МКБ-10 код Q87.4 | Синдром Марфана
ICD-10
ICD-10 is the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD), a medical classification list by the World Health Organization (WHO).
It contains codes for diseases, signs and symptoms, abnormal findings, complaints, social circumstances, and external causes of injury or diseases.
ATC
The Anatomical Therapeutic Chemical (ATC) Classification System is used for the classification of active ingredients of drugs according to the organ or system on which they act and their therapeutic, pharmacological and chemical properties.
It is controlled by the World Health Organization Collaborating Centre for Drug Statistics Methodology (WHOCC).
DDD
The defined daily dose (DDD) is a statistical measure of drug consumption, defined by the World Health Organization (WHO).
It is used to standardize the comparison of drug usage between different drugs or between different health care environments.
Синдром Марфана — Вики
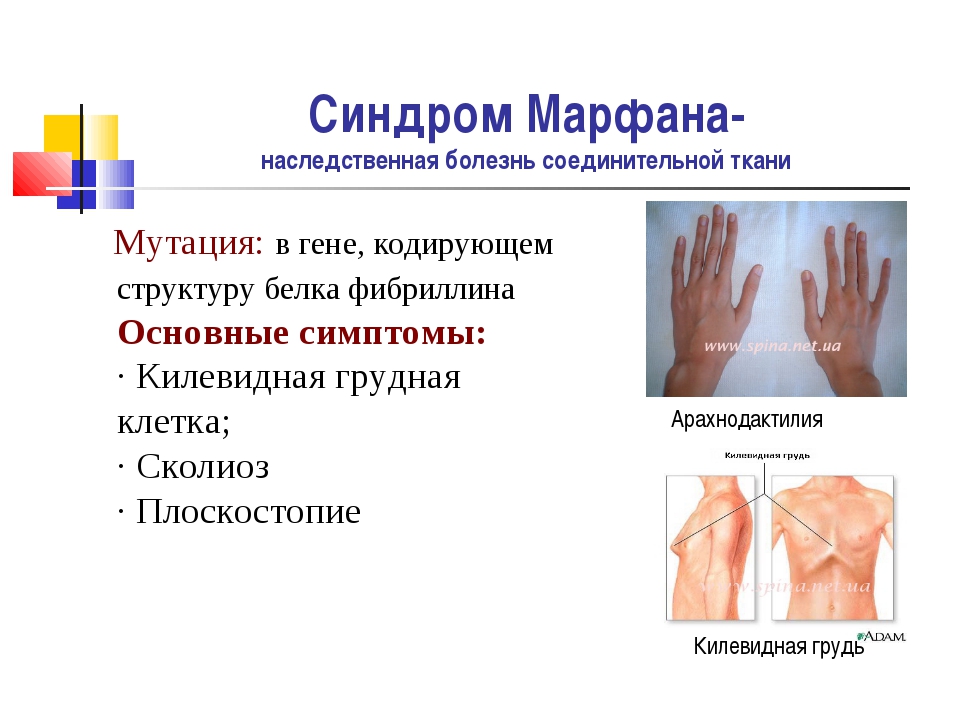
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани.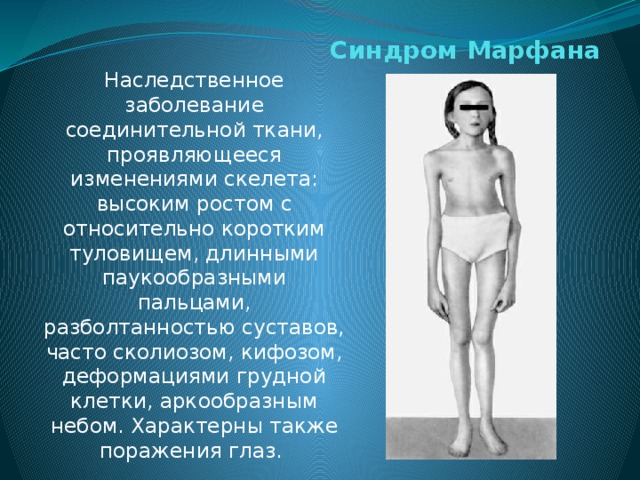 Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[2], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Наследственное заболевание.
Эпидемиология
Синдром Марфана — редкое заболевание с классическим менделевским наследованием. Распространённость в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах.
История
Впервые признаки заболевания были описаны в 1875 году американским офтальмологом Э. Вильямсом (англ. E. Williams), описавшим эктопию хрусталика у брата и сестры, которые были исключительно высокими и имели гипермобильные суставы от рождения[3]. В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном?!, который представил в 1896 году клиническое наблюдение 5-летней девочки Габриэль с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.[4]
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.[5]
Американский генетик Виктор Маккьюсик[en] открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани.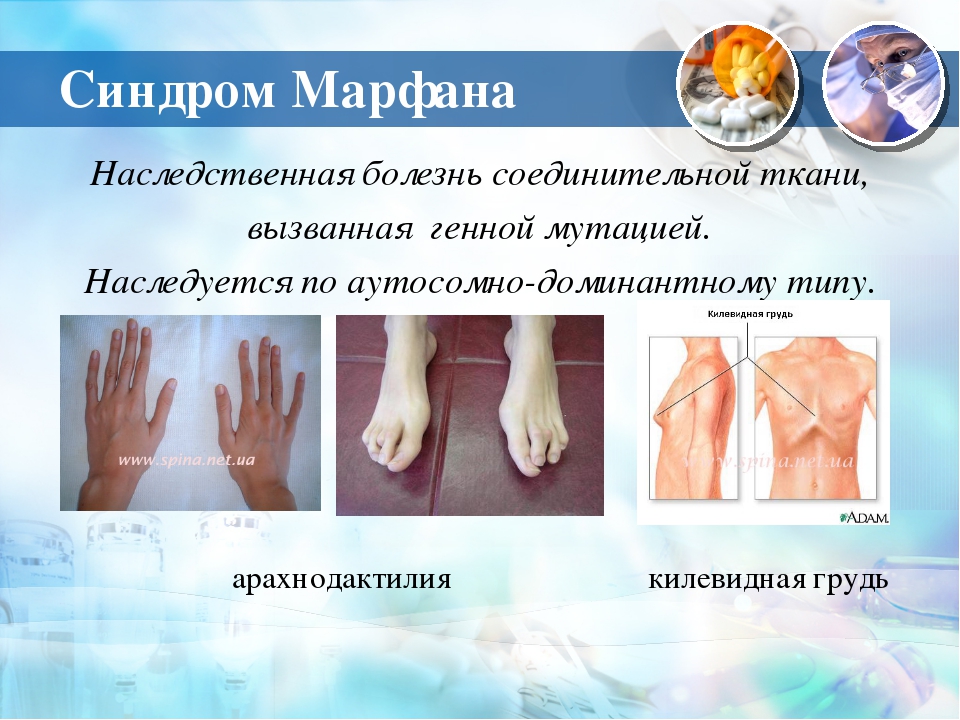 [6]
[6]
Симптомы
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.[7]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
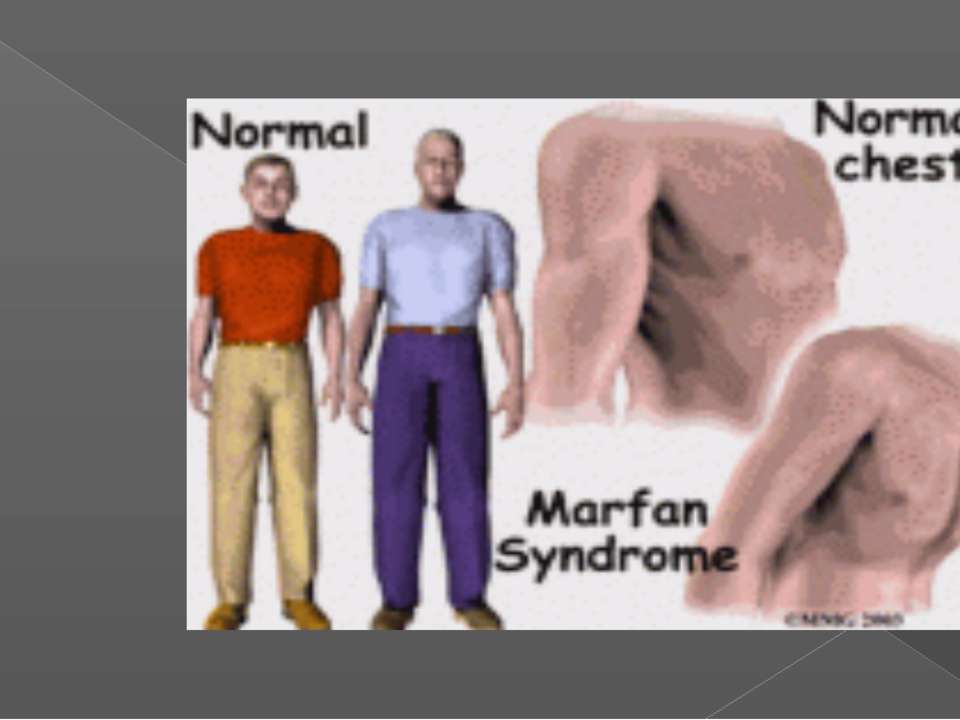
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Лечение
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.[8]
Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.[8]
Интересные факты
См. также
Примечания
- ↑ база данных Disease ontology (англ.) — 2016.
- ↑ Синдром Марфана (неопр.). Первый Московский Государственный Университет имени И.М. Сечнова (19 октября 2007). — Течение и прогноз. Дата обращения: 23 августа 2012. Архивировано 18 октября 2012 года.
- ↑ Williams E. Rare Cases, with Practical Remarks // Transactions of the American Ophthalmological Societ. — 1875. — Vol. 2. — PP. 291—301. — PMC 1361735
- ↑ Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- ↑ Hecht F., Beals R. K. «New» syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. // Pediatrics. — 1972 Apr. — Vol. 49(4). — PP. 574—579. — PMID 4552107
- ↑ McKusick V. A. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. // Circulation[en]. — 1955 Mar. — Vol. 11(3). — PP. 321—341. — PMID 14352380
- ↑ Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- ↑ Goldman’s Cecil Medicine 978-1-4557-1167-3,
Литература
- Лисиченко О. В. Синдром Марфана. Новосибирск: Наука, 1986. 164 с.
Ссылки
На русском языке
На английском языке
Терапия » Недифференцированные дисплазии соединительной ткани (проект клинических рекомендаций)
Недифференцированные дисплазии соединительной ткани (проект клинических рекомендаций)
DOI: https://dx. doi.org/10.18565/therapy.2019.7.9-42
doi.org/10.18565/therapy.2019.7.9-42
Публикуемый проект второго пересмотра клинических рекомендаций по ведению пациентов с недифференцированными дисплазиями соединительной ткани продиктован наличием обоснованных дополнений/замечаний к ранее утвержденным (в 2018 г.) клиническим рекомендациям.
Литература
- Большая медицинская энциклопедия, 3-е изд. под ред. Б.В. Петровского. Режим доступа: http://бмэ.орг/index.php/дисплазия (дата обращения: 10.10.2019).
- Яковлев В.М., Нечаева Г.И., Мартынов А.И., Викторова И.А. Дисплазия соединительной ткани в практике врачей первичного звена здравоохранения: Руководство для врачей. М.: КСТ Интерфорум. 2016; 520 с.
- Клинические рекомендации российского научного медицинского общества терапевтов по диагностике, лечению и реабилитации пациентов с дисплазиями соединительной ткани (первый пересмотр). Медицинский вестник Северного Кавказа. 2018; 1,2(13): 137–210.
- Нечаева Г.И., Мартынов А.И. Дисплазия соединительной ткани: сердечно-сосудистые изменения, современные подходы к диагностике и лечению. Москва: ООО «Медицинское информационное агентство». 2017; 400 с.
- Кадурина Т.И., Горбунова В.Н. Дисплазия соединительной ткани: рук.для врачей. СПб.: Элби, 2009; 704 с.
- Наследственные нарушения соединительной ткани в кардиологии. Диагностика и лечение. Российские рекомендации (1-й пересмотр). Российский кардиологический журнал. 2013; 1: 32.
- Merlocco A., Lacro R.V., Gauvreau K., Rabideau N., Singh M.N., Prakash A. Longitudinal changes in segmental aortic stiffness determined by cardiac magnetic resonance in children and young adults with connective tissue disorders (the Marfan, Loeys-Dietz, and Ehlers-Danlos Syndromes, and nonspecific connective tissue disorders). Am J Cardiol. 2017; 120(7):1214–19.
- Викторова И.А., Нечаева Г.И., Киселева Д.С. Калинина И.Ю. Дисплазия соединительной ткани: особенности амбулаторного ведения пациентов в различных возрастных периодах.
 Лечащий врач. 2014; 9: 76–81.
Лечащий врач. 2014; 9: 76–81. - Друк И.В., Нечаева Г.И., Осеева О.В. и др. Персонифицированная оценка риска развития неблагоприятных сердечно-сосудистых осложнений у пациентов молодого возраста с дисплазией соединительной ткани. Кардиология. 2015; 3: 75–84.
- Международная классификация болезней 10-го пересмотра (МКБ-10). Режим доступа: http://mkb-10.com/ (дата обращения: 09.10.2019).
- Конев В.П., Голошубина В.В., Московский С.Н. Особенности формулирования судебно- медицинского диагноза при синдроме дисплазии соединительной ткани. Вестник судебной медицины. 2017; 6(2): 22–26.
- Арсентьев В.Г., Баранов В.С., Шабалов Н.П. Наследственные заболевания соединительной ткани как конституциональная причина полиорганных нарушений у детей. Санкт-Петербург: СпецЛит. 2015; 231 с.
- Яковлев В.М., Нечаева Г.И. Кардиореспираторные синдромы при дисплазии соединительной ткани. Омск. 1994; 217 с.
- Клинические рекомендации Российского научного медицинского общества терапевтов. Дисплазии соединительной ткани (первый пересмотр, сокращенный вариант). Терапия. 2018; 6: 10–58.
- Кильдиярова Р.Р., Углова Д.Ф. Ассоциированная с дисплазией соединительной ткани кардиальная патология у женщин и их новорожденных детей. Российский вестник перинатологии и педиатрии. 2015; 2(60): 54–56.
- Яворская М.В., Кравцов Ю.А., Кильдиярова Р.Р., Кучеров В.А., Матвеев С.В. Критерии диагностики синдрома дисплазии соединительной ткани и задержки полового развития у детей и подростков. Уральский медицинский журнал. 2017; 8: 111–117.
- Семенова Е.В., Семенкин А.А., Чиндарева О.И., Махрова Н.В., Нечаева Г.И., Потапов В.В., Живилова Л.А., Логинова Е.Н. Оптимизация подхода к определению расширения корня аорты при недифференцированной дисплазии соединительной ткани. Кардиология: новости, мнения, обучение. 2017; 1(12): 35–39.
- Конев В.П., Московский С.Н., Коршунов А.С. и др. Использование атомно-силовой микроскопии в изучении плотных тканей орофациальной области. Казанский медицинский журнал. 2012; 6: 887–891.
- Конев В.П., Шестель И.Л., Московский С.Н. и др. Атомно-силовая микроскопия в диагностике патологии соединительной ткани: семиотика твердых тканей зубов и костной ткани. Материалы VIII Международной научно-практической конференции. Москва, 2011; 105–109.
- Тюрин А.В., Хусаинова Р.И., Лукманова Л.З., Давлетшин Р.А., Хуснутдинова Э.К. Поиск маркеров генетической предрасположенности к развитию гипермобильности суставов и остеоартрита у больных из республики Башкортостан. Молекулярная медицина. 2016; 14(6): 41–47.
- Тюрин А.В., Хусаинова Р.И., Хуснутдинова Н.Н., Давлетшин Р.А., Хуснутдинова Э.К. Поиск ассоциаций полиморфных вариантов гена рецептора витамина D (VDR) с остеоартритом и дисплазией соединительной ткани. Медицинская генетика. 2014; 13(9.147): 18–27.
- Хусаинова Р.И., Тюрин А.В., Шаповалова Д.А., Хуснутдинова Э.К. Генетические маркеры остеоартрита у женщин с недифференцированной дисплазией соединительной ткани. Генетика. 2017; 53(7): 816–826.
- Громова О.А., Торшин И.Ю., Калачева А.Г., Гришина Т.Р. О синергизме калия и магния в поддержании функции миокарда. Кардиология. 2016; 56(3): 73–80.
- Громова О.А., Торшин И.Ю., Юдина Н.В. и др. Дефицит магния и нарушения регуляции тонуса сосудов. Кардиология. 2014; 54(7): 66–72.
- Мамедов М.Н. Назначение высоких доз магния для лечения аритмий: показания, основанные на доказательствах. Клиническая фармакология и лекарственные средства. 2013; 2: 43–45.
- Трисветова T.Е. Магний в клинической практике. Фармакотерапия в кардиологии. 2012;4 (8):545–553.
- Громова О.А., Калачева А.Г., Торшин И.Ю. и др. Недостаточность магния – достоверный фактор риска коморбидных состояний: результаты крупномасштабного скрининга магниевого статуса в регионах России. Фарматека. 2013; 6(259): 115–129.
- Нечаева Г.И., Дрокина О.В., Колменкова И.В. Эффективность терапии препаратом мексикор у пациентов при вегетативно-сосудистой дисфункции с недифференцированными формами дисплазии соединительной ткани. Архив внутренней медицины. 2012; (8): 26–32.
- Бабаджанова Н.Э. Психовегетативные нарушения у больных нейроциркуляторной дистонией и возможность их коррекции. Архив внутренней медицины. 2016; 1: 110.
- Особенности психологического статуса лиц молодого возраста с дисплазией соединительной ткани. Друк И.В., Логинова Е.Н., Вершинина М.В., Лялюкова Е.А., Дрокина О.В. Омский психиатрический журнал. 2019; 3(21): 5–9.
- Pervichko E., Zinchenko Y., Martynov A., Akatova E. Assessment of psychological well-being dating and dynamics of clinical symptoms in mitral valve prolapse patients with anxiety disorders receiving long-term integrative psychotherapy. European Psychiatry. 2015; 30 (1S): 1652.
- Воробьева О.В. Полинейропатии, обусловленные соматическими заболеваниями: подходы к диагностике, основные направления лечения. Consilium Medicum. Неврология и ревматология. 2016; 01: 74–78.
- Вершинина М.В., Нечаева Г.И., Хоменя А.А., Дрокина О.В. Эффективность медицинской реабилитации при бронхолегочном синдроме у пациентов с дисплазией соединительной ткани. Медицинский вестник Северного Кавказа. 2015; 10(1): 50–55.
- Аллергология. Федеральные клинические рекомендации. Под ред. акад. РАН Р.М. Хаитов, проф. Н.И. Ильина. М.: «Фармарус Принт Медиа», 2014. 126 с.
- Кононова, Н.Ю., Чернышова Т.Е., Загртдинова Р.М. Оценка биологического возраста и темпа старения у пациенток с недифференцированной дисплазией соединительной ткани. Архив внутренней медицины. 2017; 7(4): 287–291.
- Кононова Н.Ю., Чернышева Т.Е., Стяжкина С.Н. Является ли дисплазия соединительной ткани предиктором преждевременного старения? (результаты 5-летнего мониторинга). Медицинский вестник Северного Кавказа. 2016; 11(2.2): 326–330.
- Громова О.А., Торшин И.Ю., Калачева А.Г., Гришина Т.Р. О синергизме калия и магния в поддержании функции миокарда. Кардиология. 2016; 56(3): 73–80.
- Малев Э.Г., Ким Г.И., Митрофанова Л. Б., Омельченко М.Ю., Земцовский Э.В. Функция левого желудочка при пролапсе митрального клапана, осложненном тяжелой митральной недостаточностью. Российский кардиологический журнал. 2013; 1 (99): 37–41.
- 3Пролапс митрального клапана. Клинические рекомендации. 2016; 21 с.
- Голицын С.П., Кропачева Е.С., Майков Е.Б., Миронов Н.Ю., Панченко Е.П., Соколов С.Ф., Шлевков Н.Б. Клинические рекомендации по диагностике и лечению нарушений ритма сердца и проводимости. Неотложная кардиология. 2013; 104.
- Рекомендации ESC по лечению пациентов с желудочковыми нарушениями ритма и профилактике внезапной сердечной смерти 2015. Российский кардиологический журнал. 2016; 7 (135): 5–86.
- Рекомендации ESC по лечению пациентов с фибрилляцией предсердий, разработанные совместно с EACTS Российский кардиологический журнал 2017, 7 (147): 7–86. http://dx.doi.org/10.15829/1560-4071-2017-7-7-86
- 4Guidelines for the diagnosis and management of syncope. European Heart J. 2018; VAA-21: 1–69. doi: 10.1093/eurheartj/ehy037
- Шляхто Е.В., Арутюнов Г.П., Беленков Ю.Н., Ардашев А.В. Национальные Рекомендации по определению риска и профилактике внезапной сердечной смерти. Архив внутренней медицины. 2013;(4):5–15.
- Национальные рекомендации по определению риска и профилактике внезапной сердечной смерти (2-ое издание). М.: ИД «Медиапрактика-М». 2018; 247 с.
- Kelly R.E. Jr., Mellins R.B., Shamberger R.C. et al. Multicenter study of pectus excavatum, final report: complications, static/exercise pulmonary function, and anatomic outcomes. J. Am. Coll. Surg. 2013; 217(6): 1080–89.
- Хирургическое лечение синдромальных сколиозов. Клинические рекомендации. Новосибирск, 2013; 34 с.
- Ортопедия. Клинические рекомендации под ред. С.П. Миронова. ГЭОТАР-Медиа, 2018; 784 с.
- Миопия. Клинические рекомендации. 2017; 48 с.
- Котова О.В., Акарачкова Е.С. Астенический синдром в практике невролога и семейного врача. Российский медицинский журнал. 2016; 13: 824–829.
- Нечаева Г.И., Логинова Е.Н., Вершинина М.В. Ведущие причины повышения давления в малом круге кровообращения у пациентов с дисплазией соединительной ткани. Лечащий врач. 2016; 3.
- Рекомендации ESC/ERS по диагностике и лечению легочной гипертензии 2015. Российский кардиологический журнал. 2015; 5(133): 5–64.
- Konstantinides S.V., Torbicki A., Agnelli G. et al. ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC) Endorsed by the European Respiratory Society (ERS). Russ. J. Cardiol. 2015; 8(124): 67–110.
- 2018 ЕОК/ЕОАГ Рекомендации по лечению больных с артериальной гипертензией. Российский кардиологический журнал. 2018; 23(12): 143–228.
- Меморандум экспертов Российского кардиологического общества по рекомендациям Европейского общества кардиологов/Европейского общества по артериальной гипертензии по лечению артериальной гипертензии 2018 г. Российский кардиологический журнал. 2018; 23(12): 131–142.
- Смяловский В.Э., Друк И.В., Смяловский Д.В. Особенности течения интракраниальных артериальных аневризм и артериовенозных мальформаций у пациентов с дисплазией соединительной ткани. Журнал неврологии и психиатрии им. С.С. Корсакова. 2014; 114(8): 304–305.
- Erbe R., Aboyans V., Boileau C., Bossone E., D. Bartolomeo R., Eggebrecht H., Evangelista A., Falk V., Frank H. et al. ESC Guidelines on the diagnosis and treatment of aortic diseases. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014; 35(41): 2873–926.
- Лялюкова Е.А., Нечаева Г. И., Ливзан М. А. и др. Недостаточность питания у пациентов с дисплазией соединительной ткани: роль постпрандиальных гемодинамических нарушений, подходы к терапии. Лечащий врач. 2015; 3: 67–70.
- Рекомендации Российской гастроэнтерологической ассоциации и ассоциации колопроктологов России по диагностике и лечению взрослых больных дивертикулярной болезнью ободочной кишки. Российский журнал гастроэнтерологии, гепатологии, колопроктологии. 2016; 1: 65–80.
- Нечаева Г.И., Логинова Е.Н., Цуканов А.Ю., Семенкин А.А., Фисун Н.И., Дрокина О.В. Патология почек при дисплазии соединительной ткани: междисциплинарный подход. Лечащий врач. 2016; 1: 54–57.
- Перепанова Т.С. с соавт. Антимикробная терапия и профилактика инфекций почек, мочевыводящих путей и мужских половых органов. Федеральные клинические рекомендации. М.: ООО «Прима-принт». 2015; 72 с.
- Кудинова Е.Г. Особенности беременности у пациенток с аномальным коллагенообразованием и нарушениями системы гемостаза. РМЖ. 2016; 15: 1026–32.
- Кудинова Е. Г. Риск репродуктивных нарушений у девочек-подростков с дисплазией соединительной ткани. Репродуктивное здоровье детей и подростков. 2013; 4: 31–32.
- Приказ Минздрава России от 12.11. 2012 № 572н.
- Нечаева Г.И., Друк И.В., Логинова Е.Н., Смольнова Т.Ю., Шупина М.И., Викторова И.А., Семенкин А.А., СеменоваЕ.В. Современные подходы к ведению беременности и родов у пациенток с синдромом Марфана. Медицинский вестник Северного Кавказа. 2016; 11(2): 363–368.
- Диагностика и лечение сердечно-сосудистых заболеваний при беременности. Национальные рекомендации. Российский кардиологический журнал. 2018; 3(155): 91–134.
- Смольнова Т.Ю., Адамян Л.В. Рассечение промежности в родах и леваторопластика у женщин с дисплазией соединительной ткани. Показания и противопоказания. Сб. научн. Трудов: Новые технологии в диагностике и лечении гинекологических заболеваний. Москва, 2013; 175–176.
- Смольнова Т.Ю. Пролапс гениталий и дисплазия соединительной ткани. Клиническая и экспериментальная хирургия. 2015; 2: 53–64.
- Федеральные клинические рекомендации по диагностике и лечению железодефицитной анемии. 2014; 16.
- Кудинова Е.Г., Лыдина И.В., Тараненко И.А. и др. Предикторы риска тромботических осложнений у беременных с мезенхимальной дисплазией. Проблемы клинической медицины. 2012; 4(26–29): 117–123.
- Российские клинические рекомендации по диагностике, лечению и профилактике венозных тромбоэмболических осложнений (ВТЭО). Флебология. 2015; 4(2): 1–52.
- Рекомендации ESC по диагностике и ведению пациентов с острой эмболией системы легочной артерии 2014. Российский кардиологический журнал. 2015; 8 (124): 67–110. http://dx.doi.org/10.15829/1560-4071-2015-08-67-110
- Викторова И.А., Киселева Д.С., Кульниязова Г.М. Синдром гипермобильности суставов: диагностика и лечение пациентов в амбулаторной практике диагностика и лечение пациентов в амбулаторной практике. Лечащий врач.2014; 4: 62–69.
- Остеопороз. Клинические рекомендации. 2016; 104 с.
- Кадурина Т.И., Аббакумова Л.Н. Принципы реабилитации больных с дисплазией соединительной ткани. Лечащий врач. 2010; 4: 28–31.
- Дубилей Г.С., Исаева А.С., Фомина О.А. с соавт. Немедикаментозные методы восстановительного лечения пациентов с дисплазией соединительной ткани на этапах медицинской реабилитации. Комплексная реабилитация: наука и практика. 2010; 5(13): 52–56.
- Приказ Министерства здравоохранения РФ от 13.03.2019 № 124н «Об утверждении порядка проведения профилактического медицинского осмотра и диспансеризации определенных групп взрослого населения».
- Друк И.В., Нечаева Г.И., Лялюкова Е.А., Дрокина О.В. Кардиоваскулярные синдромы дисплазии соединительной ткани у лиц молодого возраста: частота регистрации, факторы формирования. Лечащий врач. 2014; 6: 72–75.
 Лечащий врач. 2014; 9: 76–81.
Лечащий врач. 2014; 9: 76–81. Казанский медицинский журнал. 2012; 6: 887–891.
Казанский медицинский журнал. 2012; 6: 887–891. Архив внутренней медицины. 2012; (8): 26–32.
Архив внутренней медицины. 2012; (8): 26–32. Б., Омельченко М.Ю., Земцовский Э.В. Функция левого желудочка при пролапсе митрального клапана, осложненном тяжелой митральной недостаточностью. Российский кардиологический журнал. 2013; 1 (99): 37–41.
Б., Омельченко М.Ю., Земцовский Э.В. Функция левого желудочка при пролапсе митрального клапана, осложненном тяжелой митральной недостаточностью. Российский кардиологический журнал. 2013; 1 (99): 37–41. Российский медицинский журнал. 2016; 13: 824–829.
Российский медицинский журнал. 2016; 13: 824–829.
 2014; 16.
2014; 16.Синдром Марфана — Marfan syndrome
Генетическое заболевание соединительной ткани
Синдром Марфана ( MFS ) — это генетическое заболевание , поражающее соединительную ткань . Люди с этим заболеванием, как правило, высокие и худые, с длинными руками, ногами, пальцами рук и ног . У них также обычно чрезмерно гибкие суставы и сколиоз . Наиболее серьезные осложнения связаны с сердцем и аортой с повышенным риском пролапса митрального клапана и аневризмы аорты . Также часто поражаются легкие, глаза, кости и покров спинного мозга . Выраженность симптомов MFS варьируется.
У них также обычно чрезмерно гибкие суставы и сколиоз . Наиболее серьезные осложнения связаны с сердцем и аортой с повышенным риском пролапса митрального клапана и аневризмы аорты . Также часто поражаются легкие, глаза, кости и покров спинного мозга . Выраженность симптомов MFS варьируется.
MFS вызывается мутацией в FBN1 , одном из генов, вырабатывающих фибриллин , что приводит к аномальной соединительной ткани. Это аутосомно-доминантное заболевание. Примерно в 75% случаев состояние наследуется от родителя с этим заболеванием, а в 25% случаев это новая мутация. Диагноз часто основывается на критериях Гента .
Нет никакого известного лекарства от MFS. Многие из тех, кто страдает этим заболеванием, при правильном лечении имеют нормальную продолжительность жизни. Лечение часто включает использование бета-блокаторов, таких как пропранолол или атенолол, или, если они не переносятся, блокаторов кальциевых каналов или ингибиторов АПФ . Для восстановления аорты или замены сердечного клапана может потребоваться операция . Людям с этим заболеванием рекомендуется избегать физических нагрузок.
Примерно от 1 из 5 000 до 1 из 10 000 человек страдают MFS. Показатели состояния одинаковы между расами и в разных регионах мира. Он назван в честь французского педиатра Антуана Марфана , который впервые описал его в 1896 году.
Признаки и симптомы
Деформация передней грудной стенки, pectus excatum , у человека с синдромом Марфана.
Более 30 признаков и симптомов в той или иной степени связаны с синдромом Марфана. Наиболее заметные из них влияют на скелетную, сердечно-сосудистую и глазную системы, но могут быть затронуты все волокнистые соединительные ткани по всему телу.
Система скелета
Большинство легко видимых признаков связаны с костной системой . Многие люди с синдромом Марфана вырастают выше среднего, а некоторые имеют непропорционально длинные тонкие конечности с тонкими слабыми запястьями и длинными пальцами рук и ног . Помимо влияния на рост и пропорции конечностей, люди с синдромом Марфана могут иметь аномальное боковое искривление позвоночника (сколиоз), грудной лордоз , аномальное вдавливание (pectus excatum) или выпячивание (pectus carinatum) грудины , аномальную гибкость суставов , высокое дугообразное изгибание. небо с скученными зубами и неправильным прикусом, плоскостопие , молоткообразные пальцы ног , сутулые плечи и необъяснимые растяжки на коже. Это также может вызвать боль в суставах, костях и мышцах. У некоторых людей с Марфаном возникают речевые расстройства, вызванные симптомами высокого неба и маленькой челюсти. Может возникнуть ранний остеоартрит . Другие признаки включают ограниченный диапазон движений в бедрах из-за того, что головка бедра выступает в аномально глубокие тазобедренные суставы .
Помимо влияния на рост и пропорции конечностей, люди с синдромом Марфана могут иметь аномальное боковое искривление позвоночника (сколиоз), грудной лордоз , аномальное вдавливание (pectus excatum) или выпячивание (pectus carinatum) грудины , аномальную гибкость суставов , высокое дугообразное изгибание. небо с скученными зубами и неправильным прикусом, плоскостопие , молоткообразные пальцы ног , сутулые плечи и необъяснимые растяжки на коже. Это также может вызвать боль в суставах, костях и мышцах. У некоторых людей с Марфаном возникают речевые расстройства, вызванные симптомами высокого неба и маленькой челюсти. Может возникнуть ранний остеоартрит . Другие признаки включают ограниченный диапазон движений в бедрах из-за того, что головка бедра выступает в аномально глубокие тазобедренные суставы .
Глаза
При синдроме Марфана здоровье глаза может быть затронуто многими способами, но основным изменением является частичное смещение хрусталика , когда хрусталик смещается из своего нормального положения. Это происходит из-за слабости цилиарных зонул , волокон соединительной ткани, которые удерживают хрусталик внутри глаза. Мутации, ответственные за синдром Марфана, ослабляют зонулы и заставляют их растягиваться. Чаще всего растягиваются нижние зоны, в результате чего хрусталик смещается вверх и наружу, но может смещаться и в других направлениях. Близорукость (миопия) и нечеткое зрение являются обычными явлениями из-за дефектов соединительной ткани глаза. Также может возникнуть дальнозоркость, особенно если хрусталик сильно подвывих. Подвывих (частичный вывих) хрусталика может быть обнаружен клинически примерно у 60% людей с синдромом Марфана с помощью биомикроскопа с щелевой лампой . Если подвывих хрусталика незначительный, то можно использовать визуализацию с помощью ультразвуковой биомикроскопии высокого разрешения.
Другие признаки и симптомы, влияющие на глаз, включают увеличение длины по оси земного шара, миопию, плоскостность роговицы, косоглазие , экзотропию и эзотропию .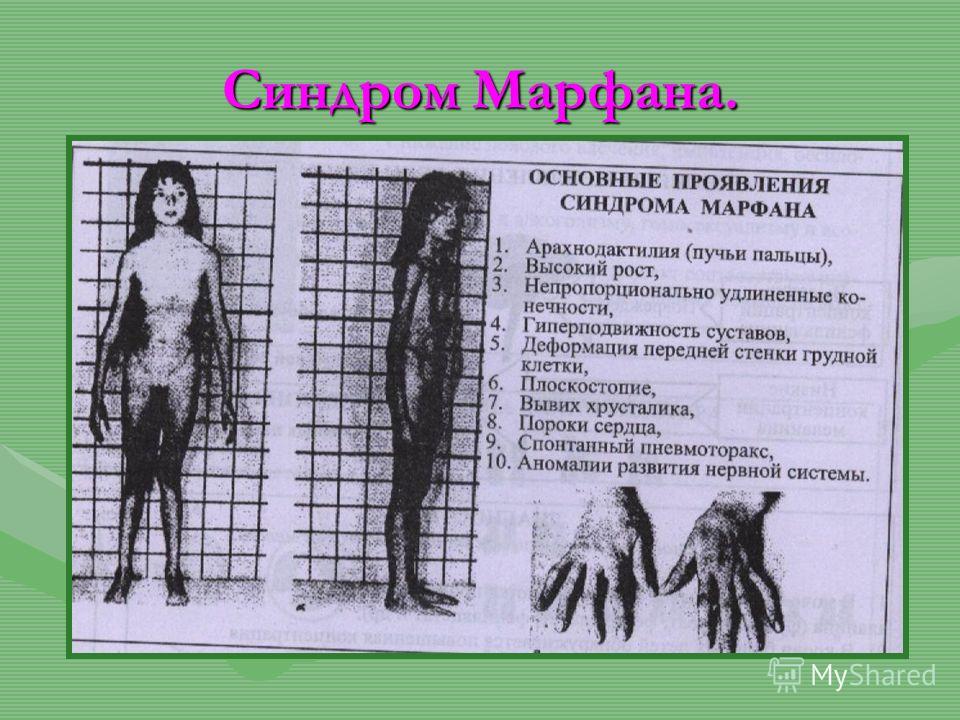 Люди с MFS также подвержены высокому риску ранней глаукомы и ранней катаракты .
Люди с MFS также подвержены высокому риску ранней глаукомы и ранней катаракты .
Сердечно-сосудистая система
Наиболее серьезные признаки и симптомы, связанные с синдромом Марфана, связаны с сердечно-сосудистой системой : чрезмерная усталость , одышка , учащенное сердцебиение , учащенное сердцебиение или боль в груди, отдающая в спину, плечо или руку. Холодные руки, кисти и стопы также могут быть связаны с MFS из-за недостаточного кровообращения. Сердце ропот , ненормальное чтение на ЭКГ или симптомы стенокардии могут указывать дальнейшее расследование. Признаки регургитации из-за пролапса митрального или аортального клапанов (которые контролируют кровоток через сердце) являются результатом кистозной медиальной дегенерации клапанов, которая обычно связана с MFS (см. Пролапс митрального клапана , аортальная регургитация ). Однако основным признаком, который заставит врача рассмотреть основное заболевание, является расширенная аорта или аневризма аорты . Иногда проблемы с сердцем не проявляются до тех пор, пока ослабление соединительной ткани (кистозная медиальная дегенерация) в восходящей аорте не вызовет аневризму аорты или расслоение аорты , что является неотложным хирургическим вмешательством. Расслоение аорты чаще всего заканчивается смертельным исходом и проявляется болью, отдающей вниз по спине, с ощущением разрываемости.
Поскольку лежащие в основе аномалии соединительной ткани вызывают MFS, частота расхождения протеза митрального клапана увеличивается. Следует проявлять осторожность, пытаясь восстановить поврежденные сердечные клапаны, а не заменять их.
Легкие
Люди с синдромом Марфана могут иметь различные проблемы с легкими. Одно исследование показало, что только 37% исследуемой выборки пациентов (средний возраст 32 ± 14 лет; M 45%) имели нормальную функцию легких. Спонтанный пневмоторакс — обычное явление. При спонтанном одностороннем пневмотораксе воздух выходит из легкого и занимает плевральное пространство между грудной стенкой и легким. Легкое частично сжимается или разрушается. Это может вызвать боль, одышку, цианоз и, если не лечить, смерть. Другие возможные легочные проявления MFS включают апноэ во сне и идиопатическую обструктивную болезнь легких. Описаны патологические изменения в легких, такие как кистозные изменения, эмфизема , пневмония , бронхоэктазы , буллы , апикальный фиброз и врожденные пороки, такие как гипоплазия средней доли.
Легкое частично сжимается или разрушается. Это может вызвать боль, одышку, цианоз и, если не лечить, смерть. Другие возможные легочные проявления MFS включают апноэ во сне и идиопатическую обструктивную болезнь легких. Описаны патологические изменения в легких, такие как кистозные изменения, эмфизема , пневмония , бронхоэктазы , буллы , апикальный фиброз и врожденные пороки, такие как гипоплазия средней доли.
Нервная система
Дуральная эктазия , ослабление соединительной ткани дурального мешка, покрывающего спинной мозг , может привести к снижению качества жизни . Он может присутствовать в течение длительного времени, не вызывая каких-либо заметных симптомов. Симптомы, которые могут возникнуть, включают боль в пояснице, боль в ногах, боль в животе, другие неврологические симптомы в нижних конечностях или головные боли — симптомы, которые обычно уменьшаются в положении лежа. Однако на рентгеновских снимках эктазия твердой мозговой оболочки не всегда видна на ранних стадиях. При ухудшении симптомов может потребоваться МРТ нижнего отдела позвоночника. Дуральная эктазия, которая прогрессировала до этой стадии, будет проявляться на МРТ как расширенный мешок, стирающийся в поясничных позвонках . Другие проблемы с позвоночником, связанные с MFS, включают остеохондроз , кисты позвоночника и дисфункцию вегетативной нервной системы .
Генетика
Каждый родитель с этим заболеванием имеет 50% -ный риск передачи генетического дефекта любому ребенку из-за его аутосомно-доминантного характера. У большинства людей с MFS есть другой пострадавший член семьи. Около 75% случаев передаются по наследству. С другой стороны, около 15–30% всех случаев вызваны генетическими мутациями de novo ; такие спонтанные мутации происходят примерно в одном случае из 20 000 рождений. Синдром Марфана также является примером доминантно-отрицательной мутации и гаплонедостаточности . Это связано с переменной выразительностью ; неполная пенетрантность окончательно не задокументирована.
Это связано с переменной выразительностью ; неполная пенетрантность окончательно не задокументирована.
Патогенез
Синдром Марфана вызван мутациями в гене FBN1 на хромосоме 15 , который кодирует фибриллин 1 , гликопротеиновый компонент внеклеточного матрикса. Фибриллин-1 необходим для правильного формирования внеклеточного матрикса, включая биогенез и поддержание эластичных волокон. Внеклеточный матрикс важен как для структурной целостности соединительной ткани, так и в качестве резервуара для факторов роста. Эластичные волокна встречаются по всему телу, но особенно много их в аорте, связках и цилиарных поясах глаза; следовательно, эти области относятся к числу наиболее пострадавших. Это также может быть вызвано рядом внутривенных обработок кристаллами у лиц, подверженных этому заболеванию.
Была создана трансгенная мышь, несущая единственную копию мутантного фибриллина-1, мутации, подобной той, которая обнаружена в гене человека, который, как известно, вызывает MFS. Эта линия мышей повторяет многие особенности болезни человека и обещает дать представление о патогенезе болезни. Снижение уровня нормального фибриллина 1 вызывает у мышей болезнь Марфана.
Трансформирующий фактор роста бета ( TGF-β ) играет важную роль в MFS. Фибриллин-1 напрямую связывает латентную форму TGF-β, удерживая ее изолированной и неспособной проявлять свою биологическую активность. Самая простая модель предполагает, что пониженные уровни фибриллина-1 позволяют уровням TGF-β повышаться из-за неадекватной секвестрации. Хотя то, как повышенные уровни TGF-β ответственны за специфическую патологию, наблюдаемую при заболевании, не доказано, известно, что имеет место воспалительная реакция, высвобождающая протеазы, которые медленно разрушают эластичные волокна и другие компоненты внеклеточного матрикса. Важность пути TGF-β была подтверждена открытием аналогичного синдрома Лойса-Дитца с участием гена TGFβR2 на хромосоме 3 , рецепторного белка TGF-β. Синдром Марфана часто путают с синдромом Лойса-Дитца из-за значительного клинического совпадения этих двух патологий.
Синдром Марфана часто путают с синдромом Лойса-Дитца из-за значительного клинического совпадения этих двух патологий.
Марфаноидно-прогероидно-липодистрофический синдром
Марфаноидно-прогероидно-липодистрофический синдром (MPL), также называемый липодистрофическим синдромом Марфана (MFLS), представляет собой вариант MFS, при котором симптомы Марфана сопровождаются особенностями, обычно связанными с неонатальным прогероидным синдромом (также называемым синдромом Видемана-Раутенштрауха) при котором уровень белой жировой ткани снижен. С 2010 года накапливаются доказательства того, что MPL вызывается мутациями около 3′-конца гена FBN1 . Было показано, что у этих людей также наблюдается дефицит аспрозина , глюко-регулирующего белкового гормона, который является продуктом C-концевого расщепления профибриллина. Уровни аспрозина у этих людей были ниже, чем ожидалось для гетерозиготного генотипа, что согласуется с доминирующим негативным эффектом.
Диагностика
УЗИ человека с синдромом Марфана, показывающее расширенный корень аорты
Диагностические критерии MFS были согласованы на международном уровне в 1996 году. Однако синдром Марфана часто трудно диагностировать у детей, поскольку они обычно не проявляют симптомов до достижения полового созревания. Диагноз основывается на семейном анамнезе и комбинации основных и второстепенных показателей расстройства, редко встречающихся в общей популяции, которые встречаются у одного человека, например: четыре скелетных признака с одним или несколькими признаками в другой системе организма, такой как глазная и сердечно-сосудистые у одного человека. Следующие состояния могут быть результатом MFS, но могут также возникать у людей без каких-либо известных основных заболеваний.
Пересмотренная гентская нозология
Знак большого пальца; верхний : нормальный, нижний : синдром Марфана
В 2010 году нозология Гента была пересмотрена, и новые диагностические критерии заменили предыдущее соглашение, заключенное в 1996 году. Семь новых критериев могут привести к постановке диагноза:
Семь новых критериев могут привести к постановке диагноза:
При отсутствии семейного анамнеза MFS:
- Z-показатель корня аорты ≥ 2 И эктопия lentis
- Z-показатель корня аорты ≥ 2 И мутация FBN1
- Z-оценка корня аорты ≥ 2 И системная оценка *> 7 баллов
- Ectopia lentis И мутация FBN1 с известной патологией аорты
При наличии семейного анамнеза MFS (как определено выше):
- Эктопия лентис
- Системный балл * ≥ 7
- Z-показатель корня аорты ≥ 2
- Очки за системный балл:
Знак большого пальца (знак Стейнберга) вызывается просьбой человека согнуть большой палец как можно дальше, а затем сомкнуть пальцы над ним. Положительный признак большого пальца — это когда вся дистальная фаланга видна за локтевым краем кисти, что вызвано сочетанием гипермобильности большого пальца, а также большого пальца, который длиннее обычного.
Знак запястья (знак Уокера-Мердока) вызывается, когда человека просят обхватить большим пальцем и пальцами одной руки другое запястье. Положительным признаком запястья является перекрытие мизинца и большого пальца из-за сочетания тонких запястий и длинных пальцев.
Дифференциальная диагностика
Многие другие расстройства могут вызывать те же характеристики тела, что и синдром Марфана. Генетическое тестирование и оценка других признаков и симптомов могут помочь их дифференцировать. Ниже приведены некоторые расстройства, которые могут проявляться как «марфаноид»:
Управление
От синдрома Марфана нет лекарств, но продолжительность жизни значительно увеличилась за последние несколько десятилетий и теперь сравнима со среднестатистическим человеком.
Рекомендуется регулярно проверять состояние сердечных клапанов и аорты . Синдром Марфана лечится путем решения каждой проблемы по мере ее возникновения и, в частности, с помощью профилактических лекарств даже для маленьких детей, чтобы замедлить прогрессирование расширения аорты. Цель этой стратегии лечения — замедлить развитие дилатации аорты и предотвратить любое повреждение сердечных клапанов за счет устранения аритмий сердца , минимизации частоты сердечных сокращений и снижения кровяного давления человека .
Цель этой стратегии лечения — замедлить развитие дилатации аорты и предотвратить любое повреждение сердечных клапанов за счет устранения аритмий сердца , минимизации частоты сердечных сокращений и снижения кровяного давления человека .
Физическая активность
Американская кардиологическая ассоциация была принята следующими рекомендации для людей с синдромом Марфан с отсутствием или слабой аортальной дилатацией:
- Вероятно допустимые занятия: боулинг, гольф, катание на коньках (но не хоккей с шайбой), снорклинг, быстрая ходьба, беговая дорожка, велотренажер, скромный пеший туризм и парный теннис.
- Промежуточный риск: баскетбол (как на корте, так и на полу), ракетбол, сквош, бег (спринт и бег трусцой), катание на лыжах (скоростной спуск и бег по пересеченной местности), футбол, одиночный теннис, сенсорный футбол (флаг), бейсбол, софтбол, езда на велосипеде , плавание на коленях, езда на мотоцикле и верховая езда.
- Высокий риск: бодибилдинг, тяжелая атлетика (несвободные и свободные веса), хоккей, скалолазание, виндсерфинг, серфинг и подводное плавание с аквалангом.
Медикамент
Лечение часто включает использование бета-блокаторов, таких как пропранолол, или, если они не переносятся, блокаторов кальциевых каналов или ингибиторов АПФ . Бета-блокаторы используются для уменьшения нагрузки на аорту и уменьшения ее расширения.
Хирургия
Если расширение аорты прогрессирует до аневризмы значительного диаметра , вызывает расслоение или разрыв либо приводит к отказу аортального или другого клапана, тогда операция (возможно, составной трансплантат аортального клапана или замена корня аорты с сохранением клапана ) становится необходимо. Хотя операция по пересадке аорты (или любая операция на сосудах) является серьезным мероприятием, она обычно бывает успешной, если проводится на выборной основе. Хирургия при остром расслоении или разрыве аорты значительно более проблематична. Плановая операция на аортальном клапане / трансплантате обычно рассматривается, когда диаметр корня аорты достигает 50 миллиметров (2,0 дюйма), но каждый случай требует особой оценки квалифицированного кардиолога. Все большее распространение получают новые хирургические методы с сохранением клапанов. По мере того как люди с синдромом Марфана живут дольше, все чаще встречаются другие способы восстановления сосудов, например, восстановление аневризм нисходящей грудной аорты и аневризм сосудов, отличных от аорты.
Плановая операция на аортальном клапане / трансплантате обычно рассматривается, когда диаметр корня аорты достигает 50 миллиметров (2,0 дюйма), но каждый случай требует особой оценки квалифицированного кардиолога. Все большее распространение получают новые хирургические методы с сохранением клапанов. По мере того как люди с синдромом Марфана живут дольше, все чаще встречаются другие способы восстановления сосудов, например, восстановление аневризм нисходящей грудной аорты и аневризм сосудов, отличных от аорты.
Скелетные и глазные проявления синдрома Марфана также могут быть серьезными, но не опасными для жизни. Эти симптомы обычно лечатся в соответствии с состоянием, например, с помощью обезболивающих или миорелаксантов . Поскольку синдром Марфана может вызывать бессимптомные аномалии позвоночника, любая операция на позвоночнике, предполагаемая на человеке Марфане, должна проводиться только после детальной визуализации и тщательного хирургического планирования, независимо от показаний к операции. Глазные осложнения MFS часто можно лечить хирургическим путем. Ectopia lentis можно лечить, поскольку искусственные линзы могут быть имплантированы хирургическим путем. Кроме того, хирургическое вмешательство может помочь в лечении глаукомы и катаракты .
Лечение спонтанного пневмоторакса зависит от объема воздуха в плевральной полости и естественного прогрессирования состояния человека. Небольшой пневмоторакс может разрешиться без активного лечения в течение одной-двух недель. Рецидивирующий пневмоторакс может потребовать хирургического вмешательства на груди. Пневмоторакс среднего размера может потребовать дренирования грудной клетки в течение нескольких дней в больнице. Большие пневмотораксы могут потребовать неотложной медицинской помощи, требующей экстренной декомпрессии.
В качестве альтернативного подхода также используются специальные опоры для корня аорты. По состоянию на 2020 год эта процедура использовалась более чем у 300 человек, первый случай произошел в 2004 году.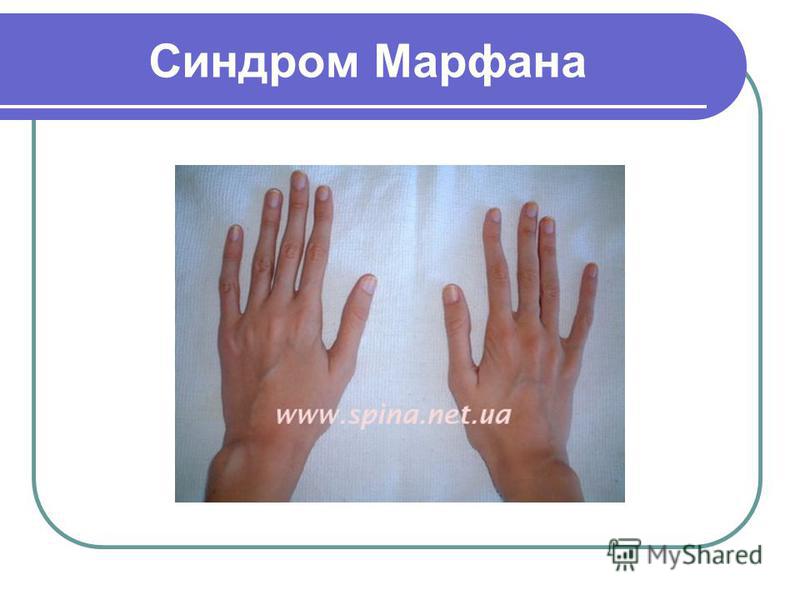
Беременность
Во время беременности, даже при отсутствии предубежденных сердечно-сосудистых аномалий, женщины с синдромом Марфана подвергаются значительному риску расслоения аорты, которое часто приводит к летальному исходу даже при быстром лечении. Таким образом, женщины с синдромом Марфана должны пройти тщательное обследование до зачатия, а эхокардиография должна выполняться каждые 6-10 недель во время беременности для оценки диаметра корня аорты. Для большинства женщин возможны безопасные вагинальные роды.
У женщин с синдромом Марфана можно провести пренатальное тестирование, чтобы определить, унаследовано ли заболевание у их ребенка. На 10–12 неделе беременности для постановки диагноза можно провести исследование участка плацентарной ткани с помощью теста, называемого забором ворсин хориона. Другой пренатальный тест может быть проведен на сроке от 16 до 18 недель, который называется амниоцентез .
Синдром Марфана выражен преобладающе. Это означает, что ребенок, у которого один из родителей является носителем гена, имеет 50% -ную вероятность получить синдром. В 1996 г. было проведено первое преимплантационное генетическое тестирование (ПГТ) для лечения Марфана; По сути, PGT означает проведение генетического теста на эмбриональных клетках ранней стадии ЭКО и отбрасывание эмбрионов, затронутых мутацией Марфана.
Прогноз
До появления современных сердечно-сосудистых хирургических методов и лекарств, таких как лозартан и метопролол , прогноз для пациентов с синдромом Марфана был неблагоприятным: ряд неизлечимых сердечно-сосудистых заболеваний был обычным явлением. Продолжительность жизни сократилась как минимум на треть, и многие умерли в подростковом и двадцатилетнем возрасте из-за сердечно-сосудистых заболеваний. Сегодня сердечно-сосудистые симптомы синдрома Марфана по-прежнему являются наиболее важной проблемой в диагностике и лечении заболевания, но адекватный профилактический мониторинг и профилактическая терапия предлагают что-то, приближающееся к нормальной продолжительности жизни, и по мере того, как все больше пациентов живут дольше, обнаруживается все больше проявлений болезни. Женщины с синдромом Марфана живут дольше мужчин.
Женщины с синдромом Марфана живут дольше мужчин.
Эпидемиология
Синдром Марфана одинаково поражает мужчин и женщин, и мутация не проявляет этнической или географической предвзятости. По оценкам, синдром Марфана страдает примерно у 1 из 5 000–10 000 человек.
История
Синдром Марфана назван в честь Антуана Марфана , французского педиатра, который впервые описал это состояние в 1896 году после того, как заметил поразительные черты у пятилетней девочки. Ген, связанный с заболеванием, был впервые идентифицирован Франческо Рамиресом в Медицинском центре горы Синай в Нью-Йорке в 1991 году.
Смотрите также
Рекомендации
внешняя ссылка
2021 Код МКБ-10-CM Q87.40 — Синдром Марфана неуточненный
Действительно для подачи
Q87.40 — это оплачиваемый диагностический код , используемый для указания медицинского диагноза синдрома Марфана, неуточненного. Код Q87.40 действителен в течение 2021 финансового года с 1 октября 2020 года по 30 сентября 2021 года для отправки транзакций, покрытых HIPAA.
Код Q87.40 МКБ-10-CM может также использоваться для определения таких состояний или терминов, как слепота, сколиоз, синдром арахнодактилии, врожденная аномалия подкожной ткани, врожденная паховая грыжа, врожденный нефрит, генетическая липодистрофия, марфаноидные фации и т. Д.Код освобождается от представления отчетов при поступлении (POA) для стационарных госпитализаций в больницы общей неотложной помощи.
Коды неуказанного диагноза, такие как Q87.40, приемлемы, когда клиническая информация о конкретном состоянии неизвестна или недоступна. Хотя более конкретный код предпочтительнее, следует использовать неопределенные коды, когда такие коды наиболее точно отражают то, что известно о состоянии пациента. Не следует использовать коды конкретных диагнозов, если они не подтверждаются медицинской картой пациента.
Синдром Марфана
Синдром Марфана — это заболевание, поражающее соединительную ткань. Соединительные ткани — это белки, которые поддерживают кожу, кости, кровеносные сосуды и другие органы. Один из этих белков — фибриллин. Проблема с геном фибриллина вызывает синдром Марфана.
Соединительные ткани — это белки, которые поддерживают кожу, кости, кровеносные сосуды и другие органы. Один из этих белков — фибриллин. Проблема с геном фибриллина вызывает синдром Марфана.
Синдром Марфана может протекать от легкой до тяжелой степени, и симптомы могут быть разными.Люди с синдромом Марфана часто бывают очень высокими, худыми и непослушными. У большинства людей с синдромом Марфана есть проблемы с сердцем и кровеносными сосудами, такие как слабость аорты или сердечные клапаны, которые протекают. У них также могут быть проблемы с костями, глазами, кожей, нервной системой и легкими.
Не существует единого теста для диагностики синдрома Марфана. Ваш врач может использовать вашу историю болезни, семейный анамнез и медицинский осмотр, чтобы поставить диагноз. Синдром Марфана неизлечим, но лечение может помочь отсрочить или предотвратить осложнения.Лечение включает в себя лекарства, хирургическое вмешательство и другие методы лечения.
NIH: Национальный институт артрита, скелетно-мышечных и кожных заболеваний
- Синдром Марфана (Медицинская энциклопедия)
[Подробнее в MedlinePlus]
Синдром Марфана
Синдром Марфана — это заболевание, поражающее соединительную ткань во многих частях тела. Соединительная ткань обеспечивает прочность и гибкость таким структурам, как кости, связки, мышцы, кровеносные сосуды и сердечные клапаны.Признаки и симптомы синдрома Марфана широко различаются по степени тяжести, времени начала и скорости прогрессирования. Поскольку соединительная ткань находится по всему телу, синдром Марфана может поражать многие системы, часто вызывая нарушения в сердце, кровеносных сосудах, глазах, костях. , и суставы. Двумя основными особенностями синдрома Марфана являются проблемы со зрением, вызванные смещением хрусталика (ectopia lentis) в одном или обоих глазах, и дефекты большого кровеносного сосуда, по которому кровь распределяется от сердца к остальному телу (аорте).Аорта может ослабнуть и растянуться, что может привести к вздутию стенки кровеносного сосуда (аневризме). Растяжение аорты может вызвать утечку аортального клапана, что может привести к внезапному разрыву слоев в стенке аорты (расслоение аорты). Аневризма и расслоение аорты могут быть опасными для жизни.Многие люди с синдромом Марфана имеют дополнительные проблемы с сердцем, включая утечку в клапане, который соединяет две из четырех камер сердца (пролапс митрального клапана), или клапан, который регулирует кровоток из сердца в аорта (регургитация аортального клапана).Утечки в этих клапанах могут вызвать одышку, утомляемость и нерегулярное сердцебиение, ощущаемое как пропущенные или лишние удары (сердцебиение). Люди с синдромом Марфана обычно высоки и стройны, имеют удлиненные пальцы рук и ног (арахнодактилия), слабые суставы и размах рук превышает их рост. Другие общие черты включают длинное и узкое лицо, скученные зубы, аномальное искривление позвоночника (сколиоз или кифоз), растяжки (стрии), не связанные с увеличением или потерей веса, а также либо впалую грудь (pectus excatum), либо выступающую грудная клетка (pectus carinatum).У некоторых людей развивается аномальное скопление воздуха в грудной полости, что может привести к коллапсу легкого (спонтанный пневмоторакс). Мембрана, называемая твердой мозговой оболочкой, которая окружает головной и спинной мозг, может быть аномально увеличена (дуральная эктазия) у людей с синдромом Марфана. Эктазия твердой мозговой оболочки может вызвать боль в спине, животе, ногах или голове. Большинство людей с синдромом Марфана в той или иной степени страдают близорукостью (миопией). Помутнение хрусталика (катаракта) может возникнуть в среднем возрасте, а повышенное внутриглазное давление (глаукома) чаще возникает у людей с синдромом Марфана, чем у людей без этого заболевания.Признаки синдрома Марфана могут проявиться в любое время между младенчеством и взрослой жизнью. В зависимости от появления и тяжести признаков и симптомов синдром Марфана может привести к летальному исходу в раннем возрасте; однако при правильном лечении у многих пораженных людей продолжительность жизни нормальная.
Растяжение аорты может вызвать утечку аортального клапана, что может привести к внезапному разрыву слоев в стенке аорты (расслоение аорты). Аневризма и расслоение аорты могут быть опасными для жизни.Многие люди с синдромом Марфана имеют дополнительные проблемы с сердцем, включая утечку в клапане, который соединяет две из четырех камер сердца (пролапс митрального клапана), или клапан, который регулирует кровоток из сердца в аорта (регургитация аортального клапана).Утечки в этих клапанах могут вызвать одышку, утомляемость и нерегулярное сердцебиение, ощущаемое как пропущенные или лишние удары (сердцебиение). Люди с синдромом Марфана обычно высоки и стройны, имеют удлиненные пальцы рук и ног (арахнодактилия), слабые суставы и размах рук превышает их рост. Другие общие черты включают длинное и узкое лицо, скученные зубы, аномальное искривление позвоночника (сколиоз или кифоз), растяжки (стрии), не связанные с увеличением или потерей веса, а также либо впалую грудь (pectus excatum), либо выступающую грудная клетка (pectus carinatum).У некоторых людей развивается аномальное скопление воздуха в грудной полости, что может привести к коллапсу легкого (спонтанный пневмоторакс). Мембрана, называемая твердой мозговой оболочкой, которая окружает головной и спинной мозг, может быть аномально увеличена (дуральная эктазия) у людей с синдромом Марфана. Эктазия твердой мозговой оболочки может вызвать боль в спине, животе, ногах или голове. Большинство людей с синдромом Марфана в той или иной степени страдают близорукостью (миопией). Помутнение хрусталика (катаракта) может возникнуть в среднем возрасте, а повышенное внутриглазное давление (глаукома) чаще возникает у людей с синдромом Марфана, чем у людей без этого заболевания.Признаки синдрома Марфана могут проявиться в любое время между младенчеством и взрослой жизнью. В зависимости от появления и тяжести признаков и симптомов синдром Марфана может привести к летальному исходу в раннем возрасте; однако при правильном лечении у многих пораженных людей продолжительность жизни нормальная.
[Подробнее в MedlinePlus]
Код МКБ-10 Q87.4 | Синдром Марфана
МКБ-10
МКБ-10 — это 10-я редакция Международной статистической классификации болезней и проблем, связанных со здоровьем (МКБ), списка медицинских классификаций Всемирной организации здравоохранения (ВОЗ).
Он содержит коды заболеваний, признаков и симптомов, отклонений от нормы, жалоб, социальных обстоятельств и внешних причин травм или заболеваний.
УВД
Анатомо-терапевтическая химическая система классификации (АТХ) используется для классификации активных ингредиентов лекарственных средств в зависимости от органа или системы, на которые они действуют, и их терапевтических, фармакологических и химических свойств.
Он контролируется Центром сотрудничества по методологии статистики лекарственных средств Всемирной организации здравоохранения (WHOCC).
DDD
Установленная суточная доза (DDD) — это статистическая мера потребления лекарств, определенная Всемирной организацией здравоохранения (ВОЗ).
Он используется для стандартизации сравнения употребления наркотиков между разными лекарствами или между различными средами здравоохранения.
For the Record: Кодирование синдрома Марфана
18 января 2010 г.
Кодирование синдрома Марфана
Для записи
Vol.22 № 1 стр. 29
Синдром Марфана (код 759.82 МКБ-9-CM) — это генетическое заболевание, поражающее соединительную ткань, в результате чего она теряет свою эластичность и прочность. Поскольку соединительная ткань расположена по всему телу, синдром может поражать множество различных систем организма, таких как сердечно-сосудистая, нервная и дыхательная.
Синдром Марфана — аутосомно-доминантное заболевание, которое имеет тенденцию к ухудшению с возрастом. Самый большой фактор риска — наличие родителя с этим заболеванием.Другая причина — изменение гена, которое происходит на раннем этапе развития в утробе матери.
Синдром присутствует при рождении, но может появиться только в более позднем возрасте. Согласно правилам кодирования: «Врожденная аномалия, хотя и присутствует при рождении, может быть обнаружена только в более позднем возрасте. Всякий раз, когда врач диагностирует заболевание, целесообразно присвоить код из кодов 740-759 »(Официальные рекомендации МКБ-9-CM по кодированию и отчетности, вступающие в силу 1 октября 2009 г., стр. 54).Поэтому у взрослого пациента уместно присвоить врожденный код 759,82.
Симптомы
Симптомы синдрома Марфана могут сильно различаться, поскольку это состояние может повлиять на многие системы организма. Характерные черты: высокое стройное телосложение; непропорционально длинные руки и ноги; длинные тонкие пальцы рук и ног; грудина, которая выступает наружу или опускается внутрь; высокое арочное небо; скученные зубы; шум в сердце; сердцебиение; миопия; сколиоз; плоскостопие; рыхлые, гибкие суставы; длинное худое лицо, глубоко посаженные глаза и маленькая нижняя челюсть; и дуральная эктазия.
Осложнения
Общие осложнения синдрома Марфана включают следующие:
• дилатация аорты;
• аневризма аорты, которая чаще всего возникает в корне аорты, с опасением, что аневризма может разорваться или разорваться;
• расслоение аорты или разрыв самого внутреннего слоя стенки аорты, который позволяет крови протискиваться между внутренним и внешним слоями стенки;
• порок развития клапана, который может привести к сердечной недостаточности;
• вывих хрусталика одного или обоих глаз;
• глаукома;
• катаракта;
• отслойка или разрыв сетчатки;
• эмфизема;
• хроническая обструктивная болезнь легких;
• коллапс легкого;
• апноэ во сне; и
• сколиоз.
Не назначайте коды для вышеуказанных осложнений, присущих синдрому Марфана ( AHA Coding Clinic для ICD-9-CM , 1993, третий квартал, стр. 11). Согласно руководящим принципам кодирования: «Когда присвоение кода специально идентифицирует врожденную аномалию, проявления, которые являются неотъемлемым компонентом аномалии, не должны кодироваться отдельно. Дополнительные коды должны быть назначены для проявлений, которые не являются неотъемлемым компонентом »(Официальные рекомендации МКБ-9-CM по кодированию и отчетности, вступающие в силу 1 октября 2009 г., стр. 53-54).
Диагноз
Чтобы диагностировать синдром Марфана, симптомы должны проявляться как группа. Некоторые диагностические тесты, выполняемые для диагностики синдрома Марфана, включают эхокардиограмму или чреспищеводную эхокардиограмму для проверки камер и клапанов сердца, электрокардиограмму для проверки сердечного ритма, МРТ для исследования аорты, компьютерную томографию для исследования аорты, щель Осмотр с лампой для проверки на наличие проблем с глазами, тест на глазное давление для проверки на глаукому, генетическое тестирование или осмотр у ортопеда для проверки суставов.
Лечение
От синдрома Марфана нет лекарства. Однако врач может устранить симптомы и попытаться предотвратить осложнения.
Лечение сердечно-сосудистых осложнений может включать в себя такие лекарства, как бета-блокаторы, ингибиторы ангиотензинпревращающего фермента, блокаторы рецепторов ангиотензина и блокаторы кальциевых каналов. Эти лекарства могут замедлить сердцебиение или снизить кровяное давление, что может уменьшить износ кровеносных сосудов.Кроме того, может потребоваться операция на сердечных клапанах или аорте.
Кодирование и секвенирование синдрома Марфана зависят от документации врача в медицинской карте и применения Официальных рекомендаций по кодированию для стационарного лечения. Кроме того, используйте специальные ссылки AHA Coding Clinic для ICD-9-CM и American Medical Association CPT Assistant , чтобы обеспечить полное и точное кодирование.
— Эта информация была подготовлена Одри Ховард, RHIA, компании 3M Consulting Services.3M Consulting Services — это бизнес компании 3M Health Information Systems, поставщика систем кодирования и классификации для более чем 4000 поставщиков медицинских услуг. Компания и ее представители не несут никакой ответственности за решения о возмещении расходов или отказы в претензиях, сделанные поставщиками или плательщиками в результате неправильного использования этой информации о кодировании. Дополнительную информацию о 3M Health Information Systems можно получить на сайте www.3mhis.com или по телефону 800-367-2447.
Заявление о принципах, касающихся синдрома Марфана No.26 от 2015 г.
Заявление о принципах
в отношении
СИНДРОМ МАРФАНА
№ 26 от 2015 г.
Закон 1986 года о правах ветеранов
и
Закон о реабилитации и компенсации военнослужащих 2004 года
Название
1. Настоящий документ можно цитировать как Заявление о принципах в отношении синдрома Марфана № 26 от 2015 года.
Настоящий документ можно цитировать как Заявление о принципах в отношении синдрома Марфана № 26 от 2015 года.
Определение
2. Медицинское управление по репатриации согласно подразделу 196B (3) и (8) из ветеранов «Закон о правах 1986 (VEA):
(a) отменяет Документ № 54 от 2007 года, касающийся синдрома Марфана; и
(b) вместо него определяет настоящее Заявление о принципах.
Вид травмы, болезни или смерти
3. (a) Настоящее Заявление о принципах касается синдрома Марфана и смерти от синдрома Марфана .
(b) Для целей настоящего Заявления о принципах «синдром Марфана» означает генетическое заболевание соединительной ткани, наследуемое как аутосомно-доминантный признак, характеризующееся аномалиями глаз, костей, сердца и кровеносных сосудов.
(c) Синдром Марфана привлекает код Q87 в МКБ-10-AM.4.
(d) При применении настоящего Заявления о принципах определение «синдром Марфана» соответствует определению, приведенному в пункте 3 (b) выше.
Основа для определения факторов
4. На основании имеющихся убедительных медицинских и научных данных Управление репатриации считает, что более вероятно, чем нет, что синдром Марфана и смерть от синдрома Марфана может быть связаны с соответствующей службой, оказываемой ветеранами или военнослужащими в рамках VEA, или военнослужащими в соответствии с Законом о военной реабилитации и компенсации 2004 года (MRCA).
Факторы, которые должны быть связаны с услугой
5. В соответствии с пунктом 7, по крайней мере, один из факторов, указанных в пункте 6, должен быть связан с соответствующей услугой, оказываемой этим лицом.
Факторы
6. Фактор, который должен существовать, прежде чем можно будет сказать, что на балансе вероятностей синдром Марфана или смерть от синдрома Марфана связана с обстоятельствами соответствующей службы человека:
(а) беременность до клинического обострения синдрома Марфана; или
(b) невозможность получить соответствующее клиническое лечение синдрома Марфана.
Факторы, относящиеся только к материальному вкладу или усугублению
7. Пункты 6 (a) и 6 (b) применяются только к материальному вкладу или обострению синдрома Марфана, если у человека был перенесен синдром Марфана, или заключенный до или во время (но не вытекающий из) соответствующего обслуживания лица.
Включение заявлений о принципах
8. В этом Заявлении о принципах, если применяется соответствующий фактор и этот фактор включает травму или заболевание, в отношении которых существует Заявление о принципах, то факторы в последнем упомянутом Заявлении о принципах Принципы применяются в соответствии с положениями этого Заявления о принципах, которое время от времени действует.
Другие определения
9. Для целей данного Заявления о принципах:
«смерть от синдрома Марфана» в отношении человека включает смерть от неизлечимого события или состояния, которому способствовал Марфан человека. синдром;
«Код МКБ-10-AM» означает номер, присвоенный определенному виду травмы или заболевания в Международной статистической классификации болезней и проблем, связанных со здоровьем, 10-я редакция, австралийская модификация (МКБ-10-AM), восьмая. Издание, вступившее в силу 1 июля 2013 г., защищено авторским правом Независимого агентства по ценообразованию в больницах и имеет ISBN 978-1-74128-213-9;
«соответствующая служба» означает:
(a) правомочную военную службу (кроме оперативной службы) в рамках VEA;
(b) служба обороны (кроме службы в опасных условиях и службы защиты от ядерных испытаний Великобритании) в рамках VEA; или
(c) служба в мирное время в рамках MRCA;
«терминальное событие» означает непосредственную или конечную причину смерти и включает:
(a) пневмонию;
(б) дыхательная недостаточность;
(c) остановка сердца;
(г) недостаточность кровообращения; или
(е) прекращение функции мозга.
Заявление
10. Этот документ применяется ко всем вопросам, к которым применяется раздел 120B VEA или раздел 339 MRCA.
Дата вступления в силу
11. Настоящий инструмент вступает в силу с 27 января 2015 года.
От девятнадцатого дня декабря 2014 года
Общая печать)
Медицинского органа по репатриации)
была проставлена по указанию of:)
ПРОФЕССОР НИКОЛАС САУНДЕРС АО
ПРЕДСЕДАТЕЛЬ
Синдром Марфана — A&I Online
ОрфанАнестезия
Л.Багирзаде
Синдром Марфана
Синдром Марфана
Schlüsselwörter
Синдром Марфана; МКБ 10: Q87.4; Синдром Марфана
Ключевые слова
Синдром Марфана; МКБ 10: Q87.4; Синдром Марфана
Zusammenfassung
Dieser Beitrag enthält keine Zusammenfassung
Резюме
Синдром Марфана — аутосомно-доминантное мультисистемное заболевание с зарегистрированной заболеваемостью от 1 до 3000–5000 человек.С синдромом Марфана ассоциируется широкий диапазон клинической степени тяжести, от отдельных признаков до неонатальных проявлений тяжелого и быстро прогрессирующего заболевания. Классические проявления включают глазные (дислокация хрусталика, миопия), сердечно-сосудистые (расширение корня аорты с аортальной регургитацией, пролапс митрального клапана с митральной регургитацией) и скелетно-мышечные аномалии (разрастание длинных костей, сколиоз, кифоз, гипермобильность суставов), однако поражение легких ( пневмоторакс), кожа (стрии) и центральная нервная система (дуральная эктазия) также распространены при синдроме Марфана.Мутации в гене (FBN1), кодирующем белок внеклеточного матрикса фибриллин-1, вызывают классический синдром Марфана. Однако до 30% случаев не затрагивают ни одного из родителей и представляют собой мутации de novo. Профилактическое лечение бета-блокаторами считается стандартом лечения взрослых (если не противопоказано) и, как было показано, снижает скорость дилатации аорты. Нет однозначных рекомендаций ни для общей, ни для регионарной анестезии. Независимо от метода анестезии следует соблюдать осторожность, чтобы предотвратить внезапное увеличение сократимости миокарда, вызывающее увеличение напряжения стенки аорты, что может привести к расслоению аорты.
Однако до 30% случаев не затрагивают ни одного из родителей и представляют собой мутации de novo. Профилактическое лечение бета-блокаторами считается стандартом лечения взрослых (если не противопоказано) и, как было показано, снижает скорость дилатации аорты. Нет однозначных рекомендаций ни для общей, ни для регионарной анестезии. Независимо от метода анестезии следует соблюдать осторожность, чтобы предотвратить внезапное увеличение сократимости миокарда, вызывающее увеличение напряжения стенки аорты, что может привести к расслоению аорты.
Экономические последствия синдрома Марфана: неэкспериментальное ретроспективное популяционное сопоставленное когортное исследование | Orphanet Journal of Rare Diseases
Keane MG, Pyeritz RE: Медицинское лечение синдрома Марфана. Тираж. 2008, 117: 2802-2813.
Артикул
PubMed
Google ученый
Судья Д.П., Дитц ХК: синдром Марфана. Ланцет. 2005, 366: 1965–1976.
CAS
Статья
PubMed
PubMed Central
Google ученый
Йетман А.Т., Борнемайер Р.А., МакКриндл Б.В.: Долгосрочный исход у пациентов с синдромом Марфана: расслоение аорты — единственная причина внезапной смерти ?. J Am Coll Cardiol. 2003, 41: 329-332.
Артикул
PubMed
Google ученый
Юань С.М., Цзин Х .: Синдром Марфана: обзор. Сан-Паулу Med J. 2010, 128: 360-366.
Артикул
PubMed
Google ученый
 org/ScholarlyArticle»> 5.
org/ScholarlyArticle»> 5.Судья Д.П., Дитц Х.С.: Терапия синдрома Марфана. Annu Rev Med. 2008, 59: 43-59.
CAS
Статья
PubMed
Google ученый
Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC: Нарушение регуляции активации TGF-бета способствует патогенезу синдрома Марфана. Нат Жене. 2003, 33: 407-411.
CAS
Статья
PubMed
Google ученый
Болар Н., Ван Лаер Л., Лойс Б.Л .: Синдром Марфана: от гена к терапии. Curr Opin Pediatr. 2012, 24: 498-504.
CAS
Статья
PubMed
Google ученый
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM: Пересмотренная гентская нозология синдрома Марфана. J Med Genet. 2010, 47: 476-485.
CAS
Статья
PubMed
Google ученый
Браун О.Р., Демотс Х., Клостер Ф.Е., Робертс А., Менаше В.Д., Билс Р.К.: Расширение корня аорты и пролапс митрального клапана при синдроме Марфана: ЭХОКАРДИОГРАФИЧЕСКОЕ исследование. Тираж. 1975, 52: 651-657.
CAS
Статья
PubMed
Google ученый
Kiotsekoglou A, Sutherland GR, Moggridge JC, Nassiri DK, Camm AJ, Child AH: Распознавание первичных нарушений миокарда при синдроме Марфана с помощью современной эхокардиографии. Сердце. 2009, 95: 1561-1566.
Сердце. 2009, 95: 1561-1566.
CAS
Статья
PubMed
Google ученый
Рыбчинский М., Бернхардт AMJ, Редер Ю., Фуистинг Б., Мейсс Л., Фосс Ю., Хаберманн С., Деттер С., Робинсон П.Н., Арслан-Кирхнер М., Шмидтке Дж., Мир Т.С., Бергер Дж., Майнерц Т., фон Кодолич Y: Спектр синдромов и проявлений у лиц, прошедших скрининг на подозрение на синдром Марфана. Am J Med Genet A. 2008, 146A: 3157-3166.
Артикул
PubMed
Google ученый
Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Бероуд К., Бонитон-Копп С., Клаустрес М., Мути С., Плаучу Х., Робинсон П.Н., Адес Л.С., Биггин А., Бенеттс Б., Бретт М., Холман К.Дж., Де Бакер Дж., Кук П., Франк Ю., Де Паэпе А. и др. и др.: Влияние типа и локализации мутации на клинический исход у 1013 пробандов с синдромом Марфана или родственными фенотипами и мутациями FBN1: международное исследование.Am J Hum Genet. 2007, 81: 454-466.
CAS
Статья
PubMed
PubMed Central
Google ученый
Marsalese DL, Moodie DS, Vacante M, Lytle BW, Gill CC, Sterba R, Cosgrove DM, Passalacqua M, Goormastic M, Kovacs A: Синдром Марфана: естественное течение и долгосрочное наблюдение сердечно-сосудистых заболеваний участие. J Am Coll Cardiol. 1989, 14: 422-428. обсуждение 429–431
CAS
Статья
PubMed
Google ученый
 org/ScholarlyArticle»> 14.
org/ScholarlyArticle»> 14.Бенедетто У., Мелина Г., Таккенберг Дж. Дж. М., Роскитано А., Ангелони Э., Синатра Р.: Хирургическое лечение болезни корня аорты при синдроме Марфана: систематический обзор и метаанализ. Сердце. 2011, 97: 955-958.
Артикул
PubMed
Google ученый
Chow K, Pyeritz RE, Litt HI: Висцеральные исследования брюшной полости у пациентов с синдромом Марфана. Genet Med. 2007, 9: 208-212.
Артикул
PubMed
Google ученый
Détaint D, Faivre L, Collod-Beroud G, Child AH, Loeys BL, Binquet C, Gautier E, Arbustini E, Mayer K, Arslan-Kirchner M, Stheneur C, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Plauchu H, Robinson PN, Kiotsekoglou A, De Backer J, Adès L, Francke U, De Paepe A, Boileau C, Jondeau G: Сердечно-сосудистые проявления у мужчин и женщин, несущих мутацию FBN1. Eur Heart J. 2010, 31: 2223-2229.
Артикул
PubMed
Google ученый
Жондо Дж., Мишель Дж. Б., Буало С: переводческая наука о синдроме Марфана. Сердце. 2011, 97: 1206-1214.
Артикул
PubMed
Google ученый
Де Паепе А., Деверо РБ, Дитц Х.С., Хеннекам Р.С., Пайериц РЭ: Пересмотренные диагностические критерии синдрома Марфана. Am J Med Genet. 1996, 62: 417-426.
CAS
Статья
PubMed
Google ученый
Сильверман Д.И., Бертон К.Дж., Грей Дж., Боснер М.С., Кушукос Н.Т., Роман М.Дж., Боксер М., Деверо Р.Б., Ципурас П.: Ожидаемая продолжительность жизни при синдроме Марфана. Am J Cardiol. 1995, 75: 157-160.
CAS
Статья
PubMed
Google ученый
Gott VL, Greene PS, Alejo DE, Cameron DE, Naftel DC, Miller DC, Gillinov AM, Laschinger JC, Borst HG, Cabrol CEA, Cooley DA, Coselli JS, David TE, Griepp RB, Kouchoukos NT , Турина М.И., Периц Р.Э .: Замена корня аорты у пациентов с синдромом Марфана.N Engl J Med. 1999, 340: 1307-1313.
CAS
Статья
PubMed
Google ученый
Kallenbach K, Karck M, Leyh RG, Hagl C, Walles T., Harringer W., Haverich A: Клапан-сохраняющая реконструкция корня аорты у пациентов со значительной аортальной недостаточностью. Ann Thorac Surg. 2002, 74: S1765-S1768. обсуждение S1792–1799
Статья
PubMed
Google ученый
Bernhardt AMJ, Treede H, Rybczynski M, Sheikzadeh S, Kersten JF, Meinertz T., von Kodolitsch Y, Reichenspurner H: Сравнение замены корня аорты у пациентов с синдромом Марфана. Eur J Cardiothorac Surg. 2011, 40: 1052-1057.
PubMed
Google ученый
Мердок Дж. Л., Уокер Б. А., Халперн Б. Л., Кузьма Дж. В., МакКусик В. А.: Ожидаемая продолжительность жизни и причины смерти при синдроме Марфана. N Engl J Med. 1972, 286: 804-808.
CAS
Статья
PubMed
Google ученый
 org/ScholarlyArticle»> 24.
org/ScholarlyArticle»> 24.Pyeritz RE: Синдром Марфана: 30 лет исследований равняются 30 годам дополнительной продолжительности жизни. Сердце. 2009, 95: 173-175.
Артикул
PubMed
Google ученый
Сильверман Д.И., Грей Дж., Роман М.Дж., Бриджес А., Бертон К., Боксер М., Деверо Р.Б., Ципурас П.: В семейном анамнезе тяжелое сердечно-сосудистое заболевание при синдроме Марфана связано с увеличением диаметра аорты и снижением выживаемости. J Am Coll Cardiol. 1995, 26: 1062-1067.
CAS
Статья
PubMed
Google ученый
Ladouceur M, Fermanian C, Lupoglazoff J-M, Edouard T, Dulac Y, Acar P, Magnier S, Jondeau G: Влияние бета-блокады на расширение восходящей аорты у детей с синдромом Марфана. Am J Cardiol. 2007, 99: 406-409.
CAS
Статья
PubMed
Google ученый
Lacro RV, Dietz HC, Wruck LM, Bradley TJ, Colan SD, Devereux RB, Klein GL, Li JS, Minich LL, Paridon SM, Pearson GD, Printz BF, Pyeritz RE, Radojewski E, Roman MJ , Saul JP, Stylianou MP, Mahony L, Pediatric Heart Network Investigators: Обоснование и дизайн рандомизированного клинического испытания терапии бета-блокаторами (атенолол) по сравнению с терапией блокаторами рецепторов ангиотензина II (лозартаном) у лиц с синдромом Марфана.Am Heart J. 2007, 154: 624-631.
CAS
Статья
PubMed
PubMed Central
Google ученый
Олдрич Х.Р., Лабарр Р.Л., Роман М.Дж., Розен С.Е., Спитцер М. С., Деверо РБ: Цветной поток и обычная эхокардиография синдрома Марфана. Эхокардиография. 1992, 9: 627-636.
С., Деверо РБ: Цветной поток и обычная эхокардиография синдрома Марфана. Эхокардиография. 1992, 9: 627-636.
CAS
Статья
PubMed
Google ученый
Сеттепани Ф., Сето Вайоминг, Пачини Д., Де Паулис Р., Кьяриелло Л., Ди Бартоломео Р., Галлотти Р., Бавария Дж. Э .: Реимплантация клапанной замены корня аорты при синдроме Марфана с использованием кондуита Вальсальвы: межконтинентальный многоцентровый изучать.Ann Thorac Surg. 2007, 83: S769-S773. обсуждение S785–790
Артикул
PubMed
Google ученый
Bernhardt AM, Treede H, Detter C, Rybczynski M, Sheikhzadeh S, Wagner FM, Von Kodolitsch Y, Reichenspurner H: Результаты современного восстановления митрального клапана у пациентов с синдромом Марфана. Thorac Cardiovasc Surg. 2014, 62: 35-41.
PubMed
Google ученый
LeMaire SA, Carter SA, Volguina IV, Laux AT, Milewicz DM, Borsato GW, Cheung CK, Bozinovski J, Markesino JM, Vaughn WK, Coselli JS: спектр операций на аорте у 300 пациентов с подтвержденным или подозреваемым синдромом Марфана. Ann Thorac Surg. 2006, 81: 2063-2078. обсуждение 2078
Статья
PubMed
Google ученый
Петерс К.Ф., Хорн Р., Конг Ф., Франкомано Калифорния, Бизекер Б. Б.: Жизнь с синдромом Марфана II. Модификация приверженности к лечению и физической активности.Clin Genet. 2001, 60: 283-292.
CAS
Статья
PubMed
Google ученый
 org/ScholarlyArticle»> 33.
org/ScholarlyArticle»> 33.Фон Кодолич Ю., Робинсон П.Н.: Синдром Марфана: обновление генетики, медицинского и хирургического лечения. Сердце. 2007, 93: 755-760.
Артикул
PubMed
PubMed Central
Google ученый
Manow ML, Paulsen N, Rybczynski M, Mir T., Bernhardt AMJ, Treede H, Ohm G, Fuisting B, Rehder U, Meier F, Vogler M, Meinertz T, Overlack K, von Kodolitsch Y: [ Анализ затрат и прибылей амбулаторной помощи пациентам с Марфаном после введения в действие новой немецкой юридической директивы (116 b SGB V)].Мед Клин (Мюнхен). 2010, 105: 529-537.
Артикул
Google ученый
Rice DP: Исследования стоимости болезней: что в них хорошего ?. Inj Prev. 2000, 6: 177-179.
CAS
Статья
PubMed
PubMed Central
Google ученый
Roll K: Влияние региональных структур здравоохранения на задержку диагностики редких заболеваний: случай синдрома Марфана.Политика здравоохранения. 2012, 105: 119-127.
Артикул
PubMed
Google ученый
Sekhon JS: Программное обеспечение для оценки многовариантности и склонности с автоматической оптимизацией баланса: пакет сопоставления для R.J. Stat Software. 2011, 42: 1-52.
Артикул
Google ученый
Mebane WR, Sekhon JS: Генетическая оптимизация с использованием производных: пакет rgenoud для R. Программное обеспечение J Stat. 2011, 42: 1-26.
Программное обеспечение J Stat. 2011, 42: 1-26.
Артикул
Google ученый
IQWIG Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen: Оценка стоимости рабочего документа (версия 1.0). 2009 г., https://www.iqwig.de/download/Working_Paper_Cost_Estimation.pdf,
Google ученый
Statistisches Bundesamt: Entwicklung der Bruttoverdienste. 2008, Бонн: Statistisches Bundesamt
Google ученый
Deutsche Krankenhausgesellschaft (Немецкая ассоциация больниц): Krankenhausstatistik. 2008 г., http://www.dkgev.de/media/file/5776.Anlage_1.pdf,
Google ученый
Statistisches Bundesamt: Gesundheitsausgabenrechnung. 2008, Бонн: Statistisches Bundesamt
Google ученый
Петерс К.Ф., Конг Ф., Хорн Р., Франкомано Калифорния, Бизекер Б.Б.: Жизнь с синдромом Марфана I.Восприятие состояния. Clin Genet. 2001, 60: 273-282.
CAS
Статья
PubMed
Google ученый
Де Би С., Де Паепе А., Дельво И., Дэвис С., Хеннекам RCM: синдром Марфана в Европе. Сообщество Genet. 2004, 7: 216-225.
Артикул
PubMed
Google ученый
 org/ScholarlyArticle»> 45.
org/ScholarlyArticle»> 45.Мальяно Л., Фиорилло А., Де Роса К., Малангоне С., майор М., Национальная рабочая группа проекта по охране психического здоровья: Бремя длительных болезней семьи: сравнительное исследование шизофрении и шизофрении.физические расстройства. Soc Sci Med. 2005, 61: 313-322.
Артикул
PubMed
Google ученый
Elixhauser A, Steiner C, Harris DR, Coffey RM: Меры коморбидности для использования с административными данными. Med Care. 1998, 36: 8-27.
CAS
Статья
PubMed
Google ученый
Лиляс Б. Как рассчитать косвенные затраты при экономической оценке. Фармакоэкономика.1998, 13 (1 Пет 1): 1-7.
CAS
Статья
PubMed
Google ученый
Купманшап М.А., Руттен Ф.Ф., ван Иневельд Б.М., ван Ройен Л.: Метод фрикционных затрат для измерения косвенных затрат на заболевание. J Health Econ. 1995, 14: 171-189.
CAS
Статья
PubMed
Google ученый
Institut für Arbeitsmarkt- und Berufsforschung (IAB): Projektion des Erwerbspersonenpotenzials bis 2050 — Annahmen und Datengrundlage.Ergebnisse aus der Projektarbeit des Instituts für Arbeitsmarkt- und Berufsforschung. 2005, Нюрнберг: Institut für Arbeitsmarkt- und Berufsforschung (IAB), Forschungsbericht Nr. 25/2005]
Google ученый
Statistisches Bundesamt: Bevölkerung und Erwerbstätigkeit — Sterbetafel Deutschland 2009/11. 2013, Висбаден: Statistisches Bundesamt
Google ученый
Statistisches Bundesamt: Reallöhne, Nettoverdienste — Reallohnindex. 2013, Висбаден: Statistisches Bundesamt
Google ученый
Smith DH, Gravelle H: Практика дисконтирования в экономических оценках медицинских вмешательств. Int J Technol Оценка здравоохранения. 2001, 17: 236-243.
CAS
Статья
PubMed
Google ученый
Хекманн М., Кеттнер А., Ребьен М: Оффене Стеллен им IV.Quartal 2008. Einbruch in der Industrie — Soziale Berufe Legen Zu. 2009, Нюрнберг: [IAB-Kurzbericht: Aktuelle Analysen und Kommentare aus dem Institut für Arbeitsmarkt- und Berufsforschung]
Google ученый
Gray JR, Bridges AB, Faed MJ, Pringle T., Baines P, Dean J, Boxer M: Установление и тяжесть синдрома Марфана у шотландского населения. J Med Genet. 1994, 31: 51-54.
CAS
Статья
PubMed
PubMed Central
Google ученый
Imbens GW: Непараметрическая оценка средних эффектов лечения в условиях экзогенности: обзор. Rev Econ Stat. 2004, 86: 4-29.
Артикул
Google ученый
Diamond A, Sekhon JS: Генетическое сопоставление для оценки причинных эффектов: общий метод многомерного сопоставления для достижения баланса в наблюдательных исследованиях. Rev Econ Stat. 2012, 95: 932-945.
Артикул
Google ученый
Abadie A, Imbens GW: Свойства большой выборки соответствующих оценок для средних эффектов лечения. Econometrica. 2006, 74: 235-267.
Артикул
Google ученый
Бертакис К.Д., Азари Р.: Гендерные различия пациентов в прогнозировании медицинских расходов. J Womens Health (Larchmt). 2010, 19: 1925-1932.
Артикул
Google ученый
Kuo RN, Dong Y-H, Liu J-P, Chang C-H, Shau W-Y, Lai M-S: Прогнозирование использования медицинских услуг с использованием метрики на основе аптек с помощью анатомо-терапевтического химического алгоритма ВОЗ.Med Care. 2011, 49: 1031-1039.
Артикул
PubMed
Google ученый
Quan H, Sundararajan V, Halfon P, Fong A, Burnand B, Luthi JC, Saunders LD, Beck CA, Feasby TE, Ghali WA: алгоритмы кодирования для определения сопутствующих заболеваний в МКБ-9-CM и ICD- 10 административных данных. Med Care. 2005, 43: 1130-1139.
Артикул
PubMed
Google ученый
Розенбаум П.Р., Рубин Д.Б .: Центральная роль шкалы предрасположенности в обсервационных исследованиях причинных эффектов.Биометрика. 1983, 70: 41-55.
Артикул
Google ученый
Austin PC: Диагностика баланса для сравнения распределения исходных ковариат между группами лечения в выборках, сопоставленных по шкале склонности. Stat Med. 2009, 28: 3083-3107.
Артикул
PubMed
PubMed Central
Google ученый
Розенбаум PR: Наблюдательные исследования. Нью-Йорк: Спрингер; 2010 г.
Google ученый
ДиПрете Т.А., Гангл М: Оценка систематической ошибки при оценке причинно-следственных связей: границы Розенбаума для сопоставления оценок и инструментальных переменных оценки с несовершенными инструментами. Sociol Methodol. 2004, 34: 271-310.
Артикул
Google ученый
Розенбаум П.Р .: Анализ чувствительности в обсервационных исследованиях. Поведенческая наука: энциклопедия статистики в; 2005 г.
Google ученый
Bundesministerium für Gesundheit (BMG) — Федеральное министерство здравоохранения: Zahlen und Fakten zur Krankenversicherung. Бонн: Bundesministerium für Gesundheit (BMG) — Федеральное министерство здравоохранения; 2012. Mitgliederstatistik KM6 — Statistik über Versicherte Gegliedert Nach Status, Alter, Wohnort und Kassenart.
Google ученый
Акобунду Э., Джу Дж., Блатт Л., Маллинс К.Д.: Исследования стоимости болезни: обзор современных методов.Фармакоэкономика. 2006, 24: 869-890.
Артикул
PubMed
Google ученый
Рыбчински М., Мир Т.С., Шейхзаде С., Бернхардт А.М.Дж., Шад С., Триде Х, Вельдхоен С., Грене Э.Ф., Кюне К., Кошик Д., Робинсон П.Н., Бергер Дж., Райхенспурнер Х., Майнерц Т, фон Кодж. Y: Частота и возрастное течение дисфункции митрального клапана при синдроме Марфана. Am J Cardiol. 2010, 106: 1048-1053.
Артикул
PubMed
Google ученый
van Karnebeek CDM, Naeff MSJ, Mulder BJM, Hennekam RCM, Offringa M: Естественная история сердечно-сосудистых проявлений при синдроме Марфана. Arch Dis Child. 2001, 84: 129-137.
CAS
Статья
PubMed
PubMed Central
Google ученый
Согава М., Озэки Х, Намура О., Хаяси Дж .: Предоперационная респираторная физиотерапия для пациента с тяжелой респираторной дисфункцией и аннулоаортальной эктазией. Ann Thorac Cardiovasc Surg.2003, 9: 266-269.
PubMed
Google ученый
Stheneur C, Faivre L, Collod-Béroud G, Gautier E, Binquet C, Bonithon-Kopp C, Claustres M, Child AH, Arbustini E, Adès LC, Francke U, Mayer K, Arslan-Kirchner M , De Paepe A, Chevallier B, Bonnet D, Jondeau G, Boileau C: Факторы прогноза у пробандов с мутацией FBN1, диагностированной в возрасте до 1 года. Pediatr Res. 2011, 69: 265-270.
Артикул
PubMed
Google ученый
Rybczynski M, Treede H, Sheikhzadeh S, Groene EF, Bernhardt AMJ, Hillebrand M, Mir TS, Kühne K, Koschyk D, Robinson PN, Berger J, Reichenspurner H, Meinertz T, von Kodolitsch Y: Предикторы исхода митрального клапана пролапс у пациентов с синдромом Марфана. Am J Cardiol. 2011, 107: 268-274.
Артикул
PubMed
Google ученый
Blankart CR, Koch T, Linder R, Verheyen F, Schreyögg J, Stargardt T: Стоимость болезни и экономическое бремя хронического лимфолейкоза.Orphanet J Rare Dis. 2013, 8: 32.
Статья
PubMed
PubMed Central
Google ученый
Канавос П., ван ден Аардвег С., Шурер В.: Расходы на диабет, бремя болезни и лечение в 5 странах ЕС. Лондон, Великобритания: LSE Health, Лондонская школа экономики, 2012: 1-105.
Google ученый
Кёстер И., Хуппертц Э., Хаунер Х., Шуберт И.: Прямые затраты на сахарный диабет в Германии — CoDiM 2000–2007.Exp Clin Endocrinol Diabetes. 2011, 119: 377-385.
Артикул
PubMed
Google ученый
Huber CA, Schneeweiss S, Signorell A, Reich O: Улучшенное прогнозирование медицинских расходов и использования здравоохранения с использованием обновленных данных о хронических заболеваниях и заявках. J Clin Epidemiol. 2013, 66: 1118-1127.
Артикул
PubMed
Google ученый
Smeets HM, de Wit NJ, Hoes AW: Данные о текущем медицинском страховании для научных исследований: потенциал и ограничения базы данных Agis Health. J Clin Epidemiol. 2011, 64: 424-430.
Артикул
PubMed
Google ученый
Робинсон PN: Классификация и кодирование редких заболеваний: обзор нашего положения, обоснование, почему это важно и что может измениться. Orphanet J Rare Dis. 2012, 7 (Дополнение 2): A10.
Артикул
PubMed Central
Google ученый
Orphanet: Orphanet: Prevalence of Rare Diseases: библиографические данные. Франция: Orphanet; 2014: 29- [Серия отчетов Orphanet — Коллекция редких заболеваний]
Google ученый
Браверман AC: Упражнение и синдром Марфана. Медико-спортивные упражнения. 1998, 30 (10 доп.): S387-S395.
CAS
Статья
PubMed
Google ученый
Петерс К., Апсе К., Блэкфорд А., МакХью Б., Михалик Д., Бизекер Б.: Жизнь с синдромом Марфана: справиться со стигмой.Clin Genet. 2005, 68: 6-14.
Артикул
PubMed
Google ученый
Брауэр ВБФ, Мердинг В.-Дж., Ламерс Л. М., Северенс Дж. Л.: Взаимосвязь между производительностью и качеством жизни, связанным со здоровьем: исследование. Фармакоэкономика. 2005, 23: 209-218.
Артикул
PubMed
Google ученый
Lamers LM, Meerding W-J, Severens JL, Brouwer WBF: Взаимосвязь между производительностью и качеством жизни, связанным со здоровьем: эмпирическое исследование людей с болями в пояснице.Qual Life Res. 2005, 14: 805-813.
Артикул
PubMed
Google ученый
Рубин ДБ: Формальный способ статистического вывода о причинных эффектах. Заключение по статистическому планированию. 1990, 25: 279-292.
Артикул
Google ученый
Тейлор К.М., Карет Франкл FE: Разработка стратегии ведения редких заболеваний. BMJ. 2012, 344: e2417.
Артикул
PubMed
Google ученый
Фон Кодолич Ю., Рагхунатх М., Ниенабер К.А.: [Синдром Марфана: стратегии междисциплинарной помощи]. Dtsch Med Wochenschr. 1998, 123: 21-25.
CAS
Статья
PubMed
Google ученый
Синдром Марфана — BINDEVEVSSYKDOMMER.NOBINDEVEVSSYKDOMMER.NO
Последнее обновление: 28.12.2020
Синдром Марфана. Иллюстрация: большой палец и мизинец встречаются при синдроме Марфана. Иллюстрация из Staufenbiel I et al.CC BY 2.0
Синдром Марфана. МКБ-10 Q87.4
Синдром Марфана — наследственное (генетическое) заболевание (аутосомно-доминантное), вызванное дефектом (неправильной укладкой) белка фибриллина-1, кодируемого геном FBN1 на хромосоме 15. Таким образом, синдром Марфана — это врожденное генетическое заболевание, которое отличается от системных аутоиммунных заболеваний соединительной ткани. Около 25% являются спонтанными мутациями без семейной предрасположенности (Big Norwegian Leksikon, A Heiberg 2009).Наряду с сосудистой формой (поражающей сосуды) синдрома Элерса-Данлоса, синдрома Лойса-Дитца и наследственной аневризмы грудной аорты (HTAD) заболевание входит в число наследственных «марфановских» заболеваний.
Распространенность: 1 среди 20 000 человек (примерно 500 в Норвегии). Таким образом, болезнь Марфана определяется как одно редкое заболевание.
Скелетная система
Глаза
Сердце и сосуды
- Наиболее серьезные осложнения, которые могут быть опасными для жизни
- Нерегулярный сердечный ритм
- Утечка клапана сердца
- Расширенный диаметр артерии аорты (аневризма аорты)
- Расслоение аорты (повреждение и утечка стенки кровеносного сосуда)
- Особое внимание при беременности важно
Легкие
Часто используемые критерии диагностики (2010 г., Гент), которые различают тех, кто болеет болезнью Марфана среди ближайших родственников, и тех, кто не имеет наследственной предрасположенности:
Без наследственности:
- Корень аорты увеличен (Z-оценка ≥2) и эктопическая линза
- Расширение корня аорты (Z-показатель ≥2) и мутация FBN1 (генный тест)
- Расширение корня аорты (Z-оценка ≥2) и системная оценка *> 7 баллов (специальная система оценки *)
- Внематочный хрусталик и мутация FBN1 с известной инфекцией аорты
С семьей с болезнью Марфана:
- Эктоптическая линза
- Системная оценка * Расширенный корень аорты ≥7
- Расширение корня аорты (Z-показатель ≥2)
* Pectus carniatum = 2 (pes excatum или асимметричная = 1), деформация лодыжки (голеностопный сустав) = 2 (плоскостопие = 1), эктазии твердой мозговой оболочки = 2, протрузия вертлужной впадины = 2, непропорциональная длина руки = 1, сколиоз или кифоз = 1, уменьшение разгибания локтя = 1, изменения лицевого скелета = 1, растяжки на коже = 1, миопия = 1, митральный пролапс = 1.
Старые критерии: Основные критерии (не менее 4):
- Уменьшение соотношения верхней / нижней части тела (0,85 против 0,93 в норме)
- Arachnodactyli (пальцы рук и ног), положительный тест большого пальца-запястья)
- Сколиоз более 20 градусов или спондилолистез
- Смещение медиальной лодыжки, приводящее к плоской подошве (пластинчатой стопе)
- Ограниченное разгибание локтей (
- Pectum carniatum (птичья грудка) (Дифференциальный диагноз: синдром Моркио, синдром Нунана, Трисоми 18, Трисоми 21, гомоцистинурия, несовершенный остеогенез, синдром множественного лентиго и синдром Слая)
- Pectus excatum (воронкообразная грудь), требующая хирургического вмешательства
- Protrusio Acetabuli (Дифференциальные диагнозы: синдром Педжета, ревматоидный артрит, анкилозирующий спондилит, остеомаляция)
До сих пор не существует лечения синдрома Марфана.Но за последние десять лет прогноз значительно улучшился. Это связано с регулярным медицинским наблюдением, которое включает осмотры кардиолога (кардиолога), анализы кровеносных сосудов для исключения или наблюдения за аневризмами и лечение пневмоторакса.
Лечение недоступно
- Если происходит расширение артерий (аневризмы), может потребоваться операция по предотвращению разрыва артерии (аортаруптур).
- У хирургов-каратэ / торакальных хирургов есть рекомендации относительно того, когда следует проводить операции.
- Такие вмешательства проводятся, среди прочего, в Рикшоспиталет в Осло.
- Измерения артериального давления важны для выявления любого повышенного артериального давления, которое может увеличить риск аневризмы аорты
- Рекомендуется обычная физическая активность. Интенсивные и тяжелые физические упражнения увеличивают давление на кровеносные сосуды, и их следует избегать
- Кортизон и другие иммуносупрессивные препараты не подходят для лечения наследственных заболеваний соединительной ткани, в отличие от аутоиммунных заболеваний соединительной ткани
- Таблетки в форме бета-блокатора могут уменьшить осложнения со стороны кровеносных сосудов и рекомендуются для многих.
- В остальном лечение зависит от каких-либо осложнений со стороны глаз, суставов, позвоночника и т. П.
Регулярный мониторинг кровеносных сосудов важен
- МРТ-ангиография позволяет исследовать основные артерии головы, шеи, груди, брюшной полости и таза
- Также включена ультразвуковая допплерография / эхокардиография, можно измерить диаметр главной артерии в сердце и дуги аорты
- Если артерия расширяется, измерения проводятся не реже одного раза в шесть месяцев, в противном случае ежегодно в течение более длительного периода
- Также с помощью компьютерной томографии можно отобразить всю главную артерию.Из-за рентгеновских лучей расследование проводится нечасто.