В России появился первый регистр пациентов с диагнозом хронический миелолейкоз
Москва, 02 ноября 2016 – Компания «Новартис» передала ЦНИИ организации и информатизации здравоохранения регистр пациентов c диагнозом хронический миелолейкоз (ХМЛ). Регистр велся на протяжении 10 лет и включает в себя информацию по 7 753 пациентам, которые проходили лечение в 111 медицинских центрах по всей стране.
Хронический миелолейкоз – третье по частоте встречаемости среди злокачественных заболеваний крови и составляет примерно 20% лейкозов. При этом пик заболеваемости приходится на трудоспособное население – людей 30-50 лет. До появления инновационного лечения, ХМЛ считалось фатальным и выживаемость редко превышала 5 лет. Сегодня прогноз больных ХМЛ зависит от стадии, на которой он был обнаружен. Около 85% пациентов к моменту постановки диагноза находятся в хронической фазе, когда клинические проявления заболевания обычно отсутствуют. В этот период правильно подобранное лечение может остановить прогрессирование болезни.
Благодаря улучшению методов диагностики и лечения ХМЛ, в стране наблюдается рост количества пациентов, что влечет за собой увеличение бремени государства в вопросах обеспечения больных. Пациенты с ХМЛ получают лечение за счет средств федерального бюджета – препарат первой линии терапии иматиниб ходит в программу «7 нозологий», и региональных бюджетов, которые закупают лекарства для пациентов, резистентных к первой линии терапии. Введение в российскую медицинскую практику регистра пациентов позволит совершенствовать льготное лекарственное обеспечение по высоко затратным нозологиям и более рационально планировать бюджет на лечение данной категории больных. Кроме того, благодаря ему будет возможно стандартизировать лечение по всей стране, а также отслеживать эффективность, безопасность и результаты терапии в условиях реальной клинической практики, вне зависимости от региона проживания пациента, что особенно актуально при лечении дорогостоящих и редких заболеваний.
«Практика ведения регистров не является новой, с 1996 года Институт занимается анализом данных и подготовкой отчетов в министерство Здравоохранения по данным канцер регистра. Однако данные по онкогематологическим заболеваниям в нем представлены не всеобъемлюще, и именно поэтому получение в дар ХМЛ регистра является для нас крайне оправданным, — говорит Ирина Ларичева, заведующая отделом создания и внедрения ИАС ФГБУ «ЦНИИ организации и информатизации» МЗ. – Регистр может стать эффективным инструментом при создании нормативных документов: министерство Здравоохранения будет иметь возможность оценить не только количество больных, нуждающихся в конкретных лекарственных препарат, но и их эффективность, своевременность переключения пациентов на вторую и последующую линии терапии. Кроме того, он будет полезен в работе региональных Минздравов и станет основой стандартизации мед помощи данной категории больных по всей стране».
Идея создания регистра пациентов с ХМЛ принадлежала «Гематологическому научному центру», и была поддержана компанией Новартис в 2006 году. Изначально это была программа, по сбору данных о диагностике ХМЛ, которая осуществлялась в сотрудничестве с 70 медицинскими центрами по всей России. С 2011 года она трансформировалась в клиническое исследование, включающее в себя данные по всем доступным в стране видам терапии ХМЛ. Сейчас в исследование вовлечены 111 центров по всей России. Регистр содержит информацию по большому количеству данных: пол, возраст, социальный статус пациентов, а также по всем видам проводимых им методов лечения, включая параметры эффективности.
«Именно наличие регистра позволило всем больным ХМЛ получать гарантированное государством лечение ингибиторами начиная с 2007 года и сегодня мы покрываем 95% от общей потребности в данном виде терапии. Мы возлагаем большие надежды на то, что и препараты второго поколения также станут доступны для всех нуждающихся пациентов», – отметила заведующая научно-консультативным отделением химиотерапии миелопролиферативных заболеваний ФГБУ «Гематологический научный центр» Минздрава РФ Анна Туркина.
С каждым годом в арсенале врачей появляются новые препараты для лечения ХМЛ. Так, сегодня российским пациентам доступен препарат нилотиниб компании Новартис, ставший первым ингибитором тирозинкиназы, продемонстрировавшем в клинических исследованиях возможность достижения ремиссии без лечения у более половины больных после относительно краткосрочного периода терапии (медиана составила ≈3,6 года) с возможностью последующей отмены терапии. И благодаря регистру пациентов можно будет определить ту категорию пациентов, которым показана данная терапия.
«Компания Новартис является не только производителем инновационных методов лечения онкогематологических заболеваний, но также стремиться стать надежным партнером российской системе здравоохранения, — заметил в своем выступлении генеральный менеджер региона развивающихся рынков Департамента онкологии компании «Новартис» Джон Кетчум. – Мы всегда открыты к конструктивному сотрудничеству и предлагаем различные проекты по диагностике и софинансированию для повышения доступности для российских пациентов самых современных методов лечения».
# # #
О группе компаний «Новартис» в России
Группа компаний «Новартис» в России предлагает решения в здравоохранении, отвечающие новым потребностям общества и пациентов. Компания обладает диверсифицированным портфелем и в настоящий момент занимает лидирующие позиции на российском рынке в области производства инновационных препаратов, брендированных дженериков и безрецептурных лекарственных средств, хирургического оборудования и препаратов для охраны зрения. Сегодня в России представлены все бизнес-подразделения компании («Новартис Фарма», «Сандоз» и «Алкон»), в которых работают более 2000 сотрудников «Новартис».
О Новартис Онкология
Новартис Онкология выделена в отдельное бизнес-подразделение группы в 2016 году после завершения слияния с портфелем онкологических препаратов компании GSK. Портфель препаратов Новартис Онкология включает в себя более 20 продуктов для лечения онкологических, гематологических и редких заболеваний. Более 30 новых молекул находятся на разных стадиях разработки. Десятилетняя стратегия компании ориентирована на лидерство в пяти ключевых направлениях – рак молочной железы, рак легкого, рак почки, рак крови и меланома. Основанная миссия компании — трансформировать онкологическую помощь ради пациентов.
Более 30 новых молекул находятся на разных стадиях разработки. Десятилетняя стратегия компании ориентирована на лидерство в пяти ключевых направлениях – рак молочной железы, рак легкого, рак почки, рак крови и меланома. Основанная миссия компании — трансформировать онкологическую помощь ради пациентов.
О ЦНИИ организации и информатизации здравоохранения
Центральный научно-исследовательский институт организации и информатизации здравоохранения Министерства здравоохранения Российской Федерации создан в 1999 году приказом Минздрава России.
Институт осуществляет свою деятельность по нескольким направлениям: научная и работа, информатизация и международное сотрудничество.
Основной целью деятельности института является разработка научных основ реализации государственной политики в сфере здравоохранения, а также научное обоснование развития системы охраны здоровья населения, организации и информатизации здравоохранения.
Контакты для СМИ
Мария Александрова
Руководитель по корпоративным
коммуникациям группы компании
«Новартис» в России
+7 495 660 75 09 (8225)
+7 985 364 02 06
[email protected]
Хронический миелолейкоз — справочник болезней — ЗдоровьеИнфо
Цель лечения хронического миелоидного лейкоза – удаление всех патологических клеток, содержащих BCR-ABL ген, который является причиной избыточного образования клеток крови. В большинстве случаев невозможно устранить все лейкозные клетки, но можно добиться долгосрочной ремиссии заболевания.
Таргетные препараты
Таргетные препараты воздействуют на специфические молекулярные механизмы роста и деления злокачественных клеток. «Мишенью» препаратов, использующихся для лечения хронического миелоидного лейкоза, является белок, кодируемый геном BCR-ABL, – тирозинкиназа. Таргетные препараты, блокирующие действие тирозинкиназы:
- Иматиниб (Гливек)
- Дазатиниб (Спрайсел)
- Нилотиниб (Тасигна)
- Бозутиниб (Бозулиф)
- Омаксетин (Синрибо)
Таргетные препараты являются в большинстве случаев препаратами первой линии. Если ответа на лечение одним таргетным препаратом не наступает, врач может назначить другой препарат или иные виды лечения. Побочными эффектами являются отеки, тошнота, мышечные судороги, сыпь на коже, слабость, диарея.
Если ответа на лечение одним таргетным препаратом не наступает, врач может назначить другой препарат или иные виды лечения. Побочными эффектами являются отеки, тошнота, мышечные судороги, сыпь на коже, слабость, диарея.
Врачи не установили, когда прекращение приема таргетных препаратов безопасно, поэтому большинство пациентов продолжает принимать их, даже когда по результатам анализов крови обнаруживается стойкая ремиссия.
Трансплантация костного мозга
Трансплантация костного мозга дает единственный шанс на окончательное излечение хронического миелогенного лейкоза, однако она остается методом резерва для пациентов, которым не помогли другие виды лечения, поскольку связана с высоким риском развития серьезных осложнений. При трансплантации используются высокие дозы химиотерапевтических препаратов для уничтожения собственного костного мозга пациента. Затем клетки крови от донора или ваши собственный, заранее заготовленные, вводятся внутривенно.
Химиотерапия
Химиотерапию обычно сочетают с другими видами лечения. Обычно химиотерапевтические препараты для лечения хронического миелоидного лейкоза принимают внутрь в виде таблеток. Побочные эффекты зависят от конкретного препарата.
Биологическая терапия
Биологическая терапия предполагает вовлечение иммунной системы в борьбу с онкологическим заболеванием. Для этого используются препараты интерферонов – синтетические аналоги веществ, вырабатывающихся иммунной системой организма. Интерфероны могут помочь замедлить размножение лейкозных клеток. Интерфероны показаны в тех случаях, когда другие методы лечения не работают или пациент не может принимать препараты, например, в связи с беременностью. Побочные эффекты интерферонов включают слабость, лихорадку, гриппоподобные симптомы и потерю веса.
Клинические исследования
В клинических испытаниях изучаются новейшие методы лечения заболеваний или новые способы использования существующих методов лечения. Участие в клинических испытаниях может дать вам возможность попробовать последнюю лечение, но не может гарантировать излечение. Поговорите со своим врачом о том, какие клинические испытания доступны для вас. Обсудите «за» и «против» участия в клинических исследованиях.
Участие в клинических испытаниях может дать вам возможность попробовать последнюю лечение, но не может гарантировать излечение. Поговорите со своим врачом о том, какие клинические испытания доступны для вас. Обсудите «за» и «против» участия в клинических исследованиях.
Образ жизни и народные средства
Многим людям приходится жить с хроническим миелоидным лейкозом в течение многих лет. Многим придется продолжать лечение иматинибом неопределенный срок. Иногда вы будете чувствовать себя больным, даже если не выглядите таковым. Иногда вы будете уставать от своего заболевания. Приведенные здесь советы помогут вам настроиться на лучшее и справиться с заболеванием:
- Обсудите с врачом возможные побочные эффекты заболевания. Мощные препараты для лечения лейкоза могут вызывать различные побочные эффекты, но вы не обязательно должны терпеть это. С побочными эффектами зачастую можно справиться с помощью других лекарственных препаратов.
- Не прекращать лечение самостоятельно. Если у вас возникли какие-либо побочные эффекты, такие как сыпь на коже или выраженная слабость, не прекращайте лечение без консультации со специалистом. Также не прекращайте принимать свои лекарства, если вы чувствуете себя лучше, и думаете, что заболевание излечено. Если вы прекратите принимать лекарства, ваше заболевание может быстро и неожиданно вернуться, даже если вы находитесь состоянии ремиссии.
- Обратитесь за помощью, если вам трудно справляться с заболеванием. Хроническое заболевание является источником стресса и эмоциональных перегрузок. Расскажите своему врачу о ваших чувствах. Попросите направление к психотерапевту или другому специалисту, с которым вы можете поговорить.
Альтернативная медицина
Ни один из методов альтернативной медицины не может вылечить хронический миелоидный лейкоз, но они могут помочь вам справляться со стрессом и побочными эффектами лечения. Обсудите с врачом такие методы, как:
- Акупунктура
- Ароматерапия
- Массаж
- Медитация
- Техники релаксации
КЛИНИЧЕСКИЙ СЛУЧАЙ ТЕРАПИИ ПАЦИЕНТА ХРОНИЧЕСКИМ МИЕЛОЛЕЙКОЗОМ С МУТАЦИЕЙ BCR-ABL Y253H И СОПУТСТВУЮЩЕЙ ПАТОЛОГИЕЙ | Сафуанова
1. Абдулкадыров К.М., Шуваев В.А., Мартынкевич И.С. и др. Хронический миелолейкоз: многолетний опыт таргетной терапии. Клиническая онкогематология. Фундаментальные исследования и клиническая практика. 2016; 9 (1): 54-60.
Абдулкадыров К.М., Шуваев В.А., Мартынкевич И.С. и др. Хронический миелолейкоз: многолетний опыт таргетной терапии. Клиническая онкогематология. Фундаментальные исследования и клиническая практика. 2016; 9 (1): 54-60.
2. Волкова М.А. Терапия хронических лейкозов в 21 веке. Эффективная фармакотерапия в онкологии, гематологии и радиологии. 2009; 2: 2-7.
3. Гематология: национальное руководство под ред. О.А. Руковицина. ГОЭТАР Медиа. 2015; 776 с.
4. Гусарова Г.А., Туркина А.Г., Воронцова А.В. и др. Отдаленные результаты терапии дазатинибом и анализ особенностей течения плеврального выпота у больных в поздней хронической фазе хронического миелолейкоза после неудачи лечения иматинибом. Бюллетень Сибирского отделения Российской академии медицинских наук. 2014; 34(6): 27-36.
5. Куцев С.И., Вельченко М.В. Значение анализа мутаций гена BCR-ABL в оптимизации таргетной терапии хронического миелолейкоза. Клиническая онкогематология. 2008; 1(3): 190-199.
6. Куцев С.И., Вельченко М.В., Морданов С.В. Роль мутаций гена BCR-ABL в развитии рефрактерности к иматинибу у пациентов с хроническим миелолейкозом. Клиническая онкогематология. 2008; 1(4): 303-309.
7. Мисюрин А.В., Мисюрина Е.Н., Тихонова В.В., и др. Частота встречаемости мутаций киназного домена гена BCR-ABL у больных хроническим миелолейкозом, резистентных к терапии иматинибом. Российский биотерапевтический журнал. 2016; 15(4): 102-109.
8. Наумова К.В.. Кривова С.П., Золотовская И. А. Хронический миелолейкоз: проблемы и перспективы (обзор литературы). Аспирантский вестник Поволжья. 2016; 1-2: 94-99.
А. Хронический миелолейкоз: проблемы и перспективы (обзор литературы). Аспирантский вестник Поволжья. 2016; 1-2: 94-99.
9. Печенкина А.А. Механизм формирования филадельфийской хромосомы и ее роль в развитии хронического миелолейкоза. Молодой ученый. 2018; 25 (211): 183-187.
10. Рябчикова Н.Р., Минниахметов И.Р, Сафуанова Г.Ш. и др. Хронический миелолейкоз: молекулярный мониторинг в клинической практике. Онкогематология. 2013; 1: 1-16.
11. Филатова Е.А. Поражение бронхолегочной системы у больных хроническим миелолейкозом. Амурский медицинский журнал. 2016; 2 (14): 59-66.
12. Тихонова В.В., Исаков М.А., Мисюрин В.А., и др. Резистентность хронического миелолейкоза к ингибиторам тирозинкиназ: 10 лет изучения профиля мутаций гена BCR-ABL В России (2006-2016 ГГ.). Клиническая онкогематология. Фундаментальные исследования и клиническая практика. 2018; 11(3): 227-233.
13. Туркина А.Г., Голенков А.К., Напсо Л.И. и др. Российский регистр по лечению хронического миелоидного лейкоза в рутинной клинической практике: итоги многолетней работы. Эффективная фармакотерапия. 2015; (10): 8-13.
14. Туркина А.Г., Зарицкий А.Ю., Шуваев В.А. и др. Клинические рекомендации по диагностике и лечению хронического миелолейкоза. Клиническая онкогематология. Фундаментальные исследования и клиническая практика. 2017; 10(3): 294-316.
15. Baccarani M., Cortes J., Pane F. et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 2009; 27:6041-6051.
J Clin Oncol 2009; 27:6041-6051.
16. Corbin A., La Rose P., Stoffregen E. et al. Several BCR-ABL kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood 2003;101(11):4611-4.
17. Ellas M.Н., Babab А.А., Azlanb А., Roslinec Н, Simd G.A., Padminie M. BCR-ABL kinase domain mutations, including 2 novel mutations inimatinib resistant Malaysian chronic myeloid leukemiapatients— Frequency and clinical outcome. Leuk. Res. 2014;38 (4):454-459
18. Monitoring after successful therapy for chronic myeloid leukemia. S. Branford [et al.] ASH Annual Meeting and Exposition. 2012; Р.105-110
19. O’Hare T., Eide C.A., Deininger M. BCR-ABL kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007; 110(7): 2242—2249.
20. Severini S. In chronic myeloid leukemia patients on second-line tyrosine kinase inhibitor therapy, deep sequencing of BCR-ABL1 at the time of warning may allow sensitive detection of emerging drugresistant mutants. BMC Cancer. 2016; 16: 572.
21. Quintas-Cardama A., Kantarjian H., Cortes J. Mechanisms of Primary and Secondary Resistance to Imatinib in Chronic Myeloid Leukemia. Cancer Control. 2009. Vol.16, № 2. 122-131.
Лечение хронического лимфобластного лейкоза. Список клиник, рейтинг, отзывы, цены
О заболевании
Хронический миелолейкоз, или ХМЛ, это один из видов рака крови. ХМЛ развивается в миелоидных клетках, ответственных за образование эритроцитов, лейкоцитов и тромбоцитов. Сначала заболевание поражает костный мозг, позже в процесс вовлекаются селезенка и другие органы. Хронический миелоидный лейкоз развивается постепенно, но может переходить в острую форму, которую, как правило, труднее лечить.
Сначала заболевание поражает костный мозг, позже в процесс вовлекаются селезенка и другие органы. Хронический миелоидный лейкоз развивается постепенно, но может переходить в острую форму, которую, как правило, труднее лечить.
Хронический миелолейкоз чаще встречается у взрослых, хотя в редких случаях диагностируется и у детей. При этом заболевании клетки крови не созревают полностью и начинают делиться беспорядочно. Такие аномальные клетки живут долго и постепенно вытесняют нормальные клетки крови. В первую очередь, от этого страдает иммунная система, человек становится более восприимчивым к инфекционным болезням.
Точные причины ХМЛ не установлены, но воздействие радиации повышает риск этой болезни. Кроме того, ХМЛ чаще встречается среди мужчин, чем среди женщин. Также риск развития заболевания повышается с возрастом.
Симптомы
- Слабость
- Утомляемость
- Потеря веса
- Боль в костях
- Тошнота
- Потливость
- Увеличение селезенки, которое определяется пальпаторно
- Склонность к инфекционным заболеваниям
- Чувство переполнения желудка после приема небольшого количества пищи
Диагностика
- Во время общего осмотра врач детально общается с пациентом, собирает анамнез и пальпирует селезенку.
- Клинический анализ крови помогает оценить количество и строение эритроцитов и лейкоцитов, выявить признаки ХМЛ.
- Трепанобиопсия костного мозга – единственная процедура, которая подтверждает диагноз и определяет тип лейкемии. Врач получает образец костного мозга, а затем исследует его в лаборатории на наличие аномальных незрелых и зрелых клеток.
- МРТ и КТ используются для оценки состояния внутренних органов, в частности, селезенки.
Виды лечения
- Химиотерапия и иммунотерапия используются для уничтожения раковых клеток и достижения ремиссии.
 Иммунотерапия используется для укрепления и стимуляции иммунной системы, которая не способна распознать раковые клетки. Обычно человеку требуется несколько курсов химиотерапии.
Иммунотерапия используется для укрепления и стимуляции иммунной системы, которая не способна распознать раковые клетки. Обычно человеку требуется несколько курсов химиотерапии. - Трансплантация стволовых клеток – это пересадка пациенту новых стволовых клеток крови. Здоровые стволовые клетки дают начало нормальным лейкоцитам, эритроцитам и тромбоцитам. Стволовые клетки берут у самого пациента (до начала химиотерапии) или же у совместимого по системе HLA-донора. Чаще всего донорами становятся родственники пациента.
 Иммунотерапия используется для укрепления и стимуляции иммунной системы, которая не способна распознать раковые клетки. Обычно человеку требуется несколько курсов химиотерапии.
Иммунотерапия используется для укрепления и стимуляции иммунной системы, которая не способна распознать раковые клетки. Обычно человеку требуется несколько курсов химиотерапии.Автор: Доктор Надежда Иванисова
Хронические миелолейкозы | Компендиум
Заболевания, относящиеся к группе хронических миелопролиферативных заболеваний (ХМПЗ), возникают в результате злокачественной трансформации полипотентной гемопоэтической стволовой клетки костного мозга и последующей клональной пролиферации клеток одной или более линий миелопоэза, сохраняющих способность к дифференцировке [1–3]. Различные формы ХМПЗ имеют ряд сходных и перекрывающихся морфологических и клинико-гематологических признаков (спленомегалия, лейкоцитоз, тромбоцитоз, увеличение количества мегакариоцитов и развитие фиброза в костном мозге). Возникновение фиброза, а в ряде случаев и явлений склероза, носит реактивный характер. Обусловлено оно пролиферацией фибробластов, являющихся основными клеточными элементами кроветворного микроокружения костного мозга, не относящихся к клону злокачественных клеток.
В то же время имеются и существенные различия клинико-лабораторных данных, на которых основывается современная классификация ХМПЗ [4–6]. При изучении мазков периферической крови и костного мозга, гистологическом исследовании трепанобиоптатов, применении цитогенетических и молекулярно-биологических методов удается выделить следующие основные формы ХМПЗ (табл. 21).
Хронический миелолейкоз
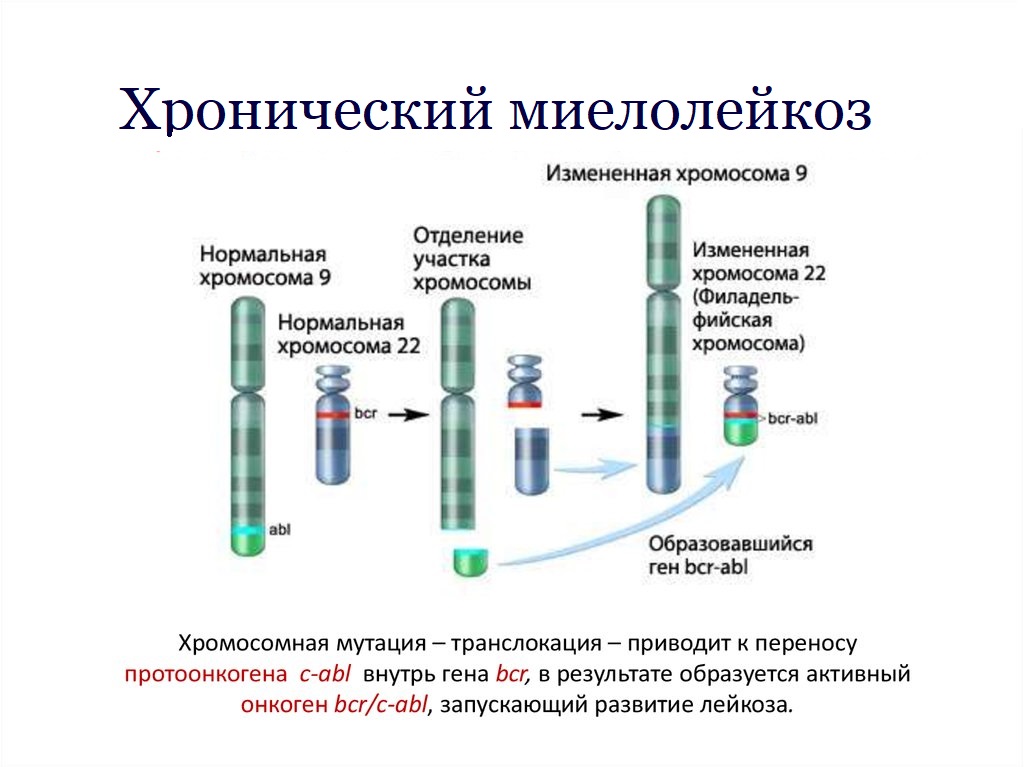
Хронический миелолейкоз (ХМЛ) является клональным злокачественным процессом. У 95% больных в клетках костного мозга и периферической крови определяется так называемая филадельфийская (Ph’) хромосома, возникающая в результате транслокации генетического материала между хромосомами 9 и 22 — t (9; 22) (q34. 1; q11.21). У 5% больных с ХМЛ Ph’ хромосома не определяется (так называемые Ph’ негативные случаи). Но при этом у некоторых больных выявляются характерные аномалии BCR-ABL [8].
1; q11.21). У 5% больных с ХМЛ Ph’ хромосома не определяется (так называемые Ph’ негативные случаи). Но при этом у некоторых больных выявляются характерные аномалии BCR-ABL [8].
На долю ХМЛ приходится около 15–20% всех случаев лейкозов у взрослых и 5% у детей. В США ежегодно регистрируется 3400 новых случаев заболевания ХМЛ [2]. Возрастной пик заболеваемости приходится на 4–5-е десятилетие жизни. Среди больных несколько преобладают лица мужского пола.
В гематологическом плане заболевание характеризуется выраженным лейкоцитозом, сочетающимся с базофилией и эозинофилией. Лейкоцитоз обусловлен увеличением в периферической крови количества зрелых и незрелых нейтрофилов [2, 3, 6].
Хроническая фаза заболевания
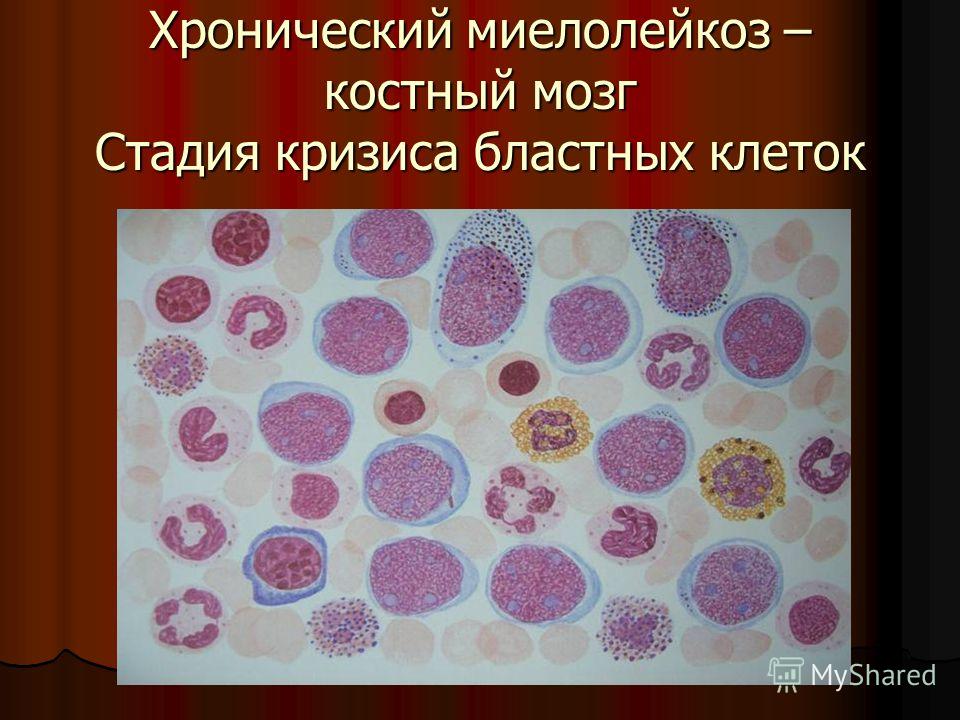
Общее количество лейкоцитов в периферической крови колеблется в широких пределах, но обычно выше 50•109/л. У 70–90% больных количество лейкоцитов превышает 100•109/л, а у 25% — выше 350•109/л [1–4, 6]. Выраженный лейкоцитоз часто наблюдается при ХМЛ у детей. В хронической фазе заболевания содержание миелобластов обычно колеблется в пределах 2–3%, а общее количество промиелоцитов и миелобластов не превышает 15–20% от общего количества лейкоцитов в периферической крови или костном мозге. Количество незрелых клеточных элементов гранулоцитарного ряда (промиелоцитов и миелоцитов) увеличивается по мере прогрессирования процесса при одновременном уменьшении процентного содержания палочкоядерных и сегментоядерных лейкоцитов. В нейтрофилах периферической крови и костного мозга в хронической фазе ХМЛ, как правило, не наблюдается диспластических изменений. У некоторых больных отмечаются выраженная эозинофилия и базофилия (так называемая эозинофило-базофильная ассоциация) и наличие многочисленных незрелых клеток эозинофильного и базофильного ряда. У большинства больных с ХМЛ определяется также абсолютный моноцитоз. Выраженный моноцитоз в ранней фазе заболевания позволяет предположить наличие хронического миеломоноцитарного лейкоза (ХММЛ). Во время установления диагноза у многих больных с ХМЛ определяется нормоцитарная нормохромная анемия, признаки анизоцитоза и пойкилоцитоза. Анемия прогрессирует по мере увеличения количества лейкоцитов в периферической крови. Одновременно в крови определяется небольшое количество ядросодержащих клеточных элементов эритробластического ряда. Почти у 50% больных с ХМЛ отмечается тромбоцитоз. Количество тромбоцитов в крови увеличивается по мере развития заболевания, нередки случаи, когда тромбоцитоз составляет 1000•109/л. В мазках периферической крови больных обнаруживаются гигантские формы тромбоцитов, пластинки с аномальной грануляцией, ядра мегакариоцитов.
Во время установления диагноза у многих больных с ХМЛ определяется нормоцитарная нормохромная анемия, признаки анизоцитоза и пойкилоцитоза. Анемия прогрессирует по мере увеличения количества лейкоцитов в периферической крови. Одновременно в крови определяется небольшое количество ядросодержащих клеточных элементов эритробластического ряда. Почти у 50% больных с ХМЛ отмечается тромбоцитоз. Количество тромбоцитов в крови увеличивается по мере развития заболевания, нередки случаи, когда тромбоцитоз составляет 1000•109/л. В мазках периферической крови больных обнаруживаются гигантские формы тромбоцитов, пластинки с аномальной грануляцией, ядра мегакариоцитов.
В периферической крови больных нередко еще за несколько месяцев до манифестации ХМЛ определяются псевдопельгеровские лейкоциты. Помимо ХМЛ, эта приобретенная аномалия гранулоцитов может обнаруживаться у больных с идиопатическим миелофиброзом, при ОМЛ, иногда при неходжкинских злокачественных лимфомах, инфекционных процессах, действии ряда токсических веществ.
В костном мозге больных с ХМЛ обнаруживается гиперклеточность, обусловленная преобладанием нейтрофилов и незрелых клеток гранулоцитарного ряда. Резко увеличено соотношение клеток гранулоцитарного и эритробластического ряда, составляющее нередко 20:1 вместо 3:1 в норме. В костном мозге определяется тот же спектр клеток гранулоцитарного ряда, что и в периферической крови, но с бо’льшим преобладанием незрелых клеток (промиелоцитов и миелоцитов). Отмечается преимущественно нормобластический эритропоэз, но у отдельных больных в хронической фазе заболевания — мегалобластический и признаки дисэритропоэза. Количество мегакариоцитов увеличено, как и при многих других миелопролиферативных заболеваниях. Палочки Ауэра в цитоплазме клеток, в отличие от ОМЛ, определяются редко. Их появление может служить предвестником развития бластного криза.
Приблизительно у 30% больных ХМЛ в костном мозге и селезенке определяются крупные клетки с пенистой цитоплазмой и эксцентрично расположенным ядром, напоминающие клетки, обнаруживаемые при болезни Гоше. Эти псевдо-Гоше-клетки изредка встречаются также при врожденной дисэритропоэтической анемии, множественной миеломе, иммунобластной лимфоме, лимфогранулематозе. Считается, что их появление обусловлено неспособностью клеточных гликоцереброзидаз расщеплять гликоцереброзиды, образующиеся в больших количествах при усилении распада лейкоцитов. Подобно истинным клеткам Гоше они PAS-положительны, окрашиваются масляным красным О, СЧВ, обладают активностью КФ [2, 3, 5].
Эти псевдо-Гоше-клетки изредка встречаются также при врожденной дисэритропоэтической анемии, множественной миеломе, иммунобластной лимфоме, лимфогранулематозе. Считается, что их появление обусловлено неспособностью клеточных гликоцереброзидаз расщеплять гликоцереброзиды, образующиеся в больших количествах при усилении распада лейкоцитов. Подобно истинным клеткам Гоше они PAS-положительны, окрашиваются масляным красным О, СЧВ, обладают активностью КФ [2, 3, 5].
В костном мозге больных ХМЛ при окраске по Романовскому—Гимзе встречаются также крупные, так называемые цвета голубого моря гистиоциты. Эти клетки с яркой сине-зеленой цитоплазмой, как и псевдо-Гоше-клетки, дающие положительную реакцию на гликоген и липиды, изредка выявляются при идиопатической тромбоцитопенической пурпуре, ОМЛ, ОЛЛ, неходжкинских злокачественных лимфомах. Возможно, что оба типа клеток в действительности представляют две разные стадии развития макрофагов, содержащих остатки фагоцитированных нейтрофилов [1].
Получение и исследование пунктатов костного мозга, как и периферической крови, не только обязательно для установления диагноза ХМЛ. Оно позволяет провести цитогенетические исследования, определить реарранжировку генов BCR и ABL, изучить характер роста клеток-предшественников в культуре in vitro [6–8].
По данным гистологического изучения трепанобиоптатов костный мозг представляется выраженно гиперклеточным за счет увеличения количества зрелых и незрелых клеток гранулоцитарного ряда и мегакариоцитов. Наибольшее количество промиелоцитов и миелоцитов располагается вблизи эндоста и периваскулярно. Мегакариоциты распределяются в срезах равномерно или образуют кластеры. По мере прогрессирования заболевания усиливается пролиферация клеток мегакариоцитарного ряда и могут выявляться многочисленные микромегакариоциты. Миелофиброз, выраженность которого усиливается по мере развития процесса, может определяться и на ранних стадиях заболевания. Увеличивается количество ретикулиновых волокон, располагающихся в виде отдельных фокусов преимущественно в периваскулярных пространствах или диффузно.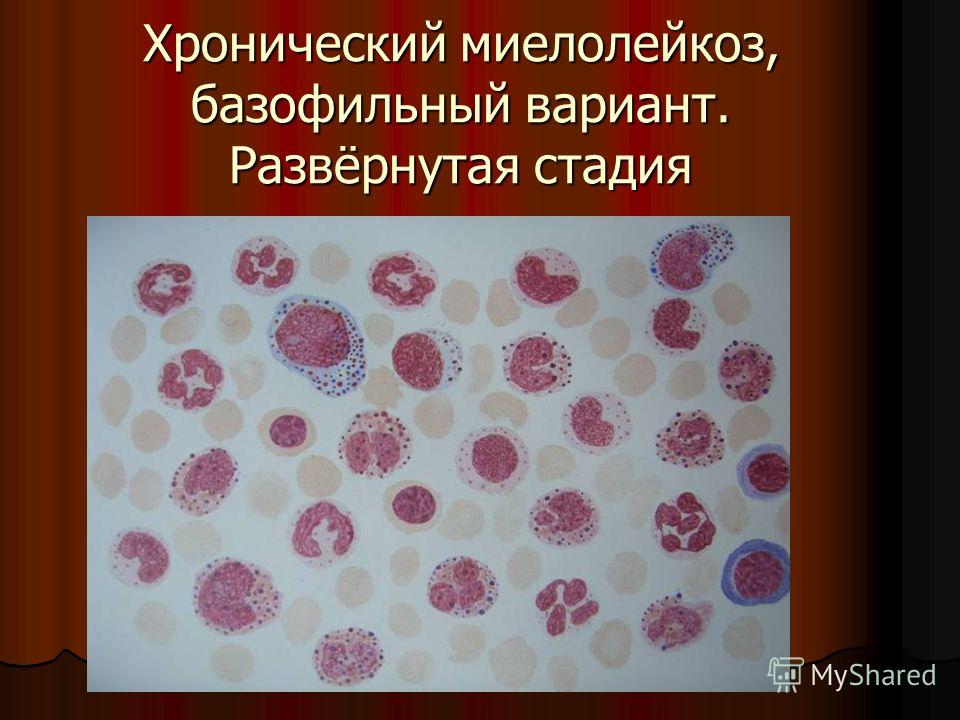 Выраженность фиброза в начальной стадии развития ХМЛ коррелирует со спленомегалией, уровнем гемоглобина, процентным содержанием бластов в костном мозге и периферической крови, дополнительными кариотипическими аномалиями. Фиброз, обусловленный наличием коллагеновых волокон, встречается реже, чем вызванный ретикулиновыми нитями. Признаки остеомиелосклероза наблюдаются еще реже. Появление фиброза, переход от очагового к диффузному считается важным прогностическим фактором [9]. Выраженный фиброз обычно ассоциируется с более короткими сроками выживаемости. Но могут быть исключения. Описаны случаи ХМЛ, когда выраженный фиброз отмечался на ранних стадиях, но заболевание имело более длительное течение [10]. У ряда больных с Ph’-положительным ХМЛ могут быть выраженный фиброз за счет разрастания коллагеновых волокон и картина крови, подобная наблюдаемой при хроническом идиопатическом миелофиброзе. Поставить правильный диагноз в этих случаях помогает низкий уровень (или практически полное отсутствие) щелочной фосфатазы в нейтрофилах [1].
Выраженность фиброза в начальной стадии развития ХМЛ коррелирует со спленомегалией, уровнем гемоглобина, процентным содержанием бластов в костном мозге и периферической крови, дополнительными кариотипическими аномалиями. Фиброз, обусловленный наличием коллагеновых волокон, встречается реже, чем вызванный ретикулиновыми нитями. Признаки остеомиелосклероза наблюдаются еще реже. Появление фиброза, переход от очагового к диффузному считается важным прогностическим фактором [9]. Выраженный фиброз обычно ассоциируется с более короткими сроками выживаемости. Но могут быть исключения. Описаны случаи ХМЛ, когда выраженный фиброз отмечался на ранних стадиях, но заболевание имело более длительное течение [10]. У ряда больных с Ph’-положительным ХМЛ могут быть выраженный фиброз за счет разрастания коллагеновых волокон и картина крови, подобная наблюдаемой при хроническом идиопатическом миелофиброзе. Поставить правильный диагноз в этих случаях помогает низкий уровень (или практически полное отсутствие) щелочной фосфатазы в нейтрофилах [1].
При ХМЛ крайне низкая активность лейкоцитарной щелочной фосфатазы (ЛЩФ) наблюдается во всех случаях — независимо от количества лейкоцитов в периферической крови и тяжести клинического течения заболевания [11]. Низкий уровень ЛЩФ обусловлен нарушением выработки фермента, а не дефектами его каталитической функции или нарушениями в образовании специфических гранул в цитоплазме лейкоцитов [12, 13]. После успешного лечения уровень ЛЩФ обычно возвращается к норме. Определение ЛЩФ, наряду с выявлением Ph’ хромосомы, используется в клинической практике для подтверждения диагноза в спорных и сомнительных случаях, когда ХМЛ приходится дифференцировать с рядом патологических процессов, сопровождающихся развитием лейкемоидных реакций миелоидного типа (инфекционные заболевания, метастазы опухолей, действие токсических веществ и др.).
К числу признаков, способствующих установлению диагноза ХМЛ (табл. 22), относится также повышенный уровень витамина В12 и витамин В12-связывающих белков в сыворотке крови больных, что, в свою очередь, обусловлено повышением содержания транскобаламинов I и II [14]. Как установлено, эти изменения, как и снижение активности ЛЩФ, увеличение количества базофилов и тромбоцитов, могут определяться за несколько лет до манифестацииХ МЛ [13].
Как установлено, эти изменения, как и снижение активности ЛЩФ, увеличение количества базофилов и тромбоцитов, могут определяться за несколько лет до манифестацииХ МЛ [13].
В табл. 23 приведены клинико-лабораторные признаки, которые могут быть использованы в дифференциальной диагностике ХМЛ и лейкемоидных реакций миелоидного типа.
Иммунофенотипический анализ антигенов поверхностных мембран гранулоцитов в хронической фазе ХМЛ позволил обнаружить некоторые отличия по сравнению с клетками соответствующей степени зрелости в норме. Отмечено уменьшение плотности HLA-DR (Ia-подобного антигена) на трансформированных клетках-предшественниках гранулоцитарного ряда. На меньшем количестве клеток обнаруживался антиген CD13, увеличенным было число незрелых клеток с двойными маркерами CD15+ CD34+ [15–17], до 50% клеток реагировало с мкАТ к CD116 [17]. В целом, изучение маркеров поверхностных мембран клеток не играет существенной роли в диагностике ХМЛ в хронической фазе заболевания. При развитии же бластного криза иммунофенотипический анализ, наряду с данными цитохимических исследований, позволяет, как и при острых лейкозах, точнее идентифицировать природу бластных клеток, определяемых в костном мозге и периферической крови.
Цитогенетический анализ. Приблизительно у 5% больных с ХМЛ в лейкозных клетках при рутинном цитогенетическом исследовании не обнаруживается t (9; 22). Однако при использовании молекулярно-биологических методов и в этих случаях может быть обнаружен гибридный ген BCR/ABL. Образующийся при этом белок, обладающий повышенной активностью тирозинкиназы, может быть ответственным за усиленную пролиферацию клеток у больных с ХМЛ [2, 18–20].
Фаза акселерации и бластной трансформации
Хроническая фаза течения ХМЛ, средняя продолжительность которой составляет 3–4 года, сменяется более агрессивной и кратковременной фазой заболевания. У большинства больных развивается бластный криз (бластная трансформация), по клинико-гематологическим проявлениям близкий к острому лейкозу, развивающемуся de novo. Он характеризуется резистентностью к применяемой терапии и средней продолжительностью жизни от 3 до 6 мес [19–21]. У 10–30% больных с ХМЛ ухудшение состояния сочетается с нарастанием изменений в костном мозге и периферической крови. Но при этом количество бластов недостаточно для диагностики бластного криза. Эта стадия развития заболевания определяется как фаза акселерации, средняя продолжительность которой составляет от 12 до 18 мес [1, 3]. Сотрудниками Международного регистра по трансплантации костного мозга разработаны критерии, позволяющие четко определить переход в эти фазы заболевания [22].
Он характеризуется резистентностью к применяемой терапии и средней продолжительностью жизни от 3 до 6 мес [19–21]. У 10–30% больных с ХМЛ ухудшение состояния сочетается с нарастанием изменений в костном мозге и периферической крови. Но при этом количество бластов недостаточно для диагностики бластного криза. Эта стадия развития заболевания определяется как фаза акселерации, средняя продолжительность которой составляет от 12 до 18 мес [1, 3]. Сотрудниками Международного регистра по трансплантации костного мозга разработаны критерии, позволяющие четко определить переход в эти фазы заболевания [22].
Фаза бластной трансформации устанавливается при наличии не менее 30% бластов в костном мозге и периферической крови. Внезапное и быстрое увеличение содержания бластных клеток сопровождается нарастанием недостаточности костномозгового кроветворения, усилением выраженности анемии и тромбоцитопении. При бластном кризе у 70% больных патологические клетки имеют миелоидную природу, у 20–25% — лимфоидную. В 5% случаев бласты имеют дифференцировочные признаки ранних клеток-предшественников эритробластического или мегакариоцитарного ряда. Прогноз благоприятнее и чувствительность к терапии несколько выше при наличии бластов лимфоидного происхождения. Для более точного определения природы клеток при бластном кризе рекомендуется применение цитохимических и иммунологических методов [1, 2, 4, 5].
При бластной трансформации миелоидного типа клетки могут иметь цитоморфологические и цитохимические признаки миелобластов, монобластов, быть сходными с бластами при ОМЛ М4 и ОМЛ М3 [3].
При бластной трансформации лимфоидного типа клетки имеют цитологические признаки, присущие бластам при ОЛЛ L1 или L2 и очень редко ОЛЛ L3 [1, 3]. В большинстве случаев при бластном кризе лимфоидного типа клетки имеют В-клеточное происхождение и очень редко являются трансформированными ранними клетками-предшественниками Т ряда. При лимфоидном бластном кризе В-клеточного подтипа ранние клетки-предшественники имеют иммунофенотип ОЛЛ «общего типа» (ОЛЛ В II) или пре-В-клеточный (ОЛЛ В III). При бластном кризе ХМЛ лимфоидного типа чаще, чем при ОЛЛ, бластные клетки по данным иммунофенотипирования имеют бифенотипические/билинейные признаки [3]. Иногда в клинику поступают больные в стадии бластного криза, у которых по разным причинам ранее не был диагностирован ХМЛ. При этом бласты костного мозга и периферической крови могут иметь цитоморфологические и цитохимические признаки миелоидных или лимфоидных клеток-предшественников. Клинико-гематологическая картина в первом случае чаще всего напоминает ОМЛ М2, но при сохранении достаточно большого количества зрелых и незрелых гранулоцитов. При бластной трансформации лимфоидного типа не наблюдается признаков созревания TdT-положительных бластов в промиелоциты. Особые трудности возникают при дифференциальной диагностике лимфоидного бластного криза ХМЛ и Ph’-положительного ОЛЛ [1].
При бластном кризе ХМЛ лимфоидного типа чаще, чем при ОЛЛ, бластные клетки по данным иммунофенотипирования имеют бифенотипические/билинейные признаки [3]. Иногда в клинику поступают больные в стадии бластного криза, у которых по разным причинам ранее не был диагностирован ХМЛ. При этом бласты костного мозга и периферической крови могут иметь цитоморфологические и цитохимические признаки миелоидных или лимфоидных клеток-предшественников. Клинико-гематологическая картина в первом случае чаще всего напоминает ОМЛ М2, но при сохранении достаточно большого количества зрелых и незрелых гранулоцитов. При бластной трансформации лимфоидного типа не наблюдается признаков созревания TdT-положительных бластов в промиелоциты. Особые трудности возникают при дифференциальной диагностике лимфоидного бластного криза ХМЛ и Ph’-положительного ОЛЛ [1].
Почти у 30% больных с ХМЛ хроническая фаза сменяется фазой акселерации, предшествующей развитию бластного криза [1, 3]. Фаза акселерации характеризуется нарастанием миелофиброза, увеличением количества базофилов в периферической крови (>20%), снижением уровня гемоглобина (<7 г/дл). Но при этом количество бластов в костном мозге и периферической крови составляет менее 30% [21, 23]. Наблюдается также увеличение количества эозинофилов в крови и костном мозге (>10%), незрелых клеток моноцитарного ряда, эритробластов. В зрелых и незрелых миелоидных клетках обнаруживаются диспластические изменения: гиперсегментация ядер и гипогрануляция цитоплазмы нейтрофилов, различные признаки дисэритропоэза.
При исследовании трепанобиоптатов костного мозга также обнаруживаются черты дезорганизации и диспластические изменения. Увеличение количества гиподольчатых мегакариоцитов сопровождается усилением ретикулинового фиброза. Может наблюдаться коллагеновый фиброз, иногда сочетающийся с развитием остеосклероза. Увеличивается количество бластных клеток, располагающихся паратрабекулярно и периваскулярно, при одновременном уменьшении количества незрелых гранулоцитов.
Ряд больных с ХМЛ поступает в стационар в фазе акселерации, в таких случаях возникает необходимость проведения дифференциальной диагностики с различными формами МДС и острых лейкозов.
В фазе акселерации и при бластной трансформации у 80% больных с ХМЛ происходит дальнейшая эволюция кариотипа. Вторичные аномалии отмечаются чаще при миелоидном типе бластного криза, чем при лимфоидной трансформации. Основными дополнительными аномалиями, которые могут быть выявлены за несколько месяцев до развития бластного криза миелоидного типа, являются i(17q)+8, t (3; 21) (q26; q22), inv(3) (q21 q26), del(13 ) (q12 q14) [24]. Возникновение лимфоидного типа бластного криза наиболее часто сочетается с del(7) (q22) и –7.
Молекулярно-генетические аномалии, связанные с развитием бластного криза ХМЛ, касаются опухолеассоциированных супрессорных генов p53, RB1, p16 и реже онкогенов ras и myc [25, 26]. Мутации генов p16 и RB1 чаще наблюдаются в кроветворных клетках у больных с лимфоидным типом бластного криза, а гена p53 — при миелоидном типе бластной трансформации [25–27].
Атипичный миелопролиферативный синдром (Ph
–/BCR– миелолейкоз)
Термины «атипичный миелопролиферативный синдром» (аМПС), «атипичный хронический лейкоз», «Ph’ отрицательный хронический миелолейкоз» употребляются для обозначения гетерогенной группы миелопролиферативных заболеваний. От ХМЛ аМПС отличается по ряду клинико-гематологических проявлений.
Основными признаками аМПС являются отсутствие Ph’-хромосомы и выявляемой реарранжировки генов BCR/ABL, гиперлейкоцитоз и нейтрофилез, наличие в периферической крови незрелых форм гранулоцитов. При аМПС, в отличие от ХМЛ, отмечаются диспластические изменения в клетках гранулоцитарного и эритробластического ряда. Количество базофилов — в пределах нормы или слегка повышено. Нечасто отмечается и эозинофилия. Количество лейкоцитов в периферической крови составляет от 20•109/л до 180•109/л. В лейкоцитарной формуле содержание промиелоцитов, миелоцитов и метамиелоцитов превышает 15%.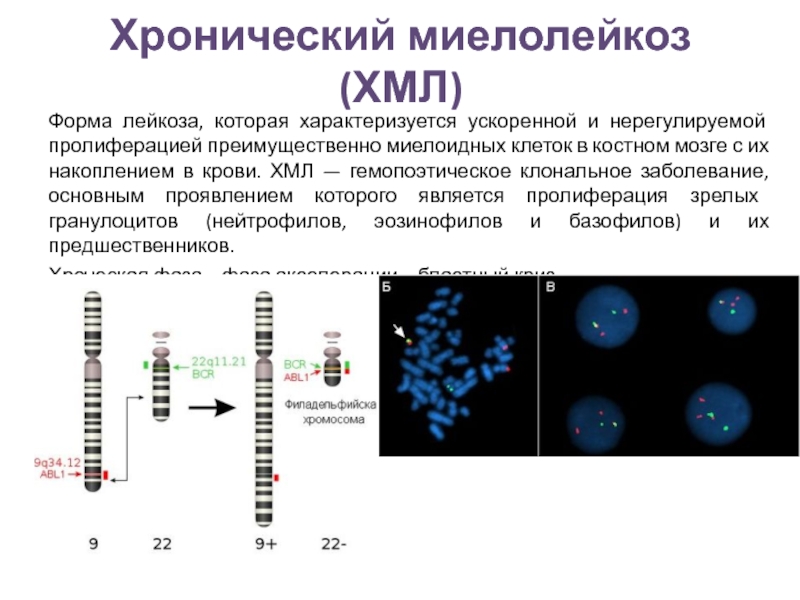
У больных с аМПС, а это в основном мужчины по возрасту на 15–20 лет старше, чем пациенты с ХМЛ, количество лейкоцитов в среднем ниже, но чаще наблюдается анемия и тромбоцитопения. Уровень гемоглобина колеблется в пределах от 3,4 до 14,2 г/дл. Анемия, наряду со спленомегалией, относится к числу основных жалоб при обращении за помощью к врачу-гематологу. Среднее количество тромбоцитов не превышает 80•109/л [1]. По сравнению с Ph’-положительным ХМЛ при аМПС в периферической крови увеличено относительное (но не более 10%) и абсолютное содержание моноцитов.
Диспластические изменения нейтрофилов проявляются неправильной формой ядер (псевдопельгеровские лейкоциты) и наличием гипогранулярной цитоплазмы у зрелых нейтрофилов, миелоцитов и метамиелоцитов. В большинстве случаев, как и при ХМЛ, снижен уровень активности ЛЩФ, хотя у отдельных больных может отмечаться повышенный показатель.
Костный мозг при аМПС, как правило, гиперклеточный за счет повышенного количества незрелых и зрелых клеток гранулоцитарного ряда. Но в отличие от ХМЛ лейко-эритроцитарное соотношение обычно менее 10:1. Нечасто увеличено количество незрелых базофилов и эозинофилов. Количество клеток моноцитарного ряда, напротив, увеличено. Количество мегакариоцитов у 30% больных уменьшено, в них, как и в клетках гранулоцитарного и эритробластического ряда, обнаруживаются диспластические признаки. Увеличено количество бластов, но оно не превышает 30%.
В терминальной стадии аМПС развивается бластный криз. Обнаруживаемые при этом в костном мозге и периферической крови бластные клетки чаще имеют миелоидную природу, реже относятся к клеткам-предшественникам лимфопоэза. Это служит еще одним доказательством того, что аМПС, как и ХМЛ, возникает в результате трансформации полипотентной стволовой кроветворной клетки.
Дифференциальную диагностику аМПС проводят с ХМЛ, различными формами МДС. Ряд сходных и перекрывающихся клинико-гематологических признаков имеют аМПС и такие миелопролиферативные процессы, как истинная полицитемия, эссенциальная тромбоцитемия и идиопатический миелофиброз в поздних стадиях заболевания.
Особенно сложной является дифференциация аМПС, ХМЛ и ХММЛ. ФАБ-группой предложены критерии, помогающие разграничить эти процессы [28]. Так, содержание базофилов в крови при аМПС и ХММЛ не превышает 2%, а при ХМЛ оно значительно выше. Количество моноцитов в крови больных с ХМЛ, как правило, ниже 3%, при аМПС — в пределах 3–10%, при ХММЛ — обычно выше 10%. Признаки дисплазии клеток гранулоцитарного ростка сильно выражены при аМПС, умеренно — при ХММЛ и практически не определяются при ХМЛ. Содержание незрелых гранулоцитов при аМПС колеблется в пределах 10–20%, при ХММЛ — ниже 10%, а при ХМЛ — выше 20%. Количество бластов в крови выше 2% в хронической фазе заболевания наблюдается только у больных с аМПС [1, 3]. Из числа цитогенетических аномалий при аМПС наиболее часто обнаруживается трисомия 8, моносомия 7 при отсутствии Ph’-хромосомы. Прогноз при аМПС значительно хуже, чем при ХМЛ и ХММЛ.
Хронический миеломоноцитарный лейкоз миелопролиферативного типа
Хронический миеломоноцитарный лейкоз — клональный процесс, обусловленный трансформацией полипотентной стволовой кроветворной клетки или клеток-предшественников, являющихся ее ближайшими потомками. Встречается преимущественно у лиц пожилого возраста, чаще у мужчин. Характеризуется совокупностью миелодиспластических и миелопролиферативных признаков. При преобладании первых, включая определяющуюся при исследовании периферической крови цитопению, заболевание классифицируется как одна из форм МДС — ХММЛ. Если заболевание по клинико-гематологическим проявлениям больше напоминает ХМЛ, оно обозначается как хронический миеломоноцитарный лейкоз миелопролиферативного типа (ХММЛ-МТ) [1]. Для данной формы заболевания присущи лейкоцитоз, нейтрофилез, повышенное содержание моноцитов (>3%) в периферической крови, наличие макроцитарной анемии и выраженной в различной степени спленомегалии. Абсолютное количество нейтрофилов в крови больных превышает 13•109/л, а моноцитов >1•109/л. В периферической крови обнаруживаются незрелые клетки нейтрофильного ряда. Обычно их содержание не превышает 10% от общего количества лейкоцитов. Бластные клетки встречаются редко. Изредка обнаруживают незрелые клеточные элементы моноцитарного ряда (промоноциты). Хотя это не является характерным признаком ХММЛ-МТ, но в сочетании с моноцитозом и отсутствием Ph’-хромосомы или гибридного гена BCR/ABL позволяет отличить данное заболевание от классического ХМЛ [29, 30]. Диспластические изменения в клетках гранулоцитарного ряда минимальные (псевдопельгеровские лейкоциты, нейтрофилы с гипогранулярной цитоплазмой и отрицательной реакцией при выявлении МПО) или отсутствуют. Уровень ЛЩФ у больных с ХММЛ-МТ снижен или находится в пределах нормы. Количество тромбоцитов обычно в пределах нормы или несколько уменьшено, в редких случаях отмечается тромбоцитоз.
Обычно их содержание не превышает 10% от общего количества лейкоцитов. Бластные клетки встречаются редко. Изредка обнаруживают незрелые клеточные элементы моноцитарного ряда (промоноциты). Хотя это не является характерным признаком ХММЛ-МТ, но в сочетании с моноцитозом и отсутствием Ph’-хромосомы или гибридного гена BCR/ABL позволяет отличить данное заболевание от классического ХМЛ [29, 30]. Диспластические изменения в клетках гранулоцитарного ряда минимальные (псевдопельгеровские лейкоциты, нейтрофилы с гипогранулярной цитоплазмой и отрицательной реакцией при выявлении МПО) или отсутствуют. Уровень ЛЩФ у больных с ХММЛ-МТ снижен или находится в пределах нормы. Количество тромбоцитов обычно в пределах нормы или несколько уменьшено, в редких случаях отмечается тромбоцитоз.
При исследовании мазков из пунктатов отмечена гиперклеточность костного мозга с увеличением количества незрелых и зрелых клеток гранулоцитарного и в меньшей степени моноцитарного ряда. Последние составляют не менее 20% всех ядросодержащих кроветворных клеток. Клетки моноцитарного ряда представлены преимущественно моноцитами и небольшим количеством промоноцитов. Их наличие подтверждается при проведении цитохимических реакций на неспецифическую -НЭ и КНЭ [5]. При ХММЛ-МТ миелобласты и монобласты составляют не более 5% всех клеточных элементов костного мозга. Количество клеток эритробластического ряда различной степени зрелости и мегакариоцитов, как правило, в пределах нормы. Могут наблюдаться признаки дисгранулоцитопоэза и дисэритропоэза, появление гигантских многоядерных мегакариоцитов. При гистологическом изучении трепанобиоптатов костного мозга почти у 30% больных выявляется миелофиброз.
Определяемые приблизительно у трети больных цитогенетические аномалии (+8, –7 и del(20q), а также точечные мутации гена ras) не являются специфически ассоциированными с ХММЛ-МТ.
Дифференциальный диагноз ХММЛ-МТ проводят с ХМЛ, ХММЛ, лейкемоидными реакциями миелоидного типа и ОМЛ М4. Как известно, при ХММЛ-МТ в кроветворных клетках не обнаруживается Ph’ хромосома или гибридный ген BCR/ABL. Сложнее отличить ХММЛ-МТ от ХМЛ с диспластическими признаками, рассматривающегося в качестве одной из форм МДС. При этом учитывается количество лейкоцитов в периферической крови, наличие нейтрофилеза и моноцитоза, другие клинико-гематологические показатели, которые детально рассматривались выше. Проводя дифференциальную диагностику с лейкемоидными реакциями, особенно сопровождающимися моноцитозом, следует обращать внимание на их возможную связь с наличием опухолей или инфекционных заболеваний.
Сложнее отличить ХММЛ-МТ от ХМЛ с диспластическими признаками, рассматривающегося в качестве одной из форм МДС. При этом учитывается количество лейкоцитов в периферической крови, наличие нейтрофилеза и моноцитоза, другие клинико-гематологические показатели, которые детально рассматривались выше. Проводя дифференциальную диагностику с лейкемоидными реакциями, особенно сопровождающимися моноцитозом, следует обращать внимание на их возможную связь с наличием опухолей или инфекционных заболеваний.
Течение заболевания характеризуется увеличением содержания незрелых клеток гранулоцитарного и моноцитарного ряда и прогрессирующей недостаточностью костномозгового кроветворения, возникновением осложнений, обусловленных развитием цитопении, или трансформацией в ОМЛ М4. Медиана выживаемости больных с ХММЛ-МТ ниже, чем у пациентов с ХМЛ.
Хронический нейтрофильный лейкоз
Хронический нейтрофильный лейкоз (ХНЛ) — редкое заболевание, встречающееся у лиц пожилого возраста (старше 60 лет), характеризуется наличием анемии, спленомегалии и иногда гепатомегалии. Диагноз ХНЛ устанавливают при проведении общего анализа крови, иногда случайно. Количество лейкоцитов в периферической крови колеблется в пределах 25–50•109/л и редко бывает выше 100•109/л. Преобладают сегментоядерные нейтрофильные лейкоциты (90–95%), но в отдельных случаях содержание палочкоядерных нейтрофилов составляет 20–50% [1, 3]. Крайне редко обнаруживаются незрелые гранулоциты (миелоциты, метамиелоциты) и ядросодержащие клетки эритробластического ряда, бласты отсутствуют. В некоторых нейтрофилах ядра имеют кольцевидную форму. В цитоплазме могут присутствовать токсическая зернистость и вакуоли. Миелодиспластические признаки (гипогрануляция цитоплазмы, псевдопельгеровские лейкоциты) не определяются. В лейкоцитах больных с ХНЛ, в отличие от ХМЛ, выявляется повышенный уровень щелочной фосфатазы. Морфологические признаки эритроцитов и тромбоцитов крови в пределах нормы. В сыворотке крови больных с ХНЛ, как и при ХМЛ, повышен уровень витамина В12 и витамин В12-связывающего белка.
При исследовании пунктатов и трепанатов костного мозга отмечается гиперклеточность, обусловленная выраженной гиперплазией нейтрофильных гранулоцитов. Представлены в основном зрелые и незрелые клеточные элементы этого ряда, количество миелобластов не увеличено. Признаки дисгранулоцитопоэза обычно отсутствуют. Может наблюдаться умеренное угнетение эритропоэза. Количество мегакариоцитов в пределах нормы или несколько повышено.
По цитохимическим и иммунофенотипическим признакам клетки крови и костного мозга не отличаются от соответствующих по степени зрелости гранулоцитов у здоровых людей. В кроветворных клетках при ХНЛ, в отличие от ХМЛ, не выявляются Ph’-хромосома и реарранжировка BCR/ABL. Описан ряд других цитогенетических аномалий, подтверждающих клональную природу ХНЛ, таких, как трисомия 8, трисомия 9, трисомия 21, 20q– и другие реарранжировки с вовлечением длинного плеча хромосомы 20 [31, 32].
О неопластической природе ХНЛ свидетельствуют также результаты исследований роста колоний в полутвердых средах. При этом гемопоэтические клетки-предшественники сохраняют ограниченную способность дифференцироваться исключительно в гранулоциты [33].
Важной является дифференциальная диагностика ХНЛ с нейтрофильными лейкемоидными реакциями, истинной полицитемией и хроническим идиопатическим миелофиброзом. Последние два заболевания могут также сопровождаться нейтрофилезом, а при ХНЛ в костном мозге некоторых больных могут отмечаться признаки фиброза и остеосклероза.
Прогноз при ХНЛ значительно хуже, чем у больных с ХМЛ. При прогрессировании ХНЛ в терминальный период в ряде случаев может происходить трансформация в ОМЛ [1, 3].
Гиперэозинофильный синдром/Хронический эозинофильный лейкоз
Диагноз гиперэозинофильного синдрома (ГЭС) или хронического эозинофильного лейкоза (ХЭЛ) очень трудно установить в ранней стадии заболевания. Подозрение на ГЭС/ХЭЛ возникает при наличии абсолютной эозинофилии (>1,5•109/л), удерживающейся на протяжении более 6 мес. При этом не удается установить причину эозинофилии (необходимо исключить заболевания, вызываемые паразитами, аллергию, коллагенозы, неходжкинские лимфомы, лимфогранулематоз, множественную миелому, метастазы). Развитие и созревание эозинофилов в костном мозге регулируется синергическим действием ГМ-КСФ, ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-5 и другими эозинофилопоэтическими факторами, вырабатываемыми Т-лимфоцитами [2].
У больных с ГЭС/ХЭЛ могут обнаруживаться анемия, тромбоцитопения, гепатомегалия, спленомегалия, лимфаденопатия, нередко симптомы, обусловленные поражением сердца и легких. В таких случаях, если на основе цитогенетических и молекулярно-биологических исследований (включая и экспрессию гена N-ras) подтверждается клональность процесса, ставится диагноз ХЭЛ. При отсутствии этих данных диагноз ХЭЛ остается предположительным.
При исследовании периферической крови у больных общее количество лейкоцитов превышает 20•109/л. Отмечается персистирующая эозинофилия c преобладанием зрелых эозинофилов и небольшим количеством незрелых клеток. В некоторых случаях отмечаются гиперсегментация ядер, вакуолизация цитоплазмы клеток и уменьшенное содержание гранул. Аномальные гранулы или включения и палочки Ауэра не определяются. У части больных может быть умеренный нейтрофилез с небольшим количеством незрелых нейтрофилов.
Костный мозг представляется гиперклеточным за счет гиперплазии эозинофильного ростка. Представлены все стадии созревания эозинофилов, однако диспропорционального увеличения количества незрелых форм или бластов не наблюдается. В ряде случаев отмечается незначительное увеличение числа клеток нейтрофильного ряда. В эозинофилах при ХЭЛ, в отличие от нейтрофилов, не выявляется активность ХАЭ. Диспластически измененные бласты или незрелые клетки гранулоцитопоэза могут быть идентифицированы как эозинофилы на основании того, что в них определяется активность МПО, устойчивой к ингибированию цианидом. Эритропоэз и мегакариоцитопоэз не претерпевают существенных изменений.
В некоторых случаях при ХЭЛ обнаруживаются клональные хромосомные аномалии, такие как трисомия 8, i(17q), t (5; 12) (q31; q13), t (1; 5) (q23; q33) [34]. Дифференциальную диагностику проводят с Ph’ положительным ХМЛ с эозинофилией, с ХММЛ с эозинофилией, ассоциированным с t (5; 12) (q33; p13) и t (8; 13) (p11; q12), с ОЛЛ, изредка сопровождающимся эозинофилией, с ОМЛ М4Эо.
Хронический базофильный лейкоз
Базофильный лейкоз — редкое заболевание. Увеличение количества базофилов может наблюдаться в ряде случаев у больных с ХМЛ в хронической фазе и в фазе акселерации. Клетки с дифференцировочными признаками данного ряда могут наблюдаться при выделяемых в соответствии с ФАБ-классификацией подвариантах ОМЛ — М2Базо и М4Базо [3]. Базофильный лейкоз с содержанием бластов в костном мозге менее 30% имеет хроническое течение. В случаях обнаружения Ph’-хромосомы они должны рассматриваться как вариант ХМЛ [3]. При истинном хроническом базофильном лейкозе не выявляются Ph’-хромосома и реарранжировка BCR/ABL. Циркулирующие в периферической крови клетки содержат базофильные гранулы, дают отрицательную реакцию при выявлении активности МПО. В клинической картине высок удельный вес симптомов, связанных с освобождением больших количеств гистамина, наблюдается ДВС-синдром.
Хронический тучноклеточный лейкоз
Заболевание может возникнуть de novo или протекать в виде терминальной стадии системного мастоцитоза. Клиническое течение чаще хроническое или подострое, в периферической крови обнаруживаются немногочисленные тучные клетки (мастоциты). Эти атипичные клетки имеют дольчатые ядра, более высокое, чем в норме ядерно-цитоплазматическое соотношение, содержат в цитоплазме гранулы, которые метахроматически окрашиваются толуидиновым синим и дают интенсивную реакцию при цитохимическом выявлении активности ХАЭ. При исследовании мазков периферической крови могут определяться анемия, моноцитоз и эозинофилия. В эозинофилах отмечаются признаки дегрануляции и вакуолизации цитоплазмы. Костный мозг гиперклеточный за счет гиперплазии клеток гранулоцитарного ряда, эозинофилов и увеличенного количества лимфоцитов. В трепанобиоптатах костного мозга наблюдается очаговая или диффузная инфильтрация тучными клетками, гиперплазия клеток гранулоцитарного и мегакариоцитарного ряда, наличие миелофиброза и остеосклероза.
Исследования показывают, что злокачественно трансформированные тучные клетки имеют фенотип CD2+ CD4+ CD11b+ CD33+ CD8– CD19– TdT– HLA-DR–.
В клинической картине, помимо гепатомегалии, спленомегалии, лимфаденопатии, на первый план выступают симптомы, обусловленные повышенной выработкой гистамина.
Истинная полицитемия
Истинная полицитемия (ИП) (эритремия, синдром Вакеза — Ослера) — клональное миелопролиферативное заболевание, характеризующееся прежде всего избыточной продукцией клеток эритробластического ряда, а также гранулоцитов и мегакариоцитов. ИП — относительно редкое заболевание, ежегодно выявляется 5–10 новых больных на 1 млн населения. Соотношение мужчин и женщин составляет 1,2:1. Средний возраст, в котором впервые выявляется заболевание, составляет 60–70 лет. В то же время ИП иногда встречается в юношеском и даже в детском возрасте.
В своем развитии ИП проходит три последовательные стадии: фазу пролиферации, основным признаком которой является увеличение массы эритроцитов; фазу стабильного течения заболевания; фазу миелоидной метаплазии [35].
Основные критерии диагностики заболевания (табл. 24) были разработаны 25 лет тому назад группой по изучению ИП (PVSG) [36].
Диагноз ИП считается правомочным при наличии трех основных критериев категории А (А1+А2+А3) или двух первых критериев (А1+А2) и любых двух критериев категории В.
У некоторых больных для подтверждения диагноза могут использоваться и некоторые дополнительные признаки, такие, как низкий или нормальный уровень эритропоэтина в сыворотке крови (при повышенном уровне ИП исключается), рост эритроидных колоний в культуре в отсутствие эритропоэтина, клональность процесса, подтвержденная результатами цитогенетического и молекулярно-биологического исследования. Ценным является также гистологическое изучение трепанобиоптатов костного мозга, позволяющее подтвердить или исключить наличие ИП в сложных для диагностики случаях.
Симптомы, наблюдающиеся в начальной стадии заболевания, обусловлены увеличенной массой эритроцитов. Больные жалуются на слабость, головную боль, нередко в виде мучительной мигрени. У больных отмечается цианоз кожи и видимых слизистых оболочек (у 90%), боль в области сердца, в костях и нижних конечностях. У многих больных основной жалобой является кожный зуд. Нарушения со стороны ЦНС колеблются от легких функциональных (на основе стазов без тромбообразования) до тяжелых, обусловленных тромбозом крупных сосудов [37]. Тромбоз сосудов (почти у 30% больных) и кровоизлияния (у 25%) относятся к числу наиболее важных и грозных осложнений при ИП.
Одним из основных клинических симптомов у больных ИП, отнесенных к категории А, является увеличение размеров селезенки. Причиной спленомегалии, выявляющейся у 80% пациентов, является ее повышенное кровенаполнение и участие в лимфопролиферативном процессе. Почти у 70% больных одновременно обнаруживаются признаки гепатомегалии, обусловленные повышенным кровенаполнением органа, миелоидной метаплазией, разрастанием фиброзной ткани (в поздних стадиях заболевания).
Из данных лабораторных исследований основным для установления диагноза ИП является увеличение массы клеток красной крови. Присущее ИП увеличение количества эритроцитов (6–7 млн в 1 мм3) и уровня гемоглобина (18–22 г/дл) сопровождается повышением показателей гематокрита. В первой, «пролиферативной», стадии заболевания на нормобластическую эритроидную гиперплазию костного мозга указывает обнаружение в периферической крови нормохромных и нормоцитарных эритроцитов. При дефиците железа, обусловленном повышенной кровоточивостью или частыми лечебными кровопусканиями, в мазках крови могут выявляться гипохромные и микроцитарные эритроциты. В периферической крови более чем у 80% больных ИП отмечается нейтрофильный лейкоцитоз с небольшим сдвигом влево. Количество лейкоцитов обычно колеблется в пределах от 10 до 20•109/л. Умеренная базофилия наблюдается у 60–70% больных. Частой является эозинофилия.
Количество тромбоцитов повышено у 50–80% больных, у 10% из них оно выше 1000•109/л [37]. В периферической крови при ИП могут обнаруживаться крупные тромбоциты и фрагменты ядер мегакариоцитов.
В мазках костного мозга определяется гиперплазия за счет увеличения числа клеточных элементов не только нормобластического эритропоэза, но и клеток других ростков миелопоэза (гранулоцитарного, мегакариоцитарного).
Наличие гиперпластических процессов при ИП подтверждается и результатами гистологического изучения трепанобиоптатов костного мозга. У большинства больных отмечается гиперклеточность костного мозга. По данным исследователей, входящих в группу по изучению ИП, кроветворные клетки различного происхождения и разной степени зрелости занимают от 37 до 100% (в среднем 80%) всей площади срезов костного мозга [38]. Наиболее выражена гиперплазия клеток эритробластического ряда. Значительным является и увеличение количества мегакариоцитов, размер которых широко варьирует — от малых до очень крупных с дольчатыми ядрами [39]. Мегакариоциты в костном мозге распределяются в виде кластеров. В 25% случаев уже в начальной стадии ИП в костном мозге определяется увеличенное количество ретикулиновых волокон. В дальнейшем оно прогрессирует, что сочетается с увеличением клеточности костного мозга.
Длительность начальной и стабильной фаз ИП составляет от 5 до 20 лет. Примерно у 10–20% больных наблюдается переход в фазу миелоидной метаплазии. Для постполицитемической миелоидной метаплазии (ППММ) характерно уменьшение массы клеток красной крови, выявляемое при радиоизотопном исследовании, увеличение степени спленомегалии, усиление фиброза костного мозга с увеличением количества ретикулиновых и коллагеновых волокон, появление очагов экстрамедуллярного гемопоэза, развитие цитопении, пойкилоцитоз и анизоцитоз [1, 6]. Могут наблюдаться диспластические изменения в клетках эритробластического и гранулоцитарного ряда, не выявлявшиеся ранее. В ряде случаев они предшествуют развитию острого лейкоза, чаще всего миелоидного происхождения. Острый лейкоз возникает у 1–2% больных ИП, не лечившихся цитотоксическими препаратами, и у 10–15% пациентов после миелосупрессивной терапии [40]. В фазе ППММ риск развития острого лейкоза у больных значительно выше (23%), чем в начальной и стабильной фазе заболевания (7%).
Цитогенетические аномалии в момент установления диагноза выявляются почти у 40–50% больных ИП и ассоциируются с менее благоприятным прогнозом [1, 6]. Частота их увеличивается по мере прогрессирования заболевания, при эволюции в миелодиспластический процесс. Наиболее часто у больных ИП встречаются не являющиеся характерными только для этого заболевания следующие аномалии кариотипа: del(20q), +8, +9 [41]. Описания каких-либо особых изменений иммунофенотипа кроветворных клеток при ИП в доступной литературе отсутствуют.
Дифференциальную диагностику ИП проводят с другими заболеваниями, сопровождающимися вторичным (симптоматическим) эритроцитозом. Различают вторичные абсолютные эритроцитозы, при которых характерны раздражение эритробластического ростка костномозгового кроветворения и увеличение массы циркулирующих в крови эритроцитов, и относительные эритроцитозы, в основе которых лежит сгущение крови, обусловленное действием различных факторов. В онкогематологической клинике особую важность приобретает дифференциальная диагностика ИП в стабильной фазе заболевания и вторичных абсолютных эритроцитозов, встречающихся у больных с гипернефромой, опухолями почек, эндокринных органов.
ППММ и развивающийся острый лейкоз не создают диагностических проблем, так как возникают у больных ИП, длительно находившихся под наблюдением [1].
Эссенциальная тромбоцитемия
Эссенциальная тромбоцитемия (ЭТ) (синонимы: первичная тромбоцитемия, геморрагическая тромбоцитемия, идиопатическая тромбоцитемия) — миелопролиферативное заболевание, характеризующееся преимущественным поражением клеток мегакариоцитарного ряда, основным проявлением которого является выраженное увеличение количества тромбоцитов. Приводится ряд доказательств клональности процесса, обусловленного повреждением на уровне стволовой кроветворной клетки костного мозга или ее ближайших потомков [5, 42]. ЭТ относится к числу относительно редких заболеваний. Показатели ежегодной заболеваемости составляют 0,7 на 100 тыс. населения [43]. Заболевают люди в возрасте 50–70 лет, но описаны случаи развития ЭТ у лиц молодого возраста и даже у детей [44]. В клинической картине на первый план выступают осложнения, обусловленные тромбозом сосудов и кровоизлияниями. Активацией тромбоцитов и микротромбозами мелких сосудов обусловлены также часто наблюдающиеся у больных ЭТ неврологические проявления. У 50% больных определяется спленомегалия, а у 15–20% — гепатомегалия [45].
Количество тормбоцитов в периферической крови увеличено и нередко превышает 1000•109/л. Минимальным же для установления диагноза ЭТ в соответствии с критериями, предложенными группой экспертов по изучению ИП, является стойко удерживающийся в течение длительного времени уровень тромбоцитов, равный 600•109/л [46]. Уровень гемоглобина в соответствии с теми же критериями у больных ЭТ может колебаться в пределах от 10 до 18,8 г/дл (в среднем 13,8 г/дл). Среднее количество лейкоцитов в периферической крови составляет 11,5•109/л, но возможны значительные колебания — от 6 до 41•109/л. Незрелые клетки гранулоцитарного ряда в мазках периферической крови обнаруживаются крайне редко. Нет характерной для ХМЛ базофилии. Уровень щелочной фосфатазы, выявляемой при цитохимическом исследовании нейтрофилов, обычно находится в пределах нормы, в редких случаях может быть повышенным или сниженным.
Тромбоциты, выявляемые в мазках крови больных, не отличаются от наблюдающихся у здоровых людей, но могут быть полиморфными, варьируя по форме и величине. В ряде случаев могут обнаруживаться крупные, атипичные, гипогранулярные формы. Изредка в крови определяются ядросодержащие фрагменты мегакариоцитов, трудно отличимые от лимфоидных клеток. В этих случаях для идентификации природы клеточных элементов приходится прибегать к иммуноцитохимическому определению линейно-специфических антигенов — маркеров клеток мегакариоцитарного ряда, Т- и В-лимфоцитов.
У 70% больных при исследовании мазков из пунктатов и трепанобиоптатов обнаруживают гиперклеточный костный мозг. У отдельных больных количество миелокариоцитов может быть в пределах нормы или несколько увеличенным. Количество мегакариоцитов значительно повышено. Мегакариоциты в мазках равномерно распределены или образуют скопления в виде групп и кластеров. Размер мегакариоцитов с дольчатыми ядрами и обширной цитоплазмой нормальный или увеличенный. Обычно отмечается также умеренная гиперплазия клеток гранулоцитарного и эритробластического ряда. У 20–50% больных преимущественно пожилого возраста в костном мозге определяется увеличенное количество ретикулиновых волокон [46, 47].
В табл. 25 приведены критерии, позволяющие отличить ЭТ от других форм миелопролиферативных заболеваний (ИП, ХМЛ, идиопатического миелофиброза) и вторичных (реактивных) тромбоцитозов. Последние нередко отмечаются у больных с опухолями, воспалительными процессами, железодефицитной анемией, после операции спленэктомии. Следует отметить, что количество пластинок при вторичных тромбоцитозах редко превышает 1000•109/л.
Крайне сложной является дифференциальная диагностика ЭТ и ИП. Выраженный тромбоцитоз может быть также ранним проявлением ХМЛ. Лишь позднее увеличивается количество лейкоцитов и наблюдается выраженный сдвиг влево. В этих случаях помогает определение Ph’-хромосомы или обнаружение реарранжировки гена BCR/ABL.
Хронический идиопатический миелофиброз, или миелосклероз с миелоидной метаплазией, характеризуется более выраженной, чем при ЭТ спленомегалией. При гистологическом исследовании биоптатов костного мозга определяется фиброз с наличием сети коллагеновых волокон, а в крови — описанные ниже изменения.
Хронический идиопатический миелофиброз с миелоидной метаплазией
Хронический идиопатический миелофиброз с миелоидной метаплазией (ХИМММ) (синонимы; «миелосклероз с миелоидной метаплазией», «агногенная миелоидная метаплазия», «идиопатический миелофиброз», «алейкемический миелоз с остеосклерозом», «остеомиелосклероз и остеомиелофиброз») возникает в результате клональной пролиферации стволовых кроветворных клеток костного мозга [48]. Характерные признаки заболевания: панмиелоз, фиброз костного мозга и нередко остеосклероз, появление очагов экстрамедуллярного гемопоэза, спленомегалия, анемия, изменения лейкоцитарной формулы крови.
ХИМММ относится к числу редких форм миелопролиферативных заболеваний. Ежегодная заболеваемость составляет 0,5 на 100 тыс. населения [48]. Встречается преимущественно у лиц пожилого возраста (60–70 лет), но изредка и у детей [49]. Среди больных несколько преобладают лица мужского пола [50].
Основными жалобами больных при поступлении в клинику являются слабость, уменьшение массы тела, лихорадка, боль в костях, иногда геморрагические симптомы. Наиболее важными признаками ХИМММ считаются спленомегалия и гепатомегалия. В то же время у части больных первоначально отсутствуют какие-либо симптомы и заболевание выявляется случайно (на основе обнаружения увеличенной селезенки и результатов анализа периферической крови).
В момент установления диагноза может наблюдаться выраженная вариабельность гематологических параметров [51]. У большинства больных обнаруживают нормохромную анемию. У 50% больных уровень гемоглобина ниже 10 г/дл, а у 20% — 8 г/дл. Постоянными являются признаки анизоцитоза и пойкилоцитоза, наличие пойкилоцитов в форме слезинки (teardrop). У многих больных в мазках периферической крови обнаруживаются ядросодержащие клетки эритробластического ряда. Количество ретикулоцитов умеренно увеличено и может колебаться в зависимости от стадии заболевания. При наличии аутоиммунного гемолиза количество ретикулоцитов увеличивается до 5–20%.
Существенно варьирует содержание лейкоцитов в периферической крови. Изредка оно может быть низким, у 40% пациентов колеблется в пределах 10–25•109/л, у некоторых превышает 40•109/л. В крови у больных с ХИМММ встречаются гипер- или гипосегментированные лейкоциты, небольшой процент незрелых клеток гранулоцитарного ряда (миелоцитов и промиелоцитов). Уровень миелобластов, выявляемых у 40% больных, обычно не превышает 1–5%. Однако даже увеличение их количества до 10% еще не служит показателем перехода заболевания в более агрессивную фазу или трансформации в острый лейкоз [39]. Показатели лейкоцитарной щелочной фосфатазы, как правило, повышены, но у ряда больных может наблюдаться нормальный уровень или даже снижение ферментативной активности [49]. При исследовании мазков периферической крови больных ХИМММ отмечается умеренная базофилия. Количество моноцитов, как правило, не изменено. Нередко наблюдается относительная лимфоцитопения [18].
Количество тромбоцитов в периферической крови при ХИМММ может быть уменьшенным (менее 150•109/л), но может отмечаться и тромбоцитоз (более 500•109/л) [50]. В мазках периферической крови выявляются атипичные, крупные и гипогранулярные тромбоциты и аномальные мегакариоциты, в том числе микромегакариоциты, промегакариоциты и ядра мегакариоцитов. Прогрессирование заболевания сопровождается увеличением в крови количества этих клеток, нарастанием лейкоцитоза и повышением уровня незрелых клеток гранулоцитарного ряда.
Анализ миелограммы при изучении мазков костного мозга, полученных при стернальной пункции, не является показательным. Результаты разнятся в зависимости от того, попала ли игла в очаг миелоидной гиперплазии или в очаги фиброза и остеосклероза. Более информативным для установления диагноза ХИМММ, как и ряда других форм хронических миелопролиферативных заболеваний, является гистологическое изучение трепанобиоптатов костного мозга. При этом практически во всех случаях определяется гиперклеточность костного мозга, как правило, диффузная и реже очаговая. Синусы костного мозга расширены и заполнены пролиферирующими кроветворными клетками. Представлены клеточные элементы всех трех основных клеточных линий миелопоэза, хотя в отдельных срезах может быть преобладание клеток того или иного типа [1, 48]. У 90% больных отмечается выраженное количество мегакариоцитов, образующих кластеры из 3–10 клеток, характерно также наличие мегакариоцитов с конденсированными и дистрофически измененными ядрами в трепанобиоптатах с выраженным коллагеновым фиброзом.
Соотношение гемопоэтических клеток и фиброзной ткани варьирует не только в пробах, полученных из различных участков костного мозга, но и в срезах одного и того же биоптата [50]. Степень выраженности фиброза также значительно колеблется. В ранней стадии заболевания у большинства больных определяется увеличенное количество ретикулиновых волокон, располагающихся преимущественно вокруг сосудов [1, 51]. Коллагеновых волокон немного. При развитии интенсивного фиброза и возникновении очагов склероза клеточность костного мозга в отдельных участках снижается. Очажки сохранившихся клеточных элементов мегакариоцитарного, гранулоцитарного и эритробластического ряда разделены волокнами соединительной ткани. В некоторых участках костного мозга кроветворные клетки практически отсутствуют, фиброзная ткань целиком заполняет межтрабекулярные пространства.
Предпринимались попытки классификации и выделения отдельных типов ХИМММ с учетом результатов изучения трепанобиоптатов костного мозга. Так, Ward, Block [52] считают возможным выделение следующих трех основных гистологических форм: пангиперплазия, миелоидная атрофия и фиброз, миелофиброз и остеосклероз.
При пангиперплазии клетки трех основных линий миелопоэза занимают более 70% площади костного мозга. Иногда наблюдается увеличение числа ретикулиновых волокон при отсутствии коллагенового фиброза. Изредка встречаются скопления лимфоцитов. Подобная картина напоминает наблюдающуюся при ИП. При миелоидной атрофии и фиброзе небольшие очаги гемопоэза разделены ретикулиновыми и коллагеновыми волокнами. В их состав входят клетки всех основных ростков миелопоэза, но преобладают мегакариоциты. Увеличено количество плазматических клеток и клеток стромы. При третьей гистологической форме на первый план выступают признаки миелофиброза и остеосклероза. Количество гемопоэтических клеток уменьшено, и они представлены в основном мегакариоцитами.
Прогностически значимым может быть и разделение больных с ХИМММ на две группы с учетом данных гистопатологического изучения костного мозга при первоначальном установлении диагноза [51]. У больных первой группы (40%) костный мозг гиперклеточный с наличием атипичных мегакариоцитов, незрелых и зрелых гранулоцитов, клеток эритробластческого ряда, располагающихся в расширенных синусах. Ретикулиновые волокна отсутствуют или их количество несколько увеличено, располагаются они преимущественно вокруг сосудов. Коллагеновые волокна не выявляются. У больных второй группы (60%) в костном мозге отмечается умеренное или значительное уменьшение количества гемопоэтических клеток и очаги выраженного фиброза и склероза. Очажки, состоящие из незрелых клеток эритроидного и гранулоцитарного ряда, разделены участками соединительной ткани. Дистрофически измененные мегакариоциты, являющиеся преобладающими клеточными элементами, находятся в виде кластеров в расширенных синусах. У таких пациентов отмечается более низкий уровень гемоглобина и уменьшенное содержание тромбоцитов в крови, более выраженная степень спленомегалии, чем у больных первой группы.
Попутно заметим, что стромальные клетки, в первую очередь фибробласты, с которыми связывается развитие фиброза и остеосклероза, не относятся к популяции неопластических клеток. Представляется, что их пролиферация может быть связана с выработкой трансформированными мегакариоцитами повышенных количеств фактора роста фибробластов [1, 5].
Очаги экстрамедуллярного гемопоэза у больных с ХИМММ могут иметь различную локализацию, но наиболее часто возникают в селезенке, печени и лимфатических узлах [53]. В селезенке и печени клетки эритробластического, гранулоцитарного и мегакариоцитарного рядов располагаются в синусах. В лимфатических узлах, наряду с подобной же локализацией клеток трех основных ростков миелопоэза, может отмечаться выраженная перифолликулярная инфильтрация.
Цитогенетические аномалии наблюдаются почти у 60% больных с ХИМММ. Наиболее частой, хотя и неспецифической, является del 13 (q13q210). Другие возможные аномалии включают –7, +8 и +9. Кариотипические аномалии, выявляемые в момент установления диагноза, относятся к числу неблагоприятных прогностических признаков. В ряде случаев эволюция кариотипа ассоциируется с трансформацией в острый лейкоз.
Дифференциальную диагностику ХИМММ проводят со многими заболеваниями, сопровождающимися развитием фиброза и остеомиелосклероза. В первую очередь это различные формы злокачественных миелопролиферативных и лимфопролиферативных процессов. К числу первых относятся ХМЛ, ИП, ЭТ, острый миелофиброз. Для разграничения ХИМММ и ХМЛ применяют определение Ph’-хромосомы и перестройки BCR/ABL. Дифференциация ХИМММ и ИП крайне затруднена и во многих случаях должна основываться на анализе клинико-гематологических признаков.
При остром миелофиброзе отмечается агрессивное клиническое течение, отсутствие или небольшая степень спленомегалии. Помимо миелофиброза отмечаются признаки вовлечения в процесс основных ростков миелопоэза с увеличением количества незрелых и бластных клеток.
Развитием фиброза сопровождается поражение костного мозга также при неходжкинских злокачественных лимфомах. Но при этом, в отличие от ХИМММ, наблюдается мономорфная инфильтрация костного мозга клетками, соответствующими той или иной форме и цитологическому варианту исходных опухолей лимфоидной ткани.
Появление очагов специфического поражения при лимфогранулематозе также сопровождается разрастанием ретикулиновых и коллагеновых волокон. В дифференциальной диагностике решающим является обнаружение специфических для лимфогранулематоза гигантских многоядерных клеток Березовского—Штернберга.
Реактивный, или вторичный фиброз, наряду с остеосклерозом и остеолитическими изменениями, наблюдается и при метастатических поражениях костного мозга у больных раком молочной железы и других органов. В этих случаях помогает выявление раковых клеток в костном мозге на основе применения иммуноцитохимических методов и мкАТ к цитокератинам, онкофетальным антигенам, антигенам мембран эпителиальных клеток [4, 5, 54].
Развитие миелофиброза возможно также при коллагенозах, диссеминированном туберкулезе, действии ионизирующей радиации [4, 5]. Указанные заболевания и состояния также необходимо принимать во внимание при установлении диагноза ХИМММ.
Медиана выживаемости больных с ХИМММ составляет 3 года — 5 лет. Основными причинами смерти являются инфекции, кровоизлияния, тромбоэмболии. Трансформация в острый лейкоз, как правило, миелоидного происхождения отмечается у 5–20% больных [1, 48].
Хронический миелопролиферативный синдром, неклассифицируемый
Термин «хронический миелопролиферативный синдром, неклассифицируемый (ХМПС-Н)» был предложен группой по изучению ИП (PVSG) для обозначения заболевания у больных со спленомегалией, с варьирующими по степени тромбоцитозом и гиперплазией клеток мегакариоцитарного ряда, у которых не наблюдается увеличения массы эритроцитов, лейкоэритробластической реакции, значительного миелофиброза и отсутствует Ph’-хромосома [7]. Безусловно, некоторые случаи, диагностируемые как ХМПС-Н, могут быть ранними стадиями процесса, который в дальнейшем по мере получения достаточной информации удастся более точно классифицировать. Эту категорию не следует также использовать для обозначения случаев, при которых полученные при клинико-лабораторных исследованиях данные недостаточны для установления более точного диагноза.
Морфологические изменения в костном мозге и периферической крови весьма гетерогенны, но, вероятно, можно выделить следующие категории или подтипы.
Первый — случаи, когда при первоначальном исследовании костного мозга обнаруживается пангиперплазия с выраженной гиперплазией клеток мегакариоцитарного ряда, в связи с чем требуется проводить дифференциальную диагностику с ХИМММ или ИП и ЭТ. Но при этом отсутствуют обычные критерии, необходимые для установления диагноза указанных заболеваний. При исследовании периферической крови у таких больных определяют лейкоцитоз и/или выраженный тромбоцитоз. Количество эритроцитов не увеличено. Нет данных, указывающих на наличие очагов экстрамедуллярного гемопоэза.
Второй — случаи, когда начальные проявления напоминают ХИМММ (спленомегалия, лекоэритробластоз, пойкилоциты в виде слезинки, фиброз костного мозга), но при этом отмечаются выраженные диспластические изменения в клетках различных линий. Количество миелобластов существенно не повышено (<5%), в костном мозге происходит обычная дифференцировка клеток всех линий. Неясно, представляют ли такие случаи трансформацию хронической фазы ХИМММ в более агрессивное заболевание или являются «переходными» нарушениями с признаками миелопролиферативных и миелодиспластических процессов. Пока же их предлагается обозначать как ХМПС-Н.
Термин этот не может использоваться для обозначения патологических состояний с признаками, обычно наблюдающимися при хронических миелопролиферативных заболеваниях, такими, как пангиперплазия с выраженной гиперплазией мегакариоцитов, но при которых отмечаются выраженный сдвиг лейкоцитарной формулы влево с увеличением количества незрелых клеток и бластов (10–30%), миелофиброз и выраженная миелодисплазия. Последние, особенно при цитопении в периферической крови и отсутствии выраженной спленомегалии, скорее должны классифицироваться как миелодиспластические синдромы с миелофиброзом (включая «острую миелодисплазию с миелофиброзом», «острый миелосклероз» или «злокачественный миелосклероз»).
При ХМПС-Н, как и при других миелопролиферативных заболеваниях, обнаруживаются такие цитогенетические аномалии, как +8, +9, del(20q). Ph’-хромосома не определяется. Клинико-лабораторные данные у больных с ХМПС-Н вариабельны. Выделение прогностических признаков станет возможным после мониторинга течения процесса у достаточного числа больных.
Ювенильный хронический миелолейкоз (ЮХМЛ) не входит в классификацию хронических миелопролиферативных заболеваний, представленных в табл. 21. Это заболевание встречается исключительно у детей [55]. ЮХМЛ, наряду с синдромом моносомии 7, в качестве самостоятельной нозологической формы включен в классификацию миелодиспластических и миелопролиферативных заболеваний детского возраста. В ФАБ-классификации МДС [56] заболевание обозначается как ХММЛ. Этим же термином пользуются и участники Европейской рабочей группы по изучению МДС у детей [57, 58].
ЮХМЛ отличается от классического Ph’-положительного лейкоза, встречающегося в основном у взрослых, но иногда диагностируемого и у детей, по ряду морфологических, цитогенетических и клинических признаков [59, 60]. Это заболевание с агрессивным острым или подострым течением, напоминающим ОМЛ. Среди больных с ЮХМЛ 95% составляют дети (в основном мальчики) до 4 лет [55]. В момент установления диагноза возраст большинства детей не превышает 2 лет [57, 61]. Риск возникновения ЮХМЛ особенно высок у детей с семейным нейрофиброматозом [62].
Основными клиническими проявлениями заболевания являются анемия, гепатомегалия, спленомегалия, лимфаденопатия (у 20% больных), экземоподобные высыпания на коже головы, обусловленные лейкемическими инфильтратами.
У больных с ЮХМЛ умеренно снижен уровень гемоглобина. Уже в самом начале заболевания значительно уменьшается количество тромбоцитов. Общее количество лейкоцитов увеличено, но ниже, чем при ХМЛ у взрослых и колеблется в пределах 25–75•109/л [63]. При анализе лейкограммы определяют большой процент миелобластов, незрелых гранулоцитов, моноцитов, лимфоцитов и более низкий, чем при Ph’-положительном ХМЛ, уровень палочко- и сегментоядерных нейтрофильных лейкоцитов. Может определяться базофилия, не являющаяся, однако, постоянным признаком. В периферической крови могут обнаруживаться ядросодержащие клетки эритроидного ряда, количество которых увеличивается по мере прогрессирования заболевания. Встречаются отдельные плазматические клетки и иммунобласты. Костный мозг гиперклеточный, содержит увеличенное количество бластов (до 10–15%), незрелых и зрелых моноцитов. Содержание мегакариоцитов, как правило, уменьшено.
Уровень щелочной фосфатазы лейкоцитов, как и при Ph’-положительном ХМЛ, снижен. Важным лабораторным признаком является резкое увеличение (на 40–55%) уровня фетального гемоглобина у большинства больных по мере прогрессирования заболевания. При ХМЛ уровень фетального гемоглобина не превышает 10%. В качестве дополнительных признаков можно указать на наблюдающиеся при ЮХМЛ снижение гемоглобина А2 и активности карбоангидразы эритроцитов, увеличение активности глюкозо-6-фосфатдегидрогеназы и экспрессии антигена і при уменьшении экспрессии антигена І. У 50% больных повышен уровень сывороточных иммуноглобулинов [59].
У большинства больных при установлении диагноза ЮХМЛ не выявляют цитогенетических нарушений. Однако в ряде случаев описаны клональные аномалии хромосом (в основном трисомия 8), выявляемые в начальный период или в фазе прогрессирования заболевания. Частыми являются мутации онкогена ras [3].
Бластный криз развивается у небольшого числа детей. Но даже у больных, у которых не происходит бластная трансформация, прогноз неблагоприятный. Особенно тяжелое течение заболевания наблюдается у детей, заболевших в возрасте 6–12 мес. Медиана выживаемости больных с ЮХМЛ составляет 10–12 мес [3].
Дифференциальную диагностику ЮХМЛ проводят с такими заболеваниями, как редкий у детей Ph’ положительный ХМЛ, ОМЛ М4, синдром, связанный с моносомией 7, с другими миелодиспластическими/миелопролиферативными нарушениями.
1. Brunning RD, McKenna RW. Tumors of the bone marrow. Washington: Armed Forces Inst Pathol 1993; 406 p.
2. Schumacher HR, Coteligham JD. Chronic leukemia. Approach to diagnosis. New York: Igaku-Shoin Med Publ 1993; 356 p.
3. Bain BJ. Leukemia Diagnosis. 2nd ed. Oxford: Blackwell Science 1999. 200 p.
4. Hoffbrand AV, Pettit JE. Color atlas of hematology. 2nd ed. London etc: Mosby-Wolfe 1998. 360 p.
5. Глузман ДФ, Абраменко ИВ, Скляренко ЛМ, Надгорная ВА. Лабораторная диагностика онкогематологических заболеваний. Киев: Морион 1998. 336 с.
6. Neoplastic Hematopathology. Ed by DM Knowles, Baltimore et al: Williams a Wilkins 1992; 1624 p.
7. Imbert M, Pierre R, Vardiman J. Chronic Myeloproliferative Disorders. Writting Committee. Chicago 1998; 1–12.
8. Morrison VA. Chronic leukemias. Ca-A Cancer J Clin 1994; 44: 353.
9. Dekmezian R, Kantarjian HM, Keating MY et al. The relevance of reticulin stain-measured fibrosis at diagnosis in chronic myelogenous leukemia. Cancer 1987; 59: 1739.
10. Clough V, Geary CG, Hashmi K et al. Myelofibrosis in chronic granulocytic leukaemia. Br J Haematol 1979; 42: 515.
11. Глузман ДФ. Диагностическая цитохимия гемобластозов. Киев: Наук думка 1978; 216 с.
12. Wachstein M. Histochemistry of enzymes in tumors. Handbuch der Histochemie, Bd 2. 1962: 73.
13. Kamada N, Uchino H. Chronologic sequence in appearance of clinical and laboratory findings characteristic of chronic myelocytic leukemia. Blood 1978; 51: 843.
14. Zittoun J, Zittoun R, Marquet J, Sultan C. The three transcobalamins in myeloproliferative disorders and acute leukaemia. Br J Haematol 1975; 31: 287.
15. Todd MB, Waldron JA, Jennings TA et al. Loss of myeloid differentiation antigens precledes blastic transformation in chronic myelogenous leukemia. Blood 1987; 70: 122.
16. Schmetzer HM, Gerhartz HH. Immunological classification of chronic phase imminent blastic transformation and acute lymphoblastic leukemia. Exp Hematol 1997; 6: 502.
17. Логинский ВЕ, Выговская ЯИ, Потемкина ГИ и др. Иммунофенотипирование мононуклеарных клеток при хронических миелопролиферативных заболеваниях. Эксперим онкология 1991; 13: 37.
18. Wardiman JW. Chronic myelogenous leukemia and myeloproliferative disorders. In: Neoplastic Hematopathology 1992: 1405.
19. Lichtman MA. Chronic myelogenous leukemia and related disorders. In: Williams Hematology. 5th ed. 1995: 298.
20. Deininger MW, Goldman JM. Chronic myeloid leukemia. Curr Opin Hematol 1998; 5: 302.
21. Katarjian HM, Deisseroth A, Kurzrock R et al. Chronic myelogenous leukemia: a coincise update. Blood 1993; 82: 691.
22. Savage DG, Szydlo RM, Chase A et al. Bone marrow transplantation for chronic myeloid leukemia: the effects of differing criteria for defining chronic phase on probabilities of survival and relapse. Br J Haematol 1997; 99: 30.
23. Muehleck SD, McKenna RW, Arthur DC et al. Transformation of chronic myelogenous leukemia: clinical, morphologic and cytogenetic features. Am J Clin Pathol 1984; 82: 1.
24. Champlin RE, Golde DW. Chronic myelogenous leukemia: recent advances. Blood 1985; 65: 1039.
25. Ahuja HG, Jat PS, Foti A et al. Abnormalities of the retinoblastoma gene in the pathogenesis of acute leukemia. Blood 1991; 78: 3259.
26. Ishikura H , Yufu Y, Yamashita S et al. Biphenotypic blast crisis of chronic myelogenous leukemia: abnormalities of p53 and retinoblastoma genes. Leuk Lymphoma 1997; 25: 573.
27. Sill H, Goldman JM, Gross NCP. Homozygous deletion of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood 1995; 85: 2013.
28. Bennet JM, Catovsky D, Daniel MT et al. The chronic myeloid leukemias: guidelines for distinguishing chronic granulocytic, atypical myeloid, and chronic myelomonocytic leukemia. Proposals by the French-American-British Cooperative group. Br J Haematol 1994; 87: 746.
29. Pugh WC, Pearson M, Vardiman JW, Rowley JD. Philadelphia chromosome-negative chronic myelogenous leukaemia: a morphologic reassessment. Br J Haematol 1985; 60: 457.
30. Kantarjian HM, Kurzrock R, Talpaz M. Philadelphia chromosome-negative chronic myelogenous leukemia and chronic myelomonocytic leukemia. Hematol Oncol Clin North Am 1990; 4: 389.
31. Hasle H, Olesen G, Kerndrup G et al. Chronic neutrophilic leukemia in adolescence and young adulthood. Br J Haematol 1996; 94: 628.
32. Matano S, Nakamura S, Kobayashi K et al. Deletion of the long arm of chromosome 20 in a patient with chronic neutrophilic leukemia: cytogenetic findings in chronic neutrophilic leukemia. Am J Hematol 1997; 54: 72.
33. Yanagisava K, Ohminami H, Sato M et al. Neoplastic involvement of granulocytic-monocytic, or erythrocytic lineage in a patient with chronic neutrophilic leukemia. Am J Hematol 1998; 57: 221.
34. Bain BJ. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br J Haematol 1996; 95: 2.
35. Beutler E. Polycythemia vera. In: Williams Hematology. 5th ed. 1995; 324.
36. Berlin NJ. Diagnosis and classification of the polycythemias. Semin Hematol 1975; 12: 339.
37. Berk PD, Goldberg JD, Donovan PB et al. Therapeutic recommendation in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol 1986; 23: 132.
38. Ellis JT, Peterson P, Geller SA, Rappaport H. Studies of the bone marrow in polycythemia vera and the evolution of myelofibrosis and secondary hematologic malignancies. Semin Hematol 1986; 23: 144.
39. Vykoupil KF, Thiele J, Stangel W et al. Histopathology, ultrastructure and cytogenetics of the bone marrow in comparison with secondary polycythemia. Virchovs Arch A 1980; 389: 307.
40. Landaw SA. Acute leukemia in polycythemia vera. Semin Hematol 1986; 23: 156.
41. Diez-Martin JL, Graham DL, Petitt RM et al. Chromosome studies in 104 patients with polycythemia vera. Mayo Clin Proc 1991; 66: 287.
42. Fialkow PY, Faquet GB, Jacobson RJ et al. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 1981; 58: 916.
43. McIntyre KJ, Hoagland HC, Silverstein MN, Petitt RM. Essential thrombocythemia in young adults. Mayo Clin Proc 1991; 66: 149.
44. Mitus AJ, Schsfer AJ. Thrombocytosis and thrombocythemia. Hematol Oncol Clin North Am 1990; 4: 157.
45. Schafer AJ. Essential (primary) trombocythemia. In: Williams Hematology 5th ed. 1995; 340.
46. Murphy S, Iland H, Rosental D, Laszlo J. Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol 1986; 23: 177.
47. Wintrobes Clinical Haematology, 10th ed. Baltimore: Williams a Wilkins 1999; 2764 p.
48. Lichtman MA. Idiopathic myelofibrosis (agnogenic myeloid metaplasia). In: Williams Hematology 5th ed. 1995; 311.
49. Weinstein IM. Idiopathic myelofibrosis: Historical review, diagnosis and management. Blood Rev 1991; 5: 98.
50. Visani G, Finelli C, Castelli U et al. Myelofibrosis with myeloid metaplasia: clinical and haematological parameters predicting survival in a series of 133 patients. Br J Haematol 1990; 75: 4.
51. Thielle J, Zankovich R, Steinberg T et al. Agnogenic myeloid metaplasia (AMM) — correlation of bone marrow lesions with laboratory data: a longitudinal clinicopathological study of 114 patients. Hematol Oncol 1989; 7: 327.
52. Ward HP, Block MH. The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with myeloproliferative syndrome. Medicine 1971; 50: 357.
53. Beckman EN, Oehrle JS. Fibrous hematopoietic tumors arising in agnogenic myeloid metaplasia. Hum Pathol 1982; 13: 804.
54. Cripe LD, Hromas RW. Malignant disorders of megakaryocytes. Semin Hematol 1998; 35: 200.
55. Passmore SJ, Hann JM, Stiller CA et al. Pediatric myelodysplasia: a study of 68 children with a new prognostic scoring system. Blood 1995; 85: 1742.
56. Bennet JM, Catovsky D, Daniel MT et al. Proposed revised criteria for the classification of acute myeloid leukemia. Ann Intern Med 1985; 103: 626.
57. Neimeyer CM, Arico M, Basso G et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood 1997; 89: 3534.
58. Neimeyer CM, Fenu S, Hasle H et al. Differentiating juvenile myelomonocytic leukemia from infections disease. Blood 1998; 91: 365.
59. Smith KL, Johnson W. Classification of chronic myelocytic leukemia in children. Cancer 1974; 34: 670.
60. Freedman MH, Estrov Z, Chan HS. Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 1988; 10: 261.
61. Arico M, Biondi A, Pui CH. Juvenile myelomonocytic leukemia. Blood 1997; 90: 479.
62. Clark RD, Hutter JJ. Familial neurofibromatosis and juvenile chronic myelogenous leukemia. Hum Genet 1982; 60: 230.
63. Hardisty RM, Speed DE, Till M. Granulocytic leukemia in childhood. Br J Haematol 1964; 10: 551.
Злокачественная кровь. Что собой представляет хронический миелолейкоз? | Здоровая жизнь | Здоровье
Хронический миелолейкоз представляет собой злокачественное образование кроветворной ткани, при котором прогрессирует поражение гранулоцитарного ростка. При этом он может длительное время протекать без каких-либо симптомов.
Гранулоциты — это разновидность лейкоцитов, т. е. белых кровяных телец. Бывают они двух видов: с гранулами и без. Клетки с гранулами ответственны за защиту организма, которая не связана с иммунитетом. Они должны поглощать чужеродные частицы и микроорганизмы, справляются с воспалениями и аллергическими реакциями. Клетки без гранул — лимфоциты, выделяющие антитела и обеспечивающие иммунитет, и моноциты, которые пожирают чужеродные вещества.
Развивается патология на фоне хромосомной мутации, когда поражаются стволовые клетки. На долю такой патологии приходится 9% от общего числа всех лейкозов в разных возрастных группах. Чаще всего такой патологии подвержены люди старше 30 лет, а пик ее диагностирования отмечается в группе 45-55 лет. Дети в возрасте до 10 лет редко оказываются у врача с таким диагнозом.
Специалисты отмечают, что у такой болезни нет какой-то зависимости от пола: она одинаково распространена как у мужчин, так и женщин. Но при этом есть исследования, показывающие, что в числе заболевших много мужчин-европейцев. Нередко патология обнаруживается случайно из-за своего длительного вялотекущего развития: бывает, что подозрение на такой диагноз появляется при сдаче анализа крови.
Течение по фазам
В процессе развития злокачественных клеток выделяют несколько фаз развития патологии. Так, первая — хроническая. Здесь говорят о том, что количество смертельно опасных клеток не более 10%. И человек ощущает себя вполне комфортно, так как у него особо нет никаких симптомов. Максимум — появление чувства утомляемости и слабости. Многие, например, на это не обращают внимания, так как списывают на работу. Иногда на ранних этапах может проявляться чувство тяжести в животе. Такая фаза может быть достаточно длительной, иногда длится до 5 лет. А это значит, что если вдруг такие симптомы проявляются, то стоит посетить врача и обследоваться.
Вторая фаза — акселерация. Тут уже количество патологически опасных клеток увеличивается: их становится 10-19%. Они распространяются в крови и костном мозге. Из-за того, что начинается нехватка тромбоцитов, усугубляется чувство усталости. Кроме того, состояние начинает дополняться симптомами анемии, падают защитные силы организма, из-за чего растут риски более частых заражений инфекционными патологиями.
Следующая фаза называется бластным кризом, здесь ускоряется процесс выработки патологических клеток: их становится больше 20%. По типичным симптомам ситуация напоминает острый миелобластный лейкоз. Тут заболевание начинает прогрессировать очень быстро. И уже начинаются характерные признаки и симптомы: повышенная температура тела, слабость усиливается, отмечается увеличение селезенки. На этом этапе болезнь угрожает жизни человека и не особенно хорошо реагирует на лечение.
Четвертая фаза — ремиссия. О ней говорят в том случае, если речь идет о прошедшем лечении. Если терапия была начата вовремя, пройдена в полном объеме и показала хороший эффект, наступает ремиссия. Снижается число патологических клеток, нормализуется уровень основных клеток крови, нет и клинических признаков патологии.
Почему появляется проблема
Естественно, многих интересует, по какой причине развивается такая патология. Провоцируют такую болезнь нередко следующие факторы:
- Наследственность — как и многие онкологические патологии, ХМЛ может развиваться у членов одной семьи. Особенно растут риски, если заболевание было обнаружено у двух близких родственников.
- Генетика — мутации хромосом могут быть врожденными. Они становятся потенциальным риском для развития болезни.
- Радиационное воздействие.
- Прохождение характерной терапии — лучевой или химио-, ее могут назначать при иных онкологических патологиях.
- Воздействие ряда лекарственных препаратов.
- Ослабленный иммунитет — по данным различных исследований, заметно, что на фоне иммунодефицитных состояний риски развития лейкоза такого рода растут.
- Заболевания кишечника — пока непонятны механизмы, но есть данные исследований, которые показывают, что воспалительные процессы в кишечнике повышают риски развития заболевания.
- Лишний вес — опять же исследования показывают, что люди с лишним весом на 25% сильнее рискуют заболеть такой патологией крови.
Симптоматика проблемы
Симптомы проявления хронического миелолейкоза напрямую зависят от фазы развития заболевания.
Итак, в хронической фазе кроме слабости и усталости могут проявляться снижение веса, повышенная потливость во сне. К числу более редких проявлений относят различные кровотечения, которые связаны с нарушением работы кровяных клеток, проблемы с кровообращением, например появление головных болей, ухудшение памяти и внимания, инфаркт, проблемы со зрением и развитие одышки.
В фазе акселерации все эти проявления лишь нарастают. Стадия бластного криза, она же терминальная, характеризуется появлением таких признаков, как значительное ухудшение самочувствия, длительные боли в костях и суставах, человек начинает потеть, причем его пот называется проливным. Может периодически повышаться температура до высоких значений (до 39 градусов), параллельно развивается сильный озноб. Развивается повышенная кровоточивость. Из-за увеличения селезенки увеличивается в размерах живот.
Определяется патология на осмотре во время сбора диагноза, прощупывании живота и лимфоузлов и на основании данных УЗИ. При необходимости берут пункцию, а также проводят цитозимическое обследование.
Варианты терапии
Специалисты могут назначить прием специальных препаратов. Также в качестве лечения больных хроническим миелолейкозом используется пересадка костного мозга. Предлагают и проведение лучевой терапии, а также удаление селезенки.
Вариант лечения — прерогатива врача, который принимает решение на основе имеющихся данных анализов и обследований, а также состояния человека. Используют и очистку крови. В любом случае решение только за врачом.
Имеются противопоказания, необходимо проконсультироваться с врачом.
МАТЕРИАЛЫ КОНГРЕССОВ И КОНФЕРЕНЦИЙ: VII РОССИЙСКАЯ ОНКОЛОГИЧЕСКАЯ КОНФЕРЕНЦИЯ
VII РОССИЙСКАЯ ОНКОЛОГИЧЕСКАЯ КОНФЕРЕНЦИЯ
50 ЛЕТ СОВРЕМЕННОЙ ТЕРАПИИ ХРОНИЧЕСКОГО МИЕЛОЛЕЙКОЗА
М.А. Волкова
ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России, Москва
В 2003 г. исполняется 50 лет со времени появления в терапии хронического миелолейкоза (ХМЛ) миелосана — первого препарата, реально изменившего картину заболевания.
Хронический миелолейкоз – первый из описанных лейкозов человека. В 1845 г. появились публикации D. Craige, а затем J.Bennett с описанием больных с увеличением селезенки и печени и с большим количеством «гнойных телец» в крови, а выдающийся немецкий патолог R. Virchow представил подробное описание клинико-морфологической картины заболевания, связав воедино изменения крови и внутренних органов.
Первые попытки лечения ХМЛ были предприняты в 1865 г., когда немецкий врач Lissauer сообщил о двух пациентах, у которых удалось добиться уменьшения размеров селезенки и улучшения самочувствия с помощью фовлерова раствора мышьяка. Мышьяк оставался единственным средством лечения ХМЛ до начала XX столетия, но его применение лишь на короткий срок уменьшало симптомы заболевания, не влияя на продолжительность жизни.
Позже делались попытки использования бензола, уретана, радиоактивного фосфора, но все эти средства давали незначительный и кратковременный эффект при очень плохой переносимости.
В 1902 г. W. Pusey впервые для лечения ХМЛ с успехом применил облучение селезенки. Сокращение размеров селезенки и исчезновение дискомфорта, нарушения функции желудочно-кишечного тракта и почек явились причиной того, что до середины ХХ столетия облучение селезенки было единственным методом лечения ХМЛ. Однако эффект всегда был кратковременным, а при повторных курсах терапии не столь полным. Больной ХМЛ в большинстве случаев уже на втором году заболевания терял работоспособность, а к третьему году становился инвалидом, нуждающимся в постороннем уходе и вынужденным большую часть времени проводить в постели.
В 1924 г. появилась работа G. Minot и соавторов, в которой было показано, что средняя продолжительность жизни больных, леченных рентгенотерапией, составила 42 мес., а больных, получавших лишь симптоматическое лечение, – 36,5 мес. В течение десятилетий эта работа цитировалась для подтверждения тезиса о невозможности с помощью имевшихся терапевтических средств увеличить продолжительности жизни больных ХМЛ (1).
Такое положение сохранялось до середины ХХ столетия, несмотря на попытки лечения ХМЛ практически каждым из появлявшихся новых лечебных средств. В 1951 г. M. Shimkin с соавт. опубликовали результаты подробного анализа течения ХМЛ за 38 лет по данным Сан-Франциско. Они показали, что, несмотря на более раннюю диагностику, совершенствование методов рентгенотерапии, применение новых препаратов (мустарген), сопроводительное лечение, медиана продолжительности жизни при ХМЛ составляла 37 мес. Лишь 15% больных доживали до 5-летнего срока (2).
Важнейшим событием в терапии ХМЛ стало появление производного дисульфоновой кислоты алкилирующего действия — препарата, получившего в разных странах различные названия, наиболее известные из которых бусульфан (США), милеран (Великобритания), миелосан (Россия).
Удобный способ применения, прекрасная переносимость, эффективность в хронической стадии практически у всех ранее не леченных больных и у большинства резистентных к рентгенотерапии пациентов, быстро сделали миелосан препаратом выбора при ХМЛ.
Главным результатом применения миелосана стало принципиальное изменение качества жизни больных. При правильной лечебной тактике (начало лечения сразу при постановке диагноза, использование сравнительно небольших доз препарата, постоянная поддерживающая терапия) на протяжении всей хронической стадии больные не чувствовали каких-либо симптомов заболевания, сохраняли трудоспособность, подавляющее большинство продолжали выполнять работу по профессии и вести образ жизни здорового человека. Достигнутые перемены позволили говорить о «миелосановой эре » в терапии ХМЛ.
Миелосан вызвал и определенное увеличение продолжительности жизни больных. В большинстве публикаций сообщалось о медиане выживаемости, равной 40-44 мес., а в отдельных сериях наблюдений она составляла 52-56 мес. Спустя 6 лет после появления миелосана в Великобритании было начато прямое сравнительное рандомизированное исследование эффективности миелосана и рентгенотерапии. Через 3 года были живы 62,5% больных, получавших миелосан, и 33,3% пациентов, получавших рентгенотерапию. Медиана выживаемости составила 30 мес. у больных, получавших рентгенотерапию, и 42 мес. – у получавших миелосан (3). После этого исследования рентгенотерапия постепенно перестала использоваться как средство лечения ХМЛ.
Позднее сравнительные рандомизированные исследования показали явные преимущества миелосана по сравнению с 6-меркаптопурином и циклофосфамидом как по скорости достижения и полноте ремиссий, так и по выживаемости больных.
Тем не менее, при лечении миелосаном через 4-5 лет неизбежно наступал переход заболевания в стадию акселерации, которая гораздо хуже поддавалась терапии и в большинстве случаев через 6-10 мес. заканчивалась бластным кризом, с момента наступления которого больной редко переживал полгода.
Уже с конца 60 гг. ХХ века стали появляться сообщения об эффективности при ХМЛ гидроксимочевины – ингибитора, необходимого для синтеза ДНК фермента рибонуклеотидазы. Миелосан оставался, тем не менее, наиболее широко используемым препаратом до тех пор, пока в 1993 г. не были опубликованы результаты рандомизированного исследования, посвященного сравнению эффективности миелосана и гидроксимочевины. Они показали явные преимущества гидроксимочевины: медиана выживаемости в группе леченных гидроксимочевиной составила 56 мес., достоверно (р=0,01) превысив медиану выживаемости в группе больных, получавших миелосан, (44 мес.). Увеличение продолжительности жизни было достигнуто за счет удлинения хронической стадии болезни, которая при лечении миелосаном составила 37 мес., а гидроксимочевиной – 47 мес. (4). Гидроксимочевина к тому же лишена тех побочных проявлений, которые свойственны миелосану, особенно при длительном применении: аменорея и аспермия, меланодермия, фиброз костного мозга, а при многолетнем использовании — нередко и легких. Эта и последующие работы определили постепенный переход на терапию гидроксимочевиной.
В эти же годы был сделан следующий принципиальный шаг вперед в лечении ХМЛ: доказана эффективность α-интерферона (ИНФ-α). К этому времени уже в основных чертах был расшифрован патогенез ХМЛ. Было доказано, что заболевание возникает в результате соматической мутации в стволовой кроветворной клетке. Результатом этой мутации является реципрокная транслокация между хромосомами 9 и 22 – t(9;22)(q34;q11) с образованием на хромосоме 22 (Ph-хромосоме) химерного гена BCR-ABL, следствием чего является активация ABL-тирозин-киназы, усиленное фосфорилирование тирозина и активация сигнального пути пролиферации в клетке независимо от действия ростовых факторов.
При использовании всех применявшихся до интерферона средств даже у больных с полной клинико-гематологической ремиссией цитогенетическое исследование выявляло Ph-хромосому во всех исследованных клетках, достигалось уменьшение размеров лейкозного клона, но он оставался доминирующим в гемопоэзе.
В 1986 г. было опубликовано первое, ставшее сенсационным, сообщение о том, что при лечении ИНФ-α в хронической стадии ХМЛ удается не только получить гематологические ремиссии у большинства больных, но и почти у половины из них уменьшить процент Ph-позитивных клеток (5). Семь рандомизированных исследований, проведенных в разных странах и включивших суммарно более 2000 больных в хронической стадии ХМЛ, подтвердили эти данные и показали достоверное увеличение продолжительности жизни при лечении ИНФ-α: медиана выживаемости при лечении миелосаном составляла 41-44 мес., гидроксимочевиной – 55-56 мес., интерфероном – 61-72 мес. Было показано, что 5-летняя выживаемость достигает 100% в группе больных с полной цитогенетической ремиссией и 92% — в группе с частичной. Даже у больных с малым цитогенетическим ответом 5-летняя выживаемость составила 59% (6).
В 1993 г. французские авторы предложили для терапии ХМЛ сочетание ИНФ-α с малыми дозами цитозин-арабинозида (Аra-C). Основанием для использования Аra-C послужили данные о том, что в культуре препарат подавляет Ph-позитивные клетки в значительно большей степени, чем соответствующие нормальные клетки-предшественники (7). Рандомизированное исследование, включившее 646 больных в хронической стадии ХМЛ, показало преимущества комбинированной терапии: медиана выживаемости в группе леченных только интерфероном составила 65 мес., при сочетании ИНФ-α с малыми дозами Аra-C – 83 мес. (8). В некоторых публикациях сообщалось о еще более высоких результатах комбинированного лечения. Проведенные на основании мета-анализа расчеты показали, что 10-летняя выживаемость при такой терапии должна составлять 40%, а у больных с полной цитогенетической ремиссией – 80% (9) Сочетание ИНФ-α с малыми дозами Аra-C стало методом выбора в терапии ХМЛ.
У части больных, однако, даже при длительном лечении ИНФ-α не удается получить цитогенетического ответа, а у большинства из тех, у кого он был получен, с течением времени наблюдается постепенное ухудшение достигнутых цитогенетических результатов. Нет данных, которые бы показывали возможность излечения заболевания с помощью интерферона, так как практически у всех больных даже при полной клинико-гематологической и цитогенетической ремиссии с помощью полимеразной цепной реакции удается обнаружить клон BCR-ABL-позитивных клеток (10).
C начала 90-х годов прошлого столетия, когда стало очевидным, что активация ABL-тирозин-киназы является пусковым механизмом развития ХМЛ, началась разработка препарата, обладающего ингибирующим тирозин-киназу эффектом. В результате B. Druker (США) был создан препарат STI 571 (Signal Trunsduction Inhibitor), получивший название иматиниб (imatinib) или Гливек (Glivec, Gleevec). Появление препарата строго направленного патогенетического действия ознаменовало начало новой эры в терапии ХМЛ.
Было показано, что Гливек вызывает дозозависимый эффект подавления роста Ph-позитивных клеток в культуре при почти полном отсутствии воздействия на Ph-негативные клетки. Механизм блокирующего активность ABL-тирозин-киназы действия Гливека сводится к встраиванию молекулы препарата в то место в молекуле ABL-тирозин-киназы, куда обычно встраивается АТФ при фосфорилировании белков, т.е. так называемый АТФ-карман. Встраивание Гливека блокирует возможность связывания тирозин-киназы с АТФ и, тем самым, прекращает фосфорилирование белка, продуцируемого BCR-ABL геном. Это приводит к блоку сигнала пролиферации в BCR-ABL-позитивных клетках, в результате чего они подвергаются апоптозу.
При лечении Гливеком гематологическая ремиссия достигается в хронической стадии болезни у 90–95% больных, у всех полная, у 70% больных в стадии акселерации, у половины полная, и у 30% — при бластном кризе, у половины с возвратом в хроническую стадию болезни (11, 12, 13). При назначении Гливека больным, которые ранее получали терапию интерфероном в течение 12 мес. без цитогенетического ответа, большой цитогенетический ответ достигается в хронической стадии у 55% больных, в стадии акселерации — у 25% и у 16% пациентов при бластном кризе (14, 15, 16).
У ранее не леченных больных в хронической стадии полная гематологическая ремиссия достигается в 97%, большой цитогенетический ответ через 6 мес. в 60% случаев, а через 12 мес. – в 80% случаев, у 40% больных — полная цитогенетическая ремиссия.
Международное рандомизированное сравнительное исследование эффективности Гливека или комбинации ИНФ-α с малыми дозами Аra-C у ранее не леченных больных в хронической стадии ХМЛ, включившее около 1200 больных, показало, что полная гематологическая ремиссия у получавших Гливек достигнута в 97% случаев, ИНФ-α и Аra-C — в 69% случаев, большой цитогенетический ответ получен и сохранился в течение полутора лет у 87% больных, получавших Гливек, и лишь у 35% больных, получавших ИНФ-α и Аra-C, а полная цитогенетическая ремиссия — у 76% леченных Гливеком и всего у 14% леченных ИНФ-α и Аra-C (р<0,001) (17).
Таким образом, очевидно, что Гливек превосходит по эффективности все когда-либо применявшиеся средства терапии ХМЛ: при лечении Гливеком большинство больных ХМЛ имеют шанс прожить 10 лет, в то время как при лечении миелосаном и гидроксимочевиной до этого срока доживали ЛИШЬ 1-5% больных, а при лечении интерфероном – не более половины.
Однако проблему терапии ХМЛ нельзя считать решенной. Часть больных оказывается мало чувствительной к Гливеку с самого начала лечения, у другой части снижение чувствительности появляется спустя некоторое время.
Исследование причин этого показало, что в большинстве случаев при терапии Гливеком достигается восстановление поликлонального кроветворения, однако, с помощью полимеразной цепной реакции все же у большой части больных удается обнаружить BCR-ABL-позитивные клетки (18).
Изучение причин резистентности к Гливеку показало, что у ряда больных с течением времени появляется значительное (до 10 раз) увеличение экспрессии гена BCR-ABL, а в части клеток обнаруживается амплификация гена (иногда определяются 2 гена BCR-ABL, что отражает появление в части клеток второй Ph-хромосомы). В некоторых клетках с течением времени появляются добавочные хромосомные изменения, которые могут быть причиной включения иных, чем активность тирозин-киназы, механизмов клеточной пролиферации или уменьшения апоптоза в связи с выключением генов-супрессоров (например, при изменении длинного плеча хромосомы 17). Частой причиной, выявленной почти у трети больных с приобретенным снижением чувствительности к Гливеку, являются точечные мутации в тирозин-киназном участке BCR-ABL с заменой одной из аминокислот в том домене, где происходит связывание с АТФ, в результате чего нарушается встраивание Гливека. На культуре клеток показано восстановление подавленной лечением активности BCR-ABL после появления точечных мутаций. Поскольку мутации не обнаруживаются до начала лечения Гливеком, очевидно, что Гливек способствует селекции нечувствительных клеток (19).
Кроме того, в некоторых клетках у больных с развившимся снижением чувствительности к Гливеку была обнаружена повышенная продукция ответственного за возникновение множественной лекарственной устойчивости белка Pgp. Наконец, в некоторых случаях в клетках больных, нечувствительных к Гливеку, было обнаружено фосфорилирование помимо BCR-ABL других белков, обеспечивающих независимый от активности BCR-ABL сигнальный путь пролиферации (20).
Выявленные механизмы резистентности показывают, что, во-первых, не следует назначать сниженные дозы Гливека, так как это может способствовать отбору нечувствительных клеток; во-вторых, комбинированная терапия может быть более рациональной с самого начала лечения. Уже существуют исследования, показавшие, что имеется синергизм действия Гливека и Аra-C, ИНФ-α, идарубицина, митоксантрона, этопозида, винкристина, кортикостероидов, сочетанный эффект при комбинации Гливека с миелосаном, дауно- и доксорубицином, флударабином и антагонизм с гидроксимочевиной, метотрексатом, кладрибином (21).
Появились первые публикации, показавшие эффективность комбинированного лечения. Комбинация Гливека со стандартными дозами Ara-C показала быструю редукцию BCR-ABL-позитивного клона, однако, вызывала выраженную миелосупрессию и развитие у многих больных оппортунистических инфекций. В то же время сочетание Гливека с малыми дозами Ara-C обладало значительно меньшей гематологической токсичностью, при этом у всех больных после 3 недель терапии была получена полная гематологическая ремиссия. После 9 мес. лечения большой цитогенетический ответ был получен у 90% больных и полный – у 80% (22).
Пока не ясно, возможно ли добиться истинного излечения с помощью Гливека или его комбинаций с другими препаратами, сколько времени должен лечиться больной после достижения полной цитогенетической и молекулярной ремиссии и не продлевает ли Гливек жизнь даже в случаях развития резистентности к нему. Однако уже сейчас в связи с результатами применения Гливека изменились подходы к трансплантации стволовых кроветворных клеток при ХМЛ.
В настоящее время применяется следующий подход к лечению ХМЛ: больным моложе 45 лет, имеющим HLA–идентичного сиблинга или больным моложе 35 лет, имеющим совместимого не родственного донора, если больной относится к группе хорошего прогноза для исхода трансплантации, должна быть рекомендована трансплантация. Для больных этой группы возможность выздоровления в результате трансплантации превышает риск послеоперационной смертности. Остальным больным следует проводить терапию Гливеком или интерфероном в сочетании с цитозин-арабинозидом в течение 12 мес. Тем из них, у кого получен цитогенетический ответ, терапию указанными препаратами следует продолжать. Тем, у кого нет цитогенетического ответа и имеется HLA-совместимый родственный или не родственный донор, при возрасте пациента до 40 лет и благоприятных для исхода трансплантации признаках, должна также быть предложена трансплантация. Риск смертности для них выше, чем в предыдущей группе, однако, отсутствие цитогенетического ответа после года терапии Гливеком или интерфероном показывает невозможность излечения без трансплантации для этой группы больных. Всем остальным больным проводится терапия Гливеком и/или интерфероном в сочетании с цитозин-арабинозидом.
Список литературы:
1. Minot GR, Bickman TE, Isaacs R. Chronic myelogenous leukemia: Age incidence, duration and benefit derived from irradiation. JAMA 1924, 82, 1489.
2. Shimkin M B, Mettier SR, Bierman HR Myelocytic leukemia: An analysis of incidence distribution and fatality, 1910-1948. Ann Intern Med 1951, 35, 194.
3. Report of the Medical Research Council’s Working Party for Therapeutic Trials in Leukemia: Comparison of radiotherapy and busulphan therapy. Br Med J 1968, 1, 201.
4. Hehlmann R, Heimpel H, Hasford J et al. Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. Blood 1993, 82,398.
5. Talpaz M, Kantarjian H, McCredie K et al. Hematological remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N Engl J Med 1986, 314, 1065.
6. Allan N, Richards S, Shepherd P et al. Medical Research Council randomized multicenter trial of interferon ? n1 for chronic myeloid leukemia. Lancet 1995, 345, 1392.
7. Sokal JE, Leong SS, Gomez GA Preferential inhibition by cytarabine of CFU-GM from patients with chronic granulocytic leukemia. Cancer 1987, 99, 197.
8. Guilhot F, Chastang C, Michallet M et al. Interferon-alfa 2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. N Engl J Med 1997, 337, 223.
9. Selected Proceedings of the First European School of Haematology Course on Chronic Myelogenous Leukemia. Biother Update 1997, 1, 1.
10. Cortes JE, Talpaz M, Kantarjian H. Chronic myelogenous leukemia: a review. Am J Med 1996, 100, 555-570.
11. Kantarjian H, Sawyers C, Hochhaus A et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 2002,346,645.
12. Talpaz M, Silver RT, Druker BJ et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: Results of a phase 2 study. Blood 2002, 99, 1928.
13. Sawyers CL, Hochhaus A, Feldman E et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myeloid leukemia in myeloid blast crisis: Results of a phase II study. Blood 2002, 99, 3530.
14. Druker BJ, Talpaz M, Resta DJ et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001, 344, 1031.
15. Druker BJ, Sawyers CL, Kantarjian H et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001, 344, 1038.
16. Glivec. Clinical Monograph, 2001, Novartis Pharma.
17. O’Brien SG, Guilhot F, Larson RA et al. The IRIS study: International Randomized study of interferon and low-dose Ara-C versus STI571 (imatinib) in patients with newly diagnosed chronic phase chronic myeloid leukemia. N Engl J Med 2003, 348, 994.
18. Hochhaus A., Lahaye T., Kreil S. et al. Interim analysis of imatinib treatment in 300 patients with chronic myelogenous leukemia (CML): evaluation of response and resistance. Program/Proceedings ASCO 2002, v 21, part 1, p 262a, no 1045.
19. Hochhaus A, Kreil S, Corbin AS et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002, 16, 2190.
20. Tipping AJ, Mahon FX, Zafiridis G et al. Drug responses of imatinib mesylate-resistant cells: synergism of imatinib with other chemotherapeutic drugs. Leukemia 2002, 16, 2349.
21. La Rosee P, O’Dwyer ME, Druker BJ. Insights from pre-clinical studies for new combination treatment regimens with the BCR-ABL kinase inhibitor imatinib mesylate (Gleevec/Glivec) in chronic myelogenous leukemia: a translational perspective. Leukemia 2002, 16, 1213.
22. Gardembas M., Rousselot Ph., Tilliez M. et al. Imatinib (Gleevec) and Cytarabine (Ara-C) Is an Effective Regimen in Philadelphia (Ph)-Positive Chronic Myelogenous Leukemia (CML) Chronic Phase (CP) Patients. Blood 2002, 100, p 95a, abstr 351.
Хронический миелогенный лейкоз (ХМЛ): причины, симптомы, лечение
Хронический миелогенный лейкоз (ХМЛ) — это рак, поражающий клетки крови и костный мозг — мягкую часть внутри ваших костей, где образуются клетки крови.
Вы также можете услышать, как ваш врач называет это хроническим миелоидным лейкозом. Это та же болезнь, только другое название.
При лечении вы можете войти в так называемую «ремиссию». Для большинства людей это не означает, что рак полностью исчез, но он менее активен, чем раньше.Вы можете находиться в ремиссии долгие годы.
ХМЛ обычно случается у людей среднего и старшего возраста. Симптомы имеют тенденцию появляться постепенно. Многие из них также могут быть признаками других болезней. Например, вы можете почувствовать усталость, похудеть, когда не пытаетесь сделать это, или иногда у вас может подняться температура.
Болезнь начинается с нарушения в генах клеток крови. Участки двух разных хромосом меняются местами и образуют новую аномальную.
Эта новая хромосома заставляет ваше тело вырабатывать белые кровяные тельца, которые не работают должным образом.Их называют лейкозными клетками, и когда они обнаруживаются в вашем кровотоке, остается меньше места для здоровых клеток крови.
Причины
Большинство людей никогда не узнают, что вызвало у них ХМЛ. Обычно это не происходит от родителей или от инфекций. Ваши привычки к курению и диета, похоже, тоже не повышают ваши шансы на это.
Единственный известный риск — это контакт с высокими уровнями радиации.
Симптомы
ХМЛ имеет три фазы: хроническую, ускоренную и бластную.Ваши симптомы зависят от того, в каком из них вы находитесь.
Хроническая фаза. Это самая ранняя стадия, и ее легче всего лечить. У вас может даже не быть симптомов.
Ускоренная фаза. В этот период количество клеток крови, которые не работают должным образом, увеличивается. У вас с большей вероятностью появятся некоторые из этих симптомов:
- Чувство сильной усталости
- Повышение температуры
- Синяки
- Ночное потоотделение
- Одышка
- Похудеть
- Не чувствовать голод
- Отек или боль в левом боку (что может быть признаком увеличения селезенки)
- Ощущение боли в костях
Другие симптомы могут включать инсульт, изменения в вашем зрении, звон в ушах, вы чувствуете себя так, как будто вы » в оцепенении — длительная эрекция.
Взрывная фаза. Лейкозные клетки размножаются и вытесняют здоровые клетки крови и тромбоциты.
На этом этапе у вас будут более серьезные симптомы, в том числе:
- Инфекции
- Кровотечение
- Кожные изменения, включая бугорки, опухоли
- Распухшие железы
- Боль в костях
Получение диагноза
Если у вас есть симптомы, ваш врач захочет узнать:
- Какие проблемы вы заметили?
- Как долго у вас проявляются симптомы?
- Ваши симптомы приходят и уходят, или они постоянны?
- Что заставляет вас чувствовать себя лучше или хуже?
- Принимаете ли вы какие-либо лекарства?
Ваш врач может провести дополнительные анализы для подтверждения диагноза, например:
Общий анализ крови. Это анализ крови, который проверяет, сколько у вас лейкоцитов, эритроцитов и тромбоцитов.
Исследование костного мозга. Это поможет вам понять, насколько далеко у вас рак. Ваш врач использует иглу, чтобы взять образец, обычно из вашей бедренной кости.
FISH-тест (флуоресцентная гибридизация in situ). Это подробный лабораторный тест ваших генов.
Ультразвук или компьютерная томография. Они могут проверить размер вашей селезенки. Ультразвук использует звуковые волны для создания изображений, которые могут прочитать врачи и другие медицинские работники.КТ — это рентгеновский снимок, который делает серию снимков внутри вашего тела.
Тест полимеразной цепной реакции. Это лабораторный тест, который ищет ген BCR-ABL, который участвует в процессе, который заставляет ваше тело вырабатывать слишком много неправильных белых кровяных телец.
Вопросы для врача
- Вы лечили кого-нибудь с ХМЛ раньше?
- Какие анализы нужно сдать, чтобы подтвердить диагноз?
- На какой стадии ХМЛ я нахожусь?
- Какое лечение вы мне порекомендуете?
- Как я буду себя чувствовать после терапии?
- Что делать, если лечение не работает?
- Как мне найти группу поддержки?
Лечение
Целью вашего лечения является уничтожение лейкозных клеток крови в вашем теле и восстановление здоровых до нормального уровня.Обычно невозможно избавиться от всех плохих ячеек.
Если вы получаете лечение на ранней, хронической стадии ХМЛ, это может помочь предотвратить переход болезни в более серьезную стадию.
Врачи обычно сначала назначают препараты, известные как ингибиторы тирозинкиназы (TKI). Они замедляют скорость, с которой ваше тело вырабатывает лейкозные клетки.
Некоторые широко используемые TKI включают:
Вы можете получить босутиниб (босулиф) и понатиниб (иклуф), если другие препараты не помогают или вызывают у вас сильное заболевание.
Продолжение
Если ваше заболевание продолжает ухудшаться после того, как вы использовали два или более TKI, ваш врач может порекомендовать лекарство под названием омацетаксина мепесукцинат (Synribo).
Другие варианты лечения ХМЛ включают химиотерапию и биологическую терапию, при которой используется лекарство под названием интерферон, которое помогает активизировать действие вашей иммунной системы — защиты вашего организма от микробов.
Пересадка стволовых клеток может вылечить некоторых пациентов. Это сложная процедура, которая обычно выполняется только тогда, когда другие ваши методы лечения не работают.О стволовых клетках много говорят в новостях, но обычно, когда вы слышите о них, они имеют в виду стволовые клетки «эмбриона», которые используются при клонировании. Стволовые клетки при трансплантации стволовых клеток разные. Это клетки, которые живут в вашем костном мозге и помогают создавать новые клетки крови.
Когда вам сделают трансплантацию стволовых клеток, донор предоставит вам новые стволовые клетки. Вам нужно будет попасть в список ожидания, чтобы найти подходящего вам донора, чтобы ваше тело не «отвергало» их.
Близкие родственники, такие как ваш брат или сестра, являются лучшими шансами на удачный брак.Если это не сработает, вам нужно попасть в список потенциальных доноров от незнакомцев. Иногда лучшие шансы на получение правильных стволовых клеток для вас будут от кого-то, кто принадлежит к той же расовой или этнической группе, что и вы.
Самообслуживание
Обязательно сообщайте врачу обо всех принимаемых вами лекарствах. Некоторые из них плохо сочетаются с лечением ХМЛ.
Следуйте плану лечения, назначенному вашим врачом, ешьте здоровую пищу и занимайтесь спортом, когда вам это удобно.
Чего ожидать
ХМЛ часто является медленно растущей формой рака.Хотя от него трудно полностью избавиться, многие люди живут с ним долгую жизнь.
После постановки диагноза вам следует обратиться к гематологу-онкологу, врачу, имеющему специальную подготовку в области болезней крови, особенно рака. Они составят для вас план лечения.
Всегда обращайтесь за консультацией к другому врачу, если чувствуете, что хотите его.
Получение поддержки
Убедитесь, что вы обращаетесь к своей семье и друзьям за необходимой эмоциональной поддержкой.Они могут оказать вам огромную помощь в борьбе с болезнью.
Также полезно поговорить с людьми, у которых есть ХМЛ. Спросите своего врача, как присоединиться к группе поддержки, где вы можете встретить людей, которые проходят через то же самое, что и вы.
Общество лейкемии и лимфомы может помочь вам найти услуги и поддержку. В нем содержится самая свежая информация о лечении и жизни с ХМЛ, включая помощь лицам, осуществляющим уход.
Хронический миелогенный лейкоз (ХМЛ): основы практики, предпосылки, патофизиология
Редакционная коллегия PDQ по лечению взрослых.Лечение хронического миелогенного лейкоза (PDQ®): версия для специалистов в области здравоохранения. 29 июля 2020 г. [Medline]. [Полный текст].
Пилы CL. Хронический миелолейкоз. N Engl J Med . 1999, 29 апреля. 340 (17): 1330-40. [Медлайн].
Druker BJ, Sawyers CL, Kantarjian H, et al. Активность специфического ингибитора тирозинкиназы BCR-ABL при бластном кризе хронического миелолейкоза и острого лимфобластного лейкоза с филадельфийской хромосомой. N Engl J Med . 2001, апрель 5. 344 (14): 1038-42. [Медлайн]. [Полный текст].
Kantarjian H, Sawyers C, Hochhaus A, et al, для Международной исследовательской группы STI571 CML. Гематологические и цитогенетические ответы на мезилат иматиниба при хроническом миелолейкозе. N Engl J Med . 2002 28 февраля. 346 (9): 645-52. [Медлайн]. [Полный текст].
Merx K, Muller MC, Kreil S, et al. Раннее снижение уровней транскрипта мРНК BCR-ABL предсказывает цитогенетический ответ у пациентов с хронической фазой ХМЛ, получавших иматиниб после неэффективности интерферона альфа. Лейкемия . 2002 Сентябрь 16 (9): 1579-83. [Медлайн]. [Полный текст].
Talpaz M, Silver RT, Druker BJ и др. Иматиниб вызывает стойкие гематологические и цитогенетические ответы у пациентов с хроническим миелолейкозом в ускоренной фазе: результаты исследования фазы 2. Кровь . 2002 15 марта. 99 (6): 1928-37. [Медлайн]. [Полный текст].
Kantarjian HM, Cortes JE, O’Brien S, et al. Терапия иматиниба мезилатом у впервые выявленных пациентов с филадельфийской хромосомно-позитивной хронической миелогенной лейкемией: высокая частота ранних полных и основных цитогенетических ответов. Кровь . 2003 г. 1 января. 101 (1): 97-100. [Медлайн]. [Полный текст].
Шах Н.П., Тран С., Ли Ф.Й. и др. Преодоление устойчивости к иматинибу с помощью нового ингибитора киназы ABL. Наука . 2004 г. 16 июля. 305 (5682): 399-401. [Медлайн].
Volpe G, Panuzzo C, Ulisciani S, Cilloni D. Устойчивость к иматинибу при ХМЛ. Раковые письма . 2009 8 февраля. 274 (1): 1-9. [Медлайн].
Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM.Биология хронического миелолейкоза. N Engl J Med . 1999 г. 15 июля. 341 (3): 164-72. [Медлайн].
Статистика рака: лейкемия — хронический миелоидный лейкоз (ХМЛ). Программа надзора, эпидемиологии и конечных результатов Национального института рака. Доступно по адресу https://seer.cancer.gov/statfacts/html/cmyl.html. Дата обращения: 24 августа 2020 г.
Рак в фактах и цифрах, 2020. Американское онкологическое общество. Доступно на https://www.cancer.org / content / dam / Cance-org / research / раковые-факты-и-статистика / годовой-рак-факты-и-цифры / 2020 / раковые-факты-и-цифры-2020.pdf. Дата обращения: 24 августа 2020 г.
Dybko J, Jaźwiec B, Haus O, Urbaniak-Kujda D, Kapelko-Słowik K, Wróbel T, et al. Оценка Хасфорда может предсказать молекулярный ответ у пациентов с хроническим миелоидным лейкозом: опыт одного учреждения. Маркеры Dis . 2016. 2016: 7531472. [Медлайн]. [Полный текст].
Gambacorti-Passerini C, Antolini L, Mahon FX, Guilhot F, Deininger M al et.Многоцентровая независимая оценка исходов у пациентов с хроническим миелолейкозом, получавших иматиниб. Национальный институт рака . 2011, 6 апреля. 103 (7): 553-61. [Медлайн].
Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP и др. Долгосрочные результаты лечения хроническим миелоидным лейкозом иматинибом. N Engl J Med . 2017 9 марта. 376 (10): 917-927. [Медлайн].
Ван В., Кортес Дж. Э., Тан Дж., Хури Дж. Д., Ван С., Буэсо-Рамос С. Е. и др.Стратификация риска хромосомных аномалий при хроническом миелолейкозе в эпоху терапии ингибиторами тирозинкиназы. Кровь . 2016 г. 22 марта. [Medline].
Редакционная коллегия PDQ по лечению взрослых. Лечение хронического миелогенного лейкоза (PDQ®): версия для пациентов. 24 апреля 2020 г. [Medline]. [Полный текст].
«Иматиниб изменил все»: будущее теперь более обнадеживающее. Медицинские новости Medscape. Доступно на http: //www.medscape.com / viewarticle / 876942. 9 марта 2017 г .; Доступ: 10 марта 2017 г.
Islamagic E, Hasic A, Kurtovic S и др. Эффективность дженерика иматиниба в качестве терапии первой и второй линии: трехлетнее наблюдение за пациентами с хроническим миелоидным лейкозом. Клин Лимфома Миелома Лейк . 2017 16 февраля [Полный текст].
Saussele S, et al; Следователи EURO-SKI. Прекращение терапии ингибиторами тирозинкиназы при хроническом миелоидном лейкозе (EURO-SKI): предварительно определенный промежуточный анализ проспективного многоцентрового нерандомизированного исследования. Ланцет Онкол . 2018 июня 19 (6): 747-757. [Медлайн].
[Рекомендации] NCCN Клинические рекомендации по онкологии (Рекомендации NCCN): Хронический миелоидный лейкоз. Национальная всеобъемлющая онкологическая сеть. Доступно на https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf. Версия 1.2021 — 14 августа 2020 г .; Дата обращения: 24 августа 2020 г.
[Рекомендации] Hochhaus A, Saussele S, Rosti G, Mahon FX, Janssen JJWM, Hjorth-Hansen H, et al.Хронический миелоидный лейкоз: Клинические рекомендации ESMO по диагностике, лечению и последующему наблюдению. Энн Онкол . 2018 г., 1. 29 (Приложение 4): iv261. [Медлайн]. [Полный текст].
[Рекомендации] Baccarani M, Deininger MW, Rosti G, et al. Рекомендации European LeukemiaNet по ведению хронического миелоидного лейкоза: 2013 г. Кровь . 2013 8 августа. 122 (6): 872-84. [Медлайн]. [Полный текст].
Барретт А.Дж., Ито С. Роль трансплантации стволовых клеток при хроническом миелолейкозе в 21 веке. Кровь . 2015 21 мая. 125 (21): 3230-5. [Медлайн].
Друкер Б.Дж., Талпаз М., Реста Д.Дж. и др. Эффективность и безопасность специфического ингибитора тирозинкиназы BCR-ABL при хроническом миелолейкозе. N Engl J Med . 2001, 5 апреля. 344 (14): 1031-7. [Медлайн]. [Полный текст].
Джаббур Э., Кантарджян Х. Хронический миелоидный лейкоз: обновленная информация о диагностике, терапии и мониторинге 2020 года. Ам Дж. Гематол . 2020 июн.95 (6): 691-709.[Медлайн].
Santos FP, Alvarado Y, Kantarjian H, Verma D, O’Brien S, Mattiuzzi G, et al. Долгосрочное прогностическое влияние использования эритропоэтических стимуляторов у пациентов с хроническим миелолейкозом в хронической фазе, получавших иматиниб. Рак . 2011 г. 1. 117 (5): 982-91. [Медлайн].
Sawyers CL, Hochhaus A, Feldman E, et al. Иматиниб вызывает гематологические и цитогенетические реакции у пациентов с хроническим миелогенным лейкозом при миелоидном бластном кризе: результаты исследования фазы II. Кровь . 2002 15 мая. 99 (10): 3530-9. [Медлайн].
Тан М., Гонен М., Квинтас-Кардама А. и др. Динамика ответа хронического миелолейкоза на длительную таргетную терапию выявляет лечебные эффекты на лейкозные стволовые клетки. Кровь . 2011 г. 11 августа. 118 (6): 1622–31. [Медлайн].
Ибрагим А.Р., Элиассон Л., Апперли Дж. Ф., Милойкович Д., Буа М., Шидло Р. и др. Плохое соблюдение режима лечения является основной причиной потери CCyR и неэффективности иматиниба у пациентов с хроническим миелолейкозом, получающих длительную терапию. Кровь . 2011, 7 апреля. 117 (14): 3733-6. [Медлайн].
Hochhaus A, Kreil S, Corbin AS, La Rosée P, Müller MC, Lahaye T. и др. Молекулярные и хромосомные механизмы устойчивости к терапии иматинибом (STI571). Лейкемия . 2002 16 ноября (11): 2190-6. [Медлайн].
Marcolino MS, Boersma E, Clementino NC, et al. Продолжительность лечения иматинибом связана со снижением расчетной скорости клубочковой фильтрации у пациентов с хроническим миелолейкозом. Энн Онкол . 2011 Сентябрь 22 (9): 2073-9. [Медлайн].
Дазатиниб. Национальный институт рака. Доступно по адресу https://www.cancer.gov/about-cancer/treatment/drugs/dasatinib. 5 июня 2019 г .; Дата обращения: 24 августа 2020 г.
Нилотиниб. Национальный институт рака. Доступно по адресу https://www.cancer.gov/about-cancer/treatment/drugs/nilotinib. 22 марта 2018 г .; Дата обращения: 24 августа 2020 г.
Босулиф (босутиниб) [вкладыш в упаковке].Нью-Йорк, Нью-Йорк: Pfizer, Inc., июнь 2020 г. Доступно на [Полный текст].
Джаббур Э., Кантарджиан Х., О’Брайен С. и др. Достижение раннего полного цитогенетического ответа является основным определяющим фактором исхода у пациентов с хроническим миелоидным лейкозом в ранней хронической фазе, получавших ингибиторы тирозинкиназы. Кровь . 2011, 27 октября, 118 (17): 4541-6; викторина 4759. [Medline].
Hughes TP, Hochhaus A, Branford S, Müller MC, Kaeda JS, Foroni L, et al.Долгосрочное прогностическое значение раннего молекулярного ответа на иматиниб при впервые диагностированном хроническом миелоидном лейкозе: анализ Международного рандомизированного исследования интерферона и STI571 (IRIS). Кровь . 11 ноября 2010 г. 116 (19): 3758-65. [Медлайн].
FDA. Sprycel (dasatinib): Сообщение о безопасности лекарств — риск легочной артериальной гипертензии. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США. Доступно по адресу https://wayback.archive-it.org/7993/20170112165154/http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm275176.htm. 11 октября 2011 г .; Дата обращения: 24 августа 2020 г.
Пилы CL. Еще лучшие ингибиторы киназ при хроническом миелолейкозе. N Engl J Med . 17 июня 2010 г. 362 (24): 2314-5. [Медлайн].
Джаббур Э., Кантарджиан Х., О’Брайен С., Шан Дж., Гарсия-Манеро Дж., Виерда В. и др. Факторы прогнозирования исхода и ответа у пациентов, получавших ингибиторы тирозинкиназы второго поколения по поводу хронического миелоидного лейкоза в хронической фазе после неэффективности иматиниба. Кровь . 28 октября 2010 г. [Medline].
Verma D, Kantarjian H, Strom SS, et al. Злокачественные новообразования, возникающие во время терапии ингибиторами тирозинкиназы (TKI) при хроническом миелоидном лейкозе (CML) и других гематологических злокачественных новообразованиях. Кровь . 2011 20 октября. 118 (16): 4353-8. [Медлайн].
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M и др. Дазатиниб в сравнении с иматинибом при впервые выявленном хроническом миелолейкозе в хронической фазе. N Engl J Med . 17 июня 2010 г. 362 (24): 2260-70. [Медлайн].
Cortes JE, Jones D, O’Brien S, Jabbour E, Ravandi F, Koller C и др. Результаты терапии дазатинибом у пациентов с хроническим миелолейкозом в ранней хронической фазе. Дж Клин Онкол . 2010 20 января. 28 (3): 398-404. [Медлайн]. [Полный текст].
Mulcahy N. FDA обновляет этикетку на дазатиниб для лечения хронического миелоидного лейкоза. Медицинские новости Medscape . 26 июня 2013 г.[Полный текст].
Джаббур Э., Кантарджиан Х.М., Саглио Дж., Стигманн Дж. Л., Шах Н. П., Боке С. и др. Ранний ответ дазатинибом или иматинибом при хроническом миелолейкозе: трехлетнее наблюдение по результатам рандомизированного исследования фазы 3 (DASISION). Кровь . 2014 23 января 123 (4): 494-500. [Медлайн]. [Полный текст].
Shah NP, Guilhot F, Cortes JE, Schiffer CA, le Coutre P, Brümmendorf TH, et al. Долгосрочные результаты применения дазатиниба после неэффективности иматиниба при хронической фазе хронического миелоидного лейкоза: последующие наблюдения за исследованием фазы 3. Кровь . 2014 25 февраля. [Medline].
Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C и др. Нилотиниб в сравнении с иматинибом при недавно диагностированном хроническом миелоидном лейкозе. N Engl J Med . 2010, 17 июня. 362 (24): 2251-9. [Медлайн].
Kantarjian HM, Hochhaus A, Saglio G, et al. Сравнение нилотиниба и иматиниба при лечении пациентов с впервые диагностированной хронической фазой, филадельфийской хромосомно-положительной хронической миелоидной лейкемией: минимальное наблюдение в течение 24 месяцев рандомизированного исследования фазы 3 ENESTnd. Ланцет Онкол . 2011 Сентябрь 12 (9): 841-51. [Медлайн].
Tasigna (нилотиниб) [вкладыш в упаковке]. Восточный Ганновер, Нью-Джерси 07936: Novartis Pharmaceuticals Corporation. Июнь 2020 г. Доступно в [Полный текст].
Хури HJ, Cortes JE, Kantarjian HM, Gambacorti-Passerini C, Baccarani M, Kim DW, et al. Босутиниб активен при хронической фазе хронического миелоидного лейкоза после неудачной терапии иматинибом и дазатинибом и / или нилотинибом. Кровь .2012 апр. 12, 119 (15): 3403-12. [Медлайн].
Cortes JE, Gambacorti-Passerini C, Deininger MW, Mauro MJ, Chuah C, Kim DW и др. Босутиниб в сравнении с иматинибом при недавно диагностированном хроническом миелоидном лейкозе: результаты рандомизированного исследования BFORE. Дж Клин Онкол . 2017 г. 1 ноября. JCO2017747162. [Медлайн]. [Полный текст].
Chustecka Z. FDA одобрило Понатиниб для лечения редких лейкозов. Медицинские новости Medscape. 14 декабря 2012 г. [Полный текст].
Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I., et al.Понатиниб при резистентных филадельфийских хромосомных лейкозах. N Engl J Med . 2012 29 ноября. 367 (22): 2075-88. [Медлайн].
Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, et al. Испытание фазы 2 понатиниба при лейкозах с положительными хромосомами в Филадельфии. N Engl J Med . 2013, 7 ноября. 369 (19): 1783-96. [Медлайн].
Сообщение FDA по безопасности лекарств. FDA просит производителя препарата Iclusig (понатиниб) от лейкемии приостановить маркетинг и продажи.Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США. Доступно по адресу https://wayback.archive-it.org/7993/20170722185758/https://www.fda.gov/Drugs/DrugSafety/ucm373040.htm. 5 ноября 2013 г .; Дата обращения: 24 августа 2020 г.
Сообщение FDA по безопасности лекарств: FDA требует нескольких новых мер безопасности для препарата Iclusig от лейкемии; Компания ожидает возобновления маркетинга. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США. Доступно по адресу https://wayback.archive-it.org/7993/20170722185750/https://www.fda.gov/Drugs/DrugSafety/ucm379554.htm. 20 декабря 2013 г .; Дата обращения: 24 августа 2020 г.
Saussele S, Haverkamp W, Lang F, Koschmieder S, Kiani A, Jentsch-Ullrich K, et al. Понатиниб в лечении хронического миелоидного лейкоза и острого лейкоза, положительного по хромосомам Филадельфии: рекомендации немецкой группы экспертов с особым вниманием к сердечно-сосудистым заболеваниям. Acta Haematol . 2020. 143 (3): 217-231. [Медлайн]. [Полный текст].
Синрибо (омацетаксин) [вставка в упаковку].Северный Уэльс, Пенсильвания: Teva Pharmaceuticals USA, Inc., октябрь 2012 г. Доступно на [Полный текст].
Симонссон Б., Гедде-Даль Т., Маркеварн Б. и др. Комбинация пегилированного IFN-a2b с иматинибом увеличивает частоту молекулярного ответа у пациентов с хроническим миелоидным лейкозом низкого или среднего риска. Кровь . 2011 22 сентября. 118 (12): 3228-35. [Медлайн].
Морб Дж., Джонсон Т., Кубилис П., Майерс Л., Облон Д., Миллер А. и др. Повышение выживаемости пациентов с хроническим миелолейкозом, перенесших аллогенную трансплантацию костного мозга. Ам Дж. Гематол . 1995 Декабрь 50 (4): 304-6. [Медлайн].
МакГлав ПБ, Битти П., Эш Р., Хоус Дж. М.. Терапия хронического миелолейкоза трансплантацией неродственного донорского костного мозга: результат 102 случая. Кровь . 1990, 15 апреля. 75 (8): 1728-32. [Медлайн].
Deininger M, Schleuning M, Greinix H, Sayer HG, Fischer T., Martinez J, et al. Влияние предшествующего воздействия иматиниба на смертность, связанную с трансплантацией. Haematologica .2006 апр. 91 (4): 452-9. [Медлайн].
Лима Л., Бернал-Мизрахи Л., Саксе Д., Манн К.П., Тигиуарт М., Ареллано М. и др. Мониторинг периферической крови при хроническом миелоидном лейкозе во время лечения иматинибом, препаратами второго ряда и другими препаратами. Рак . 2011 15 марта. 117 (6): 1245-52. [Медлайн].
Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F и др. Развивающиеся концепции лечения хронического миелоидного лейкоза: рекомендации группы экспертов от имени European LeukemiaNet. Кровь . 2006 15 сентября. 108 (6): 1809-20. [Медлайн]. [Полный текст].
Бранфорд С., Рудски З., Харпер А., Григг А., Тейлор К., Даррант С. и др. Иматиниб дает значительно лучшие молекулярные ответы по сравнению с интерфероном альфа плюс цитарабин у пациентов с впервые диагностированным хроническим миелоидным лейкозом в хронической фазе. Лейкемия . 2003 17 декабря (12): 2401-9. [Медлайн].
Кортес Дж., Талпаз М., О’Брайен С., Джонс Д., Лутра Р., Шан Дж. И др.Молекулярные ответы у пациентов с хроническим миелолейкозом в хронической фазе, получавших мезилат иматиниба. Clin Cancer Res . 2005 1 мая. 11 (9): 3425-32. [Медлайн]. [Полный текст].
Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Обнаружение мутаций BCR-ABL у пациентов с ХМЛ, получавших иматиниб, практически всегда сопровождается клинической резистентностью, а мутации в АТФ-фосфат-связывающей петле (P-петле) связаны с плохим прогнозом. Кровь . 1 июля 2003 г. 102 (1): 276-83. [Медлайн]. [Полный текст].
Press RD, Galderisi C, Yang R, Rempfer C, Willis SG, Mauro MJ, et al. Половина логарифмического увеличения BCR-ABL РНК предсказывает более высокий риск рецидива у пациентов с хроническим миелоидным лейкозом с индуцированным иматинибом полным цитогенетическим ответом. Clin Cancer Res . 2007 15 октября. 13 (20): 6136-43. [Медлайн]. [Полный текст].
Saglio G, Sharf G, Almeida A, Bogdanovic A, Bombaci F, Čugurović J, et al.Соображения по поводу ремиссии без лечения у пациентов с хроническим миелоидным лейкозом: совместная перспектива пациента и врача. Клин Лимфома Миелома Лейк . 2018 июн.18 (6): 375-379. [Медлайн]. [Полный текст].
Sweet K, Oehler V. Прекращение приема ингибиторов тирозинкиназы при хроническом миелолейкозе: когда это безопасный вариант для рассмотрения ?. Hematology Am Soc Hematol Educational Program . 2013. 2013: 184-8. [Медлайн]. [Полный текст].
Мулахи Н.Лекарство от лейкемии Понатиниб (Иклусиг) снято с рынка. Медицинские новости Medscape. Доступно на http://www.medscape.com/viewarticle/813531. 31 октября 2013 г .; Дата обращения: 24 августа 2020 г.
Нельсон Р. Понатиниб (Iclusig) возвращается на рынок с некоторыми оговорками. Медицинские новости Medscape. Доступно на http://www.medscape.com/viewarticle/818183?src=wnl_edit_specol&uac=72886PK. 20 декабря 2013 г .; Дата обращения: 24 августа 2020 г.
Хронический миелоидный лейкоз | Типы лейкемии
Также известный как хронический миелолейкоз, хронический миелоидный лейкоз (ХМЛ) — это форма рака, поражающая костный мозг и кровь.Он начинается в кроветворных клетках костного мозга, а затем со временем распространяется в кровь. Со временем болезнь распространяется на другие части тела.
Как правило, классификация как хроническая означает, что этот тип лейкемии распространяется и растет медленно. Однако ХМЛ может превратиться из медленного прогрессирования в быстрорастущую острую форму лейкемии, которая может распространиться практически на любой орган тела.
В отличие от трех других основных типов лейкемии, ХМЛ имеет существенное отличие, которое отличает его от остальных.Было показано, что CML связан с аномальной хромосомой, известной как филадельфийская хромосома (Ph-хромосома).
Хромосомы — это структуры в клетках, которые содержат гены, которые дают инструкции клеткам. Ph-хромосома — это аномалия, которая возникает, когда часть хромосомы 22 отрывается и прикрепляется к концу хромосомы 9, которая также отрывается и прикрепляется к хромосоме 22.
Разрывы в обеих хромосомах вызывают появление генов BCR и ABL, которые вместе создают ген рака.Связь между Ph-хромосомой и ХМЛ была обнаружена примерно в 1960 году.
Симптомы хронического миелоидного лейкоза
Симптомы ХМЛ включают:
- Легкое кровотечение
- Необъяснимая потеря веса
- лихорадка
- Потеря аппетита
- Ночные поты
- Бледная кожа
Варианты лечения хронического миелоидного лейкоза
Лечение ХМЛ может включать лучевую терапию, химиотерапию, трансплантацию стволовых клеток и / или иммунотерапию.Ваша интегрированная команда экспертов по лейкемии ответит на ваши вопросы и порекомендует варианты лечения, основанные на вашем уникальном диагнозе и потребностях.
Обычным химиотерапевтическим лечением хронических лейкозов является пероральная химиотерапия. Пациенты с ХМЛ могут получать Gleevec ® (иматиниб), Sprycel ® (дазатиниб) и Tasigna ® (нилотиниб).
Следующая тема: Что такое волосатоклеточный лейкоз?
Хронический миелоидный лейкоз (ХМЛ) — Фонд лейкемии
Что такое ХМЛ?
Хронический миелоидный лейкоз (ХМЛ) — это тип рака, поражающий кровь и костный мозг.При ХМЛ костный мозг производит слишком много белых клеток, называемых гранулоцитами. Эти клетки (иногда называемые бластами или лейкемическими бластами) постепенно заполняют костный мозг, нарушая нормальное производство клеток крови. Они также выходят из костного мозга и циркулируют по телу с кровотоком. Поскольку они не полностью созрели, они не могут правильно бороться с инфекциями. Со временем нехватка эритроцитов и тромбоцитов может вызвать анемию, кровотечение и / или синяки.
ХМЛ обычно развивается постепенно на ранних стадиях заболевания и медленно прогрессирует в течение недель или месяцев.Он состоит из трех фаз:
- хроническая фаза
- ускоренная фаза
- взрывная фаза.
Хроническая фаза
У большинства людей (более 90%) диагностируется ранняя хроническая фаза ХМЛ. Показатели крови остаются относительно стабильными, а доля бластных клеток в костном мозге и крови низкая (пять процентов или меньше). Большинство людей, как правило, чувствуют себя хорошо на этой стадии, и у них практически отсутствуют тревожные симптомы болезни.
Для большинства пациентов, получающих текущее лечение, достигается отличный ответ.Это называется основной молекулярной ремиссией (MMR). Пациенты, достигшие MMR, имеют нормальную продолжительность жизни.
При отсутствии лечения хроническая фаза ХМЛ в конечном итоге переходит в ускоренную и / или бластную фазу.
Ускоренная фаза
Примерно у 5% пациентов CML прогрессирует из относительно стабильного заболевания в более быстро прогрессирующее. Это известно как ускоренная фаза ХМЛ. В это время в костном мозге и циркулирующей крови может начать увеличиваться доля бластных клеток.
Во время ускоренной фазы болезни ваша лечащая бригада рассмотрит возможность перехода на более эффективный вариант лечения.
Если не лечить, ускоренная фаза ХМЛ в конечном итоге превратится в ХМЛ взрывной фазы.
Взрывная фаза
В целом для пациентов с ХМЛ риск трансформации в быстро прогрессирующее заболевание, напоминающее острый лейкоз, составляет менее 5%. Этот риск составляет менее 1% у тех, кто имеет отличный ответ на текущую медикаментозную терапию.Это известно как фаза взрыва или кризис взрыва.
Он характеризуется резким увеличением количества бластных клеток в костном мозге и крови (обычно 30% и более) и развитием более серьезных симптомов вашего заболевания.
При бластном кризе примерно в двух третях случаев ХМЛ трансформируется в заболевание, напоминающее острый миелоидный лейкоз (ОМЛ). Остальное трансформируется в заболевание, напоминающее острый лимфобластный лейкоз (ОЛЛ). Иногда говорят, что бластные клетки недифференцированы или смешаны.
Насколько распространен ХМЛ?
Ежегодно в Австралии около 330 человек заболевают ХМЛ. В целом ХМЛ — редкое заболевание, на которое приходится около 0,03% всех диагностированных онкологических заболеваний.
Кто заболевает ХМЛ?
ХМЛ может возникнуть в любом возрасте, но чаще встречается у взрослых старше 40 лет, на которые приходится почти 70% всех случаев. ХМЛ чаще встречается у мужчин, чем у женщин, и редко диагностируется у детей.
Что вызывает ХМЛ?
Мы знаем, что CML не передается по наследству (передается от поколения к поколению) и не заразен.Считается, что, как и другие типы лейкемии, ХМЛ возникает в результате приобретенной мутации (или изменения) в одном или нескольких генах , , которые обычно контролируют рост и развитие клеток крови. Это изменение или изменения приведут к ненормальному росту.
У большинства людей с диагнозом ХМЛ имеется генетическая аномалия в клетках крови, называемая филадельфийской (Ph) хромосомой. Ph-хромосома вызывает выработку фермента под названием тирозинкиназа , который приводит к CML. Почему эти мутации возникают в первую очередь, остается неизвестным, но, вероятно, это связано с рядом факторов.Все время ведутся исследования возможных причин этого ущерба, и были выявлены определенные факторы, может, , подвергнуть некоторых людей повышенному риску. К ним относятся: воздействие:
- радиация — очень высокие дозы радиации, случайно (ядерная авария) или терапевтически (для лечения других видов рака)
- химических веществ — воздействие промышленных химикатов, таких как бензол, в течение длительного периода времени или воздействие определенных типов химиотерапии для лечения других видов рака.
Каковы симптомы ХМЛ?
Поскольку ХМЛ развивается медленно, у многих людей нет никаких симптомов (особенно на ранних стадиях), и болезнь выявляется при обычном анализе крови.
По мере прогрессирования болезни симптомы возникают из-за увеличения количества аномальных клеток крови в костном мозге и крови и уменьшения количества нормальных клеток крови. Возможные симптомы могут включать:
- анемия, вызванная недостатком эритроцитов и вызывающая стойкую усталость, головокружение, бледность или одышку при физической активности
- усиление или необъяснимое кровотечение или синяк из-за очень низкого количества тромбоцитов
- частые или повторяющиеся инфекции и медленное заживление из-за отсутствия нормальных лейкоцитов
- Боль или дискомфорт под ребрами с левой стороны из-за увеличения селезенки
- чрезмерное потоотделение или непреднамеренная потеря веса.
Некоторые из описанных выше симптомов также могут наблюдаться при других заболеваниях, включая вирусные инфекции. Следовательно, у большинства людей с этими симптомами нет лейкемии. Однако важно обратиться к врачу, если у вас есть какие-либо необычные симптомы или симптомы, которые сохраняются намного дольше, чем ожидалось, чтобы вас можно было должным образом обследовать.
Последнее обновление 28 октября 2020 г.
Разработано Фондом лейкемии при консультациях с людьми, живущими с раком крови, вспомогательным персоналом Фонда лейкемии, гематологическим медперсоналом и / или австралийскими клиническими гематологами.Этот контент предоставляется только в информационных целях, и мы настоятельно рекомендуем вам всегда обращаться за советом к зарегистрированному специалисту в области здравоохранения для диагностики, лечения и ответов на ваши медицинские вопросы, включая пригодность конкретной терапии, услуги, продукта или лечения в ваших обстоятельствах. Фонд Leukemia Foundation не несет никакой ответственности ни перед кем, полагающимся на материалы, содержащиеся на этом сайте.
Лейкемия — Хронический миелоид — ХМЛ: Введение
НА ЭТОЙ СТРАНИЦЕ: Вы найдете основную информацию об этом заболевании и частях тела, на которые оно может повлиять.Это первая страница руководства Cancer.Net по хроническому миелоидному лейкозу. Используйте меню для просмотра других страниц. Думайте об этом меню как о дорожной карте для этого полного руководства.
О лейкемии
Лейкоз — это рак крови. Лейкемия начинается, когда здоровые клетки крови изменяются и выходят из-под контроля. Хронический миелоидный лейкоз (ХМЛ) — это рак кроветворных клеток, называемых миелоидными клетками, обнаруженный в костном мозге. Костный мозг — это губчатая ткань красного цвета во внутренней части крупных костей.ХМЛ чаще всего вызывает увеличение количества лейкоцитов, таких как нейтрофилы или гранулоциты, которые обычно борются с инфекцией. Его также иногда называют хроническим гранулоцитарным, хроническим миелоцитарным или хроническим миелолейкозом.
О филадельфийской хромосоме
Люди с ХМЛ имеют генетическую мутацию или изменение в клетках костного мозга. Это называется транслокацией. Транслокация — это когда часть длинной цепи генов, называемой хромосомой, отрывается и присоединяется к другой хромосоме.При ХМЛ часть хромосомы 9 отрывается и связывается с участком хромосомы 22, образуя филадельфийскую хромосому или Ph-хромосому. Хромосома Ph состоит из 2 генов, называемых BCR и ABL , которые объединяются в один гибридный ген, называемый BCR-ABL . Он находится только в кроветворных клетках, а не в других органах тела. Ген BCR-ABL заставляет миелоидные клетки вырабатывать аномально активированный фермент. В частности, это фермент тирозинкиназа.Этот аномально активированный фермент называется гибридным белком и позволяет лейкоцитам бесконтрольно расти.
Это генетическое изменение возникает в результате случайного повреждения после рождения человека. Нет никакого риска, что человек передаст этот ген своим детям.
О CML
Обычно количество лейкоцитов строго контролируется организмом — во время инфекций или стресса вырабатывается больше лейкоцитов, но затем числа возвращаются к норме, когда инфекция излечивается.При ХМЛ аномальный фермент BCR-ABL подобен переключателю, который застрял в положении «включено» — он продолжает стимулировать рост и размножение лейкоцитов. Помимо увеличения количества лейкоцитов, часто увеличивается количество тромбоцитов, которые способствуют свертыванию крови. И количество красных кровяных телец, переносящих кислород, может уменьшиться.
Ищете больше введения?
Если вы хотите получить более подробную информацию, изучите эти связанные элементы. Обратите внимание, что по этим ссылкам вы попадете в другие разделы Рака.Сеть:
Следующий раздел в этом руководстве — Статистика . Это помогает объяснить количество людей, у которых диагностирован ХМЛ, и общие показатели выживаемости. Используйте меню, чтобы выбрать другой раздел для чтения в этом руководстве.
Хронический миелоидный лейкоз — NHS
Хронический миелоидный лейкоз (ХМЛ) — это тип рака, который поражает лейкоциты и имеет тенденцию медленно прогрессировать в течение многих лет.
Это может произойти в любом возрасте, но чаще всего встречается у пожилых людей в возрасте около 60-65 лет.
При ХМЛ губчатый материал внутри некоторых костей (костный мозг) производит слишком много миелоидных клеток — незрелых лейкоцитов, которые не полностью развиты и не работают должным образом.
ХМЛ отличается от других типов лейкемии, включая хронический лимфолейкоз, острый миелоидный лейкоз и острый лимфобластный лейкоз.
Информация:
Консультации по коронавирусу
Получите консультацию о коронавирусе и раке:
Симптомы ХМЛ
ХМЛ обычно не вызывает никаких симптомов на ранних стадиях и может быть обнаружен только во время тестов, проводимых по другой причине.
По мере развития состояния симптомы могут включать:
- усталость
- потеря веса
- ночная потливость
- болезненность и припухлость в левой части живота
- чувство сытости после небольших приемов пищи
- бледность кожи и одышка
- при высокой температуре
- легко появляются синяки и кровотечения
- частые инфекции
- боли в костях
Когда обращаться за медицинской помощью
Обратитесь к терапевту, если у вас есть какие-либо постоянные симптомы, которые вас беспокоят.
У этих типов симптомов может быть много разных причин, поэтому маловероятно, что у вас ХМЛ, но рекомендуется проверить их.
Ваш терапевт может организовать анализ крови, чтобы определить возможные причины ваших симптомов. Если это обнаружит проблему, вас могут направить к специалисту больницы для дальнейших анализов.
Подробнее о диагностике ХМЛ.
Лечение ХМЛ
Лечение ХМЛ обычно начинают сразу, чтобы замедлить его прогрессирование и держать его под контролем.
Основное лечение ХМЛ — это лекарства, называемые ингибиторами тирозинкиназы, которые останавливают рост и размножение раковых клеток. Они могут помочь держать под контролем ХМЛ, если принимать их на всю жизнь.
Эти лекарства включают:
- таблеток иматиниба
- капсул нилотиниба
- таблеток дазатиниба
- таблеток босутиниба
Для проверки эффективности лекарства будут проводиться регулярные анализы крови.
Иногда можно сделать пересадку стволовых клеток.Стволовые клетки — это клетки, которые продолжают формировать другие типы клеток. В этом случае пересаживаются стволовые клетки из костного мозга, которые могут производить здоровые лейкоциты.
Трансплантация стволовых клеток потенциально может вылечить ХМЛ, хотя это очень интенсивное лечение и в большинстве случаев не подходит.
Подробнее о лечении ХМЛ.
Перспективы ХМЛ
ХМЛ — серьезное и опасное для жизни состояние, но с введением новых ингибиторов тирозинкиназы сейчас перспективы намного лучше, чем раньше.
По оценкам, около 70% мужчин и 75% женщин будут жить не менее 5 лет после постановки диагноза. Молодые люди, как правило, имеют лучшее мировоззрение, чем люди старшего возраста.
Чем раньше диагностируется ХМЛ, тем лучше прогноз.
Причины CML
CML вызывается генетическим изменением (мутацией) стволовых клеток, продуцируемых костным мозгом.
Мутация заставляет стволовые клетки производить слишком много недоразвитых лейкоцитов.Это также приводит к уменьшению количества других кровяных телец, таких как эритроциты.
Изменение касается связок ДНК, называемых хромосомами. Внутри каждой стволовой клетки участок ДНК одной хромосомы заменяется участком другой. Это изменение известно как «филадельфийская хромосома». Узнайте больше о генах и хромосомах.
Неизвестно, почему это происходит, но это не то, с чем вы родились, и вы не можете передать это своим детям.
Группы поддержки и благотворительные организации
Жить с тяжелым хроническим заболеванием, таким как ХМЛ, может быть очень сложно.
Возможно, вам будет полезно узнать как можно больше об этом заболевании и поговорить с другими людьми, страдающими от него.
Следующие группы поддержки и благотворительные организации могут предложить помощь и советы людям с ХМЛ, их семьям и опекунам:
Macmillan Cancer Support и Cancer Research UK также предоставляют информацию и поддержку по ХМЛ.
Последняя проверка страницы: 18 сентября 2019 г.
Срок следующей проверки: 18 сентября 2022 г.
Хронический миелогенный лейкоз — NORD (Национальная организация по редким заболеваниям)
Лечение
Ингибиторы тирозинкиназы (TKI) являются передовой терапией при ХМЛ.
В 2002 году FDA одобрило мезилат иматиниба (Gleevec) в качестве терапевтического средства при ХМЛ. Гливек использует метод, известный как молекулярное нацеливание, чтобы блокировать действие протеинтирозинкиназы, которая, как считается, ответственна за большинство случаев ХМЛ. Поскольку оно нацелено на конкретную причину заболевания, лечение не влияет на здоровые ткани и считается более легким для пациентов, чем другие формы лечения, такие как инъекции интерферона, химиотерапия и трансплантация костного мозга. Гливек производится Novartis Pharmaceuticals Corporation.
Другие препараты, недавно одобренные FDA, — это дазатиниб (Sprycel) и нилотиниб (Tasigna). Они действуют аналогично Гливеку, блокируя тирозинкиназу.
В июне 2010 г. Tasigna был одобрен FDA для лечения ХМЛ при первоначальном диагнозе. Tasigna производится Novartis Pharmaceuticals Corporation.
Sprycel был одобрен FDA в октябре 2010 года для лечения ХМЛ, когда другие препараты, такие как Гливек, оказались неэффективными. Sprycel производится Bristol Myers Squibb.
Босулиф (босутиниб) был одобрен FDA в 2012 году для лечения пациентов с хронической, ускоренной или бластной фазой ХМЛ с положительной филадельфийской хромосомой, которые устойчивы к другим видам лечения или которые не переносят их. Босулиф работает, блокируя сигнал тирозинкиназы, которая способствует развитию аномальных и нездоровых гранулоцитов. Босулиф производится компанией Pfizer.
Synribo (омацетаксина мепесукцинат) был одобрен FDA в 2012 году в рамках ускоренной программы одобрения для лечения взрослых с ХМЛ.Это новый вариант лечения для пациентов, которые устойчивы к другим одобренным FDA лекарствам или не переносят их при хронической или ускоренной фазе ХМЛ. Synribo блокирует определенные белки, которые способствуют развитию раковых клеток. Synribo производится Teva Pharmaceuticals.
Для лечения ХМЛ ранее использовались следующие методы лечения:
Орфанный препарат Идарубицин HCI для инъекций (Идамицин) был одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) в 1990 году для лечения ХМЛ.
Интерферон альфа-2а (роферон А), вводимый в виде инъекций, получил одобрение FDA для лечения ХМЛ в 1995 году.
Лекарства, подавляющие активность костного мозга (миелосупрессивные препараты), могут замедлить прогрессирование заболевания. Гидроксимочевина, лекарство, используемое для лечения пациентов с ХМЛ, может снизить количество лейкоцитов и, следовательно, уменьшить симптомы.
Лучевая терапия селезенки — еще один вариант лечения, применяемый лишь в относительно небольшом количестве неконтролируемых случаев, отдельно или в сочетании с химиотерапией, для замедления прогрессирования заболевания.