Клинические формы пигментного ретинита / Патологии сетчатки / Глазные болезни / Главная страница
Секторальный пигментный ретинит
Секторальный пигментный ретинит характеризуется аутосомно-доминантным типом наследования. Однако есть предположение, что не один ген является причиной патологического процесса. Заболевание двустороннее, у 15 % больных монокулярное, однако этот факт нуждается в подтверждении результатами многолетнего наблюдения. В 7 % случаев выявляют асимметричную картину поражения на двух глазах. Нижненосовые квадранты поражаются у 50 % больных, внутренняя часть — у 20 %, верхненосовые (оба внутренних или оба верхних) и верхнетемпоральные — у остальных В сетчатке отмечаются характерные для пигментного ретинита изменения- диффузная атрофия пигментного эпителия, истончение сетчатки вокруг рети-нальных сосудов, пигментные скопления в форме костных телец или кляксы.
Различают несколько подтипов секторального пигментного ретинита:
- форма с доминантным типом наследования, описанная как тип III, -атипичная секторальная пигментная дистрофия, вызываемая мутацией с неполной пенетрантностью,
-
секторальный пигментный ретинит с хроническим отеком диска зрительного нерва с раннего детства.

Острота зрения обычно хорошая. Поле зрения сужено соответственно зоне поражения сетчатки. Кинетика темновой адаптации после экспозиции к яркому свету у больных с секторальным пигментным ретинитом имеет некоторые особенности.
Предварительное исследование в относительно интактной части поля зрения до выцветания пигмента показало повышение порога на 1 лог. ед. по сравнению с нормой. После световой адаптации отмечено значительное замедление восстановления световой чувствительности. Первая часть кривой, обусловленная колбочками и палочками, соответствовала норме. Однако 1 и даже 2 ч спустя, когда чувствительность должна быть полностью восстановлена, резидуальный порог оказался повышенным на 1-2 лог. ед. по сравнению с установленным при первоначальном измерении. Для ее полного восстановления потребовалось от 80 до 120 ч. На модели заболевания у приматов установлено, что замедленное восстановление чувствительности после интенсивной световой адаптации частично может быть связано с образованием новых мембран дисков с нормальным содержанием родопсина, а не с регенерацией фотопигмента in situ.
На модельном заболевании обнаружены морфологические изменения, характерные для пигментного ретинита: укорочение наружных сегментов палочковых фоторецепторов и их отторжение на большом протяжении после интенсивного засвета. Эти исследования свидетельствуют о существовании как количественных, так и качественных различий форм пигментного ретинита, обусловленных разными мутациями гена родопсина.
При гистологическом исследовании выявляют исчезновение фоторецепторов, дегенерацию пигментного эпителия сетчатки и измененные хороикапилляры в пораженных квадрантах, отсутствие миелина в зрительном нерве. У всех больных отмечается повышение уровня витамина А в сыворотке и снижение концентрации β-каротина. Все имели дефект слуха.


Белоточечный ретинит
При одной из форм заболевания с аутосомно-рецессивным типом наследования наблюдаются функциональные нарушения, характерные для пигментного ретинита, а офтальмоскопически на глазном дне вместо пигментных костных телец отмечаются беловатые или желтовато-беловатые пятна. Первое описание белоточечного ретинита (retinitis punctata albescens) относится к 1882 г., когда было обращено внимание на заболевание с прогрессирующей ночной слепотой и изменениями на глазном дне в виде множественных мелкоточечных беловатых отложений.
Первое описание белоточечного ретинита (retinitis punctata albescens) относится к 1882 г., когда было обращено внимание на заболевание с прогрессирующей ночной слепотой и изменениями на глазном дне в виде множественных мелкоточечных беловатых отложений.
В 1910 г. Lauber выделил два подтипа заболевания со сходной офтальмоскопической картиной:
- с прогрессирующей ночной слепотой (в настоящее время известна как retinitis punctata albescens). Для неё характерны повышение палочкового и колбочкового порогов темновой адаптации, сужение поля зрения, нерегистрируемая или резко субнормальная ЭРГ и патологическая ЭОГ с отсутствием светового подъема.
- стационарной ночной слепотой (в настоящее время известна как retinitis albipunctatus). В отличие от прогрессирующей формы retinitis punctata albescens при стационарной форме retinitis albipunctatus после нескольких часов темновой адаптации палочковые пороги приходят в норму. Палочковая ЭРГ, не регистрируемая при стандартных условиях адаптации в течение 15 мин, оказывается в норме после полной темновой адаптации.
 Палочковая ЭРГ, не регистрируемая при стандартных условиях адаптации в течение 15 мин, оказывается в норме после полной темновой адаптации.
Палочковая ЭРГ, не регистрируемая при стандартных условиях адаптации в течение 15 мин, оказывается в норме после полной темновой адаптации.
На глазном дне видны дисперсно расположенные нерегулярные нежные беловатые точечные пятна.
Патологический процесс локализуется в глубине сетчатки и представляется как «ткань, изъеденная молью». Наличие на периферии пигментных включений свидетельствует о генерализованной дегенерации.
Со временем, по мере эволюции процесса, пятна блекнут и постепенно исчезают и возникают изменения фоторецепторов и атрофия пигментного эпителия сетчатки. Схожая картина описана при дефиците витамина А.
Как фенотипическое проявление одного гена прогрессирующая и стационарная формы ретинита были выявлены у членов одной семьи, и в настоящее время обе формы рассматриваются как результат мутации гена родопсина.
Пигментный ретинит без пигмента
Это название связано с отсутствием характерных для классической формы пигментного ретинита пигментных отложений в виде костных телец в сетчатке при наличии свойственных пигментному ретиниту нерегистрируемой ЭРГ, патологической ЭОГ и психофизических симптомов: нарушения цветоощущения, сужения полей зрения, повышения порога темновой адаптации и пр.
Однако в ряде случаев, когда в начальной стадии заболевания офтальмоскопически видимые изменения отсутствуют, со временем они могут появиться.
В связи с отсутствием изменений на глазном дне диагностика затруднена. Основным критерием при установлении диагноза является нерегистрируемая ЭРГ.
Инвертированный (inverse) пигментный ретинит, или прогрессирующая колбочково-палочковая дегенерация
При инвертированном (inverse) пигментном ретините (по терминологии некоторых авторов «пигментный ретинит, тип II») обычно с аутосомно-рецессивным типом наследования выявляют изменения в макулярной и парамакулярной областях, схожие с таковыми при типичной форме пигментного ретинита. Однако в связи с видимыми изменениями в макулярной области эта форма может быть ошибочно принята за колбочковую дегенерацию.
При этой форме дистрофии обнаруживают центральную скотому, соответствующую области поражения, при сохранном поле зрения и, по данным некоторых авторов, нормальной общей ЭРГ. Нельзя исключить, что эти наблюдения относятся к ранним стадиям заболевания. Порог чувствительности сетчатки в зонах вне скотомы остается нормальным.
Нельзя исключить, что эти наблюдения относятся к ранним стадиям заболевания. Порог чувствительности сетчатки в зонах вне скотомы остается нормальным.
Известна и другая форма инвертированного пигментного ретинита, при которой поражения макулярной области сочетаются с характерными функциональными симптомами генерализованного пигментного ретинита, несмотря на расположение костных телец в заднем полюсе глаза, а не на крайней периферии.
Такая форма заболевания определена как прогрессирующая колбочково-палочковая дегенерация в отличие от типичного пигментного ретинита, который все чаще называют палочково-колбочковой дистрофией, основным ранним симптомом которого является прогрессирующая ночная слепота, нерегистрируемая палочковая (скотопическая) ЭРГ и резко сниженная колбочковая (фотопическая) ЭРГ.
Колбочково-палочковые дистрофии в отличие от палочково-колбочковой характеризуются первичной и доминирующей дистрофией в колбочковой системе с типичными изменениями колбочковых компонентов ЭРГ -колбочково-палочковый паттерн. Колбочковая дистрофия поражает колбочковую функцию и проявляется в прогрессивной потере центрального зрения, дефектном цветовом зрении, фотофобии.
Колбочковая дистрофия поражает колбочковую функцию и проявляется в прогрессивной потере центрального зрения, дефектном цветовом зрении, фотофобии.
В пяти поколениях одной семьи было выявлено генетическое сцепление между прогрессирующей колбочковой дистрофией (CORD5) и генетическими маркерами на хромосоме 17 (р12-р13). Анализ сцепления дал максимальный lod-балл — 7,72 для маркера D17S938. Рекомбинантные галотипы в семье показывают, что локус колбочковой дистрофии расположен в интервале размером 25-сМ между маркерами D17S926/D17S849 и D17S804/D17S945. Рекомбинация была определена между локусом болезни и микросателлитным маркером в гене-кандидате RCV1, кодирующем ретинальный белок рековерин (recoverin). Два дополнительных кандидатных гена, кодирующих ретинальную гуанилатциклазу (GUC2D) и фактор, происходящий из пигментного эпителия (pigment epithelium-derived factor — PEDF), также локализованы на хромосоме 17р13. В той же области картированы локусы для пигментного ретинита и врожденного амавроза Лебера.
В последнее десятилетие у многих больных с пигментным ретинитом благодаря психофизическим методам исследования установлена некоторая сохранность функции палочковой системы. Так, например, по данным пространственной контрастной чувствительности (ПКЧ) выявлены незначительно сниженная ПКЧ в области низких пространственных частот, за которые ответственна парацентральная область сетчатки, значительное снижение в области средних ПЧ и полное отсутствие чувствительности в области высоких пространственных частот.
Специальные условия регистрации и стимуляции позволили выявить в ЭРГ остаточные компоненты палочковой системы. При этом ЭРГ имела вид (паттерн), характерный для колбочково-палочковой дисфункции, т.е. изолированная палочковая ЭРГ была больше сохранна, чем изолированная колбочковая ЭРГ.
У этих пациентов в раннем возрасте признаков заболевания не отмечается. С возрастом снижается острота зрения, изменятся цветоощущение, снижается или отсутствует фотопическая ЭРГ.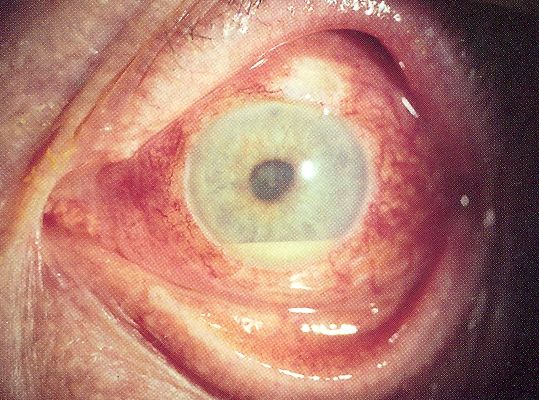 Затем поле зрения концентрически сужается, повышается порог темновой адаптации, значительно снижается или не регистрируется скотопическая ЭРГ, происходит атрофия пигментного эпителия, отмечаются типичные для пигментного ретинита отложения пигмента, хотя больные не предъявляют жалоб на ночную слепоту до того момента, пока поле зрения не сужается до 12°.
Затем поле зрения концентрически сужается, повышается порог темновой адаптации, значительно снижается или не регистрируется скотопическая ЭРГ, происходит атрофия пигментного эпителия, отмечаются типичные для пигментного ретинита отложения пигмента, хотя больные не предъявляют жалоб на ночную слепоту до того момента, пока поле зрения не сужается до 12°.
Таким образом, палочково-колбочковая дистрофия и колбочково-палочковая дистрофия — разные заболевания, которые можно дифференцировать только по ЭРГ, зарегистрированной в специальных условиях, для выделения изолированных колбочковых и палочковых компонентов. Если фотопическая b-волна редуцирована больше скотопической, имеет место паттерн колбочково-палочковой дистрофии. Если более значительно поражение палочковых компонентов, ЭРГ имеет вид паттерна палочково-колбочковой дисфункции. Именно благодаря электроретинографическим исследованиям у ряда больных с пигментным ретинитом выделен паттерн палочково-колбочковой дистрофии, а при наличии в макулярной области дистрофических изменений по типу «бычий глаз» — паттерн колбочково-палочковой дистрофии. Однако, поскольку изменения на глазном дне при эволюции колбочко-палочковой дистрофии схожи с таковыми при макулярной дистрофии типа «бычий глаз», такие макулярные изменения не являются патогномоничным признаком колбочково-палочковой дистрофии, тем более что изменения такого типа характерны, например, и для токсических ретинопатии.
Однако, поскольку изменения на глазном дне при эволюции колбочко-палочковой дистрофии схожи с таковыми при макулярной дистрофии типа «бычий глаз», такие макулярные изменения не являются патогномоничным признаком колбочково-палочковой дистрофии, тем более что изменения такого типа характерны, например, и для токсических ретинопатии.
Колбочково-палочковый вид ЭРГ отмечается не только при пигментном ретините. Описаны с разным названием колбочково-палочковая дистрофия как самостоятельная форма, синдром колбочковой дисфункции, прогрессирующая колбочковая дистрофия. Прогрессирующая колбочковая дистрофия отличается от колбочково-палочковой дистрофии относительной стабильностью и сохранностью границ поля зрения, хотя в отдельных случаях зарегистрированы центральные скотомы. Прогрессирующая же колбочково-палочковая дистрофия с пигментным ретинитом характеризуется прогрессирующим сужением границ поля зрения и наличием кольцевой скотомы.
В связи с различиями в профиле ретинальной чувствительности в ЭРГ у больных с пигментным ретинитом было выделено два типа механизмов развития дегенерации.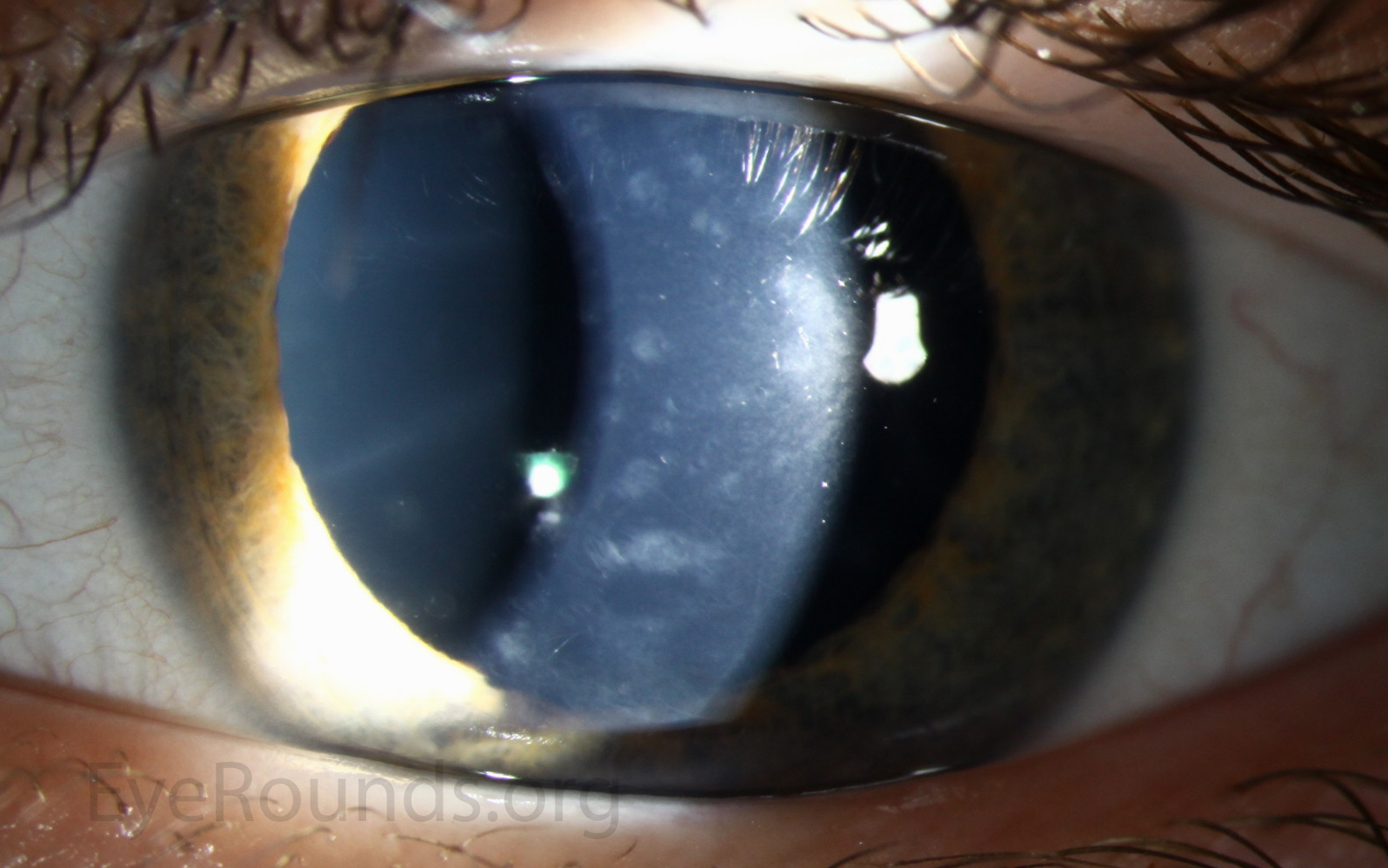
- Первый тип характеризуется диффузной потерей палочковой чувствительности, наличием симптома ночной слепоты с раннего детства.
- Для второй группы пациентов характерна регионарная комбинированная потеря или палочковой, или колбочковой чувствительности, ночная слепота появляется уже в зрелом возрасте.
Эти клинические и функциональные проявления колбочково-палочковой дистрофии и нашли свое отражение в ЭРГ у больных с фенотипическими проявлениями различных наследственных форм пигментного ретинита — аутосомно-доминантной, аутосомно-рецессивной и сцепленной с полом. Атипичная клиническая картина пигментного ретинита с различными изменениями поля зрения, типичной темпоральной атрофией зрительного нерва, телеангиэктазиями, псевдоотеком диска зрительного нерва, небольшим количеством отложений в сетчатке была неоднократно описана в литературе. Характерной чертой этой формы болезни является кольцевидная скотома, расположенная ближе к точке фиксации, чем у пациентов с палочково-колбочковой дегенерацией.
В редких случаях отмечались псевдо-альтитудинальный дефект в поле зрения, концентрическое сужение его до 10°, уменьшение величины и интенсивности изоптеры, что совпадало с демаркационной линией между нормально и ненормально функционирующей сетчаткой. У пациентов с палочково-колбочковой дегенерацией, наоборот, часто отмечались большие скачки чувствительности при проведении исследования с помощью автоматического периметра Гольдмана. У больных с периферической пигментной дегенерацией выявляли сниженную b-волну и значительное нарушение гематологического барьера.
Пациенты с фовеальными поражениями, но с нормальной или умеренно редуцированной скотопической b-волной имеют нормальные данные фотометрии. При прогрессировании процесса ЭРГ становится нерегистрируемой однако при значительном снижении палочковой функции ее порог остается достаточно хорошим, и только дальнейшее концентрическое сужение полей зрения приводит к ночной слепоте.
В поздних стадиях клиническая картина колбочково-палочковой дистрофии схожа с проявлениями палочково-колбочковой дегенерации. Однако при очевидной возможности диагностических ошибок нужно иметь в виду, что для колбочково-палочковой дегенерации характерны более медленное и более легкое течение процесса по сравнению с таковым палочково-колбочковой дегенерации и регистрируемая ЭРГ, в которой палочковые компоненты в большей степени изменены, чем колбочковые, хотя редуцированы и те, и другие. В связи с этим окончательный диагноз может быть установлен при выявлении более специфических биохимических и генетических различий, позволяющих точнее определить типы пигментного ретинита.
Однако при очевидной возможности диагностических ошибок нужно иметь в виду, что для колбочково-палочковой дегенерации характерны более медленное и более легкое течение процесса по сравнению с таковым палочково-колбочковой дегенерации и регистрируемая ЭРГ, в которой палочковые компоненты в большей степени изменены, чем колбочковые, хотя редуцированы и те, и другие. В связи с этим окончательный диагноз может быть установлен при выявлении более специфических биохимических и генетических различий, позволяющих точнее определить типы пигментного ретинита.
Регистрируемая палочково-колбочковая ЭРГ у больных этой категории дала основание определить эти формы заболевания термином «delimited» пигментный ретинит, который включает колбочковую и палочковую дисфункцию. Наблюдающаяся в ранних стадиях пигментного ретинита редуцированная b-волна при относительно нормальной или незначительно измененной колбочковой ЭРГ явилась основанием для использования термина «падочково-колбочковая дистрофия» вместо термина «пигментный ретинит».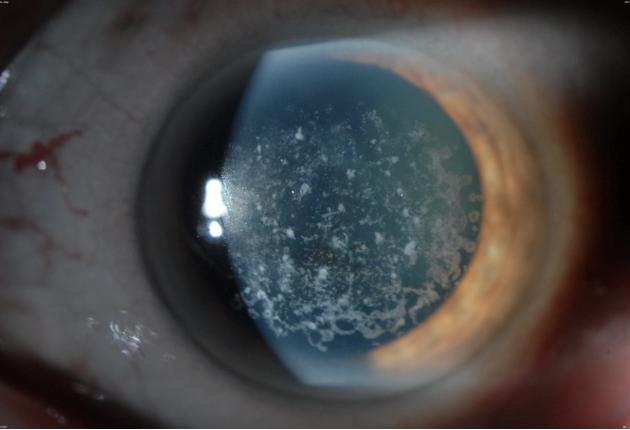 Однако имеются основания считать, что палочко-колбочковая дистрофия является определенным типом пигментного ретинита.
Однако имеются основания считать, что палочко-колбочковая дистрофия является определенным типом пигментного ретинита.
Диагноз колбочково-палочковой дистрофии или колбочкового синдрома дисфункции можно установить при наличии изменений в макулярной области типа «бычий глаз», колбочко-палочкового паттерна ЭРГ, сохранности поля зрения в течение длительного периода времени и отсутствии наследственного анамнеза пигментного ретинита. В то же время наличие увеличивающейся центральной скотомы дает основание для установления диагноза «пигментный ретинит inverse». Нужно иметь в виду, что у многих пациентов с колбочково-палочковым паттерном в анамнезе могут быть такие воспалительные заболевания, как пузырчатый глист, инфекционный мононуклеоз, заболевание, вызываемое неспецифическим вирусом. У одних пациентов течение заболевания прогрессирующее и не отличается от пигментного ретинита, в то время как у других отмечается стационарный характер процесса с не изменяющимся полем зрения.
Пигментный ретинит и дегенеративная миопия
Дегенеративная миопия нередко сочетается с другими пигментными дистрофиями, хороиретинальными дегенерациями, такими как
- пигментный ретинит,
- атрофия гирате,
- хороидеремия,
- fundus flavimaculatus,
- альбинизм,
- врожденная стационарная ночная слепота.
Она может быть частью синдромов системных поражений, например
- синдрома Штиклера (Stickler), (клинические проявления — миопия, глухота, артрит, характерные изменения лицевого черепа («плоское лицо») и расщелина верхнего неба)
- синдрома Моркио (Morquio),
- синдрома Торнера (Turner) и др..
Так, проведенные в последнее десятилетие генетические исследования при изучении семей в нескольких поколениях показали выраженную связь гена COL2A1 с синдромом Штиклера (Stickler).
В последующие годы анализ сцепленной с Х-хромосомой миопии дал положительный результат LOD для локуса F8. На основании клинических и генетических данных был выделен синдром, получивший название болезни Борнхолма (Bornholm). Этот синдром включает амблиопию, миопию и дейтеранопию, а необязательными признаками могли быть гипоплазия зрительного нерва, неспецифические аномалии пигментного эпителия сетчатки, субнормальная ритмическая ЭРГ.
Локализация гена, ответственного за развитие этого синдрома, определена в дистальной части Х-хромосомы на Xq28.
В дальнейшем проведен генетический анализ с использованием ДНК-маркеров при сочетании высокой миопии с врожденной стационарной ночной слепотой. Описана семья с Х-сцепленной врожденной стационарной ночной слепотой (полный тип — CSNBI), в которой возникли трудности при дифференциации клинических проявлений у мужчин: из двух кузенов у одного были врожденный нистагм и близорукость, в то время как у другого, как считали вначале, — пигментный ретинит с атрофией зрительного нерва.
Диагноз врожденной стационарной ночной слепоты был установлен по клиническим, электрофизиологическим и психофизическим критериям и благодаря анализу ДНК-маркеров, фланкирующих локус CSNBI. Проведенный анализ показал, что оба больных мужчины имеют один и тот же гаплотип, унаследованный от их матерей — носителей мутантного гена, исключающий возможность того, что ген миопии имеет неравновесие по сцеплению с CSNBI.
В последние годы были получены данные, которые подтвердили генетическую гетерогенность миопии. Локус для аутосомно-доминантной патологической высокой миопии был картирован в области 18р11.31. Проведен анализ восьми семей, у членов которых в двух поколениях была миопия больше 6,0 дптр. Средний возраст больных 6,8 года. Однако клинических доказательств патологии соединительной ткани не обнаружено. После геномного скрининга было показано сцепление с маркером D18S481 на хромосоме 18р, максимальный LOD-балл составил 9,59. При дальнейшем анализе гаплотипа уточнен локус миопии в интервале 7,6 сМ между маркерами D18S59 и D18S1138 на 18р11.31.
Авторы также сообщили о возможной связи высокой миопии со вторым локусом, расположенным в районе хромосомы 12q21-23. Связь локусов синдромов Штиклера типов I и II (12q13.1-q13.3 и 6р21.3), Марфана (15q21.1) и ювенильной глаукомы (хромосома 1q21-q31) с миопией в исследуемом семействе не обнаружена.
При анализе сцепления в этой же родословной максимальный LOD-балл составил 3,85 для маркеров D12S1706 и D12S327. Анализ рекомбинационных событий позволил определить маркеры фланкирующие второй ген миопии — D12S1684 и D12S1605, расстояние между которыми составило 30,1 сМ на хромосоме 12q21-23.
Анализ рекомбинационных событий позволил определить маркеры фланкирующие второй ген миопии — D12S1684 и D12S1605, расстояние между которыми составило 30,1 сМ на хромосоме 12q21-23.
Эти данные подтверждают генетическую гетерогенность миопии. Идентификация этого гена может помочь в понимании патофизиологии миопии и развития глаза.
Электрофизиологические и психофизические симптомы осложненной
Нередкое сочетание пигментного ретинита с миопией дало основание рассматривать осложненную миопию как вариант наследственной пигментной абиотрофии сетчатки. В основе видимых офтальмоскопических изменений при осложненной близорукости лежат дегенерация, гиперплазия и гипоплазия пигментного, мембраны Бруха, хориоретинальная дегенерация с нарушением хориоидальной циркуляции, неоваскуляризация, образование отверстий, сетевидная и пигментная дегенерация сетчтки с отложением пигмента на периферии, атрофией сетчатки и хориоидеи, дегенерация фоторецепторов.
Все эти дегенеративные хороиретинальные изменения включают три патогенетических процесса:
- абиотрофию,
- хороидальную ишемию и
- образование субретинальной неоваскулярной мембраны.
Степень этих клинических нарушений находит отражение в изменении психофизических и электрофизиологических тестов. Так, темновая адаптация изменяется в случае сочетания дегенеративной миопии со стационарной ночной слепотой и дегенерацией сетчатки, при этом нарушается электрогенез сетчатки.
Цветовое зрение изменяется по мере прогрессирования заболевания, отмечаются потеря цветовой чувствительности преимущественно на желто-голубой цвет, значительное снижение ее на красно-зеленый, в отдельных случаях может быть ахроматопсия.
Изменения полей зрения могут быть различными:
- гемианопсия,
- концентрическое сужение полей зрения,
- квадрантная анопсия,
- скотомы различной локализации, центральные или периферические, кольцевые, аркуатные в зависимости от величины стафиломы и дегенеративных изменений в сетчатке.

Центральная скотома развивается при наличии «пятна Фукса», сухой дистрофии, атрофии макулярной области и образовании субретинальной неоваскулярной мембраны. Причиной образования аркуатных скотом может быть ишемическое состояние диска зрительного нерва. Концентрическое сужение поля зрения, наиболее часто наблюдающееся при дегенеративной миопии, определяется степенью дегенерации сетчатки.
В немногочисленных работах, исследующих ЭРГ при осложненной миопии, отмечено снижение колбочковой функции уже на ранних стадиях заболевания и прогрессивное снижение общей ЭРГ, характеризующей функцию наружных и внутренних слоев сетчатки по мере развития дегенеративных процессов в сетчатке и хороидее. Многие авторы отмечали субнормальный характер ЭРГ при осложненной миопии. Значительно снижаются колбочковые и палочковые компоненты ЭРГ, которая при прогрессирующей выраженной пигментной дистрофии сетчатки, отмечаемой при миопии, может не регистрироваться, так же как при классической форме пигментного ретинита и синдромных поражениях органа зрения, сочетающихся с миопией. Отмечается связь между величиной миопии и степенью снижения ЭРГ.
Отмечается связь между величиной миопии и степенью снижения ЭРГ.
Постоянный потенциал глаза изменяется по-разному и не коррелирует с изменениями ЭРГ. ЭОГ, при которой регистрируется постоянный потенциал глаза, может быть нормальной, субнормальной и патологической в зависимости от доминирующих дегенеративных изменений, локализованных в различных структурах сетчатки и хороидеи. Субнормальная ЭОГ с редуцированным световым подъемом свидетельствует не только о патологии пигментного эпителия сетчатки, но также и о дегенерации фоторецепторов.
При сочетании миопии со стационарной врожденной слепотой отмечается характерная психофизическая и электрофизиологическая симптоматика. В психофизических исследованиях пространственной контрастной чувствительности при патологии палочковой системы, возможно, кривая (паттерн) палочковой дисфункции, которая характеризуется повышением ПКЧ в области высоких пространственных частот, свидетельствует о нарушении межрецепторного взаимодействия между палочками и колбочками. Однако при вовлечении в патологический процесс колбочковой системы и возникновении макулярных изменений пространственная контрастная чувствительность снижается, преимущественно в области средних и высоких пространственных частот. ЭРГ при этом имеет минус-негативный характер, а локальная ЭРГ снижается в зависимости от выраженности дистрофических изменений в макулярной области.
Однако при вовлечении в патологический процесс колбочковой системы и возникновении макулярных изменений пространственная контрастная чувствительность снижается, преимущественно в области средних и высоких пространственных частот. ЭРГ при этом имеет минус-негативный характер, а локальная ЭРГ снижается в зависимости от выраженности дистрофических изменений в макулярной области.
F. Cunier еще в 1838 г. впервые описал две принципиально различные формы врожденной стационарной ночной слепоты в семье Nougaret во Франции. В последующие годы другие поколения этой семьи были описаны Е. Nettleship (1907). Выявленные у ее членов изменения, как правило, наблюдались с рождения и не прогрессировали. Аутосомно-доминантная форма ночной слепоты типа Нугаре, так же как и Х-сцепленная форма, сочеталась с миопией. Как правило, у больных выявляли нормальное глазное дно, нормальное фотопическое поле зрения и различные типы измененной ЭРГ. Чаще описывали редукцию b-волны и в редких случаях — а-волны при регистрации ЭРГ в условиях, когда доминировала функция палочковой системы.
Отмечено сходство времени до пика b-волн ЭРГ, зарегистрированных в фотопических и скотопических условиях, между тем как в норме время до пика (implicit time) скотопического ответа меньше, чем фотопического. При этой форме стационарной ночной слепоты содержание родопсина в сетчатке было нормальным согласно результатам денситометрии, что явилось основанием для предположения об отсутствии связи имеющихся функциональных изменений ЭРГ с наружными сегментами фоторецепторов. Такого рода функциональные нарушения встречаются и у носителей гена и позволяют предположить хороший прогноз.
В связи с этим у больных с миопией при офтальмоскопически видимых изменениях в сетчатке в сочетании с другими симптомами и нарушениями зрительных функций необходимо проводить функциональные и психофизические исследования не только для оценки функционального состояния сетчатки, но и для дифференциальной диагностики тапеторетинальных дистрофий.
Что такое дистрофия Фукса и как ее лечат?
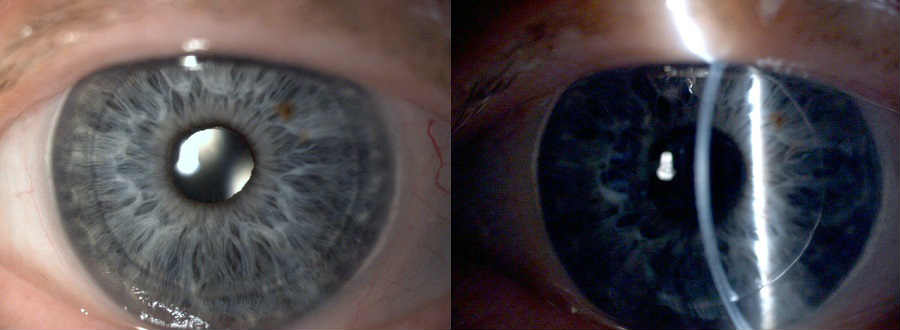
Чтобы роговица могла обеспечивать нормальную оптическую функцию, она должна быть абсолютно прозрачной.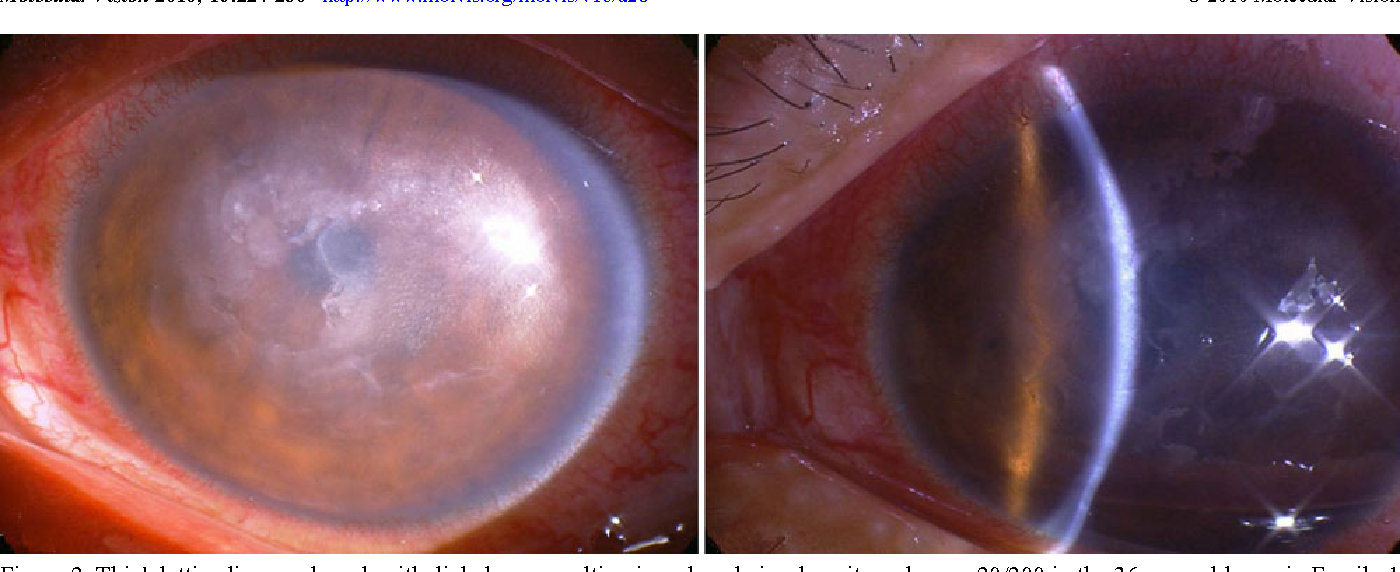 Если по какой-то причине роговая оболочка мутнеет, у человека может заметно снижаться зрение. Одним из заболеваний, при которых важная оптическая среда глаза утрачивает свою прозрачность, является дистрофия Фукса.
Если по какой-то причине роговая оболочка мутнеет, у человека может заметно снижаться зрение. Одним из заболеваний, при которых важная оптическая среда глаза утрачивает свою прозрачность, является дистрофия Фукса.
Суть заболевания
Глазная роговица — одна из важнейших оптических сред, которая позволяет нам видеть. Эта оболочка глаза состоит из пяти слоев, каждый из которых выполняет определенную функцию. Самый нижний слой называется эндотелием, или задним эпителием, и его основная задача — отводить избыток влаги от роговицы, препятствуя ее отеку и помутнению.
Дистрофия Фукса — это болезнь, при которой изменяется нормальная структура эндотелия, что ведет к накоплению влаги в роговице и, как следствие, ее помутнению. Другое название этого заболевания — эпителиально-эндотелиальная дистрофия (ЭЭД), также иногда патологию называют буллезной дистрофией.
Из-за чего может начаться ЭЭД?
Патология может носить первичный характер, в этом случае она обычно возникает у людей (чаще женщин) от 30 до 50 лет. Но нередко ЭЭД является вторичной и развивается у людей разного возраста и пола вследствие повреждения глазных тканей. Как правило, основные причины развития ЭЭД — травмы, особенно проникающие ранения, и хирургические операции по поводу катаракты с установкой искусственного хрусталика.
Но нередко ЭЭД является вторичной и развивается у людей разного возраста и пола вследствие повреждения глазных тканей. Как правило, основные причины развития ЭЭД — травмы, особенно проникающие ранения, и хирургические операции по поводу катаракты с установкой искусственного хрусталика.
При дистрофии Фукса в эндотелии начинаются структурные изменения, количество эндотелиальных клеток сокращается, и нижний слой роговицы истончается. Это приводит к попаданию большого количества жидкости в средний слой роговой оболочки, что вызывает отек и нарушает ее прозрачную структуру. Помутневшая роговица приводит к ухудшению зрения и нередко сопровождается болью в глазах. Если пациенту своевременно не оказать грамотную офтальмологическую помощь, есть риск развития слепоты.
Основные симптомы заболевания
Течение болезни может быть как скрытым — без ярко выраженных клинических проявлений, так и сопровождаться яркой симптоматикой. Основные симптомы, которые характерны для ЭЭД:
- нечеткое, мутное зрение, особенно в утренние часы;
- различные зрительные искажения;
- чрезмерная светочувствительность;
- сниженная острота зрения в темноте, ореолы вокруг световых источников;
- дискомфорт в глазах — чувство «как песка насыпали», ощущение инородного тела;
- образование пузырьков на роговой оболочке из-за накопления жидкости;
- помутнения роговицы;
- утрата зрения (является симптомом поздней стадии).

ЭЭД у разных пациентов может проявляться различными комбинациями перечисленных симптомов, за точной диагностикой следует обращаться к врачу.
Особенности лечения
Одним из методов лечения является закапывание специальных растворов, которые помогают выводить жидкость и уменьшают отек. Если на фоне заболевания развивается буллезная кератопатия, которая часто сопровождается болезненными ощущениями, врач может порекомендовать использование мягких линз. Консервативное лечение не всегда эффективно, поэтому большинству пациентов назначают пересадку роговой оболочки. Современные хирургические методики предполагают трансплантацию не всей оболочки, а лишь ее отдельных слоев. Если вовремя не провести операцию, есть риск потерять зрение.
Важнейшим методом профилактики дистрофии Фукса являются регулярные обследования у офтальмолога. Не менее важно беречь здоровье глаз, избегая любых травм.
Команда MagazinLinz.ru
Буллезная кератопатия — причины, симптомы, диагностика и лечение
Буллезная кератопатия – это патология роговицы, характеризующаяся отеком оболочки, поражением эпителиального слоя с формированием специфических «булл». Клиническая симптоматика представлена снижением остроты зрения, болевым синдромом, ощущением инородного тела в глазу, фотофобией, повышенной слезоточивостью. Для диагностики проводится биомикроскопия, кератопахиметрия, офтальмоскопия, УЗД, визометрия, тонометрия, гониоскопия. На I-II ст. показана консервативная терапия. Возможно применение контактных линз, фототерапевтической кератэктомии, кросслинкинга. Пациентам на III-V стадии выполняют послойную или сквозную кератопластику.
Клиническая симптоматика представлена снижением остроты зрения, болевым синдромом, ощущением инородного тела в глазу, фотофобией, повышенной слезоточивостью. Для диагностики проводится биомикроскопия, кератопахиметрия, офтальмоскопия, УЗД, визометрия, тонометрия, гониоскопия. На I-II ст. показана консервативная терапия. Возможно применение контактных линз, фототерапевтической кератэктомии, кросслинкинга. Пациентам на III-V стадии выполняют послойную или сквозную кератопластику.
Общие сведения
Буллезная кератопатия (вторичная эндотелиально-эпителиальная дистрофия) в 80% случаев обусловлена проведением оперативных вмешательств на переднем отделе глазного яблока. После лечения на IV-V ст. восстановить остроту зрения удается не более чем на 30%. Согласно статистическим данным, реакции отторжения трансплантата наблюдаются у 6% больных. Купировать болевой синдром удается в 91% случаев. Для 8% пациентов характерно рецидивирующее течение патологии, что проявляется образованием единичных булл и эрозивных дефектов на поверхности роговицы. Заболевание распространено повсеместно. Мужчины и женщины болеют с одинаковой частотой.
Заболевание распространено повсеместно. Мужчины и женщины болеют с одинаковой частотой.
Буллезная кератопатия
Причины буллезной кератопатии
Патология имеет приобретенный характер, однако часто удается установить наследственную предрасположенность к данной болезни. Генетические мутации и тип наследования кератопатии не изучен. Основные причины развития:
- Эндотелиальная дистрофия Фукса . Наблюдается генетически детерминированный апоптоз клеток заднего эпителия, что ведет к повышению его проницаемости и возникновению патологии.
- Инфекции глаз. Болезнь развивается при поражении роговой оболочки герпетической или сифилитической природы. Выявление специфических помутнений у новорожденных детей позволяет предположить внутриутробное инфицирование.
- Травмирование роговицы. Повреждение вследствие механической травмы или ожога провоцирует повышенную экссудацию, что становится причиной образования типичных «булл».
- Ятрогенное воздействие. Патология может возникать в раннем послеоперационном периоде после факоэмульсификации катаракты или имплантации интраокулярных линз.

Патогенез
В основе механизма развития заболевания лежит нарушение функции эндотелиального и эпителиального слоев роговой оболочки. Повышение проницаемости эндотелия приводит к пропитыванию тканей внутриглазной жидкостью из передней камеры. Из-за скопления транссудата на поверхностном слое формируются специфические пузырьки или «буллы». Хронический отек существенно нарушает трофические процессы. При присоединении воспалительного компонента организация экссудата ведет к образованию стойкого помутнения. Утолщение роговицы влечет за собой вторичное поражение нервных волокон, возникает болевой синдром. Снижение остроты зрения обусловлено, с одной стороны, нарушением проницаемости оптических сред глаза, с другой – сужением и деформацией зрачка.
Классификация
Различают врожденную и приобретенную формы. В группу риска развития патологии входят лица, у которых определяется прозрачная роговая оболочка нормальной толщины с плотностью эндотелиоцитов 320-510 кл/мм2. В клинической офтальмологии используется классификация, согласно которой выделяют следующие стадии болезни:
В группу риска развития патологии входят лица, у которых определяется прозрачная роговая оболочка нормальной толщины с плотностью эндотелиоцитов 320-510 кл/мм2. В клинической офтальмологии используется классификация, согласно которой выделяют следующие стадии болезни:
- I – визуализируются небольшие участки со сниженной прозрачностью роговицы и неравномерным расположением эндотелиальных клеток. Толщина оболочки не превышает 0,7 мм.
- II – прозрачность снижена умеренно при толщине 0,7-0,79 мм. Роговица равномерно покрыта эндотелиальным слоем благодаря выраженной гипертрофии клеток. Количество эндотелиоцитов уменьшено.
- III – значительное нарушение прозрачности. Средняя толщина превышает 0,8 миллиметров. Эндотелиальные клетки соединяются между собой только благодаря отросткам.
- IV – в среднем толщина составляет 0,97 мм. Выявляются отдельные группы эндотелиоцитов. Прозрачность резко снижена.
- V – визуализируется выраженное помутнение роговицы, толщина которой превышает 1,15 мм. Определяются единичные клетки.
Симптомы буллезной кератопатии
На первой стадии пациенты изредка отмечают у себя чувство дискомфорта в глазнице, сопровождающееся слезоточивостью. Зрительная дисфункция не возникает. Внешние изменения радужки отсутствуют. Вторая стадия характеризуется чувством инородного тела, периодическим покраснением орбитальной конъюнктивы. Отмечается незначительное снижение зрительных функций. При 3 ст. больные предъявляют жалобы на повышенное слезотечение, фотофобию. Наблюдается дискомфорт, напоминающий ощущение рези, инородного тела или «песка» в глазах и под веками. Растяжение нервных волокон приводит к выраженному болевому синдрому колющего характера. Острота зрения резко снижена.
На 4 стадии присоединяется сильная головная боль, которая иррадиирует в надбровные дуги, височные и лобные доли. Прогрессирование зрительной дисфункции приводит к появлению «тумана» или «пелены» перед глазами. Терминальная стадия сопровождается резко выраженным болевым синдромом, снижением зрительных функций вплоть до светоощущения. Пациенты отмечают, что применение контактных методов коррекции нарушения зрения не дает возможности достичь желаемых результатов. Помимо общей симптоматики за счет выраженного отека, гиперемии конъюнктивы и деформации зрачка формируется косметический дефект.
Прогрессирование зрительной дисфункции приводит к появлению «тумана» или «пелены» перед глазами. Терминальная стадия сопровождается резко выраженным болевым синдромом, снижением зрительных функций вплоть до светоощущения. Пациенты отмечают, что применение контактных методов коррекции нарушения зрения не дает возможности достичь желаемых результатов. Помимо общей симптоматики за счет выраженного отека, гиперемии конъюнктивы и деформации зрачка формируется косметический дефект.
Осложнения
При 1 стадии развитие осложнений не характерно. На 2 стадии часто диагностируется рецидивирующий ирит. 3 стадия осложняется иридоциклитом, 4 – помутнением хрусталика, ретрокорнеальной пленкой. Распространенное явление при буллезной кератопатии – поверхностный кератит. При поражении заднего сегмента глаза образуются множественные синехии, возникает отслойка сетчатой оболочки. На терминальной стадии развивается вторичная глаукома. Наиболее неблагоприятный исход заболевания – полная слепота, которая при условии выраженного болевого синдрома требует проведения энуклеации.
Диагностика
Обследование пациента с буллезной формой кератопатии предполагает проведение наружного осмотра и использование специальных методов диагностики. Визуально офтальмолог определяет стойкое помутнение переднего отдела глаз, часто в сочетании с гиперемией орбитальной конъюнктивы. Комплекс офтальмологических исследований включает:
- Биомикроскопию глаза. На начальной стадии выявляется локальный отек эндотелиального слоя, единичные складки на десцементовой мембране. 2 ст. характеризуется стойкой ограниченной отечностью, диффузным распылением пигмента, множественными складками. На следующей стадии в дополнение к вышеописанным проявлениям возникает инъекция сосудов конъюнктивы, признаки поверхностного кератита, эрозивные дефекты, усиленная неоваскуляризация. На 4 ст. отек распространяется на все слои. Для заключительной стадии характерны сосудистые помутнения разной плотности. Роговая оболочка замещается плотной рубцовой тканью с очаговыми изъязвлениями.
- Кератопахиметрию. Определяются признаки локального или диффузного отека. Толщина роговой оболочки в центральной части варьируется от 600 до 1500 мкн.
- Гониоскопию. На пятой стадии выявляется облитерация угла передней камеры, что вызвано организацией экссудата и отложением гранул пигмента. Визуализируются иридокорнеальные сращения. Водянистая влага прозрачная, содержит фибриллы стекловидного тела.
- Визометрию. Острота зрения зачастую находится в пределах от 0,1-0,3 дптр. При тяжелом поражении сохраняется лишь светоощущение.
- Офтальмоскопию. Офтальмоскопический осмотр возможен только на 1-3 стадиях заболевания. Рефлекс, полученный с глазного дна, розового, реже серого цвета. Характерны признаки отслойки сетчатки.
- УЗИ глаза. Визуализируются задние синехии и зрачковая экссудативная пленка на 3 стадии. Применяется у всех пациентов на 5 ст. из-за заращения зрачкового отверстия. Может выявляться помутнение или деструкция стекловидного тела.
- Бесконтактную тонометрию. При тотальном поражении отмечается повышение ВГД, что обусловлено нарушением оттока внутриглазной жидкости.

 Может выявляться помутнение или деструкция стекловидного тела.
Может выявляться помутнение или деструкция стекловидного тела.
Дифференциальная диагностика проводится с первичной кератопатией Фукса и кератитом. При болезни Фукса процесс двухсторонний, оперативные вмешательства в анамнезе отсутствуют, отмечается генетическая предрасположенность. При проведении УЗИ признаки поражения глазных яблок не обнаруживаются. В отличие от буллезной кератопатии при кератите отек локальный, выявляется воспалительный инфильтрат. Эпителий отсутствует только в области инфильтрата. По данным кератопахиметрии толщина роговицы не превышает 800-1000 мкн, реже определяются признаки ее истончения. Пациенты часто отмечают взаимосвязь между развитием кератита и перенесенными воспалительными заболеваниями, микроповреждениями, несоблюдением правил гигиены.
Лечение буллезной кератопатии
Терапевтическ
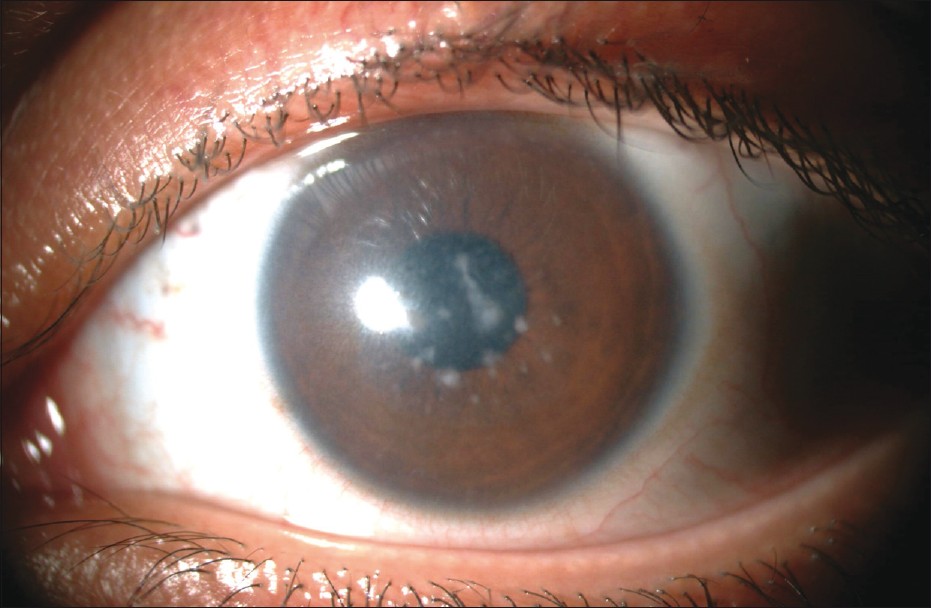
Эндотелиальная дистрофия роговицы глаза (Фукса)
Дистрофия Фукса — эндотелиальная наследственная дистрофия роговицы, при котором дегенеративные изменения происходят в эндотелии роговицы – ее самом внутреннем слое.
Эндотелиальный слой роговицы выполняет особые функции: он регулирует количество внутриглазной жидкости, проникающей к роговице вследствие внутриглазного давления. Излишнее количество внутриглазной жидкости становится причиной отека роговицы и снижения ее прозрачности, иногда до состояния «матового стекла». Особенностью эндотелиальных клеток является то, что они не размножаются делением, поэтому у больных с дистрофией Фукса их количество постоянно уменьшается. Оставшиеся эндотелиальные клетки, пытаясь занять освободившуюся площадь, распластываются, что снижает их насосную функцию. Какое-то время оставшиеся клетки эндотелия компенсируют возникающие в глазу изменения, однако довольно скоро эффективность насосной системы глаза падает, отек роговицы нарастает, что ведет к помутнению роговицы, и зрение серьезно снижается.
Признаки (симптомы) дистрофии Фукса
На ранних этапах дистрофии Фукса обычно отмечается повышенная непереносимость яркого света и засветы. По мере прогрессирования заболевания зрение ухудшается в утренние часы и несколько восстанавливается к вечеру. Обусловлено это тем, что при закрытых глазах во время сна влага с поверхности роговицы не испаряется, накапливаясь внутри. Днем открытые веки «включают» механизм выведения из роговицы внутриглазной влаги, и происходит временная нормализация состояния. Прогрессивная гибель все новых клеток эндотелия ведет к постепенному снижению зрения.
Основными признаками эндотелиальной дистрофии принято считать следующие:
- Ухудшение зрения после сна, расплывчатое зрение.
- Колебания остроты зрения.
- Засветы (ослепления) при взгляде на источник яркого света.
- Плохая переносимость интенсивного света.
- Дискомфорт и ощущение песка в глазах.
Для эндотелиальной дистрофии Фукса характерно двустороннее поражение. Заболевание чаще встречается у женщин после 30-40 лет. При серьезном ухудшении зрения, когда теряется способность самообслуживания, встает вопрос о проведении пересадки роговицы.
Диагностика
Дистрофию Фукса обнаруживают на офтальмологическом осмотре со щелевой лампой. Для уточнения диагноза и оценки толщины роговицы (выраженности отека) назначают ультразвуковую пахиметрию. Также проводят эндотелиальную микроскопию, позволяющему получить четкое изображение клеток эндотелия, что дает возможность определить методом подсчета плотность их на единицу площади.
Лечение
Излечить данное заболевание с помощью медикаментозных препаратов не представляется возможным, хотя для временного улучшения зрения могут применяться растворы, имеющие высокую осмолярность и «вытягивающие» воду из роговицы.
В настоящее время для лечения эндотелиальной дистрофии (наследственной или возникшей после операций на глазах) достаточно успешно применяют методику роговичного кросслинкинга. Кроме того, хорошие результаты показывает операция по пересадке роговицы. Однако риск осложнений такой операции относительно высок, и ее обычно назначают, когда острота зрения составляет 0,1-0,3.
В «Клинике доктора Шиловой» пациентам с эндотелиальной дистрофией роговицы может быть предложена операция задней послойной кератопластики.
Ее цель – удаление десцеметовой мембраны с пораженным эндотелием и замена их донорским трансплантатом. В зависимости от конкретной ситуации, операция может быть выполнена при помощи механического инструмента кератома или посредством фемтосекундного лазера.
В нашей клинике операции кератопластики выполняю ведущие отечественные и зарубежные специалисты, Шилова Татьяна Юрьевна и Вальтер Секундо.
Стоимость необходимых в вашем случае диагностических процедур и последующего лечения уточняйте, пожалуйста, здесь.
Узнать о стоимости всех предоставляемых в клинике услуг и записаться на консультативный прием можно по телефону в Москве +7 (495) 589-33-65 или используя форму онлайн-записи.
Эндотелиальная дистрофия роговицы, симптомы, профилактика, лечение.
Дистрофии и дегенерации роговицы
Дистрофией роговицы называют состояния, выражающиеся в перерождении, или дегенерации, а также в нарушении питания, или трофики, роговицы.
Выделяют первичные и вторичные дистрофии роговицы.
Первичные дистрофии – это врожденные, наследственно обусловленные дегенеративные поражения роговицы. Процесс, в большинстве случаев, двусторонний.
Вторичные дистрофии – приобретенные – возникают вследствие оперативных вмешательств, травм глаза или в результате длительно текущих местных воспалительных процессов. Поражается, как правило, один глаз.
Эндотелиальная дистрофия Фукса
Эндотелиальная дистрофия роговицы относится к первичным дистрофиям. Впервые была описана в самом начале 20 века австрийцем Эрнстом Фуксом, в честь которого и получила свое название. Это наследственное заболевание, которое чуть чаще встречается у женщин, чем у мужчин. Выделяют два генетически обусловленных типа дистрофии Фукса: раннюю (тип 1), которая возникает практически сразу после рождения и встречается крайне редко, и позднюю (тип 2), развивающуюся в возрасте 45–50 лет.
При дистрофии Фукса поражается самый внутренний слой роговицы — эндотелий, клетки которого не способны к делению. При операционных вмешательствах, травмах, воспалениях происходит повреждение эндотелия с гибелью части клеток. Оставшиеся клетки занимают освободившуюся площадь, как бы распластываясь по внутренней поверхности роговицы.
Эндотелий роговицы работает как насос, обеспечивая откачивание из толщи роговицы лишней жидкости и, таким образом, поддерживая прозрачность роговицы. По мере прогрессирования дистрофии Фукса происходит постепенная гибель эндотелиальных клеток. До определенного момента состояние компенсируется усиленной работой оставшихся клеток. В конце концов, происходит декомпенсация состояния. Эндотелий истончен, функция клеток ослаблена, через дефекты эндотелия влага передней камеры просачивается в строму роговицы и вызывает отек и последующую деструкцию. Роговица мутнеет. В последующем возникает и отек наружного слоя роговицы – эпителия, может развиваться буллезная кератопатия – формирование на поверхности роговицы пузырей – «булл». Разрыв буллы вызывает сильнейший дискомфорт и болевой синдром.
Потеря эндотелиальных клеток может усугубляться или ускоряться после травмы или хирургического вмешательства.
Таким образом, пациенты с дистрофией Фукса больше подвержены риску выраженного отека роговицы после глазных операций, поскольку у них изначально ослаблена функция эндотелиальных клеток.
Клинические проявления дистрофии Фукса
Эндотелиальная дистрофия роговицы клинически может быть разной степени выраженности: от бессимптомного течения до тяжелой буллезной кератопатии.
На ранних стадиях заболевания пациенты отмечают повышенную чувствительность к свету и ухудшение зрения в утренние часы с постепенным восстановлением к вечеру. Это объясняется тем, что во время сна не происходит испарения влаги с поверхности роговицы, и жидкость еще больше накапливается в роговице. Во время бодрствования за счет испарения жидкости с роговицы, отек уменьшается и зрение улучшается. По мере прогрессирования заболевания зрение остается постоянно сниженным. При вовлечении в процесс эпителия и образования булл появляется ощущение инородного тела, болевой синдром, выраженная светобоязнь.
Диагностика
Диагноз эндотелиальной дистрофии роговицы ставится на основании биомикроскопии – осмотре роговицы под щелевой лампой. При этом могут обнаружить частичное отсутствие эндотелия, отек роговицы, буллезные изменения эпителия.
Кроме того, применяется пахиметрия для оценки толщины роговицы и выраженности ее отека.
Подтвердить диагноз и оценить тяжесть процесса позволяют данные конфокальной микроскопии роговицы, в результате которой может быть получено четкое изображение эндотелия с возможностью определения плотности клеток на единицу площади и их среднего размера.
Лечение эндотелиальной дистрофии роговицы
Для симптоматического лечения применяют инстилляции гипертонических солевых раствороа, которые способны притягивать воду. Таким образом, из роговицы удаляется лишняя жидкость, и временно улучшается зрение. При развитии буллезной кератопатии для уменьшения болевого синдрома можно использовать лечебные мягкие контактные линзы.
Кардинальным лечением является пересадка роговицы. Сначала пересаживали роговицу целиком – такая кератопластика называется сквозной. В последующем была разработана послойная кератопластика, которая подразумевает пересадку определенных слоев роговицы. В последние годы при дистрофии Фукса чаще применяют так называемую DSEK – пересадку десцеметовой мембраны с эндотелиальным слоем роговицы.
Причины, симптомы, риски и лечение
Дистрофия роговицы Фукса — это заболевание передней поверхности глаза (роговицы), которое обычно поражает пожилых людей. Вот семь ключевых фактов о дистрофии Фукса, которые вы должны знать:
1. Что такое дистрофия Фукса?
Дистрофия Фукса (фукс ДИС-тру-фи) — заболевание глаз
в котором самый внутренний слой клеток роговицы подвергается
дегенеративные изменения.Этот клеточный слой, называемый эндотелием,
отвечает за поддержание необходимого количества жидкости в роговице.
Эндотелий сохраняет роговицу чистой для хорошего зрения за счет откачивания
избыток жидкости, который может вызвать отек роговицы.
Также называется Fuchs ‘
дистрофия роговицы и эндотелиальная дистрофия Фукса, заболевание обычно
влияет на оба глаза и вызывает постепенное ухудшение зрения из-за роговицы
отек (припухлость) и помутнение.
По мере прогрессирования заболевания
отек роговицы может вызвать появление волдырей на передней части роговицы
известны как эпителиальные буллы (бычий глаз).Это состояние известно как
буллезная кератопатия.
Профилактика дистрофии Фукса неизвестна.
2. Что его вызывает?
Fuchs ‘
дистрофия может иметь генетическую причину, но может возникать и без
предыдущий семейный анамнез заболевания. Во многих случаях причина в
неизвестно.
3. Каковы симптомы дистрофии Фукса?
Симптомы дистрофии Фукса включают:
Ослепление и чувствительность к свету
Боль в глазах
Затуманенное или нечеткое зрение
Видение цветных ореолов вокруг огней
9 Затруднение зрения 900
Плохое зрение после пробуждения, которое может улучшиться позже в течение дня
Ощущение, что что-то попало в глаз (ощущение инородного тела)
4.Кто больше всего подвержен этому риску?
Видение
проблемы, связанные с дистрофией роговицы Фукса, обычно возникают у людей в пожилом возрасте
50, хотя окулисты могут обнаружить ранние признаки болезни у молодых
взрослые люди. Оказывается, это чаще встречается у женщин, чем у мужчин.
Если у вашей матери или отца дистрофия Фукса, у вас есть примерно 50-процентный шанс заболеть этой болезнью.
5. Как это обнаруживается?
Для выявления дистрофии роговицы Фукса необходимо комплексное обследование зрения у оптометриста или офтальмолога.
Во время обследования окулист
будет использовать инструмент, называемый щелевой лампой, для выполнения подробных
осмотр роговицы. Во время этой процедуры он или она осмотрит
роговицу под большим увеличением, чтобы увидеть тонкие изменения
появление в эндотелии клеток, характерных для
болезнь.
Ранними клиническими признаками дистрофии Фукса являются
уменьшенное количество эндотелиальных клеток и крошечные каплевидные поражения в
Эндотелий роговицы, называемый каплевидным роговицей.
Еще одна проверка глаз
врач может измерить толщину вашей роговицы
(пахиметрия), чтобы обнаружить увеличение толщины роговицы, которое может указывать на
отек роговицы от болезни.
Кроме того, проверка остроты зрения с помощью диаграммы зрения, которая проводится во время комплексного обследования, может выявить ухудшение зрения из-за отека роговицы.
6. Какое лечение доступно для дистрофии Фукса?
Лечение
для дистрофии Фукса зависит от стадии заболевания.В начале
случаях зрение часто можно улучшить, удалив лишнюю воду из
роговица с 5% -ными глазными каплями хлорида натрия (гипертоническая).
Если у вас светобоязнь, вызванная дистрофией роговицы, очки с фотохромными линзами могут помочь снизить вашу чувствительность к солнечному свету. Кроме того, антибликовое покрытие устраняет блики в линзах очков, которые могут быть особенно неприятными для людей с дистрофией Фуха.
Если у вас эндотелиальная дистрофия и глазная гипертензия, ваш глазной врач может порекомендовать глазные капли от глаукомы для снижения внутриглазного давления (ВГД).Высокое глазное давление может повредить эндотелий роговицы, усугубив дистрофию Фукса.
По мере прогрессирования болезни эпителиальные буллы могут разрываться, вызывая болезненные ссадины роговицы и ухудшение зрения. Если это происходит или дистрофия Фукса прогрессирует до такой степени, что вызывает значительную потерю зрения, обычно требуется пересадка роговицы.
Альтернатива полнослойной трансплантации роговицы (также называемая проникающей кератопластикой,
или PK) — это глубокая ламеллярная эндотелиальная кератопластика (DLEK), которая
хирургический метод замены эндотелия, покидающего верхние слои
роговицы нетронутой.Эта процедура показала успех для
лечение дистрофии Фукса с потенциально меньшими рисками, чем
проникающая кератопластика.
В последние годы развитая форма
DLEK назвали эндотелиальный десцемет с фемтосекундным лазером для удаления эндотелия
кератопластика (FS-DSEK) показала обнадеживающие результаты в лечении
болезнь.
7. Меры предосторожности
Если вам поставили диагноз дистрофия роговицы Фукса, обязательно обсудите это со своим глазным врачом, если вы планируете LASIK или другую рефракционную операцию, или если у вас катаракта и вам требуется операция по удалению катаракты.Эти операции на глазах могут ухудшить состояние и привести к дистрофии роговицы.
часто считается противопоказанием к плановой рефракционной хирургии.
Примечания и ссылки
Офтальмология Дуэйна, 15-е изд. 2009 г.
Предварительные результаты эндотелиальной кератопластики с десцеметовой полосой с фемтосекундным лазером. Архив офтальмологии . Октябрь 2008 г.
Лазерный кератомилез in situ у пациентов с каплевидными каплями роговицы и семейным анамнезом эндотелиальной дистрофии Фукса. Журнал катарактальной и рефракционной хирургии . Декабрь 2005 г.
Хирургия катаракты у пациентов с дистрофией роговицы Фукса: Расширение рекомендаций по хирургии катаракты без одновременной кератопластики. Офтальмология . Март 2005 г.
Глубокая ламеллярная эндотелиальная кератопластика (DLEK): достижение идеальных целей эндотелиального замещения. Глаз . Ноябрь 2003 г.
Страница обновлена в августе 2017 г.
Дистрофия Фукса
Дистрофия Фукса , также известная как эндотелиальная дистрофия Фукса , это медленно прогрессирующее заболевание роговицы, которое обычно поражает оба глаза и несколько чаще встречается у женщин, чем у мужчин.Хотя врачи часто могут увидеть ранние признаки дистрофии Фукса у людей в возрасте от 30 до 40 лет, болезнь редко влияет на зрение, пока люди не достигают 50-60 лет.
Заболевание впервые было описано австрийцем Эрнстом Фуксом (1851–1930), в честь которого оно названо.
Этиология
Эндотелиальная дистрофия Фукса (ЭД) — дегенеративное заболевание эндотелия роговицы с накоплением очаговых наростов, называемых гуттами, и утолщением десцеметовой мембраны, приводящим к отеку роговицы и потере зрения.Эндотелиальные клетки роговицы являются основными «насосными» клетками роговицы, обеспечивающими прозрачность стромы. В FED мембрана Десцемета сильно утолщена за счет накопления аномального коллагена с широкими промежутками и многочисленных кишок. Эндотелиальные клетки роговицы на конечной стадии FED уменьшаются в количестве и кажутся ослабленными, вызывая прогрессирующий отек стромы. Прогрессирующая потеря эндотелиальных клеток вызывает относительный приток водянистой влаги в роговицу, что приводит к отеку (отеку стромы роговицы), что приводит к искажению зрения.В конечном итоге эпителий также становится отечным, что приводит к более серьезным нарушениям зрения. Фокальные области или волдыри отека эпителия («буллы») могут быть особенно болезненными.
Наследование FED является аутосомно-доминантным с генетическими модификаторами и факторами окружающей среды, такими как повышенная распространенность у пожилых людей и женщин. Потеря эндотелиальных клеток может усугубляться или ускоряться из-за внутриглазной травмы или хирургического вмешательства. Распространенный сценарий включает чрезмерное набухание или отек роговицы после операции по удалению катаракты или других видов хирургии глаза.Следовательно, пациенты с дистрофией Фукса в анамнезе могут подвергаться большему риску отека роговицы после операции на глазах, поскольку у них меньше функционирующих эндотелиальных клеток.
FED подразделяется на 4 стадии: от ранних признаков образования кишок до конечной стадии субэпителиального рубцевания. Диагноз ставится при биомикроскопическом исследовании; другие методы, такие как пахиметрия роговицы, конфокальная биомикроскопия и зеркальная микроскопия, могут использоваться совместно.
Точный патогенез неизвестен, но факторы включают апоптоз эндотелиальных клеток, половые гормоны, воспаление, а также поток и состав водянистой влаги.Мутации в коллагене VIII, основном компоненте десцеметовой мембраны, секретируемой эндотелиальными клетками, были связаны с ранним началом FED. [2]
Гены включают:
Признаки и симптомы
Сначала человек с дистрофией Фукса просыпается с нечетким зрением, которое постепенно проясняется в течение дня. Это происходит потому, что утром роговица обычно толще; он задерживает жидкости во время сна, которые испаряются в слезной пленке, пока мы бодрствуем.По мере развития болезни эта опухоль будет оставаться постоянной и ухудшать зрение в течение дня.
Лечение
Медицинское лечение включает применение местного гипертонического раствора, использование фена для обезвоживания прекорнеальной слезной пленки и использование мягких терапевтических контактных линз. Пациенту рекомендуется держать фен на расстоянии вытянутой руки или направлять его поперек лица, чтобы высушить эпителиальные пузыри. Это можно делать два-три раза в день. Однако окончательное лечение (особенно при увеличении отека роговицы) — хирургическое в форме трансплантации роговицы или проникающей кератопластики (PKP).
С 1998 г. Melles et al. Разработали новые хирургические методы лечения ФЭД. в Нидерландах. Эти процедуры, называемые задней ламеллярной кератопластикой или эндотелиальной кератопластикой, получили широкое распространение как глубокая ламеллярная эндотелиальная кератопластика (DLEK) и удаление десцемета с эндотелиальной кератопластикой (DSEK). DLEK и DSEK позволяют избежать хирургических осложнений PKP, таких как расхождение раны, инфекции и высокий послеоперационный астигматизм. С 2004 года DSEK стала доминирующей процедурой, потому что технически она намного проще для хирурга по сравнению с DLEK или PKP.Усовершенствованные хирургические инструменты для DSEK, такие как инъектор трансплантата DSEK, станут доступны в ближайшее время (2008 г.). Это может способствовать более быстрому восстановлению пациентов благодаря возможности выполнять DSEK через очень маленькие (3 мм) разрезы без швов.
Недавно эндотелиальная кератопластика была усовершенствована до эндотелиальной кератопластики с десцеметовой мембраной (DMEK), при которой трансплантируется только донорская десцеметовая мембрана и ее эндотелий. При использовании DMEK в 90% случаев достигается наилучшая острота зрения с оптической коррекцией 20/40 или выше, а в 60% случаев — 20/25 или лучше в течение 1-3 месяцев.
Более предположительные будущие направления в лечении FED включают in vitro расширение эндотелиальных клеток роговицы человека для трансплантации, искусственные роговицы и генетические модификации.
См. Также
Список литературы
Внешние ссылки
| |||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||
| ||||||||||||||
| ||||||||||||||||||||||||||
Дистрофия Фукса — Инфогалактика: ядро планетарного знания
Дистрофия Фукса (произносится как фукс- DIS -trə-fe ), также известная как Фукс ‘FC эндотрофия роговицы представляет собой медленно прогрессирующую дистрофию роговицы, которая обычно поражает оба глаза и несколько чаще встречается у женщин, чем у мужчин.Хотя врачи часто могут увидеть ранние признаки дистрофии Фукса у людей в возрасте от 30 до 40 лет, болезнь редко влияет на зрение, пока люди не достигают 50-60 лет.
Заболевание впервые было описано австрийским офтальмологом Эрнстом Фуксом (1851–1930), в честь которого оно названо. В 1910 году Фукс впервые сообщил о 13 случаях центрального помутнения роговицы, потере чувствительности роговицы и образовании эпителиальных булл, которые он назвал «эпителиальной дистрофией роговицы». Он характеризовался поздним началом, медленным прогрессированием, снижением остроты зрения по утрам, отсутствием воспаления, диффузным помутнением роговицы, интенсивностью в центральной части и шероховатым эпителием с везикулеподобными чертами. [2] Переход к пониманию эндотелиальной дистрофии роговицы Фукса (FCED) как болезни эндотелия роговицы произошел после ряда наблюдений в 1920-х годах. Кристаллические особенности эндотелия были отмечены Краупой в 1920 году, который предположил, что эпителиальные изменения зависят от эндотелия. Используя щелевую лампу, Фогт описал наросты, связанные с FCD, как каплевидные в 1921 году. Затем в 1924 году Грейвс предоставил чрезвычайно подробное объяснение подъемов эндотелия, видимых при биомикроскопии с щелевой лампой.Пациент с односторонней эпителиальной дистрофией и двусторонними эндотелиальными изменениями был описан Friedenwalds в 1925 г .; последующее вовлечение второго глаза заставило их подчеркнуть, что изменения эндотелия предшествуют изменениям эпителия. Поскольку только часть пациентов с эндотелиальными изменениями перешла к эпителиальному поражению, Грейвс заявил 19 октября 1925 года в Нью-Йоркской медицинской академии, что «эпителиальная дистрофия Фукса может быть очень поздним продолжением более серьезных случаев более глубокого поражения». [3]
Эпидемиология
Немногие исследования изучали распространенность FCED в крупном масштабе. Сначала Фукс оценил частоту возникновения эпителиальной дистрофии роговицы в клинических условиях, как один на каждые 2000 пациентов; скорость, которая, вероятно, отражает тех, кто прогрессирует до запущенной болезни. Поперечные исследования предполагают относительно более высокую распространенность болезни в европейских странах по сравнению с другими регионами мира. Дистрофия Фукса редко поражает людей в возрасте до 50 лет. [4]
Этиология
Эндотелиальная дистрофия роговицы Фукса (FCED) — дегенеративное заболевание эндотелия роговицы с накоплением очаговых выростов, называемых гуттами (каплями), и утолщением десцеметовой мембраны, приводящим к отеку роговицы и потере зрения. Эндотелиальные клетки роговицы являются основными «насосными» клетками роговицы, обеспечивающими прозрачность стромы. В FED мембрана Десцемета сильно утолщена за счет накопления аномального коллагена с широкими промежутками и многочисленных кишок.Эндотелиальные клетки роговицы на конечной стадии FED уменьшаются в количестве и кажутся ослабленными, вызывая прогрессирующий отек стромы. Прогрессирующая потеря эндотелиальных клеток вызывает относительный приток водянистой влаги в роговицу, что приводит к отеку (отеку стромы роговицы), что приводит к искажению зрения. В конечном итоге эпителий также становится отечным, что приводит к более серьезным нарушениям зрения. Фокальные области или волдыри отека эпителия («буллы») могут быть особенно болезненными.
Наследование FCED является аутосомно-доминантным с генетическими модификаторами и модификаторами окружающей среды, такими как повышенная распространенность у пожилых людей и женщин.Потеря эндотелиальных клеток может усугубляться или ускоряться из-за внутриглазной травмы или хирургического вмешательства. Распространенный сценарий включает чрезмерное набухание или отек роговицы после операции по удалению катаракты или других видов хирургии глаза. Следовательно, пациенты с дистрофией Фукса в анамнезе могут подвергаться большему риску отека роговицы после операции на глазах, поскольку у них меньше функционирующих эндотелиальных клеток.
FCED подразделяется на 4 стадии: от ранних признаков образования кишок до конечной стадии субэпителиального рубцевания. Диагноз ставится при биомикроскопическом исследовании.Другие методы, такие как пахиметрия роговицы, конфокальная биомикроскопия и зеркальная микроскопия, могут использоваться вместе.
Точный патогенез неизвестен, но факторы включают апоптоз эндотелиальных клеток, половые гормоны, воспаление, а также поток и состав водянистой влаги. Мутации в коллагене VIII, основном компоненте десцеметовой мембраны, секретируемой эндотелиальными клетками, были связаны с ранним началом FCED. [5]
Гены включают:
Признаки и симптомы
Сначала человек с симптоматической дистрофией Фукса просыпается с нечеткостью зрения и жалобами на блики, которые постепенно улучшаются в течение дня. [4] Это происходит потому, что обычно утром роговица толще; он удерживает жидкость во время сна из-за того, что наши веки закрыты, и эта жидкость из роговицы испаряется, пока мы бодрствуем. По мере развития болезни эта опухоль будет оставаться стойкой и ухудшать зрение в течение дня. Исследователи обнаруживают, что болезнь Фукса является генетически гетерогенной болезнью, и многие различные гены и локусы связаны с небольшим процентом общих случаев болезни Фукса.Определенные генетические поражения коррелируют с более тяжелым заболеванием и более ранним началом. [6] [7] [8] Таким образом, некоторые люди могут испытывать симптомы болезни в гораздо более раннем возрасте, в то время как другие могут не проявлять симптомов до более позднего возраста.
Лечение
Медицинское лечение FCED используется для лечения симптомов раннего заболевания. Медицинское лечение включает местное применение гипертонического раствора, использование фена для обезвоживания прекорнеальной слезной пленки и использование мягких терапевтических контактных линз.Пациенту рекомендуется держать фен на расстоянии вытянутой руки или направлять его поперек лица, чтобы высушить эпителиальные пузыри. Это можно делать два-три раза в день. Также сообщалось, что ботулотоксин может вызывать улучшение на несколько месяцев. Однако окончательное лечение (особенно при увеличении отека роговицы) — хирургическое в виде трансплантации роговицы.
С 1998 г. были разработаны новые хирургические методы лечения FCED, первоначально Г.Melles et al. в Нидерландах. Эти процедуры, называемые задней ламеллярной кератопластикой или эндотелиальной кератопластикой, изначально были популярны как глубокая ламеллярная эндотелиальная кератопластика (DLEK) и десцеметовое удаление с эндотелиальной кератопластикой (DSEK). DLEK и DSEK позволяют избежать некоторых хирургических осложнений PKP, таких как расхождение раны и высокий послеоперационный астигматизм. С 2004 года DSEK стала доминирующей процедурой для пациентов с поражением роговицы, ограниченным эндотелием. Это может быть технически проще для хирурга по сравнению с DLEK и может обеспечить превосходные визуальные результаты.При использовании DSEK пациенты должны оставаться в положении лежа на спине (положение лицом вверх) в течение 24 или более часов после процедуры, пока трансплантированная ткань прилипает к вышележащей роговице.
Усовершенствованный хирургический инструментарий для DSEK, такой как инъекторы трансплантата DSEK, и технические усовершенствования хирургической техники способствовали уменьшению осложнений и уменьшили возможность выполнения DSEK через очень маленький (3 мм) разрез без швов.
Недавно эндотелиальная кератопластика была усовершенствована до эндотелиальной кератопластики с десцеметовой мембраной (DMEK), при которой трансплантируется только донорская десцеметовая мембрана и ее эндотелий.При использовании DMEK в 90% случаев достигается наилучшая острота зрения с коррекцией на очки 20/40 или выше, а в 60% случаев — 20/25 или лучше в течение 1-3 месяцев, хотя такие осложнения, как несостоятельность и отслоение трансплантата, остаются проблемами для пациента и врач хирург. DMEK доступен в Великобритании в больницах Калдердейла и Хаддерсфилда. [9]
Более предположительные будущие направления в лечении FED включают in vitro экспансию эндотелиальных клеток роговицы человека для трансплантации, искусственные роговицы (кератопротезы) и генетические модификации.
Более глубокое понимание патофизиологии FED может помочь в будущем в разработке методов лечения для предотвращения прогрессирования заболевания. Хотя в исследовании и лечении ФЭД был достигнут значительный прогресс, еще предстоит ответить на многие вопросы. Точные причины заболевания, прогнозирование прогрессирования заболевания и предоставление точного прогноза, методы профилактики и эффективное консервативное лечение — все это вопросы, требующие ответа. Повышенное внимание следует уделять исследованиям, которые могут ответить на самые основные вопросы о том, как развивается болезнь: каков биомолекулярный путь, связанный с заболеванием, и какие генетические факторы или факторы окружающей среды способствуют его прогрессированию? Помимо формирования нашего понимания FED, идентификация этих факторов будет иметь важное значение для профилактики и лечения этого состояния. [3]
См. Также
Список литературы
Дистрофия Фукса. Причины, симптомы, лечение дистрофия Фукса
Дистрофия Фукса (произносится как Фукс) — это заболевание глаз, при котором клетки, выстилающие внутреннюю поверхность роговицы, начинают медленно отмирать. Заболевание обычно поражает оба глаза.
Причины
Дистрофия Фукса может передаваться по наследству, что означает, что она может передаваться от родителей к детям. В некоторых семьях он передается по аутосомно-доминантному типу.Это означает, что если кто-то из ваших родителей болен этим заболеванием, у вас есть 50% вероятность развития этого заболевания.
Однако это состояние может также возникать у людей, у которых нет семейной истории болезни.
Дистрофия Фукса чаще встречается у женщин, чем у мужчин. Проблемы со зрением обычно не появляются в возрасте до 50 лет, хотя врачи могут увидеть признаки болезни у пострадавших в более раннем возрасте, обычно в возрасте от 30 до 40 лет.
Дистрофия Фукса поражает тонкий слой клеток, выстилающих заднюю часть роговицы.Этот слой называется эндотелием. Заболевание возникает, когда эти клетки начинают медленно отмирать. (Причина неизвестна.) Клетки помогают откачивать лишнюю жидкость из роговицы. По мере того, как теряется все больше и больше клеток, в роговице начинает накапливаться жидкость, вызывая отек и помутнение роговицы.
Поначалу жидкость может накапливаться только во время сна, когда глаз закрыт. По мере развития болезни в эндотелии могут образовываться небольшие волдыри. Волдыри становятся больше и со временем могут сломаться, вызывая боль в глазах.Дистрофия Фукса также может вызывать изменение формы роговицы, вызывая дальнейшие проблемы со зрением.
Симптомы
- Боль в глазах
- Чувствительность глаз к свету, особенно к бликам
- Затуманенное или нечеткое зрение, сначала только по утрам
- Видеть цветные ореолы вокруг огней
- Ухудшение зрения в течение дня
Экзамены и тесты
Врач может диагностировать дистрофию Фукса при обследовании с помощью щелевой лампы.
Дополнительные тесты, которые могут быть выполнены, включают:
- Пахиметрия — измерение толщины роговицы
- Исследование под зеркальным микроскопом — позволяет врачу рассмотреть тонкий слой клеток, выстилающих заднюю часть роговицы.
- Тест на остроту зрения
Лечение
Глазные капли или мази, вытягивающие жидкость из роговицы, используются для облегчения симптомов дистрофии Фукса.
Если на роговице появляются болезненные язвы, уменьшить боль могут мягкие контактные линзы или хирургическое вмешательство по созданию лоскутов поверх язв.
Единственное лекарство от дистрофии Фукса — трансплантация роговицы. Дистрофия Фукса — одна из основных причин трансплантации роговицы в США.
Глубокая ламеллярная кератопластика (DLK) — альтернатива традиционной трансплантации. При этой процедуре донорской тканью заменяются только глубокие слои роговицы. Процедура не требует наложения швов. Время восстановления быстрее и возникает меньше осложнений, таких как отторжение.
Прогноз (прогноз)
Дистрофия Фукса со временем ухудшается.Без трансплантации роговицы пациент с тяжелой дистрофией Фукса может ослепнуть или почувствовать сильную боль и очень плохое зрение.
Легкие случаи дистрофии Фукса часто ухудшаются после операции по удалению катаракты. Хирург катаракты оценит этот риск и может изменить технику или время операции по удалению катаракты.
Возможные осложнения
Осложнения дистрофии Фукса включают:
- Светочувствительность
- От легкой до тяжелой потери зрения
- Частые сильные боли по мере обострения болезни
Когда обращаться к медицинскому работнику
Позвоните своему врачу, если у вас есть:
- Боль в глазах
- Чувствительность глаза к свету
- Ощущение, будто что-то попадает в глаз, когда там ничего нет
- Проблемы со зрением, такие как ореолы или затуманенное зрение
- Ухудшение зрения
Профилактика
Профилактика неизвестна.