Дефицит лизосомной кислой липазы | Строкова Т.В., Багаева М.Э., Матинян И.А.
Статья посвящена редкому наследственному заболеванию, связанному с дефицитом лизосомной кислой липазы
Дефицит лизосомной кислой липазы (ДЛКЛ) – редкое аутосомно-рецессивное заболевание лизосомального накопления, вызванное повреждающими мутациями гена LIPA, кодирующего синтез фермента – лизосомной кислой липазы. Ген LIPA, кодирующий лизосомную кислую липазу (гидролазу эфиров холестерина), расположен на хромосоме 10, имеет 10 экзонов. Пациенты с ДЛКЛ являются, как правило, либо гомозиготами, либо сложными гетерозиготами по мутациям гена LIPA. У некоторых пациентов могут иметь место скрытые мутации [1]. По разным источникам, частота встречаемости ДЛКЛ составляет 1:40 000–1:300 000 [2, 3]. Предполагаемая частота встречаемости в России – 1:100 000–1:150 000.
В норме после связывания с рецептором на поверхности клетки липопротеиды низкой плотности (ЛПНП), в составе которых, как известно, большая часть холестерина находится в виде эфиров холестерина, проникают в клетку, транспортируются внутри эндосомы, сливаются с лизосомой, где фермент лизосомная кислая липаза осуществляет гидролиз эфиров холестерина и триглицеридов до свободного холестерина и жирных кислот. Внутриклеточное накопление свободного холестерина посредством сложных биохимических механизмов приводит к подавляющему воздействию на рецепторы ЛПНП, уменьшению поступления в клетку и снижению синтеза холестерина, а также усилению его этерификации. Вследствие внутриклеточного накопления жирных кислот ингибируется образование фосфолипидов и триглицеридов [4, 5].
Внутриклеточное накопление свободного холестерина посредством сложных биохимических механизмов приводит к подавляющему воздействию на рецепторы ЛПНП, уменьшению поступления в клетку и снижению синтеза холестерина, а также усилению его этерификации. Вследствие внутриклеточного накопления жирных кислот ингибируется образование фосфолипидов и триглицеридов [4, 5].
При недостаточности лизосомной кислой липазы нарушается распад эфиров холестерина и триглицеридов и происходит их накопление в лизосомах клеток печени, селезенки, кровеносных сосудов, слизистой тонкого кишечника, надпочечников, на поверхности которых определяется высокая плотность рецепторов ЛПНП.
В гепатоцитах больных с ДЛКЛ увеличение синтеза холестерина приводит к увеличению продукции холестерина липопротеидов очень низкой плотности и его секреции. Это, в свою очередь, стимулирует образование холестерина ЛПНП и, таким образом, может выступать в качестве важного содействующего фактора в развитии гиперхолестеринемии при ДЛКЛ [6].
Отложение эфиров холестерина в ткани печени приводит к мелкокапельной жировой дистрофии (также называемого микровезикулярным стеатозом), что вызывает повреждение клеток печени. Маркером этого повреждения является повышенный уровень трансаминаз (АСТ, АЛТ) в сыворотке крови. Поврежденные гепатоциты замещаются соединительной тканью, развиваются фиброз и цирроз печени. Нарушение функции клеток сопровождается дислипидемией с повышенным уровнем общего холестерина в сыворотке крови, высоким уровнем холестерина ЛПНП, аполипопротеина В, низким уровнем холестерина липопротеинов высокой плотности (ЛПВП), повышенным или нормальным уровнем триглицеридов. Изменения в липидограмме соответствуют изменениям при дислипидемии IIb, что приводит к ускоренному развитию атеросклероза, сердечно-сосудистых нарушений и преждевременной смертности [2, 7].
Отложение эфиров холестерина в клетках слизистой оболочки кишечника вызывает развитие синдрома мальабсорбции, проявляющегося разной степенью выраженности диареи, стеатореи, а также синдромом избыточного бактериального роста, метеоризмом.
Увеличение селезенки может быть связано с накоплением эфиров холестерина в клетках, а также с прогрессированием заболевания, портальной гипертензией.
Возраст начала заболевания и темпы его прогрессирования в значительной степени вариабельны и могут быть связаны с природой лежащих в основе мутаций и количественным показателем остаточной ферментативной активности [8, 9].
Клиника
Исторически выделяют две клинические формы ДЛКЛ (табл. 1). Первая форма характеризуется быстро прогрессирующим течением. В 1956 г. впервые был описан случай тяжелого истощения, гепатоспленомегалии, кальцификации надпочечников с последующим летальным исходом на 1-м месяце жизни [10]. Эта форма болезни получила название болезни Вольмана. Дебют заболевания – от неонатального периода до 3–6 мес. Патологическое накопление липидов в селезенке, надпочечниках, лимфатических узлах, слизистой тонкой кишки, эндотелии сосудов и скелетных мышцах обусловливают характер патологии. Клиническими проявлениями болезни Вольмана являются персистирующая рвота, диарея, стеаторея, иногда желтуха. При осмотре обращает на себя внимание увеличенный в размере живот, что связано не только с гепатоспленомегалией, но и со вздутием кишечника, часто, по причине его паралитической непроходимости. Течение заболевания характеризуется прогрессирующей анемией, субфебрилитетом, задержкой физического и психомоторного развития. Нарастают вялость, апатия, астения, гиперрефлексия. Быстро развиваются фиброз и цирроз печени, связанные с массивным накоплением эфиров холестерина и триглицеридов [11]. При использовании визуализирующих методов исследования (рентгенологических, ультразвуковых) определяются кальцинаты надпочечников у половины больных [12]. Прогрессирующие гипотрофия, неврологические расстройства, надпочечниковая и печеночно-клеточная недостаточность, интеркуррентные инфекции приводят к летальному исходу на первом году жизни. Активность фермента составляет менее 1% от нормы [13]. Летальный исход наступает на фоне быстро прогрессирующей полиорганной недостаточности в возрасте 6–12 мес. [8].
При осмотре обращает на себя внимание увеличенный в размере живот, что связано не только с гепатоспленомегалией, но и со вздутием кишечника, часто, по причине его паралитической непроходимости. Течение заболевания характеризуется прогрессирующей анемией, субфебрилитетом, задержкой физического и психомоторного развития. Нарастают вялость, апатия, астения, гиперрефлексия. Быстро развиваются фиброз и цирроз печени, связанные с массивным накоплением эфиров холестерина и триглицеридов [11]. При использовании визуализирующих методов исследования (рентгенологических, ультразвуковых) определяются кальцинаты надпочечников у половины больных [12]. Прогрессирующие гипотрофия, неврологические расстройства, надпочечниковая и печеночно-клеточная недостаточность, интеркуррентные инфекции приводят к летальному исходу на первом году жизни. Активность фермента составляет менее 1% от нормы [13]. Летальный исход наступает на фоне быстро прогрессирующей полиорганной недостаточности в возрасте 6–12 мес. [8].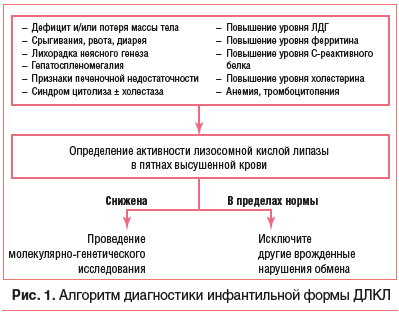
Вторая форма болезни – болезнь накопления эфиров холестерина (БНЭХ). Заболевание впервые было описано в 1963 г., когда Fredrickson [13] сообщил о случае выраженной гиперхолестеринемии, гепатомегалии и накопления эфиров холестерина при исследовании биопсийного материала печени 12-летнего мальчика. Данная форма ДЛКЛ характеризуется более поздним началом клинических проявлений, медленным прогрессированием. Возраст начала болезни варьирует в большом диапазоне – от 2-х до 25 лет, чаще – до 10 лет. Cамое позднее начало клинической манифестации заболевания выявлено у 44-летнего мужчины и 68-летней женщины [2]. Основной симптом – гепатомегалия, обнаруживаемая у подавляющего большинства пациентов [2, 14]. Клинические проявления варьируют от бессимптомного до тяжелого поражения печени. Ранний биохимический маркер поражения печени – синдром цитолиза – проявляется повышенным уровнем АСТ и АЛТ в сыворотке крови от 1,5 до 5–7 норм. При прогрессировании заболевания нарастают спленомегалия, фиброз печени, портальная гипертензия, белково-энергетическая недостаточность. Активность фермента при данной форме заболевания составляет 1–12% от нормы [15]. Заболевание можно заподозрить на основании указанной клинической симптоматики. Проводятся лабораторные и инструментальные исследования.
Активность фермента при данной форме заболевания составляет 1–12% от нормы [15]. Заболевание можно заподозрить на основании указанной клинической симптоматики. Проводятся лабораторные и инструментальные исследования.
ДЛКЛ является орфанным заболеванием, в связи с чем распознается не сразу, и многие пациенты длительный период времени наблюдаются с диагнозами: семейная гиперхолестеринемия, семейная комбинированная гиперлипидемия, неалкогольный стеатогепатит, неалкогольная жировая болезнь печени, криптогенный гепатит или цирроз печени. Поэтому знание симптомов данного заболевания, правильная интерпретация клинико-лабораторных результатов позволят в более ранние сроки установить диагноз и назначить патогенетическую терапию.
Диагностика
Обследование пациентов включает целый ряд исследований, которые помогут сориентировать врача в нужном направлении диагностического поиска.
В клиническом анализе крови обращают на себя внимание нормоцитарная анемия, переходящая в гипохромную анемию. Анемия более выражена у детей с болезнью Вольмана. У части пациентов с ДЛКЛ определяется повышенная СОЭ. При прогрессировании цирроза печени при ДЛКЛ нарастают лейко- и тромбоцитопения.
Анемия более выражена у детей с болезнью Вольмана. У части пациентов с ДЛКЛ определяется повышенная СОЭ. При прогрессировании цирроза печени при ДЛКЛ нарастают лейко- и тромбоцитопения.
Основными биохимическими характеристиками заболевания являются синдром цитолиза, гиперхолестеринемия, дислипидемия (повышение ЛПНП, триглицеридов, аполипопротеина В, снижение ЛПВП) [16]. На стадии цирроза печени о нарастании белково-синтетической недостаточности свидетельствуют гипопротеинемия, гипоальбуминемия, возможно снижение уровня холестерина до нормальных значений.
Основные диагностические маркеры ДЛКЛ, подтверждающие заболевание: дефицит активности лизосомной кислой липазы и мутации гена LIPA (однако в некоторых случаях при использовании стандартных методов клинической диагностики мутации не обнаруживаются) [2]. Уровень лизосомной кислой липазы определяется в сухом пятне крови на специальных фильтрах, применяемых, например, для неонатального скрининга. Преимуществами метода являются небольшой объем образца, транспортировка в специализированные лаборатории при температуре окружающей среды, а также возможность длительного хранения.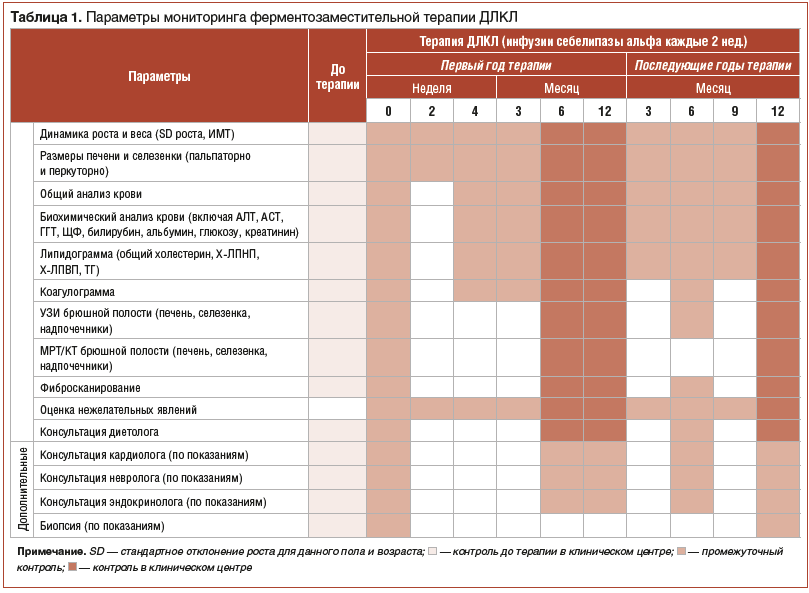
Молекулярно-генетическое исследование позволяет выявить мутации в гене LIPA кодирующих и прилегающих интронных областей. Большинство пациентов являются гомозиготами или сложными гетерозиготами по мутациям гена LIPA. У некоторых больных могут иметь место протяженные делеции, не выявляемые при проведении обычного генетического скрининга [2].
Важными визуализирующими методами диагностики являются УЗИ органов брюшной полости и забрюшинного пространства, рентгенография, МРТ.
УЗИ при подозрении на ДЛКЛ оценивает состояние печени, селезенки, надпочечников, диаметра стволов воротной и селезеночной вен. У пациентов с ДЛКЛ определяются увеличение размеров печени (верхне-нижних размеров правой и левой долей), гиперэхогенность и мелкоочаговая диффузная неоднородность паренхимы печени, у части больных – ослабление ультразвука в дистальных отделах паренхимы печени. У части пациентов отмечается увеличение селезенки, а при дальнейшем прогрессировании болезни – расширение ствола воротной и селезеночной вен, что свидетельствует о нарастании портальной гипертензии. При УЗИ можно определить кальцинаты в надпочечниках, которые говорят также в пользу болезни Вольмана.
При УЗИ можно определить кальцинаты в надпочечниках, которые говорят также в пользу болезни Вольмана.
При проведении рентгенологического исследования органов брюшной полости определяются увеличенные надпочечники полулунной или пирамидной формы с наличием точечных очагов кальцификации по всей паренхиме.
При проведении МРТ органов брюшной полости выявляют гепатоспленомегалию, при болезни Вольмана определяются гипертрофированные брыжеечные и периаортальные лимфатические узлы. Проведение МРТ необходимо для оценки степени жировой дистрофии печени, а также характера течения заболевания и эффективности проводимой патогенетической терапии [17].
При выявлении признаков портальной гипертензии, проводят эзофагогастродуоденоскопию для определения варикозного расширения вен пищевода и оценки риска возможного кровотечения.
Биопсию печени с последующим исследованием морфологической картины рекомендуют, если нет возможности применить иные, неинвазивные методы диагностики ДЛКЛ [18, 19]. При морфологическом исследовании ткани печени определяется стадия заболевания.
При морфологическом исследовании ткани печени определяется стадия заболевания.
Макропрепарат биоптата печени имеет желто-оранжевую окраску, а при проведении гистологического анализа выявляются различные степени портального и перилобулярного фиброза, а также выраженный микровезикулярный стеатоз в связи с накоплением эфиров холестерина и триглицеридов в лизосомах гепатоцитов [20]. Характерная черта – наличие в значительной степени гипертрофированных клеток Купфера и портальных макрофагов с пенистой, окрашиваемой в коричневатый цвет, резко ШИК-положительной цитоплазмой.
Результаты визуализирующих методов обследования и биопсии печени являются диагностическими в отношении ДЛКЛ. Учитывая, что ДЛКЛ – мультисистемное заболевание, при необходимости требуется проведение исследований сердечно-сосудистой системы (ЭКГ, Эхо-КГ, допплерография сосудов головы и шеи и т. д.).
Дифференциальный диагноз проводят с различными болезнями печени, нарушениями липидного обмена. Тактика дифференциального диагноза зависит от формы ДЛКЛ. При болезни Вольмана необходимо исключить все заболевания с гепатоспленомегалией, поражением печени, кишечника, мозга: болезнь Ниманна – Пика, тип IA; болезнь Гоше, тип II; GM1- ганглиозидоз; болезнь Фабера; атипичные формы галактосиалидоза; галактоземию, тип I; наследственную непереносимость фруктозы; тирозинемию, тип I. БНЭХ необходимо дифференцировать с большим количеством различных болезней, протекающих с гепатомегалией, синдромом цитолиза и нарушениями липидного обмена, а также с неалкогольной жировой болезнью печени.
При болезни Вольмана необходимо исключить все заболевания с гепатоспленомегалией, поражением печени, кишечника, мозга: болезнь Ниманна – Пика, тип IA; болезнь Гоше, тип II; GM1- ганглиозидоз; болезнь Фабера; атипичные формы галактосиалидоза; галактоземию, тип I; наследственную непереносимость фруктозы; тирозинемию, тип I. БНЭХ необходимо дифференцировать с большим количеством различных болезней, протекающих с гепатомегалией, синдромом цитолиза и нарушениями липидного обмена, а также с неалкогольной жировой болезнью печени.
Лечение
Единственным патогенетическим методом лечения является длительная ферментная заместительная терапия препаратом Себелипаза альфа. Лечение ДЛКЛ заключается в восстановлении сниженного уровня фермента для предотвращения накопления эфиров холестерина и триглицеридов и, как следствие, восстановлении нормальной функции органа. Препарат Себелипаза альфа – это рекомбинантная кислая лизосомная липаза человека. Действующее вещество попадает в лизосомы клеток за счет связывания со специфическими рецепторами, где оказывает непосредственный терапевтический эффект. Доза препарата зависит от формы болезни: при болезни Вольмана она составляет 3 мг/кг/сут, при БНЭХ – 1 мг/кг внутривенно капельно. Режим введения при БНЭХ – 1 раз в 2 нед., при болезни Вольмана, при наличии показаний, возможно более частое проведение инфузий – 1 раз в неделю. В клинических исследованиях продемонстрировано улучшение биохимических показателей (нормализация уровня трансаминаз, показателей липидограммы), морфологической картины в биоптатах печени, а также увеличение выживаемости пациентов [21]. Препарат разрешен к применению во многих европейских странах, США, Японии. На территории РФ ожидается регистрация препарата.
Доза препарата зависит от формы болезни: при болезни Вольмана она составляет 3 мг/кг/сут, при БНЭХ – 1 мг/кг внутривенно капельно. Режим введения при БНЭХ – 1 раз в 2 нед., при болезни Вольмана, при наличии показаний, возможно более частое проведение инфузий – 1 раз в неделю. В клинических исследованиях продемонстрировано улучшение биохимических показателей (нормализация уровня трансаминаз, показателей липидограммы), морфологической картины в биоптатах печени, а также увеличение выживаемости пациентов [21]. Препарат разрешен к применению во многих европейских странах, США, Японии. На территории РФ ожидается регистрация препарата.
При отсутствии патогенетической терапии проводится симптоматическая терапия. При нарушении всасывания и задержке развития, а также для коррекции липидного обмена необходима консультация диетолога в целях оптимизации питания. Клинические проявления надпочечниковой недостаточности требуют назначения заместительной гормональной терапии, выраженная анемия и тромбоцитопения – переливания крови и ее компонентов.
Прогноз зависит от формы заболевания и выраженности клинических проявлений. При своевременной диагностике заболевания и начале патогенетической терапии прогноз благоприятный. При болезни Вольмана без проведения фермент-заместительной терапии летальный исход наступает на первом году жизни.
В связи с высоким риском болезни (25%) в семьях, имеющих ребенка с ДЛКЛ, требуется обследование всех детей, т. к. заболевание в течение длительного времени может протекать без выраженной клинической симптоматики.
При наличии в семье ребенка с подтвержденным диагнозом ДЛКЛ для определения риска повторного рождения ребенка с данным заболеванием необходимо медико-генетическое консультирование. Возможна пренатальная диагностика: определение активности кислой липазы в культурах ворсин хориона и молекулярно-генетическое исследование на 9–11-й неделе беременности или исследование амниотической жидкости на 20–22-й неделе [1].
В нашем центре было обследовано 20 детей с ДЛКЛ, все дети – с БНЭХ. Мы неоднократно докладывали о результатах обследования и характере течения болезни [22]. Ниже приведен клинический случай ДЛКЛ у мальчика с установленным диагнозом ДЛКЛ.
Мы неоднократно докладывали о результатах обследования и характере течения болезни [22]. Ниже приведен клинический случай ДЛКЛ у мальчика с установленным диагнозом ДЛКЛ.
Клинический случай ДЛКЛ (БНЭХ)
Ребенок от первой беременности, протекавшей на фоне многоводия, роды первые, срочные, физиологические. Вес при рождении 3460 г, длина 52 см. Оценка по шкале Апгар 8/9 баллов. Находился на грудном вскармливании до 1 года 3-х месяцев. Профилактические прививки проведены по календарю вакцинации. Перенесенные заболевания: ОРЗ 2–3 раза в год. Семейный анамнез: у отца – артериальная гипертензия, желчнокаменная болезнь (холецистэктомия), у бабушки по линии матери – желчнокаменная болезнь, холецистэктомия.
В возрасте 3-х лет при подготовке к плановой аденомэктомии в биохимическом анализе крови было выявлено повышение АЛТ до 182 ед/л, АСТ до 159 ед/л. Печень увеличена на +2–2,5 см, селезенка увеличена на +0,5 см. Выставлен диагноз: дискинезия желчевыводящих путей. Реактивный панкреатит. Гепатомегалия. В 3,5 года при повторном обследовании получены следующие результаты: печень увеличена на +4-5 см, селезенка — на +2–3 см, АЛТ 286 ед/л, АСТ 176 ед/л. Исключены хронические вирусные гепатиты, болезнь Вильсона, дефицит a1-антитрипсина и целиакия. В дальнейшем у ребенка сохранялся синдром цитолиза, отмечалось нарастание размеров печени и селезенки. В 4,5 года: печень увеличена на +5–6 см, селезенка – на +3 см. По данным эластографии: стадия фиброза F1-F2 (по METAVIR). По данным УЗИ брюшной полости: увеличение размеров печени и селезенки, диффузная неоднородность, поглощение ультразвука на 1/4. МРТ брюшной полости: печень расположена обычно, в размерах значительно увеличена преимущественно за счет правой доли. Наибольший передне-задний размер – 130 мм (правая доля), 50 мм (левая доля), вертикальный на уровне правой доли – 156 мм, левой доли – 49 мм. Контуры ровные, четкие. Селезенка увеличена, 110×93×37 мм, контуры четкие, ровные, паренхима однородная. Желчный пузырь, поджелудочная железа, почки, надпочечники не изменены.
Гепатомегалия. В 3,5 года при повторном обследовании получены следующие результаты: печень увеличена на +4-5 см, селезенка — на +2–3 см, АЛТ 286 ед/л, АСТ 176 ед/л. Исключены хронические вирусные гепатиты, болезнь Вильсона, дефицит a1-антитрипсина и целиакия. В дальнейшем у ребенка сохранялся синдром цитолиза, отмечалось нарастание размеров печени и селезенки. В 4,5 года: печень увеличена на +5–6 см, селезенка – на +3 см. По данным эластографии: стадия фиброза F1-F2 (по METAVIR). По данным УЗИ брюшной полости: увеличение размеров печени и селезенки, диффузная неоднородность, поглощение ультразвука на 1/4. МРТ брюшной полости: печень расположена обычно, в размерах значительно увеличена преимущественно за счет правой доли. Наибольший передне-задний размер – 130 мм (правая доля), 50 мм (левая доля), вертикальный на уровне правой доли – 156 мм, левой доли – 49 мм. Контуры ровные, четкие. Селезенка увеличена, 110×93×37 мм, контуры четкие, ровные, паренхима однородная. Желчный пузырь, поджелудочная железа, почки, надпочечники не изменены.
В возрасте 5 лет печень выступала на 7–8 см, селезенка – на 2–3 см из-под края реберной дуги. Впервые проведено исследование липидограммы в 6 лет: холестерин 5,65–6,18–6,28 ммоль/л, ЛПНП 6,5 ммоль/л, ЛПВП 0,92 ммоль/л. В этом же возрасте по данным УЗИ брюшной полости выявлены: гепатоспленомегалия, диффузная неоднородность и гиперэхогенность паренхимы печени, расширение диаметра стволов воротной и селезеночной вен. При КТ брюшной полости обнаружены диффузные изменения паренхимы печени по типу жировой дистрофии. Эзофагогастродуоденоскопия: терминальный эзофагит, гастрит, дуоденит. Варикозного расширения вен пищевода не было обнаружено. Ребенку проведена пункционная биопсия печени. Результаты морфологического исследования биоптата: в срезе определяются до 6 полных и неполных портальных трактов, 4 портальных тракта фиброзированы, 3 порто-портальные септы, 2 свободно лежащие септы. Зоны перипортального перицеллюлярного фиброза, гепатоциты разделены на розетки, скудные лимфоидные инфильтраты в этих зонах. Небольшие зоны перигепатоцеллюлярного фиброза в дольках. Гепатоциты округлой формы, часть их несколько увеличены в размерах, балочное строение не определяется. Гепатоциты имеют очень светлую, мелкозернистую цитоплазму, большинство содержат вакуоли. При проведении ШИК-реакции обнаружено накопление ШИК-позитивного вещества в гепатоцитах, вакуоли остаются хорошо различимы. Заключение: морфологическая картина болезней накопления. Можно подозревать накопление эфиров холестерина. Явления хронического перипортального гепатита низкой степени активности без учета склероза, вероятно реактивного характера. Индекс гистологической активности по Knodell – 4 балла (I-А, II-C, III-B). Индекс склероза по Десмет – 2 балла (умеренный перигепатоцеллюлярный фиброз). Поэтому было проведено исследование лизосомной кислой липазы в клетках крови: ее уровень составил 27,8 нМ/мг/час (норма 30–118 нМ/мг/час, лейкоциты), что послужило поводом для проведения молекулярно-генетического исследования. Методом прямого автоматического секвенирования исследованы все кодирующие экзоны (2–10) гена LIPA, а также прилегающие интронные области.
Небольшие зоны перигепатоцеллюлярного фиброза в дольках. Гепатоциты округлой формы, часть их несколько увеличены в размерах, балочное строение не определяется. Гепатоциты имеют очень светлую, мелкозернистую цитоплазму, большинство содержат вакуоли. При проведении ШИК-реакции обнаружено накопление ШИК-позитивного вещества в гепатоцитах, вакуоли остаются хорошо различимы. Заключение: морфологическая картина болезней накопления. Можно подозревать накопление эфиров холестерина. Явления хронического перипортального гепатита низкой степени активности без учета склероза, вероятно реактивного характера. Индекс гистологической активности по Knodell – 4 балла (I-А, II-C, III-B). Индекс склероза по Десмет – 2 балла (умеренный перигепатоцеллюлярный фиброз). Поэтому было проведено исследование лизосомной кислой липазы в клетках крови: ее уровень составил 27,8 нМ/мг/час (норма 30–118 нМ/мг/час, лейкоциты), что послужило поводом для проведения молекулярно-генетического исследования. Методом прямого автоматического секвенирования исследованы все кодирующие экзоны (2–10) гена LIPA, а также прилегающие интронные области. В экзоне 7 выявлена мутация с.796G>T в гетерозиготном состоянии, приводящая к преждевременной терминации трансляции аминокислотной последовательности р.Gly266Х. В экзоне 8 выявлена мутация с.894G
В экзоне 7 выявлена мутация с.796G>T в гетерозиготном состоянии, приводящая к преждевременной терминации трансляции аминокислотной последовательности р.Gly266Х. В экзоне 8 выявлена мутация с.894G
На основании анамнеза, результатов клинико-лабораторного обследования (цитолиз, гепатоспленомегалия, гиперхолестеринемия, снижение уровня кислой липазы), а также молекулярно-генетического исследования крови был установлен диагноз: ДЛКЛ. Болезнь накопления эфиров холестерина.
По жизненным показаниям ребенку в возрасте 6 лет 10 мес. рекомендовано проведение патогенетической терапии незарегистрированным в России препаратом Себелипаза альфа в дозе 1 мг/кг внутривенно капельно 1 раз в 2 нед. Длительность терапии в настоящее время составляет 15 мес. На фоне фермент-заместительной терапии, которую ребенок переносит удовлетворительно (побочных эффектов не зарегистрировано), отмечается уменьшение размеров печени и селезенки: печень выступает на 3 см из-под края реберной дуги по правой среднеключичной линии, селезенка не пальпируется. Наблюдается снижение уровня активности трансаминаз (рис. 1). По данным УЗИ, несмотря на сохраняющуюся гепатоспленомегалию, признаков портальной гипертензии не выявлено.
Наблюдается снижение уровня активности трансаминаз (рис. 1). По данным УЗИ, несмотря на сохраняющуюся гепатоспленомегалию, признаков портальной гипертензии не выявлено.
Течение заболевания у мальчика с ДЛКЛ характеризуется наличием гепатоспленомегалии, быстрым прогрессированием с формированием признаков портальной гипертензии к 6-летнему возрасту, синдромом цитолиза, минимальным повышением уровня холестерина, более выраженным увеличением ЛПНП и хорошим ответом на проведение фермент-заместительной терапии, что требует ее продолжения. Препарат назначается для предотвращения развития тяжелых инвалидизирующих осложнений.
Заключение
Таким образом, при ДЛКЛ наблюдается прогрессирующее поражение печени с гепатомегалией, повышенным уровнем трансаминаз, микровезикулярным стеатозом. Низкая распространенность, неспецифичность клинико-лабораторных признаков болезни объясняют ее недостаточную выявляемость.
Следует помнить, что болезнь Вольмана необходимо исключить у любого ребенка первых месяцев жизни с постоянной рвотой или диареей, отставанием в развитии, гепатоспленомегалией, синдромом мальабсорбции.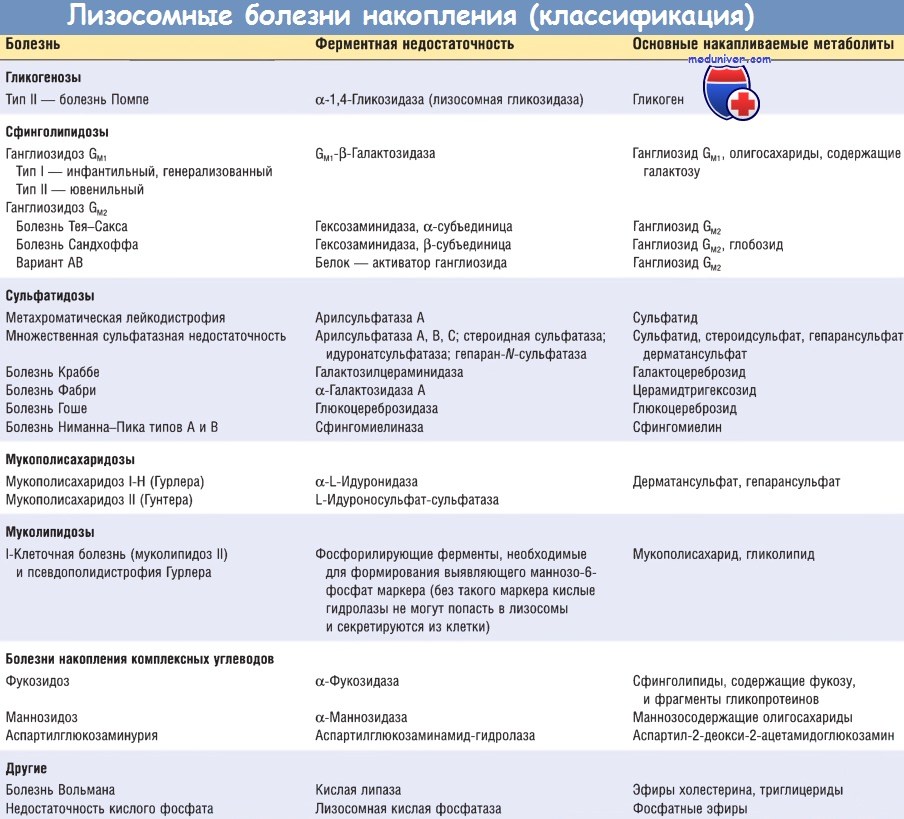 Выявление увеличенных надпочечников с кальцификацией – прямое показание к исследованию уровня кислой лизосомной липазы и молекулярно-генетическому исследованию. Идентичность липидных профилей при БНЭХ с большой группой заболеваний, особенно при выявлении синдрома цитолиза, свидетельствует о том, что необходимо исключать ДЛКЛ, если предполагаются наследственные нарушения липидного обмена.
Выявление увеличенных надпочечников с кальцификацией – прямое показание к исследованию уровня кислой лизосомной липазы и молекулярно-генетическому исследованию. Идентичность липидных профилей при БНЭХ с большой группой заболеваний, особенно при выявлении синдрома цитолиза, свидетельствует о том, что необходимо исключать ДЛКЛ, если предполагаются наследственные нарушения липидного обмена.
.
Дефицит липазы лизосомальной кислоты — Lysosomal acid lipase deficiency
Дефицит лизосомальной кислой липазы ( LAL-дефицит или LAL-D ) — это аутосомно-рецессивная врожденная ошибка метаболизма, которая приводит к тому, что организм не вырабатывает достаточно активного фермента лизосомальной кислой липазы (LAL) . Этот фермент играет важную роль в расщеплении жировых отложений ( сложных эфиров холестерина и триглицеридов ) в организме. Младенцы, дети и взрослые, страдающие дефицитом КЛЛ, испытывают ряд серьезных проблем со здоровьем. Недостаток фермента LAL может привести к накоплению жирового материала в ряде органов тела, включая печень , селезенку , кишечник , в стенках кровеносных сосудов и других важных органах.
Очень низкие уровни фермента LAL приводят к дефициту LAL. Дефицит КЛЛ обычно поражает младенцев первого года жизни. Накопление жира в стенках кишечника при раннем начале заболевания приводит к серьезным проблемам с пищеварением, включая мальабсорбцию — состояние, при котором кишечник не может усваивать питательные вещества и калории из пищи. Из-за этих пищеварительных осложнений пораженные младенцы обычно не могут расти и набирать вес с ожидаемой скоростью для их возраста ( неспособность развиваться ). По мере прогрессирования заболевания оно может вызвать опасное для жизни нарушение функции печени или печеночную недостаточность .
До 2015 года лечения не проводилось, и очень немногие дети с LAL-D доживали до первого года жизни. В 2015 году, по Фермент заместительной терапии , sebelipase альфа , был одобрен в США и ЕС. В 2016 году терапия была дополнительно одобрена в Японии .
Симптомы
Младенцы могут испытывать трудности с кормлением с частой рвотой, диареей, вздутием живота и неспособностью набрать вес, а иногда и с потерей веса.
По мере прогрессирования заболевания у младенцев увеличение накопления жира в печени приводит к другим осложнениям, включая пожелтение кожи и белков глаз ( желтуху ) и стойкую субфебрильную температуру. Ультразвуковое изучение показывает накопление мелового материала ( кальцификация ) в надпочечниках примерно в половине младенцев с LAL-D. Осложнения LAL-D со временем прогрессируют, что в конечном итоге приводит к опасным для жизни проблемам, таким как чрезвычайно низкий уровень циркулирующих эритроцитов (тяжелая анемия ), дисфункция или отказ печени и физическое истощение ( кахексия ).
Люди старшего возраста или взрослые обычно имеют широкий спектр признаков и симптомов, которые частично совпадают с другими расстройствами. У них может быть диарея, боль в животе, рвота или замедленный рост, что является признаком мальабсорбции . У них могут быть признаки проблем с желчными протоками , такие как зуд, желтуха, бледный стул или темная моча. Их кал может быть чрезмерно жирным . У них часто бывает увеличенная печень , заболевание печени, а под кожей , обычно вокруг век , могут быть желтоватые отложения жира . У взрослых заболевание часто не диагностируется. У человека в анамнезе может быть преждевременное сердечное заболевание или преждевременный инсульт.
У них часто бывает увеличенная печень , заболевание печени, а под кожей , обычно вокруг век , могут быть желтоватые отложения жира . У взрослых заболевание часто не диагностируется. У человека в анамнезе может быть преждевременное сердечное заболевание или преждевременный инсульт.
Причина
Дефицит лизосомальной кислой липазы — это генетическое заболевание, которое носит аутосомно-рецессивный характер . Это врожденная ошибка метаболизма, которая вызывает лизосомную болезнь накопления . Состояние вызвано мутацией гена LIPA, который отвечает за ген, кодирующий белок лизосомальной липазы (также называемый липазой лизосомальной кислоты или LAL), что приводит к потере нормальной функции белка. Когда КЛЛ функционирует нормально, она расщепляет холестериловые эфиры и триглицериды в частицах липопротеинов низкой плотности на свободный холестерин и свободные жирные кислоты, которые организм может повторно использовать; когда LAL не функционирует, сложные эфиры холестерина и триглицериды накапливаются в печени, селезенке и других органах. Накопление жира в стенках кишечника и других органах приводит к серьезным проблемам с пищеварением, включая мальабсорбцию , состояние, при котором кишечник не может усваивать питательные вещества и калории из пищи, постоянную и часто сильную рвоту, частую диарею, неприятный запах и т. жирный стул (стеаторея) и задержка роста.
Накопление жира в стенках кишечника и других органах приводит к серьезным проблемам с пищеварением, включая мальабсорбцию , состояние, при котором кишечник не может усваивать питательные вещества и калории из пищи, постоянную и часто сильную рвоту, частую диарею, неприятный запах и т. жирный стул (стеаторея) и задержка роста.
Дефицит лизосомальной кислой липазы возникает, когда у человека есть дефекты (мутации) в обеих копиях гена LIPA. Каждый родитель человека с дефицитом LAL несет одну копию дефектного гена LIPA. При каждой беременности родители с сыном или дочерью, страдающими дефицитом КЛЛ, имеют шанс 1 из 4 (25%) иметь еще одного больного ребенка. Человек, рожденный с дефектами обоих генов LIPA, не может производить достаточное количество фермента LAL.
Диагностика
Анализы крови могут показать анемию, и их липидные профили в целом аналогичны людям с более распространенной семейной гиперхолестеринемией , включая повышенный общий холестерин, повышенный холестерин липопротеинов низкой плотности, пониженный холестерин липопротеинов высокой плотности и повышенные сывороточные трансаминазы.
Результаты биопсии печени обычно показывают ярко-желто-оранжевый цвет, увеличенные, насыщенные липидами гепатоциты и клетки Купфера, микровезикулярный и макровезикулярный стеатоз, фиброз и цирроз. Единственные окончательные тесты — генетические, которые можно проводить любым количеством способов.
Скрининг
Поскольку дефицит КЛЛ передается по наследству, у каждого брата или сестры пораженного человека есть 25% вероятность наличия патологических мутаций в генах КЛЛ как от их матери, так и от отца, 50% вероятность наличия патологической мутации только в одном гене и 25% шанс отсутствия патологических мутаций. Генетическое тестирование членов семьи и генетическая пренатальная диагностика беременности у женщин из группы повышенного риска возможны, если выявлены члены семьи, несущие патологические мутации.
Управление
Дефицит КЛЛ можно лечить с помощью себелипазы альфа — рекомбинантной формы КЛЛ, одобренной в 2015 году в США и ЕС. Заболевание LAL поражает <0,2 из 10 000 человек в ЕС. По оценке аналитика Barclays, препарат будет стоить около 375 000 долларов США в год.
По оценке аналитика Barclays, препарат будет стоить около 375 000 долларов США в год.
Его вводят один раз в неделю внутривенно людям с быстро прогрессирующим заболеванием в первые шесть месяцев жизни. Людям с менее агрессивным заболеванием его назначают раз в две недели.
До того, как препарат был одобрен, лечение младенцев было в основном направлено на уменьшение конкретных осложнений и проводилось в специализированных центрах. Конкретные вмешательства для младенцев включали переход с молочной смеси или смеси из обычных бутылочек на специализированную смесь с низким содержанием жира, внутривенное кормление, антибиотики при инфекциях и заместительную стероидную терапию из-за опасений по поводу функции надпочечников.
Статины использовались у людей с LAL-D до утверждения себелипазы альфа; они помогли контролировать холестерин, но не замедлили повреждение печени; Трансплантация печени была необходима большинству пациентов.
Прогноз
Младенцы с дефицитом КЛЛ обычно проявляют признаки заболевания в первые недели жизни и, если их не лечить, умирают в течение 6–12 месяцев из-за полиорганной недостаточности. Дети старшего возраста или взрослые с LAL-D могут оставаться невыявленными или ошибочно диагностированными до тех пор, пока они не умрут раньше от сердечного приступа или инсульта или внезапно не умрут от печеночной недостаточности. Первая заместительная ферментная терапия была одобрена в 2015 году. В этих клинических испытаниях девять младенцев наблюдались в течение одного года; 6 из них прожили более одного года. Дети старшего возраста и взрослые наблюдались в течение 36 недель.
Дети старшего возраста или взрослые с LAL-D могут оставаться невыявленными или ошибочно диагностированными до тех пор, пока они не умрут раньше от сердечного приступа или инсульта или внезапно не умрут от печеночной недостаточности. Первая заместительная ферментная терапия была одобрена в 2015 году. В этих клинических испытаниях девять младенцев наблюдались в течение одного года; 6 из них прожили более одного года. Дети старшего возраста и взрослые наблюдались в течение 36 недель.
Эпидемиология
В зависимости от этнической принадлежности и географии распространенность оценивается от 1 на 40 000 до 1 на 300 000; на основании этих оценок болезнь может быть недооценена. Еврейские младенцы иракского или иранского происхождения, по-видимому, подвергаются наибольшему риску, согласно исследованию общины в Лос-Анджелесе, в которой показатель распространенности был равен 1 из 4200.
История
В 1956 году Моше Вольман вместе с двумя другими врачами опубликовал первое исследование дефицита КЛЛ у ребенка, родившегося от близкородственных персидских евреев; 12 лет спустя было опубликовано тематическое исследование старшего мальчика, которое оказалось первым тематическим исследованием LAL-D.
LAL-D исторически относился к 2 отдельным расстройствам:
- Болезнь Вольмана, проявляющаяся у пациентов грудного возраста
- Болезнь накопления холестерилового эфира, проявляющаяся у детей и взрослых
Примерно в 2010 году обе презентации стали известны как LAL-D, так как обе связаны с дефицитом фермента LAL.
В 2015 году фермент заместительная терапия , sebelipase альфа , был одобрен в США и ЕС для лечения LAL дефицита фермента человека. До утверждения этого препарата по состоянию на 2009 год двум самым старым в мире выжившим после ЛАЛ-Д были тогда 4 и 11 лет; оба они прошли курс лечения гемопоэтическими стволовыми клетками.
Направления исследований
Некоторым детям с LAL-D была проведена экспериментальная терапия, называемая трансплантацией гемопоэтических стволовых клеток (HSCT), также известная как трансплантация костного мозга , чтобы попытаться предотвратить обострение болезни. Данных немного, но известно, что существует высокий риск серьезных осложнений, включая смерть, болезнь трансплантат против хозяина .
Ссылки
внешние ссылки
Лизосомные болезни накопления
Концепция лизосомных болезней накопления сложилась в результате изучения
гликогеноза II типа (Помпе). Факт накопления гликогена в лизосомах вследствие
недостаточности a-глюкозидазы, а также данные, полученные при исследовании
других аномалий, позволили Эру определить врожденную лизосомную болезнь как
такое состояние, при котором:
1) определяется недостаточность какого-либо
одного лизосомного фермента
2) внутри связанных с лизосомами вакуолей
появляются необычные отложения (субстрат).
Это определение можно видоизменить,
включив в него дефекты одиночных генов, влияющие на один лизосомный фермент или
более, и тем самым распространить на такие болезни, как муколипидозы и множественная
сульфатазная недостаточность. Определение можно расширить и далее с тем, чтобы
оно распространялось на недостаточность и других белков, необходимых для
функционирования лизосом (активирующие ферменты разрушения сфинголипидов).
Данные биохимических и генетических исследований свидетельствуют о том, что эти
активирующие белки принимают участие в гидролизе некоторых субстратов.
Лизосомные болезни накопления объединяют большинство болезней накопления
липидов, мукополисахаридозы, муколипидозы, болезни накопления гликопротеинов и
другие. Недостаточность ферментов имеет аутосомно-рецессивную основу, за
исключением мукополисахаридоза II (МПС II) Хантера, который наследуется как
сцепленный с Х-хромосомой рецессивный признак, и болезни Фабри, которая сцеплена
с Х-хромосомой и часто проявляется у женщин. Органами-мишенями оказываются
обычные места разрушения той или иной макромолекулы. Например, у лиц с
нарушением процесса разрушения миелина в процесс вовлекается белое вещество
головного мозга, при нарушении процесса разрушения гликолипидов стромы
эритроцитов развивается гепатоспленомегалия, а при нарушении процесса
разрушения вездесущих мукополисахаридов — генерализованное повреждение тканей.
Накапливающийся материал часто вызывает висцеромегалию или макроцефалию, но
может развиться и вторичная атрофия, особенно мозга и мышц. Вообще симптоматика
соответствующих болезней обусловливается повреждающим действием накапливающихся
веществ, но часто неясно, каким именно образом они вызывают гибель или
дисфункцию клеток. Все эти болезни прогрессируют, и многие из них заканчиваются
смертью в детском или юношеском возрасте. Для окончательного диагноза наиболее
важны результаты определения конкретных ферментов в сыворотке, лейкоцитах или
культивируемых фибробластах кожи; соответствующие тесты выбирают, исходя из
клиники заболевания. Эти болезни имеют широкие фенотипические колебания, причем
многие из них связаны с возрастом, т. е. различают инфантильные, ювенильные и
взрослые их формы. Кроме того, при болезнях, обусловленных дефектом одиночного
гена, возможны различные сочетания висцеральных, костных и неврологических
аномалий.
Отдельные заболевания
Сфинголипозы.
Gmi-ганглиозидоз. Смгганглиозидоз обусловлен недостаточностью
р-галактозидазы. Инфантильная форма болезни проявляется уже при рождении или
вскоре после него (отставание развития, судорожные припадки, грубые черты лица,
отеки, гепатоспленомегалия, макроглоссия, вишнево-красные пятна на сетчатке и
явный мукополисахаридозоподобный множественный дизостоз). Смерть наступает
обычно в возрасте 1—2 лет. Ювенильная форма характеризуется более поздним
началом, большей продолжительностью жизни (более 5 лет), неврологическими
нарушениями и судорогами и менее тяжелыми повреждениями скелета и глаз. При
взрослой форме часто отмечают спондилоэпифизарную дисплазию, сходную с таковой
при МПС IV, помутнение роговицы и нормальный интеллект. Могут быть выражены
спастичность мышц и атаксия с незначительными костными аномалиями. Существуют
изоферменты р-галактозидазы, и разнообразие фенотипов связано с различными
мутациями одного и того же структурного гена.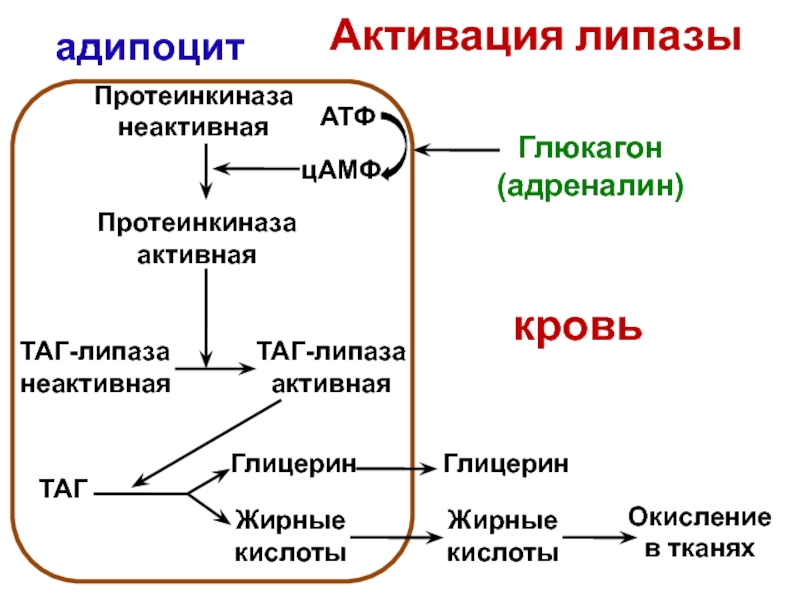 Все формы Смгганглиозидоза
Все формы Смгганглиозидоза
наследуются как аутосомный рецессивный признак.
GM2-ганглиозидоз. Болезнь Тея-Сакса — сравнительно
часто встречающаяся врожденная аномалия метаболизма: доказано уже несколько
тысяч случаев заболевания. Несмотря на то что по клинике этот синдром
напоминает болезнь Сендхоффа, генетически они различаются: в первом случае
отмечена недостаточность гексозаминидазы А, а во втором — гексозаминидаз А и В.
Еще один вид патологии (АВ-вариант GM2-ганглиозидоза)
характеризуется нормальной активностью гексозаминидаз А и В. Он обусловлен
недостаточностью белкового фактора (активатор), необходимого для реализации
активности фермента по отношению к природному субстрату. Клинические признаки
всех вариантов болезни, проявляющихся в младенчестве (инфантильные формы),
сходны и заключаются в отставании развития, что становится явным в возрасте 3—6
мес., и последующей быстро прогрессирующей неврологической симптоматике. Подозрения
на болезнь вызывают макроцефалия, судорожные припадки, вишнево-красные пятна на
сетчатке и резко выраженная реакция (чрезмерный испуг) на звук. Диагноз
подтверждается результатами определения ферментов. В большинстве случаев
недостаточность гексаминидазы, проявляющаяся позднее (ювенильная форма),
характеризуется деменцией, судорожными припадками и глазными симптомами, а у
некоторых больных развиваются атипичные дегенеративные изменения в спинном
мозге и мозжечке. У некоторых больных с ювенильной и взрослой формой появляются
признаки атрофии мышц спинального происхождения.
Болезнь Сендхоффа неаллельна болезни Тея-Сакса, тогда как ювенильные формы
недостаточности гексозаминидазы обычно аллельны последней. Болезнь Тея-Сакса — наиболее
частая форма недостаточности гексаминидазы. Все формы GM2-ганглиозидоза
наследуются как аутосомный рецессивный признак. Гексозаминидаза В состоит из
b-субъединиц, структурный ген которых расположен на 5-й хромосоме, тогда как
гексозаминидаза А включает и а-, и р-субъединицы, причем структурный ген
a-субъединицы локализуется на 15-й хромосоме. Таким образом, для синдрома Тея-Сакса
типичен дефект а-субъединицы, а при синдроме Сендхоффа — р-субъединицы.
Лейкодистрофии.
Галактозилцерамидный липидоз Краббе, или шаровидно-клеточная
лейкодистрофия, проявляется в младенчестве из-за недостаточности
галактозилцерамид-b-галактозидазы. Для него типичны начало в возрасте 2—6 мес.,
легкая возбудимость, гиперестезия, повышенная чувствительность к внешним
воздействиям, лихорадка неизвестного происхождения, атрофия зрительного нерва и
иногда судорожные припадки. Количество белка в спинномозговой жидкости обычно
увеличено. Тонус мышц и рефлексы с глубоких сухожилий вначале усилены, но затем
мышечный тонус снижается. Через 1—2 года резко усугубляется неврологическая
симптоматика и наступает смерть. Прижизненный диагноз основан на результатах
определения ферментов. Характерным и, возможно, специфическим признаком служат
шаровидные клетки в тканях нервной системы. Функция галактозилцерамид-b-галактозидазы
заключается в разрушении сульфатидов, образующихся из миелина. Повреждение
тканей настолько нарушает синтез миелина, что при аутопсии обычно не выявляют
увеличения абсолютного количества галакто-цереброзидного субстрата в тканях.
Галактозилцерамид-р-галактозидаза генетически отличается от р-галактозидазы, недостаточность
которой типична для GM1-ганглиозидоза.
Причиной метахромной лейкодистрофии (болезнь накопления липидов),
встречающейся с частотой 1:40000, служит недостаточность арилсульфатазы А (цереброзидсульфатаза).
Она проявляется в более позднем возрасте, нежели синдром Тея-Сакса или Краббе.
Больные дети начинают ходить, но в возрасте 2—5 лет походка у них часто
нарушается. Вначале мышечный тонус и рефлексы с глубоких сухожилий снижены, что
связано с повреждением периферических нервов. В первые 10 лет жизни болезнь
прогрессирует и проявляется атаксией, повышением тонуса мышц, декортикальным
или децеребральным статусом и, в конце концов, утратой всех контактов с
окружающим миром. Продолжительность жизни зависит от тщательности ухода и
кормления через носовой зонд или через гастростому.
Болезнь Нимана—Пика. Болезнь Нимана—Пика представляет собой сфингомиелиновый липидоз. При болезни типа А и В имеется явная недостаточность
сфингомиелиназы — фермента, гидролизующего сфингомиелин с образованием церамида
и фосфорилхолина. Наиболее частая форма А проявляется вскоре после рождения
гепатоспленомегалией, плохим самочувствием и неврологическими симптомами. На
сетчатке могут появиться вишнево-красные пятна, но судорожные припадки и
гиперспленизм редки. Форма В синдрома — сравнительно доброкачественный процесс,
проявляющийся гепатоспленомегалией, недостаточностью сфингомиелиназы и иногда
инфильтратами в легких; однако неврологическая симптоматика при этой форме
синдрома отсутствует. Для формы С характерен сфингомиелиновый липидоз,
прогрессирующие неврологические нарушения в детском возрасте и сохранность
(вплоть до нормы) активности сфингомиелиназы. При синдроме Нимана — Пика типа Е
определяют висцеральный сфингомиелиновый липидоз без неврологических нарушений
и недостаточности сфингомиелиназы. Биохимическая основа типов С, D и Е синдрома
не выяснена. У многих больных с синдромом гистиоцитов цвета морской волны
выявляют недостаточность сфингомиелиназы; у других больных с этим синдромом
дефекты метаболизма остаются неясными.
Болезнь Гоше. Болезнь Гоше представляет собой глюкозилцерамидный липидоз, обусловленный
недостаточностью глюкозилцерамидазы. Инфантильная форма характеризуется ранним
началом, выраженной гепатоспленомегалией и выраженными прогрессирующими
неврологическими нарушениями, приводящими к ранней смерти. Взрослая форма
относится, вероятно, к наиболее частой разновидности лизосомной болезни
накопления. Больные с ювенильной и взрослой формами встречались в одних и тех
же семьях, но они имеют разных родителей, что свидетельствует об аллельности
этих форм.
Все формы синдрома Гоше наследуются как аутосомный рецессивный признак. Несмотря
на то, что этот вариант болезни принято называть взрослой формой синдрома Гоше,
он часто проявляется еще в детстве. Критерием взрослой формы служит отсутствие
неврологических нарушений. Клинически эта форма проявляется либо случайно
обнаруживаемой спленомегалией, либо тромбоцитопенией, обусловленной
гиперспленией. Кроме того, больного могут беспокоить боли в костях или
патологические переломы, включая асептический некроз головки бедренной кости и
компрессию позвонков. Боли в костях, сопровождающиеся повышением температуры
тела, иногда называют псевдоостеомиелитом. Могут выявляться инфильтраты в
легких, легочная гипертензия и умеренное нарушение функций печени. Характерно
повышение в сыворотке уровня кислой фосфатазы. При всех формах синдрома Гоше в
костном мозге встречаются своеобразные «нагруженные» клетки, но определение
фермента всё же необходимо, так как клетки Гоше могут определяться и у больных
с гранулоцитарным лейкозом и миеломой.
Болезнь Фабри. При болезни Фабри из-за недостаточности а-галактозидазы А происходит
накопление тригексозида — галактозилгалактозилглюкозилцерамида. Синдром
наследуется как признак, сцепленный с Х-хромосомой, и особенно выражен у лиц
мужского пола. Он развивается обычно в зрелом возрасте. Если симптоматика
появляется в детском возрасте, то она, скорее всего, принимает форму болевой
нейропатии. Синдром часто диагностируют лишь после развития прогрессирующего
повреждения почек, т.е. в возрасте после 20—40 лет. Тромбозы сосудов могут
происходить в детском возрасте. Смерть чаще всего наступает от почечной недостаточности,
обычно в возрасте после 30—40 лет. У женщин — гетерозигот болезнь протекает
легче. Чаще всего у них выявляют дистрофию роговицы, хотя могут иметь место и
все другие проявления.
Недостаточность кислой липазы. Эта аномалия лежит в основе двух патологий с разным
фенотипом. Болезнь Вольмана представляет собой тяжелую аномалию с началом в
раннем возрасте, выраженной гепатоспленомегалией, анемией, рвотой, нарушением
развития и характерной кальцинацией надпочечников. Неврологическая симптоматика
минимальна по сравнению с выраженной соматической. Болезнь накопления эфиров
холестерина — это редкое состояние со сравнительно более легкой симптоматикой.
К постоянным признакам относятся гепатоспленомегалия и повышенный уровень
холестерина в плазме. Могут быть выявлены фиброз печени, варикозное расширение
вен пищевода и замедление роста. В тканях больных с недостаточностью кислой
липазы не гидролизуются ни триглицериды, ни эфиры холестерина. Не исключено,
что многие субстраты гидролизуются одним ферментом, но структура субъединиц и
гидролитические свойства различных лизосомных липаз изучены недостаточно.
Дефицит кислой липазы обусловливает нарушение процесса разрушения липопротеинов
низкой плотности и может сопровождаться преждевременным развитием
атеросклероза. Как болезнь Вольмана, так и болезнь накопления эфиров
холестерина наследуется по аутосомно-рецессивному типу.
Болезни накопления гликопротеинов. Фукозидоз, маннозидоз и аспартилглю — козаминурия
представляют собой редкие аномалии, наследуемые как аутосомные рецессивные
признаки и связанные с недостаточностью гидролаз, расщепляющих полисахаридные
связи. При фукозидозе накапливаются как гликолипиды, так и гликопротеины. Все
эти аномалии характеризуются неврологическими нарушениями и разнообразными
соматическими проявлениями. Фукозидоз и маннозидоз чаще всего приводят к смерти
в детском возрасте, тогда как аспартилглюкозаминурия проявляется как лизосомная
болезнь накопления с поздним началом, выраженной психической отсталостью и
более продолжительным течением. Для фукозидоза характерны нарушения
электролитного состава пота и кожные ангиокератомы, а для маннозидоза —
необычные круговые катаракты. При аспартилглюкозаминурии диагностическое
значение имеют результаты анализа мочи, в которой выявляют увеличение
количества аспартилглюкозамина. Заболевают чаще жители Финляндии. Под названием
сиалидоза объединяют группу фенотипов, связанных с недостаточностью
гликопротеиннейраминидазы (сиалидаза). К ним относятся взрослая форма,
характеризующаяся вишнево-красными пятнами на сетчатке и миоклонусом, инфантильная
и ювенильная формы с фенотипом, подобным мукополисахаридозу, а также врожденная
форма с водянкой плода. Во многих случаях, ранее относимых к муколипидозу I,
был выявлен маннозидоз или сиалидоз. У некоторых больных с сиалидозом
определяется недостаточность как b-галактозидазы, так и нейраминидазы.
Молекулярная основа сочетанного дефицита b-галактозидазы и нейраминидазы
остается неясной, но предполагают дефект «защитного белка». Каждую из болезней
накопления гликопротеинов можно диагностировать путем определения
соответствующих ферментов.
Мукополисахаридозы. Это общее название разнообразных нарушений,
обусловленных недостаточностью одного из группы ферментов, разрушающих
мукополисахариды трех классов: гепаран-, дерматин- и кератансульфат. Обобщенный
фенотип включает в себя грубые черты лица, помутнение роговицы,
гепатоспленомегалию, тугоподвижность суставов, грыжи, множественный дизостоз,
экскрецию мукополисахаридов с мочой и метахромное окрашивание периферических
лейкоцитов и костного мозга. Отдельные черты фенотипа мукополисахаридоза
присущи также муколипидозам, гликогенозам и другим лизосомным болезням
накопления.
Прототипом мукополисахаридоза служит синдром Гурлер, или мукополисахаридоз
IX. В этом случае имеются практически все компоненты упомянутого фенотипа,
причем они резко выражены. Ранние симптомы включают переполнение кровью сосудов
носа и макроскопически видимое помутнение роговицы. Бурный рост в первые годы
жизни по мере прогрессирования болезни замедляется. Рентгенологически выявляют
увеличение турецкого седла с характерным подковообразным дном, расширение и
укорочение длинных костей, а также гипоплазию и заостренность позвонков в
поясничной области. Последнее обусловливает усиленный кифоз или горбатость.
Смерть наступает в первые 10 лет; на секции находят гидроцефалию и поражение
сердечнососудистой системы с закупоркой коронарных артерий. Биохимический
дефект заключается в недостаточности a-идуронидазы с накоплением гепаран — и
дерматансульфата.
Мукополисахаридоз IS, или синдром Шейе, имеет клинические особенности. Он
начинается в детском возрасте, но больной выживает до зрелого возраста. Для
него характерны тугоподвижность суставов, помутнение роговицы, регургитация
аортальных клапанов и обычно ненарушенный интеллект. Удивительно, что это гораздо
более легкое заболевание также обусловлено недостаточностью a-идуронидазы; как
показывает отсутствие перекрестной коррекции активности фермента при совместном
культивировании фибробластов кожи, оно аллельно синдрому Гурлер. Встречаются
явно промежуточные между синдромами Гурлер и Шейе фенотипы. Считают, что
больные с промежуточным фенотипом — это генетические химеры с одним аллелем
синдрома Гурлер и вторым — синдрома Шейе. В любом случае это трудно отличить от
других мутаций, определяющих промежуточную тяжесть болезни.
Синдром Гунтера, или Мукополисахаридоз I, отличается от фенотипа синдрома
Гурлер отсутствием макроскопически видимого помутнения роговицы и сцепленным с
Х-хромосомой рецессивным наследованием. Инфантильная форма напоминает фенотип синдрома Гурлер, а более легкая форма
позволяет больному дожить до зрелого возраста. Тяжелая и легкая формы могут
быть аллельными, так как обе они сцеплены с Х-хромосомой и обусловлены
недостаточностью одного и того же фермента (идуронсульфатсульфатаза).
Мукополисахаридозы Санфилиппо (IIIA, IIIB, IIIC и IIID) отличаются
накоплением гепарансульфата без дерматан — или кератансульфата, а также
выраженными изменениями со стороны центральной нервной системы с более мягкой
соматической симптоматикой. Мукополисахаридоз Санфилиппо диагностируют обычно
по отставанию психического развития в детстве. Поскольку соматические
проявления выражены слабо, его можно не заметить, если нарушения со стороны
центральной нервной системы рассматривать изолированно. Смерть наступает обычно
в возрасте после 10—20 лет. Нарушения, объединяемые в группу
мукополисахаридозов III, представляют собой близкие генокопии. Другими словами,
примерно одинаковые клинические фенотипы, при которых откладывается один и тот
же продукт, обусловливаются недостаточностью четырех разных ферментов. Четыре
вида мукополисахаридоза III можно диагностировать и различить с помощью определения
ферментов.
Синдром Моркио, или
Мукополисахаридоз IV, отличается нормальным психическим
развитием и характерной дистрофией костей, которую можно классифицировать как
спондилоэпифизарную дисплазию. Резко выраженная гипоплазия зубовидного отростка
может вызывать кривошею и приводит обычно к компрессии спинного мозга той или
иной степени. Часто выявляют регургитацию аортальных клапанов. В основе
синдрома лежит недостаточность N-ацетилгалактозамин-6-сульфатсульфатазы.
Изменения костей, несколько напоминающие таковые при синдроме Моркио, могут
встречаться и при недостаточности р-галактозидазы и других формах
спондилоэпифизарной дисплазии. Синдром Марото — Лами, или мукополисахаридоз VI,
характеризуется выраженной костной патологией, помутнением роговицы и
сохранным интеллектом. Известны аллельные формы разной тяжести, но с
недостаточностью той же арилсульфатазы В (N-ацетилгексозамин-4-сульфатсульфатаза).
Мукополисахаридоз VII, или недостаточность р-глюкуронидазы, обнаружен всего у
нескольких человек с практически полным фенотипом мукополисахаридоза. Этот
синдром отличается крайним разнообразием форм: от смертельной инфантильной до
легкой взрослой.
Множественная сульфатазная недостаточность. Это необычное состояние, хотя и наследуется как аутосомный рецессивный признак, характеризуется недостаточностью пяти клеточных
сульфатаз (арилсульфатазы А и В, другие сульфатазы мукополисахаридов и нелизосомная
сульфатаза стероидов) или более. В клинической картине объединяются признаки
метахромной лейкодистрофии, фенотип мукополисахаридоза и ихтиоз. Последний
связан, вероятно, с недостаточностью сульфатазы стероидов, которая может быть
изолированной, наследуемой как признак, сцепленный с Х-хромосомой. В последнем
случае эта недостаточность проявляется нарушением родовой деятельности и
ихтиозом. Биохимические исследования при этом состоянии должны пролить
дополнительный свет на биохимическую и клиническую стороны проблемы
генетической гетерогенности.
Муколипидозы. Это общее название лизосомных болезней накопления, при которых в
определенном сочетании накапливаются мукополисахариды, гликопротеины,
олигосахариды и гликолипиды. Муколипидоз I можно, вероятно, опустить, так как
большинство лиц или все страдают в действительности той или иной болезнью
накопления гликопротеинов.
Муколипидоз II, или 1-клеточная болезнь, начинается в раннем возрасте и
проявляется отставанием психического развития и фенотипом мукополисахаридоза. К
отличительным особенностям относятся отчетливые включения в культивируемых
фибробластах кожи и резко повышенный уровень лизосомных ферментов в сыворотке.
Синдром наследуется как аутосомный рецессивный признак и, как установлено в
настоящее время, отражает дефект посттрансляционного процессинга лизосомных
ферментов. Муколипидоз III, или псевдополидистрофия Гурлер, представляет собой
более легкое заболевание с фенотипическими признаками мукополисахаридоза, в
частности множественным дизостозом. Оно проявляется в первые 10 лет жизни
тугоподвижностью суставов, что нередко заставляет думать о ревматоидном
артрите. Основные симптомы заключаются в прогрессирующей физической
инвалидизации, особенно в появлении когтеобразной деформации кистей и дисплазии
бедер. Нередко задерживается психическое развитие. К обычным признакам
относится аномалия аортальных или митрального клапанов сердца, хотя это
зачастую и не имеет функциональных последствий. Больные обычно доживают до
зрелого возраста, у них возможна стабилизация состояния, причем у мужчин
инвалидизирующие деформации выражены сильнее, чем у женщин. В культивируемых
фибробластах кожи определяются те же включения, и так же повышается уровень
лизосомных ферментов в сыворотке, что и при муколипидозе II. Это
свидетельствует об аллельности аномалий. Первичный дефект при муколипидозах II
и III заключается в недостаточности УДФ-К-ацетилглюкозамин
(GLcNAc)—гликопротеин (GLcNАс)-1-фосфотрансферазы, принимающей участие в
посттрансляционном синтезе олигосахаридной части лизосомных ферментов.
Муколипидоз IV характеризуется психической отсталостью, помутнением роговицы
и дегенерацией сетчатки без других соматических проявлений.
Другие лизосомные болезни накопления. Прототипом лизосомной болезни накопления служит гликогеноз
II типа (болезнь Помпе). Основные клинические особенности, связанные с
повреждением скелетных и сердечной мышц. Лактозилцерамидоз представляет собой,
по-видимому, вариант синдрома Нимана-Пика: гидролиз лактозилцерамида in vitro в
зависимости от условий осуществляют ферменты, недостаточность которых определяется
при ганглиозидозе gmi или синдроме Краббе. Сообщения о недостаточности
N-ацетилглюкозамин-b-сульфатсульфатазы, сопровождающейся
мукополисахаридозом VIII типа, могут быть ошибочными. Адренолейкодистрофия
представляет собой своеобразное сцепленное с Х-хромосомой заболевание,
характеризующееся накоплением в тканях эфиров холестерина с длинноцепочечными
жирными кислотами, но оно может и не представлять собой лизосомную болезнь
накопления. Выявление женщин с фенотипом синдрома Гунтера (мукополисахаридоз
II) и той же ферментной недостаточностью заставляет думать о существовании
аутосомной рецессивной формы синдрома Гунтера. Это могло бы быть в том случае,
если бы аномальный фермент состоял из неидентичных субъединиц, кодируемых одним
аутосомным и одним сцепленным с Х-хромосомой геном, или если бы были
заинтересованы регуляторные генетические элементы. С другой стороны,
фенотипические проявления у женщин могли бы вызываться разнообразными
аберрациями Х-хромосомы. Известна семья, члены которой страдают ганглиозидозом
См3. Этот синдром не представляет собой лизосомную болезнь
накопления, он отражает, вероятно, нарушение синтеза ганглиозидов. Его
клинические проявления сходны с таковыми при лизосомных болезнях накопления, но
несовпадения между сиблингами оставляют открытым вопрос о его генетической
природе. Когда-нибудь, возможно, к лизосомным болезням накопления будут
отнесены и другие нейродегенеративные синдромы, а именно ювенильный
дистонический липидоз, нейроаксональная дистрофия, синдромы Галлервордена —
Шпатца, Пелицеуса — Мерцбахера и др. Кроме того, нередко встречаются больные с
отчетливыми клиническими признаками липидоза, муколипидоза или
мукополисахаридоза, у которых не удается выявить ни одного из известных в
настоящее время биохимических нарушений. В связи с этим число лизосомных
болезней накопления будет, вероятно, увеличиваться.
лизосомный: болезни, диагноз, первые признаки
Мукополисахаридозы — группа врожденных лизосомных болезней накопления, которые наследуются по аутосомно-рецессивному типу и характеризуются нарушениями обмена глюкозаминогликанов, что приводит к их отложению в различных органах и системах.
Болезнь Волмана представляет собой наиболее тяжелое проявление дефицита лизосомальной кислой липазы. Фермент кислая липаза играет существенную роль в лизосомном гидролизе этерифицированного холестерина и триглицеридов липопротеинового происхождения.
Синдром Чедиака-Хигаси является редким тяжелым генетическим заболеванием, как правило, характеризуется частичным глазокожным альбинизмом, тяжелым иммунодефицитом, умеренными кровотечениями, неврологическими нарушениями и лимфопролиферативными расстройствами.
Нейрональный цероидный липофусциноз представляет собой группу наследственных прогрессирующих дегенеративных заболеваний мозга, характеризующихся снижением умственных способностей, эпилепсией, потерей зрения за счет дегенерации сетчатки и накоплением в нейронах цероида и липофусцина.
Ганглиозидоз GM1 является редкой лизосомной болезнью накопления, биохимически характеризуется дефицитом бета-галактозидазы, а клинически — широким диапазоном разнообразных нейровисцеральных, офтальмологических и дисморфичных симптомов.
Болезнь Ниманна-Пика является лизосомной болезнью накопления, при которой имеет место нарушение обмена сфингомиелина с накоплением его в мозге и внутренних органах, прежде всего в ретикулоэндотелиальной системе.
Врожденное нарушение гликозилирования — группа врожденных нарушений метаболизма, характеризующихся дефектной активностью ферментов, которые участвуют в гликозилировании.
Галактосиалидоз и сиалидоз — наследственные болезни хранения лизосом, характеризуются дефицитом нейраминидазы и накоплением сиалоолигосахаридов в тканях.
Дефицит миелопероксидазы является одним из наиболее распространенных наследственных дефектов фагоцитов, которое редко проявляется клиническими симптомами. Дефицит миелопероксидазы проявляется значительным снижением эффективности нейтрофилов при грибковых инфекциях.
Ганглиозидозы — наследственные лизосомные заболевания, обусловленные нарушением липидного обмена, при которых в нейронных мембранах накапливаются ганглиозиды — кислые гликосфинголипиды, содержащие нейраминовую кислоту.
Некротизирующий энтероколит характеризуется язвенно-некротическим поражением кишечника. Развивается преимущественно у глубоко недоношенных детей, предрасполагающими факторами являются пренатальная гипоксия, синдром дыхательных расстройств, катетеризация пупочной вены.
Пикнодизостоз является лизосомальным генетическим заболеванием, которое характеризуется остеосклерозом скелета, низкорослостью и хрупкостью костей.
Почечная недостаточность – патологическое состояние, при котором частично или полностью утрачивается способность почек образовывать и/или выделять мочу.
Васкулиты — большая и неоднородная группа заболеваний воспалительного характера с поражением сосудов разного анатомического строения и калибра и с вторичными патологическими изменениями различных органов и тканей. Васкулиты подразделяют на первичные, или системные, и вторичные.
Муколипидозы — группа наследственных заболеваний, обусловленных нарушением обмена липидов и мукополисахаридов. Относится к энзимопатиям и характеризуется сочетанным накоплением кислых гликозаминогликанов и сфинголипидов в нейронах и в клетках внутренних органов.
Болезнь Фарбера является редким наследственным сфинголипидозом, которая классической триадой — болезненные и прогрессивно-деформирующиеся суставы, подкожные узелки и прогрессирующая охриплость из-за вовлечения гортани.
Болезнь Краббе является наследственным лизосомальным расстройством, при котором поражается белое вещество центральной и периферической нервной системы, включает в себя детскую, подростковую и взрослые формы.
Цистиноз является метаболическим заболеванием, которое характеризуется накоплением цистина внутри лизосом, вызывая повреждение в различных органах и тканях, особенно в почках и глазах. Описаны три клинические формы заболевания: нефропатия инфантильная, нефропатия подростковая и окулярная.
Болезнь хранения глутамил рибозо-5-фосфата — гликопротеиноз. Предполагается наличие дефицита гидролазы АТФ-рибозы.
Болезнь Фабри является прогрессирующей, наследственной, мультисистемной болезнью хранения лизосом, характеризуется специфическими неврологическими, кожными, почечными, сердечно-сосудистыми, кохлео-вестибулярными и сосудисто-мозговыми проявлениями.
В зависимости от того, какой тип билирубина присутствует в сыворотке крови — неконъюгированный (непрямой) или конъюгированный (прямой), гипербилирубинемия классифицируется как неконъюгированная и конъюгированная соответственно.
Синдром Маринеску-Шегрена принадлежит к группе аутосомно-рецессивных мозжечковых атаксий. Кардинальной особенностью синдрома Маринеску-Шегрена являются мозжечковая атаксия, врожденная катаракта и задержка психомоторного развития.
Гипотиреоз — это клинический синдром, обусловленный дефицитом гормонов щитовидной железы
Аспартилглюкозаминурия является аутосомно-рецессивной болезнью хранения лизосом, принадлежит к группе олигосахаридозов (или гликопротеинозов).
Болезнь Гоше относится к болезням хранения лизосом, развивающимся в связи с недостаточностью фермента глюкоцереброзидазы, что ведет к накоплению глюкоцереброзидов в печени, селезенке, лимфатических узлах, а при церебральных формах — и в головном мозге.
Классическая гомоцистинурия — наследственное заболевание, связанное с дефицитом фермента β-цистатионинсинтазы, проявляющееся высоким уровнем метионина и гомоцистина в биологических жидкостях и характеризующийся вовлечением глаз, скелета, центральной нервной системы и сосудов.
Прогрессирующие мышечные дистрофии — гетерогенная группа наследственных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофией скелетных мышц.
что это такое, нома, что означает повышение и понижение
Из статьи вы узнаете, что означает липаза в анализе крови, нормальные показатели для взрослых и детей, причины повышения и снижения фермента.
Роль липазы в организме
Липаза – это фермент, который расщепляет и переваривает жиры в организме человека. Это его главная роль. Кроме того, липаза активно участвует в энергетическом обмене, стимулирует усваивание витаминов A, D, E, K и жирных полиненасыщенных кислот. Синтезируется фермент в поджелудочной железе, кишечнике, печени и легких. В связи с этим липаза делится на фракции, каждая из которых отвечает за переработку отдельной группы жиров. Есть еще и лингвальная липаза, которая вырабатывается только у новорожденных в ротовой полости.
Нормы липазы у взрослых и детей
У взрослых женщин и мужчин уровень липазы постоянен и одинаков, зависит только от возраста. Границы нормы – от 0 до 190 единиц вещества на миллилитр крови. У детей до 18 лет уровень липазы колеблется от 0 до 130 единиц. Определяют концентрацию фермента с помощью биохимического анализа крови. Отклонения от нормы говорят о развитии острого панкреатита, цирроза, холецистита, почечной недостаточности, непроходимости кишечника, ЯБЖ. Растет липаза и при пересадке органов и тканей.
Липаза – основной маркер поражения панкреас. При повышении уровня фермента в сочетании с ростом амилазы позволяет точно поставить диагноз панкреатита. При этом, как правило, рассматривают показатели только панкреатической липазы, нормальный уровень которой находится в диапазоне от 13 до 60 единиц на миллилитр крови.
Показания для анализа на липазу
В широком смысле слова концентрация липазы в кровотоке характеризует состояние пищеварительной трубки. Резкий рост фермента – показатель развития панкреатита, его степени тяжести, возможности осложнений. При этом в первые часы развития патологии повышение концентрации фермента достигает десятков раз, а нормализация показателей длится пару недель. Неблагоприятным диагностическим признаком считается повышение уровня липазы в 10 и больше. При этом за трое суток концентрация не снижается до трехкратной нормы.
Анализ на содержание липазы в крови – достаточно специфичен и назначается чаще всего при подозрении на острый панкреатит: пик в этом случае появляется через 48 часов с момента приступа. При хроническом панкреатите диагноз поставить сложнее, поскольку при длительном существовании воспаления органа синтез ферментов прекращается, и липаза понижается.
Показанием к сдаче анализа на липазу является паротит: показатели фермента при этом должны оставаться в нормальных пределах (увеличение уровня фермента – свидетельство нездоровья панкреас).
Причины повышения липазы у взрослых
Уже совершенно ясно, что анализ на уровень липазы в крови – это маркер патологических процессов в поджелудочной железе, то есть, основная причина повышения концентрации фермента – патология панкреас: воспаление, опухоль, киста. Кроме того, рост показателя уровня липазы дает повод для подозрения на спазм желчных протоков, нарушение обменных процессов: ожирение, подагра, сахарный диабет. Растет липаза и при ОИМ, непроходимости кишечника, травмах половых органов, перитоните, холестазе разного генеза, прободении язвы, свинке, если паротит сопровождается поражением поджелудочной железы.
Липаза может быть повышена при приеме Гепарина, Индометацина, наркотических анальгетиков, барбитуратов. Повышение уровня фермента наблюдается при переломах трубчатых костей.
Низкий уровень липазы в крови
Если липаза понижена, это свидетельствует об опухолевом процессе в организме. Исключение – рак поджелудочной. Помимо этого, низкий уровень фермента может быть при наследственной гиперлипидемии, атерогенном профиле питания с преобладанием жиров. Недостаток липазы говорит о хронизации панкреатита.
Симптомы избытка и недостатка липазы в крови
Очевидно, что при дефиците липазы, которая отвечает за переваривание жиров, развивается ферментативная недостаточность поджелудочной железы. Симптомами патологии являются:
- слабость;
- отсутствие аппетита;
- постоянное подташнивание;
- зловонный жидкий стул;
- метеоризм;
- потеря веса;
- абдоминальные боли.
При повышении уровня липазы симптомы те же, но на фоне роста температуры. Это говорит о воспалении поджелудочной.
Подготовка и проведение анализа
Если диагностика уровня липазы осуществляется по жизненным показаниям, экстренно, то специальной подготовки не требуется. В остальных случаях предподготовка к сдаче крови на липазу стандартна:
- кровь сдают натощак, до 11 утра, последний прием пищи – за 12 часов перед процедурой, при этом за несколько дней до теста прекращают прием жирной, острой пищи;
- прием лекарств согласовывают с врачом;
- накануне теста отказываются от алкоголя и сигарет, физических нагрузок, избегают стресса.
Если в день процедуры пациент проходил ФЛГ или рентгенографию, тестирование переносят на другую дату.
Забор крови проводят из вены. Очень часто липазу тестируют вместе с амилазой.
Факторы, влияющие на результат
Кроме хронических и острых соматических заболеваний, провоцировать повышение или снижение уровня липазы могут: лекарства, переломы или иные травмы – липаза растет, а при переедании жирной пищи – падает.
Как нормализовать уровень липазы в крови
Суть балансирования концентрации липазы в кровотоке – устранение первопричины патологических изменений. Чаще всего – это лекарства, которые следует либо отменить, либо заменить аналогичными, выверив дозу и кратность приема: противовоспалительные, антисекреторные, спазмолитические, антибактериальные, ферменты (Мезим, Панкреатин, Фестал, Креон).
На втором месте консервативная или оперативная терапия патологии панкреас, печени, желчного пузыря и всей билиарной системы печени, почек, сердца. Отдельную группу составляют опухоли системы пищеварения, которые подлежат удалению с последующей медикаментозной коррекцией.
Безусловно, важнейшим корректором является диета с отказом от спиртных напитков, иногда – полное голодание в лечебных целях. Суть диеты – исключение панкреас из пищеварительного процесса (нет пищи, нет и потребности в ее переваривании). На этом фоне происходит самовосстановление пораженного органа. Выход из «голодовки» осуществляют по плану диеты №5 с исключением жирных продуктов, приправ, жареных блюд. Основа – дробное питание (до 7 раз/день), исключающее появление чувства голода.
Профилактика и прогноз скачков липазы
Предупредить болезнь гораздо проще, чем потом лечить ее. Поэтому к профилактике повышения уровня липазы или ее понижения относятся очень серьезно. Врачи рекомендуют: сбалансированный рацион, отказ от вредных привычек, нормализацию веса, корректный прием препаратов, ежегодную диспансеризацию.
Прогноз такой:
- своевременная диагностика и лечение, соблюдение диеты обеспечивают понижение уровня липазы через две недели;
- рост показателя в 10 раз и выше – неблагоприятен, особенно при отсутствии результат назначенной терапии.
Запущенная болезнь может стать причиной летального исхода.
Людмила Жаворонкова
Высшее медицинское образование. 30 лет рабочего стажа в практической медицине. Подробнее об авторе
Дефицит липазы лизосомальной кислоты | Информационный центр по генетическим и редким заболеваниям (GARD) — программа NCATS
Дефицит лизосомальной кислой липазы — болезнь накопления метаболических липидов. [1] [2] Два редких состояния могут быть результатом этого дефицита (вероятно, представляют собой два конца клинического спектра): [1] [3]
- Болезнь Вольмана: раннее начало и наиболее тяжелая форма заболевания, при которой липиды накапливаются по всему телу, в основном в печени, в течение первых недель жизни.Симптомы включают увеличение печени и селезенки (гепатоспленомегалия), недостаточный набор веса, желтоватый цвет кожи и белков глаз (желтуха), рвоту, диарею, жирный стул (стеаторея) и плохое усвоение питательных веществ из пищи (мальабсорбция). ), а также отложения кальция в надпочечниках, анемия, заболевание печени (цирроз) и задержка развития. У младенцев с этой формой дефицита липазы лизосомальной кислоты развиваются недостаточность многих органов и тяжелое недоедание.
- Болезнь накопления холестерилового эфира: менее тяжелая и начинается в более позднем возрасте.Симптомы могут включать гепатоспленомегалию, заболевание печени (цирроз) и мальабсорбцию с диареей, рвотой и стеатореей.
Дефицит лизосомальной кислой липазы вызван мутациями в гене LIPA , который предоставляет инструкции по производству фермента лизосомальной кислой липазы. Когда этого фермента недостаточно, организм не может расщеплять определенные жиры, и это приводит к токсическому накоплению жировых веществ в клетках и тканях организма. [1] Наследование аутосомно-рецессивное. [2] [3] Ферментная заместительная терапия как при болезни Вольмана, так и при болезни накопления холестерилового эфира в настоящее время одобрена для использования в США, Европейском Союзе и Японии. [1]
Последнее обновление: 19.10.2017
Управление дефицитом лизосомальной кислоты и липазы
- Аптека Практика
Доступные лекарства
Соблюдение
Компаундирование
Разрешения на лекарства
ГериатрияБольница / Система здравоохранения
Вакцины в аптеке
Закон
Управление лекарствами
Новости и тенденцииВнебиржевой
Специализированная аптека
Технологии - Состояние болезни
Аллергия
Аутоиммунный
Рак молочной железы
Сердечно-сосудистые
Простуда
ДерматологияДиабет
Гастроэнтерология
Гематология
ВИЧ / СПИД
Инфекционное заболевание
Рак легкихДушевное здоровье
Неврология
Онкология
Офтальмология
Ортопедия
Контроль над болью
Дефицит липазы лизосомальной кислоты Определение и примеры
Дефицит лизосомальной кислой липазы — это тип лизосомальной болезни накопления, которая часто вызывается дефицитом лизосомальной кислой липазы, приводящей к накоплению жирового материала в различных органах тела, таких как печень, селезенка, кишечник и эндотелий.
Определение дефицита липазы лизосомальной кислоты
Дефицит лизосомальной кислой липазы — это лизосомальная болезнь накопления, вызванная дефектом гена, кодирующего фермент, лизосомальную кислотную липазу. Это одна из лизосомных болезней накопления, которые в совокупности относятся к различным метаболическим нарушениям, вызванным дефектами лизосомной функции, приводящим к аномальному накоплению токсичных материалов в клетке.
Этимология
Термин дефицит лизосомальной кислоты липазы ранее был известен как болезнь Вольмана, названная в честь израильского невропатолога Моше Вольмана.Он и его коллеги были первыми, кто сообщил о случае этого состояния в 1956 году. Были описаны два заболевания: болезнь Вольмана и болезнь накопления холестерилового эфира . В 2010 году оба этих расстройства были вместе обозначены как дефицит липазы лизосомальной кислоты . Сокращение: LAL-дефицит, LAL-D. Синонимы: болезнь Вольмана (ы).
Патофизиология
Липаза лизосомальной кислоты играет важную роль в расщеплении жирных веществ (таких как сложные эфиры холестерина и триглицериды).Без достаточного уровня этого фермента эти жирные вещества накапливаются в лизосомах в клетках различных органов, таких как печень, селезенка, кишечник и эндотелий. Это приводит к серьезным проблемам со здоровьем, особенно в раннем детстве. Это редкий семейный ксантоматоз, вызывающий смерть в раннем младенчестве. Это может привести к кальцификации надпочечников, диффузной, пунктированной кальцификации повсюду, увеличению надпочечников нормальной формы и гепатоспленомегалии.
Наследование и прогноз
Дефицит
LAL наследуется как аутосомно-рецессивный .Это означает, что у человека с двумя копиями дефектного гена LAL (каждая от двух родителей) будет заболевание и проявятся тяжелые симптомы. Например, дефицит КЛЛ у младенцев приводит к накоплению жира в стенках кишечника, что может привести к мальабсорбции и другим серьезным проблемам с пищеварением. Ребенок недоедает и не может достичь идеального диапазона веса. Состояние может ухудшаться по мере прогрессирования заболевания, что может привести к дисфункции или отказу печени, что может быть фатальным.
См. Также
Список литературы
- Райнер, Эм, Гуардаманья, О., Наир, Д., Соран, Х., Ховинг, К., Бертолини, С., Джонс, С., Чорич, М., Каландра, С., Гамильтон, Дж., Иглтон, Т., и Рос, Э. (июль 2014 г.). «Дефицит лизосомальной кислой липазы — недостаточно известная причина дислипидемии и дисфункции печени». Атеросклероз. 235 (1): 21–30.
© Биология онлайн. Контент предоставлен и модерируется Biology Online Editors
Дефицит лизосомальной кислой липазы — RightDiagnosis.com
Введение: Дефицит липазы лизосомальной кислоты
Описание дефицита липазы лизосомальной кислоты
Дефицит липазы лизосомальной кислоты (заболевание):
Редкое наследственное заболевание накопления липидов. Чем больше »
См. Также:
Болезнь Вольмана:
»Введение: болезнь Вольмана
»Симптомы болезни Вольмана
»Лечение болезни Вольмана
Дефицит лизосомальной кислой липазы: тяжелая детская форма наследственной лизосомальной болезни накопления липидов, вызванной дефицитом кислой липазы; приводит к накоплению нейтральных липидов, особенно сложных эфиров холестерина, в клетках.
Источник: База данных болезней.
Дефицит липазы лизосомальной кислоты: связанные темы
Эти темы о медицинских состояниях или симптомах могут иметь отношение к
Медицинская информация при недостаточности лизосомальной кислой липазы:
Дефицит лизосомальной кислой липазы: редкое заболевание
Отделение редких заболеваний (ORD) Национального института здоровья (NIH)
Дефицит липазы лизосомальной кислоты внесен в список «редких заболеваний» Управлением
Редкие заболевания (ORD) Национального института здоровья
(НАЦИОНАЛЬНЫЕ ИНСТИТУТЫ ЗДРАВООХРАНЕНИЯ США).Это означает, что дефицит липазы лизосомальной кислоты или подтип дефицита липазы лизосомальной кислоты,
затрагивает менее 200 000 человек среди населения США.
Источник — Национальные институты здравоохранения (NIH).
Ophanet
Ophanet, консорциум европейских партнеров,
в настоящее время определяет редкое состояние, когда он влияет на 1 человека на 2000.
Они перечисляют Дефицит липазы лизосомальной кислоты как «редкое заболевание».
Источник — Orphanet
Дефицит лизосомальной кислой липазы как заболевание
Дефицит лизосомальной кислотной липазы : Другое название болезни Вольмана (или тесной связи с заболеваниями).
»Введение: болезнь Вольмана
»Симптомы болезни Вольмана
»Лечение болезни Вольмана
Дефицит лизосомальной кислой липазы: заболевания, связанные с заболеванием
Дефицит лизосомальной кислотной липазы : Дефицит лизосомальной кислотной липазы указан в нашей базе данных как тип (или связанный с) следующих заболеваний:
Симптомы недостаточности лизосомальной кислой липазы (болезнь Вольмана)
Некоторые из симптомов недостаточности липазы лизосомальной кислоты включают:
Показать полный список из 26
симптомы недостаточности лизосомальной кислой липазы (болезнь Вольмана)
Лечение недостаточности лизосомальной кислотной липазы (болезнь Вольмана)
Лечение недостаточности лизосомальной кислотной липазы (болезнь Вольмана) включает:
- Нет специального лечения, кроме симптоматических мер:
- Гормоны надпочечников при отказе надпочечников
- Внутривенное питание
- Пересадка костного мозга вызвала ремиссию у одного пациента
- Переливание крови при развитии анемии
См. Полный список из 7
лечение дефицита липазы лизосомальной кислоты
Лечение недостаточности лизосомальной кислотной липазы: Для получения дополнительной информации о лечении недостаточности лизосомальной кислотной липазы см. Лечение болезни Вольмана (дефицит лизосомальной кислотной липазы)
Дефицит липазы лизосомальной кислоты: темы, связанные с заболеваниями
Эти медицинские темы могут быть связаны с дефицитом липазы лизосомальной кислоты:
Термины, связанные с недостаточностью липазы лизосомальной кислоты:
Термины, аналогичные дефициту липазы лизосомальной кислоты:
Источник: База данных болезней.
Источник — NIH
Внешние ссылки, связанные с: Дефицит липазы лизосомальной кислоты
Источник: База данных болезней.
Интересные медицинские статьи:
Медицинские словари:
Другие темы в медицинском словаре
Узнать больше
Найдите, чтобы узнать больше о дефиците липазы лизосомальной кислоты:
»Следующая страница: Лизосомальная альфа-глюкозидаза
Медицинские инструменты и изделия:
Инструменты и услуги:
Медицинские изделия:
Форумы и доски сообщений
- Задайте или ответьте на вопрос в досках:
Список лекарств от дефицита лизосомальной кислоты и липазы (2 сравненных)
- Процедуры
- Нарушения обмена веществ
- Дефицит лизосомальной кислоты липазы
Другие названия: Болезнь Вольмана
О дефиците лизосомальной кислой липазы
Дефицит лизосомальной кислой липазы — это генетическое заболевание, которое приводит к снижению гидролиза сложных эфиров холестерина и триглицеридов.Полная потеря липазы лизосомальной кислоты проявляется в младенчестве и называется болезнью Вулмана, а частичная потеря липазы лизосомальной кислоты проявляется в более позднем возрасте и называется болезнью накопления холестерилового эфира.
Используемые наркотики
для лечения дефицита лизосомальной кислоты липазы
Следующий список лекарств так или иначе связан с
используется при лечении этого состояния.
Легенда
| Rx | Только по рецепту |
|---|---|
| ОТС | Без рецепта |
| Rx / OTC | По рецепту или без рецепта |
| Off Label | Это лекарство не может быть одобрено FDA для лечения этого состояния. |
| Категория беременности | |
|---|---|
| A | Адекватные и хорошо контролируемые исследования не смогли продемонстрировать риск для плода в первом триместре беременности (и нет доказательств риска в более поздних триместрах). |
| B | Исследования репродукции животных не смогли продемонстрировать риск для плода, и нет адекватных и хорошо контролируемых исследований у беременных женщин. |
| С | Исследования репродукции животных показали неблагоприятное воздействие на плод, и нет адекватных и хорошо контролируемых исследований на людях, но потенциальные преимущества могут потребовать применения у беременных женщин, несмотря на потенциальные риски. |
| D | Имеются положительные доказательства риска для плода человека, основанные на данных о побочных реакциях, полученных в результате исследований или маркетингового опыта или исследований на людях, но потенциальные преимущества могут потребовать применения у беременных женщин, несмотря на потенциальные риски. |
| X | Исследования на животных или людях продемонстрировали аномалии плода и / или есть положительные доказательства риска для плода у человека, основанные на данных о побочных реакциях из исследовательского или маркетингового опыта, и риски, связанные с использованием у беременных женщин, явно перевешивают потенциальную пользу. |
| N | FDA не классифицировало препарат. |
| Закон о контролируемых веществах (CSA) Приложение | |
|---|---|
| N | Не подпадает под действие Закона о контролируемых веществах. |
| 1 | Имеет высокий потенциал для злоупотреблений. В настоящее время не применяется в медицинских целях в США. Нет общепринятых правил безопасности для использования под медицинским наблюдением. |
| 2 | Имеет высокий потенциал для злоупотреблений. В настоящее время разрешено медицинское использование для лечения в Соединенных Штатах или в настоящее время принятое медицинское использование с серьезными ограничениями. Жестокое обращение может привести к серьезной психологической или физической зависимости. |
| 3 | Имеет меньшую вероятность злоупотребления, чем те, которые указаны в таблицах 1 и 2. В настоящее время разрешено медицинское использование для лечения в Соединенных Штатах. Злоупотребление может привести к умеренной или низкой физической зависимости или высокой психологической зависимости. |
| 4 | Имеет низкий потенциал злоупотребления по сравнению с теми, которые указаны в списке 3. В настоящее время он признан в медицине для лечения в Соединенных Штатах. Злоупотребление может привести к ограниченной физической или психологической зависимости по сравнению с теми, которые указаны в таблице 3. |
| 5 | Имеет низкий потенциал злоупотребления по сравнению с теми, которые указаны в таблице 4. В настоящее время разрешено медицинское использование для лечения в Соединенных Штатах. Жестокое обращение может привести к ограниченной физической или психологической зависимости по сравнению с теми, которые указаны в таблице 4. |
| Спирт | |
|---|---|
| X | Взаимодействует с алкоголем. |
Дополнительная информация
Всегда консультируйтесь со своим врачом, чтобы убедиться, что информация, отображаемая на этой странице, применима к вашим личным обстоятельствам.
Заявление об ограничении ответственности в отношении медицинских услуг
Исследование безопасности и эффективности замены кислотной липазы (ARISE) у участников с дефицитом лизосомальной кислотной липазы — полный текст заметное снижение активности фермента липазы лизосомальной кислоты (LAL).
Спектр заболевания LAL-D варьируется от проявления у младенцев, которое быстро прогрессирует, до проявления, которое происходит в детстве, подростковом возрасте или, реже, во взрослом возрасте, при котором скорость прогрессирования заболевания более вариабельна.Независимо от того, в каком спектре заболевания находится пациент, LAL-D ассоциируется со значительным бременем болезни и сокращением продолжительности жизни у некоторых пациентов.
Неинфантильная форма заболевания, также известная как CESD, встречается как у детей, так и у взрослых и является недооцененной причиной ожирения печени с выраженным микровезикулярным стеатозом, фиброзом и циррозом. Хотя естественная история болезни недостаточно изучена, часто описываются серьезные осложнения, включая раннюю смерть, трансплантацию печени или сердечно-сосудистые нарушения.Другие осложнения включают преждевременный атеросклероз (затвердение артерий), связанный с высоким уровнем общего холестерина и холестерина липопротеидов низкой плотности (ЛПНП), часто называемого «плохим» холестерином. Уровни триглицеридов также могут быть высокими, а уровни холестерина липопротеидов высокой плотности (ЛПВП) («хороший» холестерин) обычно низкие.
В прошлом лечение в основном было сосредоточено на контроле липидных аномалий с помощью диеты и использования гиполипидемических препаратов, которые затрагивают только некоторые аспекты заболевания, хотя прогрессирование фиброза и цирроза все еще может происходить.В доклинических исследованиях и клинических исследованиях у участников с LAL-D было показано, что лечение SBC-102 (себелипаза альфа, Канума®) приводит к улучшению маркеров повреждения печени и липидных аномалий.