги, корригирующей остеотомии основной фаланги при наличии угловой деформации и восстановлении коллатеральной связки МФС (рис. 9, 10). При данном методе латеральный лоскут обеспечивает сохранение прежней чувствительности радиального края большого пальца, позволяет сформировать полноценный ногтевой валик, но исключает проведение остеотомии через сустав, зоны роста и ногтевую пластинку. Рис.
8) или, наоборот, пластинка состоит из двух раздельных частей [21]. Вторым отрицательным моментом данной методики является объем внутрисуставного вмешательства, который приводит к существенному снижению амплитуды движений в МФС, что заставило многих хирургов отказаться от этого варианта вмешательства. а б Рис. 8. Отдаленный результат (более 15 лет) операции Bilhaut-Cloquet. Деформация ногтевой пластинки (а) и двойной контур подушечки I пальца по ладонной и торцевой поверхности (б) Применяемый нами вариант заключается в формировании латерального кожного лоскута с удаляемого пальца, удалении добавочной фаланги, корригирующей остеотомии основной фаланги при наличии угловой деформации и восстановлении коллатеральной связки МФС (рис. 9, 10). При данном методе латеральный лоскут обеспечивает сохранение прежней чувствительности радиального края большого пальца, позволяет сформировать полноценный ногтевой валик, но исключает проведение остеотомии через сустав, зоны роста и ногтевую пластинку. Рис. 9. Формирование латерального лоскута на собственной пальцевой артерии со стороны удаляемой фаланги а б Рис. 10. Операция при II типе полидактилии по H.D. Wassel с формированием латерального лоскута, удалением добавочной фаланги, корригирующей остеотомией основной фаланги и остеосинтезом осевой спицей: вид до операции (а), вид после операции (б) При III типе полидактилии производится косая остеотомия сращения основных фаланг, при этом в случае искривления остающегося пальца возникает необходимость проведения корригирующей остеотомии основной фаланги. Доступ возможен как по радиальной поверхности с формированием треугольных лоскутов с удаляемого пальца, так и с формированием описанного выше латерального лоскута, имеющего осевое кровоснабжение на собственной пальцевой артерии. Тип IV является наиболее часто встречаемым, но и наиболее сложным для хирургического лечения вариантом, поскольку в большинстве случаев имеется штыкообразная деформация основного и добавочного пальцев (рис. 11). В ходе реконструкции зачастую требуется транспозиция сухожилия m.
9. Формирование латерального лоскута на собственной пальцевой артерии со стороны удаляемой фаланги а б Рис. 10. Операция при II типе полидактилии по H.D. Wassel с формированием латерального лоскута, удалением добавочной фаланги, корригирующей остеотомией основной фаланги и остеосинтезом осевой спицей: вид до операции (а), вид после операции (б) При III типе полидактилии производится косая остеотомия сращения основных фаланг, при этом в случае искривления остающегося пальца возникает необходимость проведения корригирующей остеотомии основной фаланги. Доступ возможен как по радиальной поверхности с формированием треугольных лоскутов с удаляемого пальца, так и с формированием описанного выше латерального лоскута, имеющего осевое кровоснабжение на собственной пальцевой артерии. Тип IV является наиболее часто встречаемым, но и наиболее сложным для хирургического лечения вариантом, поскольку в большинстве случаев имеется штыкообразная деформация основного и добавочного пальцев (рис. 11). В ходе реконструкции зачастую требуется транспозиция сухожилия m. abductor pollicis brevis к основанию оставляемого пальца с одновременной реконструкцией коллатеральной связки ПФС. Кроме краевой остеотомии расширенной головки пястной кости, во многих случаях необходима корригирующая остеотомия пястной кости / основной фаланги. Аномальное расположение точек прикрепления сухожилий длинных сгибателя и разгибателя требует их центрации по оси пальца для восстановления баланса и профилактики развития девиации в послеоперационном периоде. 48 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии Рис. 11. Преаксиальная полидактилия, IV тип по H.D. Wassel. Определяются штыкообразные деформации симметричных удвоенных пальцев Оперативная коррекция при V и VI типе полидактилии преследует те же задачи, что и при IV типе, однако в некоторых случаях требуется кожная пластика в первом межпальцевом промежутке для увеличения его ширины. При VII типе после определения наиболее развитого пальца выполняется удаление либо поллицизация трехфалангового пальца.
abductor pollicis brevis к основанию оставляемого пальца с одновременной реконструкцией коллатеральной связки ПФС. Кроме краевой остеотомии расширенной головки пястной кости, во многих случаях необходима корригирующая остеотомия пястной кости / основной фаланги. Аномальное расположение точек прикрепления сухожилий длинных сгибателя и разгибателя требует их центрации по оси пальца для восстановления баланса и профилактики развития девиации в послеоперационном периоде. 48 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии Рис. 11. Преаксиальная полидактилия, IV тип по H.D. Wassel. Определяются штыкообразные деформации симметричных удвоенных пальцев Оперативная коррекция при V и VI типе полидактилии преследует те же задачи, что и при IV типе, однако в некоторых случаях требуется кожная пластика в первом межпальцевом промежутке для увеличения его ширины. При VII типе после определения наиболее развитого пальца выполняется удаление либо поллицизация трехфалангового пальца. В некоторых случаях при сочетании недоразвития дистального отдела одного пальца с недоразвитием проксимального отдела другого, требуются более сложные реконструктивные вмешательства, а именно транспозиция более развитого дистального отдела одного пальца на проксимальный отдел другого (рис. 12). а б а б Рис. 12. Вариант удвоения первого луча с недоразвитием проксимального отдела ульнарно расположенного луча и дистального отдела радиально расположенного луча, внешний вид (а) и рентгенограмма (б) перед операцией. Выполнена операция транспозиции дистального отдела радиального первого луча на проксимальный отдел ульнарного первого луча с остеосинтезом спицей. Внешний вид (в) и рентгенограмма (г) в конце операции Реконструктивная хирургия 49 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 Лучевая недостаточность. Включает в себя врожденную лучевую косорукость и гипоплазию первого луча кисти. Встречается в 1 случае на 55 тыс. новорожденных [22]. Этот порок характеризуется недоразвитием структур, расположенных по преаксиальному краю конечности, основными из которых являются лучевая кость и первый луч кисти (рис.
В некоторых случаях при сочетании недоразвития дистального отдела одного пальца с недоразвитием проксимального отдела другого, требуются более сложные реконструктивные вмешательства, а именно транспозиция более развитого дистального отдела одного пальца на проксимальный отдел другого (рис. 12). а б а б Рис. 12. Вариант удвоения первого луча с недоразвитием проксимального отдела ульнарно расположенного луча и дистального отдела радиально расположенного луча, внешний вид (а) и рентгенограмма (б) перед операцией. Выполнена операция транспозиции дистального отдела радиального первого луча на проксимальный отдел ульнарного первого луча с остеосинтезом спицей. Внешний вид (в) и рентгенограмма (г) в конце операции Реконструктивная хирургия 49 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 Лучевая недостаточность. Включает в себя врожденную лучевую косорукость и гипоплазию первого луча кисти. Встречается в 1 случае на 55 тыс. новорожденных [22]. Этот порок характеризуется недоразвитием структур, расположенных по преаксиальному краю конечности, основными из которых являются лучевая кость и первый луч кисти (рис. 13). И, несмотря на то, что недоразвитие вышеописанных структур является фактически одним заболеванием, степень недоразвития первого луча и лучевой кости классифицируются отдельно, определяя выбор методики коррекции имеющихся деформаций. Существующие общие классификации лучевой недостаточности демонстрируют отсутствие корреляции между степенью недоразвития лучевой кости, костей запястья и первого луча кисти [14]. Рис. 13. Верхняя конечность ребенка с врожденной продольной лучевой недостаточностью: отсутствуют (аплазирован) первый луч кисти и лучевая кость, ткани по лучевой поверхности недоразвиты, имеется выраженная лучевая девиация кисти Наибольшее распространение в мире получила классификация лучевой косорукости по Bayne и Klug, выделяющая четыре степени. 1-я степень: лучевая кость полноценно развита, но имеется ее незначительное укорочение относительно локтевой кости. 2-я степень: лучевая кость недоразвита, укорочена. Однако представлены как проксимальный, так и дистальный ее эпифизы.
13). И, несмотря на то, что недоразвитие вышеописанных структур является фактически одним заболеванием, степень недоразвития первого луча и лучевой кости классифицируются отдельно, определяя выбор методики коррекции имеющихся деформаций. Существующие общие классификации лучевой недостаточности демонстрируют отсутствие корреляции между степенью недоразвития лучевой кости, костей запястья и первого луча кисти [14]. Рис. 13. Верхняя конечность ребенка с врожденной продольной лучевой недостаточностью: отсутствуют (аплазирован) первый луч кисти и лучевая кость, ткани по лучевой поверхности недоразвиты, имеется выраженная лучевая девиация кисти Наибольшее распространение в мире получила классификация лучевой косорукости по Bayne и Klug, выделяющая четыре степени. 1-я степень: лучевая кость полноценно развита, но имеется ее незначительное укорочение относительно локтевой кости. 2-я степень: лучевая кость недоразвита, укорочена. Однако представлены как проксимальный, так и дистальный ее эпифизы. 3-я степень: частичная аплазия лучевой кости, при которой отсутствует ее дистальный эпифиз. Дистальные, средние и даже частично проксимальные отделы могут также отсутствовать. 4-я степень: полная аплазия лучевой кости. Как правило, 1-я степень лучевой косорукости не требует оперативного лечения, во многих случаях достаточно ортезирования кисти. Ключевым моментом, определяющим показания к оперативному лечению, является отсутствие возможности у ребенка активно выводить кисть из лучевой девиации. В этом случае может потребоваться сухожильная пластика с транспозицией сухожилий лучевой группы на локтевую сторону или удлинение лучевой кости. Основным методом оперативного лечения при 2-й степени является удлинение лучевой кости дистракционным методом, хотя при более тяжелых степенях могут применяться как операции центрации или радиализации, так и различные микрохирургические реконструкции, направленные на создание стабильного сочленения между предплечьем и кистью [23]. Эти же варианты реконструкций могут применяться при 3-й степени, но основным методом оперативного лечения для 3-й и 4-й степени остаются центрация и радиализация кисти.
3-я степень: частичная аплазия лучевой кости, при которой отсутствует ее дистальный эпифиз. Дистальные, средние и даже частично проксимальные отделы могут также отсутствовать. 4-я степень: полная аплазия лучевой кости. Как правило, 1-я степень лучевой косорукости не требует оперативного лечения, во многих случаях достаточно ортезирования кисти. Ключевым моментом, определяющим показания к оперативному лечению, является отсутствие возможности у ребенка активно выводить кисть из лучевой девиации. В этом случае может потребоваться сухожильная пластика с транспозицией сухожилий лучевой группы на локтевую сторону или удлинение лучевой кости. Основным методом оперативного лечения при 2-й степени является удлинение лучевой кости дистракционным методом, хотя при более тяжелых степенях могут применяться как операции центрации или радиализации, так и различные микрохирургические реконструкции, направленные на создание стабильного сочленения между предплечьем и кистью [23]. Эти же варианты реконструкций могут применяться при 3-й степени, но основным методом оперативного лечения для 3-й и 4-й степени остаются центрация и радиализация кисти.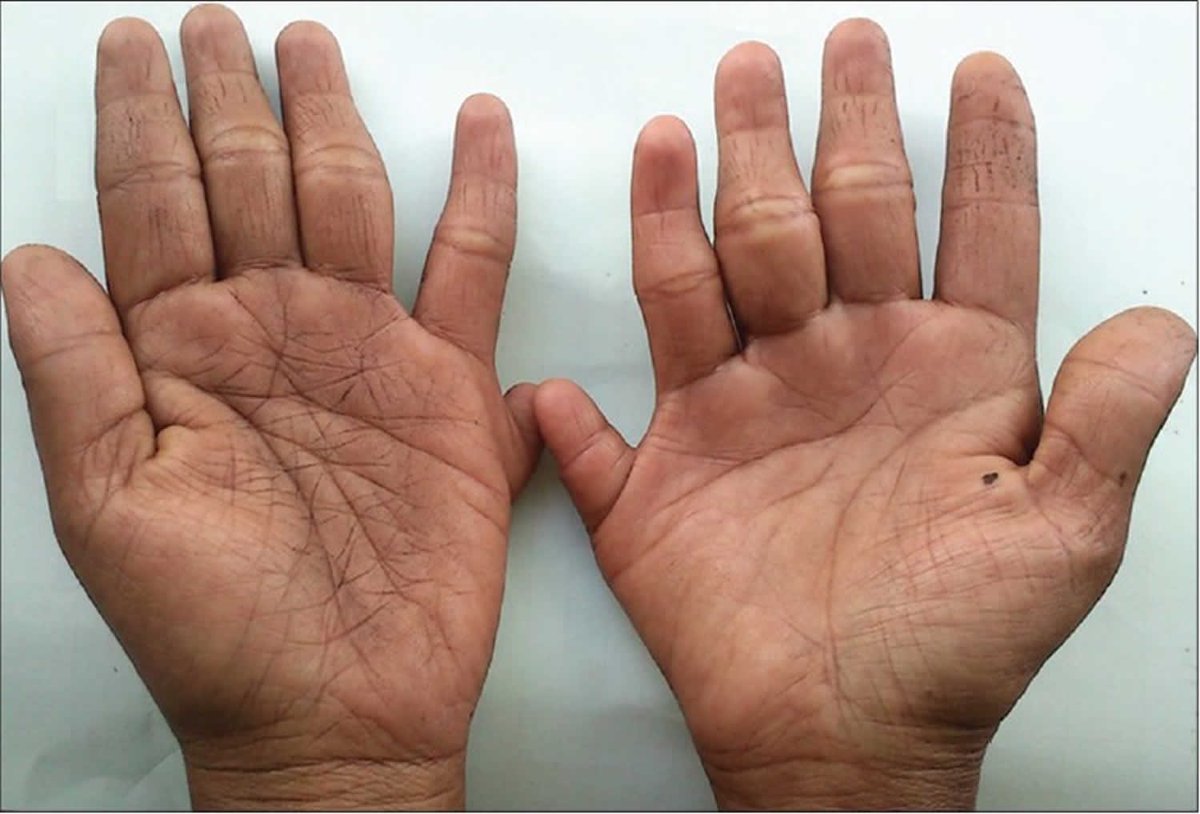 Наиболее часто используемой классификацией недоразвития I пальца является модифицированная классификация W. Blauth [24], выделяющая следующие степени: I — минимальное укорочение пальца и снижение ширины межпальцевого промежутка. Умеренное недоразвитие мышц тенара. II — cужение первого межпальцевого промежутка, аплазия или гипоплазия мышц тенара. IIIA — тип II + недоразвитие сухожилий длинных мышц I пальца, гипоплазия пястной кости при стабильном трапецио-пястном суставе. IIIB — тип II + недоразвитие сухожилий длинных мышц I пальца, частичная аплазия проксимальной части пястной кости, нестабильность трапецио-пястного сустава. IV — недоразвитый I палец соединяется с кистью мягкотканной ножкой. V — полное отсутствие первого луча кисти. Как правило, первый тип врожденной гипоплазии I пальца не требует оперативного лечения. При втором типе используются компоненты операции, направленные на восстановление отсутствующих функций I пальца. Так, гипоплазия мышц тенара с отсутствием возможности активной оппозиции корректируется за счет оппонентопластики.
Наиболее часто используемой классификацией недоразвития I пальца является модифицированная классификация W. Blauth [24], выделяющая следующие степени: I — минимальное укорочение пальца и снижение ширины межпальцевого промежутка. Умеренное недоразвитие мышц тенара. II — cужение первого межпальцевого промежутка, аплазия или гипоплазия мышц тенара. IIIA — тип II + недоразвитие сухожилий длинных мышц I пальца, гипоплазия пястной кости при стабильном трапецио-пястном суставе. IIIB — тип II + недоразвитие сухожилий длинных мышц I пальца, частичная аплазия проксимальной части пястной кости, нестабильность трапецио-пястного сустава. IV — недоразвитый I палец соединяется с кистью мягкотканной ножкой. V — полное отсутствие первого луча кисти. Как правило, первый тип врожденной гипоплазии I пальца не требует оперативного лечения. При втором типе используются компоненты операции, направленные на восстановление отсутствующих функций I пальца. Так, гипоплазия мышц тенара с отсутствием возможности активной оппозиции корректируется за счет оппонентопластики. Мы используем два варианта данной операции. При отсутствии нестабильности ПФС в качестве оппонирующей мышцы применяется abductor digitiminimi, дистальное сухожилие, которое после перемещения в подкожном тоннеле подшивается к боковой поверхности головки первой пястной кости (рис. 14). При наличии нестабильности ПФС мы применяем сухожилие поверхностного сгибателя IV пальца, которое выводится подкожно через отверстие, сделанное в карпальной связке, одна из боковых ножек проводится через канал, просверленный в пястной кости на локтевую сторону ПФС, формируя там локтевую коллатеральную связку, а вторая ножка используется для реконструкции лучевой коллатеральной связки (рис. 15). Недостаточное отведение пальца требует различных видов пластики первого 50 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии межпальцевого промежутка, начиная от простой Z-пластики при его умеренном сужении до формирования ротационного лоскута при тяжелых формах. а б Рис. 14. Гипоплазия первого пальца правой кисти, II тип по Blauth с наличием стабильного пястнофалангового сустава.
Мы используем два варианта данной операции. При отсутствии нестабильности ПФС в качестве оппонирующей мышцы применяется abductor digitiminimi, дистальное сухожилие, которое после перемещения в подкожном тоннеле подшивается к боковой поверхности головки первой пястной кости (рис. 14). При наличии нестабильности ПФС мы применяем сухожилие поверхностного сгибателя IV пальца, которое выводится подкожно через отверстие, сделанное в карпальной связке, одна из боковых ножек проводится через канал, просверленный в пястной кости на локтевую сторону ПФС, формируя там локтевую коллатеральную связку, а вторая ножка используется для реконструкции лучевой коллатеральной связки (рис. 15). Недостаточное отведение пальца требует различных видов пластики первого 50 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии межпальцевого промежутка, начиная от простой Z-пластики при его умеренном сужении до формирования ротационного лоскута при тяжелых формах. а б Рис. 14. Гипоплазия первого пальца правой кисти, II тип по Blauth с наличием стабильного пястнофалангового сустава. Определяется гипоплазия мышц тенара, отсутствие активной оппозиции (а). Выполнена оппонирующая пластика с транспозицией abductor digiti minimi, проведенного в подкожном тоннеле и подшитом к головке первой пястной кости по боковой поверхности (б) Рис. 15. Гипоплазия первого пальца правой кисти, II тип по Blauth с наличием нестабильного ПФС. Определяются гипоплазия мышц тенара, отсутствие активной оппозиции, нестабильность ПФС (а). Выполнена оппонирующая пластика с транспозицией поверхностного сгибателя IV пальца: этапы выделения сухожилия поверхностного сгибателя, выведения его в «окно» в карпальной связке (б, в), проведение к области ПФС, где одна из ножек проведена через канал в пястной кости на ульнарную поверхность сустава, вторая ножка оставлена на радиальной поверхности для формирования ульнарной и радиальной коллатеральной связки (г). Окончательный вид кисти в конце операции, I палец фиксирован в положении оппозиции осевой спицей (д) а б в г д Реконструктивная хирургия 51 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 Эта же стратегия восстановления функции I пальца применяется при типе IIIA.
Определяется гипоплазия мышц тенара, отсутствие активной оппозиции (а). Выполнена оппонирующая пластика с транспозицией abductor digiti minimi, проведенного в подкожном тоннеле и подшитом к головке первой пястной кости по боковой поверхности (б) Рис. 15. Гипоплазия первого пальца правой кисти, II тип по Blauth с наличием нестабильного ПФС. Определяются гипоплазия мышц тенара, отсутствие активной оппозиции, нестабильность ПФС (а). Выполнена оппонирующая пластика с транспозицией поверхностного сгибателя IV пальца: этапы выделения сухожилия поверхностного сгибателя, выведения его в «окно» в карпальной связке (б, в), проведение к области ПФС, где одна из ножек проведена через канал в пястной кости на ульнарную поверхность сустава, вторая ножка оставлена на радиальной поверхности для формирования ульнарной и радиальной коллатеральной связки (г). Окончательный вид кисти в конце операции, I палец фиксирован в положении оппозиции осевой спицей (д) а б в г д Реконструктивная хирургия 51 Вопросы реконструктивной и пластической хирургии № 4 (67) декабрь’ 2018 Эта же стратегия восстановления функции I пальца применяется при типе IIIA.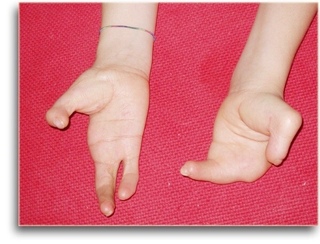 Тип IIIB является наиболее дискуссионным в плане выбора методики оперативного лечения. Существуют способы реконструкции проксимального отдела пястной кости с созданием трапецио-пястного неосустава [2], которые сочетаются обычно с большинством компонентов, используемых при I-IIIA типах. Для нас чаще всего IIIA тип гипоплазии I пальца является показанием к выполнению операции поллицизации. При IV и V типе для большинства хирургов операцией выбора является поллицизация второго луча (рис. 16). а б Рис. 16. Гипоплазия первого пальца левой кисти, V тип по Blauth (аплазия первого луча) (а), выполнена операция поллицизации второго пальца (б) Учитывая, что лучевая недостаточность является пороком, затрагивающем конечность на разных уровнях и требующим многоэтапного хирургического лечения, существует общепринятая схема последовательности этапов абилитации при данной аномалии. 1. Ортезирование и физическая терапия должны начинаться с первых дней жизни ребенка. Упражнения, направленные на растяжение недоразвитых тканей по лучевой стороне верхней конечности, проводятся ежедневно, завершаясь ношением ортеза, который этапно корректируется по достижении уменьшения лучевой девиации кисти.
Тип IIIB является наиболее дискуссионным в плане выбора методики оперативного лечения. Существуют способы реконструкции проксимального отдела пястной кости с созданием трапецио-пястного неосустава [2], которые сочетаются обычно с большинством компонентов, используемых при I-IIIA типах. Для нас чаще всего IIIA тип гипоплазии I пальца является показанием к выполнению операции поллицизации. При IV и V типе для большинства хирургов операцией выбора является поллицизация второго луча (рис. 16). а б Рис. 16. Гипоплазия первого пальца левой кисти, V тип по Blauth (аплазия первого луча) (а), выполнена операция поллицизации второго пальца (б) Учитывая, что лучевая недостаточность является пороком, затрагивающем конечность на разных уровнях и требующим многоэтапного хирургического лечения, существует общепринятая схема последовательности этапов абилитации при данной аномалии. 1. Ортезирование и физическая терапия должны начинаться с первых дней жизни ребенка. Упражнения, направленные на растяжение недоразвитых тканей по лучевой стороне верхней конечности, проводятся ежедневно, завершаясь ношением ортеза, который этапно корректируется по достижении уменьшения лучевой девиации кисти.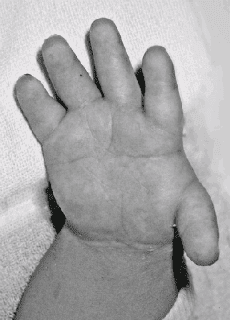 2. Установка дистракционного аппарата для выведения кисти в правильное положение и создание запаса мягких тканей позволяют не только значительно упростить проведение операции центрации или радиализации, но и существенно снизить риск рецидива деформации, а также избежать необходимости значимой резекции костей запястья и, самое главное, сохранить потенцию работы зоны роста локтевой кости за счет исключения ее компрессии после операции по стабилизации локтезапястного сочленения (рис. 17). Рис. 17. Монтаж дистракционного аппарата при лучевой косорукости для выведения кисти в правильное положение за счет растяжения мягких тканей по лучевой поверхности предплечья, как первый этап оперативного лечения перед стабилизацией локтезапястного сочленения 3. Стабилизация локтезапястного сочленения выполняется по двум методикам — операции центрации или операции радиализации, принципиальная разница между которыми продемонстрирована в статье D. Buck-Gramko (рис. 18). Еще одна методика — операция ульнаризации используется реже [25].
2. Установка дистракционного аппарата для выведения кисти в правильное положение и создание запаса мягких тканей позволяют не только значительно упростить проведение операции центрации или радиализации, но и существенно снизить риск рецидива деформации, а также избежать необходимости значимой резекции костей запястья и, самое главное, сохранить потенцию работы зоны роста локтевой кости за счет исключения ее компрессии после операции по стабилизации локтезапястного сочленения (рис. 17). Рис. 17. Монтаж дистракционного аппарата при лучевой косорукости для выведения кисти в правильное положение за счет растяжения мягких тканей по лучевой поверхности предплечья, как первый этап оперативного лечения перед стабилизацией локтезапястного сочленения 3. Стабилизация локтезапястного сочленения выполняется по двум методикам — операции центрации или операции радиализации, принципиальная разница между которыми продемонстрирована в статье D. Buck-Gramko (рис. 18). Еще одна методика — операция ульнаризации используется реже [25]. Мы в своей работе отдаем предпочтение операции радиализации, как менее агрессивной, позволяющей сохранить амплитуду движений в суставе и имеющей меньшую частоту рецидивов лучевой девиации кисти в отдаленном периоде. 4. Восстановление двустороннего схвата при гипоплазии I пальца достигается за счет сухожильно-мышечной и кожной пластики, или, при большей степени недоразвития первого луча, — за счет операции поллицизации. 5. Удлинение укороченного предплечья при лучевой косорукости выполняется последним этапом. Одним из непременных условий для начала дистракционного лечения является наличие стабильного локтезапястного сочленения, 52 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии поскольку возникающее в процессе удлинения натяжение мягких тканей может способствовать рецидиву лучевой девиации кисти. С целью профилактики данного осложнения мы всегда фиксируем кисть в дистракционном аппарате (рис. 19). а б Рис. 18. Сравнение биомеханики операций центрации (а) и радиализации (б).
Мы в своей работе отдаем предпочтение операции радиализации, как менее агрессивной, позволяющей сохранить амплитуду движений в суставе и имеющей меньшую частоту рецидивов лучевой девиации кисти в отдаленном периоде. 4. Восстановление двустороннего схвата при гипоплазии I пальца достигается за счет сухожильно-мышечной и кожной пластики, или, при большей степени недоразвития первого луча, — за счет операции поллицизации. 5. Удлинение укороченного предплечья при лучевой косорукости выполняется последним этапом. Одним из непременных условий для начала дистракционного лечения является наличие стабильного локтезапястного сочленения, 52 Заварухин В.И. № 4 (67) декабрь’ 2018 Вопросы реконструктивной и пластической хирургии поскольку возникающее в процессе удлинения натяжение мягких тканей может способствовать рецидиву лучевой девиации кисти. С целью профилактики данного осложнения мы всегда фиксируем кисть в дистракционном аппарате (рис. 19). а б Рис. 18. Сравнение биомеханики операций центрации (а) и радиализации (б). Транспозиция мышц с лучевой поверхности предплечья в сочетании с более длинным рычагом тяги по локтевой поверхности обеспечивает большую устойчивость к рецидиву лучевой девиации кисти [26] Рис. 19. Монтаж дистракционного аппарата для удлинения локтевой кости при врожденной продольной лучевой недостаточности Синдром врожденных (амниотических) перетяжек является не такой част
Транспозиция мышц с лучевой поверхности предплечья в сочетании с более длинным рычагом тяги по локтевой поверхности обеспечивает большую устойчивость к рецидиву лучевой девиации кисти [26] Рис. 19. Монтаж дистракционного аппарата для удлинения локтевой кости при врожденной продольной лучевой недостаточности Синдром врожденных (амниотических) перетяжек является не такой част
Swanson A.B. A classification for congenital limb malformations. The Journal of Hand Surgery. 1976; 1 (1): 8-22.
Шведовченко И.В. и др. Лечение детей с врожденными пороками развития верхних конечностей. В кн.: Травматология и ортопедия: руководство для врачей. СПб, 2005. Т. 2: Травмы и заболевания плечевого пояса и верхней конечности: 634-677.
Tonkin M.A. Classification of congenital anomalies of the hand and upper limb. J. Hand Surg. Eur. 2017; 42 (5): 448-456.
Lamb D.W., Wynne-Davies R., Solo L. An estimate of the population frequency of congenital malformations of the upper limb. J Hand Surg. 1982; 7: 557-62.
Watson S. The principles of management of congenital anomalies of the upper limb. 2000. March: 10-17.
Rypens F. et al. Obstetric US: Watchthe Fetal Hands. Radiographics. 2006: 811-831.
Chung M.S. Congenital Differences of the Upper Extremity: Classification and Treatment Principles. 2011: 172-177.
Oda T., Pushman A.G., Chung K.C. Treatment of common congenital hand conditions. Plast. Reconstr. Surg. 2010; 126 (3): 121e — 33e.
Kozin S.H. Syndactyly. J Am Soc Surg Hand. 2001; 1: 1-13.
Eaton C.J., Lister G.D. Syndactyly. Hand Clin. 1990; 6: 555-575.
Ireland D.C., Takayama N., Flatt A.E. Poland’ssyndrome: a reviewofforty-threecases. J Bone Joint Surg Am. 1976; 58:52: 8-33.
Hoover G.H., Flatt A.E., Weiss M.W. The hand and Apert’s syndrome. J Bone Joint Surg Am. 1970; 52: 878-895.
Guero S.J. Algorithm for treatment of Aperthand. Tech. Hand Up. Extrem. Surg. 2005; 9 (3): 126-133.
Kozin S.H. Upper-extremitycongenitalanomalies. J. Bone Joint Surg.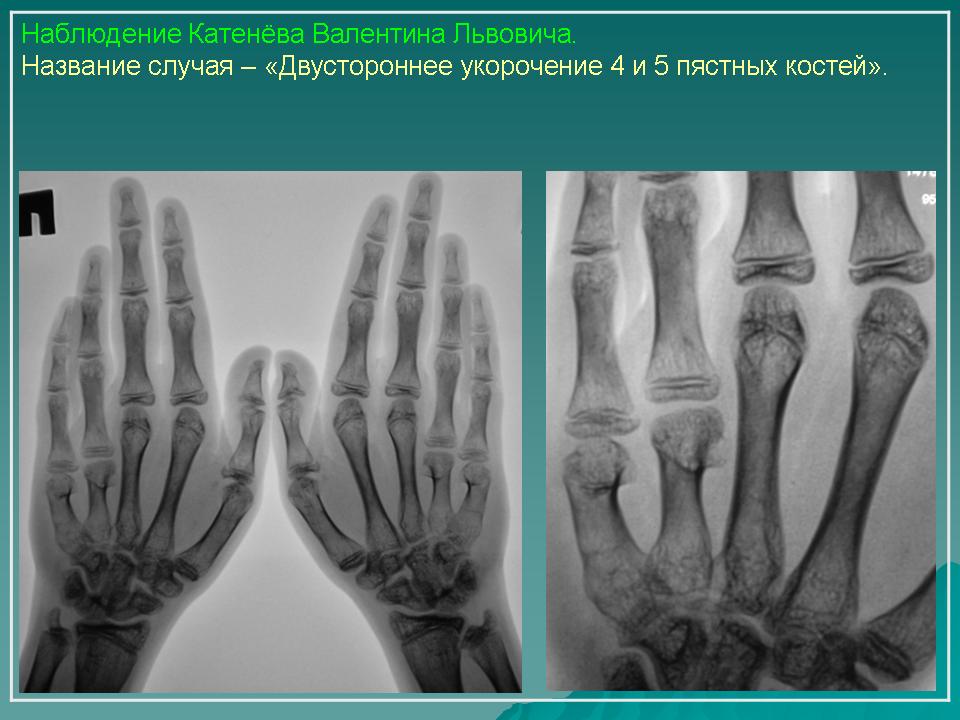 Am. 2003; 85 — A, (8): 1564-1576.
Am. 2003; 85 — A, (8): 1564-1576.
Oda T., Pushman A.G., Chung K.C. Treatment of common congenital hand conditions. Plast. Reconstr. Surg. 2010; 126 (3): 121e — 33e.
Tonkin M.A. Failure of differentiation part I: Syndactyly. Hand Clin. Elsevier Ltd, 2009; 25 (2): 171-193.
Buck-Gramcko D. Congenital malformations: syndactyly and related deformities. In: Nigst H., Buck- Gramcko D., Millesi H. et al, editors. Hand surgery. NewYork: Thieme Medical Publishers. 1988: 12-34.
Watson B.T., Hennrikus W.L. Postaxial type-B polydactyly. Prevalence and treatment. J Bone Joint Surg Am. 1997; 79:1: 65-68.
Ezaki M. Radial polydactyly. Hand Clin. 1990; 6: 577-588.
Wassel H.D. The results of surgery for polydactyly of the thumb: a review. Clin Orthop. 1969; 64: 175-193.
Comer G.C., Potter M., Ladd A.L. Polydactyly of the Hand. J. Am. Acad. Orthop. Surg. 2018; 26 (3): 75-82.
Choi M., Sharma S., Louie O. Chapter 89. Congenital Hand Abnormalities. Grabbe Smith’s Plast. Surgery, Sixth Ed. 2007: 856-863.
Surgery, Sixth Ed. 2007: 856-863.
Vilkki S.K. Distraction and microvascular epiphysis transfer for radial club hand. J Hand Surg. 1998; 23B: 445-52.
BlauthW. The hypoplastic thumb [in German]. Arch Orthop Unfallchir. 1967; 62: 225-246.
Paley D., Belthur M., Standard S. Ulnarization for the Treatment of Radial Club hand, Presented at the American Academy of Orthopedic Surgeons 75th Annual Meeting, San Francisco, CA. 2008.
Buck-Gramcko D. Radialization as a new treatment for radial club hand. J. Hand Surg. Am. American Society for Surgery of the Hand. 1985; 10 (6): 964-968.
Ogino T, Saitou Y. Congenital constriction band syndrome and transversed efficiency. J Hand Surg (Br.). 1987; 12: 343-348.
DiMeoL., MercerD.H. Single-stage correction of constriction ring syndrome. Ann Plast Surg. 1987; 19: 469-474.
Habenicht R., Hulsemann W., Lohmeyer J.A., Mann M. Ten-year experience with one-step correction of constriction rings by complete circular resection and linear circumferential skin closure. eANGrT$҄GHZr4GIl!H»:]\DDDDDDDDDDDF4-QCTNetZ PJ»:QtZKqgi»BAaV)4]եq
’pq?LzDt#h$
&y.#I%+n#R#A»:#AQʡC»Ml#!P
eANGrT$҄GHZr4GIl!H»:]\DDDDDDDDDDDF4-QCTNetZ PJ»:QtZKqgi»BAaV)4]եq
’pq?LzDt#h$
&y.#I%+n#R#A»:#AQʡC»Ml#!P
$#\»!wzA :Pj7i2)VGDb##>GqX$Eq*Coq A6I:IBYt#B»:AY쎈*$GI$쫄+GH#>B»EBDuP»>K:j0-WE!#:B»B
#/IO,s#e+B#oM$tYqV$!NAU)ը*6!)$}iA1q:Xp@ts$»>’_j+*tH$+zijDȝh»:#NVXL tIjyij»>@r:ZHAfB?زf:v»I&HNiA
j6GH»:He:#»:J (hHe 3TGI DyE$AaA|,pe3J>#+0*ҊG!G{wi jMҧDu{JWG dr
5h2(poJ(kI>5ۅ+taEф]Ft]EtaEф]FtGDt]DtGEtGDt]
E>GDx#;#x>G#|>G#xȏ̏>GDtGDx>G/>G#tYm»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»»8d|#8r
##|
9>G莈#|>G#|EtaEфqF2>G#x/菑:#:#莋菘EtGDuS9C9C9NS9C9C9NS9C9C9NSr(r)r)!qDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDDD\DDDDDDDDDDDDDDDDDDDEcOԳ2:#>GDt]V菭E%Ы,r)JUԝ&F:’G
rd6h»;8,( g.)qL
8)1xe0 hh00aC/=Xtxn;>)nI}vzn۳PPi5~>4DD5jlcjnGIN(r8OTӫ]- Du֟kҷ {\W}v’p=S{_JڭJYKlu!]7nm>N]UUc[[wOz4.`҄GV:%PDn:#~I#h!؆j! a8-CAAPPP`( )ʈeBeBeaT
9NS2DDDDDDGGK?N»a-‘Qnf8 pptdحiJNj߲kQy+VC#4]GXbb!ED»d|#tGFt]FJiXa».».»b;4Dx
&6 h5VƄaxB8cC#:#KMi}5Snn
LvӱG»`dt`фGFt]DCE’־I&M M0بD4″»a.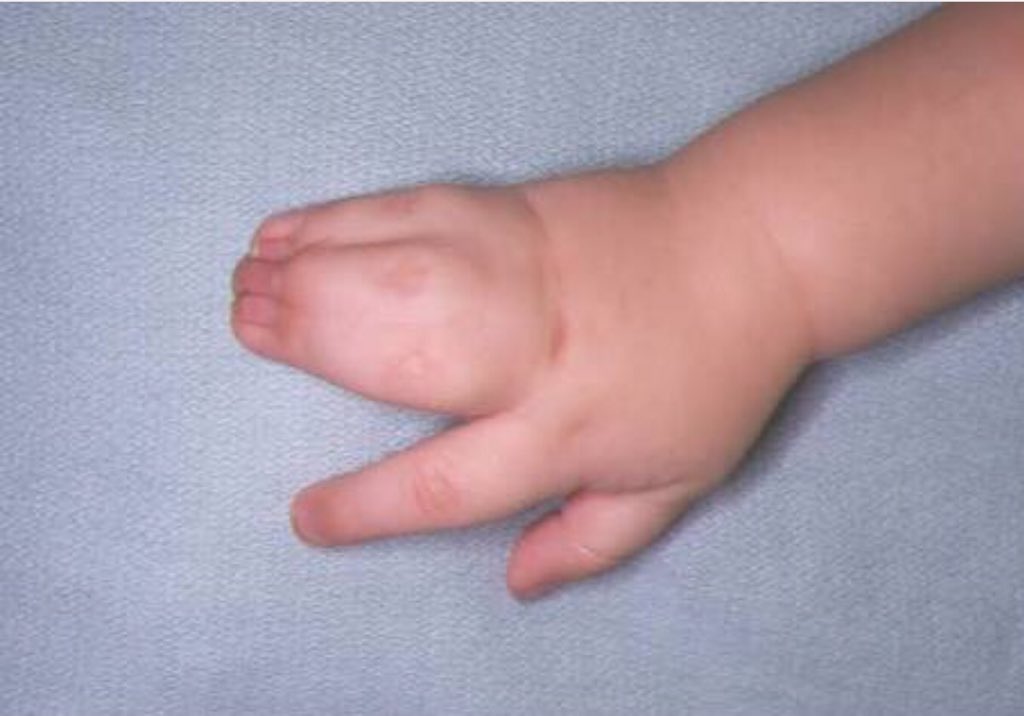 aLKUznߵ_;ЈB»»$L06!vA`J!GUuu}ª»»e)»>=vL8AA @iwKJ->B»»»»»»!m»iUd=DDQ:V hPh!lJqW~B@54Maah5T8ȰB»j9YeTU»»»1aV\nEXI eeKpD]UdX 7arBӠW-#醪*y
aLKUznߵ_;ЈB»»$L06!vA`J!GUuu}ª»»e)»>=vL8AA @iwKJ->B»»»»»»!m»iUd=DDQ:V hPh!lJqW~B@54Maah5T8ȰB»j9YeTU»»»1aV\nEXI eeKpD]UdX 7arBӠW-#醪*y
pSG2ȰGr7f=
6*I&)a`V\eȠgAN3?fIXS!M’n@7Z,4%
,`fq
{5킄jGAD
|S#»:0″:1*ua60zFpAAFhDEԈ9t;rRC( RGCA[_I60 $A»qKkDt$7tiUOWA»zVEVZ40p !gȑ/|dtGGRWE?/_tw_Aq/ii_EzAO .5mD( ЅD4″AO>]FFJGTl:8ڼ*8vǪtIh:64]0 Qg
Лечение аномалий кистей рук в Израиле
Причиной этих врожденных аномалий может быть наследственность или же другие факторы, например, они могут быть результатом побочного действия медикаментов, которые принимает женщина во время беременности. Часто употребление наркотиков, алкоголя и табака воздействует на процесс развития ребенка и может привести к упомянутым аномалиям, хотя не доказано их прямое воздействие на развитие верхних конечностей. Кроме того, плацента может обернуться вокруг кисти и пальцев, вызвав их потерю или дефект в их развитии. Возможны и другие причины, которые не известны медицине
Когда у родителей проходит первый шок, они начинают искать выход из создавшейся ситуации. И выход есть: в Центре хирургии кистей рук израильской клиники Топ Ихилов хирурги с мировым именем и высочайшей квалификацией, в совершенстве владеющие техникой пластической хирургии, микрохирургии, сосудистой хирургии и нейрохирургии, ежедневно творят настоящие чудеса в области коррекции врожденных аномалий кистей рук.
И выход есть: в Центре хирургии кистей рук израильской клиники Топ Ихилов хирурги с мировым именем и высочайшей квалификацией, в совершенстве владеющие техникой пластической хирургии, микрохирургии, сосудистой хирургии и нейрохирургии, ежедневно творят настоящие чудеса в области коррекции врожденных аномалий кистей рук.
Коррекция врождённых аномалий кисти: почему Топ Ихилов?
В клинике Топ Ихилов для операций по коррекции врожденных аномалий применяются самые современные медицинские технологии и методики, здесь работают только самые компетентные врачи, с успехом проводящие операции любой сложности.
Врожденные аномалии кисти можно исправить только хирургическим путем, и такие операции требуют от хирургов большого мастерства и опыта.
Коррекцию врождённых аномалий кисти рекомендуется проводить на первом году жизни ребенка, чтобы мышцы конечности не атрофировались вследствие нарушения функций кисти.
Специалисты ведущего в Израиле Центра хирургии кистей рук в клинике Топ Ихилов виртуозно проводят следующие восстановительные операции на кистях рук:
- Пересадка пальцев или суставов ступни на кисть в случае их нехватки
- Разделение сросшихся пальцев
- Вытягивание коротких пальцев
- Пересадка сухожилий
- Реконструкция деформированных пальцев или кисти
- Коррекция контрактур (состояние, при котором конечность не может быть полностью согнута или разогнута в одном или нескольких суставах)
- Трансплантация кожи
- Протезирование
- Наложение шин на пострадавшие части кисти
Лечение патологий кистей в Топ Ихилов: передовые технологии в руках профессионалов
Заболевания верхних конечностей в клинике Топ Ихилов проводится с использованием самых передовых достижений медицины. В силу принципа минимальной травматичности, соблюдаемого врачами в израильских клиниках, эндоскопия и микрохирургия в Израиле очень развиты, операционные оборудованы по последнему слову медицинской техники, что дает возможность врачам выполнять сложнейшие вмешательства с ювелирной точностью; эта же политика соблюдается и при лечении такого сложного органа, как кисть.
В силу принципа минимальной травматичности, соблюдаемого врачами в израильских клиниках, эндоскопия и микрохирургия в Израиле очень развиты, операционные оборудованы по последнему слову медицинской техники, что дает возможность врачам выполнять сложнейшие вмешательства с ювелирной точностью; эта же политика соблюдается и при лечении такого сложного органа, как кисть.
Особое внимание при этом уделяется такой сложной отрасли, как коррекция врожденных патологий кисти. В ней врачи Топ Ихилов добились выдающихся результатов: удаление лишних пальцев, реконструкция недостающих, разделение сросшихся, исправление различных деформаций кистей и суставов – все эти проблемы эффективно разрешаются в клинике в кратчайшие возможные сроки.
Консервативные методы лечения в клинике отлично развиты и применяются в легких случаях, однако абсолютному большинству пациентов все же требуется оперативное вмешательство. Во многом благодаря этому кистевая хирургия в Израиле развита на высочайшем уровне: врачи израильских клиник способны исправить даже те патологии, которые были признаны неоперабельными в других странах.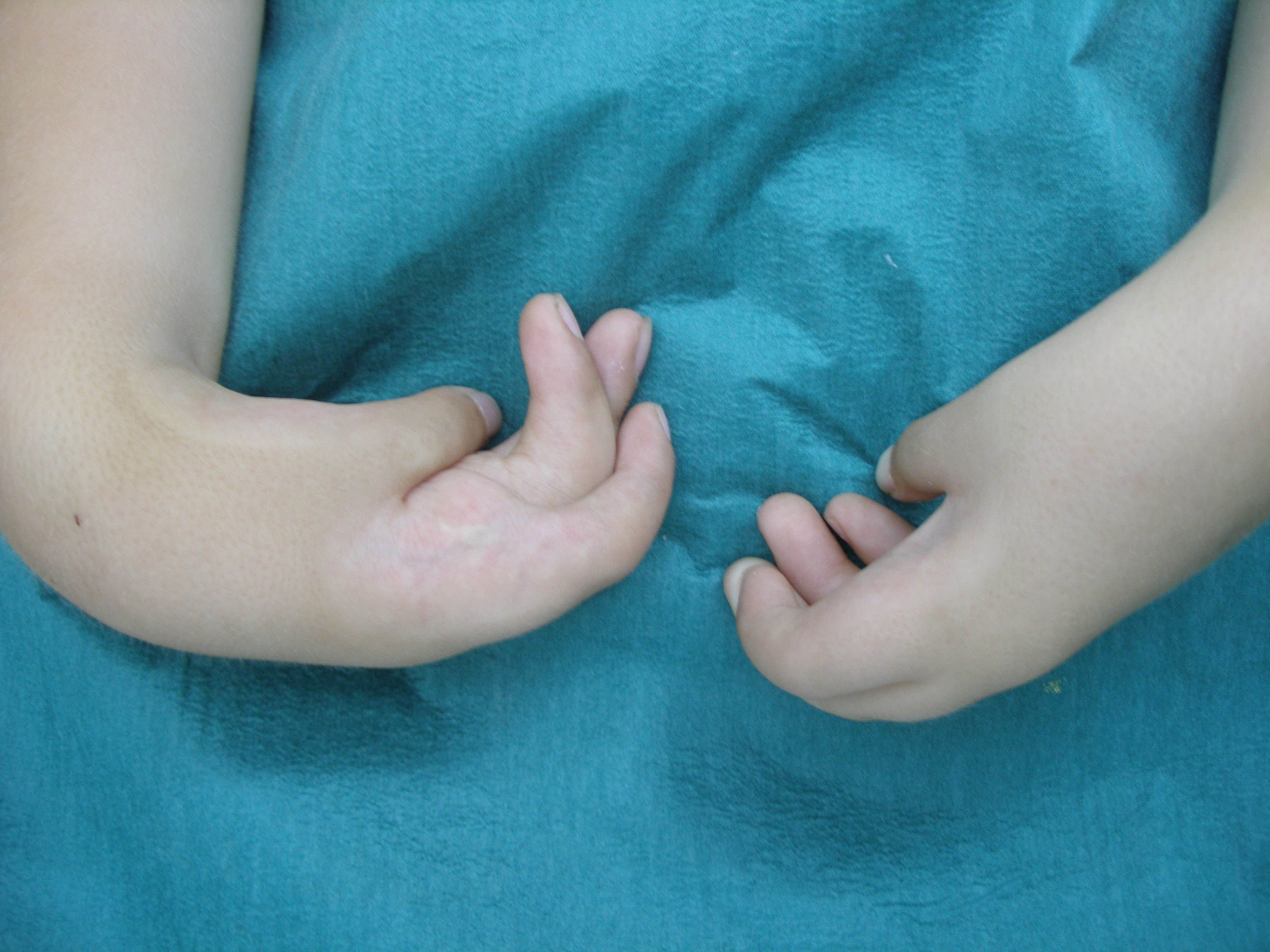 Что касается врожденных патологий, то они лечатся исключительно хирургическими методами в первые годы жизни – пока не началось атрофирование неправильно развивающейся конечности.
Что касается врожденных патологий, то они лечатся исключительно хирургическими методами в первые годы жизни – пока не началось атрофирование неправильно развивающейся конечности.
Среди методов, применяемых для коррекции врожденных аномалий развития кисти в клинике Топ Ихилов, следующие:
- трансплантация пальцев ног на кисти;
- лечение синдактилии – разделение сросшихся пальцев;
- удлинение патологически коротких фаланг пальцев;
- трансплантация поврежденных сухожилий;
- исправление деформаций как пальцев, так и кисти в целом;
- коррекция контрактур суставов.
Не смотря на всю сложность врожденных патологических состояний, медицинские технологии, применяемые в клинике Топ Ихилов, а также уровень квалификации хирургов позволяют успешно исправить их и восстановить функциональность кистей пациента.
Диагностика патологий кисти в Израиле
Ранняя и точная диагностика – основа, на которой стоит эффективность лечения в Израиле. Врачи прикладывают все усилия, чтобы собрать как можно более полную информацию о состоянии пациента перед началом лечения и применить её для максимального улучшения результатов.
Врачи прикладывают все усилия, чтобы собрать как можно более полную информацию о состоянии пациента перед началом лечения и применить её для максимального улучшения результатов.
Диагностика начинается без промедлений, в первый же день после прибытия пациента в Израиль, и занимает всего три дня.
День первый – прием у врача
Личный куратор из международного отдела встречает пациента в аэропорту и сопровождает до места проживания, а оттуда – в клинику, где уже ожидает лечащий ортопед. Врач в первый день проводит первичный осмотр и собирает анамнез, определяет, какие диагностические процедуры необходимы пациенту и выписывает направления.
День второй – диагностика
Клиника Топ Ихилов располагает передовыми моделями диагностического оборудования, которое позволяет собрать самые точные и полные данные о состоянии пациента. Среди применяемых при обследовании пациентов с патологиями кистей инструментальных и лабораторных методов следующие:
- ультрасонография кисти;
- допплерография сосудов верхних конечностей;
- рентгенография, выполняемая в нескольких различных проекциях для создания трехмерного изображения;
- компьютерная томография;
- магнитно-резонансная томография;
- артроскопия кисти;
- электромиография.
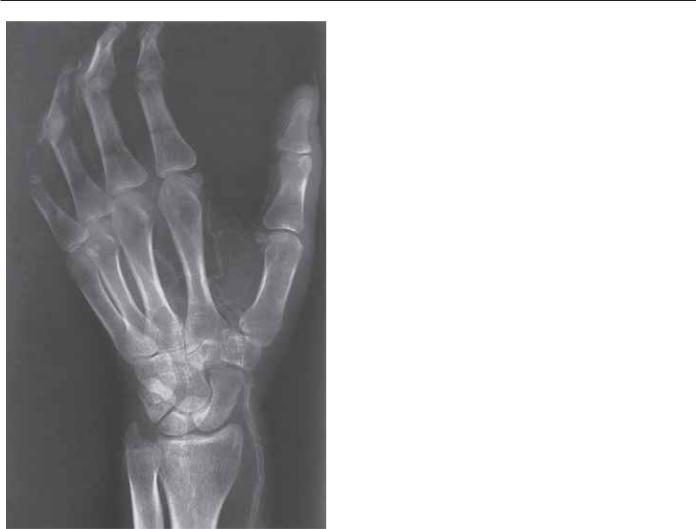
Данные методы дают врачам возможность изучить все подробности состояния больного, структуру конечностей, степень и объем поражения, и определить оптимальный метод коррекции имеющейся патологии.
День третий – план лечения
Результаты всех обследований передаются на рассмотрение медицинского консилиума, состоящего из нескольких специалистов смежных специальностей. Совместно они изучают их, подтверждают диагноз и составляют схему лечения, наилучшую для конкретного пациента, с учетом всех особенностей его состояния, возраста, сопутствующих заболеваний и других факторов, могущих повлиять на итоговый результат.
Стоимость лечения аномалий кистей рук в Израиле
Точная цена на лечение патологий кистей рук в Израиле устанавливается после того, как пациент прошел диагностику в клинике и ему было назначено лечение. Лечение каждого пациента составляется индивидуально и может включать различные процедуры, потому и стоимость лечения каждого из них будет различной.
Для получения данных об актуальных цена на лечебные процедуры в клинике Топ Ихилов оставьте заявку в форме ниже и дождитесь обратного звонка нашего сотрудника.
Запрос цены
Преимущества лечения патологий кисти в клинике Топ Ихилов
Основным преимуществом, которое предлагает клиника Топ Ихилов своим пациентом, являются, конечно же, высококвалифицированные врачи. Специалисты клиники – главная её гордость: штат укомплектован лучшими врачами Израиля, профессионалами, чей авторитет признан на международном уровне, и попасть к которым иностранцы могут только в порядке частного приема.
Отличное оборудование клиники позволяет врачам максимально реализовать свои знания и опыт в борьбе даже с самыми сложными патологиями кисти.
При этом цены на лечение в Израиле, в клинике Топ Ихилов, гораздо привлекательнее, чем в клиниках аналогичного уровня в других странах – так, итоговые счета за лечение в среднем оказываются на 30% ниже, чем в Европе, и на 50% ниже, чем американские, при том, что качество оказанных медицинских услуг на том же уровне, а иногда и выше.
- 5
- 4
- 3
- 2
- 1
(38 голосов, в среднем: 4.2 из 5)
Лечение врожденных повреждений конечностей у детей в Израиле
В клинике Топ Ихилов опытные врачи-ортопеды успешно проводят коррекцию различных врожденных патологий конечностей у детей. Хирурги ежегодно выполняют сотни операций, исправляя сложные дефекты и излечивая тяжелые повреждения рук и ног. В этом им способствует современное оснащение диагностического отделения, благодаря которому можно тщательно исследовать недуг, и инновационное оборудование операционных, которое позволяет выполнять малотравматичные операции по актуальным международным протоколам.
Сопровождение сотрудника международного отдела дает возможность сосредоточиться только на лечении ребенка – все бытовые и организационные вопросы он решит за вас.
Получить цены в клинике
Как в Топ Ихилов исправляют ошибки природы
Все врожденные патологии конечностей можно разделить на две группы:
- Аномалии развития рук – деформация суставов, гипоплазия большого пальца, косорукость, полидактилия (наличие большего количества пальцев), адактилия (отсутствие пальцев), брахидактилия (короткопалость), синдактилия (сращение пальцев), отсутствие кисти или ее расщелина и др.
- Аномалии развития ног – дисплазия тазобедренных суставов (порок развития вертлужной впадины, когда она утолщена, а головка бедренной кости недоразвита), врожденный вывих (бедра, ключицы, тазобедренного сустава), косолапость и т. д.
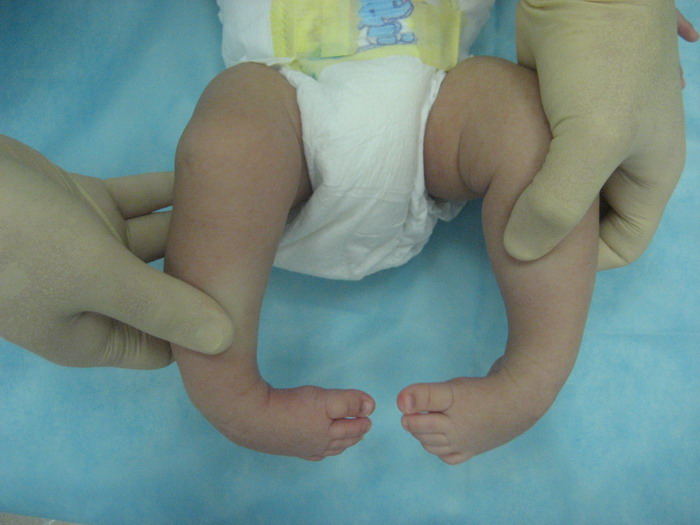
Врачи до сих пор точно не установили точные причины врожденных повреждений конечностей у детей в Израиле. Но среди возможных факторов выделяют наследственную предрасположенность, патологии внутриутробного развития, воздействие негативных факторов на плод, травмы, полученные в процессе родов, и др.
Любые симптомы врожденных повреждений конечностей у детей в Израиле врачи обнаруживают на ранних сроках беременности и устраняют профессионально и быстро, используя все доступные современной медицине методы.
- Лечебная физкультура эффективна в комплексной терапии при лечении деформации суставов, недоразвития большого пальца, косолапости и других патологий. Врачи подбирают индивидуальную программу упражнений для достижения максимальной эффективности.
- Массажи также применяются в комплексном лечении недоразвития большого пальца, врожденной косолапости, деформации суставов и др. Помогают повысить функциональную активность мышц. Являются частью реабилитационного периода после проведения операции.
- Фиксаторы правильного положения ног. При дисплазии тазобедренных суставов врачи назначают использование специальных приспособлений. Подушка Фрейка – мягкое ортопедическое приспособление, которое отличается минимальным неудобством при использовании и абсолютной безопасностью. Стремена Павлика – система ремней, которые фиксируют ноги в разведенном и согнутом положении. Сплинтер – металлическое устройство, с помощью которого ноги ребенка разводят и фиксируют в нужном положении, что способствует правильному формированию тазобедренного сустава.
- Метод Понсети применяется для лечения одного из самых сложных врожденных заболеваний – косолапости. Начинают лечение с 7-14-дневного возраста, пока сухожилия и связки еще эластичные. Состоит из трех этапов: фиксации методом гипсования с целью придать стопе правильное положение; если ожидаемый эффект не получен, прибегают к ахиллотомии – перерезанию пяточного сухожилия с целью возобновления подошвенного сгибания стопы, и, наконец, осуществляют завершающий этап – закрепление эффекта с помощь брейсов (планки-фиксатора с ботиночком).
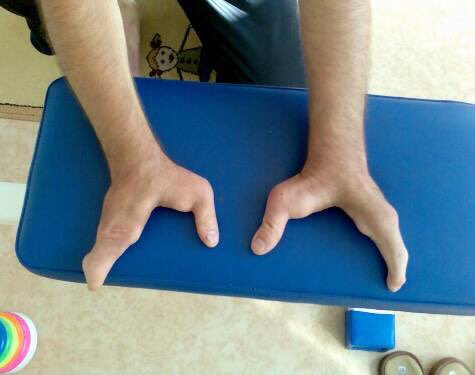
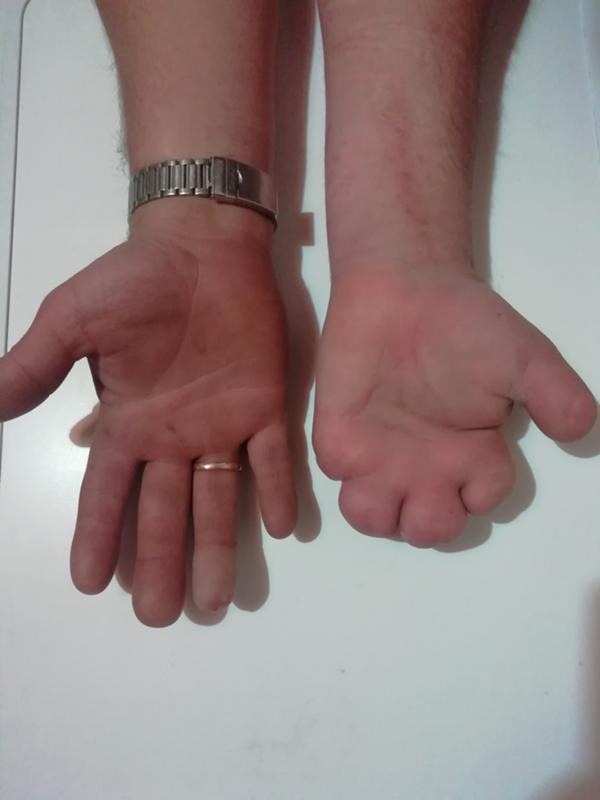 Состоит из трех этапов: фиксации методом гипсования с целью придать стопе правильное положение; если ожидаемый эффект не получен, прибегают к ахиллотомии – перерезанию пяточного сухожилия с целью возобновления подошвенного сгибания стопы, и, наконец, осуществляют завершающий этап – закрепление эффекта с помощь брейсов (планки-фиксатора с ботиночком).
Состоит из трех этапов: фиксации методом гипсования с целью придать стопе правильное положение; если ожидаемый эффект не получен, прибегают к ахиллотомии – перерезанию пяточного сухожилия с целью возобновления подошвенного сгибания стопы, и, наконец, осуществляют завершающий этап – закрепление эффекта с помощь брейсов (планки-фиксатора с ботиночком).Если патологию невозможно скорректировать с помощью методов консервативной терапии, прибегают к оперативному вмешательству. Хирургическое лечение врожденных аномалий конечностей у детей может проводиться традиционным или микрохирургическим способом. Малоинвазивные вмешательства дают возможность сократить длительность операции, уменьшить объем используемых лекарственных препаратов, снизить травматизацию тканей. Также снижается риск развития осложнений и сокращается период реабилитации.
- Пересадка соседних мышц или фиксация недоразвитых фаланг костным трансплантатом при сложной гипоплазии большого пальца для восстановления его функциональности.
- Сближение костей кисти при наличии расщелины. В ходе операции их фиксируют с помощью специальных синтетических нитей. Метод эффективен в том случае, если сохранена функциональность большого пальца.
- Удаление лишних пальцев. После операции осуществляется реконструкция тканей (кожи, связок и сухожилий).
- Разделение сросшихся пальцев выполняют малотравматичным способом – рассекают ткани в месте сращения, после чего прибегают к пластике.
- Пересадка пальцев или суставов со стопы на кисть руки обычно выполняется несколькими врачами для снижения рисков и сокращения времени проведения операции.
- Применение аппарата Илизарова. Что касается врожденных повреждений верхних конечностей, то этот внешний прибор позволяет эффективно лечить косорукость, растягивать сухожилия и мышцы предплечья, а также удлинять его недоразвитые кости. Применяется и для лечения врожденных аномалий ног (выполнения вытяжения) при деформациях костей и укорочениях конечностей. Установка происходит так: в специально сделанные в кости отверстия вставляют спицы, которые закрепляют на внешнем кольце. По необходимости выполняют рассечение кости. Разведение колец позволяет увеличивать длину конечности.
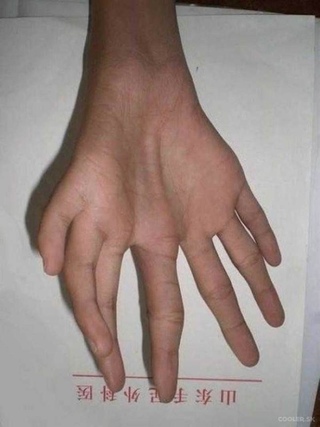
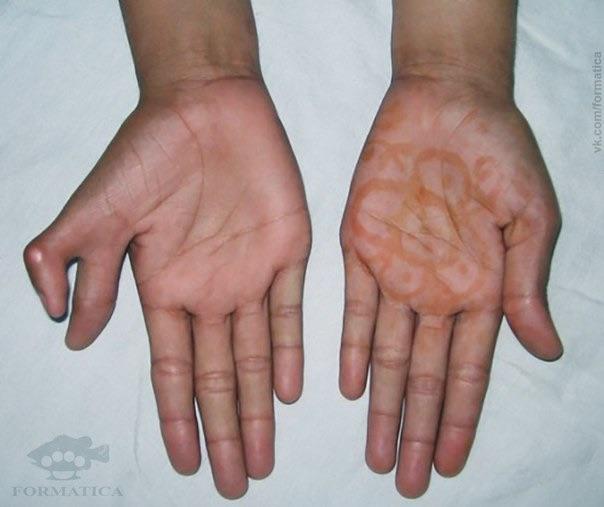 Установка происходит так: в специально сделанные в кости отверстия вставляют спицы, которые закрепляют на внешнем кольце. По необходимости выполняют рассечение кости. Разведение колец позволяет увеличивать длину конечности.
Установка происходит так: в специально сделанные в кости отверстия вставляют спицы, которые закрепляют на внешнем кольце. По необходимости выполняют рассечение кости. Разведение колец позволяет увеличивать длину конечности.Альтернативой аппарату Илизарова при деформации может быть имплантация в кость специальных металлических стержней или же выполнение фиксации с помощью металлических пластин, закрепляемых винтами и шурупами.
- Для удлинения конечностей применяют инновационный метод PreciсeNail, который состоит во введении в костномозговой канал интрамедуллярного телескопического стержня с последующей остеотомией. С помощью компьютера генерируется магнитное поле, воздействующее на магнитные полюса стержня, что заставляет его выкручиваться. Вращательные движения стержня способствуют растяжению мягких и костных тканей конечности.
- Тенотомия – перерезание сухожилия для устранения спастичности (сжатости) мышц или выравнивания конечности, например, при косолапости. Проводится с помощью специального скальпеля тенотома. Для ее выполнения не нужны разрезы кожи, операция осуществляется через прокол. Применяется при дисплазии тазобедренных суставов, варусной и вальгусной деформации стопы.
- Редукция выполняется при дисплазии тазобедренных суставов. Ее цель – вернуть головку бедренной кости в вертлужную впадину. Может быть закрытой и открытой. Во втором случае выполняют еще и тенотомию. После операции пациенту накладывают гипсовую повязку.
- Бедренная остеотомия проходит в два этапа. Сначала выполняют пересечение верхней части бедренной кости, затем осуществляют поворот ее центральной части в нужном направлении, чтобы разместить кость в правильном положении. Далее – этап фиксации с помощью металлических пластин.
- Эндопротезирование используют при дисплазии тазобедренных суставов. Его проводят в случае их выраженного недоразвития. Протезы изготавливают из современных и биосовместимых материалов. Они максимально точно имитируют естественную работу сустава и позволяют сохранить его подвижность и полноценное функционирование конечности.
- Протезирование выполняется при отсутствии кисти (аплазии), тюленеобразных верхних конечностях (фокомелии), с целью увеличения длины фаланг пальцев и др. Современные протезы подбираются в соответствии с анатомо-физиологическими особенностями пациента.
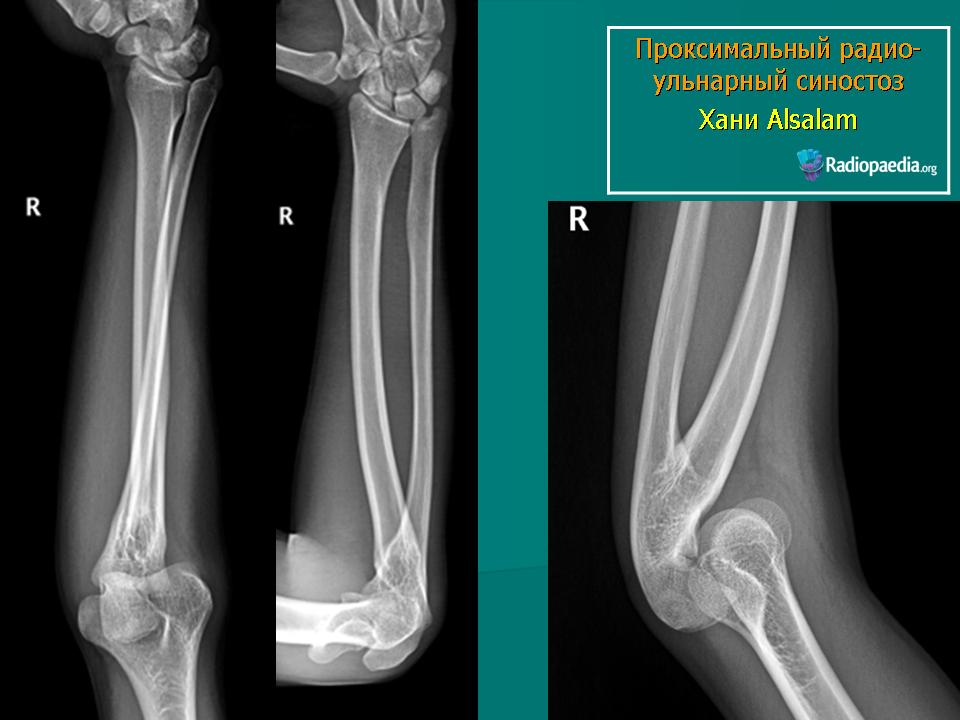 Проводится с помощью специального скальпеля тенотома. Для ее выполнения не нужны разрезы кожи, операция осуществляется через прокол. Применяется при дисплазии тазобедренных суставов, варусной и вальгусной деформации стопы.
Проводится с помощью специального скальпеля тенотома. Для ее выполнения не нужны разрезы кожи, операция осуществляется через прокол. Применяется при дисплазии тазобедренных суставов, варусной и вальгусной деформации стопы.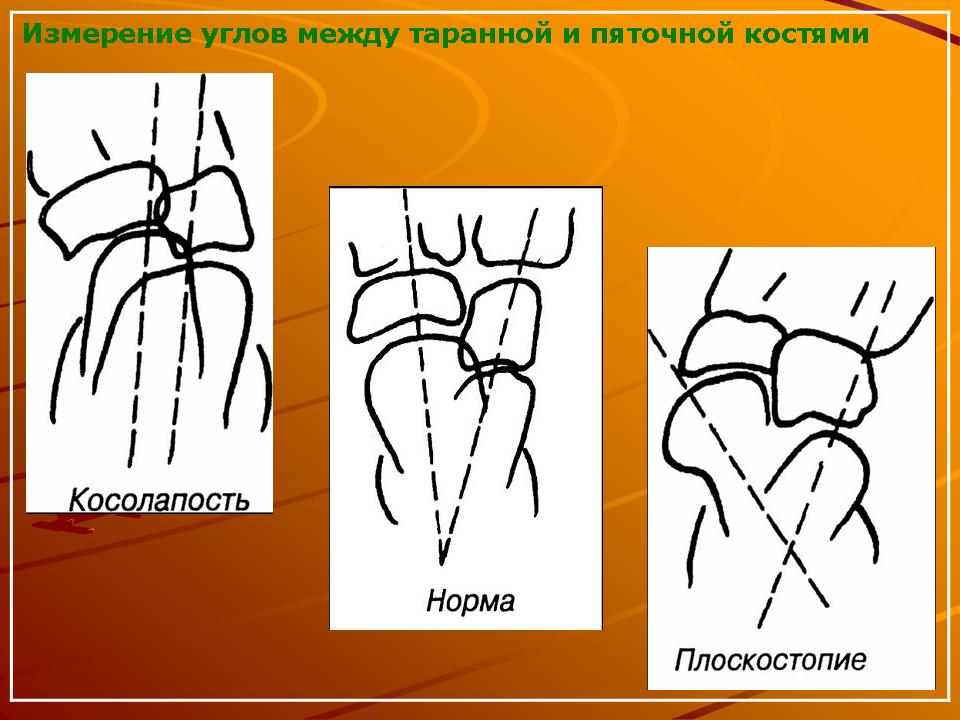 Они максимально точно имитируют естественную работу сустава и позволяют сохранить его подвижность и полноценное функционирование конечности.
Они максимально точно имитируют естественную работу сустава и позволяют сохранить его подвижность и полноценное функционирование конечности.Лучший аргумент в пользу того, насколько эффективно лечение врожденных повреждений конечностей у детей в Израиле, – отзывы родителей маленьких пациентов клиники Топ Ихилов.
Диагностика за 72 часа – точность, быстрота, качество
Лечение врожденных повреждений конечностей у детей в Израиле начинается с всестороннего обследования ребенка для выбора из всевозможных методов самый подходящий в конкретном клиническом случае.
Первый день. Консультация ортопеда
Цель консультации – собрать анамнез, изучить результаты предыдущих обследований, провести визуальный осмотр и составить программу диагностики.
Второй день. Диагностические процедуры
- Лабораторные анализы мочи и крови.
- МРТ, КТ, ПЭТ-КТ.
- УЗИ суставов.
- Допплерографическое исследование сосудов.
- Сцинтиграфия.
- Электромиография и др.
Третий день. План лечения
Лечение врожденных повреждений конечностей у детей в Израиле предусматривает комплексный подход для достижения максимальной эффективности. Поэтому стратегию терапии разрабатывает консилиум врачей разных специальностей – ортопедов, травматологов, хирургов, анестезиологов.
Как выполняют диагностические обследования, а также лечение врожденных повреждений конечностей у детей в Израиле, отзывы пациентов и другую полезную для вас информацию вы можете найти на сайте Топ Ихилов или узнать у наших консультантов после заполнения заявки.
Лечение врожденных повреждений конечностей у детей в Израиле: цены
Как известно, лечение в европейских и американских клиниках стоит очень дорого и многим оно не по карману. В Израиле цены более доступные при высоком качестве медицинского обслуживания – разница достигает 30-50%.
В Израиле цены более доступные при высоком качестве медицинского обслуживания – разница достигает 30-50%.
Узнать цены в нашей клинике можно у наших медицинских менеджеров – просто позвоните по телефону или закажите консультацию онлайн. Но помните, что окончательная цена будет сформирована только после выполнения обследования и разработки терапевтической тактики.
Получить цены в клинике
Топ Ихилов – у нас практикуют лучшие ортопеды страны
- Хирурги-ортопеды нашей клиники в совершенстве владеют современными методиками проведения операций.
- Для диагностики и лечения применяются передовые методики и медицинские технологии.
- Консилиум высококвалифицированных специалистов подбирает лечение индивидуально для каждого маленького пациента.
- Лояльная ценовая политика делает качественное медицинское обслуживание доступным.
- Представитель международного отдела позволит не думать о текущих вопросах организации лечения, поскольку спланирует пребывание в клинике и весь лечебно-диагностический процесс заранее.
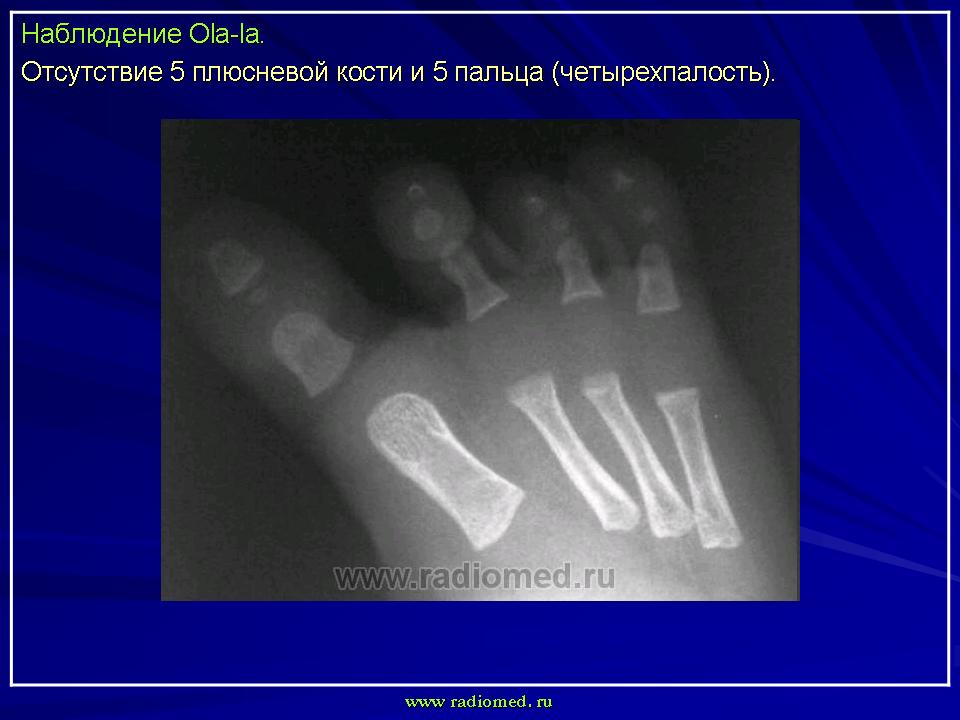
- 5
- 4
- 3
- 2
- 1
(3 голоса, в среднем: 5 из 5)
Врожденные аномалии верхней конечности, ребер и шеи — оперативное лечение > Архив — Клинические протоколы МЗ РК
Диагностические критерии
Жалобы и анамнез: деформация шеи и высокое стояние лопатки с рождения, с последующим прогрессирующим наклоном головы.
Физикальное обследование: ограничение движений в шейном отделе, вынужденный наклон головы, высокое стояние лопатки.
Лабораторные исследования: изменений в клинических, биохимических анализах при отсутствии сопутствующей патологии не наблюдается.
Инструментальные исследования: на рентгенограммах шейного отдела позвоночника аномалии развития позвонков, наклон головы, высокое стояние лопатки.
Показания для консультации специалистов: ЛОР-врача, стоматолога — для санации инфекции носоглотки, полости рта; при нарушениях ЭКГ — консультация кардиолога; при наличии ЖДА — педиатра; при вирусных гепатитах, зоонозных и в/утробных и др. инфекциях — инфекциониста; при эндокринной патологии — эндокринолога; невропатолога — при неврологической патологии.
Минимум обследования при направлении в стационар:
1. ОАМ, ОАК.
2. Трансаминазы.
3. Анализ на ВИЧ, гепатиты в случае перенесенных ранее оперативных вмешательств.
Основные диагностические мероприятия:
1. Общий анализ крови (6 параметров), гематокрит, тромбоциты, свертываемость.
2. Определение остаточного азота, мочевины, общего белка, билирубина, кальция, калия, натрия, глюкозы, АЛТ, АСТ.
3. Определение группы крови и резус-фактора.
4. Общий анализ мочи.
5. Рентгенография шейного отдела позвоночника в двух проекциях.
6. УЗИ органов брюшной полости по показаниям.
7. ЭКГ.
8. Соскоб кала.
9. ИФА на маркеры гепатитов В, С, Д, ВИЧ по показаниям.
Дополнительные диагностические мероприятия:
1. Анализ мочи по Аддису-Каковскому по показаниям.
2. Анализ мочи по Зимницкому по показаниям.
3. Посев мочи с отбором колоний по показаниям.
4. Рентгенография грудной клетки по показаниям.
5. ЭхоКГ по показаниям.
Лечение врожденных деформаций верхних конечностей
Врожденные деформации верхних конечностей — это сложные состояния, которые включают в себя поперечные дефекты, продольные дефекты предплечья и дефекты кистей и пальцев. Обычно они диагностируются во время 20-недельного анатомического УЗИ.
Семьи, направленные в наш Центр диагностики и лечения плода с пренатально диагностированной деформацией верхних конечностей, проходят полное обследование, которое включает визуализацию для подтверждения диагноза и консультации с нашими специалистами по материнско-фетальной медицине и экспертами из нашей Программы заболеваний рук и рук.
Одна из крупнейших мультидисциплинарных программ в своем роде, наша Программа по лечению заболеваний кисти и предплечья объединяет хирургов и клинический персонал из двух основных подразделений — ортопедии и пластической хирургии — которые создают индивидуальный план лечения для каждого ребенка. Объединив усилия на раннем этапе, эта группа может работать над улучшением функциональности и внешнего вида пораженной руки или руки ребенка.
Наша команда рассматривает и оценивает результаты ультразвукового исследования для подтверждения диагноза и консультирует семьи о том, чего ожидать при рождении ребенка, о влиянии этого состояния на функции и качество жизни, а также о роли протезирования, ортопедии, трудотерапии и хирургии. .Чувствительность и специфичность состояний верхних конечностей, визуализированных на УЗИ, не всегда коррелируют с точным диагнозом после рождения, и наша команда обязательно укажет на любую двусмысленность при консультировании семей.
Некоторые из редких и сложных состояний, которые консультирует наша команда во время беременности:
- Синдактилия, в которой две или более цифр сливаются вместе
- Полидактилия, в которой есть лишняя цифра
- Лучевая продольная недостаточность (RLD), при которой лучевая сторона предплечья не сформирована должным образом. Это может привести к недоразвитию большого пальца, запястья и предплечья.
 Это может привести к недоразвитию большого пальца, запястья и предплечья.
Это может привести к недоразвитию большого пальца, запястья и предплечья.График наблюдения варьируется от пациента к пациенту. Для детей, рожденных в специальном отделении для семьи Гарбозе нашей больницы, первом отделении детской больницы, разработанном специально для здоровой матери, у плода которой имеется врожденный дефект, мы встречаемся с семьями в отделении интенсивной терапии новорожденных и младенцев Харриет и Рональда Лассинов. первые пару дней после рождения осмотреть малыша и ответить на любые вопросы.
Последующее наблюдение включает оценку нашей многопрофильной командой хирургов, радиологов, медсестер, физиотерапевтов и эрготерапевтов для подтверждения заболевания кисти или руки у ребенка, оценки бимануальной активности и разработки индивидуального комплексного плана лечения, включая хирургическое вмешательство, если это необходимо. .
Ежегодно наши сертифицированные хирурги-ортопеды и пластические хирурги проводят более 1000 процедур младенцам, детям и подросткам с заболеваниями кисти или руки, в том числе:
- Реконструктивная хирургия, например поллицинизация
- Syndactyly release
- Артроскопия
- Хирургия сухожилий
- Пересадка нервов
В редких случаях, которые проявляются как в конечностях, так и в других органах, таких как VACTERL и Holt-Oram, наша команда работает вместе с другими специалистами в больнице для оказания комплексной помощи.
Врожденный порок верхней конечности: история болезни
Подавляющее большинство врожденных деформаций возникает между четвертой и восьмой неделями беременности. Врожденные дефекты обычно классифицируются как структурные или связанные с развитием, вызывающие нарушения от легких до тяжелых. Примерно 3% новорожденных в США имеют серьезные структурные или генетические врожденные дефекты. 1 По данным Центров по контролю и профилактике заболеваний (CDC), ежегодно в США рождается около 1500 детей с редукцией верхних конечностей (примерно 4 из 10 000 детей) и около 750 рождаются с редукцией нижних конечностей (примерно 2 из каждых 10000 младенцев). 2 Отсутствующая или неполная рука при рождении называется врожденным дефектом верхней конечности или врожденной ампутацией конечности или уменьшением конечности. Эти дефекты в основном объясняются первичным торможением внутриутробного развития или нарушениями, вторичными по отношению к внутриутробному разрушению нормальных эмбриональных тканей.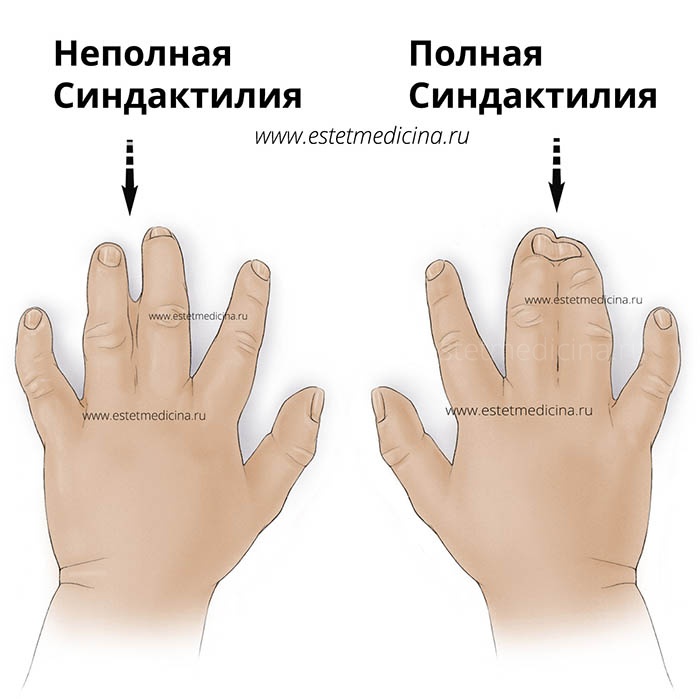
Существует несколько систем классификации врожденных деформаций верхних конечностей, таких как анатомические или топографические, эмбриологические, тератологические, генетические и различные их комбинации.Заболевания дефицита конечностей представляют собой широкую группу врожденных аномалий, характеризующихся значительной гипоплазией или аплазией 1 или более костей конечностей, которые могут возникать изолированно или ассоциироваться с другими аномалиями. 3
Дефекты конечностей могут быть продольными или поперечными. Продольные дефекты вдоль продольной оси конечности (полное или частичное отсутствие лучевой кости, локтевой, малоберцовой или большеберцовой кости) и в основном имеют генетическое или тератогенное происхождение. При поперечном дефиците все элементы ниже определенного уровня в руке пациента отсутствуют (потеря предплечья, кисти), а структуры конечностей до точки дефекта остаются нетронутыми.Это в основном вызвано ранней последовательностью разрыва амниона (амниотическая полоса). 3 Рука выглядит как культя после ампутации, которую иногда называют «врожденной ампутацией». Используемые термины могут быть от афалангии (отсутствие пальцев и фаланг) до амелии (отсутствие конечности). 4,5
3 Рука выглядит как культя после ампутации, которую иногда называют «врожденной ампутацией». Используемые термины могут быть от афалангии (отсутствие пальцев и фаланг) до амелии (отсутствие конечности). 4,5
Пример из практики
Ребенок Р. родился доношенным от матери G2 P1 в результате повторного кесарева сечения на 39 неделе беременности. Родства с родителями нет, и мать соблюдала все правила дородового ухода.У матери были отрицательные результаты на гепатиты A, B и C, вирус иммунодефицита человека (ВИЧ) и стрептококк группы B (GBS). Роды были несложными, но после родов головы и верхней части тела было отмечено, что у Baby R отсутствовало левое предплечье, несмотря на сообщенное «нормальное» пренатальное ультразвуковое исследование на 20 неделе беременности. Неонатолог поставил диагноз «разрыв околоплодных вод» и объяснил его обезумевшим родителям. Оценка APGAR составила 8 баллов на 1 минуте и 9 баллов на 5 минутах. У Baby R не было никаких других физических отклонений, сердечных шумов и дисморфических особенностей. Он прошел без осложнений и на третий день жизни был выписан домой вместе с матерью.
Он прошел без осложнений и на третий день жизни был выписан домой вместе с матерью.
Baby R находился под наблюдением в педиатрическом отделении первичной медико-санитарной помощи для планового ухода за новорожденными и был осмотрен педиатрической медсестрой (PNP) во время двухмесячного посещения по укреплению здоровья. Физическая оценка показала нормальный рост, он хорошо кормил. Физикальное обследование без особенностей, за исключением левого предплечья. На кончике деформации выше локтя левой верхней конечности отмечен небольшой валик из бугорков.У младенца были мимолетные движения конечности от плеча с плохим контролем. Правая верхняя конечность выглядела нормальной с полным диапазоном движений плеча, локтя, запястья и пальцев, с нормальным набором пальцев.
Обсуждение
Аномалии конечностей возникают на разных стадиях эмбрионального развития. Зачатки конечностей сначала появляются как небольшие возвышения вентролатерального тела на четвертой неделе беременности. На кончике зачатка каждой конечности эктодерма (самый внешний слой клеток и тканей в эмбриональном развитии) утолщается, образуя апикальный эктодермальный гребень.Взаимодействие между этим гребнем и мезенхимальными клетками, которые становятся соединительной тканью, необходимо для развития конечностей. 6 Апикальный эктодермальный гребень влияет на соединительную ткань, которая способствует росту и развитию конечностей и важна для процесса удлинения. Подобные дефекты возникают на ранних сроках беременности, поскольку остеогенез длинных костей начинается на седьмой неделе.
На кончике зачатка каждой конечности эктодерма (самый внешний слой клеток и тканей в эмбриональном развитии) утолщается, образуя апикальный эктодермальный гребень.Взаимодействие между этим гребнем и мезенхимальными клетками, которые становятся соединительной тканью, необходимо для развития конечностей. 6 Апикальный эктодермальный гребень влияет на соединительную ткань, которая способствует росту и развитию конечностей и важна для процесса удлинения. Подобные дефекты возникают на ранних сроках беременности, поскольку остеогенез длинных костей начинается на седьмой неделе.
Как и в случае с другими врожденными пороками развития, точная причина порока верхней конечности обычно не известна.Некоторые исследования показали, что воздействие на мать во время беременности различных вирусов, химических веществ, лекарств и табачного дыма может быть одним из факторов. 7 Синдром околоплодных вод возникает в результате разрыва амниотического мешка, который может вызывать различные аномалии. Это происходит из-за сужения окружающих околоплодных вод и часто может поражать конечности, лицо, суставы, живот или грудную стенку и вызывать дефекты нервной трубки. 8,9 Этот младенец был направлен в региональную больницу Шрайнерс, где в возрасте 4 месяцев ему был поставлен диагноз: поперечная недостаточность левой верхней конечности на уровне дистального отдела плечевой кости, а не амниотическая повязка.Ортопед сообщил, что дефект Baby R был результатом сбоя в функционировании эктодермального гребня, скорее всего, из-за сосудистой патологии неизвестного происхождения.
Это происходит из-за сужения окружающих околоплодных вод и часто может поражать конечности, лицо, суставы, живот или грудную стенку и вызывать дефекты нервной трубки. 8,9 Этот младенец был направлен в региональную больницу Шрайнерс, где в возрасте 4 месяцев ему был поставлен диагноз: поперечная недостаточность левой верхней конечности на уровне дистального отдела плечевой кости, а не амниотическая повязка.Ортопед сообщил, что дефект Baby R был результатом сбоя в функционировании эктодермального гребня, скорее всего, из-за сосудистой патологии неизвестного происхождения.
Когда ребенок рождается с врожденным дефектом конечности, это влияет не только на ребенка, но и на родителей и семьи. Родители переживают горе и утрату с рождением ребенка с любой деформацией, и им необходимы руководство и поддержка со стороны медицинских работников, семей и друзей. В 16-летнем проспективном исследовании пренатальной диагностики пороков развития плода авторы сообщают, что доля дефектов конечностей, диагностированных с помощью ультразвука, была низкой; при этом у 3 из 4 детей с дефектом конечности не было диагностировано внутриутробно.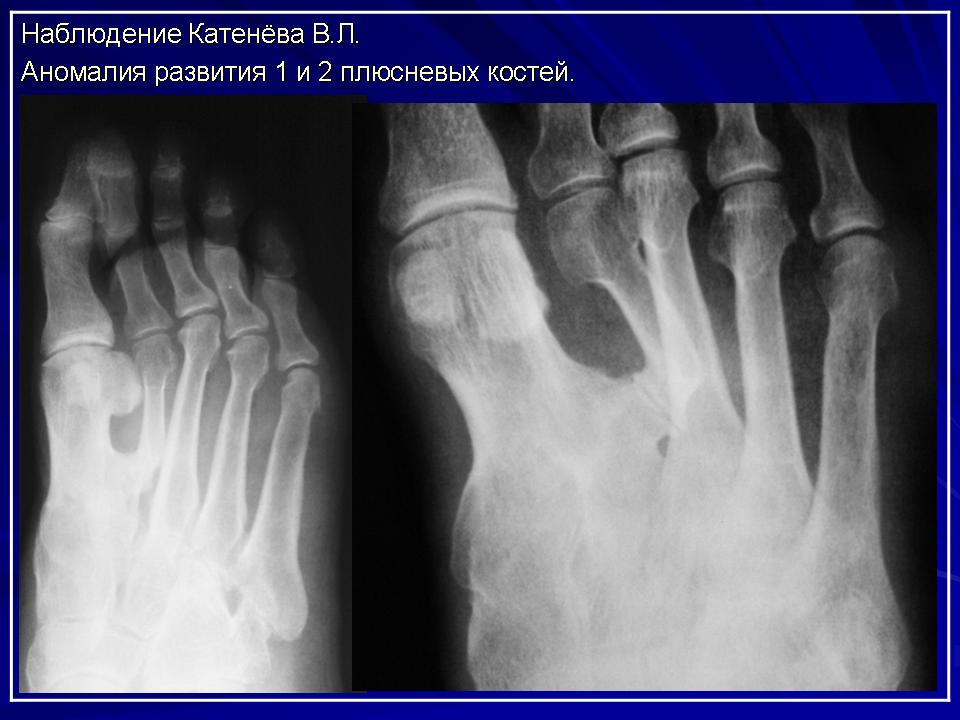 10
10
Родители детей, рожденных с (неожиданным) дефектом конечностей, часто бывают шокированы и испытывают чувство вины. Мать пытается вспомнить, сделала ли она что-нибудь не так, что способствовало возникновению дефекта. Родители могут чрезмерно защищать ребенка и относиться к нему не так, как к другим детям, из-за чувства вины, что может нанести ущерб росту и развитию ребенка. Если родители не приспосабливаются к диагнозу, это повлияет на то, как ребенок будет себя чувствовать и реагировать на свое тело.
Основная цель лечения этого дефекта — восстановление правильного функционирования конечности и внешнего вида. Лечение может варьироваться для каждого ребенка в зависимости от возраста, степени дефекта, а также предпочтений ребенка и родителей. Общие варианты лечения включают протезирование (протезы конечностей), ортезирование (шины или скобы), хирургическое вмешательство и реабилитацию (физиотерапия и трудотерапия). 9 Первый протез можно установить, как только ребенок сможет самостоятельно сидеть, потому что ребенку требуется равновесие верхней части тела для стабилизации в сидячем положении. Основные устройства включают пассивную руку, переходящую в активное устройство, которое можно использовать, когда ребенок старше и пытается удерживать объект двумя руками. 9 Разнообразие устройств может определяться функциями, стоимостью и простотой обучения и использования для родителей, детей и терапевтов, поскольку эта область постоянно развивается.
Основные устройства включают пассивную руку, переходящую в активное устройство, которое можно использовать, когда ребенок старше и пытается удерживать объект двумя руками. 9 Разнообразие устройств может определяться функциями, стоимостью и простотой обучения и использования для родителей, детей и терапевтов, поскольку эта область постоянно развивается.
Существуют разногласия по поводу возраста, подходящего для установки протеза. Несколько опубликованных отчетов показывают, что большинство детей получают первый протез в течение первого года жизни и получают некоторые инструкции по активной работе с оконечным устройством в течение второго или третьего года жизни. 9 В исследовании, проведенном в 2003 году, было проведено исследование клиник для детей с ампутированными конечностями в Северной Америке, чтобы изучить стандарты раннего приспособления детей с односторонним отсутствием конечностей ниже локтя. Большинство терапевтов / медицинских работников предпочли приспособиться к возрасту 6 месяцев, а затем добавить систему активного контроля к 18 месяцам, что было более ранним возрастом, чем сообщалось в предшествующей литературе. Считается, что ранняя установка протеза улучшает внешний вид тела ребенка, что может иметь важное значение для нормального нервно-мышечного развития. Однако более недавнее исследование показало, что возраст первой примерки не был связан с удовлетворенностью протезом, функциональным использованием протеза или моторикой. 11
Считается, что ранняя установка протеза улучшает внешний вид тела ребенка, что может иметь важное значение для нормального нервно-мышечного развития. Однако более недавнее исследование показало, что возраст первой примерки не был связан с удовлетворенностью протезом, функциональным использованием протеза или моторикой. 11
Многие дети могут адаптироваться и нормально функционировать без использования протезов, но некоторым детям может потребоваться физиотерапия, хирургическое вмешательство или другие методы лечения, чтобы лучше функционировать и быть более независимыми. 12 Дети с дефектами верхних конечностей могут испытывать как физические, так и эмоциональные проблемы. Качественное исследование показало, что дети и подростки с односторонним врожденным дефицитом ниже локтя (UCBED) испытывали как отрицательные, так и положительные чувства по поводу своего дефицита, независимо от того, носили они протез или нет. 13 Эти чувства больше всего зависели от того, как социальная среда приняла ребенка и отреагировала на недостаток. Наиболее частой общественной реакцией было пристальное внимание, а негативное отношение сказывалось на самооценке детей. Стратегии преодоления включают ношение протеза и общение со сверстниками с аналогичными недостатками. Было обнаружено, что поддержка со стороны реабилитационной бригады и родителей оказала наибольшее положительное влияние на общее психологическое функционирование.
Наиболее частой общественной реакцией было пристальное внимание, а негативное отношение сказывалось на самооценке детей. Стратегии преодоления включают ношение протеза и общение со сверстниками с аналогичными недостатками. Было обнаружено, что поддержка со стороны реабилитационной бригады и родителей оказала наибольшее положительное влияние на общее психологическое функционирование.
Обновление случая
Раннее вмешательство и многопрофильное сотрудничество необходимы, чтобы помочь этим детям и родителям.В случае с Baby R программа PNP поощряла мультидисциплинарный подход к уходу за младенцем, инициируя направление на раннее вмешательство (EI) на физиотерапию (PT) и профессиональную терапию (OT), которое начиналось в возрасте 3 месяцев. Семья Baby R прошла все медицинские осмотры, и ребенку в возрасте 6 месяцев установили протез. По настоянию матери ему был поставлен нефункциональный (косметический) протез в присутствии местного физиотерапевта в возрасте 6 месяцев.
Baby R хорошо продвинулся в течение своего первого года жизни и продолжил еженедельные ОТ и СТ.Он встречает вехи своего развития с некоторыми задержками, которые требуют бимодальной активности (например, передачи предметов из одной руки в другую), но это хорошо компенсирует. Матери была оказана значительная поддержка при послеродовой депрессии и тревоге. В настоящее время ее лечащий врач проводит амбулаторные консультации и фармакотерапию.
Советы лечащему врачу
Основные цели поставщиков первичной медико-санитарной помощи — помочь детям нормально развиваться и быть независимыми.В то время как медицинская поддержка и консультации важны для младенцев и детей с врожденным дефектом, психологическая поддержка имеет первостепенное значение для родителей и семьи. Видение «идеального ребенка» может вызвать скорбные реакции, особенно если дефект не заподозрен. В историческом исследовании детей с аномалиями рук, проведенном Грингасом, желание детей исследовать было больше ограничено тревогой матери, чем их функциональными проблемами. 14 Авторы признают, что дети могут улавливать обесценивающие отношения, в которых ребенок чувствует, что он / она не может преодолеть в одиночку.Также отмечается, что родители чрезмерно опекают и проецируют на ребенка собственные страдания, что может пагубно сказаться на его росте и развитии. Следует поощрять родителей относиться к своим детям как к нормальным и позволять им принимать решения и заботиться о себе, когда это целесообразно с точки зрения развития.
14 Авторы признают, что дети могут улавливать обесценивающие отношения, в которых ребенок чувствует, что он / она не может преодолеть в одиночку.Также отмечается, что родители чрезмерно опекают и проецируют на ребенка собственные страдания, что может пагубно сказаться на его росте и развитии. Следует поощрять родителей относиться к своим детям как к нормальным и позволять им принимать решения и заботиться о себе, когда это целесообразно с точки зрения развития.
Родители играют решающую роль в раннем развитии самосознания ребенка до того, как будут получены социальные реакции. 15 Родители, приспособившиеся к дефекту своего ребенка, смогут без проблем смотреть на руку, что побудит других сделать то же самое.По мере того как ребенок продолжает расти и развивать самосознание, родителям необходимо поддерживать осознание физических различий, но отрицать самосознание по поводу дефекта. Персонал отделения первичной медико-санитарной помощи также может моделировать поддержку и доверие семьи.
Резюме
Хотя врожденный порок конечностей не является обычным явлением, поставщики первичной медико-санитарной помощи должны постоянно осведомляться о местных и региональных ресурсах, чтобы оказывать помощь семьям и детям с дефектом конечности.Службы поддержки должны быть на месте как для ребенка, так и для семьи на протяжении всего роста и развития ребенка, чтобы способствовать достижению основных вех в пределах допустимого диапазона и обеспечивать модификации для удовлетворения потребностей развития ребенка. Междисциплинарная поддержка ребенка в школьном и подростковом возрасте будет иметь первостепенное значение для развития самосознания и положительной самооценки.
Ссылки:
1. Центры по контролю и профилактике заболеваний. Обновленная информация об общей распространенности серьезных врожденных дефектов — Атланта, Джорджия, 1978-2005 гг. MMWR Morb Mortal Wkly Rep . 2008; 57 (1): 1-5. Доступно по адресу: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5701a2. htm. На 21 февраля 2019 г.
htm. На 21 февраля 2019 г.
2. Центры по контролю и профилактике заболеваний. Врожденные дефекты: дефекты редукции верхних и нижних конечностей. Доступно по адресу: https://www.cdc.gov/ncbddd/birthdefects/ul-limbreductiondefects.html. Обновлено 20 апреля 2018 г. Проверено 21 февраля 2019 г.
3. Wilcox WR, Coulter CP, Schmitz ML. Врожденные пороки развития конечностей. Клин Перинатол .2015; 42 (2): 281-300.
4. Mattar Junior R. Deformidades congênitas do membersro superior. Бюстгальтеры Acta Ortop . 1995; 3 (2): 77-92.
5. Gold NB, Westgate MN, Holmes LB. Анатомо-этиологическая классификация врожденных пороков конечностей. Am J Med Genet A , 2011; 155A (6), 1225-1235.
6. Мур К.Л., Персо ТВН, Торчиа МГ. Развивающийся человек: клинически ориентированная эмбриология . 10-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2016.
7.Parker SE, Mai CT, Canfield MA и др .; Национальная сеть профилактики врожденных дефектов.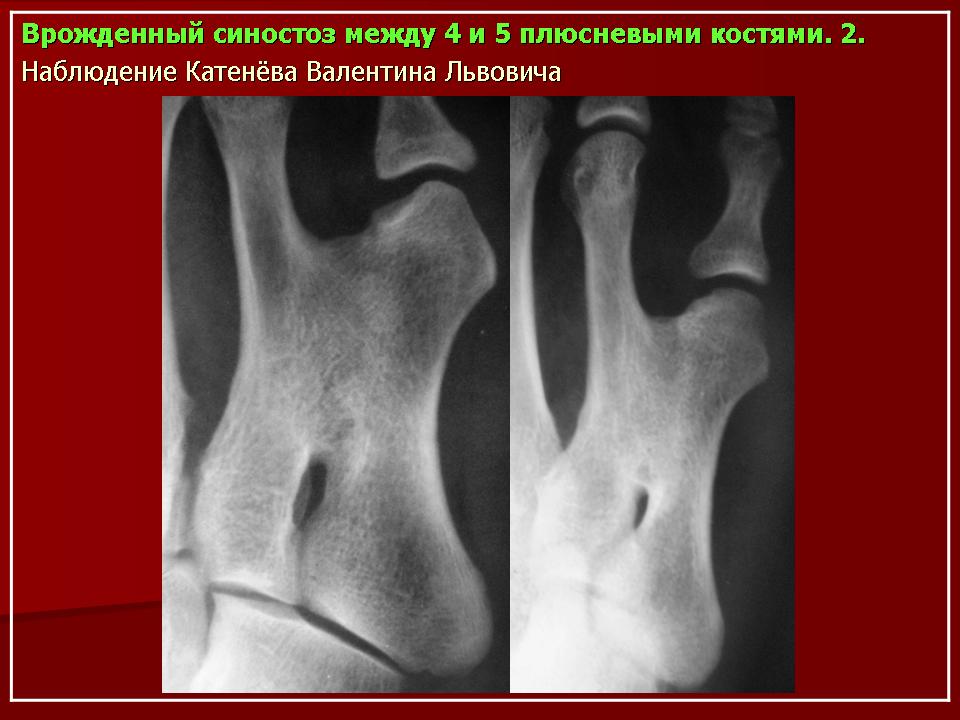 Обновленные национальные оценки распространенности рождений для отдельных врожденных дефектов в США, 2004–2006 гг. Врожденные дефекты Res A Clin Mol Teratol . 2010; 88 (12): 1008-1016.
Обновленные национальные оценки распространенности рождений для отдельных врожденных дефектов в США, 2004–2006 гг. Врожденные дефекты Res A Clin Mol Teratol . 2010; 88 (12): 1008-1016.
8. Le JT, Scott-Wyard PR. Педиатрические различия конечностей и ампутации. Phys Med Rehabil Clin N Am . 2015; 26 (1): 95-108.
9. Шаперман Дж., Ландсбергер С.Е., Сетогучи Ю. Ранняя установка протеза верхней конечности: когда и что мы подбираем. J Ортопедический протез . 2003; 15 (1): 11-17.
10. Ричмонд С., Аткинс Дж. Популяционное исследование пренатальной диагностики врожденных пороков развития в течение 16 лет. БЖОГ . 2005; 112 (10): 1349-1357.
11. Хейзинг К., Рейндерс-Месселинк Х., Маатуис К., Хаддерс-Альгра М., ван дер Слуис К.К. Возраст при первом протезировании и более поздний функциональный результат у детей и молодых людей с односторонним врожденным дефектом ниже локтя: поперечное исследование. Ортопедический протез Инт .2010; 34 (2): 166-174.
12. Американская академия хирургов-ортопедов. Врожденные отличия рук. Доступно по адресу: http://orthoinfo.aaos.org/topic.cfm?topic=A00768. Проверено в июле 2016 г. Проверено 21 февраля 2019 г.
13. de Jong IG, Reinders-Messelink HA, Janssen WG, Poelma MJ, van Wijk I., van der Sluis CK. Смешанные чувства детей и подростков с односторонним врожденным пороком ниже локтя: онлайн-исследование в фокус-группе. PLoS One . 2012; 7 (6): e37099.
14.Gingras G, Mongeau M, Moreault P, Dupuis M, Hebert B, Corriveau C. Врожденные аномалии конечностей: II. Психологические и образовательные аспекты. Кан Мед Ассо Дж. . 1964; 91 (3): 115-119.
15. Брэдбери Э. Психологические и социальные последствия обезображивания руки у детей и подростков. Dev Neurorehabil . 2007; 10 (2): 143-148.
16. Шанкар Б., Бхутла Е., Кумар Д., Кишор С., Дас С.П. Синдром Холта-Орама: редкий вариант. Иран Ж. Медицина .2017; 42 (4): 416-419.
17. Мано Х., Фудзивара С., Хага Н. Адаптивное поведение и двигательные навыки у детей с недостаточностью верхних конечностей. Ортопедический протез Инт . 2018; 42 (2) 236-240.
18. Джонсон Л., Хики А., Скуллар Б., Чондрос П. Чувство верхних конечностей у детей с врожденными пороками конечностей: последствия для функции и использования протезов. Бр. Дж. Оккуп Тер . 2002; 65 (7): 327-334.
19. Atallah R, Leijendekkers RA, Hoogeboom TJ, Frölke JP. Осложнения протезов с костной фиксацией для людей с ампутацией конечности: систематический обзор. PLoS One . 2018; 13 (8): e0201821.
20. Аллен П.Дж., Весси Дж.А., Шапиро Н.А. Первичная медицинская помощь детям с хроническими заболеваниями . 5-е изд. Сент-Луис, Миссури: Мосби Эльзевьер; 2010.
21. Нельсон В.С., Флад К.М., Брайант П.Р., Хуанг М.Э., Паскуина П.Ф., Робертс Т.Л. Дефицит конечностей и протезирование. 1. Принятие решений при назначении и лечении протезов. Arch Phys Med Rehabil . 2006; 87 (3 доп. 1): S3-S9.
22. Ольгун З.Д., Демиркиран Г., Полли Д. мл., Язычи М.Врожденное одностороннее отсутствие верхней конечности может привести к определенному типу грудопоясничной дуги. J Педиатр Ортоп B . 2018; 27 (2): 180-183.
23. Танака К.С., Лайтдейл-Мирик Н. Достижения в области создания детских протезов для лечения различий верхних конечностей, напечатанных на 3D-принтере. J Bone Joint Surg Am . 2016; 98 (15): 1320-1326.
Факты о дефектах сокращения верхних и нижних конечностей
Дефекты сокращения верхних и нижних конечностей возникают, когда часть или вся рука (верхняя конечность) или нога (нижняя конечность) плода не могут полностью сформироваться во время беременности.Дефект называют «уменьшением конечности», потому что конечность уменьшена по сравнению с нормальным размером или отсутствует.
Сколько детей рождается с дефектами редукции конечностей?
По оценкам исследователей, в США примерно 1 из 1900 детей рождается с дефектом редукции конечностей. У некоторых из этих детей будут дефекты сокращения как верхних, так и нижних конечностей. 1
Какие проблемы есть у детей с дефектами редукции конечностей?
Младенцы и дети с дефектами редукции конечностей будут сталкиваться с различными проблемами и трудностями, но степень их будет зависеть от места и размера редукции.Некоторые потенциальные трудности и проблемы включают:
- Трудности с нормальным развитием, такие как моторика
- Требуется помощь в повседневной деятельности, например, в уходе за собой
- Ограничения в отношении определенных движений, занятий спортом или занятий
- Возможные эмоциональные и социальные проблемы из-за внешнего вида
Специфическое лечение дефектов сокращения конечностей будет назначено врачом ребенка на основе таких факторов, как возраст ребенка, степень и тип дефекта, а также переносимость ребенком определенных лекарств, процедур и методов лечения.
Общая цель лечения дефектов редукции конечностей — предоставить ребенку конечность, которая имеет надлежащее функционирование и внешний вид. Лечение может быть разным для каждого ребенка. Возможные варианты лечения включают:
- Протезирование (протезы)
- Ортопедия (шины или скобы)
- Хирургия
- Реабилитация (физиотерапия или трудотерапия)
Важно помнить, что некоторые младенцы и дети с укороченными конечностями будут сталкиваться с некоторыми трудностями и ограничениями на протяжении всей жизни, но при надлежащем лечении и уходе они могут прожить долгую, здоровую и продуктивную жизнь.
Что вызывает дефекты редукции конечностей?
Причина дефекта редукции конечности неизвестна. Однако исследования показали, что определенные виды поведения или воздействия во время беременности могут увеличить риск рождения ребенка с дефектом сокращения конечностей. К ним относятся:
- Воздействие некоторых химических веществ или вирусов на мать во время беременности
- Воздействие некоторых лекарств на мать
- Возможное воздействие табакокурения на мать (хотя необходимы дополнительные исследования)
CDC работает со многими исследователями для изучения факторов риска, которые могут увеличить вероятность рождения ребенка с дефектами редукции конечностей, а также исходы рождения детей с этим дефектом.Ниже приведены примеры того, что было обнаружено в ходе этого исследования:
- Женщина, принимающая поливитамины перед беременностью, может снизить риск рождения ребенка с дефектами редукции конечностей, хотя необходимы дополнительные исследования. 2
- Определенные наборы дефектов сокращения конечностей могут быть связаны с другими врожденными дефектами, такими как пороки сердца, омфалоцеле и гастрошизис. 3
Можно ли предотвратить дефекты редукции конечностей?
Не существует известного способа предотвратить этот тип дефекта, но некоторые из проблем, с которыми в дальнейшей жизни сталкивается человек, рожденный с дефектом редукции конечности, можно предотвратить, если вылечить этот дефект на ранней стадии.
Даже в этом случае матери могут предпринять шаги до и во время беременности, чтобы иметь здоровую беременность. Эти шаги включают ежедневный прием поливитаминов с фолиевой кислотой (400 мкг), отказ от курения и употребления алкоголя во время беременности.
Список литературы
- Mai CT, Isenburg JL, Canfield MA, Meyer RE, Correa A, Alverson CJ, Lupo PJ, Riehle ‐ Colarusso T, Cho SJ, Aggarwal D, Kirby RS. Национальные популяционные оценки основных врожденных дефектов, 2010–2014 гг. Исследование врожденных дефектов. 2019; 111 (18): 1420-1435.
- Yang QH, Khoury MJ, Olney RS, & Mulinare J. Снижает ли периконцептивное употребление поливитаминов риск дефицита конечностей у потомства? Эпидемиология. 1997; 8: 157-61.
- Rosano A, Botto LD, Olney RS, Khoury MJ, Ritvanen A, Goujard J, et al. Дефекты конечностей, связанные с основными врожденными аномалиями: клиническое и эпидемиологическое исследование, проведенное Международным центром обмена информацией для систем мониторинга врожденных дефектов. Am J Med Genet. 2000; 93: 110-16.
Изображения являются общественным достоянием и, следовательно, свободны от каких-либо ограничений авторских прав.В порядке любезности мы просим, чтобы поставщик контента (Центры по контролю и профилактике заболеваний, Национальный центр по врожденным порокам и нарушениям развития) был указан и уведомлен о любом публичном или частном использовании этого изображения.
Изображения являются общественным достоянием и, следовательно, свободны от каких-либо ограничений авторских прав. В порядке любезности мы просим, чтобы поставщик контента (Центры по контролю и профилактике заболеваний, Национальный центр по врожденным дефектам и порокам развития) был указан и уведомлен о любом публичном или частном использовании этого изображения.
Врожденная недостаточность верхних конечностей — PM&R KnowledgeNow
СПРАВОЧНАЯ ИНФОРМАЦИЯ
- День H. Классификация врожденной недостаточности конечностей ISO / ISPO. Ортопедический протез Инт . 1991. 15 (2): 67–69.
- Nielsen GL, Norgard B, Puho E, et al. Риск конкретных врожденных аномалий у потомков женщин с диабетом. Diabet Med. 2005; 22: 693–696.
- Kousseff BG, Kousseff BG. Гестационный сахарный диабет (класс А): тератоген для человека? Am J Med Genet. 1999; 83: 402–408.
- Brumback BA, Cook RJ, Ryan LM. Мета-анализ исследований случай-контроль и когортных исследований с интервально-цензурированными данными воздействия: отбор проб тохориальных ворсинок. 2000 июн; 1 (2): 203-17.
- McGuirk CK, Westgate MN, Holmes LB. Недостатки конечностей у новорожденных. Педиатрия . 2001; 108: E64.
- Enjolras O, Chapot R, Merland JJ. Сосудистые аномалии и рост конечностей: обзор. J Pediatr Orthop B. 2004; 13 (6): 349–357.
- Ghabrial R, Versace P, Kourt G и др. Синдром Мебиуса: особенности и этиология. J Pediatr Ophthalmol Strabismus. 1998; 35 (6): 304–311.
- Ordal L, Keunen J, et al. Врожденные пороки конечностей сосудистой этиологии: возможная связь с тромбофилией матери. Am J Med Genet A, декабрь 2016; 170 (12): 3083-3089.
- Rijnders LJ, Boonstra AM, Groothoff JW, et al. Дети с дефицитом нижних конечностей в Нидерландах: эпидемиологические аспекты. Prosthet Orthot Int. 2000; 24: 13.
- Беннет Дж. Б., Риордан, округ Колумбия. Ulnar Hemimelia. В: Herring JA, Birch JG, ред. Ребенок с недостатком конечностей. Rosemont, IL: Американская академия хирургов-ортопедов; 1998.
- Modrcin A, McLaughlin M, Luetke M. Педиатрическая недостаточность конечностей. В Александре М, Мэтьюз Д., ред., Детская реабилитация, 5-е, , стр. 314). Нью-Йорк, штат Нью-Йорк: Demos Medical; 2015.
- ДиКоуден МАБА, Робинетт Х., Ортис О. Пособие по обучению ранней подгонке и изменению поведения с помощью оконечного устройства добровольного закрытия. Детская ортопедическая клиника J Assoc . 1987; 22: 47.
- Селедка Дж., Берч Дж. Ребенок с недостатком конечностей. Rosemont, IL: Американская академия хирургов-ортопедов; 1998.
- Goldfarb C, Shaw N, Steffen JA, et al. Распространенность врожденных аномалий кисти и верхних конечностей. J. Pedatr Orthop. 2017 Мар 37 (2) 144-148.
- Panthaki ZJ, Armstrong MB, Panthaki ZJ, et al. Патологии рук, связанные с черепно-лицевыми синдромами. J Craniofac Surg. 2003; 14 (5): 709–712.
- Gaebler-Spira D, Lipschutz R. Дефекты конечностей у детей. В: Александр М, Мэтьюз Д., ред. Детская реабилитация . 4-е изд. Нью-Йорк, штат Нью-Йорк: Demos Medical; 2010.
- Бэ, Д. С., Канисарес, М. Ф., Миллер, П. Э., Уотерс, П. М., и Гольдфарб, К. А. (2018). Функциональное влияние врожденных различий рук: ранние результаты реестра врожденных различий верхних конечностей (CoULD). Журнал хирургии кисти, 43 (4), 321-330.2018.
- Schuch CM. Принципы протезирования миоэлектрических протезов у детей. В: Herring J, Birch JG, ред. Ребенок с дефектом конечности . Роземонт, Иллинойс: Американская академия хирургов-ортопедов; 1998.
- Meurs M, Maathuis CG, Lucas C, et al. Назначение первого протеза и последующее использование у детей с врожденной односторонней недостаточностью верхних конечностей: систематический обзор. Ортопедический протез Инт . 2006. 30 (2): 165–173.
- Kuyper MA, Breedijk M, Mulders AH, et al.Протезирование детей с дефектами верхних конечностей в Нидерландах. Ортопедический протез Инт . 2001. 25 (3): 228–234.
- Baas M, Zwanenburg PR, et al. Документирование комбинированных врожденных аномалий верхних конечностей с использованием классификации Оберга, Манске и Тонкина: значение для эпидемиологических исследований и сравнения результатов. Журнал хирургии кисти. 21 марта 2018 г.
- Тонкин М.А., Толертон С.К., Квик Т.Дж., et al. Классификация врожденных аномалий кисти и верхней конечности: разработка и оценка новой системы. Journal of Hand Surgery , 38 (9) (2013), стр. 1845-1853
Библиография
Бэгли AM, Молитор F, Вагнер LV, Tomhave W, James MA. Медицина развития и ребенок. Неврология . 2006; 48: 596-575.
Фиск Дж. Терминология детской недостаточности конечностей. В: Смит Д., Майкл Дж., Боукер Дж., Ред. Атлас ампутаций и пороков конечностей: принципы хирургии, протезирования и реабилитации . 3-е изд. Роземонт, Иллинойс: Американская академия хирургов-ортопедов; 2004 г.
Лестер Д.К., Художник Г.Л., Берман А.Т., Скиннер С.Р. «Идиопатический» сколиоз, связанный с недостаточностью верхних конечностей. Clin Orthop Relat Res . 1986; (202): 205-210.
Стахели Л. Основы детской ортопедии . 4-е изд. Сиэтл, Вашингтон: Липпинкотт Уильямс и Уилкинс; 2008.
Оригинальная версия темы
Энн С. Модрчин, доктор медицины, Мэтью Маклафлин, доктор медицины. Врожденный порок верхних конечностей. 02.12.2013.
Раскрытие информации об авторе
Мэтью Маклафлин, Мэриленд
Ничего не нужно раскрывать
Сьюзан Лисенби, Мэриленд
Нечего раскрывать
Сумита Шарма, Мэриленд,
Нечего раскрывать
Энн К.Modrcin, MD
Нечего раскрывать
Успехи в понимании генетики синдромов, связанных с врожденными аномалиями верхних конечностей — Sun
Введение
Врожденные аномалии верхних конечностей (CULA) включают широкий спектр структурных аномалий, вызванных нарушением морфогенеза верхней конечности, которые затрагивают примерно 1 на 500 живорожденных (1,2). Приблизительно от 20% до 30% детей с CULA имеют по крайней мере одну связанную аномалию не конечностей (2), и примерно от 10% до 20% всех врожденных аномалий имеют поражение верхних конечностей (3).Это состояние демонстрирует крайнюю неоднородность представлений разной степени тяжести. Хотя некоторые из них приводят к легким функциональным последствиям, они также могут вызвать психологические проблемы у детей (4).
На данный момент в клинических условиях широко используются две схемы классификации, а именно: Классификация Свансона и Классификация Оберга-Манске-Тонкина (OMT). Классификация Суонсона была предложена в 1964 году и основывалась на анатомическом понимании и хирургическом подходе к лечению (5).Хотя Классификация Суонсона удовлетворительна по нескольким критериям, она не включает знания, полученные за последние 50 лет (2). Классификация OMT объединяет новейшие знания о развитии конечностей и патогенезе аномалий конечностей с использованием терминологии дисморфологии (6) и обеспечивает более практичную и легко используемую схему.
Патогенез CULA остается плохо изученным. Ограниченное количество исследований предоставляет доказательства связи воздействия химических веществ в окружающей среде и CULA, включая стойкое органическое загрязнение и загрязнение воздуха (7).Интересно, что отбор проб ворсинок хориона был идентифицирован как фактор риска синдромальной и несиндромальной CULA (8). Напротив, наблюдение семейных случаев CULA подчеркнуло вклад генетических факторов в CULA. Новые подходы, такие как массив однонуклеотидного полиморфизма (SNP), сравнительный массив геномной гибридизации (array-CGH) и секвенирование следующего поколения (NGS), позволяют идентифицировать многочисленные причинные гены CULA. Выявление генетического фона синдромальной CULA помогает расширить спектр причинных генов, определить пути, необходимые для нормального органогенеза конечностей, и объяснить механизм, лежащий в основе взаимодействия CULA и аномалий, не связанных с конечностями.В этом обзоре мы сначала познакомимся с процессом развития верхних конечностей и задействованными сигнальными путями. Затем мы даем обзор генетической основы синдромальной CULA в соответствии со связанными путями причинных генов.
Развитие верхних конечностей
Развитие конечностей позвоночных во время эмбриональной жизни — многоступенчатый процесс, который управляется сложными взаимодействиями между сигнальными центрами.У людей развитие зачатка конечности начинается примерно на четвертой неделе беременности с появления небольшой зачатка на боковой стенке тела. Зачаток конечности состоит из внутреннего мезенхимального ядра и внешнего эктодермального колпачка (9). После появления зачатка конечности формируются три оси: проксимально-дистальная (PD), переднезадняя (AP) и дорсо-вентральная (DV) оси. Фактор роста мезенхимальных фибробластов 10 (FGF10), передача сигналов костных морфогенетических белков (BMP) индуцируют образование апикального эктодермального гребня (AER), который расположен на интерфейсе DV.AER в основном опосредует формирование паттерна PD посредством регуляции сигналов FGF и WNT (10). Помимо FGF из AER, ось DV также определяется передачей сигналов ретиноевой кислоты (RA) от боковых стенок (9). Зона поляризующей активности (ZPA) находится в мезенхиме зачатка задней конечности и контролируется передачей сигналов Sonic Hedgehog (SHH). SHH, секретируемый ZPA, диффундирует вдоль зачатка конечности с образованием градиента, который контролирует формирование оси AP. Множественные сигналы, включая TBX, HAND2, RA, HOXD и GLI3R, участвуют в регуляции экспрессии SHH и дополнительно влияют на формирование паттерна AP.SHH из ZPA и FGF из AER образуют эпителиально-мезенхимальную петлю обратной связи, которая регулирует формирование PD и оси AP. Член семейства WNT 7A (WNT7A), продуцируемый презумптивной эктодермой дорсальной конечности, считается организатором оси DV. Специфическая экспрессия LIM-гомеодоменного фактора транскрипции 1 бета (LMX1B) в дорсальной мезенхиме зачатка конечности индуцируется WNT7A (6). LMX1B является необходимым и достаточным фактором в управлении формированием паттерна дорсальных конечностей (11). Передача сигналов SHH, WNT и BMP критически координируется первичной ресничкой, которая представляет собой органеллы на основе микротрубочек, которые возникают с поверхности клеток большинства типов клеток позвоночных (10).
Генетические достижения в синдромальной CULA
Aat) Аномалии не конечностей, связанные с синдромальной CULA, включают широкий спектр проявлений, например мочеполовой, сердечно-сосудистой, нервной, лицевой и т. Д. (11). Синдромные CULA составляют от 20% до 30% всех CULA (1,2), и от 10% до 20% всех врожденных аномалий имеют поражение верхних конечностей (3). Связанные с этим аномалии не конечностей могут быть опасными для жизни и, следовательно, требуют большего внимания со стороны врачей и родителей.
Как генетические мутации, так и геномные перестройки могут приводить к синдромному CULA (см. Таблицу , ). Многие гены, участвующие в патогенезе синдромальной CULA, можно отнести к нескольким биологическим путям. Здесь мы представляем роли белков Cohesin, передачу сигналов WNT, передачу сигналов Hedgehog, передачу сигналов FGF, FGFR и PI3K-AKT1-mTOR в патогенезе синдромальной CULA.
Таблица 1 Известные синдромы, связанные с CULA
Полная таблица
Белки когезина и синдром Корнелии де Ланге
Белки Cohesin важны для структурного поддержания комплексов, содержащих белок хромосом (SMC).Опосредуя организацию хроматина и дистанционное взаимодействие хроматина, белки когезии могут влиять на экспрессию генов (12). Некоторые расстройства происходят из-за мутаций в субъединицах когезина, и его ключевые регуляторы называются когезинопатиями. Синдром Корнелии де Ланге (CdLS; MIM: 122470), видный член когезинопатий, является доминантно наследуемым заболеванием, характеризующимся дисморфическими особенностями, CULA, общей задержкой развития и поражением желудочно-кишечной системы. NIPBL загружает комплекс когезина на ДНК и необходим для топологических перемещений ДНК (13).Мутации в NIPBL ответственны за значительную часть CdLS (12). В настоящее время в CdLS вовлечены шесть генов, связанных с когезинами, а именно: HDAC8 (MIM: 300882), RAD21, (MIM: 614701), NIPBL (MIM: 122470), SMC1A (MIM: 300590). ), BRD4 и SMC3 (MIM: 610759) (14). Среди них HDAC8, BRD4 и NIPBL являются ключевыми регуляторами белков когезина, тогда как SMC1A. SMC3 и RAD21 являются структурными компонентами когезинового кольца (13,15).
NIPBL индуцирует деацетилирование гистонов за счет привлечения гистондеацетилазы (HDAC) и регулирует экспрессию генов. В модели рыбок данио с нокдауном Nipbl наблюдались уменьшение размеров и дефекты формирования паттерна грудного плавника (передних конечностей) наряду с нарушенной экспрессией нескольких ключевых генов развития конечностей, включая fgfs , hand2 и несколько генов hox (16 ), что предполагает, что пороки развития конечностей при CdLS могут быть приписаны влиянию белков Cohesin на экспрессию критически важных генов развития конечностей.
WNT сигнализация
Сигналы
WNT включают разнообразную группу секретируемых белков, которые опосредуют важные процессы развития, включая формирование паттерна эмбриональной оси, спецификацию клеточной судьбы, пролиферацию и миграцию (17). Гены, кодирующие различные сигнальные молекулы, участвующие в сигнальном пути Wnt, были идентифицированы в наследуемой по менделее синдромной CULA.Рецессивно унаследованные мутации WNT7A были описаны у лиц с синдромом фокомелии Шинцеля (MIM: 276820) и синдромом Фурмана (MIM: 228930) (18). Оба синдрома имеют деформации конечностей, но синдром фокомелии Шинцеля включает более широкий набор фенотипов, включая тяжелые аномалии верхних и нижних конечностей с гипоплазией таза и гениталий. Есть много других генов в сигнальном пути Wnt, которые связаны с синдромальной CULA. Кардиоспондилокарпо-лицевой синдром (CSCF; MIM: e157800) характеризуется дефектами сердечной перегородки, синостозом позвонков, брахидактилией с запястно-предплюсневым сращением и дисморфическими чертами лица.CSCF вызывается гетерозиготной мутацией в гене MAP3K7 . MAP3K7 действует как вышестоящая киназа, регулирующая несколько путей, включая WNT, NF-κB и p38 MAPK (19). Фокальная кожная гипоплазия (MIM: 305600), вызванная мутациями в PORCN , представляет собой Х-сцепленный доминантный синдром с внутриутробной летальностью у мужчин. Общие черты этого синдрома включают явные кожные проявления, пороки развития пальцев (в основном синдактилию и полидактилию), оральные и глазные аномалии. PORCN необходим для ацетилирования и секреции лигандов WNT у мышей и людей (20).Учитывая критическую роль передачи сигналов Wnt в развитии человека, неудивительно, что эти синдромные CULA, ассоциированные с передачей сигналов Wnt, проявляют сложные фенотипы, обычно затрагивающие несколько органов и систем.
Синдром Робиноу — это генетически гетерогенная тяжелая скелетная дисплазия, характеризующаяся мезомелическим укорочением конечностей, дисморфическими особенностями, включая гипоплазию средней зоны лица, гипоплазию гениталий и позвоночные аномалии (21,22). Мутации нескольких генов, участвующих в сигнальном пути WNT, участвуют в различных подтипах синдрома Робиноу. WNT5A участвует как в каноническом, так и в неканоническом сигнальном пути Wnt (23). Доминантно унаследованные мутации WNT5A были связаны с аутосомно-доминантным синдромом Робинова-1 (DRS; MIM: 180700) и гомозиготными Wnt5a нулевых мышей с анатомическими дефектами, напоминающими людей с аутосомным DRS (24). ROR2, тирозинкиназоподобный орфанный рецептор, взаимодействует с WNT5A как функционально, так и физически (23). Связывание WNT5A с ROR2 приводит к фосфорилированию ROR2, что приводит к зависимой от Rho GTPase активации кальциевых путей WNT-JNK и WNT, т.е.е., неканонический путь WNT. Рецессивные и доминантные мутации ROR2 также участвуют в аутосомно-рецессивном синдроме Робиноу (RRS; MIM: 268310) и брахидактилии типа B1 (BDB1; MIM: 113000) соответственно. Интересно, что не все пациенты, несущие гетерозиготные нулевые мутации в ROR2 , проявляли брахидактилию. Измеряя стационарные уровни белка на модели, а также расположение на поверхности в линиях клеток, экспрессирующих вариант ROR2 , Schwarzer et al. представил гипотезу о том, что фенотипический результат может быть объяснен равновесием между внутриклеточной задержкой и экспрессией на поверхности клетки, а не простой моделью потери или увеличения функции (25). Disheveled (DVL): 13 — это внутриклеточный белок T, участвующий в канонической и неканонической передаче сигналов WNT (26). Связывание DVL с клеточной мембраной облегчает связывание Axin и GSK3β, которые впоследствии фосфорилируют LRP5 / 6 и тем самым предотвращают деградацию β-катенина. Накопление β-катенина в ядре активирует гены, отвечающие за WNT (26).В неканоническом пути WNT DVL играет ключевую роль в регуляции полярности и детерминации цитоскелета, действуя как точка ветвления для активации Rho, Rac и Cdc42 (26). Было показано, что гетерозиготные мутации DVL1 и DVL3 приводят к аутосомно-доминантному синдрому Робиноу-2 (MIM: 616331) и аутосомно-доминантному синдрому Робиноу-3 (MIM: 616894) (27). Вместе эти данные свидетельствуют о том, что сигнальный путь WNT играет важную роль в патогенезе широкого спектра гипопластических расстройств, включая CULA.
Ежик сигнальный
Сигнальный путь Hedgehog (HH) играет множество ролей в контроле клеточной пролиферации, формирования эмбрионального паттерна и развития конечностей (9). Гены Gli-kruppel (GLI) кодируют факторы транскрипции, участвующие в сигнальном пути SHH. GLI1 , GLI2 и GLI3 , члены семейства GLI, были связаны с множеством заболеваний человека, включая как синдромальные, так и несиндромальные CULA.GLI3 является фактором транскрипции и действует как модулятор пути SHH, выполняя как фасилитирующую, так и подавляющую функцию (28). Инактивация пути SHH конститутивно трансформирует GLI3 в изоформу GLI3R, которая перемещается в ядро и негативно регулирует гены-мишени SHH (29). Активированный SHH ингибирует образование изоформы GLI3R и индуцирует образование GLI2A и GLI3A (активаторов GLI). Впоследствии активаторы GLI способствуют экспрессии генов-мишеней SHH. Помимо ROR2 , GLI3 является другим геном, который участвует как в синдромальной, так и в несиндромальной CULA.Гетерозиготные мутации в гене GLI3 могут приводить к синдрому цефалополисиндактилии Грейга (GCPS; MIM: 175700), синдрому Паллистера-Холла (PHS; MIM: 146510), постаксиальной полидактилии типа A1 и B (PAP; MIM: 174200) и полидактилии преаксиального типа. IV (PPD; MIM: 174700) (28). GCPAS и PHS — это аллельные синдромы с различными клиническими проявлениями (30). Для GCPS в основном характерны полисиндактилия, макроцефалия с выступом на лбу и гипертелоризм (31). Преаксиальный отдел стопы и постаксиальный отдел кистей рук — наиболее часто определяемые формы полидактилии (31).Типичные фенотипы PHS включают центральную полидактилию, нарушение функции гипофиза, гамартому гипоталамуса и висцеральные аномалии (30). Корреляции генотип-фенотип GLI3 ассоциированных расстройств хорошо охарактеризованы (28). N-концевая часть белка GLI3 содержит домен цинкового пальца. Мутации в домене цинкового пальца в основном приводят к GCPS через механизм гапло-недостаточности. Средняя часть белка GLI3 охватывает сайт расщепления белка. Пациенты с PHS обычно несут мутации в средней части.С-конец содержит трансактивирующие домены 1 и 2. Мутации в С-концевой части приводят к различным фенотипам, включая GCPS, PAP и PPD (28). Тем не менее, механизм, лежащий в основе определенного спектра фенотипов, нуждается в дальнейших исследованиях.
SUFU, аббревиатура от Suppressor of Fused, играет критическую роль в первичных ресничках. SUFU действует как главный негативный регулятор пути SHH, образуя комплекс с GLI3 и GLI2 в первичных ресничках (32).Транспортировка SUFU к первичным ресничкам потенциально регулируется активаторами GLI, но не супрессорами (33). Известно, что рецессивные и доминантные мутации SUFU вызывают синдром Жубера 32 (MIM: 617757) и синдром базальноклеточного невуса (BCNS; MIM: 109400) соответственно (32). Синдром Жубера 32 характеризуется врожденной атаксией, постаксиальной полидактилией, гипоплазией червя мозжечка и кранио-лицевыми дисморфизмами. В то время как постаксиальная синдактилия относительно редко встречается при BCNS (32). Гетерозиготные мутации в PTCh2 и PTHC2 также могут приводить к синдрому базальноклеточного невуса.Синдром базально-клеточного невуса — это предрасполагающий к семейному раку синдром, который включает широкий спектр системных проявлений, наиболее характерными симптомами которого являются базально-клеточный рак, кератоциты челюсти и церебральные кальцификаты (34). PTCh2 и PTCh3 , кодирующие члены семейства исправленных, подавляют передачу сигналов SHH посредством ингибирования SMO. Потеря функции PTCh2 и PTCh3 приводит к аберрантному увеличению передачи сигналов SHH (34).
Регуляторная последовательность ZPA ( ZRS ), энхансер SHH, специфичный для конечностей дальнего действия, находится в интроне 5 LMBR1 (35).Недавно миссенс-мутация в p- ZRS (pre- ZRS ), некодирующей последовательности на 500 п.н. выше ZRS , была связана с синдромом полисиндактилии большого пальца трифаланга (MIM: 174500) в семье из нескольких поколений ( 36). Трансгенные мыши, несущие эту мутацию, показали эктопическую экспрессию SHH (36). Хотя регуляторные эффекты ZRS на Shh хорошо изучены (37), необходимы дальнейшие исследования для определения роли pZRS в экспрессии конечностей Shh.
Факторы роста фибробластов (FGF) и FGFR
FGF — это семейство белков, которые регулируют многие процессы развития на ранней стадии эмбрионального развития. FGFs выполняют свои функции посредством связывания с рецепторами фактора роста фибробластов (FGFR). В настоящее время известно четыре FGFR (от FGFR1 до FGFR4), которые взаимодействуют с FGFs HSGAG-зависимым образом (38).Как увеличение, так и уменьшение передачи сигналов FGF может вызывать заболевания человека. Например, мутации с усилением функции FGFR2 приводят к синдрому Аперта (MIM: 101200) (39), нарушению развития, характеризующемуся краниосиностозом, гипоплазией средней зоны лица и тяжелой синдактилией со склонностью к слиянию. Исследования показали, что около двух третей синдрома Аперта вызывается мутацией S252W в FGFR2 , а оставшаяся треть вызвана мутацией P253R в FGFR2 (40).Синдактилия рук и ног, вероятно, была более тяжелой у пациентов с мутацией P253R, в то время как волчья пасть чаще наблюдалась у пациентов, несущих мутацию P252W (40). Пролилизомераза, пептидил-пролилцис-транс-изомераза, взаимодействующая 1 ( PIN1 ), является критическим регулятором передачи сигналов FGFR, и мыши Pin1 +/- обнаруживают замедленное закрытие черепных швов (41). Путем скрещивания мышей Pin1 +/- с мышами Fgfr2 S252W / + , мышиной модели синдрома Аперта, подавление функции Pin1 ослабляло преждевременное слияние черепных и лобно-носовых швов.Хотя другие фенотипы, такие как синдактилия, не были спасены сниженной дозировкой Pin1 , этот подход действительно обеспечивает новые терапевтические идеи в отношении облегчения других фенотипов при синдроме Аперта. Мутации с повышением функции FGF10 ответственны за Lacrimoauriculodentodigital синдром (синдром LADD; MIM: 149730) (42), врожденное заболевание, в основном затрагивающее слезные железы и проток, уши, зубы и пальцы. Активирующие мутации в FGFR2 и FGFR3 также могут приводить к синдрому LADD (43).FGFR2 взаимодействует с FGF10, и условный нокаут Fgfr2 у мышей приводит к порокам конечностей и пальцев (43). Мутации в гене FGFR3 в настоящее время связаны как минимум с 10 заболеваниями человека, включая дисплазию скелета, пороки развития кожи и рак (44). Ослабление хондроцитов отвечает за скелетную дисплазию, связанную с FGFR3 (44). Недавно Bosakova et al. , обнаружили, что длительная и временная активация передачи сигналов FGF приводит к укорочению и удлинению первичных ресничек, соответственно (45).На передачу сигналов SHH впоследствии влияет нарушение функции ресничек (45), что дает представление о патогенном механизме, лежащем в основе фенотипов конечностей при расстройствах, связанных с FGFR.
PI3K-AKT1-mTOR
Передача сигналов PI3K-AKT-mTOR опосредует пролиферацию, выживание и метаболизм клеток, активация этого пути обычно участвует в онкогенезе. PIK3CA и AKT1 — часто мутировавший ген в опухолях человека (46,47).Соматический мозаицизм PIK3CA и AKT1 может приводить к синдромам избыточного роста. Генетический мозаицизм относится к процессу, происходящему постзиготически, который приводит к присутствию генетически различных клеток у одного человека. Появление глубокого NGS способствовало обнаружению нарушений развития, вызванных мозаичными мутациями. Синдром Протея (MIM: 176920), вызванный мутациями в AKT1 , характеризуется мозговыми невусами соединительной ткани и пятнистым разрастанием, чаще всего поражающим конечности.Большинство пациентов с синдромом Протея несли активную функциональную мутацию c.G49A в AKT1 (47). Соматический мозаицизм PIK3CA отвечает за широкий спектр заболеваний, называемых синдромом избыточного роста PIK3CA , включая врожденный избыточный липоматозный рост, сосудистые мальформации, эпидермальные невусы и скелетные / сколиозные / спинномозговые аномалии (синдром CLOVE-капилляра 12918; MIM: 6 порок развития нижней губы, лимфатическая деформация лица и шеи, асимметрия и частичный / генерализованный чрезмерный рост (синдром CLAPO; MIM: 613089), мегалэнцефалино-капиллярная мальформация (MCAP, MIM: 602501).Очаговые формы разрастания, связанные с PIK3CA , включают макродактилию, эпидермальные невусы, инфильтрирующий липоматоз, лимфатические мальформации и венозные мальформации (48). Наиболее часто наблюдаемым компонентом конечностей при этих заболеваниях является макродактилия, и степень чрезмерного роста варьирует.
CNV
Было идентифицировано несколько хорошо охарактеризованных CNVs, лежащих в основе синдромов с поражением конечностей.Как правило, синдром отсутствия тромбоцитопении лучевой кости (синдром TAR; MIM: 274000), синдром с множественными аномалиями, влияющими на кровообращение верхней конечности, вызван гетерозиготностью соединения для делеции 1q21.1 с участием гена RBM8A и некодирующего СНП в RBM8A (49). Влияние редкого нулевого варианта и некодирующего общего SNP также наблюдалось в TBX6 -ассоциированном врожденном сколиозе (TACS) (50,51). Распространенный гипоморфный аллель TBX6 в транс- с редким 16p11.2 или вариант TBX6 с потерей функции приводят к TACS (52). Мышиные модели гетерозиготности соединения TBX6 демонстрируют пороки развития позвоночника и предоставляют дополнительные доказательства, подтверждающие модель дозировки гена (51,52). Аномалии системы крови включают уменьшение количества тромбоцитов в крови и уменьшение количества мегакариоцитов и клеток-предшественников тромбоцитов в костном мозге. Тяжесть поражения верхней конечности колеблется от отсутствия лучевой кости до отсутствия большей части верхних конечностей, но большие пальцы всегда хорошо сохранились.Реже встречаются пороки развития позвоночника, нижних конечностей.
Другой хорошо охарактеризованный синдром, связанный с CULA, — это синдром микроделеции 2p15-p16.1 (MIM: 612513) (53). Этот синдром имеет широкий спектр фенотипов, и около 80% пациентов имеют аномалии кисти, в основном состоящие из камптодактилии (54). Размер удалений (от 0,1 до 9,5 Мб) и точек останова сильно различаются. Недавно XPO1 , REL и BCL11A были идентифицированы как гены-кандидаты для 2p15-p16.1 синдром микроделеции (54).
Существуют и другие рекуррентные CNV, связанные с синдромальной CULA, например, включая 2q37 (MIM: 600430), 7q11.2-q21.3 (MIM: 129900), 16p13.3 (MIM: 613458) и 17p11.2 (MIM: 182290). ). В большой когорте людей с несиндромальным врожденным пороком конечностей анализ CNV с высоким разрешением выявил, что 10% людей имеют CNV, относящиеся к заболеванию (55). Среди CNVs, идентифицированных в этом исследовании, 57% влияют на некодирующий cis -регуляторный геном, что предполагает вклад некодирующих областей в несиндромальную CULA.
Выводы
Последнее десятилетие стало свидетелем значительного прогресса в технологиях генетического и геномного секвенирования. Благодаря улучшениям в молекулярной генетике за последние несколько лет область генетики CULA развивается очень быстро. Эти улучшения позволили получить подробную корреляцию генотип-фенотип и генотип-прогноз для синдромов, связанных с CULA.Используя эти знания, можно принять более точный диагноз и методы лечения. Более того, ассоциация между фенотипами конечностей и специфическими генетическими / геномными вариантами привела к идентификации генов, необходимых для развития конечностей, и, следовательно, к лучшему пониманию механизмов, регулирующих развитие конечностей. Эти знания, полученные из синдромов с CULA, помогут нам расшифровать генетическую основу CULA также у несиндромальных людей.
Благодарности
Финансирование: Это исследование частично финансировалось Национальной программой ключевых исследований и разработок Китая (No.2016YFC0
1), Национальный фонд естественных наук Китая (81822030, 81772299), Beijing JST Research Funding 2019-YJ03, Beijing Jishuitan Hospital Nova Program XKXX201818 и Фонд инициативы CAMS для медицинских наук (2016-I2M-3-003, 2016-I2M- 2-006, 2017-I2M-2-001).
Конфликт интересов: Авторы не заявляют о конфликте интересов.
Этическое заявление: Авторы несут ответственность за все аспекты работы, обеспечивая надлежащее расследование и решение вопросов, связанных с точностью или целостностью любой части работы.
Список литературы
- Giele H, Giele C, Bower C и др. Заболеваемость и эпидемиология врожденных аномалий верхних конечностей: общее популяционное исследование. J Hand Surg Am 2001; 26: 628-34. [Crossref] [PubMed]
- Экблом А.Г., Лорелл Т., Арнер М. Эпидемиология врожденных аномалий верхних конечностей в Стокгольме, Швеция, 1997–2007 годы: применение классификации Оберга, Манске и Тонкина.J Hand Surg Am 2014; 39: 237-48. [Crossref] [PubMed]
- Alrabai HM, Farr A, Bettelheim D, et al. Пренатальная диагностика врожденных различий верхних конечностей: обзор современной концепции. J Matern Fetal Neonatal Med 2017; 30: 2557-63. [Crossref] [PubMed]
- Андерссон Г.Б., Гиллберг С., Фернелл Э. и др. Дети с хирургически исправленными деформациями рук и недостатками верхних конечностей: самооценка и психологическое благополучие. J Hand Surg Eur Vol 2011; 36: 795-801.[Crossref] [PubMed]
- Swanson AB. Классификация врожденных пороков развития кисти. Н. Дж. Бюлл Акад Мед, 1964; 10: 166.
- Oberg KC, Feenstra JM, Manske PR, et al. Биология развития и классификация врожденных аномалий кисти и верхней конечности. J Hand Surg Am 2010; 35: 2066-76. [Crossref] [PubMed]
- Фостер В.Г., Эванс Дж. А., Литтл Дж. И др. Воздействие на человека загрязнителей окружающей среды и врожденных аномалий: критический обзор.Crit Rev Toxicol 2017; 47: 59-84. [Crossref] [PubMed]
- Froster UG, Baird PA. Дефекты редукции конечностей и забор проб ворсинок хориона. Ланцет 1992; 339: 66. [Crossref] [PubMed]
- Зеллер Р., Лопес-Риос Дж., Зунига А. Развитие зачатков конечностей позвоночных: переход к интегративному анализу органогенеза. Нат Рев Генет 2009; 10: 845-58. [Crossref] [PubMed]
- Анвариан З., Никитин К., Мухопадхяй С. и др. Передача клеточных сигналов первичными ресничками в процессе развития, функции органа и болезни.Нат Рев Нефрол 2019; 15: 199. [Crossref] [PubMed]
- Тонкин М.А. Классификация врожденных аномалий кисти и верхней конечности. J Hand Surg Eur Vol 2017; 42: 448-56. [Crossref] [PubMed]
- Watrin E, Kaiser FJ, Wendt KS. Регуляция генов и организация хроматина: значение мутаций когезина для болезней человека. Curr Opin Genet Dev 2016; 37: 59-66. [Crossref] [PubMed]
- Olley G, Ansari M, Bengani H и др.BRD4 взаимодействует с NIPBL, а BRD4 мутирует при синдроме Корнелии де Ланге. Нат Жене 2018; 50: 329. [Crossref] [PubMed]
- Юань Б., Нейра Дж., Пехливан Д. и др. Клиническое секвенирование экзома выявляет гетерогенность локусов и фенотипическую изменчивость когезинопатий. Genet Med 2019; 21: 663-75. [Crossref] [PubMed]
- Дирдорф М.А., Уайлд Дж. Дж., Альбрехт М. и др. Мутации RAD21 вызывают когезинопатию у человека. Am J Hum Genet 2012; 90: 1014-27. [Crossref] [PubMed]
- Muto A, Ikeda S, Lopez-Burks ME, et al.Nipbl и медиатор совместно регулируют экспрессию генов для контроля развития конечностей. PLoS Genet 2014; 10: e1004671. [Crossref] [PubMed]
- Nusse R, Clevers H. Передача сигналов Wnt / β-катенина, болезнь и новые терапевтические методы. Cell 2017; 169: 985-99. [Crossref] [PubMed]
- Woods CG, Stricker S, Seemann P и др. Мутации в WNT7A вызывают ряд пороков развития конечностей, включая синдром Фурмана и синдром фокомелии Аль-Авади / Рааса-Ротшильда / Шинцеля.Am J Hum Genet 2006; 79: 402-8. [Crossref] [PubMed]
- Михали С.Р., Ниномия-Цудзи Дж., Мориока С. Контроль TAK1 клеточной гибели. Разница в клеточной смерти 2014; 21: 1667-76. [Crossref] [PubMed]
- Бичеле С., Кокберн К., Ланнер Ф. и др. Зависимая от свиней передача сигналов Wnt не требуется до гаструляции мышей. Развитие 2013; 140: 2961-71. [Crossref] [PubMed]
- White J, Mazzeu JF, Hoischen A, et al. Кластеризация мутаций сдвига рамки DVL1 в предпоследнем экзоне вызывает аутосомно-доминантный синдром Робиноу.Am J Hum Genet 2015; 96: 612-22. [Crossref] [PubMed]
- White JJ, Mazzeu JF, Hoischen A, et al. Аллели DVL3, приводящие к сдвигу кадра на -1 последнего экзона, опосредованного аутосомно-доминантным синдромом Робинова. Am J Hum Genet 2016; 98: 553-61. [Crossref] [PubMed]
- Person AD, Beiraghi S, Sieben CM, et al. Мутации WNT5A у пациентов с аутосомно-доминантным синдромом Робинова. Дев Дин 2010; 239: 327-37. [PubMed]
- Оиси И., Сузуки Х, Ониши Н. и др.Рецепторная тирозинкиназа Ror2 участвует в неканоническом сигнальном пути Wnt5a / JNK. Гены клеток 2003; 8: 645-54. [Crossref] [PubMed]
- Шварцер В., Витте Ф., Раджаб А. и др. Градиент стабильности белка ROR2 и локализация на мембране придает фенотипы брахидактилии типа B или синдрома Робинова. Hum Mol Genet 2009; 18: 4013-21. [Crossref] [PubMed]
- Sharma M, Castro-Piedras I, Simmons GE, et al. Растрепанный: мастерский проводник сложных сигналов Wnt.Cell Signal 2018; 47: 52-64. [Crossref] [PubMed]
- White JJ, Mazzeu JF, Coban-Akdemir Z, et al. Нарушения передачи сигналов WNT лежат в основе генетической гетерогенности синдрома Робиноу. Am J Hum Genet 2018; 102: 27-43. [Crossref] [PubMed]
- Демургер Ф., Ичкоу А., Мугу-Зерелли С. и др. Новые сведения о корреляции генотип-фенотип мутаций GLI3. Eur J Hum Genet 2015; 23: 92-102. [Crossref] [PubMed]
- Лопес-Риос Х.Многие жизни SHH в развитии и эволюции конечностей. Semin Cell Dev Biol 2016; 49: 116-24. [Crossref] [PubMed]
- Johnston JJ, Olivos-Glander I, Killoran C, et al. Молекулярный и клинический анализ цефалополисиндактилии Грейга и синдромов Паллистера-Холла: надежное прогнозирование фенотипа на основе типа и положения мутаций GLI3. Am J Hum Genet 2005; 76: 609-22. [Crossref] [PubMed]
- Biesecker LG. Синдром цефалополисиндактилии Грейга.Орфанет Ж. Редкие Диском 2008; 3:10. [Crossref] [PubMed]
- Де Мори Р., Романи М., Д’Арриго С. и др. Гипоморфные рецессивные варианты при SUFU нарушают путь Sonic Hedgehog и вызывают синдром Жубера с черепно-лицевыми и скелетными дефектами. Am J Hum Genet 2017; 101: 552-63. [Crossref] [PubMed]
- Zhang Z, Shen L, Law K, et al. Супрессор слитых белков-шаперонов Gli для генерации транскрипционных ответов на передачу сигналов Sonic Hedgehog. Mol Cell Biol 2017; 37: e00421-16.[Crossref] [PubMed]
- Джон А.М., Шварц РА. Синдром базально-клеточного невуса: обновленная информация о генетике и лечении. Br J Dermatol 2016; 174: 68-76. [Crossref] [PubMed]
- Фернисс Д., Леттис Л.А., Тейлор И.Б. и др. Вариант регуляторной последовательности sonic hedgehog (ZRS) связан с трехфаланговым пальцем и нарушает регуляцию экспрессии в развивающейся конечности. Хум Мол Генет 2008; 17: 2417-23. [Crossref] [PubMed]
- Potuijt JW, Baas M, Sukenik-Halevy R, et al.Точечная мутация в pre-ZRS нарушает экспрессию sonic hedgehog в зачатке конечности и приводит к трехфаланговому синдрому полисиндактилии большого пальца. Genet Med 2018; 20: 1405-13. [Crossref] [PubMed]
- Сагай Т., Хосоя М., Мизушина Ю. и др. Устранение дальнодействующего цис-регуляторного модуля вызывает полную потерю специфической для конечностей экспрессии Shh и усечение конечности мыши. Развитие 2005; 132: 797-803. [Crossref] [PubMed]
- Beenken A, Mohammadi M.Семья FGF: биология, патофизиология и терапия. Nat Rev Drug Discov 2009; 8: 235-53. [Crossref] [PubMed]
- Yu K, Herr AB, Waksman G, et al. Потеря специфичности связывания лиганда рецептора 2 фактора роста фибробластов при синдроме Аперта. Proc Natl Acad Sci 2000; 97: 14536-41. [Crossref] [PubMed]
- Слэни С.Ф., Олдридж М., Херст Дж. А. и др. Дифференциальные эффекты мутаций FGFR2 на синдактилию и волчью пасть при синдроме Аперта. Am J Hum Genet 1996; 58: 923-32.[PubMed]
- Shin HR, Bae HS, Kim BS и др. PIN1 — новая терапевтическая мишень краниосиностоза. Hum Mol Genet 2018; 27: 3827-39. [PubMed]
- Milunsky JM, Zhao G, Maher T, et al. Синдром LADD вызывается мутациями FGF10. Clin Genet 2006; 69: 349-54. [Crossref] [PubMed]
- Рохманн Э., Бруннер Х.Г., Кайсерили Х. и др. Мутации в различных компонентах передачи сигналов FGF при синдроме LADD. Нат Генет 2006; 38: 414-7.[Crossref] [PubMed]
- Foldynova-Trantirkova S, Wilcox WR, Krejci P. Шестнадцать лет и подсчет: текущее понимание передачи сигналов рецептора 3 фактора роста фибробластов (FGFR3) при дисплазиях скелета. Хум Мутат 2012; 33: 29-41. [Crossref] [PubMed]
- Кунова, Босакова М., Вареча М., Хампл М. и др. Регуляция функции ресничек посредством передачи сигналов фактора роста фибробластов идентифицирует связанные с FGFR3 расстройства ахондроплазию и танатофорную дисплазию как цилиопатии.Hum Mol Genet 2018; 27: 1093-105. [Crossref] [PubMed]
- Samuels Y, Wang Z, Bardelli A, et al. Высокая частота мутаций гена PIK3CA при раке человека. Наука 2004; 304: 554. [Crossref] [PubMed]
- Lindhurst MJ, Sapp JC, Teer JK и др. Мозаичная активирующая мутация AKT1, связанная с синдромом Протея. N Engl J Med 2011; 365: 611-9. [Crossref] [PubMed]
- Mirzaa G, Timms AE, Conti V и др.Нарушения развития, связанные с PIK3CA, демонстрируют различные классы мутаций с вариабельной экспрессией и распределением в тканях. JCI Insight 2016; 1. [Crossref] [PubMed]
- Albers CA, Paul DS, Schulze H, et al. Сложное наследование низкочастотного регуляторного SNP и редкой нулевой мутации в субъединице комплекса экзон-соединение RBM8A вызывает синдром TAR. Нат Генет 2012; 44: 435-9. [Crossref] [PubMed]
- Wu N, Ming X, Xiao J, et al. Нулевые варианты TBX6 и общий гипоморфный аллель при врожденном сколиозе.N Engl J Med 2015; 372: 341-50. [Crossref] [PubMed]
- Лю Дж., Ву Н., Ян Н. и др. TBX6-связанный врожденный сколиоз (TACS) как клинически различимый подтип врожденного сколиоза: дополнительные доказательства, подтверждающие сложную наследование и модель дозировки гена TBX6. Genet Med 2019. [Epub перед печатью]. [Crossref] [PubMed]
- Лю Дж., Чжоу Ю., Лю С. и др. Сосуществование вариаций числа копий (CNV) и однонуклеотидных полиморфизмов (SNP) в локусе может привести к искаженным расчетам значимости связывания SNP с заболеванием.Hum Genet 2018; 137: 553-67. [Crossref] [PubMed]
- de Leeuw N, Pfundt R, Koolen DA, et al. Недавно выявленный синдром микроделеции, включающий 2p15p16.1: сужение критической области путем добавления еще одного пациента, обнаруженного с помощью сравнительного анализа геномной гибридизации с массивом мозаичных путей. Журнал Мед Генет 2008; 45: 122-4. [Crossref] [PubMed]
- Багери Х., Баддук С., Цяо И и др. Идентификация генов-кандидатов для синдрома микроделеции 2p15p16.1 с использованием клинического, геномного и функционального анализа.JCI Insight 2016; 1: e85461. [Crossref] [PubMed]
- Flöttmann R, Kragesteen BK, Geuer S, et al. Некодирующие вариации числа копий связаны с врожденным пороком развития конечностей. Genet Med 2018; 20: 599-607. [Crossref] [PubMed]
doi: 10.21037 / aoj.2019.06.03
Цитируйте эту статью как: Sun L, Huang Y, Zhao S, Zhong W, Lin M, Guo Y, Yin Y, Wu N, Wu Z, Tian W. Успехи в понимании генетики синдромов, связанных с врожденными аномалиями верхних конечностей.Анн Джойнт 2019; 4:30.
Врожденные пороки конечностей | Бостонская детская больница
Врожденные дефекты конечностей возникают, когда часть или вся верхняя или нижняя конечность не формируются нормально, когда ребенок развивается в матке.
К наиболее частым врожденным дефектам конечностей относятся:
- полное или частичное отсутствие конечности (например, гемимелия малоберцовой кости или врожденное отсутствие большеберцовой кости)
- Неспособность отделить часть конечности (обычно наблюдается на пальцах рук или ног)
- Дублирование (обычно проявляется в виде лишних пальцев рук или ног)
- разрастание (конечность намного больше нормальной конечности)
- подлесок (конечность намного меньше нормальной конечности)
- Синдром околоплодных вод: ранний разрыв амниотического мешка (внутренних оболочек, покрывающих плод в утробе матери и содержащих амнионную жидкость), в результате чего связки могут запутаться в конечностях плода, вызывая иммобилизацию, сужение конечностей, ампутации, и другие уродства.
Насколько распространены врожденные пороки конечностей?
Различные врожденные дефекты конечностей встречаются чаще других. Например, синдром суженной полоски возникает у одного из каждых 10 000–15 000 рождений, в то время как лишние пальцы рук / ног возникают у одного из каждых 1000 родов.
Передаются ли врожденные пороки конечностей по наследству?
Большинство врожденных дефектов конечностей возникают спонтанно без видимой причины. Однако некоторые состояния, такие как лишние пальцы рук / ног, могут быть вызваны наследственным дефектом.
Что вызывает врожденные пороки конечностей?
Хотя мы до сих пор не знаем, что вызывает большинство врожденных дефектов конечностей, существуют определенные факторы, которые могут увеличить риск развития этих состояний, в том числе:
- Заболевания, поражающие ребенка в матке во время развития
- Воздействие химикатов или вирусов на мать во время беременности
- специальные лекарства
Некоторые врожденные дефекты конечностей, например расщелина рук, могут быть частью синдрома, который включает другие симптомы.В этих ситуациях у некоторых пациентов с расщелинами рук также могут быть заячья губа, аномалии стоп, глухота или врожденные состояния, влияющие на сердце и пищеварительную систему.
Как мы лечим врожденные пороки конечностей
Программа ортопедической хирургии кисти и ортопедической верхней конечности и Программа реконструктивной микрохирургии отделения пластической и оральной хирургии вылечили тысячи младенцев и детей с врожденными дефектами конечностей и другими проблемами рук.У нас есть опыт лечения самых разных состояний, от обычных до очень сложных, и мы можем предоставить вашему ребенку квалифицированную диагностику, лечение и уход. Мы также предлагаем преимущества некоторых из самых передовых клинических и научных исследований в мире.
Наш ортопедический центр национально известен как выдающийся центр по уходу за детьми и молодых людей с широким спектром развития, врожденная, нервно-мышечной, связанных со спортом, травматических и посттравматических проблем опорно-двигательного аппарата.
Наше отделение пластической и оральной хирургии является одним из крупнейших и наиболее опытных центров детской пластической и челюстно-лицевой хирургии в мире. Мы обеспечиваем комплексный уход и лечение широкого спектра врожденных и приобретенных заболеваний, включая деформации рук.
Деформации и недостатки конечностей | UF Health, Университет здравоохранения Флориды,
Аномалии скелетных конечностей включают в себя различные проблемы со структурой костей рук или ног (конечностей).Это может быть связано с разницей в длине конечностей или деформациями конечностей, при этом конечность может быть поражена частично или полностью. Между тем, состояние может ухудшаться по мере роста ребенка в зависимости от длины, изгиба или поворота.
Эти аномалии могут присутствовать при рождении, но они могут развиться впоследствии, если у человека есть травма или заболевание, которое влияет на структуру кости.
Независимо от того, является ли деформация или недостаток конечности врожденной или приобретенной, Центр передового опыта Shriners при UF Health, признанный на национальном уровне благодаря своей деятельности в области детской ортопедической помощи, определит тип и степень тяжести, а затем поможет вам найти решение.
Врожденные аномалии
Аномалии, которые присутствуют при рождении, являются врожденными и встречаются редко.
По данным Центров по контролю и профилактике заболеваний (CDC), около 1500 детей в США рождаются с редукцией верхних конечностей (4 из 10 000) и около 750 рождаются с редукцией нижних конечностей (2 из 10 000). Из этой популяции есть неопределенное количество, у которых есть оба типа дефектов.
Хотя точные причины врожденных аномалий неизвестны, существует несколько потенциальных факторов риска:
- Воздействие химических веществ, лекарств или вирусов до рождения
- Употребление табака беременной матерью (CDC указывает, что здесь необходимы дополнительные исследования)
- Синдром врожденного сужения полосы (полосы амниотической ткани запутываются в руках или ногах ребенка до рождения)
- Наличие других аномалий
Приобретенные отклонения от нормы
Приобретены аномалии, возникающие после рождения.
В этих случаях ребенок рождается с нормальными конечностями, но в детстве у него перелом костей, что приводит к аномальному развитию конечностей. Приобретенные аномалии также могут быть вызваны одним из следующих заболеваний, влияющих на структуру кости:
Лечение
Решение проблемы деформации или недостатка конечностей будет зависеть от нескольких факторов. Конечно, оценивается тип и тяжесть аномалии. Между тем, есть соображения, связанные с ребенком, такие как его / ее возраст, а также переносимость определенных лекарств, процедур и методов лечения.
Функционирование и внешний вид — это главное при лечении патологий конечностей. Дело каждого ребенка рассматривается с учетом его индивидуальных потребностей, обычно следуя одному из следующих четырех подходов:
• Ортопедия (шины или скобы)
• Протезирование (протезы)
• Реабилитация
• Хирургия
Чего ожидать при посещении офиса
Младенец с аномалиями конечностей обычно имеет другие симптомы и признаки, которые, взятые вместе, определяют конкретный синдром или состояние или дают ключ к разгадке причины аномалии.Диагноз основывается на семейном анамнезе, истории болезни и тщательном медицинском осмотре.
Вопросы по истории болезни могут включать:
• Есть ли у кого-нибудь в вашей семье аномалии скелета?
• Были ли проблемы во время беременности?
• Какие лекарства или лекарства принимали во время беременности?
• Какие еще симптомы или отклонения присутствуют?
Могут быть выполнены другие тесты, такие как хромосомные исследования, ферментные тесты, рентген и метаболические исследования.
Прогноз (прогноз)
Аномалии конечностей могут представлять ограничения для некоторых детей на протяжении всей жизни, но они могут жить долгой, сильной и продуктивной жизнью при соответствующем лечении и уходе.
Список литературы
Дини В.Ф., Арнольд Дж., Морленд М.С., Уорд В.Т., Дэвис Х.В. Ортопедия. В: Zitelli BJ, McIntire SC, Nowalk AJ, ред. Атлас детской физической диагностики. 6-е изд. Филадельфия, Пенсильвания: Эльзевьер Сондерс; 2012: глава 21.
Herring JA. Скелетные дисплазии. В кн .: Селедка Ж.А., под ред. Детская ортопедия Тачджяна. 5-е изд. Филадельфия, Пенсильвания: Эльзевьер Сондерс; 2014: глава 40.
Moore KL, Persaud V, Torchia MG. Система скелета. В: Мур KL, Persaud V, Torchia MG, ред. Развивающийся человек: клинически ориентированная эмбриология.10-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2016: глава 14.
Дата пересмотра
13.08.2018
Отзыв от
Доктор Лорел Блейкмор, доктор медицины, начальник отделения детской ортопедии
.