Прогрессирующая мультифокальная лейкоэнцефалопатия — причины, симптомы, диагностика и лечение
Прогрессирующая мультифокальная лейкоэнцефалопатия — редкое демиелинизирующее заболевание, обусловленное реактивацией находящегося в организме большинства людей вируса JC. Патология возникает на фоне угнетения иммунитета у больных СПИДом, гемобластозами, наследственными иммунодефицитами, у пациентов, получающих иммуносупрессивную терапию. Диагностика базируется на клинических данных, результатах томографии головного мозга, ПЦР-исследования ликвора на вирусную ДНК, гистологии церебральных биоптатов. Специфическая терапия не разработана.
Общие сведения
Прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ) ассоциирована с JC-вирусом (JCV), возникает у иммунокомпрометированных пациентов, 85% из которых составляют ВИЧ-инфицированные. Заболевание относится к оппортунистическим инфекциям, носителями вируса являются 90% человечества. До 90-х годов ХХ века заболеваемость ПМЛ не превышала 1 случая на 100 тыс. населения. С ростом числа больных СПИДом этот показатель увеличился до 1 на 20 тыс. человек. Сегодня прогрессирующая лейкоэнцефалопатия наблюдается у 5% больных СПИДом. Некоторые авторы сообщают о снижении заболеваемости за последнее десятилетие в связи с успешным применением антиретровирусной терапии. Одновременно отмечается увеличение распространённости ПМЛ среди лиц с аутоиммунными заболеваниями, что обусловлено использованием в их лечении агрессивной иммунотерапии.
До 90-х годов ХХ века заболеваемость ПМЛ не превышала 1 случая на 100 тыс. населения. С ростом числа больных СПИДом этот показатель увеличился до 1 на 20 тыс. человек. Сегодня прогрессирующая лейкоэнцефалопатия наблюдается у 5% больных СПИДом. Некоторые авторы сообщают о снижении заболеваемости за последнее десятилетие в связи с успешным применением антиретровирусной терапии. Одновременно отмечается увеличение распространённости ПМЛ среди лиц с аутоиммунными заболеваниями, что обусловлено использованием в их лечении агрессивной иммунотерапии.
Прогрессирующая мультифокальная лейкоэнцефалопатия
Причины ПМЛ
Прогрессирующая мультифокальная лейкоэнцефалопатия развивается в результате реактивации полиомавируса JC. Вирус распространён повсеместно. Источником инфекции является человек, заражение происходит воздушно-капельным, алиментарным путём. Подавляющее большинство людей заражаются в детстве, являются здоровыми носителями. В течение жизни вирус находится в латентном состоянии, персистирует в почках, селезёнке, костном мозге. Реактивация возбудителя происходит на фоне резко сниженного иммунитета. В группу риска развития заболевания входят следующие состояния:
Реактивация возбудителя происходит на фоне резко сниженного иммунитета. В группу риска развития заболевания входят следующие состояния:
Патогенез
Расстройство клеточного иммунитета провоцирует перестройку последовательности ДНК JC-вируса, приводит к его активации. Вирус обладает тропностью к клеточным элементам нейроглии (олигодендроцитам, астроцитам), поражение которых сопровождается разрушением миелина. В результате в веществе головного мозга происходит мультифокальная прогрессирующая демиелинизация с ростом и слиянием очагов поражения. Микроскопически обнаруживается увеличение астроцитов, деформация их ядер, окрашивание олигодендроцитов выявляет ядерные включения — скопления частиц JCV. Первостепенную роль в иммунной антивирусной реакции играют цитотоксические Т-лимфоциты, убивающие инфицированные активным вирусом клетки. Снижение выработки специфических Т-лимфоцитов вследствие иммунодефицита обуславливает развитие ПМЛ.
Симптомы ПМЛ
Дебют заболевания носит подострый (2-3 дня) или постепенный (1-3 недели) характер. На первый план выходит патопсихологическая симптоматика и очаговый неврологический дефицит. В типичном варианте прогрессирующая мультифокальная лейкоэнцефалопатия протекает без свойственных нейроинфекциям общемозговых симптомов, менингеального синдрома. Отмечается изменение поведения, агрессивность, эмоциональная лабильность, подозрительность, прогрессирующее ослабление когнитивной сферы (памяти, мышления, внимания). Очаговый дефицит представлен мышечной слабостью конечностей одной половины тела (гемипарезом), афазией, гемианопсией, атаксией, парестезиями в паретичных конечностях. Вначале гемипарез может отсутствовать, в дальнейшем наблюдается у 75% больных. 20% случаев протекают с пароксизмами эпилепсии. Психические расстройства отмечаются у 38% пациентов. Прогрессирование когнитивного дефицита приводит к деменции.
На первый план выходит патопсихологическая симптоматика и очаговый неврологический дефицит. В типичном варианте прогрессирующая мультифокальная лейкоэнцефалопатия протекает без свойственных нейроинфекциям общемозговых симптомов, менингеального синдрома. Отмечается изменение поведения, агрессивность, эмоциональная лабильность, подозрительность, прогрессирующее ослабление когнитивной сферы (памяти, мышления, внимания). Очаговый дефицит представлен мышечной слабостью конечностей одной половины тела (гемипарезом), афазией, гемианопсией, атаксией, парестезиями в паретичных конечностях. Вначале гемипарез может отсутствовать, в дальнейшем наблюдается у 75% больных. 20% случаев протекают с пароксизмами эпилепсии. Психические расстройства отмечаются у 38% пациентов. Прогрессирование когнитивного дефицита приводит к деменции.
В редких случаях мультифокальная лейкоэнцефалопатия протекает в атипичной форме. К атипичным вариантам относятся JC-менингоэнцефалит, JC-энцефалопатия, гранулярно-клеточная невропатия. Менингоэнцефалитическая форма характеризуется наличием менингеальных симптомов. При JC-энцефалопатии отсутствует очаговый неврологический дефицит. Клиника гранулярно-клеточного варианта представлена изолированным мозжечковым синдромом.
Менингоэнцефалитическая форма характеризуется наличием менингеальных симптомов. При JC-энцефалопатии отсутствует очаговый неврологический дефицит. Клиника гранулярно-клеточного варианта представлена изолированным мозжечковым синдромом.
Диагностика
Прогрессирующая лейкоэнцефалопатия диагностируется специалистами в области неврологии на основании клинических данных, результатов нейровизуализирующего исследования, обнаружения специфической ДНК. Диагностический алгоритм включает:
- Осмотр невролога. В классическом варианте в неврологическом статусе определяется гемипарез, гемигипестезия, шаткость, неустойчивость в позе Ромберга, дискоординация, сенсомоторная афазия, когнитивные нарушения. Наблюдается лабильность психики, психопатологические симптомы, возможно неадекватное поведение.
- Осмотр офтальмолога. У большинства пациентов диагностируют снижение зрения, периметрия выявляет гомонимную гемианопсию.
- МРТ головного мозга.
 Обнаруживается диффузная мультифокальная демиелинизация, очаги имеют различный размер, асимметрично располагаются в белом веществе, таламусе, базальных ядрах.
Обнаруживается диффузная мультифокальная демиелинизация, очаги имеют различный размер, асимметрично располагаются в белом веществе, таламусе, базальных ядрах. - ПЦР-исследование. Направлено на выявление ДНК вируса JC в цереброспинальной жидкости, полученной путём люмбальной пункции. Специфичность анализа 90-100%, чувствительность — 70-90%. Проведение антиретровирусной терапии больным СПИДом понижает чувствительность исследования до 58%, отрицательный результат не исключает наличие заболевания.
- Биопсию мозговых тканей. Инвазивная методика, проводится в диагностически затруднительных случаях. Гистологическое исследование образцов церебральных тканей позволяет подтвердить специфические для лейкоэнцефалопатии морфологические изменения.
 Обнаруживается диффузная мультифокальная демиелинизация, очаги имеют различный размер, асимметрично располагаются в белом веществе, таламусе, базальных ядрах.
Обнаруживается диффузная мультифокальная демиелинизация, очаги имеют различный размер, асимметрично располагаются в белом веществе, таламусе, базальных ядрах.
Точный диагноз «прогрессирующая мультифокальная лейкоэнцефалопатия» правомочен, когда классические клинические проявления, изменения МРТ сочетаются с положительным результатом ПЦР или имеют подтверждение по данным гистологии. Наличие только клинических и МРТ признаков позволяет трактовать диагноз как вероятный. Дифференциальная диагностика проводится с первичным нейроСПИДом, нейроревматизмом, вирусными энцефалитами.
Наличие только клинических и МРТ признаков позволяет трактовать диагноз как вероятный. Дифференциальная диагностика проводится с первичным нейроСПИДом, нейроревматизмом, вирусными энцефалитами.
Лечение ПМЛ
В настоящее время не существует препаратов для лечения прогрессирующей лейкоэнцефалопатии с доказанной эффективностью. Специфическая терапия находится в стадии разработки. Попытки лечения интерфероном, иммуностимуляторами, цитарабином, их комбинациями оказались безрезультатными. Окончились неудачей клинические испытания препарата цидофовир, показывающего анти-JC эффективность на опытах с мышами. Недавно был предложен кардинально новый метод лечения антидепрессантом миртазапином, блокирующим распространение JCV благодаря связыванию рецепторов, через которые вирус инфицирует клетки нейроглии. Способ требует клинических испытаний.
Прогноз и профилактика
Прогрессирующая мультифокальная лейкоэнцефалопатия отличается неуклонно усугубляющимся течением с исходом в кому. Продолжительность жизни варьирует от 1 мес. (острая форма) до 10-12 мес. с момента заболевания. Профилактика подразумевает меры предупреждения инфицирования ВИЧ, осторожное проведение терапии аутоиммунных заболеваний, мониторинг неврологической симптоматики у больных, получающих лечение моноклональными препаратами.
Продолжительность жизни варьирует от 1 мес. (острая форма) до 10-12 мес. с момента заболевания. Профилактика подразумевает меры предупреждения инфицирования ВИЧ, осторожное проведение терапии аутоиммунных заболеваний, мониторинг неврологической симптоматики у больных, получающих лечение моноклональными препаратами.
Вирус JC и прогрессирующая мультифокальная лейкоэнцефалопатия
?
LiveJournal
- Main
- Ratings
- Interesting
- iOS & Android
Disable ads
Login
- Login
CREATE BLOG
Join
English
Прогрессирующая мультифокальная лейкоэнцефалопатия
Прогрессирующая мультифокальная лейкоэнцефалопатия — это тяжелое прогрессирующее демиелинизирующее заболевание нервной системы, возникающее вследствие активации JC-вируса при иммунодефицитных состояниях (особенно ассоциированно со СПИДом, но также развивается у лиц, перенесших трансплантацию костного мозга или органа, страдающих онкогематологическими и онкологическими заболеваниями, воспалительными заболеваниями и имеющими изолированные CD4 лимфоцитопении). Также может быть ассоциирована с назначением натализумаба или использованием моноклональных антител. Заболевание является оппортунистической инфекцией. JC-вирус распространен повсеместно, инфицирование происходит в ранний период жизни и протекает бессимптомно.
Также может быть ассоциирована с назначением натализумаба или использованием моноклональных антител. Заболевание является оппортунистической инфекцией. JC-вирус распространен повсеместно, инфицирование происходит в ранний период жизни и протекает бессимптомно.
Клиническая картина
Клиническая картина ПМЛ неспецифична. Заболевание развивается подостро (несколько дней) или постепенно (несколько недель), формируется неврологическая и патопсихологическая симптоматика. Клинические признаки инфекции отсутствуют. Возможны расстройства чувствительности, парезы, гемианопсии, нарушения высших корковых функций, психические расстройства, деменция, эписиндром. Летальный исход наступает в течение года, в среднем от 2 до 6 месяцев.
Диагностика
Диагноз основывается на выделении ДНК вируса методом ПЦР в цереброспинальной жидкости и по данным МРТ.
Магнитно-резонансная томография
Типичной картиной по данным МРТ являются сливные двусторонние, асимметричные очаги в белом веществе головного мозга, наиболее часто поражающие теменные и затылочные доли, реже лобные, а также мозолистое тело, могут быть расположены перивентрикулярно.
Диагноз правомочен, когда имеются типичные клинические данные, картина на МРТ, выявлена ДНК вируса в ликворе или есть результаты биопсии.
Лечение
Специфическое лечение ПМЛ не разработано. При развитии данной патологии целесообразно максимально снизить дозу глюкокортикоидов и цитотоксических препаратов. Некоторые авторы предлагают сочетание плазмафереза (5 сеансов через день) с последующим приемом амино- хинолинового препарата мефлоцина и миртазипина (антидепрессант, ингибитор обратного захвата серотонина, замедляющий распространение JC-вируса путем блокирования специфических рецепторов).
Глава 1.: «Прогрессирующая мультифокальная лейкоэнцефалопатия»
Книга: «Редкие неврологические синдромы и болезни» (В.В. Пономарев)
Прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ) принадлежит к числу редких демиелинизирующих заболеваний головного мозга, развивающихся на фоне дефекта иммунной системы. Впервые ПМЛ описана в 1958 г. Э. Ричардсоном. Заболевание встречается повсеместно, в равной степени у мужчин и женщин, чаще в возрасте 50 лет и более. В настоящее время установлено, что ПМЛ вызывается несколькими штаммами вирусов группы папова JC (VJC), являющихся у человека оппортунистической инфекцией. По данным различных авторов, 70—90% населения заражены VJC еще с детства [2, 7-9]. Различные патологические состояния, сопровождающиеся явлениями иммунодефицита: лимфопролиферативные болезни (лимфомы ЦНС), миелопролиферативные болезни (лейкозы, миеломная болезнь), врожденная гипогаммаглобулинемия, канцероматозы, диффузные болезни соединительной ткани, СПИД, состояния после иммуносупрессивной терапии в связи с пересадкой органов и тканей способствуют реактивации вируса из латентного состояния [4, 10, 14]. Мишенями данной инфекции являются миелин- продуцирующие клетки ЦНС, поэтому их повреждение клинически выражается процессами демиелинизации [3, 8].
Э. Ричардсоном. Заболевание встречается повсеместно, в равной степени у мужчин и женщин, чаще в возрасте 50 лет и более. В настоящее время установлено, что ПМЛ вызывается несколькими штаммами вирусов группы папова JC (VJC), являющихся у человека оппортунистической инфекцией. По данным различных авторов, 70—90% населения заражены VJC еще с детства [2, 7-9]. Различные патологические состояния, сопровождающиеся явлениями иммунодефицита: лимфопролиферативные болезни (лимфомы ЦНС), миелопролиферативные болезни (лейкозы, миеломная болезнь), врожденная гипогаммаглобулинемия, канцероматозы, диффузные болезни соединительной ткани, СПИД, состояния после иммуносупрессивной терапии в связи с пересадкой органов и тканей способствуют реактивации вируса из латентного состояния [4, 10, 14]. Мишенями данной инфекции являются миелин- продуцирующие клетки ЦНС, поэтому их повреждение клинически выражается процессами демиелинизации [3, 8].
Клиническая картина ПМЛ характеризуется постепенным началом, прогрессирующим течением, ранним развитием деменции, нарушением речи, пирамидными и экстрапирамидными расстройствами, псевдобульбарным синдромом, эпилептическими припадками. Реже заболевание проявляется стволовым и мозжечковым синдромами [3, 13]. Описаны случаи дебюта ПМЛ с развития синдрома надъядерного паралича, фокальных двигательных расстройств. Распространение очагов демиелинизации ведет к прогрессированию симптоматики. Специфическая противовирусная терапия ПМЛ не разработана. Спонтанная стабилизация процесса отмечается редко. Большинством исследователей подчеркивается высокая летальность от ПМЛ [3].
Реже заболевание проявляется стволовым и мозжечковым синдромами [3, 13]. Описаны случаи дебюта ПМЛ с развития синдрома надъядерного паралича, фокальных двигательных расстройств. Распространение очагов демиелинизации ведет к прогрессированию симптоматики. Специфическая противовирусная терапия ПМЛ не разработана. Спонтанная стабилизация процесса отмечается редко. Большинством исследователей подчеркивается высокая летальность от ПМЛ [3].
Мы наблюдали 10 больных с ПМЛ, 50% из которых погибли спустя 4—9 месяцев от дебюта заболевания. У выживших пациентов установлена I или II группа инвалидности в связи с сохранением грубого неврологического дефицита. Приводим одно из этих наблюдений.
Больной Р., 25 лет, безработный, при поступлении в неврологическое отделение жалоб не предъявлял в связи с тяжестью состояния. Болен в течение 2 месяцев, когда на фоне перенесенных стрессов стали периодически появляться агрессивность, неадекватность поведения, двигательное беспокойство. По поводу одного из таких состояний больной был госпитализирован в психиатрический стационар с диагнозом «полиморфное психотическое расстройство». Спустя месяц на фоне терапии состояние быстро ухудшилось: развилась слабость в конечностях, нарушились речь и походка, в связи с этим переведен для дальнейшего лечения в неврологическое отделение. При переводе состояние больного было оценено как средней тяжести, АД 125/80 мм рт. ст., соматической патологии не выявлено. Неврологически: в сознании, обращенную речь понимает, на нее реагирует кивком головы, не говорит, выполняет простые инструкции. Рот самостоятельно не открывает из-за выраженного тризма мышц, язык не показывает. Глотание не нарушено, глоточный рефлекс живой. Зрачки равновеликие, реакция зрачков на свет живая, глазодвигательных нарушений нет. Положительные рефлексы орального автоматизма. Сила в левой руке достаточная, в правой руке снижена до 1—2 баллов, в правой ноге — 4 балла, мышечный тонус повышен по пирамидному типу, больше в правых конечностях. Сухожильно-периостальные рефлексы высокие, зоны расширены, D>S, патологические стопные рефлексы Бабинского, Россолимо с двух сторон.
Спустя месяц на фоне терапии состояние быстро ухудшилось: развилась слабость в конечностях, нарушились речь и походка, в связи с этим переведен для дальнейшего лечения в неврологическое отделение. При переводе состояние больного было оценено как средней тяжести, АД 125/80 мм рт. ст., соматической патологии не выявлено. Неврологически: в сознании, обращенную речь понимает, на нее реагирует кивком головы, не говорит, выполняет простые инструкции. Рот самостоятельно не открывает из-за выраженного тризма мышц, язык не показывает. Глотание не нарушено, глоточный рефлекс живой. Зрачки равновеликие, реакция зрачков на свет живая, глазодвигательных нарушений нет. Положительные рефлексы орального автоматизма. Сила в левой руке достаточная, в правой руке снижена до 1—2 баллов, в правой ноге — 4 балла, мышечный тонус повышен по пирамидному типу, больше в правых конечностях. Сухожильно-периостальные рефлексы высокие, зоны расширены, D>S, патологические стопные рефлексы Бабинского, Россолимо с двух сторон. Чувствительность достоверно проверить невозможно из-за отсутствия продуктивного контакта с больным. Координаторные пробы не выполняет, самостоятельно не садится. Менингеальных знаков не выявлено. Тазовых нарушений нет.
Чувствительность достоверно проверить невозможно из-за отсутствия продуктивного контакта с больным. Координаторные пробы не выполняет, самостоятельно не садится. Менингеальных знаков не выявлено. Тазовых нарушений нет.
При обследовании: общий анализ крови НЬ 149 г/л, лейкоциты 8,2 • 109, СОЭ 8 мм/ч, в формуле крови выявлена лимфопения 18%, остальные показатели в норме. Биохимический анализ крови без особенностей. Иммунограмма крови: IgG 6,5 г/л, IgA 0,8 г/л, IgM 1,0 г/л. ИФА на ВИЧ отрицательный. СМЖ: белок 0,25 г/л, цитоз 36 • 106 клеток/л. Иммуноферментные исследования крови и СМЖ не выявили достоверного повышения титра антител к вирусу простого герпеса. ЭЭГ: умеренные изменения биоэлектрической активности мозга на сниженном амплитудном уровне. Окулист: острота зрения OD/OS 0,1 с коррекцией 0,9/0,8, глазное дно без патологии. МРТ головного мозга (дважды): в белом веществе полушарий мозга перивентрикулярно выявлены множественные полиморфные, местами сливающиеся между собой, неоднородные, гиперинтенсивные в T2w очаги размерами от 3 до 30 мм в диаметре (рис.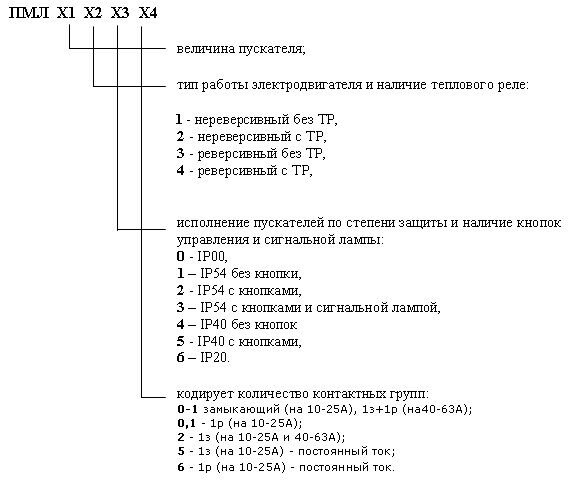 15). Базальные цистерны, желудочки мозга, кортикальные борозды диффузно расширены.
15). Базальные цистерны, желудочки мозга, кортикальные борозды диффузно расширены.
Рис. 15. МРТ в T2w режиме больного Р., 25 лет, с диагнозом «вероятный ПМЛ»:
А) субкортикально и Б) перивентрикулярно выявлены множественные, сливные, гиперинтенсивные очаги размерами от 3 до 30 мм в диаметре
Получал массивную инфузионную и противовоспалительную терапию, ноотропы, антиоксиданты (карнитин, глиатилин), миорелаксанты (баклофен), иммуноглобулин внутримышечно; проведено 5 сеансов плазмафереза. На фоне проводимой терапии состояние больного начало улучшаться: увеличилась сила, снизился мышечный тонус в конечностях, появилась возможность самостоятельной речи, жевания и глотания, а также самостоятельного передвижения с опорой на ходунки. При динамическом наблюдении в течение года у больного сохраняются умеренный псевдобульбарный, легкий акинетико-ригидный синдром, правосторонний гемипарез и выраженные когнитивные нарушения. Установлена I группа инвалидности.
Таким образом, у больного на фоне дефицита гуморального иммунитета заболевание первоначально проявилось продуктивной психотической симптоматикой с последующим присоединением поражения пирамидной, экстрапирамидной системы, псевдобульбарного синдрома и акинетического мутизма. Сочетание данных клинических признаков, результаты МРТ и были достаточно характерны для ПМЛ. К сожалению, биопсийное исследование мозга и СМЖ на VJC провести не удалось, поэтому в данном случае можно говорить лишь о «вероятном ПМЛ». Назначенное патогенетическое, иммунокорригирующее и симптоматическое лечение позволило добиться частичного регресса симптоматики с сохранением остаточного когнитивного и двигательного дефицита.
Сочетание данных клинических признаков, результаты МРТ и были достаточно характерны для ПМЛ. К сожалению, биопсийное исследование мозга и СМЖ на VJC провести не удалось, поэтому в данном случае можно говорить лишь о «вероятном ПМЛ». Назначенное патогенетическое, иммунокорригирующее и симптоматическое лечение позволило добиться частичного регресса симптоматики с сохранением остаточного когнитивного и двигательного дефицита.
Большой научный и практический интерес представляет изучение особенностей VJC-инфекции, относящейся к группе полиомавирусов. Проведенные исследования установили широкое распространение VJC среди городского населения различных географических регионов [4]. В природе вирус достаточно устойчив. Вирусные частицы остаются стабильными при 20° С более 70 дней. Проникновение VJC или вирусных ДНК в человеческий организм происходит с водой, пищей, воздушно-капельным путем [5]. После заражения вирус включается в ядра клеток. Особенностью данной инфекции является ее способность к персистенции (в почках, костном мозге, лимфоцитах крови), реактивации, хронизации и интермиттирующему течению [9, 13]. Выделено четыре генотипа VJC, имеющих географические особенности. Тип 1 (62%) преобладает в Европе. Тип 2 (9,7%) и тип 3 (8,9%) чаще встречаются в Азии и Африке. Тип 4 (5,4%) является рекомбинантным типов 1—3 и редко служит причиной ПМЛ [15]. Диссеминация вируса в головной мозг осуществляется кровью. Исследования доказали высокую специфичность VJC к миелинпродуцирующим олигодендроцитам мозга. На фоне иммуносупрессии вирус способен к реактивации из латентного состояния, вызывая деструкцию миелина и приводя к неврологическим нарушениям. Мультицентровыми исследованиями подтверждена корреляция между генотипом VJC и неблагоприятным течением заболевания. Установлено, что обнаружение типов 1 и 2 вируса — фактор риска развития ПМЛ [6]. У больных с быстро прогрессирующим течением заболевания VJC определяется в сыворотке в 75% случаев, а в мозге и ликворе в 100% [7].
Выделено четыре генотипа VJC, имеющих географические особенности. Тип 1 (62%) преобладает в Европе. Тип 2 (9,7%) и тип 3 (8,9%) чаще встречаются в Азии и Африке. Тип 4 (5,4%) является рекомбинантным типов 1—3 и редко служит причиной ПМЛ [15]. Диссеминация вируса в головной мозг осуществляется кровью. Исследования доказали высокую специфичность VJC к миелинпродуцирующим олигодендроцитам мозга. На фоне иммуносупрессии вирус способен к реактивации из латентного состояния, вызывая деструкцию миелина и приводя к неврологическим нарушениям. Мультицентровыми исследованиями подтверждена корреляция между генотипом VJC и неблагоприятным течением заболевания. Установлено, что обнаружение типов 1 и 2 вируса — фактор риска развития ПМЛ [6]. У больных с быстро прогрессирующим течением заболевания VJC определяется в сыворотке в 75% случаев, а в мозге и ликворе в 100% [7].
Патоморфологические исследования при ПМЛ выявляют множественные очаги демиелинизации, особенно в границе между серым и белым веществом, что имело место в нашем случае. В отличие от рассеянного склероза, при ПМЛ крайне редко встречаются очаги демиелинизации в субпиальных и субэпендимарных зонах. При макроскопическом исследовании обнаруживаются слабо выраженные воспалительные реакции и отек.
В отличие от рассеянного склероза, при ПМЛ крайне редко встречаются очаги демиелинизации в субпиальных и субэпендимарных зонах. При макроскопическом исследовании обнаруживаются слабо выраженные воспалительные реакции и отек.
Микроскопическое исследование указывает на типичные внутриядерные включения в олигодендроцитах, реже в астроцитах, причем последние могут сливаться между собой, образуя гигантские клетки [12].
В патогенезе демиелинизации имеют значение как непосредственное поражение олигодендроцитов, так и аутоиммунные реакции на фоне иммунодефицита. Иммунологическое исследование крови при ПМЛ свидетельствует о генерализованном вовлечении клеточного и гуморального иммунитета. Снижение гуморального иммунного ответа свидетельствует о персистенции инфекции и проявляется в снижении уровня IgG, что также встретилось у нашего больного. Изредка может наблюдаться увеличение IgM в сыворотке крови. Интратекальный синтез IgG к протеину VJC при ПМЛ обнаружен у 76% больных, в отличие от 3,2% у здоровых лиц, и, по мнению Т. Weber и соавт. [14], может использоваться как дополнительный диагностический тест. Этими же авторами установлено, что появление VJC-специфических лимфоцитов ассоциируется с благоприятным клиническим прогнозом.
Weber и соавт. [14], может использоваться как дополнительный диагностический тест. Этими же авторами установлено, что появление VJC-специфических лимфоцитов ассоциируется с благоприятным клиническим прогнозом.
Клиническая картина ПМЛ не обладает строгой специфичностью. До появления эпидемии СПИД заболевание встречалось редко, однако в настоящее время им страдают 5—10% этих больных [2, 13]. Ранее считалось, что при нормальном функционировании иммунной системы вирус не вызывает никаких клинических симптомов. В настоящее время установлено, что иммунодефицит имеет место у 85% больных, а у остальных ПМЛ может развиться при отсутствии дефекта иммунной системы [14]. Диагностика ПМЛ осуществляется на основании следующих признаков:
• Наличие многоочаговой неврологической симптоматики (головная боль и повышение внутричерепного давления не характерны).
• СМЖ чаще не изменяется.
• Типичная МРТ картина у 90% больных: высокоинтенсивные сигналы в T2w и понижение плотности в Tiw без накопления контраста и масс-эффекта. Характерно поражение аркуатных волокон (V-волокон) затылочно-теменных областей. MPT-изменения в мозге могут быть одно- или двусторонними, единичными или множественными, симметричными или асимметричными. Атипичные нарушения МРТ при ПМЛ включают фокальные геморрагии, атрофию мозга, перифокальный отек, вовлечение базальных ганглиев [11].
Характерно поражение аркуатных волокон (V-волокон) затылочно-теменных областей. MPT-изменения в мозге могут быть одно- или двусторонними, единичными или множественными, симметричными или асимметричными. Атипичные нарушения МРТ при ПМЛ включают фокальные геморрагии, атрофию мозга, перифокальный отек, вовлечение базальных ганглиев [11].
• Наличие иммунологических признаков первичного (СПИД) или вторичного иммунодефицита.
• В биоптате мозговой ткани при электронной микроскопии обнаруживают VJC, при иммуноцитохимии — антигены VJC, при полимеразной цепной реакции — вирусный геном в 95,8% случаев [7].
Дифференциальный диагноз проводится с хроническим герпетическим энцефалитом, первично-прогрессирующим течением рассеянного склероза, ВИЧ-энцефалопатией. Для последней при МРТ характерно диффузное поражение мозга, менее интенсивное на T2w и не видное на Тiw, без вовлечения аркуатных волокон. В сложных диагностических случаях применяется стерео-таксическая биопсия мозга [2].
Большинство исследователей подчеркивают прогрессирующее, чаще фатальное течение ПМЛ [1]. Сообщается о выживших больных спустя 4 года после установления диагноза ПМЛ на основании патоморфологического и вирусологического обследования [3]. Специфическая терапия ПМЛ не разработана. Описаны положительные клинико-иммунологические, гематологические результаты внутривенного и эндолюмбального введения цитарабина в сочетании с интерфероном-альфа, а также интерлейкина-2 в дозе 0,5 мЕ/м2 ежедневно [10]. В литературе встречаются сообщения о стабилизации процесса сроком на год у 36% больных после введения цитозин-арабинозида внутривенно. Подчеркивается, однако, что этот препарат обладает токсическим действием [1]. Перспективным в лечении ПМЛ является использование высокоактивной антиретровирусной терапии и цидофовира, которые будут применяться в будущем [2, 13].
Сообщается о выживших больных спустя 4 года после установления диагноза ПМЛ на основании патоморфологического и вирусологического обследования [3]. Специфическая терапия ПМЛ не разработана. Описаны положительные клинико-иммунологические, гематологические результаты внутривенного и эндолюмбального введения цитарабина в сочетании с интерфероном-альфа, а также интерлейкина-2 в дозе 0,5 мЕ/м2 ежедневно [10]. В литературе встречаются сообщения о стабилизации процесса сроком на год у 36% больных после введения цитозин-арабинозида внутривенно. Подчеркивается, однако, что этот препарат обладает токсическим действием [1]. Перспективным в лечении ПМЛ является использование высокоактивной антиретровирусной терапии и цидофовира, которые будут применяться в будущем [2, 13].
Прогрессирующая многоочаговая лейкоэнцефалопатия (ПМЛ)
Д. X. Хартер, Р. Г. Петерсдорф (D. Я. Harter, R. G. Petersdorf)
Это редкое неврологическое поражение обычно развивается у больных, страдающих
лейкозом, злокачественной лимфомой, карциноматозом, синдромом приобретенного
иммунодефицита (СПИД) и некоторыми другими хроническими заболеваниями, или у
лиц, получающих иммуносупрессивную терапию. Заболевания сопровождаются
Заболевания сопровождаются
нарушениями клеточного иммунитета и дефицитом гуморальной реакции антител (хотя
последнего может и не быть).
Болеют взрослые лица обоих полов. Длительность течения болезни от момента
возникновения симптомов до летального исхода составляет 1-6 мес и более.
Неврологические симптомы отражают диффузное асимметричное поражение полушарий
головного мозга. Характерны гемиплегия, гемианопсия, афазия или дизартрия, в
некоторых случаях — нарушения полей зрения, частичный или полный поперечный
миелит. Головная боль и судорожные припадки встречаются редко, но весьма типичны
диффузные и очаговые изменения на ЭЭГ. Очаги поражения в белом веществе могут
быть визуализированы при КТ. Эффективным методом выявления деструкции белого
вещества служит ЯМР. СМЖ без изменений. Специфический диагноз основан на
результатах биопсии мозга.
Патологоанатомические изменения характеризуются множественными участками
демиелинизации с небольшой периваскулярной инфильтрацией или без таковой и
измененными митотическими структурами в астроцитах. Присутствие отчетливых
Присутствие отчетливых
внутриядерных включений в олигодентроцитах послужило первым указанием на
вирусную этиологию заболевания. Электронно-микроскопические наблюдения показали,
что тельца внутриядерных включений состоят из плотно уложенных сфер, обладающих
физическими размерами и свойствами полиомавирусов из группы паповавирусов.
Использование тканевых культур, полученных из мозга человеческого эмбриона,
позволило выделить от больных с ПМЛ новый серотип полиомавируса человека (вирус
JC). В пораженном мозге вирусные частицы присутствуют в огромном количестве.
Быстрая идентификация вируса в мозге осуществляется с помощью окрашивания
флюоресцентными антигенами или электронно-микроскопической агглютинации с
моноспецифической гипериммунной сывороткой кролика. Серологическая диагностика с
использованием сыворотки больного дает ненадежные результаты. Вирус не был
обнаружен в других тканях, кроме мозга; болезнь не передается животным. Имеются
сообщения о клинической ремиссии на фоне терапии цитозина арабинозидом, но
излечения не наступает. Обычно больные погибают в течение 6 мес от начала
Обычно больные погибают в течение 6 мес от начала
заболевания.
Развитие ПМЛ может быть результатом активации полиомавируса, находившегося в
латентном состоянии в мозге или других тканях после перенесенной в детстве
инфекции. С другой стороны, у некоторых лиц, не приобретших иммунитета в
детстве, столкновение с вирусом происходит во время заболевания, влияющего на
развитие клеточного иммунитета. Возникающая демиелинизация может быть связана с
индуцированным вирусом повреждением олигодендроглии, клетки которой в норме
участвуют в процессах восстановления миелина.
Прогрессирующий энцефалит, вызываемый вирусом краснухи
Хронический прогрессирующий энцефалит, развивающийся у мальчиков с типичными
стигмами синдрома врожденной краснухи и имеющий некоторые общие признаки с ПСПЭ,
был впервые описан в 1974 г. Всего имеются сообщения менее чем о 20 больных.
Заболевание начинается на втором десятилетии жизни и характеризуется слабоумием,
мозжечковой атаксией, спастичностью и судорожными припадками. В СМЖ увеличены
В СМЖ увеличены
число клеток, содержание белка и IgG. Высокие титры антител к вирусу краснухи
удается выявить как в сыворотке крови, так и в СМЖ. Вирус краснухи выделяют из
мозга с помощью метода кокультивирования.
В отличие от ПСПЭ у больных с панэнцефалитом, вызванным вирусом краснухи, до
начала этого прогрессирующего заболевания находят стигмы врожденной краснухи. По
сравнению с ПСПЭ миоклонии менее постоянны и на ЭЭГ не отмечается угнетения
всплесков активности. При гистологическом исследовании мозга выявляют
минерализацию старых очагов поражений и воспалительную реакцию, но не
обнаруживают телец-включений, типичных для ПСПЭ.
Клиническая картина прогрессирующего краснушечного энцефалита сходна также с
редкими случаями юношеского паралича, наблюдающегося у больных с врожденным
сифилисом. Иммунный статус пациентов с энцефалитом, вызванным вирусом краснухи,
полностью не изучен, и патогенез заболевания остается неясным.
Персистирующие вирусные заболевания у больных с иммунодефицитом
Персистирующие, или хронические, инфекции нервной системы могут развиваться у
больных с иммунодефицитом. Состояния иммунодефицита бывают врожденными
Состояния иммунодефицита бывают врожденными
(первичными) или приобретенными. На протяжении многих лет из СМЖ больных с
первичной агаммаглобулинемией выделяют энтеровирусы, а также выявляют плеоцитоз.
Хронический или подострый энцефалит встречается также у детей с врожденной
гипогаммаглобулинемией. Ассоциация специфического вируса с данным заболеванием
не установлена.
Неврологические состояния, связанные с синдромом приобретенного иммунодефицита
(СПИД)
Поражение ЦНС при СПИДе может сопровождаться комплексными клиническими
проявлениями. Есть данные, что вызывающие СПИД вирусы иммунодефицита человека
(ВИЧ) [вирус Т-клеточной лейкемии человека (HTLV III), вирус, ассоциированный с
лимфаденопатией (LAV), или СПИД-ассоциированный ретровирус (ARV)] способны
вызывать первичное поражение нервной системы, а также иммунные сдвиги, что
позволяет другим вирусам реплицироваться в нервной ткани и повреждать ее.
В связи с нарушенной иммунологической реактивностью больной СПИДом восприимчив к
различным инфекционным агентам, способным приводить к поражению ЦНС. Среди этих
Среди этих
вирусов чаще всего встречаются герпесвирусы и паповавирусы, которые могут
оставаться латентными до возникновения иммунной дисфункции. К числу наиболее
часто изолируемых вирусов этих групп относятся вирус простого герпеса (ВПГ),
цитомегаловирус и возбудитель ПМЛ (вирус JC). Инфекции, вызываемые этими
вирусами, сопровождаются поражением нервной системы у больных СПИДом (чаще всего
это атипичный асептический менингит, острый или подострый энцефалит, ПМЛ и
вирусный миелит). Помимо вирусного энцефалита и ПМЛ, у больных СПИДом возможно
развитие токсоплазменного абсцесса мозга и первичных лимфом ЦНС.
Дифференцировать эти состояния на основании лишь данных КТ и других лабораторных
методов сложно. Поскольку лечение при этих состояниях различно, необходимо
проведение биопсии мозга.
У больного СПИДом могут развиться различные поражения спинного мозга. Это
вакуолярная миелопатия, при которой особенно сильно поражаются боковые и задние
столбы грудного отдела спинного мозга, острый вирусный миелит, обычно вызываемый
ВПГ, и восходящий миелит. При СПИДе возможно также поражение периферических
При СПИДе возможно также поражение периферических
нервов.
Есть данные, что HTLV III способен к репликации в мозге. Введение шимпанзе
суспензии мозга больных СПИДом вызывает у животных сывороточную трансформацию;
вирус удается изолировать из лейкоцитов шимпанзе. Сывороточная трансформация для
анти-HTLV III сопровождается у больных СПИДом острой энцефалопатией. ДНК и РНК
HTLV III обнаружены в мозге детей и взрослых, страдающих СПИДом, и HTLV III был
изолирован из СМЖ и невральных тканей больных СПИДом. У больных СПИДом с
неврологическими симптомами содержание HTLV III-специфичного IgG в СМЖ выше, чем
в крови, и это дает основание полагать, что синтез IgG происходит в пределах
ЦНС. HTLV III был выделен также из спинного мозга и икроножного нерва, что
позволяет рассматривать этот ретровирус в качестве возможного этиологического
фактора миелопатии и периферической нейропатии при СПИДе.
У больных СПИДом описана подострая деменция с сопутствующими двигательными
расстройствами. Данное состояние может являться непосредственной манифестацией
Данное состояние может являться непосредственной манифестацией
инфекционного поражения мо
Прогрессирующая мультифокальная лейкоэнцефалопатия (ПМЛ) и её раннее обнаружение при РС-е.
Доброго времени суток. Нашел ряд интересных исследований о прогрессирующей
мультифокальной лейкоэнцефалопатии (ПМЛ) и её раннем обнаружении при рассеянном
склерозе. Найти эту работу на английском
языке можно по ссылке https://www.ncbi.nlm.nih.gov/pubmed/30389778 , итак.
Scarpazza C, Signori A, Prosperini L, Sormani MP, Cosottini M, Capra R, Gerevini S; Группа итальянских пациентов с ПМЛ. Ранний диагноз прогрессирующей мультифокальной лейкоэнцефалопатии: продольное развитие очагов. J. Neurol Neurosurg Psychiatry. 2018 2 ноября. Pii: jnnp-2018-319208.
ЦЕЛЬ: Ранняя диагностика прогрессирующей мультифокальной лейкоэнцефалопатии, связанная с натализумабом (Тизабри, NTZ-PML), при рассеянном склерозе была признана регулирующими агентствами одним из основных приоритетов. Тем не менее это все еще не является реальностью. Настоящая работа направлена на: (1) исследовать, проходят ли пациенты с NTZ-PML через длительную предсимптомную фазу с аномалиями МРТ; (2) оценить увеличение продольного объема очагов ПМЛ во время предсимптомной фазы и (3) оценить длину предсимптомной фазы и её влияние на продолжительность терапии в качестве параметра стратификации риска.
Тем не менее это все еще не является реальностью. Настоящая работа направлена на: (1) исследовать, проходят ли пациенты с NTZ-PML через длительную предсимптомную фазу с аномалиями МРТ; (2) оценить увеличение продольного объема очагов ПМЛ во время предсимптомной фазы и (3) оценить длину предсимптомной фазы и её влияние на продолжительность терапии в качестве параметра стратификации риска.
МЕТОДЫ: В исследования были включены все итальянские пациенты, которые получили NTZ-PML (ПМЛ связанный с приемом тизабри) в период с 2009 по 2018 год. Были проанализированы доступные преддиагностические МРТ данные пациентов (n = 41, видимо их было всего 41). Подробная клиническая и нейрорадиологическая информация была доступна для каждого участника.
РЕЗУЛЬТАТЫ: (1) очаги ПМЛ были выявлены на предсимптомной стадии у 32/41 (78%) пациентов; (2) объем поражения увеличился на 62,8% за каждый месяц, преддиагностической фазы; (3) длина преддиагностической фазы составляла 150,8 ± 74,9 дня; (4) Признаки PML на MRI были обнаружены до 24-го месяца терапии у 31,7% пациентов.
ВЫВОДЫ: Учитывая задержку клинического проявления ПМЛ, во время предсимптомной фазы полезно проходить обследования МРТ каждые 3-4 месяца. Ранняя диагностика может помочь улучшить результаты для пациентов из-за взаимосвязи между объемом поражения и инфекцией вируса JC. Понимание этого исследования может также повлиять на алгоритмы стратификации риска, поскольку длительность терапии как параметра стратификации, по-видимому, нуждается в переоценке.
Таким образом, данные что у пациента ПМЛ присутствуют еще до того как заболевание проявится. Вопрос лишь в том смогут ли их увидеть?
Подача заявления на получение статуса сироты | Европейское агентство по лекарственным средствам
Спонсоры должны следовать одному из двух вариантов ниже, чтобы подать заявку на определение сироты:
- Подайте заявку напрямую в EMA через систему IRIS:
Встречи перед подачей заявки не являются обязательными, и спонсоры приветствуются отправить заявку на присвоение статуса орфанного препарата без уведомления. Однако EMA было бы признательно, если бы спонсоры могли отправить заявку за несколько дней до любого из опубликованных крайних сроков подачи, доступных на веб-сайте EMA, чтобы дать больше времени для процесса проверки и возможности вмешаться в случае технических проблем.
Однако EMA было бы признательно, если бы спонсоры могли отправить заявку за несколько дней до любого из опубликованных крайних сроков подачи, доступных на веб-сайте EMA, чтобы дать больше времени для процесса проверки и возможности вмешаться в случае технических проблем.
- Запросить встречу / телеконференцию перед подачей заявки:
EMA настоятельно рекомендует спонсорам запросить встречу с Агентством перед подачей заявки до подачи заявки. Встречи перед подачей заявки обычно проходят в режиме телеконференции, если спонсор не предпочитает лично приходить в EMA.
Если спонсор считает, что ему может быть полезно предварительное обсуждение перед подачей заявки на лекарство от орфанных лекарств, он может запросить встречу / телеконференцию перед подачей заявки не менее за два месяца до запланированной даты подачи через портал IRIS.Это должно дать достаточно времени для организации и внесения любых поправок в заявку в соответствии с рекомендациями EMA.
Спонсоры должны создать первоначальный проект заявки для обозначения сирот перед созданием заявки для обсуждения перед подачей. Это две отдельные заявки , которые необходимо подать в системе IRIS. Черновик заявки на определение сирот должен быть заполнен соответствующими данными и документами, но не представлен как минимум за неделю до даты встречи перед подачей заявки.
Встречи перед подачей заявки полезны, так как процесс оценки имеет фиксированную продолжительность 90 дней и не может быть продлен из-за отсутствия данных или других пропусков в заявке. Опыт показал, что они положительно влияют на показатель успешности приложений.
Граничные условия PML
Базовый алгоритм FDTD должен быть изменен на границах вычислительного окна
, где применяются подходящие числовые поглощающие граничные условия (ABC).
Это одна из самых сложных частей моделирования FDTD. Существует несколько вариантов
Существует несколько вариантов
для типа граничных условий. Граничные условия идеально согласованного слоя (PML)
имеют наилучшие характеристики. Наш симулятор FDTD использует версию
Anisotropic PML, или так называемую версию Un-split PML (UPML). Теория UPML
очень хорошо объяснена в некоторых из приведенных здесь ссылок. Граничные условия
UPML являются физическими, а не числовыми, поскольку их реализация основана на формулировке
Максвелла, а не на математической модели.Их поглощающие свойства
физически эквивалентны свойствам поглощающей одноосной анизотропной среды
со следующими тензорами диэлектрической проницаемости и проницаемости:
Плоская волна, падающая на полупространство, состоящее из вышеуказанной одноосной среды с границей раздела
в плоскости x = const, полностью передается в него. Безотражательное свойство
полностью не зависит от угла падения, поляризации и частоты
падающей волны.Численная реализация UPML в вычислительном окне 2D
(X-Z) требует введения таких идеально согласованных поглощающих слоев
со всех сторон. Особого внимания требуют угловые области. В этих областях
Особого внимания требуют угловые области. В этих областях
тензор из уравнения 47 должен быть изменен на:
Минимизация числового коэффициента отражения анизотропных слоев PML требует
пространственного масштабирования профиля проводимости от нуля (на границе раздела PML) до максимального значения
в конце вычислительного окна:
, где L — толщина анизотропной ПМЛ.Типичные значения параметра м
составляют от 2 до 4.
| PML | Прогрессирующая мультифокальная лейкоэнцефалопатия Медицина »Физиология — и многое другое … | Оцените: | |||
| PML | Пакистан Мусульманская лига Правительственный »Политика | Оцените: | |||
| PML | Плимутская морская лаборатория Академия и наука» Наука об океане | Оценить : | |||
| PML | Вероятный максимальный убыток Бизнес »Бухгалтерский учет | Оцените его: | |||
| PML | Язык управления принтером Вычисления» Общие вычисления | Оцените: | |||
| PML | Язык разметки Palm Вычисления »Общие вычисления | Оценить: | |||
| PML | Pine Mountain Lake Правительственный »Государственный и местный | Оцените: | |||
| PML | Pissing Myself Laughing Интернет» Chat | Оцените: | |||
| PML | Perfect Matched Layer Академия и наука »Физика | Оцените: | |||
| PML | Персонализированная рассылка Списки Интернет »Чат | Оцените его: | |||
| PML | Язык разметки продукта Вычисления »Общие вычисления | Оценить: | |||
| PML | Личный список рассылки Интернет »Чат | Оцените: | |||
| PML | Писаю сам Смеюсь Интернет» Чат | Оцените: | |||
| PML | Язык периферийного управления Вычисления »Сети | Оцените: | |||
| PML | Язык Арахисовая разметка Вычислительная техника »Общие вычисления g | Оцените: | |||
| PML | Purvis Marine Limited Бизнес »Компании и фирмы | Оцените: | |||
| PML | Список аптек и продавцов Медицина »Больницы | Оцените: | |||
| PML | Porfirio Muñoz Ledo | Оцените: | |||
| PML | Язык разметки политик Правительство »Правительство США | Оцените: | |||
| PML | Язык управления процессами 9 0002 Вычислительная техника »Общие вычисления | Оцените: | |||
| PML | Переносная ракетная установка Правительственная» Военная промышленность | Оценить: | |||
| PML | Pull My Leg Интернет »Чат | Оцените его: | |||
| PML | Playonline Markup Language Разное» Приколы | Оцените: | |||
| PML | Порт-Моллер, Аляска, США Региональные »коды аэропортов | Оцените: | |||
| PML | Уровень сопоставления страниц 9 0005 Вычислительная техника »Общие вычисления | Оцените: |
Иммунный ответ: MedlinePlus Medical Encyclopedia
Иммунная система защищает организм от потенциально вредных веществ, распознавая антигены и реагируя на них. Антигены — это вещества (обычно белки) на поверхности клеток, вирусов, грибов или бактерий. Неживые вещества, такие как токсины, химические вещества, лекарства и инородные частицы (например, заноза), также могут быть антигенами. Иммунная система распознает и уничтожает или пытается уничтожить вещества, содержащие антигены.
Антигены — это вещества (обычно белки) на поверхности клеток, вирусов, грибов или бактерий. Неживые вещества, такие как токсины, химические вещества, лекарства и инородные частицы (например, заноза), также могут быть антигенами. Иммунная система распознает и уничтожает или пытается уничтожить вещества, содержащие антигены.
В клетках вашего тела есть белки, которые являются антигенами. К ним относится группа антигенов, называемых антигенами HLA. Ваша иммунная система учится воспринимать эти антигены как нормальные и обычно не реагирует на них.
ВНУТРЕННИЙ ИММУНИТЕТ
Врожденный или неспецифический иммунитет — это система защиты, с которой вы родились. Он защищает вас от всех антигенов. Врожденный иммунитет включает в себя барьеры, препятствующие попаданию вредных веществ в ваш организм. Эти барьеры образуют первую линию защиты иммунного ответа. Примеры врожденного иммунитета:
- Рефлекс кашля
- Ферменты в слезах и кожном масле
- Слизь, задерживающая бактерии и мелкие частицы
- Кожа
- Желудочная кислота
Врожденный иммунитет также имеет химическую форму белка, называемую врожденный гуморальный иммунитет. Примеры включают систему комплемента организма и вещества, называемые интерфероном и интерлейкином-1 (вызывающим жар).
Примеры включают систему комплемента организма и вещества, называемые интерфероном и интерлейкином-1 (вызывающим жар).
Если антиген преодолевает эти барьеры, он подвергается атаке и уничтожается другими частями иммунной системы.
ПРИОБРЕТЕННЫЙ ИММУНИТЕТ
Приобретенный иммунитет — это иммунитет, который развивается при воздействии различных антигенов. Ваша иммунная система выстраивает защиту от этого специфического антигена.
ПАССИВНЫЙ ИММУНИТЕТ
Пассивный иммунитет возникает из-за антител, которые вырабатываются в организме, отличном от вашего.Младенцы обладают пассивным иммунитетом, потому что они рождаются с антителами, которые передаются через плаценту от матери. Эти антитела исчезают в возрасте от 6 до 12 месяцев.
Пассивная иммунизация также может быть вызвана инъекцией антисыворотки, которая содержит антитела, вырабатываемые другим человеком или животным. Он обеспечивает немедленную защиту от антигена, но не обеспечивает длительной защиты. Глобулин иммунной сыворотки (назначается при заражении гепатитом) и антитоксин против столбняка являются примерами пассивной иммунизации.
Глобулин иммунной сыворотки (назначается при заражении гепатитом) и антитоксин против столбняка являются примерами пассивной иммунизации.
КОМПОНЕНТЫ КРОВИ
Иммунная система включает определенные типы белых кровяных телец. Он также включает химические вещества и белки крови, такие как антитела, белки комплемента и интерферон. Некоторые из них напрямую атакуют чужеродные вещества в организме, а другие работают вместе, чтобы помочь клеткам иммунной системы.
Лимфоциты — это белые кровяные тельца. Есть лимфоциты типа B и T.
- В-лимфоциты становятся клетками, вырабатывающими антитела.Антитела прикрепляются к определенному антигену и помогают иммунным клеткам уничтожить антиген.
- Т-лимфоциты напрямую атакуют антигены и помогают контролировать иммунный ответ. Они также выделяют химические вещества, известные как цитокины, которые контролируют весь иммунный ответ.
По мере развития лимфоцитов они обычно учатся отличать ткани вашего тела от веществ, которые обычно не встречаются в вашем организме. Как только B-клетки и T-клетки образуются, некоторые из этих клеток будут размножаться и обеспечивать «память» для вашей иммунной системы.Это позволяет вашей иммунной системе реагировать быстрее и эффективнее в следующий раз, когда вы подвергнетесь воздействию того же антигена. Во многих случаях это предотвратит заболевание. Например, человек, который переболел ветряной оспой или был вакцинирован против ветряной оспы, имеет иммунитет от повторной ветряной оспы.
Как только B-клетки и T-клетки образуются, некоторые из этих клеток будут размножаться и обеспечивать «память» для вашей иммунной системы.Это позволяет вашей иммунной системе реагировать быстрее и эффективнее в следующий раз, когда вы подвергнетесь воздействию того же антигена. Во многих случаях это предотвратит заболевание. Например, человек, который переболел ветряной оспой или был вакцинирован против ветряной оспы, имеет иммунитет от повторной ветряной оспы.
ВОСПАЛЕНИЕ
Воспалительная реакция (воспаление) возникает, когда ткани повреждены бактериями, травмами, токсинами, теплом или по любой другой причине. Поврежденные клетки выделяют химические вещества, включая гистамин, брадикинин и простагландины.Эти химические вещества заставляют кровеносные сосуды пропускать жидкость в ткани, вызывая отек. Это помогает изолировать инородное вещество от дальнейшего контакта с тканями тела.
Эти химические вещества также привлекают белые кровяные тельца, называемые фагоцитами, которые «поедают» микробы и мертвые или поврежденные клетки. Этот процесс называется фагоцитозом. В конечном итоге фагоциты погибают. Гной образуется из скопления мертвых тканей, мертвых бактерий, а также живых и мертвых фагоцитов.
Этот процесс называется фагоцитозом. В конечном итоге фагоциты погибают. Гной образуется из скопления мертвых тканей, мертвых бактерий, а также живых и мертвых фагоцитов.
НАРУШЕНИЯ И АЛЛЕРГИИ ИММУННОЙ СИСТЕМЫ
Нарушения иммунной системы возникают, когда иммунный ответ направлен против тканей тела, является чрезмерным или отсутствует.Аллергия связана с иммунным ответом на вещество, которое большинство людей воспринимает как безвредное.
ИММУНИЗАЦИЯ
Вакцинация (иммунизация) — это способ вызвать иммунный ответ. Небольшие дозы антигена, например мертвых или ослабленных живых вирусов, вводятся для активации «памяти» иммунной системы (активированных В-клеток и сенсибилизированных Т-клеток). Память позволяет вашему телу быстро и эффективно реагировать на будущие воздействия.
ОСЛОЖНЕНИЯ, СВЯЗАННЫЕ С ИЗМЕНЕНИЕМ ИММУННОГО ОТВЕТА
Эффективный иммунный ответ защищает от многих болезней и расстройств.Неэффективный иммунный ответ приводит к развитию болезней. Слишком много, слишком мало или неправильный иммунный ответ вызывает нарушения иммунной системы. Сверхактивный иммунный ответ может привести к развитию аутоиммунных заболеваний, при которых образуются антитела против собственных тканей организма.
Слишком много, слишком мало или неправильный иммунный ответ вызывает нарушения иммунной системы. Сверхактивный иммунный ответ может привести к развитию аутоиммунных заболеваний, при которых образуются антитела против собственных тканей организма.
Осложнения, вызванные измененными иммунными реакциями, включают:
Новости медицинских устройств — ScienceDaily
Всего несколько часов обучения утроили уверенность врача в использовании портативных ультразвуковых устройств
Ноябрь17 января 2020 г. — Заполнив пробел в обучении, врач создал курс, ориентированный на гериатрическую медицину, для устройств УЗИ на месте (POCUS), которые удвоили количество врачей …
Черенковская люминесцентная визуализация выявляет статус хирургического края при радикальной простатэктомии
7 октября 2020 г. — Новый метод интраоперационной визуализации, люминесцентная визуализация Черенкова (CLI), может точно оценить хирургические границы во время радикальной простатэктомии, согласно первому исследованию на людях..jpg) …
…
Проницаемость сгустка связана с успехом первой попытки аспирационной тромбэктомии
11 августа 2020 г. — Многоцентровое исследование сообщает, что проницаемость или проницаемость сгустка — способность контрастного вещества, используемого во время первоначальной визуализации, просачиваться через сгусток, по оценке компьютерной томографии, — составляет …
Новый метод машинного обучения позволяет больницам обмениваться данными пациентов — конфиденциально
28 июля 2020 г. — Исследователи показали, что подход, называемый федеративным обучением, является успешным в контексте визуализации мозга, поскольку он может анализировать сканирование опухоли мозга с помощью магнитно-резонансной томографии (МРТ)…
Супервозрастные люди демонстрируют устойчивость к накоплению тау-белка и амилоида
15 июля 2020 г. — Согласно новому …
, у супервозрастных людей или людей, чьи когнитивные навыки превышают норму даже в преклонном возрасте, повышена устойчивость к тау и амилоидным белкам.
Сканирование всего тела для пациентов с травмами экономит время, проводимое в отделениях неотложной помощи
13 июля 2020 г. — Новое исследование, проведенное студентом медицинского факультета визуализации, возможно, нашло решение для облегчения разрастания больниц и уменьшения количества людей в чрезвычайных ситуациях…
«Нанорадиомика» раскрывает лечебный эффект на микросреду опухоли
13 июля 2020 г. — Исследователи разработали новый неинвазивный подход под названием нано-радиомика, который анализирует данные изображений для оценки изменений в микросреде опухоли, которые не обнаруживаются обычными …
Структурные элементы мозга при психических расстройствах
7 июля 2020 г. — Хотя ранее исследователи идентифицировали структурные признаки мозга, связанные с отдельными неврологическими заболеваниями, используя такие методы, как магнитно-резонансная томография (МРТ), команда ученых…
Врачи находят обнадеживающие результаты функциональной МРТ у неотзывчивого пациента с COVID-19
6 июля 2020 г. — Пациент с тяжелой формой COVID-19, который, несмотря на длительное отсутствие реакции и структурные аномалии мозга, продемонстрировал функционально неповрежденные мозговые связи, а затем восстановил способность следовать …
— Пациент с тяжелой формой COVID-19, который, несмотря на длительное отсутствие реакции и структурные аномалии мозга, продемонстрировал функционально неповрежденные мозговые связи, а затем восстановил способность следовать …
Исследование демонстрирует возможность использования голограммной технологии при абляции опухоли печени
15 июня 2020 г. — Данные одного из первых клинических случаев использования руководства дополненной реальности с инструментами с электромагнитным отслеживанием показывают, что эта технология может помочь врачам быстро, безопасно и точно выполнять поставленные задачи…
Миниатюрный нейростимулятор с магнитным приводом
8 июня 2020 г. — Нейроинженеры создали крошечный хирургический имплант, который может электрически стимулировать мозг и нервную систему без использования батареи или проводного питания …
Искусственный интеллект может улучшить использование изображений грудной клетки при лечении пациентов с COVID-19
3 июня 2020 г. — следует использовать искусственный интеллект (ИИ) для расширения роли рентгенографии грудной клетки — с использованием компьютерной томографии или КТ — в диагностике и оценке коронавирусной инфекции, чтобы ее можно было больше…
— следует использовать искусственный интеллект (ИИ) для расширения роли рентгенографии грудной клетки — с использованием компьютерной томографии или КТ — в диагностике и оценке коронавирусной инфекции, чтобы ее можно было больше…
«Выведение сахара с калом»? Обнаружено новое действие антидиабетического препарата
3 июня 2020 г. — Исследовательская группа обнаружила, что метформин, наиболее часто назначаемый антидиабетический препарат, вызывает выделение сахара с калом. Воспользовавшись новым аппаратом для биовизуализации ПЭТ-МРТ, они …
Визуализация выявляет неожиданные сокращения плаценты человека
28 мая 2020 г. — Согласно новому исследованию, изображения плаценты человека с высоким разрешением позволяют по-новому взглянуть на паттерны кровообращения, которые имеют решающее значение для развития плода.Эти данные улучшают наши …
Ключи к COVID-19 в мозгу, обнаруженные в новом исследовании
27 мая 2020 г. — Исследование, посвященное нейровизуализации и неврологическим симптомам у пациентов с COVID-19, может пролить свет на влияние вируса на центральную нервную систему . ..
..
Повышенное удобство использования и точность визуализации сосудов
26 мая 2020 г. — Исследователи разработали новый рентгеноконтрастный агент. Контрастное вещество проще в использовании, и он более надежно распределяется по всем кровеносным сосудам, повышая точность визуализации сосудов.Это …
Новый метод двойного контрастирования позволяет выявить небольшие опухоли на МРТ
25 мая 2020 г. — Раннее обнаружение опухолей чрезвычайно важно при лечении рака. Новая технология предлагает значительный прогресс в использовании магнитно-резонансной томографии для выделения даже очень маленьких опухолей из нормальных …
Текстура сетчатки может предоставить ранний биомаркер болезни Альцгеймера
14 мая 2020 г. — Биомедицинские инженеры разработали новое устройство визуализации, способное измерять как толщину, так и текстуру различных слоев сетчатки.Прогресс может быть использован для обнаружения биомаркера …
Признаки алкогольного синдрома плода, обнаруженные в утробе матери
13 мая 2020 г. — Новые изображения показывают самые ранние нарушения развития мозга плода приматов, отличные от человека, из-за употребления алкоголя матерью, в исследовании с участием макак-резус. Магнитно-резонансная томография показала …
— Новые изображения показывают самые ранние нарушения развития мозга плода приматов, отличные от человека, из-за употребления алкоголя матерью, в исследовании с участием макак-резус. Магнитно-резонансная томография показала …
Новый инструмент визуализации помогает исследователям увидеть степень раннего повреждения Альцгеймера
13 мая 2020 г. — Новая технология визуализации позволяет ученым увидеть широко распространенную потерю синапсов мозга на ранних стадиях болезни Альцгеймера, и это открытие может помочь в разработке лекарств…
Что такое медицина? Определение, области и отрасли
Медицина — это область здоровья и исцеления. В него входят медсестры, врачи и различные специалисты. Он охватывает диагностику, лечение и профилактику заболеваний, медицинские исследования и многие другие аспекты здоровья.
Медицина направлена на укрепление и поддержание здоровья и благополучия.
Традиционную современную медицину иногда называют аллопатической медициной. Он включает в себя использование лекарств или хирургическое вмешательство, часто подкрепленное консультированием и мерами по изменению образа жизни.
Он включает в себя использование лекарств или хирургическое вмешательство, часто подкрепленное консультированием и мерами по изменению образа жизни.
Альтернативные и дополнительные виды медицины включают акупунктуру, гомеопатию, фитотерапию, арт-терапию, традиционную китайскую медицину и многие другие.
Современная медицина имеет множество областей и аспектов. Вот некоторые из них.
Клиническая практика
Клиницист — это медицинский работник, который работает непосредственно с пациентами в больнице или другом учреждении здравоохранения. Медсестры, врачи, психотерапевты и другие специалисты — все врачи.
Не все медицинские специалисты являются клиницистами.Исследователи и лаборанты не являются клиницистами, потому что они не работают с пациентами.
Врач оценивает человека с целью диагностики, лечения и профилактики заболеваний, используя знания, полученные в результате обучения, исследований и опыта, а также клиническую оценку.
Биомедицинские исследования
Эта область науки занимается поиском способов предотвращения и лечения заболеваний, которые приводят к болезни или смерти.
Ученые-биомедики используют методы биотехнологии для изучения биологических процессов и болезней.Они стремятся разработать успешные методы лечения и лечения.
Биомедицинские исследования требуют тщательного экспериментирования, разработки и оценки. В нем участвуют биологи, химики, врачи, фармакологи и другие.
Лекарства
В этом поле рассматриваются лекарства или лекарства и способы их использования.
Врачи и другие медицинские работники используют лекарства для медицинской диагностики, лечения, лечения и профилактики заболеваний.
Хирургия
Хирургические процедуры необходимы для диагностики и лечения некоторых типов заболеваний, дефектов и травм.Они используют не лекарства, а инструментальные и ручные средства.
Хирург может провести хирургическую операцию по удалению или замене пораженной ткани или органов, или он может использовать операцию по удалению ткани для биопсии. Иногда они удаляют ненужные ткани и отправляют их на диагностику.
Медицинские устройства
Медицинские работники используют широкий спектр инструментов для диагностики и лечения заболевания или другого состояния, предотвращения ухудшения симптомов, замены поврежденной части, например бедра или колена, и т. Д.
Медицинское оборудование варьируется от пробирок до сложных сканирующих машин.
Альтернативная и дополнительная медицина
Сюда входят любые лечебные практики, которые не являются частью традиционной медицины. Методы разнятся. Они включают в себя использование трав, манипулирование «каналами» в теле, расслабление и так далее.
Альтернативное и дополнительное не имеют одинакового значения:
Альтернативная медицина : Люди используют другой вариант, отличный от традиционного, например, используют меры расслабления для облегчения головной боли, а не обезболивающие.
Дополнительная медицина : Люди добавляют еще один вариант лечения к основному лечению. Например, они могут использовать расслабление, а также обезболивающие от головной боли.
Например, они могут использовать расслабление, а также обезболивающие от головной боли.
Альтернативные и дополнительные методы лечения часто основаны на традиционных знаниях, а не на научных данных или клинических испытаниях.
Примеры включают гомеопатию, акупунктуру, аюрведу, натуропатическую медицину и традиционную китайскую медицину.
Клинические исследования
Исследователи проводят исследования, чтобы выяснить, какие заболевания присутствуют, почему они возникают, что может лечить или предотвращать их, что делает их более вероятными, а также многие другие аспекты здоровья.
Клинические испытания — один из аспектов клинических исследований. Они стремятся выяснить, является ли терапия — часто лекарственная — безопасной и эффективной для лечения конкретного состояния.
Самый эффективный способ продемонстрировать эффективность лекарственного средства или методики — это провести двойное слепое, случайное, долгосрочное крупное клиническое исследование на людях.
В этом типе исследования исследователи сравнивают эффект терапии или лекарственного препарата с плацебо, отсутствием лечения или другой терапией или лекарством.
Психотерапия
Консультации, когнитивно-поведенческая терапия (КПТ) и другие формы «лечения разговором» могут быть полезны людям с состояниями, влияющими на их психическое здоровье, от депрессии до стресса и хронической боли.
Физиотерапия и трудотерапия
Эти виды лечения не требуют приема лекарств, хотя человек может принимать лекарства одновременно с ними.
Физическая терапия может помочь улучшить прочность и гибкость в людях, которые имеют состояние, которое влияет на их костно-мышечную систему.
Трудотерапия может научить людей новым и лучшим способам делать что-то физически. Например, человеку, перенесшему инсульт, может быть полезно снова научиться ходить, используя методы, которые, возможно, они не использовали раньше.
Другие области медицины включают фармакологию и фармацию, сестринское дело, логопед, менеджмент медицинской практики и многие другие.
В медицине много отраслей. Вот некоторые из них.
Поделиться на Pinterest Анатомия — это область медицины, изучающая различные части, составляющие тело.
Анатомия : Изучение физической структуры тела.
Биохимия : Биохимик изучает химические компоненты и их влияние на организм.
Биомеханика : Основное внимание уделяется структуре биологических систем в организме и принципам их работы с использованием механического подхода.
Биостатистика : Исследователи применяют статистику к биологическим полям. Это очень важно для успешных медицинских исследований и многих областей медицинской практики.
Биофизика : использует физику, математику, химию и биологию для моделирования и понимания работы биологических систем.
Цитология : Это раздел патологии, который включает медицинское и научное микроскопическое исследование клеток.
Эмбриология : Этот раздел биологии изучает формирование, ранний рост и развитие организмов.
Эндокринология : Ученые исследуют гормоны и их влияние на организм.
Эпидемиология : исследователи отслеживают причины, распространение и контроль заболеваний среди населения.
Генетика : это исследование генов и их влияние на здоровье и организм.
Гистология : Это включает изучение формы структур под микроскопом. Это также известно как микроскопическая анатомия.
Микробиология : это исследование организмов, которые слишком малы, чтобы их можно было увидеть невооруженным глазом, известных как микроорганизмы.Аспекты микробиологии включают бактериологию, вирусологию, микологию (изучение грибов) и паразитологию.
Неврология : нейробиологи изучают нервную систему и мозг, а также исследуют заболевания нервной системы. Аспекты нейробиологии включают компьютерное моделирование и психофизику. Некоторые виды нейробиологии — это когнитивная нейробиология, клеточная нейробиология и молекулярная нейробиология.
Питание : диетологи изучают, как еда и напитки влияют на здоровье и как они могут помочь в лечении, лечении и профилактике различных заболеваний и состояний.
Поделиться на PinterestСуществуют разные типы медицинских лабораторных работников. Одни определяют причины болезней, другие изучают токсины и их действие. Иногда они имеют дело с опасными материалами.
Патология : Это исследование болезни. Патолог часто работает в лаборатории, где проводит анализы — обычно на образце крови, мочи или тканей тела — чтобы помочь диагностировать заболевания и состояния.
Фармакология : Это включает изучение фармацевтических препаратов или лекарств, откуда они берутся, как работают, как организм на них реагирует и из чего они состоят.
Радиология : Радиологи используют рентгеновские лучи и сканирующее оборудование во время диагностической процедуры, а иногда и как часть лечения.
Токсикология : токсиколог изучает яды, что они собой представляют, какое воздействие они оказывают на организм и как их обнаруживать.