Врожденный буллезный эпидермолиз > Клинические протоколы МЗ РК
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [1-4,7,8,10,12,17-19,21]
Диагностические критерии
Жалобы:
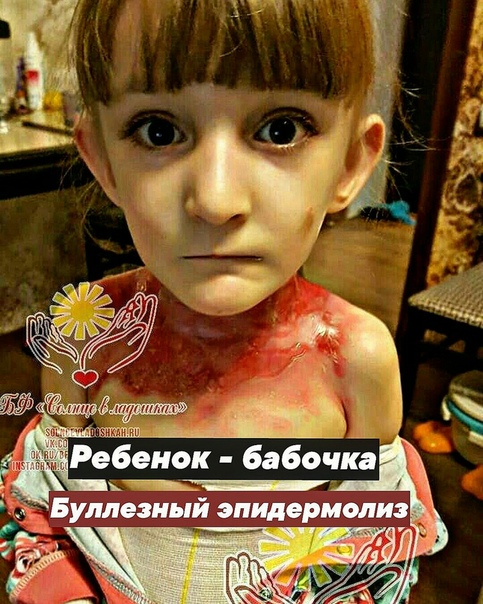
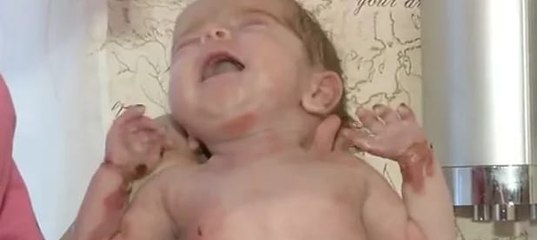
· полушаровидные напряженные пузыри на коже туловища и конечностей, а также в ротовой полости;
· длительно заживающие эрозии;
· ранимость кожных покровов, чувствительность к любым механическим воздействиям;
· уплотнение, деформация или потеря ногтей пальцев рук и ног, сращение;
· облысение;
· зуд в области пузырей.
Анамнез:
· наследственность: наличие буллёзного эпидермолиза у родственников 1-й и 2-й степени родства;
· сезонность заболевания: при простом типе буллёзного эпидермолиза наблюдается четко выраженная сезонность заболевания – обострение и ухудшение состояния кожных покровов в летний период;
· аллергоанамнез при буллёзном эпидермолизе не отягощен;
· наличие сопутствующих заболеваний и осложнений (таблица 2).
*При буллёзном эпидермолизе наблюдаются множественные внекожные проявления: микростомия, анкилоглоссия, аномалии эмали и дисплазия зубов, кариес, поражение органов зрения, патология ЛОР-органов, поражение ЖКТ и сердечно-сосудистой системы, деформации и контрактуры опорно-двигательной системы, поражение мочевыводящей системы.
Физикальное обследование:
· полушаровидные напряженные пузыри, наполненные прозрачной бесцветной жидкостью, на коже и слизистых оболочках в ротовой полости, в пищеводе и на верхних дыхательных путях, затрудняющие дыхание и питание пациента;
· длительно заживающие эрозии;
· ранимость кожных покровов, чувствительность к механическим и температурным воздействиям;
· ониходистрофия или отсутствие ногтевых пластинок;
· алопеция;
· ладонно-подошвенная кератодермия;
· милиумы;
· пигментные невусы.
Пренатальная диагностика – лучший способ профилактики тяжелых наследственных заболеваний. Она возможна только при планировании беременности задолго до ее наступления. Медико-генетическое консультирование семьи проводится с целью оценки риска появления больного ребенка в семье, информирования семьи о риске развития наследственного заболевания, о возможных диагностических и терапевтических методах. Показания для консультирования – предыдущее рождение ребенка с БЭ, наличие заболевания у одного из родителей, установленное или подозреваемое заболевание в семье. Генетический анализ крови и кожи больного позволяет уточнить тип и подтип ВБЭ, а также обнаружить мутацию соответствующего гена. При наступлении беременности в сроки 10–12 недель проводят биопсию ворсин хориона, в которых ведется поиск уже известной мутации. Быстрое получение результатов (в течение 3–4 дней после взятия материала) позволяет принять решение о прерывании беременности своевременно.
Она возможна только при планировании беременности задолго до ее наступления. Медико-генетическое консультирование семьи проводится с целью оценки риска появления больного ребенка в семье, информирования семьи о риске развития наследственного заболевания, о возможных диагностических и терапевтических методах. Показания для консультирования – предыдущее рождение ребенка с БЭ, наличие заболевания у одного из родителей, установленное или подозреваемое заболевание в семье. Генетический анализ крови и кожи больного позволяет уточнить тип и подтип ВБЭ, а также обнаружить мутацию соответствующего гена. При наступлении беременности в сроки 10–12 недель проводят биопсию ворсин хориона, в которых ведется поиск уже известной мутации. Быстрое получение результатов (в течение 3–4 дней после взятия материала) позволяет принять решение о прерывании беременности своевременно.
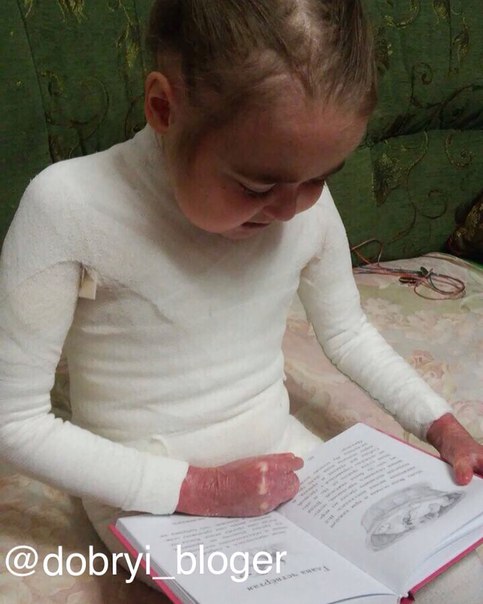
У пациента с БЭ профилактические меры в отношении появления пузырей на коже и слизистых оболочках включают ограничение возможности травмирования кожи (одежда, диета, особенности ухода, неадгезивные повязки, наружные средства, уход за полостью рта)
Профилактика развития осложнений – диспансеризация, периодический контроль лабораторных показателей для выявления и контроля анемии, полный осмотр пациентов с целью раннего выявления злокачественных опухолей кожи, своевременное лечение зубов.
Таблица 2. Основные кожные и внекожные сопутствующие заболевания и осложнения БЭ.
| Типы и под типы БЭ | Кожные осложнения | Тактика | |
| РДБЭ-АС, РДБЭ-нАС, Иногда- ПоБЭ-нХ и ДДБЭ | Плоскоклеточный рак | Агрессивный, часто первично-множественный, часто устойчив к химио- и лучевой терапии | Тщательный регулярный осмотр подозрительных незаживающих ран, раннее оперативное вмешательство |
| Все подтипы, особенно ПоБЭ-нХ | Пигментные невусы | Большие пигментные невусы от светло-бежевого до темно-коричневого и черного цвета; с возрастом светлеют; макро- и микроскопически напоминают меланому, но не перерождаются внее | Регулярные осмотры; при необходимости — биопсия (в том числе множественная) подозрительных участков кожи |
| РДБЭ | Меланома | Повышенный риск развития у детей, даже на внешне нормальных участках кожи | Регулярные осмотры; при необходимости — биопсия (в том числе множественная) подозрительных участков кожи |
| РДБЭ-АС, редко -ПоБЭ, ДДБЭ и ПБЭ | Псевдосиндактилия | Срастание пальцев на руках и ногах и деформация кистей и стоп по типу «варежки», атрофия пальцев, контрактуры большого пальца, межфаланговых и пястно-фаланговых суставов, ограничение или полная утрата функции кистей и стоп, частые послеоперационные рецидивы (хирургическое вмешательство требуется в среднем раз в 5лет) | Хирургическое вмешательство, физиотерапия, лечебная физкультура, использование напальчников или ортезов |
| Внекожные проявления и осложнения | |||
| ПоБЭ, ДБЭ | Полость рта, желудочно-кишечный тракт | Гипоплазия эмали (ПоБЭ), дисплазия зубов, тяжелый кариес, микростомия (ДБЭ), ранняя потеря зубов; стриктуры пищевода и заднего прохода (ДБЭ), дисфагия (ДБЭ), хронический запор, болезненная дефекация, задержка развития | Тщательная гигиена полости рта, ортодонтическое лечение, повторные дилатации пищевода, консультации диетолога, применение слабительных, гастростомия |
| ПоБЭ, редко РДБЭ | Дыхательные пути | Отек слизистых, пузыри, эрозии, рубцевание, охриплость голоса, стеноз гортани, острая обструкция дыхательных путей | Трахеостомия, терапия антибиотиками и глюкокортикоидами |
| ПоБЭ, РДБЭ | Глаза | Эрозии роговицы, рубцевание роговицы, симблефарон (сращение одного или обоих век с глазным яблоком), недостаточная выработка слезной жидкости, блефарит (воспаление краев век), обструкция слезных канальцев, ухудшение зрения | Обезболивающие и увлажняющие глазные капли, хирургическое вмешательство |
| ПоБЭ, ДБЭ | Мочевая система | Дизурия, гематурия, стеноз и обструкция мочевых путей, пузырно-мочеточниковый рефлюкс, гидронефроз, почечная гипертония, уросепсис, гломерулонефрит, амилоидоз, почечная недостаточность | Регулярное измерение артериального давления и анализ мочи; при необходимости катетеризация, цистоскопия, дилатация мочеиспускательного канала, меатотомия, гемодиализ, перитонеальный диализ |
| Все тяжелые формы БЭ | Метаболизм и общее состояние | Дефицит питательных веществ и белков в связи с обнажением больших участков дермы, катаболический метаболизм с увеличением потребности в калориях, отставание в росте, медленное заживление ран, рецидивирующие инфекции, хроническая анемия, плохое самочувствие | Лечебное питание, гастростомия |
Лабораторные исследования [2-4,8,21]:
Перечень основных лабораторных исследований:
1. Общий анализ крови – снижение уровня гемоглобина.
Общий анализ крови – снижение уровня гемоглобина.
2. Общий анализ мочи – повышение уровня лейкоцитов, наличие солей, глюкозы.
3. Биохимический анализ крови – изменение уровня мочевины, ферритина, общей железо-связывающей способности, сывороточного железа, электролитов, витаминов (А, Е, D).
4. Генетический анализ (молекулярная или ДНК диагностика) – обнаружениемутаций в генах, ассоциированных с буллезным эпидермолизом.
Инструментальные исследования:
1. Гистологическое исследование биоптата кожи: определение субэпидермальной полости.
2. Трансмиссионная электронная микроскопия: Простой БЭ – расслоение на уровне базального слоя эпидермиса, под ядрами клеток или выше, возможно скопление кератиновыхфиламентов. При пограничном БЭ – расслоение кожи на уровне светлой пластинки базальной мембраны, малые размеры и/или количество гемидесмосом. При дистрофическом БЭ – расслоение кожи непосредственно под плотной пластинкой базальной мембраны, малое количество или полное отсутствие крепящих фибрилл.
3. Реакция непрямой иммунофлюоресценции (нРИФ) – экспрессия структурных белков кожи в месте дермоэпидермального сочленения (присутствие, снижение или отсутствие), уровень образования пузыря (внутриэпидермально, внутри светлой пластинки базальной мембраны, под плотной пластинкой базальной мембраны).
Показания для консультации специалистов:
Для диагностики ассоциированных с ВБЭ заболеваний и внекожных поражений, а также осложнений, могут потребоваться консультации смежных специалистов:
· консультация генетика – для верификации диагноза и прогнозирования вероятности заболевания при повторных беременностях;
· консультация гастроэнтеролога – при наличии дисфагии и стриктур пищевода, рефлюкс-эзофагита, язвенной болезни желудка и двенадцатой перстной кишки, хронической диареи или запоров, ректальных кровотечений, трещин заднего прохода и другие заболевания ЖКТ;
· консультация стоматолога – при микростомии, анкилоглоссии, пузырях и эрозиях слизистой десен, аномалии эмали, дисплазии зубов, кариесе и др. ;
;
· консультация педиатра, неонатолога – при наличии пневмонии, анемии, иммунодефицитных состояний, снижение массы тела и других состояний;
· консультация хирурга, травматолога – при наличии псевдосиндактилии, деформации по типу «варежки», контрактур, скручивании дистальных фаланг, остеопорозе и остеопении, дилатация пищевода;
· консультация оториноларинголога – при наличии отита среднего уха, стеноза наружного слухового прохода, тугоухости, утолщения и рубцевания голосовых складок, пузырей и изъязвления трахеи и гортани и др.;
· консультация офтальмолога – при наличии пузырей и эрозий роговицы, рубцевания роговицы, эктропиона, обструкции слизистых канальцев и др.;
· консультация уролога-нефролога – при наличии гидронефроза, гломерулонефрита, пиелонефрита, нефропатии, стриктур и дивертикулов мочеиспускательного канала, рубцевания головки полового члена, цистите, уросепсисе, почечной недостаточности и др.;
· консультация ортопеда – для адекватного реабилитационного лечения для выполнения эффективную разработку стойких контрактур суставов, что уменьшает количество оперативных вмешательств на верхних и нижних конечностях;
· консультация нутрициолога – для оценки нутритивного статуса у детей и организация нутритивной поддержки.
Диагностический алгоритм[2,7,17-19]:
Алгоритм мониторинга пациентов с ВБЭ.
Диагностический алгоритм на уровне родильного дома
Диагностический алгоритм для определения типа и подтипа ВБЭ
Приобретенный буллезный эпидермолиз
Заболевание встречается в возрасте 40 — 66 лет (средний возраст 47 лет),чаще у женщин (2:1 в соотношении с мужчинами).Описаны единичные случаи у детей.Распространенность 0,
Этиология и патогенез точно неизвестны. Аутоантигеном является VII тип коллагена.
Циркулирующие IgG-аутоантитела направлены,
молекулы.
Приобретенный буллезный эпидермолиз может быть индуцирован лекарственными препаратами (чаще антибиотиками),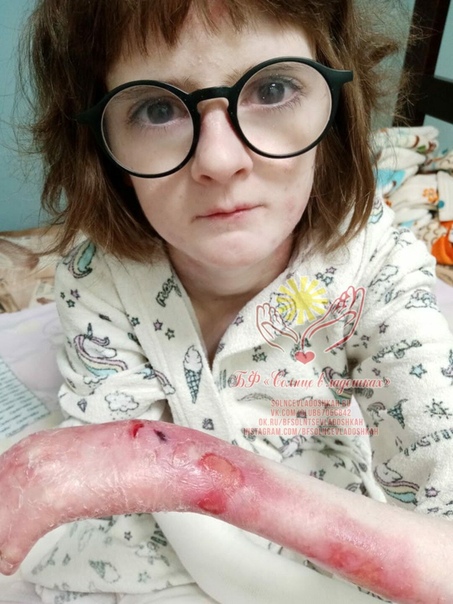 Сообщается об ассоциациях с системными заболеваниями,
Сообщается об ассоциациях с системными заболеваниями,
Классификация
- Невоспалительный (классический) приобретенный буллезный эпидермолиз
- Воспалительный приобретенный буллезный эпидермолиз (генерализованная форма)
- по типу буллезного пемфигоида
- протекающий по типу линейного IgA зависимого буллезного дерматоза
- Приобретенный буллезный эпидермолиз слизистых
- протекающий по типу рубцующего пемфигоида
- Приобретенный буллезный эпидермолиз по типу пемфигоида Бранстинга — Перри
Течение приобретенного буллезного эпидермолиза
Заболевание характеризуется хроническим течением с рецидивами и частичными ремиссиями. Могут встречаться одновременно классический и воспалительный варианты. В 30% случаев описаны вовлечения слизистых с тяжелым течением.Без лечения возможно развитие осложнений :выраженной болезненности,
Могут встречаться одновременно классический и воспалительный варианты. В 30% случаев описаны вовлечения слизистых с тяжелым течением.Без лечения возможно развитие осложнений :выраженной болезненности,
Невоспалительный приобретенный буллезный эпидермолиз
Известен также как классический вариант приобретенного буллезного эпидермолиза.Является механо-буллезным дерматозом.Характеризуется упругими напряженными пузырями на фоне внешне неизмененной кожи с прозрачным или геморрагическим содержимым и эрозиями на месте травм, Заболевание носит локализованный или распространенный характер.
|
Воспалительный приобретенный буллезный эпидермолиз по типу буллезного пемфигоида
Высыпания представлены множественными везикулами и пузырями, |
Воспалительный приобретенный буллезный эпидермолиз по типу линейного IgA зависимого буллезного дерматоза
Характеризуется появлением множественных везикул и (или) пузырей часто с кольцевидным расположенных везикул. |
 Эта клиническая форма может напоминать герпетиформный дерматит Дюринга.Рубцевание и формирование милиумов минимальное.
Эта клиническая форма может напоминать герпетиформный дерматит Дюринга.Рубцевание и формирование милиумов минимальное.Приобретенный буллезный эпидермолиз по типу рубцующего пемфигоида
Известен также как приобретенный буллезный эпидермолиз слизистых.Высыпания преимущественно локализуются на конъюнктиве глаз и/или слизистых оболочках полости рта, |
Приобретенный буллезный эпидермолиз по типу пемфигоида Бранстинга — Перри
Характерны субэпидермальные пузыри, |
Диагностические критерии Н. Roenigk (1971)
- спонтанное или в результате травмы появление пузырей
- начало заболевания у взрослых
- отсутствие семейного анамнеза
- исключение других буллезных дерматозов
Гистологическое исследование
При классическом (невоспалительном) варианте обнаруживается субэпидермальная полость,
При воспалительном варианте субэпидермальная полость может содержать нейтрофильные и единичные эозинофильные лейкоциты,
также могут образовывать микроабсцессы в сосочках дермы
В стадии разрешения могут обнаруживаться признаки рубцевания дермы и наличие мелких кист (милиумов), Кисты соединяются с волосяным фолликулом пушкового волоса или протоком эккринной потовой железы.
Кисты соединяются с волосяным фолликулом пушкового волоса или протоком эккринной потовой железы.
При прямой РИФ выявляются линейные отложения IgG (в меньшинстве случаев IgA и IgM) и СЗb,
в зоне базальной мембраны. Циркулирующие IgG — и IgA-аутоантитела к зоне базальной мембраны обнаруживаются в 50 и 20% случаев соответственно. При диагностике методами иммуноблоттинга и ИФА используются рекомбинантные NC1 домены.
Лазерная конфокальная микроскопия
Определение in vivo отложений IgG при отсутствии циркулирующих аутоантител
Буллезный эпидермолиз код мкб
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Буллезный эпидермолиз.
Буллезный эпидермолиз
Описание
Буллезный эпидермолиз. Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Дополнительные факты
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Буллезный эпидермолиз
Причины
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
• Простой буллезный эпидермолиз. Имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера — Коккейна, Кёбнера, Доулинга — Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
• Пограничный буллезный эпидермолиз. Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
• Дистрофический буллезный эпидермолиз. Имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
• Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Диагностика
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза.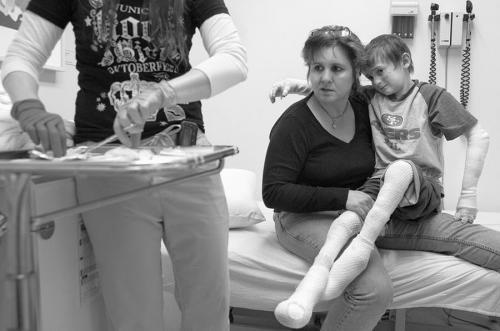 При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальная диагностика
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий.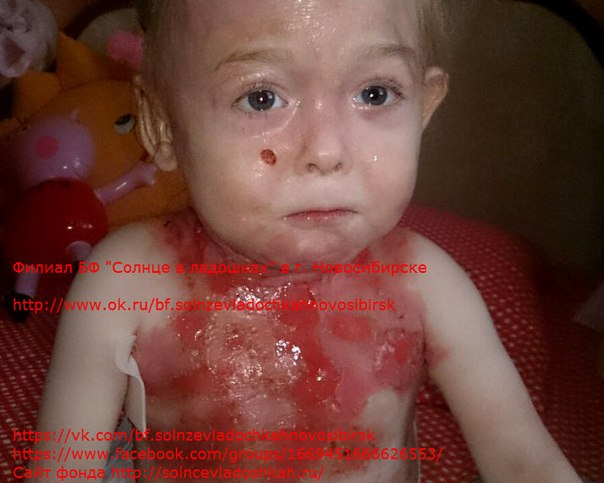 В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.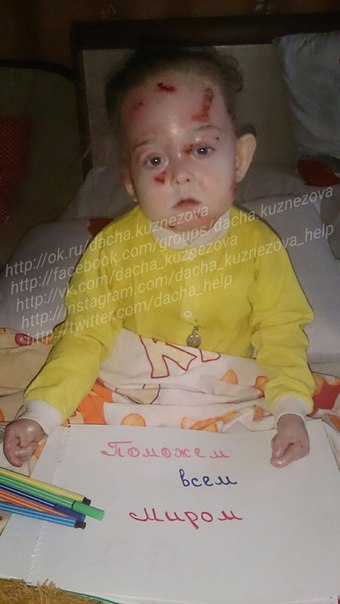
Прогноз
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Q81 Буллезный эпидермолиз.
Q81 Буллезный эпидермолиз
Описание
Буллезный эпидермолиз. Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Дополнительные факты
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Q81 Буллезный эпидермолиз
Причины
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других.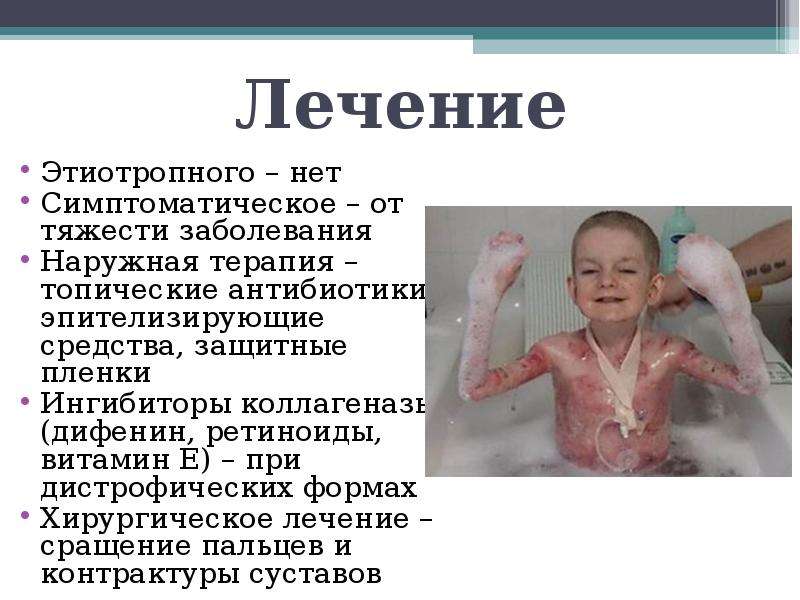 Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
• Простой буллезный эпидермолиз. Имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера — Коккейна, Кёбнера, Доулинга — Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
• Пограничный буллезный эпидермолиз. Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
• Дистрофический буллезный эпидермолиз. Имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
• Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Диагностика
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза.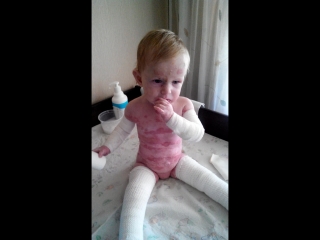 Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальная диагностика
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий.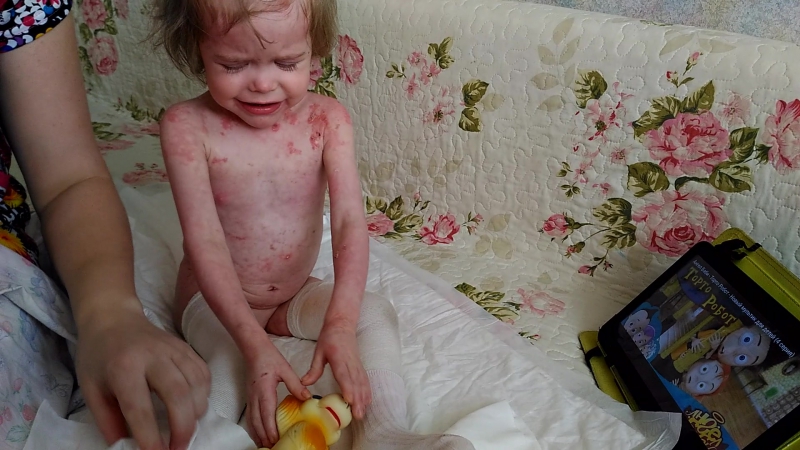 В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.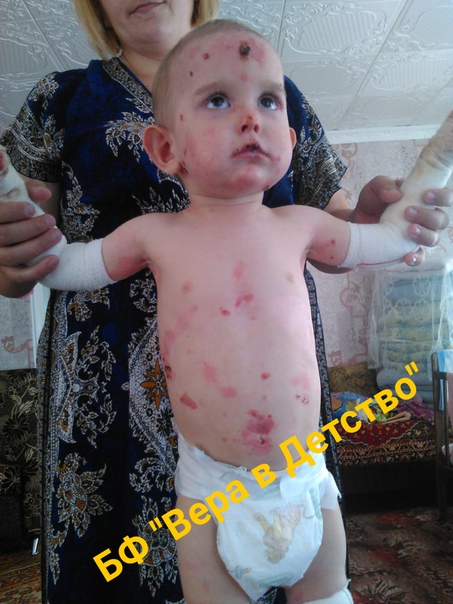
Прогноз
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Врожденный буллезный эпидермолиз код по мкб
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Буллезный эпидермолиз.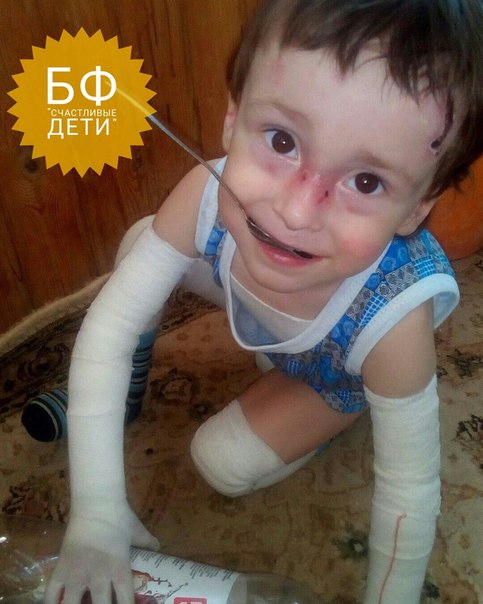
Буллезный эпидермолиз
Описание
Буллезный эпидермолиз. Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Дополнительные факты
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Буллезный эпидермолиз
Причины
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
• Простой буллезный эпидермолиз. Имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера — Коккейна, Кёбнера, Доулинга — Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
• Пограничный буллезный эпидермолиз. Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
• Дистрофический буллезный эпидермолиз. Имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
• Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Диагностика
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальная диагностика
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Q81 Буллезный эпидермолиз.
Q81 Буллезный эпидермолиз
Описание
Буллезный эпидермолиз. Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Дополнительные факты
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Q81 Буллезный эпидермолиз
Причины
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
• Простой буллезный эпидермолиз. Имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера — Коккейна, Кёбнера, Доулинга — Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
• Пограничный буллезный эпидермолиз. Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
• Дистрофический буллезный эпидермолиз. Имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
• Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Диагностика
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальная диагностика
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Эпидермолиз код по мкб
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Буллезный эпидермолиз.
Буллезный эпидермолиз
Описание
Буллезный эпидермолиз. Группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Дополнительные факты
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Буллезный эпидермолиз
Причины
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
• Простой буллезный эпидермолиз. Имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера — Коккейна, Кёбнера, Доулинга — Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
• Пограничный буллезный эпидермолиз. Делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
• Дистрофический буллезный эпидермолиз. Имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
• Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Диагностика
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальная диагностика
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: Q81.2
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q81 Буллезный эпидермолиз
Определение и общие сведения[править]
Дистрофический буллезный эпидермолиз
Синонимы: дермолитический буллезный эпидермолиз
Дистрофический буллезный эпидермолиз представляет собой форму наследственного буллезного эпидермолиза, характеризующейся хрупкостью кожи и слизистых оболочек, приводящей к образованию волдырей и эрозий на коже, которые локализуются ниже твердой пластинки (lamina densa) базальной мембраны и которые разрешаются с образованием выраженных рубцов и милиарных кист.
Дистрофический буллезный эпидермолиз включает в себя десять подтипов, три наиболее распространенных из них: генерализованный доминантный дистрофический буллезный эпидермолиз (вариант Кокейна-Турена и Пазини), тяжелый генерализованный рецессивный дистрофический буллезный эпидермолиз (вариант Аллопо-Сименса) и генерализованный рецессивный-другой дистрофический буллезный эпидермолиз.
Дистрофический буллезный эпидермолиз — вторая по распространенности форма буллезного эпидермолиза после простого буллезного эпидермолиза. Общая распространенность варьируется от 1/49 000 в Шотландии до 1/420 000 в Соединенных Штатах.
Этиология и патогенез[править]
Дистрофический буллезный эпидермолиз вызван мутациями в гене COL7A1 (3p21.31), кодирующем коллаген VII типа. Мутации этого гена изменяют функцию, уменьшают или нарушают выработку коллагена VII, что ухудшает его сборку в закрепляющие фибриллы, которые фиксируют базальную мембрану к подлежащей дерме.
Передача является аутосомно-доминантной или аутосомно-рецессивной. Спорадические случаи могут возникать из-за мутаций de novo.
Клинические проявления[править]
Тяжесть клинической картины варьируется в широких пределах. Манифестация обычно происходит при рождении, но также наблюдается отсроченной начало — в младенчестве, детстве или подростковом возрасте. Поражения кожи, возникающие спонтанно или в ответ на трение, могут иметь генерализованное или локализованное распределение, особенно на руках, на ногах или в претибеальных областях. Разрешение волдырей сопровождается образованием атрофических или, реже, гипертрофических рубцов, бело-папулоидных поражений, милиарных кист и дистрофией ногтей. Выраженное рубцевание может привести к образованию рубцовых контрактур конечностей — «деформация по типу рукавиц», типичная для генерализованного рецессивного-другого дистрофического буллезного эпидермолиза. Вовлечение слизистых оболочек происходит часто и чаще всего проявляется поражением полости рта и стриктурами пищевода. Глаза и мочеполовой тракт также затрагиваться. Участие кожи и слизистых может приводить к развитию анемии, дефициту железа и задержке роста. Пациенты с дистрофическим буллезным эпидермолизом подвержены более высокому риску возникновения плоскоклеточных карцином.
Эпидермолиз буллезный дистрофический: Диагностика[править]
Диагноз дистрофического буллезного эпидермолиза подозревается при клиническом обследовании и подтверждается биопсией образцов кожи с помощью иммунофлуоресцентного антигенного картирования и / или просвечивающей электронной микроскопии (обнаруживается плоскость сдвига пузыря под твердой пластинкой базальной мембраны) и скринингом мутаций COL7A1.
Пренатальная генетическая диагностика может быть выполнена.
Дифференциальный диагноз[править]
Дифференциальный диагноз дистрофического буллезного эпидермолиза включает другие формы ЭБ. В течение неонатального периода и в младенчестве он может также включать врожденную аплазию кожи, неонатальный герпес, эпидермолитический ихтиоз, буллезное импетиго, стафилококковой синдром обожженной кожи, линейный IgA дерматит, буллезный пемфигоид, неонатальный пемфигус и pemphigoid gestationis. Для редких форм дистрофического буллезного эпидермолиза с поздним началом дифференциальная диагностика включает приобретенные нарушения кожи, такие как плоский лишай.
Эпидермолиз буллезный дистрофический: Лечение[править]
Лечение профилактическое: защита кожи и тщательный уход за раной уменьшают образование пузырей, рубцевание и предотвращают вторичную инфекцию. Деформации рук можно купировать хирургическим путем, но они имеют склонность к рецидивированию. Стриктуры пищевода требуют проведения баллонной дилатации. Переливание крови, препараты железа и назначение эритропоэтина устраняют анемию и дефицит железа. Требуется регулярное наблюдение за развитием плоскоклеточных карцином, лечение которых является хирургическим.
Прогноз
Прогноз зависит от подтипа патологии. Пациенты с генерализованным доминантным дистрофическим буллезным эпидермолизом обычно имеют нормальную продолжительность жизни. Пациенты с тяжелым генерализованным рецессивным типом имеют высокий риск смертности в основном из-за разсития метастатической плоскоклеточной карциномы, реже от хронической почечной недостаточности и дилатационной кардиомиопатии.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Рубрика МКБ-10: L12.3
МКБ-10 / L00-L99 КЛАСС XII Болезни кожи и подкожной клетчатки / L10-L14 Буллезные нарушения / L12 Пемфигоид
Определение и общие сведения[править]
Приобретенный буллезный эпидермолиз
Синонимы: Epidermolysis bullosa acquisita
Приобретенный буллезный эпидермолиз является субэпидермальным буллезным дерматозом аутоиммунного происхождения, который получил свое название в результате его сходства с наследственными формами буллезного эпидермолиза, в первую очередь дистрофического.
Распространенность неизвестна, заболеваемость оценивается 1 на 96200 новых случаев в год.
Этиология и патогенез[править]
Один из основных патогенетических факторов приобретенного буллезного эпидермолиза — циркулирующие аутоантитела к коллагену VII типа в сыворотке крови больных. Антитела не являются специфичными для эпидермиса и взаимодействуют с антигенами базальной мембраны слизистой оболочки полости рта, пищевода и влагалища, однако не реагируют с базальной мембраной кровеносных сосудов, печени или почек. Антитела больных приобретенным буллезным эпидермолизом относятся преимущественно к иммуноглобулину класса G, титр которых может достигать 1:5120. Непрямым методом иммунофлюоресценции обнаруживают циркулирующие аутоантитела в зоне базальной мембраны эпидермиса
и дермо-эпидермальном соединении. Они выявляются в lamina densa и sublamina densa (электронно-микроскопические данные). Антигены локализуются в месте первичных деструктивных изменений. Образование подэпидермального пузыря начинается с расщепления дермо-эпидермального соединения под lamina densa или под комплексом lamina densa — якорные фибриллы в верхних отделах дермы. Непрямым методом иммунофлюоресценции фиксированные иммуноглобулины и начальные компоненты комплемента обнаруживают в зоне базальной мембраны эпидермиса и на дне подэпидермального пузыря — дифференциальнодиагностический признак приобретенного буллезного эпидермолиза.
Клинические проявления[править]
Заболевание проявляется в двух клинических формах: классической и воспалительной формах. При классической форме манифестация происходит в зрелом возрасте, буллы могут быть мягкими, напряженными или геморрагическими и они располагаются на здоровой в остальном коже. Поражения кожи, как правило, вызваны незначительными травмами и в основном локализованы на участках тела, которые легко подвергаются внешнему воздействию. Вовлечение слизистых оболочек, волос и ногтей происходит часто.
Воспалительная форма была признана совсем недавно и напоминает буллезный пемфигоид с буллами, которые возникают на эритематозных поражениях кожи, бляшках без буллезных высыпаний и диффузных поражениях, которые не ограничиваются участками тела, склонных к травмоатизации. Заболевание проявляется в детстве.
Приобретенный буллезный эпидермолиз в 10-50% случаев отмечается в сочетании с другим заболеваниеми, в первую очередь болезнью Крона, геморрагическим ректоколитом или сахарный диабетом. Нозологическая граница между приобретенным буллезным эпидермолизом и буллезной системной красной волчанкой остается в стадии обсуждения.
Приобретенный буллезный эпидермолиз: Диагностика[править]
Диагноз основывается на результатах гистологического анализа, косвенных или прямых иммунофлюоресцентных исследованиях, иммуноблоттинге и иммунной электронной микроскопии.
Дифференциальный диагноз[править]
Дифференциальный диагноз должен включать в себя другие субэпидермальные, аутоиммунные заболевания, сопровождаемые формирование буллезных высыпаний.
Приобретенный буллезный эпидермолиз: Лечение[править]
Первая линия терапия — назначение дапсона или сульфасалазина. Иммунодепресивная терапия (например, циклоспорином) может потребоваться в тяжелых случаях. Обнадеживающие результаты были получены при использовании других подходов — внутривенного иммуноглобулина, экстракорпоральной фотохимиотерапии и использовании ритуксимаба.
Прогноз
Приобретенный буллезный эпидермолиз является хроническим заболеванием, которое разрешается медленно и приводит к образованию дистрофических рубцов и милий. Вовлечение слизистых оболочек ассоциируется с более тяжелым заболеванием, которое может приводить к ухудшению функционального или даже жизненного прогноз.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова — М. : ГЭОТАР-Медиа, 2013. — https://www.rosmedlib.ru/book/ISBN9785970427101.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Буллезный эпидермолиз — Epidermolysis bullosa
| Буллезный эпидермолиз | |
|---|---|
| Другие имена | Дети-бабочки |
| Пятилетний мальчик с буллезным эпидермолизом. | |
| Специальность | Дерматология |
| Симптомы | Болезненные волдыри на коже |
| Осложнения | Сужение пищевода , плоскоклеточный рак кожи , ампутации |
| Обычное начало | При рождении |
| Продолжительность | Часто на всю жизнь |
| Типы | Врожденный буллезный эпидермолиз симплекс , дистрофический буллезный эпидермолиз , узловой буллезный эпидермолиз , синдром Киндлера |
| Причины | Генетический |
| Диагностический метод | Биопсия кожи , генетическое тестирование |
| Дифференциальный диагноз | Буллезный пемфигоид , вульгарная пузырчатка , волдыри от трения, укусы насекомых |
| лечение | Уход за ранами , обезболивание, борьба с инфекциями, нутритивная поддержка |
| Частота | c. 500 000 |
Буллезный эпидермолиз ( БЭ ) — это группа редких генетических состояний, которые приводят к легкому образованию пузырей на коже и слизистых оболочках . Волдыри возникают при незначительной травме или трении и болезненны. Его тяжесть может варьироваться от легкой до смертельной. У пациентов с легкой формой заболевания симптомы могут не развиваться, пока они не начнут ползать или ходить. Осложнения могут включать сужение пищевода , плоскоклеточный рак кожи и необходимость ампутации .
EB возникает из-за мутации по крайней мере в одном из 16 различных генов. Некоторые типы являются аутосомно-доминантными, а другие — аутосомно-рецессивными . Основным механизмом является дефект прикрепления между слоями кожи или внутри них. Есть четыре основных типа: буллезный эпидермолиз симплекс , дистрофический буллезный эпидермолиз , узловой буллезный эпидермолиз , и синдром Kindler . Диагноз подозревается на основании симптомов и подтверждается биопсией кожи или генетическим тестированием .
От этого состояния нет лекарства. Лечение включает уход за раной , обезболивание, борьбу с инфекциями , нутритивную поддержку, а также профилактику и лечение осложнений. Во всем мире пострадали около полумиллиона человек. Он одинаково часто встречается у мужчин и женщин. Пострадавшие дети могут подвергаться издевательствам со стороны других детей или получать неуместные комментарии со стороны взрослых.
Классификация
Буллезный эпидермолиз относится к группе заболеваний, которые связаны с образованием волдырей после незначительной травмы. В этом состоянии было идентифицировано более 300 мутаций. Они были разделены на следующие типы:
Простой буллезный эпидермолиз
Простой буллезный эпидермолиз — это форма буллезного эпидермолиза, при которой на месте натирания появляются волдыри. Обычно он поражает руки и ноги и обычно наследуется по аутосомно-доминантному типу, влияя на гены кератина KRT5 и KRT14 . Следовательно, происходит сбой кератинизации, что влияет на целостность и способность кожи противостоять механическим воздействиям.
Буллезный узловой эпидермолиз
Буллезный узловой эпидермолиз — наследственное заболевание, поражающее ламинин и коллаген . Это заболевание характеризуется образованием волдырей в просветной пластинке зоны базальной мембраны и наследуется по аутосомно-рецессивному типу. Он также проявляется в виде волдырей на месте трения, особенно на руках и ногах, и имеет варианты, которые могут возникнуть у детей и взрослых. По оценкам, этой формой буллезного эпидемолиза страдает менее одного человека на миллион человек.
Буллезный дистрофический эпидермолиз
Буллезный дистрофический эпидермолиз — это наследственный вариант, поражающий кожу и другие органы. Дистрофический буллезный эпидермолиз вызывается генетическими дефектами (или мутациями ) в гене COL7A1 человека, кодирующем белок коллагена VII типа (коллаген VII). Мутации, вызывающие DEB, могут быть аутосомно-доминантными или аутосомно-рецессивными. Эпидермис буллезный pruriginosa и Albopapuloid буллезный эпидермолиз (болезнь Pasini в) редкие подвиды этого заболевания.
Другие генетические
| OMIM | имя | Locus | Ген |
|---|---|---|---|
| 609638 | буллезный эпидермолиз, летальный акантолитик | 6п24 | DSP |
Приобретенный буллезный эпидермолиз
Акральный пилинг
Патофизиология
Кожа человека состоит из двух слоев: внешнего слоя, называемого эпидермисом, и нижнего слоя, называемого дермисом . У людей со здоровой кожей между этими двумя слоями есть белковые якоря, которые не позволяют им двигаться независимо друг от друга (срезание). У людей, рожденных с БЭ, два слоя кожи лишены протеиновых якорей, которые удерживают их вместе, что приводит к чрезвычайно хрупкой коже — даже незначительное механическое трение (например, трение или давление) или травма разделят слои кожи и образуют волдыри и болезненные язвы. . Больные БЭ сравнивали язвы с ожогами третьей степени. Кроме того, как осложнение хронического повреждения кожи, люди, страдающие БЭ, имеют повышенный риск злокачественных новообразований (рака) кожи.
Диагностика
Буллезный эпидермолиз может быть диагностирован либо с помощью биопсии кожи (перфорации) на краю раны с иммунофлуоресцентным картированием, либо с помощью образца крови и генетического тестирования.
лечение
Лечение буллезного эпидермолиза трансплантацией стволовых клеток, модифицированных ламинином5
Недавние исследования были сосредоточены на изменении смеси кератинов, производимых в коже. Известно 54 гена кератина, 28 из которых относятся к генам промежуточных филаментов I типа и 26 — к типу II, которые работают как гетеродимеры . Многие из этих генов имеют существенное структурное и функциональное сходство, но они специализируются на типах клеток и / или условиях, в которых они обычно образуются. Если бы баланс продукции можно было сместить от мутировавшего, дисфункционального гена кератина к интактному гену кератина, симптомы можно было бы уменьшить. Например, сульфорафан , соединение, обнаруженное в брокколи , снижает образование пузырей на модели мышей до такой степени, что пораженные детеныши не могут быть идентифицированы визуально при введении беременным мышам (5 мкмоль / день = 0,9 мг) и местному применению. новорожденные (1 мкмоль / день = 0,2 мг в масле жожоба ).
По состоянию на 2008 год в клинических исследованиях Университета Миннесоты изучалась аллогенная трансплантация костного мозга при РЗ и узловой БЭ для лечения двухлетнего ребенка, который является одним из двух братьев с БЭ. Вторая трансплантация также была сделана старшему брату ребенка, а третья трансплантация запланирована для ребенка из Калифорнии. Планируется клиническое исследование с участием 30 человек. Однако подавление иммунитета, необходимое для трансплантации костного мозга, вызывает риск серьезных инфекций с крупномасштабными волдырями и эрозиями кожи. Действительно, по меньшей мере четыре человека умерли в ходе подготовки или проведения трансплантации костного мозга от буллезного эпидермолиза из лишь небольшой группы пациентов, пролеченных на данный момент. Механизм действия этой терапии неясен, поскольку считается, что гемопоэтические стволовые клетки не вносят вклад в эпителиальные клоны. Скорее, предполагается, что перекрестная коррекция из резидентных иммунных клеток, полученных из трансплантата, вносит свой вклад в наблюдаемую клиническую пользу.
Пилотное исследование, проведенное в 2015 году, показывает, что системный фактор, стимулирующий колонии гранулоцитов (G-CSF), может способствовать ускоренному заживлению ран у людей с дистрофическим буллезным эпидермолизом. Трансплантация кожи, полученной из генетически модифицированных стволовых клеток, на раневые поверхности была изучена с отчетом об улучшениях у одного человека.
Мониторинг
Индекс активности и рубцевания буллезного эпидермолиза (EBDASI) — это система баллов, которая объективно количественно определяет тяжесть буллезного эпидермолиза. EBDASI — это инструмент для врачей и пациентов, позволяющий контролировать тяжесть заболевания. Он также был разработан для оценки реакции на новые методы лечения БЭ. EBDASI был разработан и утвержден профессором Диди Мюррелл и ее командой студентов и научных сотрудников в больнице Святого Георгия Университета Нового Южного Уэльса в Сиднее, Австралия. Он был представлен на Международном конгрессе по исследовательской дерматологии в Эдинбурге в 2013 году, а его бумажная версия была опубликована в Журнале Американской академии дерматологии в 2014 году.
Прогноз
В исследовании 2014 года случаи классифицировались на три типа — простой буллезный эпидермолиз (EBS), буллезный узловой эпидермолиз (JEB) и дистрофический буллезный эпидермолиз (DEB) — и изучалось время их смерти. Первые два типа обычно умирали в младенчестве, а последний — в раннем взрослом возрасте. При обследовании 11 семей, пострадавших от болезни, недостаточная осведомленность о болезни как со стороны населения, так и со стороны медицинских работников вызвала озабоченность по поводу оказываемой помощи, а также нечувствительные комментарии взрослых и издевательства со стороны детей.
Эпидемиология
По оценкам, у 20 на миллион живорождений диагностирована БЭ, и у 9 на миллион человек в общей популяции есть это заболевание. Из этих случаев примерно 92% представляют собой простой буллезный эпидермолиз (EBS), 5% — буллезный дистрофический эпидермолиз (DEB), 1% — буллезный соединительный эпидермолиз (JEB) и 2% — неклассифицированные. Несущая частота колеблется от 1 из 333 для JEB до 1 из 450 для DEB; предполагается, что несущая частота для EBS намного выше, чем для JEB или DEB.
Заболевание встречается у представителей всех расовых и этнических групп и затрагивает оба пола.
Общество и культура
В 2010 году Эмма Фогарти, активистка DEBRA Ireland (благотворительная организация EB), была удостоена награды « Люди года» . Актер Колин Фаррелл провел кампанию с Фогарти от имени пострадавших.
В 2014 году ведущий вокалист Pearl Jam Эдди Веддер вместе со своей женой Джилл МакКормик стал соучредителем EB Research Partnership, некоммерческой организации, занимающейся поиском лекарства от буллезного эпидермолиза. Маккормик дружит с детства с Райаном Фуллмером, чей сын Майкл родился от ЭБ. Веддер, Маккормик, Райан Фуллмер и его жена Хизер основали Heal EB. В 2014 году они объединили Heal EB с Исследовательским фондом Джексона Габриэля, чтобы создать Исследовательское партнерство EB. EBRP проводит несколько ежегодных мероприятий по сбору средств. На сегодняшний день они собрали 12 миллионов долларов на финансирование исследований по поиску лекарства. 1 марта 2019 года боксер-тяжеловес Луис Ортис был назначен почетным послом EB-сообщества Исследовательским партнерством EB. Дочь Ортиса, Лисмерседес, родилась с буллезным эпидермолизом.
Кино
Об этом заболевании стало известно в 2004 году в Великобритании в документальном фильме Channel 4 «Мальчик, у которого отвалилась кожа» , в котором рассказывается о жизни и смерти Джонни Кеннеди , англичанина с ЭБ. В Соединенных Штатах то же самое можно сказать о документальном фильме HBO « Моя плоть и кровь» 2003 года. Кроме того, в фильме « Девушка-бабочка» рассказывается о болезни Эбигейл Эванс. В Канаде отмеченный наградами документальный фильм Sports Network о Джонатане Питре привел к широкому освещению болезни мальчика, его лечения и смерти.
Другие имена
Другие термины, используемые для описания инфицированных, включают «дети-бабочки», поскольку кожа хрупкая, как крылья бабочки, «младенцы из ваты» или «дети с хрустальной кожей».
Ссылки
внешняя ссылка
Буллезный эпидермолиз | Общество дерматологов первичной медико-санитарной помощи
Создано: 28 сентября 2014 | Последнее обновление: 31 мая 2018 г.
Введение
Буллезный эпидермолиз (БЭ) включает группу генетически обусловленных состояний хрупкости кожи, характеризующихся образованием пузырей на коже и слизистых оболочках после легкой механической травмы. Три основные группы БЭ: симплекс БЭ, узловой БЭ и дистрофический БЭ. У многих пациентов состояние возникает в раннем возрасте и может быть тяжелым, но иногда БЭ проявляется у молодых людей и с гораздо более тонкими признаками, такими как волдыри на ногах или изменения ногтей. В этой главе представлен обзор необычной и сложной группы состояний, описанной ниже. Он не учитывает приобретенный буллезный эпидермолиз (EBA), редкое иммунобуллезное состояние, которое обсуждается в соответствующей главе. |
Этиология
БЭ — наследственное заболевание. При аутосомно-доминантном EB только один аномальный ген необходим для выражения состояния, т.е. один родитель должен нести ген EB. С другой стороны, аутосомно-рецессивно наследуемая EB требует двух генов EB, то есть по одному от каждого родителя. Если у человека есть один рецессивный ген EB в паре с нормальным геном, его называют носителем, и это заболевание не разовьется.Существует три основных типа БЭ, которые описаны ниже. Состояние классифицируется в зависимости от того, где в различных слоях кожи появляются пузыри.
Простой буллезный эпидермолиз (EBS)
- В эпидермисе появляются пузыри
- Это наиболее распространенный тип БЭ, на его долю приходится 70% случаев, и он, как правило, протекает легче, чем другие типы
Буллезный узловой эпидермолиз (JEB)
- Образование пузырей в зоне базальной мембраны
- JEB составляет около 5% случаев и обычно считается наиболее тяжелым типом EB
.
Буллезный дистрофический эпидермолиз (DEB)
- Волдыри возникают ниже зоны базальной мембраны в верхней части дермы
- DEB составляет около 25% случаев
Синдром Киндлера — это волдыри, развивающиеся на нескольких уровнях (внутренняя прозрачная пластинка и плотная суб-пластинка).
История
- В равной степени страдают мужчины и женщины
- EB обычно возникает при рождении или вскоре после . Иногда БЭ может быть достаточно легкой при рождении, чтобы не проявляться, и обнаруживается только тогда, когда ребенок подрастет или не достигнет совершеннолетия
- Важные общие моменты включают возраст начала; размер, частота и расположение волдырей; возможные побуждающие факторы; предыдущие попытки диагностики; предшествующие терапии; и степень боли или зуда
- Обзор системной информации , которая может быть связана с различными подтипами буллезного эпидермолиза, включает изменение роста или развития и свидетельства поражения слизистой оболочки, включая оральные, носоглоточные, глазные, мочеполовые, желудочно-кишечные или респираторные симптомы.Семейный анамнез пузырей — важный вывод для определения типа EB
.
Клинические данные
Отличительные черты кожи наследственной EB:
- Механически хрупкая кожа и легкое образование пузырей или эрозий
- Некоторые или все из следующих :
- Милия
- Дистрофия или отсутствие ногтей
- Стоматологические проблемы, такие как кариес
- Рубцы (обычно атрофические)
- Дополнительные полезные данные, если таковые имеются, включают обильную грануляционную ткань, локализованную или сливную кератодермию ладоней и подошв, а также измененную пигментацию.Нечасто наблюдаемые и крайне неспецифические кожные проявления включают уменьшение или отсутствие волос, альбопапулоидные поражения (папулы телесного цвета или гипопигментированные папулы, обычно возникающие на нижней части туловища) и гипо / гипергидроз
Из трех основных групп ЭБ насчитывается не менее 27 различных типов . Симптомы значительно различаются от одного типа к другому, что подчеркивается особенностями даже небольшого количества типов EB, как описано ниже.
- Простой локальный буллезный эпидермолиз
- Наиболее распространенная форма EBS
- Он характеризуется болезненными волдырями на ладонях рук и подошвах ног, которые появляются после легкой или умеренной физической активности, такой как ходьба, работа в саду или занятия спортом.У многих пациентов волдыри возникают только на стопах
- Симптомы обычно проявляются в раннем детстве, хотя легкие случаи могут оставаться невыявленными до раннего взрослого возраста
- Гипергидроз стоп — распространенное явление
- Волдыри обычно заживают без значительных рубцов или рубцов
- Буллезный узловой эпидермолиз не по Герлицу
- Это самая распространенная и наименее тяжелая форма JEB
- Как и все формы JEB, обычно проявляется при рождении.
- Вызывает широко распространенное образование пузырей на коже и слизистых оболочках
- Волдыри на коже головы — обычное явление, которое может привести к рубцеванию и необратимой потере волос
- Присутствие гипоплазии эмали, проявляющееся в виде локализованных или более обширных ямок наперсточника на некоторых или всех поверхностях зубов, обнаруживается при всех типах соединительной БЭ.Следовательно, это чрезвычайно полезный клинический результат, хотя его нельзя использовать в качестве диагностического инструмента до тех пор, пока не прорезываются молочные зубы.
- Другие симптомы JEB сторонних производителей включают долговременные травмы кожи и подлежащих тканей, особенно нижних конечностей, рубцевание кожи, а также деформацию или потерю ногтей рук и ног
.
- Доминантный буллезный дистрофический эпидермолиз (ДДЭБ)
- У прототипа пациента с ДДЭБ при рождении наблюдается генерализованное образование пузырей, которое со временем ассоциируется с милой, атрофическим (или, реже, гипертрофическим) рубцеванием и дистрофией ногтей.Гвозди могут вообще выпасть
- Кожа обычно менее хрупкая, чем при других формах дистрофической БЭ, волдыри обычно возникают после резких ударов или скользящих ударов, а не от легкого трения
- Рецидивирующие пузыри и эрозии пищевода, ведущие к прогрессирующей дисфагии, вторичной по отношению к образованию стриктуры пищевода, являются обычным явлением. Также часто поражается полость рта, из-за чего прием пищи или чистка зубов могут быть болезненными
- Существует несколько вариантов, включая претибиальный DDEB, который, как следует из названия, почти исключительно поражает переднюю часть голени.Отдельные поражения, которые обычно имеют вид папул или бляшек, часто имеют фиолетовую окраску, что позволяет предположить клинический диагноз красного плоского лишая. Также присутствуют пузыри и рубцы. Характерна дистрофия ногтей рук и ног
- Аутосомно-рецессивный буллезный дистрофический эпидермолиз, тип Hallopeau-Siemens (RDEB-HS)
- Больные младенцы обычно рождаются с широко распространенными волдырями и участками без кожи, что часто вызвано травмой во время родов
- Чаще всего волдыри присутствуют по всему телу и поражают слизистые оболочки, такие как влажная оболочка рта и пищеварительного тракта.По мере заживления волдырей образуются тяжелые рубцы. Рубцы во рту и пищеводе могут затруднить пережевывание и проглатывание пищи, что приводит к хроническому недоеданию и замедлению роста
- Дополнительные осложнения прогрессирующего рубцевания могут включать сращение пальцев рук и ног, потерю ногтей на руках и ногах, деформации суставов (контрактуры), ограничивающие движение, и воспаление глаз, ведущее к потере зрения
- Кроме того, молодые люди с классической формой дистрофического буллезного эпидермолиза имеют очень высокий риск развития плоскоклеточного рака, который имеет тенденцию быть необычно агрессивным и часто опасным для жизни
Изображений
Щелкните изображение, чтобы увеличить или загрузить.PCDS благодарит Dermatoweb, DermQuest (Galderma) и других, предоставивших изображения. Все названные лица и организации сохраняют авторские права на соответствующие изображения. Этот веб-сайт является некоммерческим и содержит изображения только в образовательных целях. Любое загруженное изображение должно использоваться только в учебных целях, а не в коммерческих целях. Уведомление и указание должны быть предоставлены PCDS или другому названному участнику. Пожалуйста, перейдите по этой ссылке, если у вас есть качественные изображения, которые вы можете разместить на сайте.
Рисунок: 1 EB | |
Рисунок: 2 EB — типичный невоспалительный волдырь | |
Рисунок: 3 EB — волдырь, вызванный механическим воздействием У этого пациента локализован EBS . Скопировано с любезного разрешения Dermatoweb | |
Рисунок: 4 Локализованный EBS | |
Рисунок: 5 Локализованный EBS | |
Рисунок: 6 Не-Herlitz JEB Скопировано с любезного разрешения Dermatoweb | |
Рисунок: 7 JEB — питтинг эмали | |
Рисунок: 8 Доминирующий ДЭБ Скопировано с любезного разрешения Dermatoweb | |
Рисунок: 9 Доминирующий ДЭБ — milia Скопировано с любезного разрешения Dermatoweb | |
Рисунок: 10 Доминант ДЭБ — атрофическое рубцевание | |
Рисунок: 11 Dominant DEB — d Истрофические гвозди Скопировано с любезного разрешения Dermatoweb | |
Рисунок: 12 Аутосомно-рецессивный DEB | |
Рисунок: 13 Аутосомно-рецессивный DEB Скопировано с любезного разрешения Dermatoweb |
Расследования
- Каждый основной тип БЭ диагностируется путем определения ультраструктурного уровня, в пределах которого появляются волдыри после незначительного натяжения кожи.Затем подтипы определяются на основе способа передачи, результатов иммуногистохимии и электронной микроскопии, а также клинического фенотипа
- За дополнительной информацией обращайтесь в Национальную диагностическую лабораторию буллезного эпидермолиза (EB) Гая и Сент-Томаса
.
.
Менеджмент
- Пациентов с возможным БЭ следует направить к дерматологу. Из-за редкости БЭ родители и дети обычно направляются в специализированный центр, в котором работают сотрудники, обладающие знаниями и опытом в лечении этого состояния.В Англии есть четыре специализированных центра:
- Детская больница Бирмингема
- Больница Солихалла
- Детская больница на Грейт-Ормонд-стрит, Лондон
- Больница Святого Томаса, Лондон
- Лекарства от БЭ нет — лечение симптоматическое, и его основная цель — защитить кожу и остановить образование волдырей, ускорить заживление и предотвратить осложнения. Поскольку БЭ может влиять на очень много разных частей тела, обычно требуется многопрофильный командный подход
▷ Буллезный эпидермолиз — фотографии, ожидаемая продолжительность жизни, лечение, симптомы — (2020
Что такое буллезный эпидермолиз?
Это группа состояний кожи, которая приводит к образованию волдырей по разным причинам.Существует четыре типа этого кожного заболевания, и большинство из них первоначально поражает маленьких детей и младенцев и обычно начинается при рождении или сразу после рождения.
Есть люди, которые не имеют никаких симптомов до подросткового или раннего взросления, потому что у них легкая форма буллезного эпидермолиза.
Если у человека легкая форма, она может улучшиться с возрастом, но если это тяжелая форма, она может вызвать серьезные осложнения и привести к летальному исходу.
Типы
Есть четыре типа этого состояния кожи, которые включают:
- Дистрофический буллезный эпидермолиз — этот тип может варьироваться от легкого до тяжелого.Это также обычно проявляется во время рождения или в раннем детстве.
- Простой буллезный эпидермолиз — это обычно самая легкая форма и самая распространенная. Обычно это начинается при рождении или вскоре после этого. При этом типе кожи чаще всего поражаются подошвы и ладони.
- Буллезный узловой эпидермолиз — этот тип обычно тяжелый и проявляется при рождении.
- Приобретенный буллезный эпидермолиз — это редкий вид.
Каждый из этих четырех типов также имеет различные подтипы.
Примерно один из пятидесяти тысяч рожденных детей будет иметь буллезный эпидермолиз. В общей популяции девять из миллиона страдают буллезным эпидермолизом. Это может случиться у представителей любой расы и пола.
Симптомы буллезного эпидермолиза
Основным симптомом буллезного эпидермолиза являются пузыри, наполненные жидкостью, высыпающиеся на коже. В основном это происходит на ступнях и руках, но может случиться и на других частях тела.
Насколько это серьезно и насколько широко распространено, зависит от типа буллезного эпидермолиза. В легкой форме волдыри обычно заживают без рубцов.
Симптомы, которые могут быть у человека, зависят от типа, но могут включать:
- Потеря или деформация ногтей на ногах и руках.
- Наличие внутренних пузырей, в том числе в верхних дыхательных путях, кишечнике, желудке, горле, мочевыводящих путях и пищеводе.
- Утолщение кожи на подошвах стоп и ладонях рук — гиперкератоз.
- Выпадение волос, рубцы и образование пузырей на коже черепа, называемые рубцовой алопецией.
- Кожа, которая выглядит тонкой, называется атрофическим рубцом.
- Прыщи или крошечные белые бугорки на коже, называемые милиями.
- Стоматологические аномалии, такие как кариес из-за плохо сформированной зубной эмали.
- Чрезмерное потоотделение
- Проблемы с глотанием, называемые дисфагией.
- Волдыри вокруг носа и глаз.
- Кашель, другие проблемы с дыханием или хриплый крик.
Причины
Буллезный эпидермолиз — это генетическое заболевание, поэтому в большинстве случаев оно передается по наследству. В ходе исследований было обнаружено, что существует более десяти генов, которые участвуют в формировании вашей кожи, и если какой-либо из этих генов неисправен, возможно, они могут вызвать один из типов буллезного эпидермолиза.
Существует вероятность того, что у человека может развиться это заболевание кожи из-за случайной мутации в гене во время образования сперматозоидов или яйцеклетки.При буллезном эпидермолизе образование пузырей также может быть реакцией на нагревание, трение от трения о что-то, реакцию на незначительную травму, липкую ленту или царапины.
Причинами четырех типов буллезного эпидермолиза являются:
Дистрофический буллезный эпидермолиз
Причиной этого типа являются дефектные гены, которые помогают производить определенный тип коллагена, который является типом сильного белка. и содержится в волокнах, которые скрепляют самый прочный и самый глубокий слой кожи.
Когда это происходит, волокна либо не работают, либо отсутствуют. Этот тип может быть рецессивным или доминантным.
Простой буллезный эпидермолиз
Причиной этого типа являются дефектные гены, которые участвуют в производстве кератина, который находится в верхнем слое вашей кожи и представляет собой волокнистый белок.
Наиболее вероятно, что человек унаследовал единственную копию дефектного гена от одного из родителей. Если у одного из родителей дефектный ген, вероятность того, что у их ребенка будет дефект, составляет пятьдесят процентов.
Соединительный буллезный эпидермолиз
Причина этого типа — дефектные гены, которые участвуют в образовании гемидесмосом, то есть нитевидных волокон, которые прикрепляют базальную мембрану к эпидермису. Дефектные гены возникают в результате того, что оба родителя являются носителями одной копии дефектного гена.
Хотя они передают дефектный ген, ни один из родителей не может иметь буллезный эпидермолиз. Двадцать пять процентов их детей наследуют оба дефектных гена и развивают это заболевание.
Приобретенный буллезный эпидермолиз
Этот тип не передается по наследству, и возникающие волдыри обычно являются результатом ошибочной атаки вашей иммунной системы на здоровые ткани.
Диагноз
Возможно, когда врач посмотрит на внешний вид кожи, он может заподозрить, что это буллезный эпидермолиз, но ему все равно нужно будет сделать тесты, чтобы поставить положительный диагноз. Некоторые из тестов, которые может выполнить врач, могут включать:
Биопсия кожи
Врач берет небольшой образец пораженной кожи и рассматривает его под микроскопом.Это позволит врачу узнать, какой тип буллезного эпидермолиза у них и какой слой кожи отделяется.
Генетическое тестирование
Поскольку большинство случаев передается по наследству, врач берет образец крови и отправляет его в лабораторию. Они проанализируют его, чтобы подтвердить диагноз.
Лечение
Основная цель лечения — облегчить дискомфорт от образования волдырей и помочь предотвратить осложнения в дополнение к предотвращению образования пузырей.В настоящее время нет лекарства от буллезного эпидермолиза. Некоторые из методов лечения, которые можно использовать, включают:
Для ухода за кожей
Волдыри при этом заболевании могут быть большими, и после их разрушения они становятся восприимчивыми к потере жидкости и инфекции, поэтому ваш врач может порекомендовать лечить сырую кожу и волдыри этими способами .
- Если врач просит вас проколоть волдыри, убедитесь, что вы используете стерильную иглу, чтобы волдырь не распространился дальше.Убедитесь, что вы не повредили верхнюю часть волдыря, чтобы защитить кожу под ним, но при этом позволить ей стечь.
- Нанесите вазелин, мазь с антибиотиком или какое-либо увлажняющее вещество перед тем, как наложить специальную нелипкую повязку.
- Пропитайте раны дезинфицирующим средством.
Простой буллезный эпидермолиз | IntechOpen
3.1. Кератин 5/14
Недавние блестящие обзоры были посвящены кератинам и EBS [3, 32].Здесь мы сосредоточимся на истории болезни, анализе мутаций, моделях на животных и будущих методах лечения кератин-ассоциированного EBS с точки зрения врача.
Кератин — один из самых распространенных компонентов эпителиального цитоскелета [33]. Обычно кератины I и II типа образуют гетерополимеры, которые функционируют в клетках [34]. Кератин 5 (K5) и кератин 14 (K14) специфически экспрессируются в базальных эпидермальных клетках [34, 35] (Рисунок 1). В 1980-х годах дезорганизация этих кератинов была обнаружена в базальных кератиноцитах пациентов с EBS [36, 37].На основании этих результатов было выдвинуто предположение, что пациенты с EBS имеют мутации в KRT5 или KRT14 , которые кодируют K5 или K14 соответственно. В начале 1990-х годов сообщалось, что у трансгенных мышей со сверхэкспрессией мутировавшего K14 наблюдается серьезная ломкость кожи [18]. Вскоре после этого открытия две группы исследователей определили случаи EBS с гетерозиготностью по миссенс-мутациям KRT14 [38, 39], за которыми последовала идентификация первого семейства EBS с гетерозиготной мутацией KRT5 [40].С тех пор несколько сотен пациентов с EBS были описаны как имеющие мутаций KRT5 или KRT14 и были обобщены в базе данных промежуточных волокон человека (http://www.interfil.org/) [41].
Существует несколько подтипов связанных с кератином EBS, как описано в таблице 3 [1]. Классическими и распространенными подтипами EBS, в которых признаки наследуются по аутосомно-доминантному типу, являются EBS типа Даулинга-Меары (EBS-DM), тип не Даулинга-Меара (EBS-gen-non-DM) и локализованный тип (EBS-loc), от самых суровых до самых умеренных.Ультраструктурно базальные кератиноциты EBS-DM характеризуются кератиновыми агрегатами [42]. Горячие точки мутаций в KRT5 или KRT14 расположены внутри спиральных граничных мотивов каждого кератина [41]. Миссенс-мутация в одном аллеле этих областей (которая приводит к изменению аминокислот) обычно оказывает доминантно-отрицательный эффект на организацию кератина. Выраженность клинических проявлений среди EBS-DM, EBS-gen-non-DM и EBS-loc обычно определяется местом мутации и различием между исходной и мутированной аминокислотами [32].Однако не всегда легко предсказать фенотип по лежащим в основе мутациям, и в некоторых случаях две разные аминокислотные замены в одном и том же кодоне приводят к различным клиническим проявлениям [43, 44]. Поскольку изменение одной аминокислоты не обязательно вызывает патологическое изменение, были предложены системы in vitro, и in silico, для проверки мутационных эффектов, в которых организация кератина визуализируется в клетках, трансфицированных мутированными кератинами или кератинами дикого типа [44, 45].
Патогенез развития EBS через мутации кератина также был продемонстрирован на животных моделях (Таблица 2). После открытия трансгенных мышей со сверхэкспрессией мутированного K14, описанного выше [18], у Krt5 -нулей и Krt14 -нулей было обнаружено фенотип ломкости кожи [16, 19], хотя состояние этих мышей отличалось от у большинства пациентов с EBS, где измененные аминокислоты дают доминантно-отрицательные эффекты. Вместо этого те Krt5 -null и Krt14 -null мыши обнаруживают фенотип аутосомно-рецессивного EBS (EBS-AR), у которого K5 или K14 нулевые [32].Чтобы воспроизвести доминантно-негативные эффекты мутированных кератинов в EBS человека (EBS-DM, EBS-gen-non-DM и EBS-loc), были получены мыши с индуцибельной нокаутной моделью EBS, в которых миссенс-мутация Krt14 эквивалентна введена мутация EBS человека [20]. Эта индуцируемая модель EBS воспроизводит хрупкость кожи, наблюдаемую у людей с аутосомно-доминантным EBS. Кроме того, существует один естественный крупный рогатый скот с гетерозиготной мутацией KRT5 [17]. Этот помесь фризско-джерсийского быка проявляет фенотип EBS.
Терапевтические вмешательства по поводу EBS были ограничены паллиативными методами. Однако недавние инновации в интерференции РНК привели к терапевтическим стратегиям для доминантно-негативных расстройств, включая кератин-ассоциированный EBS, когда аберрантный мутированный кератин подавляется, в то время как нормальный синтез кератина на другом аллеле остается неизменным [46]. Эта стратегия РНКи многообещающая и будет дополнительно проверена в клинических испытаниях.
3.2. Plectin
Подробный обзор был посвящен EBS и плектину [4], хотя с тех пор в этой области было сделано несколько достижений [14, 47-49].
Плектин представляет собой перекрестно связывающий белок между цитоскелетом и мембранными белками, включая гемидесмосомные компоненты (рис. 1). Известно, что плектин имеет множество изоформ транскриптов, которые отличаются друг от друга N-концевыми последовательностями на уровне белка [50]. Среди множества изоформ транскриптов плектин 1a в основном экспрессируется в эпидермальных кератиноцитах [51]. В дополнение к сложности 5’-транскрипта, плектин имеет вариант сплайсинга без стержней [52]. Существует несколько подтипов EBS, которые вызваны дефицитом плектина (таблица 4).
В середине 1990-х годов у пациентов с EBS с мышечной дистрофией (EBS-MD) были обнаружены мутации в гене, кодирующем плектин ( PLEC ) [53, 54]. С тех пор у пациентов с EBS-MD было зарегистрировано множество мутаций PLEC , в основном локализованных в области, кодирующей стержневой домен плектина [4, 47, 55].
В 2005 г. две группы независимо друг от друга сообщили о новом подтипе EBS с мутациями PLEC : EBS с атрезией привратника (EBS-PA) [56, 57]. БЭ с атрезией привратника (ПА) был известен у пациентов с мутациями ITGA6 или ITGB4 [58, 59].Однако образцы кожи этих пациентов с мутациями интегрина показывают расщепление кожи на уровне lamina lucida, что приводит к диагнозу узловой БЭ (JEB). Напротив, случаи EBS-PA с мутациями PLEC характеризовались расщеплением кожи внутри базальных эпидермальных клеток [56].
Причина, по которой мутации PLEC приводят к двум различным подтипам EBS, была выяснена только недавно. Разработка моноклональных антител против нескольких частей плектина позволила нам понять паттерны экспрессии плектина, которые различают EBS-MD и EBS-PA [47].Кожа EBS-MD обычно демонстрирует экспрессию бесстержневого плектина без экспрессии полноразмерного плектина, тогда как в коже EBS-PA не присутствует ни бесстержневой, ни полноразмерный плектин [47].
Следующий большой вопрос заключался в том, могут ли EBS-MD и EBS-PA возникать одновременно у одного пациента или эти два различных подтипа EBS являются взаимоисключающими. Недавно сообщалось об одном случае с фенотипом EBS-MD и EBS-PA (EBS-MD-PA) [48]. У пациента были мутации усечения в последнем экзоне PLEC , что привело к экспрессии уменьшенного и укороченного полноразмерного и бесстержневого плектина без промежуточного домена связывания филаментов [48].
Помимо аутосомно-рецессивных подтипов EBS, связанных с мутациями PLEC (EBS-MD, EBS-MD и EBS-MD-PA), существует один отдельный аутосомно-доминантный EBS с мутацией PLEC : EBS, Ogna (EBS- Ог). EBS-Og вызывается гетерогенной мутацией p. Arg2000Trp и характеризуется умеренным образованием пузырей без фенотипа MD или PA [4, 60]. На сегодняшний день сообщается, что 5 неродственных семейств EBS-Og имеют одну и ту же мутацию [49, 60].
Были созданы животные модели плектин-дефицитной EBS (таблица 2).У мышей Plec -null наблюдается тяжелый фенотип образования пузырей и неонатальная смерть [21], хотя желудочно-кишечные тракты не исследовались для подтверждения PA или PA-подобных поражений. У этих мышей нарушена целостность миофибрилл в скелетных и сердечных мышцах [21]. Эпидермис-специфичное удаление плектина также вызывает тяжелый фенотип образования пузырей и раннюю летальность у мышей [22]. Более того, мыши с мутацией EBS-Og, эквивалентной мышиным, демонстрируют хрупкость кожи из-за специфического для эпидермиса протеолиза мутированного плектина [14].
Соединительный и дистрофический буллезный эпидермолиз
1. Введение
Буллезный эпидермолиз (БЭ) — это врожденный генодерматоз, который поражает главным образом кожу, а иногда и другие органы [1]. Пожизненное образование пузырей и эрозия кожи и слизистой оболочки, вызванные механической травмой, угрожают пациентам с БЭ [1]. Наиболее частая причина смерти — метастазирующая плоскоклеточная карцинома [2]. EB подразделяется в основном на три категории по расположению тканевого отделения (пузыря) в зоне базальной мембраны (BMZ) на электронно-микроскопическом уровне, EB симплекс (EBS), дистрофический EB (DEB) и узловой EB (JEB) [1].Некоторые дерматологи также предлагали различать буллезный гемидесмосомный эпидермолиз [3]. В EBS волдырь располагается на уровне базальных кератиноцитов, в DEB на уровне lamina lucida и в DEB на уровне дермы [1]. БЭ вызывается в основном мутацией кератиновой нити, компонентов гемидесмосом или генов коллагена [1]. К настоящему времени идентифицировано не менее 10 различных генов как генов, вызывающих EB [1,4,5].
2. Молекулярные компоненты BMZ (Рисунок 1)
Кератин — это наиболее распространенный структурный белок, обнаруживаемый в эпителиальных клетках [6].Кератины представляют собой полимеры промежуточных филаментов типа I и типа II [6]. В базальных кератиноцитах промежуточным филаментом типа I является кератин 14 (K14), а промежуточным филаментом типа II является кератин 5 (K5) [6]. Эти два типа кератинов являются основными мутировавшими молекулами, обнаруженными в EBS [6].
Гемидесмосомы представляют собой очень плотные соединительные структуры между клетками и матриксом, которые соединяют базальные кератиноциты с базальной мембраной [7]. Гемидесмосомы прикрепляют кератиновые нити к поверхности клетки [7]. Ультраструктурно гемидемосомы включают внутренние бляшки, внешние бляшки, закрепляющие фибриллы и закрепляющие филаменты [8].На молекулярном уровне ядро каждой гемидесмосомы состоит из четырех трансмембранных белков, буллезного пемфигоидного антигена 180 кДа (BP180, коллаген XVII типа, BPAG2), интегринов α6β4 и тетраспанинового белка CD151 [7]. И BP180, и интегрин α6β4 взаимодействуют с ламинином-332 в BMZ
Рисунок 1.
Молекулярные компоненты зоны базальной мембраны.
Рис. 2.
Клинические (слева) и электронно-микроскопические (справа) проявления пациента с дистрофическим буллезным эпидермолизом.
[9]. Интегрин α6β4 является уникальным интегрином, потому что другие интегрины обычно присоединяются к актину, тогда как интегрин α6β4 прикрепляется к промежуточному филаменту, кератину [10]. Интегрин α6β4 прикрепляется к промежуточному филаменту с помощью плектина [10]. BP180 связывает кератин за счет взаимодействия с BP230 в цитоплазме [10]. И BP180, и BP230 являются мишенью для основного субэпидермального аутоиммунного буллезного заболевания, буллезного пемфигоида [11]. Базальная мембрана в основном состоит из коллагена IV [12]. Ламинин-332 и коллаген VII прикрепляются к коллагену IV [7].Все эти компоненты являются молекулами, на которые влияют пациенты с БЭ [1].
3. JEB (рис. 2)
Тип наследования у пациентов с JEB аутосомно-рецессивный [1]. Как упоминалось выше, отделение кожи при JEB происходит в lamina lucida [1]. Три подтипа JEB exsist, Herlitz JEB, не-Herlitz JEB и JEB с атрезией привратника [1]. JEB Герлица — это фатальный подтип JEB [1]. Большинство пораженных пациентов умирают от системной инфекции в результате сильной эрозии практически всей кожи [1].Herlitz JEB вызывается гомозиготной или сложной гетерозиготной мутацией преждевременного терминирующего кодона (PTC) ламинина-332 [13,14]. Не-Herlitz JEB намного мягче, чем Herlitz JEB [1]. Либо миссенс-мутация ламинина-332, либо мутация BP180 обнаруживается у пациентов, не относящихся к группе Herlitz JEB [15-17]. JEB с атрезией привратника, возможно, представляет собой опасный для жизни подтип, аналогичный JEB Герлица [1]. Тем не менее, пациенты с JEB с атрезией привратника иногда проявляют не опасный для жизни фенотип, как пациенты с JEB, не относящиеся к Herlitz [1].Мутации в генах интегрина α6 или β4 обнаруживаются при JEB у пациентов с атрезией привратника [1,18,19]. Мутации PTC генов интегрина α6 или β4 обнаруживаются при тяжелой форме JEB с пациентами с атрезией привратника, тогда как миссенс-мутации этих генов интегринов обнаруживаются при более легком подтипе JEB с пациентами с атрезией привратника [1,20].
4. DEB
Тканевое отделение DEB происходит в дерме [1]. Клинически у пациентов с DEB наблюдается образование волдырей на коже на большой площади наряду с образованием рубцов и милиумов [15].При DEB известны две модели наследования: аутосомно-доминантный и аутосомно-рецессивный [15]. Известно, что DEB вызывается мутациями в гене коллагена VII. В DEB сообщается о более чем 300 мутациях [15]. Известно, что мутация замены глицина в одном аллеле гена, кодирующего коллагеновый домен коллагена VII, тесно связана с DEB [21]. Такая мутация, вероятно, оказывает доминирующее негативное влияние на образование или сборку коллагена VII [15]. В наиболее тяжелой форме DEB, рецессивном DEB по Hallopeau-Siemens, обнаруживается мутация PTC на обоих аллелях гена, кодирующего коллаген VII [22].В наиболее легкой форме рецессивного DEB, не-Hallopeau-Siemens рецессивный DEB, мутации PTC в одном аллеле, миссенс-мутации или мутации в рамке считывания обнаруживаются в генах, кодирующих коллаген VII [23].
5. Диагностика
Отличительной чертой диагностики БЭ является анализ мутаций на основе ДНК [1]. Тем не менее, необходимо свести к минимуму усилия по уточнению возможного пораженного гена посредством сбора анамнеза, клинической оценки, гистопатологического исследования, исследования иммуномаркинга и электронно-микроскопического исследования [1].Используя эти методы, мы можем разделить пациентов по типу заболевания как минимум на три формы: EBS, JEB и DEB [1]. Образцы гистопатологии или электронной микроскопии следует брать после легкого втирания на кожу без пузырей, чтобы не ошибочно диагностировать расположение волдырей из-за дегенерации пораженной кожи [1]. Исследование иммуномартирования с использованием анти-K5, K14, интегрина α6, интегрина β4, BP180, плектина, ламинина-332 или антител к коллагену VII весьма полезно для диагностики EB, если затронутая мутация локализуется на участке эпитопа, на который нацелены эти антитела [1] .Кроме того, иммуногистохимическое исследование с использованием антител против коллагена IV также полезно для оценки части расщепления [1]. В образцах EBS или JEB положительное окрашивание коллагена IV обнаруживается на дне блистера, тогда как в образце DEB — на крыше блистера [1]. После этих тщательных оценок ставится диагноз на основе ДНК [1].
6. Лечение
Лечение БЭ в основном симптоматическое. Наиболее важной задачей является предотвращение местной инфекции, включая золотистый стафилококк, стрептококк пиогенес и синегнойную палочку.Если мы не сможем контролировать местную инфекцию, с большой вероятностью произойдет последующий сепсис. Чтобы предотвратить такую местную инфекцию, для лечения БЭ выбираются полуокклюзионные неприлипающие повязки с местными антибиотиками или без них. Кроме того, поскольку пищеводные глазные и оральные осложнения также встречаются у пациентов с БЭ, также требуется клиническая помощь при эрозиях в этих органах для предотвращения местной инфекции и последующего сепсиса.
Аллогенные кожные трансплантаты, клетки которых не происходят от пациентов, были испытаны для пациентов с БЭ.Эти аллотрансплантаты были отклонены, но могли продуцировать цитокины, способствующие заживлению ран и процессу реэпителизации.
7. Текущее лечение
Поскольку симптоматическая терапия доступна только при БЭ, будущее генно-целевой терапии весьма ожидаемо и рассматривается. Чтобы попытаться сделать это, потенциальными вариантами являются клеточная терапия с использованием фибробластов и аллогенная трансплантация костного мозга. На экспериментальном уровне такие методы лечения были успешными для JEB и DEB. Известно, что колген VII синтезируется в основном кератиноцитами и в меньшей степени фибробластами [19].Поскольку фибробласты легче культивировать и легко трансфицировать внешними генами, чем кератиноцитами, клеточная терапия с использованием фибробластов выбрана для возможной генной терапии DEB [1]. Фактически, Goto et al. успешно восстанавливает коллаген VII путем введения фибробластов, трансфицированных геном коллагена VII кожи [24]. Кроме того, клиническое исследование с участием пяти пациентов с использованием этого метода уже было успешным без каких-либо побочных эффектов [25]. Культивированные кератиноциты пациентов, трансфицированные геном ламинина β3 с помощью ретровирусной техники, были успешно перенесены и вылечили образование волдырей у одного пациента.Также была внедрена протеиновая терапия коллагеном VII, которая оказалась успешной на модели in vivo. Недостающий или дефектный белок, синтезированный рекомбинантными методами in vitro, вводится в кожу с волдырями. Успешное лечение уже было получено в случае коллагена VII [26,27].
Аллогенная трансплантация костного мозга — другой вариант. У пациентов с БЭ базальные кератиноциты продуцируют дефектный генный продукт BMZ [1]. Известно, что клетки костного мозга могут дифференцироваться в эпидермальные кератиноциты [17].Следовательно, аллогенная трансплантация костного мозга может исправить такие дефектные компоненты BMZ. На экспериментальном уровне Chino et al. оказался успешным в коррекции улучшенного содержания коллагена VII у мышей с нокаутом по коллагену VII путем аллогенной трансплантации костного мозга [28]. Более того, клинические испытания с использованием этой технологии и трансплантация пуповинной крови уже были начаты и получили успешные результаты [1].
8. Заключение
БЭ представляет собой опасное для жизни заболевание, продолжающееся всю жизнь, и до сих пор требует только симптоматического лечения.Однако целенаправленная генная терапия на основе клеток находится на пути к успеху.
[Полный текст] Дистрофический буллезный эпидермолиз: обзор
Кафедра дерматологии, Высшая школа медицины Университета Хоккайдо, Саппоро, Япония
Резюме: Дистрофический буллезный эпидермолиз — редкое наследственное заболевание с образованием волдырей, вызванное мутациями в гене COL7A Ген , кодирующий коллаген VII типа. Дефицит и / или дисфункция коллагена типа VII приводит к субэпидермальным волдырям непосредственно под плотной пластинкой, что приводит к хрупкости кожно-слизистой оболочки и осложнениям заболевания, таким как трудноизлечимые язвы, обширное рубцевание, недоедание и злокачественные новообразования.Заболевание обычно диагностируется с помощью иммунофлуоресцентного картирования и / или просвечивающей электронной микроскопии и впоследствии подразделяется на один из 14 подтипов. В этом обзоре представлены практические знания о заболевании, включая новые терапевтические стратегии.
Ключевые слова: коллаген типа VII, якорная фибрилла, подтипы, ревертантный мозаицизм, лечение, генная терапия
Введение
Буллезный эпидермолиз (БЭ) — это наследственное заболевание, характеризующееся образованием пузырей на коже и слизистых оболочках, вызванных механическим стрессом. 1 EB классифицируется на четыре основных типа, а именно: симплексный EB (EBS), узловой EB (JEB), дистрофический EB (DEB) и синдром Киндлера, на основе отличительного ультраструктурного участка расщепления кожи. 2,3 Разделение тканей происходит в эпидермисе (EBS), в lamina lucida (JEB) или в sublamina densa (DEB). Синдром Киндлера, смешанный тип, демонстрирует несколько плоскостей спайности.
Были предприняты попытки оценки распространенности и заболеваемости БЭ с использованием различных методов выборки в разных регионах мира.Есть некоторые различия в частоте DEB среди разных групп населения. 4–8 По данным нескольких регистров БЭ, общая распространенность оценивалась как 8–10 на миллион рождений (для ДЭБ: 2–6 на миллион рождений). 5,6 Соотношение мужчин и женщин в случаях EB и DEB составляет примерно 1: 1. У большинства пациентов с ДЭБ симптомы проявляются при рождении и в возрасте до 1 года.
Коллаген VII типа и патогенез буллезного дистрофического эпидермолиза
DEB вызывается мутациями в гене COL7A1 , кодирующем коллаген VII типа. 9 Коллаген VII типа является основным компонентом закрепляющих фибрилл, расположенных ниже базальной мембраны в верхней части дермы, обеспечивая стабильную дермо-эпидермальную адгезию. 3 Коллаген типа VII представляет собой гомотример, состоящий из трех цепей проα1 (VII), которые кодируются геном 32 kb COL7A1 на хромосоме 3p21. 10 Транскрипт мРНК размером примерно 8,9 т.п.н. транслируется в α 1 (VII) цепь проколлагена, состоящую из 2944 аминокислот. 11 Каждая полипептидная цепь проколлагена α 1 (VII) содержит центральный коллагеновый трехспиральный домен (145 кДа), фланкированный как большим аминоконцевым неколлагеновым доменом 1 (NC-1) (145 кДа), так и малым карбокси-концевой домен NC-2 (30 кДа). 11,12 Тройной спиральный домен состоит из повторяющейся последовательности Gly-X-Y, которая прерывается 19 неколлагеновыми областями. В середине тройного спирального домена находится неколлагеновая область из 39 аминокислот, известная как «шарнирная» область, которая восприимчива к протеолитическому перевариванию пепсином. 13 Аминоконцевой NC-1 состоит из субмодулей, гомологичных известным адгезивным белкам, включая белок хрящевого матрикса, повторы фибронектина типа III, мотив, подобный типу фактора фон Виллебранда, и короткие домены, богатые цистеином и пролином. . 14 Домен NC-1 обеспечивает прикрепление закрепляющих фибрилл к основным компонентам базальной мембраны кожи, таким как коллаген IV типа и ламинин-332. 15,16 Карбоксиконцевой домен NC-2 содержит консервативные цистеины, участвующие в образовании дисульфидных связей, которые делают возможной связь между гомотримером коллагена VII типа. 17
Коллаген VII типа синтезируется как эпидермальными кератиноцитами, так и дермальными фибробластами. 18 При синтезе полной цепи проколлагена α 1 (VII) три полипептида связываются через свои карбоксильные концы, образуя тример, коллагеновая часть которого складывается в тройное спиральное образование. 12 Затем тройные спиральные молекулы секретируются во внеклеточную среду, где две молекулы коллагена типа VII выстраиваются в антипараллельный димер «хвост к хвосту». 19 Часть домена NC-2 удаляется, и ассоциация гомотримеров стабилизируется за счет образования межмолекулярной дисульфидной связи в перекрывающихся карбоксильных концевых областях. Впоследствии большое количество этих антипараллельных димеров агрегирует латерально с образованием закрепляющих фибрилл. 12
Клинические особенности дистрофического буллезного эпидермолиза
Клинические подтипы
DEB был классифицирован в соответствии с типом наследования, клиническими признаками и уровнем экспрессии коллагена VII типа. 1 С 1988 г. было проведено четыре международных консенсусных совещания по диагностике и классификации БЭ. 1,2,20,21 За 25 лет, прошедших с момента первого консенсусного совещания, было идентифицировано несколько клинических подтипов и названия подтипов с использованием эпонимов были исключены и заменены описательными терминами. В самой последней предложенной классификации существует 14 подтипов DEB (Таблица 1). 1
Таблица 1 Клинические подтипы дистрофического буллезного эпидермолиза |
DEB имеет две модели наследования: аутосомно-доминантный (DDEB) и аутосомно-рецессивный (RDEB). DEB имеет три общих варианта, а именно «DDEB, обобщенный», «RDEB, обобщенный тяжелый» и «RDEB, обобщенный промежуточный», а также другие редкие варианты. «DDEB, претибиальный» и «RDEB, претибиальный» — это редкие формы локализованного DEB, характеризующиеся повторяющимися волдырями, язвами и гипертрофическими рубцами, возникающими в основном в претибиальной области, с различной дистрофией ногтей. 22 «DDEB, pruriginosa» и «RDEB, pruriginosa» характеризуются интенсивным кожным зудом и легионами, похожими на узелковую почесуху, локализованными преимущественно на конечностях. 23 «DDEB, только ногти» проявляется дистрофическими ногтями, но без ломкости кожи или волдырей, вызванных травмой. 24 «RDEB, inversa» характеризуется генерализованным образованием волдырей с рождения и в раннем младенчестве. 25 Позже, в младенчестве или детстве, места склонности к образованию пузырей меняются, причем изгибы тела, оси и слизистые оболочки становятся преобладающими местами образования пузырей.«RDEB, centripetalis» характеризуется медленным центростремительным прогрессированием симметричного образования пузырей и рубцов. 26 «ДДЭБ, буллезный дермолиз новорожденного» и «РДЭБ, буллезный дермолиз новорожденного» с субэпидермальными волдырями при рождении или вскоре после этого, которые имеют тенденцию спонтанно регрессировать в первые несколько месяцев жизни по аутосомно-доминантному типу и аутосомно-рецессивным образом соответственно. 27,28 Клинические особенности трех основных подтипов DEB суммированы ниже.
DDEB, обобщенный
DDEB вызывает снижение экспрессии коллагена типа VII и обычно имеет хороший прогноз. 1,29,30 Волдыри часто легкие и ограничиваются областями травмы, такими как руки, ступни, колени и локти, что обычно приводит к рубцеванию, образованию милиумов и потере ногтей (рис. 1А). Поражение слизистой оболочки встречается редко, а зубы в норме. Образование пузырей обычно начинается при рождении или вскоре после него, но активность заболевания иногда снижается с возрастом.
Рис. 1. Клинические особенности дистрофического буллезного эпидермолиза. |
РДЭБ, общая тяжелая
RDEB, генерализованный тяжелый, является наиболее тяжелым из всех подтипов DEB, потому что он вызван отсутствием или заметным снижением экспрессии коллагена типа VII. 1,29,31 Заболевание, ранее известное как тип Hallopeau – Siemens, проявляется генерализованными волдырями от рождения, которые приводят к обширным рубцам и псевдосиндактилии, а иногда и к гипо- или гиперпигментации. 29 Волосы обычно редкие, с рубцовым облысением. У пациентов возникает множество проблем в результате обширных пузырей и поражения слизистых оболочек, особенно в ротовой полости, пищеводе, глазах и анальном канале. 1 Открытие рта может стать ограниченным, а язык станет менее подвижным из-за рубцов.Поражение слизистой оболочки полости рта и стеноз пищевода затрудняют пероральное кормление. 32 Из-за плохого питания, хронического воспаления и инфекции у пациентов развиваются анемия, задержка развития, задержка полового созревания и остеопороз. 33 Поражение глаз у пациентов с DEB включает ссадины, рубцы и паннус роговицы; волдыри и выворот век; пузыри на конъюнктиве и симблефарон. 34 Поражение перианальной области часто приводит к болезненному стулу и, как следствие, к запорам.
RDEB, генерализованная тяжелая форма, связана с многочисленными внекожными проявлениями, включая поражение желудочно-кишечного тракта, мочеполовых путей, почек и сердца. 35,36 Дети с тяжелой генерализованной РДЭБ подвержены повышенному риску гломерулонефрита, амилоидоза почек, IgA-нефропатии и кардиомиопатии. 37,38 Примечательно, что пациенты имеют высокую вероятность развития агрессивной плоскоклеточной карциномы (рис. 1D). 39 Пациенты редко выживают после 30 лет из-за тяжелых почечных осложнений или агрессивной плоскоклеточной карциномы. 31
РДЭБ, общий промежуточный
RDEB, обобщенное промежуточное звено ранее называлось типом не-Hallopeau – Siemens или «RDEB, обобщенное прочее». 1,2 Клинические проявления аналогичны таковым для RDEB, генерализованная тяжелая (рис. 1B и C), но образование пузырей менее серьезное, поскольку экспрессия коллагена типа VII присутствует, хотя и снижена. 29 Пациенты обычно имеют лучший прогноз, чем у пациентов с RDEB, генерализованно тяжелым. Большинство пациентов доживают до зрелого возраста, а некоторые даже способны рожать.Риск плоскоклеточного рака также повышается при этой форме, и необходимо регулярное наблюдение. 39
Диагностика буллезного дистрофического эпидермолиза
После первоначального диагноза, основанного на тщательном изучении клинических проявлений и характера наследования, необходимо провести биопсию кожи из недавно образовавшегося волдыря, чтобы классифицировать заболевание путем определения глубины отделения ткани. 2 Световая микроскопия может различать внутриэпидермальное и субэпидермальное расщепление, но она не очень полезна для определения специфического подтипа EB, поскольку и JEB, и DEB показывают субэпидермальные пузыри.Таким образом, диагностическое тестирование и классификация БЭ начинаются с иммунофлуоресцентного картирования (IFM) и / или просвечивающей электронной микроскопии (TEM), предпочтительно на свежих блистерах. 2 После определения уровня расщепления кожи и профиля окрашивания антигеном рекомендуется провести мутационный анализ, поскольку он позволяет наиболее точную подклассификацию. Точный диагноз позволяет прогнозировать, генетическое консультирование и пренатальную диагностику. 9,29
Просвечивающая электронная микроскопия
ТЕМ и IFM эффективны при определении уровня расщепления кожи.В настоящее время ИФМ приобретает все большее значение в диагностике БЭ, поскольку ТЕА требует дорогостоящего оборудования, значительного опыта и знаний для обработки образцов биопсии кожи и точной интерпретации полученных микрофотографий. 2 Техника, однако, все еще полезна, потому что ТЕМ позволяет прямую визуализацию и обеспечивает морфологическую полуколичественную оценку структурных нарушений в зоне базальной мембраны. 3 Он также имеет то преимущество, что выявляет микроспадения и тонкие изменения дермально-эпидермального перехода при DDEB, где результаты IFM могут быть нормальными.Следовательно, ТЕА, вероятно, продолжит играть важную роль как в клинической, так и в исследовательской областях. 3
При DEB субэпидермальные пузыри постоянно возникают непосредственно под плотной пластинкой (рис. 2). RDEB, генерализованная тяжелая форма, показывает заметное снижение или отсутствие экспрессии коллагена типа VII, что ультраструктурно приводит к рудиментарным или отсутствующим якорным фибриллам. Напротив, RDEB, обобщенный промежуточный продукт, показывает уменьшенные или рудиментарные заякоренные фибриллы.В DDEB фиксирующие фибриллы обычно имеют нормальный внешний вид или немного уменьшенное количество. 3
Рис. 2 Электронно-микроскопическое изображение дистрофического буллезного эпидермолиза. |
Картирование иммунофлуоресценции
IFM на свежеиндуцированном волдыре в настоящее время рекомендуется в качестве основного метода диагностики БЭ, поскольку IFM может дать существенное представление о точном уровне расщепления ткани, а также об относительной экспрессии и распределении компонентов зоны базальной мембраны кожи и эпидермальных антигенов. 29 Как правило, степень молекулярного дефекта коррелирует с клинической серьезностью. В DEB IFM выявляет образование волдырей под плотной пластинкой, против которой реагируют антитела к коллагену IV типа. 40 В зависимости от подтипа уровень экспрессии коллагена VII типа варьирует. При RDEB, генерализованной тяжелой форме, коллаген типа VII отсутствует или значительно снижен (рис. 3A). При RDEB, генерализованном промежуточном уровне, коллаген типа VII обычно снижен в разной степени (Рисунок 3B) по сравнению с нормальной кожей (Рисунок 3C).DEB, буллезный дермолиз новорожденных, имеет характерные признаки IFM, такие как внутриэпидермальные гранулы коллагена VII типа. 41,42 Иногда IFM не может предоставить точный диагноз подтипа. В менее тяжелых подтипах DDEB коллаген VII типа снижен лишь незначительно, и кожа может казаться нормальной при IFM.
Рис. 3. Иммунофлуоресцентное картирование дистрофического буллезного эпидермолиза с антителами к коллагену VII типа. |
Мутационный анализ
Мутационный анализ может предоставить окончательный диагноз подтипа EB и способа наследования, а также может определить точное место и тип молекулярной мутации у пациента с EB. 9,43 Анализ мутаций гена COL7A1 выполняется с использованием ПЦР-амплификации всех 118 экзонов и границ экзон-интрон и последующего прямого секвенирования ДНК. В последнее время секвенирование следующего поколения стало применяться на практике и позволило провести более полный анализ мутаций с потенциальной экономией времени и денег. 44
На сегодняшний день зарегистрировано около 400 мутаций в COL7A1 . 9,45 В RDEB часто встречается сложная гетерозиготность.Различные комбинации мутаций объясняют биологический фенотип, который объясняет различную клиническую тяжесть. 29 У большинства пациентов с RDEB генерализованные тяжелые кодоны преждевременного прекращения (PTC) присутствуют на обоих аллелях гена COL7A1 . 46 Мутации PTC приводят к бессмысленному распаду мРНК или остаточной экспрессии усеченных полипептидов коллагена VII типа, которые разрушаются в клетках. 29 В результате у этих пациентов в базальной мембране наблюдается отсутствие или снижение экспрессии коллагена типа VII.В RDEB общие промежуточные гетерогенные мутации, включая миссенс-мутации, мутации в рамке считывания и сайт сплайсинга, присутствуют по крайней мере в одном аллеле COL7A1 . 29 Часто вторая мутация вызывает PTC. Эти комбинации мутаций приводят к синтезу дефектного коллагена VII типа и структурным аномалиям в закрепляющих фибриллах. DDEB обычно включает замены глицина в коллагеновом домене коллагеновой цепи α 1 (VII) только в одном аллеле, хотя в некоторых случаях могут лежать другие миссенс-варианты, небольшие вставки или делеции в рамке считывания или чередования сайтов сплайсинга. 30,47 Около 1/8 всех тримеров коллагена (AAA) типа VII в норме; 7/8 тримеров (AAa, AaA, aAA, Aaa, aAa, aaA, aaa) нарушены аномальным белком. Следовательно, замены глицина и другие мутации, вероятно, оказывают доминирующее негативное влияние на образование и сборку коллагена VII типа (A: нормальный коллаген VII типа, a: мутированный коллаген VII типа). 46
Хотя большинство мутаций COL7A1 были специфичны для отдельных семей, некоторые повторяющиеся мутации были зарегистрированы у разных пациентов. 48 В Италии были обнаружены повторяющиеся мутации, в том числе инсерционные / делеционные (497insA, 8441-14del21), сплайс-сайты (4783-1G> A, 7344G> A) и миссенс-мутации (G1664A). 43 Повторяющаяся мутация сдвига рамки считывания, 2470insG, была обнаружена только в Мексике. 49 Рецидивирующие мутации R578X, 7786delG и R2814X, по-видимому, характерны только для британской популяции. 50,51 Мутации 5818delC, 6573 + 1G> C и E2857X были расценены как повторяющиеся мутации COL7A1 , связанные с RDEB в Японии. 7,48 Высокий уровень повторения 425A> G и G2043R был обнаружен у нескольких различных этнических групп. 52
Лечение пациентов с буллезным дистрофическим эпидермолизом
Не было разработано окончательных методов лечения DEB, и симптоматическая терапия является основой клинического ведения. 53–55 Профилактика новых волдырей и язв, а также уход за ранами являются наиболее важными аспектами лечения. Новые пузыри следует осушать стерильными иглами с большим отверстием, чтобы предотвратить их расширение. 55 Крышку блистера следует оставить на месте, так как она действует как естественная повязка на рану. Сначала на антипригарную повязку, такую как Mepilex и Urgotul, наносят вазелин или другие подходящие мази, а затем поражение покрывают повязкой с покрытием. 53,56 Повязка фиксируется повязкой. После очищения кожи повязки следует менять ежедневно. Следует избегать использования адгезивов и компрессионных повязок, поскольку они вызывают образование новых пузырей.
Актуальные антибиотики и антимикробные повязки следует использовать с осторожностью у пациентов с DEB. 56,57 Бактериальная колонизация ран неизбежна, но часто не мешает заживлению. 58 В таких случаях лечение не требуется. В связи с повышенным риском бактериальной резистентности местные мази с антибиотиками и противомикробные повязки следует использовать для тех ран, которые колонизированы бактериями и не заживают, что называется «критической колонизацией». Инфекция раны, которая, по сути, является клиническим диагнозом, характеризующимся увеличением размера, экссудатом, запахом, болью, окружающей эритемой, отеком и отеком, часто требует местного или системного применения антибиотиков.В некоторых случаях местные антибиотики используются поочередно у пациентов с хроническими и / или критически колонизированными ранами. Повязки, пропитанные серебром, также эффективны в снижении бионагрузки, но есть опасения по поводу повышенного уровня серебра в плазме. 59 Как отмечалось выше, лечение ран при DEB основано на распознавании колонизации или явной инфекции.
пациентов с EB имеют повышенные потребности в калориях и белках из-за увеличения энергии, расходуемой на заживление ран. 60 В то же время поражение слизистой оболочки ротоглотки, пищевода и желудочно-кишечного тракта у некоторых пациентов с РДЭБ ограничивает их потребление, что затрудняет удовлетворение высоких потребностей в калориях. Максимальное питание имеет жизненно важное значение для стимулирования роста и развития, оптимизации заживления ран и улучшения качества жизни.
Тяжелые случаи с вторичными симптомами в различных органах требуют сотрудничества педиатров, дерматологов, терапевтов, дерматологических хирургов, стоматологов, диетологов, физиотерапевтов, психологов и других специалистов. 29
Авансы на лечение
Хронические и рецидивирующие кожные язвы у пациентов с RDEB вызывают серьезную дисфункцию, такую как сокращение рубца, и иногда приводят к агрессивной плоскоклеточной карциноме, которая часто дает метастазы и приводит к смерти. 39 Поэтому важно достичь ранней реэпителизации и предотвратить повторные раны. Различные биологические повязки, включая культивированные аутологические или аллогенные эпидермальные и / или дермальные трансплантаты, использовались для лечения трудноизлечимых язв, и они обычно эффективны в стимулировании реэпителизации. 61 Терапевтический эффект носит временный характер, и пациенты испытывают рецидив в обработанной области, потому что аллогенные трансплантаты провоцируют иммунное отторжение, а культивируемые аутотрансплантаты по своей сути имеют мутацию в COL7A1 . 62
Аллогенная трансплантация костного мозга используется при лечении лейкозов, лимфом и иммунодефицитных заболеваний. Было показано, что стволовые клетки костного мозга обладают способностью мигрировать в кожу и дифференцироваться в клетки кожи, которые производят коллаген VII типа. 63,64 Несколько групп провели терапию костного мозга для лечения мышей с RDEB-моделью, у которых отсутствует коллаген типа VII, и подтвердили, что лечение может облегчить симптомы. 63,64 Кроме того, начались клинические испытания препаратов RDEB с использованием трансплантации пуповинной крови и костного мозга. 65,66 Хотя эти методы лечения привели к улучшению фенотипа, они также были связаны со смертностью: из 20 пациентов с РДЭБ, которым была проведена трансплантация крови и костного мозга, пятеро умерли от прогрессирования болезни или осложнений трансплантации. 66
Сообщалось, что коллаген
типа VII синтезируется и секретируется кератиноцитами и фибробластами. Фибробласты намного прочнее и их легче культивировать, чем кератиноциты; следовательно, фибробласты могут быть лучшими мишенями для генной терапии на основе клеток или ex vivo. 67 Фактически, было обнаружено, что внутрикожная инъекция нормальных человеческих фибробластов или генно-скорректированных фибробластов RDEB восстанавливает синтез и стабильное отложение коллагена типа VII в дермо-эпидермальном соединении на модели мыши. 68 Пилотное исследование однократной внутрикожной инъекции аллогенных фибробластов на людях было проведено у нескольких пациентов с РДЭБ. 69 Хотя донорские фибробласты не определялись через 2 недели после инъекции, повышенная экспрессия коллагена типа VII наблюдалась у этих пациентов через 2 недели и 3 месяца. Исследователи предположили, что основным эффектом инъекции аллогенных фибробластов является повышение уровня гепарин-связывающего EGF-подобного фактора роста (HB-EGF) и увеличение собственных уровней мРНК COL7A1 реципиента, которые повышаются в кератиноцитах и фибробластах. 70 Таким образом, было показано, что внутрикожные инъекции аллогенных фибробластов обладают терапевтическим потенциалом у людей с RDEB. Внутрикожная инъекция фибробластов очень болезненна, и может потребоваться анестезия. Для практического применения потребуется дополнительная оптимизация частоты инъекций и количества клеток. 71
Другой привлекательный подход — терапия рекомбинантными белками. Исследования на мышах показали, что внутрикожные инъекции рекомбинантного человеческого коллагена VII типа приводят к отложению коллагена VII типа в дермо-эпидермальном соединении. 72 Внутривенная инъекция рекомбинантного коллагена VII типа мышам RDEB вызвала системное отложение коллагена VII типа в раненой коже. Кроме того, было обнаружено, что местный тип VII не только улучшает фенотип на мышиной модели RDEB, но также может ускорять заживление ран на коже. 73 На одну инфузию может потребоваться около 60–120 мг коллагена типа VII, чтобы перенести результаты внутривенной инъекции от модели мышей весом 35 г, которым инъецировали 60 мкг рекомбинантного белка коллагена VII типа, пациентам с РДЭБ весом 35–70 кг. 74,75 Потребуется создание крупномасштабной производственной системы и установление стандартов продукции. Кроме того, перед проведением клинических исследований безопасности рекомбинантного белка коллагена типа VII будут обязательными фармакологические и токсикологические исследования с использованием крупных животных.
Поскольку БЭ — моногенное заболевание, корригирующая генная терапия — идеальный вариант. Сообщалось о нескольких терапевтических подходах, основанных на генетике, таких как небольшие молекулы, ex vivo перенос гена COL7A1 дикого типа, транс-сплайсинг и гомологичная направленная репарация с использованием методов редактирования генов. 76–79 Аминогликозиды, которые могут индуцировать считывание PTC, восстанавливают функциональный коллаген типа VII в кератиноцитах, полученных от пациентов с RDEB, заболевание которых вызвано бессмысленными мутациями. 76 Для подходов к переносу генов ex vivo в RDEB было разработано использование ретровирусных векторов, лентивирусных векторов и невирусных векторов на основе системы интегразы бактериофага φ31 для переноса полноразмерной комплементарной ДНК COL7A1 (8,9 т.п.н.) в RDEB кератиноциты и / или фибробласты из-за большого размера генома. 80 Эти подходы представляют собой эффективные способы производства функционального коллагена VII типа в клетках, полученных от пациентов с RDEB. Фактически, коррекция гена ex vivo COL7A1 и трансплантация кератиноцитарных пластин пациентам с RDEB находится на ранней стадии исследования фазы I. 71 Однако эндогенная коррекция мутаций в геноме может дать преимущества по сравнению с использованием вирусных векторов, поскольку онкогенные события могут возникать из-за случайного инсерционного мутагенеза трансгена с помощью вирусных векторов.Кроме того, эндогенная коррекция обеспечивает физиологический транскрипционный контроль гена COL7A1 . Чтобы преодолеть эту проблему, был разработан опосредованный сплайсосомами транс-сплайсинг, в котором репарация гена осуществляется путем рекомбинации эндогенной пре-мРНК-мишени и экзогенно доставленной молекулы РНК, называемой пре-транс-сплайсинговой молекулой (PTM). Перенесенная пре-мРНК, которая короче, чем полноразмерная кДНК COL7A1 , заменяется кодирующей последовательностью дикого типа из PTM путем транс-сплайсинга в 3′- или 5′-последовательность мишени для создания новой перепрограммированной мРНК. 77 Недавно было сообщено, что сконструированные сайт-специфические эндонуклеазы в комбинации с матрицей репарации ДНК могут заменять мутантный аллель последовательностью дикого типа посредством репарации гомологичной рекомбинации. Нуклеазы «цинковые пальцы» (ZFN), эффекторные белки, подобные активаторам транскрипции (TALEN), или кластерные регуляторные промежуточные палиндромные повторы (CRISPR), основанные на управляемых РНК-эндонуклеазах ДНК, продемонстрировали способность нацеливаться на специфические ДНК-связывающие домены. 81 Следует отметить, что TALEN успешно использовались для исправления мутаций гена COL7A1 в первичных фибробластах пациентов с RDEB. 79
У некоторых пациентов с RDEB появляются участки здоровой кожи, на которых никогда не образуется пузырей, на которых показана повышенная экспрессия коллагена VII типа и восстановление закрепляющих фибрилл. 82 В этих регионах спонтанная коррекция гена COL7A1 , или «ревертантный мозаицизм», наблюдается при анализе генов, феномен, который был обнаружен при нескольких наследственных кожных заболеваниях, включая другой тип EB. 83 Клеточная терапия с использованием ревертантных кератиноцитов идеальна, потому что эти клетки не нуждаются в генной коррекции.Первоначальные попытки, которые были сосредоточены на культивировании кожи, полученной из ревертантных кератиноцитов от пациента с JEB, не увенчались успехом, поскольку степень реверсии не поддерживалась в трансплантатах. 84 Перфорация ревертантной кожи пациента с JEB с неизлечимыми язвами привела к клиническому улучшению, а иммунофлуоресценция и секвенирование ДНК показали сохранение реверсии на участках реципиента. 85
Индуцированные плюрипотентные стволовые (iPS) клетки, которые могут быть получены от пациентов, приобретают способность к мультипотенциальной дифференцировке и неограниченному самообновлению. 86 iPS-клетки способны дифференцироваться не только в кератиноциты и фибробласты, но также в гемопоэтические стволовые клетки и мезенхимальные стволовые клетки, в которых могут образовываться кожно-слизистые пузыри и дифференцироваться в кератиноциты и фибробласты. 87–90 Пациенты с РДЭБ обычно имеют труднодоступные слизистые поражения, а трансплантировать кожные трансплантаты и вводить культивированные клетки непосредственно в эти области сложно. 91 Эти стволовые клетки и дополнительные модификации, включая локальное применение рекомбинантных сигналов хоминга (таких как HMGB1), могут дополнять использование системной клеточной терапии. 89 Некоторым группам уже удалось получить iPS-клетки, полученные из ревертантных кератиноцитов пациентов с EB и из фибробластов, скорректированных с помощью генной терапии на основе гомологичной рекомбинации с использованием TALEN. 79,91,92 Следовательно, терапия iPS-клетками с использованием естественных (ревертантный мозаицизм) или искусственно (редактирование генов) обработанных генами соматических клеток, полученных от пациентов с DEB, является очень перспективной.
Заключение
Глобальные группы защиты интересов пациентов (DebRA, http: // www.debra-international.org/homepage.html) и клиническая сеть центров EB и экспертов (EB-CLINET, http://www.eb-clinet.org/home.html) были созданы, поскольку клинические проявления EB так сурово. Таким образом, EB имеет высокий авторитет не только в клинической и исследовательской областях, но и среди широкой общественности. Развитие методов лечения — актуальный вопрос. За два десятилетия, прошедшие с тех пор, как был выяснен патогенный механизм DEB, молекулярная биология и клеточная биология резко развились, и недавние данные, полученные на животных моделях, трансляционных исследованиях и начальных клинических испытаниях DEB, показывают большие перспективы.Необходимы дальнейшие исследования для практического применения новых эффективных методов лечения.
Благодарности
Эта работа была поддержана Национальным институтом биомедицинских инноваций, Япония.
Раскрытие информации
Автор сообщает об отсутствии конфликта интересов в этой работе.
Список литературы
1. | Fine JD, Bruckner-Tuderman L, Eady RA, et al. Унаследованный буллезный эпидермолиз: обновленные рекомендации по диагностике и классификации. J Am Acad Dermatol . 2014. 70 (6): 1103–1126. |
2. | Fine JD, Eady RA, Bauer EA и др. Классификация наследственного буллезного эпидермолиза (БЭ): отчет Третьего международного консенсусного совещания по диагностике и классификации БЭ. J Am Acad Dermatol . 2008. 58 (6): 931–950. |
3. | Шинкума С., Макмиллан Дж. Р., Симидзу Х. Ультраструктура и молекулярный патогенез буллезного эпидермолиза. Клин Дерматол . 2011. 29 (4): 412–419. |
4. | McKenna KE, Walsh MY, Bingham EA. Буллезный эпидермолиз в Северной Ирландии. Br J Dermatol . 1992. 127 (4): 318–321. |
5. | Kho YC, Rhodes LM, Robertson SJ, et al. Эпидемиология буллезного эпидермолиза у антиподов: Австралазийский реестр буллезного эпидермолиза с акцентом на буллезный узловой эпидермолиз Герлица. Арка Дерматол . 2010. 146 (6): 635–640. |
6. | Fine JD. Унаследованный буллезный эпидермолиз. Орфанет J Редкий Dis . 2010; 5: 12. |
7. | Shinkuma S, Natsuga K, Nishie W, Shimizu H. Буллезный эпидермолиз в Японии. Дерматол Клин . 2010; 28 (2): 431–432, xvi. |
8. | Horn HM, Priestley GC, Eady RA, Tidman MJ.Распространенность буллезного эпидермолиза в Шотландии. Br J Dermatol . 1997. 136 (4): 560–564. |
9. | Dang N, Murrell DF. Анализ мутаций и характеристика мутаций COL7A1 при дистрофическом буллезном эпидермолизе. Эксперимент Дерматол . 2008. 17 (7): 553–568. |
10. | Ryynanen M, Knowlton RG, Parente MG, Chung LC, Chu ML, Uitto J. Коллаген человека типа VII: генетическая связь гена (COL7A1) на хромосоме 3 с доминантным дистрофическим эпидермолизом Bullosa. Ам Дж. Хам Генет . 1991. 49 (4): 797–803. |
11. | Christiano AM, Greenspan DS, Lee S, Uitto J. Клонирование человеческого коллагена VII типа. Полная первичная последовательность альфа-1 (VII) цепи и идентификация внутригенных полиморфизмов. Дж. Биол. Хим. . 1994. 269 (32): 20256–20262. |
12. | Chung HJ, Uitto J. Коллаген типа VII: заякоривающий фибриллярный белок, виноватый в дистрофическом буллезном эпидермолизе. Дерматол Клин . 2010; 28 (1): 93–105. |
13. | Christiano AM, Hoffman GG, Chung-Honet LC, et al. Структурная организация гена коллагена VII типа человека (COL7A1), состоящего из большего числа экзонов, чем любой ранее охарактеризованный ген. Геномика . 1994. 21 (1): 169–179. |
14. | Christiano AM, Rosenbaum LM, Chung-Honet LC, et al. Большой неколлагеновый домен (NC-1) коллагена типа VII является аминоконцевым и химерным.Гомология с белком хрящевого матрикса, доменами III типа фибронектина и доменами A фактора фон Виллебранда. Хум Мол Генет . 1992. 1 (7): 475–481. |
15. | Чен М., Маринкович М.П., Вейс А. и др. Взаимодействие аминоконцевого неколлагенового (NC1) домена коллагена VII типа с компонентами внеклеточного матрикса. Возможная роль в адгезии между эпидермисом и кожей человека. Дж. Биол. Хим. . 1997. 272 (23): 14516–14522. |
16. | Бриттингем Р., Уитто Дж., Фертала А. Связывание с высоким сродством домена NC1 коллагена VII с ламинином 5 и коллагеном IV. Biochem Biophys Res Commun . 2006. 343 (3): 692–699. |
17. | Burgeson RE. Коллаген VII типа, закрепляющие фибриллы и буллезный эпидермолиз. Дж Инвест Дерматол . 1993. 101 (3): 252–255. |
18. | Ryynanen J, Sollberg S, Parente MG, Chung LC, Christiano AM, Uitto J. Экспрессия гена коллагена типа VII в культивируемых клетках человека и в коже плода. Обильные уровни мРНК и белка в кератиноцитах эпидермиса. Дж. Клин Инвест . 1992. 89 (1): 163–168. |
19. | Bruckner-Tuderman L, Nilssen O, Zimmermann DR, et al. Иммуногистохимический анализ и анализ мутаций демонстрируют, что проколлаген VII преобразуется в коллаген VII посредством удаления домена NC-2. Дж. Ячейка Биол . 1995. 131 (2): 551–559. |
20. | Fine JD, Bauer EA, Briggaman RA, et al. Пересмотренные клинические и лабораторные критерии подтипов наследственного буллезного эпидермолиза. Консенсусный отчет Подкомитета по диагностике и классификации Национального реестра буллезного эпидермолиза. J Am Acad Dermatol . 1991; 24 (1): 119–135. |
21. | Fine JD, Eady RA, Bauer EA и др.Пересмотренная система классификации наследственного буллезного эпидермолиза: отчет Второго международного консенсусного совещания по диагностике и классификации буллезного эпидермолиза. J Am Acad Dermatol . 2000. 42 (6): 1051–1066. |
22. | Хамада Т., Фукуда С., Исии Н. и др. Японская семья с доминантным претибиальным дистрофическим буллезным эпидермолизом: идентификация новой замены глицина в трехспиральном коллагеновом домене коллагена VII типа. J Dermatol Sci . 2009. 54 (3): 212–214. |
23. | McGrath JA, Schofield OM, Eady RA. Пруригинозный буллезный эпидермолиз: дистрофический буллезный эпидермолиз с отличительными клинико-патологическими особенностями. BrJ Дерматол . 1994. 130 (5): 617–625. |
24. | Dharma B, Moss C, McGrath JA, Mellerio JE, Ilchyshyn A. Доминирующий дистрофический буллезный эпидермолиз, проявляющийся как семейная дистрофия ногтей. Клин Эксп Дерматол . 2001. 26 (1): 93–96. |
25. | van den Akker PC, Mellerio JE, Martinez AE, et al. Обратный тип рецессивного дистрофического буллезного эпидермолиза вызывается специфическими заменами аргинина и глицина в коллагене VII типа. Дж. Мед Генет . 2011. 48 (3): 160–167. |
26. | Fine JD, Osment LS, Gay S. Буллезный дистрофический эпидермолиз. Новый вариант, характеризующийся прогрессирующим симметричным центростремительным поражением с рубцеванием. Арка Дерматол . 1985. 121 (8): 1014–1017. |
27. | Хашимото К., Мацумото М., Якобелли Д. Преходящий буллезный дермолиз новорожденного. Арка Дерматол . 1985. 121 (11): 1429–1438. |
28. | Fassihi H, Diba VC, Wessagowit V, et al. Транзиторный буллезный дермолиз новорожденных в трех поколениях. Br J Dermatol . 2005. 153 (5): 1058–1063. |
29. | Брукнер-Тудерман Л. Дистрофический буллезный эпидермолиз: патогенез и клиника. Дерматол Клин . 2010. 28 (1): 107–114. |
30. | Савамура Д., Низеки Х., Миягава С., Шинкума С., Симидзу Х. Новая мутация индел COL7A1 8068del17insGA вызывает доминирующий дистрофический буллезный эпидермолиз. Br J Dermatol . 2006. 154 (5): 995–997. |
31. | Intong LR, Murrell DF.Унаследованный буллезный эпидермолиз: новые диагностические критерии и классификация. Клин Дерматол . 2012; 30 (1): 70–77. |
32. | Файн Дж. Д., Джонсон Л.Б., Вайнер М., Сучиндран С. Желудочно-кишечные осложнения наследственного буллезного эпидермолиза: совокупный опыт Национального реестра буллезного эпидермолиза. J Педиатр Gastroenterol Nutr . 2008. 46 (2): 147–158. |
33. | Gamelli RL.Проблемы питания пациентов с острыми и хроническими ожогами. Связь с буллезным эпидермолизом. Арка Дерматол . 1988. 124 (5): 756–759. |
34. | Фигейра EC, Murrell DF, Coroneo MT. Офтальмологические поражения при наследственном буллезном эпидермолизе. Дерматол Клин . 2010. 28 (1): 143–152. |
35. | Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть I.Эпителиально связанные ткани. J Am Acad Dermatol . 2009. 61 (3): 367–384; викторина 385–366. |
36. | Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть II. Другие органы. J Am Acad Dermatol . 2009. 61 (3): 387–402; викторина 403–384. |
37. | Fine JD, Johnson LB, Weiner M, et al. Унаследованный буллезный эпидермолиз и риск смерти от почечной недостаточности: опыт Национального реестра буллезного эпидермолиза. Am J Kidney Dis . 2004. 44 (4): 651–660. |
38. | Файн Дж. Д., Холл М., Вайнер М., Ли К. П., Сучиндран С. Риск кардиомиопатии при наследственном буллезном эпидермолизе. Br J Dermatol . 2008; 159 (3): 677–682. |
39. | Файн Дж.Д., Джонсон Л. J Am Acad Dermatol . 2009. 60 (2): 203–211. |
40. | Hintner H, Stingl G, Schuler G, et al. Иммунофлуоресцентное картирование антигенных детерминант в дермо-эпидермальном соединении при механобуллезных заболеваниях. Дж Инвест Дерматол . 1981. 76 (2): 113–118. |
41. | Fine JD, Johnson LB, Cronce D, et al. Интрацитоплазматическая задержка коллагена VII типа и доминирующий дистрофический буллезный эпидермолиз: устранение дефекта после прекращения или заметного улучшения активности заболевания. Дж Инвест Дерматол . 1993. 101 (2): 232–236. |
42. | Радкевич-Браун О., Швайдер Т. Буллезный дермолиз новорожденного: четыре новых случая и клинический обзор. Педиатр дерматол . 2013. 30 (6): 736–740. |
43. | Гарделла Р., Кастилья Д., Постераро П. и др. Корреляция генотип-фенотип у итальянских пациентов с дистрофическим буллезным эпидермолизом. Дж Инвест Дерматол .2002. 119 (6): 1456–1462. |
44. | Такеичи Т., Нанда А., Лю Л. и др. Влияние секвенирования нового поколения на диагностику в клинике генетических заболеваний кожи. Эксперимент Дерматол . 2013. 22 (12): 825–831. |
45. | van den Akker PC, Jonkman MF, Rengaw T и др. Международный реестр пациентов с дистрофическим буллезным эпидермолизом: онлайн-база данных пациентов с дистрофическим буллезным эпидермолизом и их мутациями COL7A1. Хум Мутат . 2011. 32 (10): 1100–1107. |
46. | Савамура Д., Накано Х., Мацузаки Ю. Обзор буллезного эпидермолиза. Дж Дерматол . 2010. 37 (3): 214–219. |
47. | Toyonaga E, Nishie W, Komine M, et al. Пропущенный экзон в COL7A1 определяет клинические фенотипы дистрофического буллезного эпидермолиза. Br J Dermatol . 2015. 172 (4) 1141–1144. |
48. | Мурата Т., Масунага Т., Ишико А., Симидзу Х., Нисикава Т. Различия в повторяющихся мутациях COL7A1 при дистрофическом буллезном эпидермолизе: этнические и повторяющиеся во всем мире мутации. Arch Dermatol Res . 2004. 295 (10): 442–447. |
49. | Меллерио Дж. Э., Салас-Аланис Дж. К., Амая-Герра М. и др. Повторяющаяся мутация сдвига рамки считывания в экзоне 19 гена коллагена типа VII (COL7A1) у мексиканских пациентов с рецессивным дистрофическим буллезным эпидермолизом. Эксперимент Дерматол . 1999. 8 (1): 22–29. |
50. | Меллерио Дж. Э., Даннил М. Г., Эллисон В. и др. Рекуррентные мутации в гене коллагена VII типа (COL7A1) у пациентов с рецессивным дистрофическим буллезным эпидермолизом. Дж Инвест Дерматол . 1997. 109 (2): 246–249. |
51. | Mohammedi R, Mellerio JE, Ashton GH, Eady RA, McGrath JA. Повторяющаяся мутация COL7A1, R2814X, у британских пациентов с рецессивным дистрофическим буллезным эпидермолизом. Клин Эксп Дерматол . 1999. 24 (1): 37–39. |
52. | Меллерио Дж. Э., Салас-Аланис Дж. К., Таламантес М. Л. и др. Повторяющаяся мутация замещения глицина, G2043R, в гене коллагена VII типа (COL7A1) при доминантном дистрофическом буллезном эпидермолизе. Br J Dermatol . 1998. 139 (4): 730–737. |
53. | Denyer JE. Лечение ран у детей с буллезным эпидермолизом. Дерматол Клин . 2010; 28 (2): 257–264, viii – ix. |
54. | Langan SM, Williams HC. Систематический обзор рандомизированных контролируемых исследований методов лечения наследственных форм буллезного эпидермолиза. Клин Эксп Дерматол . 2009. 34 (1): 20–25. |
55. | Папа Е, Лара-Корралес I, Меллерио Дж. И др. Консенсусный подход к уходу за ранами при буллезном эпидермолизе. J Am Acad Dermatol .2012. 67 (5): 904–917. |
56. | Gonzalez ME. Обследование и лечение новорожденного с буллезным эпидермолизом. Семин Перинатол . 2013. 37 (1): 32–39. |
57. | Mellerio JE. Инфекция и колонизация при буллезном эпидермолизе. Дерматол Клин . 2010; 28 (2): 267–269, ix. |
58. | van der Kooi-Pol MM, Duipmans JC, Jonkman MF, van Dijl JM.Взаимодействие «хозяин-патоген» у пациентов с буллезным эпидермолизом, колонизированных Staphylococcus aureus. Int J Med Microbiol . 2014. 304 (2): 195–203. |
59. | Flohr C, Heague J, Leach I, English J. Местная системная аргирия, индуцированная сульфадиазином серебра, у пациента с тяжелым генерализованным дистрофическим буллезным эпидермолизом. Br J Dermatol . 2008. 159 (3): 740–741. |
60. | Haynes L.Питание детей при буллезном эпидермолизе. Дерматол Клин . 2010; 28 (2): 289–301, х. |
61. | Shinkuma S, Sawamura D, Fujita Y и др. Долгосрочное наблюдение за культивированием эпидермального аутотрансплантата у пациента с рецессивным дистрофическим буллезным эпидермолизом. Acta Derm Venereol . 2014; 94 (1): 98–99. |
62. | Wollina U, Konrad H, Fischer T. Recessive epidermolysis bullosa dystrophicans (Hallopeau-Siemens) — улучшение заживления ран с помощью аутологичных эпидермальных трансплантатов на этерифицированной мембране гиалуроновой кислоты. Дж Дерматол . 2001. 28 (4): 217–220. |
63. | Чино Т., Тамай К., Ямазаки Т. и др. Перенос клеток костного мозга в кровообращение плода может улучшить генетические кожные заболевания, обеспечивая фибробласты коже и вызывая иммунную толерантность. Ам Дж. Патол . 2008. 173 (3): 803–814. |
64. | Толар Дж., Исида-Ямамото А., Риддл М. и др. Облегчение буллезного эпидермолиза путем переноса клеток костного мозга дикого типа. Кровь . 2009. 113 (5): 1167–1174. |
65. | Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Трансплантация костного мозга при рецессивном дистрофическом буллезном эпидермолизе. N Engl J Med . 2010. 363 (7): 629–639. |
66. | Толар Дж., Вагнер Дж. Аллогенные клетки крови и костного мозга для лечения тяжелого буллезного эпидермолиза: восстановление внеклеточного матрикса. Ланцет . 2013. 382 (9899): 1214–1223. |
67. | Гото М., Савамура Д., Ито К. и др. Фибробласты обладают большим потенциалом в качестве клеток-мишеней, чем кератиноциты, в генной терапии COL7A1 дистрофического буллезного эпидермолиза. Дж Инвест Дерматол . 2006. 126 (4): 766–772. |
68. | Woodley DT, Remington J, Huang Y и др. Внутривенно введенные человеческие фибробласты домой к кожным ранам доставляют коллаген VII типа и способствуют заживлению ран. Мол тер . 2007. 15 (3): 628–635. |
69. | Вонг Т., Гаммон Л., Лю Л. и др. Возможности клеточной терапии фибробластов при рецессивном дистрофическом буллезном эпидермолизе. Дж Инвест Дерматол . 2008; 128 (9): 2179–2189. |
70. | Надь Н., Алмаани Н., Танака А. и др. HB-EGF индуцирует экспрессию COL7A1 в кератиноцитах и фибробластах: возможный механизм, лежащий в основе терапии аллогенных фибробластов при рецессивном дистрофическом буллезном эпидермолизе. Дж Инвест Дерматол . 2011. 131 (8): 1771–1774. |
71. | Hsu CK, Wang SP, Lee JY, McGrath JA. Лечение наследственного буллезного эпидермолиза: новости и перспективы на будущее. Ам Дж. Клин Дерматол . 2014; 15 (1): 1–6. |
72. | Вудли Д.Т., Кин Д.Р., Ата Т. и др. Инъекция рекомбинантного коллагена человека VII типа восстанавливает функцию коллагена при дистрофическом буллезном эпидермолизе. Нат Мед . 2004. 10 (7): 693–695. |
73. | Ван Х, Гасри П., Амир М. и др. Местное применение рекомбинантного коллагена типа VII проникает в дермо-эпидермальное соединение и способствует закрытию ран. Мол тер . 2013. 21 (7): 1335–1344. |
74. | Woodley DT, Wang X, Amir M, et al. Внутривенно введенный рекомбинантный человеческий коллаген типа VII попадает в кожные раны и восстанавливает целостность кожи при дистрофическом буллезном эпидермолизе. Дж Инвест Дерматол . 2013; 133 (7): 1910–1913. |
75. | Овнанян А. Системная белковая терапия при рецессивном дистрофическом буллезном эпидермолизе: насколько мы далеки от клинического перевода? Дж Инвест Дерматол . 2013; 133 (7): 1719–1721. |
76. | Коган Дж., Вайнштейн Дж., Ван Х и др. Аминогликозиды восстанавливают полноразмерный коллаген типа VII, преодолевая преждевременные кодоны терминации: терапевтическое значение при дистрофическом буллезном эпидермолизе. Мол тер . 2014. 22 (10): 1741–1752. |
77. | Murauer EM, Gache Y, Gratz IK, et al. Функциональная коррекция экспрессии коллагена VII типа при дистрофическом буллезном эпидермолизе. Дж Инвест Дерматол . 2011. 131 (1): 74–83. |
78. | Чен М., Касахара Н., Кин Д.Р. и др. Восстановление экспрессии и функции коллагена VII типа при буллезном дистрофическом эпидермолизе. Нат Генет . 2002. 32 (4): 670–675. |
79. | Осборн М.Дж., Старкер К.Г., МакЭлрой А.Н. и др. Генная коррекция буллезного эпидермолиза на основе TALEN. Мол тер . 2013. 21 (6): 1151–1159. |
80. | Ванден Овер М.Дж., Толар Дж. Достижения в понимании и лечении дистрофического буллезного эпидермолиза. F1000Прайм Представитель . 2014; 6: 35. |
81. | Li HL, Накано Т., Хотта А. Генетическая коррекция с использованием сконструированных нуклеаз для генной терапии. Разница в росте разработчиков . 2014; 56 (1): 63–77. |
82. | Алмаани Н., Надь Н., Лю Л. и др. Ревертантный мозаицизм при рецессивном дистрофическом буллезном эпидермолизе. Дж Инвест Дерматол . 2010. 130 (7): 1937–1940. |
83. | Йонкман М.Ф., Шеффер Х., Стулп Р. и др.Ревертантный мозаицизм при буллезном эпидермолизе, вызванный конверсией митотических генов. Ячейка . 1997; 88 (4): 543–551. |
84. | Гостынски А., Девяене ФК, Пасмуой А.М., Пас Х. Х., Йонкман МФ. Удаление адгезива для удаления эпидермиса при буллезном соединительном эпидермолизе для лечения ревертантных клеток. Br J Dermatol . 2009. 161 (2): 444–447. |
85. | Gostynski A, Pasmooij AM, Jonkman MF.Успешная терапевтическая трансплантация ревертантной кожи при буллезном эпидермолизе. J Am Acad Dermatol . 2014; 70 (1): 98–101. |
86. | Такахаши К., Яманака С. Индукция плюрипотентных стволовых клеток из культур эмбриональных и взрослых фибробластов мыши с помощью определенных факторов. Ячейка . 2006. 126 (4): 663–676. |
87. | Frobel J, Hemeda H, Lenz M, et al. Эпигенетическое омоложение мезенхимальных стромальных клеток, полученных из индуцированных плюрипотентных стволовых клеток. Репродукция стволовых клеток . 2014. 3 (3): 414–422. |
88. | Itoh M, Kiuru M, Cairo MS, Christiano AM. Генерация кератиноцитов из плюрипотентных стволовых клеток, вызванных нормальным и рецессивным дистрофическим буллезным эпидермолизом. Proc Natl Acad Sci U S A . 2011. 108 (21): 8797–8802. |
89. | Тамай К., Ямазаки Т., Чино Т. и др. PDGFRalpha-позитивные клетки в костном мозге мобилизуются высокоподвижным блоком 1 группы (HMGB1) для регенерации поврежденного эпителия. Proc Natl Acad Sci U S A . 2011. 108 (16): 6609–6614. |
90. | Ито М., Умегаки-Арао Н., Гуо З, Лю Л., Хиггинс, Калифорния, Кристиано А.М. Создание трехмерных эквивалентов кожи, полностью восстановленных из индуцированных человеком плюрипотентных стволовых клеток (ИПСК). PloS One . 2013; 8 (10): e77673. |
91. | Толар Дж., МакГрат Дж. А., Ся Л. и др. При рецессивном дистрофическом буллезном эпидермолизе специфические для пациента индуцированные естественным образом ревертированные гены плюрипотентные стволовые клетки. Дж Инвест Дерматол . 2014. 134 (5): 1246–1254. |
92. | Umegaki-Arao N, Pasmooij AM, Itoh M, et al. Индуцированные плюрипотентные стволовые клетки из ревертантных кератиноцитов человека для лечения буллезного эпидермолиза. Научный перевод . 2014; 6 (264): 264ra164. |
определение буллезного эпидермолиза и синонимов эпидермолиза-буллезного (на английском языке)
Буллезный эпидермолиз ( EB ) — это наследственное заболевание соединительной ткани, вызывающее волдыри на коже и слизистых оболочках с частотой 1/50 000.Его степень тяжести варьируется от легкой до летальной. Это вызвано мутацией в молекуле адгезии клеток интегрина α6β4 в альфа- или бета-субъединице.
В результате кожа становится очень хрупкой. Незначительное механическое трение или травма приведет к разделению слоев кожи и образованию пузырей. Люди с этим заболеванием имеют повышенный риск рака кожи, и у многих в конечном итоге он будет диагностирован как осложнение хронического повреждения кожи.
Кожа трехслойная; внешний слой называется эпидермисом, следующий слой — дерма, за ним следует подкожный слой или гиподерма.У людей со здоровой кожей между слоями есть белковые якоря из коллагена, которые не позволяют им двигаться независимо друг от друга (срезание). У людей, рожденных с БЭ, в двух верхних слоях кожи отсутствуют протеиновые якоря, которые удерживают их вместе, и любое действие, вызывающее трение между ними (например, трение или давление), приведет к образованию волдырей и болезненных язв. Больные БЭ сравнивали язвы с ожогами третьей степени. [1]
Состояние было доведено до сведения общественности Великобритании в документальном фильме канала Channel 4 «Мальчик, у которого отвалилась кожа» , в котором рассказывается о жизни и смерти Джонни Кеннеди, англичанина с ЭБ. [2] В Соединенных Штатах то же самое можно сказать о мощном документальном фильме HBO «Моя плоть и кровь» 2003 года.
«Дети-бабочки» — это термин, который часто используют для описания пациентов младшего возраста, потому что, как говорят, их кожа хрупкая, как крылья бабочки. [3]
Дети с этим заболеванием также были описаны как «дети из хлопковой ваты», [4] [5] , а в Южной Америке используется термин «дети с хрустальной кожей». [6]
Эпидемиология
Приблизительно 50 из 1 миллиона живорождений имеют диагноз БЭ, а 9 из 1 миллиона — среди населения.Из этих случаев приблизительно 92% относятся к EBS, 5% — к DEB, 1% — к JEB и 2% — к неклассифицированным. Несущая частота колеблется от 1 из 333 для Junctional до 1 из 450 для Dystrophic. Несущая частота для симплексного режима в этой статье не указана, но предполагается, что она намного выше, чем для JEB или DEB. [ необходима ссылка ]
Заболевание встречается у всех расовых и этнических групп во всем мире и затрагивает оба пола. [7] [8]
Текущее клиническое исследование в Университете Миннесоты включало трансплантацию костного мозга двухлетнему ребенку, который является одним из двух братьев с БЭ.Процедура прошла успешно, что убедительно свидетельствует о том, что лекарство могло быть найдено. Вторая трансплантация также была сделана старшему брату ребенка, а третья трансплантация запланирована для ребенка из Калифорнии. В конечном итоге клиническое испытание будет включать трансплантаты 30 пациентам. [9] Однако тяжелая иммуносупрессия, необходимая для трансплантации костного мозга, вызывает значительный риск серьезных инфекций у пациентов с крупномасштабными волдырями и эрозиями. Действительно, по крайней мере четыре пациента умерли в ходе подготовки или проведения трансплантации костного мозга от буллезного эпидермолиза из небольшой группы пациентов, лечившихся на данный момент.
Классификация
Мальчик пяти лет с врожденным буллезным эпидермолизом
Буллезный эпидермолиз относится к группе наследственных заболеваний, которые включают образование волдырей после тривиальной травмы, и которые можно разделить на следующие типы [10] [11] : 596 :
Простой буллезный эпидермолиз
Буллезный узловой эпидермолиз
Буллезный дистрофический эпидермолиз
Другой генетический
| OMIM | Имя | Локус | Ген |
|---|---|---|---|
| 609638 | буллезный эпидермолиз акантолитический летальный | 6п24 | DSP |
Другое
Лечение
Недавние исследования были сосредоточены на изменении смеси кератинов, производимых в коже.Известно 54 гена кератина, 28 генов промежуточных филаментов типа I и 26 генов типа II, которые работают как гетеродимеры. Многие из них имеют существенное структурное и функциональное сходство, но специализируются на разных типах клеток или условиях, в которых они обычно образуются. Если лекарство может изменить баланс производства в сторону неповрежденного гена кератина, симптомы могут быть уменьшены. Например, сульфорафан, соединение, обнаруженное в брокколи, снижает образование пузырей на модели мышей до такой степени, что пораженные детеныши не могут быть идентифицированы визуально при введении беременным мышам (5 мкмоль / день = 0. M Петр Маринкович, доктор медицинских наук, доцент кафедры дерматологии и программ эпителиальной биологии Медицинского центра Стэнфордского университета; Жан Поль Ортон, доктор медицины, заведующий кафедрой дерматологии, профессор больницы L’Archet, Университет Ниццы, Франция; Майкл Дж. Уэллс, доктор медицины, доцент кафедры дерматологии Центра медицинских наук Техасского технологического университета; Ван Перри, доктор медицины, доцент кафедры медицины отделения дерматологии Научного центра здравоохранения Техасского университета; Джоэл М. Гельфанд, доктор медицинских наук, MSCE, медицинский директор отдела клинических исследований, доцент кафедры дерматологии, младший научный сотрудник Центра клинической эпидемиологии и биостатистики Пенсильванского университета; Уильям Д. Джеймс, доктор медицины, профессор дерматологии Пола Р. Гросса Медицинского факультета Пенсильванского университета; Заместитель председателя, программный директор Департамента дерматологии системы здравоохранения Пенсильванского университета (3 декабря 2007 г.). Майкл Кернс et al. (4 сентября 2007 г.). «Перепрограммирование биосинтеза кератина сульфорафаном восстанавливает целостность кожи при простом буллезном эпидермолизе». Proc Natl Acad Sci U S. A. 104 (36): 14460–14465. DOI: 10.1073 / pnas.0706486104. PMC 1964870. PMID 17724334. //www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1964870.
Внешние ссылки
| ||||||||||||||||||||||||||||
.